
医薬化合物及び組成物として有用なNAALADase阻害剤
【課題】グルタメート異常、強迫性障害、前立腺疾患、痛み及び糖尿病性神経障害を治療するための化合物およびそれらを含む医薬組成物を提供する。
【解決手段】本発明は、特定の構造を有するN-アセチル化α-結合した酸性ジペプチダーゼ酵素活性阻害剤、そのような阻害剤を含有する医薬組成物、及びそれらの使用法である。
【解決手段】本発明は、特定の構造を有するN-アセチル化α-結合した酸性ジペプチダーゼ酵素活性阻害剤、そのような阻害剤を含有する医薬組成物、及びそれらの使用法である。
【発明の詳細な説明】
【技術分野】
【0001】
本件出願は1998年7月6日に出願された米国特許出願第09/110,262号、及び1999年1月12日に出願された米国特許出願第09/228,391号(これは順に1998年7月6日に出願された米国特許出願第09/110,186号の一部継続出願である)の一部継続出願である。
本発明はN-アセチル化α-結合酸性ジペプチダーゼ(NAALADase)インヒビター、このようなインヒビターを含む医薬組成物及びNAALADase酵素活性を抑制し、それにより神経細胞活性に影響し、血管形成を抑制し、グルタメート異常、強迫性障害、前立腺疾患、痛み及び糖尿病性神経障害を治療するためのそれらの使用方法に関する。
【背景技術】
【0002】
最近の研究によりグルタメート介在性障害の病因にNAALADaseが関係づけられている。筋萎縮性側索硬化症(ALS)の患者からの死後の組織に関する神経病理学研究は神経変性と関連して生じるN-アセチルアスパルテート(NAA)及びN-アセチルアスパルチルグルタメート(NAAG)組織濃度の大きい減少、並びにALSの患者からの脳脊髄液(CSF)中のNAA及びNAAGの増加を示す。一致して、異常なNAAGレベル及びNAALADase活性がまた精神分裂症患者の死後の前頭葉前部及び大脳辺縁の脳組織中で観察されていた。また、検死研究はNAAG/NAAとアルツハイマー病の間の強い相関関係を示唆する。死後の脳組織では、NAAレベル及びNAAGレベルがアルツハイマー病因により冒された脳領域(海馬及び扁桃)中で選択的に減少されることがわかった。
【0003】
グルタメートは中枢神経系(CNS)中で主たる興奮性神経伝達物質として利用できる。神経細胞は卒中又は心臓発作の如き虚血性脳障害中に起こるようにそれらが酸素を減少される場合にグルタメートを多量に放出する。グルタメートのこの過剰の放出は順にN-メチル-D-アスパルテート(NMDA)受容体、AMPA受容体、カイネート受容体及び代謝調節型グルタメート(mGlu)受容体の過剰刺激(興奮毒性)を生じる。グルタメートがこれらの受容体に結合する場合、受容体中のイオンチャンネルが開き、又は第二メッセンジャー系が刺激されて、それらの細胞膜を横切るイオンの流れ、例えば、細胞へのCa2+及びNa+流入及び細胞からのK+流出を可能にする。イオンのこれらの流れ、特にCa2+のインフラックスが神経細胞の過剰刺激を生じる。過剰刺激された神経細胞が更に多くのグルタメートを分泌し、プロテアーゼ、リパーゼ及び遊離基の生成を介して細胞死滅を最終的にもたらすと考えられるドミノ効果を生じる。
【0004】
グルタメート受容体の過剰活性化が脊髄損傷、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、糖尿病性神経障害、急性及び慢性の痛み、虚血並びに低酸素症、低血糖症、虚血、トラウマ、及び神経傷害後の神経細胞損失を含む種々の神経の疾患及び症状に関係づけらている。
特に、グルタメート作用性(glutamatergic)異常が神経分裂症と関連していた。例えば、フェニルシクリジン(PCP)及びN-メチル-D-アスパルテート(NMDA)受容体のその他のアンタゴニストは健康な個体に精神異常作用性を誘発し、精神分裂症の既存の症候を悪化し、グルタメート伝達の低下が精神分裂症に寄与し得ることを示唆する。更に、非NMDA受容体のアンタゴニスト又はグルタメート放出を減少する前処理がNMDA受容体アンタゴニストの記憶力増進効果及びその他の挙動効果を低下することが報告されていた。また、研究はmGlu受容体の或る種のサブタイプの刺激がシナプス前抑制を媒介し、グルタメートの誘発放出を減少することを示していた。1998年に、mGlu受容体アゴニストがラットでPCP誘発グルタメート外向きフラックスを減少することが報告され、アゴニストがシナプス前グルタメート作用活性を軽減することによりPCPの挙動効果を回復することが示唆された。
【0005】
また、最近の研究が強迫性障害、特に薬物依存性についてグルタメート作用の基礎を進歩していた。例えば、エタノールの神経生理学的効果及び病理学的効果がグルタメート作用系により媒介されることがわかった。詳しくは、エタノールへの急な暴露がグルタメート受容体中のチャンネル中のイオンの流れを抑制することによりグルタメート作用性神経伝達を分断し、一方、慢性暴露がグルタメート受容体の数をアップレギュレートし、それによりイオンの流れを増大する。エタノールの急性禁断症状がアップレギュレートされたチャンネルの存在下で高興奮性及び発作をもたらし、それによりシナプス後神経細胞を興奮毒性損傷を受けやすくする。
アルコール中毒患者からの組織学的に正常な脳の死後の試験は慢性アルコール中毒が前頭皮質中のグルタメート受容体のNMDAサブタイプの密度を中程度に増大することを示していた。このアップレギュレーションはエタノール誘発慢性神経毒性の段階に相当し得る。このようなものとして、中毒、禁断症状発作、振せんせん妄、ベルニケ−コルサコフ症候群及び胎児アルコール症候群を含む、アルコール中毒の神経生物学的効果が、グルタメート作用系に関するエタノールの効果の結果のスペクトルとして理解し得る。これに関して、アルコール中毒はグルタメート関連神経障害の拡大するファミリーの別の員と考えられるかもしれない。
【0006】
また、グルタメート作用系はその他の乱用薬物の挙動効果に関係づけられている。例えば、研究はグルタメート作用性アンタゴニストがアンフェタミン及びコカインにより誘発される運動刺激活性を阻止し、グルタメート作用性アゴニストがアンフェタミンにより生じられるステレオタイプと同じステレオタイプを生じることを示していた。これらの結果は精神運動刺激物質のステレオタイプ効果の発現がグルタメート作用系をともなうという薬理学的証拠に相当する。
疫学的研究は薬物依存性とその他の強迫性障害の間の強い相関関係を明らかにしていた。更に、共通の遺伝子異常がアルコール中毒、コカイン依存性、ニコチン依存性、病的ギャンブル、注意欠乏障害(ADD)、トゥレット症候群、強迫性過食症及び肥満の人々の間に見られた。このような障害は興奮毒性の効果の発現であると考えられる。
【0007】
上記知見に基づいて、本発明者らは試験し、NAALADaseインヒビターがグルタメート異常、例えば、薬物依存性、糖尿病性神経障害、痛み及び精神分裂症の薬物療法に効力があることを見出した。
現在までの殆どの研究活動及び開発活動はNMDAアンタゴニスト、グリシンアンタゴニスト、及びその他の後シナプス興奮性アミノ酸(EAA)受容体ブロッカーの如き化合物で後シナプスグルタメート受容体をブロックすることに集中していた。不運なことに、これらの薬剤は通常の条件下でさえもひどい毒性を生じ、こうしてそれらの臨床使用を制限する。如何なる一つの特別な理論にも限定されないが、NAALADaseインヒビターは後シナプスグルタメート受容体と相互作用しないでシナプス前グルタメート放出を阻止すると考えられる。NAALADaseインヒビターは基礎グルタメートレベルを変化しないことが明らかであるので、それらは後シナプスグルタメートアンタゴニストと関連する挙動毒性がないかもしれない。
【0008】
グルタメートに加えて、NAALADaseはまた前立腺特異性膜抗原(PSMA)とも関連づけられている。特に、PSMA cDNAがNAALADase活性を与えること、またNAALADase及びPSMAが少なくとも86%の相同配列同一性を示すことが示されていた。Carterら, Proc. Natl. Acad. Sci., 93巻, 749-753頁(1996)を参照のこと。PSMAの分子クローニングが潜在的な前立腺癌マーカーとして報告されており、前立腺癌の像形成方法及び細胞傷害治療方法の標的として利用できると仮定されていた。更に、PSMA抗体、特にインジウム-111標識PSMA抗体及びイットリウム標識PSMA抗体が記載されており、前立腺癌の診断及び治療について臨床試験されていた。PSMAは前立腺管上皮中で発現され、清漿、前立腺液及び尿中に存在する。
【0009】
本発明者らはNAALADaseインヒビターが前立腺疾患、特に前立腺癌を治療するのに有効であることを見出した。如何なる特別な理論にも制限されないが、NAALADaseインヒビターはPSMA活性を抑制するものと考えられる。PSMAのmAbは23の非前立腺癌を標的とすることがわかっていたので(Luiら, Science Research, 57巻, 3629-34 頁(1997))、本発明者らはNAALADaseインヒビターがまた特にNAALADaseがある組織、例えば、脳、腎臓及び睾丸中で非前立腺癌を治療するのに有効であろうと仮定する。
また、NAALADaseは新生血管系(新しい血管)中に見られた。本発明者らはNAALADaseインヒビターが新生血管系の増殖(血管形成)を抑制又は阻止し、それにより血管形成に依存する疾患を治療するのに潜在的な治療適用を与えることを発見した。血管形成依存性疾患の例として、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、及び皮膚潰瘍が挙げられるが、これらに限定されない。血管形成はまた正常な生理プロセス、例えば、成長、生殖能及び軟組織創傷治癒に必須である。
【0010】
癌は血管形成に依存性の別の疾患である。癌腫瘍細胞は内皮細胞付近で活性化する血管形成物質を分泌又は放出する。これらの内皮細胞は新しい血管の形成で極に達する挙動の細胞自律パターンを発現することにより応答する。研究は血管形成が癌腫瘍の増殖、浸潤及び転移を持続するのに必要であることを実証していたので、NAALADaseインヒビターの新生血管系抑制活性は全ての型の癌を治療する際のそれらの実用性を更に支持する。
数年前まで、わずかにいくつかのNAALADaseインヒビターが同定されているにすぎず、それらが非臨床研究に使用されていた。これらの化合物の例として、一般のメタロペプチダーゼインヒビター、例えば、o-フェナントロリン、金属キレーター、例えば、EGTA及びEDTA、並びにペプチド類似体、例えば、キスカリン酸及びβ-NAAGが挙げられる。これらの化合物は毒性副作用を有し、又は医薬有効量で投与し得ない。広範囲の潜在用途に鑑みて、新規NAALADaseインヒビター、このようなインヒビターを含む医薬組成物、及びそれらの使用の方法に対する要望がある。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は神経細胞活性に作用し、血管形成を抑制し、グルタメート異常、強迫性障害、前立腺疾患、痛み及び糖尿病性神経障害を治療するのに有益なN-アセチル化α-結合酸性ジペプチダーゼ(NAALADase)酵素インヒビター及び組成物に関する。
【課題を解決するための手段】
【0012】
更に詳しくは、本発明は一般式I
【0013】

【0014】
の化合物又は医薬上許される均等物に関する。
式中、
Xは式II、III又はIV
【0015】
である部分であり、
m及びnは独立に0、1、2、3又は4であり、
ZはSR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり、
BはN又はCR16であり、
AはO、S、CR17R18又は(CR17R18)mSであり、
R9及びR13は水素であり、
R8、R10、R11、R12、R14、R15、R16、R17及びR18は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換、又は一つ以上の置換基で置換されており、
但し、Xが式IIである部分であり、かつAがOである場合、nが2、3又は4であり;Xが式IIである部分であり、かつAがSである場合、nが2、3又は4であり;またXが式IIである部分であり、かつAが(CR17R18)mSである場合、nが0、2、3又は4であることを条件とする。
【0016】
更に、本発明は卒中の発症の60分よりも後に投与された時に哺乳類の卒中を治療するのに有効である硫黄及び酸基の両方を含む化合物に関する。
また、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のNAALADase酵素活性の抑制方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のグルタメート異常の治療方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の強迫性障害、卒中、髄鞘脱落疾患、精神分裂症、パーキンソン病及びALSからなる群から選ばれたグルタメート異常の治療方法に関する。
【0017】
また、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の神経細胞活性に作用する方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の前立腺疾患の治療方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の癌の治療方法に関する。
また、本発明は哺乳類に有効量の硫黄及び酸基の両方を含む化合物を卒中の発症の60分より後に投与することを特徴とする哺乳類の卒中の治療方法に関する。
【0018】
更に、本発明は哺乳類に有効量の硫黄及び酸基の両方を含む化合物を卒中の発症の60分より後に投与することを特徴とする哺乳類の卒中の治療方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の血管形成の抑制方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の痛みの治療方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の糖尿病性神経障害の治療方法に関する。
更に、本発明はチオラクトンを置換エステルと反応させて式VI
【0019】
【0020】
の化合物を生成する工程を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Dは(CR21R22)nであり、
nは0、1、2、3又は4であり、かつ
R20、R21及びR22は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されている。
【0021】
また、本発明は
(i)メルドラム酸を硫黄含有反応体と反応させてメルドラム酸硫黄含有誘導体を生成する工程、及び
(ii)前記メルドラム酸硫黄含有誘導体をエステルと反応させて式VII
【0022】
【0023】
の化合物を生成する工程
を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Eは硫黄含有部分であり、かつ
Fはプロピオン酸エステル含有部分である。
最後に、本発明は
(i)有効量の式Iの化合物、及び
(ii)医薬上許される担体
を含むことを特徴とする医薬組成物に関する。
【図面の簡単な説明】
【0024】
【図1A】免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図1B】アスコルビン酸による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図1C】アスコルビン酸及び化合物3による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2A】免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2B】アスコルビン酸による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2C】アスコルビン酸及び化合物2による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図3】種々の投与量の化合物3による皮下治療後の日数に対しin vivo平均LNCaP腫瘍体積をプロットするグラフである。
【図4】LNCaP細胞による注射後にビヒクル又は化合物3で皮下治療したマウス中の腫瘍:対照比をプロットする棒グラフである。
【図5】種々の投与量の化合物3による皮下治療後の日数に対しin vivo平均ダンニングG腫瘍体積をプロットするグラフである。
【図6】ダンニングG細胞による注射後にビヒクル又は化合物3で皮下治療したラット中の腫瘍:対照比をプロットする棒グラフである。
【図7】種々の投与量の化合物3による腫瘍内治療後の日数に対しin vivo平均ダンニングG腫瘍体積をプロットするグラフである。
【図8A】マウスに皮下注射され、血管形成因子の注射後にビヒクル単独で処理されたマトリゲルTMプラグの顕微鏡写真の組である。
【図8B】マウスに皮下注射され、血管形成因子の注射後に毎日3mg/kgの投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真の組である。
【図8C】マウスに皮下注射され、血管形成因子の注射後に毎日30mg/kgの投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真の組である。
【図9】マウスに皮下注射され、血管形成因子の注射後に連続投与濃度のビヒクル単独で処理されたマトリゲルTMプラグの顕微鏡写真である。
【図10】マウスに皮下注射され、血管形成因子の注射後に1μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図11】マウスに皮下注射され、血管形成因子の注射後に10μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図12】マウスに皮下注射され、血管形成因子の注射後に100μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図13】化合物2による治療後の日数に対し糖尿病ラットの逃避待ち時間をプロットするグラフである。
【図14】治療後の時間に対しビヒクル又は化合物3で治療されたラットのホルマリン誘発身もだえ挙動をプロットするグラフである。
【図15】ラットを治療したビヒクル又は化合物3の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図16】ラットを治療したビヒクル又は化合物2の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図17】ラットを治療したビヒクル又は化合物1の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図18】投薬後の日数に対しビヒクル又は化合物3で治療したラットの慢性結さつ損傷誘発痛覚過敏症をプロットする棒グラフである。
【図19A】STZによる投与後の日数に対し、ビヒクル又は化合物2で治療した非糖尿病ラットSTZ糖尿病ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図19B】STZによる投与後の日数に対し、ビヒクル又は化合物1で治療した非糖尿病ラットSTZ糖尿病ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図20】手術後の日数に対し、正常な(手術されなかった)ラット及びビヒクル又は化合物3で治療した慢性結さつ損傷誘発ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図21A】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物2で治療したSTZ糖尿病ラットの運動神経伝導速度をプロットする棒グラフである。
【図21B】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物2で治療したSTZ糖尿病ラットの知覚神経伝導速度をプロットする棒グラフである。
【図22A】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物1で治療したSTZ糖尿病ラットの運動神経伝導速度をプロットする棒グラフである。
【図22B】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物1で治療したSTZ糖尿病ラットの知覚神経伝導速度をプロットする棒グラフである。
【図23】コカインと一緒の化合物3、コカインを含む食塩水、及び食塩水を含む食塩水による治療後の日数に対しラットのコカイン(20mg/kg)自発運動をプロットするグラフである。
【図24】治療の週数に対し、非糖尿病ラット及びビヒクル、化合物1又は化合物2で治療したBB/W糖尿病ラットの逃避待ち時間をプロットするグラフである。
【図25】治療の週数に対し、非糖尿病ラット及びビヒクル、化合物1又は化合物2で治療したBB/W糖尿病ラットの神経伝達速度をプロットするグラフである。
【図26】PCP投与後の時間に対し水又は化合物2で更に治療したPCP処理ラットの平均頭部ローリングスコアーをプロットするグラフである。
【図27】PCP投与後の時間に対し水又は化合物1で更に治療したPCP処理ラットの平均頭部ローリングスコアーをプロットするグラフである。
【発明を実施するための形態】
【0025】
定義
“酸基”として、-COOH、-SO3H、-SO2HNH、-PO2H2、-CN、-PO3H2、-SH、-NHCOH、-NH2、-CONH2、-CONHOH、-CONHNHSO2H、-COHNSO2H、及び-CONHCNが挙げられるが、これらに限定されない。
“化合物1”は2-(2-スルファニルエチル)ペンタン二酸、又は実施例10により調製された化合物の純粋な形態及び不純な形態を表す。
“化合物2”は2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕-ペンタン二酸を表す。
“化合物3”は2-(ホスホノメチル)-ペンタン二酸(PMPA)を表す。
【0026】
“有効量”は所望の効果を生じるのに必要とされる量を表す。“治療有効量”はNAALADase酵素活性を抑制し、神経細胞活性に作用し、血管形成を抑制し、かつ/又はグルタメート異常、強迫性障害、前立腺疾患、痛み及び/又は糖尿病性神経障害を治療するのに必要とされる量を表す。
“IP”又は“i.p.”は腹腔内を表す。
“アイソスター”は同様の又は同じ物理的性質を有する元素、分子又はイオンを表す。典型的には、2種のアイソスター分子は同様の又は同じ嵩及び形状を有する。理想的には、アイソスター化合物は同形であるべきであり、同時結晶化することができるべきである。アイソスター化合物が通常共有するその他の物理的性質の中に、沸点、密度、粘度及び伝熱性がある。しかしながら、或る種の性質は通常異なっている:双極子モーメント、極性、偏光、サイズ及び形状。何とならば、外部軌道が異なって混成され得るからである。“アイソスター”という用語は“バイオアイソスター”を含む。
【0027】
“バイオアイソスター”は、それらの物理的類似性に加えて、幾つかの共通の生物学的性質を共有するアイソスターである。典型的には、バイオアイソスターは同じ認識部位と相互作用し、又は広く類似する生物学的効果を生じる。
“カルボン酸アイソスター”として、直接誘導体、例えば、ヒドロキサム酸、アシル−シアナミド及びアシルスルホンアミド;平面酸性複素環、例えば、テトラゾール、メルカプトアゾール、スルフィニルアゾール、スルホニルアゾール、イソオキサゾール、イソチアゾール、ヒドロキシチアジアゾール及びヒドロキシクロム;並びに非平面の硫黄又はリン誘導酸性官能基、例えば、ホスフィネート、ホスホネート、ホスホンアミド、スルホネート、スルホンアミド、及びアシルスルホンアミドが挙げられるが、これらに限定されない。
【0028】
“誘導体”は別の物質から直接に、又は修飾もしくは部分置換により生成された物質を表す。
“メルドラム酸”は2,2-ジメチル-1,3-ジオキサン-4,6-ジオンを表す。 “代謝産物”は代謝又は代謝プロセスにより生成された物質を表す。 “医薬上許される均等物”として、医薬上許される塩、水和物、代謝産物、プロドラッグ及びカルボキシルアイソスターが挙げられるが、これらに限定されない。多くの医薬上許される均等物が式I-Vの化合物と同じ又は同様のin vitro又はin vivo活性を有すると予想される。
【0029】
“医薬上許される塩”は所望の薬理学的活性を有し、かつ生物学上又はそれ以外に望ましくないことはない本発明の化合物の塩を表す。塩は無機酸により生成でき、例えば、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、硫酸水素塩、酪酸塩、クエン酸塩、ショウノウ塩、ショウノウスルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、半硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2-ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2-ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、チオシアン酸塩、トシレート及びウンデカン酸塩が挙げられる。塩基塩の例として、アンモニウム塩、アルカリ金属塩、例えば、ナトリウム塩及びカリウム塩、アルカリ土類金属塩、例えば、カルシウム塩及びマグネシウム塩、有機塩基との塩、例えば、ジシクロヘキシルアミン塩、N-メチル-D-グルカミン塩、並びにアミノ酸、例えば、アルギニン及びリシンとの塩が挙げられる。また、塩基性窒素含有基が薬剤で四級化し得る。このような薬剤として、低級アルキルハライド、例えば、メチル、エチル、プロピル、及びブチルのクロリド、ブロミド及びヨージド;ジアルキルスルフェート、例えば、ジメチルスルフェート、ジエチルスルフェート、ジブチルスルフェート及びジアミルスルフェート;長鎖ハライド、例えば、デシル、ラウリル、ミリスチル及びステアリルのクロリド、ブロミド及びヨージド;並びにアラルキルハライド、例えば、ベンジルブロミド及びフェネチルブロミドが挙げられる。
【0030】
“医薬上許されるプロドラッグ”はその一つ以上の薬理学的効果を示す前に生体内変換を受ける本発明の化合物の誘導体を表す。プロドラッグは改良された化学安定性、改良された患者許容及びコンプライアンス、改良された生物利用能、延長された作用の期間、改良された臓器選択性、改良された製剤化(例えば、増大された水溶性)、及び/又は低下された副作用(例えば、毒性)の一つ以上の目的で製剤化される。プロドラッグは当業界で知られている方法、例えば、Burger's Medicinal Chemistry and Drug Chemistry, 第5編, 1巻,172-178頁, 949-982頁(1995)により記載された方法、又は当業者に容易に明らかな方法を使用して本発明の化合物から容易に調製し得る。例えば、本発明の化合物は一つ以上のヒドロキシ基又はカルボキシ基をエステルに変換することによりプロドラッグに変換し得る。
“放射性感作物質”は電磁放射線で治療し得る疾患の治療を促進するために治療有効量で動物に投与される低分子量化合物を表す。電磁放射線で治療し得る疾患として、腫瘍性疾患、良性及び悪性の腫瘍、並びに癌細胞が挙げられる。本明細書にリストされていないその他の疾患の電磁放射線治療がまた本発明により意図されている。
【0031】
“アルキル”は指定された数の炭素原子を含む分岐又は非分岐飽和炭化水素鎖を表す。例えば、C1-C6直線状又は分岐アルキル炭化水素鎖は1〜6個の炭素原子を含み、特に示されない限り、置換基、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、t-ブチル、n-ペンチル、n-ヘキシル等が挙げられるが、これらに限定されない。
“アルケニル”は指定された数の炭素原子を含む分岐又は非分岐不飽和炭化水素鎖を表す。例えば、C2-C6直線状又は分岐アルケニル炭化水素鎖は少なくとも一つの二重結合を有する2〜6個の炭素原子を含み、特に示されない限り、置換基、例えば、エテニル、プロペニル、イソプロペニル、ブテニル、イソブテニル、t-ブテニル、n-ペンテニル、n-ヘキセニル等が挙げられるが、これらに限定されない。
【0032】
“アルコキシ”は基-OR(式中、Rは先に定義されたアルキルである)を表す。Rは1〜6個の炭素原子を含む分岐又は非分岐飽和炭化水素鎖であることが好ましい。
“ハロ”又は“ハロゲン”はフルオロ、クロロ、ブロモ及びヨードを表す。
“異性体”は同じ数及び種類の原子、ひいては同じ分子量を有する化合物を表すが、原子の配置又は立体配置に関して異なる。
“立体異性体”は同じ化学構成を有するが、空間の原子又は基の配置に関して異なる化合物を表す。
【0033】
“光学異性体”は2種の立体異性体のいずれをも表す。一つの種類は鏡像体と称される鏡像構造により表され、これらは化合物(グリセルアルデヒド、乳酸、糖、酒石酸、アミノ酸)中の一つ以上の不斉炭素原子の存在により生じる。その他の種類はジアステレオマーにより例示され、これらは鏡像ではない。これらは二つ以上の不斉炭素原子を有する化合物中で生じる。こうして、このような化合物は2n光学異性体(nは不斉炭素原子の数である)を有する。
【0034】
“鏡像体”は互いに重ならない鏡像である立体異性体を表す。
“鏡像体濃縮された”は一種の鏡像体が優勢である混合物を表す。
“ラセミ体”は個々の鏡像体の等しい部分を含む混合物を表す。
“非ラセミ体”は個々の鏡像体の等しくない部分を含む混合物を表す。
“動物”は感覚及び随意運動の力並びにその存在のために酸素及び有機食物に対する要求を有する生物を表す。例として、哺乳類、例えば、ヒト、ウマ、ブタ、ウシ、マウス、イヌ又はネコの種の員が挙げられるが、これらに限定されない。ヒトの場合、“動物”という用語はまた“患者”と称されてもよい。
“疾患”は症候及び兆候の特徴的な組により発現され、その病因、病理学、及び予後が知られていてもよく、また知られていなくてもよい生体のあらゆる部分、臓器、又は系(或いはこれらの組み合わせ)の正常な構造又は機能からのあらゆる異常又は中断を表す。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
【0035】
“障害”は機能のあらゆる障害又は異常;病的肉体状態又は精神状態を表す。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
“グルタメート異常”はグルタメートが関係するあらゆる疾患、障害又は症状を表し、上昇レベルのグルタメートを伴う病状が挙げられる。グルタメート異常の例として、脊髄損傷、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、精神分裂症、急性の痛み、慢性の痛み、虚血、神経障害及び強迫性障害が挙げられるが、これらに限定されない。
“虚血”は動脈血液の流入の閉塞のための局在化組織貧血を表す。総体虚血は心臓停止により生じるように全脳への血液の流れが或る時間の期間にわたって停止する場合に起こる。病巣虚血は脳血管の血栓塞栓閉塞、トラウマ頭部損傷、浮腫又は脳腫瘍から生じるように脳の一部がその正常な血液供給を不足している場合に起こる。たとえ一時的であっても、総体虚血及び病巣虚血の両方が広範な神経細胞損傷を生じ得る。神経組織損傷は虚血の発症の数時間後又は更には数日後に起こるが、或る種の永久の神経組織損傷が脳への血流の停止の初期の数分後に発生し得る。この損傷の多くがグルタメート毒性及び組織の再潅流の二次的な結果、例えば、損傷された内皮による血管活性生成物の放出、及び損傷された組織による細胞傷害生成物、例えば、遊離基及びロイコトリエンの放出に起因される。
【0036】
“神経機能”は、中でも、生体の内部環境及び外部環境の感知を与え、生物の種々の構造要素間で随意活動及び反射活動を可能にし、また環境変化に対する生物の応答をバランスする神経系の種々の機能を表す。
“神経障害”は神経組織へのあらゆる損傷及びそれから生じるあらゆる能力障害又は死亡を表す。神経障害の原因は代謝、毒性、神経毒性、医原性、熱又は化学薬品であってもよく、虚血、低酸素症、脳血管アクシデント、トラウマ、手術、圧力、物質作用、出血、放射線、血管痙攣、神経変性疾患、神経退化プロセス、感染症、パーキンソン病、ALS、髄鞘形成/髄鞘脱落プロセス、癲癇、認知障害、グルタメート異常及びこれらからの二次的な作用が挙げられるが、これらに限定されない。現在、神経組織損傷に有効な治療は知られていない。
“神経組織”は神経系を構成する種々の成分を表し、神経細胞、神経支持細胞、グリア、シュワン細胞、これらの構造内に含まれ、またこれらの構造に供給する血管系、中枢神経系、脳、脳幹、脊髄、末梢神経系との中枢神経系の接合部、末梢神経系及び同類の構造が挙げられるが、これらに限定されない。
【0037】
“神経保護”は神経障害を軽減、停止又は回復し、神経障害を受けた神経組織を保護、救済又は蘇生する効果を表す。
“痛み”は特殊な神経末端の刺激から生じる不快、窮迫又はアゴニーの局所感覚を表す。それは受難者がその源を除去し、又はその源から撤退することを誘発する限りにおいて、それは保護メカニズムとして利用できる。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。痛みの例として、急性、慢性、癌、やけど、切開、炎症、糖尿病性神経障害及び背部の痛みが挙げられるが、これらに限定されない。
【0038】
“精神障害”は窮迫症候又は機能の重大な障害の存在を特徴とするあらゆる臨床上重大な挙動上の又は心理学的な症候群を表す。精神障害は個体の或る種の心理学的又は器質性の機能障害から生じると推定される。その概念は個体と社会の間で本質的に相反する障害(社会的逸脱)を含まない。
“強迫性障害”は抵抗不能の強迫性挙動を特徴とするあらゆる障害を表す。強迫性障害の例として、薬物依存性、食事障害、病的ギャンブル、ADD及びトゥレット症候群が挙げられるが、これらに限定されない。
“注意欠乏障害”は活動過剰を伴い、又は伴わない、発生的に不適当な不注意及び衝動を特徴とする障害を表す。不注意は始めた仕事を完結できないこと、容易な被転導性、注意の外見上の欠如、及び持続注意を要する仕事に対する集中の難しさを意味する。衝動は考える前の行動、交替ですることの難しさ、仕事を編成する問題、及び一つの行動から別の行動に絶えずシフトすることを意味する。活動過剰は依然として着席し、また座っていることの難しさ、及び過度に走ったり、また徘徊したりすることを意味する。
【0039】
“薬物依存性”は薬物に対する心理的嗜癖又は肉体的トレランスを意味する。トレランスは初期に少量により得られた効果を生じるために用量を次第に増加しようとする要求を意味する。
“禁断症状症候群”は薬物が中断される場合又はその効果が特定のアンタゴニストにより相殺される場合に生じる不利な肉体変化を特徴とする障害を表す。
“食事障害”は強迫性過食、肥満又は重度の肥満を表す。肥満は通常の身長−体重表より20%以上の体重を意味する。重度の肥満は100%以上の過剰体重を意味する。
【0040】
“病的ギャンブル”はギャンブルによる忘我を特徴とする症状を表す。精神活性物質乱用と同様に、その効果として、次第に多額の金を賭けたい要求によるトレランスの発生、禁断症状症候、並びに家族及び職業に対するひどい負の効果にもかかわらず絶えずギャンブルすることが挙げられる。
“精神分裂症”は思考の形態及び内容(連想の散漫、妄想、幻覚)、気分(盲目化、落胆、不適当な感情)、外界に対する自己の意味及び関係(エゴ境界の損失、内閉性思考、及び自閉的孤立)、並びに挙動(奇異、外見上の無目的、及びステレオタイプの活動又は非活動)の障害を特徴とする精神障害又はグループ精神障害を表す。精神分裂症の例として、急性、外来、境界、緊張型、幼児、分裂型、破瓜病性、潜在性、核、妄想型、パラフレニー、精神病前、プロセス、偽神経症性、偽精神病性、反応性、残留、分裂−情動及び未分化の精神分裂症が挙げられるが、これらに限定されない。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
【0041】
“トゥレット症候群”は強迫性ののしり、多発性筋肉チック及び騒がしいノイズを特徴とする常染色体性多発性チック障害を表す。チックは単純でも複雑でありえる短い、迅速な、不随意運動である。それらはステレオタイプ化され、反復性であるが、周期的ではない。単純チック、例えば、まばたきはしばしば神経マンネリズムとして始まる。複雑なチックはしばしば正常な挙動の断片に似ている。
“血管形成”は新しい毛管が形成されるプロセスを表す。
“血管形成依存性疾患”として、癌、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、及び皮膚潰瘍が挙げられるが、これらに限定されない。
【0042】
血管形成の“抑制”は本発明に従って多くのパラメーターにより測定されてもよく、例えば、新生血管構造の遅延出現、新生血管構造の遅い発生、新生構造の減少された発生、血管形成依存性疾患作用の遅い、又は低下された重度、停止された血管形成的成長、又は先の血管形成的成長の回帰により評価し得る。極限では、完全抑制は本明細書では予防と称される。
血管形成又は血管形成的成長に関して、“予防”は既に発生しなかった場合には血管形成又は血管形成的成長が実質的にないことを表し、又は成長が既に発生していた場合には更なる血管形成又は血管形成的成長が実質的にないことを表す。
“癌”として、ACTH生成腫瘍、急性リンパ性白血病、急性非リンパ性白血病、副腎皮質の癌、膀胱癌、脳の癌、乳癌、子宮頸癌、慢性リンパ性白血病、慢性骨髄性白血病、結腸直腸癌、皮膚T細胞リンパ腫、子宮内膜癌、食道癌、ユーイング肉腫、胆嚢癌、ヘアリー・セル白血病、頭部及び首の癌、ホジキンリンパ腫、カポージ肉腫、腎臓癌、肝臓癌、肺癌(小細胞及び/又は非小細胞)、悪性腹腔滲出、悪性胸膜滲出、メラノーマ、中皮腫、多発性ミエローマ、ニューロブラストーマ、非ホジキンリンパ腫、骨肉腫、卵巣癌、卵巣(生殖細胞)癌、膵臓癌、ペニス癌、前立腺癌、網膜芽細胞腫、皮膚癌、軟組織肉腫、扁平上皮癌、胃癌、精巣癌、甲状腺癌、栄養膜腫瘍、子宮の癌、膣癌、外陰部の癌、及びウィルム腫瘍が挙げられるが、これらに限定されない。
【0043】
“転移”は“播殖して、非隣接部位で増殖の新しい病巣を形成する(即ち、転移を形成する)癌の細胞の能力”を表す。本明細書に引用として含まれるThe Basic Science of Oncology, Tannockら編集, McGraw-Hill, New York (1992)中のHill, R.P, 11章,“Metastasis”, 178-195頁を参照のこと。「in situ腫瘍増殖から転移性疾患への転移は一次部位の腫瘍細胞が局所組織を侵食し、組織関門を横切る腫瘍細胞の能力により特定される・・・転移プロセスを開始するために、癌細胞は最初に上皮基底膜に侵入し、次いで組織内ストローマを侵食する必要がある・・・遠位転移のために、血管内侵入は、内皮下基底膜の腫瘍細胞浸潤を必要とするが、これはまた腫瘍細胞の血管外遊出の際にも通り抜ける必要がある・・・悪性の発生はまた、原発腫瘍のエクスパンジョンを可能にするだけでなく新たに形成された血管の基底膜に欠陥があるために血管区画への容易な接近を可能にする、腫瘍誘導血管形成と関連している」。Aznavoorianら, Cancer 71: 1368-1383 (1993)(本明細書に参考として含まれる)を参照のこと。
【0044】
“電磁放射線”として、10-20〜100メートルの波長を有する放射線が挙げられるが、これらに限定されない。本発明の好ましい実施態様はγ放射線(10-20〜10-13m)、x線放射線(10-11〜10-9m)、紫外線(10nm〜400nm)、可視光線(400nm〜700nm)、赤外線(700nm〜1.0mm)、及びマイクロウェーブ放射線(1mm〜30cm)の電磁放射線を使用する。
“前立腺疾患”は前立腺を冒すあらゆる疾患を表す。前立腺疾患の例として、前立腺癌、例えば、前立腺の腺癌及び転移性癌;並びに前立腺上皮細胞の異常増殖を特徴とする症状、例えば、良性前立腺過形成が挙げられるが、これらに限定されない。
“治療”は
(i)疾患、障害及び/又は症状にかかりやすくなっているが、それを有すると未だ診断されていない動物において疾患、障害又は症状が発生することを予防する、
(ii)疾患、障害又は症状を抑制し、すなわち、その発生を停止する、かつ/又は
(iii)疾患、障害又は症状を軽減し、即ち、疾患、障害及び/又は症状を軽減するために本発明の化合物又は組成物を投与することを表す。
薬物依存性に関して、“治療”は乱用の薬物に対する心理的嗜癖又は肉体的トレランスを抑制し、かつ/又は薬物依存性から生じる禁断症状症候群を軽減し、かつ/又は予防するために本発明の化合物又は組成物を投与することを含む。
卒中に関して、“機会の治療ウインドー”又は“ウインドー”は虚血の発症と有効な治療の間の最大の遅延を表す。
【0045】
“NAAG”は、主要なインヒビター神経伝達物質γ-アミノ酪酸(GABA)に匹敵するレベルで、脳の重要なペプチド成分であるN-アセチル-アスパルチル-グルタメートを表す。NAAGは神経細胞特異性であり、シナプス小胞中に存在し、グルタメート作用性であると推定される幾つかの系中で神経刺激後に放出される。研究はNAAGが中枢神経系中で神経伝達物質及び/又は神経モジュレーターとして、又は神経伝達物質グルタメートの前駆体として機能し得ることを示唆する。
“NAALADase”はNAAGをN-アセチルアスパルテート(“NAA”)及びグルタメート(“GLU”)に異化作用する膜結合メタロペプチダーゼであるN-アセチル化α結合酸性ジペプチダーゼを表す。
【0046】
【0047】
アミノ酸配列相同性に基づいて、NAALADaseはM28ペプチダーゼファミリーに指定された。NAALADaseはまた前立腺特異性膜抗原(PSMA)又はヒトグルタメートカルボキシペプチダーゼII(GCPII)、EC番号3.4.17.21と称される。NAALADaseは補助触媒性亜鉛/亜鉛メタロペプチダーゼであると考えられる。NAALADaseは540 nMのKmでNAAGに対する高アフィニティーを示す。NAAGが生物活性ペプチドである場合、NAALADaseはNAAGのシナプス作用を不活性化するのに利用できるかもしれない。また、NAAGがグルタメートの前駆体として機能する場合、NAALADaseの一次機能はシナプスのグルタメート利用可能性を調節するのかもしれない。
“NAALADaseインヒビター”はNAALADase酵素活性を抑制するあらゆる化合物を表す。
【0048】
“抑制”は、酵素に関して、競合、未競合及び非競合の抑制の如き可逆的酵素抑制を表す。競合、未競合及び非競合の抑制は酵素の反応速度論に関するインヒビターの効果により区別し得る。競合抑制はインヒビターが活性部位で結合するために通常の基質と競合するような方法でインヒビターが酵素と可逆的に化合する場合に生じる。インヒビターと酵素の間のアフィニティーはインヒビター定数、Kiにより測定されてもよく、これは以下のように定義される。
【0049】
【0050】
式中、〔E〕は酵素の濃度であり、〔I〕はインヒビターの濃度であり、かつ〔EI〕は酵素とインヒビターの反応により生成された酵素−インヒビター複合体の濃度である。特にことわらない限り、本明細書に使用される“Ki”は本発明の化合物とNAALADaseの間のアフィニティーを表す。“IC50”は標的酵素の50%抑制を生じるのに必要とされる化合物の濃度又は量を特定するのに使用される関連用語である。
本発明の化合物
本発明は一般式I
【0051】
【0052】
の化合物又は医薬上許される均等物に関する。
式中、
Xは式II、III又はIV
【0053】
【0054】
である部分であり、
m及びnは独立に0、1、2、3又は4であり、
ZはSR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり、 BはN又はCR16であり、
AはO、S、CR17R18又は(CR17R18)mSであり、
R9及びR13は水素であり、
R8、R10、R11、R12、R14、R15、R16、R17及びR18は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されており、
但し、Xが式IIである部分であり、かつAがOである場合、nが2、3又は4であり;Xが式IIである部分であり、かつAがSである場合、nが2、3又は4であり;またXが式IIである部分であり、かつAが(CR17R18)mSである場合、nが0、2、3又は4であることを条件とする。
【0055】
前記アルケニル、シクロアルキル、シクロアルケニル、及びAr1の可能な置換基として、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、ニトロソ、ニトリロ、イソニトリロ、イミノ、アゾ、ジアゾ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル、並びに炭素環式部分及び複素環部分が挙げられるが、これらに限定されない。炭素環式部分として、脂環式構造及び芳香族構造が挙げられる。
【0056】
有益な炭素環式部分及び複素環部分の例として、フェニル、ベンジル、ナフチル、インデニル、アズレニル、フルオレニル、アントラセニル、インドリル、イソインドリル、インドリニル、ベンゾフラニル、ベンゾチオフェニル、インダゾリル、ベンズイミダゾリル、ベンゾチアゾリル、テトラヒドロフラニル、テトラヒドロピラニル、ピリジル、ピロリル、ピロリジニル、ピリジニル、ピリミジニル、プリニル、キノリニル、イソキノリニル、テトラヒドロキノリニル、キノリジニル、フリル、チオフェニル、イミダゾリル、オキサゾリル、ベンゾオキサゾリル、チアゾリル、イソオキサゾリル、イソトリアゾリル、オキサジアゾリル、トリアゾリル、チアジアゾリル、ピリダジニル、ピリミジニル、ピラジニル、トリアジニル、トリチアニル、インドリジニル、ピラゾリル、ピラゾリニル、ピラゾリジニル、チエニル、テトラヒドロイソキノリニル、シノリニル、フタラジニル、キナゾリニル、キノキサリニル、ナフチリジニル、プテリジニル、カルバゾリル、アクリジニル、フェナジニル、フェノチアジニル、及びフェノキサジニルが挙げられるが、これらに限定されない。
【0057】
Xが式IIである部分であり、nが0、1、2又は3であり、ZがSH、SO3H、SO2H、SOH又はS(NRHR14)2R15であり、かつAがO、S又はCR17R18であることが好ましい。
ZがSHであることが更に好ましい。
ZがSHである場合、R8が-(CH2)2COOHであることが最も好ましい。
【0058】
別の好ましい実施態様において、Xが式IIである部分であり、R8が-(CH2)2COOR19又は-(CH2)2CONHR19であり、AがCH2であり、nが0であり、ZがSR13である場合には、R13は水素又はCOR19ではなく、またXが式IIIである部分であり、BがNであり、かつR8が-(CH2)2COOHである場合には、R11は水素ではない。
式Iの好ましい化合物は
2-(2-スルファニルエチル)ペンタン二酸、
3-(2-スルファニルエチル)-1,3,5-ペンタントリカルボン酸、
2-(2-スルファニルプロピル)ペンタン二酸、
2-(2-スルファニルブチル)ペンタン二酸、
2-(2-スルファニル-2-フェニルエチル)ペンタン二酸、
2-(2-スルファニルヘキシル)ペンタン二酸、
2-(2-スルファニル-1-メチルエチル)ペンタン二酸、
2-〔1-(スルファニルメチル)プロピル〕ペンタン二酸、
2-(3-スルファニルペンチル)ペンタン二酸、
2-(3-スルファニルプロピル)ペンタン二酸、
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸、
2-(3-スルファニル-2-フェニルプロピル)ペンタン二酸、
2-(3-スルファニルブチル)ペンタン二酸、
2-〔3-スルファニル-2-(フェニルメチル)プロピル〕ペンタン二酸、
2-〔2-(スルファニルメチル)ブチル〕ペンタン二酸、
2-〔2-(スルファニルメチル)ペンチル〕ペンタン二酸、
2-(3-スルファニル-4-メチルペンチル)ペンタン二酸、及び
これらの医薬上許される均等物からなる群から選ばれる。
【0059】
式Iの最も好ましい化合物は2-(2-スルファニルエチル)ペンタン二酸、2-(2-スルファニルプロピル)ペンタン二酸、2-(3-スルファニルプロピル)ペンタン二酸及び医薬上許される均等物からなる群から選ばれる。理想的には、式Iの化合物は鏡像体又は鏡像体濃縮された混合物である。
Xが式IIIである部分であり、R8が-(CH2)2COOHであり、R9が水素であり、かつBがCR16である式Iの代表的な化合物として、
2-(ジチオカルボキシメチル)ペンタン二酸、
2-(1-ジチオカルボキシエチル)ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
Xが式IIIである部分であり、R8が-(CH2)2COOHであり、R9が水素であり、かつBがNである式Iの代表的な化合物として、
2-ジチオカルボキシアミノペンタン二酸、
2-〔(N-メチルジチオカルボキシ)アミノ〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
Xが式IVである部分である式Iの代表的な化合物として、
2-ベンジル-4-スルファニルブタン酸、
2-ベンジル-4-スルファニルペンタン酸、
2-(3-ピリジルメチル)-4-スルファニルペンタン酸、
2-(3-ピリジルメチル)-4-スルファニルヘキサン酸、
2-ベンジル-3-スルファニルプロパン酸、
2-ベンジル-3-スルファニルペンタン酸、
2-(4-ピリジルメチル)-3-スルファニルペンタン酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
式Iの幾つかの代表的な化合物の構造が以下に示される。
【0060】
【0061】
【0062】
【0063】
【0064】
【0065】
【0066】
【0067】
【0068】
【0069】
【0070】
【0071】
【0072】
【0073】
【0074】
【0075】
本発明の幾つかの化合物は一つ以上の不斉炭素中心を有し、こうして光学異性体の形態だけでなく、光学異性体のラセミ体混合物又は非ラセミ体混合物の形態で存在することができる。光学異性体は当業界で公知である通常の方法に従って、例えば、光学活性の酸又は塩基による処理によるジアステレオマー塩の生成によりラセミ体混合物の分割により得られる。適当な酸の例は酒石酸、ジアセチル酒石酸、ジベンゾイル酒石酸、ジトルオイル酒石酸及びショウノウスルホン酸であり、次いで結晶化によるジアステレオマーの混合物の分離、続いてこれらの塩からの光学活性塩基の放出が行われる。光学異性体の分離のための異なる方法は鏡像体の分離を最大にするために最適に選ばれたキラルクロマトグラフィーカラムの使用を伴う。更に別の利用可能な方法には、本発明の方法及び医薬組成物に使用される化合物を活性化形態の光学活性酸、光学活性ジオール又は光学活性イソシアノと反応させることにより共有結合ジアステレオマー分子、例えば、エステル、アミド、アセタール、ケタール、等の合成を含む。合成されたジアステレオマーは通常の手段、例えば、クロマトグラフィー、蒸留、結晶化又は昇華により分離でき、次いで鏡像体上純粋な化合物をデリバリするために加水分解される。或る場合には、親の光学活性薬物への加水分解は患者に投与する前に必要ではない。何とならば、化合物がプロドラッグとして挙動し得るからである。本発明の光学活性化合物は同様に光学活性出発物質を利用することにより得られる。
【0076】
本発明の化合物は光学異性体だけでなく、ラセミ体混合物及び非ラセミ体混合物を含むことが理解される。
以下に詳しく説明されるように、本発明の化合物は種々の薬理学的性質及び医薬の性質を有する。特に、本発明の化合物はNAALADase酵素活性を抑制する。NAALADase酵素活性を抑制することにより、本発明の化合物は神経変性中に生じるグルタメートの前シナプス放出を調節することが仮定される。
本発明の化合物はまたin vitroだけでなく、in vivo動物モデルで神経変性に対して保護する。幾つかの本発明の化合物は、虚血前及び後の投与されたときに、どちらも虚血の組織培養モデルで神経保護性であることが実証された。本発明の化合物の幾つかはin vitroモデルで虚血損傷の60分後までに投与された時に有意な神経保護効果を与える。
【0077】
更に、本発明の化合物の幾つかはin vivoラットMCAO卒中モデルで有意な保護を与え、また虚血後の60分、120分、180分及び360分で投与された時に保護性であることが示された。このような場合、本発明の化合物は卒中の発症後の60分以上、120分以上、180分以上、及び360分以上で投与された時に動物の卒中を治療するのに有効である。当業者はこのような化合物が卒中の発症後60分以内に投与される時にそれ以上ではないとしても同等に有効であると予想するであろう。同様に、卒中の発症後の360分以上に投与された時に卒中を治療するのに有効である化合物は、卒中の発症の360分より前のいずれの時点で投与された時に有効である化合物を包含すると期待されるであろう。神経保護を与えることに加えて、本発明の化合物は卒中後に挙動機能の回復を与えることにより卒中を治療するのに有効であることが可能である。
こうして、本発明は更に卒中の発症後に60分以上で投与された時に哺乳類の卒中を治療するのに有効である硫黄及び酸基の両方を含む化合物に関する。
化合物は卒中の発症後に120分以上で投与された時に哺乳類の卒中を治療するのに有効であることが好ましい。化合物は卒中の発症後に180分以上で投与された時に哺乳類の卒中を治療するのに有効であることが更に好ましい。化合物は卒中の発症後に360分以上で投与された時に哺乳類の卒中を治療するのに有効であることが最も好ましい。
【0078】
本発明の医薬組成物
本発明はまた
(i)有効量の式Iの化合物、及び
(ii)医薬上許される担体
を含む医薬組成物に関する。
式Iの化合物はNAALADase酵素活性を抑制し、グルタメート異常を治療し、神経活性に作用し、強迫性疾患を治療し、前立腺疾患を治療し、又は哺乳類の血管形成を抑制するのに有効な量で存在することが好ましい。
式Iの好ましい化合物は上述した。
【0079】
本発明の方法
NAALADase酵素活性の抑制方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のNAALADase酵素活性の抑制方法に関する。
グルタメート異常の治療方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のグルタメート異常の治療方法に関する。
グルタメート異常はグルタメートが関係するあらゆる疾患、障害又は症状を表し、上昇レベルのグルタメートを伴う病状が挙げられる。グルタメート異常の例として、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、精神分裂症、急性の痛み、慢性の痛み、虚血、末梢神経障害(糖尿病性神経障害を含む)、トラウマ脳損傷及び脊髄への物理的損傷が挙げられるが、これらに限定されない。好ましい実施態様において、グルタメート異常は虚血、卒中、パーキンソン病、筋萎縮性側索硬化症(ALS)及び脊髄損傷からなる群から選ばれる。
【0080】
いずれか一つの特別な理論に限定されないが、式Iの化合物はNMDA受容体により媒介される効果から上流であると仮定される貯蔵型グルタメートに作用することによりグルタメートのレベルを調節すると考えられる。
チオール官能基のラジカル除去性がまた化合物の治療効力に寄与し得る。フリーラジカル除去物質は脳及び神経組織の種々の型の急性及び慢性の病状に関連付けられている。最近の研究によりフリーラジカル除去物質が脳虚血−再潅流、興奮性アミノ酸脳損傷、ミトコンドリア機能障害、糖尿病、糖尿病性神経障害、先天性代謝異常、及び脳又は神経組織への急性又は慢性の損傷のその他の原因に神経保護効果を示すことが示されている。Krishanら, Pharmacological Research, 37巻, 1号, 23-9頁(1998年1月);Nodaら, Research Communications in Molecular Pathology and Pharmacology, 96巻, 2号, 125-36頁(1997年5月);Andersonら, Canadian Journal of Cardiology, 12巻, 10号, 1099-104頁(1996年10月);Mizunoら, General Pharmacology, 30巻, 4号, 575-8頁(1998年4月);de la Torreら, Brain Research, 779巻, 1-2号, 285-8頁(1998年1月);Yukiら, Molecular and Chemical Neuropathology, 32巻, 1-3号, 123-8頁(1997年9月−10月);Yamamotoら, Brain Research, 762巻, 1-2号, 240-2頁(1997年7月11日)を参照のこと。それ故、本発明の化合物はフリーラジカル損傷を伴う脳障害を治療するのに特に有効であり得る。
【0081】
強迫性障害の治療方法
更に、本発明はこのような治療を要する患者に有効量の式Iの化合物を投与することを特徴とする強迫性障害の治療方法に関する。
強迫性障害は抵抗不能の強迫性挙動を特徴とするあらゆる障害であってよい。本発明の方法により治療できる強迫性障害の例として、薬物依存性、食事障害、病的ギャンブル、ADD及びトゥレット症候群が挙げられる。
強迫性障害は薬物依存性であることが好ましい。依存性について潜在性を有する普通使用される薬物として、CNS抗うつ薬(オピオイド、合成麻酔薬、バルビツレート、グルテチミド、メチプリロン、エチクロルビノール、メタクアロン、アルコール);抗不安薬(ジアゼパム、クロルジアゼポキシド、アルプラゾラム、オキサゼパム、テマゼパム);興奮薬(アンフェタミン、メタムフェタミン、コカイン);及び幻覚薬(LSD、メスカリン、ペヨーテ、マリファナ)が挙げられる。
薬物依存性はアルコール依存性、ニコチン依存性、ヘロイン依存性又はコカイン依存性であることが更に好ましい。
【0082】
神経活性に作用する方法
本発明者らはまたNAALADaseの抑制が神経再生及び髄鞘形成を促進することを発見した。
それ故、更に、本発明は有効量の式Iの化合物を哺乳類に投与することを特徴とする哺乳類の神経活性に作用する方法に関する。
本発明の方法により作用される神経活性は損傷された神経細胞の刺激、神経細胞再生の促進、神経変性の予防及び神経障害の治療からなる群から選ばれてもよい。
本発明の方法により治療できる神経障害の例として、三叉神経痛;舌咽神経痛;ベル麻痺;重症筋無力症;筋ジストロフィー;筋萎縮性側索硬化症;進行性筋萎縮;進行性延髄遺伝性筋萎縮;椎間板ヘルニア症候群、断裂無脊椎板症候群又は脱出無脊椎板症候群;頸部脊椎症;神経そう障害;胸郭出口症候群;末梢神経障害、例えば、鉛、ダプソーン、チック、ポルフィリア、又はギラン・バレー症候群により生じたもの;アルツハイマー病;及びパーキンソン病が挙げられるが、これらに限定されない。
【0083】
本発明の方法は物理的損傷又は病状により生じた末梢神経障害、トラウマ脳損傷、脊髄への物理的損傷、脳損傷と関連する卒中、髄鞘脱落疾患及び神経変性に関係する神経障害からなる群から選ばれた神経障害を治療するのに特に有益である。髄鞘脱落疾患の例として、多発性硬化症並びに末梢髄鞘脱落疾患、例えば、末梢神経障害及びシャルコー−マリー歯疾患が挙げられる。神経変性に関係する神経障害の例として、アルツハイマー病、パーキンソン病、及び筋萎縮性側索硬化症(ALS)が挙げられる。
【0084】
前立腺疾患の治療方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の前立腺疾患の治療方法に関する。
好ましい実施態様において、前立腺疾患は前立腺癌、例えば、腺癌及び前立腺の転移癌、又は前立腺上皮細胞の異常増殖を特徴とする症状、例えば、良性前立腺過形成である。
癌の治療方法
前立腺癌に加えて、本発明の化合物で治療し得る癌のその他の形態として、ACTH生成腫瘍、急性リンパ性白血病、急性非リンパ性白血病、副腎皮質の癌、膀胱癌、脳の癌、乳癌、子宮頸癌、慢性リンパ性白血病、慢性骨髄性白血病、結腸直腸癌、皮膚T細胞リンパ腫、子宮内膜癌、食道癌、ユーイング肉腫、胆嚢癌、ヘアリー・セル白血病、頭部及び首の癌、ホジキンリンパ腫、カポージ肉腫、腎臓癌、肝臓癌、肺癌(小細胞及び/又は非小細胞)、悪性腹腔滲出、悪性胸膜滲出、メラノーマ、中皮腫、多発性ミエローマ、ニューロブラストーマ、非ホジキンリンパ腫、骨肉腫、卵巣癌、卵巣(生殖細胞)癌、膵臓癌、ペニス癌、網膜芽細胞腫、皮膚癌、軟組織肉腫、扁平上皮癌、胃癌、精巣癌、甲状腺癌、栄養膜腫瘍、子宮の癌、膣癌、外陰部の癌、及びウィルム腫瘍が挙げられるが、これらに限定されない。
本発明の化合物はNAALADase酵素がある組織の癌を治療するのに特に有益である。このような組織として、前立腺だけでなく、脳、腎臓及び睾丸が挙げられる。
【0085】
卒中の治療方法
更に、本発明は卒中の発症の60分以上後に有効量の硫黄及び酸基の両方を含む化合物を哺乳類に投与することを特徴とする哺乳類の卒中の治療方法に関する。
この化合物は卒中の発症の180分以上後に前記哺乳類に投与されることが好ましい。
この化合物は卒中の発症の360分以上後に前記哺乳類に投与されることが更に好ましい。
硫黄及び酸基の両方を含む化合物の例として、式Iの化合物が挙げられるが、これらに限定されない。
【0086】
血管形成の抑制方法
本発明者らはNAALADaseインヒビターがNAALADaseを含む組織中で血管形成に作用し得ることを予期せずに見出した。先の研究によりNAALADaseがシナプスプラズマ膜中で濃縮され、主として神経組織及び腎臓組織に局在化されることが示された。NAALADaseはまた前立腺及び睾丸の組織中に見られた。更に、先の知見によりNAALADaseが新生血管系中に存在することが示された。更に、NAALADaseは生体のその他の組織中で発見され続けているので、NAALADaseインヒビターはおそらくまたこれらの組織中の血管形成の抑制に効力を示すであろう。
それ故、更に本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の血管形成の抑制方法に関する。
血管形成は生殖力又は癌腫瘍の転移に必要であるかもしれず、また血管形成依存性疾患に関係しているかもしれない。従って、本発明の方法により治療できる血管形成依存性疾患として、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、並びに癌の腫瘍増殖、侵食、及び転移が挙げられるが、これらに限定されない。
本発明の方法はNAALADase酵素がある組織の癌腫瘍中の血管形成を抑制するのに特に有益である。このような組織として、脳、腎臓、前立腺、睾丸、及び血管が挙げられるが、これらに限定されない。
【0087】
痛みの治療方法
更に、本発明は有効量のNAALADaseインヒビターを哺乳類に投与することを特徴とする哺乳類の痛みの治療方法に関する。
NAALADaseインヒビターはモルヒネに対するトレランスを阻止し、痛みを治療するのに必要なモルヒネの量を減少するのに特に有効である。
本発明の方法により治療できる痛みの例として、急性、慢性、癌、やけど、切開、炎症、糖尿病性神経障害及び背部の痛みが挙げられるが、これらに限定されない。
好ましいNAALADaseインヒビターは式Iの化合物であり、これらの例は上述した。
別の好ましいNAALADaseインヒビターは式V:
【0088】
【0089】
の化合物又は医薬上許される均等物である。
式中、
YはCR3R4、NR5又はOであり、
R1は水素、C1-C9アルキル、C2-C9アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar、COOR、NR6R7及びORからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換、又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、COOR、NR6R7及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
R2は水素、C1-C6アルキル、C2-C6アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar、ハロ及びカルボキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、NR6R7及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
【0090】
R3及びR4は独立に水素又はC1-C3アルキルであり、
R5は水素又はC1-C3アルキルであり、
R、R6及びR7は水素、C1-C9アルキル、C2-C9アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル及びArからなる群から独立に選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換、又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
Arは1-ナフチル、2-ナフチル、2-インドリル、3-インドリル、4-インドリル、2-フリル、3-フリル、テトラヒドロフラニル、テトラヒドロピラニル、2-チエニル、3-チエニル、2-ピリジル、3-ピリジル、4-ピリジル及びフェニルからなる群から選ばれ、前記Arは未置換又はハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C6アルケニルオキシ、フェノキシ、ベンジルオキシ、カルボキシ及びNR1R2からなる群から独立に選ばれた一つ以上の置換基で置換されている。
【0091】
YはCH2であることが好ましい。
YがCH2である場合、R2は-(CH2)2COOHであることが更に好ましい。
YがCH2であり、かつR2が-(CH2)2COOHである場合、R1は水素、C1-C4アルキル、C2-C4アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、ベンジル、フェニル又はORであることが最も好ましく、前記アルキル、アルケニル、シクロアルキル、シクロアルケニル、ベンジル及びフェニルは未置換又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C6アルコキシ、C2-C6アルケニルオキシ、フェノキシ、ベンジルオキシ、NR6R7、ベンジル及びフェニルからなる群から独立に選ばれた一つ以上の置換基で置換されている。
【0092】
式Vの好ましい化合物は
2-(ホスホノメチル)ペンタン二酸、
2-〔〔(2-カルボキシエチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(ベンジルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔(フェニルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔((ヒドロキシ)フェニルメチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(ブチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(3-メチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、 2-〔(3-フェニルプロピルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(4-フルオロフェニル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(メチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔(フェニルエチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(4-メチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、 2-〔〔(4-フルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-メトキシベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(3-トリフルオロメチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-トリフルオロメチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-フルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、及び
医薬上許される均等物からなる群から選ばれる。
【0093】
式Vの化合物は2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸又は医薬上許される均等物であることが更に好ましい。式Vの化合物は鏡像体又は鏡像体濃縮混合物であることが最も好ましい。
R1がCOORで置換されている式Vの代表的な化合物として、
2-〔〔2-カルボキシプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-カルボキシブチル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-カルボキシペンチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-カルボキシ-3-フェニルプロピル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-カルボキシ-3-ナフチルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
【0094】
2-〔〔2-カルボキシ-3-ピリジルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-ベンジルオキシカルボニル〕-3-フェニルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-メトキシカルボニル〕-3-フェニルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(3-カルボキシ-2-メトキシカルボニル)プロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-カルボキシ-2-メトキシカルボニル)ブチル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
【0095】
R1がNR6R7で置換されている式Vの代表的な化合物として、
2-〔({〔ベンジルアミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔カルボキシアミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔ベンジルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔アセチルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔ジフェニルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔フェニルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-({〔(フェニルカルボキサミド)メチル〕(ヒドロキシホスフィニル)}メチル)ペンタン二酸、
2-({〔(フェニルスルホンアミド)メチル〕(ヒドロキシホスフィニル)}メチル)ペンタン二酸、
2-({〔(4-フルオロフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-メトキシフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-メチルフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-t-ブチルフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(チオホルムアニリド)アミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔1,3-ジオキソ-2,3-ジヒドロ-1H-2-イソインドリル〕メチル}ヒドロキシホスフィニル)メチル〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
【0096】
その他のNAALADaseインヒビターが米国特許第5,672,592号、同第5,795,877号、同第5,863,536号、同第5,880,112号及び同第5,902,817号、特許された米国特許出願第08/825,997号、同第08/833,628号、同第08/842,360号及び同第08/899,319号(これらについて、登録料が支払われた)、並びに国際特許公開番号WO 97/48399、WO 97/48400、WO 97/48409及びWO 98/53812に見られ、これらの特許、出願及び公開の全内容が参考として本明細書に含まれる。
好ましい実施態様において、NAALADaseインヒビターはモルヒネと組み合わせて投与される。
【0097】
糖尿病性神経障害の治療方法
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の糖尿病性神経障害の治療方法に関する。
有益なNAALADaseインヒビターの例が先に示されている。
【0098】
硫黄及び酸基の両方を含む化合物の調製方法
更に、本発明はチオラクトンを置換エステルと反応させて式VI
【0099】
【0100】
の化合物を生成する工程を含むことを特徴とする、硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Dは(CR21R22)nであり、
nは0、1、2、3又は4であり、かつ
R20、R21及びR22は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されている。
本方法は式VIの化合物をアルキル化剤と反応させてペンタン二酸誘導体を生成する工程を更に含むことが好ましい。
前記チオラクトンは
【0101】
【0102】
(式中、R10は先に定義されたとおりである)
であることが更に好ましい。
前記エステルは3-(ブロモ)プロピオン酸エチルエステルであることが最も好ましい。
更に、本発明は
(i)メルドラム酸を硫黄含有反応体と反応させてメルドラム酸硫黄含有誘導体を生成する工程;及び
(ii)前記メルドラム酸硫黄含有誘導体をエステルと反応させて式VII
【0103】
【0104】
(式中、
Eは硫黄含有部分であり、かつ
Fはプロピオン酸エステル含有部分である)
の化合物を生成する工程を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
本方法は式VIIの化合物を官能性誘導体化する工程を更に含むことが好ましい。
前記チオ含有反応体が3-〔(トリフェニルメチル)チオ〕プロピオン酸であることが更に好ましい。
前記エステルが3-(ブロモ)プロピオン酸メチルエステルであることが最も好ましい。
また、本発明は上記方法のいずれかにより調製された化合物に関する。このような化合物の例として、式Iの化合物が挙げられる。
【0105】
NAALADaseインヒビターの合成
本発明の方法に使用されるNAALADaseインヒビターの幾つかは、米国特許第5,672,592号、同第5,795,877号、同第5,863,536号、同第5,880,112号及び同第5,902,817号、特許された米国特許出願第08/825,997号、同第08/833,628号、同第08/842,360号及び同第08/899,319号(これらについて、登録料が支払われた)、並びに国際特許公開番号WO 97/48399、WO 97/48400、WO 97/48409及びWO 98/53812(これらの特許、出願及び公開の全内容が引用により本明細書に含まれる)に示された一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。
式VのNAALADaseインヒビターは、以下にスキームI-IXに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法、例えば、Jacksonら, J. Med. Chem., 39巻, 2号, 619-622頁(1996)及びFroestlら, J. Med. Chem., 38巻, 3313-3331頁(1995)により記載された方法により調製し得る。
【0106】
【0107】
R基を置換する方法は当業界で知られている。ホスフィン酸エステルを合成する付加的な方法がJ. Med. Chem., 31巻, 204-212頁(1988)に記載されており、以下にスキームIIに示される。
【0108】
【0109】
【0110】
上記ホスフィン酸エステルで開始して、式Vの化合物を調製する種々の経路がある。例えば、一般の経路がJ. Med. Chem., 39巻, 619-622頁(1996)に記載されており、以下にスキームIIIに示される。
【0111】
【0112】
式Vの化合物を調製する別の経路が以下にスキームIV及びスキームVに示される。スキームIV及びスキームVはホスフィン酸誘導体としての出発物質並びにスキームII及び本明細書にリストされた置換基を含む(これらに限定されない)妥当な化学置換基としてのR基を示す。
【0113】
【0114】
【0115】
式Vの化合物を調製する別の経路はR1における芳香族置換を可能にし、以下にスキームVIに示される。
【0116】
【0117】
式Vの化合物を調製する別の経路はR2位における芳香族置換を可能にし、以下にスキームVIIに示される。
【0118】
【0119】
YがNR5である式Vの化合物を調製する別の経路が以下にスキームVIIIに示される。
【0120】
【0121】
Yが酸素である式Vの化合物を調製する別の経路が以下にスキームIXに示される。
【0122】
【0123】
R1がCOORで置換されている式Vの化合物は、以下にスキームXに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法、例えば、Jacksonら, J. Med. Chem., 39巻, 2号, 619-622頁(1996)及びFroestlら, J. Med. Chem., 38巻, 3313-3331頁(1995)により記載された方法により調製し得る。
【0124】
【0125】
【0126】
【0127】
R1がNR6R7で置換されている式Vの化合物は、以下にスキームXI及びXIIに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法により調製し得る。
【0128】
【0129】
【0130】
Xが式IIである部分であり、かつAがO又はCR17R18である式Iの化合物は以下にスキームXIII-XXIIに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法により調製し得る。
【0131】
【0132】
【0133】
【0134】
【0135】
【0136】
【0137】
【0138】
【0139】
【0140】
【0141】
Xが式IIである部分であり、かつAが(CR17R18)mSである式Iの化合物は通常の合成方法、例えば、相当するチオールの酸化により容易に調製し得る。
Xが式IIである部分であり、かつAがSである化合物は通常の合成技術により容易に調製し得る。例えば、スキームXXIIが適当に置換されたチオ化合物で開始することにより変更し得る。加えて、このクラスの化合物はまたα,β-不飽和エステルへの適当に置換されたチオール誘導体のミカエル付加により調製し得る。
Xが式IIIである部分である式Iの化合物は通常の合成経路、例えば、グルタメート誘導体と二硫化炭素の反応を使用して容易に調製し得る。
【0142】
投与経路
本発明の方法において、下記の当該技術分野において有効であることが公知のいずれかの技法により化合物を投与することができる:常用の無毒の医薬として許容できる担体、補助剤及び賦形剤を含有する投与剤形において、経口的、非経口的、吸入スプレー、局所的、経直腸的、経鼻的、口腔的(baccally)、経膣的、又は植込貯蔵器。本願明細書において使用される用語「非経口」とは、皮下、静脈内、筋肉内、腹腔内、鞘内、脳室内、胸骨内、頭蓋内又は骨内への、注射及び注入技術を含む。侵襲的技法が好ましく、特に損傷した神経組織に直接投与することが好ましい。
【0143】
中枢神経系を標的として特に治療効果があるように、これらの化合物は、好ましくは末梢に投与された場合に、血液-脳関門を容易に透過するものでなければならない。血液-脳関門を容易に透過しない化合物は、脳室内経路により効果的に投与することができる。
【0144】
これらの化合物は、無菌注射用製剤、例えば無菌注射用水性又は油性懸濁剤の形で投与することもできる。これらの懸濁剤は、当該技術分野において公知の技術により、適当な分散剤又は湿潤剤及び懸濁化剤を用いて配合することができる。
無菌注射用製剤は、例えば1,3-ブタンジオール中の溶液のように、無毒の非経口的に許容できる希釈剤又は賦形剤中の無菌の注射用液剤又は懸濁剤であることもできる。水、リンゲル液及び等張塩化ナトリウム液は使用することができる許容可能な賦形剤及び溶媒のうちである。加えて、無菌の不揮発性油が、賦形剤又は懸濁媒質として通常使用される。この目的のために、合成モノ-又はジ-グリセリドのような、いずれかの無刺激の不揮発性油を使用することができる。オリーブ油及びヒマシ油を含む、オレイン酸及びそのグリセリド誘導体のような脂肪酸、特にそのポリオキシエチル化された形が、注射剤の調製において有用である。これらの油性の溶液又は懸濁液は、長鎖のアルコール希釈剤又は分散剤を含有することもできる。
【0145】
加えてこれらの化合物は、カプセル剤、錠剤、水性懸濁剤又は液剤の形状で経口投与することができる。錠剤は、乳糖及びコーンスターチのような担体、及び/又はステアリン酸マグネシウムのような滑沢剤を含有することができる。カプセル剤も、乳糖及び乾燥コーンスターチを含む希釈剤を含有することができる。水性懸濁剤は、有効成分と共に、乳化剤及び懸濁化剤を含有することができる。経口投与剤形は更に、甘味剤及び/又は香味剤及び/又は着色剤を含有することもできる。
これらの化合物は更に、坐薬の形で直腸から投与することができる。これらの組成物は、直腸で溶融し薬物を放出するために、室温では固形であるが直腸温度では液体であるような適当な非刺激性の賦形剤と、薬物を混合することにより製剤することができる。このような賦形剤は、ココアバター、ビーズワックス及びポリエチレングリコールを含む。
【0146】
更にこれらの化合物は、局所的に投与することができ、特に治療が対処する病態が、眼、皮膚又は下部腸管の神経障害を含む、局所的適用により容易に接近可能な領域又は臓器に関る場合に投与することができる。
眼への局所適用、又は眼内適用に関して、これらの化合物は、塩化ベンジルアルコニウム(benzylalkonium chloride)のような保存剤を含む又は含まずに、等張のpHを調節した無菌生理食塩水中の微小化された懸濁剤として、又は好ましくは等張のpHを調節した無菌生理食塩水中の溶液として、配合することができる。あるいは、これらの化合物は、ワセリンのような軟膏に配合することができる。
皮膚への局所適用については、これらの化合物を、例えば下記の1種以上の混合物中に懸濁又は溶解された化合物を含有する適当な軟膏に配合することができる:鉱油、液体ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化されたワックス及び水。あるいは、これらの化合物は、例えば下記の1種以上の混合物中に懸濁又は溶解された有効化合物を含有する適当なローション剤又はクリーム剤に配合することができる:鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セチアリールアルコール(cetearyl alcohol)、2-オクチルドデカノール、ベンジルアルコール及び水。
下部腸管への局所適用は、直腸坐薬製剤(前記)又は適当な浣腸製剤において作用することができる。
【0147】
本発明の化合物は、単回投与、個別の反復投与又は連続注入により投与することができる。これらの化合物は小さく、容易に拡散可能でありかつ比較的安定しているので、連続注入に良く適している。ポンプ手段、特に皮下ポンプ手段が、連続注入にとって好ましい。
【0148】
用量
有効成分である化合物の約0.1mgから約10,000mgの桁の投与量レベルが、前述の病態の治療において有用であり、好ましいレベルは、約0.1mgから約1,000mgである。特定の患者への具体的な投与量レベルは様々な要因によって左右され、これは、使用した具体的化合物の活性;患者の年齢、体重、全身の健康状態、性別及び食事;投与時間;排泄速度;併用薬;治療される特定の疾患の重症度;及び投与形態を含む。典型的には、in vitroの用量-作用の結果は、患者への投与に関する適切な投与量の有用な指針を提供している。動物モデル試験も有用である。
適当な投与量レベルを決定するための考察は、当該技術分野において周知である。
【0149】
好ましい実施態様において、これらの化合物は、凍結乾燥した形で投与される。
この場合、本発明の化合物の1〜100mgを、マンニトール及びリン酸ナトリウムのような担体及び緩衝液と共に、個別のバイアル中で凍結乾燥することができる。
この化合物は、投与前に静菌水によりバイアル中で再構成することができる。
総体虚血の治療においては、本発明の化合物は、好ましくは、経口的、経直腸的、非経口的又は局所的に、少なくとも1日1〜6回で投与され、かつより高濃度の初回ボラス用量に続けることができる。
本発明の化合物は、化学療法薬を含む1種以上の治療薬と組合せて投与することができる。表Iは、選択された化学療法薬に関する公知のメジアン用量を示す。
これらの物質及び他の治療的物質の具体的な投与量レベルは、本発明の化合物について先に確定されたものを考慮することによって変動する。
【0150】
〔表1〕
表I.
【0151】
〔表2〕
表I.つづき
【0152】
投与方式
本発明の方法に関して、薬物デリバリーの時期及び順番を調節するいずれかの投与方式が効果的な治療に必要なように使用され、かつ反復される。このような方式は、追加の治療薬の前投与及び/又は併用投与を含むことができる。
【0153】
神経傷害からの神経組織の保護を最大にするために、これらの化合物は、冒された細胞にできる限り迅速に投与されなければならない。神経傷害が予想できる状況においては、これらの化合物は、予想される神経傷害の前に投与されなければならない。このような神経傷害の可能性が増した状況としては、手術(頚動脈内膜切除術、心臓、静脈、大動脈再建(orthopedic));動脈カテーテル法のような脈管系の手技(頚動脈(cartoid)、椎骨動脈、大動脈、心臓、腎動脈、脊髄動脈、アダムキーウイク動脈);塞栓形成物質の注入;生体恒常性のためのコイル又はバルーン;脳病巣治療のための血管分布断絶;並びに、漸増性一過性虚血性発作、塞栓症、及び続発性卒中のような、病態の予備的素因を含む。卒中又は虚血の予防的治療が不可能であるか又は実践不能である場合、事象発生時又は発生後できるだけ速やかに、罹患した細胞が該化合物を得ることは重要である。卒中と卒中の間の期間の診断及び治療手技は、細胞を更なる損傷及び死から救うために最小化されなければならない。
【0154】
卒中の動物モデル及びヒトの両方において、脳虚血の影響が、脳の代謝に迅速に、分又は時単位の時間スケールで出現することは明らかである。従っていずれかの可能性のある神経保護的治療の形は、最も即効性のある経路で施されなければならず、これは実際には経静脈経路を意味する。治療投与の最適な期間及び経路は、神経保護的化合物の個々の薬物速度論的特性、薬物の有害作用プロフィール、及び卒中を生じた傷害の性質に応じて決まるであろう。卒中後のエクサイトトキシンによる損傷は、げっ歯類においては少なくとも4時間、ヒトにおいては恐らく48時間にわたって進展する。Dykerらの論文、「虚血性卒中の神経保護的治療の期間(Duration of Neuroprotective Treatment for Ischemic Stroke)」、Stroke、29:535-542 (1998)。従って、この危機的期間を通じて神経保護を提供することは望ましいであろう。理想的には、卒中治療のための如何なる化合物も、適切に血液-脳関門を通過し、かつ脳及びCSF内で十分な治療レベルを得なければならない。
【0155】
進行性でも転移性でもない前立腺癌患者については、本発明の化合物は、(i) 転移のリスクを低減するために術前又は放射線治療前に;(ii)術中又は放射線療法と併用して;及び/又は、(iii)再発のリスクを低下しかつ残存する腫瘍細胞の増殖を阻害するために術後又は放射線療法後に、投与することができる。
【0156】
進行した又は転移した前立腺癌患者については、未治療の原発性腫瘍及び存在する転移性病変の両方において腫瘍細胞の増殖を遅延化するためのホルモン途絶療法への連続補充剤(continuous supplement)として、又はその療法の代償として、本発明の化合物が投与され得る。
【0157】
本発明の方法は、隠れた(shed)細胞を外科的介入によって取り除くことができないような部位において特に有用である。本発明の方法は、術後の回復の後、このような散在細胞により惹起される腫瘍再発の機会を減少する点で有効であろう。
【0158】
他の治療との併用
a.神経傷害
神経傷害(特に急性虚血性卒中並びに水溺(drowning)及び頭部外傷により惹起された総体虚血)の治療法において、本発明の化合物は、1種以上の治療的物質、好ましくは卒中のリスクを低下することができる物質(例えばアスピリン)と、より好ましくは続発性虚血事象のリスクを低下することができる物質(例えばチクロピジン)と共に、併用投与することができる。
本発明の化合物は、1種以上の治療的物質と、(i)単一製剤中で一緒に、又は(ii)それらの各有効物質の最適放出速度となるよう設計された個々の製剤において個別にのいずれかで、併用投与することができる。各製剤は、本発明の化合物を約0.01%〜約99.99質量%、好ましくは約3.5%〜約60質量%に加え、湿潤剤、乳化剤及びpH緩衝剤のような1種以上の医薬用賦形剤を含有することができる。
【0159】
b.血管形成-依存型疾患
NAALADase阻害剤は、1種以上の治療的物質と、(i)単一製剤中一緒に、又は(ii)それらの各有効物質の最適放出速度となるよう設計された個々の製剤において個別にのいずれかで、併用投与することができる。各製剤は、NAALADase阻害剤を約0.01質量%〜約99.99質量%、好ましくは約3.5質量%〜約60質量%に加え、湿潤剤、乳化剤及びpH緩衝剤のような1種以上の医薬用賦形剤を含有することができる。
【0160】
c. 癌
手術及び放射線療法
概して、年齢70歳以下で局在化した癌を有し余命が少なくとも10年以上とみなされる患者には、潜在的に治癒力のある療法として、手術及び放射線療法が用いられる。
しかし手術のみによる治療では、多くの患者が癌の再発を経験するであろう。放射線療法も問題が多く、それは放射線療法剤が正常組織には毒性があり、かつしばしば生命を脅かすような副作用を引き起こすからである。
本発明の手術及び放射線療法との併用は、寛解を防止し、かつ毒性のある放射線療法剤の低用量レベルを可能にする。前述の統計事実を基に、手術及び/又は放射線療法との併用、又は代用として本発明を使用するかなりの機会がある。
【0161】
放射線増感剤
放射線増感剤は、電磁放射線の毒性作用に対する癌細胞の感度を増大することがわかっている。放射線増感剤の作用態様に関するいくつかの機構が文献において示されており、これは以下を含む:低酸素細胞の放射線増感剤(例えば、2-ニトロイミダゾール化合物及び二酸化ベンゾトリアジン化合物)が、低酸素の組織の再酸素化を促進し、及び/又は損傷を受けた酸素ラジカルの発生を触媒すること;DNA塩基のアナログであってよい、非低酸素細胞の放射線増感剤(例えば、ハロゲン化されたピリミジン)は癌細胞のDNAに優先的に組込まれ、それによりDNA分子の放射線誘導型の破壊を促進し及び/又は正常なDNA修復機構を妨げること;並びに、様々な他の可能性のある作用機構が、疾患の治療における放射線増感剤に関して仮定されていること。
【0162】
現在の多くの癌治療のプロトコールが、X-線の電磁放射により活性化された放射線増感剤を使用している。x-線で活性化された放射線増感剤の例は、以下を含むが、これらに限定されるものではない:メトロニダゾール、ミソニダゾール、デスメチルミソニダゾール、ピモニダゾール、エタニダゾール、ニモラゾール、マイトマイシンC、RSU 1069、SR 4233、EO9、RB 6145、ニコチンアミド、5-ブロモデオキシウリジン(BUdR)、5-ヨードデオキシウリジン(IUdR)、ブロモデオキシシチジン、フルオロデオキシウリジン(FudR)、ヒドロキシ尿素、シスプラチン、並びにこれらの治療上有効なアナログ及び誘導体である。
【0163】
光力学的療法(PDT)は、増感剤の電磁放射活性化剤として可視光を使用する。光力学的電磁放射増感剤の例は、以下を含むが、これらに限定されるものではない:ヘマトポルフィリン誘導体、フォトフリン、ベンゾポルフィリン誘導体、NFe6、エチオポルフィリンスズSnET2、フェノボルビド-a、バクテリオクロロフィル-a、ナフタロシアニン、フタロシアニン、フタロシアニン亜鉛、及びこれらの治療上有効なアナログ及び誘導体である。
【0164】
本発明の化合物は、癌細胞の電磁放射線の毒性作用に対する感度を増大するために、電磁放射線増感剤と併用投与することができる。電磁放射線増感剤と本発明の併用は、寛解を予防し、かつ低い電磁放射線線量レベルを可能にする。本発明の化合物、組成物及び方法と電磁放射線の組合せは、癌治療において、電磁放射線単独よりも効果があるはずである。
【0165】
電磁放射線増感剤と併用した場合、本発明の化合物は更に下記化合物の1種以上と併用投与することもできる:標的細胞への放射線増感剤の取込みを促進する化合物;標的細胞への治療薬、栄養及び/又は酸素の流れを制御する化合物;電磁放射線の追加の有無に関らず、腫瘍に作用する化学療法剤;又は、癌又は他の疾患を治療するためのその他の治療薬である。このような治療薬の例は、以下を含むが、これらに限定されるものではない:5-フルオロウラシル、ロイコボリン、5'-アミノ-5'デオキシチミジン、酸素、カルボゲン、赤血球輸血、過フッ化炭素(例えば、Fluosol-DA)、2,3-DFG、BW12C、カルシウムチャネルブロッカー、ペントキシフィリン、ヒドララジン、及びL-BSOである。化学療法剤の例は、表Iに列記した。
【0166】
ホルモン療法
薬物及び/又は睾丸切除術によるホルモン途絶療法は、癌の更なる増殖及び転移を促進するホルモンを遮断するために使用される。時間がたつにつれて、これらの患者の事実上全員について、原発性及び転移性腫瘍の両方がホルモン非依存性となり、治療抵抗性となる。本発明の化合物の連続補充は、この可能性のある転移-寛容状態(metastasis-permissive state)を防止又は逆行するために使用することができる。
【0167】
化学療法
化学療法は、いくつかの癌治療の形において成功している。しかし他の形の癌治療においては、化学療法は最後の手段として利用されるのみである。あらゆる症例において、化学療法は化学療法剤が正常組織にとって毒性がありかつしばしば生命を脅かすような副作用を生じるので問題が多い。加えて化学療法は高い失敗率及び/又は寛解率を有することが多い。
本発明の化学療法との併用は寛解を防止し、かつ毒性のある化学療法剤の低用量レベルを可能にする。本発明の方法と化学療法の併用は、癌治療において化学療法単独よりも効果があるはずである。
【0168】
免疫療法
本発明の化合物は、癌治療のためのモノクローナル抗体と併用することもできる。本発明は、ポリクローナル又はモノクローナル抗体-由来の薬剤を基にした免疫療法と共に使用することもできる。これらの薬剤は当該技術分野において周知であり、かつストロンチウム-89に結合されたモノクローナル抗体のような放射標識されたモノクローナル抗体を含む。
【0169】
NAALADase阻害剤のin vivo毒性
NAALADase阻害のin vivo毒性作用は、マウスにおいて試験されている。これらの結果は、NAALADase阻害剤がマウスにとって無毒であることを示しており、このことはヒトにとっても、治療有効量投与した場合に同様に無毒であることを示唆している。代表的な開示は、米国特許第5,672,592号、第5,795,877号、第5,804,602号、第5,824,662号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
【0170】
NAALADase活性のin vitro阻害
式I-Vの様々な化合物が、NAALADase活性のin vitro 阻害について試験されている。結果の一部は、米国特許第5,672,592号、第5,795,877号、第5,863,536号、第5,880,112号及び第5,902,817号、更には特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に開示されており、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。他の結果は、下記表IIに示す。
【0171】
【表3】
【0172】
【表4】
【0173】
【表5】
【0174】
【表6】
【0175】
【表7】
【0176】
【表8】
【0177】
【表9】
【表10】
【表11】
【表12】
【0178】
虚血に対するNAALADase阻害剤in vitroアッセイ
虚血時のNAALADase阻害剤のin vitro作用を調べるために、皮質細胞培養物を、虚血性発作時(シアン化カリウム及び2-デオキシグルコース)及びその後1時間、様々なNAALADase阻害剤で処理した(実験の詳細については、Vornovらの論文、J. Neurochem.、65(4):1681-1691 (1995)を参照のこと)。結果の一部については、米国特許第5,672,592号、第5,795,877号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
他の結果は、下記表IIIに示されている。神経保護作用は、EC50で表しているが、これは虚血発作後にグルタメート毒性の50%低下を引き起こすのに必要な濃度である。
【0179】
【表13】
【0180】
【表14】
【0181】
【表15】
【0182】
【表16】
【0183】
【表17】
【0184】
【表18】
【0185】
【表19】
【0186】
様々な濃度のNAALADase 阻害剤である化合物3の%毒性として測定された作用の用量-反応は、米国特許第5,672,592号、第5,795,877号、第5,804,602号、第5,824,662号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
【0187】
Sprague-DawleyラットにおけるMCAO後の脳損傷に対するNAALADase阻害剤のin vivoアッセイ
In vivoにおいて脳損傷に対するNAALADase 阻害剤の神経保護作用を試験するために、Sprague-Dawleyラットを、溶媒、及び化合物1又は2-(3-スルファニルプロピル)ペンタン二酸のいずれかで処理した。
対照群には、Hepes緩衝生理食塩水を与えた。
【0188】
4匹の薬物処置群は、化合物1を与えた。各ラットについて、中大脳動脈閉塞(MCAO)の60分後のIVボラス注射により処置を開始し、その直後に続けて4時間IV注入を0.5ml/時の速度で行った。第1群(n = 9)は、用量100mg/kgのIVボラス、その後20mg/kg/時のIV注入4時間を行なった。第2群(n = 11)は、用量30mg/kgのIVボラス、その後の6mg/kg/時のIV注入4時間を与えた。第3群(n = 9)は、用量10mg/kgのIVボラス、その後の2mg/kg/時のIV注入4時間を与えた。第3群のラットは、更に閉塞後120分、180分及び360分にも処置を受けた。第4群(n = 8)は、用量3mg/kgのIVボラス、その後の3mg/kg/時のIV注入4時間を受けた。
【0189】
2種の追加の薬物処置群は、2-(3-スルファニルプロピル)ペンタン二酸を与えた。各ラットは、中大脳動脈閉塞(MCAO)の120分後のIVボラス注射により処置を開始し、その直後に続けて4時間IV注入を0.5ml/時の速度で行った。第5群は、用量30mg/kgのIVボラス、その後6mg/kg/時のIV注入を4時間与えた。第6群は、用量10mg/kgのIVボラス、その後2mg/kg/時のIV注入を与えた。
【0190】
再灌流の22時間後、これらのラットを安楽死させ、それらの脳を摘出した。7つの対照切片(厚さ2mm)を採取し、2,3,5-トリフェニル-塩化テトラゾリウム(tetraxolium)(TTC)の1%溶液で20分間染色し、その後10%ホルマリンで固定した。最頭側脳切片の前部及び後部表面並びに残り6つの各切片の後表面を造影した。各脳の梗塞の大きさを、コンピュータを使用したデジタル画像分析システム(LOATS)を用いて、定量化した。TTC-染色を完全に欠いている脳領域は、梗塞組織を代表していると特徴づけた。各ラットの総梗塞容積を、各々連続した脳面積の数値積分(numeric integration)により算出した。
各群のラットの総梗塞容積を、下記表IV(a)及びIV(b)に示した。
【0191】
〔表20〕
表IV(a) 化合物1で処理したラット
【0192】
〔表21〕
表IV(b) 2-(3-スルファニルプロピル)ペンタン二酸で処理したラット
【0193】
溶媒で処理したラットは、平均総脳梗塞容積265±32mm3を示した。
化合物1で処理したラットは、有意に小さい梗塞サイズを示した。化合物1で処理した4群の平均の総脳梗塞容積は、第1群が123±31mm3(p = 0.014、溶媒群に対して);第2群が、141±78mm3(p = 0.002、溶媒群に対して) ;第3群が、152±32mm3(閉塞後60分で処置;p = 0.0058、溶媒群に対して);第4群が、117±22mm3(p = 0.0037、溶媒群に対して)であった。これらの結果は、ラットの卒中MCAOモデルにおいて、化合物1が、閉塞後60分、120分、180分、及び360分で投与された場合に、神経保護性であることを示している。
【0194】
2-(3-スルファニルプロピル)ペンタン二酸30mg/kgのIVボラス、その後の6mg/kg/時のIV注入4時間で処理したラットも、溶媒処理ラットよりも、有意に小さい梗塞サイズを示した。従って、2-(3-スルファニルプロピル)ペンタン二酸はこの特定の用量レベルで、閉塞後120分に投与した場合に、ラットの卒中のMCAOモデルにおいて神経保護的である。
【0195】
卒中患者は、虚血の発生と治療開始時点の間に顕著な時間的遅れを経験することが多い。従って、長期の治療ウィンドウを伴う神経保護剤の必要性がある。前述のデータは、本発明の化合物が、ラットの卒中MCAOモデルにおいて、少なくとも6時間の治療ウィンドウを有することを示している。当業者は、ヒトにおいてこの期間がより長いと予想するであろう。
【0196】
脳損傷に対するNAALADase阻害剤のin vivoアッセイプロトコール
雄のSprague-Dawleyラット(260-320g)を用いた。これらは、個飼し、自在に摂食・摂水させた。体重を維持する必要があるならば、実験の2日前に食事制限した。各ラットは、2種の手術を受けた:IV注入のための大腿静脈カニューレ挿管及びMCAO。手術時に、ラットは、吸入マスクを使って、酸素で送達される1.5%ハロタンで麻酔した。体温をモニタリングし、かつ恒温加熱システムを用い、正常温度に調節した。最初に、カテーテルを左大腿静脈から挿入した。30分後、MCAO術のためにラットを再麻酔した。MCAOは、Longらの論文、Stroke、20:84-91 (1989)に記された血管内縫合法を用いて行った。詳細に述べると、右外頚動脈(ECA)を露出し、凝着しかつ切断した。動脈切開術により、鈍端及び0.05%ポリ-1-リシンコートを伴う3-0のモノフィラメントナイロン縫合糸を、ECAの近位断端へ挿入した。これを、頚動脈分岐から、前大脳動脈の近位部へ到達するまで22mm進め、これによりMCAに起源する閉塞を生じた。ラットを覚醒させ;2時間後、ラットを再灌流のために再度麻酔し、その間ナイロン糸をECA断端へと引っ張り、血液のMCAへの再循環を可能にした。
【0197】
卒中が誘発した脳グルタメートレベルの上昇に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、卒中が誘発した細胞外グルタメート上昇を伴うラットにおいて、高グルタメート血症障害に対するその作用についてin vivo試験した。このプロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に含まれるものとする。結果は、化合物3による処置が、線条においては、卒中-誘発性細胞外グルタメートの上昇を有意に減弱し;かつ、頭頂皮質においては、同時に発生するグルタメートの変化を完全に妨げたことを示した。
【0198】
脊髄後根神経節-シュワン細胞共存培養物における髄鞘形成に対するNAALADase阻害剤のin vitroアッセイ
NAALADaseの阻害は、坐骨神経を寒冷病変形成(cryolesion)した後、溶媒処置したマウスと比べて、髄鞘形成した軸索の数の有意な増加及びミエリンの肥厚化を生じた(Soc. Neurosci. Abstr.、23(2):2302 (1997))。本発明者らは、NAALADaseが、ミエリン形成のシグナル伝達において役割を果たし、かつNAALADase阻害は髄鞘形成を促進するであろうと仮定した。この仮説を検証するために、本発明者らは、良く特徴付けられた髄鞘形成のin vitroモデルシステムにおいて、いくつかのNAALADase阻害剤の作用を試験した。脊髄後根神経節-シュワン細胞共存培養を、先に公表されているように確立した(Einheberら、J. Cell Biol.、123:1223)。共存培養の7日後髄鞘形成が始まり、その後、血清及びアスコルビン酸を、様々な用量(1nM〜10μM)のNAALADase阻害剤及びプロゲステロン(20nM;陽性対照)と共に添加した。髄鞘形成の程度を、公知のミエリンマーカーであるミエリン塩基性タンパク質(MBP)の免疫細胞化学染色を用いて、14〜21日の間試験した。
免疫染色された培養物の定量分析は、NAALADase阻害剤添加後に、髄鞘形成した軸索数が溶媒で処理した培養物中の軸索と比べて、有意に用量-反応に相関して増加したことを明らかにした。図1A-C及び2A-Cに示されたように、NAALADase阻害剤である化合物3及び2(1nM)で2週間処理すると、MBP免疫染色の有意な増加が生じた。高用量の化合物3又は化合物2(1μM)で処理された培養物は、最大用量のアスコルビン酸及びプロゲステロンで処理された培養物よりも、より大きい程度の髄鞘形成を生じた。これらの結果は、NAALADaseの阻害が、髄鞘形成を促進しかつ脱髄性疾患(demyelinating disease)の治療において臨床的に有用であり得ることを示唆している。
【0199】
坐骨神経寒冷病変(sciatic nerve cryolesion)に伴うミエリン形成に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、坐骨神経寒冷病変を形成した雄のマウスにおける神経再建及び髄鞘形成に対するその作用について、in vivoで試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に含まれるものとする。結果は、化合物3で処置されたマウスの坐骨神経が、髄鞘形成された軸索数の増大及びミエリン肥厚化を生じたことを示している。
【0200】
パーキンソン病に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、MPTPで病変形成したマウスにおけるパーキンソン病に対するその作用について、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、化合物3を与えたMPTP病変マウスが、TH-染色したドパミン作用性神経の顕著な回復を示し、このことは化合物3がMPTP-毒性に対して保護的であることを示唆している。
【0201】
ダイノルフィン誘発性脊髄損傷に対する NAALADase阻害剤のin vivoアッセイ
化合物3を、ダイノルフィンで誘発した脊髄損傷を伴うラットにおける、エクサイトトキシン性脊髄損傷に対するその作用について、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、ダイノルフィンAと同時投与した化合物3は、注射の24時間後の運動スコアにおいて有意な改善を生じ、このことは、化合物3が、ダイノルフィン-誘発性脊髄損傷に対し効果的な保護を提供することを示唆している。
【0202】
筋萎縮性側索硬化症(ALS)に対するNAALADase阻害剤のin vitroアッセイ
化合物3について、脊髄の器官培養において筋萎縮性側索硬化症(ALS)に対するその作用を、in vitroにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、化合物3が、THA-誘発毒性に対して用量-依存性の保護を発揮することを示した。
【0203】
アルコール好餌ラットにおけるエタノール消費に対するNAALADase阻害剤のin vivoアッセイ
化合物3について、アルコール好餌ラットにおけるエタノール消費に対するその作用を、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容が本願明細書に参照として組入れられている。結果は、化合物3が、体重又は24時間の摂水に影響を及ぼすことなくエタノール消費を有意に低下したことを示し、このことは、NAALADaseが、アルコール摂取行動の神経系による調節に関係していることを示唆している。
【0204】
オスのLong-Evans ラットにおけるニコチン自己投与に対するNAALADase阻害剤のin vivoアッセイ
化合物3について、ラットにおけるニコチン自己投与に対するその作用を、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容が本願明細書に参照として組入れられている。結果は、化合物3が、累積食物摂取に加え、ニコチンの自己投与を減少したことを示した。この結果は、NAALADase阻害剤以外の要因が、ニコチンの自己投与の減少に寄与し得ることを示唆しているが、ニコチン使用の神経系による調節におけるNAALADaseの関与を反証するものではない。ラットの食物摂取に対する作用は、過剰な薬物用量によって引き起こされた毒性に起因しているであろう。
【0205】
Sprague-Daw1eyラットにおけるコカインに対する行動的増感現象に対するNAALADase阻害剤のin vivoアッセイ
NAALADaseは、豊富な神経ペプチドNAAGを加水分解し、グルタメート(GLU)を遊離する。本発明者らは、NAALADaseの阻害は、このGLU供給源を妨害することにより増感(sensitization)を減弱すると仮定した。発明者らは、コカインの精神運動刺激作用を顕在化する増感現象に対する化合物3の影響を評価した。雄のSprague-Dawleyラットに、コカイン(20mg/kg/日 x 5日間;i.p.)またはセール(sale)(1.0ml/kg)を、ホームケージ(home cage)注射で与えた。注射の15分前に、ラットに化合物3を10及び50mg/kgの用量で与えた。コカイン(20mg/kg)-誘発性歩行活動を、3日後に評価した。急性のコカインは、コカイン暴露の作用を増大した(例えば増感)。化合物3をコカインと共に与えた動物においては、活動の増強は有意に低下していた。化合物3それ自身は、自発運動又は生理食塩水に対する反応を変化させなかった。結果は、図23に図示した。データは、化合物3はコカインが誘発した増感の発生を弱めることを示した。増感におけるGLUの仮定された役割を考慮すると、NAALADase阻害剤は反復コカイン投与の結果生じる行動馴化を妨げることが示された。
【0206】
癌に対するNAALADase阻害剤のin vitroアッセイ
LNCaP細胞において、化合物3及びキスカル酸の前立腺癌に対する作用を試験した。プロトコール及び結果は、米国特許第5,804,602号に開示されており、その全内容は引用により本願明細書に組入れられている。結果は、化合物3及びキスカル酸の濃度が増加するに従い、LNCaP細胞増殖が有意に減少したことを示し、これは、NAALADase阻害剤が、癌、特に前立腺癌の治療に有効であることを示唆している。
【0207】
In vitro癌アッセイのプロトコール
ウシ胎仔血清(FCS)10%を含有するRPMI 1640培地中の細胞を、24ウェルプートに播種し、かつ24時間接着させ、その後キスカル酸(10-9〜10-6)又は化合物3(10-11〜10-8)を7日間添加した。7日目に、細胞に、3H-チミジンを4時間パルスし、収穫し、放射活性を測定した。値は、各処置について6個の個別の細胞ウェルの平均±SEMで示している。全ての実験は少なくとも2回行った。
キスカル酸及び化合物3の非特異的細胞増殖抑制作用を制御するために、これらの物質を同時にNAALADase非含有前立腺癌細胞株DU145についても評価した(Carterら、Proc. Natl. Acad. Sci. USA、93:749-753 (1996))。キスカル酸及び化合物3による処置が細胞増殖に有意な作用を示さないならば、これらの物質のNAALADase阻害活性は前立腺癌細胞株に対するそれらの細胞増殖抑制作用に大いに寄与するものである。
【0208】
細胞株及び組織培養
LNCaP細胞は、Johns Hopkins School of Medicine(ボルチモア, MD)のDr. William Nelsonのご好意により得た。DU145細胞は、アメリカンタイプカルチャーコレクション(ATCC)(ロックビル、MD)から入手した。細胞は、5%熱失活したウシ胎仔血清、2mMグルタミン、100ユニット/mlペニシリン、及び100μg/mlストレプトマイシン(Paragon社)を補充した、RPMI-1640培地において、加湿したインキュベータ内で、37℃、5%CO2/95%空気大気中で培養した。
【0209】
[3H]チミジン取込みアッセイ
細胞を、RPMI-1640 培地中に1 x 103細胞/mlで懸濁し、かつ24ウェルプレートに、500μl/ウェルで播種した。24時間インキュベーションした後、様々な濃度のキスカル酸(シグマ社)又は強力なNAALADase阻害剤である化合物3(Jacksonらの方法(J. Med. Chem.、39(2):619-622)に従って合成)を、ウェルに添加し、かつプレートをインキュベータに戻した。3、5及び7日目に、培地及び薬物を新鮮なものと交換した。播種後8日目に、各ウェルを、1μCi 3H-チミジン(New England Nuclear社)で4時間パルスした。その後培地を取り除き、ウェルをリン酸緩衝生理食塩水(pH7.4)で2回洗浄した。各ウェルの内容物を、順次0.2N NaOHの250μlに可溶化し、かつシンチレーションバイアルに移した。UltimaGold (Packard社)シンチレーションカクテル5mlを添加し、放射活性を、Beckman LS6001シンチレーションカウンターを用いて測定した。
【0210】
全ての合成化合物の純度及び/又は同一性は、薄層クロマトグラフィー、高速液体クロマトグラフィー(HPLC)、質量スペクトル、及び元素分析により確認した。
核磁気共鳴(NNR)スペクトルは、Bruker分光計を用いて得た。化学シフトは、内部標準テトラメチルシランに対するppmで記した。分析用薄層クロマトグラフィー(TLC)を、積層済みのシリカゲルGHLFプレート(Analtech社、ニューアーク、DE)上で行った。プレートの視認は、UV光、リンモリブデン酸-エタノール、及び/又はヨウ化プラチナチャーリング(iodoplatinate charring)を用いて行った。フラッシュクロマトグラフィーは、Kieselgel 60、230-400メッシュ(E. Merck社、ダルムシュタット、西ドイツ)上で行った。溶媒は、試薬等級又はHPLC等級のいずれかであった。特に記さない限り、反応は、周囲温度及び窒素雰囲気下で進行させた。溶液は、Buchi回転蒸発器を用いて、減圧下で蒸発した。
【0211】
癌に対するNAALADase阻害剤のin vivoアッセイ
癌へのNAALADase阻害剤のin vivo作用を調べるために、LNCaP細胞を注射したncr雄マウス、及びDunning G細胞を注射したCopenhagan同系ラットに、化合物3を様々な用量で皮下及び/又は腫瘍内投与した。処置後の平均腫瘍容積(mm3)及び腫瘍:対照比(%T/C)を、図3-7に示した。
【0212】
結果は、LNCaP腫瘍は、化合物3の皮下投与に反応したことを示した。低用量1及び3mg/kg並びに最高用量30mg/kgは、腫瘍増殖に明らかに作用を示さなかった(図3)。用量10mg/kgは、86日目に、腫瘍増殖を、対照の24%に有意に阻害した(p = 0.006)(図4)。
Dunning G腫瘍も、化合物3の皮下投与に反応した。低用量1及び3mg/kgは、腫瘍増殖に反応を示さない一方で、2種のより高い用量10及び30g/kgは、腫瘍サイズを有意に縮小した(図5)。腫瘍サイズの縮小は、用量10mg/kgでは対照の38%(p = 0.03)に、用量30mg/kgでは対照の22%であった(図6)。
【0213】
LNCaP腫瘍は、化合物3の腫瘍内投与にも反応した。3種の低用量レベル(0.025、0.25及び2.5μg/日)は、腫瘍増殖を実質的に緩やかにし、最大の減少は用量0.025μg/日で認められた(表V)。対照群の投与42日後の腫瘍容積は807.3±197.3mm3であるのに対して、0.025μg/日処置群は465.7±176mm3であった(図7)。
【0214】
〔表22〕
表V 化合物3の抗腫瘍活性
【0215】
In vivo癌アッセイのプロトコール
皮下の薬物デリバリー
LNCaPモデル(化合物3):
雄のncrヌードマウスで、5〜6週齢のものに、Matrigel(登録商標)中の5 x 106 LNCaP細胞を右側腹部に注射した(総注射容量0.1ml)。細胞注射の2週間後、化合物3の毎日の皮下注射(s.c.)を、下記の用量で開始した:1、3、10及び30mg/kg。
対照は、毎日50mM HEPES s.c.を与えた。一旦腫瘍が触知可能となったならば、これらを週2回測定した。
DUNNING Gモデル(化合物3):
雄のCopenhagen同系ラットで、8〜10週齢のものに、107のDunning G細胞を両側腹部に注射した。細胞注射の2週間後、化合物3の毎日の皮下注射(s.c.)を、下記の用量で開始した:1、3、10及び30mg/kg。対照は、毎日50mM HEPES s.c.を与えた。腫瘍は週2回測定した。
【0216】
腫瘍内への薬物デリバリー:
LNCaPモデル(化合物3):
雄のncrヌードマウスで、5〜6週齢のものに、Matrigel(登録商標)中の107 LNCaP細胞を右側腹部に注射した(総注射容量0.1ml)。腫瘍が予定のサイズ(50〜60mm3)に達したら、各8匹のマウスを6処置群に無作為に割り当てた。化合物3を毎日腫瘍内に、下記の用量で容量0.05mlで投与した:25、2.5、0.25及び0.025μg。
対照には、毎日50mM HEPES 50μlを腫瘍内投与した。腫瘍は週2回測定した。
処置に対する反応を、2種の方法でモニタリングした。第一には、各群の平均腫瘍容積を、腫瘍:対照比(%T/C)として示し、これらの値はある時点で比較した。第二は腫瘍容積対時間をモニタリングした。
【0217】
血管形成に対するNAALADase阻害剤の日用量のin vivoアッセイ
雌のC57B1マウスで、8〜10週齢(5匹/群)のものに、Matrigel(登録商標)0.5mlと共に、血管形成因子である塩基性繊維芽細胞増殖因子(bFGF)を150ng/mL、及び化合物3の0、0.47μM又は4.7μMと共に皮下注射した。Matrigel(登録商標)注射は速やかにゲルを形成した。Matrigel(登録商標)注射と同日に、化合物3のMatrigel(登録商標)プラグ周辺への皮下注射を開始した。Matrigel(登録商標)注射の7日後、Matrigel(登録商標)プラグを摘出し、組織検査を行った。
日用量の濃度および、その時の最初のMatrigel(登録商標)プラグ組成物を、表VIに示した。
【0218】
〔表23〕
表VI NAALADase阻害剤の日用量の濃度
【0219】
図8Aに詳細に示したように、良好な血管形成反応が溶媒処置群において認められた。3mg/kg及び30mg/kgの日用量群で得られたMatrigel(登録商標)プラグ内の血管又は血管形成の減少は、各々、図8B及び図8Cにおいて示した。
【0220】
血管形成に対するNAALADase阻害剤の連続用量のin vivoアッセイ
下記表VIIに示した化合物3濃度のミニ浸透圧ポンプを、C57B1雌マウス(5匹/群)に植え込んだ。このとき、溶媒(50mM Hepes)を充填したミニポンプも植え込んだ。24時間後、マウスに、Matrigel(登録商標)0.5ml及び血管形成因子である塩基性繊維芽細胞増殖因子(bFGF)150ng/mLを、各々皮下注射した。Matrigel(登録商標)/bFGF注射後13日目に、ゲルを回収し、ホルマリンに固定し、かつ切片をトリクローム-マッソン(Trichrome-Masson)染色液で染色した。
【0221】
〔表24〕
表VII NAALADase阻害剤の連続投与濃度
【0222】
各々図9及び10に示されたように、溶媒及び1μg/日用量群において、強力な血管形成性の反応が認められた。図11及び12に示したように、化合物3の用量10μg/日及び100μg/日のデリバリーは、Matrigel(登録商標)/bFGFゲル内の血管形成を有意に減少した。
【0223】
糖尿病性ニューロパシーに対するNAALADase阻害剤のin vivoアッセイ
ストレプトゾトシン(STZ)-誘発性末梢性糖尿病性ニューロパシーモデルにおいて、NAALADase阻害剤をin vivo試験した。雄のSprague-Dawleyラットで体重200〜250gのものに、STZ 60mg/kgを尾静脈に静脈内注射し、糖尿病とした。血漿ブドウ糖レベルを、STZ投与後3週間測定した。血漿ブドウ糖レベルが>300mg/dL(17mM)のSTZ-動物のみを、本試験に用いた。灼熱痛閾値及び逃避待ち時間(withdrawal latency)を用いて、小脊髄後根神経節(DRG)知覚神経の状態を評価した。疼痛は、Ugo basil社(バレーゼ、イタリア)が製作した Basile Plantar装置を使った足底試験(Hargreavesの方法)を用いてモニタリングした。
【0224】
STZ投与の2ヶ月後、糖尿病動物は、逃避待ち時間の差で測定した場合、非糖尿病対照と比べて、痛覚過敏であった。この時点で、ラットに、NAALADase阻害剤化合物2(50mg/kg)又はビヒクルを、1日1回20日間腹腔内投与した。灼熱痛反応を、投与後3、5、12及び19日目に測定した。図13に示したように、投与5日目に、化合物2を投与された動物は、ビヒクル投与動物と比較して、逃避待ち時間において有意な増加を示した。この差は、観察期間を通じて維持された。
これらのデータは、NAALADase阻害剤が、実験的糖尿病の知覚ニューロパシーに対して保護的であり、かつ末梢性ニューロパシーの治療において有用であろうということを示唆されている。
【0225】
ホルマリン、酢酸及び慢性狭窄(Constricture)誘導(CCI)疼痛モデルにおける痛覚過敏に対するNAALADase阻害剤のin vivoアッセイ
最近の証拠は、興奮性のアミノ酸であるグルタメートが、中枢性及び末梢性の両方で媒介された疼痛感において主要な役割を果たしていることを示唆している。
神経性グルタメートのひとつの供給源は、豊富な神経ペプチドNAAGに由来し、これはNAALADaseによって加水分解されると遊離のグルタメートを放出すると考えられている。本発明者らは、NAALADaseの阻害が、このグルタメート供給源を妨害することにより、疼痛を制限することができるのではないかと仮定した。この仮説を検証するために、本発明者らは、ホルマリン-、酢酸-及び慢性狭窄性損傷(CCI;“Bennettモデル”)の疼痛モデルにおけるいくつかのNAALADase阻害剤の抗痛覚作用の可能性について試験した。ホルマリンモデルにおいて、ラットに、化合物3(50mg/kg)又はビヒクルを毎日7日間i.p.投与した。7日目に、5%ホルマリンを、ラットの後肢の背側に注射した。結果を、図14-19に図示した。化合物3の前処置は、ホルマリンモデルの早期相及び後期相の両方において、たじろぎ行動(flinching behavior)を強固に緩和した(各々、13.8±6.4を、 2.5±3へ低下、p = 0.02、及び58.0±9.8を、0.5±0.58に低下、p = 0.0001;図14)。化合物3の処置は、モルヒネ(5mg/kg)による急性前処置よりも更に強固であった。疼痛の酢酸モデルにおいては、酢酸(0.6%)が誘導した身じろぎ(writhing)は、化合物3(図15)、化合物2(図16)、化合物1(図17)を前処置したマウスにおいて、ビヒクルの対照マウスと比べて、有意に緩和された。最後に疼痛のCCI誘導モデルにおいては、動物は、術後10日目から化合物3(50mg/kg)を18日間毎日i.p.投与された。
化合物3は、灼熱痛反応で調べた場合に、坐骨神経狭窄後に、劇的に痛覚過敏を軽減した。18日目に、ラットのビヒクルによる同様の処置群と比べて、疼痛は98%緩和されていた(スコア差-0.2±1.9 vs. -4.75±2.4;p = 0.0001;図18)。これらのデータは、NAALADaseの阻害が、急性及び慢性の両方の疼痛の有用な治療モダリティであることを示している。
【0226】
ニューロパシー疼痛に対するNAALADase阻害剤のin vivoアッセイ
雄のSprague-Dawleyラット(200-225g)を、ストレプトゾトシン(STZ、リン酸緩衝生理食塩水中70mg/kg)を静脈内投与することにより、糖尿病とした。糖尿病動物を、5群に分けた:ひとつの群は、化合物2(10mg/kg又は1mg/kg)、化合物1(10mg/kg又は1mg/kg)又はビヒクルを与えた。別の動物群(非-STZ処置)を、非糖尿病対照として用いた。薬物/ビヒクル処置は、STZ投与後45日目の糖尿病動物から開始した。STZ-誘発性糖尿病ラットは、血糖値が320mg/dl以上に上昇した直後に、熱源に対する感受性について試験した(STZ後30日目)。その後ラットを、Hargreaves装置に馴化させ、かつ灼熱の疼痛感を、後肢の背側表面に向けた赤外線熱源を用いてモニタリングしたところ、動物がその肢を引っ込めるのに要した時間はほぼ0.1秒であった(方法の詳細については、Hargreavesらの論文(1998)を参照のこと)。線源の強度は、対照動物(非-STZ処置)の平均待ち時間が、およそ10秒となるよう調節した。各動物は、8回試験し、平均スコア差(非糖尿病対照の待ち時間の平均と糖尿病の待ち時間の平均の間)を、図19A及び19Bに図示した。糖尿病ラットは、非糖尿病対照と比べて痛覚過敏を示し(より短い反応待ち時間)、STZ投与後30日に始まり、これはビヒクル処置ラットでは次第に悪化していった。この痛覚過敏反応は、化合物1又は2(20mg/kg i.p.、毎日)処置を行なった糖尿病ラットでは完全に逆行した。従ってこれらの結果から、NAALADaseの阻害は、ニューロパシー疼痛を減弱することが示された。
【0227】
ニューロパシー疼痛の進行に対するNAALADase阻害剤のin vivoアッセイ
化合物3
ラットにおいて、坐骨神経の三分岐点の近位に1mm間隔で緩くこの神経の周りを縛ることで結紮を4個つくり、坐骨神経結紮を行った。この処置を行った後のラットは、熱に対する痛覚過敏及び異疼痛を示した。動物は、Hargreaves装置に慣れたので、後肢の背側表面に赤外線熱源を向け、動物がその肢を引っ込めるのに要する時間を観察した。スコアの差異(操作側対対照側について、肢が反応するまでの待ち時間の間)を決定した。動物は、術後10日目に、化合物3(50mg/kg i.p. 毎日)又はビヒクルの受取りを開始した。化合物3による処置は、連続して痛覚過敏のビヒクル-処置対照と比べて、2本の肢の間のスコアの差異を劇的に正常化した。正常な(手技を施さず)ラットは、両肢でほぼ等しい待ち時間を有した。この作用は、薬物投与の11日目に顕著に始まり、かつ本試験終了時(21日間の毎日投与)まで維持された。スコアの差異を図20に図示した。結果は、NAALADaseの阻害が、CCI-関連した痛覚過敏を緩和することを示した。
【0228】
化合物1及び2
雄のBB/Wラット(BRI, Mass)は、膵B細胞の細胞が媒介した自己免疫破壊を自発的に発症しており、その結果インスリン依存型(I型)糖尿病を発症する(Guberski、1994)。これらのラットは、特徴決定された、線維消失及び変性のような神経欠損、ヒトの糖尿病の末梢神経において認められるものに関連した変化を随伴するニューロパシーを生じることが示されている(Yagihasi、1997)。このことは、これらのラットを、この主要障害の今後の治療のための新規化合物の実験的試験にとって価値のあるものとしている。本試験において、化合物1及び化合物2は、糖尿病ニューロパシーの進行を変更するそれらの能力について試験した。ラットは、糖尿病発症(高血糖症)時に開始しその後最大6ヶ月目まで、毎日化合物1もしくは化合物2(10mg/kg i.p.)又はビヒクルの注射を行なった。ビヒクルを与えた非糖尿病ラットの別の群も試験した。全ての動物は、体重、尿量、血糖値及びグリコヘモグロビンについて連続してモニタリングした。試験の最初の月には、全ての動物を、熱に対する疼痛感について毎週試験した(Hargreaves装置)。最初の1ヶ月経過後は、2週間に1回に、その後月1回試験した。試験は、赤外線熱源をラットの後肢の背側表面に当てること、及び動物がその肢を引っ込めるのに要する時間を観察することからなる(方法の詳細については、Hargreavesらの論文(1998)を参照のこと)。各動物は8回試験し、逃避待ち時間の平均を記録した。
【0229】
これらの結果は、図24に図示した。結果は、糖尿病ラットは、非糖尿病対照と比べて、痛覚過敏(より短い反応待ち時間)を有することを示した。糖尿病薬物処置ラット(化合物1及び化合物2の両方)は、糖尿病ビヒクル処置ラットよりも、より長い逃避待ち時間を示し、これは処置の4週間後に始まり、かつ処置の6ヶ月間持続した。
【0230】
神経伝導速度も、処置開始後最初の8週間は隔週で、その後処置6ヶ月間を通じて毎月1回測定した(方法の詳細についてはDe Koningらの論文(1987)を参照のこと)。結果は図25に図示した。糖尿病動物は総じて、非糖尿病対照と比べて、神経伝導速度の遅延を示した。しかし糖尿病動物がNAALADase阻害剤(化合物1又は化合物2のいずれかを用量10mg/kg)の毎日の注射を受けると、ビヒクル投与を受けた糖尿病対照の場合と比べて、神経伝導欠損の重症度の有意な低下を示した。これは、処置8週目に明らかに始まり、かつ6ヶ月の本試験終了時までずっと同様の程度が持続した。他方で、糖尿病ビヒクルは、溶媒処置開始後6〜16週間、神経伝導速度の進行性の悪化を示し、これは6ヶ月間を通じて維持された。
【0231】
糖尿病ニューロパシーに対するNAALADase阻害剤のin vivoアッセイ
運動神経及び知覚神経の伝導速度も、STZ-糖尿病動物において、処置後4、8及び12週間に測定した(方法の詳細についてはDe Koningらの論文(1987)を参照のこと)。簡単に述べると、麻酔をかけたラットにおいて、刺激用針電極を、坐骨及び脛骨の神経に挿入し、記録用電極を遠位足筋上皮下に配置した。結果は、図21A、21B、22A及び22Bに示した。ビヒクルを与えた糖尿病動物は、非糖尿病動物と比べて、運動及び知覚神経の両方の伝導速度の有意な低下を示した。化合物2の10mg/kgによる4、8及び12週間の処置は全て、運動及び知覚神経の両方の伝導速度を改善(増大)する傾向があり、運動及び知覚神経伝導速度の顕著な改善が、各々、12週後及び8週後に認められた(図31A)。試験した低用量の化合物2(1mg/kg)は同様の作用を有した(図31B)。いずれかの用量の化合物1による動物の処置も、同じく糖尿病対照において運動及び知覚両神経の伝導速度を増大し、用量10mg/kg処置群については処置の12週間後に、1mg/kg処置群については処置後早い時期に、顕著に増大した(図32A及び32B)。従って、これらの結果は、NAALADase阻害が、糖尿病ニューロパシーの進行を変化させることを示している。
【0232】
精神分裂病に対するNAALADase阻害剤のin vivoアッセイ
ラットを、ヒトの精神病症状に相当する、半狂乱の走り及び絶え間ない頭部旋回(incessant head-turning)のような、PCP発症徴候を伴うように処置した。従って、精神分裂病に対するNAALADase阻害の作用を調べるために、PCP (5mg/kg)を受取る前のラットに、化合物1(50mg/kg)、化合物2(50mg/kg)又はビヒクル(H2O)を腹腔内投与した。頭の揺動(head rolling)スコアを、PCP注射後2時間にわたって測定した。結果を、図26及び27に図示した。これらの結果は、化合物1(図27)又は2(図26)の前投与は、PCPの歩行活動への作用を有意に低減した。PCPは、前前頭皮質においてグルタメート流出を増大することが示されており、かつPCP-誘発した歩行活動の減少は、NAALADase阻害が、シナプス前グルタメート生成活性を減弱することにより、PCPの行動に対する作用を緩和することを示唆している。従ってNAALADase阻害剤は、精神分裂病のPCPモデルにおいて有効であることを示した。
【0233】
下記実施例は、本発明を例証するものであるが、その限定を意図したものではない。特に記さない限り、全てのパーセントは最終組成物100質量%を基にしている。
【実施例1】
【0234】
実施例1
2-[[(2,3,4,5,6-ペンタフルオロベンジル)-ヒドロキシホスフィニル]-メチル]-ペンタン二酸の調製
スキームV:R1=2,3,4,5,6-ペンタフルオロベンジルヘキサメチルジシラザン(21.1mL, 100mmol)を、激しく攪拌しているホスフィン酸アンモニウム(8.30g, 100mmol)に添加し、かつ得られる懸濁液を、105℃で2時間攪拌した。その後2,3,4,5,6-ペンタフルオロ臭化ベンジル(5.0g, 27.0mmol)溶液を、懸濁液に0℃で滴下した。混合物を室温で19時間攪拌した。その後反応混合物を、ジクロロメタン(50mL)で希釈し、1N HCl(50mL)で洗浄した。有機相を分離し、Na2SO4上で乾燥し、かつ濃縮し、白色固形物4.72gを得た。これをジクロロメタン(50mL) に溶解し、かつこの溶液にベンジルアルコール(3.24g, 30mmol)を添加した。次に溶液に、1,3-ジシクロヘキシル-カルボジイミド(DCC) (6.19g, 30mmol)を0℃で添加し、懸濁液を室温で14時間攪拌した。溶媒を減圧除去し、残留物をEtOAc中に懸濁した。
得られた懸濁液をろ過し、ろ液を濃縮した。残留物を、シリカゲルクロマトグラフィー(ヘキサン:EtOAc、4:1から1:1)で精製し、2[[(2,3,4,5,6-ペンタフルオロベンジル)-ヒドロキシホスフィニル]-メチル]ペンタン二酸を、白色固形物として得た(収率34%)。
Rf 0.69 (i-PrOH:H2O, 7:3)
1H NMR (D2O):δ 1.8-2.0 (m, 3H), 2.1-2.3 (m, 1H), 2.3-2.5 (m, 2H), 2.7-2.9 (m, 1H), 3.29 (d, 2H)
元素分析
計算値:C13H12F5O6P、0.45 H2O: C, 39.20;H, 3.26
実測値:C, 39.17;H, 3.28
【実施例2】
【0235】
実施例2
2-(ホスホノメチル)ペンタンジオエートの調製
スキームIII
ジベンジル2-メチレンペンタンジオエート
ベンジルアクリレート(500g, 3.0mol)を、油浴中で100℃に加熱した。加熱を停止し、HMPT (10g, 61mmol)を滴下しながら、内部温度を140℃以下に維持した。
一旦添加を完了した後、混合物を攪拌し、室温に冷却した。シリカスラリー(5:1 ヘキサン/EtOAc)を添加し、混合物を、乾燥シリカ栓を備えたカラムに充填した。カラムを、1:1 ヘキサン/EtOAcで洗浄し、その画分を一緒にし、蒸発させ、透明な明るい金色の液体450gを得た。液体を、高真空(200μHg)下、185℃で蒸留し、無色透明の液体212g (42%)を得た。
1H NNR (CDCl3):7.3 ppm (m, 10H), 6.2 ppm (s, 1H), 5.6 ppm (s, 1H), 5.2 ppm (s, 2H), 5.1 ppm (s, 2H), 2.6 ppm (m, 4H)
【0236】
ジベンジル2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタンジオエートジクロロメタン350mLを溶媒とするジベンジルホスファイト(9.5g, 36mmol)を、0℃に冷却した。この攪拌している溶液に、トリメチルアルミニウム(18.2ml, ヘキサン中2.0M溶液, 36.4mmol)を添加した。30分後、ジクロロメタン90ml中のジベンジル2-メチレンペンタンジオエート(2, 6.0g, 37mmol)を、10分間かけて滴下した。その後無色透明の溶液を室温まで温め、一晩攪拌しながら放置した。その後混合物を、5%HClをゆっくり添加することにより反応停止した。更に1.5時間攪拌した後、下側の有機相を除去し、水相をジクロロメタン100mlで1回抽出した。有機相を一緒にし、乾燥し(MgSO4)、かつ蒸発させ、透明な明るい金色の液体を得た。液体を、シリカゲル(4cm*30cm)上でクロマトグラフィーにかけ、勾配(4:1-1:1)溶媒システム(ヘキサン/EtOAc)で溶離した。所望の生成物を含有する画分を一緒にし、蒸発させ、 ジベンジル2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタンジオエート (7.1g, 42%)を、無色透明の液体として得た。
その後液体を、Kughleror装置上で、66.7Pa(0.5mmHg)及び195〜200℃で蒸留した。留出物を廃棄し、残留している明るい金色の油分を、シリカゲル(1:1, ヘキサン/EtOAc)上でクロマトグラフィーにかけ、無色透明の油状物として2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタン二酸ジベンジル2.9gを得た。
TLC Rf 0.5 (1:1, ヘキサン/EtOAc)
1H NNR (CDCl3):7.1-7.4 (m, 20H), 5.05 (s, 2H), 4.8-5.03 (m, 6H), 2.8 (1H), 2.22-2.40 (m, 3H), 1.80-2.02 (m, 3H)
【0237】
2-(ホスホノメチル)ペンタン二酸
ベンジルペンタンジオエート (2.9g, 4.9mmol)を、10%Pd/Cを0.29g (6モル%)含有するメタノール20mlの混合物に添加した。この混合物を、Parr水素添加装置上で276kPa(40psi)で24時間水素化し、ろ過し、かつ蒸発させ、透明なわずかに金色がかった粘稠な油分(3, 1.0g, 90%)を得た。
1H-NNR (D2O):2.6-2.78 (m, 1H), 2.25-2.40 (m, 2H), 1.75-2.15 (m, 4H)
【実施例3】
【0238】
実施例3
2-[[[2-(カルボキシ)プロピル]ヒドロキシ-ホスフィニル]メチル]ペンタン二酸の調製
スキームX
ジ-t-ブチル2-メチレンペンタンジオエート
t-ブチルアクリレート(465g, 3.628mol)を、窒素下で100℃に加温し、その後ヘキサメチルリン酸トリアミド(10g, 61.2mmol)を滴下し、添加速度を反応温度100℃を維持するように調節した。反応混合物を冷却し、その後シリカ栓上に注ぎ(〜1000ml)、かつシリカを4:1 ヘキサン/酢酸エチルで完全に洗浄した。溶媒を減圧除去し、得られた油分を蒸留した。一部の物質を、高真空下室温から50℃で収集し、破棄した。その後温度を〜80℃に上昇し、生成物(300g, 65%、沸点は300μで67〜70℃)を、透明油状物として得た。
1H NMR (CDCl3):δ1.4 (m, 18H), 2.4 (t, 2H), 2.6 (t, 2H), 5.5 (s, 1H), 6.0 (s, 1H)
【0239】
ジ-t-ブチル2-[(ヒドロキシホスフィニル)メチル]-ペンタンジオエート
ホスフィン酸アンモニウム(162.6g, 1.96mol)及び1,1,1,3,3,3-ヘキサメチルジシラザン(316g, 1.96mol)の混合物を、105℃で2時間加熱した。反応混合物を、氷浴中で冷却し、かつジクロロメタン(1000ml)に溶解した2-メチレンペンタン-1,5-二酸 ジ-t-ブチル(251g, 0.979mol)を滴下した。反応混合物を一晩室温に加温した。その後反応混合物を、蒸留水(500ml)で急冷し、かつ有機相を保持した。水相を、ジクロロメタンで2回洗浄し、及び一緒にした有機相を硫酸マグネシウム上で乾燥した。その後溶媒を減圧除去し、わずかに黄色の油状物が残った(315g, 100%)。この生成物は、次の反応に使用するのに十分な純度であることが分かった。
1H NMR (CDCl3): 1.4 (m, 18H), 1.9 (m, 3H), 2.1 (m, 1H), 2.3 (m, 2H), 2.7 (m, 1H), 6.5 & 7.9 (d, 1H, P-H), 11.0 (s, 1H)
【0240】
ジ-t-ブチル2-[(t-ブトキシホスフィニル)メチル]-ペンタンジオエート
ジクロロメタン(1000ml)を溶媒とするジ-t-ブチル2-[(ヒドロキシホスフィニル)メチル]-ペンタン-1,5-ジオエート (315g, 0.977mol)を、氷浴で冷却し、かつ窒素下で、t-ブタノール(123.1g, 1.66mol)、4-ジメチルアミノピリジン(1g, 8.2mmol)、及び1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(281g, 1.47mol)を添加した。反応液を一晩攪拌した。反応混合物に水を添加し、ジクロロメタン相を保持しかつ乾燥し、溶媒を減圧下で除去した。得られる残留物を、カラムクロマトグラフィーで精製し、所望の生成物を1:1から2:3のヘキサン/酢酸エチルで溶離した。生成物を含有する画分を、減圧下で濃縮し、透明油状物が残留した(260g, 70%)。
1H NMR:(CDCl3): δ 1.4 (m, 27H), 1.8 (m, 1H), 1.9 (m, 2H), 2.1 (m, 1H), 2.3 (m, 2H), 2.7-2.8 (m, 1H), 6.7 & 8.0 (d, 1H, P-H)
【0241】
ジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]-t-ブトキシホスフィニル]メチル] ペンタンジオエート
THF(100ml)を溶媒とするジ-t-ブチル2-[(t-ブトキシホスフィニル)メチル]-ペンタン-1,5-ジオエート (13.62g, 36.0mmol)及びベンジルメタクリレート(6.35g, 36.0mmol)を窒素下で、水素化ナトリウム(0.14g, 油中60%分散体, 3.60mmol)を添加した。3時間後、反応混合物を水(300ml)に注ぎ、及びエーテル(100ml)を添加した。有機相を分離・保持し、水相を再度エーテル(100ml)で洗浄した。一緒にした有機抽出物を、硫酸マグネシウム上で乾燥し、溶媒を減圧除去した。得られた残留物を、カラムクロマトグラフィーで精製し、生成物を2:3 EtOAc/ヘキサンで溶離した。溶媒を減圧除去し、透明な油状物(10 .5g, 53%)を得た。
1H NMR (CDCl3):δ1.3 (m, 3H), 1.5 (m, 27H), 1.7 (m, 2H), 1.9 (m, 2H), 2.2 (m, 4H), 2.6 (m, 1H), 2.9 (m,1H), 5.1 (m, 2H), 7.3 (m, 5H)
【0242】
2-[[[2-(ベンジルカルボキシ)プロピル]ヒドロキシホスフィニル]-メチル]ペンタン二酸
ジクロロメタン(10ml)中のジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]t-ブトキシホスフィニル]メチル] ペンタン-1,5-ジオエート(1.6g, 2.89mmol)溶液に、窒素下で、トリフルオロ酢酸(10ml)を添加した。反応混合物を2時間攪拌し、その後減圧下で濃縮した。反応残留物に更にジクロロメタンを添加し、これを減圧下で除去した。生成物を、酢酸エチルに溶解し、水で洗浄し、その後有機相を硫酸マグネシウム上で乾燥し、溶媒を減圧除去し、透明な油状物を得た(800mg, 72%)。
1H NMR (D20):δ1.2 (m, 3H), 1.6-1.8 (m, 4H), 2.1 (m, 2H), 2.2 (m, 2H), 2.6 (m, 1H), 2.8 (m, 1H), 5.0 (m, 2H), 7.3 (m, 5H)
C17H22PO8・1.0 H2Oの分析 計算値:C, 50.50;H, 6.23
実測値:C, 50.52;H, 5.92。
【0243】
ジ-t-ブチル2-[[[2-(カルボキシ)プロピル]-t-ブトキシ-ホスフィニル]メチル]ペンタンジオエート
ジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]t-ブトキシホスフィニル]メチル] ペンタン-1,5-ジオエート (8.9g, 16.1mmol)、炭素触媒に担持されたパラジウム(10%, 1.0g)及び酢酸エチル(100ml)の溶液を、水素下(60psi)で16時間振盪した。反応混合物を、セライト上でろ過し、ろ液を減圧下で濃縮し、透明な油状分が残留した(7.5g, 100%)。
1H NMR (CDCl3):δ1.3 (d, 3H), 1.4-1.5 (m, 27H), 1.8 (m, 2H), 1.9 (m, 2H), 2.2 (m, 4H), 2.7 (m, 1H), 2.9 (m, 1H)
【0244】
2-[[[2-(カルボキシ)プロピル]ヒドロキシホスフィニル]メチル]-ペンタン二酸ジクロロメタン(10ml)を溶媒とするジ-t-ブチル2-[[[2-(カルボキシ)プロピル]t-ブトキシホスフィニル]メチル]-ペンタン-1,5-ジオエート (2.1g, 4.53mmol)に、窒素下で、トリフルオロ酢酸(10ml)を添加した。反応混合物を2時間攪拌し、その後減圧下で濃縮した。反応残留物に、追加のジクロロメタンを添加し、これを減圧下で除去した。得られた残留物をアセトニトリルで粉砕し、その後減圧下で乾燥し、濃厚な透明油状物が残留した(1.2g, 89%)。
1H NMR (D2O):δ 1.2 (d, 3H), 1.9 (m, 4H), 2.2 (m, 2H), 2.4 (m, 2H), 2.8 (m, 2H)。
C10H17PO8・0.2 CH3CNの分析の計算値:C, 41.03;H, 5.83
実測値:C, 41.05;H, 5.92。
【実施例4】
【0245】
実施例4
2-[({[ベンジルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸の調製
スキームXI
ジ-t-ブチル2-[((t-ブトキシ){[ベンジルアミノ]メチル}-ホスホリル)メチル]ペンタン-1,5-ジオエート
トルエン(200mL)を溶媒とする1,3,5-トリベンジルヘキサヒドロ-1,3,5-トリアジン(14.30g, 40.0mmol)及びジ-t-ブチル2-{[(t-ブトキシ)ホスホリル]メチル}ペンタン-1,5-ジオエート(37.85g, 100mmol)を、110℃で14時間攪拌した。溶媒を減圧除去し、残留する黄色油状物を、シリカゲルクロマトグラフィーで精製し(ヘキサン/酢酸エチル, 2/1)、明るい黄色油状物23.40gを得た(収率43%):1H NMR (CDCl3):δ 1.40-1.48 (m, 27H), 1.7-2.1 (m, 4H), 2.2-2.4 (m, 3H), 2.6-3.0 (m, 3H), 3.8-4.0 (m, 2H), 7.2-7.4 (m, 5H)。
【0246】
2-[({[ベンジルアミノ]メチル}(ヒドロキシホスフィニル))メチル]-ペンタン二酸
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-[((t-ブトキシ){[ベンジルアミノ]メチル}-ホスホリル)メチル]ペンタン-1,5-ジオエート (0.498g, 1.0mmol)溶液に、トリフルオロ酢酸(5mL)を、0℃で添加し、混合物を室温で18時間攪拌した。溶媒を減圧除去した。残留油状物を、ジクロロメタン(10mL)に溶解し、かつ濃縮した。この工程を3回繰り返し、トリフルオロ酢酸を完全に除去した。得られた油状物を、メタノールで結晶化し、白色固形物0.174gを得た(収率53%):1H NMR (D2O):δ 1.40-1.48 (m, 27H), 1.7-2.1 (m, 4H), 2.2-2.4 (m, 3H), 2.6-3.0 (m, 3H), 3.8-4.0 (m, 2H), 7.2-7.4 (m, 5H)。
【実施例5】
【0247】
実施例5
2-[({[フェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[フェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.4-1.6 (m, 1H), 1.7-1.9 (m, 3H), 2.2-2.4 (m, 2H), 2.2-2.4 (m, 2H), 2.5-2.7 (m, 1H), 3.53 (d, J = 8.8 Hz, 2H), 7.3-7.5 (m, 5H)。
【実施例6】
【0248】
実施例6
2-[({[4-フルオロフェニルアミノ]メチル}-(ヒドロキシホスフィニル)メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[4-フルオロフェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.5-1.7 (m, 1H), 1.8-2.0 (m, 3H), 2.3-2.5 (m, 2H), 2.6-2.7 (m, 1H), 3.84 (d, J = 9.0 Hz, 2H), 7.2-7.5 (4H)。
【実施例7】
【0249】
実施例7
2-[({[4-メトキシフェニルアミノ]メチル}-(ヒドロキシホスフィニル)メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[4-メトキシフェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.2-1.3 (m, 1H), 1.6-1.7 (m, 3H), 2.22-2.23 (m, 2H), 2.3-2.5 (m, 1H), 3.4 (d, J = 8.9 Hz, 2H), 3.7 (s, 314), 7.0 (d, J = 12 Hz, 2H), 7.4 (d, J= 12 Hz, 2H)。
【実施例8】
【0250】
実施例8
2-[({[フェニルスルホンアミド]メチル}-(ヒドロキシホスフィニル))メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[フェニルスルホンアミド]メチル}(ヒドロキシ-ホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.6-2.1 (m, 4H), 2.3-2.4 (m, 2H), 2.5-2.7 (m, 1H), 2.9-3.1 (m, 2H), 7.7-8.0 (m, 5H)。
【実施例9】
【0251】
実施例9
2-({[(フェニルカルボキシアミド)メチル]-(ヒドロキシホスフィニル)}メチル)ペンタン二酸の調製
スキームXII
ジ-t-ブチル2-{[(アミノメチル)(t-ブトキシ)-ホスホリル]メチル}ペンタン-1,5-ジオエート
ジ-t-ブチルエタノール(100mL)を溶媒とする2-[((t-ブトキシ)-{[ベンジルアミノ]メチル}ホスホリル)メチル]ペンタン-1,5-ジオエート (8.20g, 16.5mmol)溶液に、炭素上に担持したパラジウム(0.50g)を添加し、懸濁液を水素下で(50psi)4日間振盪した。触媒を、セライトパッドを通すろ過により除去した。ろ液を濃縮し、無色の油状物6.629g(収率99%)を得た: 1H NMR (CD3OD):δ 1.40-1.60 (m, 27H), 1.80-2.00 (m, 3H), 2.2-2.4 (m, 3H), 2.7-3.0 (m, 3H)。
【0252】
ジ-t-ブチル2-({(t-ブトキシ)[(フェニルカルボキシアミド)-メチル]ホスホリル}メチル)ペンタン-1,5-ジオエート
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-{[(アミノメチル)-(t-ブトキシ)ホスホリル]メチル}ペンタン-1,5-ジオエート (1.222g, 3.0mmol)及び塩化ベンゾイル(0.46mL, 4.0mmol)の溶液に、トリエチルアミン(0.56mL, 4.0mmol)を0℃で添加し、その後混合物を室温で16時間攪拌した。反応混合物を、ジクロロメタン(15mL)で希釈し、1N HCl(25mL)で洗浄し、Na2SO4上で乾燥し、濃縮した。粗物質を、シリカゲルクロマトグラフィー上で精製し(酢酸エチル/ヘキサン = 2/1)、無色の油状物1.259g(収率74%)を得た:1H NMR (CDCl3):δ1.30-1.60 (m, 27H), 1.60-2.00 (m, 3H), 2.20-2.40 (m, 3H), 2.70-2.90 (m, 3H), 3.5-4.2 (m, 2H), 7.0-7.3 (m, 1H), 7.4-7.6 (m, 3H), 7,8-7.9 (m, 1H)。
【0253】
2-({[(フェニルカルボキシアミド)メチル](ヒドロキシホスフィニル)}-メチル)ペンタン二酸
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-({(t-ブトキシ)-[(フェニルカルボキシアミド)メチル]ホスホリル}メチル)ペンタン-1,5-ジオエート (1.230g, 2.4mmol)の溶液に、トリフルオロ酢酸(5mL)を室温で添加し、かつ混合物を室温で18時間攪拌した。溶媒を減圧除去した。残留油状物を、ジクロロメタン(10mL)に溶解し、濃縮した。この方法を、3回繰り返し、トリフルオロ酢酸を完全に除去した。得られた油状物を、アセトニトリル-水で結晶化し、白色固形物0.620gを得た(収率75%):1H NMR (D2O):δ1.9-2.1 (m, 3H), 2.2-2.4 (m, 1H), 2.4-2.6 (m, 2H), 2.8-3.0 (m, 1H), 3.7-3.9 (m, 2H), 7.5-7.9 (m, 5H)。
【実施例10】
【0254】
実施例10
2-(2-スルファニルエチル)ペンタン二酸の調製
スキームXIII、R10=水素
3-(2-オキソテトラヒドロ-3-チオフェニル)プロパノエート
【0255】
【0256】
冷却したTHF(100ml)を溶媒とするリチウムジイソプロピルアミド(LDA) (98mmol)の溶液(-78℃)に、γ-チオブチロラクトン(10g, 98mmol)を滴下した。15分間攪拌した後、3-ブロモプロパン酸エチル(35.4g, 196mmol)を添加し、反応液を一晩室温で加温した。溶媒を減圧除去し、得られた残留物を、カラムクロマトグラフィーにかけ、透明な油状物3g(16%)を得た。1H NMR (CDCl3):δ 1.2 (t, 3H), 1.7 (m, 1H), 1.9 (m, 1H), 2.1 (m, 1H), 2.4 (t, 2H), 2.5 (m, 2H), 3.3 (t, 2H), 4.2 (q, 2H)。
【0257】
2-(2-スルファニルエチル)ペンタン二酸
【0258】
【0259】
THF(5ml)を溶媒とする3-(2-オキソテトラヒドロ-3-チオフェニル)プロパノエート(0.77g, 3.81mmol)の溶液に、水酸化ナトリウム(水中1M、5ml)を添加した。混合物を2日間攪拌し、その後THFを減圧除去し、水相をエーテルで洗浄し、その後HClでpH1とし、酢酸エチルで抽出した。一緒にした酢酸エチル抽出物を、硫酸マグネシウム上で乾燥し、溶媒を減圧除去した。得られた残留物を、カラムクロマトグラフィーで精製し、透明油状物150mgを得た(20%)。1H NMR (d6-DMSO) δ 1.7 (m, 3H), 1.8 (m, 1H), 2.2 (m, 2H), 2.3-2.5 (m, 4H)。
C7H12SO4の分析の計算値:C, 43.74;H, 6.29;S, 16.68
実測値:C, 43.61;H, 6.39;S, 16.55。
【実施例11】
【0260】
実施例11
2-(3-スルファニルプロピル)ペンタン二酸の調製
スキームXIX
2,2-ジメチル-5-[3-[(トリフェニルメチル)チオ]プロピル-1,3-ジオキサン-4,6-ジオン(I)
20mmolの3-[(トリフェニルメチル)チオ]プロピオン酸(6.9g)を、CH2Cl2の100mlを溶媒とするメルドラム(Meldrum)酸(2,2-ジメチル-1,3-ジオキサン-4,6-ジオン)の22mmol (3.2 g)及び4-ジメチルアミノピリジン31mmol (3.85g)で溶解した。
反応混合物を、-5℃に冷却し、CH2Cl2の50mlを溶媒とするジシクロヘキシルカルボジイミド(4.74g)22mmolの溶液を、1時間かけて滴下した。混合物を、温度<0℃に一晩放置し、その間ジシクロヘキシル尿素の微結晶が析出した。ろ過後、反応混合物を、4x 10% KHSO4、1x ブラインで洗浄し、MgSO4で2時間乾燥した。この溶液を、特徴決定も更なる精製もせずに第二工程に用いた。
【0261】
先の反応の溶液を、-5℃に冷却し、かつ98%酢酸13.3ml(220mmol)を添加した。
その後NaBH4の1.85g(50mmol)を、少量ずつ添加しながら、1時間以上攪拌した。
この反応混合物を一晩冷蔵庫に放置し、その後3x水及び2xブラインで洗浄した。
有機相を、MgSO4で乾燥し、ろ過し、かつ蒸発乾固した。残留物をEtOAcに溶解し、沈殿した少量のジシクロヘキシル尿素をろ過により除去し、ろ液をヘキサンを添加して結晶化した。2,2-ジメチル-5-[3-[(トリフェニルメチル)-チオ]プロピル]-1,3-ジオキサン-4,6-ジオン(I)の収量は7.5gであった(86%-2工程)。13C-NMR δ 20.0(q), 26.2(q), 27.2(t), 28.9(t), 32.0(t), 46.2(d), 67.0(s), 105.3(s), 127.0(d), 128.3(d), 130.0(d), 145.2(s), 165.6(s)。
【0262】
2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニルメチル)チオ]-プロピル] -1,3-ジオキサン-5-プロパン酸メチルエステル(II)
5mmolの2,2-ジメチル-5-[3-[(トリフェニルメチル) -チオ]-プロピル]-1,3-ジオキサン-5-プロパン酸-4,6-ジオン(I) (2.3g)を、3-ブロモプロパン酸メチル(3.34g = 2.18ml) 20mmol及びメタノール10mlを溶媒とするメトキシドナトリウム(20mmol)の4.37Nメタノール溶液4.6ml中に溶解した。反応混合物を、60℃で一晩加熱し、その後ヘキサン/酢酸エチル1:1でTLCを行ったところ、出発材料は検出されなかった。その後混合物を蒸発乾固し、10%KHSO4水溶液40mlと混合した。有機物質を、3部のEtOAcで抽出し、有機相を一緒にし、MgSO4で乾燥し、蒸発させた。残留物を、ヘキサン/酢酸エチルで結晶化し、2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニル-メチル)チオ]プロピル]-1,3-ジオキサン-5-プロパン酸メチルエステル(II)2.2g(77%)を得た。13C-NMR (CDCl3) δ 24.6, 29.4, 29.5, 29.6, 31.4, 32.6, 37.7, 51.9, 52.8, 66.8, 105.7, 126.7, 127.9, 129.5, 144.7, 168.4, 172.0。
【0263】
6-[(トリフェニルメチル)チオ]-1,3,3-ヘキサントリカルボン酸(III)
2.56mmolの2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニルメチル)チオ]プロピル]-1,3-ジオキサン-5-プロパン酸メチルエステル(II) (1.4g)を、水酸化ナトリウム18mmol(0.72g)と共に、1,4-ジオキサン5ml及び水5mlの混合物に溶解した。その後混合物を、100℃で1時間加熱し、蒸発乾固し、水に溶解し、かつ1M硫酸の添加により沈殿させた。沈殿をろ過により除去し、水で洗浄し、かつデシケータで乾燥した。6-[(トリフェニルメチル)-チオ]-1,3,3-ヘキサントリカルボン酸(III) 1.36gを得た(〜100%)。13C-NMR (MeOH) δ 25.4, 29.2, 30.7, 33.5, 33.7, 58.0, 68.3, 128.1, 129.3, 131.2, 146.7, 174.9, 176.9。
【0264】
6-[(トリフェニルメチル)チオ]-1,3-ヘキサンジカルボン酸(IV)
ジメチルスルホキシド5mlに、2.56mmolの6-[(トリフェニルメチル)チオ]-1,3,3-ヘキサントリカルボン酸(III) (1.36g)を溶解し、溶液を100℃で1時間加熱し、蒸発乾固し、水に溶解し、かつ1M硫酸の添加により沈殿した。沈殿した油状分を、超音波浴で1時間処理した後に固化した。固形物をろ過し、水で洗浄し、デシケータで乾燥した。6-[(トリフェニルメチル)-チオ]-1,3-ヘキサンジカルボン酸(IV) 1.1g (IIから2工程で89%)を得た。13C-NMR (MeOH) δ 27.9, 28.6, 33.0 (炭素2個), 33.1, 45.9, 68.1, 128.1, 129.2, 131.2, 146.8, 177.1, 179.4。
【0265】
2-(3-スルファニルプロピル)ペンタン二酸(V)
2.46mmolの6-[(トリフェニルメチル)チオ]-1,3-ヘキサンジカルボン酸(IV) (1.1g)を5mmolのトリイソプロピルシラン(0.79g)と共に、3ml CH2C13/3ml トリフルオロ酢酸の混合物に溶解し、室温で1時間静置した。その後混合物を蒸発乾固し、3x ヘキサンで洗浄した。残留している油状残留物を、水に溶解し、ろ過し、かつ凍結乾燥し、2-(3-スルファニルプロピル)ペンタン二酸(V) 0.35g (76%)を得た。13C-NMR (MeOH) δ 25.2 (t) , 28.8 (t), 32.4 (t), 33.0(t), 33.2(t), 45.9(d), 177.2(s), 179.6(s)。
【実施例12】
【0266】
実施例12
2-(4-スルファニルブチル)ペンタン二酸の調製
2-(4-スルファニルブチル)ペンタン二酸を、先に2-(3-スルファニルプロピル)ペンタン二酸について説明した方法を用いて調製した。13C-NMR (MeOH) δ 25.1(t), 27.4(t), 28.8(t), 33.0(t), 33.2(t), 35.4(t), 46.3(d), 177.2(s), 179.7(s)。
【実施例13】
【0267】
実施例13
2-(3-スルファニル-2-メチルプロピル)-ペンタン二酸の調製
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸(2種のジアステレオ異性体の混合物)を、先に2-(3-スルファニルプロピル)-ペンタン二酸について説明した方法を用いて調製した。13C-NMR (MeOH) δ 18.9(q), 19.5(q), 29.1(t), 29.6(t), 31.7(t), 32.6(t), 32.9(t), 33.0(t), 35.5(d), 35.9(d), 39.2(t), 39.7(t), 44.2(d), 44.3(d), 177.0(s), 177.1(s), 179.7(s), 179.9(s)。
【実施例14】
【0268】
実施例14
2-(2-スルファニルプロピル)ペンタン二酸及び2-(3-スルファニルプロピル)ペンタン二酸を、これらの各実施例並びに前述のin vitro及びin vivoアッセイにおいて試験した。両化合物は各々、これらの各アッセイ及び実施例においてin vitro又はin vivo活性を示すことが分かった。
【0269】
本発明はこのように説明してきたが、多くの方法で改変することができることは明らかであろう。このような改変は、本発明の精神及び範囲の逸脱とはみなされず、このような修飾は全て、上記「特許請求の範囲」の範囲に含まれることが意図されている。
【技術分野】
【0001】
本件出願は1998年7月6日に出願された米国特許出願第09/110,262号、及び1999年1月12日に出願された米国特許出願第09/228,391号(これは順に1998年7月6日に出願された米国特許出願第09/110,186号の一部継続出願である)の一部継続出願である。
本発明はN-アセチル化α-結合酸性ジペプチダーゼ(NAALADase)インヒビター、このようなインヒビターを含む医薬組成物及びNAALADase酵素活性を抑制し、それにより神経細胞活性に影響し、血管形成を抑制し、グルタメート異常、強迫性障害、前立腺疾患、痛み及び糖尿病性神経障害を治療するためのそれらの使用方法に関する。
【背景技術】
【0002】
最近の研究によりグルタメート介在性障害の病因にNAALADaseが関係づけられている。筋萎縮性側索硬化症(ALS)の患者からの死後の組織に関する神経病理学研究は神経変性と関連して生じるN-アセチルアスパルテート(NAA)及びN-アセチルアスパルチルグルタメート(NAAG)組織濃度の大きい減少、並びにALSの患者からの脳脊髄液(CSF)中のNAA及びNAAGの増加を示す。一致して、異常なNAAGレベル及びNAALADase活性がまた精神分裂症患者の死後の前頭葉前部及び大脳辺縁の脳組織中で観察されていた。また、検死研究はNAAG/NAAとアルツハイマー病の間の強い相関関係を示唆する。死後の脳組織では、NAAレベル及びNAAGレベルがアルツハイマー病因により冒された脳領域(海馬及び扁桃)中で選択的に減少されることがわかった。
【0003】
グルタメートは中枢神経系(CNS)中で主たる興奮性神経伝達物質として利用できる。神経細胞は卒中又は心臓発作の如き虚血性脳障害中に起こるようにそれらが酸素を減少される場合にグルタメートを多量に放出する。グルタメートのこの過剰の放出は順にN-メチル-D-アスパルテート(NMDA)受容体、AMPA受容体、カイネート受容体及び代謝調節型グルタメート(mGlu)受容体の過剰刺激(興奮毒性)を生じる。グルタメートがこれらの受容体に結合する場合、受容体中のイオンチャンネルが開き、又は第二メッセンジャー系が刺激されて、それらの細胞膜を横切るイオンの流れ、例えば、細胞へのCa2+及びNa+流入及び細胞からのK+流出を可能にする。イオンのこれらの流れ、特にCa2+のインフラックスが神経細胞の過剰刺激を生じる。過剰刺激された神経細胞が更に多くのグルタメートを分泌し、プロテアーゼ、リパーゼ及び遊離基の生成を介して細胞死滅を最終的にもたらすと考えられるドミノ効果を生じる。
【0004】
グルタメート受容体の過剰活性化が脊髄損傷、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、糖尿病性神経障害、急性及び慢性の痛み、虚血並びに低酸素症、低血糖症、虚血、トラウマ、及び神経傷害後の神経細胞損失を含む種々の神経の疾患及び症状に関係づけらている。
特に、グルタメート作用性(glutamatergic)異常が神経分裂症と関連していた。例えば、フェニルシクリジン(PCP)及びN-メチル-D-アスパルテート(NMDA)受容体のその他のアンタゴニストは健康な個体に精神異常作用性を誘発し、精神分裂症の既存の症候を悪化し、グルタメート伝達の低下が精神分裂症に寄与し得ることを示唆する。更に、非NMDA受容体のアンタゴニスト又はグルタメート放出を減少する前処理がNMDA受容体アンタゴニストの記憶力増進効果及びその他の挙動効果を低下することが報告されていた。また、研究はmGlu受容体の或る種のサブタイプの刺激がシナプス前抑制を媒介し、グルタメートの誘発放出を減少することを示していた。1998年に、mGlu受容体アゴニストがラットでPCP誘発グルタメート外向きフラックスを減少することが報告され、アゴニストがシナプス前グルタメート作用活性を軽減することによりPCPの挙動効果を回復することが示唆された。
【0005】
また、最近の研究が強迫性障害、特に薬物依存性についてグルタメート作用の基礎を進歩していた。例えば、エタノールの神経生理学的効果及び病理学的効果がグルタメート作用系により媒介されることがわかった。詳しくは、エタノールへの急な暴露がグルタメート受容体中のチャンネル中のイオンの流れを抑制することによりグルタメート作用性神経伝達を分断し、一方、慢性暴露がグルタメート受容体の数をアップレギュレートし、それによりイオンの流れを増大する。エタノールの急性禁断症状がアップレギュレートされたチャンネルの存在下で高興奮性及び発作をもたらし、それによりシナプス後神経細胞を興奮毒性損傷を受けやすくする。
アルコール中毒患者からの組織学的に正常な脳の死後の試験は慢性アルコール中毒が前頭皮質中のグルタメート受容体のNMDAサブタイプの密度を中程度に増大することを示していた。このアップレギュレーションはエタノール誘発慢性神経毒性の段階に相当し得る。このようなものとして、中毒、禁断症状発作、振せんせん妄、ベルニケ−コルサコフ症候群及び胎児アルコール症候群を含む、アルコール中毒の神経生物学的効果が、グルタメート作用系に関するエタノールの効果の結果のスペクトルとして理解し得る。これに関して、アルコール中毒はグルタメート関連神経障害の拡大するファミリーの別の員と考えられるかもしれない。
【0006】
また、グルタメート作用系はその他の乱用薬物の挙動効果に関係づけられている。例えば、研究はグルタメート作用性アンタゴニストがアンフェタミン及びコカインにより誘発される運動刺激活性を阻止し、グルタメート作用性アゴニストがアンフェタミンにより生じられるステレオタイプと同じステレオタイプを生じることを示していた。これらの結果は精神運動刺激物質のステレオタイプ効果の発現がグルタメート作用系をともなうという薬理学的証拠に相当する。
疫学的研究は薬物依存性とその他の強迫性障害の間の強い相関関係を明らかにしていた。更に、共通の遺伝子異常がアルコール中毒、コカイン依存性、ニコチン依存性、病的ギャンブル、注意欠乏障害(ADD)、トゥレット症候群、強迫性過食症及び肥満の人々の間に見られた。このような障害は興奮毒性の効果の発現であると考えられる。
【0007】
上記知見に基づいて、本発明者らは試験し、NAALADaseインヒビターがグルタメート異常、例えば、薬物依存性、糖尿病性神経障害、痛み及び精神分裂症の薬物療法に効力があることを見出した。
現在までの殆どの研究活動及び開発活動はNMDAアンタゴニスト、グリシンアンタゴニスト、及びその他の後シナプス興奮性アミノ酸(EAA)受容体ブロッカーの如き化合物で後シナプスグルタメート受容体をブロックすることに集中していた。不運なことに、これらの薬剤は通常の条件下でさえもひどい毒性を生じ、こうしてそれらの臨床使用を制限する。如何なる一つの特別な理論にも限定されないが、NAALADaseインヒビターは後シナプスグルタメート受容体と相互作用しないでシナプス前グルタメート放出を阻止すると考えられる。NAALADaseインヒビターは基礎グルタメートレベルを変化しないことが明らかであるので、それらは後シナプスグルタメートアンタゴニストと関連する挙動毒性がないかもしれない。
【0008】
グルタメートに加えて、NAALADaseはまた前立腺特異性膜抗原(PSMA)とも関連づけられている。特に、PSMA cDNAがNAALADase活性を与えること、またNAALADase及びPSMAが少なくとも86%の相同配列同一性を示すことが示されていた。Carterら, Proc. Natl. Acad. Sci., 93巻, 749-753頁(1996)を参照のこと。PSMAの分子クローニングが潜在的な前立腺癌マーカーとして報告されており、前立腺癌の像形成方法及び細胞傷害治療方法の標的として利用できると仮定されていた。更に、PSMA抗体、特にインジウム-111標識PSMA抗体及びイットリウム標識PSMA抗体が記載されており、前立腺癌の診断及び治療について臨床試験されていた。PSMAは前立腺管上皮中で発現され、清漿、前立腺液及び尿中に存在する。
【0009】
本発明者らはNAALADaseインヒビターが前立腺疾患、特に前立腺癌を治療するのに有効であることを見出した。如何なる特別な理論にも制限されないが、NAALADaseインヒビターはPSMA活性を抑制するものと考えられる。PSMAのmAbは23の非前立腺癌を標的とすることがわかっていたので(Luiら, Science Research, 57巻, 3629-34 頁(1997))、本発明者らはNAALADaseインヒビターがまた特にNAALADaseがある組織、例えば、脳、腎臓及び睾丸中で非前立腺癌を治療するのに有効であろうと仮定する。
また、NAALADaseは新生血管系(新しい血管)中に見られた。本発明者らはNAALADaseインヒビターが新生血管系の増殖(血管形成)を抑制又は阻止し、それにより血管形成に依存する疾患を治療するのに潜在的な治療適用を与えることを発見した。血管形成依存性疾患の例として、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、及び皮膚潰瘍が挙げられるが、これらに限定されない。血管形成はまた正常な生理プロセス、例えば、成長、生殖能及び軟組織創傷治癒に必須である。
【0010】
癌は血管形成に依存性の別の疾患である。癌腫瘍細胞は内皮細胞付近で活性化する血管形成物質を分泌又は放出する。これらの内皮細胞は新しい血管の形成で極に達する挙動の細胞自律パターンを発現することにより応答する。研究は血管形成が癌腫瘍の増殖、浸潤及び転移を持続するのに必要であることを実証していたので、NAALADaseインヒビターの新生血管系抑制活性は全ての型の癌を治療する際のそれらの実用性を更に支持する。
数年前まで、わずかにいくつかのNAALADaseインヒビターが同定されているにすぎず、それらが非臨床研究に使用されていた。これらの化合物の例として、一般のメタロペプチダーゼインヒビター、例えば、o-フェナントロリン、金属キレーター、例えば、EGTA及びEDTA、並びにペプチド類似体、例えば、キスカリン酸及びβ-NAAGが挙げられる。これらの化合物は毒性副作用を有し、又は医薬有効量で投与し得ない。広範囲の潜在用途に鑑みて、新規NAALADaseインヒビター、このようなインヒビターを含む医薬組成物、及びそれらの使用の方法に対する要望がある。
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は神経細胞活性に作用し、血管形成を抑制し、グルタメート異常、強迫性障害、前立腺疾患、痛み及び糖尿病性神経障害を治療するのに有益なN-アセチル化α-結合酸性ジペプチダーゼ(NAALADase)酵素インヒビター及び組成物に関する。
【課題を解決するための手段】
【0012】
更に詳しくは、本発明は一般式I
【0013】
【0014】
の化合物又は医薬上許される均等物に関する。
式中、
Xは式II、III又はIV
【0015】
である部分であり、
m及びnは独立に0、1、2、3又は4であり、
ZはSR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり、
BはN又はCR16であり、
AはO、S、CR17R18又は(CR17R18)mSであり、
R9及びR13は水素であり、
R8、R10、R11、R12、R14、R15、R16、R17及びR18は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換、又は一つ以上の置換基で置換されており、
但し、Xが式IIである部分であり、かつAがOである場合、nが2、3又は4であり;Xが式IIである部分であり、かつAがSである場合、nが2、3又は4であり;またXが式IIである部分であり、かつAが(CR17R18)mSである場合、nが0、2、3又は4であることを条件とする。
【0016】
更に、本発明は卒中の発症の60分よりも後に投与された時に哺乳類の卒中を治療するのに有効である硫黄及び酸基の両方を含む化合物に関する。
また、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のNAALADase酵素活性の抑制方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のグルタメート異常の治療方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の強迫性障害、卒中、髄鞘脱落疾患、精神分裂症、パーキンソン病及びALSからなる群から選ばれたグルタメート異常の治療方法に関する。
【0017】
また、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の神経細胞活性に作用する方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の前立腺疾患の治療方法に関する。
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の癌の治療方法に関する。
また、本発明は哺乳類に有効量の硫黄及び酸基の両方を含む化合物を卒中の発症の60分より後に投与することを特徴とする哺乳類の卒中の治療方法に関する。
【0018】
更に、本発明は哺乳類に有効量の硫黄及び酸基の両方を含む化合物を卒中の発症の60分より後に投与することを特徴とする哺乳類の卒中の治療方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の血管形成の抑制方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の痛みの治療方法に関する。
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の糖尿病性神経障害の治療方法に関する。
更に、本発明はチオラクトンを置換エステルと反応させて式VI
【0019】
【0020】
の化合物を生成する工程を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Dは(CR21R22)nであり、
nは0、1、2、3又は4であり、かつ
R20、R21及びR22は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されている。
【0021】
また、本発明は
(i)メルドラム酸を硫黄含有反応体と反応させてメルドラム酸硫黄含有誘導体を生成する工程、及び
(ii)前記メルドラム酸硫黄含有誘導体をエステルと反応させて式VII
【0022】
【0023】
の化合物を生成する工程
を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Eは硫黄含有部分であり、かつ
Fはプロピオン酸エステル含有部分である。
最後に、本発明は
(i)有効量の式Iの化合物、及び
(ii)医薬上許される担体
を含むことを特徴とする医薬組成物に関する。
【図面の簡単な説明】
【0024】
【図1A】免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図1B】アスコルビン酸による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図1C】アスコルビン酸及び化合物3による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2A】免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2B】アスコルビン酸による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図2C】アスコルビン酸及び化合物2による処理後の免疫染色された背根神経節−シュワン細胞同時培養液の顕微鏡写真である。
【図3】種々の投与量の化合物3による皮下治療後の日数に対しin vivo平均LNCaP腫瘍体積をプロットするグラフである。
【図4】LNCaP細胞による注射後にビヒクル又は化合物3で皮下治療したマウス中の腫瘍:対照比をプロットする棒グラフである。
【図5】種々の投与量の化合物3による皮下治療後の日数に対しin vivo平均ダンニングG腫瘍体積をプロットするグラフである。
【図6】ダンニングG細胞による注射後にビヒクル又は化合物3で皮下治療したラット中の腫瘍:対照比をプロットする棒グラフである。
【図7】種々の投与量の化合物3による腫瘍内治療後の日数に対しin vivo平均ダンニングG腫瘍体積をプロットするグラフである。
【図8A】マウスに皮下注射され、血管形成因子の注射後にビヒクル単独で処理されたマトリゲルTMプラグの顕微鏡写真の組である。
【図8B】マウスに皮下注射され、血管形成因子の注射後に毎日3mg/kgの投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真の組である。
【図8C】マウスに皮下注射され、血管形成因子の注射後に毎日30mg/kgの投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真の組である。
【図9】マウスに皮下注射され、血管形成因子の注射後に連続投与濃度のビヒクル単独で処理されたマトリゲルTMプラグの顕微鏡写真である。
【図10】マウスに皮下注射され、血管形成因子の注射後に1μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図11】マウスに皮下注射され、血管形成因子の注射後に10μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図12】マウスに皮下注射され、血管形成因子の注射後に100μg/日の連続投与量の化合物3で治療されたマトリゲルTMプラグの顕微鏡写真である。
【図13】化合物2による治療後の日数に対し糖尿病ラットの逃避待ち時間をプロットするグラフである。
【図14】治療後の時間に対しビヒクル又は化合物3で治療されたラットのホルマリン誘発身もだえ挙動をプロットするグラフである。
【図15】ラットを治療したビヒクル又は化合物3の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図16】ラットを治療したビヒクル又は化合物2の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図17】ラットを治療したビヒクル又は化合物1の投与量に対しラットの酢酸誘発身もだえをプロットする棒グラフである。
【図18】投薬後の日数に対しビヒクル又は化合物3で治療したラットの慢性結さつ損傷誘発痛覚過敏症をプロットする棒グラフである。
【図19A】STZによる投与後の日数に対し、ビヒクル又は化合物2で治療した非糖尿病ラットSTZ糖尿病ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図19B】STZによる投与後の日数に対し、ビヒクル又は化合物1で治療した非糖尿病ラットSTZ糖尿病ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図20】手術後の日数に対し、正常な(手術されなかった)ラット及びビヒクル又は化合物3で治療した慢性結さつ損傷誘発ラットの逃避待ち時間有意差スコアーをプロットする棒グラフである。
【図21A】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物2で治療したSTZ糖尿病ラットの運動神経伝導速度をプロットする棒グラフである。
【図21B】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物2で治療したSTZ糖尿病ラットの知覚神経伝導速度をプロットする棒グラフである。
【図22A】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物1で治療したSTZ糖尿病ラットの運動神経伝導速度をプロットする棒グラフである。
【図22B】STZによる投与後の日数に対し、非糖尿病ラット及びビヒクル又は化合物1で治療したSTZ糖尿病ラットの知覚神経伝導速度をプロットする棒グラフである。
【図23】コカインと一緒の化合物3、コカインを含む食塩水、及び食塩水を含む食塩水による治療後の日数に対しラットのコカイン(20mg/kg)自発運動をプロットするグラフである。
【図24】治療の週数に対し、非糖尿病ラット及びビヒクル、化合物1又は化合物2で治療したBB/W糖尿病ラットの逃避待ち時間をプロットするグラフである。
【図25】治療の週数に対し、非糖尿病ラット及びビヒクル、化合物1又は化合物2で治療したBB/W糖尿病ラットの神経伝達速度をプロットするグラフである。
【図26】PCP投与後の時間に対し水又は化合物2で更に治療したPCP処理ラットの平均頭部ローリングスコアーをプロットするグラフである。
【図27】PCP投与後の時間に対し水又は化合物1で更に治療したPCP処理ラットの平均頭部ローリングスコアーをプロットするグラフである。
【発明を実施するための形態】
【0025】
定義
“酸基”として、-COOH、-SO3H、-SO2HNH、-PO2H2、-CN、-PO3H2、-SH、-NHCOH、-NH2、-CONH2、-CONHOH、-CONHNHSO2H、-COHNSO2H、及び-CONHCNが挙げられるが、これらに限定されない。
“化合物1”は2-(2-スルファニルエチル)ペンタン二酸、又は実施例10により調製された化合物の純粋な形態及び不純な形態を表す。
“化合物2”は2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕-ペンタン二酸を表す。
“化合物3”は2-(ホスホノメチル)-ペンタン二酸(PMPA)を表す。
【0026】
“有効量”は所望の効果を生じるのに必要とされる量を表す。“治療有効量”はNAALADase酵素活性を抑制し、神経細胞活性に作用し、血管形成を抑制し、かつ/又はグルタメート異常、強迫性障害、前立腺疾患、痛み及び/又は糖尿病性神経障害を治療するのに必要とされる量を表す。
“IP”又は“i.p.”は腹腔内を表す。
“アイソスター”は同様の又は同じ物理的性質を有する元素、分子又はイオンを表す。典型的には、2種のアイソスター分子は同様の又は同じ嵩及び形状を有する。理想的には、アイソスター化合物は同形であるべきであり、同時結晶化することができるべきである。アイソスター化合物が通常共有するその他の物理的性質の中に、沸点、密度、粘度及び伝熱性がある。しかしながら、或る種の性質は通常異なっている:双極子モーメント、極性、偏光、サイズ及び形状。何とならば、外部軌道が異なって混成され得るからである。“アイソスター”という用語は“バイオアイソスター”を含む。
【0027】
“バイオアイソスター”は、それらの物理的類似性に加えて、幾つかの共通の生物学的性質を共有するアイソスターである。典型的には、バイオアイソスターは同じ認識部位と相互作用し、又は広く類似する生物学的効果を生じる。
“カルボン酸アイソスター”として、直接誘導体、例えば、ヒドロキサム酸、アシル−シアナミド及びアシルスルホンアミド;平面酸性複素環、例えば、テトラゾール、メルカプトアゾール、スルフィニルアゾール、スルホニルアゾール、イソオキサゾール、イソチアゾール、ヒドロキシチアジアゾール及びヒドロキシクロム;並びに非平面の硫黄又はリン誘導酸性官能基、例えば、ホスフィネート、ホスホネート、ホスホンアミド、スルホネート、スルホンアミド、及びアシルスルホンアミドが挙げられるが、これらに限定されない。
【0028】
“誘導体”は別の物質から直接に、又は修飾もしくは部分置換により生成された物質を表す。
“メルドラム酸”は2,2-ジメチル-1,3-ジオキサン-4,6-ジオンを表す。 “代謝産物”は代謝又は代謝プロセスにより生成された物質を表す。 “医薬上許される均等物”として、医薬上許される塩、水和物、代謝産物、プロドラッグ及びカルボキシルアイソスターが挙げられるが、これらに限定されない。多くの医薬上許される均等物が式I-Vの化合物と同じ又は同様のin vitro又はin vivo活性を有すると予想される。
【0029】
“医薬上許される塩”は所望の薬理学的活性を有し、かつ生物学上又はそれ以外に望ましくないことはない本発明の化合物の塩を表す。塩は無機酸により生成でき、例えば、酢酸塩、アジピン酸塩、アルギン酸塩、アスパラギン酸塩、安息香酸塩、ベンゼンスルホン酸塩、硫酸水素塩、酪酸塩、クエン酸塩、ショウノウ塩、ショウノウスルホン酸塩、シクロペンタンプロピオン酸塩、ジグルコン酸塩、ドデシル硫酸塩、エタンスルホン酸塩、フマル酸塩、グルコヘプタン酸塩、グリセロリン酸塩、半硫酸塩、ヘプタン酸塩、ヘキサン酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、2-ヒドロキシエタンスルホン酸塩、乳酸塩、マレイン酸塩、メタンスルホン酸塩、2-ナフタレンスルホン酸塩、ニコチン酸塩、シュウ酸塩、チオシアン酸塩、トシレート及びウンデカン酸塩が挙げられる。塩基塩の例として、アンモニウム塩、アルカリ金属塩、例えば、ナトリウム塩及びカリウム塩、アルカリ土類金属塩、例えば、カルシウム塩及びマグネシウム塩、有機塩基との塩、例えば、ジシクロヘキシルアミン塩、N-メチル-D-グルカミン塩、並びにアミノ酸、例えば、アルギニン及びリシンとの塩が挙げられる。また、塩基性窒素含有基が薬剤で四級化し得る。このような薬剤として、低級アルキルハライド、例えば、メチル、エチル、プロピル、及びブチルのクロリド、ブロミド及びヨージド;ジアルキルスルフェート、例えば、ジメチルスルフェート、ジエチルスルフェート、ジブチルスルフェート及びジアミルスルフェート;長鎖ハライド、例えば、デシル、ラウリル、ミリスチル及びステアリルのクロリド、ブロミド及びヨージド;並びにアラルキルハライド、例えば、ベンジルブロミド及びフェネチルブロミドが挙げられる。
【0030】
“医薬上許されるプロドラッグ”はその一つ以上の薬理学的効果を示す前に生体内変換を受ける本発明の化合物の誘導体を表す。プロドラッグは改良された化学安定性、改良された患者許容及びコンプライアンス、改良された生物利用能、延長された作用の期間、改良された臓器選択性、改良された製剤化(例えば、増大された水溶性)、及び/又は低下された副作用(例えば、毒性)の一つ以上の目的で製剤化される。プロドラッグは当業界で知られている方法、例えば、Burger's Medicinal Chemistry and Drug Chemistry, 第5編, 1巻,172-178頁, 949-982頁(1995)により記載された方法、又は当業者に容易に明らかな方法を使用して本発明の化合物から容易に調製し得る。例えば、本発明の化合物は一つ以上のヒドロキシ基又はカルボキシ基をエステルに変換することによりプロドラッグに変換し得る。
“放射性感作物質”は電磁放射線で治療し得る疾患の治療を促進するために治療有効量で動物に投与される低分子量化合物を表す。電磁放射線で治療し得る疾患として、腫瘍性疾患、良性及び悪性の腫瘍、並びに癌細胞が挙げられる。本明細書にリストされていないその他の疾患の電磁放射線治療がまた本発明により意図されている。
【0031】
“アルキル”は指定された数の炭素原子を含む分岐又は非分岐飽和炭化水素鎖を表す。例えば、C1-C6直線状又は分岐アルキル炭化水素鎖は1〜6個の炭素原子を含み、特に示されない限り、置換基、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、t-ブチル、n-ペンチル、n-ヘキシル等が挙げられるが、これらに限定されない。
“アルケニル”は指定された数の炭素原子を含む分岐又は非分岐不飽和炭化水素鎖を表す。例えば、C2-C6直線状又は分岐アルケニル炭化水素鎖は少なくとも一つの二重結合を有する2〜6個の炭素原子を含み、特に示されない限り、置換基、例えば、エテニル、プロペニル、イソプロペニル、ブテニル、イソブテニル、t-ブテニル、n-ペンテニル、n-ヘキセニル等が挙げられるが、これらに限定されない。
【0032】
“アルコキシ”は基-OR(式中、Rは先に定義されたアルキルである)を表す。Rは1〜6個の炭素原子を含む分岐又は非分岐飽和炭化水素鎖であることが好ましい。
“ハロ”又は“ハロゲン”はフルオロ、クロロ、ブロモ及びヨードを表す。
“異性体”は同じ数及び種類の原子、ひいては同じ分子量を有する化合物を表すが、原子の配置又は立体配置に関して異なる。
“立体異性体”は同じ化学構成を有するが、空間の原子又は基の配置に関して異なる化合物を表す。
【0033】
“光学異性体”は2種の立体異性体のいずれをも表す。一つの種類は鏡像体と称される鏡像構造により表され、これらは化合物(グリセルアルデヒド、乳酸、糖、酒石酸、アミノ酸)中の一つ以上の不斉炭素原子の存在により生じる。その他の種類はジアステレオマーにより例示され、これらは鏡像ではない。これらは二つ以上の不斉炭素原子を有する化合物中で生じる。こうして、このような化合物は2n光学異性体(nは不斉炭素原子の数である)を有する。
【0034】
“鏡像体”は互いに重ならない鏡像である立体異性体を表す。
“鏡像体濃縮された”は一種の鏡像体が優勢である混合物を表す。
“ラセミ体”は個々の鏡像体の等しい部分を含む混合物を表す。
“非ラセミ体”は個々の鏡像体の等しくない部分を含む混合物を表す。
“動物”は感覚及び随意運動の力並びにその存在のために酸素及び有機食物に対する要求を有する生物を表す。例として、哺乳類、例えば、ヒト、ウマ、ブタ、ウシ、マウス、イヌ又はネコの種の員が挙げられるが、これらに限定されない。ヒトの場合、“動物”という用語はまた“患者”と称されてもよい。
“疾患”は症候及び兆候の特徴的な組により発現され、その病因、病理学、及び予後が知られていてもよく、また知られていなくてもよい生体のあらゆる部分、臓器、又は系(或いはこれらの組み合わせ)の正常な構造又は機能からのあらゆる異常又は中断を表す。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
【0035】
“障害”は機能のあらゆる障害又は異常;病的肉体状態又は精神状態を表す。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
“グルタメート異常”はグルタメートが関係するあらゆる疾患、障害又は症状を表し、上昇レベルのグルタメートを伴う病状が挙げられる。グルタメート異常の例として、脊髄損傷、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、精神分裂症、急性の痛み、慢性の痛み、虚血、神経障害及び強迫性障害が挙げられるが、これらに限定されない。
“虚血”は動脈血液の流入の閉塞のための局在化組織貧血を表す。総体虚血は心臓停止により生じるように全脳への血液の流れが或る時間の期間にわたって停止する場合に起こる。病巣虚血は脳血管の血栓塞栓閉塞、トラウマ頭部損傷、浮腫又は脳腫瘍から生じるように脳の一部がその正常な血液供給を不足している場合に起こる。たとえ一時的であっても、総体虚血及び病巣虚血の両方が広範な神経細胞損傷を生じ得る。神経組織損傷は虚血の発症の数時間後又は更には数日後に起こるが、或る種の永久の神経組織損傷が脳への血流の停止の初期の数分後に発生し得る。この損傷の多くがグルタメート毒性及び組織の再潅流の二次的な結果、例えば、損傷された内皮による血管活性生成物の放出、及び損傷された組織による細胞傷害生成物、例えば、遊離基及びロイコトリエンの放出に起因される。
【0036】
“神経機能”は、中でも、生体の内部環境及び外部環境の感知を与え、生物の種々の構造要素間で随意活動及び反射活動を可能にし、また環境変化に対する生物の応答をバランスする神経系の種々の機能を表す。
“神経障害”は神経組織へのあらゆる損傷及びそれから生じるあらゆる能力障害又は死亡を表す。神経障害の原因は代謝、毒性、神経毒性、医原性、熱又は化学薬品であってもよく、虚血、低酸素症、脳血管アクシデント、トラウマ、手術、圧力、物質作用、出血、放射線、血管痙攣、神経変性疾患、神経退化プロセス、感染症、パーキンソン病、ALS、髄鞘形成/髄鞘脱落プロセス、癲癇、認知障害、グルタメート異常及びこれらからの二次的な作用が挙げられるが、これらに限定されない。現在、神経組織損傷に有効な治療は知られていない。
“神経組織”は神経系を構成する種々の成分を表し、神経細胞、神経支持細胞、グリア、シュワン細胞、これらの構造内に含まれ、またこれらの構造に供給する血管系、中枢神経系、脳、脳幹、脊髄、末梢神経系との中枢神経系の接合部、末梢神経系及び同類の構造が挙げられるが、これらに限定されない。
【0037】
“神経保護”は神経障害を軽減、停止又は回復し、神経障害を受けた神経組織を保護、救済又は蘇生する効果を表す。
“痛み”は特殊な神経末端の刺激から生じる不快、窮迫又はアゴニーの局所感覚を表す。それは受難者がその源を除去し、又はその源から撤退することを誘発する限りにおいて、それは保護メカニズムとして利用できる。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。痛みの例として、急性、慢性、癌、やけど、切開、炎症、糖尿病性神経障害及び背部の痛みが挙げられるが、これらに限定されない。
【0038】
“精神障害”は窮迫症候又は機能の重大な障害の存在を特徴とするあらゆる臨床上重大な挙動上の又は心理学的な症候群を表す。精神障害は個体の或る種の心理学的又は器質性の機能障害から生じると推定される。その概念は個体と社会の間で本質的に相反する障害(社会的逸脱)を含まない。
“強迫性障害”は抵抗不能の強迫性挙動を特徴とするあらゆる障害を表す。強迫性障害の例として、薬物依存性、食事障害、病的ギャンブル、ADD及びトゥレット症候群が挙げられるが、これらに限定されない。
“注意欠乏障害”は活動過剰を伴い、又は伴わない、発生的に不適当な不注意及び衝動を特徴とする障害を表す。不注意は始めた仕事を完結できないこと、容易な被転導性、注意の外見上の欠如、及び持続注意を要する仕事に対する集中の難しさを意味する。衝動は考える前の行動、交替ですることの難しさ、仕事を編成する問題、及び一つの行動から別の行動に絶えずシフトすることを意味する。活動過剰は依然として着席し、また座っていることの難しさ、及び過度に走ったり、また徘徊したりすることを意味する。
【0039】
“薬物依存性”は薬物に対する心理的嗜癖又は肉体的トレランスを意味する。トレランスは初期に少量により得られた効果を生じるために用量を次第に増加しようとする要求を意味する。
“禁断症状症候群”は薬物が中断される場合又はその効果が特定のアンタゴニストにより相殺される場合に生じる不利な肉体変化を特徴とする障害を表す。
“食事障害”は強迫性過食、肥満又は重度の肥満を表す。肥満は通常の身長−体重表より20%以上の体重を意味する。重度の肥満は100%以上の過剰体重を意味する。
【0040】
“病的ギャンブル”はギャンブルによる忘我を特徴とする症状を表す。精神活性物質乱用と同様に、その効果として、次第に多額の金を賭けたい要求によるトレランスの発生、禁断症状症候、並びに家族及び職業に対するひどい負の効果にもかかわらず絶えずギャンブルすることが挙げられる。
“精神分裂症”は思考の形態及び内容(連想の散漫、妄想、幻覚)、気分(盲目化、落胆、不適当な感情)、外界に対する自己の意味及び関係(エゴ境界の損失、内閉性思考、及び自閉的孤立)、並びに挙動(奇異、外見上の無目的、及びステレオタイプの活動又は非活動)の障害を特徴とする精神障害又はグループ精神障害を表す。精神分裂症の例として、急性、外来、境界、緊張型、幼児、分裂型、破瓜病性、潜在性、核、妄想型、パラフレニー、精神病前、プロセス、偽神経症性、偽精神病性、反応性、残留、分裂−情動及び未分化の精神分裂症が挙げられるが、これらに限定されない。Dorland's Illustrated Medical Dictionary, (W.B. Saunders Co. 第27編1988)を参照のこと。
【0041】
“トゥレット症候群”は強迫性ののしり、多発性筋肉チック及び騒がしいノイズを特徴とする常染色体性多発性チック障害を表す。チックは単純でも複雑でありえる短い、迅速な、不随意運動である。それらはステレオタイプ化され、反復性であるが、周期的ではない。単純チック、例えば、まばたきはしばしば神経マンネリズムとして始まる。複雑なチックはしばしば正常な挙動の断片に似ている。
“血管形成”は新しい毛管が形成されるプロセスを表す。
“血管形成依存性疾患”として、癌、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、及び皮膚潰瘍が挙げられるが、これらに限定されない。
【0042】
血管形成の“抑制”は本発明に従って多くのパラメーターにより測定されてもよく、例えば、新生血管構造の遅延出現、新生血管構造の遅い発生、新生構造の減少された発生、血管形成依存性疾患作用の遅い、又は低下された重度、停止された血管形成的成長、又は先の血管形成的成長の回帰により評価し得る。極限では、完全抑制は本明細書では予防と称される。
血管形成又は血管形成的成長に関して、“予防”は既に発生しなかった場合には血管形成又は血管形成的成長が実質的にないことを表し、又は成長が既に発生していた場合には更なる血管形成又は血管形成的成長が実質的にないことを表す。
“癌”として、ACTH生成腫瘍、急性リンパ性白血病、急性非リンパ性白血病、副腎皮質の癌、膀胱癌、脳の癌、乳癌、子宮頸癌、慢性リンパ性白血病、慢性骨髄性白血病、結腸直腸癌、皮膚T細胞リンパ腫、子宮内膜癌、食道癌、ユーイング肉腫、胆嚢癌、ヘアリー・セル白血病、頭部及び首の癌、ホジキンリンパ腫、カポージ肉腫、腎臓癌、肝臓癌、肺癌(小細胞及び/又は非小細胞)、悪性腹腔滲出、悪性胸膜滲出、メラノーマ、中皮腫、多発性ミエローマ、ニューロブラストーマ、非ホジキンリンパ腫、骨肉腫、卵巣癌、卵巣(生殖細胞)癌、膵臓癌、ペニス癌、前立腺癌、網膜芽細胞腫、皮膚癌、軟組織肉腫、扁平上皮癌、胃癌、精巣癌、甲状腺癌、栄養膜腫瘍、子宮の癌、膣癌、外陰部の癌、及びウィルム腫瘍が挙げられるが、これらに限定されない。
【0043】
“転移”は“播殖して、非隣接部位で増殖の新しい病巣を形成する(即ち、転移を形成する)癌の細胞の能力”を表す。本明細書に引用として含まれるThe Basic Science of Oncology, Tannockら編集, McGraw-Hill, New York (1992)中のHill, R.P, 11章,“Metastasis”, 178-195頁を参照のこと。「in situ腫瘍増殖から転移性疾患への転移は一次部位の腫瘍細胞が局所組織を侵食し、組織関門を横切る腫瘍細胞の能力により特定される・・・転移プロセスを開始するために、癌細胞は最初に上皮基底膜に侵入し、次いで組織内ストローマを侵食する必要がある・・・遠位転移のために、血管内侵入は、内皮下基底膜の腫瘍細胞浸潤を必要とするが、これはまた腫瘍細胞の血管外遊出の際にも通り抜ける必要がある・・・悪性の発生はまた、原発腫瘍のエクスパンジョンを可能にするだけでなく新たに形成された血管の基底膜に欠陥があるために血管区画への容易な接近を可能にする、腫瘍誘導血管形成と関連している」。Aznavoorianら, Cancer 71: 1368-1383 (1993)(本明細書に参考として含まれる)を参照のこと。
【0044】
“電磁放射線”として、10-20〜100メートルの波長を有する放射線が挙げられるが、これらに限定されない。本発明の好ましい実施態様はγ放射線(10-20〜10-13m)、x線放射線(10-11〜10-9m)、紫外線(10nm〜400nm)、可視光線(400nm〜700nm)、赤外線(700nm〜1.0mm)、及びマイクロウェーブ放射線(1mm〜30cm)の電磁放射線を使用する。
“前立腺疾患”は前立腺を冒すあらゆる疾患を表す。前立腺疾患の例として、前立腺癌、例えば、前立腺の腺癌及び転移性癌;並びに前立腺上皮細胞の異常増殖を特徴とする症状、例えば、良性前立腺過形成が挙げられるが、これらに限定されない。
“治療”は
(i)疾患、障害及び/又は症状にかかりやすくなっているが、それを有すると未だ診断されていない動物において疾患、障害又は症状が発生することを予防する、
(ii)疾患、障害又は症状を抑制し、すなわち、その発生を停止する、かつ/又は
(iii)疾患、障害又は症状を軽減し、即ち、疾患、障害及び/又は症状を軽減するために本発明の化合物又は組成物を投与することを表す。
薬物依存性に関して、“治療”は乱用の薬物に対する心理的嗜癖又は肉体的トレランスを抑制し、かつ/又は薬物依存性から生じる禁断症状症候群を軽減し、かつ/又は予防するために本発明の化合物又は組成物を投与することを含む。
卒中に関して、“機会の治療ウインドー”又は“ウインドー”は虚血の発症と有効な治療の間の最大の遅延を表す。
【0045】
“NAAG”は、主要なインヒビター神経伝達物質γ-アミノ酪酸(GABA)に匹敵するレベルで、脳の重要なペプチド成分であるN-アセチル-アスパルチル-グルタメートを表す。NAAGは神経細胞特異性であり、シナプス小胞中に存在し、グルタメート作用性であると推定される幾つかの系中で神経刺激後に放出される。研究はNAAGが中枢神経系中で神経伝達物質及び/又は神経モジュレーターとして、又は神経伝達物質グルタメートの前駆体として機能し得ることを示唆する。
“NAALADase”はNAAGをN-アセチルアスパルテート(“NAA”)及びグルタメート(“GLU”)に異化作用する膜結合メタロペプチダーゼであるN-アセチル化α結合酸性ジペプチダーゼを表す。
【0046】
【0047】
アミノ酸配列相同性に基づいて、NAALADaseはM28ペプチダーゼファミリーに指定された。NAALADaseはまた前立腺特異性膜抗原(PSMA)又はヒトグルタメートカルボキシペプチダーゼII(GCPII)、EC番号3.4.17.21と称される。NAALADaseは補助触媒性亜鉛/亜鉛メタロペプチダーゼであると考えられる。NAALADaseは540 nMのKmでNAAGに対する高アフィニティーを示す。NAAGが生物活性ペプチドである場合、NAALADaseはNAAGのシナプス作用を不活性化するのに利用できるかもしれない。また、NAAGがグルタメートの前駆体として機能する場合、NAALADaseの一次機能はシナプスのグルタメート利用可能性を調節するのかもしれない。
“NAALADaseインヒビター”はNAALADase酵素活性を抑制するあらゆる化合物を表す。
【0048】
“抑制”は、酵素に関して、競合、未競合及び非競合の抑制の如き可逆的酵素抑制を表す。競合、未競合及び非競合の抑制は酵素の反応速度論に関するインヒビターの効果により区別し得る。競合抑制はインヒビターが活性部位で結合するために通常の基質と競合するような方法でインヒビターが酵素と可逆的に化合する場合に生じる。インヒビターと酵素の間のアフィニティーはインヒビター定数、Kiにより測定されてもよく、これは以下のように定義される。
【0049】
【0050】
式中、〔E〕は酵素の濃度であり、〔I〕はインヒビターの濃度であり、かつ〔EI〕は酵素とインヒビターの反応により生成された酵素−インヒビター複合体の濃度である。特にことわらない限り、本明細書に使用される“Ki”は本発明の化合物とNAALADaseの間のアフィニティーを表す。“IC50”は標的酵素の50%抑制を生じるのに必要とされる化合物の濃度又は量を特定するのに使用される関連用語である。
本発明の化合物
本発明は一般式I
【0051】
【0052】
の化合物又は医薬上許される均等物に関する。
式中、
Xは式II、III又はIV
【0053】
【0054】
である部分であり、
m及びnは独立に0、1、2、3又は4であり、
ZはSR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり、 BはN又はCR16であり、
AはO、S、CR17R18又は(CR17R18)mSであり、
R9及びR13は水素であり、
R8、R10、R11、R12、R14、R15、R16、R17及びR18は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されており、
但し、Xが式IIである部分であり、かつAがOである場合、nが2、3又は4であり;Xが式IIである部分であり、かつAがSである場合、nが2、3又は4であり;またXが式IIである部分であり、かつAが(CR17R18)mSである場合、nが0、2、3又は4であることを条件とする。
【0055】
前記アルケニル、シクロアルキル、シクロアルケニル、及びAr1の可能な置換基として、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、ニトロソ、ニトリロ、イソニトリロ、イミノ、アゾ、ジアゾ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル、並びに炭素環式部分及び複素環部分が挙げられるが、これらに限定されない。炭素環式部分として、脂環式構造及び芳香族構造が挙げられる。
【0056】
有益な炭素環式部分及び複素環部分の例として、フェニル、ベンジル、ナフチル、インデニル、アズレニル、フルオレニル、アントラセニル、インドリル、イソインドリル、インドリニル、ベンゾフラニル、ベンゾチオフェニル、インダゾリル、ベンズイミダゾリル、ベンゾチアゾリル、テトラヒドロフラニル、テトラヒドロピラニル、ピリジル、ピロリル、ピロリジニル、ピリジニル、ピリミジニル、プリニル、キノリニル、イソキノリニル、テトラヒドロキノリニル、キノリジニル、フリル、チオフェニル、イミダゾリル、オキサゾリル、ベンゾオキサゾリル、チアゾリル、イソオキサゾリル、イソトリアゾリル、オキサジアゾリル、トリアゾリル、チアジアゾリル、ピリダジニル、ピリミジニル、ピラジニル、トリアジニル、トリチアニル、インドリジニル、ピラゾリル、ピラゾリニル、ピラゾリジニル、チエニル、テトラヒドロイソキノリニル、シノリニル、フタラジニル、キナゾリニル、キノキサリニル、ナフチリジニル、プテリジニル、カルバゾリル、アクリジニル、フェナジニル、フェノチアジニル、及びフェノキサジニルが挙げられるが、これらに限定されない。
【0057】
Xが式IIである部分であり、nが0、1、2又は3であり、ZがSH、SO3H、SO2H、SOH又はS(NRHR14)2R15であり、かつAがO、S又はCR17R18であることが好ましい。
ZがSHであることが更に好ましい。
ZがSHである場合、R8が-(CH2)2COOHであることが最も好ましい。
【0058】
別の好ましい実施態様において、Xが式IIである部分であり、R8が-(CH2)2COOR19又は-(CH2)2CONHR19であり、AがCH2であり、nが0であり、ZがSR13である場合には、R13は水素又はCOR19ではなく、またXが式IIIである部分であり、BがNであり、かつR8が-(CH2)2COOHである場合には、R11は水素ではない。
式Iの好ましい化合物は
2-(2-スルファニルエチル)ペンタン二酸、
3-(2-スルファニルエチル)-1,3,5-ペンタントリカルボン酸、
2-(2-スルファニルプロピル)ペンタン二酸、
2-(2-スルファニルブチル)ペンタン二酸、
2-(2-スルファニル-2-フェニルエチル)ペンタン二酸、
2-(2-スルファニルヘキシル)ペンタン二酸、
2-(2-スルファニル-1-メチルエチル)ペンタン二酸、
2-〔1-(スルファニルメチル)プロピル〕ペンタン二酸、
2-(3-スルファニルペンチル)ペンタン二酸、
2-(3-スルファニルプロピル)ペンタン二酸、
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸、
2-(3-スルファニル-2-フェニルプロピル)ペンタン二酸、
2-(3-スルファニルブチル)ペンタン二酸、
2-〔3-スルファニル-2-(フェニルメチル)プロピル〕ペンタン二酸、
2-〔2-(スルファニルメチル)ブチル〕ペンタン二酸、
2-〔2-(スルファニルメチル)ペンチル〕ペンタン二酸、
2-(3-スルファニル-4-メチルペンチル)ペンタン二酸、及び
これらの医薬上許される均等物からなる群から選ばれる。
【0059】
式Iの最も好ましい化合物は2-(2-スルファニルエチル)ペンタン二酸、2-(2-スルファニルプロピル)ペンタン二酸、2-(3-スルファニルプロピル)ペンタン二酸及び医薬上許される均等物からなる群から選ばれる。理想的には、式Iの化合物は鏡像体又は鏡像体濃縮された混合物である。
Xが式IIIである部分であり、R8が-(CH2)2COOHであり、R9が水素であり、かつBがCR16である式Iの代表的な化合物として、
2-(ジチオカルボキシメチル)ペンタン二酸、
2-(1-ジチオカルボキシエチル)ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
Xが式IIIである部分であり、R8が-(CH2)2COOHであり、R9が水素であり、かつBがNである式Iの代表的な化合物として、
2-ジチオカルボキシアミノペンタン二酸、
2-〔(N-メチルジチオカルボキシ)アミノ〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
Xが式IVである部分である式Iの代表的な化合物として、
2-ベンジル-4-スルファニルブタン酸、
2-ベンジル-4-スルファニルペンタン酸、
2-(3-ピリジルメチル)-4-スルファニルペンタン酸、
2-(3-ピリジルメチル)-4-スルファニルヘキサン酸、
2-ベンジル-3-スルファニルプロパン酸、
2-ベンジル-3-スルファニルペンタン酸、
2-(4-ピリジルメチル)-3-スルファニルペンタン酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
式Iの幾つかの代表的な化合物の構造が以下に示される。
【0060】
【0061】
【0062】
【0063】
【0064】
【0065】
【0066】
【0067】
【0068】
【0069】
【0070】
【0071】
【0072】
【0073】
【0074】
【0075】
本発明の幾つかの化合物は一つ以上の不斉炭素中心を有し、こうして光学異性体の形態だけでなく、光学異性体のラセミ体混合物又は非ラセミ体混合物の形態で存在することができる。光学異性体は当業界で公知である通常の方法に従って、例えば、光学活性の酸又は塩基による処理によるジアステレオマー塩の生成によりラセミ体混合物の分割により得られる。適当な酸の例は酒石酸、ジアセチル酒石酸、ジベンゾイル酒石酸、ジトルオイル酒石酸及びショウノウスルホン酸であり、次いで結晶化によるジアステレオマーの混合物の分離、続いてこれらの塩からの光学活性塩基の放出が行われる。光学異性体の分離のための異なる方法は鏡像体の分離を最大にするために最適に選ばれたキラルクロマトグラフィーカラムの使用を伴う。更に別の利用可能な方法には、本発明の方法及び医薬組成物に使用される化合物を活性化形態の光学活性酸、光学活性ジオール又は光学活性イソシアノと反応させることにより共有結合ジアステレオマー分子、例えば、エステル、アミド、アセタール、ケタール、等の合成を含む。合成されたジアステレオマーは通常の手段、例えば、クロマトグラフィー、蒸留、結晶化又は昇華により分離でき、次いで鏡像体上純粋な化合物をデリバリするために加水分解される。或る場合には、親の光学活性薬物への加水分解は患者に投与する前に必要ではない。何とならば、化合物がプロドラッグとして挙動し得るからである。本発明の光学活性化合物は同様に光学活性出発物質を利用することにより得られる。
【0076】
本発明の化合物は光学異性体だけでなく、ラセミ体混合物及び非ラセミ体混合物を含むことが理解される。
以下に詳しく説明されるように、本発明の化合物は種々の薬理学的性質及び医薬の性質を有する。特に、本発明の化合物はNAALADase酵素活性を抑制する。NAALADase酵素活性を抑制することにより、本発明の化合物は神経変性中に生じるグルタメートの前シナプス放出を調節することが仮定される。
本発明の化合物はまたin vitroだけでなく、in vivo動物モデルで神経変性に対して保護する。幾つかの本発明の化合物は、虚血前及び後の投与されたときに、どちらも虚血の組織培養モデルで神経保護性であることが実証された。本発明の化合物の幾つかはin vitroモデルで虚血損傷の60分後までに投与された時に有意な神経保護効果を与える。
【0077】
更に、本発明の化合物の幾つかはin vivoラットMCAO卒中モデルで有意な保護を与え、また虚血後の60分、120分、180分及び360分で投与された時に保護性であることが示された。このような場合、本発明の化合物は卒中の発症後の60分以上、120分以上、180分以上、及び360分以上で投与された時に動物の卒中を治療するのに有効である。当業者はこのような化合物が卒中の発症後60分以内に投与される時にそれ以上ではないとしても同等に有効であると予想するであろう。同様に、卒中の発症後の360分以上に投与された時に卒中を治療するのに有効である化合物は、卒中の発症の360分より前のいずれの時点で投与された時に有効である化合物を包含すると期待されるであろう。神経保護を与えることに加えて、本発明の化合物は卒中後に挙動機能の回復を与えることにより卒中を治療するのに有効であることが可能である。
こうして、本発明は更に卒中の発症後に60分以上で投与された時に哺乳類の卒中を治療するのに有効である硫黄及び酸基の両方を含む化合物に関する。
化合物は卒中の発症後に120分以上で投与された時に哺乳類の卒中を治療するのに有効であることが好ましい。化合物は卒中の発症後に180分以上で投与された時に哺乳類の卒中を治療するのに有効であることが更に好ましい。化合物は卒中の発症後に360分以上で投与された時に哺乳類の卒中を治療するのに有効であることが最も好ましい。
【0078】
本発明の医薬組成物
本発明はまた
(i)有効量の式Iの化合物、及び
(ii)医薬上許される担体
を含む医薬組成物に関する。
式Iの化合物はNAALADase酵素活性を抑制し、グルタメート異常を治療し、神経活性に作用し、強迫性疾患を治療し、前立腺疾患を治療し、又は哺乳類の血管形成を抑制するのに有効な量で存在することが好ましい。
式Iの好ましい化合物は上述した。
【0079】
本発明の方法
NAALADase酵素活性の抑制方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のNAALADase酵素活性の抑制方法に関する。
グルタメート異常の治療方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類のグルタメート異常の治療方法に関する。
グルタメート異常はグルタメートが関係するあらゆる疾患、障害又は症状を表し、上昇レベルのグルタメートを伴う病状が挙げられる。グルタメート異常の例として、癲癇、卒中、アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症(ALS)、ハンチントン病、精神分裂症、急性の痛み、慢性の痛み、虚血、末梢神経障害(糖尿病性神経障害を含む)、トラウマ脳損傷及び脊髄への物理的損傷が挙げられるが、これらに限定されない。好ましい実施態様において、グルタメート異常は虚血、卒中、パーキンソン病、筋萎縮性側索硬化症(ALS)及び脊髄損傷からなる群から選ばれる。
【0080】
いずれか一つの特別な理論に限定されないが、式Iの化合物はNMDA受容体により媒介される効果から上流であると仮定される貯蔵型グルタメートに作用することによりグルタメートのレベルを調節すると考えられる。
チオール官能基のラジカル除去性がまた化合物の治療効力に寄与し得る。フリーラジカル除去物質は脳及び神経組織の種々の型の急性及び慢性の病状に関連付けられている。最近の研究によりフリーラジカル除去物質が脳虚血−再潅流、興奮性アミノ酸脳損傷、ミトコンドリア機能障害、糖尿病、糖尿病性神経障害、先天性代謝異常、及び脳又は神経組織への急性又は慢性の損傷のその他の原因に神経保護効果を示すことが示されている。Krishanら, Pharmacological Research, 37巻, 1号, 23-9頁(1998年1月);Nodaら, Research Communications in Molecular Pathology and Pharmacology, 96巻, 2号, 125-36頁(1997年5月);Andersonら, Canadian Journal of Cardiology, 12巻, 10号, 1099-104頁(1996年10月);Mizunoら, General Pharmacology, 30巻, 4号, 575-8頁(1998年4月);de la Torreら, Brain Research, 779巻, 1-2号, 285-8頁(1998年1月);Yukiら, Molecular and Chemical Neuropathology, 32巻, 1-3号, 123-8頁(1997年9月−10月);Yamamotoら, Brain Research, 762巻, 1-2号, 240-2頁(1997年7月11日)を参照のこと。それ故、本発明の化合物はフリーラジカル損傷を伴う脳障害を治療するのに特に有効であり得る。
【0081】
強迫性障害の治療方法
更に、本発明はこのような治療を要する患者に有効量の式Iの化合物を投与することを特徴とする強迫性障害の治療方法に関する。
強迫性障害は抵抗不能の強迫性挙動を特徴とするあらゆる障害であってよい。本発明の方法により治療できる強迫性障害の例として、薬物依存性、食事障害、病的ギャンブル、ADD及びトゥレット症候群が挙げられる。
強迫性障害は薬物依存性であることが好ましい。依存性について潜在性を有する普通使用される薬物として、CNS抗うつ薬(オピオイド、合成麻酔薬、バルビツレート、グルテチミド、メチプリロン、エチクロルビノール、メタクアロン、アルコール);抗不安薬(ジアゼパム、クロルジアゼポキシド、アルプラゾラム、オキサゼパム、テマゼパム);興奮薬(アンフェタミン、メタムフェタミン、コカイン);及び幻覚薬(LSD、メスカリン、ペヨーテ、マリファナ)が挙げられる。
薬物依存性はアルコール依存性、ニコチン依存性、ヘロイン依存性又はコカイン依存性であることが更に好ましい。
【0082】
神経活性に作用する方法
本発明者らはまたNAALADaseの抑制が神経再生及び髄鞘形成を促進することを発見した。
それ故、更に、本発明は有効量の式Iの化合物を哺乳類に投与することを特徴とする哺乳類の神経活性に作用する方法に関する。
本発明の方法により作用される神経活性は損傷された神経細胞の刺激、神経細胞再生の促進、神経変性の予防及び神経障害の治療からなる群から選ばれてもよい。
本発明の方法により治療できる神経障害の例として、三叉神経痛;舌咽神経痛;ベル麻痺;重症筋無力症;筋ジストロフィー;筋萎縮性側索硬化症;進行性筋萎縮;進行性延髄遺伝性筋萎縮;椎間板ヘルニア症候群、断裂無脊椎板症候群又は脱出無脊椎板症候群;頸部脊椎症;神経そう障害;胸郭出口症候群;末梢神経障害、例えば、鉛、ダプソーン、チック、ポルフィリア、又はギラン・バレー症候群により生じたもの;アルツハイマー病;及びパーキンソン病が挙げられるが、これらに限定されない。
【0083】
本発明の方法は物理的損傷又は病状により生じた末梢神経障害、トラウマ脳損傷、脊髄への物理的損傷、脳損傷と関連する卒中、髄鞘脱落疾患及び神経変性に関係する神経障害からなる群から選ばれた神経障害を治療するのに特に有益である。髄鞘脱落疾患の例として、多発性硬化症並びに末梢髄鞘脱落疾患、例えば、末梢神経障害及びシャルコー−マリー歯疾患が挙げられる。神経変性に関係する神経障害の例として、アルツハイマー病、パーキンソン病、及び筋萎縮性側索硬化症(ALS)が挙げられる。
【0084】
前立腺疾患の治療方法
更に、本発明は哺乳類に有効量の式Iの化合物を投与することを特徴とする哺乳類の前立腺疾患の治療方法に関する。
好ましい実施態様において、前立腺疾患は前立腺癌、例えば、腺癌及び前立腺の転移癌、又は前立腺上皮細胞の異常増殖を特徴とする症状、例えば、良性前立腺過形成である。
癌の治療方法
前立腺癌に加えて、本発明の化合物で治療し得る癌のその他の形態として、ACTH生成腫瘍、急性リンパ性白血病、急性非リンパ性白血病、副腎皮質の癌、膀胱癌、脳の癌、乳癌、子宮頸癌、慢性リンパ性白血病、慢性骨髄性白血病、結腸直腸癌、皮膚T細胞リンパ腫、子宮内膜癌、食道癌、ユーイング肉腫、胆嚢癌、ヘアリー・セル白血病、頭部及び首の癌、ホジキンリンパ腫、カポージ肉腫、腎臓癌、肝臓癌、肺癌(小細胞及び/又は非小細胞)、悪性腹腔滲出、悪性胸膜滲出、メラノーマ、中皮腫、多発性ミエローマ、ニューロブラストーマ、非ホジキンリンパ腫、骨肉腫、卵巣癌、卵巣(生殖細胞)癌、膵臓癌、ペニス癌、網膜芽細胞腫、皮膚癌、軟組織肉腫、扁平上皮癌、胃癌、精巣癌、甲状腺癌、栄養膜腫瘍、子宮の癌、膣癌、外陰部の癌、及びウィルム腫瘍が挙げられるが、これらに限定されない。
本発明の化合物はNAALADase酵素がある組織の癌を治療するのに特に有益である。このような組織として、前立腺だけでなく、脳、腎臓及び睾丸が挙げられる。
【0085】
卒中の治療方法
更に、本発明は卒中の発症の60分以上後に有効量の硫黄及び酸基の両方を含む化合物を哺乳類に投与することを特徴とする哺乳類の卒中の治療方法に関する。
この化合物は卒中の発症の180分以上後に前記哺乳類に投与されることが好ましい。
この化合物は卒中の発症の360分以上後に前記哺乳類に投与されることが更に好ましい。
硫黄及び酸基の両方を含む化合物の例として、式Iの化合物が挙げられるが、これらに限定されない。
【0086】
血管形成の抑制方法
本発明者らはNAALADaseインヒビターがNAALADaseを含む組織中で血管形成に作用し得ることを予期せずに見出した。先の研究によりNAALADaseがシナプスプラズマ膜中で濃縮され、主として神経組織及び腎臓組織に局在化されることが示された。NAALADaseはまた前立腺及び睾丸の組織中に見られた。更に、先の知見によりNAALADaseが新生血管系中に存在することが示された。更に、NAALADaseは生体のその他の組織中で発見され続けているので、NAALADaseインヒビターはおそらくまたこれらの組織中の血管形成の抑制に効力を示すであろう。
それ故、更に本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の血管形成の抑制方法に関する。
血管形成は生殖力又は癌腫瘍の転移に必要であるかもしれず、また血管形成依存性疾患に関係しているかもしれない。従って、本発明の方法により治療できる血管形成依存性疾患として、慢性関節リウマチ、心血管疾患、眼の新生血管疾患、末梢血管障害、並びに癌の腫瘍増殖、侵食、及び転移が挙げられるが、これらに限定されない。
本発明の方法はNAALADase酵素がある組織の癌腫瘍中の血管形成を抑制するのに特に有益である。このような組織として、脳、腎臓、前立腺、睾丸、及び血管が挙げられるが、これらに限定されない。
【0087】
痛みの治療方法
更に、本発明は有効量のNAALADaseインヒビターを哺乳類に投与することを特徴とする哺乳類の痛みの治療方法に関する。
NAALADaseインヒビターはモルヒネに対するトレランスを阻止し、痛みを治療するのに必要なモルヒネの量を減少するのに特に有効である。
本発明の方法により治療できる痛みの例として、急性、慢性、癌、やけど、切開、炎症、糖尿病性神経障害及び背部の痛みが挙げられるが、これらに限定されない。
好ましいNAALADaseインヒビターは式Iの化合物であり、これらの例は上述した。
別の好ましいNAALADaseインヒビターは式V:
【0088】
【0089】
の化合物又は医薬上許される均等物である。
式中、
YはCR3R4、NR5又はOであり、
R1は水素、C1-C9アルキル、C2-C9アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar、COOR、NR6R7及びORからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換、又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、COOR、NR6R7及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
R2は水素、C1-C6アルキル、C2-C6アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar、ハロ及びカルボキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ、NR6R7及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
【0090】
R3及びR4は独立に水素又はC1-C3アルキルであり、
R5は水素又はC1-C3アルキルであり、
R、R6及びR7は水素、C1-C9アルキル、C2-C9アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル及びArからなる群から独立に選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは未置換、又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C9アルケニルオキシ、フェノキシ、ベンジルオキシ及びArからなる群から独立に選ばれた一つ以上の置換基で置換されており、
Arは1-ナフチル、2-ナフチル、2-インドリル、3-インドリル、4-インドリル、2-フリル、3-フリル、テトラヒドロフラニル、テトラヒドロピラニル、2-チエニル、3-チエニル、2-ピリジル、3-ピリジル、4-ピリジル及びフェニルからなる群から選ばれ、前記Arは未置換又はハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C9アルコキシ、C2-C6アルケニルオキシ、フェノキシ、ベンジルオキシ、カルボキシ及びNR1R2からなる群から独立に選ばれた一つ以上の置換基で置換されている。
【0091】
YはCH2であることが好ましい。
YがCH2である場合、R2は-(CH2)2COOHであることが更に好ましい。
YがCH2であり、かつR2が-(CH2)2COOHである場合、R1は水素、C1-C4アルキル、C2-C4アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、ベンジル、フェニル又はORであることが最も好ましく、前記アルキル、アルケニル、シクロアルキル、シクロアルケニル、ベンジル及びフェニルは未置換又はカルボキシ、C3-C8シクロアルキル、C5-C7シクロアルケニル、ハロ、ヒドロキシ、ニトロ、トリフルオロメチル、C1-C6アルキル、C2-C6アルケニル、C1-C6アルコキシ、C2-C6アルケニルオキシ、フェノキシ、ベンジルオキシ、NR6R7、ベンジル及びフェニルからなる群から独立に選ばれた一つ以上の置換基で置換されている。
【0092】
式Vの好ましい化合物は
2-(ホスホノメチル)ペンタン二酸、
2-〔〔(2-カルボキシエチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(ベンジルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔(フェニルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔((ヒドロキシ)フェニルメチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(ブチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(3-メチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、 2-〔(3-フェニルプロピルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(4-フルオロフェニル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔(メチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔(フェニルエチルヒドロキシホスフィニル)メチル〕ペンタン二酸、
2-〔〔(4-メチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、 2-〔〔(4-フルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-メトキシベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(3-トリフルオロメチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-トリフルオロメチルベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-フルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、及び
医薬上許される均等物からなる群から選ばれる。
【0093】
式Vの化合物は2-〔〔(2,3,4,5,6-ペンタフルオロベンジル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸又は医薬上許される均等物であることが更に好ましい。式Vの化合物は鏡像体又は鏡像体濃縮混合物であることが最も好ましい。
R1がCOORで置換されている式Vの代表的な化合物として、
2-〔〔2-カルボキシプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-カルボキシブチル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-カルボキシペンチル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(2-カルボキシ-3-フェニルプロピル)ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-カルボキシ-3-ナフチルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
【0094】
2-〔〔2-カルボキシ-3-ピリジルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-ベンジルオキシカルボニル〕-3-フェニルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔2-メトキシカルボニル〕-3-フェニルプロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(3-カルボキシ-2-メトキシカルボニル)プロピル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、
2-〔〔(4-カルボキシ-2-メトキシカルボニル)ブチル〕ヒドロキシホスフィニル〕メチル〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
【0095】
R1がNR6R7で置換されている式Vの代表的な化合物として、
2-〔({〔ベンジルアミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔カルボキシアミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔ベンジルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔アセチルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔ジフェニルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔フェニルアミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-({〔(フェニルカルボキサミド)メチル〕(ヒドロキシホスフィニル)}メチル)ペンタン二酸、
2-({〔(フェニルスルホンアミド)メチル〕(ヒドロキシホスフィニル)}メチル)ペンタン二酸、
2-({〔(4-フルオロフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-メトキシフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-メチルフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(4-t-ブチルフェニル)アミノ〕メチル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔(チオホルムアニリド)アミノ〕ベンジル}(ヒドロキシホスフィニル))メチル〕ペンタン二酸、
2-〔({〔1,3-ジオキソ-2,3-ジヒドロ-1H-2-イソインドリル〕メチル}ヒドロキシホスフィニル)メチル〕ペンタン二酸、及び
医薬上許される均等物が挙げられるが、これらに限定されない。
【0096】
その他のNAALADaseインヒビターが米国特許第5,672,592号、同第5,795,877号、同第5,863,536号、同第5,880,112号及び同第5,902,817号、特許された米国特許出願第08/825,997号、同第08/833,628号、同第08/842,360号及び同第08/899,319号(これらについて、登録料が支払われた)、並びに国際特許公開番号WO 97/48399、WO 97/48400、WO 97/48409及びWO 98/53812に見られ、これらの特許、出願及び公開の全内容が参考として本明細書に含まれる。
好ましい実施態様において、NAALADaseインヒビターはモルヒネと組み合わせて投与される。
【0097】
糖尿病性神経障害の治療方法
更に、本発明は哺乳類に有効量のNAALADaseインヒビターを投与することを特徴とする哺乳類の糖尿病性神経障害の治療方法に関する。
有益なNAALADaseインヒビターの例が先に示されている。
【0098】
硫黄及び酸基の両方を含む化合物の調製方法
更に、本発明はチオラクトンを置換エステルと反応させて式VI
【0099】
【0100】
の化合物を生成する工程を含むことを特徴とする、硫黄及び酸基の両方を含む化合物の調製方法に関する。
式中、
Dは(CR21R22)nであり、
nは0、1、2、3又は4であり、かつ
R20、R21及びR22は独立に水素、C1-C9直鎖又は分岐鎖アルキル、C2-C9直鎖又は分岐鎖アルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群から選ばれ、前記アルキル、アルケニル、シクロアルキル及びシクロアルケニルは独立に未置換又は一つ以上の置換基で置換されており、かつ
Ar1は炭素環式部分又は複素環部分であり、これは未置換又は一つ以上の置換基で置換されている。
本方法は式VIの化合物をアルキル化剤と反応させてペンタン二酸誘導体を生成する工程を更に含むことが好ましい。
前記チオラクトンは
【0101】
【0102】
(式中、R10は先に定義されたとおりである)
であることが更に好ましい。
前記エステルは3-(ブロモ)プロピオン酸エチルエステルであることが最も好ましい。
更に、本発明は
(i)メルドラム酸を硫黄含有反応体と反応させてメルドラム酸硫黄含有誘導体を生成する工程;及び
(ii)前記メルドラム酸硫黄含有誘導体をエステルと反応させて式VII
【0103】
【0104】
(式中、
Eは硫黄含有部分であり、かつ
Fはプロピオン酸エステル含有部分である)
の化合物を生成する工程を含むことを特徴とする硫黄及び酸基の両方を含む化合物の調製方法に関する。
本方法は式VIIの化合物を官能性誘導体化する工程を更に含むことが好ましい。
前記チオ含有反応体が3-〔(トリフェニルメチル)チオ〕プロピオン酸であることが更に好ましい。
前記エステルが3-(ブロモ)プロピオン酸メチルエステルであることが最も好ましい。
また、本発明は上記方法のいずれかにより調製された化合物に関する。このような化合物の例として、式Iの化合物が挙げられる。
【0105】
NAALADaseインヒビターの合成
本発明の方法に使用されるNAALADaseインヒビターの幾つかは、米国特許第5,672,592号、同第5,795,877号、同第5,863,536号、同第5,880,112号及び同第5,902,817号、特許された米国特許出願第08/825,997号、同第08/833,628号、同第08/842,360号及び同第08/899,319号(これらについて、登録料が支払われた)、並びに国際特許公開番号WO 97/48399、WO 97/48400、WO 97/48409及びWO 98/53812(これらの特許、出願及び公開の全内容が引用により本明細書に含まれる)に示された一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。
式VのNAALADaseインヒビターは、以下にスキームI-IXに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法、例えば、Jacksonら, J. Med. Chem., 39巻, 2号, 619-622頁(1996)及びFroestlら, J. Med. Chem., 38巻, 3313-3331頁(1995)により記載された方法により調製し得る。
【0106】
【0107】
R基を置換する方法は当業界で知られている。ホスフィン酸エステルを合成する付加的な方法がJ. Med. Chem., 31巻, 204-212頁(1988)に記載されており、以下にスキームIIに示される。
【0108】
【0109】
【0110】
上記ホスフィン酸エステルで開始して、式Vの化合物を調製する種々の経路がある。例えば、一般の経路がJ. Med. Chem., 39巻, 619-622頁(1996)に記載されており、以下にスキームIIIに示される。
【0111】
【0112】
式Vの化合物を調製する別の経路が以下にスキームIV及びスキームVに示される。スキームIV及びスキームVはホスフィン酸誘導体としての出発物質並びにスキームII及び本明細書にリストされた置換基を含む(これらに限定されない)妥当な化学置換基としてのR基を示す。
【0113】
【0114】
【0115】
式Vの化合物を調製する別の経路はR1における芳香族置換を可能にし、以下にスキームVIに示される。
【0116】
【0117】
式Vの化合物を調製する別の経路はR2位における芳香族置換を可能にし、以下にスキームVIIに示される。
【0118】
【0119】
YがNR5である式Vの化合物を調製する別の経路が以下にスキームVIIIに示される。
【0120】
【0121】
Yが酸素である式Vの化合物を調製する別の経路が以下にスキームIXに示される。
【0122】
【0123】
R1がCOORで置換されている式Vの化合物は、以下にスキームXに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法、例えば、Jacksonら, J. Med. Chem., 39巻, 2号, 619-622頁(1996)及びFroestlら, J. Med. Chem., 38巻, 3313-3331頁(1995)により記載された方法により調製し得る。
【0124】
【0125】
【0126】
【0127】
R1がNR6R7で置換されている式Vの化合物は、以下にスキームXI及びXIIに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法により調製し得る。
【0128】
【0129】
【0130】
Xが式IIである部分であり、かつAがO又はCR17R18である式Iの化合物は以下にスキームXIII-XXIIに示される一般の合成経路を使用して、有機化学の通常の技術により容易に調製し得る。前駆体化合物は当業界で知られている方法により調製し得る。
【0131】
【0132】
【0133】
【0134】
【0135】
【0136】
【0137】
【0138】
【0139】
【0140】
【0141】
Xが式IIである部分であり、かつAが(CR17R18)mSである式Iの化合物は通常の合成方法、例えば、相当するチオールの酸化により容易に調製し得る。
Xが式IIである部分であり、かつAがSである化合物は通常の合成技術により容易に調製し得る。例えば、スキームXXIIが適当に置換されたチオ化合物で開始することにより変更し得る。加えて、このクラスの化合物はまたα,β-不飽和エステルへの適当に置換されたチオール誘導体のミカエル付加により調製し得る。
Xが式IIIである部分である式Iの化合物は通常の合成経路、例えば、グルタメート誘導体と二硫化炭素の反応を使用して容易に調製し得る。
【0142】
投与経路
本発明の方法において、下記の当該技術分野において有効であることが公知のいずれかの技法により化合物を投与することができる:常用の無毒の医薬として許容できる担体、補助剤及び賦形剤を含有する投与剤形において、経口的、非経口的、吸入スプレー、局所的、経直腸的、経鼻的、口腔的(baccally)、経膣的、又は植込貯蔵器。本願明細書において使用される用語「非経口」とは、皮下、静脈内、筋肉内、腹腔内、鞘内、脳室内、胸骨内、頭蓋内又は骨内への、注射及び注入技術を含む。侵襲的技法が好ましく、特に損傷した神経組織に直接投与することが好ましい。
【0143】
中枢神経系を標的として特に治療効果があるように、これらの化合物は、好ましくは末梢に投与された場合に、血液-脳関門を容易に透過するものでなければならない。血液-脳関門を容易に透過しない化合物は、脳室内経路により効果的に投与することができる。
【0144】
これらの化合物は、無菌注射用製剤、例えば無菌注射用水性又は油性懸濁剤の形で投与することもできる。これらの懸濁剤は、当該技術分野において公知の技術により、適当な分散剤又は湿潤剤及び懸濁化剤を用いて配合することができる。
無菌注射用製剤は、例えば1,3-ブタンジオール中の溶液のように、無毒の非経口的に許容できる希釈剤又は賦形剤中の無菌の注射用液剤又は懸濁剤であることもできる。水、リンゲル液及び等張塩化ナトリウム液は使用することができる許容可能な賦形剤及び溶媒のうちである。加えて、無菌の不揮発性油が、賦形剤又は懸濁媒質として通常使用される。この目的のために、合成モノ-又はジ-グリセリドのような、いずれかの無刺激の不揮発性油を使用することができる。オリーブ油及びヒマシ油を含む、オレイン酸及びそのグリセリド誘導体のような脂肪酸、特にそのポリオキシエチル化された形が、注射剤の調製において有用である。これらの油性の溶液又は懸濁液は、長鎖のアルコール希釈剤又は分散剤を含有することもできる。
【0145】
加えてこれらの化合物は、カプセル剤、錠剤、水性懸濁剤又は液剤の形状で経口投与することができる。錠剤は、乳糖及びコーンスターチのような担体、及び/又はステアリン酸マグネシウムのような滑沢剤を含有することができる。カプセル剤も、乳糖及び乾燥コーンスターチを含む希釈剤を含有することができる。水性懸濁剤は、有効成分と共に、乳化剤及び懸濁化剤を含有することができる。経口投与剤形は更に、甘味剤及び/又は香味剤及び/又は着色剤を含有することもできる。
これらの化合物は更に、坐薬の形で直腸から投与することができる。これらの組成物は、直腸で溶融し薬物を放出するために、室温では固形であるが直腸温度では液体であるような適当な非刺激性の賦形剤と、薬物を混合することにより製剤することができる。このような賦形剤は、ココアバター、ビーズワックス及びポリエチレングリコールを含む。
【0146】
更にこれらの化合物は、局所的に投与することができ、特に治療が対処する病態が、眼、皮膚又は下部腸管の神経障害を含む、局所的適用により容易に接近可能な領域又は臓器に関る場合に投与することができる。
眼への局所適用、又は眼内適用に関して、これらの化合物は、塩化ベンジルアルコニウム(benzylalkonium chloride)のような保存剤を含む又は含まずに、等張のpHを調節した無菌生理食塩水中の微小化された懸濁剤として、又は好ましくは等張のpHを調節した無菌生理食塩水中の溶液として、配合することができる。あるいは、これらの化合物は、ワセリンのような軟膏に配合することができる。
皮膚への局所適用については、これらの化合物を、例えば下記の1種以上の混合物中に懸濁又は溶解された化合物を含有する適当な軟膏に配合することができる:鉱油、液体ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレン、ポリオキシプロピレン化合物、乳化されたワックス及び水。あるいは、これらの化合物は、例えば下記の1種以上の混合物中に懸濁又は溶解された有効化合物を含有する適当なローション剤又はクリーム剤に配合することができる:鉱油、モノステアリン酸ソルビタン、ポリソルベート60、セチルエステルワックス、セチアリールアルコール(cetearyl alcohol)、2-オクチルドデカノール、ベンジルアルコール及び水。
下部腸管への局所適用は、直腸坐薬製剤(前記)又は適当な浣腸製剤において作用することができる。
【0147】
本発明の化合物は、単回投与、個別の反復投与又は連続注入により投与することができる。これらの化合物は小さく、容易に拡散可能でありかつ比較的安定しているので、連続注入に良く適している。ポンプ手段、特に皮下ポンプ手段が、連続注入にとって好ましい。
【0148】
用量
有効成分である化合物の約0.1mgから約10,000mgの桁の投与量レベルが、前述の病態の治療において有用であり、好ましいレベルは、約0.1mgから約1,000mgである。特定の患者への具体的な投与量レベルは様々な要因によって左右され、これは、使用した具体的化合物の活性;患者の年齢、体重、全身の健康状態、性別及び食事;投与時間;排泄速度;併用薬;治療される特定の疾患の重症度;及び投与形態を含む。典型的には、in vitroの用量-作用の結果は、患者への投与に関する適切な投与量の有用な指針を提供している。動物モデル試験も有用である。
適当な投与量レベルを決定するための考察は、当該技術分野において周知である。
【0149】
好ましい実施態様において、これらの化合物は、凍結乾燥した形で投与される。
この場合、本発明の化合物の1〜100mgを、マンニトール及びリン酸ナトリウムのような担体及び緩衝液と共に、個別のバイアル中で凍結乾燥することができる。
この化合物は、投与前に静菌水によりバイアル中で再構成することができる。
総体虚血の治療においては、本発明の化合物は、好ましくは、経口的、経直腸的、非経口的又は局所的に、少なくとも1日1〜6回で投与され、かつより高濃度の初回ボラス用量に続けることができる。
本発明の化合物は、化学療法薬を含む1種以上の治療薬と組合せて投与することができる。表Iは、選択された化学療法薬に関する公知のメジアン用量を示す。
これらの物質及び他の治療的物質の具体的な投与量レベルは、本発明の化合物について先に確定されたものを考慮することによって変動する。
【0150】
〔表1〕
表I.
【0151】
〔表2〕
表I.つづき
【0152】
投与方式
本発明の方法に関して、薬物デリバリーの時期及び順番を調節するいずれかの投与方式が効果的な治療に必要なように使用され、かつ反復される。このような方式は、追加の治療薬の前投与及び/又は併用投与を含むことができる。
【0153】
神経傷害からの神経組織の保護を最大にするために、これらの化合物は、冒された細胞にできる限り迅速に投与されなければならない。神経傷害が予想できる状況においては、これらの化合物は、予想される神経傷害の前に投与されなければならない。このような神経傷害の可能性が増した状況としては、手術(頚動脈内膜切除術、心臓、静脈、大動脈再建(orthopedic));動脈カテーテル法のような脈管系の手技(頚動脈(cartoid)、椎骨動脈、大動脈、心臓、腎動脈、脊髄動脈、アダムキーウイク動脈);塞栓形成物質の注入;生体恒常性のためのコイル又はバルーン;脳病巣治療のための血管分布断絶;並びに、漸増性一過性虚血性発作、塞栓症、及び続発性卒中のような、病態の予備的素因を含む。卒中又は虚血の予防的治療が不可能であるか又は実践不能である場合、事象発生時又は発生後できるだけ速やかに、罹患した細胞が該化合物を得ることは重要である。卒中と卒中の間の期間の診断及び治療手技は、細胞を更なる損傷及び死から救うために最小化されなければならない。
【0154】
卒中の動物モデル及びヒトの両方において、脳虚血の影響が、脳の代謝に迅速に、分又は時単位の時間スケールで出現することは明らかである。従っていずれかの可能性のある神経保護的治療の形は、最も即効性のある経路で施されなければならず、これは実際には経静脈経路を意味する。治療投与の最適な期間及び経路は、神経保護的化合物の個々の薬物速度論的特性、薬物の有害作用プロフィール、及び卒中を生じた傷害の性質に応じて決まるであろう。卒中後のエクサイトトキシンによる損傷は、げっ歯類においては少なくとも4時間、ヒトにおいては恐らく48時間にわたって進展する。Dykerらの論文、「虚血性卒中の神経保護的治療の期間(Duration of Neuroprotective Treatment for Ischemic Stroke)」、Stroke、29:535-542 (1998)。従って、この危機的期間を通じて神経保護を提供することは望ましいであろう。理想的には、卒中治療のための如何なる化合物も、適切に血液-脳関門を通過し、かつ脳及びCSF内で十分な治療レベルを得なければならない。
【0155】
進行性でも転移性でもない前立腺癌患者については、本発明の化合物は、(i) 転移のリスクを低減するために術前又は放射線治療前に;(ii)術中又は放射線療法と併用して;及び/又は、(iii)再発のリスクを低下しかつ残存する腫瘍細胞の増殖を阻害するために術後又は放射線療法後に、投与することができる。
【0156】
進行した又は転移した前立腺癌患者については、未治療の原発性腫瘍及び存在する転移性病変の両方において腫瘍細胞の増殖を遅延化するためのホルモン途絶療法への連続補充剤(continuous supplement)として、又はその療法の代償として、本発明の化合物が投与され得る。
【0157】
本発明の方法は、隠れた(shed)細胞を外科的介入によって取り除くことができないような部位において特に有用である。本発明の方法は、術後の回復の後、このような散在細胞により惹起される腫瘍再発の機会を減少する点で有効であろう。
【0158】
他の治療との併用
a.神経傷害
神経傷害(特に急性虚血性卒中並びに水溺(drowning)及び頭部外傷により惹起された総体虚血)の治療法において、本発明の化合物は、1種以上の治療的物質、好ましくは卒中のリスクを低下することができる物質(例えばアスピリン)と、より好ましくは続発性虚血事象のリスクを低下することができる物質(例えばチクロピジン)と共に、併用投与することができる。
本発明の化合物は、1種以上の治療的物質と、(i)単一製剤中で一緒に、又は(ii)それらの各有効物質の最適放出速度となるよう設計された個々の製剤において個別にのいずれかで、併用投与することができる。各製剤は、本発明の化合物を約0.01%〜約99.99質量%、好ましくは約3.5%〜約60質量%に加え、湿潤剤、乳化剤及びpH緩衝剤のような1種以上の医薬用賦形剤を含有することができる。
【0159】
b.血管形成-依存型疾患
NAALADase阻害剤は、1種以上の治療的物質と、(i)単一製剤中一緒に、又は(ii)それらの各有効物質の最適放出速度となるよう設計された個々の製剤において個別にのいずれかで、併用投与することができる。各製剤は、NAALADase阻害剤を約0.01質量%〜約99.99質量%、好ましくは約3.5質量%〜約60質量%に加え、湿潤剤、乳化剤及びpH緩衝剤のような1種以上の医薬用賦形剤を含有することができる。
【0160】
c. 癌
手術及び放射線療法
概して、年齢70歳以下で局在化した癌を有し余命が少なくとも10年以上とみなされる患者には、潜在的に治癒力のある療法として、手術及び放射線療法が用いられる。
しかし手術のみによる治療では、多くの患者が癌の再発を経験するであろう。放射線療法も問題が多く、それは放射線療法剤が正常組織には毒性があり、かつしばしば生命を脅かすような副作用を引き起こすからである。
本発明の手術及び放射線療法との併用は、寛解を防止し、かつ毒性のある放射線療法剤の低用量レベルを可能にする。前述の統計事実を基に、手術及び/又は放射線療法との併用、又は代用として本発明を使用するかなりの機会がある。
【0161】
放射線増感剤
放射線増感剤は、電磁放射線の毒性作用に対する癌細胞の感度を増大することがわかっている。放射線増感剤の作用態様に関するいくつかの機構が文献において示されており、これは以下を含む:低酸素細胞の放射線増感剤(例えば、2-ニトロイミダゾール化合物及び二酸化ベンゾトリアジン化合物)が、低酸素の組織の再酸素化を促進し、及び/又は損傷を受けた酸素ラジカルの発生を触媒すること;DNA塩基のアナログであってよい、非低酸素細胞の放射線増感剤(例えば、ハロゲン化されたピリミジン)は癌細胞のDNAに優先的に組込まれ、それによりDNA分子の放射線誘導型の破壊を促進し及び/又は正常なDNA修復機構を妨げること;並びに、様々な他の可能性のある作用機構が、疾患の治療における放射線増感剤に関して仮定されていること。
【0162】
現在の多くの癌治療のプロトコールが、X-線の電磁放射により活性化された放射線増感剤を使用している。x-線で活性化された放射線増感剤の例は、以下を含むが、これらに限定されるものではない:メトロニダゾール、ミソニダゾール、デスメチルミソニダゾール、ピモニダゾール、エタニダゾール、ニモラゾール、マイトマイシンC、RSU 1069、SR 4233、EO9、RB 6145、ニコチンアミド、5-ブロモデオキシウリジン(BUdR)、5-ヨードデオキシウリジン(IUdR)、ブロモデオキシシチジン、フルオロデオキシウリジン(FudR)、ヒドロキシ尿素、シスプラチン、並びにこれらの治療上有効なアナログ及び誘導体である。
【0163】
光力学的療法(PDT)は、増感剤の電磁放射活性化剤として可視光を使用する。光力学的電磁放射増感剤の例は、以下を含むが、これらに限定されるものではない:ヘマトポルフィリン誘導体、フォトフリン、ベンゾポルフィリン誘導体、NFe6、エチオポルフィリンスズSnET2、フェノボルビド-a、バクテリオクロロフィル-a、ナフタロシアニン、フタロシアニン、フタロシアニン亜鉛、及びこれらの治療上有効なアナログ及び誘導体である。
【0164】
本発明の化合物は、癌細胞の電磁放射線の毒性作用に対する感度を増大するために、電磁放射線増感剤と併用投与することができる。電磁放射線増感剤と本発明の併用は、寛解を予防し、かつ低い電磁放射線線量レベルを可能にする。本発明の化合物、組成物及び方法と電磁放射線の組合せは、癌治療において、電磁放射線単独よりも効果があるはずである。
【0165】
電磁放射線増感剤と併用した場合、本発明の化合物は更に下記化合物の1種以上と併用投与することもできる:標的細胞への放射線増感剤の取込みを促進する化合物;標的細胞への治療薬、栄養及び/又は酸素の流れを制御する化合物;電磁放射線の追加の有無に関らず、腫瘍に作用する化学療法剤;又は、癌又は他の疾患を治療するためのその他の治療薬である。このような治療薬の例は、以下を含むが、これらに限定されるものではない:5-フルオロウラシル、ロイコボリン、5'-アミノ-5'デオキシチミジン、酸素、カルボゲン、赤血球輸血、過フッ化炭素(例えば、Fluosol-DA)、2,3-DFG、BW12C、カルシウムチャネルブロッカー、ペントキシフィリン、ヒドララジン、及びL-BSOである。化学療法剤の例は、表Iに列記した。
【0166】
ホルモン療法
薬物及び/又は睾丸切除術によるホルモン途絶療法は、癌の更なる増殖及び転移を促進するホルモンを遮断するために使用される。時間がたつにつれて、これらの患者の事実上全員について、原発性及び転移性腫瘍の両方がホルモン非依存性となり、治療抵抗性となる。本発明の化合物の連続補充は、この可能性のある転移-寛容状態(metastasis-permissive state)を防止又は逆行するために使用することができる。
【0167】
化学療法
化学療法は、いくつかの癌治療の形において成功している。しかし他の形の癌治療においては、化学療法は最後の手段として利用されるのみである。あらゆる症例において、化学療法は化学療法剤が正常組織にとって毒性がありかつしばしば生命を脅かすような副作用を生じるので問題が多い。加えて化学療法は高い失敗率及び/又は寛解率を有することが多い。
本発明の化学療法との併用は寛解を防止し、かつ毒性のある化学療法剤の低用量レベルを可能にする。本発明の方法と化学療法の併用は、癌治療において化学療法単独よりも効果があるはずである。
【0168】
免疫療法
本発明の化合物は、癌治療のためのモノクローナル抗体と併用することもできる。本発明は、ポリクローナル又はモノクローナル抗体-由来の薬剤を基にした免疫療法と共に使用することもできる。これらの薬剤は当該技術分野において周知であり、かつストロンチウム-89に結合されたモノクローナル抗体のような放射標識されたモノクローナル抗体を含む。
【0169】
NAALADase阻害剤のin vivo毒性
NAALADase阻害のin vivo毒性作用は、マウスにおいて試験されている。これらの結果は、NAALADase阻害剤がマウスにとって無毒であることを示しており、このことはヒトにとっても、治療有効量投与した場合に同様に無毒であることを示唆している。代表的な開示は、米国特許第5,672,592号、第5,795,877号、第5,804,602号、第5,824,662号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
【0170】
NAALADase活性のin vitro阻害
式I-Vの様々な化合物が、NAALADase活性のin vitro 阻害について試験されている。結果の一部は、米国特許第5,672,592号、第5,795,877号、第5,863,536号、第5,880,112号及び第5,902,817号、更には特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に開示されており、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。他の結果は、下記表IIに示す。
【0171】
【表3】
【0172】
【表4】
【0173】
【表5】
【0174】
【表6】
【0175】
【表7】
【0176】
【表8】
【0177】
【表9】
【表10】
【表11】
【表12】
【0178】
虚血に対するNAALADase阻害剤in vitroアッセイ
虚血時のNAALADase阻害剤のin vitro作用を調べるために、皮質細胞培養物を、虚血性発作時(シアン化カリウム及び2-デオキシグルコース)及びその後1時間、様々なNAALADase阻害剤で処理した(実験の詳細については、Vornovらの論文、J. Neurochem.、65(4):1681-1691 (1995)を参照のこと)。結果の一部については、米国特許第5,672,592号、第5,795,877号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
他の結果は、下記表IIIに示されている。神経保護作用は、EC50で表しているが、これは虚血発作後にグルタメート毒性の50%低下を引き起こすのに必要な濃度である。
【0179】
【表13】
【0180】
【表14】
【0181】
【表15】
【0182】
【表16】
【0183】
【表17】
【0184】
【表18】
【0185】
【表19】
【0186】
様々な濃度のNAALADase 阻害剤である化合物3の%毒性として測定された作用の用量-反応は、米国特許第5,672,592号、第5,795,877号、第5,804,602号、第5,824,662号、第5,863,536号、第5,880,112号及び第5,902,817号、並びに特許証発行料金が支払われている米国特許出願第08/825,997号、第08/833,628号、第08/842,360号及び第08/899,319号に見ることができ、これらの特許及び出願の全内容は引用により本願明細書に含まれるものとする。
【0187】
Sprague-DawleyラットにおけるMCAO後の脳損傷に対するNAALADase阻害剤のin vivoアッセイ
In vivoにおいて脳損傷に対するNAALADase 阻害剤の神経保護作用を試験するために、Sprague-Dawleyラットを、溶媒、及び化合物1又は2-(3-スルファニルプロピル)ペンタン二酸のいずれかで処理した。
対照群には、Hepes緩衝生理食塩水を与えた。
【0188】
4匹の薬物処置群は、化合物1を与えた。各ラットについて、中大脳動脈閉塞(MCAO)の60分後のIVボラス注射により処置を開始し、その直後に続けて4時間IV注入を0.5ml/時の速度で行った。第1群(n = 9)は、用量100mg/kgのIVボラス、その後20mg/kg/時のIV注入4時間を行なった。第2群(n = 11)は、用量30mg/kgのIVボラス、その後の6mg/kg/時のIV注入4時間を与えた。第3群(n = 9)は、用量10mg/kgのIVボラス、その後の2mg/kg/時のIV注入4時間を与えた。第3群のラットは、更に閉塞後120分、180分及び360分にも処置を受けた。第4群(n = 8)は、用量3mg/kgのIVボラス、その後の3mg/kg/時のIV注入4時間を受けた。
【0189】
2種の追加の薬物処置群は、2-(3-スルファニルプロピル)ペンタン二酸を与えた。各ラットは、中大脳動脈閉塞(MCAO)の120分後のIVボラス注射により処置を開始し、その直後に続けて4時間IV注入を0.5ml/時の速度で行った。第5群は、用量30mg/kgのIVボラス、その後6mg/kg/時のIV注入を4時間与えた。第6群は、用量10mg/kgのIVボラス、その後2mg/kg/時のIV注入を与えた。
【0190】
再灌流の22時間後、これらのラットを安楽死させ、それらの脳を摘出した。7つの対照切片(厚さ2mm)を採取し、2,3,5-トリフェニル-塩化テトラゾリウム(tetraxolium)(TTC)の1%溶液で20分間染色し、その後10%ホルマリンで固定した。最頭側脳切片の前部及び後部表面並びに残り6つの各切片の後表面を造影した。各脳の梗塞の大きさを、コンピュータを使用したデジタル画像分析システム(LOATS)を用いて、定量化した。TTC-染色を完全に欠いている脳領域は、梗塞組織を代表していると特徴づけた。各ラットの総梗塞容積を、各々連続した脳面積の数値積分(numeric integration)により算出した。
各群のラットの総梗塞容積を、下記表IV(a)及びIV(b)に示した。
【0191】
〔表20〕
表IV(a) 化合物1で処理したラット
【0192】
〔表21〕
表IV(b) 2-(3-スルファニルプロピル)ペンタン二酸で処理したラット
【0193】
溶媒で処理したラットは、平均総脳梗塞容積265±32mm3を示した。
化合物1で処理したラットは、有意に小さい梗塞サイズを示した。化合物1で処理した4群の平均の総脳梗塞容積は、第1群が123±31mm3(p = 0.014、溶媒群に対して);第2群が、141±78mm3(p = 0.002、溶媒群に対して) ;第3群が、152±32mm3(閉塞後60分で処置;p = 0.0058、溶媒群に対して);第4群が、117±22mm3(p = 0.0037、溶媒群に対して)であった。これらの結果は、ラットの卒中MCAOモデルにおいて、化合物1が、閉塞後60分、120分、180分、及び360分で投与された場合に、神経保護性であることを示している。
【0194】
2-(3-スルファニルプロピル)ペンタン二酸30mg/kgのIVボラス、その後の6mg/kg/時のIV注入4時間で処理したラットも、溶媒処理ラットよりも、有意に小さい梗塞サイズを示した。従って、2-(3-スルファニルプロピル)ペンタン二酸はこの特定の用量レベルで、閉塞後120分に投与した場合に、ラットの卒中のMCAOモデルにおいて神経保護的である。
【0195】
卒中患者は、虚血の発生と治療開始時点の間に顕著な時間的遅れを経験することが多い。従って、長期の治療ウィンドウを伴う神経保護剤の必要性がある。前述のデータは、本発明の化合物が、ラットの卒中MCAOモデルにおいて、少なくとも6時間の治療ウィンドウを有することを示している。当業者は、ヒトにおいてこの期間がより長いと予想するであろう。
【0196】
脳損傷に対するNAALADase阻害剤のin vivoアッセイプロトコール
雄のSprague-Dawleyラット(260-320g)を用いた。これらは、個飼し、自在に摂食・摂水させた。体重を維持する必要があるならば、実験の2日前に食事制限した。各ラットは、2種の手術を受けた:IV注入のための大腿静脈カニューレ挿管及びMCAO。手術時に、ラットは、吸入マスクを使って、酸素で送達される1.5%ハロタンで麻酔した。体温をモニタリングし、かつ恒温加熱システムを用い、正常温度に調節した。最初に、カテーテルを左大腿静脈から挿入した。30分後、MCAO術のためにラットを再麻酔した。MCAOは、Longらの論文、Stroke、20:84-91 (1989)に記された血管内縫合法を用いて行った。詳細に述べると、右外頚動脈(ECA)を露出し、凝着しかつ切断した。動脈切開術により、鈍端及び0.05%ポリ-1-リシンコートを伴う3-0のモノフィラメントナイロン縫合糸を、ECAの近位断端へ挿入した。これを、頚動脈分岐から、前大脳動脈の近位部へ到達するまで22mm進め、これによりMCAに起源する閉塞を生じた。ラットを覚醒させ;2時間後、ラットを再灌流のために再度麻酔し、その間ナイロン糸をECA断端へと引っ張り、血液のMCAへの再循環を可能にした。
【0197】
卒中が誘発した脳グルタメートレベルの上昇に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、卒中が誘発した細胞外グルタメート上昇を伴うラットにおいて、高グルタメート血症障害に対するその作用についてin vivo試験した。このプロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に含まれるものとする。結果は、化合物3による処置が、線条においては、卒中-誘発性細胞外グルタメートの上昇を有意に減弱し;かつ、頭頂皮質においては、同時に発生するグルタメートの変化を完全に妨げたことを示した。
【0198】
脊髄後根神経節-シュワン細胞共存培養物における髄鞘形成に対するNAALADase阻害剤のin vitroアッセイ
NAALADaseの阻害は、坐骨神経を寒冷病変形成(cryolesion)した後、溶媒処置したマウスと比べて、髄鞘形成した軸索の数の有意な増加及びミエリンの肥厚化を生じた(Soc. Neurosci. Abstr.、23(2):2302 (1997))。本発明者らは、NAALADaseが、ミエリン形成のシグナル伝達において役割を果たし、かつNAALADase阻害は髄鞘形成を促進するであろうと仮定した。この仮説を検証するために、本発明者らは、良く特徴付けられた髄鞘形成のin vitroモデルシステムにおいて、いくつかのNAALADase阻害剤の作用を試験した。脊髄後根神経節-シュワン細胞共存培養を、先に公表されているように確立した(Einheberら、J. Cell Biol.、123:1223)。共存培養の7日後髄鞘形成が始まり、その後、血清及びアスコルビン酸を、様々な用量(1nM〜10μM)のNAALADase阻害剤及びプロゲステロン(20nM;陽性対照)と共に添加した。髄鞘形成の程度を、公知のミエリンマーカーであるミエリン塩基性タンパク質(MBP)の免疫細胞化学染色を用いて、14〜21日の間試験した。
免疫染色された培養物の定量分析は、NAALADase阻害剤添加後に、髄鞘形成した軸索数が溶媒で処理した培養物中の軸索と比べて、有意に用量-反応に相関して増加したことを明らかにした。図1A-C及び2A-Cに示されたように、NAALADase阻害剤である化合物3及び2(1nM)で2週間処理すると、MBP免疫染色の有意な増加が生じた。高用量の化合物3又は化合物2(1μM)で処理された培養物は、最大用量のアスコルビン酸及びプロゲステロンで処理された培養物よりも、より大きい程度の髄鞘形成を生じた。これらの結果は、NAALADaseの阻害が、髄鞘形成を促進しかつ脱髄性疾患(demyelinating disease)の治療において臨床的に有用であり得ることを示唆している。
【0199】
坐骨神経寒冷病変(sciatic nerve cryolesion)に伴うミエリン形成に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、坐骨神経寒冷病変を形成した雄のマウスにおける神経再建及び髄鞘形成に対するその作用について、in vivoで試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に含まれるものとする。結果は、化合物3で処置されたマウスの坐骨神経が、髄鞘形成された軸索数の増大及びミエリン肥厚化を生じたことを示している。
【0200】
パーキンソン病に対するNAALADase阻害剤のin vivoアッセイ
化合物3を、MPTPで病変形成したマウスにおけるパーキンソン病に対するその作用について、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、化合物3を与えたMPTP病変マウスが、TH-染色したドパミン作用性神経の顕著な回復を示し、このことは化合物3がMPTP-毒性に対して保護的であることを示唆している。
【0201】
ダイノルフィン誘発性脊髄損傷に対する NAALADase阻害剤のin vivoアッセイ
化合物3を、ダイノルフィンで誘発した脊髄損傷を伴うラットにおける、エクサイトトキシン性脊髄損傷に対するその作用について、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、ダイノルフィンAと同時投与した化合物3は、注射の24時間後の運動スコアにおいて有意な改善を生じ、このことは、化合物3が、ダイノルフィン-誘発性脊髄損傷に対し効果的な保護を提供することを示唆している。
【0202】
筋萎縮性側索硬化症(ALS)に対するNAALADase阻害剤のin vitroアッセイ
化合物3について、脊髄の器官培養において筋萎縮性側索硬化症(ALS)に対するその作用を、in vitroにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容は引用により本願明細書に組入れられている。結果は、化合物3が、THA-誘発毒性に対して用量-依存性の保護を発揮することを示した。
【0203】
アルコール好餌ラットにおけるエタノール消費に対するNAALADase阻害剤のin vivoアッセイ
化合物3について、アルコール好餌ラットにおけるエタノール消費に対するその作用を、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容が本願明細書に参照として組入れられている。結果は、化合物3が、体重又は24時間の摂水に影響を及ぼすことなくエタノール消費を有意に低下したことを示し、このことは、NAALADaseが、アルコール摂取行動の神経系による調節に関係していることを示唆している。
【0204】
オスのLong-Evans ラットにおけるニコチン自己投与に対するNAALADase阻害剤のin vivoアッセイ
化合物3について、ラットにおけるニコチン自己投与に対するその作用を、in vivoにおいて試験した。プロトコール及び結果は、特許証発行料金が支払われている米国特許出願第08/899,319号に開示されており、その出願の全内容が本願明細書に参照として組入れられている。結果は、化合物3が、累積食物摂取に加え、ニコチンの自己投与を減少したことを示した。この結果は、NAALADase阻害剤以外の要因が、ニコチンの自己投与の減少に寄与し得ることを示唆しているが、ニコチン使用の神経系による調節におけるNAALADaseの関与を反証するものではない。ラットの食物摂取に対する作用は、過剰な薬物用量によって引き起こされた毒性に起因しているであろう。
【0205】
Sprague-Daw1eyラットにおけるコカインに対する行動的増感現象に対するNAALADase阻害剤のin vivoアッセイ
NAALADaseは、豊富な神経ペプチドNAAGを加水分解し、グルタメート(GLU)を遊離する。本発明者らは、NAALADaseの阻害は、このGLU供給源を妨害することにより増感(sensitization)を減弱すると仮定した。発明者らは、コカインの精神運動刺激作用を顕在化する増感現象に対する化合物3の影響を評価した。雄のSprague-Dawleyラットに、コカイン(20mg/kg/日 x 5日間;i.p.)またはセール(sale)(1.0ml/kg)を、ホームケージ(home cage)注射で与えた。注射の15分前に、ラットに化合物3を10及び50mg/kgの用量で与えた。コカイン(20mg/kg)-誘発性歩行活動を、3日後に評価した。急性のコカインは、コカイン暴露の作用を増大した(例えば増感)。化合物3をコカインと共に与えた動物においては、活動の増強は有意に低下していた。化合物3それ自身は、自発運動又は生理食塩水に対する反応を変化させなかった。結果は、図23に図示した。データは、化合物3はコカインが誘発した増感の発生を弱めることを示した。増感におけるGLUの仮定された役割を考慮すると、NAALADase阻害剤は反復コカイン投与の結果生じる行動馴化を妨げることが示された。
【0206】
癌に対するNAALADase阻害剤のin vitroアッセイ
LNCaP細胞において、化合物3及びキスカル酸の前立腺癌に対する作用を試験した。プロトコール及び結果は、米国特許第5,804,602号に開示されており、その全内容は引用により本願明細書に組入れられている。結果は、化合物3及びキスカル酸の濃度が増加するに従い、LNCaP細胞増殖が有意に減少したことを示し、これは、NAALADase阻害剤が、癌、特に前立腺癌の治療に有効であることを示唆している。
【0207】
In vitro癌アッセイのプロトコール
ウシ胎仔血清(FCS)10%を含有するRPMI 1640培地中の細胞を、24ウェルプートに播種し、かつ24時間接着させ、その後キスカル酸(10-9〜10-6)又は化合物3(10-11〜10-8)を7日間添加した。7日目に、細胞に、3H-チミジンを4時間パルスし、収穫し、放射活性を測定した。値は、各処置について6個の個別の細胞ウェルの平均±SEMで示している。全ての実験は少なくとも2回行った。
キスカル酸及び化合物3の非特異的細胞増殖抑制作用を制御するために、これらの物質を同時にNAALADase非含有前立腺癌細胞株DU145についても評価した(Carterら、Proc. Natl. Acad. Sci. USA、93:749-753 (1996))。キスカル酸及び化合物3による処置が細胞増殖に有意な作用を示さないならば、これらの物質のNAALADase阻害活性は前立腺癌細胞株に対するそれらの細胞増殖抑制作用に大いに寄与するものである。
【0208】
細胞株及び組織培養
LNCaP細胞は、Johns Hopkins School of Medicine(ボルチモア, MD)のDr. William Nelsonのご好意により得た。DU145細胞は、アメリカンタイプカルチャーコレクション(ATCC)(ロックビル、MD)から入手した。細胞は、5%熱失活したウシ胎仔血清、2mMグルタミン、100ユニット/mlペニシリン、及び100μg/mlストレプトマイシン(Paragon社)を補充した、RPMI-1640培地において、加湿したインキュベータ内で、37℃、5%CO2/95%空気大気中で培養した。
【0209】
[3H]チミジン取込みアッセイ
細胞を、RPMI-1640 培地中に1 x 103細胞/mlで懸濁し、かつ24ウェルプレートに、500μl/ウェルで播種した。24時間インキュベーションした後、様々な濃度のキスカル酸(シグマ社)又は強力なNAALADase阻害剤である化合物3(Jacksonらの方法(J. Med. Chem.、39(2):619-622)に従って合成)を、ウェルに添加し、かつプレートをインキュベータに戻した。3、5及び7日目に、培地及び薬物を新鮮なものと交換した。播種後8日目に、各ウェルを、1μCi 3H-チミジン(New England Nuclear社)で4時間パルスした。その後培地を取り除き、ウェルをリン酸緩衝生理食塩水(pH7.4)で2回洗浄した。各ウェルの内容物を、順次0.2N NaOHの250μlに可溶化し、かつシンチレーションバイアルに移した。UltimaGold (Packard社)シンチレーションカクテル5mlを添加し、放射活性を、Beckman LS6001シンチレーションカウンターを用いて測定した。
【0210】
全ての合成化合物の純度及び/又は同一性は、薄層クロマトグラフィー、高速液体クロマトグラフィー(HPLC)、質量スペクトル、及び元素分析により確認した。
核磁気共鳴(NNR)スペクトルは、Bruker分光計を用いて得た。化学シフトは、内部標準テトラメチルシランに対するppmで記した。分析用薄層クロマトグラフィー(TLC)を、積層済みのシリカゲルGHLFプレート(Analtech社、ニューアーク、DE)上で行った。プレートの視認は、UV光、リンモリブデン酸-エタノール、及び/又はヨウ化プラチナチャーリング(iodoplatinate charring)を用いて行った。フラッシュクロマトグラフィーは、Kieselgel 60、230-400メッシュ(E. Merck社、ダルムシュタット、西ドイツ)上で行った。溶媒は、試薬等級又はHPLC等級のいずれかであった。特に記さない限り、反応は、周囲温度及び窒素雰囲気下で進行させた。溶液は、Buchi回転蒸発器を用いて、減圧下で蒸発した。
【0211】
癌に対するNAALADase阻害剤のin vivoアッセイ
癌へのNAALADase阻害剤のin vivo作用を調べるために、LNCaP細胞を注射したncr雄マウス、及びDunning G細胞を注射したCopenhagan同系ラットに、化合物3を様々な用量で皮下及び/又は腫瘍内投与した。処置後の平均腫瘍容積(mm3)及び腫瘍:対照比(%T/C)を、図3-7に示した。
【0212】
結果は、LNCaP腫瘍は、化合物3の皮下投与に反応したことを示した。低用量1及び3mg/kg並びに最高用量30mg/kgは、腫瘍増殖に明らかに作用を示さなかった(図3)。用量10mg/kgは、86日目に、腫瘍増殖を、対照の24%に有意に阻害した(p = 0.006)(図4)。
Dunning G腫瘍も、化合物3の皮下投与に反応した。低用量1及び3mg/kgは、腫瘍増殖に反応を示さない一方で、2種のより高い用量10及び30g/kgは、腫瘍サイズを有意に縮小した(図5)。腫瘍サイズの縮小は、用量10mg/kgでは対照の38%(p = 0.03)に、用量30mg/kgでは対照の22%であった(図6)。
【0213】
LNCaP腫瘍は、化合物3の腫瘍内投与にも反応した。3種の低用量レベル(0.025、0.25及び2.5μg/日)は、腫瘍増殖を実質的に緩やかにし、最大の減少は用量0.025μg/日で認められた(表V)。対照群の投与42日後の腫瘍容積は807.3±197.3mm3であるのに対して、0.025μg/日処置群は465.7±176mm3であった(図7)。
【0214】
〔表22〕
表V 化合物3の抗腫瘍活性
【0215】
In vivo癌アッセイのプロトコール
皮下の薬物デリバリー
LNCaPモデル(化合物3):
雄のncrヌードマウスで、5〜6週齢のものに、Matrigel(登録商標)中の5 x 106 LNCaP細胞を右側腹部に注射した(総注射容量0.1ml)。細胞注射の2週間後、化合物3の毎日の皮下注射(s.c.)を、下記の用量で開始した:1、3、10及び30mg/kg。
対照は、毎日50mM HEPES s.c.を与えた。一旦腫瘍が触知可能となったならば、これらを週2回測定した。
DUNNING Gモデル(化合物3):
雄のCopenhagen同系ラットで、8〜10週齢のものに、107のDunning G細胞を両側腹部に注射した。細胞注射の2週間後、化合物3の毎日の皮下注射(s.c.)を、下記の用量で開始した:1、3、10及び30mg/kg。対照は、毎日50mM HEPES s.c.を与えた。腫瘍は週2回測定した。
【0216】
腫瘍内への薬物デリバリー:
LNCaPモデル(化合物3):
雄のncrヌードマウスで、5〜6週齢のものに、Matrigel(登録商標)中の107 LNCaP細胞を右側腹部に注射した(総注射容量0.1ml)。腫瘍が予定のサイズ(50〜60mm3)に達したら、各8匹のマウスを6処置群に無作為に割り当てた。化合物3を毎日腫瘍内に、下記の用量で容量0.05mlで投与した:25、2.5、0.25及び0.025μg。
対照には、毎日50mM HEPES 50μlを腫瘍内投与した。腫瘍は週2回測定した。
処置に対する反応を、2種の方法でモニタリングした。第一には、各群の平均腫瘍容積を、腫瘍:対照比(%T/C)として示し、これらの値はある時点で比較した。第二は腫瘍容積対時間をモニタリングした。
【0217】
血管形成に対するNAALADase阻害剤の日用量のin vivoアッセイ
雌のC57B1マウスで、8〜10週齢(5匹/群)のものに、Matrigel(登録商標)0.5mlと共に、血管形成因子である塩基性繊維芽細胞増殖因子(bFGF)を150ng/mL、及び化合物3の0、0.47μM又は4.7μMと共に皮下注射した。Matrigel(登録商標)注射は速やかにゲルを形成した。Matrigel(登録商標)注射と同日に、化合物3のMatrigel(登録商標)プラグ周辺への皮下注射を開始した。Matrigel(登録商標)注射の7日後、Matrigel(登録商標)プラグを摘出し、組織検査を行った。
日用量の濃度および、その時の最初のMatrigel(登録商標)プラグ組成物を、表VIに示した。
【0218】
〔表23〕
表VI NAALADase阻害剤の日用量の濃度
【0219】
図8Aに詳細に示したように、良好な血管形成反応が溶媒処置群において認められた。3mg/kg及び30mg/kgの日用量群で得られたMatrigel(登録商標)プラグ内の血管又は血管形成の減少は、各々、図8B及び図8Cにおいて示した。
【0220】
血管形成に対するNAALADase阻害剤の連続用量のin vivoアッセイ
下記表VIIに示した化合物3濃度のミニ浸透圧ポンプを、C57B1雌マウス(5匹/群)に植え込んだ。このとき、溶媒(50mM Hepes)を充填したミニポンプも植え込んだ。24時間後、マウスに、Matrigel(登録商標)0.5ml及び血管形成因子である塩基性繊維芽細胞増殖因子(bFGF)150ng/mLを、各々皮下注射した。Matrigel(登録商標)/bFGF注射後13日目に、ゲルを回収し、ホルマリンに固定し、かつ切片をトリクローム-マッソン(Trichrome-Masson)染色液で染色した。
【0221】
〔表24〕
表VII NAALADase阻害剤の連続投与濃度
【0222】
各々図9及び10に示されたように、溶媒及び1μg/日用量群において、強力な血管形成性の反応が認められた。図11及び12に示したように、化合物3の用量10μg/日及び100μg/日のデリバリーは、Matrigel(登録商標)/bFGFゲル内の血管形成を有意に減少した。
【0223】
糖尿病性ニューロパシーに対するNAALADase阻害剤のin vivoアッセイ
ストレプトゾトシン(STZ)-誘発性末梢性糖尿病性ニューロパシーモデルにおいて、NAALADase阻害剤をin vivo試験した。雄のSprague-Dawleyラットで体重200〜250gのものに、STZ 60mg/kgを尾静脈に静脈内注射し、糖尿病とした。血漿ブドウ糖レベルを、STZ投与後3週間測定した。血漿ブドウ糖レベルが>300mg/dL(17mM)のSTZ-動物のみを、本試験に用いた。灼熱痛閾値及び逃避待ち時間(withdrawal latency)を用いて、小脊髄後根神経節(DRG)知覚神経の状態を評価した。疼痛は、Ugo basil社(バレーゼ、イタリア)が製作した Basile Plantar装置を使った足底試験(Hargreavesの方法)を用いてモニタリングした。
【0224】
STZ投与の2ヶ月後、糖尿病動物は、逃避待ち時間の差で測定した場合、非糖尿病対照と比べて、痛覚過敏であった。この時点で、ラットに、NAALADase阻害剤化合物2(50mg/kg)又はビヒクルを、1日1回20日間腹腔内投与した。灼熱痛反応を、投与後3、5、12及び19日目に測定した。図13に示したように、投与5日目に、化合物2を投与された動物は、ビヒクル投与動物と比較して、逃避待ち時間において有意な増加を示した。この差は、観察期間を通じて維持された。
これらのデータは、NAALADase阻害剤が、実験的糖尿病の知覚ニューロパシーに対して保護的であり、かつ末梢性ニューロパシーの治療において有用であろうということを示唆されている。
【0225】
ホルマリン、酢酸及び慢性狭窄(Constricture)誘導(CCI)疼痛モデルにおける痛覚過敏に対するNAALADase阻害剤のin vivoアッセイ
最近の証拠は、興奮性のアミノ酸であるグルタメートが、中枢性及び末梢性の両方で媒介された疼痛感において主要な役割を果たしていることを示唆している。
神経性グルタメートのひとつの供給源は、豊富な神経ペプチドNAAGに由来し、これはNAALADaseによって加水分解されると遊離のグルタメートを放出すると考えられている。本発明者らは、NAALADaseの阻害が、このグルタメート供給源を妨害することにより、疼痛を制限することができるのではないかと仮定した。この仮説を検証するために、本発明者らは、ホルマリン-、酢酸-及び慢性狭窄性損傷(CCI;“Bennettモデル”)の疼痛モデルにおけるいくつかのNAALADase阻害剤の抗痛覚作用の可能性について試験した。ホルマリンモデルにおいて、ラットに、化合物3(50mg/kg)又はビヒクルを毎日7日間i.p.投与した。7日目に、5%ホルマリンを、ラットの後肢の背側に注射した。結果を、図14-19に図示した。化合物3の前処置は、ホルマリンモデルの早期相及び後期相の両方において、たじろぎ行動(flinching behavior)を強固に緩和した(各々、13.8±6.4を、 2.5±3へ低下、p = 0.02、及び58.0±9.8を、0.5±0.58に低下、p = 0.0001;図14)。化合物3の処置は、モルヒネ(5mg/kg)による急性前処置よりも更に強固であった。疼痛の酢酸モデルにおいては、酢酸(0.6%)が誘導した身じろぎ(writhing)は、化合物3(図15)、化合物2(図16)、化合物1(図17)を前処置したマウスにおいて、ビヒクルの対照マウスと比べて、有意に緩和された。最後に疼痛のCCI誘導モデルにおいては、動物は、術後10日目から化合物3(50mg/kg)を18日間毎日i.p.投与された。
化合物3は、灼熱痛反応で調べた場合に、坐骨神経狭窄後に、劇的に痛覚過敏を軽減した。18日目に、ラットのビヒクルによる同様の処置群と比べて、疼痛は98%緩和されていた(スコア差-0.2±1.9 vs. -4.75±2.4;p = 0.0001;図18)。これらのデータは、NAALADaseの阻害が、急性及び慢性の両方の疼痛の有用な治療モダリティであることを示している。
【0226】
ニューロパシー疼痛に対するNAALADase阻害剤のin vivoアッセイ
雄のSprague-Dawleyラット(200-225g)を、ストレプトゾトシン(STZ、リン酸緩衝生理食塩水中70mg/kg)を静脈内投与することにより、糖尿病とした。糖尿病動物を、5群に分けた:ひとつの群は、化合物2(10mg/kg又は1mg/kg)、化合物1(10mg/kg又は1mg/kg)又はビヒクルを与えた。別の動物群(非-STZ処置)を、非糖尿病対照として用いた。薬物/ビヒクル処置は、STZ投与後45日目の糖尿病動物から開始した。STZ-誘発性糖尿病ラットは、血糖値が320mg/dl以上に上昇した直後に、熱源に対する感受性について試験した(STZ後30日目)。その後ラットを、Hargreaves装置に馴化させ、かつ灼熱の疼痛感を、後肢の背側表面に向けた赤外線熱源を用いてモニタリングしたところ、動物がその肢を引っ込めるのに要した時間はほぼ0.1秒であった(方法の詳細については、Hargreavesらの論文(1998)を参照のこと)。線源の強度は、対照動物(非-STZ処置)の平均待ち時間が、およそ10秒となるよう調節した。各動物は、8回試験し、平均スコア差(非糖尿病対照の待ち時間の平均と糖尿病の待ち時間の平均の間)を、図19A及び19Bに図示した。糖尿病ラットは、非糖尿病対照と比べて痛覚過敏を示し(より短い反応待ち時間)、STZ投与後30日に始まり、これはビヒクル処置ラットでは次第に悪化していった。この痛覚過敏反応は、化合物1又は2(20mg/kg i.p.、毎日)処置を行なった糖尿病ラットでは完全に逆行した。従ってこれらの結果から、NAALADaseの阻害は、ニューロパシー疼痛を減弱することが示された。
【0227】
ニューロパシー疼痛の進行に対するNAALADase阻害剤のin vivoアッセイ
化合物3
ラットにおいて、坐骨神経の三分岐点の近位に1mm間隔で緩くこの神経の周りを縛ることで結紮を4個つくり、坐骨神経結紮を行った。この処置を行った後のラットは、熱に対する痛覚過敏及び異疼痛を示した。動物は、Hargreaves装置に慣れたので、後肢の背側表面に赤外線熱源を向け、動物がその肢を引っ込めるのに要する時間を観察した。スコアの差異(操作側対対照側について、肢が反応するまでの待ち時間の間)を決定した。動物は、術後10日目に、化合物3(50mg/kg i.p. 毎日)又はビヒクルの受取りを開始した。化合物3による処置は、連続して痛覚過敏のビヒクル-処置対照と比べて、2本の肢の間のスコアの差異を劇的に正常化した。正常な(手技を施さず)ラットは、両肢でほぼ等しい待ち時間を有した。この作用は、薬物投与の11日目に顕著に始まり、かつ本試験終了時(21日間の毎日投与)まで維持された。スコアの差異を図20に図示した。結果は、NAALADaseの阻害が、CCI-関連した痛覚過敏を緩和することを示した。
【0228】
化合物1及び2
雄のBB/Wラット(BRI, Mass)は、膵B細胞の細胞が媒介した自己免疫破壊を自発的に発症しており、その結果インスリン依存型(I型)糖尿病を発症する(Guberski、1994)。これらのラットは、特徴決定された、線維消失及び変性のような神経欠損、ヒトの糖尿病の末梢神経において認められるものに関連した変化を随伴するニューロパシーを生じることが示されている(Yagihasi、1997)。このことは、これらのラットを、この主要障害の今後の治療のための新規化合物の実験的試験にとって価値のあるものとしている。本試験において、化合物1及び化合物2は、糖尿病ニューロパシーの進行を変更するそれらの能力について試験した。ラットは、糖尿病発症(高血糖症)時に開始しその後最大6ヶ月目まで、毎日化合物1もしくは化合物2(10mg/kg i.p.)又はビヒクルの注射を行なった。ビヒクルを与えた非糖尿病ラットの別の群も試験した。全ての動物は、体重、尿量、血糖値及びグリコヘモグロビンについて連続してモニタリングした。試験の最初の月には、全ての動物を、熱に対する疼痛感について毎週試験した(Hargreaves装置)。最初の1ヶ月経過後は、2週間に1回に、その後月1回試験した。試験は、赤外線熱源をラットの後肢の背側表面に当てること、及び動物がその肢を引っ込めるのに要する時間を観察することからなる(方法の詳細については、Hargreavesらの論文(1998)を参照のこと)。各動物は8回試験し、逃避待ち時間の平均を記録した。
【0229】
これらの結果は、図24に図示した。結果は、糖尿病ラットは、非糖尿病対照と比べて、痛覚過敏(より短い反応待ち時間)を有することを示した。糖尿病薬物処置ラット(化合物1及び化合物2の両方)は、糖尿病ビヒクル処置ラットよりも、より長い逃避待ち時間を示し、これは処置の4週間後に始まり、かつ処置の6ヶ月間持続した。
【0230】
神経伝導速度も、処置開始後最初の8週間は隔週で、その後処置6ヶ月間を通じて毎月1回測定した(方法の詳細についてはDe Koningらの論文(1987)を参照のこと)。結果は図25に図示した。糖尿病動物は総じて、非糖尿病対照と比べて、神経伝導速度の遅延を示した。しかし糖尿病動物がNAALADase阻害剤(化合物1又は化合物2のいずれかを用量10mg/kg)の毎日の注射を受けると、ビヒクル投与を受けた糖尿病対照の場合と比べて、神経伝導欠損の重症度の有意な低下を示した。これは、処置8週目に明らかに始まり、かつ6ヶ月の本試験終了時までずっと同様の程度が持続した。他方で、糖尿病ビヒクルは、溶媒処置開始後6〜16週間、神経伝導速度の進行性の悪化を示し、これは6ヶ月間を通じて維持された。
【0231】
糖尿病ニューロパシーに対するNAALADase阻害剤のin vivoアッセイ
運動神経及び知覚神経の伝導速度も、STZ-糖尿病動物において、処置後4、8及び12週間に測定した(方法の詳細についてはDe Koningらの論文(1987)を参照のこと)。簡単に述べると、麻酔をかけたラットにおいて、刺激用針電極を、坐骨及び脛骨の神経に挿入し、記録用電極を遠位足筋上皮下に配置した。結果は、図21A、21B、22A及び22Bに示した。ビヒクルを与えた糖尿病動物は、非糖尿病動物と比べて、運動及び知覚神経の両方の伝導速度の有意な低下を示した。化合物2の10mg/kgによる4、8及び12週間の処置は全て、運動及び知覚神経の両方の伝導速度を改善(増大)する傾向があり、運動及び知覚神経伝導速度の顕著な改善が、各々、12週後及び8週後に認められた(図31A)。試験した低用量の化合物2(1mg/kg)は同様の作用を有した(図31B)。いずれかの用量の化合物1による動物の処置も、同じく糖尿病対照において運動及び知覚両神経の伝導速度を増大し、用量10mg/kg処置群については処置の12週間後に、1mg/kg処置群については処置後早い時期に、顕著に増大した(図32A及び32B)。従って、これらの結果は、NAALADase阻害が、糖尿病ニューロパシーの進行を変化させることを示している。
【0232】
精神分裂病に対するNAALADase阻害剤のin vivoアッセイ
ラットを、ヒトの精神病症状に相当する、半狂乱の走り及び絶え間ない頭部旋回(incessant head-turning)のような、PCP発症徴候を伴うように処置した。従って、精神分裂病に対するNAALADase阻害の作用を調べるために、PCP (5mg/kg)を受取る前のラットに、化合物1(50mg/kg)、化合物2(50mg/kg)又はビヒクル(H2O)を腹腔内投与した。頭の揺動(head rolling)スコアを、PCP注射後2時間にわたって測定した。結果を、図26及び27に図示した。これらの結果は、化合物1(図27)又は2(図26)の前投与は、PCPの歩行活動への作用を有意に低減した。PCPは、前前頭皮質においてグルタメート流出を増大することが示されており、かつPCP-誘発した歩行活動の減少は、NAALADase阻害が、シナプス前グルタメート生成活性を減弱することにより、PCPの行動に対する作用を緩和することを示唆している。従ってNAALADase阻害剤は、精神分裂病のPCPモデルにおいて有効であることを示した。
【0233】
下記実施例は、本発明を例証するものであるが、その限定を意図したものではない。特に記さない限り、全てのパーセントは最終組成物100質量%を基にしている。
【実施例1】
【0234】
実施例1
2-[[(2,3,4,5,6-ペンタフルオロベンジル)-ヒドロキシホスフィニル]-メチル]-ペンタン二酸の調製
スキームV:R1=2,3,4,5,6-ペンタフルオロベンジルヘキサメチルジシラザン(21.1mL, 100mmol)を、激しく攪拌しているホスフィン酸アンモニウム(8.30g, 100mmol)に添加し、かつ得られる懸濁液を、105℃で2時間攪拌した。その後2,3,4,5,6-ペンタフルオロ臭化ベンジル(5.0g, 27.0mmol)溶液を、懸濁液に0℃で滴下した。混合物を室温で19時間攪拌した。その後反応混合物を、ジクロロメタン(50mL)で希釈し、1N HCl(50mL)で洗浄した。有機相を分離し、Na2SO4上で乾燥し、かつ濃縮し、白色固形物4.72gを得た。これをジクロロメタン(50mL) に溶解し、かつこの溶液にベンジルアルコール(3.24g, 30mmol)を添加した。次に溶液に、1,3-ジシクロヘキシル-カルボジイミド(DCC) (6.19g, 30mmol)を0℃で添加し、懸濁液を室温で14時間攪拌した。溶媒を減圧除去し、残留物をEtOAc中に懸濁した。
得られた懸濁液をろ過し、ろ液を濃縮した。残留物を、シリカゲルクロマトグラフィー(ヘキサン:EtOAc、4:1から1:1)で精製し、2[[(2,3,4,5,6-ペンタフルオロベンジル)-ヒドロキシホスフィニル]-メチル]ペンタン二酸を、白色固形物として得た(収率34%)。
Rf 0.69 (i-PrOH:H2O, 7:3)
1H NMR (D2O):δ 1.8-2.0 (m, 3H), 2.1-2.3 (m, 1H), 2.3-2.5 (m, 2H), 2.7-2.9 (m, 1H), 3.29 (d, 2H)
元素分析
計算値:C13H12F5O6P、0.45 H2O: C, 39.20;H, 3.26
実測値:C, 39.17;H, 3.28
【実施例2】
【0235】
実施例2
2-(ホスホノメチル)ペンタンジオエートの調製
スキームIII
ジベンジル2-メチレンペンタンジオエート
ベンジルアクリレート(500g, 3.0mol)を、油浴中で100℃に加熱した。加熱を停止し、HMPT (10g, 61mmol)を滴下しながら、内部温度を140℃以下に維持した。
一旦添加を完了した後、混合物を攪拌し、室温に冷却した。シリカスラリー(5:1 ヘキサン/EtOAc)を添加し、混合物を、乾燥シリカ栓を備えたカラムに充填した。カラムを、1:1 ヘキサン/EtOAcで洗浄し、その画分を一緒にし、蒸発させ、透明な明るい金色の液体450gを得た。液体を、高真空(200μHg)下、185℃で蒸留し、無色透明の液体212g (42%)を得た。
1H NNR (CDCl3):7.3 ppm (m, 10H), 6.2 ppm (s, 1H), 5.6 ppm (s, 1H), 5.2 ppm (s, 2H), 5.1 ppm (s, 2H), 2.6 ppm (m, 4H)
【0236】
ジベンジル2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタンジオエートジクロロメタン350mLを溶媒とするジベンジルホスファイト(9.5g, 36mmol)を、0℃に冷却した。この攪拌している溶液に、トリメチルアルミニウム(18.2ml, ヘキサン中2.0M溶液, 36.4mmol)を添加した。30分後、ジクロロメタン90ml中のジベンジル2-メチレンペンタンジオエート(2, 6.0g, 37mmol)を、10分間かけて滴下した。その後無色透明の溶液を室温まで温め、一晩攪拌しながら放置した。その後混合物を、5%HClをゆっくり添加することにより反応停止した。更に1.5時間攪拌した後、下側の有機相を除去し、水相をジクロロメタン100mlで1回抽出した。有機相を一緒にし、乾燥し(MgSO4)、かつ蒸発させ、透明な明るい金色の液体を得た。液体を、シリカゲル(4cm*30cm)上でクロマトグラフィーにかけ、勾配(4:1-1:1)溶媒システム(ヘキサン/EtOAc)で溶離した。所望の生成物を含有する画分を一緒にし、蒸発させ、 ジベンジル2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタンジオエート (7.1g, 42%)を、無色透明の液体として得た。
その後液体を、Kughleror装置上で、66.7Pa(0.5mmHg)及び195〜200℃で蒸留した。留出物を廃棄し、残留している明るい金色の油分を、シリカゲル(1:1, ヘキサン/EtOAc)上でクロマトグラフィーにかけ、無色透明の油状物として2-[[ビス(ベンジルオキシ)ホスホリル]メチル]-ペンタン二酸ジベンジル2.9gを得た。
TLC Rf 0.5 (1:1, ヘキサン/EtOAc)
1H NNR (CDCl3):7.1-7.4 (m, 20H), 5.05 (s, 2H), 4.8-5.03 (m, 6H), 2.8 (1H), 2.22-2.40 (m, 3H), 1.80-2.02 (m, 3H)
【0237】
2-(ホスホノメチル)ペンタン二酸
ベンジルペンタンジオエート (2.9g, 4.9mmol)を、10%Pd/Cを0.29g (6モル%)含有するメタノール20mlの混合物に添加した。この混合物を、Parr水素添加装置上で276kPa(40psi)で24時間水素化し、ろ過し、かつ蒸発させ、透明なわずかに金色がかった粘稠な油分(3, 1.0g, 90%)を得た。
1H-NNR (D2O):2.6-2.78 (m, 1H), 2.25-2.40 (m, 2H), 1.75-2.15 (m, 4H)
【実施例3】
【0238】
実施例3
2-[[[2-(カルボキシ)プロピル]ヒドロキシ-ホスフィニル]メチル]ペンタン二酸の調製
スキームX
ジ-t-ブチル2-メチレンペンタンジオエート
t-ブチルアクリレート(465g, 3.628mol)を、窒素下で100℃に加温し、その後ヘキサメチルリン酸トリアミド(10g, 61.2mmol)を滴下し、添加速度を反応温度100℃を維持するように調節した。反応混合物を冷却し、その後シリカ栓上に注ぎ(〜1000ml)、かつシリカを4:1 ヘキサン/酢酸エチルで完全に洗浄した。溶媒を減圧除去し、得られた油分を蒸留した。一部の物質を、高真空下室温から50℃で収集し、破棄した。その後温度を〜80℃に上昇し、生成物(300g, 65%、沸点は300μで67〜70℃)を、透明油状物として得た。
1H NMR (CDCl3):δ1.4 (m, 18H), 2.4 (t, 2H), 2.6 (t, 2H), 5.5 (s, 1H), 6.0 (s, 1H)
【0239】
ジ-t-ブチル2-[(ヒドロキシホスフィニル)メチル]-ペンタンジオエート
ホスフィン酸アンモニウム(162.6g, 1.96mol)及び1,1,1,3,3,3-ヘキサメチルジシラザン(316g, 1.96mol)の混合物を、105℃で2時間加熱した。反応混合物を、氷浴中で冷却し、かつジクロロメタン(1000ml)に溶解した2-メチレンペンタン-1,5-二酸 ジ-t-ブチル(251g, 0.979mol)を滴下した。反応混合物を一晩室温に加温した。その後反応混合物を、蒸留水(500ml)で急冷し、かつ有機相を保持した。水相を、ジクロロメタンで2回洗浄し、及び一緒にした有機相を硫酸マグネシウム上で乾燥した。その後溶媒を減圧除去し、わずかに黄色の油状物が残った(315g, 100%)。この生成物は、次の反応に使用するのに十分な純度であることが分かった。
1H NMR (CDCl3): 1.4 (m, 18H), 1.9 (m, 3H), 2.1 (m, 1H), 2.3 (m, 2H), 2.7 (m, 1H), 6.5 & 7.9 (d, 1H, P-H), 11.0 (s, 1H)
【0240】
ジ-t-ブチル2-[(t-ブトキシホスフィニル)メチル]-ペンタンジオエート
ジクロロメタン(1000ml)を溶媒とするジ-t-ブチル2-[(ヒドロキシホスフィニル)メチル]-ペンタン-1,5-ジオエート (315g, 0.977mol)を、氷浴で冷却し、かつ窒素下で、t-ブタノール(123.1g, 1.66mol)、4-ジメチルアミノピリジン(1g, 8.2mmol)、及び1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(281g, 1.47mol)を添加した。反応液を一晩攪拌した。反応混合物に水を添加し、ジクロロメタン相を保持しかつ乾燥し、溶媒を減圧下で除去した。得られる残留物を、カラムクロマトグラフィーで精製し、所望の生成物を1:1から2:3のヘキサン/酢酸エチルで溶離した。生成物を含有する画分を、減圧下で濃縮し、透明油状物が残留した(260g, 70%)。
1H NMR:(CDCl3): δ 1.4 (m, 27H), 1.8 (m, 1H), 1.9 (m, 2H), 2.1 (m, 1H), 2.3 (m, 2H), 2.7-2.8 (m, 1H), 6.7 & 8.0 (d, 1H, P-H)
【0241】
ジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]-t-ブトキシホスフィニル]メチル] ペンタンジオエート
THF(100ml)を溶媒とするジ-t-ブチル2-[(t-ブトキシホスフィニル)メチル]-ペンタン-1,5-ジオエート (13.62g, 36.0mmol)及びベンジルメタクリレート(6.35g, 36.0mmol)を窒素下で、水素化ナトリウム(0.14g, 油中60%分散体, 3.60mmol)を添加した。3時間後、反応混合物を水(300ml)に注ぎ、及びエーテル(100ml)を添加した。有機相を分離・保持し、水相を再度エーテル(100ml)で洗浄した。一緒にした有機抽出物を、硫酸マグネシウム上で乾燥し、溶媒を減圧除去した。得られた残留物を、カラムクロマトグラフィーで精製し、生成物を2:3 EtOAc/ヘキサンで溶離した。溶媒を減圧除去し、透明な油状物(10 .5g, 53%)を得た。
1H NMR (CDCl3):δ1.3 (m, 3H), 1.5 (m, 27H), 1.7 (m, 2H), 1.9 (m, 2H), 2.2 (m, 4H), 2.6 (m, 1H), 2.9 (m,1H), 5.1 (m, 2H), 7.3 (m, 5H)
【0242】
2-[[[2-(ベンジルカルボキシ)プロピル]ヒドロキシホスフィニル]-メチル]ペンタン二酸
ジクロロメタン(10ml)中のジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]t-ブトキシホスフィニル]メチル] ペンタン-1,5-ジオエート(1.6g, 2.89mmol)溶液に、窒素下で、トリフルオロ酢酸(10ml)を添加した。反応混合物を2時間攪拌し、その後減圧下で濃縮した。反応残留物に更にジクロロメタンを添加し、これを減圧下で除去した。生成物を、酢酸エチルに溶解し、水で洗浄し、その後有機相を硫酸マグネシウム上で乾燥し、溶媒を減圧除去し、透明な油状物を得た(800mg, 72%)。
1H NMR (D20):δ1.2 (m, 3H), 1.6-1.8 (m, 4H), 2.1 (m, 2H), 2.2 (m, 2H), 2.6 (m, 1H), 2.8 (m, 1H), 5.0 (m, 2H), 7.3 (m, 5H)
C17H22PO8・1.0 H2Oの分析 計算値:C, 50.50;H, 6.23
実測値:C, 50.52;H, 5.92。
【0243】
ジ-t-ブチル2-[[[2-(カルボキシ)プロピル]-t-ブトキシ-ホスフィニル]メチル]ペンタンジオエート
ジ-t-ブチル2-[[[2-(ベンジルカルボキシ)プロピル]t-ブトキシホスフィニル]メチル] ペンタン-1,5-ジオエート (8.9g, 16.1mmol)、炭素触媒に担持されたパラジウム(10%, 1.0g)及び酢酸エチル(100ml)の溶液を、水素下(60psi)で16時間振盪した。反応混合物を、セライト上でろ過し、ろ液を減圧下で濃縮し、透明な油状分が残留した(7.5g, 100%)。
1H NMR (CDCl3):δ1.3 (d, 3H), 1.4-1.5 (m, 27H), 1.8 (m, 2H), 1.9 (m, 2H), 2.2 (m, 4H), 2.7 (m, 1H), 2.9 (m, 1H)
【0244】
2-[[[2-(カルボキシ)プロピル]ヒドロキシホスフィニル]メチル]-ペンタン二酸ジクロロメタン(10ml)を溶媒とするジ-t-ブチル2-[[[2-(カルボキシ)プロピル]t-ブトキシホスフィニル]メチル]-ペンタン-1,5-ジオエート (2.1g, 4.53mmol)に、窒素下で、トリフルオロ酢酸(10ml)を添加した。反応混合物を2時間攪拌し、その後減圧下で濃縮した。反応残留物に、追加のジクロロメタンを添加し、これを減圧下で除去した。得られた残留物をアセトニトリルで粉砕し、その後減圧下で乾燥し、濃厚な透明油状物が残留した(1.2g, 89%)。
1H NMR (D2O):δ 1.2 (d, 3H), 1.9 (m, 4H), 2.2 (m, 2H), 2.4 (m, 2H), 2.8 (m, 2H)。
C10H17PO8・0.2 CH3CNの分析の計算値:C, 41.03;H, 5.83
実測値:C, 41.05;H, 5.92。
【実施例4】
【0245】
実施例4
2-[({[ベンジルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸の調製
スキームXI
ジ-t-ブチル2-[((t-ブトキシ){[ベンジルアミノ]メチル}-ホスホリル)メチル]ペンタン-1,5-ジオエート
トルエン(200mL)を溶媒とする1,3,5-トリベンジルヘキサヒドロ-1,3,5-トリアジン(14.30g, 40.0mmol)及びジ-t-ブチル2-{[(t-ブトキシ)ホスホリル]メチル}ペンタン-1,5-ジオエート(37.85g, 100mmol)を、110℃で14時間攪拌した。溶媒を減圧除去し、残留する黄色油状物を、シリカゲルクロマトグラフィーで精製し(ヘキサン/酢酸エチル, 2/1)、明るい黄色油状物23.40gを得た(収率43%):1H NMR (CDCl3):δ 1.40-1.48 (m, 27H), 1.7-2.1 (m, 4H), 2.2-2.4 (m, 3H), 2.6-3.0 (m, 3H), 3.8-4.0 (m, 2H), 7.2-7.4 (m, 5H)。
【0246】
2-[({[ベンジルアミノ]メチル}(ヒドロキシホスフィニル))メチル]-ペンタン二酸
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-[((t-ブトキシ){[ベンジルアミノ]メチル}-ホスホリル)メチル]ペンタン-1,5-ジオエート (0.498g, 1.0mmol)溶液に、トリフルオロ酢酸(5mL)を、0℃で添加し、混合物を室温で18時間攪拌した。溶媒を減圧除去した。残留油状物を、ジクロロメタン(10mL)に溶解し、かつ濃縮した。この工程を3回繰り返し、トリフルオロ酢酸を完全に除去した。得られた油状物を、メタノールで結晶化し、白色固形物0.174gを得た(収率53%):1H NMR (D2O):δ 1.40-1.48 (m, 27H), 1.7-2.1 (m, 4H), 2.2-2.4 (m, 3H), 2.6-3.0 (m, 3H), 3.8-4.0 (m, 2H), 7.2-7.4 (m, 5H)。
【実施例5】
【0247】
実施例5
2-[({[フェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[フェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.4-1.6 (m, 1H), 1.7-1.9 (m, 3H), 2.2-2.4 (m, 2H), 2.2-2.4 (m, 2H), 2.5-2.7 (m, 1H), 3.53 (d, J = 8.8 Hz, 2H), 7.3-7.5 (m, 5H)。
【実施例6】
【0248】
実施例6
2-[({[4-フルオロフェニルアミノ]メチル}-(ヒドロキシホスフィニル)メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[4-フルオロフェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.5-1.7 (m, 1H), 1.8-2.0 (m, 3H), 2.3-2.5 (m, 2H), 2.6-2.7 (m, 1H), 3.84 (d, J = 9.0 Hz, 2H), 7.2-7.5 (4H)。
【実施例7】
【0249】
実施例7
2-[({[4-メトキシフェニルアミノ]メチル}-(ヒドロキシホスフィニル)メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[4-メトキシフェニルアミノ]メチル}(ヒドロキシホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.2-1.3 (m, 1H), 1.6-1.7 (m, 3H), 2.22-2.23 (m, 2H), 2.3-2.5 (m, 1H), 3.4 (d, J = 8.9 Hz, 2H), 3.7 (s, 314), 7.0 (d, J = 12 Hz, 2H), 7.4 (d, J= 12 Hz, 2H)。
【実施例8】
【0250】
実施例8
2-[({[フェニルスルホンアミド]メチル}-(ヒドロキシホスフィニル))メチル)ペンタン二酸の調製
先の実施例4において説明した方法と類似の方法で、2-[({[フェニルスルホンアミド]メチル}(ヒドロキシ-ホスフィニル))メチル]ペンタン二酸を合成した:1H NMR (D2O):δ 1.6-2.1 (m, 4H), 2.3-2.4 (m, 2H), 2.5-2.7 (m, 1H), 2.9-3.1 (m, 2H), 7.7-8.0 (m, 5H)。
【実施例9】
【0251】
実施例9
2-({[(フェニルカルボキシアミド)メチル]-(ヒドロキシホスフィニル)}メチル)ペンタン二酸の調製
スキームXII
ジ-t-ブチル2-{[(アミノメチル)(t-ブトキシ)-ホスホリル]メチル}ペンタン-1,5-ジオエート
ジ-t-ブチルエタノール(100mL)を溶媒とする2-[((t-ブトキシ)-{[ベンジルアミノ]メチル}ホスホリル)メチル]ペンタン-1,5-ジオエート (8.20g, 16.5mmol)溶液に、炭素上に担持したパラジウム(0.50g)を添加し、懸濁液を水素下で(50psi)4日間振盪した。触媒を、セライトパッドを通すろ過により除去した。ろ液を濃縮し、無色の油状物6.629g(収率99%)を得た: 1H NMR (CD3OD):δ 1.40-1.60 (m, 27H), 1.80-2.00 (m, 3H), 2.2-2.4 (m, 3H), 2.7-3.0 (m, 3H)。
【0252】
ジ-t-ブチル2-({(t-ブトキシ)[(フェニルカルボキシアミド)-メチル]ホスホリル}メチル)ペンタン-1,5-ジオエート
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-{[(アミノメチル)-(t-ブトキシ)ホスホリル]メチル}ペンタン-1,5-ジオエート (1.222g, 3.0mmol)及び塩化ベンゾイル(0.46mL, 4.0mmol)の溶液に、トリエチルアミン(0.56mL, 4.0mmol)を0℃で添加し、その後混合物を室温で16時間攪拌した。反応混合物を、ジクロロメタン(15mL)で希釈し、1N HCl(25mL)で洗浄し、Na2SO4上で乾燥し、濃縮した。粗物質を、シリカゲルクロマトグラフィー上で精製し(酢酸エチル/ヘキサン = 2/1)、無色の油状物1.259g(収率74%)を得た:1H NMR (CDCl3):δ1.30-1.60 (m, 27H), 1.60-2.00 (m, 3H), 2.20-2.40 (m, 3H), 2.70-2.90 (m, 3H), 3.5-4.2 (m, 2H), 7.0-7.3 (m, 1H), 7.4-7.6 (m, 3H), 7,8-7.9 (m, 1H)。
【0253】
2-({[(フェニルカルボキシアミド)メチル](ヒドロキシホスフィニル)}-メチル)ペンタン二酸
ジクロロメタン(10mL)を溶媒とするジ-t-ブチル2-({(t-ブトキシ)-[(フェニルカルボキシアミド)メチル]ホスホリル}メチル)ペンタン-1,5-ジオエート (1.230g, 2.4mmol)の溶液に、トリフルオロ酢酸(5mL)を室温で添加し、かつ混合物を室温で18時間攪拌した。溶媒を減圧除去した。残留油状物を、ジクロロメタン(10mL)に溶解し、濃縮した。この方法を、3回繰り返し、トリフルオロ酢酸を完全に除去した。得られた油状物を、アセトニトリル-水で結晶化し、白色固形物0.620gを得た(収率75%):1H NMR (D2O):δ1.9-2.1 (m, 3H), 2.2-2.4 (m, 1H), 2.4-2.6 (m, 2H), 2.8-3.0 (m, 1H), 3.7-3.9 (m, 2H), 7.5-7.9 (m, 5H)。
【実施例10】
【0254】
実施例10
2-(2-スルファニルエチル)ペンタン二酸の調製
スキームXIII、R10=水素
3-(2-オキソテトラヒドロ-3-チオフェニル)プロパノエート
【0255】
【0256】
冷却したTHF(100ml)を溶媒とするリチウムジイソプロピルアミド(LDA) (98mmol)の溶液(-78℃)に、γ-チオブチロラクトン(10g, 98mmol)を滴下した。15分間攪拌した後、3-ブロモプロパン酸エチル(35.4g, 196mmol)を添加し、反応液を一晩室温で加温した。溶媒を減圧除去し、得られた残留物を、カラムクロマトグラフィーにかけ、透明な油状物3g(16%)を得た。1H NMR (CDCl3):δ 1.2 (t, 3H), 1.7 (m, 1H), 1.9 (m, 1H), 2.1 (m, 1H), 2.4 (t, 2H), 2.5 (m, 2H), 3.3 (t, 2H), 4.2 (q, 2H)。
【0257】
2-(2-スルファニルエチル)ペンタン二酸
【0258】
【0259】
THF(5ml)を溶媒とする3-(2-オキソテトラヒドロ-3-チオフェニル)プロパノエート(0.77g, 3.81mmol)の溶液に、水酸化ナトリウム(水中1M、5ml)を添加した。混合物を2日間攪拌し、その後THFを減圧除去し、水相をエーテルで洗浄し、その後HClでpH1とし、酢酸エチルで抽出した。一緒にした酢酸エチル抽出物を、硫酸マグネシウム上で乾燥し、溶媒を減圧除去した。得られた残留物を、カラムクロマトグラフィーで精製し、透明油状物150mgを得た(20%)。1H NMR (d6-DMSO) δ 1.7 (m, 3H), 1.8 (m, 1H), 2.2 (m, 2H), 2.3-2.5 (m, 4H)。
C7H12SO4の分析の計算値:C, 43.74;H, 6.29;S, 16.68
実測値:C, 43.61;H, 6.39;S, 16.55。
【実施例11】
【0260】
実施例11
2-(3-スルファニルプロピル)ペンタン二酸の調製
スキームXIX
2,2-ジメチル-5-[3-[(トリフェニルメチル)チオ]プロピル-1,3-ジオキサン-4,6-ジオン(I)
20mmolの3-[(トリフェニルメチル)チオ]プロピオン酸(6.9g)を、CH2Cl2の100mlを溶媒とするメルドラム(Meldrum)酸(2,2-ジメチル-1,3-ジオキサン-4,6-ジオン)の22mmol (3.2 g)及び4-ジメチルアミノピリジン31mmol (3.85g)で溶解した。
反応混合物を、-5℃に冷却し、CH2Cl2の50mlを溶媒とするジシクロヘキシルカルボジイミド(4.74g)22mmolの溶液を、1時間かけて滴下した。混合物を、温度<0℃に一晩放置し、その間ジシクロヘキシル尿素の微結晶が析出した。ろ過後、反応混合物を、4x 10% KHSO4、1x ブラインで洗浄し、MgSO4で2時間乾燥した。この溶液を、特徴決定も更なる精製もせずに第二工程に用いた。
【0261】
先の反応の溶液を、-5℃に冷却し、かつ98%酢酸13.3ml(220mmol)を添加した。
その後NaBH4の1.85g(50mmol)を、少量ずつ添加しながら、1時間以上攪拌した。
この反応混合物を一晩冷蔵庫に放置し、その後3x水及び2xブラインで洗浄した。
有機相を、MgSO4で乾燥し、ろ過し、かつ蒸発乾固した。残留物をEtOAcに溶解し、沈殿した少量のジシクロヘキシル尿素をろ過により除去し、ろ液をヘキサンを添加して結晶化した。2,2-ジメチル-5-[3-[(トリフェニルメチル)-チオ]プロピル]-1,3-ジオキサン-4,6-ジオン(I)の収量は7.5gであった(86%-2工程)。13C-NMR δ 20.0(q), 26.2(q), 27.2(t), 28.9(t), 32.0(t), 46.2(d), 67.0(s), 105.3(s), 127.0(d), 128.3(d), 130.0(d), 145.2(s), 165.6(s)。
【0262】
2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニルメチル)チオ]-プロピル] -1,3-ジオキサン-5-プロパン酸メチルエステル(II)
5mmolの2,2-ジメチル-5-[3-[(トリフェニルメチル) -チオ]-プロピル]-1,3-ジオキサン-5-プロパン酸-4,6-ジオン(I) (2.3g)を、3-ブロモプロパン酸メチル(3.34g = 2.18ml) 20mmol及びメタノール10mlを溶媒とするメトキシドナトリウム(20mmol)の4.37Nメタノール溶液4.6ml中に溶解した。反応混合物を、60℃で一晩加熱し、その後ヘキサン/酢酸エチル1:1でTLCを行ったところ、出発材料は検出されなかった。その後混合物を蒸発乾固し、10%KHSO4水溶液40mlと混合した。有機物質を、3部のEtOAcで抽出し、有機相を一緒にし、MgSO4で乾燥し、蒸発させた。残留物を、ヘキサン/酢酸エチルで結晶化し、2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニル-メチル)チオ]プロピル]-1,3-ジオキサン-5-プロパン酸メチルエステル(II)2.2g(77%)を得た。13C-NMR (CDCl3) δ 24.6, 29.4, 29.5, 29.6, 31.4, 32.6, 37.7, 51.9, 52.8, 66.8, 105.7, 126.7, 127.9, 129.5, 144.7, 168.4, 172.0。
【0263】
6-[(トリフェニルメチル)チオ]-1,3,3-ヘキサントリカルボン酸(III)
2.56mmolの2,2-ジメチル-4,6-ジオキソ-5-[3-[(トリフェニルメチル)チオ]プロピル]-1,3-ジオキサン-5-プロパン酸メチルエステル(II) (1.4g)を、水酸化ナトリウム18mmol(0.72g)と共に、1,4-ジオキサン5ml及び水5mlの混合物に溶解した。その後混合物を、100℃で1時間加熱し、蒸発乾固し、水に溶解し、かつ1M硫酸の添加により沈殿させた。沈殿をろ過により除去し、水で洗浄し、かつデシケータで乾燥した。6-[(トリフェニルメチル)-チオ]-1,3,3-ヘキサントリカルボン酸(III) 1.36gを得た(〜100%)。13C-NMR (MeOH) δ 25.4, 29.2, 30.7, 33.5, 33.7, 58.0, 68.3, 128.1, 129.3, 131.2, 146.7, 174.9, 176.9。
【0264】
6-[(トリフェニルメチル)チオ]-1,3-ヘキサンジカルボン酸(IV)
ジメチルスルホキシド5mlに、2.56mmolの6-[(トリフェニルメチル)チオ]-1,3,3-ヘキサントリカルボン酸(III) (1.36g)を溶解し、溶液を100℃で1時間加熱し、蒸発乾固し、水に溶解し、かつ1M硫酸の添加により沈殿した。沈殿した油状分を、超音波浴で1時間処理した後に固化した。固形物をろ過し、水で洗浄し、デシケータで乾燥した。6-[(トリフェニルメチル)-チオ]-1,3-ヘキサンジカルボン酸(IV) 1.1g (IIから2工程で89%)を得た。13C-NMR (MeOH) δ 27.9, 28.6, 33.0 (炭素2個), 33.1, 45.9, 68.1, 128.1, 129.2, 131.2, 146.8, 177.1, 179.4。
【0265】
2-(3-スルファニルプロピル)ペンタン二酸(V)
2.46mmolの6-[(トリフェニルメチル)チオ]-1,3-ヘキサンジカルボン酸(IV) (1.1g)を5mmolのトリイソプロピルシラン(0.79g)と共に、3ml CH2C13/3ml トリフルオロ酢酸の混合物に溶解し、室温で1時間静置した。その後混合物を蒸発乾固し、3x ヘキサンで洗浄した。残留している油状残留物を、水に溶解し、ろ過し、かつ凍結乾燥し、2-(3-スルファニルプロピル)ペンタン二酸(V) 0.35g (76%)を得た。13C-NMR (MeOH) δ 25.2 (t) , 28.8 (t), 32.4 (t), 33.0(t), 33.2(t), 45.9(d), 177.2(s), 179.6(s)。
【実施例12】
【0266】
実施例12
2-(4-スルファニルブチル)ペンタン二酸の調製
2-(4-スルファニルブチル)ペンタン二酸を、先に2-(3-スルファニルプロピル)ペンタン二酸について説明した方法を用いて調製した。13C-NMR (MeOH) δ 25.1(t), 27.4(t), 28.8(t), 33.0(t), 33.2(t), 35.4(t), 46.3(d), 177.2(s), 179.7(s)。
【実施例13】
【0267】
実施例13
2-(3-スルファニル-2-メチルプロピル)-ペンタン二酸の調製
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸(2種のジアステレオ異性体の混合物)を、先に2-(3-スルファニルプロピル)-ペンタン二酸について説明した方法を用いて調製した。13C-NMR (MeOH) δ 18.9(q), 19.5(q), 29.1(t), 29.6(t), 31.7(t), 32.6(t), 32.9(t), 33.0(t), 35.5(d), 35.9(d), 39.2(t), 39.7(t), 44.2(d), 44.3(d), 177.0(s), 177.1(s), 179.7(s), 179.9(s)。
【実施例14】
【0268】
実施例14
2-(2-スルファニルプロピル)ペンタン二酸及び2-(3-スルファニルプロピル)ペンタン二酸を、これらの各実施例並びに前述のin vitro及びin vivoアッセイにおいて試験した。両化合物は各々、これらの各アッセイ及び実施例においてin vitro又はin vivo活性を示すことが分かった。
【0269】
本発明はこのように説明してきたが、多くの方法で改変することができることは明らかであろう。このような改変は、本発明の精神及び範囲の逸脱とはみなされず、このような修飾は全て、上記「特許請求の範囲」の範囲に含まれることが意図されている。
【特許請求の範囲】
【請求項1】
式Iの化合物又は医薬として許容できる等価体:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、R18及びR19は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;並びに
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基(複数)で置換されていて;
但し、Xが式IIである部分であり、R8が-(CH2)2COOR19又は-(CH2)2CONHR19、AがCH2、nが0、かつZがSR13であるならば、R13は水素又はCOR19でなく;Xが式IIIである部分であり、BがNであり、かつR8が-(CH2)2COOHであるならば、R11は水素ではなく;Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項2】
Xが、式IIである部分であり;
nは、0、1、2又は3であり;
Zは、SH、SO3H、SO2H、SOH、又はS(NRHR14)2R15であり;および
Aは、O、S又はCR17R18である、請求項1記載の化合物。
【請求項3】
ZがSHである、請求項2記載の化合物。
【請求項4】
R8が-(CH2)2COOHである、請求項3記載の化合物。
【請求項5】
前記式Iの化合物が、下記からなる群より選択される、請求項3記載の化合物:
2-(2-スルファニルエチル)-ペンタン二酸;
3-(2-スルファニルエチル)-1,3,5-ペンタントリカルボン酸;
2-(2-スルファニルプロピル)ペンタン二酸;
2-(2-スルファニルブチル)ペンタン二酸;
2-(2-スルファニル-2-フェニルエチル)ペンタン二酸;
2-(2-スルファニルヘキシル)ペンタン二酸;
2-(2-スルファニル-1-メチルエチル)ペンタン二酸;
2-[1-(スルファニルメチル)プロピル]ペンタン二酸;
2-(3-スルファニルペンチル)ペンタン二酸;
2-(3-スルファニルプロピル)ペンタン二酸;
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸;
2-(3-スルファニル-2-フェニルプロピル)ペンタン二酸;
2-(3-スルファニルブチル)ペンタン二酸;
2-[3-スルファニル-2-(フェニルメチル)プロピル]ペンタン二酸;
2-[2-(スルファニルメチル)ブチル]ペンタン二酸;
2-[2-(スルファニルメチル)ペンチル]ペンタン二酸;
2-(3-スルファニル-4-メチルペンチル)ペンタン二酸;および
医薬として許容できる等価体。
【請求項6】
式Iの化合物が、2-(2-スルファニルエチル)ペンタン二酸、2-(2-スルファニルプロピル)ペンタン二酸、2-(3-スルファニルプロピル)ペンタン二酸及び医薬として許容できる等価体からなる群より選択される、請求項5記載の化合物。
【請求項7】
式Iの化合物が、エナンチオマー又はエナンチオマーが豊富な混合物である、請求項6記載の化合物。
【請求項8】
卒中の発作後60分以上で投与した場合に、哺乳類において卒中を治療する際に有効であるイオウ及び酸基の両方を含む、化合物。
【請求項9】
卒中の発作後180分以上で投与した場合に、哺乳類において卒中を治療する際に有効であるイオウ及び酸根の両方を含む、請求項8記載の化合物。
【請求項10】
卒中の発作後360以上投与した場合に、哺乳類において卒中を治療する際に有効である請求項9記載の化合物。
【請求項11】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類においてNAALADase酵素活性を阻害する方法:
(式中Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;および、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項12】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項11記載の方法。
【請求項13】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類においてグルタメート異常を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立に0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分であり、BがNでありかつR8が-(CH2)2COOHであるならば、R11は水素でなく;Xが式IIである部分であり、AがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項14】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項13記載の方法。
【請求項15】
前記グルタメート異常が、虚血である、請求項13記載の方法。
【請求項16】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項15記載の方法。
【請求項17】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において、強迫性障害、卒中、脱髄疾患、精神分裂病、パーキンソン病及びALSからなる群より選択されるグルタメート異常を治療する方法:
(式中Xは、式II、III又はIVである部分であり:
m及びnは、独立に0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基sで置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項18】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項17記載の方法。
【請求項19】
前記グルタメート異常が、精神分裂病である、請求項17記載の方法。
【請求項20】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項19記載の方法。
【請求項21】
前記グルタメート異常が、強迫性障害である、請求項17記載の方法。
【請求項22】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項21記載の方法。
【請求項23】
前記強迫性障害が、薬物依存及び摂食障害からなる群より選択される、請求項21記載の方法。
【請求項24】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項23記載の方法。
【請求項25】
前記薬物依存が、アルコール依存、ニコチン依存及びコカイン依存である、請求項23記載の方法。
【請求項26】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項25記載の方法。
【請求項27】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において神経活動に影響を与える方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基(複数)で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項28】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項27記載の方法。
【請求項29】
前記神経活動が、損傷を受けた神経の刺激、神経再生の促進、神経変性の予防及び神経疾患の治療からなる群より選択される、請求項27記載の方法。
【請求項30】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項29記載の方法。
【請求項31】
前記神経障害が、物理的損傷又は病態に起因した末梢ニューロパシー、外傷性脳損傷、脊髄の物理的損壊、卒中に関連した脳損壊、脱髄疾患及び神経変性に関係した神経疾患からなる群より選択される、請求項29記載の方法。
【請求項32】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項31記載の方法。
【請求項33】
前記神経変性に関連した神経障害が、パーキンソン病及びALSからなる群より選択される、請求項31記載の方法。
【請求項34】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項33記載の方法。
【請求項35】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において前立腺疾患を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分であり、かつAがOであるならば、nは2、3又は4であり;Xが式IIである部分であり、かつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項36】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項35記載の方法。
【請求項37】
前記前立腺疾患が、前立腺癌である、請求項35記載の方法。
【請求項38】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項37記載の方法。
【請求項39】
式Iの化合物又は医薬として許容できる等価体を有効量哺乳類に投与することを含む、哺乳類において癌を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIの部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項40】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項39記載の方法。
【請求項41】
卒中発症後60分以上で、イオウ及び酸基を含有する化合物を有効量、該哺乳類に投与することを含む、哺乳類において卒中を治療する方法。
【請求項42】
前記化合物が、卒中発症後180分以上で該哺乳類に投与される、請求項41記載の方法。
【請求項43】
前記化合物が、卒中発症後360分以上で該哺乳類に投与される、請求項42記載の方法。
【請求項44】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において血管形成を阻害する方法。
【請求項45】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において疼痛を治療する方法。
【請求項46】
疼痛が糖尿病性ニューロパシー疼痛である、請求項45記載の方法。
【請求項47】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において糖尿病性ニューロパシーを治療する方法。
【請求項48】
チオラクトンと置換されたエステルを反応し、式VIの化合物を生成する工程を含む、イオウ及び酸基の両方を含む化合物を調製する方法:
(式中、Dは、(CR21R22)nであり;
nは、0、1、2、3又は4であり;及び
R20、R21及びR22は、独立して、 水素、C1-C9直鎖又は分枝鎖のアルキル、C2-C9 直鎖又は分枝鎖のアルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群より選択され、ここで該アルキル、アルケニル、シクロアルキル及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されており;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されている。)。
【請求項49】
請求項48記載の方法によって調製された化合物。
【請求項50】
下記の工程を含む、イオウ及び酸基の両方を含む化合物の調製法:
(i)メルドラム酸をイオウ含有反応体と反応させ、イオウ含有メルドラム酸誘導体を生成する工程;及び
(ii)前記イオウ含有メルドラム酸誘導体をエステルと反応させ、式VIIの化合物を生成する工程:
(式中、Eはイオウ含有部分であり;及び
Fはプロピオン酸エステル含有部分である。)。
【請求項51】
請求項50記載の方法により調製した化合物。
【請求項52】
下記を含有する医薬組成物:
(i)下記式Iの化合物又はその医薬として許容できる等価体の有効量:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。);および、
(ii)医薬として許容できる担体。
【請求項53】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項52記載の医薬組成物。
【請求項1】
式Iの化合物又は医薬として許容できる等価体:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、R18及びR19は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;並びに
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基(複数)で置換されていて;
但し、Xが式IIである部分であり、R8が-(CH2)2COOR19又は-(CH2)2CONHR19、AがCH2、nが0、かつZがSR13であるならば、R13は水素又はCOR19でなく;Xが式IIIである部分であり、BがNであり、かつR8が-(CH2)2COOHであるならば、R11は水素ではなく;Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項2】
Xが、式IIである部分であり;
nは、0、1、2又は3であり;
Zは、SH、SO3H、SO2H、SOH、又はS(NRHR14)2R15であり;および
Aは、O、S又はCR17R18である、請求項1記載の化合物。
【請求項3】
ZがSHである、請求項2記載の化合物。
【請求項4】
R8が-(CH2)2COOHである、請求項3記載の化合物。
【請求項5】
前記式Iの化合物が、下記からなる群より選択される、請求項3記載の化合物:
2-(2-スルファニルエチル)-ペンタン二酸;
3-(2-スルファニルエチル)-1,3,5-ペンタントリカルボン酸;
2-(2-スルファニルプロピル)ペンタン二酸;
2-(2-スルファニルブチル)ペンタン二酸;
2-(2-スルファニル-2-フェニルエチル)ペンタン二酸;
2-(2-スルファニルヘキシル)ペンタン二酸;
2-(2-スルファニル-1-メチルエチル)ペンタン二酸;
2-[1-(スルファニルメチル)プロピル]ペンタン二酸;
2-(3-スルファニルペンチル)ペンタン二酸;
2-(3-スルファニルプロピル)ペンタン二酸;
2-(3-スルファニル-2-メチルプロピル)ペンタン二酸;
2-(3-スルファニル-2-フェニルプロピル)ペンタン二酸;
2-(3-スルファニルブチル)ペンタン二酸;
2-[3-スルファニル-2-(フェニルメチル)プロピル]ペンタン二酸;
2-[2-(スルファニルメチル)ブチル]ペンタン二酸;
2-[2-(スルファニルメチル)ペンチル]ペンタン二酸;
2-(3-スルファニル-4-メチルペンチル)ペンタン二酸;および
医薬として許容できる等価体。
【請求項6】
式Iの化合物が、2-(2-スルファニルエチル)ペンタン二酸、2-(2-スルファニルプロピル)ペンタン二酸、2-(3-スルファニルプロピル)ペンタン二酸及び医薬として許容できる等価体からなる群より選択される、請求項5記載の化合物。
【請求項7】
式Iの化合物が、エナンチオマー又はエナンチオマーが豊富な混合物である、請求項6記載の化合物。
【請求項8】
卒中の発作後60分以上で投与した場合に、哺乳類において卒中を治療する際に有効であるイオウ及び酸基の両方を含む、化合物。
【請求項9】
卒中の発作後180分以上で投与した場合に、哺乳類において卒中を治療する際に有効であるイオウ及び酸根の両方を含む、請求項8記載の化合物。
【請求項10】
卒中の発作後360以上投与した場合に、哺乳類において卒中を治療する際に有効である請求項9記載の化合物。
【請求項11】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類においてNAALADase酵素活性を阻害する方法:
(式中Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;および、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項12】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項11記載の方法。
【請求項13】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類においてグルタメート異常を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立に0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分であり、BがNでありかつR8が-(CH2)2COOHであるならば、R11は水素でなく;Xが式IIである部分であり、AがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項14】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項13記載の方法。
【請求項15】
前記グルタメート異常が、虚血である、請求項13記載の方法。
【請求項16】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項15記載の方法。
【請求項17】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において、強迫性障害、卒中、脱髄疾患、精神分裂病、パーキンソン病及びALSからなる群より選択されるグルタメート異常を治療する方法:
(式中Xは、式II、III又はIVである部分であり:
m及びnは、独立に0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基sで置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項18】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項17記載の方法。
【請求項19】
前記グルタメート異常が、精神分裂病である、請求項17記載の方法。
【請求項20】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項19記載の方法。
【請求項21】
前記グルタメート異常が、強迫性障害である、請求項17記載の方法。
【請求項22】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項21記載の方法。
【請求項23】
前記強迫性障害が、薬物依存及び摂食障害からなる群より選択される、請求項21記載の方法。
【請求項24】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項23記載の方法。
【請求項25】
前記薬物依存が、アルコール依存、ニコチン依存及びコカイン依存である、請求項23記載の方法。
【請求項26】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項25記載の方法。
【請求項27】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において神経活動に影響を与える方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基(複数)で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項28】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項27記載の方法。
【請求項29】
前記神経活動が、損傷を受けた神経の刺激、神経再生の促進、神経変性の予防及び神経疾患の治療からなる群より選択される、請求項27記載の方法。
【請求項30】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項29記載の方法。
【請求項31】
前記神経障害が、物理的損傷又は病態に起因した末梢ニューロパシー、外傷性脳損傷、脊髄の物理的損壊、卒中に関連した脳損壊、脱髄疾患及び神経変性に関係した神経疾患からなる群より選択される、請求項29記載の方法。
【請求項32】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項31記載の方法。
【請求項33】
前記神経変性に関連した神経障害が、パーキンソン病及びALSからなる群より選択される、請求項31記載の方法。
【請求項34】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項33記載の方法。
【請求項35】
式Iの化合物又は医薬として許容できる等価体を有効量、哺乳類に投与することを含む、哺乳類において前立腺疾患を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分であり、かつAがOであるならば、nは2、3又は4であり;Xが式IIである部分であり、かつAがSであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項36】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項35記載の方法。
【請求項37】
前記前立腺疾患が、前立腺癌である、請求項35記載の方法。
【請求項38】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項37記載の方法。
【請求項39】
式Iの化合物又は医薬として許容できる等価体を有効量哺乳類に投与することを含む、哺乳類において癌を治療する方法:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;Xが式IIの部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。)。
【請求項40】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項39記載の方法。
【請求項41】
卒中発症後60分以上で、イオウ及び酸基を含有する化合物を有効量、該哺乳類に投与することを含む、哺乳類において卒中を治療する方法。
【請求項42】
前記化合物が、卒中発症後180分以上で該哺乳類に投与される、請求項41記載の方法。
【請求項43】
前記化合物が、卒中発症後360分以上で該哺乳類に投与される、請求項42記載の方法。
【請求項44】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において血管形成を阻害する方法。
【請求項45】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において疼痛を治療する方法。
【請求項46】
疼痛が糖尿病性ニューロパシー疼痛である、請求項45記載の方法。
【請求項47】
NAALADase阻害剤を有効量、該哺乳類に投与することを含む、哺乳類において糖尿病性ニューロパシーを治療する方法。
【請求項48】
チオラクトンと置換されたエステルを反応し、式VIの化合物を生成する工程を含む、イオウ及び酸基の両方を含む化合物を調製する方法:
(式中、Dは、(CR21R22)nであり;
nは、0、1、2、3又は4であり;及び
R20、R21及びR22は、独立して、 水素、C1-C9直鎖又は分枝鎖のアルキル、C2-C9 直鎖又は分枝鎖のアルケニル、C3-C8シクロアルキル、C5-C7シクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシからなる群より選択され、ここで該アルキル、アルケニル、シクロアルキル及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されており;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されている。)。
【請求項49】
請求項48記載の方法によって調製された化合物。
【請求項50】
下記の工程を含む、イオウ及び酸基の両方を含む化合物の調製法:
(i)メルドラム酸をイオウ含有反応体と反応させ、イオウ含有メルドラム酸誘導体を生成する工程;及び
(ii)前記イオウ含有メルドラム酸誘導体をエステルと反応させ、式VIIの化合物を生成する工程:
(式中、Eはイオウ含有部分であり;及び
Fはプロピオン酸エステル含有部分である。)。
【請求項51】
請求項50記載の方法により調製した化合物。
【請求項52】
下記を含有する医薬組成物:
(i)下記式Iの化合物又はその医薬として許容できる等価体の有効量:
(式中、Xは、式II、III又はIVである部分であり:
m及びnは、独立して、0、1、2、3又は4であり;
Zは、SR13、SO3R13、SO2R13、SOR13、SO(NR13)R14又はS(NR13R14)2R15であり;
Bは、N又はCR16であり;
Aは、O、S、CR17R18又は(CR17R28)mSであり;
R9及びR13は水素であり;
R8、R10、R11、R12、R14、R15、R16、R17、及びR18は、独立して、水素、C1-C9の直鎖又は分枝鎖のアルキル、C2-C9の直鎖又は分枝鎖のアルケニル、C3-C8のシクロアルキル、C5-C7のシクロアルケニル、Ar1、ヒドロキシ、カルボキシ、カルボニル、アミノ、アミド、シアノ、イソシアノ、ニトロ、スルホニル、スルホキシ、チオ、チオカルボニル、チオシアノ、ホルムアニリド、チオホルムアミド、スルフィドリル、ハロ、ハロアルキル、トリフルオロメチル又はオキシであり、ここで該アルキル、アルケニル、シクロアルキル、及びシクロアルケニルは、独立に未置換であるか又は1個以上の置換基で置換されていて;および、
Ar1は、炭素環式又はヘテロ環式部分であり、これは未置換であるか又は1個以上の置換基で置換されていて;
但し、Xが式IIである部分でありかつAがOであるならば、nは2、3又は4であり;Xが式IIである部分でありかつAがSであるならば、nは2、3又は4であり;並びに、Xが式IIである部分でありかつAが(CR17R18)mSであるならば、nは、0、2、3又は4である。);および、
(ii)医薬として許容できる担体。
【請求項53】
ZがSHであり;及び、R8が-(CH2)2COOHである、請求項52記載の医薬組成物。
【図1A】


【図1B】

【図1C】

【図2A】

【図2B】

【図2C】

【図3】
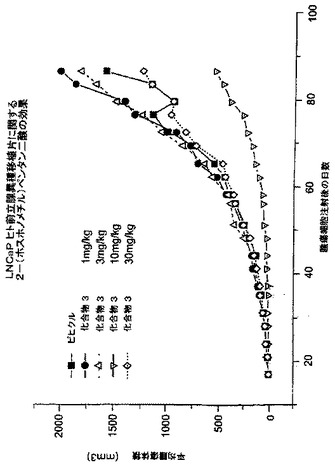
【図4】
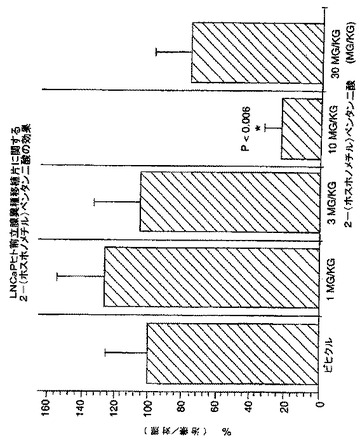
【図5】
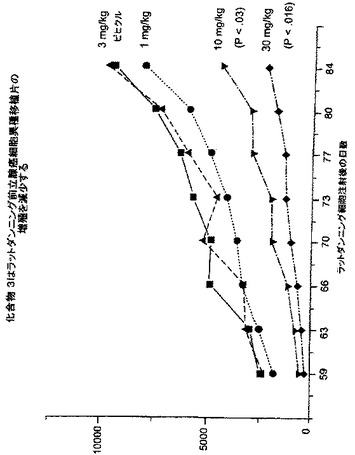
【図6】
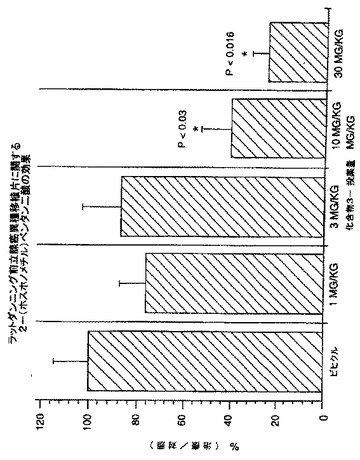
【図7】
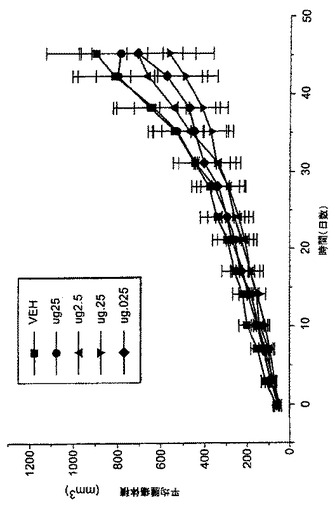
【図8A】

【図8B】

【図8C】

【図9】

【図10】

【図11】
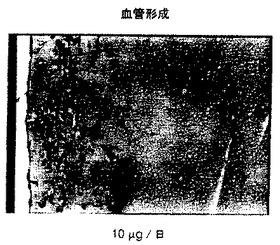
【図12】
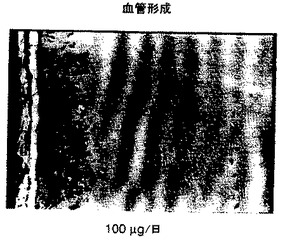
【図13】

【図14】
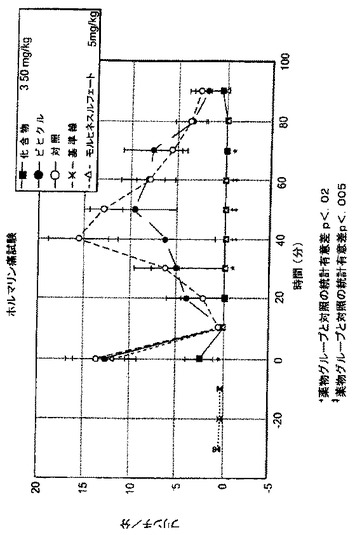
【図15】
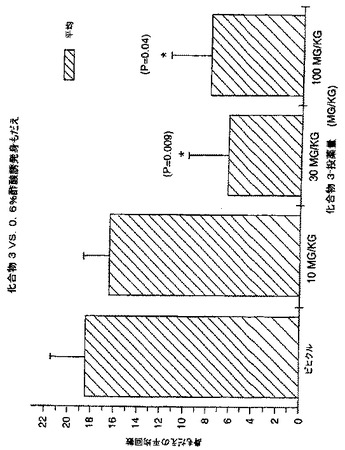
【図16】

【図17】

【図18】
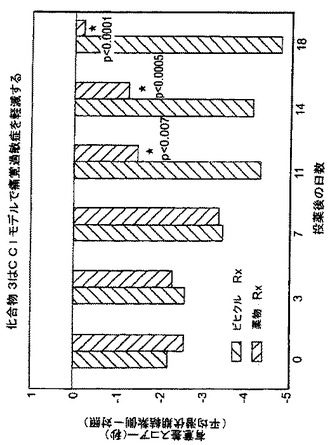
【図19A】
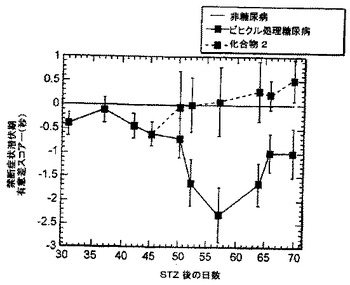
【図19B】

【図20】
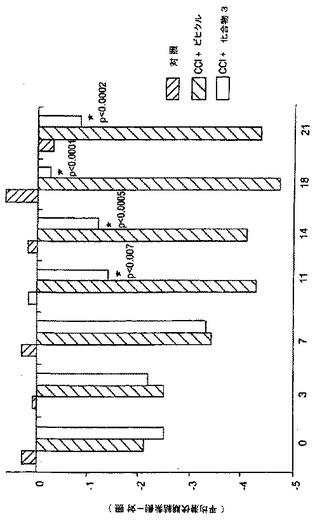
【図21A】
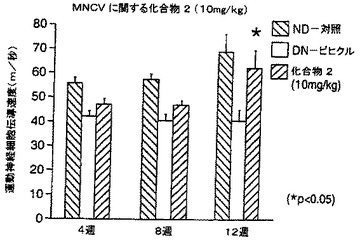
【図21B】
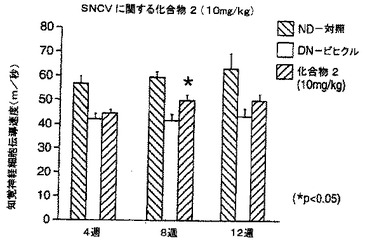
【図22A】

【図22B】

【図23】
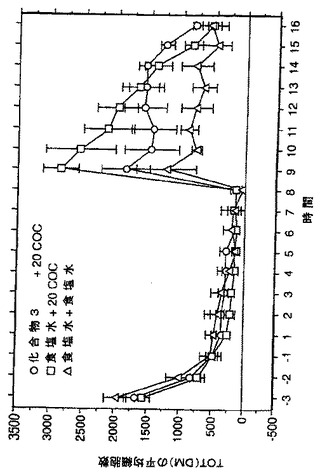
【図24】
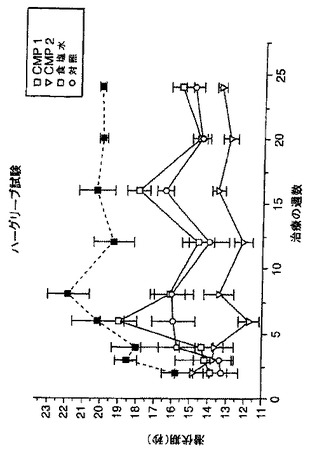
【図25】
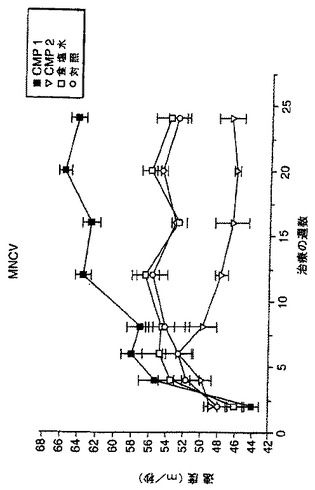
【図26】
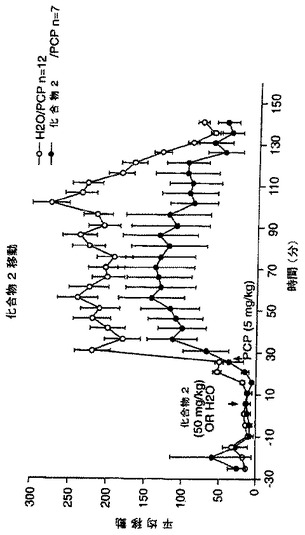
【図27】
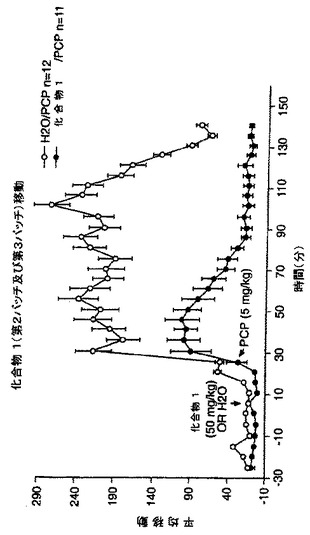
【図1B】
【図1C】
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図8C】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19A】
【図19B】
【図20】
【図21A】
【図21B】
【図22A】
【図22B】
【図23】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2010−1312(P2010−1312A)
【公開日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願番号】特願2009−228198(P2009−228198)
【出願日】平成21年9月30日(2009.9.30)
【分割の表示】特願2000−558073(P2000−558073)の分割
【原出願日】平成11年7月2日(1999.7.2)
【出願人】(509091158)エイザイ コーポレイション オブ ノース アメリカ (1)
【Fターム(参考)】
【公開日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願日】平成21年9月30日(2009.9.30)
【分割の表示】特願2000−558073(P2000−558073)の分割
【原出願日】平成11年7月2日(1999.7.2)
【出願人】(509091158)エイザイ コーポレイション オブ ノース アメリカ (1)
【Fターム(参考)】
[ Back to top ]