
医薬結晶化合物
【課題】 エラスターゼ阻害活性を有する特定化合物の結晶を得ることを目的とする。
【解決手段】 N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミドの結晶、その製造方法および該結晶を有効成分として含む医薬。
【解決手段】 N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミドの結晶、その製造方法および該結晶を有効成分として含む医薬。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は医薬、特にエラスターゼ阻害作用を有する複素環式化合物の結晶多形、その製造方法およびその複素環式化合物を有効成分とする医薬に関する。
【背景技術】
【0002】
次式で示される構造を有する、N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下、「化合物II」または「ケトン体」ということもある)
【0003】
【化1】

【0004】
は、エラスターゼ阻害剤として有用な化合物であり、特許文献1の実施例10に記載されている。
【0005】
通常、医薬品の有効成分となる化合物は、強い薬理作用を有し、且つ低毒性であるとともに品質が一定であることが条件となる。そのためには結晶状態の化合物を原薬として用いることが求められている。しかしながら、特許文献1には上記化合物IIのみならずそこに記載の他の化合物群がすべて結晶ではなく非晶質であり、さらに結晶を得るための製造方法も記載されていない。
【特許文献1】国際公報第03/066671号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、エラスターゼ阻害活性を有する化合物の結晶を得ることを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記化合物II、即ち、ケトン体の結晶を得る為に、鋭意検討を試みた。しかし、通常使用される種々の有機溶媒をもってしてもこの化合物を結晶化させることができなかった。ところが、化合物IIを特定の有機溶媒に溶解し、容器を開口状態にして放置したところ結晶化させることに成功した。しかし、分析の結果、得られた結晶は、大気中の水を吸って、水分子1個分の重量が増えていることが判明した。そこで、さらに検討を進めた結果、得られた結晶は、ケトン体のトリフルオロメチルケトン部位に1分子の水が附加したジオール体の結晶であること、およびこの結晶のエラスターゼ阻害活性は、ケトン体のそれがそのまま維持されているとの知見をえて、本発明を完成した。
【0008】
即ち、本発明は、
項1:N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下「化合物I」または「ジオール体」ということもある)
【0009】
【化2】
【0010】
の結晶を提供するものである。具体的には以下の発明を提供する。
【0011】
項2: 回折角2θで表して、約7.0°〜8.5°の位置にピークを有するX線粉末回折パターンを示す項1に記載の結晶。
【0012】
項3: 回折角2θで表して、約7.0°〜8.5°の位置に少なくとも1本の強いピークを有するX線粉末回折パターンを示す項2に記載の結晶。
【0013】
項4: 回折角2θで表して、ほぼ5.7°、6.0°、7.6°、8.0°、12.3°、12.6°および16.2°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのA型結晶。
【0014】
項5: 示差走査熱量測定において、約139℃に脱水吸熱ピークを有する項4に記載の化合物IのA型結晶。
【0015】
項6: 回折角2θで表して、ほぼ6.5°、8.0°、8.6°、9.5°、10.2°、11.0°、14.2°および20.3°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのB型結晶。
【0016】
項7: 示差走査熱量測定において、約120℃に脱水吸熱ピークを有する項6に記載の化合物IのB型結晶。
【0017】
項8: 回折角2θで表して、ほぼ6.0°、7.2°、10.6°、11.9°、14.4°、15.2°および18.0°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのC型結晶。
【0018】
項9: 示差走査熱量測定において、約128℃に脱水吸熱ピークを有する項8に記載の化合物IのC型結晶。
【0019】
項10: 項1〜9のいずれかに記載の結晶を有効成分として含む医薬。
【0020】
項11: 項1〜9のいずれかに記載の結晶を含む医薬組成物。
【0021】
項12: 化合物IのA型結晶の製造方法であって、化合物IIを、以下のいずれかの有機溶媒:
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタンからなる群から選択される単一溶媒;
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒とn−ヘキサンとの混合溶媒;
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とメチルシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ペンタンとの混合溶媒;
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ヘプタンとの混合溶媒;
トルエンとジエチルエーテルとの混合溶媒;
テトラリンとクメンとの混合溶媒;および
トルエン、n−ヘキサンおよびメチルシクロヘキサンからなる混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのA型結晶を晶析させることを特徴とする製造方法。
【0022】
項13: 化合物IのB型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
トルエンまたはクロロベンゼンである単一溶媒;または
シクロヘキサンまたはn−ヘプタンと、トルエンとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのB型結晶を晶析させることを特徴とする製造方法。
【0023】
項14: 化合物IのC型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
ジエチルエーテル;または
ジエチルエーテルとシクロペンチルメチルエーテルとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのC型結晶を晶析させることを特徴とする製造方法。
【発明の効果】
【0024】
本発明によれば、品質が安定している化合物の結晶を医薬品の原薬として供給することができる。
【発明を実施するための最良の形態】
【0025】
本発明の化合物Iの結晶は.化合物II,即ち、ケトン体を出発物質として製造することができる。このケトン体は、特許文献1に記載の方法によって製造することができる。
【0026】
本発明者らの研究によって、化合物Iの結晶として、少なくとも3種類の異なる結晶形の存在が確認された。この3種類の結晶形を、以下、A型、B型およびC型という。
【0027】
化合物Iの結晶は、X線粉末回折パターンにおいて、特徴的なピークを示す。即ち、該結晶は、回折角2θで表して、約7.0°〜8.5°の位置にピーク、具体的には、少なくとも1本の強いピークを有するX線粉末回折パターンを示す。
【0028】
以下の実施例で示す装置、条件下で測定したときのA型、B型及びC型各結晶形の特徴的な主ピークの回折角(2θ)は以下のとおりである。
【0029】
【表1】
【0030】
また、A型、B型、C型各結晶形の示差走査熱量測定(DSC)における特徴および熱重量分析(TG)における特徴は以下のとおりである。
【0031】
【表2】
【0032】
本発明の化合物IのA型結晶、B型結晶およびC型結晶は、本明細書に記載の物理化学的性質によって特定されるものであるが、上記データは、測定方法や測定器具によって多少変わるものであるから、厳密に解されるべきではない。また、A型結晶、B型結晶およびC型結晶は、純粋な結晶に限られず、未知の結晶形を含めた他の結晶形が混在しているものでもよい。
【0033】
A型結晶
化合物IのA型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、A型結晶は、化合物IIを特定溶媒に溶解し、水を添加したのち放置することにより製造できる。「放置」は、通常、ゆっくりと攪拌しながら数時間ないし数日間、通常、室温において行われる。
【0034】
ここにおける、特定溶媒としては、例えば、次の単一溶媒が挙げられる。
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタン
または、以下の組み合わせの混合溶媒が例示できる。
【0035】
(1)シクロヘキサンと下記溶媒との混合溶媒
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0036】
(2)n−ヘキサンと下記溶媒との混合溶媒
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒。
【0037】
(3)メチルシクロヘキサンと下記溶媒との混合溶媒
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0038】
(4)n−ペンタンと下記溶媒との混合溶媒
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0039】
(5)n−ヘプタンと下記溶媒との混合溶媒
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0040】
その他、トルエンとジエチルエーテルとの混合溶媒、テトラリンとクメンとの混合溶媒、または、下記実施例に示すトルエン、n−ヘキサンおよびメチルシクロヘキサンからなる3種の混合溶媒も例示できる。
【0041】
これらの溶媒の使用量は、化合物IIを溶解するに必要な最小量が好ましい。また、水の添加量は、ジオール体を形成するに必要な量であり、収率を上げるために化合物IIと当モルないしはやや過剰量の範囲から選択するのが好ましい。なお、水の添加は、人為的に行わず、大気中の水分を吸収させることによって行える場合もある。混合溶媒を用いる場合には、例えば、混合溶媒に化合物IIを溶解させて放置してもよいし、一方の有機溶媒に化合物IIを溶解させて放置し、これに他の有機溶媒を添加して放置してもよい。かくして析出した結晶は、通常、濾取後、適当な溶媒で洗浄し、乾燥される。
【0042】
また、A型結晶は下記で得られるB型結晶またはC型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0043】
B型結晶
化合物IのB型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、特定溶媒として次の単一溶媒または混合溶媒を用いるほかは、前記のA型結晶の場合と同様にして製造することができる。
【0044】
単一溶媒としては、トルエンまたはクロロベンゼンが例示できる。
【0045】
混合溶媒としてはシクロヘキサンまたはn−ヘプタンのいずれか1種の有機溶媒とトルエンとの混合溶媒が例示できる。
【0046】
また、B型結晶は、上記で得られるA型結晶または下記で得られるC型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0047】
C型結晶
化合物IのC型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、特定溶媒として次の単一溶媒または混合溶媒を用いるほかは、前記のA型結晶の場合と同様にして製造することができる。
【0048】
単一溶媒としては、ジエチルエーテルが例示できる。
【0049】
混合溶媒としては、ジエチルエーテルとシクロペンチルメチルエーテルの組み合わせが例示できる。
【0050】
また、C型結晶は、上記で得られるA型結晶またはB型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0051】
上記製法によって得られる本発明の結晶は、出発物質のトリフルオロメチルケトン部分のケトン部分がジオールに変換されている。このことは、後記に示す13C固体核磁気共鳴スペクトルによって確認されている。
【0052】
本発明の結晶化された化合物は、エラスターゼ阻害活性を有し、好中球エラスターゼが病態発症に関与していると考えられている疾患、例えば、慢性閉塞性肺疾患(肺気腫および慢性気管支炎を含む)、慢性および急性間質性肺炎、特発性間質性肺炎(IIP)、びまん性汎細気管支炎、嚢胞性肺線維症、急性肺傷害(ALI)/急性呼吸促迫症候群(ARDS)、気管支拡張症、喘息、膵炎、腎炎、肝炎(肝不全)、慢性関節リウマチ、関節硬化症、変形性関節炎、乾癬、歯周病、アテローム性動脈硬化症、臓器移植における拒絶反応、虚血・再灌流時の組織傷害、ショック、敗血症、播種性血管内凝固症(DIC)や深部静脈血栓症等を含む血液凝固異常、結膜炎、角膜炎、角膜潰瘍、クローン病、全身性エリテマトーデスの治療/または予防剤として有用であると考えられる。
【0053】
本発明の化合物Iの結晶は、好ましくは経口投与される。その投与量は、患者の症状、体重および年齢などにより大いに変動し得る。例えば、経口投与の場合、有効成分の用量は、通常1日につき体重60 kg当たり約 0.5 〜約 5000 mgの範囲内であり、好ましくは 1日につき体重60 kg当たり約5 〜 約 2000 mgの範囲内であり、最も好ましくは1日につき体重60 kg当たり15 〜 300 mgの範囲内にある。
【0054】
本発明の化合物Iの結晶は、通常の製剤の形で投与される。かかる通常の製剤としては、錠剤、カプセル剤、顆粒剤、細粒剤、散剤、水性もしくは油性の液剤などが挙げられる。これらの製剤は、化合物Iを0.01重量%以上、好ましくは0.1〜70重量%の割合で含有することができる。これらの製剤はまた、治療上有効な他の成分を含有していてもよい。
【0055】
これらの薬学上の製剤は、通常の薬学的に許容しうる製剤化成分を用い、通常の手法にて調合、製剤され得る。製剤化成分としては、製剤分野において常用され、かつ有効成分と反応しない物質が用いられる。
【実施例】
【0056】
以下、参考例および実施例を挙げて本発明の新規な結晶形について説明するが、本発明はこれに限定されるものではない。
【0057】
参考例1 N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物II)の製造
【0058】
【化3】
【0059】
工程1: N−[(1S)−2−((2S)−2−{N−[(1S,2S)−3,3,3−トリフルオロ−2−ヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物2)の製造:
工程1の出発物質である[(2S)−1−((2S)−2−アミノ−3−メチルブタノイル)ピロリジン−2−イル]−N−[(1S,2S)−3,3,3−トリフルオロ−2−ヒドロキシ−1−(メチルエチル)プロピル]カルボキサミド トシル酸塩(化合物1)は、国際公開公報WO00/52032に記載の方法に準じて製造した。
【0060】
得られた56.4g(0.10mol)の化合物1および22.2g(0.11mol)の5−メチル−1−フェニルピラゾール−4−カルボン酸を含むピリジン溶液(300ml)に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩を24.0g(0.13mol)加え、室温にて4時間攪拌したのち反応液を減圧濃縮した。
【0061】
残渣に酢酸エチルを加え、10%クエン酸水溶液で3回、飽和炭酸水素ナトリウム水溶液で2回、次いで飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、標題化合物(化合物2)37.6g(65%)を得た。
APCI−MS:552(MH+)
工程2:N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物II)の製造:
53.1g(96.3mmol)の化合物2を塩化メチレン(440ml)とジメチルスルホキシド(110ml)の混合溶媒に溶解し、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩を73.8g(0.39mol)加え、氷冷下で懸濁し、これにジクロロ酢酸の8.1ml(96.3mmol)を滴下し、氷冷下で15分間撹拌した。反応完了後、反応液に氷水を加え酢酸エチルで抽出し、飽和炭酸水素ナトリウム水溶液、氷水、次いで飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(溶出液:酢酸エチル−n−ヘキサン)で精製した。目的物を含む画分を減圧濃縮乾固し、非晶質である標題化合物(化合物II)46.3g(88%)を得た。
APCI−MS:550(MH+)
元素分析: C27H34F3N5O4・0.5H2Oとして
計算値 C 58.05 H 6.32 F 10.20 N 12.54
実験値 C 57.84 H 6.32 F 10.11 N 12.38。
【0062】
実施例1 A型結晶の製造
キシレン350mlに化合物II 88.5g(0.16mol)を加え50℃の湯浴中で溶解させ、蒸留水3.0ml(0.17mol)を加え室温で1時間緩やかに攪拌した。結晶が析出してきたところに、シクロヘキサン700mlを徐々に追加し一晩攪拌した。析出した結晶を濾取し、結晶をキシレン:シクロヘキサン=1:2(100ml)で洗浄した後、50℃で3日間送風乾燥してA型結晶を得た。収量85.7g(収率94%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.18、H 6.32、F 10.03、N 12.38。
【0063】
実施例2 A型結晶の製造
トルエン:n−ヘキサン:メチルシクロヘキサン=2:2:1の混合溶媒155mlに化合物II 15.5g(28.2mmol)を加え50℃の湯浴中で溶解させた。容器口を開口したまま室温で2日間緩やかに攪拌し、析出した結晶を濾取した。得られた結晶を40℃で一晩送風乾燥してA型結晶を得た。収量6.2g(収率39%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.25、H 6.41、F 10.00、N 12.29。
【0064】
実施例3 A型結晶の製造
シクロペンチルメチルエーテル10mlに化合物II 1.0g(1.82mmol)を加えて室温で溶解させ、メチルシクロヘキサン5mlと蒸留水33μl(1.82mmol)を加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、45℃で2日間送風乾燥してA型結晶を得た。収量0.70g(収率70%)
実施例4 B型結晶の製造
トルエン100mlに化合物II 10.0g(18.2mmol)を加えて室温で溶解させ、蒸留水0.33ml(18.2mmol)を加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、45℃で5日間送風乾燥して結晶を得た。収量8.1g(収率78%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.11、H 6.34、F 10.19、N 12.35。
【0065】
実施例5 C型結晶の製造
シクロペンチルメチルエーテル4.5mlに実施例4で得られたB型結晶1.50g(2.64mmol)を加えて120℃のホットプレート上で溶解した後、ジエチルエーテル13.5mlを加え室温で3日間緩やかに撹拌した。析出した結晶を濾取し、得られた結晶をジエチルエーテル4mlで洗浄した後、50℃で一晩送風乾燥して、結晶を得た。収量0.93g(収率62%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 56.78、H 6.33、F 9.99、N 12.27。
【0066】
実施例6 A型結晶の製造
シクロペンチルメチルエーテル16mlに実施例4で得られたB型結晶4.00g(7.05mmol)を加えて120℃のホットプレート上で溶解した後、シクロヘキサン16mlを加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、結晶をシクロペンチルメチルエーテル:シクロヘキサン=1:1(5ml)で洗浄した後、50℃で2日間送風乾燥して結晶を得た。収量3.64g(収率91%)
13C固体核磁気共鳴スペクトル
A型、B型およびC型結晶を用いて13CNMR測定を行った。
【0067】
測定条件は以下のとおりである。
装置:バリアン(Varian)社製 マーキュリープラス(mercury plus)
磁場:400MHz
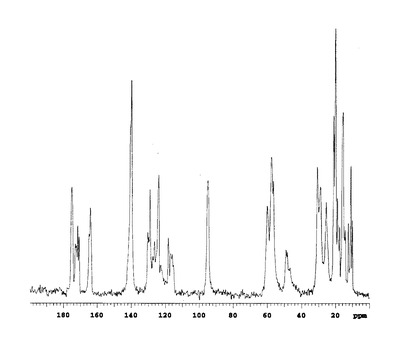
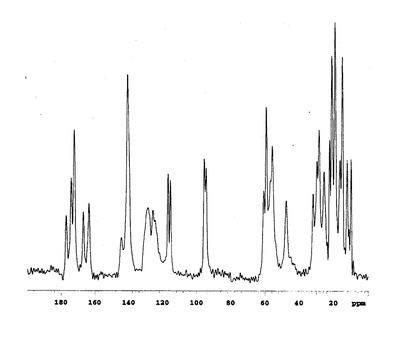
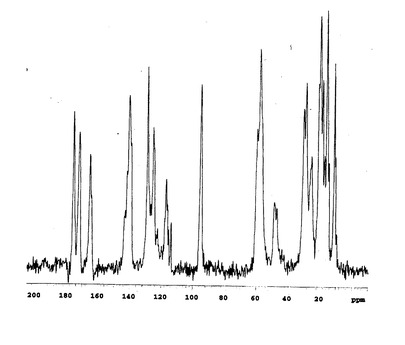
固体状態の13C核磁気共鳴スペクトルを図1(A型結晶)、図2(B型結晶)および図3(C型結晶)に示す。
【0068】
この結果、トリフルオロメチルケトンのカルボニル炭素に由来するピーク(通常190ppm付近)は見られず、ジオールの4級炭素に由来するピーク(95ppm付近)が検出され、トリフルオロメチルケトンのカルボニル部分が2本の水酸基、即ち、ケトン体からジオール体に変換されていることが確認できた。
【0069】
粉末X線結晶回折および熱分析
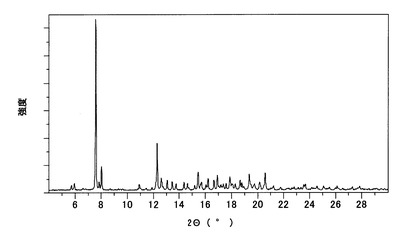
実施例で得たA型結晶、B型結晶およびC型結晶について、以下の条件により、粉末X線回折測定、示差走査熱量計(DSC)および熱重量分析計(TG)による熱分析を行った。それぞれの結果をA型結晶については図4〜図6に、B型結晶については図7〜図9に、C型結晶については図10〜図12に示す。
【0070】
(1)粉末X線回折
装置:スペクトリス(Spectris)社製 X’Pert Alpha 1
線源:CuKα 1線
管電圧: 45 kV
管電流: 40 mA
検出器:X’Celerator
ステップ:0.0167°
各ステップにおける積算時間:100秒/ステップ
ソーラースリット:0.01°。
【0071】
(2)DSCによる熱分析
装置:パーキンエルマー(Perkin Elmer)社製 Pyris 1
試料量: 3〜4 mg
試料セル: アルミニウムセル
窒素ガス流量: 20 ml/分
昇温温度: 10℃/分。
【0072】
(3)TGによる熱分析
装置:パーキンエルマー(Perkin Elmer)社製 TGA 7
試料量: 5〜10 mg
試料セル: プラチナパン
窒素ガス流量: 70 ml/分
昇温温度: 10℃/分。
【0073】
試験例1 本発明の結晶のヒト好中球エラスターゼ(HNE)誘発肺出血に対する抑制活性
ハムスターにヒト好中球エラスターゼを気管支内投与すると肺において出血が誘発される。このため、エラスターゼ投与の一定時間後に経気道的に肺を洗浄して得られる気管支肺胞洗浄液中にはヘモグロビンが検出される。本実験は、このヘモグロビン濃度を測定することにより、本発明の結晶によってこの出血がどの程度抑制するか試験した。
【0074】
ハムスタ−(Syrian系、7−10週齢の雄性)を一群あたり8匹に分け、各群について以下の処理を行った。
【0075】
(a)本発明の結晶の非投与群(病態対照群):
25 単位のヒト好中球エラスターゼ(エラスチンプロダクト社(Elastin Product Co., Inc.))を溶解した生理食塩水0.2 mlをハムスターの気管支内に投与して肺出血を誘発せしめた。エラスターゼ投与の1時間後、ハムスターを脱血により屠殺し、生理食塩液2.5 mlで経気道的に肺胞を2回洗浄し、その肺胞洗浄液5ml中のヘモグロビン濃度(414 nmの吸光度(A414))を測定した。
【0076】
(b)本発明の結晶投与群:
所定量の本発明のA型結晶に、0.5% トラガント溶液を加えて懸濁し、ヒト好中球エラスターゼ(HNE)投与の90分前にハムスターに経口投与した(投与量:10 mg/kg)。さらに前記(a)と同様の方法によりヒト好中球エラスターゼを投与した。そして、その1時間後に(a)と同様にして肺胞洗浄液中のヘモグロビン濃度を測定した。
【0077】
出血抑制率(%)は、{1−((b)でのA414値/(a)でのA414値)}×100により算出した。その結果、本発明の結晶は、76%の出血抑制率を有していた。
【産業上の利用可能性】
【0078】
本発明の結晶化された化合物は、エラスターゼ阻害活性を有することにより好中球エラスターゼが病態発症に関与していると考えられている疾患の治療/または予防剤の原薬として有用である。
【図面の簡単な説明】
【0079】
【図1】本発明のA型結晶の13C固体NMRスペクトルチャートである。
【図2】本発明のB型結晶の13C固体NMRスペクトルチャートである。
【図3】本発明のC型結晶の13C固体NMRスペクトルチャートである。
【図4】本発明のA型結晶の粉末X線回折スペクトルチャートである。
【図5】本発明のA型結晶の示差走査熱量測定(DSC)チャートである。
【図6】本発明のA型結晶の熱重量分析測定(TG)チャートである。
【図7】本発明のB型結晶の粉末X線回折スペクトルチャートである。
【図8】本発明のB型結晶の示差走査熱量測定(DSC)チャートである。
【図9】本発明のB型結晶の熱重量分析測定(TG)チャートである。
【図10】本発明のC型結晶の粉末X線回折スペクトルチャートである。
【図11】本発明のC型結晶の示差走査熱量測定(DSC)チャートである。
【図12】本発明のC型結晶の熱重量分析測定(TG)チャートである。
【技術分野】
【0001】
本発明は医薬、特にエラスターゼ阻害作用を有する複素環式化合物の結晶多形、その製造方法およびその複素環式化合物を有効成分とする医薬に関する。
【背景技術】
【0002】
次式で示される構造を有する、N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下、「化合物II」または「ケトン体」ということもある)
【0003】
【化1】
【0004】
は、エラスターゼ阻害剤として有用な化合物であり、特許文献1の実施例10に記載されている。
【0005】
通常、医薬品の有効成分となる化合物は、強い薬理作用を有し、且つ低毒性であるとともに品質が一定であることが条件となる。そのためには結晶状態の化合物を原薬として用いることが求められている。しかしながら、特許文献1には上記化合物IIのみならずそこに記載の他の化合物群がすべて結晶ではなく非晶質であり、さらに結晶を得るための製造方法も記載されていない。
【特許文献1】国際公報第03/066671号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、エラスターゼ阻害活性を有する化合物の結晶を得ることを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記化合物II、即ち、ケトン体の結晶を得る為に、鋭意検討を試みた。しかし、通常使用される種々の有機溶媒をもってしてもこの化合物を結晶化させることができなかった。ところが、化合物IIを特定の有機溶媒に溶解し、容器を開口状態にして放置したところ結晶化させることに成功した。しかし、分析の結果、得られた結晶は、大気中の水を吸って、水分子1個分の重量が増えていることが判明した。そこで、さらに検討を進めた結果、得られた結晶は、ケトン体のトリフルオロメチルケトン部位に1分子の水が附加したジオール体の結晶であること、およびこの結晶のエラスターゼ阻害活性は、ケトン体のそれがそのまま維持されているとの知見をえて、本発明を完成した。
【0008】
即ち、本発明は、
項1:N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下「化合物I」または「ジオール体」ということもある)
【0009】
【化2】
【0010】
の結晶を提供するものである。具体的には以下の発明を提供する。
【0011】
項2: 回折角2θで表して、約7.0°〜8.5°の位置にピークを有するX線粉末回折パターンを示す項1に記載の結晶。
【0012】
項3: 回折角2θで表して、約7.0°〜8.5°の位置に少なくとも1本の強いピークを有するX線粉末回折パターンを示す項2に記載の結晶。
【0013】
項4: 回折角2θで表して、ほぼ5.7°、6.0°、7.6°、8.0°、12.3°、12.6°および16.2°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのA型結晶。
【0014】
項5: 示差走査熱量測定において、約139℃に脱水吸熱ピークを有する項4に記載の化合物IのA型結晶。
【0015】
項6: 回折角2θで表して、ほぼ6.5°、8.0°、8.6°、9.5°、10.2°、11.0°、14.2°および20.3°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのB型結晶。
【0016】
項7: 示差走査熱量測定において、約120℃に脱水吸熱ピークを有する項6に記載の化合物IのB型結晶。
【0017】
項8: 回折角2θで表して、ほぼ6.0°、7.2°、10.6°、11.9°、14.4°、15.2°および18.0°の位置にピークを有するX線粉末回折パターンを示す項1〜3のいずれかに記載の化合物IのC型結晶。
【0018】
項9: 示差走査熱量測定において、約128℃に脱水吸熱ピークを有する項8に記載の化合物IのC型結晶。
【0019】
項10: 項1〜9のいずれかに記載の結晶を有効成分として含む医薬。
【0020】
項11: 項1〜9のいずれかに記載の結晶を含む医薬組成物。
【0021】
項12: 化合物IのA型結晶の製造方法であって、化合物IIを、以下のいずれかの有機溶媒:
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタンからなる群から選択される単一溶媒;
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒とn−ヘキサンとの混合溶媒;
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とメチルシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ペンタンとの混合溶媒;
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ヘプタンとの混合溶媒;
トルエンとジエチルエーテルとの混合溶媒;
テトラリンとクメンとの混合溶媒;および
トルエン、n−ヘキサンおよびメチルシクロヘキサンからなる混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのA型結晶を晶析させることを特徴とする製造方法。
【0022】
項13: 化合物IのB型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
トルエンまたはクロロベンゼンである単一溶媒;または
シクロヘキサンまたはn−ヘプタンと、トルエンとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのB型結晶を晶析させることを特徴とする製造方法。
【0023】
項14: 化合物IのC型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
ジエチルエーテル;または
ジエチルエーテルとシクロペンチルメチルエーテルとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのC型結晶を晶析させることを特徴とする製造方法。
【発明の効果】
【0024】
本発明によれば、品質が安定している化合物の結晶を医薬品の原薬として供給することができる。
【発明を実施するための最良の形態】
【0025】
本発明の化合物Iの結晶は.化合物II,即ち、ケトン体を出発物質として製造することができる。このケトン体は、特許文献1に記載の方法によって製造することができる。
【0026】
本発明者らの研究によって、化合物Iの結晶として、少なくとも3種類の異なる結晶形の存在が確認された。この3種類の結晶形を、以下、A型、B型およびC型という。
【0027】
化合物Iの結晶は、X線粉末回折パターンにおいて、特徴的なピークを示す。即ち、該結晶は、回折角2θで表して、約7.0°〜8.5°の位置にピーク、具体的には、少なくとも1本の強いピークを有するX線粉末回折パターンを示す。
【0028】
以下の実施例で示す装置、条件下で測定したときのA型、B型及びC型各結晶形の特徴的な主ピークの回折角(2θ)は以下のとおりである。
【0029】
【表1】
【0030】
また、A型、B型、C型各結晶形の示差走査熱量測定(DSC)における特徴および熱重量分析(TG)における特徴は以下のとおりである。
【0031】
【表2】
【0032】
本発明の化合物IのA型結晶、B型結晶およびC型結晶は、本明細書に記載の物理化学的性質によって特定されるものであるが、上記データは、測定方法や測定器具によって多少変わるものであるから、厳密に解されるべきではない。また、A型結晶、B型結晶およびC型結晶は、純粋な結晶に限られず、未知の結晶形を含めた他の結晶形が混在しているものでもよい。
【0033】
A型結晶
化合物IのA型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、A型結晶は、化合物IIを特定溶媒に溶解し、水を添加したのち放置することにより製造できる。「放置」は、通常、ゆっくりと攪拌しながら数時間ないし数日間、通常、室温において行われる。
【0034】
ここにおける、特定溶媒としては、例えば、次の単一溶媒が挙げられる。
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタン
または、以下の組み合わせの混合溶媒が例示できる。
【0035】
(1)シクロヘキサンと下記溶媒との混合溶媒
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0036】
(2)n−ヘキサンと下記溶媒との混合溶媒
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒。
【0037】
(3)メチルシクロヘキサンと下記溶媒との混合溶媒
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0038】
(4)n−ペンタンと下記溶媒との混合溶媒
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0039】
(5)n−ヘプタンと下記溶媒との混合溶媒
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒。
【0040】
その他、トルエンとジエチルエーテルとの混合溶媒、テトラリンとクメンとの混合溶媒、または、下記実施例に示すトルエン、n−ヘキサンおよびメチルシクロヘキサンからなる3種の混合溶媒も例示できる。
【0041】
これらの溶媒の使用量は、化合物IIを溶解するに必要な最小量が好ましい。また、水の添加量は、ジオール体を形成するに必要な量であり、収率を上げるために化合物IIと当モルないしはやや過剰量の範囲から選択するのが好ましい。なお、水の添加は、人為的に行わず、大気中の水分を吸収させることによって行える場合もある。混合溶媒を用いる場合には、例えば、混合溶媒に化合物IIを溶解させて放置してもよいし、一方の有機溶媒に化合物IIを溶解させて放置し、これに他の有機溶媒を添加して放置してもよい。かくして析出した結晶は、通常、濾取後、適当な溶媒で洗浄し、乾燥される。
【0042】
また、A型結晶は下記で得られるB型結晶またはC型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0043】
B型結晶
化合物IのB型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、特定溶媒として次の単一溶媒または混合溶媒を用いるほかは、前記のA型結晶の場合と同様にして製造することができる。
【0044】
単一溶媒としては、トルエンまたはクロロベンゼンが例示できる。
【0045】
混合溶媒としてはシクロヘキサンまたはn−ヘプタンのいずれか1種の有機溶媒とトルエンとの混合溶媒が例示できる。
【0046】
また、B型結晶は、上記で得られるA型結晶または下記で得られるC型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0047】
C型結晶
化合物IのC型結晶は、実施例または以下に記載した方法によって製造することができる。即ち、特定溶媒として次の単一溶媒または混合溶媒を用いるほかは、前記のA型結晶の場合と同様にして製造することができる。
【0048】
単一溶媒としては、ジエチルエーテルが例示できる。
【0049】
混合溶媒としては、ジエチルエーテルとシクロペンチルメチルエーテルの組み合わせが例示できる。
【0050】
また、C型結晶は、上記で得られるA型結晶またはB型結晶を上記特定溶媒に溶解させ、晶析させることによって得ることもできる。
【0051】
上記製法によって得られる本発明の結晶は、出発物質のトリフルオロメチルケトン部分のケトン部分がジオールに変換されている。このことは、後記に示す13C固体核磁気共鳴スペクトルによって確認されている。
【0052】
本発明の結晶化された化合物は、エラスターゼ阻害活性を有し、好中球エラスターゼが病態発症に関与していると考えられている疾患、例えば、慢性閉塞性肺疾患(肺気腫および慢性気管支炎を含む)、慢性および急性間質性肺炎、特発性間質性肺炎(IIP)、びまん性汎細気管支炎、嚢胞性肺線維症、急性肺傷害(ALI)/急性呼吸促迫症候群(ARDS)、気管支拡張症、喘息、膵炎、腎炎、肝炎(肝不全)、慢性関節リウマチ、関節硬化症、変形性関節炎、乾癬、歯周病、アテローム性動脈硬化症、臓器移植における拒絶反応、虚血・再灌流時の組織傷害、ショック、敗血症、播種性血管内凝固症(DIC)や深部静脈血栓症等を含む血液凝固異常、結膜炎、角膜炎、角膜潰瘍、クローン病、全身性エリテマトーデスの治療/または予防剤として有用であると考えられる。
【0053】
本発明の化合物Iの結晶は、好ましくは経口投与される。その投与量は、患者の症状、体重および年齢などにより大いに変動し得る。例えば、経口投与の場合、有効成分の用量は、通常1日につき体重60 kg当たり約 0.5 〜約 5000 mgの範囲内であり、好ましくは 1日につき体重60 kg当たり約5 〜 約 2000 mgの範囲内であり、最も好ましくは1日につき体重60 kg当たり15 〜 300 mgの範囲内にある。
【0054】
本発明の化合物Iの結晶は、通常の製剤の形で投与される。かかる通常の製剤としては、錠剤、カプセル剤、顆粒剤、細粒剤、散剤、水性もしくは油性の液剤などが挙げられる。これらの製剤は、化合物Iを0.01重量%以上、好ましくは0.1〜70重量%の割合で含有することができる。これらの製剤はまた、治療上有効な他の成分を含有していてもよい。
【0055】
これらの薬学上の製剤は、通常の薬学的に許容しうる製剤化成分を用い、通常の手法にて調合、製剤され得る。製剤化成分としては、製剤分野において常用され、かつ有効成分と反応しない物質が用いられる。
【実施例】
【0056】
以下、参考例および実施例を挙げて本発明の新規な結晶形について説明するが、本発明はこれに限定されるものではない。
【0057】
参考例1 N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物II)の製造
【0058】
【化3】
【0059】
工程1: N−[(1S)−2−((2S)−2−{N−[(1S,2S)−3,3,3−トリフルオロ−2−ヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物2)の製造:
工程1の出発物質である[(2S)−1−((2S)−2−アミノ−3−メチルブタノイル)ピロリジン−2−イル]−N−[(1S,2S)−3,3,3−トリフルオロ−2−ヒドロキシ−1−(メチルエチル)プロピル]カルボキサミド トシル酸塩(化合物1)は、国際公開公報WO00/52032に記載の方法に準じて製造した。
【0060】
得られた56.4g(0.10mol)の化合物1および22.2g(0.11mol)の5−メチル−1−フェニルピラゾール−4−カルボン酸を含むピリジン溶液(300ml)に、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩を24.0g(0.13mol)加え、室温にて4時間攪拌したのち反応液を減圧濃縮した。
【0061】
残渣に酢酸エチルを加え、10%クエン酸水溶液で3回、飽和炭酸水素ナトリウム水溶液で2回、次いで飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、標題化合物(化合物2)37.6g(65%)を得た。
APCI−MS:552(MH+)
工程2:N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(化合物II)の製造:
53.1g(96.3mmol)の化合物2を塩化メチレン(440ml)とジメチルスルホキシド(110ml)の混合溶媒に溶解し、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩を73.8g(0.39mol)加え、氷冷下で懸濁し、これにジクロロ酢酸の8.1ml(96.3mmol)を滴下し、氷冷下で15分間撹拌した。反応完了後、反応液に氷水を加え酢酸エチルで抽出し、飽和炭酸水素ナトリウム水溶液、氷水、次いで飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去し、残渣をシリカゲルカラムクロマトグラフィー(溶出液:酢酸エチル−n−ヘキサン)で精製した。目的物を含む画分を減圧濃縮乾固し、非晶質である標題化合物(化合物II)46.3g(88%)を得た。
APCI−MS:550(MH+)
元素分析: C27H34F3N5O4・0.5H2Oとして
計算値 C 58.05 H 6.32 F 10.20 N 12.54
実験値 C 57.84 H 6.32 F 10.11 N 12.38。
【0062】
実施例1 A型結晶の製造
キシレン350mlに化合物II 88.5g(0.16mol)を加え50℃の湯浴中で溶解させ、蒸留水3.0ml(0.17mol)を加え室温で1時間緩やかに攪拌した。結晶が析出してきたところに、シクロヘキサン700mlを徐々に追加し一晩攪拌した。析出した結晶を濾取し、結晶をキシレン:シクロヘキサン=1:2(100ml)で洗浄した後、50℃で3日間送風乾燥してA型結晶を得た。収量85.7g(収率94%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.18、H 6.32、F 10.03、N 12.38。
【0063】
実施例2 A型結晶の製造
トルエン:n−ヘキサン:メチルシクロヘキサン=2:2:1の混合溶媒155mlに化合物II 15.5g(28.2mmol)を加え50℃の湯浴中で溶解させた。容器口を開口したまま室温で2日間緩やかに攪拌し、析出した結晶を濾取した。得られた結晶を40℃で一晩送風乾燥してA型結晶を得た。収量6.2g(収率39%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.25、H 6.41、F 10.00、N 12.29。
【0064】
実施例3 A型結晶の製造
シクロペンチルメチルエーテル10mlに化合物II 1.0g(1.82mmol)を加えて室温で溶解させ、メチルシクロヘキサン5mlと蒸留水33μl(1.82mmol)を加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、45℃で2日間送風乾燥してA型結晶を得た。収量0.70g(収率70%)
実施例4 B型結晶の製造
トルエン100mlに化合物II 10.0g(18.2mmol)を加えて室温で溶解させ、蒸留水0.33ml(18.2mmol)を加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、45℃で5日間送風乾燥して結晶を得た。収量8.1g(収率78%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 57.11、H 6.34、F 10.19、N 12.35。
【0065】
実施例5 C型結晶の製造
シクロペンチルメチルエーテル4.5mlに実施例4で得られたB型結晶1.50g(2.64mmol)を加えて120℃のホットプレート上で溶解した後、ジエチルエーテル13.5mlを加え室温で3日間緩やかに撹拌した。析出した結晶を濾取し、得られた結晶をジエチルエーテル4mlで洗浄した後、50℃で一晩送風乾燥して、結晶を得た。収量0.93g(収率62%)
元素分析:C27H36F3N5O5として
計算値 C 57.13、H 6.39、F 10.04、N 12.34
測定値 C 56.78、H 6.33、F 9.99、N 12.27。
【0066】
実施例6 A型結晶の製造
シクロペンチルメチルエーテル16mlに実施例4で得られたB型結晶4.00g(7.05mmol)を加えて120℃のホットプレート上で溶解した後、シクロヘキサン16mlを加え室温で一晩緩やかに攪拌した。析出した結晶を濾取し、結晶をシクロペンチルメチルエーテル:シクロヘキサン=1:1(5ml)で洗浄した後、50℃で2日間送風乾燥して結晶を得た。収量3.64g(収率91%)
13C固体核磁気共鳴スペクトル
A型、B型およびC型結晶を用いて13CNMR測定を行った。
【0067】
測定条件は以下のとおりである。
装置:バリアン(Varian)社製 マーキュリープラス(mercury plus)
磁場:400MHz
固体状態の13C核磁気共鳴スペクトルを図1(A型結晶)、図2(B型結晶)および図3(C型結晶)に示す。
【0068】
この結果、トリフルオロメチルケトンのカルボニル炭素に由来するピーク(通常190ppm付近)は見られず、ジオールの4級炭素に由来するピーク(95ppm付近)が検出され、トリフルオロメチルケトンのカルボニル部分が2本の水酸基、即ち、ケトン体からジオール体に変換されていることが確認できた。
【0069】
粉末X線結晶回折および熱分析
実施例で得たA型結晶、B型結晶およびC型結晶について、以下の条件により、粉末X線回折測定、示差走査熱量計(DSC)および熱重量分析計(TG)による熱分析を行った。それぞれの結果をA型結晶については図4〜図6に、B型結晶については図7〜図9に、C型結晶については図10〜図12に示す。
【0070】
(1)粉末X線回折
装置:スペクトリス(Spectris)社製 X’Pert Alpha 1
線源:CuKα 1線
管電圧: 45 kV
管電流: 40 mA
検出器:X’Celerator
ステップ:0.0167°
各ステップにおける積算時間:100秒/ステップ
ソーラースリット:0.01°。
【0071】
(2)DSCによる熱分析
装置:パーキンエルマー(Perkin Elmer)社製 Pyris 1
試料量: 3〜4 mg
試料セル: アルミニウムセル
窒素ガス流量: 20 ml/分
昇温温度: 10℃/分。
【0072】
(3)TGによる熱分析
装置:パーキンエルマー(Perkin Elmer)社製 TGA 7
試料量: 5〜10 mg
試料セル: プラチナパン
窒素ガス流量: 70 ml/分
昇温温度: 10℃/分。
【0073】
試験例1 本発明の結晶のヒト好中球エラスターゼ(HNE)誘発肺出血に対する抑制活性
ハムスターにヒト好中球エラスターゼを気管支内投与すると肺において出血が誘発される。このため、エラスターゼ投与の一定時間後に経気道的に肺を洗浄して得られる気管支肺胞洗浄液中にはヘモグロビンが検出される。本実験は、このヘモグロビン濃度を測定することにより、本発明の結晶によってこの出血がどの程度抑制するか試験した。
【0074】
ハムスタ−(Syrian系、7−10週齢の雄性)を一群あたり8匹に分け、各群について以下の処理を行った。
【0075】
(a)本発明の結晶の非投与群(病態対照群):
25 単位のヒト好中球エラスターゼ(エラスチンプロダクト社(Elastin Product Co., Inc.))を溶解した生理食塩水0.2 mlをハムスターの気管支内に投与して肺出血を誘発せしめた。エラスターゼ投与の1時間後、ハムスターを脱血により屠殺し、生理食塩液2.5 mlで経気道的に肺胞を2回洗浄し、その肺胞洗浄液5ml中のヘモグロビン濃度(414 nmの吸光度(A414))を測定した。
【0076】
(b)本発明の結晶投与群:
所定量の本発明のA型結晶に、0.5% トラガント溶液を加えて懸濁し、ヒト好中球エラスターゼ(HNE)投与の90分前にハムスターに経口投与した(投与量:10 mg/kg)。さらに前記(a)と同様の方法によりヒト好中球エラスターゼを投与した。そして、その1時間後に(a)と同様にして肺胞洗浄液中のヘモグロビン濃度を測定した。
【0077】
出血抑制率(%)は、{1−((b)でのA414値/(a)でのA414値)}×100により算出した。その結果、本発明の結晶は、76%の出血抑制率を有していた。
【産業上の利用可能性】
【0078】
本発明の結晶化された化合物は、エラスターゼ阻害活性を有することにより好中球エラスターゼが病態発症に関与していると考えられている疾患の治療/または予防剤の原薬として有用である。
【図面の簡単な説明】
【0079】
【図1】本発明のA型結晶の13C固体NMRスペクトルチャートである。
【図2】本発明のB型結晶の13C固体NMRスペクトルチャートである。
【図3】本発明のC型結晶の13C固体NMRスペクトルチャートである。
【図4】本発明のA型結晶の粉末X線回折スペクトルチャートである。
【図5】本発明のA型結晶の示差走査熱量測定(DSC)チャートである。
【図6】本発明のA型結晶の熱重量分析測定(TG)チャートである。
【図7】本発明のB型結晶の粉末X線回折スペクトルチャートである。
【図8】本発明のB型結晶の示差走査熱量測定(DSC)チャートである。
【図9】本発明のB型結晶の熱重量分析測定(TG)チャートである。
【図10】本発明のC型結晶の粉末X線回折スペクトルチャートである。
【図11】本発明のC型結晶の示差走査熱量測定(DSC)チャートである。
【図12】本発明のC型結晶の熱重量分析測定(TG)チャートである。
【特許請求の範囲】
【請求項1】
N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下「化合物I」という。)の結晶。
【請求項2】
回折角2θで表して、約7.0°〜8.5°の位置にピークを有するX線粉末回折パターンを示す請求項1に記載の結晶。
【請求項3】
回折角2θで表して、約7.0°〜8.5°の位置に少なくとも1本の強いピークを有するX線粉末回折パターンを示す請求項2に記載の結晶。
【請求項4】
回折角2θで表して、ほぼ5.7°、6.0°、7.6°、8.0°、12.3°、12.6°および16.2°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのA型結晶。
【請求項5】
示差走査熱量測定において、約139℃に脱水吸熱ピークを有する請求項4に記載の化合物IのA型結晶。
【請求項6】
回折角2θで表して、ほぼ6.5°、8.0°、8.6°、9.5°、10.2°、11.0°、14.2°および20.3°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのB型結晶。
【請求項7】
示差走査熱量測定において、約120℃に脱水吸熱ピークを有する請求項6に記載の化合物IのB型結晶。
【請求項8】
回折角2θで表して、ほぼ6.0°、7.2°、10.6°、11.9°、14.4°、15.2°および18.0°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのC型結晶。
【請求項9】
示差走査熱量測定において、約128℃に脱水吸熱ピークを有する請求項8に記載の化合物IのC型結晶。
【請求項10】
請求項1〜9のいずれかに記載の結晶を有効成分として含む医薬。
【請求項11】
請求項1〜9のいずれかに記載の結晶を含む医薬組成物。
【請求項12】
化合物IのA型結晶の製造方法であって、N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下、「化合物II」)を、以下のいずれかの有機溶媒:
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタンからなる群から選択される単一溶媒;
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒とn−ヘキサンとの混合溶媒;
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とメチルシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ペンタンとの混合溶媒;
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ヘプタンとの混合溶媒;
トルエンとジエチルエーテルとの混合溶媒;
テトラリンとクメンとの混合溶媒;および
トルエン、n−ヘキサンおよびメチルシクロヘキサンからなる混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのA型結晶を晶析させることを特徴とする製造方法。
【請求項13】
化合物IのB型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
トルエンまたはクロロベンゼンである単一溶媒;または
シクロヘキサンまたはn−ヘプタンと、トルエンとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのB型結晶を晶析させることを特徴とする製造方法。
【請求項14】
化合物IのC型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
ジエチルエーテル;または
ジエチルエーテルとシクロペンチルメチルエーテルとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのC型結晶を晶析させることを特徴とする製造方法。
【請求項1】
N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−2,2−ジヒドロキシ−1−(メチルエチル)プロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下「化合物I」という。)の結晶。
【請求項2】
回折角2θで表して、約7.0°〜8.5°の位置にピークを有するX線粉末回折パターンを示す請求項1に記載の結晶。
【請求項3】
回折角2θで表して、約7.0°〜8.5°の位置に少なくとも1本の強いピークを有するX線粉末回折パターンを示す請求項2に記載の結晶。
【請求項4】
回折角2θで表して、ほぼ5.7°、6.0°、7.6°、8.0°、12.3°、12.6°および16.2°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのA型結晶。
【請求項5】
示差走査熱量測定において、約139℃に脱水吸熱ピークを有する請求項4に記載の化合物IのA型結晶。
【請求項6】
回折角2θで表して、ほぼ6.5°、8.0°、8.6°、9.5°、10.2°、11.0°、14.2°および20.3°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのB型結晶。
【請求項7】
示差走査熱量測定において、約120℃に脱水吸熱ピークを有する請求項6に記載の化合物IのB型結晶。
【請求項8】
回折角2θで表して、ほぼ6.0°、7.2°、10.6°、11.9°、14.4°、15.2°および18.0°の位置にピークを有するX線粉末回折パターンを示す請求項1〜3のいずれかに記載の化合物IのC型結晶。
【請求項9】
示差走査熱量測定において、約128℃に脱水吸熱ピークを有する請求項8に記載の化合物IのC型結晶。
【請求項10】
請求項1〜9のいずれかに記載の結晶を有効成分として含む医薬。
【請求項11】
請求項1〜9のいずれかに記載の結晶を含む医薬組成物。
【請求項12】
化合物IのA型結晶の製造方法であって、N−[(1S)−2−((2S)−2−{N−[(1S)−3,3,3−トリフルオロ−1−(メチルエチル)−2−オキソプロピル]カルバモイル}ピロリジニル)−1−(メチルエチル)−2−オキソエチル]−(5−メチル−1−フェニルピラゾル−4−イル)カルボキサミド(以下、「化合物II」)を、以下のいずれかの有機溶媒:
キシレン、テトラリン、ジフェニルエーテル、シクロヘキサン、n−ヘキサン、メチルシクロヘキサンおよびn−ヘプタンからなる群から選択される単一溶媒;
キシレン、テトラリン、ジフェニルエーテル、シクロペンチルメチルエーテル、酢酸エチル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリンおよびシクロペンチルメチルエーテルからなる群から選択される1種の有機溶媒とn−ヘキサンとの混合溶媒;
キシレン、シクロペンチルメチルエーテル、酢酸エチル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とメチルシクロヘキサンとの混合溶媒;
キシレン、トルエン、テトラリン、酢酸イソプロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ペンタンとの混合溶媒;
キシレン、テトラリン、シクロペンチルメチルエーテル、酢酸イソプロピル、酢酸n−プロピル、酢酸イソブチルおよび酢酸n−ブチルからなる群から選択される1種の有機溶媒とn−ヘプタンとの混合溶媒;
トルエンとジエチルエーテルとの混合溶媒;
テトラリンとクメンとの混合溶媒;および
トルエン、n−ヘキサンおよびメチルシクロヘキサンからなる混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのA型結晶を晶析させることを特徴とする製造方法。
【請求項13】
化合物IのB型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
トルエンまたはクロロベンゼンである単一溶媒;または
シクロヘキサンまたはn−ヘプタンと、トルエンとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのB型結晶を晶析させることを特徴とする製造方法。
【請求項14】
化合物IのC型結晶の製造方法であって、化合物IIを以下のいずれかの溶媒:
ジエチルエーテル;または
ジエチルエーテルとシクロペンチルメチルエーテルとの混合溶媒
に溶解させ、これに水を添加した溶液から化合物IのC型結晶を晶析させることを特徴とする製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
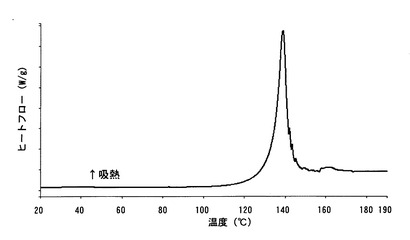
【図6】
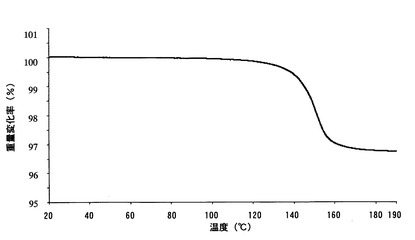
【図7】
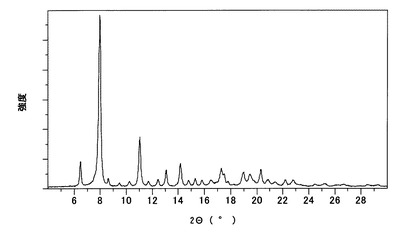
【図8】
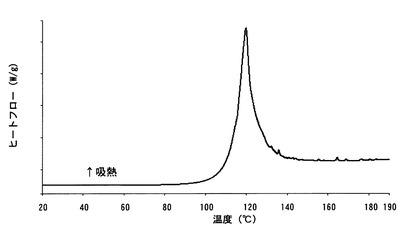
【図9】
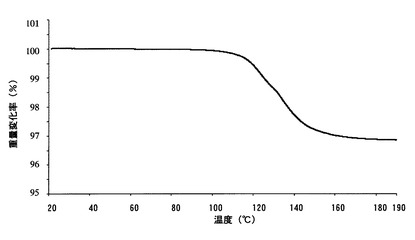
【図10】
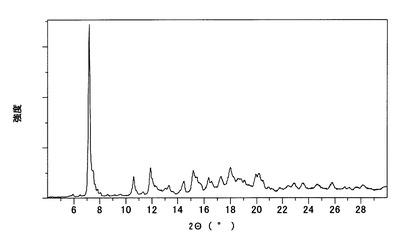
【図11】
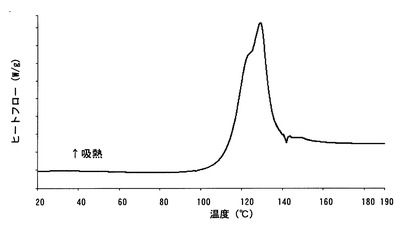
【図12】
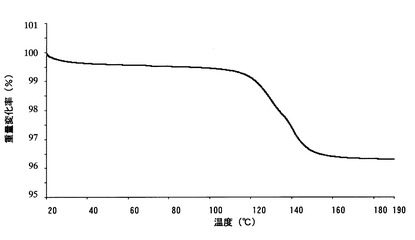
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2007−332028(P2007−332028A)
【公開日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願番号】特願2004−251716(P2004−251716)
【出願日】平成16年8月31日(2004.8.31)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
【公開日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願日】平成16年8月31日(2004.8.31)
【出願人】(000002912)大日本住友製薬株式会社 (332)
【Fターム(参考)】
[ Back to top ]