
単一プローブ分子素子及び単一プローブ分子素子の製造方法
【課題】単一分子レベルで生体分子の高精度及び高感度検出が可能な生体分子検出素子を提供する。
【解決手段】表面に単一プローブ分子を固定した金属微粒子を製造するとともに,該金属微粒子を担体基板表面に固定した単一プローブ分子素子を製造する。
【解決手段】表面に単一プローブ分子を固定した金属微粒子を製造するとともに,該金属微粒子を担体基板表面に固定した単一プローブ分子素子を製造する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は,生体分子を検出するための単一プローブ分子素子及びその製造方法ならびに生体分子の検出方法に関するものである。
【背景技術】
【0002】
近年,核酸やタンパク質などの生体分子を特異的かつ網羅的に解析するための方法として,様々な生体分子検出素子の研究開発が進んでいる。こうした生体分子検出素子の重要な例として,ナノメートルからマイクロメートルサイズの微粒子の表面に生体分子を検出するためのプローブ分子を固定したプローブ分子固定微粒子がある。これまでに,プローブ分子固定微粒子を用いた様々な生体分子検出方法が報告されている。プローブ分子固定微粒子のうち,金属微粒子の表面にプローブ分子を固定したプローブ分子固定金属微粒子に着目すると,例えば特許文献1には,担体基板に固定した解析対象分子に対して,プローブ分子固定金ナノ粒子を結合し,該金ナノ粒子を電子顕微鏡などで検出することにより解析対象分子の識別及び定量評価を行う方法が開示されている。また,特許文献2には,表面にオリゴヌクレオチドを固定した金ナノ粒子がその凝集状態に応じて異なる色を呈することを利用した生体分子の分析方法が開示されている。
【0003】
プローブ分子固定金属微粒子を用いて生体分子の定量的な解析を行うためには,金属微粒子表面に固定されたプローブ分子の個数が正確にわかっていることが重要である。さらに,生体分子の単一分子レベルでの定量的な解析を実現するためには,表面に単一のプローブ分子を固定した金属微粒子の利用が必要不可欠である。従来の公知例において使用されているプローブ分子固定金属微粒子は,金属微粒子及び金属微粒子と強く相互作用する官能基を持つプローブ分子を所定の混合比にて混合し反応させることにより製造されている。従って,この方法により製造されるプローブ分子固定金属微粒子は,表面に固定されたプローブ分子の個数に分布を持つ金属微粒子の混合物である。単一プローブ分子固定金属微粒子を得るためには,該金属微粒子の混合物からの分離・精製を行う必要がある。
【0004】
単一プローブ分子固定金属微粒子を得るための方法として,ゲル電気泳動を利用する方法が非特許文献1に開示されている。この方法では,金ナノ粒子及び5’末端にチオール基をもつプローブDNAを所定のモル比にて混合・反応し,金ナノ粒子の表面にプローブDNAを固定する。このとき,表面に固定されたプローブDNAの個数が0個,1個,2個,3個,及び3個以上である金ナノ粒子の混合物が得られる。この金ナノ粒子の混合物のゲル電気泳動を行うと,固定DNA数が少ない金ナノ粒子から順にゲル中での泳動距離が大きくなる。従って,固定DNA数に応じて金ナノ粒子を分離することができ,単一プローブDNA固定金ナノ粒子の分離・回収が可能である。しかしながら,この方法における単一プローブDNA固定金ナノ粒子の分離・回収操作は非常に煩雑であるとともに,単一プローブDNA固定金ナノ粒子の回収率が非常に低いことが問題である。
【0005】
また,ポリマー分子を介して金属微粒子の表面にプローブDNAを固定する方法が非特許文献2に開示されている。この方法では,官能基として複数のジスルフィド基を持つデキストラン分子を合成し,このデキストラン分子にプローブDNAを結合する。ジスルフィド基を利用してプローブDNAが結合したデキストラン分子を金ナノ粒子の表面に固定する。この方法ではデキストラン分子を介して金ナノ粒子の表面にプローブDNAを固定することが可能であるが,デキストラン1分子に結合するプローブDNAの個数に分布があるという問題がある。従って,表面に単一プローブ分子を固定した金属微粒子を簡便かつ高収率で製造することのできる新規の方法が必要である。
【0006】
一方,生体分子を単一分子レベルで検出する技術のうち,非特許文献3に開示されているような単一DNA分子レベルのシーケンシングによる遺伝子配列解析技術の開発が活発に進められている。この方式では,担体基板表面に検体となる膨大な数のポリヌクレオチドを固定し,これを鋳型としてポリメラーゼによる一塩基伸長反応を行い,該ポリヌクレオチドの相補鎖を形成する。一塩基伸長反応過程で必要な4種類のヌクレオチドを異なる蛍光色素で標識しておき,一塩基が伸長するたびに蛍光検出を行い,導入されたヌクレオチドを識別する。担体基板上に固定された各ポリヌクレオチドに対してこの蛍光検出を繰り返し行うことにより,該ポリヌクレオチドの塩基配列を網羅的に解読することができる。この方式では一塩基伸長反応の際に単一分子蛍光検出を行う必要があり,高精度の結果を得るためには,担体基板上に解析対象となるポリヌクレオチドを1分子ずつ固定する方法,及び蛍光検出を高感度化する方法が必要である。なお,DNAシーケンシングにおいて実際の検体となるポリヌクレオチドは,ゲノムDNAの転写産物であるメッセンジャーRNA(mRNA)である。
【0007】
蛍光の検出感度を向上させる方法の一例として,金属微粒子による蛍光増強効果を利用する方法が非特許文献4に報告されている。この報告例では,担体基板上に銀ナノ粒子を規則的に配列し,この表面にプローブDNAを結合している。このプローブDNAを蛍光標識された検体と反応させ,担体基板に励起光を照射すると,銀ナノ粒子の自由電子が共鳴振動(局在プラズモン共鳴)を起こすため,蛍光が増強する。この蛍光増強効果により,検体の高感度検出が可能であるとしている。この報告例では蛍光増強が起こることが示されているが,単一分子蛍光検出を狙ったものではないため,銀ナノ粒子表面には多数のプローブDNAが固定されている。従って,この方法を利用して高精度の単一分子蛍光検出を実現するためには,表面に単一プローブ分子を固定した金属微粒子を利用することが必要である。
【0008】
【特許文献1】特開2006-153826号公報
【特許文献2】特開2006-288406号公報
【非特許文献1】Nano Letters Vol.1, No.1, p32 (2001)
【非特許文献2】Chem. Commun., p1156 (2004)
【非特許文献3】Proc.Natl.Acad.Sci.USA, Vol.100(7), p3960,2003
【非特許文献4】Biochem. Biophys. Res. Comm. 306 p213 (2003)
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の目的は,生体分子を蛍光法により単一分子レベルで高感度かつ高精度に検出することである。この目的を達成するための課題は,生体分子検出素子上に解析対象である生体分子を1分子ずつ固定すること,及び生体分子検出素子の蛍光検出感度を増大することである。
【課題を解決するための手段】
【0010】
本発明ではまず,解析対象である生体分子を1分子ずつ捕捉するために,表面に1個のプローブ分子を化学結合により固定した金属微粒子,すなわち単一プローブ分子固定金属微粒子を製造する。金属微粒子の表面と化学結合しうる複数の官能基を有するプローブ分子を使用することにより,単一プローブ分子固定金属微粒子を高収率で製造する。次に,単一プローブ分子固定金属微粒子を担体基板上に固定し,単一プローブ分子素子を製造する。この単一プローブ分子素子は,単一プローブ分子固定金属微粒子の単一プローブ分子を介して,担体基板表面に解析対象である生体分子を1分子ずつ捕捉することが可能である。また,プローブ分子近傍での蛍光検出の際の励起光による金属微粒子のプラズモン共鳴効果を活用して蛍光強度を増強し,単一分子の蛍光を高感度かつ高精度に検出することが可能である。
【発明の効果】
【0011】
本発明によれば,生体分子を蛍光法により単一分子レベルで高感度かつ高精度に検出することのできる単一プローブ分子素子を提供することができる。単一プローブ分子素子では,担体基板表面に固定した金属微粒子の表面にプローブ分子が1分子ずつ固定されているため,そのプローブ分子を利用して解析対象である生体分子を担体基板表面に1分子ずつ捕捉することが可能である。蛍光検出の際には励起光と金属微粒子内自由電子の共鳴振動に基づく蛍光の増強が起こり,単一蛍光分子からの蛍光を高感度かつ高精度に検出することが可能である。
【発明を実施するための最良の形態】
【0012】
以下,図面を参照して本発明の実施の形態を説明する。
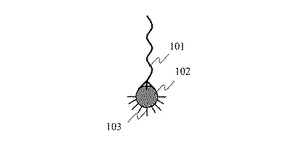
図1に,本発明による単一プローブ分子固定金属微粒子の一例を示す。解析対象となる生体分子を1分子だけ捕捉するための単一プローブ分子101が金属微粒子102の表面に複数個の化学結合により固定されている。図1では単一プローブ分子101が3個の化学結合により金属微粒子102の表面に固定されている。また,金属微粒子102の表面のうち,単一プローブ分子101が固定されていない部分には,様々な生体分子の非特異的な吸着を防止するために,吸着阻害分子103が化学結合により固定されている。なお,図1では金属微粒子として球形の微粒子を示したが,金属微粒子102の形状は円柱,円錐,角柱,角錐などでもよい。
【0013】
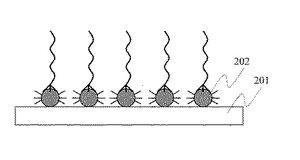
図2に,本発明による単一プローブ分子素子の一例を示す。担体基板201の表面に,化学結合により単一プローブ分子固定金属微粒子202を固定する。
【0014】
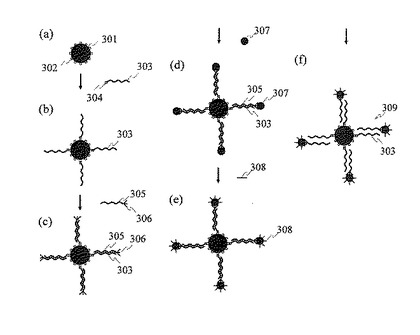
本発明による単一プローブ分子固定金属微粒子の製造プロセスの一例を,図3及び図4を用いて説明する。単一プローブ分子固定金属微粒子の製造プロセスは下記の6工程から成る。ここではプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した金属微粒子を製造することが可能である。
(1)磁性微粒子表面への固定官能基の結合
(2)磁性微粒子表面へのDNAの結合
(3)磁性微粒子表面での二本鎖DNAの形成
(4)プローブDNAの末端官能基への金属微粒子の結合
(5)金属微粒子表面への吸着阻害分子の結合
(6)単一プローブDNA固定金属微粒子の分離・回収
【0015】
各工程について以下に説明する。
(1)磁性微粒子表面への固定官能基の結合(図3(a))
微小な磁性微粒子301の表面に,プローブDNAと相補的な塩基配列を持つDNAを結合するための固定官能基302を結合する。磁性微粒子301は中心部に強い磁化を持つ磁性材料を含むポリマーブレンドのコアがあり,このコアの周囲をポリスチレン,ポリプロピレンなどのポリマーが取り囲む構造を持つ。プローブDNAと相補的なDNAを固定するための固定官能基302の例としてカルボキシル基,ストレプトアビジン,エポキシ基,アミノ基などがある。この固定官能基に結合されるDNAで起こるハイブリダイゼーションなどの化学反応が,磁性微粒子の表面上で隣接する他のDNAからの立体的な相互作用により妨げられることのないよう,磁性微粒子表面に結合する固定官能基302の密度を適切に調整し,固定官能基間の距離を十分にとる。
【0016】
(2)磁性微粒子表面へのDNAの結合(図3(b))
磁性微粒子表面の固定官能基302にプローブDNAと相補的な塩基配列を持つDNA303を結合する。反応容器中で磁性微粒子301を含む溶液及びDNA303を含む溶液を混合して反応する。このDNA303の一方の末端は,磁性微粒子表面の固定官能基302と結合可能な末端官能基304にて修飾する。例えば,磁性微粒子表面の固定官能基302がカルボキシル基の場合,プローブDNAと相補的なDNA303の末端官能基304はアミノ基が好適である。このとき,DNA303はアミド結合により磁性微粒子の表面に結合する。また,磁性微粒子表面の固定官能基302がストレプトアビジンの場合,プローブDNAと相補的なDNA303の末端官能基304はビオチンが好適である。このとき,DNA303はアビジン−ビオチン結合により磁性微粒子の表面に結合する。反応後,磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面付近に集合しておき,磁性微粒子表面の固定官能基に結合しなかった余剰のDNA303を適切なバッファにより洗浄除去する。
【0017】
(3)磁性微粒子表面での二本鎖DNAの形成(図3(c))
磁性微粒子の表面に結合したDNA303とプローブDNA305をハイブリダイズし,二本鎖DNAを形成する。プローブDNAの一方の末端には,金属微粒子を結合するための末端官能基306を導入してある。プローブDNAの末端官能基306の例としてチオール基,ビオチンなどが挙げられる。反応後,磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面付近に集合しておき,磁性微粒子表面で二本鎖DNAを形成しなかった余剰のプローブDNA305を適切なバッファにより洗浄除去する。
【0018】
(4)プローブDNAの末端官能基への金属微粒子の結合(図3(d))
磁性微粒子に結合したDNA303とハイブリダイズしたプローブDNA305の末端官能基306に,単一の金属微粒子307を結合する。例えばプローブDNAの末端官能基がチオール基である場合,金属微粒子として金ナノ粒子が好適である。また,プローブDNAの末端官能基がビオチンである場合,金属微粒子として表面にストレプトアビジンが結合した金ナノ粒子が好適である。反応後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合しておき,プローブDNAの末端官能基306に結合しなかった金属微粒子307を適切なバッファにより洗浄除去する。以上の工程により,金属微粒子307の表面に単一のプローブDNA305が固定された単一プローブDNA固定金属微粒子を製作する。
【0019】
なお,金属微粒子307の材料としては貴金属類である金,銀,白金,パラジウム,ロジウム,イリジウム,ルテニウム,オスミウム,あるいはこれらの合金を使用することができる。あるいは,これらの貴金属類で作られた微粒子の表面に他の貴金属類がコーティングされたもの,例えば銀微粒子の表面に金がコーティングされた金属微粒子を使用してもよい。金属微粒子のサイズは,基板表面への固定安定性の観点から10nm以上が望ましい。一方,蛍光増強の観点からは,金属微粒子のサイズは蛍光増強効果を得ることができる10nm以上1m以下が望ましい。
【0020】
なお,図3ではプローブ分子305の例として金属微粒子と結合する官能基306を3個持つものを示したが,金属微粒子と結合する官能基を複数個持つプローブ分子を使用してもよい。例えば図4(a)には分岐した分子構造を持ち,それぞれの分岐鎖の末端に金属微粒子401と結合することのできる官能基402を持つプローブ分子403を示した。また,図4(b)には分岐した分子構造を持ち,それぞれの分岐鎖の途中及び末端に金属微粒子404と結合することのできる官能基405を複数個持つプローブ分子406を示した。図4(a)及び図4(b)に示したプローブ分子は金属微粒子と結合することのできる官能基を複数個持つため,金属微粒子表面により強固に結合することが可能となる。
【0021】
また,ここでは工程(3)に示す磁性微粒子表面での二本鎖DNAの形成のあとに工程(4)に示すプローブDNAの末端官能基への金属微粒子の結合を行ったが,工程(4)のあとに工程(3)を行ってもよい。すなわち,磁性微粒子301を含む溶液の入った反応容器とは別の反応容器内で,プローブDNA305及び金属微粒子307を混合し反応させる。このとき,金属微粒子のモル数に対するプローブDNAのモル数の比率を1以下とし,プローブDNAが希薄な状態で反応させることにより,単一プローブDNA固定金属微粒子を含む溶液を調製する。この溶液を,表面にプローブDNAと相補的なDNA303が結合した磁性微粒子301を含む溶液と混合する。このとき,磁性微粒子の表面に結合したDNA303と単一プローブDNA固定金属微粒子のプローブDNAが二本鎖を形成する。従って,磁性微粒子の表面に単一プローブDNA固定金属微粒子を結合し回収することができる。
【0022】
(5)金属微粒子表面への吸着阻害分子の結合(図3(e))
工程(4)で製作された単一プローブDNA固定金属微粒子では,プローブ分子が固定していない金属微粒子の表面部分に生体分子などが非特異的に吸着する可能性がある。そこで,金属微粒子表面の単一プローブ分子が結合していない部分に吸着阻害分子308を結合する。金属微粒子として金ナノ粒子を用いた場合,吸着阻害分子として1−メルカプトヘキサノール,2−メルカプトエタノール,ビス−(p−(スルホナトフェニル)フェニルホスフィン(bis-(p-sulfonatophenyl)phenylphosphine)などを使用することができる。反応後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合しておき,金属微粒子の表面に結合しなかった吸着阻害分子308を適切なバッファにより洗浄除去する。なお,ここでは工程(4)に示すプローブDNAの末端官能基への金属微粒子の結合のあとに工程(5)に示す金属微粒子表面への吸着阻害分子の結合を行ったが,工程(5)のあとに工程(4)を行ってもよい。すなわち,予め表面に吸着阻害分子を結合した金属微粒子を,プローブDNAの末端官能基に結合してもよい。
【0023】
(6)単一プローブDNA固定金属微粒子の分離・回収(図3(f))
加温,又はアルカリ処理などのDNAアニールの処理を行い,磁性微粒子の表面に形成された二本鎖DNAを解離し,一本鎖DNAにする。このとき,単一プローブDNA固定金属微粒子309と磁性微粒子の混合物が得られる。磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面に集合しておき,上澄み液中に含まれる単一プローブDNA固定金属微粒子を分離・回収する。
【0024】
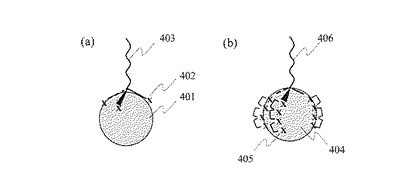
次に,工程(1)から工程(6)によって得られた単一プローブDNA固定金属微粒子の固定DNA数を評価する方法を,図5を用いて説明する。金属微粒子表面に固定されたDNA数の評価方法としてゲル電気泳動を利用する方法,光吸収スペクトルを利用する方法などがある。ここでは非特許文献1に開示されている方法を利用し,ゲル電気泳動により単一プローブDNA固定金属微粒子の固定DNA数を評価する。
【0025】
(7)単一プローブDNA固定金属微粒子の固定DNA数の評価
工程(1)から工程(6)により調製した単一プローブDNA固定金属微粒子を含む溶液をアガロース,アクリルアミドなどで製作した電気泳動用ゲル501に注入し,ゲル電気泳動を行う。比較対照用サンプルとして,金属微粒子及びこの金属微粒子の表面に結合しうる官能基を末端に有するプローブDNAを混合・反応した溶液を調製する。このとき,金属微粒子とプローブDNAのモル比がそれぞれ1:0,1:1,1:2及び1:3となるように混合・反応した4種類の溶液を調製する。これらの比較対象用サンプルには,表面にプローブDNAが固定された金属微粒子が含まれる。
【0026】
単一プローブDNA固定金属微粒子を含む溶液を電気泳動用ゲル501のウェル502に,また,金属微粒子とプローブDNAのモル比がそれぞれ1:0,1:1,1:2及び1:3となるように混合し反応させた溶液をそれぞれウェル503,504,505,及び506に注入し,ゲル電気泳動を開始する。金属微粒子及びプローブDNAは表面に負電荷を持つため,電気泳動を開始すると各サンプル中に含まれる金属微粒子は電気泳動ゲル中を負極側507から正極側508へと,泳動方向509の向きに泳動する。また,プローブ固定金属微粒子では固定DNA数の増加とともにゲル電気泳動時の有効体積が増加するため,泳動開始位置であるウェルからの泳動距離が短くなる。
【0027】
従って,比較対照用サンプルを泳動させると,表面にプローブDNAが固定されていない金属微粒子510,表面にプローブDNAが1個固定された金属微粒子511,表面にプローブDNAが2個固定された金属微粒子512,及び表面にプローブDNAが3個固定された金属微粒子513に対応する電気泳動バンドが観察される。ここで工程(1)から工程(6)により製造された単一プローブDNA固定金属微粒子を含む溶液では,表面にプローブDNAが1個固定された金属微粒子に対応する電気泳動バンド514だけが観察される。この観察結果から,工程(1)から工程(6)により,単一プローブDNA固定金属微粒子が高純度で製作されていることを確認することができる。
【0028】
なお,上記の説明では,表面にプローブDNAを固定した金属微粒子のゲル電気泳動を行った。しかしながらプローブDNAの塩基長が短い場合,金属微粒子の表面に固定したプローブDNA数が増加しても,ゲル電気泳動時のプローブ固定金属微粒子の有効体積の増加は小さい。従って,固定プローブDNA数の変化に対応する泳動距離の変化が小さくなり,プローブ固定金属微粒子を分離することが困難となる。このような場合,プローブDNA固定金属微粒子を,プローブDNAと相補的に結合することのできる分子量の大きい相補的分子と反応させた後にゲル電気泳動を行ってもよい。前記相補的分子としてはプローブDNAと相補的な塩基配列を持つ長鎖DNAなどを使用することができる。プローブ固定金属微粒子を分子量の大きい相補的分子と反応させると,金属微粒子表面の各プローブDNAに相補的分子が結合する。すなわち,表面にプローブDNAを1個,2個,3個固定した金属微粒子にはそれぞれ相補的分子が1個,2個,3個結合する。結合した相補的分子数が増加すると,プローブDNA固定金属微粒子のゲル電気泳動時の有効体積が大きく増加し,泳動距離が大きく減少する。従って,相補的分子の結合数,すなわち固定プローブDNA数に応じてプローブDNA固定金属微粒子の泳動距離が大きく変化し,電気泳動ゲル中で分離することが可能となる。
【0029】
次に,本発明の単一プローブ分子固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造プロセスの一例を図6を用いて説明する。ここでもプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した金属微粒子を担体基板表面に固定した単一プローブ分子素子を製造することが可能である。単一プローブDNA固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造プロセスは下記の3工程からなる。
(8)担体基板表面のリンカー分子層の形成
(9)担体基板表面への単一プローブDNA固定金属微粒子の固定
(10)担体基板表面の吸着阻害分子層の形成
【0030】
各工程について以下に説明する。
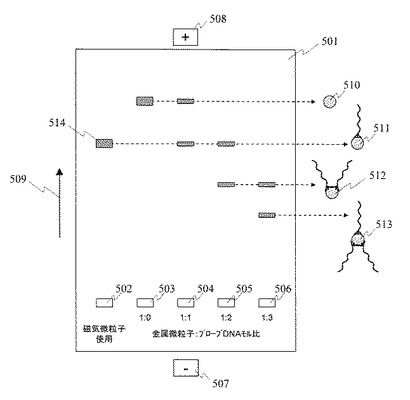
(8)担体基板表面のリンカー分子層の形成(図6(a)及び図6(b))
洗浄した担体基板601をリンカー分子Aの溶液に浸漬し,担体基板表面にリンカー分子Aを反応させる。基板としてガラス基板,石英基板などを使用する。また,リンカー分子Aとして,基板表面と結合する官能基及び金属微粒子と結合する官能基を併せ持つ分子を使用する。例えば,3−アミノプロピルトリメトキシシラン,3−アミノプロピルトリエトキシシラン,3−メルカプトプロピルトリメトキシシランなどを使用する。溶媒はメタノール,エタノール,トルエン,水などを使用する。リンカー分子Aは基板表面のシラノール基と結合するとともに,アミノ基,チオール基などの官能基が基板と反対側に露出し,次の工程にて単一プローブDNA固定金属微粒子と結合する。
【0031】
(9)担体基板表面への単一プローブDNA固定金属微粒子の固定(図6(c))
担体基板表面に露出した官能基と金属微粒子の相互作用により,担体基板表面に単一プローブDNA固定金属微粒子602を固定する。担体基板表面に露出した官能基は,単一プローブDNA固定金属微粒子の金属微粒子表面に固定された吸着阻害分子と置換し金属微粒子表面に結合するか,又は吸着阻害分子と化学結合を形成することにより,担体基板表面に単一プローブDNA固定金属微粒子を固定する。具体的には単一プローブDNA固定金属微粒子を含む溶液を担体基板表面に滴下し,20〜80℃にて0.5〜50時間反応する。担体基板表面に滴下する単一プローブDNA固定金属微粒子を含む溶液の濃度を調整することにより,担体基板表面に固定される単一プローブDNA固定金属微粒子の密度を調整する。
【0032】
(10)担体基板表面の吸着阻害分子層の形成(図6(d))
担体基板表面のうち,単一プローブDNA固定金属微粒子が固定されていない表面には,その後のプロセスで生体分子などが非特異的に吸着する可能性がある。担体基板表面に非特異的に吸着した生体分子などは検出ノイズとなるため,これを抑制する必要がある。そこで担体基板表面で単一プローブDNA固定金属微粒子が固定されなかった領域に吸着阻害分子Bを固定する。吸着阻害分子としてはNHS(N−ヒドロスクシンイミジル)エステルを末端に持つポリエチレングリコール(NHS-PEG)などを使用することができる。
【0033】
次に,工程(1)から工程(10)を経て製作した単一プローブDNA固定金属微粒子を担体基板表面に固定した単一プローブ分子素子をDNAシーケンシングに使用する例について説明する。
【0034】
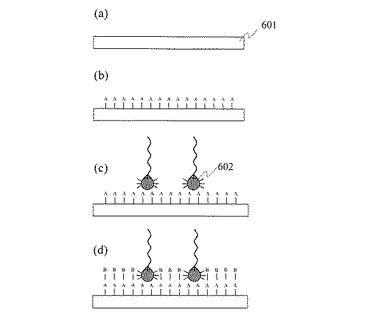
(11)DNAシーケンシング評価
まず,工程(1)から工程(10)によって,図7に示すように単一プローブDNA固定金属微粒子701を担体基板702の表面に固定する。次に,シーケンシング対象の単一DNA分子703を基板上の金属微粒子表面に固定したプローブDNA704と1:1で反応させ,基板表面に1分子ずつ固定する。具体的には,シーケンシング対象のDNA分子を塩化ナトリウム(NaCl)などの塩を含む溶液に溶解し,この溶液を単一プローブDNA固定金属微粒子を固定した基板702の表面に滴下する。反応温度は20℃〜80℃であり,反応時間は1時間から24時間である。
【0035】
DNAシーケンシングにおいて実際の解析対象となるのは,ゲノムDNAの転写産物であるメッセンジャーRNA(mRNA)である。mRNAの特徴は3’末端にアデニン(A)が平均約200個連続した配列(PolyAテイル)を持つことである。シーケンシング対象の単一mRNA分子と金属微粒子の表面に固定したプローブDNAを反応させるためには,プローブDNAが単一mRNA分子との反応サイトを持つ必要がある。従ってシーケンシング対象が単一mRNA分子である場合,プローブDNAとしてPolyAテイルと相補的な塩基配列であるPolyT配列を有するものを使用する。
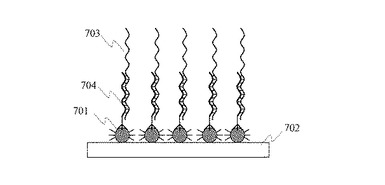
【0036】
次に図8に示すように,金属微粒子801に固定したプローブDNA802をプライマーとして,ポリメラーゼ反応によりプローブDNAの先端にヌクレオチドを一塩基伸長させる。なお,図8ではDNAが4種類の塩基(アデニン(A),チミン(T),グアニン(G),シトシン(C))のうちのいずれか1つを持つヌクレオチドが連続して結合した分子であることを示すため,DNAをヌクレオチドを表す正方形が直線状に多数連なった構造として表現している。図8に示す反応では,塩化マグネシウム(MgCl2)などの塩を含む溶液にヌクレオチド804を溶解した溶液を基板と接触させる。伸長したヌクレオチド804には蛍光分子805が結合している。この蛍光分子からの蛍光を検出することにより,伸長したヌクレオチドの種類を識別することができる。図中,803はポリメラーゼである。
【0037】
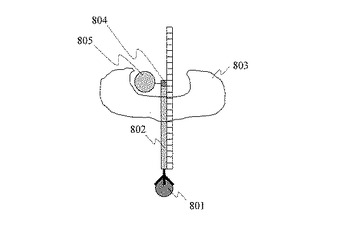
図9に,DNAシーケンシング読取システムを示す。このシステムでは,励起レーザー光901を担体基板902の裏面から石英プリズム903を通して全反射条件で入射させる。全反射条件で入射した励起レーザー光のうち,解析対象である単一DNA分子906が固定された面に染み出したエバネッセント光によって,一塩基伸長したヌクレオチド908に結合した蛍光分子909を励起する。蛍光分子909から発せられた蛍光910を基板上面から高感度のCCDカメラ911によって測定する。ヌクレオチドが伸長した後,ヌクレオチドに結合した蛍光分子を除去する。例えばヌクレオチドのリン酸基の末端に蛍光分子が結合している場合,リン酸基の切断により蛍光分子が単一DNA分子から除去される。引き続き,別の種類のヌクレオチドを反応させる。図中,907はポリメラーゼである。
【0038】
例えば,まずヌクレオチドAを含む溶液と基板を接触させ,ポリメラーゼ反応によってAが一塩基伸長した場合,上記の読取システムによって蛍光が検出される。ヌクレオチドAが一塩基伸長しなかった場合には蛍光は検出されない。この測定結果から,蛍光が検出された金属微粒子では,解析対象の単一DNA分子の解析対象部分にAと相補的であるTを持つことがわかる。また,蛍光が検出されなかった金属微粒子では,解析対象部分にTを持たないことがわかる。次に例えばヌクレオチドCを含む溶液を基板と接触させた後に蛍光を検出し,各金属微粒子において単一DNA分子の解析対象部分にCと相補的なGを有するか否かを判別する。引き続きヌクレオチドT及びGを反応させる。これらの工程を繰り返すことにより,各金属微粒子904に固定した単一DNA分子905の塩基配列を読み取る。ここでは一塩基伸長に使用する蛍光物質としてヌクレオチドのリン酸部分に蛍光色素が結合した試薬を使用したが,ヌクレオチドのプリン又はピリミジン塩基に蛍光色素が結合した試薬,あるいはヌクレオチドの3’OH末端に蛍光色素が結合した試薬を使用してもよい。
【0039】
なお,工程(8)から工程(11)では,工程(1)から工程(7)により製造した単一プローブ分子固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造方法,及びこれを用いた生体分子検出方法を示した。一方,工程(1)から工程(7)により製造した単一プローブ分子固定金属微粒子を担体基板表面に固定することなく,溶液中において生体分子検出用プローブ微粒子,生体分子捕捉用微粒子,及び生体分子搬送用微粒子などとして使用することもできる。例えば,単一プローブ分子固定金属微粒子を細胞に取り込ませ,所望の分量のプローブ分子を細胞内の標的部位に搬送する。また,細胞内での金属微粒子の軌跡を追跡することにより,細胞内でのプローブ分子の搬送経路を追跡することが可能である。
【0040】
次に,本発明の単一プローブ分子固定金属微粒子の製造方法,この金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造方法,及びこの単一プローブ分子素子を利用したDNAシーケンシングの例を実施例1により詳細に説明する。
【実施例1】
【0041】
(1)磁性微粒子表面への固定官能基の結合
本実施例では,表面に固定官能基が結合されている市販の磁性微粒子を使用した。具体的には,市販のカルボキシル基結合型磁性微粒子を使用した。この磁性微粒子の平均直径は1.05μmであった。この磁性微粒子は中心部に強い磁化をもつ磁性材料であるγFe2O3とFe3O4が一様に分布したポリマーブレンドのコアを持ち,このコアを外側のポリスチレン層で被覆した構造である。
【0042】
(2)磁性微粒子表面へのDNAの結合
本実施例ではプローブDNAとして,塩基配列が5’末端から,TTTTTTTTTTTTTTTTTTTT(PolyT20, 塩基長20)であるDNAを選択した。従って,このプローブDNAと相補的なDNAの塩基配列は,5’末端側からAAAAAAAAAAAAAAAAAAAA(PolyA 20, 塩基長20)である。そこで,磁性微粒子表面にPolyT20を結合させた。
【0043】
具体的にはまず,磁性微粒子を25mM MES(2−(N−モルホリノ)エタン スルホニック アシド,2-(N-morpholino)ethane sulfonic acid)バッファ(pH6)により洗浄した。またEDC(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド ハイドロクロライド,1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride)及びNHS(N−ヒドロキシスクシンイミジル,N-hydroxysuccinimidyl)エステルを25mM MESバッファにより溶解し,それぞれ濃度を50mg/mlに調製した。反応容器中にMESバッファで洗浄した磁性微粒子を含む溶液を取り,さらにEDC及びNHSを添加し30分間撹拌した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合させ,上澄み液を除去した。次に,5’末端にアミノ基を有するPolyA20をMESバッファに溶解して濃度を1nM〜1μMに調製し,これを前記磁性微粒子を含む溶液と混合・撹拌し,25℃にて30分間反応した。この反応により,PolyA20がアミド結合を介して磁性微粒子表面に結合した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合させ,上澄み液を除去した。最後に,磁性微粒子表面のカルボキシル基のうち,PolyT20が結合しなかったものをキャッピングするため,磁性微粒子をPBS(リン酸バッファ,Phosphate Buffered Saline)で調製した50mMエタノールアミン(pH8.0)で25℃,1時間処理した。
【0044】
(3)磁性微粒子表面での二本鎖DNAの形成
磁性微粒子の表面にプローブDNAが非特異的に吸着することを防止するため,表面にPolyA20を固定した磁性微粒子を0.1〜0.5%BSA(ウシ血清アルブミン,Bovine Serum Albumin)及び0.1%界面活性剤(Tween-20又はTriton X-100)を添加したPBSで洗浄した。次に,DNAのハイブリダイゼーション溶液である5×SSC(クエン酸バッファ,Standard Saline Citrate), 0.5%SDS(ドデシル硫酸ナトリウム,Sodium Dodecyl Sulfate)にてプローブDNAであるPolyT20の溶液(濃度1nM〜1μM)を調製した。なお,PolyT20の5’末端には,次の工程(4)にて金属微粒子を結合するためにチオール基を導入した。表面にPolyA20を結合した磁性微粒子を含む溶液とPolyT20の溶液を混合し,42℃の恒温オーブン中にて20時間ハイブリダイゼーションを行った。これにより,磁性微粒子の表面にPolyA20 とPolyT20から成る二本鎖DNAが形成された。ハイブリダイゼーション終了後,磁石を有する治具を反応容器に接近し,反応容器壁面に磁性微粒子を集合し,上澄み液を除去することにより磁性微粒子表面に結合したPolyA20とハイブリダイゼーションしなかった余剰のPolyT20を除去した。さらに5×SSC, 0.5%SDSにより磁性微粒子を洗浄した。
【0045】
(4)プローブDNAの末端官能基への金属微粒子の結合
磁性微粒子表面で二本鎖DNAを形成しているPolyT20の5’末端にあるチオール基に,金属微粒子である金ナノ粒子を固定した。本実施例では粒子径30nmの金ナノ粒子クエン酸溶液(濃度0.007% Weight/Volume)を使用した。5×SSC, 0.5%SDS溶液にて濃度を1nM〜1μMに調製した金ナノ粒子を磁性微粒子と混合し,25℃にて16〜24時間反応した。これにより,磁性微粒子表面で二本鎖DNAを形成しているPolyT20の5’末端に,チオール基を介して金ナノ粒子を固定した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合して上澄み液を除去することにより,PolyT20の末端チオール基に結合しなかった余剰の金ナノ粒子を除去した。
【0046】
(5)金属微粒子表面への吸着阻害分子の結合
単一プローブDNA固定金ナノ粒子において,金ナノ粒子の表面のうちプローブDNAであるPolyTが固定されていない部分に,吸着阻害分子であるビス−(p−(スルホナトフェニル)フェニルホスフィン(bis-(p-sulfonatophenyl)phenylphosphine)を結合した。具体的には工程(1)から工程(4)で調製した磁気微粒子を含む溶液にビス−(p−(スルホナトフェニル)フェニルホスフィンの水溶液を添加し,反応溶液中のビス−(p−(スルホナトフェニル)フェニルホスフィンの終濃度を1mg/mlに調整したのち,25℃で16〜24時間反応した。
【0047】
(6)単一プローブDNA固定金属微粒子の分離・回収
工程(1)から工程(5)により調製された磁性微粒子を含む溶液を70℃に加温した。このとき,工程(4)において磁性微粒子表面に形成された二本鎖DNAが解離し,磁性微粒子表面に固定したPolyA20及び金ナノ粒子表面に固定したPolyT20がそれぞれ一本鎖DNAとなった。従って反応溶液中には単一PolyT20固定金ナノ粒子,及び表面にPolyA20が固定した磁性微粒子が遊離した。磁石を含む治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合し,上澄み液を回収することにより単一PolyT20固定金ナノ粒子を回収した。
【0048】
(7)単一プローブDNA固定金属微粒子に固定されたDNA数の評価
工程(6)では,表面に単一PolyT20分子が固定された粒子径30nmの金ナノ粒子が回収された。この金ナノ粒子では,金ナノ粒子が占める体積に対してPolyT20が占める体積が小さいため,表面に固定したPolyT20の数に応じて金ナノ粒子をゲル電気泳動により分離することは困難であった。そこで,金ナノ粒子の表面に固定したPolyT20に対して,さらに塩基長の長いDNAをハイブリダイズした後にゲル電気泳動を行った。本実施例では金ナノ粒子表面に固定したPolyT20に対して,3’末端にPolyA20配列を持つ70merDNAをハイブリダイズさせた。70merDNAの塩基配列は3’末端から5’末端に向かって
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG(70mer)
である。具体的には単一PolyT20固定金ナノ粒子と70merDNAを5×SSC, 0.5%SDS中で混合し,42℃の恒温オーブン中にて20時間ハイブリダイゼーションを行った。
【0049】
上記70merDNAとハイブリダイズした単一PolyT20固定金ナノ粒子を含む溶液を3%アガロースゲルのウェルに注入した。比較対照用サンプルとして,金ナノ粒子と5’末端をチオール基で修飾したPolyT20をモル比1:0,1:1,1:2及び1:3にて反応させた溶液を調製した。これらの比較対照用サンプルも上記70merDNAとハイブリダイズさせたのち,それぞれ前記アガロースゲルのウェルに注入した。このアガロースゲルに一定電圧(1〜10V/cm)を印加し,70merDNAとハイブリダイズしたPolyT20固定金ナノ粒子のゲル電気泳動を行った。金ナノ粒子及び金ナノ粒子表面で形成されたPolyT20及び70merDNAから成る二本鎖DNAは負電荷を有するため,電気泳動を開始すると各サンプル中に含まれるPolyT20固定金ナノ粒子はゲル中を負極側から正極側に泳動した。PolyT20固定金ナノ粒子は表面に固定されたPolyT20の数が増加するに伴い,より多くの70merDNAと二本鎖DNAを形成する。これによりPolyT20固定金ナノ粒子のゲル電気泳動時の有効体積が増大し,泳動開始位置からの泳動距離が短くなった。従って,金ナノ粒子表面に固定されたPolyT20の数の違いによる泳動距離の差がより大きくなった。工程(1)から工程(6)によって製造された単一PolyT20固定金ナノ粒子の電気泳動では,表面にPolyT20が1個固定された金ナノ粒子に対応する1本の泳動バンドが観察され,その他のバンドは観察されなかった。従って,本実施例で開示した方法により,単一PolyT20固定金ナノ粒子が高純度で製造されたことが確認された。
【0050】
(8)石英基板表面へのリンカー分子層の形成
洗浄した石英基板をシランカップリング剤である3−アミノプロピルトリメトキシシランのメタノール溶液に5分間浸漬した。続いて基板をメタノールで洗浄したのち,80℃で2時間アニールを行った。この工程により,石英基板表面にアミノ基が導入された。
【0051】
(9)石英基板表面への単一プローブDNA固定金属微粒子の固定
アミノ基を導入した石英基板表面に,単一PolyT20固定金ナノ粒子の溶液を滴下し,25℃にて16時間反応した。基板に滴下する単一PolyT20固定金ナノ粒子溶液の濃度を0.01〜100nMに調整することにより,石英基板表面に固定される単一PolyT20固定金ナノ粒子の密度を調整した。
【0052】
(10)基板表面の吸着阻害分子層の形成
担体基板上で単一PolyT20固定金ナノ粒子が固定されなかった領域に,吸着阻害分子であるNHSエステルを末端に有するポリエチレングリコール(NHS-PEG)を結合した。具体的にはトリエタノールアミン(pH8.0)で4mMに調製したNHS-PEG溶液に担体基板を浸漬し,25℃で1時間反応させた。反応終了後,担体基板を純水で十分に洗浄した。
【0053】
(11)DNAシーケンシング(図10,図11,図12)
工程(1)から工程(10)に従って製造した基板を用いて, DNAシーケンシングを行った。石英基板1001に固定された金ナノ粒子1002表面に固定された単一PolyT20分子1003に,シーケンシング対象である単一DNA分子1004をハイブリダイズさせた。本実施例ではシーケンシング対象の単一DNA分子として3’末端からA配列を連続して持つDNAをハイブリダイズさせた。この単一DNA分子1004の塩基配列を3’末端から5’末端の方向に示す。
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG(70mer)
【0054】
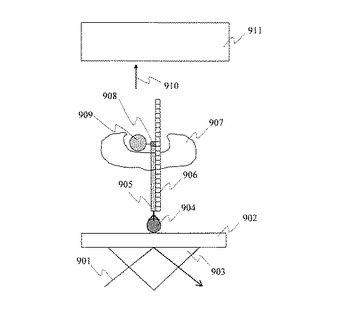
次に,プローブDNAである単一PolyT20分子1003をプライマーとして,単一DNA分子1004のシーケンシングを行った。製作した単一PolyT20固定金ナノ粒子を固定した基板とヌクレオチド3リン酸のリン酸基末端に蛍光分子Cy3を持つ4種類のヌクレオチド(A, T, G, C)を,ポリメラーゼ反応により反応させた。反応溶液は10mM トリス−塩酸(Tris-HCl), 5mM 塩化マグネシウム(MgCl2)溶液にて調製した1μM ヌクレオチド溶液である。ポリメラーゼ反応によって単一DNA分子1004の塩基配列と相補的な塩基を持つヌクレオチドが単一PolyT20分子に結合する。本実施例ではヌクレオチドC(図10において1005で示す)の溶液を石英基板1001と反応させた場合,単一PolyT20分子にヌクレオチドCが結合する。これは,シーケンシング対象である単一DNA分子においてPolyA配列に隣接するGにヌクレオチドCが結合するからである。
【0055】
図9に示した蛍光検出系を用いて,ポリメラーゼ反応中の蛍光画像を取得する。蛍光強度は石英基板上面から高感度CCDカメラで測定した。励起光を全反射条件で照射することにより,蛍光分子Cy3(図10において1006で示す)が結合したヌクレオチドCが結合した金ナノ粒子1002からの蛍光が検出される。ヌクレオチドが単一PolyT20分子に結合した後,このヌクレオチドのリン酸基が切断される。これによって,リン酸基の末端に結合していた蛍光分子Cy3が単一PolyT20分子から除去される。次に,ヌクレオチドAの溶液を基板と反応させると,シーケンシング対象である単一DNA分子1004のヌクレオチドGの隣のTにヌクレオチドAが結合する。このとき,蛍光が検出される。これらの手順を繰り返すことにより,各金ナノ粒子において単一DNA分子の塩基配列を読み取ることが可能であった。
【0056】
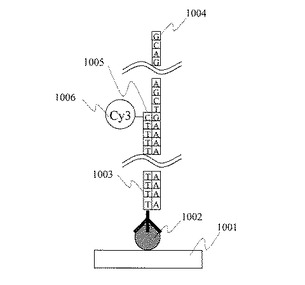
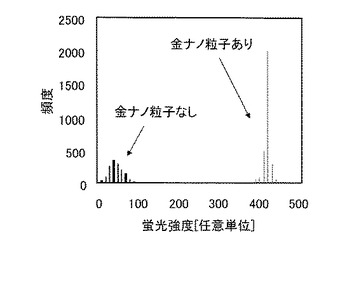
表面に単一PolyT20固定金ナノ粒子を固定した石英基板と比較対照するため,表面にPolyT20分子を直接固定した石英基板を製作した。本実施例で製作した,表面に単一PolyT20固定金ナノ粒子を固定した石英基板の概念図を図11(a)に,また表面にPolyT20分子を直接固定した石英基板の概念図を図11(b)に示した。これらの基板を用いて,単一DNA分子のシーケンシングを行うために,単一PolyT20分子に含まれるある1塩基の伸長反応を行った。このときに得られた単一PolyT20分子の蛍光強度のヒストグラムを図12に示した。
【0057】
表面に単一PolyT20固定金ナノ粒子を固定した石英基板(金ナノ粒子あり,図11(a)に対応する),ならびに表面にPolyT20分子を直接固定した石英基板(金ナノ粒子なし,図11(b)に対応する)のそれぞれについて,約3,000μm2の面積に含まれる蛍光輝点の強度を数値化した。その結果,金ナノ粒子がない基板では蛍光輝点の総数は約1,400個であったのに対し,金ナノ粒子がある基板では約3,000個の蛍光輝点が検出された。また,金ナノ粒子がない基板と比較して,金ナノ粒子がある場合には蛍光強度の平均値が約10倍であるとともに,蛍光強度の標準偏差も半減した。金ナノ粒子なしで基板に直接固定したPolyT20分子はランダムに固定されており,隣接するPolyT20分子間の距離も一定ではなく,蛍光強度のばらつきが大きくなった。一方,金ナノ粒子の表面に固定された単一PolyT20分子は石英基板表面に1分子ずつ,かつ距離が十分に離れて固定されており,より均質な反応条件下に置かれていることから,蛍光強度のバラツキが小さくなった。さらに,金ナノ粒子による蛍光増強効果により蛍光強度が増大することも確認された。従って,本実施例において表面に単一プローブDNA固定金ナノ粒子を固定した基板では,DNAシーケンシングを高精度かつ高感度で実施できることが示された。
【実施例2】
【0058】
次に,本発明で開示する単一プローブ分子素子において,金属微粒子の表面に2個以上の化学結合を介して固定されるプローブ分子の化学構造及び該プローブ分子を表面に固定した単一プローブ分子固定金属微粒子の製造プロセスの一例を実施例2に示す。ここでもプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した単一プローブ分子固定金属微粒子を製造することが可能である。
【0059】
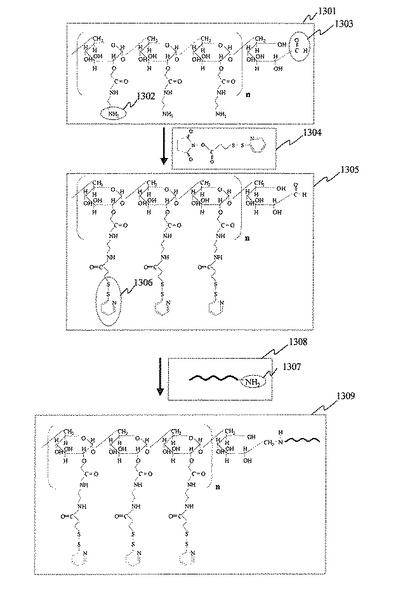
アミノデキストラン(Aminodextran)構造を有するプローブDNA分子の化学構造及び製造方法を図13に示す。アミノデキストラン1301はアミノ基1302を有するD−グルコースの縮重合ポリマーである。また,アミノデキストラン1301は還元末端に1個だけアルデヒド基1303を有する。D−グルコースの重合度が異なる様々な分子量のアミノデキストランを合成することが可能である。本実施例では平均分子量70,000のアミノデキストラン構造を有するプローブDNA分子の製造プロセスを説明する。
【0060】
(1)アミノデキストランへのジスルフィド基の導入
アミノデキストラン1301(平均分子量70,000)を1×PBS(リン酸バッファ,Phosphate Buffered Saline,pH7.0〜9.0)に溶解した。また,SPDP分子1304(3-(2-Pyridyldithio)propionic acid N-hydrosuccinimide ester)をジメチルスルホキシド(Dimethylsulfoxide)に溶解し,濃度を10mM〜500mMに調製した。アミノデキストランの溶液にSPDPの溶液を混合し,室温で撹拌しながら0.5〜20時間反応した。この反応により,ジスルフィド基1306が導入されたアミノデキストラン1305を得た。反応終了後,反応液の透析又はゲルろ過により,反応液に含まれる未反応のSPDP分子1304を除去した。
【0061】
(2)アミノデキストランと単一プローブDNA分子の結合
末端がアミノ基1307で修飾されたプローブDNA分子1308を,1×PBS(リン酸バッファ,Phosphate Buffered Saline ,pH7.0〜9.0)に溶解した。前記ジスルフィド基が導入されたアミノデキストラン1305の溶液と,前記プローブDNA分子1308の溶液を混合して反応液を調製した。ジスルフィド基が導入されたアミノデキストラン1305と末端がアミノ基で修飾されたプローブDNA分子1308の混合モル比は,1:1〜1:100の範囲とした。さらに前記反応液に1M NaOH(水酸化ナトリウム,Sodium hydroxide)に溶解したNaCNBH3(Soduim cyanoborohydride)の溶液を添加し,室温で撹拌しながら2〜20時間反応した。この反応により,ジスルフィド基が導入されたアミノデキストラン1305の還元末端にあるアルデヒド基とプローブDNA分子1308の末端アミノ基1307がアミド結合を形成し,アミノデキストランとプローブDNA分子が1:1で結合したデキストラン型プローブDNA分子1309を得た。反応終了後,反応液の透析又はゲルろ過により,反応液に含まれる未反応のプローブDNA分子1308を除去した。
【0062】
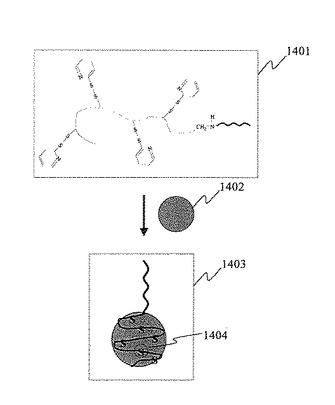
次に,デキストラン型プローブDNA分子を表面に固定した単一プローブDNA分子固定金属微粒子の模式図を図14に示す。本実施例では,実施例1の工程(1)〜(4)に示した方法に従い,デキストラン型プローブDNA分子1401(1401は1309を簡略化して示したものである)を粒子径10nmの金ナノ粒子1402の表面に結合し,単一プローブ分子固定金ナノ粒子1403を得た。本実施例では,デキストラン型プローブDNA分子1401の多数のジスルフィド結合が切断し,多数の硫黄原子1404が金ナノ粒子1402の表面に結合する。
【0063】
本実施例で示したデキストラン型プローブDNA分子1401のように多数の化学結合を介して金属微粒子の表面に固定されるプローブ分子は,1個の化学結合を介して金属微粒子の表面に固定されるプローブ分子と比較して以下のような利点がある。
(a)使用する金属微粒子の粒子径に対応してプローブ分子のポリマー部分(本実施例ではアミノデキストラン)の分子量を適正化することにより,金属微粒子とプローブ分子が1:1で結合した単一プローブ分子固定金属微粒子を高収率で得ることが可能である。
(b)プローブ分子と金属微粒子が多数の化学結合を介して強固に結合しているため,溶液の温度,イオン強度,pHなどが変化してもプローブ分子と金属微粒子が解離しにくく安定である。
(c)プローブ分子のポリマー部分が多数の親水性官能基(本実施例ではアミノデキストランのヒドロキシル基)を有する場合,単一プローブ分子固定金属微粒子の表面は多数の親水性官能基で被覆されるため,生体分子などの非特異的な吸着が抑制される。
【産業上の利用可能性】
【0064】
本発明の素子は,核酸の網羅的定量解析を行うDNAマイクロアレイや核酸の塩基配列を解読するDNAシーケンサにおいて,単一分子レベルで高精度かつ高感度の検出を行うための生体分子検出素子として適用できる。また,本発明の素子は,タンパク質や糖鎖などの生体分子,及び様々な化学物質を蛍光法を用いて高精度かつ高感度に検出するための検出素子として利用することができる。
【図面の簡単な説明】
【0065】
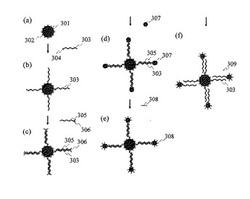
【図1】単一プローブ分子固定金属微粒子を示す模式図。
【図2】単一プローブ分子固定金属微粒子を担体基板表面に固定した生体分子検出素子を示す模式図。
【図3】磁性微粒子を用いて単一プローブ分子固定金属微粒子を製造する方法を示す模式図。
【図4】複数の官能基を介して金属微粒子の表面に結合した単一プローブ分子を示す模式図。
【図5】ゲル電気泳動によりプローブ分子固定金属微粒子を分離する方法を示す模式図。
【図6】単一プローブ分子固定金属微粒子を担体基板表面に固定して生体分子検出素子を製造する方法を示す模式図。
【図7】単一プローブDNA固定金属微粒子を担体基板表面に固定した生体分子検出素子にシーケンシング対象である単一DNA分子を固定した状態を示す模式図。
【図8】DNAシーケンシングの単一プローブDNA及びシーケンシング対象である単一DNA分子を示す模式図。
【図9】DNAシーケンシングの読取装置を示す模式図。
【図10】DNAシーケンシングの一塩基伸長反応を示す模式図。
【図11】(a)単一PolyT20固定金ナノ粒子を石英基板表面に固定した生体分子検出素子,及び(b)PolyT20分子を石英基板表面に直接固定した生体分子検出素子を示す模式図。
【図12】DNAシーケンシングの一塩基伸長反応中に検出した,各単一PolyT20分子の蛍光強度のヒストグラム。
【図13】アミノデキストラン構造を有するプローブDNA分子の化学構造及び製造方法を示す模式図。
【図14】デキストラン型プローブDNA分子を表面に固定した単一プローブDNA分子固定金属微粒子を示す模式図。
【符号の説明】
【0066】
101:単一プローブ分子,102:金属微粒子,103:吸着阻害分子,201:担体基板,202:単一プローブ分子固定金属微粒子,301:磁性微粒子,302:固定官能基,303:プローブDNAと相補的な塩基配列を持つDNA,304:末端官能基,305:プローブDNA,306:末端官能基,307:金属微粒子,308:吸着阻害分子,309:単一プローブDNA固定金属微粒子,401:金属微粒子,402:金属微粒子と結合する官能基,403:単一プローブ分子,404:金属微粒子,405:金属微粒子と結合する官能基,406:単一プローブ分子,501:電気泳動用ゲル,502:電気泳動用ゲルのウェル,503:電気泳動用ゲルのウェル,504:電気泳動用ゲルのウェル,505:電気泳動用ゲルのウェル,506:電気泳動用ゲルのウェル,507:電気泳動用ゲルの負極側,508:電気泳動用ゲルの正極側,509:電気泳動の泳動方向,510:表面にDNAが固定されていない金属微粒子,511:表面にDNAが1個固定された金属微粒子,512:表面にDNAが2個固定された金属微粒子,513:表面にDNAが3個固定された金属微粒子,514:単一プローブDNA固定金属微粒子に対応する電気泳動バンド,601:担体基板,602:単一プローブDNA固定金属微粒子,701:金属微粒子,702:担体基板,703:シーケンシング対象である単一DNA分子,704:単一プローブDNA,801:金属微粒子,802:単一プローブDNA,803:ポリメラーゼ,804:ヌクレオチド,805:蛍光分子,901:励起レーザー光,902:担体基板,903:石英プリズム,904:金属微粒子,905:単一プローブDNA,906:シーケンシング対象である単一DNA分子,907:ポリメラーゼ,908:ヌクレオチド,909:蛍光分子,910:蛍光分子から発した蛍光,911:高感度CCDカメラ,1001:担体基板,1002:金属微粒子,1003:単一PolyT20分子,1004:シーケンシング対象である単一DNA分子,1005:ヌクレオチドC,1006:蛍光分子Cy3,1101:単一PolyT20固定金ナノ粒子,1102:石英基板,1103:単一PolyT20分子,1104:石英基板,1301:アミノデキストラン,1302:アミノ基,1303:アルデヒド基,1304:SPDP分子,1305:ジスルフィド基が導入されたアミノデキストラン,1306:ジスルフィド基,1307:アミノ基,1308:末端がアミノ基で修飾されたプローブDNA分子,1309:デキストラン型プローブDNA分子,1401:デキストラン型プローブDNA分子,1402:金ナノ粒子,1403:単一プローブ分子固定金ナノ粒子,1404:硫黄原子。
【技術分野】
【0001】
本発明は,生体分子を検出するための単一プローブ分子素子及びその製造方法ならびに生体分子の検出方法に関するものである。
【背景技術】
【0002】
近年,核酸やタンパク質などの生体分子を特異的かつ網羅的に解析するための方法として,様々な生体分子検出素子の研究開発が進んでいる。こうした生体分子検出素子の重要な例として,ナノメートルからマイクロメートルサイズの微粒子の表面に生体分子を検出するためのプローブ分子を固定したプローブ分子固定微粒子がある。これまでに,プローブ分子固定微粒子を用いた様々な生体分子検出方法が報告されている。プローブ分子固定微粒子のうち,金属微粒子の表面にプローブ分子を固定したプローブ分子固定金属微粒子に着目すると,例えば特許文献1には,担体基板に固定した解析対象分子に対して,プローブ分子固定金ナノ粒子を結合し,該金ナノ粒子を電子顕微鏡などで検出することにより解析対象分子の識別及び定量評価を行う方法が開示されている。また,特許文献2には,表面にオリゴヌクレオチドを固定した金ナノ粒子がその凝集状態に応じて異なる色を呈することを利用した生体分子の分析方法が開示されている。
【0003】
プローブ分子固定金属微粒子を用いて生体分子の定量的な解析を行うためには,金属微粒子表面に固定されたプローブ分子の個数が正確にわかっていることが重要である。さらに,生体分子の単一分子レベルでの定量的な解析を実現するためには,表面に単一のプローブ分子を固定した金属微粒子の利用が必要不可欠である。従来の公知例において使用されているプローブ分子固定金属微粒子は,金属微粒子及び金属微粒子と強く相互作用する官能基を持つプローブ分子を所定の混合比にて混合し反応させることにより製造されている。従って,この方法により製造されるプローブ分子固定金属微粒子は,表面に固定されたプローブ分子の個数に分布を持つ金属微粒子の混合物である。単一プローブ分子固定金属微粒子を得るためには,該金属微粒子の混合物からの分離・精製を行う必要がある。
【0004】
単一プローブ分子固定金属微粒子を得るための方法として,ゲル電気泳動を利用する方法が非特許文献1に開示されている。この方法では,金ナノ粒子及び5’末端にチオール基をもつプローブDNAを所定のモル比にて混合・反応し,金ナノ粒子の表面にプローブDNAを固定する。このとき,表面に固定されたプローブDNAの個数が0個,1個,2個,3個,及び3個以上である金ナノ粒子の混合物が得られる。この金ナノ粒子の混合物のゲル電気泳動を行うと,固定DNA数が少ない金ナノ粒子から順にゲル中での泳動距離が大きくなる。従って,固定DNA数に応じて金ナノ粒子を分離することができ,単一プローブDNA固定金ナノ粒子の分離・回収が可能である。しかしながら,この方法における単一プローブDNA固定金ナノ粒子の分離・回収操作は非常に煩雑であるとともに,単一プローブDNA固定金ナノ粒子の回収率が非常に低いことが問題である。
【0005】
また,ポリマー分子を介して金属微粒子の表面にプローブDNAを固定する方法が非特許文献2に開示されている。この方法では,官能基として複数のジスルフィド基を持つデキストラン分子を合成し,このデキストラン分子にプローブDNAを結合する。ジスルフィド基を利用してプローブDNAが結合したデキストラン分子を金ナノ粒子の表面に固定する。この方法ではデキストラン分子を介して金ナノ粒子の表面にプローブDNAを固定することが可能であるが,デキストラン1分子に結合するプローブDNAの個数に分布があるという問題がある。従って,表面に単一プローブ分子を固定した金属微粒子を簡便かつ高収率で製造することのできる新規の方法が必要である。
【0006】
一方,生体分子を単一分子レベルで検出する技術のうち,非特許文献3に開示されているような単一DNA分子レベルのシーケンシングによる遺伝子配列解析技術の開発が活発に進められている。この方式では,担体基板表面に検体となる膨大な数のポリヌクレオチドを固定し,これを鋳型としてポリメラーゼによる一塩基伸長反応を行い,該ポリヌクレオチドの相補鎖を形成する。一塩基伸長反応過程で必要な4種類のヌクレオチドを異なる蛍光色素で標識しておき,一塩基が伸長するたびに蛍光検出を行い,導入されたヌクレオチドを識別する。担体基板上に固定された各ポリヌクレオチドに対してこの蛍光検出を繰り返し行うことにより,該ポリヌクレオチドの塩基配列を網羅的に解読することができる。この方式では一塩基伸長反応の際に単一分子蛍光検出を行う必要があり,高精度の結果を得るためには,担体基板上に解析対象となるポリヌクレオチドを1分子ずつ固定する方法,及び蛍光検出を高感度化する方法が必要である。なお,DNAシーケンシングにおいて実際の検体となるポリヌクレオチドは,ゲノムDNAの転写産物であるメッセンジャーRNA(mRNA)である。
【0007】
蛍光の検出感度を向上させる方法の一例として,金属微粒子による蛍光増強効果を利用する方法が非特許文献4に報告されている。この報告例では,担体基板上に銀ナノ粒子を規則的に配列し,この表面にプローブDNAを結合している。このプローブDNAを蛍光標識された検体と反応させ,担体基板に励起光を照射すると,銀ナノ粒子の自由電子が共鳴振動(局在プラズモン共鳴)を起こすため,蛍光が増強する。この蛍光増強効果により,検体の高感度検出が可能であるとしている。この報告例では蛍光増強が起こることが示されているが,単一分子蛍光検出を狙ったものではないため,銀ナノ粒子表面には多数のプローブDNAが固定されている。従って,この方法を利用して高精度の単一分子蛍光検出を実現するためには,表面に単一プローブ分子を固定した金属微粒子を利用することが必要である。
【0008】
【特許文献1】特開2006-153826号公報
【特許文献2】特開2006-288406号公報
【非特許文献1】Nano Letters Vol.1, No.1, p32 (2001)
【非特許文献2】Chem. Commun., p1156 (2004)
【非特許文献3】Proc.Natl.Acad.Sci.USA, Vol.100(7), p3960,2003
【非特許文献4】Biochem. Biophys. Res. Comm. 306 p213 (2003)
【発明の開示】
【発明が解決しようとする課題】
【0009】
本発明の目的は,生体分子を蛍光法により単一分子レベルで高感度かつ高精度に検出することである。この目的を達成するための課題は,生体分子検出素子上に解析対象である生体分子を1分子ずつ固定すること,及び生体分子検出素子の蛍光検出感度を増大することである。
【課題を解決するための手段】
【0010】
本発明ではまず,解析対象である生体分子を1分子ずつ捕捉するために,表面に1個のプローブ分子を化学結合により固定した金属微粒子,すなわち単一プローブ分子固定金属微粒子を製造する。金属微粒子の表面と化学結合しうる複数の官能基を有するプローブ分子を使用することにより,単一プローブ分子固定金属微粒子を高収率で製造する。次に,単一プローブ分子固定金属微粒子を担体基板上に固定し,単一プローブ分子素子を製造する。この単一プローブ分子素子は,単一プローブ分子固定金属微粒子の単一プローブ分子を介して,担体基板表面に解析対象である生体分子を1分子ずつ捕捉することが可能である。また,プローブ分子近傍での蛍光検出の際の励起光による金属微粒子のプラズモン共鳴効果を活用して蛍光強度を増強し,単一分子の蛍光を高感度かつ高精度に検出することが可能である。
【発明の効果】
【0011】
本発明によれば,生体分子を蛍光法により単一分子レベルで高感度かつ高精度に検出することのできる単一プローブ分子素子を提供することができる。単一プローブ分子素子では,担体基板表面に固定した金属微粒子の表面にプローブ分子が1分子ずつ固定されているため,そのプローブ分子を利用して解析対象である生体分子を担体基板表面に1分子ずつ捕捉することが可能である。蛍光検出の際には励起光と金属微粒子内自由電子の共鳴振動に基づく蛍光の増強が起こり,単一蛍光分子からの蛍光を高感度かつ高精度に検出することが可能である。
【発明を実施するための最良の形態】
【0012】
以下,図面を参照して本発明の実施の形態を説明する。
図1に,本発明による単一プローブ分子固定金属微粒子の一例を示す。解析対象となる生体分子を1分子だけ捕捉するための単一プローブ分子101が金属微粒子102の表面に複数個の化学結合により固定されている。図1では単一プローブ分子101が3個の化学結合により金属微粒子102の表面に固定されている。また,金属微粒子102の表面のうち,単一プローブ分子101が固定されていない部分には,様々な生体分子の非特異的な吸着を防止するために,吸着阻害分子103が化学結合により固定されている。なお,図1では金属微粒子として球形の微粒子を示したが,金属微粒子102の形状は円柱,円錐,角柱,角錐などでもよい。
【0013】
図2に,本発明による単一プローブ分子素子の一例を示す。担体基板201の表面に,化学結合により単一プローブ分子固定金属微粒子202を固定する。
【0014】
本発明による単一プローブ分子固定金属微粒子の製造プロセスの一例を,図3及び図4を用いて説明する。単一プローブ分子固定金属微粒子の製造プロセスは下記の6工程から成る。ここではプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した金属微粒子を製造することが可能である。
(1)磁性微粒子表面への固定官能基の結合
(2)磁性微粒子表面へのDNAの結合
(3)磁性微粒子表面での二本鎖DNAの形成
(4)プローブDNAの末端官能基への金属微粒子の結合
(5)金属微粒子表面への吸着阻害分子の結合
(6)単一プローブDNA固定金属微粒子の分離・回収
【0015】
各工程について以下に説明する。
(1)磁性微粒子表面への固定官能基の結合(図3(a))
微小な磁性微粒子301の表面に,プローブDNAと相補的な塩基配列を持つDNAを結合するための固定官能基302を結合する。磁性微粒子301は中心部に強い磁化を持つ磁性材料を含むポリマーブレンドのコアがあり,このコアの周囲をポリスチレン,ポリプロピレンなどのポリマーが取り囲む構造を持つ。プローブDNAと相補的なDNAを固定するための固定官能基302の例としてカルボキシル基,ストレプトアビジン,エポキシ基,アミノ基などがある。この固定官能基に結合されるDNAで起こるハイブリダイゼーションなどの化学反応が,磁性微粒子の表面上で隣接する他のDNAからの立体的な相互作用により妨げられることのないよう,磁性微粒子表面に結合する固定官能基302の密度を適切に調整し,固定官能基間の距離を十分にとる。
【0016】
(2)磁性微粒子表面へのDNAの結合(図3(b))
磁性微粒子表面の固定官能基302にプローブDNAと相補的な塩基配列を持つDNA303を結合する。反応容器中で磁性微粒子301を含む溶液及びDNA303を含む溶液を混合して反応する。このDNA303の一方の末端は,磁性微粒子表面の固定官能基302と結合可能な末端官能基304にて修飾する。例えば,磁性微粒子表面の固定官能基302がカルボキシル基の場合,プローブDNAと相補的なDNA303の末端官能基304はアミノ基が好適である。このとき,DNA303はアミド結合により磁性微粒子の表面に結合する。また,磁性微粒子表面の固定官能基302がストレプトアビジンの場合,プローブDNAと相補的なDNA303の末端官能基304はビオチンが好適である。このとき,DNA303はアビジン−ビオチン結合により磁性微粒子の表面に結合する。反応後,磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面付近に集合しておき,磁性微粒子表面の固定官能基に結合しなかった余剰のDNA303を適切なバッファにより洗浄除去する。
【0017】
(3)磁性微粒子表面での二本鎖DNAの形成(図3(c))
磁性微粒子の表面に結合したDNA303とプローブDNA305をハイブリダイズし,二本鎖DNAを形成する。プローブDNAの一方の末端には,金属微粒子を結合するための末端官能基306を導入してある。プローブDNAの末端官能基306の例としてチオール基,ビオチンなどが挙げられる。反応後,磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面付近に集合しておき,磁性微粒子表面で二本鎖DNAを形成しなかった余剰のプローブDNA305を適切なバッファにより洗浄除去する。
【0018】
(4)プローブDNAの末端官能基への金属微粒子の結合(図3(d))
磁性微粒子に結合したDNA303とハイブリダイズしたプローブDNA305の末端官能基306に,単一の金属微粒子307を結合する。例えばプローブDNAの末端官能基がチオール基である場合,金属微粒子として金ナノ粒子が好適である。また,プローブDNAの末端官能基がビオチンである場合,金属微粒子として表面にストレプトアビジンが結合した金ナノ粒子が好適である。反応後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合しておき,プローブDNAの末端官能基306に結合しなかった金属微粒子307を適切なバッファにより洗浄除去する。以上の工程により,金属微粒子307の表面に単一のプローブDNA305が固定された単一プローブDNA固定金属微粒子を製作する。
【0019】
なお,金属微粒子307の材料としては貴金属類である金,銀,白金,パラジウム,ロジウム,イリジウム,ルテニウム,オスミウム,あるいはこれらの合金を使用することができる。あるいは,これらの貴金属類で作られた微粒子の表面に他の貴金属類がコーティングされたもの,例えば銀微粒子の表面に金がコーティングされた金属微粒子を使用してもよい。金属微粒子のサイズは,基板表面への固定安定性の観点から10nm以上が望ましい。一方,蛍光増強の観点からは,金属微粒子のサイズは蛍光増強効果を得ることができる10nm以上1m以下が望ましい。
【0020】
なお,図3ではプローブ分子305の例として金属微粒子と結合する官能基306を3個持つものを示したが,金属微粒子と結合する官能基を複数個持つプローブ分子を使用してもよい。例えば図4(a)には分岐した分子構造を持ち,それぞれの分岐鎖の末端に金属微粒子401と結合することのできる官能基402を持つプローブ分子403を示した。また,図4(b)には分岐した分子構造を持ち,それぞれの分岐鎖の途中及び末端に金属微粒子404と結合することのできる官能基405を複数個持つプローブ分子406を示した。図4(a)及び図4(b)に示したプローブ分子は金属微粒子と結合することのできる官能基を複数個持つため,金属微粒子表面により強固に結合することが可能となる。
【0021】
また,ここでは工程(3)に示す磁性微粒子表面での二本鎖DNAの形成のあとに工程(4)に示すプローブDNAの末端官能基への金属微粒子の結合を行ったが,工程(4)のあとに工程(3)を行ってもよい。すなわち,磁性微粒子301を含む溶液の入った反応容器とは別の反応容器内で,プローブDNA305及び金属微粒子307を混合し反応させる。このとき,金属微粒子のモル数に対するプローブDNAのモル数の比率を1以下とし,プローブDNAが希薄な状態で反応させることにより,単一プローブDNA固定金属微粒子を含む溶液を調製する。この溶液を,表面にプローブDNAと相補的なDNA303が結合した磁性微粒子301を含む溶液と混合する。このとき,磁性微粒子の表面に結合したDNA303と単一プローブDNA固定金属微粒子のプローブDNAが二本鎖を形成する。従って,磁性微粒子の表面に単一プローブDNA固定金属微粒子を結合し回収することができる。
【0022】
(5)金属微粒子表面への吸着阻害分子の結合(図3(e))
工程(4)で製作された単一プローブDNA固定金属微粒子では,プローブ分子が固定していない金属微粒子の表面部分に生体分子などが非特異的に吸着する可能性がある。そこで,金属微粒子表面の単一プローブ分子が結合していない部分に吸着阻害分子308を結合する。金属微粒子として金ナノ粒子を用いた場合,吸着阻害分子として1−メルカプトヘキサノール,2−メルカプトエタノール,ビス−(p−(スルホナトフェニル)フェニルホスフィン(bis-(p-sulfonatophenyl)phenylphosphine)などを使用することができる。反応後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合しておき,金属微粒子の表面に結合しなかった吸着阻害分子308を適切なバッファにより洗浄除去する。なお,ここでは工程(4)に示すプローブDNAの末端官能基への金属微粒子の結合のあとに工程(5)に示す金属微粒子表面への吸着阻害分子の結合を行ったが,工程(5)のあとに工程(4)を行ってもよい。すなわち,予め表面に吸着阻害分子を結合した金属微粒子を,プローブDNAの末端官能基に結合してもよい。
【0023】
(6)単一プローブDNA固定金属微粒子の分離・回収(図3(f))
加温,又はアルカリ処理などのDNAアニールの処理を行い,磁性微粒子の表面に形成された二本鎖DNAを解離し,一本鎖DNAにする。このとき,単一プローブDNA固定金属微粒子309と磁性微粒子の混合物が得られる。磁石を有する治具を反応容器に接近し,磁性微粒子を反応容器の壁面に集合しておき,上澄み液中に含まれる単一プローブDNA固定金属微粒子を分離・回収する。
【0024】
次に,工程(1)から工程(6)によって得られた単一プローブDNA固定金属微粒子の固定DNA数を評価する方法を,図5を用いて説明する。金属微粒子表面に固定されたDNA数の評価方法としてゲル電気泳動を利用する方法,光吸収スペクトルを利用する方法などがある。ここでは非特許文献1に開示されている方法を利用し,ゲル電気泳動により単一プローブDNA固定金属微粒子の固定DNA数を評価する。
【0025】
(7)単一プローブDNA固定金属微粒子の固定DNA数の評価
工程(1)から工程(6)により調製した単一プローブDNA固定金属微粒子を含む溶液をアガロース,アクリルアミドなどで製作した電気泳動用ゲル501に注入し,ゲル電気泳動を行う。比較対照用サンプルとして,金属微粒子及びこの金属微粒子の表面に結合しうる官能基を末端に有するプローブDNAを混合・反応した溶液を調製する。このとき,金属微粒子とプローブDNAのモル比がそれぞれ1:0,1:1,1:2及び1:3となるように混合・反応した4種類の溶液を調製する。これらの比較対象用サンプルには,表面にプローブDNAが固定された金属微粒子が含まれる。
【0026】
単一プローブDNA固定金属微粒子を含む溶液を電気泳動用ゲル501のウェル502に,また,金属微粒子とプローブDNAのモル比がそれぞれ1:0,1:1,1:2及び1:3となるように混合し反応させた溶液をそれぞれウェル503,504,505,及び506に注入し,ゲル電気泳動を開始する。金属微粒子及びプローブDNAは表面に負電荷を持つため,電気泳動を開始すると各サンプル中に含まれる金属微粒子は電気泳動ゲル中を負極側507から正極側508へと,泳動方向509の向きに泳動する。また,プローブ固定金属微粒子では固定DNA数の増加とともにゲル電気泳動時の有効体積が増加するため,泳動開始位置であるウェルからの泳動距離が短くなる。
【0027】
従って,比較対照用サンプルを泳動させると,表面にプローブDNAが固定されていない金属微粒子510,表面にプローブDNAが1個固定された金属微粒子511,表面にプローブDNAが2個固定された金属微粒子512,及び表面にプローブDNAが3個固定された金属微粒子513に対応する電気泳動バンドが観察される。ここで工程(1)から工程(6)により製造された単一プローブDNA固定金属微粒子を含む溶液では,表面にプローブDNAが1個固定された金属微粒子に対応する電気泳動バンド514だけが観察される。この観察結果から,工程(1)から工程(6)により,単一プローブDNA固定金属微粒子が高純度で製作されていることを確認することができる。
【0028】
なお,上記の説明では,表面にプローブDNAを固定した金属微粒子のゲル電気泳動を行った。しかしながらプローブDNAの塩基長が短い場合,金属微粒子の表面に固定したプローブDNA数が増加しても,ゲル電気泳動時のプローブ固定金属微粒子の有効体積の増加は小さい。従って,固定プローブDNA数の変化に対応する泳動距離の変化が小さくなり,プローブ固定金属微粒子を分離することが困難となる。このような場合,プローブDNA固定金属微粒子を,プローブDNAと相補的に結合することのできる分子量の大きい相補的分子と反応させた後にゲル電気泳動を行ってもよい。前記相補的分子としてはプローブDNAと相補的な塩基配列を持つ長鎖DNAなどを使用することができる。プローブ固定金属微粒子を分子量の大きい相補的分子と反応させると,金属微粒子表面の各プローブDNAに相補的分子が結合する。すなわち,表面にプローブDNAを1個,2個,3個固定した金属微粒子にはそれぞれ相補的分子が1個,2個,3個結合する。結合した相補的分子数が増加すると,プローブDNA固定金属微粒子のゲル電気泳動時の有効体積が大きく増加し,泳動距離が大きく減少する。従って,相補的分子の結合数,すなわち固定プローブDNA数に応じてプローブDNA固定金属微粒子の泳動距離が大きく変化し,電気泳動ゲル中で分離することが可能となる。
【0029】
次に,本発明の単一プローブ分子固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造プロセスの一例を図6を用いて説明する。ここでもプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した金属微粒子を担体基板表面に固定した単一プローブ分子素子を製造することが可能である。単一プローブDNA固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造プロセスは下記の3工程からなる。
(8)担体基板表面のリンカー分子層の形成
(9)担体基板表面への単一プローブDNA固定金属微粒子の固定
(10)担体基板表面の吸着阻害分子層の形成
【0030】
各工程について以下に説明する。
(8)担体基板表面のリンカー分子層の形成(図6(a)及び図6(b))
洗浄した担体基板601をリンカー分子Aの溶液に浸漬し,担体基板表面にリンカー分子Aを反応させる。基板としてガラス基板,石英基板などを使用する。また,リンカー分子Aとして,基板表面と結合する官能基及び金属微粒子と結合する官能基を併せ持つ分子を使用する。例えば,3−アミノプロピルトリメトキシシラン,3−アミノプロピルトリエトキシシラン,3−メルカプトプロピルトリメトキシシランなどを使用する。溶媒はメタノール,エタノール,トルエン,水などを使用する。リンカー分子Aは基板表面のシラノール基と結合するとともに,アミノ基,チオール基などの官能基が基板と反対側に露出し,次の工程にて単一プローブDNA固定金属微粒子と結合する。
【0031】
(9)担体基板表面への単一プローブDNA固定金属微粒子の固定(図6(c))
担体基板表面に露出した官能基と金属微粒子の相互作用により,担体基板表面に単一プローブDNA固定金属微粒子602を固定する。担体基板表面に露出した官能基は,単一プローブDNA固定金属微粒子の金属微粒子表面に固定された吸着阻害分子と置換し金属微粒子表面に結合するか,又は吸着阻害分子と化学結合を形成することにより,担体基板表面に単一プローブDNA固定金属微粒子を固定する。具体的には単一プローブDNA固定金属微粒子を含む溶液を担体基板表面に滴下し,20〜80℃にて0.5〜50時間反応する。担体基板表面に滴下する単一プローブDNA固定金属微粒子を含む溶液の濃度を調整することにより,担体基板表面に固定される単一プローブDNA固定金属微粒子の密度を調整する。
【0032】
(10)担体基板表面の吸着阻害分子層の形成(図6(d))
担体基板表面のうち,単一プローブDNA固定金属微粒子が固定されていない表面には,その後のプロセスで生体分子などが非特異的に吸着する可能性がある。担体基板表面に非特異的に吸着した生体分子などは検出ノイズとなるため,これを抑制する必要がある。そこで担体基板表面で単一プローブDNA固定金属微粒子が固定されなかった領域に吸着阻害分子Bを固定する。吸着阻害分子としてはNHS(N−ヒドロスクシンイミジル)エステルを末端に持つポリエチレングリコール(NHS-PEG)などを使用することができる。
【0033】
次に,工程(1)から工程(10)を経て製作した単一プローブDNA固定金属微粒子を担体基板表面に固定した単一プローブ分子素子をDNAシーケンシングに使用する例について説明する。
【0034】
(11)DNAシーケンシング評価
まず,工程(1)から工程(10)によって,図7に示すように単一プローブDNA固定金属微粒子701を担体基板702の表面に固定する。次に,シーケンシング対象の単一DNA分子703を基板上の金属微粒子表面に固定したプローブDNA704と1:1で反応させ,基板表面に1分子ずつ固定する。具体的には,シーケンシング対象のDNA分子を塩化ナトリウム(NaCl)などの塩を含む溶液に溶解し,この溶液を単一プローブDNA固定金属微粒子を固定した基板702の表面に滴下する。反応温度は20℃〜80℃であり,反応時間は1時間から24時間である。
【0035】
DNAシーケンシングにおいて実際の解析対象となるのは,ゲノムDNAの転写産物であるメッセンジャーRNA(mRNA)である。mRNAの特徴は3’末端にアデニン(A)が平均約200個連続した配列(PolyAテイル)を持つことである。シーケンシング対象の単一mRNA分子と金属微粒子の表面に固定したプローブDNAを反応させるためには,プローブDNAが単一mRNA分子との反応サイトを持つ必要がある。従ってシーケンシング対象が単一mRNA分子である場合,プローブDNAとしてPolyAテイルと相補的な塩基配列であるPolyT配列を有するものを使用する。
【0036】
次に図8に示すように,金属微粒子801に固定したプローブDNA802をプライマーとして,ポリメラーゼ反応によりプローブDNAの先端にヌクレオチドを一塩基伸長させる。なお,図8ではDNAが4種類の塩基(アデニン(A),チミン(T),グアニン(G),シトシン(C))のうちのいずれか1つを持つヌクレオチドが連続して結合した分子であることを示すため,DNAをヌクレオチドを表す正方形が直線状に多数連なった構造として表現している。図8に示す反応では,塩化マグネシウム(MgCl2)などの塩を含む溶液にヌクレオチド804を溶解した溶液を基板と接触させる。伸長したヌクレオチド804には蛍光分子805が結合している。この蛍光分子からの蛍光を検出することにより,伸長したヌクレオチドの種類を識別することができる。図中,803はポリメラーゼである。
【0037】
図9に,DNAシーケンシング読取システムを示す。このシステムでは,励起レーザー光901を担体基板902の裏面から石英プリズム903を通して全反射条件で入射させる。全反射条件で入射した励起レーザー光のうち,解析対象である単一DNA分子906が固定された面に染み出したエバネッセント光によって,一塩基伸長したヌクレオチド908に結合した蛍光分子909を励起する。蛍光分子909から発せられた蛍光910を基板上面から高感度のCCDカメラ911によって測定する。ヌクレオチドが伸長した後,ヌクレオチドに結合した蛍光分子を除去する。例えばヌクレオチドのリン酸基の末端に蛍光分子が結合している場合,リン酸基の切断により蛍光分子が単一DNA分子から除去される。引き続き,別の種類のヌクレオチドを反応させる。図中,907はポリメラーゼである。
【0038】
例えば,まずヌクレオチドAを含む溶液と基板を接触させ,ポリメラーゼ反応によってAが一塩基伸長した場合,上記の読取システムによって蛍光が検出される。ヌクレオチドAが一塩基伸長しなかった場合には蛍光は検出されない。この測定結果から,蛍光が検出された金属微粒子では,解析対象の単一DNA分子の解析対象部分にAと相補的であるTを持つことがわかる。また,蛍光が検出されなかった金属微粒子では,解析対象部分にTを持たないことがわかる。次に例えばヌクレオチドCを含む溶液を基板と接触させた後に蛍光を検出し,各金属微粒子において単一DNA分子の解析対象部分にCと相補的なGを有するか否かを判別する。引き続きヌクレオチドT及びGを反応させる。これらの工程を繰り返すことにより,各金属微粒子904に固定した単一DNA分子905の塩基配列を読み取る。ここでは一塩基伸長に使用する蛍光物質としてヌクレオチドのリン酸部分に蛍光色素が結合した試薬を使用したが,ヌクレオチドのプリン又はピリミジン塩基に蛍光色素が結合した試薬,あるいはヌクレオチドの3’OH末端に蛍光色素が結合した試薬を使用してもよい。
【0039】
なお,工程(8)から工程(11)では,工程(1)から工程(7)により製造した単一プローブ分子固定金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造方法,及びこれを用いた生体分子検出方法を示した。一方,工程(1)から工程(7)により製造した単一プローブ分子固定金属微粒子を担体基板表面に固定することなく,溶液中において生体分子検出用プローブ微粒子,生体分子捕捉用微粒子,及び生体分子搬送用微粒子などとして使用することもできる。例えば,単一プローブ分子固定金属微粒子を細胞に取り込ませ,所望の分量のプローブ分子を細胞内の標的部位に搬送する。また,細胞内での金属微粒子の軌跡を追跡することにより,細胞内でのプローブ分子の搬送経路を追跡することが可能である。
【0040】
次に,本発明の単一プローブ分子固定金属微粒子の製造方法,この金属微粒子を担体基板表面に固定した単一プローブ分子素子の製造方法,及びこの単一プローブ分子素子を利用したDNAシーケンシングの例を実施例1により詳細に説明する。
【実施例1】
【0041】
(1)磁性微粒子表面への固定官能基の結合
本実施例では,表面に固定官能基が結合されている市販の磁性微粒子を使用した。具体的には,市販のカルボキシル基結合型磁性微粒子を使用した。この磁性微粒子の平均直径は1.05μmであった。この磁性微粒子は中心部に強い磁化をもつ磁性材料であるγFe2O3とFe3O4が一様に分布したポリマーブレンドのコアを持ち,このコアを外側のポリスチレン層で被覆した構造である。
【0042】
(2)磁性微粒子表面へのDNAの結合
本実施例ではプローブDNAとして,塩基配列が5’末端から,TTTTTTTTTTTTTTTTTTTT(PolyT20, 塩基長20)であるDNAを選択した。従って,このプローブDNAと相補的なDNAの塩基配列は,5’末端側からAAAAAAAAAAAAAAAAAAAA(PolyA 20, 塩基長20)である。そこで,磁性微粒子表面にPolyT20を結合させた。
【0043】
具体的にはまず,磁性微粒子を25mM MES(2−(N−モルホリノ)エタン スルホニック アシド,2-(N-morpholino)ethane sulfonic acid)バッファ(pH6)により洗浄した。またEDC(1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド ハイドロクロライド,1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride)及びNHS(N−ヒドロキシスクシンイミジル,N-hydroxysuccinimidyl)エステルを25mM MESバッファにより溶解し,それぞれ濃度を50mg/mlに調製した。反応容器中にMESバッファで洗浄した磁性微粒子を含む溶液を取り,さらにEDC及びNHSを添加し30分間撹拌した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合させ,上澄み液を除去した。次に,5’末端にアミノ基を有するPolyA20をMESバッファに溶解して濃度を1nM〜1μMに調製し,これを前記磁性微粒子を含む溶液と混合・撹拌し,25℃にて30分間反応した。この反応により,PolyA20がアミド結合を介して磁性微粒子表面に結合した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合させ,上澄み液を除去した。最後に,磁性微粒子表面のカルボキシル基のうち,PolyT20が結合しなかったものをキャッピングするため,磁性微粒子をPBS(リン酸バッファ,Phosphate Buffered Saline)で調製した50mMエタノールアミン(pH8.0)で25℃,1時間処理した。
【0044】
(3)磁性微粒子表面での二本鎖DNAの形成
磁性微粒子の表面にプローブDNAが非特異的に吸着することを防止するため,表面にPolyA20を固定した磁性微粒子を0.1〜0.5%BSA(ウシ血清アルブミン,Bovine Serum Albumin)及び0.1%界面活性剤(Tween-20又はTriton X-100)を添加したPBSで洗浄した。次に,DNAのハイブリダイゼーション溶液である5×SSC(クエン酸バッファ,Standard Saline Citrate), 0.5%SDS(ドデシル硫酸ナトリウム,Sodium Dodecyl Sulfate)にてプローブDNAであるPolyT20の溶液(濃度1nM〜1μM)を調製した。なお,PolyT20の5’末端には,次の工程(4)にて金属微粒子を結合するためにチオール基を導入した。表面にPolyA20を結合した磁性微粒子を含む溶液とPolyT20の溶液を混合し,42℃の恒温オーブン中にて20時間ハイブリダイゼーションを行った。これにより,磁性微粒子の表面にPolyA20 とPolyT20から成る二本鎖DNAが形成された。ハイブリダイゼーション終了後,磁石を有する治具を反応容器に接近し,反応容器壁面に磁性微粒子を集合し,上澄み液を除去することにより磁性微粒子表面に結合したPolyA20とハイブリダイゼーションしなかった余剰のPolyT20を除去した。さらに5×SSC, 0.5%SDSにより磁性微粒子を洗浄した。
【0045】
(4)プローブDNAの末端官能基への金属微粒子の結合
磁性微粒子表面で二本鎖DNAを形成しているPolyT20の5’末端にあるチオール基に,金属微粒子である金ナノ粒子を固定した。本実施例では粒子径30nmの金ナノ粒子クエン酸溶液(濃度0.007% Weight/Volume)を使用した。5×SSC, 0.5%SDS溶液にて濃度を1nM〜1μMに調製した金ナノ粒子を磁性微粒子と混合し,25℃にて16〜24時間反応した。これにより,磁性微粒子表面で二本鎖DNAを形成しているPolyT20の5’末端に,チオール基を介して金ナノ粒子を固定した。反応終了後,磁石を有する治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合して上澄み液を除去することにより,PolyT20の末端チオール基に結合しなかった余剰の金ナノ粒子を除去した。
【0046】
(5)金属微粒子表面への吸着阻害分子の結合
単一プローブDNA固定金ナノ粒子において,金ナノ粒子の表面のうちプローブDNAであるPolyTが固定されていない部分に,吸着阻害分子であるビス−(p−(スルホナトフェニル)フェニルホスフィン(bis-(p-sulfonatophenyl)phenylphosphine)を結合した。具体的には工程(1)から工程(4)で調製した磁気微粒子を含む溶液にビス−(p−(スルホナトフェニル)フェニルホスフィンの水溶液を添加し,反応溶液中のビス−(p−(スルホナトフェニル)フェニルホスフィンの終濃度を1mg/mlに調整したのち,25℃で16〜24時間反応した。
【0047】
(6)単一プローブDNA固定金属微粒子の分離・回収
工程(1)から工程(5)により調製された磁性微粒子を含む溶液を70℃に加温した。このとき,工程(4)において磁性微粒子表面に形成された二本鎖DNAが解離し,磁性微粒子表面に固定したPolyA20及び金ナノ粒子表面に固定したPolyT20がそれぞれ一本鎖DNAとなった。従って反応溶液中には単一PolyT20固定金ナノ粒子,及び表面にPolyA20が固定した磁性微粒子が遊離した。磁石を含む治具を反応容器に接近し,反応容器の壁面に磁性微粒子を集合し,上澄み液を回収することにより単一PolyT20固定金ナノ粒子を回収した。
【0048】
(7)単一プローブDNA固定金属微粒子に固定されたDNA数の評価
工程(6)では,表面に単一PolyT20分子が固定された粒子径30nmの金ナノ粒子が回収された。この金ナノ粒子では,金ナノ粒子が占める体積に対してPolyT20が占める体積が小さいため,表面に固定したPolyT20の数に応じて金ナノ粒子をゲル電気泳動により分離することは困難であった。そこで,金ナノ粒子の表面に固定したPolyT20に対して,さらに塩基長の長いDNAをハイブリダイズした後にゲル電気泳動を行った。本実施例では金ナノ粒子表面に固定したPolyT20に対して,3’末端にPolyA20配列を持つ70merDNAをハイブリダイズさせた。70merDNAの塩基配列は3’末端から5’末端に向かって
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG(70mer)
である。具体的には単一PolyT20固定金ナノ粒子と70merDNAを5×SSC, 0.5%SDS中で混合し,42℃の恒温オーブン中にて20時間ハイブリダイゼーションを行った。
【0049】
上記70merDNAとハイブリダイズした単一PolyT20固定金ナノ粒子を含む溶液を3%アガロースゲルのウェルに注入した。比較対照用サンプルとして,金ナノ粒子と5’末端をチオール基で修飾したPolyT20をモル比1:0,1:1,1:2及び1:3にて反応させた溶液を調製した。これらの比較対照用サンプルも上記70merDNAとハイブリダイズさせたのち,それぞれ前記アガロースゲルのウェルに注入した。このアガロースゲルに一定電圧(1〜10V/cm)を印加し,70merDNAとハイブリダイズしたPolyT20固定金ナノ粒子のゲル電気泳動を行った。金ナノ粒子及び金ナノ粒子表面で形成されたPolyT20及び70merDNAから成る二本鎖DNAは負電荷を有するため,電気泳動を開始すると各サンプル中に含まれるPolyT20固定金ナノ粒子はゲル中を負極側から正極側に泳動した。PolyT20固定金ナノ粒子は表面に固定されたPolyT20の数が増加するに伴い,より多くの70merDNAと二本鎖DNAを形成する。これによりPolyT20固定金ナノ粒子のゲル電気泳動時の有効体積が増大し,泳動開始位置からの泳動距離が短くなった。従って,金ナノ粒子表面に固定されたPolyT20の数の違いによる泳動距離の差がより大きくなった。工程(1)から工程(6)によって製造された単一PolyT20固定金ナノ粒子の電気泳動では,表面にPolyT20が1個固定された金ナノ粒子に対応する1本の泳動バンドが観察され,その他のバンドは観察されなかった。従って,本実施例で開示した方法により,単一PolyT20固定金ナノ粒子が高純度で製造されたことが確認された。
【0050】
(8)石英基板表面へのリンカー分子層の形成
洗浄した石英基板をシランカップリング剤である3−アミノプロピルトリメトキシシランのメタノール溶液に5分間浸漬した。続いて基板をメタノールで洗浄したのち,80℃で2時間アニールを行った。この工程により,石英基板表面にアミノ基が導入された。
【0051】
(9)石英基板表面への単一プローブDNA固定金属微粒子の固定
アミノ基を導入した石英基板表面に,単一PolyT20固定金ナノ粒子の溶液を滴下し,25℃にて16時間反応した。基板に滴下する単一PolyT20固定金ナノ粒子溶液の濃度を0.01〜100nMに調整することにより,石英基板表面に固定される単一PolyT20固定金ナノ粒子の密度を調整した。
【0052】
(10)基板表面の吸着阻害分子層の形成
担体基板上で単一PolyT20固定金ナノ粒子が固定されなかった領域に,吸着阻害分子であるNHSエステルを末端に有するポリエチレングリコール(NHS-PEG)を結合した。具体的にはトリエタノールアミン(pH8.0)で4mMに調製したNHS-PEG溶液に担体基板を浸漬し,25℃で1時間反応させた。反応終了後,担体基板を純水で十分に洗浄した。
【0053】
(11)DNAシーケンシング(図10,図11,図12)
工程(1)から工程(10)に従って製造した基板を用いて, DNAシーケンシングを行った。石英基板1001に固定された金ナノ粒子1002表面に固定された単一PolyT20分子1003に,シーケンシング対象である単一DNA分子1004をハイブリダイズさせた。本実施例ではシーケンシング対象の単一DNA分子として3’末端からA配列を連続して持つDNAをハイブリダイズさせた。この単一DNA分子1004の塩基配列を3’末端から5’末端の方向に示す。
AAAAAAAAAAAAAAAAAAAAAGTCGAGCGGTAGCACAGAGAGCTTGCTCTCGGGTGACGAGCGGCGGACG(70mer)
【0054】
次に,プローブDNAである単一PolyT20分子1003をプライマーとして,単一DNA分子1004のシーケンシングを行った。製作した単一PolyT20固定金ナノ粒子を固定した基板とヌクレオチド3リン酸のリン酸基末端に蛍光分子Cy3を持つ4種類のヌクレオチド(A, T, G, C)を,ポリメラーゼ反応により反応させた。反応溶液は10mM トリス−塩酸(Tris-HCl), 5mM 塩化マグネシウム(MgCl2)溶液にて調製した1μM ヌクレオチド溶液である。ポリメラーゼ反応によって単一DNA分子1004の塩基配列と相補的な塩基を持つヌクレオチドが単一PolyT20分子に結合する。本実施例ではヌクレオチドC(図10において1005で示す)の溶液を石英基板1001と反応させた場合,単一PolyT20分子にヌクレオチドCが結合する。これは,シーケンシング対象である単一DNA分子においてPolyA配列に隣接するGにヌクレオチドCが結合するからである。
【0055】
図9に示した蛍光検出系を用いて,ポリメラーゼ反応中の蛍光画像を取得する。蛍光強度は石英基板上面から高感度CCDカメラで測定した。励起光を全反射条件で照射することにより,蛍光分子Cy3(図10において1006で示す)が結合したヌクレオチドCが結合した金ナノ粒子1002からの蛍光が検出される。ヌクレオチドが単一PolyT20分子に結合した後,このヌクレオチドのリン酸基が切断される。これによって,リン酸基の末端に結合していた蛍光分子Cy3が単一PolyT20分子から除去される。次に,ヌクレオチドAの溶液を基板と反応させると,シーケンシング対象である単一DNA分子1004のヌクレオチドGの隣のTにヌクレオチドAが結合する。このとき,蛍光が検出される。これらの手順を繰り返すことにより,各金ナノ粒子において単一DNA分子の塩基配列を読み取ることが可能であった。
【0056】
表面に単一PolyT20固定金ナノ粒子を固定した石英基板と比較対照するため,表面にPolyT20分子を直接固定した石英基板を製作した。本実施例で製作した,表面に単一PolyT20固定金ナノ粒子を固定した石英基板の概念図を図11(a)に,また表面にPolyT20分子を直接固定した石英基板の概念図を図11(b)に示した。これらの基板を用いて,単一DNA分子のシーケンシングを行うために,単一PolyT20分子に含まれるある1塩基の伸長反応を行った。このときに得られた単一PolyT20分子の蛍光強度のヒストグラムを図12に示した。
【0057】
表面に単一PolyT20固定金ナノ粒子を固定した石英基板(金ナノ粒子あり,図11(a)に対応する),ならびに表面にPolyT20分子を直接固定した石英基板(金ナノ粒子なし,図11(b)に対応する)のそれぞれについて,約3,000μm2の面積に含まれる蛍光輝点の強度を数値化した。その結果,金ナノ粒子がない基板では蛍光輝点の総数は約1,400個であったのに対し,金ナノ粒子がある基板では約3,000個の蛍光輝点が検出された。また,金ナノ粒子がない基板と比較して,金ナノ粒子がある場合には蛍光強度の平均値が約10倍であるとともに,蛍光強度の標準偏差も半減した。金ナノ粒子なしで基板に直接固定したPolyT20分子はランダムに固定されており,隣接するPolyT20分子間の距離も一定ではなく,蛍光強度のばらつきが大きくなった。一方,金ナノ粒子の表面に固定された単一PolyT20分子は石英基板表面に1分子ずつ,かつ距離が十分に離れて固定されており,より均質な反応条件下に置かれていることから,蛍光強度のバラツキが小さくなった。さらに,金ナノ粒子による蛍光増強効果により蛍光強度が増大することも確認された。従って,本実施例において表面に単一プローブDNA固定金ナノ粒子を固定した基板では,DNAシーケンシングを高精度かつ高感度で実施できることが示された。
【実施例2】
【0058】
次に,本発明で開示する単一プローブ分子素子において,金属微粒子の表面に2個以上の化学結合を介して固定されるプローブ分子の化学構造及び該プローブ分子を表面に固定した単一プローブ分子固定金属微粒子の製造プロセスの一例を実施例2に示す。ここでもプローブ分子としてDNAを用いて説明するが,本製造プロセスによりDNAやRNAなどの核酸分子だけではなく,タンパク質や糖鎖などの生体分子検出用プローブ分子を1分子だけ表面に固定した単一プローブ分子固定金属微粒子を製造することが可能である。
【0059】
アミノデキストラン(Aminodextran)構造を有するプローブDNA分子の化学構造及び製造方法を図13に示す。アミノデキストラン1301はアミノ基1302を有するD−グルコースの縮重合ポリマーである。また,アミノデキストラン1301は還元末端に1個だけアルデヒド基1303を有する。D−グルコースの重合度が異なる様々な分子量のアミノデキストランを合成することが可能である。本実施例では平均分子量70,000のアミノデキストラン構造を有するプローブDNA分子の製造プロセスを説明する。
【0060】
(1)アミノデキストランへのジスルフィド基の導入
アミノデキストラン1301(平均分子量70,000)を1×PBS(リン酸バッファ,Phosphate Buffered Saline,pH7.0〜9.0)に溶解した。また,SPDP分子1304(3-(2-Pyridyldithio)propionic acid N-hydrosuccinimide ester)をジメチルスルホキシド(Dimethylsulfoxide)に溶解し,濃度を10mM〜500mMに調製した。アミノデキストランの溶液にSPDPの溶液を混合し,室温で撹拌しながら0.5〜20時間反応した。この反応により,ジスルフィド基1306が導入されたアミノデキストラン1305を得た。反応終了後,反応液の透析又はゲルろ過により,反応液に含まれる未反応のSPDP分子1304を除去した。
【0061】
(2)アミノデキストランと単一プローブDNA分子の結合
末端がアミノ基1307で修飾されたプローブDNA分子1308を,1×PBS(リン酸バッファ,Phosphate Buffered Saline ,pH7.0〜9.0)に溶解した。前記ジスルフィド基が導入されたアミノデキストラン1305の溶液と,前記プローブDNA分子1308の溶液を混合して反応液を調製した。ジスルフィド基が導入されたアミノデキストラン1305と末端がアミノ基で修飾されたプローブDNA分子1308の混合モル比は,1:1〜1:100の範囲とした。さらに前記反応液に1M NaOH(水酸化ナトリウム,Sodium hydroxide)に溶解したNaCNBH3(Soduim cyanoborohydride)の溶液を添加し,室温で撹拌しながら2〜20時間反応した。この反応により,ジスルフィド基が導入されたアミノデキストラン1305の還元末端にあるアルデヒド基とプローブDNA分子1308の末端アミノ基1307がアミド結合を形成し,アミノデキストランとプローブDNA分子が1:1で結合したデキストラン型プローブDNA分子1309を得た。反応終了後,反応液の透析又はゲルろ過により,反応液に含まれる未反応のプローブDNA分子1308を除去した。
【0062】
次に,デキストラン型プローブDNA分子を表面に固定した単一プローブDNA分子固定金属微粒子の模式図を図14に示す。本実施例では,実施例1の工程(1)〜(4)に示した方法に従い,デキストラン型プローブDNA分子1401(1401は1309を簡略化して示したものである)を粒子径10nmの金ナノ粒子1402の表面に結合し,単一プローブ分子固定金ナノ粒子1403を得た。本実施例では,デキストラン型プローブDNA分子1401の多数のジスルフィド結合が切断し,多数の硫黄原子1404が金ナノ粒子1402の表面に結合する。
【0063】
本実施例で示したデキストラン型プローブDNA分子1401のように多数の化学結合を介して金属微粒子の表面に固定されるプローブ分子は,1個の化学結合を介して金属微粒子の表面に固定されるプローブ分子と比較して以下のような利点がある。
(a)使用する金属微粒子の粒子径に対応してプローブ分子のポリマー部分(本実施例ではアミノデキストラン)の分子量を適正化することにより,金属微粒子とプローブ分子が1:1で結合した単一プローブ分子固定金属微粒子を高収率で得ることが可能である。
(b)プローブ分子と金属微粒子が多数の化学結合を介して強固に結合しているため,溶液の温度,イオン強度,pHなどが変化してもプローブ分子と金属微粒子が解離しにくく安定である。
(c)プローブ分子のポリマー部分が多数の親水性官能基(本実施例ではアミノデキストランのヒドロキシル基)を有する場合,単一プローブ分子固定金属微粒子の表面は多数の親水性官能基で被覆されるため,生体分子などの非特異的な吸着が抑制される。
【産業上の利用可能性】
【0064】
本発明の素子は,核酸の網羅的定量解析を行うDNAマイクロアレイや核酸の塩基配列を解読するDNAシーケンサにおいて,単一分子レベルで高精度かつ高感度の検出を行うための生体分子検出素子として適用できる。また,本発明の素子は,タンパク質や糖鎖などの生体分子,及び様々な化学物質を蛍光法を用いて高精度かつ高感度に検出するための検出素子として利用することができる。
【図面の簡単な説明】
【0065】
【図1】単一プローブ分子固定金属微粒子を示す模式図。
【図2】単一プローブ分子固定金属微粒子を担体基板表面に固定した生体分子検出素子を示す模式図。
【図3】磁性微粒子を用いて単一プローブ分子固定金属微粒子を製造する方法を示す模式図。
【図4】複数の官能基を介して金属微粒子の表面に結合した単一プローブ分子を示す模式図。
【図5】ゲル電気泳動によりプローブ分子固定金属微粒子を分離する方法を示す模式図。
【図6】単一プローブ分子固定金属微粒子を担体基板表面に固定して生体分子検出素子を製造する方法を示す模式図。
【図7】単一プローブDNA固定金属微粒子を担体基板表面に固定した生体分子検出素子にシーケンシング対象である単一DNA分子を固定した状態を示す模式図。
【図8】DNAシーケンシングの単一プローブDNA及びシーケンシング対象である単一DNA分子を示す模式図。
【図9】DNAシーケンシングの読取装置を示す模式図。
【図10】DNAシーケンシングの一塩基伸長反応を示す模式図。
【図11】(a)単一PolyT20固定金ナノ粒子を石英基板表面に固定した生体分子検出素子,及び(b)PolyT20分子を石英基板表面に直接固定した生体分子検出素子を示す模式図。
【図12】DNAシーケンシングの一塩基伸長反応中に検出した,各単一PolyT20分子の蛍光強度のヒストグラム。
【図13】アミノデキストラン構造を有するプローブDNA分子の化学構造及び製造方法を示す模式図。
【図14】デキストラン型プローブDNA分子を表面に固定した単一プローブDNA分子固定金属微粒子を示す模式図。
【符号の説明】
【0066】
101:単一プローブ分子,102:金属微粒子,103:吸着阻害分子,201:担体基板,202:単一プローブ分子固定金属微粒子,301:磁性微粒子,302:固定官能基,303:プローブDNAと相補的な塩基配列を持つDNA,304:末端官能基,305:プローブDNA,306:末端官能基,307:金属微粒子,308:吸着阻害分子,309:単一プローブDNA固定金属微粒子,401:金属微粒子,402:金属微粒子と結合する官能基,403:単一プローブ分子,404:金属微粒子,405:金属微粒子と結合する官能基,406:単一プローブ分子,501:電気泳動用ゲル,502:電気泳動用ゲルのウェル,503:電気泳動用ゲルのウェル,504:電気泳動用ゲルのウェル,505:電気泳動用ゲルのウェル,506:電気泳動用ゲルのウェル,507:電気泳動用ゲルの負極側,508:電気泳動用ゲルの正極側,509:電気泳動の泳動方向,510:表面にDNAが固定されていない金属微粒子,511:表面にDNAが1個固定された金属微粒子,512:表面にDNAが2個固定された金属微粒子,513:表面にDNAが3個固定された金属微粒子,514:単一プローブDNA固定金属微粒子に対応する電気泳動バンド,601:担体基板,602:単一プローブDNA固定金属微粒子,701:金属微粒子,702:担体基板,703:シーケンシング対象である単一DNA分子,704:単一プローブDNA,801:金属微粒子,802:単一プローブDNA,803:ポリメラーゼ,804:ヌクレオチド,805:蛍光分子,901:励起レーザー光,902:担体基板,903:石英プリズム,904:金属微粒子,905:単一プローブDNA,906:シーケンシング対象である単一DNA分子,907:ポリメラーゼ,908:ヌクレオチド,909:蛍光分子,910:蛍光分子から発した蛍光,911:高感度CCDカメラ,1001:担体基板,1002:金属微粒子,1003:単一PolyT20分子,1004:シーケンシング対象である単一DNA分子,1005:ヌクレオチドC,1006:蛍光分子Cy3,1101:単一PolyT20固定金ナノ粒子,1102:石英基板,1103:単一PolyT20分子,1104:石英基板,1301:アミノデキストラン,1302:アミノ基,1303:アルデヒド基,1304:SPDP分子,1305:ジスルフィド基が導入されたアミノデキストラン,1306:ジスルフィド基,1307:アミノ基,1308:末端がアミノ基で修飾されたプローブDNA分子,1309:デキストラン型プローブDNA分子,1401:デキストラン型プローブDNA分子,1402:金ナノ粒子,1403:単一プローブ分子固定金ナノ粒子,1404:硫黄原子。
【特許請求の範囲】
【請求項1】
基板と,
前記基板の表面に固定された複数の金属微粒子と,
1個の金属微粒子に1個ずつ,表面に化学結合を介して固定されたプローブ分子と
を有することを特徴とする単一プローブ分子素子。
【請求項2】
請求項1に記載の単一プローブ分子素子において,各プローブ分子は2個以上の化学結合を介して1個の金属微粒子の表面に固定されていることを特徴とする単一プローブ分子素子。
【請求項3】
請求項1に記載の単一プローブ分子素子において,前記金属微粒子の表面は,前記プローブ分子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子素子。
【請求項4】
請求項1に記載の単一プローブ分子素子において,前記基板の表面は,前記金属微粒子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子素子。
【請求項5】
請求項1に記載の単一プローブ分子素子において,前記金属微粒子は,貴金属類に属する金属,貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものから成ることを特徴とする単一プローブ分子素子。
【請求項6】
請求項1に記載の単一プローブ分子素子において,前記プローブ分子は核酸であることを特徴とする単一プローブ分子素子。
【請求項7】
金属微粒子と,
前記金属微粒子の表面に化学結合を介して固定された1個のプローブ分子と
を有することを特徴とする単一プローブ分子固定金属微粒子。
【請求項8】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記プローブ分子は2個以上の化学結合を介して前記金属微粒子の表面に固定されていることを特徴とする単一プローブ分子固定金属微粒子。
【請求項9】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記金属微粒子の表面は,前記プローブ分子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子固定金属微粒子。
【請求項10】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記金属微粒子は,貴金属類に属する金属,貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものから成ることを特徴とする単一プローブ分子固定金属微粒子。
【請求項11】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記プローブ分子は核酸であることを特徴とする単一プローブ分子固定金属微粒子。
【請求項12】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
前記相補的分子に対し,金属微粒子と結合することのできる前記プローブ分子を相補的に結合する工程と,
前記相補的分子に結合した前記プローブ分子に金属微粒子を結合させる工程と,
前記磁性微粒子表面に結合した前記相補的分子と前記プローブ分子との相補的結合を解離する工程と,
表面に単一の前記プローブ分子が固定された金属微粒子を回収する工程と,
担体基板に前記回収された金属微粒子を固定する工程と,
前記担体基板の表面のうち,前記金属微粒子が固定されていない領域に吸着阻害分子を固定する工程と
を有することを特徴とする,基板表面に固定した複数の金属微粒子のそれぞれに単一のプローブ分子が固定された単一プローブ分子素子の製造方法。
【請求項13】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
プローブ分子が金属微粒子に1個だけ結合する条件下で前記プローブ分子と金属微粒子を混合し反応させる工程と,
前記磁性微粒子の表面に結合した前記相補的分子と前記金属微粒子の表面に固定した前記プローブ分子とを相補的に結合させ,単一のプローブ分子が固定された金属微粒子を前記磁性微粒子の表面に回収する工程と,
前記磁性微粒子の表面に結合した相補的分子と前記金属微粒子の表面に固定した前記プローブ分子との相補的結合を解離する工程と,
表面に単一のプローブ分子が固定した金属微粒子を前記磁性微粒子から分離・回収する工程と,
担体基板に前記分離・回収した金属微粒子を固定する工程と,
前記担体基板の表面のうち,前記金属微粒子が固定されていない領域に吸着阻害分子を固定する工程と
を有することを特徴とする,基板表面に固定した複数の金属微粒子にそれぞれ単一のプローブ分子が固定された単一プローブ分子素子の製造方法。
【請求項14】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
前記相補的分子に対し,金属微粒子と結合することのできる前記プローブ分子を相補的に結合する工程と,
前記相補的分子に結合した前記プローブ分子に金属微粒子を結合させる工程と,
前記磁性微粒子表面に結合した前記相補的分子と前記プローブ分子との相補的結合を解離する工程と,
表面に単一の前記プローブ分子を固定した金属微粒子を回収する工程と,
を有することを特徴とする単一のプローブ分子を固定した金属微粒子の製造方法。
【請求項15】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
プローブ分子が金属微粒子に1個だけ結合する条件下で前記プローブ分子と金属微粒子を混合し反応させる工程と,
前記磁性微粒子の表面に結合した前記相補的分子と前記金属微粒子の表面に固定した前記プローブ分子とを相補的に結合させ,単一のプローブ分子が固定された金属微粒子を前記磁性微粒子の表面に回収する工程と,
前記磁性微粒子の表面に結合した相補的分子と前記金属微粒子の表面に固定した前記プローブ分子との相補的結合を解離する工程と,
表面に単一のプローブ分子が固定した金属微粒子を前記磁性微粒子から分離・回収する工程と,
を有することを特徴とする単一のプローブ分子を固定した金属微粒子の製造方法。
【請求項16】
基板表面に固定した複数の金属微粒子にそれぞれ単一のプローブ核酸分子が固定された単一プローブ分子素子の前記プローブ核酸分子とシーケンシング対象である核酸分子とを反応させる工程と,
前記プローブ核酸分子の一塩基伸長反応を行い,シーケンシング対象である核酸分子に蛍光標識されたヌクレオチドを反応させる工程と,
前記単一プローブ分子素子に励起光を照射する工程と,
前記蛍光標識されたヌクレオチドから発生する蛍光を検出する工程と
を有することを特徴とする核酸分子のシーケンシング方法。
【請求項1】
基板と,
前記基板の表面に固定された複数の金属微粒子と,
1個の金属微粒子に1個ずつ,表面に化学結合を介して固定されたプローブ分子と
を有することを特徴とする単一プローブ分子素子。
【請求項2】
請求項1に記載の単一プローブ分子素子において,各プローブ分子は2個以上の化学結合を介して1個の金属微粒子の表面に固定されていることを特徴とする単一プローブ分子素子。
【請求項3】
請求項1に記載の単一プローブ分子素子において,前記金属微粒子の表面は,前記プローブ分子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子素子。
【請求項4】
請求項1に記載の単一プローブ分子素子において,前記基板の表面は,前記金属微粒子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子素子。
【請求項5】
請求項1に記載の単一プローブ分子素子において,前記金属微粒子は,貴金属類に属する金属,貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものから成ることを特徴とする単一プローブ分子素子。
【請求項6】
請求項1に記載の単一プローブ分子素子において,前記プローブ分子は核酸であることを特徴とする単一プローブ分子素子。
【請求項7】
金属微粒子と,
前記金属微粒子の表面に化学結合を介して固定された1個のプローブ分子と
を有することを特徴とする単一プローブ分子固定金属微粒子。
【請求項8】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記プローブ分子は2個以上の化学結合を介して前記金属微粒子の表面に固定されていることを特徴とする単一プローブ分子固定金属微粒子。
【請求項9】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記金属微粒子の表面は,前記プローブ分子が固定された領域以外の領域が化学結合を介して固定された吸着阻害分子により被覆されていることを特徴とする単一プローブ分子固定金属微粒子。
【請求項10】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記金属微粒子は,貴金属類に属する金属,貴金属類に属する金属の合金又は貴金属類に属する金属を積層したものから成ることを特徴とする単一プローブ分子固定金属微粒子。
【請求項11】
請求項7に記載の単一プローブ分子固定金属微粒子において,前記プローブ分子は核酸であることを特徴とする単一プローブ分子固定金属微粒子。
【請求項12】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
前記相補的分子に対し,金属微粒子と結合することのできる前記プローブ分子を相補的に結合する工程と,
前記相補的分子に結合した前記プローブ分子に金属微粒子を結合させる工程と,
前記磁性微粒子表面に結合した前記相補的分子と前記プローブ分子との相補的結合を解離する工程と,
表面に単一の前記プローブ分子が固定された金属微粒子を回収する工程と,
担体基板に前記回収された金属微粒子を固定する工程と,
前記担体基板の表面のうち,前記金属微粒子が固定されていない領域に吸着阻害分子を固定する工程と
を有することを特徴とする,基板表面に固定した複数の金属微粒子のそれぞれに単一のプローブ分子が固定された単一プローブ分子素子の製造方法。
【請求項13】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
プローブ分子が金属微粒子に1個だけ結合する条件下で前記プローブ分子と金属微粒子を混合し反応させる工程と,
前記磁性微粒子の表面に結合した前記相補的分子と前記金属微粒子の表面に固定した前記プローブ分子とを相補的に結合させ,単一のプローブ分子が固定された金属微粒子を前記磁性微粒子の表面に回収する工程と,
前記磁性微粒子の表面に結合した相補的分子と前記金属微粒子の表面に固定した前記プローブ分子との相補的結合を解離する工程と,
表面に単一のプローブ分子が固定した金属微粒子を前記磁性微粒子から分離・回収する工程と,
担体基板に前記分離・回収した金属微粒子を固定する工程と,
前記担体基板の表面のうち,前記金属微粒子が固定されていない領域に吸着阻害分子を固定する工程と
を有することを特徴とする,基板表面に固定した複数の金属微粒子にそれぞれ単一のプローブ分子が固定された単一プローブ分子素子の製造方法。
【請求項14】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
前記相補的分子に対し,金属微粒子と結合することのできる前記プローブ分子を相補的に結合する工程と,
前記相補的分子に結合した前記プローブ分子に金属微粒子を結合させる工程と,
前記磁性微粒子表面に結合した前記相補的分子と前記プローブ分子との相補的結合を解離する工程と,
表面に単一の前記プローブ分子を固定した金属微粒子を回収する工程と,
を有することを特徴とする単一のプローブ分子を固定した金属微粒子の製造方法。
【請求項15】
プローブ分子と相補的に結合する相補的分子を磁性微粒子の表面に結合する工程と,
プローブ分子が金属微粒子に1個だけ結合する条件下で前記プローブ分子と金属微粒子を混合し反応させる工程と,
前記磁性微粒子の表面に結合した前記相補的分子と前記金属微粒子の表面に固定した前記プローブ分子とを相補的に結合させ,単一のプローブ分子が固定された金属微粒子を前記磁性微粒子の表面に回収する工程と,
前記磁性微粒子の表面に結合した相補的分子と前記金属微粒子の表面に固定した前記プローブ分子との相補的結合を解離する工程と,
表面に単一のプローブ分子が固定した金属微粒子を前記磁性微粒子から分離・回収する工程と,
を有することを特徴とする単一のプローブ分子を固定した金属微粒子の製造方法。
【請求項16】
基板表面に固定した複数の金属微粒子にそれぞれ単一のプローブ核酸分子が固定された単一プローブ分子素子の前記プローブ核酸分子とシーケンシング対象である核酸分子とを反応させる工程と,
前記プローブ核酸分子の一塩基伸長反応を行い,シーケンシング対象である核酸分子に蛍光標識されたヌクレオチドを反応させる工程と,
前記単一プローブ分子素子に励起光を照射する工程と,
前記蛍光標識されたヌクレオチドから発生する蛍光を検出する工程と
を有することを特徴とする核酸分子のシーケンシング方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2009−85840(P2009−85840A)
【公開日】平成21年4月23日(2009.4.23)
【国際特許分類】
【出願番号】特願2007−257894(P2007−257894)
【出願日】平成19年10月1日(2007.10.1)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
【公開日】平成21年4月23日(2009.4.23)
【国際特許分類】
【出願日】平成19年10月1日(2007.10.1)
【出願人】(501387839)株式会社日立ハイテクノロジーズ (4,325)
【Fターム(参考)】
[ Back to top ]