
原虫の増殖阻害剤を検出する方法
【課題】トリパノソーマ・ブルースの中枢神経症状をきたす患者に有効かつ安全な薬を提供する。
【解決手段】(1)特定のアミノ酸配列からなるタンパク質等を用意するステップと、(2)試験化合物と前記ステップ(1)で用意されたタンパク質とを含む反応混合液のキナーゼ活性を測定するステップと、(3)前記試験化合物の非存在下で前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップを含む方法からなる。
【解決手段】(1)特定のアミノ酸配列からなるタンパク質等を用意するステップと、(2)試験化合物と前記ステップ(1)で用意されたタンパク質とを含む反応混合液のキナーゼ活性を測定するステップと、(3)前記試験化合物の非存在下で前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップを含む方法からなる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、原虫の増殖阻害剤を検出する方法と、AKB14−3−3−1タンパク質等の配列特異的発現抑制剤と、AKB14−3−3−1タンパク質と、トリパノソーマ14−3−3−IIのヘテロ2量体との会合を阻害するホスホペプチド等と、これらを含む原虫感染症の治療用組成物とに関する。
【背景技術】
【0002】
アフリカ睡眠病は、ヒトのみならず家畜にも感染する人畜共通感染症である。現在は、患者数は減少傾向(WHOによる推定5万人−7万人)にあるが、アンゴラ、コンゴ、スーダン、中央アフリカ共和国、チャド、コートジボワール、ギニア、マラウィ、ウガンダ及びタンザニアなどでは、2005年にアウトブレイクが見られた。治療しなければ死亡する疾患で、特に脳血液関門を通過した原虫が引き起こす脳神経性症状は重篤である。
【0003】
感染初期では、ペンタミジン及びスラミンが使われる。ペンタミジンには副作用があるが、患者が耐えうる程度である。スラミンは色素化合物であり、アレルギー、尿路系の副作用がある。中枢神経期には、メラルソプロル及びエフロルニチンが使われる。メラルソプロルにはさまざまな副作用があり、特に3−10%が副作用の反応性脳症にて死亡する。さらに耐性が報告されている。エフロルニチンは、メラルソプロルより毒性がすくないが、トリパノソーマ・ブルース・ガンビエンセ(Trypanosoma brucei gambiense)には有効であるが、トリパノソーマ・ブルース・ローデシイ(Trypanosoma brucei rhodesie)のように週ないし月単位で急激に中枢神経に移行する原虫には効かない。そして、現在この中枢神経症状をきたす患者に有効かつ安全な薬は存在しない。
【0004】
発明者らは、トリパノソーマ症の予防・治療薬の開発に携わってきた(特許文献1)が、近年、多機能分子14−3−3の研究から、トリパノソーマ・ブルースの14−3−3ホモログ(以下、「Tb14−3−3」という。)としてI型及びII型アイソフォームを単離し、これらが、細胞周期及び細胞骨格の調節に関与し、I型及びII型がともに細胞分裂に必須であることを証明した(非特許文献1)。
【0005】
これまでの本発明の発明者らの研究から、14−3−3分子は、モード1あるいはモード2のモチーフには、ほとんど結合を示さず、モード3には、ヒト14−3−3よりも弱いながらも結合を示すことを明らかにし、さらにアフィニティの高い新規モード3ペプチドを世界に先駆けて同定した。しかしながら(1)普遍的なモチーフへの結合が弱いこと、(2)ペプチドタグによる14−3−3タンパクのヘテロ2量体形成不全、(3)Tb14−3−3の標的タンパクへの結合が弱いこと、(4)従来のほ乳類の阻害剤の組成だとトリパノソーマ・ブルースの脱リン酸化酵素の活性を抑えられないこと等の理由により、Tb14−3−3と結合するタンパク質の同定は、困難をきわめた。さらに、弱い結合が細胞内で果たす機能の意義を評価することも困難である。
【0006】
このような状況の中で、本発明の発明者らは、Tb14−3−3のヘテロ2量体と特異的に結合するタンパク質の網羅的解析を行って、AKB14−3−3−1と命名された1種類のタンパク質の発現抑制及び過剰発現が、Tb14−3−3の発現抑制と同様に細胞分裂異常を伴う細胞増殖停止をもたらすことを発見した。本発明は、かかる新発見にもとづいて想到された。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2007−297280
【非特許文献】
【0008】
【非特許文献1】Inoue,M.ら、J.Biol.Chem.、280:14085、(2005)
【発明の開示】
【発明が解決しようとする課題】
【0009】
そこで、トリパノソーマ・ブルースの中枢神経症状をきたす患者に有効かつ安全な薬を開発する必要がある。
【課題を解決するための手段】
【0010】
本発明は、原虫の増殖阻害剤を検出する方法を提供する。本発明の方法は、(1)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、(2)前記ステップ(1)で用意されたタンパク質と、試験化合物とを含む反応混合液のキナーゼ活性を測定するステップと、(3)前記試験化合物の非存在下で、前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップとを含む。
【0011】
本明細書において、特異的結合タグとは、所望の機能を有するポリペプチドを遺伝子組み換えによって調製する際に、前記所望の機能を有するポリペプチドとペプチド結合で連結した融合タンパク質として発現させることにより、形質転換体からの発現タンパク質の分離、精製又は検出をより容易に行うことを可能にするために、他のタンパク質、多糖類、糖脂質、核酸及びこれらの誘導体、樹脂、シリコン等と特異的に結合するポリペプチドをいう。前記特異的結合タグは、水溶液中に溶解した物質又は固体支持体と結合する場合がある。前記特異的結合タグには、Hisタグ、Mycタグ、HAタグ、インテインタグ、MBP、GSTその他これらに類するポリペプチドが含まれるが、これらに限定されない。前記特異的結合タグは、Hisタグ、mycタグ、HAタグ、インテインタグ、MBP又はGSTからなるグループから選択される場合がある。
【0012】
本明細書において、原虫とは、トリパノソーマ・ブルース、トリパノソーマ・クルーズ及びリーシュマニアを含むが、これらに限定されないキネトプラスト科原虫をいう。
【0013】
本発明の原虫の増殖阻害剤において、前記ステップ(1)で用意されたタンパク質には、配列番号1に列挙されるアミノ酸配列からなるAKB14−3−3−1タンパク質のトリパノソーマ・クルーズ又はリーシュマニアにおけるホモログの場合がある。
【0014】
本発明は、(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤を提供する。
【0015】
本明細書において、あるタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤とは、当該タンパク質のアミノ酸配列をエンコードするヌクレオチド配列に相補的なヌクレオチド配列を含み、当該タンパク質の発現を一過性に抑制するものをいう。本明細書における配列特異的遺伝子発現抑制剤には、siRNA、miRNA、リボザイム、アンチセンス核酸及びDNA/RNAキメラポリヌクレオチドを含むが、これらに限定されない。本明細書のsiRNAとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する21−23塩基対程度の2本鎖RNAであって、該標的とする遺伝子のmRNAの分解を引き起こすことにより前記標的とする遺伝子の発現を特異的に抑制する、2本鎖RNAをいう。本明細書のmiRNAとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する1本鎖RNAであって、該標的とする遺伝子のmRNAの分解又は翻訳阻害を引き起こすことにより前記標的とする遺伝子の発現を特異的に抑制し、長い(数百塩基以上の)転写産物として精製された後、切断、プロセシングを受けて短い(20−25塩基程度の)オリゴリボヌクレオチドとなり機能する、1本鎖RNAをいう。本明細書のリボザイムとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有するRNAであって、該標的とする遺伝子のmRNAの分解する触媒として働くことにより前記標的とする遺伝子の発現を特異的に抑制するRNAをいう。本明細書のアンチセンス核酸とは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する20塩基程度の1本鎖DNA又はRNAであって、該標的とする遺伝子のmRNAと2本鎖を形成することで分解又は翻訳阻害を引き起こすことにより、前記標的とする遺伝子の発現を特異的に抑制する、1本鎖DNA又はRNAをいう。これらの配列特異的遺伝子発現抑制剤は、標的とする遺伝子のヌクレオチド配列に相補的な配列を有するポリヌクレオチドであって、デオキシリボヌクレオチドとリボヌクレオチドとが混在するように重合された、DNA/RNAキメラポリヌクレオチドの場合がある。前記DNA/RNAキメラポリヌクレオチドは、核酸分子の安定性を高める目的等で使用される。
【0016】
本発明は、配列番号2に列挙されるアミノ酸配列を含み、該アミノ酸配列のカルボキシル末端から2番目のセリン残基がリン酸化され、配列番号1に列挙されるアミノ酸配列からなるタンパク質と、トリパノソーマ・ブルースの14−3−3−I及び14−3−3−IIのヘテロ2量体との会合を阻害する、ホスホペプチドを提供する。
【0017】
前記ホスホペプチドは、細胞膜透過ペプチド、特異的結合タグを含むが、これらに限定されない、他の機能を有するポリペプチド鎖が、配列番号2に列挙されるアミノ酸配列からなるポリペプチドのアミノ末端及び/又はカルボキシル末端に連結された融合タンパク質の場合がある。
【0018】
本発明は、本発明のホスホペプチドと、細胞膜透過性ペプチドとの融合ペプチドを提供する。
【0019】
本明細書において、細胞膜透過ペプチドとは、他のペプチド、ポリペプチド又はタンパク質との融合タンパク質を細胞外に添加すると、細胞膜を透過して前記融合タンパク質を細胞内に移行させることができるペプチドをいい、ヒト免疫不全ウイルス1型(HIV−1)のTatタンパク質RNA 結合領域(48−60位)由来のアルギニンに富む塩基性ペプチド(TATペプチド)や、アルギニン残基が6ないし12個連続したオリゴホモペプチドや、ショウジョウバエ由来の転写因子Antennapediaタンパク質のDNA 結合領域由来の塩基性両親媒性ヘリックス構造を有するペプチド(penetratin)が含まれるが、これらに限定されない。
【0020】
本発明は、本発明の配列特異的発現抑制剤か、あるいは、本発明のホスホペプチドかを含む、原虫感染症の治療用組成物を提供する。
【0021】
本明細書において、原虫感染症とは、アフリカ睡眠病、Chagas病及びリーシュマニア症を含むが、これらに限定されない、キネトプラスト科原虫を病原体とする感染症をいう。
【0022】
本発明の原虫感染症の治療用組成物は、原虫の増殖阻害効果を損なわないことを条件として、さらに、他の疾患の治療に有効な成分及び/又は薬学的に許容される添加物を1種類又は2種類以上含む場合がある。前記添加物は、希釈剤及び膨張剤と、結合剤及び接着剤と、滑剤と、流動促進剤と、可塑剤と、崩壊剤と、担体溶媒と、緩衝剤と、着色料と、香料と、甘味料と、防腐剤及び安定化剤と、吸着剤と、当業者に知られたその他の医薬品添加剤とを含むが、これらに限られない。
【0023】
本発明は、原虫感染症治療薬候補を選択するスクリーニング方法を提供する。本発明のスクリーニング方法は、(1)試験化合物を用意するステップと、(2)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、(3)前記試験化合物の存在下で、前記ステップ(2)で用意されたタンパク質のキナーゼ活性を測定するステップと、(4)前記試験化合物の非存在下で前記ポリペプチドのキナーゼ活性とを測定するステップと、(5)前記ステップ(3)のキナーゼ活性が、前記ステップ(4)のキナーゼ活性より低いとき、前記試験化合物をトリパノソーマ・ブルース感染症治療薬候補として選択するステップとを含む。
【0024】
本発明のスクリーニング方法において、前記試験化合物は、配列番号1に列挙されるアミノ酸配列からなるAKB14−3−3−1タンパク質が、原虫、特に、血流型トリパノソーマ・ブルース原虫において発現することを抑制する場合がある。
【0025】
本発明のスクリーニング方法において、前記試験化合物は、AKB14−3−3−1タンパク質と、14−3−3のヘテロ2量体との会合を阻害する場合がある。
【0026】
本発明において、ある特定のアミノ酸配列を含むペプチド、ポリペプチド又はタンパク質とは、当該ペプチド、ポリペプチド又はタンパク質の機能を損なわないことを条件として、当該ペプチド、ポリペプチド又はタンパク質のアミノ末端及び/又はカルボキシル末端に、細胞膜透過ペプチドと、特異的結合タグとを含むが、これらに限定されない、他の機能を有するペプチド、ポリペプチド又はタンパク質が融合したものをいう。
【図面の簡単な説明】
【0027】
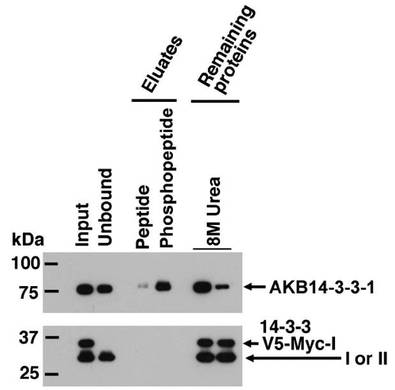
【図1】AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性を検討した結果を示すウェスタンブロット図。
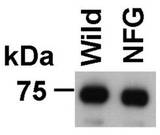
【図2】AKB14−3−3−1の野生型と、突然変異型(とのV5タグ付きのタンパク質との精製収量を示すブロット図。
【図3】原虫細胞内で発現され、精製された、AKB14−3−3−1の野生型及び突然変異型タンパク質を用いたキナーゼアッセイの結果を示す、蛍光染色された電気泳動図。
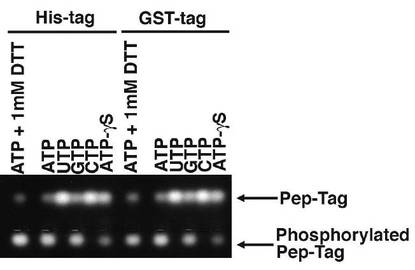
【図4】AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図。
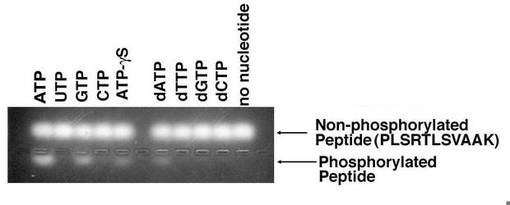
【図5】AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図。
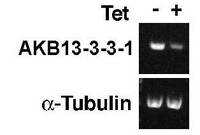
【図6】PCF細胞B5クローンを、テトラサイクリン存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図。
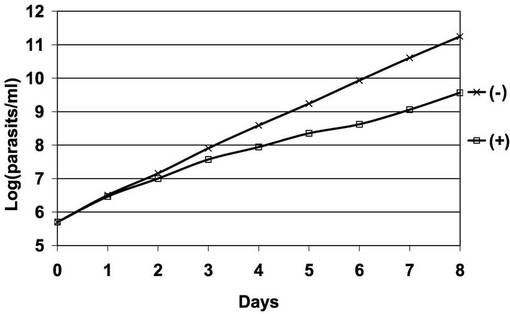
【図7】PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
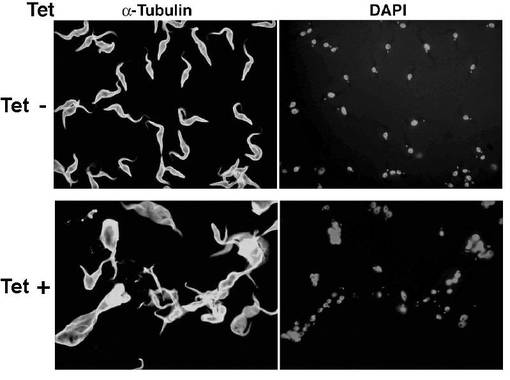
【図8】PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真。
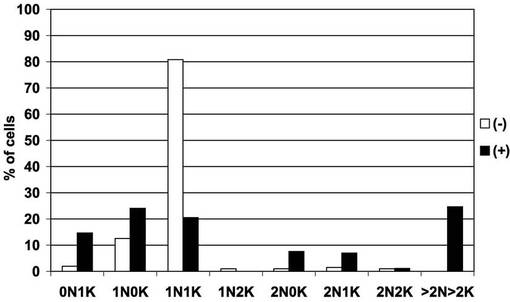
【図9】図8で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
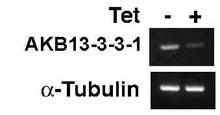
【図10】BSF細胞A5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図。
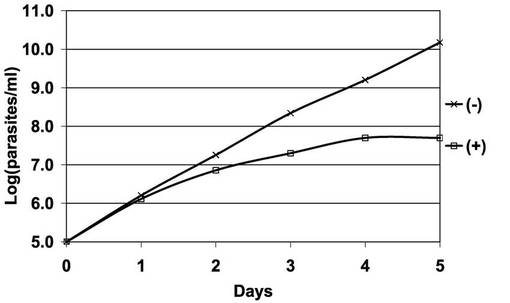
【図11】BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
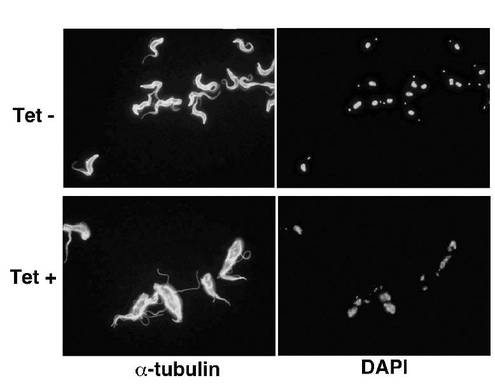
【図12】BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真。
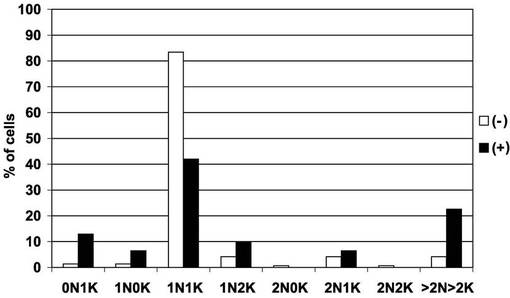
【図13】図12で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【図14】PCF細胞A11クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1の発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図。
【図15】テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞A11クローンの典型的なDAPI染色蛍光顕微鏡写真。
【図16】PCF細胞のA11クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図17】図15で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【図18】PCF細胞B10クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1NFGの発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図。
【図19】テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞B10クローンの典型的なDAPI染色蛍光顕微鏡写真。
【図20】PCF細胞のB10クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図21】図19で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【発明を実施するための形態】
【0028】
以下に説明する本発明の実施例は例示のみを目的とし、本発明の技術的範囲を限定するものではない。本発明の技術的範囲は特許請求の範囲の記載によってのみ限定される。本発明の趣旨を逸脱しないことを条件として、本発明の変更、例えば、本発明の構成要件の追加、削除及び置換を行うことができる。
【実施例1】
【0029】
トリパノソーマ14−3−3タンパク質ヘテロ2量体と結合する可能性のあるタンパク質の同定
1.トリパノソーマ14−3−3タンパク質の発現ベクターの構築
トリパノソーマにおいて外来遺伝子を構成的に発現できるpLew82ベクターは、G.Cross博士より恵与され、発明者らの研究室で、T7プロモーターが追加され、pLew82T7phleoと命名された。pLew82T7は、さらに、V−5タグ配列、3Cプロテアーゼ切断配列及び制限酵素Hpa I切断部位が追加され、フレオマイシン耐性遺伝子がピューロマイシン耐性遺伝子に置換され、pLew82T7puroN−V5−3Cと命名された。トリパノソーマ・ブルース14−3−3のI型アイソフォーム(Inoue,M.ら、J.Biol.Chem.、280:14085、(2005)、GenBankアクセッション番号AB059827)は、mycタグ配列が追加され、それぞれ、pLew82T7puroN−V5−3Cにサブクローニングされ、pLew82T7puroN−V5−3C−Myc−14−3−3Iと命名された。トリパノソーマ・ブルース14−3−3のII型アイソフォーム(GenBankアクセッション番号AB066565)は、pLew82T7phleoにサブクローニングされ、pLew82T7phleo−14−3−3IIと命名された。
【0030】
2.トリパノソーマ14−3−3タンパク質を発現するトリパノソーマ細胞の調製
29−13細胞は、ハイグロマイシン(50μg/mL)、G418(25μg/mL)及び10%ウシ胎仔血清が添加されたSDM−79培地で維持される。以下では、SDM−79培地にはハイグロマイシン及びG418が含まれる。29−13細胞は、pLew82T7phleo−14−3−3IIが29−13細胞に常法に従ってトランスフェクションされ、フレオマイシン(2.5μg/mL)が添加されたSDM−79培地で選択された。得られたフレオマイシン耐性クローンに、pLew82T7puroN−V5−3C−Myc−14−3−3Iがさらにトランスフェクションされ、二重にトランスフェクションされたクローンが、フレオマイシン(2.5μg/mL)及びピューロマイシン(1μg/mL)、が添加されたSDM−79培地で選択され、維持された。前記二重にトランスフェクションされたクローンについて、トリパノソーマ・ブルース14−3−3のI型及びII型アイソフォームのヘテロ2量体の存在量が、抗V5タグ抗体を不動化したアガロースビーズを各クローンの細胞の溶解液に懸濁して、前記アガロースビーズに吸着したタンパク質を抗トリパノソーマ・ブルース14−3−3I抗体及び抗トリパノソーマ・ブルース14−3−3II抗体(Inoue,M.ら、J.Biol.Chem.、280:14085、(2005))を用いるウェスタンブロッティング法で分析されたその結果、前記二重にトランスフェクションされた細胞のクローンのうち、I型及びII型アイソフォームのヘテロ2量体を最も大量に産生するものとして、A6クローンの細胞が選ばれた。
【0031】
3.トリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質の回収
2×106個/mLの前記A6クローンの細胞懸濁液250mLはテトラサイクリン(1μg/mL)が添加されたSDM−79培地で26時間培養された。A6細胞のペレットはPBS(−)で2回洗浄された後、1mLのPMEM細胞溶解バッファー(0.1M PIPES pH6.9、2mM EGTA、1mM MgSO4、0.1mM EDTA、200μM ジギトニン、プロテアーゼ阻害剤混合液(シグマ、カタログ番号P8340)、10mM E64、50mM NaF及び2.5mM NaVO4)とともに氷上で30分間インキュベーションされて溶解された。細胞溶解液は、5000rpm、4°Cで2分間遠心され、上清はさらに100,000g、4°Cで1時間遠心され、その上清が回収された。前記上清は、抗V5タグ抗体を不動化したアガロースビーズ(シグマ、カタログ番号A7345)100μLとともに4°Cで3.5時間回転しながらインキュベーションされた。前記アガロースビーズは、50mM NaF及び2.5mM NaVO4が添加された前記PMEM細胞溶解バッファーの2倍希釈液で3回洗浄され、前記PMEM細胞溶解バッファーの1倍液で1回洗浄された後、スピン−Xカラム(Costar、カタログ番号8160)に移された。前記アガロースビーズに結合したタンパク質は、HVSGLKRRRSV(配列番号2)からなり、カルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチド100μg/mLを含む前記PMEM細胞溶解バッファーの1倍液210μLで溶出された。溶出されたタンパク質は、氷冷されたメタノール、クロロホルム及び脱イオン水で沈殿され、15,000rpm、4°Cで1時間遠心され、ペレットは、100mM Tris−HCl、pH8.5中の8M尿素溶液10μLに再懸濁された。
【0032】
4.質量分析によるトリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質の解析
前記8M尿素溶液に0.5μLの0.1M DTTが添加されて、60°C、20分間インキュベーションされ、その後、0.5μLの0.2M ヨウ化アセトアミドが添加されて、室温、1時間インキュベーションによりチオール基がカルボキシメチル化された。そして反応混合液に1μLの0.5mg/mLリシル・エンドペプチダーゼ(和光純薬株式会社、カタログ番号125−05061)が添加され、37°Cで3時間インキュベーションされた。その後、前記反応混合液は40μLの100mM Tris−HCl、pH8.5で希釈され、125ngのトリプシン(MSグレード、和光純薬株式会社、カタログ番号202−1591)が添加され、37°C、終夜インキュベーションされた。前記反応混合液は減圧乾燥の後、0.3% ギ酸(和光純薬株式会社、カタログ番号067−04531)に再懸濁され、日立液体クロマトグラフ質量分析計(日立ハイテク株式会社、NanoFrontierLD)によるナノLC−MS/MS分析に供された。
【0033】
5.結果
トリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質のトリプシン消化断片ペプチドの質量分析ピーク値から、トリパノソーマ・ブルースのゲノムにエンコードされるタンパク質が75個同定された。
【実施例2】
【0034】
トリパノソーマ14−3−3タンパク質ヘテロ2量体と結合する可能性のあるタンパク質の予備的な特徴付け
1.全長cDNAのクローニング
実施例1で同定されたトリパノソーマ・ブルースのタンパク質75個のうち、14−3−3のI型及びII型アイソフォームと、アミノ酸の相同性から機能が推測できないものと、熱ショックタンパク質、リボソームタンパク質、カルモジュリン等の14−3−3ヘテロ2量体との特異的結合が考えにくいタンパク質を除いて、MASCOT検索(マトリックスサイエンス株式会社)のスコアの高いタンパク質4個(Tb10.61.1900、Tb927.10.7780、Tb927.5.4300及びTb11.47.0035)について、全長cDNAクローンがTb427プロサイクリック型(昆虫感染型)細胞由来cDNAライブラリからPCR法で単離された。Tb10.61.1900と、Tb927.10.7780と、Tb927.5.4300と、Tb11.47.0035とは、それぞれ、推定キナーゼと、マイトジェン活性化キナーゼと、プロリリルオリゴペプチダーゼ、推定、セリンペプチダーゼと、カルパイン様システインペプチダーゼとしてアノテーションがされている。
【0035】
2.誘導可能なノックダウンベクターの構築と、トリパノソーマ細胞への導入
前記全長cDNA4個は、それぞれ、pQuadra3bsrテトラサイクリン依存性ノックダウンベクター(Inoue,M.ら、J.Biol.Chem.、280:14085(2005))にサブクローニングされ、常法に従って29−13プロサイクリック型(以下、「PCF」ともいう。)細胞株及び90−13血流型(以下、「BSF」ともいう。)細胞株にトランスフェクションされた。PCF細胞は、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で選択された。BSF細胞は、ブラストサイジン(5μg/mL)、20%ウシ胎仔血清、ハイグロマイシ(50μg/mL)及びG418(25μg/mL)を含むHMI−9培地で選択された。
【0036】
3.原虫細胞のキネトプラスト、核及び鞭毛の形態学的解析
トリパノソーマ・ブルース細胞のDNA及びα−チューブリンの検出は常法にしたがって行われた。簡潔には、トリパノソーマ・ブルース細胞は、室温で、4% パラメータホルムアルデヒドにより固定され、0.2% Triton−Xでパーメアライズ処理された。さらに、1M グリシンで自家蛍光を消光する処理を行った後、BlockAce(DSファーマバイオメディカル株式会社、カタログ番号UK1380)でブロッキングされた。それから、前記細胞は、FITC抗α−チューブリン抗体(Sigma、DM−1A、カタログ番号F2168−2ML)と反応され、さらに、DAPIで染色された。
【0037】
4.結果
前記ノックダウンベクター4種類がトランスフェクションされた細胞のうち、Tb10.61.1900の2本鎖RNAがテトラサイクリン存在下で誘導される細胞では、キネトプラスト、核及び鞭毛が過剰形成されたり、あるいは、全くなかったり、というように細胞分裂およびサイトキネシスに異常をきたしたモンスター様細胞(monster−like cell with multikinetoplasts、Multinucleus and multiflagella)が観察された。かかるモンスター様細胞は、トリパノソーマ・ブルースの14−3−3のI型及びII型アイソフォームを両方とも過剰発現させるか、あるいは、両方とも発現抑制させることによっても観察された。そこで、Tb10.61.1900の遺伝子座タグ(locus tag)にエンコードされるタンパク質はトリパノソーマ・ブルースの14−3−3の下流で機能することが示唆された。
【0038】
なお、残りのノックダウンベクター3種類がトランスフェクションされた細胞については、テトラサイクリン存在下で2本鎖RNAを発現させたとき、キネトプラスト、核及び鞭毛のいずれにも影響が見られなかった。そこで以下では、Tb10.61.1900の遺伝子座タグ(locus tag)にエンコードされるタンパク質は、AKB14−3−3−1(Associated Kinase of Trypabnosoma brucei 14−3−3 Type I)という。
【実施例3】
【0039】
AKB14−3−3−1の特徴付け(1)
1.AKB14−3−3−1のトリパノソーマ発現ベクターの構築
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列は配列番号1に示される。前記全長アミノ酸配列をエンコードするポリヌクレオチドが、前記pLew82T7puroN−V5ベクターにサブクローニングされて、V5タグが連結されたAKB14−3−3−1の融合タンパク質を原虫細胞内で過剰に発現するベクターが構築された。
【0040】
2.AKB14−3−3−1の大腸菌発現ベクターの構築
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列は配列番号1に示される。前記全長アミノ酸配列をエンコードするポリヌクレオチドが、pET47b及びpGEX4T2にサブクローニングされて、それぞれHisタグ及びGSTと連結した融合タンパク質(His−AKB−14−3−3−1及びGST−AKB−14−3−3−1)を大腸菌で発現するベクターpET47b−His−AKB−14−3−3−1及びpGEX4T2−AKB14−3−3−1が構築された。
【0041】
2.AKB14−3−3−1に対する特異抗体の作成
GST−AKB−14−3−3−1は、常法に従って、pGEX4T2−AKB14−3−3−1ベクターで形質転換された大腸菌T7Express lysY/Iq株(ニュー・イングランド・バイオラボ・ジャパン株式会社、カタログ番号#C3013H)を0.4mM IPTG存在下で終夜培養した後リゾチームで溶菌させて得られた溶菌液からグルタチオン−セファロース・カラム(GEヘルスサイエンス株式会社、カタログ番号17−0756−01)を用いて精製された。得られた精製GST−AKB−14−3−3−1は1回に150μgずつフロイントアジュバントに添加してウィスター系ラットに感作された。4回の免疫感作後の前記ラットの血清が抗AKB14−3−3−1抗体として用いられた。
【0042】
3.抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法による原虫細胞でのAKB14−3−3−1の検出
トリパノソーマ・ブルースのPCF細胞2×106個又はBSF細胞5×106個は、5%のβ−メルカプトエタノールが添加された2×SDSローディングバッファー(バイオ・ラッド ラボラトリーズ株式会社)に溶解され、常法にしたがって、SDS−ポリアクリルアミドゲル電気泳動法により分離され、電気泳動後のゲルからPVDFメンブレンに電気的に転写された。抗AKB14−3−3−1抗体として、前記抗AKB14−3−3−1ラット血清が2000倍に希釈して用いられた。トリパノソーマ・ブルース14−3−3I及びIIを検出するためには、抗トリパノソーマ・ブルース14−3−3I及びII血清が2:1の割合で混合された血清混合液(以下、「抗Tb14−3−3混合血清」という。)が1000倍に希釈して用いられた。
【0043】
4.AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性の検討
実施例1で説明されたのと同様に、テトラサイクリン(1μg/mL)が添加されたSDM−79培地で終夜培養された約7.5x106個/mLの前記A6クローンの細胞懸濁液250mLに由来する細胞溶解液の上清(以下、「Input」という。)が抗V5タグ抗体を不動化したアガロースビーズと反応され、スピン−Xカラムに移された。前記上清との反応の後、前記アガロースビーズから分離された溶液(以下、「Unbound」という。)が除去され、その後前記スピン−Xカラム中のアガロースビーズに、HVSGLKRRRSV(配列番号2)からなるペプチド(100μg/mL)が添加されて得られた溶出液(以下、「Peptide」という。)と、該ペプチドのカルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチド(100μg/mL)が添加されて得られた溶出液(以下、「Phosphopetide」という。)とが回収された。さらに、それぞれのペプチドによる処理後の前記アガロースビーズに、8M尿素が添加され、溶出液が回収された。Inputの細胞溶解液上清と、Unboundの溶液と、Peptide又はPhosphopetideの溶出液と、前記Peptide又はPhosphopetideによる溶出後の前記アガロースビーズから8M尿素で溶出された溶出液とが、抗Tb14−3−3混合血清又は抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法によって解析された。
【0044】
5.結果
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列(配列番号1)には、全長640個のアミノ酸からなり、そのうち40−333番目には、推定セリン/スレオニン タンパク質キナーゼの触媒ドメインが含まれ、また、571−638番目には、細胞骨格との会合に関与するCap−Glyドメインが含まれる。
【0045】
図1は、AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性を検討した結果を示すウェスタンブロット図である。図1の上半分は抗AKB14−3−3−1抗体を用いるウェスタンブロッティングの結果であり、矢印はAKB14−3−3−1のバンドの位置を示す。図1の下半分は抗Tb14−3−3混合血清を用いるウェスタンブロッティングの結果である。図1から明らかなとおり、AKB14−3−3−1はリン酸化ペプチドによる溶出液(Phosphopeptide)のレーンでは検出されたが、リン酸化されないペプチドによる溶出液(Peptide)のレーンではほとんど検出されなかった。また、8M Ureaの左側のレーンは、リン酸化されないペプチドで溶離後、8M 尿素によって溶離された溶出液のレーンであり、大量のAKB14−3−3−1が、V5−3C−Myc−14−3−3I(図1では「14−3−3V5−Myc−I」と表される。)と、タグが連結しない14−3−3I及び14−3−3IIとともに検出された。これに対し、8M Ureaの右側のレーンは、リン酸化されたペプチドで溶離後、8M 尿素によって溶離された溶出液のレーンであり、V5−Myc−14−3−3Iと、タグが連結しない14−3−3I及び14−3−3IIとは、8M Ureaの左側のレーンとほぼ同じ量が出てされたが、AKB14−3−3−1はわずかしか検出されなかった。
【0046】
図1の結果から、AKB14−3−3−1タンパク質と、トリパノソーマ・ブルース14−3−3ヘテロ2量体との結合は、HVSGLKRRRSV(配列番号2)からなるペプチドでは競合阻害されないが、該ペプチドのカルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチドでは競合阻害されることが示された。したがって、本発明のAKB14−3−3−1タンパク質は原虫細胞内でリン酸化され、14−3−3ヘテロ2量体と、特異的に結合していることが示唆された。
【実施例4】
【0047】
AKB14−3−3−1の特徴付け(2)
1.AKB14−3−3−1タンパク質のキナーゼ活性突然変異体の作成
AKB−14−3−3−1の推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異体タンパク質はAKB−14−3−3−1NFGと命名され、そのHisタグ又はGSTとの融合タンパク質を大腸菌で発現するベクターが構築された。
【0048】
2.AKB14−3−3のキナーゼ活性のアッセイ
大腸菌で発現され、精製された、His−AKB−14−3−3−1及びGST−AKB−14−3−3−1と、これらの突然変異体タンパク質(NFG)のキナーゼ活性は、PepTag(登録商標)非放射性PKCアッセイ(プロメガ株式会社、カタログ番号V5330)を用いて確認された。すなわち、1回の反応あたり0.1又は0.5μgのHis−AKB−14−3−3−1又はGST−AKB−14−3−3−1が、0.08mg/mLのPepTag C1ペプチド(PLSRTLSVAAK(配列番号3)のアミノ酸配列からなる蛍光標識されたCキナーゼ基質ペプチド)と、1mMのATP、UTP、GTP、CTP又はATP−γSか、1mMのATP及び1mMのDTTかとが、反応混合液(20mM Tris−HCl、pH7.4及び10mM MgCl2)に添加され、30°Cで、GST−Tagが0.1μgのとき30分間、His−Tag又はGST−Tagが0.5μgのとき1時間インキュベーションされた後、製造者の指示に従って、アガロース電気泳動法により、リン酸化された基質が未反応の基質から分離され、蛍光により検出された。
【0049】
2.結果
図2は、AKB14−3−3−1の野生型(図2及び3では「Wild」と表される。)と、推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異型(図2及び3では「NFG」と表される。)とのV5タグ付きのタンパク質とが、原虫細胞内で発現され、抗V5タグ抗体を固定化したアガロースビーズにより精製され、遊離のV5タグペプチドにより溶離された溶出液が抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法で解析された結果を示すブロット図である。図2から明らかなとおり、突然変異型、原虫細胞から精製された量には違いが見られなかった。
【0050】
図3は、原虫細胞内で発現され、精製された、AKB14−3−3−1の野生型及び突然変異型タンパク質を用いたキナーゼアッセイの結果を示す、蛍光染色された電気泳動図である。図3から明らかなとおり、野生型タンパク質にはキナーゼ活性が認められたが、突然変異タンパク質にはキナーゼ活性は認められなかった。したがって、AKB14−3−3−1タンパク質のキナーゼ活性は、精製が不十分なために他の酵素が混入したためではないことが示された。
【0051】
図4及び5は、AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図である。図4では、His−AKB−14−3−3−1(図4では「His−tag」と表される。)と、GST−AKB−14−3−3−1(図4では「GST−tag」と表される。)とが、さまざまなリボモノヌクレオチド存在下で、基質のPepTag C1ペプチドをリン酸化するかどうかが調べられた。その結果、Hisタグとの融合タンパク質でも、GSTとの融合タンパク質でも、AKB14−3−3−1は、ATPの他、GTP、及びATP−γSは利用可能であるが、UTP及びCTPは利用できないことが示された。さらに図5では、GST−AKB−14−3−3−1について、さまざまなリボモノヌクレオチドだけでなく、さまざまなデオキシリボモノヌクレオチドの存在下で、基質のPepTag C1ペプチドとのキナーゼ反応に供された。図4及び5から、AKB14−3−3−1タンパク質は、リン酸基の供与体として、ATPの他、GTP、ATP−γS及びdATPは利用可能であるが、UTP、CTP、dTTP、dGTP及びdCTPは利用できないことが示された。
【実施例5】
【0052】
AKB14−3−3−1の特徴付け(3)
【0053】
1.原虫細胞におけるAKB14−3−3−1遺伝子発現量の解析
トリパノソーマ・ブルース細胞の全RNAは、RNeasy Plus Mini kit(株式会社キアゲン、カタログ番号74134)を用いて抽出された。100ngの全RNAがスーパースクリプトVILO cDNA合成キット(ライフテクノロジーズジャパン株式会社、インビトロジェン、カタログ番号11754050)を用いて逆転写された。全RNA10ngから合成されたcDNAが1回のPCR反応に供された。AKB14−3−3−1をエンコードするcDNAを増幅するためのPCRプライマーは、配列番号4及び配列番号5が用いられ、α−チューブリンをエンコードするcDNAを増幅するためのPCRプライマーは、配列番号6及び配列番号7が用いられた。AKB14−3−3−1をエンコードするcDNAを増幅するためには、95°C1分間の後、95°C15秒間、55°C15秒間、72°C15秒間を22ないし23回繰り返して、最後に72°C5分間という反応条件が用いられた。α−チューブリンをエンコードするcDNAを増幅するためには、95°C1分間の後、95°C15秒間、55°C15秒間、72°C15秒間を20回繰り返して、最後に72°C5分間という反応条件が用いられた。PCR産物は、2%TAE−アガロースゲル電気泳動による分離され、GelRed(Biotium、ナカライテスク株式会社)により染色された。
【0054】
2.原虫細胞の増殖の解析
細胞数は、血球計算盤により測定された。細胞密度を一定範囲内に保つために、PCF細胞の培養では毎日5倍に希釈して播種され、BSF細胞の培養では毎日10倍に希釈して播種された。pQuadra3bsrAKB14−3−3−1ベクターでdsRNAiによるAKB14−3−3−1の発現抑制を行う場合には、テトラサイクリン(1μg/mL)が添加されたSDM−79培地が用いられた。なお、pQuadra3bsrAKB14−3−3−1がトランスフェクションされたBSF細胞をテトラサイクリン存在下で培養する場合には、細胞密度が低下するため、希釈して播種する作業は第4日で中止された。
【0055】
3.PCFでの結果
PCF細胞(29−13株)にpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で、安定的にpQuadra3bsrAKB14−3−3−1ベクターがインテグレーションされたPCF細胞のクローンB5が選択された。図6は、かかるPCF細胞B5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図である。図6から明らかなとおり、PCF細胞B5クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1のmRNA量が顕著に少なかった。これに対して、対照実験のα−チューブリンのmRNA量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞B5クローンでは、テトラサイクリンによりAKB14−3−3−1遺伝子の発現が抑制されることが示された。
【0056】
図7は、図6に用いられた、PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図7から明らかなとおり、テトラサイクリンによってAKB14−3−3−1の遺伝子発現が抑制されたPCF細胞では、遺伝子発現抑制が起こらないときと比較して、細胞の増殖が10分の1程度に低下した。したがって、AKB14−3−3−1遺伝子の発現抑制は細胞増殖の低減をもたらすことが示された。
【0057】
図8は、図6及び7に用いられた、PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真である。テトラサイクリン非存在下(Tet−)及び存在下(+)で3日間培養された細胞を、それぞれ、抗α−チューブリン抗体又はDAPIで染色後、蛍光顕微鏡で観察したところ、テトラサイクリン存在下で培養されたPCF細胞ではサイトキネシスの異常が観察された。
【0058】
図9は、図8で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図8及び9から明らかなとおり、PCF細胞におけるAKB14−3−3−1遺伝子の発現抑制は、細胞分裂及びサイトキネシスの異常をもたらした。
【0059】
4.BSFでの結果
BSF細胞(90−13株)にpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたHMI−9培地で、安定的にpQuadra3bsrAKB14−3−3−1ベクターがインテグレーションされたBSF細胞のA5クローンが選択された。図10は、BSF細胞A5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図である。図10から明らかなとおり、BSF細胞A5クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1のmRNA量が顕著に少なかった。これに対して、対照実験のα−チューブリンのmRNA量は、テトラサイクリンの有無によって違いは認められなかった。したがって、このpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされたBSF細胞のクローンでは、テトラサイクリンによりAKB14−3−3−1遺伝子の発現が抑制されることが示された。なお、図6と図10とを比較すると、BSF細胞での遺伝子発現の抑制は、PCF細胞よりはやや弱かった。
【0060】
図11は、図10に用いられた、BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記BSF細胞のクローンの増殖曲線である。図11から明らかなとおり、テトラサイクリンによってAKB14−3−3−1の遺伝子発現が抑制されたBSF細胞では、遺伝子発現抑制が起こらないときと比較して、細胞の増殖が100分の1程度に低下した。また、培養4日目以降は細胞増殖がほぼ完全に停止した。したがって、AKB14−3−3−1遺伝子の発現抑制はBSF細胞の細胞増殖の阻害をもたらすことが示された。
【0061】
図12は、図10及び11に用いられた、BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真である。テトラサイクリン非存在下(Tet−)及び存在下(+)で3日間培養された細胞を、それぞれ、抗α−チューブリン抗体又はDAPIで染色後、蛍光顕微鏡で観察したところ、テトラサイクリン存在下で培養されたPCF細胞ではサイトキネシスの異常が観察された。
【0062】
図13は、図12で観察された細胞について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図12及び13から明らかなとおり、BSF細胞におけるAKB14−3−3−1遺伝子の発現抑制は、細胞分裂及びサイトキネシスをもたらした。
【0063】
以上詳しく説明したとおり、AKB14−3−3−1タンパク質には、正常な細胞分裂及びサイトキネシスに必須の機能があることが証明された。
【実施例6】
【0064】
AKB14−3−3−1の特徴付け(3)
1.AKB14−3−3−1タンパク質の野生型及び突然変異体の原虫細胞内での過剰発現
AKB−14−3−3−1と、AKB−14−3−3−1の推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異体タンパク質(AKB−14−3−3−1NFG)とが、テトラサイクリン依存性発現ベクターのpLew82T7bsrにサブクローニングされ、それぞれ、pLew82T7bsr AKB−14−3−3−1及びpLew82T7bsr AKB−14−3−3−1NFGと命名された。pLew82T7bsr AKB−14−3−3−1及びpLew82T7bsr AKB−14−3−3−1NFGが、PCF細胞(29−13株)にトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で、安定的に過剰発現ベクターがインテグレーションされたPCF細胞のクローンが選択された。
【0065】
2.結果
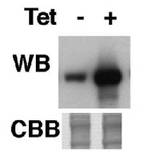
図14は、かかるpLew82T7bsrAKB14−3−3−1ベクターがトランスフェクションされたPCF細胞A11クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1の発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図である。図14の上半分に示されるとおり、PCF細胞A11クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1タンパク質の量が顕著に多かった。図14の下半分は、前記ウェスタンブロッティング法のサンプルの電気泳動ゲルのクーマジーブルー染色写真のうち、AKB14−3−3−1タンパク質のバンドに相当する移動度の部分を示したものである。図14の下半分に示されるとおり、AKB14−3−3−1タンパク質以外の原虫細胞のタンパク質の量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞のA11クローンでは、テトラサイクリンによりAKB14−3−3−1タンパク質の発現が特異的に誘導されることが示された。
【0066】
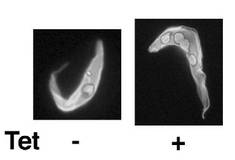
図15は、テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞A11クローンの典型的なDAPI染色蛍光顕微鏡写真である。図15の左側に示すとおり、テトラサイクリン非存在下では、PCF細胞は1個の核と1個のキネトプラストを有する。これに対し、図15の右側に示すとおり、テトラサイクリン存在下では、PCF細胞は2個以上の核と2個以上のキネトプラストを有する。
【0067】
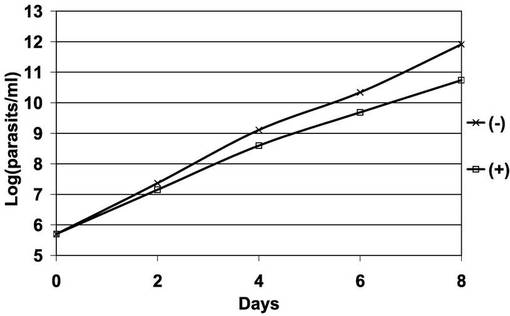
図16は、PCF細胞のA11クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図16から明らかなとおり、PCF細胞A11クローンでは、過剰発現が起こらないときと比較して、7日間で細胞の増殖が10分の1程度に低下した。したがって、AKB14−3−3−1の過剰発現はPCF細胞の細胞増殖の低下をもたらすことが示された。
【0068】
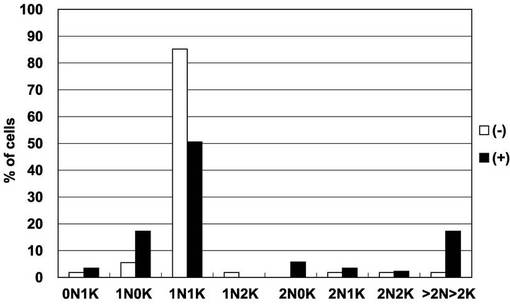
図17は、図15で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図17から明らかなとおり、PCF細胞におけるAKB14−3−3−1の過剰発現は、細胞分裂及びサイトキネシスの異常をもたらした。
【0069】
図18は、pLew82T7bsr AKB−14−3−3−1NFGがトランスフェクションされたPCF細胞B10クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1NFGの発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図である。図18の上半分に示されるとおり、PCF細胞B10クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1NFGタンパク質の量が顕著に多かった。図18の下半分は、前記ウェスタンブロッティング法のサンプルの電気泳動ゲルのクーマジーブルー染色写真のうち、AKB14−3−3−1NFGタンパク質のバンドに相当する移動度の部分を示したものである。図18の下半分に示されるとおり、AKB14−3−3−1NFGタンパク質以外の原虫細胞のタンパク質の量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞のB10クローンでは、テトラサイクリンによりAKB14−3−3−1NFGタンパク質の発現が特異的に誘導されることが示された。
【0070】
図19は、テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞B10クローンの典型的なDAPI染色蛍光顕微鏡写真である。図19に示すとおり、テトラサイクリン存在下でも非存在下でも、PCF細胞は1個の核と1個のキネトプラストを有する。ただし、核及びキネトプラストの位置関係が正常なPCF細胞と異なる細胞が8.5%認められた。これらは、エピマスチゴート型というトリパノソーマ・ブルースの生活史でPCFとは異なるステージの細胞に変化したことを示唆する。
【0071】
図20は、PCF細胞のB10クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図20から明らかなとおり、テトラサイクリンによってAKB14−3−3−1NFGが過剰に発現されたPCF細胞では、過剰発現が起こらないときと比較して、7日間の細胞の増殖はほとんど違いが認められなかった。したがって、AKB14−3−3−1NFGの過剰発現はPCF細胞の細胞増殖には影響を与えないことが示された。
【0072】
図21は、図19で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図21から明らかなとおり、PCF細胞におけるAKB14−3−3−1NFGの過剰発現は、サイトキネシスの異常をもたらさなかった。よって、AKB14−3−3−1が細胞分裂、サイトキネシスを調節する機能を発揮するためには、キナーゼ活性が必要であることが示唆された。
【0073】
これらの実施例から、本発明のAKB14−3−3−1タンパク質は、トリパノソーマ・ブルースの14−3−3ヘテロ2量体と結合するタンパク質キナーゼであって、14−3−3と同様に、細胞増殖及びサイトキネシスに関与することが示された。とくに、図11に示されるとおり、本発明のAKB14−3−3−1遺伝子発現を抑制すると、BSF細胞の増殖が完全に阻害される。BSFすなわち血流型はヒトや家畜に感染した状態を反映するため、本発明のAKB14−3−3−1タンパク質の機能を阻害することによって、ヒトや家畜に感染したトリパノソーマ・ブルースの増殖を阻害できると考えられる。つまり、本発明のAKB14−3−3−1タンパク質の機能を阻害する物質は、アフリカ睡眠病の有効な治療薬となることが期待できる。
【技術分野】
【0001】
本発明は、原虫の増殖阻害剤を検出する方法と、AKB14−3−3−1タンパク質等の配列特異的発現抑制剤と、AKB14−3−3−1タンパク質と、トリパノソーマ14−3−3−IIのヘテロ2量体との会合を阻害するホスホペプチド等と、これらを含む原虫感染症の治療用組成物とに関する。
【背景技術】
【0002】
アフリカ睡眠病は、ヒトのみならず家畜にも感染する人畜共通感染症である。現在は、患者数は減少傾向(WHOによる推定5万人−7万人)にあるが、アンゴラ、コンゴ、スーダン、中央アフリカ共和国、チャド、コートジボワール、ギニア、マラウィ、ウガンダ及びタンザニアなどでは、2005年にアウトブレイクが見られた。治療しなければ死亡する疾患で、特に脳血液関門を通過した原虫が引き起こす脳神経性症状は重篤である。
【0003】
感染初期では、ペンタミジン及びスラミンが使われる。ペンタミジンには副作用があるが、患者が耐えうる程度である。スラミンは色素化合物であり、アレルギー、尿路系の副作用がある。中枢神経期には、メラルソプロル及びエフロルニチンが使われる。メラルソプロルにはさまざまな副作用があり、特に3−10%が副作用の反応性脳症にて死亡する。さらに耐性が報告されている。エフロルニチンは、メラルソプロルより毒性がすくないが、トリパノソーマ・ブルース・ガンビエンセ(Trypanosoma brucei gambiense)には有効であるが、トリパノソーマ・ブルース・ローデシイ(Trypanosoma brucei rhodesie)のように週ないし月単位で急激に中枢神経に移行する原虫には効かない。そして、現在この中枢神経症状をきたす患者に有効かつ安全な薬は存在しない。
【0004】
発明者らは、トリパノソーマ症の予防・治療薬の開発に携わってきた(特許文献1)が、近年、多機能分子14−3−3の研究から、トリパノソーマ・ブルースの14−3−3ホモログ(以下、「Tb14−3−3」という。)としてI型及びII型アイソフォームを単離し、これらが、細胞周期及び細胞骨格の調節に関与し、I型及びII型がともに細胞分裂に必須であることを証明した(非特許文献1)。
【0005】
これまでの本発明の発明者らの研究から、14−3−3分子は、モード1あるいはモード2のモチーフには、ほとんど結合を示さず、モード3には、ヒト14−3−3よりも弱いながらも結合を示すことを明らかにし、さらにアフィニティの高い新規モード3ペプチドを世界に先駆けて同定した。しかしながら(1)普遍的なモチーフへの結合が弱いこと、(2)ペプチドタグによる14−3−3タンパクのヘテロ2量体形成不全、(3)Tb14−3−3の標的タンパクへの結合が弱いこと、(4)従来のほ乳類の阻害剤の組成だとトリパノソーマ・ブルースの脱リン酸化酵素の活性を抑えられないこと等の理由により、Tb14−3−3と結合するタンパク質の同定は、困難をきわめた。さらに、弱い結合が細胞内で果たす機能の意義を評価することも困難である。
【0006】
このような状況の中で、本発明の発明者らは、Tb14−3−3のヘテロ2量体と特異的に結合するタンパク質の網羅的解析を行って、AKB14−3−3−1と命名された1種類のタンパク質の発現抑制及び過剰発現が、Tb14−3−3の発現抑制と同様に細胞分裂異常を伴う細胞増殖停止をもたらすことを発見した。本発明は、かかる新発見にもとづいて想到された。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2007−297280
【非特許文献】
【0008】
【非特許文献1】Inoue,M.ら、J.Biol.Chem.、280:14085、(2005)
【発明の開示】
【発明が解決しようとする課題】
【0009】
そこで、トリパノソーマ・ブルースの中枢神経症状をきたす患者に有効かつ安全な薬を開発する必要がある。
【課題を解決するための手段】
【0010】
本発明は、原虫の増殖阻害剤を検出する方法を提供する。本発明の方法は、(1)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、(2)前記ステップ(1)で用意されたタンパク質と、試験化合物とを含む反応混合液のキナーゼ活性を測定するステップと、(3)前記試験化合物の非存在下で、前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップとを含む。
【0011】
本明細書において、特異的結合タグとは、所望の機能を有するポリペプチドを遺伝子組み換えによって調製する際に、前記所望の機能を有するポリペプチドとペプチド結合で連結した融合タンパク質として発現させることにより、形質転換体からの発現タンパク質の分離、精製又は検出をより容易に行うことを可能にするために、他のタンパク質、多糖類、糖脂質、核酸及びこれらの誘導体、樹脂、シリコン等と特異的に結合するポリペプチドをいう。前記特異的結合タグは、水溶液中に溶解した物質又は固体支持体と結合する場合がある。前記特異的結合タグには、Hisタグ、Mycタグ、HAタグ、インテインタグ、MBP、GSTその他これらに類するポリペプチドが含まれるが、これらに限定されない。前記特異的結合タグは、Hisタグ、mycタグ、HAタグ、インテインタグ、MBP又はGSTからなるグループから選択される場合がある。
【0012】
本明細書において、原虫とは、トリパノソーマ・ブルース、トリパノソーマ・クルーズ及びリーシュマニアを含むが、これらに限定されないキネトプラスト科原虫をいう。
【0013】
本発明の原虫の増殖阻害剤において、前記ステップ(1)で用意されたタンパク質には、配列番号1に列挙されるアミノ酸配列からなるAKB14−3−3−1タンパク質のトリパノソーマ・クルーズ又はリーシュマニアにおけるホモログの場合がある。
【0014】
本発明は、(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤を提供する。
【0015】
本明細書において、あるタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤とは、当該タンパク質のアミノ酸配列をエンコードするヌクレオチド配列に相補的なヌクレオチド配列を含み、当該タンパク質の発現を一過性に抑制するものをいう。本明細書における配列特異的遺伝子発現抑制剤には、siRNA、miRNA、リボザイム、アンチセンス核酸及びDNA/RNAキメラポリヌクレオチドを含むが、これらに限定されない。本明細書のsiRNAとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する21−23塩基対程度の2本鎖RNAであって、該標的とする遺伝子のmRNAの分解を引き起こすことにより前記標的とする遺伝子の発現を特異的に抑制する、2本鎖RNAをいう。本明細書のmiRNAとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する1本鎖RNAであって、該標的とする遺伝子のmRNAの分解又は翻訳阻害を引き起こすことにより前記標的とする遺伝子の発現を特異的に抑制し、長い(数百塩基以上の)転写産物として精製された後、切断、プロセシングを受けて短い(20−25塩基程度の)オリゴリボヌクレオチドとなり機能する、1本鎖RNAをいう。本明細書のリボザイムとは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有するRNAであって、該標的とする遺伝子のmRNAの分解する触媒として働くことにより前記標的とする遺伝子の発現を特異的に抑制するRNAをいう。本明細書のアンチセンス核酸とは、標的とする遺伝子のヌクレオチド配列に相補的な配列を有する20塩基程度の1本鎖DNA又はRNAであって、該標的とする遺伝子のmRNAと2本鎖を形成することで分解又は翻訳阻害を引き起こすことにより、前記標的とする遺伝子の発現を特異的に抑制する、1本鎖DNA又はRNAをいう。これらの配列特異的遺伝子発現抑制剤は、標的とする遺伝子のヌクレオチド配列に相補的な配列を有するポリヌクレオチドであって、デオキシリボヌクレオチドとリボヌクレオチドとが混在するように重合された、DNA/RNAキメラポリヌクレオチドの場合がある。前記DNA/RNAキメラポリヌクレオチドは、核酸分子の安定性を高める目的等で使用される。
【0016】
本発明は、配列番号2に列挙されるアミノ酸配列を含み、該アミノ酸配列のカルボキシル末端から2番目のセリン残基がリン酸化され、配列番号1に列挙されるアミノ酸配列からなるタンパク質と、トリパノソーマ・ブルースの14−3−3−I及び14−3−3−IIのヘテロ2量体との会合を阻害する、ホスホペプチドを提供する。
【0017】
前記ホスホペプチドは、細胞膜透過ペプチド、特異的結合タグを含むが、これらに限定されない、他の機能を有するポリペプチド鎖が、配列番号2に列挙されるアミノ酸配列からなるポリペプチドのアミノ末端及び/又はカルボキシル末端に連結された融合タンパク質の場合がある。
【0018】
本発明は、本発明のホスホペプチドと、細胞膜透過性ペプチドとの融合ペプチドを提供する。
【0019】
本明細書において、細胞膜透過ペプチドとは、他のペプチド、ポリペプチド又はタンパク質との融合タンパク質を細胞外に添加すると、細胞膜を透過して前記融合タンパク質を細胞内に移行させることができるペプチドをいい、ヒト免疫不全ウイルス1型(HIV−1)のTatタンパク質RNA 結合領域(48−60位)由来のアルギニンに富む塩基性ペプチド(TATペプチド)や、アルギニン残基が6ないし12個連続したオリゴホモペプチドや、ショウジョウバエ由来の転写因子Antennapediaタンパク質のDNA 結合領域由来の塩基性両親媒性ヘリックス構造を有するペプチド(penetratin)が含まれるが、これらに限定されない。
【0020】
本発明は、本発明の配列特異的発現抑制剤か、あるいは、本発明のホスホペプチドかを含む、原虫感染症の治療用組成物を提供する。
【0021】
本明細書において、原虫感染症とは、アフリカ睡眠病、Chagas病及びリーシュマニア症を含むが、これらに限定されない、キネトプラスト科原虫を病原体とする感染症をいう。
【0022】
本発明の原虫感染症の治療用組成物は、原虫の増殖阻害効果を損なわないことを条件として、さらに、他の疾患の治療に有効な成分及び/又は薬学的に許容される添加物を1種類又は2種類以上含む場合がある。前記添加物は、希釈剤及び膨張剤と、結合剤及び接着剤と、滑剤と、流動促進剤と、可塑剤と、崩壊剤と、担体溶媒と、緩衝剤と、着色料と、香料と、甘味料と、防腐剤及び安定化剤と、吸着剤と、当業者に知られたその他の医薬品添加剤とを含むが、これらに限られない。
【0023】
本発明は、原虫感染症治療薬候補を選択するスクリーニング方法を提供する。本発明のスクリーニング方法は、(1)試験化合物を用意するステップと、(2)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、(3)前記試験化合物の存在下で、前記ステップ(2)で用意されたタンパク質のキナーゼ活性を測定するステップと、(4)前記試験化合物の非存在下で前記ポリペプチドのキナーゼ活性とを測定するステップと、(5)前記ステップ(3)のキナーゼ活性が、前記ステップ(4)のキナーゼ活性より低いとき、前記試験化合物をトリパノソーマ・ブルース感染症治療薬候補として選択するステップとを含む。
【0024】
本発明のスクリーニング方法において、前記試験化合物は、配列番号1に列挙されるアミノ酸配列からなるAKB14−3−3−1タンパク質が、原虫、特に、血流型トリパノソーマ・ブルース原虫において発現することを抑制する場合がある。
【0025】
本発明のスクリーニング方法において、前記試験化合物は、AKB14−3−3−1タンパク質と、14−3−3のヘテロ2量体との会合を阻害する場合がある。
【0026】
本発明において、ある特定のアミノ酸配列を含むペプチド、ポリペプチド又はタンパク質とは、当該ペプチド、ポリペプチド又はタンパク質の機能を損なわないことを条件として、当該ペプチド、ポリペプチド又はタンパク質のアミノ末端及び/又はカルボキシル末端に、細胞膜透過ペプチドと、特異的結合タグとを含むが、これらに限定されない、他の機能を有するペプチド、ポリペプチド又はタンパク質が融合したものをいう。
【図面の簡単な説明】
【0027】
【図1】AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性を検討した結果を示すウェスタンブロット図。
【図2】AKB14−3−3−1の野生型と、突然変異型(とのV5タグ付きのタンパク質との精製収量を示すブロット図。
【図3】原虫細胞内で発現され、精製された、AKB14−3−3−1の野生型及び突然変異型タンパク質を用いたキナーゼアッセイの結果を示す、蛍光染色された電気泳動図。
【図4】AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図。
【図5】AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図。
【図6】PCF細胞B5クローンを、テトラサイクリン存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図。
【図7】PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図8】PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真。
【図9】図8で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【図10】BSF細胞A5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図。
【図11】BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図12】BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真。
【図13】図12で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【図14】PCF細胞A11クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1の発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図。
【図15】テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞A11クローンの典型的なDAPI染色蛍光顕微鏡写真。
【図16】PCF細胞のA11クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図17】図15で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【図18】PCF細胞B10クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1NFGの発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図。
【図19】テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞B10クローンの典型的なDAPI染色蛍光顕微鏡写真。
【図20】PCF細胞のB10クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフ。
【図21】図19で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラム。
【発明を実施するための形態】
【0028】
以下に説明する本発明の実施例は例示のみを目的とし、本発明の技術的範囲を限定するものではない。本発明の技術的範囲は特許請求の範囲の記載によってのみ限定される。本発明の趣旨を逸脱しないことを条件として、本発明の変更、例えば、本発明の構成要件の追加、削除及び置換を行うことができる。
【実施例1】
【0029】
トリパノソーマ14−3−3タンパク質ヘテロ2量体と結合する可能性のあるタンパク質の同定
1.トリパノソーマ14−3−3タンパク質の発現ベクターの構築
トリパノソーマにおいて外来遺伝子を構成的に発現できるpLew82ベクターは、G.Cross博士より恵与され、発明者らの研究室で、T7プロモーターが追加され、pLew82T7phleoと命名された。pLew82T7は、さらに、V−5タグ配列、3Cプロテアーゼ切断配列及び制限酵素Hpa I切断部位が追加され、フレオマイシン耐性遺伝子がピューロマイシン耐性遺伝子に置換され、pLew82T7puroN−V5−3Cと命名された。トリパノソーマ・ブルース14−3−3のI型アイソフォーム(Inoue,M.ら、J.Biol.Chem.、280:14085、(2005)、GenBankアクセッション番号AB059827)は、mycタグ配列が追加され、それぞれ、pLew82T7puroN−V5−3Cにサブクローニングされ、pLew82T7puroN−V5−3C−Myc−14−3−3Iと命名された。トリパノソーマ・ブルース14−3−3のII型アイソフォーム(GenBankアクセッション番号AB066565)は、pLew82T7phleoにサブクローニングされ、pLew82T7phleo−14−3−3IIと命名された。
【0030】
2.トリパノソーマ14−3−3タンパク質を発現するトリパノソーマ細胞の調製
29−13細胞は、ハイグロマイシン(50μg/mL)、G418(25μg/mL)及び10%ウシ胎仔血清が添加されたSDM−79培地で維持される。以下では、SDM−79培地にはハイグロマイシン及びG418が含まれる。29−13細胞は、pLew82T7phleo−14−3−3IIが29−13細胞に常法に従ってトランスフェクションされ、フレオマイシン(2.5μg/mL)が添加されたSDM−79培地で選択された。得られたフレオマイシン耐性クローンに、pLew82T7puroN−V5−3C−Myc−14−3−3Iがさらにトランスフェクションされ、二重にトランスフェクションされたクローンが、フレオマイシン(2.5μg/mL)及びピューロマイシン(1μg/mL)、が添加されたSDM−79培地で選択され、維持された。前記二重にトランスフェクションされたクローンについて、トリパノソーマ・ブルース14−3−3のI型及びII型アイソフォームのヘテロ2量体の存在量が、抗V5タグ抗体を不動化したアガロースビーズを各クローンの細胞の溶解液に懸濁して、前記アガロースビーズに吸着したタンパク質を抗トリパノソーマ・ブルース14−3−3I抗体及び抗トリパノソーマ・ブルース14−3−3II抗体(Inoue,M.ら、J.Biol.Chem.、280:14085、(2005))を用いるウェスタンブロッティング法で分析されたその結果、前記二重にトランスフェクションされた細胞のクローンのうち、I型及びII型アイソフォームのヘテロ2量体を最も大量に産生するものとして、A6クローンの細胞が選ばれた。
【0031】
3.トリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質の回収
2×106個/mLの前記A6クローンの細胞懸濁液250mLはテトラサイクリン(1μg/mL)が添加されたSDM−79培地で26時間培養された。A6細胞のペレットはPBS(−)で2回洗浄された後、1mLのPMEM細胞溶解バッファー(0.1M PIPES pH6.9、2mM EGTA、1mM MgSO4、0.1mM EDTA、200μM ジギトニン、プロテアーゼ阻害剤混合液(シグマ、カタログ番号P8340)、10mM E64、50mM NaF及び2.5mM NaVO4)とともに氷上で30分間インキュベーションされて溶解された。細胞溶解液は、5000rpm、4°Cで2分間遠心され、上清はさらに100,000g、4°Cで1時間遠心され、その上清が回収された。前記上清は、抗V5タグ抗体を不動化したアガロースビーズ(シグマ、カタログ番号A7345)100μLとともに4°Cで3.5時間回転しながらインキュベーションされた。前記アガロースビーズは、50mM NaF及び2.5mM NaVO4が添加された前記PMEM細胞溶解バッファーの2倍希釈液で3回洗浄され、前記PMEM細胞溶解バッファーの1倍液で1回洗浄された後、スピン−Xカラム(Costar、カタログ番号8160)に移された。前記アガロースビーズに結合したタンパク質は、HVSGLKRRRSV(配列番号2)からなり、カルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチド100μg/mLを含む前記PMEM細胞溶解バッファーの1倍液210μLで溶出された。溶出されたタンパク質は、氷冷されたメタノール、クロロホルム及び脱イオン水で沈殿され、15,000rpm、4°Cで1時間遠心され、ペレットは、100mM Tris−HCl、pH8.5中の8M尿素溶液10μLに再懸濁された。
【0032】
4.質量分析によるトリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質の解析
前記8M尿素溶液に0.5μLの0.1M DTTが添加されて、60°C、20分間インキュベーションされ、その後、0.5μLの0.2M ヨウ化アセトアミドが添加されて、室温、1時間インキュベーションによりチオール基がカルボキシメチル化された。そして反応混合液に1μLの0.5mg/mLリシル・エンドペプチダーゼ(和光純薬株式会社、カタログ番号125−05061)が添加され、37°Cで3時間インキュベーションされた。その後、前記反応混合液は40μLの100mM Tris−HCl、pH8.5で希釈され、125ngのトリプシン(MSグレード、和光純薬株式会社、カタログ番号202−1591)が添加され、37°C、終夜インキュベーションされた。前記反応混合液は減圧乾燥の後、0.3% ギ酸(和光純薬株式会社、カタログ番号067−04531)に再懸濁され、日立液体クロマトグラフ質量分析計(日立ハイテク株式会社、NanoFrontierLD)によるナノLC−MS/MS分析に供された。
【0033】
5.結果
トリパノソーマ14−3−3ヘテロ2量体と結合するタンパク質のトリプシン消化断片ペプチドの質量分析ピーク値から、トリパノソーマ・ブルースのゲノムにエンコードされるタンパク質が75個同定された。
【実施例2】
【0034】
トリパノソーマ14−3−3タンパク質ヘテロ2量体と結合する可能性のあるタンパク質の予備的な特徴付け
1.全長cDNAのクローニング
実施例1で同定されたトリパノソーマ・ブルースのタンパク質75個のうち、14−3−3のI型及びII型アイソフォームと、アミノ酸の相同性から機能が推測できないものと、熱ショックタンパク質、リボソームタンパク質、カルモジュリン等の14−3−3ヘテロ2量体との特異的結合が考えにくいタンパク質を除いて、MASCOT検索(マトリックスサイエンス株式会社)のスコアの高いタンパク質4個(Tb10.61.1900、Tb927.10.7780、Tb927.5.4300及びTb11.47.0035)について、全長cDNAクローンがTb427プロサイクリック型(昆虫感染型)細胞由来cDNAライブラリからPCR法で単離された。Tb10.61.1900と、Tb927.10.7780と、Tb927.5.4300と、Tb11.47.0035とは、それぞれ、推定キナーゼと、マイトジェン活性化キナーゼと、プロリリルオリゴペプチダーゼ、推定、セリンペプチダーゼと、カルパイン様システインペプチダーゼとしてアノテーションがされている。
【0035】
2.誘導可能なノックダウンベクターの構築と、トリパノソーマ細胞への導入
前記全長cDNA4個は、それぞれ、pQuadra3bsrテトラサイクリン依存性ノックダウンベクター(Inoue,M.ら、J.Biol.Chem.、280:14085(2005))にサブクローニングされ、常法に従って29−13プロサイクリック型(以下、「PCF」ともいう。)細胞株及び90−13血流型(以下、「BSF」ともいう。)細胞株にトランスフェクションされた。PCF細胞は、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で選択された。BSF細胞は、ブラストサイジン(5μg/mL)、20%ウシ胎仔血清、ハイグロマイシ(50μg/mL)及びG418(25μg/mL)を含むHMI−9培地で選択された。
【0036】
3.原虫細胞のキネトプラスト、核及び鞭毛の形態学的解析
トリパノソーマ・ブルース細胞のDNA及びα−チューブリンの検出は常法にしたがって行われた。簡潔には、トリパノソーマ・ブルース細胞は、室温で、4% パラメータホルムアルデヒドにより固定され、0.2% Triton−Xでパーメアライズ処理された。さらに、1M グリシンで自家蛍光を消光する処理を行った後、BlockAce(DSファーマバイオメディカル株式会社、カタログ番号UK1380)でブロッキングされた。それから、前記細胞は、FITC抗α−チューブリン抗体(Sigma、DM−1A、カタログ番号F2168−2ML)と反応され、さらに、DAPIで染色された。
【0037】
4.結果
前記ノックダウンベクター4種類がトランスフェクションされた細胞のうち、Tb10.61.1900の2本鎖RNAがテトラサイクリン存在下で誘導される細胞では、キネトプラスト、核及び鞭毛が過剰形成されたり、あるいは、全くなかったり、というように細胞分裂およびサイトキネシスに異常をきたしたモンスター様細胞(monster−like cell with multikinetoplasts、Multinucleus and multiflagella)が観察された。かかるモンスター様細胞は、トリパノソーマ・ブルースの14−3−3のI型及びII型アイソフォームを両方とも過剰発現させるか、あるいは、両方とも発現抑制させることによっても観察された。そこで、Tb10.61.1900の遺伝子座タグ(locus tag)にエンコードされるタンパク質はトリパノソーマ・ブルースの14−3−3の下流で機能することが示唆された。
【0038】
なお、残りのノックダウンベクター3種類がトランスフェクションされた細胞については、テトラサイクリン存在下で2本鎖RNAを発現させたとき、キネトプラスト、核及び鞭毛のいずれにも影響が見られなかった。そこで以下では、Tb10.61.1900の遺伝子座タグ(locus tag)にエンコードされるタンパク質は、AKB14−3−3−1(Associated Kinase of Trypabnosoma brucei 14−3−3 Type I)という。
【実施例3】
【0039】
AKB14−3−3−1の特徴付け(1)
1.AKB14−3−3−1のトリパノソーマ発現ベクターの構築
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列は配列番号1に示される。前記全長アミノ酸配列をエンコードするポリヌクレオチドが、前記pLew82T7puroN−V5ベクターにサブクローニングされて、V5タグが連結されたAKB14−3−3−1の融合タンパク質を原虫細胞内で過剰に発現するベクターが構築された。
【0040】
2.AKB14−3−3−1の大腸菌発現ベクターの構築
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列は配列番号1に示される。前記全長アミノ酸配列をエンコードするポリヌクレオチドが、pET47b及びpGEX4T2にサブクローニングされて、それぞれHisタグ及びGSTと連結した融合タンパク質(His−AKB−14−3−3−1及びGST−AKB−14−3−3−1)を大腸菌で発現するベクターpET47b−His−AKB−14−3−3−1及びpGEX4T2−AKB14−3−3−1が構築された。
【0041】
2.AKB14−3−3−1に対する特異抗体の作成
GST−AKB−14−3−3−1は、常法に従って、pGEX4T2−AKB14−3−3−1ベクターで形質転換された大腸菌T7Express lysY/Iq株(ニュー・イングランド・バイオラボ・ジャパン株式会社、カタログ番号#C3013H)を0.4mM IPTG存在下で終夜培養した後リゾチームで溶菌させて得られた溶菌液からグルタチオン−セファロース・カラム(GEヘルスサイエンス株式会社、カタログ番号17−0756−01)を用いて精製された。得られた精製GST−AKB−14−3−3−1は1回に150μgずつフロイントアジュバントに添加してウィスター系ラットに感作された。4回の免疫感作後の前記ラットの血清が抗AKB14−3−3−1抗体として用いられた。
【0042】
3.抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法による原虫細胞でのAKB14−3−3−1の検出
トリパノソーマ・ブルースのPCF細胞2×106個又はBSF細胞5×106個は、5%のβ−メルカプトエタノールが添加された2×SDSローディングバッファー(バイオ・ラッド ラボラトリーズ株式会社)に溶解され、常法にしたがって、SDS−ポリアクリルアミドゲル電気泳動法により分離され、電気泳動後のゲルからPVDFメンブレンに電気的に転写された。抗AKB14−3−3−1抗体として、前記抗AKB14−3−3−1ラット血清が2000倍に希釈して用いられた。トリパノソーマ・ブルース14−3−3I及びIIを検出するためには、抗トリパノソーマ・ブルース14−3−3I及びII血清が2:1の割合で混合された血清混合液(以下、「抗Tb14−3−3混合血清」という。)が1000倍に希釈して用いられた。
【0043】
4.AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性の検討
実施例1で説明されたのと同様に、テトラサイクリン(1μg/mL)が添加されたSDM−79培地で終夜培養された約7.5x106個/mLの前記A6クローンの細胞懸濁液250mLに由来する細胞溶解液の上清(以下、「Input」という。)が抗V5タグ抗体を不動化したアガロースビーズと反応され、スピン−Xカラムに移された。前記上清との反応の後、前記アガロースビーズから分離された溶液(以下、「Unbound」という。)が除去され、その後前記スピン−Xカラム中のアガロースビーズに、HVSGLKRRRSV(配列番号2)からなるペプチド(100μg/mL)が添加されて得られた溶出液(以下、「Peptide」という。)と、該ペプチドのカルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチド(100μg/mL)が添加されて得られた溶出液(以下、「Phosphopetide」という。)とが回収された。さらに、それぞれのペプチドによる処理後の前記アガロースビーズに、8M尿素が添加され、溶出液が回収された。Inputの細胞溶解液上清と、Unboundの溶液と、Peptide又はPhosphopetideの溶出液と、前記Peptide又はPhosphopetideによる溶出後の前記アガロースビーズから8M尿素で溶出された溶出液とが、抗Tb14−3−3混合血清又は抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法によって解析された。
【0044】
5.結果
AKB14−3−3−1、すなわち、Tb10.61.1900の遺伝子座タグにエンコードされるタンパク質の配列(配列番号1)には、全長640個のアミノ酸からなり、そのうち40−333番目には、推定セリン/スレオニン タンパク質キナーゼの触媒ドメインが含まれ、また、571−638番目には、細胞骨格との会合に関与するCap−Glyドメインが含まれる。
【0045】
図1は、AKB14−3−3−1と14−3−3ヘテロ2量体との結合の特異性を検討した結果を示すウェスタンブロット図である。図1の上半分は抗AKB14−3−3−1抗体を用いるウェスタンブロッティングの結果であり、矢印はAKB14−3−3−1のバンドの位置を示す。図1の下半分は抗Tb14−3−3混合血清を用いるウェスタンブロッティングの結果である。図1から明らかなとおり、AKB14−3−3−1はリン酸化ペプチドによる溶出液(Phosphopeptide)のレーンでは検出されたが、リン酸化されないペプチドによる溶出液(Peptide)のレーンではほとんど検出されなかった。また、8M Ureaの左側のレーンは、リン酸化されないペプチドで溶離後、8M 尿素によって溶離された溶出液のレーンであり、大量のAKB14−3−3−1が、V5−3C−Myc−14−3−3I(図1では「14−3−3V5−Myc−I」と表される。)と、タグが連結しない14−3−3I及び14−3−3IIとともに検出された。これに対し、8M Ureaの右側のレーンは、リン酸化されたペプチドで溶離後、8M 尿素によって溶離された溶出液のレーンであり、V5−Myc−14−3−3Iと、タグが連結しない14−3−3I及び14−3−3IIとは、8M Ureaの左側のレーンとほぼ同じ量が出てされたが、AKB14−3−3−1はわずかしか検出されなかった。
【0046】
図1の結果から、AKB14−3−3−1タンパク質と、トリパノソーマ・ブルース14−3−3ヘテロ2量体との結合は、HVSGLKRRRSV(配列番号2)からなるペプチドでは競合阻害されないが、該ペプチドのカルボキシル末端から2番目のセリン残基がリン酸化されたホスホペプチドでは競合阻害されることが示された。したがって、本発明のAKB14−3−3−1タンパク質は原虫細胞内でリン酸化され、14−3−3ヘテロ2量体と、特異的に結合していることが示唆された。
【実施例4】
【0047】
AKB14−3−3−1の特徴付け(2)
1.AKB14−3−3−1タンパク質のキナーゼ活性突然変異体の作成
AKB−14−3−3−1の推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異体タンパク質はAKB−14−3−3−1NFGと命名され、そのHisタグ又はGSTとの融合タンパク質を大腸菌で発現するベクターが構築された。
【0048】
2.AKB14−3−3のキナーゼ活性のアッセイ
大腸菌で発現され、精製された、His−AKB−14−3−3−1及びGST−AKB−14−3−3−1と、これらの突然変異体タンパク質(NFG)のキナーゼ活性は、PepTag(登録商標)非放射性PKCアッセイ(プロメガ株式会社、カタログ番号V5330)を用いて確認された。すなわち、1回の反応あたり0.1又は0.5μgのHis−AKB−14−3−3−1又はGST−AKB−14−3−3−1が、0.08mg/mLのPepTag C1ペプチド(PLSRTLSVAAK(配列番号3)のアミノ酸配列からなる蛍光標識されたCキナーゼ基質ペプチド)と、1mMのATP、UTP、GTP、CTP又はATP−γSか、1mMのATP及び1mMのDTTかとが、反応混合液(20mM Tris−HCl、pH7.4及び10mM MgCl2)に添加され、30°Cで、GST−Tagが0.1μgのとき30分間、His−Tag又はGST−Tagが0.5μgのとき1時間インキュベーションされた後、製造者の指示に従って、アガロース電気泳動法により、リン酸化された基質が未反応の基質から分離され、蛍光により検出された。
【0049】
2.結果
図2は、AKB14−3−3−1の野生型(図2及び3では「Wild」と表される。)と、推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異型(図2及び3では「NFG」と表される。)とのV5タグ付きのタンパク質とが、原虫細胞内で発現され、抗V5タグ抗体を固定化したアガロースビーズにより精製され、遊離のV5タグペプチドにより溶離された溶出液が抗AKB14−3−3−1抗体を用いるウェスタンブロッティング法で解析された結果を示すブロット図である。図2から明らかなとおり、突然変異型、原虫細胞から精製された量には違いが見られなかった。
【0050】
図3は、原虫細胞内で発現され、精製された、AKB14−3−3−1の野生型及び突然変異型タンパク質を用いたキナーゼアッセイの結果を示す、蛍光染色された電気泳動図である。図3から明らかなとおり、野生型タンパク質にはキナーゼ活性が認められたが、突然変異タンパク質にはキナーゼ活性は認められなかった。したがって、AKB14−3−3−1タンパク質のキナーゼ活性は、精製が不十分なために他の酵素が混入したためではないことが示された。
【0051】
図4及び5は、AKB14−3−3−1タンパク質のキナーゼアッセイの結果を示す、蛍光標識ペプチド基質の電気泳動図である。図4では、His−AKB−14−3−3−1(図4では「His−tag」と表される。)と、GST−AKB−14−3−3−1(図4では「GST−tag」と表される。)とが、さまざまなリボモノヌクレオチド存在下で、基質のPepTag C1ペプチドをリン酸化するかどうかが調べられた。その結果、Hisタグとの融合タンパク質でも、GSTとの融合タンパク質でも、AKB14−3−3−1は、ATPの他、GTP、及びATP−γSは利用可能であるが、UTP及びCTPは利用できないことが示された。さらに図5では、GST−AKB−14−3−3−1について、さまざまなリボモノヌクレオチドだけでなく、さまざまなデオキシリボモノヌクレオチドの存在下で、基質のPepTag C1ペプチドとのキナーゼ反応に供された。図4及び5から、AKB14−3−3−1タンパク質は、リン酸基の供与体として、ATPの他、GTP、ATP−γS及びdATPは利用可能であるが、UTP、CTP、dTTP、dGTP及びdCTPは利用できないことが示された。
【実施例5】
【0052】
AKB14−3−3−1の特徴付け(3)
【0053】
1.原虫細胞におけるAKB14−3−3−1遺伝子発現量の解析
トリパノソーマ・ブルース細胞の全RNAは、RNeasy Plus Mini kit(株式会社キアゲン、カタログ番号74134)を用いて抽出された。100ngの全RNAがスーパースクリプトVILO cDNA合成キット(ライフテクノロジーズジャパン株式会社、インビトロジェン、カタログ番号11754050)を用いて逆転写された。全RNA10ngから合成されたcDNAが1回のPCR反応に供された。AKB14−3−3−1をエンコードするcDNAを増幅するためのPCRプライマーは、配列番号4及び配列番号5が用いられ、α−チューブリンをエンコードするcDNAを増幅するためのPCRプライマーは、配列番号6及び配列番号7が用いられた。AKB14−3−3−1をエンコードするcDNAを増幅するためには、95°C1分間の後、95°C15秒間、55°C15秒間、72°C15秒間を22ないし23回繰り返して、最後に72°C5分間という反応条件が用いられた。α−チューブリンをエンコードするcDNAを増幅するためには、95°C1分間の後、95°C15秒間、55°C15秒間、72°C15秒間を20回繰り返して、最後に72°C5分間という反応条件が用いられた。PCR産物は、2%TAE−アガロースゲル電気泳動による分離され、GelRed(Biotium、ナカライテスク株式会社)により染色された。
【0054】
2.原虫細胞の増殖の解析
細胞数は、血球計算盤により測定された。細胞密度を一定範囲内に保つために、PCF細胞の培養では毎日5倍に希釈して播種され、BSF細胞の培養では毎日10倍に希釈して播種された。pQuadra3bsrAKB14−3−3−1ベクターでdsRNAiによるAKB14−3−3−1の発現抑制を行う場合には、テトラサイクリン(1μg/mL)が添加されたSDM−79培地が用いられた。なお、pQuadra3bsrAKB14−3−3−1がトランスフェクションされたBSF細胞をテトラサイクリン存在下で培養する場合には、細胞密度が低下するため、希釈して播種する作業は第4日で中止された。
【0055】
3.PCFでの結果
PCF細胞(29−13株)にpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で、安定的にpQuadra3bsrAKB14−3−3−1ベクターがインテグレーションされたPCF細胞のクローンB5が選択された。図6は、かかるPCF細胞B5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図である。図6から明らかなとおり、PCF細胞B5クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1のmRNA量が顕著に少なかった。これに対して、対照実験のα−チューブリンのmRNA量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞B5クローンでは、テトラサイクリンによりAKB14−3−3−1遺伝子の発現が抑制されることが示された。
【0056】
図7は、図6に用いられた、PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図7から明らかなとおり、テトラサイクリンによってAKB14−3−3−1の遺伝子発現が抑制されたPCF細胞では、遺伝子発現抑制が起こらないときと比較して、細胞の増殖が10分の1程度に低下した。したがって、AKB14−3−3−1遺伝子の発現抑制は細胞増殖の低減をもたらすことが示された。
【0057】
図8は、図6及び7に用いられた、PCF細胞B5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真である。テトラサイクリン非存在下(Tet−)及び存在下(+)で3日間培養された細胞を、それぞれ、抗α−チューブリン抗体又はDAPIで染色後、蛍光顕微鏡で観察したところ、テトラサイクリン存在下で培養されたPCF細胞ではサイトキネシスの異常が観察された。
【0058】
図9は、図8で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図8及び9から明らかなとおり、PCF細胞におけるAKB14−3−3−1遺伝子の発現抑制は、細胞分裂及びサイトキネシスの異常をもたらした。
【0059】
4.BSFでの結果
BSF細胞(90−13株)にpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたHMI−9培地で、安定的にpQuadra3bsrAKB14−3−3−1ベクターがインテグレーションされたBSF細胞のA5クローンが選択された。図10は、BSF細胞A5クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1及びα−チューブリンの遺伝子発現がそれぞれのmRNAのRT−PCR法によって分析された結果の電気泳動図である。図10から明らかなとおり、BSF細胞A5クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1のmRNA量が顕著に少なかった。これに対して、対照実験のα−チューブリンのmRNA量は、テトラサイクリンの有無によって違いは認められなかった。したがって、このpQuadra3bsrAKB14−3−3−1ベクターがトランスフェクションされたBSF細胞のクローンでは、テトラサイクリンによりAKB14−3−3−1遺伝子の発現が抑制されることが示された。なお、図6と図10とを比較すると、BSF細胞での遺伝子発現の抑制は、PCF細胞よりはやや弱かった。
【0060】
図11は、図10に用いられた、BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記BSF細胞のクローンの増殖曲線である。図11から明らかなとおり、テトラサイクリンによってAKB14−3−3−1の遺伝子発現が抑制されたBSF細胞では、遺伝子発現抑制が起こらないときと比較して、細胞の増殖が100分の1程度に低下した。また、培養4日目以降は細胞増殖がほぼ完全に停止した。したがって、AKB14−3−3−1遺伝子の発現抑制はBSF細胞の細胞増殖の阻害をもたらすことが示された。
【0061】
図12は、図10及び11に用いられた、BSF細胞A5クローンのテトラサイクリンの存在下又は非存在下での形態を示す蛍光顕微鏡写真である。テトラサイクリン非存在下(Tet−)及び存在下(+)で3日間培養された細胞を、それぞれ、抗α−チューブリン抗体又はDAPIで染色後、蛍光顕微鏡で観察したところ、テトラサイクリン存在下で培養されたPCF細胞ではサイトキネシスの異常が観察された。
【0062】
図13は、図12で観察された細胞について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図12及び13から明らかなとおり、BSF細胞におけるAKB14−3−3−1遺伝子の発現抑制は、細胞分裂及びサイトキネシスをもたらした。
【0063】
以上詳しく説明したとおり、AKB14−3−3−1タンパク質には、正常な細胞分裂及びサイトキネシスに必須の機能があることが証明された。
【実施例6】
【0064】
AKB14−3−3−1の特徴付け(3)
1.AKB14−3−3−1タンパク質の野生型及び突然変異体の原虫細胞内での過剰発現
AKB−14−3−3−1と、AKB−14−3−3−1の推定ATP結合部位の1つである第186番目のアスパラギン酸残基がアスパラギン残基に置換された突然変異体タンパク質(AKB−14−3−3−1NFG)とが、テトラサイクリン依存性発現ベクターのpLew82T7bsrにサブクローニングされ、それぞれ、pLew82T7bsr AKB−14−3−3−1及びpLew82T7bsr AKB−14−3−3−1NFGと命名された。pLew82T7bsr AKB−14−3−3−1及びpLew82T7bsr AKB−14−3−3−1NFGが、PCF細胞(29−13株)にトランスフェクションされ、ブラストサイジン(10μg/mL)が添加されたSDM−79培地で、安定的に過剰発現ベクターがインテグレーションされたPCF細胞のクローンが選択された。
【0065】
2.結果
図14は、かかるpLew82T7bsrAKB14−3−3−1ベクターがトランスフェクションされたPCF細胞A11クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1の発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図である。図14の上半分に示されるとおり、PCF細胞A11クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1タンパク質の量が顕著に多かった。図14の下半分は、前記ウェスタンブロッティング法のサンプルの電気泳動ゲルのクーマジーブルー染色写真のうち、AKB14−3−3−1タンパク質のバンドに相当する移動度の部分を示したものである。図14の下半分に示されるとおり、AKB14−3−3−1タンパク質以外の原虫細胞のタンパク質の量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞のA11クローンでは、テトラサイクリンによりAKB14−3−3−1タンパク質の発現が特異的に誘導されることが示された。
【0066】
図15は、テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞A11クローンの典型的なDAPI染色蛍光顕微鏡写真である。図15の左側に示すとおり、テトラサイクリン非存在下では、PCF細胞は1個の核と1個のキネトプラストを有する。これに対し、図15の右側に示すとおり、テトラサイクリン存在下では、PCF細胞は2個以上の核と2個以上のキネトプラストを有する。
【0067】
図16は、PCF細胞のA11クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図16から明らかなとおり、PCF細胞A11クローンでは、過剰発現が起こらないときと比較して、7日間で細胞の増殖が10分の1程度に低下した。したがって、AKB14−3−3−1の過剰発現はPCF細胞の細胞増殖の低下をもたらすことが示された。
【0068】
図17は、図15で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図17から明らかなとおり、PCF細胞におけるAKB14−3−3−1の過剰発現は、細胞分裂及びサイトキネシスの異常をもたらした。
【0069】
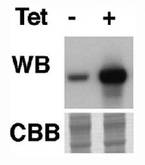
図18は、pLew82T7bsr AKB−14−3−3−1NFGがトランスフェクションされたPCF細胞B10クローンを、テトラサイクリン(Tet)存在下又は非存在下で1日間培養した後、AKB14−3−3−1NFGの発現が抗AKB14−3−3−1抗体を用いたウェスタンブロッティング法で解析された結果のブロット図である。図18の上半分に示されるとおり、PCF細胞B10クローンは、テトラサイクリン存在下で培養された細胞(Tet+)では、テトラサイクリン非存在下で培養された細胞(Tet−)と比較すると、AKB14−3−3−1NFGタンパク質の量が顕著に多かった。図18の下半分は、前記ウェスタンブロッティング法のサンプルの電気泳動ゲルのクーマジーブルー染色写真のうち、AKB14−3−3−1NFGタンパク質のバンドに相当する移動度の部分を示したものである。図18の下半分に示されるとおり、AKB14−3−3−1NFGタンパク質以外の原虫細胞のタンパク質の量は、テトラサイクリンの有無によって違いは認められなかった。したがって、PCF細胞のB10クローンでは、テトラサイクリンによりAKB14−3−3−1NFGタンパク質の発現が特異的に誘導されることが示された。
【0070】
図19は、テトラサイクリン存在下(+)又は非存在下(−)で培養された、PCF細胞B10クローンの典型的なDAPI染色蛍光顕微鏡写真である。図19に示すとおり、テトラサイクリン存在下でも非存在下でも、PCF細胞は1個の核と1個のキネトプラストを有する。ただし、核及びキネトプラストの位置関係が正常なPCF細胞と異なる細胞が8.5%認められた。これらは、エピマスチゴート型というトリパノソーマ・ブルースの生活史でPCFとは異なるステージの細胞に変化したことを示唆する。
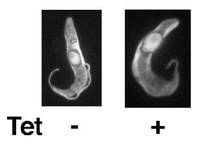
【0071】
図20は、PCF細胞のB10クローンのテトラサイクリンの存在下又は非存在下での増殖曲線を示す片対数直線グラフである。「□」が結ばれたグラフは、テトラサイクリン存在下での前記PCF細胞のクローンの増殖曲線である。「×」が結ばれたグラフはテトラサイクリン非存在下での前記PCF細胞のクローンの増殖曲線である。図20から明らかなとおり、テトラサイクリンによってAKB14−3−3−1NFGが過剰に発現されたPCF細胞では、過剰発現が起こらないときと比較して、7日間の細胞の増殖はほとんど違いが認められなかった。したがって、AKB14−3−3−1NFGの過剰発現はPCF細胞の細胞増殖には影響を与えないことが示された。
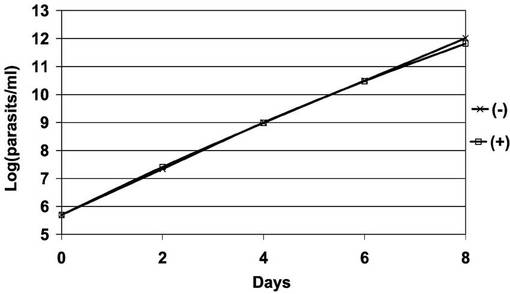
【0072】
図21は、図19で観察された細胞100個について、細胞ごとの核及びキネトプラストの個数をプロットしたヒストグラムである。黒及び白の棒グラフは、それぞれ、テトラサイクリン存在下(+)及び非存在下(−)での核の個数が、0個、1個、2個、3個又はそれ以上(「0N」、「1N」、「2N」又は「>2N」)で、キネトプラストの個数が0個、1個、2個、3個又はそれ以上(「0K」、「1K」、「2K」又は「>2K」)の細胞の百分率を表す。図21から明らかなとおり、PCF細胞におけるAKB14−3−3−1NFGの過剰発現は、サイトキネシスの異常をもたらさなかった。よって、AKB14−3−3−1が細胞分裂、サイトキネシスを調節する機能を発揮するためには、キナーゼ活性が必要であることが示唆された。
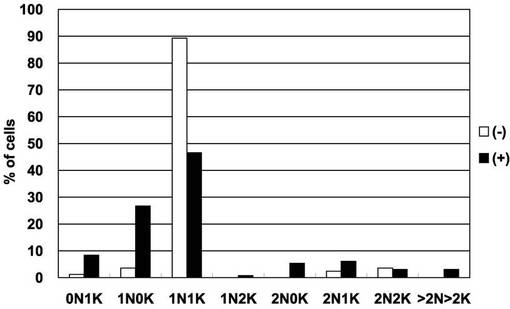
【0073】
これらの実施例から、本発明のAKB14−3−3−1タンパク質は、トリパノソーマ・ブルースの14−3−3ヘテロ2量体と結合するタンパク質キナーゼであって、14−3−3と同様に、細胞増殖及びサイトキネシスに関与することが示された。とくに、図11に示されるとおり、本発明のAKB14−3−3−1遺伝子発現を抑制すると、BSF細胞の増殖が完全に阻害される。BSFすなわち血流型はヒトや家畜に感染した状態を反映するため、本発明のAKB14−3−3−1タンパク質の機能を阻害することによって、ヒトや家畜に感染したトリパノソーマ・ブルースの増殖を阻害できると考えられる。つまり、本発明のAKB14−3−3−1タンパク質の機能を阻害する物質は、アフリカ睡眠病の有効な治療薬となることが期待できる。
【特許請求の範囲】
【請求項1】
(1)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、
(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、
(2)前記ステップ(1)で用意されたタンパク質と、試験化合物とを含む反応混合液のキナーゼ活性を測定するステップと、
(3)前記試験化合物の非存在下で、前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、
(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップとを含むことを特徴とする、原虫の増殖阻害剤を検出する方法。
【請求項2】
(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、
(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤。
【請求項3】
配列番号2に列挙されるアミノ酸配列を含み、該アミノ酸配列のカルボキシル末端から2番目のセリン残基がリン酸化され、配列番号1に列挙されるアミノ酸配列からなるタンパク質と、トリパノソーマ・ブルースの14−3−3−I及び14−3−3−IIのヘテロ2量体との会合を阻害することを特徴とする、ホスホペプチド。
【請求項4】
請求項3に記載のホスホペプチドと、細胞膜透過性ペプチドとの融合ペプチド。
【請求項5】
請求項2に記載の配列特異的発現抑制剤と、請求項3に記載のホスホペプチドと、請求項4に記載の融合ペプチドとからなるグループから選択される少なくとも1つを含むことを特徴とする、原虫感染症の治療用組成物。
【請求項1】
(1)(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、
(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を用意するステップと、
(2)前記ステップ(1)で用意されたタンパク質と、試験化合物とを含む反応混合液のキナーゼ活性を測定するステップと、
(3)前記試験化合物の非存在下で、前記ステップ(1)で用意されたタンパク質を含む反応混合液のキナーゼ活性を測定するステップと、
(4)前記ステップ(2)のキナーゼ活性と、前記ステップ(3)のキナーゼ活性とを比較するステップとを含むことを特徴とする、原虫の増殖阻害剤を検出する方法。
【請求項2】
(a)配列番号1に列挙されるアミノ酸配列からなるタンパク質と、
(b)配列番号1に列挙されるアミノ酸配列と70%以上の相同性を示すアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(c)配列番号1に列挙されるアミノ酸配列に1個若しくは数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列からなり、かつ、タンパク質キナーゼ活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのポリペプチドに連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質の血流型トリパノソーマ・ブルース原虫における発現を抑制する配列特異的発現抑制剤。
【請求項3】
配列番号2に列挙されるアミノ酸配列を含み、該アミノ酸配列のカルボキシル末端から2番目のセリン残基がリン酸化され、配列番号1に列挙されるアミノ酸配列からなるタンパク質と、トリパノソーマ・ブルースの14−3−3−I及び14−3−3−IIのヘテロ2量体との会合を阻害することを特徴とする、ホスホペプチド。
【請求項4】
請求項3に記載のホスホペプチドと、細胞膜透過性ペプチドとの融合ペプチド。
【請求項5】
請求項2に記載の配列特異的発現抑制剤と、請求項3に記載のホスホペプチドと、請求項4に記載の融合ペプチドとからなるグループから選択される少なくとも1つを含むことを特徴とする、原虫感染症の治療用組成物。
【図1】


【図2】
【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2012−80804(P2012−80804A)
【公開日】平成24年4月26日(2012.4.26)
【国際特許分類】
【出願番号】特願2010−228186(P2010−228186)
【出願日】平成22年10月8日(2010.10.8)
【出願人】(599045903)学校法人 久留米大学 (72)
【出願人】(504205521)国立大学法人 長崎大学 (226)
【Fターム(参考)】
【公開日】平成24年4月26日(2012.4.26)
【国際特許分類】
【出願日】平成22年10月8日(2010.10.8)
【出願人】(599045903)学校法人 久留米大学 (72)
【出願人】(504205521)国立大学法人 長崎大学 (226)
【Fターム(参考)】
[ Back to top ]