
古細菌アンプラウイルスABV耐熱性DNAポリメラーゼ及びその用途
本発明は、古細菌アンプラウイルスABV(好酸性瓶状ウイルス)の熱安定性DNAポリメラーゼタンパク質及び前記DNAポリメラーゼをコードする核酸に向けられる。本発明はまた、前記DNAポリメラーゼタンパク質をコードする核酸の合成方法、増幅方法又は配列決定方法、並びに前記DNAポリメラーゼタンパク質を含んで成るキット又は装置にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、古細菌アンプラウイルスABV(Acidianus Bottle-shaped Virus)の熱安定性DNAポリメラーゼタンパク質及び前記DNAポリメラーゼをコードする核酸に向けられる。本発明は、前記DNAポリメラーゼタンパク質をコードする核酸の合成、増幅又は配列決定方法、並びに前記DNAポリメラーゼタンパク質を含んで成るキット又は装置にも関する。
【背景技術】
【0002】
超好熱性菌クレン古細菌(Crenarchaeota)の二本鎖(ds)DNAウイルスは、著しく多様な形態とゲノム構造を示し、それらの特徴に基づいて、既に6つの新規ウイルスファミリーに分類されている:紡錘形フセロウイルス科(Fuselloviridae)、繊維状リポスリクスウイルス科(Lipothrixdviridae)、棒状ルディウイルス科(Rudiviridae)、液滴状グタウイルス科(Guttaviridae)、球状グローブロウイルス科(Globuloviridae)及び二本の鞭毛を持つビコウダウイルス科(Bicaudaviridae)(Prangishvili他、2001に概説;Prangishvili & Garrett, 2004, 2005)。最近、ユニークな瓶形の形態を有する新種ウイルスが発見され、新ファミリーであるアンプラウイルス科(Ampullaviridae)に指定された(Haring他、2005a)。
【0003】
核酸分析及び核酸増幅のための手段として開発された様々な核酸増幅技術が、遺伝病や感染症の臨床診断に好結果に利用されている。増幅技術は温度循環を必要とするもの(PCRとリガーゼ連鎖反応)と等温系のもの〔増幅系(3SRやNASBA)、鎖置換増幅法、及びQβ複製系〕に分類することができる。次の二点がそれらの手法におけるよくある警告である:合成の忠実性と増幅生成物の長さ。
【0004】
ファージphi29(φ29)のDNA複製機構に頼る増幅方法の開発が刊行物や特許文書の主題となっている(Dean他、Genome Res. 2001 Jun, 11(6):1095-9 ; Mendez他、EMBO J., 1997, 1, 16(9):2519-27 ; Hutchison他、Proc. Natl. Acad. Sci. USA, 2005, 102(48):17332-6 ; Mamone, Innovations Forum: GenomiPhi DNA amplification, Life Sciences News 14, 2003 Amersham Biosciences; Blanco他、1994, EP 0 862 656又はUS 5,001,050)。
【0005】
phi29 DNAポリメラーゼは、非常に効率的な等温系DNA増幅を可能にする、強力な鎖置換活性を特徴とする高処理能力ポリメラーゼである(Blanco他、Proc. Natl. Acad. Sci. USA, 81, 5325-5329, 1984及びJ. Biol. Chem., 264, 8935-8940, 1989)。phi29 DNAポリメラーゼは一本鎖DNA上で優先的に作用する3′→5′エキソヌクレアーゼ(校正)活性も有する(Garmendia,J. Biol. Chem., 267, 2594-2599, 1992)。
【0006】
上記特徴の中で、既知DNAポリメラーゼのうち最高の処理能力と鎖置換活性、即ち、70kb以上の長鎖DNA片を合成できること(Blanco他、1989)、その高度に正確なDNA合成(Esteban他、J. Biol. Chem., 268, 4, 2719-2726, 1993)、微量の鋳型からであっても高収率の増幅DNAが得られること、及び増幅生成物が下流用途(PCR、制限消化、SNP遺伝子型分析、他)に直接利用できることを言及することができる。この特定のDNAポリメラーゼに関して多数の特別な用途が発見された。例えば、ローリングサークル増幅(RCA)(Lizardi他、Nat. Genet., 19, 225-232, 1998 ; Dean他、Genome Res., 11, 1095-1099, 2001 ; Baner他、Nucleic. Acids Res., 26, 5073-5078, 1998)。多重置換増幅(MDA)(Dean他、Proc. Natl. Acad. Sci. USA, 99, 5261-5266, 2002)、全ゲノムの不偏増幅又は配列決定用のDNA鋳型調製。この系は、今日利用できる増幅系を使って得られるサイズ限界を大幅に上回る、70 kbより長いDNA分子の正確な増幅(Blanco他、1989)に適しているだろう。この等温TP感作増幅(“TP”は末端タンパク質)方法は、phi29 DNAポリメラーゼの特定性質:(i)プライマーとしてタンパク質を使用できること;(ii) 本質的な高処理能力(>70 kb);及び(iii) DNA合成と協同した鎖置換、を有効に利用する。このphi29 DNAポリメラーゼの特異活性は30℃の温度で発揮され、そしてこのphi29 DNAポリメラーゼは65℃で不活性化すると推測される。
【発明の開示】
【発明が解決しようとする課題】
【0007】
30℃より有意に高い温度で作用でき、且つ60℃で完全には不活性化されない、phi29 DNAポリメラーゼのようなタンパク質感作型DNAポリメラーゼファミリーに属する新規DNAポリメラーゼが現在必要とされている。
これが本発明の目的である。
【課題を解決するための手段】
【0008】
ウイルスABV(Acidianus Bottled-shaped Virus)感染アシディアヌス(Acidianus)属好熱性古細菌の全ゲノム配列を配列決定しそして確認した後、本発明者らはDNA依存性DNAポリメラーゼをコードする核酸配列を発見した。驚くべきことに、該タンパク質配列の分析は、それがタンパク質感作型DNAポリメラーゼファミリーに属することを示した。該DNAポリメラーゼ遺伝子がE.コリ(E. coli;大腸菌)中で異種発現され、該組換えタンパク質のDNA重合活性が確認された。この新規酵素は、既知のウイルスDNAポリメラーゼと同様、高処理能力を有し、自給自足型で、補助タンパク質を必要としない。それらの特徴のため、該酵素はPCRによるDNA増幅のための手段として有利に利用することができる。タンパク質感作される熱安定性ウイルス酵素であるため、それは一本鎖又は二本鎖直線状DNAの指数的増幅(即ちAmersham社により開発されたGenomiPhi法による)において、現在この方法で利用されている中温性タンパク質感作酵素のバクテリオファージPhi29 DNAポリメラーゼよりもずっと有効であり得る。Amersham社のGenomiPhi増幅キットは、少数の細胞又は限定量の貴重な試料から、無制限のDNA試験を実施できるようにし、全ゲノムを典型的に増幅する容易なゲノムDNA増幅法である。
【発明を実施するための最良の形態】
【0009】
よって、第一の観点において、本発明は、
a) 配列番号1のアミノ酸配列を有するポリペプチド;
b) DNAポリメラーゼ活性を有するa)の断片;
c) a)の前記DNAポリメラーゼのDNAポリメラーゼ活性を有する、配列番号1断片を少なくとも含んで成るキメラポリペプチド;
d) 配列番号1のアミノ酸配列を有するポリペプチドに比較して有意に低い検出可能なエキソヌクレアーゼ活性を有するか又は全く持たないDNAポリメラーゼポリペプチドを生じるように、図4に示されるようなエキソヌクレアーゼ部位ExoI, Exo II及びExoIIIが変異又は削除されている、配列番号1のアミノ酸配列を有するポリペプチド;
e) 配列番号1の配列又はb)〜d)に定義したような配列との最適整列後に少なくとも80%同一である配列を有するポリペプチドであって、好ましくは50℃の温度で又は50℃より高い温度でDNAポリメラーゼ活性を有するポリペプチド;
から成るポリペプチド群より選択された、単離されたDNAポリメラーゼに向けられる。
【0010】
好ましい態様では、前記DNAポリメラーゼ活性を有する断片は、少なくとも50,100,150,200,250,300,350,400,450,500又は600アミノ酸を有する。
【0011】
好ましい態様では、本発明に係るDNAポリメラーゼは、ABVから又はDBAポリメラーゼをコードするABV遺伝子から単離される。
【0012】
より好ましい態様では、本発明のDNAポリメラーゼは、少なくとも図4に示されるような配列番号1のPol I, Pol IIa, Pol IIb, Pol III及びPol IV断片を含んで成る。
【0013】
図4に関して、DNAポリメラーゼ活性は保存されているが、配列番号1の配列を有するポリペプチドよりも欠損した又は有意に低いエキソヌクレアーゼ活性を有する本発明のポリペプチドは、別のポリメラーゼとのアミノ酸配列相同性及びDNAポリメラーゼのエキソヌクレアーゼ活性を減少させることが知られているそれらの変異(Derbyshire他、Science, 1988, Apr 8, 240(4849):199-201)を考慮することにより、選択することができる。一般に、図4中Exo I, Exo II及び/又はExo IIIとして示されている位置のアミノ酸を、削除するか又は別のアミノ酸と置換することができる。それらのExo I, Exo II及びExo III位置でのアミノ酸の大型除去又は多重置換も実施できる。配列番号1の配列を有するポリペプチドを変異誘発させた後、エキソヌクレアーゼ活性レベルを測定し、そしてDNAポリメラーゼ活性の量を求め、それが本発明における使用に十分であることを確認する。
【0014】
用語「5′エキソヌクレアーゼ活性」とは、オリゴヌクレオチドの5′末端からヌクレオチドを除去することができるタンパク質の活性の存在を言う。5′エキソヌクレアーゼ活性は本明細書中に提供されるアッセイのいずれかを使って測定することができる。
【0015】
本発明のDNAポリメラーゼは、そのポリメラーゼのエキソヌクレアーゼ活性を減少させるように遺伝的に改変されているポリペプチド、並びに実質上同一の(少なくとも80%の同一性)天然に存在するABV DNAポリメラーゼ又はその改変ポリメラーゼであるもの、又は上記に列挙された同等の酵素へと遺伝的に改変されているポリペプチドを包含する。それらの酵素の各々は、ABV DNAポリメラーゼのものと同様な性質を有するように改変することができる。該酵素をABVウイルス感染細胞から直接単離することもできるが、好ましくはそれを過剰生産(組換え発現)する細胞から単離する。
【0016】
用語「エキソヌクレアーゼ活性」とは、或るオリゴヌクレオチドの3′末端又は5′末端からヌクレオチドを除去することができるタンパク質の活性の存在を言う。そのようなエキソヌクレアーゼ活性は当業者により周知である任意のエキソヌクレアーゼ活性アッセイを使って測定することができる。
【0017】
用語「DNAポリメラーゼ活性」とは、デオキシヌクレオシド三リン酸の取り込みにより新規DNA鎖を合成する酵素的ポリペプチドの能力を言う。下記実施例4は、DNAポリメラーゼ活性の測定のためのアッセイの一例を提供する。そのようなDNAポリメラーゼ活性は、当業者に周知のDNAポリメラーゼ活性アッセイのいずれかを使って測定することができる。鋳型依存性方式でのデオキシヌクレオシド三リン酸の取込みにより新規DNA鎖の合成(DNA合成)を指令することができるタンパク質は、「DNAポリメラーゼ活性を発揮できる」と言われる。
【0018】
本明細書では、ポリペプチド、ポリペプチド配列、ペプチド及びタンパク質という用語は相互交換可能である。
【0019】
2以上のポリペプチド配列の概念において、「同一」又は「同一性」%という語は、規定値(デフォルト)パラメーターを用いるBLASTもしくはBLAST 2.0配列比較演算法を使って、又は手法整列及び目視検査(例えばNCBIウエブサイトを参照のこと)により測定した時に、同一であるか又は特定比率の同一アミノ酸(即ち、比較ウィンドウまたは指定領域に渡り最大一致について比較・整列した時に、或る特定領域に関して約80%同一性、好ましくは85%、90%、95%、98%、99%もしくはそれより高い同一性)を有する2以上の配列又は部分配列のことを言う。この定義は、欠失及び/又は付加を有する配列、並びに置換を有する配列も包含する。後述するように、好ましい演算法はギャップ等を明らかにすることができる。好ましくは、少なくとも長さ約25アミノ酸である領域、より好ましくは長さ25〜75アミノ酸である領域に渡り同一性が存在する。
【0020】
配列比較では、典型的には、或る1つの配列が比較参照配列として働き、それに対して試験配列が比較される。配列比較演算法を使った場合、試験配列と参照配列をコンピューターに入力し、必要であれば部分配列候補をデザインし、そして配列演算プログラムパラメーターを設定する。好ましくは、規定値プログラムパラメーターを使用することができ、又は代替パラメーターを設定することができる。次いで配列比較演算法がそのプログラムパラメーターに基づいて参照配列に対する試験配列の配列同一性を算出する。
【0021】
比較のための配列の整列方法は当業者に周知である。配列同一性%及び配列相同性を求めるのに好ましい演算法の例はBLAST及びBLAST 2.0演算法であり、それらはAltschul他、Nuc. Acids Res. 25:3389-3402 (1977) 及びAltschul他、J. Mol. Biol. 215:403-410 (1990) に記載されている。
【0022】
例えば、サイトhttp://www.ncbi.nlm.gov/gorf/bl2.html上で入手可能なBLASTプログラム“BLAST 2配列”(Tatusova他、“Blast 2 sequences - a new tool for comparing protein and nucleotide sequences”, FEMS Microbiol Lett. 174:247-250)を使うことが可能であり、使用されるパラメーターは、規定値(デフォルト)により与えられたものであり(特に“オープンギャップペナルティ”:5、“伸長ギャップペナルティ”:2;選択されるマトリクスは、該プログラムにより提唱されたマトリクス“BLOSUM 62”である)、比較される2つの配列間の同一性%は該プログラムにより直接算出される。
【0023】
参照アミノ酸配列に対して少なくとも80%、好ましくは85%、90%、95%、98%、99%又はそれより高い同一性を有するアミノ酸配列とは、参照配列に関して、ある種の変更、特に少なくとも1つのアミノ酸の欠失もしくは付加又は置換、先端切除又は伸長を有するものが好ましい。1又は複数の保存的もしくは非保存的アミノ酸置換の場合、置換されるアミノ酸が「等価の」アミノ酸により置換される置換が好ましい。「等価アミノ酸」という表現は、本明細書中では、参照ポリペプチドのDNAポリメラーゼ活性を本質的に改変することなく、基本構造のアミノ酸の1つにより置換されることができる任意アミノ酸を示し、そしてそのようなアミノ酸は下記、特に実施例4の最終段落において定義されるだろう。
【0024】
このような等価アミノ酸は、それらが置き換わるアミノ酸との構造的相同性を頼みにするか、別のポリペプチド間でのDNAポリメラーゼ活性の比較試験の結果を頼みにすることにより、決定することができる。一例として、対応する改変されたポリペプチドのDNAポリメラーゼ活性の顕著な変更を引き起こすことなく実施することができる置換の可能性が挙げられる。従って、ロイシンをバリン又はイソロイシンに置換、アスパラギン酸をグルタミン酸に置換、グルタミンをアスパラギンに置換、アルギニンをリジンに置換することが可能であり、その逆の置換も同条件下で普通に予想できる。
【0025】
よって、第二の観点では、本発明は、本発明に係るDNAポリメラーゼポリペプチドをコードする核酸、特に配列番号2の配列を有するか又は最適整列後に配列番号2の配列と少なくとも80%同一性である配列を有する核酸を提供し、前記核酸によりコードされるポリペプチドは、好ましくは50℃又は50℃より高い温度でDNAポリメラーゼ活性を有する。
【0026】
本明細書中では、核酸、ポリヌクレオチド、オリゴヌクレオチド、又は核酸配列もしくはヌクレオチド配列という語は相互交換可能に用いられる。
【0027】
別の観点では、本発明は、本発明の核酸を含んで成るベクター、好ましくはクローニング又は発現ベクターに関する。
好ましい態様では、本発明に係るベクターは、前記核酸がプロモーターに作用可能に連結されていることを特徴とする。
【0028】
本発明は特に、本発明に係るヌクレオチド配列を含有するクローニング及び/又は発現ベクターを目的とする。
【0029】
本発明に係るベクターは、好ましくは、所定の宿主細胞中でのヌクレオチド配列の発現及び/又は分泌を可能にする要素を含有する。従って、該ベクターは、プロモーター、翻訳開始及び終結のシグナル、並びに適当な転写調節領域を含有しなければならない。それは宿主細胞中で安定した方法で維持され、そして場合により翻訳されたタンパク質の分泌を指令する特定シグナルを有することができる。それらの種々の要素は、用いられる宿主の関数として当業者により選択されそして最適化される。この結果により、本発明に係るヌクレオチド配列は、選択された宿主中で自律複製ベクター中に挿入することができるか、又は選択された宿主の組込み型ベクターであることができる。
【0030】
そのようなベクターは当業者により現在使用されている方法により調製され、そして生じたクローンは標準法、例えばリポフェクション、エレクトロポレーション、熱ショック法又は化学的方法により、適当な宿主に導入することができる。
【0031】
本発明に係るベクターは、例えば、プラスミド又はウイルス起源のベクターである。それらは、本発明に係るヌクレオチド配列をクローニングするため又は発現させるために宿主細胞を形質転換せしめるのに有用である。
【0032】
好ましい態様では、本発明のベクターは、ブタペスト条約に従って2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(Collection Nationale de Cultures de Microorganismes, フランス国パリのパスツール研究所)に寄託されている細菌中に含まれるプラスミドベクターである。
【0033】
このクローン化プラスミドベクターは、本発明のDNAポリメラーゼの核酸配列がpET30aプラスミドのNdeI部位とXbaI部位の間に挿入されているベクターpET30aである。
【0034】
用語「発現ベクター」とは、所望の核酸コード配列と特定の宿主細胞中での作用可能に連結された該コード配列の発現に必要な適当な核酸配列とを含有する組換えDNA分子を言う。原核生物中での発現に必要な核酸配列は、一般に、しばしば別の配列と共に、プロモーター、オペレーター(任意)、及びリボソーム結合部位を含んで成る。真核細胞はプロモーター、エンハンサー、並びに終止及びポリアデニル化シグナルを利用することが知られている。
【0035】
別の観点では、本発明は、本発明に係るベクターを含んで成る宿主細胞、特にブタペスト条約に従って2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(Collection Nationale de Cultures de Microorganismes, フランス国パリのパスツール研究所)に寄託されている組換え細菌に関する。
【0036】
本発明のDNAポリメラーゼポリペプチドは、原核又は真核宿主細胞のいずれにおいても発現させることができる。本発明のDNAポリメラーゼポリペプチドをコードする核酸は、塩化カルシウム処理により形質転換に許容性にされた細菌細胞の形質転換又はエレクトロポレーションをはじめとする多数の手法により、細菌宿主細胞中に導入することができる。本発明のDNAポリメラーゼポリペプチドを真核宿主細胞中で発現させるつもりであれば、本発明のDNAポリメラーゼポリペプチドをコードする核酸を、リン酸カルシウム共沈、スフェロプラスト融合、エレクトロポレーション等といった多数の手法により、真核宿主細胞中に導入することができる。真核宿主細胞が酵母細胞である時、形質転換は酢酸リチウムによる宿主細胞の処理又はエレクトロポレーションにより、あるいは当業界で既知の任意の別法により、実施することができる。本発明のペプチドもしくはタンパク質又はそれらの断片を生産するのに任意の宿主細胞が有用であると予想される。
【0037】
本発明に従って形質転換された細胞は、本発明の組換えポリペプチドの調製方法において使用することができる。本発明のベクター及び/又はベクターにより形質転換された細胞を使用することを特徴とする、組換え形での本発明ポリペプチドの調製方法は、それ自体本発明に含まれる。
【0038】
好ましくは、本発明のベクターにより形質転換された細胞を、前記ポリペプチドの発現を可能にする条件下で培養し、そして前記組換えポリペプチドを回収する。
【0039】
別の観点では、本発明は、DNAポリメラーゼの生産方法であって、
(a) 前記核酸の発現に適当な条件下で本発明の宿主細胞を培養する段階;及び
(b) 前記宿主細胞から前記DNAポリメラーゼを単離する段階
を含んで成る方法に関する。
【0040】
前記宿主細胞は原核細胞または真核細胞であることができる。
【0041】
既に言及したように、宿主細胞は原核系又は真核系から選択できる。特に、そのような原核系又は真核系における分泌を促進するヌクレオチド配列を使用することができる。従って、そのような配列を担持する本発明に係るベクターは、分泌させようとする組換えタンパク質の生産に有意に利用することができる。実際に、それらの組換えタンパク質が宿主細胞の内側よりもむしろ細胞培養物の上清中に存在するという事実により、着目のそれらの組換えタンパク質の精製が容易になるだろう。
【0042】
別の観点では、本発明は、二本鎖DNA分子を合成する方法であって、
(a) 第一のDNA分子にプライマーをハイブリダイズさせる段階;及び
(b) 段階(a)の前記DNA分子を1又は複数のデオキシリボヌクレオシド三リン酸又はその類似体及び本発明のポリペプチドの存在下で、前記第一のDNA分子の全部又は一部に相補的な第二のDNA分子を合成するのに十分な条件下でインキュベートする段階
を含んで成る方法に関する。
【0043】
別の観点では、本発明は、一本鎖DNA分子を合成する方法であって、
(a) 本発明の方法により二本鎖DNA分子を合成する段階;
(b) 段階(a)で得られた二本鎖DNA分子を変性させる段階;及び
(c) 段階(b)で得られた一本鎖DNA分子を回収する段階
を含んで成る方法に関する。
【0044】
別の観点では、本発明は、長さ10キロ塩基より大きいDNA分子の生産方法であって、
段階(a)における鋳型として働く第一のDNA分子が10キロ塩基より大きい、本発明に係る方法を含んで成る方法に関する。
【0045】
本発明に係る方法において、前記デオキシリボヌクレオシド三リン酸はdATP, dCTP, dGTP及びdTTPから成る群より選択される。
【0046】
別の観点において、本発明は、二本鎖DNA分子を増幅させる方法であって、
(a) 第一及び第二プライマーを用意し、ここで前記第一プライマーは前記DNA分子の第一鎖の3′末端の所又はその付近の配列に相補的であり、そして前記第二プライマーは前記DNA分子の第二鎖の3′末端の所又はその付近の配列に相補的である段階;
(b) 前記第一鎖に相補的な核酸と前記第二鎖に相補的な核酸が合成されるような条件下で、本発明に係るポリペプチドの存在下で前記第一プライマーを前記第一鎖にそして前記第二プライマーを前記第二鎖にハイブリダイズせしめる段階;
(c) 前記第一鎖とその相補鎖:及び
前記第二鎖とその相補鎖
を変性させる段階;及び
(d) 段階(a)から(c)を1回又は複数回繰り返す段階
を含んで成る方法に関する。
【0047】
前記増幅段階が、DNAポリメラーゼ活性を有するポリペプチド(DNAポリメラーゼポリペプチド)を包含するPCR、PCR様法又はRT−PCR反応により実施されることも好ましい。
【0048】
「PCR」とは、DNA試料中の特定遺伝子への、プライマーの配列を基準としたハイブリダイゼーション、それに続く熱安定性ポリメラーゼを使ったアニーリング(ハイブリダイゼーション)、伸長及び変性の反復を伴う増幅のことを言う。
【0049】
「RT−PCR」は逆転写ポリメラーゼ連鎖反応の略語である。mRNAを逆転写酵素にかけると、該mRNAの塩基配列に相補的であるcDNAが生成する。次いで熱安定性DNAポリメラーゼの作用を当てにしたポリメラーゼ連鎖反応によって、多量の特定cDNAを製造することができる。
【0050】
「PCR様」とは、核酸配列の直接又は間接再生を使う全ての方法、或いは標識系が増殖されている方法を意味し、それらの技術はもちろん既知であり、一般にそれらはポリメラーゼによるDNAの増幅を伴う。元の試料がRNAである時、前もって逆転写を実施することが望ましい。この増幅を可能にする多数の方法が現存し、例えばいわゆるNASBA(核酸配列ベース増幅“Nucleic Acid Sequence Based Amplification”)、TAS(転写ベース増幅系“Transcription based Amplification System”)、LCR(リガーゼ連鎖反応(“Ligase Chain Reaction”)、ERA(エンドラン増幅“Endo Run Amplification”)、CPR(循環プローブ反応“Cycling Probe Reaction”)、及びSDA(鎖置換増幅“Strand Displacement Amplification”)があり、それらの方法は当業者に周知である。
【0051】
mRNAを用いる場合、該方法は、逆転写酵素を使った標準法(RT−PCR)に従って、単離されたmRNAをcDNAに変換することにより実施することができる。
【0052】
別の観点では、本発明は、mRNAからcDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマーまたは別の相補的プライマーと接触させてハイブリッドを形成せしめる段階;及び
(b) 段階(a)で形成した前記ハイブリッドを、本発明に係るDNAポリメラーゼポリペプチド並びにdATP, dCTP, dGTP及びdTTPと接触させ、それによりcDNA−RNAハイブリッドが得られる段階
を含んで成る方法に関する。
【0053】
本発明は更に、mRNAからdsDNA(二本鎖DNA)を調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触させてハイブリッドを形成せしめる段階;及び
(b) 段階(a)で形成した前記ハイブリッドを、本発明に係るポリペプチド、dATP, dCTP, dGTP及びdTTP、並びに前記第一鎖cDNAに相補的であるオリゴヌクレオチド又はプライマーと接触させ、そしてによりdsDNAが得られる段階
を含んで成る方法にも向けられる。
【0054】
別の観点では、本発明は、DNA分子のヌクレオチド塩基配列を決定する方法であって、
(a) 前記DNA分子を前記DNA分子にハイブリダイズできるプライマー分子と接触させる段階;
(b) 段階(a)で形成した前記ハイブリッドを、4種の異なるデオキシヌクレオシド三リン酸、本発明に係るDNAポリメラーゼポリペプチド、及び特定のヌクレオチド塩基の所でDNA合成を終結させる1又は複数のDNA合成終結剤を含有する容器の中でインキュベートする段階であって、各々の終結剤は異なるヌクレオチド塩基の所でDNA合成を終結させる段階;及び
(c) 前記インキュベーション反応のDNA生成物をサイズに従って分離する段階であって、ここでそれにより前記DNAヌクレオチド塩基配列の少なくとも一部分を決定することができる段階
を含んで成る方法に関する。
【0055】
好ましい態様では、前記終結剤がジデオキシヌクレオシド三リン酸である。
【0056】
特定のヌクレオチド塩基の所でDNA合成を終結させるDNA合成終結剤とは、2′,3′−ジデオキシ構造を有するジデオキシヌクレオシド(例えばddATP, ddCTP, ddGTP及びddTTP)化合物を言うが、それに限定されない。特定の塩基の所でDNA配列合成反応を特異的に終結させることができるどんな化合物でもDNA合成終結剤として使用できる。
【0057】
別の観点では、本発明はDNA分子の増幅方法であって、次の段階:
(a) 前記DNA分子を本発明に係るDNAポリメラーゼ活性を有するポリペプチド、古細菌ABVの末端タンパク質、及び異なるデオキシヌクレオシド三リン酸の混合物の存在下でインキュベートする段階
を含んで成る方法に関する。
【0058】
好ましい態様では、本発明に係るDNA分子の増幅方法が、前記DNA分子の一端の所に前記ABVの複製開始点を含有する断片が共有結合されていることにより特徴付けられる。
【0059】
実際、ABVはphi29のものと同様な方法で複製を開始するようであり、逆方向末端反復配列(ITR)とその周囲領域の配列が複製開始に関係するだろう。配列番号5(左側)と配列番号6(右側)の配列は、ITRを含む2つのゲノム末端の配列である。
【0060】
好ましい態様では、前記ABVの複製開始点を含む断片の配列が、配列番号5(左側)と配列番号6(右側)の配列を含んで成る。
【0061】
更に別の観点では、本発明は、DNA分子を配列決定するためのキットであって、
(a) 本発明のポリペプチドを含んで成る第一のコンテナー手段;
(b) 1又は複数のジデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段;
(c) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第三のコンテナー手段
を含んで成るキットに向けられる。
【0062】
本発明はまた、DNA分子を増幅するためのキットであって、
(a) 本発明のポリペプチドを含んで成る第一のコンテナー手段;及び
(b) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段
を含んで成るキットにも関する。
【0063】
より好ましい態様では、本発明に係るDNA分子を増幅するためのキットは、ABVゲノムのORF163(配列番号4)によりコードされる配列番号3を有するポリペプチドに相当する、古細菌アンプラウイルスABVの単離された末端タンパク質を更に含んで成る。
【0064】
本発明は、ローリングサークル増幅、多重置換増幅、又はタンパク質感作増幅法を実施するための本発明に係るポリペプチドの使用も含んで成る。
【0065】
それらの特定方法は、当業者に周知であり且つ例えば次の文献に記載されている:
等温ローリングサークル増幅法については、Lizardi他、1998;Baner他、1998;Dean他、Genome Res., 11, 1095-1099, 2001;Larsson他、Nature methods 1, 227-232, 2004;
多重置換増幅法については、Dean他、2002;及び
タンパク質感作増幅法については、Blanco他、1994。
【0066】
好ましい態様では、本発明に係る方法、本発明に係るキット又は本発明に係る使用は、本発明のDNAポリメラーゼポリペプチドが、DNAポリメラーゼ活性と欠損エキソヌクレアーゼ活性(野生型ABVのDNAポリメラーゼが通常有する該活性の少なくとも1%未満、好ましくは0.1%未満)を有するポリペプチドであることを特徴とする。
【0067】
本発明のDNAポリメラーゼポリペプチドに関するエキソヌクレアーゼ活性は、DNA配列決定、合成又は増幅反応におけるDNAポリメラーゼの使用を有意に妨害することはない。しかしながら、エキソヌクレアーゼ活性のレベルが、天然のABVに感染した細胞より単離されたDNAポリメラーゼが通常有する活性又は配列番号1の配列の活性の10%未満又は1%未満、好ましくは0.1%未満であるレベルにまで減少することが好ましい。
【0068】
本発明は、DNA配列決定又は増幅のための装置であって、本発明のDNAポリメラーゼポリペプチドを含んで成るリアクターを有する装置にも向けられる。
【0069】
本発明は、本発明の抗DNAポリメラーゼポリペプチドを生産する方法であって、免疫寛容細胞を有する動物を、本発明のポリペプチド又は本発明のポリペプチドの少なくとも抗原性部分(決定基)を含んで成る免疫原に、前記免疫寛容細胞が前記本発明のポリペプチドに対して特異的に向けられた抗体又はそのエピトープ部分を生産するような条件下で暴露することを含んで成る。一態様では、該方法が前記抗体を収集する段階を更に含んで成る。別の態様では、該方法が、ハイブリドーマが生産されるような条件下で前記免疫寛容細胞を不死化細胞系と融合せしめる段階を更に含んで成る。
【0070】
そのような抗体は、別の成分が存在する試料中の本発明ポリペプチドを精製するために特に利用することができる。
【0071】
下記実施例は、本発明の種々の態様を例示する目的で提供され、どのような形にせよ本発明を限定するものではない。
【実施例】
【0072】
実施例1:材料と方法
ヌクレオチド、プライマー及び酵素
(γ−32P)ATP、dNTP及び酵素はPharmacia社より得られた。オリゴヌクレオチドpolF1(5’-CCTCCCTATTTGATAGGC-3’、配列番号8)を、(γ−32P)ATP及びT4ポリヌクレオチドキナーゼで5′標識し、そして8M尿素−20%ポリアクリルアミドゲル上で電気泳動により精製した。標識されたpolF1をpolF1c+24(5’-AGGTAAGCATGCATCAGTTAATACGCCTATCAAATAGGGAGG-3’、配列番号9)と混合し、該混合物を重合アッセイ(下記参照)におけるプライマー鋳型DNA分子として使用した。
【0073】
ウイルスの精製及びウイルスDNAの調製
以前記載された通り(Haring他、2005a)に、イタリアのポッツオーリにあるSoffatara 火山の噴火口中の87〜93℃でpH 1.5-2の貯蔵水から採取した試料より、好酸性の濃厚培養物を調製した。それらを75℃、pH 3で増殖させた。ウイルス粒子をCsCl浮遊密度勾配中での遠心分離により精製し、室温で1時間1%(w/v)SDSで破壊した後、以前に記載された通りに(Haring他、2005a)DNAを抽出し沈澱させた。
【0074】
ゲノムDNAの配列決定
合計わずか約200 ngの精製済ウイルスDNAしか最初のプロジェクトで入手できなかったことを考慮して、GenomiPhi増幅キット(Amersham Biotech, Amersham)を使って約1 ngのDNAを試験管内で増幅させて数μgを得た。次いでpUC18のSmaI部位中にクローニングしたサイズ範囲1.5〜4 kbpの音波処理DNA断片から、ショットガンライブラリーを作製した。該ライブラリーは高度に偏性のゲノムを生成した。
【0075】
増幅ライブラリーから得られた集成体と配列を、約50 ngの元のABV DNAと好酸性Acidianus属ベータリポスリクスウイルスbetalipothrixvirus(Vestergaard他、印刷中)から抽出した1μgのDNAを使って、混合ショットガンライブラリーを調製することにより確認した。該ライブラリーは、より長鎖の音波処理DNA断片を2〜6.5 kbpの範囲でクローニングすること以外は上述した通りに調製した。PCR反応を実施して、第二ライブラリーによりカバーされなかった領域を確かめ、そして少数のクローンをプライマーウオーキング(Peng他、2001)により更に配列決定した。
【0076】
配列分析
NCBIデータベースに対してBlastP検索を実施した。SMART及びMotifScanを使って保存された領域、プロフィール又はパターンを検出した。コイルドコイル、二次構造及び膜貫通ヘリックスは、ExPASy Proteomics Tools(http://expasy.org/tools/)中のプログラムにより検出した。
【0077】
ポリメラーゼ(ORF653)のクローニング及び精製
元のウイルスDNAから、NdeIとXhoI制限部位をそれぞれ含有する2つのプライマー(5’-TATTTTTACATATGCTACAAATCCT-3’配列番号10、及び5’-TATAACTCGAGTGAGAGAATACTATTTAAGTC-3’配列番号11)を使ってORF653をPCR増幅せしめた。PCR生成物をまずpGEM-Tベクター(Promega)中にクローニングし、そしてORF653挿入断片を含有する精製プラスミドを、NdeIとXhoIで消化した。付着性NdeI及びXhoI末端を持つORF653を含有するDNA断片を、低融点アガロース(Promega)ゲル電気泳動によりpGEM-T断片から分離し、そしてゲル抽出キット(Quiagen)を使って精製した。精製断片を次いで、NdeIとXhoIで消化しそして仔牛腸ホスファターゼにより処理したpET30-aベクター中にクローニングした。生じた構成物は、ORF653の生成物のC末端の後方に6個のヒスチジン残基をコードする配列を含有する。発現宿主細胞Posetta(Novagen)中に形質転換する前に、該構成物を配列決定して挿入されたORF653の配列を確認した。
【0078】
Rosetta形質転換体の単一コロニーを、25μg/mlのカナマイシンとクロラムフェニコールを含有する5 mlのLB培地に接種し、OD値が0.5に達するまで37℃にてインキュベートした。次いで前記5 ml培養物を、同じ抗生物質を含む250 mlのLB培地に移した。0.1 mM IPTG及び1%エタノールの存在下で30℃にて一晩増殖させた後、細胞を収穫した。製造会社(Novagen)により提供されたプロトコルに従って、Ni-NTA His.Bind樹脂を使ってヒスチジン標識タンパク質を精製し、そしてSDS-PAGEにより確認した。
【0079】
重合アッセイ
ハイブリッド分子polF1/polF1c+24(前掲)は、24ヌクレオチド長さの5′突出末端を含むので、DNA重合のためのプライマー−鋳型として使用することができる。反応混合物は、10μl中に、25 mM Tris-HCl (pH 7.6)、1 mM ジチオトレイトール、10 mM MgCl2、250μMの4つのdNTPを各々250μM、0.1μMのプライマー−鋳型DNA分子、及び増加する濃度の組換えポリメラーゼを含有した。50℃で20分間インキュベートした後、5μlの負荷緩衝液(80%ホルムアミド、10 mM EDTA, 50 μg/mlのブロモフェノールブルー)を添加し、80℃にて3分間加熱することにより、反応を停止させた。試料を8 M尿素−20%PAGEとオートラジオグラフィーにより分析した。5′標識プライマー鎖(polF1)のサイズの増加として重合を検出した。
【0080】
実施例2:ゲノム配列及び組織化
核酸をABVウイルス粒子から単離し、RNase Aには不感受性であるがII型制限エンドヌクレアーゼにより消化可能であることが示され、このことはそれがdsDNAであることと一致する。入手できるのが少量のゲノムDNAであることを考えて(<200 ng精製DNA)、本発明者らは二段階ゲノム配列決定法を適用した。
【0081】
第一に、GenomiPhi増幅キット(Amersham Biotech)を使って、約1 ng DNAを試験管内で増幅して約2 μgのDNAを得た。次いで以前に同じ手順により増幅させた古細菌ルジウイルスSIRV1のゲノムDNAについて観察されたのと同様に(Peng他、2004)、高偏向ゲノムカバレージを生成するショットガンライブラリーを作製した(材料と方法の項目を参照のこと)。高レベルのキメラクローンも作製された。ゲノムを平均20倍被覆率で配列決定し、そしてコンティグ中のそれらの低頻度によりキメラクローンの配列を同定し、ライブラリーからそれらを除外した。
【0082】
増幅DNAライブラリーから得られた配列集成体を確かめるために、約50 ngの元のABV DNAをアシディアヌス(Acidianus)属リポスリクスウイルス(Vestergaard他、2005)から抽出された1μgDNAと混合し、そしてより大型のクローン挿入断片(2〜6.5 kbp)を用いて混合ショットガンライブラリーを調製した。第一ライブラリーのものへのそれらABVクローンの会合及び配列決定は、2つのライブラリーからの配列が完全に一致することを示した。更に、第二ライブラリーによりカバーされない領域の配列を、それらの領域のPCR増幅後に、又はプライマーウオーキングの後に確認した。それら両方ともウイルスDNA及び/又は大型挿入断片クローンに対して実施した。加えて、左端の所の少数クローンの配列を、約22kbの最終コンティグを与えるプライマーウオーキングにより伸長させた。
【0083】
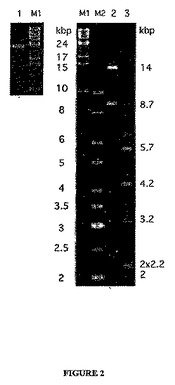
ゲノム会合を確認するため、約40 ngのウイルスDNAを制限酵素AgeIとAflIIで消化した。生成物を完全なウイルスDNAと一緒にアガロースゲル電気泳動により分画し、バンドをSYBRゴールド(Invitrogen)により染色した。断片サイズは会合したコンティグの配列と一致し、完全なウイルスDNAのバンドは約23.8 kbのゲノムサイズを示した(図2)。
【0084】
制限消化からのサイズ推定値と単一コンティグサイズとの間の食い違いは、恐らく直線状ウイルスゲノムの末端領域がクローンライブラリー中に表示されないことを反映するのであろう(Haring他、2004;Haring他、2005b)。従って、本発明者らは増幅されたDNAに対するプライマーウオーキングにより、コンティグ末端から更に配列決定することを試みた。これは、常に終結と解読される配列の後方に約2kbの追加配列を与えた。得られた全配列は23,794 bpであり、これは制限断片消化の推定値と一致した。G+C含量は35%であった。
【0085】
該ゲノムは580 bpの逆方向末端反復配列(ITRs)を示し、それはルジウイルスのゲノムのもの(Peng他、2001)よりも小さいが、古細菌ベータリポスリクスウイルスのもの(Vestergaard他、出版準備中)と同様であった。
【0086】
該ゲノムが環化するかどうかを試験するために、ゲノムの各末端付近にアニールする幾つかの異なるプライマー対を使ってPCR実験を行ったが、それらのいずれも増幅生成物を生じなかった(データは示してない)。従って、本発明者らはABVのゲノムが直鎖状であると推測した。
【0087】
実施例3:ゲノム含量
ゲノムに注釈をつけ、開始コドン(88%AUG,6%GUG及び6%UUG)、TATA様プロモーターモチーフ及び/又はShine-Dalgarnoモチーフを以前に記載された通りに(Bettstetter他、2003, 増補表1S)割り当てた。57〜653アミノ酸サイズに及ぶ59の推定ORFを含有する全ウイルスゲノムについての地図を図3に与える。3つのORF 653, 103及び257は、Shine-Dalgarnoモチーフにより前置きされる内部開始コドンを含有する。内部ORFはよって推定遺伝子として割り当てられる(図3及び表1)。1つを除く全てのORFが一方の鎖上の位置8.5 kbと右端の間に位置する。残りのORFのうち、3つ(ORF247、ORF53a及びORF156)を除く全てが他方の鎖上の左端と位置8.5 kbの間に位置する(図3)。図3に示したORFの約49%が推定プロモーター配列により前置きされ、そして68%が推定Shine-Dalgarnoモチーフにより前置きされる。更に、ORFの約11%がTに富む下流の推定ターミネーターを示す。該ORFの約85%が推定オペロン中に配列され、そして該遺伝子の約25%がリーダー無しであるか又は非常に短いリーダーを担持するかのいずれかである転写物を生成すると推定される。ORF間の距離は一般に非常に短くそしてORFの24%が上流ORFとオーバーラップしており、このことはゲノムがコンパクトであることを示す。非常に驚くべきことに、位置10 kb〜21 kbの間に位置する29個のORFが1つの単一大型オペロンを形成するように思われる(図3及び表1S)。
【0088】
わずか3つのORFだけに公表配列データベース内での相同性検索に基づいて明確な機能を割り当てることができる(材料と方法を参照のこと)。ORF653は、タンパク質感作ポリメラーゼに最大の一致を有するファミリーBのDNAポリメラーゼと有意な配列相同性を示した。更に、ORF156はチミジレートキナーゼとして同定され、そしてORF315は推定グリコシルトランスフェラーゼとして同定された。
【0089】
全ての遺伝子注釈を表1に要約する。配列決定されたクレン古細菌ウイルスゲノムの大部分が、クレン古細菌ウイルス中で最も一般的な遺伝子産物である少なくとも1つのリボン−ヘリックス−ヘリックス(RHH)領域タンパク質をコードするけれども、ABVゲノム中には全くRHH領域は検出されなかった。しかしながら、ORF56は、薬剤やストレスに対する耐性に関与する幾つかの原核性転写調節因子中に存在するtetR型ヘリックス−ターン−ヘリックス領域に対して、限られた類似性を示す。別の3つのORFは、転写調節に関係するようであるロイシンジッパーパターンを含有する(図3及び表1)。ORF188は有意なEFハンド・カルシウム結合部位を含み、そしてII型プロテインキナーゼAの調節サブユニットに対して限定された類似性を示す。従って、それはCa++依存性プロテインキナーゼをコードするかもしれない。ORF133の配列中に、タンパク質−タンパク質相互作用に関係するかもしれない推定apaG領域プロフィールが検出された。ORF133タンパク質配列の推定二次構造は、約90%の伸長鎖とランダムコイルの存在を明らかにした。これは、別のapaGタンパク質の三次構造におけるβシートの高含有量と相関する。3つの隣接ORF(112、166及び346)は少数の膜貫通ヘリックスを含み、推定膜タンパク質又は膜結合タンパク質であるように見える。特に着目されるのはORF346であり、それは推定上の原核性膜リポタンパク質−脂質付着部位とEGF様領域を担持する。後者は一般に膜結合タンパク質の細胞外領域中又は分泌タンパク質中に存在する(表1)。それらの性質は、宿主膜タンパク質と相互作用するウイルス外皮タンパク質、又は宿主細胞からのウイルス粒子の放出を促進する膜貫通タンパク質を構成するORF346と一致する。2つの上流ORF(112と166)によりコードされる推定膜貫通タンパク質も同じ過程に関係するかもしれない。ORF346と下流のORF470の両者のC末端配列は、別のクレン古細菌ウイルスゲノム中の数個の大型ORFにおいて観察されるような(例えばHaring他、2005;Neumann & Zillig, 1990)低複雑性を示す。該タンパク質の機能は未知である。
【0090】
3つのORFが明確な機能を指定されそして幾つかは推定上の保存されたモチーフまたはパターンを担持する(表1)一方で、ABVと別のクレン古細菌ウイルスの間で共有される唯一の遺伝子は、グリコシルトランスフェラーゼ遺伝子のORF315である。以前、異なるクレン古細菌ウイルスゲノム間で共有される遺伝子が全くないか又はごく少数しかないことを比較ゲノム学は示した(Haring他、2004;Peng他、2001;Bettstetter他、2003)。この研究からの結果は、クレン古細菌ウイルスが非常に多様なグループを形成することを強調する。
【0091】
【表1】

【0092】
実施例4:DNA複製
ルジウイルスを例外として、本発明者らは古細菌ウイルスゲノムの複製機構にほとんど見識がない。しかしながら、ABVはそのゲノムが推定上のタンパク質感作DNAポリメラーゼをコードするという点で特別である。それらの酵素は、共有結合した末端タンパク質と共にITRを担持している直鎖状dsDNAゲノム中に常にコードされ、そしてバクテリオファージφ29と真核性アデノウイルスの両方において特徴付けられている(Salasにより概説、1991)。それらのウイルスについての複製開始モデルは、DNAポリメラーゼによりヘテロ二量体を形成しそしてウイルスDNA結合末端タンパク質及び該ゲノムのいずれかの末端の所の特定ヌクレオチド配列を介して複製開始点と相互作用する、遊離末端タンパク質を含む。末端タンパク質中のセリン、スレオニン又はチロシンのヒドロキシル基が第一ヌクレオチドのための受容部位として働く。更に、多数の直鎖状dsDNAプラスミドやミトコンドリアゲノムが、ITRとタンパク質感作DNAポリメラーゼを提示し、そして同様な方法で複製すると思われる末端タンパク質を担持している(Salas,1991)。タンパク質感作DNAポリメラーゼのサブファミリーは、DNA依存性DNAポリメラーゼファミリーBに属し、そして2つの挿入断片TPR-1とTPR-2を有する(Blasco他、2000;Dufour他、2000;Rodriguez他、2005)。
【0093】
ORF653とφ29 DNAポリメラーゼとの配列整列(図4)は、ORF653が、ファミリーBのDNAポリメラーゼに特徴的である(Blanco他、1991;Rohe他、1992)、N末端領域に3つのエキソヌクレアーゼ領域(Exo I, II及びIII)を含みそしてC末端部分に5つの保存された合成領域(pol I, IIa, IIb, III及びIV)を含むことを示す。更に、挿入断片TPR-1とTPR-2もORF653中に存在する(図4)。TPR-1は、ヒトアデノウイルスによりコードされるものを含む(Dufour他、2000)、全てのタンパク質感作DNAポリメラーゼのものとサイズが類似しており(50 aa)、一方でモチーフpol IIaとIIbの間に位置するTPR-2は、28 aa(φ29)から118 aa(アデノウイルス2型)までに及ぶ挿入断片の既知サイズ範囲(Bois他、1999)に比較して、短縮されている。φ29DNAポリメラーゼについては、TPR-1は末端の感作タンパク質との相互作用に関与することが分かっており(Dufour他、2000)、一方でTPR-2は該ポリメラーゼの高処理能力と鎖置換活性に必要であることが示されている(Rodriguez他、2005)。より保存的であるTPR-1の機能は、全てのタンパク質感作DNAポリメラーゼに一般的であるようであるが、これがより可変性のTPR-2にも当てはまるかどうかは不明のままである。
【0094】
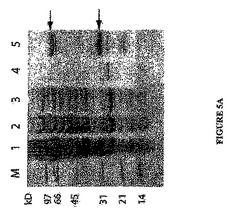
ORF653がDNAポリメラーゼであることを確かめるために、該遺伝子をPCRによりウイルスゲノムから増幅し、E.コリ(E.coli)発現ベクター中にクローニングした。適度な量の可溶性組換えタンパク質が、該タンパク質のC末端断片と一緒に精製された(図5A)。重合活性を試験するために、該タンパク質を標識プライマー−鋳型DNAと共に50℃でインキュベートした。図5Bは、重合活性の指標である、プライマー(18nt)が鋳型のサイズ(42nt)へと伸長したことを明らかに示す。dNTPsの代わりにNTPsを用いた時、全く重合は検出されなかった(データは示してない)。これは、ORF653の生成物が確かにDNAポリメラーゼであることを確証する。
【0095】
実施例5:末端タンパク質
ABVの直鎖状ゲノム中のITR及び推定上タンパク質感作DNAポリメラーゼをコードする遺伝子の存在は、各5′末端が末端タンパク質に共有結合していることを強く示唆する。これは、生産されるウイルス粒子の収量が非常に低いために、実験的に確かめることは困難である。従って、本発明者らは、関連のバクテリオファージ、直鎖状プラスミド及びヒトアデノウイルスからの末端タンパク質配列を分析し、それらのタンパク質の保存された特徴に関する識見を得た。ポリメラーゼは比較的保存されているが、末端タンパク質は非常に低い保存を示した。例えば、大腸菌バクテリオファージPRD1の末端タンパク質は、別の既知末端タンパク質と全く有意な配列相同性を示さず(Savilahti他、1987)、そしてφ29の末端タンパク質と白色腐朽菌Pleurotus ostreatusの直鎖状ミトコンドリアプラスミド(Kim他、2000)の間には13/48%の同一性/類似性しか観察されなかった。しかしながら、末端タンパク質の遺伝子座は高度に保存されている。例えば、バクテリオファージφ29、PRD1, GA-1及びCP-1(受入番号P03681, P09009, X96987及びZ47794)並びにヒトアデノウイルスでは、該遺伝子は常にDNAポリメラーゼ遺伝子のすぐ上流に常に位置し、一方で該タンパク質のサイズはバクテリオファージでは230 aaと266 aaの間であり、そしてアデノウイルス2型(AC_000007)では671 aaである。DNA複製は直鎖状プラスミドについてはあまり研究されておらず、そのうち2つは融合したN末端の末端タンパク質とC末端のDNAポリメラーゼをコードすることが分かっている(Kim他、2000;Takeda他、1996)。DNAポリメラーゼの配列整列は、別の直鎖状プラスミド中のN末端部分における大きな配列伸長も明らかにし(Bois他、1999)、このことはポリメラーゼの遺伝子と末端タンパク質の遺伝子が一般に融合している可能性があることを示す。更に、転写物マッピングは、DNAポリメラーゼ遺伝子と末端タンパク質遺伝子が、CP-1(Martin他、1996)及びφ29ファミリーファージ(Meijer他により概説、2001)を含む研究されたウイルス全てにおいて、単一のmRNA中に同時転写されることも示した。従って、ポリメラーゼと末端タンパク質は遺伝子編成と機能の両方に関して密接に関連している。ABVの場合、ポリメラーゼの上流の遺伝子は、バクテリオファージ末端タンパク質のサイズの三分の二である163 aaをコードする。しかしながら、ClustalW(EMBL-EBI)を使った配列整列は、PRD1のTPとφ29のTPとの間よりもORF163とPRD1からのTPとの間に高いスコアを示し(データは示していない)、これはORF163が末端タンパク質をコードするかもしれないことを示唆する。
【0096】
実施例6:ウイルスDNAパッケージング
以前に、ABVのウイルス粒子構造が別の既知クレン古細菌クレンアキオータ門ウイルスと比較して非常に複雑であることが示された。瓶形のウイルス粒子は細い末端に「ストッパー」を、そして広い方の末端に20本の短いフィラメントを有するディスク又はリングを含有する(Haring他、2005)。本体は複雑なコアを包み込む二層から構成され、核タンパク質フィラメントがコンパクトに本体の内部にパッケージされている。DNAパッケージング機構は複雑であると思われる。
【0097】
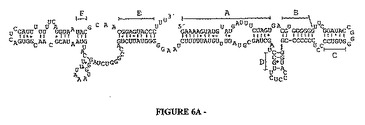
ゲノムDNAのパッケージングは、様々なバクテリオファージや直鎖状ゲノムを有する真核性ウイルスについて研究されている。その機構は、予備形成されたプロカプシド中に長鎖DNA分子をトランスロケートするのに、一対の非キャプシドタンパク質とエネルギー源ATPの関与といった、幾つかの共通の特徴を共有する(Guo他により概説、2005)。φ29パッケージング機構の必須成分は、プロカプシドとATPに結合しそしてパッケージングタンパク質と協力することによりDNAトランスロケーションに積極的に参加する174 nt RNAのpRNAである(Guo、2005)。pRNAはφ29ゲノムの一端のITRに隣接してコードされ、それは高レベルの二次構造を示し、そして保存された二次構造モチーフは全ての既知φ29関連バクテリオファージを形成する(Meijer他により概説、2001)。更に、ITRに隣接する対応領域も、アデノウイルスDNAのパッケージングに重要であることが示された(Grable & Hearing, 1990)。ABVゲノム中の対応領域の調査は、左ITRに隣接した、転写解読枠を欠いている600 bp領域を明らかにし、その領域は中心部が比較的G+C豊富であった。200 bpのG+C豊富配列の推定二次構造は、バクテリオファージφ29からのpRNA及びCP-1のものと高い相同性を示す(図6)。A〜Fと記された7つのヘリックスがバクテリオファージの全てのpRNA中で高度に保存されている。違いはヘリックスFの左側の領域中にのみ存在し、そこには推定ABV RNAでは追加のヘアピンループが存在し、一方でバクテリオファージpRNAでは小さなループが存在する(図6)。ABV RNAの転写は、該要素の20 bp上流に置かれたプロモーター様配列ATTTAATの所で開始される可能性がある。保存されたゲノム位置、類似の二次構造、高いG+C含量、及び推定プロモーターの存在はいずれも、この非コード領域が恐らくウイルスDNAパッケージングに関与するであろうRNA分子をコードすることを強く暗示する。
【0098】
φ29のDNAパッケージングに関与する別の重要な成分は、該DNAをプロヘッドにトランスロケートするために回転すると提唱されたコネクターである(Meijer他、2001)。ABVの一般的形態学はφ29のそれとは異なるけれども、「ストッパー」はφ29のコネクターと類似しており、それもまたボトルネック形状を有し、その広い方の末端は同じくプロヘッドの内側に埋没している(Meiyer他、2000)。更に、該ストッパーの広い方の末端は核タンパク質フィラメントに連結される(Haring他、2005)。従って、コネクターもABVのパッケージングに関係するのかもしれない。
【0099】
現在、直鎖状DNAを有する古細菌ウイルスのパッケージングについては殆ど知られていない。人は、クレンアーキオータ(クレン古細菌)門ルジウイルス(rudivirus)と糸状ウイルスが、1〜3つのタンパク質を含有する、長く且つ「直鎖状」の構造(棒状又は糸状)に配列されたスーパーコイル状ゲノムDNAを有する方がよりたやすいと推測する傾向にある。従って、それらは予備形成された構造にゲノムDNAをパッケージングしないように思われる。よりコンパクトな構造と特に非常に長い直線状ゲノムを有するABVとPSVについては、それらが包括的カプシド化又はパッケージング機構を必要とすると推測するだろう。
【0100】
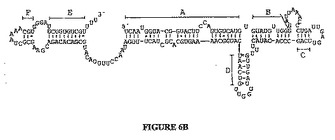
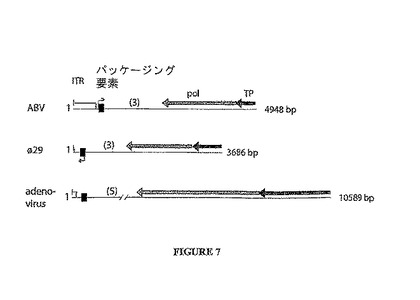
ABV、バクテリオファージφ29及び真核性アデノウイルスの左端の所のゲノム内容物を図7に示し、その3種のウイルス間での高い類似性を示す。ABVはタンパク質感作DNAポリメラーゼを含むことが報告された最初の古細菌ウイルスである。生命に係る重大な3領域からの3つの形態学的に異なるウイルス中の該ポリメラーゼの存在は、タンパク質感作DNA複製機構が古代のものであり、恐らく生命に係る3領域の分岐よりも以前に存在していたことを強力に指摘する。
【図面の簡単な説明】
【0101】
【図1】3%酢酸ウラニルでのネガティブ染色後のABV粒子の電子顕微鏡写真。目盛:100 nm。
【0102】
【図2】そのままのウイルスDNA(左側パネル)及び制限酵素消化したウイルスDNAをアガロースゲル中で泳動することによるゲノムサイズの推定。レーン1:そのままのウイルスDNA;レーン2と3:それぞれAgeI及びAflIIで消化したDNA;M1:New England BiolabsからのλDNA−モノカット混合物サイズマーカー;M2:AmershamからのラッダーDNAサイズマーカー。
【0103】
【図3】2本のDNA鎖上に存在する推定遺伝子の位置及びサイズを示すABVのゲノム地図。大部分の遺伝子が右向きの矢印により示される一方の鎖上に表示され、そして少数の遺伝子が左向きの矢印により示される相補鎖上に表示される。黒色の矢印は、機能が特定されたORFを示し、仮定の遺伝子は灰色の矢印により示される。3つの内部ORFは白抜きの矢印により指摘され、それらのサイズは括弧内に示される。この地図はMacPlasmap 2.05及びAdobe Illustratorを使って作製した。
【0104】
【図4】全て既知のタンパク質感作DNAポリメラーゼ配列に特異的である2つの挿入断片(TPRI及びII)を示す、ORF653(配列番号1)とPhi29 DNAポリメラーゼ〔DNAポリメラーゼB型ファミリーに相当する配列番号7(Genbank受入番号1XI1B)から入手〕との配列の整列。エキソヌクレアーゼ活性(Exo I, II, III)及び重合(Pol I, IIa, IIb, III及びIV)に関係する保存されたモチーフが示される。数字は配列間のアミノ酸長さを示す。
【0105】
【図5】図5AはE.コリからのORF653によりコードされる組換えポリメラーゼの精製。レーン1:タンパク質サイズマーカー;2:誘導された細胞の全粗製物;3:音波処理と遠心後の上清;4:Ni-NTAアガロース樹脂へのHis標識タンパク質の結合後の素通り液;5:樹脂カラムの洗浄液;6:精製済タンパク質。マーカー中のポリペプチドのサイズ(kD)が左側に示される。2つの矢印はそのままの(上側)及び断片化した(下側)ポリメラーゼの位置を示す。 図5Bはポリメラーゼアッセイを示す。反応中のポリメラーゼの濃度が最上部に示され、そして18 ntプライマーと伸長分子(42nt)の位置が左側に示される。
【0106】
【図6】図6AはABVパッケージングに関係する推定RNA要素の二次構造を示す。 図6Bはphi29のプロヘッドRNAの二次構造を示す。バクテリオファージpRNA中に保存されている7つのヘリックスは、5′末端から3′末端の方にA〜Fで表示され、一方で最初の命名は幹の長さに対応して行われた(Bailey他、1990)。
【0107】
【図7】ABD、phi29及びアデノウイルス(5型)の左端におけるゲノム含有物の描写。ITRの長さはそれぞれ580、6及び103 bpである。黒色ボックスはパッケージングに関与する領域を表し、一方ABVとphi29についての転写方向が小さい矢印により示される。ポリメラーゼ(pol)と末端タンパク質(TP)をコードする遺伝子は、それぞれ薄灰色と濃灰色の矢印により与えられる。polとパッケージング要素の間に存在するORFの数が括弧内に示される。
【0108】
参考文献
Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller & D.J. Lipmann. 1997. “Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.” Nucl. Acids Res. 25:3389-3402.
Arnold, H.P., W. Zillig, U.Ziese, I. Holz, M. Crosby, T. Utterback, J.F. Weidmann, J. Kristjansson, H.P. Klenk, K.E. Nelson & C.M. Fraser. 2000. “A novel lipothrixvirus, SIFV, of the extremely thermophilic crenarchaeon Sulfolobus.” Virology 267:252-266.
Barthelemy, I., M. Salas & R.P. Mellado. 1986. “In vitro transcription of bacteriophage φ29 DNA: transcription initiation sites.” J. Virol. 60:874-879.
Bettstetter, M., X. Peng, R.A. Garrett & D. Prangishvili. 2003. “AFV1,a novel virus infecting hyperthermophilic archaea of the genus Acidianus.” Virology 315:68-79.
【0109】
Blum, H., W. Zillig, S. Mallock, H. Domday & D. Prangishvili. 2001. “The genome of the archaeal virus SIRV1 has features in common with genomes of eukaryal viruses.” Virology 281:6-9.
Blanco L., Lazaro J.M., de Vega M., Bonnin A., Salas M. “Terminal protein-primed DNA amplification.” Proc. Natl. Acad. Sci. USA 1994 Dec 6; 91(25):12198-202.
Bois F., Barroso G., Gonzalez P., Labarere J. 1999. “Molecular cloning, sequence and expression of Aa-polB, a mitochondrial gene encoding a family B DNA polymerase from the edible basidiomycete Agrocybe aegerita.”Mol. Gen. Genet. 261:508-513.
【0110】
Bravo, A., B. Illana & M. Salas. 2000. “Compartmentalization of phage φ29 DNA replication : interaction between the primer terminal protein and the membrane-associated protein p1.” EMBO J. 19:5575-5584.
Brugger, K., P. Redder & M. Skovgaard. 2003. “MUTAGEN: Multi User Tool for Annotating GENomes.” Bioinformatics 19:2480-2481.
Dufour, E., J. Mendez, J.M. Lazaro, M. de Vega, L. Blanco & M. Salas. 2000. “An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein.” J. Mol. Biol. 304:289-300.
Guo.P. 2005. “Bacterial virus phi29 DNA-packaging motor and its potential applications in gene therapy and nanotechnology.” Methods Mol. Biol. 300:285-324.
【0111】
Grable M., Hearig P. 1990. “Adenovirus type 5 packaging domain is composed of a repeated element that is functionally redundant.” J. Viol. 64:2047-56.
Hatfield, L. & P. Hearing. 1993. “The NFIII/OCT-1 binding site stimulates adenovirus DNA replication in vitro and is functionally redundant with adjacent sequences.” J. Virol. 67:3931-3939.
Haring, M., X. Peng, K. Brugger, R. Rachel, K.O. Stetter, R.A. Garrett & D. Prangishvili. 2004. “Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae.” Virology 323:233-242.
【0112】
Haring M., R. Rachel, X. Peng, R.A. Garrett & D. Prangishvili 2005a. “Diverse viruses in hot spring of Pozzuoli, Italy, including a unique bottled-shaped archaeal virus ABV from a new family, the Ampullaviridae.” J. Virol. 79:9904-9911.
Haring, M., Vestergaard, G., Brugger, K., Rachel, R,m Garrett, R.A. & Prangishvili, D. 2005b. “Structure and genome organization of AFV2, a novel filamentous archaeal virus with unusual terminal structures.” J. Bacteriol. 187:3855-3858.
Haring, M.他 Nature 印刷中。
【0113】
Janekovic, D., S. Wunderl, I. Holtz, W. Zillig, A. & H. Neumann. 1983. TTV1, TTV2 and TTV3, a family of viruses of the extremely thermophilic, anaerobic sulfur reducing archaebacterium Thermoproteus tenax.” Mol. Gen. Genet. 192:39-45.
Kim, E.K., J.H. Jeong, H.S. Youn, Y.B. Koo & J.H. Roe. 2000. “The terminal protein of a linear mitochondrial plasmid is encoded in the N-terminus of the DNA polymerase gene in white-rot fungus Pleurotus ostreatus. Curr. Genet. 38:283-290.
Martin, A.C., R. Lopez & P. Garcia. 1996. “Analysis of the complete nucleotide sequence and functional organization of the genome of Streptococcus pneumoniae bacteriophage Cp-1.” J. Virol. 70:3678-3687.
【0114】
Meijer, W.J., J.A. Horcajadas & M. Salas. 2001. Phi29 family of phages.” Microbiol. Mol. Biol. Rev. 65:261-287.
Peng, X., H. Blum, Q. She, S. Mallok, K. Brugger, R.A. Garrett, W. Zillig & D. Prangishvili. 2001. “Sequences and replication of genomes of the archaeal rudiviruses SIRV1 and SIRV2: Relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology 291:226-234.
Peng, X. A. Kessler, H. Phan, R.A. Garrett & D. Prangishvili. 2004. “Multiple variants of the archael DNA rudivirus SIRV1 in a single host and a novel mechanism of genome variation.” Mol. Microbiol. 54:366-375.
Picardeau, M., J.R. Lobry & B.J. Hinnebusch. 1999. “Physical mapping of an origin of bidirectional replication at the centre of the Borrelia burgdorferi linear chromosome.” Mol. Microbiol. 32:437-445.
【0115】
Prangichvili, D., K.M. Stedman & W. Zillig, 2001. “Viruses of the extremely thermophilic archaeon Sulcolobus.” Trends Microbiol. 9:39-42.
Prangishvili, D. & Garrett R.A., 2005. “Vuruses of hyperthermophilic Crenarchaea.” Trends Microbiol. 13:535-542.
Rachel, R., M. Bettstetter, B.P. Hedlund, M. Haring, A. Kessler, K.O. Stetter & D. Prangishvili. 2002. “Remarkable morphological diversity of viruses and virus-like particles in terrestrial hot environments.” Arch. Virol. 147:2419-2429.
Rodriguez, I., J.M. Lazaro, L. Blanco, S. Kamtekar, A.J. Berman, J. Wang, T.A. Steitz, M. Salas & M. de Vega. 2005. “A specific subdomain in phi29 DNA polymerase confers both processivity and strand-displacement capacity.” Proc. Natil. Acad. Sci. USA. 102:6407-12.
【0116】
Salas, M. 1991. “Protein-priming of DNA replication.” Annu. Rev. Biochem. 60:39-71.
Savilahti H. & D.H. Bamford. 1987. The complete nucleotide sequence of the left very early region of Escherichia coli bacteriophage PRD1 coding for the terminal protein and the DNA polymerase.” Gene 57:121-130.
Takeda M., H. Hiraishi, T. Takesako, S. Tanase & N. Gunge. 1996. “The terminal protein of the linear DNA plasmid pGKL2 shares an N-terminal domain of the plasmid-encoded DNA polymerase.” Yeast 12:241-246.
Torarinsson, E., H.-P. Klenk & R.A. Garrett. 2005. “Divergent transcriptional and translational signals in Archaea.” Environ. Microbiol. 7:47-54.
【0117】
Vestergaard, G., M. Haering, X. Peng, R. Rachel, R.A. Garrett & D. Prangishvili. 2005. ARV1, a rudivirus infecting the hyperthermophilic archaeal genus Acidianus.” Virology, 336:83-92.
Zillig, W., D. Prangishvilli, C. Schleper, M. Elferink, I. Holz, S. Albers, D. Janekovic & D. Gotz. 1996. “Viruses, plasmids and other genetic elements of thermophilic and hyperthermophilic Archaea.” FEMS Microbiol. Rev. 18:225-236.
【技術分野】
【0001】
本発明は、古細菌アンプラウイルスABV(Acidianus Bottle-shaped Virus)の熱安定性DNAポリメラーゼタンパク質及び前記DNAポリメラーゼをコードする核酸に向けられる。本発明は、前記DNAポリメラーゼタンパク質をコードする核酸の合成、増幅又は配列決定方法、並びに前記DNAポリメラーゼタンパク質を含んで成るキット又は装置にも関する。
【背景技術】
【0002】
超好熱性菌クレン古細菌(Crenarchaeota)の二本鎖(ds)DNAウイルスは、著しく多様な形態とゲノム構造を示し、それらの特徴に基づいて、既に6つの新規ウイルスファミリーに分類されている:紡錘形フセロウイルス科(Fuselloviridae)、繊維状リポスリクスウイルス科(Lipothrixdviridae)、棒状ルディウイルス科(Rudiviridae)、液滴状グタウイルス科(Guttaviridae)、球状グローブロウイルス科(Globuloviridae)及び二本の鞭毛を持つビコウダウイルス科(Bicaudaviridae)(Prangishvili他、2001に概説;Prangishvili & Garrett, 2004, 2005)。最近、ユニークな瓶形の形態を有する新種ウイルスが発見され、新ファミリーであるアンプラウイルス科(Ampullaviridae)に指定された(Haring他、2005a)。
【0003】
核酸分析及び核酸増幅のための手段として開発された様々な核酸増幅技術が、遺伝病や感染症の臨床診断に好結果に利用されている。増幅技術は温度循環を必要とするもの(PCRとリガーゼ連鎖反応)と等温系のもの〔増幅系(3SRやNASBA)、鎖置換増幅法、及びQβ複製系〕に分類することができる。次の二点がそれらの手法におけるよくある警告である:合成の忠実性と増幅生成物の長さ。
【0004】
ファージphi29(φ29)のDNA複製機構に頼る増幅方法の開発が刊行物や特許文書の主題となっている(Dean他、Genome Res. 2001 Jun, 11(6):1095-9 ; Mendez他、EMBO J., 1997, 1, 16(9):2519-27 ; Hutchison他、Proc. Natl. Acad. Sci. USA, 2005, 102(48):17332-6 ; Mamone, Innovations Forum: GenomiPhi DNA amplification, Life Sciences News 14, 2003 Amersham Biosciences; Blanco他、1994, EP 0 862 656又はUS 5,001,050)。
【0005】
phi29 DNAポリメラーゼは、非常に効率的な等温系DNA増幅を可能にする、強力な鎖置換活性を特徴とする高処理能力ポリメラーゼである(Blanco他、Proc. Natl. Acad. Sci. USA, 81, 5325-5329, 1984及びJ. Biol. Chem., 264, 8935-8940, 1989)。phi29 DNAポリメラーゼは一本鎖DNA上で優先的に作用する3′→5′エキソヌクレアーゼ(校正)活性も有する(Garmendia,J. Biol. Chem., 267, 2594-2599, 1992)。
【0006】
上記特徴の中で、既知DNAポリメラーゼのうち最高の処理能力と鎖置換活性、即ち、70kb以上の長鎖DNA片を合成できること(Blanco他、1989)、その高度に正確なDNA合成(Esteban他、J. Biol. Chem., 268, 4, 2719-2726, 1993)、微量の鋳型からであっても高収率の増幅DNAが得られること、及び増幅生成物が下流用途(PCR、制限消化、SNP遺伝子型分析、他)に直接利用できることを言及することができる。この特定のDNAポリメラーゼに関して多数の特別な用途が発見された。例えば、ローリングサークル増幅(RCA)(Lizardi他、Nat. Genet., 19, 225-232, 1998 ; Dean他、Genome Res., 11, 1095-1099, 2001 ; Baner他、Nucleic. Acids Res., 26, 5073-5078, 1998)。多重置換増幅(MDA)(Dean他、Proc. Natl. Acad. Sci. USA, 99, 5261-5266, 2002)、全ゲノムの不偏増幅又は配列決定用のDNA鋳型調製。この系は、今日利用できる増幅系を使って得られるサイズ限界を大幅に上回る、70 kbより長いDNA分子の正確な増幅(Blanco他、1989)に適しているだろう。この等温TP感作増幅(“TP”は末端タンパク質)方法は、phi29 DNAポリメラーゼの特定性質:(i)プライマーとしてタンパク質を使用できること;(ii) 本質的な高処理能力(>70 kb);及び(iii) DNA合成と協同した鎖置換、を有効に利用する。このphi29 DNAポリメラーゼの特異活性は30℃の温度で発揮され、そしてこのphi29 DNAポリメラーゼは65℃で不活性化すると推測される。
【発明の開示】
【発明が解決しようとする課題】
【0007】
30℃より有意に高い温度で作用でき、且つ60℃で完全には不活性化されない、phi29 DNAポリメラーゼのようなタンパク質感作型DNAポリメラーゼファミリーに属する新規DNAポリメラーゼが現在必要とされている。
これが本発明の目的である。
【課題を解決するための手段】
【0008】
ウイルスABV(Acidianus Bottled-shaped Virus)感染アシディアヌス(Acidianus)属好熱性古細菌の全ゲノム配列を配列決定しそして確認した後、本発明者らはDNA依存性DNAポリメラーゼをコードする核酸配列を発見した。驚くべきことに、該タンパク質配列の分析は、それがタンパク質感作型DNAポリメラーゼファミリーに属することを示した。該DNAポリメラーゼ遺伝子がE.コリ(E. coli;大腸菌)中で異種発現され、該組換えタンパク質のDNA重合活性が確認された。この新規酵素は、既知のウイルスDNAポリメラーゼと同様、高処理能力を有し、自給自足型で、補助タンパク質を必要としない。それらの特徴のため、該酵素はPCRによるDNA増幅のための手段として有利に利用することができる。タンパク質感作される熱安定性ウイルス酵素であるため、それは一本鎖又は二本鎖直線状DNAの指数的増幅(即ちAmersham社により開発されたGenomiPhi法による)において、現在この方法で利用されている中温性タンパク質感作酵素のバクテリオファージPhi29 DNAポリメラーゼよりもずっと有効であり得る。Amersham社のGenomiPhi増幅キットは、少数の細胞又は限定量の貴重な試料から、無制限のDNA試験を実施できるようにし、全ゲノムを典型的に増幅する容易なゲノムDNA増幅法である。
【発明を実施するための最良の形態】
【0009】
よって、第一の観点において、本発明は、
a) 配列番号1のアミノ酸配列を有するポリペプチド;
b) DNAポリメラーゼ活性を有するa)の断片;
c) a)の前記DNAポリメラーゼのDNAポリメラーゼ活性を有する、配列番号1断片を少なくとも含んで成るキメラポリペプチド;
d) 配列番号1のアミノ酸配列を有するポリペプチドに比較して有意に低い検出可能なエキソヌクレアーゼ活性を有するか又は全く持たないDNAポリメラーゼポリペプチドを生じるように、図4に示されるようなエキソヌクレアーゼ部位ExoI, Exo II及びExoIIIが変異又は削除されている、配列番号1のアミノ酸配列を有するポリペプチド;
e) 配列番号1の配列又はb)〜d)に定義したような配列との最適整列後に少なくとも80%同一である配列を有するポリペプチドであって、好ましくは50℃の温度で又は50℃より高い温度でDNAポリメラーゼ活性を有するポリペプチド;
から成るポリペプチド群より選択された、単離されたDNAポリメラーゼに向けられる。
【0010】
好ましい態様では、前記DNAポリメラーゼ活性を有する断片は、少なくとも50,100,150,200,250,300,350,400,450,500又は600アミノ酸を有する。
【0011】
好ましい態様では、本発明に係るDNAポリメラーゼは、ABVから又はDBAポリメラーゼをコードするABV遺伝子から単離される。
【0012】
より好ましい態様では、本発明のDNAポリメラーゼは、少なくとも図4に示されるような配列番号1のPol I, Pol IIa, Pol IIb, Pol III及びPol IV断片を含んで成る。
【0013】
図4に関して、DNAポリメラーゼ活性は保存されているが、配列番号1の配列を有するポリペプチドよりも欠損した又は有意に低いエキソヌクレアーゼ活性を有する本発明のポリペプチドは、別のポリメラーゼとのアミノ酸配列相同性及びDNAポリメラーゼのエキソヌクレアーゼ活性を減少させることが知られているそれらの変異(Derbyshire他、Science, 1988, Apr 8, 240(4849):199-201)を考慮することにより、選択することができる。一般に、図4中Exo I, Exo II及び/又はExo IIIとして示されている位置のアミノ酸を、削除するか又は別のアミノ酸と置換することができる。それらのExo I, Exo II及びExo III位置でのアミノ酸の大型除去又は多重置換も実施できる。配列番号1の配列を有するポリペプチドを変異誘発させた後、エキソヌクレアーゼ活性レベルを測定し、そしてDNAポリメラーゼ活性の量を求め、それが本発明における使用に十分であることを確認する。
【0014】
用語「5′エキソヌクレアーゼ活性」とは、オリゴヌクレオチドの5′末端からヌクレオチドを除去することができるタンパク質の活性の存在を言う。5′エキソヌクレアーゼ活性は本明細書中に提供されるアッセイのいずれかを使って測定することができる。
【0015】
本発明のDNAポリメラーゼは、そのポリメラーゼのエキソヌクレアーゼ活性を減少させるように遺伝的に改変されているポリペプチド、並びに実質上同一の(少なくとも80%の同一性)天然に存在するABV DNAポリメラーゼ又はその改変ポリメラーゼであるもの、又は上記に列挙された同等の酵素へと遺伝的に改変されているポリペプチドを包含する。それらの酵素の各々は、ABV DNAポリメラーゼのものと同様な性質を有するように改変することができる。該酵素をABVウイルス感染細胞から直接単離することもできるが、好ましくはそれを過剰生産(組換え発現)する細胞から単離する。
【0016】
用語「エキソヌクレアーゼ活性」とは、或るオリゴヌクレオチドの3′末端又は5′末端からヌクレオチドを除去することができるタンパク質の活性の存在を言う。そのようなエキソヌクレアーゼ活性は当業者により周知である任意のエキソヌクレアーゼ活性アッセイを使って測定することができる。
【0017】
用語「DNAポリメラーゼ活性」とは、デオキシヌクレオシド三リン酸の取り込みにより新規DNA鎖を合成する酵素的ポリペプチドの能力を言う。下記実施例4は、DNAポリメラーゼ活性の測定のためのアッセイの一例を提供する。そのようなDNAポリメラーゼ活性は、当業者に周知のDNAポリメラーゼ活性アッセイのいずれかを使って測定することができる。鋳型依存性方式でのデオキシヌクレオシド三リン酸の取込みにより新規DNA鎖の合成(DNA合成)を指令することができるタンパク質は、「DNAポリメラーゼ活性を発揮できる」と言われる。
【0018】
本明細書では、ポリペプチド、ポリペプチド配列、ペプチド及びタンパク質という用語は相互交換可能である。
【0019】
2以上のポリペプチド配列の概念において、「同一」又は「同一性」%という語は、規定値(デフォルト)パラメーターを用いるBLASTもしくはBLAST 2.0配列比較演算法を使って、又は手法整列及び目視検査(例えばNCBIウエブサイトを参照のこと)により測定した時に、同一であるか又は特定比率の同一アミノ酸(即ち、比較ウィンドウまたは指定領域に渡り最大一致について比較・整列した時に、或る特定領域に関して約80%同一性、好ましくは85%、90%、95%、98%、99%もしくはそれより高い同一性)を有する2以上の配列又は部分配列のことを言う。この定義は、欠失及び/又は付加を有する配列、並びに置換を有する配列も包含する。後述するように、好ましい演算法はギャップ等を明らかにすることができる。好ましくは、少なくとも長さ約25アミノ酸である領域、より好ましくは長さ25〜75アミノ酸である領域に渡り同一性が存在する。
【0020】
配列比較では、典型的には、或る1つの配列が比較参照配列として働き、それに対して試験配列が比較される。配列比較演算法を使った場合、試験配列と参照配列をコンピューターに入力し、必要であれば部分配列候補をデザインし、そして配列演算プログラムパラメーターを設定する。好ましくは、規定値プログラムパラメーターを使用することができ、又は代替パラメーターを設定することができる。次いで配列比較演算法がそのプログラムパラメーターに基づいて参照配列に対する試験配列の配列同一性を算出する。
【0021】
比較のための配列の整列方法は当業者に周知である。配列同一性%及び配列相同性を求めるのに好ましい演算法の例はBLAST及びBLAST 2.0演算法であり、それらはAltschul他、Nuc. Acids Res. 25:3389-3402 (1977) 及びAltschul他、J. Mol. Biol. 215:403-410 (1990) に記載されている。
【0022】
例えば、サイトhttp://www.ncbi.nlm.gov/gorf/bl2.html上で入手可能なBLASTプログラム“BLAST 2配列”(Tatusova他、“Blast 2 sequences - a new tool for comparing protein and nucleotide sequences”, FEMS Microbiol Lett. 174:247-250)を使うことが可能であり、使用されるパラメーターは、規定値(デフォルト)により与えられたものであり(特に“オープンギャップペナルティ”:5、“伸長ギャップペナルティ”:2;選択されるマトリクスは、該プログラムにより提唱されたマトリクス“BLOSUM 62”である)、比較される2つの配列間の同一性%は該プログラムにより直接算出される。
【0023】
参照アミノ酸配列に対して少なくとも80%、好ましくは85%、90%、95%、98%、99%又はそれより高い同一性を有するアミノ酸配列とは、参照配列に関して、ある種の変更、特に少なくとも1つのアミノ酸の欠失もしくは付加又は置換、先端切除又は伸長を有するものが好ましい。1又は複数の保存的もしくは非保存的アミノ酸置換の場合、置換されるアミノ酸が「等価の」アミノ酸により置換される置換が好ましい。「等価アミノ酸」という表現は、本明細書中では、参照ポリペプチドのDNAポリメラーゼ活性を本質的に改変することなく、基本構造のアミノ酸の1つにより置換されることができる任意アミノ酸を示し、そしてそのようなアミノ酸は下記、特に実施例4の最終段落において定義されるだろう。
【0024】
このような等価アミノ酸は、それらが置き換わるアミノ酸との構造的相同性を頼みにするか、別のポリペプチド間でのDNAポリメラーゼ活性の比較試験の結果を頼みにすることにより、決定することができる。一例として、対応する改変されたポリペプチドのDNAポリメラーゼ活性の顕著な変更を引き起こすことなく実施することができる置換の可能性が挙げられる。従って、ロイシンをバリン又はイソロイシンに置換、アスパラギン酸をグルタミン酸に置換、グルタミンをアスパラギンに置換、アルギニンをリジンに置換することが可能であり、その逆の置換も同条件下で普通に予想できる。
【0025】
よって、第二の観点では、本発明は、本発明に係るDNAポリメラーゼポリペプチドをコードする核酸、特に配列番号2の配列を有するか又は最適整列後に配列番号2の配列と少なくとも80%同一性である配列を有する核酸を提供し、前記核酸によりコードされるポリペプチドは、好ましくは50℃又は50℃より高い温度でDNAポリメラーゼ活性を有する。
【0026】
本明細書中では、核酸、ポリヌクレオチド、オリゴヌクレオチド、又は核酸配列もしくはヌクレオチド配列という語は相互交換可能に用いられる。
【0027】
別の観点では、本発明は、本発明の核酸を含んで成るベクター、好ましくはクローニング又は発現ベクターに関する。
好ましい態様では、本発明に係るベクターは、前記核酸がプロモーターに作用可能に連結されていることを特徴とする。
【0028】
本発明は特に、本発明に係るヌクレオチド配列を含有するクローニング及び/又は発現ベクターを目的とする。
【0029】
本発明に係るベクターは、好ましくは、所定の宿主細胞中でのヌクレオチド配列の発現及び/又は分泌を可能にする要素を含有する。従って、該ベクターは、プロモーター、翻訳開始及び終結のシグナル、並びに適当な転写調節領域を含有しなければならない。それは宿主細胞中で安定した方法で維持され、そして場合により翻訳されたタンパク質の分泌を指令する特定シグナルを有することができる。それらの種々の要素は、用いられる宿主の関数として当業者により選択されそして最適化される。この結果により、本発明に係るヌクレオチド配列は、選択された宿主中で自律複製ベクター中に挿入することができるか、又は選択された宿主の組込み型ベクターであることができる。
【0030】
そのようなベクターは当業者により現在使用されている方法により調製され、そして生じたクローンは標準法、例えばリポフェクション、エレクトロポレーション、熱ショック法又は化学的方法により、適当な宿主に導入することができる。
【0031】
本発明に係るベクターは、例えば、プラスミド又はウイルス起源のベクターである。それらは、本発明に係るヌクレオチド配列をクローニングするため又は発現させるために宿主細胞を形質転換せしめるのに有用である。
【0032】
好ましい態様では、本発明のベクターは、ブタペスト条約に従って2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(Collection Nationale de Cultures de Microorganismes, フランス国パリのパスツール研究所)に寄託されている細菌中に含まれるプラスミドベクターである。
【0033】
このクローン化プラスミドベクターは、本発明のDNAポリメラーゼの核酸配列がpET30aプラスミドのNdeI部位とXbaI部位の間に挿入されているベクターpET30aである。
【0034】
用語「発現ベクター」とは、所望の核酸コード配列と特定の宿主細胞中での作用可能に連結された該コード配列の発現に必要な適当な核酸配列とを含有する組換えDNA分子を言う。原核生物中での発現に必要な核酸配列は、一般に、しばしば別の配列と共に、プロモーター、オペレーター(任意)、及びリボソーム結合部位を含んで成る。真核細胞はプロモーター、エンハンサー、並びに終止及びポリアデニル化シグナルを利用することが知られている。
【0035】
別の観点では、本発明は、本発明に係るベクターを含んで成る宿主細胞、特にブタペスト条約に従って2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(Collection Nationale de Cultures de Microorganismes, フランス国パリのパスツール研究所)に寄託されている組換え細菌に関する。
【0036】
本発明のDNAポリメラーゼポリペプチドは、原核又は真核宿主細胞のいずれにおいても発現させることができる。本発明のDNAポリメラーゼポリペプチドをコードする核酸は、塩化カルシウム処理により形質転換に許容性にされた細菌細胞の形質転換又はエレクトロポレーションをはじめとする多数の手法により、細菌宿主細胞中に導入することができる。本発明のDNAポリメラーゼポリペプチドを真核宿主細胞中で発現させるつもりであれば、本発明のDNAポリメラーゼポリペプチドをコードする核酸を、リン酸カルシウム共沈、スフェロプラスト融合、エレクトロポレーション等といった多数の手法により、真核宿主細胞中に導入することができる。真核宿主細胞が酵母細胞である時、形質転換は酢酸リチウムによる宿主細胞の処理又はエレクトロポレーションにより、あるいは当業界で既知の任意の別法により、実施することができる。本発明のペプチドもしくはタンパク質又はそれらの断片を生産するのに任意の宿主細胞が有用であると予想される。
【0037】
本発明に従って形質転換された細胞は、本発明の組換えポリペプチドの調製方法において使用することができる。本発明のベクター及び/又はベクターにより形質転換された細胞を使用することを特徴とする、組換え形での本発明ポリペプチドの調製方法は、それ自体本発明に含まれる。
【0038】
好ましくは、本発明のベクターにより形質転換された細胞を、前記ポリペプチドの発現を可能にする条件下で培養し、そして前記組換えポリペプチドを回収する。
【0039】
別の観点では、本発明は、DNAポリメラーゼの生産方法であって、
(a) 前記核酸の発現に適当な条件下で本発明の宿主細胞を培養する段階;及び
(b) 前記宿主細胞から前記DNAポリメラーゼを単離する段階
を含んで成る方法に関する。
【0040】
前記宿主細胞は原核細胞または真核細胞であることができる。
【0041】
既に言及したように、宿主細胞は原核系又は真核系から選択できる。特に、そのような原核系又は真核系における分泌を促進するヌクレオチド配列を使用することができる。従って、そのような配列を担持する本発明に係るベクターは、分泌させようとする組換えタンパク質の生産に有意に利用することができる。実際に、それらの組換えタンパク質が宿主細胞の内側よりもむしろ細胞培養物の上清中に存在するという事実により、着目のそれらの組換えタンパク質の精製が容易になるだろう。
【0042】
別の観点では、本発明は、二本鎖DNA分子を合成する方法であって、
(a) 第一のDNA分子にプライマーをハイブリダイズさせる段階;及び
(b) 段階(a)の前記DNA分子を1又は複数のデオキシリボヌクレオシド三リン酸又はその類似体及び本発明のポリペプチドの存在下で、前記第一のDNA分子の全部又は一部に相補的な第二のDNA分子を合成するのに十分な条件下でインキュベートする段階
を含んで成る方法に関する。
【0043】
別の観点では、本発明は、一本鎖DNA分子を合成する方法であって、
(a) 本発明の方法により二本鎖DNA分子を合成する段階;
(b) 段階(a)で得られた二本鎖DNA分子を変性させる段階;及び
(c) 段階(b)で得られた一本鎖DNA分子を回収する段階
を含んで成る方法に関する。
【0044】
別の観点では、本発明は、長さ10キロ塩基より大きいDNA分子の生産方法であって、
段階(a)における鋳型として働く第一のDNA分子が10キロ塩基より大きい、本発明に係る方法を含んで成る方法に関する。
【0045】
本発明に係る方法において、前記デオキシリボヌクレオシド三リン酸はdATP, dCTP, dGTP及びdTTPから成る群より選択される。
【0046】
別の観点において、本発明は、二本鎖DNA分子を増幅させる方法であって、
(a) 第一及び第二プライマーを用意し、ここで前記第一プライマーは前記DNA分子の第一鎖の3′末端の所又はその付近の配列に相補的であり、そして前記第二プライマーは前記DNA分子の第二鎖の3′末端の所又はその付近の配列に相補的である段階;
(b) 前記第一鎖に相補的な核酸と前記第二鎖に相補的な核酸が合成されるような条件下で、本発明に係るポリペプチドの存在下で前記第一プライマーを前記第一鎖にそして前記第二プライマーを前記第二鎖にハイブリダイズせしめる段階;
(c) 前記第一鎖とその相補鎖:及び
前記第二鎖とその相補鎖
を変性させる段階;及び
(d) 段階(a)から(c)を1回又は複数回繰り返す段階
を含んで成る方法に関する。
【0047】
前記増幅段階が、DNAポリメラーゼ活性を有するポリペプチド(DNAポリメラーゼポリペプチド)を包含するPCR、PCR様法又はRT−PCR反応により実施されることも好ましい。
【0048】
「PCR」とは、DNA試料中の特定遺伝子への、プライマーの配列を基準としたハイブリダイゼーション、それに続く熱安定性ポリメラーゼを使ったアニーリング(ハイブリダイゼーション)、伸長及び変性の反復を伴う増幅のことを言う。
【0049】
「RT−PCR」は逆転写ポリメラーゼ連鎖反応の略語である。mRNAを逆転写酵素にかけると、該mRNAの塩基配列に相補的であるcDNAが生成する。次いで熱安定性DNAポリメラーゼの作用を当てにしたポリメラーゼ連鎖反応によって、多量の特定cDNAを製造することができる。
【0050】
「PCR様」とは、核酸配列の直接又は間接再生を使う全ての方法、或いは標識系が増殖されている方法を意味し、それらの技術はもちろん既知であり、一般にそれらはポリメラーゼによるDNAの増幅を伴う。元の試料がRNAである時、前もって逆転写を実施することが望ましい。この増幅を可能にする多数の方法が現存し、例えばいわゆるNASBA(核酸配列ベース増幅“Nucleic Acid Sequence Based Amplification”)、TAS(転写ベース増幅系“Transcription based Amplification System”)、LCR(リガーゼ連鎖反応(“Ligase Chain Reaction”)、ERA(エンドラン増幅“Endo Run Amplification”)、CPR(循環プローブ反応“Cycling Probe Reaction”)、及びSDA(鎖置換増幅“Strand Displacement Amplification”)があり、それらの方法は当業者に周知である。
【0051】
mRNAを用いる場合、該方法は、逆転写酵素を使った標準法(RT−PCR)に従って、単離されたmRNAをcDNAに変換することにより実施することができる。
【0052】
別の観点では、本発明は、mRNAからcDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマーまたは別の相補的プライマーと接触させてハイブリッドを形成せしめる段階;及び
(b) 段階(a)で形成した前記ハイブリッドを、本発明に係るDNAポリメラーゼポリペプチド並びにdATP, dCTP, dGTP及びdTTPと接触させ、それによりcDNA−RNAハイブリッドが得られる段階
を含んで成る方法に関する。
【0053】
本発明は更に、mRNAからdsDNA(二本鎖DNA)を調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触させてハイブリッドを形成せしめる段階;及び
(b) 段階(a)で形成した前記ハイブリッドを、本発明に係るポリペプチド、dATP, dCTP, dGTP及びdTTP、並びに前記第一鎖cDNAに相補的であるオリゴヌクレオチド又はプライマーと接触させ、そしてによりdsDNAが得られる段階
を含んで成る方法にも向けられる。
【0054】
別の観点では、本発明は、DNA分子のヌクレオチド塩基配列を決定する方法であって、
(a) 前記DNA分子を前記DNA分子にハイブリダイズできるプライマー分子と接触させる段階;
(b) 段階(a)で形成した前記ハイブリッドを、4種の異なるデオキシヌクレオシド三リン酸、本発明に係るDNAポリメラーゼポリペプチド、及び特定のヌクレオチド塩基の所でDNA合成を終結させる1又は複数のDNA合成終結剤を含有する容器の中でインキュベートする段階であって、各々の終結剤は異なるヌクレオチド塩基の所でDNA合成を終結させる段階;及び
(c) 前記インキュベーション反応のDNA生成物をサイズに従って分離する段階であって、ここでそれにより前記DNAヌクレオチド塩基配列の少なくとも一部分を決定することができる段階
を含んで成る方法に関する。
【0055】
好ましい態様では、前記終結剤がジデオキシヌクレオシド三リン酸である。
【0056】
特定のヌクレオチド塩基の所でDNA合成を終結させるDNA合成終結剤とは、2′,3′−ジデオキシ構造を有するジデオキシヌクレオシド(例えばddATP, ddCTP, ddGTP及びddTTP)化合物を言うが、それに限定されない。特定の塩基の所でDNA配列合成反応を特異的に終結させることができるどんな化合物でもDNA合成終結剤として使用できる。
【0057】
別の観点では、本発明はDNA分子の増幅方法であって、次の段階:
(a) 前記DNA分子を本発明に係るDNAポリメラーゼ活性を有するポリペプチド、古細菌ABVの末端タンパク質、及び異なるデオキシヌクレオシド三リン酸の混合物の存在下でインキュベートする段階
を含んで成る方法に関する。
【0058】
好ましい態様では、本発明に係るDNA分子の増幅方法が、前記DNA分子の一端の所に前記ABVの複製開始点を含有する断片が共有結合されていることにより特徴付けられる。
【0059】
実際、ABVはphi29のものと同様な方法で複製を開始するようであり、逆方向末端反復配列(ITR)とその周囲領域の配列が複製開始に関係するだろう。配列番号5(左側)と配列番号6(右側)の配列は、ITRを含む2つのゲノム末端の配列である。
【0060】
好ましい態様では、前記ABVの複製開始点を含む断片の配列が、配列番号5(左側)と配列番号6(右側)の配列を含んで成る。
【0061】
更に別の観点では、本発明は、DNA分子を配列決定するためのキットであって、
(a) 本発明のポリペプチドを含んで成る第一のコンテナー手段;
(b) 1又は複数のジデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段;
(c) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第三のコンテナー手段
を含んで成るキットに向けられる。
【0062】
本発明はまた、DNA分子を増幅するためのキットであって、
(a) 本発明のポリペプチドを含んで成る第一のコンテナー手段;及び
(b) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段
を含んで成るキットにも関する。
【0063】
より好ましい態様では、本発明に係るDNA分子を増幅するためのキットは、ABVゲノムのORF163(配列番号4)によりコードされる配列番号3を有するポリペプチドに相当する、古細菌アンプラウイルスABVの単離された末端タンパク質を更に含んで成る。
【0064】
本発明は、ローリングサークル増幅、多重置換増幅、又はタンパク質感作増幅法を実施するための本発明に係るポリペプチドの使用も含んで成る。
【0065】
それらの特定方法は、当業者に周知であり且つ例えば次の文献に記載されている:
等温ローリングサークル増幅法については、Lizardi他、1998;Baner他、1998;Dean他、Genome Res., 11, 1095-1099, 2001;Larsson他、Nature methods 1, 227-232, 2004;
多重置換増幅法については、Dean他、2002;及び
タンパク質感作増幅法については、Blanco他、1994。
【0066】
好ましい態様では、本発明に係る方法、本発明に係るキット又は本発明に係る使用は、本発明のDNAポリメラーゼポリペプチドが、DNAポリメラーゼ活性と欠損エキソヌクレアーゼ活性(野生型ABVのDNAポリメラーゼが通常有する該活性の少なくとも1%未満、好ましくは0.1%未満)を有するポリペプチドであることを特徴とする。
【0067】
本発明のDNAポリメラーゼポリペプチドに関するエキソヌクレアーゼ活性は、DNA配列決定、合成又は増幅反応におけるDNAポリメラーゼの使用を有意に妨害することはない。しかしながら、エキソヌクレアーゼ活性のレベルが、天然のABVに感染した細胞より単離されたDNAポリメラーゼが通常有する活性又は配列番号1の配列の活性の10%未満又は1%未満、好ましくは0.1%未満であるレベルにまで減少することが好ましい。
【0068】
本発明は、DNA配列決定又は増幅のための装置であって、本発明のDNAポリメラーゼポリペプチドを含んで成るリアクターを有する装置にも向けられる。
【0069】
本発明は、本発明の抗DNAポリメラーゼポリペプチドを生産する方法であって、免疫寛容細胞を有する動物を、本発明のポリペプチド又は本発明のポリペプチドの少なくとも抗原性部分(決定基)を含んで成る免疫原に、前記免疫寛容細胞が前記本発明のポリペプチドに対して特異的に向けられた抗体又はそのエピトープ部分を生産するような条件下で暴露することを含んで成る。一態様では、該方法が前記抗体を収集する段階を更に含んで成る。別の態様では、該方法が、ハイブリドーマが生産されるような条件下で前記免疫寛容細胞を不死化細胞系と融合せしめる段階を更に含んで成る。
【0070】
そのような抗体は、別の成分が存在する試料中の本発明ポリペプチドを精製するために特に利用することができる。
【0071】
下記実施例は、本発明の種々の態様を例示する目的で提供され、どのような形にせよ本発明を限定するものではない。
【実施例】
【0072】
実施例1:材料と方法
ヌクレオチド、プライマー及び酵素
(γ−32P)ATP、dNTP及び酵素はPharmacia社より得られた。オリゴヌクレオチドpolF1(5’-CCTCCCTATTTGATAGGC-3’、配列番号8)を、(γ−32P)ATP及びT4ポリヌクレオチドキナーゼで5′標識し、そして8M尿素−20%ポリアクリルアミドゲル上で電気泳動により精製した。標識されたpolF1をpolF1c+24(5’-AGGTAAGCATGCATCAGTTAATACGCCTATCAAATAGGGAGG-3’、配列番号9)と混合し、該混合物を重合アッセイ(下記参照)におけるプライマー鋳型DNA分子として使用した。
【0073】
ウイルスの精製及びウイルスDNAの調製
以前記載された通り(Haring他、2005a)に、イタリアのポッツオーリにあるSoffatara 火山の噴火口中の87〜93℃でpH 1.5-2の貯蔵水から採取した試料より、好酸性の濃厚培養物を調製した。それらを75℃、pH 3で増殖させた。ウイルス粒子をCsCl浮遊密度勾配中での遠心分離により精製し、室温で1時間1%(w/v)SDSで破壊した後、以前に記載された通りに(Haring他、2005a)DNAを抽出し沈澱させた。
【0074】
ゲノムDNAの配列決定
合計わずか約200 ngの精製済ウイルスDNAしか最初のプロジェクトで入手できなかったことを考慮して、GenomiPhi増幅キット(Amersham Biotech, Amersham)を使って約1 ngのDNAを試験管内で増幅させて数μgを得た。次いでpUC18のSmaI部位中にクローニングしたサイズ範囲1.5〜4 kbpの音波処理DNA断片から、ショットガンライブラリーを作製した。該ライブラリーは高度に偏性のゲノムを生成した。
【0075】
増幅ライブラリーから得られた集成体と配列を、約50 ngの元のABV DNAと好酸性Acidianus属ベータリポスリクスウイルスbetalipothrixvirus(Vestergaard他、印刷中)から抽出した1μgのDNAを使って、混合ショットガンライブラリーを調製することにより確認した。該ライブラリーは、より長鎖の音波処理DNA断片を2〜6.5 kbpの範囲でクローニングすること以外は上述した通りに調製した。PCR反応を実施して、第二ライブラリーによりカバーされなかった領域を確かめ、そして少数のクローンをプライマーウオーキング(Peng他、2001)により更に配列決定した。
【0076】
配列分析
NCBIデータベースに対してBlastP検索を実施した。SMART及びMotifScanを使って保存された領域、プロフィール又はパターンを検出した。コイルドコイル、二次構造及び膜貫通ヘリックスは、ExPASy Proteomics Tools(http://expasy.org/tools/)中のプログラムにより検出した。
【0077】
ポリメラーゼ(ORF653)のクローニング及び精製
元のウイルスDNAから、NdeIとXhoI制限部位をそれぞれ含有する2つのプライマー(5’-TATTTTTACATATGCTACAAATCCT-3’配列番号10、及び5’-TATAACTCGAGTGAGAGAATACTATTTAAGTC-3’配列番号11)を使ってORF653をPCR増幅せしめた。PCR生成物をまずpGEM-Tベクター(Promega)中にクローニングし、そしてORF653挿入断片を含有する精製プラスミドを、NdeIとXhoIで消化した。付着性NdeI及びXhoI末端を持つORF653を含有するDNA断片を、低融点アガロース(Promega)ゲル電気泳動によりpGEM-T断片から分離し、そしてゲル抽出キット(Quiagen)を使って精製した。精製断片を次いで、NdeIとXhoIで消化しそして仔牛腸ホスファターゼにより処理したpET30-aベクター中にクローニングした。生じた構成物は、ORF653の生成物のC末端の後方に6個のヒスチジン残基をコードする配列を含有する。発現宿主細胞Posetta(Novagen)中に形質転換する前に、該構成物を配列決定して挿入されたORF653の配列を確認した。
【0078】
Rosetta形質転換体の単一コロニーを、25μg/mlのカナマイシンとクロラムフェニコールを含有する5 mlのLB培地に接種し、OD値が0.5に達するまで37℃にてインキュベートした。次いで前記5 ml培養物を、同じ抗生物質を含む250 mlのLB培地に移した。0.1 mM IPTG及び1%エタノールの存在下で30℃にて一晩増殖させた後、細胞を収穫した。製造会社(Novagen)により提供されたプロトコルに従って、Ni-NTA His.Bind樹脂を使ってヒスチジン標識タンパク質を精製し、そしてSDS-PAGEにより確認した。
【0079】
重合アッセイ
ハイブリッド分子polF1/polF1c+24(前掲)は、24ヌクレオチド長さの5′突出末端を含むので、DNA重合のためのプライマー−鋳型として使用することができる。反応混合物は、10μl中に、25 mM Tris-HCl (pH 7.6)、1 mM ジチオトレイトール、10 mM MgCl2、250μMの4つのdNTPを各々250μM、0.1μMのプライマー−鋳型DNA分子、及び増加する濃度の組換えポリメラーゼを含有した。50℃で20分間インキュベートした後、5μlの負荷緩衝液(80%ホルムアミド、10 mM EDTA, 50 μg/mlのブロモフェノールブルー)を添加し、80℃にて3分間加熱することにより、反応を停止させた。試料を8 M尿素−20%PAGEとオートラジオグラフィーにより分析した。5′標識プライマー鎖(polF1)のサイズの増加として重合を検出した。
【0080】
実施例2:ゲノム配列及び組織化
核酸をABVウイルス粒子から単離し、RNase Aには不感受性であるがII型制限エンドヌクレアーゼにより消化可能であることが示され、このことはそれがdsDNAであることと一致する。入手できるのが少量のゲノムDNAであることを考えて(<200 ng精製DNA)、本発明者らは二段階ゲノム配列決定法を適用した。
【0081】
第一に、GenomiPhi増幅キット(Amersham Biotech)を使って、約1 ng DNAを試験管内で増幅して約2 μgのDNAを得た。次いで以前に同じ手順により増幅させた古細菌ルジウイルスSIRV1のゲノムDNAについて観察されたのと同様に(Peng他、2004)、高偏向ゲノムカバレージを生成するショットガンライブラリーを作製した(材料と方法の項目を参照のこと)。高レベルのキメラクローンも作製された。ゲノムを平均20倍被覆率で配列決定し、そしてコンティグ中のそれらの低頻度によりキメラクローンの配列を同定し、ライブラリーからそれらを除外した。
【0082】
増幅DNAライブラリーから得られた配列集成体を確かめるために、約50 ngの元のABV DNAをアシディアヌス(Acidianus)属リポスリクスウイルス(Vestergaard他、2005)から抽出された1μgDNAと混合し、そしてより大型のクローン挿入断片(2〜6.5 kbp)を用いて混合ショットガンライブラリーを調製した。第一ライブラリーのものへのそれらABVクローンの会合及び配列決定は、2つのライブラリーからの配列が完全に一致することを示した。更に、第二ライブラリーによりカバーされない領域の配列を、それらの領域のPCR増幅後に、又はプライマーウオーキングの後に確認した。それら両方ともウイルスDNA及び/又は大型挿入断片クローンに対して実施した。加えて、左端の所の少数クローンの配列を、約22kbの最終コンティグを与えるプライマーウオーキングにより伸長させた。
【0083】
ゲノム会合を確認するため、約40 ngのウイルスDNAを制限酵素AgeIとAflIIで消化した。生成物を完全なウイルスDNAと一緒にアガロースゲル電気泳動により分画し、バンドをSYBRゴールド(Invitrogen)により染色した。断片サイズは会合したコンティグの配列と一致し、完全なウイルスDNAのバンドは約23.8 kbのゲノムサイズを示した(図2)。
【0084】
制限消化からのサイズ推定値と単一コンティグサイズとの間の食い違いは、恐らく直線状ウイルスゲノムの末端領域がクローンライブラリー中に表示されないことを反映するのであろう(Haring他、2004;Haring他、2005b)。従って、本発明者らは増幅されたDNAに対するプライマーウオーキングにより、コンティグ末端から更に配列決定することを試みた。これは、常に終結と解読される配列の後方に約2kbの追加配列を与えた。得られた全配列は23,794 bpであり、これは制限断片消化の推定値と一致した。G+C含量は35%であった。
【0085】
該ゲノムは580 bpの逆方向末端反復配列(ITRs)を示し、それはルジウイルスのゲノムのもの(Peng他、2001)よりも小さいが、古細菌ベータリポスリクスウイルスのもの(Vestergaard他、出版準備中)と同様であった。
【0086】
該ゲノムが環化するかどうかを試験するために、ゲノムの各末端付近にアニールする幾つかの異なるプライマー対を使ってPCR実験を行ったが、それらのいずれも増幅生成物を生じなかった(データは示してない)。従って、本発明者らはABVのゲノムが直鎖状であると推測した。
【0087】
実施例3:ゲノム含量
ゲノムに注釈をつけ、開始コドン(88%AUG,6%GUG及び6%UUG)、TATA様プロモーターモチーフ及び/又はShine-Dalgarnoモチーフを以前に記載された通りに(Bettstetter他、2003, 増補表1S)割り当てた。57〜653アミノ酸サイズに及ぶ59の推定ORFを含有する全ウイルスゲノムについての地図を図3に与える。3つのORF 653, 103及び257は、Shine-Dalgarnoモチーフにより前置きされる内部開始コドンを含有する。内部ORFはよって推定遺伝子として割り当てられる(図3及び表1)。1つを除く全てのORFが一方の鎖上の位置8.5 kbと右端の間に位置する。残りのORFのうち、3つ(ORF247、ORF53a及びORF156)を除く全てが他方の鎖上の左端と位置8.5 kbの間に位置する(図3)。図3に示したORFの約49%が推定プロモーター配列により前置きされ、そして68%が推定Shine-Dalgarnoモチーフにより前置きされる。更に、ORFの約11%がTに富む下流の推定ターミネーターを示す。該ORFの約85%が推定オペロン中に配列され、そして該遺伝子の約25%がリーダー無しであるか又は非常に短いリーダーを担持するかのいずれかである転写物を生成すると推定される。ORF間の距離は一般に非常に短くそしてORFの24%が上流ORFとオーバーラップしており、このことはゲノムがコンパクトであることを示す。非常に驚くべきことに、位置10 kb〜21 kbの間に位置する29個のORFが1つの単一大型オペロンを形成するように思われる(図3及び表1S)。
【0088】
わずか3つのORFだけに公表配列データベース内での相同性検索に基づいて明確な機能を割り当てることができる(材料と方法を参照のこと)。ORF653は、タンパク質感作ポリメラーゼに最大の一致を有するファミリーBのDNAポリメラーゼと有意な配列相同性を示した。更に、ORF156はチミジレートキナーゼとして同定され、そしてORF315は推定グリコシルトランスフェラーゼとして同定された。
【0089】
全ての遺伝子注釈を表1に要約する。配列決定されたクレン古細菌ウイルスゲノムの大部分が、クレン古細菌ウイルス中で最も一般的な遺伝子産物である少なくとも1つのリボン−ヘリックス−ヘリックス(RHH)領域タンパク質をコードするけれども、ABVゲノム中には全くRHH領域は検出されなかった。しかしながら、ORF56は、薬剤やストレスに対する耐性に関与する幾つかの原核性転写調節因子中に存在するtetR型ヘリックス−ターン−ヘリックス領域に対して、限られた類似性を示す。別の3つのORFは、転写調節に関係するようであるロイシンジッパーパターンを含有する(図3及び表1)。ORF188は有意なEFハンド・カルシウム結合部位を含み、そしてII型プロテインキナーゼAの調節サブユニットに対して限定された類似性を示す。従って、それはCa++依存性プロテインキナーゼをコードするかもしれない。ORF133の配列中に、タンパク質−タンパク質相互作用に関係するかもしれない推定apaG領域プロフィールが検出された。ORF133タンパク質配列の推定二次構造は、約90%の伸長鎖とランダムコイルの存在を明らかにした。これは、別のapaGタンパク質の三次構造におけるβシートの高含有量と相関する。3つの隣接ORF(112、166及び346)は少数の膜貫通ヘリックスを含み、推定膜タンパク質又は膜結合タンパク質であるように見える。特に着目されるのはORF346であり、それは推定上の原核性膜リポタンパク質−脂質付着部位とEGF様領域を担持する。後者は一般に膜結合タンパク質の細胞外領域中又は分泌タンパク質中に存在する(表1)。それらの性質は、宿主膜タンパク質と相互作用するウイルス外皮タンパク質、又は宿主細胞からのウイルス粒子の放出を促進する膜貫通タンパク質を構成するORF346と一致する。2つの上流ORF(112と166)によりコードされる推定膜貫通タンパク質も同じ過程に関係するかもしれない。ORF346と下流のORF470の両者のC末端配列は、別のクレン古細菌ウイルスゲノム中の数個の大型ORFにおいて観察されるような(例えばHaring他、2005;Neumann & Zillig, 1990)低複雑性を示す。該タンパク質の機能は未知である。
【0090】
3つのORFが明確な機能を指定されそして幾つかは推定上の保存されたモチーフまたはパターンを担持する(表1)一方で、ABVと別のクレン古細菌ウイルスの間で共有される唯一の遺伝子は、グリコシルトランスフェラーゼ遺伝子のORF315である。以前、異なるクレン古細菌ウイルスゲノム間で共有される遺伝子が全くないか又はごく少数しかないことを比較ゲノム学は示した(Haring他、2004;Peng他、2001;Bettstetter他、2003)。この研究からの結果は、クレン古細菌ウイルスが非常に多様なグループを形成することを強調する。
【0091】
【表1】
【0092】
実施例4:DNA複製
ルジウイルスを例外として、本発明者らは古細菌ウイルスゲノムの複製機構にほとんど見識がない。しかしながら、ABVはそのゲノムが推定上のタンパク質感作DNAポリメラーゼをコードするという点で特別である。それらの酵素は、共有結合した末端タンパク質と共にITRを担持している直鎖状dsDNAゲノム中に常にコードされ、そしてバクテリオファージφ29と真核性アデノウイルスの両方において特徴付けられている(Salasにより概説、1991)。それらのウイルスについての複製開始モデルは、DNAポリメラーゼによりヘテロ二量体を形成しそしてウイルスDNA結合末端タンパク質及び該ゲノムのいずれかの末端の所の特定ヌクレオチド配列を介して複製開始点と相互作用する、遊離末端タンパク質を含む。末端タンパク質中のセリン、スレオニン又はチロシンのヒドロキシル基が第一ヌクレオチドのための受容部位として働く。更に、多数の直鎖状dsDNAプラスミドやミトコンドリアゲノムが、ITRとタンパク質感作DNAポリメラーゼを提示し、そして同様な方法で複製すると思われる末端タンパク質を担持している(Salas,1991)。タンパク質感作DNAポリメラーゼのサブファミリーは、DNA依存性DNAポリメラーゼファミリーBに属し、そして2つの挿入断片TPR-1とTPR-2を有する(Blasco他、2000;Dufour他、2000;Rodriguez他、2005)。
【0093】
ORF653とφ29 DNAポリメラーゼとの配列整列(図4)は、ORF653が、ファミリーBのDNAポリメラーゼに特徴的である(Blanco他、1991;Rohe他、1992)、N末端領域に3つのエキソヌクレアーゼ領域(Exo I, II及びIII)を含みそしてC末端部分に5つの保存された合成領域(pol I, IIa, IIb, III及びIV)を含むことを示す。更に、挿入断片TPR-1とTPR-2もORF653中に存在する(図4)。TPR-1は、ヒトアデノウイルスによりコードされるものを含む(Dufour他、2000)、全てのタンパク質感作DNAポリメラーゼのものとサイズが類似しており(50 aa)、一方でモチーフpol IIaとIIbの間に位置するTPR-2は、28 aa(φ29)から118 aa(アデノウイルス2型)までに及ぶ挿入断片の既知サイズ範囲(Bois他、1999)に比較して、短縮されている。φ29DNAポリメラーゼについては、TPR-1は末端の感作タンパク質との相互作用に関与することが分かっており(Dufour他、2000)、一方でTPR-2は該ポリメラーゼの高処理能力と鎖置換活性に必要であることが示されている(Rodriguez他、2005)。より保存的であるTPR-1の機能は、全てのタンパク質感作DNAポリメラーゼに一般的であるようであるが、これがより可変性のTPR-2にも当てはまるかどうかは不明のままである。
【0094】
ORF653がDNAポリメラーゼであることを確かめるために、該遺伝子をPCRによりウイルスゲノムから増幅し、E.コリ(E.coli)発現ベクター中にクローニングした。適度な量の可溶性組換えタンパク質が、該タンパク質のC末端断片と一緒に精製された(図5A)。重合活性を試験するために、該タンパク質を標識プライマー−鋳型DNAと共に50℃でインキュベートした。図5Bは、重合活性の指標である、プライマー(18nt)が鋳型のサイズ(42nt)へと伸長したことを明らかに示す。dNTPsの代わりにNTPsを用いた時、全く重合は検出されなかった(データは示してない)。これは、ORF653の生成物が確かにDNAポリメラーゼであることを確証する。
【0095】
実施例5:末端タンパク質
ABVの直鎖状ゲノム中のITR及び推定上タンパク質感作DNAポリメラーゼをコードする遺伝子の存在は、各5′末端が末端タンパク質に共有結合していることを強く示唆する。これは、生産されるウイルス粒子の収量が非常に低いために、実験的に確かめることは困難である。従って、本発明者らは、関連のバクテリオファージ、直鎖状プラスミド及びヒトアデノウイルスからの末端タンパク質配列を分析し、それらのタンパク質の保存された特徴に関する識見を得た。ポリメラーゼは比較的保存されているが、末端タンパク質は非常に低い保存を示した。例えば、大腸菌バクテリオファージPRD1の末端タンパク質は、別の既知末端タンパク質と全く有意な配列相同性を示さず(Savilahti他、1987)、そしてφ29の末端タンパク質と白色腐朽菌Pleurotus ostreatusの直鎖状ミトコンドリアプラスミド(Kim他、2000)の間には13/48%の同一性/類似性しか観察されなかった。しかしながら、末端タンパク質の遺伝子座は高度に保存されている。例えば、バクテリオファージφ29、PRD1, GA-1及びCP-1(受入番号P03681, P09009, X96987及びZ47794)並びにヒトアデノウイルスでは、該遺伝子は常にDNAポリメラーゼ遺伝子のすぐ上流に常に位置し、一方で該タンパク質のサイズはバクテリオファージでは230 aaと266 aaの間であり、そしてアデノウイルス2型(AC_000007)では671 aaである。DNA複製は直鎖状プラスミドについてはあまり研究されておらず、そのうち2つは融合したN末端の末端タンパク質とC末端のDNAポリメラーゼをコードすることが分かっている(Kim他、2000;Takeda他、1996)。DNAポリメラーゼの配列整列は、別の直鎖状プラスミド中のN末端部分における大きな配列伸長も明らかにし(Bois他、1999)、このことはポリメラーゼの遺伝子と末端タンパク質の遺伝子が一般に融合している可能性があることを示す。更に、転写物マッピングは、DNAポリメラーゼ遺伝子と末端タンパク質遺伝子が、CP-1(Martin他、1996)及びφ29ファミリーファージ(Meijer他により概説、2001)を含む研究されたウイルス全てにおいて、単一のmRNA中に同時転写されることも示した。従って、ポリメラーゼと末端タンパク質は遺伝子編成と機能の両方に関して密接に関連している。ABVの場合、ポリメラーゼの上流の遺伝子は、バクテリオファージ末端タンパク質のサイズの三分の二である163 aaをコードする。しかしながら、ClustalW(EMBL-EBI)を使った配列整列は、PRD1のTPとφ29のTPとの間よりもORF163とPRD1からのTPとの間に高いスコアを示し(データは示していない)、これはORF163が末端タンパク質をコードするかもしれないことを示唆する。
【0096】
実施例6:ウイルスDNAパッケージング
以前に、ABVのウイルス粒子構造が別の既知クレン古細菌クレンアキオータ門ウイルスと比較して非常に複雑であることが示された。瓶形のウイルス粒子は細い末端に「ストッパー」を、そして広い方の末端に20本の短いフィラメントを有するディスク又はリングを含有する(Haring他、2005)。本体は複雑なコアを包み込む二層から構成され、核タンパク質フィラメントがコンパクトに本体の内部にパッケージされている。DNAパッケージング機構は複雑であると思われる。
【0097】
ゲノムDNAのパッケージングは、様々なバクテリオファージや直鎖状ゲノムを有する真核性ウイルスについて研究されている。その機構は、予備形成されたプロカプシド中に長鎖DNA分子をトランスロケートするのに、一対の非キャプシドタンパク質とエネルギー源ATPの関与といった、幾つかの共通の特徴を共有する(Guo他により概説、2005)。φ29パッケージング機構の必須成分は、プロカプシドとATPに結合しそしてパッケージングタンパク質と協力することによりDNAトランスロケーションに積極的に参加する174 nt RNAのpRNAである(Guo、2005)。pRNAはφ29ゲノムの一端のITRに隣接してコードされ、それは高レベルの二次構造を示し、そして保存された二次構造モチーフは全ての既知φ29関連バクテリオファージを形成する(Meijer他により概説、2001)。更に、ITRに隣接する対応領域も、アデノウイルスDNAのパッケージングに重要であることが示された(Grable & Hearing, 1990)。ABVゲノム中の対応領域の調査は、左ITRに隣接した、転写解読枠を欠いている600 bp領域を明らかにし、その領域は中心部が比較的G+C豊富であった。200 bpのG+C豊富配列の推定二次構造は、バクテリオファージφ29からのpRNA及びCP-1のものと高い相同性を示す(図6)。A〜Fと記された7つのヘリックスがバクテリオファージの全てのpRNA中で高度に保存されている。違いはヘリックスFの左側の領域中にのみ存在し、そこには推定ABV RNAでは追加のヘアピンループが存在し、一方でバクテリオファージpRNAでは小さなループが存在する(図6)。ABV RNAの転写は、該要素の20 bp上流に置かれたプロモーター様配列ATTTAATの所で開始される可能性がある。保存されたゲノム位置、類似の二次構造、高いG+C含量、及び推定プロモーターの存在はいずれも、この非コード領域が恐らくウイルスDNAパッケージングに関与するであろうRNA分子をコードすることを強く暗示する。
【0098】
φ29のDNAパッケージングに関与する別の重要な成分は、該DNAをプロヘッドにトランスロケートするために回転すると提唱されたコネクターである(Meijer他、2001)。ABVの一般的形態学はφ29のそれとは異なるけれども、「ストッパー」はφ29のコネクターと類似しており、それもまたボトルネック形状を有し、その広い方の末端は同じくプロヘッドの内側に埋没している(Meiyer他、2000)。更に、該ストッパーの広い方の末端は核タンパク質フィラメントに連結される(Haring他、2005)。従って、コネクターもABVのパッケージングに関係するのかもしれない。
【0099】
現在、直鎖状DNAを有する古細菌ウイルスのパッケージングについては殆ど知られていない。人は、クレンアーキオータ(クレン古細菌)門ルジウイルス(rudivirus)と糸状ウイルスが、1〜3つのタンパク質を含有する、長く且つ「直鎖状」の構造(棒状又は糸状)に配列されたスーパーコイル状ゲノムDNAを有する方がよりたやすいと推測する傾向にある。従って、それらは予備形成された構造にゲノムDNAをパッケージングしないように思われる。よりコンパクトな構造と特に非常に長い直線状ゲノムを有するABVとPSVについては、それらが包括的カプシド化又はパッケージング機構を必要とすると推測するだろう。
【0100】
ABV、バクテリオファージφ29及び真核性アデノウイルスの左端の所のゲノム内容物を図7に示し、その3種のウイルス間での高い類似性を示す。ABVはタンパク質感作DNAポリメラーゼを含むことが報告された最初の古細菌ウイルスである。生命に係る重大な3領域からの3つの形態学的に異なるウイルス中の該ポリメラーゼの存在は、タンパク質感作DNA複製機構が古代のものであり、恐らく生命に係る3領域の分岐よりも以前に存在していたことを強力に指摘する。
【図面の簡単な説明】
【0101】
【図1】3%酢酸ウラニルでのネガティブ染色後のABV粒子の電子顕微鏡写真。目盛:100 nm。
【0102】
【図2】そのままのウイルスDNA(左側パネル)及び制限酵素消化したウイルスDNAをアガロースゲル中で泳動することによるゲノムサイズの推定。レーン1:そのままのウイルスDNA;レーン2と3:それぞれAgeI及びAflIIで消化したDNA;M1:New England BiolabsからのλDNA−モノカット混合物サイズマーカー;M2:AmershamからのラッダーDNAサイズマーカー。
【0103】
【図3】2本のDNA鎖上に存在する推定遺伝子の位置及びサイズを示すABVのゲノム地図。大部分の遺伝子が右向きの矢印により示される一方の鎖上に表示され、そして少数の遺伝子が左向きの矢印により示される相補鎖上に表示される。黒色の矢印は、機能が特定されたORFを示し、仮定の遺伝子は灰色の矢印により示される。3つの内部ORFは白抜きの矢印により指摘され、それらのサイズは括弧内に示される。この地図はMacPlasmap 2.05及びAdobe Illustratorを使って作製した。
【0104】
【図4】全て既知のタンパク質感作DNAポリメラーゼ配列に特異的である2つの挿入断片(TPRI及びII)を示す、ORF653(配列番号1)とPhi29 DNAポリメラーゼ〔DNAポリメラーゼB型ファミリーに相当する配列番号7(Genbank受入番号1XI1B)から入手〕との配列の整列。エキソヌクレアーゼ活性(Exo I, II, III)及び重合(Pol I, IIa, IIb, III及びIV)に関係する保存されたモチーフが示される。数字は配列間のアミノ酸長さを示す。
【0105】
【図5】図5AはE.コリからのORF653によりコードされる組換えポリメラーゼの精製。レーン1:タンパク質サイズマーカー;2:誘導された細胞の全粗製物;3:音波処理と遠心後の上清;4:Ni-NTAアガロース樹脂へのHis標識タンパク質の結合後の素通り液;5:樹脂カラムの洗浄液;6:精製済タンパク質。マーカー中のポリペプチドのサイズ(kD)が左側に示される。2つの矢印はそのままの(上側)及び断片化した(下側)ポリメラーゼの位置を示す。 図5Bはポリメラーゼアッセイを示す。反応中のポリメラーゼの濃度が最上部に示され、そして18 ntプライマーと伸長分子(42nt)の位置が左側に示される。
【0106】
【図6】図6AはABVパッケージングに関係する推定RNA要素の二次構造を示す。 図6Bはphi29のプロヘッドRNAの二次構造を示す。バクテリオファージpRNA中に保存されている7つのヘリックスは、5′末端から3′末端の方にA〜Fで表示され、一方で最初の命名は幹の長さに対応して行われた(Bailey他、1990)。
【0107】
【図7】ABD、phi29及びアデノウイルス(5型)の左端におけるゲノム含有物の描写。ITRの長さはそれぞれ580、6及び103 bpである。黒色ボックスはパッケージングに関与する領域を表し、一方ABVとphi29についての転写方向が小さい矢印により示される。ポリメラーゼ(pol)と末端タンパク質(TP)をコードする遺伝子は、それぞれ薄灰色と濃灰色の矢印により与えられる。polとパッケージング要素の間に存在するORFの数が括弧内に示される。
【0108】
参考文献
Altschul, S.F., T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller & D.J. Lipmann. 1997. “Gapped BLAST and PSI-BLAST: a new generation of protein database search programs.” Nucl. Acids Res. 25:3389-3402.
Arnold, H.P., W. Zillig, U.Ziese, I. Holz, M. Crosby, T. Utterback, J.F. Weidmann, J. Kristjansson, H.P. Klenk, K.E. Nelson & C.M. Fraser. 2000. “A novel lipothrixvirus, SIFV, of the extremely thermophilic crenarchaeon Sulfolobus.” Virology 267:252-266.
Barthelemy, I., M. Salas & R.P. Mellado. 1986. “In vitro transcription of bacteriophage φ29 DNA: transcription initiation sites.” J. Virol. 60:874-879.
Bettstetter, M., X. Peng, R.A. Garrett & D. Prangishvili. 2003. “AFV1,a novel virus infecting hyperthermophilic archaea of the genus Acidianus.” Virology 315:68-79.
【0109】
Blum, H., W. Zillig, S. Mallock, H. Domday & D. Prangishvili. 2001. “The genome of the archaeal virus SIRV1 has features in common with genomes of eukaryal viruses.” Virology 281:6-9.
Blanco L., Lazaro J.M., de Vega M., Bonnin A., Salas M. “Terminal protein-primed DNA amplification.” Proc. Natl. Acad. Sci. USA 1994 Dec 6; 91(25):12198-202.
Bois F., Barroso G., Gonzalez P., Labarere J. 1999. “Molecular cloning, sequence and expression of Aa-polB, a mitochondrial gene encoding a family B DNA polymerase from the edible basidiomycete Agrocybe aegerita.”Mol. Gen. Genet. 261:508-513.
【0110】
Bravo, A., B. Illana & M. Salas. 2000. “Compartmentalization of phage φ29 DNA replication : interaction between the primer terminal protein and the membrane-associated protein p1.” EMBO J. 19:5575-5584.
Brugger, K., P. Redder & M. Skovgaard. 2003. “MUTAGEN: Multi User Tool for Annotating GENomes.” Bioinformatics 19:2480-2481.
Dufour, E., J. Mendez, J.M. Lazaro, M. de Vega, L. Blanco & M. Salas. 2000. “An aspartic acid residue in TPR-1, a specific region of protein-priming DNA polymerases, is required for the functional interaction with primer terminal protein.” J. Mol. Biol. 304:289-300.
Guo.P. 2005. “Bacterial virus phi29 DNA-packaging motor and its potential applications in gene therapy and nanotechnology.” Methods Mol. Biol. 300:285-324.
【0111】
Grable M., Hearig P. 1990. “Adenovirus type 5 packaging domain is composed of a repeated element that is functionally redundant.” J. Viol. 64:2047-56.
Hatfield, L. & P. Hearing. 1993. “The NFIII/OCT-1 binding site stimulates adenovirus DNA replication in vitro and is functionally redundant with adjacent sequences.” J. Virol. 67:3931-3939.
Haring, M., X. Peng, K. Brugger, R. Rachel, K.O. Stetter, R.A. Garrett & D. Prangishvili. 2004. “Morphology and genome organization of the virus PSV of the hyperthermophilic archaeal genera Pyrobaculum and Thermoproteus: a novel virus family, the Globuloviridae.” Virology 323:233-242.
【0112】
Haring M., R. Rachel, X. Peng, R.A. Garrett & D. Prangishvili 2005a. “Diverse viruses in hot spring of Pozzuoli, Italy, including a unique bottled-shaped archaeal virus ABV from a new family, the Ampullaviridae.” J. Virol. 79:9904-9911.
Haring, M., Vestergaard, G., Brugger, K., Rachel, R,m Garrett, R.A. & Prangishvili, D. 2005b. “Structure and genome organization of AFV2, a novel filamentous archaeal virus with unusual terminal structures.” J. Bacteriol. 187:3855-3858.
Haring, M.他 Nature 印刷中。
【0113】
Janekovic, D., S. Wunderl, I. Holtz, W. Zillig, A. & H. Neumann. 1983. TTV1, TTV2 and TTV3, a family of viruses of the extremely thermophilic, anaerobic sulfur reducing archaebacterium Thermoproteus tenax.” Mol. Gen. Genet. 192:39-45.
Kim, E.K., J.H. Jeong, H.S. Youn, Y.B. Koo & J.H. Roe. 2000. “The terminal protein of a linear mitochondrial plasmid is encoded in the N-terminus of the DNA polymerase gene in white-rot fungus Pleurotus ostreatus. Curr. Genet. 38:283-290.
Martin, A.C., R. Lopez & P. Garcia. 1996. “Analysis of the complete nucleotide sequence and functional organization of the genome of Streptococcus pneumoniae bacteriophage Cp-1.” J. Virol. 70:3678-3687.
【0114】
Meijer, W.J., J.A. Horcajadas & M. Salas. 2001. Phi29 family of phages.” Microbiol. Mol. Biol. Rev. 65:261-287.
Peng, X., H. Blum, Q. She, S. Mallok, K. Brugger, R.A. Garrett, W. Zillig & D. Prangishvili. 2001. “Sequences and replication of genomes of the archaeal rudiviruses SIRV1 and SIRV2: Relationships to the archaeal lipothrixvirus SIFV and some eukaryal viruses. Virology 291:226-234.
Peng, X. A. Kessler, H. Phan, R.A. Garrett & D. Prangishvili. 2004. “Multiple variants of the archael DNA rudivirus SIRV1 in a single host and a novel mechanism of genome variation.” Mol. Microbiol. 54:366-375.
Picardeau, M., J.R. Lobry & B.J. Hinnebusch. 1999. “Physical mapping of an origin of bidirectional replication at the centre of the Borrelia burgdorferi linear chromosome.” Mol. Microbiol. 32:437-445.
【0115】
Prangichvili, D., K.M. Stedman & W. Zillig, 2001. “Viruses of the extremely thermophilic archaeon Sulcolobus.” Trends Microbiol. 9:39-42.
Prangishvili, D. & Garrett R.A., 2005. “Vuruses of hyperthermophilic Crenarchaea.” Trends Microbiol. 13:535-542.
Rachel, R., M. Bettstetter, B.P. Hedlund, M. Haring, A. Kessler, K.O. Stetter & D. Prangishvili. 2002. “Remarkable morphological diversity of viruses and virus-like particles in terrestrial hot environments.” Arch. Virol. 147:2419-2429.
Rodriguez, I., J.M. Lazaro, L. Blanco, S. Kamtekar, A.J. Berman, J. Wang, T.A. Steitz, M. Salas & M. de Vega. 2005. “A specific subdomain in phi29 DNA polymerase confers both processivity and strand-displacement capacity.” Proc. Natil. Acad. Sci. USA. 102:6407-12.
【0116】
Salas, M. 1991. “Protein-priming of DNA replication.” Annu. Rev. Biochem. 60:39-71.
Savilahti H. & D.H. Bamford. 1987. The complete nucleotide sequence of the left very early region of Escherichia coli bacteriophage PRD1 coding for the terminal protein and the DNA polymerase.” Gene 57:121-130.
Takeda M., H. Hiraishi, T. Takesako, S. Tanase & N. Gunge. 1996. “The terminal protein of the linear DNA plasmid pGKL2 shares an N-terminal domain of the plasmid-encoded DNA polymerase.” Yeast 12:241-246.
Torarinsson, E., H.-P. Klenk & R.A. Garrett. 2005. “Divergent transcriptional and translational signals in Archaea.” Environ. Microbiol. 7:47-54.
【0117】
Vestergaard, G., M. Haering, X. Peng, R. Rachel, R.A. Garrett & D. Prangishvili. 2005. ARV1, a rudivirus infecting the hyperthermophilic archaeal genus Acidianus.” Virology, 336:83-92.
Zillig, W., D. Prangishvilli, C. Schleper, M. Elferink, I. Holz, S. Albers, D. Janekovic & D. Gotz. 1996. “Viruses, plasmids and other genetic elements of thermophilic and hyperthermophilic Archaea.” FEMS Microbiol. Rev. 18:225-236.
【特許請求の範囲】
【請求項1】
次のポリペプチド群:
a) 配列番号1のアミノ酸配列を有するポリペプチド;
b) DNAポリメラーゼ活性を有するa)の断片;
c) a)の前記DNAポリメラーゼのDNAポリメラーゼ活性を可能にする配列番号1断片を少なくとも含んで成るポリペプチド;
d) 配列番号1のアミノ酸配列を有するポリペプチドであって、生成するDNAポリメラーゼペプチドが配列番号1のアミノ酸配列を有するポリペプチドに比較して検出可能なエキソヌクレアーゼ活性を有意に少ないか又は全く持たない程度に、図4に指定されたエキソヌクレアーゼ部位Exo I, Exo II及び/又はExoIIIが変異されているか又は削除されている前記ポリペプチド;
e) 配列番号1の配列との最適整列後に、少なくとも80%の同一性である配列を有するポリペプチドであって、DNAポリメラーゼ活性を有する前記ポリペプチド
より選択された単離されたDNAポリメラーゼ。
【請求項2】
古細菌アンプラウイルスABVから単離される、請求項1記載のDNAポリメラーゼ。
【請求項3】
図4に同定された配列番号1のPol I, Pol IIa, Pol IIb, Pol III及びPol IV断片を少なくとも含んで成る、請求項1又は2記載のDNAポリメラーゼ。
【請求項4】
請求項1〜3のいずれか一項に記載のDNAポリメラーゼポリペプチドをコードする核酸。
【請求項5】
請求項4の核酸を含んで成るベクター。
【請求項6】
前記核酸がプロモーターに作用可能に連結されている、請求項5記載のベクター。
【請求項7】
2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(フランス国パリのパスツール研究所の微生物寄託機関Nationale de Culture de Microorganismes)に寄託されている、請求項5記載のベクター。
【請求項8】
請求項5〜7記載のベクターを含んで成る宿主細胞。
【請求項9】
2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(フランス国パリのパスツール研究所の微生物寄託機関Nationale de Culture de Microorganismes)に寄託されている、請求項8記載の宿主細胞。
【請求項10】
DNAポリメラーゼを生産する方法であって、
(a) 前記核酸の発現に適した条件下で請求項8又は9記載の宿主細胞を培養する段階;及び
(b) 前記宿主細胞から前記DNAポリメラーゼを単離する段階
を含んで成る方法。
【請求項11】
前記宿主細胞が原核細胞又は真核細胞である、請求項10記載の方法。
【請求項12】
二本鎖DNA分子を合成する方法であって、
(a) 第一のDNA分子にプライマーをハイブリダイズせしめる段階;及び
(b) 段階(a) の前記DNA分子を、1又は複数のデオキシリボヌクレオシド三リン酸又はその類似体並びに請求項1〜3記載のポリペプチドの存在下で、前記第一のDNA分子の全部又は一部に相補的な第二のDNA分子を合成するのに十分な条件下でインキュベートする段階
を含んで成る方法。
【請求項13】
一本鎖DNA分子を合成する方法であって、
(a) 請求項12記載の方法により二本鎖DNA分子を合成する段階;
(b) 段階(a)で得られた二本鎖DNA分子を変性させる段階;及び
(c) 段階(b)で得られた一本鎖DNA分子を回収する段階
を含んで成る方法。
【請求項14】
請求項12又は13の方法を含んで成る10キロ塩基より大きいDNA分子の製造方法であって、段階(a)において鋳型として働く前記第一のDNA分子が10キロ塩基より大きい方法。
【請求項15】
前記デオキシリボヌクレオシド三リン酸がdATP, dCTP, dGTP及びdTTPから成る群より選ばれる、請求項12〜14のいずれか一項記載の方法。
【請求項16】
二本鎖DNA分子を増幅する方法であって、
(a) 第一及び第二プライマーを用意する段階であって、前記第一プライマーが前記DNA分子の第一鎖の3′末端の所又はその付近の配列に相補的であり、そして前記第二プライマーが前記DNA分子の第二鎖の3′末端の所又はその付近の配列に相補的であるという段階;
(b) 請求項1〜3記載のポリペプチドの存在下で、前記第一鎖に相補的な核酸と前記第二鎖に相補的な核酸が合成されるような条件下で、前記第一プライマーを前記第一鎖にそして前記第二プライマーを前記第二鎖にハイブリダイズせしめる段階;
(c) 前記第一鎖とそれの相補的鎖;及び
前記第二鎖とそれの相補的鎖
を変性せしめる段階;そして
(d) 段階(a)〜(c)を1回又は複数回繰り返す段階
を含んで成る方法。
【請求項17】
mRNAからcDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触せしめてハイブリッドを形成する段階;及び
(b) 段階(a)で形成された前記ハイブリッドを、請求項1〜3記載のDNAポリメラーゼ、並びにdATP, dCTP, dGTP及びdTTPと接触せしめ、それによりcDNA−RNAハイブリッドを得る段階
を含んで成る方法。
【請求項18】
mRNAからdsDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触せしめてハイブリッドを形成する段階;及び
(b) 段階(a)で形成された前記ハイブリッドを、請求項1〜3記載のポリペプチド、dATP, dCTP, dGTP及びdTTP、並びに第一鎖cDNAに相補的であるオリゴヌクレオチド又はプライマーと接触せしめ、それによりdsDNAが得られる段階
を含んで成る方法。
【請求項19】
DNA分子のヌクレオチド塩基配列を決定する方法であって、
(a) 前記DNA分子を、前記DNA分子にハイブリダイズできるプライマー分子と接触せしめる段階;
(b) 段階(a)で形成された前記ハイブリッドを、4種類のデオキシヌクレオシド三リン酸、請求項1〜3記載のDNAポリメラーゼポリペプチド、及び特定のヌクレオチド塩基の所でDNA合成を終了させる1又は複数のDNA合成終結剤を含有する容器の中でインキュベートし、ここで前記各々の剤は異なるヌクレオチド塩基のところでDNA合成を終了させるという段階;及び
(c) 前記インキュベーション反応のDNA生成物をサイズに従って分離し、それにより前記DNAのヌクレオチド塩基配列の少なくとも一部を決定することができるという段階
を含んで成る方法。
【請求項20】
前記終結剤がジデオキヌクレオシド三リン酸である、請求項19記載の方法。
【請求項21】
DNA分子の増幅方法であって、
(a) 前記DNA分子を、請求項1〜3記載のDNAポリメラーゼを有するポリペプチド、古細菌アンプラウイルスABVの末端タンパク質、及び異なるデオキシヌクレオシド三リン酸の混合物の存在下でインキュベートする段階
を含んで成る方法。
【請求項22】
前記DNA分子の一端の所に、前記ABVの複製開始点を含有する断片が共有結合される、請求項21記載のDNA分子の増幅方法。
【請求項23】
DNA分子を配列決定するためのキットであって、
(a) 請求項1〜3記載のポリペプチドを含んで成る第一のコンテナー手段;
(b) 1又は複数のジデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段;及び
(c) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第三のコンテナー手段
を含んで成るキット。
【請求項24】
DNA分子を増幅するためのキットであって、
(a) 請求項1〜3記載のポリペプチドを含んで成る第一のコンテナー手段;及び
(b) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段
を含んで成るキット。
【請求項25】
配列番号3の配列を有する古細菌アンプラウイルスABVの単離された又は組換え末端タンパク質を更に含んで成る、請求項24記載のキット。
【請求項26】
ローリングサークル増幅、多重置換増幅又はタンパク質感作増幅のための、請求項1〜3記載のポリペプチドの使用。
【請求項27】
請求項1〜3記載のポリペプチドが、d) 欠損したエキソヌクレアーゼ活性とDNAポリメラーゼ活性を有するd)に定義されたポリペプチドである、請求項10〜22記載の方法、請求項23〜25記載のキット、又は請求項26記載の使用。
【請求項28】
請求項1〜3記載のDNAポリメラーゼポリペプチドを含んで成るリアクターを有する、DNA配列決定又は増幅のための装置。
【請求項1】
次のポリペプチド群:
a) 配列番号1のアミノ酸配列を有するポリペプチド;
b) DNAポリメラーゼ活性を有するa)の断片;
c) a)の前記DNAポリメラーゼのDNAポリメラーゼ活性を可能にする配列番号1断片を少なくとも含んで成るポリペプチド;
d) 配列番号1のアミノ酸配列を有するポリペプチドであって、生成するDNAポリメラーゼペプチドが配列番号1のアミノ酸配列を有するポリペプチドに比較して検出可能なエキソヌクレアーゼ活性を有意に少ないか又は全く持たない程度に、図4に指定されたエキソヌクレアーゼ部位Exo I, Exo II及び/又はExoIIIが変異されているか又は削除されている前記ポリペプチド;
e) 配列番号1の配列との最適整列後に、少なくとも80%の同一性である配列を有するポリペプチドであって、DNAポリメラーゼ活性を有する前記ポリペプチド
より選択された単離されたDNAポリメラーゼ。
【請求項2】
古細菌アンプラウイルスABVから単離される、請求項1記載のDNAポリメラーゼ。
【請求項3】
図4に同定された配列番号1のPol I, Pol IIa, Pol IIb, Pol III及びPol IV断片を少なくとも含んで成る、請求項1又は2記載のDNAポリメラーゼ。
【請求項4】
請求項1〜3のいずれか一項に記載のDNAポリメラーゼポリペプチドをコードする核酸。
【請求項5】
請求項4の核酸を含んで成るベクター。
【請求項6】
前記核酸がプロモーターに作用可能に連結されている、請求項5記載のベクター。
【請求項7】
2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(フランス国パリのパスツール研究所の微生物寄託機関Nationale de Culture de Microorganismes)に寄託されている、請求項5記載のベクター。
【請求項8】
請求項5〜7記載のベクターを含んで成る宿主細胞。
【請求項9】
2006年4月28日に寄託番号I-3601のもとにC.N.C.M.(フランス国パリのパスツール研究所の微生物寄託機関Nationale de Culture de Microorganismes)に寄託されている、請求項8記載の宿主細胞。
【請求項10】
DNAポリメラーゼを生産する方法であって、
(a) 前記核酸の発現に適した条件下で請求項8又は9記載の宿主細胞を培養する段階;及び
(b) 前記宿主細胞から前記DNAポリメラーゼを単離する段階
を含んで成る方法。
【請求項11】
前記宿主細胞が原核細胞又は真核細胞である、請求項10記載の方法。
【請求項12】
二本鎖DNA分子を合成する方法であって、
(a) 第一のDNA分子にプライマーをハイブリダイズせしめる段階;及び
(b) 段階(a) の前記DNA分子を、1又は複数のデオキシリボヌクレオシド三リン酸又はその類似体並びに請求項1〜3記載のポリペプチドの存在下で、前記第一のDNA分子の全部又は一部に相補的な第二のDNA分子を合成するのに十分な条件下でインキュベートする段階
を含んで成る方法。
【請求項13】
一本鎖DNA分子を合成する方法であって、
(a) 請求項12記載の方法により二本鎖DNA分子を合成する段階;
(b) 段階(a)で得られた二本鎖DNA分子を変性させる段階;及び
(c) 段階(b)で得られた一本鎖DNA分子を回収する段階
を含んで成る方法。
【請求項14】
請求項12又は13の方法を含んで成る10キロ塩基より大きいDNA分子の製造方法であって、段階(a)において鋳型として働く前記第一のDNA分子が10キロ塩基より大きい方法。
【請求項15】
前記デオキシリボヌクレオシド三リン酸がdATP, dCTP, dGTP及びdTTPから成る群より選ばれる、請求項12〜14のいずれか一項記載の方法。
【請求項16】
二本鎖DNA分子を増幅する方法であって、
(a) 第一及び第二プライマーを用意する段階であって、前記第一プライマーが前記DNA分子の第一鎖の3′末端の所又はその付近の配列に相補的であり、そして前記第二プライマーが前記DNA分子の第二鎖の3′末端の所又はその付近の配列に相補的であるという段階;
(b) 請求項1〜3記載のポリペプチドの存在下で、前記第一鎖に相補的な核酸と前記第二鎖に相補的な核酸が合成されるような条件下で、前記第一プライマーを前記第一鎖にそして前記第二プライマーを前記第二鎖にハイブリダイズせしめる段階;
(c) 前記第一鎖とそれの相補的鎖;及び
前記第二鎖とそれの相補的鎖
を変性せしめる段階;そして
(d) 段階(a)〜(c)を1回又は複数回繰り返す段階
を含んで成る方法。
【請求項17】
mRNAからcDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触せしめてハイブリッドを形成する段階;及び
(b) 段階(a)で形成された前記ハイブリッドを、請求項1〜3記載のDNAポリメラーゼ、並びにdATP, dCTP, dGTP及びdTTPと接触せしめ、それによりcDNA−RNAハイブリッドを得る段階
を含んで成る方法。
【請求項18】
mRNAからdsDNAを調製する方法であって、
(a) mRNAをオリゴ(dT)プライマー又は別の相補的プライマーと接触せしめてハイブリッドを形成する段階;及び
(b) 段階(a)で形成された前記ハイブリッドを、請求項1〜3記載のポリペプチド、dATP, dCTP, dGTP及びdTTP、並びに第一鎖cDNAに相補的であるオリゴヌクレオチド又はプライマーと接触せしめ、それによりdsDNAが得られる段階
を含んで成る方法。
【請求項19】
DNA分子のヌクレオチド塩基配列を決定する方法であって、
(a) 前記DNA分子を、前記DNA分子にハイブリダイズできるプライマー分子と接触せしめる段階;
(b) 段階(a)で形成された前記ハイブリッドを、4種類のデオキシヌクレオシド三リン酸、請求項1〜3記載のDNAポリメラーゼポリペプチド、及び特定のヌクレオチド塩基の所でDNA合成を終了させる1又は複数のDNA合成終結剤を含有する容器の中でインキュベートし、ここで前記各々の剤は異なるヌクレオチド塩基のところでDNA合成を終了させるという段階;及び
(c) 前記インキュベーション反応のDNA生成物をサイズに従って分離し、それにより前記DNAのヌクレオチド塩基配列の少なくとも一部を決定することができるという段階
を含んで成る方法。
【請求項20】
前記終結剤がジデオキヌクレオシド三リン酸である、請求項19記載の方法。
【請求項21】
DNA分子の増幅方法であって、
(a) 前記DNA分子を、請求項1〜3記載のDNAポリメラーゼを有するポリペプチド、古細菌アンプラウイルスABVの末端タンパク質、及び異なるデオキシヌクレオシド三リン酸の混合物の存在下でインキュベートする段階
を含んで成る方法。
【請求項22】
前記DNA分子の一端の所に、前記ABVの複製開始点を含有する断片が共有結合される、請求項21記載のDNA分子の増幅方法。
【請求項23】
DNA分子を配列決定するためのキットであって、
(a) 請求項1〜3記載のポリペプチドを含んで成る第一のコンテナー手段;
(b) 1又は複数のジデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段;及び
(c) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第三のコンテナー手段
を含んで成るキット。
【請求項24】
DNA分子を増幅するためのキットであって、
(a) 請求項1〜3記載のポリペプチドを含んで成る第一のコンテナー手段;及び
(b) 1又は複数のデオキシリボヌクレオシド三リン酸を含んで成る第二のコンテナー手段
を含んで成るキット。
【請求項25】
配列番号3の配列を有する古細菌アンプラウイルスABVの単離された又は組換え末端タンパク質を更に含んで成る、請求項24記載のキット。
【請求項26】
ローリングサークル増幅、多重置換増幅又はタンパク質感作増幅のための、請求項1〜3記載のポリペプチドの使用。
【請求項27】
請求項1〜3記載のポリペプチドが、d) 欠損したエキソヌクレアーゼ活性とDNAポリメラーゼ活性を有するd)に定義されたポリペプチドである、請求項10〜22記載の方法、請求項23〜25記載のキット、又は請求項26記載の使用。
【請求項28】
請求項1〜3記載のDNAポリメラーゼポリペプチドを含んで成るリアクターを有する、DNA配列決定又は増幅のための装置。
【図1】


【図2】
【図3】

【図4】

【図5A】
【図5B】

【図6A】
【図6B】
【図7】
【図2】
【図3】
【図4】
【図5A】
【図5B】
【図6A】
【図6B】
【図7】
【公表番号】特表2009−536820(P2009−536820A)
【公表日】平成21年10月22日(2009.10.22)
【国際特許分類】
【出願番号】特願2009−508542(P2009−508542)
【出願日】平成19年5月14日(2007.5.14)
【国際出願番号】PCT/IB2007/002319
【国際公開番号】WO2007/132358
【国際公開日】平成19年11月22日(2007.11.22)
【出願人】(591282984)アンスティテュ パストゥール (17)
【氏名又は名称原語表記】INSTITUT PASTEUR
【出願人】(508335820)ユニバーシティ オブ コペンハーゲン (1)
【Fターム(参考)】
【公表日】平成21年10月22日(2009.10.22)
【国際特許分類】
【出願日】平成19年5月14日(2007.5.14)
【国際出願番号】PCT/IB2007/002319
【国際公開番号】WO2007/132358
【国際公開日】平成19年11月22日(2007.11.22)
【出願人】(591282984)アンスティテュ パストゥール (17)
【氏名又は名称原語表記】INSTITUT PASTEUR
【出願人】(508335820)ユニバーシティ オブ コペンハーゲン (1)
【Fターム(参考)】
[ Back to top ]