
可変遺伝子間配列を使用する発現の制御
【課題】可変遺伝子間配列を使用するジシストロン性メッセージを提供する。
【解決手段】上流側シストロンが、特定の抗体分子の重鎖及び軽鎖のどちらかをコードするDNAを含み、下流側シストロンが、それぞれ対応する軽鎖または重鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性メッセージであって、2つのシストロンが最適化された遺伝子間配列によって連結されているジシストロン性メッセージからなる。
【解決手段】上流側シストロンが、特定の抗体分子の重鎖及び軽鎖のどちらかをコードするDNAを含み、下流側シストロンが、それぞれ対応する軽鎖または重鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性メッセージであって、2つのシストロンが最適化された遺伝子間配列によって連結されているジシストロン性メッセージからなる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定の抗体分子の重鎖及び軽鎖が、2つのシストロンが最適化された遺伝子間配列によって連結されているジシストロン性(dicistronic)メッセージ中に存在するDNAによってコードされる抗体の産生方法に関する。
【背景技術】
【0002】
抗体は、特定の抗原を認識する能力によって、医学及び生物工学において非常に有用で効果的なツールとなっている。選択されたある種の細胞上の抗原に特異的な抗体は、これらの抗体を、選択した細胞に向けるために使用されてきた。いくつかのケースでは、細胞上のレセプターに抗体が結合すると、細胞の機能に影響を及ぼすことがわかっている。また、治療剤及び診断剤は、選択した細胞に、これらの薬剤を特異的に送り込むために、これらに抗体を結合させることが行なわれている。この技術は、特に癌細胞のターゲティングに使用されている。また、抗体は、ウイルス又は細菌に感染した細胞上の抗原を標的とするため、TNFαなどの他の分子を標的とするため、又は分析用に使用されている。生物工学では、抗体は、精製及び触媒反応において、プローブなどの使用に多く利用されている。
【0003】
すべての完全な抗体分子は、4つのポリペプチド鎖、すなわち、2つの同一の重鎖及び2つの同一の軽鎖から構成される。各鎖は、可変ドメインと定常ドメインを含んでいる。軽鎖は、2つのドメイン、つまり、VL及びCLを含み、重鎖は、少なくとも5つのドメイン、VH、CH1、ヒンジ、CH2、及びCH3を含み、場合によっては、更にCH4を含んでいる。4つの鎖は、いつでもおおよそ同じ様式で組織されている。すなわち、2つの重鎖が、少なくとも1つのジスルフィド結合で互いに連結され、各重鎖はまた、軽鎖のうちの1本とジスルフィド結合で連結され、2つの軽鎖はそれぞれ別の重鎖に連結する。
【0004】
抗体分子全体は、ほぼY字型で、基本的に2つの主機能部から構成される。第1の機能部は、特異的抗原を認識する役割を担い、Yの腕の上部を形成する。各部の抗原結合領域は、1つのVHドメインと1つのVLドメインを含む。各可変ドメインは、3つの超可変領域を含み、他方の鎖の3つの超可変領域と互いに抗原結合部位を形成する。これら超可変領域は、相補性決定領域(CDR1,CDR2,CDR3)として知られる。これらCDRはループを形成し、フレームワーク領域上に保持されている。VH及びVLを含む領域は、抗体ごとに可変であるため、「V」領域として知られる。
【0005】
第2の機能部は、抗体に認識された抗原を処理する他の細胞のエフェクター機能を誘発ーする役割を担い、Yの腕の下部とステムから形成される。この領域は、比較的定常であるため、定常「C」領域として知られる。この領域は、CLドメインと重鎖のCドメインすべてを含む。
【0006】
軽鎖には、κ及びλの2種類があり、重鎖には、α、δ、γ、ε及びμの5種類がある。抗体のクラスは、重鎖の型によって決定され、それぞれ、IgA、IgD、IgE、IgG及びIgMである。
【0007】
酵素切断によってその定常領域の一部が除去された抗体断片も、医学及び生物工学で使用される。これらには、Fab、Fab’、F(ab’)2及びFv断片が含まれる。
【0008】
抗体を産生する脾臓細胞と骨髄腫細胞を融合させて、ハイブリドーマを得ることによって、大量のモノクローナル抗体(特別な抗体特異性を有する)を直接に産生することが知られている(Kohler,G.and Milstein,C.「所期の特異性を有する融合細胞分泌抗体の連続培養」(Continuous cultures of fused cells secreting antibody of predefined specificity)Nature、256、495−497(1975))。しかし、このような抗体は、ヒトにおいて免疫原性でないため、ヒトの治療に使用することは適切ではない。多くのモノクローナル抗体は、非ヒト細胞で産生される。
【0009】
複数の抗体の有用な特性を組み合わせることを可能にして、1つの新しい抗体を作る組み換えDNA技術が開発されている。1つの抗体の完全な可変領域を含む抗体結合部位を、別の抗体の定常領域に連結させてキメラ抗体を産生することが、欧州特許第0120694号(Celltech Limited)、欧州特許第0125023号(Genentech Inc.and City of Hope)、欧州特許第0171496号(Research Development Corporation,Japan)、欧州特許第0173494号(Stanford University)及び特許国際公開第86/01533号(Celltech Limited)に記載されている。
【0010】
たとえば、特許国際公開第86/01533号は、ネズミ可変領域をヒトの定常領域と結合させたキメラ抗体の調製を記載している。しかし、キメラ非ヒト/ヒト抗体には非ヒトドナー由来の残基をかなり比率で含むので、特に長期間が投与すると、抗体が潜在的に有害な免疫応答を誘発する可能性がある(Begentら、Br.J.Cancer,62,487(1990))。
【0011】
抗体の「ヒト化」とは、非ヒト/ヒトキメラ抗体を、ヒト免疫系に対してヒト抗体により似たものにする技術で、上記に記載した望ましくない免疫反応を解消するために開発された。欧州特許第0239400号(Winter)は、完全なネズミの可変領域を使用する代わりに、ネズミのモノクローナル抗体のCDRをヒト抗体の可変ドメインのフレームワーク領域上に移植する方法について記載している。したがって、ネズミ由来であるのは、抗原結合ドメイン自体を形成するCDRのみで、その他の残基がヒト由来である。
【0012】
Reichmannら(「治療用ヒト抗体の再形成」”Reshaping human anitbodies for therapy”、Nature、332、323−324、1988)は、可変ドメインにある他のヒト残基を非ヒトドナーの等価物に変換して、抗体結合活性を改善することが有益であることを見出した。このような残基は、ヒト重鎖のポジション27に見つかった。ヒトのセリンから対応するラットの残基(フェニールアラニン)に変換すると、抗体結合能力の改善がもたらされた。このような残基が別にポジション30で見出された。しかし、上記に記載されたポシション27の変更に加えて、重鎖のポジション30にヒトのセリンからラットのチロシンへの変更を含む構築物は、セリンからフェニールアラニンへの変更をポジション27のみに有するヒト化された抗体に比べて、優位に結合活性を変更しなかった。
【0013】
重鎖残基27及び30は、CDR1近隣の構造ループ内にある。Queenら(特許国際公開90/07861)は、CDRと相互作用する他の残基もまた、抗体結合親和性を決定する際に重要であると推測した。このことを念頭において、Queenらは、ヒト化抗体を設計する際、どの残基がドナーに由来し、どれがアクセプタに由来すべきかを決定するための4つの判定基準を提案した。より決定的な分析で、Adairら(特許国際公開91/09967)が、ヒト化抗体の設計を可能にする残基番号の優先度を開示した。
【0014】
Vaughanら(Nature Biotechnology、16、535−539,1998)を含めてCDRを移植された抗体について論じた数多くの論文が公表されている。
【0015】
定常領域が、治療剤又は診断剤として作用可能なエフェクター又はレポーター分子と融合した抗体複合体も記載されている(特許国際公開第95/01155号、米国特許第3927193号、第4331647号、第4348376号、第4361544号、第468457号、第4444744号、第4460459号及び第4460561号並びに以下の総説:Waldmann,T.A.、Science;252、1657(1991);Koppel,G.A.、Bioconjug.Chem.、1、13(1990);Oeltmann,T.N.and Frankel,A.E.、FASEB J.、5、2334(1991);及びvan den Bergh,H.E.、Chemistry in Britain、May 1986、430−439)。
【0016】
適切なホスト細胞を、1つは求める抗体の重鎖をコードするDNA配列を含み、1つはその抗体の軽鎖をコードするDNA配列を含む2つの発現ベクターでトランスフェクトして、正常、キメラ又はヒト化された抗体を産生することが知られている(特許国際公開第91/09967号)。また、適切なホスト細胞を、求める抗体の重鎖をコードするDNA配列とその抗体の軽鎖をコードするDNA配列との両方を含む発現ベクターでトランスフェクトすることも知られている。後者の例では、重鎖及び軽鎖をコードするDNA配列はどちらも、それ自体の個々のプロモータの制御下(特許国際公開第91/09967号)にあるか、ジシストロン性メッセージ中(Better,M.、Paul Chang,C.、Robinson,R.R.及びHorwitz,A.H.Escherichia coli:「活性キメラ抗体断片の分泌」(Secretion of an Active Chimeric Antibody Fragment.Science、240、1041−1043(1988))に存在する。
【0017】
ジシストロン性メッセージは、多くのヒトの癌の表面上に発現しているガングリオシド抗原に対するキメラマウスL6Fab抗体を産生するためにBetterらによって使用された。このケースでは、一部を切り詰めた重鎖(Fd)とκ軽鎖とが、確実に、物理的に近接して翻訳され、それらが正しく組み合わさり、分泌されることを企図して、ジシストロン性メッセージが選択された。ジシストロン性メッセージは、細菌中で機能できるのみであり、哺乳類及び細菌中においては、「1遺伝子1プロモータ」という概念が機能する。ジシストロン性メッセージでは、プロモータは第1のシストロンにのみ連結している。第2のシストロンは、ポリメラーゼによって第2のシストロンまで「読み通され」て転写され、2つのシストロンが単一のRNA分子で表されることになる。2つのコードDNAシストロンは、「遺伝子間配列」又は「IGS」として知られる、ある長さのDNAによって分離される。このIGS領域は、DNAから転写されるRNA分子中にも存在する。
【0018】
転写開始速度を最適化することが、大腸菌で分泌される異種タンパクを高度に発現させるためには必要不可欠であることが、しばらく前から認識されている(Simmons,L.及びYansura,D.Nature Biotech、14、629−634(1996))。しかし、Fabが異なると、フレームワークとCDR配列が異なり、それによって大腸菌ペリプラズム内での折りたたまれ易さ(又は速度)の差を含めて異なる特性が分子に付与される。(Knappik,A.及びPluckthun,A.Prot Eng 8、81−89(1995))。たとえば、大腸菌のペリプラズム内で容易かつ迅速に折りたたまれて生来のコンフォーメーションをとるFab’は、高度の「耐容性をもつ」と思われ、迅速な翻訳に適応し、高度な蓄積を達成することができる。折りたたみがより不十分なFab’は、迅速に翻訳された場合、ホスト細菌による耐容性が不十分になると思われ、ホストの折りたたみ/分泌機構を飽和させ、ホスト細胞の生理に有害な影響を与える可能性がある。
【0019】
したがって、ホスト細胞発現ベクターシステムを使用して抗体を産生する場合、特定の抗体における高レベル発現は耐容性があるが、抗体が異なると、高レベルの発現は、おそらく分泌又は折りたたみの効率が異なるという理由で、細胞に対して毒性となる可能性がある。
【0020】
個々の抗体の発現レベルは、重鎖及び軽鎖の存在する量によって決まる。最大量の産生を得るためには、重鎖と軽鎖の比を、たとえば、両方の量が等しくするなどバランスさせなければならない。しかし、重鎖と軽鎖のどちらか一方の量が少ないと、その量が産生される抗体の量を制限することになる。過剰の重鎖蓄積は耐容性が特に不十分となる可能性がある。
【0021】
ジシストロン性メッセージによってコードされる抗体の場合、上流側シストロンは、特定の抗原特異性を有する抗体の重鎖と軽鎖のどちらか一方をコードするDNAを含むことができる。その場合、下流側シストロンは、それぞれ、上流側シストロンの重鎖又は軽鎖のパートナーをコードする。特定の抗体を所望の発現レベルで産生させるには、ジシストロン性メッセージの下流側シストロンでコードDNAに対応する抗体鎖の発現レベルを調節できれば有利である。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】欧州特許第0120694号
【特許文献2】欧州特許第0125023号
【特許文献3】欧州特許第0171496号
【特許文献4】欧州特許第0173494号
【特許文献5】国際公開第86/01533号パンフレット
【特許文献6】欧州特許第0239400号
【特許文献7】国際公開90/07861号パンフレット
【特許文献8】国際公開91/09967号パンフレット
【特許文献9】国際公開第95/01155号パンフレット
【特許文献10】米国特許第3927193号
【特許文献11】米国特許第4331647号
【特許文献12】米国特許第4348376号
【特許文献13】米国特許第4361544号
【特許文献14】米国特許第468457号
【特許文献15】米国特許第4444744号
【特許文献16】米国特許第4460459号
【特許文献17】米国特許第4460561号
【特許文献18】国際公開第91/09967号パンフレット
【特許文献19】国際公開第91/09967号パンフレット
【非特許文献】
【0023】
【非特許文献1】Kohler,G.and Milstein,C.(Continuous cultures of fused cells secreting antibody of predefined specificity)Nature、256、495−497(1975)
【非特許文献2】Begentら、Br.J.Cancer,62,487(1990))
【非特許文献3】Reichmannら ”Reshaping human anitbodies for therapy”、Nature、332、323−324、1988)
【非特許文献4】Vaughanら(Nature Biotechnology、16、535−539,1998)
【非特許文献5】Waldmann,T.A.、Science;252、1657(1991)
【非特許文献6】Koppel,G.A.、Bioconjug.Chem.、1、13(1990)
【非特許文献7】Oeltmann,T.N.and Frankel,A.E.、FASEB J.、5、2334(1991)
【非特許文献8】van den Bergh,H.E.、Chemistry in Britain、May 1986、430−439
【非特許文献9】Better,M.、Paul Chang,C.、Robinson,R.R.及びHorwitz,A.H.Escherichia coli:Secretion of an Active Chimeric Antibody Fragment.Science、240、1041−1043(1988)
【非特許文献10】Simmons,L.及びYansura,D.Nature Biotech、14、629−634(1996)
【非特許文献11】Knappik,A.及びPluckthun,A.Prot Eng 8、81−89(1995)
【発明の概要】
【課題を解決するための手段】
【0024】
第1の態様において、本発明は、上流側シストロンが、抗体の重鎖と軽鎖のどちらかをコードするDNAを含み、下流側シストロンが、それぞれ対応する軽鎖又は重鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性メッセージであって、2つのシストロンが、最適化された遺伝子間配列(IGS)によって連結されており、産生される抗体が、抗TNFα抗体(たとえば、特許国際出願GBO1/02477参照)でもなく、抗ヒトキナーゼ挿入ドメインを含むレセプタ(抗KDR)抗体(たとえば、特許国際出願GB02/004619参照)でもないことを特徴とする、ジシストロン性メッセージを提供する。
【0025】
好ましくは、上流側シストロンは、抗体の軽鎖をコードし、下流側シストロンは対応するその重鎖をコードする。
【0026】
特定の抗体について所望の発現レベルを達成するために、IGSは、ジシストロン性メッセージにおける下流側シストロンに対応する抗体鎖の発現レベルが調節されるように最適化されている。
【0027】
本明細書では、IGSの長さを、上流側シストロンの終結コドンと下流側シストロン(両端を含まない)の開始コドンとの間のヌクレオチドの数であると定義する。IGSの長さは、下流側シストロンに対応するRNA部分の翻訳速度の決定に関係する。たとえば、遺伝子間配列が短いと、重鎖と軽鎖の間の翻訳のカップリングが非常に緊密になり、上流側シストロンによってコードされる抗体鎖の合成が終了した後、下流側シストロンによってコードされる抗体鎖の合成が開始される前に、翻訳を行うリボソームが、mRNAから十分に解離しなくなる可能性がある(Adhin,M.and van Duin、J.J.Mol.Biol.、213、811−818(1990);Andre,A.ら、FEBS Letts.、468、73−78(2000))。このような過程は、翻訳の再開始と呼ばれ、上流の遺伝子の終結コドンと下流遺伝子の開始コドンが非常に接近している場合にのみ生じる。速度論的に考察すると、このような過程により、翻訳において、IGSで別のリボソームがmRNA分子と結合する必要がなくなるので、下流側シストロンによってコードされるポリペプチドの発現が増加することになる。
【0028】
2つの遺伝子間が離れすぎていると、上流側シストロンの合成が終了した後にリボソームが解離するので、下流側シストロンの翻訳を開始するには新しいリボソームが必要となる。そのため、通常、翻訳開始効率が低下することになる。
【0029】
IGSにはシャイン−ダルガルノ(SD)リボソーム結合部位(16SrRNAと相補的である)が存在しているので、リボソームがIGS配列に結合し、下流側シストロンを翻訳することができる。翻訳の再開始が可能になるのに十分なほどシストロン間の距離が短い場合、上流側シストロン内にSD部位が存在すると、下流側シストロンの翻訳開始効率が上がることを示唆する証拠がいくつかある(Spanjaard,R.A.and van Duin,J.Nucl.Acids Res.、17、5501−5507(1989))。
【0030】
SD部位、及びSD部位の端とAUG開始コドンの間のヌクレオチド配列には、翻訳開始の強さに影響を与えるいくつかの特徴がある(Makrides,S.Microbiol.Revs.、60、512−538(1996)に総説される)。下流側シストロンの、SD部位とAUG開始コドンの間の距離は、そのような特徴の1つであり、SD部位自体の「効力(strength)」もその特徴の1つである。SD部位に結合する16SrRNA配列と100%相補的なSD部位は、通常、16SrRNA配列と部分的にしか相補的でないSD部位の場合よりも、発現は大きくなる。
【0031】
SD配列と開始コドン間領域の配列は、下流側シストロンの翻訳速度を決定する別の重要な要素である。距離及び配列は、開始コドンの周りでmRNAのとり得る2次構造に影響する(Makrides,S.Microbiol.Revs.、60、512−538(1996)参照)。開始コドンは、「ループ」中に存在するはずで、「ステム」内には閉じ込められていないが、SD配列はその逆である。したがって、IGSの配列と長さを変更することによって、翻訳カップリングの程度及び/又は翻訳開始の強さを変更することができ、それによって、下流側シストロンの翻訳のレベル、及びそれをコードする抗体鎖のその後の蓄積速度を変更することができる。
【0032】
本発明のジシストロン性メッセージのIGSは、2つのシストロンの最適な翻訳カップリングが実現されるように、長さ、配列及び2次構造が変更されている。
【0033】
本発明に使用される最適なIGS配列は、以下の方法を使用して実験的に決定することができる。この方法は、抗体分子をテスト用に挿入することが可能な一連のIGS変異体を含む一連の適切な発現ベクターを構築することを含む。各抗体の各IGS配列の実験を、発現ベクターで適切なホストを形質転換して、抗体の発現及び収率を分析することによって行うことができる。本発明に記載された、長さと配列の異なる1組のIGS配列を使用して、かかるベクターを構築することができ、その中から特定の抗体分子の最適なIGS配列を選択することができる。この配列には、IGS配列1から4が含まれる。
【図面の簡単な説明】
【0034】
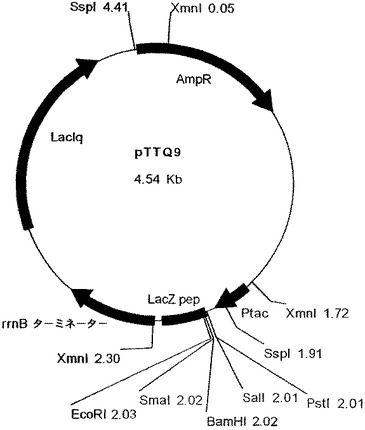
【図1】ベクターpTTQ9を示す図である。
【図2】OmpAポリリンカー領域の配列を示す図である。
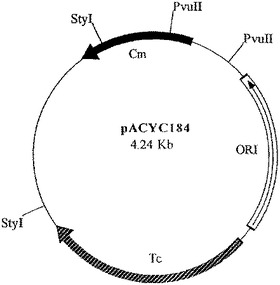
【図3】ベクターpACYC184を示す図である。
【図4】ベクターpTTO−1を示す図である。
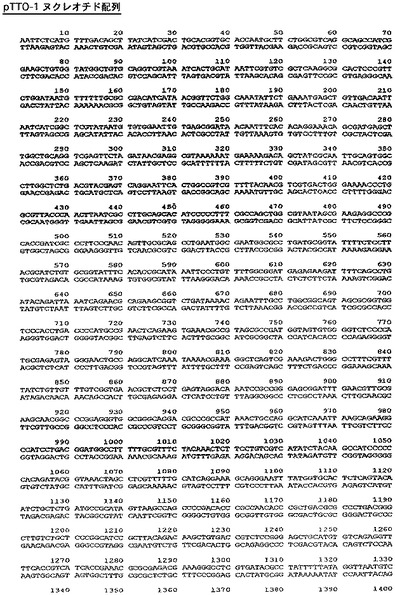
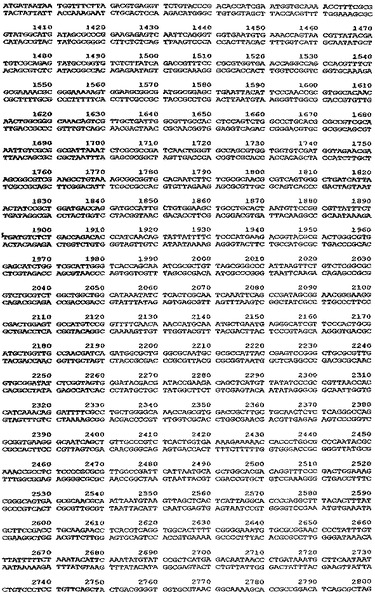
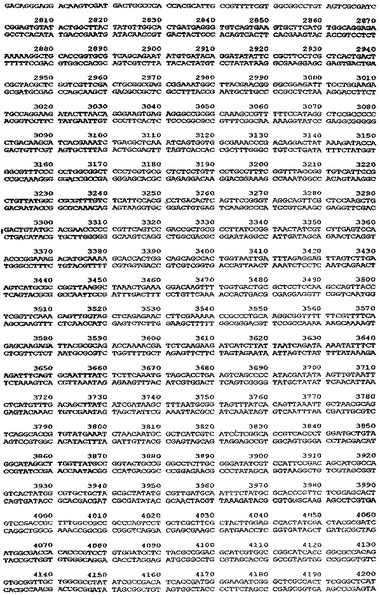
【図5】ベクターpTTO−1の完全なDNA配列を示す図である。
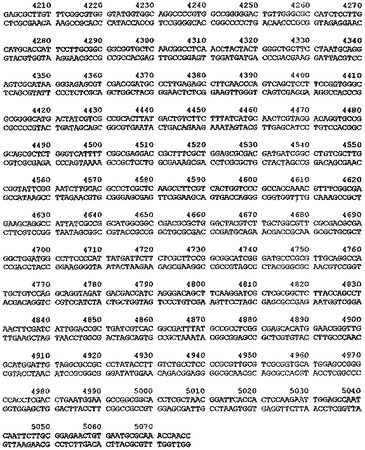
【図6】ベクターpTTO−2を示す図である。
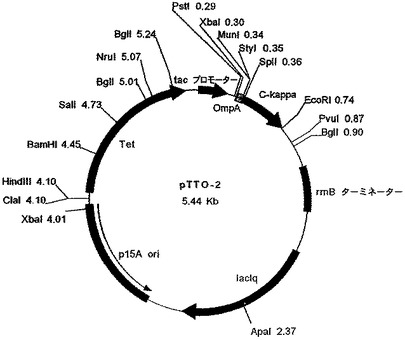
【図7】ベクターpDNAbEngG1を示す図である。
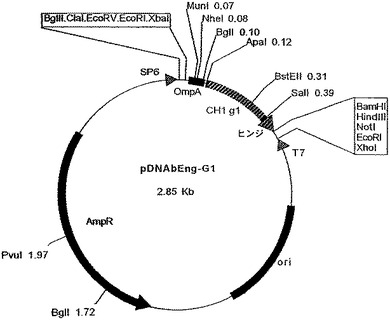
【図8】ベクターpTTO(TNFα)を示す図である。
【図9】大腸菌Fab’発現用の、4つの異なる遺伝子間配列をコードするオリゴヌクレオチドカセットを示す図である。
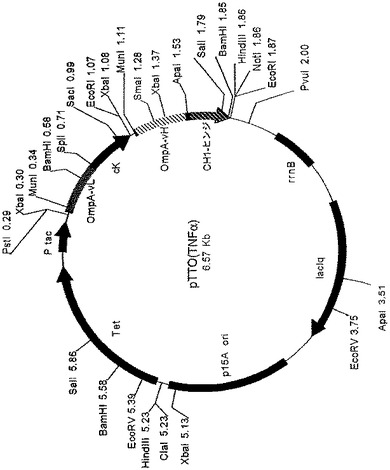
【図10】振盪フラスコ分析の結果として、IGS変異体pTTO(TNFαIGS−1、pTTO(TNFαIGS−2)、pTTO(TNFαIGS−3)及びpTTO(TNFαIGS−4)のペリプラズムFab’の蓄積を示す図である。
【図11】Fab’g163の4つのIGS変異体を生成するために使用する4通りの連結を示す図である。
【図12】振盪フラスコ分析の結果として、IGS変異体pTTO(g163 IGS−1)、pTTO(gl63 IGS−2)、pTTO(gl63 IGS−3)及びpTTO(g163 IGS−4)の可溶性Fab’の蓄積を示す図である。
【図13】ベクターpTTOD(g163 IGS−3)の制限マップを示す図である。
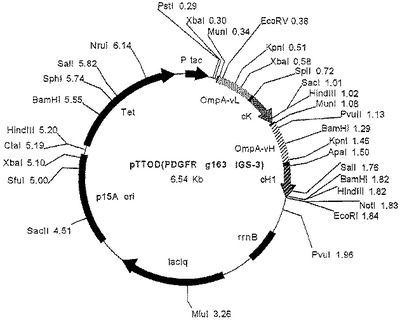
【図14】振盪フラスコ分析の結果として、IGS変異体pTTOD(g165 IGS−1)、pTTOD(g165 IGS−2)及びpTTOD(g165 IGS−3)の可溶性Fab’の蓄積を示す図である。
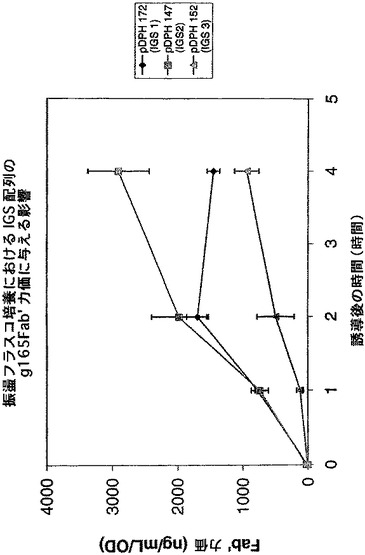
【図15】発酵槽比較分析の結果として、IGS変異体、pTTOD(gl65 IGS−1)、pTTOD(gl65 IGS−2)、pTTOD(gl65 IGS−3)及びpTTOD(g165 IGS−4)の可溶性Fab’蓄積を示す図である。
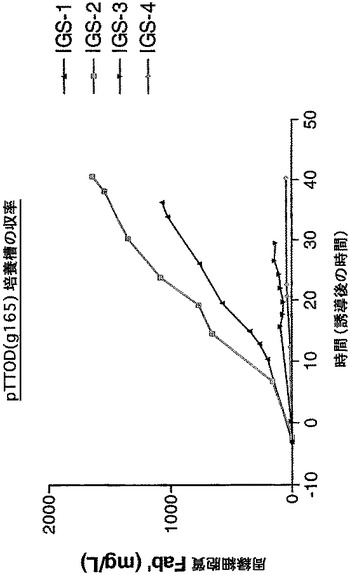
【図16】発酵槽比較分析の結果として、IGS変異体pTTOD(gA33 IGS−2)及びpTTOD(gA33 IGS−3)の可溶性Fab’蓄積を示す図である。
【発明を実施するための形態】
【0035】
好ましい一実施形態では、下流側シストロンによってコードされる抗体鎖が最高に発現されるように、IGSが最適化されている。これにより、下流側シストロンによってコードされる抗体鎖の量には制限がなく、特定の抗体の最高レベルの発現がもたらされる。
【0036】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度ができるだけ速くなるように、IGSが最適化される。
【0037】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度が、達成可能なもっとも大きな速度にならないように、IGSが最適化される。
【0038】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度が低い速度となるように、IGSが最適化される。
【0039】
本発明のジシストロン性メッセージは、特定の抗体分子の重鎖及び軽鎖をコードしている。抗体は、抗体全体でもFab又はFab’断片など特定の断片でもよい。抗体はまた、キメラ又はヒト化された抗体であってもよい。
【0040】
本発明のジシストロン性メッセージは、合成DNA、cDNA又はゲノムDNAもしくはいかなる組合せを含んでいてもよい。
【0041】
特定の抗体のコードDNA配列は、当分野の技術者に周知の方法で得られる。たとえば、抗体重鎖及び軽鎖の一部又は全部をコードするDNA配列は、所望により、決定されているDNA配列から又は対応するアミノ酸配列に基づいて合成できる。
【0042】
本発明のジシストロン性メッセージでは、IGSによって連結される、特異的抗体分子の重鎖又は軽鎖をコードするDNA配列を調製するために、分子生物学の標準技法が使用できる。所望のDNA配列は、オリゴヌクレオチド合成技法を使用して、完全又は部分的に合成できる。部位特異的突然変異及びポリメラーゼ連鎖反応(PCR)技術が、適宜に使用できる。
【0043】
本発明のジシストロン性メッセージは、抗体鎖の1つをコードするDNA配列と融合させたエフェクター又はレポーター蛋白をコードするDNA配列を含むことができる。
【0044】
本発明のジシストロン性メッセージには、抗体鎖の1つをコードするDNA配列と融合されたペプチド結合鎖をコードするDNA配列も含むことができ、それによって、その後にエフェクター又はレポーター蛋白もしくは分子をジシストロン性メッセージから発現される抗体に付加することが可能になる。
【0045】
本発明のジシストロン性メッセージには、抗体鎖を、ペリプラズム又は細胞外部を標的として送ることができるように、抗体鎖の1つ又は両方をコードするDNA配列の上流に融合された分泌シグナル配列を含んでいてもよい。
【0046】
好ましくは、分泌シグナル配列は、OmpAペプチド配列である。
【0047】
第2の態様において、本発明は、本発明の第1の態様によるジシストロン性メッセージを含む発現ベクターを提供する。
【0048】
好ましくは、発現ベクターのフレームワークをなすのは、pTTOである。pTTO発現ベクターは、大腸菌中で組み換えタンパクが可溶性でペリプラズムに蓄積するように設計されている。このベクターは、以下の主な特徴を有する。
【0049】
(i)テトラサイクリン耐性マーカー:耐性遺伝子の産物によって不活性化されない抗生物質、したがってプラスミドを含む細胞の選択が維持される;
(ii)低コピー数:colE1由来のレプリコンを含むプラスミドと整合性があるプラスミドp15A由来の複製起点;
(iii)クローン遺伝子の転写のための強力で誘導性のtacプロモータ;
(iv)lacIq遺伝子:IPTG/アロラクトースで誘導されるまでtacプロモータを抑制状態に維持するLacリプレッサー蛋白の構成的発現をもたらす;
(v)OmpAシグナル配列:クローン化された遺伝子のペリプラズムでの分泌をもたらす;
(vi)短いLacZペプチドに対してOmpAシグナル配列を翻訳カップリングして、効率的な転写開始をもたらす。
【0050】
第3の態様において、本発明は、本発明の第1態様によるジシストロン性メッセージを含むクローニングベクターを提供する。
【0051】
発現及びクローニングベクターを構築する一般的方法、トランスフェクション法、及び培養法は、当分野の技術者に周知の方法である。
【0052】
第4の態様において、本発明はまた、特定の抗体分子をコードするDNAの発現をもたらすのに適した条件下で、本発明の発現ベクターで形質転換された細菌ホスト細胞を培養し、その抗体分子を単離することを含む特定の抗体分子を産生する方法であって、抗体の発現レベルが最適化されている方法を提供する。
【0053】
適切な細菌ホスト細胞であれば何なる細胞であっても、本発明によるジシストロン性メッセージによってコードされる特定の抗体分子の重鎖及び軽鎖を発現させるのに使用することができる。しかし、好ましくは、大腸菌のホスト細胞を使用する。他の微生物系を使用してもよい。
【0054】
抗体分子は、細胞から分泌されることができ、また、適切なシグナル配列によってペリプラズムを標的とすることができる。あるいは、抗体分子は、細胞の細胞質内に蓄積されることもできる。産生された抗体及び使用された方法に応じて、抗体分子が再び折りたたまれることを可能として、機能的なコンフォーメーションを形成することが望ましい。抗体分子を再び折りたたませることを可能とする手順は、当分野の技術者に周知である。
【0055】
本発明によるジシストロン性メッセージによって産生される抗体分子は、特定の抗体が製薬上許容される賦形剤、希釈剤、又は担体と共に含まれる治療用又は診断用組成物を作製するために使用することができる。
【0056】
抗体分子は、治療用組成物又は診断用組成物中で唯一の有効成分であっても、たとえば、抗T細胞、抗IFNγ又は抗LPS抗体などの他の抗体成分、あるいはキサンチンなどの非抗体成分を含む1つ又は複数の成分が一緒であってもよい。
【0057】
本発明によって産生された特定の抗体分子は、その抗体分子を使用する治療法に従って任意の適切な形態及び量で投与することができる。
【0058】
適切な投与形態には、非経口的投与に適する形態、たとえば注射又は点滴、たとえばボーラス注射又は連続点滴が挙げられる。産物を注射又は点滴投与する場合、油性又は水溶性媒体中で、懸濁液、溶液又は乳濁剤の形をとってもよいし、懸濁剤、保存剤、安定剤、及び/又は分散剤などの製剤用添加剤(formulatory agent)を含んでいてもよい。
【0059】
あるいは、抗体分子は、使用する前に適当な滅菌液で溶解して使用されるような乾燥形態であってもよい。
【0060】
抗体分子が経口投与に適している場合、たとえば、抗体断片の場合、配合物は、有効成分の他に経口投与組成物の配合に使用される適切な添加剤を含んでいてもよい。
【0061】
治療用及び診断用組成物は、単位剤形であってもよく、その場合、各単位投与量は、有効量の特定の抗体分子を含む。また投与量は、年齢及び患者の状態によって選択される。
【0062】
抗体分子の半減期が短い場合(たとえば、2から10時間)、1日に1回又は複数回の投与が必要となる。一方、抗体分子の半減期が長い場合(たとえば、2から15日)、1日に1回、1週間に1回の投与、又は1カ月又は2カ月ごとに1回の投与しか必要でないこともある。
【0063】
本発明を、さらに、添付の図面を参照して以下の実施例で単に例として説明する。
【実施例】
【0064】
抗TNFα抗体をコードするジシストロン性メッセージ
本発明のジシストロン性メッセージを使用して、抗TNFαFab’抗体断片の高レベル発現を達成した。上流側シストロンは、抗体の軽鎖をコードし、下流側シストロンは、抗体の重鎖をコードしていた。OmpAシグナルペプチドをコードするDNA配列を、軽鎖及び重鎖の各々をコードするDNAの5’末端に融合させて、ペリプラズムに対する効率的な分泌を可能にした。
【0065】
重鎖の発現のレベルを変えるため、種々のIGSの範囲をコードする一連のオリゴヌクレオチドカセットをジシストロン性メッセージに使用した。異なるカセットを使用して、重鎖の翻訳開始速度を変更した結果、翻訳された重鎖産物の蓄積が、ある範囲の速度で生じた。
【0066】
最適配列を実験的に決定することが可能になるように一連の4つのIGSを設計した。最高量の可溶性Fab’の蓄積を生じるIGS変異体を、振盪させたフラスコを誘導装置として、又は発酵槽発現を決定手段として使用して、実験的に選択した。驚くべきことに、異なるFab’に対して異なるIGSが選択された。これは、それぞれ新しいFab’断片を発現させるには、実験的な選定が必要であることを示唆する。
【0067】
(実験)
材料と方法
一般的微生物学及びDNA操作技法
形質転換及び慣用培養物増殖用に、大腸菌株INVαF’(Invitrogen、De Schelp、Netherlands)を使用し、発現の研究には、大腸菌株W3110(ATCC # 27325)を使用した。DNA制限酵素及び修飾酵素は、ベーリンガーマンハイム(Boehringer Mannheim(Lewes、East Sussex、UK))及びニューイングランドバイオラボ(New England Biolabs(Hitcher、Herts、UK))から入手した。プラスミドの調製は、プラスミド精製キット(QIAGEN、Crawley、West Sussex、UK)を使用して行った。DNA断片の精製は、キアゲン社のスピンカラムを使用して行った。DNA断片は、GeneCleanプロトコル(BIO 101)を使用してアガロースから精製した。オリゴヌクレオチドは、Oswel Oligonucleotide Serviceから供給され、40nMのスケールで合成した。PCRは、推奨のように、パーキンエルマー(Perkin Elmer)のAmplitaqを使用して行った。DNAシークエンス反応は、ABI Prism DyeDeoxyチェーンターミネーターキットを使用して行い、ABI373A 自動シークエンサー(PE Applied Biosystems、Warrington、Cheshire、UK)で実施した。データをプログラム:オートアセンブラー(PE Applied Biosystems)を使用して分析した。
【0068】
振盪フラスコによる誘導
大腸菌W3110培養物を、テトラサイクリン(7.5μg/ml)を補給した液体ブロス中で生育させた。誘導するために、一晩培養した新鮮な培養物(30℃で生育させた)を、2Lのバッフル付きフラスコ中、液体ブロス200mlで、OD600=0.1まで希釈し、オービタルインキュベータ中30℃で増殖させた。OD600=0.5で、IPTGを200μMまで加えた。サンプル(OD用に正規化)を間隔をおいて採取した。
【0069】
ペリプラズム抽出
培養物サンプルを氷上で(5分)冷却し、次いで細胞を遠心分離によって採集した。抽出緩衝液で懸濁した(100mM Tris HCl、10mM EDTA;pH7.4)後に、サンプルを一晩30℃でインキュベートし、遠心分離で清澄させた。
【0070】
アッセンブリーアッセイ
Fab’の濃度をELISAで求めた。プレートを4℃一晩、抗ヒトFd6045(コーティング緩衝液、生理的食塩水中2μg/ml、ウエルあたり100μ1)(欧州特許第491031号参照)でコートした。洗浄後、ウエルあたり100μlのサンプルを充填し、精製されたA5B7γ−1 Fab’(欧州特許第491031号参照)を、初期濃度2μg/mlで標準として使用した。サンプルをプレートの端から端までサンプルコンジュゲート緩衝液(リットル当たり:トリス アミノメタン6.05g、塩化ナトリウム2.92g、ツイーン20を0.1ml、カゼイン(0.2%)を1ml)で2倍に連続希釈し、プレートを1時間室温で攪拌しながらインキュベートした。プレートを洗浄、乾燥し、次いで、抗ヒトCκ(GD12)ペルオキシダーゼ100μl(サンプルコンジュゲート緩衝液中で希釈したもの)を加えた。インキュベーションを室温で1時間、攪拌しながら行った。プレートを洗浄、乾燥し、次いで基質溶液100μl(10ml酢酸ナトリウム/クエン酸溶液(0.1M、pH6)、過酸化水素水溶液100μl、テトラメチルベンジジン溶液(ジメチルスルホキシド中10mg/ml)を100μ1)を加えた。基質添加の4から6分後に、630nmで吸光度を読み取った。
【0071】
発酵
大腸菌W3110培養物を、振盪フラスコで、テトラサイクリン(7.5μl/ml)を補給した液体ブロス中で、30℃で、OD600=1.0になるまで生育させ、この培養物100mlを使用して、1.5Lのバッチ供給式培養発酵槽に入れたSM6培地(+グリセロール)(欧州特許第651803号)1Lに播種した。pHを50%アンモニウムと5%硫酸とで7.0に調節した。溶存酸素濃度を可変式攪拌で30%に維持した。テトラサイクリンは、発酵培地に含まれていなかった。初期のグリセロール濃度は3%w/vであり、発酵中、培養物がOD600≒60に達した後、グリセロールが利用可能でなくなるような場合がもう一回あった時には、さらにグリセロールを供給した。培養物を30℃で、OD600=55まで生育させ、50%ラクトースを120ml添加した:利用可能なグルコースが利用された後、ラクトースが誘導される。20時間後、さらにラクトース60mlを一度に添加し、誘導後、25から30時間発酵をモニターし、サンプル(OD用に正規化されたもの)を間隔を置いて採取した。
【0072】
(結果)
プラスミドpTTO−1及びpTTO−2の構築
プラスミドpTTQ9をアマーシャム(Amersham)から入手した。これを図1に示す。アリコット(2μg)を制限酵素SalI及びEcoRIで消化し、消化物を1%アガロースゲルにかけ、大きなDNA断片(4520bp)を精製した。2つのオリゴヌクレオチドを合成し、これを互いにアニールしたところ、図2に示すOmpAポリリンカー領域をコードしていた。この配列は、pTTQ9の制限酵素消化によって得られたSalI及びEcoRI末端と適合する付着端を有する。このオリゴヌクレオチドの「カセット」をpTTQ9ベクター中に挿入した。これによって、SalI部位は再生されないが、EcoRI部位は保持される。このカセットは、大腸菌外膜タンパクOmp−Aのシグナル配列の最初の13アミノ酸をコードし、その前にOmpA遺伝子のシャインダルガノリボソーム結合部位がある。これに加えて、酵素XbaI、MunI、StyI及びSplIの制限部位が存在する。MunI及びStyI部位は、OmpAシグナル配列のコード領域内にあり、遺伝子の挿入用の5’クローニング部位として意図される。このカセットを構成する2つのオリゴヌクレオチドを、5pモル/μlの濃度で混合し、湯浴で95℃まで3分かけて加熱し、室温までゆっくり冷却することによって、互いにアニールさせた。次に、アニールした配列を、SalI/EcoRIで切断したpTTQ9に連結した。得られたプラスミド中間産物は、pTQOmpと呼ばれ、DNAシーケンスで検証した。
【0073】
中間産物のアリコットを、次に、SspI及びEcoRI(2350bp精製断片)並びにEcoRI及びXmnI(350bp精製断片)で切断した。2350bp断片は、翻訳終結領域及びlacIq遺伝子をコードし、また350bp断片はtacプロモータ、OmpAシグナル配列、及びマルチクローニング部位をコードしている。プラスミドpACYC184(New England Biolabs、図3)を、StyIで消化し、大豆ヌクレアーゼで処理して、平滑末端を生成し、次にPvuIIで消化した(2348bp精製断片−この断片はテトラサイクリン耐性マーカー及びp15A複製起点をコードする)。この断片をアルカリホスファターゼで処理して、(自己連結を防止するために)5’末端のリン酸を除去し、他の精製断片と連結した。得られたプラスミドをpTTO−1と名付けた。これを図4のマップに示す。図5はpTTO−1の完全なDNA配列を示す。プラスミドpHC132由来のSplI−EcoRI断片であるヒトIg軽鎖κ定常ドメインを挿入すると、pTTO−2が得られた(図6)。
【0074】
Fab’可変領域のpTTO−2への挿入
Fab’TNFα可変領域遺伝子は、国際出願GBOl/02477号の配列番号8由来の配列を含む抗体全体を哺乳類細胞発現用ベクターからPCR増幅することによって生成した。OmpAシグナル配列をコードし、pTTO−2中にフレームを合わせて挿入するためのMunI制限酵素部位を含むDNAを、各遺伝子の5’末端に付加し、その生来のIgリーダーと置き換えた。
【0075】
次に、精製されたVL遺伝子(MunI/SplI)を、pTTO−2のMunI/SplI部位に挿入して、軽鎖中間産物pTTO(TNFαL)を得た。
【0076】
重鎖VH遺伝子を、中間ベクターpDNAbEng−G1(図7)によって、MunI部位とApaI部位の間に挿入し、pDNAbEng(TNFαH)を得た。重鎖遺伝子を、EcoRI断片として、このプラスミドからpTTO(TNFαL)のEcoRI部位に挿入することによって、大腸菌発現プラスミドpTTO(TNFα)(図8)を得た。
【0077】
pTTO(TNFα)のIGS変異体の構築
一連の4つの遺伝子間配列(IGS)変異体を、TNFαFab’用の最適IGSの実験的決定が可能になるように設計した(図9)。IGS1及びIGS2は、非常に短い遺伝子間配列(それぞれ、−1及び+1)を有し、緊密に繋がって翻訳されることが予想され、それらのSD配列(下線)がわずかに異なっている。これらの2つの配列が、高度の翻訳開始レベルをもたらす可能性が高い。IGS3及びIGS4は、終結コドンと開始コドンの距離(+13)が長く、配列の構成が異なる。IGS3のSD配列は、IGS1、2又は4のSD配列より働きが強い。全配列を2次構造について(m/foldプログラム(Jaeger,J.A.ら、Methods Enzymology、183:281−306(1990);part of GCG Wisconsin Package、Accelrys)を使用して)調べ、できるだけ「最適化」した。しかし、2つの鎖が翻訳上緊密にカップリングしているので、リボソームが解離しない。これは、mRNAが「裸」でない可能性があり、2次構造の形成を妨げることを意味する。
【0078】
IGS変異体は、pTTO(TNFα)のSacI−NotI消化によって調製したベクター中に2つの断片を連結することによって構築した。pDNAbEng(TNFα)のMunI−NotI消化によって調製した挿入断片を精製し、各IGSカセット(SacI−MunI)と一緒にベクター中に連結し、4つの発現プラスミドpTTO(TNFα IGS−1)、pTTO(TNFα IGS−2)、pTTO(TNFα IGS−3)及びpTTO(TNFα IGS−4)を得た。
【0079】
TNFαIGS変異体の振盪フラスコ発現分析
4つのIGS変異体ベクター及び元の発現ベクターPTTO(TNFα)をそれぞれ使用して、大腸菌株W3110を形質転換した。次に形質転換された大腸菌を、Fab’の発現について、上記に記載したように振盪フラスコで分析した。典型的実験結果を図10に示す。
【0080】
異なる遺伝子間配列は、異なる発現プロフィルをもたらした。IGS1及びIGS2は、ペリプラズムFab’を迅速に蓄積し、誘導から1時間後がピークであり、その後、回復レベルが落ち込んだ。IGS1については、ピークが大きくなるほど急激な落ち込みとなった。こうした結果は、これら構築物では翻訳カップリングが緊密であることから予想されるように、合成のレベルが高いことと整合していた。IGS1は、明らかに、IGS2よりも高いレベルの重鎖発現をもたらした。この場合、1時間後のピークの後に、ペリプラズム発現レベルが落ちるので、高レベルの発現に対する耐容性が不十分と思われる。これは、IGS1培養物の増殖プロフィル(図示せず)で観察され、誘導から1時間後にピークに達した後落ち込み、細胞死及び溶菌が示唆された。
【0081】
IGS3はFab’をよりゆっくりと蓄積し、誘導2時間後に、高いピーク値(325ng/ml/OD)を有するピークを示した後、レベルが落ち込んだ。この培養物の増殖は、誘導の3時間後まで続き、より高い生物体量に達した(図示せず)。これは、重鎖の合成レベルがより低いことと整合する。
【0082】
IGS4は、物質をやはりよりゆっくりした速度で蓄積し、他の3つの構築物の示す高い生産性のピークに達することはなかった。
【0083】
IGS変異体は全て、オリジナルのpTTO(TNFα)ベクターより著しい成績を収めた。異なるIGS配列は、異なる翻訳開始速度をもたらすという仮説が、これらの実験結果から裏付けられた。TNFαFab’については、高い重鎖の翻訳開始速度に対する耐容性が不十分であり、したがって最適ではないと思われる。IGS3によってもたらされるようなより遅い速度では、より良好な増殖特性がもたらされ、その結果、時間がたつに連れ、より良い収率で蓄積される。
【0084】
発酵槽(図示せず)での生産性を比較した後、IGS3構築物が最高の収量であったのでこれを選択した。
【0085】
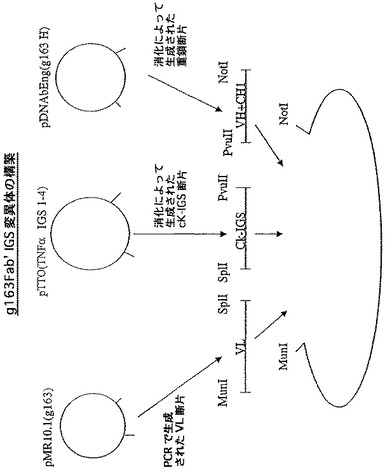
異なるFab’抗体をコードするジシストロン性メッセージ:g163Fab’IGS変異体の作成
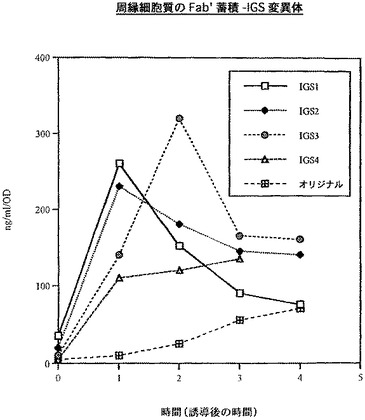
上記に記載の1組のpTTO/IGSベクターを使用して、もう1つのFab’の発現分析を行った。最初に、ヒト化Fab’g163を使用した。4通りの連結の仕方を使用して、Fab’g163の4つのIGS変異体(図11)を生成した。g163のVH遺伝子(哺乳類発現ベクターpγ−4からPCRによって得た)のMunI−ApaI断片をプラスミドpDNAbEngGIに挿入して、重鎖の中間産物を生成した。この中間産物をPvuIIとNotIで切断し、g163VH+CH1断片を生成した。プラスミドpTTO(TNFα)をMunIとNotIで切断し、大きな断片を精製してベクター断片を得た。PCRによって、哺乳類発現ベクターpMR10.1(gl63)からMunI−SplI VL断片を生成した。最後に、上記の4pTTO(TNFα)IGS変異体のSplI−PvuII消化によって、Cκ−IGS断片を生成した。次に、別々の4通りの連結を行い、構築物pTTO(gl63 IGS−1)、pTTO(g163 IGS−2)、pTTO(g163 IGS−3)、及びpTTO(gl63 IGS−4)を生成した。これら構築物の振盪フラスコ分析の結果を図12に示す。
【0086】
IGS−1及び−2が迅速なFab’蓄積をもたらし、誘導の1時間後にピークとなり、その後急激に低下するという点で、この発現プロフィルは、Fab’TNFαで見られる発現プロフィルと非常に類似していた。IGS−3は、より遅い蓄積開始速度をもたらし、より遅い時間(誘導開始2から3時間後)にピークに達し、その後より遅い速度で低下する。IGS−4構築物は、誘導の経過時間を通してゆっくり立ち上がり、この場合もやはり他の構築物の示す高い収率に達することはなかった。IGS−1及び−2では、やはり誘導のわずか1時間後(図示せず)に、生物体量がピークに達することが発現プロフィル及び増殖プロフィルで示されるため、IGS−3を選択し、発酵槽で高い収率(図示せず)まで発現させた。
【0087】
プラスミドpTTODの構築
Fab’のコード戦略を簡略化するために、プラスミドpTTO−1からPvuII(3部位)、EcoRV(2部位)及びApaI(1部位)に対するフレームワークの制限部位を取り除くことによって、プラスミドpTTODを得た。これらに変更を加える際、lacIq遺伝子及びテトラサイクリン耐性遺伝子の蛋白コード配列を変えずに、遺伝情報を発現しない「サイレント」な変化をDNAレベルで加えた。PCR戦略を使用して、「サイレント」変化(これらの制限部位を除去した)を有するプライマーを設計し、これを使用して親プラスミド(pTTO−1)の一部分を増幅した。フランキング制限部位(未変更)を使用して、親プラスミド中の配列をこれらの修飾配列で置き換えた。プラスミドpTTODをこの多段階方法で作成した。ベクターpTTO内の既存のg163Fab’遺伝子を、遺伝子に隣接するユニークなPstI及びEcoRI部位を使用してpTTOD中にトランスファーし、pTTOD(g163)IGS変異体1から4を作成した。pTTOD(g163IGS−3)の制限マップを図13に示す。
【0088】
IGS変異体としての他のFab’の発現
TNFα及びg163の他に、さらにいくつかのFab’が、2個以上のIGS変異体として発現した。それには、g165及びgA33と名付けられたFab’が含まれる。図14は、振盪フラスコで比較されたpTTOD(gl65)IGS−1から−3を示す。TNFα及びg163とは対照的に、IGS−2及びIGS−1は、IGS−3より成績がよい。このFab’の発現は、たとえ迅速な速度でも、ホスト細胞による十分な耐容性があるようである。IGS−2を発現する培養物は、誘導期間中(図示せず)ずっと増殖し続けた。図15は、このFab’を有するIGS−1から4の発酵槽での比較を示し、これは、基本的に振盪フラスコで行われた観測を再現している。IGS−2は、収率が最高の変異体であることが確認された。発酵槽中で、Fab’gA33、pTTOD(gA33)IGS−2及び−3を比較すると、両者は同様の収率を示した(図16)。したがって、最適の収率を得るためには、Fab’が異なれば異なるIGS配列が必要となる。
【0089】
振盪フラスコ対発酵槽分析
振盪フラスコでは、プロモータの脱抑制を、発現の迅速な誘導をもたらすIPTGによって行った。発酵槽における誘導方式は異なり、ラクトース(細菌によってアロラクトースに変換される)を使用して、より穏やかにプロモータのスイッチを入れる。こうした誘導の速度論が異なるにもかかわらず、振盪フラスコでは、発酵槽中で構築物がどのような対比を示すかの示唆を与えることが明らかになった(図14及び15参照)。
【0090】
最適な収率を得るためには、Fab’が異なれば異なるIGSが必要になるという原理がはっきり証明されている。本明細書に記載した本システムの新規性は、高レベルのFab’の発現を達成する手段として、ジシストロン性メッセージの第2遺伝子の翻訳開始速度を最適にするために、IGSカセットを使用することにある。上記に記載したシステムでは、軽鎖の発現は変わらないままであり、重鎖の翻訳開始速度のみが最適化される。この戦略がなぜ成功したかについて、2つの可能な説があげられる。
【0091】
(i)もし十分な軽鎖が合成されて、重鎖が過剰になることを防ぐとしても、軽鎖の発現がFab’の全蓄積レベルに影響を与えることはほとんどない。過剰の軽鎖は通常何の問題もなく許容される。可溶性発現の効率を規定するのは重鎖の翻訳レベルである;あるいは
【0092】
(ii)重鎖の最適化は、実際、軽鎖発現速度が一定になるように調節され、したがって、2つの鎖のレベルのバランスがとられる。可溶性発現の効率を規定するのは、重鎖の折りたたみ/分泌速度それ自体ではなく、2つの異種鎖の発現のバランスである。
【0093】
上記に記載された実施例は単なる模範例にすぎず、以下の特許請求の範囲に定義される本発明の範囲を限定するものではないことを理解されたい。
【技術分野】
【0001】
本発明は、特定の抗体分子の重鎖及び軽鎖が、2つのシストロンが最適化された遺伝子間配列によって連結されているジシストロン性(dicistronic)メッセージ中に存在するDNAによってコードされる抗体の産生方法に関する。
【背景技術】
【0002】
抗体は、特定の抗原を認識する能力によって、医学及び生物工学において非常に有用で効果的なツールとなっている。選択されたある種の細胞上の抗原に特異的な抗体は、これらの抗体を、選択した細胞に向けるために使用されてきた。いくつかのケースでは、細胞上のレセプターに抗体が結合すると、細胞の機能に影響を及ぼすことがわかっている。また、治療剤及び診断剤は、選択した細胞に、これらの薬剤を特異的に送り込むために、これらに抗体を結合させることが行なわれている。この技術は、特に癌細胞のターゲティングに使用されている。また、抗体は、ウイルス又は細菌に感染した細胞上の抗原を標的とするため、TNFαなどの他の分子を標的とするため、又は分析用に使用されている。生物工学では、抗体は、精製及び触媒反応において、プローブなどの使用に多く利用されている。
【0003】
すべての完全な抗体分子は、4つのポリペプチド鎖、すなわち、2つの同一の重鎖及び2つの同一の軽鎖から構成される。各鎖は、可変ドメインと定常ドメインを含んでいる。軽鎖は、2つのドメイン、つまり、VL及びCLを含み、重鎖は、少なくとも5つのドメイン、VH、CH1、ヒンジ、CH2、及びCH3を含み、場合によっては、更にCH4を含んでいる。4つの鎖は、いつでもおおよそ同じ様式で組織されている。すなわち、2つの重鎖が、少なくとも1つのジスルフィド結合で互いに連結され、各重鎖はまた、軽鎖のうちの1本とジスルフィド結合で連結され、2つの軽鎖はそれぞれ別の重鎖に連結する。
【0004】
抗体分子全体は、ほぼY字型で、基本的に2つの主機能部から構成される。第1の機能部は、特異的抗原を認識する役割を担い、Yの腕の上部を形成する。各部の抗原結合領域は、1つのVHドメインと1つのVLドメインを含む。各可変ドメインは、3つの超可変領域を含み、他方の鎖の3つの超可変領域と互いに抗原結合部位を形成する。これら超可変領域は、相補性決定領域(CDR1,CDR2,CDR3)として知られる。これらCDRはループを形成し、フレームワーク領域上に保持されている。VH及びVLを含む領域は、抗体ごとに可変であるため、「V」領域として知られる。
【0005】
第2の機能部は、抗体に認識された抗原を処理する他の細胞のエフェクター機能を誘発ーする役割を担い、Yの腕の下部とステムから形成される。この領域は、比較的定常であるため、定常「C」領域として知られる。この領域は、CLドメインと重鎖のCドメインすべてを含む。
【0006】
軽鎖には、κ及びλの2種類があり、重鎖には、α、δ、γ、ε及びμの5種類がある。抗体のクラスは、重鎖の型によって決定され、それぞれ、IgA、IgD、IgE、IgG及びIgMである。
【0007】
酵素切断によってその定常領域の一部が除去された抗体断片も、医学及び生物工学で使用される。これらには、Fab、Fab’、F(ab’)2及びFv断片が含まれる。
【0008】
抗体を産生する脾臓細胞と骨髄腫細胞を融合させて、ハイブリドーマを得ることによって、大量のモノクローナル抗体(特別な抗体特異性を有する)を直接に産生することが知られている(Kohler,G.and Milstein,C.「所期の特異性を有する融合細胞分泌抗体の連続培養」(Continuous cultures of fused cells secreting antibody of predefined specificity)Nature、256、495−497(1975))。しかし、このような抗体は、ヒトにおいて免疫原性でないため、ヒトの治療に使用することは適切ではない。多くのモノクローナル抗体は、非ヒト細胞で産生される。
【0009】
複数の抗体の有用な特性を組み合わせることを可能にして、1つの新しい抗体を作る組み換えDNA技術が開発されている。1つの抗体の完全な可変領域を含む抗体結合部位を、別の抗体の定常領域に連結させてキメラ抗体を産生することが、欧州特許第0120694号(Celltech Limited)、欧州特許第0125023号(Genentech Inc.and City of Hope)、欧州特許第0171496号(Research Development Corporation,Japan)、欧州特許第0173494号(Stanford University)及び特許国際公開第86/01533号(Celltech Limited)に記載されている。
【0010】
たとえば、特許国際公開第86/01533号は、ネズミ可変領域をヒトの定常領域と結合させたキメラ抗体の調製を記載している。しかし、キメラ非ヒト/ヒト抗体には非ヒトドナー由来の残基をかなり比率で含むので、特に長期間が投与すると、抗体が潜在的に有害な免疫応答を誘発する可能性がある(Begentら、Br.J.Cancer,62,487(1990))。
【0011】
抗体の「ヒト化」とは、非ヒト/ヒトキメラ抗体を、ヒト免疫系に対してヒト抗体により似たものにする技術で、上記に記載した望ましくない免疫反応を解消するために開発された。欧州特許第0239400号(Winter)は、完全なネズミの可変領域を使用する代わりに、ネズミのモノクローナル抗体のCDRをヒト抗体の可変ドメインのフレームワーク領域上に移植する方法について記載している。したがって、ネズミ由来であるのは、抗原結合ドメイン自体を形成するCDRのみで、その他の残基がヒト由来である。
【0012】
Reichmannら(「治療用ヒト抗体の再形成」”Reshaping human anitbodies for therapy”、Nature、332、323−324、1988)は、可変ドメインにある他のヒト残基を非ヒトドナーの等価物に変換して、抗体結合活性を改善することが有益であることを見出した。このような残基は、ヒト重鎖のポジション27に見つかった。ヒトのセリンから対応するラットの残基(フェニールアラニン)に変換すると、抗体結合能力の改善がもたらされた。このような残基が別にポジション30で見出された。しかし、上記に記載されたポシション27の変更に加えて、重鎖のポジション30にヒトのセリンからラットのチロシンへの変更を含む構築物は、セリンからフェニールアラニンへの変更をポジション27のみに有するヒト化された抗体に比べて、優位に結合活性を変更しなかった。
【0013】
重鎖残基27及び30は、CDR1近隣の構造ループ内にある。Queenら(特許国際公開90/07861)は、CDRと相互作用する他の残基もまた、抗体結合親和性を決定する際に重要であると推測した。このことを念頭において、Queenらは、ヒト化抗体を設計する際、どの残基がドナーに由来し、どれがアクセプタに由来すべきかを決定するための4つの判定基準を提案した。より決定的な分析で、Adairら(特許国際公開91/09967)が、ヒト化抗体の設計を可能にする残基番号の優先度を開示した。
【0014】
Vaughanら(Nature Biotechnology、16、535−539,1998)を含めてCDRを移植された抗体について論じた数多くの論文が公表されている。
【0015】
定常領域が、治療剤又は診断剤として作用可能なエフェクター又はレポーター分子と融合した抗体複合体も記載されている(特許国際公開第95/01155号、米国特許第3927193号、第4331647号、第4348376号、第4361544号、第468457号、第4444744号、第4460459号及び第4460561号並びに以下の総説:Waldmann,T.A.、Science;252、1657(1991);Koppel,G.A.、Bioconjug.Chem.、1、13(1990);Oeltmann,T.N.and Frankel,A.E.、FASEB J.、5、2334(1991);及びvan den Bergh,H.E.、Chemistry in Britain、May 1986、430−439)。
【0016】
適切なホスト細胞を、1つは求める抗体の重鎖をコードするDNA配列を含み、1つはその抗体の軽鎖をコードするDNA配列を含む2つの発現ベクターでトランスフェクトして、正常、キメラ又はヒト化された抗体を産生することが知られている(特許国際公開第91/09967号)。また、適切なホスト細胞を、求める抗体の重鎖をコードするDNA配列とその抗体の軽鎖をコードするDNA配列との両方を含む発現ベクターでトランスフェクトすることも知られている。後者の例では、重鎖及び軽鎖をコードするDNA配列はどちらも、それ自体の個々のプロモータの制御下(特許国際公開第91/09967号)にあるか、ジシストロン性メッセージ中(Better,M.、Paul Chang,C.、Robinson,R.R.及びHorwitz,A.H.Escherichia coli:「活性キメラ抗体断片の分泌」(Secretion of an Active Chimeric Antibody Fragment.Science、240、1041−1043(1988))に存在する。
【0017】
ジシストロン性メッセージは、多くのヒトの癌の表面上に発現しているガングリオシド抗原に対するキメラマウスL6Fab抗体を産生するためにBetterらによって使用された。このケースでは、一部を切り詰めた重鎖(Fd)とκ軽鎖とが、確実に、物理的に近接して翻訳され、それらが正しく組み合わさり、分泌されることを企図して、ジシストロン性メッセージが選択された。ジシストロン性メッセージは、細菌中で機能できるのみであり、哺乳類及び細菌中においては、「1遺伝子1プロモータ」という概念が機能する。ジシストロン性メッセージでは、プロモータは第1のシストロンにのみ連結している。第2のシストロンは、ポリメラーゼによって第2のシストロンまで「読み通され」て転写され、2つのシストロンが単一のRNA分子で表されることになる。2つのコードDNAシストロンは、「遺伝子間配列」又は「IGS」として知られる、ある長さのDNAによって分離される。このIGS領域は、DNAから転写されるRNA分子中にも存在する。
【0018】
転写開始速度を最適化することが、大腸菌で分泌される異種タンパクを高度に発現させるためには必要不可欠であることが、しばらく前から認識されている(Simmons,L.及びYansura,D.Nature Biotech、14、629−634(1996))。しかし、Fabが異なると、フレームワークとCDR配列が異なり、それによって大腸菌ペリプラズム内での折りたたまれ易さ(又は速度)の差を含めて異なる特性が分子に付与される。(Knappik,A.及びPluckthun,A.Prot Eng 8、81−89(1995))。たとえば、大腸菌のペリプラズム内で容易かつ迅速に折りたたまれて生来のコンフォーメーションをとるFab’は、高度の「耐容性をもつ」と思われ、迅速な翻訳に適応し、高度な蓄積を達成することができる。折りたたみがより不十分なFab’は、迅速に翻訳された場合、ホスト細菌による耐容性が不十分になると思われ、ホストの折りたたみ/分泌機構を飽和させ、ホスト細胞の生理に有害な影響を与える可能性がある。
【0019】
したがって、ホスト細胞発現ベクターシステムを使用して抗体を産生する場合、特定の抗体における高レベル発現は耐容性があるが、抗体が異なると、高レベルの発現は、おそらく分泌又は折りたたみの効率が異なるという理由で、細胞に対して毒性となる可能性がある。
【0020】
個々の抗体の発現レベルは、重鎖及び軽鎖の存在する量によって決まる。最大量の産生を得るためには、重鎖と軽鎖の比を、たとえば、両方の量が等しくするなどバランスさせなければならない。しかし、重鎖と軽鎖のどちらか一方の量が少ないと、その量が産生される抗体の量を制限することになる。過剰の重鎖蓄積は耐容性が特に不十分となる可能性がある。
【0021】
ジシストロン性メッセージによってコードされる抗体の場合、上流側シストロンは、特定の抗原特異性を有する抗体の重鎖と軽鎖のどちらか一方をコードするDNAを含むことができる。その場合、下流側シストロンは、それぞれ、上流側シストロンの重鎖又は軽鎖のパートナーをコードする。特定の抗体を所望の発現レベルで産生させるには、ジシストロン性メッセージの下流側シストロンでコードDNAに対応する抗体鎖の発現レベルを調節できれば有利である。
【先行技術文献】
【特許文献】
【0022】
【特許文献1】欧州特許第0120694号
【特許文献2】欧州特許第0125023号
【特許文献3】欧州特許第0171496号
【特許文献4】欧州特許第0173494号
【特許文献5】国際公開第86/01533号パンフレット
【特許文献6】欧州特許第0239400号
【特許文献7】国際公開90/07861号パンフレット
【特許文献8】国際公開91/09967号パンフレット
【特許文献9】国際公開第95/01155号パンフレット
【特許文献10】米国特許第3927193号
【特許文献11】米国特許第4331647号
【特許文献12】米国特許第4348376号
【特許文献13】米国特許第4361544号
【特許文献14】米国特許第468457号
【特許文献15】米国特許第4444744号
【特許文献16】米国特許第4460459号
【特許文献17】米国特許第4460561号
【特許文献18】国際公開第91/09967号パンフレット
【特許文献19】国際公開第91/09967号パンフレット
【非特許文献】
【0023】
【非特許文献1】Kohler,G.and Milstein,C.(Continuous cultures of fused cells secreting antibody of predefined specificity)Nature、256、495−497(1975)
【非特許文献2】Begentら、Br.J.Cancer,62,487(1990))
【非特許文献3】Reichmannら ”Reshaping human anitbodies for therapy”、Nature、332、323−324、1988)
【非特許文献4】Vaughanら(Nature Biotechnology、16、535−539,1998)
【非特許文献5】Waldmann,T.A.、Science;252、1657(1991)
【非特許文献6】Koppel,G.A.、Bioconjug.Chem.、1、13(1990)
【非特許文献7】Oeltmann,T.N.and Frankel,A.E.、FASEB J.、5、2334(1991)
【非特許文献8】van den Bergh,H.E.、Chemistry in Britain、May 1986、430−439
【非特許文献9】Better,M.、Paul Chang,C.、Robinson,R.R.及びHorwitz,A.H.Escherichia coli:Secretion of an Active Chimeric Antibody Fragment.Science、240、1041−1043(1988)
【非特許文献10】Simmons,L.及びYansura,D.Nature Biotech、14、629−634(1996)
【非特許文献11】Knappik,A.及びPluckthun,A.Prot Eng 8、81−89(1995)
【発明の概要】
【課題を解決するための手段】
【0024】
第1の態様において、本発明は、上流側シストロンが、抗体の重鎖と軽鎖のどちらかをコードするDNAを含み、下流側シストロンが、それぞれ対応する軽鎖又は重鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性メッセージであって、2つのシストロンが、最適化された遺伝子間配列(IGS)によって連結されており、産生される抗体が、抗TNFα抗体(たとえば、特許国際出願GBO1/02477参照)でもなく、抗ヒトキナーゼ挿入ドメインを含むレセプタ(抗KDR)抗体(たとえば、特許国際出願GB02/004619参照)でもないことを特徴とする、ジシストロン性メッセージを提供する。
【0025】
好ましくは、上流側シストロンは、抗体の軽鎖をコードし、下流側シストロンは対応するその重鎖をコードする。
【0026】
特定の抗体について所望の発現レベルを達成するために、IGSは、ジシストロン性メッセージにおける下流側シストロンに対応する抗体鎖の発現レベルが調節されるように最適化されている。
【0027】
本明細書では、IGSの長さを、上流側シストロンの終結コドンと下流側シストロン(両端を含まない)の開始コドンとの間のヌクレオチドの数であると定義する。IGSの長さは、下流側シストロンに対応するRNA部分の翻訳速度の決定に関係する。たとえば、遺伝子間配列が短いと、重鎖と軽鎖の間の翻訳のカップリングが非常に緊密になり、上流側シストロンによってコードされる抗体鎖の合成が終了した後、下流側シストロンによってコードされる抗体鎖の合成が開始される前に、翻訳を行うリボソームが、mRNAから十分に解離しなくなる可能性がある(Adhin,M.and van Duin、J.J.Mol.Biol.、213、811−818(1990);Andre,A.ら、FEBS Letts.、468、73−78(2000))。このような過程は、翻訳の再開始と呼ばれ、上流の遺伝子の終結コドンと下流遺伝子の開始コドンが非常に接近している場合にのみ生じる。速度論的に考察すると、このような過程により、翻訳において、IGSで別のリボソームがmRNA分子と結合する必要がなくなるので、下流側シストロンによってコードされるポリペプチドの発現が増加することになる。
【0028】
2つの遺伝子間が離れすぎていると、上流側シストロンの合成が終了した後にリボソームが解離するので、下流側シストロンの翻訳を開始するには新しいリボソームが必要となる。そのため、通常、翻訳開始効率が低下することになる。
【0029】
IGSにはシャイン−ダルガルノ(SD)リボソーム結合部位(16SrRNAと相補的である)が存在しているので、リボソームがIGS配列に結合し、下流側シストロンを翻訳することができる。翻訳の再開始が可能になるのに十分なほどシストロン間の距離が短い場合、上流側シストロン内にSD部位が存在すると、下流側シストロンの翻訳開始効率が上がることを示唆する証拠がいくつかある(Spanjaard,R.A.and van Duin,J.Nucl.Acids Res.、17、5501−5507(1989))。
【0030】
SD部位、及びSD部位の端とAUG開始コドンの間のヌクレオチド配列には、翻訳開始の強さに影響を与えるいくつかの特徴がある(Makrides,S.Microbiol.Revs.、60、512−538(1996)に総説される)。下流側シストロンの、SD部位とAUG開始コドンの間の距離は、そのような特徴の1つであり、SD部位自体の「効力(strength)」もその特徴の1つである。SD部位に結合する16SrRNA配列と100%相補的なSD部位は、通常、16SrRNA配列と部分的にしか相補的でないSD部位の場合よりも、発現は大きくなる。
【0031】
SD配列と開始コドン間領域の配列は、下流側シストロンの翻訳速度を決定する別の重要な要素である。距離及び配列は、開始コドンの周りでmRNAのとり得る2次構造に影響する(Makrides,S.Microbiol.Revs.、60、512−538(1996)参照)。開始コドンは、「ループ」中に存在するはずで、「ステム」内には閉じ込められていないが、SD配列はその逆である。したがって、IGSの配列と長さを変更することによって、翻訳カップリングの程度及び/又は翻訳開始の強さを変更することができ、それによって、下流側シストロンの翻訳のレベル、及びそれをコードする抗体鎖のその後の蓄積速度を変更することができる。
【0032】
本発明のジシストロン性メッセージのIGSは、2つのシストロンの最適な翻訳カップリングが実現されるように、長さ、配列及び2次構造が変更されている。
【0033】
本発明に使用される最適なIGS配列は、以下の方法を使用して実験的に決定することができる。この方法は、抗体分子をテスト用に挿入することが可能な一連のIGS変異体を含む一連の適切な発現ベクターを構築することを含む。各抗体の各IGS配列の実験を、発現ベクターで適切なホストを形質転換して、抗体の発現及び収率を分析することによって行うことができる。本発明に記載された、長さと配列の異なる1組のIGS配列を使用して、かかるベクターを構築することができ、その中から特定の抗体分子の最適なIGS配列を選択することができる。この配列には、IGS配列1から4が含まれる。
【図面の簡単な説明】
【0034】
【図1】ベクターpTTQ9を示す図である。
【図2】OmpAポリリンカー領域の配列を示す図である。
【図3】ベクターpACYC184を示す図である。
【図4】ベクターpTTO−1を示す図である。
【図5】ベクターpTTO−1の完全なDNA配列を示す図である。
【図6】ベクターpTTO−2を示す図である。
【図7】ベクターpDNAbEngG1を示す図である。
【図8】ベクターpTTO(TNFα)を示す図である。
【図9】大腸菌Fab’発現用の、4つの異なる遺伝子間配列をコードするオリゴヌクレオチドカセットを示す図である。
【図10】振盪フラスコ分析の結果として、IGS変異体pTTO(TNFαIGS−1、pTTO(TNFαIGS−2)、pTTO(TNFαIGS−3)及びpTTO(TNFαIGS−4)のペリプラズムFab’の蓄積を示す図である。
【図11】Fab’g163の4つのIGS変異体を生成するために使用する4通りの連結を示す図である。
【図12】振盪フラスコ分析の結果として、IGS変異体pTTO(g163 IGS−1)、pTTO(gl63 IGS−2)、pTTO(gl63 IGS−3)及びpTTO(g163 IGS−4)の可溶性Fab’の蓄積を示す図である。
【図13】ベクターpTTOD(g163 IGS−3)の制限マップを示す図である。
【図14】振盪フラスコ分析の結果として、IGS変異体pTTOD(g165 IGS−1)、pTTOD(g165 IGS−2)及びpTTOD(g165 IGS−3)の可溶性Fab’の蓄積を示す図である。
【図15】発酵槽比較分析の結果として、IGS変異体、pTTOD(gl65 IGS−1)、pTTOD(gl65 IGS−2)、pTTOD(gl65 IGS−3)及びpTTOD(g165 IGS−4)の可溶性Fab’蓄積を示す図である。
【図16】発酵槽比較分析の結果として、IGS変異体pTTOD(gA33 IGS−2)及びpTTOD(gA33 IGS−3)の可溶性Fab’蓄積を示す図である。
【発明を実施するための形態】
【0035】
好ましい一実施形態では、下流側シストロンによってコードされる抗体鎖が最高に発現されるように、IGSが最適化されている。これにより、下流側シストロンによってコードされる抗体鎖の量には制限がなく、特定の抗体の最高レベルの発現がもたらされる。
【0036】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度ができるだけ速くなるように、IGSが最適化される。
【0037】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度が、達成可能なもっとも大きな速度にならないように、IGSが最適化される。
【0038】
別の実施形態では、下流側シストロンの翻訳の翻訳開始速度が低い速度となるように、IGSが最適化される。
【0039】
本発明のジシストロン性メッセージは、特定の抗体分子の重鎖及び軽鎖をコードしている。抗体は、抗体全体でもFab又はFab’断片など特定の断片でもよい。抗体はまた、キメラ又はヒト化された抗体であってもよい。
【0040】
本発明のジシストロン性メッセージは、合成DNA、cDNA又はゲノムDNAもしくはいかなる組合せを含んでいてもよい。
【0041】
特定の抗体のコードDNA配列は、当分野の技術者に周知の方法で得られる。たとえば、抗体重鎖及び軽鎖の一部又は全部をコードするDNA配列は、所望により、決定されているDNA配列から又は対応するアミノ酸配列に基づいて合成できる。
【0042】
本発明のジシストロン性メッセージでは、IGSによって連結される、特異的抗体分子の重鎖又は軽鎖をコードするDNA配列を調製するために、分子生物学の標準技法が使用できる。所望のDNA配列は、オリゴヌクレオチド合成技法を使用して、完全又は部分的に合成できる。部位特異的突然変異及びポリメラーゼ連鎖反応(PCR)技術が、適宜に使用できる。
【0043】
本発明のジシストロン性メッセージは、抗体鎖の1つをコードするDNA配列と融合させたエフェクター又はレポーター蛋白をコードするDNA配列を含むことができる。
【0044】
本発明のジシストロン性メッセージには、抗体鎖の1つをコードするDNA配列と融合されたペプチド結合鎖をコードするDNA配列も含むことができ、それによって、その後にエフェクター又はレポーター蛋白もしくは分子をジシストロン性メッセージから発現される抗体に付加することが可能になる。
【0045】
本発明のジシストロン性メッセージには、抗体鎖を、ペリプラズム又は細胞外部を標的として送ることができるように、抗体鎖の1つ又は両方をコードするDNA配列の上流に融合された分泌シグナル配列を含んでいてもよい。
【0046】
好ましくは、分泌シグナル配列は、OmpAペプチド配列である。
【0047】
第2の態様において、本発明は、本発明の第1の態様によるジシストロン性メッセージを含む発現ベクターを提供する。
【0048】
好ましくは、発現ベクターのフレームワークをなすのは、pTTOである。pTTO発現ベクターは、大腸菌中で組み換えタンパクが可溶性でペリプラズムに蓄積するように設計されている。このベクターは、以下の主な特徴を有する。
【0049】
(i)テトラサイクリン耐性マーカー:耐性遺伝子の産物によって不活性化されない抗生物質、したがってプラスミドを含む細胞の選択が維持される;
(ii)低コピー数:colE1由来のレプリコンを含むプラスミドと整合性があるプラスミドp15A由来の複製起点;
(iii)クローン遺伝子の転写のための強力で誘導性のtacプロモータ;
(iv)lacIq遺伝子:IPTG/アロラクトースで誘導されるまでtacプロモータを抑制状態に維持するLacリプレッサー蛋白の構成的発現をもたらす;
(v)OmpAシグナル配列:クローン化された遺伝子のペリプラズムでの分泌をもたらす;
(vi)短いLacZペプチドに対してOmpAシグナル配列を翻訳カップリングして、効率的な転写開始をもたらす。
【0050】
第3の態様において、本発明は、本発明の第1態様によるジシストロン性メッセージを含むクローニングベクターを提供する。
【0051】
発現及びクローニングベクターを構築する一般的方法、トランスフェクション法、及び培養法は、当分野の技術者に周知の方法である。
【0052】
第4の態様において、本発明はまた、特定の抗体分子をコードするDNAの発現をもたらすのに適した条件下で、本発明の発現ベクターで形質転換された細菌ホスト細胞を培養し、その抗体分子を単離することを含む特定の抗体分子を産生する方法であって、抗体の発現レベルが最適化されている方法を提供する。
【0053】
適切な細菌ホスト細胞であれば何なる細胞であっても、本発明によるジシストロン性メッセージによってコードされる特定の抗体分子の重鎖及び軽鎖を発現させるのに使用することができる。しかし、好ましくは、大腸菌のホスト細胞を使用する。他の微生物系を使用してもよい。
【0054】
抗体分子は、細胞から分泌されることができ、また、適切なシグナル配列によってペリプラズムを標的とすることができる。あるいは、抗体分子は、細胞の細胞質内に蓄積されることもできる。産生された抗体及び使用された方法に応じて、抗体分子が再び折りたたまれることを可能として、機能的なコンフォーメーションを形成することが望ましい。抗体分子を再び折りたたませることを可能とする手順は、当分野の技術者に周知である。
【0055】
本発明によるジシストロン性メッセージによって産生される抗体分子は、特定の抗体が製薬上許容される賦形剤、希釈剤、又は担体と共に含まれる治療用又は診断用組成物を作製するために使用することができる。
【0056】
抗体分子は、治療用組成物又は診断用組成物中で唯一の有効成分であっても、たとえば、抗T細胞、抗IFNγ又は抗LPS抗体などの他の抗体成分、あるいはキサンチンなどの非抗体成分を含む1つ又は複数の成分が一緒であってもよい。
【0057】
本発明によって産生された特定の抗体分子は、その抗体分子を使用する治療法に従って任意の適切な形態及び量で投与することができる。
【0058】
適切な投与形態には、非経口的投与に適する形態、たとえば注射又は点滴、たとえばボーラス注射又は連続点滴が挙げられる。産物を注射又は点滴投与する場合、油性又は水溶性媒体中で、懸濁液、溶液又は乳濁剤の形をとってもよいし、懸濁剤、保存剤、安定剤、及び/又は分散剤などの製剤用添加剤(formulatory agent)を含んでいてもよい。
【0059】
あるいは、抗体分子は、使用する前に適当な滅菌液で溶解して使用されるような乾燥形態であってもよい。
【0060】
抗体分子が経口投与に適している場合、たとえば、抗体断片の場合、配合物は、有効成分の他に経口投与組成物の配合に使用される適切な添加剤を含んでいてもよい。
【0061】
治療用及び診断用組成物は、単位剤形であってもよく、その場合、各単位投与量は、有効量の特定の抗体分子を含む。また投与量は、年齢及び患者の状態によって選択される。
【0062】
抗体分子の半減期が短い場合(たとえば、2から10時間)、1日に1回又は複数回の投与が必要となる。一方、抗体分子の半減期が長い場合(たとえば、2から15日)、1日に1回、1週間に1回の投与、又は1カ月又は2カ月ごとに1回の投与しか必要でないこともある。
【0063】
本発明を、さらに、添付の図面を参照して以下の実施例で単に例として説明する。
【実施例】
【0064】
抗TNFα抗体をコードするジシストロン性メッセージ
本発明のジシストロン性メッセージを使用して、抗TNFαFab’抗体断片の高レベル発現を達成した。上流側シストロンは、抗体の軽鎖をコードし、下流側シストロンは、抗体の重鎖をコードしていた。OmpAシグナルペプチドをコードするDNA配列を、軽鎖及び重鎖の各々をコードするDNAの5’末端に融合させて、ペリプラズムに対する効率的な分泌を可能にした。
【0065】
重鎖の発現のレベルを変えるため、種々のIGSの範囲をコードする一連のオリゴヌクレオチドカセットをジシストロン性メッセージに使用した。異なるカセットを使用して、重鎖の翻訳開始速度を変更した結果、翻訳された重鎖産物の蓄積が、ある範囲の速度で生じた。
【0066】
最適配列を実験的に決定することが可能になるように一連の4つのIGSを設計した。最高量の可溶性Fab’の蓄積を生じるIGS変異体を、振盪させたフラスコを誘導装置として、又は発酵槽発現を決定手段として使用して、実験的に選択した。驚くべきことに、異なるFab’に対して異なるIGSが選択された。これは、それぞれ新しいFab’断片を発現させるには、実験的な選定が必要であることを示唆する。
【0067】
(実験)
材料と方法
一般的微生物学及びDNA操作技法
形質転換及び慣用培養物増殖用に、大腸菌株INVαF’(Invitrogen、De Schelp、Netherlands)を使用し、発現の研究には、大腸菌株W3110(ATCC # 27325)を使用した。DNA制限酵素及び修飾酵素は、ベーリンガーマンハイム(Boehringer Mannheim(Lewes、East Sussex、UK))及びニューイングランドバイオラボ(New England Biolabs(Hitcher、Herts、UK))から入手した。プラスミドの調製は、プラスミド精製キット(QIAGEN、Crawley、West Sussex、UK)を使用して行った。DNA断片の精製は、キアゲン社のスピンカラムを使用して行った。DNA断片は、GeneCleanプロトコル(BIO 101)を使用してアガロースから精製した。オリゴヌクレオチドは、Oswel Oligonucleotide Serviceから供給され、40nMのスケールで合成した。PCRは、推奨のように、パーキンエルマー(Perkin Elmer)のAmplitaqを使用して行った。DNAシークエンス反応は、ABI Prism DyeDeoxyチェーンターミネーターキットを使用して行い、ABI373A 自動シークエンサー(PE Applied Biosystems、Warrington、Cheshire、UK)で実施した。データをプログラム:オートアセンブラー(PE Applied Biosystems)を使用して分析した。
【0068】
振盪フラスコによる誘導
大腸菌W3110培養物を、テトラサイクリン(7.5μg/ml)を補給した液体ブロス中で生育させた。誘導するために、一晩培養した新鮮な培養物(30℃で生育させた)を、2Lのバッフル付きフラスコ中、液体ブロス200mlで、OD600=0.1まで希釈し、オービタルインキュベータ中30℃で増殖させた。OD600=0.5で、IPTGを200μMまで加えた。サンプル(OD用に正規化)を間隔をおいて採取した。
【0069】
ペリプラズム抽出
培養物サンプルを氷上で(5分)冷却し、次いで細胞を遠心分離によって採集した。抽出緩衝液で懸濁した(100mM Tris HCl、10mM EDTA;pH7.4)後に、サンプルを一晩30℃でインキュベートし、遠心分離で清澄させた。
【0070】
アッセンブリーアッセイ
Fab’の濃度をELISAで求めた。プレートを4℃一晩、抗ヒトFd6045(コーティング緩衝液、生理的食塩水中2μg/ml、ウエルあたり100μ1)(欧州特許第491031号参照)でコートした。洗浄後、ウエルあたり100μlのサンプルを充填し、精製されたA5B7γ−1 Fab’(欧州特許第491031号参照)を、初期濃度2μg/mlで標準として使用した。サンプルをプレートの端から端までサンプルコンジュゲート緩衝液(リットル当たり:トリス アミノメタン6.05g、塩化ナトリウム2.92g、ツイーン20を0.1ml、カゼイン(0.2%)を1ml)で2倍に連続希釈し、プレートを1時間室温で攪拌しながらインキュベートした。プレートを洗浄、乾燥し、次いで、抗ヒトCκ(GD12)ペルオキシダーゼ100μl(サンプルコンジュゲート緩衝液中で希釈したもの)を加えた。インキュベーションを室温で1時間、攪拌しながら行った。プレートを洗浄、乾燥し、次いで基質溶液100μl(10ml酢酸ナトリウム/クエン酸溶液(0.1M、pH6)、過酸化水素水溶液100μl、テトラメチルベンジジン溶液(ジメチルスルホキシド中10mg/ml)を100μ1)を加えた。基質添加の4から6分後に、630nmで吸光度を読み取った。
【0071】
発酵
大腸菌W3110培養物を、振盪フラスコで、テトラサイクリン(7.5μl/ml)を補給した液体ブロス中で、30℃で、OD600=1.0になるまで生育させ、この培養物100mlを使用して、1.5Lのバッチ供給式培養発酵槽に入れたSM6培地(+グリセロール)(欧州特許第651803号)1Lに播種した。pHを50%アンモニウムと5%硫酸とで7.0に調節した。溶存酸素濃度を可変式攪拌で30%に維持した。テトラサイクリンは、発酵培地に含まれていなかった。初期のグリセロール濃度は3%w/vであり、発酵中、培養物がOD600≒60に達した後、グリセロールが利用可能でなくなるような場合がもう一回あった時には、さらにグリセロールを供給した。培養物を30℃で、OD600=55まで生育させ、50%ラクトースを120ml添加した:利用可能なグルコースが利用された後、ラクトースが誘導される。20時間後、さらにラクトース60mlを一度に添加し、誘導後、25から30時間発酵をモニターし、サンプル(OD用に正規化されたもの)を間隔を置いて採取した。
【0072】
(結果)
プラスミドpTTO−1及びpTTO−2の構築
プラスミドpTTQ9をアマーシャム(Amersham)から入手した。これを図1に示す。アリコット(2μg)を制限酵素SalI及びEcoRIで消化し、消化物を1%アガロースゲルにかけ、大きなDNA断片(4520bp)を精製した。2つのオリゴヌクレオチドを合成し、これを互いにアニールしたところ、図2に示すOmpAポリリンカー領域をコードしていた。この配列は、pTTQ9の制限酵素消化によって得られたSalI及びEcoRI末端と適合する付着端を有する。このオリゴヌクレオチドの「カセット」をpTTQ9ベクター中に挿入した。これによって、SalI部位は再生されないが、EcoRI部位は保持される。このカセットは、大腸菌外膜タンパクOmp−Aのシグナル配列の最初の13アミノ酸をコードし、その前にOmpA遺伝子のシャインダルガノリボソーム結合部位がある。これに加えて、酵素XbaI、MunI、StyI及びSplIの制限部位が存在する。MunI及びStyI部位は、OmpAシグナル配列のコード領域内にあり、遺伝子の挿入用の5’クローニング部位として意図される。このカセットを構成する2つのオリゴヌクレオチドを、5pモル/μlの濃度で混合し、湯浴で95℃まで3分かけて加熱し、室温までゆっくり冷却することによって、互いにアニールさせた。次に、アニールした配列を、SalI/EcoRIで切断したpTTQ9に連結した。得られたプラスミド中間産物は、pTQOmpと呼ばれ、DNAシーケンスで検証した。
【0073】
中間産物のアリコットを、次に、SspI及びEcoRI(2350bp精製断片)並びにEcoRI及びXmnI(350bp精製断片)で切断した。2350bp断片は、翻訳終結領域及びlacIq遺伝子をコードし、また350bp断片はtacプロモータ、OmpAシグナル配列、及びマルチクローニング部位をコードしている。プラスミドpACYC184(New England Biolabs、図3)を、StyIで消化し、大豆ヌクレアーゼで処理して、平滑末端を生成し、次にPvuIIで消化した(2348bp精製断片−この断片はテトラサイクリン耐性マーカー及びp15A複製起点をコードする)。この断片をアルカリホスファターゼで処理して、(自己連結を防止するために)5’末端のリン酸を除去し、他の精製断片と連結した。得られたプラスミドをpTTO−1と名付けた。これを図4のマップに示す。図5はpTTO−1の完全なDNA配列を示す。プラスミドpHC132由来のSplI−EcoRI断片であるヒトIg軽鎖κ定常ドメインを挿入すると、pTTO−2が得られた(図6)。
【0074】
Fab’可変領域のpTTO−2への挿入
Fab’TNFα可変領域遺伝子は、国際出願GBOl/02477号の配列番号8由来の配列を含む抗体全体を哺乳類細胞発現用ベクターからPCR増幅することによって生成した。OmpAシグナル配列をコードし、pTTO−2中にフレームを合わせて挿入するためのMunI制限酵素部位を含むDNAを、各遺伝子の5’末端に付加し、その生来のIgリーダーと置き換えた。
【0075】
次に、精製されたVL遺伝子(MunI/SplI)を、pTTO−2のMunI/SplI部位に挿入して、軽鎖中間産物pTTO(TNFαL)を得た。
【0076】
重鎖VH遺伝子を、中間ベクターpDNAbEng−G1(図7)によって、MunI部位とApaI部位の間に挿入し、pDNAbEng(TNFαH)を得た。重鎖遺伝子を、EcoRI断片として、このプラスミドからpTTO(TNFαL)のEcoRI部位に挿入することによって、大腸菌発現プラスミドpTTO(TNFα)(図8)を得た。
【0077】
pTTO(TNFα)のIGS変異体の構築
一連の4つの遺伝子間配列(IGS)変異体を、TNFαFab’用の最適IGSの実験的決定が可能になるように設計した(図9)。IGS1及びIGS2は、非常に短い遺伝子間配列(それぞれ、−1及び+1)を有し、緊密に繋がって翻訳されることが予想され、それらのSD配列(下線)がわずかに異なっている。これらの2つの配列が、高度の翻訳開始レベルをもたらす可能性が高い。IGS3及びIGS4は、終結コドンと開始コドンの距離(+13)が長く、配列の構成が異なる。IGS3のSD配列は、IGS1、2又は4のSD配列より働きが強い。全配列を2次構造について(m/foldプログラム(Jaeger,J.A.ら、Methods Enzymology、183:281−306(1990);part of GCG Wisconsin Package、Accelrys)を使用して)調べ、できるだけ「最適化」した。しかし、2つの鎖が翻訳上緊密にカップリングしているので、リボソームが解離しない。これは、mRNAが「裸」でない可能性があり、2次構造の形成を妨げることを意味する。
【0078】
IGS変異体は、pTTO(TNFα)のSacI−NotI消化によって調製したベクター中に2つの断片を連結することによって構築した。pDNAbEng(TNFα)のMunI−NotI消化によって調製した挿入断片を精製し、各IGSカセット(SacI−MunI)と一緒にベクター中に連結し、4つの発現プラスミドpTTO(TNFα IGS−1)、pTTO(TNFα IGS−2)、pTTO(TNFα IGS−3)及びpTTO(TNFα IGS−4)を得た。
【0079】
TNFαIGS変異体の振盪フラスコ発現分析
4つのIGS変異体ベクター及び元の発現ベクターPTTO(TNFα)をそれぞれ使用して、大腸菌株W3110を形質転換した。次に形質転換された大腸菌を、Fab’の発現について、上記に記載したように振盪フラスコで分析した。典型的実験結果を図10に示す。
【0080】
異なる遺伝子間配列は、異なる発現プロフィルをもたらした。IGS1及びIGS2は、ペリプラズムFab’を迅速に蓄積し、誘導から1時間後がピークであり、その後、回復レベルが落ち込んだ。IGS1については、ピークが大きくなるほど急激な落ち込みとなった。こうした結果は、これら構築物では翻訳カップリングが緊密であることから予想されるように、合成のレベルが高いことと整合していた。IGS1は、明らかに、IGS2よりも高いレベルの重鎖発現をもたらした。この場合、1時間後のピークの後に、ペリプラズム発現レベルが落ちるので、高レベルの発現に対する耐容性が不十分と思われる。これは、IGS1培養物の増殖プロフィル(図示せず)で観察され、誘導から1時間後にピークに達した後落ち込み、細胞死及び溶菌が示唆された。
【0081】
IGS3はFab’をよりゆっくりと蓄積し、誘導2時間後に、高いピーク値(325ng/ml/OD)を有するピークを示した後、レベルが落ち込んだ。この培養物の増殖は、誘導の3時間後まで続き、より高い生物体量に達した(図示せず)。これは、重鎖の合成レベルがより低いことと整合する。
【0082】
IGS4は、物質をやはりよりゆっくりした速度で蓄積し、他の3つの構築物の示す高い生産性のピークに達することはなかった。
【0083】
IGS変異体は全て、オリジナルのpTTO(TNFα)ベクターより著しい成績を収めた。異なるIGS配列は、異なる翻訳開始速度をもたらすという仮説が、これらの実験結果から裏付けられた。TNFαFab’については、高い重鎖の翻訳開始速度に対する耐容性が不十分であり、したがって最適ではないと思われる。IGS3によってもたらされるようなより遅い速度では、より良好な増殖特性がもたらされ、その結果、時間がたつに連れ、より良い収率で蓄積される。
【0084】
発酵槽(図示せず)での生産性を比較した後、IGS3構築物が最高の収量であったのでこれを選択した。
【0085】
異なるFab’抗体をコードするジシストロン性メッセージ:g163Fab’IGS変異体の作成
上記に記載の1組のpTTO/IGSベクターを使用して、もう1つのFab’の発現分析を行った。最初に、ヒト化Fab’g163を使用した。4通りの連結の仕方を使用して、Fab’g163の4つのIGS変異体(図11)を生成した。g163のVH遺伝子(哺乳類発現ベクターpγ−4からPCRによって得た)のMunI−ApaI断片をプラスミドpDNAbEngGIに挿入して、重鎖の中間産物を生成した。この中間産物をPvuIIとNotIで切断し、g163VH+CH1断片を生成した。プラスミドpTTO(TNFα)をMunIとNotIで切断し、大きな断片を精製してベクター断片を得た。PCRによって、哺乳類発現ベクターpMR10.1(gl63)からMunI−SplI VL断片を生成した。最後に、上記の4pTTO(TNFα)IGS変異体のSplI−PvuII消化によって、Cκ−IGS断片を生成した。次に、別々の4通りの連結を行い、構築物pTTO(gl63 IGS−1)、pTTO(g163 IGS−2)、pTTO(g163 IGS−3)、及びpTTO(gl63 IGS−4)を生成した。これら構築物の振盪フラスコ分析の結果を図12に示す。
【0086】
IGS−1及び−2が迅速なFab’蓄積をもたらし、誘導の1時間後にピークとなり、その後急激に低下するという点で、この発現プロフィルは、Fab’TNFαで見られる発現プロフィルと非常に類似していた。IGS−3は、より遅い蓄積開始速度をもたらし、より遅い時間(誘導開始2から3時間後)にピークに達し、その後より遅い速度で低下する。IGS−4構築物は、誘導の経過時間を通してゆっくり立ち上がり、この場合もやはり他の構築物の示す高い収率に達することはなかった。IGS−1及び−2では、やはり誘導のわずか1時間後(図示せず)に、生物体量がピークに達することが発現プロフィル及び増殖プロフィルで示されるため、IGS−3を選択し、発酵槽で高い収率(図示せず)まで発現させた。
【0087】
プラスミドpTTODの構築
Fab’のコード戦略を簡略化するために、プラスミドpTTO−1からPvuII(3部位)、EcoRV(2部位)及びApaI(1部位)に対するフレームワークの制限部位を取り除くことによって、プラスミドpTTODを得た。これらに変更を加える際、lacIq遺伝子及びテトラサイクリン耐性遺伝子の蛋白コード配列を変えずに、遺伝情報を発現しない「サイレント」な変化をDNAレベルで加えた。PCR戦略を使用して、「サイレント」変化(これらの制限部位を除去した)を有するプライマーを設計し、これを使用して親プラスミド(pTTO−1)の一部分を増幅した。フランキング制限部位(未変更)を使用して、親プラスミド中の配列をこれらの修飾配列で置き換えた。プラスミドpTTODをこの多段階方法で作成した。ベクターpTTO内の既存のg163Fab’遺伝子を、遺伝子に隣接するユニークなPstI及びEcoRI部位を使用してpTTOD中にトランスファーし、pTTOD(g163)IGS変異体1から4を作成した。pTTOD(g163IGS−3)の制限マップを図13に示す。
【0088】
IGS変異体としての他のFab’の発現
TNFα及びg163の他に、さらにいくつかのFab’が、2個以上のIGS変異体として発現した。それには、g165及びgA33と名付けられたFab’が含まれる。図14は、振盪フラスコで比較されたpTTOD(gl65)IGS−1から−3を示す。TNFα及びg163とは対照的に、IGS−2及びIGS−1は、IGS−3より成績がよい。このFab’の発現は、たとえ迅速な速度でも、ホスト細胞による十分な耐容性があるようである。IGS−2を発現する培養物は、誘導期間中(図示せず)ずっと増殖し続けた。図15は、このFab’を有するIGS−1から4の発酵槽での比較を示し、これは、基本的に振盪フラスコで行われた観測を再現している。IGS−2は、収率が最高の変異体であることが確認された。発酵槽中で、Fab’gA33、pTTOD(gA33)IGS−2及び−3を比較すると、両者は同様の収率を示した(図16)。したがって、最適の収率を得るためには、Fab’が異なれば異なるIGS配列が必要となる。
【0089】
振盪フラスコ対発酵槽分析
振盪フラスコでは、プロモータの脱抑制を、発現の迅速な誘導をもたらすIPTGによって行った。発酵槽における誘導方式は異なり、ラクトース(細菌によってアロラクトースに変換される)を使用して、より穏やかにプロモータのスイッチを入れる。こうした誘導の速度論が異なるにもかかわらず、振盪フラスコでは、発酵槽中で構築物がどのような対比を示すかの示唆を与えることが明らかになった(図14及び15参照)。
【0090】
最適な収率を得るためには、Fab’が異なれば異なるIGSが必要になるという原理がはっきり証明されている。本明細書に記載した本システムの新規性は、高レベルのFab’の発現を達成する手段として、ジシストロン性メッセージの第2遺伝子の翻訳開始速度を最適にするために、IGSカセットを使用することにある。上記に記載したシステムでは、軽鎖の発現は変わらないままであり、重鎖の翻訳開始速度のみが最適化される。この戦略がなぜ成功したかについて、2つの可能な説があげられる。
【0091】
(i)もし十分な軽鎖が合成されて、重鎖が過剰になることを防ぐとしても、軽鎖の発現がFab’の全蓄積レベルに影響を与えることはほとんどない。過剰の軽鎖は通常何の問題もなく許容される。可溶性発現の効率を規定するのは重鎖の翻訳レベルである;あるいは
【0092】
(ii)重鎖の最適化は、実際、軽鎖発現速度が一定になるように調節され、したがって、2つの鎖のレベルのバランスがとられる。可溶性発現の効率を規定するのは、重鎖の折りたたみ/分泌速度それ自体ではなく、2つの異種鎖の発現のバランスである。
【0093】
上記に記載された実施例は単なる模範例にすぎず、以下の特許請求の範囲に定義される本発明の範囲を限定するものではないことを理解されたい。
【特許請求の範囲】
【請求項1】
上流側シストロンが、抗体の重鎖をコードするDNAを含み、下流側シストロンが、それに対応する軽鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性配列からなる核酸であって、
2つのシストロンが、配列番号4によって表わされる遺伝子間配列1(IGS1)、配列番号7によって表わされる遺伝子間配列2(IGS2)、配列番号10によって表わされる遺伝子間配列3(IGS3)、又は配列番号13によって表わされる遺伝子間配列4(IGS4)によって連結されており、産生抗体が抗TNFα抗体でもなく、抗KDR抗体でもないことを特徴とする、核酸。
【請求項2】
請求項1に記載のジシストロン性配列を含む、発現ベクター。
【請求項3】
前記発現ベクターが、配列番号3のヌクレオチド配列を有するベクターである、請求項2に記載の発現ベクター。
【請求項4】
請求項2又は3に記載の発現ベクターで形質転換した、ホスト細胞。
【請求項5】
前記ホスト細胞が大腸菌である、請求項4に記載のホスト細胞。
【請求項6】
請求項4又は5に記載のホスト細胞を培養することを含む、抗体分子の産生方法。
【請求項7】
請求項6に記載の方法によって産生される抗体分子。
【請求項1】
上流側シストロンが、抗体の重鎖をコードするDNAを含み、下流側シストロンが、それに対応する軽鎖をコードするDNAを含む、特定の抗原結合特異性を有する抗体分子を産生するためのジシストロン性配列からなる核酸であって、
2つのシストロンが、配列番号4によって表わされる遺伝子間配列1(IGS1)、配列番号7によって表わされる遺伝子間配列2(IGS2)、配列番号10によって表わされる遺伝子間配列3(IGS3)、又は配列番号13によって表わされる遺伝子間配列4(IGS4)によって連結されており、産生抗体が抗TNFα抗体でもなく、抗KDR抗体でもないことを特徴とする、核酸。
【請求項2】
請求項1に記載のジシストロン性配列を含む、発現ベクター。
【請求項3】
前記発現ベクターが、配列番号3のヌクレオチド配列を有するベクターである、請求項2に記載の発現ベクター。
【請求項4】
請求項2又は3に記載の発現ベクターで形質転換した、ホスト細胞。
【請求項5】
前記ホスト細胞が大腸菌である、請求項4に記載のホスト細胞。
【請求項6】
請求項4又は5に記載のホスト細胞を培養することを含む、抗体分子の産生方法。
【請求項7】
請求項6に記載の方法によって産生される抗体分子。
【図1】


【図2】

【図3】
【図4】

【図5−1】
【図5−2】
【図5−3】
【図5−4】
【図6】
【図7】
【図8】
【図9】

【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】

【図2】
【図3】
【図4】
【図5−1】
【図5−2】
【図5−3】
【図5−4】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2010−207241(P2010−207241A)
【公開日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願番号】特願2010−116444(P2010−116444)
【出願日】平成22年5月20日(2010.5.20)
【分割の表示】特願2003−549395(P2003−549395)の分割
【原出願日】平成14年12月5日(2002.12.5)
【出願人】(507073918)ユセベ ファルマ ソシエテ アノニム (70)
【Fターム(参考)】
【公開日】平成22年9月24日(2010.9.24)
【国際特許分類】
【出願日】平成22年5月20日(2010.5.20)
【分割の表示】特願2003−549395(P2003−549395)の分割
【原出願日】平成14年12月5日(2002.12.5)
【出願人】(507073918)ユセベ ファルマ ソシエテ アノニム (70)
【Fターム(参考)】
[ Back to top ]