
可溶性ポリペプチドの単離方法

【課題】物物理学的特性(可溶性、高い発現及び/又は安定性)が改良されたポリペプチド、特に抗体フラグメントを同定するハイスループットスクリーニング方法を提供する。
【解決手段】任意のポリペプチド配列を発現することができるファージディスプレイライブラリを得る工程、ライブラリファージによる細菌層の感染を可能にする工程、及び細菌層上の平均よりも大きいプラークを形成するファージを同定する工程を包含する、望ましい生物物理学的特性を有するポリペプチド(単量体のヒトVH及びヒトVLを含む)を同定する方法。前記、望ましい生物物理学的特性を有するポリペプチドの、免疫治療、又は診断用薬剤としての使用。
【解決手段】任意のポリペプチド配列を発現することができるファージディスプレイライブラリを得る工程、ライブラリファージによる細菌層の感染を可能にする工程、及び細菌層上の平均よりも大きいプラークを形成するファージを同定する工程を包含する、望ましい生物物理学的特性を有するポリペプチド(単量体のヒトVH及びヒトVLを含む)を同定する方法。前記、望ましい生物物理学的特性を有するポリペプチドの、免疫治療、又は診断用薬剤としての使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリペプチド、特に単量体のヒト抗体フラグメントの単離、同定及び操作に関する。
【背景技術】
【0002】
脊椎動物の抗体は通常、一対の重(H)鎖及び軽(L)鎖から構成される。組み合わされたH鎖及びL鎖の最初のドメインであるVH及びVLは配列においてより可変的であり、抗原を認識し、抗原に結合する抗体部分である。VHドメイン及びVLドメインは対で抗原を認識する。
【0003】
ラクダ科(ラクダ、ヒトコブラクダ及びラマ)の免疫レパートリは、重鎖抗体と呼ばれる変わった種類の抗体を保有するという点で、特殊である(Hamers, Casterman C. et al., 1993)。これらの抗体は軽鎖を欠いており、したがってこれらの組み合わせ部位はVH
Hと呼ばれる1個のドメインから構成される。
【0004】
組み換え型VHHの単一ドメイン抗体(sdAb)は、従来の四本鎖抗体由来の一本鎖Fv(scFv)フラグメントに勝る幾つかの利点を提供する。sdAbの親和性はscFvの親和性と同程度であるが、sdAbは、可溶性、安定性、凝集耐性、リフォールディング性、発現収率及びDNA操作の易操作性、ライブラリ構築並びに三次元構造決定といった点でscFvより優れている。抗体を伴う適用において、上記のVHH sdAbの特性の多くが望ましい。
【0005】
しかし、VHHの非ヒト性質によって、免疫原性によるヒトの免疫療法におけるVHHの利用が制限される。この点において、ヒトのVH及びVL sdAbの免疫原性が最小であると考えられるので、免疫療法への適用に対する理想的な候補である。
【0006】
しかし、ヒトのVH及びVLは従来の抗体由来のVH及びVLに共通の特徴である凝集の傾向が強い(Davies, J. et al., 1994; Tanha, J. et al., 2001; Ward, E. S. et al., 1989)。このように抗体用途に好適な単量体のヒトのVH及びVLを得る試みが行われ
てきた。このようなVH及びVLは、高い発現収率、高いリフォールディング性及び凝集耐性等のVHHに一般的な他の有用な特性も示す。これらのVH及びVLでライブラリ骨格として構築される合成ライブラリは、治療用の有望なタンパク質源として役立つ可能性がある。
【0007】
ラクダ及びラマのVHHからそれぞれ、ヒトのVH又はVLに組み込まれる鍵となる可溶性残余物を伴うラクダ化(camelization)並びにラマ化(llamination)を利用して、単量
体のヒトのVH及びVLを生成した。これらのVH及びVLに基づき構築され、CDRランダム化により生成される合成sdAbライブラリは、様々な抗原との結合因子を得るといった点で機能的であることが示された(Davies, J. et al., 1995; Tanha, J. et al., 2001)。
【0008】
別の手法において、上記の技術的手段を用いることなく、完全なヒトの単量体のVH及びVLをヒトの合成VH及びVLライブラリから単離した。一実験において、ヒトVHライブラリを鶏卵リゾチームに対してパンニングした(panned)時に単量体のヒトVHを発見した(Jesperps, L. et al., 2004b)。さらに最近では、可逆的非フォールディング及び親和性基準に基づく選択方法によって、合成ヒトVHライブラリから多くの単量体のVHが得られた(Jespers, L. et al., 2004a)。この知見は、適切な選択方法が望ましい生物物
理学的特性を有する珍しい単量体のヒトVHを効率的に捕捉する鍵となるという事実を明確に示していた。
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明の第1の目的は、生物物理学的特性(可溶性、高い発現及び/又は安定性(熱変性後の高いリフォールディング、化学変性剤に対する高い耐性、及びプロテアーゼ、特にトリプシン等の胃腸プロテアーゼに対する高い耐性等)を含む)が改良されたポリペプチド、特に抗体フラグメントを同定するハイスループットスクリーニング方法を提供することである。
【0010】
本発明の第2の目的は、単量体のヒトVH及びヒトVLを同定するハイスループットスクリーニング方法を提供することである。
【0011】
本発明の第3の目的は、単量体のヒトVH及びヒトVLを同定、単離及び特徴付けすることである。
【0012】
本発明の第4の目的は、抗体フラグメント、特に単量体のヒトVH及びヒトVLの多量体を構築及び特徴付けることである。
【0013】
本発明の第5の目的は、ポリペプチド、特に抗体フラグメント及び最も特定的には単量体のヒトVH及びヒトVLからディスプレイライブラリを構築することである。
【0014】
本発明の第6の目的は、ポリペプチド、特に抗体フラグメント及び最も特定的には生物物理的な特性が改良された単量体のヒトVH及びヒトVLを生成するDNAシャッフリング法を提供することである。
【課題を解決するための手段】
【0015】
発明の要約
ポリペプチド、好ましくは抗体フラグメント及び最も好ましくは望ましい生物物理学的特性(可溶性、安定性、高い発現性、単量体性、非凝集性、結合特異性)を有するヒトVH及びヒトVLを単離する方法が提供される。この方法は、様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得る工程、ライブラリファージによる細菌層の感染を可能にする工程、及び細菌層上の平均よりも大きいプラークを形成するファージを同定する工程を包含する。それからファージを単離し、シークエンシングか、又はそうでなければポリペプチド配列を特定するこれらの工程を用いる。
【0016】
本発明は、上記の方法で同定されるポリペプチド、特に単量体のヒトVH及びヒトVLも提供し、これは免疫療法に及び/又は診断用薬剤もしくは検出用薬剤として有用であり得る。単量体のヒトVH及びヒトVLは組合せて、二量体、三量体、五量体又は他の多量体を形成してもよく、これは免疫療法に及び/又は診断用薬剤もしくは検出用薬剤として有用であり得る。
【0017】
上記の方法によって同定されたポリペプチド(ヒトVH及びヒトVLを含む)を、可溶性、安定性、単量体性、高発現性、結合特異性、及びヒト起源等の改良された生物物理学的特性のために選択するDNAシャッフリング等の方法によって操作することができる。
【0018】
上記の方法によって同定されたポリペプチド(ヒトVH及びヒトVLを含む)は、さらにディスプレイライブラリを生成するのにも使用することがあり、これを同様に使用して、上記の方法によってさらにポリペプチドを単離することができる。
【0019】
第1の態様において、本発明は、標的ポリペプチドを同定する方法であって、a)様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得ること、b)ライブラリファージによって、細菌層の感染を可能にすること、c)細菌層上の平均よりも大きいプラークを形成するファージを同定することを含む、方法を提供する。
【0020】
第2の態様において、本発明は、配列番号8〜配列番号54から成る群より選択されるアミノ酸配列を有するポリペプチドを提供する。
【0021】
第3の態様において、本発明は、配列番号8〜配列番号22から成る群より選択される少なくとも1個のアミノ酸配列を含むVH抗体フラグメントを提供する。
【0022】
第4の態様において、本発明は、配列番号23〜配列番号54から成る群より選択される少なくとも1個のアミノ酸配列を含むVL抗体フラグメントを提供する。
【0023】
第5の態様において、本発明は、望ましい生物物理学的特性を有するポリペプチドを生成する方法であって、a)請求項41、42、44、45、47、48、59若しくは70に記載の抗体フラグメントをコードするか、又は請求項24、27、37若しくは39に記載のポリペプチド配列をコードし、且つ第1の望ましい特性を有する少なくとも1個の第1の核酸配列を提供する工程と、b)第2の望ましい特性を有する抗体フラグメントをコードする少なくとも1個の第2の核酸配列を提供する工程と、c)少なくとも1個の第1の核酸配列及び少なくとも1個の第2の核酸配列をランダムなフラグメントに切断する工程と、d)ランダムなフラグメントを再び集合化させる工程と、e)ランダムなフラグメントを発現する工程と、f)発現されたランダムなフラグメントを第1の望ましい特性及び前記第2の望ましい特性に関してスクリーニングする工程とを含む、方法を提供する。
【発明を実施するための形態】
【0024】
発明の詳細な説明
ヒト起源であり、可溶性、安定性、凝集に対する耐性、リフォールディング性、高発現性、及びDNAレベルでの易操作性があり、ライブラリ構築及び三次元構造決定に理想的であるポリペプチド、特に抗体フラグメントを同定することが望ましい。このような抗体フラグメントは、多種の免疫療法への適用、並びに診断用薬剤及び検出用薬剤としても有用である。ヒトの単量体のVH抗体及びVL抗体は上記の特性の多くを有しているとされるので、特に興味深い。
【0025】
上記の特性を有するポリペプチドは、様々なポリペプチド配列を発現することができるライブラリのハイスループットスクリーニングによって同定することができる。例えば、ファージディスプレイライブラリ(好ましくは、M13又はfd等の繊維状ファージ)は、ファージの影響を受けやすい細菌の領域(細菌層)をファージで感染させ、それからプラークとして知られるはっきりした無菌領域を探すことにより、どのファージが細菌を首尾よく溶解させるかを決定することによりスクリーニングすることができる。単量体のラマ化VH及びVLが、凝集傾向のある完全なヒトVHを示すファージよりも大きいプラークを細菌層上に形成する。したがって、ヒトVHレパートリから珍しく自然発生的な単量体のVH及びVLを同定する手段として、プラークサイズを使用することができる。
【0026】
本明細書中に記載の方法は、可溶性、安定性(安定性には、多くの特徴(高熱リフォールディング効率、高い融解温度、37℃での長期(数日)のインキュベート後の機能性の維持、化学変性剤に対する耐性、プロテアーゼに対する耐性、0℃未満、4℃未満及び室温での長期の保存期間、細胞内環境における機能性の維持、並びに血流中等のヒトの体内
の機能性の維持を含むが、これらに限定されない)が含まれる)、及び様々な起源の高発現性のタンパク質を同定するのにも有用であり、これらのタンパク質としては、
1.VH、VL、Fab、scFv及びより詳細にはヒト抗体であるIgG等の全抗体、
2.非抗体スカフォールドの一本鎖T細胞受容体、T細胞受容体ドメイン、トランスフェリン、リポカリン、kunitzドメイン、アンキリン反復、及び細胞傷害性Tリンパ球関連抗原(CTLA−4)に基づくタンパク質変異体(ヒトのものを含む)、
3.ウイルス性及び細菌性のタンパク質ワクチン等のワクチン、
4.治療用タンパク質(例えば、インスリン、成長ホルモン、エリスロポエチン)
5.タンパク質性の診断用試薬及び生化学試薬(例えば、プロテインA、プロテインG)
が挙げられる。
【0027】
ポリペプチドをこの方法で同定されれば、このポリペプチドを使用して、さらなるライブラリを構築することができる。例えば、VHの核酸配列を選択することによって、これを行うことができる。ランダムなコドンを有するオリゴヌクレオチドを作り、VH配列に組み込む。このようにして、それぞれの特殊なオリゴヌクレオチドをVH遺伝子に組み込み、修飾されたVH遺伝子によってわずかに変異のある配列のライブラリが構成される。一般的に、VHのCDR又はループをランダム化させるように、オリゴヌクレオチドを設計する。例えば、1個、2個又は3個全てのVH CDRをランダム化させることができる。それから、使用するライブラリの種類に応じて、VHライブラリを適切なベクターにクローニングさせ、核酸配列をポリペプチドとして発現させる。一般的にパンニング(panning)によって、ライブラリポリペプチドと結合する分子に関してライブラリをスクリー
ニングする。ライブラリはファージディスプレイライブラリ、又はリボソームディスプレイ及び酵母ディスプレイ等の他のディスプレイライブラリであり得る。
【0028】
本明細書中に記載の方法で同定したポリペプチドを例えば、単量体の架橋により二量体、三量体、五量体及び他の多量体を形成し、免疫療法に使用することができる。このことにより、抗原分子におけるより良好な親和性及び幾つかの抗原におけるより緩やかな拡散速度がもたらされ得る。別の可能性のある手法は、ポリペプチドを様々な機能を有する種々の分子と結合又は融合させることである。例えば、抗体フラグメントは、特定の細胞又は分子を標的、破壊又は修飾するために、放射性核種、細胞傷害性薬剤、トキシン、ペプチド、タンパク質、酵素、リポソーム、脂質、T細胞超抗原又はウイルスと結合することができる。
【0029】
本明細書中に記載の選択方法で同定したVH又はVLを単離すれば、VH又はVLをさらに操作し、可溶性、安定性、単量体性、結合特異性、ヒト起源又は高発現性等の改良された生物物理学的特性に関して選択することができる。DNAシャッフリング又は交互伸長法等のin vitro組み換え技法で、これを達成することができる。DNAシャッフリングは、抗体フラグメント等の第1のポリペプチド(ドナー)及び第2のポリペプチド(アクセプタ)の核酸配列をランダムなフラグメントに切断し、PCR様反応によりランダムなフラグメントを再び集合化させることを含む。それから、再集合化されたフラグメントをスクリーニングし、所望の特性に関して選択する。
【0030】
例えば、高い安定性のある1つ又は複数のVH(ドナー)を十分な安定性を欠いている1つ又は複数のVH(アクセプタ)と混合し、DNAシャッフリングにかけることができる。このことにより、ドナーVHから安定性残基を組み込んだアクセプタVHの突然変異型が発生する。本明細書に記載の方法によって、又はリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ及びファージディスプレイ等の他の発展的なタンパク質スクリーニングシステムによって、新たな安定性のある突然変異型を同定することができ
る。同様に、この技法を使用して、可溶性、単量体性、及び高発現性等の望ましい特質を形質転換することができる。
【0031】
ドナーVH及びアクセプタVHが望ましい特性を有する場合に、この技法を使用して両方の特性を有するVHを生成することができる。例えば、重要な治療用リガンド又は診断用リガンドと結合する不安定なドナーVHを安定性のあるアクセプタVHでシャッフルすることができる。新たに生成した安定性のあるVHもリガンドと結合する能力があることを確実にするために、スクリーニングシステムは、リガンド結合工程を伴い得る。
【0032】
DNAシャッフリングは、ラクダ科の重鎖抗体の可変領域並びにナースシャーク及びテンジクザメの可変領域等の非ヒトVH、又は治療標的と結合する非ヒトVLをヒト化するのにも有用であり得る。可溶性、安定性、単量体性、及び高発現性等の望ましい特性を有するヒトVH及びヒトVLをドナーとして使用することができる。例えば、良好な安定性のある1つ又は複数のヒトVH(ドナー)を1つ又は複数の治療用の非ヒトVH(アクセプタ)と混合し、DNAシャッフリングにかけることができる。これにより、安定性があり、且つヒト化されたアクセプタVHの突然変異型が発生する。本明細書に記載の方法によって、又はリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ及びファージディスプレイ等の他の発展的なタンパク質スクリーニングシステムによって、新たに発生したヒト化され、且つ安定性がある変異型を同定することができる。さらなる実施例では、アクセプタVHは治療用VHH(ラクダ科の重鎖抗体の可変ドメイン)であり得る。
【0033】
さらに、この技法はVH及びVL以外のポリペプチドの望ましい特性を選択するのにも有用である。上記で示されるように、ドナーポリペプチド及びアクセプタポリペプチドは、両方ともヒトであり得るか、又はドナーがヒトであり、アセプタが非ヒトであり得る。
【0034】
可溶性、単量体性、高発現性、又は安定性をVH及びVLに与える可能性のある手法は、アクセプタVH及びVL上の相補性決定領域(CDR)をグラフト化することによるものであり得る。CDRは、単一ドメイン抗体の可溶性及び安定性、及びそれに応じた本明細書中に記載の方法で単離したVH及びVL由来のCDR等のこれらの領域のグラフト化を伴うことが知られているので、可溶性及び/又は安定性をアクセプタVH及びVLに与えることができる。
ヒトの単量体のVH及びVL
異なる生殖細胞及び全配列を有する幾つかの単量体のヒトVHが、ファージプラークサイズに基づくこの選択方法によって、ナイーブヒトVHファージディスプレイライブラリから同定された(図1及び配列番号8〜配列番号22を参照のこと)。VHは、37℃でのトリプシン処理、37℃での数週間のインキュベート、又は4℃での数ヶ月の保存後でも機能性及び単量体を維持し、熱リフォールディング効率が高く、大腸菌において良好な収率で生成され、プロテインA結合活性を有する。
【0035】
さらに、幾つかの単量体のヒトVLが同定された(図6及び配列番号23〜配列番号54を参照のこと)。VLも大腸菌において良好な収率で生成され、プロテインL結合活性を有する。
【0036】
また、スカフォールドとして上記のVHを利用する合成ライブラリからのVHによって、このような特性が明らかになる。したがって、このようなライブラリによって、生理学的温度での良好な有効性、長い保存期間、及び費用効率が高い生産を有する治療用VH又は診断用VHが得られ得る。高温リフォールディング効率の特徴によって、VH結合因子が一時的に高温に曝された後に、その活性を維持するように要求される場合に対するこれらのライブラリの生物工学的適用をさらに拡大させる。VHは、その望ましい生物物理学
的特性により、体内適用に非常に適しているはずである。プロテインA結合特性は、診断試験におけるVH精製及びVH検出、免疫ブロット法及び免疫細胞化学を単純化し、ライブラリから非機能性のVHを取り除くことによって、ライブラリ性能を高めるために利用することができる。同様に、スカフォールドとしてVLを利用するライブラリによって、同様に望ましい特性を有する治療用のVLS又は診断用のVLSが得られる。VLがプロテインLと結合するので、このプロテインL結合特性を利用することにより、VL精製及びVL検出が単純化される。
【0037】
本発明のVH及びVLで構成されたディスプレイライブラリは、診断用薬剤及び検出用薬剤の有用な供給源でもあり得る。
【0038】
これまでに報告された有益な生物物理学的特性を有する完全なヒトVHは、単一のV生殖細胞系配列:DP−47に基づいていた(Jespers, L. et al., 2004b; Jespers, L. et
al., 2004a)。この研究における単量体のヒトVHが、DP−47を含む6個の異なる生殖細胞系配列から起因しているという見解によって、安定性のあるVHは生殖細胞系遺伝子の利用に関して限定されないことが実証される。事実、本発明者らがファミリーの単量体のVHを単離し、本明細書で記載するものとは異なる生殖細胞系起源は、プロテインA結合活性のあるVH3ファミリーのVHのサブセットに対する本発明者らの選択を制限させない可能性が非常に高い。VHにおいて複数の突然変異が発生すること、及びCDR3はsdAbの生物物理学的プロファイルの形成にも関与することが知られているという事実により、本発明のVHの観察された生物物理学な挙動に関与するアミノ酸突然変異(第1表)を特定することは不可能である。しかし、sdAb安定性及び可溶性に重要であることが知られている位置(例えば、HVHP423及びHVHP44BにおけるV37F)での突然変異又は同じ位置(例えば、9個のVHにおけるL5V/Q及びV5Q)で複数回起こる突然変異は、VHの生物物理学的特性を決定するのに役立つ。ライブラリ構築に関して、ライブラリ安定性を危うくすることなくCDRランダム化が行われるように、本発明のVHの単量体性は、CDR、特にCDR3には依存しないことが望ましい。これに関して、安定性に関してCDR3への依存性が小さいので、より小さいCDR3を有するVH(例えばHVHB82)が好ましいスカフォールドであり得る。
【0039】
全体の配列及びCDR3の長さに関する本発明のVH及びVLの多様性によって、より良好な性能のライブラリの構築が可能になる。合成VHライブラリが単一スカフォールドで構築されている。レパートリ形成へのこのような手法は、多様なスカフォールドを利用する天然のin vivoでの「手法」とはかなり異なっている。本明細書中に報告されている配列に基づいて、多様な組のVH及びVLの可用性を利用し、複数のVH及びVLのスカフォールドに基づくライブラリを作ることができる。このようなライブラリは、in vivoでのレパートリのより良好な模倣であり、したがってより最適な複雑性を有する。sdAbにおける3個のCDRの中で、CDR3は一般的に最も有意にレパートリの多様性に寄与し、この理由から一般的にCDR3の長さを同時に変えることによって、VH及びVLのスカフォールドにおけるCDR3ランダム化が達成される。この有意性がライブラリの複雑性を改良するが、親スカフォールドCDR3の長さを壊すことによって、ライブラリの安定性をも危うくする可能性がある。CDR3の長さに関して本明細書中で開示されるVH及びVLの不均一性によって、良好な複雑性、良好な安定性及び良好な生物物理学的な特徴の両方を有するライブラリの形成を可能にする。このようなライブラリは、好ましくはサブライブラリから成り、それぞれのサブライブラリは、親CDR3の長さを壊すことなく、単一のVH、及びVLスカフォールド上のCDR3のランダム化(及び必要に応じてCDR1及び/又はCDR2のランダム化)によって作られる。
【0040】
治療標的に特異的なVHH、VH及びVLをヒト化することに関して、最適なVH及びVLフレームワークを選択するといった点でも、本発明のVH及びVLの多用途性は有益
である。比較的容易に免疫性、非免疫性又は合成VHHライブラリから治療標的に対する親和性の高いラクダ科のVHHを得て、その後ヒト化(CDRグラフト化、再サーフェイシング(resurfacing)、脱免疫化)を行い、可能性のあるVHH免疫原性を除去すること
ができる。それにより、治療用VHの生成に対するヒトVHライブラリの手法の代替方法を提供する。後者の手法による親和性の高い治療用VHの生成は、最初の合成ヒトVHライブラリから選択される主要な結合因子(複数可)の面倒で、且つ時間のかかるさらなるin vitroの親和性突然変異を要求することが度々あり得る。
【0041】
比較的容易に免疫性、非免疫性又は合成VHライブラリから治療標的に対する非ヒトVHを得て、その後ヒト化(CDRグラフト化、再サーフェイシング、脱免疫化)を行い、非ヒトVH免疫原性を除去することができる。それにより、治療用VHの生成に対するヒトVHライブラリの手法の代替方法を提供する。
【0042】
比較的容易に免疫性、非免疫性又は合成VHHライブラリから治療標的に対する非ヒトVLを得て、その後ヒト化(CDRグラフト化、再サーフェイシング、脱免疫化)を行い、VHH免疫原性を除去することができる。それにより、治療用VLの生成に対するヒトVLライブラリの手法の代替方法を提供する。
【0043】
改良された生物物理学的特性を有するタンパク質の選択に対する多くの発展的な手法が記載されている(Forrer, P. et al., 1999; Waldo, G. S., 2003)、(Jespers, L. et al., 2004a; Jung, S. et al., 1999; Matsuura, T. et al., 2003)。一般的に確実にライブラリ集合から不安定又は安定性の低い変異型よりも安定性のある変異型を優先的に選択するために、安定性圧力が必要とされる。例えば関連の研究で、凝集耐性のあるVHを選択するために、VHファージディスプレイライブラリの熱処理が要求された(Jespers, L. et al., 2004a)。ファージディスプレイを含む発展的な選択の手法の例としては、従来の
ファージディスプレイ、選択的感染ファージ及びタンパク質分解の手法が挙げられる。最初の2個の手法において、安定性のあるタンパク質が不安定なものよりもそのリガンドに対してより良好な結合特性を有するという仮定に基づき、親和性の選択を使用して、ライブラリから安定性のある種を選択する。しかし、安定性選択工程がさらに包含されていても、これらの手法は主に安定性よりも親和性をより高める(Jung, S. et al., 1999)。結
合工程が要求されることによって、既知のリガンドを有するタンパク質に対するこれらの手法の適用性も制限される。第3のタンパク質分解の手法は、不安定なタンパク質がプロテアーゼに対して耐性がないのに対して、安定性のあるタンパク質は一般的に小さく、したがってプロテアーゼに対して耐性があるという事実に基づいている。ファージディスプレイフォーマットは、表示されたタンパク質のプロテアーゼ安定性が、ファージ感染性に変換されるように操作される。このように、変異型のファージディスプレイライブラリをプロテアーゼで処理する場合、安定性のあるタンパク質を表示するファージのみがこれらの感染性を維持し、その後、大腸菌宿主を感染させることにより選択することができる。この手法はリガンド結合と独立しているので、一般的に有用である。しかし、安定であり、且つ十分にフォールディングしたタンパク質であってもプロテアーゼ感受性部位(例えば、ループ及びリンカー)を有し、これによりタンパク質分解の手法における安定性のある種の選択が妨げられることもあり得る(Bai, Y. et al., 2004)。
【0044】
これに対して、本発明の発展的な手法において、優れた生物物理学的特性を有するタンパク質は肉眼で簡単に判別される。この手法は、リガンド結合工程、タンパク質分解工程又は脱安定化工程を要求せず、したがって報告された選択的手法において直面する可能性のある複雑性が回避される。結合工程に対する要求がないことは、この手法が一般的に有用であることも意味する。オプションとして、結合工程は選択されたタンパク質が機能的であることを確実にすることが含まれてもよい。しかし、本発明の手法の(プラーク視覚化のための)プレート培養に対する依存性によって、操作することができ、小さいライブ
ラリに対する適用を制限し得るプレートの数に関して、可能性のある運搬上の制限が導入される。それにもかかわらず、ライブラリを最初に取り扱いが可能な大きさに縮小する場合、現在の手法の有用性は大きなライブラリにまで拡大することが可能である。例えば、選択システムに大きい集合の不安定な種を取り除き(例えば、プロテインA表面上又は疎水性相互作用カラム上のライブラリ吸収)、これにより露出した疎水性表面を有する不完全にフォールディングしたタンパク質を除去する、取り除く工程を組み込むことにより、これを行うことができる(Matsuura, T. et al., 2003)。本明細書中において、この手法
を使用して、非常に不安定なVH及びVLのバックグラウンドにおいて良好な生物物理学的特性を有するVH及びVLを選択した。しかし、適度に良好な安定性を有するタンパク質で集合化された突然変異型のライブラリから「最良の」種を選択することはより困難であり得る。この場合、より短期間のインキュベート時間でのプラーク形成速度に基づくか、又はプラークサイズ及び頻度基準(frequency criteria)に基づいて、主要な変異型を同定することができる。
【0045】
本発明の選択手法は、プロテインL、プロテインA又は任意のリガンドを伴う結合工程の選択システムにおける任意選択の包含を有するscFv及びFab等の安定性があり、且つ十分フォールディングした抗体フラグメント、並びに安定性のある非抗体スカフォールド及びこれらの突然変異型の同定にまで拡大することができる。さらに、ファージプラークサイズとVH発現収率との間に見られる相関関係は、本発明の手法を利用しなければ変異型のファージディスプレイライブラリから不完全であるか、又は不十分な発現を有するタンパク質の高発現性がある変形を得るために本発明の手法を利用することができることを意味する。ブースト化タンパク質の発現がタンパク質の生産コストを有意に埋め合わせる治療用タンパク質又は高価で不完全な発現性のタンパク質試薬の場合、この適用は特に魅力的である。
五量体の結合分析
VL及びVHの両方が五量体化の影響を受けやすく、五量体化を使用して、親和性の低いVL又はVHの単量体を親和性の高いVL又はVHの五量体に迅速に変換することができる。このような五量体は貴重な診断用薬剤及び検出用薬剤である。このような用途において、VL又はVHの五量体のその標的との結合は、酵素(例えば、セイヨウワサビペルオキシダーゼ又はアルカリ性のホスファターゼ)等のレポーター分子又は五量体と結合する蛍光分子で検出することができる。代替的に、五量体の結合は、レポーター分子と結合する二次分子で検出することができる。二次分子は、五量体自体又は6Hisタグ若しくはc−Mycタグ等のタグに特異的であり得る。例えば、一般的な二次分子はイムノグロブリンである。
【0046】
プロテインAを有するVHとプロテインLを有するVLとの間の相互作用は、これらの標的抗原を有するVHとVLとの間の相互作用とは基本的に異なっている。VH又はVLの抗原結合は、抗体ドメインの結合部位を形成する3個の抗原結合ループを伴う。プロテインA結合活性を有するVH及びプロテインL結合活性を有するVLのプロテインA結合は、抗体結合部位とは全体的に異なる抗体ドメイン上に結合部位及び残基を伴う。このように、プロテインA結合活性を有するVHは、同時にプロテインA及びその標的抗原と結合することができ、プロテインL結合活性を有するVLは、同時にプロテインL及びその標的抗原と結合することができる。本発明のVHとVLはそれぞれ、プロテインA及びプロテインLに対する親和性を有するので、上記の検出用途及び診断用途のための二次分子として、プロテインA及びプロテインLを使用することができる。ヒトVH及びヒトVLの五量体は治療のためにも使用することができる。
五量体による病原体検出
VHのプロテインA結合活性及びVLのプロテインL結合活性を使用して、表面上にプロテインA及び/又はプロテインLを有する細菌を検出することができる。プロテインAは、病原菌である黄色ブドウ球菌の表面上に存在する。このように、本明細書に記載のも
の等のプロテインA結合活性を有するVHを使用して、黄色ブドウ球菌を検出することができる。同様に、細胞表面上にプロテインLを有する細菌、特にペプトストレプトコッカス属マグヌス(Peptostreptococcus magnus)等の病原菌の検出のために、プロテインL結
合活性を有するVL単量体及びVL五量体を使用することができる。
【0047】
プロテインLは、ヒトにおけるP.magnus(Ricci, S. et al., 2001)の発症の毒性因子に関連する。膣炎において、プロテインLは、架橋表面結合性のIgEによりその効果を発揮すると考えられる。プロテインL結合活性を有するVL単量体及び/又はVL五量体がプロテインLのIgE架橋作用を阻害する可能性があるので、これらは治療剤として潜在性がある。
【0048】
プロテインAは、ヒトにおける黄色ブドウ球菌(Fournier, B. et al., 2004)の発症の
毒性因子に関連する。その毒性は、抗体と結合することを含む宿主成分と相互作用する能力に起因している。プロテインA結合活性を有するVH単量体及び/又はVH五量体が宿主成分を有するプロテインAの相互作用を阻害する可能性があるので、これらは治療剤として潜在性がある。
【実施例】
【0049】
単量体のヒトVHの同定及び配列分析
完全なヒトVHライブラリ及びラマ化したヒトVHライブラリの構築の間に、単量体のラマ化したVHを表示するファージが、凝集傾向のある完全なヒトVHを表示するファージよりも細菌層上でより大きいプラークを形成したことを確認した。このように、ヒトVHレパートリから珍しく自然発生的な単量体のVHを同定する手段として、プラークサイズを使用した(図1)。この目的で、サイズが6×108であるヒトVHを示すファージライブラリを構築し、寒天プレート上でプラークとして増殖させた。タイタープレート上で、ライブラリは本質的に、幾つかの大きいプラークが散在する小さいプラークから成る。20個のクローンにおけるPCRによって、小さいプラークがVH表示ファージに対応し、一方で大きいプラークは野性型のファージ(すなわち、VH配列挿入を欠くファージ)を表すことが示された。VH表示ファージは、大きいプラーク形態では見出されなかった。このことは、ヒトのレパートリ及び大きいサイズのライブラリにおける単量体のVHの不足により予想外のことではなかった。単量体のVHの同定を容易にするために、VH3ファミリー由来のヒトVHのサブセットと結合するプロテインAに対してライブラリをパンニングすることにより、ライブラリを取り扱いが可能な大きさまで縮小し、プラークサイズ形態が大きくなるのを阻害する野性型のファージを取り除くことを決めた。
【0050】
数回のパンニングの後、ライブラリはファージを生成する大きいプラークに関して富化され、110個を超えるこのようなプラークのPCR及びシークエンシングにより、全てがVHオープンリーディングフレームを完成させたことを示した。分析のために選択された大きいプラークのサイズは図1で示す。シークエンシングによって、VH3ファミリーに属する15個の異なるVHが示され、DP−38、DP−47、V3−49、V3−53、YAC−5又は8−1Bの生殖細胞系Vセグメントを利用した(第1表、図2)。DP−38及びDP−47の生殖細胞系配列は、これまではプロテインA結合に関係していた。さらに、それらのプロテインA結合活性と一致して、全てのVHは、57位でThr残基を有していた(図2)。最も頻繁に利用された生殖細胞系Vセグメントは、VHの50%を越えて発生するDP−47であったが、最も高頻度なクローン(すなわち、HVHP428、相対頻度46%)はV3−49生殖細胞系Vセグメントを利用した。DP−47生殖細胞系配列を有するHVHP429は相対頻度が21%である2番目に豊富なVHであった(図2)。VH CDR3の長さは、HVHB82における4個のアミノ酸からHVHP430における16個のアミノ酸の範囲であり、HVHP430はCDR3において一対のCys残基を有していた。親の生殖細胞系Vセグメント(残基1〜94)及び
FR4(残基103〜113)配列に対するアミノ酸突然変異は全てのVHで見られ、HVHP44(L5V及びQ105R)及びHVHB82(E1Q及びL5Q)に対する2個の突然変異からHVHP426に対する16個の突然変異までの範囲であった(第1表)。突然変異はVセグメントに集中し、105位及び108位の2個の突然変異のみが15個のFR4全てで検出された。HVHP44及びHVHB82は、その両方がGlnの代わりに105位で正に荷電したアミノ酸を有するという点で他のVHとは異なっていた(第1表、図2)。しかし、HVHP44において正に荷電したアミノ酸が突然変異によって得られたが、HVHB82における正に荷電したアミノ酸は生殖細胞系でコードされていた。HVHP423及びHVHP44Bを除いて、残りのVHは鍵となる可溶性位置で生殖細胞系残基を有していた:37V/44G/45L/47W又は37F/44G/45L/47W(HVHP428)、HVHP423及びHVHP44BはV37F突然変異を有していた。VH可溶性に重要であることが示されるか、又は仮定される他の位置での突然変異には、7個のE6Q、3個のS35T/H、1個のR83G及び1個のK83R、1個のA84P及び1個のT84A及び1個のM108Lが包含されていた。11個のE1Q、8個のL5V/Q及び1個のV5Q突然変異を包含した高頻度の突然変異も1位及び5位で観察された。
ヒトVHの生物物理学的特徴
HVHP44B(HVHP423と本質的に同じであった)を除いて、全てのVHはc−Myc−His5タグとの融合における培養液量1Lの大腸菌株TG1で発現し、固定化金属親和性クロマトグラフィ(IMAC)によって、ペリプラズム抽出物から均一に精製した。発現収率は、振盪フラスコ内で細菌培養液1L当たりで精製タンパク質1.8〜62.1mgの範囲であり、VHのほとんどは数mgの収率があった(第2表)。HVHP423及びHVHP430の例において、「明らかに」同じ発現条件下での別の試験によって、62.1及び23.7mgに対してそれぞれ、2.4及び6.4mgの収率が得られた。このことは、本明細書中に記載の多くのVHに対して、大した努力をせずに最適な発現条件を達成でき、それにより第2表で報告された値より有意に高い発現収率がもたらされることを意味している。予想通り、全てのVHは表面プラズモン共鳴(SPR)分析(KDは0.2〜3μMである)においてプロテインAと結合していて、範囲及び規模はプロテインA結合活性を有するラマVHH変異型についてこれまでに報告されていた範囲及び規模に匹敵していた。VHはFab基準面と結合しなかった。
【0051】
ゲルろ過クロマトグラフィ及びNMRによるオリゴマー化状態に関して、ヒトVHの凝集傾向を評価した(第2表)。全てのVHをSuperdex75ゲルろ過クロマトグラフィにかけた。ラマVHH(すなわち、H11C7)と同様に、全てのVHにおいて、単量体で期待される溶離量で対称な単一のピークが与えられ、凝集体は実質的に全く存在しなかった(図3AのHVHP428における例を参照のこと)。これに対して、一般的なヒトVH(すなわち、BT32/A6)はかなりの量の凝集体を形成した。3個のVHに関しては、VH二量体で期待される移動度を有する微小ピークも観察された。微小ピークのSPR分析によって、二量体のVHと一致して、単量体VHに対する値よりも有意に緩やかな解離速度値(off-rate value)が与えられた。二量体のピークは、ラマVHH(H11C7)の場合にも観察された。高濃度のVHのフォールディング状態及びオリゴマー化状態をさらにNMR分析で研究した。表IIに示すように、全てのVHタンパク質の研究によって、比較的可溶性であると考えられ、十分にフォールディングされた三次元構造が推測された。VHフラグメントの一次元NMRスペクトル(図3B)によって、VHドメインの構造フォールディング特徴が示された。HVHP414フラグメント及びHVHP414の2個のイソ型(c−Myc配列を有する及び有さないVH14及びVH14−cMyc−)に対するPFG−NMR拡散実験の使用によっても、タンパク質の凝集状態を評価した。VH14は、c−Myc N132E突然変異を有し、且つさらにN末端にメチオニン残基を有するHVHP414の改変バージョンである。簡単に述べると、PFG−NMRデータ(示されず)によって、全てのタンパク質サンプルが、NMR実験に使用
した比較的高いタンパク質濃度であっても予想される単量体の分子量を有することが示された。
【0052】
一貫して37℃でのトリプシンに対する耐性に関して、VHの安定性をさらに研究し、その後37℃で長期的にインキュベートを行った。トリプシンは、C末端のArg又はLys残基でポリペプチドのアミド骨格を切断する。ヒトVHにおいて9〜13個のArg及びLys残基が存在する(図2)。トリプシンによる消化の影響を受けやすいC末端のc−MycタグにおいてさらなるLys残基も存在する。図4aは、トリプシン消化の間のHVHP414のSDS−PAGE分析である。1時間以内に、元のバンドは、c−Myc−His5タグを有しないVHで期待される移動度があった単一の生成物に完全に変換された。トリプシンによる1時間のインキュベート後に12個の他のVHにおいて、同じ結果が得られた。トリプシン処理したVHのランダムに選択されたサンプル(すなわち、HVHP414、HVHP419、HVHP420、HVHP423、HVHP429、HVHP430及びHVHM81)における質量分析によって、それぞれの場合に、消化した生成物の分子質量がC末端残基としてのc−Myc Lysを有するVHに対応していたことが確認された。HVHM41において、消化時の残りよりも有意に短いフラグメントが与えられ、この場合質量分析実験によって、CDR3におけるArg99に対する切断部位がマッピングされた(データ図示せず)。
【0053】
0.32mg/ml(HVHP428)〜3.2mg/ml(HVHP420)の濃度に及ぶ11個のVHを37℃で17日間インキュベートした。その後、オリゴマー化状態及びプロテインA結合に関して、これらの安定性を決定した。ゲルろ過クロマトグラフィによって示されるように、37℃でのVHの処理は凝集体形成を全く誘導しなかった:全てのVHは、未処理のVHのものと実質的に同一であったクロマトグラムプロファイルを与え、本質的に単量体として存在していた(HVHP420についての例、図4cを参照のこと)。VHが37℃での処理後の自然のフォールディングを維持したことを確実するために、2個のVH(すなわち、HVHP414(1.2mg/ml)及びHVHP420(3.2mg/ml))をランダムに選択し、SPRによってプロテインAと結合するこれらのKDを求め(HVHP420に関して示されるデータ、図4cの挿入図)、未処理のVHに対して得られたKDと比較した(第2表)。熱処理したVHに関して算出したKDは1.4μMであり、HVHP414及びHVHP420に関して算出したKDはそれぞれ1.0μMであった。これらの値は未処理のVHの対応する値と本質的に同一であり(第2表)、このことはVHの37℃での処理がこれらの天然フォールディング(native fold)に影響を与えなかったことを示している。VHが37℃のインキュベート期間で
余り小さくなく、且つ非天然であり、ゲルろ過実験及びSPR実験の間室温まで戻した時にこれらの天然のフォールディングを再開する可能性は、十分にフォールディングした天然のタンパク質に一般的に関連する特性であるVHが37℃でトリプシンに対して耐性があった(上記を参照のこと)という事実を鑑みて疑わしい。
【0054】
天然のVH(KDn)及び熱処理して、リフォールディングしたVH(KDref)のプロテインAとの結合のKDを比較することによって、ヒトVHのリフォールディング効率(RE)を研究した(Tanha, J. et al., 2002)。VH分画が熱処理によって不活性化される場合、このパラメータはフォールディングした、すなわち活性のある抗体フラグメントの濃度に基づいているので、測定KDは高くなる。このように、KDnとKDrefとの比によって、VHREの測定値が与えられる。図5は、幾つかの選択されたVH濃度での天然状態(太線)及びリフォールディング状態(細線)における固定化されたプロテインAとのHVHP423の結合に対するセンサーグラムを比較している。示され得るように、リフォールディングしたVHのプロテインAとの結合は全ての例でより小さく、これにより非フォールディングは完全に可逆ではないことが示される。14個のVHそれぞれにおいて、天然状態及びリフォールディング状態におけるプロテインAの結合を幾つかの
濃度で測定し、KD及びその後のREを求めた(第2表、KDref値は示されていない)。2個の抗イディオタイプのラマVHH(H11F9及びH11B2)のKD及びREも求め、これらは参照として使用された。4個のVHは、H11F9及びH11B2のそれぞれの、95%及び100%のREと同程度の92%〜95%の範囲のREであった。別の5個のVHは、84%〜88%の範囲のREであり、3個は70%を超えていた。2個のVHだけが有意に低いREであった:HVHP413(52%)及びHVHP421(14%)。これまでに調べられた幾つかの公開されたVHHは約50%のREであった(van der Linden, R. H. et al., 1999)。
ヒトVHファージディスプレイライブラリ構築及びパンニング
ランダムヘキサヌクレオチドプライマー及びFirst Strand cDNA(商標)キット(GE Healthcare, Baie d'Urfe, QC, Canada)を使用して、ヒト脾臓mRNAからcDNAを合成した。鋳型としてcDNAを使用して、VHフレームワーク領域1(FR1)に特異的なプライマー及びイムノグロブリンM特異的なプライマー(de Haard, H. J. et al., 1999)を使用した9個の別々の反応におけるポリメラーゼ連鎖反応(PCR)によって、CH配列をフランキングしたVH遺伝子を増幅した。生成物をゲル精製し、2回目のPCRにおける鋳型として使用することにより、クローニング目的のためにフランキングしたApal I及びNot I制限部位も導入したFR1特異的なプライマー及びFR4特異的なプライマー(de Haard, H. J. et al., 1999)を使用して、VH遺伝子を構築した。得られたVHレパートリDNAをfd−tetGIIIDファージベクターにクローニングし、VHファージディスプレイライブラリを構築した(Tanha, J. et al., 2001)。プロテインAに対するパンニング(Amersham Biosciences Inc.)を記載のように行
った(Tanha, J. et al., 2001)。DNAPLOTソフトウェアバージョン2.0.1及びV BASEバージョン1.0(http://vbase.dnaplot.de/cgi-bin/vbase/vsearch.pl)を使用して、選択されたVHの生殖細胞系配列帰属を行った。記載のようにH11 scFvに対してパンニングすることによって、ラマVHHファージディスプレイライブラリからラマVHH H11C7、H11F9及びH11B2を単離した(Tanha, J. et al., 2002)。
VHの発現及び精製
標準的なクローニング技法により、VHをpSJF2発現ベクターにクローニングした(Sambrook, J. Fritsch E. F. and Maniatis T, 1989)。sdAbのペリプラズム発現及
びその後の固定化金属親和性クロマトグラフィ(IMAC)による精製を記載のように行った(Muruganandam, A. et al., 2002)。それぞれのタンパク質において算出したモル吸
収係数を使用したA280測定によって、タンパク質濃度を測定した(Pace, C. N. et al., 1995)。記載のように、Superdex75カラム(GE Healthcare)において、精製
したVHのゲルろ過クロマトグラフィを行った(Deng, S. J. et al., 1995)。
結合及びリフォールディング効率の実験
VH/VHHの平衡解離定数(KD)及びリフォールディング効率(RE)は、BIACORE3000バイオセンサーシステム(Biacore Inc., Piscataway, NJ)で回収した表面プラズモン共鳴(SPR)データから得られた。プロテインAとのVHの結合を測定するために、プロテインAの2000個の共鳴単位(RU)又は基準となる抗原結合フラグメント(Fab)を研究等級のCM5センサーチップ(Biacore Inc.)上で固定化した。製造業者によって提供されるアミンカップリングキットを使用して、10mMの酢酸ナトリウム緩衝液(pH4.5)中の25μg/ml(プロテインA)又は50μg/ml(Fab)の濃度で固定化を行った。抗イディオタイプのラマVHHのH11scFvとの結合を測定するために、上記のように、50μg/mlのH11 scFv 4100RU又は10μg/mlのSe155−4 IgG基準 3000RUを固定化した。全ての例において、流量40μl/分で、150mMのNaCl、3mMのEDTA及び0.005%のP20を含む10mMのHEPES(pH7.4)中、25℃で分析を行い、表面を洗浄緩衝液で洗浄することにより再生させた。リフォールディングタンパク質の結合活性を測定するために、10μg/mlの濃度、85℃で20分間インキュベートするこ
とによって、VH又はVHHを変性させた。それから、リフォールディングするためにタンパク質サンプルを30分間室温まで冷却し、その後室温で5分間14000rpmでマイクロフュージで遠心分離して、任意のタンパク質沈殿物を除去した。上清を回収し、上記のようにSPRによって結合活性について分析した。BIAevaluation4.1ソフトウェア(Biacore Inc.)を同時に使用して、フォールディングしたタンパク質及びリフォールディングタンパク質両方に関するデータを1:1の相互作用モデルに適合させ、その後KDを求めた。REは、
【0055】
【化1】

【0056】
(式中、KDnは天然タンパク質のKDであり、KDrefはリフォールディングタンパク質のKDである)から求めた。
トリプシン消化実験
1mMのHCl中の新たに調製した0.1μg/μlのシークエンシング等級のトリプシン(Hoffmann-La Roche Ltd., Mississauga, ON, Canada)3μlを100mMのTri
s−HCl緩衝液(pH7.8)中のVH60μgに加えた。37℃で1時間、全量60μlで消化反応を行い、0.1μg/μlのトリプシン阻害剤(Sigma, Oakville, ON, Canada)5μlを加えることにより停止させた。消化の完了後、5μlを取り除き、SDS
−PAGEで分析して、ZipTipC4(Millipore, Nepean, ON, Canada)を使用して
残りを脱塩し、50:50のメタノール:水の1%酢酸で溶離して、MALDI質量分析によってVHの質量を求めた。
37℃でのタンパク質安定性研究
37℃で17日間PBS緩衝液において0.32〜3.2mg/mlの濃度での単一ドメイン抗体(sdAb)をインキュベートした。インキュベートの後、視覚的に凝集体の形成が全く見られなくても、5分間最大速度でミクロフュージにおいてタンパク質サンプルを沈降させた。それから、サンプルをSuperdex75サイズ排除カラム(GE Healthcare)に適用させ、プロテインAに対するSPR分析における単量体のピークを回収し
た。プロテインA500RU又は基準Fabを固定化したこと、及び50μg/mlの濃度で固定化を行ったことを除いて、上記のようにSPR分析を行った。
NMR実験
NMR分析におけるVHサンプルを10mMのリン酸ナトリウム、150mMのNaCl、0.5mMのEDTA及び0.02%のNaN3(pH7.0)中に溶解した。タンパク質濃度は40μM〜1.0mMであった。全てのNMR実験は、Bruker Avance−800NMRスペクトロメータ又はBruker Avance−500NMRスペクトロメータにおいて298Kで行った。一次元(1D)1H NMRスペクトルを16384個のデータ点で記録し、スペクトル幅はそれぞれ、500MHzで8992.81Hz及び800MHzで17605.63Hzであった。2048×400のデータ点の二次元1H−1H NOESYスペクトルがスペクトル幅11990.04Hz及び混合時間120msのBruker Avance−800NMRスペクトロメータにおいて得られた。全てのNMR実験において、3−9−19パルス列によって行われたWATERGATE法(Piotto, M. et al., 1992; Sklenar, V. et al., 1993)を使用して
水抑制を達成した。NMRデータを処理し、Bruker XWINNMRソフトウェアパッケージを使用して分析した。3軸勾配がある三重共鳴プローブを備えるBruker
Avance−500 NMRスペクトルメータで水抑制LED配列(Altieri, A. S. et al., 1995)によって、全てのPFG−NMR拡散測定を行った。一次元プロトンスペ
クトルを処理し、Bruker Xwinnmrソフトウェアパッケージを使用して分析
した。全ての所与のPFG強度で均一に全てのNMRシグナルが弱まった場合に、メチル及びメチレンプロトン領域(2.3ppm〜−0.3ppm)におけるNMRスペクトルを統合することにより、NMRシグナル強度を得た。
ヒトVLファージディスプレイライブラリ構築及びパンニング
ヒトVHに関して上記のように、cDNAをヒト脾臓mRNAから合成した。cDNAをPCRの鋳型として使用し、6個のVκバックプライマー、11個のVλバックプライマー(de Haard, H. J. et al., 1999)、4個のVκフォワードプライマー及び2個のVλフォワードプライマー(Sblattero, D. et al., 1998)を使用して、反応体積50μlでVL遺伝子を増幅させた。バックプライマー及びフォワードプライマーを修飾して、その後のクローニング目的のためにそれぞれ、Apa LI制限部位及びNot I制限部位をフランキングさせた。これらの縮重の程度に反映された比で共にフォワードプライマーがプールされた。プールされたVλフォワードプライマー及び11個の個々のVλバックプライマーを使用して、11個の別々の反応でVλ遺伝子をPCRした。同様に、プールされたVκフォワードプライマー及び6個の個々のVλバックプライマーを使用して、6個の別々の反応でVλ遺伝子を増幅させた。PCR生成物をプールし、ゲル精製して、Apa LI制限エンドヌクレアーゼ及びNot I制限エンドヌクレアーゼで消化した。ヒトVHに関して上記のように、ライブラリを構築した。個々のライブラリコロニーにおいてプラークPCRを行い、増幅したVL遺伝子を記載のようにシークエンシングした(Tanha, J. et al., 2003)。ヒトVHライブラリに関して上記のように、プロテインL(Biolynx Inc., Brockville, ON, Canada)に対するパンニング及び選択されたVLの生殖細胞系の配列帰属を行った。
VLの発現及び精製
「VHの発現及び精製」においてVHに関して上記のように、VLの発現、精製、濃度決定及びゲルろ過クロマトグラフィを行った。
VL及びVH五量体の発現及び精製
標準的なPCRにおいて特異的なプライマーを使用して、HVHP328 VH及びHVLP335 VL遺伝子を増幅させた。標準的なクローニング技法を使用して、発現ベクターにおいてVT1B五量体化ドメイン遺伝子と融合して、HVHP328及びHVLP335遺伝子をクローニングして、HVHP328PVT2及びHVLP335PTV2五量体を得た(Zhang, J. et al. 2004)。五量体を記載のように発現及び精製した(Zhang, J. et al., 2004)。上記のようにタンパク質濃度を決定した。
VLの表面プラズモン共鳴
BIACORE3000バイオセンサーシステム(Biacore, Inc., Piscataway, NJ)を
使用したSPRによって、VLのプロテインLとの相互作用に対する結合反応速度を測定した。プロテインL680RU又は基準Fab 870RUを研究等級のCM5センサーチップ(Biacore)上で固定化させた。製造業者によって供給されたアミンカップリングキ
ットを使用して、10mMの酢酸緩衝液(pH4.5)において50μg/mlのタンパク質濃度で固定化を行った。50μl/分又は100μl/分の流量で150mMのNaCl、3mMのEDTA及び0.005%のP20を含む10mMのHEPES緩衝液(pH7.4)中、25℃で全ての測定を行った。洗浄緩衝液で洗浄することにより表面を再生した。BlAevaluation4.1ソフトウェア(Biacore, Inc.)を使用して
、データを評価した。
五量体VL及びVHの表面プラズモン共鳴
SPRによって、HVHP328PVT2のプロテインAとの相互作用及びHVLP335PTV2のプロテインLとの相互作用に対する結合反応速度を決定した。520RUのプロテインA又は基準Fabを上記のように固定化した。VL五量体に関して、上記のように調製した同様の表面を使用した。上記のように測定を行ったが、流量は20μl/分であった。3秒間、50mMのHClで洗浄することによって、表面を再生した。単量体に関して記載のようにデータを評価した。
細胞の微小凝集
BHIプレート由来の黄色ブドウ球菌の単一コロニーを使用して、BHI培地15mLを接種した。細菌を37℃、200rpmで一晩成長させた。翌朝、培養液を回転バケット、Sorvall RT6000B冷凍遠心器において4000rpmで10分間遠心分離し(spun down)、上清を除去して、細胞ペレットをPBS緩衝液で再懸濁させた。
細胞を再び遠心分離し、上清を除去して、細胞ペレットを再びPBS緩衝液で再懸濁させた。細胞をA600が1.0になるまで希釈し、段階希釈した細胞を37℃のBHIプレート上に広げて、一晩成長させた。翌朝、細胞力価を測定した。A600 1.0が1.5×109細胞/mlに対応する。同一の工程を行い、成長培地が2xYTであったことを除いて、その後の微小凝集アッセイのために大腸菌株TG1細胞を調製した。生菌数は同様であった、A600 1.0=2.1×109細胞/ml。
【0057】
微小凝集アッセイを行うために、マイクロタイタープレートにおけるウェル1〜ウェル11でPBSにおいてHVHP328PVT2の2倍希釈を行った。ウェル12(ブランク)はPBSのみを含んでいた。それぞれのウェルの全量は50μlであった。その後、PBS50μlにおける1×108個の黄色ブドウ球菌細胞を全てのウェルに加えて、プレートを4℃で一晩インキュベートした。結果を永続的に記録するために、翌朝にプレートの写真を撮った。五量体の対照実験のために、HVHP328PVT2をVL五量体、HVLP335PTV2に置き換えた。細胞の対照実験において、同じ2個の組の実験を大腸菌TG1細胞で繰り返した。
単量体のヒトVLの同定及び配列分析
ヒトVHファージディスプレイライブラリから可溶性のVHを単離するために用いられた本質的に同じ選択方法を、可溶性で単量体のVLを単離するためにヒトVLライブラリに適用した。大きさが3×106であるヒトVLライブラリを構築した。ライブラリタイタープレートから24個のプラークを選別し、これらのVL遺伝子をPCR及びシークエンシングした。配列は、生殖細胞系起源に関して異なるが、VLの75%はVλ起源であった(データ図示せず)。プロテインLに対する3回のパンニングによって、大きいプラークにおける富化がもたらされた。39個の大きいプラークをシークエンシングして、32個の特殊な配列を同定した(図6)。HVLP325、HVLP335及びHVLP351はそれぞれ、3、4及び2の頻度で起こった。λクラス(サブグループVλ1、生殖細胞系1b)であるHVLP389を除いて、残りの31個のVLはVκクラスに属していた。31個のκVLの中で、24個はVκIIIサブグループに含まれ、7個はVκ1サブグループに含まれていた。24個のVκIII配列の16個は、残りの利用されるA27、L2及びL6生殖細胞系配列を有するL6生殖細胞系配列を利用する。Vκ1サブグループのVLはO2/O12又はA30生殖細胞系配列に由来する。顕著な突然変異は96位で起こった。この位置での生殖細胞系アミノ酸は芳香族及び疎水性アミノ酸、κVLに対するTrp、Phe、Tyr、Leu又はIle、及びλVLに対するTyr、Val又はAlaである。しかし、κVLの選択プールにおける31個の中の5個だけが96位でこれらの生殖細胞系アミノ酸を有している:HVLP325、HVLP349、HVLP388、HVLP3109及びHVLP393。96位での21個のアミノ酸を帯電させ、この中の20個が正に帯電する:Arg、Lys又はHis。2個のアミノ酸はPro、1個はGln、1個はSer、及び1個はThrである。単量体化のためにゲルろ過クロマトグラフィで分析された7個のκVLの中で96位がArg又はLysである6個が単量体であった一方で、96位が生殖細胞系アミノ酸LeuであるHVLP325が凝集体を形成した(以下を参照のこと)。同様に、λクラスであり、且つSerに対する生殖細胞系突然変異を有していたHVLP389も単量体であった(以下を参照のこと)。これらのデータは単量体化等のVLの生物物理学的特性が改良された96位での生殖細胞系アミノ酸からの偏差に相関がある(32個の中の27個)。
【0058】
κクラスの18個のVLは、λVLでのみ見出されるアミノ酸Thr、Val及びLeuに置き換えられたこれらの最後の3残基(105〜107)を有していた。これらの置
換はκVLの生物物理学的特性を改良する役割を果たしている可能性があり、これにより105〜107位の元のκ残基を有する親クローンを超えた上記のVLの選択がもたらされる。
ヒトVLの特徴付け
異なるV生殖細胞系起源を有する選択VLの中の8個は、培養液1L中の大腸菌で発現し、精製された:HVLP324、HVLP325、HVLP335、HVLP342、HVLP351、HVLP364、HVLP389及びHVLP3103(第6表)。全てがHVLP324に対する6.2mgからHVLP335及びHVLP364に対する約75mgまでに及ぶ良好な収率で発現した。
【0059】
ゲルろ過クロマトグラフィによるこれらのオリゴマー化状態に関するヒトVLの凝集傾向を評価した。VLを0.6mg/mlの濃度でSuperdex75ゲルろ過クロマトグラフィにかけた。HVLP325を除いた全てが凝集体を本質的に含んでおらず、平均見掛けの分子質量が12.7kDa(範囲、6.2〜19.2kDa)である対称の単一ピークを得た(図7A及び第3表)。これは単量体のVLに関する期待分子質量(13.4〜13.8kDa)と一致している。単一ドメインの抗体に関する見掛けの分子質量の変動はこれまでに報告されている(Jespers, L. et al., 2004a; Stevens, F. J. et al.,
1980)。HVLP325に関して、凝集体が全タンパク質(凝集体+単量体)の11%を構成していた。HVLP351、HVLP342、HVLP335及びHVLP3103は、これらの利用可能な最も高い濃度、すなわちそれぞれ、0.89mg/ml、1.0mg/ml、4.9mg/ml及び5.9mg/mlで試験した場合、依然として単量体であった(図7B)。
【0060】
BIACORE分析前に、VLをSuperdex−75クロマトグラフィにかけて、物質の凝集に何ら確証がなかったとしても精製した単量体のピークを回収した。SPR分析において、全ての選択されたVLは、プロテインLと結合した(図8)。VLがプロテインLに対するパンニングによって単離されたので、このことは予想外のことではなかった。全てに関して、プロテインLとの結合のKDは、0.6〜3μMであった(第3表)。HVLP324及びHVLP342はKDがそれぞれさらに10nM及び40nM小さかった。VκIサブグループのVLのプロテインLとの低い親和性及び高い親和性の結合が、これまでに報告されている(参考文献)。HVLP324及びHVLP342の両方がVκIサブグループに属している(第3表)。予想通りに、速度論データ及び平衡データは、実際に単量体である単量体ピークと一致していた。
五量体の結合分析
表面プラズモン共鳴によって、HVHP328PVT2五量体のプロテインAとの結合及びHVLP335PTV2五量体のプロテインLとの結合を測定した(図9)。会合速度をkobs対濃度のプロットから独立して算出した。五量体の集合の間の多価結合における不均一性によって、1を超えた解離速度(kd)を算出することができる。したがって、1を超えた平衡解離定数(KD)を得ることができる。HVHP328PTV2及びHVLP335PTV2はそれぞれ、2nM及び200pMの最小KDを有していた(第4表)。kdがより遅い場合、HVHP328PTV2及びHVLP335PTV2はそれぞれ、900pM及び90pMまで低いKDを有していた。
VL及びVHによる病原菌検出
VHのプロテインA結合活性及びVLのプロテインL結合活性を使用して、これらの表面上にプロテインA及び/又はプロテインLを有する細菌を検出することができる。本明細書中のVH及びVL等のVH及びVLが可溶性であり、且つ単量体である(凝集傾向が失われている)場合、このことは可能である。ラクダ科の重鎖抗体又はナースシャーク及びテンジクザメのIgNAR等の軽鎖が欠失した抗体由来の可変ドメインは、自然発生的に可溶性且つ単量体である。このことから、プロテインA結合活性及びプロテインL結合活性を有するものを使用して、これらの表面上にプロテインA及び/又はプロテインLを
有する細菌を検出することもできる。プロテインAは、病原菌である黄色ブドウ球菌の表面上に存在する。このように、本明細書中に記載のもの等のプロテインA結合活性のあるVHを使用して、黄色ブドウ球菌を検出することができる。本発明者らは微小凝集アッセイを行い、HVHP328PVT2 VH五量体の黄色ブドウ球菌と結合する能力を検出した。マイクロタイターウェル(ウェル1〜ウェル11)において2倍希釈したHVHP328PVT2で一定数の細菌細胞をインキュベートした(図10)。ウェル12は五量体の代わりに緩衝液を含んでいた。VHが細菌細胞と結合する場合、したがってその多量体の性質により、五量体は細胞を架橋し、その結果、細胞凝集をもたらすことが可能であるはずである。凝集細胞がマイクロタイターウェルにおける拡散細胞として現れる(図10)。何の結合もなければ、凝集は起こるはずがなく、したがって凝集体は存在せず、細胞はウェルの底部に点として現れる。図10で示されるように、細胞の凝集体が存在するので、五量体は黄色ブドウ球菌と結合する。凝集体は最大ウェル7まで観察される。ウェル7より先は、五量体の濃度が結合するには低すぎるので、凝集体は存在しない。対照のVL五量体は、全く凝集を示さず、これによりVH五量体の黄色ブドウ球菌に対する特異性が実証される(図10)。予想通りVH五量体が大腸菌(TG1株)又はサルモネラ細胞を凝集させないので、結合は細胞特異的でもある(データは示さず)。同様に、これらの細胞表面上にプロテインLを有する細菌、特にペプトストレプトコッカス属マグヌス等の病原菌の検出にプロテインL結合活性を有するVL単量体及びVL五量体を使用することができる。
【0061】
上記で記載の実施例は、本発明の実際の範囲を限定する役割を果たすものでは決してなく、むしろ例示目的のために表されていることが理解される。
参考文献リスト
【0062】
【表1】
【0063】
【表2】
【0064】
【表3】
【0065】
【表4】
【0066】
【表5】
【0067】
【表6】
【図面の簡単な説明】
【0068】
【図1】選択された実施例の結果(可溶性VH(HVHP428)を示すファージと不溶性VH(BT32/A6)を示すファージとの間のプラークサイズの対比)の図的記述を示す。この写真はプラークの可視化を高めるために拡大した細菌層の寒天プレートの一部を示す。プレートは、それぞれ同じ数の2個のプラーク型を含んでいたが、この写真は基本的に大きいHVHP428プラークを含んでいる。BT32/A6プラークの大多数は小さすぎて写真において鮮明ではっきりした画像を得ることができなかった。矢印で示されたプラークは、この画像で可視化させるのに十分大きかった小さい割合のBT32/A6ファージを示している。アスタリスクは、HVHP428ファージの代表的なプラークサイズを示す。プラークの同一性は、DNAシークエンシングによって決定された。
【図2】プロテインAに対する親和性及びプラークサイズに基づき選択されたヒトVHのアミノ酸配列。入力配列(sequence entries)の点は、HVHP2M10又はHVHP44とアミノ酸が同一であることを示す。ダッシュ記号は配列アラインメントのために含まれている。鍵となる可溶性部分の残基及びプロテインA結合特性を有するVH/VHHに関連する57T残基は太字である。カバットナンバリングシステムを使用する。「頻度」の値の総計は114である。CDRは相補性決定領域であり、FRはフレームワーク領域であり、gln seqは生殖細胞系の配列である。
【図3A】ヒトVHの凝集傾向。ゲルろ過クロマトグラムによって、この研究で単離したヒトVH(HVHP428)のオリゴマー化状態をラマVHH(H11C7)及び典型的なヒトVH(BT32/A6)のものと比較する。それぞれのクロマトグラムの最後に溶離したピークは単量体のVHに対応する。二量体のH11C7のピークは矢印によって示される。
【図3B】(i)800MHzでのHVHP414、(ii)500MHzのHVHP423、及び800MHzのHVHP428の一次元の1H NMRスペクトル。左側のパネルのスペクトルは、低強度のシグナルのより良好な視覚化を可能にするように2倍に拡大されている。
【図4A】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。21kDaのマーカーに対して未処理のHVHP414VHと15、30及び60分でトリプシン処理したHVHP414VHとの移動度を比較するSDS−PAGE。HVHP414−cMycは、c−Mycを欠失したHVHP414VHを示す。
【図4B】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。未処理のHVHP414VH及びトリプシン処理(60分)HVHP414VHの質量分析法によって得られた分子質量プロファイル。処理済VHの質量分析プロファイルを未処理のVHのものの上に重ねて、より良好な視覚的比較を行う。未処理のVHの実験の分子質量は、14967.6Daであり、これは期待分子質量14967.7Daとほぼ同一である。トリプシン処理したVHで観察された分子質量(13368.5Da)は、c−MycタグにおけるK(Lys)での切断によるC末端での13アミノ酸の欠失を示し、期待分子質量13368.0Daを与える。トリプシン切断部位は、HVHP414のアミノ酸配列上の垂直な矢印によって示される。
【図4C】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。37℃で処理したHVHP420VHのオリゴマー化状態(上方のプロファイル)と未処理のVH(下方のプロファイル)のものとの比較をするゲルろ過クロマトグラム。重ねるとクロマトグラムが区別できないので、このクロマトグラムを垂直に移動させた。それぞれのクロマトグラムの主なピーク及び微小ピークは、それぞれ単量体のVH及び二量体のVHに対応する。二量体のVHは、全タンパク質の3%を構成する。挿入図は、様々な濃度でのプロテインAとの37℃で処理したHVHP420の結合に関するセンサーグラムオーバーレイを示す。温度安定性試験に使用されるVHは、既に4℃で数ヶ月間調製したストック由来であった。
【図5】天然HVHP423(太線)及びリフォールディングしたHVHP423(細線)の75、100、150及び200nMの濃度で固定化されたプロテインAとの結合を示すセンサーグラムオーバーレイ。KDn及びKDrefがそれぞれのセンサーグラムから算出され、以下で記載のようにREを求めるのに使用した。
【図6】プロテインLに対する親和性及びプラークサイズに基づき選択されたヒトVLのアミノ酸配列。入力配列における点は、HVLP333とアミノ酸が同一であることを示す。ダッシュ記号は配列アラインメントのために含まれる。配列ナンバリング及びCDR指定に関するV BASE(http://vbase.mrc-cpe.cam.ac.uk/index.php?module=pagemaster&PAGE_user_op=view_page&PAGE_id=7&MMN_position=5:5)を参照されたい。L6、A27、L2、L16、O2/O12、A30及び1bは、V生殖細胞系設計である。J生殖細胞系設計は括弧内である。NFは見当たらない。
【図7A】ヒトVLドメインのサイズ排除クロマトグラム。VLを0.6mg/mlの濃度で添加した。「#」及び「*」は、それぞれ凝集体及び単量体のピークを示す。凝集体は排除体積で溶出する。
【図7B】ヒトVLドメインのサイズ排除クロマトグラム。VLを利用可能な最も高い濃度(1.0mg/mlのHVLP342、5.9mg/mlのHVLP3103、4.9mg/mlのHVLP335、0.89mg/mlのHVLP351)で適用した。「#」及び「*」は、それぞれ凝集体及び単量体のピークを示す。凝集体は排除体積で溶出する。HVLP342パネルにおける矢印で示されたピークは、前の測定からのキャリーオーバーである。
【図8】0.2、0.5、0.75、1、2、3、5及び10μM(HVLP389、HVLP351及びHVLP364);1、2、3、5、7.5及び10nM(HVLP342);0.2、0.5、1、2、3、5及び10μM(HVLP335);0.2、0.5、1、1.5、2及び5μM(HVLP325);0.2、0.5、0.75、1、1.5、2、3及び5μM(HVLP3103);並びに1、2、3、4、5及び6nM(HVLP324)の濃度で固定化されたプロテインLとのVLの結合を示すセンサーグラムオーバーレイ。プロテインLの低親和性部位と結合するHVLP324及びHVLP342に関するセンサーグラムは含まれないが、算出されたKDは第3表に記録される。
【図9A】表面プラズモン共鳴実験におけるプロテインAとHVHP328PTV2との結合を示す図である。1、2、3、4、6、8及び10nMの濃度で固定化したプロテインAとHVH28PTV2との結合を示すセンサーグラムオーバーレイ。結合データは第4表に記録される。
【図9B】表面プラズモン共鳴実験におけるプロテインLとHVLP335PTV2との結合を示す図である。1、2、2.5、3、3.5、4及び4.5nMの濃度で固定化したプロテインLとHVH335PTV2との結合を示すセンサーグラムオーバーレイ。結合データは第4表に記録される。
【図10】黄色ブドウ球菌による微小凝集実験の結果を示す図である。五量体の濃度は、ウェル1からウェル11まで2分の1ずつに減少し、ウェル12は五量体がPBS緩衝液で置き換えられている。上の行のウェルはHVHP328PTV2五量体を含み、下の行はHVLP335PTV2五量体を含む。ウェル1〜6の五量体の濃度はそれぞれ、215、108、54、27、13及び7μg/mlである。
【技術分野】
【0001】
本発明は、ポリペプチド、特に単量体のヒト抗体フラグメントの単離、同定及び操作に関する。
【背景技術】
【0002】
脊椎動物の抗体は通常、一対の重(H)鎖及び軽(L)鎖から構成される。組み合わされたH鎖及びL鎖の最初のドメインであるVH及びVLは配列においてより可変的であり、抗原を認識し、抗原に結合する抗体部分である。VHドメイン及びVLドメインは対で抗原を認識する。
【0003】
ラクダ科(ラクダ、ヒトコブラクダ及びラマ)の免疫レパートリは、重鎖抗体と呼ばれる変わった種類の抗体を保有するという点で、特殊である(Hamers, Casterman C. et al., 1993)。これらの抗体は軽鎖を欠いており、したがってこれらの組み合わせ部位はVH
Hと呼ばれる1個のドメインから構成される。
【0004】
組み換え型VHHの単一ドメイン抗体(sdAb)は、従来の四本鎖抗体由来の一本鎖Fv(scFv)フラグメントに勝る幾つかの利点を提供する。sdAbの親和性はscFvの親和性と同程度であるが、sdAbは、可溶性、安定性、凝集耐性、リフォールディング性、発現収率及びDNA操作の易操作性、ライブラリ構築並びに三次元構造決定といった点でscFvより優れている。抗体を伴う適用において、上記のVHH sdAbの特性の多くが望ましい。
【0005】
しかし、VHHの非ヒト性質によって、免疫原性によるヒトの免疫療法におけるVHHの利用が制限される。この点において、ヒトのVH及びVL sdAbの免疫原性が最小であると考えられるので、免疫療法への適用に対する理想的な候補である。
【0006】
しかし、ヒトのVH及びVLは従来の抗体由来のVH及びVLに共通の特徴である凝集の傾向が強い(Davies, J. et al., 1994; Tanha, J. et al., 2001; Ward, E. S. et al., 1989)。このように抗体用途に好適な単量体のヒトのVH及びVLを得る試みが行われ
てきた。このようなVH及びVLは、高い発現収率、高いリフォールディング性及び凝集耐性等のVHHに一般的な他の有用な特性も示す。これらのVH及びVLでライブラリ骨格として構築される合成ライブラリは、治療用の有望なタンパク質源として役立つ可能性がある。
【0007】
ラクダ及びラマのVHHからそれぞれ、ヒトのVH又はVLに組み込まれる鍵となる可溶性残余物を伴うラクダ化(camelization)並びにラマ化(llamination)を利用して、単量
体のヒトのVH及びVLを生成した。これらのVH及びVLに基づき構築され、CDRランダム化により生成される合成sdAbライブラリは、様々な抗原との結合因子を得るといった点で機能的であることが示された(Davies, J. et al., 1995; Tanha, J. et al., 2001)。
【0008】
別の手法において、上記の技術的手段を用いることなく、完全なヒトの単量体のVH及びVLをヒトの合成VH及びVLライブラリから単離した。一実験において、ヒトVHライブラリを鶏卵リゾチームに対してパンニングした(panned)時に単量体のヒトVHを発見した(Jesperps, L. et al., 2004b)。さらに最近では、可逆的非フォールディング及び親和性基準に基づく選択方法によって、合成ヒトVHライブラリから多くの単量体のVHが得られた(Jespers, L. et al., 2004a)。この知見は、適切な選択方法が望ましい生物物
理学的特性を有する珍しい単量体のヒトVHを効率的に捕捉する鍵となるという事実を明確に示していた。
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明の第1の目的は、生物物理学的特性(可溶性、高い発現及び/又は安定性(熱変性後の高いリフォールディング、化学変性剤に対する高い耐性、及びプロテアーゼ、特にトリプシン等の胃腸プロテアーゼに対する高い耐性等)を含む)が改良されたポリペプチド、特に抗体フラグメントを同定するハイスループットスクリーニング方法を提供することである。
【0010】
本発明の第2の目的は、単量体のヒトVH及びヒトVLを同定するハイスループットスクリーニング方法を提供することである。
【0011】
本発明の第3の目的は、単量体のヒトVH及びヒトVLを同定、単離及び特徴付けすることである。
【0012】
本発明の第4の目的は、抗体フラグメント、特に単量体のヒトVH及びヒトVLの多量体を構築及び特徴付けることである。
【0013】
本発明の第5の目的は、ポリペプチド、特に抗体フラグメント及び最も特定的には単量体のヒトVH及びヒトVLからディスプレイライブラリを構築することである。
【0014】
本発明の第6の目的は、ポリペプチド、特に抗体フラグメント及び最も特定的には生物物理的な特性が改良された単量体のヒトVH及びヒトVLを生成するDNAシャッフリング法を提供することである。
【課題を解決するための手段】
【0015】
発明の要約
ポリペプチド、好ましくは抗体フラグメント及び最も好ましくは望ましい生物物理学的特性(可溶性、安定性、高い発現性、単量体性、非凝集性、結合特異性)を有するヒトVH及びヒトVLを単離する方法が提供される。この方法は、様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得る工程、ライブラリファージによる細菌層の感染を可能にする工程、及び細菌層上の平均よりも大きいプラークを形成するファージを同定する工程を包含する。それからファージを単離し、シークエンシングか、又はそうでなければポリペプチド配列を特定するこれらの工程を用いる。
【0016】
本発明は、上記の方法で同定されるポリペプチド、特に単量体のヒトVH及びヒトVLも提供し、これは免疫療法に及び/又は診断用薬剤もしくは検出用薬剤として有用であり得る。単量体のヒトVH及びヒトVLは組合せて、二量体、三量体、五量体又は他の多量体を形成してもよく、これは免疫療法に及び/又は診断用薬剤もしくは検出用薬剤として有用であり得る。
【0017】
上記の方法によって同定されたポリペプチド(ヒトVH及びヒトVLを含む)を、可溶性、安定性、単量体性、高発現性、結合特異性、及びヒト起源等の改良された生物物理学的特性のために選択するDNAシャッフリング等の方法によって操作することができる。
【0018】
上記の方法によって同定されたポリペプチド(ヒトVH及びヒトVLを含む)は、さらにディスプレイライブラリを生成するのにも使用することがあり、これを同様に使用して、上記の方法によってさらにポリペプチドを単離することができる。
【0019】
第1の態様において、本発明は、標的ポリペプチドを同定する方法であって、a)様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得ること、b)ライブラリファージによって、細菌層の感染を可能にすること、c)細菌層上の平均よりも大きいプラークを形成するファージを同定することを含む、方法を提供する。
【0020】
第2の態様において、本発明は、配列番号8〜配列番号54から成る群より選択されるアミノ酸配列を有するポリペプチドを提供する。
【0021】
第3の態様において、本発明は、配列番号8〜配列番号22から成る群より選択される少なくとも1個のアミノ酸配列を含むVH抗体フラグメントを提供する。
【0022】
第4の態様において、本発明は、配列番号23〜配列番号54から成る群より選択される少なくとも1個のアミノ酸配列を含むVL抗体フラグメントを提供する。
【0023】
第5の態様において、本発明は、望ましい生物物理学的特性を有するポリペプチドを生成する方法であって、a)請求項41、42、44、45、47、48、59若しくは70に記載の抗体フラグメントをコードするか、又は請求項24、27、37若しくは39に記載のポリペプチド配列をコードし、且つ第1の望ましい特性を有する少なくとも1個の第1の核酸配列を提供する工程と、b)第2の望ましい特性を有する抗体フラグメントをコードする少なくとも1個の第2の核酸配列を提供する工程と、c)少なくとも1個の第1の核酸配列及び少なくとも1個の第2の核酸配列をランダムなフラグメントに切断する工程と、d)ランダムなフラグメントを再び集合化させる工程と、e)ランダムなフラグメントを発現する工程と、f)発現されたランダムなフラグメントを第1の望ましい特性及び前記第2の望ましい特性に関してスクリーニングする工程とを含む、方法を提供する。
【発明を実施するための形態】
【0024】
発明の詳細な説明
ヒト起源であり、可溶性、安定性、凝集に対する耐性、リフォールディング性、高発現性、及びDNAレベルでの易操作性があり、ライブラリ構築及び三次元構造決定に理想的であるポリペプチド、特に抗体フラグメントを同定することが望ましい。このような抗体フラグメントは、多種の免疫療法への適用、並びに診断用薬剤及び検出用薬剤としても有用である。ヒトの単量体のVH抗体及びVL抗体は上記の特性の多くを有しているとされるので、特に興味深い。
【0025】
上記の特性を有するポリペプチドは、様々なポリペプチド配列を発現することができるライブラリのハイスループットスクリーニングによって同定することができる。例えば、ファージディスプレイライブラリ(好ましくは、M13又はfd等の繊維状ファージ)は、ファージの影響を受けやすい細菌の領域(細菌層)をファージで感染させ、それからプラークとして知られるはっきりした無菌領域を探すことにより、どのファージが細菌を首尾よく溶解させるかを決定することによりスクリーニングすることができる。単量体のラマ化VH及びVLが、凝集傾向のある完全なヒトVHを示すファージよりも大きいプラークを細菌層上に形成する。したがって、ヒトVHレパートリから珍しく自然発生的な単量体のVH及びVLを同定する手段として、プラークサイズを使用することができる。
【0026】
本明細書中に記載の方法は、可溶性、安定性(安定性には、多くの特徴(高熱リフォールディング効率、高い融解温度、37℃での長期(数日)のインキュベート後の機能性の維持、化学変性剤に対する耐性、プロテアーゼに対する耐性、0℃未満、4℃未満及び室温での長期の保存期間、細胞内環境における機能性の維持、並びに血流中等のヒトの体内
の機能性の維持を含むが、これらに限定されない)が含まれる)、及び様々な起源の高発現性のタンパク質を同定するのにも有用であり、これらのタンパク質としては、
1.VH、VL、Fab、scFv及びより詳細にはヒト抗体であるIgG等の全抗体、
2.非抗体スカフォールドの一本鎖T細胞受容体、T細胞受容体ドメイン、トランスフェリン、リポカリン、kunitzドメイン、アンキリン反復、及び細胞傷害性Tリンパ球関連抗原(CTLA−4)に基づくタンパク質変異体(ヒトのものを含む)、
3.ウイルス性及び細菌性のタンパク質ワクチン等のワクチン、
4.治療用タンパク質(例えば、インスリン、成長ホルモン、エリスロポエチン)
5.タンパク質性の診断用試薬及び生化学試薬(例えば、プロテインA、プロテインG)
が挙げられる。
【0027】
ポリペプチドをこの方法で同定されれば、このポリペプチドを使用して、さらなるライブラリを構築することができる。例えば、VHの核酸配列を選択することによって、これを行うことができる。ランダムなコドンを有するオリゴヌクレオチドを作り、VH配列に組み込む。このようにして、それぞれの特殊なオリゴヌクレオチドをVH遺伝子に組み込み、修飾されたVH遺伝子によってわずかに変異のある配列のライブラリが構成される。一般的に、VHのCDR又はループをランダム化させるように、オリゴヌクレオチドを設計する。例えば、1個、2個又は3個全てのVH CDRをランダム化させることができる。それから、使用するライブラリの種類に応じて、VHライブラリを適切なベクターにクローニングさせ、核酸配列をポリペプチドとして発現させる。一般的にパンニング(panning)によって、ライブラリポリペプチドと結合する分子に関してライブラリをスクリー
ニングする。ライブラリはファージディスプレイライブラリ、又はリボソームディスプレイ及び酵母ディスプレイ等の他のディスプレイライブラリであり得る。
【0028】
本明細書中に記載の方法で同定したポリペプチドを例えば、単量体の架橋により二量体、三量体、五量体及び他の多量体を形成し、免疫療法に使用することができる。このことにより、抗原分子におけるより良好な親和性及び幾つかの抗原におけるより緩やかな拡散速度がもたらされ得る。別の可能性のある手法は、ポリペプチドを様々な機能を有する種々の分子と結合又は融合させることである。例えば、抗体フラグメントは、特定の細胞又は分子を標的、破壊又は修飾するために、放射性核種、細胞傷害性薬剤、トキシン、ペプチド、タンパク質、酵素、リポソーム、脂質、T細胞超抗原又はウイルスと結合することができる。
【0029】
本明細書中に記載の選択方法で同定したVH又はVLを単離すれば、VH又はVLをさらに操作し、可溶性、安定性、単量体性、結合特異性、ヒト起源又は高発現性等の改良された生物物理学的特性に関して選択することができる。DNAシャッフリング又は交互伸長法等のin vitro組み換え技法で、これを達成することができる。DNAシャッフリングは、抗体フラグメント等の第1のポリペプチド(ドナー)及び第2のポリペプチド(アクセプタ)の核酸配列をランダムなフラグメントに切断し、PCR様反応によりランダムなフラグメントを再び集合化させることを含む。それから、再集合化されたフラグメントをスクリーニングし、所望の特性に関して選択する。
【0030】
例えば、高い安定性のある1つ又は複数のVH(ドナー)を十分な安定性を欠いている1つ又は複数のVH(アクセプタ)と混合し、DNAシャッフリングにかけることができる。このことにより、ドナーVHから安定性残基を組み込んだアクセプタVHの突然変異型が発生する。本明細書に記載の方法によって、又はリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ及びファージディスプレイ等の他の発展的なタンパク質スクリーニングシステムによって、新たな安定性のある突然変異型を同定することができ
る。同様に、この技法を使用して、可溶性、単量体性、及び高発現性等の望ましい特質を形質転換することができる。
【0031】
ドナーVH及びアクセプタVHが望ましい特性を有する場合に、この技法を使用して両方の特性を有するVHを生成することができる。例えば、重要な治療用リガンド又は診断用リガンドと結合する不安定なドナーVHを安定性のあるアクセプタVHでシャッフルすることができる。新たに生成した安定性のあるVHもリガンドと結合する能力があることを確実にするために、スクリーニングシステムは、リガンド結合工程を伴い得る。
【0032】
DNAシャッフリングは、ラクダ科の重鎖抗体の可変領域並びにナースシャーク及びテンジクザメの可変領域等の非ヒトVH、又は治療標的と結合する非ヒトVLをヒト化するのにも有用であり得る。可溶性、安定性、単量体性、及び高発現性等の望ましい特性を有するヒトVH及びヒトVLをドナーとして使用することができる。例えば、良好な安定性のある1つ又は複数のヒトVH(ドナー)を1つ又は複数の治療用の非ヒトVH(アクセプタ)と混合し、DNAシャッフリングにかけることができる。これにより、安定性があり、且つヒト化されたアクセプタVHの突然変異型が発生する。本明細書に記載の方法によって、又はリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ及びファージディスプレイ等の他の発展的なタンパク質スクリーニングシステムによって、新たに発生したヒト化され、且つ安定性がある変異型を同定することができる。さらなる実施例では、アクセプタVHは治療用VHH(ラクダ科の重鎖抗体の可変ドメイン)であり得る。
【0033】
さらに、この技法はVH及びVL以外のポリペプチドの望ましい特性を選択するのにも有用である。上記で示されるように、ドナーポリペプチド及びアクセプタポリペプチドは、両方ともヒトであり得るか、又はドナーがヒトであり、アセプタが非ヒトであり得る。
【0034】
可溶性、単量体性、高発現性、又は安定性をVH及びVLに与える可能性のある手法は、アクセプタVH及びVL上の相補性決定領域(CDR)をグラフト化することによるものであり得る。CDRは、単一ドメイン抗体の可溶性及び安定性、及びそれに応じた本明細書中に記載の方法で単離したVH及びVL由来のCDR等のこれらの領域のグラフト化を伴うことが知られているので、可溶性及び/又は安定性をアクセプタVH及びVLに与えることができる。
ヒトの単量体のVH及びVL
異なる生殖細胞及び全配列を有する幾つかの単量体のヒトVHが、ファージプラークサイズに基づくこの選択方法によって、ナイーブヒトVHファージディスプレイライブラリから同定された(図1及び配列番号8〜配列番号22を参照のこと)。VHは、37℃でのトリプシン処理、37℃での数週間のインキュベート、又は4℃での数ヶ月の保存後でも機能性及び単量体を維持し、熱リフォールディング効率が高く、大腸菌において良好な収率で生成され、プロテインA結合活性を有する。
【0035】
さらに、幾つかの単量体のヒトVLが同定された(図6及び配列番号23〜配列番号54を参照のこと)。VLも大腸菌において良好な収率で生成され、プロテインL結合活性を有する。
【0036】
また、スカフォールドとして上記のVHを利用する合成ライブラリからのVHによって、このような特性が明らかになる。したがって、このようなライブラリによって、生理学的温度での良好な有効性、長い保存期間、及び費用効率が高い生産を有する治療用VH又は診断用VHが得られ得る。高温リフォールディング効率の特徴によって、VH結合因子が一時的に高温に曝された後に、その活性を維持するように要求される場合に対するこれらのライブラリの生物工学的適用をさらに拡大させる。VHは、その望ましい生物物理学
的特性により、体内適用に非常に適しているはずである。プロテインA結合特性は、診断試験におけるVH精製及びVH検出、免疫ブロット法及び免疫細胞化学を単純化し、ライブラリから非機能性のVHを取り除くことによって、ライブラリ性能を高めるために利用することができる。同様に、スカフォールドとしてVLを利用するライブラリによって、同様に望ましい特性を有する治療用のVLS又は診断用のVLSが得られる。VLがプロテインLと結合するので、このプロテインL結合特性を利用することにより、VL精製及びVL検出が単純化される。
【0037】
本発明のVH及びVLで構成されたディスプレイライブラリは、診断用薬剤及び検出用薬剤の有用な供給源でもあり得る。
【0038】
これまでに報告された有益な生物物理学的特性を有する完全なヒトVHは、単一のV生殖細胞系配列:DP−47に基づいていた(Jespers, L. et al., 2004b; Jespers, L. et
al., 2004a)。この研究における単量体のヒトVHが、DP−47を含む6個の異なる生殖細胞系配列から起因しているという見解によって、安定性のあるVHは生殖細胞系遺伝子の利用に関して限定されないことが実証される。事実、本発明者らがファミリーの単量体のVHを単離し、本明細書で記載するものとは異なる生殖細胞系起源は、プロテインA結合活性のあるVH3ファミリーのVHのサブセットに対する本発明者らの選択を制限させない可能性が非常に高い。VHにおいて複数の突然変異が発生すること、及びCDR3はsdAbの生物物理学的プロファイルの形成にも関与することが知られているという事実により、本発明のVHの観察された生物物理学な挙動に関与するアミノ酸突然変異(第1表)を特定することは不可能である。しかし、sdAb安定性及び可溶性に重要であることが知られている位置(例えば、HVHP423及びHVHP44BにおけるV37F)での突然変異又は同じ位置(例えば、9個のVHにおけるL5V/Q及びV5Q)で複数回起こる突然変異は、VHの生物物理学的特性を決定するのに役立つ。ライブラリ構築に関して、ライブラリ安定性を危うくすることなくCDRランダム化が行われるように、本発明のVHの単量体性は、CDR、特にCDR3には依存しないことが望ましい。これに関して、安定性に関してCDR3への依存性が小さいので、より小さいCDR3を有するVH(例えばHVHB82)が好ましいスカフォールドであり得る。
【0039】
全体の配列及びCDR3の長さに関する本発明のVH及びVLの多様性によって、より良好な性能のライブラリの構築が可能になる。合成VHライブラリが単一スカフォールドで構築されている。レパートリ形成へのこのような手法は、多様なスカフォールドを利用する天然のin vivoでの「手法」とはかなり異なっている。本明細書中に報告されている配列に基づいて、多様な組のVH及びVLの可用性を利用し、複数のVH及びVLのスカフォールドに基づくライブラリを作ることができる。このようなライブラリは、in vivoでのレパートリのより良好な模倣であり、したがってより最適な複雑性を有する。sdAbにおける3個のCDRの中で、CDR3は一般的に最も有意にレパートリの多様性に寄与し、この理由から一般的にCDR3の長さを同時に変えることによって、VH及びVLのスカフォールドにおけるCDR3ランダム化が達成される。この有意性がライブラリの複雑性を改良するが、親スカフォールドCDR3の長さを壊すことによって、ライブラリの安定性をも危うくする可能性がある。CDR3の長さに関して本明細書中で開示されるVH及びVLの不均一性によって、良好な複雑性、良好な安定性及び良好な生物物理学的な特徴の両方を有するライブラリの形成を可能にする。このようなライブラリは、好ましくはサブライブラリから成り、それぞれのサブライブラリは、親CDR3の長さを壊すことなく、単一のVH、及びVLスカフォールド上のCDR3のランダム化(及び必要に応じてCDR1及び/又はCDR2のランダム化)によって作られる。
【0040】
治療標的に特異的なVHH、VH及びVLをヒト化することに関して、最適なVH及びVLフレームワークを選択するといった点でも、本発明のVH及びVLの多用途性は有益
である。比較的容易に免疫性、非免疫性又は合成VHHライブラリから治療標的に対する親和性の高いラクダ科のVHHを得て、その後ヒト化(CDRグラフト化、再サーフェイシング(resurfacing)、脱免疫化)を行い、可能性のあるVHH免疫原性を除去すること
ができる。それにより、治療用VHの生成に対するヒトVHライブラリの手法の代替方法を提供する。後者の手法による親和性の高い治療用VHの生成は、最初の合成ヒトVHライブラリから選択される主要な結合因子(複数可)の面倒で、且つ時間のかかるさらなるin vitroの親和性突然変異を要求することが度々あり得る。
【0041】
比較的容易に免疫性、非免疫性又は合成VHライブラリから治療標的に対する非ヒトVHを得て、その後ヒト化(CDRグラフト化、再サーフェイシング、脱免疫化)を行い、非ヒトVH免疫原性を除去することができる。それにより、治療用VHの生成に対するヒトVHライブラリの手法の代替方法を提供する。
【0042】
比較的容易に免疫性、非免疫性又は合成VHHライブラリから治療標的に対する非ヒトVLを得て、その後ヒト化(CDRグラフト化、再サーフェイシング、脱免疫化)を行い、VHH免疫原性を除去することができる。それにより、治療用VLの生成に対するヒトVLライブラリの手法の代替方法を提供する。
【0043】
改良された生物物理学的特性を有するタンパク質の選択に対する多くの発展的な手法が記載されている(Forrer, P. et al., 1999; Waldo, G. S., 2003)、(Jespers, L. et al., 2004a; Jung, S. et al., 1999; Matsuura, T. et al., 2003)。一般的に確実にライブラリ集合から不安定又は安定性の低い変異型よりも安定性のある変異型を優先的に選択するために、安定性圧力が必要とされる。例えば関連の研究で、凝集耐性のあるVHを選択するために、VHファージディスプレイライブラリの熱処理が要求された(Jespers, L. et al., 2004a)。ファージディスプレイを含む発展的な選択の手法の例としては、従来の
ファージディスプレイ、選択的感染ファージ及びタンパク質分解の手法が挙げられる。最初の2個の手法において、安定性のあるタンパク質が不安定なものよりもそのリガンドに対してより良好な結合特性を有するという仮定に基づき、親和性の選択を使用して、ライブラリから安定性のある種を選択する。しかし、安定性選択工程がさらに包含されていても、これらの手法は主に安定性よりも親和性をより高める(Jung, S. et al., 1999)。結
合工程が要求されることによって、既知のリガンドを有するタンパク質に対するこれらの手法の適用性も制限される。第3のタンパク質分解の手法は、不安定なタンパク質がプロテアーゼに対して耐性がないのに対して、安定性のあるタンパク質は一般的に小さく、したがってプロテアーゼに対して耐性があるという事実に基づいている。ファージディスプレイフォーマットは、表示されたタンパク質のプロテアーゼ安定性が、ファージ感染性に変換されるように操作される。このように、変異型のファージディスプレイライブラリをプロテアーゼで処理する場合、安定性のあるタンパク質を表示するファージのみがこれらの感染性を維持し、その後、大腸菌宿主を感染させることにより選択することができる。この手法はリガンド結合と独立しているので、一般的に有用である。しかし、安定であり、且つ十分にフォールディングしたタンパク質であってもプロテアーゼ感受性部位(例えば、ループ及びリンカー)を有し、これによりタンパク質分解の手法における安定性のある種の選択が妨げられることもあり得る(Bai, Y. et al., 2004)。
【0044】
これに対して、本発明の発展的な手法において、優れた生物物理学的特性を有するタンパク質は肉眼で簡単に判別される。この手法は、リガンド結合工程、タンパク質分解工程又は脱安定化工程を要求せず、したがって報告された選択的手法において直面する可能性のある複雑性が回避される。結合工程に対する要求がないことは、この手法が一般的に有用であることも意味する。オプションとして、結合工程は選択されたタンパク質が機能的であることを確実にすることが含まれてもよい。しかし、本発明の手法の(プラーク視覚化のための)プレート培養に対する依存性によって、操作することができ、小さいライブ
ラリに対する適用を制限し得るプレートの数に関して、可能性のある運搬上の制限が導入される。それにもかかわらず、ライブラリを最初に取り扱いが可能な大きさに縮小する場合、現在の手法の有用性は大きなライブラリにまで拡大することが可能である。例えば、選択システムに大きい集合の不安定な種を取り除き(例えば、プロテインA表面上又は疎水性相互作用カラム上のライブラリ吸収)、これにより露出した疎水性表面を有する不完全にフォールディングしたタンパク質を除去する、取り除く工程を組み込むことにより、これを行うことができる(Matsuura, T. et al., 2003)。本明細書中において、この手法
を使用して、非常に不安定なVH及びVLのバックグラウンドにおいて良好な生物物理学的特性を有するVH及びVLを選択した。しかし、適度に良好な安定性を有するタンパク質で集合化された突然変異型のライブラリから「最良の」種を選択することはより困難であり得る。この場合、より短期間のインキュベート時間でのプラーク形成速度に基づくか、又はプラークサイズ及び頻度基準(frequency criteria)に基づいて、主要な変異型を同定することができる。
【0045】
本発明の選択手法は、プロテインL、プロテインA又は任意のリガンドを伴う結合工程の選択システムにおける任意選択の包含を有するscFv及びFab等の安定性があり、且つ十分フォールディングした抗体フラグメント、並びに安定性のある非抗体スカフォールド及びこれらの突然変異型の同定にまで拡大することができる。さらに、ファージプラークサイズとVH発現収率との間に見られる相関関係は、本発明の手法を利用しなければ変異型のファージディスプレイライブラリから不完全であるか、又は不十分な発現を有するタンパク質の高発現性がある変形を得るために本発明の手法を利用することができることを意味する。ブースト化タンパク質の発現がタンパク質の生産コストを有意に埋め合わせる治療用タンパク質又は高価で不完全な発現性のタンパク質試薬の場合、この適用は特に魅力的である。
五量体の結合分析
VL及びVHの両方が五量体化の影響を受けやすく、五量体化を使用して、親和性の低いVL又はVHの単量体を親和性の高いVL又はVHの五量体に迅速に変換することができる。このような五量体は貴重な診断用薬剤及び検出用薬剤である。このような用途において、VL又はVHの五量体のその標的との結合は、酵素(例えば、セイヨウワサビペルオキシダーゼ又はアルカリ性のホスファターゼ)等のレポーター分子又は五量体と結合する蛍光分子で検出することができる。代替的に、五量体の結合は、レポーター分子と結合する二次分子で検出することができる。二次分子は、五量体自体又は6Hisタグ若しくはc−Mycタグ等のタグに特異的であり得る。例えば、一般的な二次分子はイムノグロブリンである。
【0046】
プロテインAを有するVHとプロテインLを有するVLとの間の相互作用は、これらの標的抗原を有するVHとVLとの間の相互作用とは基本的に異なっている。VH又はVLの抗原結合は、抗体ドメインの結合部位を形成する3個の抗原結合ループを伴う。プロテインA結合活性を有するVH及びプロテインL結合活性を有するVLのプロテインA結合は、抗体結合部位とは全体的に異なる抗体ドメイン上に結合部位及び残基を伴う。このように、プロテインA結合活性を有するVHは、同時にプロテインA及びその標的抗原と結合することができ、プロテインL結合活性を有するVLは、同時にプロテインL及びその標的抗原と結合することができる。本発明のVHとVLはそれぞれ、プロテインA及びプロテインLに対する親和性を有するので、上記の検出用途及び診断用途のための二次分子として、プロテインA及びプロテインLを使用することができる。ヒトVH及びヒトVLの五量体は治療のためにも使用することができる。
五量体による病原体検出
VHのプロテインA結合活性及びVLのプロテインL結合活性を使用して、表面上にプロテインA及び/又はプロテインLを有する細菌を検出することができる。プロテインAは、病原菌である黄色ブドウ球菌の表面上に存在する。このように、本明細書に記載のも
の等のプロテインA結合活性を有するVHを使用して、黄色ブドウ球菌を検出することができる。同様に、細胞表面上にプロテインLを有する細菌、特にペプトストレプトコッカス属マグヌス(Peptostreptococcus magnus)等の病原菌の検出のために、プロテインL結
合活性を有するVL単量体及びVL五量体を使用することができる。
【0047】
プロテインLは、ヒトにおけるP.magnus(Ricci, S. et al., 2001)の発症の毒性因子に関連する。膣炎において、プロテインLは、架橋表面結合性のIgEによりその効果を発揮すると考えられる。プロテインL結合活性を有するVL単量体及び/又はVL五量体がプロテインLのIgE架橋作用を阻害する可能性があるので、これらは治療剤として潜在性がある。
【0048】
プロテインAは、ヒトにおける黄色ブドウ球菌(Fournier, B. et al., 2004)の発症の
毒性因子に関連する。その毒性は、抗体と結合することを含む宿主成分と相互作用する能力に起因している。プロテインA結合活性を有するVH単量体及び/又はVH五量体が宿主成分を有するプロテインAの相互作用を阻害する可能性があるので、これらは治療剤として潜在性がある。
【実施例】
【0049】
単量体のヒトVHの同定及び配列分析
完全なヒトVHライブラリ及びラマ化したヒトVHライブラリの構築の間に、単量体のラマ化したVHを表示するファージが、凝集傾向のある完全なヒトVHを表示するファージよりも細菌層上でより大きいプラークを形成したことを確認した。このように、ヒトVHレパートリから珍しく自然発生的な単量体のVHを同定する手段として、プラークサイズを使用した(図1)。この目的で、サイズが6×108であるヒトVHを示すファージライブラリを構築し、寒天プレート上でプラークとして増殖させた。タイタープレート上で、ライブラリは本質的に、幾つかの大きいプラークが散在する小さいプラークから成る。20個のクローンにおけるPCRによって、小さいプラークがVH表示ファージに対応し、一方で大きいプラークは野性型のファージ(すなわち、VH配列挿入を欠くファージ)を表すことが示された。VH表示ファージは、大きいプラーク形態では見出されなかった。このことは、ヒトのレパートリ及び大きいサイズのライブラリにおける単量体のVHの不足により予想外のことではなかった。単量体のVHの同定を容易にするために、VH3ファミリー由来のヒトVHのサブセットと結合するプロテインAに対してライブラリをパンニングすることにより、ライブラリを取り扱いが可能な大きさまで縮小し、プラークサイズ形態が大きくなるのを阻害する野性型のファージを取り除くことを決めた。
【0050】
数回のパンニングの後、ライブラリはファージを生成する大きいプラークに関して富化され、110個を超えるこのようなプラークのPCR及びシークエンシングにより、全てがVHオープンリーディングフレームを完成させたことを示した。分析のために選択された大きいプラークのサイズは図1で示す。シークエンシングによって、VH3ファミリーに属する15個の異なるVHが示され、DP−38、DP−47、V3−49、V3−53、YAC−5又は8−1Bの生殖細胞系Vセグメントを利用した(第1表、図2)。DP−38及びDP−47の生殖細胞系配列は、これまではプロテインA結合に関係していた。さらに、それらのプロテインA結合活性と一致して、全てのVHは、57位でThr残基を有していた(図2)。最も頻繁に利用された生殖細胞系Vセグメントは、VHの50%を越えて発生するDP−47であったが、最も高頻度なクローン(すなわち、HVHP428、相対頻度46%)はV3−49生殖細胞系Vセグメントを利用した。DP−47生殖細胞系配列を有するHVHP429は相対頻度が21%である2番目に豊富なVHであった(図2)。VH CDR3の長さは、HVHB82における4個のアミノ酸からHVHP430における16個のアミノ酸の範囲であり、HVHP430はCDR3において一対のCys残基を有していた。親の生殖細胞系Vセグメント(残基1〜94)及び
FR4(残基103〜113)配列に対するアミノ酸突然変異は全てのVHで見られ、HVHP44(L5V及びQ105R)及びHVHB82(E1Q及びL5Q)に対する2個の突然変異からHVHP426に対する16個の突然変異までの範囲であった(第1表)。突然変異はVセグメントに集中し、105位及び108位の2個の突然変異のみが15個のFR4全てで検出された。HVHP44及びHVHB82は、その両方がGlnの代わりに105位で正に荷電したアミノ酸を有するという点で他のVHとは異なっていた(第1表、図2)。しかし、HVHP44において正に荷電したアミノ酸が突然変異によって得られたが、HVHB82における正に荷電したアミノ酸は生殖細胞系でコードされていた。HVHP423及びHVHP44Bを除いて、残りのVHは鍵となる可溶性位置で生殖細胞系残基を有していた:37V/44G/45L/47W又は37F/44G/45L/47W(HVHP428)、HVHP423及びHVHP44BはV37F突然変異を有していた。VH可溶性に重要であることが示されるか、又は仮定される他の位置での突然変異には、7個のE6Q、3個のS35T/H、1個のR83G及び1個のK83R、1個のA84P及び1個のT84A及び1個のM108Lが包含されていた。11個のE1Q、8個のL5V/Q及び1個のV5Q突然変異を包含した高頻度の突然変異も1位及び5位で観察された。
ヒトVHの生物物理学的特徴
HVHP44B(HVHP423と本質的に同じであった)を除いて、全てのVHはc−Myc−His5タグとの融合における培養液量1Lの大腸菌株TG1で発現し、固定化金属親和性クロマトグラフィ(IMAC)によって、ペリプラズム抽出物から均一に精製した。発現収率は、振盪フラスコ内で細菌培養液1L当たりで精製タンパク質1.8〜62.1mgの範囲であり、VHのほとんどは数mgの収率があった(第2表)。HVHP423及びHVHP430の例において、「明らかに」同じ発現条件下での別の試験によって、62.1及び23.7mgに対してそれぞれ、2.4及び6.4mgの収率が得られた。このことは、本明細書中に記載の多くのVHに対して、大した努力をせずに最適な発現条件を達成でき、それにより第2表で報告された値より有意に高い発現収率がもたらされることを意味している。予想通り、全てのVHは表面プラズモン共鳴(SPR)分析(KDは0.2〜3μMである)においてプロテインAと結合していて、範囲及び規模はプロテインA結合活性を有するラマVHH変異型についてこれまでに報告されていた範囲及び規模に匹敵していた。VHはFab基準面と結合しなかった。
【0051】
ゲルろ過クロマトグラフィ及びNMRによるオリゴマー化状態に関して、ヒトVHの凝集傾向を評価した(第2表)。全てのVHをSuperdex75ゲルろ過クロマトグラフィにかけた。ラマVHH(すなわち、H11C7)と同様に、全てのVHにおいて、単量体で期待される溶離量で対称な単一のピークが与えられ、凝集体は実質的に全く存在しなかった(図3AのHVHP428における例を参照のこと)。これに対して、一般的なヒトVH(すなわち、BT32/A6)はかなりの量の凝集体を形成した。3個のVHに関しては、VH二量体で期待される移動度を有する微小ピークも観察された。微小ピークのSPR分析によって、二量体のVHと一致して、単量体VHに対する値よりも有意に緩やかな解離速度値(off-rate value)が与えられた。二量体のピークは、ラマVHH(H11C7)の場合にも観察された。高濃度のVHのフォールディング状態及びオリゴマー化状態をさらにNMR分析で研究した。表IIに示すように、全てのVHタンパク質の研究によって、比較的可溶性であると考えられ、十分にフォールディングされた三次元構造が推測された。VHフラグメントの一次元NMRスペクトル(図3B)によって、VHドメインの構造フォールディング特徴が示された。HVHP414フラグメント及びHVHP414の2個のイソ型(c−Myc配列を有する及び有さないVH14及びVH14−cMyc−)に対するPFG−NMR拡散実験の使用によっても、タンパク質の凝集状態を評価した。VH14は、c−Myc N132E突然変異を有し、且つさらにN末端にメチオニン残基を有するHVHP414の改変バージョンである。簡単に述べると、PFG−NMRデータ(示されず)によって、全てのタンパク質サンプルが、NMR実験に使用
した比較的高いタンパク質濃度であっても予想される単量体の分子量を有することが示された。
【0052】
一貫して37℃でのトリプシンに対する耐性に関して、VHの安定性をさらに研究し、その後37℃で長期的にインキュベートを行った。トリプシンは、C末端のArg又はLys残基でポリペプチドのアミド骨格を切断する。ヒトVHにおいて9〜13個のArg及びLys残基が存在する(図2)。トリプシンによる消化の影響を受けやすいC末端のc−MycタグにおいてさらなるLys残基も存在する。図4aは、トリプシン消化の間のHVHP414のSDS−PAGE分析である。1時間以内に、元のバンドは、c−Myc−His5タグを有しないVHで期待される移動度があった単一の生成物に完全に変換された。トリプシンによる1時間のインキュベート後に12個の他のVHにおいて、同じ結果が得られた。トリプシン処理したVHのランダムに選択されたサンプル(すなわち、HVHP414、HVHP419、HVHP420、HVHP423、HVHP429、HVHP430及びHVHM81)における質量分析によって、それぞれの場合に、消化した生成物の分子質量がC末端残基としてのc−Myc Lysを有するVHに対応していたことが確認された。HVHM41において、消化時の残りよりも有意に短いフラグメントが与えられ、この場合質量分析実験によって、CDR3におけるArg99に対する切断部位がマッピングされた(データ図示せず)。
【0053】
0.32mg/ml(HVHP428)〜3.2mg/ml(HVHP420)の濃度に及ぶ11個のVHを37℃で17日間インキュベートした。その後、オリゴマー化状態及びプロテインA結合に関して、これらの安定性を決定した。ゲルろ過クロマトグラフィによって示されるように、37℃でのVHの処理は凝集体形成を全く誘導しなかった:全てのVHは、未処理のVHのものと実質的に同一であったクロマトグラムプロファイルを与え、本質的に単量体として存在していた(HVHP420についての例、図4cを参照のこと)。VHが37℃での処理後の自然のフォールディングを維持したことを確実するために、2個のVH(すなわち、HVHP414(1.2mg/ml)及びHVHP420(3.2mg/ml))をランダムに選択し、SPRによってプロテインAと結合するこれらのKDを求め(HVHP420に関して示されるデータ、図4cの挿入図)、未処理のVHに対して得られたKDと比較した(第2表)。熱処理したVHに関して算出したKDは1.4μMであり、HVHP414及びHVHP420に関して算出したKDはそれぞれ1.0μMであった。これらの値は未処理のVHの対応する値と本質的に同一であり(第2表)、このことはVHの37℃での処理がこれらの天然フォールディング(native fold)に影響を与えなかったことを示している。VHが37℃のインキュベート期間で
余り小さくなく、且つ非天然であり、ゲルろ過実験及びSPR実験の間室温まで戻した時にこれらの天然のフォールディングを再開する可能性は、十分にフォールディングした天然のタンパク質に一般的に関連する特性であるVHが37℃でトリプシンに対して耐性があった(上記を参照のこと)という事実を鑑みて疑わしい。
【0054】
天然のVH(KDn)及び熱処理して、リフォールディングしたVH(KDref)のプロテインAとの結合のKDを比較することによって、ヒトVHのリフォールディング効率(RE)を研究した(Tanha, J. et al., 2002)。VH分画が熱処理によって不活性化される場合、このパラメータはフォールディングした、すなわち活性のある抗体フラグメントの濃度に基づいているので、測定KDは高くなる。このように、KDnとKDrefとの比によって、VHREの測定値が与えられる。図5は、幾つかの選択されたVH濃度での天然状態(太線)及びリフォールディング状態(細線)における固定化されたプロテインAとのHVHP423の結合に対するセンサーグラムを比較している。示され得るように、リフォールディングしたVHのプロテインAとの結合は全ての例でより小さく、これにより非フォールディングは完全に可逆ではないことが示される。14個のVHそれぞれにおいて、天然状態及びリフォールディング状態におけるプロテインAの結合を幾つかの
濃度で測定し、KD及びその後のREを求めた(第2表、KDref値は示されていない)。2個の抗イディオタイプのラマVHH(H11F9及びH11B2)のKD及びREも求め、これらは参照として使用された。4個のVHは、H11F9及びH11B2のそれぞれの、95%及び100%のREと同程度の92%〜95%の範囲のREであった。別の5個のVHは、84%〜88%の範囲のREであり、3個は70%を超えていた。2個のVHだけが有意に低いREであった:HVHP413(52%)及びHVHP421(14%)。これまでに調べられた幾つかの公開されたVHHは約50%のREであった(van der Linden, R. H. et al., 1999)。
ヒトVHファージディスプレイライブラリ構築及びパンニング
ランダムヘキサヌクレオチドプライマー及びFirst Strand cDNA(商標)キット(GE Healthcare, Baie d'Urfe, QC, Canada)を使用して、ヒト脾臓mRNAからcDNAを合成した。鋳型としてcDNAを使用して、VHフレームワーク領域1(FR1)に特異的なプライマー及びイムノグロブリンM特異的なプライマー(de Haard, H. J. et al., 1999)を使用した9個の別々の反応におけるポリメラーゼ連鎖反応(PCR)によって、CH配列をフランキングしたVH遺伝子を増幅した。生成物をゲル精製し、2回目のPCRにおける鋳型として使用することにより、クローニング目的のためにフランキングしたApal I及びNot I制限部位も導入したFR1特異的なプライマー及びFR4特異的なプライマー(de Haard, H. J. et al., 1999)を使用して、VH遺伝子を構築した。得られたVHレパートリDNAをfd−tetGIIIDファージベクターにクローニングし、VHファージディスプレイライブラリを構築した(Tanha, J. et al., 2001)。プロテインAに対するパンニング(Amersham Biosciences Inc.)を記載のように行
った(Tanha, J. et al., 2001)。DNAPLOTソフトウェアバージョン2.0.1及びV BASEバージョン1.0(http://vbase.dnaplot.de/cgi-bin/vbase/vsearch.pl)を使用して、選択されたVHの生殖細胞系配列帰属を行った。記載のようにH11 scFvに対してパンニングすることによって、ラマVHHファージディスプレイライブラリからラマVHH H11C7、H11F9及びH11B2を単離した(Tanha, J. et al., 2002)。
VHの発現及び精製
標準的なクローニング技法により、VHをpSJF2発現ベクターにクローニングした(Sambrook, J. Fritsch E. F. and Maniatis T, 1989)。sdAbのペリプラズム発現及
びその後の固定化金属親和性クロマトグラフィ(IMAC)による精製を記載のように行った(Muruganandam, A. et al., 2002)。それぞれのタンパク質において算出したモル吸
収係数を使用したA280測定によって、タンパク質濃度を測定した(Pace, C. N. et al., 1995)。記載のように、Superdex75カラム(GE Healthcare)において、精製
したVHのゲルろ過クロマトグラフィを行った(Deng, S. J. et al., 1995)。
結合及びリフォールディング効率の実験
VH/VHHの平衡解離定数(KD)及びリフォールディング効率(RE)は、BIACORE3000バイオセンサーシステム(Biacore Inc., Piscataway, NJ)で回収した表面プラズモン共鳴(SPR)データから得られた。プロテインAとのVHの結合を測定するために、プロテインAの2000個の共鳴単位(RU)又は基準となる抗原結合フラグメント(Fab)を研究等級のCM5センサーチップ(Biacore Inc.)上で固定化した。製造業者によって提供されるアミンカップリングキットを使用して、10mMの酢酸ナトリウム緩衝液(pH4.5)中の25μg/ml(プロテインA)又は50μg/ml(Fab)の濃度で固定化を行った。抗イディオタイプのラマVHHのH11scFvとの結合を測定するために、上記のように、50μg/mlのH11 scFv 4100RU又は10μg/mlのSe155−4 IgG基準 3000RUを固定化した。全ての例において、流量40μl/分で、150mMのNaCl、3mMのEDTA及び0.005%のP20を含む10mMのHEPES(pH7.4)中、25℃で分析を行い、表面を洗浄緩衝液で洗浄することにより再生させた。リフォールディングタンパク質の結合活性を測定するために、10μg/mlの濃度、85℃で20分間インキュベートするこ
とによって、VH又はVHHを変性させた。それから、リフォールディングするためにタンパク質サンプルを30分間室温まで冷却し、その後室温で5分間14000rpmでマイクロフュージで遠心分離して、任意のタンパク質沈殿物を除去した。上清を回収し、上記のようにSPRによって結合活性について分析した。BIAevaluation4.1ソフトウェア(Biacore Inc.)を同時に使用して、フォールディングしたタンパク質及びリフォールディングタンパク質両方に関するデータを1:1の相互作用モデルに適合させ、その後KDを求めた。REは、
【0055】
【化1】
【0056】
(式中、KDnは天然タンパク質のKDであり、KDrefはリフォールディングタンパク質のKDである)から求めた。
トリプシン消化実験
1mMのHCl中の新たに調製した0.1μg/μlのシークエンシング等級のトリプシン(Hoffmann-La Roche Ltd., Mississauga, ON, Canada)3μlを100mMのTri
s−HCl緩衝液(pH7.8)中のVH60μgに加えた。37℃で1時間、全量60μlで消化反応を行い、0.1μg/μlのトリプシン阻害剤(Sigma, Oakville, ON, Canada)5μlを加えることにより停止させた。消化の完了後、5μlを取り除き、SDS
−PAGEで分析して、ZipTipC4(Millipore, Nepean, ON, Canada)を使用して
残りを脱塩し、50:50のメタノール:水の1%酢酸で溶離して、MALDI質量分析によってVHの質量を求めた。
37℃でのタンパク質安定性研究
37℃で17日間PBS緩衝液において0.32〜3.2mg/mlの濃度での単一ドメイン抗体(sdAb)をインキュベートした。インキュベートの後、視覚的に凝集体の形成が全く見られなくても、5分間最大速度でミクロフュージにおいてタンパク質サンプルを沈降させた。それから、サンプルをSuperdex75サイズ排除カラム(GE Healthcare)に適用させ、プロテインAに対するSPR分析における単量体のピークを回収し
た。プロテインA500RU又は基準Fabを固定化したこと、及び50μg/mlの濃度で固定化を行ったことを除いて、上記のようにSPR分析を行った。
NMR実験
NMR分析におけるVHサンプルを10mMのリン酸ナトリウム、150mMのNaCl、0.5mMのEDTA及び0.02%のNaN3(pH7.0)中に溶解した。タンパク質濃度は40μM〜1.0mMであった。全てのNMR実験は、Bruker Avance−800NMRスペクトロメータ又はBruker Avance−500NMRスペクトロメータにおいて298Kで行った。一次元(1D)1H NMRスペクトルを16384個のデータ点で記録し、スペクトル幅はそれぞれ、500MHzで8992.81Hz及び800MHzで17605.63Hzであった。2048×400のデータ点の二次元1H−1H NOESYスペクトルがスペクトル幅11990.04Hz及び混合時間120msのBruker Avance−800NMRスペクトロメータにおいて得られた。全てのNMR実験において、3−9−19パルス列によって行われたWATERGATE法(Piotto, M. et al., 1992; Sklenar, V. et al., 1993)を使用して
水抑制を達成した。NMRデータを処理し、Bruker XWINNMRソフトウェアパッケージを使用して分析した。3軸勾配がある三重共鳴プローブを備えるBruker
Avance−500 NMRスペクトルメータで水抑制LED配列(Altieri, A. S. et al., 1995)によって、全てのPFG−NMR拡散測定を行った。一次元プロトンスペ
クトルを処理し、Bruker Xwinnmrソフトウェアパッケージを使用して分析
した。全ての所与のPFG強度で均一に全てのNMRシグナルが弱まった場合に、メチル及びメチレンプロトン領域(2.3ppm〜−0.3ppm)におけるNMRスペクトルを統合することにより、NMRシグナル強度を得た。
ヒトVLファージディスプレイライブラリ構築及びパンニング
ヒトVHに関して上記のように、cDNAをヒト脾臓mRNAから合成した。cDNAをPCRの鋳型として使用し、6個のVκバックプライマー、11個のVλバックプライマー(de Haard, H. J. et al., 1999)、4個のVκフォワードプライマー及び2個のVλフォワードプライマー(Sblattero, D. et al., 1998)を使用して、反応体積50μlでVL遺伝子を増幅させた。バックプライマー及びフォワードプライマーを修飾して、その後のクローニング目的のためにそれぞれ、Apa LI制限部位及びNot I制限部位をフランキングさせた。これらの縮重の程度に反映された比で共にフォワードプライマーがプールされた。プールされたVλフォワードプライマー及び11個の個々のVλバックプライマーを使用して、11個の別々の反応でVλ遺伝子をPCRした。同様に、プールされたVκフォワードプライマー及び6個の個々のVλバックプライマーを使用して、6個の別々の反応でVλ遺伝子を増幅させた。PCR生成物をプールし、ゲル精製して、Apa LI制限エンドヌクレアーゼ及びNot I制限エンドヌクレアーゼで消化した。ヒトVHに関して上記のように、ライブラリを構築した。個々のライブラリコロニーにおいてプラークPCRを行い、増幅したVL遺伝子を記載のようにシークエンシングした(Tanha, J. et al., 2003)。ヒトVHライブラリに関して上記のように、プロテインL(Biolynx Inc., Brockville, ON, Canada)に対するパンニング及び選択されたVLの生殖細胞系の配列帰属を行った。
VLの発現及び精製
「VHの発現及び精製」においてVHに関して上記のように、VLの発現、精製、濃度決定及びゲルろ過クロマトグラフィを行った。
VL及びVH五量体の発現及び精製
標準的なPCRにおいて特異的なプライマーを使用して、HVHP328 VH及びHVLP335 VL遺伝子を増幅させた。標準的なクローニング技法を使用して、発現ベクターにおいてVT1B五量体化ドメイン遺伝子と融合して、HVHP328及びHVLP335遺伝子をクローニングして、HVHP328PVT2及びHVLP335PTV2五量体を得た(Zhang, J. et al. 2004)。五量体を記載のように発現及び精製した(Zhang, J. et al., 2004)。上記のようにタンパク質濃度を決定した。
VLの表面プラズモン共鳴
BIACORE3000バイオセンサーシステム(Biacore, Inc., Piscataway, NJ)を
使用したSPRによって、VLのプロテインLとの相互作用に対する結合反応速度を測定した。プロテインL680RU又は基準Fab 870RUを研究等級のCM5センサーチップ(Biacore)上で固定化させた。製造業者によって供給されたアミンカップリングキ
ットを使用して、10mMの酢酸緩衝液(pH4.5)において50μg/mlのタンパク質濃度で固定化を行った。50μl/分又は100μl/分の流量で150mMのNaCl、3mMのEDTA及び0.005%のP20を含む10mMのHEPES緩衝液(pH7.4)中、25℃で全ての測定を行った。洗浄緩衝液で洗浄することにより表面を再生した。BlAevaluation4.1ソフトウェア(Biacore, Inc.)を使用して
、データを評価した。
五量体VL及びVHの表面プラズモン共鳴
SPRによって、HVHP328PVT2のプロテインAとの相互作用及びHVLP335PTV2のプロテインLとの相互作用に対する結合反応速度を決定した。520RUのプロテインA又は基準Fabを上記のように固定化した。VL五量体に関して、上記のように調製した同様の表面を使用した。上記のように測定を行ったが、流量は20μl/分であった。3秒間、50mMのHClで洗浄することによって、表面を再生した。単量体に関して記載のようにデータを評価した。
細胞の微小凝集
BHIプレート由来の黄色ブドウ球菌の単一コロニーを使用して、BHI培地15mLを接種した。細菌を37℃、200rpmで一晩成長させた。翌朝、培養液を回転バケット、Sorvall RT6000B冷凍遠心器において4000rpmで10分間遠心分離し(spun down)、上清を除去して、細胞ペレットをPBS緩衝液で再懸濁させた。
細胞を再び遠心分離し、上清を除去して、細胞ペレットを再びPBS緩衝液で再懸濁させた。細胞をA600が1.0になるまで希釈し、段階希釈した細胞を37℃のBHIプレート上に広げて、一晩成長させた。翌朝、細胞力価を測定した。A600 1.0が1.5×109細胞/mlに対応する。同一の工程を行い、成長培地が2xYTであったことを除いて、その後の微小凝集アッセイのために大腸菌株TG1細胞を調製した。生菌数は同様であった、A600 1.0=2.1×109細胞/ml。
【0057】
微小凝集アッセイを行うために、マイクロタイタープレートにおけるウェル1〜ウェル11でPBSにおいてHVHP328PVT2の2倍希釈を行った。ウェル12(ブランク)はPBSのみを含んでいた。それぞれのウェルの全量は50μlであった。その後、PBS50μlにおける1×108個の黄色ブドウ球菌細胞を全てのウェルに加えて、プレートを4℃で一晩インキュベートした。結果を永続的に記録するために、翌朝にプレートの写真を撮った。五量体の対照実験のために、HVHP328PVT2をVL五量体、HVLP335PTV2に置き換えた。細胞の対照実験において、同じ2個の組の実験を大腸菌TG1細胞で繰り返した。
単量体のヒトVLの同定及び配列分析
ヒトVHファージディスプレイライブラリから可溶性のVHを単離するために用いられた本質的に同じ選択方法を、可溶性で単量体のVLを単離するためにヒトVLライブラリに適用した。大きさが3×106であるヒトVLライブラリを構築した。ライブラリタイタープレートから24個のプラークを選別し、これらのVL遺伝子をPCR及びシークエンシングした。配列は、生殖細胞系起源に関して異なるが、VLの75%はVλ起源であった(データ図示せず)。プロテインLに対する3回のパンニングによって、大きいプラークにおける富化がもたらされた。39個の大きいプラークをシークエンシングして、32個の特殊な配列を同定した(図6)。HVLP325、HVLP335及びHVLP351はそれぞれ、3、4及び2の頻度で起こった。λクラス(サブグループVλ1、生殖細胞系1b)であるHVLP389を除いて、残りの31個のVLはVκクラスに属していた。31個のκVLの中で、24個はVκIIIサブグループに含まれ、7個はVκ1サブグループに含まれていた。24個のVκIII配列の16個は、残りの利用されるA27、L2及びL6生殖細胞系配列を有するL6生殖細胞系配列を利用する。Vκ1サブグループのVLはO2/O12又はA30生殖細胞系配列に由来する。顕著な突然変異は96位で起こった。この位置での生殖細胞系アミノ酸は芳香族及び疎水性アミノ酸、κVLに対するTrp、Phe、Tyr、Leu又はIle、及びλVLに対するTyr、Val又はAlaである。しかし、κVLの選択プールにおける31個の中の5個だけが96位でこれらの生殖細胞系アミノ酸を有している:HVLP325、HVLP349、HVLP388、HVLP3109及びHVLP393。96位での21個のアミノ酸を帯電させ、この中の20個が正に帯電する:Arg、Lys又はHis。2個のアミノ酸はPro、1個はGln、1個はSer、及び1個はThrである。単量体化のためにゲルろ過クロマトグラフィで分析された7個のκVLの中で96位がArg又はLysである6個が単量体であった一方で、96位が生殖細胞系アミノ酸LeuであるHVLP325が凝集体を形成した(以下を参照のこと)。同様に、λクラスであり、且つSerに対する生殖細胞系突然変異を有していたHVLP389も単量体であった(以下を参照のこと)。これらのデータは単量体化等のVLの生物物理学的特性が改良された96位での生殖細胞系アミノ酸からの偏差に相関がある(32個の中の27個)。
【0058】
κクラスの18個のVLは、λVLでのみ見出されるアミノ酸Thr、Val及びLeuに置き換えられたこれらの最後の3残基(105〜107)を有していた。これらの置
換はκVLの生物物理学的特性を改良する役割を果たしている可能性があり、これにより105〜107位の元のκ残基を有する親クローンを超えた上記のVLの選択がもたらされる。
ヒトVLの特徴付け
異なるV生殖細胞系起源を有する選択VLの中の8個は、培養液1L中の大腸菌で発現し、精製された:HVLP324、HVLP325、HVLP335、HVLP342、HVLP351、HVLP364、HVLP389及びHVLP3103(第6表)。全てがHVLP324に対する6.2mgからHVLP335及びHVLP364に対する約75mgまでに及ぶ良好な収率で発現した。
【0059】
ゲルろ過クロマトグラフィによるこれらのオリゴマー化状態に関するヒトVLの凝集傾向を評価した。VLを0.6mg/mlの濃度でSuperdex75ゲルろ過クロマトグラフィにかけた。HVLP325を除いた全てが凝集体を本質的に含んでおらず、平均見掛けの分子質量が12.7kDa(範囲、6.2〜19.2kDa)である対称の単一ピークを得た(図7A及び第3表)。これは単量体のVLに関する期待分子質量(13.4〜13.8kDa)と一致している。単一ドメインの抗体に関する見掛けの分子質量の変動はこれまでに報告されている(Jespers, L. et al., 2004a; Stevens, F. J. et al.,
1980)。HVLP325に関して、凝集体が全タンパク質(凝集体+単量体)の11%を構成していた。HVLP351、HVLP342、HVLP335及びHVLP3103は、これらの利用可能な最も高い濃度、すなわちそれぞれ、0.89mg/ml、1.0mg/ml、4.9mg/ml及び5.9mg/mlで試験した場合、依然として単量体であった(図7B)。
【0060】
BIACORE分析前に、VLをSuperdex−75クロマトグラフィにかけて、物質の凝集に何ら確証がなかったとしても精製した単量体のピークを回収した。SPR分析において、全ての選択されたVLは、プロテインLと結合した(図8)。VLがプロテインLに対するパンニングによって単離されたので、このことは予想外のことではなかった。全てに関して、プロテインLとの結合のKDは、0.6〜3μMであった(第3表)。HVLP324及びHVLP342はKDがそれぞれさらに10nM及び40nM小さかった。VκIサブグループのVLのプロテインLとの低い親和性及び高い親和性の結合が、これまでに報告されている(参考文献)。HVLP324及びHVLP342の両方がVκIサブグループに属している(第3表)。予想通りに、速度論データ及び平衡データは、実際に単量体である単量体ピークと一致していた。
五量体の結合分析
表面プラズモン共鳴によって、HVHP328PVT2五量体のプロテインAとの結合及びHVLP335PTV2五量体のプロテインLとの結合を測定した(図9)。会合速度をkobs対濃度のプロットから独立して算出した。五量体の集合の間の多価結合における不均一性によって、1を超えた解離速度(kd)を算出することができる。したがって、1を超えた平衡解離定数(KD)を得ることができる。HVHP328PTV2及びHVLP335PTV2はそれぞれ、2nM及び200pMの最小KDを有していた(第4表)。kdがより遅い場合、HVHP328PTV2及びHVLP335PTV2はそれぞれ、900pM及び90pMまで低いKDを有していた。
VL及びVHによる病原菌検出
VHのプロテインA結合活性及びVLのプロテインL結合活性を使用して、これらの表面上にプロテインA及び/又はプロテインLを有する細菌を検出することができる。本明細書中のVH及びVL等のVH及びVLが可溶性であり、且つ単量体である(凝集傾向が失われている)場合、このことは可能である。ラクダ科の重鎖抗体又はナースシャーク及びテンジクザメのIgNAR等の軽鎖が欠失した抗体由来の可変ドメインは、自然発生的に可溶性且つ単量体である。このことから、プロテインA結合活性及びプロテインL結合活性を有するものを使用して、これらの表面上にプロテインA及び/又はプロテインLを
有する細菌を検出することもできる。プロテインAは、病原菌である黄色ブドウ球菌の表面上に存在する。このように、本明細書中に記載のもの等のプロテインA結合活性のあるVHを使用して、黄色ブドウ球菌を検出することができる。本発明者らは微小凝集アッセイを行い、HVHP328PVT2 VH五量体の黄色ブドウ球菌と結合する能力を検出した。マイクロタイターウェル(ウェル1〜ウェル11)において2倍希釈したHVHP328PVT2で一定数の細菌細胞をインキュベートした(図10)。ウェル12は五量体の代わりに緩衝液を含んでいた。VHが細菌細胞と結合する場合、したがってその多量体の性質により、五量体は細胞を架橋し、その結果、細胞凝集をもたらすことが可能であるはずである。凝集細胞がマイクロタイターウェルにおける拡散細胞として現れる(図10)。何の結合もなければ、凝集は起こるはずがなく、したがって凝集体は存在せず、細胞はウェルの底部に点として現れる。図10で示されるように、細胞の凝集体が存在するので、五量体は黄色ブドウ球菌と結合する。凝集体は最大ウェル7まで観察される。ウェル7より先は、五量体の濃度が結合するには低すぎるので、凝集体は存在しない。対照のVL五量体は、全く凝集を示さず、これによりVH五量体の黄色ブドウ球菌に対する特異性が実証される(図10)。予想通りVH五量体が大腸菌(TG1株)又はサルモネラ細胞を凝集させないので、結合は細胞特異的でもある(データは示さず)。同様に、これらの細胞表面上にプロテインLを有する細菌、特にペプトストレプトコッカス属マグヌス等の病原菌の検出にプロテインL結合活性を有するVL単量体及びVL五量体を使用することができる。
【0061】
上記で記載の実施例は、本発明の実際の範囲を限定する役割を果たすものでは決してなく、むしろ例示目的のために表されていることが理解される。
参考文献リスト
【0062】
【表1】
【0063】
【表2】
【0064】
【表3】
【0065】
【表4】
【0066】
【表5】
【0067】
【表6】
【図面の簡単な説明】
【0068】
【図1】選択された実施例の結果(可溶性VH(HVHP428)を示すファージと不溶性VH(BT32/A6)を示すファージとの間のプラークサイズの対比)の図的記述を示す。この写真はプラークの可視化を高めるために拡大した細菌層の寒天プレートの一部を示す。プレートは、それぞれ同じ数の2個のプラーク型を含んでいたが、この写真は基本的に大きいHVHP428プラークを含んでいる。BT32/A6プラークの大多数は小さすぎて写真において鮮明ではっきりした画像を得ることができなかった。矢印で示されたプラークは、この画像で可視化させるのに十分大きかった小さい割合のBT32/A6ファージを示している。アスタリスクは、HVHP428ファージの代表的なプラークサイズを示す。プラークの同一性は、DNAシークエンシングによって決定された。
【図2】プロテインAに対する親和性及びプラークサイズに基づき選択されたヒトVHのアミノ酸配列。入力配列(sequence entries)の点は、HVHP2M10又はHVHP44とアミノ酸が同一であることを示す。ダッシュ記号は配列アラインメントのために含まれている。鍵となる可溶性部分の残基及びプロテインA結合特性を有するVH/VHHに関連する57T残基は太字である。カバットナンバリングシステムを使用する。「頻度」の値の総計は114である。CDRは相補性決定領域であり、FRはフレームワーク領域であり、gln seqは生殖細胞系の配列である。
【図3A】ヒトVHの凝集傾向。ゲルろ過クロマトグラムによって、この研究で単離したヒトVH(HVHP428)のオリゴマー化状態をラマVHH(H11C7)及び典型的なヒトVH(BT32/A6)のものと比較する。それぞれのクロマトグラムの最後に溶離したピークは単量体のVHに対応する。二量体のH11C7のピークは矢印によって示される。
【図3B】(i)800MHzでのHVHP414、(ii)500MHzのHVHP423、及び800MHzのHVHP428の一次元の1H NMRスペクトル。左側のパネルのスペクトルは、低強度のシグナルのより良好な視覚化を可能にするように2倍に拡大されている。
【図4A】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。21kDaのマーカーに対して未処理のHVHP414VHと15、30及び60分でトリプシン処理したHVHP414VHとの移動度を比較するSDS−PAGE。HVHP414−cMycは、c−Mycを欠失したHVHP414VHを示す。
【図4B】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。未処理のHVHP414VH及びトリプシン処理(60分)HVHP414VHの質量分析法によって得られた分子質量プロファイル。処理済VHの質量分析プロファイルを未処理のVHのものの上に重ねて、より良好な視覚的比較を行う。未処理のVHの実験の分子質量は、14967.6Daであり、これは期待分子質量14967.7Daとほぼ同一である。トリプシン処理したVHで観察された分子質量(13368.5Da)は、c−MycタグにおけるK(Lys)での切断によるC末端での13アミノ酸の欠失を示し、期待分子質量13368.0Daを与える。トリプシン切断部位は、HVHP414のアミノ酸配列上の垂直な矢印によって示される。
【図4C】37℃でのトリプシンに対する耐性及び37℃での長期のインキュベート後の完全性に関するヒトVHの安定性。37℃で処理したHVHP420VHのオリゴマー化状態(上方のプロファイル)と未処理のVH(下方のプロファイル)のものとの比較をするゲルろ過クロマトグラム。重ねるとクロマトグラムが区別できないので、このクロマトグラムを垂直に移動させた。それぞれのクロマトグラムの主なピーク及び微小ピークは、それぞれ単量体のVH及び二量体のVHに対応する。二量体のVHは、全タンパク質の3%を構成する。挿入図は、様々な濃度でのプロテインAとの37℃で処理したHVHP420の結合に関するセンサーグラムオーバーレイを示す。温度安定性試験に使用されるVHは、既に4℃で数ヶ月間調製したストック由来であった。
【図5】天然HVHP423(太線)及びリフォールディングしたHVHP423(細線)の75、100、150及び200nMの濃度で固定化されたプロテインAとの結合を示すセンサーグラムオーバーレイ。KDn及びKDrefがそれぞれのセンサーグラムから算出され、以下で記載のようにREを求めるのに使用した。
【図6】プロテインLに対する親和性及びプラークサイズに基づき選択されたヒトVLのアミノ酸配列。入力配列における点は、HVLP333とアミノ酸が同一であることを示す。ダッシュ記号は配列アラインメントのために含まれる。配列ナンバリング及びCDR指定に関するV BASE(http://vbase.mrc-cpe.cam.ac.uk/index.php?module=pagemaster&PAGE_user_op=view_page&PAGE_id=7&MMN_position=5:5)を参照されたい。L6、A27、L2、L16、O2/O12、A30及び1bは、V生殖細胞系設計である。J生殖細胞系設計は括弧内である。NFは見当たらない。
【図7A】ヒトVLドメインのサイズ排除クロマトグラム。VLを0.6mg/mlの濃度で添加した。「#」及び「*」は、それぞれ凝集体及び単量体のピークを示す。凝集体は排除体積で溶出する。
【図7B】ヒトVLドメインのサイズ排除クロマトグラム。VLを利用可能な最も高い濃度(1.0mg/mlのHVLP342、5.9mg/mlのHVLP3103、4.9mg/mlのHVLP335、0.89mg/mlのHVLP351)で適用した。「#」及び「*」は、それぞれ凝集体及び単量体のピークを示す。凝集体は排除体積で溶出する。HVLP342パネルにおける矢印で示されたピークは、前の測定からのキャリーオーバーである。
【図8】0.2、0.5、0.75、1、2、3、5及び10μM(HVLP389、HVLP351及びHVLP364);1、2、3、5、7.5及び10nM(HVLP342);0.2、0.5、1、2、3、5及び10μM(HVLP335);0.2、0.5、1、1.5、2及び5μM(HVLP325);0.2、0.5、0.75、1、1.5、2、3及び5μM(HVLP3103);並びに1、2、3、4、5及び6nM(HVLP324)の濃度で固定化されたプロテインLとのVLの結合を示すセンサーグラムオーバーレイ。プロテインLの低親和性部位と結合するHVLP324及びHVLP342に関するセンサーグラムは含まれないが、算出されたKDは第3表に記録される。
【図9A】表面プラズモン共鳴実験におけるプロテインAとHVHP328PTV2との結合を示す図である。1、2、3、4、6、8及び10nMの濃度で固定化したプロテインAとHVH28PTV2との結合を示すセンサーグラムオーバーレイ。結合データは第4表に記録される。
【図9B】表面プラズモン共鳴実験におけるプロテインLとHVLP335PTV2との結合を示す図である。1、2、2.5、3、3.5、4及び4.5nMの濃度で固定化したプロテインLとHVH335PTV2との結合を示すセンサーグラムオーバーレイ。結合データは第4表に記録される。
【図10】黄色ブドウ球菌による微小凝集実験の結果を示す図である。五量体の濃度は、ウェル1からウェル11まで2分の1ずつに減少し、ウェル12は五量体がPBS緩衝液で置き換えられている。上の行のウェルはHVHP328PTV2五量体を含み、下の行はHVLP335PTV2五量体を含む。ウェル1〜6の五量体の濃度はそれぞれ、215、108、54、27、13及び7μg/mlである。
【特許請求の範囲】
【請求項1】
標的ポリペプチドを同定する方法であって、
(a)様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得ること、
(b)前記ライブラリファージによって、細菌層(bacterial lawn)の感染を可能にすること、
(c)前記細菌層上の平均よりも大きいプラークを形成するファージを同定すること
を含む、標的ポリペプチドを同定する方法。
【請求項2】
前記標的ポリペプチドが可溶性である、請求項1に記載の方法。
【請求項3】
前記標的ポリペプチドが単量体である、請求項1に記載の方法。
【請求項4】
前記標的ポリペプチドが非凝集性である、請求項1に記載の方法。
【請求項5】
前記標的ポリペプチドが高発現性である、請求項1に記載の方法。
【請求項6】
前記標的ポリペプチドが安定である、請求項1に記載の方法。
【請求項7】
前記ファージが繊維状ファージである、請求項1に記載の方法。
【請求項8】
前記ファージがM13又はfdである、請求項7に記載の方法。
【請求項9】
前記安定なポリペプチドが比較的高い熱リフォールディング効率を有する、請求項6に記載の方法。
【請求項10】
前記安定なポリペプチドが比較的高い融解温度を有する、請求項6に記載の方法。
【請求項11】
前記安定なポリペプチドが37℃での長期のインキュベート後にこれらの機能性を維持する、請求項6に記載の方法。
【請求項12】
前記安定なポリペプチドが化学変性剤に比較的耐性がある、請求項6に記載の方法。
【請求項13】
前記安定なポリペプチドがプロテアーゼに比較的耐性がある、請求項6に記載の方法。
【請求項14】
前記安定なポリペプチドが室温以上で、4℃、又は0℃未満で比較的長期の保存期間を有する、請求項6に記載の方法。
【請求項15】
前記安定性のポリペプチドが、細胞内環境において機能的である、請求項6に記載の方法。
【請求項16】
前記安定なポリペプチドが、ヒト体内に投与される場合に機能的である、請求項6に記載の方法。
【請求項17】
(d)工程(c)の前記より大きいプラークファージを単離する工程と、
(e)前記より大きいプラークファージにより発現される前記ポリペプチドの配列又は他の特徴を決定する工程と
をさらに含む、請求項1に記載の方法。
【請求項18】
前記標的ポリペプチドが抗体又は抗体のフラグメントである、請求項1に記載の方法。
【請求項19】
前記標的ポリペプチドがヒトVH抗体フラグメント又はヒトVL抗体フラグメントである、請求項18に記載の方法。
【請求項20】
前記標的ポリペプチドがワクチンである、請求項1に記載の方法。
【請求項21】
前記標的ポリペプチドが治療用タンパク質である、請求項1に記載の方法。
【請求項22】
前記標的ポリペプチドが、一本鎖T細胞受容体、T細胞受容体ドメイン、トランスフェリン、リポカリン、kunitzドメイン、アンキリン反復、及び細胞傷害性Tリンパ球関連抗原から成る群より選択される、請求項1に記載の方法。
【請求項23】
前記標的ポリペプチドがタンパク質性の診断用試薬及び生化学試薬である、請求項1に記載の方法。
【請求項24】
配列番号8〜配列番号54から成る群より選択されるアミノ酸配列を有するポリペプチド。
【請求項25】
請求項24に記載のアミノ酸配列と実質的に同一であるアミノ酸配列を有するポリペプチド。
【請求項26】
請求項24又は25に記載のポリペプチドをコードする核酸配列。
【請求項27】
請求項24に記載のアミノ酸配列のCDR3部分を含むポリペプチド。
【請求項28】
請求項27に記載のポリペプチドをコードする核酸配列。
【請求項29】
請求項24に記載のアミノ酸配列のFR1部分を含むポリペプチド。
【請求項30】
請求項29に記載のポリペプチドをコードする核酸配列。
【請求項31】
請求項24に記載のアミノ酸配列のFR2部分を含むポリペプチド。
【請求項32】
請求項31に記載のポリペプチドをコードする核酸配列。
【請求項33】
請求項24に記載のアミノ酸配列のFR3部分を含むポリペプチド。
【請求項34】
請求項33に記載のポリペプチドをコードする核酸配列。
【請求項35】
請求項24に記載のアミノ酸配列のFR4部分を含むポリペプチド。
【請求項36】
請求項35に記載のポリペプチドをコードする核酸配列。
【請求項37】
請求項24に記載のアミノ酸配列のCDR2部分を含むポリペプチド。
【請求項38】
請求項37に記載のポリペプチドをコードする核酸配列。
【請求項39】
請求項24に記載のアミノ酸配列のCDR1部分を含むポリペプチド。
【請求項40】
請求項39に記載のポリペプチドをコードする核酸配列。
【請求項41】
配列番号8〜配列番号22から成る群より選択される少なくとも1個のアミノ酸配列を含むVH抗体フラグメント。
【請求項42】
配列番号23〜配列番号54から成る群より選択される少なくとも1個のアミノ酸配列を含むVL抗体フラグメント。
【請求項43】
請求項41に記載のVH抗体フラグメント又は請求項42に記載のVL抗体フラグメントをコードする核酸配列。
【請求項44】
請求項41に記載の少なくとも2個のアミノ酸配列を含むVH抗体フラグメント。
【請求項45】
請求項42に記載の少なくとも2個のアミノ酸配列を含むVL抗体フラグメント。
【請求項46】
請求項44に記載のVH抗体フラグメント又は請求項45に記載のVL抗体フラグメントをコードする核酸配列。
【請求項47】
請求項41に記載の少なくとも3個のアミノ酸配列を含むVH抗体フラグメント。
【請求項48】
請求項42に記載の少なくとも3個のアミノ酸配列を含むVL抗体フラグメント。
【請求項49】
請求項47に記載のVH抗体フラグメント又は請求項48に記載のVL抗体フラグメントをコードする核酸配列。
【請求項50】
請求項41、44又は47に記載の少なくとも2個のVH抗体フラグメントを含む多量体。
【請求項51】
請求項42、45又は48に記載の少なくとも2個のVL抗体フラグメントを含む多量体。
【請求項52】
請求項41、44又は47に記載の少なくとも1個のVH抗体フラグメント及び請求項42、45又は48に記載の少なくとも1個のVL抗体フラグメントを含む多量体。
【請求項53】
請求項41、44又は47に記載の2個のVH抗体フラグメントを含む二量体。
【請求項54】
請求項42、45又は48に記載の2個のVL抗体フラグメントを含む二量体。
【請求項55】
請求項41、44又は47に記載の3個のVH抗体フラグメントを含む三量体。
【請求項56】
請求項42、45又は48に記載の3個のVL抗体フラグメントを含む三量体。
【請求項57】
請求項41、44又は47に記載の5個のVH抗体フラグメントを含む五量体。
【請求項58】
請求項42、45又は48に記載の5個のVL抗体フラグメントを含む五量体。
【請求項59】
請求項1に記載の方法で同定されるVH抗体フラグメント。
【請求項60】
ヒト起源のものである、請求項59に記載のVH抗体フラグメント。
【請求項61】
第1表に示される対応する親の生殖細胞系(germline)配列由来のアミノ酸配列を有する、請求項59に記載のVH抗体フラグメント。
【請求項62】
VH3ファミリーに属する、請求項59に記載のVH抗体フラグメント。
【請求項63】
DP47、V3−49、DP−38、V3−53、YAC−5及び8−1Bから成る群より選択されるV生殖細胞系に属する、請求項59に記載のVH抗体フラグメント。
【請求項64】
JH4b、JH6c、JH3b、JH4、JH3a及びJH1から成る群より選択されるJ生殖細胞系に属する、請求項59に記載のVH抗体フラグメント。
【請求項65】
そのアミノ酸配列の37位にバリン以外の残基を有する、請求項59に記載のVH抗体フラグメント。
【請求項66】
そのアミノ酸配列の37位にフェニルアラニン残基を有する、請求項63に記載のVH抗体フラグメント。
【請求項67】
そのアミノ酸配列の37位にチロシン残基を有する、請求項63に記載のVH抗体フラグメント。
【請求項68】
そのアミノ酸配列の1、5、6、35、83、84、84a及び108位から成る群より選択される位置で突然変異を有する、請求項59に記載のVH抗体フラグメント。
【請求項69】
プロテインAと結合する、請求項59に記載のVH抗体フラグメント。
【請求項70】
請求項1に記載の方法で同定されるVL抗体フラグメント。
【請求項71】
ヒト起源のものである、請求項70に記載のVL抗体フラグメント。
【請求項72】
Vκ1、Vκ3又はVλ1のサブグループに属する、請求項70に記載のVL抗体フラグメント。
【請求項73】
κクラスに属する、請求項70に記載のVL抗体フラグメント。
【請求項74】
L6、A27、L2、L16、O2/O12、A30及び1bから成る群より選択されるV生殖細胞系に属する、請求項70に記載のVL抗体フラグメント。
【請求項75】
Jκ1、Jκ4、Jκ2及びJλ3bから成る群より選択されるJ生殖細胞系に属する、請求項70に記載のVL抗体フラグメント。
【請求項76】
そのアミノ酸配列の96位にアルギニン、プロリン、リシン、スレオニン、ロイシン、セリン、チロシン、グルタミン酸、グルタミン又はヒスチジン残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項77】
そのアミノ酸配列の96位に荷電アミノ酸残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項78】
プロテインLと結合する、請求項70に記載のVL抗体フラグメント。
【請求項79】
そのアミノ酸配列の105、106及び107位にスレオニン、バリン又はロイシンの
アミノ酸残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項80】
請求項1に記載の方法で同定されるポリペプチド配列。
【請求項81】
請求項80に記載のポリペプチド配列をコードする核酸配列。
【請求項82】
請求項80に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項83】
請求項24、59又は70に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項84】
請求項41、42、44、45、47又は48に記載のVH抗体フラグメント又はVL抗体フラグメントを使用して構築されるディスプレイライブラリ。
【請求項85】
請求項27、37又は39に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項86】
請求項24、59又は70に記載のポリペプチドのループ(L1、L2、L3、H1、H2又はH3)領域を使用して構築されるディスプレイライブラリ。
【請求項87】
ファージディスプレイライブラリである、請求項82〜86のいずれか一項に記載のディスプレイライブラリ。
【請求項88】
リボソームディスプレイ、ARMリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ又はin vitro区画化ライブラリである、請求項82〜86のいずれか一項に記載のディスプレイライブラリ。
【請求項89】
望ましい生物物理学的特性を有するポリペプチドを調製する方法であって、
a)請求項41、42、44、45、47、48、59若しくは70に記載の抗体フラグメントをコードするか、又は請求項24、27、37若しくは39に記載のポリペプチド配列をコードし、且つ第1の望ましい特性を有する少なくとも1個の第1の核酸配列を提供する工程と、
b)第2の望ましい特性を有する抗体フラグメントをコードする少なくとも1個の第2の核酸配列を提供する工程と、
c)前記少なくとも1個の第1の核酸配列及び前記少なくとも1個の第2の核酸配列をランダムなフラグメントに切断する工程と、
d)前記ランダムなフラグメントを再び集合化させる工程と、
e)前記ランダムなフラグメントを発現する工程と、
f)前記発現されたランダムなフラグメントを前記第1の望ましい特性及び前記第2の望ましい特性に関してスクリーニングする工程と
を含む、方法。
【請求項90】
前記第1の望ましい特性が、可溶性、安定性、単量体性(monomericity)、高発現性、ヒト起源及び結合特異性から成る群より選択される、請求項89に記載の方法。
【請求項91】
前記第2の望ましい特性が、可溶性、安定性、単量体性、高発現性、ヒト起源及び結合特異性から成る群より選択される、請求項89に記載の方法。
【請求項92】
前記少なくとも1個の第2の抗体フラグメントが非ヒト起源のものである、請求項89に記載の方法。
【請求項93】
前記少なくとも1個の第2の抗体フラグメントがヒト起源のものである、請求項89に記載の方法。
【請求項94】
前記少なくとも1個の第2の抗体フラグメントがヒトVH又はヒトVLである、請求項89に記載の方法。
【請求項95】
前記再び集合化されたランダムなフラグメントのスクリーニングが、リボソームディスプレイ、酵母ディスプレイ、ファージディスプレイ、細菌細胞ディスプレイ、ARMリボソームディスプレイ又はin vitro区画化によって達成される、請求項89に記載の方法。
【請求項96】
請求項59又は70に記載の抗体フラグメント及び薬学的に好適な薬剤を含む薬学的組成物。
【請求項97】
請求項24又は80に記載のポリペプチド配列及び薬学的に好適な薬剤を含む薬学的組成物。
【請求項98】
請求項26又は81に記載の核酸配列を含む組換えベクター。
【請求項99】
請求項98に記載の組み換えベクターで形質転換される宿主細胞。
【請求項1】
標的ポリペプチドを同定する方法であって、
(a)様々なポリペプチド配列を発現することができるファージディスプレイライブラリを得ること、
(b)前記ライブラリファージによって、細菌層(bacterial lawn)の感染を可能にすること、
(c)前記細菌層上の平均よりも大きいプラークを形成するファージを同定すること
を含む、標的ポリペプチドを同定する方法。
【請求項2】
前記標的ポリペプチドが可溶性である、請求項1に記載の方法。
【請求項3】
前記標的ポリペプチドが単量体である、請求項1に記載の方法。
【請求項4】
前記標的ポリペプチドが非凝集性である、請求項1に記載の方法。
【請求項5】
前記標的ポリペプチドが高発現性である、請求項1に記載の方法。
【請求項6】
前記標的ポリペプチドが安定である、請求項1に記載の方法。
【請求項7】
前記ファージが繊維状ファージである、請求項1に記載の方法。
【請求項8】
前記ファージがM13又はfdである、請求項7に記載の方法。
【請求項9】
前記安定なポリペプチドが比較的高い熱リフォールディング効率を有する、請求項6に記載の方法。
【請求項10】
前記安定なポリペプチドが比較的高い融解温度を有する、請求項6に記載の方法。
【請求項11】
前記安定なポリペプチドが37℃での長期のインキュベート後にこれらの機能性を維持する、請求項6に記載の方法。
【請求項12】
前記安定なポリペプチドが化学変性剤に比較的耐性がある、請求項6に記載の方法。
【請求項13】
前記安定なポリペプチドがプロテアーゼに比較的耐性がある、請求項6に記載の方法。
【請求項14】
前記安定なポリペプチドが室温以上で、4℃、又は0℃未満で比較的長期の保存期間を有する、請求項6に記載の方法。
【請求項15】
前記安定性のポリペプチドが、細胞内環境において機能的である、請求項6に記載の方法。
【請求項16】
前記安定なポリペプチドが、ヒト体内に投与される場合に機能的である、請求項6に記載の方法。
【請求項17】
(d)工程(c)の前記より大きいプラークファージを単離する工程と、
(e)前記より大きいプラークファージにより発現される前記ポリペプチドの配列又は他の特徴を決定する工程と
をさらに含む、請求項1に記載の方法。
【請求項18】
前記標的ポリペプチドが抗体又は抗体のフラグメントである、請求項1に記載の方法。
【請求項19】
前記標的ポリペプチドがヒトVH抗体フラグメント又はヒトVL抗体フラグメントである、請求項18に記載の方法。
【請求項20】
前記標的ポリペプチドがワクチンである、請求項1に記載の方法。
【請求項21】
前記標的ポリペプチドが治療用タンパク質である、請求項1に記載の方法。
【請求項22】
前記標的ポリペプチドが、一本鎖T細胞受容体、T細胞受容体ドメイン、トランスフェリン、リポカリン、kunitzドメイン、アンキリン反復、及び細胞傷害性Tリンパ球関連抗原から成る群より選択される、請求項1に記載の方法。
【請求項23】
前記標的ポリペプチドがタンパク質性の診断用試薬及び生化学試薬である、請求項1に記載の方法。
【請求項24】
配列番号8〜配列番号54から成る群より選択されるアミノ酸配列を有するポリペプチド。
【請求項25】
請求項24に記載のアミノ酸配列と実質的に同一であるアミノ酸配列を有するポリペプチド。
【請求項26】
請求項24又は25に記載のポリペプチドをコードする核酸配列。
【請求項27】
請求項24に記載のアミノ酸配列のCDR3部分を含むポリペプチド。
【請求項28】
請求項27に記載のポリペプチドをコードする核酸配列。
【請求項29】
請求項24に記載のアミノ酸配列のFR1部分を含むポリペプチド。
【請求項30】
請求項29に記載のポリペプチドをコードする核酸配列。
【請求項31】
請求項24に記載のアミノ酸配列のFR2部分を含むポリペプチド。
【請求項32】
請求項31に記載のポリペプチドをコードする核酸配列。
【請求項33】
請求項24に記載のアミノ酸配列のFR3部分を含むポリペプチド。
【請求項34】
請求項33に記載のポリペプチドをコードする核酸配列。
【請求項35】
請求項24に記載のアミノ酸配列のFR4部分を含むポリペプチド。
【請求項36】
請求項35に記載のポリペプチドをコードする核酸配列。
【請求項37】
請求項24に記載のアミノ酸配列のCDR2部分を含むポリペプチド。
【請求項38】
請求項37に記載のポリペプチドをコードする核酸配列。
【請求項39】
請求項24に記載のアミノ酸配列のCDR1部分を含むポリペプチド。
【請求項40】
請求項39に記載のポリペプチドをコードする核酸配列。
【請求項41】
配列番号8〜配列番号22から成る群より選択される少なくとも1個のアミノ酸配列を含むVH抗体フラグメント。
【請求項42】
配列番号23〜配列番号54から成る群より選択される少なくとも1個のアミノ酸配列を含むVL抗体フラグメント。
【請求項43】
請求項41に記載のVH抗体フラグメント又は請求項42に記載のVL抗体フラグメントをコードする核酸配列。
【請求項44】
請求項41に記載の少なくとも2個のアミノ酸配列を含むVH抗体フラグメント。
【請求項45】
請求項42に記載の少なくとも2個のアミノ酸配列を含むVL抗体フラグメント。
【請求項46】
請求項44に記載のVH抗体フラグメント又は請求項45に記載のVL抗体フラグメントをコードする核酸配列。
【請求項47】
請求項41に記載の少なくとも3個のアミノ酸配列を含むVH抗体フラグメント。
【請求項48】
請求項42に記載の少なくとも3個のアミノ酸配列を含むVL抗体フラグメント。
【請求項49】
請求項47に記載のVH抗体フラグメント又は請求項48に記載のVL抗体フラグメントをコードする核酸配列。
【請求項50】
請求項41、44又は47に記載の少なくとも2個のVH抗体フラグメントを含む多量体。
【請求項51】
請求項42、45又は48に記載の少なくとも2個のVL抗体フラグメントを含む多量体。
【請求項52】
請求項41、44又は47に記載の少なくとも1個のVH抗体フラグメント及び請求項42、45又は48に記載の少なくとも1個のVL抗体フラグメントを含む多量体。
【請求項53】
請求項41、44又は47に記載の2個のVH抗体フラグメントを含む二量体。
【請求項54】
請求項42、45又は48に記載の2個のVL抗体フラグメントを含む二量体。
【請求項55】
請求項41、44又は47に記載の3個のVH抗体フラグメントを含む三量体。
【請求項56】
請求項42、45又は48に記載の3個のVL抗体フラグメントを含む三量体。
【請求項57】
請求項41、44又は47に記載の5個のVH抗体フラグメントを含む五量体。
【請求項58】
請求項42、45又は48に記載の5個のVL抗体フラグメントを含む五量体。
【請求項59】
請求項1に記載の方法で同定されるVH抗体フラグメント。
【請求項60】
ヒト起源のものである、請求項59に記載のVH抗体フラグメント。
【請求項61】
第1表に示される対応する親の生殖細胞系(germline)配列由来のアミノ酸配列を有する、請求項59に記載のVH抗体フラグメント。
【請求項62】
VH3ファミリーに属する、請求項59に記載のVH抗体フラグメント。
【請求項63】
DP47、V3−49、DP−38、V3−53、YAC−5及び8−1Bから成る群より選択されるV生殖細胞系に属する、請求項59に記載のVH抗体フラグメント。
【請求項64】
JH4b、JH6c、JH3b、JH4、JH3a及びJH1から成る群より選択されるJ生殖細胞系に属する、請求項59に記載のVH抗体フラグメント。
【請求項65】
そのアミノ酸配列の37位にバリン以外の残基を有する、請求項59に記載のVH抗体フラグメント。
【請求項66】
そのアミノ酸配列の37位にフェニルアラニン残基を有する、請求項63に記載のVH抗体フラグメント。
【請求項67】
そのアミノ酸配列の37位にチロシン残基を有する、請求項63に記載のVH抗体フラグメント。
【請求項68】
そのアミノ酸配列の1、5、6、35、83、84、84a及び108位から成る群より選択される位置で突然変異を有する、請求項59に記載のVH抗体フラグメント。
【請求項69】
プロテインAと結合する、請求項59に記載のVH抗体フラグメント。
【請求項70】
請求項1に記載の方法で同定されるVL抗体フラグメント。
【請求項71】
ヒト起源のものである、請求項70に記載のVL抗体フラグメント。
【請求項72】
Vκ1、Vκ3又はVλ1のサブグループに属する、請求項70に記載のVL抗体フラグメント。
【請求項73】
κクラスに属する、請求項70に記載のVL抗体フラグメント。
【請求項74】
L6、A27、L2、L16、O2/O12、A30及び1bから成る群より選択されるV生殖細胞系に属する、請求項70に記載のVL抗体フラグメント。
【請求項75】
Jκ1、Jκ4、Jκ2及びJλ3bから成る群より選択されるJ生殖細胞系に属する、請求項70に記載のVL抗体フラグメント。
【請求項76】
そのアミノ酸配列の96位にアルギニン、プロリン、リシン、スレオニン、ロイシン、セリン、チロシン、グルタミン酸、グルタミン又はヒスチジン残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項77】
そのアミノ酸配列の96位に荷電アミノ酸残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項78】
プロテインLと結合する、請求項70に記載のVL抗体フラグメント。
【請求項79】
そのアミノ酸配列の105、106及び107位にスレオニン、バリン又はロイシンの
アミノ酸残基を有する、請求項70に記載のVL抗体フラグメント。
【請求項80】
請求項1に記載の方法で同定されるポリペプチド配列。
【請求項81】
請求項80に記載のポリペプチド配列をコードする核酸配列。
【請求項82】
請求項80に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項83】
請求項24、59又は70に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項84】
請求項41、42、44、45、47又は48に記載のVH抗体フラグメント又はVL抗体フラグメントを使用して構築されるディスプレイライブラリ。
【請求項85】
請求項27、37又は39に記載のポリペプチド配列を使用して構築されるディスプレイライブラリ。
【請求項86】
請求項24、59又は70に記載のポリペプチドのループ(L1、L2、L3、H1、H2又はH3)領域を使用して構築されるディスプレイライブラリ。
【請求項87】
ファージディスプレイライブラリである、請求項82〜86のいずれか一項に記載のディスプレイライブラリ。
【請求項88】
リボソームディスプレイ、ARMリボソームディスプレイ、酵母ディスプレイ、細菌細胞ディスプレイ又はin vitro区画化ライブラリである、請求項82〜86のいずれか一項に記載のディスプレイライブラリ。
【請求項89】
望ましい生物物理学的特性を有するポリペプチドを調製する方法であって、
a)請求項41、42、44、45、47、48、59若しくは70に記載の抗体フラグメントをコードするか、又は請求項24、27、37若しくは39に記載のポリペプチド配列をコードし、且つ第1の望ましい特性を有する少なくとも1個の第1の核酸配列を提供する工程と、
b)第2の望ましい特性を有する抗体フラグメントをコードする少なくとも1個の第2の核酸配列を提供する工程と、
c)前記少なくとも1個の第1の核酸配列及び前記少なくとも1個の第2の核酸配列をランダムなフラグメントに切断する工程と、
d)前記ランダムなフラグメントを再び集合化させる工程と、
e)前記ランダムなフラグメントを発現する工程と、
f)前記発現されたランダムなフラグメントを前記第1の望ましい特性及び前記第2の望ましい特性に関してスクリーニングする工程と
を含む、方法。
【請求項90】
前記第1の望ましい特性が、可溶性、安定性、単量体性(monomericity)、高発現性、ヒト起源及び結合特異性から成る群より選択される、請求項89に記載の方法。
【請求項91】
前記第2の望ましい特性が、可溶性、安定性、単量体性、高発現性、ヒト起源及び結合特異性から成る群より選択される、請求項89に記載の方法。
【請求項92】
前記少なくとも1個の第2の抗体フラグメントが非ヒト起源のものである、請求項89に記載の方法。
【請求項93】
前記少なくとも1個の第2の抗体フラグメントがヒト起源のものである、請求項89に記載の方法。
【請求項94】
前記少なくとも1個の第2の抗体フラグメントがヒトVH又はヒトVLである、請求項89に記載の方法。
【請求項95】
前記再び集合化されたランダムなフラグメントのスクリーニングが、リボソームディスプレイ、酵母ディスプレイ、ファージディスプレイ、細菌細胞ディスプレイ、ARMリボソームディスプレイ又はin vitro区画化によって達成される、請求項89に記載の方法。
【請求項96】
請求項59又は70に記載の抗体フラグメント及び薬学的に好適な薬剤を含む薬学的組成物。
【請求項97】
請求項24又は80に記載のポリペプチド配列及び薬学的に好適な薬剤を含む薬学的組成物。
【請求項98】
請求項26又は81に記載の核酸配列を含む組換えベクター。
【請求項99】
請求項98に記載の組み換えベクターで形質転換される宿主細胞。
【図1】


【図2】
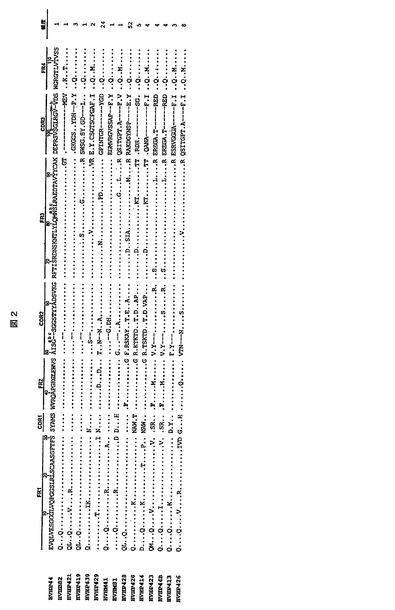
【図3A】
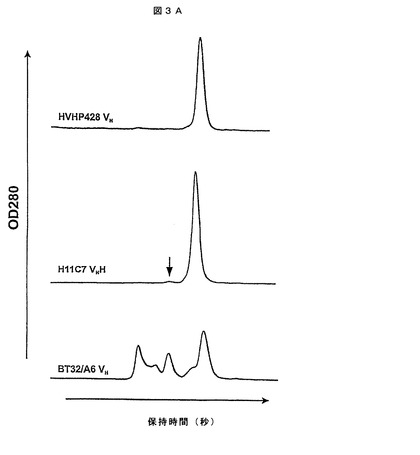
【図3B】
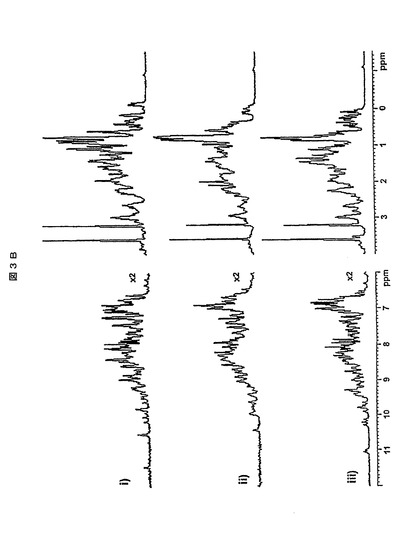
【図4A】

【図4B】
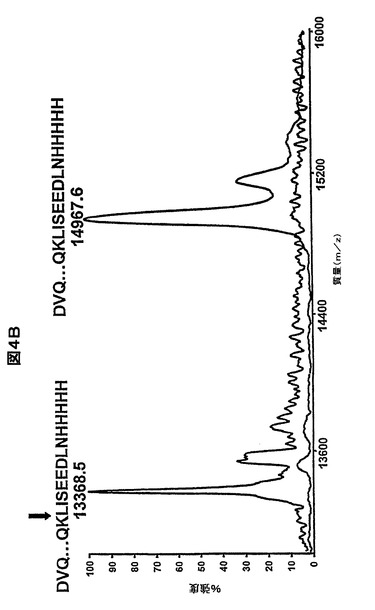
【図4C】
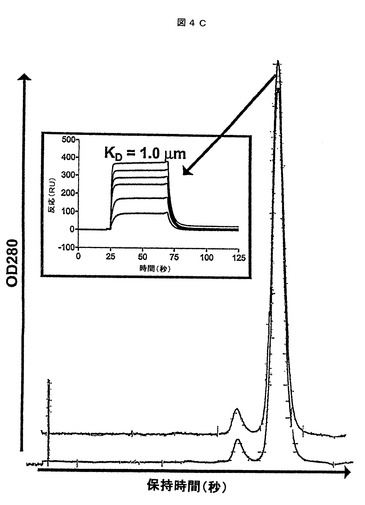
【図5】
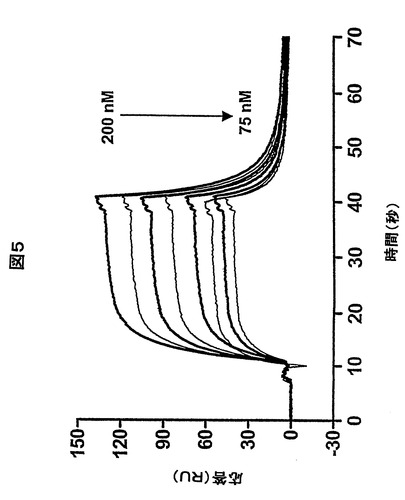
【図6】

【図7A】
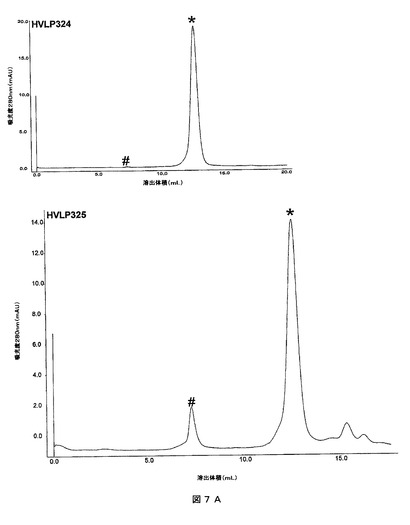
【図7B】
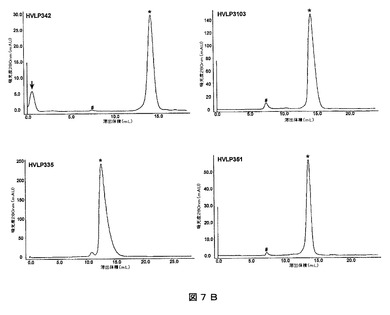
【図8】
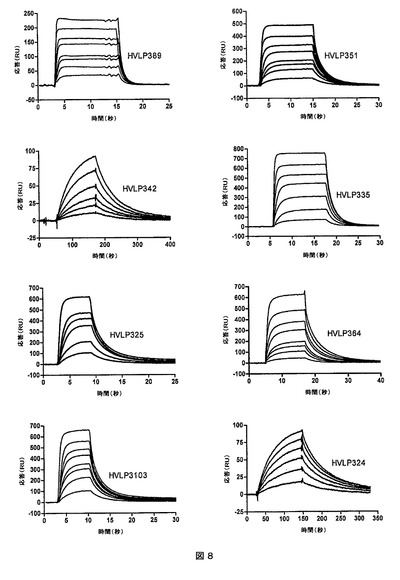
【図9A】
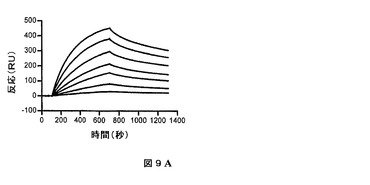
【図9B】
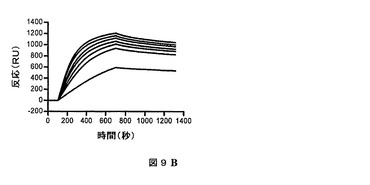
【図10】
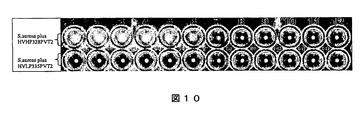
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図9A】
【図9B】
【図10】
【公開番号】特開2012−70748(P2012−70748A)
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−254988(P2011−254988)
【出願日】平成23年11月22日(2011.11.22)
【分割の表示】特願2008−502207(P2008−502207)の分割
【原出願日】平成18年3月24日(2006.3.24)
【出願人】(506175792)ナショナル リサーチ カウンシル オブ カナダ (9)
【Fターム(参考)】
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願番号】特願2011−254988(P2011−254988)
【出願日】平成23年11月22日(2011.11.22)
【分割の表示】特願2008−502207(P2008−502207)の分割
【原出願日】平成18年3月24日(2006.3.24)
【出願人】(506175792)ナショナル リサーチ カウンシル オブ カナダ (9)
【Fターム(参考)】
[ Back to top ]