
合成システム、タイヤ用ゴム薬品、タイヤ用合成ゴム及び空気入りタイヤ
【課題】アニリン及び/又はスチレンを効率良く合成できる合成システム、ブタジエン(1,3−ブタジエン)を効率良く合成できる合成システム、該合成システムから得られたアニリンを原料として合成されたタイヤ用ゴム薬品、該合成システムから得られたスチレン及び/又はブタジエンを原料として合成されたタイヤ用合成ゴム、及び該タイヤ用ゴム薬品及び/又は該タイヤ用合成ゴムを用いた空気入りタイヤを提供する。
【解決手段】炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システムに関する。
【解決手段】炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システムに関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アニリン及び/又はスチレンを効率良く合成できる合成システム、ブタジエン(1,3−ブタジエン)を効率良く合成できる合成システム、該合成システムから得られたアニリンを原料として合成されたタイヤ用ゴム薬品、該合成システムから得られたスチレン及び/又はブタジエンを原料として合成されたタイヤ用合成ゴム、及び該タイヤ用ゴム薬品及び/又は該タイヤ用合成ゴムを用いた空気入りタイヤに関する。
【背景技術】
【0002】
老化防止剤、チアゾール系加硫促進剤、スルフェンアミド系加硫促進剤などのゴム薬品の原料であるアニリンや、スチレンブタジエンゴム、ブタジエンゴムなどの合成ゴムの原料であるスチレン、ブタジエンは、通常、石油を原料として合成されている。しかし、石油や天然ガスなどの化石燃料は枯渇しつつあり、将来の価格高騰が予想されることから、収率の向上や、化石燃料からバイオマス資源への代替により、化石燃料の使用量を削減することが求められている。
【0003】
天然資源の利用という観点から、天然油脂を加水分解して得られる飽和又は不飽和脂肪酸を還元アミノ化して合成された天然由来の長鎖アミンを原料とし、加硫促進剤を合成する方法が知られている。しかし、製造過程でメルカプトベンゾチアゾール類やジベンゾチアゾリルジスルフィドが使用される合成方法であり、これらの物質が天然資源から生産されているという記載はない。
【0004】
バイオマス資源を原料とした合成の例として、バイオガスに含まれるメタンなどの低級炭化水素を原料とし、ベンゼンなどの芳香族化合物を合成する方法が知られているが、原料が気体であり、ハンドリングし難いという点で改善の余地がある。また、他の例として、バイオメタノールを原料とする方法も知られているが、原料の毒性が高いという点で改善の余地がある。更に、これらの方法に共通して、十分な収率を確保することが困難であるという点でも改善の余地がある。
【0005】
特許文献1及び2には、グルコースを原料とし、微生物によってアニリンを合成する方法が開示されている。しかし、生産速度や生産スケール等の観点で改善が必要な場合があり、その他の手法が求められている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2010−17176号公報
【特許文献2】特開2008−274225号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、前記課題を解決し、アニリン及び/又はスチレンを効率良く合成できる合成システム、ブタジエン(1,3−ブタジエン)を効率良く合成できる合成システム、該合成システムから得られたアニリンを原料として合成されたタイヤ用ゴム薬品、該合成システムから得られたスチレン及び/又はブタジエンを原料として合成されたタイヤ用合成ゴム、及び該タイヤ用ゴム薬品及び/又は該タイヤ用合成ゴムを用いた空気入りタイヤを提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明は、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システムに関する。
【0009】
上記アルコールがエタノールであることが好ましい。
【0010】
上記エタノールがバイオエタノールであることが好ましい。
【0011】
上記芳香族化合物がベンゼンであることが好ましい。
【0012】
上記ベンゼンがトルエン及び/又はキシレンを経由して合成されたものであることが好ましい。
【0013】
上記芳香族化合物がアルケンを経由して合成されたものであることが好ましい。
【0014】
上記合成システムでは、上記アルコールを固体酸触媒に接触反応させることが好ましい。
【0015】
上記固体酸触媒が、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸であることが好ましい。
【0016】
上記固体酸触媒がMFI型ゼオライトであることが好ましい。
【0017】
上記合成システムでは、上記アルコールを固体酸触媒に接触反応させ、得られた生成物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0018】
上記合成システムでは、上記生成物を蒸留し、目的物以外の化合物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0019】
上記合成システムでは、上記生成物を蒸留し、得られた蒸留物をベンゼンの融点以下に冷却してベンゼンを回収し、ベンゼン以外の化合物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0020】
上記合成システムでは、上記循環が繰返し行われることが好ましい。
【0021】
本発明はまた、炭素数2以上のアルコールを原料としてブタジエンを合成する合成システムに関する。
【0022】
本発明はまた、上記合成システムで得られたアニリンを原料として合成されたタイヤ用ゴム薬品に関する。
【0023】
本発明はまた、上記合成システムで得られたスチレン及び/又は上記合成システムで得られたブタジエンを原料として合成されたタイヤ用合成ゴムに関する。
【0024】
本発明はまた、上記タイヤ用ゴム薬品及び/又は上記タイヤ用合成ゴムを用いた空気入りタイヤに関する。
【発明の効果】
【0025】
本発明によれば、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム、及び、炭素数2以上のアルコールを原料としてブタジエン(1,3−ブタジエン)を合成する合成システムであるので、アニリン、スチレン及びブタジエンを効率良く合成することができる。従って、上記合成システムで合成されたアニリン、スチレン及びブタジエンからなる群より選択される少なくとも1種を用いることで、タイヤ用ゴム薬品、タイヤ用合成ゴム及び空気入りタイヤの製造時における化石資源の使用量を削減することができる。
【図面の簡単な説明】
【0026】
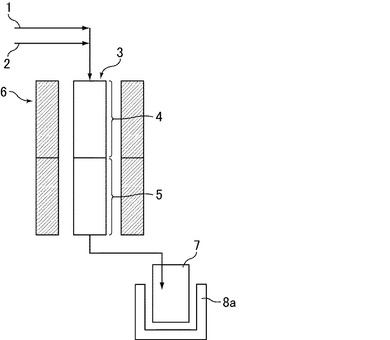
【図1】アルコールから芳香族化合物を直接合成する装置の一形態を示す模式図である。
【図2】アルコールから芳香族化合物を直接合成する装置の一形態を示す模式図である(循環型)。
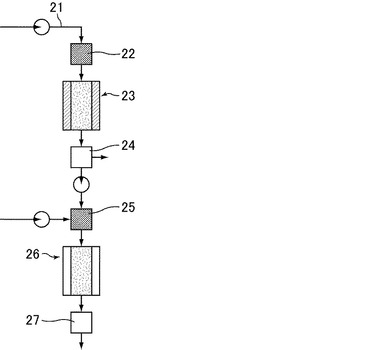
【図3】アルコールからアルケンを経由して芳香族化合物を合成する装置の一形態を示す模式図である。
【図4】アルコールからアルケンを経由して芳香族化合物を合成する装置の一形態を示す模式図である(循環型)。
【発明を実施するための形態】
【0027】
本発明は、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム(合成方法)、及び、炭素数2以上のアルコールを原料としてブタジエンを合成する合成システム(合成方法)である。
【0028】
炭素数2以上のアルコールとしては特に限定されず、一般的なものを使用できるが、毒性が低い点、輸送が容易である点、低コストである点から、炭素数2〜8のアルコールが好ましく、エタノールがより好ましい。また、化石資源に依存せずに製造でき、芳香族化合物、アルケンの収率の向上も期待できるという点から、エタノールとしては、バイオマス資源から合成されたバイオエタノールを好適に使用できる。
【0029】
以下、バイオエタノールの製造方法について説明する。
バイオエタノールは、バイオマス資源(トウモロコシ、サトウキビ、バガス、ケナフ、マメ科植物、藁、麦わら、籾殻、間伐材、廃木材、古紙、廃パルプ、有機系都市ごみなど)を低分子化し(工程1)、得られた糖類をエタノール発酵させ(工程2)、分離精製することにより得られる(工程3)。
【0030】
(工程1)では、バイオマス資源から、六炭糖、五炭糖などの糖類や、でんぷん、セルロース、ヘミセルロース、リグニンなどを生成する。これらはそのまま、又は選別されて(工程2)のエタノール発酵に用いられ、でんぷん、セルロース、ヘミセルロースは蒸煮、加水分解、酵素分解などの処理により糖化してから(工程2)のエタノール発酵に用いられる。
【0031】
(工程2)では、(工程1)で得られた単糖類などから微生物を利用してエタノールが生成される。用いられる微生物としては、酵母、大腸菌、ザイモモナス属細菌などの野生株や、これらを形質変換したものが挙げられる。
【0032】
(工程3)では、発酵液中の固体成分と液層を分離した後、蒸留工程で蒸発と凝縮を繰り返してエタノールの濃縮を行う。また、脱水剤や分離膜を用いてさらに濃縮する方法も用いられる。
【0033】
上記アルコールから芳香族化合物及び/又はアルケンを合成する方法の好適な例として、上記アルコールと触媒とを接触反応させる方法が挙げられる。反応温度は、好ましくは280〜500℃、より好ましくは300〜460℃である。反応圧力は、常圧、加圧のいずれでもよい(好ましくは0.3〜3.0MPaG)。アルコールの供給速度は、LHSV換算で好ましくは0.1〜3.0/hr、より好ましくは0.5〜1.5/hrである。
【0034】
上記触媒としては、ゼオライト、アルミナ、チタン化合物、硫酸イオン担持ジルコニア、WO3担持ジルコニアなどの固体酸を使用することができ、なかでも、反応効率を高めることが出来るという点で、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸が好ましく、ゼオライト単独、又は、アルミナ及びゼオライトの併用がより好ましい。
【0035】
アルコールから芳香族化合物を合成する場合は、特にゼオライトが好ましく、更に後述する様なSiO2とAl2O3とのモル比や細孔径を持つゼオライトが、目的とするベンゼン等の芳香族化合物を選択的に合成出来るという理由から好ましい。
【0036】
アルミナ及びゼオライトを併用してアルコールから芳香族化合物を合成する場合、後述する様に一段目にアルミナ及び/又はゼオライト等でアルケンを合成し、更に得られたアルケンをゼオライト等に接触反応させることで、より経済的、より高効率で芳香族化合物を合成することが出来る。
【0037】
また、アルコールからエチレン等のアルケン、及び/又はブタジエンを合成する場合は、アルミナ及び/又はゼオライトが好ましい。
【0038】
ゼオライトは、細孔構造を有する結晶性のアルミノケイ酸塩であり、その具体例としては、A型ゼオライト、L型ゼオライト、X型ゼオライト、Y型ゼオライト、MFI型ゼオライト、MWW型ゼオライト、β型ゼオライト、モルデナイト、フェリエライト、エリオナイトなどが挙げられる。また、ゼオライト骨格に含まれるアルミニウム原子がGa、Ti、Fe、Mn、Zn、B、Cu、Pt、Re、Mo、Gd、Nb、Y、Nd、W、La、Pなどのアルミニウム以外の金属又はその化合物で置換されたものであってもよい。なかでも、ベンゼンを選択的に精製し、さらなるアルキル化等の副反応を最小限にとどめるという点から、MFI型のZSM−5及びMWW型のMCM−22が好ましい。
【0039】
MFI型ゼオライトとしては、ZSM−5、ZSM−8、ゼータ1、ゼータ3、Nu−4、Nu−5、TZ−1、TPZ−1、TS−1等のMFI(Mobilfive)構造を有するものが挙げられ、この中でも選択性の高さ、反応効率の点からZSM−5型が特に好ましい。
【0040】
ゼオライトのイオン交換可能なカチオンサイトに占有されているカチオンは特に限定されず、水素イオン(プロトン);リチウムイオン、ナトリウムイオン、カリウムイオンなどのアルカリ金属イオン;マグネシウムイオン、カルシウムイオン、ストロンチウムイオン、バリウムイオンなどのアルカリ土類金属イオン;鉄イオン、銀イオンなどの遷移金属イオン;1〜4級アンモニウムイオンなどが挙げられる。なかでも、表面活性を高くして反応効率を上げることが出来るという点から、水素イオン(プロトン)が好ましい。該カチオンは、1種であってもよいし、2種以上であってもよい。
上記ゼオライトのうち、MFI構造を有し、プロトン型のH−ZSM−5が特に好ましい。
【0041】
ゼオライトの結晶構造中のSiO2とAl2O3とのモル比(SiO2/Al2O3)は、反応装置、温度及び原料の不純物によっても異なるが、好ましくは5〜2000、より好ましくは10〜500、更に好ましくは12〜70、特に好ましくは15〜35である。上記範囲内であれば、生成したベンゼンのさらなるアルキル化等の副反応を最小限に抑えることができる。同様の理由から、ゼオライトの結晶の大きさは、(0.001〜50)μm×(0.01〜100)μmが好ましい。また、ゼオライトの粒子の大きさは、0.1〜50μmが好ましく、1〜20μmがより好ましい。更に、ゼオライトの窒素吸着比表面積は、10〜1000m2/gが好ましく、100〜500m2/gがより好ましい。
【0042】
炭素数2以上のアルコールから合成される芳香族化合物としては、ベンゼン、トルエン、キシレン、エチルベンゼン、ジエチルベンゼン、ブチルベンゼンなどが挙げられる。なかでも、アニリン、スチレンを効率よく合成できるという点から、ベンゼン、エチルベンゼンが好ましく、ベンゼンがより好ましい。なお、ベンゼンは、トルエン、キシレンなどを経由して合成されるものであってもよいし、エチレンなどのアルケンを経由して合成されるものであってもよい。
【0043】
上記芳香族化合物を合成する装置としては特に限定されず、例えば、触媒を保持する反応管等に、加熱装置、原料供給システムが付属した装置を使用できる。また、上記装置に、反応生成物を蒸留して目的物を分離し、蒸留されなかった高沸点生成物及び気体生成物を循環させて更に触媒との反応に供する循環システムを有しているものが好ましい。目的物がベンゼンである場合、生成したベンゼンを融点(5.5℃)以下に冷却して取得できるシステムであれば、ベンゼン変換効率の観点からより好ましい。また、アルケンを経由して芳香族化合物を合成する場合は、反応カラムを2本連結し、1本目でアルコールの脱水反応を行ってアルケン類を生成させ、2本目のカラムで芳香族化合物を合成するシステムが、ベンゼン収率と触媒寿命維持の観点から好ましい。
【0044】
芳香族化合物からアニリンを合成する方法としては特に限定されず、公知の方法を使用でき、例えば、ベンゼンを濃硝酸及び濃硫酸の混酸と反応させ、得られたニトロベンゼンをBechamp還元法や接触還元法などで還元する方法が挙げられる。
【0045】
同様に、芳香族化合物からスチレンを合成する方法についても、公知の方法を使用でき、例えば、ベンゼンをフリーデル・クラフツ反応などによりエチル化し、得られたエチルベンゼンを鉄触媒などで脱水素する方法が挙げられる。フリーデル・クラフツ反応で使用するエチレンは、例えば、バイオエタノールを脱水反応することにより製造可能であるため、石油資源によらずにスチレンを製造できる。
また、芳香族化合物としてエチルベンゼンが直接合成される場合は、それをそのまま使用して脱水素し、スチレンを合成することが出来る。
【0046】
上記で調製されたアニリンを用いることで、老化防止剤、加硫促進剤などのタイヤ用ゴム薬品の製造時における石油資源の使用量を削減することができ、また、石油資源を使用せずに該タイヤ用ゴム薬品を製造することも可能となる。
【0047】
老化防止剤としては、p−フェニレンジアミン系老化防止剤として、N−(1,3−ジメチルブチル)−N’−フェニル−p−フェニレンジアミン、キノリン系老化防止剤として、2,2,4−トリメチル−1,2−ジヒドロキノリン重合物が挙げられる。
【0048】
たとえば、N−(1,3−ジメチルブチル)−N’−フェニル−p−フェニレンジアミンは、アニリンを原料として、後述の方法で製造できる。ここで、中間体のアミンに加えるメチルイソブチルケトンは、次の方法で合成出来る。たとえば後述の方法で合成したアセトン2分子のアルドール縮合により合成できるジアセトンアルコールが、容易に脱水されてメシチルオキシドに変わり、このメシチルオキシドをパラジウム触媒等で水素添加することでメチルイソブチルケトンとなる。この方法により、石油資源によらずに老化防止剤を製造できる。
【0049】
また、2,2,4−トリメチル−1,2−ジヒドロキノリン重合物は、アニリンを原料として、酸性触媒存在下140℃でアセトンを随時供給し続けることで合成できる。なお、アセトンは、以下の方法で製造可能であるため、石油資源によらずに該重合物を製造できる。
【0050】
上記の老化防止剤の合成に必要なアセトンは、例えば、バイオマスを原料として微生物によりアセトン・ブタノール発酵を行うと、ブタノール、アセトン等の混合溶媒が得られるので、これを蒸留することで合成できる。上記バイオマス原料としては、セルロース、農作物及びその廃棄物、糖類等が用いられるが、糖類が特に好ましい。アセトン・ブタノール発酵を行う微生物は特に限定されないが、野生型、変異体、または組換え体である、エシュリヒア(Escherichia)、ジモモナス(Zymomonas)、カンジダ(Candida)、サッカロミセス(Saccharomyces)、ピキア(Pichia)、ストレプトマイセス(Streptomyces)、バチルス(Bacillus)、ラクトバチルス(Lactobacillus)、コリネ(Coryne)およびクロストリジウム(Clostridium)からなる群より選択される属が好ましい。なかでも、クロストリジウム属がより好ましく、Clostridium acetobutylicum、Clostridium beijerinckii、Clostridium saccharobutylicum、およびClostridium saccharoperbutylacetonicumが特に好ましい。
また上記クロストリジウム属のアセトアセテートデカルボキシラーゼ(EC4.1.1.4)、コエンザイムAトランスフェラーゼ、チオラーゼをコードする遺伝子を組み込んだ微生物であっても構わない。
【0051】
また、木材を乾留して得られる木酢液をさらに分留、または液体クロマトグラフィー等での分取などによりアセトンを取得することもできる。
また、バイオエタノールをZr−Fe触媒の存在下で400℃以上に加熱することで合成できる。また、糖質原料由来のバイオエタノールを脱水反応させてエチレンを合成する工程、石油化学で汎用されている手法でエチレンからプロピレンを合成する工程、水和反応によりプロピレンからイソプロパノールを調製し、更に脱水素反応させる工程を経てアセトンを合成できる。
また、木質原料中のセルロースを熱分解して得られた酢酸を水酸化カルシウムで中和して酢酸カルシウムを得、次いで熱分解することでアセトンを合成できる。バイオエタノールの合成における発酵過程でエタノールが酸化されることで酢酸が生成するので、その酢酸を利用し、上記と同様のプロセスを経ることでも合成できる。更に、糖質原料由来のバイオエタノールを、ZnO/CaO触媒などで転換反応を進行させることでアセトンを合成できる。
【0052】
加硫促進剤としては、2−メルカプトベンゾチアゾール、ジベンゾチアジルジスルフィドなどのチアゾール系加硫促進剤、N−シクロヘキシル−2−ベンゾチアジルスルフェンアミド、N,N−ジシクロヘキシル−2−ベンゾチアジルスルフェンアミド、N−tert−ブチル−2−ベンゾチアジルスルフェンアミドなどのスルフェンアミド系加硫促進剤などが挙げられる。
【0053】
2−メルカプトベンゾチアゾールは、アニリンを原料として、下記合成方法により製造できる。ここで、二硫化炭素は、たとえば、からし菜に約0.4%含まれるからし油に硫化水素を反応させることで分離生成させることができる。この方法によれば、石油資源によらずに加硫促進剤を製造できる。また、そのようにして製造された2−メルカプトベンゾチアゾールを酸化することにより、ジベンゾチアジルジスルフィドを合成できる。
【0054】
【化1】

【0055】
【化2】
【0056】
また、上記で調製されたスチレン、1,3−ブタジエンを用いて、タイヤ用合成ゴムを石油資源を使用することなく製造できる。
【0057】
タイヤ用合成ゴムとしては、スチレンブタジエンゴム(SBR)、ブタジエンゴム(BR)などが挙げられる。SBRは、スチレンと1,3−ブタジエンとの共重合により製造でき、BRは、1,3−ブタジエンの重合により製造できる。ここで、1,3−ブタジエンは、たとえば前述する様なゼオライト、アルミナ、チタン化合物、硫酸イオン担持ジルコニア、WO3担持ジルコニア等の固体酸触媒存在下で、バイオエタノールを高温で反応させる方法や、バイオエタノールを酸化してアセトアルデヒドとした後、タンタル/二酸化ケイ素の触媒下でバイオエタノールを加えて加熱する方法等で製造可能であるため、石油資源によらずにタイヤ用合成ゴムを製造できる。
【0058】
以上で得られたタイヤ用ゴム薬品、タイヤ用合成ゴムは、タイヤ用ゴム組成物(トレッド、サイドウォールなど)に使用できる。
【0059】
上記ゴム組成物には、上記成分以外に、カーボンブラック、シリカ、クレー、水酸化アルミニウム、炭酸カルシウムなどの無機充填剤、シランカップリング剤、プロセスオイル、軟化剤、加硫剤、加硫促進助剤など、通常のゴム工業で使用される配合剤が適宜配合される。また、通常の石油等化石資源由来の老化防止剤、加硫促進剤、合成ゴムを一部含んでいてもよい。
【0060】
上記ゴム組成物の製造方法としては、公知の方法を用いることができ、例えば、前記各成分をオープンロール、バンバリーミキサー、密閉式混練機などのゴム混練装置を用いて混練し、その後加硫する方法等により製造できる。
【0061】
本発明の空気入りタイヤは、上記ゴム組成物を用いて通常の方法によって製造される。すなわち、必要に応じて各成分を配合したゴム組成物を、未加硫の段階でタイヤの各部材の形状に合わせて押し出し加工し、タイヤ成型機上にて通常の方法にて成形することで未加硫タイヤを形成した後、加硫機中で加熱加圧してタイヤを製造できる。
【実施例】
【0062】
実施例に基づいて、本発明を具体的に説明するが、本発明はこれらのみに限定されるものではない。
【0063】
(アルコールからのベンゼン合成)
(実施例1)
原料のアルコールとしては、石油由来のエチレンの水和反応によって得られた工業用エチルアルコール(石油由来エタノール)を用いた。
アルコールからのベンゼン合成は、ガス導入管1と、アルコール導入管(原料導入管)2と、アルコール気化層(原料気化層)4及び触媒層(反応層)5を有する反応管3と、該反応管3を加熱する加熱装置(電気炉)6と、触媒層5を経て生成された生成物を収集する生成物トラップ7と、冷却装置8aとを備える流通型反応装置(図1参照)を用いて行った。生成物トラップ7は冷却装置8aにより−15℃に冷却した。
触媒層5の内部の石英ウール上に、ゼオライト触媒H−ZSM−5(東ソー(株)製の840HOA(840NHA(SiO2/Al2O3=40(モル比)、窒素吸着比表面積:330m2/g、結晶の大きさ:2μm×4μm、粒子の大きさ:10μm)を焼成処理したもの))を10.0gとり、上記ガス導入管1より窒素ガスを供給した。窒素ガスの供給速度はLHSV換算で1/hrとした。加熱装置6によって反応管3を、所定温度まで昇温した後、アルコール導入管2より石油由来エタノールを所定量供給した。その時の反応条件は、反応温度500℃、反応圧力は常圧、石油由来エタノールの供給速度はLHSV換算で1/hr、石油由来エタノールと窒素とのモル比(石油由来エタノール/窒素)は50/50とした。反応時間は2時間とした。生成物は反応管3に連結された生成物トラップ7に集められた。
【0064】
生成物はガスクロマトグラフを用いて分析した。カラム充填剤として、ガス成分の分析には、PORAPAK P(登録商標、GLサイエンス社)、その他の分析には、SUPELCOWAX(登録商標、SUPELCO社)を用いた。
【0065】
石油由来エタノールの転化率は100%であり、得られた生成物は炭素モル数比で、ベンゼン12.0%、トルエン14.2%、キシレン7.6%、その他66.2%であった。
【0066】
上記生成物を蒸留し、ベンゼンを回収した。還流比は2、蒸気流速は0.2m/sとした。上記生成モル数比での蒸留での回収効率が90%であった。これより、以下の式により算出したベンゼンの合計収率は11%であった。
合計収率(%)=(単位時間に生成したベンゼンの炭素モル数)/(単位時間に供給したエタノールの炭素モル数)×蒸留での回収効率×100
【0067】
(実施例2)
実施例1と同様の方法により、ベンゼンを合成した。このとき、副生成物であるトルエン、キシレンを、目的物で回収し、再度、実施例1で使用したゼオライト触媒とともに反応させた。ベンゼンの合計収率は17%であった。
【0068】
(実施例3)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例1と同様の方法により、ベンゼンを合成した。用いたバイオエタノールはトウモロコシ由来のものであり、水分約20%、アセトアルデヒド等他の成分約8%が含まれるものを用いた。このバイオエタノールはろ過したのみで、蒸留精製せずに用いた。ベンゼンの合計収率は13%であった。
【0069】
(実施例4)
東ソー(株)製の840HOAの代わりにゼオライト触媒H−ZSM−5:(東ソー(株)製の820HOA(820NHA(SiO2/Al2O3=23(モル比)、窒素吸着比表面積:350m2/g、結晶の大きさ:0.03μm×0.1μm、粒子の大きさ:5μm)を焼成処理したもの))を使用した以外は実施例3と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は21%であった。
【0070】
(実施例5)
東ソー(株)製の840HOAの代わりにゼオライト触媒H−モルデナイト(ゼオリスト社製のCBV90A(SiO2/Al2O3=90))を使用した以外は実施例1と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は1%であった。
【0071】
(比較例1)
石油由来エタノールの代わりに石炭由来メタノール(石炭の部分酸化で製造した一酸化炭素(CO)に、酸化銅−酸化亜鉛/アルミナ複合酸化物を触媒として、50〜100気圧、240〜260℃で水素を反応させることにより得られた工業用メタノール)を使用した以外は実施例5と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は0.5%であった。
【0072】
(実施例6)
石油由来エタノールを原料とし、実施例4で使用したゼオライト触媒を用いてベンゼンを合成した。合成は、図1で示した装置の生成物トラップ7に、加熱装置8b、分留装置(分留管)9、蒸留物トラップ(目的物トラップ)10、反応物再循環ライン12a及び12bを取り付け、触媒反応により生成した反応混合物を分留して低沸点生成物を分取した後、気化成分と高沸点生成物を連続的に反応管3に供給できるシステム(循環型反応装置)で行った(図2参照)。生成物トラップ7は内温90℃になるように加熱装置8bにより加熱した。
上記装置を用いて実施例4の条件にて反応させ、分留管9を通じて反応生成物を連続的に蒸留した。その後、冷却装置11で−15℃に冷却した蒸留物トラップ10を用いて、蒸留物中のベンゼンを固化させて回収した。固化もしくは液化しなかった気体生成物と蒸留されなかった高沸点生成物は反応物再循環ライン12a及び12bより連続的に反応管3に供給されるようにした。
実施例4と同様の条件で石油由来エタノールを供給した後、供給を終了し、14時間同様の加熱条件で循環反応を継続した。ベンゼンの合計収率は31%であった。
【0073】
(実施例7)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例6と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は39%であった。
【0074】
(アルコールからのベンゼン合成例の考察)
実施例1及び2を比較することにより、副生成物について触媒反応を繰り返し行うことによりベンゼンの収率が向上することがわかる。これは、副生成物のトルエン、キシレンが触媒によりベンゼンに変換されることによると思われ、本プロセスにおいて触媒反応工程を繰り返すことの有用性を示すものである。
【0075】
実施例3及び4を比較することにより、ゼオライト触媒のSi/Ai比(SiO2/Al2O3比)が異なると反応性、選択性が異なることが判明した。しかし、反応温度、装置の形態にも依存すると考えられ、適用する反応装置によりSi/Al比の最適化を図ることも場合によっては必要と考えられる。
【0076】
実施例3より、水分を主とした副生成物を含んだバイオエタノールを用いても、本発明の反応が進行することが判明した。実施例1及び3のベンゼンの収率をエタノール成分比を考慮して炭素モル数換算で比較すると、バイオエタノールを用いた実施例3の方が実施例1よりもわずかに収率が高かった。
【0077】
実施例1、2、4及び7を比較することにより、副反応生成物を触媒層に複数回もしくは連続的に接触することにより、ベンゼンの収率が大幅に向上することが判明した。
【0078】
実施例6及び7をエタノール成分比を考慮して炭素モル換算で比較すると、驚くべきことに、バイオエタノールを用いた実施例7の方が石油由来エタノールを用いた実施例6よりもベンゼンの収率が高かった。これは、バイオエタノールに含まれる水分が反応を大きく阻害しないだけでなく、他の不純物がベンゼンに変換されたり、該不純物が変換反応や触媒を活性化したりする作用を発揮しているためではないかと推測される。
【0079】
(アルコールからアルケンを経由したベンゼン合成)
(実施例8)
アルコール導入管(原料導入管)21と、導入されたアルコールを気化させる加熱装置22と、該アルコールを脱水反応させる脱水反応用カラム23と、該脱水反応で得られた生成物を冷却してアルケンを回収するための冷却装置24と、該アルケンを気化させる加熱装置25と、該アルケンから芳香族化合物を合成する芳香族化合物合成用カラム26と、生成した芳香族化合物を回収するための冷却装置27(図3参照)を用いて、アルコールを原料として、アルケンを経由して芳香族化合物を合成した。脱水反応用カラム23は、触媒として酸化アルミニウム(メルク(株)製の101095100)を10g充填し、300℃に加熱した。芳香族化合物合成用カラム26は、図1の反応管3と同じ構成とした。原料は石油由来エタノールを使用し、実施例1と同様の供給条件で脱水反応用カラム23に供給し、得られたエチレンを芳香族合成用カラム26で反応させ、得られた生成物を、蒸留、精製し、ベンゼンを27%の合計収率で得た。
【0080】
(実施例9)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例8と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は36%であった。
【0081】
(実施例10)
芳香族合成用カラム26で生成した副生成物を回収する反応物再循環ライン28を追加した以外は図3の装置と同様の構成を有する装置(図4参照)を用いて、ベンゼンを合成した。脱水反応用カラム23を300℃に、芳香族化合物合成用カラム26を500℃に加熱し、実施例8と同じ条件で石油由来エタノールを供給し、ベンゼンを合成した。循環反応の時間は、実施例6同様14時間とした。得られた生成物を蒸留、生成してベンゼンを得た。ベンゼンの合計収率は82%であった。
【0082】
(実施例11)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例10と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は90%であった。
【0083】
(ベンゼンからのアニリンの合成)
上記方法により得られたベンゼンを用い、下記の方法によりアニリンを合成した。
ベンゼンを含むクロロホルム溶液に硫酸を入れ、次いで硝酸を加えて50℃で5時間加熱した。反応終了後、有機層を5%炭酸カリウム水溶液で中和し、次いで水洗し、硫酸マグネシウムで乾燥した。溶媒を留去して得られた白色固体を石油由来エタノールから再結晶し、ニトロベンゼンを得た。得られたニトロベンゼンを、ニッケル触媒の存在化、200℃で水素ガスと反応させ、アニリンを得た。
【0084】
(アセトンの石油資源外調達方法)
(アセトンの石油資源外調達方法1−1)
300mlの発酵槽(DASGIP)にSoni et al(Soni et al,1987,Appl.Microbiol.Biotechnol.27:1−5)に記載の250mlの合成培地を満たし、窒素で30分スパージした。そこにClostridium acetobutylicum(ATCC824)を嫌気性条件下で、接種した。培養温度は35℃に一定維持し、pHはNH4OH溶液を用い、常に5.5に調節した。発酵期間中、嫌気性条件を維持し、振盪速度は300rpmで維持した。5日間培養後、培養液を蒸留し、従来より周知となっているイオン交換樹脂法により分離して、アセトンを得た。
【0085】
(アセトンの石油資源外調達方法1−2)
上記調達方法1−1のClostridium acetobutylicumを菌株IFP903(ATCC39057)に変更した以外は同様にして培養、分離し、アセトンを得た。
【0086】
(アセトンの石油資源外調達方法2)
冷却管付き煙誘導管を備えたオートクレーブに木材チップを入れ、400℃に加熱し、発生した木酢液を集めた。得られた木酢液より沈殿したタール分を除去し、ジエチルエーテルにより抽出した。抽出分を炭酸水素ナトリウム溶液にて洗浄した後、分留を繰り返してアセトンを得た。
【0087】
(アニリンからの老化防止剤の製造例1(表1の老化防止剤TMDQ−1の合成方法))
アセトン導入装置、蒸留装置、温度計、および攪拌機を備えたフラスコに、前記(ベンゼンからのアニリンの合成)で得られたアニリン190g(2.0モル)と、酸性触媒として塩酸(0.20モル)を加え、140℃まで加熱した。その後140℃で保温しながら、6時間にわたり前記(アセトンの石油資源外調達方法1−2)で得られたアセトン580g(10モル)を反応系に連続的に供給した。留出する未反応のアセトンやアニリンは、随時反応系に戻した。2,2,4−トリメチル−1,2−ジヒドロキノリンの重合物180.7g(収率約30%)を得た。重合度は2〜4であった。なお、未反応のアニリン、および2,2,4−トリメチル−1,2−ジヒドロキノリンのモノマーは、減圧蒸留により回収した。140℃で未反応のアニリンが留出し、その後190℃まで昇温することにより、モノマーが留出した。モノマーの収量は19.1gであり、収率は6.9%であった。
【0088】
(アニリンからの老化防止剤の製造例1(表1の老化防止剤6PPD−1の合成方法))
前記(アセトンの石油資源外調達方法1−2)により合成したアセトン2分子をアルドール縮合により反応させて、ジアセトンアルコールを合成し更に、かかるジアセトンアルコールが容易に脱水されてメシチルオキシドに変わった。このメシチルオキシドをパラジウム触媒で水素添加することでメチルイソブチルケトンを合成した。
【0089】
上記の方法で得たバイオマス由来アニリン、及びその途中で生成するニトロベンゼンを用い下記反応を行った。なお、バイオマス由来アニリンの一部を公知の方法で酸化することによりニトロベンゼンを合成してもよい。
25%水酸化テトラメチルアンモニウム水溶液(TMAOH)187gを、温度55℃、圧力75mbarで蒸留濃縮して、35%溶液を得た。上記バイオマス由来アニリン269mlを添加後、アニリン/水共沸混合物を、水:塩基のモル比が、約4:1になるまで、温度75℃、圧力75mbarで溜去し、次いで、上記ニトロベンゼン60gを加え、混合液を、さらに4時間撹拌した。この間、水/アニリン共沸混合物の蒸留を継続した。Pt/C触媒(5%Pt)2.2gと水120mlを、この粗混合液に添加した。次に、温度80℃において、水素を用いて圧力を最大15barまで上昇させ、そして反応混合液を、水素のさらなる吸収が認められなくなるまで撹拌した。トルエン100mlを添加し、触媒を濾別し、有機相と水相を分液ロートにて分離した。次に、有機相を、分留によって精製することにより、4−アミノジフェニルアミンを91%の収率で得た。
攪拌式オートクレーブに、4−アミノジフェニルアミン129.3g、上記により合成されたメチルイソブチルケトン120.2g、白金触媒(エヌ・イーケムキャット社製、5%Ptカーボンサルファイド粉末含水品;水分55.26質量%)0.77g、および活性炭(二村化学工業(株)製、太閤活性炭S)0.65gを入れ、水素雰囲気下とした後、約1時間かけて内温を室温から150℃まで上昇させた。次いで、水素を30kgf/cm2(2.94MPa)に加圧し、消費された水素を補給しながら、同温度、同圧力を保持して反応を行った。
水素加圧開始から2時間後に、オートクレーブから水素を抜いて常圧に戻すと共に、反応液を室温まで冷却した。反応液を濾過して触媒と活性炭を濾別し、反応生成物を高速液体クロマトグラフィーにて分取することにより、4−(1,3−ジメチルブチルアミノ)ジフェニルアミン(老化防止剤6PPD−1)を99.4%の収率で得た。
【0090】
(二硫化炭素の石油資源外調達方法)
二硫化炭素は、からし菜に約0.4%含まれるからし油に硫化水素を反応させること、または木炭と硫黄を900℃で加熱することによって得た。
【0091】
(アニリンからの加硫促進剤MBTの製造例(表1の加硫促進剤MBT−1の合成方法))
300ml加圧反応器内に、前記製造例により得られたアニリン93g(1.0モル)、前記(二硫化炭素の石油資源外調達方法)により得られた二硫化炭素80g(1.1モル)、および硫黄16g(1.0モル)を投入し、250℃、10MPaの条件で2時間反応させた後、180℃まで冷却し、2−メルカプトベンゾチアゾール粗生成物を調製した。収量は130g(収率87%)であった。更に、得られた2−メルカプトベンゾチアゾールの粗生成物(純度:79%)をイソプロパノール中に沸騰温度において不活性気体としての窒素下で溶解させた。次に混合物を室温で放置して冷却した。沈澱した生成物を濾別し、イソプロパノールで洗浄し、そして乾燥した。薄黄色の生成物(高純度2−メルカプトベンゾチアゾール(融点:180.1〜181.1℃、純度:98.1%))が得られた。
【0092】
(アニリンからの加硫促進剤CBSの製造例(表1の加硫促進剤CBS−1の合成方法))
前記で得られた2−メルカプトベンゾチアゾール粗生成物を水酸化ナトリウム水溶液に溶かし、メルカプトベンゾチアゾールのナトリウム塩の20%水溶液を作製した。この水溶液に等モル量のシクロヘキシルアミンを加え、更に40℃でメタノール100mLに混合した。これに次亜塩素酸ナトリウム13%溶液をメルカプトベンゾチアゾールのナトリウム塩に対して1.2倍モルとなる様に作用させて、1時間撹拌した。反応後、水分と有機溶媒を除去することで、N−シクロヘキシル−ベンゾチアゾリルスルフェンアミドの油状物を得た(収率93%)。
【0093】
(ベンゼンからのスチレンの合成)
ベンゼンと、上記実施例9、11でバイオエタノールを脱水反応して得たエチレンとを用いて、塩化アルミニウムの存在下、反応温度320℃、ベンゼン/エチレン(モル比)10の条件で反応させ、エチルベンゼンを得た。得られたエチルベンゼンを鉄触媒下で脱水素し、スチレンを得た。
【0094】
(1,3−ブタジエンの石油資源外調達方法)
バイオエタノールを酸化してアセトアルデヒドとした後、タンタル/二酸化ケイ素の触媒下でバイオエタノールを加えて加熱し、1,3−ブタジエンを得た。なお、上記実施例9、11でバイオエタノールを脱水反応した場合にも少量得られるので、それを分別して使用してもよい。
【0095】
(SBRの合成)
上記合成例で得られたスチレン、1,3−ブタジエンを用いて、下記の方法によりSBRを重合した。
【0096】
(溶液重合SBRの合成例(表1のS−SBR−1の合成方法))
内容積20リットルのステンレス製重合反応器内を洗浄、乾燥し、乾燥窒素で置換し、ヘキサン(比重0.68g/cm3)10.2kg、上記(1,3−ブタジエンの石油資源外調達方法)により得た1,3−ブタジエン547g、上記(ベンゼンからのスチレンの合成)により得たスチレン173g、テトラヒドロフラン6.1ml、エチレングリコールジエチルエーテル5.0mlを重合反応容器内に投入した。次に、n−ブチルリチウム13.1mmolをn−ヘキサン溶液として投入し、重合を開始した。
撹拌速度を130rpm、重合反応器内温度を65℃とし、単量体を重合反応容器内に連続的に供給しながら、1,3−ブタジエンとスチレンの共重合を3時間行った。全重合での1,3−ブタジエンの供給量は821g、スチレンの供給量は259gであった。
次に、重合体溶液にメタノール0.54mlを含むヘキサン溶液20mlを加えて、更に重合体溶液を5分間撹拌した。
【0097】
重合体溶液に3−(N,N−ジメチルアミノプロピル)−トリメトキシシラン(アヅマックス(株)製)を13.1mmol加え、次に、スチームストリッピングによって重合体溶液から溶液重合SBR(S−SBR−1)を回収した。
【0098】
(乳化重合SBRの合成例(表1のE−SBR−1の合成方法))
撹拌機付き耐圧反応器に水2000g、ロジン酸石鹸(ハリマ化成(株)製)45g、脂肪酸石鹸(和光純薬工業(株)製)1.5g、リン酸ナトリウム(和光純薬工業(株)製)8g、上記(ベンゼンからのスチレンの合成)により得たスチレン250g、上記(1,3−ブタジエンの石油資源外調達方法)で合成した1,3−ブタジエン750g及びtert−ドデシルメルカプタン(和光純薬工業(株)製)2gを仕込んだ。反応器温度を5℃とし、パラメンタンヒドロペルオキシド(日油(株)製)1g及びソディウム・ホルムアルデヒド・スルホキシレート(和光純薬工業(株)製)1.5gを溶解した水溶液と、エチレンジアミン四酢酸ナトリウム(和光純薬工業(株)製)0.7g及び硫酸第二鉄(和光純薬工業(株)製)0.5gを溶解した水溶液とを反応器に添加して重合を開始した。重合開始から5時間後、N,N’−ジメチルジチオカルバメート(和光純薬工業(株)製)2gを添加して反応を停止させ、ラテックスを得た。
【0099】
得られたラテックスから、水蒸気蒸留により、未反応単量体を除去した。その後、該ラテックスをアルコールに添加し、飽和塩化ナトリウム水溶液又はギ酸でpH3〜5になるように調整しながら、凝固させ、クラム状の重合体を得た。該重合体を40℃の減圧乾燥機で乾燥し、固形ゴム(乳化重合SBR(E−SBR−1))を得た。
【0100】
(BRの合成例)
(表1のBR−1の合成方法)
上記(1,3−ブタジエンの石油資源外調達方法)で得られた1,3−ブタジエンを用いて、下記の方法によりBR(表1のBR−1)を重合した。
反応釜(3Lの耐圧ステンレス容器)を窒素置換し、窒素雰囲気を保持しながらシクロヘキサンを1800ml、(1,3−ブタジエンの石油資源外調達方法)で得られた1,3−ブタジエンを150g、THF1.5mlを投入し、撹拌を開始した。次に容器内温度を40℃に昇温し、ブチルリチウム溶液を1ml投入し、重合を開始させた。3時間撹拌後、シラン溶液(1)(信越化学工業(株)製のビス(ジメチルアミノ)メチルビニルシラン3mlと無水シクロヘキサン7.5mlの混合溶液)を1ml、ビス(ジメチルアミノ)メチルビニルシランを1.49mmol添加し、15分撹拌した。重合体溶液にIPA0.5ml及びBHT溶液1mlを添加し、さらに重合体溶液を5分間撹拌した。さらに重合体溶液を取り出し、3Lメタノール中に添加し、重合物を凝固させ、一晩風乾し、24時間減圧乾燥を行い、重合体(表1のBR−1)を得た。収率は96%であった。分析の結果、得られた重合体のMwは26.2×104、Mw/Mnは1.29であった。また、重合体のビニル結合量は、共役ジエン単位の含有量100モル%中11.4モル%であった。
なお、投入量から計算される、重合体中の下記式(I)で表される構成単位の含有量は、0.01mmol/g重合体であった。ジエン系共重合体中の共役ジエンに基づく構成単位の含有量は、98.4質量%であった。
【化3】
[式中、X1、X2及びX3は、それぞれ独立に、下式(Ia)で表される基、水酸基、ヒドロカルビル基又は置換ヒドロカルビル基を表し、X1、X2及びX3の少なくとも1つが、下式(Ia)で表される基又は水酸基である。]
【化4】
[式中、R1及びR2は、それぞれ独立に、炭素原子数が1〜6のヒドロカルビル基、炭素原子数が1〜6の置換ヒドロカルビル基、シリル基又は置換シリル基を表し、R1及びR2は結合して窒素原子と共に環構造を形成していてもよい。]
【0101】
(表1のBR−2の合成方法)
比較として、通常の化石資源由来の1,3−ブタジエンを用いて、下記の方法によりBR(表1のBR−2)を重合した。
反応釜(3Lの耐圧ステンレス容器)を窒素置換し、窒素雰囲気を保持しながらシクロヘキサンを1800ml、化石資源由来の1,3−ブタジエン(高千穂化学工業(株)製)を150g、THF1.5mlを投入し、撹拌を開始した。次に容器内温度を40℃に昇温し、ブチルリチウム溶液を1ml投入し、重合を開始させた。3時間撹拌後、上記シラン溶液(1)を1ml、ビス(ジメチルアミノ)メチルビニルシランを1.49mmol添加し、15分撹拌した。重合体溶液にIPA0.5ml及びBHT溶液1mlを添加し、さらに重合体溶液を5分間撹拌した。さらに重合体溶液を取り出し、3Lメタノール中に添加し、重合物を凝固させ、一晩風乾し、24時間減圧乾燥を行い、重合体(表1のBR−2)を得た。収率は96%であった。分析の結果、得られた重合体のMwは26.1×104、Mw/Mnは1.30であった。また、重合体のビニル結合量は、共役ジエン単位の含有量100モル%中11.3モル%であった。
なお、投入量から計算される、重合体中の上記式(I)で表される構成単位の含有量は、0.01mmol/g重合体であった。ジエン系共重合体中の共役ジエンに基づく構成単位の含有量は、98.4質量%であった。
【0102】
(トレッド用ゴム組成物の作成)
バンバリーミキサーを用いて、表1の工程1に示す配合量の薬品を投入して、排出温度が約150℃となる様に5分間混練した。その後、工程1で得られた混練り物に、工程2に示す配合量の硫黄及び加硫促進剤を加え、バンバリーミキサーを用いて、排出温度が100℃となるように約3分間混練して、未加硫ゴム組成物を得た。得られた未加硫ゴム組成物をトレッド形状に成形して、他のタイヤ部材とはりあわせ、170℃で20分間加硫することにより、試験用タイヤを作製した。
また、各未加硫ゴム組成物を170℃で20分間加硫することにより加硫ゴムシートを作製した。
【0103】
得られた未加硫ゴム組成物、加硫ゴムシート、試験用タイヤを使用して、下記の評価を行った。それぞれの試験結果を表1に示す。
【0104】
なお、上記で使用した各種薬品は、以下のとおりである。
S−SBR−1:上記方法で合成(下記式(II)で表される化合物で末端が変性、結合スチレン量:25質量%、ビニル量:59質量%)
【化5】
(R3、R4及びR5=−OCH3、R6及びR7=−C2H5、n=3)
S−SBR−2:住友化学(株)製のSE0190(上記式(II)で表される化合物で末端が変性、結合スチレン量:25質量%、ビニル量:59質量%)
E−SBR−1:上記方法で合成
E−SBR−2:JSR(株)製のSBR1502
BR−1:上記方法で合成
BR−2:上記方法で合成
NR:RSS#3
シリカ:Degussa社製のウルトラジルVN2(BET比表面積:125m2/g)
カーボンブラック:新日化カーボン(株)製のニテロン#55S(石炭系重質油を原料としたカーボンブラック、N2SA:28×103m2/kg)
シランカップリング剤:Degussa社製のSi69
ミネラルオイル:出光興産(株)製のPS−32
ステアリン酸:日油(株)製の桐
酸化亜鉛:三井金属鉱業(株)製の酸化亜鉛2種
老化防止剤6PPD−1:上記方法で合成
老化防止剤6PPD−2:大内新興化学工業(株)製のノクラック6C
老化防止剤TMDQ−1:上記方法で合成
老化防止剤TMDQ−2:大内新興化学工業(株)製のノクラック224
ワックス:大内新興化学工業(株)製のサンノックワックス
硫黄:鶴見化学工業(株)製の粉末硫黄
加硫促進剤CBS−1:上記方法で合成
加硫促進剤CBS−2:大内新興化学工業(株)製のノクセラーCZ
加硫促進剤MBT−1:上記方法で合成
加硫促進剤MBT−2:大内新興化学工業(株)製のノクセラーM
【0105】
得られた未加硫ゴム組成物、加硫ゴムシート、試験用タイヤを使用して、下記の評価を行った。それぞれの試験結果を表1に示す。
(加硫試験)
JSR製キュラストメータW型を用い、JIS規格の「振動式加硫試験機による加硫試験」の「ダイ加硫試験A法」に従い、上記未加硫ゴム組成物に破壊しない程度の低振幅(ここでは、1°)の正弦波振動を与え、試験片から上ダイスに伝わるトルクを未加硫から過加硫に至るまで測定し、170℃における未加硫ゴム組成物の加硫曲線を得た。
(1)トルク上昇値
最大トルク(MH)値から最低トルク(ML)値を引いたトルク上昇値を算出した。基準配合(比較例)のトルク上昇値を100として、各配合のトルク上昇値を指数表示した。指数は架橋効率の指標として用いられ、指数が大きいほど架橋効率が高く、良好といえる。
(2)加硫時間
最適加硫時間の指標となるtc(95)(95%トルク上昇点:t95)[分]を算出した。(1)と同じく、基準配合(比較例)のtcを100として、各配合のtcを指数表示した。指数が小さいほど、加硫速度が早いことを示す。
【0106】
(破壊エネルギー指数)
JIS K 6251「加硫ゴム及び熱可塑性ゴム−引張特性の求め方」に従って、各加硫ゴムシートの引張強度と破断伸びを測定した。更に、引張強度×破断伸び/2により破壊エネルギーを計算し、下記式にて、破壊エネルギー指数を計算した。破壊エネルギー指数が大きいほど、力学強度に優れることを示す。
(破壊エネルギー指数)=(各配合の破壊エネルギー)/(基準配合(比較例)の破壊エネルギー)×100
【0107】
(耐摩耗性試験(摩耗試験))
製造した試験用タイヤを車に装着し、市街地を8000km走行後の溝深さの減少量を測定し、溝深さが1mm減少するときの走行距離を算出した。更に、基準比較例の耐摩耗性指数を100とし、下記計算式により、各配合の溝深さの減少量を指数表示した。なお、耐摩耗性指数が大きいほど、耐摩耗性に優れることを示す。
(耐摩耗性指数)=(各配合で1mm溝深さが減るときの走行距離)/(基準配合(比較例)のタイヤの溝が1mm減るときの走行距離)×100
【0108】
(転がり抵抗試験)
2mm×130mm×130mmの加硫ゴムシートを作製し、そこから測定用試験片を切り出し、粘弾性スペクトロメーターVES((株)岩本製作所製)を用いて、温度50℃、初期歪10%、動歪2%、周波数10Hzの条件下で、各試験片のtanδを測定した。基準比較例の転がり抵抗指数を100として、下記計算式により、転がり抵抗特性をそれぞれ指数表示した。指数が小さいほど、転がり抵抗が低く、優れることを示す。
(転がり抵抗指数)=(各配合のtanδ/基準配合(比較例)のtanδ)×100
【0109】
(ウェットグリップ性能)
アンチロックブレーキシステム(ABS)評価試験により得られた制動性能をもとにして、グリップ性能を評価した。すなわち、1800cc級のABSが装備された乗用車に、前記試験用タイヤを装着して、アスファルト路面(ウェット路面状態、スキッドナンバー約50)を実車走行させ、時速100km/hの時点でブレーキをかけ、乗用車が停止するまでの減速度を算出した。ここで、減速度とは、乗用車が停止するまでの距離である。
そして、基準配合(比較例)のウェットグリップ性能指数を100とし、下記計算式により、各配合の減速度をウェットグリップ性能指数として示した。なお、ウェットグリップ性能指数が大きいほど制動性能が良好であり、ウェットグリップ性能に優れることを示す。
(ウェットグリップ性能指数)=(基準配合(比較例)の減速度)/(各配合の減速度)×100
【0110】
(ドライグリップ性能)
上記試験用タイヤを乗用車に装置してドライアスファルト路面のテストコースを走行し、ハンドル応答性、剛性感、グリップ等に関する特性をドライバーの官能評価により評価した。結果は、基準配合(比較例)を100とする指数で表示している。数値が大きい程良好であり、ドライグリップ性能、操縦安定性に優れていることを示す。
【0111】
【表1】
【0112】
実施例では、加硫特性、破壊エネルギー指数の各ゴム物性、耐摩耗性、転がり抵抗特性、ウエット・ドライのグリップ特性の各タイヤ特性とも、現在の化石資源から合成した加硫促進剤、老化防止剤、各種合成ゴムを用いた比較例と同等であった。このことから、実用上全く問題なく、化石資源の枯渇に対応出来ることを示す。
【符号の説明】
【0113】
1 ガス導入管
2 アルコール導入管(原料導入管)
3 反応管
4 アルコール気化層(原料気化層)
5 触媒層(反応層)
6 加熱装置(電気炉)
7 生成物トラップ
8a 冷却装置
8b 加熱装置
9 分留装置(分留管)
10 蒸留物トラップ(目的物トラップ)
11 冷却装置
12a、12b 反応物再循環ライン
21 アルコール導入管(原料導入管)
22 加熱装置
23 脱水反応用カラム
24 冷却装置
25 加熱装置
26 芳香族化合物合成用カラム
27 冷却装置
28 反応物再循環ライン
【技術分野】
【0001】
本発明は、アニリン及び/又はスチレンを効率良く合成できる合成システム、ブタジエン(1,3−ブタジエン)を効率良く合成できる合成システム、該合成システムから得られたアニリンを原料として合成されたタイヤ用ゴム薬品、該合成システムから得られたスチレン及び/又はブタジエンを原料として合成されたタイヤ用合成ゴム、及び該タイヤ用ゴム薬品及び/又は該タイヤ用合成ゴムを用いた空気入りタイヤに関する。
【背景技術】
【0002】
老化防止剤、チアゾール系加硫促進剤、スルフェンアミド系加硫促進剤などのゴム薬品の原料であるアニリンや、スチレンブタジエンゴム、ブタジエンゴムなどの合成ゴムの原料であるスチレン、ブタジエンは、通常、石油を原料として合成されている。しかし、石油や天然ガスなどの化石燃料は枯渇しつつあり、将来の価格高騰が予想されることから、収率の向上や、化石燃料からバイオマス資源への代替により、化石燃料の使用量を削減することが求められている。
【0003】
天然資源の利用という観点から、天然油脂を加水分解して得られる飽和又は不飽和脂肪酸を還元アミノ化して合成された天然由来の長鎖アミンを原料とし、加硫促進剤を合成する方法が知られている。しかし、製造過程でメルカプトベンゾチアゾール類やジベンゾチアゾリルジスルフィドが使用される合成方法であり、これらの物質が天然資源から生産されているという記載はない。
【0004】
バイオマス資源を原料とした合成の例として、バイオガスに含まれるメタンなどの低級炭化水素を原料とし、ベンゼンなどの芳香族化合物を合成する方法が知られているが、原料が気体であり、ハンドリングし難いという点で改善の余地がある。また、他の例として、バイオメタノールを原料とする方法も知られているが、原料の毒性が高いという点で改善の余地がある。更に、これらの方法に共通して、十分な収率を確保することが困難であるという点でも改善の余地がある。
【0005】
特許文献1及び2には、グルコースを原料とし、微生物によってアニリンを合成する方法が開示されている。しかし、生産速度や生産スケール等の観点で改善が必要な場合があり、その他の手法が求められている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2010−17176号公報
【特許文献2】特開2008−274225号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、前記課題を解決し、アニリン及び/又はスチレンを効率良く合成できる合成システム、ブタジエン(1,3−ブタジエン)を効率良く合成できる合成システム、該合成システムから得られたアニリンを原料として合成されたタイヤ用ゴム薬品、該合成システムから得られたスチレン及び/又はブタジエンを原料として合成されたタイヤ用合成ゴム、及び該タイヤ用ゴム薬品及び/又は該タイヤ用合成ゴムを用いた空気入りタイヤを提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明は、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システムに関する。
【0009】
上記アルコールがエタノールであることが好ましい。
【0010】
上記エタノールがバイオエタノールであることが好ましい。
【0011】
上記芳香族化合物がベンゼンであることが好ましい。
【0012】
上記ベンゼンがトルエン及び/又はキシレンを経由して合成されたものであることが好ましい。
【0013】
上記芳香族化合物がアルケンを経由して合成されたものであることが好ましい。
【0014】
上記合成システムでは、上記アルコールを固体酸触媒に接触反応させることが好ましい。
【0015】
上記固体酸触媒が、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸であることが好ましい。
【0016】
上記固体酸触媒がMFI型ゼオライトであることが好ましい。
【0017】
上記合成システムでは、上記アルコールを固体酸触媒に接触反応させ、得られた生成物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0018】
上記合成システムでは、上記生成物を蒸留し、目的物以外の化合物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0019】
上記合成システムでは、上記生成物を蒸留し、得られた蒸留物をベンゼンの融点以下に冷却してベンゼンを回収し、ベンゼン以外の化合物を循環させて更に上記固体酸触媒に接触反応させることが好ましい。
【0020】
上記合成システムでは、上記循環が繰返し行われることが好ましい。
【0021】
本発明はまた、炭素数2以上のアルコールを原料としてブタジエンを合成する合成システムに関する。
【0022】
本発明はまた、上記合成システムで得られたアニリンを原料として合成されたタイヤ用ゴム薬品に関する。
【0023】
本発明はまた、上記合成システムで得られたスチレン及び/又は上記合成システムで得られたブタジエンを原料として合成されたタイヤ用合成ゴムに関する。
【0024】
本発明はまた、上記タイヤ用ゴム薬品及び/又は上記タイヤ用合成ゴムを用いた空気入りタイヤに関する。
【発明の効果】
【0025】
本発明によれば、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム、及び、炭素数2以上のアルコールを原料としてブタジエン(1,3−ブタジエン)を合成する合成システムであるので、アニリン、スチレン及びブタジエンを効率良く合成することができる。従って、上記合成システムで合成されたアニリン、スチレン及びブタジエンからなる群より選択される少なくとも1種を用いることで、タイヤ用ゴム薬品、タイヤ用合成ゴム及び空気入りタイヤの製造時における化石資源の使用量を削減することができる。
【図面の簡単な説明】
【0026】
【図1】アルコールから芳香族化合物を直接合成する装置の一形態を示す模式図である。
【図2】アルコールから芳香族化合物を直接合成する装置の一形態を示す模式図である(循環型)。
【図3】アルコールからアルケンを経由して芳香族化合物を合成する装置の一形態を示す模式図である。
【図4】アルコールからアルケンを経由して芳香族化合物を合成する装置の一形態を示す模式図である(循環型)。
【発明を実施するための形態】
【0027】
本発明は、炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム(合成方法)、及び、炭素数2以上のアルコールを原料としてブタジエンを合成する合成システム(合成方法)である。
【0028】
炭素数2以上のアルコールとしては特に限定されず、一般的なものを使用できるが、毒性が低い点、輸送が容易である点、低コストである点から、炭素数2〜8のアルコールが好ましく、エタノールがより好ましい。また、化石資源に依存せずに製造でき、芳香族化合物、アルケンの収率の向上も期待できるという点から、エタノールとしては、バイオマス資源から合成されたバイオエタノールを好適に使用できる。
【0029】
以下、バイオエタノールの製造方法について説明する。
バイオエタノールは、バイオマス資源(トウモロコシ、サトウキビ、バガス、ケナフ、マメ科植物、藁、麦わら、籾殻、間伐材、廃木材、古紙、廃パルプ、有機系都市ごみなど)を低分子化し(工程1)、得られた糖類をエタノール発酵させ(工程2)、分離精製することにより得られる(工程3)。
【0030】
(工程1)では、バイオマス資源から、六炭糖、五炭糖などの糖類や、でんぷん、セルロース、ヘミセルロース、リグニンなどを生成する。これらはそのまま、又は選別されて(工程2)のエタノール発酵に用いられ、でんぷん、セルロース、ヘミセルロースは蒸煮、加水分解、酵素分解などの処理により糖化してから(工程2)のエタノール発酵に用いられる。
【0031】
(工程2)では、(工程1)で得られた単糖類などから微生物を利用してエタノールが生成される。用いられる微生物としては、酵母、大腸菌、ザイモモナス属細菌などの野生株や、これらを形質変換したものが挙げられる。
【0032】
(工程3)では、発酵液中の固体成分と液層を分離した後、蒸留工程で蒸発と凝縮を繰り返してエタノールの濃縮を行う。また、脱水剤や分離膜を用いてさらに濃縮する方法も用いられる。
【0033】
上記アルコールから芳香族化合物及び/又はアルケンを合成する方法の好適な例として、上記アルコールと触媒とを接触反応させる方法が挙げられる。反応温度は、好ましくは280〜500℃、より好ましくは300〜460℃である。反応圧力は、常圧、加圧のいずれでもよい(好ましくは0.3〜3.0MPaG)。アルコールの供給速度は、LHSV換算で好ましくは0.1〜3.0/hr、より好ましくは0.5〜1.5/hrである。
【0034】
上記触媒としては、ゼオライト、アルミナ、チタン化合物、硫酸イオン担持ジルコニア、WO3担持ジルコニアなどの固体酸を使用することができ、なかでも、反応効率を高めることが出来るという点で、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸が好ましく、ゼオライト単独、又は、アルミナ及びゼオライトの併用がより好ましい。
【0035】
アルコールから芳香族化合物を合成する場合は、特にゼオライトが好ましく、更に後述する様なSiO2とAl2O3とのモル比や細孔径を持つゼオライトが、目的とするベンゼン等の芳香族化合物を選択的に合成出来るという理由から好ましい。
【0036】
アルミナ及びゼオライトを併用してアルコールから芳香族化合物を合成する場合、後述する様に一段目にアルミナ及び/又はゼオライト等でアルケンを合成し、更に得られたアルケンをゼオライト等に接触反応させることで、より経済的、より高効率で芳香族化合物を合成することが出来る。
【0037】
また、アルコールからエチレン等のアルケン、及び/又はブタジエンを合成する場合は、アルミナ及び/又はゼオライトが好ましい。
【0038】
ゼオライトは、細孔構造を有する結晶性のアルミノケイ酸塩であり、その具体例としては、A型ゼオライト、L型ゼオライト、X型ゼオライト、Y型ゼオライト、MFI型ゼオライト、MWW型ゼオライト、β型ゼオライト、モルデナイト、フェリエライト、エリオナイトなどが挙げられる。また、ゼオライト骨格に含まれるアルミニウム原子がGa、Ti、Fe、Mn、Zn、B、Cu、Pt、Re、Mo、Gd、Nb、Y、Nd、W、La、Pなどのアルミニウム以外の金属又はその化合物で置換されたものであってもよい。なかでも、ベンゼンを選択的に精製し、さらなるアルキル化等の副反応を最小限にとどめるという点から、MFI型のZSM−5及びMWW型のMCM−22が好ましい。
【0039】
MFI型ゼオライトとしては、ZSM−5、ZSM−8、ゼータ1、ゼータ3、Nu−4、Nu−5、TZ−1、TPZ−1、TS−1等のMFI(Mobilfive)構造を有するものが挙げられ、この中でも選択性の高さ、反応効率の点からZSM−5型が特に好ましい。
【0040】
ゼオライトのイオン交換可能なカチオンサイトに占有されているカチオンは特に限定されず、水素イオン(プロトン);リチウムイオン、ナトリウムイオン、カリウムイオンなどのアルカリ金属イオン;マグネシウムイオン、カルシウムイオン、ストロンチウムイオン、バリウムイオンなどのアルカリ土類金属イオン;鉄イオン、銀イオンなどの遷移金属イオン;1〜4級アンモニウムイオンなどが挙げられる。なかでも、表面活性を高くして反応効率を上げることが出来るという点から、水素イオン(プロトン)が好ましい。該カチオンは、1種であってもよいし、2種以上であってもよい。
上記ゼオライトのうち、MFI構造を有し、プロトン型のH−ZSM−5が特に好ましい。
【0041】
ゼオライトの結晶構造中のSiO2とAl2O3とのモル比(SiO2/Al2O3)は、反応装置、温度及び原料の不純物によっても異なるが、好ましくは5〜2000、より好ましくは10〜500、更に好ましくは12〜70、特に好ましくは15〜35である。上記範囲内であれば、生成したベンゼンのさらなるアルキル化等の副反応を最小限に抑えることができる。同様の理由から、ゼオライトの結晶の大きさは、(0.001〜50)μm×(0.01〜100)μmが好ましい。また、ゼオライトの粒子の大きさは、0.1〜50μmが好ましく、1〜20μmがより好ましい。更に、ゼオライトの窒素吸着比表面積は、10〜1000m2/gが好ましく、100〜500m2/gがより好ましい。
【0042】
炭素数2以上のアルコールから合成される芳香族化合物としては、ベンゼン、トルエン、キシレン、エチルベンゼン、ジエチルベンゼン、ブチルベンゼンなどが挙げられる。なかでも、アニリン、スチレンを効率よく合成できるという点から、ベンゼン、エチルベンゼンが好ましく、ベンゼンがより好ましい。なお、ベンゼンは、トルエン、キシレンなどを経由して合成されるものであってもよいし、エチレンなどのアルケンを経由して合成されるものであってもよい。
【0043】
上記芳香族化合物を合成する装置としては特に限定されず、例えば、触媒を保持する反応管等に、加熱装置、原料供給システムが付属した装置を使用できる。また、上記装置に、反応生成物を蒸留して目的物を分離し、蒸留されなかった高沸点生成物及び気体生成物を循環させて更に触媒との反応に供する循環システムを有しているものが好ましい。目的物がベンゼンである場合、生成したベンゼンを融点(5.5℃)以下に冷却して取得できるシステムであれば、ベンゼン変換効率の観点からより好ましい。また、アルケンを経由して芳香族化合物を合成する場合は、反応カラムを2本連結し、1本目でアルコールの脱水反応を行ってアルケン類を生成させ、2本目のカラムで芳香族化合物を合成するシステムが、ベンゼン収率と触媒寿命維持の観点から好ましい。
【0044】
芳香族化合物からアニリンを合成する方法としては特に限定されず、公知の方法を使用でき、例えば、ベンゼンを濃硝酸及び濃硫酸の混酸と反応させ、得られたニトロベンゼンをBechamp還元法や接触還元法などで還元する方法が挙げられる。
【0045】
同様に、芳香族化合物からスチレンを合成する方法についても、公知の方法を使用でき、例えば、ベンゼンをフリーデル・クラフツ反応などによりエチル化し、得られたエチルベンゼンを鉄触媒などで脱水素する方法が挙げられる。フリーデル・クラフツ反応で使用するエチレンは、例えば、バイオエタノールを脱水反応することにより製造可能であるため、石油資源によらずにスチレンを製造できる。
また、芳香族化合物としてエチルベンゼンが直接合成される場合は、それをそのまま使用して脱水素し、スチレンを合成することが出来る。
【0046】
上記で調製されたアニリンを用いることで、老化防止剤、加硫促進剤などのタイヤ用ゴム薬品の製造時における石油資源の使用量を削減することができ、また、石油資源を使用せずに該タイヤ用ゴム薬品を製造することも可能となる。
【0047】
老化防止剤としては、p−フェニレンジアミン系老化防止剤として、N−(1,3−ジメチルブチル)−N’−フェニル−p−フェニレンジアミン、キノリン系老化防止剤として、2,2,4−トリメチル−1,2−ジヒドロキノリン重合物が挙げられる。
【0048】
たとえば、N−(1,3−ジメチルブチル)−N’−フェニル−p−フェニレンジアミンは、アニリンを原料として、後述の方法で製造できる。ここで、中間体のアミンに加えるメチルイソブチルケトンは、次の方法で合成出来る。たとえば後述の方法で合成したアセトン2分子のアルドール縮合により合成できるジアセトンアルコールが、容易に脱水されてメシチルオキシドに変わり、このメシチルオキシドをパラジウム触媒等で水素添加することでメチルイソブチルケトンとなる。この方法により、石油資源によらずに老化防止剤を製造できる。
【0049】
また、2,2,4−トリメチル−1,2−ジヒドロキノリン重合物は、アニリンを原料として、酸性触媒存在下140℃でアセトンを随時供給し続けることで合成できる。なお、アセトンは、以下の方法で製造可能であるため、石油資源によらずに該重合物を製造できる。
【0050】
上記の老化防止剤の合成に必要なアセトンは、例えば、バイオマスを原料として微生物によりアセトン・ブタノール発酵を行うと、ブタノール、アセトン等の混合溶媒が得られるので、これを蒸留することで合成できる。上記バイオマス原料としては、セルロース、農作物及びその廃棄物、糖類等が用いられるが、糖類が特に好ましい。アセトン・ブタノール発酵を行う微生物は特に限定されないが、野生型、変異体、または組換え体である、エシュリヒア(Escherichia)、ジモモナス(Zymomonas)、カンジダ(Candida)、サッカロミセス(Saccharomyces)、ピキア(Pichia)、ストレプトマイセス(Streptomyces)、バチルス(Bacillus)、ラクトバチルス(Lactobacillus)、コリネ(Coryne)およびクロストリジウム(Clostridium)からなる群より選択される属が好ましい。なかでも、クロストリジウム属がより好ましく、Clostridium acetobutylicum、Clostridium beijerinckii、Clostridium saccharobutylicum、およびClostridium saccharoperbutylacetonicumが特に好ましい。
また上記クロストリジウム属のアセトアセテートデカルボキシラーゼ(EC4.1.1.4)、コエンザイムAトランスフェラーゼ、チオラーゼをコードする遺伝子を組み込んだ微生物であっても構わない。
【0051】
また、木材を乾留して得られる木酢液をさらに分留、または液体クロマトグラフィー等での分取などによりアセトンを取得することもできる。
また、バイオエタノールをZr−Fe触媒の存在下で400℃以上に加熱することで合成できる。また、糖質原料由来のバイオエタノールを脱水反応させてエチレンを合成する工程、石油化学で汎用されている手法でエチレンからプロピレンを合成する工程、水和反応によりプロピレンからイソプロパノールを調製し、更に脱水素反応させる工程を経てアセトンを合成できる。
また、木質原料中のセルロースを熱分解して得られた酢酸を水酸化カルシウムで中和して酢酸カルシウムを得、次いで熱分解することでアセトンを合成できる。バイオエタノールの合成における発酵過程でエタノールが酸化されることで酢酸が生成するので、その酢酸を利用し、上記と同様のプロセスを経ることでも合成できる。更に、糖質原料由来のバイオエタノールを、ZnO/CaO触媒などで転換反応を進行させることでアセトンを合成できる。
【0052】
加硫促進剤としては、2−メルカプトベンゾチアゾール、ジベンゾチアジルジスルフィドなどのチアゾール系加硫促進剤、N−シクロヘキシル−2−ベンゾチアジルスルフェンアミド、N,N−ジシクロヘキシル−2−ベンゾチアジルスルフェンアミド、N−tert−ブチル−2−ベンゾチアジルスルフェンアミドなどのスルフェンアミド系加硫促進剤などが挙げられる。
【0053】
2−メルカプトベンゾチアゾールは、アニリンを原料として、下記合成方法により製造できる。ここで、二硫化炭素は、たとえば、からし菜に約0.4%含まれるからし油に硫化水素を反応させることで分離生成させることができる。この方法によれば、石油資源によらずに加硫促進剤を製造できる。また、そのようにして製造された2−メルカプトベンゾチアゾールを酸化することにより、ジベンゾチアジルジスルフィドを合成できる。
【0054】
【化1】
【0055】
【化2】
【0056】
また、上記で調製されたスチレン、1,3−ブタジエンを用いて、タイヤ用合成ゴムを石油資源を使用することなく製造できる。
【0057】
タイヤ用合成ゴムとしては、スチレンブタジエンゴム(SBR)、ブタジエンゴム(BR)などが挙げられる。SBRは、スチレンと1,3−ブタジエンとの共重合により製造でき、BRは、1,3−ブタジエンの重合により製造できる。ここで、1,3−ブタジエンは、たとえば前述する様なゼオライト、アルミナ、チタン化合物、硫酸イオン担持ジルコニア、WO3担持ジルコニア等の固体酸触媒存在下で、バイオエタノールを高温で反応させる方法や、バイオエタノールを酸化してアセトアルデヒドとした後、タンタル/二酸化ケイ素の触媒下でバイオエタノールを加えて加熱する方法等で製造可能であるため、石油資源によらずにタイヤ用合成ゴムを製造できる。
【0058】
以上で得られたタイヤ用ゴム薬品、タイヤ用合成ゴムは、タイヤ用ゴム組成物(トレッド、サイドウォールなど)に使用できる。
【0059】
上記ゴム組成物には、上記成分以外に、カーボンブラック、シリカ、クレー、水酸化アルミニウム、炭酸カルシウムなどの無機充填剤、シランカップリング剤、プロセスオイル、軟化剤、加硫剤、加硫促進助剤など、通常のゴム工業で使用される配合剤が適宜配合される。また、通常の石油等化石資源由来の老化防止剤、加硫促進剤、合成ゴムを一部含んでいてもよい。
【0060】
上記ゴム組成物の製造方法としては、公知の方法を用いることができ、例えば、前記各成分をオープンロール、バンバリーミキサー、密閉式混練機などのゴム混練装置を用いて混練し、その後加硫する方法等により製造できる。
【0061】
本発明の空気入りタイヤは、上記ゴム組成物を用いて通常の方法によって製造される。すなわち、必要に応じて各成分を配合したゴム組成物を、未加硫の段階でタイヤの各部材の形状に合わせて押し出し加工し、タイヤ成型機上にて通常の方法にて成形することで未加硫タイヤを形成した後、加硫機中で加熱加圧してタイヤを製造できる。
【実施例】
【0062】
実施例に基づいて、本発明を具体的に説明するが、本発明はこれらのみに限定されるものではない。
【0063】
(アルコールからのベンゼン合成)
(実施例1)
原料のアルコールとしては、石油由来のエチレンの水和反応によって得られた工業用エチルアルコール(石油由来エタノール)を用いた。
アルコールからのベンゼン合成は、ガス導入管1と、アルコール導入管(原料導入管)2と、アルコール気化層(原料気化層)4及び触媒層(反応層)5を有する反応管3と、該反応管3を加熱する加熱装置(電気炉)6と、触媒層5を経て生成された生成物を収集する生成物トラップ7と、冷却装置8aとを備える流通型反応装置(図1参照)を用いて行った。生成物トラップ7は冷却装置8aにより−15℃に冷却した。
触媒層5の内部の石英ウール上に、ゼオライト触媒H−ZSM−5(東ソー(株)製の840HOA(840NHA(SiO2/Al2O3=40(モル比)、窒素吸着比表面積:330m2/g、結晶の大きさ:2μm×4μm、粒子の大きさ:10μm)を焼成処理したもの))を10.0gとり、上記ガス導入管1より窒素ガスを供給した。窒素ガスの供給速度はLHSV換算で1/hrとした。加熱装置6によって反応管3を、所定温度まで昇温した後、アルコール導入管2より石油由来エタノールを所定量供給した。その時の反応条件は、反応温度500℃、反応圧力は常圧、石油由来エタノールの供給速度はLHSV換算で1/hr、石油由来エタノールと窒素とのモル比(石油由来エタノール/窒素)は50/50とした。反応時間は2時間とした。生成物は反応管3に連結された生成物トラップ7に集められた。
【0064】
生成物はガスクロマトグラフを用いて分析した。カラム充填剤として、ガス成分の分析には、PORAPAK P(登録商標、GLサイエンス社)、その他の分析には、SUPELCOWAX(登録商標、SUPELCO社)を用いた。
【0065】
石油由来エタノールの転化率は100%であり、得られた生成物は炭素モル数比で、ベンゼン12.0%、トルエン14.2%、キシレン7.6%、その他66.2%であった。
【0066】
上記生成物を蒸留し、ベンゼンを回収した。還流比は2、蒸気流速は0.2m/sとした。上記生成モル数比での蒸留での回収効率が90%であった。これより、以下の式により算出したベンゼンの合計収率は11%であった。
合計収率(%)=(単位時間に生成したベンゼンの炭素モル数)/(単位時間に供給したエタノールの炭素モル数)×蒸留での回収効率×100
【0067】
(実施例2)
実施例1と同様の方法により、ベンゼンを合成した。このとき、副生成物であるトルエン、キシレンを、目的物で回収し、再度、実施例1で使用したゼオライト触媒とともに反応させた。ベンゼンの合計収率は17%であった。
【0068】
(実施例3)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例1と同様の方法により、ベンゼンを合成した。用いたバイオエタノールはトウモロコシ由来のものであり、水分約20%、アセトアルデヒド等他の成分約8%が含まれるものを用いた。このバイオエタノールはろ過したのみで、蒸留精製せずに用いた。ベンゼンの合計収率は13%であった。
【0069】
(実施例4)
東ソー(株)製の840HOAの代わりにゼオライト触媒H−ZSM−5:(東ソー(株)製の820HOA(820NHA(SiO2/Al2O3=23(モル比)、窒素吸着比表面積:350m2/g、結晶の大きさ:0.03μm×0.1μm、粒子の大きさ:5μm)を焼成処理したもの))を使用した以外は実施例3と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は21%であった。
【0070】
(実施例5)
東ソー(株)製の840HOAの代わりにゼオライト触媒H−モルデナイト(ゼオリスト社製のCBV90A(SiO2/Al2O3=90))を使用した以外は実施例1と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は1%であった。
【0071】
(比較例1)
石油由来エタノールの代わりに石炭由来メタノール(石炭の部分酸化で製造した一酸化炭素(CO)に、酸化銅−酸化亜鉛/アルミナ複合酸化物を触媒として、50〜100気圧、240〜260℃で水素を反応させることにより得られた工業用メタノール)を使用した以外は実施例5と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は0.5%であった。
【0072】
(実施例6)
石油由来エタノールを原料とし、実施例4で使用したゼオライト触媒を用いてベンゼンを合成した。合成は、図1で示した装置の生成物トラップ7に、加熱装置8b、分留装置(分留管)9、蒸留物トラップ(目的物トラップ)10、反応物再循環ライン12a及び12bを取り付け、触媒反応により生成した反応混合物を分留して低沸点生成物を分取した後、気化成分と高沸点生成物を連続的に反応管3に供給できるシステム(循環型反応装置)で行った(図2参照)。生成物トラップ7は内温90℃になるように加熱装置8bにより加熱した。
上記装置を用いて実施例4の条件にて反応させ、分留管9を通じて反応生成物を連続的に蒸留した。その後、冷却装置11で−15℃に冷却した蒸留物トラップ10を用いて、蒸留物中のベンゼンを固化させて回収した。固化もしくは液化しなかった気体生成物と蒸留されなかった高沸点生成物は反応物再循環ライン12a及び12bより連続的に反応管3に供給されるようにした。
実施例4と同様の条件で石油由来エタノールを供給した後、供給を終了し、14時間同様の加熱条件で循環反応を継続した。ベンゼンの合計収率は31%であった。
【0073】
(実施例7)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例6と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は39%であった。
【0074】
(アルコールからのベンゼン合成例の考察)
実施例1及び2を比較することにより、副生成物について触媒反応を繰り返し行うことによりベンゼンの収率が向上することがわかる。これは、副生成物のトルエン、キシレンが触媒によりベンゼンに変換されることによると思われ、本プロセスにおいて触媒反応工程を繰り返すことの有用性を示すものである。
【0075】
実施例3及び4を比較することにより、ゼオライト触媒のSi/Ai比(SiO2/Al2O3比)が異なると反応性、選択性が異なることが判明した。しかし、反応温度、装置の形態にも依存すると考えられ、適用する反応装置によりSi/Al比の最適化を図ることも場合によっては必要と考えられる。
【0076】
実施例3より、水分を主とした副生成物を含んだバイオエタノールを用いても、本発明の反応が進行することが判明した。実施例1及び3のベンゼンの収率をエタノール成分比を考慮して炭素モル数換算で比較すると、バイオエタノールを用いた実施例3の方が実施例1よりもわずかに収率が高かった。
【0077】
実施例1、2、4及び7を比較することにより、副反応生成物を触媒層に複数回もしくは連続的に接触することにより、ベンゼンの収率が大幅に向上することが判明した。
【0078】
実施例6及び7をエタノール成分比を考慮して炭素モル換算で比較すると、驚くべきことに、バイオエタノールを用いた実施例7の方が石油由来エタノールを用いた実施例6よりもベンゼンの収率が高かった。これは、バイオエタノールに含まれる水分が反応を大きく阻害しないだけでなく、他の不純物がベンゼンに変換されたり、該不純物が変換反応や触媒を活性化したりする作用を発揮しているためではないかと推測される。
【0079】
(アルコールからアルケンを経由したベンゼン合成)
(実施例8)
アルコール導入管(原料導入管)21と、導入されたアルコールを気化させる加熱装置22と、該アルコールを脱水反応させる脱水反応用カラム23と、該脱水反応で得られた生成物を冷却してアルケンを回収するための冷却装置24と、該アルケンを気化させる加熱装置25と、該アルケンから芳香族化合物を合成する芳香族化合物合成用カラム26と、生成した芳香族化合物を回収するための冷却装置27(図3参照)を用いて、アルコールを原料として、アルケンを経由して芳香族化合物を合成した。脱水反応用カラム23は、触媒として酸化アルミニウム(メルク(株)製の101095100)を10g充填し、300℃に加熱した。芳香族化合物合成用カラム26は、図1の反応管3と同じ構成とした。原料は石油由来エタノールを使用し、実施例1と同様の供給条件で脱水反応用カラム23に供給し、得られたエチレンを芳香族合成用カラム26で反応させ、得られた生成物を、蒸留、精製し、ベンゼンを27%の合計収率で得た。
【0080】
(実施例9)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例8と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は36%であった。
【0081】
(実施例10)
芳香族合成用カラム26で生成した副生成物を回収する反応物再循環ライン28を追加した以外は図3の装置と同様の構成を有する装置(図4参照)を用いて、ベンゼンを合成した。脱水反応用カラム23を300℃に、芳香族化合物合成用カラム26を500℃に加熱し、実施例8と同じ条件で石油由来エタノールを供給し、ベンゼンを合成した。循環反応の時間は、実施例6同様14時間とした。得られた生成物を蒸留、生成してベンゼンを得た。ベンゼンの合計収率は82%であった。
【0082】
(実施例11)
石油由来エタノールの代わりにバイオエタノールを使用した以外は実施例10と同様の方法により、ベンゼンを合成した。ベンゼンの合計収率は90%であった。
【0083】
(ベンゼンからのアニリンの合成)
上記方法により得られたベンゼンを用い、下記の方法によりアニリンを合成した。
ベンゼンを含むクロロホルム溶液に硫酸を入れ、次いで硝酸を加えて50℃で5時間加熱した。反応終了後、有機層を5%炭酸カリウム水溶液で中和し、次いで水洗し、硫酸マグネシウムで乾燥した。溶媒を留去して得られた白色固体を石油由来エタノールから再結晶し、ニトロベンゼンを得た。得られたニトロベンゼンを、ニッケル触媒の存在化、200℃で水素ガスと反応させ、アニリンを得た。
【0084】
(アセトンの石油資源外調達方法)
(アセトンの石油資源外調達方法1−1)
300mlの発酵槽(DASGIP)にSoni et al(Soni et al,1987,Appl.Microbiol.Biotechnol.27:1−5)に記載の250mlの合成培地を満たし、窒素で30分スパージした。そこにClostridium acetobutylicum(ATCC824)を嫌気性条件下で、接種した。培養温度は35℃に一定維持し、pHはNH4OH溶液を用い、常に5.5に調節した。発酵期間中、嫌気性条件を維持し、振盪速度は300rpmで維持した。5日間培養後、培養液を蒸留し、従来より周知となっているイオン交換樹脂法により分離して、アセトンを得た。
【0085】
(アセトンの石油資源外調達方法1−2)
上記調達方法1−1のClostridium acetobutylicumを菌株IFP903(ATCC39057)に変更した以外は同様にして培養、分離し、アセトンを得た。
【0086】
(アセトンの石油資源外調達方法2)
冷却管付き煙誘導管を備えたオートクレーブに木材チップを入れ、400℃に加熱し、発生した木酢液を集めた。得られた木酢液より沈殿したタール分を除去し、ジエチルエーテルにより抽出した。抽出分を炭酸水素ナトリウム溶液にて洗浄した後、分留を繰り返してアセトンを得た。
【0087】
(アニリンからの老化防止剤の製造例1(表1の老化防止剤TMDQ−1の合成方法))
アセトン導入装置、蒸留装置、温度計、および攪拌機を備えたフラスコに、前記(ベンゼンからのアニリンの合成)で得られたアニリン190g(2.0モル)と、酸性触媒として塩酸(0.20モル)を加え、140℃まで加熱した。その後140℃で保温しながら、6時間にわたり前記(アセトンの石油資源外調達方法1−2)で得られたアセトン580g(10モル)を反応系に連続的に供給した。留出する未反応のアセトンやアニリンは、随時反応系に戻した。2,2,4−トリメチル−1,2−ジヒドロキノリンの重合物180.7g(収率約30%)を得た。重合度は2〜4であった。なお、未反応のアニリン、および2,2,4−トリメチル−1,2−ジヒドロキノリンのモノマーは、減圧蒸留により回収した。140℃で未反応のアニリンが留出し、その後190℃まで昇温することにより、モノマーが留出した。モノマーの収量は19.1gであり、収率は6.9%であった。
【0088】
(アニリンからの老化防止剤の製造例1(表1の老化防止剤6PPD−1の合成方法))
前記(アセトンの石油資源外調達方法1−2)により合成したアセトン2分子をアルドール縮合により反応させて、ジアセトンアルコールを合成し更に、かかるジアセトンアルコールが容易に脱水されてメシチルオキシドに変わった。このメシチルオキシドをパラジウム触媒で水素添加することでメチルイソブチルケトンを合成した。
【0089】
上記の方法で得たバイオマス由来アニリン、及びその途中で生成するニトロベンゼンを用い下記反応を行った。なお、バイオマス由来アニリンの一部を公知の方法で酸化することによりニトロベンゼンを合成してもよい。
25%水酸化テトラメチルアンモニウム水溶液(TMAOH)187gを、温度55℃、圧力75mbarで蒸留濃縮して、35%溶液を得た。上記バイオマス由来アニリン269mlを添加後、アニリン/水共沸混合物を、水:塩基のモル比が、約4:1になるまで、温度75℃、圧力75mbarで溜去し、次いで、上記ニトロベンゼン60gを加え、混合液を、さらに4時間撹拌した。この間、水/アニリン共沸混合物の蒸留を継続した。Pt/C触媒(5%Pt)2.2gと水120mlを、この粗混合液に添加した。次に、温度80℃において、水素を用いて圧力を最大15barまで上昇させ、そして反応混合液を、水素のさらなる吸収が認められなくなるまで撹拌した。トルエン100mlを添加し、触媒を濾別し、有機相と水相を分液ロートにて分離した。次に、有機相を、分留によって精製することにより、4−アミノジフェニルアミンを91%の収率で得た。
攪拌式オートクレーブに、4−アミノジフェニルアミン129.3g、上記により合成されたメチルイソブチルケトン120.2g、白金触媒(エヌ・イーケムキャット社製、5%Ptカーボンサルファイド粉末含水品;水分55.26質量%)0.77g、および活性炭(二村化学工業(株)製、太閤活性炭S)0.65gを入れ、水素雰囲気下とした後、約1時間かけて内温を室温から150℃まで上昇させた。次いで、水素を30kgf/cm2(2.94MPa)に加圧し、消費された水素を補給しながら、同温度、同圧力を保持して反応を行った。
水素加圧開始から2時間後に、オートクレーブから水素を抜いて常圧に戻すと共に、反応液を室温まで冷却した。反応液を濾過して触媒と活性炭を濾別し、反応生成物を高速液体クロマトグラフィーにて分取することにより、4−(1,3−ジメチルブチルアミノ)ジフェニルアミン(老化防止剤6PPD−1)を99.4%の収率で得た。
【0090】
(二硫化炭素の石油資源外調達方法)
二硫化炭素は、からし菜に約0.4%含まれるからし油に硫化水素を反応させること、または木炭と硫黄を900℃で加熱することによって得た。
【0091】
(アニリンからの加硫促進剤MBTの製造例(表1の加硫促進剤MBT−1の合成方法))
300ml加圧反応器内に、前記製造例により得られたアニリン93g(1.0モル)、前記(二硫化炭素の石油資源外調達方法)により得られた二硫化炭素80g(1.1モル)、および硫黄16g(1.0モル)を投入し、250℃、10MPaの条件で2時間反応させた後、180℃まで冷却し、2−メルカプトベンゾチアゾール粗生成物を調製した。収量は130g(収率87%)であった。更に、得られた2−メルカプトベンゾチアゾールの粗生成物(純度:79%)をイソプロパノール中に沸騰温度において不活性気体としての窒素下で溶解させた。次に混合物を室温で放置して冷却した。沈澱した生成物を濾別し、イソプロパノールで洗浄し、そして乾燥した。薄黄色の生成物(高純度2−メルカプトベンゾチアゾール(融点:180.1〜181.1℃、純度:98.1%))が得られた。
【0092】
(アニリンからの加硫促進剤CBSの製造例(表1の加硫促進剤CBS−1の合成方法))
前記で得られた2−メルカプトベンゾチアゾール粗生成物を水酸化ナトリウム水溶液に溶かし、メルカプトベンゾチアゾールのナトリウム塩の20%水溶液を作製した。この水溶液に等モル量のシクロヘキシルアミンを加え、更に40℃でメタノール100mLに混合した。これに次亜塩素酸ナトリウム13%溶液をメルカプトベンゾチアゾールのナトリウム塩に対して1.2倍モルとなる様に作用させて、1時間撹拌した。反応後、水分と有機溶媒を除去することで、N−シクロヘキシル−ベンゾチアゾリルスルフェンアミドの油状物を得た(収率93%)。
【0093】
(ベンゼンからのスチレンの合成)
ベンゼンと、上記実施例9、11でバイオエタノールを脱水反応して得たエチレンとを用いて、塩化アルミニウムの存在下、反応温度320℃、ベンゼン/エチレン(モル比)10の条件で反応させ、エチルベンゼンを得た。得られたエチルベンゼンを鉄触媒下で脱水素し、スチレンを得た。
【0094】
(1,3−ブタジエンの石油資源外調達方法)
バイオエタノールを酸化してアセトアルデヒドとした後、タンタル/二酸化ケイ素の触媒下でバイオエタノールを加えて加熱し、1,3−ブタジエンを得た。なお、上記実施例9、11でバイオエタノールを脱水反応した場合にも少量得られるので、それを分別して使用してもよい。
【0095】
(SBRの合成)
上記合成例で得られたスチレン、1,3−ブタジエンを用いて、下記の方法によりSBRを重合した。
【0096】
(溶液重合SBRの合成例(表1のS−SBR−1の合成方法))
内容積20リットルのステンレス製重合反応器内を洗浄、乾燥し、乾燥窒素で置換し、ヘキサン(比重0.68g/cm3)10.2kg、上記(1,3−ブタジエンの石油資源外調達方法)により得た1,3−ブタジエン547g、上記(ベンゼンからのスチレンの合成)により得たスチレン173g、テトラヒドロフラン6.1ml、エチレングリコールジエチルエーテル5.0mlを重合反応容器内に投入した。次に、n−ブチルリチウム13.1mmolをn−ヘキサン溶液として投入し、重合を開始した。
撹拌速度を130rpm、重合反応器内温度を65℃とし、単量体を重合反応容器内に連続的に供給しながら、1,3−ブタジエンとスチレンの共重合を3時間行った。全重合での1,3−ブタジエンの供給量は821g、スチレンの供給量は259gであった。
次に、重合体溶液にメタノール0.54mlを含むヘキサン溶液20mlを加えて、更に重合体溶液を5分間撹拌した。
【0097】
重合体溶液に3−(N,N−ジメチルアミノプロピル)−トリメトキシシラン(アヅマックス(株)製)を13.1mmol加え、次に、スチームストリッピングによって重合体溶液から溶液重合SBR(S−SBR−1)を回収した。
【0098】
(乳化重合SBRの合成例(表1のE−SBR−1の合成方法))
撹拌機付き耐圧反応器に水2000g、ロジン酸石鹸(ハリマ化成(株)製)45g、脂肪酸石鹸(和光純薬工業(株)製)1.5g、リン酸ナトリウム(和光純薬工業(株)製)8g、上記(ベンゼンからのスチレンの合成)により得たスチレン250g、上記(1,3−ブタジエンの石油資源外調達方法)で合成した1,3−ブタジエン750g及びtert−ドデシルメルカプタン(和光純薬工業(株)製)2gを仕込んだ。反応器温度を5℃とし、パラメンタンヒドロペルオキシド(日油(株)製)1g及びソディウム・ホルムアルデヒド・スルホキシレート(和光純薬工業(株)製)1.5gを溶解した水溶液と、エチレンジアミン四酢酸ナトリウム(和光純薬工業(株)製)0.7g及び硫酸第二鉄(和光純薬工業(株)製)0.5gを溶解した水溶液とを反応器に添加して重合を開始した。重合開始から5時間後、N,N’−ジメチルジチオカルバメート(和光純薬工業(株)製)2gを添加して反応を停止させ、ラテックスを得た。
【0099】
得られたラテックスから、水蒸気蒸留により、未反応単量体を除去した。その後、該ラテックスをアルコールに添加し、飽和塩化ナトリウム水溶液又はギ酸でpH3〜5になるように調整しながら、凝固させ、クラム状の重合体を得た。該重合体を40℃の減圧乾燥機で乾燥し、固形ゴム(乳化重合SBR(E−SBR−1))を得た。
【0100】
(BRの合成例)
(表1のBR−1の合成方法)
上記(1,3−ブタジエンの石油資源外調達方法)で得られた1,3−ブタジエンを用いて、下記の方法によりBR(表1のBR−1)を重合した。
反応釜(3Lの耐圧ステンレス容器)を窒素置換し、窒素雰囲気を保持しながらシクロヘキサンを1800ml、(1,3−ブタジエンの石油資源外調達方法)で得られた1,3−ブタジエンを150g、THF1.5mlを投入し、撹拌を開始した。次に容器内温度を40℃に昇温し、ブチルリチウム溶液を1ml投入し、重合を開始させた。3時間撹拌後、シラン溶液(1)(信越化学工業(株)製のビス(ジメチルアミノ)メチルビニルシラン3mlと無水シクロヘキサン7.5mlの混合溶液)を1ml、ビス(ジメチルアミノ)メチルビニルシランを1.49mmol添加し、15分撹拌した。重合体溶液にIPA0.5ml及びBHT溶液1mlを添加し、さらに重合体溶液を5分間撹拌した。さらに重合体溶液を取り出し、3Lメタノール中に添加し、重合物を凝固させ、一晩風乾し、24時間減圧乾燥を行い、重合体(表1のBR−1)を得た。収率は96%であった。分析の結果、得られた重合体のMwは26.2×104、Mw/Mnは1.29であった。また、重合体のビニル結合量は、共役ジエン単位の含有量100モル%中11.4モル%であった。
なお、投入量から計算される、重合体中の下記式(I)で表される構成単位の含有量は、0.01mmol/g重合体であった。ジエン系共重合体中の共役ジエンに基づく構成単位の含有量は、98.4質量%であった。
【化3】
[式中、X1、X2及びX3は、それぞれ独立に、下式(Ia)で表される基、水酸基、ヒドロカルビル基又は置換ヒドロカルビル基を表し、X1、X2及びX3の少なくとも1つが、下式(Ia)で表される基又は水酸基である。]
【化4】
[式中、R1及びR2は、それぞれ独立に、炭素原子数が1〜6のヒドロカルビル基、炭素原子数が1〜6の置換ヒドロカルビル基、シリル基又は置換シリル基を表し、R1及びR2は結合して窒素原子と共に環構造を形成していてもよい。]
【0101】
(表1のBR−2の合成方法)
比較として、通常の化石資源由来の1,3−ブタジエンを用いて、下記の方法によりBR(表1のBR−2)を重合した。
反応釜(3Lの耐圧ステンレス容器)を窒素置換し、窒素雰囲気を保持しながらシクロヘキサンを1800ml、化石資源由来の1,3−ブタジエン(高千穂化学工業(株)製)を150g、THF1.5mlを投入し、撹拌を開始した。次に容器内温度を40℃に昇温し、ブチルリチウム溶液を1ml投入し、重合を開始させた。3時間撹拌後、上記シラン溶液(1)を1ml、ビス(ジメチルアミノ)メチルビニルシランを1.49mmol添加し、15分撹拌した。重合体溶液にIPA0.5ml及びBHT溶液1mlを添加し、さらに重合体溶液を5分間撹拌した。さらに重合体溶液を取り出し、3Lメタノール中に添加し、重合物を凝固させ、一晩風乾し、24時間減圧乾燥を行い、重合体(表1のBR−2)を得た。収率は96%であった。分析の結果、得られた重合体のMwは26.1×104、Mw/Mnは1.30であった。また、重合体のビニル結合量は、共役ジエン単位の含有量100モル%中11.3モル%であった。
なお、投入量から計算される、重合体中の上記式(I)で表される構成単位の含有量は、0.01mmol/g重合体であった。ジエン系共重合体中の共役ジエンに基づく構成単位の含有量は、98.4質量%であった。
【0102】
(トレッド用ゴム組成物の作成)
バンバリーミキサーを用いて、表1の工程1に示す配合量の薬品を投入して、排出温度が約150℃となる様に5分間混練した。その後、工程1で得られた混練り物に、工程2に示す配合量の硫黄及び加硫促進剤を加え、バンバリーミキサーを用いて、排出温度が100℃となるように約3分間混練して、未加硫ゴム組成物を得た。得られた未加硫ゴム組成物をトレッド形状に成形して、他のタイヤ部材とはりあわせ、170℃で20分間加硫することにより、試験用タイヤを作製した。
また、各未加硫ゴム組成物を170℃で20分間加硫することにより加硫ゴムシートを作製した。
【0103】
得られた未加硫ゴム組成物、加硫ゴムシート、試験用タイヤを使用して、下記の評価を行った。それぞれの試験結果を表1に示す。
【0104】
なお、上記で使用した各種薬品は、以下のとおりである。
S−SBR−1:上記方法で合成(下記式(II)で表される化合物で末端が変性、結合スチレン量:25質量%、ビニル量:59質量%)
【化5】
(R3、R4及びR5=−OCH3、R6及びR7=−C2H5、n=3)
S−SBR−2:住友化学(株)製のSE0190(上記式(II)で表される化合物で末端が変性、結合スチレン量:25質量%、ビニル量:59質量%)
E−SBR−1:上記方法で合成
E−SBR−2:JSR(株)製のSBR1502
BR−1:上記方法で合成
BR−2:上記方法で合成
NR:RSS#3
シリカ:Degussa社製のウルトラジルVN2(BET比表面積:125m2/g)
カーボンブラック:新日化カーボン(株)製のニテロン#55S(石炭系重質油を原料としたカーボンブラック、N2SA:28×103m2/kg)
シランカップリング剤:Degussa社製のSi69
ミネラルオイル:出光興産(株)製のPS−32
ステアリン酸:日油(株)製の桐
酸化亜鉛:三井金属鉱業(株)製の酸化亜鉛2種
老化防止剤6PPD−1:上記方法で合成
老化防止剤6PPD−2:大内新興化学工業(株)製のノクラック6C
老化防止剤TMDQ−1:上記方法で合成
老化防止剤TMDQ−2:大内新興化学工業(株)製のノクラック224
ワックス:大内新興化学工業(株)製のサンノックワックス
硫黄:鶴見化学工業(株)製の粉末硫黄
加硫促進剤CBS−1:上記方法で合成
加硫促進剤CBS−2:大内新興化学工業(株)製のノクセラーCZ
加硫促進剤MBT−1:上記方法で合成
加硫促進剤MBT−2:大内新興化学工業(株)製のノクセラーM
【0105】
得られた未加硫ゴム組成物、加硫ゴムシート、試験用タイヤを使用して、下記の評価を行った。それぞれの試験結果を表1に示す。
(加硫試験)
JSR製キュラストメータW型を用い、JIS規格の「振動式加硫試験機による加硫試験」の「ダイ加硫試験A法」に従い、上記未加硫ゴム組成物に破壊しない程度の低振幅(ここでは、1°)の正弦波振動を与え、試験片から上ダイスに伝わるトルクを未加硫から過加硫に至るまで測定し、170℃における未加硫ゴム組成物の加硫曲線を得た。
(1)トルク上昇値
最大トルク(MH)値から最低トルク(ML)値を引いたトルク上昇値を算出した。基準配合(比較例)のトルク上昇値を100として、各配合のトルク上昇値を指数表示した。指数は架橋効率の指標として用いられ、指数が大きいほど架橋効率が高く、良好といえる。
(2)加硫時間
最適加硫時間の指標となるtc(95)(95%トルク上昇点:t95)[分]を算出した。(1)と同じく、基準配合(比較例)のtcを100として、各配合のtcを指数表示した。指数が小さいほど、加硫速度が早いことを示す。
【0106】
(破壊エネルギー指数)
JIS K 6251「加硫ゴム及び熱可塑性ゴム−引張特性の求め方」に従って、各加硫ゴムシートの引張強度と破断伸びを測定した。更に、引張強度×破断伸び/2により破壊エネルギーを計算し、下記式にて、破壊エネルギー指数を計算した。破壊エネルギー指数が大きいほど、力学強度に優れることを示す。
(破壊エネルギー指数)=(各配合の破壊エネルギー)/(基準配合(比較例)の破壊エネルギー)×100
【0107】
(耐摩耗性試験(摩耗試験))
製造した試験用タイヤを車に装着し、市街地を8000km走行後の溝深さの減少量を測定し、溝深さが1mm減少するときの走行距離を算出した。更に、基準比較例の耐摩耗性指数を100とし、下記計算式により、各配合の溝深さの減少量を指数表示した。なお、耐摩耗性指数が大きいほど、耐摩耗性に優れることを示す。
(耐摩耗性指数)=(各配合で1mm溝深さが減るときの走行距離)/(基準配合(比較例)のタイヤの溝が1mm減るときの走行距離)×100
【0108】
(転がり抵抗試験)
2mm×130mm×130mmの加硫ゴムシートを作製し、そこから測定用試験片を切り出し、粘弾性スペクトロメーターVES((株)岩本製作所製)を用いて、温度50℃、初期歪10%、動歪2%、周波数10Hzの条件下で、各試験片のtanδを測定した。基準比較例の転がり抵抗指数を100として、下記計算式により、転がり抵抗特性をそれぞれ指数表示した。指数が小さいほど、転がり抵抗が低く、優れることを示す。
(転がり抵抗指数)=(各配合のtanδ/基準配合(比較例)のtanδ)×100
【0109】
(ウェットグリップ性能)
アンチロックブレーキシステム(ABS)評価試験により得られた制動性能をもとにして、グリップ性能を評価した。すなわち、1800cc級のABSが装備された乗用車に、前記試験用タイヤを装着して、アスファルト路面(ウェット路面状態、スキッドナンバー約50)を実車走行させ、時速100km/hの時点でブレーキをかけ、乗用車が停止するまでの減速度を算出した。ここで、減速度とは、乗用車が停止するまでの距離である。
そして、基準配合(比較例)のウェットグリップ性能指数を100とし、下記計算式により、各配合の減速度をウェットグリップ性能指数として示した。なお、ウェットグリップ性能指数が大きいほど制動性能が良好であり、ウェットグリップ性能に優れることを示す。
(ウェットグリップ性能指数)=(基準配合(比較例)の減速度)/(各配合の減速度)×100
【0110】
(ドライグリップ性能)
上記試験用タイヤを乗用車に装置してドライアスファルト路面のテストコースを走行し、ハンドル応答性、剛性感、グリップ等に関する特性をドライバーの官能評価により評価した。結果は、基準配合(比較例)を100とする指数で表示している。数値が大きい程良好であり、ドライグリップ性能、操縦安定性に優れていることを示す。
【0111】
【表1】
【0112】
実施例では、加硫特性、破壊エネルギー指数の各ゴム物性、耐摩耗性、転がり抵抗特性、ウエット・ドライのグリップ特性の各タイヤ特性とも、現在の化石資源から合成した加硫促進剤、老化防止剤、各種合成ゴムを用いた比較例と同等であった。このことから、実用上全く問題なく、化石資源の枯渇に対応出来ることを示す。
【符号の説明】
【0113】
1 ガス導入管
2 アルコール導入管(原料導入管)
3 反応管
4 アルコール気化層(原料気化層)
5 触媒層(反応層)
6 加熱装置(電気炉)
7 生成物トラップ
8a 冷却装置
8b 加熱装置
9 分留装置(分留管)
10 蒸留物トラップ(目的物トラップ)
11 冷却装置
12a、12b 反応物再循環ライン
21 アルコール導入管(原料導入管)
22 加熱装置
23 脱水反応用カラム
24 冷却装置
25 加熱装置
26 芳香族化合物合成用カラム
27 冷却装置
28 反応物再循環ライン
【特許請求の範囲】
【請求項1】
炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム。
【請求項2】
前記アルコールがエタノールである請求項1記載の合成システム。
【請求項3】
前記エタノールがバイオエタノールである請求項2記載の合成システム。
【請求項4】
前記芳香族化合物がベンゼンである請求項1〜3のいずれかに記載の合成システム。
【請求項5】
前記ベンゼンがトルエン及び/又はキシレンを経由して合成されたものである請求項4記載の合成システム。
【請求項6】
前記芳香族化合物がアルケンを経由して合成されたものである請求項1〜5のいずれかに記載の合成システム。
【請求項7】
前記アルコールを固体酸触媒に接触反応させる請求項1〜6のいずれかに記載の合成システム。
【請求項8】
前記固体酸触媒が、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸である請求項7記載の合成システム。
【請求項9】
前記固体酸触媒がMFI型ゼオライトである請求項7又は8記載の合成システム。
【請求項10】
前記アルコールを固体酸触媒に接触反応させ、得られた生成物を循環させて更に前記固体酸触媒に接触反応させる請求項1〜9のいずれかに記載の合成システム。
【請求項11】
前記生成物を蒸留し、目的物以外の化合物を循環させて更に前記固体酸触媒に接触反応させる請求項10記載の合成システム。
【請求項12】
前記生成物を蒸留し、得られた蒸留物をベンゼンの融点以下に冷却してベンゼンを回収し、ベンゼン以外の化合物を循環させて更に前記固体酸触媒に接触反応させる請求項10又は11記載の合成システム。
【請求項13】
前記循環が繰返し行われる請求項10〜12のいずれかに記載の合成システム。
【請求項14】
炭素数2以上のアルコールを原料としてブタジエンを合成する合成システム。
【請求項15】
請求項1〜13のいずれかに記載の合成システムで得られたアニリンを原料として合成されたタイヤ用ゴム薬品。
【請求項16】
請求項1〜13のいずれかに記載の合成システムで得られたスチレン及び/又は請求項14記載の合成システムで得られたブタジエンを原料として合成されたタイヤ用合成ゴム。
【請求項17】
請求項15記載のタイヤ用ゴム薬品及び/又は請求項16記載のタイヤ用合成ゴムを用いた空気入りタイヤ。
【請求項1】
炭素数2以上のアルコールを原料として、芳香族化合物を経由してアニリン及び/又はスチレンを合成する合成システム。
【請求項2】
前記アルコールがエタノールである請求項1記載の合成システム。
【請求項3】
前記エタノールがバイオエタノールである請求項2記載の合成システム。
【請求項4】
前記芳香族化合物がベンゼンである請求項1〜3のいずれかに記載の合成システム。
【請求項5】
前記ベンゼンがトルエン及び/又はキシレンを経由して合成されたものである請求項4記載の合成システム。
【請求項6】
前記芳香族化合物がアルケンを経由して合成されたものである請求項1〜5のいずれかに記載の合成システム。
【請求項7】
前記アルコールを固体酸触媒に接触反応させる請求項1〜6のいずれかに記載の合成システム。
【請求項8】
前記固体酸触媒が、ゼオライト、アルミナ及びチタン化合物からなる群より選択される少なくとも1種の固体酸である請求項7記載の合成システム。
【請求項9】
前記固体酸触媒がMFI型ゼオライトである請求項7又は8記載の合成システム。
【請求項10】
前記アルコールを固体酸触媒に接触反応させ、得られた生成物を循環させて更に前記固体酸触媒に接触反応させる請求項1〜9のいずれかに記載の合成システム。
【請求項11】
前記生成物を蒸留し、目的物以外の化合物を循環させて更に前記固体酸触媒に接触反応させる請求項10記載の合成システム。
【請求項12】
前記生成物を蒸留し、得られた蒸留物をベンゼンの融点以下に冷却してベンゼンを回収し、ベンゼン以外の化合物を循環させて更に前記固体酸触媒に接触反応させる請求項10又は11記載の合成システム。
【請求項13】
前記循環が繰返し行われる請求項10〜12のいずれかに記載の合成システム。
【請求項14】
炭素数2以上のアルコールを原料としてブタジエンを合成する合成システム。
【請求項15】
請求項1〜13のいずれかに記載の合成システムで得られたアニリンを原料として合成されたタイヤ用ゴム薬品。
【請求項16】
請求項1〜13のいずれかに記載の合成システムで得られたスチレン及び/又は請求項14記載の合成システムで得られたブタジエンを原料として合成されたタイヤ用合成ゴム。
【請求項17】
請求項15記載のタイヤ用ゴム薬品及び/又は請求項16記載のタイヤ用合成ゴムを用いた空気入りタイヤ。
【図1】

【図2】

【図3】
【図4】

【図2】
【図3】
【図4】
【公開番号】特開2012−153654(P2012−153654A)
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願番号】特願2011−14494(P2011−14494)
【出願日】平成23年1月26日(2011.1.26)
【出願人】(000183233)住友ゴム工業株式会社 (3,458)
【Fターム(参考)】
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願日】平成23年1月26日(2011.1.26)
【出願人】(000183233)住友ゴム工業株式会社 (3,458)
【Fターム(参考)】
[ Back to top ]