
合成ヘパラナーゼ分子及びその使用
本発明は、高収率異種発現系で発現させることができる、合成的に産生され、酵素的活性を有するヘパラナーゼ核酸分子及び該分子によりコードされるポリペプチドに関する。本明細書において、また、先行技術の方法と比較して高収率で生物学的活性を有するヘパラナーゼが産生される、異種発現系において哺乳類ヘパラナーゼを発現させる方法も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、高収率異種発現系で発現可能である、合成的に産生され、酵素的活性を有するヘパラナーゼ分子に関する。また、本明細書において、異種発現系で哺乳類ヘパラナーゼを発現させる方法も提供する。
【背景技術】
【0002】
ヘパラン硫酸プロテオグリカン(HSPGs)は、細胞外マトリックス(ECM)及び細胞表面上で見られる遍在性の高分子であり、細胞−細胞及び細胞−ECM相互作用の維持に寄与する。HSPGsは、タンパク質コアに共有結合するいくつかのヘパラン硫酸(HS)鎖から構成される。ヘパラン硫酸は、フィブロネクチン、ラミニン及びコラーゲンなどの構造的ECMタンパク質の、細胞表面及び他のECMタンパク質との結合を促進するが、このことから、自己集合及びECM成分の不溶性、細胞接着及び移動運動においてこのグリコサミノグリカンが果たす役割が示唆される。正しい細胞−細胞及び細胞−ECM相互作用の維持は重要であるため、細胞外環境においてHSPGsは、重大な構造的及び調整的役割を果たし、胚形成、形態形成及び発生から、炎症、血管新生及び癌転移にわたる範囲の重要な正常及び病的プロセスの調節を行う。
【0003】
上述の構造的及び細胞マトリックスのアンカー的役割に加えて、HSの構造多様性(Eskoら、J.Clin.Invest.108:169−173(2001);Turnbullら、Trends Cell Biol.11:75−82(2001))により、HSPGsは、増殖因子、酵素及びケモカインなどの様々な細胞外シグナル伝達タンパク質と相互作用することが可能となる。繊維芽細胞増殖因子(FGF1及びFGF2)、血管内皮細胞増殖因子(VEGF)、肝細胞増殖因子、形質転換成長因子β及び血小板由来増殖因子などの増殖因子は、腫瘍成長、侵襲及び血管新生において重要な役割を果たす。これらのシグナル伝達分子の拠点としての作用、これらの活性化又は安定化に加えて、HSPGsは、FGF受容体の多様なアイソフォームへのFGF2の結合など、リガンド−受容体相互作用に関与し得る(Changら、FASEB J.14:137−144(2000))。
【0004】
ヘパラン硫酸は、血小板、胎盤栄養膜細胞及び白血球により放出されるエンドβ−D−グルクロニダーゼヘパラナーゼにより分解される。ヘパラナーゼは、加水分解酵素メカニズムを介してグリコシド結合を切断することにより、特異的にヘパラン硫酸を分解する。この分解の結果、bFGF、ウロキナーゼプラスミノーゲン活性化因子(uPA)及び組織プラスミノーゲン活性化因子(tPA)が放出されるが、これらにより、血管新生が開始され得るか、又はECM分解が可能となり得る。さらに、ヘパラナーゼによるHS切断により、基底膜(BM)を介して細胞が移動し、ECM障壁を横断できるようになる。HS分解は、細胞外の変化に細胞が素早く反応することができるようにすることにより、多数の生理学的プロセスにおいて重要な役割を果たす。従って、ヘパラナーゼ活性の阻害は、炎症、転移及び自己免疫疾患など、細胞移動の変化に関係する病変に影響を及ぼし得る。
【0005】
このような極めて重要な役割のために、ヘパラナーゼは、抗腫瘍、抗転移又は抗炎症剤の開発のための、有力な新規ターゲットとなる。ヘパラナーゼは、単一遺伝子産物であり、関連タンパク質の複合ファミリーの一部ではないと思われるので、薬物開発の目的に対して、ECM−調節酵素でもあるマトリックスメタロプロテアーゼを凌ぐ顕著な利点を有する。信頼できる高スループットのアッセイが不足していることと、その複雑な生合成との両方により、現在、薬物ターゲットとしてヘパラナーゼを活用することには障害が多く、活性タンパク質を大量に産生させることが困難となっている。
【0006】
ヒトヘパラナーゼcDNAは、小胞体(ER)への移動の際にシグナルペプチダーゼにより除去されるシグナルペプチド配列を有する、プレプロタンパク質として最初に合成されるタンパク質をコードする。得られる65kDaのプロ型は、さらに、157個のN−末端アミノ酸を除去することによりプロセシングされ、成熟型の50kDヘパラナーゼとなる。この50kDタンパク質は、非プロセシング型の65kDa前駆体よりも少なくとも100倍高い特異的活性を有する(Vlodavskyら、Nat.Med.5:793−802(1999))。興味深いことに、この50kDaタンパク質は、哺乳類細胞などで発現される場合、不活性である(Hulettら、Nat.Med.5:803−809(1999))。この酵素の活性型が、未同定のタンパク質分解酵素による6kDaの介在ペプチドの除去により生じる、50kDa断片と8kDa断片との間のヘテロ二量体から構成されることが提案された(Fairbanksら、J.Biol.Chem.274:29587−29590(1999))。この仮説と一致して、McKenzieら(Biochem J.373:423−435(2003))は、昆虫細胞において活性ヘテロ二量体ヘパラナーゼを産生させ、8kDaサブユニットがヘパラナーゼ活性に必要であることを確認した。
【0007】
内因性ヘパラナーゼは、様々なソースから精製することができる。しかし、ヘパラナーゼ発現レベルが低い場合は、煩雑で費用の高い精製法が必要となる。例えば、Toyoshima及びNakajima(J.Biol.Chem.274:24153−24160(1999))は、4つの異なるクロマトグラフィー段階を必要とし、少なくとも5日間かかる、血小板からの内因性ヒトヘパラナーゼの精製プロセスについて述べた。
【0008】
内因性ヘパラナーゼの精製に対する他の欠点は、全体的な収率が特徴的に低いことである。例えば、Fairbanksら(J.Biol.Chem.274,29587−29590(1999))は、血小板からヘパラナーゼをたった22μgのみ、6%の回収率で精製したことについて報告している。同様に、Fuks及び共同研究者ら(米国特許第5,362,641号)は、ヒトヘパトーマ細胞株Sk−Hep−1由来のタンパク質 1.4kgからヘパラナーゼを4000倍精製し、1.9%の回収率で、精製ヘパラナーゼタンパク質をわずか6.5μgのみ調製したことについて述べている。同じ細胞株からのヘパラナーゼの240,000倍精製が、Peckerら(米国特許第5,968,822号)により開示された。しかし、このプロセスには、細胞培養物 500リットル超が必要であった。
【0009】
ヒトヘパラナーゼ遺伝子の同定及びクローニング(Vlodavksyら、Nature Med.5:793−802(1999);Hulettら、Nature Med.5:803−809(1999);Toyoshima及びNakajima,J.Biol.Chem.274:24153−24160(1999))により、異種発現系におけるヘパラナーゼタンパク質の組み換え発現が可能となった。しかし、そのような異種発現系について、ヘパラナーゼ産生に関して、重大な欠点が知られている。例えば、Ben−Artziら(WO99/57244)は、細菌、哺乳類、酵母及び昆虫細胞における組み換えヒトヘパラナーゼの発現について述べている。ヘパラナーゼ発現は達成されたが、E.コリが宿主細胞であった場合、その組み換えタンパク質に付随する酵素活性は検出されず、酵母Pichia pastoris(ピキア パストリス)でヘパラナーゼを発現させた場合、70kDaの非プロセシング前駆体のみが検出された。
【0010】
Ben−Artzi及び共同研究者ら(前出)はまた、哺乳類細胞、すなわち、ヒト腎臓繊維芽細胞(293)、仔ハムスター腎臓細胞(BHL21)及びチャイニーズハムスター卵巣細胞(CHO)における組み換えヘパラナーゼの発現について述べている。しかし、これらの発現系は、効率が低く、必要なコストが高いことが知られている。さらに、活性を有する成熟型タンパク質を得るための組み換え全長前駆体のプロセシングがこれらの細胞で観察されるという事実にもかかわらず、このプロセシング反応が効率的でないため、均質的にプロセシングされたタンパク質は得られない。さらに、ヘパラナーゼの分泌を促進する発現ベクターを使用しても、CHO細胞の馴化培地において組み換えヘパラナーゼの産生は導かれず、ヘパラナーゼを分泌するために、カルシウムイオノフォア又はPMAを添加することにより、さらに刺激を行う必要がある。この系で正しくプロセシングされるのは、分泌されたタンパク質のごく一部のみであると思われる。
【0011】
Sf21又はHigh Five(ハイファイブ)細胞などの昆虫細胞発現系におけるヘパラナーゼの産生が、この技術分野において述べられている(WO99/57244、WO99/11798、米国特許第5,968,822号;米国特許第6,348,344号;及び米国特許第6,190,875号)。しかし、増殖培地へと効率的に分泌されることがこのような方法で観察されるにもかかわらず、この酵素の特異的活性は非常に低く、正しいプロセシングは観察されなかった。例えば、Ben−Artziら(WO99/57244)は、昆虫細胞発現系で正しくプロセシングされたヘパラナーゼを生成させるために、そのヘパラナーゼタンパク質の位置119又は157の下流にプロテアーゼ切断部位を導入することについて述べている。しかし、これらのコンストラクトは、酵素的に活性であることが示されなかった。
【0012】
McKenzieら(前出)は、昆虫細胞における活性を有するヘテロ二量体ヘパラナーゼの産生について述べた。しかし、この系は、2種類の異なる組み換えタンパク質(8kDa及び50kDaサブユニット)を同時に生成させる必要があるという欠点を有する。単離した8kDa及び50kDaドメインの混合の結果、ヘパラナーゼは活性化されないので、同時発現により活性を有するヘテロ二量体を成功裡に回収できるか否かは、おそらく、そのヘテロ二量体複合体の共翻訳的な形成に依存する。この複合体をグリカナーゼで処理することにより、それが解離し、50kDaサブユニットが沈降するが、このことから、安定性及び溶解性に乏しいことが示唆される。
【0013】
活性又は不活性型でヘパラナーゼを得るための上述の方法にもかかわらず、高収率、低コストの異種発現系において発現させることができる、生物学的に活性を有するヘパラナーゼ分子を生成させることは利益があると思われる。前記分子は、転移成長及び/又は炎症を阻害及び/又は治療する、治療又は医薬品の開発のための阻害剤スクリーニングアッセイにおいて使用することができる。
【発明の開示】
【0014】
本発明は、高収率の異種発現系で発現させることができる、生物学的に活性を有する哺乳類ヘパラナーゼをコードする合成核酸分子を提供する。本明細書で提供される合成ヘパラナーゼ分子は、哺乳類系で発現が低レベルであり異種発現系で正しくプロセシングされない野生型ヘパラナーゼを凌ぐ顕著な長所を与える。本発明の合成分子は、転移成長、自己免疫疾患及び/又は炎症を阻害及び/又は治療するための治療又は医薬品開発のための、阻害剤スクリーニングアッセイにおいて使用することができる。
【0015】
本発明のある態様において、上記の合成核酸分子は、哺乳類ヘパラナーゼタンパク質をコードするヌクレオチド配列を含有し、このヌクレオチド配列は、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、この切断部位は、本ヘパラナーゼタンパク質の残基100及び168をコードするヌクレオチドの間に位置する。前記核酸分子は、適切な酵素とともにインキュベーションすると生物学的活性が可能となる、ヘパラナーゼタンパク質をコードする。
【0016】
本発明はさらに、哺乳類ヘパラナーゼタンパク質をコードする部分を含有し、このタンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチド配列と、から構成され、該N−末端及びC−末端断片が、野生型ヘパラナーゼ断片と実質的に同様であるタンパク質断片をコードし、該コードされるヘパラナーゼタンパク質が構成的に活性を有する、合成哺乳類ヘパラナーゼ核酸分子に関する。
【0017】
合成的に産生される、生物学的に活性を有する哺乳類ヘパラナーゼポリペプチド、及び適切な酵素とインキュベーションすると生物学的活性が可能となる、エンドプロテアーゼコンセンサス切断部位を含むヘパラナーゼポリペプチドもまた本明細書で提供する。
【0018】
本発明はさらに、結果として生物学的に活性を有するヘパラナーゼが高レベルで発現される、異種発現系において哺乳類ヘパラナーゼを発現させるための方法を提供する。
【0019】
本明細書を通して、及び添付の請求項で使用される場合、単数形「a」、「an」及び「the」は、他に文脈が明確に指示を与えない限り、複数形の意味を含む。
【0020】
本明細書を通して、及び添付の請求項で使用される場合、次の定義及び略語を適用する。
【0021】
「保存的アミノ酸置換」は、あるアミノ酸残基を別の化学的に同等のアミノ酸残基に置換することを意味する。そのような保存的置換の例は、ある疎水性残基(イソロイシン、ロイシン、バリン又はメチオニン)を別のものへ置換すること;ある極性残基を同様の電荷の別の極性残基へ置換すること(例えば、リジンに対するアルギニン;アスパラギン酸に対するグルタミン酸)である。
【0022】
「哺乳類」という用語は、ヒトを含むあらゆる哺乳動物を意味する。
【0023】
「治療」という用語は、治療的処置及び予防的(Prophylactic又はPreventative)手段の両方を意味する。治療を必要とする者には、既に疾患に罹患している者ならびにその疾患に罹患する傾向がある者又はその疾患が予防されるべきである者が含まれる。「疾患」とは、本明細書中に記載の核酸分子及びポリペプチドを用いて同定した分子による治療が役立つあらゆる状態である。このような疾患には、これらに限定されないが、癌、炎症及び自己免疫疾患が含まれる。
【0024】
「ベクター」という用語は、DNA断片を宿主生物又は宿主組織に導入することができるある手段を意味する。プラスミド、ウイルス(アデノウイルスを含む。)、バクテリオファージ及びコスミドをはじめとした様々な種類のベクターがある。
【0025】
「生物学的に活性を有する」とは、タンパク質が、天然に生じる分子又はそのアイソフォームに付随する構造的、調整的又は生物化学的機能を有することを意味する。ヘパラナーゼとの関連において、「生物学的に活性を有する」タンパク質は、ヘパラナーゼ酵素活性を含む。
【0026】
「実質的に同様」とは、ある配列が参照配列と、少なくとも80%、好ましくは90%、より好ましくは95%及びさらにより好ましくは99%の相同性を共有することを意味する。本発明において、この参照配列は、この文章の文脈で示される場合、全長ヒトヘパラナーゼヌクレオチドもしくはアミノ酸配列又は8kDa(配列番号15)もしくは50kDa(配列番号16)ヘパラナーゼフラグメントのヌクレオチドもしくはアミノ酸配列であり得る。このように、8kDaヒトヘパラナーゼ断片(配列番号15)と「実質的に同様」であるヘパラナーゼタンパク質配列は、この8kDaヒトヘパラナーゼ断片と、少なくとも80%の相同性、好ましくは90%の相同性、より好ましくは95%の相同性、さらにより好ましくは99%の相同性を共有することとなる。あるヘパラナーゼタンパク質又はヌクレオチド配列が参照配列に対して「実質的に同様」であるか否かは、例えば、University of Wisconsin Genetics Computer Group(UWGCG)から入手できるGAPコンピュータープログラム、バージョン6.0などの配列解析ソフトウェアを用いた比較配列情報により決定することができる。GAPプログラムは、Smith及びWaterman(Adv.Appl.Math.2:482,1981)により改訂された、Needleman及びWunsch(J.Mol.Biol.48:443,1970)のアラインメント法を利用する。
【0027】
「遺伝子」は、そのヌクレオチド配列がポリペプチド分子をコードする核酸分子を意味する。遺伝子は、中断されないヌクレオチド配列であり得るか、又は、イントロン、プロモーター領域、スプライシング部位及び反復配列などの介在部分を含み得る。遺伝子は、RNAであってもDNAであってもよい。好ましい遺伝子は、本発明のペプチドをコードするものである。
【0028】
「核酸」又は「核酸分子」という用語は、リボ核酸(RNA)又はデオキシリボ核酸(DNA)、プローブ、オリゴヌクレオチド、断片又はそれらの一部及びプライマーを意味するものとする。DNAは、相補的DNA(cDNA)又はゲノムDNA、例えば、本発明のペプチドをコードする遺伝子のいずれでもあり得る。
【0029】
「野生型ヘパラナーゼ」又は「野生型タンパク質」もしくは「wtタンパク質」は、アミノ酸又はそれらの変異型の天然に生じる配列を含むタンパク質を意味する。野生型ヒトヘパラナーゼのアミノ酸配列は、本分野において入手可能である(Vlodavksyら、Nature Med.5:793−802(1999);Hulettら、Nature Med.5:803−809(1999);Toyoshima及びNakajima,J.Biol.Chem.274(34):24153−24160(1999);(参照により、全体を本明細書に組込む。))。
【0030】
「野生型ヘパラナーゼ遺伝子」は、ヒト由来のタンパク質又は、ショウジョウバエ(Drosophila)などの昆虫、ツメガエル(Xenopus)などの両生類及びラット、マウス及びアカゲザルなどの哺乳類を含むがこれらに限定されない、別の生物から得られたタンパク質をはじめとする、天然に生じるヘパラナーゼタンパク質をコードするヌクレオチド配列を含有する遺伝子を意味する。本ヒトヘパラナーゼ遺伝子のヌクレオチド配列は、本分野で入手可能である(Genbank Accession No.AF155510;Toyoshima及びNakajima、前出(参照により、全体を本明細書に組込む。))。
【0031】
「実質的に他のタンパク質不含である」又は「実質的に精製された」とは、少なくとも90%、好ましくは95%、より好ましくは99%及びさらにより好ましくは99.9%、他のタンパク質が含まれていないことを意味する。したがって、実質的に他のタンパク質不含のヘパラナーゼタンパク質調製物は、その総タンパク質のパーセンテージとして、非ヘパラナーゼタンパク質が、10%を超えない、好ましくは5%を超えない、より好ましくは1%を超えない、さらにより好ましくは0.1%を超えない。あるヘパラナーゼタンパク質調製物が実質的に他のタンパク質不含であるか否かは、例えば、適切な検出方法(例えば銀染色又は免疫ブロッティングなど)と組み合わせたドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS−PAGE)などの従来のタンパク質純度評価技術により決定することができる。
【発明を実施するための最良の形態】
【0032】
ヘパラナーゼは、加水分解酵素メカニズムを介してグリコシド結合を切断することによりヘパラン硫酸(HS)を分解する哺乳類酵素である。HS分解は、細胞−細胞及び細胞−ECM相互作用を変更することで細胞外の変化に対して細胞が素早く反応できるようにすることによって、多くの生理学的プロセスにおいて重要な役割を果たす。これらの相互作用は重要なので、ヘパラナーゼ活性の阻害は、腫瘍細胞転移、T細胞介在遅延型過敏及び自己免疫疾患などのいくつかの病態に影響を及ぼし得る。
【0033】
いくつかの証拠から、ヘパラナーゼが腫瘍細胞転移に関与することが示唆される。第一に、ヘパラナーゼの発現レベルは、いくつかの腫瘍及び腫瘍細胞株の転移能と相関がある。第二に、侵襲性の強い転移性疾患の患者では、その尿中において測定可能なヘパラナーゼ活性を有する。この所見は、全ての癌患者で見られるものではない。さらに、非−抗凝血剤ヘパリン誘導体によるヘパラナーゼ活性の阻害により、B16メラノーマ、ルイス肺癌及び乳癌細胞による転移発生率が低下した。最後に、ヒトヘパラナーゼ遺伝子を用いた非転移性のマウス細胞に対する形質移入の結果、2種類のマウスモデルにおいて、死亡率及び転移発生が増加した。
【0034】
ヒトヘパラナーゼは、いかなる他の既知のタンパク質とも実質的な相同性を共有しない。その発見時において、ヘパラナーゼ遺伝子が遺伝子ファミリーのメンバーではなく、むしろ、HSPG分解に関与する単一遺伝子又は少なくとも主要なエンドグルクロニダーゼであることが示唆された。アミノ酸レベルで35%の相同性を共有する第二のヘパラナーゼ(hpa2)が後に同定されたが、しかし、hpa2は、その組織分布に基づき、異なる機能を果たすと思われる。転移成長におけるヘパラナーゼの役割を示す上記の証拠と合わせて、類似の作用を遂行する密接に関連するタンパク質が欠如していることにより、へパラナーゼはこれらの領域における治療法の開発のための優れたターゲットとなる。
【0035】
図1は、ヒトヘパラナーゼの生合成を示す。簡潔に述べると、ヘパラナーゼcDNAは、ERへの移動時にシグナルペプチダーゼにより除去されるシグナルペプチド配列(残基Met1−Ala35)を有するプレプロタンパク質として最初に合成されるタンパク質をコードする。その結果生じる65kDaプロ形態は、さらに、157のN−末端アミノ酸を除去することによりプロセシングされ、成熟型の50kDヘパラナーゼ(配列番号16)となる。この50kDタンパク質は、非プロセシング型の65kDa前駆体よりも少なくとも100倍高い特異的活性を有する(Vlodavskyら、Nat.Med.5:793−802(1999))。この酵素の活性型が、未同定のタンパク質分解酵素により6kDaの介在ペプチド(残基Glu109−Gln157)(以下、「介在断片」又は「6kDa断片」と呼ぶ。)を除去することで生じる、50kDa断片と8kDa断片(配列番号15)との間のヘテロ二量体であることが提案された(Fairbanksら、J.Biol.Chem.274:29587−29590(1999))。
【0036】
最近の証拠により8kDaサブユニット(配列番号15)がヘパラナーゼ活性に必要であることが示されているにもかかわらず(McKenzieら、Biochem J.373:423−435(2003))、ヘパラナーゼの活性化プロセスにおけるこの8kDaサブユニットの役割は、本明細書中で開示される研究以前は、不明確なままであった:これは、不可欠なサブユニットとして働く、又は、あるいは、シャペロンとして作用する可能性があり、この機能が遂行された後は不必要となり得る。また、8kDaサブユニット以外の他の成分がヘパラナーゼ活性の誘導に必要であるか否かも明確ではなかった。
【0037】
多配列アラインメント及び二次構造予想から、いくつかのグリコシダーゼで見られるように、そのタンパク質がTIMバレルフォールドを取ることによるヒトヘパラナーゼのモデルが導かれる(Hulettら、Biochemistry 39:15659−15667(2000))。この共通フォールドモチーフは、通常、8個の交互のα−へリックス及びβ−鎖からなる。50kDa断片内で、明確な相同性が観察されるのは、TIMバレルフォールドの3番目のα/βユニットからのみであるが、このことから、ヘパラナーゼが6個のみのα/βユニットからなる新規フォールドを取るか、又は、このタンパク質の他の部分が、欠落ユニットに寄与するか、のいずれかが示唆される。8kDa断片がこの欠落構造エレメントに寄与し得ることが主張された(Hulettら、前出)。
【0038】
この仮説に従い、本明細書中で述べるように、一緒に共有結合する8kDa及び50kDaサブユニットを有する1本鎖ヘパラナーゼ分子を設計するために、多配列アラインメント(図3A及び3B)に基づきヘパラナーゼの二次構造のモデルを確立した。本発明は、8kDa及び50kDa断片のリンカーとの連結の結果、タンパク質分解的プロセシングを必要としない構成的に活性を有する1本鎖ヘパラナーゼ分子が得られることを示す。本発明の典型的な実施形態において、Hirudinaria manillensis(ヒルディナリア マニレンシス)ヒアルロニダーゼ由来のループを移植することにより、又は、3つのグリシン−セリンリピートを含有するリンカーを用いて、この2個の断片を連結した。
【0039】
また、6kDa介在断片のN及びC−末端周囲のエンドプロテアーゼ切断部位を改変することにより、少なくとも部分的に精製されたタンパク質の両部位におけるタンパク質分解性のプロセシングによって、他の成分なしで、ヘパラナーゼ活性化が導かれることが、本明細書において示された。本発明のこの態様の典型的実施形態において、タバコエッチウイルスプロテアーゼ切断部位をこの6kDa介在断片のN及びC−末端に付加し、その結果、コードされるタンパク質の精製又は部分精製及びそれに続く適切な酵素とのインキュベーション後に、活性を有するヘパラナーゼが得られる。本発明は、ヒトヘパラナーゼが標準的なTIMバレルフォールドを取ることの証拠を提供し、有利に、特異的な阻害剤の同定のための活性酵素分子の容易な産生方法を提供する。
【0040】
異種発現系、特に昆虫細胞において生物学的に活性を有するヘパラナーゼを発現させるための、本発明の改変タンパク質、核酸分子及び方法は、特徴的に、0.5mg/lから5.0mg/lの回収率となる。さらに、これらのタンパク質は、効率的に、増殖培地に分泌され、哺乳類細胞においては、細胞内部又は細胞膜に会合して、真正のヒト酵素が主に得られる(Vlodavskyら、Semin.Cancer Biol.12:121−129(2002))。
【0041】
従って、本発明は、活性を有する哺乳類ヘパラナーゼをコードする合成核酸分子に関し、その核酸分子は、高収率異種発現系で発現させることができる。本明細書で提供される合成ヘパラナーゼ分子は、哺乳類系において発現レベルが低く、異種発現系において正しくプロセシングされない野生型ヘパラナーゼを凌ぐ顕著な長所を与える。本発明の合成分子は、転移成長及び/又は炎症を阻害及び/又は治療する治療又は医薬品の開発のための阻害剤スクリーニングアッセイにおいて使用することができる。前記合成分子はまた、自己免疫疾患の治療及び/又は予防のための治療又は医薬品開発においても有用である。
【0042】
本発明のある態様において、哺乳類ヘパラナーゼタンパク質をコードするヌクレオチド配列を含有する合成核酸分子が提供され、該ヌクレオチド配列は、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、それらの切断部位は、本ヘパラナーゼタンパク質の残基100及び168をコードするヌクレオチドの間に位置する。本発明のこの態様は、野生型ヘパラナーゼの生合成と同様の、本ヘパラナーゼタンパク質のタンパク質分解性プロセシングを遂行し、結果として生物学的に活性を有する酵素を得るための方法において使用することができる合成核酸分子を提供する。
【0043】
上記核酸分子によりコードされる実質的に純粋なポリペプチドもまた、本明細書で提供する。
【0044】
本発明の好ましい実施形態において、本哺乳類ヘパラナーゼタンパク質は、ヒトヘパラナーゼである。
【0045】
この2個のコンセンサス切断部位は、コードされるタンパク質の精製又は部分精製及び適切な酵素とのインキュベーション後に、その結果得られる断片が、野生型8kDa断片(配列番号15)と実質的に同様の少なくとも1つの断片と、野生型50kDa断片(配列番号16)と実質的に同様の少なくとも1つの断片と、を含有するという条件において、本ヘパラナーゼタンパク質の残基100と168との間のいずれかの位置に導入することができる。本発明の好ましい実施形態において、本コンセンサス切断部位は、本ヒトヘパラナーゼタンパク質の残基G110及びK158の前に位置し、その結果、コードされるタンパク質の精製又は部分精製及びそれに続く適切な酵素とのインキュベーション後に、8kDaの第一の断片、6kDaの第二の「介在」断片及び50kDaの第三の断片が得られる。
【0046】
活性を有するヘテロ二量体ヘパラナーゼを得るために、これらに限定されないが、タバコエッチウイルス、ピコルナウイルスの3Cプロテアーゼ、トロンビン、血液凝固因子Xa及びエンテロキナーゼからの切断部位を含む、何らかのエンドプロテアーゼに対応する切断部位を、ヘパラナーゼ分子に導入することができることは、当業者により理解される。本発明の好ましい実施形態において、前記切断部位は、タバコエッチウイルス由来である。
【0047】
本発明の別の態様において、哺乳類ヘパラナーゼタンパク質をコードする部分を含有し、そのタンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチド配列とから構成される、構成的に活性を有する1本鎖哺乳類ヘパラナーゼ核酸分子が提供される。本発明のこの態様は、タンパク質分解性プロセシングなしで構成的に活性を有するヘパラナーゼをコードし、その6kDa「介在」断片が実質的に除去されるように改変され、その介在断片がより小さいリンカーで置換される、合成遺伝子を提供する。
【0048】
本発明のこの態様の好ましい実施形態において、本哺乳類ヘパラナーゼタンパク質は、ヒトヘパラナーゼである。
【0049】
上記の構成的に活性を有する1本鎖哺乳類ヘパラナーゼ遺伝子によりコードされる精製された合成ヘパラナーゼタンパク質もまた、本明細書で提供される。
【0050】
本発明のこの態様において、約1残基から約67残基からなるペプチドをコードする何らかの配列を、リンカーとして使用することができる。前記リンカーは、合成するか、又は天然に生じるソースから単離することができる。本発明の典型的な実施形態において、本リンカーは、ヒアルロニダーゼタンパク質の中央ループ領域をコードするヌクレオチド配列を含む。このヒアルロニダーゼは、H.マニレンシス(H.manillensis)由来であることがが好ましい。他の実施形態において、本リンカーは、(GlySer)3リンカーをコードするヌクレオチド配列を含む。
【0051】
本発明はさらに、本明細書を通じて開示される合成核酸分子を含有する組み換えベクターに関する。これらのベクターは、DNA又はRNAからなるものであり得る。殆どのクローニング目的に対して、DNAベクターが好ましい。代表的なベクターには、組み換えヘパラナーゼタンパク質をコードすることができる、プラスミド、改変ウイルス、バキュロウイルス、バクテリオファージ、コスミド、酵母人工染色体及びエピソーム性のDNAもしくは統合DNAの他の型が含まれる。特定の遺伝子導入又は他の使用のための適切なベクターを決定することは、十分に熟練者の範囲内にある。
【0052】
本明細書を通じて開示される本合成核酸分子を含有する発現ベクターは、組み換え宿主細胞における哺乳類ヘパラナーゼの高レベル発現に使用し得る。発現ベクターには、これらに限定されないが、クローニングベクター、改変クローニングベクター、特に設計したプラスミド又はウイルスが含まれ得る。また、様々な細菌発現ベクターも、必要ならば、細菌細胞において組み換えヘパラナーゼを発現させるために使用し得る。さらに、真菌細胞において組み換えヘパラナーゼを発現させるために、様々な真菌細胞発現ベクターを使用し得る。さらに、昆虫細胞において組み換えタンパク質を発現させるために、様々な昆虫細胞発現ベクターを使用し得る。本発明の好ましい実施形態において、ベクターは、バキュロウイルスベクターである。
【0053】
本発明はまた、本発明の合成核酸分子を含有するベクターを用いて形質転換又は形質移入を行った宿主細胞にも関する。組み換え宿主細胞は、原核又は真核であり得、これらに限定されないが、E.コリなどの細菌、これらに限定されないがPichia pastoris(ピキア パストリス)、Hansenula polymorpha(ハンセヌラ ポリモルファ)及びSaccharomyces cervisiae(サッカロミセス セルビシエ)を含む酵母などの真菌細胞及び、これらに限定されないがショウジョウバエ(Drosophila)及びカイコ由来の細胞株を含む昆虫細胞が含まれる。このような組み換え宿主細胞は、哺乳類ヘパラナーゼ又は生物学的に同等の型を高レベルで産生させるための適切な条件下で培養することができる。本明細書中で定義する場合、「宿主細胞」という用語は、トランスジェニックヒト、トランスジェニックヒト胎児又はトランスジェニックヒト胚の身体における宿主細胞を含むものではない。
【0054】
上記のごとく、本発明の合成分子は、高収率異種発現系で発現させることができるので、先行技術を凌ぐ顕著な長所を提供する。本明細書で提供される合成分子によりコードされるヘパラナーゼタンパク質は、正しくプロセシングされ、酵素的に活性を有し、高レベルで発現される。従って、本発明の好ましい実施形態において、選択される宿主細胞は、これらに限定されないが、昆虫細胞、細菌細胞及び酵母細胞を含む高収率異種発現系の一部である。本発明の特に好ましい実施形態において、宿主細胞は、昆虫細胞である。
【0055】
本発明はまた、本明細書を通じて開示される核酸分子を含有する、組み換えベクター及び組み換え宿主細胞(両者とも原核及び真核である。)に関する。本発明の合成核酸分子、関連ベクター及び宿主は、癌、炎症及び/又は自己免疫の治療のために役立つ、ヘパラナーゼ活性の阻害剤を同定するためのスクリーニングアッセイにおいて有用である。
【0056】
本発明の別の態様において、(a)哺乳類ヘパラナーゼタンパク質をコードし、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、該切断部位が該ヘパラナーゼタンパク質の残基100及び残基168をコードするヌクレオチドの間に位置するヌクレオチド配列を含有するベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で宿主細胞を培養することと;
(c)前記細胞を崩壊させて、前記ヘパラナーゼタンパク質を少なくとも部分的に精製することと;
(d)少なくとも部分的に精製したタンパク質をエンドプロテアーゼに曝露し、該ヘパラナーゼタンパク質をコンセンサス切断部位で切断することと、を含む、非哺乳類細胞において哺乳類ヘパラナーゼを発現させる方法が提供される。
【0057】
本発明はまた、上記の方法により産生された、実質的に精製されたタンパク質も提供する。
【0058】
本発明のこの態様の好ましい実施形態において、本哺乳類ヘパラナーゼは、ヒトヘパラナーゼである。さらに好ましい実施形態において、本コンセンサス切断部位は、ヒトヘパラナーゼの残基G110及びK158の前に位置する。
【0059】
別の好ましい実施形態において、本切断部位は、タバコエッチ(tobacco etch)タンパク質切断部位である。
【0060】
本明細書において、(a)ヘパラナーゼタンパク質をコードする部分を含有し、該タンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーをコードするヌクレオチド配列と、約50kDaのC−末端断片をコードするヌクレオチド配列とから構成される、合成哺乳類ヘパラナーゼ遺伝子を含有するベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で宿主細胞を培養することと、を含む、非哺乳類細胞において、1本鎖の、構成的に活性を有する哺乳類ヘパラナーゼを発現させる方法もまた提供される。
【0061】
本明細書において、上記の方法により産生された、実質的に精製されたタンパク質もまた提供される。本発明のさらなる実施形態において、本タンパク質は、野生型ヘパラナーゼに特異的な抗体に結合することができる。
【0062】
本発明のこの態様の好ましい実施形態において、本リンカーは、ヒアルロニダーゼタンパク質の中央ループ領域を含有する。別の好ましい実施形態において、本リンカーは、(GlySer)3ペプチドを含有する。
【0063】
本明細書中で言及した公表物は全て、本発明に関連して使用され得る方法及び材料を説明し開示する目的のために、参照により本明細書に組込む。明細書のいかなる開示内容も、本発明が先行技術の効力によってこのような開示に対して先行する権利を与えられていないことを認めるものではない。
【0064】
付随する図面を参照して本発明の好ましい実施形態を説明してきたが、本発明がこれらの明確な実施形態に限定されず、添付の請求項において定義される本発明の範囲又は精神から逸脱することなく、当業者によりそれらにおいて様々な変更及び改変が行われ得ることを理解されたい。
【0065】
次の実施例は、説明するものであり、本発明を限定するものではない。
【実施例1】
【0066】
ヒト胎盤cDNAライブラリーからのヘパラナーゼのクローニング
TaKaRa Taqポリメラーゼ(TaKaRa Bio Inc.,Otsu,Shiga,Japan)を用いて、PCRにより、ヒトヘパラナーゼ(Accession No.AF155510)を正常ヒト胎盤cDNAライブラリー(Invitrogen Corp.,Carlsbad CA)から増幅した。緩衝液条件は、供給者により示されたものであった。cDNAテンプレートのPCR増幅は、94℃、1分間を1サイクル、その後、94℃にて30秒間、57℃にて30秒間及び68℃にて110秒間を35サイクルで行うように構成した。増幅した断片をゲル精製し、リン酸化し、充填後、pFAST BAC1のBamHI部位(バキュロウイルス発現)、又は、BamHI/EcoRI消化したpcDNA3ベクター(哺乳類細胞発現)のいずれかにクローニングした。PCR増幅及びKozak配列の同時最適化のために次のプライマーを使用した:hHEP1−24 BamHI opti
【0067】
【化1】
;及びhHEP rev 1632
【0068】
【化2】
【実施例2】
【0069】
1本鎖ヘパラナーゼ分子の構築
次のコンストラクト(hepWT、hep109(配列番号19、タンパク質配列番号20に対応)、hep106(配列番号18、タンパク質配列番号17に対応)、hepGS3(配列番号22、タンパク質配列番号21に対応)、hepGS6(配列番号24、タンパク質配列番号26に対応)、hepGS4(配列番号23、タンパク質配列番号25に対応)及びhepHyaluro(配列番号28、タンパク質配列番号27に対応)、直接的に8及び50kDaサブユニットを共有結合で連結しているもの(hep109及びhep106)、これらのサブユニットをグリシン−セリンスペーサーを介して連結しているもの(hepGS3、hepGS4及びhepGS6)又は酵素ヒアルロニダーゼからのループ領域を移植することにより連結しているもの(Hyaluro))を、指示したプライマーを用いて標準的なPCR突然変異誘発により生成させた:
hHEP1−24 BamHI opti(配列番号1)及びhHEP rev 1632(配列番号2)
hep109 M1_E109−Q157_I543
突然変異誘発プライマー:hHEP 304/504
【0070】
【化3】
hep106 M1_P106−K158_I543
突然変異誘発プライマー:hHEP 291/504 bis
【0071】
【化4】
hepGS3 M1_E109−(GS)3−Q157_I543
突然変異誘発プライマー:hHEP 304(GS3)504
【0072】
【化5】
hepGS6 M1_E109−(GS)6−Q157_I543
突然変異誘発プライマー:hHEP 304(GS6 Ala)
【0073】
【化6】
hepGS4 M1_W118−(GS)4−E143_I543
突然変異誘発プライマー:hHEP329(GS4 Ala)
【0074】
【化7】
hepHyaluro M1_W118−(AFKDKPT)(配列番号8)−E143_I543
【0075】
突然変異誘発プライマー:hHEP Hyaluro 5’−
【0076】
【化8】
【実施例3】
【0077】
改変プロテアーゼ切断部位を有するヘパラナーゼ分子の構築
アミノ酸E109とG110との間の、GSリピート(E109−GSGSENLYFQ−GSG−G110(配列番号10)、QとGとの間に切断されやすい結合がある。)に隣接する、タバコエッチウイルス(TEV)プロテアーゼに対するコンセンサス切断部位を挿入する改変ヘパラナーゼ分子を構築するために、テンプレートとしてwtヘパラナーゼを用い、プライマー、hHEP1−24 BamHI opti(配列番号1)及びhHEP rev 1632(配列番号2)及び突然変異誘発プライマー、TEV 110bis
【0078】
【化9】
を用いてPCR突然変異誘発を行った。
【0079】
残基E109/G110の間及びQ157/K158の間の両方にTEV−切断部位を有する改変ヘパラナーゼを構築するために、TEV110コンストラクト(配列番号30、タンパク質配列番号31に対応)をテンプレートとして使用し、PCR突然変異誘発により、突然変異誘発プライマー、TEV158 ter
【0080】
【化10】
を用いて、配列
【0081】
【化11】
を挿入し、hepTEV110/158(配列番号29及び32)を作製した。
【0082】
コンストラクトは全て、PCRにより突然変異が導入されていないことを確認するために両鎖において配列決定を行い、上述のようにpFASTBAC1にクローニングした。
【実施例4】
【0083】
COS7細胞におけるヘパラナーゼ分子の一時的発現。
【0084】
ダルベッコMEM(Gibco BRL、Gaithersburg、MD)中で細胞を増殖させた。全てのコンストラクトを、真核細胞発現プラスミドpcDNA3(Invitrogen)にクローニングした。各コンストラクトの形質移入効率を調べるために、レポーター遺伝子 β−ラクタマーゼ(BLA)をコードするベクターを共形質移入した。同程度の形質移入効率にするために形質移入した各ベクターの量を調整した。fuGENE6形質移入試薬(Roche,Basel,Switzerland)を用いて、製造者の説明書に従い、COS7細胞の一時的形質移入を行った。形質移入から24時間後、BLA−陽性細胞の蛍光検出により効率を評価した。形質移入から96時間後、細胞を回収し、完全プロテアーゼ阻害剤カクテル(Roche)を含有する細胞溶解緩衝液(50mM Tris−HCl pH7.5、150mM NaCl、0.5% Triton)中で再懸濁した。氷上で30分間、細胞溶解を行った。14000rpmで30分間遠心した後、ヘパラナーゼを含有する上清を回収し、下記で概説するように部分精製を行った。
【0085】
一時的形質移入により、ヘパラナーゼコンストラクトを、内因性へパラナーゼ活性を欠くCOS7細胞で発現させた。ヘパリンアフィニティークロマトグラフィーにより細胞溶解液からヘパラナーゼを抽出し、ウェスタンブロットで定量した。並行して、放射性又は蛍光分析のいずれかでヘパラナーゼ酵素活性を測定した(図5)。ウェスタンブロット分析から、wtヘパラナーゼならびに1本鎖コンストラクトGS3及びhyaluroが効率的に発現され、プロセシングされるが、一方で、コンストラクト106及びGS4は、発現されるがプロセシングされないと結論付けた。コンストラクト109及びGS6の発現レベルはきわめて低く、ウェスタンブロット分析により辛うじて検出できる程度であった。wt、GS3及びhyaluroコンストラクトのみが酵素活性を示した。発明者らは、1本鎖コンストラクト106及びGS4は不活性であり、一方、109及びGS6は、おそらく不安定であると結論付けた。GS3及びhyaluroは活性を有するが、切断部位に変更を導入したにもかかわらずプロセシングされるので、前駆体の固有の活性については結論を導くことはできなかった。従って、発明者らは、ヘパラナーゼプロセシングに関与する酵素を欠く細胞において発現を進めた。
【実施例5】
【0086】
昆虫細胞におけるヘパラナーゼ分子の発現。
【0087】
Bac to Bac発現系(Invitrogen)を用いて、ヘパラナーゼコンストラクトを含有する組み換えバキュロウイルスを作製した。10%FBSを含有するGrace昆虫培地で増殖させたSf9昆虫細胞(50x106細胞/T−175フラスコ)を感染させるために、組み換えバキュロウイルスを使用した。感染から48時間後に細胞を回収し、500gで5分間遠心した。COS7に対して使用した150mM NaClのかわりに、可溶性分画におけるタンパク質の量を増加させる、500mM NaClを含有する細胞溶解緩衝液を使用すること以外、上述のようにして、細胞溶解液を調製した。
【0088】
COS7細胞で産生した場合に酵素活性を示した3種類のヘパラナーゼコンストラクトをバキュロウイルス発現系に導入した。Sf9細胞においてタンパク質を発現させ、ヘパリンアフィニティークロマトグラフィーにより精製した。ウェスタンブロット分析により、COS7細胞で観察されたものと対照的に、wt又は突然変異ヘパラナーゼのプロセシングがこの発現系では起こらなかったことが示された。蛍光活性分析による精製1本鎖タンパク質の酵素活性の分析から、非プロセシングwt酵素活性は非常に低く、一方、非プロセシングGS3及びHyaluroタンパク質は、COS7細胞で産生された正しくプロセシングされた野生型酵素で観察されたものに匹敵する特異的活性を有し、活性が高いことが分かった。
【0089】
GS3及びhyaluroは、酵素活性のpH及びイオン強度依存性の点に関してCOS7細胞から抽出された野生型組み換え酵素と、又はHCT−116細胞から部分精製された真正のwt酵素と非常に似ており、ヘパリンにより同様の効力で阻害された。
【0090】
位置109/110及び109/110+157/158においてTEV切断部位を有するコンストラクトを発現させ、ヘパリンアフィニティーカラムで精製し、室温にて一晩、50mM Mes pH6.0、10% グリセロール、0.5mM EDTA中のTEVプロテアーゼ(0.5μM)を用いて消化した。両ケースにおいて完全なプロセシングが観察されたが、しかし、両方の切断ジャンクションにおいてTEV配列を有する二重変異体のみが、この処理により活性化され、このことから、E109/G110ジャンクションにおけるプロセシングのみでは、ヘパラナーゼ活性化を誘導するには十分でないことが示された。
【実施例6】
【0091】
ヘパリンセファロースアフィニティークロマトグラフィーによる組み換えヘパラナーゼコンストラクトの精製。
【0092】
COS7又はSf9昆虫細胞からの細胞溶解液を重力により500μl ヘパリンセファロース CL−6B(Amersham,Piscataway,NJ)に通した。細胞溶解緩衝液2mLでそのカラムを洗浄し、次に、50mM Tris−HCl、pH7.5、500mM NaCl 2mLで洗浄し、50mM Tris−HCl pH7.5、1M NaCl 2mLでヘパラナーゼを溶出し、Biomax−30K遠心分離濃縮装置(Millipore,Bedford,MA)で約5倍に濃縮した。10%グリセロールを添加し、−80℃にて、そのタンパク質を分注して保存した。BIO−RADタンパク質アッセイによりタンパク質濃度を測定した。
【実施例7】
【0093】
大規模発現及び精製
Sf21(又はSf9)細胞を血清不含培地(Sf−900 II SFM、Invitrogen)中での増殖に順応させた。1から10の間の様々な感染の多重度でヘパラナーゼコンストラクトをコードする組み換えバキュロウイルスにより細胞を感染させた。滅菌空気の一定気流状態下、27℃にて、感染細胞3Lをスピナーフラスコ中で増殖させた。感染48時間から96時間後、細胞を回収し、遠心により培地から分離した。合成及びwtヘパラナーゼが細胞ペレットと上清の両方で見られた。細胞ペレットから合成ヘパラナーゼを抽出するために、上記で概説したようにして細胞を溶解した。細胞溶解液又は未精製培地上清を0.22μフィルターで濾過し、50mM Tris−HCl pH7.5、150mM NaClで平衡化した20ml−HyperD Heparin カラム(Biosepra Inc.,Marlboro,MA)に載せた。50mM Tris−HCl pH7.5中の0.15Mから1M NaClの直線的グラディエントを用いて、合成又はwtヘパラナーゼを溶出した。組み換えタンパク質は、>500mMのNaCl濃度で溶出した。集めたヘパラナーゼ含有Heparin−カラム分画を一晩、50mM HEPES pH7.5に対して透析し、同じ緩衝液で平衡化したSource Sカラム(Amersham)に載せた。ヘパラナーゼコンストラクトは、400mMから600mM NaClで溶出した。15/30 Superdex 75サイズ排除カラムでのさらなるクロマトグラフィー段階により、タンパク質を均質になるように精製した。精製したタンパク質を分注し、液体窒素中で瞬時に冷凍し、−80℃で保存した。
【実施例8】
【0094】
ウェスタンブロッティング
50kDaサブユニット内に含有されるペプチド(アミノ酸225から241に対応し、そのC−末端にさらなる配列GGCを含有する、EPNSFLKKADIFINGSQ(配列番号14))に対して、ウサギポリクローナル抗体を作製した。チオプロピルセファロースレジン(Amersham)に固定化した免疫原ペプチドを用いて抗血清を免疫精製した。ヘパリンカラムから溶出したタンパク質10μlを10% SDS−ポリアクリルアミドゲル電気泳動に供し、Protran BA 83 Cellulosenitrate膜(Schleicher&Schuell Bioscience,Keene,NH)に転写した。5%ミルクにより非特異的結合を飽和させた後、4℃にて一晩、5%ミルク、TBS及び0.05% Tween 20中で1:500希釈した上記のポリクローナル抗体とともにその膜をインキュベーションした。洗浄後、1:5000に希釈した抗ウサギホースラディッシュペルオキシダーゼ結合抗体とともに、室温にて30分間、その膜をインキュベーションした。SuperSignal West Pico Chemiluminescent Substrate(Pierce Biotechnology,Rockford,IL)により、免疫反応のあるバンドを検出した。最後に、BIOMAX MRフィルム(Kodak)に10秒間、その膜を曝露した。
【実施例9】
【0095】
ヘパラン硫酸の蛍光標識
既に述べられているようにしてフルオレセインイソチオシアネート(FITC)でウシ腎臓由来のヘパラン硫酸ナトリウム塩(Sigma−Aldrich Corp.,St.Louis,MO)を標識した(Toyoshima及びNakajima,J.,Biol.Chem.274:24153−24160(1999))。ヘパラン硫酸 5mgとFITC5mgとを、0.1M Na2CO3 pH9.5 1mLに溶解し、4℃にて一晩、暗所でインキュベーションした。次に、未処理ヘパラン硫酸からFITC標識ヘパラン硫酸(FITC−HS)を分離するために、MicroSpin G−25カラムにこの溶液を載せた。高分子量のヘパラン硫酸種を分離するために、150mM NaCl、25mM Tris−HCl pH=7.5緩衝液中のSephacryl S−300を介した第一のゲル濾過クロマトグラフィー段階にこのFITC−HSを供した。着色した分画を集め、Biomax−10K遠心濃縮装置(Millipore)で濃縮し、一様な分子量のヘパラン硫酸種を得るために、Sephacryl S−300(上記)で再びクロマトグラフィーを行った。HPLC Superdex 75TM(Pharmacia Biotech)クロマトグラフィーシステムにより、溶出した分画を分析した。L−7485蛍光検出装置(Merck Hitachi)により、各分画の蛍光を測定した。発明者らは、分子量が異なるヘパラン硫酸産物を含む4個の主要な分画を得た。Blyscan Glycosaminoglycan Assay(Biocolor Ltd.,Belfast,Northern Ireland)により各分画におけるFITC−HSの量を測定した。
【実施例10】
【0096】
蛍光分析。
【0097】
このアッセイは、HPLCサイズ排除クロマトグラフィーによりモニターするFITC−HSの分解に基づく。50mM MES pH6、10% グリセロール(ヘパラナーセ活性緩衝液、HAB) 50μl中で、FITC−HS 5μlとともに精製ヘパラナーゼ 8μをインキュベーションした。この反応混合物を室温にて一定の時間インキュベーションし、ヘパリン 50μgを添加することにより反応を停止させた。次に、この混合物をUltrafree−MC遠心濾過装置(Millipore)を用いて濾過した。50mM Hepes pH7.5 150mM Na2SO4緩衝液中で平衡化し、Merck−Hitachi HPLCシステムに連結したSuperdex 75TM(Pharmacia Biotech)カラムに20μlを注入した。L−7485蛍光検出装置により蛍光ヘパラン硫酸分解産物を検出した。そのままのFITC−HSに対する、低分子量のヘパラン硫酸種における増加をモニタリングすることにより、ヘパラナーゼ活性を評価し、ピーク領域の積分により定量した。
【実施例11】
【0098】
ヘパラン硫酸の還元末端における放射性標識及びビオチン化
ウシ腎臓由来のヘパラン硫酸ナトリウム塩 10mg(Sigma)を部分的に、N−デ−アセチル化し、既に述べられているようにして[3H]無水酢酸で再びアセチル化した(Freeman及びParish,Biochem.J.325:229−237(1997))。次に、トリチウム化したヘパラン硫酸を、述べたように還元末端で還元的アミノ化した。トリチウム化し、還元的にアミノ化したヘパラン硫酸を、EZ−Link Sulfo−NHS−LC−Biotin(Pierce)を用いて、さらにビオチンに結合させた。このビオチン類似体は、ヘパラン硫酸分子の還元末端で生成されるアミノ基と反応し得るN−ヒドロキシスクシンイミドエステル部分を有する。発明者らは、還元的にアミノ化し、H2O 1mLに再懸濁したトリチウム化ヘパラン硫酸 約5mg(ヘパラン硫酸分子量の平均値を500kDaとして、100M 推定最終濃度)において、回収率を計算した。この溶液 100μlに対して、EZ−LinkSulfo−NHS−LC−Biotin 1mg(約100倍モル過剰)及びリン酸緩衝液 pH7.5 20μlを添加した。この反応混合液を室温にて一晩インキュベーションした。次に、ビオチン化したトリチウム化ヘパラン硫酸を未反応ビオチンから分離するために、この反応混合物をPD−10脱塩カラムにかけた。発明者らは、最終的に、4個の分画(各1mL)を得て、Reacti−Bind Streptavidin High Binding Capacity Coated Plates(Pierce)に固定化するために、それらの活性について試験した。
【実施例12】
【0099】
放射性アッセイ
このアッセイは、マイクロプレートに固定化したトリチウム化ヘパラン硫酸の分解に基づく。Reacti−Bind Streptavidin High Binding Capacity Coated Platesの各ウェルを、製造者の説明書に従い前処理した。最初に、PD−10脱塩カラム後に得られた、トリチウム化したビオチン化ヘパラン硫酸の各分画の様々な量を、PBS中で、最終体積100μlとなるように、各ウェルに添加(デュプリケート)した。100x103d.p.m.に対応する分画2の体積で最大結合が得られることを見極めた後、いつもこの量を使用した。室温にて一晩この結合を行った。次にウェルをPBSで3回洗浄し、HABで2回洗浄した。精製ヘパラナーゼ 10μlを各ウェルにHAB中で添加し、最終体積を100μlとした。このウェルを室温にて2時間から24時間インキュベーションした。最終的に、各ウェルにおいてヘパラナーゼにより生じるトリチウム化ヘパラン硫酸産物による遊離放射性活性を測定し、緩衝液ブランクに対して正規化した。
【実施例13】
【0100】
ヘパラナーゼコンストラクトの特異的活性の測定。
【0101】
COS7細胞において一時的に発現させた、又はバキュロウイルス系において発現させたいずれかのヘパラナーゼコンストラクトの特異的活性を次のように測定した:
特異的活性=正規化活性(d.p.m./μl)/正規化した濃度測定量(densitometric volume)(測定量/μl)
詳しく説明すると、放射性アッセイにおいて、直線的な用量−活性関係が見られるような方法において各調製物を滴定することにより部分精製ヘパラナーゼコンストラクトの活性を測定した。各調製物を用いてこれらの滴定を3回繰り返し、平均、正規化活性(d.p.m./μl)を計算した。ウェスタンブロッティング実験によりタンパク質発現を調べ:デンシトメトリーにより化学発光の読み出しを定量した。再び、実験を3回繰り返し、平均値を決定した。正規化した濃度測定量(densitometric volume)(測定量/μl)で正規化した活性(d.p.m./μl)を割ることにより、特異的活性を得た。
【図面の簡単な説明】
【0102】
【図1】図1は、哺乳類細胞におけるヒトヘパラナーゼの生合成を示す。
【図2A】図2パネルAは、改変TEV切断部位を有するヘパラナーゼコンストラクトの概略図を示す。
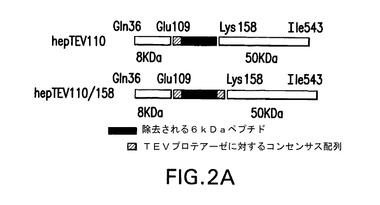
【図2B】パネルB(左)は、COS7細胞(レーン1)、hepTEV110(レーン2)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110(レーン3)又は0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110(レーン4)、hepTEV110/158(レーン5)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110/158(レーン6)、0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110/158(レーン7)において発現された、正しくプロセシングされたwtヘパラナーゼのウェスタンブロット分析の結果を示す。パネルB(右)は、hepTEV110(カラム1)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110(カラム2)又は0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110(カラム3)、hepTEV110/158(カラム4)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110/158(カラム5)、0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110/158(カラム6)のヘパラナーゼ活性を示す。蛍光法を使用して、これらの試料のヘパラナーゼ活性を評価した。
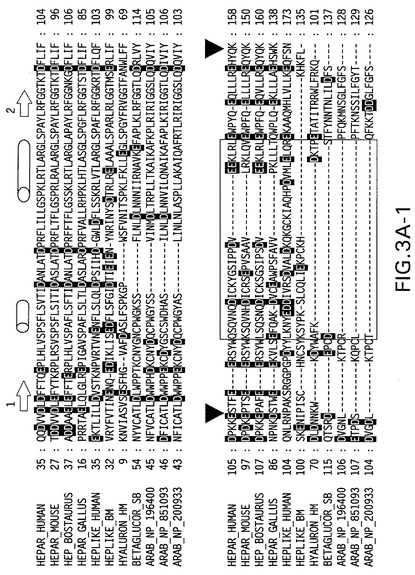
【図3A−1】パネルA:関連配列に対する、ヘパラナーゼの多配列アラインメント。予想される二次構造エレメントをアラインメントの上に示す(矢印=β鎖、円柱=へリックス)。この2つの切断部位の位置は、黒い三角印により示す。ヒアルロニダーゼ断片により置換される、除去されるヘパラナーゼ部分の領域を、灰色のボックスで囲む。
【図3A−2】パネルA:関連配列に対する、ヘパラナーゼの多配列アラインメント。予想される二次構造エレメントをアラインメントの上に示す(矢印=β鎖、円柱=へリックス)。この2つの切断部位の位置は、黒い三角印により示す。ヒアルロニダーゼ断片により置換される、除去されるヘパラナーゼ部分の領域を、灰色のボックスで囲む。
【図3B】パネルB:TIMバレル構造の概略図。除去されるヘパラナーゼ部分の位置は、三角印で表す切断位置により示す。存在する場合は、その部分は、β/αユニット1及び2により基質(灰色の矢印)の結合を妨げる可能性が高い。より短いループ(点線)を設計することにより、この抑制を解除し、これにより、活性を有する酵素が得られ、同時に、その酵素の構造的完全性が維持される。
【図4A】パネルA:本明細書で述べた1本鎖ヘパラナーゼコンストラクトの概略図。
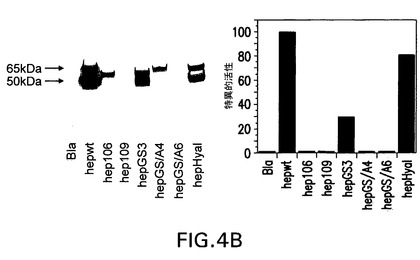
【図4B】パネルB、左:COS7細胞において発現させたwtヘパラナーゼ又は1本鎖コンストラクトのウェスタンブロッティング分析。Blaは、レポーター遺伝子β−ラクタマーゼ(材料と方法セクションを参照のこと。)をコードするベクターのみを形質移入したCOS7細胞の部分精製細胞溶解液に相当する対照である。右:放射性アッセイを用いた同じ試料のヘパラナーゼ活性。全ての1本鎖コンストラクトの特異的活性は、wtヘパラナーゼの特異的活性に対して正規化した。
【図5】左、COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼ又はSf9細胞で発現されたwtヘパラナーゼ及び1本鎖コンストラクトのウェスタンブロット分析。右:放射性アッセイを用いた同じ試料のヘパラナーゼ活性。Sf9細胞で発現されたwtヘパラナーゼ及び1本鎖コンストラクトの特異的活性は、COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼの特異的活性に対して正規化した。
【図6】COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼ(黒丸)及び非プロセシングFITC−HS(白丸)と比較した、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖タンパク質とともに6時間インキュベーションを行った後に得られたFITC−HS分解産物のサイズ排除クロマトグラフィー。
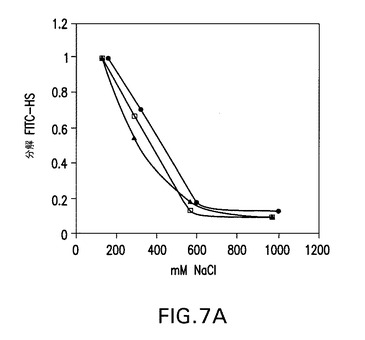
【図7A】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、イオン強度依存性(パネルA)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
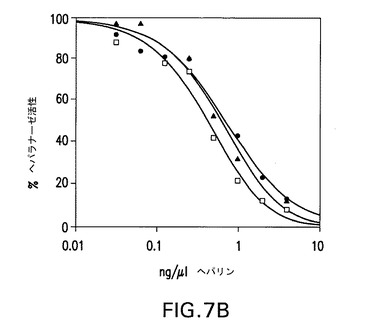
【図7B】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、ヘパリンによる阻害(パネルB)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
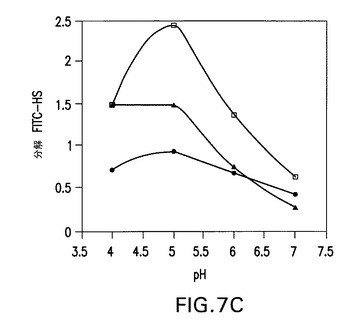
【図7C】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、pH依存性(パネルC)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
【技術分野】
【0001】
本発明は、高収率異種発現系で発現可能である、合成的に産生され、酵素的活性を有するヘパラナーゼ分子に関する。また、本明細書において、異種発現系で哺乳類ヘパラナーゼを発現させる方法も提供する。
【背景技術】
【0002】
ヘパラン硫酸プロテオグリカン(HSPGs)は、細胞外マトリックス(ECM)及び細胞表面上で見られる遍在性の高分子であり、細胞−細胞及び細胞−ECM相互作用の維持に寄与する。HSPGsは、タンパク質コアに共有結合するいくつかのヘパラン硫酸(HS)鎖から構成される。ヘパラン硫酸は、フィブロネクチン、ラミニン及びコラーゲンなどの構造的ECMタンパク質の、細胞表面及び他のECMタンパク質との結合を促進するが、このことから、自己集合及びECM成分の不溶性、細胞接着及び移動運動においてこのグリコサミノグリカンが果たす役割が示唆される。正しい細胞−細胞及び細胞−ECM相互作用の維持は重要であるため、細胞外環境においてHSPGsは、重大な構造的及び調整的役割を果たし、胚形成、形態形成及び発生から、炎症、血管新生及び癌転移にわたる範囲の重要な正常及び病的プロセスの調節を行う。
【0003】
上述の構造的及び細胞マトリックスのアンカー的役割に加えて、HSの構造多様性(Eskoら、J.Clin.Invest.108:169−173(2001);Turnbullら、Trends Cell Biol.11:75−82(2001))により、HSPGsは、増殖因子、酵素及びケモカインなどの様々な細胞外シグナル伝達タンパク質と相互作用することが可能となる。繊維芽細胞増殖因子(FGF1及びFGF2)、血管内皮細胞増殖因子(VEGF)、肝細胞増殖因子、形質転換成長因子β及び血小板由来増殖因子などの増殖因子は、腫瘍成長、侵襲及び血管新生において重要な役割を果たす。これらのシグナル伝達分子の拠点としての作用、これらの活性化又は安定化に加えて、HSPGsは、FGF受容体の多様なアイソフォームへのFGF2の結合など、リガンド−受容体相互作用に関与し得る(Changら、FASEB J.14:137−144(2000))。
【0004】
ヘパラン硫酸は、血小板、胎盤栄養膜細胞及び白血球により放出されるエンドβ−D−グルクロニダーゼヘパラナーゼにより分解される。ヘパラナーゼは、加水分解酵素メカニズムを介してグリコシド結合を切断することにより、特異的にヘパラン硫酸を分解する。この分解の結果、bFGF、ウロキナーゼプラスミノーゲン活性化因子(uPA)及び組織プラスミノーゲン活性化因子(tPA)が放出されるが、これらにより、血管新生が開始され得るか、又はECM分解が可能となり得る。さらに、ヘパラナーゼによるHS切断により、基底膜(BM)を介して細胞が移動し、ECM障壁を横断できるようになる。HS分解は、細胞外の変化に細胞が素早く反応することができるようにすることにより、多数の生理学的プロセスにおいて重要な役割を果たす。従って、ヘパラナーゼ活性の阻害は、炎症、転移及び自己免疫疾患など、細胞移動の変化に関係する病変に影響を及ぼし得る。
【0005】
このような極めて重要な役割のために、ヘパラナーゼは、抗腫瘍、抗転移又は抗炎症剤の開発のための、有力な新規ターゲットとなる。ヘパラナーゼは、単一遺伝子産物であり、関連タンパク質の複合ファミリーの一部ではないと思われるので、薬物開発の目的に対して、ECM−調節酵素でもあるマトリックスメタロプロテアーゼを凌ぐ顕著な利点を有する。信頼できる高スループットのアッセイが不足していることと、その複雑な生合成との両方により、現在、薬物ターゲットとしてヘパラナーゼを活用することには障害が多く、活性タンパク質を大量に産生させることが困難となっている。
【0006】
ヒトヘパラナーゼcDNAは、小胞体(ER)への移動の際にシグナルペプチダーゼにより除去されるシグナルペプチド配列を有する、プレプロタンパク質として最初に合成されるタンパク質をコードする。得られる65kDaのプロ型は、さらに、157個のN−末端アミノ酸を除去することによりプロセシングされ、成熟型の50kDヘパラナーゼとなる。この50kDタンパク質は、非プロセシング型の65kDa前駆体よりも少なくとも100倍高い特異的活性を有する(Vlodavskyら、Nat.Med.5:793−802(1999))。興味深いことに、この50kDaタンパク質は、哺乳類細胞などで発現される場合、不活性である(Hulettら、Nat.Med.5:803−809(1999))。この酵素の活性型が、未同定のタンパク質分解酵素による6kDaの介在ペプチドの除去により生じる、50kDa断片と8kDa断片との間のヘテロ二量体から構成されることが提案された(Fairbanksら、J.Biol.Chem.274:29587−29590(1999))。この仮説と一致して、McKenzieら(Biochem J.373:423−435(2003))は、昆虫細胞において活性ヘテロ二量体ヘパラナーゼを産生させ、8kDaサブユニットがヘパラナーゼ活性に必要であることを確認した。
【0007】
内因性ヘパラナーゼは、様々なソースから精製することができる。しかし、ヘパラナーゼ発現レベルが低い場合は、煩雑で費用の高い精製法が必要となる。例えば、Toyoshima及びNakajima(J.Biol.Chem.274:24153−24160(1999))は、4つの異なるクロマトグラフィー段階を必要とし、少なくとも5日間かかる、血小板からの内因性ヒトヘパラナーゼの精製プロセスについて述べた。
【0008】
内因性ヘパラナーゼの精製に対する他の欠点は、全体的な収率が特徴的に低いことである。例えば、Fairbanksら(J.Biol.Chem.274,29587−29590(1999))は、血小板からヘパラナーゼをたった22μgのみ、6%の回収率で精製したことについて報告している。同様に、Fuks及び共同研究者ら(米国特許第5,362,641号)は、ヒトヘパトーマ細胞株Sk−Hep−1由来のタンパク質 1.4kgからヘパラナーゼを4000倍精製し、1.9%の回収率で、精製ヘパラナーゼタンパク質をわずか6.5μgのみ調製したことについて述べている。同じ細胞株からのヘパラナーゼの240,000倍精製が、Peckerら(米国特許第5,968,822号)により開示された。しかし、このプロセスには、細胞培養物 500リットル超が必要であった。
【0009】
ヒトヘパラナーゼ遺伝子の同定及びクローニング(Vlodavksyら、Nature Med.5:793−802(1999);Hulettら、Nature Med.5:803−809(1999);Toyoshima及びNakajima,J.Biol.Chem.274:24153−24160(1999))により、異種発現系におけるヘパラナーゼタンパク質の組み換え発現が可能となった。しかし、そのような異種発現系について、ヘパラナーゼ産生に関して、重大な欠点が知られている。例えば、Ben−Artziら(WO99/57244)は、細菌、哺乳類、酵母及び昆虫細胞における組み換えヒトヘパラナーゼの発現について述べている。ヘパラナーゼ発現は達成されたが、E.コリが宿主細胞であった場合、その組み換えタンパク質に付随する酵素活性は検出されず、酵母Pichia pastoris(ピキア パストリス)でヘパラナーゼを発現させた場合、70kDaの非プロセシング前駆体のみが検出された。
【0010】
Ben−Artzi及び共同研究者ら(前出)はまた、哺乳類細胞、すなわち、ヒト腎臓繊維芽細胞(293)、仔ハムスター腎臓細胞(BHL21)及びチャイニーズハムスター卵巣細胞(CHO)における組み換えヘパラナーゼの発現について述べている。しかし、これらの発現系は、効率が低く、必要なコストが高いことが知られている。さらに、活性を有する成熟型タンパク質を得るための組み換え全長前駆体のプロセシングがこれらの細胞で観察されるという事実にもかかわらず、このプロセシング反応が効率的でないため、均質的にプロセシングされたタンパク質は得られない。さらに、ヘパラナーゼの分泌を促進する発現ベクターを使用しても、CHO細胞の馴化培地において組み換えヘパラナーゼの産生は導かれず、ヘパラナーゼを分泌するために、カルシウムイオノフォア又はPMAを添加することにより、さらに刺激を行う必要がある。この系で正しくプロセシングされるのは、分泌されたタンパク質のごく一部のみであると思われる。
【0011】
Sf21又はHigh Five(ハイファイブ)細胞などの昆虫細胞発現系におけるヘパラナーゼの産生が、この技術分野において述べられている(WO99/57244、WO99/11798、米国特許第5,968,822号;米国特許第6,348,344号;及び米国特許第6,190,875号)。しかし、増殖培地へと効率的に分泌されることがこのような方法で観察されるにもかかわらず、この酵素の特異的活性は非常に低く、正しいプロセシングは観察されなかった。例えば、Ben−Artziら(WO99/57244)は、昆虫細胞発現系で正しくプロセシングされたヘパラナーゼを生成させるために、そのヘパラナーゼタンパク質の位置119又は157の下流にプロテアーゼ切断部位を導入することについて述べている。しかし、これらのコンストラクトは、酵素的に活性であることが示されなかった。
【0012】
McKenzieら(前出)は、昆虫細胞における活性を有するヘテロ二量体ヘパラナーゼの産生について述べた。しかし、この系は、2種類の異なる組み換えタンパク質(8kDa及び50kDaサブユニット)を同時に生成させる必要があるという欠点を有する。単離した8kDa及び50kDaドメインの混合の結果、ヘパラナーゼは活性化されないので、同時発現により活性を有するヘテロ二量体を成功裡に回収できるか否かは、おそらく、そのヘテロ二量体複合体の共翻訳的な形成に依存する。この複合体をグリカナーゼで処理することにより、それが解離し、50kDaサブユニットが沈降するが、このことから、安定性及び溶解性に乏しいことが示唆される。
【0013】
活性又は不活性型でヘパラナーゼを得るための上述の方法にもかかわらず、高収率、低コストの異種発現系において発現させることができる、生物学的に活性を有するヘパラナーゼ分子を生成させることは利益があると思われる。前記分子は、転移成長及び/又は炎症を阻害及び/又は治療する、治療又は医薬品の開発のための阻害剤スクリーニングアッセイにおいて使用することができる。
【発明の開示】
【0014】
本発明は、高収率の異種発現系で発現させることができる、生物学的に活性を有する哺乳類ヘパラナーゼをコードする合成核酸分子を提供する。本明細書で提供される合成ヘパラナーゼ分子は、哺乳類系で発現が低レベルであり異種発現系で正しくプロセシングされない野生型ヘパラナーゼを凌ぐ顕著な長所を与える。本発明の合成分子は、転移成長、自己免疫疾患及び/又は炎症を阻害及び/又は治療するための治療又は医薬品開発のための、阻害剤スクリーニングアッセイにおいて使用することができる。
【0015】
本発明のある態様において、上記の合成核酸分子は、哺乳類ヘパラナーゼタンパク質をコードするヌクレオチド配列を含有し、このヌクレオチド配列は、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、この切断部位は、本ヘパラナーゼタンパク質の残基100及び168をコードするヌクレオチドの間に位置する。前記核酸分子は、適切な酵素とともにインキュベーションすると生物学的活性が可能となる、ヘパラナーゼタンパク質をコードする。
【0016】
本発明はさらに、哺乳類ヘパラナーゼタンパク質をコードする部分を含有し、このタンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチド配列と、から構成され、該N−末端及びC−末端断片が、野生型ヘパラナーゼ断片と実質的に同様であるタンパク質断片をコードし、該コードされるヘパラナーゼタンパク質が構成的に活性を有する、合成哺乳類ヘパラナーゼ核酸分子に関する。
【0017】
合成的に産生される、生物学的に活性を有する哺乳類ヘパラナーゼポリペプチド、及び適切な酵素とインキュベーションすると生物学的活性が可能となる、エンドプロテアーゼコンセンサス切断部位を含むヘパラナーゼポリペプチドもまた本明細書で提供する。
【0018】
本発明はさらに、結果として生物学的に活性を有するヘパラナーゼが高レベルで発現される、異種発現系において哺乳類ヘパラナーゼを発現させるための方法を提供する。
【0019】
本明細書を通して、及び添付の請求項で使用される場合、単数形「a」、「an」及び「the」は、他に文脈が明確に指示を与えない限り、複数形の意味を含む。
【0020】
本明細書を通して、及び添付の請求項で使用される場合、次の定義及び略語を適用する。
【0021】
「保存的アミノ酸置換」は、あるアミノ酸残基を別の化学的に同等のアミノ酸残基に置換することを意味する。そのような保存的置換の例は、ある疎水性残基(イソロイシン、ロイシン、バリン又はメチオニン)を別のものへ置換すること;ある極性残基を同様の電荷の別の極性残基へ置換すること(例えば、リジンに対するアルギニン;アスパラギン酸に対するグルタミン酸)である。
【0022】
「哺乳類」という用語は、ヒトを含むあらゆる哺乳動物を意味する。
【0023】
「治療」という用語は、治療的処置及び予防的(Prophylactic又はPreventative)手段の両方を意味する。治療を必要とする者には、既に疾患に罹患している者ならびにその疾患に罹患する傾向がある者又はその疾患が予防されるべきである者が含まれる。「疾患」とは、本明細書中に記載の核酸分子及びポリペプチドを用いて同定した分子による治療が役立つあらゆる状態である。このような疾患には、これらに限定されないが、癌、炎症及び自己免疫疾患が含まれる。
【0024】
「ベクター」という用語は、DNA断片を宿主生物又は宿主組織に導入することができるある手段を意味する。プラスミド、ウイルス(アデノウイルスを含む。)、バクテリオファージ及びコスミドをはじめとした様々な種類のベクターがある。
【0025】
「生物学的に活性を有する」とは、タンパク質が、天然に生じる分子又はそのアイソフォームに付随する構造的、調整的又は生物化学的機能を有することを意味する。ヘパラナーゼとの関連において、「生物学的に活性を有する」タンパク質は、ヘパラナーゼ酵素活性を含む。
【0026】
「実質的に同様」とは、ある配列が参照配列と、少なくとも80%、好ましくは90%、より好ましくは95%及びさらにより好ましくは99%の相同性を共有することを意味する。本発明において、この参照配列は、この文章の文脈で示される場合、全長ヒトヘパラナーゼヌクレオチドもしくはアミノ酸配列又は8kDa(配列番号15)もしくは50kDa(配列番号16)ヘパラナーゼフラグメントのヌクレオチドもしくはアミノ酸配列であり得る。このように、8kDaヒトヘパラナーゼ断片(配列番号15)と「実質的に同様」であるヘパラナーゼタンパク質配列は、この8kDaヒトヘパラナーゼ断片と、少なくとも80%の相同性、好ましくは90%の相同性、より好ましくは95%の相同性、さらにより好ましくは99%の相同性を共有することとなる。あるヘパラナーゼタンパク質又はヌクレオチド配列が参照配列に対して「実質的に同様」であるか否かは、例えば、University of Wisconsin Genetics Computer Group(UWGCG)から入手できるGAPコンピュータープログラム、バージョン6.0などの配列解析ソフトウェアを用いた比較配列情報により決定することができる。GAPプログラムは、Smith及びWaterman(Adv.Appl.Math.2:482,1981)により改訂された、Needleman及びWunsch(J.Mol.Biol.48:443,1970)のアラインメント法を利用する。
【0027】
「遺伝子」は、そのヌクレオチド配列がポリペプチド分子をコードする核酸分子を意味する。遺伝子は、中断されないヌクレオチド配列であり得るか、又は、イントロン、プロモーター領域、スプライシング部位及び反復配列などの介在部分を含み得る。遺伝子は、RNAであってもDNAであってもよい。好ましい遺伝子は、本発明のペプチドをコードするものである。
【0028】
「核酸」又は「核酸分子」という用語は、リボ核酸(RNA)又はデオキシリボ核酸(DNA)、プローブ、オリゴヌクレオチド、断片又はそれらの一部及びプライマーを意味するものとする。DNAは、相補的DNA(cDNA)又はゲノムDNA、例えば、本発明のペプチドをコードする遺伝子のいずれでもあり得る。
【0029】
「野生型ヘパラナーゼ」又は「野生型タンパク質」もしくは「wtタンパク質」は、アミノ酸又はそれらの変異型の天然に生じる配列を含むタンパク質を意味する。野生型ヒトヘパラナーゼのアミノ酸配列は、本分野において入手可能である(Vlodavksyら、Nature Med.5:793−802(1999);Hulettら、Nature Med.5:803−809(1999);Toyoshima及びNakajima,J.Biol.Chem.274(34):24153−24160(1999);(参照により、全体を本明細書に組込む。))。
【0030】
「野生型ヘパラナーゼ遺伝子」は、ヒト由来のタンパク質又は、ショウジョウバエ(Drosophila)などの昆虫、ツメガエル(Xenopus)などの両生類及びラット、マウス及びアカゲザルなどの哺乳類を含むがこれらに限定されない、別の生物から得られたタンパク質をはじめとする、天然に生じるヘパラナーゼタンパク質をコードするヌクレオチド配列を含有する遺伝子を意味する。本ヒトヘパラナーゼ遺伝子のヌクレオチド配列は、本分野で入手可能である(Genbank Accession No.AF155510;Toyoshima及びNakajima、前出(参照により、全体を本明細書に組込む。))。
【0031】
「実質的に他のタンパク質不含である」又は「実質的に精製された」とは、少なくとも90%、好ましくは95%、より好ましくは99%及びさらにより好ましくは99.9%、他のタンパク質が含まれていないことを意味する。したがって、実質的に他のタンパク質不含のヘパラナーゼタンパク質調製物は、その総タンパク質のパーセンテージとして、非ヘパラナーゼタンパク質が、10%を超えない、好ましくは5%を超えない、より好ましくは1%を超えない、さらにより好ましくは0.1%を超えない。あるヘパラナーゼタンパク質調製物が実質的に他のタンパク質不含であるか否かは、例えば、適切な検出方法(例えば銀染色又は免疫ブロッティングなど)と組み合わせたドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS−PAGE)などの従来のタンパク質純度評価技術により決定することができる。
【発明を実施するための最良の形態】
【0032】
ヘパラナーゼは、加水分解酵素メカニズムを介してグリコシド結合を切断することによりヘパラン硫酸(HS)を分解する哺乳類酵素である。HS分解は、細胞−細胞及び細胞−ECM相互作用を変更することで細胞外の変化に対して細胞が素早く反応できるようにすることによって、多くの生理学的プロセスにおいて重要な役割を果たす。これらの相互作用は重要なので、ヘパラナーゼ活性の阻害は、腫瘍細胞転移、T細胞介在遅延型過敏及び自己免疫疾患などのいくつかの病態に影響を及ぼし得る。
【0033】
いくつかの証拠から、ヘパラナーゼが腫瘍細胞転移に関与することが示唆される。第一に、ヘパラナーゼの発現レベルは、いくつかの腫瘍及び腫瘍細胞株の転移能と相関がある。第二に、侵襲性の強い転移性疾患の患者では、その尿中において測定可能なヘパラナーゼ活性を有する。この所見は、全ての癌患者で見られるものではない。さらに、非−抗凝血剤ヘパリン誘導体によるヘパラナーゼ活性の阻害により、B16メラノーマ、ルイス肺癌及び乳癌細胞による転移発生率が低下した。最後に、ヒトヘパラナーゼ遺伝子を用いた非転移性のマウス細胞に対する形質移入の結果、2種類のマウスモデルにおいて、死亡率及び転移発生が増加した。
【0034】
ヒトヘパラナーゼは、いかなる他の既知のタンパク質とも実質的な相同性を共有しない。その発見時において、ヘパラナーゼ遺伝子が遺伝子ファミリーのメンバーではなく、むしろ、HSPG分解に関与する単一遺伝子又は少なくとも主要なエンドグルクロニダーゼであることが示唆された。アミノ酸レベルで35%の相同性を共有する第二のヘパラナーゼ(hpa2)が後に同定されたが、しかし、hpa2は、その組織分布に基づき、異なる機能を果たすと思われる。転移成長におけるヘパラナーゼの役割を示す上記の証拠と合わせて、類似の作用を遂行する密接に関連するタンパク質が欠如していることにより、へパラナーゼはこれらの領域における治療法の開発のための優れたターゲットとなる。
【0035】
図1は、ヒトヘパラナーゼの生合成を示す。簡潔に述べると、ヘパラナーゼcDNAは、ERへの移動時にシグナルペプチダーゼにより除去されるシグナルペプチド配列(残基Met1−Ala35)を有するプレプロタンパク質として最初に合成されるタンパク質をコードする。その結果生じる65kDaプロ形態は、さらに、157のN−末端アミノ酸を除去することによりプロセシングされ、成熟型の50kDヘパラナーゼ(配列番号16)となる。この50kDタンパク質は、非プロセシング型の65kDa前駆体よりも少なくとも100倍高い特異的活性を有する(Vlodavskyら、Nat.Med.5:793−802(1999))。この酵素の活性型が、未同定のタンパク質分解酵素により6kDaの介在ペプチド(残基Glu109−Gln157)(以下、「介在断片」又は「6kDa断片」と呼ぶ。)を除去することで生じる、50kDa断片と8kDa断片(配列番号15)との間のヘテロ二量体であることが提案された(Fairbanksら、J.Biol.Chem.274:29587−29590(1999))。
【0036】
最近の証拠により8kDaサブユニット(配列番号15)がヘパラナーゼ活性に必要であることが示されているにもかかわらず(McKenzieら、Biochem J.373:423−435(2003))、ヘパラナーゼの活性化プロセスにおけるこの8kDaサブユニットの役割は、本明細書中で開示される研究以前は、不明確なままであった:これは、不可欠なサブユニットとして働く、又は、あるいは、シャペロンとして作用する可能性があり、この機能が遂行された後は不必要となり得る。また、8kDaサブユニット以外の他の成分がヘパラナーゼ活性の誘導に必要であるか否かも明確ではなかった。
【0037】
多配列アラインメント及び二次構造予想から、いくつかのグリコシダーゼで見られるように、そのタンパク質がTIMバレルフォールドを取ることによるヒトヘパラナーゼのモデルが導かれる(Hulettら、Biochemistry 39:15659−15667(2000))。この共通フォールドモチーフは、通常、8個の交互のα−へリックス及びβ−鎖からなる。50kDa断片内で、明確な相同性が観察されるのは、TIMバレルフォールドの3番目のα/βユニットからのみであるが、このことから、ヘパラナーゼが6個のみのα/βユニットからなる新規フォールドを取るか、又は、このタンパク質の他の部分が、欠落ユニットに寄与するか、のいずれかが示唆される。8kDa断片がこの欠落構造エレメントに寄与し得ることが主張された(Hulettら、前出)。
【0038】
この仮説に従い、本明細書中で述べるように、一緒に共有結合する8kDa及び50kDaサブユニットを有する1本鎖ヘパラナーゼ分子を設計するために、多配列アラインメント(図3A及び3B)に基づきヘパラナーゼの二次構造のモデルを確立した。本発明は、8kDa及び50kDa断片のリンカーとの連結の結果、タンパク質分解的プロセシングを必要としない構成的に活性を有する1本鎖ヘパラナーゼ分子が得られることを示す。本発明の典型的な実施形態において、Hirudinaria manillensis(ヒルディナリア マニレンシス)ヒアルロニダーゼ由来のループを移植することにより、又は、3つのグリシン−セリンリピートを含有するリンカーを用いて、この2個の断片を連結した。
【0039】
また、6kDa介在断片のN及びC−末端周囲のエンドプロテアーゼ切断部位を改変することにより、少なくとも部分的に精製されたタンパク質の両部位におけるタンパク質分解性のプロセシングによって、他の成分なしで、ヘパラナーゼ活性化が導かれることが、本明細書において示された。本発明のこの態様の典型的実施形態において、タバコエッチウイルスプロテアーゼ切断部位をこの6kDa介在断片のN及びC−末端に付加し、その結果、コードされるタンパク質の精製又は部分精製及びそれに続く適切な酵素とのインキュベーション後に、活性を有するヘパラナーゼが得られる。本発明は、ヒトヘパラナーゼが標準的なTIMバレルフォールドを取ることの証拠を提供し、有利に、特異的な阻害剤の同定のための活性酵素分子の容易な産生方法を提供する。
【0040】
異種発現系、特に昆虫細胞において生物学的に活性を有するヘパラナーゼを発現させるための、本発明の改変タンパク質、核酸分子及び方法は、特徴的に、0.5mg/lから5.0mg/lの回収率となる。さらに、これらのタンパク質は、効率的に、増殖培地に分泌され、哺乳類細胞においては、細胞内部又は細胞膜に会合して、真正のヒト酵素が主に得られる(Vlodavskyら、Semin.Cancer Biol.12:121−129(2002))。
【0041】
従って、本発明は、活性を有する哺乳類ヘパラナーゼをコードする合成核酸分子に関し、その核酸分子は、高収率異種発現系で発現させることができる。本明細書で提供される合成ヘパラナーゼ分子は、哺乳類系において発現レベルが低く、異種発現系において正しくプロセシングされない野生型ヘパラナーゼを凌ぐ顕著な長所を与える。本発明の合成分子は、転移成長及び/又は炎症を阻害及び/又は治療する治療又は医薬品の開発のための阻害剤スクリーニングアッセイにおいて使用することができる。前記合成分子はまた、自己免疫疾患の治療及び/又は予防のための治療又は医薬品開発においても有用である。
【0042】
本発明のある態様において、哺乳類ヘパラナーゼタンパク質をコードするヌクレオチド配列を含有する合成核酸分子が提供され、該ヌクレオチド配列は、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、それらの切断部位は、本ヘパラナーゼタンパク質の残基100及び168をコードするヌクレオチドの間に位置する。本発明のこの態様は、野生型ヘパラナーゼの生合成と同様の、本ヘパラナーゼタンパク質のタンパク質分解性プロセシングを遂行し、結果として生物学的に活性を有する酵素を得るための方法において使用することができる合成核酸分子を提供する。
【0043】
上記核酸分子によりコードされる実質的に純粋なポリペプチドもまた、本明細書で提供する。
【0044】
本発明の好ましい実施形態において、本哺乳類ヘパラナーゼタンパク質は、ヒトヘパラナーゼである。
【0045】
この2個のコンセンサス切断部位は、コードされるタンパク質の精製又は部分精製及び適切な酵素とのインキュベーション後に、その結果得られる断片が、野生型8kDa断片(配列番号15)と実質的に同様の少なくとも1つの断片と、野生型50kDa断片(配列番号16)と実質的に同様の少なくとも1つの断片と、を含有するという条件において、本ヘパラナーゼタンパク質の残基100と168との間のいずれかの位置に導入することができる。本発明の好ましい実施形態において、本コンセンサス切断部位は、本ヒトヘパラナーゼタンパク質の残基G110及びK158の前に位置し、その結果、コードされるタンパク質の精製又は部分精製及びそれに続く適切な酵素とのインキュベーション後に、8kDaの第一の断片、6kDaの第二の「介在」断片及び50kDaの第三の断片が得られる。
【0046】
活性を有するヘテロ二量体ヘパラナーゼを得るために、これらに限定されないが、タバコエッチウイルス、ピコルナウイルスの3Cプロテアーゼ、トロンビン、血液凝固因子Xa及びエンテロキナーゼからの切断部位を含む、何らかのエンドプロテアーゼに対応する切断部位を、ヘパラナーゼ分子に導入することができることは、当業者により理解される。本発明の好ましい実施形態において、前記切断部位は、タバコエッチウイルス由来である。
【0047】
本発明の別の態様において、哺乳類ヘパラナーゼタンパク質をコードする部分を含有し、そのタンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチド配列とから構成される、構成的に活性を有する1本鎖哺乳類ヘパラナーゼ核酸分子が提供される。本発明のこの態様は、タンパク質分解性プロセシングなしで構成的に活性を有するヘパラナーゼをコードし、その6kDa「介在」断片が実質的に除去されるように改変され、その介在断片がより小さいリンカーで置換される、合成遺伝子を提供する。
【0048】
本発明のこの態様の好ましい実施形態において、本哺乳類ヘパラナーゼタンパク質は、ヒトヘパラナーゼである。
【0049】
上記の構成的に活性を有する1本鎖哺乳類ヘパラナーゼ遺伝子によりコードされる精製された合成ヘパラナーゼタンパク質もまた、本明細書で提供される。
【0050】
本発明のこの態様において、約1残基から約67残基からなるペプチドをコードする何らかの配列を、リンカーとして使用することができる。前記リンカーは、合成するか、又は天然に生じるソースから単離することができる。本発明の典型的な実施形態において、本リンカーは、ヒアルロニダーゼタンパク質の中央ループ領域をコードするヌクレオチド配列を含む。このヒアルロニダーゼは、H.マニレンシス(H.manillensis)由来であることがが好ましい。他の実施形態において、本リンカーは、(GlySer)3リンカーをコードするヌクレオチド配列を含む。
【0051】
本発明はさらに、本明細書を通じて開示される合成核酸分子を含有する組み換えベクターに関する。これらのベクターは、DNA又はRNAからなるものであり得る。殆どのクローニング目的に対して、DNAベクターが好ましい。代表的なベクターには、組み換えヘパラナーゼタンパク質をコードすることができる、プラスミド、改変ウイルス、バキュロウイルス、バクテリオファージ、コスミド、酵母人工染色体及びエピソーム性のDNAもしくは統合DNAの他の型が含まれる。特定の遺伝子導入又は他の使用のための適切なベクターを決定することは、十分に熟練者の範囲内にある。
【0052】
本明細書を通じて開示される本合成核酸分子を含有する発現ベクターは、組み換え宿主細胞における哺乳類ヘパラナーゼの高レベル発現に使用し得る。発現ベクターには、これらに限定されないが、クローニングベクター、改変クローニングベクター、特に設計したプラスミド又はウイルスが含まれ得る。また、様々な細菌発現ベクターも、必要ならば、細菌細胞において組み換えヘパラナーゼを発現させるために使用し得る。さらに、真菌細胞において組み換えヘパラナーゼを発現させるために、様々な真菌細胞発現ベクターを使用し得る。さらに、昆虫細胞において組み換えタンパク質を発現させるために、様々な昆虫細胞発現ベクターを使用し得る。本発明の好ましい実施形態において、ベクターは、バキュロウイルスベクターである。
【0053】
本発明はまた、本発明の合成核酸分子を含有するベクターを用いて形質転換又は形質移入を行った宿主細胞にも関する。組み換え宿主細胞は、原核又は真核であり得、これらに限定されないが、E.コリなどの細菌、これらに限定されないがPichia pastoris(ピキア パストリス)、Hansenula polymorpha(ハンセヌラ ポリモルファ)及びSaccharomyces cervisiae(サッカロミセス セルビシエ)を含む酵母などの真菌細胞及び、これらに限定されないがショウジョウバエ(Drosophila)及びカイコ由来の細胞株を含む昆虫細胞が含まれる。このような組み換え宿主細胞は、哺乳類ヘパラナーゼ又は生物学的に同等の型を高レベルで産生させるための適切な条件下で培養することができる。本明細書中で定義する場合、「宿主細胞」という用語は、トランスジェニックヒト、トランスジェニックヒト胎児又はトランスジェニックヒト胚の身体における宿主細胞を含むものではない。
【0054】
上記のごとく、本発明の合成分子は、高収率異種発現系で発現させることができるので、先行技術を凌ぐ顕著な長所を提供する。本明細書で提供される合成分子によりコードされるヘパラナーゼタンパク質は、正しくプロセシングされ、酵素的に活性を有し、高レベルで発現される。従って、本発明の好ましい実施形態において、選択される宿主細胞は、これらに限定されないが、昆虫細胞、細菌細胞及び酵母細胞を含む高収率異種発現系の一部である。本発明の特に好ましい実施形態において、宿主細胞は、昆虫細胞である。
【0055】
本発明はまた、本明細書を通じて開示される核酸分子を含有する、組み換えベクター及び組み換え宿主細胞(両者とも原核及び真核である。)に関する。本発明の合成核酸分子、関連ベクター及び宿主は、癌、炎症及び/又は自己免疫の治療のために役立つ、ヘパラナーゼ活性の阻害剤を同定するためのスクリーニングアッセイにおいて有用である。
【0056】
本発明の別の態様において、(a)哺乳類ヘパラナーゼタンパク質をコードし、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含有し、該切断部位が該ヘパラナーゼタンパク質の残基100及び残基168をコードするヌクレオチドの間に位置するヌクレオチド配列を含有するベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で宿主細胞を培養することと;
(c)前記細胞を崩壊させて、前記ヘパラナーゼタンパク質を少なくとも部分的に精製することと;
(d)少なくとも部分的に精製したタンパク質をエンドプロテアーゼに曝露し、該ヘパラナーゼタンパク質をコンセンサス切断部位で切断することと、を含む、非哺乳類細胞において哺乳類ヘパラナーゼを発現させる方法が提供される。
【0057】
本発明はまた、上記の方法により産生された、実質的に精製されたタンパク質も提供する。
【0058】
本発明のこの態様の好ましい実施形態において、本哺乳類ヘパラナーゼは、ヒトヘパラナーゼである。さらに好ましい実施形態において、本コンセンサス切断部位は、ヒトヘパラナーゼの残基G110及びK158の前に位置する。
【0059】
別の好ましい実施形態において、本切断部位は、タバコエッチ(tobacco etch)タンパク質切断部位である。
【0060】
本明細書において、(a)ヘパラナーゼタンパク質をコードする部分を含有し、該タンパク質コード部分が、基本的に、約8kDaのN−末端断片をコードするヌクレオチド配列と、リンカーをコードするヌクレオチド配列と、約50kDaのC−末端断片をコードするヌクレオチド配列とから構成される、合成哺乳類ヘパラナーゼ遺伝子を含有するベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で宿主細胞を培養することと、を含む、非哺乳類細胞において、1本鎖の、構成的に活性を有する哺乳類ヘパラナーゼを発現させる方法もまた提供される。
【0061】
本明細書において、上記の方法により産生された、実質的に精製されたタンパク質もまた提供される。本発明のさらなる実施形態において、本タンパク質は、野生型ヘパラナーゼに特異的な抗体に結合することができる。
【0062】
本発明のこの態様の好ましい実施形態において、本リンカーは、ヒアルロニダーゼタンパク質の中央ループ領域を含有する。別の好ましい実施形態において、本リンカーは、(GlySer)3ペプチドを含有する。
【0063】
本明細書中で言及した公表物は全て、本発明に関連して使用され得る方法及び材料を説明し開示する目的のために、参照により本明細書に組込む。明細書のいかなる開示内容も、本発明が先行技術の効力によってこのような開示に対して先行する権利を与えられていないことを認めるものではない。
【0064】
付随する図面を参照して本発明の好ましい実施形態を説明してきたが、本発明がこれらの明確な実施形態に限定されず、添付の請求項において定義される本発明の範囲又は精神から逸脱することなく、当業者によりそれらにおいて様々な変更及び改変が行われ得ることを理解されたい。
【0065】
次の実施例は、説明するものであり、本発明を限定するものではない。
【実施例1】
【0066】
ヒト胎盤cDNAライブラリーからのヘパラナーゼのクローニング
TaKaRa Taqポリメラーゼ(TaKaRa Bio Inc.,Otsu,Shiga,Japan)を用いて、PCRにより、ヒトヘパラナーゼ(Accession No.AF155510)を正常ヒト胎盤cDNAライブラリー(Invitrogen Corp.,Carlsbad CA)から増幅した。緩衝液条件は、供給者により示されたものであった。cDNAテンプレートのPCR増幅は、94℃、1分間を1サイクル、その後、94℃にて30秒間、57℃にて30秒間及び68℃にて110秒間を35サイクルで行うように構成した。増幅した断片をゲル精製し、リン酸化し、充填後、pFAST BAC1のBamHI部位(バキュロウイルス発現)、又は、BamHI/EcoRI消化したpcDNA3ベクター(哺乳類細胞発現)のいずれかにクローニングした。PCR増幅及びKozak配列の同時最適化のために次のプライマーを使用した:hHEP1−24 BamHI opti
【0067】
【化1】
;及びhHEP rev 1632
【0068】
【化2】
【実施例2】
【0069】
1本鎖ヘパラナーゼ分子の構築
次のコンストラクト(hepWT、hep109(配列番号19、タンパク質配列番号20に対応)、hep106(配列番号18、タンパク質配列番号17に対応)、hepGS3(配列番号22、タンパク質配列番号21に対応)、hepGS6(配列番号24、タンパク質配列番号26に対応)、hepGS4(配列番号23、タンパク質配列番号25に対応)及びhepHyaluro(配列番号28、タンパク質配列番号27に対応)、直接的に8及び50kDaサブユニットを共有結合で連結しているもの(hep109及びhep106)、これらのサブユニットをグリシン−セリンスペーサーを介して連結しているもの(hepGS3、hepGS4及びhepGS6)又は酵素ヒアルロニダーゼからのループ領域を移植することにより連結しているもの(Hyaluro))を、指示したプライマーを用いて標準的なPCR突然変異誘発により生成させた:
hHEP1−24 BamHI opti(配列番号1)及びhHEP rev 1632(配列番号2)
hep109 M1_E109−Q157_I543
突然変異誘発プライマー:hHEP 304/504
【0070】
【化3】
hep106 M1_P106−K158_I543
突然変異誘発プライマー:hHEP 291/504 bis
【0071】
【化4】
hepGS3 M1_E109−(GS)3−Q157_I543
突然変異誘発プライマー:hHEP 304(GS3)504
【0072】
【化5】
hepGS6 M1_E109−(GS)6−Q157_I543
突然変異誘発プライマー:hHEP 304(GS6 Ala)
【0073】
【化6】
hepGS4 M1_W118−(GS)4−E143_I543
突然変異誘発プライマー:hHEP329(GS4 Ala)
【0074】
【化7】
hepHyaluro M1_W118−(AFKDKPT)(配列番号8)−E143_I543
【0075】
突然変異誘発プライマー:hHEP Hyaluro 5’−
【0076】
【化8】
【実施例3】
【0077】
改変プロテアーゼ切断部位を有するヘパラナーゼ分子の構築
アミノ酸E109とG110との間の、GSリピート(E109−GSGSENLYFQ−GSG−G110(配列番号10)、QとGとの間に切断されやすい結合がある。)に隣接する、タバコエッチウイルス(TEV)プロテアーゼに対するコンセンサス切断部位を挿入する改変ヘパラナーゼ分子を構築するために、テンプレートとしてwtヘパラナーゼを用い、プライマー、hHEP1−24 BamHI opti(配列番号1)及びhHEP rev 1632(配列番号2)及び突然変異誘発プライマー、TEV 110bis
【0078】
【化9】
を用いてPCR突然変異誘発を行った。
【0079】
残基E109/G110の間及びQ157/K158の間の両方にTEV−切断部位を有する改変ヘパラナーゼを構築するために、TEV110コンストラクト(配列番号30、タンパク質配列番号31に対応)をテンプレートとして使用し、PCR突然変異誘発により、突然変異誘発プライマー、TEV158 ter
【0080】
【化10】
を用いて、配列
【0081】
【化11】
を挿入し、hepTEV110/158(配列番号29及び32)を作製した。
【0082】
コンストラクトは全て、PCRにより突然変異が導入されていないことを確認するために両鎖において配列決定を行い、上述のようにpFASTBAC1にクローニングした。
【実施例4】
【0083】
COS7細胞におけるヘパラナーゼ分子の一時的発現。
【0084】
ダルベッコMEM(Gibco BRL、Gaithersburg、MD)中で細胞を増殖させた。全てのコンストラクトを、真核細胞発現プラスミドpcDNA3(Invitrogen)にクローニングした。各コンストラクトの形質移入効率を調べるために、レポーター遺伝子 β−ラクタマーゼ(BLA)をコードするベクターを共形質移入した。同程度の形質移入効率にするために形質移入した各ベクターの量を調整した。fuGENE6形質移入試薬(Roche,Basel,Switzerland)を用いて、製造者の説明書に従い、COS7細胞の一時的形質移入を行った。形質移入から24時間後、BLA−陽性細胞の蛍光検出により効率を評価した。形質移入から96時間後、細胞を回収し、完全プロテアーゼ阻害剤カクテル(Roche)を含有する細胞溶解緩衝液(50mM Tris−HCl pH7.5、150mM NaCl、0.5% Triton)中で再懸濁した。氷上で30分間、細胞溶解を行った。14000rpmで30分間遠心した後、ヘパラナーゼを含有する上清を回収し、下記で概説するように部分精製を行った。
【0085】
一時的形質移入により、ヘパラナーゼコンストラクトを、内因性へパラナーゼ活性を欠くCOS7細胞で発現させた。ヘパリンアフィニティークロマトグラフィーにより細胞溶解液からヘパラナーゼを抽出し、ウェスタンブロットで定量した。並行して、放射性又は蛍光分析のいずれかでヘパラナーゼ酵素活性を測定した(図5)。ウェスタンブロット分析から、wtヘパラナーゼならびに1本鎖コンストラクトGS3及びhyaluroが効率的に発現され、プロセシングされるが、一方で、コンストラクト106及びGS4は、発現されるがプロセシングされないと結論付けた。コンストラクト109及びGS6の発現レベルはきわめて低く、ウェスタンブロット分析により辛うじて検出できる程度であった。wt、GS3及びhyaluroコンストラクトのみが酵素活性を示した。発明者らは、1本鎖コンストラクト106及びGS4は不活性であり、一方、109及びGS6は、おそらく不安定であると結論付けた。GS3及びhyaluroは活性を有するが、切断部位に変更を導入したにもかかわらずプロセシングされるので、前駆体の固有の活性については結論を導くことはできなかった。従って、発明者らは、ヘパラナーゼプロセシングに関与する酵素を欠く細胞において発現を進めた。
【実施例5】
【0086】
昆虫細胞におけるヘパラナーゼ分子の発現。
【0087】
Bac to Bac発現系(Invitrogen)を用いて、ヘパラナーゼコンストラクトを含有する組み換えバキュロウイルスを作製した。10%FBSを含有するGrace昆虫培地で増殖させたSf9昆虫細胞(50x106細胞/T−175フラスコ)を感染させるために、組み換えバキュロウイルスを使用した。感染から48時間後に細胞を回収し、500gで5分間遠心した。COS7に対して使用した150mM NaClのかわりに、可溶性分画におけるタンパク質の量を増加させる、500mM NaClを含有する細胞溶解緩衝液を使用すること以外、上述のようにして、細胞溶解液を調製した。
【0088】
COS7細胞で産生した場合に酵素活性を示した3種類のヘパラナーゼコンストラクトをバキュロウイルス発現系に導入した。Sf9細胞においてタンパク質を発現させ、ヘパリンアフィニティークロマトグラフィーにより精製した。ウェスタンブロット分析により、COS7細胞で観察されたものと対照的に、wt又は突然変異ヘパラナーゼのプロセシングがこの発現系では起こらなかったことが示された。蛍光活性分析による精製1本鎖タンパク質の酵素活性の分析から、非プロセシングwt酵素活性は非常に低く、一方、非プロセシングGS3及びHyaluroタンパク質は、COS7細胞で産生された正しくプロセシングされた野生型酵素で観察されたものに匹敵する特異的活性を有し、活性が高いことが分かった。
【0089】
GS3及びhyaluroは、酵素活性のpH及びイオン強度依存性の点に関してCOS7細胞から抽出された野生型組み換え酵素と、又はHCT−116細胞から部分精製された真正のwt酵素と非常に似ており、ヘパリンにより同様の効力で阻害された。
【0090】
位置109/110及び109/110+157/158においてTEV切断部位を有するコンストラクトを発現させ、ヘパリンアフィニティーカラムで精製し、室温にて一晩、50mM Mes pH6.0、10% グリセロール、0.5mM EDTA中のTEVプロテアーゼ(0.5μM)を用いて消化した。両ケースにおいて完全なプロセシングが観察されたが、しかし、両方の切断ジャンクションにおいてTEV配列を有する二重変異体のみが、この処理により活性化され、このことから、E109/G110ジャンクションにおけるプロセシングのみでは、ヘパラナーゼ活性化を誘導するには十分でないことが示された。
【実施例6】
【0091】
ヘパリンセファロースアフィニティークロマトグラフィーによる組み換えヘパラナーゼコンストラクトの精製。
【0092】
COS7又はSf9昆虫細胞からの細胞溶解液を重力により500μl ヘパリンセファロース CL−6B(Amersham,Piscataway,NJ)に通した。細胞溶解緩衝液2mLでそのカラムを洗浄し、次に、50mM Tris−HCl、pH7.5、500mM NaCl 2mLで洗浄し、50mM Tris−HCl pH7.5、1M NaCl 2mLでヘパラナーゼを溶出し、Biomax−30K遠心分離濃縮装置(Millipore,Bedford,MA)で約5倍に濃縮した。10%グリセロールを添加し、−80℃にて、そのタンパク質を分注して保存した。BIO−RADタンパク質アッセイによりタンパク質濃度を測定した。
【実施例7】
【0093】
大規模発現及び精製
Sf21(又はSf9)細胞を血清不含培地(Sf−900 II SFM、Invitrogen)中での増殖に順応させた。1から10の間の様々な感染の多重度でヘパラナーゼコンストラクトをコードする組み換えバキュロウイルスにより細胞を感染させた。滅菌空気の一定気流状態下、27℃にて、感染細胞3Lをスピナーフラスコ中で増殖させた。感染48時間から96時間後、細胞を回収し、遠心により培地から分離した。合成及びwtヘパラナーゼが細胞ペレットと上清の両方で見られた。細胞ペレットから合成ヘパラナーゼを抽出するために、上記で概説したようにして細胞を溶解した。細胞溶解液又は未精製培地上清を0.22μフィルターで濾過し、50mM Tris−HCl pH7.5、150mM NaClで平衡化した20ml−HyperD Heparin カラム(Biosepra Inc.,Marlboro,MA)に載せた。50mM Tris−HCl pH7.5中の0.15Mから1M NaClの直線的グラディエントを用いて、合成又はwtヘパラナーゼを溶出した。組み換えタンパク質は、>500mMのNaCl濃度で溶出した。集めたヘパラナーゼ含有Heparin−カラム分画を一晩、50mM HEPES pH7.5に対して透析し、同じ緩衝液で平衡化したSource Sカラム(Amersham)に載せた。ヘパラナーゼコンストラクトは、400mMから600mM NaClで溶出した。15/30 Superdex 75サイズ排除カラムでのさらなるクロマトグラフィー段階により、タンパク質を均質になるように精製した。精製したタンパク質を分注し、液体窒素中で瞬時に冷凍し、−80℃で保存した。
【実施例8】
【0094】
ウェスタンブロッティング
50kDaサブユニット内に含有されるペプチド(アミノ酸225から241に対応し、そのC−末端にさらなる配列GGCを含有する、EPNSFLKKADIFINGSQ(配列番号14))に対して、ウサギポリクローナル抗体を作製した。チオプロピルセファロースレジン(Amersham)に固定化した免疫原ペプチドを用いて抗血清を免疫精製した。ヘパリンカラムから溶出したタンパク質10μlを10% SDS−ポリアクリルアミドゲル電気泳動に供し、Protran BA 83 Cellulosenitrate膜(Schleicher&Schuell Bioscience,Keene,NH)に転写した。5%ミルクにより非特異的結合を飽和させた後、4℃にて一晩、5%ミルク、TBS及び0.05% Tween 20中で1:500希釈した上記のポリクローナル抗体とともにその膜をインキュベーションした。洗浄後、1:5000に希釈した抗ウサギホースラディッシュペルオキシダーゼ結合抗体とともに、室温にて30分間、その膜をインキュベーションした。SuperSignal West Pico Chemiluminescent Substrate(Pierce Biotechnology,Rockford,IL)により、免疫反応のあるバンドを検出した。最後に、BIOMAX MRフィルム(Kodak)に10秒間、その膜を曝露した。
【実施例9】
【0095】
ヘパラン硫酸の蛍光標識
既に述べられているようにしてフルオレセインイソチオシアネート(FITC)でウシ腎臓由来のヘパラン硫酸ナトリウム塩(Sigma−Aldrich Corp.,St.Louis,MO)を標識した(Toyoshima及びNakajima,J.,Biol.Chem.274:24153−24160(1999))。ヘパラン硫酸 5mgとFITC5mgとを、0.1M Na2CO3 pH9.5 1mLに溶解し、4℃にて一晩、暗所でインキュベーションした。次に、未処理ヘパラン硫酸からFITC標識ヘパラン硫酸(FITC−HS)を分離するために、MicroSpin G−25カラムにこの溶液を載せた。高分子量のヘパラン硫酸種を分離するために、150mM NaCl、25mM Tris−HCl pH=7.5緩衝液中のSephacryl S−300を介した第一のゲル濾過クロマトグラフィー段階にこのFITC−HSを供した。着色した分画を集め、Biomax−10K遠心濃縮装置(Millipore)で濃縮し、一様な分子量のヘパラン硫酸種を得るために、Sephacryl S−300(上記)で再びクロマトグラフィーを行った。HPLC Superdex 75TM(Pharmacia Biotech)クロマトグラフィーシステムにより、溶出した分画を分析した。L−7485蛍光検出装置(Merck Hitachi)により、各分画の蛍光を測定した。発明者らは、分子量が異なるヘパラン硫酸産物を含む4個の主要な分画を得た。Blyscan Glycosaminoglycan Assay(Biocolor Ltd.,Belfast,Northern Ireland)により各分画におけるFITC−HSの量を測定した。
【実施例10】
【0096】
蛍光分析。
【0097】
このアッセイは、HPLCサイズ排除クロマトグラフィーによりモニターするFITC−HSの分解に基づく。50mM MES pH6、10% グリセロール(ヘパラナーセ活性緩衝液、HAB) 50μl中で、FITC−HS 5μlとともに精製ヘパラナーゼ 8μをインキュベーションした。この反応混合物を室温にて一定の時間インキュベーションし、ヘパリン 50μgを添加することにより反応を停止させた。次に、この混合物をUltrafree−MC遠心濾過装置(Millipore)を用いて濾過した。50mM Hepes pH7.5 150mM Na2SO4緩衝液中で平衡化し、Merck−Hitachi HPLCシステムに連結したSuperdex 75TM(Pharmacia Biotech)カラムに20μlを注入した。L−7485蛍光検出装置により蛍光ヘパラン硫酸分解産物を検出した。そのままのFITC−HSに対する、低分子量のヘパラン硫酸種における増加をモニタリングすることにより、ヘパラナーゼ活性を評価し、ピーク領域の積分により定量した。
【実施例11】
【0098】
ヘパラン硫酸の還元末端における放射性標識及びビオチン化
ウシ腎臓由来のヘパラン硫酸ナトリウム塩 10mg(Sigma)を部分的に、N−デ−アセチル化し、既に述べられているようにして[3H]無水酢酸で再びアセチル化した(Freeman及びParish,Biochem.J.325:229−237(1997))。次に、トリチウム化したヘパラン硫酸を、述べたように還元末端で還元的アミノ化した。トリチウム化し、還元的にアミノ化したヘパラン硫酸を、EZ−Link Sulfo−NHS−LC−Biotin(Pierce)を用いて、さらにビオチンに結合させた。このビオチン類似体は、ヘパラン硫酸分子の還元末端で生成されるアミノ基と反応し得るN−ヒドロキシスクシンイミドエステル部分を有する。発明者らは、還元的にアミノ化し、H2O 1mLに再懸濁したトリチウム化ヘパラン硫酸 約5mg(ヘパラン硫酸分子量の平均値を500kDaとして、100M 推定最終濃度)において、回収率を計算した。この溶液 100μlに対して、EZ−LinkSulfo−NHS−LC−Biotin 1mg(約100倍モル過剰)及びリン酸緩衝液 pH7.5 20μlを添加した。この反応混合液を室温にて一晩インキュベーションした。次に、ビオチン化したトリチウム化ヘパラン硫酸を未反応ビオチンから分離するために、この反応混合物をPD−10脱塩カラムにかけた。発明者らは、最終的に、4個の分画(各1mL)を得て、Reacti−Bind Streptavidin High Binding Capacity Coated Plates(Pierce)に固定化するために、それらの活性について試験した。
【実施例12】
【0099】
放射性アッセイ
このアッセイは、マイクロプレートに固定化したトリチウム化ヘパラン硫酸の分解に基づく。Reacti−Bind Streptavidin High Binding Capacity Coated Platesの各ウェルを、製造者の説明書に従い前処理した。最初に、PD−10脱塩カラム後に得られた、トリチウム化したビオチン化ヘパラン硫酸の各分画の様々な量を、PBS中で、最終体積100μlとなるように、各ウェルに添加(デュプリケート)した。100x103d.p.m.に対応する分画2の体積で最大結合が得られることを見極めた後、いつもこの量を使用した。室温にて一晩この結合を行った。次にウェルをPBSで3回洗浄し、HABで2回洗浄した。精製ヘパラナーゼ 10μlを各ウェルにHAB中で添加し、最終体積を100μlとした。このウェルを室温にて2時間から24時間インキュベーションした。最終的に、各ウェルにおいてヘパラナーゼにより生じるトリチウム化ヘパラン硫酸産物による遊離放射性活性を測定し、緩衝液ブランクに対して正規化した。
【実施例13】
【0100】
ヘパラナーゼコンストラクトの特異的活性の測定。
【0101】
COS7細胞において一時的に発現させた、又はバキュロウイルス系において発現させたいずれかのヘパラナーゼコンストラクトの特異的活性を次のように測定した:
特異的活性=正規化活性(d.p.m./μl)/正規化した濃度測定量(densitometric volume)(測定量/μl)
詳しく説明すると、放射性アッセイにおいて、直線的な用量−活性関係が見られるような方法において各調製物を滴定することにより部分精製ヘパラナーゼコンストラクトの活性を測定した。各調製物を用いてこれらの滴定を3回繰り返し、平均、正規化活性(d.p.m./μl)を計算した。ウェスタンブロッティング実験によりタンパク質発現を調べ:デンシトメトリーにより化学発光の読み出しを定量した。再び、実験を3回繰り返し、平均値を決定した。正規化した濃度測定量(densitometric volume)(測定量/μl)で正規化した活性(d.p.m./μl)を割ることにより、特異的活性を得た。
【図面の簡単な説明】
【0102】
【図1】図1は、哺乳類細胞におけるヒトヘパラナーゼの生合成を示す。
【図2A】図2パネルAは、改変TEV切断部位を有するヘパラナーゼコンストラクトの概略図を示す。
【図2B】パネルB(左)は、COS7細胞(レーン1)、hepTEV110(レーン2)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110(レーン3)又は0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110(レーン4)、hepTEV110/158(レーン5)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110/158(レーン6)、0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110/158(レーン7)において発現された、正しくプロセシングされたwtヘパラナーゼのウェスタンブロット分析の結果を示す。パネルB(右)は、hepTEV110(カラム1)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110(カラム2)又は0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110(カラム3)、hepTEV110/158(カラム4)、0.5μM TEVプロテアーゼとの16時間インキュベーション後のhepTEV110/158(カラム5)、0.5μM TEVプロテアーゼ無しでの16時間インキュベーション後のhepTEV110/158(カラム6)のヘパラナーゼ活性を示す。蛍光法を使用して、これらの試料のヘパラナーゼ活性を評価した。
【図3A−1】パネルA:関連配列に対する、ヘパラナーゼの多配列アラインメント。予想される二次構造エレメントをアラインメントの上に示す(矢印=β鎖、円柱=へリックス)。この2つの切断部位の位置は、黒い三角印により示す。ヒアルロニダーゼ断片により置換される、除去されるヘパラナーゼ部分の領域を、灰色のボックスで囲む。
【図3A−2】パネルA:関連配列に対する、ヘパラナーゼの多配列アラインメント。予想される二次構造エレメントをアラインメントの上に示す(矢印=β鎖、円柱=へリックス)。この2つの切断部位の位置は、黒い三角印により示す。ヒアルロニダーゼ断片により置換される、除去されるヘパラナーゼ部分の領域を、灰色のボックスで囲む。
【図3B】パネルB:TIMバレル構造の概略図。除去されるヘパラナーゼ部分の位置は、三角印で表す切断位置により示す。存在する場合は、その部分は、β/αユニット1及び2により基質(灰色の矢印)の結合を妨げる可能性が高い。より短いループ(点線)を設計することにより、この抑制を解除し、これにより、活性を有する酵素が得られ、同時に、その酵素の構造的完全性が維持される。
【図4A】パネルA:本明細書で述べた1本鎖ヘパラナーゼコンストラクトの概略図。
【図4B】パネルB、左:COS7細胞において発現させたwtヘパラナーゼ又は1本鎖コンストラクトのウェスタンブロッティング分析。Blaは、レポーター遺伝子β−ラクタマーゼ(材料と方法セクションを参照のこと。)をコードするベクターのみを形質移入したCOS7細胞の部分精製細胞溶解液に相当する対照である。右:放射性アッセイを用いた同じ試料のヘパラナーゼ活性。全ての1本鎖コンストラクトの特異的活性は、wtヘパラナーゼの特異的活性に対して正規化した。
【図5】左、COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼ又はSf9細胞で発現されたwtヘパラナーゼ及び1本鎖コンストラクトのウェスタンブロット分析。右:放射性アッセイを用いた同じ試料のヘパラナーゼ活性。Sf9細胞で発現されたwtヘパラナーゼ及び1本鎖コンストラクトの特異的活性は、COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼの特異的活性に対して正規化した。
【図6】COS7細胞で産生された、正しくプロセシングされたwtヘパラナーゼ(黒丸)及び非プロセシングFITC−HS(白丸)と比較した、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖タンパク質とともに6時間インキュベーションを行った後に得られたFITC−HS分解産物のサイズ排除クロマトグラフィー。
【図7A】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、イオン強度依存性(パネルA)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
【図7B】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、ヘパリンによる阻害(パネルB)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
【図7C】COS7細胞で産生された野生型ヘパラナーゼ(黒丸)、昆虫細胞で産生されたhepG3(白四角)及びhepHyal(黒三角)1本鎖コンストラクトの、蛍光活性分析を用いた、pH依存性(パネルC)。ヘパリンにおいて、滴定実験で、次のIC50値が得られた:hepwt、0.9ng/μl;hepGS3、1.1ng/μl;hepHyal、1.5ng/μl。
【特許請求の範囲】
【請求項1】
哺乳類ヘパラナーゼタンパク質をコードするヌクレオチドの配列を含み、該ヌクレオチドの配列が、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含み、該切断部位が、前記ヘパラナーゼタンパク質の残基100及び残基168をコードするヌクレオチド間に位置する、合成核酸分子。
【請求項2】
請求項1に記載の核酸分子を含む、ベクター。
【請求項3】
ベクターがバキュロウイルスベクターである、請求項2に記載のベクター。
【請求項4】
請求項3に記載のベクターを含む、宿主細胞。
【請求項5】
宿主細胞が、昆虫細胞である、請求項4に記載の宿主細胞。
【請求項6】
宿主細胞が、酵母細胞である、請求項4に記載の宿主細胞。
【請求項7】
酵母が、Pichia pastoris(ピキア パストリス)、Hansenula polymorpha(ハンセヌラ ポリモルファ)及びSaccharomyces cervisiae(サッカロミセス セルビシエ)からなる群から選択される、請求項6に記載の宿主細胞。
【請求項8】
ヘパラナーゼタンパク質が、ヒトヘパラナーゼである、請求項1に記載の合成核酸分子。
【請求項9】
コンセンサス切断部位が、ヒトヘパラナーゼタンパク質の残基G110及びK158の前に位置する、請求項8に記載の合成核酸分子。
【請求項10】
コンセンサス切断部位が、タバコエッチウイルス(TEV)プロテアーゼ切断部位、ピコルナウイルスの3Cプロテアーゼ切断部位、トロンビンプロテアーゼ切断部位、エンテロキナーゼ切断部位及び因子Xa切断部位からなる群から選択される、請求項8に記載の合成核酸分子。
【請求項11】
哺乳類ヘパラナーゼタンパク質をコードする部分を含み、該タンパク質コード部分が、約8kDaのN−末端断片をコードするヌクレオチドの配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチドの配列と、から実質的になり、前記N−末端及びC−末端断片が、野生型ヘパラナーゼ断片と実質的に同様であるタンパク質断片をコードし、該コードされるヘパラナーゼタンパク質が構成的に活性である、合成哺乳類ヘパラナーゼ核酸分子。
【請求項12】
タンパク質コード部分が、ヒトヘパラナーゼをコードする、請求項11に記載の遺伝子。
【請求項13】
リンカーが、ヒアルロニダーゼタンパク質の中央ループ領域をコードするヌクレオチドの配列を含む、請求項11に記載の遺伝子。
【請求項14】
ヒアルロニダーゼが、H.マニレンシス(H.manillensis)由来である、請求項13に記載の遺伝子。
【請求項15】
リンカーが、(GlySer)3リンカーをコードするヌクレオチドの配列を含む、請求項12に記載の遺伝子。
【請求項16】
請求項12に記載の遺伝子を含むベクター。
【請求項17】
請求項16に記載のベクターを含む宿主細胞。
【請求項18】
昆虫細胞又は酵母細胞である、請求項17に記載の宿主細胞。
【請求項19】
請求項12に記載の遺伝子によりコードされる、精製された合成ヘパラナーゼタンパク質。
【請求項20】
(a)哺乳類ヘパラナーゼタンパク質をコードし、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含み、該切断部位が前記ヘパラナーゼタンパク質の残基100と残基168との間に位置するヌクレオチドの配列を含むベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で前記宿主細胞を培養することと;
(c)前記細胞を崩壊させて、少なくとも部分的に前記ヘパラナーゼタンパク質を精製することと;
(d)少なくとも部分的に精製したヘパラナーゼタンパク質をエンドプロテアーゼに曝露し、前記ヘパラナーゼタンパク質を前記コンセンサス切断部位で切断することと、を含む、非哺乳類細胞において哺乳類ヘパラナーゼを発現させる方法。
【請求項21】
ヘパラナーゼがヒトである、請求項20に記載の方法。
【請求項22】
(a)ヘパラナーゼタンパク質をコードする部分を含み、該タンパク質コード部分が、約8kDaのN−末端断片をコードするヌクレオチドの配列と、リンカーをコードするヌクレオチドの配列と、約50kDaのC−末端断片をコードするヌクレオチドの配列と、から実質的になり、前記N−末端及びC−末端断片が、野生型断片と実質的に同様であるタンパク質断片をコードする、合成哺乳類ヘパラナーゼ遺伝子を含むベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で前記宿主細胞を培養することと、を含む、非哺乳類細胞において、1本鎖の、構成的に活性な哺乳類ヘパラナーゼを発現させる方法。
【請求項23】
リンカーが、ヒアルロニダーゼタンパク質の中央ループ領域を含む、請求項22に記載の方法。
【請求項24】
リンカーが、中央(GlySer)3を含む、請求項22に記載の方法。
【請求項25】
請求項22に記載の方法により産生される実質的に純粋なタンパク質。
【請求項1】
哺乳類ヘパラナーゼタンパク質をコードするヌクレオチドの配列を含み、該ヌクレオチドの配列が、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含み、該切断部位が、前記ヘパラナーゼタンパク質の残基100及び残基168をコードするヌクレオチド間に位置する、合成核酸分子。
【請求項2】
請求項1に記載の核酸分子を含む、ベクター。
【請求項3】
ベクターがバキュロウイルスベクターである、請求項2に記載のベクター。
【請求項4】
請求項3に記載のベクターを含む、宿主細胞。
【請求項5】
宿主細胞が、昆虫細胞である、請求項4に記載の宿主細胞。
【請求項6】
宿主細胞が、酵母細胞である、請求項4に記載の宿主細胞。
【請求項7】
酵母が、Pichia pastoris(ピキア パストリス)、Hansenula polymorpha(ハンセヌラ ポリモルファ)及びSaccharomyces cervisiae(サッカロミセス セルビシエ)からなる群から選択される、請求項6に記載の宿主細胞。
【請求項8】
ヘパラナーゼタンパク質が、ヒトヘパラナーゼである、請求項1に記載の合成核酸分子。
【請求項9】
コンセンサス切断部位が、ヒトヘパラナーゼタンパク質の残基G110及びK158の前に位置する、請求項8に記載の合成核酸分子。
【請求項10】
コンセンサス切断部位が、タバコエッチウイルス(TEV)プロテアーゼ切断部位、ピコルナウイルスの3Cプロテアーゼ切断部位、トロンビンプロテアーゼ切断部位、エンテロキナーゼ切断部位及び因子Xa切断部位からなる群から選択される、請求項8に記載の合成核酸分子。
【請求項11】
哺乳類ヘパラナーゼタンパク質をコードする部分を含み、該タンパク質コード部分が、約8kDaのN−末端断片をコードするヌクレオチドの配列と、リンカーと、約50kDaのC−末端断片をコードするヌクレオチドの配列と、から実質的になり、前記N−末端及びC−末端断片が、野生型ヘパラナーゼ断片と実質的に同様であるタンパク質断片をコードし、該コードされるヘパラナーゼタンパク質が構成的に活性である、合成哺乳類ヘパラナーゼ核酸分子。
【請求項12】
タンパク質コード部分が、ヒトヘパラナーゼをコードする、請求項11に記載の遺伝子。
【請求項13】
リンカーが、ヒアルロニダーゼタンパク質の中央ループ領域をコードするヌクレオチドの配列を含む、請求項11に記載の遺伝子。
【請求項14】
ヒアルロニダーゼが、H.マニレンシス(H.manillensis)由来である、請求項13に記載の遺伝子。
【請求項15】
リンカーが、(GlySer)3リンカーをコードするヌクレオチドの配列を含む、請求項12に記載の遺伝子。
【請求項16】
請求項12に記載の遺伝子を含むベクター。
【請求項17】
請求項16に記載のベクターを含む宿主細胞。
【請求項18】
昆虫細胞又は酵母細胞である、請求項17に記載の宿主細胞。
【請求項19】
請求項12に記載の遺伝子によりコードされる、精製された合成ヘパラナーゼタンパク質。
【請求項20】
(a)哺乳類ヘパラナーゼタンパク質をコードし、エンドプロテアーゼにより認識される2個のコンセンサス切断部位を含み、該切断部位が前記ヘパラナーゼタンパク質の残基100と残基168との間に位置するヌクレオチドの配列を含むベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で前記宿主細胞を培養することと;
(c)前記細胞を崩壊させて、少なくとも部分的に前記ヘパラナーゼタンパク質を精製することと;
(d)少なくとも部分的に精製したヘパラナーゼタンパク質をエンドプロテアーゼに曝露し、前記ヘパラナーゼタンパク質を前記コンセンサス切断部位で切断することと、を含む、非哺乳類細胞において哺乳類ヘパラナーゼを発現させる方法。
【請求項21】
ヘパラナーゼがヒトである、請求項20に記載の方法。
【請求項22】
(a)ヘパラナーゼタンパク質をコードする部分を含み、該タンパク質コード部分が、約8kDaのN−末端断片をコードするヌクレオチドの配列と、リンカーをコードするヌクレオチドの配列と、約50kDaのC−末端断片をコードするヌクレオチドの配列と、から実質的になり、前記N−末端及びC−末端断片が、野生型断片と実質的に同様であるタンパク質断片をコードする、合成哺乳類ヘパラナーゼ遺伝子を含むベクターを用いて、非哺乳類細胞に対して形質転換又は形質移入を行うことと;
(b)前記ヘパラナーゼタンパク質の発現を可能にする条件下で前記宿主細胞を培養することと、を含む、非哺乳類細胞において、1本鎖の、構成的に活性な哺乳類ヘパラナーゼを発現させる方法。
【請求項23】
リンカーが、ヒアルロニダーゼタンパク質の中央ループ領域を含む、請求項22に記載の方法。
【請求項24】
リンカーが、中央(GlySer)3を含む、請求項22に記載の方法。
【請求項25】
請求項22に記載の方法により産生される実質的に純粋なタンパク質。
【図1】


【図2A】
【図2B】

【図3A−1】
【図3A−2】
【図3B】

【図4A】

【図4B】
【図5】

【図6】

【図7A】
【図7B】
【図7C】
【図2A】
【図2B】
【図3A−1】
【図3A−2】
【図3B】
【図4A】
【図4B】
【図5】
【図6】
【図7A】
【図7B】
【図7C】
【公表番号】特表2007−506416(P2007−506416A)
【公表日】平成19年3月22日(2007.3.22)
【国際特許分類】
【出願番号】特願2006−527328(P2006−527328)
【出願日】平成16年9月17日(2004.9.17)
【国際出願番号】PCT/EP2004/010517
【国際公開番号】WO2005/030962
【国際公開日】平成17年4月7日(2005.4.7)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
【公表日】平成19年3月22日(2007.3.22)
【国際特許分類】
【出願日】平成16年9月17日(2004.9.17)
【国際出願番号】PCT/EP2004/010517
【国際公開番号】WO2005/030962
【国際公開日】平成17年4月7日(2005.4.7)
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
[ Back to top ]