
吸水剤およびその製造方法、並びに、衛生材料
【課題】 無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性のバランスに優れるとともに、製造の際のロットごと、あるいは各ロット中における生理食塩水流れ誘導性の値の振れが小さい、物性の安定した吸水剤を短時間で製造する方法および吸水剤を提供する。
【解決手段】 本発明にかかる吸水剤の製造方法の1つは、酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする。
【解決手段】 本発明にかかる吸水剤の製造方法の1つは、酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、吸水剤およびその製造方法に関する。さらに詳しくは、本発明は、吸水性樹脂を架橋剤で改質することで得られる吸水剤であって、無加圧下でも加圧下でも高い吸収倍率、さらに高い生理食塩水流れ誘導性を示す吸水剤およびその製造方法に関する。
【背景技術】
【0002】
近年、紙オムツや生理用ナプキン、いわゆる失禁パット等の衛生材料には、その構成材として、体液を吸収させることを目的とした吸水性樹脂(吸水剤)が幅広く使用されている。
上記の吸水性樹脂としては、例えば、ポリアクリル酸部分中和物架橋体、澱粉−アクリル酸グラフト重合体の加水分解物、酢酸ビニル−アクリル酸エステル共重合体ケン化物、アクリロニトリル共重合体若しくはアクリルアミド共重合体の加水分解物またはこれらの架橋体、及びカチオン性モノマーの架橋重合体等が知られている。
上記の吸水性樹脂が備えるべき特性として、従来より体液等の水性液体に接した際の優れた吸液量や吸水速度、ゲル強度、ゲル通液性、水性液体を含んだ基材から水を吸い上げる吸引力等が唱えられている。そして、従来よりこれらの特性を複数併せ持ち、紙オムツや生理用ナプキン等の衛生材料に用いられた場合に、優れた性能(吸収特性)を示す吸水性樹脂(吸水剤)が種々提案されている。
【0003】
例えば、吸水性樹脂の無加圧下吸収倍率および加圧下吸収倍率等の吸収特性をバランス良く改良する方法として、吸水性樹脂の表面近傍を架橋する技術が知られており、これまでに様々な方法が開示されている。
その例として、多価アルコールを用いる方法(特許文献1、特許文献2)、多価グリシジル化合物、多価アジリジン化合物、多価アミン化合物、多価イソシアネート化合物を用いる方法(特許文献3)、グリオキサールを用いる方法(特許文献4)、多価金属を用いる方法(特許文献5、特許文献6、特許文献7)、シランカップリング剤を用いる方法(特許文献8、特許文献9、特許文献10)、アルキレンカーボネートを用いる方法(特許文献11)、多価ヘテロ環カーボネートを用いる方法(特許文献12)、オキサゾリジノンを用いる方法(特許文献13)、多価オキサゾリジノンを用いる方法(特許文献14)、オキサジンを用いる方法(特許文献15)、オキサゾリン化合物を用いる方法(特許文献16)等が知られている。
【0004】
さらに、上記架橋剤によって吸収特性の向上を行う際に、更なる性能向上のために添加剤(不活性混合助剤、酸触媒、塩基)を用いる方法も知られている。すなわち、添加剤として不活性混合助剤を用いる方法(1)として、不活性無機粉末を存在させる方法(特許文献17、特許文献18)、多価金属の塩および/または水酸化物を含む水を存在させる方法(特許文献7)、二価アルコールを存在させる方法(特許文献19)、水とエーテル化合物を存在させる方法(特許文献20)、水溶性ポリマーを存在させる方法(特許文献21)、1価アルコールのアルキレンオキサイド付加物、有機酸の1価塩、またはラクタム類を存在させる方法(特許文献22、特許文献23)、一価金属塩を存在させる方法(特許文献24)、カチオンを存在させる方法(特許文献25、特許文献26)等が知られている。
【0005】
さらに、添加剤として酸触媒を用いる方法(2)として、リン酸を存在させる方法(特許文献27)、無機酸または有機酸を存在させる方法(特許文献28)等が知られている。
また、添加剤として塩基を用いる方法(3)として、水溶性アルカリ化合物を存在させる方法(特許文献29)等も知られている。
これら(1)、(2)、(3)の方法で用いる添加剤が架橋剤と共に存在することにより、架橋剤単独に比べて吸水剤の吸収特性のバランスをある程度向上させることもできるが、まだまだ十分なものとは言い難いものであった。
【0006】
すなわち、(1)の方法で用いるような添加剤(不活性混合助剤)では、使用する吸水性樹脂に微紛が多く含まれる等の場合には混合助剤としての働きによりその効果が現れるものの、一方ではその存在により架橋剤の吸水性樹脂粉末への浸透性の過度の低下や架橋反応の阻害等により吸収特性の改善がほとんど見られなかったり、改善するにしても架橋剤の使用量が増加する、反応時間が長くなる、反応温度を上昇させなければならない、等という問題点もあった。
(2)の方法で用いるような添加剤(酸触媒)では、架橋剤の反応を促進する触媒としての効果が期待できるものの、ある程度の効果を得るための量を添加すると架橋剤溶液のpHの極度な低下と、酸基を含有する部分中和型の吸水性樹脂の場合などには特に表面の酸性化が起こって架橋剤の浸透性の制御が困難となる。また、表面の酸性化は吸水性樹脂の粒子間の接着性を増加し、凝集体の形成につながる傾向にあるので好ましくない。その結果、所望する吸水性樹脂粒子表面層の架橋密度が得られず性能的に満足できるものが得難いという問題点がある。
【0007】
(3)の方法には、添加剤(塩基)とカルボキシル基と容易に反応する官能基を2個以上有する化合物(多価金属塩、ポリエポキシ化合物、ポリアジリジニル化合物、ポリイソシアネート化合物)との組み合わせによる表面架橋が開示され、ゲル強度や比較的低荷重(20g/cm2)での加圧下吸収倍率の改善が図られている。しかし、特許文献29の方法では、いまだ表面架橋剤の混合性改良や吸水性樹脂の物性改良に不十分であり、特に、近年求められているSFCや高荷重下(4.83kPa、約50g/cm2)での加圧下吸収倍率(AAP)(いずれも後述)を向上させることは困難であった。
また、典型的な吸水性樹脂としては、その高物性やコスト面から、アクリル酸の部分中和塩架橋体からなるアクリル酸系吸水性樹脂が挙げられる。そして、かかるアクリル酸系吸水性樹脂の製造方法としては、予め所定中和率に中和したアクリル酸およびその塩を重合する方法(以下、中和重合法と呼ぶ)と、未中和ないし低中和のアクリル酸を重合してから重合ゲルを後中和する方法(以下、酸型重合法)の2つが一般的に行われている。
【0008】
中和重合法に比べて、後者の酸型重合法では高吸収倍率で低可溶分の吸水性樹脂が得られる傾向にはあるが、重合後の含水ゲル状架橋重合体を均一に中和するには長時間を要する上に技術的に非常に困難であり、得られる吸水性樹脂粉末の個々の粒子の中和率が不均一になる場合がある。このような場合、酸型重合法の吸水性樹脂は高吸収倍率で低可溶分にもかかわらず、表面架橋処理を行っても十分な吸水剤の性能が得られないことが、特許文献30(欧州特許公開0882502A1)で開示されている。
すなわち、従来、吸水性樹脂を表面架橋する場合にはその中和率の違いにより必要とされる表面架橋処理が異なり、ある中和率で最適な架橋処理でも他の中和率では所望する吸水剤の性能が得られなかったり、特に吸水性樹脂粉末の個々の粒子の中和率が多岐に混在する場合には、所望する吸水剤の性能が得られないことがあった。
【0009】
以上述べたように、従来の技術によっては表面架橋処理を均一に行うことができておらず、その結果、得られる吸水性樹脂の各種物性(後述のCRC、AAP、SFCなど、特にSFC)のバランスが悪くなるとともに、SFCにバラツキが生じていた。このため、例えば、オムツの性能のロット振れや、オムツ1枚の中でも性能に大きな差が生じていた。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭58−180233号公報
【特許文献2】特開昭61−16903号公報
【特許文献3】特開昭59−189103号公報
【特許文献4】特開昭52−117393号公報
【特許文献5】特開昭51−136588号公報
【特許文献6】特開昭61−257235号公報
【特許文献7】特開昭62−7745号公報
【特許文献8】特開昭61−211305号公報
【特許文献9】特開昭61−252212号公報
【特許文献10】特開昭61−264006号公報
【特許文献11】独国特許第4020780号公報
【特許文献12】特開平11−315216号公報
【特許文献13】WO99/42494号公報
【特許文献14】WO99/43720号公報
【特許文献15】WO00/31153号公報
【特許文献16】特開2000−197818号公報
【特許文献17】特開昭60−163956号公報
【特許文献18】特開昭60−255814号公報
【特許文献19】特開平1−292004号公報
【特許文献20】特開平2−153903号公報
【特許文献21】特開平3−126730号公報
【特許文献22】特公平6−74331号公報
【特許文献23】特開平7−33818号公報
【特許文献24】WO98/49221号公報
【特許文献25】WO00/53664号公報
【特許文献26】WO00/53644号公報
【特許文献27】WO94/15651号公報
【特許文献28】特開平7−278225号公報
【特許文献29】特開平6−298841号公報
【特許文献30】特開平10−101735号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、上記従来の問題点に鑑みなされたものである。すなわち、本発明の課題は、架橋処理時に、混合助剤としての効果を示しながらも架橋反応を阻害せず、場合によっては反応触媒としての効果も併せ持ち、かつ部分中和重合された吸水性樹脂の中和率の違いや酸型重合の後中和操作に起因する中和率の均一性にほとんど関わりなく、均一な表面架橋が発現できる吸水剤の製造方法を提供することにある。さらに本発明の課題は、無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性のバランスに優れるとともに、製造の際のロットごと、あるいは各ロット中における生理食塩水流れ誘導性の値の振れ(バラツキ)が小さい、物性の安定した吸水剤を短時間で製造する方法および吸水剤を提供することにもある。
【課題を解決するための手段】
【0012】
本発明者は、吸収特性に優れた吸水剤を鋭意検討した結果、特定の添加剤を吸水性樹脂の特定の架橋剤と共に用いることで上記課題が解決することを見出し、本発明を完成させるに至った。
すなわち、本発明にかかる吸水剤は、
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤に関する。
【0013】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
また、本発明にかかる別の吸水剤は
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤に関する。
【0014】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
さらに、本発明にかかる別の吸水剤は、
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、連続生産された吸水剤に関する。
【0015】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
さらに、本発明にかかる別の吸水剤は、
酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上である。
【0016】
また、本発明にかかる吸水剤の製造方法は、
酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする。
さらに、本発明にかかる別の吸水剤の製造方法は、
酸基を含有し且つ重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる架橋剤(c)を混合し、前記吸水性樹脂粉末(a1)を架橋処理することを特徴とする。
【0017】
また、本発明の衛生材料は、本発明にかかる吸水剤を含む。
【発明の効果】
【0018】
本発明によれば、架橋処理時に、混合助剤としての効果を示しながらも架橋反応を阻害せず、場合によっては反応触媒としての効果も併せ持ち、かつ部分中和重合された吸水性樹脂の中和率の違いや酸型重合の後中和操作に起因する中和率の均一性にほとんど関わりなく、均一な表面架橋が発現でき、無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性のバランスに優れるとともに、製造の際のロットごと、あるいは各ロット中における生理食塩水流れ誘導性の値の振れ(バラツキ)が小さい、物性の安定した吸水剤を短時間で製造する方法および吸水剤を提供することができる。
【図面の簡単な説明】
【0019】
【図1】生理食塩水流れ誘導性の測定に用いる測定装置の概略の断面図。
【図2】機械的ダメージ試験に用いるガラス製容器の側面概略図(a)と平面概略図(b)。
【図3】機械的ダメージ試験に用いる分散機の概略図。
【発明を実施するための形態】
【0020】
以下、本発明について詳細に説明する。なお、以下、本発明で吸水剤とは、架橋構造を有する吸水性樹脂(以下、単に吸水性樹脂と呼ぶ)を主成分(好ましくは50〜100重量%、より好ましくは80〜100重量%、さらに好ましくは90〜100重量%)とし、吸水性樹脂をさらに架橋剤で改質(好ましくは表面改質、特に表面架橋)されたものを吸水剤と呼ぶ。
(吸水性樹脂の製造方法)
以下、本発明において、酸基含有の吸水性樹脂粉末を吸水性樹脂粉末(a)と呼び、かかる吸水性樹脂粉末(a)の中でも、さらに粒度を制御したもの、例えば、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下のものを吸水性樹脂粉末(a1)と呼ぶ。
【0021】
本発明の吸水性樹脂とは、従来から知られている吸水性樹脂のことであり、例えばイオン交換水中において、必須に自重の5倍以上、好ましくは、50倍から1000倍という多量の水を吸収し、アニオン性、ノニオン性、またはカチオン性の水不溶性ヒドロゲルを形成する従来公知の架橋重合体のことである。
これらは、一般に、不飽和単量体成分(好ましくは酸基、特に、カルボキシル基含有不飽和単量体)を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、単量体溶液の状態で重合され、必要に応じて該重合体を乾燥し、乾燥の前および/または後で通常粉砕して得られたものである。このような吸水性樹脂としては、ポリアクリル酸部分中和物重合体、デンプン−アクリロニトリルグラフト重合体の加水分解物、デンプン−アクリル酸グラフト重合体、酢酸ビニル−アクリル酸エステル共重合体のケン化物、アクリロニトリル共重合体もしくはアクリルアミド共重合体の加水分解物、またはこれらの架橋体、カルボキシル基含有架橋ポリビニルアルコール変性物、架橋イソブチレンー無水マレイン酸共重合体等の1種または2種以上を挙げることができる。
【0022】
これらの吸水性樹脂は、1種または混合物でも用いられるが、中でも酸基含有の吸水性樹脂、さらには、カルボン酸またはその塩であるカルボキシル基含有の吸水性樹脂の1種またはその混合物が好ましく、典型的にはアクリル酸及び/又はその塩(中和物)を主成分とする単量体を重合・架橋することにより得られる重合体、すなわち、必要によりグラフト成分を含むポリアクリル酸塩架橋重合体が主成分とされる。
また、上記吸水性樹脂としては、水膨潤性水不溶性であることが必須であり、該吸水性樹脂中の未架橋の水可溶性成分(水溶性高分子)は、好ましくは50重量%以下、より好ましくは25重量%以下、さらに好ましくは20重量%以下、さらにより好ましくは15重量%以下、特に好ましくは10重量%以下のものが用いられる。
【0023】
上記アクリル酸塩としては、アクリル酸のナトリウム、カリウム、リチウム等のアルカリ金属塩、アンモニウム塩及びアミン塩等を例示することができる。上記吸水性樹脂は、その構成単位としてアクリル酸0〜50モル%およびアクリル酸塩100〜50モル%(但し、両者の合計量は100モル%以下とする)の範囲にあるものが好ましく、アクリル酸10〜40モル%およびアクリル酸塩90〜60モル%(但し、両者の合計量は100モル%以下とする)の範囲にあるものがより好ましい。なお、この酸と塩とのモル比を中和率と呼ぶ。上記塩を形成させるための吸水性樹脂の中和は重合前に単量体の状態で行っても良いし、あるいは重合途中や重合後に重合体の状態で行っても良いし、それらを併用してもよい。
【0024】
一般に、未中和ないし低中和の単量体を重合し重合体の状態で中和を行う場合(酸型重合法)には高吸収倍率で低可溶分の吸水性樹脂が得られる傾向にはあるが、吸水性樹脂の個々の粒子の均一な中和にはかなりの労力、設備と時間を要する(特許文献30)。しかし、本発明の方法を用いることで、吸水性樹脂の中和状態や製造方法の如何にかかわらず、すべての吸水性樹脂を良好に表面架橋などに使用することができ、よって、吸水剤の物性と生産性を大幅に向上することが可能となる。
本発明で用いる吸水性樹脂を得るための単量体は、必要に応じて上記アクリル酸(塩)以外の単量体を含有していてもよい。アクリル酸(塩)以外の単量体としては、特に限定されるものではないが、具体的には、例えば、メタクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸等のアニオン性不飽和単量体及びその塩;アクリルアミド、メタアクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン、N−ビニルアセトアミド等のノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミド、及びこれらの四級塩等のカチオン性不飽和単量体等が挙げられる。これら単量体は、単独で用いてもよく、適宜2種類以上を混合して用いてもよい。
【0025】
本発明において、アクリル酸(塩)以外の単量体を用いる場合には、該アクリル酸(塩)以外の単量体は、主成分として用いるアクリル酸及びその塩との合計量に対して、好ましくは30モル%以下、より好ましくは10モル%以下の割合である。上記アクリル酸(塩)以外の単量体を上記の割合で用いることにより、最終的に得られる吸水性樹脂(吸水剤)の吸収特性がより一層向上すると共に、吸水性樹脂(吸水剤)をより一層安価に得ることができる。
本発明に用いられる吸水性樹脂を得るために上述の単量体を重合するに際しては、バルク重合や沈殿重合を行うことが可能であるが、性能面や重合の制御の容易さ、さらに膨潤ゲルの吸収特性の観点から、上記単量体を水溶液とすることによる水溶液重合や逆相懸濁重合を行うことが好ましい。尚、上記単量体を水溶液とする場合の該水溶液(以下、単量体水溶液と称する)中の単量体の濃度は、水溶液の温度や単量体によって決まり、特に限定されるものではないが、10〜70重量%の範囲内が好ましく、20〜60重量%の範囲内がさらに好ましい。また、上記水溶液重合を行う際には、水以外の溶媒を必要に応じて併用してもよく、併用して用いられる溶媒の種類は、特に限定されるものではない。
【0026】
水溶液重合の方法としては、双腕型ニーダー中で単量体水溶液を、得られる含水ゲルを砕きながら重合したり、所定の容器中や駆動するベルト上に単量体水溶液を供給し、重合して得られたゲルをミートチョッパー等で粉砕する方法等が挙げられる。
上記の重合を開始させる際には、例えば過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2’−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤や、2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−オン等の光重合開始剤を用いることができる。
さらに、これら重合開始剤の分解を促進する還元剤を併用し、両者を組み合わせることによりレドックス系開始剤とすることもできる。上記の還元剤としては、例えば、亜硫酸ナトリウム、亜硫酸水素ナトリウム等の(重)亜硫酸(塩)、L−アスコルビン酸(塩)、第一鉄塩等の還元性金属(塩)、アミン類等が挙げられるが、特に限定されるものではない。
【0027】
これら重合開始剤の使用量は、通常0.001〜2モル%、好ましくは0.01〜0.1モル%である。これら重合開始剤の使用量が0.001モル%未満の場合には、未反応の単量体が多くなり、従って、得られる吸水性樹脂や吸水剤中の残存単量体量が増加するので好ましくない。一方、これら重合開始剤の使用量が2モル%を超える場合には、得られる吸水性樹脂や吸水剤中の水可溶成分量が増加するので好ましくない場合がある。
また、反応系に放射線、電子線、紫外線等の活性エネルギー線を照射することにより重合反応の開始を行ってもよいし、さらに、上記重合開始剤を併用してもよい。尚、上記重合反応における反応温度は、特に限定されるものではないが、15〜130℃の範囲が好ましく、20〜120℃の範囲内がより好ましい。また、反応時間や重合圧力も特に限定されるものではなく、単量体や重合開始剤の種類、反応温度等に応じて適宜設定すればよい。
【0028】
前記吸水性樹脂としては、架橋剤を使用しない自己架橋型のものであってもよいが、一分子中に、2個以上の重合性不飽和基や、2個以上の反応性基を有する架橋剤(吸水性樹脂の内部架橋剤)を共重合又は反応させたものがさらに好ましい。
これら内部架橋剤の具体例としては、例えば、N,N’−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチルロールプロパントリ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、エチレンカーボネート、プロピレンカーボネート、ポリエチレンイミン、グリシジル(メタ)アクリレート等を挙げることができる
これら内部架橋剤は、単独で用いてもよく、適宜2種類以上を混合して用いてもよい。また、これら内部架橋剤は、反応系に一括添加してもよく、分割添加してもよい。少なくとも1種または2種類以上の内部架橋剤を使用する場合には、最終的に得られる吸水性樹脂や吸水剤の吸収特性等を考慮して、2個以上の重合性不飽和基を有する化合物を重合時に必須に用いることが好ましい。
【0029】
これら内部架橋剤の使用量は前記単量体(架橋剤を除く)に対して、好ましくは0.001〜2モル%、より好ましくは0.005〜0.5モル%、さらに好ましくは0.01〜0.2モル%、特に好ましくは0.03〜0.15モル%の範囲内とされる。上記内部架橋剤の使用量が0.001モル%よりも少ない場合、並びに、2モル%よりも多い場合には、充分な吸収特性が得られないおそれがある。
上記内部架橋剤を用いて架橋構造を重合体内部に導入する場合には、上記内部架橋剤を、上記単量体の重合前あるいは重合途中、あるいは重合後、または中和後に反応系に添加するようにすればよい。
【0030】
尚、上記重合に際しては、反応系に、澱粉・セルロース、澱粉・セルロースの誘導体、ポリビニルアルコール、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子0〜50重量%(対単量体)や、その他0〜10重量%の、炭酸(水素)塩、二酸化炭素、アゾ化合物、不活性有機溶媒等の各種発泡剤;各種界面活性剤;キレート剤;次亜燐酸(塩)等の連鎖移動剤などを添加してもよい。
上記架橋重合体が水溶液重合で得られたものでゲル状である場合、すなわち含水ゲル状架橋重合体である場合、該架橋重合体は、必要に応じて乾燥し、乾燥の前および/または後で通常粉砕されて吸水性樹脂とする。また、乾燥は通常60℃〜250℃、好ましくは100℃〜220℃、より好ましくは120℃〜200℃の温度範囲で行われる。乾燥時間は、重合体の表面積、含水率、および乾燥機の種類に依存し、目的とする含水率になるよう選択される。
【0031】
本発明に用いることのできる吸水性樹脂や吸水剤の含水率(吸水性樹脂や吸水剤中に含まれる水分量で規定/180℃で3時間の乾燥減量で測定)は特に限定されないが、得られる吸水剤の物性面から室温でも流動性を示す粉末であり、より好ましくは0.2〜30重量%、さらに好ましくは0.3〜15重量%、特に好ましくは0.5〜10重量%の粉末状態である。
また本発明の製造方法に用いることのできる吸水性樹脂としては、粉末状のものを挙げることができる。吸水性樹脂の粒子は重合反応により得られた乾燥粉砕前のゲル状の平均粒径が1000μmを超えるようなものも使用できるが、通常、粉末とならないため、必要により(好ましくは)乾燥・粉砕・分級をすることにより目的に応じた粉末粒径に調整される。
【0032】
吸水性樹脂粉末や吸水剤の粒径としては、重量平均粒子径が10〜2000μm、好ましくは100〜1000μm、より好ましくは200〜700μm、さらに好ましくは300〜600μm、特に好ましくは400〜550μmの範囲が好適に用いられる。さらに好ましくは、吸水性樹脂粉末や吸水剤中の微粉末(例えば100μm以下、好ましくは150μm以下)の微粉末は少ない方が好ましく、具体的には10重量%以下、さらには5重量%以下、特に1重量%以下であることが好ましい。また、吸水性樹脂粉末や吸水剤は好ましくは実質1000μm以上、さらに好ましくは850μm以上の粒子が5重量%以下、さらには1重量%以下とされる。
【0033】
このようにして得られた吸水性樹脂や吸水剤の粒子形状は、球状、破砕状、不定形状等特に限定されるものではないが、粉砕工程を経て得られた不定形破砕状のものが好ましく使用できる。さらに、その嵩比重(JIS K−3362で規定)は、吸水剤の優れた物性から好ましくは0.40〜0.80g/ml、より好ましくは0.50〜0.75g/ml、さらに好ましくは0.60〜0.73g/mlの範囲である。
上記の方法により得られた吸水性樹脂は、通常、無加圧下での生理食塩水に対する飽和吸収倍率が10〜100g/g程度を有し、この吸収倍率などの物性は目的に応じて適宜調整される。
【0034】
(水溶性無機塩基(b1))
本発明では上記吸水性樹脂粉末(a)あるいは(a1)に対して、非架橋性の水溶性無機塩基(b1)、すなわち、好ましくは、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物などからなる群から選ばれる水溶性無機塩基(b1)(以下、水溶性無機塩基(b1)と呼ぶ)、および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、架橋剤(c)あるいは脱水反応性架橋剤(c1)が添加されるが、以下、水溶性無機塩基(b1)について説明する。
すなわち、本発明で水溶性無機塩基とは、水溶液中で解離することで水あるいは該塩基よりOH−を生じ、酸基を中和して塩を生じる無機化合物(炭酸塩や重炭酸塩を含む)を指す。本発明で用いられる水溶性無機塩基(b1)は、好ましくは、アルカリ金属塩,アンモニウム塩,アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物からなる群から選ばれ、これらは、通常、実質的に非架橋性の水溶性無機塩基である。(なお、酸基含有の吸水性樹脂に対して架橋性の水溶性無機塩基として、水酸化カルシウムや水酸化アルミニウムに代表される多価金属の水酸化物が例示されるが、一般に、これら多価金属は本発明の水溶性無機塩基には含まれない)。
【0035】
水溶性無機塩基(b1)として、得られる吸水剤の物性面から水溶性であることが必須であり、室温の水100g当たり通常5g以上、好ましくは20g以上、より好ましくは50g以上、さらに好ましくは100g以上の溶解性を示すものが用いられる。なお、本発明で非水溶性無機塩基、有機塩基や架橋性無機塩基(多価金属の水酸化物)などの併用は排除しないが、非架橋性の水溶性無機塩基(b1)を用いない場合、得られた吸水剤の物性が低い。
具体的に水溶性無機塩基(b1)としては、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸ナトリウムカリウム、炭酸セシウム、炭酸ルビジム、炭酸アンモニウム等のアルカリ金属塩および/またはアンモニウム塩を含む炭酸化合物やその水和物(十水塩、七水塩、一.五水塩、一水塩など)、重炭酸リチウム、重炭酸ナトリウム、重炭酸カリウム、重炭酸セシウム、重炭酸ルビジウム、重炭酸アンモニウム等のアルカリ金属および/またはアンモニウムを含む重炭酸塩、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化ルビジウム、水酸化アンモニウム、水ガラス等のアルカリ金属およびアンモニウムを含む水酸化化合物、燐酸水素2ナトリウム、燐酸水素2カリウム、燐酸水素2リチウム、燐酸水素2ルビジウム、燐酸水素2セシウムなどの燐酸水素化合物、セスキ炭酸ナトリウム(Na2CO3・NaHCO3・2H2O)などの複合塩が挙げられ、これらの2種以上を混合ないし併用しても良い。また、これら水溶性無機塩基(b1)は、粉末やその水和物、ペレットや水溶液として購入・保存ないし使用されるが、その形態に制限はない。
【0036】
水溶性無機塩基(b1)の中でも物性面や溶解性から、好ましくはアルカリ金属塩、さらに好ましくはリチウム塩、ナトリウム塩ないしカリウム塩、より好ましくはナトリウム塩が用いられる。また、化合物(b1)の中でも物性面から、好ましくは、炭酸塩/炭酸水素塩/水酸化物、さらに好ましくは、水酸物およびまたは炭酸水素塩、特に好ましくは水酸化物が用いられる。すなわち、具体的には水溶性無機塩基(b1)として好ましくは、炭酸水素ナトリウムおよび/または水酸化ナトリウム、さらに好ましくは、水酸化ナトリウムが用いられる。
本発明における水溶性無機塩基(b1)の使用量は、後述の非還元性アルカリ金属塩pH緩衝剤(b2)と併用しない場合には、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。上記の範囲内で用いることにより、尿や汗、経血等の体液(水性液体)に対する吸収特性をさらに一層向上させることができる。使用量が0.001重量部未満では、吸水性樹脂の表面近傍の官能基の中和率を適度に調整することができず、吸収特性が向上しない場合がある。水溶性無機塩基(b1)の使用量が10重量部より多い場合には過剰となり、不経済であるとともに、吸収倍率が向上しない恐れがある。
【0037】
なお、水溶性無機塩基(b1)を、後述する非還元性アルカリ金属塩pH緩衝剤(b2)と併用する場合には、上記と同様の理由により、それらの合計使用量が、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。ただし、本発明で水溶性無機塩基(b1)と後述の非還元性アルカリ金属塩pH緩衝剤(b2)とを併用する場合、少なくとも何れか一方の働きを示す範囲で(b1)と(b2)が適宜併用される。
この吸収特性の向上機構は明らかではないが、以下2つの理由((1)表面中和率の均一化による均一な表面架橋層の形成、(2)塩濃度の変化による混合と浸透の最適化)と推定される。
【0038】
すなわち、理由(1)として、一般に吸水性樹脂粉末では重合後の後中和の有無(前述の中和重合法か酸型重合法)にかかわらず、吸水性樹脂粉末の一粒一粒の中和率や、同じ一粒の粉末であっても微小には粉末表面の中和率が異なっており、従来の表面処理においては、これらの理由による粉末の架橋反応や架橋剤の混合に不均一が生じ、物性の低下が発生していた。そこで、本発明においては水溶性無機塩基(b1)を架橋剤(c)と併用することで、吸水性樹脂粉末の一粒一粒の中和率や、一粒の粉末の微小な表面中和率の差をなくすことにより、架橋に関与する表面近傍のカルボキシル基の中和率が一様に最適化でき、その結果均一に表面架橋を行うことが可能となった。例えば、70モル%中和のポリアクリル酸系吸水性樹脂および0.01〜2重量部の水酸化ナトリウムより得られた吸水剤は0.025〜5モル%の範囲で吸水剤の中和率は高められ、さらにその粒子の表面近傍において選択的に高中和率である。さらには、本発明の水溶性無機塩基(b1)は、架橋剤の反応触媒としても作用することが吸収特性向上に起因していると推測される。
【0039】
また、理由(2)として、水溶性無機塩基(b1)は、架橋剤の混合時には架橋剤溶液中の高い塩濃度由来で吸水性樹脂への浸透を制御し混合性を改良しているが、混合後には吸水性樹脂のカルボキシル基と中和反応してアルカリ金属塩およびアンモニウム塩となる事で架橋剤溶液中から消失するため、架橋剤の浸透を促進する働きをなしていると推定される。これは従来の添加剤(イソプロパノールなどの親水性有機溶媒)では、不活性な有機溶媒由来の吸水性樹脂への浸透が制御され混合性は改良されるが、混合後にも有機溶媒が架橋剤溶液中に残存して架橋剤の表面内部への浸透を妨げてしまうと推測される。
また、水溶性無機塩基(b1)と異なり、アルミニウムのような多価金属塩を使用した場合、多価金属イオンによる架橋反応が進行してしまい、無加圧もしくは加圧下での吸収倍率を低下させている事が推測される。さらに、多価金属イオンによる架橋はイオン結合を形成するため非常に弱く、また水膨潤状態では多価金属イオンが粒子内部に移動して架橋を形成するためより一層の物性の低下をもたらす事も推測される。
【0040】
以上のような現象から起こる吸水剤の架橋層の厚みの不足と物性が低下するという従来の欠点を、本願の水溶性無機塩基(b1)では改良していると推定される。さらに脱水反応性架橋剤(c1)を用いることにより、脱水架橋反応から生じる水によってさらに架橋剤が表面近傍に浸透し、架橋層の厚みがより増しているとも推定される。
(非還元性アルカリ金属塩pH緩衝剤(b2))
本発明では上記吸水性樹脂粉末(a)あるいは(a1)に対して、非架橋性の水溶性無機塩基(b1)、すなわち、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物などからなる群から選ばれる水溶性無機塩基(b1)、および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、架橋剤(c)あるいは脱水反応性架橋剤(c1)が添加されるが、以下、非還元性アルカリ金属塩pH緩衝剤(b2)について説明する。
【0041】
本発明における非還元性アルカリ金属塩pH緩衝剤(b2)は、溶液中である程度の酸や塩基の添加の消失でも、ほぼ一定の水素イオン濃度を維持するものであり、必須に非還元性で、好ましくはさらに非酸化性のアルカリ金属塩が用いられる(例えば、リンや硫黄を含むアルカリ金属塩pH緩衝剤の場合、リン原子の酸化数が+5/硫黄原子の酸化数が+6、ならば該pH緩衝剤は非酸化性非還元性を示す)。
pH緩衝剤が還元性を有したり、アルカリ金属塩でない場合、架橋の阻害となる恐れがあり、本発明の目的を十分には達成できない。本発明にはpH緩衝剤として働くアルカリ金属塩であり、種々の酸、塩基、または塩の組み合わせから作成されるpH緩衝剤が適用される。また、吸水性樹脂への混合性や浸透性から、pH緩衝剤の分子量は50〜1000、さらには60〜800、特に70〜500のものが用いられる。
【0042】
本発明でいうpH緩衝剤(b2)として働くアルカリ金属塩とは、代表的には炭酸水素塩、リン酸二水素塩、リン酸水素塩の1種または2種以上が例示される。
具体的な例としては、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、リン酸二水素ナトリウム、リン酸二水素カリウム、リン酸水素二ナトリウム、リン酸水素二カリウムといった無機多塩基酸の部分アルカリ金属塩;クエン酸二水素カリウム、クエン酸二水素ナトリウム、クエン酸水素二ナトリウム、クエン酸水素二カリウム、酒石酸水素ナトリウム、酒石酸水素カリウム、フマル酸一ナトリウム、フタル酸水素カリウムといった有機多価カルボン酸の部分アルカリ金属塩、特にナトリウム塩ないしカリウム塩、さらにはリチウム塩が挙げられる。
【0043】
また、上記pH緩衝剤(b2)のほかに、本発明でいう種々の酸、塩基、または塩の組み合わせから作成されるpH緩衝剤(b2)とは、代表的には従来公知の緩衝液を作成する際に用いられる化合物の組み合わせから作られる緩衝剤が例示される。緩衝液としては、緩衝剤の溶液、特に、弱酸と強塩基からなる塩または強酸と弱塩基からなる塩、またはそれらの塩の混合物の水溶液である。カルボキシル基などの酸基を含有する吸水性樹脂の場合には、好ましくは弱酸と強塩基からなる塩の混合物としての緩衝剤が用いられ、より好ましくは無機塩を用いたものである。
具体的な例としては、化学便覧(日本化学会編、II−355、356)に記載の緩衝液に使用されている化合物の組み合わせが緩衝剤として挙げられる。例えば、Clark−Lubsの緩衝液(塩化カリウム/塩酸;pH1.0〜2.2、フタル酸水素カリウム/塩酸;pH2.2〜3.8、フタル酸水素カリウム/水酸化ナトリウム;pH4.0〜6.2、リン酸二水素カリウム/水酸化ナトリウム;pH5.8〜8.0、ホウ酸/塩化カリウム/水酸化ナトリウム;pH7.8〜10.0)、Sφrensenの緩衝液(グリシン+塩化ナトリウム/塩酸;pH1.1〜4.6、グリシン+塩化ナトリウム/塩酸;pH8.6〜13.0、クエン酸ナトリウム/塩酸;pH1.1〜4.9、クエン酸ナトリウム/水酸化ナトリウム;pH5.0〜6.7、四ホウ酸ナトリウム/塩酸;pH7.6〜9.2、四ホウ酸ナトリウム/水酸化ナトリウム;pH9.3〜12.4、リン酸二水素カリウム/リン酸水素二ナトリウム;pH5.3〜8.0)、Kolthoffの緩衝液(クエン酸カリウム/クエン酸;pH2.2〜3.6、クエン酸二水素カリウム/塩酸;pH2.2〜3.6、クエン酸二水素カリウム/水酸化ナトリウム;pH3.8〜6.0、コハク酸/四ホウ酸ナトリウム;pH3.0〜5.8、クエン酸二水素カリウム/四ホウ酸ナトリウム;pH3.8〜6.0、リン酸二水素カリウム/四ホウ酸ナトリウム;pH5.8〜9.2、四ホウ酸ナトリウム/炭酸ナトリウム;pH9.2〜11.0、塩酸/炭酸ナトリウム;pH10.2〜11.2、リン酸水素二ナトリウム/水酸化ナトリウム;pH11.0〜12.0)、Michaelisの緩衝液(酒石酸/酒石酸ナトリウム;pH1.4〜4.5、乳酸/乳酸ナトリウム;pH2.3〜5.3、酢酸/酢酸ナトリウム;pH3.2〜6.2、リン酸二水素カリウム/リン酸水素二ナトリウム;pH5.2〜8.3、ジエチルバルビツル酸ナトリウム+酢酸ナトリウム/塩酸;pH2.6〜9.2、ジエチルバルビツル酸ナトリウム/塩酸;pH6.8〜9.6、N,N−ジメチルグリシンナトリウム塩/塩酸;pH8.6〜10.6)、Mcilvaineの広域緩衝液(リン酸水素二ナトリウム/クエン酸;pH2.2〜8.0)、Britton−Robinsonの広域緩衝液(クエン酸+リン酸二水素カリウム+ホウ酸+ジエチルバルビツル酸/リン酸三ナトリウム)、Carmodyの広域緩衝液(ホウ酸+クエン酸/リン酸三ナトリウム;pH2.0〜12.0)、Gomoriの緩衝液(2,4,6−トリメチルピリジン/塩酸;pH6.4〜8.4、トリス(ヒドロキシメチル)アミノメタン/塩酸;pH7.2〜9.1、2−アミノ−2−メチル−1,3−プロパンジオール/塩酸;pH7.8〜9.7)、Bates−BowerのTris緩衝液(トリス(ヒドロキシメチル)アミノメタン/塩酸;pH7.0〜9.0)、Delory−King緩衝液(炭酸塩/炭酸水素塩;pH9.2〜10.7)、等が挙げられる。そして、使用される緩衝剤のpHおよび濃度は、吸水性樹脂の中和率や用いる表面架橋剤の種類にもよるが、好ましくは緩衝剤を添加することにより該表面架橋剤溶液のpHが1.5〜10.0の範囲に調整される。
【0044】
上記の内でも、性能、安定性、一成分系での使用、コスト、等の点から無機多塩基酸の部分中和塩が好ましく、リン酸、炭酸の部分アルカリ金属中和塩がより好ましい。
本発明における上記pH緩衝剤(b2)の使用量は、前述の水溶性無機塩基(b1)と併用しない場合には、吸水性樹脂の固形分100重量部に対して0.005〜10重量部の範囲内が好ましく、0.05〜5重量部の範囲内がより好ましい。上記の範囲内で用いることにより、尿や汗、経血等の体液(水性液体)に対する吸収特性をさらに一層向上させることができる。使用量が0.005重量部未満では、吸水性樹脂の表面近傍の官能基の中和率を適度に調整することができず、吸収特性が向上しない場合がある。pH緩衝剤(b2)の使用量が10重量部より多い場合には、該添加剤が過剰となり、不経済であるとともに、吸収倍率が向上しない恐れがある。
【0045】
なお、非還元性アルカリ金属塩pH緩衝剤(b2)を、前述の水溶性無機塩基(b1)と併用する場合には、上記と同様の理由により、それらの合計使用量が、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。ただし、本発明で水溶性無機塩基(b1)と前述の非還元性アルカリ金属塩pH緩衝剤(b2)と併用する場合、少なくとも何れか一方の働きを示す範囲で(b1)と(b2)が適宜併用される。
この吸収特性の向上機構は明らかではないが、以下2つの理由((1)表面中和率の均一化、(2)塩濃度の変化による混合と浸透の最適化)とも推定される。
【0046】
すなわち、理由(1)として、吸水性樹脂粉末では重合後の後中和の有無(前述の中和重合法か酸型重合法)にかかわらず、吸水性樹脂粉末の一粒一粒の中和率や、同じ一粒の粉末であっても微小には粉末表面の中和率が異なっており、粉末の架橋の反応や架橋剤の混合に不均一を生じて物性を低下されていたものが、本発明のpH緩衝剤(a)を架橋剤(b)と併用することで、吸水性樹脂粉末の一粒一粒の中和率や、一粒の粉末の微小な表面中和率の差をなくしているため、吸水性樹脂粉末の中和率や後述の中和指数の如何に関わらず、本発明では、架橋に関与する表面近傍のカルボキシル基の中和率が一様に最適化できるためと考えられる。その結果、本発明のpH緩衝剤は、吸水性樹脂粉末への架橋剤の浸透を妨げられることなく、混合助剤としても作用し、さらには、架橋剤の反応触媒としても作用することが吸収特性向上に起因していると推測される。
【0047】
また、理由(2)として、炭酸水素塩などpH緩衝剤は、架橋剤の混合時には架橋剤溶液中のアルカリ金属塩として存在するために、高い塩濃度由来の吸水性樹脂への浸透を制御し混合性を改良しているが、混合後には吸水性樹脂のカルボキシル基と中和反応してpH緩衝剤のアルカリ金属塩が架橋剤溶液中から消失するため、混合後には架橋剤の浸透を阻害した塩が消失して架橋剤の浸透を促進する働きをなしていると推定される。これは従来の添加剤(イソプロパノールなどの親水性有機溶媒)では、不活性な有機溶媒由来の吸水性樹脂への浸透が制御され混合性は改良されるが、混合後にも有機溶媒が架橋剤溶液中に残存して架橋剤の表面内部への浸透を妨げてしまい、吸水剤の架橋層の厚みが不足している従来の欠点を、本願のpH緩衝剤では改良していると推定される。
【0048】
(架橋剤(c)およびその混合と架橋処理)
本発明では、酸基と反応しうる架橋剤(c)として、好ましくは表面架橋剤、さらには、脱水反応性架橋剤(c1)が好ましく用いられる。なお、本発明で脱水反応性とは、吸水性樹脂の官能基(特に表面近傍の官能基)と架橋剤とが脱水反応、好ましくは、脱水エステル化および/または脱水アミド化、さらに好ましくが、脱水エステル化で架橋する架橋剤である。
具体的に吸水性樹脂がカルボキシル基を含有する場合、多価アルコールなどのヒドロキシル基含有の架橋剤、多価アミンなどのアミノ基含有の架橋剤、さらには、アルキレンカーボネートやモノ、ジまたはポリのオキサゾリジノン化合物;3−メチル−3−オキセタンメタノール等のオキセタン化合物などの環状架橋剤であって、その環状架橋剤の開環反応に伴ってヒドロキシル基やアミノ基を生成し該ヒドロキシル基やアミノ基が架橋反応を行う環状架橋剤、などが脱水反応性を示す架橋剤(c1)として例示される。脱水反応性架橋剤(c1)の1種または2種以上が用いられるが、さらに、非脱水反応性の架橋剤、例えば、多価金属なども併用してもよい。
【0049】
具体的に、本発明に用いることのできる脱水反応性架橋剤(c1)としては、吸水性樹脂の官能基と反応しうる架橋剤ならば制限なく使用され、通常、該用途に用いられている架橋剤(表面架橋剤)のことである。例えば、エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、ジプロピレングリコール、ポリプロピレングリコール、1,3−プロパンジオール、2,2,4−トリメチル−1,3−ペンタンジオール、グリセリン、ジグリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレンオキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトールなどの多価アルコール化合物;エチレンジアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ポリアミドポリアミン、ポリアリルアミン、ポリエチレンイミン等の多価アミン化合物、並びに、それら多価アミンとハロエポキシ化合物との縮合物;1,3−ジオキソラン−2−オン、4−メチル−1,3−ジオキソラン−2−オン、4、5−ジメチル−1,3−ジオキソラン−2−オン、4、4−ジメチル−1,3−ジオキソラン−2−オン、4−エチル−1,3−ジオキソラン−2−オン、4−ヒドロキシメチル−1,3−ジオキソラン−2−オン、1,3−ジオキサン−2−オン、4−メチル−1,3−ジオキサン−2−オン、4、6−ジメチル−1,3−ジオキサン−2−オン、1,3−ジオキソパン−2−オン等のアルキレンカーボネート化合物、並びに、エチレングリコールビス(4−メチレン−1,3−ジオキソラン−2−オン)エーテル等の多価アルキレンカーボネート化合物;モノ、ジまたはポリのオキサゾリジノン化合物;3−メチル−3−オキセタンメタノール等のオキセタン化合物ならびに多価オキセタン化合物;等より選ばれる1種または2種以上のものが例示できる。
【0050】
これら脱水反応性架橋剤の中でも、多価アルコール、アルキレンカーボネート、オキサゾリジノン化合物、(多価)オキセタン化合物から選ばれた1種以上が好ましく、少なくとも多価アルコールを用いることが特に好ましい。
架橋剤(c)としては、これら脱水反応性架橋剤(c1)に加えて、さらに、エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、ポリエチレンジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリシドール、γ−グリシドキシプロピルトリメトキシシラン等のエポキシ化合物;2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート等の多価イソシアネート化合物;1,2−エチレンビスオキサゾリン等の多価オキサゾリン化合物;γ−グリシドキシプロピルトリメトキシシラン、γ−アミノプロピルトリメトキシシラン等のシランカップリング剤;2,2−ビスヒドロキシメチルブタノール−トリス[3−(1−アジリジニル)プロピオネート]などの多価アジリジン化合物、ベリリウム、マグネシウム、カルシウム、ストロンチウム、亜鉛、アルミニウム、鉄、クロム、マンガン、チタン、ジルコニウムなどの多価金属が例示される。
【0051】
なお、本発明で脱水反応性架橋剤(c1)、および、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)および/または粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含む吸水性樹脂粉末の両方を用いない場合、一般に得られる吸水剤の物性が低く、好ましくは、該脱水反応性架橋剤(c1)、および、該特定粒度の吸水性樹脂粉末(a1)の両方が本発明で使用される。
【0052】
本発明において、吸水性樹脂粉末(a)あるいは(a1)と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合する場合には、水を用いることが好ましい。この際、使用される水の量は、使用する吸水性樹脂の含水率にもよるが、通常、吸水性樹脂100重量部に対し、0.5〜20重量部、好ましくは0.5〜10重量部の範囲である。水の使用量が20重量部を越えると吸収倍率が低下してしまうことがある。0.5重量部よりも少ないと効果が現れにくくなり、加圧下吸収倍率を向上させることができなくなる恐れがある。
また、本発明において、吸水性樹脂粉末(a)あるいは(a1)と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合する場合には、親水性有機溶媒を用いてもよい。用いられる親水性有機溶媒としては、メチルアルコール、エチルアルコール、プロピルアルコール、イソプロピルアルコール、ブチルアルコール、イソブチルアルコール、t−ブチルアルコール等のアルコール;アセトン、メチルエチルケトン等のケトン類;ジオキサン、アルコキシ(ポリ)エチレングリコール、テトラヒドロフラン等のエーテル類;ε−カプロラクタム、N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキサイド等のスルホキサイド類;エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、グリセロリン酸、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール類が挙げられる。
【0053】
使用される親水性有機溶媒の量は、吸水性樹脂の種類や粒度によって異なるが、通常、吸水性樹脂100重量部に対し0〜10重量部、好ましくは0〜5重量部、より好ましくは0〜3重量部の範囲である。親水性有機溶媒の使用量が10重量部以上の場合、上記添加剤の溶解性が低下し吸収特性が向上しない恐れがある。なお、上記多価アルコールは反応条件(加熱温度や時間、含水率など)によって吸水性樹脂と反応させて架橋剤としてもよいし、反応させずに溶媒としてもよいし、それらの働きを併用させてもよい。
さらに、本発明において吸水性樹脂粉末(a)あるいは(a1)と、上記添加剤(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)とを混合する場合、水や親水性有機溶媒以外の物質として、本発明の効果を妨げない範囲で界面活性剤や不活性無機微粒子粉末を用いてもよい。用いられる界面活性剤や不活性無機微粒子粉末は、米国特許第5164459号公報、欧州特許第827753号公報、欧州特許第349240号公報、欧州特許第761241号公報などに例示される。
【0054】
本発明において、吸水性樹脂粉末(a)あるいは(a1)と、上記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)の混合は、親水性有機溶媒またはシクロヘキサン、ペンタン等の有機溶媒中に該吸水性樹脂を分散させた状態で行ってもよいが、水、架橋剤、添加剤の混合物を数回に分けて添加してもよく、混合方法は特に限定されるものではない。また、上記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)、さらに必要に応じて用いられる水や親水性有機溶媒、無機粉末などは、吸水性樹脂に対して別々に混合してもよいし、一括で混合してもよいし、数回に分けて混合してもよいが、好ましくは、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を予め混合した後に吸水性樹脂に添加させ、その際、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)は水溶液とさせることがより好ましい。また、この際の水溶液の温度は混合性や安定性から0℃〜沸点、好ましくは5〜50℃、さらには10〜30℃にさせる。また、混合前の吸水性樹脂粉末(a)あるいは(a1)の温度は、混合性から好ましくは0〜80℃、さらには40〜70℃の範囲である。
【0055】
さらに、本発明では種々の混合方法の中で、必要により水および/または親水性有機溶媒と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)とを予め混合した後、次いで、その水溶液を吸水性樹脂粉末(a)に噴霧あるいは滴下混合する方法が好ましく、噴霧する方法がより好ましい。噴霧される液滴の大きさは、300μm以下が好ましく、200μm以下がより好ましい。また混合に際し、本発明の効果を妨げない範囲で水不溶性微粒子粉体や界面活性剤を共存させてもよい。
前記混合に用いられる好適な混合装置は、均一な混合を確実にするため大きな混合力を生み出せることが必要である。本発明に用いることのできる混合装置としては、例えば、円筒型混合機、二重壁円錐型混合機、高速攪拌型混合機、V字型混合機、リボン型混合機、スクリュー型混合機、流動型炉ロータリーディスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機、スクリュー型押出機等が好適である。
【0056】
本発明の吸水剤の製造方法は、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合し、前記吸水性樹脂粉末(a)あるいは(a1)を架橋処理することを特徴とする。
本発明の吸水剤の製造方法は、好ましくは、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合した後、吸水性樹脂の表面近傍を架橋させる際に、加熱処理を行う。
【0057】
本発明で加熱処理を行う場合、処理時間は、1分〜180分が好ましく、3分〜120分がより好ましく、5分〜100分がさらに好ましい。加熱処理温度(熱媒温度ないし材料温度で規定)は100〜250℃の範囲が好ましく、140〜240℃の範囲がより好ましく、150〜230℃の範囲がさらに好ましく、160〜220℃の範囲がさらにより好ましい。加熱温度が100℃未満では、加熱処理や脱水反応に時間がかかり生産性の低下を引き起こすのみならず、均一な架橋が達成されず、優れた吸水剤が得られなくなる恐れがある。また処理温度が250℃を越えると、得られる吸水剤がダメージを受け、物性に優れたものが得られにくいとことがある。
【0058】
加熱処理は通常の乾燥機または加熱炉を用いて行うことができ、溝型混合乾燥機、ロータリー乾燥機、ディスク乾燥機、流動層乾燥機、気流型乾燥機、および赤外線乾燥機が例示される。
上記の本発明に係る吸水剤の製造方法においては、さらに、必要に応じて、消臭剤、抗菌剤、香料、二酸化珪素や酸化チタン等の無機粉末、発泡剤、顔料、染料、親水性短繊維、可塑剤、粘着剤、界面活性剤、肥料、酸化剤、還元剤、水、塩類、キレート剤、殺菌剤、ポリエチレングリコールやポリエチレンイミンなどの親水性高分子、パラフィンなどの疎水性高分子、ポリエチレンやポリプロピレンなどの熱可塑性樹脂、ポリエステル樹脂やユリア樹脂などの熱硬化性樹脂等を添加する等、吸水剤や吸水性樹脂に種々の機能を付与する工程を含んでいてもよい。これら添加剤の使用量は吸水剤100重量部に対して0〜10重量、好ましくは0〜1重量部の範囲で用いられる。
【0059】
本発明で吸水剤に用いられるカチオン性高分子化合物は、吸水剤の衛生材料への固定性などを向上でき、好ましくは重量平均分子量が2000以上で、さらに好ましくは5000以上、最も好ましくは重量平均分子量が10000以上である。また、その使用量は、好ましくは吸水性樹脂100重量部に対し0.01〜10重量部、より好ましくは0.05〜5重量部、さらに好ましくは0.1〜3重量部である。カチオン性高分子化合物の混合は、単独あるいは溶液(水溶液)で添加され、好ましくは、表面架橋後に添加される。カチオン性高分子化合物の具体例としては、ポリエチレンイミン、ポリビニルアミン、ポリアリルアミン、ポリアミドアミンとエピクロルヒドリンの縮合物、ポリアミジン、ポリ(N−ビニルホルムアルデヒド)の部分加水分解物またはこれらの塩などが例示される。
【0060】
水不溶性微粒子を用いてさらに吸水剤の通液性や吸湿時の耐ブロッキング性などを改善することができる。用いられる微粒子としては、好ましくは10μm以下、さらには1μm以下、特に0.1μm以下の無機または有機の水不溶性微粒子が用いられ、具体的には酸化珪素(商品名、Aerosil、日本アエロジル社製)、酸化チタン、酸化アルミ、などが用いられる。混合には粉体混合(Dry−Blend)やスラリー混合が用いられ、その使用量は吸水剤100重量部に対して10重量部以下、さらには0.001〜5重量部、好ましくは0.01〜2重量部用いられる。
(吸水剤およびそれを用いた衛生材料)
本発明においては、好ましくは、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合し、前記吸水性樹脂粉末(a)あるいは(a1)を架橋処理することによって、前述の本発明の効果に起因する高物性の新規な吸水剤を提供する。
【0061】
本発明にかかる吸水剤は、好ましくは、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤である。
(1)酸重合する場合、得られる吸水剤
本発明にかかる吸水剤は、酸重合で吸水性樹脂を得る場合、好ましくは、酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した吸水剤である。より好ましくは、前記の後中和して得られた吸水性樹脂の中和指数が15以上、さらに好ましくは17以上、特に好ましくは20以上である。また、好ましくは、前記の表面架橋した吸水剤の中和指数が15以上、さらに好ましくは17以上、特に好ましくは20以上である。表面架橋効果を高めるために中和指数を低くする場合、中和に長時間や複雑な工程を要するが、本発明では不均一な中和でも簡便に優れた表面架橋が達成できる。
【0062】
従来、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた吸水性樹脂は高吸収倍率で低可溶分だが、得られた吸水性樹脂の中和の不均一のため、一般に加圧下吸収倍率が向上しにくいものであった。かかる問題を解決するため、特許文献30(欧州特許公開0882502号)では高度に吸水性樹脂の一粒一粒の粒子の中和率の差(中和指数)を制御する技術が知られている。
かかる中和指数を制御する方法では、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた低可溶分の吸水性樹脂であって、従来にない高い加圧下吸収倍率を達成するが、中和指数の制御に非常に手間を必要とするものであった。しかし、本発明のアルカリ金属pH緩衝剤(b2)を用いる本発明の方法では、中和指数の高度な制御を必要とせず、簡便な不均一な後中和でも高い加圧下吸収倍率を与えるので非常に好ましい。なお、勿論、本発明は、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた、酸型重合法による低可溶分の吸水性樹脂に限定されるものでなく、後述の実施例などにも示すように、後中和工程を含まない中和重合法による吸水性樹脂にも好適に適用される。
【0063】
(2)5つの物性を併せ持つ新規な吸水剤
また、酸重合する場合しない場合を含めて、本発明の吸水剤は下記物性が好ましく、特に本発明では、下記特性の5つの物性(粒度、CRC、AAP、SFCの4つに加えて、さらにSFC変化指数、SFC変動係数、SFC変動率、連続生産系SFC標準偏差、表層可溶分などの1つ以上、好ましくは2つ以上、より好ましくは3つ以上、特に好ましくは4つ以上)を併せ持った新規な吸水剤を与える。
(a)粒度
本発明にかかる吸水剤の平均粒子径や嵩比重は、前記吸水性樹脂の範囲、すわわち、重量平均粒子径300〜600μmで150μm以下の微粉末が10重量%以下であることが好ましい。より好ましくは5重量%以下、さらに好ましくは3重量%以下、特に2重量%以下である。
【0064】
本発明にかかる吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、さらに好ましくは3種以上、より好ましくは4種を含む。
本発明にかかる吸水剤は、好ましくは、粒子径が850μm未満で150μm以上の粒子を全粒子の95重量%以上含み、より好ましくは97重量%以上、さらに好ましくは98重量%以上である。かかる特定の粒度に制御されることで、衛生材料で高物性を達成する。
【0065】
本発明にかかる吸水剤は、好ましくは、前記粒子A1からA4までの4種をそれぞれ0.1重量%以上含み、より好ましくは1重量%以上、さらに好ましくは3重量%以上である。この場合、上限値は特に限定されないが、好ましくは、前記粒子A1からA4までの4種がそれぞれ99重量%以下、より好ましくは90重量%以下、さらに好ましくは80重量%以下である。各粒度を一定以上含有する事で粒子の表面積に依存する吸水速度がバランスよく制御される。
(b)CRC
本発明にかかる吸水剤は、0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(Centrifuge Retention Capacity/CRC)が31g/g以上であることが好ましい。CRCが31g/g以上となることによって、吸水剤を用いた衛生材料の吸収に臨界的に優れ、コンパクトな衛生材料を達成でき、さらに吸水体(なお、吸水体とは、吸水剤と必要により繊維などの他の吸水材料を含む体液吸水体を意味する)のコスト低減にもなる。CRCは、より好ましくは32g/g以上、さらに好ましくは33g/g以上、さらにより好ましくは34g/g以上、特に好ましくは35g/g以上、特により好ましくは36g/g以上である。0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率が31g/gよりも小さいと、吸水体に吸収されうる尿の総量が小さくなり、吸水体に吸収された尿がおむつの表面への戻りが非常に大きくなる。さらに吸水体に求められる尿の吸液量を維持しようとする場合、吸水体に使用する吸水剤の量が多くなり、衛生材料が嵩高く重いものになり、吸水体のコストアップに繋がることになる点で好ましくない。
【0066】
(c)AAP
本発明にかかる吸水剤は、0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(Absorbency Against Pressure/AAP)が20g/g以上であることが好ましい。AAPが20g/g以上となることによって、本発明の吸水剤を紙おむつの吸水体の一部に使用した場合、吸水体に吸収された尿がおむつの表面への戻りを防ぐ効果が非常に大きくなる。0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率は、より好ましくは22g/g以上、さらに好ましくは24g/g以上、さらにより好ましくは25g/g以上、特に好ましくは26g/g、特により好ましくは27g/g以上である。0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率が20g/gよりも小さいと、吸水体に吸収された尿がおむつの表面への戻りを防ぐ効果が非常に小さくなる点で好ましくない。
【0067】
(d)SFC
本発明にかかる吸水剤は、0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上であることが好ましい。SFCは、本発明で得られる吸水剤の膨潤後の通液性に、非常に大きな影響を与える。つまり、例えば、本発明の吸水剤を紙おむつの吸水体の一部に使用した場合の、通液性を良好にし、吸水体に液を十分に行き渡らせ、吸液量を増大させ、液の漏れを防止するという効果が著しく向上する。SFCは、より好ましくは25(単位:10−7×cm3×s×g−1)以上、さらに好ましくは30(単位:10−7×cm3×s×g−1)以上、さらにより好ましくは35(単位:10−7×cm3×s×g−1)以上、特に好ましくは40(単位:10−7×cm3×s×g−1)以上、特により好ましくは50(単位:10−7×cm3×s×g−1)以上である。SFCが20(単位:10−7×cm3×s×g−1)よりも小さいと、例えば、紙おむつの吸水体に使用した場合の通液性が低下し、吸水体に液が局在化し、吸液量が減少し、液の漏れが多くなり、吸水体としての性能が著しく低下する点で好ましくない。
【0068】
すなわち、本発明の吸水剤は衛生材料に用いるには粒度に加えて、下記3つの物性をバランスよく併せ持つことが好ましい。すなわち、CRC、AAP、SFCの1つまたは2つが高物性だけでは、衛生材料に十分に好適ではないことが見出された。これらの3つの物性は上記酸重合した特定の中和指数の吸水剤のみならず、後中和工程を含まない中和重合法による吸水性樹脂にも好適に適用される。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaでの加圧下吸水倍率(AAP)が24g/g以上。
【0069】
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
(e)SFC変化指数
また、本発明者らは、従来粒子全体(Bulk)で物性を評価および管理されていた吸水性樹脂(吸水剤)粒子において、吸水性樹脂(吸水剤)粒子の粒度ごとの物性が大きく異なり、その粒度ごとの物性の違いが衛生材料での物性低下を引き起こしている事実を見出した。衛生材料に含まれる吸水剤粒子は微視的には粒度にバラツキが見られるため、その粒度由来の個々の衛生材料の物性の違いや衛生材料の部分での物性の違いが、衛生材料の物性低下を引き起こすと推定される。
【0070】
そこで、本発明者らは粒度ごとの物性の差、特に粒度ごとのSFCのバラツキ小さい吸水剤を提供する。
本発明にかかる吸水剤は、下記式(1)で規定されるSFC変化指数が0〜25%であることが好ましい。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
このSFC変化指数や後述のSFC変動係数において、粒子A1からA4のSFCの標準偏差は、吸水剤粒子を分級して、吸水剤中で粒子A1からA4のうちで存在する2〜4種類の粒子を得て、次いで得られた各粒度ごとのSFCを1回ずつ測定して、その2〜4種類のSFC値から標準偏差を計算して求められる。また、その2〜4種類のSFC値から平均値も同様に計算される。
【0071】
このSFC変化指数は、粒度ごとのSFCのバラツキを表し、SFC変化指数が0〜25%となることによって、吸水体がより均一な通液性を持つようになり、吸水体全体に液が分散し易く、液の漏れを防止するという効果となる。SFC変化指数は、より好ましくは0〜23%、さらに好ましくは0〜20%、さらにより好ましくは0〜18%、特に好ましくは0〜15%、特により好ましくは0〜10%である。SFC変化指数が25%よりも大きいと、例えば吸水剤を紙おむつの吸水体に使用した場合、吸水体に通液性のばらつきが生じ、吸水体全体に液が分散し難くなり、液の漏れの発生や、吸水体の吸液量が低減する点で好ましくない。
【0072】
(f)SFC変動係数
本発明にかかる吸水剤は、下記式(2)で規定されるSFC変動係数が0〜0.25であることが好ましい。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
このSFC変動係数や下記のSFC変動率も同様に粒度ごとのSFCの差の少なさを表し、SFC変動係数が0〜0.25となることによって、粒度ごと(例えばA1からA4)のSFCのばらつきの度合いが小さくなり、例えば本発明の吸水剤を用いて紙おむつの吸水体を製造する場合、吸水体内での通液性が均一になる上に、吸水体の各々を比較しても、紙おむつの性能にばらつきがなくなり、安定した品質の紙おむつを製造できることとなる。SFC変動係数は、より好ましくは0〜0.23、さらに好ましくは0〜0.20、さらにより好ましくは0〜0.18、特に好ましくは0〜0.15、特により好ましくは0〜0.10である。SFC変動係数が0.25よりも大きい場合および/またはSFC変動率が0.65未満である場合、吸水剤を用いた衛生材料(特に紙おむつ)の吸水体内での通液性が不均一になる上に、吸水体の各々を比較しても、紙おむつの性能のばらつきが大きくなり、安定した品質の紙おむつを製造できなくなる点で好ましくない。
【0073】
(g)SFC変動率
すなわち、本発明にかかる吸水剤は、下記式(3)で規定されるSFC変動率が0.65〜1.00であることが好ましい。
SFC変動率=(粒子A1からA4のSFCの中で最小のSFC)/(粒子A1からA4のSFCの中で最大のSFC) (3)
このSFC変動率も同様に粒度ごとのSFCの差の少なさを表し、好ましくは0.70〜1.00、さらにより好ましくは0.75〜1.00、特に好ましくは0.80〜1.00である。
【0074】
(h)表層可溶分量
本発明の吸水剤は、表層可溶分量が6.0重量%以下(対吸水剤)であることが好ましい。この表層可溶分量は、好ましくは5.5重量%以下、より好ましくは5.0重量%以下、さらに好ましくは4.5重量%以下、特に好ましくは4.0重量%以下である。
本願の測定法で規定される表層可溶分は衛生材料への実使用に対応した可溶分量であり、より衛生材料での吸水能に優れる。すなわち、従来、水可溶分の測定(US Re 32649やEDANA法など)は多く知られていたが、過剰の液で長時間の測定のため、実使用において溶出してくると考えられる可溶分の量よりも過剰の量の可溶分を測定していることがわかった。そして、本発明の方法が衛生材料での実使用に最もモデルとなっていることが見出された。
【0075】
(i)連続生産系SFC標準偏差
本発明にかかる吸水剤は、下記式(4)で規定される連続生産系SFC標準偏差が5.0以下であることが好ましい。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、各Lotは20kg以上。Lot数は10以上。)
上記連続生産系とは、1ラインで、24時間以上、若しくは10t以上、連続して吸水剤を生産することを示す。また、吸水剤の製造方法の基本工程である、重合、乾燥、粉砕、表面処理等は、連続生産系においては、各工程は連続式またはバッチ式を問わないが(例えば、重合工程においては連続重合とバッチ重合など)、各工程の間隔は24時間以下、好ましくは12時間以下、さらに好ましくは6時間以下、特に好ましくは3時間以下で行うものとする。
【0076】
各Lotは、好ましくは20kg〜100t、より好ましくは0.1t〜50t、さらに好ましくは0.5t〜25tである。
Lot数は、好ましくは20以上、より好ましくは30以上、さらに好ましくは50以上、特に好ましくは100以上である。
この連続生産系SFC標準偏差は、吸水剤の連続生産において各LotのSFCの値のばらつきを表し、連続生産系SFC標準偏差が5.0以下となることによって、各Lot毎のSFCのばらつきが小さくなり、例えば、本発明の吸水剤を用いて紙おむつの吸水体を製造する場合、安定した品質の紙おむつを製造できることとなる。連続生産系SFC標準偏差は、より好ましくは4.5以下、さらに好ましくは4.3以下、さらにより好ましくは4.0以下、特に好ましくは3.5以下、特により好ましくは3.0以下、最も好ましくは2.5以下である。連続生産系SFC標準偏差が5.0よりも大きい場合、安定した品質の紙おむつを製造できなくなる点で好ましくない。
【0077】
なお、上記した各種物性は、吸水剤が衛生材料として使用される場合、その実使用の結果(おむつの吸収量や漏れなど)と相関する重要な物性であり、本発明の吸水剤は上記各種物性に特に優れている。これら測定法は実施例に記載する。
本発明にかかる吸水剤は、以上に述べた種々の特徴を有することが好ましく、特に好ましい構成として、上記の式(1)、(2)、(4)のいずれか1つ以上、より好ましくは2つ以上、さらに好ましくは3つ全てを満たす吸水剤である。
すなわち、本発明にかかる吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0078】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
また、本発明にかかる別の吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0079】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
また、本発明にかかるさらに別の吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0080】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
本発明にかかるさらに別の吸水剤は、酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上であることを特徴とする、吸水剤である。
【0081】
上記吸水剤において、CRC、AAP、SFCやSFC変化指数、SFC変動係数などは好ましくは前述の範囲であり、本発明の新規な吸水剤は、例えば、前述の本発明の吸水剤の製造方法で得られる。本発明の吸水剤はCRC、AAP、SFCが非常に高く高物性をバランスよく維持している上で、さらに粒度ごとの物性の変化も従来になく少ないため、いかなる衛生材料の使用条件でも高物性を発揮する優れた吸水剤である。
本発明によれば、無加圧下の吸収倍率、加圧下の吸収倍率、生理食塩水流れ誘導性のバランスに優れた良好な吸収特性を備えた吸水剤を簡便に製造することができ、農園芸保水剤、工業用保水剤、吸湿剤、除湿剤、建材、などで広く用いられるが、その吸水剤は紙おむつ、生理用ナプキンなどの、糞、尿ないし血液の吸収用衛生材料に特に好適に用いられる。さらに、本発明の吸水剤は上記各種物性にバランスよく優れるため、衛生材料は一般に吸水剤の濃度(吸水剤および繊維基材の合計に対する吸水剤の重量比)として高濃度、例えば30〜100重量%、好ましくは40〜100重量%の範囲、さらに好ましくは50〜95重量%で使用可能である。また、衛生材料中の吸水体の構造は、一般の吸水性物品に用いられる構造であれば特に制限はなく、例えば、シート状に成形した親水性繊維材料の間に吸水剤を配する、いわゆるサンドイッチ構造の吸水体や、親水性繊維材料と吸水剤を混合したものを成形した、いわゆるブレンド構造の吸水体が挙げられる。
【実施例】
【0082】
以下、実施例および比較例により、本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。なお、吸水性樹脂ないし吸水剤の諸性能は、以下の方法で測定した。
なお、衛生材料などの最終製品として使用された吸水剤の場合は、吸水剤は吸湿しているので、適宜、吸水剤を最終製品から分離して減圧低温乾燥後(例えば、1mmHg以下、60℃で12時間)に測定すればよい。また、本発明の実施例および比較例において使用された吸水性樹脂の含水率はすべて6重量%以下であった。
さらに、後述の(4)で粒度別のSFCを測定する場合、衛生材料などの最終製品として使用された吸水剤では、上記操作ののちにSFCが測定されるが、使用前の吸水剤、すなわち、実験室で製造された吸水剤を測定する場合、実生産や実使用のダメージに相関させるため、すべて後述(5)の機械的ダメージを与えた後の吸水剤で、粒度別のSFC、分級する前のSFCを測定するものとする。
【0083】
(1)無加圧下吸収倍率(0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率/CRC)
室温(20〜25℃)、湿度50RH%の条件下で、吸水性樹脂ないし吸水剤0.20gを不織布製の袋(60mm×60mm)に均一に入れてシールした後、室温で0.9重量%生理食塩水中に浸漬した。30分後に袋を引き上げ、遠心分離機(株式会社コクサン社製、遠心機:型式H−122)を用いて250Gで3分間水切りを行った後、袋の重量W1(g)を測定した。また、同様の操作を吸水性樹脂あるいは吸水剤を用いずに行い、その時の重量W0(g)を測定した。そして、これらW1、W0から、次式に従って無加圧下吸収倍率(g/g)を算出した。
【0084】
無加圧下吸収倍率(g/g)=(W1(g)−W0(g))/吸水性樹脂ないし吸水剤の重量(g)
(2)加圧下吸収倍率(0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率/AAP)
内径60mmのプラスチックの支持円筒の底に、ステンレス製400メッシュの金網(目の大きさ38μm)を融着させ、室温(20〜25℃)、湿度50RH%の条件下で、該網上に吸水剤0.90gを均一に散布し、その上に、吸水剤に対して4.83kPa(0.7psi)の荷重を均一に加えることができるよう調整された、外径が60mmよりわずかに小さく支持円筒との隙間が生じず、かつ上下の動きが妨げられないピストンと荷重とをこの順に載置し、この測定装置一式の重量Wa(g)を測定した。
【0085】
直径150mmのペトリ皿の内側に直径90mmのガラスフィルター(株式会社相互理化学硝子製作所社製、細孔直径:100〜120μm)を置き、0.90重量%生理食塩水(20〜25℃)をガラスフィルターの上面と同じレベルになるように加えた。その上に、直径90mmの濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を載せ、表面が全て濡れるようにし、かつ過剰の液を除いた。
上記測定装置一式を前記湿った濾紙上に載せ、液を荷重下で吸収させた。1時間後、測定装置一式を持ち上げ、その重量Wb(g)を測定した。そして、Wa、Wbから、次式に従って加圧下吸収倍率(g/g)を算出した。
【0086】
加圧下吸収倍率(g/g)
=(Wa(g)−Wb(g))/吸水剤の重量((0.9)g)
(3)重量平均粒子径
吸水性樹脂粉末ないし吸水剤を目開き850μm、600μm、500μm、425μm、300μm、212μm、150μm、106μm、75μmなどのJIS標準ふるいで篩い分けし、残留百分率Rを対数確率紙にプロットした。これにより、重量平均粒子径(D50)を読み取った。
篩い分け、および、後述の粒度別のSFCを測定する際の分級方法は、吸水性樹脂粉末ないし吸水剤10.0gを、室温(20〜25℃)、湿度50RH%の条件下で、目開き850μm、600μm、500μm、300μm、150μmのJIS標準ふるい(THE IIDA TESTING SIEVE:径8cm)に仕込み、振動分級器(IIDA SIEVE SHAKER、TYPE:ES−65型、SER.No.0501)により、10分間、分級を行った。
【0087】
(4)0.69重量%生理食塩水流れ誘導性(SFC)
特表平9−509591の生理食塩水流れ誘導性(SFC)試験に準じて行った。
図1に示す装置を用い、容器40に均一に入れた吸水剤(0.900g)を人工尿(1)中で0.3psi(2.07kPa)の加圧下、60分間膨潤させ、ゲル44のゲル層の高さを記録し、次に0.3psi(2.07kPa)の加圧下、0.69重量%生理食塩水33を、一定の静水圧でタンク31から膨潤したゲル層を通液させる。このSFC試験は室温(20〜25℃)で行った。コンピューターと天秤を用い、時間の関数として20秒間隔でゲル層を通過する液体量を10分間記録する。膨潤したゲル44(の主に粒子間)を通過する流速Fs(t)は増加重量(g)を増加時間(s)で割ることによりg/sの単位で決定する。一定の静水圧と安定した流速が得られた時間をtsとし、tsと10分間の間に得たデータだけを流速計算に使用して、tsと10分間の間に得た流速を使用してFs(t=0)の値、つまりゲル層を通る最初の流速を計算する。Fs(t=0)はFs(t)対時間の最小2乗法の結果をt=0に外挿することにより計算される。
【0088】
生理食塩水流れ誘導性=(Fs(t=0)×L0)/(ρ×A×ΔP)
=(Fs(t=0)×L0)/139506
ここで、
Fs(t=0):g/sで表した流速
L0:cmで表したゲル層の高さ
ρ:NaCl溶液の密度(1.003g/cm3)
A:セル41中のゲル層上側の面積(28.27cm2)
ΔP:ゲル層にかかる静水圧(4920dyne/cm2)
およびSFC値の単位は(10−7×cm3×s×g−1)である。
【0089】
図1に示す装置としては、タンク31には、ガラス管32が挿入されており、ガラス管32の下端は、0.69重量%生理食塩水33をセル41中の膨潤ゲル44の底部から、5cm上の高さに維持できるように配置した。タンク31中の0.69重量%生理食塩水33は、コック付きL字管34を通じてセル41へ供給された。セル41の下には、通過した液を補集する容器48が配置されており、補集容器48は上皿天秤49の上に設置されていた。セル41の内径は6cmであり、下部の底面にはNo.400ステンレス製金網(目開き38μm)42が設置されていた。ピストン46の下部には液が通過するのに十分な穴47があり、底部には吸水剤あるいはその膨潤ゲルが、穴47へ入り込まないように透過性の良いガラスフィルター45が取り付けてあった。セル41は、セルを乗せるための台の上に置かれ、セルと接する台の面は、液の透過を妨げないステンレス製の金網43の上に設置した。
【0090】
人工尿(1)は、塩化カルシウムの2水和物0.25g、塩化カリウム2.0g、塩化マグネシウムの6水和物0.50g、硫酸ナトリウム2.0g、りん酸2水素アンモニウム0.85g、りん酸水素2アンモニウム0.15g、および、純水994.25gを加えたものを用いた。
(5)機械的ダメージ試験
図2に示すガラス製容器(山村硝子社製マヨネーズ瓶、商品名:A−29)に吸水剤30gとガラスビーズ(玉径約6mmの精密分留充填用ソーダ石灰ガラスビーズ)10gを入れた。これを図3に示す分散機(東洋精機製作所社製、No.488試験用分散機)に備えられたクランプ間に挟み固定し、100V/60Hzで振動回転数750cpmの振動を10分間与えた。これにより、上記分散機に固定された容器は、分散機における上記クランプの取り付け面に対して左右に各々12.5°(合計25°)傾斜運動すると同時に、前後に各々8mm(合計16mm)振動することにより、容器内部の吸水剤に衝撃を与える。上記衝撃は、吸水剤の製造工程中に吸水剤が受ける衝撃力を代表するものとして経験的に定められた力であるが、製造後の輸送や吸水体製造時のダメージにも広く適用できるものである。本発明において、機械的ダメージを与える場合は、特に、吸水剤の製造時、吸水体の製造時を想定したものである。さらに、SFC変化指数、SFC変動係数、SFC変動率は、吸水体内部における吸水剤の性能のばらつきを測る指標である。よって粒度別の物性を測定する場合は、実験室(1回の製造工程で得られる吸水剤の量が20kg以下)のスケールにおいて製造された吸水剤は、すべて前記機械的ダメージを与えなければならない。
【0091】
(6)可溶分(水可溶成分)量
250ml容量の蓋付きプラスチック容器に0.9重量%生理食塩水溶液(生理食塩水)の184.3gを測り取り、その水溶液中に吸水性樹脂1.00gを加え16時間攪拌することにより樹脂中の可溶分を抽出した。この抽出液を濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を用いて濾過することにより得られた濾液の50.0gを測り取り測定溶液とした。
はじめに生理食塩水だけを、まず、0.1NのNaOH水溶液でpH10まで滴定を行い、その後、0.1NのHCl水溶液でpH2.7まで滴定して空滴定量([bNaOH]ml、[bHCl]ml)を得た。
【0092】
同様の滴定操作を測定溶液についても行うことにより滴定量([NaOH]ml、[HCl]ml)を求めた。
例えば既知量のアクリル酸とそのナトリウム塩からなる吸水性樹脂の場合、そのモノマーの平均分子量と上記操作により得られた滴定量をもとに、吸水性樹脂中の可溶分量を以下の計算式により算出することができる。未知量の場合は滴定により求めた中和率を用いてモノマーの平均分子量を算出する。
可溶分(重量%)=0.1×(平均分子量)×184.3×100×([HCl]−[bHCl])/1000/1.0/50.0
中和率(mol%)=(1−([NaOH]−[bNaOH])/([HCl]−[bHCl]))×100
(7)表層可溶分(水可溶成分)量
250ml容量の蓋付きプラスチック容器に0.50重量%生理食塩水溶液の100gを測り取り、その水溶液中に吸水剤0.50gを加え、1時間吸水剤中の表層可溶分を抽出した。この抽出操作において、攪拌は初期の吸水剤の分散および最後の液の均一化のみを目的に攪拌し、前記以外は攪拌を行わない。具体的には、表層可溶分の抽出は静置で行い、抽出前後の1分間のみ攪拌(攪拌速度:400rpm、攪拌子:テフロン製(2.5cm))を行う。この抽出液を濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を用いて濾過することにより得られた濾液の50.0gを測り取り測定溶液とした。
【0093】
はじめに0.5重量%NaCl水溶液だけを、まず、0.1NのNaOH水溶液でpH10まで滴定を行い、その後、0.1NのHCl水溶液でpH2.7まで滴定して空滴定量([bNaOH]ml、[bHCl]ml)を得た。
同様の滴定操作を測定溶液についても行うことにより滴定量([NaOH]ml、[HCl]ml)を求めた。
例えば既知量のアクリル酸とそのナトリウム塩からなる吸水剤の場合、そのモノマーの平均分子量と上記操作により得られた滴定量をもとに、吸水剤中の表層可溶分量を以下の計算式により算出することができる。未知量の場合は滴定により求めた中和率を用いてモノマーの平均分子量を算出する。
【0094】
表層可溶分(重量%)=0.1×(平均分子量)×100×100×([HCl]−[bHCl])/1000/0.5/50.0
中和率(mol%)=(1−([NaOH]−[bNaOH])/([HCl]−[bHCl]))×100
(8)中和指数
特許文献30およびその請求項3(欧州特許公開0882502号およびその請求項2〜4)に従い、以下、吸水性樹脂粉末および吸水剤の中和指数を求めた。
すなわち、JIS標準篩で300〜600μmに分級した吸水性樹脂粉末ないし吸水剤の粒子200粒を、カバーガラスを貼りつけた20mm×20mmの開口部を有する厚さ1.6mmのプラスチックプレートに入れ、0.2mlの脱イオン水を添加する。さらに、上記膨潤ゲルにブルムチモールブルー(BTB)0.1重量%エタノール溶液とメチルレッド(MR)0.1重量%エタノール溶液との1.5:1混合溶液0.05mlをマイクロシリンジで添加して、pH指示薬によって粒子200粒の着色を観察した。こうして吸水性樹脂ないし吸水剤の平均中和率から20モル%を越えて不均一に中和された粒子の個数(200個中で何個か)を求めて、この不均一な中和粒子の個数を中和指数(上記特許では第一中和指数/請求項3)として求めた。もちろん、中和指数が大きいほど、吸水性樹脂粉末の中和は不均一である。詳しくは、上記特許を参照。
【0095】
(参考例1):吸水性樹脂粉末(A)の製造/中和重合
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度38重量%、モノマーの平均分子量88.5)にポリエチレングリコールジアクリレート(n=9)3.70gを溶解させて反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に10重量%過硫酸ナトリウム水溶液28.3gおよび1重量%L−アスコルビン酸水溶液2.1gを攪拌しながら添加したところ、およそ1分後に重合が開始した。そして、生成したゲルを粉砕しながら、20〜95℃で重合を行い、重合が開始して30分後に含水ゲル状架橋重合体(1)を取り出した。
【0096】
得られた含水ゲル状架橋重合体(1)は、その径が約5mm以下に細分化されていた。この細分化された含水ゲル状架橋重合体(1)を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き850μmおよび106μmのJIS標準篩で分級することで大部分の粒子が850μm〜106μmの範囲にある吸水性樹脂粉末(A)を得た。
得られた吸水性樹脂粉末(A)の無加圧下の吸収倍率は49(g/g)、可溶分量は23重量%、重量平均粒径(D50)は330μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
【0097】
(参考例2):吸水性樹脂粉末(B)の製造/中和重合
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、71モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度41重量%、モノマーの平均分子量87.7)にポリエチレングリコールジアクリレート(n=9)8.05gを溶解させて反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に10重量%過硫酸ナトリウム水溶液30.8gおよび1重量%L−アスコルビン酸水溶液2.57gを攪拌しながら添加したところ、およそ1分後に重合が開始した。そして、生成したゲルを粉砕しながら、20〜95℃で重合を行い、重合が開始して30分後に含水ゲル状架橋重合体(2)を取り出した。
【0098】
得られた含水ゲル状架橋重合体(2)は、その径が約5mm以下に細分化されていた。この細分化された含水ゲル状架橋重合体(2)を50メッシュ(目開き300μm)の金網上に広げ、180℃で50分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き850μmのJIS標準篩で分級することで大部分の粒子が850μm以下の範囲にある吸水性樹脂粉末(B)を得た。
得られた吸水性樹脂粉末(B)の無加圧下の吸収倍率は36(g/g)、可溶分量は10重量%、重量平均粒径(D50)は450μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
【0099】
(参考例3):吸水性樹脂粉末(C)の製造例/中和重合
参考例2において、ポリエチレングリコールジアクリレート5.01gに変更した以外は参考例2と同様にして重合、乾燥、粉砕および分級を行い、吸水性樹脂粉末(C)を得た。得られた吸水性樹脂粉末(C)の無加圧下の吸収倍率は39(g/g)、可溶分量は13重量%、重量平均粒径(D50)は450μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
(参考例4):吸水性樹脂粉末(D)の製造例/中和重合
参考例2において、ポリエチレングリコールジアクリレート5.01gに変更した以外は参考例2と同様にして重合、乾燥、および粉砕を行った。得られた粉砕物をさらに目開き600μmおよび300μmのJIS標準篩で分級することで、600μm〜300μmの範囲にある吸水性樹脂粉末(D)を得た。得られた吸水性樹脂粉末(D)の無加圧下の吸収倍率は40(g/g)、可溶分量は9重量%、重量平均粒径(D50)は450μmであった。
【0100】
(実施例1):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基あり
参考例1で得られた吸水性樹脂粉末(A)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.027g、プロピレングリコール0.9g、水2.7g及び炭酸水素ナトリウム0.18gの混合液からなる表面処理剤を混合した後、混合物を210℃で35分間加熱処理することにより、吸水剤(1)を得た。得られた吸水剤(1)も粉末状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例2):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基あり
実施例1において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウムを0.09g用いた以外は同様にして、吸水剤(2)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0101】
(比較例1):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基なし
実施例1において、炭酸水素ナトリウムを用いる代わりに、イソプロピルアルコール0.81gを用いた以外は同様にして、比較用吸水剤(1)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例2):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基なし
実施例1において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(2)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0102】
(実施例3):吸水性樹脂粉末(B)の架橋処理/水溶性無機塩基あり
参考例2で得られた吸水性樹脂粉末(B)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で30分間加熱処理することにより、吸水剤(3)を得た。得られた吸水剤(3)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例3):吸水性樹脂粉末(B)の架橋処理/水溶性無機塩基なし
実施例3において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(3)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0103】
(実施例4):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および炭酸水素ナトリウム0.24gの混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(4)を得た。得られた吸水剤(4)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例5):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウム0.12gを用いた以外は同様にして、吸水剤(5)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0104】
(実施例6):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、炭酸水素ナトリウムを用いる代わりに、24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)を用いた以外は同様にして、吸水剤(6)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例4):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例4において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(4)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0105】
(実施例7):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、加熱処理時間を30分間にした以外は同様にして、吸水剤(7)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例8):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例7において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウム0.12gを用いた以外は同様にして、吸水剤(8)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0106】
(実施例9):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例7において、炭酸水素ナトリウムを用いる代わりに、24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)を用いた以外は同様にして、吸水剤(9)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。また、吸水剤(9)の表層可溶分は3.8重量%であった。
(比較例5):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例7において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(5)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。また、比較用吸水剤(5)の表層可溶分は6.5重量%であった。
【0107】
(比較例6):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例9において、24重量%水酸化ナトリウム水溶液の代わりに、特許文献25に準じて、硫酸アルミニウム14〜18水和物を0.455g用いた以外は同様にして、比較用吸水剤(6)を得た。その結果を表1に示した。
(実施例10):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.784g、水4.0g、および24重量%水酸化ナトリウム水溶液0.5g(固形分0.12g)の混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(10)を得た。得られた吸水剤(10)も粉末形状であり、結果を表1に示した。
【0108】
(比較例7):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例10において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(7)を得た。その結果を表1に示した。
(実施例11):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gに3−エチル−3−オキセタンメタノール0.4g、水3.0g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(11)を得た。得られた吸水剤(11)も粉末形状であり、その結果を表1に示した。
【0109】
(比較例8):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例11において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(8)を得た。その結果を表1に示した。
(実施例12):吸水性樹脂粉末(C)の架橋処理/pH緩衝剤あり
参考例3で得られた吸水性樹脂粉末(C)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成製)0.15g、プロピレングリコール1.0g、水5.0g、およびリン酸2水素ナトリウム・2水和物0.5gの混合液からなる表面処理剤を混合した後、混合物を150℃で30分間加熱処理することにより、吸水剤(12)を得た。得られた吸水剤(12)も粉末形状であり、その結果を表1に示した。
【0110】
(比較例9):吸水性樹脂粉末(C)の架橋処理/pH緩衝剤なし
実施例12において、リン酸二水素ナトリウム二水和物を用いなかった以外は同様にして、比較用吸水剤(9)を得た。その結果を表1に示した。
(実施例13):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例9において、同様の手法を用いて、吸水剤(13)を得た。得られた吸水剤(13)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(13)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた吸水剤(13)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(13−a)、粒子径が600μm未満で500μm以上の粒子(13−b)、粒子径が500μm未満で300μm以上の粒子(13−c)、粒子径が300μm未満で150μm以上の粒子(13−d)を得た。得られた吸水剤(13−a)、(13−b)、(13−c)、(13−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0111】
(実施例14):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例9において、加熱時間を20分間にした以外は同様の手法をもちいて吸水剤(14)を得た。得られた吸水剤(14)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(14)を測定した結果を表2に示した。さらに得られた吸水剤(14)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(14−a)、粒子径が600μm未満で500μm以上の粒子(14−b)、粒子径が500μm未満で300μm以上の粒子(14−c)、粒子径が300μm未満で150μm以上の粒子(14−d)を得た。得られた吸水剤(14−a)、(14−b)、(14−c)、(14−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0112】
(比較例10):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
比較例5において、同様の手法を用いて、比較用吸水剤(10)を得た。得られた比較用吸水剤(10)に機械的ダメージを10分間与えた後、得られた比較用吸水剤(10)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた比較用吸水剤(10)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(10−a)、粒子径が600μm未満で500μm以上の粒子(10−b)、粒子径が500μm未満で300μm以上の粒子(10−c)、粒子径が300μm未満で150μm以上の粒子(10−d)を得た。得られた比較用吸水剤(10−a)、(10−b)、(10−c)、(10−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0113】
(比較例11):市販品の吸水剤
ドイツで市販されているPampers Acitve Fit(P&G社製、2001年12月5日購入)のおむつから吸水剤を取り出し、比較用吸水剤(11)を得た。得られた比較用吸水剤(11)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた比較用吸水剤(11)を、JIS標準ふるい(目開き850、600、500、300、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(11−a)、粒子径が600μm未満で500μm以上の粒子(11−b)、粒子径が500μm未満で300μm以上の粒子(11−c)、粒子径が300μm未満で150μm以上の粒子(11−d)を得た。得られた吸水剤(11−a)、(11−b)、(11−c)、(11−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0114】
(比較例12):市販品の吸水剤
日本で市販されていた「パンパースさらさらケア」(P&G社製、1997年12月購入)のおむつから吸水剤を取り出し、比較用吸水剤(12)とした。比較吸水剤(12)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。
さらに得られた比較用吸水剤(12)をJIS標準ふるい(目開き850、600、500、300、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(12−a)、粒子径が600μm未満で500μm以上の粒子(12−b)、粒子径が500μm未満で300μm以上の粒子(12−c)、粒子径が300μm未満で150μm以上の粒子(12−d)を得た。得られた吸水剤(12−a)、(12−b)、(12−c)、(12−d)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0115】
(実施例15):吸水性樹脂粉末(D)の架橋処理/水溶性無機塩基あり
参考例4で得られた吸水性樹脂粉末(D)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で20分間加熱処理することにより、吸水剤(15)を得た。得られた吸水剤(15)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(15)を測定した結果を表2に示した。さらに得られた吸水剤(15)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、粒子径が600μm未満で500μm以上の粒子(15−b)、粒子径が500μm未満で300μm以上の粒子(15−c)を得た。得られた吸水剤(15−b)、(15−c)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0116】
(実施例16):吸水性樹脂粉末(D)の架橋処理/水溶性無機塩基あり
実施例15において、加熱時間を40分間にした以外は同様にして、吸水剤(16)を得た。得られた吸水剤(16)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(16)を測定した結果を表2に示した。さらに得られた吸水剤(16)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、粒子径が600μm未満で500μm以上の粒子(16−b)、粒子径が500μm未満で300μm以上の粒子(16−c)を得た。得られた吸水剤(16−b)、(16−c)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0117】
(比較例13〜16):市販品の吸水剤
実使用されている吸水剤(吸水性樹脂)として、2001年に市販の紙おむつより吸水性樹脂を取り出し比較吸水剤(13)〜(16)とした。比較吸水剤(13)〜(16)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表4に示した。
(参考例5):吸水性樹脂粉末(E)の製造/酸型重合で後中和
2Lのプラスチック容器にアクリル酸252.2g、N,N’−メチレンビスアクリルアミド1.1g、水998.4gを混合し反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に15重量%2,2’−アゾビス(2−アミジノプロパン)二塩酸塩5.1g、10重量%L−アスコルビン酸水溶液0.63g、および7重量%過酸化水素水溶液3.6gを加えて重合を開始させた。16〜74℃で重合を行い、重合が開始してから3時間後に含水ゲル状架橋重合体(5)を取り出した。得られた重合体(5)を約5cm角に切断してからその1000gについて、中和率が65モル%となる量の48重量%の水酸化ナトリウム水溶液15.0gを混合し、さらにダイス径9.5mmのミートチョッパーに通して細分化された粉砕ゲルとした。得られた粉砕ゲルを50メッシュ(目開き300μm)の金網上に広げ、170℃で40分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き500μmおよび300μmのJIS標準網で分級することで大部分の粒子が500μm〜300μmの範囲にある吸水性樹脂粉末(E)を得た。得られた吸水性樹脂粉末(E)の無加圧下の吸収倍率は49(g/g)、可溶分量は6重量%であった。
【0118】
さらに得られた粒子の中和率の均一性を調べるために、200個の粒子を膨潤させ、さらに、pH指示薬であるブロモチモールブルーとメチルレッドの0.1重量%エタノール溶液を添加し中和指数(特許文献30の請求項3)を測定した。その結果、参考例1、6、7で得られた中和重合から得られた吸水性樹脂粉末(A)、(F)、(G)は200個の粒子が一様に黄色(中和指数が0)であるのに対して、吸水性樹脂粉末(E)の粒子は濃緑色から赤色まで様々な色に発色する粒子が混在し、個々の粒子の中和率が非常に不均一(中和指数が15以上)であった。
(参考例6):吸水性樹脂粉末(F)の製造例/中和重合
参考例1において、単量体濃度を39重量%、中和率を71モル%、ポリエチレングリコールジアクリレート9.6gに変更した以外は参考例1と同様にして重合、乾燥および粉砕を行った。得られた粉砕物をさらに目開き500μmおよび300μmのJIS標準篩で分級することで大部分の粒子が500μm〜300μmの範囲にある吸水性樹脂粉末(F)を得た。得られた吸水性樹脂粉末(F)の無加圧下の吸収倍率は32(g/g)、可溶分量は10重量%であった。
【0119】
(参考例7):吸水性樹脂粉末(G)の製造例/中和重合
参考例1において、単量体濃度を41重量%、中和率を71モル%、ポリエチレングリコールジアクリレート5.47g、過硫酸ナトリウム水溶液30.8g、L−アスコルビン酸水溶液2.57g、熱風乾燥温度180℃、熱風乾燥時間50分間に変更した以外は参考例1と同様にして重合、乾燥および粉砕を行った。得られた粉砕物をさらに目開き850μmのJIS標準篩で分級することで大部分の粒子が850μm以下の範囲にある吸水性樹脂粉末(G)を得た。
得られた吸水性樹脂粉末(G)の無加圧下の吸収倍率は38(g/g)、可溶分量は13重量%、重量平均粒径(D50)は400μmであった。
【0120】
(実施例17):吸水性樹脂粉末(E)の架橋処理/pH緩衝剤あり
参考例5で得られた吸水性樹脂粉末(E)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.5g、プロピレングリコール1.0g、中性リン酸塩pH標準液(リン酸二水素カリウム/リン酸水素二ナトリウム;pH6.86)6.0g、およびイソプロピルアルコール1.0gの混合液からなる表面処理剤を混合した後、混合物を120℃で30分間加熱処理することにより、吸水剤(17)を得た。得られた吸水剤(17)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0121】
(比較例17):吸水性樹脂粉末(E)の架橋処理/pH緩衝剤なし
実施例17において、中性リン酸塩pH標準液の代わりに水6.0gを用いた以外は同様にして比較用吸水剤(17)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定し、その結果を表5に示した。
(実施例18):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤あり
参考例6で得られた吸水性樹脂粉末(F)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、リン酸二水素ナトリウム二水和物1.2gの混合液からなる表面処理剤を混合した後、混合物を197℃で10分加熱処理することにより吸水剤(18)を得た。得られた吸水剤(18)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0122】
(比較例18):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤なし
実施例18において、リン酸二水素ナトリウム二水和物の代わりにリン酸(85重量%)0.6gを用いた以外は同様にして比較用吸水剤(18)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(比較例19):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤なし
実施例18において、リン酸二水素ナトリウム二水和物を用いなかった以外は同様にして比較用吸水剤(19)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0123】
(実施例19):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤あり
参考例7で得られた吸水性樹脂粉末(G)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、および炭酸水素ナトリウム0.2gの混合液からなる表面処理剤溶液を混合した後、混合物を212℃で25分間加熱処理することにより、吸水剤(19)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(実施例20):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤あり
参考例7で得られた吸水性樹脂粉末(G)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、および炭酸水素カリウム0.24gの混合液からなる表面処理剤溶液を混合した後、混合物を212℃で25分間加熱処理することにより、吸水剤(20)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0124】
(比較例20):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤なし
実施例19において、炭酸水素ナトリウムを用いなかった以外は同様にして比較用吸水剤(20)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(実施例21):連続生産系
71モル%が中和されたアクリル酸部分ナトリウム塩とポリエチレングリコールジアクリレート(n=9)を参考例3の比率で、連続的に水溶液重合(ベルト滞留時間:約30分、厚み:約25mm)し、得られた吸水性樹脂の含水ゲル状架橋重合体をミートチョッパーで粒子状に粗砕し、これをバンド乾燥機の多孔板上に薄く広げて載せ、180℃で30分間連続熱風乾燥した。乾燥機出口でブロック状の乾燥重合体が得られた。この乾燥重合体を取り出したと同時に解砕し、得られた粒子状乾燥物を1000kg/hで3段ロールグラニュレーター(ロールギャップが上から1.0mm/0.55mm/0.42mm)に連続供給することで粉砕した。得られた約60℃の粒子状吸水性樹脂の粉末を、網目開き850μmのふるい網を有する篩い分け装置で分級し、90重量%以上が850μm未満で150μm以上のサイズの吸水性樹脂粉末(H)(平均粒子径:430〜460μm)を得た。得られた吸水性樹脂粉末(H)の無加圧下の平均吸収倍率(CRC)は40g/g、可溶分量の平均は11重量%であった。なお、CRCおよび可溶分量の物性は2時間ごと(2000kg/Lot)に測定した平均値である。
【0125】
さらに、吸水性樹脂粉末(H)を、高速連続混合機(タービュライザー/1000rpm)に1000kg/hで連続供給して、さらに、吸水性樹脂粉末(H)に1,4−ブタンジオール/プロピレングリコール/水/24%水酸化ナトリウム水溶液=0.384/0.63/3.39/0.3(重量%/対粉末)からなる表面架橋剤水溶液を、約200μmの液滴になるスプレーで噴霧し混合した。次いで、得られた混合物を195℃で40分間、パドルドライヤーにより連続的に加熱処理することで吸水剤粉末を得た。さらに得られた吸水剤粉末を、網目開き850μmのふるい網を有する篩い分け装置で分級して、850μm未満で150μm以上のサイズの粒子が90重量%以上の吸水剤(21)を得た。
【0126】
前記の一連の工程(重合、乾燥、粉砕、加熱処理)を連続で24時間で行い、2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:11点)を測定した結果、無加圧下の吸収倍率の平均値は31.1g/g、加圧下での吸収倍率の平均値は25.5g/g、生理食塩水流れ誘導性の平均値は30(Lot毎のSFCの値:32、32、28、28、30、27、27、32、31、32、28)、そのSFCの標準偏差の値は2.1であった。
(実施例22):連続生産系
実施例21において、24時間を10日間に変えた以外は同様の方法で連続生産を行い、吸水剤(22)を得た。2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:110点)を測定した結果、無加圧下の吸収倍率の平均値は31.3g/g、加圧下での吸収倍率の平均値は25.2g/g、生理食塩水流れ誘導性の平均値は30(Lot毎のSFCの値は省略)、そのSFCの標準偏差の値は3.9であった。
【0127】
(比較例21):連続生産系
実施例21において、24%水酸化ナトリウム水溶液を使わなかった以外は同様の方法で連続生産を行い、比較用吸水剤(21)を得た。2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:11点)を測定した結果、無加圧下の吸収倍率の平均値は31.1g/g、加圧下での吸収倍率の平均値は24.2g/g、生理食塩水流れ誘導性の平均値は20(Lot毎のSFCの値:26、17、11、17、17、18、20、17、16、28、28)、そのSFCの標準偏差の値は5.5であった。
実施例21および実施例22に記載の吸水剤(21)、(22)は、比較例21に記載の比較用吸水剤(21)に比べて、SFCの標準偏差が小さくなり、本発明の吸水剤を連続生産する場合、不良品(低SFC品など)の発生もなく、かつ、品質(SFC)のばらつきが低減されることを示している。
【0128】
【表1】

【0129】
【表2】
【0130】
【表3】
【0131】
実施例1および実施例2に記載の吸水剤(1)、(2)は、同じく表面架橋された比較用吸水剤(1)、(2)と比べて、加圧下吸収倍率が大きく、無加圧下吸収倍率と加圧下吸収倍率のバランスに優れている。
実施例3に記載の吸水剤(3)は、比較例3に記載の比較用吸水剤(3)と比べて、無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスに優れている。
さらに、実施例4および実施例5および実施例6に記載の吸水剤(4)、(5)、(6)は、比較例4に記載の比較用吸水剤(4)と比べて、無加圧下吸収倍率はほぼ同じであるが生理食塩水流れ誘導性は高い値を示している。
【0132】
同様に、実施例7および実施例8および実施例9に記載の吸水剤(7)(8)(9)においても、比較例5および比較例6に記載の比較用吸水剤(5)および比較用吸水剤(6)と比べて、無加圧下吸収倍率はほぼ同じであるが、加圧下吸水倍率と生理食塩水流れ誘導性は高い値を示している。
また、実施例10、実施例11、実施例12に記載の吸水剤(10)、(11)、(12)は、それぞれ比較例7、比較例8、比較例9に記載の比較用吸水剤(7)、(8)、(9)と比べて、無加圧下吸収倍率はほぼ同じであるが、生理食塩水流れ誘導性は高い値を示している。
【0133】
実施例13および実施例14に記載の吸水剤(13)、(14)は、比較例10および比較例11および比較例12に記載の比較用吸水剤(10)、(11)、(12)と比べて、SFC変化指数およびSFC変動係数は小さく、SFC変動率は大きくなりいずれもSFCのばらつきが低減されている。
また、実施例15および実施例16に記載の吸水剤(15)、(16)も、SFC変化指数およびSFC変動指数は小さく、SFC変動率は大きくなり、いずれもSFCのばらつきが低減されている。
このように、本発明の方法で製造される吸水剤は、無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスに優れ、良好な性能を備えたものである。さらに、粒度毎のSFCのばらつきも少ない事から、おむつの性能を安定させる上で非常に優れた吸水剤である。
【0134】
【表4】
【0135】
比較例13、比較例14、比較例15、比較例16に記載の比較用吸水剤(13)、(14)、(15)、(16)は、いずれも無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスが悪いものであった。
【0136】
【表5】
【0137】
実施例17記載の吸水剤(17)は、比較例17記載の比較用吸水剤(17)と比べて、無加圧下吸収倍率と加圧下吸収倍率のバランスやその合計に優れている。
また、実施例18記載の吸水剤(18)は、リン酸を加えた比較例18記載の比較用吸水剤(18)と比べて、同様に10分という短い時間で架橋反応が進行し、無加圧下吸収倍率は同じであるが加圧下吸収倍率は高い値を示している。さらに、何も加えなかった比較例19記載の比較用吸水剤(19)については、10分という短時間では十分な架橋反応が進行せず、無加圧下吸収倍率の低下がほとんど見られず、加圧下吸収倍率も低いものとなっている。
【0138】
このように、本発明の方法で製造される吸水剤は、無加圧下吸収倍率と加圧下吸収倍率のバランスやその合計に優れ、短い反応時間でも良好な性能を備えたものである。
【符号の説明】
【0139】
31 タンク
32 ガラス管
33 0.69重量%塩化ナトリウム水溶液
34 コック付きL字管
35 コック
40 容器
41 セル
42 ステンレス製金網
43 ステンレス製金網
44 膨潤ゲル
45 ガラスフィルター
46 ピストン
47 ピストン中の穴
48 補集容器
49 上皿天秤
51 ガラス容器
52 分散機
53 上側クランプ
54 下側クランプ
【技術分野】
【0001】
本発明は、吸水剤およびその製造方法に関する。さらに詳しくは、本発明は、吸水性樹脂を架橋剤で改質することで得られる吸水剤であって、無加圧下でも加圧下でも高い吸収倍率、さらに高い生理食塩水流れ誘導性を示す吸水剤およびその製造方法に関する。
【背景技術】
【0002】
近年、紙オムツや生理用ナプキン、いわゆる失禁パット等の衛生材料には、その構成材として、体液を吸収させることを目的とした吸水性樹脂(吸水剤)が幅広く使用されている。
上記の吸水性樹脂としては、例えば、ポリアクリル酸部分中和物架橋体、澱粉−アクリル酸グラフト重合体の加水分解物、酢酸ビニル−アクリル酸エステル共重合体ケン化物、アクリロニトリル共重合体若しくはアクリルアミド共重合体の加水分解物またはこれらの架橋体、及びカチオン性モノマーの架橋重合体等が知られている。
上記の吸水性樹脂が備えるべき特性として、従来より体液等の水性液体に接した際の優れた吸液量や吸水速度、ゲル強度、ゲル通液性、水性液体を含んだ基材から水を吸い上げる吸引力等が唱えられている。そして、従来よりこれらの特性を複数併せ持ち、紙オムツや生理用ナプキン等の衛生材料に用いられた場合に、優れた性能(吸収特性)を示す吸水性樹脂(吸水剤)が種々提案されている。
【0003】
例えば、吸水性樹脂の無加圧下吸収倍率および加圧下吸収倍率等の吸収特性をバランス良く改良する方法として、吸水性樹脂の表面近傍を架橋する技術が知られており、これまでに様々な方法が開示されている。
その例として、多価アルコールを用いる方法(特許文献1、特許文献2)、多価グリシジル化合物、多価アジリジン化合物、多価アミン化合物、多価イソシアネート化合物を用いる方法(特許文献3)、グリオキサールを用いる方法(特許文献4)、多価金属を用いる方法(特許文献5、特許文献6、特許文献7)、シランカップリング剤を用いる方法(特許文献8、特許文献9、特許文献10)、アルキレンカーボネートを用いる方法(特許文献11)、多価ヘテロ環カーボネートを用いる方法(特許文献12)、オキサゾリジノンを用いる方法(特許文献13)、多価オキサゾリジノンを用いる方法(特許文献14)、オキサジンを用いる方法(特許文献15)、オキサゾリン化合物を用いる方法(特許文献16)等が知られている。
【0004】
さらに、上記架橋剤によって吸収特性の向上を行う際に、更なる性能向上のために添加剤(不活性混合助剤、酸触媒、塩基)を用いる方法も知られている。すなわち、添加剤として不活性混合助剤を用いる方法(1)として、不活性無機粉末を存在させる方法(特許文献17、特許文献18)、多価金属の塩および/または水酸化物を含む水を存在させる方法(特許文献7)、二価アルコールを存在させる方法(特許文献19)、水とエーテル化合物を存在させる方法(特許文献20)、水溶性ポリマーを存在させる方法(特許文献21)、1価アルコールのアルキレンオキサイド付加物、有機酸の1価塩、またはラクタム類を存在させる方法(特許文献22、特許文献23)、一価金属塩を存在させる方法(特許文献24)、カチオンを存在させる方法(特許文献25、特許文献26)等が知られている。
【0005】
さらに、添加剤として酸触媒を用いる方法(2)として、リン酸を存在させる方法(特許文献27)、無機酸または有機酸を存在させる方法(特許文献28)等が知られている。
また、添加剤として塩基を用いる方法(3)として、水溶性アルカリ化合物を存在させる方法(特許文献29)等も知られている。
これら(1)、(2)、(3)の方法で用いる添加剤が架橋剤と共に存在することにより、架橋剤単独に比べて吸水剤の吸収特性のバランスをある程度向上させることもできるが、まだまだ十分なものとは言い難いものであった。
【0006】
すなわち、(1)の方法で用いるような添加剤(不活性混合助剤)では、使用する吸水性樹脂に微紛が多く含まれる等の場合には混合助剤としての働きによりその効果が現れるものの、一方ではその存在により架橋剤の吸水性樹脂粉末への浸透性の過度の低下や架橋反応の阻害等により吸収特性の改善がほとんど見られなかったり、改善するにしても架橋剤の使用量が増加する、反応時間が長くなる、反応温度を上昇させなければならない、等という問題点もあった。
(2)の方法で用いるような添加剤(酸触媒)では、架橋剤の反応を促進する触媒としての効果が期待できるものの、ある程度の効果を得るための量を添加すると架橋剤溶液のpHの極度な低下と、酸基を含有する部分中和型の吸水性樹脂の場合などには特に表面の酸性化が起こって架橋剤の浸透性の制御が困難となる。また、表面の酸性化は吸水性樹脂の粒子間の接着性を増加し、凝集体の形成につながる傾向にあるので好ましくない。その結果、所望する吸水性樹脂粒子表面層の架橋密度が得られず性能的に満足できるものが得難いという問題点がある。
【0007】
(3)の方法には、添加剤(塩基)とカルボキシル基と容易に反応する官能基を2個以上有する化合物(多価金属塩、ポリエポキシ化合物、ポリアジリジニル化合物、ポリイソシアネート化合物)との組み合わせによる表面架橋が開示され、ゲル強度や比較的低荷重(20g/cm2)での加圧下吸収倍率の改善が図られている。しかし、特許文献29の方法では、いまだ表面架橋剤の混合性改良や吸水性樹脂の物性改良に不十分であり、特に、近年求められているSFCや高荷重下(4.83kPa、約50g/cm2)での加圧下吸収倍率(AAP)(いずれも後述)を向上させることは困難であった。
また、典型的な吸水性樹脂としては、その高物性やコスト面から、アクリル酸の部分中和塩架橋体からなるアクリル酸系吸水性樹脂が挙げられる。そして、かかるアクリル酸系吸水性樹脂の製造方法としては、予め所定中和率に中和したアクリル酸およびその塩を重合する方法(以下、中和重合法と呼ぶ)と、未中和ないし低中和のアクリル酸を重合してから重合ゲルを後中和する方法(以下、酸型重合法)の2つが一般的に行われている。
【0008】
中和重合法に比べて、後者の酸型重合法では高吸収倍率で低可溶分の吸水性樹脂が得られる傾向にはあるが、重合後の含水ゲル状架橋重合体を均一に中和するには長時間を要する上に技術的に非常に困難であり、得られる吸水性樹脂粉末の個々の粒子の中和率が不均一になる場合がある。このような場合、酸型重合法の吸水性樹脂は高吸収倍率で低可溶分にもかかわらず、表面架橋処理を行っても十分な吸水剤の性能が得られないことが、特許文献30(欧州特許公開0882502A1)で開示されている。
すなわち、従来、吸水性樹脂を表面架橋する場合にはその中和率の違いにより必要とされる表面架橋処理が異なり、ある中和率で最適な架橋処理でも他の中和率では所望する吸水剤の性能が得られなかったり、特に吸水性樹脂粉末の個々の粒子の中和率が多岐に混在する場合には、所望する吸水剤の性能が得られないことがあった。
【0009】
以上述べたように、従来の技術によっては表面架橋処理を均一に行うことができておらず、その結果、得られる吸水性樹脂の各種物性(後述のCRC、AAP、SFCなど、特にSFC)のバランスが悪くなるとともに、SFCにバラツキが生じていた。このため、例えば、オムツの性能のロット振れや、オムツ1枚の中でも性能に大きな差が生じていた。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開昭58−180233号公報
【特許文献2】特開昭61−16903号公報
【特許文献3】特開昭59−189103号公報
【特許文献4】特開昭52−117393号公報
【特許文献5】特開昭51−136588号公報
【特許文献6】特開昭61−257235号公報
【特許文献7】特開昭62−7745号公報
【特許文献8】特開昭61−211305号公報
【特許文献9】特開昭61−252212号公報
【特許文献10】特開昭61−264006号公報
【特許文献11】独国特許第4020780号公報
【特許文献12】特開平11−315216号公報
【特許文献13】WO99/42494号公報
【特許文献14】WO99/43720号公報
【特許文献15】WO00/31153号公報
【特許文献16】特開2000−197818号公報
【特許文献17】特開昭60−163956号公報
【特許文献18】特開昭60−255814号公報
【特許文献19】特開平1−292004号公報
【特許文献20】特開平2−153903号公報
【特許文献21】特開平3−126730号公報
【特許文献22】特公平6−74331号公報
【特許文献23】特開平7−33818号公報
【特許文献24】WO98/49221号公報
【特許文献25】WO00/53664号公報
【特許文献26】WO00/53644号公報
【特許文献27】WO94/15651号公報
【特許文献28】特開平7−278225号公報
【特許文献29】特開平6−298841号公報
【特許文献30】特開平10−101735号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、上記従来の問題点に鑑みなされたものである。すなわち、本発明の課題は、架橋処理時に、混合助剤としての効果を示しながらも架橋反応を阻害せず、場合によっては反応触媒としての効果も併せ持ち、かつ部分中和重合された吸水性樹脂の中和率の違いや酸型重合の後中和操作に起因する中和率の均一性にほとんど関わりなく、均一な表面架橋が発現できる吸水剤の製造方法を提供することにある。さらに本発明の課題は、無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性のバランスに優れるとともに、製造の際のロットごと、あるいは各ロット中における生理食塩水流れ誘導性の値の振れ(バラツキ)が小さい、物性の安定した吸水剤を短時間で製造する方法および吸水剤を提供することにもある。
【課題を解決するための手段】
【0012】
本発明者は、吸収特性に優れた吸水剤を鋭意検討した結果、特定の添加剤を吸水性樹脂の特定の架橋剤と共に用いることで上記課題が解決することを見出し、本発明を完成させるに至った。
すなわち、本発明にかかる吸水剤は、
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤に関する。
【0013】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
また、本発明にかかる別の吸水剤は
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤に関する。
【0014】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
さらに、本発明にかかる別の吸水剤は、
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、連続生産された吸水剤に関する。
【0015】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
さらに、本発明にかかる別の吸水剤は、
酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上である。
【0016】
また、本発明にかかる吸水剤の製造方法は、
酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする。
さらに、本発明にかかる別の吸水剤の製造方法は、
酸基を含有し且つ重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる架橋剤(c)を混合し、前記吸水性樹脂粉末(a1)を架橋処理することを特徴とする。
【0017】
また、本発明の衛生材料は、本発明にかかる吸水剤を含む。
【発明の効果】
【0018】
本発明によれば、架橋処理時に、混合助剤としての効果を示しながらも架橋反応を阻害せず、場合によっては反応触媒としての効果も併せ持ち、かつ部分中和重合された吸水性樹脂の中和率の違いや酸型重合の後中和操作に起因する中和率の均一性にほとんど関わりなく、均一な表面架橋が発現でき、無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性のバランスに優れるとともに、製造の際のロットごと、あるいは各ロット中における生理食塩水流れ誘導性の値の振れ(バラツキ)が小さい、物性の安定した吸水剤を短時間で製造する方法および吸水剤を提供することができる。
【図面の簡単な説明】
【0019】
【図1】生理食塩水流れ誘導性の測定に用いる測定装置の概略の断面図。
【図2】機械的ダメージ試験に用いるガラス製容器の側面概略図(a)と平面概略図(b)。
【図3】機械的ダメージ試験に用いる分散機の概略図。
【発明を実施するための形態】
【0020】
以下、本発明について詳細に説明する。なお、以下、本発明で吸水剤とは、架橋構造を有する吸水性樹脂(以下、単に吸水性樹脂と呼ぶ)を主成分(好ましくは50〜100重量%、より好ましくは80〜100重量%、さらに好ましくは90〜100重量%)とし、吸水性樹脂をさらに架橋剤で改質(好ましくは表面改質、特に表面架橋)されたものを吸水剤と呼ぶ。
(吸水性樹脂の製造方法)
以下、本発明において、酸基含有の吸水性樹脂粉末を吸水性樹脂粉末(a)と呼び、かかる吸水性樹脂粉末(a)の中でも、さらに粒度を制御したもの、例えば、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下のものを吸水性樹脂粉末(a1)と呼ぶ。
【0021】
本発明の吸水性樹脂とは、従来から知られている吸水性樹脂のことであり、例えばイオン交換水中において、必須に自重の5倍以上、好ましくは、50倍から1000倍という多量の水を吸収し、アニオン性、ノニオン性、またはカチオン性の水不溶性ヒドロゲルを形成する従来公知の架橋重合体のことである。
これらは、一般に、不飽和単量体成分(好ましくは酸基、特に、カルボキシル基含有不飽和単量体)を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、単量体溶液の状態で重合され、必要に応じて該重合体を乾燥し、乾燥の前および/または後で通常粉砕して得られたものである。このような吸水性樹脂としては、ポリアクリル酸部分中和物重合体、デンプン−アクリロニトリルグラフト重合体の加水分解物、デンプン−アクリル酸グラフト重合体、酢酸ビニル−アクリル酸エステル共重合体のケン化物、アクリロニトリル共重合体もしくはアクリルアミド共重合体の加水分解物、またはこれらの架橋体、カルボキシル基含有架橋ポリビニルアルコール変性物、架橋イソブチレンー無水マレイン酸共重合体等の1種または2種以上を挙げることができる。
【0022】
これらの吸水性樹脂は、1種または混合物でも用いられるが、中でも酸基含有の吸水性樹脂、さらには、カルボン酸またはその塩であるカルボキシル基含有の吸水性樹脂の1種またはその混合物が好ましく、典型的にはアクリル酸及び/又はその塩(中和物)を主成分とする単量体を重合・架橋することにより得られる重合体、すなわち、必要によりグラフト成分を含むポリアクリル酸塩架橋重合体が主成分とされる。
また、上記吸水性樹脂としては、水膨潤性水不溶性であることが必須であり、該吸水性樹脂中の未架橋の水可溶性成分(水溶性高分子)は、好ましくは50重量%以下、より好ましくは25重量%以下、さらに好ましくは20重量%以下、さらにより好ましくは15重量%以下、特に好ましくは10重量%以下のものが用いられる。
【0023】
上記アクリル酸塩としては、アクリル酸のナトリウム、カリウム、リチウム等のアルカリ金属塩、アンモニウム塩及びアミン塩等を例示することができる。上記吸水性樹脂は、その構成単位としてアクリル酸0〜50モル%およびアクリル酸塩100〜50モル%(但し、両者の合計量は100モル%以下とする)の範囲にあるものが好ましく、アクリル酸10〜40モル%およびアクリル酸塩90〜60モル%(但し、両者の合計量は100モル%以下とする)の範囲にあるものがより好ましい。なお、この酸と塩とのモル比を中和率と呼ぶ。上記塩を形成させるための吸水性樹脂の中和は重合前に単量体の状態で行っても良いし、あるいは重合途中や重合後に重合体の状態で行っても良いし、それらを併用してもよい。
【0024】
一般に、未中和ないし低中和の単量体を重合し重合体の状態で中和を行う場合(酸型重合法)には高吸収倍率で低可溶分の吸水性樹脂が得られる傾向にはあるが、吸水性樹脂の個々の粒子の均一な中和にはかなりの労力、設備と時間を要する(特許文献30)。しかし、本発明の方法を用いることで、吸水性樹脂の中和状態や製造方法の如何にかかわらず、すべての吸水性樹脂を良好に表面架橋などに使用することができ、よって、吸水剤の物性と生産性を大幅に向上することが可能となる。
本発明で用いる吸水性樹脂を得るための単量体は、必要に応じて上記アクリル酸(塩)以外の単量体を含有していてもよい。アクリル酸(塩)以外の単量体としては、特に限定されるものではないが、具体的には、例えば、メタクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸等のアニオン性不飽和単量体及びその塩;アクリルアミド、メタアクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン、N−ビニルアセトアミド等のノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミド、及びこれらの四級塩等のカチオン性不飽和単量体等が挙げられる。これら単量体は、単独で用いてもよく、適宜2種類以上を混合して用いてもよい。
【0025】
本発明において、アクリル酸(塩)以外の単量体を用いる場合には、該アクリル酸(塩)以外の単量体は、主成分として用いるアクリル酸及びその塩との合計量に対して、好ましくは30モル%以下、より好ましくは10モル%以下の割合である。上記アクリル酸(塩)以外の単量体を上記の割合で用いることにより、最終的に得られる吸水性樹脂(吸水剤)の吸収特性がより一層向上すると共に、吸水性樹脂(吸水剤)をより一層安価に得ることができる。
本発明に用いられる吸水性樹脂を得るために上述の単量体を重合するに際しては、バルク重合や沈殿重合を行うことが可能であるが、性能面や重合の制御の容易さ、さらに膨潤ゲルの吸収特性の観点から、上記単量体を水溶液とすることによる水溶液重合や逆相懸濁重合を行うことが好ましい。尚、上記単量体を水溶液とする場合の該水溶液(以下、単量体水溶液と称する)中の単量体の濃度は、水溶液の温度や単量体によって決まり、特に限定されるものではないが、10〜70重量%の範囲内が好ましく、20〜60重量%の範囲内がさらに好ましい。また、上記水溶液重合を行う際には、水以外の溶媒を必要に応じて併用してもよく、併用して用いられる溶媒の種類は、特に限定されるものではない。
【0026】
水溶液重合の方法としては、双腕型ニーダー中で単量体水溶液を、得られる含水ゲルを砕きながら重合したり、所定の容器中や駆動するベルト上に単量体水溶液を供給し、重合して得られたゲルをミートチョッパー等で粉砕する方法等が挙げられる。
上記の重合を開始させる際には、例えば過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2’−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤や、2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−オン等の光重合開始剤を用いることができる。
さらに、これら重合開始剤の分解を促進する還元剤を併用し、両者を組み合わせることによりレドックス系開始剤とすることもできる。上記の還元剤としては、例えば、亜硫酸ナトリウム、亜硫酸水素ナトリウム等の(重)亜硫酸(塩)、L−アスコルビン酸(塩)、第一鉄塩等の還元性金属(塩)、アミン類等が挙げられるが、特に限定されるものではない。
【0027】
これら重合開始剤の使用量は、通常0.001〜2モル%、好ましくは0.01〜0.1モル%である。これら重合開始剤の使用量が0.001モル%未満の場合には、未反応の単量体が多くなり、従って、得られる吸水性樹脂や吸水剤中の残存単量体量が増加するので好ましくない。一方、これら重合開始剤の使用量が2モル%を超える場合には、得られる吸水性樹脂や吸水剤中の水可溶成分量が増加するので好ましくない場合がある。
また、反応系に放射線、電子線、紫外線等の活性エネルギー線を照射することにより重合反応の開始を行ってもよいし、さらに、上記重合開始剤を併用してもよい。尚、上記重合反応における反応温度は、特に限定されるものではないが、15〜130℃の範囲が好ましく、20〜120℃の範囲内がより好ましい。また、反応時間や重合圧力も特に限定されるものではなく、単量体や重合開始剤の種類、反応温度等に応じて適宜設定すればよい。
【0028】
前記吸水性樹脂としては、架橋剤を使用しない自己架橋型のものであってもよいが、一分子中に、2個以上の重合性不飽和基や、2個以上の反応性基を有する架橋剤(吸水性樹脂の内部架橋剤)を共重合又は反応させたものがさらに好ましい。
これら内部架橋剤の具体例としては、例えば、N,N’−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチルロールプロパントリ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、エチレンカーボネート、プロピレンカーボネート、ポリエチレンイミン、グリシジル(メタ)アクリレート等を挙げることができる
これら内部架橋剤は、単独で用いてもよく、適宜2種類以上を混合して用いてもよい。また、これら内部架橋剤は、反応系に一括添加してもよく、分割添加してもよい。少なくとも1種または2種類以上の内部架橋剤を使用する場合には、最終的に得られる吸水性樹脂や吸水剤の吸収特性等を考慮して、2個以上の重合性不飽和基を有する化合物を重合時に必須に用いることが好ましい。
【0029】
これら内部架橋剤の使用量は前記単量体(架橋剤を除く)に対して、好ましくは0.001〜2モル%、より好ましくは0.005〜0.5モル%、さらに好ましくは0.01〜0.2モル%、特に好ましくは0.03〜0.15モル%の範囲内とされる。上記内部架橋剤の使用量が0.001モル%よりも少ない場合、並びに、2モル%よりも多い場合には、充分な吸収特性が得られないおそれがある。
上記内部架橋剤を用いて架橋構造を重合体内部に導入する場合には、上記内部架橋剤を、上記単量体の重合前あるいは重合途中、あるいは重合後、または中和後に反応系に添加するようにすればよい。
【0030】
尚、上記重合に際しては、反応系に、澱粉・セルロース、澱粉・セルロースの誘導体、ポリビニルアルコール、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子0〜50重量%(対単量体)や、その他0〜10重量%の、炭酸(水素)塩、二酸化炭素、アゾ化合物、不活性有機溶媒等の各種発泡剤;各種界面活性剤;キレート剤;次亜燐酸(塩)等の連鎖移動剤などを添加してもよい。
上記架橋重合体が水溶液重合で得られたものでゲル状である場合、すなわち含水ゲル状架橋重合体である場合、該架橋重合体は、必要に応じて乾燥し、乾燥の前および/または後で通常粉砕されて吸水性樹脂とする。また、乾燥は通常60℃〜250℃、好ましくは100℃〜220℃、より好ましくは120℃〜200℃の温度範囲で行われる。乾燥時間は、重合体の表面積、含水率、および乾燥機の種類に依存し、目的とする含水率になるよう選択される。
【0031】
本発明に用いることのできる吸水性樹脂や吸水剤の含水率(吸水性樹脂や吸水剤中に含まれる水分量で規定/180℃で3時間の乾燥減量で測定)は特に限定されないが、得られる吸水剤の物性面から室温でも流動性を示す粉末であり、より好ましくは0.2〜30重量%、さらに好ましくは0.3〜15重量%、特に好ましくは0.5〜10重量%の粉末状態である。
また本発明の製造方法に用いることのできる吸水性樹脂としては、粉末状のものを挙げることができる。吸水性樹脂の粒子は重合反応により得られた乾燥粉砕前のゲル状の平均粒径が1000μmを超えるようなものも使用できるが、通常、粉末とならないため、必要により(好ましくは)乾燥・粉砕・分級をすることにより目的に応じた粉末粒径に調整される。
【0032】
吸水性樹脂粉末や吸水剤の粒径としては、重量平均粒子径が10〜2000μm、好ましくは100〜1000μm、より好ましくは200〜700μm、さらに好ましくは300〜600μm、特に好ましくは400〜550μmの範囲が好適に用いられる。さらに好ましくは、吸水性樹脂粉末や吸水剤中の微粉末(例えば100μm以下、好ましくは150μm以下)の微粉末は少ない方が好ましく、具体的には10重量%以下、さらには5重量%以下、特に1重量%以下であることが好ましい。また、吸水性樹脂粉末や吸水剤は好ましくは実質1000μm以上、さらに好ましくは850μm以上の粒子が5重量%以下、さらには1重量%以下とされる。
【0033】
このようにして得られた吸水性樹脂や吸水剤の粒子形状は、球状、破砕状、不定形状等特に限定されるものではないが、粉砕工程を経て得られた不定形破砕状のものが好ましく使用できる。さらに、その嵩比重(JIS K−3362で規定)は、吸水剤の優れた物性から好ましくは0.40〜0.80g/ml、より好ましくは0.50〜0.75g/ml、さらに好ましくは0.60〜0.73g/mlの範囲である。
上記の方法により得られた吸水性樹脂は、通常、無加圧下での生理食塩水に対する飽和吸収倍率が10〜100g/g程度を有し、この吸収倍率などの物性は目的に応じて適宜調整される。
【0034】
(水溶性無機塩基(b1))
本発明では上記吸水性樹脂粉末(a)あるいは(a1)に対して、非架橋性の水溶性無機塩基(b1)、すなわち、好ましくは、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物などからなる群から選ばれる水溶性無機塩基(b1)(以下、水溶性無機塩基(b1)と呼ぶ)、および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、架橋剤(c)あるいは脱水反応性架橋剤(c1)が添加されるが、以下、水溶性無機塩基(b1)について説明する。
すなわち、本発明で水溶性無機塩基とは、水溶液中で解離することで水あるいは該塩基よりOH−を生じ、酸基を中和して塩を生じる無機化合物(炭酸塩や重炭酸塩を含む)を指す。本発明で用いられる水溶性無機塩基(b1)は、好ましくは、アルカリ金属塩,アンモニウム塩,アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物からなる群から選ばれ、これらは、通常、実質的に非架橋性の水溶性無機塩基である。(なお、酸基含有の吸水性樹脂に対して架橋性の水溶性無機塩基として、水酸化カルシウムや水酸化アルミニウムに代表される多価金属の水酸化物が例示されるが、一般に、これら多価金属は本発明の水溶性無機塩基には含まれない)。
【0035】
水溶性無機塩基(b1)として、得られる吸水剤の物性面から水溶性であることが必須であり、室温の水100g当たり通常5g以上、好ましくは20g以上、より好ましくは50g以上、さらに好ましくは100g以上の溶解性を示すものが用いられる。なお、本発明で非水溶性無機塩基、有機塩基や架橋性無機塩基(多価金属の水酸化物)などの併用は排除しないが、非架橋性の水溶性無機塩基(b1)を用いない場合、得られた吸水剤の物性が低い。
具体的に水溶性無機塩基(b1)としては、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸ナトリウムカリウム、炭酸セシウム、炭酸ルビジム、炭酸アンモニウム等のアルカリ金属塩および/またはアンモニウム塩を含む炭酸化合物やその水和物(十水塩、七水塩、一.五水塩、一水塩など)、重炭酸リチウム、重炭酸ナトリウム、重炭酸カリウム、重炭酸セシウム、重炭酸ルビジウム、重炭酸アンモニウム等のアルカリ金属および/またはアンモニウムを含む重炭酸塩、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化セシウム、水酸化ルビジウム、水酸化アンモニウム、水ガラス等のアルカリ金属およびアンモニウムを含む水酸化化合物、燐酸水素2ナトリウム、燐酸水素2カリウム、燐酸水素2リチウム、燐酸水素2ルビジウム、燐酸水素2セシウムなどの燐酸水素化合物、セスキ炭酸ナトリウム(Na2CO3・NaHCO3・2H2O)などの複合塩が挙げられ、これらの2種以上を混合ないし併用しても良い。また、これら水溶性無機塩基(b1)は、粉末やその水和物、ペレットや水溶液として購入・保存ないし使用されるが、その形態に制限はない。
【0036】
水溶性無機塩基(b1)の中でも物性面や溶解性から、好ましくはアルカリ金属塩、さらに好ましくはリチウム塩、ナトリウム塩ないしカリウム塩、より好ましくはナトリウム塩が用いられる。また、化合物(b1)の中でも物性面から、好ましくは、炭酸塩/炭酸水素塩/水酸化物、さらに好ましくは、水酸物およびまたは炭酸水素塩、特に好ましくは水酸化物が用いられる。すなわち、具体的には水溶性無機塩基(b1)として好ましくは、炭酸水素ナトリウムおよび/または水酸化ナトリウム、さらに好ましくは、水酸化ナトリウムが用いられる。
本発明における水溶性無機塩基(b1)の使用量は、後述の非還元性アルカリ金属塩pH緩衝剤(b2)と併用しない場合には、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。上記の範囲内で用いることにより、尿や汗、経血等の体液(水性液体)に対する吸収特性をさらに一層向上させることができる。使用量が0.001重量部未満では、吸水性樹脂の表面近傍の官能基の中和率を適度に調整することができず、吸収特性が向上しない場合がある。水溶性無機塩基(b1)の使用量が10重量部より多い場合には過剰となり、不経済であるとともに、吸収倍率が向上しない恐れがある。
【0037】
なお、水溶性無機塩基(b1)を、後述する非還元性アルカリ金属塩pH緩衝剤(b2)と併用する場合には、上記と同様の理由により、それらの合計使用量が、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。ただし、本発明で水溶性無機塩基(b1)と後述の非還元性アルカリ金属塩pH緩衝剤(b2)とを併用する場合、少なくとも何れか一方の働きを示す範囲で(b1)と(b2)が適宜併用される。
この吸収特性の向上機構は明らかではないが、以下2つの理由((1)表面中和率の均一化による均一な表面架橋層の形成、(2)塩濃度の変化による混合と浸透の最適化)と推定される。
【0038】
すなわち、理由(1)として、一般に吸水性樹脂粉末では重合後の後中和の有無(前述の中和重合法か酸型重合法)にかかわらず、吸水性樹脂粉末の一粒一粒の中和率や、同じ一粒の粉末であっても微小には粉末表面の中和率が異なっており、従来の表面処理においては、これらの理由による粉末の架橋反応や架橋剤の混合に不均一が生じ、物性の低下が発生していた。そこで、本発明においては水溶性無機塩基(b1)を架橋剤(c)と併用することで、吸水性樹脂粉末の一粒一粒の中和率や、一粒の粉末の微小な表面中和率の差をなくすことにより、架橋に関与する表面近傍のカルボキシル基の中和率が一様に最適化でき、その結果均一に表面架橋を行うことが可能となった。例えば、70モル%中和のポリアクリル酸系吸水性樹脂および0.01〜2重量部の水酸化ナトリウムより得られた吸水剤は0.025〜5モル%の範囲で吸水剤の中和率は高められ、さらにその粒子の表面近傍において選択的に高中和率である。さらには、本発明の水溶性無機塩基(b1)は、架橋剤の反応触媒としても作用することが吸収特性向上に起因していると推測される。
【0039】
また、理由(2)として、水溶性無機塩基(b1)は、架橋剤の混合時には架橋剤溶液中の高い塩濃度由来で吸水性樹脂への浸透を制御し混合性を改良しているが、混合後には吸水性樹脂のカルボキシル基と中和反応してアルカリ金属塩およびアンモニウム塩となる事で架橋剤溶液中から消失するため、架橋剤の浸透を促進する働きをなしていると推定される。これは従来の添加剤(イソプロパノールなどの親水性有機溶媒)では、不活性な有機溶媒由来の吸水性樹脂への浸透が制御され混合性は改良されるが、混合後にも有機溶媒が架橋剤溶液中に残存して架橋剤の表面内部への浸透を妨げてしまうと推測される。
また、水溶性無機塩基(b1)と異なり、アルミニウムのような多価金属塩を使用した場合、多価金属イオンによる架橋反応が進行してしまい、無加圧もしくは加圧下での吸収倍率を低下させている事が推測される。さらに、多価金属イオンによる架橋はイオン結合を形成するため非常に弱く、また水膨潤状態では多価金属イオンが粒子内部に移動して架橋を形成するためより一層の物性の低下をもたらす事も推測される。
【0040】
以上のような現象から起こる吸水剤の架橋層の厚みの不足と物性が低下するという従来の欠点を、本願の水溶性無機塩基(b1)では改良していると推定される。さらに脱水反応性架橋剤(c1)を用いることにより、脱水架橋反応から生じる水によってさらに架橋剤が表面近傍に浸透し、架橋層の厚みがより増しているとも推定される。
(非還元性アルカリ金属塩pH緩衝剤(b2))
本発明では上記吸水性樹脂粉末(a)あるいは(a1)に対して、非架橋性の水溶性無機塩基(b1)、すなわち、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物などからなる群から選ばれる水溶性無機塩基(b1)、および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、架橋剤(c)あるいは脱水反応性架橋剤(c1)が添加されるが、以下、非還元性アルカリ金属塩pH緩衝剤(b2)について説明する。
【0041】
本発明における非還元性アルカリ金属塩pH緩衝剤(b2)は、溶液中である程度の酸や塩基の添加の消失でも、ほぼ一定の水素イオン濃度を維持するものであり、必須に非還元性で、好ましくはさらに非酸化性のアルカリ金属塩が用いられる(例えば、リンや硫黄を含むアルカリ金属塩pH緩衝剤の場合、リン原子の酸化数が+5/硫黄原子の酸化数が+6、ならば該pH緩衝剤は非酸化性非還元性を示す)。
pH緩衝剤が還元性を有したり、アルカリ金属塩でない場合、架橋の阻害となる恐れがあり、本発明の目的を十分には達成できない。本発明にはpH緩衝剤として働くアルカリ金属塩であり、種々の酸、塩基、または塩の組み合わせから作成されるpH緩衝剤が適用される。また、吸水性樹脂への混合性や浸透性から、pH緩衝剤の分子量は50〜1000、さらには60〜800、特に70〜500のものが用いられる。
【0042】
本発明でいうpH緩衝剤(b2)として働くアルカリ金属塩とは、代表的には炭酸水素塩、リン酸二水素塩、リン酸水素塩の1種または2種以上が例示される。
具体的な例としては、炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウム、リン酸二水素ナトリウム、リン酸二水素カリウム、リン酸水素二ナトリウム、リン酸水素二カリウムといった無機多塩基酸の部分アルカリ金属塩;クエン酸二水素カリウム、クエン酸二水素ナトリウム、クエン酸水素二ナトリウム、クエン酸水素二カリウム、酒石酸水素ナトリウム、酒石酸水素カリウム、フマル酸一ナトリウム、フタル酸水素カリウムといった有機多価カルボン酸の部分アルカリ金属塩、特にナトリウム塩ないしカリウム塩、さらにはリチウム塩が挙げられる。
【0043】
また、上記pH緩衝剤(b2)のほかに、本発明でいう種々の酸、塩基、または塩の組み合わせから作成されるpH緩衝剤(b2)とは、代表的には従来公知の緩衝液を作成する際に用いられる化合物の組み合わせから作られる緩衝剤が例示される。緩衝液としては、緩衝剤の溶液、特に、弱酸と強塩基からなる塩または強酸と弱塩基からなる塩、またはそれらの塩の混合物の水溶液である。カルボキシル基などの酸基を含有する吸水性樹脂の場合には、好ましくは弱酸と強塩基からなる塩の混合物としての緩衝剤が用いられ、より好ましくは無機塩を用いたものである。
具体的な例としては、化学便覧(日本化学会編、II−355、356)に記載の緩衝液に使用されている化合物の組み合わせが緩衝剤として挙げられる。例えば、Clark−Lubsの緩衝液(塩化カリウム/塩酸;pH1.0〜2.2、フタル酸水素カリウム/塩酸;pH2.2〜3.8、フタル酸水素カリウム/水酸化ナトリウム;pH4.0〜6.2、リン酸二水素カリウム/水酸化ナトリウム;pH5.8〜8.0、ホウ酸/塩化カリウム/水酸化ナトリウム;pH7.8〜10.0)、Sφrensenの緩衝液(グリシン+塩化ナトリウム/塩酸;pH1.1〜4.6、グリシン+塩化ナトリウム/塩酸;pH8.6〜13.0、クエン酸ナトリウム/塩酸;pH1.1〜4.9、クエン酸ナトリウム/水酸化ナトリウム;pH5.0〜6.7、四ホウ酸ナトリウム/塩酸;pH7.6〜9.2、四ホウ酸ナトリウム/水酸化ナトリウム;pH9.3〜12.4、リン酸二水素カリウム/リン酸水素二ナトリウム;pH5.3〜8.0)、Kolthoffの緩衝液(クエン酸カリウム/クエン酸;pH2.2〜3.6、クエン酸二水素カリウム/塩酸;pH2.2〜3.6、クエン酸二水素カリウム/水酸化ナトリウム;pH3.8〜6.0、コハク酸/四ホウ酸ナトリウム;pH3.0〜5.8、クエン酸二水素カリウム/四ホウ酸ナトリウム;pH3.8〜6.0、リン酸二水素カリウム/四ホウ酸ナトリウム;pH5.8〜9.2、四ホウ酸ナトリウム/炭酸ナトリウム;pH9.2〜11.0、塩酸/炭酸ナトリウム;pH10.2〜11.2、リン酸水素二ナトリウム/水酸化ナトリウム;pH11.0〜12.0)、Michaelisの緩衝液(酒石酸/酒石酸ナトリウム;pH1.4〜4.5、乳酸/乳酸ナトリウム;pH2.3〜5.3、酢酸/酢酸ナトリウム;pH3.2〜6.2、リン酸二水素カリウム/リン酸水素二ナトリウム;pH5.2〜8.3、ジエチルバルビツル酸ナトリウム+酢酸ナトリウム/塩酸;pH2.6〜9.2、ジエチルバルビツル酸ナトリウム/塩酸;pH6.8〜9.6、N,N−ジメチルグリシンナトリウム塩/塩酸;pH8.6〜10.6)、Mcilvaineの広域緩衝液(リン酸水素二ナトリウム/クエン酸;pH2.2〜8.0)、Britton−Robinsonの広域緩衝液(クエン酸+リン酸二水素カリウム+ホウ酸+ジエチルバルビツル酸/リン酸三ナトリウム)、Carmodyの広域緩衝液(ホウ酸+クエン酸/リン酸三ナトリウム;pH2.0〜12.0)、Gomoriの緩衝液(2,4,6−トリメチルピリジン/塩酸;pH6.4〜8.4、トリス(ヒドロキシメチル)アミノメタン/塩酸;pH7.2〜9.1、2−アミノ−2−メチル−1,3−プロパンジオール/塩酸;pH7.8〜9.7)、Bates−BowerのTris緩衝液(トリス(ヒドロキシメチル)アミノメタン/塩酸;pH7.0〜9.0)、Delory−King緩衝液(炭酸塩/炭酸水素塩;pH9.2〜10.7)、等が挙げられる。そして、使用される緩衝剤のpHおよび濃度は、吸水性樹脂の中和率や用いる表面架橋剤の種類にもよるが、好ましくは緩衝剤を添加することにより該表面架橋剤溶液のpHが1.5〜10.0の範囲に調整される。
【0044】
上記の内でも、性能、安定性、一成分系での使用、コスト、等の点から無機多塩基酸の部分中和塩が好ましく、リン酸、炭酸の部分アルカリ金属中和塩がより好ましい。
本発明における上記pH緩衝剤(b2)の使用量は、前述の水溶性無機塩基(b1)と併用しない場合には、吸水性樹脂の固形分100重量部に対して0.005〜10重量部の範囲内が好ましく、0.05〜5重量部の範囲内がより好ましい。上記の範囲内で用いることにより、尿や汗、経血等の体液(水性液体)に対する吸収特性をさらに一層向上させることができる。使用量が0.005重量部未満では、吸水性樹脂の表面近傍の官能基の中和率を適度に調整することができず、吸収特性が向上しない場合がある。pH緩衝剤(b2)の使用量が10重量部より多い場合には、該添加剤が過剰となり、不経済であるとともに、吸収倍率が向上しない恐れがある。
【0045】
なお、非還元性アルカリ金属塩pH緩衝剤(b2)を、前述の水溶性無機塩基(b1)と併用する場合には、上記と同様の理由により、それらの合計使用量が、吸水性樹脂100重量部に対して0.001〜10重量部の範囲内が好ましく、0.01〜5重量部の範囲内がより好ましく、さらに好ましくは0.01〜2重量部の範囲である。ただし、本発明で水溶性無機塩基(b1)と前述の非還元性アルカリ金属塩pH緩衝剤(b2)と併用する場合、少なくとも何れか一方の働きを示す範囲で(b1)と(b2)が適宜併用される。
この吸収特性の向上機構は明らかではないが、以下2つの理由((1)表面中和率の均一化、(2)塩濃度の変化による混合と浸透の最適化)とも推定される。
【0046】
すなわち、理由(1)として、吸水性樹脂粉末では重合後の後中和の有無(前述の中和重合法か酸型重合法)にかかわらず、吸水性樹脂粉末の一粒一粒の中和率や、同じ一粒の粉末であっても微小には粉末表面の中和率が異なっており、粉末の架橋の反応や架橋剤の混合に不均一を生じて物性を低下されていたものが、本発明のpH緩衝剤(a)を架橋剤(b)と併用することで、吸水性樹脂粉末の一粒一粒の中和率や、一粒の粉末の微小な表面中和率の差をなくしているため、吸水性樹脂粉末の中和率や後述の中和指数の如何に関わらず、本発明では、架橋に関与する表面近傍のカルボキシル基の中和率が一様に最適化できるためと考えられる。その結果、本発明のpH緩衝剤は、吸水性樹脂粉末への架橋剤の浸透を妨げられることなく、混合助剤としても作用し、さらには、架橋剤の反応触媒としても作用することが吸収特性向上に起因していると推測される。
【0047】
また、理由(2)として、炭酸水素塩などpH緩衝剤は、架橋剤の混合時には架橋剤溶液中のアルカリ金属塩として存在するために、高い塩濃度由来の吸水性樹脂への浸透を制御し混合性を改良しているが、混合後には吸水性樹脂のカルボキシル基と中和反応してpH緩衝剤のアルカリ金属塩が架橋剤溶液中から消失するため、混合後には架橋剤の浸透を阻害した塩が消失して架橋剤の浸透を促進する働きをなしていると推定される。これは従来の添加剤(イソプロパノールなどの親水性有機溶媒)では、不活性な有機溶媒由来の吸水性樹脂への浸透が制御され混合性は改良されるが、混合後にも有機溶媒が架橋剤溶液中に残存して架橋剤の表面内部への浸透を妨げてしまい、吸水剤の架橋層の厚みが不足している従来の欠点を、本願のpH緩衝剤では改良していると推定される。
【0048】
(架橋剤(c)およびその混合と架橋処理)
本発明では、酸基と反応しうる架橋剤(c)として、好ましくは表面架橋剤、さらには、脱水反応性架橋剤(c1)が好ましく用いられる。なお、本発明で脱水反応性とは、吸水性樹脂の官能基(特に表面近傍の官能基)と架橋剤とが脱水反応、好ましくは、脱水エステル化および/または脱水アミド化、さらに好ましくが、脱水エステル化で架橋する架橋剤である。
具体的に吸水性樹脂がカルボキシル基を含有する場合、多価アルコールなどのヒドロキシル基含有の架橋剤、多価アミンなどのアミノ基含有の架橋剤、さらには、アルキレンカーボネートやモノ、ジまたはポリのオキサゾリジノン化合物;3−メチル−3−オキセタンメタノール等のオキセタン化合物などの環状架橋剤であって、その環状架橋剤の開環反応に伴ってヒドロキシル基やアミノ基を生成し該ヒドロキシル基やアミノ基が架橋反応を行う環状架橋剤、などが脱水反応性を示す架橋剤(c1)として例示される。脱水反応性架橋剤(c1)の1種または2種以上が用いられるが、さらに、非脱水反応性の架橋剤、例えば、多価金属なども併用してもよい。
【0049】
具体的に、本発明に用いることのできる脱水反応性架橋剤(c1)としては、吸水性樹脂の官能基と反応しうる架橋剤ならば制限なく使用され、通常、該用途に用いられている架橋剤(表面架橋剤)のことである。例えば、エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、ジプロピレングリコール、ポリプロピレングリコール、1,3−プロパンジオール、2,2,4−トリメチル−1,3−ペンタンジオール、グリセリン、ジグリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレンオキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトールなどの多価アルコール化合物;エチレンジアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ポリアミドポリアミン、ポリアリルアミン、ポリエチレンイミン等の多価アミン化合物、並びに、それら多価アミンとハロエポキシ化合物との縮合物;1,3−ジオキソラン−2−オン、4−メチル−1,3−ジオキソラン−2−オン、4、5−ジメチル−1,3−ジオキソラン−2−オン、4、4−ジメチル−1,3−ジオキソラン−2−オン、4−エチル−1,3−ジオキソラン−2−オン、4−ヒドロキシメチル−1,3−ジオキソラン−2−オン、1,3−ジオキサン−2−オン、4−メチル−1,3−ジオキサン−2−オン、4、6−ジメチル−1,3−ジオキサン−2−オン、1,3−ジオキソパン−2−オン等のアルキレンカーボネート化合物、並びに、エチレングリコールビス(4−メチレン−1,3−ジオキソラン−2−オン)エーテル等の多価アルキレンカーボネート化合物;モノ、ジまたはポリのオキサゾリジノン化合物;3−メチル−3−オキセタンメタノール等のオキセタン化合物ならびに多価オキセタン化合物;等より選ばれる1種または2種以上のものが例示できる。
【0050】
これら脱水反応性架橋剤の中でも、多価アルコール、アルキレンカーボネート、オキサゾリジノン化合物、(多価)オキセタン化合物から選ばれた1種以上が好ましく、少なくとも多価アルコールを用いることが特に好ましい。
架橋剤(c)としては、これら脱水反応性架橋剤(c1)に加えて、さらに、エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、ポリエチレンジグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリシドール、γ−グリシドキシプロピルトリメトキシシラン等のエポキシ化合物;2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート等の多価イソシアネート化合物;1,2−エチレンビスオキサゾリン等の多価オキサゾリン化合物;γ−グリシドキシプロピルトリメトキシシラン、γ−アミノプロピルトリメトキシシラン等のシランカップリング剤;2,2−ビスヒドロキシメチルブタノール−トリス[3−(1−アジリジニル)プロピオネート]などの多価アジリジン化合物、ベリリウム、マグネシウム、カルシウム、ストロンチウム、亜鉛、アルミニウム、鉄、クロム、マンガン、チタン、ジルコニウムなどの多価金属が例示される。
【0051】
なお、本発明で脱水反応性架橋剤(c1)、および、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)および/または粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含む吸水性樹脂粉末の両方を用いない場合、一般に得られる吸水剤の物性が低く、好ましくは、該脱水反応性架橋剤(c1)、および、該特定粒度の吸水性樹脂粉末(a1)の両方が本発明で使用される。
【0052】
本発明において、吸水性樹脂粉末(a)あるいは(a1)と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合する場合には、水を用いることが好ましい。この際、使用される水の量は、使用する吸水性樹脂の含水率にもよるが、通常、吸水性樹脂100重量部に対し、0.5〜20重量部、好ましくは0.5〜10重量部の範囲である。水の使用量が20重量部を越えると吸収倍率が低下してしまうことがある。0.5重量部よりも少ないと効果が現れにくくなり、加圧下吸収倍率を向上させることができなくなる恐れがある。
また、本発明において、吸水性樹脂粉末(a)あるいは(a1)と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合する場合には、親水性有機溶媒を用いてもよい。用いられる親水性有機溶媒としては、メチルアルコール、エチルアルコール、プロピルアルコール、イソプロピルアルコール、ブチルアルコール、イソブチルアルコール、t−ブチルアルコール等のアルコール;アセトン、メチルエチルケトン等のケトン類;ジオキサン、アルコキシ(ポリ)エチレングリコール、テトラヒドロフラン等のエーテル類;ε−カプロラクタム、N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキサイド等のスルホキサイド類;エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、グリセロリン酸、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール類が挙げられる。
【0053】
使用される親水性有機溶媒の量は、吸水性樹脂の種類や粒度によって異なるが、通常、吸水性樹脂100重量部に対し0〜10重量部、好ましくは0〜5重量部、より好ましくは0〜3重量部の範囲である。親水性有機溶媒の使用量が10重量部以上の場合、上記添加剤の溶解性が低下し吸収特性が向上しない恐れがある。なお、上記多価アルコールは反応条件(加熱温度や時間、含水率など)によって吸水性樹脂と反応させて架橋剤としてもよいし、反応させずに溶媒としてもよいし、それらの働きを併用させてもよい。
さらに、本発明において吸水性樹脂粉末(a)あるいは(a1)と、上記添加剤(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)とを混合する場合、水や親水性有機溶媒以外の物質として、本発明の効果を妨げない範囲で界面活性剤や不活性無機微粒子粉末を用いてもよい。用いられる界面活性剤や不活性無機微粒子粉末は、米国特許第5164459号公報、欧州特許第827753号公報、欧州特許第349240号公報、欧州特許第761241号公報などに例示される。
【0054】
本発明において、吸水性樹脂粉末(a)あるいは(a1)と、上記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)の混合は、親水性有機溶媒またはシクロヘキサン、ペンタン等の有機溶媒中に該吸水性樹脂を分散させた状態で行ってもよいが、水、架橋剤、添加剤の混合物を数回に分けて添加してもよく、混合方法は特に限定されるものではない。また、上記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)、さらに必要に応じて用いられる水や親水性有機溶媒、無機粉末などは、吸水性樹脂に対して別々に混合してもよいし、一括で混合してもよいし、数回に分けて混合してもよいが、好ましくは、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を予め混合した後に吸水性樹脂に添加させ、その際、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)は水溶液とさせることがより好ましい。また、この際の水溶液の温度は混合性や安定性から0℃〜沸点、好ましくは5〜50℃、さらには10〜30℃にさせる。また、混合前の吸水性樹脂粉末(a)あるいは(a1)の温度は、混合性から好ましくは0〜80℃、さらには40〜70℃の範囲である。
【0055】
さらに、本発明では種々の混合方法の中で、必要により水および/または親水性有機溶媒と、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)とを予め混合した後、次いで、その水溶液を吸水性樹脂粉末(a)に噴霧あるいは滴下混合する方法が好ましく、噴霧する方法がより好ましい。噴霧される液滴の大きさは、300μm以下が好ましく、200μm以下がより好ましい。また混合に際し、本発明の効果を妨げない範囲で水不溶性微粒子粉体や界面活性剤を共存させてもよい。
前記混合に用いられる好適な混合装置は、均一な混合を確実にするため大きな混合力を生み出せることが必要である。本発明に用いることのできる混合装置としては、例えば、円筒型混合機、二重壁円錐型混合機、高速攪拌型混合機、V字型混合機、リボン型混合機、スクリュー型混合機、流動型炉ロータリーディスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機、スクリュー型押出機等が好適である。
【0056】
本発明の吸水剤の製造方法は、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合し、前記吸水性樹脂粉末(a)あるいは(a1)を架橋処理することを特徴とする。
本発明の吸水剤の製造方法は、好ましくは、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合した後、吸水性樹脂の表面近傍を架橋させる際に、加熱処理を行う。
【0057】
本発明で加熱処理を行う場合、処理時間は、1分〜180分が好ましく、3分〜120分がより好ましく、5分〜100分がさらに好ましい。加熱処理温度(熱媒温度ないし材料温度で規定)は100〜250℃の範囲が好ましく、140〜240℃の範囲がより好ましく、150〜230℃の範囲がさらに好ましく、160〜220℃の範囲がさらにより好ましい。加熱温度が100℃未満では、加熱処理や脱水反応に時間がかかり生産性の低下を引き起こすのみならず、均一な架橋が達成されず、優れた吸水剤が得られなくなる恐れがある。また処理温度が250℃を越えると、得られる吸水剤がダメージを受け、物性に優れたものが得られにくいとことがある。
【0058】
加熱処理は通常の乾燥機または加熱炉を用いて行うことができ、溝型混合乾燥機、ロータリー乾燥機、ディスク乾燥機、流動層乾燥機、気流型乾燥機、および赤外線乾燥機が例示される。
上記の本発明に係る吸水剤の製造方法においては、さらに、必要に応じて、消臭剤、抗菌剤、香料、二酸化珪素や酸化チタン等の無機粉末、発泡剤、顔料、染料、親水性短繊維、可塑剤、粘着剤、界面活性剤、肥料、酸化剤、還元剤、水、塩類、キレート剤、殺菌剤、ポリエチレングリコールやポリエチレンイミンなどの親水性高分子、パラフィンなどの疎水性高分子、ポリエチレンやポリプロピレンなどの熱可塑性樹脂、ポリエステル樹脂やユリア樹脂などの熱硬化性樹脂等を添加する等、吸水剤や吸水性樹脂に種々の機能を付与する工程を含んでいてもよい。これら添加剤の使用量は吸水剤100重量部に対して0〜10重量、好ましくは0〜1重量部の範囲で用いられる。
【0059】
本発明で吸水剤に用いられるカチオン性高分子化合物は、吸水剤の衛生材料への固定性などを向上でき、好ましくは重量平均分子量が2000以上で、さらに好ましくは5000以上、最も好ましくは重量平均分子量が10000以上である。また、その使用量は、好ましくは吸水性樹脂100重量部に対し0.01〜10重量部、より好ましくは0.05〜5重量部、さらに好ましくは0.1〜3重量部である。カチオン性高分子化合物の混合は、単独あるいは溶液(水溶液)で添加され、好ましくは、表面架橋後に添加される。カチオン性高分子化合物の具体例としては、ポリエチレンイミン、ポリビニルアミン、ポリアリルアミン、ポリアミドアミンとエピクロルヒドリンの縮合物、ポリアミジン、ポリ(N−ビニルホルムアルデヒド)の部分加水分解物またはこれらの塩などが例示される。
【0060】
水不溶性微粒子を用いてさらに吸水剤の通液性や吸湿時の耐ブロッキング性などを改善することができる。用いられる微粒子としては、好ましくは10μm以下、さらには1μm以下、特に0.1μm以下の無機または有機の水不溶性微粒子が用いられ、具体的には酸化珪素(商品名、Aerosil、日本アエロジル社製)、酸化チタン、酸化アルミ、などが用いられる。混合には粉体混合(Dry−Blend)やスラリー混合が用いられ、その使用量は吸水剤100重量部に対して10重量部以下、さらには0.001〜5重量部、好ましくは0.01〜2重量部用いられる。
(吸水剤およびそれを用いた衛生材料)
本発明においては、好ましくは、吸水性樹脂粉末(a)あるいは(a1)に、水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および架橋剤(c)あるいは(c1)を混合し、前記吸水性樹脂粉末(a)あるいは(a1)を架橋処理することによって、前述の本発明の効果に起因する高物性の新規な吸水剤を提供する。
【0061】
本発明にかかる吸水剤は、好ましくは、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤である。
(1)酸重合する場合、得られる吸水剤
本発明にかかる吸水剤は、酸重合で吸水性樹脂を得る場合、好ましくは、酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した吸水剤である。より好ましくは、前記の後中和して得られた吸水性樹脂の中和指数が15以上、さらに好ましくは17以上、特に好ましくは20以上である。また、好ましくは、前記の表面架橋した吸水剤の中和指数が15以上、さらに好ましくは17以上、特に好ましくは20以上である。表面架橋効果を高めるために中和指数を低くする場合、中和に長時間や複雑な工程を要するが、本発明では不均一な中和でも簡便に優れた表面架橋が達成できる。
【0062】
従来、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた吸水性樹脂は高吸収倍率で低可溶分だが、得られた吸水性樹脂の中和の不均一のため、一般に加圧下吸収倍率が向上しにくいものであった。かかる問題を解決するため、特許文献30(欧州特許公開0882502号)では高度に吸水性樹脂の一粒一粒の粒子の中和率の差(中和指数)を制御する技術が知られている。
かかる中和指数を制御する方法では、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた低可溶分の吸水性樹脂であって、従来にない高い加圧下吸収倍率を達成するが、中和指数の制御に非常に手間を必要とするものであった。しかし、本発明のアルカリ金属pH緩衝剤(b2)を用いる本発明の方法では、中和指数の高度な制御を必要とせず、簡便な不均一な後中和でも高い加圧下吸収倍率を与えるので非常に好ましい。なお、勿論、本発明は、酸基含有単量体(塩)を含む単量体を重合しさらに後中和して得られた、酸型重合法による低可溶分の吸水性樹脂に限定されるものでなく、後述の実施例などにも示すように、後中和工程を含まない中和重合法による吸水性樹脂にも好適に適用される。
【0063】
(2)5つの物性を併せ持つ新規な吸水剤
また、酸重合する場合しない場合を含めて、本発明の吸水剤は下記物性が好ましく、特に本発明では、下記特性の5つの物性(粒度、CRC、AAP、SFCの4つに加えて、さらにSFC変化指数、SFC変動係数、SFC変動率、連続生産系SFC標準偏差、表層可溶分などの1つ以上、好ましくは2つ以上、より好ましくは3つ以上、特に好ましくは4つ以上)を併せ持った新規な吸水剤を与える。
(a)粒度
本発明にかかる吸水剤の平均粒子径や嵩比重は、前記吸水性樹脂の範囲、すわわち、重量平均粒子径300〜600μmで150μm以下の微粉末が10重量%以下であることが好ましい。より好ましくは5重量%以下、さらに好ましくは3重量%以下、特に2重量%以下である。
【0064】
本発明にかかる吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、さらに好ましくは3種以上、より好ましくは4種を含む。
本発明にかかる吸水剤は、好ましくは、粒子径が850μm未満で150μm以上の粒子を全粒子の95重量%以上含み、より好ましくは97重量%以上、さらに好ましくは98重量%以上である。かかる特定の粒度に制御されることで、衛生材料で高物性を達成する。
【0065】
本発明にかかる吸水剤は、好ましくは、前記粒子A1からA4までの4種をそれぞれ0.1重量%以上含み、より好ましくは1重量%以上、さらに好ましくは3重量%以上である。この場合、上限値は特に限定されないが、好ましくは、前記粒子A1からA4までの4種がそれぞれ99重量%以下、より好ましくは90重量%以下、さらに好ましくは80重量%以下である。各粒度を一定以上含有する事で粒子の表面積に依存する吸水速度がバランスよく制御される。
(b)CRC
本発明にかかる吸水剤は、0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(Centrifuge Retention Capacity/CRC)が31g/g以上であることが好ましい。CRCが31g/g以上となることによって、吸水剤を用いた衛生材料の吸収に臨界的に優れ、コンパクトな衛生材料を達成でき、さらに吸水体(なお、吸水体とは、吸水剤と必要により繊維などの他の吸水材料を含む体液吸水体を意味する)のコスト低減にもなる。CRCは、より好ましくは32g/g以上、さらに好ましくは33g/g以上、さらにより好ましくは34g/g以上、特に好ましくは35g/g以上、特により好ましくは36g/g以上である。0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率が31g/gよりも小さいと、吸水体に吸収されうる尿の総量が小さくなり、吸水体に吸収された尿がおむつの表面への戻りが非常に大きくなる。さらに吸水体に求められる尿の吸液量を維持しようとする場合、吸水体に使用する吸水剤の量が多くなり、衛生材料が嵩高く重いものになり、吸水体のコストアップに繋がることになる点で好ましくない。
【0066】
(c)AAP
本発明にかかる吸水剤は、0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(Absorbency Against Pressure/AAP)が20g/g以上であることが好ましい。AAPが20g/g以上となることによって、本発明の吸水剤を紙おむつの吸水体の一部に使用した場合、吸水体に吸収された尿がおむつの表面への戻りを防ぐ効果が非常に大きくなる。0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率は、より好ましくは22g/g以上、さらに好ましくは24g/g以上、さらにより好ましくは25g/g以上、特に好ましくは26g/g、特により好ましくは27g/g以上である。0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率が20g/gよりも小さいと、吸水体に吸収された尿がおむつの表面への戻りを防ぐ効果が非常に小さくなる点で好ましくない。
【0067】
(d)SFC
本発明にかかる吸水剤は、0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上であることが好ましい。SFCは、本発明で得られる吸水剤の膨潤後の通液性に、非常に大きな影響を与える。つまり、例えば、本発明の吸水剤を紙おむつの吸水体の一部に使用した場合の、通液性を良好にし、吸水体に液を十分に行き渡らせ、吸液量を増大させ、液の漏れを防止するという効果が著しく向上する。SFCは、より好ましくは25(単位:10−7×cm3×s×g−1)以上、さらに好ましくは30(単位:10−7×cm3×s×g−1)以上、さらにより好ましくは35(単位:10−7×cm3×s×g−1)以上、特に好ましくは40(単位:10−7×cm3×s×g−1)以上、特により好ましくは50(単位:10−7×cm3×s×g−1)以上である。SFCが20(単位:10−7×cm3×s×g−1)よりも小さいと、例えば、紙おむつの吸水体に使用した場合の通液性が低下し、吸水体に液が局在化し、吸液量が減少し、液の漏れが多くなり、吸水体としての性能が著しく低下する点で好ましくない。
【0068】
すなわち、本発明の吸水剤は衛生材料に用いるには粒度に加えて、下記3つの物性をバランスよく併せ持つことが好ましい。すなわち、CRC、AAP、SFCの1つまたは2つが高物性だけでは、衛生材料に十分に好適ではないことが見出された。これらの3つの物性は上記酸重合した特定の中和指数の吸水剤のみならず、後中和工程を含まない中和重合法による吸水性樹脂にも好適に適用される。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaでの加圧下吸水倍率(AAP)が24g/g以上。
【0069】
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
(e)SFC変化指数
また、本発明者らは、従来粒子全体(Bulk)で物性を評価および管理されていた吸水性樹脂(吸水剤)粒子において、吸水性樹脂(吸水剤)粒子の粒度ごとの物性が大きく異なり、その粒度ごとの物性の違いが衛生材料での物性低下を引き起こしている事実を見出した。衛生材料に含まれる吸水剤粒子は微視的には粒度にバラツキが見られるため、その粒度由来の個々の衛生材料の物性の違いや衛生材料の部分での物性の違いが、衛生材料の物性低下を引き起こすと推定される。
【0070】
そこで、本発明者らは粒度ごとの物性の差、特に粒度ごとのSFCのバラツキ小さい吸水剤を提供する。
本発明にかかる吸水剤は、下記式(1)で規定されるSFC変化指数が0〜25%であることが好ましい。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
このSFC変化指数や後述のSFC変動係数において、粒子A1からA4のSFCの標準偏差は、吸水剤粒子を分級して、吸水剤中で粒子A1からA4のうちで存在する2〜4種類の粒子を得て、次いで得られた各粒度ごとのSFCを1回ずつ測定して、その2〜4種類のSFC値から標準偏差を計算して求められる。また、その2〜4種類のSFC値から平均値も同様に計算される。
【0071】
このSFC変化指数は、粒度ごとのSFCのバラツキを表し、SFC変化指数が0〜25%となることによって、吸水体がより均一な通液性を持つようになり、吸水体全体に液が分散し易く、液の漏れを防止するという効果となる。SFC変化指数は、より好ましくは0〜23%、さらに好ましくは0〜20%、さらにより好ましくは0〜18%、特に好ましくは0〜15%、特により好ましくは0〜10%である。SFC変化指数が25%よりも大きいと、例えば吸水剤を紙おむつの吸水体に使用した場合、吸水体に通液性のばらつきが生じ、吸水体全体に液が分散し難くなり、液の漏れの発生や、吸水体の吸液量が低減する点で好ましくない。
【0072】
(f)SFC変動係数
本発明にかかる吸水剤は、下記式(2)で規定されるSFC変動係数が0〜0.25であることが好ましい。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
このSFC変動係数や下記のSFC変動率も同様に粒度ごとのSFCの差の少なさを表し、SFC変動係数が0〜0.25となることによって、粒度ごと(例えばA1からA4)のSFCのばらつきの度合いが小さくなり、例えば本発明の吸水剤を用いて紙おむつの吸水体を製造する場合、吸水体内での通液性が均一になる上に、吸水体の各々を比較しても、紙おむつの性能にばらつきがなくなり、安定した品質の紙おむつを製造できることとなる。SFC変動係数は、より好ましくは0〜0.23、さらに好ましくは0〜0.20、さらにより好ましくは0〜0.18、特に好ましくは0〜0.15、特により好ましくは0〜0.10である。SFC変動係数が0.25よりも大きい場合および/またはSFC変動率が0.65未満である場合、吸水剤を用いた衛生材料(特に紙おむつ)の吸水体内での通液性が不均一になる上に、吸水体の各々を比較しても、紙おむつの性能のばらつきが大きくなり、安定した品質の紙おむつを製造できなくなる点で好ましくない。
【0073】
(g)SFC変動率
すなわち、本発明にかかる吸水剤は、下記式(3)で規定されるSFC変動率が0.65〜1.00であることが好ましい。
SFC変動率=(粒子A1からA4のSFCの中で最小のSFC)/(粒子A1からA4のSFCの中で最大のSFC) (3)
このSFC変動率も同様に粒度ごとのSFCの差の少なさを表し、好ましくは0.70〜1.00、さらにより好ましくは0.75〜1.00、特に好ましくは0.80〜1.00である。
【0074】
(h)表層可溶分量
本発明の吸水剤は、表層可溶分量が6.0重量%以下(対吸水剤)であることが好ましい。この表層可溶分量は、好ましくは5.5重量%以下、より好ましくは5.0重量%以下、さらに好ましくは4.5重量%以下、特に好ましくは4.0重量%以下である。
本願の測定法で規定される表層可溶分は衛生材料への実使用に対応した可溶分量であり、より衛生材料での吸水能に優れる。すなわち、従来、水可溶分の測定(US Re 32649やEDANA法など)は多く知られていたが、過剰の液で長時間の測定のため、実使用において溶出してくると考えられる可溶分の量よりも過剰の量の可溶分を測定していることがわかった。そして、本発明の方法が衛生材料での実使用に最もモデルとなっていることが見出された。
【0075】
(i)連続生産系SFC標準偏差
本発明にかかる吸水剤は、下記式(4)で規定される連続生産系SFC標準偏差が5.0以下であることが好ましい。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、各Lotは20kg以上。Lot数は10以上。)
上記連続生産系とは、1ラインで、24時間以上、若しくは10t以上、連続して吸水剤を生産することを示す。また、吸水剤の製造方法の基本工程である、重合、乾燥、粉砕、表面処理等は、連続生産系においては、各工程は連続式またはバッチ式を問わないが(例えば、重合工程においては連続重合とバッチ重合など)、各工程の間隔は24時間以下、好ましくは12時間以下、さらに好ましくは6時間以下、特に好ましくは3時間以下で行うものとする。
【0076】
各Lotは、好ましくは20kg〜100t、より好ましくは0.1t〜50t、さらに好ましくは0.5t〜25tである。
Lot数は、好ましくは20以上、より好ましくは30以上、さらに好ましくは50以上、特に好ましくは100以上である。
この連続生産系SFC標準偏差は、吸水剤の連続生産において各LotのSFCの値のばらつきを表し、連続生産系SFC標準偏差が5.0以下となることによって、各Lot毎のSFCのばらつきが小さくなり、例えば、本発明の吸水剤を用いて紙おむつの吸水体を製造する場合、安定した品質の紙おむつを製造できることとなる。連続生産系SFC標準偏差は、より好ましくは4.5以下、さらに好ましくは4.3以下、さらにより好ましくは4.0以下、特に好ましくは3.5以下、特により好ましくは3.0以下、最も好ましくは2.5以下である。連続生産系SFC標準偏差が5.0よりも大きい場合、安定した品質の紙おむつを製造できなくなる点で好ましくない。
【0077】
なお、上記した各種物性は、吸水剤が衛生材料として使用される場合、その実使用の結果(おむつの吸収量や漏れなど)と相関する重要な物性であり、本発明の吸水剤は上記各種物性に特に優れている。これら測定法は実施例に記載する。
本発明にかかる吸水剤は、以上に述べた種々の特徴を有することが好ましく、特に好ましい構成として、上記の式(1)、(2)、(4)のいずれか1つ以上、より好ましくは2つ以上、さらに好ましくは3つ全てを満たす吸水剤である。
すなわち、本発明にかかる吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0078】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
また、本発明にかかる別の吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0079】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
また、本発明にかかるさらに別の吸水剤は、不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤である。
【0080】
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
本発明にかかるさらに別の吸水剤は、酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上であることを特徴とする、吸水剤である。
【0081】
上記吸水剤において、CRC、AAP、SFCやSFC変化指数、SFC変動係数などは好ましくは前述の範囲であり、本発明の新規な吸水剤は、例えば、前述の本発明の吸水剤の製造方法で得られる。本発明の吸水剤はCRC、AAP、SFCが非常に高く高物性をバランスよく維持している上で、さらに粒度ごとの物性の変化も従来になく少ないため、いかなる衛生材料の使用条件でも高物性を発揮する優れた吸水剤である。
本発明によれば、無加圧下の吸収倍率、加圧下の吸収倍率、生理食塩水流れ誘導性のバランスに優れた良好な吸収特性を備えた吸水剤を簡便に製造することができ、農園芸保水剤、工業用保水剤、吸湿剤、除湿剤、建材、などで広く用いられるが、その吸水剤は紙おむつ、生理用ナプキンなどの、糞、尿ないし血液の吸収用衛生材料に特に好適に用いられる。さらに、本発明の吸水剤は上記各種物性にバランスよく優れるため、衛生材料は一般に吸水剤の濃度(吸水剤および繊維基材の合計に対する吸水剤の重量比)として高濃度、例えば30〜100重量%、好ましくは40〜100重量%の範囲、さらに好ましくは50〜95重量%で使用可能である。また、衛生材料中の吸水体の構造は、一般の吸水性物品に用いられる構造であれば特に制限はなく、例えば、シート状に成形した親水性繊維材料の間に吸水剤を配する、いわゆるサンドイッチ構造の吸水体や、親水性繊維材料と吸水剤を混合したものを成形した、いわゆるブレンド構造の吸水体が挙げられる。
【実施例】
【0082】
以下、実施例および比較例により、本発明をさらに詳細に説明するが、本発明はこれら実施例に限定されるものではない。なお、吸水性樹脂ないし吸水剤の諸性能は、以下の方法で測定した。
なお、衛生材料などの最終製品として使用された吸水剤の場合は、吸水剤は吸湿しているので、適宜、吸水剤を最終製品から分離して減圧低温乾燥後(例えば、1mmHg以下、60℃で12時間)に測定すればよい。また、本発明の実施例および比較例において使用された吸水性樹脂の含水率はすべて6重量%以下であった。
さらに、後述の(4)で粒度別のSFCを測定する場合、衛生材料などの最終製品として使用された吸水剤では、上記操作ののちにSFCが測定されるが、使用前の吸水剤、すなわち、実験室で製造された吸水剤を測定する場合、実生産や実使用のダメージに相関させるため、すべて後述(5)の機械的ダメージを与えた後の吸水剤で、粒度別のSFC、分級する前のSFCを測定するものとする。
【0083】
(1)無加圧下吸収倍率(0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率/CRC)
室温(20〜25℃)、湿度50RH%の条件下で、吸水性樹脂ないし吸水剤0.20gを不織布製の袋(60mm×60mm)に均一に入れてシールした後、室温で0.9重量%生理食塩水中に浸漬した。30分後に袋を引き上げ、遠心分離機(株式会社コクサン社製、遠心機:型式H−122)を用いて250Gで3分間水切りを行った後、袋の重量W1(g)を測定した。また、同様の操作を吸水性樹脂あるいは吸水剤を用いずに行い、その時の重量W0(g)を測定した。そして、これらW1、W0から、次式に従って無加圧下吸収倍率(g/g)を算出した。
【0084】
無加圧下吸収倍率(g/g)=(W1(g)−W0(g))/吸水性樹脂ないし吸水剤の重量(g)
(2)加圧下吸収倍率(0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率/AAP)
内径60mmのプラスチックの支持円筒の底に、ステンレス製400メッシュの金網(目の大きさ38μm)を融着させ、室温(20〜25℃)、湿度50RH%の条件下で、該網上に吸水剤0.90gを均一に散布し、その上に、吸水剤に対して4.83kPa(0.7psi)の荷重を均一に加えることができるよう調整された、外径が60mmよりわずかに小さく支持円筒との隙間が生じず、かつ上下の動きが妨げられないピストンと荷重とをこの順に載置し、この測定装置一式の重量Wa(g)を測定した。
【0085】
直径150mmのペトリ皿の内側に直径90mmのガラスフィルター(株式会社相互理化学硝子製作所社製、細孔直径:100〜120μm)を置き、0.90重量%生理食塩水(20〜25℃)をガラスフィルターの上面と同じレベルになるように加えた。その上に、直径90mmの濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を載せ、表面が全て濡れるようにし、かつ過剰の液を除いた。
上記測定装置一式を前記湿った濾紙上に載せ、液を荷重下で吸収させた。1時間後、測定装置一式を持ち上げ、その重量Wb(g)を測定した。そして、Wa、Wbから、次式に従って加圧下吸収倍率(g/g)を算出した。
【0086】
加圧下吸収倍率(g/g)
=(Wa(g)−Wb(g))/吸水剤の重量((0.9)g)
(3)重量平均粒子径
吸水性樹脂粉末ないし吸水剤を目開き850μm、600μm、500μm、425μm、300μm、212μm、150μm、106μm、75μmなどのJIS標準ふるいで篩い分けし、残留百分率Rを対数確率紙にプロットした。これにより、重量平均粒子径(D50)を読み取った。
篩い分け、および、後述の粒度別のSFCを測定する際の分級方法は、吸水性樹脂粉末ないし吸水剤10.0gを、室温(20〜25℃)、湿度50RH%の条件下で、目開き850μm、600μm、500μm、300μm、150μmのJIS標準ふるい(THE IIDA TESTING SIEVE:径8cm)に仕込み、振動分級器(IIDA SIEVE SHAKER、TYPE:ES−65型、SER.No.0501)により、10分間、分級を行った。
【0087】
(4)0.69重量%生理食塩水流れ誘導性(SFC)
特表平9−509591の生理食塩水流れ誘導性(SFC)試験に準じて行った。
図1に示す装置を用い、容器40に均一に入れた吸水剤(0.900g)を人工尿(1)中で0.3psi(2.07kPa)の加圧下、60分間膨潤させ、ゲル44のゲル層の高さを記録し、次に0.3psi(2.07kPa)の加圧下、0.69重量%生理食塩水33を、一定の静水圧でタンク31から膨潤したゲル層を通液させる。このSFC試験は室温(20〜25℃)で行った。コンピューターと天秤を用い、時間の関数として20秒間隔でゲル層を通過する液体量を10分間記録する。膨潤したゲル44(の主に粒子間)を通過する流速Fs(t)は増加重量(g)を増加時間(s)で割ることによりg/sの単位で決定する。一定の静水圧と安定した流速が得られた時間をtsとし、tsと10分間の間に得たデータだけを流速計算に使用して、tsと10分間の間に得た流速を使用してFs(t=0)の値、つまりゲル層を通る最初の流速を計算する。Fs(t=0)はFs(t)対時間の最小2乗法の結果をt=0に外挿することにより計算される。
【0088】
生理食塩水流れ誘導性=(Fs(t=0)×L0)/(ρ×A×ΔP)
=(Fs(t=0)×L0)/139506
ここで、
Fs(t=0):g/sで表した流速
L0:cmで表したゲル層の高さ
ρ:NaCl溶液の密度(1.003g/cm3)
A:セル41中のゲル層上側の面積(28.27cm2)
ΔP:ゲル層にかかる静水圧(4920dyne/cm2)
およびSFC値の単位は(10−7×cm3×s×g−1)である。
【0089】
図1に示す装置としては、タンク31には、ガラス管32が挿入されており、ガラス管32の下端は、0.69重量%生理食塩水33をセル41中の膨潤ゲル44の底部から、5cm上の高さに維持できるように配置した。タンク31中の0.69重量%生理食塩水33は、コック付きL字管34を通じてセル41へ供給された。セル41の下には、通過した液を補集する容器48が配置されており、補集容器48は上皿天秤49の上に設置されていた。セル41の内径は6cmであり、下部の底面にはNo.400ステンレス製金網(目開き38μm)42が設置されていた。ピストン46の下部には液が通過するのに十分な穴47があり、底部には吸水剤あるいはその膨潤ゲルが、穴47へ入り込まないように透過性の良いガラスフィルター45が取り付けてあった。セル41は、セルを乗せるための台の上に置かれ、セルと接する台の面は、液の透過を妨げないステンレス製の金網43の上に設置した。
【0090】
人工尿(1)は、塩化カルシウムの2水和物0.25g、塩化カリウム2.0g、塩化マグネシウムの6水和物0.50g、硫酸ナトリウム2.0g、りん酸2水素アンモニウム0.85g、りん酸水素2アンモニウム0.15g、および、純水994.25gを加えたものを用いた。
(5)機械的ダメージ試験
図2に示すガラス製容器(山村硝子社製マヨネーズ瓶、商品名:A−29)に吸水剤30gとガラスビーズ(玉径約6mmの精密分留充填用ソーダ石灰ガラスビーズ)10gを入れた。これを図3に示す分散機(東洋精機製作所社製、No.488試験用分散機)に備えられたクランプ間に挟み固定し、100V/60Hzで振動回転数750cpmの振動を10分間与えた。これにより、上記分散機に固定された容器は、分散機における上記クランプの取り付け面に対して左右に各々12.5°(合計25°)傾斜運動すると同時に、前後に各々8mm(合計16mm)振動することにより、容器内部の吸水剤に衝撃を与える。上記衝撃は、吸水剤の製造工程中に吸水剤が受ける衝撃力を代表するものとして経験的に定められた力であるが、製造後の輸送や吸水体製造時のダメージにも広く適用できるものである。本発明において、機械的ダメージを与える場合は、特に、吸水剤の製造時、吸水体の製造時を想定したものである。さらに、SFC変化指数、SFC変動係数、SFC変動率は、吸水体内部における吸水剤の性能のばらつきを測る指標である。よって粒度別の物性を測定する場合は、実験室(1回の製造工程で得られる吸水剤の量が20kg以下)のスケールにおいて製造された吸水剤は、すべて前記機械的ダメージを与えなければならない。
【0091】
(6)可溶分(水可溶成分)量
250ml容量の蓋付きプラスチック容器に0.9重量%生理食塩水溶液(生理食塩水)の184.3gを測り取り、その水溶液中に吸水性樹脂1.00gを加え16時間攪拌することにより樹脂中の可溶分を抽出した。この抽出液を濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を用いて濾過することにより得られた濾液の50.0gを測り取り測定溶液とした。
はじめに生理食塩水だけを、まず、0.1NのNaOH水溶液でpH10まで滴定を行い、その後、0.1NのHCl水溶液でpH2.7まで滴定して空滴定量([bNaOH]ml、[bHCl]ml)を得た。
【0092】
同様の滴定操作を測定溶液についても行うことにより滴定量([NaOH]ml、[HCl]ml)を求めた。
例えば既知量のアクリル酸とそのナトリウム塩からなる吸水性樹脂の場合、そのモノマーの平均分子量と上記操作により得られた滴定量をもとに、吸水性樹脂中の可溶分量を以下の計算式により算出することができる。未知量の場合は滴定により求めた中和率を用いてモノマーの平均分子量を算出する。
可溶分(重量%)=0.1×(平均分子量)×184.3×100×([HCl]−[bHCl])/1000/1.0/50.0
中和率(mol%)=(1−([NaOH]−[bNaOH])/([HCl]−[bHCl]))×100
(7)表層可溶分(水可溶成分)量
250ml容量の蓋付きプラスチック容器に0.50重量%生理食塩水溶液の100gを測り取り、その水溶液中に吸水剤0.50gを加え、1時間吸水剤中の表層可溶分を抽出した。この抽出操作において、攪拌は初期の吸水剤の分散および最後の液の均一化のみを目的に攪拌し、前記以外は攪拌を行わない。具体的には、表層可溶分の抽出は静置で行い、抽出前後の1分間のみ攪拌(攪拌速度:400rpm、攪拌子:テフロン製(2.5cm))を行う。この抽出液を濾紙1枚(ADVANTEC東洋株式会社、品名:(JIS P 3801、No.2)、厚さ0.26mm、保留粒子径5μm)を用いて濾過することにより得られた濾液の50.0gを測り取り測定溶液とした。
【0093】
はじめに0.5重量%NaCl水溶液だけを、まず、0.1NのNaOH水溶液でpH10まで滴定を行い、その後、0.1NのHCl水溶液でpH2.7まで滴定して空滴定量([bNaOH]ml、[bHCl]ml)を得た。
同様の滴定操作を測定溶液についても行うことにより滴定量([NaOH]ml、[HCl]ml)を求めた。
例えば既知量のアクリル酸とそのナトリウム塩からなる吸水剤の場合、そのモノマーの平均分子量と上記操作により得られた滴定量をもとに、吸水剤中の表層可溶分量を以下の計算式により算出することができる。未知量の場合は滴定により求めた中和率を用いてモノマーの平均分子量を算出する。
【0094】
表層可溶分(重量%)=0.1×(平均分子量)×100×100×([HCl]−[bHCl])/1000/0.5/50.0
中和率(mol%)=(1−([NaOH]−[bNaOH])/([HCl]−[bHCl]))×100
(8)中和指数
特許文献30およびその請求項3(欧州特許公開0882502号およびその請求項2〜4)に従い、以下、吸水性樹脂粉末および吸水剤の中和指数を求めた。
すなわち、JIS標準篩で300〜600μmに分級した吸水性樹脂粉末ないし吸水剤の粒子200粒を、カバーガラスを貼りつけた20mm×20mmの開口部を有する厚さ1.6mmのプラスチックプレートに入れ、0.2mlの脱イオン水を添加する。さらに、上記膨潤ゲルにブルムチモールブルー(BTB)0.1重量%エタノール溶液とメチルレッド(MR)0.1重量%エタノール溶液との1.5:1混合溶液0.05mlをマイクロシリンジで添加して、pH指示薬によって粒子200粒の着色を観察した。こうして吸水性樹脂ないし吸水剤の平均中和率から20モル%を越えて不均一に中和された粒子の個数(200個中で何個か)を求めて、この不均一な中和粒子の個数を中和指数(上記特許では第一中和指数/請求項3)として求めた。もちろん、中和指数が大きいほど、吸水性樹脂粉末の中和は不均一である。詳しくは、上記特許を参照。
【0095】
(参考例1):吸水性樹脂粉末(A)の製造/中和重合
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度38重量%、モノマーの平均分子量88.5)にポリエチレングリコールジアクリレート(n=9)3.70gを溶解させて反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に10重量%過硫酸ナトリウム水溶液28.3gおよび1重量%L−アスコルビン酸水溶液2.1gを攪拌しながら添加したところ、およそ1分後に重合が開始した。そして、生成したゲルを粉砕しながら、20〜95℃で重合を行い、重合が開始して30分後に含水ゲル状架橋重合体(1)を取り出した。
【0096】
得られた含水ゲル状架橋重合体(1)は、その径が約5mm以下に細分化されていた。この細分化された含水ゲル状架橋重合体(1)を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き850μmおよび106μmのJIS標準篩で分級することで大部分の粒子が850μm〜106μmの範囲にある吸水性樹脂粉末(A)を得た。
得られた吸水性樹脂粉末(A)の無加圧下の吸収倍率は49(g/g)、可溶分量は23重量%、重量平均粒径(D50)は330μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
【0097】
(参考例2):吸水性樹脂粉末(B)の製造/中和重合
シグマ型羽根を2本有する内容積10リットルのジャケット付きステンレス型双腕型ニーダーに蓋を付けて形成した反応器中で、71モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度41重量%、モノマーの平均分子量87.7)にポリエチレングリコールジアクリレート(n=9)8.05gを溶解させて反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に10重量%過硫酸ナトリウム水溶液30.8gおよび1重量%L−アスコルビン酸水溶液2.57gを攪拌しながら添加したところ、およそ1分後に重合が開始した。そして、生成したゲルを粉砕しながら、20〜95℃で重合を行い、重合が開始して30分後に含水ゲル状架橋重合体(2)を取り出した。
【0098】
得られた含水ゲル状架橋重合体(2)は、その径が約5mm以下に細分化されていた。この細分化された含水ゲル状架橋重合体(2)を50メッシュ(目開き300μm)の金網上に広げ、180℃で50分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き850μmのJIS標準篩で分級することで大部分の粒子が850μm以下の範囲にある吸水性樹脂粉末(B)を得た。
得られた吸水性樹脂粉末(B)の無加圧下の吸収倍率は36(g/g)、可溶分量は10重量%、重量平均粒径(D50)は450μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
【0099】
(参考例3):吸水性樹脂粉末(C)の製造例/中和重合
参考例2において、ポリエチレングリコールジアクリレート5.01gに変更した以外は参考例2と同様にして重合、乾燥、粉砕および分級を行い、吸水性樹脂粉末(C)を得た。得られた吸水性樹脂粉末(C)の無加圧下の吸収倍率は39(g/g)、可溶分量は13重量%、重量平均粒径(D50)は450μm、850μm〜150μmの範囲にある吸水性樹脂粉末は97重量%であった。
(参考例4):吸水性樹脂粉末(D)の製造例/中和重合
参考例2において、ポリエチレングリコールジアクリレート5.01gに変更した以外は参考例2と同様にして重合、乾燥、および粉砕を行った。得られた粉砕物をさらに目開き600μmおよび300μmのJIS標準篩で分級することで、600μm〜300μmの範囲にある吸水性樹脂粉末(D)を得た。得られた吸水性樹脂粉末(D)の無加圧下の吸収倍率は40(g/g)、可溶分量は9重量%、重量平均粒径(D50)は450μmであった。
【0100】
(実施例1):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基あり
参考例1で得られた吸水性樹脂粉末(A)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.027g、プロピレングリコール0.9g、水2.7g及び炭酸水素ナトリウム0.18gの混合液からなる表面処理剤を混合した後、混合物を210℃で35分間加熱処理することにより、吸水剤(1)を得た。得られた吸水剤(1)も粉末状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例2):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基あり
実施例1において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウムを0.09g用いた以外は同様にして、吸水剤(2)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0101】
(比較例1):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基なし
実施例1において、炭酸水素ナトリウムを用いる代わりに、イソプロピルアルコール0.81gを用いた以外は同様にして、比較用吸水剤(1)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例2):吸水性樹脂粉末(A)の架橋処理/水溶性無機塩基なし
実施例1において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(2)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0102】
(実施例3):吸水性樹脂粉末(B)の架橋処理/水溶性無機塩基あり
参考例2で得られた吸水性樹脂粉末(B)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で30分間加熱処理することにより、吸水剤(3)を得た。得られた吸水剤(3)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例3):吸水性樹脂粉末(B)の架橋処理/水溶性無機塩基なし
実施例3において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(3)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0103】
(実施例4):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および炭酸水素ナトリウム0.24gの混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(4)を得た。得られた吸水剤(4)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例5):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウム0.12gを用いた以外は同様にして、吸水剤(5)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0104】
(実施例6):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、炭酸水素ナトリウムを用いる代わりに、24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)を用いた以外は同様にして、吸水剤(6)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(比較例4):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例4において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(4)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0105】
(実施例7):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例4において、加熱処理時間を30分間にした以外は同様にして、吸水剤(7)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
(実施例8):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例7において、炭酸水素ナトリウムを用いる代わりに、炭酸ナトリウム0.12gを用いた以外は同様にして、吸水剤(8)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。
【0106】
(実施例9):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例7において、炭酸水素ナトリウムを用いる代わりに、24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)を用いた以外は同様にして、吸水剤(9)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。また、吸水剤(9)の表層可溶分は3.8重量%であった。
(比較例5):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例7において、炭酸水素ナトリウムを用いなかった以外は同様にして、比較用吸水剤(5)を得た。無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表1に示した。また、比較用吸水剤(5)の表層可溶分は6.5重量%であった。
【0107】
(比較例6):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例9において、24重量%水酸化ナトリウム水溶液の代わりに、特許文献25に準じて、硫酸アルミニウム14〜18水和物を0.455g用いた以外は同様にして、比較用吸水剤(6)を得た。その結果を表1に示した。
(実施例10):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.784g、水4.0g、および24重量%水酸化ナトリウム水溶液0.5g(固形分0.12g)の混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(10)を得た。得られた吸水剤(10)も粉末形状であり、結果を表1に示した。
【0108】
(比較例7):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例10において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(7)を得た。その結果を表1に示した。
(実施例11):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
参考例3で得られた吸水性樹脂粉末(C)100gに3−エチル−3−オキセタンメタノール0.4g、水3.0g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で40分間加熱処理することにより、吸水剤(11)を得た。得られた吸水剤(11)も粉末形状であり、その結果を表1に示した。
【0109】
(比較例8):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
実施例11において、24重量%水酸化ナトリウム水溶液を用いなかった以外は同様にして、比較用吸水剤(8)を得た。その結果を表1に示した。
(実施例12):吸水性樹脂粉末(C)の架橋処理/pH緩衝剤あり
参考例3で得られた吸水性樹脂粉末(C)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成製)0.15g、プロピレングリコール1.0g、水5.0g、およびリン酸2水素ナトリウム・2水和物0.5gの混合液からなる表面処理剤を混合した後、混合物を150℃で30分間加熱処理することにより、吸水剤(12)を得た。得られた吸水剤(12)も粉末形状であり、その結果を表1に示した。
【0110】
(比較例9):吸水性樹脂粉末(C)の架橋処理/pH緩衝剤なし
実施例12において、リン酸二水素ナトリウム二水和物を用いなかった以外は同様にして、比較用吸水剤(9)を得た。その結果を表1に示した。
(実施例13):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例9において、同様の手法を用いて、吸水剤(13)を得た。得られた吸水剤(13)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(13)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた吸水剤(13)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(13−a)、粒子径が600μm未満で500μm以上の粒子(13−b)、粒子径が500μm未満で300μm以上の粒子(13−c)、粒子径が300μm未満で150μm以上の粒子(13−d)を得た。得られた吸水剤(13−a)、(13−b)、(13−c)、(13−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0111】
(実施例14):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基あり
実施例9において、加熱時間を20分間にした以外は同様の手法をもちいて吸水剤(14)を得た。得られた吸水剤(14)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(14)を測定した結果を表2に示した。さらに得られた吸水剤(14)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(14−a)、粒子径が600μm未満で500μm以上の粒子(14−b)、粒子径が500μm未満で300μm以上の粒子(14−c)、粒子径が300μm未満で150μm以上の粒子(14−d)を得た。得られた吸水剤(14−a)、(14−b)、(14−c)、(14−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0112】
(比較例10):吸水性樹脂粉末(C)の架橋処理/水溶性無機塩基なし
比較例5において、同様の手法を用いて、比較用吸水剤(10)を得た。得られた比較用吸水剤(10)に機械的ダメージを10分間与えた後、得られた比較用吸水剤(10)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた比較用吸水剤(10)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(10−a)、粒子径が600μm未満で500μm以上の粒子(10−b)、粒子径が500μm未満で300μm以上の粒子(10−c)、粒子径が300μm未満で150μm以上の粒子(10−d)を得た。得られた比較用吸水剤(10−a)、(10−b)、(10−c)、(10−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0113】
(比較例11):市販品の吸水剤
ドイツで市販されているPampers Acitve Fit(P&G社製、2001年12月5日購入)のおむつから吸水剤を取り出し、比較用吸水剤(11)を得た。得られた比較用吸水剤(11)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。さらに得られた比較用吸水剤(11)を、JIS標準ふるい(目開き850、600、500、300、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(11−a)、粒子径が600μm未満で500μm以上の粒子(11−b)、粒子径が500μm未満で300μm以上の粒子(11−c)、粒子径が300μm未満で150μm以上の粒子(11−d)を得た。得られた吸水剤(11−a)、(11−b)、(11−c)、(11−d)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0114】
(比較例12):市販品の吸水剤
日本で市販されていた「パンパースさらさらケア」(P&G社製、1997年12月購入)のおむつから吸水剤を取り出し、比較用吸水剤(12)とした。比較吸水剤(12)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。
さらに得られた比較用吸水剤(12)をJIS標準ふるい(目開き850、600、500、300、150μm)でふるい、それぞれ、850μm未満で600μm以上の粒子(12−a)、粒子径が600μm未満で500μm以上の粒子(12−b)、粒子径が500μm未満で300μm以上の粒子(12−c)、粒子径が300μm未満で150μm以上の粒子(12−d)を得た。得られた吸水剤(12−a)、(12−b)、(12−c)、(12−d)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0115】
(実施例15):吸水性樹脂粉末(D)の架橋処理/水溶性無機塩基あり
参考例4で得られた吸水性樹脂粉末(D)100gに1,4−ブタンジオール0.384g、プロピレングリコール0.6g、水3.28g、および24重量%水酸化ナトリウム水溶液0.3g(固形分0.072g)の混合液からなる表面処理剤を混合した後、混合物を212℃で20分間加熱処理することにより、吸水剤(15)を得た。得られた吸水剤(15)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(15)を測定した結果を表2に示した。さらに得られた吸水剤(15)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、粒子径が600μm未満で500μm以上の粒子(15−b)、粒子径が500μm未満で300μm以上の粒子(15−c)を得た。得られた吸水剤(15−b)、(15−c)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0116】
(実施例16):吸水性樹脂粉末(D)の架橋処理/水溶性無機塩基あり
実施例15において、加熱時間を40分間にした以外は同様にして、吸水剤(16)を得た。得られた吸水剤(16)に前記の機械的ダメージを10分間与えた後、得られた吸水剤(16)を測定した結果を表2に示した。さらに得られた吸水剤(16)を、JIS標準ふるい(目開き850μm、600μm、500μm、300μm、150μm)でふるい、それぞれ、粒子径が600μm未満で500μm以上の粒子(16−b)、粒子径が500μm未満で300μm以上の粒子(16−c)を得た。得られた吸水剤(16−b)、(16−c)を測定した結果を表2に示した。また、SFC変化指数、SFC変動係数、SFC変動率を算出した結果を表3に示した。
【0117】
(比較例13〜16):市販品の吸水剤
実使用されている吸水剤(吸水性樹脂)として、2001年に市販の紙おむつより吸水性樹脂を取り出し比較吸水剤(13)〜(16)とした。比較吸水剤(13)〜(16)の無加圧下吸収倍率、加圧下吸収倍率、生理食塩水流れ誘導性を測定した結果を表4に示した。
(参考例5):吸水性樹脂粉末(E)の製造/酸型重合で後中和
2Lのプラスチック容器にアクリル酸252.2g、N,N’−メチレンビスアクリルアミド1.1g、水998.4gを混合し反応液とした。次にこの反応液を窒素ガス雰囲気下で、30分間脱気した。続いて、反応液に15重量%2,2’−アゾビス(2−アミジノプロパン)二塩酸塩5.1g、10重量%L−アスコルビン酸水溶液0.63g、および7重量%過酸化水素水溶液3.6gを加えて重合を開始させた。16〜74℃で重合を行い、重合が開始してから3時間後に含水ゲル状架橋重合体(5)を取り出した。得られた重合体(5)を約5cm角に切断してからその1000gについて、中和率が65モル%となる量の48重量%の水酸化ナトリウム水溶液15.0gを混合し、さらにダイス径9.5mmのミートチョッパーに通して細分化された粉砕ゲルとした。得られた粉砕ゲルを50メッシュ(目開き300μm)の金網上に広げ、170℃で40分間熱風乾燥した。次いで、ロールミルを用いて粉砕し、さらに目開き500μmおよび300μmのJIS標準網で分級することで大部分の粒子が500μm〜300μmの範囲にある吸水性樹脂粉末(E)を得た。得られた吸水性樹脂粉末(E)の無加圧下の吸収倍率は49(g/g)、可溶分量は6重量%であった。
【0118】
さらに得られた粒子の中和率の均一性を調べるために、200個の粒子を膨潤させ、さらに、pH指示薬であるブロモチモールブルーとメチルレッドの0.1重量%エタノール溶液を添加し中和指数(特許文献30の請求項3)を測定した。その結果、参考例1、6、7で得られた中和重合から得られた吸水性樹脂粉末(A)、(F)、(G)は200個の粒子が一様に黄色(中和指数が0)であるのに対して、吸水性樹脂粉末(E)の粒子は濃緑色から赤色まで様々な色に発色する粒子が混在し、個々の粒子の中和率が非常に不均一(中和指数が15以上)であった。
(参考例6):吸水性樹脂粉末(F)の製造例/中和重合
参考例1において、単量体濃度を39重量%、中和率を71モル%、ポリエチレングリコールジアクリレート9.6gに変更した以外は参考例1と同様にして重合、乾燥および粉砕を行った。得られた粉砕物をさらに目開き500μmおよび300μmのJIS標準篩で分級することで大部分の粒子が500μm〜300μmの範囲にある吸水性樹脂粉末(F)を得た。得られた吸水性樹脂粉末(F)の無加圧下の吸収倍率は32(g/g)、可溶分量は10重量%であった。
【0119】
(参考例7):吸水性樹脂粉末(G)の製造例/中和重合
参考例1において、単量体濃度を41重量%、中和率を71モル%、ポリエチレングリコールジアクリレート5.47g、過硫酸ナトリウム水溶液30.8g、L−アスコルビン酸水溶液2.57g、熱風乾燥温度180℃、熱風乾燥時間50分間に変更した以外は参考例1と同様にして重合、乾燥および粉砕を行った。得られた粉砕物をさらに目開き850μmのJIS標準篩で分級することで大部分の粒子が850μm以下の範囲にある吸水性樹脂粉末(G)を得た。
得られた吸水性樹脂粉末(G)の無加圧下の吸収倍率は38(g/g)、可溶分量は13重量%、重量平均粒径(D50)は400μmであった。
【0120】
(実施例17):吸水性樹脂粉末(E)の架橋処理/pH緩衝剤あり
参考例5で得られた吸水性樹脂粉末(E)100gにエチレングリコールジグリシジルエーテル(デナコールEX−810、ナガセ化成(株)社製)0.5g、プロピレングリコール1.0g、中性リン酸塩pH標準液(リン酸二水素カリウム/リン酸水素二ナトリウム;pH6.86)6.0g、およびイソプロピルアルコール1.0gの混合液からなる表面処理剤を混合した後、混合物を120℃で30分間加熱処理することにより、吸水剤(17)を得た。得られた吸水剤(17)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0121】
(比較例17):吸水性樹脂粉末(E)の架橋処理/pH緩衝剤なし
実施例17において、中性リン酸塩pH標準液の代わりに水6.0gを用いた以外は同様にして比較用吸水剤(17)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定し、その結果を表5に示した。
(実施例18):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤あり
参考例6で得られた吸水性樹脂粉末(F)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、リン酸二水素ナトリウム二水和物1.2gの混合液からなる表面処理剤を混合した後、混合物を197℃で10分加熱処理することにより吸水剤(18)を得た。得られた吸水剤(18)も粉末形状であり、その無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0122】
(比較例18):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤なし
実施例18において、リン酸二水素ナトリウム二水和物の代わりにリン酸(85重量%)0.6gを用いた以外は同様にして比較用吸水剤(18)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(比較例19):吸水性樹脂粉末(F)の架橋処理/pH緩衝剤なし
実施例18において、リン酸二水素ナトリウム二水和物を用いなかった以外は同様にして比較用吸水剤(19)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0123】
(実施例19):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤あり
参考例7で得られた吸水性樹脂粉末(G)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、および炭酸水素ナトリウム0.2gの混合液からなる表面処理剤溶液を混合した後、混合物を212℃で25分間加熱処理することにより、吸水剤(19)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(実施例20):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤あり
参考例7で得られた吸水性樹脂粉末(G)100gに1,4−ブタンジオール0.32g、プロピレングリコール0.5g、水2.73g、および炭酸水素カリウム0.24gの混合液からなる表面処理剤溶液を混合した後、混合物を212℃で25分間加熱処理することにより、吸水剤(20)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
【0124】
(比較例20):吸水性樹脂粉末(G)の架橋処理/pH緩衝剤なし
実施例19において、炭酸水素ナトリウムを用いなかった以外は同様にして比較用吸水剤(20)を得た。無加圧下吸収倍率、加圧下吸収倍率を測定した結果を表5に示した。
(実施例21):連続生産系
71モル%が中和されたアクリル酸部分ナトリウム塩とポリエチレングリコールジアクリレート(n=9)を参考例3の比率で、連続的に水溶液重合(ベルト滞留時間:約30分、厚み:約25mm)し、得られた吸水性樹脂の含水ゲル状架橋重合体をミートチョッパーで粒子状に粗砕し、これをバンド乾燥機の多孔板上に薄く広げて載せ、180℃で30分間連続熱風乾燥した。乾燥機出口でブロック状の乾燥重合体が得られた。この乾燥重合体を取り出したと同時に解砕し、得られた粒子状乾燥物を1000kg/hで3段ロールグラニュレーター(ロールギャップが上から1.0mm/0.55mm/0.42mm)に連続供給することで粉砕した。得られた約60℃の粒子状吸水性樹脂の粉末を、網目開き850μmのふるい網を有する篩い分け装置で分級し、90重量%以上が850μm未満で150μm以上のサイズの吸水性樹脂粉末(H)(平均粒子径:430〜460μm)を得た。得られた吸水性樹脂粉末(H)の無加圧下の平均吸収倍率(CRC)は40g/g、可溶分量の平均は11重量%であった。なお、CRCおよび可溶分量の物性は2時間ごと(2000kg/Lot)に測定した平均値である。
【0125】
さらに、吸水性樹脂粉末(H)を、高速連続混合機(タービュライザー/1000rpm)に1000kg/hで連続供給して、さらに、吸水性樹脂粉末(H)に1,4−ブタンジオール/プロピレングリコール/水/24%水酸化ナトリウム水溶液=0.384/0.63/3.39/0.3(重量%/対粉末)からなる表面架橋剤水溶液を、約200μmの液滴になるスプレーで噴霧し混合した。次いで、得られた混合物を195℃で40分間、パドルドライヤーにより連続的に加熱処理することで吸水剤粉末を得た。さらに得られた吸水剤粉末を、網目開き850μmのふるい網を有する篩い分け装置で分級して、850μm未満で150μm以上のサイズの粒子が90重量%以上の吸水剤(21)を得た。
【0126】
前記の一連の工程(重合、乾燥、粉砕、加熱処理)を連続で24時間で行い、2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:11点)を測定した結果、無加圧下の吸収倍率の平均値は31.1g/g、加圧下での吸収倍率の平均値は25.5g/g、生理食塩水流れ誘導性の平均値は30(Lot毎のSFCの値:32、32、28、28、30、27、27、32、31、32、28)、そのSFCの標準偏差の値は2.1であった。
(実施例22):連続生産系
実施例21において、24時間を10日間に変えた以外は同様の方法で連続生産を行い、吸水剤(22)を得た。2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:110点)を測定した結果、無加圧下の吸収倍率の平均値は31.3g/g、加圧下での吸収倍率の平均値は25.2g/g、生理食塩水流れ誘導性の平均値は30(Lot毎のSFCの値は省略)、そのSFCの標準偏差の値は3.9であった。
【0127】
(比較例21):連続生産系
実施例21において、24%水酸化ナトリウム水溶液を使わなかった以外は同様の方法で連続生産を行い、比較用吸水剤(21)を得た。2時間ごと(2000kg/Lot)に吸水剤の物性(Lot数:11点)を測定した結果、無加圧下の吸収倍率の平均値は31.1g/g、加圧下での吸収倍率の平均値は24.2g/g、生理食塩水流れ誘導性の平均値は20(Lot毎のSFCの値:26、17、11、17、17、18、20、17、16、28、28)、そのSFCの標準偏差の値は5.5であった。
実施例21および実施例22に記載の吸水剤(21)、(22)は、比較例21に記載の比較用吸水剤(21)に比べて、SFCの標準偏差が小さくなり、本発明の吸水剤を連続生産する場合、不良品(低SFC品など)の発生もなく、かつ、品質(SFC)のばらつきが低減されることを示している。
【0128】
【表1】
【0129】
【表2】
【0130】
【表3】
【0131】
実施例1および実施例2に記載の吸水剤(1)、(2)は、同じく表面架橋された比較用吸水剤(1)、(2)と比べて、加圧下吸収倍率が大きく、無加圧下吸収倍率と加圧下吸収倍率のバランスに優れている。
実施例3に記載の吸水剤(3)は、比較例3に記載の比較用吸水剤(3)と比べて、無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスに優れている。
さらに、実施例4および実施例5および実施例6に記載の吸水剤(4)、(5)、(6)は、比較例4に記載の比較用吸水剤(4)と比べて、無加圧下吸収倍率はほぼ同じであるが生理食塩水流れ誘導性は高い値を示している。
【0132】
同様に、実施例7および実施例8および実施例9に記載の吸水剤(7)(8)(9)においても、比較例5および比較例6に記載の比較用吸水剤(5)および比較用吸水剤(6)と比べて、無加圧下吸収倍率はほぼ同じであるが、加圧下吸水倍率と生理食塩水流れ誘導性は高い値を示している。
また、実施例10、実施例11、実施例12に記載の吸水剤(10)、(11)、(12)は、それぞれ比較例7、比較例8、比較例9に記載の比較用吸水剤(7)、(8)、(9)と比べて、無加圧下吸収倍率はほぼ同じであるが、生理食塩水流れ誘導性は高い値を示している。
【0133】
実施例13および実施例14に記載の吸水剤(13)、(14)は、比較例10および比較例11および比較例12に記載の比較用吸水剤(10)、(11)、(12)と比べて、SFC変化指数およびSFC変動係数は小さく、SFC変動率は大きくなりいずれもSFCのばらつきが低減されている。
また、実施例15および実施例16に記載の吸水剤(15)、(16)も、SFC変化指数およびSFC変動指数は小さく、SFC変動率は大きくなり、いずれもSFCのばらつきが低減されている。
このように、本発明の方法で製造される吸水剤は、無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスに優れ、良好な性能を備えたものである。さらに、粒度毎のSFCのばらつきも少ない事から、おむつの性能を安定させる上で非常に優れた吸水剤である。
【0134】
【表4】
【0135】
比較例13、比較例14、比較例15、比較例16に記載の比較用吸水剤(13)、(14)、(15)、(16)は、いずれも無加圧下吸収倍率と加圧下吸収倍率と生理食塩水流れ誘導性の3つのバランスが悪いものであった。
【0136】
【表5】
【0137】
実施例17記載の吸水剤(17)は、比較例17記載の比較用吸水剤(17)と比べて、無加圧下吸収倍率と加圧下吸収倍率のバランスやその合計に優れている。
また、実施例18記載の吸水剤(18)は、リン酸を加えた比較例18記載の比較用吸水剤(18)と比べて、同様に10分という短い時間で架橋反応が進行し、無加圧下吸収倍率は同じであるが加圧下吸収倍率は高い値を示している。さらに、何も加えなかった比較例19記載の比較用吸水剤(19)については、10分という短時間では十分な架橋反応が進行せず、無加圧下吸収倍率の低下がほとんど見られず、加圧下吸収倍率も低いものとなっている。
【0138】
このように、本発明の方法で製造される吸水剤は、無加圧下吸収倍率と加圧下吸収倍率のバランスやその合計に優れ、短い反応時間でも良好な性能を備えたものである。
【符号の説明】
【0139】
31 タンク
32 ガラス管
33 0.69重量%塩化ナトリウム水溶液
34 コック付きL字管
35 コック
40 容器
41 セル
42 ステンレス製金網
43 ステンレス製金網
44 膨潤ゲル
45 ガラスフィルター
46 ピストン
47 ピストン中の穴
48 補集容器
49 上皿天秤
51 ガラス容器
52 分散機
53 上側クランプ
54 下側クランプ
【特許請求の範囲】
【請求項1】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
【請求項2】
前記吸水剤の粒度分布が、粒子A1からA4をそれぞれ0.1重量%以上含む、請求項1に記載の吸水剤。
【請求項3】
下記式(2)で規定されるSFC変動係数が0〜0.25である、請求項1または2に記載の吸水剤。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
【請求項4】
下記式(3)で規定されるSFC変動率が0.65〜1.00である、請求項1から3までのいずれかに記載の吸水剤。
SFC変動率=(粒子A1からA4のSFCの中で最小のSFC)/(粒子A1からA4のSFCの中で最大のSFC) (3)
【請求項5】
前記吸水剤がさらにカチオン性高分子化合物および/または水不溶性微粒子を含む、請求項1から4までのいずれかに記載の吸水剤。
【請求項6】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
【請求項7】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、連続生産された吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
【請求項8】
酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上であることを特徴とする、吸水剤。
【請求項9】
酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする、吸水剤の製造方法。
【請求項10】
酸基を含有し且つ重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる架橋剤(c)を混合し、前記吸水性樹脂粉末(a1)を架橋処理することを特徴とする、吸水剤の製造方法。
【請求項11】
前記吸水性樹脂粉末(a)が、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)である、請求項9に記載の吸水剤の製造方法。
【請求項12】
前記水溶性無機塩基(b1)が、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、アンモニアあるいはその水酸化物からなる群から選ばれる少なくとも1種である、請求項9から11までの何れかに記載の吸水剤の製造方法。
【請求項13】
前記アルカリ金属塩pH緩衝剤(b2)が、弱酸と強塩基からなる無機塩である、請求項9から12までの何れかに記載の吸水剤の製造方法。
【請求項14】
前記アルカリ金属塩pH緩衝剤(b2)が、多塩基酸の部分アルカリ金属中和塩である、請求項9から13までの何れかに記載の吸水剤の製造方法。
【請求項15】
前記架橋剤(c)あるいは脱水反応性架橋剤(c1)が、多価アルコール化合物であることを特徴とする、請求項9から14までの何れかに記載の吸水剤の製造方法。
【請求項16】
前記架橋剤(c)あるいは脱水反応性架橋剤(c1)に、前記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)を予め混合して水溶液としたのち、前記吸水性樹脂粉末(a)あるいは(a1)に対して該水溶液が噴霧添加される、請求項9から15までの何れかに記載の吸水剤の製造方法。
【請求項17】
架橋処理が140〜240℃の加熱処理でなされる、請求項9から16までの何れかに記載の吸水剤の製造方法。
【請求項18】
請求項9から17までの何れかに記載の製造方法で得られた吸水剤。
【請求項19】
請求項1から8および請求項18の何れかに記載の吸水剤を含む衛生材料。
【請求項1】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(1)で規定されるSFC変化指数が0〜25%。
SFC変化指数(%)=[(粒子A1からA4のSFCの標準偏差)/(粒子状吸水剤全体のSFC)]×100 (1)
【請求項2】
前記吸水剤の粒度分布が、粒子A1からA4をそれぞれ0.1重量%以上含む、請求項1に記載の吸水剤。
【請求項3】
下記式(2)で規定されるSFC変動係数が0〜0.25である、請求項1または2に記載の吸水剤。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
【請求項4】
下記式(3)で規定されるSFC変動率が0.65〜1.00である、請求項1から3までのいずれかに記載の吸水剤。
SFC変動率=(粒子A1からA4のSFCの中で最小のSFC)/(粒子A1からA4のSFCの中で最大のSFC) (3)
【請求項5】
前記吸水剤がさらにカチオン性高分子化合物および/または水不溶性微粒子を含む、請求項1から4までのいずれかに記載の吸水剤。
【請求項6】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(2)で規定されるSFC変動係数が0〜0.25。
SFC変動係数=(粒子A1からA4のSFCの標準偏差)/(粒子A1からA4のSFCの平均値) (2)
【請求項7】
不飽和単量体成分を重合して得られる架橋構造を有する吸水性樹脂を主成分とする粒子状吸水剤であって、
前記粒子状吸水剤は、粒子径が850μm未満で150μm以上の粒子を全粒子の90重量%以上含み、かつ、粒子径が850μm未満で600μm以上の粒子(A1)、粒子径が600μm未満で500μm以上の粒子(A2)、粒子径が500μm未満で300μm以上の粒子(A3)、粒子径が300μm未満で150μm以上の粒子(A4)から選ばれる少なくとも2種以上を含み、
さらに、下記物性を満たすことを特徴とする、連続生産された吸水剤。
0.90重量%生理食塩水に対する無加圧下で30分の吸収倍率(CRC)が31g/g以上。
0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が24g/g以上。
0.69重量%生理食塩水流れ誘導性(SFC)が20(単位:10−7×cm3×s×g−1)以上。
下記式(4)で規定される連続生産系SFC標準偏差が5.0以下。
連続生産系SFC標準偏差=各LotのSFCの標準偏差 (4)
(ただし、CRC、AAP、SFCはLot平均であり、各Lotは20kg以上。Lot数は10以上。)
【請求項8】
酸基含有単量体(塩)を含む単量体を重合し、さらに後中和して得られた吸水性樹脂を表面架橋した粒子状吸水剤であって、
前記粒子状吸水剤または吸水性樹脂の中和指数が15以上で、かつ、表面架橋後の0.90重量%生理食塩水に対する4.83kPaで60分の加圧下吸収倍率(AAP)が20(g/g)以上であることを特徴とする、吸水剤。
【請求項9】
酸基含有の吸水性樹脂粉末(a)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる脱水反応性架橋剤(c1)を混合し、前記吸水性樹脂粉末(a)を架橋処理することを特徴とする、吸水剤の製造方法。
【請求項10】
酸基を含有し且つ重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)に、非架橋性の水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)、および、該酸基と反応しうる架橋剤(c)を混合し、前記吸水性樹脂粉末(a1)を架橋処理することを特徴とする、吸水剤の製造方法。
【請求項11】
前記吸水性樹脂粉末(a)が、重量平均粒子径300〜600μmで150μm以下の微粉が10重量%以下の吸水性樹脂粉末(a1)である、請求項9に記載の吸水剤の製造方法。
【請求項12】
前記水溶性無機塩基(b1)が、アルカリ金属塩、アンモニウム塩、アルカリ金属水酸化物、アンモニアあるいはその水酸化物からなる群から選ばれる少なくとも1種である、請求項9から11までの何れかに記載の吸水剤の製造方法。
【請求項13】
前記アルカリ金属塩pH緩衝剤(b2)が、弱酸と強塩基からなる無機塩である、請求項9から12までの何れかに記載の吸水剤の製造方法。
【請求項14】
前記アルカリ金属塩pH緩衝剤(b2)が、多塩基酸の部分アルカリ金属中和塩である、請求項9から13までの何れかに記載の吸水剤の製造方法。
【請求項15】
前記架橋剤(c)あるいは脱水反応性架橋剤(c1)が、多価アルコール化合物であることを特徴とする、請求項9から14までの何れかに記載の吸水剤の製造方法。
【請求項16】
前記架橋剤(c)あるいは脱水反応性架橋剤(c1)に、前記水溶性無機塩基(b1)および/または非還元性のアルカリ金属塩pH緩衝剤(b2)を予め混合して水溶液としたのち、前記吸水性樹脂粉末(a)あるいは(a1)に対して該水溶液が噴霧添加される、請求項9から15までの何れかに記載の吸水剤の製造方法。
【請求項17】
架橋処理が140〜240℃の加熱処理でなされる、請求項9から16までの何れかに記載の吸水剤の製造方法。
【請求項18】
請求項9から17までの何れかに記載の製造方法で得られた吸水剤。
【請求項19】
請求項1から8および請求項18の何れかに記載の吸水剤を含む衛生材料。
【図1】

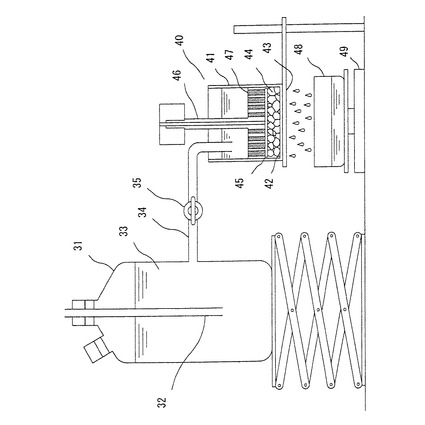
【図2】
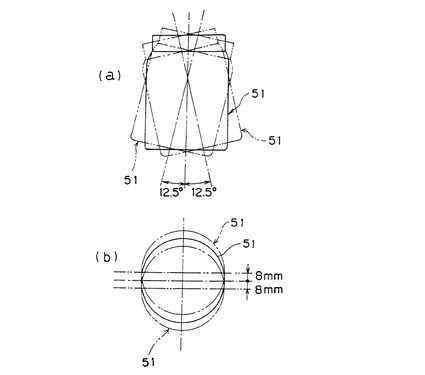
【図3】
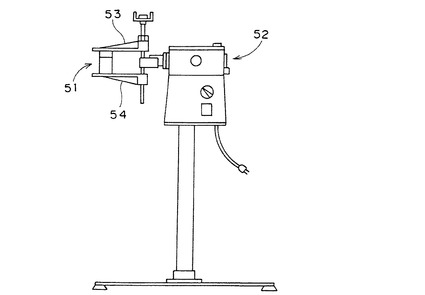
【図2】
【図3】
【公開番号】特開2009−209373(P2009−209373A)
【公開日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願番号】特願2009−117974(P2009−117974)
【出願日】平成21年5月14日(2009.5.14)
【分割の表示】特願2002−166178(P2002−166178)の分割
【原出願日】平成14年6月6日(2002.6.6)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願日】平成21年5月14日(2009.5.14)
【分割の表示】特願2002−166178(P2002−166178)の分割
【原出願日】平成14年6月6日(2002.6.6)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]