
吸水性樹脂粒子とその製造方法、吸水性樹脂粒子組成物、ならびに用途
【課題】 静置状態の水性液全体を速やかにゲル化することが可能な、新規な吸水性樹脂粒子および吸水性樹脂粒子組成物を提供する。吸水性樹脂粒子の製造方法、吸水性樹脂粒子や吸水性樹脂粒子組成物の用途をも提供する。
【解決手段】 本発明にかかる吸水性樹脂粒子は、低発塵性の吸水性樹脂粒子であり、親水性と疎水性とを有し重量平均分子量が10000〜1000000である熱可塑性樹脂および/または無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、静置水ゲル化時間が120秒以内であることを特徴とする。
【解決手段】 本発明にかかる吸水性樹脂粒子は、低発塵性の吸水性樹脂粒子であり、親水性と疎水性とを有し重量平均分子量が10000〜1000000である熱可塑性樹脂および/または無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、静置水ゲル化時間が120秒以内であることを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定の表面処理層を有する吸水性樹脂粒子とその製造方法、その吸水性樹脂粒子を用いて得られる吸水性樹脂粒子組成物、ならびにこれら吸水性樹脂粒子と吸水性樹脂粒子組成物の用途に関する。用途としては特に、静置水ゲル化剤、およびそれを用いた水性廃液の処理が好ましく挙げられる。
【背景技術】
【0002】
塗料廃液、メッキ廃液、放射性廃液、医療廃液などの水性廃液を処理する方法は、環境問題が重視される昨今、これらを扱う分野において極めて重要な技術であり、これまでに様々な方法が知られている(例えば、特許文献1など参照)。
本発明者は、これまでに、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料に好適な吸水性樹脂について長年研究を重ねてきた。今回、本発明者は、従来はそれ自身に水性液を吸収させる(すなわち、基材等に固定化された吸水性樹脂に外部から水性液を吸収させる)ことを主たる目的とした吸水性樹脂についての知見を活かし、逆に、水性廃液の貯留容器中に吸水性樹脂を粒子の状態で添加することによって水性廃液全体を速やかにゲル化させて廃棄することができないか考えた。特に、これら水性廃液は人体に有害な物質(ウイルスなど)を含んでいることが多いので、攪拌等による飛散を防ぐために、水性廃液を「静置状態」で吸水性樹脂粒子を添加して水性廃液全体を速やかにゲル化させる技術について検討を開始した。
【0003】
まず、従来から良く知られているいくつかの吸水性樹脂粒子(特に、衛生材料に好適とされる吸水性樹脂粒子)について、静置状態の水性廃液が入った貯留容器中に添加したところ、(1)吸水性樹脂粒子のほとんど全てがすぐに容器下部に沈降してしまい、容器下部から水性廃液の50〜80%ほどまでは比較的速やかにゲル化するものの、液面までは完全にゲル化が進行しないという問題や、(2)液面に吸水性樹脂粒子のほとんど全てが浮いた状態になって液面で吸水性樹脂粒子同士のゲルブロッキングが起こり、吸水性樹脂粒子が容器下部にまで行き渡らなくなってしまい、容器下部〜中部までは完全にゲル化が進行しないという問題が生じた。これらの問題は、貯留容器が縦長(容器の平均断面積(円形換算直径)に対する鉛直方向の長さの比が大きい)になればなるほど顕著に現れた。
【特許文献1】特開2002−119853号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明が解決しようとする課題は、静置状態の水性液全体を速やかにゲル化することが可能な、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな自己分散による高吸収速度とを両立した新規な吸水性樹脂粒子および吸水性樹脂粒子組成物を提供することにある。また、そのような吸水性樹脂粒子の製造方法、吸水性樹脂粒子や吸水性樹脂粒子組成物の用途をも提供することにある。さらに、特定処理により微粉末状態とした吸水性樹脂粉末を表面処理することによる、高いVortex吸収速度を実現する、低発塵性の吸水性樹脂粒子を提供することにある。
【課題を解決するための手段】
【0005】
吸水性樹脂粒子同士のゲルブロッキングを防止するための表面処理技術として、吸水性樹脂粉末表面を熱可塑性樹脂で被覆する技術(例えば、特開平2−242858号公報、特開2003−301019号公報など参照)が知られている。しかし、従来公知のこれらの方法においては、ゲルブロッキング防止を達成するために吸収速度を犠牲にしており、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とは両立できていないことが判った。
本発明者は、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立するための技術手段について、鋭意検討を行った。
【0006】
その結果、原料となる吸水性樹脂粉末の表面を、極めて少量の、親水性と疎水性を有する特定の熱可塑性樹脂および必要により無機粒子で処理して、特定の表面処理層を形成させることにより、得られる吸水性樹脂粒子の表面上において適度な親水性と適度な疎水性との両立が実現でき、適度なゲルブロッキング抑制が発現できるとともに静置状態の水性液に添加した場合の適度な沈降速度を発現でき、その結果、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立できることを見出した。
さらに、熱可塑性樹脂としてアルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種を用いると、適度な疎水性によってゲルブロックが適度に抑制でき、適度な親水性によって過度なゲルブロック抑制を防止でき、従って、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度との両立がより一層達成できることも見出した。
【0007】
すなわち、本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
前記表面処理された吸水性樹脂粒子は、水分含有量が1〜15重量%であることが好ましい。
【0008】
前記表面処理された吸水性樹脂粒子は、前記ゲル化するまでの時間(静置水ゲル化時間)が80秒以内であることが好ましい。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記表面処理前の原料吸水性樹脂粉末の粒度が、目開き600μmの篩、好ましくは目開き500μmの篩を通過するものであり、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%である。
【0009】
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であり、得られた上記表面処理された吸水性樹脂粒子の静置水ゲル化時間が原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂がアルコール可溶性であり、重量平均分子量が20000〜500000である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂が、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種である。
【0010】
本発明にかかる吸水性樹脂粒子組成物は、本発明の表面処理された吸水性樹脂粒子30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子あるいは本発明の吸水性樹脂粒子組成物を含む。
【0011】
本発明にかかる吸水性樹脂粒子の製造方法は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩、好ましくは目開き500μm以下の篩を通して解砕する。
本発明にかかる水性廃液の処理方法は、水性廃液の入った貯槽に本発明の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる。
本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、前記表面処理前の原料吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、Vortex吸収速度が5秒以下であることを特徴とする。
【0012】
本発明にかかるケーブル用止水テープは、本発明の吸水性樹脂粒子を用いる。
本発明にかかる衛生材料用吸水性樹脂は、目開き212〜125μmの少なくとも1つの篩を通過しない吸水性樹脂粒子と、本発明の吸水性樹脂粒子を混合してなる。
なお、本発明の吸水性樹脂粒子は吸水性樹脂粒子組成物と呼ぶことがある。本発明の吸水性樹脂粒子以外の吸水性樹脂粒子、その他の添加剤との混合物も吸水性樹脂粒子組成物と呼ぶことがある。
【発明の効果】
【0013】
本発明によれば、静置状態の水性液全体を速やかにゲル化することが可能な、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな自己分散による高吸収速度とを両立した新規な吸水性樹脂粒子および吸水性樹脂粒子組成物を提供でき、また、そのような吸水性樹脂粒子の製造方法、吸水性樹脂粒子や吸水性樹脂粒子組成物の用途をも提供できる。さらに、篩により分級して微粉末状態とした原料吸水性樹脂粉末を表面処理することによる、高いVortex吸収速度を実現する、低発塵性の吸水性樹脂粒子を提供することにある。また、吸水性樹脂微粉の回収にも有用である。
【発明を実施するための最良の形態】
【0014】
以下、本発明について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更実施し得る。
〔吸水性樹脂粉末〕
本発明にかかる吸水性樹脂粒子は、特定の熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた表面処理層を有してなる。
本発明で用いることができる吸水性樹脂粉末は、親水性単量体を重合して得ることができる水不溶性水膨潤性ヒドロゲル形成性重合体(以下、吸水性樹脂とも言う)の粉体であって、少なくとも生理食塩水の吸収倍率が10倍以上、好ましくは20〜100倍、より好ましくは30〜80倍である(JIS K7223−1996)、球形或いは不定形の粒子形状のものである。なお、本発明においては、吸水性樹脂粉末を単に吸水性樹脂と称することもある。
【0015】
水不溶性水膨潤性ヒドロゲル形成性重合体の具体例としては、部分中和架橋ポリアクリル酸重合体(米国特許第4625001号、米国特許第4654039号、米国特許第5250640号、米国特許第5275773号、欧州特許第456136号等)、架橋され部分的に中和された澱粉−アクリル酸グラフトポリマー(米国特許第4076663号)、イソブチレン−マレイン酸共重合体(米国特許第4389513号)、酢酸ビニル−アクリル酸共重合体のケン化物(米国特許第4124748号)、アクリルアミド(共)重合体の加水分解物(米国特許第3959569号)、アクリロニトリル重合体の加水分解物(米国特許第3935099号)等が挙げられるが、本発明で用いることができる吸水性樹脂粉末はアクリル酸および/またはその塩を含む単量体を重合して得られるポリアクリル酸(塩)系架橋重合体からなる吸水性樹脂粉末であることが好ましい。
【0016】
本発明において、ポリアクリル酸(塩)系架橋重合体とは、重合に用いる重合性単量体の総量を100モル%として、アクリル酸および/またはその塩を50モル%以上、好ましくは70モル%以上、より好ましくは90モル%以上含む単量体を重合して得られる架橋重合体である。また、重合体中の酸基は、その50〜90モル%が中和されていることが好ましく、60〜80モル%が中和されていることがより好ましく、塩としてはナトリウム、カリウム、リチウム等のアルカリ金属塩、アンモニウム塩、アミン塩などを例示する事ができる。塩を形成させるための吸水性樹脂の中和は重合前に単量体の状態で行っても良いし、あるいは重合途中や重合後に重合体の状態で行っても良いし、それらを併用してもよい。
【0017】
本発明に好ましく用いられる吸水性樹脂であるポリアクリル酸(塩)系架橋重合体としては、主成分として用いられる単量体(アクリル酸および/またはその塩)に併用して、必要により他の単量体を共重合させたものであってもよい。他の単量体の具体例としては、メタアクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸などのアニオン性不飽和単量体およびその塩;アクリルアミド、メタアクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン、N−ビニルアセトアミドなどのノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミドおよびそれらの四級塩などのカチオン性不飽和単量体などを挙げることができる。これらアクリル酸および/またはその塩以外の単量体の使用量は、全単量体(単量体成分と称することがある)中、0〜30モル%が好ましく、より好ましくは0〜10モル%である。
【0018】
また、上記ノニオン性の親水基含有不飽和単量体が主要成分である耐塩型の吸水性樹脂を組み合わせて使用してもよい。
本発明で用いることができる吸水性樹脂粉末は、内部架橋構造を有する架橋重合体である。
本発明に用いられる吸水性樹脂粉末に内部架橋構造を導入する方法として、架橋剤を使用しない自己架橋によって導入する方法や、1分子中に2個以上の重合性不飽和基および/または2個以上の反応性基を有する内部架橋剤を共重合または反応させて導入する方法等を例示できる。好ましくは、内部架橋剤を共重合または反応させて導入する方法である。
【0019】
これらの内部架橋剤の具体例としては、例えば、N,N′−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリト−ルテトラ(メタ)アクリレ−ト、ジペンタエリスリト−ルヘキサ(メタ)アクリレ−ト、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、ポリエチレンイミン、グリシジル(メタ)アクリレートなどを挙げることが出来る。これらの内部架橋剤は1種のみ用いてもよいし2種以上使用してもよい。中でも、得られる吸水性樹脂の吸水特性などから、2個以上の重合性不飽和基を有する化合物を内部架橋剤として必須に用いることが好ましく、その使用量としては、全単量体に対して0.005〜3モル%が好ましく、より好ましくは0.01〜1.5モル%である。
【0020】
重合に際しては、澱粉−セルロ−ス、澱粉−セルロ−スの誘導体、ポリビニルアルコ−ル、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子や、次亜リン酸(塩)等の連鎖移動剤を添加してもよい。
本発明に用いることができる吸水性樹脂粉末を得るために、上記したアクリル酸および/またはその塩を主成分とする単量体を重合するに際しては、バルク重合、逆相懸濁重合、沈澱重合を行うことも可能であるが、性能面や重合の制御の容易さから、単量体を水溶液として、水溶液重合を行うことが好ましい。かかる重合方法は、例えば、米国特許第4625001号明細書、米国特許第4769427号明細書、米国特許第4873299号明細書、米国特許第4093776号明細書、米国特許第4367323号明細書、米国特許第4446261号明細書、米国特許第4683274号明細書、米国特許第4690996号明細書、米国特許第4721647号明細書、米国特許第4738867号明細書、米国特許第4748076号明細書、欧州特許第1178059号明細書などに記載されている。
【0021】
重合を行うにあたり、過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2′−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤、紫外線や電子線などの活性エネルギー線等を用いることができる。また、ラジカル重合開始剤を用いる場合、亜硫酸ナトリウム、亜硫酸水素ナトリウム、硫酸第一鉄、L−アスコルビン酸等の還元剤を併用してレドックス重合としても良い。これらの重合開始剤の使用量は、全単量体に対して、0.001〜2モル%が好ましく、より好ましくは0.01〜0.5モル%である。
上記の重合および乾燥により得られた吸水性樹脂の形状は、一般には、不定形破砕状、球状、繊維状、棒状、略球状、偏平状等であるが、逆相懸濁重合で得られた球状のものやその凝集体であるブドウの房状のものも好適に用いることができる。また、本発明に用いられる吸水性樹脂は粒子状が望ましく、乾燥後に粉砕して得られるような不定形破砕状のものを用いると、より本発明の効果が大きくなるので好ましい。
【0022】
本発明で用いることができる吸水性樹脂は、その表面近傍が表面架橋剤でさらに表面架橋処理されていてもよいし、表面架橋処理されていなくてもよい。また、モノマーとしてアクリルアミドが用いられた吸水性樹脂の場合は、熱表面架橋されたものでもよい。
表面架橋処理に用いることの出来る表面架橋剤としては、吸水性樹脂の有する官能基、特に、カルボキシル基と反応し得る官能基を2個以上有する有機表面架橋剤や多価金属化合物が挙げられる。例えば、エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール化合物;エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリシドール等のエポキシ化合物;エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ポリエチレンイミン等の多価アミン化合物や、それらの無機塩ないし有機塩(例えば、アゼチジニウム塩等);2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート等の多価イソシアネート化合物;1,2−エチレンビスオキサゾリン等の多価オキサゾリン化合物;環状尿素、2−オキサゾリジノン等の炭酸誘導体;1,3−ジオキソラン−2−オン、4−メチル−1,3−ジオキソラン−2−オン、4,5−ジメチル−1,3−ジオキソラン−2−オン、4,4−ジメチル−1,3−ジオキソラン−2−オン、4−エチル−1,3−ジオキソラン−2−オン、4−ヒドロキシメチル−1,3−ジオキソラン−2−オン、1,3−ジオキサン−2−オン、4−メチル−1,3−ジオキサン−2−オン、4,6−ジメチル−1,3−ジオキサン−2−オン、1,3−ジオキソパン−2−オン等のアルキレンカーボネート化合物;エピクロロヒドリン、エピブロムヒドリン、α−メチルエピクロロヒドリン等のハロエポキシ化合物、および、その多価アミン付加物(例えばハーキュレス製カイメン:登録商標);γ−グリシドキシプロピルトリメトキシシラン、γーアミノプロピルトリエトキシシラン等のシランカップリング剤;3−メチル−3−オキセタンメタノール、3−エチル−3−オキセタンメタノール、3−ブチル−3−オキセタンメタノール、3−メチル−3−オキセタンエタノール、3−エチル−3−オキセタンエタノール、3−ブチル−3−オキセタンエタノール、3−クロロメチル−3−メチルオキセタン、3−クロロメチル−3−エチルオキセタン、多価オキセタン化合物などのオキセタン化合物;亜鉛、カルシウム、マグネシウム、アルミニウム、鉄、ジルコニウム等の水酸化物又は塩化物等の多価金属化合物等が挙げられる。これら表面架橋剤は、1種のみ用いてもよいし、2種以上を併用してもよい。
【0023】
表面架橋剤の使用量は、吸水性樹脂の固形分100重量部に対して0.001〜5重量部が好ましい。
表面架橋剤と吸水性樹脂との混合の際には水を用いてもよい。水の使用量は、吸水性樹脂の固形分100重量部に対して、0〜15重量部の範囲内が好ましく、0.5を越え10重量部以下の範囲内がより好ましく、1〜5重量部の範囲内がさらに好ましい。
表面架橋剤やその水溶液を混合する際には、親水性有機溶媒や、それ以外の第三物質を混合助剤として用いてもよい。
親水性有機溶媒を用いる場合には、例えば、メチルアルコール、エチルアルコール、n−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、イソブチルアルコール、t−ブチルアルコール等の低級アルコール類;アセトン等のケトン類;ジオキサン、テトラヒドロフラン、メトキシ(ポリ)エチレングリコール等のエーテル類;ε−カプロラクタム、N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類;エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール類等が挙げられる。親水性有機溶媒の使用量は、吸水性樹脂の種類や粒径、含水率等にもよるが、吸水性樹脂の固形分100重量部に対して、0〜10重量部の範囲内が好ましく、0.1〜5重量部の範囲内がより好ましい。また、第三物質として欧州特許第0668080号公報に示された無機酸、有機酸、ポリアミノ酸等を存在させてもよい。これらの混合助剤は表面架橋剤として作用しても良いが、表面架橋後に吸水性樹脂の吸水性能を低下させないものが好ましい。特に沸点が150℃未満の揮発性アルコール類は表面架橋処理時に揮発してしまうので、残存物が残らず望ましい。
【0024】
吸水性樹脂と表面架橋剤とをより均一に混合するため、非架橋性の水溶性無機塩基類(好ましくは、アルカリ金属塩,アンモニウム塩,アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物)や、非還元性アルカリ金属塩pH緩衝剤(好ましくは炭酸水素塩、リン酸二水素塩、リン酸水素塩等)を、吸水性樹脂と表面架橋剤とを混合する際に共存させても良い。これらの使用量は、吸水性樹脂の種類や粒径等にもよるが、吸水性樹脂の固形分100重量部に対して0〜15重量部の範囲内が好ましく、0.005〜10重量部の範囲内がより好ましく、0.05〜5重量部の範囲内がさらに好ましい。
吸水性樹脂と表面架橋剤とを混合する混合方法は特に限定されないが、たとえば吸水性樹脂を親水性有機溶剤に浸漬し、必要に応じて水および/または親水性有機溶媒に溶解させた表面架橋剤を混合する方法、吸水性樹脂に直接、水および/または親水性有機溶媒に溶解させた表面架橋剤を噴霧若しくは滴下して混合する方法等が例示できる。
【0025】
吸水性樹脂と表面架橋剤とを混合した後、通常、加熱処理を行い、架橋反応を遂行させる。上記加熱処理温度は、用いる表面架橋剤にもよるが、材料温度で、40〜250℃が好ましく、150〜250℃がより好ましい。処理温度が40℃未満の場合には、加圧下の吸収倍率等の吸収特性が十分に改善されない場合がある。処理温度が250℃を越える場合には、吸水性樹脂の劣化を引き起こし、性能が低下する場合があり注意を要する。加熱処理時間は、好ましくは1分〜2時間、より好ましくは5分〜1時間である。
本発明で用いることができる吸水性樹脂粉末は、目開き600μmの篩を通過するものであることが好ましい。より好ましくは、目開き500μmの篩を通過するものである。これは、篩による分級や気流分級等によって得ることができる。
【0026】
本発明で用いることができる原料吸水性樹脂粉末は、後述の重量平均粒子径が500μm以下であることが好ましく、より好ましくは400μm以下であり、さらに好ましくは300μm以下である。
また、微粉末が少なく高吸収速度の吸水性樹脂組成物を得るためには、好ましくは目開き212〜125μmの篩から選択された少なくとも1つの篩を通過したもの、より好ましくは目開き150〜125μmの篩を通過したものを使用した場合には、Vortex吸収速度(JIS K7224−1996)が5秒以下の吸水性樹脂粒子を得ることが可能となる。
【0027】
本発明で用いることができる吸水性樹脂粉末としては、粒子径300μm以下の粉末を表面架橋処理および/または造粒し粒度調整したものを用いても良い。また、粉砕して得られる一次粒子の不定形破砕状の粒子に対し、微粉の造粒物を、好ましくは5重量%以上、より好ましくは10重量%以上、さらに好ましくは15重量%以上、混合した吸水性樹脂を用いても良い。
微粉造粒物の作成方法としては、微粉を再生する公知の技術が使用可能である。例えば、温水と吸水性樹脂の微粉を混合し乾燥する方法(米国特許第6228930号)や、吸水性樹脂の微粉を単量体水溶液と混合し重合する方法(米国特許第5264495号)、吸水性樹脂の微粉に水を加え特定の面圧以上で造粒する方法(欧州特許第844270号)、吸水性樹脂の微粉を十分に湿潤させ非晶質のゲルを形成し乾燥・粉砕する方法(米国特許第4950692号)、吸水性樹脂の微粉と重合ゲルを混合する方法(米国特許第5478879号)などを用いることが可能であるが、好ましくは前記の温水と吸水性樹脂の微粉を混合し乾燥する方法が用いられる。なお、粒子径は分級される篩目径で示される。
【0028】
〔吸水性樹脂粒子〕
本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
【0029】
本発明にかかる吸水性樹脂粒子は、水分含有量が1〜15重量%であることが好ましい。
なお、本発明における表面処理とは、表面へのコーティング、吸水性樹脂粉末間の接着造粒などが含まれる。
また、当該表面処理された吸水性樹脂粒子が本発明の所望の効果を有する範囲において、上記表面処理は部分コーティングないし付着であってもよい。
表面処理前の吸水性樹脂粉末は、前述した通りである。
本発明にかかる吸水性樹脂粒子は、表面処理された吸水性樹脂粒子であり、特定の熱可塑性樹脂および必要により無機粒子で処理された表面処理層を有してなる。すなわち、本発明にかかる吸水性樹脂粒子は、表面処理された吸水性樹脂粒子であり、特定の熱可塑性樹脂および無機粒子で処理された表面処理層を有してなるものでもよいし、特定の熱可塑性樹脂のみで処理された表面処理層を有してなるものでもよい。
【0030】
前記熱可塑性樹脂は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である。
前記熱可塑性樹脂の重量平均分子量は、好ましくは20000〜500000、より好ましくは40000〜200000である。
前記熱可塑性樹脂は、親水性と疎水性とを有する。親水性とは「親水性を有する構造」を有することを意味し、疎水性とは「疎水性を有する構造」を有することを意味する。具体的には、親水性とは、例えば、カルボン酸基、水酸基、アミノ基、アミド基、スルホン酸基、リン酸基、アルキレングリコール基、N−メトキシアミノ基等の親水性基を有する構造が挙げられ、疎水性とは、例えば、メチル基、エチル基、プロピル基、ブチル基等のアルキル基やフェニル基、アセチル基などの疎水性基を有する構造が挙げられる。
【0031】
親水性と疎水性とを有する熱可塑性樹脂を用いて表面処理されることにより、(i)親水性の吸水性樹脂粉末表面への処理時に、熱可塑性樹脂の親水性基が吸水性樹脂粉末表面への付着性を向上させているので、極めて少量の熱可塑性樹脂の適用が可能になったものと考えられ、(ii)そのために、表面の熱可塑性樹脂による膨潤阻害が起こり難く、(iii)また、熱可塑性樹脂の疎水性基が吸水性樹脂粉末表面に存在することにより、静置水と接触した時点での吸水性樹脂粒子の膨潤ゲル粒子間の粘着・付着力が低減されているために膨潤ゲル粒子が順次液面から離れて沈降するものと考えられる。
前記熱可塑性樹脂としては、アルコール可溶性であり、重量平均分子量が20000〜500000であることが好ましい。ここでいうアルコール可溶性とは、常圧下、40℃でメタノールまたはエタノールに少なくとも0.5重量%以上、好ましくは2重量%以上、さらに好ましくは5重量%以上の濃度で溶解して溶液となることをいう。上限は、70重量%以下、好ましくは50重量%以下である。
【0032】
さらに、アルコール可溶性で水不溶性であるものが好ましい。これは適度の疎水性と適度の親水性を持った熱可塑性樹脂を少量使用することにより、疎水性基による過度の撥水性により吸水速度が抑制されることもなく、水性液と接触した場合にも水性液に溶解せず、接触液を増粘させないため、通液性を確保して全体のゲル化を促進するのに好ましい。
前記熱可塑性樹脂としては、例えば、親水性モノマーと疎水性アクリルエステル単量体の共重合体、低ゲル化度のポリビニルアルコール、アルコール可溶性ナイロンなどが挙げられ、これらの1種のみ用いてもよいし、2種以上を併用してもよい。好ましくは、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種であり、特に好ましくは、アルコール可溶性であり水不溶性であるアルカリ水可溶性樹脂である。
【0033】
アルカリ水可溶性樹脂とは、常温、常圧で、水には不溶であるがイオン交換水に水酸化ナトリウムを0.4重量%の割合で溶解することによってアルカリ性にしたアルカリ水に溶解する樹脂である。
上記アルカリ水可溶性樹脂とは、特許第3224533号(特開2002−060695号公報)等に記載されるアルカリ水可溶性樹脂を用いることができる。アルカリ水可溶性樹脂としては、酸性または中性を呈する水には溶解せず、アルカリ性を呈する水には溶解する樹脂を意味する。ここで、「中性を呈する水」とは、pH値が6〜8の範囲内の水であり、「酸性を呈する水」とは、pH値が該中性の範囲未満の水であり、「アルカリ性を呈する水」とは、pH値が該中性の範囲よりも大きい水である。
【0034】
なお、上記アルカリ水可溶性樹脂としては、アルカリ水への溶解性の程度として、下記評価試験によって求められる減少率が50〜100%のものが好ましく、60〜100%のものがより好ましく、70〜100%のものがさらに好ましい。
(アルカリ水への溶解性の評価試験)
二軸押出機を用いて得ることができるバインダー樹脂を、直径5mm、長さ5mmの円筒状のペレット形状に成形したものを用いて測定する。この成形体10gを、1Lのビーカーに入れた0.4重量%濃度のNaOHの水溶液500gに投入し、25℃にて、直径が40mm、4枚はねを用い、300rpmで24時間攪拌を行う。その後のバインダー樹脂の成形体におけるアルカリ水へ溶解した重量の、もとの成形体からの減少率で評価する。すなわち、24時間攪拌後に溶解せずに残った樹脂分について、濾別等を行い、水で洗浄し、乾燥後の重量を求め、溶解性試験にかける前における元のバインダー樹脂の重量からの減少率(%):(もとの重量−溶解性試験後の重量)/(もとの重量)で評価する。また、ペレット化されていなくても、5mm角以下の任意の形状の成形品であっても、アルカリ水への溶解性を示す場合には、上記アルカリ水可溶性樹脂の範囲である。
【0035】
アルカリ水可溶性樹脂を用いることにより、適度な疎水性によってゲルブロックが適度に抑制でき、適度な親水性によって過度なゲルブロック抑制を防止でき、従って、本発明の効果が十分に発現できる。
アルカリ水可溶性樹脂としては、例えば、カルボン酸基、スルホン酸基、ホスホン酸基等の置換基を有する樹脂;フェノール性ヒドロキシル基を含むノボラック樹脂;ポリビニルフェノール樹脂;等の1種または2種以上を用いることができる。中でも、アルカリ水に対する溶解性や経済性、樹脂組成物の各種物性等に優れる点で、α,β−不飽和カルボン酸系単量体とα,β−不飽和カルボン酸系単量体以外のビニル系単量体とを共重合して得られる樹脂が好適である。なお、カルボン酸基を有する樹脂であるヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、セルロースアセテートヘキサヒドロフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースヘキサヒドロフタレート等のセルロース誘導体を用いることもできる。
【0036】
α,β−不飽和カルボン酸系単量体としては、具体的には、例えば、アクリル酸、メタクリル酸等のα,β−不飽和モノカルボン酸;イタコン酸、マレイン酸、フマル酸等のα,β−不飽和ジカルボン酸;無水マレイン酸、無水イタコン酸等のα,β−不飽和ジカルボン酸無水物;マレイン酸モノエステル、フマル酸モノエステル、イタコン酸モノエステル等のα,β−不飽和ジカルボン酸モノエステル;等が挙げられるが、特に限定されない。これらα,β−不飽和カルボン酸系単量体は、1種のみを用いてもよいし、2種以上を併用してもよい。上記例示のα,β−不飽和カルボン酸系単量体のうち、アクリル酸、メタクリル酸が、本発明の効果を十分に発現させる点で特に好ましい。
【0037】
ビニル系単量体としては、具体的には、例えば、アクリル酸メチル、アクリル酸エチル、アクリル酸プロピル、アクリル酸ブチル、アクリル酸ステアリル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸ブチル、メタクリル酸ステアリル等の、炭素数1〜18の1価アルコールと(メタ)アクリル酸とのエステル;アクリロニトリル、メタクリロニトリル等のニトリル基含有ビニル系単量体;アクリルアミド、メタクリルアミド等のアミド基含有ビニル系単量体;アクリル酸ヒドロキシエチル、メタクリル酸ヒドロキシプロピル等の水酸基含有ビニル系単量体;スチレン、α−メチルスチレン等の芳香族ビニル系単量体;酢酸ビニル等の脂肪族ビニル系単量体;アリルエーテル類;マレイン酸モノアルキルエステル、マレイン酸ジアルキルエステル等のマレイン酸誘導体;フマル酸モノアルキルエステル、フマル酸ジアルキルエステル等のフマル酸誘導体;マレイミド、N−メチルマレイミド、N−ステアリルマレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド等のマレイミド誘導体;イタコン酸モノアルキルエステル、イタコン酸ジアルキルエステル、イタコンアミド類、イタコンイミド類、イタコンアミドエステル類等のイタコン酸誘導体;エチレン、プロピレン等のアルケン類;ブタジエン、イソプレン等のジエン類;等が挙げられるが、特に限定されない。これらビニル系単量体は、1種のみを用いてもよいし、2種以上を併用してもよい。上記例示のビニル系単量体のうち、(メタ)アクリル酸エステルが、本発明の効果を十分に発現させる点で特に好ましい。
【0038】
α,β−不飽和カルボン酸系単量体およびビニル系単量体の合計量に占めるα,β−不飽和カルボン酸系単量体の割合は、9重量%以上であることがより好ましく、9重量%〜40重量%の範囲内がさらに好ましい。α,β−不飽和カルボン酸系単量体の割合を9重量%以上にすることにより、本発明の効果を十分に発現させることができる。
α,β−不飽和カルボン酸系単量体とビニル系単量体とを共重合することにより、アルカリ水可溶性樹脂が得られる。共重合方法、つまり、アルカリ水可溶性樹脂の製造方法は、特に限定されず、公知の方法を適用すればよい。
アルカリ水可溶性樹脂の酸価は、本発明の効果を十分に発現させる点で、15mgKOH/g以上であることが好ましく、30mgKOH/g以上であることがより好ましく、50mgKOH/g以上であることがさらに好ましく、70mgKOH/g以上であることが特に好ましく、70mgKOH/g〜500mgKOH/gの範囲内であることが最も好ましい。
【0039】
アルカリ水可溶性樹脂は、示差走査熱量測定(DSC(differential scanning calorimetry))によって測定されるガラス転移温度が、−80℃〜120℃の範囲内であることが好ましく、−30℃〜20℃の範囲内に低温側のガラス転移温度を有する一方、40℃〜100℃の範囲内に高温側のガラス転移温度を有していることがさらに好ましい。
疎水性ポリビニルアルコールとしては、例えば、ケン化度45モル%以下のクラレLMポリマー、LM−10HD、LM−15、LM−20、LM−25などが挙げられる。これらは、ポリビニルアルコール濃度2重量%で40℃で攪拌するとき、メタノールやエタノールに可溶である。
【0040】
アルコール可溶性ナイロンとしては、例えばナガセケムテックス製のアルコール可溶性ナイロンとして販売されているトレジンF30K、MF−30、EF−30T等のN−メトキシメチル化ナイロンなどが挙げられる。
前記無機粒子としては、例えば、日産化学製のスノーテックスC等のコロイダルシリカ分散液、日本アエロジル製のアエロジル#200等のシリカ微粒子、二酸化チタン微粒子、疎水性シリカR972などが挙げられ、本発明の効果を十分に発現させる点で、好ましくは、コロイダルシリカ分散液である。このような無機粒子を熱可塑性樹脂と併用することにより、本発明の吸水性樹脂粒子を静置水ゲル化剤として用いた場合に、吸水性樹脂粒子の沈降性制御が容易になり、静置水のゲル化が速やかに起こる。なお、このような無機粒子は、比重調整成分として作用することもある。
【0041】
熱可塑性樹脂および無機粒子の使用量は、熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜1.0重量%が好ましく、より好ましくは0.05〜0.5重量%、さらに好ましくは0.1〜0.3重量%であり、無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%であることが好ましい。より好ましくは、熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.05〜0.5重量%であり、無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%である。このような量を用いることにより、十分な吸収速度を維持でき、例えば、Vortex吸収速度(JIS K7224−1996)を十分に発現できる。Vortex吸収速度の測定に関する詳細は後述する。
【0042】
本発明にかかる吸水性樹脂粒子が有する表面処理層は、前記熱可塑性樹脂および必要により前記無機粒子で処理して得られるが、かかる表面処理は、前記熱可塑性樹脂および必要により前記無機粒子を含む水性液(表面処理剤)によってなされることが好ましい。
本発明にかかる吸水性樹脂粒子は、上記表面処理層を有するので、通常の吸水性樹脂粉末において良く行われる表面架橋処理は必ずしも必要ではなく、表面架橋処理を行っていない吸水性樹脂粉末を用いた場合には、もとの吸水性樹脂粉末が有する吸収性能が損なわれにくいという利点がある。
なお、ここでいう表面処理層とは、必ずしも吸水性樹脂粒子が前記熱可塑性樹脂および無機粒子で均一に覆われている場合のみではなく、部分的に覆われていたり、付着していたり、粒子同士が結合造粒されている場合も含む。
【0043】
本発明にかかる吸水性樹脂粒子は、本発明の効果を十分に発現させるため、好ましくは水分含有量が0〜15重量%であり、より好ましくは水分含有量が1〜15重量%であり、さらに好ましくは2〜12重量%、特に好ましくは3〜9重量%である。なお、水分含有量の測定は、105℃で3時間の乾燥減量によって求めた。
本発明にかかる吸水性樹脂粒子は、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である。静置水ゲル化時間の測定に関する詳細は後述する。
【0044】
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が、好ましくは1秒を超えて100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が、3600秒以内に測定できる限りにおいて、表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下であることが好ましく、より好ましくは60%以下、さらに好ましくは50%以下、特に好ましくは40%以下である。静置水ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して70%を超えると、不十分な処理状態となり、静置水ゲル化時間に大きな改善が認められないために好ましくない。
【0045】
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して80%以下である場合、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、好ましくは0.05〜0.5重量%であり、より好ましくは0.1〜0.4重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であることがより好ましい。さらに好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.1〜0.3重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.1〜4重量%である。これにより、本発明の効果をより一層発揮させることができる。
【0046】
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が、120秒以内であることが好ましい。静置水45度傾斜ゲル化時間の測定に関する詳細は後述する。
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水45度傾斜ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
本発明にかかる吸水性樹脂粒子は、静置水ゲル化時間が、3600秒以内に測定できる限りにおいて、静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して0.1%を超えて80%以下であることが好ましく、より好ましくは60%以下、さらに好ましくは50%以下、特に好ましくは40%以下である。静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して80%を超えると、不十分な処理状態となり、静置水45度傾斜ゲル化時間に大きな改善が認められないために好ましくない。
【0047】
つまり、処理後のゲル化時間が、少なくとも20%短縮されるということである。また、測定ゲル化時間を3600秒以内に限定したのは、便宜的なもので、3600秒以上のものは、3600秒としてゲル化時間の短縮割合を求めても構わない。
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して80%以下である場合、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、好ましくは0.05〜0.5重量%であり、より好ましくは0.1〜0.4重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であることがより好ましい。さらに好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.1〜0.3重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.1〜4重量%である。これにより、本発明の効果をより一層発揮させることができる。
【0048】
本発明にかかる吸水性樹脂粒子は、前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内であることが好ましく、より好ましくは5秒以内、さらに好ましくは1秒以内である。静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が10秒を超えると、完全ゲル化時間とみかけのゲル化時間との差が大きくなってしまうので好ましくない。
本発明にかかる低発塵性で高吸収速度の吸水性樹脂粒子は、また、前記表面処理前の吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、Vortex吸収速度が5秒以下であることが好ましい。
【0049】
〔吸水性樹脂粒子の製造方法〕
本発明にかかる吸水性樹脂粒子の製造方法は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩を通して解砕する。好ましくは、目開き500μm以下の篩を通して解砕する。
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および無機粒子については、前述した通りである。
【0050】
水性液状態とは、前記熱可塑性樹脂および必要により無機粒子を水のみと共に水性液状態とする形態でもよいし、前記熱可塑性樹脂および必要により無機粒子を親水性溶媒のみと共に水性液状態とする形態でもよいし、前記熱可塑性樹脂および必要により無機粒子を親水性溶媒と水とともに水性液状態とする形態でもよい。また、可塑剤として不揮発性の親水性有機溶媒を併用してもよい。
親水性溶媒としては、例えば、メタノール、エタノール、プロパノール、イソプロパノール、アセトン、メチルエチルケトンなどが挙げられる。特に、メタノール、エタノール、メチルエチルケトンが好ましい。
【0051】
可塑剤としての不揮発性の親水性有機溶媒としては、例えば、プロピレングリコール、ジプロピレングリコール、エチレングリコール、ジエチレングリコール、これらのモノアルキルエーテル、液状ポリエチレングリコールやそのアルキルエーテル、グリセリン、ジグリセリン、ポリグリセリンなどが挙げられる。特に、プロピレングリコール、グリセリンが好ましい。可塑剤としての不揮発性の親水性有機溶媒を併用することにより、本発明の吸水性樹脂粒子を静置水ゲル化剤として用いた場合に、静置水のゲル化が速やかに起こる。
上記全水性液中の前記熱可塑性樹脂および必要により無機粒子の含有割合は、好ましくは1〜60重量%、より好ましくは5〜50重量%、さらに好ましくは10〜40重量%である。水の割合は、好ましくは0〜90重量%、より好ましくは10〜80重量%、さらに好ましくは40〜70重量%である。
【0052】
前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理する形態としては、特に限定されないが、例えば、
(1)熱可塑性樹脂と親水性溶媒とを含む水性液を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(2)熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(3)無機粒子と親水性溶媒とを含む水性液を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(4)無機粒子と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(5)熱可塑性樹脂と無機粒子と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(6)熱可塑性樹脂と無機粒子と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(7)熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加し、溶媒除去後に無機粒子を添加する形態(水を水性液中に添加してもよいし、吸水性樹脂粉末の添加後に添加してもよいし、無機粒子の添加後に添加してもよい)、
(8)吸水性樹脂粉末に無機粒子を添加混合後、熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
が、好ましい形態として挙げられる。
【0053】
表面処理の際の温度は、特に限定されないが、好ましくは材料温度で室温〜100℃、より好ましくは室温〜80℃、さらに好ましくは室温〜50℃である。
本発明にかかる吸水性樹脂粒子の製造方法においては、前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させる。静置させる際の温度は、材料温度で好ましくは20〜100℃、より好ましくは40〜100℃、さらに好ましくは40〜80℃である。
本発明にかかる吸水性樹脂粒子の製造方法においては、前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩、好ましくは500μm以下の篩ないし相当品を通るまで解砕する。解砕する際に通す篩の目開きは、粒子径と目的とする用途によって適宜選択すればよい。
【0054】
〔吸水性樹脂粒子組成物〕
本発明にかかる吸水性樹脂粒子組成物は、本発明の吸水性樹脂粒子(表面処理された吸水性樹脂粒子)30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
本発明にかかる吸水性樹脂粒子組成物は、静置水ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
【0055】
本発明にかかる吸水性樹脂粒子組成物は、静置水45度傾斜ゲル化時間が120秒以内であることが好ましい。
本発明にかかる吸水性樹脂粒子組成物は、静置水45度傾斜ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水45度傾斜ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
吸水性樹脂粒子組成物中の吸水性樹脂粒子および他の吸水性樹脂粒子の含有割合が上記範囲を外れると、静置水45度傾斜ゲル化時間が長くなり、且つ、静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が大きくなるために好ましくない。
【0056】
吸水性樹脂粒子組成物中の本発明の吸水性樹脂粒子の含有割合は、好ましくは30〜99重量%、より好ましくは40〜99重量%、さらに好ましくは60〜99重量%、特に好ましくは80〜99重量%である。
吸水性樹脂粒子組成物中の他の吸水性樹脂粒子の含有割合は、好ましくは1〜60重量%、より好ましくは1〜40重量%、さらに好ましくは1〜20重量%である。
上記割合は、吸水性樹脂粒子組成物を100重量%としたときの比率である。なお、本発明の物性を損なわない範囲において、添加物等の他の物質を、例えば、0〜20重量%配合することができる。添加物としては、公知の消臭剤、抗菌剤、フラボノイド類、キレート剤等が挙げられる。
【0057】
他の吸水性樹脂粒子としては、例えば、市販の衛生材料用吸水性樹脂であり、日本触媒製の「アクアリックCA」、住友精化製の「アクアキープ」、ストックハウゼン社製の「Favor」、ダウ社製の「ドライテック」などが挙げられる。
本発明にかかる吸水性樹脂粒子組成物の特徴、物性等は、前述した本発明の吸水性樹脂粒子と同様である。
〔静置水ゲル化剤〕
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物を含む。
【0058】
本発明にかかる静置水ゲル化剤中の、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物の含有割合は、好ましくは80〜100重量%、より好ましくは90〜100重量%、さらに好ましくは95〜100重量%である。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物以外に、その他の成分として、0〜20重量%の範囲で、キレート剤、殺菌剤、殺ウイルス剤、タンパク質分解酵素、血液凝固阻害剤などを含んでいても良い。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物を含むことにより、固化性能の向上、安全性の向上といった優れた効果を発現できる。
【0059】
なお、本発明の静置水ゲル化剤の使用対象としては、例えば、手術時の血液や血液と洗浄液との混合液等の医療廃液、重金属等を含有する廃液がある。
〔水性廃液の処理方法〕
本発明にかかる水性廃液の処理方法は、水性廃液の入った貯槽に本発明の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる。
本発明の静置水ゲル化剤は、前述の通り、静置水に一括添加した時の静置水ゲル化時間が短く、静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が短く、容器中の水性液を速やかにゲル化するといった優れた効果を発現できるので、水性廃液の処理に好適である。
【0060】
水性廃液としては、例えば、手術時の血液と洗浄液との混合液等の医療廃液、重金属等を含有する廃液などが挙げられる。
水性廃液の処理に用いる本発明の静置水ゲル化剤の使用量は、水性廃液全量に対して、好ましくは1〜100重量%、より好ましくは2〜20重量%、さらに好ましくは2.5〜5重量%である。水性廃液の処理に用いる本発明の静置水ゲル化剤の使用量が1重量%より少ないと、ゲル化が十分でなかったり、ゲル化しても全体としてのゲル化後の強度が低く、流動性が出やすかったりという問題があり、100重量%より多いと、経済的でないという問題がある。
【0061】
〔その他の用途〕
本発明にかかる吸水性樹脂粒子、本発明にかかる吸水性樹脂粒子組成物は、上述のような構成を有するため、ゲルブロッキングが効果的に防止でき、また、表面架橋の有無に関わらず高い吸収性能(例えば、Vortex吸収速度など)を発揮できるので、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料にも好ましく適用可能である。
また、目開き212〜125μmの篩から選択された少なくとも1つの篩、好ましくは150〜125μmの篩を通過させることにより微粒子化した原料吸水性樹脂(A)を使用した本発明の吸水性樹脂粒子は、Vortex吸収速度に優れ、テープ化工程時の粉塵の飛散等の問題がなくなり、光ケーブルや電力ケーブル用などのケーブル用止水テープに好適に用いることができる。
【0062】
ケーブル用止水テープ用には、本発明の処理後の吸水性樹脂は、目開き125μm以上850μm以下で、好ましくは、目開き150μm以上600μm以下、より好ましくは、目開き150μm以上500μm以下の篩を通過するように解砕して使用することができる。
本発明における表面処理を施した吸水性樹脂粒子(A)を、例えば、850μmの篩を通過するように解砕したものと、本発明における表面処理を施さなかった目開き212〜125μmの篩に相当する少なくとも1つの篩を通過しなかった吸水性樹脂粒子(B)とを混合することにより、低発塵性の衛生材料用吸水性樹脂を得ることができる。このようにして原料吸水性樹脂の微粒子を有効に活用することができる。この場合、原料吸水性樹脂としては、表面架橋は施されているものでも施されていないものでもよい。また、吸水性樹脂(A)と吸水性樹脂粒子(B)のうちの片方が表面架橋処理されていてもよい。表面架橋処理されたものを用いた方が、衛生材料用吸水性樹脂としては好ましい。このように、本発明は、粉砕工程を経て製造される吸水性樹脂の微粉の回収方法としても有用である。
【0063】
なお、本明細書において、篩の目開きについては、JIS標準篩を基準として、相当するASTMに定められた篩や目開きがそれに相当する篩であれば特注品であってもよい。
【実施例】
【0064】
以下に、実施例および比較例によって本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。以下では、便宜上、「重量部」を単に「部」と、「リットル」を単に「L」と記すことがある。また、「重量%」を「wt%」と記すことがある。
実施例および比較例における、測定方法および評価方法を以下に示す。
<静置水ゲル化時間および静置水45度傾斜ゲル化時間の測定>
22℃の0.90重量%塩化ナトリウム水溶液(生理食塩水)100gをガラス製管(マルエムスクリュー管No.8(40X120):40mmφ、高さ120mm)に入れ、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面から2cm上部に設置したものを使用して、試料3.3gを一気に投入し、目視でガラス製管を横から観察してゲル化によって透明な液体部分がなくなるまでの時間を測定し、静置水ゲル化時間(秒)とした。
【0065】
次にガラス製管を45度傾けても流動化しなくなるまでの時間(秒)を測定し、静置水45度傾斜ゲル化時間とした。ここで、流動化しなくなるとは、45度傾けてもガラス管内のゲル最上面が流動あるいは変化せず、そのままの状態であることをいう。
全体がゲル化固定化する時間は、静置水45度傾斜ゲル化時間で表される。
静置水ゲル化時間は、見かけの全体ゲル化時間であり、十分な静置水中での垂直分散性を持たないものであれば、45度傾けた時に流動性が認められる。しかし、最も好ましく改質された垂直分散性を持つものを用いた場合には、静置水ゲル化時間と静置水45度傾斜ゲル化時間とはほぼ一致する。
【0066】
<Vortex吸収速度>
0.90重量%塩化ナトリウム水溶液(生理食塩水)に1000重量部に食品添加物である食用青色1号0.02重量部を添加し、液温22℃に調整した。その生理食塩水50mlを100mlビーカーに計り取り、長さ40mmで太さ8mmの円筒型攪拌子で600rpmで攪拌する中に、試料2.0gを投入し、Vortex吸収速度(秒)を測定した。
終点は、JIS K 7224−1996「高吸水性樹脂の吸水速度試験方法 解説」に記載されている基準に準じ、試料(吸水性樹脂粒子や吸水性樹脂粒子組成物など)が生理食塩水を吸液してスターラーチップを試験液で覆うまでの時間を吸収速度(秒)として測定した。
【0067】
Vortex吸収速度は、撹拌下でのゲル化時間を測定するものである。
<発塵性試験>
試料5gを100mlの円筒形透明ガラス製容器(マルエムサンプル管:内径37mm、高さ102mm)に入れてふたを締め、手で持ち上下に約50cmの巾で激しく10〜20秒で30回振った後に机上に置いた。置いた直後の粉立ちの程度を観察した後、1日放置後のガラス製容器内壁への微粉の付着程度をデジタルカメラ(フラッシュON)で近接撮影し、判定を行った。
判定は以下の5段階で行った。
【0068】
極めて多量(図1参照)
多量(図2参照)
中等度(図3参照)
軽微(図4参照)
ほとんどなし(図5参照)
<含水率>
105℃のオーブン中で3時間乾燥させたときの減量により求めた。
<重量平均粒子径(D50)>
吸水性樹脂粒子や吸水性樹脂粒子組成物を、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmなどのJIS標準ふるいで篩い分けし、残留百分率Rを対数確率紙にプロットした。これにより、R=50重量%に相当する粒子径を重量平均粒子径(D50)として読み取った。
【0069】
重量平均粒子径(D50)を測定する際の分級方法は、吸水性樹脂粒子や吸水性樹脂粒子組成物10.0gを、室温(20〜25℃)、湿度50RH%の条件下で、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmのJIS標準ふるい(THE IIDA TESTING SIEVE:径8cm)に仕込み、振動分級器(IIDA SIEVE SHAKER、TYPE:ES−65型、SER.No.0501)により、5分間、分級を行った。
〔合成例1:アルカリ水可溶性樹脂の調製〕
アルカリ水可溶性樹脂を以下の方法で調製した。
【0070】
すなわち、温度計、攪拌翼、還流冷却器、および滴下装置を備えた容量100Lの槽型反応器に、アクリル酸1.8kg、アクリル酸エチル10.2kg、重合開始剤である2,2´−アゾビス−(2,4−ジメチルバレロニトリル)24g、および、溶媒であるメチルアルコール28kgを仕込んだ。また、滴下装置に、アクリル酸2.7kg、アクリル酸メチル5.4kg、メタクリル酸メチル9.9kg、2,2´−アゾビス−(2,4−ジメチルバレロニトリル)66g、およびメチルアルコール2kgからなる混合溶液を仕込んだ。
上記のメチルアルコール溶液を、窒素ガス雰囲気下、攪拌しながら65℃に加熱し、20分間反応させた。これにより、内容物の重合率を72%に調節した。続いて、内温を65℃に保ちながら、滴下装置から上記の混合溶液を2時間かけて均等に滴下した。滴下終了後、内容物にメチルエチルケトン60kgを混合することにより、アルカリ水可溶性樹脂の25重量%溶液を得た。以下、このアルカリ水可溶性樹脂を「ASP」と称することがある。
【0071】
得られたアルカリ水可溶性樹脂の酸価は117mgKOH/gであった。また、該アルカリ水可溶性樹脂の重量平均分子量Mwは156000、数平均分子量Mnは69000であった。さらに、示差走査熱量機で測定したところ、ガラス転移温度(Tg)が10℃と67℃とに観測された。また、上記樹脂の、25℃のイオン交換水への溶解性は、0.5%未満であり、また、アルカリ水可溶性は100%であった。
〔実施例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液8.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて試料(1)を作製した。重量平均粒子径は210μm、45μm以下の微粉は1.1重量%であり、試料(1)の発塵性試験による判定は「軽微」であった。また、含水率は2.9重量%であった。
【0072】
試料(1)の静置水ゲル化時間は51秒、静置水45度傾斜ゲル化時間は55秒、Vortex吸収速度は26秒であった。
ゲル化時間測定時、試料(1)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gと脱イオン水2gを混合したものを滴下混合した。
【0073】
60℃の恒温器中で1時間熟成させ、500μmの篩を通過させて試料(2)を作製した。重量平均粒子径は205μm、45μm以下の微粉は1.6重量%であり、試料(2)の発塵性試験による判定は「軽微」であった。また、含水率は3.8重量%であった。
試料(2)の静置水ゲル化時間は64秒、静置水45度傾斜ゲル化時間は68秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(2)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの速やかに上部液面まで全体がゲル化した。
〔実施例3〕
脱イオン水の量を6gとした以外は実施例2と同様にして、試料(3)を作製した。
【0074】
試料(3)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は5.7重量%であった。
試料(3)の静置水ゲル化時間は62秒、静置水45度傾斜ゲル化時間は65秒、Vortex吸収速度は26秒であった。
ゲル化時間測定時、試料(3)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの速やかに上部液面まで全体がゲル化した。
〔実施例4〕
脱イオン水の量を10gとした以外は実施例2と同様にして、試料(4)を作製した。
【0075】
試料(4)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は7.6重量%であった。
試料(4)の静置水ゲル化時間は52秒、静置水45度傾斜ゲル化時間は57秒、Vortex吸収速度は25秒であった。
ゲル化時間測定時、試料(4)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの、速やかに上部液面まで全体がゲル化した。
〔実施例5〕
脱イオン水を入れない以外は実施例2と同様にして、試料(5)を作製した。
【0076】
試料(5)の発塵性試験による判定は「軽微」であった。また、含水率は2.8重量%であった。
試料(5)の静置水ゲル化時間は108秒、静置水45度傾斜ゲル化時間は111秒、Vortex吸収速度は28秒であった。
ゲル化時間測定時、試料(5)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの、速やかに上部液面まで全体がゲル化した。
〔比較例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)(比較試料(1))をそのまま測定をした。
【0077】
比較試料(1)の発塵性試験による判定は「中等度」であった。また、含水率は2.6重量%であった。
比較試料(1)の静置水ゲル化時間は228秒、静置水45度傾斜ゲル化時間は300秒、Vortex吸収速度は19秒であった。
ゲル化時間測定時、比較試料(1)は、全量が一旦沈降した後、ゆっくりと上部液面方向へのゲル化が進行したが、全体がゲル化するのに時間を要した。
〔比較例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)(比較試料(2))をそのまま測定した。
【0078】
比較試料(2)の発塵性試験による判定は「中等度」であった。また、含水率は2.7重量%であった。
比較試料(2)の静置水ゲル化時間は135秒、静置水45度傾斜ゲル化時間は147秒、Vortex吸収速度は24秒であった。
ゲル化時間測定時、比較試料(2)は、全量が一旦沈降した後、ゆっくりと上部液面方向へのゲル化が進行したが、全体がゲル化するのに時間を要した。
〔比較例3〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液64.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて比較試料(3)を作製した。
【0079】
比較試料(3)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は2.7重量%であった。
比較試料(3)の静置水ゲル化時間は240秒、静置水45度傾斜ゲル化時間は420秒、Vortex吸収速度は35秒であった。
ゲル化時間測定時、比較試料(3)は、液面に浮いてゲルブロッキングを起こしている状態ではなく非常にゆっくりとではあるが少しずつ膨潤した部分から沈降ゲル化したが、全体のゲル化に時間を要した。
〔実施例6〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%シリカ水溶液)20gを混合し、さらに撹拌混合した。40℃の恒温器中で1時間熟成させ、500μmの篩を通過させて試料(6)を作製した。
【0080】
試料(6)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は11.5重量%であった。
試料(6)の静置水ゲル化時間は35秒、静置水45度傾斜ゲル化時間は35秒、Vortex吸収速度は20秒であった。
ゲル化時間測定時、試料(6)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例7〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%シリカ水溶液)20gを混合し、さらに撹拌混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を0.5g添加して均一に混合し、試料(7)を作製した。
【0081】
試料(7)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は10.8重量%であった。
試料(7)の静置水ゲル化時間は57秒、静置水45度傾斜ゲル化時間は57秒、Vortex吸収速度は14秒であった。
ゲル化時間測定時、試料(7)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例8〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を1.0g添加して均一に混合し、試料(8)を作製した。
【0082】
試料(8)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は3.3重量%であった。
試料(8)の静置水ゲル化時間は66秒、静置水45度傾斜ゲル化時間は66秒、Vortex吸収速度は15秒であった。
ゲル化時間測定時、試料(8)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔参考例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液2.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を0.5g添加して均一に混合し、参考試料(1)を作製した。
【0083】
参考試料(1)の発塵性試験による判定は「軽微」であった。また、含水率は3.1重量%であった。
参考試料(1)の静置水ゲル化時間は126秒、静置水45度傾斜ゲル化時間は156秒、Vortex吸収速度は14秒であった。
ゲル化時間測定時、参考試料(1)は、全量が一旦沈降しゆっくりと液面へ向かって膨潤し全体がゲル化した。
上記参考例1は、熱可塑性樹脂の使用量が少ない場合の一例である。
〔実施例9〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)と耐塩性吸水性ポリマー(アクアリックCS−6、(株)日本触媒製)との1:1混合物200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を1.0g添加して均一に混合し、試料(9)を作製した。
【0084】
試料(9)の発塵性試験による判定は「軽微」であった。また、含水率は4.1重量%であった。
試料(9)の静置水ゲル化時間は36秒、静置水45度傾斜ゲル化時間は36秒、Vortex吸収速度は6秒であった。
ゲル化時間測定時、試料(9)は、液面に浮いてから下部の膨潤した部分から順次膨潤沈降し、速やかに速やかに全体がゲル化した。
〔参考例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)と耐塩性吸水性ポリマー(アクアリックCS−6、(株)日本触媒製)との1:1混合物200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて参考試料(2)を作製した(アエロジル200を1.0g添加しなかった以外は実施例9に同じ)。
【0085】
参考試料(2)の発塵性試験による判定は「軽微」であった。また、含水率は4.0重量%であった。
参考試料(2)の静置水ゲル化時間は594秒、静置水45度傾斜ゲル化時間は594秒、Vortex吸収速度は11秒であった。
ゲル化時間測定時、参考試料(2)は、全量が液面に浮いてからその下部の膨潤した部分から非常にゆっくりと膨潤沈降しゲル化した。
〔製造例1〕
75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度33重量%)に、トリメチロールプロパントリアクリレート1.2gを溶解し、反応液とした。次に、この反応液を窒素ガス雰囲気下で30分間脱気した。ついで、シグマ型羽根を2本有する内容積10Lのジャケット付きステンレス製双腕型ニーダーに蓋を付けて形成した反応器に、上記反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。続いて、反応液を撹拌しながら、過硫酸ナトリウム2.46gおよびL−アスコルビン酸0.10gを添加したところ、およそ1分後に重合が開始した。そして、30〜80℃で重合を行い、重合開始から60分後に含水ゲル状重合体を取り出した。得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を目開き500μmのステンレス製ネット上に広げ、150℃で90分間熱風乾燥した。ついで、乾燥物をハンマーミルを用いて粉砕し、さらに目開き500μmの篩で分級することにより、不定形破砕状の吸水性樹脂前駆体(a)を得た。
【0086】
得られた吸水性樹脂前駆体(a)100重量部に、グリセリン0.5重量部と、水2重量部と、イソプロピルアルコール1重量部とからなる表面架橋剤を混合した。上記の混合物を210℃で45分間加熱処理することにより吸水性樹脂粉末(1)を得た。重量平均粒子径は169μm、45μm以下の微粉は10.5重量%であった。
〔実施例10〕
製造例1で得られた吸水性樹脂粉末(1)200gをクッキングカッターに入れ、撹拌混合しながら、下記に示す処理液Aの30gを滴下混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(10)を得た。重量平均粒子径は210μm、45μm以下の微粉は1.8重量%であった。
【0087】
試料(10)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は6.5重量%であった。
試料(10)の静置水ゲル化時間は105秒、静置水45度傾斜ゲル化時間は105秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(10)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
処理液A:
25%濃度のASPのメタノール・(MEK)溶液 3.0g
1規定NaOH水溶液 2.0g
脱イオン水 5.0g
プロピレングリコール 5.0g
スノーテックスC(日産化学製コロイダルシリカ分散液) 15.0g
〔実施例11〕
製造例1で得られた吸水性樹脂粉末(1)200gをクッキングカッターに入れ、撹拌混合しながら、下記に示す処理液Bの25.8gを混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(11)を得た。
【0088】
試料(11)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は5.6重量%であった。
試料(11)の静置水ゲル化時間は114秒、静置水45度傾斜ゲル化時間は114秒、Vortex吸収速度は32秒であった。
ゲル化時間測定時、試料(11)は、液面に浮いてから膨潤した部分から順次沈降しゲル化した。
処理液B:
25%濃度のASPのメタノール・MEK溶液 1.92g
1規定NaOH水溶液 1.28g
脱イオン水 3.2g
プロピレングリコール 3.8g
スノーテックスC 15.6g
〔実施例12〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら、下記に示す処理液Cの280gを噴霧混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機で1時間熟成した後、目開き500μmのメッシュを通過させて試料(12)を得た。
【0089】
試料(12)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は3.1重量%であった。
試料(12)の静置水ゲル化時間は112秒、静置水45度傾斜ゲル化時間は112秒、Vortex吸収速度は32秒であった。
ゲル化時間測定時、試料(12)は、液面に浮いてから順次膨潤沈降し全体がゲル化した。
処理液C:
25%濃度のASPのメタノール・MEK溶液 19.2g
1規定NaOH水溶液 12.8g
脱イオン水 32g
グリセリン 60g
スノーテックスC 156g
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、試料(12)の33gを投入し、全体がゲル化固定化する時間を測定したところ、135秒であった。
【0090】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、試料(12)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、190秒および132秒であった。
〔実施例13〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら、下記に示す処理液Dの155gを噴霧混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(13)を得た。
【0091】
試料(13)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は1.9重量%であった。
試料(13)の静置水ゲル化時間は98秒、静置水45度傾斜ゲル化時間は98秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(13)は、液面に浮いてから順次膨潤沈降し全体がゲル化した。
処理液D:
25%濃度のASPのメタノール・MEK溶液 11.5g
1規定NaOH水溶液 7.7g
脱イオン水 19.2g
プロピレングリコール 23.0g
スノーテックスC 93.6g
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、試料(13)の33gを投入し、全体がゲル化固定化する時間を測定したところ、105秒であった。
【0092】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、試料(13)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、147秒および119秒であった。
〔比較例4〕
製造例1で得られた吸水性樹脂粉末(1)をそのまま使用し、比較試料(4)とした。
比較試料(4)の発塵性試験による判定は「多量」であった。また、含水率は0.3重量%であった。
【0093】
比較試料(4)の静置水ゲル化時間は約1400秒、静置水45度傾斜ゲル化時間は約3900秒、Vortex吸収速度は31秒であった。
ゲル化時間測定時、比較試料(4)は、一旦全量が容器底部に沈降してから極めてゆっくりと上部液面へとゲル化が進行し全体がゲル化した。
〔比較例5〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら脱イオン水200gを噴霧した。生成物を取り出しバットにいれて70℃で相対湿度60%の恒温高湿機中で2時間熟成した。これを目開き500μmのメッシュを通過させて比較試料(5)を得た。重量平均粒子径は193μm、45μm以下の微粉は6.6重量%であった。
【0094】
比較試料(5)の発塵性試験による判定は「中等度」であった。また、含水率は2.5重量%であった。
比較試料(5)の静置水ゲル化時間は252秒、静置水45度傾斜ゲル化時間は335秒、Vortex吸収速度は18秒であった。
ゲル化時間測定時、比較試料(5)は、一旦全量が容器底部に沈降してから非常にゆっくりと上部液面へとゲル化が進行し全体がゲル化した。
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、比較試料(5)の33gを投入し、全体がゲル化固定化する時間を測定したところ、535秒であった。
【0095】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、比較試料(5)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、639秒および411秒であった。
〔実施例14〕
実施例6で得られた試料(6)70gに市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)30gを混合することにより、試料(14)を作製した。
【0096】
試料(14)の発塵性試験による判定は「軽微」であった。また、含水率は8.2重量%であった。
試料(14)の静置水ゲル化時間は92秒、静置水45度傾斜ゲル化時間は92秒、Vortex吸収速度は22秒であった。
ゲル化時間測定時、試料(14)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例15〕
実施例6で得られた試料(6)80gに市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−H3、(株)日本触媒製)20gを混合することにより、試料(15)を作製した。
【0097】
試料(15)の発塵性試験による判定は「軽微」であった。また、含水率は7.9重量%であった。
試料(15)の静置水ゲル化時間は85秒、静置水45度傾斜ゲル化時間は88秒、Vortex吸収速度は29秒であった。
ゲル化時間測定時、試料(15)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔製造例2〕
75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度33重量%)に、ポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数10)5.44gを溶解し反応液とした。次に、この反応液を窒素ガス雰囲気下で30分間脱気した。ついで、シグマ型羽根を2本有する内容積10Lのジャケット付きステンレス製双腕型ニーダーに蓋を付けて形成した反応器に、上記反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。続いて、反応液を撹拌しながら、過硫酸ナトリウム2.46gおよびL−アスコルビン酸0.10gを添加したところ、およそ1分後に重合が開始した。そして、30〜80℃で重合を行い、重合開始から60分後に含水ゲル状重合体を取り出した。得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を目開き500μmのステンレス製ネット上に広げ、150℃で90分間熱風乾燥した。ついで、乾燥物をロールミルを用いて粉砕し、目開き850μmの篩を通過する吸水性樹脂粉末(2)を得た。重量平均粒子径は410μm、45μm以下の微粉は0.5重量%であった。さらに目開き150μmの篩で分級することにより、重量平均粒子径115μmの不定形破砕状の吸水性樹脂粉末(3)を得た。45μm以下の微粉は、7.6重量%であった。目開き150μmの篩上に残った吸水性樹脂粉末100重量部に、グリセリン0.5重量部と、水2重量部と、イソプロピルアルコール1重量部とからなる表面架橋剤を混合した。上記の混合物を210℃で45分間加熱処理することにより吸水性樹脂粉末(4)を得た。重量平均粒子径は436μm、45μm以下の微粉は0.1重量%であった。
【0098】
〔比較例6〕
製造例2で得られた吸水性樹脂粉末(3)をそのまま比較試料(6)として使用し、測定した。
比較試料(6)の静置水ゲル化時間も静置水45度傾斜ゲル化時間も共に観測できなかった(86400秒以上、つまり24時間後も、ママコができており部分的にしかゲル化せず流動状態であった)。Vortex吸収速度は15秒(ただしママコ生成)であった。比較試料(6)の発塵性試験による判定は「多量」であった。また、含水率は2.9重量%であった。
【0099】
〔実施例16〕
製造例2で得られた吸水性樹脂粉末(3)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%水溶液)30gを混合しさらに撹拌混合した。60℃の恒温器中で1時間熟成させ、850μmの篩を通過させて試料(16)を作製した。重量平均粒子径は415μm、45μm以下の微粉は0.0重量%であった。
試料(16)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は7.4重量%であった。
【0100】
試料(16)の静置水ゲル化時間は30秒、静置水45度傾斜ゲル化時間は30秒、Vortex吸収速度は3秒であった。静置水ゲル化時間、Vortex吸収速度、ともに、比較例8に比べて著しい改善を示した。
ゲル化時間測定時、試料(16)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例17〕
実施例16で得られた試料(16)20gを、ブチルゴム20%含有のトルエン溶液に分散させて混合し、不織布基材に塗布し、止水テープを作成した。
【0101】
比較試料(6)を使用して同様に塗布した場合には、試料の添加時に粉塵が発生したが、試料(16)を使用した場合には、試料の添加時に粉塵の飛散が全くなかった。
純水を用いた止水性能も良好であった。
〔実施例18〕
実施例16で得られた試料(16)10gに製造例2で得られた表面架橋された吸水性樹脂粉末(4)90gを混合して試料(18)を得た。重量平均粒子径は432μm、45μm以下の微粉は0.0重量%であった。発塵性試験による判定も「ほとんどなし」であった。微粉の極めて少ない良好な衛生材料用吸水性樹脂が得られた。
【0102】
【表1】

【0103】
【表2】
【0104】
【表3】
【産業上の利用可能性】
【0105】
本発明にかかる吸水性樹脂粒子や吸水性樹脂粒子組成物は、静置状態の水性液全体を速やかにゲル化することが可能、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立することができるので、塗料廃液、メッキ廃液、放射性廃液、医療廃液などの水性廃液のゲル化剤に好適である。また、ケーブル用止水剤(止水テープなど)や、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料にも好ましく適用可能である。
【図面の簡単な説明】
【0106】
【図1】発塵性試験における「極めて多量」の判定となったサンプルの一例を示す写真図。
【図2】発塵性試験における「多量」の判定となったサンプルの一例を示す写真図。
【図3】発塵性試験における「中等度」の判定となったサンプルの一例を示す写真図。
【図4】発塵性試験における「軽微」の判定となったサンプルの一例を示す写真図。
【図5】発塵性試験における「ほとんどなし」の判定となったサンプルの一例を示す写真図。
【技術分野】
【0001】
本発明は、特定の表面処理層を有する吸水性樹脂粒子とその製造方法、その吸水性樹脂粒子を用いて得られる吸水性樹脂粒子組成物、ならびにこれら吸水性樹脂粒子と吸水性樹脂粒子組成物の用途に関する。用途としては特に、静置水ゲル化剤、およびそれを用いた水性廃液の処理が好ましく挙げられる。
【背景技術】
【0002】
塗料廃液、メッキ廃液、放射性廃液、医療廃液などの水性廃液を処理する方法は、環境問題が重視される昨今、これらを扱う分野において極めて重要な技術であり、これまでに様々な方法が知られている(例えば、特許文献1など参照)。
本発明者は、これまでに、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料に好適な吸水性樹脂について長年研究を重ねてきた。今回、本発明者は、従来はそれ自身に水性液を吸収させる(すなわち、基材等に固定化された吸水性樹脂に外部から水性液を吸収させる)ことを主たる目的とした吸水性樹脂についての知見を活かし、逆に、水性廃液の貯留容器中に吸水性樹脂を粒子の状態で添加することによって水性廃液全体を速やかにゲル化させて廃棄することができないか考えた。特に、これら水性廃液は人体に有害な物質(ウイルスなど)を含んでいることが多いので、攪拌等による飛散を防ぐために、水性廃液を「静置状態」で吸水性樹脂粒子を添加して水性廃液全体を速やかにゲル化させる技術について検討を開始した。
【0003】
まず、従来から良く知られているいくつかの吸水性樹脂粒子(特に、衛生材料に好適とされる吸水性樹脂粒子)について、静置状態の水性廃液が入った貯留容器中に添加したところ、(1)吸水性樹脂粒子のほとんど全てがすぐに容器下部に沈降してしまい、容器下部から水性廃液の50〜80%ほどまでは比較的速やかにゲル化するものの、液面までは完全にゲル化が進行しないという問題や、(2)液面に吸水性樹脂粒子のほとんど全てが浮いた状態になって液面で吸水性樹脂粒子同士のゲルブロッキングが起こり、吸水性樹脂粒子が容器下部にまで行き渡らなくなってしまい、容器下部〜中部までは完全にゲル化が進行しないという問題が生じた。これらの問題は、貯留容器が縦長(容器の平均断面積(円形換算直径)に対する鉛直方向の長さの比が大きい)になればなるほど顕著に現れた。
【特許文献1】特開2002−119853号公報
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明が解決しようとする課題は、静置状態の水性液全体を速やかにゲル化することが可能な、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな自己分散による高吸収速度とを両立した新規な吸水性樹脂粒子および吸水性樹脂粒子組成物を提供することにある。また、そのような吸水性樹脂粒子の製造方法、吸水性樹脂粒子や吸水性樹脂粒子組成物の用途をも提供することにある。さらに、特定処理により微粉末状態とした吸水性樹脂粉末を表面処理することによる、高いVortex吸収速度を実現する、低発塵性の吸水性樹脂粒子を提供することにある。
【課題を解決するための手段】
【0005】
吸水性樹脂粒子同士のゲルブロッキングを防止するための表面処理技術として、吸水性樹脂粉末表面を熱可塑性樹脂で被覆する技術(例えば、特開平2−242858号公報、特開2003−301019号公報など参照)が知られている。しかし、従来公知のこれらの方法においては、ゲルブロッキング防止を達成するために吸収速度を犠牲にしており、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とは両立できていないことが判った。
本発明者は、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立するための技術手段について、鋭意検討を行った。
【0006】
その結果、原料となる吸水性樹脂粉末の表面を、極めて少量の、親水性と疎水性を有する特定の熱可塑性樹脂および必要により無機粒子で処理して、特定の表面処理層を形成させることにより、得られる吸水性樹脂粒子の表面上において適度な親水性と適度な疎水性との両立が実現でき、適度なゲルブロッキング抑制が発現できるとともに静置状態の水性液に添加した場合の適度な沈降速度を発現でき、その結果、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立できることを見出した。
さらに、熱可塑性樹脂としてアルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種を用いると、適度な疎水性によってゲルブロックが適度に抑制でき、適度な親水性によって過度なゲルブロック抑制を防止でき、従って、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度との両立がより一層達成できることも見出した。
【0007】
すなわち、本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
前記表面処理された吸水性樹脂粒子は、水分含有量が1〜15重量%であることが好ましい。
【0008】
前記表面処理された吸水性樹脂粒子は、前記ゲル化するまでの時間(静置水ゲル化時間)が80秒以内であることが好ましい。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記表面処理前の原料吸水性樹脂粉末の粒度が、目開き600μmの篩、好ましくは目開き500μmの篩を通過するものであり、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%である。
【0009】
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であり、得られた上記表面処理された吸水性樹脂粒子の静置水ゲル化時間が原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂がアルコール可溶性であり、重量平均分子量が20000〜500000である。
本発明にかかる吸水性樹脂粒子は、好ましくは、前記熱可塑性樹脂が、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種である。
【0010】
本発明にかかる吸水性樹脂粒子組成物は、本発明の表面処理された吸水性樹脂粒子30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子あるいは本発明の吸水性樹脂粒子組成物を含む。
【0011】
本発明にかかる吸水性樹脂粒子の製造方法は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩、好ましくは目開き500μm以下の篩を通して解砕する。
本発明にかかる水性廃液の処理方法は、水性廃液の入った貯槽に本発明の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる。
本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、前記表面処理前の原料吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、Vortex吸収速度が5秒以下であることを特徴とする。
【0012】
本発明にかかるケーブル用止水テープは、本発明の吸水性樹脂粒子を用いる。
本発明にかかる衛生材料用吸水性樹脂は、目開き212〜125μmの少なくとも1つの篩を通過しない吸水性樹脂粒子と、本発明の吸水性樹脂粒子を混合してなる。
なお、本発明の吸水性樹脂粒子は吸水性樹脂粒子組成物と呼ぶことがある。本発明の吸水性樹脂粒子以外の吸水性樹脂粒子、その他の添加剤との混合物も吸水性樹脂粒子組成物と呼ぶことがある。
【発明の効果】
【0013】
本発明によれば、静置状態の水性液全体を速やかにゲル化することが可能な、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな自己分散による高吸収速度とを両立した新規な吸水性樹脂粒子および吸水性樹脂粒子組成物を提供でき、また、そのような吸水性樹脂粒子の製造方法、吸水性樹脂粒子や吸水性樹脂粒子組成物の用途をも提供できる。さらに、篩により分級して微粉末状態とした原料吸水性樹脂粉末を表面処理することによる、高いVortex吸収速度を実現する、低発塵性の吸水性樹脂粒子を提供することにある。また、吸水性樹脂微粉の回収にも有用である。
【発明を実施するための最良の形態】
【0014】
以下、本発明について詳しく説明するが、本発明の範囲はこれらの説明に拘束されることはなく、以下の例示以外についても、本発明の趣旨を損なわない範囲で適宜変更実施し得る。
〔吸水性樹脂粉末〕
本発明にかかる吸水性樹脂粒子は、特定の熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた表面処理層を有してなる。
本発明で用いることができる吸水性樹脂粉末は、親水性単量体を重合して得ることができる水不溶性水膨潤性ヒドロゲル形成性重合体(以下、吸水性樹脂とも言う)の粉体であって、少なくとも生理食塩水の吸収倍率が10倍以上、好ましくは20〜100倍、より好ましくは30〜80倍である(JIS K7223−1996)、球形或いは不定形の粒子形状のものである。なお、本発明においては、吸水性樹脂粉末を単に吸水性樹脂と称することもある。
【0015】
水不溶性水膨潤性ヒドロゲル形成性重合体の具体例としては、部分中和架橋ポリアクリル酸重合体(米国特許第4625001号、米国特許第4654039号、米国特許第5250640号、米国特許第5275773号、欧州特許第456136号等)、架橋され部分的に中和された澱粉−アクリル酸グラフトポリマー(米国特許第4076663号)、イソブチレン−マレイン酸共重合体(米国特許第4389513号)、酢酸ビニル−アクリル酸共重合体のケン化物(米国特許第4124748号)、アクリルアミド(共)重合体の加水分解物(米国特許第3959569号)、アクリロニトリル重合体の加水分解物(米国特許第3935099号)等が挙げられるが、本発明で用いることができる吸水性樹脂粉末はアクリル酸および/またはその塩を含む単量体を重合して得られるポリアクリル酸(塩)系架橋重合体からなる吸水性樹脂粉末であることが好ましい。
【0016】
本発明において、ポリアクリル酸(塩)系架橋重合体とは、重合に用いる重合性単量体の総量を100モル%として、アクリル酸および/またはその塩を50モル%以上、好ましくは70モル%以上、より好ましくは90モル%以上含む単量体を重合して得られる架橋重合体である。また、重合体中の酸基は、その50〜90モル%が中和されていることが好ましく、60〜80モル%が中和されていることがより好ましく、塩としてはナトリウム、カリウム、リチウム等のアルカリ金属塩、アンモニウム塩、アミン塩などを例示する事ができる。塩を形成させるための吸水性樹脂の中和は重合前に単量体の状態で行っても良いし、あるいは重合途中や重合後に重合体の状態で行っても良いし、それらを併用してもよい。
【0017】
本発明に好ましく用いられる吸水性樹脂であるポリアクリル酸(塩)系架橋重合体としては、主成分として用いられる単量体(アクリル酸および/またはその塩)に併用して、必要により他の単量体を共重合させたものであってもよい。他の単量体の具体例としては、メタアクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸などのアニオン性不飽和単量体およびその塩;アクリルアミド、メタアクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン、N−ビニルアセトアミドなどのノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミドおよびそれらの四級塩などのカチオン性不飽和単量体などを挙げることができる。これらアクリル酸および/またはその塩以外の単量体の使用量は、全単量体(単量体成分と称することがある)中、0〜30モル%が好ましく、より好ましくは0〜10モル%である。
【0018】
また、上記ノニオン性の親水基含有不飽和単量体が主要成分である耐塩型の吸水性樹脂を組み合わせて使用してもよい。
本発明で用いることができる吸水性樹脂粉末は、内部架橋構造を有する架橋重合体である。
本発明に用いられる吸水性樹脂粉末に内部架橋構造を導入する方法として、架橋剤を使用しない自己架橋によって導入する方法や、1分子中に2個以上の重合性不飽和基および/または2個以上の反応性基を有する内部架橋剤を共重合または反応させて導入する方法等を例示できる。好ましくは、内部架橋剤を共重合または反応させて導入する方法である。
【0019】
これらの内部架橋剤の具体例としては、例えば、N,N′−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキサイド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリト−ルテトラ(メタ)アクリレ−ト、ジペンタエリスリト−ルヘキサ(メタ)アクリレ−ト、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、ポリエチレンイミン、グリシジル(メタ)アクリレートなどを挙げることが出来る。これらの内部架橋剤は1種のみ用いてもよいし2種以上使用してもよい。中でも、得られる吸水性樹脂の吸水特性などから、2個以上の重合性不飽和基を有する化合物を内部架橋剤として必須に用いることが好ましく、その使用量としては、全単量体に対して0.005〜3モル%が好ましく、より好ましくは0.01〜1.5モル%である。
【0020】
重合に際しては、澱粉−セルロ−ス、澱粉−セルロ−スの誘導体、ポリビニルアルコ−ル、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子や、次亜リン酸(塩)等の連鎖移動剤を添加してもよい。
本発明に用いることができる吸水性樹脂粉末を得るために、上記したアクリル酸および/またはその塩を主成分とする単量体を重合するに際しては、バルク重合、逆相懸濁重合、沈澱重合を行うことも可能であるが、性能面や重合の制御の容易さから、単量体を水溶液として、水溶液重合を行うことが好ましい。かかる重合方法は、例えば、米国特許第4625001号明細書、米国特許第4769427号明細書、米国特許第4873299号明細書、米国特許第4093776号明細書、米国特許第4367323号明細書、米国特許第4446261号明細書、米国特許第4683274号明細書、米国特許第4690996号明細書、米国特許第4721647号明細書、米国特許第4738867号明細書、米国特許第4748076号明細書、欧州特許第1178059号明細書などに記載されている。
【0021】
重合を行うにあたり、過硫酸カリウム、過硫酸アンモニウム、過硫酸ナトリウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2′−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤、紫外線や電子線などの活性エネルギー線等を用いることができる。また、ラジカル重合開始剤を用いる場合、亜硫酸ナトリウム、亜硫酸水素ナトリウム、硫酸第一鉄、L−アスコルビン酸等の還元剤を併用してレドックス重合としても良い。これらの重合開始剤の使用量は、全単量体に対して、0.001〜2モル%が好ましく、より好ましくは0.01〜0.5モル%である。
上記の重合および乾燥により得られた吸水性樹脂の形状は、一般には、不定形破砕状、球状、繊維状、棒状、略球状、偏平状等であるが、逆相懸濁重合で得られた球状のものやその凝集体であるブドウの房状のものも好適に用いることができる。また、本発明に用いられる吸水性樹脂は粒子状が望ましく、乾燥後に粉砕して得られるような不定形破砕状のものを用いると、より本発明の効果が大きくなるので好ましい。
【0022】
本発明で用いることができる吸水性樹脂は、その表面近傍が表面架橋剤でさらに表面架橋処理されていてもよいし、表面架橋処理されていなくてもよい。また、モノマーとしてアクリルアミドが用いられた吸水性樹脂の場合は、熱表面架橋されたものでもよい。
表面架橋処理に用いることの出来る表面架橋剤としては、吸水性樹脂の有する官能基、特に、カルボキシル基と反応し得る官能基を2個以上有する有機表面架橋剤や多価金属化合物が挙げられる。例えば、エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール化合物;エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、グリシドール等のエポキシ化合物;エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、ポリエチレンイミン等の多価アミン化合物や、それらの無機塩ないし有機塩(例えば、アゼチジニウム塩等);2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート等の多価イソシアネート化合物;1,2−エチレンビスオキサゾリン等の多価オキサゾリン化合物;環状尿素、2−オキサゾリジノン等の炭酸誘導体;1,3−ジオキソラン−2−オン、4−メチル−1,3−ジオキソラン−2−オン、4,5−ジメチル−1,3−ジオキソラン−2−オン、4,4−ジメチル−1,3−ジオキソラン−2−オン、4−エチル−1,3−ジオキソラン−2−オン、4−ヒドロキシメチル−1,3−ジオキソラン−2−オン、1,3−ジオキサン−2−オン、4−メチル−1,3−ジオキサン−2−オン、4,6−ジメチル−1,3−ジオキサン−2−オン、1,3−ジオキソパン−2−オン等のアルキレンカーボネート化合物;エピクロロヒドリン、エピブロムヒドリン、α−メチルエピクロロヒドリン等のハロエポキシ化合物、および、その多価アミン付加物(例えばハーキュレス製カイメン:登録商標);γ−グリシドキシプロピルトリメトキシシラン、γーアミノプロピルトリエトキシシラン等のシランカップリング剤;3−メチル−3−オキセタンメタノール、3−エチル−3−オキセタンメタノール、3−ブチル−3−オキセタンメタノール、3−メチル−3−オキセタンエタノール、3−エチル−3−オキセタンエタノール、3−ブチル−3−オキセタンエタノール、3−クロロメチル−3−メチルオキセタン、3−クロロメチル−3−エチルオキセタン、多価オキセタン化合物などのオキセタン化合物;亜鉛、カルシウム、マグネシウム、アルミニウム、鉄、ジルコニウム等の水酸化物又は塩化物等の多価金属化合物等が挙げられる。これら表面架橋剤は、1種のみ用いてもよいし、2種以上を併用してもよい。
【0023】
表面架橋剤の使用量は、吸水性樹脂の固形分100重量部に対して0.001〜5重量部が好ましい。
表面架橋剤と吸水性樹脂との混合の際には水を用いてもよい。水の使用量は、吸水性樹脂の固形分100重量部に対して、0〜15重量部の範囲内が好ましく、0.5を越え10重量部以下の範囲内がより好ましく、1〜5重量部の範囲内がさらに好ましい。
表面架橋剤やその水溶液を混合する際には、親水性有機溶媒や、それ以外の第三物質を混合助剤として用いてもよい。
親水性有機溶媒を用いる場合には、例えば、メチルアルコール、エチルアルコール、n−プロピルアルコール、イソプロピルアルコール、n−ブチルアルコール、イソブチルアルコール、t−ブチルアルコール等の低級アルコール類;アセトン等のケトン類;ジオキサン、テトラヒドロフラン、メトキシ(ポリ)エチレングリコール等のエーテル類;ε−カプロラクタム、N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類;エチレングリコール、ジエチレングリコール、プロピレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール、1,3−プロパンジオール、ジプロピレングリコール、2,2,4−トリメチル−1,3−ペンタンジオール、ポリプロピレングリコール、グリセリン、ポリグリセリン、2−ブテン−1,4−ジオール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、1,2−シクロヘキサンジメタノール、1,2−シクロヘキサノール、トリメチロールプロパン、ジエタノールアミン、トリエタノールアミン、ポリオキシプロピレン、オキシエチレン−オキシプロピレンブロック共重合体、ペンタエリスリトール、ソルビトール等の多価アルコール類等が挙げられる。親水性有機溶媒の使用量は、吸水性樹脂の種類や粒径、含水率等にもよるが、吸水性樹脂の固形分100重量部に対して、0〜10重量部の範囲内が好ましく、0.1〜5重量部の範囲内がより好ましい。また、第三物質として欧州特許第0668080号公報に示された無機酸、有機酸、ポリアミノ酸等を存在させてもよい。これらの混合助剤は表面架橋剤として作用しても良いが、表面架橋後に吸水性樹脂の吸水性能を低下させないものが好ましい。特に沸点が150℃未満の揮発性アルコール類は表面架橋処理時に揮発してしまうので、残存物が残らず望ましい。
【0024】
吸水性樹脂と表面架橋剤とをより均一に混合するため、非架橋性の水溶性無機塩基類(好ましくは、アルカリ金属塩,アンモニウム塩,アルカリ金属水酸化物、および、アンモニアあるいはその水酸化物)や、非還元性アルカリ金属塩pH緩衝剤(好ましくは炭酸水素塩、リン酸二水素塩、リン酸水素塩等)を、吸水性樹脂と表面架橋剤とを混合する際に共存させても良い。これらの使用量は、吸水性樹脂の種類や粒径等にもよるが、吸水性樹脂の固形分100重量部に対して0〜15重量部の範囲内が好ましく、0.005〜10重量部の範囲内がより好ましく、0.05〜5重量部の範囲内がさらに好ましい。
吸水性樹脂と表面架橋剤とを混合する混合方法は特に限定されないが、たとえば吸水性樹脂を親水性有機溶剤に浸漬し、必要に応じて水および/または親水性有機溶媒に溶解させた表面架橋剤を混合する方法、吸水性樹脂に直接、水および/または親水性有機溶媒に溶解させた表面架橋剤を噴霧若しくは滴下して混合する方法等が例示できる。
【0025】
吸水性樹脂と表面架橋剤とを混合した後、通常、加熱処理を行い、架橋反応を遂行させる。上記加熱処理温度は、用いる表面架橋剤にもよるが、材料温度で、40〜250℃が好ましく、150〜250℃がより好ましい。処理温度が40℃未満の場合には、加圧下の吸収倍率等の吸収特性が十分に改善されない場合がある。処理温度が250℃を越える場合には、吸水性樹脂の劣化を引き起こし、性能が低下する場合があり注意を要する。加熱処理時間は、好ましくは1分〜2時間、より好ましくは5分〜1時間である。
本発明で用いることができる吸水性樹脂粉末は、目開き600μmの篩を通過するものであることが好ましい。より好ましくは、目開き500μmの篩を通過するものである。これは、篩による分級や気流分級等によって得ることができる。
【0026】
本発明で用いることができる原料吸水性樹脂粉末は、後述の重量平均粒子径が500μm以下であることが好ましく、より好ましくは400μm以下であり、さらに好ましくは300μm以下である。
また、微粉末が少なく高吸収速度の吸水性樹脂組成物を得るためには、好ましくは目開き212〜125μmの篩から選択された少なくとも1つの篩を通過したもの、より好ましくは目開き150〜125μmの篩を通過したものを使用した場合には、Vortex吸収速度(JIS K7224−1996)が5秒以下の吸水性樹脂粒子を得ることが可能となる。
【0027】
本発明で用いることができる吸水性樹脂粉末としては、粒子径300μm以下の粉末を表面架橋処理および/または造粒し粒度調整したものを用いても良い。また、粉砕して得られる一次粒子の不定形破砕状の粒子に対し、微粉の造粒物を、好ましくは5重量%以上、より好ましくは10重量%以上、さらに好ましくは15重量%以上、混合した吸水性樹脂を用いても良い。
微粉造粒物の作成方法としては、微粉を再生する公知の技術が使用可能である。例えば、温水と吸水性樹脂の微粉を混合し乾燥する方法(米国特許第6228930号)や、吸水性樹脂の微粉を単量体水溶液と混合し重合する方法(米国特許第5264495号)、吸水性樹脂の微粉に水を加え特定の面圧以上で造粒する方法(欧州特許第844270号)、吸水性樹脂の微粉を十分に湿潤させ非晶質のゲルを形成し乾燥・粉砕する方法(米国特許第4950692号)、吸水性樹脂の微粉と重合ゲルを混合する方法(米国特許第5478879号)などを用いることが可能であるが、好ましくは前記の温水と吸水性樹脂の微粉を混合し乾燥する方法が用いられる。なお、粒子径は分級される篩目径で示される。
【0028】
〔吸水性樹脂粒子〕
本発明にかかる吸水性樹脂粒子は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
【0029】
本発明にかかる吸水性樹脂粒子は、水分含有量が1〜15重量%であることが好ましい。
なお、本発明における表面処理とは、表面へのコーティング、吸水性樹脂粉末間の接着造粒などが含まれる。
また、当該表面処理された吸水性樹脂粒子が本発明の所望の効果を有する範囲において、上記表面処理は部分コーティングないし付着であってもよい。
表面処理前の吸水性樹脂粉末は、前述した通りである。
本発明にかかる吸水性樹脂粒子は、表面処理された吸水性樹脂粒子であり、特定の熱可塑性樹脂および必要により無機粒子で処理された表面処理層を有してなる。すなわち、本発明にかかる吸水性樹脂粒子は、表面処理された吸水性樹脂粒子であり、特定の熱可塑性樹脂および無機粒子で処理された表面処理層を有してなるものでもよいし、特定の熱可塑性樹脂のみで処理された表面処理層を有してなるものでもよい。
【0030】
前記熱可塑性樹脂は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である。
前記熱可塑性樹脂の重量平均分子量は、好ましくは20000〜500000、より好ましくは40000〜200000である。
前記熱可塑性樹脂は、親水性と疎水性とを有する。親水性とは「親水性を有する構造」を有することを意味し、疎水性とは「疎水性を有する構造」を有することを意味する。具体的には、親水性とは、例えば、カルボン酸基、水酸基、アミノ基、アミド基、スルホン酸基、リン酸基、アルキレングリコール基、N−メトキシアミノ基等の親水性基を有する構造が挙げられ、疎水性とは、例えば、メチル基、エチル基、プロピル基、ブチル基等のアルキル基やフェニル基、アセチル基などの疎水性基を有する構造が挙げられる。
【0031】
親水性と疎水性とを有する熱可塑性樹脂を用いて表面処理されることにより、(i)親水性の吸水性樹脂粉末表面への処理時に、熱可塑性樹脂の親水性基が吸水性樹脂粉末表面への付着性を向上させているので、極めて少量の熱可塑性樹脂の適用が可能になったものと考えられ、(ii)そのために、表面の熱可塑性樹脂による膨潤阻害が起こり難く、(iii)また、熱可塑性樹脂の疎水性基が吸水性樹脂粉末表面に存在することにより、静置水と接触した時点での吸水性樹脂粒子の膨潤ゲル粒子間の粘着・付着力が低減されているために膨潤ゲル粒子が順次液面から離れて沈降するものと考えられる。
前記熱可塑性樹脂としては、アルコール可溶性であり、重量平均分子量が20000〜500000であることが好ましい。ここでいうアルコール可溶性とは、常圧下、40℃でメタノールまたはエタノールに少なくとも0.5重量%以上、好ましくは2重量%以上、さらに好ましくは5重量%以上の濃度で溶解して溶液となることをいう。上限は、70重量%以下、好ましくは50重量%以下である。
【0032】
さらに、アルコール可溶性で水不溶性であるものが好ましい。これは適度の疎水性と適度の親水性を持った熱可塑性樹脂を少量使用することにより、疎水性基による過度の撥水性により吸水速度が抑制されることもなく、水性液と接触した場合にも水性液に溶解せず、接触液を増粘させないため、通液性を確保して全体のゲル化を促進するのに好ましい。
前記熱可塑性樹脂としては、例えば、親水性モノマーと疎水性アクリルエステル単量体の共重合体、低ゲル化度のポリビニルアルコール、アルコール可溶性ナイロンなどが挙げられ、これらの1種のみ用いてもよいし、2種以上を併用してもよい。好ましくは、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種であり、特に好ましくは、アルコール可溶性であり水不溶性であるアルカリ水可溶性樹脂である。
【0033】
アルカリ水可溶性樹脂とは、常温、常圧で、水には不溶であるがイオン交換水に水酸化ナトリウムを0.4重量%の割合で溶解することによってアルカリ性にしたアルカリ水に溶解する樹脂である。
上記アルカリ水可溶性樹脂とは、特許第3224533号(特開2002−060695号公報)等に記載されるアルカリ水可溶性樹脂を用いることができる。アルカリ水可溶性樹脂としては、酸性または中性を呈する水には溶解せず、アルカリ性を呈する水には溶解する樹脂を意味する。ここで、「中性を呈する水」とは、pH値が6〜8の範囲内の水であり、「酸性を呈する水」とは、pH値が該中性の範囲未満の水であり、「アルカリ性を呈する水」とは、pH値が該中性の範囲よりも大きい水である。
【0034】
なお、上記アルカリ水可溶性樹脂としては、アルカリ水への溶解性の程度として、下記評価試験によって求められる減少率が50〜100%のものが好ましく、60〜100%のものがより好ましく、70〜100%のものがさらに好ましい。
(アルカリ水への溶解性の評価試験)
二軸押出機を用いて得ることができるバインダー樹脂を、直径5mm、長さ5mmの円筒状のペレット形状に成形したものを用いて測定する。この成形体10gを、1Lのビーカーに入れた0.4重量%濃度のNaOHの水溶液500gに投入し、25℃にて、直径が40mm、4枚はねを用い、300rpmで24時間攪拌を行う。その後のバインダー樹脂の成形体におけるアルカリ水へ溶解した重量の、もとの成形体からの減少率で評価する。すなわち、24時間攪拌後に溶解せずに残った樹脂分について、濾別等を行い、水で洗浄し、乾燥後の重量を求め、溶解性試験にかける前における元のバインダー樹脂の重量からの減少率(%):(もとの重量−溶解性試験後の重量)/(もとの重量)で評価する。また、ペレット化されていなくても、5mm角以下の任意の形状の成形品であっても、アルカリ水への溶解性を示す場合には、上記アルカリ水可溶性樹脂の範囲である。
【0035】
アルカリ水可溶性樹脂を用いることにより、適度な疎水性によってゲルブロックが適度に抑制でき、適度な親水性によって過度なゲルブロック抑制を防止でき、従って、本発明の効果が十分に発現できる。
アルカリ水可溶性樹脂としては、例えば、カルボン酸基、スルホン酸基、ホスホン酸基等の置換基を有する樹脂;フェノール性ヒドロキシル基を含むノボラック樹脂;ポリビニルフェノール樹脂;等の1種または2種以上を用いることができる。中でも、アルカリ水に対する溶解性や経済性、樹脂組成物の各種物性等に優れる点で、α,β−不飽和カルボン酸系単量体とα,β−不飽和カルボン酸系単量体以外のビニル系単量体とを共重合して得られる樹脂が好適である。なお、カルボン酸基を有する樹脂であるヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、セルロースアセテートヘキサヒドロフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースヘキサヒドロフタレート等のセルロース誘導体を用いることもできる。
【0036】
α,β−不飽和カルボン酸系単量体としては、具体的には、例えば、アクリル酸、メタクリル酸等のα,β−不飽和モノカルボン酸;イタコン酸、マレイン酸、フマル酸等のα,β−不飽和ジカルボン酸;無水マレイン酸、無水イタコン酸等のα,β−不飽和ジカルボン酸無水物;マレイン酸モノエステル、フマル酸モノエステル、イタコン酸モノエステル等のα,β−不飽和ジカルボン酸モノエステル;等が挙げられるが、特に限定されない。これらα,β−不飽和カルボン酸系単量体は、1種のみを用いてもよいし、2種以上を併用してもよい。上記例示のα,β−不飽和カルボン酸系単量体のうち、アクリル酸、メタクリル酸が、本発明の効果を十分に発現させる点で特に好ましい。
【0037】
ビニル系単量体としては、具体的には、例えば、アクリル酸メチル、アクリル酸エチル、アクリル酸プロピル、アクリル酸ブチル、アクリル酸ステアリル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸ブチル、メタクリル酸ステアリル等の、炭素数1〜18の1価アルコールと(メタ)アクリル酸とのエステル;アクリロニトリル、メタクリロニトリル等のニトリル基含有ビニル系単量体;アクリルアミド、メタクリルアミド等のアミド基含有ビニル系単量体;アクリル酸ヒドロキシエチル、メタクリル酸ヒドロキシプロピル等の水酸基含有ビニル系単量体;スチレン、α−メチルスチレン等の芳香族ビニル系単量体;酢酸ビニル等の脂肪族ビニル系単量体;アリルエーテル類;マレイン酸モノアルキルエステル、マレイン酸ジアルキルエステル等のマレイン酸誘導体;フマル酸モノアルキルエステル、フマル酸ジアルキルエステル等のフマル酸誘導体;マレイミド、N−メチルマレイミド、N−ステアリルマレイミド、N−フェニルマレイミド、N−シクロヘキシルマレイミド等のマレイミド誘導体;イタコン酸モノアルキルエステル、イタコン酸ジアルキルエステル、イタコンアミド類、イタコンイミド類、イタコンアミドエステル類等のイタコン酸誘導体;エチレン、プロピレン等のアルケン類;ブタジエン、イソプレン等のジエン類;等が挙げられるが、特に限定されない。これらビニル系単量体は、1種のみを用いてもよいし、2種以上を併用してもよい。上記例示のビニル系単量体のうち、(メタ)アクリル酸エステルが、本発明の効果を十分に発現させる点で特に好ましい。
【0038】
α,β−不飽和カルボン酸系単量体およびビニル系単量体の合計量に占めるα,β−不飽和カルボン酸系単量体の割合は、9重量%以上であることがより好ましく、9重量%〜40重量%の範囲内がさらに好ましい。α,β−不飽和カルボン酸系単量体の割合を9重量%以上にすることにより、本発明の効果を十分に発現させることができる。
α,β−不飽和カルボン酸系単量体とビニル系単量体とを共重合することにより、アルカリ水可溶性樹脂が得られる。共重合方法、つまり、アルカリ水可溶性樹脂の製造方法は、特に限定されず、公知の方法を適用すればよい。
アルカリ水可溶性樹脂の酸価は、本発明の効果を十分に発現させる点で、15mgKOH/g以上であることが好ましく、30mgKOH/g以上であることがより好ましく、50mgKOH/g以上であることがさらに好ましく、70mgKOH/g以上であることが特に好ましく、70mgKOH/g〜500mgKOH/gの範囲内であることが最も好ましい。
【0039】
アルカリ水可溶性樹脂は、示差走査熱量測定(DSC(differential scanning calorimetry))によって測定されるガラス転移温度が、−80℃〜120℃の範囲内であることが好ましく、−30℃〜20℃の範囲内に低温側のガラス転移温度を有する一方、40℃〜100℃の範囲内に高温側のガラス転移温度を有していることがさらに好ましい。
疎水性ポリビニルアルコールとしては、例えば、ケン化度45モル%以下のクラレLMポリマー、LM−10HD、LM−15、LM−20、LM−25などが挙げられる。これらは、ポリビニルアルコール濃度2重量%で40℃で攪拌するとき、メタノールやエタノールに可溶である。
【0040】
アルコール可溶性ナイロンとしては、例えばナガセケムテックス製のアルコール可溶性ナイロンとして販売されているトレジンF30K、MF−30、EF−30T等のN−メトキシメチル化ナイロンなどが挙げられる。
前記無機粒子としては、例えば、日産化学製のスノーテックスC等のコロイダルシリカ分散液、日本アエロジル製のアエロジル#200等のシリカ微粒子、二酸化チタン微粒子、疎水性シリカR972などが挙げられ、本発明の効果を十分に発現させる点で、好ましくは、コロイダルシリカ分散液である。このような無機粒子を熱可塑性樹脂と併用することにより、本発明の吸水性樹脂粒子を静置水ゲル化剤として用いた場合に、吸水性樹脂粒子の沈降性制御が容易になり、静置水のゲル化が速やかに起こる。なお、このような無機粒子は、比重調整成分として作用することもある。
【0041】
熱可塑性樹脂および無機粒子の使用量は、熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜1.0重量%が好ましく、より好ましくは0.05〜0.5重量%、さらに好ましくは0.1〜0.3重量%であり、無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%であることが好ましい。より好ましくは、熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.05〜0.5重量%であり、無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%である。このような量を用いることにより、十分な吸収速度を維持でき、例えば、Vortex吸収速度(JIS K7224−1996)を十分に発現できる。Vortex吸収速度の測定に関する詳細は後述する。
【0042】
本発明にかかる吸水性樹脂粒子が有する表面処理層は、前記熱可塑性樹脂および必要により前記無機粒子で処理して得られるが、かかる表面処理は、前記熱可塑性樹脂および必要により前記無機粒子を含む水性液(表面処理剤)によってなされることが好ましい。
本発明にかかる吸水性樹脂粒子は、上記表面処理層を有するので、通常の吸水性樹脂粉末において良く行われる表面架橋処理は必ずしも必要ではなく、表面架橋処理を行っていない吸水性樹脂粉末を用いた場合には、もとの吸水性樹脂粉末が有する吸収性能が損なわれにくいという利点がある。
なお、ここでいう表面処理層とは、必ずしも吸水性樹脂粒子が前記熱可塑性樹脂および無機粒子で均一に覆われている場合のみではなく、部分的に覆われていたり、付着していたり、粒子同士が結合造粒されている場合も含む。
【0043】
本発明にかかる吸水性樹脂粒子は、本発明の効果を十分に発現させるため、好ましくは水分含有量が0〜15重量%であり、より好ましくは水分含有量が1〜15重量%であり、さらに好ましくは2〜12重量%、特に好ましくは3〜9重量%である。なお、水分含有量の測定は、105℃で3時間の乾燥減量によって求めた。
本発明にかかる吸水性樹脂粒子は、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である。静置水ゲル化時間の測定に関する詳細は後述する。
【0044】
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が、好ましくは1秒を超えて100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が、3600秒以内に測定できる限りにおいて、表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下であることが好ましく、より好ましくは60%以下、さらに好ましくは50%以下、特に好ましくは40%以下である。静置水ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して70%を超えると、不十分な処理状態となり、静置水ゲル化時間に大きな改善が認められないために好ましくない。
【0045】
本発明にかかる吸水性樹脂粒子(表面処理された吸水性樹脂粒子)は、静置水ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水ゲル化時間に対して80%以下である場合、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、好ましくは0.05〜0.5重量%であり、より好ましくは0.1〜0.4重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であることがより好ましい。さらに好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.1〜0.3重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.1〜4重量%である。これにより、本発明の効果をより一層発揮させることができる。
【0046】
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が、120秒以内であることが好ましい。静置水45度傾斜ゲル化時間の測定に関する詳細は後述する。
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水45度傾斜ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
本発明にかかる吸水性樹脂粒子は、静置水ゲル化時間が、3600秒以内に測定できる限りにおいて、静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して0.1%を超えて80%以下であることが好ましく、より好ましくは60%以下、さらに好ましくは50%以下、特に好ましくは40%以下である。静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して80%を超えると、不十分な処理状態となり、静置水45度傾斜ゲル化時間に大きな改善が認められないために好ましくない。
【0047】
つまり、処理後のゲル化時間が、少なくとも20%短縮されるということである。また、測定ゲル化時間を3600秒以内に限定したのは、便宜的なもので、3600秒以上のものは、3600秒としてゲル化時間の短縮割合を求めても構わない。
本発明にかかる吸水性樹脂粒子は、静置水45度傾斜ゲル化時間が表面処理前の原料吸水性樹脂粉末の静置水45度傾斜ゲル化時間に対して80%以下である場合、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、好ましくは0.05〜0.5重量%であり、より好ましくは0.1〜0.4重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であることがより好ましい。さらに好ましくは、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.1〜0.3重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.1〜4重量%である。これにより、本発明の効果をより一層発揮させることができる。
【0048】
本発明にかかる吸水性樹脂粒子は、前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内であることが好ましく、より好ましくは5秒以内、さらに好ましくは1秒以内である。静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が10秒を超えると、完全ゲル化時間とみかけのゲル化時間との差が大きくなってしまうので好ましくない。
本発明にかかる低発塵性で高吸収速度の吸水性樹脂粒子は、また、前記表面処理前の吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、Vortex吸収速度が5秒以下であることが好ましい。
【0049】
〔吸水性樹脂粒子の製造方法〕
本発明にかかる吸水性樹脂粒子の製造方法は、親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩を通して解砕する。好ましくは、目開き500μm以下の篩を通して解砕する。
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および無機粒子については、前述した通りである。
【0050】
水性液状態とは、前記熱可塑性樹脂および必要により無機粒子を水のみと共に水性液状態とする形態でもよいし、前記熱可塑性樹脂および必要により無機粒子を親水性溶媒のみと共に水性液状態とする形態でもよいし、前記熱可塑性樹脂および必要により無機粒子を親水性溶媒と水とともに水性液状態とする形態でもよい。また、可塑剤として不揮発性の親水性有機溶媒を併用してもよい。
親水性溶媒としては、例えば、メタノール、エタノール、プロパノール、イソプロパノール、アセトン、メチルエチルケトンなどが挙げられる。特に、メタノール、エタノール、メチルエチルケトンが好ましい。
【0051】
可塑剤としての不揮発性の親水性有機溶媒としては、例えば、プロピレングリコール、ジプロピレングリコール、エチレングリコール、ジエチレングリコール、これらのモノアルキルエーテル、液状ポリエチレングリコールやそのアルキルエーテル、グリセリン、ジグリセリン、ポリグリセリンなどが挙げられる。特に、プロピレングリコール、グリセリンが好ましい。可塑剤としての不揮発性の親水性有機溶媒を併用することにより、本発明の吸水性樹脂粒子を静置水ゲル化剤として用いた場合に、静置水のゲル化が速やかに起こる。
上記全水性液中の前記熱可塑性樹脂および必要により無機粒子の含有割合は、好ましくは1〜60重量%、より好ましくは5〜50重量%、さらに好ましくは10〜40重量%である。水の割合は、好ましくは0〜90重量%、より好ましくは10〜80重量%、さらに好ましくは40〜70重量%である。
【0052】
前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理する形態としては、特に限定されないが、例えば、
(1)熱可塑性樹脂と親水性溶媒とを含む水性液を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(2)熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(3)無機粒子と親水性溶媒とを含む水性液を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(4)無機粒子と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(5)熱可塑性樹脂と無機粒子と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(6)熱可塑性樹脂と無機粒子と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
(7)熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を吸水性樹脂粉末に添加し、溶媒除去後に無機粒子を添加する形態(水を水性液中に添加してもよいし、吸水性樹脂粉末の添加後に添加してもよいし、無機粒子の添加後に添加してもよい)、
(8)吸水性樹脂粉末に無機粒子を添加混合後、熱可塑性樹脂と可塑剤としての不揮発性の親水性有機溶媒と親水性溶媒とを含む水性液(添加順序は問わない)を添加する形態(水を水性液中に添加してもよいし、表面処理後に添加してもよい)、
が、好ましい形態として挙げられる。
【0053】
表面処理の際の温度は、特に限定されないが、好ましくは材料温度で室温〜100℃、より好ましくは室温〜80℃、さらに好ましくは室温〜50℃である。
本発明にかかる吸水性樹脂粒子の製造方法においては、前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させる。静置させる際の温度は、材料温度で好ましくは20〜100℃、より好ましくは40〜100℃、さらに好ましくは40〜80℃である。
本発明にかかる吸水性樹脂粒子の製造方法においては、前記熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩、好ましくは500μm以下の篩ないし相当品を通るまで解砕する。解砕する際に通す篩の目開きは、粒子径と目的とする用途によって適宜選択すればよい。
【0054】
〔吸水性樹脂粒子組成物〕
本発明にかかる吸水性樹脂粒子組成物は、本発明の吸水性樹脂粒子(表面処理された吸水性樹脂粒子)30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であることを特徴とする。
本発明にかかる吸水性樹脂粒子組成物は、静置水ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
【0055】
本発明にかかる吸水性樹脂粒子組成物は、静置水45度傾斜ゲル化時間が120秒以内であることが好ましい。
本発明にかかる吸水性樹脂粒子組成物は、静置水45度傾斜ゲル化時間が、好ましくは100秒以内、より好ましくは80秒以内、さらに好ましくは60秒以内である。静置水45度傾斜ゲル化時間が120秒を超えると、静置水全体のゲル化に要する時間が長くなるために好ましくない。
吸水性樹脂粒子組成物中の吸水性樹脂粒子および他の吸水性樹脂粒子の含有割合が上記範囲を外れると、静置水45度傾斜ゲル化時間が長くなり、且つ、静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が大きくなるために好ましくない。
【0056】
吸水性樹脂粒子組成物中の本発明の吸水性樹脂粒子の含有割合は、好ましくは30〜99重量%、より好ましくは40〜99重量%、さらに好ましくは60〜99重量%、特に好ましくは80〜99重量%である。
吸水性樹脂粒子組成物中の他の吸水性樹脂粒子の含有割合は、好ましくは1〜60重量%、より好ましくは1〜40重量%、さらに好ましくは1〜20重量%である。
上記割合は、吸水性樹脂粒子組成物を100重量%としたときの比率である。なお、本発明の物性を損なわない範囲において、添加物等の他の物質を、例えば、0〜20重量%配合することができる。添加物としては、公知の消臭剤、抗菌剤、フラボノイド類、キレート剤等が挙げられる。
【0057】
他の吸水性樹脂粒子としては、例えば、市販の衛生材料用吸水性樹脂であり、日本触媒製の「アクアリックCA」、住友精化製の「アクアキープ」、ストックハウゼン社製の「Favor」、ダウ社製の「ドライテック」などが挙げられる。
本発明にかかる吸水性樹脂粒子組成物の特徴、物性等は、前述した本発明の吸水性樹脂粒子と同様である。
〔静置水ゲル化剤〕
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物を含む。
【0058】
本発明にかかる静置水ゲル化剤中の、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物の含有割合は、好ましくは80〜100重量%、より好ましくは90〜100重量%、さらに好ましくは95〜100重量%である。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物以外に、その他の成分として、0〜20重量%の範囲で、キレート剤、殺菌剤、殺ウイルス剤、タンパク質分解酵素、血液凝固阻害剤などを含んでいても良い。
本発明にかかる静置水ゲル化剤は、本発明の吸水性樹脂粒子および/または本発明の吸水性樹脂粒子組成物を含むことにより、固化性能の向上、安全性の向上といった優れた効果を発現できる。
【0059】
なお、本発明の静置水ゲル化剤の使用対象としては、例えば、手術時の血液や血液と洗浄液との混合液等の医療廃液、重金属等を含有する廃液がある。
〔水性廃液の処理方法〕
本発明にかかる水性廃液の処理方法は、水性廃液の入った貯槽に本発明の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる。
本発明の静置水ゲル化剤は、前述の通り、静置水に一括添加した時の静置水ゲル化時間が短く、静置水45度傾斜ゲル化時間と静置水ゲル化時間との差(静置水45度傾斜ゲル化時間−静置水ゲル化時間)が短く、容器中の水性液を速やかにゲル化するといった優れた効果を発現できるので、水性廃液の処理に好適である。
【0060】
水性廃液としては、例えば、手術時の血液と洗浄液との混合液等の医療廃液、重金属等を含有する廃液などが挙げられる。
水性廃液の処理に用いる本発明の静置水ゲル化剤の使用量は、水性廃液全量に対して、好ましくは1〜100重量%、より好ましくは2〜20重量%、さらに好ましくは2.5〜5重量%である。水性廃液の処理に用いる本発明の静置水ゲル化剤の使用量が1重量%より少ないと、ゲル化が十分でなかったり、ゲル化しても全体としてのゲル化後の強度が低く、流動性が出やすかったりという問題があり、100重量%より多いと、経済的でないという問題がある。
【0061】
〔その他の用途〕
本発明にかかる吸水性樹脂粒子、本発明にかかる吸水性樹脂粒子組成物は、上述のような構成を有するため、ゲルブロッキングが効果的に防止でき、また、表面架橋の有無に関わらず高い吸収性能(例えば、Vortex吸収速度など)を発揮できるので、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料にも好ましく適用可能である。
また、目開き212〜125μmの篩から選択された少なくとも1つの篩、好ましくは150〜125μmの篩を通過させることにより微粒子化した原料吸水性樹脂(A)を使用した本発明の吸水性樹脂粒子は、Vortex吸収速度に優れ、テープ化工程時の粉塵の飛散等の問題がなくなり、光ケーブルや電力ケーブル用などのケーブル用止水テープに好適に用いることができる。
【0062】
ケーブル用止水テープ用には、本発明の処理後の吸水性樹脂は、目開き125μm以上850μm以下で、好ましくは、目開き150μm以上600μm以下、より好ましくは、目開き150μm以上500μm以下の篩を通過するように解砕して使用することができる。
本発明における表面処理を施した吸水性樹脂粒子(A)を、例えば、850μmの篩を通過するように解砕したものと、本発明における表面処理を施さなかった目開き212〜125μmの篩に相当する少なくとも1つの篩を通過しなかった吸水性樹脂粒子(B)とを混合することにより、低発塵性の衛生材料用吸水性樹脂を得ることができる。このようにして原料吸水性樹脂の微粒子を有効に活用することができる。この場合、原料吸水性樹脂としては、表面架橋は施されているものでも施されていないものでもよい。また、吸水性樹脂(A)と吸水性樹脂粒子(B)のうちの片方が表面架橋処理されていてもよい。表面架橋処理されたものを用いた方が、衛生材料用吸水性樹脂としては好ましい。このように、本発明は、粉砕工程を経て製造される吸水性樹脂の微粉の回収方法としても有用である。
【0063】
なお、本明細書において、篩の目開きについては、JIS標準篩を基準として、相当するASTMに定められた篩や目開きがそれに相当する篩であれば特注品であってもよい。
【実施例】
【0064】
以下に、実施例および比較例によって本発明をより具体的に説明するが、本発明はこれらに限定されるものではない。以下では、便宜上、「重量部」を単に「部」と、「リットル」を単に「L」と記すことがある。また、「重量%」を「wt%」と記すことがある。
実施例および比較例における、測定方法および評価方法を以下に示す。
<静置水ゲル化時間および静置水45度傾斜ゲル化時間の測定>
22℃の0.90重量%塩化ナトリウム水溶液(生理食塩水)100gをガラス製管(マルエムスクリュー管No.8(40X120):40mmφ、高さ120mm)に入れ、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面から2cm上部に設置したものを使用して、試料3.3gを一気に投入し、目視でガラス製管を横から観察してゲル化によって透明な液体部分がなくなるまでの時間を測定し、静置水ゲル化時間(秒)とした。
【0065】
次にガラス製管を45度傾けても流動化しなくなるまでの時間(秒)を測定し、静置水45度傾斜ゲル化時間とした。ここで、流動化しなくなるとは、45度傾けてもガラス管内のゲル最上面が流動あるいは変化せず、そのままの状態であることをいう。
全体がゲル化固定化する時間は、静置水45度傾斜ゲル化時間で表される。
静置水ゲル化時間は、見かけの全体ゲル化時間であり、十分な静置水中での垂直分散性を持たないものであれば、45度傾けた時に流動性が認められる。しかし、最も好ましく改質された垂直分散性を持つものを用いた場合には、静置水ゲル化時間と静置水45度傾斜ゲル化時間とはほぼ一致する。
【0066】
<Vortex吸収速度>
0.90重量%塩化ナトリウム水溶液(生理食塩水)に1000重量部に食品添加物である食用青色1号0.02重量部を添加し、液温22℃に調整した。その生理食塩水50mlを100mlビーカーに計り取り、長さ40mmで太さ8mmの円筒型攪拌子で600rpmで攪拌する中に、試料2.0gを投入し、Vortex吸収速度(秒)を測定した。
終点は、JIS K 7224−1996「高吸水性樹脂の吸水速度試験方法 解説」に記載されている基準に準じ、試料(吸水性樹脂粒子や吸水性樹脂粒子組成物など)が生理食塩水を吸液してスターラーチップを試験液で覆うまでの時間を吸収速度(秒)として測定した。
【0067】
Vortex吸収速度は、撹拌下でのゲル化時間を測定するものである。
<発塵性試験>
試料5gを100mlの円筒形透明ガラス製容器(マルエムサンプル管:内径37mm、高さ102mm)に入れてふたを締め、手で持ち上下に約50cmの巾で激しく10〜20秒で30回振った後に机上に置いた。置いた直後の粉立ちの程度を観察した後、1日放置後のガラス製容器内壁への微粉の付着程度をデジタルカメラ(フラッシュON)で近接撮影し、判定を行った。
判定は以下の5段階で行った。
【0068】
極めて多量(図1参照)
多量(図2参照)
中等度(図3参照)
軽微(図4参照)
ほとんどなし(図5参照)
<含水率>
105℃のオーブン中で3時間乾燥させたときの減量により求めた。
<重量平均粒子径(D50)>
吸水性樹脂粒子や吸水性樹脂粒子組成物を、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmなどのJIS標準ふるいで篩い分けし、残留百分率Rを対数確率紙にプロットした。これにより、R=50重量%に相当する粒子径を重量平均粒子径(D50)として読み取った。
【0069】
重量平均粒子径(D50)を測定する際の分級方法は、吸水性樹脂粒子や吸水性樹脂粒子組成物10.0gを、室温(20〜25℃)、湿度50RH%の条件下で、目開き850μm、710μm、600μm、500μm、425μm、300μm、212μm、150μm、45μmのJIS標準ふるい(THE IIDA TESTING SIEVE:径8cm)に仕込み、振動分級器(IIDA SIEVE SHAKER、TYPE:ES−65型、SER.No.0501)により、5分間、分級を行った。
〔合成例1:アルカリ水可溶性樹脂の調製〕
アルカリ水可溶性樹脂を以下の方法で調製した。
【0070】
すなわち、温度計、攪拌翼、還流冷却器、および滴下装置を備えた容量100Lの槽型反応器に、アクリル酸1.8kg、アクリル酸エチル10.2kg、重合開始剤である2,2´−アゾビス−(2,4−ジメチルバレロニトリル)24g、および、溶媒であるメチルアルコール28kgを仕込んだ。また、滴下装置に、アクリル酸2.7kg、アクリル酸メチル5.4kg、メタクリル酸メチル9.9kg、2,2´−アゾビス−(2,4−ジメチルバレロニトリル)66g、およびメチルアルコール2kgからなる混合溶液を仕込んだ。
上記のメチルアルコール溶液を、窒素ガス雰囲気下、攪拌しながら65℃に加熱し、20分間反応させた。これにより、内容物の重合率を72%に調節した。続いて、内温を65℃に保ちながら、滴下装置から上記の混合溶液を2時間かけて均等に滴下した。滴下終了後、内容物にメチルエチルケトン60kgを混合することにより、アルカリ水可溶性樹脂の25重量%溶液を得た。以下、このアルカリ水可溶性樹脂を「ASP」と称することがある。
【0071】
得られたアルカリ水可溶性樹脂の酸価は117mgKOH/gであった。また、該アルカリ水可溶性樹脂の重量平均分子量Mwは156000、数平均分子量Mnは69000であった。さらに、示差走査熱量機で測定したところ、ガラス転移温度(Tg)が10℃と67℃とに観測された。また、上記樹脂の、25℃のイオン交換水への溶解性は、0.5%未満であり、また、アルカリ水可溶性は100%であった。
〔実施例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液8.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて試料(1)を作製した。重量平均粒子径は210μm、45μm以下の微粉は1.1重量%であり、試料(1)の発塵性試験による判定は「軽微」であった。また、含水率は2.9重量%であった。
【0072】
試料(1)の静置水ゲル化時間は51秒、静置水45度傾斜ゲル化時間は55秒、Vortex吸収速度は26秒であった。
ゲル化時間測定時、試料(1)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gと脱イオン水2gを混合したものを滴下混合した。
【0073】
60℃の恒温器中で1時間熟成させ、500μmの篩を通過させて試料(2)を作製した。重量平均粒子径は205μm、45μm以下の微粉は1.6重量%であり、試料(2)の発塵性試験による判定は「軽微」であった。また、含水率は3.8重量%であった。
試料(2)の静置水ゲル化時間は64秒、静置水45度傾斜ゲル化時間は68秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(2)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの速やかに上部液面まで全体がゲル化した。
〔実施例3〕
脱イオン水の量を6gとした以外は実施例2と同様にして、試料(3)を作製した。
【0074】
試料(3)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は5.7重量%であった。
試料(3)の静置水ゲル化時間は62秒、静置水45度傾斜ゲル化時間は65秒、Vortex吸収速度は26秒であった。
ゲル化時間測定時、試料(3)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの速やかに上部液面まで全体がゲル化した。
〔実施例4〕
脱イオン水の量を10gとした以外は実施例2と同様にして、試料(4)を作製した。
【0075】
試料(4)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は7.6重量%であった。
試料(4)の静置水ゲル化時間は52秒、静置水45度傾斜ゲル化時間は57秒、Vortex吸収速度は25秒であった。
ゲル化時間測定時、試料(4)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの、速やかに上部液面まで全体がゲル化した。
〔実施例5〕
脱イオン水を入れない以外は実施例2と同様にして、試料(5)を作製した。
【0076】
試料(5)の発塵性試験による判定は「軽微」であった。また、含水率は2.8重量%であった。
試料(5)の静置水ゲル化時間は108秒、静置水45度傾斜ゲル化時間は111秒、Vortex吸収速度は28秒であった。
ゲル化時間測定時、試料(5)は、全量がゆっくりと膨潤しながら容器下半に沈降したものの、速やかに上部液面まで全体がゲル化した。
〔比較例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)(比較試料(1))をそのまま測定をした。
【0077】
比較試料(1)の発塵性試験による判定は「中等度」であった。また、含水率は2.6重量%であった。
比較試料(1)の静置水ゲル化時間は228秒、静置水45度傾斜ゲル化時間は300秒、Vortex吸収速度は19秒であった。
ゲル化時間測定時、比較試料(1)は、全量が一旦沈降した後、ゆっくりと上部液面方向へのゲル化が進行したが、全体がゲル化するのに時間を要した。
〔比較例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)(比較試料(2))をそのまま測定した。
【0078】
比較試料(2)の発塵性試験による判定は「中等度」であった。また、含水率は2.7重量%であった。
比較試料(2)の静置水ゲル化時間は135秒、静置水45度傾斜ゲル化時間は147秒、Vortex吸収速度は24秒であった。
ゲル化時間測定時、比較試料(2)は、全量が一旦沈降した後、ゆっくりと上部液面方向へのゲル化が進行したが、全体がゲル化するのに時間を要した。
〔比較例3〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液64.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて比較試料(3)を作製した。
【0079】
比較試料(3)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は2.7重量%であった。
比較試料(3)の静置水ゲル化時間は240秒、静置水45度傾斜ゲル化時間は420秒、Vortex吸収速度は35秒であった。
ゲル化時間測定時、比較試料(3)は、液面に浮いてゲルブロッキングを起こしている状態ではなく非常にゆっくりとではあるが少しずつ膨潤した部分から沈降ゲル化したが、全体のゲル化に時間を要した。
〔実施例6〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4SP、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%シリカ水溶液)20gを混合し、さらに撹拌混合した。40℃の恒温器中で1時間熟成させ、500μmの篩を通過させて試料(6)を作製した。
【0080】
試料(6)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は11.5重量%であった。
試料(6)の静置水ゲル化時間は35秒、静置水45度傾斜ゲル化時間は35秒、Vortex吸収速度は20秒であった。
ゲル化時間測定時、試料(6)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例7〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%シリカ水溶液)20gを混合し、さらに撹拌混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を0.5g添加して均一に混合し、試料(7)を作製した。
【0081】
試料(7)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は10.8重量%であった。
試料(7)の静置水ゲル化時間は57秒、静置水45度傾斜ゲル化時間は57秒、Vortex吸収速度は14秒であった。
ゲル化時間測定時、試料(7)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例8〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を1.0g添加して均一に混合し、試料(8)を作製した。
【0082】
試料(8)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は3.3重量%であった。
試料(8)の静置水ゲル化時間は66秒、静置水45度傾斜ゲル化時間は66秒、Vortex吸収速度は15秒であった。
ゲル化時間測定時、試料(8)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔参考例1〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液2.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を0.5g添加して均一に混合し、参考試料(1)を作製した。
【0083】
参考試料(1)の発塵性試験による判定は「軽微」であった。また、含水率は3.1重量%であった。
参考試料(1)の静置水ゲル化時間は126秒、静置水45度傾斜ゲル化時間は156秒、Vortex吸収速度は14秒であった。
ゲル化時間測定時、参考試料(1)は、全量が一旦沈降しゆっくりと液面へ向かって膨潤し全体がゲル化した。
上記参考例1は、熱可塑性樹脂の使用量が少ない場合の一例である。
〔実施例9〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)と耐塩性吸水性ポリマー(アクアリックCS−6、(株)日本触媒製)との1:1混合物200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて粉末試料を得た後、アエロジル200を1.0g添加して均一に混合し、試料(9)を作製した。
【0084】
試料(9)の発塵性試験による判定は「軽微」であった。また、含水率は4.1重量%であった。
試料(9)の静置水ゲル化時間は36秒、静置水45度傾斜ゲル化時間は36秒、Vortex吸収速度は6秒であった。
ゲル化時間測定時、試料(9)は、液面に浮いてから下部の膨潤した部分から順次膨潤沈降し、速やかに速やかに全体がゲル化した。
〔参考例2〕
市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)と耐塩性吸水性ポリマー(アクアリックCS−6、(株)日本触媒製)との1:1混合物200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液12.0gを滴下混合した。室温で放置し溶媒を揮散させた後、500μmの篩を通過させて参考試料(2)を作製した(アエロジル200を1.0g添加しなかった以外は実施例9に同じ)。
【0085】
参考試料(2)の発塵性試験による判定は「軽微」であった。また、含水率は4.0重量%であった。
参考試料(2)の静置水ゲル化時間は594秒、静置水45度傾斜ゲル化時間は594秒、Vortex吸収速度は11秒であった。
ゲル化時間測定時、参考試料(2)は、全量が液面に浮いてからその下部の膨潤した部分から非常にゆっくりと膨潤沈降しゲル化した。
〔製造例1〕
75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度33重量%)に、トリメチロールプロパントリアクリレート1.2gを溶解し、反応液とした。次に、この反応液を窒素ガス雰囲気下で30分間脱気した。ついで、シグマ型羽根を2本有する内容積10Lのジャケット付きステンレス製双腕型ニーダーに蓋を付けて形成した反応器に、上記反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。続いて、反応液を撹拌しながら、過硫酸ナトリウム2.46gおよびL−アスコルビン酸0.10gを添加したところ、およそ1分後に重合が開始した。そして、30〜80℃で重合を行い、重合開始から60分後に含水ゲル状重合体を取り出した。得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を目開き500μmのステンレス製ネット上に広げ、150℃で90分間熱風乾燥した。ついで、乾燥物をハンマーミルを用いて粉砕し、さらに目開き500μmの篩で分級することにより、不定形破砕状の吸水性樹脂前駆体(a)を得た。
【0086】
得られた吸水性樹脂前駆体(a)100重量部に、グリセリン0.5重量部と、水2重量部と、イソプロピルアルコール1重量部とからなる表面架橋剤を混合した。上記の混合物を210℃で45分間加熱処理することにより吸水性樹脂粉末(1)を得た。重量平均粒子径は169μm、45μm以下の微粉は10.5重量%であった。
〔実施例10〕
製造例1で得られた吸水性樹脂粉末(1)200gをクッキングカッターに入れ、撹拌混合しながら、下記に示す処理液Aの30gを滴下混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(10)を得た。重量平均粒子径は210μm、45μm以下の微粉は1.8重量%であった。
【0087】
試料(10)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は6.5重量%であった。
試料(10)の静置水ゲル化時間は105秒、静置水45度傾斜ゲル化時間は105秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(10)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
処理液A:
25%濃度のASPのメタノール・(MEK)溶液 3.0g
1規定NaOH水溶液 2.0g
脱イオン水 5.0g
プロピレングリコール 5.0g
スノーテックスC(日産化学製コロイダルシリカ分散液) 15.0g
〔実施例11〕
製造例1で得られた吸水性樹脂粉末(1)200gをクッキングカッターに入れ、撹拌混合しながら、下記に示す処理液Bの25.8gを混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(11)を得た。
【0088】
試料(11)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は5.6重量%であった。
試料(11)の静置水ゲル化時間は114秒、静置水45度傾斜ゲル化時間は114秒、Vortex吸収速度は32秒であった。
ゲル化時間測定時、試料(11)は、液面に浮いてから膨潤した部分から順次沈降しゲル化した。
処理液B:
25%濃度のASPのメタノール・MEK溶液 1.92g
1規定NaOH水溶液 1.28g
脱イオン水 3.2g
プロピレングリコール 3.8g
スノーテックスC 15.6g
〔実施例12〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら、下記に示す処理液Cの280gを噴霧混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機で1時間熟成した後、目開き500μmのメッシュを通過させて試料(12)を得た。
【0089】
試料(12)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は3.1重量%であった。
試料(12)の静置水ゲル化時間は112秒、静置水45度傾斜ゲル化時間は112秒、Vortex吸収速度は32秒であった。
ゲル化時間測定時、試料(12)は、液面に浮いてから順次膨潤沈降し全体がゲル化した。
処理液C:
25%濃度のASPのメタノール・MEK溶液 19.2g
1規定NaOH水溶液 12.8g
脱イオン水 32g
グリセリン 60g
スノーテックスC 156g
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、試料(12)の33gを投入し、全体がゲル化固定化する時間を測定したところ、135秒であった。
【0090】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、試料(12)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、190秒および132秒であった。
〔実施例13〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら、下記に示す処理液Dの155gを噴霧混合した。それをバットにひろげて40℃で相対湿度60%の恒温高湿機中で1時間熟成した後、目開き500μmのメッシュを通過させて試料(13)を得た。
【0091】
試料(13)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は1.9重量%であった。
試料(13)の静置水ゲル化時間は98秒、静置水45度傾斜ゲル化時間は98秒、Vortex吸収速度は30秒であった。
ゲル化時間測定時、試料(13)は、液面に浮いてから順次膨潤沈降し全体がゲル化した。
処理液D:
25%濃度のASPのメタノール・MEK溶液 11.5g
1規定NaOH水溶液 7.7g
脱イオン水 19.2g
プロピレングリコール 23.0g
スノーテックスC 93.6g
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、試料(13)の33gを投入し、全体がゲル化固定化する時間を測定したところ、105秒であった。
【0092】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、試料(13)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、147秒および119秒であった。
〔比較例4〕
製造例1で得られた吸水性樹脂粉末(1)をそのまま使用し、比較試料(4)とした。
比較試料(4)の発塵性試験による判定は「多量」であった。また、含水率は0.3重量%であった。
【0093】
比較試料(4)の静置水ゲル化時間は約1400秒、静置水45度傾斜ゲル化時間は約3900秒、Vortex吸収速度は31秒であった。
ゲル化時間測定時、比較試料(4)は、一旦全量が容器底部に沈降してから極めてゆっくりと上部液面へとゲル化が進行し全体がゲル化した。
〔比較例5〕
製造例1で得られた吸水性樹脂粉末(1)4000gをレディゲミキサーに入れ、撹拌しながら脱イオン水200gを噴霧した。生成物を取り出しバットにいれて70℃で相対湿度60%の恒温高湿機中で2時間熟成した。これを目開き500μmのメッシュを通過させて比較試料(5)を得た。重量平均粒子径は193μm、45μm以下の微粉は6.6重量%であった。
【0094】
比較試料(5)の発塵性試験による判定は「中等度」であった。また、含水率は2.5重量%であった。
比較試料(5)の静置水ゲル化時間は252秒、静置水45度傾斜ゲル化時間は335秒、Vortex吸収速度は18秒であった。
ゲル化時間測定時、比較試料(5)は、一旦全量が容器底部に沈降してから非常にゆっくりと上部液面へとゲル化が進行し全体がゲル化した。
1Lのパックエース((株)テラオカ製)に1000gの生理食塩水を入れて静置し、プラスチック(PP)製の粉末ロート(足径15mm)(東京ガラス器械(株)の「科学機器総合カタログ2002/2003」品番167)の足先端部を液面上4cm上に設置しておいて、比較試料(5)の33gを投入し、全体がゲル化固定化する時間を測定したところ、535秒であった。
【0095】
さらに、3Lの容量のTOP社製ガラスビーカー(内径145mm、高さ220mm)に3000gの生理食塩水を入れて静置し、同様に設置したプラスチックロートから、比較試料(5)の99gおよび150gを投入し、全体がゲル化固定化する時間をそれぞれ測定したところ、639秒および411秒であった。
〔実施例14〕
実施例6で得られた試料(6)70gに市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−W4S、(株)日本触媒製)30gを混合することにより、試料(14)を作製した。
【0096】
試料(14)の発塵性試験による判定は「軽微」であった。また、含水率は8.2重量%であった。
試料(14)の静置水ゲル化時間は92秒、静置水45度傾斜ゲル化時間は92秒、Vortex吸収速度は22秒であった。
ゲル化時間測定時、試料(14)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例15〕
実施例6で得られた試料(6)80gに市販のアクリル酸塩系吸水性ポリマー(アクアリックCA−H3、(株)日本触媒製)20gを混合することにより、試料(15)を作製した。
【0097】
試料(15)の発塵性試験による判定は「軽微」であった。また、含水率は7.9重量%であった。
試料(15)の静置水ゲル化時間は85秒、静置水45度傾斜ゲル化時間は88秒、Vortex吸収速度は29秒であった。
ゲル化時間測定時、試料(15)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔製造例2〕
75モル%の中和率を有するアクリル酸ナトリウムの水溶液5500g(単量体濃度33重量%)に、ポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数10)5.44gを溶解し反応液とした。次に、この反応液を窒素ガス雰囲気下で30分間脱気した。ついで、シグマ型羽根を2本有する内容積10Lのジャケット付きステンレス製双腕型ニーダーに蓋を付けて形成した反応器に、上記反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。続いて、反応液を撹拌しながら、過硫酸ナトリウム2.46gおよびL−アスコルビン酸0.10gを添加したところ、およそ1分後に重合が開始した。そして、30〜80℃で重合を行い、重合開始から60分後に含水ゲル状重合体を取り出した。得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を目開き500μmのステンレス製ネット上に広げ、150℃で90分間熱風乾燥した。ついで、乾燥物をロールミルを用いて粉砕し、目開き850μmの篩を通過する吸水性樹脂粉末(2)を得た。重量平均粒子径は410μm、45μm以下の微粉は0.5重量%であった。さらに目開き150μmの篩で分級することにより、重量平均粒子径115μmの不定形破砕状の吸水性樹脂粉末(3)を得た。45μm以下の微粉は、7.6重量%であった。目開き150μmの篩上に残った吸水性樹脂粉末100重量部に、グリセリン0.5重量部と、水2重量部と、イソプロピルアルコール1重量部とからなる表面架橋剤を混合した。上記の混合物を210℃で45分間加熱処理することにより吸水性樹脂粉末(4)を得た。重量平均粒子径は436μm、45μm以下の微粉は0.1重量%であった。
【0098】
〔比較例6〕
製造例2で得られた吸水性樹脂粉末(3)をそのまま比較試料(6)として使用し、測定した。
比較試料(6)の静置水ゲル化時間も静置水45度傾斜ゲル化時間も共に観測できなかった(86400秒以上、つまり24時間後も、ママコができており部分的にしかゲル化せず流動状態であった)。Vortex吸収速度は15秒(ただしママコ生成)であった。比較試料(6)の発塵性試験による判定は「多量」であった。また、含水率は2.9重量%であった。
【0099】
〔実施例16〕
製造例2で得られた吸水性樹脂粉末(3)200gをクッキングカッターに入れ、撹拌混合しながら、アルカリ水可溶性樹脂(ASP)2.5重量%を溶解しているメタノール92.5重量%/5重量%メチルエチルケトン溶液16.0gを滴下混合した。続いてそこにスノーテックスC(日産化学社製:20%水溶液)30gを混合しさらに撹拌混合した。60℃の恒温器中で1時間熟成させ、850μmの篩を通過させて試料(16)を作製した。重量平均粒子径は415μm、45μm以下の微粉は0.0重量%であった。
試料(16)の発塵性試験による判定は「ほとんどなし」であった。また、含水率は7.4重量%であった。
【0100】
試料(16)の静置水ゲル化時間は30秒、静置水45度傾斜ゲル化時間は30秒、Vortex吸収速度は3秒であった。静置水ゲル化時間、Vortex吸収速度、ともに、比較例8に比べて著しい改善を示した。
ゲル化時間測定時、試料(16)は、液面に浮いてから膨潤した部分から順次あるいは部分的に一団となってちぎれて膨潤沈降し、速やかに上部液面と容器下部の両方からゲル化し速やかに全体がゲル化した。
〔実施例17〕
実施例16で得られた試料(16)20gを、ブチルゴム20%含有のトルエン溶液に分散させて混合し、不織布基材に塗布し、止水テープを作成した。
【0101】
比較試料(6)を使用して同様に塗布した場合には、試料の添加時に粉塵が発生したが、試料(16)を使用した場合には、試料の添加時に粉塵の飛散が全くなかった。
純水を用いた止水性能も良好であった。
〔実施例18〕
実施例16で得られた試料(16)10gに製造例2で得られた表面架橋された吸水性樹脂粉末(4)90gを混合して試料(18)を得た。重量平均粒子径は432μm、45μm以下の微粉は0.0重量%であった。発塵性試験による判定も「ほとんどなし」であった。微粉の極めて少ない良好な衛生材料用吸水性樹脂が得られた。
【0102】
【表1】
【0103】
【表2】
【0104】
【表3】
【産業上の利用可能性】
【0105】
本発明にかかる吸水性樹脂粒子や吸水性樹脂粒子組成物は、静置状態の水性液全体を速やかにゲル化することが可能、すなわち、吸収初期のゲルブロッキングの効果的な防止と速やかな吸収速度とを両立することができるので、塗料廃液、メッキ廃液、放射性廃液、医療廃液などの水性廃液のゲル化剤に好適である。また、ケーブル用止水剤(止水テープなど)や、紙おむつ、生理用ナプキン、失禁パッド等の衛生材料にも好ましく適用可能である。
【図面の簡単な説明】
【0106】
【図1】発塵性試験における「極めて多量」の判定となったサンプルの一例を示す写真図。
【図2】発塵性試験における「多量」の判定となったサンプルの一例を示す写真図。
【図3】発塵性試験における「中等度」の判定となったサンプルの一例を示す写真図。
【図4】発塵性試験における「軽微」の判定となったサンプルの一例を示す写真図。
【図5】発塵性試験における「ほとんどなし」の判定となったサンプルの一例を示す写真図。
【特許請求の範囲】
【請求項1】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である、
ことを特徴とする、表面処理された吸水性樹脂粒子。
【請求項2】
前記ゲル化するまでの時間(静置水ゲル化時間)が80秒以内である、請求項1に記載の表面処理された吸水性樹脂粒子。
【請求項3】
前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内である、請求項1または2に記載の吸水性樹脂粒子。
【請求項4】
前記表面処理前の原料吸水性樹脂粉末の粒度が、目開き600μmの篩を通過するものであり、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%である、請求項1から3までのいずれかに記載の吸水性樹脂粒子。
【請求項5】
前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であり、得られた上記表面処理された吸水性樹脂粒子の静置水ゲル化時間が原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下である、請求項1から4までのいずれかに記載の吸水性樹脂粒子。
【請求項6】
前記熱可塑性樹脂がアルコール可溶性であり、重量平均分子量が20000〜500000である、請求項1から5までのいずれかに記載の吸水性樹脂粒子。
【請求項7】
前記アルコール可溶性の熱可塑性樹脂が、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種である、請求項1から6までのいずれかに記載の吸水性樹脂粒子。
【請求項8】
請求項1から7までのいずれかに記載の表面処理された吸水性樹脂粒子30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である、
ことを特徴とする、吸水性樹脂粒子組成物。
【請求項9】
請求項1から7までのいずれかに記載の吸水性樹脂粒子を含む、静置水ゲル化剤。
【請求項10】
請求項8に記載の吸水性樹脂粒子組成物を含む、静置水ゲル化剤。
【請求項11】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩を通して解砕する、吸水性樹脂粒子の製造方法。
【請求項12】
水性廃液の入った貯槽に請求項9または10に記載の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる、水性廃液の処理方法。
【請求項13】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、
前記表面処理前の原料吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、
Vortex吸収速度が5秒以下である、
ことを特徴とする、表面処理された吸水性樹脂粒子。
【請求項14】
請求項13に記載の吸水性樹脂粒子を用いた、ケーブル用止水テープ。
【請求項15】
目開き212〜125μmの少なくとも1つの篩を通過しない吸水性樹脂粒子と、請求項13に記載の吸水性樹脂粒子を混合してなる、衛生材料用吸水性樹脂。
【請求項1】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で原料吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である、
ことを特徴とする、表面処理された吸水性樹脂粒子。
【請求項2】
前記ゲル化するまでの時間(静置水ゲル化時間)が80秒以内である、請求項1に記載の表面処理された吸水性樹脂粒子。
【請求項3】
前記ガラス製容器を45度傾けてもゲル最上面が流動しなくなるまでの時間を静置水45度傾斜ゲル化時間として、(静置水45度傾斜ゲル化時間−静置水ゲル化時間)で表される2種のゲル化時間の差が10秒以内である、請求項1または2に記載の吸水性樹脂粒子。
【請求項4】
前記表面処理前の原料吸水性樹脂粉末の粒度が、目開き600μmの篩を通過するものであり、前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0〜5重量%である、請求項1から3までのいずれかに記載の吸水性樹脂粒子。
【請求項5】
前記熱可塑性樹脂の使用量が、前記吸水性樹脂粉末に対して0.01〜0.7重量%であり、前記無機粒子の使用量が、前記吸水性樹脂粉末に対して0.02〜5重量%であり、得られた上記表面処理された吸水性樹脂粒子の静置水ゲル化時間が原料吸水性樹脂粉末の静置水ゲル化時間に対して0.1%を超えて80%以下である、請求項1から4までのいずれかに記載の吸水性樹脂粒子。
【請求項6】
前記熱可塑性樹脂がアルコール可溶性であり、重量平均分子量が20000〜500000である、請求項1から5までのいずれかに記載の吸水性樹脂粒子。
【請求項7】
前記アルコール可溶性の熱可塑性樹脂が、アルカリ水可溶性樹脂、疎水性ポリビニルアルコール、アルコール可溶性ナイロンから選ばれる少なくとも1種である、請求項1から6までのいずれかに記載の吸水性樹脂粒子。
【請求項8】
請求項1から7までのいずれかに記載の表面処理された吸水性樹脂粒子30〜99重量%とそれ以外の吸水性樹脂粒子1〜70重量%を含む吸水性樹脂粒子組成物であって、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内である、
ことを特徴とする、吸水性樹脂粒子組成物。
【請求項9】
請求項1から7までのいずれかに記載の吸水性樹脂粒子を含む、静置水ゲル化剤。
【請求項10】
請求項8に記載の吸水性樹脂粒子組成物を含む、静置水ゲル化剤。
【請求項11】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子を水性液状態として用いて吸水性樹脂粉末を表面処理した後、室温〜100℃で静置して全体を固結させ、さらに、目開き600μm以下の篩を通して解砕する、吸水性樹脂粒子の製造方法。
【請求項12】
水性廃液の入った貯槽に請求項9または10に記載の静置水ゲル化剤を一括添加することにより前記廃液をゲル化させる、水性廃液の処理方法。
【請求項13】
親水性と疎水性とを有し、かつ、重量平均分子量が10000〜1000000である熱可塑性樹脂および必要により無機粒子で吸水性樹脂粉末の表面を処理して得られた、表面処理された吸水性樹脂粒子であって、
前記表面処理前の原料吸水性樹脂粉末が、目開き212〜125μmの篩から選択された少なくとも1つの篩を通過した粉末であり、
鉛直方向に置かれた透明の円筒形ガラス製容器(内径37mm、高さ102mm)内に静置されている22℃の生理食塩水100mlの液面に前記吸水性樹脂粒子3.3gを一括添加したときに、該添加時から前記生理食塩水全体がゲル化するまでの時間(静置水ゲル化時間)が120秒以内であり、
Vortex吸収速度が5秒以下である、
ことを特徴とする、表面処理された吸水性樹脂粒子。
【請求項14】
請求項13に記載の吸水性樹脂粒子を用いた、ケーブル用止水テープ。
【請求項15】
目開き212〜125μmの少なくとも1つの篩を通過しない吸水性樹脂粒子と、請求項13に記載の吸水性樹脂粒子を混合してなる、衛生材料用吸水性樹脂。
【図1】

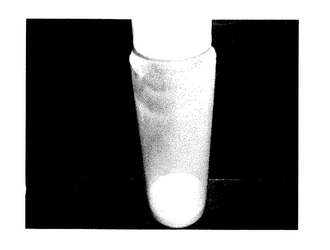
【図2】

【図3】

【図4】

【図5】

【図2】
【図3】
【図4】
【図5】
【公開番号】特開2006−143972(P2006−143972A)
【公開日】平成18年6月8日(2006.6.8)
【国際特許分類】
【出願番号】特願2004−339486(P2004−339486)
【出願日】平成16年11月24日(2004.11.24)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成18年6月8日(2006.6.8)
【国際特許分類】
【出願日】平成16年11月24日(2004.11.24)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]