
吸水材、表面架橋吸水性樹脂の製造方法および吸水材の評価方法
表面架橋が均一な表面架橋吸水性樹脂およびその製造方法を提供する。
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で2−35%である。使用する表面架橋吸水性樹脂は、特定粒子径の吸水性樹脂に、水濃度が特定範囲の表面架橋剤で表面架橋することで製造できる。
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で2−35%である。使用する表面架橋吸水性樹脂は、特定粒子径の吸水性樹脂に、水濃度が特定範囲の表面架橋剤で表面架橋することで製造できる。
【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本発明は吸水材に関するものである。更に詳しくは、特定範囲の平均粒子径を有し、かつ粒度分布の狭い表面が均一な表面架橋層および所定の表面硬さを有し、吸収特性、取り扱い性に優れる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材、該表面架橋吸水性樹脂の製造方法、および吸水材の評価方法に関する。
【背景技術】
【0002】
背景技術
近年、体液を吸収させることを目的とし、紙オムツ、生理用ナプキンなどの衛生材料の構成材料の一つとして吸水性樹脂が幅広く利用されている。吸水性樹脂に望まれる特性としては、以下の3つの特性を挙げることができる。
【0003】
(1)吸収特性に優れること、すなわち水性液体に接した際に吸水倍率が高く、加圧下吸水倍率が高く、吸収速度が速く、水性液体を含んだ基材から水性液体を吸い上げる能力が高いこと。
【0004】
(2)吸収対象が広いこと、すなわち水や尿のみならず、血液や経血等の体液に対して親和性が高いこと。
【0005】
(3)樹脂の取り扱い性に優れること、すなわち高湿下における吸湿性が低く流動性が高いこと、また、微粒子(例えば106μm未満)の含有量が少なく、発塵性が低いこと。
【0006】
しかしながら、吸収特性に関し、上記(1)に記載の各性能間の関係は必ずしも正の相関関係を示さず、例えば、吸水倍率の高いものほど吸収速度、ゲル強度の物性は低下してしまう。また、吸水性樹脂の平均粒子径が大きくなるほど通液性は高くなるが、吸収速度が低下してしまう。
【0007】
このような吸水性樹脂の吸水諸特性をバランス良く改良する方法として、吸水性樹脂の表面近傍を架橋する技術が知られており、これまで様々な方法が提案されている。例えば、吸水性樹脂100質量部に対して架橋剤として多価アルコール化合物などの不活性溶媒0.2〜20質量部と、グリシジルエーテル化合物、ハロエポキシ化合物、アルデヒド化合物およびイソシアネート化合物よりなる群より選ばれた1種の架橋剤0.005〜5.0質量部を使用し、表面架橋前吸水性樹脂1質量部に対して水0.01〜1.3質量部を存在させ、温度40〜150℃で架橋させる方法が開示されている(US−A−4,507,438およびUS−A−4,541,871)。架橋密度を高くすると吸収速度は高くなるが吸水能が低下するため、特定の量の水で膨潤させた吸水性樹脂を不活性溶媒中に分散させ、これに架橋剤を反応させ、水への分散性に優れかつ高い吸収速度の吸水性樹脂を得る、というものである。
【0008】
また、無加圧下での吸水倍率に加えて加圧下吸水倍率に優れた吸水性樹脂を得る目的で、吸水性樹脂100質量部に対して、SP値12.5(cal/cm3)以上の第1架橋剤0.1〜5質量部、SP値12.5以下の第2架橋剤0.005〜0.5質量部、および水20質量部以下を用いて、温度160℃以上で加熱処理して不定形破砕状の吸水性樹脂を製造する方法が開示されている(US−A−5,422,405)。第1架橋剤としては、エチレングリコールやプロピレングリコールが例示され、第2架橋剤としてはジエチレングリコール、1,3−ブタンジオール、ポリエチレングリコールジグリシジルエーテル、エチレンジアミン、2,4−トリレンジイソシアネート、エピクロロヒドリンなどが例示されている。実施例では、吸水性樹脂100質量部に対して、水3〜5質量部の水の存在下に架橋剤を作用させている。なお、使用する吸水性樹脂の平均粒子径は最も好ましくは300〜600μmである。
【0009】
また、生理食塩水の平衡膨潤吸収量が40g/g以上であり、所定方法で測定した生理食塩水5mlの液通過時間が40秒以下である高吸収性ポリマーもある(GB−B−2,267,094)。該ポリマーは、カルボキシル基またはカルボキシレート基を有する水不溶性親水性架橋重合体の表面の架橋密度を高めることによって得られる。実施例では、カルボキシル基またはカルボキシレート基を有する水不溶性親水性架橋重合体を用い、水濃度20〜35質量%の条件下で表面架橋剤を2,500ppm添加し、前記架橋重合体粒子の表面の架橋密度を高めている。
【0010】
更に、樹脂濃度の高い衛生材料に好適な吸水性樹脂として、表面架橋層の厚さをサブミクロン単位に薄くし、吸水性樹脂全体に対する表面架橋層の質量%を特定範囲に制御した吸水性樹脂が開示されている(JP−A−2001−192,464)。表面架橋層が厚すぎると吸水性樹脂の吸水能力を妨げたり、膨潤時や膨潤後に表面架橋層中にクラックが入るため、表面架橋層の厚さを50nm以上とし、および表面架橋層の質量%を0.3〜3質量%に制限している。このような吸水性樹脂は、好ましくは多価アルコールを表面架橋剤として使用し、吸水性樹脂の温度を5〜20℃にし、および/または表面架橋剤含有液の温度を0〜20℃にして得られる。実施例では、表面架橋前吸水性樹脂100質量部に対して水2〜4質量部を存在させて表面架橋を行っている。
【0011】
一方、吸水性樹脂の平均粒子径を100〜600μm、粒度分布の対数標準偏差σζ0.35以下に調整することで、表面架橋剤の吸水性樹脂表面への均一分散と表面近傍への適度な浸透を図り、粉体同士の会合を防止して均一な表面架橋を達成する方法もある(US−A−4,973,632、US−A−5,026,800、US−A−5,244,735およびUS−A−6,087,002)。該表面架橋前の吸水性樹脂は、好ましくはブルックフィールド回転粘度計による25℃、0.6rpmにおける粘度が15cps以上の水溶性エチレン性不飽和単量体水溶液を用い、分散剤としてショ糖脂肪酸エステルおよび/またはポリグリセリン脂肪酸エステルを用いて、重合不活性な疎水性有機溶剤中に分散・懸濁させ、ラジカル重合開始剤で重合させて得ている。次いで、含水率10%未満になるまで乾燥した吸水性樹脂粉末を、表面架橋剤0.005〜20質量%、水0.1〜5質量%および親水性有機溶媒0.01〜6質量%と混合した後に加熱反応させ、表面架橋吸水性樹脂を得ている。
【発明の開示】
【発明が解決しようとする課題】
【0012】
しかしながら、上記のような従来公知の表面架橋技術によって得られる表面架橋吸水性樹脂では上記(1)に記載の加圧下吸水倍率、吸収速度が向上する効果があるものの、無加圧下で測定される吸水倍率が大きく低下してしまうという問題があった。また、上記(2)に記載の血液に対する親和性、上記(3)に記載の高湿下での吸湿性が全く改善されないか、むしろ悪化する傾向にあった。
【0013】
血液に対する親和性や、高湿下での吸湿性は吸水性樹脂表面の特性に依存するものと考えられるが、従来公知の報告はそのような表面特性になんら着目することなく、吸収特性の評価のみに終始している場合がほとんど多数を占め、表面架橋処理に伴なう架橋の均一性、不均一性、または表面の化学特性の差異を表現しようとした取り組みはほとんど皆無であった。このような架橋の均一性、不均一性、または表面の化学特性の差異を簡便に、かつ明確に明らかにする手法が求められている。
【0014】
さらに、近年、吸収性製品の薄型化、薄型化に伴なう加工技術の進歩に伴ない、吸水倍率が高く、平均粒子径が小さく、吸収速度の速い吸水性樹脂であって、かつ高い総吸収量の高い吸水性樹脂が要求されている。このような吸水倍率が高く平均粒子径が小さい吸水性樹脂に従来法を行うと、吸水性樹脂の吸収速度が速いため、表面架橋剤水溶液を吸水性樹脂粒子表面に均一に塗布することができず、均一な表面架橋処理を行なうことができなかった。
【0015】
したがって、本発明は、高い吸水倍率と速い吸収速度を確保しつつ、高い総吸収量を示す、優れた吸水材とその製造方法を提供することを目的とする。
【0016】
また、血液、経血に対する親和性を有し、かつ取り扱い性にも優れる表面架橋吸水性樹脂からなる吸水材とこのような表面架橋吸水性樹脂の製造方法を提供することを目的とする。
【0017】
さらに、吸水材の表面特性(架橋特性、化学特性)を調べるための簡便でかつ明確な評価方法を提供することを目的とする。
【0018】
発明の開示
我々は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる従来の表面架橋吸水性樹脂、特に重量平均粒子径が200〜300μmの微細な表面架橋吸水性樹脂について、その架橋表面をX線光電子分光法(ESCAまたはXPS)を用いて詳細に検討したところ、表面架橋された吸水性樹脂であっても、カルボキシル基(−COOH)やその中和物であるカルボキシル塩(−COOM:Mは金属原子を示す。)が吸水性樹脂表面に検出されることを見出した。さらに、上記表面架橋吸水性樹脂をArイオン放電研磨しつつ経時的にX線光電子分光法による分析を行ない、金属原子濃度を測定すると、経時的に吸水性樹脂粒子の金属原子濃度が変化して行き、やがては表面架橋処理していない吸水性樹脂と同様の金属濃度に収束していくことを見出した。このことは上記測定方法により、表面架橋処理による表面架橋層の硬さや厚さを推定することが可能であるということを示している。
【0019】
さらに、我々は、上記知見に基づき、表面架橋吸水性樹脂の表面に残っているカルボキシル基またはカルボキシル塩が血液や経血に対する親和性を悪化させ、さらには高湿下での吸湿性を引き起こしているという仮設を立て、表面架橋吸水性樹脂の表面の金属原子濃度と血液吸収性や吸湿性を比較したところ、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂であって、該樹脂を加圧電圧500VでArイオン放電研磨して測定される該樹脂表面の金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で2〜35%であれば、高い吸水倍率、速い吸収速度、高い総吸収量を示しつつ、劇的に血液に対する親和性が向上し、また、劇的に高湿下での吸湿性が低下するという効果を見出した。このような表面架橋吸水性樹脂は吸水材として使用することができる。
【0020】
さらに、上記のような表面に残存するカルボキシル基またはカルボキシル塩あるいは表面に残存する金属原子濃度の低い表面架橋吸水性樹脂を得るための一例として、重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む、表面架橋吸水性樹脂の製造方法を見出し、本発明を完成させるに至った。
【0021】
本発明の吸水材は、重量平均粒子径が200〜300μmと微細であっても、均一な表面架橋層および所定の硬度を有する。このため、高吸水倍率、血液吸収率、吸収速度等の吸収特性、低吸湿性などの取り扱い性に優れる。該吸水材は、吸収量が多く性能が良好である。特に、水性液体を素早く吸収することができ、かつ、高い液拡散性を維持することができる。また、上記吸水材を構成する表面架橋吸水性樹脂は、特定組成の表面架橋剤含有溶液を使用することで、簡便に調製することができる。
【0022】
図面の簡単な説明
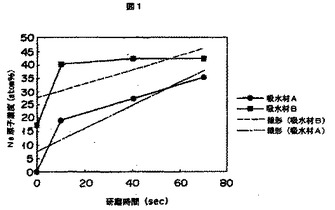
図1は、Arイオン放電研磨による吸水材Aと吸水材Bにおける金属原子濃度(Na濃度)を示す図である。
【0023】
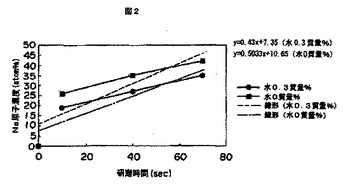
図2は、表面架橋時水0.3質量%添加して調製した表面架橋吸水性樹脂からなる吸水材と、水無添加で調製した表面架橋吸水性樹脂からなる吸水材におけるArイオン放電研磨による金属原子濃度(Na濃度)を示す図である。
【発明を実施するための最良の形態】
【0024】
発明を実施するための最良の形態
本発明の第一は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物よりなる不定形破砕粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で35%以下である、吸水材である。
【0025】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物は、樹脂の全体にわたってカルボキシル基(−COOH)がアルカリ性物質によって中和されたカルボキシル塩(−COOM:Mは金属原子を示す。)を中和度に応じて含有する。このような吸水性樹脂に多価アルコール類、多価グリシジル化合物類などのカルボキシル基と反応しうる架橋剤を使用し、樹脂表面層にあるカルボキシル基やカルボキシル塩と反応させて表面架橋を形成すると、カルボキシル基やカルボキシル塩が存在しない表面架橋層が形成される。一方、表面架橋が不十分であったり、均一でない場合には、樹脂内部のカルボキシル塩が架橋表面に残存する。したがって、得られた表面架橋吸水性樹脂の表面層のカルボキシル塩濃度、ひいては金属原子濃度を測定すれば、表面架橋処理の均一性を知ることができる。また、樹脂表面を研磨しながら中心に向かって金属原子濃度を測定すれば、濃度の変化によって表面架橋層の厚さを知ることができる。研磨の程度は樹脂の硬度と相関するため、所定の研磨力で樹脂を研磨しながら金属原子濃度を測定すると、樹脂表面の硬度を知ることができる。
【0026】
本発明は、このような原理に基づき、表面架橋吸水性樹脂からなる吸水材について、研磨方法としてArイオン放電研磨を採用し、加圧電圧500Vで研磨した場合の研磨0秒値での金属原子濃度が0〜10%であり、研磨10秒値での該金属原子濃度が2〜35%である吸水材を提供するものである。このような特性を有する吸水材は、表面架橋が均一に行われ、適度な表面硬度を有するため、吸収特性にも優れる。なお、本発明において「吸水材表面の金属原子濃度」とは、吸水材をArイオン放電研磨して得た吸水材表面層を構成する原子数に対する金属原子数の百分率である。研磨0秒値の金属原子濃度とは、研磨未処理の状態での架橋表面最外層を構成する原子数に対する金属原子数の百分率である。以下、本発明を詳細に説明する。
【0027】
本発明の吸水材は、加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の金属原子濃度で評価される。アルゴンイオンスパッタにより吸水材を研磨し、研磨の経過に伴う金属原子に由来するピーク比率を測定する。具体的には、吸水材約0.1gを導電性粘着テープ上に固定し、加圧電圧500Vで連続的にアルゴンイオンを放電して研磨し、研磨によって露出した表面の金属原子濃度を経時的に測定する。従って、得られた金属原子濃度は、測定した吸水材の平均値となる。
【0028】
図1に、Arイオン放電研磨装置としてJEOL社製:JPS−9000MXを使用し、MgKα:10kV、10mA、積算10回の条件で、吸水材Aと吸水材Bとを、それぞれ加圧電圧500Vにて70秒間研磨した結果を示す。吸水材Aの実測値は、研磨0秒、10秒、40秒、70秒値において金属原子濃度0%、19%、27%、35%を示し、吸水材Bは17%、40%、42%、42%である。
【0029】
図1より、吸水材Bは、研磨0秒値の金属原子濃度が17%であるため表面架橋が形成されていないか、または表面架橋層が不均一であることを示す。一方、吸水材Aは研磨0秒値の該金属原子濃度が0%であるから、吸水材に表面架橋層が形成されていることを示す。また、吸水材Bは、研磨10秒値以降は、40〜42%で該金属原子濃度が一定するため研磨10秒値で吸水材内部に到達し、吸水材表面が柔らかく、または架橋表面層が薄いことを示す。一方、吸水材Aは該金属原子濃度が研磨70秒値まで漸増していることから吸水材Bよりも耐研磨性が高く表面架橋層が硬くまたは厚いことを示し、吸水材Aは、吸水材Bよりも均一な表面架橋層を有するため、加圧下吸水倍率に優れると推定できる。なお、加圧電圧500V、Arイオン放電によって、モデル材としてSiO2を約30〜40nm/min、PMMAを55nm/min削れることが分かっている。なお、本発明におけるArイオン放電研磨して測定される該吸水材表面の金属原子濃度は、後記する実施例に記載する方法で測定した数値とする。
【0030】
本発明の吸水材は表面架橋吸水性樹脂からなり、アクリル酸またはその金属塩を主成分とするが他の共重合成分を排除するものではない。このため、カルボキシル基以外にも官能基が存在しうる。このような官能基は吸水性樹脂の中和用化合物と反応すると、金属原子として樹脂中に残存する。従って、本発明において、Arイオン放電研磨によって測定する金属原子としては、吸水性樹脂に含まれるアクリル酸や他の官能基の中和塩となる金属原子であり、リチウム、ナトリウム、カリウム、ルビジウムなどの第1族の原子、ベリリウム、マグネシウム、カルシウムなどの第2族の原子が該当する。例えば、水酸化ナトリウムと水酸化カリウムによってアクリル酸や吸水性樹脂が中和されている場合には、カルボキシル基は、ナトリウム塩やカリウム塩となって存在するが、この場合には研磨によって露出したナトリウム原子とカリウム原子との総和の濃度で評価する。
【0031】
このようにArイオン放電研磨によって吸水材表面に現れた金属原子濃度を測定すると、吸水材の表面架橋層の状態を知ることができる。本発明は、研磨0秒値での金属原子濃度が0〜10%、好ましくは0〜7%、特に好ましくは0〜5%であり、さらに好ましくは0〜2%であり、研磨10秒値での金属原子濃度が2〜35%、好ましくは10〜35%、さらに好ましくは10〜30%である吸水材である。研磨0秒値の金属原子濃度が10%を超えると、表面架橋の均一性が不足し、吸収特性が低下する場合がある。なお、研磨10秒時の金属原子濃度は、架橋表面の最外層近傍の架橋状態をよく示す。研磨10秒値の金属原子濃度が35%を超えると、架橋密度が低下し、吸収特性に劣る場合がある。
【0032】
一方、上記Arイオン放電研磨による金属原子濃度の測定によって、研磨の進行に伴う吸水材架橋表面層と吸水材内部との金属原子濃度の推移を評価することができる。本発明では、上記Arイオン放電研磨による吸水材の金属原子濃度の、研磨0秒、10秒、40秒、70秒値における金属原子濃度から得られる研磨時間Xと金属原子濃度(%)Yとの間に下記式で示す関係を有するものがより好ましい。
【0033】
【数1】

【0034】
ここで、0.30≦a≦0.60、0<b<20であり、
Xは、時間(0、10、40、70秒)である。
【0035】
上記式は、経験値に基づくものであり、右辺のbは吸水材の最表面の金属原子濃度を、aは、吸水材表面から吸水材内部に向かって増加する金属原子濃度の増加の程度を示す。本発明の吸水材は、上記Arイオン放電研磨法による研磨0秒、10秒、40秒および70秒値の4つの測定時の金属原子濃度から算出される式:Y=aX+bにおいて、上記関係を満たせばよい。aの傾きが0.30〜0.60、好ましくは0.40〜0.60、bが0<b<20、好ましくは5<b<20である吸水材である。研磨0秒値にYが20%を超えることは表面架橋が不均一であることを意味する。aの傾きが小さい場合や研磨0秒値にYが0%未満や数値が小さい場合には、表面架橋は均一であるが架橋層が硬い。または分厚すぎるために、ポリマー自身に由来する浸透圧の機能を損なう可能性がある。研磨時間Xと金属原子濃度(%)Yとが上記関係を満たす場合には、表面架橋層の均一性、ならびに表面架橋層の硬度などが吸水材として至適であり、均一な表面架橋が形成されることで、高い血液吸収率、溶液分散性、吸水性なども確保される。なお、図1に吸水材Aの研磨時間Xと金属原子濃度(%)Yとの関係式は、Y=0.43X+7.35であり、吸水材BはY=0.2633X+27.35である。得られた直線を図1に併せて示す。
【0036】
吸水材の特性は、吸水材最表面に存在する官能基の種類や量に依存する。このような官能基の中でも、吸水材表面に存在するカルボキシル基やカルボキシル塩は、保存時の着色の原因となり、流動性や粘着性、取扱い性低下を招く。このようなカルボキシル基(金属塩)は、表面架橋が均一に行われない場合や表面架橋が不十分な場合に、吸水材の架橋表面に残存する。このため架橋表面に存在するカルボキシル基量がわかると、吸水材表面の特性を知る指標とできる。本発明では、アクリル酸またはその金属塩を主成分とし、部分中和率が50〜90モル%の吸水性樹脂を表面架橋してなる表面架橋吸水性樹脂からなる吸水材であって、該吸水材にある−COOHを−COOCH2CF3に変換した該表面近傍の炭素原子数に対するフッ素原子比(F/C比)が0.03以下、より好ましくは0.02〜0.01であり、特には0〜0.01である。本来、アクリル酸(H2C=CHCOOH:炭素数3)におけるカルボキシル基(−COOH:炭素数1)を構成する炭素の比は1/3=0.33である。また、アクリル酸の中和率が50〜90モル%の吸水材の内部では、−COOHの10〜50%がそのまま−COOHとなって残存する。従って、表面層の架橋が不十分な場合には−COOHの官能基が表面層から突出する。しかしながら、F/C比が0.03以下であれば、これらの官能基が少なく表面架橋層の均一さが確保され、高吸水倍率、血液吸収率、吸収速度などの吸収特性に優れ、かつ低吸湿性に優れる。なお、該F/C比は、後記する実施例で記載する方法で測定した値である。
【0037】
本発明の吸水材は、重量平均粒子径が200〜300μmであることが好ましい。より好ましくは220〜280μm、特に好ましくは240〜260μmである。この範囲であれば、粒子径が細かく粒度分布がシャープであるため、水性液体の保持能力といった吸水性特性に優れ、また大きな粒子を含まないため、紙オムツ等に加工したときの手触りが良く、異物感を発生しない。近年これら紙おむつや生理用ナプキン等の衛生材料は、高機能かつ薄型化が進み、衛生材料一枚あたりの吸水性樹脂の使用量や、吸水性樹脂と親水性繊維等からなる吸収体全体に対する吸水性樹脂の重量%が増加する傾向にある。つまり、かさ比重の小さい親水性繊維を少なくし、吸水性に優れかつかさ比重の大きい吸水性樹脂を多く使用することにより、吸収体における吸水性樹脂の比率を高め、これにより吸水量を低下させることなく衛生材料の薄型化を図っている。しかしながら、液体の通液・拡散を考えた場合には、多量の吸水性樹脂は吸水により柔らかいゲル状となり、いわゆるゲルブロッキングという液の拡散を妨げる現象をひき起こす。従って、薄型化物に使用される吸水材には吸水性樹脂の物理的形状に由来する吸収特性と、表面架橋処理することで発現するポリマー自身の浸透圧に由来する吸収特性とのバランスが非常に重要であると考えられる。本発明の重量平均粒子径が200〜300μmの範囲にある吸水材は、このような薄型化された紙オムツ等の吸収物品に特に好適である。
【0038】
また、106μm未満の吸水材の割合は0〜10質量%であることが好ましく、0〜7質量%であることがより好ましく、0〜5質量%であることがさらに好ましく、0〜3質量%であることが最も好ましい。106μm未満の吸水材の割合が上記範囲内にあれば、紙オムツ等の吸収性物品での高い総吸収量が確保される上、取り扱い中に粉塵の発生を抑制でき、作業環境が好適に確保される。また、粒子径の分散性を示す対数標準偏差値σζは、0.25〜0.45の範囲にあることが好ましく、0.25〜0.42の範囲にあることがさらに好ましく、0.25〜0.40の範囲であることが最も好ましく、0.25〜0.38の範囲にあることがさらに好ましい。上記範囲に粒子径の分散性を示す対数標準偏差値σζを制御することにより粒子の偏析を抑制でき、連続的に紙オムツ等の吸収性物品を作成する場合に各製品の性能の変動が少なく、安定して吸収性物品を製造することができる。
【0039】
すなわち、吸水材をArイオン放電研磨で研磨しつつ金属原子濃度を測定した場合、架橋表面層の厚さや吸水材内部に至る金属原子濃度の勾配が同じでも、吸水材の平均粒子径が異なれば吸収特性は変化する。しかしながら、吸水材の重量平均粒子径および粒子径106μm未満の吸水材の割合が上記範囲にあり、かつ上記Arイオン放電研磨による吸水材表面の該金属原子濃度の条件を満たすものは、吸収特性や取り扱い性に優れる。更に、上記特性を満たす本発明の吸水材は、表面架橋が均一かつ吸水材として至適な表面架橋層を有するため、生理食塩水に対する吸水倍率が35(g/g)以上となり、35〜60g/g、特に好ましくは35〜50g/g、さらに好ましくは40〜50g/gである。吸水倍率は高ければ高いほどよいが、物性バランスやコストなどから選択すればよい。血液吸収率が70〜100%、より好ましくは80〜100%となる。なお、吸水倍率および血液吸収率は後記する実施例で測定する方法によるものである。
【0040】
本発明の吸水材は、残存エポキシ化合物の含有量が5ppm以下であることが好ましい。表面架橋剤として温度が低くて表面架橋を形成し得る点でエポキシ系表面架橋剤が多用されているが、残存する反応性の高いエポキシによって吸水材表面が劣化したり表面特性が変化する。本発明では、吸水材の残存エポキシ化合物の含有量は、0〜5ppm、好ましくは0〜1ppm、特に好ましくは0〜0.5ppmである。また、吸収速度(Vortex)は、1〜30秒、好ましくは1〜25秒であり、吸湿率は3%以下、好ましくは2%以下である。
【0041】
このような吸水材を構成する表面架橋吸水性樹脂の製造方法に特に制限はないが、本発明の第二によって製造することができる。
【0042】
本発明の第二は、重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む、表面架橋吸水性樹脂の製造方法である。
【0043】
吸水性樹脂は、水を含む表面架橋剤と接触させると直ちに吸水および膨潤するため、表面架橋処理剤中の水の含有量が多い場合には不均一な表面架橋層が形成されやすい。このため、従来から2.0質量%を超える水分の存在下で表面架橋処理を行うことが一般的であった。しかしながら、本発明では、樹脂固形分100質量部に対して水分を2質量%以下に制限しても、溶解度パラメータが11.5(cal/cm3)1/2未満の第二架橋剤が0.015〜0.5質量部の範囲であり、同時に溶解度パラメータが11.5(cal/cm3)1/2以上の第一架橋剤を0.1〜1質量部配合することで、極めて表面均一性の高い表面架橋処理が行えることを見出した。特に、重量平均粒子径が200〜300μmであるような吸水性樹脂は微細であるため、水を多量に含有する表面架橋剤溶液に均一に分散させることは困難であるが、上記組成であれば吸水性樹脂への分散性にも優れるため、均一かつ樹脂内部に向かって適度な厚みを有する表面架橋吸水性樹脂を調製することができる。
【0044】
このような表面架橋処理に際して、吸水性樹脂100質量部に対して表面架橋剤に水を0.3質量部添加して表面架橋した樹脂と、水を添加せずに表面架橋した樹脂について、Arイオン放電研磨によって測定される該樹脂表面の金属原子濃度の推移を図2に示す。水0.3質量%で調製した樹脂の実測値は、研磨0秒、10秒、40秒、70秒値において金属原子濃度0%、19%、27%、35%を示し、水0質量%で調製した樹脂は0%、26%、35%、42%である。樹脂(水0.3質量%)と、樹脂(水0質量%)とは共に研磨0秒値での金属原子濃度が0%であり、表面架橋が均一に行われていることを示している。一方、樹脂(水0.3質量%)では、研磨10秒値に金属原子濃度19%、樹脂(水0質量%)は26%と大きく異なった。なお、研磨10秒値から研磨70秒値までの金属原子濃度の増加傾向は両樹脂において並行している。この結果から明らかなように、表面架橋剤に含まれる水濃度は、架橋表面時の最表面層の金属原子濃度および表面架橋状態に大きな影響を与えている。
【0045】
本発明で使用できる表面架橋前の吸水性樹脂としては、アクリル酸またはその金属塩を主成分とする親水性不飽和単量体を重合させることによって得られる。一般には、イオン交換水に対する吸水倍率が20〜1,000g/g、好ましくは50〜1,000g/g、より好ましくは100〜1,000g/gの、水膨潤性かつ水不溶性(US Re 32649に規定する可溶分(抽出性含量)が0〜50質量%、好ましくは0〜30質量%のものを指す。)の親水性架橋重合体が挙げられる。親水性架橋重合体は該架橋重合体中の酸基のうち、例えば、50〜90モル%、好ましくは60〜80モル%、特には65〜75モル%が、例えば、アルカリ金属塩やアンモニウム塩、アミン塩等によって中和されていることがより好ましい。このアクリル酸基の中和は該架橋重合体を得る前の親水性不飽和単量体を調製する段階で予め中和しておいてから重合反応を開始してもよく、また、重合中あるいは重合反応終了後に得られた該架橋重合体の酸基を中和してもよいし、それらを併用してもよい。「アクリル酸またはその金属塩を主成分とする」とは、親水性不飽和単量体中にアクリル酸またはその金属塩を50〜100モル%含有することを意味する。したがって、上記親水性不飽和単量体は、必要に応じて、アクリル酸またはその金属塩以外の不飽和単量体を含有していてもよい。
【0046】
他の単量体としては、具体的には、例えば、メタクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸等の、アニオン性不飽和単量体およびその塩;アクリルアミド、メタクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン等の、ノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミド、および、これらの四級塩等の、カチオン性不飽和単量体;等が挙げられるが、特に限定されるものではない。
【0047】
該吸水性樹脂を得る際には、内部架橋剤を用いて架橋構造を内部に導入することが望ましい。上記の内部架橋剤は、重合性不飽和基および/またはカルボキシル基と反応し得る反応性基を一分子中に複数有する化合物であればよく、特に限定されるものではない。つまり、内部架橋剤は、親水性不飽和単量体と共重合および/またはカルボキシル基と反応する置換基を一分子中に複数有する化合物であればよい。尚、親水性不飽和単量体は、内部架橋剤を用いなくとも架橋構造が形成される自己架橋型の化合物からなっていてもよい。
【0048】
内部架橋剤としては、具体的には、例えば、N,N’−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキシド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、ポリエチレンイミン、グリシジル(メタ)アクリレート等が挙げられるが、特に限定されるものではない。これらの内部架橋剤は、一種類のみを用いてもよく、また、二種類以上を併用してもよい。そして、上記例示の内部架橋剤のうち、重合性不飽和基を一分子中に複数有する内部架橋剤を用いることにより、得られる吸水性樹脂の吸収特性等をより一層向上させることができる。
【0049】
内部架橋剤の使用量は、親水性不飽和単量体に対して、0.0001〜3モル%、さらに好ましくは0.001〜2モル%、特には0.05〜1モル%の範囲内が好ましく、親水性不飽和単量体を重合させて吸水性樹脂を得る際には、反応系に、デンプン、デンプンの誘導体、セルロース、セルロースの誘導体、ポリビニルアルコール、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子;次亜リン酸(塩)等の連鎖移動剤;水溶性もしくは水分散性の界面活性剤等を0〜100質量%、より好ましくは0〜10質量%の範囲で添加してもよい。
【0050】
親水性不飽和単量体の重合方法は、特に限定されるものではなく、例えば、水溶液重合、バルク重合、沈殿重合等の公知の方法を採用することができる。性能面や重合の制御の容易さから、単量体を水溶液として、水溶液重合を行うことが好ましい。また、反応温度や反応時間等の反応条件は、用いる単量体成分の組成等に応じて適宜設定すればよく、特に限定されるものではない。
【0051】
親水性不飽和単量体を重合させる際には、例えば、過硫酸カリウム、過硫酸ナトリウム、過硫酸アンモニウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2’−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤;2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−オン等のラジカル系光重合開始剤;紫外線や電子線等の活性エネルギー線;等を用いることができる。また、酸化性ラジカル重合開始剤を用いる場合には、例えば、亜硫酸ナトリウム、亜硫酸水素ナトリウム、硫酸第一鉄、L−アスコルビン酸等の還元剤を併用して、レドックス重合を行ってもよい。これら重合開始剤の使用量は、0.001〜2モル%の範囲内が好ましく、0.01〜0.5モル%の範囲内がより好ましい。
【0052】
次いで、上記の重合方法で得られた含水ゲル状重合体を乾燥する。乾燥には、通常の乾燥機や加熱炉を用いることができる。例えば、薄型撹拌乾燥機、回転乾燥機、円盤乾燥機、流動層乾燥機、気流乾燥機、赤外線乾燥機等である。
【0053】
上記乾燥によって得られた乾燥物は、そのまま吸水性樹脂として用いることもできるが、粉砕、分級して平均粒子径が200〜300μm、106μm未満の樹脂の割合を0〜10質量%、好ましくは0〜7質量%、より好ましくは0〜5質量%、特に好ましくは0〜3質量%とすることができる。本発明では、対数標準偏差値σζに限定はないが、0.25〜0.45の範囲でも特定の表面架橋剤を使用することで均一な表面架橋を行うことができる。その他、(1)吸水性樹脂を特定の粉砕機で粉砕する方法、(2)微粉末を造粒して整粒する方法によっても、重量平均粒子径200〜300μm、106μm未満の樹脂の割合が0〜10質量%の吸水性樹脂を調製することができる。
【0054】
(1)粉砕機
使用する粉砕機としては、粉体工学便覧(粉体工学会編、初版)の表1.10で分類されている粉砕機種名のうち、せん断粗砕機、衝撃破砕機、高速回転式粉砕機に分類されるものであり、切断、せん断、衝撃、摩擦という粉砕機構の1つ以上の機構を有するものが好ましく使用でき、特に切断、せん断機構が主機構である粉砕機が好ましい。また、ロール転動型、ロールミル(ロール回転形)に分類されるものであり、粉砕機構として圧縮機構も有するものであっても、せん断、切断効果が強い場合には使用できる。上記した好ましい粉砕機の内でも、複数の回転刃と固定刃のせん断により粉砕する装置であることが好ましい。その回転刃の周速は、3.0〜200m/秒、より好ましくは5.0〜150m/秒である。このような高速回転刃による粉砕でも微粉をほとんど発生せず、粉砕効率も高く、生産性に優れる。回転刃の周速が3.0m/秒未満であると極端な処理量低下とともに、粉砕粒子径が増大し、材料の練られも増えて可溶分の増加が起こる場合がある。一方、200m/秒よりも速いと処理量は多くなるものの装置コストが高くなる場合がある。
【0055】
本発明においては、含水率10質量%以上30質量%以下の含水ゲル状重合体を粉砕機で粉砕することにより、分級操作を経ない粉砕後または分級操作を経ない粉砕・乾燥後の粒径分布において、粒径150μm以上850μm以下の微細粒子が75質量%以上である粒子状吸水性樹脂を高生産性で得ることができる。それに加え、粒径150μm未満の微細粒子が15質量%以下および/または粒径106μm未満の超微細粒子が10質量%未満となる粒子状吸水性樹脂を高生産性で得ることができる。さらに、粉砕し、粒径150μm以上500μm以下という狭い粒径範囲にある粒子が50質量%以上でかつ粒径150μm未満の微細粒子が15質量%以下、および/または粒径150μm以上500μm以下の微細粒子が50質量%以上で粒径106μm未満の微細粒子が10質量%以下である、粒子状吸水性樹脂を高生産性で得ることが容易にできる。
【0056】
(2)微粉末の造粒
水性液をバインダーとして使用し、該水性液と吸水性樹脂粉末中に含まれる微粉末とを混合して粘結造流体を形成しこれを造粒すると、微粉末を除去でき、かつ所定範囲に粒度を調整することができる。なお、水性液の使用量が多いために造粒体の表面が粘着性を有する場合には、該造粒体を一定時間放置したり、加熱することで粘着性を低減させることができる。加熱温度は50〜200℃で3分〜12時間が好適であり、さらに好ましくは70〜120℃、10分〜2時間である。なお、乾燥は必ずしも必要でない。使用できる水性液としては、水単独、水と混和性のある有機溶剤との混合液があり、水の配合量は、水50〜100質量%、より好ましくは水70〜100質量%である。水との混和性のある有機溶剤としては、低級アルコール、テトラヒドロフラン、アセトン等を挙げることができる。上記水性バインダーには、更に各種化合物や混合物を溶解または分解させてもよい。このような化合物や混合物としては、JP−A−61−97333に記載された消臭剤、植物生育助剤の他に、微粒子状シリカのスラリー等を挙げることができる。なお、微粉末に添加する水性液量は特に限定されず広い範囲で使用できるが、少量では顕著な造粒効果が得られ難く、多量使用の場合に造粒後の乾燥工程がないと吸水性能の低下を招く場合がある。好ましくは、吸水性樹脂100質量部に対して、水性液を1〜50質量部、より好ましくは3〜35質量部とする。
【0057】
上記のようにして得られた吸水性樹脂は、不定形破砕状、顆粒状、棒状、繊維状、鱗片状等の形状であってもよい。一般には、不定形破砕状粒子は、均一な表面架橋が困難と考えられるが、本発明においては、特定の表面架橋剤を使用することで、このような形状の吸水性樹脂粒子でも均一な表面架橋層を有する吸水性樹脂とすることができる。なお、本発明における吸水性樹脂の「重量平均粒子径」は、試験用ふるい(JIS Z8801−2000)目開き500μm、425μm、355μm、300μm、150μm、106μm、75μmの篩を用いて重合体粉体を篩分級した後、残留百分率Rを対数確率紙にプロットし、R=50%に相当する粒子径を重量平均粒子径とした。また、粒度分布は、その指標として下記の式であらわされる対数標準偏差値σζを用いた。ここでは、σζの値が小さいほど粒度分布が狭いことを意味する。ふるい目開きを適宜変更することで、下式を満たすことができる。
【0058】
【数2】
【0059】
(X1はR=84.1%、X2はR=15.9%、近傍2点で線形近似するときのそれぞれの粒子径)
上記によって得られた含水ゲル状重合体、すなわち表面架橋前の吸水性樹脂は、含水率(180℃、3時間乾燥条件)が10質量%未満、好ましくは7質量%未満、さらに好ましくは5質量%未満である。吸水性樹脂の含水率が10質量%を超えると、本発明に用いる第一架橋剤と第二架橋剤とが吸水性樹脂の表面に均一に分散できず、架橋表面にカルボキシル基やカルボキシル塩が残存する場合がある。
【0060】
本発明に用いることのできる第一架橋剤は、溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で水性樹脂前駆体が有するカルボキシル基と反応可能な化合物である。溶解度パラメータ(SP値)は化合物の極性を現すファクタ−として一般に用いられており本発明ではポリマ−ハンドブック第3版(WILEY INTERSCIENCE社発行)VII−527〜539に記載されている溶媒の溶解度パラメータ(SP値)δ[(cal/cm3)1/2]の値を適用するものとし、この表に記載されていない架橋剤に関しては同ハンドブックのVII−524のSmallの式にVII−525に記載されているHoyの凝集エネルギ−定数を代入して導いた値δ[(cal/cm3)1/2]を適用するものとする。
【0061】
このような溶解度パラメータ(SP値)11.5(cal/cm3)1/2以上でカルボキシル基と反応し得る第一架橋剤としては、エチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、ソルビトール、エチレンカ−ボネイト、プロピレンカ−ボネイト、ジエチレングリコール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタジオール、2,4−ペンタジオール、1,6−ヘキサンジオール、2,5−ヘンキサンジオール、トリメチロールプロパン、ジエタノールアミン、4,5−ジメチル−1,3−ジオキソラン−2−オン等が例示され、これらの群より選ばれる1種または2種以上を用いることができる。好ましくは溶解度パラメータ(SP値)13.0(cal/cm3)1/2以上のものである。
【0062】
本発明に用いることのできる第二架橋剤は、溶解度パラメータ(SP値)11.5(cal/cm3)1/2未満で水性樹脂前駆体が有するカルボキシル基と反応可能な化合物であり、具体的には、トリエチレングリコール、テトラエチレングリコール、ジプロピレングリコール、トリプロピレングリコール、トリエタノールアミン、エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート、エピクロロヒドリン、エピブロモヒドリン等が例示できこれらの群より選ばれる1種または2種以上を用いることができる。より好ましくは、溶解度パラメータ(SP値)9.5〜11.0(cal/cm3)1/2の範囲にあるものである。
【0063】
またさらに、第一および第二架橋剤を選択するにあたっては、第一架橋剤の溶解度パラメータ(SP値)と第二架橋剤の溶解度パラメータ(SP値)の差が2(cal/cm3)1/2以上になるように選ぶことが好ましい。溶解度パラメータ(SP値)の差が2(cal/cm3)1/2未満の場合には二種類の架橋剤を用いた効果が現れにくく得られた吸水性樹脂の加圧下の吸収特性が向上しにくいことがある。さらに好ましくは第一架橋剤の溶解度パラメータ(SP値)と第二架橋剤の溶解度パラメータ(SP値)の差が2.5(cal/cm3)1/2以上、特に好ましくは3.0(cal/cm3)1/2以上であるように選択される。
【0064】
本発明において用いることのできる架橋剤としては、重合体の有する官能基と反応し得る官能基を2個以上有する化合物であれば特に制限はないが、好ましくは親水性、より好ましくは水溶性の化合物であり、例えば第一架橋剤としては、多価アルコール類、第二架橋剤としては多価グリシジル化合物類が好ましい。
【0065】
本発明では架橋剤として、上記した第一架橋剤の群より選ばれる1種以上と第二架橋剤の群より選ばれる1種以上に加えて水を用いることが必要である。これらの使用量は、吸水性樹脂100質量部に対し、第一架橋剤0.1〜1質量部、第二架橋剤0.015〜0.5質量部、水0〜2質量部であり、より好ましくは第一架橋剤0.1〜0.5質量部、第二架橋剤0.015〜0.3質量部、水0〜1質量部であり、特に好ましくは第一架橋剤0.1〜0.3質量部、第二架橋剤0.015〜0.1質量部、水0〜0.5質量部である。特に、重量平均粒子径が200〜300μmである前記吸水性樹脂に表面架橋を行う際に、第一架橋剤のみを使用した場合には本発明で規定する特定の表面特性を有する表面架橋がされない恐れがあり好ましくない。また、第一架橋剤の使用量が、吸水性樹脂100質量部に対して0.1質量部を下回ると、同時に配合する第二架橋剤が樹脂表面に均一に分散されず表面架橋が不均一となる場合がある。一方、第一架橋剤および第二架橋剤の使用量が上記範囲を超えると、残存表面架橋剤量が増加し、品質が低下する場合がある。また、水の配合量を2質量部以内に限定すると、表面架橋剤の吸水性樹脂内部への浸透を防止でき、樹脂表面をより均一に架橋することができる。このため、表面にカルボキシル基やカルボキシル塩の残存量が少ない表面架橋吸水性樹脂を得ることができる。なお、従来から、重量平均粒子径が200〜300μmと微細な吸水性樹脂を表面架橋剤などの液状組成物に混合する際に、粒度分布が広いと吸水性樹脂同士が合一して大きな塊を生じるため、均一な表面架橋が困難であるとされていた。しかしながら、本発明では、上記する特定組成の表面架橋剤を使用することで、吸水性樹脂の粒度分布の対数標準偏差値σζが0.25〜0.45と範囲が広い場合であっても、表面架橋層が均一かつ硬度の高い表面架橋吸水性樹脂を得ることができる。
【0066】
本発明で使用する表面架橋剤溶液は、上記第一架橋剤、第二架橋剤および水を上記範囲で含むものであるが、更に、吸水性樹脂の固形分100質量部に対して0.015〜1.0質量部、好ましくは0.1〜0.4質量部の範囲で、親水性有機溶媒を添加してもよい。用いられる親水性有機溶媒としては、メチルアルコール、エチルアルコール、n−プロピルアルコール、iso−プロピルアルコール、n−ブチルアルコール、iso−ブチルアルコール、t−ブチルアルコール等の低級アルコール類;アセトン等のケトン類;ジオキサン、テトラヒドロフラン等のエーテル類;N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類を挙げることができる。
【0067】
本発明では、第一および第二架橋剤および水を含む表面架橋剤溶液を直接、吸水性樹脂に噴霧あるいは滴下し混合する。好適な混合装置は、均一な混合を確実にするため大きな混合力を生み出せる事が必要である。本発明に用いることのできる混合装置としては例えば、円筒型混合機、二重壁円錐型混合機、V字型混合機、リボン型混合機、スクリュ−型混合機、流動型炉ロータリーデスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機およびスクリュー型押出機等があげられる。
【0068】
本発明では、ベースポリマーとしての吸水性樹脂と第一および第二架橋剤とを混合した後、更に加熱を行い、表面近傍を架橋させる。
【0069】
本発明で架橋剤を添加した後、加熱処理を行う場合、加熱処理温度(熱媒温度または材料温度)は100〜300℃、好ましくは160〜250℃、より好ましくは180〜250℃である。加熱処理温度が100℃未満では、加熱処理に時間がかかり生産性の低下をひき起こすのみならず、第一および第二架橋剤両者による均一な架橋が達成されず、本発明の目的とする吸収特性の高い樹脂が得られない。
【0070】
加熱処理は通常の乾燥機または加熱炉を用いて行うことができ、溝型混合乾燥機、ロータリー乾燥機、デスク乾燥機、流動層乾燥機、気流型乾燥機、および赤外線乾燥機等が例示できる。
【0071】
従来無加圧下の吸水倍率を高めると、加圧下での吸水倍率が低下してしまい、また逆に加圧下での吸水倍率を高めると、無加圧下での吸水倍率が低下してしまうという問題が常にあったが、本発明で得られる吸水性樹脂は無加圧下吸水倍率、加圧下吸水倍率および血液吸収率に優れ、吸収速度が高く、低吸湿性を示すなど極めて優れた吸収特性を示す。この原因については定かではないが、吸水性樹脂を溶解度パラメータの異なる二種類以上の架橋剤および特定範囲の水分量の表面架橋剤と混合し、特定の温度範囲で架橋することによって、吸水性樹脂の表面近傍における両架橋剤の分布が最適化されかつ両者が速やかに架橋反応することにより、これまでの方法では達成されることのなかったような特異な密度勾配をもつ架橋が粒子表面近傍において形成されるためではないかと推測される。また表面架橋前の吸水性樹脂の重量平均粒子径と粒度分布を先に述べたように限定することで、上記優れた吸収特性に加え、表面特性が一定化するため分散性が良好であり、残存表面架橋剤が少なく、2次添加物の添加が容易、かつ官能基との反応性が低いため、混合性(パルプとのブレンド)に優れ、不織布などに均一に塗布しうる表面特性に優れる、という特徴を有する。このように、従来の表面架橋吸水性樹脂に比べて、一度に多量の水性液体が添加されても、該水性液体を素早く吸収することができ、かつ、液拡散性や吸収量等の吸収特性に優れるため、衛生材料等の吸収性物品を提供することができる。
【0072】
本発明の吸水材は、上記表面架橋吸水性樹脂をそのまま吸水材として使用することができ、例えば、高機能化かつ薄型化が望まれている紙オムツや生理用ナプキン、いわゆる失禁パット等の衛生材料等の吸収性物品に特に好適に用いることができる。また、上記表面架橋吸水性樹脂に、他の添加剤、例えば機能性向上剤、消臭剤、香料、無機粉末、発泡剤、顔料、染料、親水性短繊維、肥料、酸化剤、還元剤、水、塩類等を0〜30質量%添加して新たな機能を附加させたものであってもよい。
【0073】
本発明では、例えば、上記の様にして得られたカルボキシル基を有する吸水性樹脂に対して、イオン封鎖剤とカルボキシル基と反応し得る表面架橋剤を混合することにより、耐尿性の優れた吸水剤を得ることができる。本発明に用いられるイオン封鎖剤としては、以下の化合物が挙げられる。
【0074】
(1)アミノカルボン酸及びその塩、(2)クエン酸モノアルキルアミド及びクエン酸モノアルケニルアミド及びそれらの塩、(3)マロン酸モノアルキルアミド及びマロン酸モノアルケニルアミド及びそれらの塩、(4)モノアルキルリン酸エステル及びモノアルケニルリン酸エステル及びそれらの塩、(5)N−アシル化グルタミン酸及びN−アシル化アスパラギン酸及びそれらの塩、(6)β―ジケトン誘導体、(7)トロポロン誘導体、(8)有機リン酸化合物。
【0075】
(1)アミノカルボン酸及びその塩としてはカルボキシル基を3個以上有するアミノカルボン酸及びその塩がイオン封鎖能の点で好ましい。具体的には、ニトリロトリ酢酸、エチレンジアミンテトラ酢酸、ジエチレントリアミンペンタ酢酸、トリエチレンテトラアミンヘキサ酢酸、シクロヘキサンー1,2−ジアミンテトラ酢酸、N−ヒドロキシエチルエチレンジアミントリ酢酸、エチレングリコールジエチルエーテルジアミンテトラ酢酸、エチレンジアミンテトラプロピオン酸、N−アルキルーN’−カルボキシメチルアスパラギン酸、N−アルケニルーN’−カルボキシメチルアスパラギン酸、及びこれらのアルカリ金属塩、アルカリ土類金属塩、アンモニウム塩もしくはアミン塩が挙げられる。
【0076】
(2)クエン酸モノアルキルアミド及びクエン酸モノアルケニルアミド及びそれらの塩は、例えばアルコールとクエン酸の脱水縮合により得られる。
【0077】
(3)マロン酸モノアルキルアミド及びマロン酸モノアルケニルアミド及びそれらの塩は、例えば、α―オレフィンをマロン酸メチルに付加せしめた後加水分解することにより得られる。
【0078】
(4)モノアルキルリン酸エステル及びモノアルケニルリン酸エステル及びそれらの塩としては、ラウリルリン酸、ステアリルリン酸等が挙げられる。
【0079】
(5)N−アシル化グルタミン酸及びN−アシル化アスパラギン酸及びそれらの塩としては、例えば(株)味の素より市販されているアミソフトHS−11やGS−11等が挙げられる。
【0080】
(6)β―ジケトン誘導体としては、アセチルアセトン、ベンゾイルアセトン等が挙げられる。
【0081】
(7)トロポロン誘導体としてはトロポロン、β―ツヤプリシン、γ―ツヤプリシン等が挙げられる。
【0082】
(8)有機リン酸化合物としてはエチリデンホスホン酸;1−ヒドロキシエチリデン−1,1−ジホスホン酸;アミノトリメチレンホスホン酸;エチレンジアミンテトラ(メチレンホスホン酸);ジエチレントリアミンペンタ(メチレンホスホン酸)等を挙げることができるが、特に好ましいものは1−ヒドロキシエチリデン−1,1−ジホスホン酸;エチレンジアミンテトラ(メチレンホスホン酸);ジエチレントリアミンペンタ(メチレンホスホン酸)である。塩として好ましいものは、Na塩、K塩等のアルカリ金属塩、アンモニウム塩、アミン塩を挙げることができる。これらの化合物は、金属封鎖剤の一種として知られているものである。
【0083】
これらイオン封鎖剤の中でも好ましくはカルボキシル基を3個以上有するアミノカルボン酸及びその塩であり、中でもジエチレントリアミンペンタ酢酸、トリエチレンテトラアミンヘキサ酢酸、シクロヘキサンー1,2−ジアミノテトラ酢酸、N−ヒドロキシエチルエチレンジアミントリ酢酸及びその塩が、耐尿性の点で最も好ましい。
【0084】
本発明において上記イオン封鎖剤の使用量は、表面近傍の架橋に用いる表面架橋剤(添加時期については特に制限はないが、重合工程、表面処理工程、造粒工程、微粉回収工程のいずれかに添加する方法がある。)によって異なるが、通常吸水性樹脂の固形分100重量部に対して0.0001〜10重量部、好ましくは0.0002〜5重量部の範囲である。使用量が10重量部を越えると、使用に見合う効果が得られず不経済になるばかりか、吸収量が低下するなどの問題が生じる。また、0.0001重量部よりも少ないと耐尿性向上の効果が得られない。
【0085】
本発明で使用される機能性向上剤として、例えば、親水性の無機化合物があり、水不溶性親水性の無機微粒子や水溶性の多価金属塩などが好ましく用いられる。なお、本発明でいう親水性無機微粒子とは、例えば、EP−B−0629411に記載されている親水性度70%以上のものが例示できる。また、US−A−5797893のカラム11に例示されるカチオン性高分子化合物や疎水性の無機微粒子なども機能性向上剤として使用でき、液透過性を向上させ得る。例えば、タルク、カオリン、フラー土、ベントナイト、活性白土、重晶石、天然アスファルタム、ストロンチウム鉱石、イルメナイト、パーライトなどの鉱産物;硫酸アルミニウム14〜18水塩(または無水物)、硫酸カリウムアルミニウム12水塩、硫酸ナトリウムアルミニウム12水塩、硫酸アンモニウムアルミニウム12水塩、塩化アルミニウム、ポリ塩化アルミニウム、酸化アルミニウムなどのアルミニウム化合物類;その他の多価金属塩、多価金属酸化物および多価金属水酸化物;親水性のアモルファスシリカ(例、乾式法:トクヤマ社 ReolosilQS−20、沈殿法:DEGUSSA社 Sipernat22S, Sipernat2200)類;酸化ケイ素・酸化アルミ・酸化マグネシウム複合体(例、ENGELHARD社.Attagel#50)、酸化ケイ素・酸化アルミニウム複合体、酸化ケイ素・酸化マグネシウム複合体などの酸化物複合体類;などが例示できる。また、US−A−5164459、EP−A−761241に例示されたものも使用することができる。これらの粒子の中から親水性の粒子(例えば、硫酸アルミニウム14〜18水塩や親水性のアモルファスシリカ)を選択して使用するのが好ましいが、粒子の親水性が低い場合は、粒子表面を親水性化合物で処理して親水化したものを使用してもよい。本発明では、これらのいずれか1種を単独で用いてもよいし、2種以上を併用してもよい。なお、添加できる機能性向上剤(例えば、粉体流動性向上剤、吸湿プロッキング性向上剤、通液性向上剤等)の粒径は、無機微粒子の場合には、好ましくは5μm以下、より好ましくは1μm以下、最も好ましくは0.1μm以下である。なお、凝集体や造粒物を形成する場合には、それらを構成する1次粒子の粒子径を上記範囲とすることが好ましい。
【0086】
このような機能向上剤の混合方法は、硫酸アルミニウム等の水溶性多価金属塩やカチオン性高分子化合物を水溶液として混合する方法やスラリー状として混合する方法、粉体として混合する方法などある。好ましくは粉体として混合する方法である。吸水性樹脂粒子に対する添加量は、0.01〜5質量%が好ましく、0.05〜3質量%がより好ましい。添加量が5質量%より多くなると吸収倍率の低下を招き、0.01質量%より少ないと添加の効果が得られない場合がある。混合装置は、特に大きな混合力を備える必要はなく、例えば、解砕機や篩機などで混合されてもよい。上記の混合装置としては、例えば、円筒型混合機、二重壁円錐型混合機、V字型混合機、リボン型混合機、スクリュー型混合機、流動型炉ロータリーデスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機、スクリュー型押出機、スタティックミキサー等が好適である。また、添加の時期は前記製法において、吸水剤が得られる前、製造中、製造後のいずれでもよいが、好ましくは表面架橋後である。
【0087】
本発明の第三は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕粒子を表面架橋した吸水性樹脂から構成される吸水材の評価方法であって、該吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材の前記金属原子濃度を経時的に計測する工程を含む、吸水材の評価方法である。ここに、「不定形破砕状の粒子」とは、水溶液重合等によって得られる含水重合物を乾燥後、粉砕して得られる粒子であり、粉砕に伴う破断面(平滑面)と角が電子顕微鏡または光学顕微鏡によって確認できる粒子である。
【0088】
上記第一の発明で説明したように、Arイオン放電研磨して測定される該樹脂粒子表面の金属原子濃度を経時的に計測すると、各測定時における特定の金属種の原子濃度から吸水材の表面特性を知ることができる。例えば、上記したように研磨0秒、10秒、40秒、70秒において、研磨時間Xと金属原子濃度(%)Yとの関係式:Y=aX+bにおいて、0.30≦a≦0.60、0<b<20を満たす場合には、表面架橋層の均一性、ならびに表面架橋層の硬度、血液吸収率、凝集性、溶液分散性、吸水性などとの相関が観察される。このことは、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂からなる吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材粒子表面の金属原子濃度を経時的に計測すると、吸水材を評価できることを意味する。この際、加圧電圧やArイオン放電研磨などは、対象とする吸水材の種類や粒子径などに応じて適宜選択すればよく、金属原子濃度の測定時も粒子径などに応じて適宜選択すればよい。また、測定結果と評価項目との関係は予め対象とする吸水材の有する特性と相関させることで評価することができる。本発明においては、特に吸水材の架橋表面層の硬度、表面架橋層の均一性、吸水性、血液吸収率、凝集性、および溶液分散性などの評価を行うことができる。従来から、吸水材の特性は、実際に保水条件で行うことが一般的であったが、保水させることなく特性を評価できることは画期的なことである。本発明の評価方法によれば、吸収特性、機能性の評価、基本物性等の解析などに有用である。
【実施例】
【0089】
次に実施例を挙げて本発明を具体的に説明する。ただし、これらの実施例は何ら本発明を制限するものではない。以下に、表面架橋吸水性樹脂または吸収性物品を対象として測定方法を説明するが、これらに代えて吸水材を使用すれば、吸水材の諸特性を評価することができる。なお、本発明においては、吸収特性を評価するために用いる溶液は0.9質量%塩化ナトリウム水溶液であり、それを略称して生理食塩水、または、0.9質量%生理食塩水と呼ぶことがあるが、それらは全て同じものである。市販のおむつに含まれる吸水性樹脂を評価する場合には、流通過程での吸湿を考慮して、吸水性樹脂を乾燥(例えば、60℃での減圧乾燥で16時間)して平衡含水状態(例えば、5質量%前後)とした後に物性を測定する。なお、本発明で得られる表面架橋吸水性樹脂は、実質水不溶性であり、含水率がいずれも7質量%以下(固形分93%以上)、残存モノマー量が400質量ppm以下であった。
【0090】
(1) 吸水倍率(CRC/Centrifuge Retention Capacity)
表面架橋吸水性樹脂約0.20g(Wp1)を正確に秤量し、不織布製の袋(60×60mm)に均一に入れてヒートシールしたのち、25℃±2℃に調温した0.9質量%塩化ナトリウム水溶液中に浸漬した。60分後に袋を引き上げ、遠心分離機(株式会社コクサン製、型式H−122小型遠心分離機)を用いて250G(250×9.81m/s2)で3分間水切りを行った後重量Wa(g)を測定した。また、同様の操作を、吸水性樹脂を用いないで行い、そのときの重量Wb(g)を測定した。そして、これらの重量Wp1、WaおよびWbから、次式に従って表面架橋吸水性樹脂の吸水倍率(g/g)を算出した。なお、イオン交換水の場合には、吸水材は0.02gで測定した。
【0091】
【数3】
【0092】
(2) 粒度分布・重量平均粒子径
表面架橋吸水性樹脂10.0gを、目開き500μm、425μm、355μm、300μm、150μm、106μm、75μmの篩を用いて重合体粉体を仕込み、ロータップ型ふるい振盪機(株式会社飯田製作所製、ES−65型ふるい振盪機)により5分間分級した後、残留百分率Rを対数確率紙にプロットし、R=50%に相当する粒子径を重量平均粒子径とした。また、粒度分布は、その指標として下記の式であらわされる対数標準偏差値σζを用いた。
【0093】
【数4】
【0094】
(X1はR=84.1%、X2はR=15.9%、近傍2点で線形近似するときのそれぞれの粒子径)
(3) 吸湿率
表面架橋吸水性樹脂約1.0gを正確に秤量し、アルミカップ(底面の直径52mm、高さ22mm)上に均一に散布し、25℃、50%RHの恒温恒湿室に24時間放置した。そのときの重量Wc(g)を測定した。そして、含水した吸水性樹脂を、180℃3時間乾燥を行い、絶乾重量Wdを測定し、次式に従って吸水性樹脂の吸湿率(%)を算出した。(ただし、アルミカップの重量は除いて算出する。)
【0095】
【数5】
【0096】
(4) 吸収体、吸収性物品の製造例と吸収性物品(モデルおむつ)の性能評価方法
4−1.吸収体の作成
液体非通液透過性の裏面シートとして大きさ14×40cmの長方形ポリエチレンフィルム(坪量18g/m2)の上に、吸水性樹脂粒子を12×38cmの面積に16.4g散布した(吸水性樹脂の散布量360g/m2)。その上に液獲得部材として子供用おむつに使用する綿状パルプ3gを大きさ8×24cm(密度0.04g/cm3で坪量160g/m2)を積層して吸収体を構成した。最後に液透過性表面材として12×40cmの長方形ポリエステル不織布(坪量20g/m2)を載置し、サイド部分をシールして、モデル吸収性物品を得た。
【0097】
4−2.通液時間・総吸収量
上記吸収性物品を机上にテープで平面状に固定し、その上に、12×40cmのアクリル板(中央部分に液注入のための直径70mmの円筒が具備されている)および、1.3kgの荷重をかけた。
【0098】
37℃に調整した生理食塩水75mlを円筒の中に注ぎ、表面シートから液が吸収性物品内部に吸収し終わった時間(通液時間1回目)を測定した。
【0099】
この操作を4回、60分毎に繰り返し、通液時間を測定(sec.)した。
【0100】
また、上記した吸収性物品を、生理食塩水に1時間浸漬を行った。そして、吸収性物品を引き上げ、液透過性表面材を下方に2つ折で10分間水切りを行った。10分間水切り後の重量を求め、以下の式で総吸収量(g)を求めた。
【0101】
【数6】
【0102】
(5) 血液吸収率
内径60mmのシャーレ中に吸水性樹脂0.5g(175g/m2)を均一に散布し、羊血(脱繊血;緬羊脱繊維血液「日本バイオテスト研究所」)10gをシャーレの中心点から約10秒かけて滴下し添加し、静置下で10分間放置後、未吸収液を0.7psi下、1分間ろ紙10枚(戻り量により適宜変更)(ADVANTEC 東洋濾紙株式会社製、品名 JIS P 3801 No.2)により吸着させ戻り量(We g)を測定する。添加液量10gより戻り量を差し引いて吸収量(g)を測定し、下記式に従い、羊血10gの全量を吸液できた場合の最大吸収量20(=10g/0.5g)に対する血液吸収率を算出した。
【0103】
【数7】
【0104】
(6) Arイオン放電研磨による金属原子濃度
吸水性樹脂を、目開き300μmパス、150μmオン(試料収集が困難な場合、425〜75μmの範囲内で適宜交換する)を110℃、約4時間乾燥させる。この試料を用いて加速イオンを各試料に照射し、イオン衝突により粒子表面を研磨した。この試料を用いてX線光電子分光ESCA(Electron Spectroscopy for Chemical Analysis)で以下に従い表面架橋吸水性樹脂の官能基を分析した。
【0105】
1.試料調製
試料台(試料を載せる部分は約6cm×1cmの長方形)に、導電性テープを約1cm角に切断して貼り、その上に試料を散布して固定する。最大6試料載せうるが、スパッタする場合は4個までとする。導電性テープ上に試料を適当量散布し、テープに付かなかった分は窒素ガス吹き飛ばし、目視で隙間がほとんどなくなる程度に固定する。なお、ESCAは表面状態を評価するものであり、導電性テープ(日新EM株式会社製)に隙間なく試料が付着していることが重要である。
【0106】
2.ESCA測定条件
装置:JEOL JPS−9000MX
条件:MgをターゲットにしKα線使用、加速電圧:10kV、エミュション電流:10mA、積算:10回、
3.スパッタ条件
装置:Arイオンガン(熱陰極電子衝撃型)
条件:イオンビーム電流:50mA、イオンビーム径:1.5mm、Arイオンの加速電圧:500Vで加速電流:8.5mA、Arガス圧:3×10−2Pa
4.試料測定
(1)試料をESCAの予備排気室に入れ、約24時間予備排気する。
【0107】
(2)試料をESCAの測定用の試料室に移動し、ESCA測定してゼロ秒データとする。
【0108】
(3)試料を予備排気室に移動し、スパッタを指定時間(a秒時)行なう。Arガスを排気後、試料を測定用の試料室に移動してESCA測定し、a秒データとする。(0秒値)
(4)同じ試料について、スパッタを指定時間(b秒時)行ないESCA測定し、a+b秒データとする(X秒値)。
【0109】
(5)必要な時間スパッタと測定を繰り返す。
【0110】
5.原子濃度(原子%)の算出
得られた光電子スペクトル(ESCAスペクトル)を装置付属の解析ソフトを用いて、以下の手順で計算処理を実施した。
【0111】
(1)得られたスペクトルに対しスムージングを行ない、微細なノイズを除去する。参照点数は11点で実施した。
【0112】
(2)スペクトルのエネルギー軸を炭素の1s準位を基準(285eV)として、帯電補正を行なう。なお、この操作は定量計算には影響しない。
【0113】
(3)スペクトルからバックグランド補正計算により、バックグランド・スペクトルを除去する。バックグランド補正計算は、Shirly法で行なう。バックグランド補正計算を行なうための開始・終点の指定は、画面上でピーク形状を確認して手動で行なった。ただし、この作業は、定量計算結果に影響を与える。特に、含有比率が小さく、ピーク形状が明瞭でない場合は、誤差を生ずる要因となる。
【0114】
(4)スペクトルから得られる面積値(eV*cps)を対象に、装置付属の解析ソフトに備わった相対感度因子を用いて定量補正計算を行ない、各原子の相対原子比率(C、Oおよび目的金属原子の総数を100%とした場合の目的金属原子の比率(原子%))を原子濃度(原子%)とする。対象原子がナトリウムであれば、Na原子濃度が求められる。
【0115】
(7)F/C比
各試料を110℃、約4時間乾燥させた。この試料45mgを10mlサンプル管に秤りとり、2,2,2−トリフルオロエタノール(TFE)/ジシクロヘキシルカルボジイミド(DCC)/ピリジンの0.02/0.001/0.04モル比の混合液約0.3gを添加してから密閉し、60℃、8時間加熱した。ついで、ピリジンを加えて攪拌し、濾液をろ紙で吸い上げ除去を3回繰り返して洗浄した。その後、70℃、約100Torrで16時間、更に約1Torrで8時間真空乾燥した。該反応によって、表面架橋吸水性樹脂の表面にある−COOHが−COOCH2CF3に変換される。該反応を下記に示す。
【0116】
【化1】
【0117】
この試料を用いて上記(7)に記載するESCA測定条件で表面架橋吸水性樹脂の官能基を分析した。F/C比は下式で示され、Fの相対原子比率をCの相対原子比率で除したものである。
【0118】
【数8】
【0119】
(8)残存表面架橋剤量(エポキシ化合物の場合)
吸水材2.0gを100mlのビーカーに加え、メタノール/水=2/1質量比からなる組成液2mlを加え、蓋をして1時間放置する。メタノール5mlを上記ビーカーに加え濾過し、濾液1.0gを50mlのナスフラスコに入れ、12質量%のニコチンアミド水溶液0.05mlを添加する。ナスフラスコに空冷管をつけ沸騰したウォーターバスで30分間加熱する。反応液を、濾紙を用いて濾過し、濾過液を高性能液体クロマトグラフィーで分析する。一方、吸水材を用いず既知量の架橋剤を加えて同様の操作を行ない、得られた検量線を外部標準とし、濾過液の希釈倍率を考慮して、吸水材中の残存表面架橋剤量(ppm)を求める。
【0120】
(9)吸収速度(Vortex)
予め調整された0.90質量%塩化ナトリウム水溶液(生理食塩水)1,000質量部に食品添加物である食用青色1号(CAS番号:3844−45−9)0.02質量部を添加し、液温30℃に調整した。その生理食塩水50mlを胴径55mm、高さ70mmの容量100mlのビーカー(例えば相互理化学硝子製作所が販売するJISR−3503に準拠したビーカー)に計り取り、長さ40mm、太さ8mmの円筒型テフロン(登録商標)製マグネット式撹拌子(例えば、相互理化学ガラス製作所が販売するS型)で600rpmで撹拌する中に、後述する実施例または比較例で得られた吸水性樹脂2.0gを投入し、吸収速度(秒)を測定する。始点、終点は、JIS K 7224(1996年度)「高吸水性樹脂の吸水速度試験方法 解説」に記載されている基準に準じ、吸水性樹脂が生理食塩水を吸液してスターラーチップを試験液で覆うまでの時間を測定し吸収速度(秒)として評価する。
【0121】
(実施例1)
中和率75モル%のアクリル酸ナトリウム37質量%水溶液5,500gに、内部架橋剤としてのポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数:8モル)2.8g(0.025mol%)を溶解させて反応液とした。次に、この反応液を、窒素ガス雰囲気下で30分間脱気した。次いで、開閉可能な蓋付きのシグマ型羽根を2本有するジャケット付きステンレス製双腕型ニーダーに、上記の反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。
【0122】
続いて、反応液を攪拌しながら、過硫酸ナトリウム2.8gおよびL−アスコルビン酸0.12gを添加したところ、およそ1分後に重合が開始した。そして、開始からピーク温度30℃〜95℃で重合を行い、重合を開始してから60分後に反応を終了して含水ゲル状重合体を取り出した。
【0123】
得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を、振動ミルを用いて粉砕し、さらに30メッシュ(目開き500μm)の金網で分級することにより、重量平均粒子径が240μmで、しかも、粒子径が106μm未満の粒子の割合が5質量%の不定形破砕状の吸水性樹脂を得た。このものの吸水倍率は52g/gであった。
【0124】
得られた吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))0.3質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))0.1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理することにより、表面架橋吸水性樹脂を得た。得られた表面架橋吸水性樹脂の重量平均粒子径は249μmであり、粒子径が106μm未満の粒子の割合は1質量%であった。
【0125】
この表面架橋吸水性樹脂について、金属原子濃度(0秒値、10秒値)、F/C比、吸水倍率、吸収速度、吸湿率、残存表面架橋剤量などを測定した。結果を表1に記載した。また、この表面架橋吸水性樹脂を前記吸体吸収性物品の製造例および吸収性物品の性能評価方法に従って、吸収性物品とし、性能を評価した。各実施例および比較例の結果を表1に記載した。
【0126】
(実施例2)
上記の実施例1で得た表面架橋吸水性樹脂100gに、更に微粒子状の親水性二酸化ケイ素(商品名・アエロジル200;日本アエロジル株式会社製)0.3gを添加・混合したものを吸水性樹脂とした。
【0127】
(実施例3)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))0.3質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))0.1質量部と、水0.3質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂の重量平均粒子径は258μmであり、粒子径が106μm未満の粒子の割合は1質量%であった。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0128】
(実施例4)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール0.5質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル0.1質量部と、水1.5質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0129】
(比較例1)
上記の実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール0.8質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル0.02質量部と、水3質量部と、イソプロピルアルコール1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で30分間加熱処理することにより、表面架橋が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0130】
(比較例2)
上記の実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))0.3質量部と、プロピレングリコール0.5質量部、水2質量部とからなる表面架橋剤組成液を混合した。上記の混合物を195℃で40分間加熱処理することにより、比較用吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0131】
(比較例3)
実施例1で得られた含水ゲル重合体を、目開き500μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を振動ミルを用いて粉砕し、さらに18.5メッシュ(目開き850μm)の金網で分級することにより、重量平均粒子径が480μmで、しかも、粒子径が106μm未満の粒子の割合が5質量%の不定形破砕状の吸水性樹脂を得た。
【0132】
得られた不定形破砕状の吸水性樹脂100質量部に、エチレングリコールジグリシジルエーテル0.1質量部と、プロピレングリコール0.3質量部、水5質量部とからなる表面架橋剤組成液を混合した。上記の混合物を210℃で30分間加熱処理することにより、比較用吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0133】
(実施例5)
中和率75モル%のアクリル酸ナトリウム37質量%水溶液5,500gに、内部架橋剤としてのポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数:8モル)6.72g(0.06mol%)を溶解させて反応液とした。次に、この反応液を、窒素ガス雰囲気下で30分間脱気した。次いで、開閉可能な蓋付きのシグマ型羽根を2本有するジャケット付きステンレス製双腕型ニーダーに、上記の反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。
【0134】
続いて、反応液を攪拌しながら、過硫酸ナトリウム2.8gおよびL−アスコルビン酸0.12gを添加したところ、およそ1分後に重合が開始した。そして、30℃〜95℃で重合を行い、重合を開始してから60分後に反応を終了して含水ゲル状重合体を取り出した。
【0135】
得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を、振動ミルを用いて粉砕し、さらに30メッシュ(目開き500μm)の金網で分級することにより、重量平均粒子径が240μmで、しかも、粒子径が106μm未満の粒子の割合が2質量%の不定形破砕状の吸水性樹脂を得た。このものの吸水倍率は40g/gであった。
【0136】
得られた吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))を0.2質量部、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))を0.2質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))を0.1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を210℃で30分間加熱処理することにより、表面処理が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0137】
(実施例6)
実施例5で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))を0.2質量部、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))を0.2質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))を0.1質量部と水0.3質量%からなる表面架橋剤溶液を混合した。上記の混合物を210℃で30分間加熱処理することにより、表面処理が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0138】
(実施例7)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてプロピレングリコール0.1質量部と、第二架橋剤としてエチレングリコールジグリシジルエーテル0.02質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0139】
(比較例4)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、パラフィン(融点60−62℃ 和光純薬工業(株))の破砕物1.0質量部を混合した。上記の混合物を80℃で10分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0140】
【表1−1】
【0141】
【表1−2】
【0142】
(実施例8)
実施例3において、吸水性樹脂100質量部に対する表面架橋時に添加する水量を表2に示す配合量で使用した以外は実施例3と同様に操作して吸水性樹脂を得た。各吸水性樹脂のF/C比、金属原子濃度(Na濃度)、混合性を評価した。なお、混合性は、表面架橋剤溶液と吸水性樹脂との同量を混合した際の評価であり、○は混合性良好を示し、△は一部凝集物生成を示し、×は凝集物生成または一体化を示す。結果を表2に合わせて示す。
【0143】
【表2】
【産業上の利用可能性】
【0144】
本発明によれば、吸収特性に優れる吸水材が提供され、紙おむつなどの吸収材として有用である。
【図面の簡単な説明】
【0145】
【図1】図1は、Arイオン放電研磨による吸水材Aと吸水材Bにおける金属原子濃度(Na濃度)を示す図である。
【図2】図2は、表面架橋時水0.3質量%添加して調製した表面架橋吸水性樹脂からなる吸水材と、水無添加で調製した表面架橋吸水性樹脂からなる吸水材におけるArイオン放電研磨による金属原子濃度(Na濃度)を示す図である。
【技術分野】
【0001】
技術分野
本発明は吸水材に関するものである。更に詳しくは、特定範囲の平均粒子径を有し、かつ粒度分布の狭い表面が均一な表面架橋層および所定の表面硬さを有し、吸収特性、取り扱い性に優れる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材、該表面架橋吸水性樹脂の製造方法、および吸水材の評価方法に関する。
【背景技術】
【0002】
背景技術
近年、体液を吸収させることを目的とし、紙オムツ、生理用ナプキンなどの衛生材料の構成材料の一つとして吸水性樹脂が幅広く利用されている。吸水性樹脂に望まれる特性としては、以下の3つの特性を挙げることができる。
【0003】
(1)吸収特性に優れること、すなわち水性液体に接した際に吸水倍率が高く、加圧下吸水倍率が高く、吸収速度が速く、水性液体を含んだ基材から水性液体を吸い上げる能力が高いこと。
【0004】
(2)吸収対象が広いこと、すなわち水や尿のみならず、血液や経血等の体液に対して親和性が高いこと。
【0005】
(3)樹脂の取り扱い性に優れること、すなわち高湿下における吸湿性が低く流動性が高いこと、また、微粒子(例えば106μm未満)の含有量が少なく、発塵性が低いこと。
【0006】
しかしながら、吸収特性に関し、上記(1)に記載の各性能間の関係は必ずしも正の相関関係を示さず、例えば、吸水倍率の高いものほど吸収速度、ゲル強度の物性は低下してしまう。また、吸水性樹脂の平均粒子径が大きくなるほど通液性は高くなるが、吸収速度が低下してしまう。
【0007】
このような吸水性樹脂の吸水諸特性をバランス良く改良する方法として、吸水性樹脂の表面近傍を架橋する技術が知られており、これまで様々な方法が提案されている。例えば、吸水性樹脂100質量部に対して架橋剤として多価アルコール化合物などの不活性溶媒0.2〜20質量部と、グリシジルエーテル化合物、ハロエポキシ化合物、アルデヒド化合物およびイソシアネート化合物よりなる群より選ばれた1種の架橋剤0.005〜5.0質量部を使用し、表面架橋前吸水性樹脂1質量部に対して水0.01〜1.3質量部を存在させ、温度40〜150℃で架橋させる方法が開示されている(US−A−4,507,438およびUS−A−4,541,871)。架橋密度を高くすると吸収速度は高くなるが吸水能が低下するため、特定の量の水で膨潤させた吸水性樹脂を不活性溶媒中に分散させ、これに架橋剤を反応させ、水への分散性に優れかつ高い吸収速度の吸水性樹脂を得る、というものである。
【0008】
また、無加圧下での吸水倍率に加えて加圧下吸水倍率に優れた吸水性樹脂を得る目的で、吸水性樹脂100質量部に対して、SP値12.5(cal/cm3)以上の第1架橋剤0.1〜5質量部、SP値12.5以下の第2架橋剤0.005〜0.5質量部、および水20質量部以下を用いて、温度160℃以上で加熱処理して不定形破砕状の吸水性樹脂を製造する方法が開示されている(US−A−5,422,405)。第1架橋剤としては、エチレングリコールやプロピレングリコールが例示され、第2架橋剤としてはジエチレングリコール、1,3−ブタンジオール、ポリエチレングリコールジグリシジルエーテル、エチレンジアミン、2,4−トリレンジイソシアネート、エピクロロヒドリンなどが例示されている。実施例では、吸水性樹脂100質量部に対して、水3〜5質量部の水の存在下に架橋剤を作用させている。なお、使用する吸水性樹脂の平均粒子径は最も好ましくは300〜600μmである。
【0009】
また、生理食塩水の平衡膨潤吸収量が40g/g以上であり、所定方法で測定した生理食塩水5mlの液通過時間が40秒以下である高吸収性ポリマーもある(GB−B−2,267,094)。該ポリマーは、カルボキシル基またはカルボキシレート基を有する水不溶性親水性架橋重合体の表面の架橋密度を高めることによって得られる。実施例では、カルボキシル基またはカルボキシレート基を有する水不溶性親水性架橋重合体を用い、水濃度20〜35質量%の条件下で表面架橋剤を2,500ppm添加し、前記架橋重合体粒子の表面の架橋密度を高めている。
【0010】
更に、樹脂濃度の高い衛生材料に好適な吸水性樹脂として、表面架橋層の厚さをサブミクロン単位に薄くし、吸水性樹脂全体に対する表面架橋層の質量%を特定範囲に制御した吸水性樹脂が開示されている(JP−A−2001−192,464)。表面架橋層が厚すぎると吸水性樹脂の吸水能力を妨げたり、膨潤時や膨潤後に表面架橋層中にクラックが入るため、表面架橋層の厚さを50nm以上とし、および表面架橋層の質量%を0.3〜3質量%に制限している。このような吸水性樹脂は、好ましくは多価アルコールを表面架橋剤として使用し、吸水性樹脂の温度を5〜20℃にし、および/または表面架橋剤含有液の温度を0〜20℃にして得られる。実施例では、表面架橋前吸水性樹脂100質量部に対して水2〜4質量部を存在させて表面架橋を行っている。
【0011】
一方、吸水性樹脂の平均粒子径を100〜600μm、粒度分布の対数標準偏差σζ0.35以下に調整することで、表面架橋剤の吸水性樹脂表面への均一分散と表面近傍への適度な浸透を図り、粉体同士の会合を防止して均一な表面架橋を達成する方法もある(US−A−4,973,632、US−A−5,026,800、US−A−5,244,735およびUS−A−6,087,002)。該表面架橋前の吸水性樹脂は、好ましくはブルックフィールド回転粘度計による25℃、0.6rpmにおける粘度が15cps以上の水溶性エチレン性不飽和単量体水溶液を用い、分散剤としてショ糖脂肪酸エステルおよび/またはポリグリセリン脂肪酸エステルを用いて、重合不活性な疎水性有機溶剤中に分散・懸濁させ、ラジカル重合開始剤で重合させて得ている。次いで、含水率10%未満になるまで乾燥した吸水性樹脂粉末を、表面架橋剤0.005〜20質量%、水0.1〜5質量%および親水性有機溶媒0.01〜6質量%と混合した後に加熱反応させ、表面架橋吸水性樹脂を得ている。
【発明の開示】
【発明が解決しようとする課題】
【0012】
しかしながら、上記のような従来公知の表面架橋技術によって得られる表面架橋吸水性樹脂では上記(1)に記載の加圧下吸水倍率、吸収速度が向上する効果があるものの、無加圧下で測定される吸水倍率が大きく低下してしまうという問題があった。また、上記(2)に記載の血液に対する親和性、上記(3)に記載の高湿下での吸湿性が全く改善されないか、むしろ悪化する傾向にあった。
【0013】
血液に対する親和性や、高湿下での吸湿性は吸水性樹脂表面の特性に依存するものと考えられるが、従来公知の報告はそのような表面特性になんら着目することなく、吸収特性の評価のみに終始している場合がほとんど多数を占め、表面架橋処理に伴なう架橋の均一性、不均一性、または表面の化学特性の差異を表現しようとした取り組みはほとんど皆無であった。このような架橋の均一性、不均一性、または表面の化学特性の差異を簡便に、かつ明確に明らかにする手法が求められている。
【0014】
さらに、近年、吸収性製品の薄型化、薄型化に伴なう加工技術の進歩に伴ない、吸水倍率が高く、平均粒子径が小さく、吸収速度の速い吸水性樹脂であって、かつ高い総吸収量の高い吸水性樹脂が要求されている。このような吸水倍率が高く平均粒子径が小さい吸水性樹脂に従来法を行うと、吸水性樹脂の吸収速度が速いため、表面架橋剤水溶液を吸水性樹脂粒子表面に均一に塗布することができず、均一な表面架橋処理を行なうことができなかった。
【0015】
したがって、本発明は、高い吸水倍率と速い吸収速度を確保しつつ、高い総吸収量を示す、優れた吸水材とその製造方法を提供することを目的とする。
【0016】
また、血液、経血に対する親和性を有し、かつ取り扱い性にも優れる表面架橋吸水性樹脂からなる吸水材とこのような表面架橋吸水性樹脂の製造方法を提供することを目的とする。
【0017】
さらに、吸水材の表面特性(架橋特性、化学特性)を調べるための簡便でかつ明確な評価方法を提供することを目的とする。
【0018】
発明の開示
我々は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる従来の表面架橋吸水性樹脂、特に重量平均粒子径が200〜300μmの微細な表面架橋吸水性樹脂について、その架橋表面をX線光電子分光法(ESCAまたはXPS)を用いて詳細に検討したところ、表面架橋された吸水性樹脂であっても、カルボキシル基(−COOH)やその中和物であるカルボキシル塩(−COOM:Mは金属原子を示す。)が吸水性樹脂表面に検出されることを見出した。さらに、上記表面架橋吸水性樹脂をArイオン放電研磨しつつ経時的にX線光電子分光法による分析を行ない、金属原子濃度を測定すると、経時的に吸水性樹脂粒子の金属原子濃度が変化して行き、やがては表面架橋処理していない吸水性樹脂と同様の金属濃度に収束していくことを見出した。このことは上記測定方法により、表面架橋処理による表面架橋層の硬さや厚さを推定することが可能であるということを示している。
【0019】
さらに、我々は、上記知見に基づき、表面架橋吸水性樹脂の表面に残っているカルボキシル基またはカルボキシル塩が血液や経血に対する親和性を悪化させ、さらには高湿下での吸湿性を引き起こしているという仮設を立て、表面架橋吸水性樹脂の表面の金属原子濃度と血液吸収性や吸湿性を比較したところ、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂であって、該樹脂を加圧電圧500VでArイオン放電研磨して測定される該樹脂表面の金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で2〜35%であれば、高い吸水倍率、速い吸収速度、高い総吸収量を示しつつ、劇的に血液に対する親和性が向上し、また、劇的に高湿下での吸湿性が低下するという効果を見出した。このような表面架橋吸水性樹脂は吸水材として使用することができる。
【0020】
さらに、上記のような表面に残存するカルボキシル基またはカルボキシル塩あるいは表面に残存する金属原子濃度の低い表面架橋吸水性樹脂を得るための一例として、重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む、表面架橋吸水性樹脂の製造方法を見出し、本発明を完成させるに至った。
【0021】
本発明の吸水材は、重量平均粒子径が200〜300μmと微細であっても、均一な表面架橋層および所定の硬度を有する。このため、高吸水倍率、血液吸収率、吸収速度等の吸収特性、低吸湿性などの取り扱い性に優れる。該吸水材は、吸収量が多く性能が良好である。特に、水性液体を素早く吸収することができ、かつ、高い液拡散性を維持することができる。また、上記吸水材を構成する表面架橋吸水性樹脂は、特定組成の表面架橋剤含有溶液を使用することで、簡便に調製することができる。
【0022】
図面の簡単な説明
図1は、Arイオン放電研磨による吸水材Aと吸水材Bにおける金属原子濃度(Na濃度)を示す図である。
【0023】
図2は、表面架橋時水0.3質量%添加して調製した表面架橋吸水性樹脂からなる吸水材と、水無添加で調製した表面架橋吸水性樹脂からなる吸水材におけるArイオン放電研磨による金属原子濃度(Na濃度)を示す図である。
【発明を実施するための最良の形態】
【0024】
発明を実施するための最良の形態
本発明の第一は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物よりなる不定形破砕粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で35%以下である、吸水材である。
【0025】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物は、樹脂の全体にわたってカルボキシル基(−COOH)がアルカリ性物質によって中和されたカルボキシル塩(−COOM:Mは金属原子を示す。)を中和度に応じて含有する。このような吸水性樹脂に多価アルコール類、多価グリシジル化合物類などのカルボキシル基と反応しうる架橋剤を使用し、樹脂表面層にあるカルボキシル基やカルボキシル塩と反応させて表面架橋を形成すると、カルボキシル基やカルボキシル塩が存在しない表面架橋層が形成される。一方、表面架橋が不十分であったり、均一でない場合には、樹脂内部のカルボキシル塩が架橋表面に残存する。したがって、得られた表面架橋吸水性樹脂の表面層のカルボキシル塩濃度、ひいては金属原子濃度を測定すれば、表面架橋処理の均一性を知ることができる。また、樹脂表面を研磨しながら中心に向かって金属原子濃度を測定すれば、濃度の変化によって表面架橋層の厚さを知ることができる。研磨の程度は樹脂の硬度と相関するため、所定の研磨力で樹脂を研磨しながら金属原子濃度を測定すると、樹脂表面の硬度を知ることができる。
【0026】
本発明は、このような原理に基づき、表面架橋吸水性樹脂からなる吸水材について、研磨方法としてArイオン放電研磨を採用し、加圧電圧500Vで研磨した場合の研磨0秒値での金属原子濃度が0〜10%であり、研磨10秒値での該金属原子濃度が2〜35%である吸水材を提供するものである。このような特性を有する吸水材は、表面架橋が均一に行われ、適度な表面硬度を有するため、吸収特性にも優れる。なお、本発明において「吸水材表面の金属原子濃度」とは、吸水材をArイオン放電研磨して得た吸水材表面層を構成する原子数に対する金属原子数の百分率である。研磨0秒値の金属原子濃度とは、研磨未処理の状態での架橋表面最外層を構成する原子数に対する金属原子数の百分率である。以下、本発明を詳細に説明する。
【0027】
本発明の吸水材は、加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の金属原子濃度で評価される。アルゴンイオンスパッタにより吸水材を研磨し、研磨の経過に伴う金属原子に由来するピーク比率を測定する。具体的には、吸水材約0.1gを導電性粘着テープ上に固定し、加圧電圧500Vで連続的にアルゴンイオンを放電して研磨し、研磨によって露出した表面の金属原子濃度を経時的に測定する。従って、得られた金属原子濃度は、測定した吸水材の平均値となる。
【0028】
図1に、Arイオン放電研磨装置としてJEOL社製:JPS−9000MXを使用し、MgKα:10kV、10mA、積算10回の条件で、吸水材Aと吸水材Bとを、それぞれ加圧電圧500Vにて70秒間研磨した結果を示す。吸水材Aの実測値は、研磨0秒、10秒、40秒、70秒値において金属原子濃度0%、19%、27%、35%を示し、吸水材Bは17%、40%、42%、42%である。
【0029】
図1より、吸水材Bは、研磨0秒値の金属原子濃度が17%であるため表面架橋が形成されていないか、または表面架橋層が不均一であることを示す。一方、吸水材Aは研磨0秒値の該金属原子濃度が0%であるから、吸水材に表面架橋層が形成されていることを示す。また、吸水材Bは、研磨10秒値以降は、40〜42%で該金属原子濃度が一定するため研磨10秒値で吸水材内部に到達し、吸水材表面が柔らかく、または架橋表面層が薄いことを示す。一方、吸水材Aは該金属原子濃度が研磨70秒値まで漸増していることから吸水材Bよりも耐研磨性が高く表面架橋層が硬くまたは厚いことを示し、吸水材Aは、吸水材Bよりも均一な表面架橋層を有するため、加圧下吸水倍率に優れると推定できる。なお、加圧電圧500V、Arイオン放電によって、モデル材としてSiO2を約30〜40nm/min、PMMAを55nm/min削れることが分かっている。なお、本発明におけるArイオン放電研磨して測定される該吸水材表面の金属原子濃度は、後記する実施例に記載する方法で測定した数値とする。
【0030】
本発明の吸水材は表面架橋吸水性樹脂からなり、アクリル酸またはその金属塩を主成分とするが他の共重合成分を排除するものではない。このため、カルボキシル基以外にも官能基が存在しうる。このような官能基は吸水性樹脂の中和用化合物と反応すると、金属原子として樹脂中に残存する。従って、本発明において、Arイオン放電研磨によって測定する金属原子としては、吸水性樹脂に含まれるアクリル酸や他の官能基の中和塩となる金属原子であり、リチウム、ナトリウム、カリウム、ルビジウムなどの第1族の原子、ベリリウム、マグネシウム、カルシウムなどの第2族の原子が該当する。例えば、水酸化ナトリウムと水酸化カリウムによってアクリル酸や吸水性樹脂が中和されている場合には、カルボキシル基は、ナトリウム塩やカリウム塩となって存在するが、この場合には研磨によって露出したナトリウム原子とカリウム原子との総和の濃度で評価する。
【0031】
このようにArイオン放電研磨によって吸水材表面に現れた金属原子濃度を測定すると、吸水材の表面架橋層の状態を知ることができる。本発明は、研磨0秒値での金属原子濃度が0〜10%、好ましくは0〜7%、特に好ましくは0〜5%であり、さらに好ましくは0〜2%であり、研磨10秒値での金属原子濃度が2〜35%、好ましくは10〜35%、さらに好ましくは10〜30%である吸水材である。研磨0秒値の金属原子濃度が10%を超えると、表面架橋の均一性が不足し、吸収特性が低下する場合がある。なお、研磨10秒時の金属原子濃度は、架橋表面の最外層近傍の架橋状態をよく示す。研磨10秒値の金属原子濃度が35%を超えると、架橋密度が低下し、吸収特性に劣る場合がある。
【0032】
一方、上記Arイオン放電研磨による金属原子濃度の測定によって、研磨の進行に伴う吸水材架橋表面層と吸水材内部との金属原子濃度の推移を評価することができる。本発明では、上記Arイオン放電研磨による吸水材の金属原子濃度の、研磨0秒、10秒、40秒、70秒値における金属原子濃度から得られる研磨時間Xと金属原子濃度(%)Yとの間に下記式で示す関係を有するものがより好ましい。
【0033】
【数1】
【0034】
ここで、0.30≦a≦0.60、0<b<20であり、
Xは、時間(0、10、40、70秒)である。
【0035】
上記式は、経験値に基づくものであり、右辺のbは吸水材の最表面の金属原子濃度を、aは、吸水材表面から吸水材内部に向かって増加する金属原子濃度の増加の程度を示す。本発明の吸水材は、上記Arイオン放電研磨法による研磨0秒、10秒、40秒および70秒値の4つの測定時の金属原子濃度から算出される式:Y=aX+bにおいて、上記関係を満たせばよい。aの傾きが0.30〜0.60、好ましくは0.40〜0.60、bが0<b<20、好ましくは5<b<20である吸水材である。研磨0秒値にYが20%を超えることは表面架橋が不均一であることを意味する。aの傾きが小さい場合や研磨0秒値にYが0%未満や数値が小さい場合には、表面架橋は均一であるが架橋層が硬い。または分厚すぎるために、ポリマー自身に由来する浸透圧の機能を損なう可能性がある。研磨時間Xと金属原子濃度(%)Yとが上記関係を満たす場合には、表面架橋層の均一性、ならびに表面架橋層の硬度などが吸水材として至適であり、均一な表面架橋が形成されることで、高い血液吸収率、溶液分散性、吸水性なども確保される。なお、図1に吸水材Aの研磨時間Xと金属原子濃度(%)Yとの関係式は、Y=0.43X+7.35であり、吸水材BはY=0.2633X+27.35である。得られた直線を図1に併せて示す。
【0036】
吸水材の特性は、吸水材最表面に存在する官能基の種類や量に依存する。このような官能基の中でも、吸水材表面に存在するカルボキシル基やカルボキシル塩は、保存時の着色の原因となり、流動性や粘着性、取扱い性低下を招く。このようなカルボキシル基(金属塩)は、表面架橋が均一に行われない場合や表面架橋が不十分な場合に、吸水材の架橋表面に残存する。このため架橋表面に存在するカルボキシル基量がわかると、吸水材表面の特性を知る指標とできる。本発明では、アクリル酸またはその金属塩を主成分とし、部分中和率が50〜90モル%の吸水性樹脂を表面架橋してなる表面架橋吸水性樹脂からなる吸水材であって、該吸水材にある−COOHを−COOCH2CF3に変換した該表面近傍の炭素原子数に対するフッ素原子比(F/C比)が0.03以下、より好ましくは0.02〜0.01であり、特には0〜0.01である。本来、アクリル酸(H2C=CHCOOH:炭素数3)におけるカルボキシル基(−COOH:炭素数1)を構成する炭素の比は1/3=0.33である。また、アクリル酸の中和率が50〜90モル%の吸水材の内部では、−COOHの10〜50%がそのまま−COOHとなって残存する。従って、表面層の架橋が不十分な場合には−COOHの官能基が表面層から突出する。しかしながら、F/C比が0.03以下であれば、これらの官能基が少なく表面架橋層の均一さが確保され、高吸水倍率、血液吸収率、吸収速度などの吸収特性に優れ、かつ低吸湿性に優れる。なお、該F/C比は、後記する実施例で記載する方法で測定した値である。
【0037】
本発明の吸水材は、重量平均粒子径が200〜300μmであることが好ましい。より好ましくは220〜280μm、特に好ましくは240〜260μmである。この範囲であれば、粒子径が細かく粒度分布がシャープであるため、水性液体の保持能力といった吸水性特性に優れ、また大きな粒子を含まないため、紙オムツ等に加工したときの手触りが良く、異物感を発生しない。近年これら紙おむつや生理用ナプキン等の衛生材料は、高機能かつ薄型化が進み、衛生材料一枚あたりの吸水性樹脂の使用量や、吸水性樹脂と親水性繊維等からなる吸収体全体に対する吸水性樹脂の重量%が増加する傾向にある。つまり、かさ比重の小さい親水性繊維を少なくし、吸水性に優れかつかさ比重の大きい吸水性樹脂を多く使用することにより、吸収体における吸水性樹脂の比率を高め、これにより吸水量を低下させることなく衛生材料の薄型化を図っている。しかしながら、液体の通液・拡散を考えた場合には、多量の吸水性樹脂は吸水により柔らかいゲル状となり、いわゆるゲルブロッキングという液の拡散を妨げる現象をひき起こす。従って、薄型化物に使用される吸水材には吸水性樹脂の物理的形状に由来する吸収特性と、表面架橋処理することで発現するポリマー自身の浸透圧に由来する吸収特性とのバランスが非常に重要であると考えられる。本発明の重量平均粒子径が200〜300μmの範囲にある吸水材は、このような薄型化された紙オムツ等の吸収物品に特に好適である。
【0038】
また、106μm未満の吸水材の割合は0〜10質量%であることが好ましく、0〜7質量%であることがより好ましく、0〜5質量%であることがさらに好ましく、0〜3質量%であることが最も好ましい。106μm未満の吸水材の割合が上記範囲内にあれば、紙オムツ等の吸収性物品での高い総吸収量が確保される上、取り扱い中に粉塵の発生を抑制でき、作業環境が好適に確保される。また、粒子径の分散性を示す対数標準偏差値σζは、0.25〜0.45の範囲にあることが好ましく、0.25〜0.42の範囲にあることがさらに好ましく、0.25〜0.40の範囲であることが最も好ましく、0.25〜0.38の範囲にあることがさらに好ましい。上記範囲に粒子径の分散性を示す対数標準偏差値σζを制御することにより粒子の偏析を抑制でき、連続的に紙オムツ等の吸収性物品を作成する場合に各製品の性能の変動が少なく、安定して吸収性物品を製造することができる。
【0039】
すなわち、吸水材をArイオン放電研磨で研磨しつつ金属原子濃度を測定した場合、架橋表面層の厚さや吸水材内部に至る金属原子濃度の勾配が同じでも、吸水材の平均粒子径が異なれば吸収特性は変化する。しかしながら、吸水材の重量平均粒子径および粒子径106μm未満の吸水材の割合が上記範囲にあり、かつ上記Arイオン放電研磨による吸水材表面の該金属原子濃度の条件を満たすものは、吸収特性や取り扱い性に優れる。更に、上記特性を満たす本発明の吸水材は、表面架橋が均一かつ吸水材として至適な表面架橋層を有するため、生理食塩水に対する吸水倍率が35(g/g)以上となり、35〜60g/g、特に好ましくは35〜50g/g、さらに好ましくは40〜50g/gである。吸水倍率は高ければ高いほどよいが、物性バランスやコストなどから選択すればよい。血液吸収率が70〜100%、より好ましくは80〜100%となる。なお、吸水倍率および血液吸収率は後記する実施例で測定する方法によるものである。
【0040】
本発明の吸水材は、残存エポキシ化合物の含有量が5ppm以下であることが好ましい。表面架橋剤として温度が低くて表面架橋を形成し得る点でエポキシ系表面架橋剤が多用されているが、残存する反応性の高いエポキシによって吸水材表面が劣化したり表面特性が変化する。本発明では、吸水材の残存エポキシ化合物の含有量は、0〜5ppm、好ましくは0〜1ppm、特に好ましくは0〜0.5ppmである。また、吸収速度(Vortex)は、1〜30秒、好ましくは1〜25秒であり、吸湿率は3%以下、好ましくは2%以下である。
【0041】
このような吸水材を構成する表面架橋吸水性樹脂の製造方法に特に制限はないが、本発明の第二によって製造することができる。
【0042】
本発明の第二は、重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む、表面架橋吸水性樹脂の製造方法である。
【0043】
吸水性樹脂は、水を含む表面架橋剤と接触させると直ちに吸水および膨潤するため、表面架橋処理剤中の水の含有量が多い場合には不均一な表面架橋層が形成されやすい。このため、従来から2.0質量%を超える水分の存在下で表面架橋処理を行うことが一般的であった。しかしながら、本発明では、樹脂固形分100質量部に対して水分を2質量%以下に制限しても、溶解度パラメータが11.5(cal/cm3)1/2未満の第二架橋剤が0.015〜0.5質量部の範囲であり、同時に溶解度パラメータが11.5(cal/cm3)1/2以上の第一架橋剤を0.1〜1質量部配合することで、極めて表面均一性の高い表面架橋処理が行えることを見出した。特に、重量平均粒子径が200〜300μmであるような吸水性樹脂は微細であるため、水を多量に含有する表面架橋剤溶液に均一に分散させることは困難であるが、上記組成であれば吸水性樹脂への分散性にも優れるため、均一かつ樹脂内部に向かって適度な厚みを有する表面架橋吸水性樹脂を調製することができる。
【0044】
このような表面架橋処理に際して、吸水性樹脂100質量部に対して表面架橋剤に水を0.3質量部添加して表面架橋した樹脂と、水を添加せずに表面架橋した樹脂について、Arイオン放電研磨によって測定される該樹脂表面の金属原子濃度の推移を図2に示す。水0.3質量%で調製した樹脂の実測値は、研磨0秒、10秒、40秒、70秒値において金属原子濃度0%、19%、27%、35%を示し、水0質量%で調製した樹脂は0%、26%、35%、42%である。樹脂(水0.3質量%)と、樹脂(水0質量%)とは共に研磨0秒値での金属原子濃度が0%であり、表面架橋が均一に行われていることを示している。一方、樹脂(水0.3質量%)では、研磨10秒値に金属原子濃度19%、樹脂(水0質量%)は26%と大きく異なった。なお、研磨10秒値から研磨70秒値までの金属原子濃度の増加傾向は両樹脂において並行している。この結果から明らかなように、表面架橋剤に含まれる水濃度は、架橋表面時の最表面層の金属原子濃度および表面架橋状態に大きな影響を与えている。
【0045】
本発明で使用できる表面架橋前の吸水性樹脂としては、アクリル酸またはその金属塩を主成分とする親水性不飽和単量体を重合させることによって得られる。一般には、イオン交換水に対する吸水倍率が20〜1,000g/g、好ましくは50〜1,000g/g、より好ましくは100〜1,000g/gの、水膨潤性かつ水不溶性(US Re 32649に規定する可溶分(抽出性含量)が0〜50質量%、好ましくは0〜30質量%のものを指す。)の親水性架橋重合体が挙げられる。親水性架橋重合体は該架橋重合体中の酸基のうち、例えば、50〜90モル%、好ましくは60〜80モル%、特には65〜75モル%が、例えば、アルカリ金属塩やアンモニウム塩、アミン塩等によって中和されていることがより好ましい。このアクリル酸基の中和は該架橋重合体を得る前の親水性不飽和単量体を調製する段階で予め中和しておいてから重合反応を開始してもよく、また、重合中あるいは重合反応終了後に得られた該架橋重合体の酸基を中和してもよいし、それらを併用してもよい。「アクリル酸またはその金属塩を主成分とする」とは、親水性不飽和単量体中にアクリル酸またはその金属塩を50〜100モル%含有することを意味する。したがって、上記親水性不飽和単量体は、必要に応じて、アクリル酸またはその金属塩以外の不飽和単量体を含有していてもよい。
【0046】
他の単量体としては、具体的には、例えば、メタクリル酸、マレイン酸、ビニルスルホン酸、スチレンスルホン酸、2−(メタ)アクリルアミド−2−メチルプロパンスルホン酸、2−(メタ)アクリロイルエタンスルホン酸、2−(メタ)アクリロイルプロパンスルホン酸等の、アニオン性不飽和単量体およびその塩;アクリルアミド、メタクリルアミド、N−エチル(メタ)アクリルアミド、N−n−プロピル(メタ)アクリルアミド、N−イソプロピル(メタ)アクリルアミド、N,N−ジメチル(メタ)アクリルアミド、2−ヒドロキシエチル(メタ)アクリレート、2−ヒドロキシプロピル(メタ)アクリレート、メトキシポリエチレングリコール(メタ)アクリレート、ポリエチレングリコールモノ(メタ)アクリレート、ビニルピリジン、N−ビニルピロリドン、N−アクリロイルピペリジン、N−アクリロイルピロリジン等の、ノニオン性の親水基含有不飽和単量体;N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリレート、N,N−ジメチルアミノプロピル(メタ)アクリルアミド、および、これらの四級塩等の、カチオン性不飽和単量体;等が挙げられるが、特に限定されるものではない。
【0047】
該吸水性樹脂を得る際には、内部架橋剤を用いて架橋構造を内部に導入することが望ましい。上記の内部架橋剤は、重合性不飽和基および/またはカルボキシル基と反応し得る反応性基を一分子中に複数有する化合物であればよく、特に限定されるものではない。つまり、内部架橋剤は、親水性不飽和単量体と共重合および/またはカルボキシル基と反応する置換基を一分子中に複数有する化合物であればよい。尚、親水性不飽和単量体は、内部架橋剤を用いなくとも架橋構造が形成される自己架橋型の化合物からなっていてもよい。
【0048】
内部架橋剤としては、具体的には、例えば、N,N’−メチレンビス(メタ)アクリルアミド、(ポリ)エチレングリコールジ(メタ)アクリレート、(ポリ)プロピレングリコールジ(メタ)アクリレート、トリメチロールプロパントリ(メタ)アクリレート、トリメチロールプロパンジ(メタ)アクリレート、グリセリントリ(メタ)アクリレート、グリセリンアクリレートメタクリレート、エチレンオキシド変性トリメチロールプロパントリ(メタ)アクリレート、ペンタエリスリトールテトラ(メタ)アクリレート、ジペンタエリスリトールヘキサ(メタ)アクリレート、トリアリルシアヌレート、トリアリルイソシアヌレート、トリアリルホスフェート、トリアリルアミン、ポリ(メタ)アリロキシアルカン、(ポリ)エチレングリコールジグリシジルエーテル、グリセロールジグリシジルエーテル、エチレングリコール、ポリエチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、エチレンジアミン、ポリエチレンイミン、グリシジル(メタ)アクリレート等が挙げられるが、特に限定されるものではない。これらの内部架橋剤は、一種類のみを用いてもよく、また、二種類以上を併用してもよい。そして、上記例示の内部架橋剤のうち、重合性不飽和基を一分子中に複数有する内部架橋剤を用いることにより、得られる吸水性樹脂の吸収特性等をより一層向上させることができる。
【0049】
内部架橋剤の使用量は、親水性不飽和単量体に対して、0.0001〜3モル%、さらに好ましくは0.001〜2モル%、特には0.05〜1モル%の範囲内が好ましく、親水性不飽和単量体を重合させて吸水性樹脂を得る際には、反応系に、デンプン、デンプンの誘導体、セルロース、セルロースの誘導体、ポリビニルアルコール、ポリアクリル酸(塩)、ポリアクリル酸(塩)架橋体等の親水性高分子;次亜リン酸(塩)等の連鎖移動剤;水溶性もしくは水分散性の界面活性剤等を0〜100質量%、より好ましくは0〜10質量%の範囲で添加してもよい。
【0050】
親水性不飽和単量体の重合方法は、特に限定されるものではなく、例えば、水溶液重合、バルク重合、沈殿重合等の公知の方法を採用することができる。性能面や重合の制御の容易さから、単量体を水溶液として、水溶液重合を行うことが好ましい。また、反応温度や反応時間等の反応条件は、用いる単量体成分の組成等に応じて適宜設定すればよく、特に限定されるものではない。
【0051】
親水性不飽和単量体を重合させる際には、例えば、過硫酸カリウム、過硫酸ナトリウム、過硫酸アンモニウム、t−ブチルハイドロパーオキサイド、過酸化水素、2,2’−アゾビス(2−アミジノプロパン)二塩酸塩等のラジカル重合開始剤;2−ヒドロキシ−2−メチル−1−フェニル−プロパン−1−オン等のラジカル系光重合開始剤;紫外線や電子線等の活性エネルギー線;等を用いることができる。また、酸化性ラジカル重合開始剤を用いる場合には、例えば、亜硫酸ナトリウム、亜硫酸水素ナトリウム、硫酸第一鉄、L−アスコルビン酸等の還元剤を併用して、レドックス重合を行ってもよい。これら重合開始剤の使用量は、0.001〜2モル%の範囲内が好ましく、0.01〜0.5モル%の範囲内がより好ましい。
【0052】
次いで、上記の重合方法で得られた含水ゲル状重合体を乾燥する。乾燥には、通常の乾燥機や加熱炉を用いることができる。例えば、薄型撹拌乾燥機、回転乾燥機、円盤乾燥機、流動層乾燥機、気流乾燥機、赤外線乾燥機等である。
【0053】
上記乾燥によって得られた乾燥物は、そのまま吸水性樹脂として用いることもできるが、粉砕、分級して平均粒子径が200〜300μm、106μm未満の樹脂の割合を0〜10質量%、好ましくは0〜7質量%、より好ましくは0〜5質量%、特に好ましくは0〜3質量%とすることができる。本発明では、対数標準偏差値σζに限定はないが、0.25〜0.45の範囲でも特定の表面架橋剤を使用することで均一な表面架橋を行うことができる。その他、(1)吸水性樹脂を特定の粉砕機で粉砕する方法、(2)微粉末を造粒して整粒する方法によっても、重量平均粒子径200〜300μm、106μm未満の樹脂の割合が0〜10質量%の吸水性樹脂を調製することができる。
【0054】
(1)粉砕機
使用する粉砕機としては、粉体工学便覧(粉体工学会編、初版)の表1.10で分類されている粉砕機種名のうち、せん断粗砕機、衝撃破砕機、高速回転式粉砕機に分類されるものであり、切断、せん断、衝撃、摩擦という粉砕機構の1つ以上の機構を有するものが好ましく使用でき、特に切断、せん断機構が主機構である粉砕機が好ましい。また、ロール転動型、ロールミル(ロール回転形)に分類されるものであり、粉砕機構として圧縮機構も有するものであっても、せん断、切断効果が強い場合には使用できる。上記した好ましい粉砕機の内でも、複数の回転刃と固定刃のせん断により粉砕する装置であることが好ましい。その回転刃の周速は、3.0〜200m/秒、より好ましくは5.0〜150m/秒である。このような高速回転刃による粉砕でも微粉をほとんど発生せず、粉砕効率も高く、生産性に優れる。回転刃の周速が3.0m/秒未満であると極端な処理量低下とともに、粉砕粒子径が増大し、材料の練られも増えて可溶分の増加が起こる場合がある。一方、200m/秒よりも速いと処理量は多くなるものの装置コストが高くなる場合がある。
【0055】
本発明においては、含水率10質量%以上30質量%以下の含水ゲル状重合体を粉砕機で粉砕することにより、分級操作を経ない粉砕後または分級操作を経ない粉砕・乾燥後の粒径分布において、粒径150μm以上850μm以下の微細粒子が75質量%以上である粒子状吸水性樹脂を高生産性で得ることができる。それに加え、粒径150μm未満の微細粒子が15質量%以下および/または粒径106μm未満の超微細粒子が10質量%未満となる粒子状吸水性樹脂を高生産性で得ることができる。さらに、粉砕し、粒径150μm以上500μm以下という狭い粒径範囲にある粒子が50質量%以上でかつ粒径150μm未満の微細粒子が15質量%以下、および/または粒径150μm以上500μm以下の微細粒子が50質量%以上で粒径106μm未満の微細粒子が10質量%以下である、粒子状吸水性樹脂を高生産性で得ることが容易にできる。
【0056】
(2)微粉末の造粒
水性液をバインダーとして使用し、該水性液と吸水性樹脂粉末中に含まれる微粉末とを混合して粘結造流体を形成しこれを造粒すると、微粉末を除去でき、かつ所定範囲に粒度を調整することができる。なお、水性液の使用量が多いために造粒体の表面が粘着性を有する場合には、該造粒体を一定時間放置したり、加熱することで粘着性を低減させることができる。加熱温度は50〜200℃で3分〜12時間が好適であり、さらに好ましくは70〜120℃、10分〜2時間である。なお、乾燥は必ずしも必要でない。使用できる水性液としては、水単独、水と混和性のある有機溶剤との混合液があり、水の配合量は、水50〜100質量%、より好ましくは水70〜100質量%である。水との混和性のある有機溶剤としては、低級アルコール、テトラヒドロフラン、アセトン等を挙げることができる。上記水性バインダーには、更に各種化合物や混合物を溶解または分解させてもよい。このような化合物や混合物としては、JP−A−61−97333に記載された消臭剤、植物生育助剤の他に、微粒子状シリカのスラリー等を挙げることができる。なお、微粉末に添加する水性液量は特に限定されず広い範囲で使用できるが、少量では顕著な造粒効果が得られ難く、多量使用の場合に造粒後の乾燥工程がないと吸水性能の低下を招く場合がある。好ましくは、吸水性樹脂100質量部に対して、水性液を1〜50質量部、より好ましくは3〜35質量部とする。
【0057】
上記のようにして得られた吸水性樹脂は、不定形破砕状、顆粒状、棒状、繊維状、鱗片状等の形状であってもよい。一般には、不定形破砕状粒子は、均一な表面架橋が困難と考えられるが、本発明においては、特定の表面架橋剤を使用することで、このような形状の吸水性樹脂粒子でも均一な表面架橋層を有する吸水性樹脂とすることができる。なお、本発明における吸水性樹脂の「重量平均粒子径」は、試験用ふるい(JIS Z8801−2000)目開き500μm、425μm、355μm、300μm、150μm、106μm、75μmの篩を用いて重合体粉体を篩分級した後、残留百分率Rを対数確率紙にプロットし、R=50%に相当する粒子径を重量平均粒子径とした。また、粒度分布は、その指標として下記の式であらわされる対数標準偏差値σζを用いた。ここでは、σζの値が小さいほど粒度分布が狭いことを意味する。ふるい目開きを適宜変更することで、下式を満たすことができる。
【0058】
【数2】
【0059】
(X1はR=84.1%、X2はR=15.9%、近傍2点で線形近似するときのそれぞれの粒子径)
上記によって得られた含水ゲル状重合体、すなわち表面架橋前の吸水性樹脂は、含水率(180℃、3時間乾燥条件)が10質量%未満、好ましくは7質量%未満、さらに好ましくは5質量%未満である。吸水性樹脂の含水率が10質量%を超えると、本発明に用いる第一架橋剤と第二架橋剤とが吸水性樹脂の表面に均一に分散できず、架橋表面にカルボキシル基やカルボキシル塩が残存する場合がある。
【0060】
本発明に用いることのできる第一架橋剤は、溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で水性樹脂前駆体が有するカルボキシル基と反応可能な化合物である。溶解度パラメータ(SP値)は化合物の極性を現すファクタ−として一般に用いられており本発明ではポリマ−ハンドブック第3版(WILEY INTERSCIENCE社発行)VII−527〜539に記載されている溶媒の溶解度パラメータ(SP値)δ[(cal/cm3)1/2]の値を適用するものとし、この表に記載されていない架橋剤に関しては同ハンドブックのVII−524のSmallの式にVII−525に記載されているHoyの凝集エネルギ−定数を代入して導いた値δ[(cal/cm3)1/2]を適用するものとする。
【0061】
このような溶解度パラメータ(SP値)11.5(cal/cm3)1/2以上でカルボキシル基と反応し得る第一架橋剤としては、エチレングリコール、プロピレングリコール、グリセリン、ペンタエリスリトール、ソルビトール、エチレンカ−ボネイト、プロピレンカ−ボネイト、ジエチレングリコール、1,3−ブタンジオール、1,4−ブタンジオール、1,5−ペンタジオール、2,4−ペンタジオール、1,6−ヘキサンジオール、2,5−ヘンキサンジオール、トリメチロールプロパン、ジエタノールアミン、4,5−ジメチル−1,3−ジオキソラン−2−オン等が例示され、これらの群より選ばれる1種または2種以上を用いることができる。好ましくは溶解度パラメータ(SP値)13.0(cal/cm3)1/2以上のものである。
【0062】
本発明に用いることのできる第二架橋剤は、溶解度パラメータ(SP値)11.5(cal/cm3)1/2未満で水性樹脂前駆体が有するカルボキシル基と反応可能な化合物であり、具体的には、トリエチレングリコール、テトラエチレングリコール、ジプロピレングリコール、トリプロピレングリコール、トリエタノールアミン、エチレングリコールジグリシジルエーテル、ポリエチレングリコールジグリシジルエーテル、グリセロールポリグリシジルエーテル、ジグリセロールポリグリシジルエーテル、ポリグリセロールポリグリシジルエーテル、プロピレングリコールジグリシジルエーテル、ポリプロピレングリコールジグリシジルエーテル、エチレンジアミン、ジエチレントリアミン、トリエチレンテトラミン、2,4−トリレンジイソシアネート、ヘキサメチレンジイソシアネート、エピクロロヒドリン、エピブロモヒドリン等が例示できこれらの群より選ばれる1種または2種以上を用いることができる。より好ましくは、溶解度パラメータ(SP値)9.5〜11.0(cal/cm3)1/2の範囲にあるものである。
【0063】
またさらに、第一および第二架橋剤を選択するにあたっては、第一架橋剤の溶解度パラメータ(SP値)と第二架橋剤の溶解度パラメータ(SP値)の差が2(cal/cm3)1/2以上になるように選ぶことが好ましい。溶解度パラメータ(SP値)の差が2(cal/cm3)1/2未満の場合には二種類の架橋剤を用いた効果が現れにくく得られた吸水性樹脂の加圧下の吸収特性が向上しにくいことがある。さらに好ましくは第一架橋剤の溶解度パラメータ(SP値)と第二架橋剤の溶解度パラメータ(SP値)の差が2.5(cal/cm3)1/2以上、特に好ましくは3.0(cal/cm3)1/2以上であるように選択される。
【0064】
本発明において用いることのできる架橋剤としては、重合体の有する官能基と反応し得る官能基を2個以上有する化合物であれば特に制限はないが、好ましくは親水性、より好ましくは水溶性の化合物であり、例えば第一架橋剤としては、多価アルコール類、第二架橋剤としては多価グリシジル化合物類が好ましい。
【0065】
本発明では架橋剤として、上記した第一架橋剤の群より選ばれる1種以上と第二架橋剤の群より選ばれる1種以上に加えて水を用いることが必要である。これらの使用量は、吸水性樹脂100質量部に対し、第一架橋剤0.1〜1質量部、第二架橋剤0.015〜0.5質量部、水0〜2質量部であり、より好ましくは第一架橋剤0.1〜0.5質量部、第二架橋剤0.015〜0.3質量部、水0〜1質量部であり、特に好ましくは第一架橋剤0.1〜0.3質量部、第二架橋剤0.015〜0.1質量部、水0〜0.5質量部である。特に、重量平均粒子径が200〜300μmである前記吸水性樹脂に表面架橋を行う際に、第一架橋剤のみを使用した場合には本発明で規定する特定の表面特性を有する表面架橋がされない恐れがあり好ましくない。また、第一架橋剤の使用量が、吸水性樹脂100質量部に対して0.1質量部を下回ると、同時に配合する第二架橋剤が樹脂表面に均一に分散されず表面架橋が不均一となる場合がある。一方、第一架橋剤および第二架橋剤の使用量が上記範囲を超えると、残存表面架橋剤量が増加し、品質が低下する場合がある。また、水の配合量を2質量部以内に限定すると、表面架橋剤の吸水性樹脂内部への浸透を防止でき、樹脂表面をより均一に架橋することができる。このため、表面にカルボキシル基やカルボキシル塩の残存量が少ない表面架橋吸水性樹脂を得ることができる。なお、従来から、重量平均粒子径が200〜300μmと微細な吸水性樹脂を表面架橋剤などの液状組成物に混合する際に、粒度分布が広いと吸水性樹脂同士が合一して大きな塊を生じるため、均一な表面架橋が困難であるとされていた。しかしながら、本発明では、上記する特定組成の表面架橋剤を使用することで、吸水性樹脂の粒度分布の対数標準偏差値σζが0.25〜0.45と範囲が広い場合であっても、表面架橋層が均一かつ硬度の高い表面架橋吸水性樹脂を得ることができる。
【0066】
本発明で使用する表面架橋剤溶液は、上記第一架橋剤、第二架橋剤および水を上記範囲で含むものであるが、更に、吸水性樹脂の固形分100質量部に対して0.015〜1.0質量部、好ましくは0.1〜0.4質量部の範囲で、親水性有機溶媒を添加してもよい。用いられる親水性有機溶媒としては、メチルアルコール、エチルアルコール、n−プロピルアルコール、iso−プロピルアルコール、n−ブチルアルコール、iso−ブチルアルコール、t−ブチルアルコール等の低級アルコール類;アセトン等のケトン類;ジオキサン、テトラヒドロフラン等のエーテル類;N,N−ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類を挙げることができる。
【0067】
本発明では、第一および第二架橋剤および水を含む表面架橋剤溶液を直接、吸水性樹脂に噴霧あるいは滴下し混合する。好適な混合装置は、均一な混合を確実にするため大きな混合力を生み出せる事が必要である。本発明に用いることのできる混合装置としては例えば、円筒型混合機、二重壁円錐型混合機、V字型混合機、リボン型混合機、スクリュ−型混合機、流動型炉ロータリーデスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機およびスクリュー型押出機等があげられる。
【0068】
本発明では、ベースポリマーとしての吸水性樹脂と第一および第二架橋剤とを混合した後、更に加熱を行い、表面近傍を架橋させる。
【0069】
本発明で架橋剤を添加した後、加熱処理を行う場合、加熱処理温度(熱媒温度または材料温度)は100〜300℃、好ましくは160〜250℃、より好ましくは180〜250℃である。加熱処理温度が100℃未満では、加熱処理に時間がかかり生産性の低下をひき起こすのみならず、第一および第二架橋剤両者による均一な架橋が達成されず、本発明の目的とする吸収特性の高い樹脂が得られない。
【0070】
加熱処理は通常の乾燥機または加熱炉を用いて行うことができ、溝型混合乾燥機、ロータリー乾燥機、デスク乾燥機、流動層乾燥機、気流型乾燥機、および赤外線乾燥機等が例示できる。
【0071】
従来無加圧下の吸水倍率を高めると、加圧下での吸水倍率が低下してしまい、また逆に加圧下での吸水倍率を高めると、無加圧下での吸水倍率が低下してしまうという問題が常にあったが、本発明で得られる吸水性樹脂は無加圧下吸水倍率、加圧下吸水倍率および血液吸収率に優れ、吸収速度が高く、低吸湿性を示すなど極めて優れた吸収特性を示す。この原因については定かではないが、吸水性樹脂を溶解度パラメータの異なる二種類以上の架橋剤および特定範囲の水分量の表面架橋剤と混合し、特定の温度範囲で架橋することによって、吸水性樹脂の表面近傍における両架橋剤の分布が最適化されかつ両者が速やかに架橋反応することにより、これまでの方法では達成されることのなかったような特異な密度勾配をもつ架橋が粒子表面近傍において形成されるためではないかと推測される。また表面架橋前の吸水性樹脂の重量平均粒子径と粒度分布を先に述べたように限定することで、上記優れた吸収特性に加え、表面特性が一定化するため分散性が良好であり、残存表面架橋剤が少なく、2次添加物の添加が容易、かつ官能基との反応性が低いため、混合性(パルプとのブレンド)に優れ、不織布などに均一に塗布しうる表面特性に優れる、という特徴を有する。このように、従来の表面架橋吸水性樹脂に比べて、一度に多量の水性液体が添加されても、該水性液体を素早く吸収することができ、かつ、液拡散性や吸収量等の吸収特性に優れるため、衛生材料等の吸収性物品を提供することができる。
【0072】
本発明の吸水材は、上記表面架橋吸水性樹脂をそのまま吸水材として使用することができ、例えば、高機能化かつ薄型化が望まれている紙オムツや生理用ナプキン、いわゆる失禁パット等の衛生材料等の吸収性物品に特に好適に用いることができる。また、上記表面架橋吸水性樹脂に、他の添加剤、例えば機能性向上剤、消臭剤、香料、無機粉末、発泡剤、顔料、染料、親水性短繊維、肥料、酸化剤、還元剤、水、塩類等を0〜30質量%添加して新たな機能を附加させたものであってもよい。
【0073】
本発明では、例えば、上記の様にして得られたカルボキシル基を有する吸水性樹脂に対して、イオン封鎖剤とカルボキシル基と反応し得る表面架橋剤を混合することにより、耐尿性の優れた吸水剤を得ることができる。本発明に用いられるイオン封鎖剤としては、以下の化合物が挙げられる。
【0074】
(1)アミノカルボン酸及びその塩、(2)クエン酸モノアルキルアミド及びクエン酸モノアルケニルアミド及びそれらの塩、(3)マロン酸モノアルキルアミド及びマロン酸モノアルケニルアミド及びそれらの塩、(4)モノアルキルリン酸エステル及びモノアルケニルリン酸エステル及びそれらの塩、(5)N−アシル化グルタミン酸及びN−アシル化アスパラギン酸及びそれらの塩、(6)β―ジケトン誘導体、(7)トロポロン誘導体、(8)有機リン酸化合物。
【0075】
(1)アミノカルボン酸及びその塩としてはカルボキシル基を3個以上有するアミノカルボン酸及びその塩がイオン封鎖能の点で好ましい。具体的には、ニトリロトリ酢酸、エチレンジアミンテトラ酢酸、ジエチレントリアミンペンタ酢酸、トリエチレンテトラアミンヘキサ酢酸、シクロヘキサンー1,2−ジアミンテトラ酢酸、N−ヒドロキシエチルエチレンジアミントリ酢酸、エチレングリコールジエチルエーテルジアミンテトラ酢酸、エチレンジアミンテトラプロピオン酸、N−アルキルーN’−カルボキシメチルアスパラギン酸、N−アルケニルーN’−カルボキシメチルアスパラギン酸、及びこれらのアルカリ金属塩、アルカリ土類金属塩、アンモニウム塩もしくはアミン塩が挙げられる。
【0076】
(2)クエン酸モノアルキルアミド及びクエン酸モノアルケニルアミド及びそれらの塩は、例えばアルコールとクエン酸の脱水縮合により得られる。
【0077】
(3)マロン酸モノアルキルアミド及びマロン酸モノアルケニルアミド及びそれらの塩は、例えば、α―オレフィンをマロン酸メチルに付加せしめた後加水分解することにより得られる。
【0078】
(4)モノアルキルリン酸エステル及びモノアルケニルリン酸エステル及びそれらの塩としては、ラウリルリン酸、ステアリルリン酸等が挙げられる。
【0079】
(5)N−アシル化グルタミン酸及びN−アシル化アスパラギン酸及びそれらの塩としては、例えば(株)味の素より市販されているアミソフトHS−11やGS−11等が挙げられる。
【0080】
(6)β―ジケトン誘導体としては、アセチルアセトン、ベンゾイルアセトン等が挙げられる。
【0081】
(7)トロポロン誘導体としてはトロポロン、β―ツヤプリシン、γ―ツヤプリシン等が挙げられる。
【0082】
(8)有機リン酸化合物としてはエチリデンホスホン酸;1−ヒドロキシエチリデン−1,1−ジホスホン酸;アミノトリメチレンホスホン酸;エチレンジアミンテトラ(メチレンホスホン酸);ジエチレントリアミンペンタ(メチレンホスホン酸)等を挙げることができるが、特に好ましいものは1−ヒドロキシエチリデン−1,1−ジホスホン酸;エチレンジアミンテトラ(メチレンホスホン酸);ジエチレントリアミンペンタ(メチレンホスホン酸)である。塩として好ましいものは、Na塩、K塩等のアルカリ金属塩、アンモニウム塩、アミン塩を挙げることができる。これらの化合物は、金属封鎖剤の一種として知られているものである。
【0083】
これらイオン封鎖剤の中でも好ましくはカルボキシル基を3個以上有するアミノカルボン酸及びその塩であり、中でもジエチレントリアミンペンタ酢酸、トリエチレンテトラアミンヘキサ酢酸、シクロヘキサンー1,2−ジアミノテトラ酢酸、N−ヒドロキシエチルエチレンジアミントリ酢酸及びその塩が、耐尿性の点で最も好ましい。
【0084】
本発明において上記イオン封鎖剤の使用量は、表面近傍の架橋に用いる表面架橋剤(添加時期については特に制限はないが、重合工程、表面処理工程、造粒工程、微粉回収工程のいずれかに添加する方法がある。)によって異なるが、通常吸水性樹脂の固形分100重量部に対して0.0001〜10重量部、好ましくは0.0002〜5重量部の範囲である。使用量が10重量部を越えると、使用に見合う効果が得られず不経済になるばかりか、吸収量が低下するなどの問題が生じる。また、0.0001重量部よりも少ないと耐尿性向上の効果が得られない。
【0085】
本発明で使用される機能性向上剤として、例えば、親水性の無機化合物があり、水不溶性親水性の無機微粒子や水溶性の多価金属塩などが好ましく用いられる。なお、本発明でいう親水性無機微粒子とは、例えば、EP−B−0629411に記載されている親水性度70%以上のものが例示できる。また、US−A−5797893のカラム11に例示されるカチオン性高分子化合物や疎水性の無機微粒子なども機能性向上剤として使用でき、液透過性を向上させ得る。例えば、タルク、カオリン、フラー土、ベントナイト、活性白土、重晶石、天然アスファルタム、ストロンチウム鉱石、イルメナイト、パーライトなどの鉱産物;硫酸アルミニウム14〜18水塩(または無水物)、硫酸カリウムアルミニウム12水塩、硫酸ナトリウムアルミニウム12水塩、硫酸アンモニウムアルミニウム12水塩、塩化アルミニウム、ポリ塩化アルミニウム、酸化アルミニウムなどのアルミニウム化合物類;その他の多価金属塩、多価金属酸化物および多価金属水酸化物;親水性のアモルファスシリカ(例、乾式法:トクヤマ社 ReolosilQS−20、沈殿法:DEGUSSA社 Sipernat22S, Sipernat2200)類;酸化ケイ素・酸化アルミ・酸化マグネシウム複合体(例、ENGELHARD社.Attagel#50)、酸化ケイ素・酸化アルミニウム複合体、酸化ケイ素・酸化マグネシウム複合体などの酸化物複合体類;などが例示できる。また、US−A−5164459、EP−A−761241に例示されたものも使用することができる。これらの粒子の中から親水性の粒子(例えば、硫酸アルミニウム14〜18水塩や親水性のアモルファスシリカ)を選択して使用するのが好ましいが、粒子の親水性が低い場合は、粒子表面を親水性化合物で処理して親水化したものを使用してもよい。本発明では、これらのいずれか1種を単独で用いてもよいし、2種以上を併用してもよい。なお、添加できる機能性向上剤(例えば、粉体流動性向上剤、吸湿プロッキング性向上剤、通液性向上剤等)の粒径は、無機微粒子の場合には、好ましくは5μm以下、より好ましくは1μm以下、最も好ましくは0.1μm以下である。なお、凝集体や造粒物を形成する場合には、それらを構成する1次粒子の粒子径を上記範囲とすることが好ましい。
【0086】
このような機能向上剤の混合方法は、硫酸アルミニウム等の水溶性多価金属塩やカチオン性高分子化合物を水溶液として混合する方法やスラリー状として混合する方法、粉体として混合する方法などある。好ましくは粉体として混合する方法である。吸水性樹脂粒子に対する添加量は、0.01〜5質量%が好ましく、0.05〜3質量%がより好ましい。添加量が5質量%より多くなると吸収倍率の低下を招き、0.01質量%より少ないと添加の効果が得られない場合がある。混合装置は、特に大きな混合力を備える必要はなく、例えば、解砕機や篩機などで混合されてもよい。上記の混合装置としては、例えば、円筒型混合機、二重壁円錐型混合機、V字型混合機、リボン型混合機、スクリュー型混合機、流動型炉ロータリーデスク型混合機、気流型混合機、双腕型ニーダー、内部混合機、粉砕型ニーダー、回転式混合機、スクリュー型押出機、スタティックミキサー等が好適である。また、添加の時期は前記製法において、吸水剤が得られる前、製造中、製造後のいずれでもよいが、好ましくは表面架橋後である。
【0087】
本発明の第三は、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕粒子を表面架橋した吸水性樹脂から構成される吸水材の評価方法であって、該吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材の前記金属原子濃度を経時的に計測する工程を含む、吸水材の評価方法である。ここに、「不定形破砕状の粒子」とは、水溶液重合等によって得られる含水重合物を乾燥後、粉砕して得られる粒子であり、粉砕に伴う破断面(平滑面)と角が電子顕微鏡または光学顕微鏡によって確認できる粒子である。
【0088】
上記第一の発明で説明したように、Arイオン放電研磨して測定される該樹脂粒子表面の金属原子濃度を経時的に計測すると、各測定時における特定の金属種の原子濃度から吸水材の表面特性を知ることができる。例えば、上記したように研磨0秒、10秒、40秒、70秒において、研磨時間Xと金属原子濃度(%)Yとの関係式:Y=aX+bにおいて、0.30≦a≦0.60、0<b<20を満たす場合には、表面架橋層の均一性、ならびに表面架橋層の硬度、血液吸収率、凝集性、溶液分散性、吸水性などとの相関が観察される。このことは、アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂からなる吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材粒子表面の金属原子濃度を経時的に計測すると、吸水材を評価できることを意味する。この際、加圧電圧やArイオン放電研磨などは、対象とする吸水材の種類や粒子径などに応じて適宜選択すればよく、金属原子濃度の測定時も粒子径などに応じて適宜選択すればよい。また、測定結果と評価項目との関係は予め対象とする吸水材の有する特性と相関させることで評価することができる。本発明においては、特に吸水材の架橋表面層の硬度、表面架橋層の均一性、吸水性、血液吸収率、凝集性、および溶液分散性などの評価を行うことができる。従来から、吸水材の特性は、実際に保水条件で行うことが一般的であったが、保水させることなく特性を評価できることは画期的なことである。本発明の評価方法によれば、吸収特性、機能性の評価、基本物性等の解析などに有用である。
【実施例】
【0089】
次に実施例を挙げて本発明を具体的に説明する。ただし、これらの実施例は何ら本発明を制限するものではない。以下に、表面架橋吸水性樹脂または吸収性物品を対象として測定方法を説明するが、これらに代えて吸水材を使用すれば、吸水材の諸特性を評価することができる。なお、本発明においては、吸収特性を評価するために用いる溶液は0.9質量%塩化ナトリウム水溶液であり、それを略称して生理食塩水、または、0.9質量%生理食塩水と呼ぶことがあるが、それらは全て同じものである。市販のおむつに含まれる吸水性樹脂を評価する場合には、流通過程での吸湿を考慮して、吸水性樹脂を乾燥(例えば、60℃での減圧乾燥で16時間)して平衡含水状態(例えば、5質量%前後)とした後に物性を測定する。なお、本発明で得られる表面架橋吸水性樹脂は、実質水不溶性であり、含水率がいずれも7質量%以下(固形分93%以上)、残存モノマー量が400質量ppm以下であった。
【0090】
(1) 吸水倍率(CRC/Centrifuge Retention Capacity)
表面架橋吸水性樹脂約0.20g(Wp1)を正確に秤量し、不織布製の袋(60×60mm)に均一に入れてヒートシールしたのち、25℃±2℃に調温した0.9質量%塩化ナトリウム水溶液中に浸漬した。60分後に袋を引き上げ、遠心分離機(株式会社コクサン製、型式H−122小型遠心分離機)を用いて250G(250×9.81m/s2)で3分間水切りを行った後重量Wa(g)を測定した。また、同様の操作を、吸水性樹脂を用いないで行い、そのときの重量Wb(g)を測定した。そして、これらの重量Wp1、WaおよびWbから、次式に従って表面架橋吸水性樹脂の吸水倍率(g/g)を算出した。なお、イオン交換水の場合には、吸水材は0.02gで測定した。
【0091】
【数3】
【0092】
(2) 粒度分布・重量平均粒子径
表面架橋吸水性樹脂10.0gを、目開き500μm、425μm、355μm、300μm、150μm、106μm、75μmの篩を用いて重合体粉体を仕込み、ロータップ型ふるい振盪機(株式会社飯田製作所製、ES−65型ふるい振盪機)により5分間分級した後、残留百分率Rを対数確率紙にプロットし、R=50%に相当する粒子径を重量平均粒子径とした。また、粒度分布は、その指標として下記の式であらわされる対数標準偏差値σζを用いた。
【0093】
【数4】
【0094】
(X1はR=84.1%、X2はR=15.9%、近傍2点で線形近似するときのそれぞれの粒子径)
(3) 吸湿率
表面架橋吸水性樹脂約1.0gを正確に秤量し、アルミカップ(底面の直径52mm、高さ22mm)上に均一に散布し、25℃、50%RHの恒温恒湿室に24時間放置した。そのときの重量Wc(g)を測定した。そして、含水した吸水性樹脂を、180℃3時間乾燥を行い、絶乾重量Wdを測定し、次式に従って吸水性樹脂の吸湿率(%)を算出した。(ただし、アルミカップの重量は除いて算出する。)
【0095】
【数5】
【0096】
(4) 吸収体、吸収性物品の製造例と吸収性物品(モデルおむつ)の性能評価方法
4−1.吸収体の作成
液体非通液透過性の裏面シートとして大きさ14×40cmの長方形ポリエチレンフィルム(坪量18g/m2)の上に、吸水性樹脂粒子を12×38cmの面積に16.4g散布した(吸水性樹脂の散布量360g/m2)。その上に液獲得部材として子供用おむつに使用する綿状パルプ3gを大きさ8×24cm(密度0.04g/cm3で坪量160g/m2)を積層して吸収体を構成した。最後に液透過性表面材として12×40cmの長方形ポリエステル不織布(坪量20g/m2)を載置し、サイド部分をシールして、モデル吸収性物品を得た。
【0097】
4−2.通液時間・総吸収量
上記吸収性物品を机上にテープで平面状に固定し、その上に、12×40cmのアクリル板(中央部分に液注入のための直径70mmの円筒が具備されている)および、1.3kgの荷重をかけた。
【0098】
37℃に調整した生理食塩水75mlを円筒の中に注ぎ、表面シートから液が吸収性物品内部に吸収し終わった時間(通液時間1回目)を測定した。
【0099】
この操作を4回、60分毎に繰り返し、通液時間を測定(sec.)した。
【0100】
また、上記した吸収性物品を、生理食塩水に1時間浸漬を行った。そして、吸収性物品を引き上げ、液透過性表面材を下方に2つ折で10分間水切りを行った。10分間水切り後の重量を求め、以下の式で総吸収量(g)を求めた。
【0101】
【数6】
【0102】
(5) 血液吸収率
内径60mmのシャーレ中に吸水性樹脂0.5g(175g/m2)を均一に散布し、羊血(脱繊血;緬羊脱繊維血液「日本バイオテスト研究所」)10gをシャーレの中心点から約10秒かけて滴下し添加し、静置下で10分間放置後、未吸収液を0.7psi下、1分間ろ紙10枚(戻り量により適宜変更)(ADVANTEC 東洋濾紙株式会社製、品名 JIS P 3801 No.2)により吸着させ戻り量(We g)を測定する。添加液量10gより戻り量を差し引いて吸収量(g)を測定し、下記式に従い、羊血10gの全量を吸液できた場合の最大吸収量20(=10g/0.5g)に対する血液吸収率を算出した。
【0103】
【数7】
【0104】
(6) Arイオン放電研磨による金属原子濃度
吸水性樹脂を、目開き300μmパス、150μmオン(試料収集が困難な場合、425〜75μmの範囲内で適宜交換する)を110℃、約4時間乾燥させる。この試料を用いて加速イオンを各試料に照射し、イオン衝突により粒子表面を研磨した。この試料を用いてX線光電子分光ESCA(Electron Spectroscopy for Chemical Analysis)で以下に従い表面架橋吸水性樹脂の官能基を分析した。
【0105】
1.試料調製
試料台(試料を載せる部分は約6cm×1cmの長方形)に、導電性テープを約1cm角に切断して貼り、その上に試料を散布して固定する。最大6試料載せうるが、スパッタする場合は4個までとする。導電性テープ上に試料を適当量散布し、テープに付かなかった分は窒素ガス吹き飛ばし、目視で隙間がほとんどなくなる程度に固定する。なお、ESCAは表面状態を評価するものであり、導電性テープ(日新EM株式会社製)に隙間なく試料が付着していることが重要である。
【0106】
2.ESCA測定条件
装置:JEOL JPS−9000MX
条件:MgをターゲットにしKα線使用、加速電圧:10kV、エミュション電流:10mA、積算:10回、
3.スパッタ条件
装置:Arイオンガン(熱陰極電子衝撃型)
条件:イオンビーム電流:50mA、イオンビーム径:1.5mm、Arイオンの加速電圧:500Vで加速電流:8.5mA、Arガス圧:3×10−2Pa
4.試料測定
(1)試料をESCAの予備排気室に入れ、約24時間予備排気する。
【0107】
(2)試料をESCAの測定用の試料室に移動し、ESCA測定してゼロ秒データとする。
【0108】
(3)試料を予備排気室に移動し、スパッタを指定時間(a秒時)行なう。Arガスを排気後、試料を測定用の試料室に移動してESCA測定し、a秒データとする。(0秒値)
(4)同じ試料について、スパッタを指定時間(b秒時)行ないESCA測定し、a+b秒データとする(X秒値)。
【0109】
(5)必要な時間スパッタと測定を繰り返す。
【0110】
5.原子濃度(原子%)の算出
得られた光電子スペクトル(ESCAスペクトル)を装置付属の解析ソフトを用いて、以下の手順で計算処理を実施した。
【0111】
(1)得られたスペクトルに対しスムージングを行ない、微細なノイズを除去する。参照点数は11点で実施した。
【0112】
(2)スペクトルのエネルギー軸を炭素の1s準位を基準(285eV)として、帯電補正を行なう。なお、この操作は定量計算には影響しない。
【0113】
(3)スペクトルからバックグランド補正計算により、バックグランド・スペクトルを除去する。バックグランド補正計算は、Shirly法で行なう。バックグランド補正計算を行なうための開始・終点の指定は、画面上でピーク形状を確認して手動で行なった。ただし、この作業は、定量計算結果に影響を与える。特に、含有比率が小さく、ピーク形状が明瞭でない場合は、誤差を生ずる要因となる。
【0114】
(4)スペクトルから得られる面積値(eV*cps)を対象に、装置付属の解析ソフトに備わった相対感度因子を用いて定量補正計算を行ない、各原子の相対原子比率(C、Oおよび目的金属原子の総数を100%とした場合の目的金属原子の比率(原子%))を原子濃度(原子%)とする。対象原子がナトリウムであれば、Na原子濃度が求められる。
【0115】
(7)F/C比
各試料を110℃、約4時間乾燥させた。この試料45mgを10mlサンプル管に秤りとり、2,2,2−トリフルオロエタノール(TFE)/ジシクロヘキシルカルボジイミド(DCC)/ピリジンの0.02/0.001/0.04モル比の混合液約0.3gを添加してから密閉し、60℃、8時間加熱した。ついで、ピリジンを加えて攪拌し、濾液をろ紙で吸い上げ除去を3回繰り返して洗浄した。その後、70℃、約100Torrで16時間、更に約1Torrで8時間真空乾燥した。該反応によって、表面架橋吸水性樹脂の表面にある−COOHが−COOCH2CF3に変換される。該反応を下記に示す。
【0116】
【化1】
【0117】
この試料を用いて上記(7)に記載するESCA測定条件で表面架橋吸水性樹脂の官能基を分析した。F/C比は下式で示され、Fの相対原子比率をCの相対原子比率で除したものである。
【0118】
【数8】
【0119】
(8)残存表面架橋剤量(エポキシ化合物の場合)
吸水材2.0gを100mlのビーカーに加え、メタノール/水=2/1質量比からなる組成液2mlを加え、蓋をして1時間放置する。メタノール5mlを上記ビーカーに加え濾過し、濾液1.0gを50mlのナスフラスコに入れ、12質量%のニコチンアミド水溶液0.05mlを添加する。ナスフラスコに空冷管をつけ沸騰したウォーターバスで30分間加熱する。反応液を、濾紙を用いて濾過し、濾過液を高性能液体クロマトグラフィーで分析する。一方、吸水材を用いず既知量の架橋剤を加えて同様の操作を行ない、得られた検量線を外部標準とし、濾過液の希釈倍率を考慮して、吸水材中の残存表面架橋剤量(ppm)を求める。
【0120】
(9)吸収速度(Vortex)
予め調整された0.90質量%塩化ナトリウム水溶液(生理食塩水)1,000質量部に食品添加物である食用青色1号(CAS番号:3844−45−9)0.02質量部を添加し、液温30℃に調整した。その生理食塩水50mlを胴径55mm、高さ70mmの容量100mlのビーカー(例えば相互理化学硝子製作所が販売するJISR−3503に準拠したビーカー)に計り取り、長さ40mm、太さ8mmの円筒型テフロン(登録商標)製マグネット式撹拌子(例えば、相互理化学ガラス製作所が販売するS型)で600rpmで撹拌する中に、後述する実施例または比較例で得られた吸水性樹脂2.0gを投入し、吸収速度(秒)を測定する。始点、終点は、JIS K 7224(1996年度)「高吸水性樹脂の吸水速度試験方法 解説」に記載されている基準に準じ、吸水性樹脂が生理食塩水を吸液してスターラーチップを試験液で覆うまでの時間を測定し吸収速度(秒)として評価する。
【0121】
(実施例1)
中和率75モル%のアクリル酸ナトリウム37質量%水溶液5,500gに、内部架橋剤としてのポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数:8モル)2.8g(0.025mol%)を溶解させて反応液とした。次に、この反応液を、窒素ガス雰囲気下で30分間脱気した。次いで、開閉可能な蓋付きのシグマ型羽根を2本有するジャケット付きステンレス製双腕型ニーダーに、上記の反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。
【0122】
続いて、反応液を攪拌しながら、過硫酸ナトリウム2.8gおよびL−アスコルビン酸0.12gを添加したところ、およそ1分後に重合が開始した。そして、開始からピーク温度30℃〜95℃で重合を行い、重合を開始してから60分後に反応を終了して含水ゲル状重合体を取り出した。
【0123】
得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を、振動ミルを用いて粉砕し、さらに30メッシュ(目開き500μm)の金網で分級することにより、重量平均粒子径が240μmで、しかも、粒子径が106μm未満の粒子の割合が5質量%の不定形破砕状の吸水性樹脂を得た。このものの吸水倍率は52g/gであった。
【0124】
得られた吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))0.3質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))0.1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理することにより、表面架橋吸水性樹脂を得た。得られた表面架橋吸水性樹脂の重量平均粒子径は249μmであり、粒子径が106μm未満の粒子の割合は1質量%であった。
【0125】
この表面架橋吸水性樹脂について、金属原子濃度(0秒値、10秒値)、F/C比、吸水倍率、吸収速度、吸湿率、残存表面架橋剤量などを測定した。結果を表1に記載した。また、この表面架橋吸水性樹脂を前記吸体吸収性物品の製造例および吸収性物品の性能評価方法に従って、吸収性物品とし、性能を評価した。各実施例および比較例の結果を表1に記載した。
【0126】
(実施例2)
上記の実施例1で得た表面架橋吸水性樹脂100gに、更に微粒子状の親水性二酸化ケイ素(商品名・アエロジル200;日本アエロジル株式会社製)0.3gを添加・混合したものを吸水性樹脂とした。
【0127】
(実施例3)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))0.3質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))0.1質量部と、水0.3質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂の重量平均粒子径は258μmであり、粒子径が106μm未満の粒子の割合は1質量%であった。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0128】
(実施例4)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール0.5質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル0.1質量部と、水1.5質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0129】
(比較例1)
上記の実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール0.8質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル0.02質量部と、水3質量部と、イソプロピルアルコール1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で30分間加熱処理することにより、表面架橋が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0130】
(比較例2)
上記の実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))0.3質量部と、プロピレングリコール0.5質量部、水2質量部とからなる表面架橋剤組成液を混合した。上記の混合物を195℃で40分間加熱処理することにより、比較用吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0131】
(比較例3)
実施例1で得られた含水ゲル重合体を、目開き500μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を振動ミルを用いて粉砕し、さらに18.5メッシュ(目開き850μm)の金網で分級することにより、重量平均粒子径が480μmで、しかも、粒子径が106μm未満の粒子の割合が5質量%の不定形破砕状の吸水性樹脂を得た。
【0132】
得られた不定形破砕状の吸水性樹脂100質量部に、エチレングリコールジグリシジルエーテル0.1質量部と、プロピレングリコール0.3質量部、水5質量部とからなる表面架橋剤組成液を混合した。上記の混合物を210℃で30分間加熱処理することにより、比較用吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0133】
(実施例5)
中和率75モル%のアクリル酸ナトリウム37質量%水溶液5,500gに、内部架橋剤としてのポリエチレングリコールジアクリレート(エチレンオキシドの平均付加モル数:8モル)6.72g(0.06mol%)を溶解させて反応液とした。次に、この反応液を、窒素ガス雰囲気下で30分間脱気した。次いで、開閉可能な蓋付きのシグマ型羽根を2本有するジャケット付きステンレス製双腕型ニーダーに、上記の反応液を供給し、反応液を30℃に保ちながら系を窒素ガス置換した。
【0134】
続いて、反応液を攪拌しながら、過硫酸ナトリウム2.8gおよびL−アスコルビン酸0.12gを添加したところ、およそ1分後に重合が開始した。そして、30℃〜95℃で重合を行い、重合を開始してから60分後に反応を終了して含水ゲル状重合体を取り出した。
【0135】
得られた含水ゲル状重合体は、その径が約5mmに細分化されていた。この細分化された含水ゲル状重合体を50メッシュ(目開き300μm)の金網上に広げ、150℃で90分間熱風乾燥した。次いで、乾燥物を、振動ミルを用いて粉砕し、さらに30メッシュ(目開き500μm)の金網で分級することにより、重量平均粒子径が240μmで、しかも、粒子径が106μm未満の粒子の割合が2質量%の不定形破砕状の吸水性樹脂を得た。このものの吸水倍率は40g/gであった。
【0136】
得られた吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))を0.2質量部、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))を0.2質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))を0.1質量部とからなる表面架橋剤溶液を混合した。上記の混合物を210℃で30分間加熱処理することにより、表面処理が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0137】
(実施例6)
実施例5で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてのプロピレングリコール(SP値:δ=12.6(cal/cm3)1/2(2.58×104(J/m3)1/2))を0.2質量部、1,4−ブタンジオール(SP値:δ=12.1(cal/cm3)1/2(2.47×104(J/m3)1/2))を0.2質量部と、第二架橋剤としてのエチレングリコールジグリシジルエーテル(SP値:δ=10.2(cal/cm3)1/2(2.09×104(J/m3)1/2))を0.1質量部と水0.3質量%からなる表面架橋剤溶液を混合した。上記の混合物を210℃で30分間加熱処理することにより、表面処理が施された吸水性樹脂を得た。この表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0138】
(実施例7)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、第一架橋剤としてプロピレングリコール0.1質量部と、第二架橋剤としてエチレングリコールジグリシジルエーテル0.02質量部とからなる表面架橋剤溶液を混合した。上記の混合物を195℃で40分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0139】
(比較例4)
実施例1で得られた不定形破砕状の吸水性樹脂100質量部に、パラフィン(融点60−62℃ 和光純薬工業(株))の破砕物1.0質量部を混合した。上記の混合物を80℃で10分間加熱処理した。得られた表面架橋吸水性樹脂について実施例1と同様の評価を行った。
【0140】
【表1−1】
【0141】
【表1−2】
【0142】
(実施例8)
実施例3において、吸水性樹脂100質量部に対する表面架橋時に添加する水量を表2に示す配合量で使用した以外は実施例3と同様に操作して吸水性樹脂を得た。各吸水性樹脂のF/C比、金属原子濃度(Na濃度)、混合性を評価した。なお、混合性は、表面架橋剤溶液と吸水性樹脂との同量を混合した際の評価であり、○は混合性良好を示し、△は一部凝集物生成を示し、×は凝集物生成または一体化を示す。結果を表2に合わせて示す。
【0143】
【表2】
【産業上の利用可能性】
【0144】
本発明によれば、吸収特性に優れる吸水材が提供され、紙おむつなどの吸収材として有用である。
【図面の簡単な説明】
【0145】
【図1】図1は、Arイオン放電研磨による吸水材Aと吸水材Bにおける金属原子濃度(Na濃度)を示す図である。
【図2】図2は、表面架橋時水0.3質量%添加して調製した表面架橋吸水性樹脂からなる吸水材と、水無添加で調製した表面架橋吸水性樹脂からなる吸水材におけるArイオン放電研磨による金属原子濃度(Na濃度)を示す図である。
【特許請求の範囲】
【請求項1】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で35%以下である吸水材。
【請求項2】
前記吸水材は、加圧電圧500VでのArイオン放電研磨法による研磨表面の前記金属原子濃度Yと研磨時間X(研磨0秒、10秒、40秒および70秒時)との間に下記式で示す関係を有するものである請求項1記載の吸水材。
【数1】
この際、0.30≦a≦0.60、0<b<20である
【請求項3】
該研磨10秒値が30%以下である請求項1または2記載の吸水材。
【請求項4】
前記金属原子濃度Yと研磨時間Xとの間の関係式が、次のとおりである請求項1〜3のいずれか一つに記載の吸水材。
【数2】
この際、0.30≦a、0<b<15である
【請求項5】
アクリル酸またはその金属塩を主成分とし、部分中和率が50〜90モル%の吸水性樹脂からなる表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材表面にある−COOHを−OOCH2CF3に変換した該表面近傍の炭素原子数に対するフッ素原子比(F/C比)を有する、請求項1〜4のいずれか一つに記載の吸水材。
【請求項6】
重量平均粒子径が200〜300μm、かつ、粒子径106μm未満の吸水材の割合が10質量%以下である請求項1〜5のいずれか一つに記載の吸水材。
【請求項7】
前記吸水材の残存エポキシ化合物の含有量が5ppm以下である請求項1〜6のいずれか一つに記載の吸水材。
【請求項8】
生理食塩水に対する吸水倍率が35g/g以上、血液吸収率が70〜100%である請求項1〜7のいずれか一つに記載の吸水材。
【請求項9】
生理食塩水に対する吸収速度が1〜30秒である請求項1〜8のいずれか一つに記載の吸水材。
【請求項10】
請求項1〜9のいずれか一つに記載の吸水材を含む吸収性物品。
【請求項11】
重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部および、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む表面架橋吸水性樹脂の製造方法。
【請求項12】
該第二架橋剤の量が0.05〜0.5質量部である請求項11記載の方法。
【請求項13】
前記アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を、切断、せん断、衝撃、摩擦という粉砕機構の1つ以上の機構によって粉砕する工程を含む請求項11または12記載の方法。
【請求項14】
前記吸水性樹脂に前記表面架橋剤溶液を混合した後、100〜300℃で加熱する工程を含む請求項11〜13のいずれか一つに記載の方法。
【請求項15】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂からなる吸水材の評価方法であって、
該吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材の前記金属原子濃度を経時的に計測する工程を含む吸水材の評価方法。
【請求項1】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材を加圧電圧500VでArイオン放電研磨して測定される該吸水材表面の前記金属原子濃度が、研磨0秒値で0〜10%であり、研磨10秒値で35%以下である吸水材。
【請求項2】
前記吸水材は、加圧電圧500VでのArイオン放電研磨法による研磨表面の前記金属原子濃度Yと研磨時間X(研磨0秒、10秒、40秒および70秒時)との間に下記式で示す関係を有するものである請求項1記載の吸水材。
【数1】
この際、0.30≦a≦0.60、0<b<20である
【請求項3】
該研磨10秒値が30%以下である請求項1または2記載の吸水材。
【請求項4】
前記金属原子濃度Yと研磨時間Xとの間の関係式が、次のとおりである請求項1〜3のいずれか一つに記載の吸水材。
【数2】
この際、0.30≦a、0<b<15である
【請求項5】
アクリル酸またはその金属塩を主成分とし、部分中和率が50〜90モル%の吸水性樹脂からなる表面架橋した吸水性樹脂から構成される吸水材であって、該吸水材表面にある−COOHを−OOCH2CF3に変換した該表面近傍の炭素原子数に対するフッ素原子比(F/C比)を有する、請求項1〜4のいずれか一つに記載の吸水材。
【請求項6】
重量平均粒子径が200〜300μm、かつ、粒子径106μm未満の吸水材の割合が10質量%以下である請求項1〜5のいずれか一つに記載の吸水材。
【請求項7】
前記吸水材の残存エポキシ化合物の含有量が5ppm以下である請求項1〜6のいずれか一つに記載の吸水材。
【請求項8】
生理食塩水に対する吸水倍率が35g/g以上、血液吸収率が70〜100%である請求項1〜7のいずれか一つに記載の吸水材。
【請求項9】
生理食塩水に対する吸収速度が1〜30秒である請求項1〜8のいずれか一つに記載の吸水材。
【請求項10】
請求項1〜9のいずれか一つに記載の吸水材を含む吸収性物品。
【請求項11】
重量平均粒子径が200〜300μmであるアクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物からなる不定形破砕状粒子を、樹脂固形分100質量部に対し、
a)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]以上で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第一架橋剤0.1〜1質量部、
b)溶解度パラメータ(SP値)が11.5(cal/cm3)1/2[2.35×104(J/m3)1/2]未満で吸水性樹脂が有するカルボキシル基と反応可能な化合物である第二架橋剤0.015〜0.5質量部および、
c)水0〜2質量部、
を含む表面架橋剤溶液と混合する工程を含む表面架橋吸水性樹脂の製造方法。
【請求項12】
該第二架橋剤の量が0.05〜0.5質量部である請求項11記載の方法。
【請求項13】
前記アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を、切断、せん断、衝撃、摩擦という粉砕機構の1つ以上の機構によって粉砕する工程を含む請求項11または12記載の方法。
【請求項14】
前記吸水性樹脂に前記表面架橋剤溶液を混合した後、100〜300℃で加熱する工程を含む請求項11〜13のいずれか一つに記載の方法。
【請求項15】
アクリル酸またはその金属塩を主成分とする吸水性樹脂の部分中和物または完全中和物を表面架橋してなる表面架橋吸水性樹脂からなる吸水材の評価方法であって、
該吸水材を所定の加圧電圧(V)でArイオン放電研磨して測定される該吸水材の前記金属原子濃度を経時的に計測する工程を含む吸水材の評価方法。
【図1】

【図2】
【図2】
【公表番号】特表2008−516007(P2008−516007A)
【公表日】平成20年5月15日(2008.5.15)
【国際特許分類】
【出願番号】特願2007−511110(P2007−511110)
【出願日】平成17年8月31日(2005.8.31)
【国際出願番号】PCT/JP2005/016360
【国際公開番号】WO2006/025586
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公表日】平成20年5月15日(2008.5.15)
【国際特許分類】
【出願日】平成17年8月31日(2005.8.31)
【国際出願番号】PCT/JP2005/016360
【国際公開番号】WO2006/025586
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]