
哺乳動物型糖質構造を操作するための方法
【課題】改変された脂質連結オリゴ糖を有する宿主細胞を提供すること。
【解決手段】本発明は、改変された脂質連結オリゴ糖を有する宿主細胞に関する。この脂質連結オリゴ糖は、グリコシルトランスフェラーゼ、糖トランスポーターおよびマンノシダーゼのセットの異種発現によってさらに改変されて、哺乳動物(例えば、ヒト)の治療用糖タンパク質の産生のための宿主系となり得る。このプロセスは、操作された宿主細胞を提供し、この宿主細胞を使用して、グリコシル化に関与する任意の望ましい遺伝子を発現させ得、標的化し得る。改変された脂質連結オリゴ糖を有する宿主細胞が、産生または選択される。
【解決手段】本発明は、改変された脂質連結オリゴ糖を有する宿主細胞に関する。この脂質連結オリゴ糖は、グリコシルトランスフェラーゼ、糖トランスポーターおよびマンノシダーゼのセットの異種発現によってさらに改変されて、哺乳動物(例えば、ヒト)の治療用糖タンパク質の産生のための宿主系となり得る。このプロセスは、操作された宿主細胞を提供し、この宿主細胞を使用して、グリコシル化に関与する任意の望ましい遺伝子を発現させ得、標的化し得る。改変された脂質連結オリゴ糖を有する宿主細胞が、産生または選択される。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、米国仮出願番号60/344,169(2001年12月27日)(本明細
書中に参考としてその全体を援用する)に優先権を主張する。
【0002】
(発明の分野)
本発明は、一般的に、真菌または他の低級真核生物において発現される組換えタンパク
質のグリコシル化構造(より詳細には高等哺乳動物(特にヒト)のタンパク質のグリコシ
ル化)の改変に関する。
【背景技術】
【0003】
(発明の背景)
DNAが転写されそしてタンパク質へと翻訳された後の、さらなる翻訳後プロセッシン
グは、リコシル化として公知のプロセスである糖残基の結合を含む。異なる生物は、異な
るグリコシル化酵素(グリコシルトランスフェラーゼおよびグリコシダーゼ)を生産し、
そして利用可能な異なる基質(ヌクレオチド糖)を有し、その結果、個々のオリゴ糖のグ
リコシル化パターンならびに組成物は、1つでも同じタンパク質でさえも、特定のタンパ
ク質が発現される宿主系に依存して異なる。細菌は、代表的に、タンパク質をグリコシル
化せず、そうであれば、非常に非特異的な様式である(Moens,1997)。低級真
核生物(例えば、糸状菌および酵母)は、主なマンノースおよびリン酸マンノシル(ma
nnosylphosphate)糖を添加するのに対して、昆虫細胞(例えば、Sf9
細胞)は、さらなる別の方法でタンパク質をグリコシル化する。例えば、(Bretth
auer,1999;Martinet,1998;Weikert,1999;Mal
issard,2000;Jarvis,1998;およびTakeuchi,1997
)を参照のこと。
【0004】
一連の反応からなる哺乳動物型オリゴ糖構造の合成は、その過程において、糖残基が、
タンパク質が、宿主生物における分泌経路に沿って移動する間に、添加されそして除去さ
れる。宿主生物または細胞のグリコシル化経路に沿って存在する酵素は、分泌されたタン
パク質の得られたグリコシル化パターンを決定する。あいにく、低級真核生物宿主細胞で
発現されるタンパク質の得られたグリコシル化パターンは、高級真核生物(例えば、ヒト
および他の哺乳動物)において見られるグリコシル化とは実質的に異なる(Bretth
auer,1999)。さらに、非常に異なるグリコシル化パターンは、いくつかの場合
において、ヒトにおけるこれらのタンパク質の免疫原性の増加およびそれらの半減期の減
少を示している(Takeuchi,1997)。非ヒト宿主細胞(特に、低級真核生物
細胞)中でヒト様糖タンパク質を生産することが所望される。
【0005】
ヒトグリコシル化の初期段階は、2つの異なる相に分けられ得る:(i)脂質連結Gl
c3Man9GlcNAc2オリゴ糖は、小胞体(ER)の膜で反応の経時的なセットによ
ってアセンブリされ、そして(ii)脂質アンカードリコール(dolichyl)ピロ
リン酸から新規に合成されたタンパク質上へのこのオリゴ糖の移動。特異的な移動部位は
、配列Asn−Xaa−Ser/Thr(図1を参照のこと)中のアスパラギン(Asn
)残基によって規定され、Xaaは、プロリンを除く任意のアミノ酸であり得る(Gav
el,1990)。グルコシダーゼおよびマンノシダーゼによるさらにプロセッシングは
、ER中で生じ、その後、新生の糖タンパク質が、初期のゴルジ装置に移動され、さらな
るマンノース残基は、ゴルジ特異的α(α)−1,2−マンノシダーゼによって除去され
る。プロセッシングは、ゴルジを通って進むタンパク質として続く。内側のゴルジにおい
て、多くの改変酵素(N−アセチルグルコサミニルトランスフェラーゼ(GnT I、G
nT II、GnT III、GnT IV GnT V GnT VI)、マンノシダ
ーゼIIおよびフコシルトランスフェラーゼを含む)は、特定の糖残基を付加し、そして
除去する(例えば、図2および図3を参照のこと)。最後に、trans−ゴルジにおい
て、ガラクトシルトランスフェラーゼおよびシアリルトランスフェラーゼは、ゴルジから
放出される糖タンパク質構造を生産する。これらの構造において、ジアンテナ構造(an
tennary structure)、トリアンテナ構造およびテトラアンテナ構造(
ガラクトース、フコース、N−アセチルグルコサミンおよび糖タンパク質にそれらのヒト
特徴付けを与える末端シアル酸の高い程度を含む)によって特徴付けされる。
【0006】
ほとんど全ての真核生物において、糖タンパク質は、一般的なコアオリゴ糖前駆体Gl
c3Man9GlcNAc2−PP−Dolから誘導され、このPP−Dolは、ドリコー
ル−ピロリン酸を意味する(図1)。小胞体において、オリゴ糖に結合するドリコールピ
ロリン酸の合成およびプロセッシングは、周知の真核生物の間で同一である。しかし、酵
母によるコアオリゴ糖のさらなるプロセッシングは、一旦ERを離れそしてゴルジに侵入
するペプチドに移動されると、分泌経路に沿って移動し、そしていくつかのマンノース糖
の添加に関与するので、ヒトと有意に異なる。
【0007】
酵母において、これらの工程は、Ochlp、MntlpおよびMnnlpのようなマ
ンノシルトランスフェラーゼに存在するゴルジによって触媒され、コアオリゴ糖にマンノ
ース糖を連続して添加する。得られた構造は、ヒューマノイドタンパク質の生産を所望せ
ず、従って、マンノシルトランスフェラーゼ活性を減少するか除去することを所望する。
S.cerevisiaeの変異体(マンノシルトランスフェラーゼ活性を欠損する)(
例えば、och1変異体またはmnn9変異体)は、非致死であり、そして酵母糖タンパ
ク質のオリゴ糖中の減少したマンノース含量を提示することを示す。他のオリゴ糖プロセ
ッシング酵素(例えば、リン酸マンノシルトランスフェラーゼ)はまた、宿主の特定の内
因的なグリコシル化パターンに依存して除去され得る。
【0008】
(脂質連結オリゴ糖前駆体)
本発明の特定の興味深いことは、N−グリコシル化の初期段階である(図1および図2
)。Glc3Man9GlcNAc2−PP−Dolの生合成が不完全であるalg(アス
パラギン連結グリコシル化)変異体の研究は、N−グリコシル化の開始段階を明らかにす
ることを助ける。
【0009】
S.cerevisiaeのALG3遺伝子は、首尾よくクローン化され、そして欠失
によってノックアウトされている(Aebi,1996)。ALG3は、酵素Dol−P
−Man:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼをコード
することが示され、これは、ERの管腔側で、Man5GlcNAc2−PP−Dolから
Man6GlcNAc2−PP−Dolまでの第1のDol−P−Man依存性マンノシル
化工程に関与する(Sharma,2001)(図1および2)。漏出alg3−1変異
を有するS.cerevisiae細胞は、Man5GlcNAc2−PP−Dol(構造
I)を集積する(Huffaker,1983)。
【0010】
【化1】

Man5GlcNAc2(構造I)およびMan8GlcNAc2は、och1 mnn1
alg3変異体の全細胞マンノタンパク質を集積する(Nakanishi−Shind
o,1993)。このS.cerevisiae och1、mnn1、alg3変異体
は、生存可能であるが、温度感受性であり、そしてα−1,6ポリマンノース外鎖(ou
ter chain)を欠損することが示された。
【0011】
別の研究において、alg3を欠損した株(Δalg3バックグラウンド)中で発現さ
れた分泌タンパク質は、Endo−β−N−アセチルグルコサミニダーゼ H(Endo
H)に対する耐性について研究された(Aebi,1996)。以前の観察は、Man
5GlcNAc2より長いこれらのオリゴ糖のみが、Endo Hによって切断されやすい
ことを示している(Hubbard,1980)。alg3−1表現型において、いくつ
かのグリコフォームは、その漏出(leakiness)を確認するEndo H切断に
感受性であるが、Δalg3変異体において、全てのグリコフォームは耐性であるようで
あり、かつMan5−型であり(Aebi,1996)、密接な表現型および発生期のポ
リペプチド鎖上にMan5GlcNAc2オリゴ糖構造の移動を示唆する。明らかな表現型
は、ALG3遺伝子の不活性化と関係しない(Aebi,1996)。Saccharo
myces cerevisiae alg3変異体中で生産された、分泌エクソグルコ
ナーゼ(exogluconase)は、35〜44%の間で過小グリコシル化(und
erglycosylate)形態および非グリコシル化形態ならびEndo H処理に
耐性のままである約50%のみの移動オリゴ糖を含むことが見出された(Cueva,1
996)。エキソグルカナーゼ(Exg)(Asn165およびAsn325での2つの潜在的
なN−グリコシル化部位を含む酵素)は、より詳細に分析された。2つのオリゴ糖を受け
取るExg分子について、これは、第1のN−グリコシル化部位(Asn165)が、短縮
型残基中で富化されたが、第2(Asn325)は、規則的なオリゴ糖中で富化されたこと
が示された。35〜44%の分泌エキソグルカナーゼは、非グリコシル化または過小グル
コシル化され、そして約73〜78%の全ての利用可能なN−グリコシル化部位は、短縮
型オリゴ糖または規則的なオリゴ糖のいずれかで占められた(Cueva,1996)。
【0012】
(グルコシル化脂質連結オリゴ糖の移動)
証拠は、哺乳動物細胞において、グルコシル化脂質連結オリゴ糖のみが、新生タンパク
質に移動される(Turco,1977)が、酵母alg5変異体、alg6変異体、お
よびdpg1変異体においては、非グルコシル化オリゴ糖が移動され得る(Ballou
,1986;Runge,1984)ことを示唆する。Saccharomyces c
erevisiae alg8変異体において、過小グリコシル化のGlcMan9Gl
cNAc2が移動される(Runge,1986)。Verostekおよび共同研究者
らは、alg3変異体、secl8変異体、glsl変異体を研究し、そしてMan5G
lcNAc2構造(構造I、上記)のグルコシル化は、脂質連結Man9構造のグルコシル
化と比較して、比較的遅いことを提案する。さらに、このMan5GlcNAc2構造のタ
ンパク質への移動は、Glc3Man5GlcNAc2へのグルコシル化よりも約5倍効率
的であるようである。Man5GlcNAc2グルコシル化の減少した速度は、比較的早い
速度のMan5構造と組み合わせて、alg3変異体酵母中で観察された非グルコシル化
Man5構造の蓄積の原因であると考えられる新生タンパク質上に移動する(Veros
tek−a,1993;Verostek−b,1993)。
【0013】
上記の研究に先行する研究は、グルコシル化オリゴ糖が、その非グルコシル化対応物よ
りも非常に早い速度で移動され、従って、単離するのがより困難であるという結果を認め
る、任意の脂質連結グルコシル化オリゴ糖を明らかにしなかった(Orlean,199
0;Huffaker,1983)。最近の研究は、非グルコシル化オリゴ糖および低グ
ルコシル化オリゴ糖を有する酵母株の作製および研究を可能にし、そしてオリゴサッカリ
ルトランスフェラーゼ複合体による基質認識のための脂質連結オリゴ糖のアンテナに対す
るグルコースの付加の重要性をさらに確認した(Reiss,1996;Staglja
r,1994;Burda,1998)。alg3変異体における脂質連結Man5オリ
ゴ糖のグルコシル化の減少程度は、新生タンパク質上の脂質連結オリゴ糖の移動の動力学
にネガティブに影響し、そしてこれは、alg3ノックアウト株中の分泌タンパク質の強
い過小グリコシル化を引き起こすと考えられる(Aebi,1996)。
【0014】
上記のように、脂質連結核オリゴ糖Man9GlcNAc2のアセンブリは、小胞体(e
ndoplasmatic reticulum)の膜で生じる。3つのグルコース単位
の、脂質連結オリゴ糖のα−1,3−アンテナへの付加は、オリゴ糖アセンブリの最終反
応である。最初のα−1,4グルコース残基が付加され、その後別のα−1,3グルコー
ス残基および末端α−1,2グルコース残基を付加される。ドリコール連結Man9Gl
cNAc2を蓄積する変異体は、ALG6座中で欠損していることが示され、そしてAl
g6pは、Alg8pに対して類似性を有し、α−1,3−グルコシルトランスフェラー
ゼは、第2のα−1,3−連結グルコースの付加を触媒する(Reiss,1996)。
欠損ALG8座を有する細胞は、ドリコール連結Glc1Man9GlcNAc2を蓄積す
る(Runge,1986;Stagljar,1994)。このALG10座は、Gl
c2Man9GlcNAc2−PP−Dolへの単一の末端グルコースの付加に応答性であ
るα−1,2グルコシルトランスフェラーゼをコードする(Burda,1998)。
【0015】
(局在化した酵素活性の連続的なN−グリカンのプロセシング)
糖トランスフェラーゼおよびマンノシダーゼは、ERおよびゴルジ装置の内側(管腔)
表面に並び、それによって糖タンパク質の連続的なプロセシングを可能にする「触媒」表
面を提供する。なぜなら、糖タンパク質は、ERおよびゴルジのネットワークを介して進
むからである。実際、これらのシス、中間、およびトランスのゴルジならびにトランス−
ゴルジネットワーク(TGN)の複数の区画は、順番に並べられたグリコシル化反応が起
こり得る、異なる局所性を提供する。糖タンパク質は、ER中の合成から、後期のゴルジ
またはTGNにおける完全な成熟まで進められるので、異なるグリコシダーゼ、マンノシ
ダーゼおよびグリコシルトランスフェラーゼに順番に曝され、その結果、特異的糖質構造
が合成され得る。多くの研究は、これらの酵素がそれぞれのオルガネラに保持およびアン
カーされる、正確な機構を明らかにすることに奉げられていつ。画像を展開することは複
雑であるが、証拠は、幹領域、膜貫通領域および細胞質テイルが、個々にかまたは協力し
て、個々のオルガネラの膜に酵素を指向し、それによって関連する触媒ドメインをその座
に局在化する。
【0016】
いくつかの場合において、これらの特異的な相互作用は、種をまたいで機能することが
見出された。例えば、ラット由来のα2,6−STの膜貫通ドメインは、動物のトランス
−ゴルジに局在化されることが知られている酵素であり、酵母ゴルジ中のレポーター遺伝
子(インベルターゼ)も局在化することが示された(Schwientek,1995)
。しかし、全長α2,6STの一部と非常に同じ膜貫通ドメインは、ER中に保持され、
そして酵母のゴルジにさらに輸送されない(Krezdorn,1994)。ヒト由来の
全長Gal−Trは、明らかに高い転写レベルにも関わらず、酵母中でさえも合成されな
い。一方で、インベルターゼレポーターに融合されるGalTと同じヒトの膜貫通領域は
、低い生成レベルにも関わらず、酵母ゴルジに直接局在化され得た。Schwiente
kおよび共同研究者は、ヒトGalTの触媒ドメインに対する酵母マンノシルトランスフ
ェラーゼ(Mnt1)、細胞質テイルを含む領域、膜貫通領域の28個のアミノ酸および
幹領域の8個のアミノ酸が、活性GalTのゴルジ局在化のために十分であることを示し
ている。他のガラクトシルトランスフェラーゼは、酵素残基(特に、オルガネラ)との相
互作用に依存するようである。なぜなら、これらの膜貫通領域の除去後、これらは、適切
に局在化され得るからである。現在、より低級真核生物中で特定の非相同的に発現された
グリコシルトランスフェラーゼまたはマンノシダーゼが、(1)十分に翻訳されているか
、(2)触媒的に活性か、または(3)分泌経路内の適切なオルガネラに位置しているか
どうかを予測する信頼性の高い方法は存在しない。これらの3つ全てが、低級真核生物中
のグリコシル化パターンに影響するのに必要であり、所望の触媒機能および予測ツールの
非存在下での酵素の適切な保持を達成するための全体的なスキームは、現在利用可能では
なく、設計段階である。
【0017】
(治療用糖タンパク質の製造)
ヒトまたは動物から単離される有意な数のタンパク質は、最も有意な改変の1つである
グリコシル化によって翻訳後に改変される。全ての治療用タンパク質の推定で70%がグ
リコシル化され、従って、ヒトと類似の様式でグリコシル化し得る産生系(すなわち、宿
主細胞)に依存している。現在、多くの糖タンパク質は、哺乳動物宿主系で作製される。
いくつかの研究は、グリコシル化は、(1)免疫原性、(2)薬物動態学特性、(3)ト
ラフィック、および(4)治療用タンパク質の効果を決定するのに重要な役割を果たすこ
とが示されている。従って、製薬産業の実質的な労力は、可能であれば「ヒューマノイド
」または「ヒト様」のようなプロセスを開発し、糖タンパク質を得ることに向けられてい
る。これは、このような哺乳動物細胞を遺伝子操作して、細胞によって発現されたタンパ
ク質のシアリル化(すなわち、シアル酸の末端付加)(これは、このようなタンパク質の
薬物動態学特性を改善することが知られている)の程度を増加することを含み得る。ある
いは、公知のグリコシルトランスフェラーゼおよびそれぞれのヌクレオチド糖(例えば、
2,3シアリルトランスフェラーゼおよびCMP−シアル酸)を使用する、このような糖
のインビトロでの付加によってシアリル化の程度を改善し得る。
【0018】
さらなる調査は、特定の糖形態の生物学的有意性および治療的有意性を明らかにし得、
従って、そのような望ましい特定の糖形態を生成するための能力を与える。現在までに、
十分に特徴付けられた糖化パターンを有するタンパク質を生成すること、および以下の高
等真核生物のタンパク質発現系の1つにおいて、そのようなタンパク質をコードするcD
NAを発現することに、努力が費やされている。:
1.高等真核生物(例えば、チャイニーズハムスターの卵細胞(CHO)、マウス線維
芽細胞およびマウス骨髄腫細胞(Werner、1998);
2.トランスジェニック動物(例えば、ヤギ、ヒツジ、マウスおよその他(Dente
、1988);(Cole、1994);(McGarvey、1995);(Bard
or、1999);
3.植物(Arabidopsis、thaliana,tobaccoなど)(St
aub、2000);(McGarvey、1995);(Bardor、1999);
4.昆虫細胞(組換えバキュロウイルス(例えば、鱗翅目細胞に感染するAutogr
apha california多核ポリヒドローシスウイルス(Altmann,19
99))との組み合わせで、Spodoptera frugiperda Sf9,S
f21,Trichoplusia niなど)。
【0019】
最も高等な真核生物が、ヒトにおいて認められる糖化反応に類似する糖化反応を起こす
一方、上述の宿主系において発現される組換えヒトタンパク質は、不変的にそれらの「天
然の」ヒトの対応部分と異なる(Raju、2000)。従って、さらなる開発研究が、
これらの発現系において生成されるタンパク質の「ヒト特徴」を改善する方法を発見する
ことに向けられている。これは、発酵条件の最適化および、ヒト様糖形態の形成に関する
酵素をコードする遺伝子を導入することによる、タンパク質発現宿主の遺伝的改変を含む
(Werner、1998);(Weikert、1999);(Andersen、1
994);(Yang、2000)。全ての哺乳動物の発現系に関連する固有の問題は、
解決されていない。
【0020】
例えば、哺乳動物の細胞培養に基づく発酵プロセス(例えば、CHO細胞、マウス細胞
、またはヒト細胞)は、非常に遅い傾向があり(1週間を越える発酵時間は、まれではな
い)、しばしば、低い生成価を生じ、高価な栄養および補因子(例えば、ウシ胎児血清)
を要求し、プログラムされた細胞死(アポトーシス)によって制限され、しばしば、特定
の治療的価値のあるタンパク質の発現を不可能にする。より重要なことには、哺乳動物細
胞は、ヒト病原体となる能力を有するウイルスに感受性でありまた、厳密な質の制御が、
生成物安全を保証するために要求される。これは、多くのそのようなプロセスが、動物(
例えば、ウシ血清)由来の、複雑かつ温度感受性な培地成分の添加を必要とするので、特
定の関心事の内であり、ヒトに対する病原性因子(例えば、牛海綿状脳症(BSE)プリ
オンまたはウイルス)を保有し得る。さらに、治療的化合物の生成は、好ましくは、十分
に制御された滅菌環境において、実施される。動物飼育は、(どんなに清潔を保っていて
も)そのような環境を構築せず、従って、多量の治療的タンパク質を製造するためのトラ
ンスジェニック動物の使用におけるさらなる問題を与える。
【0021】
そのため、全てではないにしても、最近産生された治療的糖タンパク質は、哺乳動物細
胞において発現され、また、これらの組換えタンパク質の糖化パターンを改善すること(
すなわち。「ヒト化」すること)に多くの努力が、向けられている。培地組成物における
変化ならびにヒト糖化に関する酵素をコードする遺伝子の共発現が、継続的に行われてい
る(例えば、Weikert、1999を参照のこと)。
【0022】
ヒト対応部分に類似する組換えタンパク質が、哺乳動物発現系において作製され得、下
等真核生物(菌類および酵母)におけるヒト様糖化パターンを有するタンパク質を作製す
ることは、近年不可能である。小包体中で、タンパク質に移行される、核となるオリゴ糖
構造は、基本的には、哺乳動物および下等な真核生物において、同一であり、実質的な差
異は、次のプロセッシング反応において認められ、それは、菌類および哺乳動物のゴルジ
体において起こる。実際、異なる下等な真核生物間でさえ、多くの種々の糖化構造が、存
在する。これは、以下の哺乳動物発現系にわたって別に留意すべき利点を除いて、組換え
ヒト糖タンパク質の生成のための宿主として、下等真核生物の使用を妨げている。例えば
、:(1)一般的に高い産生力価、(2)短い発酵時間、(3)哺乳動物細胞において乏
しく発現されるタンパク質のための代替を有すること、(4)化学的に規定された無タン
パク質培地中での増殖し、従って、複雑な動物由来の培地成分を要求しない能力、(5)
およびウイルスの非存在(特に、そのような宿主のロタウイルス感染の非存在)。
【0023】
種々のメチル栄養性酵母(例えば、Pichia pastoris、Pichia
methanolicaおよびHansenula polymorphaは、真核生物
発現系として、特に重要な役割を果たす。なぜなら、それらは、高い細胞密度まで増殖し
得、大量の組換えタンパク質を分泌するためである。しかし、上記のように、下等真核生
物(例えば、酵母)は、高等動物のように、タンパク質を糖化しない。例えば、小包体(
ER)中の異種性マンノシダーゼを開示するMartinetら、(1998)Biot
echnol Let.20巻、12号を参照のこと。
【0024】
Chibaら、(1998)は、S.cerevisiaeが、1,6マンノシルトラ
ンスフェラーゼ(OCH1)、1,3マンノシルトランスフェラーゼ(MNN1)および
マンノシルトランスフェラーゼ(MNN4)の調節因子を取り除くことによって、ならび
に、ER検索配列(Chiba,1998)を用いてS.cerevisiaeのERに
Aspergillus saitoi由来のα−1,2−マンノシダーゼIの酵素作用
ドメインを標的することによって、Man8GlcNAc2構造からMan5GlcNAc2
構造の範囲の構造を提供するために操作され得ることを示した。しかし、この試みは、所
望のMan5GlcNAc2(例えば、インビボで生成され、また、GnT1についての基
質(ヒト様グリカン構造を作製する上での次の工程)として働き得るもの)。のわずかな
生成か、または全く生成しないことを生じる。Chibaら、(1998)は、P.pa
storisが、内在的に有用な量(5%より多くの)の、炭化水素を受け取るGlcN
AcトランスフェラーゼIを生成し得えない。
【0025】
Marasおよび同僚は、T.reeseiにおいて、「十分な濃度のアクセプター基
質(すなわち、Man5GlcNAc2)が存在する」ことを主張するが、このアクセプタ
ー基質をGlcNAcMan5GlcNAc2に、インビボで2%未満で転換することを試
みる場合、従って複合体Nグリカン形態のための適切な前駆体でないMan5GlcNA
c2構造の存在を示しことで変換された(Maras,1997;Maras,1999
)。現在まで、下等真核生物において、商業的に十分な量のGlcNAcMan5Glc
NAc2の生成が可能である開示は存在しない。
【発明の概要】
【発明が解決しようとする課題】
【0026】
このため、非ヒト宿主細胞において発現される組換え糖タンパク質の糖化をヒト化する
ためのシステムおよび方法を提供することが、本発明の目的である。
【課題を解決するための手段】
【0027】
(発明の要旨)
本発明は、改変された脂質連結オリゴ糖を有する菌株のような宿主細胞に関し、この宿
主細胞は、グリコシルトランスフェラーゼのセット、糖輸送体、およびマンノシダーゼの
異種発現によってされに改変され、哺乳動物の生成物(例えば、ヒト治療的糖タンパク質
)についての宿主株とされ得る。タンパク質生成方法は、以下を用いて開発されている;
(1)下等な真核生物宿主(例えば、単細胞真菌または繊維状真菌)または(2)ヒト由
来の異なる糖化パターンを有する、あらゆる非ヒト真核生物(ヒトタンパク質中に見出さ
れる炭化水素構造により類似するように、宿主生物(「宿主細胞」)中で生成される糖化
の組成およびタンパク質の構造が改変される。このプロセスは、科学文献およびタンパク
質発現の当業者に公知の、十分確立された方法によって、糖化に関する任意の所望の遺伝
子を発現および標的化するために使用され得る、操作された宿主細胞を得ることを可能に
する。本明細書に記載のように、改変された脂質連結オリゴ糖を有する宿主細胞は、作成
または選択される。操作された宿主細胞中のN−グリカンは、GlcNAcMan3Gl
cNAc2コア構造を有し、次いで、これは1つ以上の酵素(例えば、グリコシルトラン
スフェラーゼ、糖輸送体およびマンノシダーゼ)の異種発現によって、さらに改変され、
ヒト様糖タンパク質を産生し得る。治療的タンパク質の生成のために、この方法が、あら
ゆる所望の糖化構造を得ることができる細胞株を操作するために適合され得る。
【図面の簡単な説明】
【0028】
【図1】図1は、ドリチルピロホスフェート結合オリゴ糖の構造の概略図を示す。
【図2】図2は、alg3活性、alg9活性またはalg12活性を欠く真菌宿主細胞由来のGlcNAc2Man3GlcNAc2N−グリカンの生成の概略図を示す。
【図3】図3は、alg3、och1遺伝子型を有する真菌宿主細胞中の哺乳動物型のオリゴ糖構造を生成するために要求されるプロセッシング反応の概略図を示す。
【図4−1】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−2】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−3】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−4】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図5】図5は、S.cerevisiae Alg3およびAlg3p配列を示す。
【図6】図6は、P.pastoris Alg3およびAlg3p配列を示す。
【図7−1】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図7−2】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図7−3】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図8】図8は、K.lactis Alg3およびAlg3p配列を示す。
【図9】図9は、K.lactis Alg3配列比較(Blast)を示す。
【図10】図10は、S.cerevisiae Alg9およびAlg9p配列を示す。
【図11】図11は、P.pastoris Alg9およびAlg9p配列を示す。
【図12−1】図12は、P.pastoris Alg9配列比較(Blast)を示す。
【図12−2】図12は、P.pastoris Alg9配列比較(Blast)を示す。
【図13】図13は、S.cerevisiae Alg12およびAlg12p配列を示す。
【図14】図14は、P.pastoris Alg12およびAlg12p配列を示す。
【図15−1】図15は、P.pastoris Alg12配列比較(Blast)を示す。
【図15−2】図15は、P.pastoris Alg12配列比較(Blast)を示す。
【図16】図16は、優位なN−グリカンが、GlcNAcMan5GlcNAc2であることを示すP.pastoris中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図17】図17は、図16の優位なN−グリカンが、GlcNAcMan3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man5GlcNAc2に一致するピーク)を用いて処理されるP.pastoris(図16)中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図18】図18は、優位なN−グリカンが、GlcNAcMan3GlcNAc2およびGlcNAcMan4GlcNAc2であること示すP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図19】図19は、図18のGlcNAcMan4GlcNAc2が、GlcNAcMan3GlcNAc2に変換されることを示す、α1,2マンノシダーゼを用いて処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図20】図20は、図19のN−グリカンが、GlcNAcMan3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man3GlcNAc2に一致するピーク)を用いて処理される図19のN−グリカンのMALDI−TOF−MS分析である。
【図21】図21は、図19のGlcNAcMan3GlcNAc2が、GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図22】図22は、図21のN−グリカンが、GlcNAc2Man3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man3GlcNAc2に一致するピーク)を用いて処理される図21のN−グリカンのMALDI−TOF−MS分析である。
【図23】図23は、図21のGlcNAc2Man3GlcNAc2が、Gal2GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて、UDP−ガラクトースおよびβ1,4−ガラクトシルトランスフェラーゼの存在下で処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図24】図24は、Gal2GlcNAc2Man3GlcNAc2が、NANA2Gal2GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて、UDP−ガラクトースおよびβ1,4−ガラクトシルトランスフェラーゼの存在下で処理され、さらにCMP−N−アセチルノイラミン酸およびシアリルトランスフェラーゼを用いて処理される、P.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図25】図25は、S.cerevisiaeのAlg6配列およびAlg 6p配列を示す。
【図26】図26は、P.pastorisのAlg6配列およびAlg 6p配列を示す。
【図27−1】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−2】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−3】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−4】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図28】図28は、K.lactisのAlg6配列およびAlg 6p配列を示す。
【図29−1】図29は、K.lactis Alg 6配列の比較(Blast)を示す。
【図29−2】図29は、K.lactis Alg 6配列の比較(Blast)を示す。
【図30】図30 IgG免疫グロブリンのモデル。重鎖および軽鎖は、類似の二次構造および三次構造に基づいて、ドメインに細分される。2つの重鎖(ドメインVH、CH1、CH2およびCH3)は、3つのジスルフィド架橋により連結される。軽鎖(ドメインVLおよびCL)は、別のジスルフィド架橋により重鎖のCH1部分に連結され、そしてCH1フラグメントおよびVHフラグメントと共に、Fab領域を構成する。抗原は、Fab領域の末端部分に結合する。エフェクター機能(例えば、Fc−γ−レセプター結合)は、ヒンジ領域の直ぐ下流のCH2ドメインに局在化し、そして重鎖におけるアスパラギン297のN−グリコシル化により影響を受ける。
【図31】図31は、モジュラーIgG1発現ベクターの模式図である。
【図32】図32は、M.musculis GnT IIIの核酸配列およびアミノ酸配列を示す。
【図33】図33は、H.sapiens GnT IVの核酸配列およびアミノ酸配列を示す。
【図34−1】図34は、M.musculis GnT Vの核酸配列およびアミノ酸配列を示す。
【図34−2】図34は、M.musculis GnT Vの核酸配列およびアミノ酸配列を示す。
【発明を実施するための形態】
【0029】
(発明の詳細な説明)
本明細書中で他に規定されない限り、本発明に関連して使用される科学的用語および技
術用語は、当業者により一般的に理解される意味を有する。さらに、文脈により他に必要
とされなければ、単数形の用語は、複数形を含み、そして複数形の用語は、単数形を含む
。本発明の方法および技術は、当該分野において周知の従来の方法に従って一般的に実施
される。一般的に、本明細書中に記載される生化学、酵素学、分子生物学および細胞生物
学、微生物学、遺伝学ならびにタンパク質化学および核酸化学、ならびにハイブリダイゼ
ーションに関連して使用される名称、およびこれらの技術は、周知のものであり、そして
当該分野で一般的に使用される。本発明の方法および技術は、他に示されなければ、一般
的に、当該分野で周知の従来の方法に従って、そして本明細書の全体にわたって引用され
そして考察される種々の一般的な参考文献およびより具体的な参考文献に記載されるとお
りに実施される。例えば、Sambrookら、Molecular Cloning:
A Laboratory Manual,第2版,Cold Spring Harb
or Laboratory Press,Cold Spring Harbor,N
.Y.(1989);Ausubel et al.,Current Protoco
ls in Molecular Biology,Greene Publishin
g Associates(1992および2002までの補遺);Harlow an
d Lane Antibodies:A Laboratory Manual Co
ld Spring Harbor Laboratory Press,Cold S
pring Harbor,N.Y.(1990);Introduction to
Glycobiology,Maureen E.Taylor,KurtDricka
mer,Oxford Univ.Press (2003);Worthington
Enzyme Manual,Worthington Biochemical C
orp.Freehold,NJ;Handbook of Biochemistry
:Section A Proteins VolI 1976 CRC Press;
Handbook of Biochemistry:Section A Prote
ins Vol II 1976 CRC Press;Essentials of
Glycobiology,Cold Spring Harbor Laborato
ry Press (1999)を参照のこと。本明細書中に記載される生化学および分
子生物学に関連して使用される名称、ならびにこれらの研究室手順および技術は、当該分
野で周知でありかつ一般的に使用されるものである。
【0030】
本明細書中に記述される全ての刊行物、特許および他の参考文献は、参考として援用さ
れる。
【0031】
以下の用語は、他に示されなければ、以下の意味を有すると解釈される。
【0032】
本明細書中において使用される場合、用語「N−グリカン」とは、N−連結オリゴ糖を
いい、例えば、アスパラギン−N−アセチルグルコサミン結合でポリペプチドのアスパラ
ギン残基に結合されるものである。N−グリカンは、Man3GlcNAc2(「Man」
は、マンノースを指し;「Glc」は、グルコースを指し;そして「NAc」は、N−ア
セチルを指し;GlcNAcは、N−アセチルグルコサミンを指す)の共通のペンタサッ
カリドコアを有する。N−グリカンは、Man3GlcNAc2(「Man3」)コア構造
に付加される抹消糖(例えば、フコースおよびシアル酸)を含む分枝(アンテナ)の数に
関して異なる。N−グリカンは、それらの分枝構造(高マンノース、複合(comple
x)またはハイブリッド)に従って分類される。「高マンノース」型N−グリカンは、5
個以上のマンノース残基を有する。「複合」型N−グリカンは、代表的には、1,3マン
ノースアームに結合した少なくとも1つのGlcNAc、および「トリマンノース」コア
の1,6マンノースアームに結合した少なくとも1つのGlcNAcを有する。「トリマ
ンノースコア」は、Man3構造を有するペンタサッカリドコアである。複合N−グリカ
ンはまた、必要に応じてシアル酸または誘導体(「NeuAc」、ここで「Neu」は、
ノイラミン酸を指し、「Ac」は、アセチルを指す)で改変されたガラクトース(「Ga
l」)残基を有し得る。複合N−グリカンはまた、「分岐する(bisecting)」
GlcNAcおよびコアフコース(「Fuc」)を含む鎖内置換を有し得る。「ハイブリ
ッド」N−グリカンは、トリマンノースコアの1,3マンノースアームの末端に少なくと
も1つのGlcNAc、およびトリマンノースコアの1,6マンノースアームの0以上の
マンノースを有する。
【0033】
本明細書中で使用される略語は、当該分野で共通して利用される。例えば、上記の糖の
略語を参照のこと。他の共通の略語としては、以下が挙げられる:「PNGase」、こ
れは、ペプチドN−グリコシダーゼF(EC 3.2.2.18)を指す;「GlcNA
c Tr(I−III)」、3つのN−アセチルグルコサミニルトランスフェラーゼ酵素
のうちの1つを指す;「NANA」は、N−アセチルノイラミン酸を指す。
【0034】
本明細書中で使用される場合、用語「分泌経路」とは、細胞質から小胞体および(ER)およびゴルジ装置の区画への発生期ポリペプチド鎖の分子フローに沿って、脂質連結オリゴ糖前駆体およびN−グリカン基質が、連続的に暴露される、種々のグリコシル化酵素の集合ラインをいう。酵素は、この経路に沿って局在化されるといわれる。酵素Yの前に、脂質連結グリカンまたはN−グリカンに対して作用する酵素Xは、酵素Yの「上流」にある、または「上流」で作用するといわれる;同様に、酵素Yは、酵素Xから「下流」で作用する。
【0035】
本明細書中で使用される場合、用語「alg X活性」とは、「alg X」遺伝子に
よりコードされる酵素活性、および相同遺伝子もしくは遺伝子産物(以下を参照のこと)
または非関連遺伝子もしくは遺伝子産物によりコードされる酵素活性を有する酵素をいう
。
【0036】
本明細書中で使用される場合、用語「抗体」とは、完全な抗体(2つの重鎖および2つ
の軽鎖からなる)またはそのフラグメントをいう。このようなフラグメントとしては、種
々のプロテアーゼで消化することにより産生されるフラグメント、化学的切断および/ま
たは化学的解離、ならびにそのフラグメントが抗原に特異的に結合し得るままである限り
、組み換えにより産生されるフラグメントが挙げられるが、これらに限定されない。これ
らのフラグメントには、Fabフラグメント、Fab’フラグメント、F(ab’)2フ
ラグメントおよび単鎖Fv(scFv)フラグメントがある。用語「抗体」の範囲内には
、配列を改変されているが、抗原に特異的に結合し得るままである抗体もある。改変され
た抗体の例は、種間キメラ抗体およびヒト化抗体;抗体融合物;およびヘテロマー抗体複
合体(例えば、二価抗体(diabody)(二重特異性抗体)、単鎖二価抗体、および
細胞内発現抗体(intrabody))(例えば、Marasco(編),Intra
cellular Antibodies:Research and Disease
Applications,Springer−Verlag New York,I
nc.(1998)(ISBN:3540641513)(その開示は、本明細書中にそ
の全体が参考として援用される)を参照のこと)。
【0037】
本明細書中で使用される場合、用語「変異」とは、遺伝子産物(例えば、グリコシル化
関連酵素)の核酸配列またはアミノ酸配列における任意の変化をいう。
【0038】
用語「ポリヌクレオチド」または「核酸分子」とは、少なくとも10塩基長のヌクレオ
チドのポリマー形態をいう。この用語は、DNA分子(例えば、cDNAまたはゲノムD
NAもしくは合成DNA)およびRNA分子(例えば、mRNAまたは合成RNA)、な
らびにDNAまたはRNA含有非天然ヌクレオチドアナログ、非ネイティブヌクレオシド
間結合、またはその両方を含む。核酸は、任意のトポロジー的コンホメーションである得
る。例えば、核酸は、一本鎖、二本鎖、三本鎖、四重、部分的に二本鎖、分枝、ヘアピン
、環状または南京錠型のコンホメーションであり得る。この用語は、DNAの一本鎖形態
および二本鎖形態を含む。
【0039】
他に示さなければ、「配列番号Xを含む核酸」とは、その核酸の少なくとも一部が、(
i)配列番号Xの配列、または(ii)配列番号Xに相補的な配列のいずれかを有する核
酸をいう。これら2つの間の選択は、状況により必然的に決められる。例えば、核酸がプ
ローブとして使用される場合、2つの間の選択は、そのプローブが所望の標的に相補的で
ある必要性により必然的に決められる。
【0040】
「単離された」または「実質的に純粋な」核酸またはポリヌクレオチド(例えば、RN
A、DNAまたは混合ポリマー)は、天然の宿主細胞においてネイティブポリヌクレオチ
ドに自然に付随する他の細胞成分(例えば、リボソーム、ポリメラーゼ、およびそれが天
然で関連するゲノム配列)から実質的に分離された、核酸またはポリヌクレオチドをいう
。この用語は、(1)その天然に存在する環境から除去されたか、(2)その「単離され
たポリヌクレオチド」が天然に見出されるポリヌクレオチドの全てまたは一部を伴わない
か、(3)天然では連結されていないポリヌクレオチドに作動可能に連結されているか、
または(4)天然には存在しない、核酸またはポリヌクレオチドを包含する。用語「単離
された」または「実質的に純粋な」はまた、組み換え体もしくはクローン化されたDNA
単離物、化学的に合成されたポリヌクレオチドアナログ、または異種系により生物学的に
合成されたポリヌクレオチドアナログを言及する際に使用され得る。
【0041】
しかし、「単離された」は、そのように記載される核酸またはポリヌクレオチド自体が
、そのネイティブ環境から物理的に除去されたことを必ずしも必要とするわけではない。
例えば、生物のゲノム中の内因性核酸配列は、異種配列(すなわち、この内因性核酸配列
に天然では隣接していない配列)が、内因性核酸配列に隣接して配置されて、その結果こ
の内因性核酸配列の発現が変更される場合に、本明細書中で「単離された」とみなされる
。例として、非ネイティブプロモーター配列は、ヒト細胞のゲノムにおいて遺伝子のネイ
ティブプロモーターと(例えば、相同組み換えにより)置換され得、その結果、この遺伝
子が変更された発現パターンを有する。この遺伝子は、ここで「単離された」となる。な
ぜなら、これは、天然で隣接する配列の少なくとも一部から分離されているからである。
【0042】
核酸はまた、ゲノム中の対応する核酸に対する、天然に存在しない任意の改変を含む場
合に「単離された」とみなされる。例えば、内因性コード配列は、例えばヒトの介入によ
り人工的に導入された、挿入、欠失または点変異を含む場合に、「単離された」とみなさ
れる。「単離された核酸」はまた、異種部位において宿主細胞染色体に組み込まれた核酸
、エピソームとして存在する核酸構築物を含む。さらに、「単離された核酸」は、他の細
胞物質を実質的に含み得ないか、または組み換え技術により産生された場合に培養培地を
実質的に含み得ないか、または化学的に合成された場合に化学的前駆体もしくは他の化学
物質を実質的に含み得ない。
【0043】
本明細書中で使用される場合、用語、参照核酸配列の「縮重改変体」は、標準の遺伝子
コードに従って翻訳され得、参照核酸配列から翻訳されるアミノ酸配列と同一のアミノ酸
配列を提供する核酸配列を包含する。
【0044】
核酸配列の関係で、用語「パーセント配列同一性」または「同一な」は、最大一致で整
列された場合に同一である2つの配列中の残基をいう。配列同一性比較の長さは、少なく
とも約9ヌクレオチド、通常は少なくとも約20ヌクレオチド、より通常は少なくとも約
24ヌクレオチド、代表的には少なくとも約28ヌクレオチド、より代表的には少なくと
も約32ヌクレオチド、および好ましくは少なくとも約36以上のヌクレオチドのストレ
ッチにわたり得る。ヌクレオチド配列同一性を測定するのに使用され得る多くの異なるア
ルゴリズムが、当該分野で公知である。例えば、ポリヌクレオチド配列は、FASTA、
Gap、またはBestfitを用いて比較され得、これらは、Wisconsin P
ackage Version 10.0,Genetics Computer Gr
oup(GCG),Madison,Wisconsinのプログラムである。FAST
Aは、問い合わせ(query)配列と検索(search)配列との間の最良の重複領
域のアラインメントおよびパーセント配列同一性を提供する(Pearson,1990
(本明細書中で参考として援用される))。例えば、核酸配列間のパーセント配列同一性
は、本明細書中で参考として援用される、GCGバージョン6.1に提供されるように、
FASTAを用いて(そのデフォルトパラメーター(ワードサイズ6およびスコアリング
マトリクスについてのNOPAM因子)を用いて)決定され得るかまたはGapを用いて
(そのデフォルトパラメーターを用いて)決定され得る。
【0045】
用語「実質的に相同」または「実質的に同一」は、上記のような任意の周知の配列同一
性アルゴリズム(例えば、FASTA、BLAST、またはGap)により測定される場
合、核酸またはそのフラグメントを参照する場合、別の核酸(またはその相補鎖)と、適
切なヌクレオチド挿入または欠失と共に最適に整列される場合、少なくとも約50%、よ
り好ましくは60%のヌクレオチド塩基、通常は少なくとも約70%、より通常は少なく
とも約80%、好ましくは少なくとも約90%、およびより好ましくは少なくとも約95
%、96%、97%、98%または99%のヌクレオチド塩基においてヌクレオチド配列
同一性が存在することを示す。
【0046】
あるいは、実質的な相同性または類似性は、核酸またはそのフラグメントが、別の核酸
、別の核酸の鎖、またはその相補鎖に、ストリンジェントなハイブリダイゼーション条件
下でハイブリダイズする場合に存在する。核酸のハイブリダイゼーション実験の文脈にお
ける、「ストリンジェントなハイブリダイゼーション条件」および「ストリンジェントな
洗浄条件」は、多数の異なる物理的パラメーターに依存する。核酸ハイブリダイゼーショ
ンは、当業者によって容易に理解されるように、塩濃度、温度、溶媒、ハイブリダイズす
る種の塩基組成、相補領域の長さ、およびハイブリダイズする核酸の間のヌクレオチド塩
基のミスマッチの数のような条件によって、影響を受ける。当業者は、どのようにこのよ
うなパラメーターを変更して、特定のストリンジェンシーのハイブリダイゼーションを達
成するかを知っている。
【0047】
一般に、「ストリンジェントなハイブリダイゼーション」は、特定のDNAハイブリッ
ドについての熱的融点(Tm)より約25℃低い温度で、特定のセットの条件下で実施さ
れる。「ストリンジェントな洗浄」は、特定のDNAハイブリッドについてのTmより約
5℃低い温度で、特定のセットの条件下で実施される。Tmとは、標的配列の50%が、
完全にマッチするプローブにハイブリダイズする温度である。Sambrookら(前出
)、9.51頁(本明細書中に参考として援用される)を参照のこと。本明細書中での目
的のために、「高ストリンジェンシーの条件」とは、溶液相でのハイブリダイゼーション
について、6×SSC(ここで、20×SSCは、3.0M NaClおよび0.3Mク
エン酸ナトリウムを含有する)、1%SDS中65℃で8〜12時間の水性ハイブリダイ
ゼーション(すなわち、ホルムアミドを含まない)、続いて0.2×SSC、0.1%S
DS中65℃で20分間の2回の洗浄と定義される。65℃でのハイブリダイゼーション
は、ハイブリダイズする配列の長さおよび同一性の百分率を含む、多数の要因に依存して
、異なる速度で起こることが、当業者によって理解される。
【0048】
本発明の核酸(ポリヌクレオチドともまた称される)は、RNA、cDNA、ゲノムD
NA、ならびに上記のものの合成形態および混合ポリマーの、センス鎖とアンチセンス鎖
との両方を包含し得る。これらは、当業者によって容易に理解されるように、化学的にか
もしくは生化学的に改変され得るか、または非天然もしくは誘導体化されたヌクレオチド
塩基を含み得る。このような改変としては、例えば、標識、メチル化、1つ以上の天然に
存在するヌクレオチドのアナログでの置換、ヌクレオチド間改変(例えば、非荷電結合(
例えば、メチルホスホネート、ホスホトリエステル、ホスホロアミデート、カルバミドな
ど)、荷電結合(例えば、ホスホロチオエート、ホスホロジチオエートなど)、ペンダン
ト部分(例えば、ポリペプチド)、インターカレーター(例えば、アクリジン、ソラレン
など)、キレーター、アルキレーター、および改変された結合(例えば、αアノマー核酸
など))が挙げられる。水素結合および他の化学的相互作用を介して、指定された配列に
結合する能力において、ポリヌクレオチドを模倣する合成分子もまた含まれる。このよう
な分子は、当該分野において公知であり、そして例えば、分子の骨格においてペプチド結
合がホスフェート結合を置換する分子が挙げられる。
【0049】
用語「変異した」とは、核酸配列に対して適用される場合、核酸配列におけるヌクレオ
チドが、参照核酸配列と比較して、挿入、欠失、または変化し得ることを意味する。単一
の変化が、遺伝子座においてなされ得るか(点変異)、または複数のヌクレオチドが、単
一の遺伝子座において、挿入、欠失、または変化され得る。さらに、1つ以上の変更が、
核酸配列における任意の数の遺伝子座においてなされ得る。核酸配列は、当該分野におい
て公知の任意の方法によって変異され得、この方法としては、変異誘発技術(例えば、「
誤りやすいPCR」(DNAポリメラーゼのコピーの忠実度が低く、その結果、高い割合
の点変異が、PCR産物の全長に沿って得られる条件下でPCRを実施するためのプロセ
ス、例えば、Leung,D.W.ら,Technique,1,11−15頁(198
9)およびCaldwell,R.C.およびJoyce G.F.,PCR Meth
ods Applic.,2,28−33頁(1992)を参照のこと);および「オリ
ゴヌクレオチド特異的変異誘発」(目的の任意のクローニングされたDNAセグメントに
おける部位特異的変異の発生を可能にするプロセス。例えば、Reidhaar−Ols
on,J.F.およびSauer,R.T.ら、Science,241,53−57頁
(1988)を参照のこと))が挙げられるが、これらに限定されない。
【0050】
用語「ベクター」とは、本明細書中で使用される場合、核酸分子が結合していた別の核
酸を移動し得る核酸分子をいうことが意図される。1つの型のベクターは、「プラスミド
」であり、これは、内部にさらなるDNAセグメントが連結され得る、環状の二重鎖DN
Aループをいう。他のベクターとしては、コスミド、細菌性人工染色体(BAC)および
酵母人工染色体(YAC)が挙げられる。別の型のベクターは、ウイルスベクターであり
、ここで、さらなるDNAセグメントが、ウイルスゲノム内に連結され得る(以下にさら
に詳細に議論される)。特定のベクターは、それらが導入される宿主細胞において、自律
的に複製し得る(例えば、宿主細胞において機能する複製起点を有するベクター)。他の
ベクターは、宿主細胞への導入の際に、宿主細胞のゲノムに統合され得、そしてこれによ
って、宿主ゲノムとともに複製され得る。さらに、特定の好ましいベクターは、それらが
作動可能に連結した遺伝子の発現を導き得る。このようなベクターは、本明細書中で、「
組換え発現ベクター」(または単に、「発現ベクター」)と称される。
【0051】
「作動可能に連結された」発現制御配列とは、発現制御配列が、目的の遺伝子を制御す
るために、この目的の遺伝子と連続的である結合、およびこの目的の遺伝子を制御するた
めに、トランスでかまたはある距離で働く発現制御配列をいう。
【0052】
用語「発現制御配列」とは、本明細書中で使用される場合、それらが作動可能に連結し
たコード配列の発現に影響を与えるために必要な、ポリヌクレオチド配列をいう。発現制
御配列とは、核酸配列の転写、転写後事象および翻訳を制御する配列をいう。発現制御配
列は、適切な転写開始配列、終結配列、プロモーター配列およびエンハンサー配列;スプ
ライシングおよびポリアデニル化シグナルのような、効率的なRNAプロセシングシグナ
ル;細胞質mRNAを安定化する配列;翻訳効率を増強する配列(例えば、リボソーム結
合部位);タンパク質安定性を増強する配列;ならびに望ましい場合、タンパク質分泌を
増強する配列を含む。このような制御配列の性質は、宿主生物に依存して異なる;原核生
物においては、このような制御配列は、一般に、プロモーター、リボソーム結合部位、お
よび転写終結配列を含む。用語「制御配列」は、最低でも、その存在が発現のために必須
である全ての成分を含むことを意図され、そしてまた、その存在が有利であるさらなる成
分(例えば、リーダー配列および融合パートナー配列)を含み得る。
【0053】
用語「組換え宿主細胞」(または単に「宿主細胞」)とは、本明細書中で使用される場
合、組換えベクターが導入された細胞をいうことが意図される。このような用語は、特定
の被験体細胞のみでなく、そのような細胞の子孫をもまたいうことが意図されることが、
理解されるべきである。特定の改変が、変異または環境の影響のいずれかに起因して、引
き続く世代において起こり得るので、このような子孫は、実際に、親細胞と同一ではない
かもしれないが、本明細書中において使用される場合の用語「宿主細胞」の範囲内に依然
として含まれる。組換え宿主細胞は、培養中で増殖された、単離された細胞または細胞株
であり得か、あるいは生存組織または器官内に存在する細胞であり得る。
【0054】
用語「ペプチド」とは、本明細書中で使用される場合、短いポリペプチド(例えば、代
表的に約50アミノ酸長未満、そしてより代表的には約30アミノ酸長未満のポリペプチ
ド)をいう。この用語は、本明細書中において使用される場合、構造および従って生物学
的機能を模倣する、アナログおよび模倣物を包含する。
【0055】
用語「ポリペプチド」は、天然に存在するタンパク質および天然には存在しないタンパ
ク質の両方、ならびにそのフラグメント、変異体、誘導体およびアナログを包含する。ポ
リペプチドは、モノマーであってもポリマーであってもよい。さらに、ポリペプチドは、
多数の異なるドメインを含み得、これらのドメインの各々は、1つ以上の異なる活性を有
する。
【0056】
用語「単離されたタンパク質」または「単離されたポリペプチド」とは、その誘導の起
源または供給源のおかげで、(1)ネイティブな状態で付随する天然に会合する成分と会
合していないか、(2)天然には見出されない純度で存在する場合、純度が他の細胞材料
の存在に関して調節され得る(例えば、同じ種由来の他のタンパク質を含まない)か、(
3)異なる種由来の細胞によって発現されるか、あるいは(4)天然には存在しない(例
えば、天然に見出されるポリペプチドのフラグメントであるか、または天然には見出され
ないアミノ酸のアナログもしくは誘導体または標準的なペプチド結合以外の結合を含む)
タンパク質またはポリペプチドである。従って、化学的に合成されるかまたは天然に起源
とする細胞とは異なる細胞系において合成されるポリペプチドは、その天然に会合する成
分から「単離される」。ポリペプチドまたはタンパク質はまた、当該分野において周知の
タンパク質精製技術を使用する単離によって、天然に会合する成分を実質的に含まなくさ
れ得る。このように定義される場合、「単離された」とは、このように記載されるタンパ
ク質、ポリペプチド、ペプチドまたはオリゴペプチドが、そのネイティブの環境から物理
的に取り出されていることを、必ずしも必要としない。
【0057】
用語「ポリペプチドフラグメント」とは、本明細書中において使用される場合、全長ポ
リペプチドと比較して、アミノ末端および/またはカルボキシ末端の欠失を有するポリペ
プチドをいう。好ましい実施形態において、ポリペプチドフラグメントとは、そのフラグ
メントのアミノ酸配列が、天然に存在する配列における対応する部分と同一である、連続
的な配列である。フラグメントは、代表的に、少なくとも5アミノ酸長、6アミノ酸長、
7アミノ酸長、8アミノ酸長、9アミノ酸長、または10アミノ酸長であり、好ましくは
、少なくとも12アミノ酸長、14アミノ酸長、16アミノ酸長または18アミノ酸長で
あり、最も好ましくは、少なくとも20アミノ酸長であり、より好ましくは、少なくとも
25アミノ酸長、30アミノ酸長、35アミノ酸長、40アミノ酸長または45アミノ酸
長であり、なおより好ましくは、少なくとも50アミノ酸長または60アミノ酸長であり
、そしてなおより好ましくは、少なくとも70アミノ酸長である。
【0058】
「改変された誘導体」とは、一次構造の配列において実質的に相同であるが、例えば、
インビボまたはインビトロで、化学的改変および生化学的改変を含む、ポリペプチドまた
はそのフラグメント、あるいはネイティブなポリペプチドにおいては見出されないアミノ
酸を組み込むポリペプチドまたはそのフラグメントをいう。このような改変としては、例
えば、当業者によって容易に理解されるような、アセチル化、カルボキシル化、ホスホリ
ル化、グリコシル化、ユビキチン化、標識(例えば、放射性核種を用いる)、および種々
の酵素的改変が挙げられる。ポリペプチドを標識するための種々の方法、およびこのよう
な目的に有用な置換基または標識は、当該分野において周知であり、そして放射性同位体
(例えば、125I、32P、35S、および3H)、標識された抗リガンドに結合するリガンド
(例えば、抗体)、発蛍光団、化学発光剤、酵素、および標識されたリガンドに対する特
異的結合対メンバーとして働き得る抗リガンドが挙げられる。標識の選択は、必要とされ
る感度、プライマーとの結合体化の容易さ、安定性の要件、および利用可能な機器に依存
する。ポリペプチドを標識するための方法は、当該分野において周知である。Ausub
elら、1992(本明細書中に参考として援用される)を参照のこと。
【0059】
用語「融合タンパク質」とは、異種アミノ酸配列に結合したポリペプチドまたはフラグ
メントを含む、ポリペプチドをいう。融合タンパク質は、有用である。なぜなら、これら
は、2つ以上の異なるタンパク質由来の2つ以上の所望の機能的エレメントを含むように
構築され得るからである。融合タンパク質は、目的のポリペプチド由来の少なくとも10
個の連続したアミノ酸、より好ましくは、少なくとも20個または30個のアミノ酸、な
おより好ましくは、少なくとも40個、50個、または60個のアミノ酸、なおより好ま
しくは、少なくとも75個、100個、または125個のアミノ酸を含む。融合タンパク
質は、ポリペプチドまたはそのフラグメントを、異なるタンパク質またはペプチドをコー
ドする核酸配列とインフレームでコードする核酸配列を構築し、次いで、融合タンパク質
を発現させることによって、組換え的に産生され得る。あるいは、融合タンパク質は、ポ
リペプチドまたはそのフラグメントを、別のタンパク質に架橋させることによって、化学
的に産生され得る。
【0060】
用語「非ペプチドアナログ」とは、参照ポリペプチドの特性に類似の特性を有する化合
物をいう。非ペプチド化合物はまた、「ペプチド模倣物(peptide mimeti
c)」または「ペプチド模倣物(peptidomimetic)」と称され得る。例え
ば、Jones(1992)Amino Acid and Peptide Synt
hesis,Oxford University Press;Jung(1997)
Combinatorial Peptide and Nonpeptide Lib
raries:A Handbook John Wiley;Badanszkyら(
1993)Peptide Chemistry−−A Practical Text
book,Springer Verlag;「Synthetic Peptides
:A Users Guide」、G.A.Grant編、W.H.Freeman a
nd Co.,1992;Evansら、J.Med.Chem.30:1229(19
87);Fauchere,J.Adv.Drug Res.15:29(1986);
VeberおよびFreidinger TINS 392頁(1985);ならびに上
記のものの各々において引用される参考文献(これらは、本明細書中に参考として援用さ
れる)を参照のこと。このような化合物は、しばしば、コンピュータ化された分子モデリ
ングの補助によって開発される。本発明の有用なペプチドに構造的に類似のペプチド模倣
物を使用して、等価な効果を生じ得、従って、これらのペプチド模倣物は、本発明の一部
であると意図される。
【0061】
「ポリペプチド変異型」または「ムテイン」は、ネイティブタンパク質または野生型タ
ンパク質のアミノ酸配列に比べて、1つ以上のアミノ酸の挿入、重複、欠失、再配置また
は置換を含むポリペプチド配列をいう。ムテインは、アミノ末端またはカルボキシ末端の
いずれかまたは両方で、天然に存在するタンパク質および/またはアミノ酸配列の短縮型
の配列において、1つの位置で単一アミノ酸が、別のアミノ酸に変化される1つ以上のア
ミノ酸点変異を有し得、1つ以上のアミノ酸が、それぞれ、挿入されるか、もしくは欠失
される1つ以上の挿入および/または欠失を有し得る。ムテインは、天然に存在するタン
パク質に比べて、同じ生物学的活性を有し得るが、おそらく異なる生物学的活性を有し得
る。例えば、ムテインは、増加したニューロン結合活性もしくは減少したニューロン結合
活性またはNgR結合活性を有し得る。本発明の好ましい実施形態において、(例えば、
MAG Ig様ドメイン5中の)ムテインであるMAG誘導体は、内因性MAGまたは可
溶性野生型MAGに比べて、減少したニューロン成長阻害活性を有する。
【0062】
ムテインは、その野生型対応物に対して少なくとも70%の全配列相同性を有する。野
生型タンパク質に対して80%、85%または90%の全配列相同性を有するムテインは
、さらにより好ましい。さらにより好ましい実施形態において、ムテインは、95%の配
列同一性、なおより好ましくは97%の、さらにより好ましくは98%の、およびさらに
より好ましくは99%の全配列同一性を示す。配列相同性は、任意の共通配列分析アルゴ
リズム(例えば、GapまたはBestfit)によって測定され得る。
【0063】
好ましいアミノ酸置換は、以下のものである:(1)タンパク質分解に対する感受性を
減少する置換、(2)酸化に対する感受性を減少する置換、(3)タンパク質複合体を形
成するための結合親和性を変更する置換、(4)結合親和性または酵素活性を変更する置
換、および(5)このようなアナログの他の物理化学的特徴または機能的特徴を付与する
かまたは改変する置換。
【0064】
本明細書中で使用する場合、20の従来のアミノ酸およびこれらの省略形は、従来の慣
例に従う。Immunology−A Synthesis(第2版,E.S.Golu
bおよびD.R.Gren編,Sinauer Associates,Sunderl
and,Mass.(1991))(これは、参考として本明細書中に援用される)を参
照のこと。20の従来のアミノ酸、非天然アミノ酸(例えば、α−,α−二置換アミノ酸
、N−アルキルアミノ酸)、および他の非従来的なアミノ酸の立体異性体(例えば、D−
アミノ酸)はまた、本発明のポリペプチドのための適切な成分であり得る。非従来的なア
ミノ酸の例としては、以下:4−ヒドロキシプロリン、γ−カルボキシグルタミン酸、ε
−N,N,N−トリメチルリジン、ε−N−アセチルリジン、O−ホスホセリン、N−ア
セチルセリン、N−ホルミルメチオニン、3−メチルヒスチジン、5−ヒドロキシリジン
、s−N−メチルアルギニン、および他の類似のアミノ酸およびイミノ酸(例えば、4−
ヒドロキシプロリン)が挙げられる。本明細書中で使用されるポリペプチド表記において
、標準的な使用および慣例に従って、左手方向は、アミノ末端方向であり、右手方向は、
カルボキシ末端方向である。
【0065】
タンパク質は、このタンパク質をコードする核酸配列が、第2のタンパク質をコードす
る核酸配列に類似する配列を有する場合、「相同性」を有するか、または第2のタンパク
質に対して「相同」である。あるいは、タンパク質は、2つのタンパク質が、「類似する
」アミノ酸配列を有する場合、第2のタンパク質に対して相同性を有する。(従って、用
語「相同性タンパク質」は、2つのタンパク質が、類似するアミノ酸配列を有することを
意味するように規定される)。好ましい実施形態において、相同タンパク質は、野生型タ
ンパク質に対して60%の配列相同性を示し、より好ましくは、70%の配列相同性を示
す。野生型タンパク質に対して80%、85%または90%の配列相同性を示す相同タン
パク質が、さらにより好ましい。さらにより好ましい実施形態において、相同タンパク質
は、95%、97%、98%または99%の配列同一性を示す。本明細書中で使用される
場合、アミノ酸配列の2つの領域の間の相同性(特に予測された構造類似性に関して)は
、機能が類似することを意味するとして解釈される。
【0066】
「相同」が、タンパク質またはペプチドに関して使用される場合、同一ではない残基位
置が、保存的アミノ酸置換によってしばしば異なることが理解される。「保存的アミノ酸
置換」は、類似する化学的特性(例えば、電荷または疎水性度)を有する側鎖(R基)を
有する別のアミノ酸残基によって置換される置換である。一般的に、保存的アミノ酸置換
は、タンパク質の機能特性を実質的には変更しない。2つ以上のアミノ酸配列が、保存的
置換によって互いに異なる場合において、パーセント配列同一性または相同性の程度は、
置換の保存的性質を修正するように上方に調整され得る。この調整をするための手段は、
当業者に周知である(例えば、Pearsonら、1994(本明細書中に参考として援
用される)を参照のこと)。
【0067】
以下の6つの群は、各々互いに対して保存的な置換であるアミノ酸を含む:1)セリン
(S)、スレオニン(T);2)アスパラギン酸(D)、グルタミン酸(E);3)アス
パラギン(N)、グルタミン(Q);4)アルギニン(R)、リジン(K);5)イソロ
イシン(I)、ロイシン(L)、メチオニン(M)、アラニン(A)、バリン(V)、お
よび6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W)。
【0068】
ポリペプチドに対する配列相同性(パーセント配列同一性としても参照される)は、代
表的に、配列分析ソフトウェアを使用して決定される。例えば、Sequence An
alysis Software Package of the Genetics
Computer Group(GCG),University of Wiscon
sin Biotechnology Center,910 University
Avenue,Madison,Wisconsin 53705を参照のこと。タンパ
ク質分析ソフトウェアは、保存的アミノ酸置換を含む、種々の置換、欠失および他の改変
に対して割り当てられる相同性の尺度を使用して類似の配列を符合させる。例えば、GC
Gは、「Gap」および「Bestfit」のようなプログラムを備え、これは、デフォ
ルトパラメーターと共に使用され、密接に関連するポリペプチド(例えば、異なる生物種
からの相同性ポリペプチド)の間、あるいは野生型タンパク質とその変異型との間の配列
相同性または配列同一性を決定し得る。例えば、GCG Version 6.1を参照
のこと。
【0069】
阻害性分子配列を異なる生物由来の膨大な配列を含むデータベースと比較する場合、好
ましいアルゴリズムは、コンピュータープログラムBLAST(Altschul,S.
F.ら(1990)J.Mol.Biol.215:403−410;GishおよびS
tates(1993)Nature Genet.3:266−272;Madden
,T.L.ら(1996)Meth.Enzymol.266:131−141;Alt
schul,S.F.ら(1997)Nucleic Acids Res.25:33
89−3402;Zhang,J.およびMadden,T.L.(1997)Geno
me Res.7:649−656)、特に、blastpまたはtblatn(Alt
schulら、1997)である。BLASTpについての好ましいパラメーターは、
期待値:10(デフォルト)
フィルター:seg(デフォルト)
ギャップを開くためのコスト:11(デフォルト)
ギャップを伸長するためのコスト:1(デフォルト)
最大整列:100(デフォルト)
ワードサイズ:11(デフォルト)
記述の数:100(デフォルト)
ペナルティーマトリクス:BLOWSUM62
である。
【0070】
相同性について比較されるポリペプチド配列の長さは、一般的に、少なくとも約16ア
ミノ酸残基であり、通常には、少なくとも約20残基であり、より通常には、少なくとも
約24残基であり、代表的には、少なくとも約28残基であり、好ましくは、約35残基
を超える。膨大な数の異なる生物由来の配列を含むデータベースを検索する場合、アミノ
酸配列を比較することが好ましい。使用するアミノ酸配列を検索するデータベースは、当
該分野で公知のblastp以外のアルゴリズムによって決定され得る。例えば、ポリペ
プチド配列は、GCG Version 6.1中のプログラムFASTAを使用して比
較され得る。FASTAは、アライメントおよび質問配列と検索配列との間の最良の重複
の領域のアライメントおよびパーセント配列同一性を提供する(Pearson,199
0(参考として本明細書中に援用される))。例えば、アミノ酸配列間の間のパーセント
配列同一性は、GCG version 6.1(本明細書中に参考として援用される)
に提供されるようなそのデフォルトパラメーター(2のワードサイズおよびPAM250
スコアリングマトリクス)を用いてFASTAを使用して決定され得る。
【0071】
「特異的な結合」は、その環境における他の分子に対する結合に優先して、互いに結合
する2つの分子の能力をいう。代表的には、「特異的結合」は、少なくとも2倍、より代
表的には少なくとも10倍、しばしば少なくとも100倍、反応中の偶然の結合に対して
区別する。代表的には、特異的な結合反応の親和性または親和力は、少なくとも約10-7
M(例えば、少なくとも約10-8Mまたは10-9M)である。
【0072】
用語「領域」は、本明細書中で使用される場合、生体分子の一次構造の物理的に連続し
た部分をいう。タンパク質の場合において、領域は、このタンパク質のアミノ酸配列の連
続的領域によって規定される。
【0073】
用語「ドメイン」は、本明細書中で使用される場合、生体分子の公知の機能または推測
の機能に寄与する生体分子の構造をいう。ドメインは、その領域または部分と同程度に広
がり得;ドメインはまた、生体分子の別個の非連続的な領域を含み得る。タンパク質ドメ
インの例としては、Igドメイン、細胞外ドメイン、膜貫通ドメイン、および細胞質ドメ
インが挙げられるがこれらに限定されない。
【0074】
本明細書中で使用される場合、用語「分子」は、任意の化合物を意味し、これとしては
、低分子、ペプチド、タンパク質、糖、ヌクレオチド、核酸、脂質などが挙げられるがこ
れらに限定されず、このような化合物は、天然であっても合成であってもよい。
【0075】
他に定義されない場合、本明細書中で使用される全ての技術用語および科学用語は、本
発明に関する分野の当業者によって通常理解される意味と同じ意味を有する。例示的な方
法および材料は、以下に示されるが、本明細書中に記載される方法および材料と類似する
かまたは等価な方法および材料はまた、本発明の実施において使用され得、そして、これ
らは、当業者に明らかである。全ての刊行物および本明細書中に記載される他の参考は、
その全体を参考として援用される。矛盾する場合、定義を含む本明細書は、制御する。材
料、方法、および実施例は、例示のみであり、そして、限定を意図されない。
【0076】
本明細書および特許請求の範囲の全体にわたって、単語「含む(comprise)」
またはバリエーション(例えば、「含む(comprises)」または「含む(com
prising)」)は、示された整数または整数の群の含入を意味するが、任意の他の
整数または整数の群の排除を意図されないことが理解される。
【0077】
(ヒト様N−グリカンの生成に対して改変された脂質連結オリゴ糖を有する宿主の操作
または選択)
本発明は、非ヒト真核生物宿主細胞において、ヒト様糖タンパク質を産生するための方
法を提供する。本発明は、alg遺伝子活性(すなわち、alg活性(非真菌宿主細胞中
の等価な酵素活性を含む))を低下するかまたは激減する非ヒト真核生物宿主細胞を作製
する工程または使用する工程、および宿主細胞に少なくとも1つのグリコシダーゼ活性を
導入する工程を包含する。好ましい実施例において、グリコオシダーゼ活性は、宿主細胞
内の1つ以上のマンノシダーゼ活性の発現によって、例えば、マンノシダーゼ活性の活性
化によって、または宿主細胞におけるマンノシダーゼ活性の核酸分子からの発現によって
、導入される。
【0078】
別の実施形態において、本方法は、糖残基を、脂質連結オリゴ糖前駆体の1,6アーム
に転位する1つ以上の酵素の活性を低下されたか、または激減された宿主細胞を作製する
工程またはそれを使用する工程を包含する(図1)。本発明の宿主細胞は、糖残基(例え
ば、マンノシレート)を、脂質連結オリゴ糖前駆体の1,6アームに転位する酵素をコー
ドする1つ以上の遺伝子中に変異を導入することについて選択されるかまたはそのことに
よって操作される。糖残基は、より好ましくはマンノースであり、好ましくはグルコース
、GlcNAc、ガラクトース、シアル酸、フコースまたはGlcNAcリン酸残基であ
る。好ましい実施形態において、脂質連結オリゴ糖前駆体の1,6アームをマンノシル化
する1つ以上の酵素の活性は、低下するかまたは激減する。本方法は、少なくとも1つの
グリコシダーゼ活性を宿主細胞に導入する工程をさらに包含し得る(以下を参照のこと)
。
【0079】
なお別の実施形態において、本発明は、非ヒト宿主においてヒト様糖タンパク質を産生
するための方法を提供し、ここで、糖タンパク質は、トリマンノースコア構造に結合され
る少なくとも2つのGlcNAcを有するN−グリカンを含む。
【0080】
上記の各々の実施形態において、本方法は、宿主細胞を作製する工程に関し、ここで、
脂質連結オリゴ糖前駆体は、ManXGlcNAc2構造中で濃縮され、ここで、Xは、3
、4、または5である(図2)。これらの構造は、宿主細胞のER中で、オリゴサッカリ
ルトランスフェラーゼによって新生ポリペプチド鎖上に転位され、次いで、グリコシダー
ゼ(例えば、α−マンノシダーゼ)およびグリコシルトランスフェラーゼ(例えば、Gn
T1)での処理によってプロセスされ、GlcNAcManXGlcNAc2コア構造を有
するN−グリカンを生成し、ここで、Xは、3、4または5であり、そして好ましくは3
である(図2および図3)。図2に示されるように、GlcNAcManXGlcNAc2
コア構造を有し、Xが3を超えるN−グリカンは、適用可能な場合、例えば、α−1,3
マンノシダーゼ活性および/またはα−1,2−1,3マンノシダーゼ活性で処理するこ
とによって、GlcNAcMan3GlcNAc2に転換され得る。
【0081】
グリコシルトランスフェラーゼ(例えば、GnTII)での処理によるGlcNAcM
an3GlcNAc2のさらなるプロセシングは、GlcNAc2Man3GlcNAc2コ
ア構造を生成し、これは次いで、所望の場合、例えば、エキソビボ処理によって、または
一連のグリコシル化酵素(グリコシルトランスフェラーゼ、糖トランスポーターおよびマ
ンノシダーゼを含む(以下を参照のこと))の宿主細胞内での異種発現によって、改変さ
れて、ヒト様N−グリカンになり得る。本発明に従って生成され得る好ましいヒト様糖タ
ンパク質であって、7個以下、または3個以下のマンノース残基を有するN−グリカンを
含む糖タンパク質は、ガラクトース、GlcNAc、シアル酸およびフコースからなる群
から選択される1つ以上の糖を含み;そしてNeuNAc−Gal−GlcNAc−Ma
nを含む少なくとも1つのオリゴ糖分枝を含む。
【0082】
一実施形態において、宿主細胞は、減退されたかまたは減少されたDol−P−Man
:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼ活性を有し、この
活性は、ERのルミナル(luminal)側におけるMan5GlcNAc2−PP−D
olからMan6GlcNAc2−PP−Dolへの第1のマンノシル化工程に関与する(
例えば、ALG3 図1;図2)。S.cerevisiaeにおいて、この酵素は、A
LG3遺伝子によってコードされる。上記のように、漏出alg3−1変異を有するS.
cerevisiae細胞は、Man5GlcNAc2−PP−Dolを蓄積し、そしてa
lg3における欠失を有するS.serevisiae細胞は、Man5GlcNAc2構
造をER内の新生ポリペプチド鎖上に移動させるようである。従って、この実施形態にお
いて、宿主細胞は、Man5GlcNAc2構造に富化されたN−グリカンを蓄積し、これ
は次いで、グリコシダーゼを用いる(例えば、α−1,2−マンノシダーゼ、α−1,3
マンノシダーゼ、またはα−1,2−1,3マンノシダーゼ活性を用いる)処理によって
GlcNAc2Man3GlcNAc2に変換され得る(図2) 実施例1に記載されるよ
うに、縮重プライマーを、S.cerevisiae、D.melanogasterお
よびヒト(H.sapiens)由来のAlg3タンパク質配列の整列に基づいて設計し
(図4および5)、そしてこれを使用して、P.pastorisゲノムDNA由来の産
物を増幅した。得られたPCR産物を、プローブとして使用して、S.cerevisi
ae ALG3遺伝子に対して35%の全配列同一性および53%の配列類似性を有する
タンパク質をコードするオープンリーディングフレーム(ORF)を含むP.pasto
risゲノムクローンを同定しそして単離した(図6および7)。このP.pastor
is遺伝子を、本明細書中で、「PpALG3」と呼ぶ。ALG3遺伝子を、同様に同定
し、そしてK.lactisから単離した(実施例1;図8および9)。
【0083】
従って、別の実施形態において、本発明は、P.pastoris ALG3遺伝子(
図6)およびK.lactis ALG3遺伝子(図8)ならびにそれらのホモログ、改
変体および誘導体の、少なくとも45、好ましくは少なくとも50、より好ましくは少な
くとも60、最も好ましくは75以上のヌクレオチド残基を含むか、これらからなる核酸
配列を有する単離された核酸分子を提供する。本発明はまた、ストリンジェントな条件下
で、上記の核酸分子にハイブリダイズする核酸分子を提供する。同様に、本発明の核酸分
子によってコードされる単離されたポリペプチド(ムテイン、対立遺伝子改変体、フラグ
メント、誘導体およびアナログを含む)が、提供される(P.pastorisおよびK
.lactisのALG3遺伝子産物は、図6および8に示される)。さらに、本明細書
中にさらに記載されるように、本発明の核酸分子を含むベクター(発現ベクターを含む)
もまた提供される。
【0084】
遺伝子特異的プライマーを使用して、構築物を作製して、P.pastorisのゲノ
ムからPpALG3遺伝子を欠失させた(実施例1)。この株を使用して、Dol−P−
Man:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼ活性を欠失
した宿主細胞を作製し、そして脂質連結Man5GlcNAc2−PP−Dol前駆体を生
成した。この脂質連結Man5GlcNAc2−PP−Dol前駆体は、新生ポリペプチド
鎖上に移動され、Man5GlcNAc2糖質構造を有するN−グリカンを生成する。
【0085】
実施例2に記載されるように、このような宿主細胞は、所望のMan3GlcNAc2コ
ア糖質構造を有するN−グリカンを生成するように、適切なマンノシダーゼの発現によっ
て操作され得る。宿主細胞におけるGnTの発現(例えば、下記のように、核酸分子また
は核酸分子のライブラリーを標的化することによって)により、改変された宿主細胞は、
Man3コア構造(すなわち、GlcNAc1Man3GlcNAc2またはGlcNAc2
Man3GlcNAc2;図3を参照のこと)の各アームに結合した1つまたは2つのGl
cNAc構造を有するN−グリカンを生成し得る。これらの構造は、本発明の方法を使用
してさらに処理されて、宿主細胞の分泌経路に入るヒト様N−グリカンまたはタンパク質
を生成し得る。
【0086】
別の実施形態において、宿主細胞は、減退されたかまたは欠失されたドリチル(dol
ichyl)−P−Man:Man6GlcNAc2−PP−ドリチルα1,2−マンノシ
ルトランスフェラーゼ活性を有し、この活性は、ERのルミナル側においてMan6Gl
cNAc2−PP−DolをMan7GlcNAc2−PP−Dolへ変換するマンノシル
化工程に関与する(上記ならびに図1および図2を参照のこと)。S.cerevisi
aeにおいて、この酵素は、ALG9遺伝子によってコードされる。alg9変異を有す
る細胞は、Man6GlcNAc2−PP−Dolを蓄積し(図2)、そしてMan6Gl
cNAc2構造をER内の新生ポリペプチド鎖上に移動させる。従って、この実施形態に
おいて、宿主細胞は、Man6GlcNAc2構造に富化されたN−グリカンを蓄積し、こ
れは次いで、α−1,2−マンノシダーゼおよびα−1,3マンノシダーゼを用いる処理
によってコアMan3構造にプロセスダウンされ得る(図3ならびに実施例3および4を
参照のこと)。
【0087】
alg9遺伝子(または等価な活性をコードする遺伝子)が欠失された宿主細胞を構築
する(実施例3を参照のこと)。ALG9(または、ALG12;以下を参照のこと)の
欠失は、1,6アーム上の、それぞれ、1つまたは2つのさらなるマンノースを有するN
−グリカンを生成する宿主細胞を作製する(図2)。N−アセチルグルコサミニルトラン
スフェラーゼII(Gn TII)に接近可能な1,6−コアマンノースを作製するため
に、これらのマンノースは、グリコシダーゼによって除去される必要がある。ERマンノ
シダーゼは、代表的に、1,6アーム上の末端1,2マンノースを除去し、続いてマンノ
シダーゼII(α1−3,6マンノシダーゼ)または他のマンノシダーゼ(例えば、α1
,2マンノシダーゼ、α1,3マンノシダーゼまたはα1−2,3マンノシダーゼ(例え
ば、Xanthomonas manihotis由来;実施例4を参照のこと))が、
1,6アームに作用し得、続いて、GnTIIが、N−アセチルグルコサミンを移動させ
て、GlcNAc2Man3を生成する(図2)。
【0088】
alg9p活性を欠く得られた宿主細胞を、1つ以上の酵素由来のα1,2−マンノシ
ダーゼ活性およびα1,3マンノシダーゼ活性を発現するように、好ましくは、宿主細胞
に導入される核酸分子からの発現によって、操作され、そしてこれは、好ましい細胞成分
に標的化される酵素を発現する(以下を参照のこと)。実施例4は、Xanthomon
as manihotis由来のこのような酵素のクローニングおよび発現を記載する。
【0089】
別の実施形態において、宿主細胞は、減退されたかまたは欠失されたドリチル−P−M
an:Man7GlcNAc2−PP−ドリチルα1,6−マンノシルトランスフェラーゼ
活性を有し、この活性は、ERのルミナル側においてMan7GlcNAc2−PP−Do
lをMan8GlcNAc2−PP−Dol(これはコアマンノース構造の1,6アーム上
のα−1,6マンノースをマンノシル化する)へ変換するマンノシル化工程に関与する(
上記ならびに図1および図2を参照のこと)。S.cerevisiaeにおいて、この
酵素は、ALG12遺伝子によってコードされる。alg12変異を有する細胞は、Ma
n7GlcNAc2−PP−Dolを蓄積し(図2)、そしてMan7GlcNAc2構造を
ER内の新生ポリペプチド鎖上に移動させる。従って、この実施形態において、宿主細胞
は、Man7GlcNAc2構造に富化されたN−グリカンを蓄積し、これは次いで、α−
1,2−マンノシダーゼおよびα−1,3マンノシダーゼを用いる処理によってコアMa
n3構造にプロセスダウンされ得る(図3ならびに実施例3および4を参照
のこと)。
【0090】
alg9変異体宿主について上で記載されるように、alg12p活性を欠く得られた
宿主細胞を、(例えば、1つ以上の酵素由来の)α1,2−マンノシダーゼ活性およびα
1,3マンノシダーゼ活性を発現するように、好ましくは、宿主細胞に導入される1つ以
上の核酸分子からの発現によって、操作され、そしてこれは、好ましい非細胞成分に標的
化される酵素活性を発現する(以下を参照のこと)。
【0091】
(必要に応じて減少した開始α−1,6マンノシルトランスフェラーゼ活性を有する宿
主の操作または選択)
好ましい実施形態において、本発明の方法は、(a)alg遺伝子の活性またはMan
3GlcNAc2(「Man3」)コア糖質構造上のα−1,6アームのN−グリカンをマ
ンノシル化する1つ以上の活性が減退または減少しており、かつ(b)(Man3コア構
造のα−1,3アーム上の)開始α−1,6−マンノシルトランスフェラーゼ(すなわち
、外側鎖マンノシル化を開始する開始特異的酵素)の活性が減退または欠失している、宿
主細胞を作製または使用することを包含する。S.cerevisiaeにおいて、この
酵素は、OCH1遺伝子によってコードされる。S.cerevisiaeにおけるoc
h1遺伝子の破壊は、N連結糖がポリマンノース外側鎖を完全に欠く表現型を生じる。真
菌株における哺乳動物型グリコシル化を得るための以前のアプローチは、OCH1の不活
性化を必要とした(例えば、Chiba,1998を参照のこと)。しかし、Och1p
酵素が、有効なマンノース外側鎖開始についてインタクトなMan8GlcNAcを必要
とする場合、本発明の宿主細胞における開始α−1,6−マンノシルトランスフェラーゼ
活性の破壊は、任意である(選択された宿主細胞に依存して)。従って、本発明に従って
選択または生成された宿主細胞(これは、7個以下のマンノース残基を有する脂質連結オ
リゴ糖を蓄積する)は、移動の後、Och1pに対する乏しい基質の可能性のある低グリ
コシル化N−グリカンを生成する(例えば、Nakayama,1997を参照のこと)
。
【0092】
(増加したグリコシルトランスフェラーゼ活性を有する宿主の操作または選択)
上記のように、グリコシル化オリゴ糖は、それらの非グリコシル化対応物よりもはるか
に高い割合で新生ポリペプチド鎖に移動されると考えられる。オリゴ糖トランスフェラー
ゼ複合体による基質認識は、脂質連結オリゴ糖のアンテナへのグルコースの付加によって
増大するようである。従って、脂質連結オリゴ糖を最適にグリコシル化し得る宿主細胞を
作製または選択することが望まれる。このような宿主細胞において、過小グリコシル化(
underglycosylation)は、グリコシル化Man5構造の新生ポリペプ
チド鎖上へのより速くより効率的な移動に起因して、実質的に減少されるかまたは消滅さ
れさえする。
【0093】
従って、本発明の別の実施形態において、この方法は、脂質連結Nグリカン前駆体がE
R中の新生ポリペプチド鎖に効率的に移動される宿主細胞を作製することに関する。好ま
しい実施形態において、移動は、脂質連結オリゴ糖の分枝上のグリコシル化レベルを増加
することによって増大され、このことにより、このオリゴ糖は、オリゴサッカリルトラン
スフェラーゼに対するより良い基質となる。
【0094】
1つの好ましい実施形態において、本発明は、ヒト様糖タンパク質を作製するための方
法を提供し、この方法は、ER中の脂質連結オリゴ糖のグリコシル化を担う1つ以上の酵
素が増加した活性を有する宿主細胞を使用する。脂質連結オリゴ糖のグリコシル化の程度
を増大するための1つの方法は、脂質連結オリゴ糖のアンテナ上へのグルコース残基の移
動を担う1つ以上の酵素を過剰発現させることである。特に、α−1,3グルコシルトラ
ンスフェラーゼ活性の増加により、グルコシル化脂質連結Man5構造の量が増加し、そ
して分泌タンパク質の過小グリコシル化を減少させるかまたは排除する。S.cerev
isiaeにおいて、この酵素は、ALG6遺伝子によってコードされる。
【0095】
Saccharomyces cerevisiae ALG6およびそのヒト対応物
は、クローニングされている(Imbach,1999;Reiss,1996)。グリ
コシル化の初期工程の進化的保存に起因して、ALG6座は、種間で相同であると予測さ
れ、そして当業者によって、配列類似性に基づいてクローニングされ得る(同じことが、
異なる種由来のALG8およびALG10座のクローニングおよび同定にあてはまる)。
さらに、異なる種由来の異なるグルコシルトランスフェラーゼが、次いで、最適な活性を
有するグルコシルトランスフェラーゼを同定するために試験され得る。
【0096】
強力なプロモーター(例えば、GAPDHプロモーター)の制御下でのALG6遺伝子
のさらなるコピーの導入および/またはALG6の発現は、グルコシル化脂質連結オリゴ
糖の程度を増加するためのいくつかの方法の1つである。P.pastoris由来のA
LG6遺伝子を、クローニングしそして発現する(実施例5)。ALG6核酸およびアミ
ノ酸配列を、図25(S.cerevisiae)および図26(P.pastoris
)について示す。これらの配列を、図27の他の真核生物ALG6配列と比較する。
【0097】
従って、本発明の別の実施形態は、脂質連結オリゴ糖のグリコシル化の程度を増大する
方法を提供し、この方法は、宿主細胞におけるα−1,3グルコシルトランスフェラーゼ
活性を増大する工程を包含する。この活性の増加は、例えば、この活性をコードする核酸
を1つ以上の異種発現制御配列と作動可能に連結することによって、この活性をコードす
る核酸配列を過剰発現することによって、達成され得る。好ましい発現制御配列としては
、転写開始配列、転写終結配列、プロモーター配列およびエンハンサー配列;RNAスプ
ライスドナーおよびポリアデニル化シグナル;mRNA安定化配列;リボソーム結合部位
;タンパク質安定化配列;ならびにタンパク質分泌配列が挙げられる。
【0098】
別の実施形態において、α−1,3−グルコシルトランスフェラーゼ活性の増加は、当
業者に周知の技術を使用して、マルチコピープラスミド上に、活性をコードする核酸分子
を導入することによって達成される。さらに別の実施形態において、脂質連結オリゴ糖の
グルコシル化の程度は、宿主細胞中のオリゴサッカリルトランスフェラーゼ(oligo
saccharyl transferase)活性の基質特異性を低下することを含む
。このことは、例えば、活性をコードする少なくとも1つの核酸を、遺伝子シャッフリン
グ、インビトロ変異誘発、および誤りがちのポリメラーゼ連鎖反応のような技術(これら
の全ては、当業者に周知である)に供することによって達成される。当然、ALG8およ
びALG10は、宿主細胞中で過剰発現され、同様の様式で試験され得る。
【0099】
従って、好ましい実施形態において、本発明は、ER中の脂質連結オリゴ糖のグリコシ
ル化を担う1つ以上の酵素が、活性を増加するように操作されたかまたは選択された宿主
細胞を使用してヒト様糖タンパク質を作製するための方法を提供する。より好ましい実施
形態において、本発明は、以下:
(a)1つ以上のalg遺伝子活性またはMan3GlcNAc2(「Man3」)コア糖
質構造のα−1,6アームで、N−グリカンをマンノシル化する活性の活性が低下または
激減し、かつ(b)ER中での脂質連結オリゴ糖のグリコシル化を担う1つ以上の酵素が
活性を増加するように操作されるかまたは選択される宿主細胞を使用する。しかし、al
g3変異型バックグラウンドにおいて見出される脂質連結Man5構造は、Alg6pに
対する好ましい基質ではない。従って、当業者は、例えば、当業者に周知の分子生物学お
よび遺伝子選択技術を使用して、基質特異性が増加した酵素をコードする核酸を、1以上
のラウンドの遺伝子シャッフリング、誤りがちのPCR、またはインビトロ変異誘発アプ
ローチに供することによって、そして目的の宿主細胞中で基質特異性の上昇について選択
することによって、基質特異性が上昇したAlg6p、Alg8pおよびAlg10pを
同定し得る(Gibbs,2001)。選択された宿主株において酵素基質特異性を改善
するためのこのような技術が、本発明のこの特定の実施形態に限定されないが、非ヒト宿
主細胞においてヒト様N−グリカンの産生をさらに最適化する任意の実施形態において使
用され得ることは、当業者に理解される。
【0100】
記載されるように、Man5は、新生ポリペプチド鎖に移されると、適切なα−1,2
−マンノシダーゼの発現は、本発明によって提供されるように、Man5GlcNAc2構
造をさらに小さくし、所望のコアMan3GlcNAc2構造を生じる。α−1,2−マン
ノシダーゼは、末端α−1,2−結合マンノース残基のみを除去し、alg3、alg9
およびalg12変異型宿主細胞およびこれらの遺伝子に対するホモログが変異された宿
主細胞中で作られたMan5GlcNAc2−Man7GlcNAc2特異的構造を認識する
と予想される。
【0101】
図3に概略的に示されるように、適切なUDP−糖−輸送体およびUDP−糖−トラン
スフェラーゼの共発現は、末端α−1,6残基およびα−1,3残基をGlcNAcでキ
ャップし、このことは、哺乳動物型複合体およびハイブリッドN−グリコシル化に必要な
前駆体(GlcNAc2Man5GlcNAc2)を生じる。次いで、ペプチドが結合した
N−連結オリゴ糖鎖(GlcNAc2Man5GlcNAc2(図3)は、哺乳動物型オリ
ゴ糖構造へのさらなる改変のための前駆体として働く。続いてのガラクトシルトランスフ
ェラーゼの発現およびシアル酸(sialylic acid)を転位する能力を遺伝子
操作することは、哺乳動物型(例えば、ヒト様)N−グリカン構造を生じる。
【0102】
本発明に従う所望の宿主細胞は、1つの酵素または1つを超える酵素を同時に操作し得
る。さらに、潜在的に有用な酵素をコードする遺伝子のライブラリーが、作製され得、最
適な活性を有する1つ以上の酵素を有する株または最も「ヒト様」糖タンパク質を産生す
る株は、ライブラリーの1つ以上のメンバーを用いて標的宿主細胞を形質転換することに
よって選択される。コアN−グリカンMan3GlcNAc2を有する糖タンパク質を産生
し得る下等真核生物は、遺伝子操作の実施の容易さ、ならびに安全性および効率特性に起
因して、特に有用である。好ましい実施形態において、少なくとも1つのさらなるグリコ
シル化反応が、エキソビボまたはインビボで実施され、ヒト様N−グリカンを産生する。
より好ましい実施形態において、グリコシル化酵素の活性型は、宿主細胞の小胞体および
/またはゴルジ装置において発現され、所望のヒト様糖タンパク質を産生する。
【0103】
(宿主細胞)
本発明の好ましい非ヒト宿主細胞は、下等真核生物細胞(例えば、単細胞真菌または線
維状真菌)であり、これは、1つ以上のalg遺伝子活性(alg活性に対して相同であ
るか、または等価である酵素活性を含む)が減少しているか、または激減している。本発
明の別の好ましい宿主細胞は、脂質連結オリゴ糖構造のα−1,6アームをマンノシル化
する1つ以上の酵素の活性(alg活性以外)が低下されるか、または激減する。
【0104】
下等真核生物宿主細胞が、好ましくある、一方で、前述の特徴を有する広く種々の宿主
細胞は、本発明の方法において有用であるとして構想される。例えば、植物細胞は、本発
明に従って、ヒト様糖タンパク質を発現するように操作され得る。同様に、種々の非ヒト
哺乳動物宿主細胞は、本発明の方法を使用して、よりヒト様の糖タンパク質を発現するよ
うに変更され得る。適切な宿主細胞が、操作され得るか、または酵母において既に記載さ
れる多くのこのような変異型の1つが、使用され得る。本明細書中に例示される場合、本
発明の好ましい宿主細胞は、Pichia Pastoris中の過剰マンノシル化欠損
(OCH1)変異型であり、この変異型は、alg3遺伝子をさらに改変し、欠失する。
他の好ましい宿主は、och1変異およびalg9変異またはalg12変異を有するP
ichia pastoris変異型である。
【0105】
(複合N−グリカンの形成)
改変された、新生N−グリカン構造に糖を連続的に添加することは、グルコシルトラン
スフェラーゼのゴルジ装置への首尾よい標的化および首尾よい発現を含む。このプロセス
は、例えば、機能的発現(例えば、初期ゴルジ装置または中期ゴルジ装置におけるGnT
Iの機能的発現)を必要とし、UDP−GlcNAc(例えば、UDP−GlcNAc輸
送体の発現による)の十分な供給を保証する。
【0106】
糖タンパク質を特徴づけ、そして所望されるグリコシル化を確認するために、糖タンパ
ク質を精製し、N−グリカンを、PNGase−F放出し、次いで、MALDI−TOF
−MSによって分析した(実施例2)。ヒトプラスミノゲンのクリングル3ドメインを、
レポータータンパク質として使用した。この可溶性糖タンパク質を、alg3、och1
ノックアウトのバックグラウンドで、P.pastoris中で産生した(実施例2)。
【0107】
GlcNAcMan5GlcNAc2を、ヒトGnTIおよび図16のK.lactis
UDP−GlcNac輸送体の添加後、優勢なN−グリカンとして提供した(実施例2
)。このN−グリカンの質量は、1463(m/z)であるGlcNAcMan5Glc
NAc2質量と一致する。Man5GlcNAc2へのGlcNAcの付加を確認するため
に、β−N−ヘキソサミニダーゼ消化を実施し、これは、Man5GlcNAc2の質量と
一致する1260(m/z)のピークを明らかにした(図17)。
【0108】
1つの株PBP3(実施例2)中のalg3 och1欠失由来のN−グリカンは、1
138(m/z)および1300(m/z)に、2つの異なるピークを提供した。これは
、構造(GlcNAcMan3GlcNAc2およびGlcNAcMan4GlcNAc2)
に一致する(図18)。多くのマンノースに対するインビトロα1,2−マンノシダーゼ
消化の後、ピークは、1138(m/z)に溶出された。これは、GlcNAcMan3
GlcNAc2と一致する(図19)。GlcNAcのMan3GlcNAc2構造に対す
る付加を確認するために、β−N−ヘキソサミニダーゼ消化を実施し、これは、934(
m/z)にピークを示し、Man3GlcNAc2の質量と一致した(図20)。
【0109】
GlcNAcのGlcNAcMan3GlcNAc2構造に対する第2の付加を、図21
に示す。1357(m/z)のピークは、GlcNAc2Man3GlcNAc2に対応す
る。コアマンノース構造(Man3GlcNAc2)への2つのGlcNAcの付加を確認
するために、別のβ−N−ヘキソサミニダーゼ消化を、実施し、これは、934(m/z
)にピークを示し、Man3GlcNAc2の質量と一致した(図22)。これは、酵母細
胞中で作られる複合型糖タンパク質を示す決定的なデータである。
【0110】
GlcNAc2Man3GlcNAc2に対するUDP−ガラクトースおよびβ1,4−
ガラクトシルトランスフェラーゼのインビトロ付加は、1664(m/z)にピークを生
じた。これは、Gal2GlcNAc2Man3GlcNAc2の質量と一致する(図23)
。最終的に、CMP−N−アセチルノイラミン酸およびシアリルトランスフェラーゼのイ
ンビトロ付加は、2248(m/z)にピークを生じた。これは、NANA2Gal2Gl
cNAc2Man3GlcNAc2の質量と一致する(図24)。上記のデータは、複雑な
ヒト様糖タンパク質を産生し得る非哺乳動物宿主細胞の使用を支持する。
【0111】
(グルコシルトランスフェラーゼおよびガラクトシルトランスフェラーゼの特定の細胞
小器官への標的化)
多くの研究が、これらの酵素が、これらのそれぞれの細胞小器官に保持され、そして固
定される正確な機構を示すために、提供されてきた。複雑ではあるが、証拠は、幹領域、
膜貫通領域、および細胞質テイルが、個々にまたは一斉に、酵素を個々の細胞小器官の膜
に指向し、それによって、結合する触媒領域が、その位置に局在化することを示唆する。
【0112】
活性なグリコシルトランスフェラーゼが、発現され得、そして適切な細胞小器官に指向
され得、その結果、連続した順番の反応が生じ得、複合N−グリカン形成を導く方法は、
以下の通りである:
(A)分泌経路中の特定の位置(ER、ゴルジおよびトランスゴルジネットワーク)への
局在化を媒介するタンパク質/ペプチドをコードすることが既知である領域のDNAライ
ブラリーを確立すること。これらの酵素の限られた選択物およびこれらのそれぞれの位置
を、表1に示す。これらの配列は、操作される宿主および他の関連する生物または関連し
ない生物から選択され得る。一般的に、このような配列は、以下の3つの範疇に分類され
る:
(1)細胞質テイル(ct)、膜貫通ドメイン(tmd)およびいくらかよりあいまい
に規定された幹領域(sr)の一部をコードするN−末端配列。これらは、一緒にかまた
は個々にタンパク質をゴルジの内(内腔)膜に固定する、
(2)一般的にC−末端で見出される回収シグナル(例えば、HDELまたはKDEL
テトラペプチド)、および
(3)膜貫通ヌクレオチド糖輸送体(これは、ゴルジに局在化することが公知である)
。
第1の場合において、局在化領域が種々の要素(ct、tmdおよびsr)からなる場合
、ライブラリーは、ct、tmdおよび幹領域の種々の部分が示されるように設計される
。これは、細胞質領域をコードするDNAの5’末端に結合するPCRプライマーを使用
すること、および幹領域の種々の部分に結合する対向する一連のプライマーを使用するこ
とによって達成され得る。さらに、糖ヌクレオチド輸送体および公知の回収シグナルをコ
ードする融合タンパク質構築物を作製する。
(B)第2工程は、一連の融合タンパク質構築物の作製を包含し、この構築物は、上記の
局在化配列およびこのような局在化配列にインフレームでクローン化された特定のグリコ
シルトランスフェラーゼの触媒ドメインをコードする(例えば、GnTI、GalT、フ
コシルトランスフェラーゼまたはST)。触媒ドメインに融合された糖ヌクレオチド輸送
体の場合において、このような構築物を設計し、その結果、触媒ドメイン(例えば、Gn
TI)が、得られたポリペプチドのN−末端またはC−末端のいずれかに存在し得る。局
在化配列と同様に、触媒ドメインは、種々の異なる供給源に由来し得る。このような触媒
ドメインの選択は、触媒ドメインが活性である特定の環境の知見によって左右される。例
えば、特定のグリコシルトランスフェラーゼが、後期ゴルジにおいて活性である場合、宿
主生物の後期ゴルジ中の全ての公知の酵素が、7.0の最適pHを有する場合、または後
期ゴルジが、特定のpHを有することが公知である場合、そのpHで最適の活性を有する
触媒ドメインを選択することを試行する。特定の宿主において、このような酵素の活性に
対する現存するインビボデータはまた、有用であり得る。例えば、Schwientek
および共同研究者らは、GalT活性をS.cerevisiaeのゴルジに操作し得る
ことを示し、そしてこのような活性が、S.cerevisiaeのalg変異型におい
て、いくつかのGalの存在するGlcNAc2への転位を示すことによって存在したこ
とを示した。さらに、数ラウンドの遺伝子シャッフリングまたは誤りがちのPCRを実施
し、融合構築物のプール内に大きな多様性を得る。なぜなら、単一アミノ酸変異は、糖タ
ンパク質保有酵素の活性を劇的に変更し得ることが示されたからである(Romeroら
、2000)。グリコシルトランスフェラーゼおよびこれらの内因性固定配列(anch
oring sequence)の全長配列がまた、使用され得る。好ましい実施形態に
おいて、このような局在化/触媒ドメインライブラリーは、より高等な真核生物における
グリコシル化反応の一連の性質に対する現存する情報を組込むように設計される。言い換
えると、糖タンパク質のプロセシングの過程の初期に起こることが公知である反応は、こ
のような反応を触媒する酵素の、ゴルジまたはERの初期部分への標的化を必要とする。
例えば、Man8GlcNAc2からMan5GlcNAc2へのトリミングは、複合N−グ
リカンの形成における初期段階である。タンパク質プロセシングが、ERにおいて開始し
、次いで、初期ゴルジ、中期ゴルジおよび後期ゴルジの間進行するので、ERまたは初期
ゴルジにおいて起こるこのトリミング反応を有することが、所望される。マンノシダーゼ
I局在化についてのライブラリーを設計する場合、マンノシダーゼIの触媒ドメインを有
するER標的化シグナルと初期ゴルジ標的化シグナルとを一致することを試行する。
【0113】
融合構築物ライブラリーを用いて宿主株を形質転換する際に、選択プロセスを使用して
、どの特定の局在化配列と触媒ドメインとの組み合わせが、実際に、そのような宿主株に
おいて見出される糖質構造に対して最大の効果を有するかを、同定する。そのような選択
は、多数のアッセイ方法または検出方法に基づき得る。それらの方法は、手動により行わ
れ得るか、または高スループットスクリーニング機器を使用することを介して自動化され
得る。
【0114】
別の例において、GnTI活性が、複合N−グリカンの成熟のために必要である。なぜ
なら、末端α1,3マンノース残基へのGlcNAcの付加後のみ、そのような構造から
続く中間体GlcNAcMan3GlcNAc2構造へのさらなる調整が生じ得るからであ
る。マンノシダーゼIIは、末端β1,2−GlcNAcの非存在下で末端α1,3−マ
ンノース残基およびα1,6−マンノース残基を多分除去することができず、従って、複
合N−グリカンの形成は、GnTI活性の非存在下では進行しない(Schachter
,1991)。あるいは、充分な量の本発明に記載されるMan5GlcNAc2を生じる
株を、Alg3P活性が欠損した株を操作または選択することにより、まず操作または選
択し得る。十分なUDP−GlcNAcトランスポーター活性の存在下で、このようなU
DP−GlcNAcトランスポーター活性を有する株を操作または選択することにより達
成し得るように、GlcNAcが、GnTIにより末端α−1,3残基に付加され得る。
なぜなら、インビトロでのMan3構造は、ラット肝臓GnTIにより認識されるからで
ある(Moller,1992)。
【0115】
別のアプローチにおいて、上記のライブラリー中にUDP−GlcNAcトランスポー
ターの発現を組込み得、望ましい構築物が、以下を含むようにする:(1)形質転換構築
物が細胞中に維持される領域(例えば、複製起点、または染色体組み込みを媒介する領域
)、(2)形質転換された細胞の選択を可能にするマーカー遺伝子(対比選択可能で再利
用可能なマーカー(例えば、ura3もしくはT−urf13(Soderholm,2
001)、または充分に特徴付けられた他の選択マーカー(例えば、his4、bla、
Sh bleなど)を含む)、(3)UDP−GlcNAcトランスポーターをコードす
る遺伝子(例えば、K.lactis由来(Abeijon,1996)またはH.sa
piens由来(Ishida,1996)、および(4)上記局在化ドメイン/触媒ド
メイン融合構築物ライブラリーの発現を活性化するプロモーター。
【0116】
上記の融合構築物のライブラリーを用いる宿主の形質転換の後、細胞表面上に最高濃度
の末端GlcNAcを有する細胞、または最高の末端GlcNAc含量を有するタンパク
質を分泌する細胞について、スクリーニングし得る。そのようなスクリーニングは、視覚
的方法(染色手順など)、特異的末端GlcNAc結合抗体に結合する能力、またはマー
カーに結合体化したレクチンに結合する能力(そのようなレクチンは、E.Y.Labo
ratories Inc.,San Mateo,CAから入手可能である)、特定の
レクチンが末端マンノース残基に結合する能力の減少、インビトロで放射性標識糖を組込
む能力、色素もしくは荷電表面への結合の変化に基づき得るか、または発蛍光団標識レク
チンもしくは抗体と組み合わせて蛍光補助細胞ソーティング(FACS)デバイスを使用
することにより達成され得る(Guillen,1998)。形質転換集団内の特定の表
現型を、細胞傷害性レクチンを用いて富化することは、有利であり得る。米国特許第5,
595,900号は、望ましい細胞外糖質構造を有する細胞が同定され得るいくつかの方
法を教示する。このストラテジーを繰返し実行することにより、下等真核生物においてま
すます複雑なグリカンを連続して操作することが可能である。
【0117】
形質転換後、インビトロアッセイにおいて、GlcNAcトランスフェラーゼIIによ
るUDP−GlcNAcからのGlcNAcの最も効率的な移動を可能にする形質転換体
について、選択し得る。このスクリーニングは、上記形質転換ライブラリーを保有する細
胞を、寒天プレート上にて選択圧下で増殖させること、そして個々のコロニーを96ウェ
ルマイクロタイタープレートへと移すことによって、実行され得る。細胞を増殖させた後
、その細胞を遠心分離し、その細胞を緩衝液中に再懸濁し、そしてUDP−GlcNAc
およびGnT Vを添加した後、UDPの放出を、HPLCか、またはUDPについての
酵素結合アッセイのいずれかによって、測定する。あるいは、放射性標識したUDP−G
lcNAcおよびGnT Vを使用し、その細胞を洗浄し、その後、N−アセチルグルコ
サミニダーゼによる放射性GlcNAcの放出を探索し得る。これはすべて、手動により
行われ得るか、または高スループットスクリーニング機器の使用を介して自動化され得る
。
【0118】
第1のアッセイにおいてより多くのUDPを放出するかまたは第2のアッセイにおいて
より多くの放射標識GlcNAcを放出する形質転換体は、その表面上により高い程度の
GlcNAcMan3GlcNAc2を有し(図3)、従って、望ましい表現型を構成する
と、予測される。あるいは、形質転換細胞の表面上の変化したグリコシル化パターンを明
らかにし得る、レクチン結合アッセイのような他の任意の適切なスクリーニングを使用し
得る。この場合、末端マンノースに特異的なレクチンの結合の減少が、適切な選択ツール
であり得る。Galantus nivalisレクチンは、末端α−1,3マンノース
に特異的に結合し、このことは、ゴルジ装置中に充分なマンノシダーゼII活性が存在す
る場合には、減少すると予期される。高末端マンノース含量を含む細胞の除去を可能にす
るクロマトグラフィー分離工程を実行することによってもまた、望ましい形質転換体を富
化し得る。この分離工程は、低末端マンノース含量を有する細胞よりも、高末端マンノー
ス含量を有する細胞に特異的に結合するレクチンカラム(例えば、アガロースに結合した
Galantus nivalisレクチン、Sigma,St.Louis,MO)を
用いて、実行される。さらに、このような融合タンパク質構築物を直接作製し得る。なぜ
なら、種々の下等真核生物宿主における活性糖質改変酵素の局在化に関するさらなる情報
が、科学文献にて利用可能であるからである。例えば、先行技術は、ヒトβ1,4−Ga
lTrが、MNT(S.cerevisiae由来のマンノシルトランスフェラーゼ)の
膜ドメインに融合され得、かつその触媒活性を保持しながらゴルジ装置へと局在化され得
ることを、本発明者らに教示する(Schwientekら、1995)。S.cere
visiaeまたは関連生物が、操作されるべき宿主である場合、そのような宿主由来の
複合N−グリカンを得るために、全体的ストラテジーにこのような知見を直接組込み得る
。S.cerevisiae中のグリコシルトランスフェラーゼに関連する、P.pas
toris中のいくつかのそのような遺伝子フラグメントが、同定されており、従って、
その目的のために使用され得る。
【0119】
(表1)
【0120】
【表1】
(組み込み部位)
この遺伝子操作の試みの1つの最終的な目的は、工業的発酵プロセスにおいて充分に実
施可能である頑丈なタンパク質産生株であるので、宿主(例えば、真菌)染色体中に複数
の遺伝子を組込むことは、注意深い計画を伴う。操作された株は、多分、一定範囲の異な
る遺伝子で形質転換される必要があり、これらの遺伝子は、望ましい活性が発酵プロセス
全体にわたり維持されるのを確保するために、安定な様式で形質転換される必要がある。
以下の酵素活性の任意の組み合わせが、タンパク質発現真菌宿主中に操作される必要があ
る:シアリルトランスフェラーゼ、マンノシダーゼ、フコシルトランスフェラーゼ、ガラ
クトシルトランスフェラーゼ、グルコシルトランスフェラーゼ、GlcNAcトランスフ
ェラーゼ、ERおよびゴルジ特異的トランスポーター(例えば、UPD−ガラクトースお
よび他の前駆体についての共輸送トランスポーターおよび対向輸送トランスポーター)、
オリゴ糖のプロセシングに関与する他の酵素、ならびに活性化オリゴ糖前駆体(例えば、
UDP−ガラクトース、CMP−N−アセチルノイラミン酸)の合成に関与する酵素。同
時に、非ヒトグリコシル化反応の特徴であることが公知である酵素をコードする多数の遺
伝子が、欠失されなければならない。そのような遺伝子および対応するタンパク質は、多
数の下等真核生物(例えば、S.cerevisiae、T.reesei、A.nid
ulansなど)において広範に特徴付けられており、それにより、下等真核生物におけ
る公知のグリコシルトランスフェラーゼ、その活性、およびその個々の遺伝子配列のリス
トが提供される。これらの遺伝子は、マンノシルトランスフェラーゼの群(例えば、1,
3−マンノシルトランスフェラーゼ(例えば、S.cerevisiae中のMNN1)
(Graham,1991)、1,2−マンノシルトランスフェラーゼ(例えば、S.c
erevisiae由来のKTR/KREファミリー)、1,6−マンノシルトランスフ
ェラーゼ(S.cerevisiae由来のOCH1)、マンノシルホスフェートトラン
スフェラーゼ(S.cerevisiae由来のMNN4およびMNN6)、および異所
性(すなわち、非ヒト)グリコシル化反応に関与するさらなる酵素)から選択される。こ
れらの遺伝子の多くは、実際に、欠失されており、変化したグリコシル化プロフィールを
有する生存可能な表現型を個別に生じる。例が、表2に示される。
【0121】
(表2)
【0122】
【表2】
下等真核生物中への複合N−グリカンの形成を操作するためのどのストラテジーも、グリ
コシルトランスフェラーゼ活性の排除および付加の両方を包含するので、包括的スキーム
は、両方の要件を協調することを試みる。望ましくない酵素をコードする遺伝子は、望ま
しい遺伝子について可能な組み込み部位として役立つ。例えば、1,6−マンノシルトラ
ンスフェラーゼ活性は、公知の多くの下等真核生物におけるグリコシル化の特徴である。
α−1,6−マンノシルトランスフェラーゼ(OCH1)をコードする遺伝子は、S.c
erevisiaeからクローン化されており、その遺伝子中の変異は、マンノシル化が
減少した生存可能な表現型を生じる。そのα−1,6−マンノシルトランスフェラーゼ活
性をコードする遺伝子座は、グリコシルトランスフェラーゼ活性をコードする遺伝子を組
込むための主要な標的である。同様な様式で、その座における遺伝子破壊事象に基づいて
、(1)よりヒトと類似する様式でグリコシル化する細胞能力を改善し、(2)タンパク
質を分泌する細胞能力を改善し、(3)外来タンパク質のタンパク質分解を減少し、そし
て(4)精製または発酵プロセス自体を促進するプロセスの他の特徴を改善することが予
測される、他の染色体組み込み部位の範囲を選択し得る。
【0123】
(糖ヌクレオチド前駆体の提供)
高等真核生物グリコシル化の特徴は、糖タンパク質におけるガラクトース、フコース、
および高い程度の末端シアル酸の存在である。これらの糖は、酵母および繊維状真菌にお
いて生成される糖タンパク質においては一般的に見出されず、上記の方法により、望まし
いオルガネラにおいてグリコシルトランスフェラーゼが局在化する株の操作が可能である
。複合N−グリカン合成の形成は、特定の糖残基が除去されそしてコアオリゴ糖構造に結
合される、連続的プロセスである。高等真核生物において、これは、基質を、種々の処理
酵素に連続的に曝露させることによって達成される。これらの酵素は、処理カスケード全
体における特定の位置に依存して、特定の反応を行う。この「アセンブリライン」は、E
R、初期ゴルジ、中期ゴルジ、および後期ゴルジ、ならびにトランスゴルジネットワーク
すべてと、その特定の処理環境からなる。下等真核生物のゴルジおよびERにおいてヒト
糖タンパク質の処理を再生するために、多数の酵素(例えば、グリコシルトランスフェラ
ーゼ、グリコシダーゼ、ホスファターゼ、およびトランスポーター)が、発現され、これ
らのオルガネラに特異的に標的付けられなければならず、好ましくは、それらがその環境
およびその経路中の他の酵素と関連して最も効率的に機能するような位置に、標的付けら
れなければならない。いくつかの個々のグリコシルトランスフェラーゼが、クローン化さ
れ、そしてS.cerevisiae(GalT、GnT I)、Aspergillu
s nidulans(GnT I)および他の真菌において発現されているが、その生
物のグリコシル化パターンにおける望ましい「ヒト化」の結果は示さない(Yoshid
a,1995;Schwientek,1995;Kalsner,1995)。このよ
うなグリコシルトランスフェラーゼの作用により糖を受容するのに必要な糖質構造は、充
分な量存在しないと、推測された。このことは、おそらく、複合N−グリカン形成の欠如
に寄与し、一方、Man5GlcNAc2構造を供給し、GnT I活性を有し、そして真
菌注に操作されたUDP−Gnトランスポーター活性を有する真菌についての報告は現在
存在しない。ハイブリッドな複合N−グリカン形成のために必要なのは、これらの3つの
生化学的事象の組み合わせである。
【0124】
ヒトにおいて、ヌクレオチド糖前駆体(例えば、UDP−N−アセチルグルコサミン、
UDP−N−アセチルガラクトサミン、CMP−N−アセチルノイラミン酸、UDP−ガ
ラクトースなど)の全範囲は、一般的に、サイトゾルにおいて合成され、そしてゴルジに
輸送され、このゴルジにおいて、これらは、グリコシルトランスフェラーゼによりコアオ
リゴ糖に結合される。下等真核生物においてこのプロセスを繰り返すために、糖ヌクレオ
シドに特異的な輸送体は、適切なレベルのヌクレオシド糖前駆体を確実にするために、ゴ
ルジにおいて発現されなければならない(Sommers,1981;Sommers,
1982;Perez,1987)。この反応の副生成物は、ヌクレオシドジホスフェー
トまたはヌクレオシドモノホスフェートのいずれかである。モノホスフェートが、交互輸
送機構によるヌクレオシドトリホスフェート糖の交換において直接的に輸送され得るが、
ジホスホヌクレオシド(例えば、GDP)は、輸送される前に、ヌクレオシドモノホスフ
ェートおよび無機ホスフェートを生じるために、ホスファターゼ(例えば、GDPase
)によって切断されなければならない。この反応は、S.cerevisiae由来のG
DPaseが、マンノシル化に必要であることが見出されているので、効率的なグリコシ
ル化に重要であるようである。しかし、酵素のみが、UDPに対して10%の活性を有す
る(Berninsone,1994)。下等真核生物は、しばしば、ゴルジにおいてU
DP特異的ジホスファターゼ活性を有さない。なぜなら、これらは、ゴルジにおける糖タ
ンパク質合成のためにUDP−糖前駆体を利用しないからである。
【0125】
Schizosaccharomyces pombe(細胞壁多糖類へガラクトース
残基を付加することが見出された酵母(UDP−ガラクトース由来))は、このような酵
素に対する要件をさらに示唆する特異的なUDPase活性を有することを見出した(B
eminsoneら、1994)。UDPは、グリコシルトランスフェラーゼの強力なイ
ンヒビターであることが公知であり、このグリコシル化副産物の除去は、ゴルジの内腔に
おけるグリコシルトランスフェラーゼ阻害を妨げるために重要である。(Khatara
ら,1974)。従って、UDPの除去を提供する必要があり得、これは、このような操
作された株のゴルジにおいて蓄積することが予期される(Berninsone,199
5;Beaudet,1998)。
【0126】
別の例において、2,3シアリルトランスフェラーゼおよび2,6シアリルトランスフ
ェラーゼは、ヒトのトランス−ゴルジおよびTGNにおいてシアル酸でガラクトース残基
をキャップし、糖タンパク質の成熟形態を導く。代謝的に操作された酵母または真菌に、
このプロセシング工程を再操作することは、(1)2,3−シアリルトランスフェラーゼ
活性および(2)酵母の後期(late)ゴルジにおけるCMP−N−アセチルノイラミ
ン酸の十分な供給を必要とする。後期ゴルジにおける十分な2,3−シアリルトランスフ
ェラーゼ活性を得るために、公知のシアリルトランスフェラーゼ(例えば、ヒト由来)の
触媒性ドメインは、真菌において後期ゴルジに指向されなければならない(上記を参照の
こと)。同様に、輸送体は、後期ゴルジへのCMP−N−アセチルノイラミン酸の輸送を
可能にするように操作されなければならない。現在、真菌が、十分な量のCMP−N−ア
セチルノイラミン酸を合成することを指摘するものはなく、このような糖−ヌクレオチド
のゴルジへの輸送を言及しない。結果として、対応するグリコシルトランスフェラーゼに
対する基質の適切な供給を確実にするために、真菌に対してCMP−シアル酸の産生を代
謝的に操作しなければならない。
【0127】
(ゴルジ装置に糖ヌクレオチド前駆体を提供するための方法)
(UDP−N−アセチル−グルコサミン)
ヒトUDP−N−アセチルグルコサミン輸送体のcDNA(これは、発現配列タグデー
タベース(dbEST)における相同性検索によって認識された)は、Ishidaおよ
び共同研究者(Ishida,1999)によってクローニングされた。Guillen
および共同研究者は、上記ヌクレオチド糖のゴルジ輸送体において欠損した、最近特徴付
けられたKluyveromyces lactis変異体のイヌ腎細胞(MDCK)由
来のcDNAとの表現型相関によって、UDP−N−アセチルグルコサミンに対する哺乳
動物ゴルジ膜輸送体をクローニングした(Guillen、1998)。それらの結果は
、哺乳動物ゴルジ UDP−N−GlcNAc輸送体遺伝子が、このタンパク質が、発現
しそして酵母のゴルジ装置に機能的に標的化されるのに必要な情報の全てを有すること、
および非常に異なるアミノ酸配列を有する2つのタンパク質が、同じゴルジ膜内に同じ溶
質を輸送し得ることを実証する。((Guillen、1998)。
【0128】
(GDP−フコース)
ラット肝臓ゴルジ膜GDP−フコース輸送体が、Puglielli,L.およびC.
B.Hirschberg(Puglielli,1999)によって同定および精製さ
れた。対応する遺伝子は、同定されていないが、N−末端配列決定を、対応する遺伝子に
特異的なオリゴヌクレオチドプローブの設計のために使用し得る。これらのオリゴヌクレ
オチドをプローブとして使用して、GDP−フコース輸送体をコードする遺伝子をクロー
ニングし得る。
【0129】
(UDP−ガラクトース)
2つの異種遺伝子(Schizosaccharomyces pombe由来のα1
,2−ガラクトシルトランスフェラーゼ(α1,2 GalT)をコードするgmal2
(+)およびヒトUDP−ガラクトース(UDP−Gal)輸送体をコードする(hUG
T2))は、ガラクトシル化に必要とされる細胞内条件を調べるために、S. cere
visiaeにおいて機能的に発現された。タンパク質ガラクトシル化とUDP−ガラク
トース輸送体活性との間の相関は、α1,2−GalTよりも、UDP−Gal輸送体の
外因的な供給が、S.cerevisiaeにおける効率的なガラクトシル化に重要な役
割を果たすことを示した(Kainuma,1999)。同様に、S.pombe由来の
UDP−ガラクトース輸送体は、クローニングされた(Aoki,1999;Segaw
a,1999)。
【0130】
(CMP−N−アセチルノイラミン酸(CMP−シアル酸))
ヒトCMP−シアル酸輸送体(hCST)をクローニングし、そしてLec 8 CH
O細胞において発現させた(Aoki,1999;Eckhardt,1997)。マウ
スCMP−シアル酸輸送体の機能的発現は、Saccharomyces cerevi
siaeにおいて達成された(Berninsone,1997)。シアル酸は、いくら
かの真菌において見出されたが、選択された宿主系が、十分なレベルのCMP−シアル酸
を供給し得るか否かは、明確でない。シアル酸は、培地において供給され得るか、あるい
は、シアル酸合成に関係する真菌経路がまた宿主ゲノム内に組み込まれ得るかのいずれか
である。
【0131】
(ジホスファターゼ)
糖が糖タンパク質上に輸送される場合、ヌクレオシドジホスフェートまたはヌクレオシ
ドモノホスフェートのいずれかが、糖ヌクレオチド前駆体から放出される。モノホスフェ
ートが交互輸送機構によるヌクレオシドトリホスフェート糖の交換において直接的に輸送
され得るが、ジホスホノモノヌクレオシド(例えば、GDP)は、輸送される前に、ヌク
レオシドモノホスフェートおよび無機ホスフェートを生じるために、ホスファターゼ(例
えば、GDPase)によって切断されなければならない。この反応は、S.cerev
isiae由来のGDPaseが、マンノシル化に必要であることが見出されているので
、効率的なグリコシル化に重要であるようである。しかし、酵素のみが、UDPに対して
10%の活性を有する(Berninsone,1994)。下等真核生物は、しばしば
、ゴルジにおいてUDP特異的ジホスファターゼ活性を有さない。なぜなら、これらは、
ゴルジにおける糖タンパク質合成のためにUDP−糖前駆体を利用しないからである。S
chizosaccharomyces pombe(細胞壁多糖類へガラクトース残基
を付加することが見出された酵母(UDP−ガラクトース由来))は、このような酵素に
対する要件をさらに示唆する特異的なUDPase活性を有することを見出した(Bem
insoneら、1994)。UDPは、グリコシルトランスフェラーゼの強力なインヒ
ビターであることが公知であり、このグリコシル化副産物の除去は、ゴルジの内腔におけ
るグリコシルトランスフェラーゼ阻害を妨げるために重要である。(Khataraら,
1974)。
【0132】
(複合体N−グリカンを産生するためのGnTの発現)
(抗体機能をブーストするためのGnT−IIIの発現)
N−アセチルグルコサミニルトランスフェラーゼIIおよびIIIによるGlcNAc
1Man3GlcNAc2構造へのN−アセチルグルコサミンの付加は、いわゆる分岐した
N−グリカンGlcNAc3Man3GlcNAc2を生じる(図3)。この構造は、より
大きな抗体依存性の細胞性の細胞傷害(ADCC)に関係している(Umanaら、19
99)。哺乳動物細胞によって発現される免疫グロブリンの糖形態を再操作することは、
冗漫で厄介な作業である。特に、GnT III(この酵素の過剰発現は、増殖阻害に関
係している)の場合、調節された(誘導性の)遺伝子の発現を含む方法が、分岐したN−
グリカンを有する免疫グロブリンを作製するために使用されなければならい(Umana
ら、1999a、1999b)。
【0133】
従って、別の実施形態において、本発明は、コアマンノース構造上に、分岐したN−ア
セチルグルコサミン(GlcNAc)を有するヒト様N−グリカンを作製するためのシス
テムおよび方法を提供する。好ましい実施形態において、本発明は、分岐したN−グリカ
ンを有する免疫グロブリンを産生するためのシステムおよび方法を提供する。本明細書中
に記載されるシステムおよび方法は、先の問題(例えば、GnT IIIまたはADCC
の過剰発現に関連する細胞傷害性)を被らない。なぜなら、本発明の宿主細胞は、実質的
に改変されたヒト型糖形態(例えば、GlcNAc2Man3GlcNAc2)を有するN
−グリカンを産生する、生存可能であり好ましくは、頑強な細胞であるように操作および
選択されるからである。従って、本発明の宿主細胞における分岐したN−アセチルグルコ
サミンの付加は、増殖−表現型またはそれらの宿主細胞の生存能力に対して、無視し得る
効果を有する。
【0134】
さらに、以前の研究(Umana)は、GnT III発現レベルとADCCの程度と
の間に直線的な関係がないことを示した。哺乳動物細胞における最適な発現レベルを見つ
け出すこと、およびFDAが許可した発酵プロセス全体にわたってそれを維持することは
、挑戦であるようである。しかし、本発明の細胞(例えば、真菌細胞)において、頑強で
、信頼性があり、かつ最適なGnT III発現レベルを確立するための適切な強度のプ
ロモーターを発見することは、当業者には比較的簡単な作業である。
【0135】
分岐したN−グリカンを用いて糖タンパク質を産生し得る酵母菌株のような宿主細胞は
、宿主細胞内にGnT III活性を導入することによって、本発明に従って操作される
(実施例6)。好ましくは、宿主細胞は、GnT III(例えば、図32を参照のこと
)をコードする核酸または酵素活性を有するそのドメイン((例えば、本発明のライブラ
リーおよび関連する方法を使用して)必要に応じて、異種細胞シグナル標的化ペプチドに
融合される)を用いて形質転換される。GnT IIIを発現するように操作された宿主
細胞は、哺乳動物細胞が可能であるよりも高い抗体力価を生じる。これらはまた、ADC
Cに関してより高い効力を有する抗体を産生する。
【0136】
GnT IIIをトランスフェクトされた哺乳動物細胞株によって産生された抗体は、
非トランスフェクト細胞株によって産生された抗体と同様に効果的であるが、10〜20
倍低い濃度であることが示された(Daviesら、2001)。本発明の産生ビヒクル
の生産性の、哺乳動物系に対する20倍の増加、および10倍の効力の増加は、200倍
の正味の生産性の改善を生じる。従って、本発明は、高い効力(例えば、現在生産され得
るものよりも数桁より強力)を有する高い力価の抗体を作製するための系および方法を提
供する。この系および方法は、安全であり、高い効力の抗体を短い時間で低コストで提供
する。本発明に従って、GnT IIIを発現するように操作された宿主細胞は、少なく
とも50mg/リットル/日〜少なくとも500mg/リットル/日の速度で分岐した(
bisected) N−グリカンを有する免疫グロブリンを産生する。さらに、各免疫
グロブリン(Ig)分子(分岐したGlcNAcを含む)は、分岐したGlcNAcを含
まないで産生された同じIg分子よりも強力である。
【0137】
(GnT−IVおよびGnT−Vのクローニングおよび発現)
複合体N−グリカンの全ての分枝構造を、N−アセチルグルコサミントランスフェラー
ゼ(GnT)−I〜−VIにより、共通のコア五糖(Man3GlcNAc2またはMan
α−6(Man α1−3)Man β1−4 GlcNAc β1−4 GlcNAc
β1−4またはMan3GlcNAc2)上に合成した(Schachter Hら(1
989)Methods Enzyme;179:351−97)。より高度に分枝した
N−グリカンの生合成ついての現在の理解は、GnT IIの作用(GlcNAc2Ma
n3GlcNAc2構造の作製)の後に、GnTIVが、GlcNAcを、β1,4−連結
のUDP−GlcNAcからGlcNAc2Man3GlcNAc2 N−グリカンのMa
n α1,3 Man β1,4アームに移動させ(Allen SDら(1984)J
Biol C11em.Jun 10;259(11):6984−90;ならびにG
leeson PAおよびSchachter H.J(1983);J.Biol C
hem 25;258 (10):6162−73)、トリアンテナリアガラクト(tr
iantennary agalacto)糖鎖を生じることを示唆する。このN−グリ
カン(GlcNAc β1−2 Man α1−6(GlcNAc β1−2 Man
α1−3)Man β1−4 GlcNAc β1−4 GlcNAc β1,4 As
n)は、GnT−IIIおよびGnT−Vに対する共通の基質であり、分岐したトリアテ
ナリー構造およびテトラアテナリー構造の合成を導く。GnT IIIの作用が、分岐し
たN−グリカンを生じる場合、およびGnTVが、β1−6GlcNAcのα1−6マン
ノシルコアへの付加を触媒する場合、β1−6分枝を生じる。ガラクトースおよびシアル
酸のこれらの分枝への付加は、完全にシアル化された複合N−グリカンの生成を導く。
【0138】
分枝した複合型N−グリカンは、治療タンパク質の生理学的活性に関連している(例え
ば、ヒトエリスロポエチン(hEPO))。バイアンテナリー構造を有するヒトEPOは
、低い活性を有することが示されているのに対し、テトラアンテナリー構造を有するhE
POは、血流からのよりゆっくりとしたクリアランスを生じ、従って、より高い活性を生
じた(Misaizu Tら(1995)Blood Dec 1;86(11):40
97−104)。
【0139】
DNA配列情報を用いて、当業者は、GnT IVおよび/またはGnT V活性をコ
ードするDNA分子をクローニングし得る(実施例6;図33および34)。当業者に周
知の標準的な技術を使用して、GnT IVまたはGnT Vをコードする(または触媒
的に活性なそのフラグメントをコードする)核酸分子は、本発明の選択された宿主細胞(
例えば、本明細書中に記載のPichia sp.、Kluyveromyces sp
.およびAspergillus sp.のような真菌宿主)において転写を駆動し得る
プロモーターおよび他の発現制御配列の転写制御下にある適切な発現ベクターに挿入され
得、その結果、これらの哺乳動物GnT酵素のうちの1つ以上が、ヒト様複合型糖タンパ
ク質の生成のために選択された宿主細胞において活性に発現され得る。
【0140】
以下は、本発明の組成物および方法を例示する実施例である。これらの実施例は、限定
として解釈されるべきではなく、この実施例は、単に例示目的で含まれる。
【実施例】
【0141】
(実施例1:P.pastorisおよびK.lactisにおけるALG3遺伝子の
同定、クローニングおよび欠失)
縮重プライマーを、S.cerevisiae、H.sapiens、およびD.me
lanogaster由来のAlg3タンパク質配列のアラインメントに基づいて作製し
、P.pastorisゲノムDNA由来の83bp産物を増幅するために使用した:
5’−GGTGTTTTGTTTTCTAGATCTTTGCAYTAYCARTT−3
’および
5’−AGAATTTGGTGGGTAAGAATTCCARCACCAYTCRTG−
3’。得られたPCR産物を、pCR2.1ベクター(Invitrogen,Carl
sbad,CA)にクローニングしたところ、配列分析により、公知のALG3/RHK
1/NOT56ホモログ(Genbank NC_001134.2、AF309689
、NC_003424.1)に対する相同性が示された。その後、始めのPCR産物の1
929bp上流および2738bp下流を、以下の内部オリゴヌクレオチド:
【0142】
【化2】
を使用して、P.pastorisゲノムDNAライブラリーから増幅した(Boehm
,T.Yeast 1999 May;15(7):563−72)。得られたフラグメ
ントを、pCR2.1−TOPOベクター(Invitrogen)にクローニングし、
配列決定した。この配列から、S.cerevisiae ALG3遺伝子に対して35
%同一性および53%類似性を有するタンパク質をコードする1395bp ORFを、
(BLASTプログラムを使用して)同定した。この遺伝子を、PpALG3と名付けた
。
【0143】
PpALG3の配列を使用して、プライマーのセットを作製し、PpALG3遺伝子の欠失構築物を、PCR重複(Davidsonら,2002 Microbiol.148(Pt 8):2607−15)により生成した。以下のプライマーを使用して、PpALG3 ORFおよびKANR遺伝子の5’側および3’側の1kb領域を、それぞれ増幅した:
【0144】
【化3】
引き続いて、プライマーRCD142およびRCD147を使用して、3つの得られた
PCR産物を、単一の3.6kb alg3::KANRの欠失対立遺伝子に重ねた。
【0145】
(K.lactisにおけるALG3遺伝子の同定、クローニングおよび欠失)
S.cerevisiae、Drosophila melanogaster、Ho
mo sapiensなどに由来するALG3p配列を、K.lactis配列(PEN
DANT ESTデータベース)と整列させた。共通ホモログ中に存在するが、それらの
ホモログにおいて正確な配列は異なる、高い相同性の領域を使用して、株MG1/2(B
ianchiら,1987)に由来するゲノムDNAに対して指向する縮重プライマーの
対を作製した。ALG3の場合において、プライマー:
【0146】
【化4】
を用いるPCR増幅は、クローニングされ、配列決定された産物を生じ、推定翻訳産物は
、Alg3pタンパク質(S.cerevisiae Alg3pに対して>50%)に
対して高い程度の相同性を有することが示された。
【0147】
そのPCR産物を使用して、K.lactis株(MG1/2)由来のゲノムDNAの
サザンブロットを、高いストリンジェンシー下でプローブした(Sambrookら,1
989)。ハイブリダイゼーションは、単一の遺伝子と一致したパターンで観察された。
このサザンブロットを使用して、ゲノム遺伝子座をマッピングした。ゲノムフラグメント
を、ゲノムDNAを消化し、適切なサイズ範囲にあるこれらのフラグメントをpUCl9
に連結することによってクローニングして、K.lactisサブゲノムライブラリーを
作製した。このサブゲノムライブラリーを、E.coliに形質転換し、数百のクローン
を、プライマーKAL−1およびKAL−2を使用するコロニーPCRによって試験した
。推定KlALG3遺伝子および推定KlALG61遺伝子を含むクローンを、配列決定
し、オープンリーディングフレームを同定した。
【0148】
PCR重複法(Davidsonら,2002)を使用する、alg3::NATR欠
失対立遺伝子の構築物のためのプライマーを設計し、得られた欠失対立遺伝子を、2つの
K.lactis株に形質転換し、NAT耐性コロニーを選択した。これらのコロニーを
、PCRによってスクリーニングし、ALG3 ORFがoch1::NATR変異対立
遺伝子で置換された形質転換体を得た。
【0149】
(実施例2:ヒト様糖タンパク質の生成のための、α−1,2−マンノシダーゼ、Gn
TIおよびGnTIIを発現するalg3/ochl変異株の生成)
P.pastoris OCH1遺伝子の1215bpオープンリーディングフレーム
、ならびに2685bp上流および1175bp下流を、PCRによって増幅し(B.K
.Choiら(Proc.Natl.Acad.Sci.USA 2002に投稿);W
O02/00879もまた参照のこと;これらの各々は、本明細書中に参考として援用さ
れる)、pCR2.1−TOPOベクター(Invitrogen)にクローニングし、
pBK9と名付けた。複数の栄養要求マーカーを含むoch1ノックアウト株を作製する
ために、100μgのpJN329(SfiI制限部位と隣接したoch1::URA3
変異対立遺伝子を含むプラスミド)を、SfiIで消化し、これを使用して、P.pas
toris株JC308(Cereghinoら、Gene 263(2001)159
−169)をエレクトロポレーションにより形質転換した。ウラシルを欠く規定の培地上
で10日間室温でインキュベートした後、1000個のコロニーを拾い上げ、再び画線し
た。37℃で増殖できないが、室温では増殖するURA+クローンを、コロニーPCRに
供して、och1::URA3変異対立遺伝子の正確な組み込みについて試験した。予測
されるPCRパターンを示した1つのクローンを、YJN153と名付けた。ヒトプラス
ミノゲンのKringle 3ドメイン(K3)を、モデルタンパク質として使用した。
K3遺伝子を含むNeoRマーカーを挿入した(NeoR marked)プラスミドを、
YJN153株に形質転換し、K3を発現する得られた株を、BK64−1と名付けた(
B.K.Choiら,Proc.Natl.Acad.Sci.USA 2002に投稿
)。
【0150】
ゴルジUDP−N−アセチルグルコサミン輸送体をコードするプラスミドpPB103
(Kluyveromyces lactis MNN2−2遺伝子を含む)を、ベクタ
ーpDL02(Abeijonら(1996)Proc.Natl.Acad.Sci.
U.S.A.93:5963−5968)由来の平滑BglII−HidIIIフラグメ
ントを、P.pastoris ADE1遺伝子を含む、BglIIおよびBamHI消
化した平滑末端のpBLADE−SX(Cereghinoら(2001)Gene 2
63:159−169)にクローニングすることによって構築した。このプラスミドをE
coNIで線状にし、エレクトロポレーションによりBK64−1株に形質転換し、PC
R分析によりMNN2−2を含むことが確認された1つの株を、PBP1と名付けた。
【0151】
ヒト、マウス、ハエ、線虫(worm)および酵母供給源由来のいくつかのα−1,2−マンノシダーゼ遺伝子の触媒ドメインと融合した、S.cerevisiaeおよびP.pastoris由来のいくつかのI型またはII型膜タンパク質のリーダードメインのインフレーム融合物を含むマンノシダーゼ構築物のライブラリーを、作製した(例えば、WO02/00879(本明細書中に参考として援用される)を参照のこと)。このライブラリーを、P.pastoris HIS4組み込みベクター中に作製し、SalIで線状にし、PBP1株にエレクトロポレーションにより形質転換し、K3レポータータンパク質から放出されたグリカンを分析することにより、スクリーニングした。選択した1つの活性な構築物は、マウスα−1,2−マンノシダーゼIA(MmMannIA)遺伝子のN末端欠失物(これは、187個のヌクレオチドを欠いている)と融合した、酵母SEC12遺伝子の988〜1296のヌクレオチド(C末端)のキメラであった。この構築物を発現するP.pastoris株を、PBP2と名付けた。
【0152】
ヒト、線虫、カエルおよびハエ供給源由来のGnTI遺伝子の触媒ドメインを有する同
じリーダーライブラリーのインフレーム融合物を含むGnTI構築物のライブラリーを作
製した(WO02/00879)。このライブラリーを、P.pastoris ARG
4組み込みベクター中に作製し、AatIIで線状にし、PBP2株へのエレクトロポレ
ーションにより形質転換し、K3から放出されたグリカンを分析することによって、スク
リーニングした。選択した1つの活性な構築物は、ヒトGnTI遺伝子の欠失物(これは
、最初の154bpを欠いている)に融合したS.cerevisiae MNN9遺伝
子の最初の120bpのキメラであった。この構築物を発現するP.pastoris株
を、PBP3と名付けた。
【0153】
引き続いて、P.pastoris alg3::KANR欠失構築物を、上記のよう
に作製した。得られたPCR産物の約5μgを、PBP3株に形質転換し、コロニーを、
200μg/ml G418を含むYPD培地上で選択した。PCRによってスクリーニ
ングした20株のうちの1株が、alg3::KANR変異対立遺伝子の正確な組み込み
を含み、野生型対立遺伝子を欠くことを確認した。この株を、RDP27と名付けた。
【0154】
最後に、ヒトおよびラット供給源由来のGnTII遺伝子の触媒ドメインを有するリー
ダーライブラリーのインフレーム融合物から構成されるGnTII構築物のライブラリー
を作製した(WO02/00879)。このライブラリーを、薬物ノールセオスリシン(
nourseothricin)に対する耐性を付与するNSTR遺伝子を含むP.pa
storis組み込みベクター中に作製した。このライブラリープラスミドを、EcoR
Iで線状にし、RDP27株にエレクトロポレーションにより形質転換し、得られた株を
、精製K3から放出されたグリカンの分析によりスクリーニングした。
【0155】
(材料)
MOPS(カコジル酸ナトリウム)、塩化マンガン、UDP−ガラクトースおよびCM
P−N−アセチルノイラミン酸は、シグマ製であった。TFAは、Aldrich製であ
った。Spodoptera frugiperda由来の組換えラットα2,6−シア
リルトランスフェラーゼおよびウシ乳汁由来のβ1,4−ガラクトシルトランスフェラー
ゼは、Calbiochem製であった。タンパク質N−グリコシダーゼF、マンノシダ
ーゼ、およびオリゴ糖は、Glyko(San Rafael,CA)製であった。DE
AE ToyoPearl樹脂は、TosoHaas製であった。金属キレート「His
Bind」樹脂は、Novagen(Madison,WI)製であった。96ウェル溶
解液除去プレートは、Promega(Madison,WI)製であった。タンパク質
結合96ウェルプレートは、Millipore(Bedford,MA)製であった。
塩および緩衝剤は、Sigma(St.Louis,MO)製であった。MALDIマト
リクスは、Aldrich(Milwaukee,WI)製であった。
【0156】
(タンパク質精製)
Kringle3を、Beckman BioMek 2000サンプル処理ロボット
(Beckman/Coulter Ranch Cucamonga,CA)で、96
ウェル形式を使用して精製した。Kringle3を、C末端ヘキサヒスチジンタグを使
用して、発現培地から精製した。このロボット精製は、そのHisBind樹脂について
Novagenによって提供されたプロトコルの適応である。簡潔には、150uL(μ
L)の固定容積の樹脂を、96ウェルの溶解物結合プレートのウェル中に注ぎ、3容積の
水で洗浄し、そして5容積の50mM NiSO4を充填し、そして3容積の結合緩衝液
(5mMイミダゾール、0.5M NaCl、20mM Tris−HCL(pH7.9
))で洗浄する。タンパク質発現培地を、培地/PBS(60mM PO4、16mM
KC1、822mM NaCl(pH7.4))で3:2希釈し、そしてカラムに充填し
た。吸引後、カラムを10容積の結合緩衝液および6容積の洗浄緩衝液(30mMイミダ
ゾール、0.5M NaCl、20mM Tris−HCl(pH7.9))で洗浄し、
そしてタンパク質を、6容積の溶出緩衝液(1Mイミダゾール、0.5M NaCl、2
0mM Tris−HCl(pH7.9))で溶出する。溶出された糖タンパク質を、凍
結乾燥によって、乾燥するまでエバポレートする。
【0157】
(N連結グリカンの放出)
グリカンを、以前に報告された方法(Papacら、A.J.S.(1998)Glycobiology 8,445−454)の改変によって、糖タンパク質から放出および分離する。96ウェルのMultiScreen IP(Immobilon−Pメンブレン)プレート(Millipore)のウェルを、100uLのメタノールで湿らせ、3×150uLの水および50uLのRCM緩衝液(8M尿素、360mM Tris、3.2mM EDTA(pH8.6))で洗浄し、各添加後に、穏やかな減圧で吸引する。乾燥したタンパク質サンプルを、30uLのRCM緩衝液中に溶解し、そして10uLのRCM緩衝液を含むウェルに移す。ウェルを吸引し、そしてRCM緩衝液で2回洗浄する。タンパク質を、60uLの、RCM緩衝液中の0.1M DTTを、37℃で1時間に亘って添加して、還元した。ウェルを、300uLの水で3回洗浄し、そして60uLの0.1Mヨード酢酸を、暗中での室温にて30分間で添加して、カルボキシメチル化する。ウェルを、水で再度3回洗浄し、そしてメンブレンを、100uLの水中1%PVP 360の、室温で1時間の添加によって、ブロッキングする。ウェルを吸引し、そして300uLの水で3回洗浄し、そして30uLの10mM NH4HCO3(pH8.3)(1ミリ単位のN−グリカナーゼ(glycanase)(Glyko)を含む)の添加によって、脱グリコシル化する。37℃16時間の後、グリカンを含む溶液を、遠心分離によって取り出し、そして乾燥するまで蒸発させる。
【0158】
(飛行質量分析の、マトリクス補助レーザー脱離イオン化時間)
グリカンの分子量を、遅延抽出を使用して、Voyager DE PRO線形MAL
DI−TOF(Applied Biosciences)質量分析機を用いて決定した
。各ウェル由来の乾燥グリカンを、15uLの水中に溶解し、そして0.5uLを、ステ
ンレス鋼サンプルプレート上にスポットし、そして0.5uLのS−DHBマトリクス(
9mg/mLのジヒドロキシ安息香酸、1mg/mLの5−メトキシサリチル酸(1:1
の水/アセトニトリル 0.1% TFA中))と混合し、そして乾燥させた。
【0159】
パルス窒素レーザー(337nm)を、4nsのパルス時間で照射することによって、
イオンを発生させた。この機器を、125nsの遅延を用いる遅延抽出モードおよび20
kVの加速電圧で操作した。格子電圧は、93.00%であり、ガイドワイヤ電圧は、0
.10%であり、内部圧力は、5×10-7トル未満であり、そして低質量ゲートは、87
5Daであった。スペクトルを、合計100〜200のレーザーパルスから発生させ、そ
して2GHzのディジタイザーを用いて獲得した。Man5オリゴ糖を、外部分子量標準
として使用した。全てのスペクトルを、陽イオンモードでこの機器を用いて発生させた。
このスペクトルの概算の質量精度は、0.5%であった。
【0160】
(材料)
MOPS、カコジル酸ナトリウム、塩化マグネシウム、UDP−ガラクトースおよびC
MP−N−アセチルノイラミン酸は、Sigma,Saint Louis,MO製であ
った。トリフルオロ酢酸(TFA)は、Sigma/Aldrich,Saint Lo
uis,MO製であった。Spodoptera frugiperda由来の組換えラ
ットα−2,6−シアリルトランスフェラーゼおよびウシ乳汁由来のβ−1,4−ガラク
トシルトランスフェラーゼは、Calbiochem,San Diego,CA製であ
った。
【0161】
(β−N−アセチルヘキソサミニナーゼ設計)
グリカンを、以前に報告された方法(Papacら、A.J.S.(1998)Gly
cobiology 8,445−454)の改変によって、糖タンパク質から放出およ
び分離した。タンパク質を還元およびカルボキシメチル化し、そしてメンブレンをブロッ
キングした後、ウェルを、水で3回洗浄した。タンパク質を、30μlの10mM NH
4HCO3(pH8.3)(1ミリ単位のN−グリカナーゼ(glycanase)(Gl
yko,Novato,CA)を含む)の添加によって、脱グリコシル化した。37℃1
6時間の後、グリカンを含む溶液を、遠心分離によって取り出し、そして乾燥するまでエ
バポレートした。次いで、グリカンを、SC210Aスピード減圧(Thermo Sa
vant,Halbrook,NY)中で乾燥させた。乾燥したグリカンを、50mM
NH4Ac(pH5.0)中に37℃で一晩置き、そして1mUのヘキソース(hexo
s)(Glyko,Novato,CA)を添加した。
【0162】
(ガラクトシルトランスフェラーゼ反応)
約2mgのタンパク質(r−K3:hPg[PBP6−5])を、ニッケル親和性クロ
マトグラフィーによって精製し、0.1% TFAに対して広範に透析し、そして乾燥す
るまで凍結乾燥した。このタンパク質を、150μlの50mM MOPS、20mM
MnC12(pH7.4)中に再溶解した。32.5μg(533nmol)のUDP−
ガラクトースおよび4mUのβ 1,4−ガラクトシルトランスフェラーゼの添加後、サ
ンプルを、37℃で18時間インキュベートした。次いで、これらのサンプルを、MAL
DITOF質量分析による分析のために、0.1% TFAに対して透析した。
【0163】
ガラクトシルトランスフェラーゼと反応したタンパク質のスペクトルは、酵素なしでイ
ンキュベートした類似のタンパク質サンプルのスペクトルと比較して、2つのガラクトー
ス部分の付加と一致する、質量の増加を示した。次に、タンパク質サンプルを還元し、カ
ルボキシメチル化し、そしてPNGase Fで脱グリコシル化した。回収したN−グリ
カンを、MALDI−TOF質量分析によって分析した。タンパク質と反応したガラクト
シルトランスフェラーゼ由来の優勢なグリカンの質量は、2つのガラクトース部分の付加
と一致する質量分(325.4Da)だけ、コントロールグリカンの質量より多かった。
【0164】
(シアリルトランスフェラーゼ反応)
(ガラクトシルトランスフェラーゼ反応した)タンパク質を、10μlの50mM カ
コジル酸ナトリウム緩衝液(pH6.0)中に再懸濁した後、15μLの同じ緩衝液中に
溶解された300μg(488nmol)のCMP−N−アセチルノイラミン酸(CMP
−NANA)および5μl(2mU)の組換えα−2,6 シアリルトランスフェラーゼ
を添加した。37℃で15時間のインキュベート後、さらに200μgのCMP−NAN
Aおよび1mUのシアリルトランスフェラーゼを添加した。これらのタンパク質サンプル
を、さらに8時間インキュベートし、次いで、透析し、上記のようにMALDI−TOF
−MSによって分析した。
【0165】
シアリルトランスフェラーゼと反応した糖タンパク質のスペクトルは、出発物質(ガラ
クトシルトランスフェラーゼ反応後のタンパク質)の質量と比較して、質量の増加を示し
た。N−グリカンを、上記のように放出および分析した。優勢なグリカンの2つのイオン
付加物の質量の増加は、2つのシアル酸残基の付加と一致した(580Daおよび583
Da)。
【0166】
(実施例3)
(P.pastorisにおけるALG9遺伝子およびALG12遺伝子の同定、クロ
ーニングおよび欠失)
実施例1と同様に、ALG9p配列およびALG12配列(それぞれ、S.cerev
isiae、Drosophila melanogaster、Homo sapie
nsなどに由来する)を整列し、そして高い相同性の領域を、縮重プライマーを設計する
ために使用する。これらのプライマーを、P.pastoris由来のゲノムDNAに対
するPCR反応において使用する。得られた最初のPCR産物をサブクローニングし、配
列決定し、そして高いストリンジェンシーで、P.pastoris由来のゲノムDNA
のサザンブロットをプローブするために使用した(Sambrookら、1989)。ハ
イブリダイゼーションを観察する。このサザンブロットを使用して、遺伝子座位のマッピ
ングを行った。ゲノムフラグメントを、ゲノムDNAを消化し、そして適切なサイズ範囲
のフラグメントをpUCl9に連結して、P.pastorisサブゲノムライブラリー
を作製することによって、クローニングした。このサブゲノムライブラリーを、E.co
li中に形質転換し、そして数百個のクローンを、最初のPCR産物の配列に基づいて設
計されたプライマーを使用して、コロニーPCRによって試験する。予測された遺伝子を
含むクローンを配列決定し、そしてオープンリーディングフレームを同定する。alg9
::NAIR欠失対立遺伝子の構築のためのプライマーを、PCR重複法(Davids
onら、2002)を使用して設計する。得られた欠失対立遺伝子を、2つのP.pas
toris株に形質転換し、そしてNAT耐性コロニーを選択する。これらのコロニーを
、PCRによってスクリーニングし、そしてALG9 ORFがochl::NATR変
異体対立遺伝子で置換された形質転換体を得る。一般に、以下を参照のこと:Cipol
loら、Glycobiology 2002(12)11:749−762;Chan
tretら、J.Biol.Chem.Jul.12,2002(277)28:258
15−25822;Cipolloら、J.Biol.Chem.Feb.11,200
0(275)6:4267−4277;Burdaら、Proc.Natl.Acad.
Sci.U.S.A.July 1996(93):7160−7165;Karaog
luら、Biochemistyy 2001,40,12193−12206;Gri
mmeら、J.Biol.Chem.July 20,2001(276)29:277
31−27739;Verostekら、J.Biol.Chem.June 5,19
93(268)16:12095−12103;Huffakerら、Proc.Nat
l.Acad.Sci.U.S.A.Dec.1983(80):7466−7470。
【0167】
(実施例4)
(Xanthomonas Manihotis由来のα1,2−3マンノシダーゼの
同定、クローニングおよび発現)
Xanthomonas Manihotis由来のα1,2−3マンノシダーゼは、
2つの活性(α−1,2マンノシダーゼおよびα−1,3 マンノシダーゼ)を有する。
本発明の方法はまた、これらの活性を有する2つの独立したマンノシダーゼを使用し得、
これらは、選択された目的の生物から同様に同定およびクローニングされ得る。
【0168】
Landryら、によって記載されるように、α−マンノシダーゼは、Xanthom
onas sp.(例えば、Xanthomonas manihotis)から精製さ
れ得る。X.manihotisは、American Type Culture C
ollection(ATCCカタログ番号49764)(Xanthomonas a
xonopodis StarrおよびXanthomonas manihotis
(Arthaud−Berthet)Starrとして寄託されたGarces pat
hovar manihotis)から購入され得る。酵素を、粗細胞抽出物から、以前
に記載されたように精製する(Wong−Madden,S.T.およびLandry,
D.(1995)Purification and characterizatio
n of novel glycosidases from the bacteri
al genus Xanthomonas;およびLandry,D.米国特許第6,
300,113 B1号、Isolation and composition of
novel Glycosidases)。マンノシダーゼの精製後、いくつかの方法
のうちの1つを使用して、ペプチド配列タグを得る(例えば、W.Quadroni M
ら、(2000).A method for the chemical gener
ation of N−terminal peptide sequence tag
s for rapid protein identification.Anal
Chem(2000)Mar 1;72(5):1006−14;Wilkins MR
ら、Rapid protein identification using N−t
erminal”sequence tag”and amino acid anal
ysis.Biochem Biophys Res Commun.(1996)Ap
r 25;221(3):609−13;およびTsugita A.(1987)De
velopments in protein microsequencing.Ad
v Biophys(1987)23:81−113を参照のこと)。
【0169】
次いで、上記方法を使用して作製された配列タグを使用して、当業者に周知の方法を使
用して、縮重プライマーのセットを作製する。縮重プライマーを使用して、ポリメラーゼ
連鎖反応(例えば、Taqポリメラーゼキットを、製造者の指示に従って使用する)にお
いてDNA増幅をプライムして、DNAフラグメントを増幅する。増幅されたDNAフラ
グメントをプローブとして使用して、例えば、目的の酵素をコードするゲノム片を同定お
よび単離(クローニング)するための、標準的なサザンDNAハイブリダイゼーション技
術を使用して、所望のマンノシダーゼをコードする遺伝子を含むDNA分子を単離する。
ゲノムDNA分子を配列決定し、推定オープンリーディングフレームおよびコード配列を
同定する。次いで、目的のグリコシダーゼをコードする適切な発現構築物を、本明細書中
に記載される方法および当該分野で周知の方法を使用して、作製し得る。
【0170】
α1,2−3 マンノシダーゼ活性(またはその触媒的に活性なフラグメント)をコー
ドする配列を含む核酸フラグメントを、本明細書中に示される方法に従って、適切な宿主
細胞でのこれらの活性の発現(好ましくは、標的化された発現)のために、適切な発現ベ
クター中にクローニングする。
【0171】
(実施例5)
(P.pastorisにおけるALG6遺伝子の同定、クローニング、および発現)
実施例1と同様に、S.cerevisiae、Drosophia melanog
aster、Homo sapiensなどに由来するALG6p配列を整列し、、そし
て高い相同性の領域を変性プライマーを産生するために使用する。これらのプライマーは
、P.pastoris由来のゲノムDNAにおけるPCR反応に用いられる。得られた
初めのPCR産物をサブクローン化し、配列を決定して、そして高ストリンジェンシーを
有するP.pastoris由来のゲノムDNAのサザンブロットのプローブとして使用
する(Sambrookら、1989)。ハイブリダイゼーションが観察される。このサ
ザンブロットを、ゲノム座位をマップするために使用する。ゲノムDNAを消化し、そし
てそれらのフラグメントを、適切なサイズ範囲で、pUC19中にライゲーションして、
ゲノムフラグメントをクローン化することで、P.pastorisサブゲノムライブラ
リーを産生する。このサブゲノムライブラリーを、E.coliに形質転換し、そして、
初めのPCR産物の配列に基づいて設計されたプライマーを用いて、コロニーPCRによ
って、数百クローンをテストする。予想した遺伝子を含むクローンの配列を決定し、そし
てオープンリーディングフレームを同定する。PCR重複法(Davidsonら、20
02)を用いて、alg6::NATR欠失アレルの構築のためのプライマーを設計し、
その結果得られた欠失対立遺伝子を2つのP.pastoris株および選択されたNA
T抵抗性コロニー中に形質転換した。これらのコロニーを、PCRによってスクリーニン
グし、ALG6 ORFが,och1::NATR変異対立遺伝子に置換される形質転換
体を得る。例えば、Imbachら、Proc.Natl.Acad.Sci.USA
June 1999(96)6982−698を参照のこと。
【0172】
Alg6p(またはその触媒活性フラグメント)をコ−ドする配列を含む核酸フラグメ
ントが、本明細書中に記載された方法で、適切な宿主細胞におけるこれらの活性の発現お
よび、好ましくは標的発現のための適切な発現ベクター中にクローン化する。クローン化
ALG6遺伝子は、任意の適切なプロモーターの制御で得られ、過剰発現を達成し得る。
その遺伝子自体のプロモーターの制御下にある遺伝子の発現さえも可能である。マルチコ
ピープラスミドの発現は、高レベルの発現(「過剰発現」)を生じる。
【0173】
(実施例6)
(抗体機能性を高める分岐したGlcNAcsを産生するためのGnTIIIのクロー
ニングおよび発現)
(A.背景)
N−アセチルグルコサミルトランスフェラーゼIIIによるGlcNAc2Man3Gl
cNAc2構造へのN−アセチルグルコサミンの付加によって、いわゆる分岐したNグリ
カンを生じる(図3を参照のこと)。この構造は、より大きな抗体依存性細胞傷害性(A
DCC)に関係する(Umanaら、1999)。
【0174】
宿主細胞(例えば、分岐したNグリカンを用いて糖タンパク質を産生できる酵母株)は
、宿主細胞中にGnTIII活性を導入することによって、本発明に従って操作される。
好ましくは、宿主細胞は、異種の細胞シグナル標的ペプチドに(例えば、本発明のライブ
ラリーおよび関連した方法を使用して必要に応じて融合されたGnTIII活性(例えば
、図32に示したマウスGnTIIIのような哺乳動物)またはその酵素活性を有するド
メインコードする核酸を用いて、形質転換される。
【0175】
IgGは、3つのジスルフィド架橋を介するヒンジ領域で相互結合した、2つの重鎖(
図30におけるVH、CH1、CH2およびCH3)、および2つの軽鎖(図30におけるV
L、CL)からなる。軽鎖(ドメインVLおよびドメインCL)は、別のジスルフィド架橋に
よって、重鎖のCH1部分に結合し、そしてCH1フラグメントおよびVHフラグメントと
一緒になって、いわゆるFab領域を形成する。抗原は、Fab領域の末端部分に結合す
る。IgGのFc領域は、CH3、CH2およびヒンジ領域からなり、いわゆるエフェクタ
ー機能の発揮に関連する(下記を参照のこと)。
【0176】
抗体の第1機能は抗原との結合である。しかし、抗原との結合が、その抗原を直接的に
不活化しない限り(例えば、細菌毒素の場合)、いわゆるエフェクター機能が増強されな
ければ、単なる結合は意味を成さない。IgGサブクラスの抗体は、以下の2つの主要な
エフェクター効果を発揮する:補体系の活性化および貪食作用の導入。補体系は、貪食作
用の活性化および細胞膜の溶解による破壊において、炎症の制御に関係する血清タンパク
質の複雑な群からなる。補体活性化は、C1複合体の近接した2つのIgGのFc部分と
の結合で開始する。C1は、1つの分子(C1q)および2つの分子(C1rおよびC1
s)からなる。貪食作用は、IgGのFcフラグメントとFc−γ−レセプター(図30
におけるFcγRI、IIおよびIII)との間における相互作用を介して起こる。Fc
レセプターは、第1に免疫システムのエフェクター細胞表面、特にマクロファージ、単核
細胞、骨髄細胞および樹状細胞の表面に、主に発現する。
【0177】
CH2部分は、アスパラギン297(Asp297)における保存されたNグリコシレ
ーション部位を有する。Asp297 Nグリカンは、高度に異種性であり、Fcレセプ
ター結合および補体活性に影響することが知られている。IgGの少数のみ(すなわち、
約15%〜20%)が、ジシアリル化を受け、そしてIgGの3%〜10%がモノシアリ
ル化したNグリカンを有する(Jefferis,R.,Glycosylation
of human IgG Antibodies.BioOhra,2001の総説を
参照のこと)。興味深いことに、補体活性およびFcレセプター結合が可能な十分機能的
な抗体にとって必要であることが示された最小のNグリカン構造は、末端Nアセチルグル
コサミン残基(GlcNAc2Man3)を有する五単糖類である(Jefferis,R
.,Glycosylation of human IgG Antibodies.
BioPharm,2001の総説を参照のこと)。GlcNAc2Man3Nグリカン
以下またはAsp297の位置にNグリコシル化を有さない抗体は、それでも抗原と結合
することが可能なようであるが、重要な下流の作用(例えば、貪食作用および補体活性化
)を活性化しない可能性がある。さらに、Asp297に結合した真菌型のNグリカンを
有する抗体は、おそらく哺乳動物において免疫応答を誘導し、治療的糖タンパク質として
その抗体を役に立たないものにする。
【0178】
(B.GnTIIIのクローニングおよび発現)
TMドメインを欠くマウスGnTIIIタンパク質の一部をコードするDNAフラグメ
ントは、以下のプライマーを用いて、マウス(あるいは他の哺乳動物)のゲノムDNAか
らPCR増幅される:順方向 5’−TCCTGGCGCGCCTTCCCGAGAGA
ACTGGCCTCCCTC−3’および5’−AATTAATTAACCCTAGCC
CTCCGCTGTATCCAACTTG−3’逆方向プライマー。それらのプライマー
は、リーダーライブラリーとの融合に適したベクターへのクローニングに使用されるAs
cIおよびPacI制限部位を含む。マウスGnTIIIの核酸配列およびアミノ酸配列
を。図32に示す。
【0179】
(C.免疫グロブリンコード配列のクローニング)
抗体の可変領域(プライマー配列を含む)のクローニングのためのプロトコルは、以前
公開されている。抗体およびコ−ド遺伝子の供給源は、とりわけ、インビトロ免疫化ヒト
B細胞(例えば、Borreback,C.A.ら(1988)Proc.Natl.A
cad.Sci.USA85,3995−3999、末梢血リンパ球細胞または単一ヒト
B細胞(例えば、Lagerkvist,A.Cら(1995) Biotechniq
ues 18,862−869;およびTerness,P.ら(1997) Hum.
Immunol.56,17−27を参照のこと)またはハイブリドーマ細胞株の産生
を可能にする、ヒト免疫グロブリン部位を含むトランスジェニックマウスであり得る。
【0180】
標準的な組み換えDNA技術を用いて、抗体をコードする核酸配列がクローン化され得
る。目的の免疫グロブリンをコードする遺伝情報の供給源は、代表的に、目的の細胞(例
えば、血液リンパ球細胞またはハイブリドーマ細胞株)由来の総RNA調製物である。例
えば、特異的プライマーを用いたプロトコルに基づいたPCRを用いて、可変領域は、I
gGCH1ドメインとハイブリダイズする配列特異的プライマー、およびマウスκ定常領
域のアミノ酸111〜118をコードする第2プライマーから開始される逆転写を解して
クローン化され得る。さらに、DNAをコードするVHおよびVkは、以前公開された方
法で増幅される(例えば、Graziano,R.Fら、(1995) J Immun
ol. 155(10):p.4996−5002;Welschof,Mら(1995
) J. Immunol. Methods 179,203−214;および Or
landi,Rら(1988 Proc.Natl.Acad Sci.USA 86:
3833)を参照のこと)。全免疫グロブリンのクローニング手順(重鎖および軽鎖)も
また公開されている(例えば、Buckel,P.ら(1987) Gene 51:1
3−19;Recinos A3rdら(1994) Gene 149:385−386
;(1995) GeneJun 9;158(2):311−2;および Recin
os A3rdら(1994) Gene Nov 18;149(2)385−6を参照
のこと)。抗体フラグメントおよび抗体発現構築物のクローニングおよび産生のさらなる
プロトコルは、Antibody Engineering,R.Kontermann
およびS. Dubel(2001),Editors,Springer Verla
g:Berlin Heidelberg New Yorkに記載されている。
【0181】
免疫グロブリンの重鎖および軽鎖をコードする真菌発現プラスミドは、記載されている
(例えば、Abdel−Salam,H.A.ら(2001)Appl. Microb
iol.Biotechnol. 56:157−164;および Ogunijimi
,A.Aら(1999) Biotechnology Letters 21:561
−567を参照のこと)。従って、免疫グロブリンの定常領域を有する発現プラスミドを
産生し得る。これらの発現ベクター中への可変領域のクローニングを促進するために、適
切な制限部位が可変領域の末端に非常に近い位置に配置され得る。定常領域は、単一制限
酵素消化/ライゲーション実験によって定常領域にインフレームで融合するような様式で
、可変領域を構築され得る。図31は、そのような発現構築の体系的概要を示す。その発
現構築は、プロモーター、転写ターミネーター、組込み標的ドメイン、および選択マーカ
ーさえも容易に変更できるように、非常に修正可能な方法で設計された。
【0182】
図31に示すように、選択したVLドメインおよびVHドメインは、それぞれ、CL領域
およびCH領域に、インフレームで容易にクローン化され得る。第1の組込みは、P.p
astoris AOX部位(または別の真菌細胞の相同部位)に標的化され、そしてメ
タノール誘導性AOXプロモーターは発現を誘導する。あるいは、任意の他の所望の構成
的または誘導的プロモーターカセットが使用され得る。このように、所望の場合、5’A
OX領域および3’AOX領域、ならびに転写ターミネーター(TT)フラグメントが、
異なったTT、プロモーターおよび組込み標的ドメインで容易に置換され、発現を最適化
し得る。第1に、標準KEXプロテアーゼ部位を有するα因子分泌シグナルは、重鎖およ
び軽鎖の分泌を促進するために使用される。さらに、発現ベクターの特性は、標準技術を
用いてさらに精製され得る。
【0183】
Ig発現ベクター(例えば、上述のベクター)は、好ましくは宿主細胞のゴルジ装置内
にGnTIIIを発現する本発明の宿主細胞中に導入される。そのような宿主細胞におい
て発現されるIg分子は、分岐したGlcNacsを有するNグリカンを含む。
【0184】
(実施例7)
GnT−IV(UDP−GlcNAc:α−1,3−D−マンノシドβ−1,4−N−
アセチルグルコサミニルトランスフェラーゼIV)およびGnT−V(β−1−6−N−
アセチルグルコサミニルトランスフェラーゼのクローニングおよび発現)
GnTIVをコードするcDNAを、ウシおよびヒト細胞から単離した(Minowa
、M.T.ら(1998) J.Biol.Chem.273(19),11556−1
1562;)およびYoshida,A.ら(1999) Glycobiology
9(3),303−310)。TMドメインを欠くヒトGnT−IVタンパク質(図33
)の全長および一部をコードするDNAフラグメントを、各々以下のプライマーを用いて
、cDNAライブラリーからPCR増幅した:順方向 5’−AATGAGATGAGG
CTCCGCAATGGAACTG−3’,5’−CTGATTGCTTATCAACG
AGAATTCCTTG−3’,および逆方向5’−TGTTGGTTTCTCAGAT
GATCAGTTGGTG−3’プライマー。
その結果生じるPCR産物をクローン化し、配列を決定した。
【0185】
同様に、GnT−Vたんぱく質をコードする遺伝子は、いくつかの哺乳動物(マウスを
含む)から単離された(例えば、Alverez,Kら Glycobiology 1
2(7),389−394(2002)を参照のこと)。TMドメインを欠くマウスGn
T−Vたんぱく質(図34)の全長および一部をコードするDNAフラグメントを、各々
以下のプライマーを用いて、cDNAライブラリーからPCR増幅した:順方向5’−A
GAGAGAGATGGCTTTCTTTTCTCCCTGG−3’,5’−AAATC
AAGTGGATGAAGGACATGTGGC−3’、および逆方向5’−AGCGA
TGCTATAGGCAGTCTTTGCAGAG−3プライマー。’その結果生じるP
CR産物をクローン化し、配列を決定した。
【0186】
GnT IVまたはV(またはその触媒活性フラグメント)をコードする配列を含む核
酸フラグメントは、本明細書中に記載された方法で、適切な宿主細胞におけるこれらの活
性の発現および、好ましくは標的発現のための適切な発現ベクター中にクローン化する。
【0187】
【化5】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、米国仮出願番号60/344,169(2001年12月27日)(本明細
書中に参考としてその全体を援用する)に優先権を主張する。
【0002】
(発明の分野)
本発明は、一般的に、真菌または他の低級真核生物において発現される組換えタンパク
質のグリコシル化構造(より詳細には高等哺乳動物(特にヒト)のタンパク質のグリコシ
ル化)の改変に関する。
【背景技術】
【0003】
(発明の背景)
DNAが転写されそしてタンパク質へと翻訳された後の、さらなる翻訳後プロセッシン
グは、リコシル化として公知のプロセスである糖残基の結合を含む。異なる生物は、異な
るグリコシル化酵素(グリコシルトランスフェラーゼおよびグリコシダーゼ)を生産し、
そして利用可能な異なる基質(ヌクレオチド糖)を有し、その結果、個々のオリゴ糖のグ
リコシル化パターンならびに組成物は、1つでも同じタンパク質でさえも、特定のタンパ
ク質が発現される宿主系に依存して異なる。細菌は、代表的に、タンパク質をグリコシル
化せず、そうであれば、非常に非特異的な様式である(Moens,1997)。低級真
核生物(例えば、糸状菌および酵母)は、主なマンノースおよびリン酸マンノシル(ma
nnosylphosphate)糖を添加するのに対して、昆虫細胞(例えば、Sf9
細胞)は、さらなる別の方法でタンパク質をグリコシル化する。例えば、(Bretth
auer,1999;Martinet,1998;Weikert,1999;Mal
issard,2000;Jarvis,1998;およびTakeuchi,1997
)を参照のこと。
【0004】
一連の反応からなる哺乳動物型オリゴ糖構造の合成は、その過程において、糖残基が、
タンパク質が、宿主生物における分泌経路に沿って移動する間に、添加されそして除去さ
れる。宿主生物または細胞のグリコシル化経路に沿って存在する酵素は、分泌されたタン
パク質の得られたグリコシル化パターンを決定する。あいにく、低級真核生物宿主細胞で
発現されるタンパク質の得られたグリコシル化パターンは、高級真核生物(例えば、ヒト
および他の哺乳動物)において見られるグリコシル化とは実質的に異なる(Bretth
auer,1999)。さらに、非常に異なるグリコシル化パターンは、いくつかの場合
において、ヒトにおけるこれらのタンパク質の免疫原性の増加およびそれらの半減期の減
少を示している(Takeuchi,1997)。非ヒト宿主細胞(特に、低級真核生物
細胞)中でヒト様糖タンパク質を生産することが所望される。
【0005】
ヒトグリコシル化の初期段階は、2つの異なる相に分けられ得る:(i)脂質連結Gl
c3Man9GlcNAc2オリゴ糖は、小胞体(ER)の膜で反応の経時的なセットによ
ってアセンブリされ、そして(ii)脂質アンカードリコール(dolichyl)ピロ
リン酸から新規に合成されたタンパク質上へのこのオリゴ糖の移動。特異的な移動部位は
、配列Asn−Xaa−Ser/Thr(図1を参照のこと)中のアスパラギン(Asn
)残基によって規定され、Xaaは、プロリンを除く任意のアミノ酸であり得る(Gav
el,1990)。グルコシダーゼおよびマンノシダーゼによるさらにプロセッシングは
、ER中で生じ、その後、新生の糖タンパク質が、初期のゴルジ装置に移動され、さらな
るマンノース残基は、ゴルジ特異的α(α)−1,2−マンノシダーゼによって除去され
る。プロセッシングは、ゴルジを通って進むタンパク質として続く。内側のゴルジにおい
て、多くの改変酵素(N−アセチルグルコサミニルトランスフェラーゼ(GnT I、G
nT II、GnT III、GnT IV GnT V GnT VI)、マンノシダ
ーゼIIおよびフコシルトランスフェラーゼを含む)は、特定の糖残基を付加し、そして
除去する(例えば、図2および図3を参照のこと)。最後に、trans−ゴルジにおい
て、ガラクトシルトランスフェラーゼおよびシアリルトランスフェラーゼは、ゴルジから
放出される糖タンパク質構造を生産する。これらの構造において、ジアンテナ構造(an
tennary structure)、トリアンテナ構造およびテトラアンテナ構造(
ガラクトース、フコース、N−アセチルグルコサミンおよび糖タンパク質にそれらのヒト
特徴付けを与える末端シアル酸の高い程度を含む)によって特徴付けされる。
【0006】
ほとんど全ての真核生物において、糖タンパク質は、一般的なコアオリゴ糖前駆体Gl
c3Man9GlcNAc2−PP−Dolから誘導され、このPP−Dolは、ドリコー
ル−ピロリン酸を意味する(図1)。小胞体において、オリゴ糖に結合するドリコールピ
ロリン酸の合成およびプロセッシングは、周知の真核生物の間で同一である。しかし、酵
母によるコアオリゴ糖のさらなるプロセッシングは、一旦ERを離れそしてゴルジに侵入
するペプチドに移動されると、分泌経路に沿って移動し、そしていくつかのマンノース糖
の添加に関与するので、ヒトと有意に異なる。
【0007】
酵母において、これらの工程は、Ochlp、MntlpおよびMnnlpのようなマ
ンノシルトランスフェラーゼに存在するゴルジによって触媒され、コアオリゴ糖にマンノ
ース糖を連続して添加する。得られた構造は、ヒューマノイドタンパク質の生産を所望せ
ず、従って、マンノシルトランスフェラーゼ活性を減少するか除去することを所望する。
S.cerevisiaeの変異体(マンノシルトランスフェラーゼ活性を欠損する)(
例えば、och1変異体またはmnn9変異体)は、非致死であり、そして酵母糖タンパ
ク質のオリゴ糖中の減少したマンノース含量を提示することを示す。他のオリゴ糖プロセ
ッシング酵素(例えば、リン酸マンノシルトランスフェラーゼ)はまた、宿主の特定の内
因的なグリコシル化パターンに依存して除去され得る。
【0008】
(脂質連結オリゴ糖前駆体)
本発明の特定の興味深いことは、N−グリコシル化の初期段階である(図1および図2
)。Glc3Man9GlcNAc2−PP−Dolの生合成が不完全であるalg(アス
パラギン連結グリコシル化)変異体の研究は、N−グリコシル化の開始段階を明らかにす
ることを助ける。
【0009】
S.cerevisiaeのALG3遺伝子は、首尾よくクローン化され、そして欠失
によってノックアウトされている(Aebi,1996)。ALG3は、酵素Dol−P
−Man:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼをコード
することが示され、これは、ERの管腔側で、Man5GlcNAc2−PP−Dolから
Man6GlcNAc2−PP−Dolまでの第1のDol−P−Man依存性マンノシル
化工程に関与する(Sharma,2001)(図1および2)。漏出alg3−1変異
を有するS.cerevisiae細胞は、Man5GlcNAc2−PP−Dol(構造
I)を集積する(Huffaker,1983)。
【0010】
【化1】
Man5GlcNAc2(構造I)およびMan8GlcNAc2は、och1 mnn1
alg3変異体の全細胞マンノタンパク質を集積する(Nakanishi−Shind
o,1993)。このS.cerevisiae och1、mnn1、alg3変異体
は、生存可能であるが、温度感受性であり、そしてα−1,6ポリマンノース外鎖(ou
ter chain)を欠損することが示された。
【0011】
別の研究において、alg3を欠損した株(Δalg3バックグラウンド)中で発現さ
れた分泌タンパク質は、Endo−β−N−アセチルグルコサミニダーゼ H(Endo
H)に対する耐性について研究された(Aebi,1996)。以前の観察は、Man
5GlcNAc2より長いこれらのオリゴ糖のみが、Endo Hによって切断されやすい
ことを示している(Hubbard,1980)。alg3−1表現型において、いくつ
かのグリコフォームは、その漏出(leakiness)を確認するEndo H切断に
感受性であるが、Δalg3変異体において、全てのグリコフォームは耐性であるようで
あり、かつMan5−型であり(Aebi,1996)、密接な表現型および発生期のポ
リペプチド鎖上にMan5GlcNAc2オリゴ糖構造の移動を示唆する。明らかな表現型
は、ALG3遺伝子の不活性化と関係しない(Aebi,1996)。Saccharo
myces cerevisiae alg3変異体中で生産された、分泌エクソグルコ
ナーゼ(exogluconase)は、35〜44%の間で過小グリコシル化(und
erglycosylate)形態および非グリコシル化形態ならびEndo H処理に
耐性のままである約50%のみの移動オリゴ糖を含むことが見出された(Cueva,1
996)。エキソグルカナーゼ(Exg)(Asn165およびAsn325での2つの潜在的
なN−グリコシル化部位を含む酵素)は、より詳細に分析された。2つのオリゴ糖を受け
取るExg分子について、これは、第1のN−グリコシル化部位(Asn165)が、短縮
型残基中で富化されたが、第2(Asn325)は、規則的なオリゴ糖中で富化されたこと
が示された。35〜44%の分泌エキソグルカナーゼは、非グリコシル化または過小グル
コシル化され、そして約73〜78%の全ての利用可能なN−グリコシル化部位は、短縮
型オリゴ糖または規則的なオリゴ糖のいずれかで占められた(Cueva,1996)。
【0012】
(グルコシル化脂質連結オリゴ糖の移動)
証拠は、哺乳動物細胞において、グルコシル化脂質連結オリゴ糖のみが、新生タンパク
質に移動される(Turco,1977)が、酵母alg5変異体、alg6変異体、お
よびdpg1変異体においては、非グルコシル化オリゴ糖が移動され得る(Ballou
,1986;Runge,1984)ことを示唆する。Saccharomyces c
erevisiae alg8変異体において、過小グリコシル化のGlcMan9Gl
cNAc2が移動される(Runge,1986)。Verostekおよび共同研究者
らは、alg3変異体、secl8変異体、glsl変異体を研究し、そしてMan5G
lcNAc2構造(構造I、上記)のグルコシル化は、脂質連結Man9構造のグルコシル
化と比較して、比較的遅いことを提案する。さらに、このMan5GlcNAc2構造のタ
ンパク質への移動は、Glc3Man5GlcNAc2へのグルコシル化よりも約5倍効率
的であるようである。Man5GlcNAc2グルコシル化の減少した速度は、比較的早い
速度のMan5構造と組み合わせて、alg3変異体酵母中で観察された非グルコシル化
Man5構造の蓄積の原因であると考えられる新生タンパク質上に移動する(Veros
tek−a,1993;Verostek−b,1993)。
【0013】
上記の研究に先行する研究は、グルコシル化オリゴ糖が、その非グルコシル化対応物よ
りも非常に早い速度で移動され、従って、単離するのがより困難であるという結果を認め
る、任意の脂質連結グルコシル化オリゴ糖を明らかにしなかった(Orlean,199
0;Huffaker,1983)。最近の研究は、非グルコシル化オリゴ糖および低グ
ルコシル化オリゴ糖を有する酵母株の作製および研究を可能にし、そしてオリゴサッカリ
ルトランスフェラーゼ複合体による基質認識のための脂質連結オリゴ糖のアンテナに対す
るグルコースの付加の重要性をさらに確認した(Reiss,1996;Staglja
r,1994;Burda,1998)。alg3変異体における脂質連結Man5オリ
ゴ糖のグルコシル化の減少程度は、新生タンパク質上の脂質連結オリゴ糖の移動の動力学
にネガティブに影響し、そしてこれは、alg3ノックアウト株中の分泌タンパク質の強
い過小グリコシル化を引き起こすと考えられる(Aebi,1996)。
【0014】
上記のように、脂質連結核オリゴ糖Man9GlcNAc2のアセンブリは、小胞体(e
ndoplasmatic reticulum)の膜で生じる。3つのグルコース単位
の、脂質連結オリゴ糖のα−1,3−アンテナへの付加は、オリゴ糖アセンブリの最終反
応である。最初のα−1,4グルコース残基が付加され、その後別のα−1,3グルコー
ス残基および末端α−1,2グルコース残基を付加される。ドリコール連結Man9Gl
cNAc2を蓄積する変異体は、ALG6座中で欠損していることが示され、そしてAl
g6pは、Alg8pに対して類似性を有し、α−1,3−グルコシルトランスフェラー
ゼは、第2のα−1,3−連結グルコースの付加を触媒する(Reiss,1996)。
欠損ALG8座を有する細胞は、ドリコール連結Glc1Man9GlcNAc2を蓄積す
る(Runge,1986;Stagljar,1994)。このALG10座は、Gl
c2Man9GlcNAc2−PP−Dolへの単一の末端グルコースの付加に応答性であ
るα−1,2グルコシルトランスフェラーゼをコードする(Burda,1998)。
【0015】
(局在化した酵素活性の連続的なN−グリカンのプロセシング)
糖トランスフェラーゼおよびマンノシダーゼは、ERおよびゴルジ装置の内側(管腔)
表面に並び、それによって糖タンパク質の連続的なプロセシングを可能にする「触媒」表
面を提供する。なぜなら、糖タンパク質は、ERおよびゴルジのネットワークを介して進
むからである。実際、これらのシス、中間、およびトランスのゴルジならびにトランス−
ゴルジネットワーク(TGN)の複数の区画は、順番に並べられたグリコシル化反応が起
こり得る、異なる局所性を提供する。糖タンパク質は、ER中の合成から、後期のゴルジ
またはTGNにおける完全な成熟まで進められるので、異なるグリコシダーゼ、マンノシ
ダーゼおよびグリコシルトランスフェラーゼに順番に曝され、その結果、特異的糖質構造
が合成され得る。多くの研究は、これらの酵素がそれぞれのオルガネラに保持およびアン
カーされる、正確な機構を明らかにすることに奉げられていつ。画像を展開することは複
雑であるが、証拠は、幹領域、膜貫通領域および細胞質テイルが、個々にかまたは協力し
て、個々のオルガネラの膜に酵素を指向し、それによって関連する触媒ドメインをその座
に局在化する。
【0016】
いくつかの場合において、これらの特異的な相互作用は、種をまたいで機能することが
見出された。例えば、ラット由来のα2,6−STの膜貫通ドメインは、動物のトランス
−ゴルジに局在化されることが知られている酵素であり、酵母ゴルジ中のレポーター遺伝
子(インベルターゼ)も局在化することが示された(Schwientek,1995)
。しかし、全長α2,6STの一部と非常に同じ膜貫通ドメインは、ER中に保持され、
そして酵母のゴルジにさらに輸送されない(Krezdorn,1994)。ヒト由来の
全長Gal−Trは、明らかに高い転写レベルにも関わらず、酵母中でさえも合成されな
い。一方で、インベルターゼレポーターに融合されるGalTと同じヒトの膜貫通領域は
、低い生成レベルにも関わらず、酵母ゴルジに直接局在化され得た。Schwiente
kおよび共同研究者は、ヒトGalTの触媒ドメインに対する酵母マンノシルトランスフ
ェラーゼ(Mnt1)、細胞質テイルを含む領域、膜貫通領域の28個のアミノ酸および
幹領域の8個のアミノ酸が、活性GalTのゴルジ局在化のために十分であることを示し
ている。他のガラクトシルトランスフェラーゼは、酵素残基(特に、オルガネラ)との相
互作用に依存するようである。なぜなら、これらの膜貫通領域の除去後、これらは、適切
に局在化され得るからである。現在、より低級真核生物中で特定の非相同的に発現された
グリコシルトランスフェラーゼまたはマンノシダーゼが、(1)十分に翻訳されているか
、(2)触媒的に活性か、または(3)分泌経路内の適切なオルガネラに位置しているか
どうかを予測する信頼性の高い方法は存在しない。これらの3つ全てが、低級真核生物中
のグリコシル化パターンに影響するのに必要であり、所望の触媒機能および予測ツールの
非存在下での酵素の適切な保持を達成するための全体的なスキームは、現在利用可能では
なく、設計段階である。
【0017】
(治療用糖タンパク質の製造)
ヒトまたは動物から単離される有意な数のタンパク質は、最も有意な改変の1つである
グリコシル化によって翻訳後に改変される。全ての治療用タンパク質の推定で70%がグ
リコシル化され、従って、ヒトと類似の様式でグリコシル化し得る産生系(すなわち、宿
主細胞)に依存している。現在、多くの糖タンパク質は、哺乳動物宿主系で作製される。
いくつかの研究は、グリコシル化は、(1)免疫原性、(2)薬物動態学特性、(3)ト
ラフィック、および(4)治療用タンパク質の効果を決定するのに重要な役割を果たすこ
とが示されている。従って、製薬産業の実質的な労力は、可能であれば「ヒューマノイド
」または「ヒト様」のようなプロセスを開発し、糖タンパク質を得ることに向けられてい
る。これは、このような哺乳動物細胞を遺伝子操作して、細胞によって発現されたタンパ
ク質のシアリル化(すなわち、シアル酸の末端付加)(これは、このようなタンパク質の
薬物動態学特性を改善することが知られている)の程度を増加することを含み得る。ある
いは、公知のグリコシルトランスフェラーゼおよびそれぞれのヌクレオチド糖(例えば、
2,3シアリルトランスフェラーゼおよびCMP−シアル酸)を使用する、このような糖
のインビトロでの付加によってシアリル化の程度を改善し得る。
【0018】
さらなる調査は、特定の糖形態の生物学的有意性および治療的有意性を明らかにし得、
従って、そのような望ましい特定の糖形態を生成するための能力を与える。現在までに、
十分に特徴付けられた糖化パターンを有するタンパク質を生成すること、および以下の高
等真核生物のタンパク質発現系の1つにおいて、そのようなタンパク質をコードするcD
NAを発現することに、努力が費やされている。:
1.高等真核生物(例えば、チャイニーズハムスターの卵細胞(CHO)、マウス線維
芽細胞およびマウス骨髄腫細胞(Werner、1998);
2.トランスジェニック動物(例えば、ヤギ、ヒツジ、マウスおよその他(Dente
、1988);(Cole、1994);(McGarvey、1995);(Bard
or、1999);
3.植物(Arabidopsis、thaliana,tobaccoなど)(St
aub、2000);(McGarvey、1995);(Bardor、1999);
4.昆虫細胞(組換えバキュロウイルス(例えば、鱗翅目細胞に感染するAutogr
apha california多核ポリヒドローシスウイルス(Altmann,19
99))との組み合わせで、Spodoptera frugiperda Sf9,S
f21,Trichoplusia niなど)。
【0019】
最も高等な真核生物が、ヒトにおいて認められる糖化反応に類似する糖化反応を起こす
一方、上述の宿主系において発現される組換えヒトタンパク質は、不変的にそれらの「天
然の」ヒトの対応部分と異なる(Raju、2000)。従って、さらなる開発研究が、
これらの発現系において生成されるタンパク質の「ヒト特徴」を改善する方法を発見する
ことに向けられている。これは、発酵条件の最適化および、ヒト様糖形態の形成に関する
酵素をコードする遺伝子を導入することによる、タンパク質発現宿主の遺伝的改変を含む
(Werner、1998);(Weikert、1999);(Andersen、1
994);(Yang、2000)。全ての哺乳動物の発現系に関連する固有の問題は、
解決されていない。
【0020】
例えば、哺乳動物の細胞培養に基づく発酵プロセス(例えば、CHO細胞、マウス細胞
、またはヒト細胞)は、非常に遅い傾向があり(1週間を越える発酵時間は、まれではな
い)、しばしば、低い生成価を生じ、高価な栄養および補因子(例えば、ウシ胎児血清)
を要求し、プログラムされた細胞死(アポトーシス)によって制限され、しばしば、特定
の治療的価値のあるタンパク質の発現を不可能にする。より重要なことには、哺乳動物細
胞は、ヒト病原体となる能力を有するウイルスに感受性でありまた、厳密な質の制御が、
生成物安全を保証するために要求される。これは、多くのそのようなプロセスが、動物(
例えば、ウシ血清)由来の、複雑かつ温度感受性な培地成分の添加を必要とするので、特
定の関心事の内であり、ヒトに対する病原性因子(例えば、牛海綿状脳症(BSE)プリ
オンまたはウイルス)を保有し得る。さらに、治療的化合物の生成は、好ましくは、十分
に制御された滅菌環境において、実施される。動物飼育は、(どんなに清潔を保っていて
も)そのような環境を構築せず、従って、多量の治療的タンパク質を製造するためのトラ
ンスジェニック動物の使用におけるさらなる問題を与える。
【0021】
そのため、全てではないにしても、最近産生された治療的糖タンパク質は、哺乳動物細
胞において発現され、また、これらの組換えタンパク質の糖化パターンを改善すること(
すなわち。「ヒト化」すること)に多くの努力が、向けられている。培地組成物における
変化ならびにヒト糖化に関する酵素をコードする遺伝子の共発現が、継続的に行われてい
る(例えば、Weikert、1999を参照のこと)。
【0022】
ヒト対応部分に類似する組換えタンパク質が、哺乳動物発現系において作製され得、下
等真核生物(菌類および酵母)におけるヒト様糖化パターンを有するタンパク質を作製す
ることは、近年不可能である。小包体中で、タンパク質に移行される、核となるオリゴ糖
構造は、基本的には、哺乳動物および下等な真核生物において、同一であり、実質的な差
異は、次のプロセッシング反応において認められ、それは、菌類および哺乳動物のゴルジ
体において起こる。実際、異なる下等な真核生物間でさえ、多くの種々の糖化構造が、存
在する。これは、以下の哺乳動物発現系にわたって別に留意すべき利点を除いて、組換え
ヒト糖タンパク質の生成のための宿主として、下等真核生物の使用を妨げている。例えば
、:(1)一般的に高い産生力価、(2)短い発酵時間、(3)哺乳動物細胞において乏
しく発現されるタンパク質のための代替を有すること、(4)化学的に規定された無タン
パク質培地中での増殖し、従って、複雑な動物由来の培地成分を要求しない能力、(5)
およびウイルスの非存在(特に、そのような宿主のロタウイルス感染の非存在)。
【0023】
種々のメチル栄養性酵母(例えば、Pichia pastoris、Pichia
methanolicaおよびHansenula polymorphaは、真核生物
発現系として、特に重要な役割を果たす。なぜなら、それらは、高い細胞密度まで増殖し
得、大量の組換えタンパク質を分泌するためである。しかし、上記のように、下等真核生
物(例えば、酵母)は、高等動物のように、タンパク質を糖化しない。例えば、小包体(
ER)中の異種性マンノシダーゼを開示するMartinetら、(1998)Biot
echnol Let.20巻、12号を参照のこと。
【0024】
Chibaら、(1998)は、S.cerevisiaeが、1,6マンノシルトラ
ンスフェラーゼ(OCH1)、1,3マンノシルトランスフェラーゼ(MNN1)および
マンノシルトランスフェラーゼ(MNN4)の調節因子を取り除くことによって、ならび
に、ER検索配列(Chiba,1998)を用いてS.cerevisiaeのERに
Aspergillus saitoi由来のα−1,2−マンノシダーゼIの酵素作用
ドメインを標的することによって、Man8GlcNAc2構造からMan5GlcNAc2
構造の範囲の構造を提供するために操作され得ることを示した。しかし、この試みは、所
望のMan5GlcNAc2(例えば、インビボで生成され、また、GnT1についての基
質(ヒト様グリカン構造を作製する上での次の工程)として働き得るもの)。のわずかな
生成か、または全く生成しないことを生じる。Chibaら、(1998)は、P.pa
storisが、内在的に有用な量(5%より多くの)の、炭化水素を受け取るGlcN
AcトランスフェラーゼIを生成し得えない。
【0025】
Marasおよび同僚は、T.reeseiにおいて、「十分な濃度のアクセプター基
質(すなわち、Man5GlcNAc2)が存在する」ことを主張するが、このアクセプタ
ー基質をGlcNAcMan5GlcNAc2に、インビボで2%未満で転換することを試
みる場合、従って複合体Nグリカン形態のための適切な前駆体でないMan5GlcNA
c2構造の存在を示しことで変換された(Maras,1997;Maras,1999
)。現在まで、下等真核生物において、商業的に十分な量のGlcNAcMan5Glc
NAc2の生成が可能である開示は存在しない。
【発明の概要】
【発明が解決しようとする課題】
【0026】
このため、非ヒト宿主細胞において発現される組換え糖タンパク質の糖化をヒト化する
ためのシステムおよび方法を提供することが、本発明の目的である。
【課題を解決するための手段】
【0027】
(発明の要旨)
本発明は、改変された脂質連結オリゴ糖を有する菌株のような宿主細胞に関し、この宿
主細胞は、グリコシルトランスフェラーゼのセット、糖輸送体、およびマンノシダーゼの
異種発現によってされに改変され、哺乳動物の生成物(例えば、ヒト治療的糖タンパク質
)についての宿主株とされ得る。タンパク質生成方法は、以下を用いて開発されている;
(1)下等な真核生物宿主(例えば、単細胞真菌または繊維状真菌)または(2)ヒト由
来の異なる糖化パターンを有する、あらゆる非ヒト真核生物(ヒトタンパク質中に見出さ
れる炭化水素構造により類似するように、宿主生物(「宿主細胞」)中で生成される糖化
の組成およびタンパク質の構造が改変される。このプロセスは、科学文献およびタンパク
質発現の当業者に公知の、十分確立された方法によって、糖化に関する任意の所望の遺伝
子を発現および標的化するために使用され得る、操作された宿主細胞を得ることを可能に
する。本明細書に記載のように、改変された脂質連結オリゴ糖を有する宿主細胞は、作成
または選択される。操作された宿主細胞中のN−グリカンは、GlcNAcMan3Gl
cNAc2コア構造を有し、次いで、これは1つ以上の酵素(例えば、グリコシルトラン
スフェラーゼ、糖輸送体およびマンノシダーゼ)の異種発現によって、さらに改変され、
ヒト様糖タンパク質を産生し得る。治療的タンパク質の生成のために、この方法が、あら
ゆる所望の糖化構造を得ることができる細胞株を操作するために適合され得る。
【図面の簡単な説明】
【0028】
【図1】図1は、ドリチルピロホスフェート結合オリゴ糖の構造の概略図を示す。
【図2】図2は、alg3活性、alg9活性またはalg12活性を欠く真菌宿主細胞由来のGlcNAc2Man3GlcNAc2N−グリカンの生成の概略図を示す。
【図3】図3は、alg3、och1遺伝子型を有する真菌宿主細胞中の哺乳動物型のオリゴ糖構造を生成するために要求されるプロセッシング反応の概略図を示す。
【図4−1】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−2】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−3】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図4−4】図4は、S.cerevisiae Alg3配列比較(Blast)を示す。
【図5】図5は、S.cerevisiae Alg3およびAlg3p配列を示す。
【図6】図6は、P.pastoris Alg3およびAlg3p配列を示す。
【図7−1】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図7−2】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図7−3】図7は、P.pastoris Alg3配列比較(Blast)を示す。
【図8】図8は、K.lactis Alg3およびAlg3p配列を示す。
【図9】図9は、K.lactis Alg3配列比較(Blast)を示す。
【図10】図10は、S.cerevisiae Alg9およびAlg9p配列を示す。
【図11】図11は、P.pastoris Alg9およびAlg9p配列を示す。
【図12−1】図12は、P.pastoris Alg9配列比較(Blast)を示す。
【図12−2】図12は、P.pastoris Alg9配列比較(Blast)を示す。
【図13】図13は、S.cerevisiae Alg12およびAlg12p配列を示す。
【図14】図14は、P.pastoris Alg12およびAlg12p配列を示す。
【図15−1】図15は、P.pastoris Alg12配列比較(Blast)を示す。
【図15−2】図15は、P.pastoris Alg12配列比較(Blast)を示す。
【図16】図16は、優位なN−グリカンが、GlcNAcMan5GlcNAc2であることを示すP.pastoris中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図17】図17は、図16の優位なN−グリカンが、GlcNAcMan3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man5GlcNAc2に一致するピーク)を用いて処理されるP.pastoris(図16)中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図18】図18は、優位なN−グリカンが、GlcNAcMan3GlcNAc2およびGlcNAcMan4GlcNAc2であること示すP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図19】図19は、図18のGlcNAcMan4GlcNAc2が、GlcNAcMan3GlcNAc2に変換されることを示す、α1,2マンノシダーゼを用いて処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図20】図20は、図19のN−グリカンが、GlcNAcMan3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man3GlcNAc2に一致するピーク)を用いて処理される図19のN−グリカンのMALDI−TOF−MS分析である。
【図21】図21は、図19のGlcNAcMan3GlcNAc2が、GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図22】図22は、図21のN−グリカンが、GlcNAc2Man3GlcNAc2であること確認するために、β−N−ヘキサミニダーゼ(Man3GlcNAc2に一致するピーク)を用いて処理される図21のN−グリカンのMALDI−TOF−MS分析である。
【図23】図23は、図21のGlcNAc2Man3GlcNAc2が、Gal2GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて、UDP−ガラクトースおよびβ1,4−ガラクトシルトランスフェラーゼの存在下で処理されるP.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図24】図24は、Gal2GlcNAc2Man3GlcNAc2が、NANA2Gal2GlcNAc2Man3GlcNAc2に変換されることを示す、α1,2マンノシダーゼおよびGnTIIを用いて、UDP−ガラクトースおよびβ1,4−ガラクトシルトランスフェラーゼの存在下で処理され、さらにCMP−N−アセチルノイラミン酸およびシアリルトランスフェラーゼを用いて処理される、P.pastoris alg3欠損変異体中に生成されるkringle3糖タンパク質から単離されるN−グリカンのMALDI−TOF−MS分析である。
【図25】図25は、S.cerevisiaeのAlg6配列およびAlg 6p配列を示す。
【図26】図26は、P.pastorisのAlg6配列およびAlg 6p配列を示す。
【図27−1】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−2】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−3】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図27−4】図27は、P.pastoris Alg 6配列の比較(Blast)を示す。
【図28】図28は、K.lactisのAlg6配列およびAlg 6p配列を示す。
【図29−1】図29は、K.lactis Alg 6配列の比較(Blast)を示す。
【図29−2】図29は、K.lactis Alg 6配列の比較(Blast)を示す。
【図30】図30 IgG免疫グロブリンのモデル。重鎖および軽鎖は、類似の二次構造および三次構造に基づいて、ドメインに細分される。2つの重鎖(ドメインVH、CH1、CH2およびCH3)は、3つのジスルフィド架橋により連結される。軽鎖(ドメインVLおよびCL)は、別のジスルフィド架橋により重鎖のCH1部分に連結され、そしてCH1フラグメントおよびVHフラグメントと共に、Fab領域を構成する。抗原は、Fab領域の末端部分に結合する。エフェクター機能(例えば、Fc−γ−レセプター結合)は、ヒンジ領域の直ぐ下流のCH2ドメインに局在化し、そして重鎖におけるアスパラギン297のN−グリコシル化により影響を受ける。
【図31】図31は、モジュラーIgG1発現ベクターの模式図である。
【図32】図32は、M.musculis GnT IIIの核酸配列およびアミノ酸配列を示す。
【図33】図33は、H.sapiens GnT IVの核酸配列およびアミノ酸配列を示す。
【図34−1】図34は、M.musculis GnT Vの核酸配列およびアミノ酸配列を示す。
【図34−2】図34は、M.musculis GnT Vの核酸配列およびアミノ酸配列を示す。
【発明を実施するための形態】
【0029】
(発明の詳細な説明)
本明細書中で他に規定されない限り、本発明に関連して使用される科学的用語および技
術用語は、当業者により一般的に理解される意味を有する。さらに、文脈により他に必要
とされなければ、単数形の用語は、複数形を含み、そして複数形の用語は、単数形を含む
。本発明の方法および技術は、当該分野において周知の従来の方法に従って一般的に実施
される。一般的に、本明細書中に記載される生化学、酵素学、分子生物学および細胞生物
学、微生物学、遺伝学ならびにタンパク質化学および核酸化学、ならびにハイブリダイゼ
ーションに関連して使用される名称、およびこれらの技術は、周知のものであり、そして
当該分野で一般的に使用される。本発明の方法および技術は、他に示されなければ、一般
的に、当該分野で周知の従来の方法に従って、そして本明細書の全体にわたって引用され
そして考察される種々の一般的な参考文献およびより具体的な参考文献に記載されるとお
りに実施される。例えば、Sambrookら、Molecular Cloning:
A Laboratory Manual,第2版,Cold Spring Harb
or Laboratory Press,Cold Spring Harbor,N
.Y.(1989);Ausubel et al.,Current Protoco
ls in Molecular Biology,Greene Publishin
g Associates(1992および2002までの補遺);Harlow an
d Lane Antibodies:A Laboratory Manual Co
ld Spring Harbor Laboratory Press,Cold S
pring Harbor,N.Y.(1990);Introduction to
Glycobiology,Maureen E.Taylor,KurtDricka
mer,Oxford Univ.Press (2003);Worthington
Enzyme Manual,Worthington Biochemical C
orp.Freehold,NJ;Handbook of Biochemistry
:Section A Proteins VolI 1976 CRC Press;
Handbook of Biochemistry:Section A Prote
ins Vol II 1976 CRC Press;Essentials of
Glycobiology,Cold Spring Harbor Laborato
ry Press (1999)を参照のこと。本明細書中に記載される生化学および分
子生物学に関連して使用される名称、ならびにこれらの研究室手順および技術は、当該分
野で周知でありかつ一般的に使用されるものである。
【0030】
本明細書中に記述される全ての刊行物、特許および他の参考文献は、参考として援用さ
れる。
【0031】
以下の用語は、他に示されなければ、以下の意味を有すると解釈される。
【0032】
本明細書中において使用される場合、用語「N−グリカン」とは、N−連結オリゴ糖を
いい、例えば、アスパラギン−N−アセチルグルコサミン結合でポリペプチドのアスパラ
ギン残基に結合されるものである。N−グリカンは、Man3GlcNAc2(「Man」
は、マンノースを指し;「Glc」は、グルコースを指し;そして「NAc」は、N−ア
セチルを指し;GlcNAcは、N−アセチルグルコサミンを指す)の共通のペンタサッ
カリドコアを有する。N−グリカンは、Man3GlcNAc2(「Man3」)コア構造
に付加される抹消糖(例えば、フコースおよびシアル酸)を含む分枝(アンテナ)の数に
関して異なる。N−グリカンは、それらの分枝構造(高マンノース、複合(comple
x)またはハイブリッド)に従って分類される。「高マンノース」型N−グリカンは、5
個以上のマンノース残基を有する。「複合」型N−グリカンは、代表的には、1,3マン
ノースアームに結合した少なくとも1つのGlcNAc、および「トリマンノース」コア
の1,6マンノースアームに結合した少なくとも1つのGlcNAcを有する。「トリマ
ンノースコア」は、Man3構造を有するペンタサッカリドコアである。複合N−グリカ
ンはまた、必要に応じてシアル酸または誘導体(「NeuAc」、ここで「Neu」は、
ノイラミン酸を指し、「Ac」は、アセチルを指す)で改変されたガラクトース(「Ga
l」)残基を有し得る。複合N−グリカンはまた、「分岐する(bisecting)」
GlcNAcおよびコアフコース(「Fuc」)を含む鎖内置換を有し得る。「ハイブリ
ッド」N−グリカンは、トリマンノースコアの1,3マンノースアームの末端に少なくと
も1つのGlcNAc、およびトリマンノースコアの1,6マンノースアームの0以上の
マンノースを有する。
【0033】
本明細書中で使用される略語は、当該分野で共通して利用される。例えば、上記の糖の
略語を参照のこと。他の共通の略語としては、以下が挙げられる:「PNGase」、こ
れは、ペプチドN−グリコシダーゼF(EC 3.2.2.18)を指す;「GlcNA
c Tr(I−III)」、3つのN−アセチルグルコサミニルトランスフェラーゼ酵素
のうちの1つを指す;「NANA」は、N−アセチルノイラミン酸を指す。
【0034】
本明細書中で使用される場合、用語「分泌経路」とは、細胞質から小胞体および(ER)およびゴルジ装置の区画への発生期ポリペプチド鎖の分子フローに沿って、脂質連結オリゴ糖前駆体およびN−グリカン基質が、連続的に暴露される、種々のグリコシル化酵素の集合ラインをいう。酵素は、この経路に沿って局在化されるといわれる。酵素Yの前に、脂質連結グリカンまたはN−グリカンに対して作用する酵素Xは、酵素Yの「上流」にある、または「上流」で作用するといわれる;同様に、酵素Yは、酵素Xから「下流」で作用する。
【0035】
本明細書中で使用される場合、用語「alg X活性」とは、「alg X」遺伝子に
よりコードされる酵素活性、および相同遺伝子もしくは遺伝子産物(以下を参照のこと)
または非関連遺伝子もしくは遺伝子産物によりコードされる酵素活性を有する酵素をいう
。
【0036】
本明細書中で使用される場合、用語「抗体」とは、完全な抗体(2つの重鎖および2つ
の軽鎖からなる)またはそのフラグメントをいう。このようなフラグメントとしては、種
々のプロテアーゼで消化することにより産生されるフラグメント、化学的切断および/ま
たは化学的解離、ならびにそのフラグメントが抗原に特異的に結合し得るままである限り
、組み換えにより産生されるフラグメントが挙げられるが、これらに限定されない。これ
らのフラグメントには、Fabフラグメント、Fab’フラグメント、F(ab’)2フ
ラグメントおよび単鎖Fv(scFv)フラグメントがある。用語「抗体」の範囲内には
、配列を改変されているが、抗原に特異的に結合し得るままである抗体もある。改変され
た抗体の例は、種間キメラ抗体およびヒト化抗体;抗体融合物;およびヘテロマー抗体複
合体(例えば、二価抗体(diabody)(二重特異性抗体)、単鎖二価抗体、および
細胞内発現抗体(intrabody))(例えば、Marasco(編),Intra
cellular Antibodies:Research and Disease
Applications,Springer−Verlag New York,I
nc.(1998)(ISBN:3540641513)(その開示は、本明細書中にそ
の全体が参考として援用される)を参照のこと)。
【0037】
本明細書中で使用される場合、用語「変異」とは、遺伝子産物(例えば、グリコシル化
関連酵素)の核酸配列またはアミノ酸配列における任意の変化をいう。
【0038】
用語「ポリヌクレオチド」または「核酸分子」とは、少なくとも10塩基長のヌクレオ
チドのポリマー形態をいう。この用語は、DNA分子(例えば、cDNAまたはゲノムD
NAもしくは合成DNA)およびRNA分子(例えば、mRNAまたは合成RNA)、な
らびにDNAまたはRNA含有非天然ヌクレオチドアナログ、非ネイティブヌクレオシド
間結合、またはその両方を含む。核酸は、任意のトポロジー的コンホメーションである得
る。例えば、核酸は、一本鎖、二本鎖、三本鎖、四重、部分的に二本鎖、分枝、ヘアピン
、環状または南京錠型のコンホメーションであり得る。この用語は、DNAの一本鎖形態
および二本鎖形態を含む。
【0039】
他に示さなければ、「配列番号Xを含む核酸」とは、その核酸の少なくとも一部が、(
i)配列番号Xの配列、または(ii)配列番号Xに相補的な配列のいずれかを有する核
酸をいう。これら2つの間の選択は、状況により必然的に決められる。例えば、核酸がプ
ローブとして使用される場合、2つの間の選択は、そのプローブが所望の標的に相補的で
ある必要性により必然的に決められる。
【0040】
「単離された」または「実質的に純粋な」核酸またはポリヌクレオチド(例えば、RN
A、DNAまたは混合ポリマー)は、天然の宿主細胞においてネイティブポリヌクレオチ
ドに自然に付随する他の細胞成分(例えば、リボソーム、ポリメラーゼ、およびそれが天
然で関連するゲノム配列)から実質的に分離された、核酸またはポリヌクレオチドをいう
。この用語は、(1)その天然に存在する環境から除去されたか、(2)その「単離され
たポリヌクレオチド」が天然に見出されるポリヌクレオチドの全てまたは一部を伴わない
か、(3)天然では連結されていないポリヌクレオチドに作動可能に連結されているか、
または(4)天然には存在しない、核酸またはポリヌクレオチドを包含する。用語「単離
された」または「実質的に純粋な」はまた、組み換え体もしくはクローン化されたDNA
単離物、化学的に合成されたポリヌクレオチドアナログ、または異種系により生物学的に
合成されたポリヌクレオチドアナログを言及する際に使用され得る。
【0041】
しかし、「単離された」は、そのように記載される核酸またはポリヌクレオチド自体が
、そのネイティブ環境から物理的に除去されたことを必ずしも必要とするわけではない。
例えば、生物のゲノム中の内因性核酸配列は、異種配列(すなわち、この内因性核酸配列
に天然では隣接していない配列)が、内因性核酸配列に隣接して配置されて、その結果こ
の内因性核酸配列の発現が変更される場合に、本明細書中で「単離された」とみなされる
。例として、非ネイティブプロモーター配列は、ヒト細胞のゲノムにおいて遺伝子のネイ
ティブプロモーターと(例えば、相同組み換えにより)置換され得、その結果、この遺伝
子が変更された発現パターンを有する。この遺伝子は、ここで「単離された」となる。な
ぜなら、これは、天然で隣接する配列の少なくとも一部から分離されているからである。
【0042】
核酸はまた、ゲノム中の対応する核酸に対する、天然に存在しない任意の改変を含む場
合に「単離された」とみなされる。例えば、内因性コード配列は、例えばヒトの介入によ
り人工的に導入された、挿入、欠失または点変異を含む場合に、「単離された」とみなさ
れる。「単離された核酸」はまた、異種部位において宿主細胞染色体に組み込まれた核酸
、エピソームとして存在する核酸構築物を含む。さらに、「単離された核酸」は、他の細
胞物質を実質的に含み得ないか、または組み換え技術により産生された場合に培養培地を
実質的に含み得ないか、または化学的に合成された場合に化学的前駆体もしくは他の化学
物質を実質的に含み得ない。
【0043】
本明細書中で使用される場合、用語、参照核酸配列の「縮重改変体」は、標準の遺伝子
コードに従って翻訳され得、参照核酸配列から翻訳されるアミノ酸配列と同一のアミノ酸
配列を提供する核酸配列を包含する。
【0044】
核酸配列の関係で、用語「パーセント配列同一性」または「同一な」は、最大一致で整
列された場合に同一である2つの配列中の残基をいう。配列同一性比較の長さは、少なく
とも約9ヌクレオチド、通常は少なくとも約20ヌクレオチド、より通常は少なくとも約
24ヌクレオチド、代表的には少なくとも約28ヌクレオチド、より代表的には少なくと
も約32ヌクレオチド、および好ましくは少なくとも約36以上のヌクレオチドのストレ
ッチにわたり得る。ヌクレオチド配列同一性を測定するのに使用され得る多くの異なるア
ルゴリズムが、当該分野で公知である。例えば、ポリヌクレオチド配列は、FASTA、
Gap、またはBestfitを用いて比較され得、これらは、Wisconsin P
ackage Version 10.0,Genetics Computer Gr
oup(GCG),Madison,Wisconsinのプログラムである。FAST
Aは、問い合わせ(query)配列と検索(search)配列との間の最良の重複領
域のアラインメントおよびパーセント配列同一性を提供する(Pearson,1990
(本明細書中で参考として援用される))。例えば、核酸配列間のパーセント配列同一性
は、本明細書中で参考として援用される、GCGバージョン6.1に提供されるように、
FASTAを用いて(そのデフォルトパラメーター(ワードサイズ6およびスコアリング
マトリクスについてのNOPAM因子)を用いて)決定され得るかまたはGapを用いて
(そのデフォルトパラメーターを用いて)決定され得る。
【0045】
用語「実質的に相同」または「実質的に同一」は、上記のような任意の周知の配列同一
性アルゴリズム(例えば、FASTA、BLAST、またはGap)により測定される場
合、核酸またはそのフラグメントを参照する場合、別の核酸(またはその相補鎖)と、適
切なヌクレオチド挿入または欠失と共に最適に整列される場合、少なくとも約50%、よ
り好ましくは60%のヌクレオチド塩基、通常は少なくとも約70%、より通常は少なく
とも約80%、好ましくは少なくとも約90%、およびより好ましくは少なくとも約95
%、96%、97%、98%または99%のヌクレオチド塩基においてヌクレオチド配列
同一性が存在することを示す。
【0046】
あるいは、実質的な相同性または類似性は、核酸またはそのフラグメントが、別の核酸
、別の核酸の鎖、またはその相補鎖に、ストリンジェントなハイブリダイゼーション条件
下でハイブリダイズする場合に存在する。核酸のハイブリダイゼーション実験の文脈にお
ける、「ストリンジェントなハイブリダイゼーション条件」および「ストリンジェントな
洗浄条件」は、多数の異なる物理的パラメーターに依存する。核酸ハイブリダイゼーショ
ンは、当業者によって容易に理解されるように、塩濃度、温度、溶媒、ハイブリダイズす
る種の塩基組成、相補領域の長さ、およびハイブリダイズする核酸の間のヌクレオチド塩
基のミスマッチの数のような条件によって、影響を受ける。当業者は、どのようにこのよ
うなパラメーターを変更して、特定のストリンジェンシーのハイブリダイゼーションを達
成するかを知っている。
【0047】
一般に、「ストリンジェントなハイブリダイゼーション」は、特定のDNAハイブリッ
ドについての熱的融点(Tm)より約25℃低い温度で、特定のセットの条件下で実施さ
れる。「ストリンジェントな洗浄」は、特定のDNAハイブリッドについてのTmより約
5℃低い温度で、特定のセットの条件下で実施される。Tmとは、標的配列の50%が、
完全にマッチするプローブにハイブリダイズする温度である。Sambrookら(前出
)、9.51頁(本明細書中に参考として援用される)を参照のこと。本明細書中での目
的のために、「高ストリンジェンシーの条件」とは、溶液相でのハイブリダイゼーション
について、6×SSC(ここで、20×SSCは、3.0M NaClおよび0.3Mク
エン酸ナトリウムを含有する)、1%SDS中65℃で8〜12時間の水性ハイブリダイ
ゼーション(すなわち、ホルムアミドを含まない)、続いて0.2×SSC、0.1%S
DS中65℃で20分間の2回の洗浄と定義される。65℃でのハイブリダイゼーション
は、ハイブリダイズする配列の長さおよび同一性の百分率を含む、多数の要因に依存して
、異なる速度で起こることが、当業者によって理解される。
【0048】
本発明の核酸(ポリヌクレオチドともまた称される)は、RNA、cDNA、ゲノムD
NA、ならびに上記のものの合成形態および混合ポリマーの、センス鎖とアンチセンス鎖
との両方を包含し得る。これらは、当業者によって容易に理解されるように、化学的にか
もしくは生化学的に改変され得るか、または非天然もしくは誘導体化されたヌクレオチド
塩基を含み得る。このような改変としては、例えば、標識、メチル化、1つ以上の天然に
存在するヌクレオチドのアナログでの置換、ヌクレオチド間改変(例えば、非荷電結合(
例えば、メチルホスホネート、ホスホトリエステル、ホスホロアミデート、カルバミドな
ど)、荷電結合(例えば、ホスホロチオエート、ホスホロジチオエートなど)、ペンダン
ト部分(例えば、ポリペプチド)、インターカレーター(例えば、アクリジン、ソラレン
など)、キレーター、アルキレーター、および改変された結合(例えば、αアノマー核酸
など))が挙げられる。水素結合および他の化学的相互作用を介して、指定された配列に
結合する能力において、ポリヌクレオチドを模倣する合成分子もまた含まれる。このよう
な分子は、当該分野において公知であり、そして例えば、分子の骨格においてペプチド結
合がホスフェート結合を置換する分子が挙げられる。
【0049】
用語「変異した」とは、核酸配列に対して適用される場合、核酸配列におけるヌクレオ
チドが、参照核酸配列と比較して、挿入、欠失、または変化し得ることを意味する。単一
の変化が、遺伝子座においてなされ得るか(点変異)、または複数のヌクレオチドが、単
一の遺伝子座において、挿入、欠失、または変化され得る。さらに、1つ以上の変更が、
核酸配列における任意の数の遺伝子座においてなされ得る。核酸配列は、当該分野におい
て公知の任意の方法によって変異され得、この方法としては、変異誘発技術(例えば、「
誤りやすいPCR」(DNAポリメラーゼのコピーの忠実度が低く、その結果、高い割合
の点変異が、PCR産物の全長に沿って得られる条件下でPCRを実施するためのプロセ
ス、例えば、Leung,D.W.ら,Technique,1,11−15頁(198
9)およびCaldwell,R.C.およびJoyce G.F.,PCR Meth
ods Applic.,2,28−33頁(1992)を参照のこと);および「オリ
ゴヌクレオチド特異的変異誘発」(目的の任意のクローニングされたDNAセグメントに
おける部位特異的変異の発生を可能にするプロセス。例えば、Reidhaar−Ols
on,J.F.およびSauer,R.T.ら、Science,241,53−57頁
(1988)を参照のこと))が挙げられるが、これらに限定されない。
【0050】
用語「ベクター」とは、本明細書中で使用される場合、核酸分子が結合していた別の核
酸を移動し得る核酸分子をいうことが意図される。1つの型のベクターは、「プラスミド
」であり、これは、内部にさらなるDNAセグメントが連結され得る、環状の二重鎖DN
Aループをいう。他のベクターとしては、コスミド、細菌性人工染色体(BAC)および
酵母人工染色体(YAC)が挙げられる。別の型のベクターは、ウイルスベクターであり
、ここで、さらなるDNAセグメントが、ウイルスゲノム内に連結され得る(以下にさら
に詳細に議論される)。特定のベクターは、それらが導入される宿主細胞において、自律
的に複製し得る(例えば、宿主細胞において機能する複製起点を有するベクター)。他の
ベクターは、宿主細胞への導入の際に、宿主細胞のゲノムに統合され得、そしてこれによ
って、宿主ゲノムとともに複製され得る。さらに、特定の好ましいベクターは、それらが
作動可能に連結した遺伝子の発現を導き得る。このようなベクターは、本明細書中で、「
組換え発現ベクター」(または単に、「発現ベクター」)と称される。
【0051】
「作動可能に連結された」発現制御配列とは、発現制御配列が、目的の遺伝子を制御す
るために、この目的の遺伝子と連続的である結合、およびこの目的の遺伝子を制御するた
めに、トランスでかまたはある距離で働く発現制御配列をいう。
【0052】
用語「発現制御配列」とは、本明細書中で使用される場合、それらが作動可能に連結し
たコード配列の発現に影響を与えるために必要な、ポリヌクレオチド配列をいう。発現制
御配列とは、核酸配列の転写、転写後事象および翻訳を制御する配列をいう。発現制御配
列は、適切な転写開始配列、終結配列、プロモーター配列およびエンハンサー配列;スプ
ライシングおよびポリアデニル化シグナルのような、効率的なRNAプロセシングシグナ
ル;細胞質mRNAを安定化する配列;翻訳効率を増強する配列(例えば、リボソーム結
合部位);タンパク質安定性を増強する配列;ならびに望ましい場合、タンパク質分泌を
増強する配列を含む。このような制御配列の性質は、宿主生物に依存して異なる;原核生
物においては、このような制御配列は、一般に、プロモーター、リボソーム結合部位、お
よび転写終結配列を含む。用語「制御配列」は、最低でも、その存在が発現のために必須
である全ての成分を含むことを意図され、そしてまた、その存在が有利であるさらなる成
分(例えば、リーダー配列および融合パートナー配列)を含み得る。
【0053】
用語「組換え宿主細胞」(または単に「宿主細胞」)とは、本明細書中で使用される場
合、組換えベクターが導入された細胞をいうことが意図される。このような用語は、特定
の被験体細胞のみでなく、そのような細胞の子孫をもまたいうことが意図されることが、
理解されるべきである。特定の改変が、変異または環境の影響のいずれかに起因して、引
き続く世代において起こり得るので、このような子孫は、実際に、親細胞と同一ではない
かもしれないが、本明細書中において使用される場合の用語「宿主細胞」の範囲内に依然
として含まれる。組換え宿主細胞は、培養中で増殖された、単離された細胞または細胞株
であり得か、あるいは生存組織または器官内に存在する細胞であり得る。
【0054】
用語「ペプチド」とは、本明細書中で使用される場合、短いポリペプチド(例えば、代
表的に約50アミノ酸長未満、そしてより代表的には約30アミノ酸長未満のポリペプチ
ド)をいう。この用語は、本明細書中において使用される場合、構造および従って生物学
的機能を模倣する、アナログおよび模倣物を包含する。
【0055】
用語「ポリペプチド」は、天然に存在するタンパク質および天然には存在しないタンパ
ク質の両方、ならびにそのフラグメント、変異体、誘導体およびアナログを包含する。ポ
リペプチドは、モノマーであってもポリマーであってもよい。さらに、ポリペプチドは、
多数の異なるドメインを含み得、これらのドメインの各々は、1つ以上の異なる活性を有
する。
【0056】
用語「単離されたタンパク質」または「単離されたポリペプチド」とは、その誘導の起
源または供給源のおかげで、(1)ネイティブな状態で付随する天然に会合する成分と会
合していないか、(2)天然には見出されない純度で存在する場合、純度が他の細胞材料
の存在に関して調節され得る(例えば、同じ種由来の他のタンパク質を含まない)か、(
3)異なる種由来の細胞によって発現されるか、あるいは(4)天然には存在しない(例
えば、天然に見出されるポリペプチドのフラグメントであるか、または天然には見出され
ないアミノ酸のアナログもしくは誘導体または標準的なペプチド結合以外の結合を含む)
タンパク質またはポリペプチドである。従って、化学的に合成されるかまたは天然に起源
とする細胞とは異なる細胞系において合成されるポリペプチドは、その天然に会合する成
分から「単離される」。ポリペプチドまたはタンパク質はまた、当該分野において周知の
タンパク質精製技術を使用する単離によって、天然に会合する成分を実質的に含まなくさ
れ得る。このように定義される場合、「単離された」とは、このように記載されるタンパ
ク質、ポリペプチド、ペプチドまたはオリゴペプチドが、そのネイティブの環境から物理
的に取り出されていることを、必ずしも必要としない。
【0057】
用語「ポリペプチドフラグメント」とは、本明細書中において使用される場合、全長ポ
リペプチドと比較して、アミノ末端および/またはカルボキシ末端の欠失を有するポリペ
プチドをいう。好ましい実施形態において、ポリペプチドフラグメントとは、そのフラグ
メントのアミノ酸配列が、天然に存在する配列における対応する部分と同一である、連続
的な配列である。フラグメントは、代表的に、少なくとも5アミノ酸長、6アミノ酸長、
7アミノ酸長、8アミノ酸長、9アミノ酸長、または10アミノ酸長であり、好ましくは
、少なくとも12アミノ酸長、14アミノ酸長、16アミノ酸長または18アミノ酸長で
あり、最も好ましくは、少なくとも20アミノ酸長であり、より好ましくは、少なくとも
25アミノ酸長、30アミノ酸長、35アミノ酸長、40アミノ酸長または45アミノ酸
長であり、なおより好ましくは、少なくとも50アミノ酸長または60アミノ酸長であり
、そしてなおより好ましくは、少なくとも70アミノ酸長である。
【0058】
「改変された誘導体」とは、一次構造の配列において実質的に相同であるが、例えば、
インビボまたはインビトロで、化学的改変および生化学的改変を含む、ポリペプチドまた
はそのフラグメント、あるいはネイティブなポリペプチドにおいては見出されないアミノ
酸を組み込むポリペプチドまたはそのフラグメントをいう。このような改変としては、例
えば、当業者によって容易に理解されるような、アセチル化、カルボキシル化、ホスホリ
ル化、グリコシル化、ユビキチン化、標識(例えば、放射性核種を用いる)、および種々
の酵素的改変が挙げられる。ポリペプチドを標識するための種々の方法、およびこのよう
な目的に有用な置換基または標識は、当該分野において周知であり、そして放射性同位体
(例えば、125I、32P、35S、および3H)、標識された抗リガンドに結合するリガンド
(例えば、抗体)、発蛍光団、化学発光剤、酵素、および標識されたリガンドに対する特
異的結合対メンバーとして働き得る抗リガンドが挙げられる。標識の選択は、必要とされ
る感度、プライマーとの結合体化の容易さ、安定性の要件、および利用可能な機器に依存
する。ポリペプチドを標識するための方法は、当該分野において周知である。Ausub
elら、1992(本明細書中に参考として援用される)を参照のこと。
【0059】
用語「融合タンパク質」とは、異種アミノ酸配列に結合したポリペプチドまたはフラグ
メントを含む、ポリペプチドをいう。融合タンパク質は、有用である。なぜなら、これら
は、2つ以上の異なるタンパク質由来の2つ以上の所望の機能的エレメントを含むように
構築され得るからである。融合タンパク質は、目的のポリペプチド由来の少なくとも10
個の連続したアミノ酸、より好ましくは、少なくとも20個または30個のアミノ酸、な
おより好ましくは、少なくとも40個、50個、または60個のアミノ酸、なおより好ま
しくは、少なくとも75個、100個、または125個のアミノ酸を含む。融合タンパク
質は、ポリペプチドまたはそのフラグメントを、異なるタンパク質またはペプチドをコー
ドする核酸配列とインフレームでコードする核酸配列を構築し、次いで、融合タンパク質
を発現させることによって、組換え的に産生され得る。あるいは、融合タンパク質は、ポ
リペプチドまたはそのフラグメントを、別のタンパク質に架橋させることによって、化学
的に産生され得る。
【0060】
用語「非ペプチドアナログ」とは、参照ポリペプチドの特性に類似の特性を有する化合
物をいう。非ペプチド化合物はまた、「ペプチド模倣物(peptide mimeti
c)」または「ペプチド模倣物(peptidomimetic)」と称され得る。例え
ば、Jones(1992)Amino Acid and Peptide Synt
hesis,Oxford University Press;Jung(1997)
Combinatorial Peptide and Nonpeptide Lib
raries:A Handbook John Wiley;Badanszkyら(
1993)Peptide Chemistry−−A Practical Text
book,Springer Verlag;「Synthetic Peptides
:A Users Guide」、G.A.Grant編、W.H.Freeman a
nd Co.,1992;Evansら、J.Med.Chem.30:1229(19
87);Fauchere,J.Adv.Drug Res.15:29(1986);
VeberおよびFreidinger TINS 392頁(1985);ならびに上
記のものの各々において引用される参考文献(これらは、本明細書中に参考として援用さ
れる)を参照のこと。このような化合物は、しばしば、コンピュータ化された分子モデリ
ングの補助によって開発される。本発明の有用なペプチドに構造的に類似のペプチド模倣
物を使用して、等価な効果を生じ得、従って、これらのペプチド模倣物は、本発明の一部
であると意図される。
【0061】
「ポリペプチド変異型」または「ムテイン」は、ネイティブタンパク質または野生型タ
ンパク質のアミノ酸配列に比べて、1つ以上のアミノ酸の挿入、重複、欠失、再配置また
は置換を含むポリペプチド配列をいう。ムテインは、アミノ末端またはカルボキシ末端の
いずれかまたは両方で、天然に存在するタンパク質および/またはアミノ酸配列の短縮型
の配列において、1つの位置で単一アミノ酸が、別のアミノ酸に変化される1つ以上のア
ミノ酸点変異を有し得、1つ以上のアミノ酸が、それぞれ、挿入されるか、もしくは欠失
される1つ以上の挿入および/または欠失を有し得る。ムテインは、天然に存在するタン
パク質に比べて、同じ生物学的活性を有し得るが、おそらく異なる生物学的活性を有し得
る。例えば、ムテインは、増加したニューロン結合活性もしくは減少したニューロン結合
活性またはNgR結合活性を有し得る。本発明の好ましい実施形態において、(例えば、
MAG Ig様ドメイン5中の)ムテインであるMAG誘導体は、内因性MAGまたは可
溶性野生型MAGに比べて、減少したニューロン成長阻害活性を有する。
【0062】
ムテインは、その野生型対応物に対して少なくとも70%の全配列相同性を有する。野
生型タンパク質に対して80%、85%または90%の全配列相同性を有するムテインは
、さらにより好ましい。さらにより好ましい実施形態において、ムテインは、95%の配
列同一性、なおより好ましくは97%の、さらにより好ましくは98%の、およびさらに
より好ましくは99%の全配列同一性を示す。配列相同性は、任意の共通配列分析アルゴ
リズム(例えば、GapまたはBestfit)によって測定され得る。
【0063】
好ましいアミノ酸置換は、以下のものである:(1)タンパク質分解に対する感受性を
減少する置換、(2)酸化に対する感受性を減少する置換、(3)タンパク質複合体を形
成するための結合親和性を変更する置換、(4)結合親和性または酵素活性を変更する置
換、および(5)このようなアナログの他の物理化学的特徴または機能的特徴を付与する
かまたは改変する置換。
【0064】
本明細書中で使用する場合、20の従来のアミノ酸およびこれらの省略形は、従来の慣
例に従う。Immunology−A Synthesis(第2版,E.S.Golu
bおよびD.R.Gren編,Sinauer Associates,Sunderl
and,Mass.(1991))(これは、参考として本明細書中に援用される)を参
照のこと。20の従来のアミノ酸、非天然アミノ酸(例えば、α−,α−二置換アミノ酸
、N−アルキルアミノ酸)、および他の非従来的なアミノ酸の立体異性体(例えば、D−
アミノ酸)はまた、本発明のポリペプチドのための適切な成分であり得る。非従来的なア
ミノ酸の例としては、以下:4−ヒドロキシプロリン、γ−カルボキシグルタミン酸、ε
−N,N,N−トリメチルリジン、ε−N−アセチルリジン、O−ホスホセリン、N−ア
セチルセリン、N−ホルミルメチオニン、3−メチルヒスチジン、5−ヒドロキシリジン
、s−N−メチルアルギニン、および他の類似のアミノ酸およびイミノ酸(例えば、4−
ヒドロキシプロリン)が挙げられる。本明細書中で使用されるポリペプチド表記において
、標準的な使用および慣例に従って、左手方向は、アミノ末端方向であり、右手方向は、
カルボキシ末端方向である。
【0065】
タンパク質は、このタンパク質をコードする核酸配列が、第2のタンパク質をコードす
る核酸配列に類似する配列を有する場合、「相同性」を有するか、または第2のタンパク
質に対して「相同」である。あるいは、タンパク質は、2つのタンパク質が、「類似する
」アミノ酸配列を有する場合、第2のタンパク質に対して相同性を有する。(従って、用
語「相同性タンパク質」は、2つのタンパク質が、類似するアミノ酸配列を有することを
意味するように規定される)。好ましい実施形態において、相同タンパク質は、野生型タ
ンパク質に対して60%の配列相同性を示し、より好ましくは、70%の配列相同性を示
す。野生型タンパク質に対して80%、85%または90%の配列相同性を示す相同タン
パク質が、さらにより好ましい。さらにより好ましい実施形態において、相同タンパク質
は、95%、97%、98%または99%の配列同一性を示す。本明細書中で使用される
場合、アミノ酸配列の2つの領域の間の相同性(特に予測された構造類似性に関して)は
、機能が類似することを意味するとして解釈される。
【0066】
「相同」が、タンパク質またはペプチドに関して使用される場合、同一ではない残基位
置が、保存的アミノ酸置換によってしばしば異なることが理解される。「保存的アミノ酸
置換」は、類似する化学的特性(例えば、電荷または疎水性度)を有する側鎖(R基)を
有する別のアミノ酸残基によって置換される置換である。一般的に、保存的アミノ酸置換
は、タンパク質の機能特性を実質的には変更しない。2つ以上のアミノ酸配列が、保存的
置換によって互いに異なる場合において、パーセント配列同一性または相同性の程度は、
置換の保存的性質を修正するように上方に調整され得る。この調整をするための手段は、
当業者に周知である(例えば、Pearsonら、1994(本明細書中に参考として援
用される)を参照のこと)。
【0067】
以下の6つの群は、各々互いに対して保存的な置換であるアミノ酸を含む:1)セリン
(S)、スレオニン(T);2)アスパラギン酸(D)、グルタミン酸(E);3)アス
パラギン(N)、グルタミン(Q);4)アルギニン(R)、リジン(K);5)イソロ
イシン(I)、ロイシン(L)、メチオニン(M)、アラニン(A)、バリン(V)、お
よび6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W)。
【0068】
ポリペプチドに対する配列相同性(パーセント配列同一性としても参照される)は、代
表的に、配列分析ソフトウェアを使用して決定される。例えば、Sequence An
alysis Software Package of the Genetics
Computer Group(GCG),University of Wiscon
sin Biotechnology Center,910 University
Avenue,Madison,Wisconsin 53705を参照のこと。タンパ
ク質分析ソフトウェアは、保存的アミノ酸置換を含む、種々の置換、欠失および他の改変
に対して割り当てられる相同性の尺度を使用して類似の配列を符合させる。例えば、GC
Gは、「Gap」および「Bestfit」のようなプログラムを備え、これは、デフォ
ルトパラメーターと共に使用され、密接に関連するポリペプチド(例えば、異なる生物種
からの相同性ポリペプチド)の間、あるいは野生型タンパク質とその変異型との間の配列
相同性または配列同一性を決定し得る。例えば、GCG Version 6.1を参照
のこと。
【0069】
阻害性分子配列を異なる生物由来の膨大な配列を含むデータベースと比較する場合、好
ましいアルゴリズムは、コンピュータープログラムBLAST(Altschul,S.
F.ら(1990)J.Mol.Biol.215:403−410;GishおよびS
tates(1993)Nature Genet.3:266−272;Madden
,T.L.ら(1996)Meth.Enzymol.266:131−141;Alt
schul,S.F.ら(1997)Nucleic Acids Res.25:33
89−3402;Zhang,J.およびMadden,T.L.(1997)Geno
me Res.7:649−656)、特に、blastpまたはtblatn(Alt
schulら、1997)である。BLASTpについての好ましいパラメーターは、
期待値:10(デフォルト)
フィルター:seg(デフォルト)
ギャップを開くためのコスト:11(デフォルト)
ギャップを伸長するためのコスト:1(デフォルト)
最大整列:100(デフォルト)
ワードサイズ:11(デフォルト)
記述の数:100(デフォルト)
ペナルティーマトリクス:BLOWSUM62
である。
【0070】
相同性について比較されるポリペプチド配列の長さは、一般的に、少なくとも約16ア
ミノ酸残基であり、通常には、少なくとも約20残基であり、より通常には、少なくとも
約24残基であり、代表的には、少なくとも約28残基であり、好ましくは、約35残基
を超える。膨大な数の異なる生物由来の配列を含むデータベースを検索する場合、アミノ
酸配列を比較することが好ましい。使用するアミノ酸配列を検索するデータベースは、当
該分野で公知のblastp以外のアルゴリズムによって決定され得る。例えば、ポリペ
プチド配列は、GCG Version 6.1中のプログラムFASTAを使用して比
較され得る。FASTAは、アライメントおよび質問配列と検索配列との間の最良の重複
の領域のアライメントおよびパーセント配列同一性を提供する(Pearson,199
0(参考として本明細書中に援用される))。例えば、アミノ酸配列間の間のパーセント
配列同一性は、GCG version 6.1(本明細書中に参考として援用される)
に提供されるようなそのデフォルトパラメーター(2のワードサイズおよびPAM250
スコアリングマトリクス)を用いてFASTAを使用して決定され得る。
【0071】
「特異的な結合」は、その環境における他の分子に対する結合に優先して、互いに結合
する2つの分子の能力をいう。代表的には、「特異的結合」は、少なくとも2倍、より代
表的には少なくとも10倍、しばしば少なくとも100倍、反応中の偶然の結合に対して
区別する。代表的には、特異的な結合反応の親和性または親和力は、少なくとも約10-7
M(例えば、少なくとも約10-8Mまたは10-9M)である。
【0072】
用語「領域」は、本明細書中で使用される場合、生体分子の一次構造の物理的に連続し
た部分をいう。タンパク質の場合において、領域は、このタンパク質のアミノ酸配列の連
続的領域によって規定される。
【0073】
用語「ドメイン」は、本明細書中で使用される場合、生体分子の公知の機能または推測
の機能に寄与する生体分子の構造をいう。ドメインは、その領域または部分と同程度に広
がり得;ドメインはまた、生体分子の別個の非連続的な領域を含み得る。タンパク質ドメ
インの例としては、Igドメイン、細胞外ドメイン、膜貫通ドメイン、および細胞質ドメ
インが挙げられるがこれらに限定されない。
【0074】
本明細書中で使用される場合、用語「分子」は、任意の化合物を意味し、これとしては
、低分子、ペプチド、タンパク質、糖、ヌクレオチド、核酸、脂質などが挙げられるがこ
れらに限定されず、このような化合物は、天然であっても合成であってもよい。
【0075】
他に定義されない場合、本明細書中で使用される全ての技術用語および科学用語は、本
発明に関する分野の当業者によって通常理解される意味と同じ意味を有する。例示的な方
法および材料は、以下に示されるが、本明細書中に記載される方法および材料と類似する
かまたは等価な方法および材料はまた、本発明の実施において使用され得、そして、これ
らは、当業者に明らかである。全ての刊行物および本明細書中に記載される他の参考は、
その全体を参考として援用される。矛盾する場合、定義を含む本明細書は、制御する。材
料、方法、および実施例は、例示のみであり、そして、限定を意図されない。
【0076】
本明細書および特許請求の範囲の全体にわたって、単語「含む(comprise)」
またはバリエーション(例えば、「含む(comprises)」または「含む(com
prising)」)は、示された整数または整数の群の含入を意味するが、任意の他の
整数または整数の群の排除を意図されないことが理解される。
【0077】
(ヒト様N−グリカンの生成に対して改変された脂質連結オリゴ糖を有する宿主の操作
または選択)
本発明は、非ヒト真核生物宿主細胞において、ヒト様糖タンパク質を産生するための方
法を提供する。本発明は、alg遺伝子活性(すなわち、alg活性(非真菌宿主細胞中
の等価な酵素活性を含む))を低下するかまたは激減する非ヒト真核生物宿主細胞を作製
する工程または使用する工程、および宿主細胞に少なくとも1つのグリコシダーゼ活性を
導入する工程を包含する。好ましい実施例において、グリコオシダーゼ活性は、宿主細胞
内の1つ以上のマンノシダーゼ活性の発現によって、例えば、マンノシダーゼ活性の活性
化によって、または宿主細胞におけるマンノシダーゼ活性の核酸分子からの発現によって
、導入される。
【0078】
別の実施形態において、本方法は、糖残基を、脂質連結オリゴ糖前駆体の1,6アーム
に転位する1つ以上の酵素の活性を低下されたか、または激減された宿主細胞を作製する
工程またはそれを使用する工程を包含する(図1)。本発明の宿主細胞は、糖残基(例え
ば、マンノシレート)を、脂質連結オリゴ糖前駆体の1,6アームに転位する酵素をコー
ドする1つ以上の遺伝子中に変異を導入することについて選択されるかまたはそのことに
よって操作される。糖残基は、より好ましくはマンノースであり、好ましくはグルコース
、GlcNAc、ガラクトース、シアル酸、フコースまたはGlcNAcリン酸残基であ
る。好ましい実施形態において、脂質連結オリゴ糖前駆体の1,6アームをマンノシル化
する1つ以上の酵素の活性は、低下するかまたは激減する。本方法は、少なくとも1つの
グリコシダーゼ活性を宿主細胞に導入する工程をさらに包含し得る(以下を参照のこと)
。
【0079】
なお別の実施形態において、本発明は、非ヒト宿主においてヒト様糖タンパク質を産生
するための方法を提供し、ここで、糖タンパク質は、トリマンノースコア構造に結合され
る少なくとも2つのGlcNAcを有するN−グリカンを含む。
【0080】
上記の各々の実施形態において、本方法は、宿主細胞を作製する工程に関し、ここで、
脂質連結オリゴ糖前駆体は、ManXGlcNAc2構造中で濃縮され、ここで、Xは、3
、4、または5である(図2)。これらの構造は、宿主細胞のER中で、オリゴサッカリ
ルトランスフェラーゼによって新生ポリペプチド鎖上に転位され、次いで、グリコシダー
ゼ(例えば、α−マンノシダーゼ)およびグリコシルトランスフェラーゼ(例えば、Gn
T1)での処理によってプロセスされ、GlcNAcManXGlcNAc2コア構造を有
するN−グリカンを生成し、ここで、Xは、3、4または5であり、そして好ましくは3
である(図2および図3)。図2に示されるように、GlcNAcManXGlcNAc2
コア構造を有し、Xが3を超えるN−グリカンは、適用可能な場合、例えば、α−1,3
マンノシダーゼ活性および/またはα−1,2−1,3マンノシダーゼ活性で処理するこ
とによって、GlcNAcMan3GlcNAc2に転換され得る。
【0081】
グリコシルトランスフェラーゼ(例えば、GnTII)での処理によるGlcNAcM
an3GlcNAc2のさらなるプロセシングは、GlcNAc2Man3GlcNAc2コ
ア構造を生成し、これは次いで、所望の場合、例えば、エキソビボ処理によって、または
一連のグリコシル化酵素(グリコシルトランスフェラーゼ、糖トランスポーターおよびマ
ンノシダーゼを含む(以下を参照のこと))の宿主細胞内での異種発現によって、改変さ
れて、ヒト様N−グリカンになり得る。本発明に従って生成され得る好ましいヒト様糖タ
ンパク質であって、7個以下、または3個以下のマンノース残基を有するN−グリカンを
含む糖タンパク質は、ガラクトース、GlcNAc、シアル酸およびフコースからなる群
から選択される1つ以上の糖を含み;そしてNeuNAc−Gal−GlcNAc−Ma
nを含む少なくとも1つのオリゴ糖分枝を含む。
【0082】
一実施形態において、宿主細胞は、減退されたかまたは減少されたDol−P−Man
:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼ活性を有し、この
活性は、ERのルミナル(luminal)側におけるMan5GlcNAc2−PP−D
olからMan6GlcNAc2−PP−Dolへの第1のマンノシル化工程に関与する(
例えば、ALG3 図1;図2)。S.cerevisiaeにおいて、この酵素は、A
LG3遺伝子によってコードされる。上記のように、漏出alg3−1変異を有するS.
cerevisiae細胞は、Man5GlcNAc2−PP−Dolを蓄積し、そしてa
lg3における欠失を有するS.serevisiae細胞は、Man5GlcNAc2構
造をER内の新生ポリペプチド鎖上に移動させるようである。従って、この実施形態にお
いて、宿主細胞は、Man5GlcNAc2構造に富化されたN−グリカンを蓄積し、これ
は次いで、グリコシダーゼを用いる(例えば、α−1,2−マンノシダーゼ、α−1,3
マンノシダーゼ、またはα−1,2−1,3マンノシダーゼ活性を用いる)処理によって
GlcNAc2Man3GlcNAc2に変換され得る(図2) 実施例1に記載されるよ
うに、縮重プライマーを、S.cerevisiae、D.melanogasterお
よびヒト(H.sapiens)由来のAlg3タンパク質配列の整列に基づいて設計し
(図4および5)、そしてこれを使用して、P.pastorisゲノムDNA由来の産
物を増幅した。得られたPCR産物を、プローブとして使用して、S.cerevisi
ae ALG3遺伝子に対して35%の全配列同一性および53%の配列類似性を有する
タンパク質をコードするオープンリーディングフレーム(ORF)を含むP.pasto
risゲノムクローンを同定しそして単離した(図6および7)。このP.pastor
is遺伝子を、本明細書中で、「PpALG3」と呼ぶ。ALG3遺伝子を、同様に同定
し、そしてK.lactisから単離した(実施例1;図8および9)。
【0083】
従って、別の実施形態において、本発明は、P.pastoris ALG3遺伝子(
図6)およびK.lactis ALG3遺伝子(図8)ならびにそれらのホモログ、改
変体および誘導体の、少なくとも45、好ましくは少なくとも50、より好ましくは少な
くとも60、最も好ましくは75以上のヌクレオチド残基を含むか、これらからなる核酸
配列を有する単離された核酸分子を提供する。本発明はまた、ストリンジェントな条件下
で、上記の核酸分子にハイブリダイズする核酸分子を提供する。同様に、本発明の核酸分
子によってコードされる単離されたポリペプチド(ムテイン、対立遺伝子改変体、フラグ
メント、誘導体およびアナログを含む)が、提供される(P.pastorisおよびK
.lactisのALG3遺伝子産物は、図6および8に示される)。さらに、本明細書
中にさらに記載されるように、本発明の核酸分子を含むベクター(発現ベクターを含む)
もまた提供される。
【0084】
遺伝子特異的プライマーを使用して、構築物を作製して、P.pastorisのゲノ
ムからPpALG3遺伝子を欠失させた(実施例1)。この株を使用して、Dol−P−
Man:Man5GlcNAc2−PP−Dolマンノシルトランスフェラーゼ活性を欠失
した宿主細胞を作製し、そして脂質連結Man5GlcNAc2−PP−Dol前駆体を生
成した。この脂質連結Man5GlcNAc2−PP−Dol前駆体は、新生ポリペプチド
鎖上に移動され、Man5GlcNAc2糖質構造を有するN−グリカンを生成する。
【0085】
実施例2に記載されるように、このような宿主細胞は、所望のMan3GlcNAc2コ
ア糖質構造を有するN−グリカンを生成するように、適切なマンノシダーゼの発現によっ
て操作され得る。宿主細胞におけるGnTの発現(例えば、下記のように、核酸分子また
は核酸分子のライブラリーを標的化することによって)により、改変された宿主細胞は、
Man3コア構造(すなわち、GlcNAc1Man3GlcNAc2またはGlcNAc2
Man3GlcNAc2;図3を参照のこと)の各アームに結合した1つまたは2つのGl
cNAc構造を有するN−グリカンを生成し得る。これらの構造は、本発明の方法を使用
してさらに処理されて、宿主細胞の分泌経路に入るヒト様N−グリカンまたはタンパク質
を生成し得る。
【0086】
別の実施形態において、宿主細胞は、減退されたかまたは欠失されたドリチル(dol
ichyl)−P−Man:Man6GlcNAc2−PP−ドリチルα1,2−マンノシ
ルトランスフェラーゼ活性を有し、この活性は、ERのルミナル側においてMan6Gl
cNAc2−PP−DolをMan7GlcNAc2−PP−Dolへ変換するマンノシル
化工程に関与する(上記ならびに図1および図2を参照のこと)。S.cerevisi
aeにおいて、この酵素は、ALG9遺伝子によってコードされる。alg9変異を有す
る細胞は、Man6GlcNAc2−PP−Dolを蓄積し(図2)、そしてMan6Gl
cNAc2構造をER内の新生ポリペプチド鎖上に移動させる。従って、この実施形態に
おいて、宿主細胞は、Man6GlcNAc2構造に富化されたN−グリカンを蓄積し、こ
れは次いで、α−1,2−マンノシダーゼおよびα−1,3マンノシダーゼを用いる処理
によってコアMan3構造にプロセスダウンされ得る(図3ならびに実施例3および4を
参照のこと)。
【0087】
alg9遺伝子(または等価な活性をコードする遺伝子)が欠失された宿主細胞を構築
する(実施例3を参照のこと)。ALG9(または、ALG12;以下を参照のこと)の
欠失は、1,6アーム上の、それぞれ、1つまたは2つのさらなるマンノースを有するN
−グリカンを生成する宿主細胞を作製する(図2)。N−アセチルグルコサミニルトラン
スフェラーゼII(Gn TII)に接近可能な1,6−コアマンノースを作製するため
に、これらのマンノースは、グリコシダーゼによって除去される必要がある。ERマンノ
シダーゼは、代表的に、1,6アーム上の末端1,2マンノースを除去し、続いてマンノ
シダーゼII(α1−3,6マンノシダーゼ)または他のマンノシダーゼ(例えば、α1
,2マンノシダーゼ、α1,3マンノシダーゼまたはα1−2,3マンノシダーゼ(例え
ば、Xanthomonas manihotis由来;実施例4を参照のこと))が、
1,6アームに作用し得、続いて、GnTIIが、N−アセチルグルコサミンを移動させ
て、GlcNAc2Man3を生成する(図2)。
【0088】
alg9p活性を欠く得られた宿主細胞を、1つ以上の酵素由来のα1,2−マンノシ
ダーゼ活性およびα1,3マンノシダーゼ活性を発現するように、好ましくは、宿主細胞
に導入される核酸分子からの発現によって、操作され、そしてこれは、好ましい細胞成分
に標的化される酵素を発現する(以下を参照のこと)。実施例4は、Xanthomon
as manihotis由来のこのような酵素のクローニングおよび発現を記載する。
【0089】
別の実施形態において、宿主細胞は、減退されたかまたは欠失されたドリチル−P−M
an:Man7GlcNAc2−PP−ドリチルα1,6−マンノシルトランスフェラーゼ
活性を有し、この活性は、ERのルミナル側においてMan7GlcNAc2−PP−Do
lをMan8GlcNAc2−PP−Dol(これはコアマンノース構造の1,6アーム上
のα−1,6マンノースをマンノシル化する)へ変換するマンノシル化工程に関与する(
上記ならびに図1および図2を参照のこと)。S.cerevisiaeにおいて、この
酵素は、ALG12遺伝子によってコードされる。alg12変異を有する細胞は、Ma
n7GlcNAc2−PP−Dolを蓄積し(図2)、そしてMan7GlcNAc2構造を
ER内の新生ポリペプチド鎖上に移動させる。従って、この実施形態において、宿主細胞
は、Man7GlcNAc2構造に富化されたN−グリカンを蓄積し、これは次いで、α−
1,2−マンノシダーゼおよびα−1,3マンノシダーゼを用いる処理によってコアMa
n3構造にプロセスダウンされ得る(図3ならびに実施例3および4を参照
のこと)。
【0090】
alg9変異体宿主について上で記載されるように、alg12p活性を欠く得られた
宿主細胞を、(例えば、1つ以上の酵素由来の)α1,2−マンノシダーゼ活性およびα
1,3マンノシダーゼ活性を発現するように、好ましくは、宿主細胞に導入される1つ以
上の核酸分子からの発現によって、操作され、そしてこれは、好ましい非細胞成分に標的
化される酵素活性を発現する(以下を参照のこと)。
【0091】
(必要に応じて減少した開始α−1,6マンノシルトランスフェラーゼ活性を有する宿
主の操作または選択)
好ましい実施形態において、本発明の方法は、(a)alg遺伝子の活性またはMan
3GlcNAc2(「Man3」)コア糖質構造上のα−1,6アームのN−グリカンをマ
ンノシル化する1つ以上の活性が減退または減少しており、かつ(b)(Man3コア構
造のα−1,3アーム上の)開始α−1,6−マンノシルトランスフェラーゼ(すなわち
、外側鎖マンノシル化を開始する開始特異的酵素)の活性が減退または欠失している、宿
主細胞を作製または使用することを包含する。S.cerevisiaeにおいて、この
酵素は、OCH1遺伝子によってコードされる。S.cerevisiaeにおけるoc
h1遺伝子の破壊は、N連結糖がポリマンノース外側鎖を完全に欠く表現型を生じる。真
菌株における哺乳動物型グリコシル化を得るための以前のアプローチは、OCH1の不活
性化を必要とした(例えば、Chiba,1998を参照のこと)。しかし、Och1p
酵素が、有効なマンノース外側鎖開始についてインタクトなMan8GlcNAcを必要
とする場合、本発明の宿主細胞における開始α−1,6−マンノシルトランスフェラーゼ
活性の破壊は、任意である(選択された宿主細胞に依存して)。従って、本発明に従って
選択または生成された宿主細胞(これは、7個以下のマンノース残基を有する脂質連結オ
リゴ糖を蓄積する)は、移動の後、Och1pに対する乏しい基質の可能性のある低グリ
コシル化N−グリカンを生成する(例えば、Nakayama,1997を参照のこと)
。
【0092】
(増加したグリコシルトランスフェラーゼ活性を有する宿主の操作または選択)
上記のように、グリコシル化オリゴ糖は、それらの非グリコシル化対応物よりもはるか
に高い割合で新生ポリペプチド鎖に移動されると考えられる。オリゴ糖トランスフェラー
ゼ複合体による基質認識は、脂質連結オリゴ糖のアンテナへのグルコースの付加によって
増大するようである。従って、脂質連結オリゴ糖を最適にグリコシル化し得る宿主細胞を
作製または選択することが望まれる。このような宿主細胞において、過小グリコシル化(
underglycosylation)は、グリコシル化Man5構造の新生ポリペプ
チド鎖上へのより速くより効率的な移動に起因して、実質的に減少されるかまたは消滅さ
れさえする。
【0093】
従って、本発明の別の実施形態において、この方法は、脂質連結Nグリカン前駆体がE
R中の新生ポリペプチド鎖に効率的に移動される宿主細胞を作製することに関する。好ま
しい実施形態において、移動は、脂質連結オリゴ糖の分枝上のグリコシル化レベルを増加
することによって増大され、このことにより、このオリゴ糖は、オリゴサッカリルトラン
スフェラーゼに対するより良い基質となる。
【0094】
1つの好ましい実施形態において、本発明は、ヒト様糖タンパク質を作製するための方
法を提供し、この方法は、ER中の脂質連結オリゴ糖のグリコシル化を担う1つ以上の酵
素が増加した活性を有する宿主細胞を使用する。脂質連結オリゴ糖のグリコシル化の程度
を増大するための1つの方法は、脂質連結オリゴ糖のアンテナ上へのグルコース残基の移
動を担う1つ以上の酵素を過剰発現させることである。特に、α−1,3グルコシルトラ
ンスフェラーゼ活性の増加により、グルコシル化脂質連結Man5構造の量が増加し、そ
して分泌タンパク質の過小グリコシル化を減少させるかまたは排除する。S.cerev
isiaeにおいて、この酵素は、ALG6遺伝子によってコードされる。
【0095】
Saccharomyces cerevisiae ALG6およびそのヒト対応物
は、クローニングされている(Imbach,1999;Reiss,1996)。グリ
コシル化の初期工程の進化的保存に起因して、ALG6座は、種間で相同であると予測さ
れ、そして当業者によって、配列類似性に基づいてクローニングされ得る(同じことが、
異なる種由来のALG8およびALG10座のクローニングおよび同定にあてはまる)。
さらに、異なる種由来の異なるグルコシルトランスフェラーゼが、次いで、最適な活性を
有するグルコシルトランスフェラーゼを同定するために試験され得る。
【0096】
強力なプロモーター(例えば、GAPDHプロモーター)の制御下でのALG6遺伝子
のさらなるコピーの導入および/またはALG6の発現は、グルコシル化脂質連結オリゴ
糖の程度を増加するためのいくつかの方法の1つである。P.pastoris由来のA
LG6遺伝子を、クローニングしそして発現する(実施例5)。ALG6核酸およびアミ
ノ酸配列を、図25(S.cerevisiae)および図26(P.pastoris
)について示す。これらの配列を、図27の他の真核生物ALG6配列と比較する。
【0097】
従って、本発明の別の実施形態は、脂質連結オリゴ糖のグリコシル化の程度を増大する
方法を提供し、この方法は、宿主細胞におけるα−1,3グルコシルトランスフェラーゼ
活性を増大する工程を包含する。この活性の増加は、例えば、この活性をコードする核酸
を1つ以上の異種発現制御配列と作動可能に連結することによって、この活性をコードす
る核酸配列を過剰発現することによって、達成され得る。好ましい発現制御配列としては
、転写開始配列、転写終結配列、プロモーター配列およびエンハンサー配列;RNAスプ
ライスドナーおよびポリアデニル化シグナル;mRNA安定化配列;リボソーム結合部位
;タンパク質安定化配列;ならびにタンパク質分泌配列が挙げられる。
【0098】
別の実施形態において、α−1,3−グルコシルトランスフェラーゼ活性の増加は、当
業者に周知の技術を使用して、マルチコピープラスミド上に、活性をコードする核酸分子
を導入することによって達成される。さらに別の実施形態において、脂質連結オリゴ糖の
グルコシル化の程度は、宿主細胞中のオリゴサッカリルトランスフェラーゼ(oligo
saccharyl transferase)活性の基質特異性を低下することを含む
。このことは、例えば、活性をコードする少なくとも1つの核酸を、遺伝子シャッフリン
グ、インビトロ変異誘発、および誤りがちのポリメラーゼ連鎖反応のような技術(これら
の全ては、当業者に周知である)に供することによって達成される。当然、ALG8およ
びALG10は、宿主細胞中で過剰発現され、同様の様式で試験され得る。
【0099】
従って、好ましい実施形態において、本発明は、ER中の脂質連結オリゴ糖のグリコシ
ル化を担う1つ以上の酵素が、活性を増加するように操作されたかまたは選択された宿主
細胞を使用してヒト様糖タンパク質を作製するための方法を提供する。より好ましい実施
形態において、本発明は、以下:
(a)1つ以上のalg遺伝子活性またはMan3GlcNAc2(「Man3」)コア糖
質構造のα−1,6アームで、N−グリカンをマンノシル化する活性の活性が低下または
激減し、かつ(b)ER中での脂質連結オリゴ糖のグリコシル化を担う1つ以上の酵素が
活性を増加するように操作されるかまたは選択される宿主細胞を使用する。しかし、al
g3変異型バックグラウンドにおいて見出される脂質連結Man5構造は、Alg6pに
対する好ましい基質ではない。従って、当業者は、例えば、当業者に周知の分子生物学お
よび遺伝子選択技術を使用して、基質特異性が増加した酵素をコードする核酸を、1以上
のラウンドの遺伝子シャッフリング、誤りがちのPCR、またはインビトロ変異誘発アプ
ローチに供することによって、そして目的の宿主細胞中で基質特異性の上昇について選択
することによって、基質特異性が上昇したAlg6p、Alg8pおよびAlg10pを
同定し得る(Gibbs,2001)。選択された宿主株において酵素基質特異性を改善
するためのこのような技術が、本発明のこの特定の実施形態に限定されないが、非ヒト宿
主細胞においてヒト様N−グリカンの産生をさらに最適化する任意の実施形態において使
用され得ることは、当業者に理解される。
【0100】
記載されるように、Man5は、新生ポリペプチド鎖に移されると、適切なα−1,2
−マンノシダーゼの発現は、本発明によって提供されるように、Man5GlcNAc2構
造をさらに小さくし、所望のコアMan3GlcNAc2構造を生じる。α−1,2−マン
ノシダーゼは、末端α−1,2−結合マンノース残基のみを除去し、alg3、alg9
およびalg12変異型宿主細胞およびこれらの遺伝子に対するホモログが変異された宿
主細胞中で作られたMan5GlcNAc2−Man7GlcNAc2特異的構造を認識する
と予想される。
【0101】
図3に概略的に示されるように、適切なUDP−糖−輸送体およびUDP−糖−トラン
スフェラーゼの共発現は、末端α−1,6残基およびα−1,3残基をGlcNAcでキ
ャップし、このことは、哺乳動物型複合体およびハイブリッドN−グリコシル化に必要な
前駆体(GlcNAc2Man5GlcNAc2)を生じる。次いで、ペプチドが結合した
N−連結オリゴ糖鎖(GlcNAc2Man5GlcNAc2(図3)は、哺乳動物型オリ
ゴ糖構造へのさらなる改変のための前駆体として働く。続いてのガラクトシルトランスフ
ェラーゼの発現およびシアル酸(sialylic acid)を転位する能力を遺伝子
操作することは、哺乳動物型(例えば、ヒト様)N−グリカン構造を生じる。
【0102】
本発明に従う所望の宿主細胞は、1つの酵素または1つを超える酵素を同時に操作し得
る。さらに、潜在的に有用な酵素をコードする遺伝子のライブラリーが、作製され得、最
適な活性を有する1つ以上の酵素を有する株または最も「ヒト様」糖タンパク質を産生す
る株は、ライブラリーの1つ以上のメンバーを用いて標的宿主細胞を形質転換することに
よって選択される。コアN−グリカンMan3GlcNAc2を有する糖タンパク質を産生
し得る下等真核生物は、遺伝子操作の実施の容易さ、ならびに安全性および効率特性に起
因して、特に有用である。好ましい実施形態において、少なくとも1つのさらなるグリコ
シル化反応が、エキソビボまたはインビボで実施され、ヒト様N−グリカンを産生する。
より好ましい実施形態において、グリコシル化酵素の活性型は、宿主細胞の小胞体および
/またはゴルジ装置において発現され、所望のヒト様糖タンパク質を産生する。
【0103】
(宿主細胞)
本発明の好ましい非ヒト宿主細胞は、下等真核生物細胞(例えば、単細胞真菌または線
維状真菌)であり、これは、1つ以上のalg遺伝子活性(alg活性に対して相同であ
るか、または等価である酵素活性を含む)が減少しているか、または激減している。本発
明の別の好ましい宿主細胞は、脂質連結オリゴ糖構造のα−1,6アームをマンノシル化
する1つ以上の酵素の活性(alg活性以外)が低下されるか、または激減する。
【0104】
下等真核生物宿主細胞が、好ましくある、一方で、前述の特徴を有する広く種々の宿主
細胞は、本発明の方法において有用であるとして構想される。例えば、植物細胞は、本発
明に従って、ヒト様糖タンパク質を発現するように操作され得る。同様に、種々の非ヒト
哺乳動物宿主細胞は、本発明の方法を使用して、よりヒト様の糖タンパク質を発現するよ
うに変更され得る。適切な宿主細胞が、操作され得るか、または酵母において既に記載さ
れる多くのこのような変異型の1つが、使用され得る。本明細書中に例示される場合、本
発明の好ましい宿主細胞は、Pichia Pastoris中の過剰マンノシル化欠損
(OCH1)変異型であり、この変異型は、alg3遺伝子をさらに改変し、欠失する。
他の好ましい宿主は、och1変異およびalg9変異またはalg12変異を有するP
ichia pastoris変異型である。
【0105】
(複合N−グリカンの形成)
改変された、新生N−グリカン構造に糖を連続的に添加することは、グルコシルトラン
スフェラーゼのゴルジ装置への首尾よい標的化および首尾よい発現を含む。このプロセス
は、例えば、機能的発現(例えば、初期ゴルジ装置または中期ゴルジ装置におけるGnT
Iの機能的発現)を必要とし、UDP−GlcNAc(例えば、UDP−GlcNAc輸
送体の発現による)の十分な供給を保証する。
【0106】
糖タンパク質を特徴づけ、そして所望されるグリコシル化を確認するために、糖タンパ
ク質を精製し、N−グリカンを、PNGase−F放出し、次いで、MALDI−TOF
−MSによって分析した(実施例2)。ヒトプラスミノゲンのクリングル3ドメインを、
レポータータンパク質として使用した。この可溶性糖タンパク質を、alg3、och1
ノックアウトのバックグラウンドで、P.pastoris中で産生した(実施例2)。
【0107】
GlcNAcMan5GlcNAc2を、ヒトGnTIおよび図16のK.lactis
UDP−GlcNac輸送体の添加後、優勢なN−グリカンとして提供した(実施例2
)。このN−グリカンの質量は、1463(m/z)であるGlcNAcMan5Glc
NAc2質量と一致する。Man5GlcNAc2へのGlcNAcの付加を確認するため
に、β−N−ヘキソサミニダーゼ消化を実施し、これは、Man5GlcNAc2の質量と
一致する1260(m/z)のピークを明らかにした(図17)。
【0108】
1つの株PBP3(実施例2)中のalg3 och1欠失由来のN−グリカンは、1
138(m/z)および1300(m/z)に、2つの異なるピークを提供した。これは
、構造(GlcNAcMan3GlcNAc2およびGlcNAcMan4GlcNAc2)
に一致する(図18)。多くのマンノースに対するインビトロα1,2−マンノシダーゼ
消化の後、ピークは、1138(m/z)に溶出された。これは、GlcNAcMan3
GlcNAc2と一致する(図19)。GlcNAcのMan3GlcNAc2構造に対す
る付加を確認するために、β−N−ヘキソサミニダーゼ消化を実施し、これは、934(
m/z)にピークを示し、Man3GlcNAc2の質量と一致した(図20)。
【0109】
GlcNAcのGlcNAcMan3GlcNAc2構造に対する第2の付加を、図21
に示す。1357(m/z)のピークは、GlcNAc2Man3GlcNAc2に対応す
る。コアマンノース構造(Man3GlcNAc2)への2つのGlcNAcの付加を確認
するために、別のβ−N−ヘキソサミニダーゼ消化を、実施し、これは、934(m/z
)にピークを示し、Man3GlcNAc2の質量と一致した(図22)。これは、酵母細
胞中で作られる複合型糖タンパク質を示す決定的なデータである。
【0110】
GlcNAc2Man3GlcNAc2に対するUDP−ガラクトースおよびβ1,4−
ガラクトシルトランスフェラーゼのインビトロ付加は、1664(m/z)にピークを生
じた。これは、Gal2GlcNAc2Man3GlcNAc2の質量と一致する(図23)
。最終的に、CMP−N−アセチルノイラミン酸およびシアリルトランスフェラーゼのイ
ンビトロ付加は、2248(m/z)にピークを生じた。これは、NANA2Gal2Gl
cNAc2Man3GlcNAc2の質量と一致する(図24)。上記のデータは、複雑な
ヒト様糖タンパク質を産生し得る非哺乳動物宿主細胞の使用を支持する。
【0111】
(グルコシルトランスフェラーゼおよびガラクトシルトランスフェラーゼの特定の細胞
小器官への標的化)
多くの研究が、これらの酵素が、これらのそれぞれの細胞小器官に保持され、そして固
定される正確な機構を示すために、提供されてきた。複雑ではあるが、証拠は、幹領域、
膜貫通領域、および細胞質テイルが、個々にまたは一斉に、酵素を個々の細胞小器官の膜
に指向し、それによって、結合する触媒領域が、その位置に局在化することを示唆する。
【0112】
活性なグリコシルトランスフェラーゼが、発現され得、そして適切な細胞小器官に指向
され得、その結果、連続した順番の反応が生じ得、複合N−グリカン形成を導く方法は、
以下の通りである:
(A)分泌経路中の特定の位置(ER、ゴルジおよびトランスゴルジネットワーク)への
局在化を媒介するタンパク質/ペプチドをコードすることが既知である領域のDNAライ
ブラリーを確立すること。これらの酵素の限られた選択物およびこれらのそれぞれの位置
を、表1に示す。これらの配列は、操作される宿主および他の関連する生物または関連し
ない生物から選択され得る。一般的に、このような配列は、以下の3つの範疇に分類され
る:
(1)細胞質テイル(ct)、膜貫通ドメイン(tmd)およびいくらかよりあいまい
に規定された幹領域(sr)の一部をコードするN−末端配列。これらは、一緒にかまた
は個々にタンパク質をゴルジの内(内腔)膜に固定する、
(2)一般的にC−末端で見出される回収シグナル(例えば、HDELまたはKDEL
テトラペプチド)、および
(3)膜貫通ヌクレオチド糖輸送体(これは、ゴルジに局在化することが公知である)
。
第1の場合において、局在化領域が種々の要素(ct、tmdおよびsr)からなる場合
、ライブラリーは、ct、tmdおよび幹領域の種々の部分が示されるように設計される
。これは、細胞質領域をコードするDNAの5’末端に結合するPCRプライマーを使用
すること、および幹領域の種々の部分に結合する対向する一連のプライマーを使用するこ
とによって達成され得る。さらに、糖ヌクレオチド輸送体および公知の回収シグナルをコ
ードする融合タンパク質構築物を作製する。
(B)第2工程は、一連の融合タンパク質構築物の作製を包含し、この構築物は、上記の
局在化配列およびこのような局在化配列にインフレームでクローン化された特定のグリコ
シルトランスフェラーゼの触媒ドメインをコードする(例えば、GnTI、GalT、フ
コシルトランスフェラーゼまたはST)。触媒ドメインに融合された糖ヌクレオチド輸送
体の場合において、このような構築物を設計し、その結果、触媒ドメイン(例えば、Gn
TI)が、得られたポリペプチドのN−末端またはC−末端のいずれかに存在し得る。局
在化配列と同様に、触媒ドメインは、種々の異なる供給源に由来し得る。このような触媒
ドメインの選択は、触媒ドメインが活性である特定の環境の知見によって左右される。例
えば、特定のグリコシルトランスフェラーゼが、後期ゴルジにおいて活性である場合、宿
主生物の後期ゴルジ中の全ての公知の酵素が、7.0の最適pHを有する場合、または後
期ゴルジが、特定のpHを有することが公知である場合、そのpHで最適の活性を有する
触媒ドメインを選択することを試行する。特定の宿主において、このような酵素の活性に
対する現存するインビボデータはまた、有用であり得る。例えば、Schwientek
および共同研究者らは、GalT活性をS.cerevisiaeのゴルジに操作し得る
ことを示し、そしてこのような活性が、S.cerevisiaeのalg変異型におい
て、いくつかのGalの存在するGlcNAc2への転位を示すことによって存在したこ
とを示した。さらに、数ラウンドの遺伝子シャッフリングまたは誤りがちのPCRを実施
し、融合構築物のプール内に大きな多様性を得る。なぜなら、単一アミノ酸変異は、糖タ
ンパク質保有酵素の活性を劇的に変更し得ることが示されたからである(Romeroら
、2000)。グリコシルトランスフェラーゼおよびこれらの内因性固定配列(anch
oring sequence)の全長配列がまた、使用され得る。好ましい実施形態に
おいて、このような局在化/触媒ドメインライブラリーは、より高等な真核生物における
グリコシル化反応の一連の性質に対する現存する情報を組込むように設計される。言い換
えると、糖タンパク質のプロセシングの過程の初期に起こることが公知である反応は、こ
のような反応を触媒する酵素の、ゴルジまたはERの初期部分への標的化を必要とする。
例えば、Man8GlcNAc2からMan5GlcNAc2へのトリミングは、複合N−グ
リカンの形成における初期段階である。タンパク質プロセシングが、ERにおいて開始し
、次いで、初期ゴルジ、中期ゴルジおよび後期ゴルジの間進行するので、ERまたは初期
ゴルジにおいて起こるこのトリミング反応を有することが、所望される。マンノシダーゼ
I局在化についてのライブラリーを設計する場合、マンノシダーゼIの触媒ドメインを有
するER標的化シグナルと初期ゴルジ標的化シグナルとを一致することを試行する。
【0113】
融合構築物ライブラリーを用いて宿主株を形質転換する際に、選択プロセスを使用して
、どの特定の局在化配列と触媒ドメインとの組み合わせが、実際に、そのような宿主株に
おいて見出される糖質構造に対して最大の効果を有するかを、同定する。そのような選択
は、多数のアッセイ方法または検出方法に基づき得る。それらの方法は、手動により行わ
れ得るか、または高スループットスクリーニング機器を使用することを介して自動化され
得る。
【0114】
別の例において、GnTI活性が、複合N−グリカンの成熟のために必要である。なぜ
なら、末端α1,3マンノース残基へのGlcNAcの付加後のみ、そのような構造から
続く中間体GlcNAcMan3GlcNAc2構造へのさらなる調整が生じ得るからであ
る。マンノシダーゼIIは、末端β1,2−GlcNAcの非存在下で末端α1,3−マ
ンノース残基およびα1,6−マンノース残基を多分除去することができず、従って、複
合N−グリカンの形成は、GnTI活性の非存在下では進行しない(Schachter
,1991)。あるいは、充分な量の本発明に記載されるMan5GlcNAc2を生じる
株を、Alg3P活性が欠損した株を操作または選択することにより、まず操作または選
択し得る。十分なUDP−GlcNAcトランスポーター活性の存在下で、このようなU
DP−GlcNAcトランスポーター活性を有する株を操作または選択することにより達
成し得るように、GlcNAcが、GnTIにより末端α−1,3残基に付加され得る。
なぜなら、インビトロでのMan3構造は、ラット肝臓GnTIにより認識されるからで
ある(Moller,1992)。
【0115】
別のアプローチにおいて、上記のライブラリー中にUDP−GlcNAcトランスポー
ターの発現を組込み得、望ましい構築物が、以下を含むようにする:(1)形質転換構築
物が細胞中に維持される領域(例えば、複製起点、または染色体組み込みを媒介する領域
)、(2)形質転換された細胞の選択を可能にするマーカー遺伝子(対比選択可能で再利
用可能なマーカー(例えば、ura3もしくはT−urf13(Soderholm,2
001)、または充分に特徴付けられた他の選択マーカー(例えば、his4、bla、
Sh bleなど)を含む)、(3)UDP−GlcNAcトランスポーターをコードす
る遺伝子(例えば、K.lactis由来(Abeijon,1996)またはH.sa
piens由来(Ishida,1996)、および(4)上記局在化ドメイン/触媒ド
メイン融合構築物ライブラリーの発現を活性化するプロモーター。
【0116】
上記の融合構築物のライブラリーを用いる宿主の形質転換の後、細胞表面上に最高濃度
の末端GlcNAcを有する細胞、または最高の末端GlcNAc含量を有するタンパク
質を分泌する細胞について、スクリーニングし得る。そのようなスクリーニングは、視覚
的方法(染色手順など)、特異的末端GlcNAc結合抗体に結合する能力、またはマー
カーに結合体化したレクチンに結合する能力(そのようなレクチンは、E.Y.Labo
ratories Inc.,San Mateo,CAから入手可能である)、特定の
レクチンが末端マンノース残基に結合する能力の減少、インビトロで放射性標識糖を組込
む能力、色素もしくは荷電表面への結合の変化に基づき得るか、または発蛍光団標識レク
チンもしくは抗体と組み合わせて蛍光補助細胞ソーティング(FACS)デバイスを使用
することにより達成され得る(Guillen,1998)。形質転換集団内の特定の表
現型を、細胞傷害性レクチンを用いて富化することは、有利であり得る。米国特許第5,
595,900号は、望ましい細胞外糖質構造を有する細胞が同定され得るいくつかの方
法を教示する。このストラテジーを繰返し実行することにより、下等真核生物においてま
すます複雑なグリカンを連続して操作することが可能である。
【0117】
形質転換後、インビトロアッセイにおいて、GlcNAcトランスフェラーゼIIによ
るUDP−GlcNAcからのGlcNAcの最も効率的な移動を可能にする形質転換体
について、選択し得る。このスクリーニングは、上記形質転換ライブラリーを保有する細
胞を、寒天プレート上にて選択圧下で増殖させること、そして個々のコロニーを96ウェ
ルマイクロタイタープレートへと移すことによって、実行され得る。細胞を増殖させた後
、その細胞を遠心分離し、その細胞を緩衝液中に再懸濁し、そしてUDP−GlcNAc
およびGnT Vを添加した後、UDPの放出を、HPLCか、またはUDPについての
酵素結合アッセイのいずれかによって、測定する。あるいは、放射性標識したUDP−G
lcNAcおよびGnT Vを使用し、その細胞を洗浄し、その後、N−アセチルグルコ
サミニダーゼによる放射性GlcNAcの放出を探索し得る。これはすべて、手動により
行われ得るか、または高スループットスクリーニング機器の使用を介して自動化され得る
。
【0118】
第1のアッセイにおいてより多くのUDPを放出するかまたは第2のアッセイにおいて
より多くの放射標識GlcNAcを放出する形質転換体は、その表面上により高い程度の
GlcNAcMan3GlcNAc2を有し(図3)、従って、望ましい表現型を構成する
と、予測される。あるいは、形質転換細胞の表面上の変化したグリコシル化パターンを明
らかにし得る、レクチン結合アッセイのような他の任意の適切なスクリーニングを使用し
得る。この場合、末端マンノースに特異的なレクチンの結合の減少が、適切な選択ツール
であり得る。Galantus nivalisレクチンは、末端α−1,3マンノース
に特異的に結合し、このことは、ゴルジ装置中に充分なマンノシダーゼII活性が存在す
る場合には、減少すると予期される。高末端マンノース含量を含む細胞の除去を可能にす
るクロマトグラフィー分離工程を実行することによってもまた、望ましい形質転換体を富
化し得る。この分離工程は、低末端マンノース含量を有する細胞よりも、高末端マンノー
ス含量を有する細胞に特異的に結合するレクチンカラム(例えば、アガロースに結合した
Galantus nivalisレクチン、Sigma,St.Louis,MO)を
用いて、実行される。さらに、このような融合タンパク質構築物を直接作製し得る。なぜ
なら、種々の下等真核生物宿主における活性糖質改変酵素の局在化に関するさらなる情報
が、科学文献にて利用可能であるからである。例えば、先行技術は、ヒトβ1,4−Ga
lTrが、MNT(S.cerevisiae由来のマンノシルトランスフェラーゼ)の
膜ドメインに融合され得、かつその触媒活性を保持しながらゴルジ装置へと局在化され得
ることを、本発明者らに教示する(Schwientekら、1995)。S.cere
visiaeまたは関連生物が、操作されるべき宿主である場合、そのような宿主由来の
複合N−グリカンを得るために、全体的ストラテジーにこのような知見を直接組込み得る
。S.cerevisiae中のグリコシルトランスフェラーゼに関連する、P.pas
toris中のいくつかのそのような遺伝子フラグメントが、同定されており、従って、
その目的のために使用され得る。
【0119】
(表1)
【0120】
【表1】
(組み込み部位)
この遺伝子操作の試みの1つの最終的な目的は、工業的発酵プロセスにおいて充分に実
施可能である頑丈なタンパク質産生株であるので、宿主(例えば、真菌)染色体中に複数
の遺伝子を組込むことは、注意深い計画を伴う。操作された株は、多分、一定範囲の異な
る遺伝子で形質転換される必要があり、これらの遺伝子は、望ましい活性が発酵プロセス
全体にわたり維持されるのを確保するために、安定な様式で形質転換される必要がある。
以下の酵素活性の任意の組み合わせが、タンパク質発現真菌宿主中に操作される必要があ
る:シアリルトランスフェラーゼ、マンノシダーゼ、フコシルトランスフェラーゼ、ガラ
クトシルトランスフェラーゼ、グルコシルトランスフェラーゼ、GlcNAcトランスフ
ェラーゼ、ERおよびゴルジ特異的トランスポーター(例えば、UPD−ガラクトースお
よび他の前駆体についての共輸送トランスポーターおよび対向輸送トランスポーター)、
オリゴ糖のプロセシングに関与する他の酵素、ならびに活性化オリゴ糖前駆体(例えば、
UDP−ガラクトース、CMP−N−アセチルノイラミン酸)の合成に関与する酵素。同
時に、非ヒトグリコシル化反応の特徴であることが公知である酵素をコードする多数の遺
伝子が、欠失されなければならない。そのような遺伝子および対応するタンパク質は、多
数の下等真核生物(例えば、S.cerevisiae、T.reesei、A.nid
ulansなど)において広範に特徴付けられており、それにより、下等真核生物におけ
る公知のグリコシルトランスフェラーゼ、その活性、およびその個々の遺伝子配列のリス
トが提供される。これらの遺伝子は、マンノシルトランスフェラーゼの群(例えば、1,
3−マンノシルトランスフェラーゼ(例えば、S.cerevisiae中のMNN1)
(Graham,1991)、1,2−マンノシルトランスフェラーゼ(例えば、S.c
erevisiae由来のKTR/KREファミリー)、1,6−マンノシルトランスフ
ェラーゼ(S.cerevisiae由来のOCH1)、マンノシルホスフェートトラン
スフェラーゼ(S.cerevisiae由来のMNN4およびMNN6)、および異所
性(すなわち、非ヒト)グリコシル化反応に関与するさらなる酵素)から選択される。こ
れらの遺伝子の多くは、実際に、欠失されており、変化したグリコシル化プロフィールを
有する生存可能な表現型を個別に生じる。例が、表2に示される。
【0121】
(表2)
【0122】
【表2】
下等真核生物中への複合N−グリカンの形成を操作するためのどのストラテジーも、グリ
コシルトランスフェラーゼ活性の排除および付加の両方を包含するので、包括的スキーム
は、両方の要件を協調することを試みる。望ましくない酵素をコードする遺伝子は、望ま
しい遺伝子について可能な組み込み部位として役立つ。例えば、1,6−マンノシルトラ
ンスフェラーゼ活性は、公知の多くの下等真核生物におけるグリコシル化の特徴である。
α−1,6−マンノシルトランスフェラーゼ(OCH1)をコードする遺伝子は、S.c
erevisiaeからクローン化されており、その遺伝子中の変異は、マンノシル化が
減少した生存可能な表現型を生じる。そのα−1,6−マンノシルトランスフェラーゼ活
性をコードする遺伝子座は、グリコシルトランスフェラーゼ活性をコードする遺伝子を組
込むための主要な標的である。同様な様式で、その座における遺伝子破壊事象に基づいて
、(1)よりヒトと類似する様式でグリコシル化する細胞能力を改善し、(2)タンパク
質を分泌する細胞能力を改善し、(3)外来タンパク質のタンパク質分解を減少し、そし
て(4)精製または発酵プロセス自体を促進するプロセスの他の特徴を改善することが予
測される、他の染色体組み込み部位の範囲を選択し得る。
【0123】
(糖ヌクレオチド前駆体の提供)
高等真核生物グリコシル化の特徴は、糖タンパク質におけるガラクトース、フコース、
および高い程度の末端シアル酸の存在である。これらの糖は、酵母および繊維状真菌にお
いて生成される糖タンパク質においては一般的に見出されず、上記の方法により、望まし
いオルガネラにおいてグリコシルトランスフェラーゼが局在化する株の操作が可能である
。複合N−グリカン合成の形成は、特定の糖残基が除去されそしてコアオリゴ糖構造に結
合される、連続的プロセスである。高等真核生物において、これは、基質を、種々の処理
酵素に連続的に曝露させることによって達成される。これらの酵素は、処理カスケード全
体における特定の位置に依存して、特定の反応を行う。この「アセンブリライン」は、E
R、初期ゴルジ、中期ゴルジ、および後期ゴルジ、ならびにトランスゴルジネットワーク
すべてと、その特定の処理環境からなる。下等真核生物のゴルジおよびERにおいてヒト
糖タンパク質の処理を再生するために、多数の酵素(例えば、グリコシルトランスフェラ
ーゼ、グリコシダーゼ、ホスファターゼ、およびトランスポーター)が、発現され、これ
らのオルガネラに特異的に標的付けられなければならず、好ましくは、それらがその環境
およびその経路中の他の酵素と関連して最も効率的に機能するような位置に、標的付けら
れなければならない。いくつかの個々のグリコシルトランスフェラーゼが、クローン化さ
れ、そしてS.cerevisiae(GalT、GnT I)、Aspergillu
s nidulans(GnT I)および他の真菌において発現されているが、その生
物のグリコシル化パターンにおける望ましい「ヒト化」の結果は示さない(Yoshid
a,1995;Schwientek,1995;Kalsner,1995)。このよ
うなグリコシルトランスフェラーゼの作用により糖を受容するのに必要な糖質構造は、充
分な量存在しないと、推測された。このことは、おそらく、複合N−グリカン形成の欠如
に寄与し、一方、Man5GlcNAc2構造を供給し、GnT I活性を有し、そして真
菌注に操作されたUDP−Gnトランスポーター活性を有する真菌についての報告は現在
存在しない。ハイブリッドな複合N−グリカン形成のために必要なのは、これらの3つの
生化学的事象の組み合わせである。
【0124】
ヒトにおいて、ヌクレオチド糖前駆体(例えば、UDP−N−アセチルグルコサミン、
UDP−N−アセチルガラクトサミン、CMP−N−アセチルノイラミン酸、UDP−ガ
ラクトースなど)の全範囲は、一般的に、サイトゾルにおいて合成され、そしてゴルジに
輸送され、このゴルジにおいて、これらは、グリコシルトランスフェラーゼによりコアオ
リゴ糖に結合される。下等真核生物においてこのプロセスを繰り返すために、糖ヌクレオ
シドに特異的な輸送体は、適切なレベルのヌクレオシド糖前駆体を確実にするために、ゴ
ルジにおいて発現されなければならない(Sommers,1981;Sommers,
1982;Perez,1987)。この反応の副生成物は、ヌクレオシドジホスフェー
トまたはヌクレオシドモノホスフェートのいずれかである。モノホスフェートが、交互輸
送機構によるヌクレオシドトリホスフェート糖の交換において直接的に輸送され得るが、
ジホスホヌクレオシド(例えば、GDP)は、輸送される前に、ヌクレオシドモノホスフ
ェートおよび無機ホスフェートを生じるために、ホスファターゼ(例えば、GDPase
)によって切断されなければならない。この反応は、S.cerevisiae由来のG
DPaseが、マンノシル化に必要であることが見出されているので、効率的なグリコシ
ル化に重要であるようである。しかし、酵素のみが、UDPに対して10%の活性を有す
る(Berninsone,1994)。下等真核生物は、しばしば、ゴルジにおいてU
DP特異的ジホスファターゼ活性を有さない。なぜなら、これらは、ゴルジにおける糖タ
ンパク質合成のためにUDP−糖前駆体を利用しないからである。
【0125】
Schizosaccharomyces pombe(細胞壁多糖類へガラクトース
残基を付加することが見出された酵母(UDP−ガラクトース由来))は、このような酵
素に対する要件をさらに示唆する特異的なUDPase活性を有することを見出した(B
eminsoneら、1994)。UDPは、グリコシルトランスフェラーゼの強力なイ
ンヒビターであることが公知であり、このグリコシル化副産物の除去は、ゴルジの内腔に
おけるグリコシルトランスフェラーゼ阻害を妨げるために重要である。(Khatara
ら,1974)。従って、UDPの除去を提供する必要があり得、これは、このような操
作された株のゴルジにおいて蓄積することが予期される(Berninsone,199
5;Beaudet,1998)。
【0126】
別の例において、2,3シアリルトランスフェラーゼおよび2,6シアリルトランスフ
ェラーゼは、ヒトのトランス−ゴルジおよびTGNにおいてシアル酸でガラクトース残基
をキャップし、糖タンパク質の成熟形態を導く。代謝的に操作された酵母または真菌に、
このプロセシング工程を再操作することは、(1)2,3−シアリルトランスフェラーゼ
活性および(2)酵母の後期(late)ゴルジにおけるCMP−N−アセチルノイラミ
ン酸の十分な供給を必要とする。後期ゴルジにおける十分な2,3−シアリルトランスフ
ェラーゼ活性を得るために、公知のシアリルトランスフェラーゼ(例えば、ヒト由来)の
触媒性ドメインは、真菌において後期ゴルジに指向されなければならない(上記を参照の
こと)。同様に、輸送体は、後期ゴルジへのCMP−N−アセチルノイラミン酸の輸送を
可能にするように操作されなければならない。現在、真菌が、十分な量のCMP−N−ア
セチルノイラミン酸を合成することを指摘するものはなく、このような糖−ヌクレオチド
のゴルジへの輸送を言及しない。結果として、対応するグリコシルトランスフェラーゼに
対する基質の適切な供給を確実にするために、真菌に対してCMP−シアル酸の産生を代
謝的に操作しなければならない。
【0127】
(ゴルジ装置に糖ヌクレオチド前駆体を提供するための方法)
(UDP−N−アセチル−グルコサミン)
ヒトUDP−N−アセチルグルコサミン輸送体のcDNA(これは、発現配列タグデー
タベース(dbEST)における相同性検索によって認識された)は、Ishidaおよ
び共同研究者(Ishida,1999)によってクローニングされた。Guillen
および共同研究者は、上記ヌクレオチド糖のゴルジ輸送体において欠損した、最近特徴付
けられたKluyveromyces lactis変異体のイヌ腎細胞(MDCK)由
来のcDNAとの表現型相関によって、UDP−N−アセチルグルコサミンに対する哺乳
動物ゴルジ膜輸送体をクローニングした(Guillen、1998)。それらの結果は
、哺乳動物ゴルジ UDP−N−GlcNAc輸送体遺伝子が、このタンパク質が、発現
しそして酵母のゴルジ装置に機能的に標的化されるのに必要な情報の全てを有すること、
および非常に異なるアミノ酸配列を有する2つのタンパク質が、同じゴルジ膜内に同じ溶
質を輸送し得ることを実証する。((Guillen、1998)。
【0128】
(GDP−フコース)
ラット肝臓ゴルジ膜GDP−フコース輸送体が、Puglielli,L.およびC.
B.Hirschberg(Puglielli,1999)によって同定および精製さ
れた。対応する遺伝子は、同定されていないが、N−末端配列決定を、対応する遺伝子に
特異的なオリゴヌクレオチドプローブの設計のために使用し得る。これらのオリゴヌクレ
オチドをプローブとして使用して、GDP−フコース輸送体をコードする遺伝子をクロー
ニングし得る。
【0129】
(UDP−ガラクトース)
2つの異種遺伝子(Schizosaccharomyces pombe由来のα1
,2−ガラクトシルトランスフェラーゼ(α1,2 GalT)をコードするgmal2
(+)およびヒトUDP−ガラクトース(UDP−Gal)輸送体をコードする(hUG
T2))は、ガラクトシル化に必要とされる細胞内条件を調べるために、S. cere
visiaeにおいて機能的に発現された。タンパク質ガラクトシル化とUDP−ガラク
トース輸送体活性との間の相関は、α1,2−GalTよりも、UDP−Gal輸送体の
外因的な供給が、S.cerevisiaeにおける効率的なガラクトシル化に重要な役
割を果たすことを示した(Kainuma,1999)。同様に、S.pombe由来の
UDP−ガラクトース輸送体は、クローニングされた(Aoki,1999;Segaw
a,1999)。
【0130】
(CMP−N−アセチルノイラミン酸(CMP−シアル酸))
ヒトCMP−シアル酸輸送体(hCST)をクローニングし、そしてLec 8 CH
O細胞において発現させた(Aoki,1999;Eckhardt,1997)。マウ
スCMP−シアル酸輸送体の機能的発現は、Saccharomyces cerevi
siaeにおいて達成された(Berninsone,1997)。シアル酸は、いくら
かの真菌において見出されたが、選択された宿主系が、十分なレベルのCMP−シアル酸
を供給し得るか否かは、明確でない。シアル酸は、培地において供給され得るか、あるい
は、シアル酸合成に関係する真菌経路がまた宿主ゲノム内に組み込まれ得るかのいずれか
である。
【0131】
(ジホスファターゼ)
糖が糖タンパク質上に輸送される場合、ヌクレオシドジホスフェートまたはヌクレオシ
ドモノホスフェートのいずれかが、糖ヌクレオチド前駆体から放出される。モノホスフェ
ートが交互輸送機構によるヌクレオシドトリホスフェート糖の交換において直接的に輸送
され得るが、ジホスホノモノヌクレオシド(例えば、GDP)は、輸送される前に、ヌク
レオシドモノホスフェートおよび無機ホスフェートを生じるために、ホスファターゼ(例
えば、GDPase)によって切断されなければならない。この反応は、S.cerev
isiae由来のGDPaseが、マンノシル化に必要であることが見出されているので
、効率的なグリコシル化に重要であるようである。しかし、酵素のみが、UDPに対して
10%の活性を有する(Berninsone,1994)。下等真核生物は、しばしば
、ゴルジにおいてUDP特異的ジホスファターゼ活性を有さない。なぜなら、これらは、
ゴルジにおける糖タンパク質合成のためにUDP−糖前駆体を利用しないからである。S
chizosaccharomyces pombe(細胞壁多糖類へガラクトース残基
を付加することが見出された酵母(UDP−ガラクトース由来))は、このような酵素に
対する要件をさらに示唆する特異的なUDPase活性を有することを見出した(Bem
insoneら、1994)。UDPは、グリコシルトランスフェラーゼの強力なインヒ
ビターであることが公知であり、このグリコシル化副産物の除去は、ゴルジの内腔におけ
るグリコシルトランスフェラーゼ阻害を妨げるために重要である。(Khataraら,
1974)。
【0132】
(複合体N−グリカンを産生するためのGnTの発現)
(抗体機能をブーストするためのGnT−IIIの発現)
N−アセチルグルコサミニルトランスフェラーゼIIおよびIIIによるGlcNAc
1Man3GlcNAc2構造へのN−アセチルグルコサミンの付加は、いわゆる分岐した
N−グリカンGlcNAc3Man3GlcNAc2を生じる(図3)。この構造は、より
大きな抗体依存性の細胞性の細胞傷害(ADCC)に関係している(Umanaら、19
99)。哺乳動物細胞によって発現される免疫グロブリンの糖形態を再操作することは、
冗漫で厄介な作業である。特に、GnT III(この酵素の過剰発現は、増殖阻害に関
係している)の場合、調節された(誘導性の)遺伝子の発現を含む方法が、分岐したN−
グリカンを有する免疫グロブリンを作製するために使用されなければならい(Umana
ら、1999a、1999b)。
【0133】
従って、別の実施形態において、本発明は、コアマンノース構造上に、分岐したN−ア
セチルグルコサミン(GlcNAc)を有するヒト様N−グリカンを作製するためのシス
テムおよび方法を提供する。好ましい実施形態において、本発明は、分岐したN−グリカ
ンを有する免疫グロブリンを産生するためのシステムおよび方法を提供する。本明細書中
に記載されるシステムおよび方法は、先の問題(例えば、GnT IIIまたはADCC
の過剰発現に関連する細胞傷害性)を被らない。なぜなら、本発明の宿主細胞は、実質的
に改変されたヒト型糖形態(例えば、GlcNAc2Man3GlcNAc2)を有するN
−グリカンを産生する、生存可能であり好ましくは、頑強な細胞であるように操作および
選択されるからである。従って、本発明の宿主細胞における分岐したN−アセチルグルコ
サミンの付加は、増殖−表現型またはそれらの宿主細胞の生存能力に対して、無視し得る
効果を有する。
【0134】
さらに、以前の研究(Umana)は、GnT III発現レベルとADCCの程度と
の間に直線的な関係がないことを示した。哺乳動物細胞における最適な発現レベルを見つ
け出すこと、およびFDAが許可した発酵プロセス全体にわたってそれを維持することは
、挑戦であるようである。しかし、本発明の細胞(例えば、真菌細胞)において、頑強で
、信頼性があり、かつ最適なGnT III発現レベルを確立するための適切な強度のプ
ロモーターを発見することは、当業者には比較的簡単な作業である。
【0135】
分岐したN−グリカンを用いて糖タンパク質を産生し得る酵母菌株のような宿主細胞は
、宿主細胞内にGnT III活性を導入することによって、本発明に従って操作される
(実施例6)。好ましくは、宿主細胞は、GnT III(例えば、図32を参照のこと
)をコードする核酸または酵素活性を有するそのドメイン((例えば、本発明のライブラ
リーおよび関連する方法を使用して)必要に応じて、異種細胞シグナル標的化ペプチドに
融合される)を用いて形質転換される。GnT IIIを発現するように操作された宿主
細胞は、哺乳動物細胞が可能であるよりも高い抗体力価を生じる。これらはまた、ADC
Cに関してより高い効力を有する抗体を産生する。
【0136】
GnT IIIをトランスフェクトされた哺乳動物細胞株によって産生された抗体は、
非トランスフェクト細胞株によって産生された抗体と同様に効果的であるが、10〜20
倍低い濃度であることが示された(Daviesら、2001)。本発明の産生ビヒクル
の生産性の、哺乳動物系に対する20倍の増加、および10倍の効力の増加は、200倍
の正味の生産性の改善を生じる。従って、本発明は、高い効力(例えば、現在生産され得
るものよりも数桁より強力)を有する高い力価の抗体を作製するための系および方法を提
供する。この系および方法は、安全であり、高い効力の抗体を短い時間で低コストで提供
する。本発明に従って、GnT IIIを発現するように操作された宿主細胞は、少なく
とも50mg/リットル/日〜少なくとも500mg/リットル/日の速度で分岐した(
bisected) N−グリカンを有する免疫グロブリンを産生する。さらに、各免疫
グロブリン(Ig)分子(分岐したGlcNAcを含む)は、分岐したGlcNAcを含
まないで産生された同じIg分子よりも強力である。
【0137】
(GnT−IVおよびGnT−Vのクローニングおよび発現)
複合体N−グリカンの全ての分枝構造を、N−アセチルグルコサミントランスフェラー
ゼ(GnT)−I〜−VIにより、共通のコア五糖(Man3GlcNAc2またはMan
α−6(Man α1−3)Man β1−4 GlcNAc β1−4 GlcNAc
β1−4またはMan3GlcNAc2)上に合成した(Schachter Hら(1
989)Methods Enzyme;179:351−97)。より高度に分枝した
N−グリカンの生合成ついての現在の理解は、GnT IIの作用(GlcNAc2Ma
n3GlcNAc2構造の作製)の後に、GnTIVが、GlcNAcを、β1,4−連結
のUDP−GlcNAcからGlcNAc2Man3GlcNAc2 N−グリカンのMa
n α1,3 Man β1,4アームに移動させ(Allen SDら(1984)J
Biol C11em.Jun 10;259(11):6984−90;ならびにG
leeson PAおよびSchachter H.J(1983);J.Biol C
hem 25;258 (10):6162−73)、トリアンテナリアガラクト(tr
iantennary agalacto)糖鎖を生じることを示唆する。このN−グリ
カン(GlcNAc β1−2 Man α1−6(GlcNAc β1−2 Man
α1−3)Man β1−4 GlcNAc β1−4 GlcNAc β1,4 As
n)は、GnT−IIIおよびGnT−Vに対する共通の基質であり、分岐したトリアテ
ナリー構造およびテトラアテナリー構造の合成を導く。GnT IIIの作用が、分岐し
たN−グリカンを生じる場合、およびGnTVが、β1−6GlcNAcのα1−6マン
ノシルコアへの付加を触媒する場合、β1−6分枝を生じる。ガラクトースおよびシアル
酸のこれらの分枝への付加は、完全にシアル化された複合N−グリカンの生成を導く。
【0138】
分枝した複合型N−グリカンは、治療タンパク質の生理学的活性に関連している(例え
ば、ヒトエリスロポエチン(hEPO))。バイアンテナリー構造を有するヒトEPOは
、低い活性を有することが示されているのに対し、テトラアンテナリー構造を有するhE
POは、血流からのよりゆっくりとしたクリアランスを生じ、従って、より高い活性を生
じた(Misaizu Tら(1995)Blood Dec 1;86(11):40
97−104)。
【0139】
DNA配列情報を用いて、当業者は、GnT IVおよび/またはGnT V活性をコ
ードするDNA分子をクローニングし得る(実施例6;図33および34)。当業者に周
知の標準的な技術を使用して、GnT IVまたはGnT Vをコードする(または触媒
的に活性なそのフラグメントをコードする)核酸分子は、本発明の選択された宿主細胞(
例えば、本明細書中に記載のPichia sp.、Kluyveromyces sp
.およびAspergillus sp.のような真菌宿主)において転写を駆動し得る
プロモーターおよび他の発現制御配列の転写制御下にある適切な発現ベクターに挿入され
得、その結果、これらの哺乳動物GnT酵素のうちの1つ以上が、ヒト様複合型糖タンパ
ク質の生成のために選択された宿主細胞において活性に発現され得る。
【0140】
以下は、本発明の組成物および方法を例示する実施例である。これらの実施例は、限定
として解釈されるべきではなく、この実施例は、単に例示目的で含まれる。
【実施例】
【0141】
(実施例1:P.pastorisおよびK.lactisにおけるALG3遺伝子の
同定、クローニングおよび欠失)
縮重プライマーを、S.cerevisiae、H.sapiens、およびD.me
lanogaster由来のAlg3タンパク質配列のアラインメントに基づいて作製し
、P.pastorisゲノムDNA由来の83bp産物を増幅するために使用した:
5’−GGTGTTTTGTTTTCTAGATCTTTGCAYTAYCARTT−3
’および
5’−AGAATTTGGTGGGTAAGAATTCCARCACCAYTCRTG−
3’。得られたPCR産物を、pCR2.1ベクター(Invitrogen,Carl
sbad,CA)にクローニングしたところ、配列分析により、公知のALG3/RHK
1/NOT56ホモログ(Genbank NC_001134.2、AF309689
、NC_003424.1)に対する相同性が示された。その後、始めのPCR産物の1
929bp上流および2738bp下流を、以下の内部オリゴヌクレオチド:
【0142】
【化2】
を使用して、P.pastorisゲノムDNAライブラリーから増幅した(Boehm
,T.Yeast 1999 May;15(7):563−72)。得られたフラグメ
ントを、pCR2.1−TOPOベクター(Invitrogen)にクローニングし、
配列決定した。この配列から、S.cerevisiae ALG3遺伝子に対して35
%同一性および53%類似性を有するタンパク質をコードする1395bp ORFを、
(BLASTプログラムを使用して)同定した。この遺伝子を、PpALG3と名付けた
。
【0143】
PpALG3の配列を使用して、プライマーのセットを作製し、PpALG3遺伝子の欠失構築物を、PCR重複(Davidsonら,2002 Microbiol.148(Pt 8):2607−15)により生成した。以下のプライマーを使用して、PpALG3 ORFおよびKANR遺伝子の5’側および3’側の1kb領域を、それぞれ増幅した:
【0144】
【化3】
引き続いて、プライマーRCD142およびRCD147を使用して、3つの得られた
PCR産物を、単一の3.6kb alg3::KANRの欠失対立遺伝子に重ねた。
【0145】
(K.lactisにおけるALG3遺伝子の同定、クローニングおよび欠失)
S.cerevisiae、Drosophila melanogaster、Ho
mo sapiensなどに由来するALG3p配列を、K.lactis配列(PEN
DANT ESTデータベース)と整列させた。共通ホモログ中に存在するが、それらの
ホモログにおいて正確な配列は異なる、高い相同性の領域を使用して、株MG1/2(B
ianchiら,1987)に由来するゲノムDNAに対して指向する縮重プライマーの
対を作製した。ALG3の場合において、プライマー:
【0146】
【化4】
を用いるPCR増幅は、クローニングされ、配列決定された産物を生じ、推定翻訳産物は
、Alg3pタンパク質(S.cerevisiae Alg3pに対して>50%)に
対して高い程度の相同性を有することが示された。
【0147】
そのPCR産物を使用して、K.lactis株(MG1/2)由来のゲノムDNAの
サザンブロットを、高いストリンジェンシー下でプローブした(Sambrookら,1
989)。ハイブリダイゼーションは、単一の遺伝子と一致したパターンで観察された。
このサザンブロットを使用して、ゲノム遺伝子座をマッピングした。ゲノムフラグメント
を、ゲノムDNAを消化し、適切なサイズ範囲にあるこれらのフラグメントをpUCl9
に連結することによってクローニングして、K.lactisサブゲノムライブラリーを
作製した。このサブゲノムライブラリーを、E.coliに形質転換し、数百のクローン
を、プライマーKAL−1およびKAL−2を使用するコロニーPCRによって試験した
。推定KlALG3遺伝子および推定KlALG61遺伝子を含むクローンを、配列決定
し、オープンリーディングフレームを同定した。
【0148】
PCR重複法(Davidsonら,2002)を使用する、alg3::NATR欠
失対立遺伝子の構築物のためのプライマーを設計し、得られた欠失対立遺伝子を、2つの
K.lactis株に形質転換し、NAT耐性コロニーを選択した。これらのコロニーを
、PCRによってスクリーニングし、ALG3 ORFがoch1::NATR変異対立
遺伝子で置換された形質転換体を得た。
【0149】
(実施例2:ヒト様糖タンパク質の生成のための、α−1,2−マンノシダーゼ、Gn
TIおよびGnTIIを発現するalg3/ochl変異株の生成)
P.pastoris OCH1遺伝子の1215bpオープンリーディングフレーム
、ならびに2685bp上流および1175bp下流を、PCRによって増幅し(B.K
.Choiら(Proc.Natl.Acad.Sci.USA 2002に投稿);W
O02/00879もまた参照のこと;これらの各々は、本明細書中に参考として援用さ
れる)、pCR2.1−TOPOベクター(Invitrogen)にクローニングし、
pBK9と名付けた。複数の栄養要求マーカーを含むoch1ノックアウト株を作製する
ために、100μgのpJN329(SfiI制限部位と隣接したoch1::URA3
変異対立遺伝子を含むプラスミド)を、SfiIで消化し、これを使用して、P.pas
toris株JC308(Cereghinoら、Gene 263(2001)159
−169)をエレクトロポレーションにより形質転換した。ウラシルを欠く規定の培地上
で10日間室温でインキュベートした後、1000個のコロニーを拾い上げ、再び画線し
た。37℃で増殖できないが、室温では増殖するURA+クローンを、コロニーPCRに
供して、och1::URA3変異対立遺伝子の正確な組み込みについて試験した。予測
されるPCRパターンを示した1つのクローンを、YJN153と名付けた。ヒトプラス
ミノゲンのKringle 3ドメイン(K3)を、モデルタンパク質として使用した。
K3遺伝子を含むNeoRマーカーを挿入した(NeoR marked)プラスミドを、
YJN153株に形質転換し、K3を発現する得られた株を、BK64−1と名付けた(
B.K.Choiら,Proc.Natl.Acad.Sci.USA 2002に投稿
)。
【0150】
ゴルジUDP−N−アセチルグルコサミン輸送体をコードするプラスミドpPB103
(Kluyveromyces lactis MNN2−2遺伝子を含む)を、ベクタ
ーpDL02(Abeijonら(1996)Proc.Natl.Acad.Sci.
U.S.A.93:5963−5968)由来の平滑BglII−HidIIIフラグメ
ントを、P.pastoris ADE1遺伝子を含む、BglIIおよびBamHI消
化した平滑末端のpBLADE−SX(Cereghinoら(2001)Gene 2
63:159−169)にクローニングすることによって構築した。このプラスミドをE
coNIで線状にし、エレクトロポレーションによりBK64−1株に形質転換し、PC
R分析によりMNN2−2を含むことが確認された1つの株を、PBP1と名付けた。
【0151】
ヒト、マウス、ハエ、線虫(worm)および酵母供給源由来のいくつかのα−1,2−マンノシダーゼ遺伝子の触媒ドメインと融合した、S.cerevisiaeおよびP.pastoris由来のいくつかのI型またはII型膜タンパク質のリーダードメインのインフレーム融合物を含むマンノシダーゼ構築物のライブラリーを、作製した(例えば、WO02/00879(本明細書中に参考として援用される)を参照のこと)。このライブラリーを、P.pastoris HIS4組み込みベクター中に作製し、SalIで線状にし、PBP1株にエレクトロポレーションにより形質転換し、K3レポータータンパク質から放出されたグリカンを分析することにより、スクリーニングした。選択した1つの活性な構築物は、マウスα−1,2−マンノシダーゼIA(MmMannIA)遺伝子のN末端欠失物(これは、187個のヌクレオチドを欠いている)と融合した、酵母SEC12遺伝子の988〜1296のヌクレオチド(C末端)のキメラであった。この構築物を発現するP.pastoris株を、PBP2と名付けた。
【0152】
ヒト、線虫、カエルおよびハエ供給源由来のGnTI遺伝子の触媒ドメインを有する同
じリーダーライブラリーのインフレーム融合物を含むGnTI構築物のライブラリーを作
製した(WO02/00879)。このライブラリーを、P.pastoris ARG
4組み込みベクター中に作製し、AatIIで線状にし、PBP2株へのエレクトロポレ
ーションにより形質転換し、K3から放出されたグリカンを分析することによって、スク
リーニングした。選択した1つの活性な構築物は、ヒトGnTI遺伝子の欠失物(これは
、最初の154bpを欠いている)に融合したS.cerevisiae MNN9遺伝
子の最初の120bpのキメラであった。この構築物を発現するP.pastoris株
を、PBP3と名付けた。
【0153】
引き続いて、P.pastoris alg3::KANR欠失構築物を、上記のよう
に作製した。得られたPCR産物の約5μgを、PBP3株に形質転換し、コロニーを、
200μg/ml G418を含むYPD培地上で選択した。PCRによってスクリーニ
ングした20株のうちの1株が、alg3::KANR変異対立遺伝子の正確な組み込み
を含み、野生型対立遺伝子を欠くことを確認した。この株を、RDP27と名付けた。
【0154】
最後に、ヒトおよびラット供給源由来のGnTII遺伝子の触媒ドメインを有するリー
ダーライブラリーのインフレーム融合物から構成されるGnTII構築物のライブラリー
を作製した(WO02/00879)。このライブラリーを、薬物ノールセオスリシン(
nourseothricin)に対する耐性を付与するNSTR遺伝子を含むP.pa
storis組み込みベクター中に作製した。このライブラリープラスミドを、EcoR
Iで線状にし、RDP27株にエレクトロポレーションにより形質転換し、得られた株を
、精製K3から放出されたグリカンの分析によりスクリーニングした。
【0155】
(材料)
MOPS(カコジル酸ナトリウム)、塩化マンガン、UDP−ガラクトースおよびCM
P−N−アセチルノイラミン酸は、シグマ製であった。TFAは、Aldrich製であ
った。Spodoptera frugiperda由来の組換えラットα2,6−シア
リルトランスフェラーゼおよびウシ乳汁由来のβ1,4−ガラクトシルトランスフェラー
ゼは、Calbiochem製であった。タンパク質N−グリコシダーゼF、マンノシダ
ーゼ、およびオリゴ糖は、Glyko(San Rafael,CA)製であった。DE
AE ToyoPearl樹脂は、TosoHaas製であった。金属キレート「His
Bind」樹脂は、Novagen(Madison,WI)製であった。96ウェル溶
解液除去プレートは、Promega(Madison,WI)製であった。タンパク質
結合96ウェルプレートは、Millipore(Bedford,MA)製であった。
塩および緩衝剤は、Sigma(St.Louis,MO)製であった。MALDIマト
リクスは、Aldrich(Milwaukee,WI)製であった。
【0156】
(タンパク質精製)
Kringle3を、Beckman BioMek 2000サンプル処理ロボット
(Beckman/Coulter Ranch Cucamonga,CA)で、96
ウェル形式を使用して精製した。Kringle3を、C末端ヘキサヒスチジンタグを使
用して、発現培地から精製した。このロボット精製は、そのHisBind樹脂について
Novagenによって提供されたプロトコルの適応である。簡潔には、150uL(μ
L)の固定容積の樹脂を、96ウェルの溶解物結合プレートのウェル中に注ぎ、3容積の
水で洗浄し、そして5容積の50mM NiSO4を充填し、そして3容積の結合緩衝液
(5mMイミダゾール、0.5M NaCl、20mM Tris−HCL(pH7.9
))で洗浄する。タンパク質発現培地を、培地/PBS(60mM PO4、16mM
KC1、822mM NaCl(pH7.4))で3:2希釈し、そしてカラムに充填し
た。吸引後、カラムを10容積の結合緩衝液および6容積の洗浄緩衝液(30mMイミダ
ゾール、0.5M NaCl、20mM Tris−HCl(pH7.9))で洗浄し、
そしてタンパク質を、6容積の溶出緩衝液(1Mイミダゾール、0.5M NaCl、2
0mM Tris−HCl(pH7.9))で溶出する。溶出された糖タンパク質を、凍
結乾燥によって、乾燥するまでエバポレートする。
【0157】
(N連結グリカンの放出)
グリカンを、以前に報告された方法(Papacら、A.J.S.(1998)Glycobiology 8,445−454)の改変によって、糖タンパク質から放出および分離する。96ウェルのMultiScreen IP(Immobilon−Pメンブレン)プレート(Millipore)のウェルを、100uLのメタノールで湿らせ、3×150uLの水および50uLのRCM緩衝液(8M尿素、360mM Tris、3.2mM EDTA(pH8.6))で洗浄し、各添加後に、穏やかな減圧で吸引する。乾燥したタンパク質サンプルを、30uLのRCM緩衝液中に溶解し、そして10uLのRCM緩衝液を含むウェルに移す。ウェルを吸引し、そしてRCM緩衝液で2回洗浄する。タンパク質を、60uLの、RCM緩衝液中の0.1M DTTを、37℃で1時間に亘って添加して、還元した。ウェルを、300uLの水で3回洗浄し、そして60uLの0.1Mヨード酢酸を、暗中での室温にて30分間で添加して、カルボキシメチル化する。ウェルを、水で再度3回洗浄し、そしてメンブレンを、100uLの水中1%PVP 360の、室温で1時間の添加によって、ブロッキングする。ウェルを吸引し、そして300uLの水で3回洗浄し、そして30uLの10mM NH4HCO3(pH8.3)(1ミリ単位のN−グリカナーゼ(glycanase)(Glyko)を含む)の添加によって、脱グリコシル化する。37℃16時間の後、グリカンを含む溶液を、遠心分離によって取り出し、そして乾燥するまで蒸発させる。
【0158】
(飛行質量分析の、マトリクス補助レーザー脱離イオン化時間)
グリカンの分子量を、遅延抽出を使用して、Voyager DE PRO線形MAL
DI−TOF(Applied Biosciences)質量分析機を用いて決定した
。各ウェル由来の乾燥グリカンを、15uLの水中に溶解し、そして0.5uLを、ステ
ンレス鋼サンプルプレート上にスポットし、そして0.5uLのS−DHBマトリクス(
9mg/mLのジヒドロキシ安息香酸、1mg/mLの5−メトキシサリチル酸(1:1
の水/アセトニトリル 0.1% TFA中))と混合し、そして乾燥させた。
【0159】
パルス窒素レーザー(337nm)を、4nsのパルス時間で照射することによって、
イオンを発生させた。この機器を、125nsの遅延を用いる遅延抽出モードおよび20
kVの加速電圧で操作した。格子電圧は、93.00%であり、ガイドワイヤ電圧は、0
.10%であり、内部圧力は、5×10-7トル未満であり、そして低質量ゲートは、87
5Daであった。スペクトルを、合計100〜200のレーザーパルスから発生させ、そ
して2GHzのディジタイザーを用いて獲得した。Man5オリゴ糖を、外部分子量標準
として使用した。全てのスペクトルを、陽イオンモードでこの機器を用いて発生させた。
このスペクトルの概算の質量精度は、0.5%であった。
【0160】
(材料)
MOPS、カコジル酸ナトリウム、塩化マグネシウム、UDP−ガラクトースおよびC
MP−N−アセチルノイラミン酸は、Sigma,Saint Louis,MO製であ
った。トリフルオロ酢酸(TFA)は、Sigma/Aldrich,Saint Lo
uis,MO製であった。Spodoptera frugiperda由来の組換えラ
ットα−2,6−シアリルトランスフェラーゼおよびウシ乳汁由来のβ−1,4−ガラク
トシルトランスフェラーゼは、Calbiochem,San Diego,CA製であ
った。
【0161】
(β−N−アセチルヘキソサミニナーゼ設計)
グリカンを、以前に報告された方法(Papacら、A.J.S.(1998)Gly
cobiology 8,445−454)の改変によって、糖タンパク質から放出およ
び分離した。タンパク質を還元およびカルボキシメチル化し、そしてメンブレンをブロッ
キングした後、ウェルを、水で3回洗浄した。タンパク質を、30μlの10mM NH
4HCO3(pH8.3)(1ミリ単位のN−グリカナーゼ(glycanase)(Gl
yko,Novato,CA)を含む)の添加によって、脱グリコシル化した。37℃1
6時間の後、グリカンを含む溶液を、遠心分離によって取り出し、そして乾燥するまでエ
バポレートした。次いで、グリカンを、SC210Aスピード減圧(Thermo Sa
vant,Halbrook,NY)中で乾燥させた。乾燥したグリカンを、50mM
NH4Ac(pH5.0)中に37℃で一晩置き、そして1mUのヘキソース(hexo
s)(Glyko,Novato,CA)を添加した。
【0162】
(ガラクトシルトランスフェラーゼ反応)
約2mgのタンパク質(r−K3:hPg[PBP6−5])を、ニッケル親和性クロ
マトグラフィーによって精製し、0.1% TFAに対して広範に透析し、そして乾燥す
るまで凍結乾燥した。このタンパク質を、150μlの50mM MOPS、20mM
MnC12(pH7.4)中に再溶解した。32.5μg(533nmol)のUDP−
ガラクトースおよび4mUのβ 1,4−ガラクトシルトランスフェラーゼの添加後、サ
ンプルを、37℃で18時間インキュベートした。次いで、これらのサンプルを、MAL
DITOF質量分析による分析のために、0.1% TFAに対して透析した。
【0163】
ガラクトシルトランスフェラーゼと反応したタンパク質のスペクトルは、酵素なしでイ
ンキュベートした類似のタンパク質サンプルのスペクトルと比較して、2つのガラクトー
ス部分の付加と一致する、質量の増加を示した。次に、タンパク質サンプルを還元し、カ
ルボキシメチル化し、そしてPNGase Fで脱グリコシル化した。回収したN−グリ
カンを、MALDI−TOF質量分析によって分析した。タンパク質と反応したガラクト
シルトランスフェラーゼ由来の優勢なグリカンの質量は、2つのガラクトース部分の付加
と一致する質量分(325.4Da)だけ、コントロールグリカンの質量より多かった。
【0164】
(シアリルトランスフェラーゼ反応)
(ガラクトシルトランスフェラーゼ反応した)タンパク質を、10μlの50mM カ
コジル酸ナトリウム緩衝液(pH6.0)中に再懸濁した後、15μLの同じ緩衝液中に
溶解された300μg(488nmol)のCMP−N−アセチルノイラミン酸(CMP
−NANA)および5μl(2mU)の組換えα−2,6 シアリルトランスフェラーゼ
を添加した。37℃で15時間のインキュベート後、さらに200μgのCMP−NAN
Aおよび1mUのシアリルトランスフェラーゼを添加した。これらのタンパク質サンプル
を、さらに8時間インキュベートし、次いで、透析し、上記のようにMALDI−TOF
−MSによって分析した。
【0165】
シアリルトランスフェラーゼと反応した糖タンパク質のスペクトルは、出発物質(ガラ
クトシルトランスフェラーゼ反応後のタンパク質)の質量と比較して、質量の増加を示し
た。N−グリカンを、上記のように放出および分析した。優勢なグリカンの2つのイオン
付加物の質量の増加は、2つのシアル酸残基の付加と一致した(580Daおよび583
Da)。
【0166】
(実施例3)
(P.pastorisにおけるALG9遺伝子およびALG12遺伝子の同定、クロ
ーニングおよび欠失)
実施例1と同様に、ALG9p配列およびALG12配列(それぞれ、S.cerev
isiae、Drosophila melanogaster、Homo sapie
nsなどに由来する)を整列し、そして高い相同性の領域を、縮重プライマーを設計する
ために使用する。これらのプライマーを、P.pastoris由来のゲノムDNAに対
するPCR反応において使用する。得られた最初のPCR産物をサブクローニングし、配
列決定し、そして高いストリンジェンシーで、P.pastoris由来のゲノムDNA
のサザンブロットをプローブするために使用した(Sambrookら、1989)。ハ
イブリダイゼーションを観察する。このサザンブロットを使用して、遺伝子座位のマッピ
ングを行った。ゲノムフラグメントを、ゲノムDNAを消化し、そして適切なサイズ範囲
のフラグメントをpUCl9に連結して、P.pastorisサブゲノムライブラリー
を作製することによって、クローニングした。このサブゲノムライブラリーを、E.co
li中に形質転換し、そして数百個のクローンを、最初のPCR産物の配列に基づいて設
計されたプライマーを使用して、コロニーPCRによって試験する。予測された遺伝子を
含むクローンを配列決定し、そしてオープンリーディングフレームを同定する。alg9
::NAIR欠失対立遺伝子の構築のためのプライマーを、PCR重複法(Davids
onら、2002)を使用して設計する。得られた欠失対立遺伝子を、2つのP.pas
toris株に形質転換し、そしてNAT耐性コロニーを選択する。これらのコロニーを
、PCRによってスクリーニングし、そしてALG9 ORFがochl::NATR変
異体対立遺伝子で置換された形質転換体を得る。一般に、以下を参照のこと:Cipol
loら、Glycobiology 2002(12)11:749−762;Chan
tretら、J.Biol.Chem.Jul.12,2002(277)28:258
15−25822;Cipolloら、J.Biol.Chem.Feb.11,200
0(275)6:4267−4277;Burdaら、Proc.Natl.Acad.
Sci.U.S.A.July 1996(93):7160−7165;Karaog
luら、Biochemistyy 2001,40,12193−12206;Gri
mmeら、J.Biol.Chem.July 20,2001(276)29:277
31−27739;Verostekら、J.Biol.Chem.June 5,19
93(268)16:12095−12103;Huffakerら、Proc.Nat
l.Acad.Sci.U.S.A.Dec.1983(80):7466−7470。
【0167】
(実施例4)
(Xanthomonas Manihotis由来のα1,2−3マンノシダーゼの
同定、クローニングおよび発現)
Xanthomonas Manihotis由来のα1,2−3マンノシダーゼは、
2つの活性(α−1,2マンノシダーゼおよびα−1,3 マンノシダーゼ)を有する。
本発明の方法はまた、これらの活性を有する2つの独立したマンノシダーゼを使用し得、
これらは、選択された目的の生物から同様に同定およびクローニングされ得る。
【0168】
Landryら、によって記載されるように、α−マンノシダーゼは、Xanthom
onas sp.(例えば、Xanthomonas manihotis)から精製さ
れ得る。X.manihotisは、American Type Culture C
ollection(ATCCカタログ番号49764)(Xanthomonas a
xonopodis StarrおよびXanthomonas manihotis
(Arthaud−Berthet)Starrとして寄託されたGarces pat
hovar manihotis)から購入され得る。酵素を、粗細胞抽出物から、以前
に記載されたように精製する(Wong−Madden,S.T.およびLandry,
D.(1995)Purification and characterizatio
n of novel glycosidases from the bacteri
al genus Xanthomonas;およびLandry,D.米国特許第6,
300,113 B1号、Isolation and composition of
novel Glycosidases)。マンノシダーゼの精製後、いくつかの方法
のうちの1つを使用して、ペプチド配列タグを得る(例えば、W.Quadroni M
ら、(2000).A method for the chemical gener
ation of N−terminal peptide sequence tag
s for rapid protein identification.Anal
Chem(2000)Mar 1;72(5):1006−14;Wilkins MR
ら、Rapid protein identification using N−t
erminal”sequence tag”and amino acid anal
ysis.Biochem Biophys Res Commun.(1996)Ap
r 25;221(3):609−13;およびTsugita A.(1987)De
velopments in protein microsequencing.Ad
v Biophys(1987)23:81−113を参照のこと)。
【0169】
次いで、上記方法を使用して作製された配列タグを使用して、当業者に周知の方法を使
用して、縮重プライマーのセットを作製する。縮重プライマーを使用して、ポリメラーゼ
連鎖反応(例えば、Taqポリメラーゼキットを、製造者の指示に従って使用する)にお
いてDNA増幅をプライムして、DNAフラグメントを増幅する。増幅されたDNAフラ
グメントをプローブとして使用して、例えば、目的の酵素をコードするゲノム片を同定お
よび単離(クローニング)するための、標準的なサザンDNAハイブリダイゼーション技
術を使用して、所望のマンノシダーゼをコードする遺伝子を含むDNA分子を単離する。
ゲノムDNA分子を配列決定し、推定オープンリーディングフレームおよびコード配列を
同定する。次いで、目的のグリコシダーゼをコードする適切な発現構築物を、本明細書中
に記載される方法および当該分野で周知の方法を使用して、作製し得る。
【0170】
α1,2−3 マンノシダーゼ活性(またはその触媒的に活性なフラグメント)をコー
ドする配列を含む核酸フラグメントを、本明細書中に示される方法に従って、適切な宿主
細胞でのこれらの活性の発現(好ましくは、標的化された発現)のために、適切な発現ベ
クター中にクローニングする。
【0171】
(実施例5)
(P.pastorisにおけるALG6遺伝子の同定、クローニング、および発現)
実施例1と同様に、S.cerevisiae、Drosophia melanog
aster、Homo sapiensなどに由来するALG6p配列を整列し、、そし
て高い相同性の領域を変性プライマーを産生するために使用する。これらのプライマーは
、P.pastoris由来のゲノムDNAにおけるPCR反応に用いられる。得られた
初めのPCR産物をサブクローン化し、配列を決定して、そして高ストリンジェンシーを
有するP.pastoris由来のゲノムDNAのサザンブロットのプローブとして使用
する(Sambrookら、1989)。ハイブリダイゼーションが観察される。このサ
ザンブロットを、ゲノム座位をマップするために使用する。ゲノムDNAを消化し、そし
てそれらのフラグメントを、適切なサイズ範囲で、pUC19中にライゲーションして、
ゲノムフラグメントをクローン化することで、P.pastorisサブゲノムライブラ
リーを産生する。このサブゲノムライブラリーを、E.coliに形質転換し、そして、
初めのPCR産物の配列に基づいて設計されたプライマーを用いて、コロニーPCRによ
って、数百クローンをテストする。予想した遺伝子を含むクローンの配列を決定し、そし
てオープンリーディングフレームを同定する。PCR重複法(Davidsonら、20
02)を用いて、alg6::NATR欠失アレルの構築のためのプライマーを設計し、
その結果得られた欠失対立遺伝子を2つのP.pastoris株および選択されたNA
T抵抗性コロニー中に形質転換した。これらのコロニーを、PCRによってスクリーニン
グし、ALG6 ORFが,och1::NATR変異対立遺伝子に置換される形質転換
体を得る。例えば、Imbachら、Proc.Natl.Acad.Sci.USA
June 1999(96)6982−698を参照のこと。
【0172】
Alg6p(またはその触媒活性フラグメント)をコ−ドする配列を含む核酸フラグメ
ントが、本明細書中に記載された方法で、適切な宿主細胞におけるこれらの活性の発現お
よび、好ましくは標的発現のための適切な発現ベクター中にクローン化する。クローン化
ALG6遺伝子は、任意の適切なプロモーターの制御で得られ、過剰発現を達成し得る。
その遺伝子自体のプロモーターの制御下にある遺伝子の発現さえも可能である。マルチコ
ピープラスミドの発現は、高レベルの発現(「過剰発現」)を生じる。
【0173】
(実施例6)
(抗体機能性を高める分岐したGlcNAcsを産生するためのGnTIIIのクロー
ニングおよび発現)
(A.背景)
N−アセチルグルコサミルトランスフェラーゼIIIによるGlcNAc2Man3Gl
cNAc2構造へのN−アセチルグルコサミンの付加によって、いわゆる分岐したNグリ
カンを生じる(図3を参照のこと)。この構造は、より大きな抗体依存性細胞傷害性(A
DCC)に関係する(Umanaら、1999)。
【0174】
宿主細胞(例えば、分岐したNグリカンを用いて糖タンパク質を産生できる酵母株)は
、宿主細胞中にGnTIII活性を導入することによって、本発明に従って操作される。
好ましくは、宿主細胞は、異種の細胞シグナル標的ペプチドに(例えば、本発明のライブ
ラリーおよび関連した方法を使用して必要に応じて融合されたGnTIII活性(例えば
、図32に示したマウスGnTIIIのような哺乳動物)またはその酵素活性を有するド
メインコードする核酸を用いて、形質転換される。
【0175】
IgGは、3つのジスルフィド架橋を介するヒンジ領域で相互結合した、2つの重鎖(
図30におけるVH、CH1、CH2およびCH3)、および2つの軽鎖(図30におけるV
L、CL)からなる。軽鎖(ドメインVLおよびドメインCL)は、別のジスルフィド架橋に
よって、重鎖のCH1部分に結合し、そしてCH1フラグメントおよびVHフラグメントと
一緒になって、いわゆるFab領域を形成する。抗原は、Fab領域の末端部分に結合す
る。IgGのFc領域は、CH3、CH2およびヒンジ領域からなり、いわゆるエフェクタ
ー機能の発揮に関連する(下記を参照のこと)。
【0176】
抗体の第1機能は抗原との結合である。しかし、抗原との結合が、その抗原を直接的に
不活化しない限り(例えば、細菌毒素の場合)、いわゆるエフェクター機能が増強されな
ければ、単なる結合は意味を成さない。IgGサブクラスの抗体は、以下の2つの主要な
エフェクター効果を発揮する:補体系の活性化および貪食作用の導入。補体系は、貪食作
用の活性化および細胞膜の溶解による破壊において、炎症の制御に関係する血清タンパク
質の複雑な群からなる。補体活性化は、C1複合体の近接した2つのIgGのFc部分と
の結合で開始する。C1は、1つの分子(C1q)および2つの分子(C1rおよびC1
s)からなる。貪食作用は、IgGのFcフラグメントとFc−γ−レセプター(図30
におけるFcγRI、IIおよびIII)との間における相互作用を介して起こる。Fc
レセプターは、第1に免疫システムのエフェクター細胞表面、特にマクロファージ、単核
細胞、骨髄細胞および樹状細胞の表面に、主に発現する。
【0177】
CH2部分は、アスパラギン297(Asp297)における保存されたNグリコシレ
ーション部位を有する。Asp297 Nグリカンは、高度に異種性であり、Fcレセプ
ター結合および補体活性に影響することが知られている。IgGの少数のみ(すなわち、
約15%〜20%)が、ジシアリル化を受け、そしてIgGの3%〜10%がモノシアリ
ル化したNグリカンを有する(Jefferis,R.,Glycosylation
of human IgG Antibodies.BioOhra,2001の総説を
参照のこと)。興味深いことに、補体活性およびFcレセプター結合が可能な十分機能的
な抗体にとって必要であることが示された最小のNグリカン構造は、末端Nアセチルグル
コサミン残基(GlcNAc2Man3)を有する五単糖類である(Jefferis,R
.,Glycosylation of human IgG Antibodies.
BioPharm,2001の総説を参照のこと)。GlcNAc2Man3Nグリカン
以下またはAsp297の位置にNグリコシル化を有さない抗体は、それでも抗原と結合
することが可能なようであるが、重要な下流の作用(例えば、貪食作用および補体活性化
)を活性化しない可能性がある。さらに、Asp297に結合した真菌型のNグリカンを
有する抗体は、おそらく哺乳動物において免疫応答を誘導し、治療的糖タンパク質として
その抗体を役に立たないものにする。
【0178】
(B.GnTIIIのクローニングおよび発現)
TMドメインを欠くマウスGnTIIIタンパク質の一部をコードするDNAフラグメ
ントは、以下のプライマーを用いて、マウス(あるいは他の哺乳動物)のゲノムDNAか
らPCR増幅される:順方向 5’−TCCTGGCGCGCCTTCCCGAGAGA
ACTGGCCTCCCTC−3’および5’−AATTAATTAACCCTAGCC
CTCCGCTGTATCCAACTTG−3’逆方向プライマー。それらのプライマー
は、リーダーライブラリーとの融合に適したベクターへのクローニングに使用されるAs
cIおよびPacI制限部位を含む。マウスGnTIIIの核酸配列およびアミノ酸配列
を。図32に示す。
【0179】
(C.免疫グロブリンコード配列のクローニング)
抗体の可変領域(プライマー配列を含む)のクローニングのためのプロトコルは、以前
公開されている。抗体およびコ−ド遺伝子の供給源は、とりわけ、インビトロ免疫化ヒト
B細胞(例えば、Borreback,C.A.ら(1988)Proc.Natl.A
cad.Sci.USA85,3995−3999、末梢血リンパ球細胞または単一ヒト
B細胞(例えば、Lagerkvist,A.Cら(1995) Biotechniq
ues 18,862−869;およびTerness,P.ら(1997) Hum.
Immunol.56,17−27を参照のこと)またはハイブリドーマ細胞株の産生
を可能にする、ヒト免疫グロブリン部位を含むトランスジェニックマウスであり得る。
【0180】
標準的な組み換えDNA技術を用いて、抗体をコードする核酸配列がクローン化され得
る。目的の免疫グロブリンをコードする遺伝情報の供給源は、代表的に、目的の細胞(例
えば、血液リンパ球細胞またはハイブリドーマ細胞株)由来の総RNA調製物である。例
えば、特異的プライマーを用いたプロトコルに基づいたPCRを用いて、可変領域は、I
gGCH1ドメインとハイブリダイズする配列特異的プライマー、およびマウスκ定常領
域のアミノ酸111〜118をコードする第2プライマーから開始される逆転写を解して
クローン化され得る。さらに、DNAをコードするVHおよびVkは、以前公開された方
法で増幅される(例えば、Graziano,R.Fら、(1995) J Immun
ol. 155(10):p.4996−5002;Welschof,Mら(1995
) J. Immunol. Methods 179,203−214;および Or
landi,Rら(1988 Proc.Natl.Acad Sci.USA 86:
3833)を参照のこと)。全免疫グロブリンのクローニング手順(重鎖および軽鎖)も
また公開されている(例えば、Buckel,P.ら(1987) Gene 51:1
3−19;Recinos A3rdら(1994) Gene 149:385−386
;(1995) GeneJun 9;158(2):311−2;および Recin
os A3rdら(1994) Gene Nov 18;149(2)385−6を参照
のこと)。抗体フラグメントおよび抗体発現構築物のクローニングおよび産生のさらなる
プロトコルは、Antibody Engineering,R.Kontermann
およびS. Dubel(2001),Editors,Springer Verla
g:Berlin Heidelberg New Yorkに記載されている。
【0181】
免疫グロブリンの重鎖および軽鎖をコードする真菌発現プラスミドは、記載されている
(例えば、Abdel−Salam,H.A.ら(2001)Appl. Microb
iol.Biotechnol. 56:157−164;および Ogunijimi
,A.Aら(1999) Biotechnology Letters 21:561
−567を参照のこと)。従って、免疫グロブリンの定常領域を有する発現プラスミドを
産生し得る。これらの発現ベクター中への可変領域のクローニングを促進するために、適
切な制限部位が可変領域の末端に非常に近い位置に配置され得る。定常領域は、単一制限
酵素消化/ライゲーション実験によって定常領域にインフレームで融合するような様式で
、可変領域を構築され得る。図31は、そのような発現構築の体系的概要を示す。その発
現構築は、プロモーター、転写ターミネーター、組込み標的ドメイン、および選択マーカ
ーさえも容易に変更できるように、非常に修正可能な方法で設計された。
【0182】
図31に示すように、選択したVLドメインおよびVHドメインは、それぞれ、CL領域
およびCH領域に、インフレームで容易にクローン化され得る。第1の組込みは、P.p
astoris AOX部位(または別の真菌細胞の相同部位)に標的化され、そしてメ
タノール誘導性AOXプロモーターは発現を誘導する。あるいは、任意の他の所望の構成
的または誘導的プロモーターカセットが使用され得る。このように、所望の場合、5’A
OX領域および3’AOX領域、ならびに転写ターミネーター(TT)フラグメントが、
異なったTT、プロモーターおよび組込み標的ドメインで容易に置換され、発現を最適化
し得る。第1に、標準KEXプロテアーゼ部位を有するα因子分泌シグナルは、重鎖およ
び軽鎖の分泌を促進するために使用される。さらに、発現ベクターの特性は、標準技術を
用いてさらに精製され得る。
【0183】
Ig発現ベクター(例えば、上述のベクター)は、好ましくは宿主細胞のゴルジ装置内
にGnTIIIを発現する本発明の宿主細胞中に導入される。そのような宿主細胞におい
て発現されるIg分子は、分岐したGlcNacsを有するNグリカンを含む。
【0184】
(実施例7)
GnT−IV(UDP−GlcNAc:α−1,3−D−マンノシドβ−1,4−N−
アセチルグルコサミニルトランスフェラーゼIV)およびGnT−V(β−1−6−N−
アセチルグルコサミニルトランスフェラーゼのクローニングおよび発現)
GnTIVをコードするcDNAを、ウシおよびヒト細胞から単離した(Minowa
、M.T.ら(1998) J.Biol.Chem.273(19),11556−1
1562;)およびYoshida,A.ら(1999) Glycobiology
9(3),303−310)。TMドメインを欠くヒトGnT−IVタンパク質(図33
)の全長および一部をコードするDNAフラグメントを、各々以下のプライマーを用いて
、cDNAライブラリーからPCR増幅した:順方向 5’−AATGAGATGAGG
CTCCGCAATGGAACTG−3’,5’−CTGATTGCTTATCAACG
AGAATTCCTTG−3’,および逆方向5’−TGTTGGTTTCTCAGAT
GATCAGTTGGTG−3’プライマー。
その結果生じるPCR産物をクローン化し、配列を決定した。
【0185】
同様に、GnT−Vたんぱく質をコードする遺伝子は、いくつかの哺乳動物(マウスを
含む)から単離された(例えば、Alverez,Kら Glycobiology 1
2(7),389−394(2002)を参照のこと)。TMドメインを欠くマウスGn
T−Vたんぱく質(図34)の全長および一部をコードするDNAフラグメントを、各々
以下のプライマーを用いて、cDNAライブラリーからPCR増幅した:順方向5’−A
GAGAGAGATGGCTTTCTTTTCTCCCTGG−3’,5’−AAATC
AAGTGGATGAAGGACATGTGGC−3’、および逆方向5’−AGCGA
TGCTATAGGCAGTCTTTGCAGAG−3プライマー。’その結果生じるP
CR産物をクローン化し、配列を決定した。
【0186】
GnT IVまたはV(またはその触媒活性フラグメント)をコードする配列を含む核
酸フラグメントは、本明細書中に記載された方法で、適切な宿主細胞におけるこれらの活
性の発現および、好ましくは標的発現のための適切な発現ベクター中にクローン化する。
【0187】
【化5】
【特許請求の範囲】
【請求項1】
非ヒト真核生物宿主細胞においてヒト様糖タンパク質を産生するための方法であって、該方法は、糖残基を脂質連結オリゴ糖構造の1,6アームに移動させる宿主細胞において、1種以上の酵素の活性を減退させるかまたは減少させる工程を包含する、方法。
【請求項2】
前記宿主細胞に少なくとも1つのグリコシダーゼ活性を導入する工程をさらに包含する、請求項1に記載の方法。
【請求項3】
前記少なくとも1つのグリコシダーゼ活性は、マンノシダーゼ活性である、請求項2に記載の方法。
【請求項4】
N−グリカンを産生する工程をさらに包含する、請求項1に記載の方法。
【請求項5】
前記N−グリカンは、GlcNAcManxGlcNAc2構造を有し、ここでXは、3、4または5である、請求項4に記載の方法。
【請求項6】
請求項5に記載の方法であって、前記宿主細胞内で1種以上の酵素活性を発現させて、GlcNAc2Man3GlcNAc2構造を作製する工程をさらに包含し、該酵素活性は、グリコシダーゼ活性およびグリコシルトランスフェラーゼ活性から選択される、方法。
【請求項7】
前記活性は、α−1,2マンノシダーゼ活性、α−1,3マンノシダーゼ活性およびGnTII活性から選択される、請求項6に記載の方法。
【請求項8】
請求項1に記載の方法であって、少なくとも1種の減退または減少された酵素は、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を有する酵素;ドリチル−P−Man:Man6GlcNAc2−PP−ドリチルα−1,2マンノシルトランスフェラーゼ活性を有する酵素、およびドリチル−P−Man:Man7GlcNAc2−PP−ドリチルα−1,6マンノシルトランスフェラーゼ活性を有する酵素からなる群より選択される、方法。
【請求項9】
請求項1に記載の方法であって、前記減退または減少された酵素は、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を有する、方法。
【請求項10】
前記酵素は、該酵素活性をコードする宿主細胞遺伝子の変異によって減退されるかまたは減少される、請求項1に記載の方法。
【請求項11】
前記変異は、前記酵素活性をコードする宿主細胞遺伝子の部分的な欠損または全体的な欠損である、請求項10に記載の方法。
【請求項12】
前記糖タンパク質は、7個またはそれより少ないマンノース残基を有するN−グリカンを含む、請求項1に記載の方法。
【請求項13】
前記糖タンパク質は、3個またはそれより少ないマンノース残基を有するN−グリカンを含む、請求項1に記載の方法。
【請求項14】
前記糖タンパク質は、ガラクトース、GlcNAc、シアル酸、およびフコースからなる群より選択される1種以上の糖を含む、請求項1に記載の方法。
【請求項15】
前記糖タンパク質は、構造NeuNAc−Gal−GlcNAc−Manを含む少なくとも1つのオリゴ糖分枝を含む、請求項1に記載の方法。
【請求項16】
前記宿主は、下等真核生物細胞である、請求項1に記載の方法。
【請求項17】
請求項1に記載の方法であって、前記宿主細胞は、以下:Pichia pastoris、Pichia finlandica、Pichia trehalophila、Pichia koclamae、Pichia membranaefaciens、Pichia opuntiae、Pichia thermotolerans、Pichia salictaria、Pichia guercuum、Pichia pijperi、Pichia stiptis、Pichia methanolica、Pichia sp.、Saccharomyces cerevisiae、Saccharomyces sp.、Hansenula polymorpha、Kluyveromyces sp.、Candida albicans、Aspergillus nidulans、Aspergillus niger、Aspergillus oryzae、Trichoderma reesei、Chrysosporium lucknowense、Fusarium sp.、Fusarium gramineum、Fusarium venenatumおよびNeurospora crassaからなる群より選択される、方法。
【請求項18】
前記宿主細胞は、開始α−1,6マンノシルトランスフェラーゼ活性の発現をさらに欠損している、請求項1に記載の方法。
【請求項19】
前記宿主細胞は、P.pastorisのOCH1変異体である、請求項18に記載の方法。
【請求項20】
前記宿主細胞は、GnTIおよびUDP−GlcNAcトランスポーター活性を発現する、請求項1に記載の方法。
【請求項21】
前記宿主細胞は、UDP特異的ジホスファターゼジホスファターゼ活性またはGDP特異的ジホスファターゼジホスファターゼ活性を発現する、請求項1に記載の方法。
【請求項22】
前記宿主から前記糖タンパク質を単離する工程をさらに包含する、請求項1に記載の方法。
【請求項23】
請求項22に記載の方法であって、インビトロにおいて、前記単離された糖タンパク質を、少なくとも1種のさらなるグリコシル化反応に供し、続いて前記宿主からの単離に供する工程をさらに包含する、方法。
【請求項24】
請求項1に記載の方法であって、GlcNAcMan3GlcNAc2またはGlcNAc2Man3GlcNAc2の産生に関与する1種以上の酵素をコードする核酸分子を、前記宿主に導入する工程をさらに包含する、方法。
【請求項25】
前記少なくとも1種の酵素は、マンノシダーゼ活性を有する、請求項24に記載の方法。
【請求項26】
請求項25に記載の方法であって、前記酵素は、α−1,2−マンノシダーゼ活性を有し、かつマウス、ヒト、Lepidoptera、Aspergillus nidulans、C.elegans、D.melanogaster、またはBacillus sp.に由来する、方法。
【請求項27】
前記酵素は、α−1,3−マンノシダーゼ活性を有する、請求項25に記載の方法。
【請求項28】
前記少なくとも1種の酵素は、グリコシルトランスフェラーゼ活性を有する、請求項24に記載の方法。
【請求項29】
前記グリコシルトランスフェラーゼ活性は、GnTIおよびGnTIIからなる群より選択される、請求項28に記載の方法。
【請求項30】
少なくとも1種の酵素は、該酵素の触媒ドメインと細胞標的シグナルペプチドとの間で融合ペプチドを形成することによって局在化される、請求項24に記載の方法。
【請求項31】
請求項30に記載の方法であって、前記融合ペプチドは、少なくとも1つの遺伝子構築物によってコードされ、該遺伝子構築物は、細胞標的シグナルペプチドをコードするDNAフラグメントと、グリコシル化酵素またはその触媒活性フラグメントをコードするDNAフラグメントとのインフレームでの結合によって形成される、方法。
【請求項32】
請求項31に記載の方法であって、前記コードされた標的シグナルペプチドは、マンノシルトランスフェラーゼ、ジホスファターゼ、プロテアーゼ、GnT I、GnT II、GnT III、GnT IV、GnT V、GnT VI、GalT、FT、およびSTからなる群のメンバーに由来する、方法。
【請求項33】
請求項31に記載の方法であって、前記触媒ドメインは、GnT I、GnT II、GnT III、GnT IV、GnT V、GnT VI、GalT、FucosylトランスフェラーゼおよびSTからなる群のメンバーに由来する、グリコシダーゼまたはグリコシルトランスフェラーゼをコードし、かつ該触媒ドメインは、小器官内の他の代表的な酵素の平均最適pHの、1.4pH単位内に最適pHを有し、該小器官おいて、該酵素は、局在化されるかまたは5.1と8.0との間のpHにて最適な活性を有する、方法。
【請求項34】
請求項31に記載の方法であって、前記核酸分子は、UDP−GlcNAcトランスフェラーゼ、UDP−ガラクトシルトランスフェラーゼ、GDP−フコシルトランスフェラーゼ、CMP−シアリルトランスフェラーゼ、UDP−GlcNAcトランスポーター、UDP−ガラクトーストランスポーター、GDP−フコーストランスポーター、CMP−シアル酸トランスポーター、およびヌクレオチドジホスファターゼからなる群より選択される1種以上の酵素をコードする、方法。
【請求項35】
前記宿主は、GnTIおよびUDP−GlcNAcトランスポーター活性を発現する、請求項31に記載の方法。
【請求項36】
前記宿主は、UDP特異的ジホスファターゼ活性またはGDP特異的ジホスファターゼ活性を発現する、請求項31に記載の方法。
【請求項37】
請求項1に記載の方法であって、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を欠損している宿主に、GlcNAcMan4GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項38】
請求項1に記載の方法であって、ドリチル−P−Man:Man6GlcNAc2−PP−ドリチルα−1,2マンノシルトランスフェラーゼまたはドリチル−P−Man:Man7GlcNAc2−PP−ドリチルα−1,6マンノシルトランスフェラーゼ活性を欠損している宿主に、GlcNAcMan4GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項39】
前記核酸分子は、α−1,2マンノシダーゼ、UDP GlcNAcトランスポーターおよびGnTlからなる群より選択される少なくとも1種の酵素をコードする、請求項37または38に記載の方法。
【請求項40】
請求項39に記載の方法であって、前記欠損している宿主細胞に、GlcNAclMan4GlcNAc2構造をGlcNAclMan3GlcNAc2構造に転換するためのα−1,3マンノシダーゼ活性またはα−1,2/α−1,3マンノシダーゼ活性をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項41】
請求項1に記載の方法であって、前記宿主に、GlcNAc2Man3GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項42】
前記少なくとも1種の酵素は、GnTIIである、請求項41に記載の方法。
【請求項43】
請求項1に記載の方法であって、前記宿主細胞に、グリコシルトランスフェラーゼ、フコシルトランスフェラーゼ、ガラクトシルトランスフェラーゼ、N−アセチルガラクトサミニルトランスフェラーゼ、N−アセチルグリコサミニルトランスフェラーゼおよびスルホトランスフェラーゼからなる群より選択される少なくとも1種の哺乳動物グリコシル化酵素をコードする少なくとも1種の核酸分子を導入する工程をさらに包含する、方法。
【請求項44】
請求項1に記載の方法であって、宿主細胞をDNAライブラリーで形質転換して、該ライブラリーから誘導された少なくとも1種のグルコシル化酵素を発現する、遺伝学的に混合された細胞集団を作製する工程をさらに包含し、該ライブラリーは、少なくとも2つの異なる遺伝子構築物を含み、該構築物のうちの少なくとも一方は、細胞標的シグナルペプチドをコードするDNAフラグメントを含み、該細胞標的シグナルペプチドは、グリコシル化酵素またはその触媒活性フラグメントをコードするDNAフラグメントと、インフレームで結合される、方法。
【請求項45】
請求項1または44に記載の方法によって産生される、宿主細胞。
【請求項46】
請求項1または44に記載の方法によって産生される、ヒト様糖タンパク質。
【請求項47】
図6(P.pastoris ALG 3遺伝子)の少なくとも45個連続するヌクレオチド残基を含むかまたはそれらからなる、核酸分子。
【請求項48】
請求項47に記載の核酸分子を含む、ベクター。
【請求項49】
請求項47に記載の核酸分子を含む、宿主細胞。
【請求項50】
P.pastoris細胞であって、ここで、図6(P.pastoris ALG 3遺伝子)の配列は変異しており、それによって、該細胞のグリコシル化が変更される、P.pastoris細胞。
【請求項51】
脂質連結オリゴ糖のグリコシル化の程度を高めるための方法であって、該方法は、宿主細胞においてα−1,3グリコシルトランスフェラーゼ活性を増加させる工程を包含する、方法。
【請求項52】
脂質連結オリゴ糖のグリコシル化の程度を高めるための方法であって、
該方法は、宿主細胞においてオリゴ糖トランスフェラーゼ活性の基質特異性を減退させる工程を包含する、方法。
【請求項53】
非哺乳動物宿主細胞において、分岐したGlcNAcを含むN−グリカンを有する免疫グロブリンポリペプチドを産生するための方法であって、該方法は、該宿主細胞においてGnTIII活性を発現させる工程を包含する、方法。
【請求項54】
分岐したGlcNAcを含むN−グリカンを有する免疫グロブリンを産生する、非哺乳動物宿主細胞。
【請求項55】
請求項54に記載の宿主細胞によって産生される、免疫グロブリン。
【請求項56】
非哺乳動物宿主細胞において、分岐したGlcNAcを含むN−グリカンを有するポリペプチドを産生するための方法であって、該方法は、該宿主細胞においてGnTIII活性を発現させる工程を包含する、方法。
【請求項57】
分岐したGlcNAcを含むN−グリカンを有するポリペプチドを産生する、非ヒト宿主細胞。
【請求項58】
請求項57に記載の宿主細胞によって産生される、ポリペプチド。
【請求項59】
非ヒト真核生物宿主細胞においてヒト様糖タンパク質を産生するための方法であって、該方法は、該宿主細胞から、alg遺伝子活性を減退または減少させる工程、および該宿主細胞に少なくとも1種のグリコシダーゼ活性を導入する工程を包含する、方法。
【請求項60】
トリマンノースコアに結合した少なくとも2つのGlcNAcを含むN−グリカンを有するヒト様糖タンパク質を産生するための、方法。
【請求項1】
非ヒト真核生物宿主細胞においてヒト様糖タンパク質を産生するための方法であって、該方法は、糖残基を脂質連結オリゴ糖構造の1,6アームに移動させる宿主細胞において、1種以上の酵素の活性を減退させるかまたは減少させる工程を包含する、方法。
【請求項2】
前記宿主細胞に少なくとも1つのグリコシダーゼ活性を導入する工程をさらに包含する、請求項1に記載の方法。
【請求項3】
前記少なくとも1つのグリコシダーゼ活性は、マンノシダーゼ活性である、請求項2に記載の方法。
【請求項4】
N−グリカンを産生する工程をさらに包含する、請求項1に記載の方法。
【請求項5】
前記N−グリカンは、GlcNAcManxGlcNAc2構造を有し、ここでXは、3、4または5である、請求項4に記載の方法。
【請求項6】
請求項5に記載の方法であって、前記宿主細胞内で1種以上の酵素活性を発現させて、GlcNAc2Man3GlcNAc2構造を作製する工程をさらに包含し、該酵素活性は、グリコシダーゼ活性およびグリコシルトランスフェラーゼ活性から選択される、方法。
【請求項7】
前記活性は、α−1,2マンノシダーゼ活性、α−1,3マンノシダーゼ活性およびGnTII活性から選択される、請求項6に記載の方法。
【請求項8】
請求項1に記載の方法であって、少なくとも1種の減退または減少された酵素は、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を有する酵素;ドリチル−P−Man:Man6GlcNAc2−PP−ドリチルα−1,2マンノシルトランスフェラーゼ活性を有する酵素、およびドリチル−P−Man:Man7GlcNAc2−PP−ドリチルα−1,6マンノシルトランスフェラーゼ活性を有する酵素からなる群より選択される、方法。
【請求項9】
請求項1に記載の方法であって、前記減退または減少された酵素は、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を有する、方法。
【請求項10】
前記酵素は、該酵素活性をコードする宿主細胞遺伝子の変異によって減退されるかまたは減少される、請求項1に記載の方法。
【請求項11】
前記変異は、前記酵素活性をコードする宿主細胞遺伝子の部分的な欠損または全体的な欠損である、請求項10に記載の方法。
【請求項12】
前記糖タンパク質は、7個またはそれより少ないマンノース残基を有するN−グリカンを含む、請求項1に記載の方法。
【請求項13】
前記糖タンパク質は、3個またはそれより少ないマンノース残基を有するN−グリカンを含む、請求項1に記載の方法。
【請求項14】
前記糖タンパク質は、ガラクトース、GlcNAc、シアル酸、およびフコースからなる群より選択される1種以上の糖を含む、請求項1に記載の方法。
【請求項15】
前記糖タンパク質は、構造NeuNAc−Gal−GlcNAc−Manを含む少なくとも1つのオリゴ糖分枝を含む、請求項1に記載の方法。
【請求項16】
前記宿主は、下等真核生物細胞である、請求項1に記載の方法。
【請求項17】
請求項1に記載の方法であって、前記宿主細胞は、以下:Pichia pastoris、Pichia finlandica、Pichia trehalophila、Pichia koclamae、Pichia membranaefaciens、Pichia opuntiae、Pichia thermotolerans、Pichia salictaria、Pichia guercuum、Pichia pijperi、Pichia stiptis、Pichia methanolica、Pichia sp.、Saccharomyces cerevisiae、Saccharomyces sp.、Hansenula polymorpha、Kluyveromyces sp.、Candida albicans、Aspergillus nidulans、Aspergillus niger、Aspergillus oryzae、Trichoderma reesei、Chrysosporium lucknowense、Fusarium sp.、Fusarium gramineum、Fusarium venenatumおよびNeurospora crassaからなる群より選択される、方法。
【請求項18】
前記宿主細胞は、開始α−1,6マンノシルトランスフェラーゼ活性の発現をさらに欠損している、請求項1に記載の方法。
【請求項19】
前記宿主細胞は、P.pastorisのOCH1変異体である、請求項18に記載の方法。
【請求項20】
前記宿主細胞は、GnTIおよびUDP−GlcNAcトランスポーター活性を発現する、請求項1に記載の方法。
【請求項21】
前記宿主細胞は、UDP特異的ジホスファターゼジホスファターゼ活性またはGDP特異的ジホスファターゼジホスファターゼ活性を発現する、請求項1に記載の方法。
【請求項22】
前記宿主から前記糖タンパク質を単離する工程をさらに包含する、請求項1に記載の方法。
【請求項23】
請求項22に記載の方法であって、インビトロにおいて、前記単離された糖タンパク質を、少なくとも1種のさらなるグリコシル化反応に供し、続いて前記宿主からの単離に供する工程をさらに包含する、方法。
【請求項24】
請求項1に記載の方法であって、GlcNAcMan3GlcNAc2またはGlcNAc2Man3GlcNAc2の産生に関与する1種以上の酵素をコードする核酸分子を、前記宿主に導入する工程をさらに包含する、方法。
【請求項25】
前記少なくとも1種の酵素は、マンノシダーゼ活性を有する、請求項24に記載の方法。
【請求項26】
請求項25に記載の方法であって、前記酵素は、α−1,2−マンノシダーゼ活性を有し、かつマウス、ヒト、Lepidoptera、Aspergillus nidulans、C.elegans、D.melanogaster、またはBacillus sp.に由来する、方法。
【請求項27】
前記酵素は、α−1,3−マンノシダーゼ活性を有する、請求項25に記載の方法。
【請求項28】
前記少なくとも1種の酵素は、グリコシルトランスフェラーゼ活性を有する、請求項24に記載の方法。
【請求項29】
前記グリコシルトランスフェラーゼ活性は、GnTIおよびGnTIIからなる群より選択される、請求項28に記載の方法。
【請求項30】
少なくとも1種の酵素は、該酵素の触媒ドメインと細胞標的シグナルペプチドとの間で融合ペプチドを形成することによって局在化される、請求項24に記載の方法。
【請求項31】
請求項30に記載の方法であって、前記融合ペプチドは、少なくとも1つの遺伝子構築物によってコードされ、該遺伝子構築物は、細胞標的シグナルペプチドをコードするDNAフラグメントと、グリコシル化酵素またはその触媒活性フラグメントをコードするDNAフラグメントとのインフレームでの結合によって形成される、方法。
【請求項32】
請求項31に記載の方法であって、前記コードされた標的シグナルペプチドは、マンノシルトランスフェラーゼ、ジホスファターゼ、プロテアーゼ、GnT I、GnT II、GnT III、GnT IV、GnT V、GnT VI、GalT、FT、およびSTからなる群のメンバーに由来する、方法。
【請求項33】
請求項31に記載の方法であって、前記触媒ドメインは、GnT I、GnT II、GnT III、GnT IV、GnT V、GnT VI、GalT、FucosylトランスフェラーゼおよびSTからなる群のメンバーに由来する、グリコシダーゼまたはグリコシルトランスフェラーゼをコードし、かつ該触媒ドメインは、小器官内の他の代表的な酵素の平均最適pHの、1.4pH単位内に最適pHを有し、該小器官おいて、該酵素は、局在化されるかまたは5.1と8.0との間のpHにて最適な活性を有する、方法。
【請求項34】
請求項31に記載の方法であって、前記核酸分子は、UDP−GlcNAcトランスフェラーゼ、UDP−ガラクトシルトランスフェラーゼ、GDP−フコシルトランスフェラーゼ、CMP−シアリルトランスフェラーゼ、UDP−GlcNAcトランスポーター、UDP−ガラクトーストランスポーター、GDP−フコーストランスポーター、CMP−シアル酸トランスポーター、およびヌクレオチドジホスファターゼからなる群より選択される1種以上の酵素をコードする、方法。
【請求項35】
前記宿主は、GnTIおよびUDP−GlcNAcトランスポーター活性を発現する、請求項31に記載の方法。
【請求項36】
前記宿主は、UDP特異的ジホスファターゼ活性またはGDP特異的ジホスファターゼ活性を発現する、請求項31に記載の方法。
【請求項37】
請求項1に記載の方法であって、ドリチル−P−Man:Man5GlcNAc2−PP−ドリチルα−1,3マンノシルトランスフェラーゼ活性を欠損している宿主に、GlcNAcMan4GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項38】
請求項1に記載の方法であって、ドリチル−P−Man:Man6GlcNAc2−PP−ドリチルα−1,2マンノシルトランスフェラーゼまたはドリチル−P−Man:Man7GlcNAc2−PP−ドリチルα−1,6マンノシルトランスフェラーゼ活性を欠損している宿主に、GlcNAcMan4GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項39】
前記核酸分子は、α−1,2マンノシダーゼ、UDP GlcNAcトランスポーターおよびGnTlからなる群より選択される少なくとも1種の酵素をコードする、請求項37または38に記載の方法。
【請求項40】
請求項39に記載の方法であって、前記欠損している宿主細胞に、GlcNAclMan4GlcNAc2構造をGlcNAclMan3GlcNAc2構造に転換するためのα−1,3マンノシダーゼ活性またはα−1,2/α−1,3マンノシダーゼ活性をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項41】
請求項1に記載の方法であって、前記宿主に、GlcNAc2Man3GlcNAc2糖質構造を作製するための1種以上の酵素をコードする核酸分子を導入する工程をさらに包含する、方法。
【請求項42】
前記少なくとも1種の酵素は、GnTIIである、請求項41に記載の方法。
【請求項43】
請求項1に記載の方法であって、前記宿主細胞に、グリコシルトランスフェラーゼ、フコシルトランスフェラーゼ、ガラクトシルトランスフェラーゼ、N−アセチルガラクトサミニルトランスフェラーゼ、N−アセチルグリコサミニルトランスフェラーゼおよびスルホトランスフェラーゼからなる群より選択される少なくとも1種の哺乳動物グリコシル化酵素をコードする少なくとも1種の核酸分子を導入する工程をさらに包含する、方法。
【請求項44】
請求項1に記載の方法であって、宿主細胞をDNAライブラリーで形質転換して、該ライブラリーから誘導された少なくとも1種のグルコシル化酵素を発現する、遺伝学的に混合された細胞集団を作製する工程をさらに包含し、該ライブラリーは、少なくとも2つの異なる遺伝子構築物を含み、該構築物のうちの少なくとも一方は、細胞標的シグナルペプチドをコードするDNAフラグメントを含み、該細胞標的シグナルペプチドは、グリコシル化酵素またはその触媒活性フラグメントをコードするDNAフラグメントと、インフレームで結合される、方法。
【請求項45】
請求項1または44に記載の方法によって産生される、宿主細胞。
【請求項46】
請求項1または44に記載の方法によって産生される、ヒト様糖タンパク質。
【請求項47】
図6(P.pastoris ALG 3遺伝子)の少なくとも45個連続するヌクレオチド残基を含むかまたはそれらからなる、核酸分子。
【請求項48】
請求項47に記載の核酸分子を含む、ベクター。
【請求項49】
請求項47に記載の核酸分子を含む、宿主細胞。
【請求項50】
P.pastoris細胞であって、ここで、図6(P.pastoris ALG 3遺伝子)の配列は変異しており、それによって、該細胞のグリコシル化が変更される、P.pastoris細胞。
【請求項51】
脂質連結オリゴ糖のグリコシル化の程度を高めるための方法であって、該方法は、宿主細胞においてα−1,3グリコシルトランスフェラーゼ活性を増加させる工程を包含する、方法。
【請求項52】
脂質連結オリゴ糖のグリコシル化の程度を高めるための方法であって、
該方法は、宿主細胞においてオリゴ糖トランスフェラーゼ活性の基質特異性を減退させる工程を包含する、方法。
【請求項53】
非哺乳動物宿主細胞において、分岐したGlcNAcを含むN−グリカンを有する免疫グロブリンポリペプチドを産生するための方法であって、該方法は、該宿主細胞においてGnTIII活性を発現させる工程を包含する、方法。
【請求項54】
分岐したGlcNAcを含むN−グリカンを有する免疫グロブリンを産生する、非哺乳動物宿主細胞。
【請求項55】
請求項54に記載の宿主細胞によって産生される、免疫グロブリン。
【請求項56】
非哺乳動物宿主細胞において、分岐したGlcNAcを含むN−グリカンを有するポリペプチドを産生するための方法であって、該方法は、該宿主細胞においてGnTIII活性を発現させる工程を包含する、方法。
【請求項57】
分岐したGlcNAcを含むN−グリカンを有するポリペプチドを産生する、非ヒト宿主細胞。
【請求項58】
請求項57に記載の宿主細胞によって産生される、ポリペプチド。
【請求項59】
非ヒト真核生物宿主細胞においてヒト様糖タンパク質を産生するための方法であって、該方法は、該宿主細胞から、alg遺伝子活性を減退または減少させる工程、および該宿主細胞に少なくとも1種のグリコシダーゼ活性を導入する工程を包含する、方法。
【請求項60】
トリマンノースコアに結合した少なくとも2つのGlcNAcを含むN−グリカンを有するヒト様糖タンパク質を産生するための、方法。
【図32】


【図33】

【図34−1】

【図34−2】

【図1】
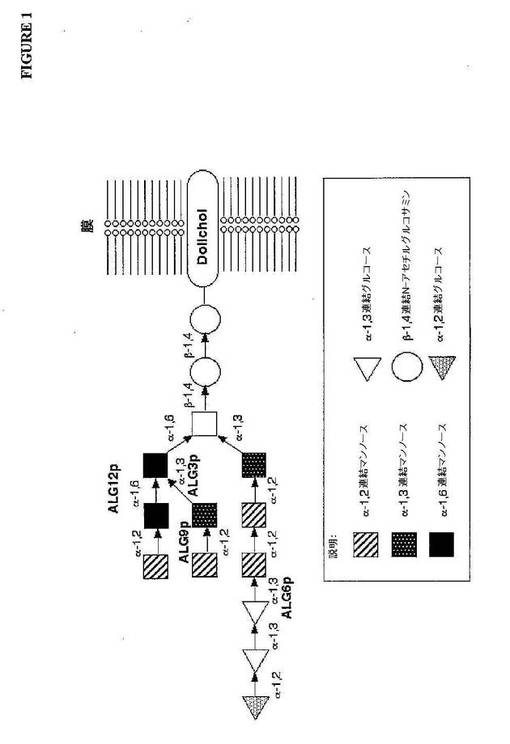
【図2】

【図3】
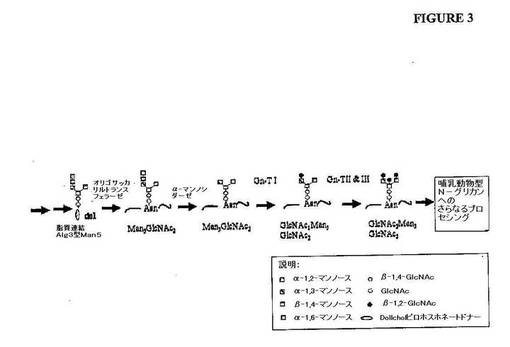
【図4−1】

【図4−2】

【図4−3】

【図4−4】

【図5】

【図6】

【図7−1】

【図7−2】

【図7−3】

【図8】

【図9】

【図10】

【図11】

【図12−1】

【図12−2】

【図13】

【図14】

【図15−1】

【図15−2】

【図16】

【図17】
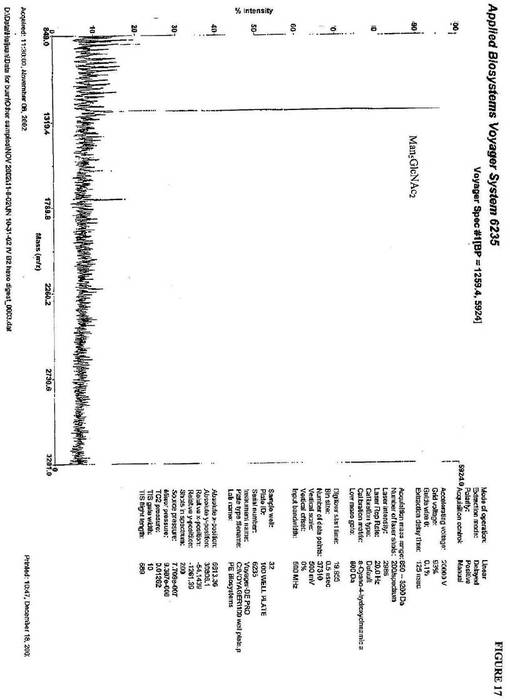
【図18】

【図19】

【図20】

【図21】

【図22】

【図23】
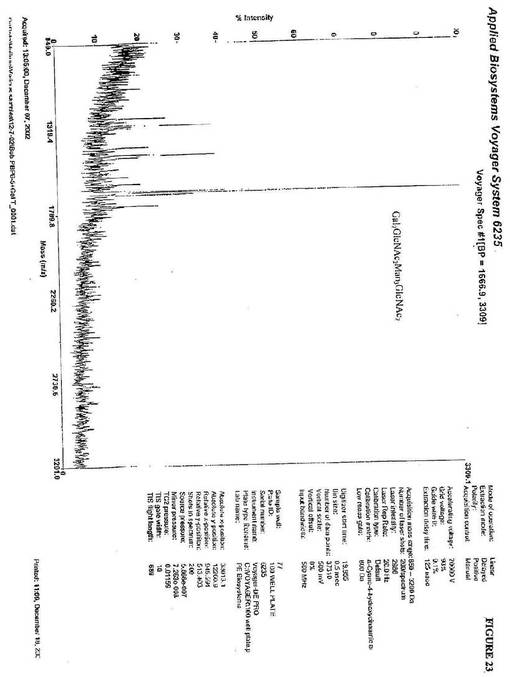
【図24】
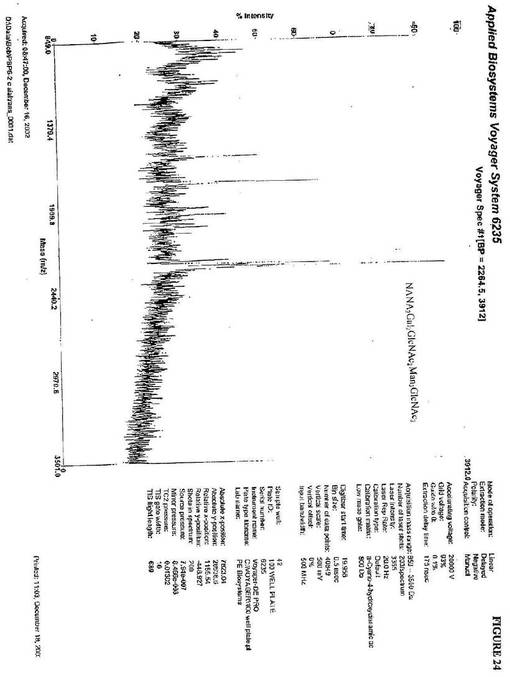
【図25】

【図26】

【図27−1】

【図27−2】

【図27−3】

【図27−4】

【図28】

【図29−1】

【図29−2】

【図30】

【図31】

【図33】
【図34−1】
【図34−2】
【図1】
【図2】
【図3】
【図4−1】
【図4−2】
【図4−3】
【図4−4】
【図5】
【図6】
【図7−1】
【図7−2】
【図7−3】
【図8】
【図9】
【図10】
【図11】
【図12−1】
【図12−2】
【図13】
【図14】
【図15−1】
【図15−2】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27−1】
【図27−2】
【図27−3】
【図27−4】
【図28】
【図29−1】
【図29−2】
【図30】
【図31】
【公開番号】特開2011−78431(P2011−78431A)
【公開日】平成23年4月21日(2011.4.21)
【国際特許分類】
【出願番号】特願2010−282293(P2010−282293)
【出願日】平成22年12月17日(2010.12.17)
【分割の表示】特願2003−557288(P2003−557288)の分割
【原出願日】平成14年12月24日(2002.12.24)
【出願人】(503007287)グライコフィ, インコーポレイテッド (37)
【Fターム(参考)】
【公開日】平成23年4月21日(2011.4.21)
【国際特許分類】
【出願日】平成22年12月17日(2010.12.17)
【分割の表示】特願2003−557288(P2003−557288)の分割
【原出願日】平成14年12月24日(2002.12.24)
【出願人】(503007287)グライコフィ, インコーポレイテッド (37)
【Fターム(参考)】
[ Back to top ]