
哺乳動物細胞におけるIi発現の阻害
本発明は抗原提示経路を改変する目的で、細胞のIi発現の阻害が関与する組成物および方法を対象とする。より詳細には開示するのは、正常な状況下ではMHCクラスII分子と結合して提示されない抗原性エピトープのMHCクラスII分子提示に関係する組成物および方法である。本発明は通常はMHCクラスII分子を発現する細胞、ならびにMHCクラスII分子を発現するように誘導できる細胞における提示に関する。

【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
特定の抗原に対する免疫応答はTリンパ球によるそれら抗原のペプチド断片の認識により調節される。抗原提示細胞中で、プロセシングされた抗原のペプチド断片は主要組織適合遺伝子複合体(MHC)分子の抗原ペプチド結合部位に結合するようになる。次いでこれらペプチド−MHC複合体は、ヘルパーもしくは細胞傷害性Tリンパ球上のT細胞レセプターによる(外来ペプチドおよび提示するMHC分子の隣接表面の両方の)認識のために細胞表面に輸送される。ペプチドを送達する2つのクラスのMHC分子、MHCクラスIおよびMHCクラスIIが存在する。
【0002】
MHCクラスI分子は抗原をCD8−陽性細胞傷害性Tリンパ球に提示し、次いでこれは活性化されるようになり、そして抗原提示細胞を直接殺すことができる。クラスI MHC分子はそれらのほぼ合成時に、小胞体で感染性ウイルスのような内的に合成されるタンパク質からのペプチドを排他的に受ける。
【0003】
MHCクラスII分子は抗原をCD4−陽性ヘルパーTリンパ球(Tヘルパー細胞)に提示する。いったん活性化されると、Tヘルパー細胞は物理的接触およびサイトカインの放出を介して細胞傷害性Tリンパ球(Tキラー細胞)およびBリンパ球の活性化に貢献する。MHCクラスI分子とは異なり、MHCクラスII分子は非特異的または特異的エンドサイトーシスを介してインターナリゼーションした外因性抗原に結合する。ほぼ合成時にMHCクラスII分子は、代わりにインバリアント(invariant)鎖タンパク質(Ii)に結合することにより内因性抗原への結合が遮断される。これらMHCクラスII−Iiタンパク質複合体は小胞体から後ゴルジ区画(post−Golgi compartment)に輸送され、ここでIiがタンパク質分解により放出され、そして外因性の抗原ペプチドが結合する(非特許文献1;非特許文献2)。
【0004】
MHCクラスIおよびクラスII分子は、細胞中で明確な分布を有する。ほとんどすべての有核細胞がMHCクラスI分子を発現するが、その発現レベルは細胞型の間で変動する。免疫系の細胞はその表面上に豊富なMHCクラスIを発現する一方、肝臓細胞は比較的低レベルで発現する。核を持たない細胞はMHCクラスIをほとんど発現しないか、または全く発現しない。MHCクラスII分子はBリンパ球およびマクロファージ上に高度に発現されるが、他の組織の細胞上では発現されない。しかし多くの他の細胞型はサイトカインに対する暴露によりMHCクラスII分子を発現するように誘導することができる。
【0005】
正常な条件下で、(自己決定基により自己免疫疾患を導く可能性がある)内因性ペプチドはIiタンパク質が新生MHCクラスII分子と常に同時合成されるので、MHCクラスII分子に結合しない。自己決定基ペプチドおよびMHCクラスII分子を含む複合体は身体の免疫監視系により見いだされることはなく、これらの決定基に対する免疫寛容は生じない。MHCクラスII分子が発生した個体でIiにより阻害されない場合、内因性の自己決定基がMHCクラスII分子により提示されるようになり、これら内因性抗原に対する自己免疫応答を開始する。それらは特定の自己免疫疾患の場合である。そのような効果を悪性細胞で操作することにより、腫瘍の内因性抗原に対する「自己免疫応答」は、腫瘍細胞の増殖を制限もしくは排除するいずれかのために治療的に使用することができる。
【0006】
Iiタンパク質が同時に上昇しない、MHCクラスII分子の上昇した発現の治療効果は、MHCクラスII−陰性、Ii−陰性腫瘍で実証された(非特許文献3;非特許文献4;非特許文献5;非特許文献6;および非特許文献7)。これらの研究において、MHCクラスII分子の遺伝子のMHCクラスII陰性マウス肉腫へのトランスフェクションにより、MHCクラスII−陽性であるが、Ii−陰性腫瘍細胞株を生じた。これらの細胞のMHC適合性宿主への注入は、親腫瘍の増殖の遅延を導いた。Iiタンパク質の遺伝子を、MHCクラスII遺伝子と一緒に肉腫細胞株にコトランスフェクションすると、Ii鎖が内因性の腫瘍抗原の提示を遮断したので、MHCクラスII遺伝子の腫瘍治療効果を阻害した。類似の結果がマウス黒色腫でも生じた(非特許文献8)。
【0007】
この治療的取り組みの成功には、樹状細胞の天然の活性が関与すると考えられる。樹状細胞は専門的なスカベンジャーであり、これは外来抗原をペプチドに処理し、そしてそれらをTリンパ球にそれらの細胞表面上でMHC抗原から提示する。樹状細胞は抗原をMHCクラスIおよびクラスII分子を介して提示する能力を有し、それらがTヘルパーおよびTキラー細胞の両方を活性化できるようにする。効果的なTヘルパー細胞応答には強力なTキラー細胞応答の誘導が必要であり、そして樹状細胞により生じた活性化の組み合わせが最高の抗−腫瘍応答を導くと考えられている(非特許文献9;非特許文献10)。マクロファージ系統の樹状細胞は腫瘍細胞を見つけると、腫瘍特異的および腫瘍関連抗原の両方を摂取し、そして処理する。次いで樹状細胞は腫瘍部位を排出するリンパ節に移動し、そして新たなT細胞が成長し始めるリンパ節皮質(node cortex)近傍のリンパ節に在住する。リンパ節皮質では、樹状細胞上の腫瘍決定基を認識する休止Tキラー細胞が活性化し、そして増殖するようになり、そして続いて有能な抗−腫瘍キラーT細胞として循環に放出される。
【0008】
T−ヘルパー細胞との相互作用は、樹状細胞が抗原をMHCクラスI分子を介して提示するように活性化または「ライセンスを与え(licence)」、すなわちTキラー細胞を活性化するが、Tヘルパー細胞とTキラー細胞との同時相互作用は必要ではなく;活性化された樹状細胞は時折、Tヘルパー細胞媒介型活性化の後に、Tキラー細胞を刺激する能力を維持している。MHCクラスIIまたはMHCクラスI決定基のいずれかにより提示されるようになる個々の抗原性ペプチドは、1つの抗原性タンパク質に由来する必要は無く、悪性細胞に由来する2以上の抗原が1つの樹状細胞により処理そして提示され得る。したがって恐らく腫瘍特異的ではない1つの決定基に対するライセンスは、他の恐らく腫瘍特異的な決定基に対するTキラー細胞の活性化にライセンスを与える力を持つ。そのような「マイナーな」または「隠れた(cryptic)」決定基が様々な治療目的に使用されてきた(非特許文献11)。
【0009】
MHCクラスII抗原提示の実験的改変は、これらマイナーな決定基に対する免疫応答を拡大すると考えられる。通常はMHCクラスII分子への装填に利用できない一連のペプチドが、MHCクラスII提示について変動するペプチドの豊かな供給源を提供する。この一連の決定基の活用により、反応性Tヘルパー細胞集団の拡大を導く。そのような拡大された集団は樹状細胞のライセンシングを誘導し、その中には腫瘍特異的および腫瘍関連決定基に向けられるものもある。正常な細胞は潜在的に腫瘍細胞決定基を共有するが、正常細胞に対しては細胞の傷害がわずかしか生じない。これは抗−腫瘍応答の多くのエフェクター応答(キラーT細胞の密集、周辺の活性化サイトカイン、食作用を行うマクロファージ、およびそれらの産物等)が正常細胞には向かわないからである。
【0010】
正常なMHCクラスII抗原提示は、MHCクラスII分子とIiタンパク質との相互作用を阻害することにより改変することができる。これは全Iiタンパク質を減少させることにより(例えば発現を減少させることにより)、またはIiの免疫調節機能を妨害することにより達成することができる。Ii発現の阻害は種々のアンチセンス技法を使用してなされてきた。Iiタンパク質のmRNAのAUG部位と相互作用するアンチセンスオリゴヌクレオチドは、外因性抗原のMHCクラスII提示を低下させることが記載された(非特許文献12)。しかしIiタンパク質の発現およびMHCクラスII分子による内因性抗原の提示に及ぼす効果は調査されなかった。さらに最近では、Humphreys et al.、特許文献1が、MHCクラスII分子を発現する抗原提示細胞に導入すると、Iiタンパク質発現の効果的な抑制を現す3種のアンチセンスオリゴヌクレオチドおよびリバース遺伝子構築物(reverse gene construct)を同定した。このメカニズムにより、Iiが抑制された腫瘍細胞を接種したマウスは、未処置の親腫瘍細胞を接種したマウスよりも有意に長く生存することが示された。この観察はIiタンパク質の抑制が抗原決定基提示の範囲の増大を生じ、腫瘍細胞に対してより効果的な免疫応答を惹起することを示す。
【参考文献】
【0011】
【特許文献1】米国特許第5,726,020号明細書
【非特許文献1】Daibata et al.,Molecular Immunology 31:255−260(1994)
【非特許文献2】Xu et al.,Molecular Immunology 31:723−731(1994)
【非特許文献3】Ostrand−Rosenberg et al.,Journal of Immunol.144:4068−4071(1990)
【非特許文献4】Clements et al.,Journal of Immunol.149:2391−2396(1992)
【非特許文献5】Basker et al.,Cell.Immunol.155:123−133(1994)
【非特許文献6】Baskar et al.,J.Exp.Med.181:619−629(1995)
【非特許文献7】Armstrong et al.,Proc.Natl.Acad.Sci.USA 94:6886−6891(1997)
【非特許文献8】Chen and Ananthaswamy,Journal of Immunolgy.151:244−255(1993)
【非特許文献9】Ridge et al.,Nature 193:474−477(1998)
【非特許文献10】Schoenberger et al.,Nature 193:480−483(1998)
【非特許文献11】Mougdil et al.,J.Immunol.159:2574−2579(1997)
【非特許文献12】Bertolino et al.,Internat.Immunology 3:435−443(1991)
【発明の開示】
【0012】
発明の要約
本発明は抗原提示経路を改変する目的のために、細胞中のIi発現の阻害が関与する組成物および方法を対象とする。1つの観点では、本発明はRNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物に関する。また本発明はそのような発現可能なリバース遺伝子構築物を含む哺乳動物細胞に関する。好適な態様では、RNA分子はコード配列の翻訳開始部位(translation initiation start site)および約435ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である。
【0013】
前に示したように、Ii発現の抑制は抗原提示経路の改変を意図している。より具体的にはIi発現の阻害は、通常はこの内容では提示されない抗原性エピトープのMHCクラスII分子への装填を促進することを意図する。すなわち別の観点では、本発明はMHCクラスII分子−陰性細胞をMHCクラスII分子−陽性細胞に転換することに関する。この転換は例えば、MHCクラスII分子−陰性細胞をタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターでトランスフェクトし、そのトランスフェクションがMHCクラスII分子−陰性細胞中でトランスフェクトされた細胞の表面上にMHCクラスII分子の誘導をもたらすことによる。
【0014】
別の観点では、本発明はIiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上で目的の抗原性エピトープを展示する方法に関する。この方法は:a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そしてb)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、ことを含む。
【0015】
別の観点では、本発明は免疫学的応答のために動物の細胞の型を標的化する方法に関し、細胞の型は識別抗原の発現を特徴とする。この方法では個体に由来する末梢血単核細胞のカルチャーが準備され、このカルチャーは抗原提示細胞を含む。Ii発現のインヒビターをカルチャーの抗原提示細胞に導入し、同様に識別抗原をコードする発現可能な核酸配列を発現に適する条件下でカルチャー中の細胞に導入する。
【0016】
多数の関連する観点を以下の章で詳細に記載する。
発明の詳細な説明
本発明は、1つの観点では個体における病状に特異的な調節、または免疫応答の標的化のための組成物および方法に関する。本明細書で使用する調節(modulation)という用語は、個体において抗原に対する免疫系の増大した感受性もしくは低下した感受性(免疫寛容)を称することを意味する。使用する標的化という用語は、抗原性エピトープに対して増大した感受性を指すことを意図する。
【0017】
本開示のすべての観点に関連して必要な要素は、細胞中でのIi合成の阻害である。発明の背景の章で検討したように、Iiはタンパク質であり、これはMHCクラスII分子と同時に調節されている(co−regulated)。IiはMHCクラスII分子に結合し、これにより内部で合成された抗原(すなわちMHCクラスII分子−発現細胞内で合成された抗原)のMHCクラスII分子への接近が遮断される。MHCクラスII分子/Ii複合体は、小胞体から後ゴルジ区画へ輸送され、そこでIiが段階的開裂プロセスにより放出され、これにより外因性抗原(すなわち抗原提示細胞内で合成されず、そして食作用、オプソニン作用、細胞表面抗体認識、補体レセプター認識およびFcレセプター認識のようなメカニズムにより抗原提示細胞に取り込まれるために選択された抗原)の装填が可能となる。
【0018】
複合体を形成したIiタンパク質の存在により小胞体中のMHCクラスII分子への結合から排除される抗原のクラスは、内的に合成された抗原と呼ぶことができる。そのような抗原は、プロテオソームにより消化され、そして抗原性ペプチド(TAP)の輸送体によりペプチドとして小胞体に輸送された細胞質タンパク質の調査(survey)を含んでなる。そのような内部で合成された抗原は通常、小胞体中でMHCクラスI分子に結合する。そのような抗原断片はIiタンパク質が抗原性ペプチド結合部位を遮断するので、通常、小胞体内でMHCクラスII分子に結合しない。
【0019】
Iiタンパク質の発現を抑制することにより、MHCクラスIに結合するために小胞体に輸送され、そして続いてCD8+Tリンパ球に提示されるこの膨大なペプチドのレパートリーは、続いてCD4+T免疫調節細胞に提示され、そしてそれを活性化するためにMHCクラスII分子に結合することができる。そのようなCD4+T免疫調節細胞は免疫応答の種々の経路と調和してヘルパーまたはサプレッサー機能のいずれかを有することができる。T免疫調節細胞は、物理的接触およびサイトカインの放出を介して細胞傷害性Tリンパ球(Tキラー細胞)、Bリンパ球および樹状細胞のような他の細胞の活性化に貢献している。
【0020】
本明細書で使用する「目的の抗原性エピトープ」という用語は、抗原提示が起こる細胞の中で生産されるタンパク質に由来するペプチドに存在する抗原性エピトープを指す。本明細書で使用するようにこの用語は、既知もしくは未知の抗原性エピトープを包含することを意図する。すなわち「目的の(of interest)」という修飾語は、エピトープが事前に決定されることを意味しない。「目的の」抗原性エピトープは、単に抗原提示が起こる細胞の細胞質内で合成されるタンパク質に含まれるという事実による。
【0021】
治療的介入の好機を与える重要な生物学的結果は、MHCクラスI分子により小胞体中で結合するためにそこに輸送されたペプチドのレパートリーに由来するペプチドのMHCクラスII分子による結合後に続く。しばしばIi抑制の存在下でMHCクラスII分子に結合するエピトープは「隠れた」エピトープであるので、そのようなエピトープは抗原提示の古典的経路によりMHCクラスII分子と結合する以外は免疫系に提示されない。隠れたエピトープは、試験抗原のアミノ酸配列が重複する合成ペプチドのライブラリーを分析することにより実験的に明らかにすることができる。試験抗原で免疫感作したマウスの一種の動物がライブラリーに由来する1組のペプチドに応答することが分かった(「優性エピトープ(dominant epitope)」)。しかしそうではない同一マウスをライブラリーの1つのペプチドで免疫感作した時、これまでに同定されなかったサブセット(免疫感作するペプチド中の優性エピトープに加えて)が、免疫学的エピトープを含むことが分かる。これらの以前に同定されなかったエピトープは、1組の隠れたエピトープを含んでなる。
【0022】
本発明の方法は、優性および隠れたエピトープの両方に対する免疫を促進するが、幾つかの臨床的状況においては隠れたエピトープに対する免疫応答の強化が治療効果に特別な役割を果たす。例えばガンに関連する抗原性エピトープに対する治療的応答を追加免疫感作する場合、サプレッサーT細胞応答がこれまでに生じなかった隠れたエピトープに対するTヘルパー細胞応答は、効果的な樹状細胞ライセンシングをさらに提供するようであり、これは次いで強固な細胞傷害性Tリンパ球抗−腫瘍応答を生成する。ガンに関連する抗原の優性エピトープに対するサプレッシングT細胞応答の発生は、腫瘍の微小ガン組織転移の成長に役割を果たしていることが示された。したがって本発明の重要な用途は、推定される隠れたガン関連決定基に対するTヘルパー細胞応答の促進である。
【0023】
自己免疫応答の別の観点では、自己免疫疾患の場合は自己免疫疾患に関連する抗原の優性エピトープに対する応答が、そのような疾患の病原を促進する。ここで選択的、例えば新しい隠れたエピトープに対する免疫応答の経路の抑制を活用することは、治療的に有用となり得る。これらの概念は同様に、アレルギー、移植片拒絶ならびに感染性および心血管疾患のようなさらなる医学的状態の治療に応用することができる。そのような状態の患者の診断、処置的モニタリングおよび治療に応用する化合物の開発において必須かつ有用な第1段階は、Iiタンパク質抑制の条件下で抗原提示細胞により提示されるようになるMHCクラスIIエピトープの同定である。そのようなエピトープには優性および隠れたエピトープの両方を含む。隠れたエピトープは例えばガンまたは感染性疾患の場合、免疫抑制応答がそれらに向かって発生することはなく、しかも自己免疫疾患または移植片拒絶の場合には応答の活性化が発生しなければ特に有用となるだろう。したがって医師は上記の病状の場合に状況次第で新たにTh1またはTh2を誘導し、応答を活性化または抑制するだろう。そのようなエピトープを生成し、単離し、そして特性決定する方法は、本発明の主題である。
【0024】
別の観点では本発明は、Iiタンパク質の不存在下で小胞体中のMHCクラスII分子に結合する抗原性エピトープを含む個体のペプチドの提示、単離および同定を提供する。そのようなペプチドを合成し、そして個別に、またはそれらが生じた疾患に由来する抗原に対する免疫応答を強化または抑制するためのワクチン応用と組み合わせて使用することができる。そのようなエピトープペプチドを単離し、そして特性決定するための方法は、引用により本明細書に編入する米国特許第5,827,526号、同第5,874,531号および同5,880,103号明細書に提示された。
【0025】
「内部で合成される(endogenously synthesized)」クラス(これは通常、MHCクラスII分子提示から排除される)に入る種々の抗原は、特に特定の病状を伴う。例えば腫瘍細胞または他の悪性細胞を考えることができる。そのような細胞はガンに特異的な、そしてガンに関連するタンパク質を合成し、これらは治療に有用なMHCクラスIIエピトープを含む。しかしこれらのタンパク質は抗原提示細胞内で合成されるので、そのようなタンパク質の抗原性エピトープは同じ細胞のMHCクラスII分子との結合での提示からは排除される。中で抗原性タンパク質が合成される細胞による抗原性エピトープの提示に関する制限は、ウイルスに感染した細胞の場合にもある。ウイルスに特異的な抗原はウイルスに感染した細胞のMHCクラスII分子との結合での提示から排除されるが、そのような抗原は同じ細胞のMHCクラスI分子との結合で提示され得る。他の外因性病原体(例えばバクテリアまたは寄生体)が細胞を占有し、そして細胞機構を利用して病状に特異的なタンパク質を合成しても、成り行きは同じである。病原に特異的な抗原をMHCクラスII分子との結合で提示することによる事象の正常な過程を改変する能力は、新規なMHCクラスII抗原性エピトープにより開始される応答の強化をもたらす。
【0026】
多数の治療的モダリティが本発明の範囲に入る。特許請求できる組成物は、これら多くの治療的モダリティに関連する。治療的取り組みには、インビボおよびエクスビボの態様を含む。Ii阻害を標的とする細胞はMHCクラスII分子−陽性細胞(例えば樹状細胞、マクロファージまたはBリンパ球のような自然に存在する抗原提示細胞)、あるいはMHCクラスII分子を発現するように誘導されたMHCクラスII−陰性細胞(例えば腫瘍細胞)のいずれかであることができる。本明細書で使用する「MHCクラスII分子−陰性」という表現は、具体的には細胞表面上にMHCクラスII分子を発現しない細胞を含むだけでなく、自然に存在する抗原提示細胞(例えば樹状細胞)のような陽性対照細胞の表面上のMHCクラスII分子の数と比べた時、細胞表面上に比較的少数のMHCクラスII分子を含む細胞も含む。本内容において、用語「比較的少数」とはMHCクラスII分子−陽性対照細胞(例えば自然に存在する抗原提示細胞)上に見いだされる数のわずか約25%以下の数のMHCクラスII分子をそれらの細胞表面上に含むと見積もられる細胞を含むことを意味する。MHCクラスII分子の存在度(abundance)の予測は、例えば当該技術分野で周知な免疫蛍光技法を使用して行うことができる。
【0027】
出願人は以前に、免疫応答を調節する目的のIi阻害を開示する特許出願を出願し、そして要求した。これらの出願は具体的には細胞に導入され、そしてIi mRNAに結合することによりIi合成を直接阻害する阻害コポリマー、ならびに核酸構築物として細胞に導入され、続いてRNA分子に転写され、これが特異的ハイブリダイゼーション後にIi発現を阻害するリバース遺伝子構築物を開示する。これらの以前に出願された特許出願には、米国特許出願第08/661,627号および同第09/205,995号明細書を含み、それらは引用により本明細書に編入する。米国特許出願第08/661,627号明細書は、米国特許第5,726,020号として発効され、そして米国特許出願第09/205,995号の発効手数料が最近、米国特許庁に支払われた。
【0028】
上に簡単に述べたように、米国特許出願09/205,995号明細書は約10〜約50ヌクレオチド塩基を含む化学的に合成されたコポリマーに関する徹底的な開示を含む。
【0029】
これらのコポリマーはRNA分子の標的部分に相補的であるか、あるいはアンチセンス配列として知られているヌクレオチド塩基配列を含む。そのようなコポリマーの1例は、アンチセンスオリゴヌクレオチドである。コポリマーはRNAからのタンパク質翻訳を2つのメカニズムにより阻害する。1つの方法はリボソーム、スプライソソームまたはRNAの成熟もしくは翻訳に必須な他の因子と相互作用しなければならないRNAの部分への接近を遮断する。第2の方法は、DNAにハイブリダイズするRNAの配列を開裂する酵素であるリボヌクレアーゼHの強化が関与する。すなわちDNAもしくはDNA様のコポリマーの対応するRNA中のセグメントへの結合は、コポリマー結合部位でRNAの開裂を導く。
【0030】
コポリマーはワトソン−クリック塩基対合により標的RNAにハイブリダイズする。コポリマーの配列は、標的RNAの相補的配列により定められる。コポリマーは通常、標的RNAの少なくとも6個の相補的ヌクレオチドに広がるヌクレオチド配列長を用いて化学的に合成されるが、12〜25ヌクレオチド長が最も普通である。統計的に、約15ヌクレオチドの配列は細胞内のすべてのRNAの群中で独特であり、特定のRNAを高度な特異性で標的化できるようにする。RNAへの結合はまた、20塩基対を包含するコポリマーについて約10−7M付近のKd値で大変安定である。
【0031】
幾つかの場合では、カルチャー中の細胞は有用な効果を達成するために十分な量で自然にコポリマーを取り込む。そのような取り込みは生物化学的エネルギーおよび特定の細胞表面タンパク質の参加を要する能動的なプロセスになるように思われる。取り込みは飲作用によっても起こり得る。この経路は細胞をコポリマーを含有する高張性培地にインキュベーションし、続いて細胞をわずかに低張性培地に再懸濁して細胞内の飲作用性小胞の破裂を誘導することにより強化される。別の場合では、取り込みは脂質、リポソーム、またはポリアルキルオキシコポリマーの使用により、電気穿孔により、または細胞膜を透過するストレプトリシンO処理により援助することができる。しばしばインビボで細胞は培養した細胞よりも容易にコポリマーを取り込む。コポリマーの電気穿孔による細胞の取り込みに関する最適な条件は、米国特許出願第09/205,995号明細書の実施例2に提供されている。
【0032】
標的RNAの潜在的部位は、タンパク質の機能的複合体の結合について開いた部位、およびそうではなくコポリマーの結合について開いた部位である。そのような部位はDNAにハイブリダイズするRNAを開裂する酵素であるリボヌクレアーゼH(RNaseH)を使用して同定することができる。DNAオリゴヌクレオチドを単独で、または混合物中で5’−放射性リン標識RNAにリボヌクレアーゼHの存在下で結合することにより、オリゴヌクレオチドおよび他のコポリマーがハイブリダイズするRNA上の部位は、RNAのゲル電気泳動およびオートラジオグラフィー後に同定される。本発明でRNaseH開裂について最も開いていることが分かったIiのRNA部位は、AUG開始コドンの領域およびプレ−mRNAの第1スプライス部位の領域であった。
【0033】
本発明に関して「オリゴヌクレオチド」という用語は、自然に存在する塩基およびホスホジエステル結合で連結したペントフラノシル糖で形成されたヌクレオチド単位を含んでなるポリヌクレオチドを称する。用語「コポリマー」にはオリゴヌクレオチド、およびまた自然には存在しないか、もしくは修飾されたオリゴヌクレオチドのサブユニットから形成された構造的に関連する分子を含む。これらの修飾はヌクレオチドの塩基部分、ヌクレオチドの糖部分、またはヌクレオチド間連結基のいずれかで生じる。さらなる連結基はしばしば、天然のオリゴヌクレオチドの糖およびホスフェート骨格が置換されて、以下でより詳細に検討するコポリマーを生成する。
【0034】
生成されるそのようなオリゴヌクレオチド修飾および特性は、当業者には容易に利用可能である。例示的修飾は、引用により本明細書に編入する米国特許第4,469,863号明細書(1984);米国特許第5,216,141号明細書(1993);米国特許第5,264,564号明細書(1993);米国特許第5,514,786号明細書(1996);米国特許第5,587,300号明細書(1996);米国特許第5,587,469号明細書(1996);米国特許第5,602,240号明細書(1997);米国特許第5,610,289号明細書(1997);米国特許第5,614,617号明細書(1997);米国特許第5,623,065号明細書(1997);米国特許第5,623,070号明細書(1997);米国特許第5,700,922号明細書(1997);および米国特許第5,726,297号明細書(1998)に与えられている。
【0035】
オリゴヌクレオチドが相補的RNAにハイブリダイズする能力は、化学的修飾に対して大変抵抗がある。したがって多くの異なる機能的コポリマーが可能である。特に糖ホスフェート骨格は、ワトソン−クリック塩基対を形成する能力を失うことなく徹底的に改変することができる。定義によりヌクレオチドは糖、窒素複素環およびホスフェート部分を含んでなる。糖もしくはホスフェート基のいずれかまたは両方を欠くオリゴヌクレオチドの幾つかの合成類似体は、それでもアンチセンスオリゴヌクレオチドと同様にワトソン−クリック塩基対によりハイブリダイズすることができ、そしてこの目的に使用することができる。ヌクレオチド塩基を含有するこれらのコポリマーは、RNAへのハイブリダイズにおいてオリゴヌクレオチドの機能的均等物である。以下の要約はアンチセンスの応用のためにオリゴヌクレオチドの特性を変え、そして改善するそれらに対する幾つかの修飾である。
【0036】
多数の具体的修飾が開示され、それらには非−架橋酸素原子の置換、架橋酸素原子の置換、ヌクレオシド間ホスフェート基の置換、糖環の回りの立体化学に対する変化、リボフラノシル環構造の修飾、ヌクレオチド連結の修飾、およびペプチド骨格による糖ホスフェート骨格の置換によりペプチジル核酸(pna)の生成を含む。
【0037】
引用した特許出願の手続処理は以下の請求項に特許査定をもたらした。以下は代表的な独立請求項である。
1.哺乳動物のB7分子をコードする外因性の構築物を含まず、そしてIiタンパク質発現の特異的レギュレーターもしくは免疫調節機能を含むMHCクラスII−陽性抗原提示細胞であって、オリゴヌクレオチドCTCGGTACCTACTGG(配列番号11)が特別に除外され、特異的レギュレーターが10〜50ヌクレオチド塩基のコポリマーから本質的になり、コポリマーは生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、ここで特異的レギュレーターはIi発現を阻害する能力を特徴とする上記のMHCクラスII−陽性抗原提示細胞。
【0038】
本開示の内容では、出願人は以前に記載した種類のコポリマーが新規な内容で使用される態様を開示し、そして特許請求する。あるいはより徹底的な支持実験が本明細書で開示される場合、本出願で請求する主題は先に出願した特許出願に開示した主題に実質的に類似するかもしれない。
【0039】
関連する背景情報を提供し、そしてさらなる請求項の限定を支持する目的で、上で説明した請求項において少なくとも2つの具体的な限定を、従来技術を開示するために含んだ点に注目されたい。例えばオリゴヌクレオチドCTCGGTACCTACTGG(配列番号11)の特別な排除は、Bertolino et al.,Int.Immunol.3(5)435−443(1991)の開示に照らして包含した。哺乳動物のB7分子をコードする外因性構築物を対象とした限定は、Ostrand−Rosenberg(米国特許第5,858,776号明細書)の開示に照らして導入した。
【0040】
これまでに公開されていない本開示の重要な要素は、ヒトの細胞中でIiの発現を阻害するためのリバース遺伝子構築物の使用である。ヒトIi配列は以前に報告されたが(Strubin et al.,EMBO J.3:869−872(1984))、この配列の少なくとも一部を含むリバース遺伝子構築物の使用は報告されたことがない。さらに例えばマウスIi配列とヒトIi配列との間の有意な保存が報告されたが、非ヒトリバース遺伝子構築物はヒト細胞中でIiの翻訳を阻害するために使用するには効果的ではなかった。
【0041】
このように1つの観点では、本発明はRNA分子がmRNA分子にハイブリダイズする能力を有し、これによりヒト細胞中でmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物に関する。本発明のこの観点は以下に続く実施例の章で具体的に証明する。より具体的にはcDNA挿入物を含む発現構築物が、ヒトのリンパ腫細胞株でIi発現を阻害するのに効果的であることが証明された。このアッセイで効果的であった構築物は、Ii mRNAの5’非翻訳領域の一部に相補的なcDNA挿入物を含み、そして翻訳開始コドンを含んだ。効果的な構築物は、約435ヌクレオチド長までの阻害RNAをコードした。
【0042】
ヒトIi mRNAの部分に完全に相補的なRNAをコードするリバース遺伝子構築物の使用に加えて、当業者は野生型ヒト配列からある程度の逸脱が許容されると認識するだろう。本発明の範囲は、日常的な実験により経験的に決定され得るそのようなバリアントを包含することを意図する(すなわちそれらはヒト細胞中でIi発現を阻害する能力により特徴づけられる)。特に有用で、しかもヒト細胞中でIi発現の阻害に効果的であることが証明された野生型からの変異の例は、ヒトIi mRNAに相補的な長い半減期のアンチセンスRNA(野生型アンチセンスRNAに比べて)の作成に関する。長い半減期の種では、アンチセンスRNAのリーデイングフレイムは、開始コドンAUGに同じリーディングフレイム内で短く/直ちに終止コドンが続くことを回避するように設計されている。この状況を回避するために、例えばリーディングフレイム1中の終止コドンの直前にあるAUGに関して、新たなAUGを設計し、そしてリーディングフレイム1のAUGの前、リーディングフレイム2またはリーディングフレイム3のどちらかの中に(ただし終止コドンはそのような修飾後のリーディングフレイムには無い)導入する。
【0043】
阻害コポリマーおよびIiリバース遺伝子構築物の使用に加えて、当業者はIi発現の他のインヒビターが容易に設計され、そして構築されると認識するだろう。例えば二本鎖の小さい干渉性のRNA(siRNA)およびこれらの分子をコードする遺伝子をRNA干渉に使用することができ、この方法により二本鎖RNA(dsRNA)はその相補的配列を持つ遺伝子の発現を特異的に抑制する(Moss,Curr,Biol.11(19):R772−5(2001);Elbashir,Genes Dev.15(2):188−200(2001))。二本鎖RNAは相補的mRNAに結合し、そして縮重を導く21〜23塩基対の断片に処理される(Bernstein,Nature 409(6818):363−6(2001)および国際公開第0175164号パンフレット)。siRNAは配列特異的な翻訳後遺伝子サイレンシングを誘導する。そのような分子は細胞に導入されて、国際公開第0175164号パンフレットに記載されているような治療用もしくは予防用の目的で遺伝子発現を抑制することができる。
【0044】
広い種類の送達系を、本発明のリバース遺伝子構築物の標的細胞へのインビトロおよびインビボ送達の使用に利用することができる。これらの送達系はIiリバース遺伝子構築物に応用できるだけでなく、本発明と関連して検討する発現可能な核酸配列にも応用することができる。そのような送達系には、例えばウイルスおよび非ウイルス系を含む。適当なウイルス系の例には、例えばアデノウイルスベクター、アデノ随伴ウイルス、レトロウイルスベクター、ワクシニア、単純ヘルペスウイルス、HIV、マウスの微小ウイルス(minute virus)、B型肝炎ウイルスおよびインフルエンザウイルスを含む。非ウイルス送達系も使用でき、例えば非複合化DNA、DNA−リポソーム複合体、DNA−タンパク質複合体およびDNA−コート金粒子、サルモネラのようなバクテリアベクター、およびVP22輸送タンパク質、Co−X−遺伝子およびレプリコンベクターが関与するような他の技術を使用する。
【0045】
動物細胞中で目的の核酸配列を発現させるための1つの選択は、アデノウイルス系である。以下の実施例の章では、アデノウイルス系の使用を具体的に開示する。アデノウイルスは二本鎖DNAゲノムを保有し、そして宿主細胞分裂とは独立して複製する。アデノウイルスベクターは、発現可能な構築物を細胞に導入するための別の選択的方法と比べて、様々な利点を提供する。例えばアデノウイルスベクターは広範囲のヒト組織に形質導入させることができ、そして高レベルの遺伝子発現を分裂および非分裂細胞で得ることができる。アデノウイルスベクターは免疫系のクリアランスにより比較的短期間の導入遺伝子発現および標的細胞分裂中に希釈損失を特徴とする。静脈内、胆嚢内、腹腔内、嚢内、頭蓋内およびクモ膜下腔内注射、および標的器官または組織の直接注射を含む幾つかの投与経路を使用することができる。このように当該技術分野では解剖学的境界に基づく標的化を達成することができると認識されている。
【0046】
アデノウイルスゲノムは約15種のタンパク質をコードし、そして感染には細胞表面レセプターに結合する繊維タンパク質が関与する。このレセプター相互作用はウイルスのインターナリゼーションをもたらす。ウイルスDNAは感染した細胞の核に入り、そして転写が細胞分裂の不存在下で開始される。発現および複製はE1AおよびE1B遺伝子の制御下にある(Horwitz,M.S.,In Virology,2.sup.nded,1990,pp1723−1740を参照にされたい)。E1遺伝子の除去によりウイルスを複製−不能(replication−incompetent)とする。
【0047】
アデノウイルス血清型2および5は、ベクターの構築に広範囲に使用された。Bett et al.(Proc.Nat.Acad.Sci.U.S.A.91:8802−8806(1994))は、E1およびE3アデノウイルス遺伝子の欠失があるアデノウイルス5型ベクター系を使用した。293ヒト胚性腎細胞株をE1タンパク質を発現するように操作し、そしてこのようにE1−欠失ウイルスゲノムをトランス相補することができる。ウイルスは293細胞培地から単離し、そして限界希釈プラークアッセイにより精製することができる(Graham and Prevek、分子生物学の方法:遺伝子転移および発現プロトコールで(In Methods in Molecular Biology:Gene Transfer and Expression Protocols)、ヒューマナ(Humana)出版 1991、第109〜128頁)。組換えウイルスを293細胞株のカルチャー中で成長させ、そして感染した細胞を溶解することにより単離し、そして塩化セシウム密度遠心により精製することができる。組換えアデノウイルスの製造について293細胞に付随する問題は、E1遺伝子のさらなるフランキング領域により、それらがウイルス粒子の生産中に複製可能(replication−competent)なアデノウイルス(RCA)を生じるかもしれない点である。この材料は野生型アデノウイルスだけであり、そして複製可能な組換えウイルスではないので、所望するアデノウイルス材料の最終的な収量に有意な影響を有し、そして製造経費の増大、製造作業の品質管理の問題および臨床的使用のためのバッチの容認を導く。あるいは293細胞よりも明確なE1遺伝子組込みを有するPER.C6のような細胞株は(すなわちフランキングウイルス配列を含まない)が開発され、これはRCAを生じる組換え反応を可能とせず、すなわち上記のウイルス生産の問題を克服する可能性を有する。
【0048】
アデノ随伴ウイルス(AAV)(Kotin,R.M.Hum.Gene Ther.5:793−801(1994))は、大変広範な宿主の非分裂細胞のゲノムに組込むことができる1本鎖DNAの非自律複製パルボウイルスである。AAVはヒトの疾患に関連することが示されておらず、そして免疫応答を誘導しない。
【0049】
AAVは2つの明確な生活環の相を有する。野生型ウイルスは宿主細胞に感染し、組込まれ、そして潜伏する。アデノウイルスの存在下で初期アデノウイルス遺伝子の発現に依存するウイルスの溶菌相が誘導され、そして活発なウイルス複製を導く。AAVゲノムは、逆方向末端反復(ITR)配列により挟まれた2つのオープンリーディングフレイム(repおよびcapと呼ぶ)からなる。rep領域はAAV複製、ウイルスDNA転写および宿主ゲノムの組込みに使用されるエンドヌクレアーゼ機能を媒介する4種のタンパク質をコードする。rep遺伝子はウイルス複製に必要なAAV配列だけである。cap配列はウイルスキャプシドを形成する構造的タンパク質をコードする。ITRsは複製のウイルス起点を含み、そして莢膜形成(encapsidation)シグナルを提供し、そしてウイルスDNAの組込みに参加する。遺伝子治療用に開発された組越えの複製欠損ウイルスは、repおよびcap配列を欠いている。複製−欠損AAVは、AAV複製に必要な別々の要素を許容293細胞株にコトランスフェクションすることにより生産することができる。米国特許第4,797,368号明細書は関連する開示を含み、そしてそのような開示は引用により本明細書に編入する。
【0050】
レトロウイルスベクターは分割している細胞を感染させるために有用であり、そして宿主細胞膜およびウイルスタンパク質に由来するエンベロップにパッケージングされるRNAゲノムからなる。レトロウイルス遺伝子発現には逆転写段階が関与し、ここでその陽性−鎖RNAゲノムが鋳型として使用されて二本鎖DNAの合成を支配し、これは次いで宿主細胞DNAに組み込まれる。組み込まれたプロウイルスは遺伝子発現のために宿主細胞の機構を使用することができる。
【0051】
マウス白血病ウイルスは、一般に使用されているレトロウイルス種である(Miller et al.,Methods Enzymol.217:581−599(1993))。レトロウイルスベクターは典型的にはgag、polおよびenv遺伝子の欠失により構築されている。これらの配列の欠失により目的の核酸配列の挿入能力を提供し、そしてウイルスの複製機能を排除する。抗生物質耐性をコードする遺伝子はしばしば選択の手段として含まれる。プロモーターおよびエンハンサー機能も、例えばインビボ投与後の組織特異的発現を提供するために含むことができる。長い末端反復配列に含まれるプロモーターおよびエンハンサー機能も使用することができる。
【0052】
そのようなウイルス、および目的の外因性核酸配列を運ぶそのようなウイルスの修飾は、ウイルスパッケージング細胞株でのみ生成することができる。パッケージング細胞株は欠失ウイルス遺伝子(gag、polおよびenv)を、組換えを防止するために異なる染色体上にそれらが存在するように細胞に安定に挿入することにより構築することができる。パッケージング細胞株を使用して、組換えプロウイルスDNAを挿入することにより目的の核酸配列を含む複製欠損レトロウイルスを生成する生産細胞株を構築する。莢膜形成配列を含むgag遺伝子の小さい部分を挟む長い末端反復配列および目的の遺伝子を含むプラスミドDNAは、DNAの転移および取り込みのための標準的技法(電気穿孔、カルシウム沈殿等)を使用してパッケージング細胞株にトランスフェクトされる。この取り組みの変形は、複製可能なウイルス生産の見込みを下げるために採用された(Jolly,D.,Cancer Gene Therapy 1:51−64(1994))。このウイルスの宿主細胞範囲は、エンベロープ遺伝子(env)により決定され、そして異なる細胞特異性をもつenv遺伝子の置換を使用することができる。エンベロープタンパク質へ適切なリガンドと包含させることも、標的化に使用することができる。
【0053】
組換えレトロウイルスベクターの投与は、適当な技術により達成することができる。そのような技術には例えば、患者の細胞のエクスビボ形質導入、組織へのウイルスの直接注射を含み、そしてレトロウイルス生産細胞の投与による。エクスビボの取り組みには患者の細胞の単離および組織培養での維持が必要である。この内容において、細胞を標的とするために高比率のウイルス粒子を達成し、すなわち形質導入効率を向上させる(例えば引用により本明細書に編入する米国特許第5,399,346号明細書を参照にされたい)。米国特許第4,650,764号明細書はレトロウイルス発現系の使用に関する開示を含み、そしてこの引用した特許は参照により本明細書に編入する。
【0054】
場合により、インビボでウイルスの直接的導入が必要であり、または好ましい。レトロウイルスは脳腫瘍を処置するために使用され、ここではレトロウイルスが分裂している細胞(腫瘍細胞)のみに感染する能力が特に有利となる。
【0055】
レトロウイルス生産細胞株の患者の脳腫瘍への直接投与も提案された(例えば、Oldfield et al.,Hum.Gene Ther.4:39−69(1993))。そのような生産細胞は数日間、脳腫瘍内で生存し、そして周囲の脳腫瘍を形質導入することができるレトロウイルスを分泌する。
【0056】
発現用のポックスウイルスに基づく系が記載された(Moss and Flexner,Annu.Rev.Immunol.5:305−324(1987);Moss,B.In Virology,1990,pp.2079−2111)。例えばワクシニアは、感染した細胞の細胞質中で複製する大きな、エンベロープを持つDNAウイルスである。多くの異なる組織に由来する非分裂および分裂細胞が感染し、そして非組込みゲノムからの遺伝子発現が観察される。組換えウイルスは導入遺伝子をワクシニアに由来するプラスミドに挿入し、そしてこのDNAをワクシニアに感染した細胞(ここでDNAの相同的組換えがウイルス生産を導く)にトランスフェクトすることにより生産することができる。重要な欠点は、これが150〜200のウイルスにコードされるタンパク質に対して宿主免疫応答を誘導し、反復投与が問題となる点である。
【0057】
単純ヘルペスウイルスは、感染した細胞の核内で複製する大きな、二本鎖DNAウイルスである。このウイルスは外因性の核酸配列と関連した使用に適合可能である(Kennedy and Steiner,Q.J.Med.86:697−702(1993)を参照にされたい)。利点には広い宿主細胞範囲、分裂および非分裂細胞の感染を含み、そして外来DNAの大きな配列を相同的組換えによりウイルスゲノムに挿入することができる。欠点は複製可能なウイルスを含まないウイルス調製物とすることが難しいこと、および強い免疫応答である。ウイルスのチミジンキナーゼ遺伝子の欠失は、低レベルのチミジンキナーゼを持つ細胞中でウイルスを複製欠損とする。活発な細胞分裂を受けている細胞(例えば腫瘍細胞)は、複製を可能にする十分なチミジンキナーゼ活性を保有する。
【0058】
HIV、マウスの微小ウイルス、B型肝炎ウイルスおよびインフルエンザウイルスを含む他の種々のウイルスが、遺伝子転移用のベクターとして開示された(Jolly,D.,Cancer Gene Therapy 1:51−64(1994)を参照にされたい)。
【0059】
非ウイルス性のDNA送達法も応用できる。これらのDNA送達法は非複合化プラスミドDNA、DNA−脂質複合体、DNA−リポソーム複合体、DNA−タンパク質複合体、DNA−コート金粒子およびDNA−コートポリラクチドコグリコライド粒子に関する。精製した核酸は組織に直接注入することができ、そして例えば筋肉組織、特に筋肉の再生に効果的な一過性の遺伝子発現をもたらす(Wolff et al.,Science 247:1465−1468(1990))。Davis et al.,(Hum.Gene Ther.4:733−740(1993))は、成熟した筋肉(骨格筋が一般に好適である)へのDNAの直接注入について公開した。
【0060】
金粒子上のプラスミドDNAは遺伝子銃を使用して細胞(例えば表皮または黒色腫)に「発射(fired)」され得る。DNAは金粒子上に共沈殿し、そして推進剤として電気的スパークまたは加圧ガスを使用して発射される(Fynan et al.,Proc.Natl.Acad.Sci.U.S.A.90:11478−11482(1993))。電気穿孔も多くの針のアレイおよびパルスをかけた、回転する電場を採用した電気穿孔プローブを使用して、DNAを充実性腫瘍に転移できるようにするために使用された(Nishi et al.,Cancer Res.56:1050−1055(1996))。有意な細胞トランスフェクションの強化および腫瘍内注入法に優る良好な分布特性の、皮下腫瘍への高効率遺伝子転移が特許請求された。
【0061】
脂質が媒介するトランスフェクションはインビトロおよびインビボのトランスフェクションの両方に好適である(Horton et al.,J.Immunology 162:6378(1999))。脂質−DNA複合体は、DMRIE−C試薬のような市販されている脂質を使用してDNAおよび脂質を注入の1〜5分前に混合することにより形成された。
【0062】
リポソームは細胞に入り易くするために、親水性分子を疎水性分子で取り囲むことにより作用する。リポソームは脂質により作られた単層または多層の球である。脂質組成および製造工程は、リポソーム構造に影響を及ぼす。他の分子は脂質膜に包含することができる。リポソームはアニオン性またはカチオン性でよい。Nicolau et al.(Proc.Natl.Acad.Sci.U.S.A.80:1068−1072(1983))は、ラットに注入されたアニオン性リポソームからのインスリン発現に関する研究を公開した。アニオン性リポソームは他を標的としない限り、主に肝臓の網内細胞を主に標的とする。分子はリポソームの表面に包含されてそれらの挙動、例えば細胞−選択的送達を改変させることができる(Wu and Wu,J.Biol.Chem.262:4429−4432(1987))。
【0063】
Felgner et al.(Proc.Natl.Acad.Sci.U.S.A.84:7413−7417(1987))はカチオン性リポソームに関する研究を公開し、静電的相互作用によるそれらの核酸の結合を示し、そして細胞へ入ることを示した。カチオン性リポソームの静脈内注入は、器官への導入性の血液供給の注入でほとんどの器官に導入遺伝子の発現を導く。カチオン性リポソームは肺上皮を標的とするためにエーロゾルにより投与することができる(Brigham et al., Am.J.Med.Sci.298:278−281(1989))。カチオン性リポソームの導入遺伝子送達を用いたインビボ実験が公開された(例えば、Nabel et al.,Rev.Hum.Gene.Ther.5:79−92(1994);Hyde et al.,Nature 362:250−255(1993)および;Conary et al.,J.Clin.Invest.93:1834−1840(1994))。
【0064】
ミクロ粒子は食作用細胞にDNAを送達するための系として実験され、そのような取り組みはパンゲア ファーマシューティカル(Pangaea Pharmaceuticals)により報告された。そのようなDNAミクロカプセル化送達系は、微小球を摂取するマクロファージのような食細胞のさらに効率的な形質導入を行うために使用された。微小球は、潜在的に免疫原性のペプチドをコードするプラスミドDNAをカプセル化し、このDNAは発現した時、そのようなペプチドおよび同じエピトープを含むタンパク質配列に対する免疫応答を刺激することができる細胞表面上のMHC分子を介してペプチド展示を導く。この取り組みは現在、抗腫瘍および病原ワクチン開発における有力な役割を目的としているが、他にも可能な遺伝子治療の応用を有することができる。
【0065】
ウイルス−様粒子(VLPs)内で相同的自己集合を行うことができる天然のウイルスコートタンパク質も、送達用にDNAをパッケージングするために使用された。ヒトのポリオーマウイルスの主要な構造コートタンパク質(VP1)は組換えタンパク質として発現することができ、そしてVLPへの自己集合中にプラスミドDNAをパッケージングすることができる。生成した粒子は続いて種々の細胞系を形質導入するために使用することができる。
【0066】
DNAベクターにおける改善もなされ、そして多くの非ウイルス送達系に応用できると思われる。これらにはスーパーコイル化されたミニサークル(これはバクテリアの複製起点も抗生物質耐性遺伝子も持たず、すなわち高レベルの生物学的封じ込めを示す時、潜在的により安全である)、エピソーム発現ベクター(複製エピソーム発現系、これはプラスミドが核内で、しかし染色体外で増幅し、すなわちゲノムへの組込み反応(event)を回避する)、およびT7系(厳密な細胞質発現ベクターであり、ここでベクター自体はファージT7RNAポリメラーゼを発現し、そして第1プロモーターにより生成されたポリメラーゼを使用して治療用遺伝子が第2T7プロモーターにより駆動される)を含む。DNAベクター技術に対する他のより一般的な改善には、高レベルの発現を行うためのシス作用エレメント、細胞のサイクル毎に1回の複製を供給するためのアルフォイド反復DNAに由来する配列および核標的配列の使用を含む。
【0067】
上で検討したように、本発明は種々の動物細胞型においてインビボもしくはエクスビボのいずれかでIiの阻害に関する。この考察に関連する動物細胞型間の広い分類を、MHCクラスII分子発現の状態に基づき作成することができる。この広い分類をここで簡単に紹介し、そして具体的な治療的取り組みの内容の中で再検討する。
【0068】
自然に存在する抗原提示細胞(時折、専門的な抗原提示細胞と呼ぶ)は、獲得した免疫応答に参加する。樹状細胞、マクロファージ、Bリンパ球および特定の他の単核細胞を含むこれらの細胞は、MHCクラスII分子−陽性である。加えてTリンパ球のような幾つかの細胞は、休止状態ではMHCクラスII分子を発現しないが、適切な活性化でMHCクラスII分子を発現するように誘導することができる。抗原性ペプチドのMHCクラスII−拘束提示の機能に対してインビボまたはエクスビボで誘導することができるそのような細胞は、自然に存在する抗原提示細胞の範疇に含まれる。細胞は多形核細胞について記載されたように(Rasdak,Immunol.101(4):521−30(2000))、自己血清、IFN−GM−CSFとの共存培養を介してMHCクラスII分子を発現するように誘導することができる。T細胞もMHCクラスII分子を発現するように誘導し、そしてマイトジェンおよび異種APCsと培養した時に抗原提示細胞の機能性を予測することができる(Patel,J.Immunol.163(10):5201−10(1999))。
【0069】
以下の章でさらに詳細に検討するように、そのような細胞に目的の抗原性エピトープをコードする発現可能な核酸配列を導入することが可能である。このエピトープ発現をIi発現と組み合わせた時、目的の抗原性エピトープはMHCクラスII分子と結合して抗原提示細胞の表面上に展示される。
【0070】
自然に存在する抗原提示細胞は身体全体、そして末梢リンパ組織を通って循環する。末梢リンパ組織は身体の2つの流体系、血液およびリンパを取り巻いて組織されている。これら2つの流体系は接触している。リンパは血液から組織の中および回りの空間に運ばれた流体により形成される。これらの細胞外空間からリンパは薄い壁のリンパ管に流れ、ここでリンパは大きな中央集合管にゆっくりと移動する。最終的にリンパは静脈に戻され、ここでリンパは血液に再度入る。血液では、リンパ球が有核細胞の20〜30パーセントを構成し;リンパではそれが99パーセントを構成する。これらの流体系内を循環している抗原提示細胞はリンパ節および脾臓の小胞中心(follicle center)を通る。身体のリンパ節中の高濃度Tリンパ球およびBリンパ球および脾臓の小胞中心は、細胞の相互作用およびクローン増殖を促す。
【0071】
典型的にはMHCクラスII分子をほとんど発現しないか、または発現しない目的の他の細胞には、ほとんどの悪性およびウイルスが感染した細胞を含む。特に通常はMHCクラスII分子−陰性であると考えられている数種の腫瘍は、幾つか、もしくはすべての細胞上に低レベルのMHCクラスII分子を発現することが報告されたことに注目されたい。これらには例えば、胸部、肺または結腸癌腫を含む。これらの細胞は、病原に特異的な抗原を発現することができるが、MHCクラスII分子の不存在もしくは比較的低い存在度を与え、そのような細胞によるそのような抗原に由来するペプチドの有意な程度のMHCクラスII提示は無い。これらの細胞では、MHCクラスII分子の発現を誘導し、ならびにIi発現を阻害する両方が可能である(Ii発現およびMHCクラスII発現は同時調節される)。この組み合わせ介入は、病原に関連した、抗原性エピトープを含むペプチドをMHCクラスII分子と結合して細胞の表面に展示することをもたらす。
【0072】
目的とする他のクラスの細胞は、悪性の、ウイルスに感染したものでも、自然に存在する抗原提示細胞でもない。そのような細胞の例には、繊維芽細胞、ケラチノサイトおよび筋肉細胞を含む。この細胞はMHCクラスII分子−陰性であり、そして自然に存在する抗原提示細胞には分類されない。そのような細胞はインビボまたはエクスビボのいずれかでワクチン接種法と関連して有用である。例えばMHCクラスII分子と結合した抗原提示のために、筋肉細胞が標的とされるインビボの内容を考慮されたい。目的の抗原性エピトープおよびMHCクラスII分子のインデューサーをコードする発現可能な核酸配列を、筋肉組織に注入することができる。そのような配列は組織中の筋肉細胞に取り込まれ、そして発現される。注入領域内のある割合の筋肉細胞が、最終的にMHCクラスII分子と結合して目的の抗原性エピトープを細胞表面上に発現するだろう。そのような提示による刺激に応答できる(competent)細胞(例えばヘルパーT細胞)は、リンパを循環する刺激に応答できる細胞として提示細胞と接触する。上で述べたように、リンパ球は循環しているリンパ中の有核細胞の99%を構成する。刺激された抗原提示細胞は脾臓のリンパ節のTリンパ球およびBリンパ球と共同作用し、ここで細胞濃度および他の因子が相互作用を促し、そしてクローン選択を拡大する。分泌されるBリンパ球およびそれらの成熟子孫、血漿細胞により生産される抗体は、リンパの節を去り、そして血液に輸送される。
【0073】
直前の章は限定された文脈上の考察であるが、Ii抑制のための目的の細胞型を紹介するのに役立つ。以下に続く考察は、これらの紹介した細胞型および関連する方法をさらに詳細に調査する。
【0074】
Ii抑制治療は、腫瘍性疾患と関連して示される。これらには例えば、限定された原発性部位を有するガン、ならびに未知の原発性部位の転移性ガンを含む。前者のクラスには胸部ガン、頭および首の悪性腫瘍、卵巣の癌腫、精巣ガンおよび他の栄養膜疾患、皮膚ガンおよび黒色腫および他の色素沈着皮膚損傷を含む。
【0075】
Ii抑制治療はPAI−1を過剰発現する特定の細胞にも示され、そしてMHCクラスII分子を発現するように誘導された。そのような細胞は冠状のアテローム硬化性プラーク、頸動脈、腎動脈、静脈およびガン細胞に見いだされる。PAI−1過剰発現は、腫瘍の侵襲、新脈管形成(neoangiogenesis)および転移、ならびに心筋梗塞、アテローム硬化症、再狭窄および血栓塞栓疾患に関連している(米国特許第6,224,865号明細書;Gunther,J.Surg.Res.103(1):68−78(2002);Harbeck,J.Clin.Oncol.20(4):1000−7(2002);DeYoung,Circulation 104(16):1972−land(2001);Rerolle,Nephrologie 22(1):5−13(2001)。プラスミノーゲンアクチベーターインヒビター1型(PAI−1)は糖尿病患者の動脈壁で増加し、アテローム硬化症の加速および糖尿病患者で臨床的に観察されるプラークの進行に貢献する(Pandolfi,Arterioscler.Thromb.Vasc.Biol.21(8):1378−82(2001))。PAI−1活性は特異抗体、ペプチド拮抗薬、アンチセンスおよびおとり(decoy)オリゴヌクレオチドの使用を介して抑制された(Rerolle,Arterioscler.Thromb.Vasc.Biol.21(8):1378−82(2001))。
【0076】
Ii抑制治療は感染性疾患との関連でも示される。これらにはウイルス疾患(DNAおよびRNAウイルス)、バクテリア疾患(グラム−陽性およびグラム−陰性)、マイコバクテリア疾患、スピロヘータ疾患、リケッチア疾患、マイコプラズマおよびクラジミア疾患、真菌感染、原生生物および寄生生物感染および外部寄生生物感染を含む。
【0077】
自然に存在する抗原提示細胞に関連して、インビボおよびエクスビボ応用が含まれる。本開示において、用語「標的化」は時折、抗原性タンパク質または抗原性タンパク質中の特定の抗原性エピトープに免疫応答を向けることを記載するために使用する。この免疫応答は一部は、応答の内容に依存してTh1またはTh2またはTh3細胞で変動することができるTヘルパー細胞またはTサプレッサー細胞のようなT免疫調節細胞の活性化を特徴とする。例えばTh1応答は、腫瘍抗原に応答するCTL応答の発生に関連するヘルパー応答であり、この応答は腫瘍細胞を殺すことを導く。しかしアレルゲンに対するTh1応答は、Th2応答からアレルゲンに対する応答の免疫逸脱(immunodeviating)に関して、機能的には抑制反応となることができ、これは病原性IgE抗体の生産を導く。加えて標的化の概念は、新規もしく増加した量のMHCクラスII提示エピトープの提示により刺激される免疫応答の初期部分だけでなく、T免疫調節細胞に及ぼす初期作用により誘導または調節される下流のエフェクター応答も含む。このように例えば標的化には本明細書で教示する標的化法により開始され得るCTL−抗ガン応答または免疫グロブリン抗ウイルス応答を含む。
【0078】
標的化には、抗原が特定されているか、もしくは未知であるか、またはたとえ過度な実験無しには同定することができなくても、免疫応答が抗原に向けられるという概念を含む。例えば標的化は、各々が免疫応答の生成に貢献し得る多数の抗原を発現することができる細胞に向けることができる。細胞中のどの特定の抗原が免疫応答に参加するかは、個体の遺伝的構成に依存して人毎に異なり得る。遺伝的因子に対する免疫応答の感受性は、十分に記載されている。その結果、有用な治療的または診断的目的のために標的化法を使用するにあたり、細胞の特異的抗原成分は特定される必要はなく、そしてしばしば特定することはできない。
【0079】
標的化のプロセスには、インビボまたはインビトロのいずれかで起こるプロセスを含む。例えばインビボでは、腫瘍細胞または樹状細胞のいずれかであるMHCクラスII−陽性細胞により提示された抗原に対する免疫調節T細胞の活性化は、非−腫瘍場所または浸潤している腫瘍のいずれかで起こることができる。免疫応答のエフェクター部分の拡大も同様にインビボまたはインビトロのいずれかで起こり得る。インビトロ応答の場合、個体または別の選択した個体に再導入され得る産物を生成して、治療的応答を行うことができる。そのような産物の例は、樹状細胞調製物、細胞傷害性T細胞調製物、およびそのようなインビトロ標的化カルチャーからB細胞をクローニングした後に生産されるであろう抗体(例えばB細胞ハイブリドーマの生産後)を含む。
【0080】
この目的に向けて、末梢血単核球を得た個体に導入することを望む治療用産物に依存して、元のカルチャーを所望の細胞集団、例えば樹状細胞もしくはTリンパ球について濃縮するために分画する。さらにここで教示する標的化プロセスを行った後のカルチャーを、所望の細胞集団、例えば樹状細胞もしくはTリンパ球について濃縮するために分画することができる。単離後すぐに、および本発明の標的化前に、あるいはその標的化が行われた後のいずれかで個体から得た細胞の分画に、確立された方法を利用することができる。さらに確立された手法は、そのような産物を末梢血単核細胞が元々得られた個体に導入するために利用することができる。このために標的化に関する本発明の方法は、末梢血単核細胞に限定されず、口腔咽頭部または他の領域に由来する粘膜細胞、気管支もしくは胃洗浄後に得た細胞、腫瘍組織または正常組織、例えば肝臓、膵臓、前立腺、骨格筋、脂肪、皮膚のような任意の器官の生検もしくは切除により得た細胞を含め、個体から得られるすべての細胞調製物を含む。
【0081】
すべての場合で、目的は自然に存在する抗原提示細胞に、処置する病状に特異的な目的の抗原性エピトープならびにIi発現のサプレッサーを導入することである。腫瘍またはウイルス遺伝子でトランスフェクトした樹状細胞は、強力な抗−腫瘍または抗−ウイルス免疫応答を誘導する。そのような抗原遺伝子でトランスフェクトした樹状細胞中でのIiタンパク質発現の阻害は、そのようなDNAワクチン接種の効力を強化する。自然に存在する抗原提示細胞が関与するインビボおよびエクスビボ態様の両方の場合で、目的の抗原性エピトープをコードする発現可能な核酸配列、およびIiのRNAインヒビターをコードするリバース遺伝子構築物を導入することが好ましい。
【0082】
用語「発現可能な核酸配列」は、翻訳可能(competent)なRNA種をコードする転写可能なDNA構築物、ならびに導入前に転写される翻訳可能なmRNA種を包含することを意図する。当業者は転写および翻訳能力(competency)を付与するために必要な分子シグナルについて精通している。
【0083】
これらの必要な要素の両方を、1つの分子構築物として提供することが可能である(例えば、エピトープおよびIiインヒビターの両方をコードする核酸を受ける十分な能力を有するウイルスベクター送達系を使用して)。あるいは別個の発現構築物を使用して各要素を運ぶことができる。独立した様式で送達される別個の構築物の場合、2つの構築物の各々を取り込む1つの抗原提示細胞の見込みは、統計的確率の問題である。さらに1つのウイルス粒子中に1より多くの構築物をパッケージングすることは、Ii抑制の治療に効果的な誘導を最大にすること、そして示される場合には、有害である免疫応答を生成するウイルスタンパク質の合成に対して、MHCクラスIi誘導および/または所望するタンパク質抗原の合成の誘導に用途を有する。そのような抗−ウイルス免疫応答は、例えばそのような治療的介入が可能な頻度を制限することになる。
【0084】
非−ウイルス送達系による誘導にも特別な配慮が必要である。非−ウイルス送達系を使用して、非複合化DNA、DNA−リポソーム複合体、DNA−タンパク質複合体およびDNA−コート金粒子を細胞に送達することができる。これらの各方法は、特定の病状に関して選択を制御する利点および欠点を与える。複合化DNA(例えばDNA−リポソーム複合体、DNA−タンパク質複合体、DNA−コート金粒子およびポリラクチド コグリコライド粒子中へのマイクロカプセル化)の使用は、1つの細胞へエピトープをコードする核酸配列およびIi発現のインヒビターをコードする核酸配列の両方の送達を確実とする傾向がある。たとえ異なる分子種によりコードされても、それらは「パッケージされる」ので(例えばリポソーム中へのカプセル化、または金粒子上への被覆のどちらか)、両種が1つの細胞に送達される傾向がある。
【0085】
DNA−コート金粒子は一般に、いわゆる「遺伝子銃」技法を使用した弾道法(ballistic method)により送達される。この技術を使用して、金粒子は皮膚または筋肉組織に発射され、そして細胞を貫通するために使用される。貫通された細胞はこの様式で導入された核酸配列を発現することが示された。樹状細胞は、この技法を使用して効果的にトランスフェクトされる自然に存在する抗原提示細胞である。そのような発現構築物は1つの樹状細胞に導入された時、例えば抗原提示細胞の表面上にMHCクラスII分子と結合した目的の抗原性エピトープの展示を生じるだろう。抗原提示細胞の表面上のこのエピトープ/MHCクラスII分子複合体の展示はさらに免疫細胞を刺激して、高まった免疫応答を提供するするだろう。
【0086】
あるいは限定された解剖学的場所(例えば原発性腫瘍または腫瘍性疾患の転移の幾つか)を有する病状に取り組む時、限定された解剖学的場所への直接的注入を示すことができる。そのような部位は樹状細胞のような抗原提示細胞の濃度が高い傾向がある。腫瘍はそのような導入の局所的部位の1例である。関連する発現可能な核酸構築物の細胞への導入を行うための手段が適当である。そのような構築物を局在化した腫瘍部位に導入する時、MHCクラスII分子の発現を刺激するタンパク質をコードするさらなる発現可能な核酸配列を含むことが好ましい。この第3成分の包含は、腫瘍細胞自体を意図している。Ii発現を阻害する構築物、およびMHCクラスII分子生産の発現可能なインデューサーが病状を現している細胞(例えば腫瘍細胞)に取り込まれる場合、細胞は病状に特異的なエピトープをMHCクラスII分子と結合してその細胞表面に展示する。これらの細胞はTヘルパー細胞およびBリンパ球も刺激する。このように局在化した病状に向けられた治療と合わせたこれら3つの発現可能な要素の直接注入は、正常な抗原提示細胞および病状を現すMHCクラスII分子−陰性細胞を標的化する併用療法とみることができる。
【0087】
MHCクラスII分子の生産を誘導するための発現可能な核酸配列の使用に加えて、当業者は本内容および関連する内容において核転移の方法の応用性を認識するだろう(Wolf,Arch.Med.Res.32(6):609−13(2001);Wakayama,Science 292(5517):740−3(2001))。さらに抗原提示細胞は脱分化するように誘導された体細胞に由来してもよく、これによりガン−胚性抗原を発現する(Rohrer,J.Immunol.162(111):6880−92(1999))。そのような細胞は目的の抗原性エピトープに免疫的攻撃を誘導するために使用できる。完全に分化した細胞は、インビボで未成熟(premature)状態に脱分化するように誘導して、器官の再生を行うことができる(Abbate,Am,J.Physiol.277(3Pt2):F454−63(1999))。これらの細胞は異所性の細胞への免疫学的攻撃を刺激するために抗原提示細胞としても機能することができる(Fu,Lancet 358(9287):1067−8(2001))。
【0088】
抗原提示細胞がインビボで標的にされる本発明の別の態様では、正常組織がサイトカイン(例えばGM−CSF)の皮下注射により刺激される。この皮下の「プライミング(priming)」は、樹状細胞をその領域に誘引する。プライミング注射に続いて目的の抗原性エピトープをコードする発現可能な核酸配列、ならびにIi合成のインヒビター(例えばリバース遺伝子構築物)が注射される。腫瘍性細胞は病状に特異的なペプチドの生産体であるが、一般にそれらの表面にMHCクラスII分子と結合して提示しない。そのような細胞では、MHCクラスII分子発現を誘導し、ならびにIi発現を阻害する両方が可能である。例えばMHCクラスII分子の発現は、MHCクラスII分子−陰性細胞に、MHCクラスII分子の生産を刺激するタンパク質をコードするcDNAを導入することにより誘導することができる。そのようなタンパク質には例えば、CIITAまたはインターフェロンガンマを含む。この併用介入は病状に特異的な、抗原性エピトープを含むペプチドのMHCクラスII分子と結合した細胞表面上での展示をもたらす。前に検討したように、発現可能な核酸配列の導入は、これらの目標を達成するために好適な方法である。
【0089】
上で検討したように、直接注入は病状が限定された原発性の場所(腫瘍のような)に存在する場合に示される。場合により、目的の抗原性エピトープをコードする発現可能な核酸配列を、その領域の抗原提示細胞を標的にするために注入材料に含めてもよい。ここでも抗原提示細胞に送達するための目標は、Ii抑制および目的の抗原性エピトープである。病気の細胞にはこの送達目標は、IiサプレッサーおよびMHCクラスII分子インデューサーである。
【0090】
腫瘍内注入と関連して、続く特異的プロトコールは、例えばサイトカインをコードする核酸配列またはサイトカインの腫瘍内注射が関与する確立された治療的プロトコールに基づく。そのようなプロトコールは例えば:
【0091】
【表1】
【0092】
を含む多くの刊行物に記載されている。
【0093】
エクスビボの応用に関して、腫瘍細胞は個体から単離され、そしてエクスビボ培養が樹立される。そのような培養は付随する正常細胞の分離を行うか、行わずに個体から得られた悪性細胞の非選択集団から樹立することができ、あるいは細胞はそのような細胞株に由来する細胞株もしくはクローンから得ることができる。あるいはそのような細胞は無関係な患者の樹立された悪性細胞株、あるいは新鮮な悪性組織(例えば結腸もしくは卵巣癌腫)の外植片として得ることができる。
【0094】
IiサプレッサーおよびMHCクラスII分子インデューサーを培養した細胞に導入すると、所望するMHCクラスII分子が結合した腫瘍特異的もしくは腫瘍関連抗原性エピトープの提示を生じる。MHCクラスII分子発現のインデューサーは当該技術分野では周知であり、そして例えばMHCクラスII分子トランス活性化因子(CIITA)、インターフェロンガンマ遺伝子およびインターフェロンガンマサイトカインを含む。この様式で処理した細胞は複製不能(incompetent)とされ(例えば照射もしくは固定により)、そして通例の免疫感作プロトコール(例えば皮下、静脈内、腹腔内もしくは筋肉内免疫感作による)で使用される。全細胞製剤に加えて、それらの他の誘導体を免疫感作製剤に使用してもよい。
【0095】
関連する腫瘍細胞のほとんどはMHCクラスII分子およびIi−陰性であるが、幾つかの腫瘍(例えば胸部、肺および結腸に影響するある種のリンパ腫、黒色腫および腺ガン)は、MHCクラスII分子−陽性およびIi−陽性である。MHCクラスII分子を発現するこのサブセットでは、所望の免疫刺激を達成するためにIiサプレッサーのみの導入で十分である。そのような細胞にMHCクラスII分子インデューサーの包含は、MHCクラスII分子−結合抗原とTヘルパー細胞との相互作用の見込みが増すことにより、所望の刺激を強化するために役立つと認識されるだろう。
【0096】
興味深い別の細胞のクラスは、悪性の、ウイルスに感染したものではなく、自然に存在する抗原提示細胞でもない。発現可能な核酸配列は、個体の組織の間隙空間に送達される。そのような組織には例えば、筋肉、皮膚、脳、肺、肝臓、脾臓、骨髄、胸腺、心臓、リンパ、血液、骨、軟骨、膵臓、腎臓、胆嚢、胃、小腸、精巣、卵巣、子宮、直腸、神経系、目、腺および結合組織を含む。組織の間隙空間は細胞間、流体、器官組織の網状繊維間のムコ多糖マトリックス、脈管または室の壁中の弾性繊維、繊維状組織のコラーゲン繊維、あるいは筋肉細胞を覆う結合組織内、または骨の空隙中のそのマトリックスを含んでなる。循環している血漿およびリンパ経路のリンパ液により占められる空間も同様である。
【0097】
筋肉細胞はインビボで、発現可能な核酸配列を取り込み、そして発現するそれらの能力が特にある(competent)ことが報告された(例えば引用により本明細書に編入する米国特許第6,214,804号明細書を参照にされたい)。この送達の利点は、多核細胞、筋小胞体およびT管系を含んでなる筋肉の無類の組織構造によることができる。発現可能な核酸配列は、細胞外流体を含み、そして筋肉細胞の深部に広がるT管系を通って筋肉に入る。またそのような発現可能な核酸配列は傷害を受けた筋肉細胞に入り、次いで修復することも可能である。
【0098】
動物は皮膚を通す直接注射により都合よく到達する、比例して大きな筋肉塊を有するので、筋肉は治療的応用において発現可能な核酸配列を送達するための部位としても有利に使用される。この理由から、比較的大用量の発現可能な核酸配列が多回および反復注射により筋肉中に蓄積され得る。治療は長期間に延長することができ、そして特別な技術および装置無しで安全かつ容易に行われる。これらの筋肉以外の組織、および発現可能な核酸配列の低い効率の取り込みおよびまたは発現を特徴とする組織も、注射部位に使用することができる。
【0099】
本発明と関連して、標的細胞中のIi合成を阻害し、そして目的の抗原性エピトープも発現することが望ましい(抗原性エピトープは処置する病状に特異的に関連する)。当該技術分野では知られているように、発現可能な核酸配列の効果的な投薬用量は、一般に約0.05マイクログラム/kg体重〜約50mg/kg体重の範囲に入る(通常、約0.005mg/kg体重〜約5mg/kg)。効果的な投薬用量は関連する因子の数に依存して変動することができると認識されるだろう。
【0100】
上記の種類の病状に関連した抗原を生産し、そして細胞の表面上にMHCクラスII分子と結合した抗原を展示する細胞の別の生成法は細胞融合法である。より具体的にはこれは目的の病状を現す細胞(例えば腫瘍細胞)を用いて、MHCクラスII分子を自然に生産する細胞(例えば樹状細胞のような自然に存在する抗原提示細胞)の融合体を生産する日常的な実験の事柄である。そのような融合細胞では、腫瘍特異的抗原は融合細胞の表面上でMHCクラスII分子と結合して展示される。ほとんどの場合、産物は樹状細胞、マクロファージ、Bリンパ球のような自然に存在する抗原提示細胞、または特定の多機能性細胞の種類と、目的の抗原性エピトープを発現する細胞との融合体である。目的の抗原性エピトープを発現するような細胞には、例えば悪性細胞、ウイルスに感染した細胞または形質転換した細胞、自己免疫応答の誘導に関連する細胞、および自己免疫応答を調節する細胞を含む。後者の種類にはそれらの影響を抗−イディオタイプネットワークメカニズムを介して発揮する細胞を含む(例えば慢性関節リウマチにおいて病原に関連性のあるT細胞レセプターを発現する)。この型の細胞融合体は、エクスビボで生産される。Ii抑制およびワクチン接種は本開示のいたるところに記載するように行う。
【0101】
当業者は本発明の方法を、処置する細胞またはそれらの局所的環境へのサイトカイン治療(すなわちサイトカインをコードする核酸配列、またはサイトカイン自体の導入)と組み合わせることができると認識するだろう。他の免疫同時−刺激(immune co−stimulatory)分子も使用することができる(Akiyama,Y.,Gene Ther.7(24):2113−21(2000):Miller,P.W.,Hum.Gene.Ther.11(1):53−65(2000);J.Neurosurg.94(2):287−292(2001);Jantscheff,P.,Cancer Immunol.Immunother.48(6):321−30(1999);Kikuchi,T.,Blood 96(1):91−9(2000);Melero,I.,Gene Ther.7(14):1167−70(2000);Lei,H,Zhongua Zhong Liu Za Zhi20(3):174−7(1998))。
【0102】
前に述べたように、当業者は過度な実験を行うことなく、強化された免疫応答を提供する、MHCクラスII分子が結合して展示する病原に特異的な抗原を同定することができる。ここでもすべての場合で、Ii阻害はMHCクラスII分子に結合したそのような抗原の展示を行うために必要である。以下の一覧はこのクラスに入る抗原の非限定的、非排他的な例の列挙であることを意図している:HIVgp120(Barouch et al.,J.Immunol.15:168:562−8(2002));HIVgag(Shingh et al.,Vaccine 20:594−602(2001));インフルエンザM1およびM2(Okuda et al.,Vaccine 19:3681−91(2001));B型肝炎表面抗原およびコア抗原(Musacchio et al.,Biochem.Biophys,Res.Commun.282:442−6(2001));ヒトテロメラーゼ逆転写酵素(hTERT)(Heiser et al.,Cancer Res.61:3388−93(2001));Gp75TRP−1(Bowne,Cytokines Cell Mol.Ther.5:217−25(1999));TRP−2およびgp100(Xiang,Proc.Natl.Acad.Sci.USA 97:5492−7(2000));PSA(Kim,Oncogene 20(33):4497−506(2001));CEA(von Mehren et al.,Clin.Cancer Res.7:1181−91(2001));Erb2/Neu(Pilon et al.,Immunol.167:3201−6(2001)およびTuting,Gene Ther.6:629−36(1999))。
【実施例】
【0103】
実施例1
CIITA cDNAを含むアデノウイルスベクターの構築
この実験の最初の目標は、MHCクラスII分子陰性細胞(例えばMC−38およびRenca)にMHCクラスII分子の効率的誘導のためのアデノウイルスベクターを構築することであった。CMVプロモーターおよびポリAテイルを含むCIITA遺伝子構築物は、CIITAを含有するpCEP4ベクター(L.Glimcher博士から得た)から、Sal1を使用して切り出した。この断片をpBluescriptに連結して、pBlue/CIITAを作成した。次いでpBlue/CIITAをEcoRVおよびXhoIで消化して、CMVプロモーター、CIITA cDNAおよびポリAシグナルを含むDNA断片を放出し、これをpQBI/BN(クウォンタム(Quantum)、モントリオール、カナダ)に連結して、pQBI/BN/CIITAを作成した。
【0104】
このベクターを293Aアデノウイルスパッケージング細胞に、Cla1消化アデノウイルスDNA(ウイルスの左腕は削除されてバックグラウンドを下げた)と一緒に、製造元の指示に従いコトランスフェクトした。コトランスフェクションから3週間後、生成したプラークは、CIITA cDNAの−7から+12および+751から+769に位置する2つのDNAプライマーを使用したPCRによりスクリーニングして、CIITA遺伝子の存在を確実にした。1つのクローンを使用して2つのマウス腫瘍細胞株:MC−38結腸腺ガンおよびRenca腎臓細胞腺ガンにおいて、MHCクラスII分子の導入を試験した。MHCクラスII分子の導入に関するタイムコースは、アデノ/CIITA組換えアデノウイルスベクターでの感染後にこれらの細胞株でアッセイした。CIITA挿入物を欠く点を除いて同一のアデノウイルスベクターを対照として使用した。MHCクラスII分子は感染から48〜72時間後に>95%の細胞で強く誘導されることが証明された。
【0105】
実施例2
アデノ/CIITAでの感染に加えてIiアンチセンスオリゴヌクレオチドでの処置によるMHCクラスII+/Ii−表現型の生成
この実施例は、細胞のアデノ/CIITAによる感染および定めたIiアンチセンスオリゴヌクレオチドによるIi発現の阻害により、MHCクラスII+/Ii−表現型を発現する細胞の生成を示す。Iiアンチセンスオリゴヌクレオチドは効果的であることが以前に証明された(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。対照実験には:a)処置無し;b)アデノ/CIITA構築物のみ;c)センス対照オリゴヌクレオチドと一緒のアデノ/CIITA構築物;およびd)4つのヌクレオチド誤対合対照アンチセンスオリゴヌクレオチドと一緒のアデノ/CIITA構築物を含んだ。簡単に説明すると、1.5×106個のMC−38細胞を25cm2のフラスコにオリゴの電気穿孔の24時間前にまき、そして1.5mlのウイルスストック溶液(1.26×106PFU/ml)を含む5mlの総容量で48時間感染させた。感染から最初の24時間後、10mlの新鮮な培地を加え、そして細胞をさらに24時間インキュベーションした。次いで細胞をトリプシン処理し、そしてアンチセンス、センスまたは誤対合オリゴヌクレオチドでの電気穿孔前に洗浄した。電気穿孔の条件は以下の通りであった:3〜5×106個の細胞を、50μMのオリゴヌクレオチドを含有する0.5mlのRPMI1640中の電気穿孔用のキュベットに加えた。次いで細胞を氷上で10分間インキュベーションし、そしてBTX600エレクトロポレーターを使用して200ボルト/1250μFにかけた。次いでキュベットをさらに10分間、氷上でインキュベーションし、その後に細胞を1回洗浄し、新しい25cm2フラスコにまき、そして24時間インキュベーションした。この時点で細胞をトリプシン処理し、そして以前に記載されたように(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))、MHCクラスII分子およびIiタンパク質に関する染色後にフロサイトメトリーにより分析した。
【0106】
図1に示す典型的な実験では、Iiアンチセンスで処理し、そしてアデノ/CIITA感染した細胞が、MHCクラスII分子の発現にほとんど影響しないか、または影響せずにIiの良好な選択的阻害を示した。対照のオリゴヌクレオチド処理した細胞(すなわち誤対合またはセンス配列を使用した)は、Iiの阻害を示さず、そしてアデノ/CIITA感染細胞に関してMHCクラスII分子に匹敵する発現があった。
【0107】
動物実験の予測および大量にMHCクラスII+/Ii−細胞を生成する必要性から、上記実験をスケールアップした系で繰り返した。5×106個のMC−38細胞を75cm2のフラスコに感染の18〜24時間前にまいた。細胞は5mlのウイルスストック溶液(1.26×106PFU/ml)で90分間感染させ、そして20mlの新しい培地を加えた。次いで細胞を48時間インキュベーションし、そして上記のようにオリゴヌクレオチド(50mM)を送達するために電気穿孔にかけた。次いで細胞をプールし、そして新しい75cm2フラスコ中でさらに24時間インキュベーションし、その後に培地を交換し、そして細胞をさらに3時間インキュベーションした。次いで細胞をMHCクラスII分子およびIiタンパク質の発現、およびマウスの免疫感作について分析した。Iiタンパク質発現の配列特異的阻害は、以前の実験で観察されたようにアデノ/CIITAで感染させ、そしてIiアンチセンスで処理した細胞中でのみ得られた。
【0108】
実施例3
MHCクラスII+/Ii−腫瘍ワクチンによる腫瘍防御
これらの実験のために、MC−38腫瘍ワクチン細胞を上記のように調製し、そして6〜7週齢の、メスのC57BL/6マウス(ジャクソンラボズ:Jackson Labs)を接種するために使用した。具体的にはMC−38細胞は上記のようにアデノ/CIITAで感染させ、4群に分け、そして電気穿孔により:a)無し;b)50mM Iiアンチセンスオリゴヌクレオチド;c)50mM 誤対合対照オリゴヌクレオチド;またはd)50mMセンス対照オリゴヌクレオチドで処理し、そしてフラスコに播種した。24時間後、新しい培地を加え、そして細胞をさらに3時間インキュベーションした。次いで細胞をトリプシン処理し、50Gy(セシウム源)で致死的に照射し、そして1.2×106個の細胞/マウスをマウスに接種した。5週間後、マウスを5×105個の親MC−38細胞でチャレンジし、そして腫瘍の出現について監視した。図2に示すように、Iiアンチセンス処置したアデノ/CIITA感染MC−38細胞の接種は腫瘍の成長に対して、すべての他の対照群よりも良い防御を提供した。これらのデータはCIITAで安定にトランスフェクトし、そしてIiアンチセンスで処置したMC−38細胞を使用した我々の以前の実験と一致する(図3)。見れば分かるように、安定にCIITAでトランスフェクトしたMC−38またはIiアンチセンスで処置した一時的なアデノ/CIITA感染細胞のいずれかを使用した防御のレベルは、匹敵するレベルの防御を与える。
【0109】
実施例4
MHCクラスII+/Ii−腫瘍ワクチンおよびGM−CSFによる腫瘍防御
別の組の動物実験では、後の親のMC−38細胞の成長に及ぼすGM−CSF処理と一緒のIi−阻害MC−38ワクチンの役割を調査した。これらの実験に関して、樹状細胞を誘引するためのMC−38細胞の免疫感作の1日前に、マウスに18mgのGM−CSF(R&Dシステム、ミネアポリス、ミネソタ州)を右後脚にs.c.注射した。またマウスを免疫感作するために使用したMC−38細胞の数は3×105個/マウスであり、以前の実験で使用した数より4倍少なかった。図4に示すように、GM−CSFはクラスII+/Ii−MC−38細胞により誘導される防御効果を強化する。わずか3×105個のクラスII+/Ii−細胞を接種されたマウスは、GM−CSFの不存在下で1.2×106個のクラスII+/Ii−MC−38細胞により誘導された時と同様に親細胞の成長を阻害した。以前の実験では、IFN−γにより誘導されるMHCがCIITAによりもはるかに強い抗−腫瘍免疫応答の誘導を与えることが示された(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。
【0110】
これらの実験はサイトカインとIi阻害の相乗的効果が可能であることを示した。さらなる実験は、GM−CSFおよびIFN−γの使用とIiアンチセンス法とを組み合わせ、そしてこの免疫感作プロトコールを至適化し、そして増幅させるために計画する。
【0111】
実施例5
IFN−γ cDNAを含むアデノウイルスベクターの構築
IFN−γは免疫応答の方向の調節に重要な役割を果たし、そしてMHCクラスII分子およびIiを種々の組織および幾つかの悪性細胞を含む細胞で誘導する。IFN−γ構築物でのトランスフェクト、およびアンチセンスオリゴヌクレオチドによるIi阻害により作成されたMHCクラスII+/Ii−腫瘍ワクチンは、MHCクラスII分子の発現がCIITAトランスフェクションにより誘導されること以外は同一の腫瘍ワクチンに比べて、増大した免疫原性を有する(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。発現可能なマウスIFN−γ配列がアデノウイルスにクローン化された。MHCクラスII分子およびIiタンパク質の両方の発現が、大変低濃度のアデノ/IFN−γの感染後に誘導された(図5を参照にされたい)(わずか1のMOIでも。データは示さず)。アデノ/IFN−γ構築物を作成するために、マウスIFN−γcDNA(Chen et al.,J.Immunol.151:244−55(1993))が、IFN−γcDNAの開始および終止コドンを含む領域に相補的な2つの特異的オリゴヌクレオチドを用いたPCRにより増幅された。このIFN−γ断片は、特異的に設計したエンドヌクレアーゼ消化部位を使用してpCDNA(3+)プラスミドにクローン化され、そしてシークエンシングにより確認された。CMVプロモーター、IFN−γおよびポリAシグナルは、適切なオリゴヌクレオチドでさらにPCR増幅され、次いで適切な制限部位を使用してpQBI/Ad/BNにクローン化された。アデノ/IFN−γ組換えウイルスの生成は、実施例1に記載したものと同じ手順により行った。
【0112】
実施例6
Ii−RGCを含むアデノウイルスベクターの構築
数個のIiリバース遺伝子構築物を、RSV.5およびpcDNA(3+)発現ベクターにクローン化した。構築物のサブサットは、古典的なトランスフェクション法(例えばリポフェクチン)を使用してMHCクラスII分子−陽性細胞(A20)中のIiを阻害する能力を有することが示された。Iiアンチセンスオリゴヌクレオチドも効果的であることが示されたが、それらは電気穿孔法または有意な毒性を伴う他の方法を必要とする。またIi発現の有意な阻害を示すのは、オリゴヌクレオチドで処理した細胞の30〜70%より多くはない。対照的に(そしてアデノ/CIITA構築物を使用して示されるように)、遺伝子送達用のアデノウイルスベクターの使用により、すべての細胞へのほぼ100%の送達、所望する表現型の変化およびほぼ皆無の毒性がもたらされる。MHCクラスII+/Ii−表現型のより良い誘導のために、幾つかのIi−RGCがアデノウイルスにクローン化された。Ii−RGCを含む組換えアデノウイルスを作成するために、RSV(またはCMV)プロモーター、Iiリバース遺伝子断片およびポリAシグナルからなる発現カセットをPCRにより増幅し、そしてNotIおよびXhoIまたは他の適当な制限酵素部位を使用してpQBI/BNベクターにクローン化し、pQBI/BN/Ii−RGCを作成した。アデノ/Ii−RGCの最終的な構造は、実施例1に記載したものと同じ手順により完成した。MHCクラスII+/Ii−表現型の誘導実験では、アデノ/Ii−RGCの濃度がアデノ/CIITAの濃度の4倍に増加した時、Iiが>95%の細胞で阻害されたが、MHCクラスII分子の発現にはほとんど影響しないことが観察された(図6)。
【0113】
実施例7
IFN−γおよびIi−RGCを含むアデノウイルスベクターの構築
感染を簡略化するために、アデノ/IFN−γ/Ii−RGC構築物を生成した。プロモーター、Ii−RGC断片およびポリAシグナルは、適切なオリゴヌクレオチドを用いてPCRにより増幅し、そしてpQBI/Ad/BN/IFN−γにクローン化してpQBI/Ad/BN/IFN−γ/Ii−RGCを作成し、続いてこれを使用してアデノ/IFN−γ/Ii−RGCを作成した。アデノ/IFN−γ/Ii−RGCでの感染によるMHCクラスII+/Ii−表現型の誘導実験では、pQBI/Ad/BN/IFN−γ/Ii−RGC構築物(アデノ/IFN−γ/Ii−RGC(−92,+97))の1つの感染により、感染から96時間後にMHCクラスII+/Ii−表現型がMC/38細胞中に生成したことが観察された。
【0114】
実施例8
多数のIi−RGCを含むアデノウイルスベクターの構築
Ii−RGCの効率を最大にするために、幾つかのIi−RGCを1つのアデノウイルスベクターにクローン化した。PCR増幅および他の適切な分子生物学的方法を使用して、種々のIi−RGCの組み合わせを含むpQBI/Ad/BN構築物を生成した。そのような構築物の例には、以下に示す組を含む。マウスIi挿入物(−92,+97)、(+32,+136)、 (+314,+458)のヌクレオチド配列は、それぞれ配列番号1、2および3として配列表に提示する。
【0115】
【表2】
【0116】
Ii−RGCの幾つかは、以下に示す組を含め、IFN−γも含んでクローン化された。
【0117】
【表3】
【0118】
続いてIi−RGCの効果を最大にするための努力では、各々が異なるプロモーターにより駆動されるIi−RGCの多コピーを含むプラスミドを作成した。これらのプラスミドを以下に記載する。
【0119】
【表4】
【0120】
プロモーターの略号:RSV(ラウス肉腫ウイルスプロモーター)、EF−1a(ヒト延長因子−aプロモーター)、UbC(ユビキチンCプロモーター)、CMV(サイトメガロウイルスプロモーター)。
【0121】
実施例9
ヒトIi−RGCを含むプラスミドの構築
ヒトIi遺伝子配列に由来するヒトIi−RGC(hIi−RGC)によるヒトIi発現の阻害を本明細書に開示する。ヒト細胞中のIi発現を阻害するhIi−RGCを使用した実験の結果を、表2に示す。ヒトIi cDNA配列(Strubin et al.,EMBO J.3:869−72(1984))は、Eric Long博士から贈られた。異なる長さのIi遺伝子の断片は、適切なオリゴヌクレオチドを使用してPCRにより生成した。すべてのIi断片が多数のAUG開始および終止コドンを含む。いかなるリーディングフレイム中でもAUGの直後に終止コドンが続くことを回避するためにすべて設計し、アンチセンスRNAの半減期を延ばした。このために、AUGを作成して、異なるリーディングフレイム中の終止コドンに重ねた(override)。これらIi PCR断片は、適切な制限部位によりpcDNA3(+)発現ベクターにクローン化した。ヒトリンパ腫細胞株であるRajiを使用して、これらhIi−RGCを使用したIi阻害を測定した。Raji細胞は、Polyfectトランスフェクション試薬(キアゲン:Qiagen)で1μgのhIi−RGCプラスミドDNAを使用して、製造元の指示に従い一時的にトランスフェクトされた。48時間のインキュベーション後、細胞はIiおよびMHCクラスIIおよびIiの発現について、細胞を抗−ヒトIi抗体、LN2(ファミンゲン:Pharmingen)および抗−DR抗体(ファミンゲン)で染色することにより染色され、続いてフロサイトメトリーにかけられた。
【0122】
Ii−発現が一部の細胞で阻害さる(4〜9%の細胞はバックグラウンドより上であった(図8を参照にされたい)ことが観察された。この阻害は高度に再現性があった。さらにそのような一時的なトランスフェクションアッセイでは、典型的にはカルチャー中のわずか10%以下の細胞が実際に加えたDNA構築物を取り込む。すなわちバックグラウンドより上の4〜9%の細胞は、このアッセイ系の実際のトランスフェクション効率を反映している。これらの細胞では、MHCクラスII分子の発現に対する観察可能な影響は無かった。
【0123】
【表5】
【0124】
hIi−RGCの活性を最大にする試みでは、多コピーhIi−RGC(1つのプラスミド中に数コピーのhIi−RGC)が作成され、ここで各発現カセットは異なるプロモーターにより駆動される。これらのプラスミドを以下に列挙する。
【0125】
【表6】
【0126】
実施例10
MHCクラスII+/Ii−表現型および治療効力を誘導するために、IL−2と一緒のIi−RGCベクターの腫瘍内注射
BALB/cマウスに105個のRenca細胞をs.c.注射した。1:6(重量:重量)のCIITA:Ii−RGC DNA比で、CIITA cDNA遺伝子、CIITA cDNA遺伝子およびIi−RGC(−92,97)を含むプラスミド、またはトリプルIi−RGC(−92,97/32,136/314,459)を含むプラスミドを加えたCIITA遺伝子を、サイズが0.05〜0.2cm3のRenca腫瘍に注射した。注射の1〜5分前に、25mgの全DNAをDMRIE/C(1,2−ジメリスチルオキシプロピル−3−ジメチルヒドロキシ エチル アンモニウム ブロミド/コレステロール)(ギブコ:GIBCO)と、1:1(重量:重量)比でインキュベーションした。DNA注射から5日後、スライドを摘出した腫瘍の凍結切片から作成した。スライドはマウスMHCクラスIIおよびIiに対する抗体で染色して、腫瘍細胞のクラスII+/Ii−表現型を決定した。腫瘍中のクラスII+細胞がT細胞、B細胞またはマクロファージを表すかもしれない可能性を排除するために、染色はCD4、CD8、CD3、CD19(B細胞用)およびMAC(マクロファージ)に対する抗体を用いても行った。結果(データは示さず)は、CIITAのみを注射した腫瘍では類似のMHCクラスIIおよびIi染色が示されたが、CIITA/Ii−RGC(−92,97)またはCIITAプラスミドにトリプルIi−RGCを含むプラスミドを加えたもののいずれかを注射した腫瘍において、Ii抑制の証拠があった。CD4、CD8およびCD3染色は大変少ない陽性細胞を示し、腫瘍内のクラスII+細胞がT細胞を浸潤してないことを示す。B細胞およびマクロファージ染色も、クラスII+細胞がB細胞またはマクロファージでないことを除外した。同時に、脾臓サンプルのスライドは陽性対照として上記抗体のすべてで染色された。
【0127】
Ii抑制の治療効力を調査する実験のために、BALB/cマウスにRenca腎腺ガン細胞をs.c.注射し、そしてIL−2(2mg)、CIITA(3mg)およびIi−RGC(−92,97)(18mg)を含んでなる種々のプラスミド調製物を1日に腫瘍内注射し、そしてCIITAを含まない同じ調製物を2〜4日に注射することにより処置した。対照マウスは2μgのIL−2と一緒に連続して4日間、空ベクターを受けた。次いで腫瘍は2または3日毎に測定された。マウスは31日間追跡され、そして腫瘍サイズが1000cm3に達した時に終了した。結果はCIITAおよびIL−2と一緒にIi−RGCを含むベクターで処置したマウスが腫瘍の成長に劇的な減少を現す一方、IL−2のみまたは対照ベクターを受けたマウスは進行性であり、そしてマウスのターミネーション(termination)が必要であることを示した(図9を参照にされたい)。
【図面の簡単な説明】
【0128】
【図1】相対的蛍光強度を表す図である。MHCクラスII+/Ii−表現型は、マウス結腸腺ガン細胞(MC−38)のアデノ/CIITAアデノウイルスベクターでの感染、そして続いてIiアンチセンスオリゴヌクレオチドでの処理により生成した。A)親のMC−38細胞(処置無し);B)アデノ/CIITAに感染したMC38細胞;C)アデノ/CIITAに感染し、そしてセンス対照オリゴヌクレオチドで処理したMC38細胞;D)アデノ/CIITAに感染し、そして誤対合対照オリゴヌクレオチドで処理したMC38細胞;E)アデノ/CIITAに感染し、そしてIiアンチセンスオリゴヌクレオチドで処理したMC38細胞。
【図2】MHCクラスII+/Ii−細胞をワクチン接種したマウスでのMC−38結腸腺ガンの成長の阻害を表す図である。符号:(丸)MC−38細胞で免疫感作したマウス;(三角)アデノ/CIITAおよび誤対合対照オリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス;(菱形)アデノ/CIITAおよびセンス対照オリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス;および(四角)アデノ/CIITAおよびIiアンチセンスオリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス。
【図3】CIITAで安定にトランスフェクトされ、そしてIiアンチセンスを使用してIi発現が阻害された致死的に照射したMC−38細胞を接種したマウスにおける親腫瘍の成長の阻害を表す図である。マウスにPBS(三角)、センスオリゴヌクレオチド(丸)またはIiアンチセンス(四角)で処理したCIITAトランスフェクトMC−38細胞を接種した(5マウス/群)。
【図4】MHCクラスII+/Ii−細胞をワクチン接種し、そしてGM−CSFで処理したマウスでのMC−38結腸腺ガンの成長の阻害を表す図である。符号:(三角)親のMC−38細胞で免疫感作したマウス;(丸)MC−38細胞およびGM−CSFで免疫感作したマウス;(白抜き四角)CIITA、センス対照オリゴヌクレオチドで処理したMC−38細胞およびGM−CSFで免疫感作したマウス;および(菱形)CIITA、Iiアンチセンスオリゴヌクレオチドで処理したMC−38細胞およびGM−CSFで免疫感作したマウス。
【図5】MC−38細胞中のアデノ/IFN−γによるMHCクラスII分子およびIi誘導を表す図である。MC−38細胞はアデノ/IFN−γ(3MOI)で示した時期に感染させ、次いで抗−MHCクラスII分子またはIi抗体で染色し、そしてフローサイトメトリーで分析した。
【図6】相対的蛍光強度を表す図である。MHCクラスII+/Ii−表現型は、細胞をアデノ/CIITAおよびアデノ/Ii−RGC(Ii−92,97)で同時感染させることによりRenca細胞に生成した。Renca細胞は異なる比率のアデノ/CIITA対アデノ/Ii−RGCで同時感染させ、72時間インキュベーションし、そしてMHCクラスII分子またはIiタンパク質発現について染色した。親のRenca細胞をAに示し;アデノ/CIITに感染した細胞をBに示し;1:2の比率のアデノ/CIITA対アデノ/Ii−RGCに同時感染したものをCに示し;そして1:4の比率のアデノ/CIITA対アデノ/Ii−RGCに同時感染したものをDに示す。
【図7】MHCクラスII+/Ii−表現型が、細胞をアデノ/IFN−γ/Ii−RGC(mIi−92,97)に感染させることによりMC−38細胞に生成されるタイムコース実験を表す図である。Ii−しかしII+表現型はアデノ/IFN−γ/Ii−RGC(mIi−92,97)(左)の120時間後に作成されたが、アデノ/IFN−γの感染のみではMC−38細胞にMHCクラスII+/Ii−表現型を生成しなかった(右)。
【図8】一時的にトランスフェクトされたRaji細胞(MHCクラスII+/Ii+)、ヒトB−リンパ腫細胞株におけるIi抑制を表す図である。細胞は12−ウェルプレートに一晩、1.25×105細胞/ウェルでまき、そしてヒトIi−リバース遺伝子構築物(hIi−RGC)でトランスフェクトしてIi発現を阻害した。エフェクテン(effecten)トランスフェクション試薬(25μl、キアゲン)を濃縮hIi−RGCプラスミドDNA(1ug)とインキュベーションして、培地と混合したエフェクテンDNA複合体を生成し、これを細胞に直接加えた。48時間のインキュベーション後、細胞はIiおよびMHCクラスII分子発現について、抗−ヒトIi抗体、LN2(ファミンゲン)およびMHCクラスII分子(ファミンゲン)の染色用の抗−HLA−DR抗体での免疫染色により分析された。見れば分かるように、Ii発現は使用したIi−RGC配列に依存して、陽性対照細胞(左パネル)に比べて細胞の4%および9%で阻害されたが、MHCクラスII分子の発現には効果が無かった(右パネル)。
【図9】アデノ/Ii−RGCベクターのインビボ投与による腫瘍成長の阻害、およびMHCクラスII+/Ii−表現型の生成を表す図である。BALB/cマウスに、5×105個のRenca腎腺ガン細胞を皮下注射した。腫瘍が50から200mm3の間のサイズに達した時、腫瘍細胞注射から約10日後、腫瘍に種々のベクターの組み合わせをDMRIE/cと連続して4日間、毎日注射した。次いで腫瘍は2〜3日毎にサイズを測定した。マウスは腫瘍サイズが1000mm3に達した時にターミネーションした。左パネルのデータは、腫瘍に2mgのIL−2、3mgのアデノ/BN/CIITAおよび18mgのアデノ/BN/Ii−RGC(−92,97)を1日に、続いて2mgのIL−2、18mgのアデノ/BN/Ii−RGC(−92,97)および3mgの空プラスミド(アデノ/BN)(CIITA無し)を2〜4日に注射した4匹のマウスを表す。IL−2と共にCIITAおよびIi−RGCを含むベクターで処置したマウスが、腫瘍の成長に劇的な減少を表す一方、IL−2および対照ベクターのみを受けたマウスの腫瘍の成長は進行的であり、マウスのターミネーションが必要であったことは明らかである。
【配列表】
【背景技術】
【0001】
発明の背景
特定の抗原に対する免疫応答はTリンパ球によるそれら抗原のペプチド断片の認識により調節される。抗原提示細胞中で、プロセシングされた抗原のペプチド断片は主要組織適合遺伝子複合体(MHC)分子の抗原ペプチド結合部位に結合するようになる。次いでこれらペプチド−MHC複合体は、ヘルパーもしくは細胞傷害性Tリンパ球上のT細胞レセプターによる(外来ペプチドおよび提示するMHC分子の隣接表面の両方の)認識のために細胞表面に輸送される。ペプチドを送達する2つのクラスのMHC分子、MHCクラスIおよびMHCクラスIIが存在する。
【0002】
MHCクラスI分子は抗原をCD8−陽性細胞傷害性Tリンパ球に提示し、次いでこれは活性化されるようになり、そして抗原提示細胞を直接殺すことができる。クラスI MHC分子はそれらのほぼ合成時に、小胞体で感染性ウイルスのような内的に合成されるタンパク質からのペプチドを排他的に受ける。
【0003】
MHCクラスII分子は抗原をCD4−陽性ヘルパーTリンパ球(Tヘルパー細胞)に提示する。いったん活性化されると、Tヘルパー細胞は物理的接触およびサイトカインの放出を介して細胞傷害性Tリンパ球(Tキラー細胞)およびBリンパ球の活性化に貢献する。MHCクラスI分子とは異なり、MHCクラスII分子は非特異的または特異的エンドサイトーシスを介してインターナリゼーションした外因性抗原に結合する。ほぼ合成時にMHCクラスII分子は、代わりにインバリアント(invariant)鎖タンパク質(Ii)に結合することにより内因性抗原への結合が遮断される。これらMHCクラスII−Iiタンパク質複合体は小胞体から後ゴルジ区画(post−Golgi compartment)に輸送され、ここでIiがタンパク質分解により放出され、そして外因性の抗原ペプチドが結合する(非特許文献1;非特許文献2)。
【0004】
MHCクラスIおよびクラスII分子は、細胞中で明確な分布を有する。ほとんどすべての有核細胞がMHCクラスI分子を発現するが、その発現レベルは細胞型の間で変動する。免疫系の細胞はその表面上に豊富なMHCクラスIを発現する一方、肝臓細胞は比較的低レベルで発現する。核を持たない細胞はMHCクラスIをほとんど発現しないか、または全く発現しない。MHCクラスII分子はBリンパ球およびマクロファージ上に高度に発現されるが、他の組織の細胞上では発現されない。しかし多くの他の細胞型はサイトカインに対する暴露によりMHCクラスII分子を発現するように誘導することができる。
【0005】
正常な条件下で、(自己決定基により自己免疫疾患を導く可能性がある)内因性ペプチドはIiタンパク質が新生MHCクラスII分子と常に同時合成されるので、MHCクラスII分子に結合しない。自己決定基ペプチドおよびMHCクラスII分子を含む複合体は身体の免疫監視系により見いだされることはなく、これらの決定基に対する免疫寛容は生じない。MHCクラスII分子が発生した個体でIiにより阻害されない場合、内因性の自己決定基がMHCクラスII分子により提示されるようになり、これら内因性抗原に対する自己免疫応答を開始する。それらは特定の自己免疫疾患の場合である。そのような効果を悪性細胞で操作することにより、腫瘍の内因性抗原に対する「自己免疫応答」は、腫瘍細胞の増殖を制限もしくは排除するいずれかのために治療的に使用することができる。
【0006】
Iiタンパク質が同時に上昇しない、MHCクラスII分子の上昇した発現の治療効果は、MHCクラスII−陰性、Ii−陰性腫瘍で実証された(非特許文献3;非特許文献4;非特許文献5;非特許文献6;および非特許文献7)。これらの研究において、MHCクラスII分子の遺伝子のMHCクラスII陰性マウス肉腫へのトランスフェクションにより、MHCクラスII−陽性であるが、Ii−陰性腫瘍細胞株を生じた。これらの細胞のMHC適合性宿主への注入は、親腫瘍の増殖の遅延を導いた。Iiタンパク質の遺伝子を、MHCクラスII遺伝子と一緒に肉腫細胞株にコトランスフェクションすると、Ii鎖が内因性の腫瘍抗原の提示を遮断したので、MHCクラスII遺伝子の腫瘍治療効果を阻害した。類似の結果がマウス黒色腫でも生じた(非特許文献8)。
【0007】
この治療的取り組みの成功には、樹状細胞の天然の活性が関与すると考えられる。樹状細胞は専門的なスカベンジャーであり、これは外来抗原をペプチドに処理し、そしてそれらをTリンパ球にそれらの細胞表面上でMHC抗原から提示する。樹状細胞は抗原をMHCクラスIおよびクラスII分子を介して提示する能力を有し、それらがTヘルパーおよびTキラー細胞の両方を活性化できるようにする。効果的なTヘルパー細胞応答には強力なTキラー細胞応答の誘導が必要であり、そして樹状細胞により生じた活性化の組み合わせが最高の抗−腫瘍応答を導くと考えられている(非特許文献9;非特許文献10)。マクロファージ系統の樹状細胞は腫瘍細胞を見つけると、腫瘍特異的および腫瘍関連抗原の両方を摂取し、そして処理する。次いで樹状細胞は腫瘍部位を排出するリンパ節に移動し、そして新たなT細胞が成長し始めるリンパ節皮質(node cortex)近傍のリンパ節に在住する。リンパ節皮質では、樹状細胞上の腫瘍決定基を認識する休止Tキラー細胞が活性化し、そして増殖するようになり、そして続いて有能な抗−腫瘍キラーT細胞として循環に放出される。
【0008】
T−ヘルパー細胞との相互作用は、樹状細胞が抗原をMHCクラスI分子を介して提示するように活性化または「ライセンスを与え(licence)」、すなわちTキラー細胞を活性化するが、Tヘルパー細胞とTキラー細胞との同時相互作用は必要ではなく;活性化された樹状細胞は時折、Tヘルパー細胞媒介型活性化の後に、Tキラー細胞を刺激する能力を維持している。MHCクラスIIまたはMHCクラスI決定基のいずれかにより提示されるようになる個々の抗原性ペプチドは、1つの抗原性タンパク質に由来する必要は無く、悪性細胞に由来する2以上の抗原が1つの樹状細胞により処理そして提示され得る。したがって恐らく腫瘍特異的ではない1つの決定基に対するライセンスは、他の恐らく腫瘍特異的な決定基に対するTキラー細胞の活性化にライセンスを与える力を持つ。そのような「マイナーな」または「隠れた(cryptic)」決定基が様々な治療目的に使用されてきた(非特許文献11)。
【0009】
MHCクラスII抗原提示の実験的改変は、これらマイナーな決定基に対する免疫応答を拡大すると考えられる。通常はMHCクラスII分子への装填に利用できない一連のペプチドが、MHCクラスII提示について変動するペプチドの豊かな供給源を提供する。この一連の決定基の活用により、反応性Tヘルパー細胞集団の拡大を導く。そのような拡大された集団は樹状細胞のライセンシングを誘導し、その中には腫瘍特異的および腫瘍関連決定基に向けられるものもある。正常な細胞は潜在的に腫瘍細胞決定基を共有するが、正常細胞に対しては細胞の傷害がわずかしか生じない。これは抗−腫瘍応答の多くのエフェクター応答(キラーT細胞の密集、周辺の活性化サイトカイン、食作用を行うマクロファージ、およびそれらの産物等)が正常細胞には向かわないからである。
【0010】
正常なMHCクラスII抗原提示は、MHCクラスII分子とIiタンパク質との相互作用を阻害することにより改変することができる。これは全Iiタンパク質を減少させることにより(例えば発現を減少させることにより)、またはIiの免疫調節機能を妨害することにより達成することができる。Ii発現の阻害は種々のアンチセンス技法を使用してなされてきた。Iiタンパク質のmRNAのAUG部位と相互作用するアンチセンスオリゴヌクレオチドは、外因性抗原のMHCクラスII提示を低下させることが記載された(非特許文献12)。しかしIiタンパク質の発現およびMHCクラスII分子による内因性抗原の提示に及ぼす効果は調査されなかった。さらに最近では、Humphreys et al.、特許文献1が、MHCクラスII分子を発現する抗原提示細胞に導入すると、Iiタンパク質発現の効果的な抑制を現す3種のアンチセンスオリゴヌクレオチドおよびリバース遺伝子構築物(reverse gene construct)を同定した。このメカニズムにより、Iiが抑制された腫瘍細胞を接種したマウスは、未処置の親腫瘍細胞を接種したマウスよりも有意に長く生存することが示された。この観察はIiタンパク質の抑制が抗原決定基提示の範囲の増大を生じ、腫瘍細胞に対してより効果的な免疫応答を惹起することを示す。
【参考文献】
【0011】
【特許文献1】米国特許第5,726,020号明細書
【非特許文献1】Daibata et al.,Molecular Immunology 31:255−260(1994)
【非特許文献2】Xu et al.,Molecular Immunology 31:723−731(1994)
【非特許文献3】Ostrand−Rosenberg et al.,Journal of Immunol.144:4068−4071(1990)
【非特許文献4】Clements et al.,Journal of Immunol.149:2391−2396(1992)
【非特許文献5】Basker et al.,Cell.Immunol.155:123−133(1994)
【非特許文献6】Baskar et al.,J.Exp.Med.181:619−629(1995)
【非特許文献7】Armstrong et al.,Proc.Natl.Acad.Sci.USA 94:6886−6891(1997)
【非特許文献8】Chen and Ananthaswamy,Journal of Immunolgy.151:244−255(1993)
【非特許文献9】Ridge et al.,Nature 193:474−477(1998)
【非特許文献10】Schoenberger et al.,Nature 193:480−483(1998)
【非特許文献11】Mougdil et al.,J.Immunol.159:2574−2579(1997)
【非特許文献12】Bertolino et al.,Internat.Immunology 3:435−443(1991)
【発明の開示】
【0012】
発明の要約
本発明は抗原提示経路を改変する目的のために、細胞中のIi発現の阻害が関与する組成物および方法を対象とする。1つの観点では、本発明はRNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物に関する。また本発明はそのような発現可能なリバース遺伝子構築物を含む哺乳動物細胞に関する。好適な態様では、RNA分子はコード配列の翻訳開始部位(translation initiation start site)および約435ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である。
【0013】
前に示したように、Ii発現の抑制は抗原提示経路の改変を意図している。より具体的にはIi発現の阻害は、通常はこの内容では提示されない抗原性エピトープのMHCクラスII分子への装填を促進することを意図する。すなわち別の観点では、本発明はMHCクラスII分子−陰性細胞をMHCクラスII分子−陽性細胞に転換することに関する。この転換は例えば、MHCクラスII分子−陰性細胞をタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターでトランスフェクトし、そのトランスフェクションがMHCクラスII分子−陰性細胞中でトランスフェクトされた細胞の表面上にMHCクラスII分子の誘導をもたらすことによる。
【0014】
別の観点では、本発明はIiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上で目的の抗原性エピトープを展示する方法に関する。この方法は:a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そしてb)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、ことを含む。
【0015】
別の観点では、本発明は免疫学的応答のために動物の細胞の型を標的化する方法に関し、細胞の型は識別抗原の発現を特徴とする。この方法では個体に由来する末梢血単核細胞のカルチャーが準備され、このカルチャーは抗原提示細胞を含む。Ii発現のインヒビターをカルチャーの抗原提示細胞に導入し、同様に識別抗原をコードする発現可能な核酸配列を発現に適する条件下でカルチャー中の細胞に導入する。
【0016】
多数の関連する観点を以下の章で詳細に記載する。
発明の詳細な説明
本発明は、1つの観点では個体における病状に特異的な調節、または免疫応答の標的化のための組成物および方法に関する。本明細書で使用する調節(modulation)という用語は、個体において抗原に対する免疫系の増大した感受性もしくは低下した感受性(免疫寛容)を称することを意味する。使用する標的化という用語は、抗原性エピトープに対して増大した感受性を指すことを意図する。
【0017】
本開示のすべての観点に関連して必要な要素は、細胞中でのIi合成の阻害である。発明の背景の章で検討したように、Iiはタンパク質であり、これはMHCクラスII分子と同時に調節されている(co−regulated)。IiはMHCクラスII分子に結合し、これにより内部で合成された抗原(すなわちMHCクラスII分子−発現細胞内で合成された抗原)のMHCクラスII分子への接近が遮断される。MHCクラスII分子/Ii複合体は、小胞体から後ゴルジ区画へ輸送され、そこでIiが段階的開裂プロセスにより放出され、これにより外因性抗原(すなわち抗原提示細胞内で合成されず、そして食作用、オプソニン作用、細胞表面抗体認識、補体レセプター認識およびFcレセプター認識のようなメカニズムにより抗原提示細胞に取り込まれるために選択された抗原)の装填が可能となる。
【0018】
複合体を形成したIiタンパク質の存在により小胞体中のMHCクラスII分子への結合から排除される抗原のクラスは、内的に合成された抗原と呼ぶことができる。そのような抗原は、プロテオソームにより消化され、そして抗原性ペプチド(TAP)の輸送体によりペプチドとして小胞体に輸送された細胞質タンパク質の調査(survey)を含んでなる。そのような内部で合成された抗原は通常、小胞体中でMHCクラスI分子に結合する。そのような抗原断片はIiタンパク質が抗原性ペプチド結合部位を遮断するので、通常、小胞体内でMHCクラスII分子に結合しない。
【0019】
Iiタンパク質の発現を抑制することにより、MHCクラスIに結合するために小胞体に輸送され、そして続いてCD8+Tリンパ球に提示されるこの膨大なペプチドのレパートリーは、続いてCD4+T免疫調節細胞に提示され、そしてそれを活性化するためにMHCクラスII分子に結合することができる。そのようなCD4+T免疫調節細胞は免疫応答の種々の経路と調和してヘルパーまたはサプレッサー機能のいずれかを有することができる。T免疫調節細胞は、物理的接触およびサイトカインの放出を介して細胞傷害性Tリンパ球(Tキラー細胞)、Bリンパ球および樹状細胞のような他の細胞の活性化に貢献している。
【0020】
本明細書で使用する「目的の抗原性エピトープ」という用語は、抗原提示が起こる細胞の中で生産されるタンパク質に由来するペプチドに存在する抗原性エピトープを指す。本明細書で使用するようにこの用語は、既知もしくは未知の抗原性エピトープを包含することを意図する。すなわち「目的の(of interest)」という修飾語は、エピトープが事前に決定されることを意味しない。「目的の」抗原性エピトープは、単に抗原提示が起こる細胞の細胞質内で合成されるタンパク質に含まれるという事実による。
【0021】
治療的介入の好機を与える重要な生物学的結果は、MHCクラスI分子により小胞体中で結合するためにそこに輸送されたペプチドのレパートリーに由来するペプチドのMHCクラスII分子による結合後に続く。しばしばIi抑制の存在下でMHCクラスII分子に結合するエピトープは「隠れた」エピトープであるので、そのようなエピトープは抗原提示の古典的経路によりMHCクラスII分子と結合する以外は免疫系に提示されない。隠れたエピトープは、試験抗原のアミノ酸配列が重複する合成ペプチドのライブラリーを分析することにより実験的に明らかにすることができる。試験抗原で免疫感作したマウスの一種の動物がライブラリーに由来する1組のペプチドに応答することが分かった(「優性エピトープ(dominant epitope)」)。しかしそうではない同一マウスをライブラリーの1つのペプチドで免疫感作した時、これまでに同定されなかったサブセット(免疫感作するペプチド中の優性エピトープに加えて)が、免疫学的エピトープを含むことが分かる。これらの以前に同定されなかったエピトープは、1組の隠れたエピトープを含んでなる。
【0022】
本発明の方法は、優性および隠れたエピトープの両方に対する免疫を促進するが、幾つかの臨床的状況においては隠れたエピトープに対する免疫応答の強化が治療効果に特別な役割を果たす。例えばガンに関連する抗原性エピトープに対する治療的応答を追加免疫感作する場合、サプレッサーT細胞応答がこれまでに生じなかった隠れたエピトープに対するTヘルパー細胞応答は、効果的な樹状細胞ライセンシングをさらに提供するようであり、これは次いで強固な細胞傷害性Tリンパ球抗−腫瘍応答を生成する。ガンに関連する抗原の優性エピトープに対するサプレッシングT細胞応答の発生は、腫瘍の微小ガン組織転移の成長に役割を果たしていることが示された。したがって本発明の重要な用途は、推定される隠れたガン関連決定基に対するTヘルパー細胞応答の促進である。
【0023】
自己免疫応答の別の観点では、自己免疫疾患の場合は自己免疫疾患に関連する抗原の優性エピトープに対する応答が、そのような疾患の病原を促進する。ここで選択的、例えば新しい隠れたエピトープに対する免疫応答の経路の抑制を活用することは、治療的に有用となり得る。これらの概念は同様に、アレルギー、移植片拒絶ならびに感染性および心血管疾患のようなさらなる医学的状態の治療に応用することができる。そのような状態の患者の診断、処置的モニタリングおよび治療に応用する化合物の開発において必須かつ有用な第1段階は、Iiタンパク質抑制の条件下で抗原提示細胞により提示されるようになるMHCクラスIIエピトープの同定である。そのようなエピトープには優性および隠れたエピトープの両方を含む。隠れたエピトープは例えばガンまたは感染性疾患の場合、免疫抑制応答がそれらに向かって発生することはなく、しかも自己免疫疾患または移植片拒絶の場合には応答の活性化が発生しなければ特に有用となるだろう。したがって医師は上記の病状の場合に状況次第で新たにTh1またはTh2を誘導し、応答を活性化または抑制するだろう。そのようなエピトープを生成し、単離し、そして特性決定する方法は、本発明の主題である。
【0024】
別の観点では本発明は、Iiタンパク質の不存在下で小胞体中のMHCクラスII分子に結合する抗原性エピトープを含む個体のペプチドの提示、単離および同定を提供する。そのようなペプチドを合成し、そして個別に、またはそれらが生じた疾患に由来する抗原に対する免疫応答を強化または抑制するためのワクチン応用と組み合わせて使用することができる。そのようなエピトープペプチドを単離し、そして特性決定するための方法は、引用により本明細書に編入する米国特許第5,827,526号、同第5,874,531号および同5,880,103号明細書に提示された。
【0025】
「内部で合成される(endogenously synthesized)」クラス(これは通常、MHCクラスII分子提示から排除される)に入る種々の抗原は、特に特定の病状を伴う。例えば腫瘍細胞または他の悪性細胞を考えることができる。そのような細胞はガンに特異的な、そしてガンに関連するタンパク質を合成し、これらは治療に有用なMHCクラスIIエピトープを含む。しかしこれらのタンパク質は抗原提示細胞内で合成されるので、そのようなタンパク質の抗原性エピトープは同じ細胞のMHCクラスII分子との結合での提示からは排除される。中で抗原性タンパク質が合成される細胞による抗原性エピトープの提示に関する制限は、ウイルスに感染した細胞の場合にもある。ウイルスに特異的な抗原はウイルスに感染した細胞のMHCクラスII分子との結合での提示から排除されるが、そのような抗原は同じ細胞のMHCクラスI分子との結合で提示され得る。他の外因性病原体(例えばバクテリアまたは寄生体)が細胞を占有し、そして細胞機構を利用して病状に特異的なタンパク質を合成しても、成り行きは同じである。病原に特異的な抗原をMHCクラスII分子との結合で提示することによる事象の正常な過程を改変する能力は、新規なMHCクラスII抗原性エピトープにより開始される応答の強化をもたらす。
【0026】
多数の治療的モダリティが本発明の範囲に入る。特許請求できる組成物は、これら多くの治療的モダリティに関連する。治療的取り組みには、インビボおよびエクスビボの態様を含む。Ii阻害を標的とする細胞はMHCクラスII分子−陽性細胞(例えば樹状細胞、マクロファージまたはBリンパ球のような自然に存在する抗原提示細胞)、あるいはMHCクラスII分子を発現するように誘導されたMHCクラスII−陰性細胞(例えば腫瘍細胞)のいずれかであることができる。本明細書で使用する「MHCクラスII分子−陰性」という表現は、具体的には細胞表面上にMHCクラスII分子を発現しない細胞を含むだけでなく、自然に存在する抗原提示細胞(例えば樹状細胞)のような陽性対照細胞の表面上のMHCクラスII分子の数と比べた時、細胞表面上に比較的少数のMHCクラスII分子を含む細胞も含む。本内容において、用語「比較的少数」とはMHCクラスII分子−陽性対照細胞(例えば自然に存在する抗原提示細胞)上に見いだされる数のわずか約25%以下の数のMHCクラスII分子をそれらの細胞表面上に含むと見積もられる細胞を含むことを意味する。MHCクラスII分子の存在度(abundance)の予測は、例えば当該技術分野で周知な免疫蛍光技法を使用して行うことができる。
【0027】
出願人は以前に、免疫応答を調節する目的のIi阻害を開示する特許出願を出願し、そして要求した。これらの出願は具体的には細胞に導入され、そしてIi mRNAに結合することによりIi合成を直接阻害する阻害コポリマー、ならびに核酸構築物として細胞に導入され、続いてRNA分子に転写され、これが特異的ハイブリダイゼーション後にIi発現を阻害するリバース遺伝子構築物を開示する。これらの以前に出願された特許出願には、米国特許出願第08/661,627号および同第09/205,995号明細書を含み、それらは引用により本明細書に編入する。米国特許出願第08/661,627号明細書は、米国特許第5,726,020号として発効され、そして米国特許出願第09/205,995号の発効手数料が最近、米国特許庁に支払われた。
【0028】
上に簡単に述べたように、米国特許出願09/205,995号明細書は約10〜約50ヌクレオチド塩基を含む化学的に合成されたコポリマーに関する徹底的な開示を含む。
【0029】
これらのコポリマーはRNA分子の標的部分に相補的であるか、あるいはアンチセンス配列として知られているヌクレオチド塩基配列を含む。そのようなコポリマーの1例は、アンチセンスオリゴヌクレオチドである。コポリマーはRNAからのタンパク質翻訳を2つのメカニズムにより阻害する。1つの方法はリボソーム、スプライソソームまたはRNAの成熟もしくは翻訳に必須な他の因子と相互作用しなければならないRNAの部分への接近を遮断する。第2の方法は、DNAにハイブリダイズするRNAの配列を開裂する酵素であるリボヌクレアーゼHの強化が関与する。すなわちDNAもしくはDNA様のコポリマーの対応するRNA中のセグメントへの結合は、コポリマー結合部位でRNAの開裂を導く。
【0030】
コポリマーはワトソン−クリック塩基対合により標的RNAにハイブリダイズする。コポリマーの配列は、標的RNAの相補的配列により定められる。コポリマーは通常、標的RNAの少なくとも6個の相補的ヌクレオチドに広がるヌクレオチド配列長を用いて化学的に合成されるが、12〜25ヌクレオチド長が最も普通である。統計的に、約15ヌクレオチドの配列は細胞内のすべてのRNAの群中で独特であり、特定のRNAを高度な特異性で標的化できるようにする。RNAへの結合はまた、20塩基対を包含するコポリマーについて約10−7M付近のKd値で大変安定である。
【0031】
幾つかの場合では、カルチャー中の細胞は有用な効果を達成するために十分な量で自然にコポリマーを取り込む。そのような取り込みは生物化学的エネルギーおよび特定の細胞表面タンパク質の参加を要する能動的なプロセスになるように思われる。取り込みは飲作用によっても起こり得る。この経路は細胞をコポリマーを含有する高張性培地にインキュベーションし、続いて細胞をわずかに低張性培地に再懸濁して細胞内の飲作用性小胞の破裂を誘導することにより強化される。別の場合では、取り込みは脂質、リポソーム、またはポリアルキルオキシコポリマーの使用により、電気穿孔により、または細胞膜を透過するストレプトリシンO処理により援助することができる。しばしばインビボで細胞は培養した細胞よりも容易にコポリマーを取り込む。コポリマーの電気穿孔による細胞の取り込みに関する最適な条件は、米国特許出願第09/205,995号明細書の実施例2に提供されている。
【0032】
標的RNAの潜在的部位は、タンパク質の機能的複合体の結合について開いた部位、およびそうではなくコポリマーの結合について開いた部位である。そのような部位はDNAにハイブリダイズするRNAを開裂する酵素であるリボヌクレアーゼH(RNaseH)を使用して同定することができる。DNAオリゴヌクレオチドを単独で、または混合物中で5’−放射性リン標識RNAにリボヌクレアーゼHの存在下で結合することにより、オリゴヌクレオチドおよび他のコポリマーがハイブリダイズするRNA上の部位は、RNAのゲル電気泳動およびオートラジオグラフィー後に同定される。本発明でRNaseH開裂について最も開いていることが分かったIiのRNA部位は、AUG開始コドンの領域およびプレ−mRNAの第1スプライス部位の領域であった。
【0033】
本発明に関して「オリゴヌクレオチド」という用語は、自然に存在する塩基およびホスホジエステル結合で連結したペントフラノシル糖で形成されたヌクレオチド単位を含んでなるポリヌクレオチドを称する。用語「コポリマー」にはオリゴヌクレオチド、およびまた自然には存在しないか、もしくは修飾されたオリゴヌクレオチドのサブユニットから形成された構造的に関連する分子を含む。これらの修飾はヌクレオチドの塩基部分、ヌクレオチドの糖部分、またはヌクレオチド間連結基のいずれかで生じる。さらなる連結基はしばしば、天然のオリゴヌクレオチドの糖およびホスフェート骨格が置換されて、以下でより詳細に検討するコポリマーを生成する。
【0034】
生成されるそのようなオリゴヌクレオチド修飾および特性は、当業者には容易に利用可能である。例示的修飾は、引用により本明細書に編入する米国特許第4,469,863号明細書(1984);米国特許第5,216,141号明細書(1993);米国特許第5,264,564号明細書(1993);米国特許第5,514,786号明細書(1996);米国特許第5,587,300号明細書(1996);米国特許第5,587,469号明細書(1996);米国特許第5,602,240号明細書(1997);米国特許第5,610,289号明細書(1997);米国特許第5,614,617号明細書(1997);米国特許第5,623,065号明細書(1997);米国特許第5,623,070号明細書(1997);米国特許第5,700,922号明細書(1997);および米国特許第5,726,297号明細書(1998)に与えられている。
【0035】
オリゴヌクレオチドが相補的RNAにハイブリダイズする能力は、化学的修飾に対して大変抵抗がある。したがって多くの異なる機能的コポリマーが可能である。特に糖ホスフェート骨格は、ワトソン−クリック塩基対を形成する能力を失うことなく徹底的に改変することができる。定義によりヌクレオチドは糖、窒素複素環およびホスフェート部分を含んでなる。糖もしくはホスフェート基のいずれかまたは両方を欠くオリゴヌクレオチドの幾つかの合成類似体は、それでもアンチセンスオリゴヌクレオチドと同様にワトソン−クリック塩基対によりハイブリダイズすることができ、そしてこの目的に使用することができる。ヌクレオチド塩基を含有するこれらのコポリマーは、RNAへのハイブリダイズにおいてオリゴヌクレオチドの機能的均等物である。以下の要約はアンチセンスの応用のためにオリゴヌクレオチドの特性を変え、そして改善するそれらに対する幾つかの修飾である。
【0036】
多数の具体的修飾が開示され、それらには非−架橋酸素原子の置換、架橋酸素原子の置換、ヌクレオシド間ホスフェート基の置換、糖環の回りの立体化学に対する変化、リボフラノシル環構造の修飾、ヌクレオチド連結の修飾、およびペプチド骨格による糖ホスフェート骨格の置換によりペプチジル核酸(pna)の生成を含む。
【0037】
引用した特許出願の手続処理は以下の請求項に特許査定をもたらした。以下は代表的な独立請求項である。
1.哺乳動物のB7分子をコードする外因性の構築物を含まず、そしてIiタンパク質発現の特異的レギュレーターもしくは免疫調節機能を含むMHCクラスII−陽性抗原提示細胞であって、オリゴヌクレオチドCTCGGTACCTACTGG(配列番号11)が特別に除外され、特異的レギュレーターが10〜50ヌクレオチド塩基のコポリマーから本質的になり、コポリマーは生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、ここで特異的レギュレーターはIi発現を阻害する能力を特徴とする上記のMHCクラスII−陽性抗原提示細胞。
【0038】
本開示の内容では、出願人は以前に記載した種類のコポリマーが新規な内容で使用される態様を開示し、そして特許請求する。あるいはより徹底的な支持実験が本明細書で開示される場合、本出願で請求する主題は先に出願した特許出願に開示した主題に実質的に類似するかもしれない。
【0039】
関連する背景情報を提供し、そしてさらなる請求項の限定を支持する目的で、上で説明した請求項において少なくとも2つの具体的な限定を、従来技術を開示するために含んだ点に注目されたい。例えばオリゴヌクレオチドCTCGGTACCTACTGG(配列番号11)の特別な排除は、Bertolino et al.,Int.Immunol.3(5)435−443(1991)の開示に照らして包含した。哺乳動物のB7分子をコードする外因性構築物を対象とした限定は、Ostrand−Rosenberg(米国特許第5,858,776号明細書)の開示に照らして導入した。
【0040】
これまでに公開されていない本開示の重要な要素は、ヒトの細胞中でIiの発現を阻害するためのリバース遺伝子構築物の使用である。ヒトIi配列は以前に報告されたが(Strubin et al.,EMBO J.3:869−872(1984))、この配列の少なくとも一部を含むリバース遺伝子構築物の使用は報告されたことがない。さらに例えばマウスIi配列とヒトIi配列との間の有意な保存が報告されたが、非ヒトリバース遺伝子構築物はヒト細胞中でIiの翻訳を阻害するために使用するには効果的ではなかった。
【0041】
このように1つの観点では、本発明はRNA分子がmRNA分子にハイブリダイズする能力を有し、これによりヒト細胞中でmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物に関する。本発明のこの観点は以下に続く実施例の章で具体的に証明する。より具体的にはcDNA挿入物を含む発現構築物が、ヒトのリンパ腫細胞株でIi発現を阻害するのに効果的であることが証明された。このアッセイで効果的であった構築物は、Ii mRNAの5’非翻訳領域の一部に相補的なcDNA挿入物を含み、そして翻訳開始コドンを含んだ。効果的な構築物は、約435ヌクレオチド長までの阻害RNAをコードした。
【0042】
ヒトIi mRNAの部分に完全に相補的なRNAをコードするリバース遺伝子構築物の使用に加えて、当業者は野生型ヒト配列からある程度の逸脱が許容されると認識するだろう。本発明の範囲は、日常的な実験により経験的に決定され得るそのようなバリアントを包含することを意図する(すなわちそれらはヒト細胞中でIi発現を阻害する能力により特徴づけられる)。特に有用で、しかもヒト細胞中でIi発現の阻害に効果的であることが証明された野生型からの変異の例は、ヒトIi mRNAに相補的な長い半減期のアンチセンスRNA(野生型アンチセンスRNAに比べて)の作成に関する。長い半減期の種では、アンチセンスRNAのリーデイングフレイムは、開始コドンAUGに同じリーディングフレイム内で短く/直ちに終止コドンが続くことを回避するように設計されている。この状況を回避するために、例えばリーディングフレイム1中の終止コドンの直前にあるAUGに関して、新たなAUGを設計し、そしてリーディングフレイム1のAUGの前、リーディングフレイム2またはリーディングフレイム3のどちらかの中に(ただし終止コドンはそのような修飾後のリーディングフレイムには無い)導入する。
【0043】
阻害コポリマーおよびIiリバース遺伝子構築物の使用に加えて、当業者はIi発現の他のインヒビターが容易に設計され、そして構築されると認識するだろう。例えば二本鎖の小さい干渉性のRNA(siRNA)およびこれらの分子をコードする遺伝子をRNA干渉に使用することができ、この方法により二本鎖RNA(dsRNA)はその相補的配列を持つ遺伝子の発現を特異的に抑制する(Moss,Curr,Biol.11(19):R772−5(2001);Elbashir,Genes Dev.15(2):188−200(2001))。二本鎖RNAは相補的mRNAに結合し、そして縮重を導く21〜23塩基対の断片に処理される(Bernstein,Nature 409(6818):363−6(2001)および国際公開第0175164号パンフレット)。siRNAは配列特異的な翻訳後遺伝子サイレンシングを誘導する。そのような分子は細胞に導入されて、国際公開第0175164号パンフレットに記載されているような治療用もしくは予防用の目的で遺伝子発現を抑制することができる。
【0044】
広い種類の送達系を、本発明のリバース遺伝子構築物の標的細胞へのインビトロおよびインビボ送達の使用に利用することができる。これらの送達系はIiリバース遺伝子構築物に応用できるだけでなく、本発明と関連して検討する発現可能な核酸配列にも応用することができる。そのような送達系には、例えばウイルスおよび非ウイルス系を含む。適当なウイルス系の例には、例えばアデノウイルスベクター、アデノ随伴ウイルス、レトロウイルスベクター、ワクシニア、単純ヘルペスウイルス、HIV、マウスの微小ウイルス(minute virus)、B型肝炎ウイルスおよびインフルエンザウイルスを含む。非ウイルス送達系も使用でき、例えば非複合化DNA、DNA−リポソーム複合体、DNA−タンパク質複合体およびDNA−コート金粒子、サルモネラのようなバクテリアベクター、およびVP22輸送タンパク質、Co−X−遺伝子およびレプリコンベクターが関与するような他の技術を使用する。
【0045】
動物細胞中で目的の核酸配列を発現させるための1つの選択は、アデノウイルス系である。以下の実施例の章では、アデノウイルス系の使用を具体的に開示する。アデノウイルスは二本鎖DNAゲノムを保有し、そして宿主細胞分裂とは独立して複製する。アデノウイルスベクターは、発現可能な構築物を細胞に導入するための別の選択的方法と比べて、様々な利点を提供する。例えばアデノウイルスベクターは広範囲のヒト組織に形質導入させることができ、そして高レベルの遺伝子発現を分裂および非分裂細胞で得ることができる。アデノウイルスベクターは免疫系のクリアランスにより比較的短期間の導入遺伝子発現および標的細胞分裂中に希釈損失を特徴とする。静脈内、胆嚢内、腹腔内、嚢内、頭蓋内およびクモ膜下腔内注射、および標的器官または組織の直接注射を含む幾つかの投与経路を使用することができる。このように当該技術分野では解剖学的境界に基づく標的化を達成することができると認識されている。
【0046】
アデノウイルスゲノムは約15種のタンパク質をコードし、そして感染には細胞表面レセプターに結合する繊維タンパク質が関与する。このレセプター相互作用はウイルスのインターナリゼーションをもたらす。ウイルスDNAは感染した細胞の核に入り、そして転写が細胞分裂の不存在下で開始される。発現および複製はE1AおよびE1B遺伝子の制御下にある(Horwitz,M.S.,In Virology,2.sup.nded,1990,pp1723−1740を参照にされたい)。E1遺伝子の除去によりウイルスを複製−不能(replication−incompetent)とする。
【0047】
アデノウイルス血清型2および5は、ベクターの構築に広範囲に使用された。Bett et al.(Proc.Nat.Acad.Sci.U.S.A.91:8802−8806(1994))は、E1およびE3アデノウイルス遺伝子の欠失があるアデノウイルス5型ベクター系を使用した。293ヒト胚性腎細胞株をE1タンパク質を発現するように操作し、そしてこのようにE1−欠失ウイルスゲノムをトランス相補することができる。ウイルスは293細胞培地から単離し、そして限界希釈プラークアッセイにより精製することができる(Graham and Prevek、分子生物学の方法:遺伝子転移および発現プロトコールで(In Methods in Molecular Biology:Gene Transfer and Expression Protocols)、ヒューマナ(Humana)出版 1991、第109〜128頁)。組換えウイルスを293細胞株のカルチャー中で成長させ、そして感染した細胞を溶解することにより単離し、そして塩化セシウム密度遠心により精製することができる。組換えアデノウイルスの製造について293細胞に付随する問題は、E1遺伝子のさらなるフランキング領域により、それらがウイルス粒子の生産中に複製可能(replication−competent)なアデノウイルス(RCA)を生じるかもしれない点である。この材料は野生型アデノウイルスだけであり、そして複製可能な組換えウイルスではないので、所望するアデノウイルス材料の最終的な収量に有意な影響を有し、そして製造経費の増大、製造作業の品質管理の問題および臨床的使用のためのバッチの容認を導く。あるいは293細胞よりも明確なE1遺伝子組込みを有するPER.C6のような細胞株は(すなわちフランキングウイルス配列を含まない)が開発され、これはRCAを生じる組換え反応を可能とせず、すなわち上記のウイルス生産の問題を克服する可能性を有する。
【0048】
アデノ随伴ウイルス(AAV)(Kotin,R.M.Hum.Gene Ther.5:793−801(1994))は、大変広範な宿主の非分裂細胞のゲノムに組込むことができる1本鎖DNAの非自律複製パルボウイルスである。AAVはヒトの疾患に関連することが示されておらず、そして免疫応答を誘導しない。
【0049】
AAVは2つの明確な生活環の相を有する。野生型ウイルスは宿主細胞に感染し、組込まれ、そして潜伏する。アデノウイルスの存在下で初期アデノウイルス遺伝子の発現に依存するウイルスの溶菌相が誘導され、そして活発なウイルス複製を導く。AAVゲノムは、逆方向末端反復(ITR)配列により挟まれた2つのオープンリーディングフレイム(repおよびcapと呼ぶ)からなる。rep領域はAAV複製、ウイルスDNA転写および宿主ゲノムの組込みに使用されるエンドヌクレアーゼ機能を媒介する4種のタンパク質をコードする。rep遺伝子はウイルス複製に必要なAAV配列だけである。cap配列はウイルスキャプシドを形成する構造的タンパク質をコードする。ITRsは複製のウイルス起点を含み、そして莢膜形成(encapsidation)シグナルを提供し、そしてウイルスDNAの組込みに参加する。遺伝子治療用に開発された組越えの複製欠損ウイルスは、repおよびcap配列を欠いている。複製−欠損AAVは、AAV複製に必要な別々の要素を許容293細胞株にコトランスフェクションすることにより生産することができる。米国特許第4,797,368号明細書は関連する開示を含み、そしてそのような開示は引用により本明細書に編入する。
【0050】
レトロウイルスベクターは分割している細胞を感染させるために有用であり、そして宿主細胞膜およびウイルスタンパク質に由来するエンベロップにパッケージングされるRNAゲノムからなる。レトロウイルス遺伝子発現には逆転写段階が関与し、ここでその陽性−鎖RNAゲノムが鋳型として使用されて二本鎖DNAの合成を支配し、これは次いで宿主細胞DNAに組み込まれる。組み込まれたプロウイルスは遺伝子発現のために宿主細胞の機構を使用することができる。
【0051】
マウス白血病ウイルスは、一般に使用されているレトロウイルス種である(Miller et al.,Methods Enzymol.217:581−599(1993))。レトロウイルスベクターは典型的にはgag、polおよびenv遺伝子の欠失により構築されている。これらの配列の欠失により目的の核酸配列の挿入能力を提供し、そしてウイルスの複製機能を排除する。抗生物質耐性をコードする遺伝子はしばしば選択の手段として含まれる。プロモーターおよびエンハンサー機能も、例えばインビボ投与後の組織特異的発現を提供するために含むことができる。長い末端反復配列に含まれるプロモーターおよびエンハンサー機能も使用することができる。
【0052】
そのようなウイルス、および目的の外因性核酸配列を運ぶそのようなウイルスの修飾は、ウイルスパッケージング細胞株でのみ生成することができる。パッケージング細胞株は欠失ウイルス遺伝子(gag、polおよびenv)を、組換えを防止するために異なる染色体上にそれらが存在するように細胞に安定に挿入することにより構築することができる。パッケージング細胞株を使用して、組換えプロウイルスDNAを挿入することにより目的の核酸配列を含む複製欠損レトロウイルスを生成する生産細胞株を構築する。莢膜形成配列を含むgag遺伝子の小さい部分を挟む長い末端反復配列および目的の遺伝子を含むプラスミドDNAは、DNAの転移および取り込みのための標準的技法(電気穿孔、カルシウム沈殿等)を使用してパッケージング細胞株にトランスフェクトされる。この取り組みの変形は、複製可能なウイルス生産の見込みを下げるために採用された(Jolly,D.,Cancer Gene Therapy 1:51−64(1994))。このウイルスの宿主細胞範囲は、エンベロープ遺伝子(env)により決定され、そして異なる細胞特異性をもつenv遺伝子の置換を使用することができる。エンベロープタンパク質へ適切なリガンドと包含させることも、標的化に使用することができる。
【0053】
組換えレトロウイルスベクターの投与は、適当な技術により達成することができる。そのような技術には例えば、患者の細胞のエクスビボ形質導入、組織へのウイルスの直接注射を含み、そしてレトロウイルス生産細胞の投与による。エクスビボの取り組みには患者の細胞の単離および組織培養での維持が必要である。この内容において、細胞を標的とするために高比率のウイルス粒子を達成し、すなわち形質導入効率を向上させる(例えば引用により本明細書に編入する米国特許第5,399,346号明細書を参照にされたい)。米国特許第4,650,764号明細書はレトロウイルス発現系の使用に関する開示を含み、そしてこの引用した特許は参照により本明細書に編入する。
【0054】
場合により、インビボでウイルスの直接的導入が必要であり、または好ましい。レトロウイルスは脳腫瘍を処置するために使用され、ここではレトロウイルスが分裂している細胞(腫瘍細胞)のみに感染する能力が特に有利となる。
【0055】
レトロウイルス生産細胞株の患者の脳腫瘍への直接投与も提案された(例えば、Oldfield et al.,Hum.Gene Ther.4:39−69(1993))。そのような生産細胞は数日間、脳腫瘍内で生存し、そして周囲の脳腫瘍を形質導入することができるレトロウイルスを分泌する。
【0056】
発現用のポックスウイルスに基づく系が記載された(Moss and Flexner,Annu.Rev.Immunol.5:305−324(1987);Moss,B.In Virology,1990,pp.2079−2111)。例えばワクシニアは、感染した細胞の細胞質中で複製する大きな、エンベロープを持つDNAウイルスである。多くの異なる組織に由来する非分裂および分裂細胞が感染し、そして非組込みゲノムからの遺伝子発現が観察される。組換えウイルスは導入遺伝子をワクシニアに由来するプラスミドに挿入し、そしてこのDNAをワクシニアに感染した細胞(ここでDNAの相同的組換えがウイルス生産を導く)にトランスフェクトすることにより生産することができる。重要な欠点は、これが150〜200のウイルスにコードされるタンパク質に対して宿主免疫応答を誘導し、反復投与が問題となる点である。
【0057】
単純ヘルペスウイルスは、感染した細胞の核内で複製する大きな、二本鎖DNAウイルスである。このウイルスは外因性の核酸配列と関連した使用に適合可能である(Kennedy and Steiner,Q.J.Med.86:697−702(1993)を参照にされたい)。利点には広い宿主細胞範囲、分裂および非分裂細胞の感染を含み、そして外来DNAの大きな配列を相同的組換えによりウイルスゲノムに挿入することができる。欠点は複製可能なウイルスを含まないウイルス調製物とすることが難しいこと、および強い免疫応答である。ウイルスのチミジンキナーゼ遺伝子の欠失は、低レベルのチミジンキナーゼを持つ細胞中でウイルスを複製欠損とする。活発な細胞分裂を受けている細胞(例えば腫瘍細胞)は、複製を可能にする十分なチミジンキナーゼ活性を保有する。
【0058】
HIV、マウスの微小ウイルス、B型肝炎ウイルスおよびインフルエンザウイルスを含む他の種々のウイルスが、遺伝子転移用のベクターとして開示された(Jolly,D.,Cancer Gene Therapy 1:51−64(1994)を参照にされたい)。
【0059】
非ウイルス性のDNA送達法も応用できる。これらのDNA送達法は非複合化プラスミドDNA、DNA−脂質複合体、DNA−リポソーム複合体、DNA−タンパク質複合体、DNA−コート金粒子およびDNA−コートポリラクチドコグリコライド粒子に関する。精製した核酸は組織に直接注入することができ、そして例えば筋肉組織、特に筋肉の再生に効果的な一過性の遺伝子発現をもたらす(Wolff et al.,Science 247:1465−1468(1990))。Davis et al.,(Hum.Gene Ther.4:733−740(1993))は、成熟した筋肉(骨格筋が一般に好適である)へのDNAの直接注入について公開した。
【0060】
金粒子上のプラスミドDNAは遺伝子銃を使用して細胞(例えば表皮または黒色腫)に「発射(fired)」され得る。DNAは金粒子上に共沈殿し、そして推進剤として電気的スパークまたは加圧ガスを使用して発射される(Fynan et al.,Proc.Natl.Acad.Sci.U.S.A.90:11478−11482(1993))。電気穿孔も多くの針のアレイおよびパルスをかけた、回転する電場を採用した電気穿孔プローブを使用して、DNAを充実性腫瘍に転移できるようにするために使用された(Nishi et al.,Cancer Res.56:1050−1055(1996))。有意な細胞トランスフェクションの強化および腫瘍内注入法に優る良好な分布特性の、皮下腫瘍への高効率遺伝子転移が特許請求された。
【0061】
脂質が媒介するトランスフェクションはインビトロおよびインビボのトランスフェクションの両方に好適である(Horton et al.,J.Immunology 162:6378(1999))。脂質−DNA複合体は、DMRIE−C試薬のような市販されている脂質を使用してDNAおよび脂質を注入の1〜5分前に混合することにより形成された。
【0062】
リポソームは細胞に入り易くするために、親水性分子を疎水性分子で取り囲むことにより作用する。リポソームは脂質により作られた単層または多層の球である。脂質組成および製造工程は、リポソーム構造に影響を及ぼす。他の分子は脂質膜に包含することができる。リポソームはアニオン性またはカチオン性でよい。Nicolau et al.(Proc.Natl.Acad.Sci.U.S.A.80:1068−1072(1983))は、ラットに注入されたアニオン性リポソームからのインスリン発現に関する研究を公開した。アニオン性リポソームは他を標的としない限り、主に肝臓の網内細胞を主に標的とする。分子はリポソームの表面に包含されてそれらの挙動、例えば細胞−選択的送達を改変させることができる(Wu and Wu,J.Biol.Chem.262:4429−4432(1987))。
【0063】
Felgner et al.(Proc.Natl.Acad.Sci.U.S.A.84:7413−7417(1987))はカチオン性リポソームに関する研究を公開し、静電的相互作用によるそれらの核酸の結合を示し、そして細胞へ入ることを示した。カチオン性リポソームの静脈内注入は、器官への導入性の血液供給の注入でほとんどの器官に導入遺伝子の発現を導く。カチオン性リポソームは肺上皮を標的とするためにエーロゾルにより投与することができる(Brigham et al., Am.J.Med.Sci.298:278−281(1989))。カチオン性リポソームの導入遺伝子送達を用いたインビボ実験が公開された(例えば、Nabel et al.,Rev.Hum.Gene.Ther.5:79−92(1994);Hyde et al.,Nature 362:250−255(1993)および;Conary et al.,J.Clin.Invest.93:1834−1840(1994))。
【0064】
ミクロ粒子は食作用細胞にDNAを送達するための系として実験され、そのような取り組みはパンゲア ファーマシューティカル(Pangaea Pharmaceuticals)により報告された。そのようなDNAミクロカプセル化送達系は、微小球を摂取するマクロファージのような食細胞のさらに効率的な形質導入を行うために使用された。微小球は、潜在的に免疫原性のペプチドをコードするプラスミドDNAをカプセル化し、このDNAは発現した時、そのようなペプチドおよび同じエピトープを含むタンパク質配列に対する免疫応答を刺激することができる細胞表面上のMHC分子を介してペプチド展示を導く。この取り組みは現在、抗腫瘍および病原ワクチン開発における有力な役割を目的としているが、他にも可能な遺伝子治療の応用を有することができる。
【0065】
ウイルス−様粒子(VLPs)内で相同的自己集合を行うことができる天然のウイルスコートタンパク質も、送達用にDNAをパッケージングするために使用された。ヒトのポリオーマウイルスの主要な構造コートタンパク質(VP1)は組換えタンパク質として発現することができ、そしてVLPへの自己集合中にプラスミドDNAをパッケージングすることができる。生成した粒子は続いて種々の細胞系を形質導入するために使用することができる。
【0066】
DNAベクターにおける改善もなされ、そして多くの非ウイルス送達系に応用できると思われる。これらにはスーパーコイル化されたミニサークル(これはバクテリアの複製起点も抗生物質耐性遺伝子も持たず、すなわち高レベルの生物学的封じ込めを示す時、潜在的により安全である)、エピソーム発現ベクター(複製エピソーム発現系、これはプラスミドが核内で、しかし染色体外で増幅し、すなわちゲノムへの組込み反応(event)を回避する)、およびT7系(厳密な細胞質発現ベクターであり、ここでベクター自体はファージT7RNAポリメラーゼを発現し、そして第1プロモーターにより生成されたポリメラーゼを使用して治療用遺伝子が第2T7プロモーターにより駆動される)を含む。DNAベクター技術に対する他のより一般的な改善には、高レベルの発現を行うためのシス作用エレメント、細胞のサイクル毎に1回の複製を供給するためのアルフォイド反復DNAに由来する配列および核標的配列の使用を含む。
【0067】
上で検討したように、本発明は種々の動物細胞型においてインビボもしくはエクスビボのいずれかでIiの阻害に関する。この考察に関連する動物細胞型間の広い分類を、MHCクラスII分子発現の状態に基づき作成することができる。この広い分類をここで簡単に紹介し、そして具体的な治療的取り組みの内容の中で再検討する。
【0068】
自然に存在する抗原提示細胞(時折、専門的な抗原提示細胞と呼ぶ)は、獲得した免疫応答に参加する。樹状細胞、マクロファージ、Bリンパ球および特定の他の単核細胞を含むこれらの細胞は、MHCクラスII分子−陽性である。加えてTリンパ球のような幾つかの細胞は、休止状態ではMHCクラスII分子を発現しないが、適切な活性化でMHCクラスII分子を発現するように誘導することができる。抗原性ペプチドのMHCクラスII−拘束提示の機能に対してインビボまたはエクスビボで誘導することができるそのような細胞は、自然に存在する抗原提示細胞の範疇に含まれる。細胞は多形核細胞について記載されたように(Rasdak,Immunol.101(4):521−30(2000))、自己血清、IFN−GM−CSFとの共存培養を介してMHCクラスII分子を発現するように誘導することができる。T細胞もMHCクラスII分子を発現するように誘導し、そしてマイトジェンおよび異種APCsと培養した時に抗原提示細胞の機能性を予測することができる(Patel,J.Immunol.163(10):5201−10(1999))。
【0069】
以下の章でさらに詳細に検討するように、そのような細胞に目的の抗原性エピトープをコードする発現可能な核酸配列を導入することが可能である。このエピトープ発現をIi発現と組み合わせた時、目的の抗原性エピトープはMHCクラスII分子と結合して抗原提示細胞の表面上に展示される。
【0070】
自然に存在する抗原提示細胞は身体全体、そして末梢リンパ組織を通って循環する。末梢リンパ組織は身体の2つの流体系、血液およびリンパを取り巻いて組織されている。これら2つの流体系は接触している。リンパは血液から組織の中および回りの空間に運ばれた流体により形成される。これらの細胞外空間からリンパは薄い壁のリンパ管に流れ、ここでリンパは大きな中央集合管にゆっくりと移動する。最終的にリンパは静脈に戻され、ここでリンパは血液に再度入る。血液では、リンパ球が有核細胞の20〜30パーセントを構成し;リンパではそれが99パーセントを構成する。これらの流体系内を循環している抗原提示細胞はリンパ節および脾臓の小胞中心(follicle center)を通る。身体のリンパ節中の高濃度Tリンパ球およびBリンパ球および脾臓の小胞中心は、細胞の相互作用およびクローン増殖を促す。
【0071】
典型的にはMHCクラスII分子をほとんど発現しないか、または発現しない目的の他の細胞には、ほとんどの悪性およびウイルスが感染した細胞を含む。特に通常はMHCクラスII分子−陰性であると考えられている数種の腫瘍は、幾つか、もしくはすべての細胞上に低レベルのMHCクラスII分子を発現することが報告されたことに注目されたい。これらには例えば、胸部、肺または結腸癌腫を含む。これらの細胞は、病原に特異的な抗原を発現することができるが、MHCクラスII分子の不存在もしくは比較的低い存在度を与え、そのような細胞によるそのような抗原に由来するペプチドの有意な程度のMHCクラスII提示は無い。これらの細胞では、MHCクラスII分子の発現を誘導し、ならびにIi発現を阻害する両方が可能である(Ii発現およびMHCクラスII発現は同時調節される)。この組み合わせ介入は、病原に関連した、抗原性エピトープを含むペプチドをMHCクラスII分子と結合して細胞の表面に展示することをもたらす。
【0072】
目的とする他のクラスの細胞は、悪性の、ウイルスに感染したものでも、自然に存在する抗原提示細胞でもない。そのような細胞の例には、繊維芽細胞、ケラチノサイトおよび筋肉細胞を含む。この細胞はMHCクラスII分子−陰性であり、そして自然に存在する抗原提示細胞には分類されない。そのような細胞はインビボまたはエクスビボのいずれかでワクチン接種法と関連して有用である。例えばMHCクラスII分子と結合した抗原提示のために、筋肉細胞が標的とされるインビボの内容を考慮されたい。目的の抗原性エピトープおよびMHCクラスII分子のインデューサーをコードする発現可能な核酸配列を、筋肉組織に注入することができる。そのような配列は組織中の筋肉細胞に取り込まれ、そして発現される。注入領域内のある割合の筋肉細胞が、最終的にMHCクラスII分子と結合して目的の抗原性エピトープを細胞表面上に発現するだろう。そのような提示による刺激に応答できる(competent)細胞(例えばヘルパーT細胞)は、リンパを循環する刺激に応答できる細胞として提示細胞と接触する。上で述べたように、リンパ球は循環しているリンパ中の有核細胞の99%を構成する。刺激された抗原提示細胞は脾臓のリンパ節のTリンパ球およびBリンパ球と共同作用し、ここで細胞濃度および他の因子が相互作用を促し、そしてクローン選択を拡大する。分泌されるBリンパ球およびそれらの成熟子孫、血漿細胞により生産される抗体は、リンパの節を去り、そして血液に輸送される。
【0073】
直前の章は限定された文脈上の考察であるが、Ii抑制のための目的の細胞型を紹介するのに役立つ。以下に続く考察は、これらの紹介した細胞型および関連する方法をさらに詳細に調査する。
【0074】
Ii抑制治療は、腫瘍性疾患と関連して示される。これらには例えば、限定された原発性部位を有するガン、ならびに未知の原発性部位の転移性ガンを含む。前者のクラスには胸部ガン、頭および首の悪性腫瘍、卵巣の癌腫、精巣ガンおよび他の栄養膜疾患、皮膚ガンおよび黒色腫および他の色素沈着皮膚損傷を含む。
【0075】
Ii抑制治療はPAI−1を過剰発現する特定の細胞にも示され、そしてMHCクラスII分子を発現するように誘導された。そのような細胞は冠状のアテローム硬化性プラーク、頸動脈、腎動脈、静脈およびガン細胞に見いだされる。PAI−1過剰発現は、腫瘍の侵襲、新脈管形成(neoangiogenesis)および転移、ならびに心筋梗塞、アテローム硬化症、再狭窄および血栓塞栓疾患に関連している(米国特許第6,224,865号明細書;Gunther,J.Surg.Res.103(1):68−78(2002);Harbeck,J.Clin.Oncol.20(4):1000−7(2002);DeYoung,Circulation 104(16):1972−land(2001);Rerolle,Nephrologie 22(1):5−13(2001)。プラスミノーゲンアクチベーターインヒビター1型(PAI−1)は糖尿病患者の動脈壁で増加し、アテローム硬化症の加速および糖尿病患者で臨床的に観察されるプラークの進行に貢献する(Pandolfi,Arterioscler.Thromb.Vasc.Biol.21(8):1378−82(2001))。PAI−1活性は特異抗体、ペプチド拮抗薬、アンチセンスおよびおとり(decoy)オリゴヌクレオチドの使用を介して抑制された(Rerolle,Arterioscler.Thromb.Vasc.Biol.21(8):1378−82(2001))。
【0076】
Ii抑制治療は感染性疾患との関連でも示される。これらにはウイルス疾患(DNAおよびRNAウイルス)、バクテリア疾患(グラム−陽性およびグラム−陰性)、マイコバクテリア疾患、スピロヘータ疾患、リケッチア疾患、マイコプラズマおよびクラジミア疾患、真菌感染、原生生物および寄生生物感染および外部寄生生物感染を含む。
【0077】
自然に存在する抗原提示細胞に関連して、インビボおよびエクスビボ応用が含まれる。本開示において、用語「標的化」は時折、抗原性タンパク質または抗原性タンパク質中の特定の抗原性エピトープに免疫応答を向けることを記載するために使用する。この免疫応答は一部は、応答の内容に依存してTh1またはTh2またはTh3細胞で変動することができるTヘルパー細胞またはTサプレッサー細胞のようなT免疫調節細胞の活性化を特徴とする。例えばTh1応答は、腫瘍抗原に応答するCTL応答の発生に関連するヘルパー応答であり、この応答は腫瘍細胞を殺すことを導く。しかしアレルゲンに対するTh1応答は、Th2応答からアレルゲンに対する応答の免疫逸脱(immunodeviating)に関して、機能的には抑制反応となることができ、これは病原性IgE抗体の生産を導く。加えて標的化の概念は、新規もしく増加した量のMHCクラスII提示エピトープの提示により刺激される免疫応答の初期部分だけでなく、T免疫調節細胞に及ぼす初期作用により誘導または調節される下流のエフェクター応答も含む。このように例えば標的化には本明細書で教示する標的化法により開始され得るCTL−抗ガン応答または免疫グロブリン抗ウイルス応答を含む。
【0078】
標的化には、抗原が特定されているか、もしくは未知であるか、またはたとえ過度な実験無しには同定することができなくても、免疫応答が抗原に向けられるという概念を含む。例えば標的化は、各々が免疫応答の生成に貢献し得る多数の抗原を発現することができる細胞に向けることができる。細胞中のどの特定の抗原が免疫応答に参加するかは、個体の遺伝的構成に依存して人毎に異なり得る。遺伝的因子に対する免疫応答の感受性は、十分に記載されている。その結果、有用な治療的または診断的目的のために標的化法を使用するにあたり、細胞の特異的抗原成分は特定される必要はなく、そしてしばしば特定することはできない。
【0079】
標的化のプロセスには、インビボまたはインビトロのいずれかで起こるプロセスを含む。例えばインビボでは、腫瘍細胞または樹状細胞のいずれかであるMHCクラスII−陽性細胞により提示された抗原に対する免疫調節T細胞の活性化は、非−腫瘍場所または浸潤している腫瘍のいずれかで起こることができる。免疫応答のエフェクター部分の拡大も同様にインビボまたはインビトロのいずれかで起こり得る。インビトロ応答の場合、個体または別の選択した個体に再導入され得る産物を生成して、治療的応答を行うことができる。そのような産物の例は、樹状細胞調製物、細胞傷害性T細胞調製物、およびそのようなインビトロ標的化カルチャーからB細胞をクローニングした後に生産されるであろう抗体(例えばB細胞ハイブリドーマの生産後)を含む。
【0080】
この目的に向けて、末梢血単核球を得た個体に導入することを望む治療用産物に依存して、元のカルチャーを所望の細胞集団、例えば樹状細胞もしくはTリンパ球について濃縮するために分画する。さらにここで教示する標的化プロセスを行った後のカルチャーを、所望の細胞集団、例えば樹状細胞もしくはTリンパ球について濃縮するために分画することができる。単離後すぐに、および本発明の標的化前に、あるいはその標的化が行われた後のいずれかで個体から得た細胞の分画に、確立された方法を利用することができる。さらに確立された手法は、そのような産物を末梢血単核細胞が元々得られた個体に導入するために利用することができる。このために標的化に関する本発明の方法は、末梢血単核細胞に限定されず、口腔咽頭部または他の領域に由来する粘膜細胞、気管支もしくは胃洗浄後に得た細胞、腫瘍組織または正常組織、例えば肝臓、膵臓、前立腺、骨格筋、脂肪、皮膚のような任意の器官の生検もしくは切除により得た細胞を含め、個体から得られるすべての細胞調製物を含む。
【0081】
すべての場合で、目的は自然に存在する抗原提示細胞に、処置する病状に特異的な目的の抗原性エピトープならびにIi発現のサプレッサーを導入することである。腫瘍またはウイルス遺伝子でトランスフェクトした樹状細胞は、強力な抗−腫瘍または抗−ウイルス免疫応答を誘導する。そのような抗原遺伝子でトランスフェクトした樹状細胞中でのIiタンパク質発現の阻害は、そのようなDNAワクチン接種の効力を強化する。自然に存在する抗原提示細胞が関与するインビボおよびエクスビボ態様の両方の場合で、目的の抗原性エピトープをコードする発現可能な核酸配列、およびIiのRNAインヒビターをコードするリバース遺伝子構築物を導入することが好ましい。
【0082】
用語「発現可能な核酸配列」は、翻訳可能(competent)なRNA種をコードする転写可能なDNA構築物、ならびに導入前に転写される翻訳可能なmRNA種を包含することを意図する。当業者は転写および翻訳能力(competency)を付与するために必要な分子シグナルについて精通している。
【0083】
これらの必要な要素の両方を、1つの分子構築物として提供することが可能である(例えば、エピトープおよびIiインヒビターの両方をコードする核酸を受ける十分な能力を有するウイルスベクター送達系を使用して)。あるいは別個の発現構築物を使用して各要素を運ぶことができる。独立した様式で送達される別個の構築物の場合、2つの構築物の各々を取り込む1つの抗原提示細胞の見込みは、統計的確率の問題である。さらに1つのウイルス粒子中に1より多くの構築物をパッケージングすることは、Ii抑制の治療に効果的な誘導を最大にすること、そして示される場合には、有害である免疫応答を生成するウイルスタンパク質の合成に対して、MHCクラスIi誘導および/または所望するタンパク質抗原の合成の誘導に用途を有する。そのような抗−ウイルス免疫応答は、例えばそのような治療的介入が可能な頻度を制限することになる。
【0084】
非−ウイルス送達系による誘導にも特別な配慮が必要である。非−ウイルス送達系を使用して、非複合化DNA、DNA−リポソーム複合体、DNA−タンパク質複合体およびDNA−コート金粒子を細胞に送達することができる。これらの各方法は、特定の病状に関して選択を制御する利点および欠点を与える。複合化DNA(例えばDNA−リポソーム複合体、DNA−タンパク質複合体、DNA−コート金粒子およびポリラクチド コグリコライド粒子中へのマイクロカプセル化)の使用は、1つの細胞へエピトープをコードする核酸配列およびIi発現のインヒビターをコードする核酸配列の両方の送達を確実とする傾向がある。たとえ異なる分子種によりコードされても、それらは「パッケージされる」ので(例えばリポソーム中へのカプセル化、または金粒子上への被覆のどちらか)、両種が1つの細胞に送達される傾向がある。
【0085】
DNA−コート金粒子は一般に、いわゆる「遺伝子銃」技法を使用した弾道法(ballistic method)により送達される。この技術を使用して、金粒子は皮膚または筋肉組織に発射され、そして細胞を貫通するために使用される。貫通された細胞はこの様式で導入された核酸配列を発現することが示された。樹状細胞は、この技法を使用して効果的にトランスフェクトされる自然に存在する抗原提示細胞である。そのような発現構築物は1つの樹状細胞に導入された時、例えば抗原提示細胞の表面上にMHCクラスII分子と結合した目的の抗原性エピトープの展示を生じるだろう。抗原提示細胞の表面上のこのエピトープ/MHCクラスII分子複合体の展示はさらに免疫細胞を刺激して、高まった免疫応答を提供するするだろう。
【0086】
あるいは限定された解剖学的場所(例えば原発性腫瘍または腫瘍性疾患の転移の幾つか)を有する病状に取り組む時、限定された解剖学的場所への直接的注入を示すことができる。そのような部位は樹状細胞のような抗原提示細胞の濃度が高い傾向がある。腫瘍はそのような導入の局所的部位の1例である。関連する発現可能な核酸構築物の細胞への導入を行うための手段が適当である。そのような構築物を局在化した腫瘍部位に導入する時、MHCクラスII分子の発現を刺激するタンパク質をコードするさらなる発現可能な核酸配列を含むことが好ましい。この第3成分の包含は、腫瘍細胞自体を意図している。Ii発現を阻害する構築物、およびMHCクラスII分子生産の発現可能なインデューサーが病状を現している細胞(例えば腫瘍細胞)に取り込まれる場合、細胞は病状に特異的なエピトープをMHCクラスII分子と結合してその細胞表面に展示する。これらの細胞はTヘルパー細胞およびBリンパ球も刺激する。このように局在化した病状に向けられた治療と合わせたこれら3つの発現可能な要素の直接注入は、正常な抗原提示細胞および病状を現すMHCクラスII分子−陰性細胞を標的化する併用療法とみることができる。
【0087】
MHCクラスII分子の生産を誘導するための発現可能な核酸配列の使用に加えて、当業者は本内容および関連する内容において核転移の方法の応用性を認識するだろう(Wolf,Arch.Med.Res.32(6):609−13(2001);Wakayama,Science 292(5517):740−3(2001))。さらに抗原提示細胞は脱分化するように誘導された体細胞に由来してもよく、これによりガン−胚性抗原を発現する(Rohrer,J.Immunol.162(111):6880−92(1999))。そのような細胞は目的の抗原性エピトープに免疫的攻撃を誘導するために使用できる。完全に分化した細胞は、インビボで未成熟(premature)状態に脱分化するように誘導して、器官の再生を行うことができる(Abbate,Am,J.Physiol.277(3Pt2):F454−63(1999))。これらの細胞は異所性の細胞への免疫学的攻撃を刺激するために抗原提示細胞としても機能することができる(Fu,Lancet 358(9287):1067−8(2001))。
【0088】
抗原提示細胞がインビボで標的にされる本発明の別の態様では、正常組織がサイトカイン(例えばGM−CSF)の皮下注射により刺激される。この皮下の「プライミング(priming)」は、樹状細胞をその領域に誘引する。プライミング注射に続いて目的の抗原性エピトープをコードする発現可能な核酸配列、ならびにIi合成のインヒビター(例えばリバース遺伝子構築物)が注射される。腫瘍性細胞は病状に特異的なペプチドの生産体であるが、一般にそれらの表面にMHCクラスII分子と結合して提示しない。そのような細胞では、MHCクラスII分子発現を誘導し、ならびにIi発現を阻害する両方が可能である。例えばMHCクラスII分子の発現は、MHCクラスII分子−陰性細胞に、MHCクラスII分子の生産を刺激するタンパク質をコードするcDNAを導入することにより誘導することができる。そのようなタンパク質には例えば、CIITAまたはインターフェロンガンマを含む。この併用介入は病状に特異的な、抗原性エピトープを含むペプチドのMHCクラスII分子と結合した細胞表面上での展示をもたらす。前に検討したように、発現可能な核酸配列の導入は、これらの目標を達成するために好適な方法である。
【0089】
上で検討したように、直接注入は病状が限定された原発性の場所(腫瘍のような)に存在する場合に示される。場合により、目的の抗原性エピトープをコードする発現可能な核酸配列を、その領域の抗原提示細胞を標的にするために注入材料に含めてもよい。ここでも抗原提示細胞に送達するための目標は、Ii抑制および目的の抗原性エピトープである。病気の細胞にはこの送達目標は、IiサプレッサーおよびMHCクラスII分子インデューサーである。
【0090】
腫瘍内注入と関連して、続く特異的プロトコールは、例えばサイトカインをコードする核酸配列またはサイトカインの腫瘍内注射が関与する確立された治療的プロトコールに基づく。そのようなプロトコールは例えば:
【0091】
【表1】
【0092】
を含む多くの刊行物に記載されている。
【0093】
エクスビボの応用に関して、腫瘍細胞は個体から単離され、そしてエクスビボ培養が樹立される。そのような培養は付随する正常細胞の分離を行うか、行わずに個体から得られた悪性細胞の非選択集団から樹立することができ、あるいは細胞はそのような細胞株に由来する細胞株もしくはクローンから得ることができる。あるいはそのような細胞は無関係な患者の樹立された悪性細胞株、あるいは新鮮な悪性組織(例えば結腸もしくは卵巣癌腫)の外植片として得ることができる。
【0094】
IiサプレッサーおよびMHCクラスII分子インデューサーを培養した細胞に導入すると、所望するMHCクラスII分子が結合した腫瘍特異的もしくは腫瘍関連抗原性エピトープの提示を生じる。MHCクラスII分子発現のインデューサーは当該技術分野では周知であり、そして例えばMHCクラスII分子トランス活性化因子(CIITA)、インターフェロンガンマ遺伝子およびインターフェロンガンマサイトカインを含む。この様式で処理した細胞は複製不能(incompetent)とされ(例えば照射もしくは固定により)、そして通例の免疫感作プロトコール(例えば皮下、静脈内、腹腔内もしくは筋肉内免疫感作による)で使用される。全細胞製剤に加えて、それらの他の誘導体を免疫感作製剤に使用してもよい。
【0095】
関連する腫瘍細胞のほとんどはMHCクラスII分子およびIi−陰性であるが、幾つかの腫瘍(例えば胸部、肺および結腸に影響するある種のリンパ腫、黒色腫および腺ガン)は、MHCクラスII分子−陽性およびIi−陽性である。MHCクラスII分子を発現するこのサブセットでは、所望の免疫刺激を達成するためにIiサプレッサーのみの導入で十分である。そのような細胞にMHCクラスII分子インデューサーの包含は、MHCクラスII分子−結合抗原とTヘルパー細胞との相互作用の見込みが増すことにより、所望の刺激を強化するために役立つと認識されるだろう。
【0096】
興味深い別の細胞のクラスは、悪性の、ウイルスに感染したものではなく、自然に存在する抗原提示細胞でもない。発現可能な核酸配列は、個体の組織の間隙空間に送達される。そのような組織には例えば、筋肉、皮膚、脳、肺、肝臓、脾臓、骨髄、胸腺、心臓、リンパ、血液、骨、軟骨、膵臓、腎臓、胆嚢、胃、小腸、精巣、卵巣、子宮、直腸、神経系、目、腺および結合組織を含む。組織の間隙空間は細胞間、流体、器官組織の網状繊維間のムコ多糖マトリックス、脈管または室の壁中の弾性繊維、繊維状組織のコラーゲン繊維、あるいは筋肉細胞を覆う結合組織内、または骨の空隙中のそのマトリックスを含んでなる。循環している血漿およびリンパ経路のリンパ液により占められる空間も同様である。
【0097】
筋肉細胞はインビボで、発現可能な核酸配列を取り込み、そして発現するそれらの能力が特にある(competent)ことが報告された(例えば引用により本明細書に編入する米国特許第6,214,804号明細書を参照にされたい)。この送達の利点は、多核細胞、筋小胞体およびT管系を含んでなる筋肉の無類の組織構造によることができる。発現可能な核酸配列は、細胞外流体を含み、そして筋肉細胞の深部に広がるT管系を通って筋肉に入る。またそのような発現可能な核酸配列は傷害を受けた筋肉細胞に入り、次いで修復することも可能である。
【0098】
動物は皮膚を通す直接注射により都合よく到達する、比例して大きな筋肉塊を有するので、筋肉は治療的応用において発現可能な核酸配列を送達するための部位としても有利に使用される。この理由から、比較的大用量の発現可能な核酸配列が多回および反復注射により筋肉中に蓄積され得る。治療は長期間に延長することができ、そして特別な技術および装置無しで安全かつ容易に行われる。これらの筋肉以外の組織、および発現可能な核酸配列の低い効率の取り込みおよびまたは発現を特徴とする組織も、注射部位に使用することができる。
【0099】
本発明と関連して、標的細胞中のIi合成を阻害し、そして目的の抗原性エピトープも発現することが望ましい(抗原性エピトープは処置する病状に特異的に関連する)。当該技術分野では知られているように、発現可能な核酸配列の効果的な投薬用量は、一般に約0.05マイクログラム/kg体重〜約50mg/kg体重の範囲に入る(通常、約0.005mg/kg体重〜約5mg/kg)。効果的な投薬用量は関連する因子の数に依存して変動することができると認識されるだろう。
【0100】
上記の種類の病状に関連した抗原を生産し、そして細胞の表面上にMHCクラスII分子と結合した抗原を展示する細胞の別の生成法は細胞融合法である。より具体的にはこれは目的の病状を現す細胞(例えば腫瘍細胞)を用いて、MHCクラスII分子を自然に生産する細胞(例えば樹状細胞のような自然に存在する抗原提示細胞)の融合体を生産する日常的な実験の事柄である。そのような融合細胞では、腫瘍特異的抗原は融合細胞の表面上でMHCクラスII分子と結合して展示される。ほとんどの場合、産物は樹状細胞、マクロファージ、Bリンパ球のような自然に存在する抗原提示細胞、または特定の多機能性細胞の種類と、目的の抗原性エピトープを発現する細胞との融合体である。目的の抗原性エピトープを発現するような細胞には、例えば悪性細胞、ウイルスに感染した細胞または形質転換した細胞、自己免疫応答の誘導に関連する細胞、および自己免疫応答を調節する細胞を含む。後者の種類にはそれらの影響を抗−イディオタイプネットワークメカニズムを介して発揮する細胞を含む(例えば慢性関節リウマチにおいて病原に関連性のあるT細胞レセプターを発現する)。この型の細胞融合体は、エクスビボで生産される。Ii抑制およびワクチン接種は本開示のいたるところに記載するように行う。
【0101】
当業者は本発明の方法を、処置する細胞またはそれらの局所的環境へのサイトカイン治療(すなわちサイトカインをコードする核酸配列、またはサイトカイン自体の導入)と組み合わせることができると認識するだろう。他の免疫同時−刺激(immune co−stimulatory)分子も使用することができる(Akiyama,Y.,Gene Ther.7(24):2113−21(2000):Miller,P.W.,Hum.Gene.Ther.11(1):53−65(2000);J.Neurosurg.94(2):287−292(2001);Jantscheff,P.,Cancer Immunol.Immunother.48(6):321−30(1999);Kikuchi,T.,Blood 96(1):91−9(2000);Melero,I.,Gene Ther.7(14):1167−70(2000);Lei,H,Zhongua Zhong Liu Za Zhi20(3):174−7(1998))。
【0102】
前に述べたように、当業者は過度な実験を行うことなく、強化された免疫応答を提供する、MHCクラスII分子が結合して展示する病原に特異的な抗原を同定することができる。ここでもすべての場合で、Ii阻害はMHCクラスII分子に結合したそのような抗原の展示を行うために必要である。以下の一覧はこのクラスに入る抗原の非限定的、非排他的な例の列挙であることを意図している:HIVgp120(Barouch et al.,J.Immunol.15:168:562−8(2002));HIVgag(Shingh et al.,Vaccine 20:594−602(2001));インフルエンザM1およびM2(Okuda et al.,Vaccine 19:3681−91(2001));B型肝炎表面抗原およびコア抗原(Musacchio et al.,Biochem.Biophys,Res.Commun.282:442−6(2001));ヒトテロメラーゼ逆転写酵素(hTERT)(Heiser et al.,Cancer Res.61:3388−93(2001));Gp75TRP−1(Bowne,Cytokines Cell Mol.Ther.5:217−25(1999));TRP−2およびgp100(Xiang,Proc.Natl.Acad.Sci.USA 97:5492−7(2000));PSA(Kim,Oncogene 20(33):4497−506(2001));CEA(von Mehren et al.,Clin.Cancer Res.7:1181−91(2001));Erb2/Neu(Pilon et al.,Immunol.167:3201−6(2001)およびTuting,Gene Ther.6:629−36(1999))。
【実施例】
【0103】
実施例1
CIITA cDNAを含むアデノウイルスベクターの構築
この実験の最初の目標は、MHCクラスII分子陰性細胞(例えばMC−38およびRenca)にMHCクラスII分子の効率的誘導のためのアデノウイルスベクターを構築することであった。CMVプロモーターおよびポリAテイルを含むCIITA遺伝子構築物は、CIITAを含有するpCEP4ベクター(L.Glimcher博士から得た)から、Sal1を使用して切り出した。この断片をpBluescriptに連結して、pBlue/CIITAを作成した。次いでpBlue/CIITAをEcoRVおよびXhoIで消化して、CMVプロモーター、CIITA cDNAおよびポリAシグナルを含むDNA断片を放出し、これをpQBI/BN(クウォンタム(Quantum)、モントリオール、カナダ)に連結して、pQBI/BN/CIITAを作成した。
【0104】
このベクターを293Aアデノウイルスパッケージング細胞に、Cla1消化アデノウイルスDNA(ウイルスの左腕は削除されてバックグラウンドを下げた)と一緒に、製造元の指示に従いコトランスフェクトした。コトランスフェクションから3週間後、生成したプラークは、CIITA cDNAの−7から+12および+751から+769に位置する2つのDNAプライマーを使用したPCRによりスクリーニングして、CIITA遺伝子の存在を確実にした。1つのクローンを使用して2つのマウス腫瘍細胞株:MC−38結腸腺ガンおよびRenca腎臓細胞腺ガンにおいて、MHCクラスII分子の導入を試験した。MHCクラスII分子の導入に関するタイムコースは、アデノ/CIITA組換えアデノウイルスベクターでの感染後にこれらの細胞株でアッセイした。CIITA挿入物を欠く点を除いて同一のアデノウイルスベクターを対照として使用した。MHCクラスII分子は感染から48〜72時間後に>95%の細胞で強く誘導されることが証明された。
【0105】
実施例2
アデノ/CIITAでの感染に加えてIiアンチセンスオリゴヌクレオチドでの処置によるMHCクラスII+/Ii−表現型の生成
この実施例は、細胞のアデノ/CIITAによる感染および定めたIiアンチセンスオリゴヌクレオチドによるIi発現の阻害により、MHCクラスII+/Ii−表現型を発現する細胞の生成を示す。Iiアンチセンスオリゴヌクレオチドは効果的であることが以前に証明された(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。対照実験には:a)処置無し;b)アデノ/CIITA構築物のみ;c)センス対照オリゴヌクレオチドと一緒のアデノ/CIITA構築物;およびd)4つのヌクレオチド誤対合対照アンチセンスオリゴヌクレオチドと一緒のアデノ/CIITA構築物を含んだ。簡単に説明すると、1.5×106個のMC−38細胞を25cm2のフラスコにオリゴの電気穿孔の24時間前にまき、そして1.5mlのウイルスストック溶液(1.26×106PFU/ml)を含む5mlの総容量で48時間感染させた。感染から最初の24時間後、10mlの新鮮な培地を加え、そして細胞をさらに24時間インキュベーションした。次いで細胞をトリプシン処理し、そしてアンチセンス、センスまたは誤対合オリゴヌクレオチドでの電気穿孔前に洗浄した。電気穿孔の条件は以下の通りであった:3〜5×106個の細胞を、50μMのオリゴヌクレオチドを含有する0.5mlのRPMI1640中の電気穿孔用のキュベットに加えた。次いで細胞を氷上で10分間インキュベーションし、そしてBTX600エレクトロポレーターを使用して200ボルト/1250μFにかけた。次いでキュベットをさらに10分間、氷上でインキュベーションし、その後に細胞を1回洗浄し、新しい25cm2フラスコにまき、そして24時間インキュベーションした。この時点で細胞をトリプシン処理し、そして以前に記載されたように(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))、MHCクラスII分子およびIiタンパク質に関する染色後にフロサイトメトリーにより分析した。
【0106】
図1に示す典型的な実験では、Iiアンチセンスで処理し、そしてアデノ/CIITA感染した細胞が、MHCクラスII分子の発現にほとんど影響しないか、または影響せずにIiの良好な選択的阻害を示した。対照のオリゴヌクレオチド処理した細胞(すなわち誤対合またはセンス配列を使用した)は、Iiの阻害を示さず、そしてアデノ/CIITA感染細胞に関してMHCクラスII分子に匹敵する発現があった。
【0107】
動物実験の予測および大量にMHCクラスII+/Ii−細胞を生成する必要性から、上記実験をスケールアップした系で繰り返した。5×106個のMC−38細胞を75cm2のフラスコに感染の18〜24時間前にまいた。細胞は5mlのウイルスストック溶液(1.26×106PFU/ml)で90分間感染させ、そして20mlの新しい培地を加えた。次いで細胞を48時間インキュベーションし、そして上記のようにオリゴヌクレオチド(50mM)を送達するために電気穿孔にかけた。次いで細胞をプールし、そして新しい75cm2フラスコ中でさらに24時間インキュベーションし、その後に培地を交換し、そして細胞をさらに3時間インキュベーションした。次いで細胞をMHCクラスII分子およびIiタンパク質の発現、およびマウスの免疫感作について分析した。Iiタンパク質発現の配列特異的阻害は、以前の実験で観察されたようにアデノ/CIITAで感染させ、そしてIiアンチセンスで処理した細胞中でのみ得られた。
【0108】
実施例3
MHCクラスII+/Ii−腫瘍ワクチンによる腫瘍防御
これらの実験のために、MC−38腫瘍ワクチン細胞を上記のように調製し、そして6〜7週齢の、メスのC57BL/6マウス(ジャクソンラボズ:Jackson Labs)を接種するために使用した。具体的にはMC−38細胞は上記のようにアデノ/CIITAで感染させ、4群に分け、そして電気穿孔により:a)無し;b)50mM Iiアンチセンスオリゴヌクレオチド;c)50mM 誤対合対照オリゴヌクレオチド;またはd)50mMセンス対照オリゴヌクレオチドで処理し、そしてフラスコに播種した。24時間後、新しい培地を加え、そして細胞をさらに3時間インキュベーションした。次いで細胞をトリプシン処理し、50Gy(セシウム源)で致死的に照射し、そして1.2×106個の細胞/マウスをマウスに接種した。5週間後、マウスを5×105個の親MC−38細胞でチャレンジし、そして腫瘍の出現について監視した。図2に示すように、Iiアンチセンス処置したアデノ/CIITA感染MC−38細胞の接種は腫瘍の成長に対して、すべての他の対照群よりも良い防御を提供した。これらのデータはCIITAで安定にトランスフェクトし、そしてIiアンチセンスで処置したMC−38細胞を使用した我々の以前の実験と一致する(図3)。見れば分かるように、安定にCIITAでトランスフェクトしたMC−38またはIiアンチセンスで処置した一時的なアデノ/CIITA感染細胞のいずれかを使用した防御のレベルは、匹敵するレベルの防御を与える。
【0109】
実施例4
MHCクラスII+/Ii−腫瘍ワクチンおよびGM−CSFによる腫瘍防御
別の組の動物実験では、後の親のMC−38細胞の成長に及ぼすGM−CSF処理と一緒のIi−阻害MC−38ワクチンの役割を調査した。これらの実験に関して、樹状細胞を誘引するためのMC−38細胞の免疫感作の1日前に、マウスに18mgのGM−CSF(R&Dシステム、ミネアポリス、ミネソタ州)を右後脚にs.c.注射した。またマウスを免疫感作するために使用したMC−38細胞の数は3×105個/マウスであり、以前の実験で使用した数より4倍少なかった。図4に示すように、GM−CSFはクラスII+/Ii−MC−38細胞により誘導される防御効果を強化する。わずか3×105個のクラスII+/Ii−細胞を接種されたマウスは、GM−CSFの不存在下で1.2×106個のクラスII+/Ii−MC−38細胞により誘導された時と同様に親細胞の成長を阻害した。以前の実験では、IFN−γにより誘導されるMHCがCIITAによりもはるかに強い抗−腫瘍免疫応答の誘導を与えることが示された(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。
【0110】
これらの実験はサイトカインとIi阻害の相乗的効果が可能であることを示した。さらなる実験は、GM−CSFおよびIFN−γの使用とIiアンチセンス法とを組み合わせ、そしてこの免疫感作プロトコールを至適化し、そして増幅させるために計画する。
【0111】
実施例5
IFN−γ cDNAを含むアデノウイルスベクターの構築
IFN−γは免疫応答の方向の調節に重要な役割を果たし、そしてMHCクラスII分子およびIiを種々の組織および幾つかの悪性細胞を含む細胞で誘導する。IFN−γ構築物でのトランスフェクト、およびアンチセンスオリゴヌクレオチドによるIi阻害により作成されたMHCクラスII+/Ii−腫瘍ワクチンは、MHCクラスII分子の発現がCIITAトランスフェクションにより誘導されること以外は同一の腫瘍ワクチンに比べて、増大した免疫原性を有する(Qiu et al.,Cancer Immunol.Immunother.48:499−506(1999))。発現可能なマウスIFN−γ配列がアデノウイルスにクローン化された。MHCクラスII分子およびIiタンパク質の両方の発現が、大変低濃度のアデノ/IFN−γの感染後に誘導された(図5を参照にされたい)(わずか1のMOIでも。データは示さず)。アデノ/IFN−γ構築物を作成するために、マウスIFN−γcDNA(Chen et al.,J.Immunol.151:244−55(1993))が、IFN−γcDNAの開始および終止コドンを含む領域に相補的な2つの特異的オリゴヌクレオチドを用いたPCRにより増幅された。このIFN−γ断片は、特異的に設計したエンドヌクレアーゼ消化部位を使用してpCDNA(3+)プラスミドにクローン化され、そしてシークエンシングにより確認された。CMVプロモーター、IFN−γおよびポリAシグナルは、適切なオリゴヌクレオチドでさらにPCR増幅され、次いで適切な制限部位を使用してpQBI/Ad/BNにクローン化された。アデノ/IFN−γ組換えウイルスの生成は、実施例1に記載したものと同じ手順により行った。
【0112】
実施例6
Ii−RGCを含むアデノウイルスベクターの構築
数個のIiリバース遺伝子構築物を、RSV.5およびpcDNA(3+)発現ベクターにクローン化した。構築物のサブサットは、古典的なトランスフェクション法(例えばリポフェクチン)を使用してMHCクラスII分子−陽性細胞(A20)中のIiを阻害する能力を有することが示された。Iiアンチセンスオリゴヌクレオチドも効果的であることが示されたが、それらは電気穿孔法または有意な毒性を伴う他の方法を必要とする。またIi発現の有意な阻害を示すのは、オリゴヌクレオチドで処理した細胞の30〜70%より多くはない。対照的に(そしてアデノ/CIITA構築物を使用して示されるように)、遺伝子送達用のアデノウイルスベクターの使用により、すべての細胞へのほぼ100%の送達、所望する表現型の変化およびほぼ皆無の毒性がもたらされる。MHCクラスII+/Ii−表現型のより良い誘導のために、幾つかのIi−RGCがアデノウイルスにクローン化された。Ii−RGCを含む組換えアデノウイルスを作成するために、RSV(またはCMV)プロモーター、Iiリバース遺伝子断片およびポリAシグナルからなる発現カセットをPCRにより増幅し、そしてNotIおよびXhoIまたは他の適当な制限酵素部位を使用してpQBI/BNベクターにクローン化し、pQBI/BN/Ii−RGCを作成した。アデノ/Ii−RGCの最終的な構造は、実施例1に記載したものと同じ手順により完成した。MHCクラスII+/Ii−表現型の誘導実験では、アデノ/Ii−RGCの濃度がアデノ/CIITAの濃度の4倍に増加した時、Iiが>95%の細胞で阻害されたが、MHCクラスII分子の発現にはほとんど影響しないことが観察された(図6)。
【0113】
実施例7
IFN−γおよびIi−RGCを含むアデノウイルスベクターの構築
感染を簡略化するために、アデノ/IFN−γ/Ii−RGC構築物を生成した。プロモーター、Ii−RGC断片およびポリAシグナルは、適切なオリゴヌクレオチドを用いてPCRにより増幅し、そしてpQBI/Ad/BN/IFN−γにクローン化してpQBI/Ad/BN/IFN−γ/Ii−RGCを作成し、続いてこれを使用してアデノ/IFN−γ/Ii−RGCを作成した。アデノ/IFN−γ/Ii−RGCでの感染によるMHCクラスII+/Ii−表現型の誘導実験では、pQBI/Ad/BN/IFN−γ/Ii−RGC構築物(アデノ/IFN−γ/Ii−RGC(−92,+97))の1つの感染により、感染から96時間後にMHCクラスII+/Ii−表現型がMC/38細胞中に生成したことが観察された。
【0114】
実施例8
多数のIi−RGCを含むアデノウイルスベクターの構築
Ii−RGCの効率を最大にするために、幾つかのIi−RGCを1つのアデノウイルスベクターにクローン化した。PCR増幅および他の適切な分子生物学的方法を使用して、種々のIi−RGCの組み合わせを含むpQBI/Ad/BN構築物を生成した。そのような構築物の例には、以下に示す組を含む。マウスIi挿入物(−92,+97)、(+32,+136)、 (+314,+458)のヌクレオチド配列は、それぞれ配列番号1、2および3として配列表に提示する。
【0115】
【表2】
【0116】
Ii−RGCの幾つかは、以下に示す組を含め、IFN−γも含んでクローン化された。
【0117】
【表3】
【0118】
続いてIi−RGCの効果を最大にするための努力では、各々が異なるプロモーターにより駆動されるIi−RGCの多コピーを含むプラスミドを作成した。これらのプラスミドを以下に記載する。
【0119】
【表4】
【0120】
プロモーターの略号:RSV(ラウス肉腫ウイルスプロモーター)、EF−1a(ヒト延長因子−aプロモーター)、UbC(ユビキチンCプロモーター)、CMV(サイトメガロウイルスプロモーター)。
【0121】
実施例9
ヒトIi−RGCを含むプラスミドの構築
ヒトIi遺伝子配列に由来するヒトIi−RGC(hIi−RGC)によるヒトIi発現の阻害を本明細書に開示する。ヒト細胞中のIi発現を阻害するhIi−RGCを使用した実験の結果を、表2に示す。ヒトIi cDNA配列(Strubin et al.,EMBO J.3:869−72(1984))は、Eric Long博士から贈られた。異なる長さのIi遺伝子の断片は、適切なオリゴヌクレオチドを使用してPCRにより生成した。すべてのIi断片が多数のAUG開始および終止コドンを含む。いかなるリーディングフレイム中でもAUGの直後に終止コドンが続くことを回避するためにすべて設計し、アンチセンスRNAの半減期を延ばした。このために、AUGを作成して、異なるリーディングフレイム中の終止コドンに重ねた(override)。これらIi PCR断片は、適切な制限部位によりpcDNA3(+)発現ベクターにクローン化した。ヒトリンパ腫細胞株であるRajiを使用して、これらhIi−RGCを使用したIi阻害を測定した。Raji細胞は、Polyfectトランスフェクション試薬(キアゲン:Qiagen)で1μgのhIi−RGCプラスミドDNAを使用して、製造元の指示に従い一時的にトランスフェクトされた。48時間のインキュベーション後、細胞はIiおよびMHCクラスIIおよびIiの発現について、細胞を抗−ヒトIi抗体、LN2(ファミンゲン:Pharmingen)および抗−DR抗体(ファミンゲン)で染色することにより染色され、続いてフロサイトメトリーにかけられた。
【0122】
Ii−発現が一部の細胞で阻害さる(4〜9%の細胞はバックグラウンドより上であった(図8を参照にされたい)ことが観察された。この阻害は高度に再現性があった。さらにそのような一時的なトランスフェクションアッセイでは、典型的にはカルチャー中のわずか10%以下の細胞が実際に加えたDNA構築物を取り込む。すなわちバックグラウンドより上の4〜9%の細胞は、このアッセイ系の実際のトランスフェクション効率を反映している。これらの細胞では、MHCクラスII分子の発現に対する観察可能な影響は無かった。
【0123】
【表5】
【0124】
hIi−RGCの活性を最大にする試みでは、多コピーhIi−RGC(1つのプラスミド中に数コピーのhIi−RGC)が作成され、ここで各発現カセットは異なるプロモーターにより駆動される。これらのプラスミドを以下に列挙する。
【0125】
【表6】
【0126】
実施例10
MHCクラスII+/Ii−表現型および治療効力を誘導するために、IL−2と一緒のIi−RGCベクターの腫瘍内注射
BALB/cマウスに105個のRenca細胞をs.c.注射した。1:6(重量:重量)のCIITA:Ii−RGC DNA比で、CIITA cDNA遺伝子、CIITA cDNA遺伝子およびIi−RGC(−92,97)を含むプラスミド、またはトリプルIi−RGC(−92,97/32,136/314,459)を含むプラスミドを加えたCIITA遺伝子を、サイズが0.05〜0.2cm3のRenca腫瘍に注射した。注射の1〜5分前に、25mgの全DNAをDMRIE/C(1,2−ジメリスチルオキシプロピル−3−ジメチルヒドロキシ エチル アンモニウム ブロミド/コレステロール)(ギブコ:GIBCO)と、1:1(重量:重量)比でインキュベーションした。DNA注射から5日後、スライドを摘出した腫瘍の凍結切片から作成した。スライドはマウスMHCクラスIIおよびIiに対する抗体で染色して、腫瘍細胞のクラスII+/Ii−表現型を決定した。腫瘍中のクラスII+細胞がT細胞、B細胞またはマクロファージを表すかもしれない可能性を排除するために、染色はCD4、CD8、CD3、CD19(B細胞用)およびMAC(マクロファージ)に対する抗体を用いても行った。結果(データは示さず)は、CIITAのみを注射した腫瘍では類似のMHCクラスIIおよびIi染色が示されたが、CIITA/Ii−RGC(−92,97)またはCIITAプラスミドにトリプルIi−RGCを含むプラスミドを加えたもののいずれかを注射した腫瘍において、Ii抑制の証拠があった。CD4、CD8およびCD3染色は大変少ない陽性細胞を示し、腫瘍内のクラスII+細胞がT細胞を浸潤してないことを示す。B細胞およびマクロファージ染色も、クラスII+細胞がB細胞またはマクロファージでないことを除外した。同時に、脾臓サンプルのスライドは陽性対照として上記抗体のすべてで染色された。
【0127】
Ii抑制の治療効力を調査する実験のために、BALB/cマウスにRenca腎腺ガン細胞をs.c.注射し、そしてIL−2(2mg)、CIITA(3mg)およびIi−RGC(−92,97)(18mg)を含んでなる種々のプラスミド調製物を1日に腫瘍内注射し、そしてCIITAを含まない同じ調製物を2〜4日に注射することにより処置した。対照マウスは2μgのIL−2と一緒に連続して4日間、空ベクターを受けた。次いで腫瘍は2または3日毎に測定された。マウスは31日間追跡され、そして腫瘍サイズが1000cm3に達した時に終了した。結果はCIITAおよびIL−2と一緒にIi−RGCを含むベクターで処置したマウスが腫瘍の成長に劇的な減少を現す一方、IL−2のみまたは対照ベクターを受けたマウスは進行性であり、そしてマウスのターミネーション(termination)が必要であることを示した(図9を参照にされたい)。
【図面の簡単な説明】
【0128】
【図1】相対的蛍光強度を表す図である。MHCクラスII+/Ii−表現型は、マウス結腸腺ガン細胞(MC−38)のアデノ/CIITAアデノウイルスベクターでの感染、そして続いてIiアンチセンスオリゴヌクレオチドでの処理により生成した。A)親のMC−38細胞(処置無し);B)アデノ/CIITAに感染したMC38細胞;C)アデノ/CIITAに感染し、そしてセンス対照オリゴヌクレオチドで処理したMC38細胞;D)アデノ/CIITAに感染し、そして誤対合対照オリゴヌクレオチドで処理したMC38細胞;E)アデノ/CIITAに感染し、そしてIiアンチセンスオリゴヌクレオチドで処理したMC38細胞。
【図2】MHCクラスII+/Ii−細胞をワクチン接種したマウスでのMC−38結腸腺ガンの成長の阻害を表す図である。符号:(丸)MC−38細胞で免疫感作したマウス;(三角)アデノ/CIITAおよび誤対合対照オリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス;(菱形)アデノ/CIITAおよびセンス対照オリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス;および(四角)アデノ/CIITAおよびIiアンチセンスオリゴヌクレオチドで処理したMC−38細胞で免疫感作したマウス。
【図3】CIITAで安定にトランスフェクトされ、そしてIiアンチセンスを使用してIi発現が阻害された致死的に照射したMC−38細胞を接種したマウスにおける親腫瘍の成長の阻害を表す図である。マウスにPBS(三角)、センスオリゴヌクレオチド(丸)またはIiアンチセンス(四角)で処理したCIITAトランスフェクトMC−38細胞を接種した(5マウス/群)。
【図4】MHCクラスII+/Ii−細胞をワクチン接種し、そしてGM−CSFで処理したマウスでのMC−38結腸腺ガンの成長の阻害を表す図である。符号:(三角)親のMC−38細胞で免疫感作したマウス;(丸)MC−38細胞およびGM−CSFで免疫感作したマウス;(白抜き四角)CIITA、センス対照オリゴヌクレオチドで処理したMC−38細胞およびGM−CSFで免疫感作したマウス;および(菱形)CIITA、Iiアンチセンスオリゴヌクレオチドで処理したMC−38細胞およびGM−CSFで免疫感作したマウス。
【図5】MC−38細胞中のアデノ/IFN−γによるMHCクラスII分子およびIi誘導を表す図である。MC−38細胞はアデノ/IFN−γ(3MOI)で示した時期に感染させ、次いで抗−MHCクラスII分子またはIi抗体で染色し、そしてフローサイトメトリーで分析した。
【図6】相対的蛍光強度を表す図である。MHCクラスII+/Ii−表現型は、細胞をアデノ/CIITAおよびアデノ/Ii−RGC(Ii−92,97)で同時感染させることによりRenca細胞に生成した。Renca細胞は異なる比率のアデノ/CIITA対アデノ/Ii−RGCで同時感染させ、72時間インキュベーションし、そしてMHCクラスII分子またはIiタンパク質発現について染色した。親のRenca細胞をAに示し;アデノ/CIITに感染した細胞をBに示し;1:2の比率のアデノ/CIITA対アデノ/Ii−RGCに同時感染したものをCに示し;そして1:4の比率のアデノ/CIITA対アデノ/Ii−RGCに同時感染したものをDに示す。
【図7】MHCクラスII+/Ii−表現型が、細胞をアデノ/IFN−γ/Ii−RGC(mIi−92,97)に感染させることによりMC−38細胞に生成されるタイムコース実験を表す図である。Ii−しかしII+表現型はアデノ/IFN−γ/Ii−RGC(mIi−92,97)(左)の120時間後に作成されたが、アデノ/IFN−γの感染のみではMC−38細胞にMHCクラスII+/Ii−表現型を生成しなかった(右)。
【図8】一時的にトランスフェクトされたRaji細胞(MHCクラスII+/Ii+)、ヒトB−リンパ腫細胞株におけるIi抑制を表す図である。細胞は12−ウェルプレートに一晩、1.25×105細胞/ウェルでまき、そしてヒトIi−リバース遺伝子構築物(hIi−RGC)でトランスフェクトしてIi発現を阻害した。エフェクテン(effecten)トランスフェクション試薬(25μl、キアゲン)を濃縮hIi−RGCプラスミドDNA(1ug)とインキュベーションして、培地と混合したエフェクテンDNA複合体を生成し、これを細胞に直接加えた。48時間のインキュベーション後、細胞はIiおよびMHCクラスII分子発現について、抗−ヒトIi抗体、LN2(ファミンゲン)およびMHCクラスII分子(ファミンゲン)の染色用の抗−HLA−DR抗体での免疫染色により分析された。見れば分かるように、Ii発現は使用したIi−RGC配列に依存して、陽性対照細胞(左パネル)に比べて細胞の4%および9%で阻害されたが、MHCクラスII分子の発現には効果が無かった(右パネル)。
【図9】アデノ/Ii−RGCベクターのインビボ投与による腫瘍成長の阻害、およびMHCクラスII+/Ii−表現型の生成を表す図である。BALB/cマウスに、5×105個のRenca腎腺ガン細胞を皮下注射した。腫瘍が50から200mm3の間のサイズに達した時、腫瘍細胞注射から約10日後、腫瘍に種々のベクターの組み合わせをDMRIE/cと連続して4日間、毎日注射した。次いで腫瘍は2〜3日毎にサイズを測定した。マウスは腫瘍サイズが1000mm3に達した時にターミネーションした。左パネルのデータは、腫瘍に2mgのIL−2、3mgのアデノ/BN/CIITAおよび18mgのアデノ/BN/Ii−RGC(−92,97)を1日に、続いて2mgのIL−2、18mgのアデノ/BN/Ii−RGC(−92,97)および3mgの空プラスミド(アデノ/BN)(CIITA無し)を2〜4日に注射した4匹のマウスを表す。IL−2と共にCIITAおよびIi−RGCを含むベクターで処置したマウスが、腫瘍の成長に劇的な減少を表す一方、IL−2および対照ベクターのみを受けたマウスの腫瘍の成長は進行的であり、マウスのターミネーションが必要であったことは明らかである。
【配列表】
【特許請求の範囲】
【請求項1】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物。
【請求項2】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項3】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項4】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項5】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項4に記載のリバース遺伝子構築物。
【請求項6】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を含む哺乳動物細胞。
【請求項7】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項6に記載の哺乳動物細胞。
【請求項8】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項6に記載の哺乳動物細胞。
【請求項9】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項6に記載の哺乳動物細胞。
【請求項10】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項9に記載の哺乳動物細胞。
【請求項11】
MHCクラスII分子−陽性細胞である請求項6に記載の哺乳動物細胞。
【請求項12】
操作前に個体中に存在する時にはMHCクラスII分子−陰性である請求項6に記載の哺乳動物細胞。
【請求項13】
タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを含み、そのトランスフェクションがMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項12に記載の哺乳動物細胞。
【請求項14】
悪性細胞である、請求項6に記載の哺乳動物細胞。
【請求項15】
ウイルスに感染した細胞である請求項6に記載の哺乳動物細胞。
【請求項16】
自然に存在する抗原提示細胞である請求項6に記載の哺乳動物細胞。
【請求項17】
樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項16に記載の哺乳動物細胞。
【請求項18】
さらに目的の抗原を含んでなる請求項16に記載の哺乳動物細胞。
【請求項19】
目的の抗原が、目的の抗原をコードする発現可能な核酸配列から細胞内で合成される請求項18に記載の哺乳動物細胞。
【請求項20】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的の抗原性エピトープを展示する方法であって:
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そして
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項21】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項20に記載の方法。
【請求項22】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項20に記載の方法。
【請求項23】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項22に記載の方法。
【請求項24】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項22に記載の方法。
【請求項25】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項20に記載の方法。
【請求項26】
目的の抗原性エピトープがガン細胞抗原である請求項20に記載の方法。
【請求項27】
目的の抗原性エピトープがウイルス抗原である請求項20に記載の方法。
【請求項28】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項20に記載の方法。
【請求項29】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項20に記載の方法。
【請求項30】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項29に記載の方法。
【請求項31】
Iiタンパク質発現が抑制されているMHCクラスII分子陽性細胞の表面上の目的の抗原性エピトープに向けられる、哺乳動物における免疫応答を刺激する方法であって:
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
c)哺乳動物を、工程b)の細胞または工程b)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで免疫感作する、
ことを含んでなる上記方法。
【請求項32】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項31に記載の方法。
【請求項33】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項31に記載の方法。
【請求項34】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項33に記載の方法。
【請求項35】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項33に記載の方法。
【請求項36】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項31に記載の方法。
【請求項37】
目的の抗原性エピトープがガン細胞抗原である請求項31に記載の方法。
【請求項38】
目的の抗原性エピトープがウイルス抗原である請求項31に記載の方法。
【請求項39】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項31に記載の方法。
【請求項40】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項31に記載の方法。
【請求項41】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項40に記載の方法。
【請求項42】
識別抗原の発現により細胞型が特徴付けられる、免疫学的応答のために個体の特定の型の細胞を標的化する方法であって:
a)カルチャー中に抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;そして
c)識別抗原をコードする発現可能な核酸配列を、発現に適する条件下でカルチャー中の細胞に導入する、
ことを含んでなる上記方法。
【請求項43】
抗原提示細胞が樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項42に記載の方法。
【請求項44】
Iiの発現が、細胞カルチャーの細胞に、生理学的条件下で哺乳動物Iiタンパク質をコードするmRNA分子の領域に特異的にハイブリダイズする能力を特徴とする約10〜約50ヌクレオチド塩基のコポリマーを導入することにより阻害され、ここでコポリマーがIi発現を阻害する能力を特徴とする、請求項42に記載の方法。
【請求項45】
Iiの発現が、細胞カルチャーの細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入することにより阻害される、請求項42に記載の方法。
【請求項46】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項45に記載の方法。
【請求項47】
発現可能な核酸配列がプラスミドベクター中に生成される、請求項42に記載の方法。
【請求項48】
プラスミドベクターが、脂質、リポソーム、金粒子、ポリラクチドコグリコライド粒子およびポリアルキルオキシドコポリマーからなる群から選択される媒介物を使用する方法により導入される、請求項47に記載の方法。
【請求項49】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項42に記載の方法。
【請求項50】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項49に記載の方法。
【請求項51】
工程c)の細胞が治療効果のために個体に導入される、請求項42に記載の方法。
【請求項52】
工程a)の末梢血単核細胞が抗原提示細胞を濃縮するために分画される、請求項42に記載の方法。
【請求項53】
工程c)の細胞が治療効果のために個体に導入される、請求項52に記載の方法。
【請求項54】
工程c)の抗原提示細胞を含むカルチャー中に分画された細胞のサブポピュレーションが得られる、請求項42に記載の方法。
【請求項55】
工程c)の細胞が治療効果のために個体に導入される、請求項54に記載の方法。
【請求項56】
識別抗原がガン細胞抗原である、請求項42に記載の方法。
【請求項57】
識別抗原がウイルス抗原である、請求項42に記載の方法。
【請求項58】
免疫応答のために個体の抗原を標的化する方法であって:
a)カルチャー中に自然に存在する抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;そして
c)工程a)の自然に存在する抗原提示細胞のカルチャーに、免疫学的応答のために標的化される抗原をコードする発現可能な核酸配列をに導入する、
ことを含んでなる上記方法。
【請求項59】
抗原提示細胞が樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項58に記載の方法。
【請求項60】
工程c)の細胞が治療効果のために個体に導入される、請求項58に記載の方法。
【請求項61】
工程a)の末梢血単核細胞が抗原提示細胞を濃縮するために分画される、請求項58に記載の方法。
【請求項62】
工程c)の細胞が治療効果のために個体に導入される、請求項61に記載の方法。
【請求項63】
工程c)の抗原提示細胞を含むカルチャー中に分画された細胞のサブポピュレーションが得られる、請求項58に記載の方法。
【請求項64】
工程c)の細胞が治療効果のために個体に導入される、請求項63に記載の方法。
【請求項65】
Iiの発現が、細胞カルチャーの細胞に、生理学的条件下で哺乳動物Iiタンパク質をコードするRNA分子の標的に特異的にハイブリダイズする能力を特徴とする約10〜約50ヌクレオチド塩基のコポリマーを導入することにより阻害され、ここでコポリマーがIi発現を阻害する能力を特徴とする、請求項58に記載の方法。
【請求項66】
Iiの発現が、細胞カルチャーの細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入することにより阻害される、請求項58に記載の方法。
【請求項67】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である請求項51に記載の発現可能なリバース遺伝子構築物である請求項66に記載の方法。
【請求項68】
発現可能な核酸配列がプラスミドベクター中に生成される、請求項58に記載の方法。
【請求項69】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項58に記載の方法。
【請求項70】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項69に記載の方法。
【請求項71】
識別抗原がガン細胞抗原である、請求項58に記載の方法。
【請求項72】
識別抗原がウイルス性である請求項58に記載の方法。
【請求項73】
MHCクラスII分子−陽性細胞であり、そして10〜50ヌクレオチド塩基のコポリマーを含む細胞であって、コポリマーが生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、上記細胞。
【請求項74】
ガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞のいずれかである請求項73に記載の細胞。
【請求項75】
MHCクラスII分子を発現し、そして細胞表面に展示するように誘導されたMHCクラスII分子−陰性細胞であって、細胞がタンパク質をコードする発現可能な核酸配列を含んでなり、その発現がMHCクラスII分子−陰性細胞中で細胞表面上のMHCクラスII分子の誘導をもたらし、細胞がさらに10〜50ヌクレオチド塩基のコポリマーを含んでなり、コポリマーが生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、上記細胞。
【請求項76】
発現可能な核酸配列が、ウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項75に記載の細胞。
【請求項77】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項76に記載の細胞。
【請求項78】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項75に記載の細胞。
【請求項79】
ガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項75に記載の細胞。
【請求項80】
コポリマーが電気穿孔により導入される、請求項75に記載の細胞。
【請求項81】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的の抗原性エピトープを展示する方法であって、
a)MHCクラスII分子−陽性細胞またはMHCクラスII分子を発現するように誘導される細胞のいずれかであり、そして目的の抗原性エピトープを発現する細胞を準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;そして
c)工程a)の細胞に10〜50ヌクレオチド塩基のコポリマーを導入し、このコポリマーは生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、
ことを含んでなる上記方法。
【請求項82】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項81に記載の方法。
【請求項83】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項82に記載の方法。
【請求項84】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項81に記載の方法。
【請求項85】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項81に記載の方法。
【請求項86】
コポリマーが電気穿孔、脂質が媒介する輸送、リポソームおよびストレプトリシンO−媒介型細胞透過からなる群から選択される方法により導入される、請求項81に記載の方法。
【請求項87】
目的の抗原性エピトープがガン細胞抗原に由来する、請求項81に記載の方法。
【請求項88】
目的の抗原性エピトープがウイルス抗原に由来する、請求項81に記載の方法。
【請求項89】
Iiタンパク質発現が抑制されているMHCクラスII分子−陽性細胞の表面上の目的の抗原性エピトープに向けられる、哺乳動物の免疫応答を刺激する方法であって;
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞、または目的の抗原性エピトープを発現し、そしてMHCクラスII分子をその細胞表面に発現するように誘導されたMHCクラスII分子−陰性細胞のいずれかを準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えアデノウイルスゲノムを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;
c)工程a)の細胞に、生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する10〜50ヌクレオチド塩基のコポリマーを導入し;そして
d)哺乳動物に、工程c)の細胞または工程c)の細胞に由来する目的の抗原性エピトープを結合した、MHCクラスII分子を導入する、
ことを含んでなる上記方法。
【請求項90】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項89に記載の方法。
【請求項91】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項90に記載の方法。
【請求項92】
タンパク質がMHCクラスIIトランスアクチベーターまたはインターフェロンガンマからなる群から選択される、請求項89に記載の方法。
【請求項93】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項89に記載の方法。
【請求項94】
コポリマーが電気穿孔により導入される、請求項89に記載の方法。
【請求項95】
目的の抗原性エピトープがガン細胞抗原である、請求項89にに記載の方法。
【請求項96】
目的の抗原性エピトープがウイルス抗原である、請求項89にに記載の方法。
【請求項97】
MHCクラスII分子−陽性細胞の表面上に抗原性エピトープを展示する方法であって、
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そして
b)工程a)の細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNAに相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項98】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項97に記載の方法。
【請求項99】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中でトランスフェクトされた細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項97に記載の方法。
【請求項100】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項99に記載の方法。
【請求項101】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項99に記載の方法。
【請求項102】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項97に記載の方法。
【請求項103】
目的の抗原性エピトープがガン細胞抗原である、請求項97に記載の方法。
【請求項104】
目的の抗原性エピトープがウイルスエピトープである、請求項97に記載の方法。
【請求項105】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項97に記載の方法。
【請求項106】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項97に記載の方法。
【請求項107】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項106に記載の方法。
【請求項108】
疾患プロセスの診断または治療のために、目的の抗原性エピトープを単離する方法であって:
a)抗原提示細胞を含むカルチャーを準備し;
b)カルチャー中の抗原提示細胞に、診断または治療が望まれる疾患プロセスと特異的に関連するタンパク質またはタンパク質の部分をコードする発現可能な核酸配列を導入し、;そして
c)工程b)の抗原提示細胞のMHCクラスII分子から抗原性エピトープペプチドを単離する、
ことを含んでなる上記方法。
【請求項109】
疾患プロセスがガン、感染、自己免疫疾患、アレルギーおよび移植片の拒絶からなる群から選択される、請求項108に記載の方法。
【請求項110】
請求項108に記載の方法により単離されるペプチド。
【請求項111】
工程c)の抗原性エピトープペプチドの配列を決定することをさらに含んでなる、請求項108に記載の方法。
【請求項112】
病状に特異的に関連するタンパク質またはポリペプチドを発現する、病状を現すMHCクラスII分子−陰性細胞に融合した抗原提示細胞を含んでなる細胞融合物であって、細胞融合物がさらにIi合成の阻害を含んでなる上記細胞融合物。
【請求項113】
Iiタンパク質発現が抑制されている細胞表面上の目的のガン細胞特異的抗原性エピトープに向けられる哺乳動物の免疫応答を刺激する方法であって;
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陰性細胞を準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;
c)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
d)工程c)の細胞、または工程c)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで哺乳動物を免疫感作する、
ことを含んでなる上記方法。
【請求項114】
a)MHCクラスII分子−陰性細胞中での発現が細胞の表面上にMHCクラスII分子の誘導をもたらす、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクター;および
b)RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物、
を含んでなる悪性哺乳動物細胞。
【請求項115】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的のガン細胞特異的抗原性エピトープを展示する方法であって;
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項116】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的のウイルス特異的抗原性エピトープを展示する方法であって;
a)目的のウイルス特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項117】
識別抗原の発現により細胞型が特徴付けられる、免疫学的応答のために個体の特定の型の細胞を標的化する方法であって:
a)カルチャー中に抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;
c)識別抗原をコードする発現可能な核酸配列を、発現に適する条件下でカルチャー中の細胞に導入し;そして
d)工程c)の細胞を個体に再導入する、
ことを含んでなる上記方法。
【請求項118】
細胞にIi合成のインヒビターを導入することを含んでなる、細胞の小胞体中のMHCクラスII分子への目的の抗原性エピトープの装填を促進する方法。
【請求項119】
Iiタンパク質発現が抑制されている細胞の表面上の目的のガン細胞特異的抗原性エピトープに向けられる、哺乳動物における免疫応答を刺激する方法であって:
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
c)哺乳動物を、工程b)の細胞または工程b)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで免疫感作する、
ことを含んでなる上記方法。
【請求項120】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を含むことを含んでなる、悪性哺乳動物細胞。
【請求項1】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物。
【請求項2】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項3】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項4】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項1に記載の発現可能なリバース遺伝子構築物。
【請求項5】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項4に記載のリバース遺伝子構築物。
【請求項6】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を含む哺乳動物細胞。
【請求項7】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項6に記載の哺乳動物細胞。
【請求項8】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項6に記載の哺乳動物細胞。
【請求項9】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項6に記載の哺乳動物細胞。
【請求項10】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項9に記載の哺乳動物細胞。
【請求項11】
MHCクラスII分子−陽性細胞である請求項6に記載の哺乳動物細胞。
【請求項12】
操作前に個体中に存在する時にはMHCクラスII分子−陰性である請求項6に記載の哺乳動物細胞。
【請求項13】
タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを含み、そのトランスフェクションがMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項12に記載の哺乳動物細胞。
【請求項14】
悪性細胞である、請求項6に記載の哺乳動物細胞。
【請求項15】
ウイルスに感染した細胞である請求項6に記載の哺乳動物細胞。
【請求項16】
自然に存在する抗原提示細胞である請求項6に記載の哺乳動物細胞。
【請求項17】
樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項16に記載の哺乳動物細胞。
【請求項18】
さらに目的の抗原を含んでなる請求項16に記載の哺乳動物細胞。
【請求項19】
目的の抗原が、目的の抗原をコードする発現可能な核酸配列から細胞内で合成される請求項18に記載の哺乳動物細胞。
【請求項20】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的の抗原性エピトープを展示する方法であって:
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そして
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項21】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項20に記載の方法。
【請求項22】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項20に記載の方法。
【請求項23】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項22に記載の方法。
【請求項24】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項22に記載の方法。
【請求項25】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項20に記載の方法。
【請求項26】
目的の抗原性エピトープがガン細胞抗原である請求項20に記載の方法。
【請求項27】
目的の抗原性エピトープがウイルス抗原である請求項20に記載の方法。
【請求項28】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項20に記載の方法。
【請求項29】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項20に記載の方法。
【請求項30】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項29に記載の方法。
【請求項31】
Iiタンパク質発現が抑制されているMHCクラスII分子陽性細胞の表面上の目的の抗原性エピトープに向けられる、哺乳動物における免疫応答を刺激する方法であって:
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
c)哺乳動物を、工程b)の細胞または工程b)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで免疫感作する、
ことを含んでなる上記方法。
【請求項32】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項31に記載の方法。
【請求項33】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中で細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項31に記載の方法。
【請求項34】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項33に記載の方法。
【請求項35】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項33に記載の方法。
【請求項36】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項31に記載の方法。
【請求項37】
目的の抗原性エピトープがガン細胞抗原である請求項31に記載の方法。
【請求項38】
目的の抗原性エピトープがウイルス抗原である請求項31に記載の方法。
【請求項39】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項31に記載の方法。
【請求項40】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項31に記載の方法。
【請求項41】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項40に記載の方法。
【請求項42】
識別抗原の発現により細胞型が特徴付けられる、免疫学的応答のために個体の特定の型の細胞を標的化する方法であって:
a)カルチャー中に抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;そして
c)識別抗原をコードする発現可能な核酸配列を、発現に適する条件下でカルチャー中の細胞に導入する、
ことを含んでなる上記方法。
【請求項43】
抗原提示細胞が樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項42に記載の方法。
【請求項44】
Iiの発現が、細胞カルチャーの細胞に、生理学的条件下で哺乳動物Iiタンパク質をコードするmRNA分子の領域に特異的にハイブリダイズする能力を特徴とする約10〜約50ヌクレオチド塩基のコポリマーを導入することにより阻害され、ここでコポリマーがIi発現を阻害する能力を特徴とする、請求項42に記載の方法。
【請求項45】
Iiの発現が、細胞カルチャーの細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入することにより阻害される、請求項42に記載の方法。
【請求項46】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項45に記載の方法。
【請求項47】
発現可能な核酸配列がプラスミドベクター中に生成される、請求項42に記載の方法。
【請求項48】
プラスミドベクターが、脂質、リポソーム、金粒子、ポリラクチドコグリコライド粒子およびポリアルキルオキシドコポリマーからなる群から選択される媒介物を使用する方法により導入される、請求項47に記載の方法。
【請求項49】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項42に記載の方法。
【請求項50】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項49に記載の方法。
【請求項51】
工程c)の細胞が治療効果のために個体に導入される、請求項42に記載の方法。
【請求項52】
工程a)の末梢血単核細胞が抗原提示細胞を濃縮するために分画される、請求項42に記載の方法。
【請求項53】
工程c)の細胞が治療効果のために個体に導入される、請求項52に記載の方法。
【請求項54】
工程c)の抗原提示細胞を含むカルチャー中に分画された細胞のサブポピュレーションが得られる、請求項42に記載の方法。
【請求項55】
工程c)の細胞が治療効果のために個体に導入される、請求項54に記載の方法。
【請求項56】
識別抗原がガン細胞抗原である、請求項42に記載の方法。
【請求項57】
識別抗原がウイルス抗原である、請求項42に記載の方法。
【請求項58】
免疫応答のために個体の抗原を標的化する方法であって:
a)カルチャー中に自然に存在する抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;そして
c)工程a)の自然に存在する抗原提示細胞のカルチャーに、免疫学的応答のために標的化される抗原をコードする発現可能な核酸配列をに導入する、
ことを含んでなる上記方法。
【請求項59】
抗原提示細胞が樹状細胞、マクロファージ、Bリンパ球およびTリンパ球からなる群から選択される、請求項58に記載の方法。
【請求項60】
工程c)の細胞が治療効果のために個体に導入される、請求項58に記載の方法。
【請求項61】
工程a)の末梢血単核細胞が抗原提示細胞を濃縮するために分画される、請求項58に記載の方法。
【請求項62】
工程c)の細胞が治療効果のために個体に導入される、請求項61に記載の方法。
【請求項63】
工程c)の抗原提示細胞を含むカルチャー中に分画された細胞のサブポピュレーションが得られる、請求項58に記載の方法。
【請求項64】
工程c)の細胞が治療効果のために個体に導入される、請求項63に記載の方法。
【請求項65】
Iiの発現が、細胞カルチャーの細胞に、生理学的条件下で哺乳動物Iiタンパク質をコードするRNA分子の標的に特異的にハイブリダイズする能力を特徴とする約10〜約50ヌクレオチド塩基のコポリマーを導入することにより阻害され、ここでコポリマーがIi発現を阻害する能力を特徴とする、請求項58に記載の方法。
【請求項66】
Iiの発現が、細胞カルチャーの細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入することにより阻害される、請求項58に記載の方法。
【請求項67】
RNA分子が、コード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である請求項51に記載の発現可能なリバース遺伝子構築物である請求項66に記載の方法。
【請求項68】
発現可能な核酸配列がプラスミドベクター中に生成される、請求項58に記載の方法。
【請求項69】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項58に記載の方法。
【請求項70】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項69に記載の方法。
【請求項71】
識別抗原がガン細胞抗原である、請求項58に記載の方法。
【請求項72】
識別抗原がウイルス性である請求項58に記載の方法。
【請求項73】
MHCクラスII分子−陽性細胞であり、そして10〜50ヌクレオチド塩基のコポリマーを含む細胞であって、コポリマーが生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、上記細胞。
【請求項74】
ガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞のいずれかである請求項73に記載の細胞。
【請求項75】
MHCクラスII分子を発現し、そして細胞表面に展示するように誘導されたMHCクラスII分子−陰性細胞であって、細胞がタンパク質をコードする発現可能な核酸配列を含んでなり、その発現がMHCクラスII分子−陰性細胞中で細胞表面上のMHCクラスII分子の誘導をもたらし、細胞がさらに10〜50ヌクレオチド塩基のコポリマーを含んでなり、コポリマーが生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、上記細胞。
【請求項76】
発現可能な核酸配列が、ウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項75に記載の細胞。
【請求項77】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項76に記載の細胞。
【請求項78】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項75に記載の細胞。
【請求項79】
ガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を導く感染源を含む細胞である請求項75に記載の細胞。
【請求項80】
コポリマーが電気穿孔により導入される、請求項75に記載の細胞。
【請求項81】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的の抗原性エピトープを展示する方法であって、
a)MHCクラスII分子−陽性細胞またはMHCクラスII分子を発現するように誘導される細胞のいずれかであり、そして目的の抗原性エピトープを発現する細胞を準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;そして
c)工程a)の細胞に10〜50ヌクレオチド塩基のコポリマーを導入し、このコポリマーは生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する、
ことを含んでなる上記方法。
【請求項82】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項81に記載の方法。
【請求項83】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項82に記載の方法。
【請求項84】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項81に記載の方法。
【請求項85】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項81に記載の方法。
【請求項86】
コポリマーが電気穿孔、脂質が媒介する輸送、リポソームおよびストレプトリシンO−媒介型細胞透過からなる群から選択される方法により導入される、請求項81に記載の方法。
【請求項87】
目的の抗原性エピトープがガン細胞抗原に由来する、請求項81に記載の方法。
【請求項88】
目的の抗原性エピトープがウイルス抗原に由来する、請求項81に記載の方法。
【請求項89】
Iiタンパク質発現が抑制されているMHCクラスII分子−陽性細胞の表面上の目的の抗原性エピトープに向けられる、哺乳動物の免疫応答を刺激する方法であって;
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞、または目的の抗原性エピトープを発現し、そしてMHCクラスII分子をその細胞表面に発現するように誘導されたMHCクラスII分子−陰性細胞のいずれかを準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えアデノウイルスゲノムを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;
c)工程a)の細胞に、生理学的条件下で哺乳動物のIiタンパク質をコードするRNA分子の標的領域に特異的にハイブリダイズする能力を特徴とし、これによりIi発現を阻害する10〜50ヌクレオチド塩基のコポリマーを導入し;そして
d)哺乳動物に、工程c)の細胞または工程c)の細胞に由来する目的の抗原性エピトープを結合した、MHCクラスII分子を導入する、
ことを含んでなる上記方法。
【請求項90】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項89に記載の方法。
【請求項91】
ウイルス発現ベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項90に記載の方法。
【請求項92】
タンパク質がMHCクラスIIトランスアクチベーターまたはインターフェロンガンマからなる群から選択される、請求項89に記載の方法。
【請求項93】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項89に記載の方法。
【請求項94】
コポリマーが電気穿孔により導入される、請求項89に記載の方法。
【請求項95】
目的の抗原性エピトープがガン細胞抗原である、請求項89にに記載の方法。
【請求項96】
目的の抗原性エピトープがウイルス抗原である、請求項89にに記載の方法。
【請求項97】
MHCクラスII分子−陽性細胞の表面上に抗原性エピトープを展示する方法であって、
a)目的の抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;そして
b)工程a)の細胞に、RNA分子がmRNA分子とハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNAに相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項98】
RNA分子がコード配列の翻訳開始部位および約425ヌクレオチドまでを含んでなるmRNA分子の部分に相補的である、請求項97に記載の方法。
【請求項99】
MHCクラスII分子の発現が、工程a)の細胞にタンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入することにより誘導され、その発現がMHCクラスII分子−陰性細胞中でトランスフェクトされた細胞の表面上にMHCクラスII分子の誘導をもたらす、請求項97に記載の方法。
【請求項100】
発現可能な核酸配列がウイルスまたは非ウイルス発現ベクターにより運ばれる、請求項99に記載の方法。
【請求項101】
タンパク質がMHCクラスIIトランスアクチベーターおよびインターフェロンガンマからなる群から選択される、請求項99に記載の方法。
【請求項102】
工程a)の細胞がガン細胞、または感染源の存在がなければ細胞には存在しないタンパク質の合成を支配する感染源を含む細胞である請求項97に記載の方法。
【請求項103】
目的の抗原性エピトープがガン細胞抗原である、請求項97に記載の方法。
【請求項104】
目的の抗原性エピトープがウイルスエピトープである、請求項97に記載の方法。
【請求項105】
リバース遺伝子構築物がプラスミドベクター中に生成される、請求項97に記載の方法。
【請求項106】
リバース遺伝子構築物がウイルスベクター中に生成される、請求項97に記載の方法。
【請求項107】
ウイルスベクターがアデノウイルス、アデノ随伴ウイルス、レンチウイルス、ポックスウイルスおよびレトロウイルスからなる群から選択される、請求項106に記載の方法。
【請求項108】
疾患プロセスの診断または治療のために、目的の抗原性エピトープを単離する方法であって:
a)抗原提示細胞を含むカルチャーを準備し;
b)カルチャー中の抗原提示細胞に、診断または治療が望まれる疾患プロセスと特異的に関連するタンパク質またはタンパク質の部分をコードする発現可能な核酸配列を導入し、;そして
c)工程b)の抗原提示細胞のMHCクラスII分子から抗原性エピトープペプチドを単離する、
ことを含んでなる上記方法。
【請求項109】
疾患プロセスがガン、感染、自己免疫疾患、アレルギーおよび移植片の拒絶からなる群から選択される、請求項108に記載の方法。
【請求項110】
請求項108に記載の方法により単離されるペプチド。
【請求項111】
工程c)の抗原性エピトープペプチドの配列を決定することをさらに含んでなる、請求項108に記載の方法。
【請求項112】
病状に特異的に関連するタンパク質またはポリペプチドを発現する、病状を現すMHCクラスII分子−陰性細胞に融合した抗原提示細胞を含んでなる細胞融合物であって、細胞融合物がさらにIi合成の阻害を含んでなる上記細胞融合物。
【請求項113】
Iiタンパク質発現が抑制されている細胞表面上の目的のガン細胞特異的抗原性エピトープに向けられる哺乳動物の免疫応答を刺激する方法であって;
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陰性細胞を準備し;
b)工程a)の細胞に、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクターを導入し、その発現がMHCクラスII分子−陰性細胞中で細胞の表面にMHCクラスII分子の誘導をもたらし;
c)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害するヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
d)工程c)の細胞、または工程c)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで哺乳動物を免疫感作する、
ことを含んでなる上記方法。
【請求項114】
a)MHCクラスII分子−陰性細胞中での発現が細胞の表面上にMHCクラスII分子の誘導をもたらす、タンパク質をコードする発現可能な核酸配列を含んでなる組換えベクター;および
b)RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物、
を含んでなる悪性哺乳動物細胞。
【請求項115】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的のガン細胞特異的抗原性エピトープを展示する方法であって;
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項116】
Iiタンパク質発現が抑制されるMHCクラスII分子−陽性細胞の表面上に目的のウイルス特異的抗原性エピトープを展示する方法であって;
a)目的のウイルス特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入する、
ことを含んでなる上記方法。
【請求項117】
識別抗原の発現により細胞型が特徴付けられる、免疫学的応答のために個体の特定の型の細胞を標的化する方法であって:
a)カルチャー中に抗原提示細胞を含む個体の末梢血単核細胞を準備し;
b)工程a)のカルチャーの抗原提示細胞に、Ii合成のインヒビターを導入し;
c)識別抗原をコードする発現可能な核酸配列を、発現に適する条件下でカルチャー中の細胞に導入し;そして
d)工程c)の細胞を個体に再導入する、
ことを含んでなる上記方法。
【請求項118】
細胞にIi合成のインヒビターを導入することを含んでなる、細胞の小胞体中のMHCクラスII分子への目的の抗原性エピトープの装填を促進する方法。
【請求項119】
Iiタンパク質発現が抑制されている細胞の表面上の目的のガン細胞特異的抗原性エピトープに向けられる、哺乳動物における免疫応答を刺激する方法であって:
a)目的のガン細胞特異的抗原性エピトープを発現するMHCクラスII分子−陽性細胞を準備し;
b)工程a)の細胞に、RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を導入し;そして
c)哺乳動物を、工程b)の細胞または工程b)の細胞に由来する目的の抗原性エピトープと複合体を形成したMHCクラスII分子のいずれかで免疫感作する、
ことを含んでなる上記方法。
【請求項120】
RNA分子がmRNA分子にハイブリダイズする能力を有し、これによりmRNA分子の翻訳を阻害する、ヒトIiタンパク質をコードするmRNA分子に相補的なRNA分子をコードするDNA分子を含んでなる発現可能なリバース遺伝子構築物を含むことを含んでなる、悪性哺乳動物細胞。
【図1】

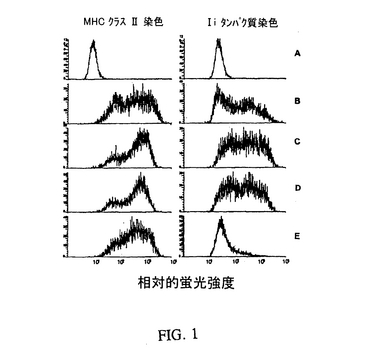
【図2】
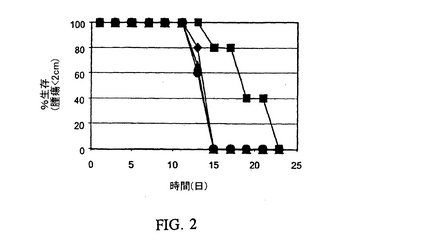
【図3】
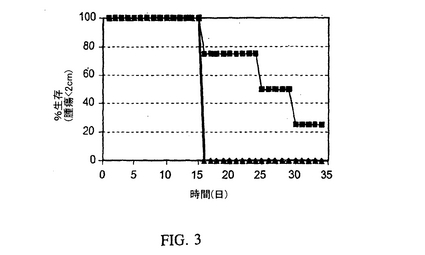
【図4】
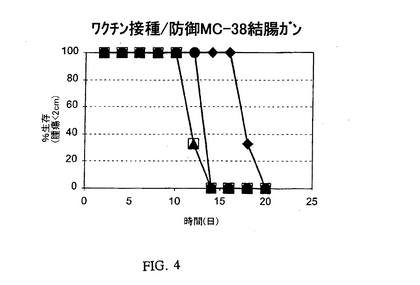
【図5】
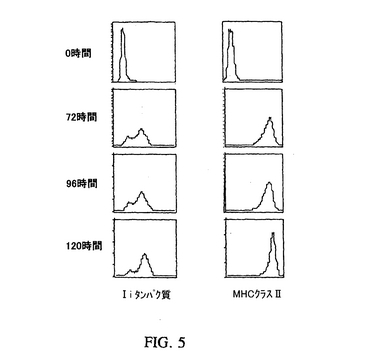
【図6】
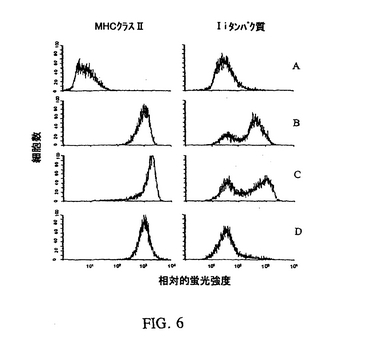
【図7】
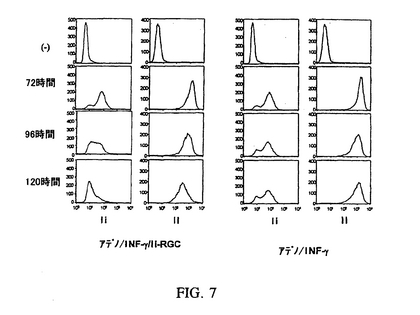
【図8】
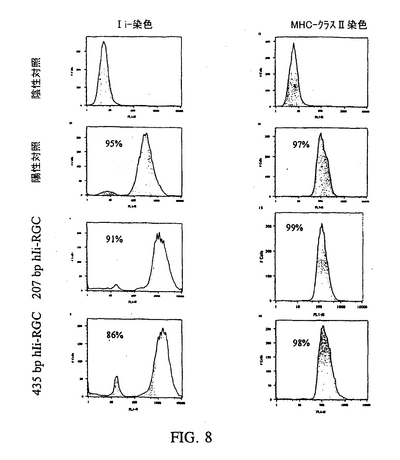
【図9】
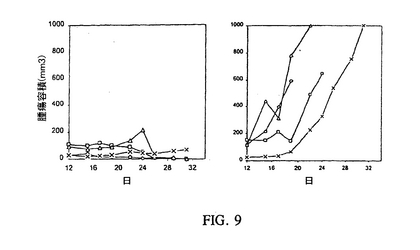
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2006−506949(P2006−506949A)
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願番号】特願2003−586173(P2003−586173)
【出願日】平成15年4月18日(2003.4.18)
【国際出願番号】PCT/US2003/012159
【国際公開番号】WO2003/089453
【国際公開日】平成15年10月30日(2003.10.30)
【出願人】(504386565)アンテイジエン・エクスプレス・インコーポレーテツド (3)
【Fターム(参考)】
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願日】平成15年4月18日(2003.4.18)
【国際出願番号】PCT/US2003/012159
【国際公開番号】WO2003/089453
【国際公開日】平成15年10月30日(2003.10.30)
【出願人】(504386565)アンテイジエン・エクスプレス・インコーポレーテツド (3)
【Fターム(参考)】
[ Back to top ]