
哺乳類βアクチンプロモーターを利用した発現系
【課題】組換えタンパク質を効率よく動物細胞に産生させる技術において、動物細胞発現プロモーターよりも強力なプロモーターと、それを利用したベクターの提供。
【解決手段】CMV(サイトメガウイルス)エンハンサーと哺乳類βアクチンプロモーター、もしくはWoodchuck Hepatitis Virusゲノム配列の転写後調節領域(WPRE)と哺乳類βアクチンプロモーターを組み合わせたハイブリッドプロモーター、さらに、このβアクチンプロモーターは、トランスアクティベーターである癌遺伝子産物のRasの同時発現によって、活性が高められる。
【解決手段】CMV(サイトメガウイルス)エンハンサーと哺乳類βアクチンプロモーター、もしくはWoodchuck Hepatitis Virusゲノム配列の転写後調節領域(WPRE)と哺乳類βアクチンプロモーターを組み合わせたハイブリッドプロモーター、さらに、このβアクチンプロモーターは、トランスアクティベーターである癌遺伝子産物のRasの同時発現によって、活性が高められる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、組換えタンパク質の産生における、より強力なプロモーターおよび、それらを保持するベクターの開発に関する。
【背景技術】
【0002】
近年、多くのバイオ製剤が輩出されているが、こうした医薬品の多くが動物細胞に遺伝子を導入し、組換えタンパク質として産生させたものである。こうした組換えタンパク質を効率よく動物細胞に産生させる技術は、バイオ製剤の低コスト化につながり、患者への安定な供給を約束する。
【0003】
従来は、EF1α(elongation factor 1)プロモーターやSRαプロモーターなどが、そうした発現ベクターに用いられてきた(特許文献1, 非特許文献1,2)。また、最近ではさらに発現の改良が進み、CAGプロモーター(ヒトCMVエンハンサーにニワトリのβアクチンプロモーターを組み合わせたもの)や、CEFプロモーター(ヒトCMVエンハンサーにヒトEF1αプロモーター)などの強力なプロモーターが作られ、こうした用途に使われるようになってきた(非特許文献3〜5)。
【0004】
しかしながら、さらに強力なプロモーターを保持するベクターの開発の必要性はいまなお存在し、これによりバイオ製剤のより一層の低コスト化や安定な供給に大きく貢献できるものと考えられる。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】WO92/19759
【非特許文献】
【0006】
【非特許文献1】Mol. Cell. Biol., Vol.8 (1), p.466-472, 1988
【非特許文献2】Gene, Vol.87(2), p.291-294, 1990
【非特許文献3】Gene, Vol.272, p.149-156, 2001
【非特許文献4】Analytical Biochemistry, Vol.247, p.179-181, 1997
【非特許文献5】Gene, Vol.108, p.193-200, 1991
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、このような状況に鑑みてなされたものであり、その目的は既存の動物細胞発現プロモーターよりも強力なプロモーターと、それを利用したベクターの提供である。また、本発明は、該ベクターを利用した、所望タンパク質の製造方法および所望のDNAの発現方法の開発をも目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記の課題を解決すべく、様々な遺伝子のプロモーターやエンハンサーの組み合わせによるプロモーター活性の比較、検討を行った。
【0009】
まず、NCBI、及びJackson laboratoryで公開されているマウスゲノム情報より、マウスβアクチンのシーケンス情報を得て、PCRによってマウスβアクチンの5'領域を増幅した。増幅したPCR産物をpGEM-T-Easy vectorにクローニングし、シーケンスすることによって塩基配列を確認した。その後、PCRによりマウスβアクチンのプロモーター領域のみを増幅し、pGL3-BasicのBgl II-Hind IIIサイトに組み込んだ。さらにこのベクターに、ネオマイシン耐性遺伝子を導入し、pmAct-Luc-neoとした。
【0010】
Woodchuck hepatitis virusゲノム配列の転写後調節領域(WPRE)をPCRにより増幅し、反応産物をpGEM-T-Easy vectorにクローニングし、塩基配列を決定した。さらにこのpGEM-T/WPREをXbaIで消化し、得られたWPRE断片をpmAct-Luc-neoのXbaI部位に挿入し、pmAct-WPRE-Luc-neoを作製した。
【0011】
次にpGL3-BasicベクターのMCS(multi cloning site : 配列5位〜53位)にCMVエンハンサー領域を組み込むことによって発現ベクターのpmAct-Luc-neoを、あるいはさらにヒトCMVエンハンサー-マウスβアクチンプロモーターを組み込むことによって発現ベクターのphCMV-mAct-Luc-neoを完成させた(図1)。
【0012】
比較対象となるベクターとして、ヒトEF1αプロモーターはDHFR-ΔE-RVh-PM1-f(参考文献 WO92/19759)よりCMVエンハンサーと共にpGLNベクターのMCSに組み込むことで、pCEF-Luc-neoを作製した(図1)。
【0013】
これらのベクターをそれぞれCHO細胞へ導入し、CO2インキュベーター内で2日間培養して、ルシフェラーゼ活性をLuciferase Assay Systemを用いて測定した。これらの測定の結果より、作製したpmAct-Luc-neoが他のベクターよりも有意に活性が高いことが明らかになった(図2)。以上のことから、本発明であるマウスβアクチンプロモーターは、既存のCEFプロモーターよりも強力であることが示された。さらに、WPREエレメントの付加や、あるいはCMVエンハンサーを付加することによって、マウスβアクチンのプロモーター活性が有意に亢進することも明らかになった(図3)。
【0014】
さらに、トランスアクティベーターを同時に発現させた場合の効果について検討した。マウスc-H-ras遺伝子、マウス活性型c-H-ras及びヒト活性型K-rasのクローニングを行い、pCXNベクターに組み込んで、それぞれをpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型K-ras)とした。様々な割合で2種類、あるいは3種類のベクターをCHO細胞に導入し、ルシフェラーゼ活性を測定した。これらの結果より、pmAct-Luc-neoがpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型c-K-ras)の存在によって、プロモーター活性が亢進することが明らかになった(図4)。以上のことから、本発明のマウスβアクチンプロモーターは、癌遺伝子産物のRas(活性型、野生型、H-Ras、K-Rasを問わない)によってより強力な活性を付与されることが示された。
【0015】
即ち、本発明者らは、CMVエンハンサーと哺乳類βアクチンプロモーター、もしくはWoodchuck Hepatitis Virusゲノム配列の転写後調節領域(WPRE)と哺乳類βアクチンプロモーターの組み合わせが、強力なプロモーターになりうることを見出した。さらに、これらのプロモーターを保持した発現系は、トランスアクティベーターである癌遺伝子産物のRasの同時発現によって、さらに発現活性が高められることを見出し、本発明の完成に至った。
【0016】
本発明は、より具体的には、以下の〔1〕〜〔37〕に関する。
〔1〕 エンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物。
〔2〕 エンハンサーがサイトメガロウイルス(CMV)である〔1〕に記載のDNA構築物。
〔3〕 エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である〔1〕に記載のDNA構築物。
〔4〕 哺乳類βアクチンプロモーターがげっ歯類βアクチンプロモーターである、〔1〕から〔3〕のいずれかに記載のDNA構築物。
〔5〕 CMVエンハンサーが配列番号:4に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、〔2〕に記載のDNA構築物。
〔6〕 Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)が配列番号:3に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、〔3〕に記載のDNA構築物。
〔7〕 〔1〕から〔6〕のいずれかに記載のDNA構築物を含むベクター。
〔8〕 哺乳類βアクチンプロモーターの下流に所望のDNAが機能的に結合されたDNAを有する〔7〕に記載のベクター。
〔9〕 トランスアクティベーターをコードするDNAを発現可能に保持する、〔7〕または〔8〕に記載のベクター。
〔10〕 トランスアクティベーターが癌遺伝子産物である〔9〕に記載のベクター。
〔11〕 癌遺伝子産物がRasである、〔10〕に記載のベクター。
〔12〕 所望のDNAが、所望のタンパク質をコードするDNAである、〔8〕から〔11〕のいずれかに記載のベクター。
〔13〕 〔8〕から〔12〕のいずれかに記載のベクターを保持する細胞。
〔14〕 〔8〕から〔12〕のいずれかに記載のベクターを保持し、癌遺伝子が活性化している細胞。
〔15〕 癌遺伝子が活性化している細胞が、トランスアクティベーターをコードする遺伝子を含むベクターが導入された細胞である〔14〕記載の細胞。
〔16〕 癌遺伝子が活性化している細胞が、癌化している細胞である〔14〕記載の細胞。
〔17〕 哺乳動物細胞である、〔13〕から〔16〕のいずれかに記載の細胞。
〔18〕 げっ歯類細胞である、〔17〕に記載の細胞。
〔19〕 βアクチンプロモーターと同じ動物目であることを特徴とする〔13〕から〔18〕のいずれかに記載の細胞。
〔20〕 βアクチンプロモーターと同じ動物種であることを特徴とする〔19〕に記載の細胞。
〔21〕 〔8〕から〔12〕のいずれかに記載のベクターが導入されたトランスジェニック非ヒト動物。
〔22〕 〔8〕から〔12〕のいずれかに記載のベクターが導入された全能性細胞。
〔23〕 〔12〕に記載のベクターを保持する細胞を培養し、培養した細胞又は培地から発現させたタンパク質を回収することを含む、所望のタンパク質の製造方法。
〔24〕 培地にトランスアクティベーターを添加することを含む、〔23〕に記載の方法。
〔25〕 〔8〕から〔12〕のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
〔26〕 〔8〕から〔12〕のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
〔27〕 〔8〕に記載のベクターおよびトランスアクティベーターをコードするDNAを発現可能に保持するベクターを、〔8〕に記載のベクター上のβアクチンプロモーターと同種由来の宿主細胞に導入することを含む、宿主細胞における所望のDNAを発現させる方法。
〔28〕 宿主細胞が哺乳動物細胞である、〔25〕から〔27〕のいずれかに記載の方法。
〔29〕 宿主細胞がげっ歯類細胞である、〔25〕から〔27〕のいずれかに記載の方法。
〔30〕 宿主細胞と同じ動物目由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
〔31〕 宿主細胞と同じ動物種由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
〔32〕 エンハンサーをさらに組み込むことを特徴とする〔30〕もしくは〔31〕に記載の方法。
〔33〕 エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である〔32〕に記載の方法。
〔34〕 エンハンサーがCMVエンハンサーである〔32〕に記載の方法。
〔35〕 トランスアクティベーターをコードする遺伝子を組み込むことを特徴とする〔30〕から〔34〕のいずれかに記載の方法。
〔36〕 宿主細胞が哺乳動物細胞である〔30〕から〔35〕いずれかに記載の方法。
〔37〕 宿主細胞がげっ歯類細胞である〔30〕から〔35〕いずれかに記載の方法。
【図面の簡単な説明】
【0017】
【図1】実験に使用したベクターを示した図である。
【図2】既存のプロモーターであるCEFプロモーターとマウスβアクチンプロモーターを比較した図である。
【図3】マウスβアクチンプロモーターに対するWPRE、CMVエンハンサーの効果を示した図である。
【図4】マウスRas蛋白質のマウスβアクチンプロモーターに対する効果を示した図である。
【発明を実施するための形態】
【0018】
本発明はエンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物、及び該DNA構築物を含むベクターを提供する。本発明のDNA構築物やベクターは、所望のDNAの発現(例えば、所望のタンパク質をコードするDNAの発現、アンチセンスDNAの発現、dsRNAをコードするDNAの発現など)に用いることができる。
【0019】
本発明において、哺乳類βアクチンプロモーターとエンハンサーとが「機能的に結合した」とは、プロモーター活性が高まるように、本発明のプロモーター活性を有する哺乳類βアクチンプロモーターとエンハンサーとが結合していることをいう。従って、哺乳類βアクチンプロモーターとエンハンサーの距離が離れていたり、間に他の遺伝子が挿入された場合であっても、哺乳類βアクチンプロモーターのプロモーター活性が高まるのであれば、上記の「機能的に結合した」の意に含まれる。エンハンサーは哺乳類βアクチンプロモーターの上流下流どちらに存在していても良い。
【0020】
また、哺乳類βアクチンプロモーターと所望のDNAが「機能的に結合した」とは、哺乳類βアクチンプロモーターの活性化により、所望のDNAの発現が誘導されるように、哺乳類βアクチンプロモーターと所望のDNAとが結合していることをいう。該DNAと哺乳類βアクチンプロモーターとの間には、該DNAの発現の誘導が起こり得る限り、任意のDNA配列を有していても良い。本発明における「DNAの発現」には、転写および翻訳の双方が含まれる。また、所望のDNAが、所望のタンパク質をコードするDNAであってもよい。
【0021】
本発明においてエンハンサーとは、転写により生成するメッセンジャーRNA(mRNA)の量を結果的に増加させるものであれば限定されない。エンハンサーはプロモーターの作用を促進する効果を持つ塩基配列であり、一般的には100bp前後からなるものが多い。エンハンサーは配列の向きにかかわらず転写を促進することができる。又、エンハンサー自体はプロモーター活性を持っていないのであるが、エンハンサーは数千塩基対も離れた所から転写を活性化することができる。さらに、エンハンサーは転写される遺伝子領域の上流、下流、および遺伝子内にも存在し、転写を活性化することができる。また、上述したように本発明のエンハンサーは、転写により生成するmRNAの量を増加させるものであれば限定されないので、mRNAの転写後にmRNAの分解を阻害したり、mRNAを安定化することによって、細胞内に存在するmRNAの量を増加させ、アミノ酸の翻訳効率を上げるものも本発明のエンハンサーに含まれる。
【0022】
本発明で用いられるエンハンサーは1種類でもよく、又、2つ以上の同一のエンハンサーを複数用いたり、あるいは異なる複数のエンハンサーを組み合わせて用いても良い。
【0023】
本発明において用いられるエンハンサーとしては、WPRE、CMVエンハンサー、HTLV-IのLTRに存在するR-U5'セグメント(Mol. Cell. Biol., Vol.8(1), p.466-472, 1988 )、SV40エンハンサー、ウサギβグロビンの第二エクソンと第三エクソン間に存在するイントロン配列(Proc. Natl. Acad. Sci. USA., Vol.78(3), p.1527-31, 1981)、ヒト成長ホルモンのゲノム領域(J Immunol., Vol. 155(3), p.1286-95, 1995)などを挙げることができるが、より好ましいものはWPREまたはCMVエンハンサーである。
【0024】
WPREは、Woodchuck hepatitis virusゲノム配列(GenBank Accession No. J04514)に存在するHBVPRE(human hepatitis B virus posttranscriptional reguratory element)に類似した領域であり、ゲノム配列の1093番目から1684番目の 592塩基が転写後調節領域であることが報告されている(Journal of Virology, Vol.72, p.5085-5092, 1998)。その後、レトロウイルスベクターを用いた解析により、WPREは目的遺伝子3'末端の非翻訳領域に挿入することにより、5〜8倍高い蛋白産生が得られることが示された。また、WPREの導入により、mRNAの分解が抑制されることも報告された(Journal of Virology, Vol.73, p.2886-2892, 1999)。WPREのようにmRNAの安定化により、アミノ酸の翻訳効率を上げるものも広義のエンハンサーと考えられる。本発明において用いられるWPREとしては、配列番号:3に記載の塩基配列からなるDNAが、より好ましい。
【0025】
また、CMVエンハンサーもWPREと同様の目的で用いることができる。本発明で用いられるサイトメガロウイルスはヒトに感染するサイトメガロウイルス(ヒトサイトメガロウイルス)でもよく、又、非ヒト動物に感染するサイトメガロウイルスでもよい。非ヒト動物に感染するサイトメガロウイルスとしては、例えば、げっ歯類に感染するサイトメガロウイルス(例えば、マウスサイトメガロウイルスなど)を用いることが可能である。CMVエンハンサーは公知の配列を用いることが可能であり、例えばGenBank Accession No. X17403(ヒトサイトメガロウイルス)、GenBank Accession No. L06816(マウスサイトメガロウイルス)に記載のCMVエンハンサーを用いることが可能である。また、CMVエンハンサーは市販されている発現ベクター(ex. ストラタジーン社のpCMV-Script、インビトロジェン社のpcDNA3.1)の多くに、CMVプロモーターの一部として組み込まれている。本発明において用いられるCMVエンハンサーとしては、配列番号:4に記載の塩基配列からなるDNAが、より好ましい。
【0026】
プロモーターはDNAを鋳型に転写を開始するDNA上の特定の塩基配列であり、一般的に、共通の配列を持つ。例えば、大腸菌などの原核生物では、通常、転写開始部位の-10塩基対部位にTATAATG配列を、-35塩基対部位にTTGACA配列を持つ。又、真核生物では、通常、-20塩基対部位にTATAボックスを持つ。
【0027】
本発明で用いられるβアクチンプロモーターとしては、哺乳類βアクチンプロモーターを用いることができる。哺乳類βアクチンプロモーターは、ヒトβアクチンプロモーターやげっ歯類βアクチンプロモーターなど、どのような哺乳類由来のβアクチンプロモーターを用いてもよいが、好ましくはげっ歯類βアクチンプロモーターを用いることができる。げっ歯類βアクチンプロモーターとしては、例えば、マウスβアクチンプロモーター(GenBank Accession No. NT_039317)、ラットβアクチンプロモーター(GenBank Accession No. NW_042778)などを用いることができる。本発明において用いられる哺乳類βアクチンプロモーターとしては、配列番号:1もしくは2に記載の塩基配列からなるDNAが、より好ましい。
【0028】
げっ歯類としては、ネズミ下目、リス下目、ビーバー下目、パラミス下目、デバネズミ下目、ヤマアラシ下目、デンジクネズミ下目などに属する動物を挙げることができ、より具体的には、マウス、ラット、ハムスターなどを挙げることができる。
【0029】
ベクターは、一般的には、所望の遺伝子を宿主に導入して、所望の遺伝子を増幅や発現させる為の運搬体DNA分子のことをいい、好ましくは、公知の制限酵素部位を持ち、栄養要求性であり、宿主内で複製できる能力を持つ。ベクターは通常、プロモーター、エンハンサー、ターミネーター、SD配列、翻訳開始コドン、翻訳終止コドン、複製開始点などを含むことができる。ベクターには、必要に応じて、ベクターが導入された細胞を選択し得る選択マーカーなどをさらに保持させることができる。選択マーカーとしては、アンピシリン、テトラサイクリン、カナマイシン、クロラムフェニコール、ネオマイシン、ハイグロマイシン、ピューロマイシン、ゼオシンのような薬剤耐性遺伝子、ガラクトシダーゼなどの酵素活性を指標に選択し得るマーカー、あるいは、GFPなどの蛍光発光などを指標に選択し得るマーカーなどが挙げられる。また、EGFレセプター、B7-2などの表面抗原を指標に選択し得る選択マーカーなども用いてもよい。このように選択マーカーを用いることにより、該ベクターが導入された細胞、すなわち、本発明のベクターが導入された細胞のみを選択することが可能となる。又、ベクターには、ポリペプチド分泌のためのシグナル配列が含まれていてもよい。ポリペプチド分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(J. Bacteriol., Vol.169(9), p.4379-4383, 1987)を使用すればよい。
【0030】
本発明で用いられるベクターは特に制限されず、いかなるベクターを用いてもよい。
具体的には、哺乳動物由来のベクター(例えば、pcDNA3 (インビトロジェン社製)や、pEGF-BOS (Nucleic Acids. Res., 18(17), p.5322, 1990)、pEF 、pCDM8、pCXN、昆虫細胞由来のベクター(例えば「Bac-to-BAC baculovairus expression system」(インビトロジェン社製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来のベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来のベクター(例えば、pZIPneo)、酵母由来のベクター(例えば、「Pichia Expression Kit」(インビトロジェン社製)、pNV11 、SP-Q01)、枯草菌由来のベクター(例えば、pPL608、pKTH50)、大腸菌ベクター(M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Script)、などが挙げることができる。本発明においては、哺乳動物細胞内で発現可能なベクターを用いることが好ましく、又、発現ベクターを用いることが好ましい。
【0031】
宿主細胞がCHO細胞である場合、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする為に、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pCHOIなど)を導入し、メトトレキセート(MTX)により増幅させることができる。
【0032】
さらに本発明のベクターには、トランスアクティベーターをコードするDNAが含まれていてもよい。トランスアクティベーターは遺伝子の転写を活性化するトランス作動性因子であり、トランス活性化因子ともいわれる。トランスアクティベーターには、DNA結合能を持ちシスエレメントへの結合を介して直接転写を促進するものや、DNA結合能を持たず、他の因子を活性化することで間接的に転写を促進するもの、等が知られている。本発明で使用されるトランスアクティベーターは特に限定されず、どのようなトランスアクティベーターを用いてもよい。トランスアクティベーターの具体的な例としては、アデノウイルスのE1A、ヒト免疫不全ウイルスのTat、ヒトT細胞白血病ウイルスのTax、癌遺伝子産物などを挙げることができるが、本発明においては癌遺伝産物を用いることが好ましい。癌遺伝子は動物の正常細胞に内在し、その活性化突然変異により細胞の癌化を誘導する遺伝子群の総称である。ウイルスがもつ癌遺伝子はウイルス性癌遺伝子、その由来である正常細胞の遺伝子は細胞性癌遺伝子と呼ばれている。癌遺伝子としては例えば、src、yes、fgr、fms、erbB、abl、kit、crk、mos、raf、sis、ras、myc、fos、junなどを挙げることができる。ヒト活性型H-Rasがヒトβアクチンプロモーターを組み込んだ発現ベクターの発現効率を亢進することが報告されているが(Cytotechnology, Vol.16, p.167-178, 1994)、本発明においてもRasを用いることが好ましい。RasにはK-ras、H-ras、N-ras、c-rasがあり、さらに、rho遺伝子ファミリー、rab遺伝子ファミリーを含む場合には、rasスーパーファミリーと呼ばれている。本発明においてはH-rasを好適に用いることができる。トランスアクティベーターとしてRasを用いる場合、Rasは野生型でも活性型でもよい。本発明において用いられるH-rasとしては、配列番号:5又は6のいずれかのDNA又は配列番号:7又は8のアミノ酸配列をコードするDNAが、より好ましい。
【0033】
さらに、本発明のベクターを、時期特異的に遺伝子を発現するよう設計してもよい。時期特異的に遺伝子を発現させる方法としては、例えば、Cre-loxPシステムなどの部位特異的組換えを用いた方法を挙げることができる。
【0034】
Creは、2個のloxP配列間の部位特異的組換えを仲介する、E.coliバクテリオファージP1由来の組換え酵素である。LoxP配列は、Creが結合するための認識配列として働く2個の13bpの逆方向反復配列に両側を挟まれた8bpのスペーサー領域からなる。所望の遺伝子の発現が抑制されるようにloxP配列をベクターに導入しておけば、Creを投与してloxP配列を除去することにより、好きな時期に所望の遺伝子を発現させることが可能である。loxP配列の挿入部位は、プロモーターと所望の遺伝子の間など、所望の遺伝子の発現が抑制される部位であればよい。又、Creの投与は、Cre自体を投与してもよいし、Creをコードする遺伝子を投与してもよい。トランスジェニック動物などの動物でCre-loxPシステムを用いる場合には、例えば、Creを発現するアデノウイルスを感染させることなどにより、所望の遺伝子を発現させることができる(Nucleic. Acids. Res., Vol.23(19), p.3816-3821, 1995)。このような酵素を利用した部位特異的組換えとしては、他にFlp-FRT、Zygosaccharomyces rouxii pSR1、レソルバーゼ-rfsF、ファージMu Ginなどを挙げることができる。
【0035】
さらに、上述した時期特異的発現システム以外に、テトラサイクリンなどの抗生物質を利用した方法や、エクジソンなどのホルモンを利用した方法などを用いることが可能である。これらの方法は当業者に公知であり、市販されているキットを用いることが可能である(エクジソン発現システム(インビトロジェン社)、テトラサイクリン発現システム(クロンテック社)、など)。
【0036】
また、本発明は、本発明のベクターが導入された宿主細胞を提供する。本発明の宿主細胞は、例えば、所望のタンパク質の製造や発現のための産生系として使用することができる。本発明のベクターが導入される宿主細胞としては特に制限はなく、株化された細胞、初代継代細胞、動物個体、受精卵など細胞種を問わない。本発明の宿主細胞としては、例えば、種々の動物細胞(例えば、哺乳動物細胞)や大腸菌などを用いることが可能である。真核細胞を使用する場合、例えば、動物細胞、植物細胞、真菌細胞を宿主に用いることができる。動物細胞としては、ヒト、マウス、ハムスターなどの哺乳類細胞(例えば、CHO、COS、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero)、両生類細胞(例えばアフリカツメガエル卵母細胞(Nature, Vol.291, p.358-360, 1981))、あるいは昆虫細胞(例えば、Sf9、Sf21、Tn5)などが知られている。CHO細胞としては、特に、DHFR遺伝子を欠損したCHO細胞であるdhfr-CHO(Proc. Natl. Acad. Sci. USA, Vol.77, p.4216-4220, 1980)やCHO K-1(Proc. Natl. Acad. Sci. USA, Vol.60, p.1275, 1968)を好適に使用することができる。植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞がポリペプチド生産系として知られており、これをカルス培養すればよい。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属、例えば、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、糸状菌、例えば、アスペルギルス(Aspergillus)属、例えば、アスペルギルス・ニガー(Aspergillus niger)が知られている。原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、大腸菌(E. coli)、例えば、JM109、DH5α、HB101等が挙げられ、その他、枯草菌が知られている。本発明において好ましい宿主細胞としては、動物細胞を挙げることができ、さらに好ましくは哺乳動物細胞、特に好ましくはげっ歯類細胞(例えばCHO細胞)を挙げることができる。
【0037】
又、本発明の宿主細胞として、癌遺伝子が活性化している細胞を用いることも可能である。癌遺伝子が活性化している細胞とは、正常細胞と比較して癌遺伝子が多量に発現している細胞、または変異した癌遺伝子が発現している細胞などをいう。
【0038】
癌遺伝子が活性化している細胞は、人為的に癌遺伝子が活性化された細胞でも非人為的に癌遺伝子が活性化された細胞でもよい。人為的に癌遺伝子が活性化された細胞の具体的な例としては、癌遺伝子(野生型または活性型)を含むベクターが導入された細胞や、癌遺伝子に人為的に変異を導入した細胞などを挙げることができる。非人為的に癌遺伝子が活性化された細胞の具体的な例としては、癌化した細胞(ex. T24ヒト膀胱癌細胞 Nature, Vol.302, p.33-37, 1983)などを挙げることができる。癌遺伝子の活性化のメカニズムとしては、プロモーター・インサーション、点突然変異、遺伝子増幅、転座などによると考えられている。
【0039】
また、本発明のベクターの上記細胞への導入方法は、当業者においては、細胞の種類により適宜選択することができる。例えば、哺乳動物細胞への導入では、リン酸カルシウム法(Virology, Vol.52, p.456, 1973)、DEAEデキストラン法、カチオニックリボソームDOTAP(ロッシュ・ダイアグノスティックス社製)を用いた方法、エレクトロポレーション法(Nucleic Acids Res., Vol.15, p.1311, 1987) 、リポフェクション法(J. Clin. Biochem. Nutr., Vol.7, p.175, 1989)、ウィルスによる感染導入方法(Sci.Am., p.34, 1994) 、パーティクルガンなどから選択することができ、植物細胞への導入では、エレクトロポレーション法(Nature, Vol.319, p.791, 1986) 、ポリエチレングリコール法(EMBO J., Vol.3, p.2717, 1984) 、パーティクルガン法(Proc. Natl. Acad. Sci. USA, Vo l.85, p.8502, 1988) 、アグロバクテリュウムを介した方法(Nucleic. Acids Res., Vol.12, p.8711, 1984) 等により行うことができる。
【0040】
また、Trans IT(タカラ社)、PolyFect Transfection Reagent(キアゲン社)、LipofectAMINE(インビトロジェン社)などのキットを利用してもよい。さらに、本発明の宿主細胞は、本発明のベクター以外に、トランスアクティベーターをコードするDNAを有するベクターをさらに含んでいてもよい。
【0041】
本発明はさらに本発明のベクターを用いた所望のタンパク質の製造方法及び所望のDNAを発現させる方法に関する。タンパク質製造のための産生系は、in vitroおよびin vivo の産生系がある。in vitroの産生系としては、真核細胞を使用する産生系や原核細胞を使用する産生系が挙げられる。例えば、上述した宿主細胞をin vitroで培養することにより所望のタンパク質が得られる。培養は、公知の方法に従い行うことができる。例えば、動物細胞の培養液として、例えば、DMEM、MEM、RPMI1640、IMDM、F10培地、F12培地などを使用することができる。その際、牛胎児血清(FCS)等の血清補液を併用することもできるし、無血清培養してもよい。さらに、トランスアクティベーターを培地に添加してもよい。培養時のpHは、約6〜8であるのが好ましい。培養は、通常、約30〜40℃で約15〜200時間行い、必要に応じて培地の交換、通気、攪拌を加える。培養条件は使用する細胞の種類によって異なるので、当業者は適宜好適な条件を決定することができる。例えばCHO細胞であれば通常、気相のCO2濃度が0−40%、好ましくは、2−10%の雰囲気下、30−39℃、好ましくは、37℃程度で、1−14日間培養すればよい。また、動物細胞培養用の各種の培養装置としては、例えば発酵槽型タンク培養装置、エアーリフト型培養装置、カルチャーフラスコ型培養装置、スピンナーフラスコ型培養装置、マイクロキャリアー型培養装置、流動層型培養装置、ホロファイバー型培養装置、ローラーボトル型培養装置、充填槽型培養装置等を用いて培養することができる。
【0042】
一方、in vivoでタンパク質を産生させる系としては、例えば、動物を使用する産生系や植物を使用する産生系が挙げられる。これらの動物又は植物に目的とするDNAを導入し、動物又は植物の体内でポリペプチドを産生させ、回収する。本発明における「宿主」とは、これらの動物、植物を包含する。動物を使用する場合、哺乳類動物、昆虫を用いる産生系がある。哺乳類動物としては、ヤギ、ブタ、ヒツジ、マウス、ウシを用いることができる(Vicki Glaser, SPECTRUM Biotechnology Applications, 1993)。また、哺乳類動物を用いる場合、トランスジェニック動物を用いることができる。例えば、所望のタンパク質をコードするDNAを、ヤギβカゼインのような乳汁中に固有に産生されるポリペプチドをコードする遺伝子との融合遺伝子として調製する。次いで、この融合遺伝子を含むDNA断片をヤギの胚へ注入し、この胚を雌のヤギへ移植する。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、所望のタンパク質を得ることができる。トランスジェニックヤギから産生されるポリペプチドを含む乳汁量を増加させるために、適宜ホルモンをトランスジェニックヤギに使用してもよい(Bio/Technology, Vol.12, p.699-702, 1994)。また、昆虫としては、例えばカイコを用いることができる。カイコを用いる場合、所望のタンパク質をコードするDNAを挿入したバキュロウィルスをカイコに感染させることにより、このカイコの体液から所望のタンパク質を得ることができる(Nature, Vol.315, p.592-594, 1985)。
【0043】
さらに、植物を使用する場合、例えばタバコを用いることができる。タバコを用いる場合、所望のタンパク質をコードするDNAを植物発現用ベクター、例えばpMON 530に挿入し、このベクターをアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)のようなバクテリアに導入する。このバクテリアをタバコ、例えば、ニコチアナ・タバカム(Nicotiana tabacum)に感染させ、本タバコの葉より所望のタンパク質を得ることができる(Eur. J. Immunol., Vol.24, p.131-138, 1994)。
【0044】
本発明のタンパク質製造方法において、遺伝子の発現は一過性発現でもよい。遺伝子の一過性の発現を目的とする場合には、例えば、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。さらに、遺伝子治療などのように所望の遺伝子を生体内で発現させる場合には、所望の遺伝子をベクターに組み込み、例えば、レトロウイルス法、リポソーム法、カチオニックリポソーム法、アデノウイルス法などにより生体内に導入する方法などが挙げられる。用いられるベクターとしては、例えば、アデノウイルスベクター(例えばpAdexlcw)やレトロウイルスベクター(例えばpZIPneo)などが挙げられるが、これらに制限されない。ベクターへの所望の遺伝子の挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning, 5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0045】
本発明により得られた所望のタンパク質は、宿主細胞内または細胞外(培地など)から単離し、実質的に純粋で均一なタンパク質として精製することができる。タンパク質の分離、精製は、通常のタンパク質の精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせればタンパク質を分離、精製することができる。クロマトグラフィーとしては、例えばアフィニティークロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al., Cold Spring Harbor Laboratory Press, 1996)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。なお、タンパク質を精製前又は精製後に適当なタンパク質修飾酵素を作用させることにより、任意に修飾を加えたり、部分的にペプチドを除去することもできる。タンパク質修飾酵素としては、例えば、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼなどが用いられる。
【0046】
本発明のベクター、DNA構築物または宿主細胞は、所望のタンパク質を製造する際に用いることができる。本発明において、所望のタンパク質とはいかなるタンパク質でもよく、タンパク質断片やペプチドなども含まれる。所望のタンパク質の具体例としては、抗体、エリスロポイエチン、コロニー刺激因子(顆粒球、マクロファージ、顆粒球マクロファージ)、インターロイキン1〜31、インターフェロン、RANTES、リンホトキシンβ、Fasリガンド、flt−3リガンド、NF−κBの受容体活性化因子のリガンド(RANKL)、TNF関連アポトーシス誘導リガンド(TRAIL)、CD40リガンド、OX40リガンド、4−1BBリガンド(およびTNFファミリーの他のメンバー)、胸腺ストロマ由来リンホポエチン、マスト細胞増殖因子、幹細胞増殖因子、上皮増殖因子、成長ホルモン、腫瘍壊死因子、白血病阻害因子、オンコスタチンM、トロンボポエチンのような造血因子などのサイトカイン、成長因子、などを挙げることができる。
【0047】
さらに、本発明のベクター又はDNA構築物はトランスジェニック動物の作製に用いることも可能である。トランスジェニック動物の作製は公知の方法、例えば、以下のようにして作製することが可能である。まず、トランスジェニック動物内で発現させたい目的遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入する。該ベクター、該DNA構築物、又はそれらから切り出した発現カセットを全能性細胞に導入する。全能性細胞としては、受精卵、初期胚、胚性幹細胞(ES細胞)等を用いることが可能である。全能性細胞への導入は、静電パルス法、リポソーム法、リン酸カルシウム法、マイクロインジェクション法、あるいはレトロウイルスを感染させるなど、通常用いられる方法により行うことができる。上記処理を行った全能性細胞を仮親の卵管に移植し、産仔を出生させ、その産仔の中から、目的遺伝子を有する個体を選抜する。目的遺伝子を有する個体であるか否かは、目的遺伝子に特異的なプライマーを用いたサザンブロッティングやPCRによる確認などで判定することができる。
【0048】
トランスジェニック動物としては、ヒト以外の非ヒト動物であれば特に制限されず、マウス、ラット、ハムスター、モルモット、ウサギ、ブタ、ミニブタ、ウシ、ヒツジ、ネコ、イヌ等の哺乳動物、ニワトリ等の鳥類、魚類、昆虫類、線虫類などを挙げることができる。操作上の点などから、好ましくはげっ歯類であり、特に好ましくはマウスである。
【0049】
又、本発明のベクター又はDNA構築物は、アンチセンス法やRNAiなどに用いることも可能である。アンチセンス法は、標的遺伝子に対して相補的な配列を持つオリゴヌクレオチドが細胞内に存在すると、該アンチセンス(アンチセンスDNA、アンチセンスRNAなど)が標的遺伝子(標的mRNA、標的DNA、など)と対合することにより、翻訳・転写を阻害し、標的遺伝子の発現を抑制することをいう。従って、アンチセンスオリゴヌクレオチドを、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターで細胞を形質転換することにより、細胞内での標的DNAの発現を阻害することが可能となる。
【0050】
又、RNA干渉(RNA interfearenece:RNAi)を用いて標的遺伝子を阻害することも可能である。RNAiは、二本鎖RNA(dsRNA)を細胞内に導入した際に、そのRNA配列に対応する細胞内のmRNAが特異的に分解され、蛋白質として発現されなくなる現象をいう。RNAiの場合、通常、二本鎖RNAが用いられるが、自己相補的な一本鎖RNA中で形成される二本鎖を用いることも可能である。二本鎖を形成する領域は、全ての領域において二本鎖を形成していてもよいし、一部の領域(例えば両末端又は片方の末端など)が一本鎖等になっていてもよい。RNAiに用いられるオリゴRNAは10〜100bpのRNAが用いられることが多く、通常19〜23bpのRNAが用いられる。従って、細胞内で二本鎖RNAを形成するように設計された遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターで細胞を形質転換することにより、細胞内での標的遺伝子の発現を阻害することが可能となる。RNAi法は、Nature, Vol.391, p.806, 1998、Proc.Natl.Acsd.Sci.USA, Vol.95, p.15502, 1998、Nature, Vol.395, p.854, 1998、Proc.Natl.Acsd.Sci.USA, Vol.96, p.5049, 1999、Cell, Vol.95, p.1017, 1998、Proc.Natl.Acsd.Sci.USA, Vol.96, p.1451, 1999、Proc.Natl.Acsd.Sci.USA, Vol.95, p.13959, 1998、Nature Cell Biol., Vol.2, p.70, 2000等の記載に従って行うことができる。
【0051】
さらに、本発明のベクター又はDNA構築物は、遺伝子治療に用いることも可能である。遺伝子治療は、変異した遺伝子を補正することを目的に、外部から患者の細胞に正常な遺伝子を導入し、細胞の表現型を変化させることにより、病気の治療を行う方法である。又、遺伝子治療は、遺伝子病の治療のみならず、エイズや癌などの他の疾患に対しても有効であると考えられている。遺伝子治療は、生体に直接遺伝子を導入し、細胞に遺伝子を組み込む方法(in vivo法)と、患者の細胞を採取し、体外で細胞に遺伝子を導入した後、該細胞を再度、患者に移植する方法(ex vivo法)に分けられる。
【0052】
遺伝子の投与は如何なる方法を用いてもよく、マイクロインジェクション、リン酸カルシウム法、エレクトロポレーション法、パーティクルガン、レトロウイルスなどのウイルスベクターを用いた方法などにより投与することが可能である。現在、遺伝子治療にはウイルスベクターが広範に用いられている。ウイルスベクターとして用いられるウイルスの例としては、アデノウイルス、アデノ関連ウイルス、ヘルペスウイルス、ワクシニアウイルス、レトロウイルス、ポリオーマウイルス、パピローマウイルス、レンチウイルスなどを挙げることができる。
【0053】
又、パーティクルガンでの導入のように、遺伝子を直接導入する方法も確立されている。in vivo法で遺伝子を導入する場合、その投与経路は限定されず、静脈内、筋肉、皮下、皮内、粘膜、心臓や肝臓などの臓器に直接投与、など如何なる経路で投与してもよい。このように、患者の細胞に導入したい遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターを、in vivo法又はex vivo法により患者の細胞内に導入することにより、遺伝子治療が可能となる。遺伝子治療の具体的な方法は、別冊実験医学 遺伝子治療の基礎技術(羊土社)、別冊実験医学 遺伝子導入と発現解析実験法(羊土社)、日本遺伝子治療学会編遺伝子治療開発研究ハンドブック(エヌ・ティー・エス)などを参考に行うことができる。
【0054】
さらに、本発明のベクター又はDNA構築物は、DNAワクチンの発現の為に用いることも可能である。DNAワクチンは、抗原DNAを体内で発現させ、その抗原に対する免疫反応を誘起する方法と、CpGの繰り返し配列を免疫増強物質として接種する方法の二つがある。DNAワクチンを接種すると、体内で抗原タンパク質が合成され、ウイルスなどの自然感染と同様の免疫反応を誘導する。DNAワクチン発現の為のベクター、投与方法、投与経路などは遺伝子治療と同様の方法により行うことが可能である。
【0055】
さらに、本発明のベクター又はDNA構築物は、免疫原として抗体作製に用いることも可能である(J. Virol., Vol.70(9), p.6119-25, 1996)。免疫原となるタンパク質又はペプチドを発現するよう、本発明のDNA構築物又はベクターを構築し、該ベクター又はDNA構築物を免疫動物に投与することにより、免疫動物の体内で免疫原となるタンパク質又はペプチドが発現し、該タンパク質又はペプチドに対する抗体が産生される。より具体的には、例えば、以下のような方法により抗体を作製することが可能である。
【0056】
まず、免疫原となるタンパク質又はペプチドをコードする遺伝子を、本発明のDNA構築物又はベクターに挿入する。該DNA構築物またはベクターを免疫動物(非ヒト動物)に投与する。投与は如何なる方法を用いてもよく、ベクターをそのまま投与する方法以外に、例えば、エレクトロポレーション法、パーティクルガン、レトロウイルスなどのウイルスベクターを用いた方法などにより投与することが可能である。又、化学物質(ブピバカインなど)など、他の物質と一緒に投与してもよい。投与経路は静脈内、筋肉、皮下、皮内、粘膜、心臓や肝臓などの臓器に直接投与など、如何なる経路で投与してもよい。感作抗原で免疫される哺乳動物としては、特に限定されるものではないが、細胞融合に使用する親細胞との適合性を考慮して選択するのが好ましく、一般的にはげっ歯類の動物、例えば、マウス、ラット、ハムスター、あるいはウサギ、サル等が使用される。このように哺乳動物を免疫し、血清中に所望の抗体レベルが上昇するのを確認した後に、哺乳動物から免疫細胞を採取し、細胞融合に付されるが、好ましい免疫細胞としては、特に脾細胞が挙げられる。
【0057】
前記免疫細胞と融合される他方の親細胞として、哺乳動物のミエローマ細胞を用いる。このミエローマ細胞は、公知の種々の細胞株、例えば、P3(P3x63Ag8.653)(J. Immnol., Vol.123, p.1548-1550, 1979)、 P3x63Ag8U.1(Current Topics in Microbiology and Immunology, Vol.81, p.1-7, 1978)、 NS-1 (Eur. J. Immunol., Vol.6, p.511-519, 1976)、MPC-11(Cell, Vol.8, p.405-415, 1976)、SP2/0 (Nature, Vol.276, p.269-270, 1978)、FO(J. Immunol. Methods, Vol.35, p.1-21, 1980)、S194(Trowbridge, I. S. J. Exp. Med. Vol.148, p.313-323, 1978)、R210(Nature, Vol.277, p.131-133, 1979)等が好適に使用される。前記免疫細胞とミエローマ細胞との細胞融合は、基本的には公知の方法、たとえば、ケーラーとミルステインらの方法(Methods Enzymol., Vol.73, p.3-46, 1981)等に準じて行うことができる。より具体的には、前記細胞融合は、例えば細胞融合促進剤の存在下に通常の栄養培養液中で実施される。融合促進剤としては、例えばポリエチレングリコール(PEG)、センダイウイルス(HVJ)等が使用され、更に所望により融合効率を高めるためにジメチルスルホキシド等の補助剤を添加使用することもできる。
【0058】
免疫細胞とミエローマ細胞との使用割合は任意に設定することができる。例えば、ミエローマ細胞に対して免疫細胞を1-10倍とするのが好ましい。前記細胞融合に用いる培養液としては、例えば、前記ミエローマ細胞株の増殖に好適なRPMI1640培養液、MEM培養液、その他、この種の細胞培養に用いられる通常の培養液が使用可能であり、さらに、牛胎児血清(FCS)等の血清補液を併用することもできる。細胞融合は、前記免疫細胞とミエローマ細胞との所定量を前記培養液中でよく混合し、予め37℃程度に加温したPEG溶液(例えば平均分子量1000-6000程度)を通常30-60%(w/v)の濃度で添加し、混合することによって目的とする融合細胞(ハイブリドーマ)を形成する。続いて、適当な培養液を逐次添加し、遠心して上清を除去する操作を繰り返すことによりハイブリドーマの生育に好ましくない細胞融合剤等を除去する。
【0059】
このようにして得られたハイブリドーマは、通常の選択培養液、例えばHAT培養液(ヒポキサンチン、アミノプテリンおよびチミジンを含む培養液)で培養することにより選択される。上記HAT培養液での培養は、目的とするハイブリドーマ以外の細胞(非融合細胞)が死滅するのに十分な時間(通常、数日〜数週間)継続する。ついで、通常の限界希釈法を実施し、目的とする抗体を産生するハイブリドーマのスクリーニングおよび単一クローニングを行う。
【0060】
また、ヒト以外の動物に抗原を免疫して上記ハイブリドーマを得る他に、ヒトリンパ球をin vitroで感作し、感作リンパ球をヒト由来の永久分裂能を有するミエローマ細胞と融合させ、結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878号公報参照)。さらに、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物に抗原を投与して抗体産生細胞を取得し、これを不死化させた細胞から抗原に対するヒト抗体を取得してもよい(国際特許出願公開番号WO 94/25585 号公報、WO 93/12227 号公報、WO92/03918 号公報、WO 94/02602 号公報参照)。このようにして作製されるモノクローナル抗体を産生するハイブリドーマは、通常の培養液中で継代培養することが可能であり、また、液体窒素中で長期保存することが可能である。当該ハイブリドーマからモノクローナル抗体を取得するには、当該ハイブリドーマを通常の方法にしたがい培養し、その培養上清として得る方法、あるいはハイブリドーマをこれと適合性がある哺乳動物に投与して増殖させ、その腹水として得る方法などが採用される。前者の方法は、高純度の抗体を得るのに適しており、一方、後者の方法は、抗体の大量生産に適している。
【0061】
本発明では、モノクローナル抗体として、抗体遺伝子をハイブリドーマからクローニングし、適当なベクターに組み込んで、これを宿主に導入し、遺伝子組換え技術を用いて産生させた組換え型のものを用いることができる(例えば、Eur. J. Biochem., Vol.192, p.767-775, 1990参照)。
【0062】
具体的には、抗体を産生するハイブリドーマから、抗体の可変(V)領域をコードするmRNAを単離する。mRNAの単離は、公知の方法、例えば、グアニジン超遠心法(Biochemistry, Vol.18, p.5294-5299, 1979)、AGPC法(Anal. Biochem., Vol.162, p.156-159, 1987)等により行って全RNAを調製し、mRNA Purification Kit (ファルマシア製)等を使用して目的のmRNAを調製する。また、QuickPrep mRNA Purification Kit (ファルマシア製)を用いることによりmRNAを直接調製することもできる。得られたmRNAから逆転写酵素を用いて抗体V領域のcDNAを合成する。cDNAの合成は、AMV Reverse Transcriptase First-strand cDNA Synthesis Kit(生化学工業社製)等を用いて行う。また、cDNAの合成および増幅を行うには、5'-Ampli FINDER RACE Kit(クロンテック製)およびPCRを用いた5'-RACE法(Proc. Natl. Acad. Sci. USA, Vol.85, p.8998-9002, 1988、Nucleic Acids Res., Vol.17, p.2919-2932, 1989)等を使用することができる。得られたPCR産物から目的とするDNA断片を精製し、ベクターDNAと連結する。さらに、これより組換えベクターを作製し、大腸菌等に導入してコロニーを選択して所望の組換えベクターを調製する。そして、目的とするDNAの塩基配列を公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法等により確認する。目的とする抗体のV領域をコードするDNAを得たのち、これを、所望の抗体定常領域(C領域)をコードするDNAを含有する発現ベクターへ組み込む。
【0063】
抗体を製造するには、通常、抗体遺伝子を発現制御領域、例えば、エンハンサー、プロモーターの制御のもとで発現するよう発現ベクターに組み込む。次に、この発現ベクターにより、宿主細胞を形質転換し、抗体を発現させる。抗体遺伝子の発現は、抗体重鎖(H鎖)または軽鎖(L鎖)をコードするDNAを別々に発現ベクターに組み込んで宿主細胞を同時形質転換させてもよいし、あるいはH鎖およびL鎖をコードするDNAを単一の発現ベクターに組み込んで宿主細胞を形質転換させてもよい(WO 94/11523 号公報参照)。
【0064】
以上のように、本発明のDNA構築物又はベクターは、所望のタンパク質などの製造、アンチセンス法、RNAi法、遺伝子治療、DNAワクチン、トランスジェニック動物、抗体の作製などに適用することが可能である。
【0065】
さらに本発明は、本発明のベクターを、該ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法に関する。
【0066】
動物目とは、生物分類のリンネ式階層分類において、綱の次下位、科階級群の上位におかれる基本的階級あるいは、その階級にあるタクソンのことをいう。具体的な動物目としては、例えば、ネズミ目、ウサギ目、ハネジネズミ目、ツパイ目などを挙げることができるが、ネズミ目が好ましい。ネズミ目には、ハムスター、ラット、マウス、モルモット、リス、ビーバーなどが含まれる。宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0067】
又本発明は、本発明のベクターを、該ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法に関する。
【0068】
同じ動物種由来とは、宿主細胞がヒト細胞の場合にはヒトβアクチンプロモーターを用い、宿主細胞がマウス細胞の場合にはマウスβアクチンプロモーターを用い、宿主細胞がハムスター細胞の場合にはハムスターβアクチンプロモーターを用いることをいう。本方法によって、宿主細胞と同種由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0069】
さらに本発明は、宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることを特徴とする所望の遺伝子の発現量を増加させる方法に関する。動物目とは、上記に記載の定義による。宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0070】
又、本発明は、宿主細胞と同じ動物種由来のβアクチンプロモーターを用いることを特徴とする所望の遺伝子の発現量を増加させる方法に関する。同じ動物種由来とは、上記に記載の定義による。宿主細胞と同じ動物種由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0071】
又、本発明においては所望のDNAの発現量を増加させる目的で、トランスアクティベーターを用いてもよい。トランスアクティベーターは、トランスアクティベーターをコードするDNAを含むベクターを宿主細胞に導入し、培養時にトランスアクティベーターが発現するようにしてもよいし、トランスアクティベーターを培地に添加してもよい。トランスアクティベーターをコードするDNAは、所望のタンパク質をコードするDNAと同一のベクター上に組み込んでもよいし、異なるベクターに組み込んで両方のベクターを細胞に導入してもよい。
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0072】
以下、本発明を実施例により、さらに具体的に説明するが本発明はこれら実施例に制限されるものではない。
【0073】
〔実施例1〕 発現ベクターpmAct-Luc-neoの構築
(1)マウスβアクチンプロモーターのクローニング
NCBI(http://www.ncbi.nlm.nih.gov/)、及びJackson laboratory(http://www.jax.org/)で公開されているマウスゲノム情報より、マウスβアクチンのシーケンス情報を得た。以下に示す配列のプライマーmAct5-F1(5'- GGGAGTGACTCTCTGTCCATTCAATCC -3'/配列番号:9)およびmAcr5-R1(5'- TTGTCGACGACCAGCGCAGCGATATCG -3'/配列番号:10)を合成し(エスペックオリゴ社)、PCRによってマウスβアクチンのプロモーター領域(1,577 bp)を増幅した。試薬としてはタカラバイオ社のTaKaRa LA Taq with GC Buffer(cat. RR02AG)、鋳型DNAにはクロンテック社のMouse Genomic DNA(cat. 6650-1)を使用してPCRを行った。PCR反応溶液の組成は、鋳型DNA(100ng/ml) 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mAct5-F1(10μM)2.0μl、mAcr5-R1(10μM)2.0μl、H2O 11.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で1分、「95℃で30秒、60℃で30秒、72℃で90秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、1,577bpのバンドを切り出し、Mag Extractor(東洋紡社、cat.NPK-601)で精製した。これをpGEM-T-Easy vector(プロメガ社、cat.A1360)にクローニングし、シーケンスすることによって塩基配列を確認した。クローニングしたマウスβアクチン5'領域を配列番号:1に示す。
【0074】
公共のデータベースの配列では、305番目から始まるTは13個の連続であるが、クローニングした領域は記載した配列の通り14個の連続したTであった。データベースと配列が異なるが、これをそのまま実験に用いることにした。また、上記の配列中、斜体で示した414番目から始まるCAAT、及び475番目から始まるTATAAはhouse keeping遺伝子のプロモーター領域によく見出される配列であり、得られた領域はマウスβアクチンのプロモーター領域であることが推察された。
【0075】
クローニングした領域はマウスベータアクチンの開始コドン(上記配列中の1543番目から始まるATG)を含むこと、及びその後のベクター構築のために、プライマーmAct5-BG(5'- AGATCTGGGAGTGACTCTCTGTCCAT -3'/配列番号:11、Bgl IIサイトを含む)およびmAct5-HN(5'- AAGCTTGGCGAACTATCAAGACACAA -3'/配列番号:12、Hind IIIサイトを含む)を合成し(エスペックオリゴ社)、再度PCRすることによって必要な領域(1〜1542/配列番号:2)にBgl II(5'末)、Hind III(3'末)サイトを付加した。
【0076】
PCR反応溶液の組成は、鋳型DNA(約10ng/ml) 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mAct5-HN(10μM)1.0μl、mAct5-BG(10μM)1.0μl、H2O 13.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で30秒、「95℃で30秒、60℃で30秒、72℃で60秒」を30サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は前述のようにpGEM-T-Easy vectorにクローニングし、シーケンス確認を行った。こうして作製したマウスβアクチンフラグメントをpGL3-Basic(プロメガ社、cat. E1751)のBgl II-Hind IIIサイトに組み込んだ。さらに、pGL3-Basicはベクターにおける配列4650位のNot IサイトをBamH Iサイトに変換し、配列2004位のBamH Iサイトとの間のベクター骨格を、pCXNベクターのベクター骨格(BamH I- BamH Iフラグメント)と変換することで、ネオマイシン耐性遺伝子を導入し、pmAct-Luc-neoとした(図1)。なお、pGL3-Basicのベクター骨格を変換したベクターを、以下pGLNベクターと称する。
【0077】
(2)WPREのクローニングとpmAct-WPRE-Luc-neoの作製
Woodchuck hepatitis virusゲノム配列(GenBank Accession No. J04514)の1093番目から1684番目の 592塩基が転写後調節領域(WPRE)は、以下のようにAssemble PCR法を用いて増幅し、クローニングした。タカラバイオ社のTaKaRa Ex Taq (cat. RR001B) を使用して、以下のような二つの条件系でPCRを行った。
【0078】
<PCR反応1>
まず、反応溶液の組成:合成DNA(WP-1〜WP-6,各10uM, 各1uL) 6.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、H2O 28.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃で5分、「94℃で2分、55℃で2分、72℃で2分」を2サイクル、によりPCR反応を行った。さらに、プライマーWP-f (10uM) 1.0μl、WP-r2 (10uM) 1.0μlを添加し総量を50,0μlとし、反応条件:「94℃で30秒、55℃で30秒、72℃で1分」で30サイクル、72℃ で5分、によりPCR反応を行った。
【0079】
<PCR反応2>
まず、反応溶液の組成:合成DNA(WP-5〜WP-17,各10uM, 各1uL) 13.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、H2O 21.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃で5分、「94℃で2分、55℃で2分、72℃で2分」を2サイクル、によりPCR反応を行った。さらに、プライマーWP-f2 (10uM) 1.0μl、WP-r (10uM) 1.0uLを添加し総量を50,0μlとし、反応条件:「94℃ 30秒、55℃ 30秒、72℃ 1分」で30サイクル、72℃ 5分、によりPCR反応を行った。
【0080】
PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、反応1は約200bp、反応2は約400bpのバンドを切り出し、QIAquick Gel Extraction Kit(キアゲン社、cat.28704)で精製した。
【0081】
各反応産物を用いて、さらにPCRを行った。試薬は、タカラバイオ社のTaKaRa Ex Taq (cat. RR001B) を使用した。反応溶液の組成:反応1産物 1.0μl、反応2産物 1.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、WP-f (10uM) 1.0μl、WP-r (10uM) 1.0μl、H2O 32.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃ 30秒、「94℃ 15秒、72℃ 2分」を5サイクル、「94℃ 15秒、70℃ 2分」を5サイクル、「94℃ 15秒、68℃ 2分」を28サイクル、72℃ 5分、によりPCR反応を行った。
【0082】
増幅したPCR産物は1% アガロースゲルに電気泳動した後に、約600bpのバンドを切り出し、QIAquick Gel Extraction Kit(キアゲン社、cat.28704)で精製した。反応産物をpGEM-T-Easy vector(プロメガ社、cat.A1360)にクローニングし、塩基配列を決定した。GenBank Accession No. J04514の1093番目から1684番目の配列と同じ配列であることを確認した。pGEM-T/WPREをXbaIで消化し、得られたWPRE断片をpmAct-Luc-neoのXbaI部位に挿入し、pmAct-WPRE-Luc-neoを作製した。配列番号:3にクローニングしたWPRE配列および使用した合成DNAを、使用した合成DNA(配列番号:13から33)を以下に示す。
<使用した合成DNA>
WP-1: AATCAACCTCTGGATTACAAAATTTGTGAAAGATTGACTGGTATTCTTAA(配列番号:13)
WP-2: GCGTATCCACATAGCGTAAAAGGAGCAACATAGTTAAGAATACCAGTCAA(配列番号:14)
WP-3: ACGCTATGTGGATACGCTGCTTTAATGCCTTTGTATCATGCTATTGCTTC(配列番号:15)
WP-4: TTATACAAGGAGGAGAAAATGAAAGCCATACGGGAAGCAATAGCATGATA(配列番号:16)
WP-5: TTCTCCTCCTTGTATAAATCCTGGTTGCTGTCTCTTTATGAGGAGTTGTG(配列番号:17)
WP-6: GTGCACACCACGCCACGTTGCCTGACAACGGGCCACAACTCCTCATAAAG(配列番号:18)
WP-7: CGTGGCGTGGTGTGCACTGTGTTTGCTGACGCAACCCCCACTGGTTGGGG(配列番号:19)
WP-8: GTCCCGGAAAGGAGCTGACAGGTGGTGGCAATGCCCCAACCAGTGGGGGT(配列番号:20)
WP-9: CAGCTCCTTTCCGGGACTTTCGCTTTCCCCCTCCCTATTGCCACGGCGGA(配列番号:21)
WP-10: TGTCCAGCAGCGGGCAAGGCAGGCGGCGATGAGTTCCGCCGTGGCAATAG(配列番号:22)
WP-11: TTGCCCGCTGCTGGACAGGGGCTCGGCTGTTGGGCACTGACAATTCCGTG(配列番号:23)
WP-12: CCATGGAAAGGACGTCAGCTTCCCCGACAACACCACGGAATTGTCAGTGC(配列番号:24)
WP-13: TGACGTCCTTTCCATGGCTGCTCGCCTGTGTTGCCACCTGGATTCTGCGC(配列番号:25)
WP-14: GAGGGCCGAAGGGACGTAGCAGAAGGACGTCCCGCGCAGAATCCAGGTGG(配列番号:26)
WP-15: ACGTCCCTTCGGCCCTCAATCCAGCGGACCTTCCTTCCCGCGGCCTGCTG(配列番号:27)
WP-16: GAGGGCGAAGGCGAAGACGCGGAAGAGGCCGCAGAGCCGGCAGCAGGCCGCGGGAAG(配列番号:28)
WP-17: GTCTTCGCCTTCGCCCTCAGACGAGTCGGATCTCCCTTTGGGCCGCCTCCCCGCCTG(配列番号:29)
WP-f: TCTAGAAATCAACCTCTGGATTACAAAATT(配列番号:30)
WP-r: TCTAGAAGGCGGGGAGGCGGCCCAAA(配列番号:31)
WP-f2: ATCCTGGTTGCTGTCTCTTTATGAG(配列番号:32)
WP-r2: GTGCACACCACGCCACGTTGCC(配列番号:33)
【0083】
(3)phCMV-mAct-Luc-neoの作製
CMVエンハンサー領域(配列番号:4)をpmAct-Luc-neoベクターの EcoR Iサイトにクローニングし、phCMV-mAct-Luc-neoを完成させた(図1)。
【0084】
(4)比較対象となるベクターの構築
ヒトEF1αプロモーターはDHFR-ΔE-RVh-PM1-f(参考文献 WO92/19759)よりCMVエンハンサーと共にpGLNベクターのMCSに組み込むことで、pCEF-Luc-neoを作製した(図1)。実験に用いた全てのベクターは、キアゲン社のEndoFree Plasmid Maxi Kit (cat.12362)によって精製した。
【0085】
〔実施例2〕ベクターのCHO細胞への導入および発現測定
(1)ベクターのCHO細胞への導入
2μgのベクターをそれぞれ20μlのPLUS(tm) Reagent (インビトロジェン社、Cat. No. 11514-015 )と混合し、OPTI-MEM I Reduced-Serum Medium (インビトロジェン社、Cat. No. 11058-021)で容量を200μlに調整し、室温で15分間保温した。ここに、LipofectAMINE (インビトロジェン社、Cat. No. 18324-012 )を20μl、OPTI-MEM I Reduced-Serum Medium を180μl加え、室温でさらに15分間保温した。CHO細胞は96well Cell Culture Cluster (コーニング社、Cat. No. 3595)で1ウェルあたり2×104 cells、50μlのOPTI-MEM I Reduced-Serum Mediumで調整した。上記で作製したDNA溶液20μlをCHO細胞に添加し、37℃、3時間、CO2インキュベーター内で培養することで、ベクターをCHO細胞に導入した。その後、培養上清を静かに除き、CHO用の培地を加えた。CHO用の培地は、CHO-S-SFMII培地(インビトロジェン社、Cat. No. 12052-098)に1/100量のHT Supplement (100×), liquid(インビトロジェン社、Cat. No. 11067-030)と、1/100量のPenicillin-Streptomycin (インビトロジェン社製、Cat. No. 15140-122)を添加することで作製したものを使用した。この状態のまま、37℃、CO2インキュベーター内で2日間培養した。
【0086】
(2)ルシフェラーゼ活性の測定
ルシフェラーゼ活性は、Luciferase Assay System (プロメガ社、Cat. No. E1501)を用いて測定した。培地を除き、1ウェルあたりキットに添付されている5× Passive lysis bufferを5倍に薄めたものを100 μl加え、振とうすることで細胞を溶解した。その細胞溶解液をAssay Plate Tissue Culture Treated White with Clear Bottom(コーニング社、Cat No. 437842)に10μlずつ移した。測定はMicroLuMAT(バートホールド社)を用いて行い、データはソフトWinGlow-Control Program LB96PV ver.1.24を用いて取り込んだ。
【0087】
以上の測定の結果より、作製したpmAct-Luc-neoがpCEF-Luc-neoよりも有意に(p<0.0009:対応のないt検定)活性が高いことが明らかになった(図2)。以上のことから、本発明であるマウスβアクチンプロモーターは、既存のCEFプロモーターよりも強力であることが示された。さらに、WPREエレメントの付加や、あるいはCMVエンハンサーを付加することによって、マウスβアクチンのプロモーター活性が有意に(pmAct-WPRE-Luc-neoとpmAct-Luc-neoの比較: p<0.0005、phCMV-mAct-Luc-neoとpmAct-Luc-neoの比較: p<0.0007:対応のないt検定)亢進することも明らかになった(図3)。
【0088】
〔実施例3〕 マウスc-H-ras遺伝子のクローニングおよび改変
(1)マウスc-H-ras遺伝子のクローニング
ヒト活性型H-Rasがヒトβアクチンプロモーターを組み込んだ発現ベクターの発現効率を亢進することは既に報告されている(Cytotechnology, Vol. 16, p.167-178, 1994)。しかし、マウスβアクチンプロモーターに対する活性型H-Ras、あるいは野生型のH-Ras、活性型K-Rasの効果に関する報告は無いことから、マウスH-Ras(活性型、野生型)、ヒト活性型K-Rasのマウスβアクチンプロモーターに対する効果について検討を行った。
【0089】
NCBI(http://www.ncbi.nlm.nih.gov/)で公開されているGenBank Accession No. M30733の情報より、プライマーmRas-F1(5'- TCCTGGATTGGCAGCCGCTGTAGAAGC -3'/配列番号:34)およびmRas-R1(5'- GTTCATCTGGCTAGCTGAGGTCACTGC -3'/配列番号:35)を合成し(エスペックオリゴ社)、PCRによってマウスc-H-ras遺伝子を増幅した。試薬としてはタカラバイオ社のTaKaRa LA Taq with GC Buffer、鋳型cDNAにはクロンテック社のEmbryo Marathon-Ready DNA day15(cat. 7459-1)を使用してPCRを行った。
【0090】
PCR反応溶液の組成は、鋳型DNA 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mRas-F1(10μM)2.0μl、mRas-R1(10μM)2.0μl、H2O 11.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で60秒、「95℃で30秒、60℃で30秒、72℃で60秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、660bpのバンドを切り出し、Mag Extractor(東洋紡社)で精製した。これをpGEM-T-Easy vector(プロメガ社)にクローニングし、シーケンスすることによって塩基配列を確認した。確認したマウスc-H-rasのcDNA配列を配列番号:5に示す。データベースでは62番目の塩基がAであったが、クローニングしたc-H-rasはGあった。NCBIのマウスゲノムデータベースで検索したところ、この部分の配列はGであったので、そのまま実験に使用することにした。
【0091】
(2)マウスc-H-rasの改変
CHO細胞で効率よくマウスc-H-Rasを発現させるために、プライマーRAS-ATG(5'- GCCACCATGACAGAATACAAGCTT -3'/配列番号:36)およびRAS-R2(5'- TCAGGACAGCACACATTTGC -3'/配列番号:37)を用いたPCRによって、開始コドンの上流の配列をKozak rule(Nicleic Acids Research, Vol.15(20), p.8125-8148, 1987)に従った配列に変換するとともに、タンパク質がコードされている3'UTRを除去した。試薬としてはタカラバイオ社のTaKaRa LA Taq(cat. RR002A)、鋳型cDNAには先にpGEM-T-Easyベクターにクローニングしたマウスc-H-rasを鋳型に用いて以下のような条件系でPCRを行った。
【0092】
PCR反応溶液の組成は、鋳型DNA 1.0μl、10×LA PCR buffer II(Mg2+free) 5.0μl、dNTP Mixture 5.0μl、MgCl2(25mM) 5.0μl、RAS-ATG(10μM)2.0μl、RAS-R2(10μM)2.0μl、H2O 29.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で60秒、「95℃で30秒、60℃で30秒、72℃で60秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、576bpのバンドを切り出し、Mag Extractor(東洋紡社)で精製した。これをpGEM-T-Easy vector(プロメガ社)にクローニングし、シーケンスすることによって塩基配列を確認した。改変したマウスc-H-rasのcDNA配列を配列番号:6に示す。
【0093】
(3)マウス活性型H-Rasの作製
ストラタジーン社のQuikChangeTM Site-Directe Mutagenesis Kit(cat.#200518)を用いて、上記のマウスc-H-rasに点変異(上の配列の下線を引いたGをTに変換)を入れることによって12番目のアミノ酸をバリンに変換することで活性型H-Rasを作製した。
使用したプライマーは、mRasV12-F(5'- GTGGTGGGCGCTGTAGGCGTGGGA AAG-3'/配列番号:38)および、mRasV12-R(5'- CTTTCCCACGCCTACAGCGCCCACCAC-3'/配列番号:39)であり、反応溶液の組成:鋳型DNA、H-Ras/pGEM-T-Easy(10ng/μl) 2.0μl、10×reaction buffer 5.0μl、dNTP Mix 1.0μl、mRasV12-F(100ng/μl) 1.25μl、mRasV12-R(100ng/μl) 1.25μl、H2O 38.5μl、PfuTurbo DNA polymerase(2.5U/μl) 1.0μl、反応条件:95℃で30秒、「95℃で30秒、55℃で1分、68℃で7分20秒」で12サイクルにてPCR反応を行った。PCRにはGene Amp PCR System 2400を用いた。増幅したPCR産物をキットのマニュアルに従って、Dpn I処理した後に大腸菌にトランスフォームした。そして、得られた大腸菌からプラスミドを回収してシーケンスすることによって変異が導入されていることを確認した。マウス活性型H-Rasのアミノ酸配列を配列番号:7に示す。
【0094】
同様にヒト活性型K-Rasを準備した。実験に使用したK-Rasのアミノ酸配列を配列番号:8に示す。
これらの遺伝子は、すべてpCXNベクター(Gene, Vol.108, p.193-200, 1991)に組み込んで、それぞれをpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型K-ras)とした。
【0095】
〔実施例4〕癌遺伝子産物Rasのプロモーター活性への影響
(1)ベクターのCHO細胞への導入
CHO細胞に導入する2μgのベクターは、以下のように2種類、あるいは3種類のベクターを、様々な割合で混合することで調整した(表1)。実施例2と同じ方法でCHO細胞に導入し、ルシフェラーゼ活性を測定した。
【0096】
【表1】

【0097】
この結果より、pmAct-Luc-neoがpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型c-K-ras)の存在によって、プロモーター活性が亢進することが明らかになった(図4)。以上のことから、本発明であるマウスβアクチンプロモーターは、癌遺伝子産物のRas(活性型、野生型、H-Ras、K-Rasを問わない)によってより強力な活性を付与されることが示された。
【産業上の利用可能性】
【0098】
これまでは、動物細胞でタンパク質を産生させるために使用されていたベクターには、EF1αプロモーター(WO92/19759)やCMVプロモーターが使用されてきた。近年、医薬品におけるバイオ製剤のシェアが大きくなり、特に多くの抗体医薬品が上市されるようになってきた。抗体医薬品は、従来のエリスロポエチン、G-CSF、インターフェロンなどのサイトカインをバイオ製剤としていたものと比較して、大量の投与が必要とされている。従って安価な抗体医薬品の安定な供給には、動物細胞で大量にタンパク質を発現させることが必須となっている。本発明の方法は、従来の技術と比較してタンパク質をより大量に産生させることを可能としたことから、産業上の観点から有用性があるものと考えられる。また、抗体医薬の作製においては、抗原を動物に免疫する過程が必要であるが、こうした発現ベクターに抗原の遺伝子を組み込み、ベクターそのものを動物に免疫することが可能であることが明らかになってきた。本方法のメリットは、抗原をタンパク質として精製する過程を省略できることにある。免疫過程には、いかに多くの抗原がベクターを投与された動物個体内で産生されるかが重要であり、従って発現ベクターが強力なものであるほど好ましい。モノクローナル抗体作製には、マウスやラットが一般に用いられているが、本発明のベクターはこうした用途に好適であると考えられる。さらに、ヒトにおいても遺伝子治療に使用されるベクターなどに組み込むことで、臨床に応用することも可能である。
【技術分野】
【0001】
本発明は、組換えタンパク質の産生における、より強力なプロモーターおよび、それらを保持するベクターの開発に関する。
【背景技術】
【0002】
近年、多くのバイオ製剤が輩出されているが、こうした医薬品の多くが動物細胞に遺伝子を導入し、組換えタンパク質として産生させたものである。こうした組換えタンパク質を効率よく動物細胞に産生させる技術は、バイオ製剤の低コスト化につながり、患者への安定な供給を約束する。
【0003】
従来は、EF1α(elongation factor 1)プロモーターやSRαプロモーターなどが、そうした発現ベクターに用いられてきた(特許文献1, 非特許文献1,2)。また、最近ではさらに発現の改良が進み、CAGプロモーター(ヒトCMVエンハンサーにニワトリのβアクチンプロモーターを組み合わせたもの)や、CEFプロモーター(ヒトCMVエンハンサーにヒトEF1αプロモーター)などの強力なプロモーターが作られ、こうした用途に使われるようになってきた(非特許文献3〜5)。
【0004】
しかしながら、さらに強力なプロモーターを保持するベクターの開発の必要性はいまなお存在し、これによりバイオ製剤のより一層の低コスト化や安定な供給に大きく貢献できるものと考えられる。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】WO92/19759
【非特許文献】
【0006】
【非特許文献1】Mol. Cell. Biol., Vol.8 (1), p.466-472, 1988
【非特許文献2】Gene, Vol.87(2), p.291-294, 1990
【非特許文献3】Gene, Vol.272, p.149-156, 2001
【非特許文献4】Analytical Biochemistry, Vol.247, p.179-181, 1997
【非特許文献5】Gene, Vol.108, p.193-200, 1991
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、このような状況に鑑みてなされたものであり、その目的は既存の動物細胞発現プロモーターよりも強力なプロモーターと、それを利用したベクターの提供である。また、本発明は、該ベクターを利用した、所望タンパク質の製造方法および所望のDNAの発現方法の開発をも目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記の課題を解決すべく、様々な遺伝子のプロモーターやエンハンサーの組み合わせによるプロモーター活性の比較、検討を行った。
【0009】
まず、NCBI、及びJackson laboratoryで公開されているマウスゲノム情報より、マウスβアクチンのシーケンス情報を得て、PCRによってマウスβアクチンの5'領域を増幅した。増幅したPCR産物をpGEM-T-Easy vectorにクローニングし、シーケンスすることによって塩基配列を確認した。その後、PCRによりマウスβアクチンのプロモーター領域のみを増幅し、pGL3-BasicのBgl II-Hind IIIサイトに組み込んだ。さらにこのベクターに、ネオマイシン耐性遺伝子を導入し、pmAct-Luc-neoとした。
【0010】
Woodchuck hepatitis virusゲノム配列の転写後調節領域(WPRE)をPCRにより増幅し、反応産物をpGEM-T-Easy vectorにクローニングし、塩基配列を決定した。さらにこのpGEM-T/WPREをXbaIで消化し、得られたWPRE断片をpmAct-Luc-neoのXbaI部位に挿入し、pmAct-WPRE-Luc-neoを作製した。
【0011】
次にpGL3-BasicベクターのMCS(multi cloning site : 配列5位〜53位)にCMVエンハンサー領域を組み込むことによって発現ベクターのpmAct-Luc-neoを、あるいはさらにヒトCMVエンハンサー-マウスβアクチンプロモーターを組み込むことによって発現ベクターのphCMV-mAct-Luc-neoを完成させた(図1)。
【0012】
比較対象となるベクターとして、ヒトEF1αプロモーターはDHFR-ΔE-RVh-PM1-f(参考文献 WO92/19759)よりCMVエンハンサーと共にpGLNベクターのMCSに組み込むことで、pCEF-Luc-neoを作製した(図1)。
【0013】
これらのベクターをそれぞれCHO細胞へ導入し、CO2インキュベーター内で2日間培養して、ルシフェラーゼ活性をLuciferase Assay Systemを用いて測定した。これらの測定の結果より、作製したpmAct-Luc-neoが他のベクターよりも有意に活性が高いことが明らかになった(図2)。以上のことから、本発明であるマウスβアクチンプロモーターは、既存のCEFプロモーターよりも強力であることが示された。さらに、WPREエレメントの付加や、あるいはCMVエンハンサーを付加することによって、マウスβアクチンのプロモーター活性が有意に亢進することも明らかになった(図3)。
【0014】
さらに、トランスアクティベーターを同時に発現させた場合の効果について検討した。マウスc-H-ras遺伝子、マウス活性型c-H-ras及びヒト活性型K-rasのクローニングを行い、pCXNベクターに組み込んで、それぞれをpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型K-ras)とした。様々な割合で2種類、あるいは3種類のベクターをCHO細胞に導入し、ルシフェラーゼ活性を測定した。これらの結果より、pmAct-Luc-neoがpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型c-K-ras)の存在によって、プロモーター活性が亢進することが明らかになった(図4)。以上のことから、本発明のマウスβアクチンプロモーターは、癌遺伝子産物のRas(活性型、野生型、H-Ras、K-Rasを問わない)によってより強力な活性を付与されることが示された。
【0015】
即ち、本発明者らは、CMVエンハンサーと哺乳類βアクチンプロモーター、もしくはWoodchuck Hepatitis Virusゲノム配列の転写後調節領域(WPRE)と哺乳類βアクチンプロモーターの組み合わせが、強力なプロモーターになりうることを見出した。さらに、これらのプロモーターを保持した発現系は、トランスアクティベーターである癌遺伝子産物のRasの同時発現によって、さらに発現活性が高められることを見出し、本発明の完成に至った。
【0016】
本発明は、より具体的には、以下の〔1〕〜〔37〕に関する。
〔1〕 エンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物。
〔2〕 エンハンサーがサイトメガロウイルス(CMV)である〔1〕に記載のDNA構築物。
〔3〕 エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である〔1〕に記載のDNA構築物。
〔4〕 哺乳類βアクチンプロモーターがげっ歯類βアクチンプロモーターである、〔1〕から〔3〕のいずれかに記載のDNA構築物。
〔5〕 CMVエンハンサーが配列番号:4に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、〔2〕に記載のDNA構築物。
〔6〕 Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)が配列番号:3に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、〔3〕に記載のDNA構築物。
〔7〕 〔1〕から〔6〕のいずれかに記載のDNA構築物を含むベクター。
〔8〕 哺乳類βアクチンプロモーターの下流に所望のDNAが機能的に結合されたDNAを有する〔7〕に記載のベクター。
〔9〕 トランスアクティベーターをコードするDNAを発現可能に保持する、〔7〕または〔8〕に記載のベクター。
〔10〕 トランスアクティベーターが癌遺伝子産物である〔9〕に記載のベクター。
〔11〕 癌遺伝子産物がRasである、〔10〕に記載のベクター。
〔12〕 所望のDNAが、所望のタンパク質をコードするDNAである、〔8〕から〔11〕のいずれかに記載のベクター。
〔13〕 〔8〕から〔12〕のいずれかに記載のベクターを保持する細胞。
〔14〕 〔8〕から〔12〕のいずれかに記載のベクターを保持し、癌遺伝子が活性化している細胞。
〔15〕 癌遺伝子が活性化している細胞が、トランスアクティベーターをコードする遺伝子を含むベクターが導入された細胞である〔14〕記載の細胞。
〔16〕 癌遺伝子が活性化している細胞が、癌化している細胞である〔14〕記載の細胞。
〔17〕 哺乳動物細胞である、〔13〕から〔16〕のいずれかに記載の細胞。
〔18〕 げっ歯類細胞である、〔17〕に記載の細胞。
〔19〕 βアクチンプロモーターと同じ動物目であることを特徴とする〔13〕から〔18〕のいずれかに記載の細胞。
〔20〕 βアクチンプロモーターと同じ動物種であることを特徴とする〔19〕に記載の細胞。
〔21〕 〔8〕から〔12〕のいずれかに記載のベクターが導入されたトランスジェニック非ヒト動物。
〔22〕 〔8〕から〔12〕のいずれかに記載のベクターが導入された全能性細胞。
〔23〕 〔12〕に記載のベクターを保持する細胞を培養し、培養した細胞又は培地から発現させたタンパク質を回収することを含む、所望のタンパク質の製造方法。
〔24〕 培地にトランスアクティベーターを添加することを含む、〔23〕に記載の方法。
〔25〕 〔8〕から〔12〕のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
〔26〕 〔8〕から〔12〕のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
〔27〕 〔8〕に記載のベクターおよびトランスアクティベーターをコードするDNAを発現可能に保持するベクターを、〔8〕に記載のベクター上のβアクチンプロモーターと同種由来の宿主細胞に導入することを含む、宿主細胞における所望のDNAを発現させる方法。
〔28〕 宿主細胞が哺乳動物細胞である、〔25〕から〔27〕のいずれかに記載の方法。
〔29〕 宿主細胞がげっ歯類細胞である、〔25〕から〔27〕のいずれかに記載の方法。
〔30〕 宿主細胞と同じ動物目由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
〔31〕 宿主細胞と同じ動物種由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
〔32〕 エンハンサーをさらに組み込むことを特徴とする〔30〕もしくは〔31〕に記載の方法。
〔33〕 エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である〔32〕に記載の方法。
〔34〕 エンハンサーがCMVエンハンサーである〔32〕に記載の方法。
〔35〕 トランスアクティベーターをコードする遺伝子を組み込むことを特徴とする〔30〕から〔34〕のいずれかに記載の方法。
〔36〕 宿主細胞が哺乳動物細胞である〔30〕から〔35〕いずれかに記載の方法。
〔37〕 宿主細胞がげっ歯類細胞である〔30〕から〔35〕いずれかに記載の方法。
【図面の簡単な説明】
【0017】
【図1】実験に使用したベクターを示した図である。
【図2】既存のプロモーターであるCEFプロモーターとマウスβアクチンプロモーターを比較した図である。
【図3】マウスβアクチンプロモーターに対するWPRE、CMVエンハンサーの効果を示した図である。
【図4】マウスRas蛋白質のマウスβアクチンプロモーターに対する効果を示した図である。
【発明を実施するための形態】
【0018】
本発明はエンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物、及び該DNA構築物を含むベクターを提供する。本発明のDNA構築物やベクターは、所望のDNAの発現(例えば、所望のタンパク質をコードするDNAの発現、アンチセンスDNAの発現、dsRNAをコードするDNAの発現など)に用いることができる。
【0019】
本発明において、哺乳類βアクチンプロモーターとエンハンサーとが「機能的に結合した」とは、プロモーター活性が高まるように、本発明のプロモーター活性を有する哺乳類βアクチンプロモーターとエンハンサーとが結合していることをいう。従って、哺乳類βアクチンプロモーターとエンハンサーの距離が離れていたり、間に他の遺伝子が挿入された場合であっても、哺乳類βアクチンプロモーターのプロモーター活性が高まるのであれば、上記の「機能的に結合した」の意に含まれる。エンハンサーは哺乳類βアクチンプロモーターの上流下流どちらに存在していても良い。
【0020】
また、哺乳類βアクチンプロモーターと所望のDNAが「機能的に結合した」とは、哺乳類βアクチンプロモーターの活性化により、所望のDNAの発現が誘導されるように、哺乳類βアクチンプロモーターと所望のDNAとが結合していることをいう。該DNAと哺乳類βアクチンプロモーターとの間には、該DNAの発現の誘導が起こり得る限り、任意のDNA配列を有していても良い。本発明における「DNAの発現」には、転写および翻訳の双方が含まれる。また、所望のDNAが、所望のタンパク質をコードするDNAであってもよい。
【0021】
本発明においてエンハンサーとは、転写により生成するメッセンジャーRNA(mRNA)の量を結果的に増加させるものであれば限定されない。エンハンサーはプロモーターの作用を促進する効果を持つ塩基配列であり、一般的には100bp前後からなるものが多い。エンハンサーは配列の向きにかかわらず転写を促進することができる。又、エンハンサー自体はプロモーター活性を持っていないのであるが、エンハンサーは数千塩基対も離れた所から転写を活性化することができる。さらに、エンハンサーは転写される遺伝子領域の上流、下流、および遺伝子内にも存在し、転写を活性化することができる。また、上述したように本発明のエンハンサーは、転写により生成するmRNAの量を増加させるものであれば限定されないので、mRNAの転写後にmRNAの分解を阻害したり、mRNAを安定化することによって、細胞内に存在するmRNAの量を増加させ、アミノ酸の翻訳効率を上げるものも本発明のエンハンサーに含まれる。
【0022】
本発明で用いられるエンハンサーは1種類でもよく、又、2つ以上の同一のエンハンサーを複数用いたり、あるいは異なる複数のエンハンサーを組み合わせて用いても良い。
【0023】
本発明において用いられるエンハンサーとしては、WPRE、CMVエンハンサー、HTLV-IのLTRに存在するR-U5'セグメント(Mol. Cell. Biol., Vol.8(1), p.466-472, 1988 )、SV40エンハンサー、ウサギβグロビンの第二エクソンと第三エクソン間に存在するイントロン配列(Proc. Natl. Acad. Sci. USA., Vol.78(3), p.1527-31, 1981)、ヒト成長ホルモンのゲノム領域(J Immunol., Vol. 155(3), p.1286-95, 1995)などを挙げることができるが、より好ましいものはWPREまたはCMVエンハンサーである。
【0024】
WPREは、Woodchuck hepatitis virusゲノム配列(GenBank Accession No. J04514)に存在するHBVPRE(human hepatitis B virus posttranscriptional reguratory element)に類似した領域であり、ゲノム配列の1093番目から1684番目の 592塩基が転写後調節領域であることが報告されている(Journal of Virology, Vol.72, p.5085-5092, 1998)。その後、レトロウイルスベクターを用いた解析により、WPREは目的遺伝子3'末端の非翻訳領域に挿入することにより、5〜8倍高い蛋白産生が得られることが示された。また、WPREの導入により、mRNAの分解が抑制されることも報告された(Journal of Virology, Vol.73, p.2886-2892, 1999)。WPREのようにmRNAの安定化により、アミノ酸の翻訳効率を上げるものも広義のエンハンサーと考えられる。本発明において用いられるWPREとしては、配列番号:3に記載の塩基配列からなるDNAが、より好ましい。
【0025】
また、CMVエンハンサーもWPREと同様の目的で用いることができる。本発明で用いられるサイトメガロウイルスはヒトに感染するサイトメガロウイルス(ヒトサイトメガロウイルス)でもよく、又、非ヒト動物に感染するサイトメガロウイルスでもよい。非ヒト動物に感染するサイトメガロウイルスとしては、例えば、げっ歯類に感染するサイトメガロウイルス(例えば、マウスサイトメガロウイルスなど)を用いることが可能である。CMVエンハンサーは公知の配列を用いることが可能であり、例えばGenBank Accession No. X17403(ヒトサイトメガロウイルス)、GenBank Accession No. L06816(マウスサイトメガロウイルス)に記載のCMVエンハンサーを用いることが可能である。また、CMVエンハンサーは市販されている発現ベクター(ex. ストラタジーン社のpCMV-Script、インビトロジェン社のpcDNA3.1)の多くに、CMVプロモーターの一部として組み込まれている。本発明において用いられるCMVエンハンサーとしては、配列番号:4に記載の塩基配列からなるDNAが、より好ましい。
【0026】
プロモーターはDNAを鋳型に転写を開始するDNA上の特定の塩基配列であり、一般的に、共通の配列を持つ。例えば、大腸菌などの原核生物では、通常、転写開始部位の-10塩基対部位にTATAATG配列を、-35塩基対部位にTTGACA配列を持つ。又、真核生物では、通常、-20塩基対部位にTATAボックスを持つ。
【0027】
本発明で用いられるβアクチンプロモーターとしては、哺乳類βアクチンプロモーターを用いることができる。哺乳類βアクチンプロモーターは、ヒトβアクチンプロモーターやげっ歯類βアクチンプロモーターなど、どのような哺乳類由来のβアクチンプロモーターを用いてもよいが、好ましくはげっ歯類βアクチンプロモーターを用いることができる。げっ歯類βアクチンプロモーターとしては、例えば、マウスβアクチンプロモーター(GenBank Accession No. NT_039317)、ラットβアクチンプロモーター(GenBank Accession No. NW_042778)などを用いることができる。本発明において用いられる哺乳類βアクチンプロモーターとしては、配列番号:1もしくは2に記載の塩基配列からなるDNAが、より好ましい。
【0028】
げっ歯類としては、ネズミ下目、リス下目、ビーバー下目、パラミス下目、デバネズミ下目、ヤマアラシ下目、デンジクネズミ下目などに属する動物を挙げることができ、より具体的には、マウス、ラット、ハムスターなどを挙げることができる。
【0029】
ベクターは、一般的には、所望の遺伝子を宿主に導入して、所望の遺伝子を増幅や発現させる為の運搬体DNA分子のことをいい、好ましくは、公知の制限酵素部位を持ち、栄養要求性であり、宿主内で複製できる能力を持つ。ベクターは通常、プロモーター、エンハンサー、ターミネーター、SD配列、翻訳開始コドン、翻訳終止コドン、複製開始点などを含むことができる。ベクターには、必要に応じて、ベクターが導入された細胞を選択し得る選択マーカーなどをさらに保持させることができる。選択マーカーとしては、アンピシリン、テトラサイクリン、カナマイシン、クロラムフェニコール、ネオマイシン、ハイグロマイシン、ピューロマイシン、ゼオシンのような薬剤耐性遺伝子、ガラクトシダーゼなどの酵素活性を指標に選択し得るマーカー、あるいは、GFPなどの蛍光発光などを指標に選択し得るマーカーなどが挙げられる。また、EGFレセプター、B7-2などの表面抗原を指標に選択し得る選択マーカーなども用いてもよい。このように選択マーカーを用いることにより、該ベクターが導入された細胞、すなわち、本発明のベクターが導入された細胞のみを選択することが可能となる。又、ベクターには、ポリペプチド分泌のためのシグナル配列が含まれていてもよい。ポリペプチド分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(J. Bacteriol., Vol.169(9), p.4379-4383, 1987)を使用すればよい。
【0030】
本発明で用いられるベクターは特に制限されず、いかなるベクターを用いてもよい。
具体的には、哺乳動物由来のベクター(例えば、pcDNA3 (インビトロジェン社製)や、pEGF-BOS (Nucleic Acids. Res., 18(17), p.5322, 1990)、pEF 、pCDM8、pCXN、昆虫細胞由来のベクター(例えば「Bac-to-BAC baculovairus expression system」(インビトロジェン社製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来のベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来のベクター(例えば、pZIPneo)、酵母由来のベクター(例えば、「Pichia Expression Kit」(インビトロジェン社製)、pNV11 、SP-Q01)、枯草菌由来のベクター(例えば、pPL608、pKTH50)、大腸菌ベクター(M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Script)、などが挙げることができる。本発明においては、哺乳動物細胞内で発現可能なベクターを用いることが好ましく、又、発現ベクターを用いることが好ましい。
【0031】
宿主細胞がCHO細胞である場合、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする為に、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pCHOIなど)を導入し、メトトレキセート(MTX)により増幅させることができる。
【0032】
さらに本発明のベクターには、トランスアクティベーターをコードするDNAが含まれていてもよい。トランスアクティベーターは遺伝子の転写を活性化するトランス作動性因子であり、トランス活性化因子ともいわれる。トランスアクティベーターには、DNA結合能を持ちシスエレメントへの結合を介して直接転写を促進するものや、DNA結合能を持たず、他の因子を活性化することで間接的に転写を促進するもの、等が知られている。本発明で使用されるトランスアクティベーターは特に限定されず、どのようなトランスアクティベーターを用いてもよい。トランスアクティベーターの具体的な例としては、アデノウイルスのE1A、ヒト免疫不全ウイルスのTat、ヒトT細胞白血病ウイルスのTax、癌遺伝子産物などを挙げることができるが、本発明においては癌遺伝産物を用いることが好ましい。癌遺伝子は動物の正常細胞に内在し、その活性化突然変異により細胞の癌化を誘導する遺伝子群の総称である。ウイルスがもつ癌遺伝子はウイルス性癌遺伝子、その由来である正常細胞の遺伝子は細胞性癌遺伝子と呼ばれている。癌遺伝子としては例えば、src、yes、fgr、fms、erbB、abl、kit、crk、mos、raf、sis、ras、myc、fos、junなどを挙げることができる。ヒト活性型H-Rasがヒトβアクチンプロモーターを組み込んだ発現ベクターの発現効率を亢進することが報告されているが(Cytotechnology, Vol.16, p.167-178, 1994)、本発明においてもRasを用いることが好ましい。RasにはK-ras、H-ras、N-ras、c-rasがあり、さらに、rho遺伝子ファミリー、rab遺伝子ファミリーを含む場合には、rasスーパーファミリーと呼ばれている。本発明においてはH-rasを好適に用いることができる。トランスアクティベーターとしてRasを用いる場合、Rasは野生型でも活性型でもよい。本発明において用いられるH-rasとしては、配列番号:5又は6のいずれかのDNA又は配列番号:7又は8のアミノ酸配列をコードするDNAが、より好ましい。
【0033】
さらに、本発明のベクターを、時期特異的に遺伝子を発現するよう設計してもよい。時期特異的に遺伝子を発現させる方法としては、例えば、Cre-loxPシステムなどの部位特異的組換えを用いた方法を挙げることができる。
【0034】
Creは、2個のloxP配列間の部位特異的組換えを仲介する、E.coliバクテリオファージP1由来の組換え酵素である。LoxP配列は、Creが結合するための認識配列として働く2個の13bpの逆方向反復配列に両側を挟まれた8bpのスペーサー領域からなる。所望の遺伝子の発現が抑制されるようにloxP配列をベクターに導入しておけば、Creを投与してloxP配列を除去することにより、好きな時期に所望の遺伝子を発現させることが可能である。loxP配列の挿入部位は、プロモーターと所望の遺伝子の間など、所望の遺伝子の発現が抑制される部位であればよい。又、Creの投与は、Cre自体を投与してもよいし、Creをコードする遺伝子を投与してもよい。トランスジェニック動物などの動物でCre-loxPシステムを用いる場合には、例えば、Creを発現するアデノウイルスを感染させることなどにより、所望の遺伝子を発現させることができる(Nucleic. Acids. Res., Vol.23(19), p.3816-3821, 1995)。このような酵素を利用した部位特異的組換えとしては、他にFlp-FRT、Zygosaccharomyces rouxii pSR1、レソルバーゼ-rfsF、ファージMu Ginなどを挙げることができる。
【0035】
さらに、上述した時期特異的発現システム以外に、テトラサイクリンなどの抗生物質を利用した方法や、エクジソンなどのホルモンを利用した方法などを用いることが可能である。これらの方法は当業者に公知であり、市販されているキットを用いることが可能である(エクジソン発現システム(インビトロジェン社)、テトラサイクリン発現システム(クロンテック社)、など)。
【0036】
また、本発明は、本発明のベクターが導入された宿主細胞を提供する。本発明の宿主細胞は、例えば、所望のタンパク質の製造や発現のための産生系として使用することができる。本発明のベクターが導入される宿主細胞としては特に制限はなく、株化された細胞、初代継代細胞、動物個体、受精卵など細胞種を問わない。本発明の宿主細胞としては、例えば、種々の動物細胞(例えば、哺乳動物細胞)や大腸菌などを用いることが可能である。真核細胞を使用する場合、例えば、動物細胞、植物細胞、真菌細胞を宿主に用いることができる。動物細胞としては、ヒト、マウス、ハムスターなどの哺乳類細胞(例えば、CHO、COS、3T3、ミエローマ、BHK(baby hamster kidney)、HeLa、Vero)、両生類細胞(例えばアフリカツメガエル卵母細胞(Nature, Vol.291, p.358-360, 1981))、あるいは昆虫細胞(例えば、Sf9、Sf21、Tn5)などが知られている。CHO細胞としては、特に、DHFR遺伝子を欠損したCHO細胞であるdhfr-CHO(Proc. Natl. Acad. Sci. USA, Vol.77, p.4216-4220, 1980)やCHO K-1(Proc. Natl. Acad. Sci. USA, Vol.60, p.1275, 1968)を好適に使用することができる。植物細胞としては、例えば、ニコチアナ・タバカム(Nicotiana tabacum)由来の細胞がポリペプチド生産系として知られており、これをカルス培養すればよい。真菌細胞としては、酵母、例えば、サッカロミセス(Saccharomyces)属、例えば、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)、糸状菌、例えば、アスペルギルス(Aspergillus)属、例えば、アスペルギルス・ニガー(Aspergillus niger)が知られている。原核細胞を使用する場合、細菌細胞を用いる産生系がある。細菌細胞としては、大腸菌(E. coli)、例えば、JM109、DH5α、HB101等が挙げられ、その他、枯草菌が知られている。本発明において好ましい宿主細胞としては、動物細胞を挙げることができ、さらに好ましくは哺乳動物細胞、特に好ましくはげっ歯類細胞(例えばCHO細胞)を挙げることができる。
【0037】
又、本発明の宿主細胞として、癌遺伝子が活性化している細胞を用いることも可能である。癌遺伝子が活性化している細胞とは、正常細胞と比較して癌遺伝子が多量に発現している細胞、または変異した癌遺伝子が発現している細胞などをいう。
【0038】
癌遺伝子が活性化している細胞は、人為的に癌遺伝子が活性化された細胞でも非人為的に癌遺伝子が活性化された細胞でもよい。人為的に癌遺伝子が活性化された細胞の具体的な例としては、癌遺伝子(野生型または活性型)を含むベクターが導入された細胞や、癌遺伝子に人為的に変異を導入した細胞などを挙げることができる。非人為的に癌遺伝子が活性化された細胞の具体的な例としては、癌化した細胞(ex. T24ヒト膀胱癌細胞 Nature, Vol.302, p.33-37, 1983)などを挙げることができる。癌遺伝子の活性化のメカニズムとしては、プロモーター・インサーション、点突然変異、遺伝子増幅、転座などによると考えられている。
【0039】
また、本発明のベクターの上記細胞への導入方法は、当業者においては、細胞の種類により適宜選択することができる。例えば、哺乳動物細胞への導入では、リン酸カルシウム法(Virology, Vol.52, p.456, 1973)、DEAEデキストラン法、カチオニックリボソームDOTAP(ロッシュ・ダイアグノスティックス社製)を用いた方法、エレクトロポレーション法(Nucleic Acids Res., Vol.15, p.1311, 1987) 、リポフェクション法(J. Clin. Biochem. Nutr., Vol.7, p.175, 1989)、ウィルスによる感染導入方法(Sci.Am., p.34, 1994) 、パーティクルガンなどから選択することができ、植物細胞への導入では、エレクトロポレーション法(Nature, Vol.319, p.791, 1986) 、ポリエチレングリコール法(EMBO J., Vol.3, p.2717, 1984) 、パーティクルガン法(Proc. Natl. Acad. Sci. USA, Vo l.85, p.8502, 1988) 、アグロバクテリュウムを介した方法(Nucleic. Acids Res., Vol.12, p.8711, 1984) 等により行うことができる。
【0040】
また、Trans IT(タカラ社)、PolyFect Transfection Reagent(キアゲン社)、LipofectAMINE(インビトロジェン社)などのキットを利用してもよい。さらに、本発明の宿主細胞は、本発明のベクター以外に、トランスアクティベーターをコードするDNAを有するベクターをさらに含んでいてもよい。
【0041】
本発明はさらに本発明のベクターを用いた所望のタンパク質の製造方法及び所望のDNAを発現させる方法に関する。タンパク質製造のための産生系は、in vitroおよびin vivo の産生系がある。in vitroの産生系としては、真核細胞を使用する産生系や原核細胞を使用する産生系が挙げられる。例えば、上述した宿主細胞をin vitroで培養することにより所望のタンパク質が得られる。培養は、公知の方法に従い行うことができる。例えば、動物細胞の培養液として、例えば、DMEM、MEM、RPMI1640、IMDM、F10培地、F12培地などを使用することができる。その際、牛胎児血清(FCS)等の血清補液を併用することもできるし、無血清培養してもよい。さらに、トランスアクティベーターを培地に添加してもよい。培養時のpHは、約6〜8であるのが好ましい。培養は、通常、約30〜40℃で約15〜200時間行い、必要に応じて培地の交換、通気、攪拌を加える。培養条件は使用する細胞の種類によって異なるので、当業者は適宜好適な条件を決定することができる。例えばCHO細胞であれば通常、気相のCO2濃度が0−40%、好ましくは、2−10%の雰囲気下、30−39℃、好ましくは、37℃程度で、1−14日間培養すればよい。また、動物細胞培養用の各種の培養装置としては、例えば発酵槽型タンク培養装置、エアーリフト型培養装置、カルチャーフラスコ型培養装置、スピンナーフラスコ型培養装置、マイクロキャリアー型培養装置、流動層型培養装置、ホロファイバー型培養装置、ローラーボトル型培養装置、充填槽型培養装置等を用いて培養することができる。
【0042】
一方、in vivoでタンパク質を産生させる系としては、例えば、動物を使用する産生系や植物を使用する産生系が挙げられる。これらの動物又は植物に目的とするDNAを導入し、動物又は植物の体内でポリペプチドを産生させ、回収する。本発明における「宿主」とは、これらの動物、植物を包含する。動物を使用する場合、哺乳類動物、昆虫を用いる産生系がある。哺乳類動物としては、ヤギ、ブタ、ヒツジ、マウス、ウシを用いることができる(Vicki Glaser, SPECTRUM Biotechnology Applications, 1993)。また、哺乳類動物を用いる場合、トランスジェニック動物を用いることができる。例えば、所望のタンパク質をコードするDNAを、ヤギβカゼインのような乳汁中に固有に産生されるポリペプチドをコードする遺伝子との融合遺伝子として調製する。次いで、この融合遺伝子を含むDNA断片をヤギの胚へ注入し、この胚を雌のヤギへ移植する。胚を受容したヤギから生まれるトランスジェニックヤギ又はその子孫が産生する乳汁から、所望のタンパク質を得ることができる。トランスジェニックヤギから産生されるポリペプチドを含む乳汁量を増加させるために、適宜ホルモンをトランスジェニックヤギに使用してもよい(Bio/Technology, Vol.12, p.699-702, 1994)。また、昆虫としては、例えばカイコを用いることができる。カイコを用いる場合、所望のタンパク質をコードするDNAを挿入したバキュロウィルスをカイコに感染させることにより、このカイコの体液から所望のタンパク質を得ることができる(Nature, Vol.315, p.592-594, 1985)。
【0043】
さらに、植物を使用する場合、例えばタバコを用いることができる。タバコを用いる場合、所望のタンパク質をコードするDNAを植物発現用ベクター、例えばpMON 530に挿入し、このベクターをアグロバクテリウム・ツメファシエンス(Agrobacterium tumefaciens)のようなバクテリアに導入する。このバクテリアをタバコ、例えば、ニコチアナ・タバカム(Nicotiana tabacum)に感染させ、本タバコの葉より所望のタンパク質を得ることができる(Eur. J. Immunol., Vol.24, p.131-138, 1994)。
【0044】
本発明のタンパク質製造方法において、遺伝子の発現は一過性発現でもよい。遺伝子の一過性の発現を目的とする場合には、例えば、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。さらに、遺伝子治療などのように所望の遺伝子を生体内で発現させる場合には、所望の遺伝子をベクターに組み込み、例えば、レトロウイルス法、リポソーム法、カチオニックリポソーム法、アデノウイルス法などにより生体内に導入する方法などが挙げられる。用いられるベクターとしては、例えば、アデノウイルスベクター(例えばpAdexlcw)やレトロウイルスベクター(例えばpZIPneo)などが挙げられるが、これらに制限されない。ベクターへの所望の遺伝子の挿入などの一般的な遺伝子操作は、常法に従って行うことが可能である(Molecular Cloning, 5.61-5.63)。生体内への投与は、ex vivo法であっても、in vivo法であってもよい。
【0045】
本発明により得られた所望のタンパク質は、宿主細胞内または細胞外(培地など)から単離し、実質的に純粋で均一なタンパク質として精製することができる。タンパク質の分離、精製は、通常のタンパク質の精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせればタンパク質を分離、精製することができる。クロマトグラフィーとしては、例えばアフィニティークロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al., Cold Spring Harbor Laboratory Press, 1996)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。なお、タンパク質を精製前又は精製後に適当なタンパク質修飾酵素を作用させることにより、任意に修飾を加えたり、部分的にペプチドを除去することもできる。タンパク質修飾酵素としては、例えば、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼなどが用いられる。
【0046】
本発明のベクター、DNA構築物または宿主細胞は、所望のタンパク質を製造する際に用いることができる。本発明において、所望のタンパク質とはいかなるタンパク質でもよく、タンパク質断片やペプチドなども含まれる。所望のタンパク質の具体例としては、抗体、エリスロポイエチン、コロニー刺激因子(顆粒球、マクロファージ、顆粒球マクロファージ)、インターロイキン1〜31、インターフェロン、RANTES、リンホトキシンβ、Fasリガンド、flt−3リガンド、NF−κBの受容体活性化因子のリガンド(RANKL)、TNF関連アポトーシス誘導リガンド(TRAIL)、CD40リガンド、OX40リガンド、4−1BBリガンド(およびTNFファミリーの他のメンバー)、胸腺ストロマ由来リンホポエチン、マスト細胞増殖因子、幹細胞増殖因子、上皮増殖因子、成長ホルモン、腫瘍壊死因子、白血病阻害因子、オンコスタチンM、トロンボポエチンのような造血因子などのサイトカイン、成長因子、などを挙げることができる。
【0047】
さらに、本発明のベクター又はDNA構築物はトランスジェニック動物の作製に用いることも可能である。トランスジェニック動物の作製は公知の方法、例えば、以下のようにして作製することが可能である。まず、トランスジェニック動物内で発現させたい目的遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入する。該ベクター、該DNA構築物、又はそれらから切り出した発現カセットを全能性細胞に導入する。全能性細胞としては、受精卵、初期胚、胚性幹細胞(ES細胞)等を用いることが可能である。全能性細胞への導入は、静電パルス法、リポソーム法、リン酸カルシウム法、マイクロインジェクション法、あるいはレトロウイルスを感染させるなど、通常用いられる方法により行うことができる。上記処理を行った全能性細胞を仮親の卵管に移植し、産仔を出生させ、その産仔の中から、目的遺伝子を有する個体を選抜する。目的遺伝子を有する個体であるか否かは、目的遺伝子に特異的なプライマーを用いたサザンブロッティングやPCRによる確認などで判定することができる。
【0048】
トランスジェニック動物としては、ヒト以外の非ヒト動物であれば特に制限されず、マウス、ラット、ハムスター、モルモット、ウサギ、ブタ、ミニブタ、ウシ、ヒツジ、ネコ、イヌ等の哺乳動物、ニワトリ等の鳥類、魚類、昆虫類、線虫類などを挙げることができる。操作上の点などから、好ましくはげっ歯類であり、特に好ましくはマウスである。
【0049】
又、本発明のベクター又はDNA構築物は、アンチセンス法やRNAiなどに用いることも可能である。アンチセンス法は、標的遺伝子に対して相補的な配列を持つオリゴヌクレオチドが細胞内に存在すると、該アンチセンス(アンチセンスDNA、アンチセンスRNAなど)が標的遺伝子(標的mRNA、標的DNA、など)と対合することにより、翻訳・転写を阻害し、標的遺伝子の発現を抑制することをいう。従って、アンチセンスオリゴヌクレオチドを、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターで細胞を形質転換することにより、細胞内での標的DNAの発現を阻害することが可能となる。
【0050】
又、RNA干渉(RNA interfearenece:RNAi)を用いて標的遺伝子を阻害することも可能である。RNAiは、二本鎖RNA(dsRNA)を細胞内に導入した際に、そのRNA配列に対応する細胞内のmRNAが特異的に分解され、蛋白質として発現されなくなる現象をいう。RNAiの場合、通常、二本鎖RNAが用いられるが、自己相補的な一本鎖RNA中で形成される二本鎖を用いることも可能である。二本鎖を形成する領域は、全ての領域において二本鎖を形成していてもよいし、一部の領域(例えば両末端又は片方の末端など)が一本鎖等になっていてもよい。RNAiに用いられるオリゴRNAは10〜100bpのRNAが用いられることが多く、通常19〜23bpのRNAが用いられる。従って、細胞内で二本鎖RNAを形成するように設計された遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターで細胞を形質転換することにより、細胞内での標的遺伝子の発現を阻害することが可能となる。RNAi法は、Nature, Vol.391, p.806, 1998、Proc.Natl.Acsd.Sci.USA, Vol.95, p.15502, 1998、Nature, Vol.395, p.854, 1998、Proc.Natl.Acsd.Sci.USA, Vol.96, p.5049, 1999、Cell, Vol.95, p.1017, 1998、Proc.Natl.Acsd.Sci.USA, Vol.96, p.1451, 1999、Proc.Natl.Acsd.Sci.USA, Vol.95, p.13959, 1998、Nature Cell Biol., Vol.2, p.70, 2000等の記載に従って行うことができる。
【0051】
さらに、本発明のベクター又はDNA構築物は、遺伝子治療に用いることも可能である。遺伝子治療は、変異した遺伝子を補正することを目的に、外部から患者の細胞に正常な遺伝子を導入し、細胞の表現型を変化させることにより、病気の治療を行う方法である。又、遺伝子治療は、遺伝子病の治療のみならず、エイズや癌などの他の疾患に対しても有効であると考えられている。遺伝子治療は、生体に直接遺伝子を導入し、細胞に遺伝子を組み込む方法(in vivo法)と、患者の細胞を採取し、体外で細胞に遺伝子を導入した後、該細胞を再度、患者に移植する方法(ex vivo法)に分けられる。
【0052】
遺伝子の投与は如何なる方法を用いてもよく、マイクロインジェクション、リン酸カルシウム法、エレクトロポレーション法、パーティクルガン、レトロウイルスなどのウイルスベクターを用いた方法などにより投与することが可能である。現在、遺伝子治療にはウイルスベクターが広範に用いられている。ウイルスベクターとして用いられるウイルスの例としては、アデノウイルス、アデノ関連ウイルス、ヘルペスウイルス、ワクシニアウイルス、レトロウイルス、ポリオーマウイルス、パピローマウイルス、レンチウイルスなどを挙げることができる。
【0053】
又、パーティクルガンでの導入のように、遺伝子を直接導入する方法も確立されている。in vivo法で遺伝子を導入する場合、その投与経路は限定されず、静脈内、筋肉、皮下、皮内、粘膜、心臓や肝臓などの臓器に直接投与、など如何なる経路で投与してもよい。このように、患者の細胞に導入したい遺伝子を、本発明のDNA構築物又は本発明のベクターに挿入し、該DNA構築物または該ベクターを、in vivo法又はex vivo法により患者の細胞内に導入することにより、遺伝子治療が可能となる。遺伝子治療の具体的な方法は、別冊実験医学 遺伝子治療の基礎技術(羊土社)、別冊実験医学 遺伝子導入と発現解析実験法(羊土社)、日本遺伝子治療学会編遺伝子治療開発研究ハンドブック(エヌ・ティー・エス)などを参考に行うことができる。
【0054】
さらに、本発明のベクター又はDNA構築物は、DNAワクチンの発現の為に用いることも可能である。DNAワクチンは、抗原DNAを体内で発現させ、その抗原に対する免疫反応を誘起する方法と、CpGの繰り返し配列を免疫増強物質として接種する方法の二つがある。DNAワクチンを接種すると、体内で抗原タンパク質が合成され、ウイルスなどの自然感染と同様の免疫反応を誘導する。DNAワクチン発現の為のベクター、投与方法、投与経路などは遺伝子治療と同様の方法により行うことが可能である。
【0055】
さらに、本発明のベクター又はDNA構築物は、免疫原として抗体作製に用いることも可能である(J. Virol., Vol.70(9), p.6119-25, 1996)。免疫原となるタンパク質又はペプチドを発現するよう、本発明のDNA構築物又はベクターを構築し、該ベクター又はDNA構築物を免疫動物に投与することにより、免疫動物の体内で免疫原となるタンパク質又はペプチドが発現し、該タンパク質又はペプチドに対する抗体が産生される。より具体的には、例えば、以下のような方法により抗体を作製することが可能である。
【0056】
まず、免疫原となるタンパク質又はペプチドをコードする遺伝子を、本発明のDNA構築物又はベクターに挿入する。該DNA構築物またはベクターを免疫動物(非ヒト動物)に投与する。投与は如何なる方法を用いてもよく、ベクターをそのまま投与する方法以外に、例えば、エレクトロポレーション法、パーティクルガン、レトロウイルスなどのウイルスベクターを用いた方法などにより投与することが可能である。又、化学物質(ブピバカインなど)など、他の物質と一緒に投与してもよい。投与経路は静脈内、筋肉、皮下、皮内、粘膜、心臓や肝臓などの臓器に直接投与など、如何なる経路で投与してもよい。感作抗原で免疫される哺乳動物としては、特に限定されるものではないが、細胞融合に使用する親細胞との適合性を考慮して選択するのが好ましく、一般的にはげっ歯類の動物、例えば、マウス、ラット、ハムスター、あるいはウサギ、サル等が使用される。このように哺乳動物を免疫し、血清中に所望の抗体レベルが上昇するのを確認した後に、哺乳動物から免疫細胞を採取し、細胞融合に付されるが、好ましい免疫細胞としては、特に脾細胞が挙げられる。
【0057】
前記免疫細胞と融合される他方の親細胞として、哺乳動物のミエローマ細胞を用いる。このミエローマ細胞は、公知の種々の細胞株、例えば、P3(P3x63Ag8.653)(J. Immnol., Vol.123, p.1548-1550, 1979)、 P3x63Ag8U.1(Current Topics in Microbiology and Immunology, Vol.81, p.1-7, 1978)、 NS-1 (Eur. J. Immunol., Vol.6, p.511-519, 1976)、MPC-11(Cell, Vol.8, p.405-415, 1976)、SP2/0 (Nature, Vol.276, p.269-270, 1978)、FO(J. Immunol. Methods, Vol.35, p.1-21, 1980)、S194(Trowbridge, I. S. J. Exp. Med. Vol.148, p.313-323, 1978)、R210(Nature, Vol.277, p.131-133, 1979)等が好適に使用される。前記免疫細胞とミエローマ細胞との細胞融合は、基本的には公知の方法、たとえば、ケーラーとミルステインらの方法(Methods Enzymol., Vol.73, p.3-46, 1981)等に準じて行うことができる。より具体的には、前記細胞融合は、例えば細胞融合促進剤の存在下に通常の栄養培養液中で実施される。融合促進剤としては、例えばポリエチレングリコール(PEG)、センダイウイルス(HVJ)等が使用され、更に所望により融合効率を高めるためにジメチルスルホキシド等の補助剤を添加使用することもできる。
【0058】
免疫細胞とミエローマ細胞との使用割合は任意に設定することができる。例えば、ミエローマ細胞に対して免疫細胞を1-10倍とするのが好ましい。前記細胞融合に用いる培養液としては、例えば、前記ミエローマ細胞株の増殖に好適なRPMI1640培養液、MEM培養液、その他、この種の細胞培養に用いられる通常の培養液が使用可能であり、さらに、牛胎児血清(FCS)等の血清補液を併用することもできる。細胞融合は、前記免疫細胞とミエローマ細胞との所定量を前記培養液中でよく混合し、予め37℃程度に加温したPEG溶液(例えば平均分子量1000-6000程度)を通常30-60%(w/v)の濃度で添加し、混合することによって目的とする融合細胞(ハイブリドーマ)を形成する。続いて、適当な培養液を逐次添加し、遠心して上清を除去する操作を繰り返すことによりハイブリドーマの生育に好ましくない細胞融合剤等を除去する。
【0059】
このようにして得られたハイブリドーマは、通常の選択培養液、例えばHAT培養液(ヒポキサンチン、アミノプテリンおよびチミジンを含む培養液)で培養することにより選択される。上記HAT培養液での培養は、目的とするハイブリドーマ以外の細胞(非融合細胞)が死滅するのに十分な時間(通常、数日〜数週間)継続する。ついで、通常の限界希釈法を実施し、目的とする抗体を産生するハイブリドーマのスクリーニングおよび単一クローニングを行う。
【0060】
また、ヒト以外の動物に抗原を免疫して上記ハイブリドーマを得る他に、ヒトリンパ球をin vitroで感作し、感作リンパ球をヒト由来の永久分裂能を有するミエローマ細胞と融合させ、結合活性を有する所望のヒト抗体を得ることもできる(特公平1-59878号公報参照)。さらに、ヒト抗体遺伝子の全てのレパートリーを有するトランスジェニック動物に抗原を投与して抗体産生細胞を取得し、これを不死化させた細胞から抗原に対するヒト抗体を取得してもよい(国際特許出願公開番号WO 94/25585 号公報、WO 93/12227 号公報、WO92/03918 号公報、WO 94/02602 号公報参照)。このようにして作製されるモノクローナル抗体を産生するハイブリドーマは、通常の培養液中で継代培養することが可能であり、また、液体窒素中で長期保存することが可能である。当該ハイブリドーマからモノクローナル抗体を取得するには、当該ハイブリドーマを通常の方法にしたがい培養し、その培養上清として得る方法、あるいはハイブリドーマをこれと適合性がある哺乳動物に投与して増殖させ、その腹水として得る方法などが採用される。前者の方法は、高純度の抗体を得るのに適しており、一方、後者の方法は、抗体の大量生産に適している。
【0061】
本発明では、モノクローナル抗体として、抗体遺伝子をハイブリドーマからクローニングし、適当なベクターに組み込んで、これを宿主に導入し、遺伝子組換え技術を用いて産生させた組換え型のものを用いることができる(例えば、Eur. J. Biochem., Vol.192, p.767-775, 1990参照)。
【0062】
具体的には、抗体を産生するハイブリドーマから、抗体の可変(V)領域をコードするmRNAを単離する。mRNAの単離は、公知の方法、例えば、グアニジン超遠心法(Biochemistry, Vol.18, p.5294-5299, 1979)、AGPC法(Anal. Biochem., Vol.162, p.156-159, 1987)等により行って全RNAを調製し、mRNA Purification Kit (ファルマシア製)等を使用して目的のmRNAを調製する。また、QuickPrep mRNA Purification Kit (ファルマシア製)を用いることによりmRNAを直接調製することもできる。得られたmRNAから逆転写酵素を用いて抗体V領域のcDNAを合成する。cDNAの合成は、AMV Reverse Transcriptase First-strand cDNA Synthesis Kit(生化学工業社製)等を用いて行う。また、cDNAの合成および増幅を行うには、5'-Ampli FINDER RACE Kit(クロンテック製)およびPCRを用いた5'-RACE法(Proc. Natl. Acad. Sci. USA, Vol.85, p.8998-9002, 1988、Nucleic Acids Res., Vol.17, p.2919-2932, 1989)等を使用することができる。得られたPCR産物から目的とするDNA断片を精製し、ベクターDNAと連結する。さらに、これより組換えベクターを作製し、大腸菌等に導入してコロニーを選択して所望の組換えベクターを調製する。そして、目的とするDNAの塩基配列を公知の方法、例えば、ジデオキシヌクレオチドチェインターミネーション法等により確認する。目的とする抗体のV領域をコードするDNAを得たのち、これを、所望の抗体定常領域(C領域)をコードするDNAを含有する発現ベクターへ組み込む。
【0063】
抗体を製造するには、通常、抗体遺伝子を発現制御領域、例えば、エンハンサー、プロモーターの制御のもとで発現するよう発現ベクターに組み込む。次に、この発現ベクターにより、宿主細胞を形質転換し、抗体を発現させる。抗体遺伝子の発現は、抗体重鎖(H鎖)または軽鎖(L鎖)をコードするDNAを別々に発現ベクターに組み込んで宿主細胞を同時形質転換させてもよいし、あるいはH鎖およびL鎖をコードするDNAを単一の発現ベクターに組み込んで宿主細胞を形質転換させてもよい(WO 94/11523 号公報参照)。
【0064】
以上のように、本発明のDNA構築物又はベクターは、所望のタンパク質などの製造、アンチセンス法、RNAi法、遺伝子治療、DNAワクチン、トランスジェニック動物、抗体の作製などに適用することが可能である。
【0065】
さらに本発明は、本発明のベクターを、該ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法に関する。
【0066】
動物目とは、生物分類のリンネ式階層分類において、綱の次下位、科階級群の上位におかれる基本的階級あるいは、その階級にあるタクソンのことをいう。具体的な動物目としては、例えば、ネズミ目、ウサギ目、ハネジネズミ目、ツパイ目などを挙げることができるが、ネズミ目が好ましい。ネズミ目には、ハムスター、ラット、マウス、モルモット、リス、ビーバーなどが含まれる。宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0067】
又本発明は、本発明のベクターを、該ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法に関する。
【0068】
同じ動物種由来とは、宿主細胞がヒト細胞の場合にはヒトβアクチンプロモーターを用い、宿主細胞がマウス細胞の場合にはマウスβアクチンプロモーターを用い、宿主細胞がハムスター細胞の場合にはハムスターβアクチンプロモーターを用いることをいう。本方法によって、宿主細胞と同種由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0069】
さらに本発明は、宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることを特徴とする所望の遺伝子の発現量を増加させる方法に関する。動物目とは、上記に記載の定義による。宿主細胞と同じ動物目由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0070】
又、本発明は、宿主細胞と同じ動物種由来のβアクチンプロモーターを用いることを特徴とする所望の遺伝子の発現量を増加させる方法に関する。同じ動物種由来とは、上記に記載の定義による。宿主細胞と同じ動物種由来のβアクチンプロモーターを用いることにより、所望の遺伝子の発現量を増加させることが可能となる。
【0071】
又、本発明においては所望のDNAの発現量を増加させる目的で、トランスアクティベーターを用いてもよい。トランスアクティベーターは、トランスアクティベーターをコードするDNAを含むベクターを宿主細胞に導入し、培養時にトランスアクティベーターが発現するようにしてもよいし、トランスアクティベーターを培地に添加してもよい。トランスアクティベーターをコードするDNAは、所望のタンパク質をコードするDNAと同一のベクター上に組み込んでもよいし、異なるベクターに組み込んで両方のベクターを細胞に導入してもよい。
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0072】
以下、本発明を実施例により、さらに具体的に説明するが本発明はこれら実施例に制限されるものではない。
【0073】
〔実施例1〕 発現ベクターpmAct-Luc-neoの構築
(1)マウスβアクチンプロモーターのクローニング
NCBI(http://www.ncbi.nlm.nih.gov/)、及びJackson laboratory(http://www.jax.org/)で公開されているマウスゲノム情報より、マウスβアクチンのシーケンス情報を得た。以下に示す配列のプライマーmAct5-F1(5'- GGGAGTGACTCTCTGTCCATTCAATCC -3'/配列番号:9)およびmAcr5-R1(5'- TTGTCGACGACCAGCGCAGCGATATCG -3'/配列番号:10)を合成し(エスペックオリゴ社)、PCRによってマウスβアクチンのプロモーター領域(1,577 bp)を増幅した。試薬としてはタカラバイオ社のTaKaRa LA Taq with GC Buffer(cat. RR02AG)、鋳型DNAにはクロンテック社のMouse Genomic DNA(cat. 6650-1)を使用してPCRを行った。PCR反応溶液の組成は、鋳型DNA(100ng/ml) 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mAct5-F1(10μM)2.0μl、mAcr5-R1(10μM)2.0μl、H2O 11.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で1分、「95℃で30秒、60℃で30秒、72℃で90秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、1,577bpのバンドを切り出し、Mag Extractor(東洋紡社、cat.NPK-601)で精製した。これをpGEM-T-Easy vector(プロメガ社、cat.A1360)にクローニングし、シーケンスすることによって塩基配列を確認した。クローニングしたマウスβアクチン5'領域を配列番号:1に示す。
【0074】
公共のデータベースの配列では、305番目から始まるTは13個の連続であるが、クローニングした領域は記載した配列の通り14個の連続したTであった。データベースと配列が異なるが、これをそのまま実験に用いることにした。また、上記の配列中、斜体で示した414番目から始まるCAAT、及び475番目から始まるTATAAはhouse keeping遺伝子のプロモーター領域によく見出される配列であり、得られた領域はマウスβアクチンのプロモーター領域であることが推察された。
【0075】
クローニングした領域はマウスベータアクチンの開始コドン(上記配列中の1543番目から始まるATG)を含むこと、及びその後のベクター構築のために、プライマーmAct5-BG(5'- AGATCTGGGAGTGACTCTCTGTCCAT -3'/配列番号:11、Bgl IIサイトを含む)およびmAct5-HN(5'- AAGCTTGGCGAACTATCAAGACACAA -3'/配列番号:12、Hind IIIサイトを含む)を合成し(エスペックオリゴ社)、再度PCRすることによって必要な領域(1〜1542/配列番号:2)にBgl II(5'末)、Hind III(3'末)サイトを付加した。
【0076】
PCR反応溶液の組成は、鋳型DNA(約10ng/ml) 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mAct5-HN(10μM)1.0μl、mAct5-BG(10μM)1.0μl、H2O 13.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で30秒、「95℃で30秒、60℃で30秒、72℃で60秒」を30サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は前述のようにpGEM-T-Easy vectorにクローニングし、シーケンス確認を行った。こうして作製したマウスβアクチンフラグメントをpGL3-Basic(プロメガ社、cat. E1751)のBgl II-Hind IIIサイトに組み込んだ。さらに、pGL3-Basicはベクターにおける配列4650位のNot IサイトをBamH Iサイトに変換し、配列2004位のBamH Iサイトとの間のベクター骨格を、pCXNベクターのベクター骨格(BamH I- BamH Iフラグメント)と変換することで、ネオマイシン耐性遺伝子を導入し、pmAct-Luc-neoとした(図1)。なお、pGL3-Basicのベクター骨格を変換したベクターを、以下pGLNベクターと称する。
【0077】
(2)WPREのクローニングとpmAct-WPRE-Luc-neoの作製
Woodchuck hepatitis virusゲノム配列(GenBank Accession No. J04514)の1093番目から1684番目の 592塩基が転写後調節領域(WPRE)は、以下のようにAssemble PCR法を用いて増幅し、クローニングした。タカラバイオ社のTaKaRa Ex Taq (cat. RR001B) を使用して、以下のような二つの条件系でPCRを行った。
【0078】
<PCR反応1>
まず、反応溶液の組成:合成DNA(WP-1〜WP-6,各10uM, 各1uL) 6.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、H2O 28.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃で5分、「94℃で2分、55℃で2分、72℃で2分」を2サイクル、によりPCR反応を行った。さらに、プライマーWP-f (10uM) 1.0μl、WP-r2 (10uM) 1.0μlを添加し総量を50,0μlとし、反応条件:「94℃で30秒、55℃で30秒、72℃で1分」で30サイクル、72℃ で5分、によりPCR反応を行った。
【0079】
<PCR反応2>
まず、反応溶液の組成:合成DNA(WP-5〜WP-17,各10uM, 各1uL) 13.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、H2O 21.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃で5分、「94℃で2分、55℃で2分、72℃で2分」を2サイクル、によりPCR反応を行った。さらに、プライマーWP-f2 (10uM) 1.0μl、WP-r (10uM) 1.0uLを添加し総量を50,0μlとし、反応条件:「94℃ 30秒、55℃ 30秒、72℃ 1分」で30サイクル、72℃ 5分、によりPCR反応を行った。
【0080】
PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、反応1は約200bp、反応2は約400bpのバンドを切り出し、QIAquick Gel Extraction Kit(キアゲン社、cat.28704)で精製した。
【0081】
各反応産物を用いて、さらにPCRを行った。試薬は、タカラバイオ社のTaKaRa Ex Taq (cat. RR001B) を使用した。反応溶液の組成:反応1産物 1.0μl、反応2産物 1.0μl、10x Ex Taq buffer 5.0μl、dNTP Mixture 8.0μl、WP-f (10uM) 1.0μl、WP-r (10uM) 1.0μl、H2O 32.5μl、Ex Taq(5U/μl) 0.5μl、反応条件:94℃ 30秒、「94℃ 15秒、72℃ 2分」を5サイクル、「94℃ 15秒、70℃ 2分」を5サイクル、「94℃ 15秒、68℃ 2分」を28サイクル、72℃ 5分、によりPCR反応を行った。
【0082】
増幅したPCR産物は1% アガロースゲルに電気泳動した後に、約600bpのバンドを切り出し、QIAquick Gel Extraction Kit(キアゲン社、cat.28704)で精製した。反応産物をpGEM-T-Easy vector(プロメガ社、cat.A1360)にクローニングし、塩基配列を決定した。GenBank Accession No. J04514の1093番目から1684番目の配列と同じ配列であることを確認した。pGEM-T/WPREをXbaIで消化し、得られたWPRE断片をpmAct-Luc-neoのXbaI部位に挿入し、pmAct-WPRE-Luc-neoを作製した。配列番号:3にクローニングしたWPRE配列および使用した合成DNAを、使用した合成DNA(配列番号:13から33)を以下に示す。
<使用した合成DNA>
WP-1: AATCAACCTCTGGATTACAAAATTTGTGAAAGATTGACTGGTATTCTTAA(配列番号:13)
WP-2: GCGTATCCACATAGCGTAAAAGGAGCAACATAGTTAAGAATACCAGTCAA(配列番号:14)
WP-3: ACGCTATGTGGATACGCTGCTTTAATGCCTTTGTATCATGCTATTGCTTC(配列番号:15)
WP-4: TTATACAAGGAGGAGAAAATGAAAGCCATACGGGAAGCAATAGCATGATA(配列番号:16)
WP-5: TTCTCCTCCTTGTATAAATCCTGGTTGCTGTCTCTTTATGAGGAGTTGTG(配列番号:17)
WP-6: GTGCACACCACGCCACGTTGCCTGACAACGGGCCACAACTCCTCATAAAG(配列番号:18)
WP-7: CGTGGCGTGGTGTGCACTGTGTTTGCTGACGCAACCCCCACTGGTTGGGG(配列番号:19)
WP-8: GTCCCGGAAAGGAGCTGACAGGTGGTGGCAATGCCCCAACCAGTGGGGGT(配列番号:20)
WP-9: CAGCTCCTTTCCGGGACTTTCGCTTTCCCCCTCCCTATTGCCACGGCGGA(配列番号:21)
WP-10: TGTCCAGCAGCGGGCAAGGCAGGCGGCGATGAGTTCCGCCGTGGCAATAG(配列番号:22)
WP-11: TTGCCCGCTGCTGGACAGGGGCTCGGCTGTTGGGCACTGACAATTCCGTG(配列番号:23)
WP-12: CCATGGAAAGGACGTCAGCTTCCCCGACAACACCACGGAATTGTCAGTGC(配列番号:24)
WP-13: TGACGTCCTTTCCATGGCTGCTCGCCTGTGTTGCCACCTGGATTCTGCGC(配列番号:25)
WP-14: GAGGGCCGAAGGGACGTAGCAGAAGGACGTCCCGCGCAGAATCCAGGTGG(配列番号:26)
WP-15: ACGTCCCTTCGGCCCTCAATCCAGCGGACCTTCCTTCCCGCGGCCTGCTG(配列番号:27)
WP-16: GAGGGCGAAGGCGAAGACGCGGAAGAGGCCGCAGAGCCGGCAGCAGGCCGCGGGAAG(配列番号:28)
WP-17: GTCTTCGCCTTCGCCCTCAGACGAGTCGGATCTCCCTTTGGGCCGCCTCCCCGCCTG(配列番号:29)
WP-f: TCTAGAAATCAACCTCTGGATTACAAAATT(配列番号:30)
WP-r: TCTAGAAGGCGGGGAGGCGGCCCAAA(配列番号:31)
WP-f2: ATCCTGGTTGCTGTCTCTTTATGAG(配列番号:32)
WP-r2: GTGCACACCACGCCACGTTGCC(配列番号:33)
【0083】
(3)phCMV-mAct-Luc-neoの作製
CMVエンハンサー領域(配列番号:4)をpmAct-Luc-neoベクターの EcoR Iサイトにクローニングし、phCMV-mAct-Luc-neoを完成させた(図1)。
【0084】
(4)比較対象となるベクターの構築
ヒトEF1αプロモーターはDHFR-ΔE-RVh-PM1-f(参考文献 WO92/19759)よりCMVエンハンサーと共にpGLNベクターのMCSに組み込むことで、pCEF-Luc-neoを作製した(図1)。実験に用いた全てのベクターは、キアゲン社のEndoFree Plasmid Maxi Kit (cat.12362)によって精製した。
【0085】
〔実施例2〕ベクターのCHO細胞への導入および発現測定
(1)ベクターのCHO細胞への導入
2μgのベクターをそれぞれ20μlのPLUS(tm) Reagent (インビトロジェン社、Cat. No. 11514-015 )と混合し、OPTI-MEM I Reduced-Serum Medium (インビトロジェン社、Cat. No. 11058-021)で容量を200μlに調整し、室温で15分間保温した。ここに、LipofectAMINE (インビトロジェン社、Cat. No. 18324-012 )を20μl、OPTI-MEM I Reduced-Serum Medium を180μl加え、室温でさらに15分間保温した。CHO細胞は96well Cell Culture Cluster (コーニング社、Cat. No. 3595)で1ウェルあたり2×104 cells、50μlのOPTI-MEM I Reduced-Serum Mediumで調整した。上記で作製したDNA溶液20μlをCHO細胞に添加し、37℃、3時間、CO2インキュベーター内で培養することで、ベクターをCHO細胞に導入した。その後、培養上清を静かに除き、CHO用の培地を加えた。CHO用の培地は、CHO-S-SFMII培地(インビトロジェン社、Cat. No. 12052-098)に1/100量のHT Supplement (100×), liquid(インビトロジェン社、Cat. No. 11067-030)と、1/100量のPenicillin-Streptomycin (インビトロジェン社製、Cat. No. 15140-122)を添加することで作製したものを使用した。この状態のまま、37℃、CO2インキュベーター内で2日間培養した。
【0086】
(2)ルシフェラーゼ活性の測定
ルシフェラーゼ活性は、Luciferase Assay System (プロメガ社、Cat. No. E1501)を用いて測定した。培地を除き、1ウェルあたりキットに添付されている5× Passive lysis bufferを5倍に薄めたものを100 μl加え、振とうすることで細胞を溶解した。その細胞溶解液をAssay Plate Tissue Culture Treated White with Clear Bottom(コーニング社、Cat No. 437842)に10μlずつ移した。測定はMicroLuMAT(バートホールド社)を用いて行い、データはソフトWinGlow-Control Program LB96PV ver.1.24を用いて取り込んだ。
【0087】
以上の測定の結果より、作製したpmAct-Luc-neoがpCEF-Luc-neoよりも有意に(p<0.0009:対応のないt検定)活性が高いことが明らかになった(図2)。以上のことから、本発明であるマウスβアクチンプロモーターは、既存のCEFプロモーターよりも強力であることが示された。さらに、WPREエレメントの付加や、あるいはCMVエンハンサーを付加することによって、マウスβアクチンのプロモーター活性が有意に(pmAct-WPRE-Luc-neoとpmAct-Luc-neoの比較: p<0.0005、phCMV-mAct-Luc-neoとpmAct-Luc-neoの比較: p<0.0007:対応のないt検定)亢進することも明らかになった(図3)。
【0088】
〔実施例3〕 マウスc-H-ras遺伝子のクローニングおよび改変
(1)マウスc-H-ras遺伝子のクローニング
ヒト活性型H-Rasがヒトβアクチンプロモーターを組み込んだ発現ベクターの発現効率を亢進することは既に報告されている(Cytotechnology, Vol. 16, p.167-178, 1994)。しかし、マウスβアクチンプロモーターに対する活性型H-Ras、あるいは野生型のH-Ras、活性型K-Rasの効果に関する報告は無いことから、マウスH-Ras(活性型、野生型)、ヒト活性型K-Rasのマウスβアクチンプロモーターに対する効果について検討を行った。
【0089】
NCBI(http://www.ncbi.nlm.nih.gov/)で公開されているGenBank Accession No. M30733の情報より、プライマーmRas-F1(5'- TCCTGGATTGGCAGCCGCTGTAGAAGC -3'/配列番号:34)およびmRas-R1(5'- GTTCATCTGGCTAGCTGAGGTCACTGC -3'/配列番号:35)を合成し(エスペックオリゴ社)、PCRによってマウスc-H-ras遺伝子を増幅した。試薬としてはタカラバイオ社のTaKaRa LA Taq with GC Buffer、鋳型cDNAにはクロンテック社のEmbryo Marathon-Ready DNA day15(cat. 7459-1)を使用してPCRを行った。
【0090】
PCR反応溶液の組成は、鋳型DNA 1.0μl、2×GC buffer I 25.0μl、dNTP Mixture 8.0μl、mRas-F1(10μM)2.0μl、mRas-R1(10μM)2.0μl、H2O 11.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で60秒、「95℃で30秒、60℃で30秒、72℃で60秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、660bpのバンドを切り出し、Mag Extractor(東洋紡社)で精製した。これをpGEM-T-Easy vector(プロメガ社)にクローニングし、シーケンスすることによって塩基配列を確認した。確認したマウスc-H-rasのcDNA配列を配列番号:5に示す。データベースでは62番目の塩基がAであったが、クローニングしたc-H-rasはGあった。NCBIのマウスゲノムデータベースで検索したところ、この部分の配列はGであったので、そのまま実験に使用することにした。
【0091】
(2)マウスc-H-rasの改変
CHO細胞で効率よくマウスc-H-Rasを発現させるために、プライマーRAS-ATG(5'- GCCACCATGACAGAATACAAGCTT -3'/配列番号:36)およびRAS-R2(5'- TCAGGACAGCACACATTTGC -3'/配列番号:37)を用いたPCRによって、開始コドンの上流の配列をKozak rule(Nicleic Acids Research, Vol.15(20), p.8125-8148, 1987)に従った配列に変換するとともに、タンパク質がコードされている3'UTRを除去した。試薬としてはタカラバイオ社のTaKaRa LA Taq(cat. RR002A)、鋳型cDNAには先にpGEM-T-Easyベクターにクローニングしたマウスc-H-rasを鋳型に用いて以下のような条件系でPCRを行った。
【0092】
PCR反応溶液の組成は、鋳型DNA 1.0μl、10×LA PCR buffer II(Mg2+free) 5.0μl、dNTP Mixture 5.0μl、MgCl2(25mM) 5.0μl、RAS-ATG(10μM)2.0μl、RAS-R2(10μM)2.0μl、H2O 29.5μl、LA Taq(5U/μl)0.5μlである。またPCRの条件は、95℃で60秒、「95℃で30秒、60℃で30秒、72℃で60秒」を35サイクル、72℃で7分にて終結させる条件である。PCRにはGene Amp PCR System 2400(アプライドバイオシステム社)を用いた。増幅したPCR産物は1% アガロースゲルに電気泳動した後に、576bpのバンドを切り出し、Mag Extractor(東洋紡社)で精製した。これをpGEM-T-Easy vector(プロメガ社)にクローニングし、シーケンスすることによって塩基配列を確認した。改変したマウスc-H-rasのcDNA配列を配列番号:6に示す。
【0093】
(3)マウス活性型H-Rasの作製
ストラタジーン社のQuikChangeTM Site-Directe Mutagenesis Kit(cat.#200518)を用いて、上記のマウスc-H-rasに点変異(上の配列の下線を引いたGをTに変換)を入れることによって12番目のアミノ酸をバリンに変換することで活性型H-Rasを作製した。
使用したプライマーは、mRasV12-F(5'- GTGGTGGGCGCTGTAGGCGTGGGA AAG-3'/配列番号:38)および、mRasV12-R(5'- CTTTCCCACGCCTACAGCGCCCACCAC-3'/配列番号:39)であり、反応溶液の組成:鋳型DNA、H-Ras/pGEM-T-Easy(10ng/μl) 2.0μl、10×reaction buffer 5.0μl、dNTP Mix 1.0μl、mRasV12-F(100ng/μl) 1.25μl、mRasV12-R(100ng/μl) 1.25μl、H2O 38.5μl、PfuTurbo DNA polymerase(2.5U/μl) 1.0μl、反応条件:95℃で30秒、「95℃で30秒、55℃で1分、68℃で7分20秒」で12サイクルにてPCR反応を行った。PCRにはGene Amp PCR System 2400を用いた。増幅したPCR産物をキットのマニュアルに従って、Dpn I処理した後に大腸菌にトランスフォームした。そして、得られた大腸菌からプラスミドを回収してシーケンスすることによって変異が導入されていることを確認した。マウス活性型H-Rasのアミノ酸配列を配列番号:7に示す。
【0094】
同様にヒト活性型K-Rasを準備した。実験に使用したK-Rasのアミノ酸配列を配列番号:8に示す。
これらの遺伝子は、すべてpCXNベクター(Gene, Vol.108, p.193-200, 1991)に組み込んで、それぞれをpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型K-ras)とした。
【0095】
〔実施例4〕癌遺伝子産物Rasのプロモーター活性への影響
(1)ベクターのCHO細胞への導入
CHO細胞に導入する2μgのベクターは、以下のように2種類、あるいは3種類のベクターを、様々な割合で混合することで調整した(表1)。実施例2と同じ方法でCHO細胞に導入し、ルシフェラーゼ活性を測定した。
【0096】
【表1】
【0097】
この結果より、pmAct-Luc-neoがpCXN-H-Ras(マウスc-H-ras)、pCXN-A-H-Ras(マウス活性型c-H-ras)、及びpCXN-A-K-Ras(ヒト活性型c-K-ras)の存在によって、プロモーター活性が亢進することが明らかになった(図4)。以上のことから、本発明であるマウスβアクチンプロモーターは、癌遺伝子産物のRas(活性型、野生型、H-Ras、K-Rasを問わない)によってより強力な活性を付与されることが示された。
【産業上の利用可能性】
【0098】
これまでは、動物細胞でタンパク質を産生させるために使用されていたベクターには、EF1αプロモーター(WO92/19759)やCMVプロモーターが使用されてきた。近年、医薬品におけるバイオ製剤のシェアが大きくなり、特に多くの抗体医薬品が上市されるようになってきた。抗体医薬品は、従来のエリスロポエチン、G-CSF、インターフェロンなどのサイトカインをバイオ製剤としていたものと比較して、大量の投与が必要とされている。従って安価な抗体医薬品の安定な供給には、動物細胞で大量にタンパク質を発現させることが必須となっている。本発明の方法は、従来の技術と比較してタンパク質をより大量に産生させることを可能としたことから、産業上の観点から有用性があるものと考えられる。また、抗体医薬の作製においては、抗原を動物に免疫する過程が必要であるが、こうした発現ベクターに抗原の遺伝子を組み込み、ベクターそのものを動物に免疫することが可能であることが明らかになってきた。本方法のメリットは、抗原をタンパク質として精製する過程を省略できることにある。免疫過程には、いかに多くの抗原がベクターを投与された動物個体内で産生されるかが重要であり、従って発現ベクターが強力なものであるほど好ましい。モノクローナル抗体作製には、マウスやラットが一般に用いられているが、本発明のベクターはこうした用途に好適であると考えられる。さらに、ヒトにおいても遺伝子治療に使用されるベクターなどに組み込むことで、臨床に応用することも可能である。
【特許請求の範囲】
【請求項1】
エンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物。
【請求項2】
エンハンサーがサイトメガロウイルス(CMV)である請求項1に記載のDNA構築物。
【請求項3】
エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である請求項1に記載のDNA構築物。
【請求項4】
哺乳類βアクチンプロモーターがげっ歯類βアクチンプロモーターである、請求項1から3のいずれかに記載のDNA構築物。
【請求項5】
CMVエンハンサーが配列番号:4に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、請求項2に記載のDNA構築物。
【請求項6】
Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)が配列番号:3に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、請求項3に記載のDNA構築物。
【請求項7】
請求項1から6のいずれかに記載のDNA構築物を含むベクター。
【請求項8】
哺乳類βアクチンプロモーターの下流に所望のDNAが機能的に結合されたDNAを有する請求項7に記載のベクター。
【請求項9】
トランスアクティベーターをコードするDNAを発現可能に保持する、請求項7または8に記載のベクター。
【請求項10】
トランスアクティベーターが癌遺伝子産物である請求項9に記載のベクター。
【請求項11】
癌遺伝子産物がRasである、請求項10に記載のベクター。
【請求項12】
所望のDNAが、所望のタンパク質をコードするDNAである、請求項8から11のいずれかに記載のベクター。
【請求項13】
請求項8から12のいずれかに記載のベクターを保持する細胞。
【請求項14】
請求項8から12のいずれかに記載のベクターを保持し、癌遺伝子が活性化している細胞。
【請求項15】
癌遺伝子が活性化している細胞が、トランスアクティベーターをコードする遺伝子を含むベクターが導入された細胞である請求項14に記載の細胞。
【請求項16】
癌遺伝子が活性化している細胞が、癌化している細胞である請求項14に記載の細胞。
【請求項17】
哺乳動物細胞である、請求項13から16のいずれかに記載の細胞。
【請求項18】
げっ歯類細胞である、請求項17に記載の細胞。
【請求項19】
βアクチンプロモーターと同じ動物目であることを特徴とする請求項13から18のいずれかに記載の細胞。
【請求項20】
βアクチンプロモーターと同じ動物種であることを特徴とする請求項19に記載の細胞。
【請求項21】
請求項8から12のいずれかに記載のベクターが導入されたトランスジェニック非ヒト動物。
【請求項22】
請求項8から12のいずれかに記載のベクターが導入された全能性細胞。
【請求項23】
請求項12に記載のベクターを保持する細胞を培養し、培養した細胞又は培地から発現させたタンパク質を回収することを含む、所望のタンパク質の製造方法。
【請求項24】
培地にトランスアクティベーターを添加することを含む、請求項23に記載の方法。
【請求項25】
請求項8から12のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
【請求項26】
請求項8から12のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
【請求項27】
請求項8に記載のベクターおよびトランスアクティベーターをコードするDNAを発現可能に保持するベクターを、請求項8に記載のベクター上のβアクチンプロモーターと同種由来の宿主細胞に導入することを含む、宿主細胞における所望のDNAを発現させる方法。
【請求項28】
宿主細胞が哺乳動物細胞である、請求項25から27のいずれかに記載の方法。
【請求項29】
宿主細胞がげっ歯類細胞である、請求項25から27のいずれかに記載の方法。
【請求項30】
宿主細胞と同じ動物目由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
【請求項31】
宿主細胞と同じ動物種由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
【請求項32】
エンハンサーをさらに組み込むことを特徴とする請求項30もしくは31に記載の方法。
【請求項33】
エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である請求項32記載の方法。
【請求項34】
エンハンサーがCMVエンハンサーである請求項32に記載の方法。
【請求項35】
トランスアクティベーターをコードする遺伝子を組み込むことを特徴とする請求項30から34のいずれかに記載の方法。
【請求項36】
宿主細胞が哺乳動物細胞である請求項30から35のいずれかに記載の方法。
【請求項37】
宿主細胞がげっ歯類細胞である請求項30から35のいずれかに記載の方法。
【請求項1】
エンハンサーに哺乳類βアクチンプロモーターが機能的に結合したDNA構築物。
【請求項2】
エンハンサーがサイトメガロウイルス(CMV)である請求項1に記載のDNA構築物。
【請求項3】
エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である請求項1に記載のDNA構築物。
【請求項4】
哺乳類βアクチンプロモーターがげっ歯類βアクチンプロモーターである、請求項1から3のいずれかに記載のDNA構築物。
【請求項5】
CMVエンハンサーが配列番号:4に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、請求項2に記載のDNA構築物。
【請求項6】
Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)が配列番号:3に記載の塩基配列からなり、哺乳類βアクチンプロモーターが配列番号:2に記載の塩基配列からなる、請求項3に記載のDNA構築物。
【請求項7】
請求項1から6のいずれかに記載のDNA構築物を含むベクター。
【請求項8】
哺乳類βアクチンプロモーターの下流に所望のDNAが機能的に結合されたDNAを有する請求項7に記載のベクター。
【請求項9】
トランスアクティベーターをコードするDNAを発現可能に保持する、請求項7または8に記載のベクター。
【請求項10】
トランスアクティベーターが癌遺伝子産物である請求項9に記載のベクター。
【請求項11】
癌遺伝子産物がRasである、請求項10に記載のベクター。
【請求項12】
所望のDNAが、所望のタンパク質をコードするDNAである、請求項8から11のいずれかに記載のベクター。
【請求項13】
請求項8から12のいずれかに記載のベクターを保持する細胞。
【請求項14】
請求項8から12のいずれかに記載のベクターを保持し、癌遺伝子が活性化している細胞。
【請求項15】
癌遺伝子が活性化している細胞が、トランスアクティベーターをコードする遺伝子を含むベクターが導入された細胞である請求項14に記載の細胞。
【請求項16】
癌遺伝子が活性化している細胞が、癌化している細胞である請求項14に記載の細胞。
【請求項17】
哺乳動物細胞である、請求項13から16のいずれかに記載の細胞。
【請求項18】
げっ歯類細胞である、請求項17に記載の細胞。
【請求項19】
βアクチンプロモーターと同じ動物目であることを特徴とする請求項13から18のいずれかに記載の細胞。
【請求項20】
βアクチンプロモーターと同じ動物種であることを特徴とする請求項19に記載の細胞。
【請求項21】
請求項8から12のいずれかに記載のベクターが導入されたトランスジェニック非ヒト動物。
【請求項22】
請求項8から12のいずれかに記載のベクターが導入された全能性細胞。
【請求項23】
請求項12に記載のベクターを保持する細胞を培養し、培養した細胞又は培地から発現させたタンパク質を回収することを含む、所望のタンパク質の製造方法。
【請求項24】
培地にトランスアクティベーターを添加することを含む、請求項23に記載の方法。
【請求項25】
請求項8から12のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物目由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
【請求項26】
請求項8から12のいずれかに記載のベクターを、ベクター上のβアクチンプロモーターと同じ動物種由来の宿主細胞に導入することを含む、宿主細胞において所望のDNAを発現させる方法。
【請求項27】
請求項8に記載のベクターおよびトランスアクティベーターをコードするDNAを発現可能に保持するベクターを、請求項8に記載のベクター上のβアクチンプロモーターと同種由来の宿主細胞に導入することを含む、宿主細胞における所望のDNAを発現させる方法。
【請求項28】
宿主細胞が哺乳動物細胞である、請求項25から27のいずれかに記載の方法。
【請求項29】
宿主細胞がげっ歯類細胞である、請求項25から27のいずれかに記載の方法。
【請求項30】
宿主細胞と同じ動物目由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
【請求項31】
宿主細胞と同じ動物種由来のβアクチンプロモーターを所望のDNAの上流に組み込むことを特徴とする、所望のDNAの発現量を増加させる方法。
【請求項32】
エンハンサーをさらに組み込むことを特徴とする請求項30もしくは31に記載の方法。
【請求項33】
エンハンサーがWoodchuck Hepatitis Virus Posttranscriptional Regulatory Element(WPRE)である請求項32記載の方法。
【請求項34】
エンハンサーがCMVエンハンサーである請求項32に記載の方法。
【請求項35】
トランスアクティベーターをコードする遺伝子を組み込むことを特徴とする請求項30から34のいずれかに記載の方法。
【請求項36】
宿主細胞が哺乳動物細胞である請求項30から35のいずれかに記載の方法。
【請求項37】
宿主細胞がげっ歯類細胞である請求項30から35のいずれかに記載の方法。
【図1】

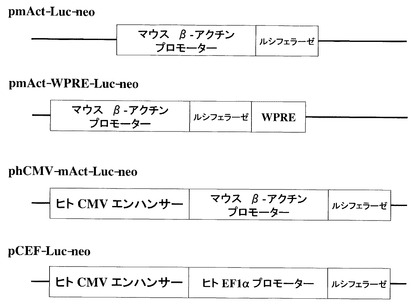
【図2】
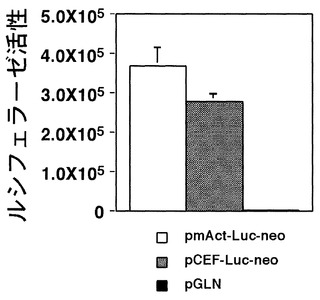
【図3】
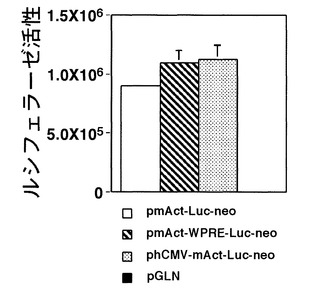
【図4】
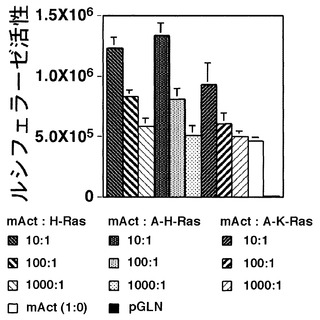
【図2】
【図3】
【図4】
【公開番号】特開2012−70744(P2012−70744A)
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願番号】特願2011−247207(P2011−247207)
【出願日】平成23年11月11日(2011.11.11)
【分割の表示】特願2005−515978(P2005−515978)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
【公開日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願日】平成23年11月11日(2011.11.11)
【分割の表示】特願2005−515978(P2005−515978)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
[ Back to top ]