
固体内服医薬組成物及びその製造方法
【課題】低温保存後でもイブプロフェンの溶出性が維持された、固体内服医薬組成物及びその製造方法を提供する。
【解決手段】(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
【解決手段】(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、低温保存後もイブプロフェンの溶出性が維持される、イブプロフェンを含む固体内服医薬組成物及びその製造方法に関するものである。
【背景技術】
【0002】
カルボキシル基を有する非ステロイド性抗炎症薬であるイブプロフェンは、水への溶解性が低い難溶性薬物である。この溶解性を向上させ、溶出速度を高める技術として、難溶性薬物と高分子化合物とを含有する組成物が知られている(特許文献1:特開平8−291063号公報、特許文献2:特開平3−83922号公報、特許文献3:特開2011−132219号公報)。
【0003】
例えば、特許文献1:特開平8−291063号公報には、カルボキシル基を含有する難溶性薬物、特定のアミノ基含有高分子化合物及び賦形剤を含む易吸収性製剤が提案されている。しかしながら、この易吸収性製剤は、経時的に、特に低温下での保存において難溶性薬物の結晶化が進み、溶解性の低下が生じる。このことから、プロピオン酸系又は酢酸系の非ステロイド性抗炎症薬が、低温保存時において結晶化することを抑制し、溶出性が低温保存後でも維持される低温安定化が望まれていた。
【0004】
また、特許文献3では、イブプロフェンとアミノアルキルメタクリレートE、ケイ酸カルシウムを用いてイブプロフェンの溶出性を改善することが提示されている。しかしながら、加熱温度や加熱時間の記載はなく、イブプロフェン混合物に造粒溶媒としてエタノールを添加するため、固形製剤を製造するためにはエタノールを留去する必要が生じる。また、製造後、製剤化するためには組成物を粉砕する必要があるが、イブプロフェンとアミノアルキルメタクリレートEに、ケイ酸カルシウムを添加しただけでは、粉砕機への付着が激しく粉砕することが困難であった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平8−291063号公報
【特許文献2】特開平3−83922号公報
【特許文献3】特開2011−132219号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は上記事情に鑑みなされたもので、イブプロフェンが低温保存時において再結晶化することを抑制し、低温保存後でも上記成分の溶出性が維持された固体内服医薬組成物及び製造方法を提供することを目的とする。また、さらなる課題としては、粉砕性及び装置非付着性が良好な製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記目的を達成するため鋭意検討した結果、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間(hour))との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)となるように加熱し、得られた加熱混合物を粉砕する固体内服医薬組成物とすることで、上記課題を解決できることを知見し、本発明をなすに至ったものである。
【0008】
従って、本発明は、下記固体内服医薬組成物及びその製造方法を提供する。
[1].(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
[2].(B)/(A)で表される(A)成分と(B)成分との配合質量比が0.5以上である[1]記載の固体内服医薬組成物の製造方法。
[3].(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比が0.05〜3である[1]又は[2]記載の固体内服医薬組成物の製造方法。
[4].上記加熱混合物を(D)粉砕助剤と共に粉砕することを特徴とする[1]〜[3]のいずれかに記載の固体内服医薬組成物の製造方法。
[5].(C)賦型剤が、結晶セルロース、マンニトール及び二酸化ケイ素から選ばれる1種または2種以上である[1]〜[4]のいずれかに記載の固体内服医薬組成物の製造方法。
[6].(D)/[(A)+(B)+(C)]で表される、(A)〜(D)成分の配合質量比が0.05以上である[4]又は[5]記載の固体内服医薬組成物の製造方法。
[7].(D)成分が、二酸化珪素である[4]〜[6]のいずれかに記載の固体内服医薬組成物の製造方法。
[8].(C)賦型剤が、見掛比重120g/L以下である二酸化ケイ素である[1]〜[7]のいずれかに記載の固体内服医薬組成物の製造方法。
[9].(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる固体内服医薬組成物。
[10].加熱温度(X℃)と加熱時間(Y時間)との関係が、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)である[9]記載の固体内服医薬組成物。
[11].上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなる[9]又は[10]記載の固体内服医薬組成物。
【発明の効果】
【0009】
本発明によれば、低温保存後でもイブプロフェンの溶出性が維持された、固形内服医薬組成物及びその製造方法を提供することができる。さらに、特定の(C)成分の選択や、加熱混合物を(D)粉砕助剤と共に粉砕することで、粉砕性及び装置非付着性が良好な製造方法を提供することができる。
【図面の簡単な説明】
【0010】
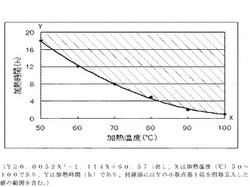
【図1】本発明の加熱温度(X℃)と加熱時間(Y時間(h))との関係を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明について詳細に説明する。
本発明の固体内服医薬組成物は、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる。
【0012】
[固体内服医薬組成物]
(A)イブプロフェン
イブプロフェンは解熱鎮痛薬であり、水難溶性の薬物で、溶解性の改善が望まれている。(A)成分の含有量は内服薬への配合許容範囲内(医薬品承認基準量)であれば、特に限定されない。OTC医薬品とする場合、例えば、イブプロフェン1日量として200〜600mgが好ましく、390〜450mgがより好ましい。また、固体内服医薬組成物中に1〜50質量%が好ましく、5〜50質量%がより好ましい。
【0013】
(B)アミノアルキルメタクリレートコポリマーE
アミノアルキルメタクリレートコポリマーEは、化学名:メタアクリル酸メチル・メタアクリル酸ブチル・メタアクリル酸ジメチルアミノエチルコポリマーであり、医薬品添加物規格又は日本薬局方外医薬品成分規格に記載された成分である。上記(B)成分としては、市販のものを用いることができ、例えば、エボニック社のオイドラギットE100、オイドラギットEPO(モノマーモル比;メタクリル酸メチル1:メタクリル酸ブチル1:メタクリル酸ジメチルアミノエチル2、いずれも商品名)等が挙げられ、1種単独で又は2種以上を適宜組み合わせて用いることができる。
【0014】
(A)成分は水難溶性の薬物で、溶出性の改善が課題であるが、(A)成分と(B)成分とを混合し複合化することで、(A)成分の溶出性が向上する。そのメカニズムは不明であるが、(A)イブプロフェンのカルボキシ基と、(B)成分の(メタアクリル酸ジメチルアミノエチル)のアミノ基とで、(A)成分と(B)成分とが複合化するものと予想される。複合化は(A)成分が非晶質化(非結晶化)することにより確認することができる。(A)成分の非晶質化は、例えば、DSCやXRD等での(A)成分のピークにより確認することができる。
【0015】
(B)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては60〜1800mgが好ましく、100〜1200mgがより好ましく、120〜900mgがさらに好ましい。また、(B)/(A)で表される、(A)成分に対する(B)成分の配合質量比が0.5以上が好ましく、0.5〜1.5がより好ましい。この質量比を0.5以上とすることで、溶出性がより向上する。なお、この比率が1.5を超えても、それ以上の溶出性向上がみられない。なお、本発明においては、(A)成分に対する(B)成分の含有質量比の影響が大きく、(B)成分の含有量は特に限定されないが、固体内服医薬組成物中に0.5〜60質量%が好ましく、1〜50質量%がより好ましく、2.5〜50質量%がさらに好ましく、10〜50質量%が特に好ましい。
【0016】
(C)賦型剤
(C)成分を配合することで、(A)成分と(B)成分との複合化による、前記組成物の固化を防止することができ、目的とする粉砕物からなる固体内服医薬組成物を得ることができる。
【0017】
賦型剤としては、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素、結晶セルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、クロスカルメロースナトリウム、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、クロスポピドン、乳糖、コーンスターチ、タルク、粉糖、マンニトール等が挙げられる。粉砕する工程において粉砕性が向上し、上記加熱混合物の装置非付着性が向上する点から、結晶セルロース、マンニトール及び二酸化ケイ素が好ましい。前記結晶セルロースとしては、旭化成ケミカルズのセオラスUF−711、セオラスPH−101、セオラスKG1000等が挙げられる。なお、賦型剤としてケイ酸カルシウムを用いると、粉砕性が不十分になるおそれがある。なお、「粉砕性」とは、上記加熱混合物を粉砕する際の粉砕のし易さ(装置への付着や粉砕時の凝集がなく粉砕可能)をいう。「装置非付着性」とは、上記加熱混合物が装置に付着せず、取り出しやすいことをいう。
【0018】
さらに、(C)賦型剤が、見掛比重が小さい、例えば120g/L以下、好ましくは60g/L以下、より好ましくは40g/L以下である二酸化ケイ素を用いると、(D)成分の粉砕助剤を添加しなくても、粉砕性及び装置非付着性がより向上する。前記見掛比重が小さい二酸化珪素としては、日本エアロジルのアエロジル200CF(見掛比重:30)、アエロジル300CF(1次粒子の平均粒径見掛比重 30)アエロジル200(見掛比重 50)等が挙げられる。なお、見掛比重は、ISO787/XIで測定したつき固め密度である。
【0019】
(C)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては50〜3000mgが好ましい。なお、(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比は0.05以上が好ましく、この範囲とすることで粉砕性及び装置非付着性がより向上する。上記比率は、0.1以上が好ましく、0.15以上がさらに好ましい。一方、(C)成分の量が多すぎても、一定以上の効果が得られず、錠剤にした場合に、錠剤の体積が大きくなってしまうため、(C)/[(A)+(B)]の質量比は3以下が好ましく、2以下がより好ましい。なお、本発明においては、上記配合比率の影響が大きく、(C)成分の含有量は特に限定されないが、固体内服医薬組成物中に1〜70質量%が好ましく、5〜50質量%がより好ましく、10〜50質量%がさらに好ましい。
【0020】
本発明の固体内服医薬組成物は、上記加熱温度と加熱時間とすることで、低温保存後でも、イブプロフェンの溶出性を維持することができる。
【0021】
(D)粉砕助剤
本発明の固体内服医薬組成物には、粉砕助剤を配合することが好ましく、上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなることが好ましい。このことにより、上記加熱混合物の粉砕性及び装置非付着性が向上する。粉砕助剤としては、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素、炭酸マグネシウム、炭酸カルシウム、炭酸亜鉛、石灰等のアルカリ土類金属炭酸塩、マグネシア、硫酸カルシウム、酸化亜鉛、酸化鉄、酸化チタン等の金属酸化物、珪灰石等が挙げられる。中でも、粉砕性の点から、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素が好ましい。二酸化珪素としては、富士シリシア化学製のサイリシア、サイロスフェア等が挙げられる。
【0022】
(D)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては1〜3000mgが好ましい。なお、(D)/[(A)+(B)+(C)]で表される含有質量比は0.05以上が好ましい。この比率を0.05以上とすることで、粉砕性がより向上する。一方、上記比率が0.5を超えると、製剤の結合力が不足して打錠できなくなったり、粉っぽくなって服用性に課題が生じたりする場合があるため、0.5以下が好ましい。なお、本発明においては、上記配合比率の影響が大きく、(D)成分の含有量は特に限定されないが、固体内服医薬組成物中に0.1〜50質量%が好ましく、1〜50質量%がより好ましい。
【0023】
[固体内服医薬組成物の製造方法]
本発明の固形内服医薬組成物は、例えば、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)となるように加熱し、得られた加熱混合物を粉砕するものである。
【0024】
この式は、後述する実施例1〜6に記載の加熱温度(X℃)と加熱時間(Y時間(h))をグラフにプロットし、2次の多項式近似より算出して得られたものである。
Y≧0.0052X2−1.114X+60.57
また、実施例7の結果からも明らかであるように、この2次の多項式近似より算出して得られた値以上の加熱時間(Y時間、但しYは小数点第1位を四捨五入した値の範囲を含む)であれば、本発明の効果が得られるものである。加熱時間(Y)の上限は特に限定されないが、50時間、30時間、20時間としてもよい。上記範囲で、加熱温度及び加熱時間を選択することで、低温保存後でもイブプロフェンの溶出性が維持される。例えば、50℃であれば18時間以上、60℃であれば12時間以上、70℃であれば8時間以上、80℃であれば5時間以上、90℃であれば2時間以上、100℃であれば1時間以上が好ましい。なお、図1の斜線部の領域であれば特に問題はないが、加熱時間を長くしても、一定以上の溶解性向上はみられない。
【0025】
実施例のデータと上記計算式で得られた値は表1となり、実施例で得られた加熱温度(X℃)と加熱時間(Y時間)のプロットと、上記式をグラフにすると図1のようになる。この場合は、本発明の加熱温度(X℃)と加熱時間(Y時間)との関係は、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)となる。なお、図1の50℃・18時間以上、60℃・12時間以上、70℃・8時間、80℃・5時間、90℃・2時間、100℃・1時間のプロットを直線で結ぶ線の斜線部とすることもできる。
【0026】
【表1】

【0027】
なお、加熱温度(X℃)と加熱時間(Y時間)との関係は、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)であるが、Y≧0.0052X2−1.114X+(60.57−0.25)の範囲とすることもでき、Y≧0.0052X2−1.114X+(60.57−0.5)の範囲とすることもできる。
【0028】
本発明においては、上記加熱温度(X℃)及び加熱時間(Y時間)が重要であり、その混合方法は特に限定されず、(A)〜(C)成分を粉体混合すればよい。混合はビニール袋に上記成分を入れて振り混ぜてもよいし、適当な容器に混合して、モーターで撹拌してもよく、ボーレコンテナミキサー等の混合機を用いてもよい。湿式造粒や乾式造粒も選択できるが、(A)〜(C)成分をエタノール等の溶媒に溶解して造粒を行うと、エタノールを除去する工程を設ける必要がある。その際、混合物の凝集が激しく、内部にエタノールが残存すると、保存時にエタノール蒸気が発生し、製剤の安定性が保てない場合もある。
【0029】
得られた固体内服医薬組成物は、(A)成分が非晶質化したもので、(A)成分と(B)成分とが複合した複合物である。本発明の固形医薬組成物の構造は、例えば、(A)成分が非晶質化し、共溶融した(A)成分と(B)成分とからなる複合物の表面に(C)成分が付着したり、複合物中に(C)成分が混在したり、その製法によって異なる。
【0030】
加熱後は冷却するが、冷却方法は特に限定されず、室温まで冷却すればよい。
【0031】
さらに、(D)成分を配合する場合は、上記で得られた加熱混合物を、(D)粉砕助剤と共に粉砕することにより得ることができる。粉砕の条件としては特に限定されず、卓上粉砕機、フィオーレ、コーミル等の粉砕機、整粒機等の公知の方法を選択できる。
【0032】
固体内服医薬組成物の平均粒径は20〜1000μmが好ましく、50〜850μmがより好ましく、80〜600μmがさらに好ましい。なお、平均粒径はレーザー回折散乱式粒度分布測定器(乾式)による体積メジアン径(D50)である。
【0033】
本発明の固体内服医薬組成物には、本発明の効果を損なわない範囲で、1種単独で又は2種以上の各種任意成分を配合することができる。
【0034】
具体的には、生理活性成分としては、アスピリン、アセトアミノフェン、エトドラック、メフェナミック、メクロフェナミック、ピロキシカム、イソプロピルアンチピリン、トラネキサム酸等の(A)成分以外の抗炎症剤;ニトラゼパム、トリアゾラム、フェノバルビタール、アミバルビタ−ル、アリルイソプロピルアセチル尿素、ブロムワレニル尿素等の催眠・鎮静剤;フェニトイン、メタルビタール、プリミドン、クロナゼパム、カルバマゼピン、バルプロ酸等の抗てんかん剤;塩酸メクリジン、ジメンヒドリナート等の鎮うん剤;イミプラニン、ノキシプチリン、フェネルジン等の抗うつ剤;ハロペリドール、メプロバメート、クロルジアゼポキシド、ジアゼバム、オキサゼバム、スルピリド等の精神神経用剤;パパベリン、アトロピン、エトミドリン等の鎮けい剤;ジゴキシン、ジギトキシン、メチルジゴキシン、ユビデカレノン等の強心剤;ピンドロール、アジマリン、ジソピラミド等の不整脈剤;ヒドロクロロチアジド、スピロノラクトン、トリアムテレン、フロセミド、ブメタニド等の利尿剤;レセルピン、メシル酸ジヒドロエルゴトキシン、塩酸プラゾシン、メトプロロール、プロプラノロール、アテノロール等の抗高血圧剤;ニトログリセリン、硝酸イソソルビド、ジルチアゼム、ニフェジピン、ジピリダモール等の冠血管拡張剤;ノスカピン、サルブタモール、プロカテロール、ツロプテロール、トラニラスト、臭化水素酸デキストロメトルファン、リン酸ジヒドロコデイン等の鎮咳剤;ブロムヘキシン塩酸塩、アンブロキソール塩酸塩、グアイフェネシン等の去痰剤;ニカルジピン、ピンポセチン等の脳循環改善剤;塩酸メチルエフェドリン等の交感神経興奮剤;エリスロマイシン、ジョサマイシン、クロラムフェニコール、テトラサイクリン、リファンピシン、グリセオフルビン等の抗生物質;ジフェンヒドラミン、プロメタジン、メキタジン、クレマスチンフマル酸塩等の抗ヒスタミン剤;トリアムシノロン、デキサメタゾン、ベタメタゾン、プレドニソロン、ダナゾール、メチルテストステロン、酢酸クロルマジノン等のステロイド剤;ビタミンA類、ビタミンB類、ビタミンC類(アスコルビン酸等)、ビタミンD類、ビタミンE類、ビタミンK類、葉酸(ビタミンM類)等のビタミン剤;ジメチコン、ファモチジン、ラニチジン、シメチジン、ニザチジン、メトクロプラミド、ファモチジン、オメプラゾール、スルピリド、トレピブトン、スクラルファート、制酸剤(合成ヒドロタルサイト、酸化マグネシウム等)等の消化器系疾患治療剤;カフェイン、ジクマロール、シンナリジン、クロフィブラート、ゲファルナート、ブロベネシド、メルカプトプリン、メトトレキサート、ウルソデスオキシコール酸、メシル酸ジヒドロエルゴタミン、グルクロノラクトン、γ−アミノ酪酸、コンドロイチン、コンドロイチン硫酸ナトリウム、ラクトフェリン、乳性タンパク、システイン、コラーゲン等が挙げられる。
【0035】
結合剤としては、例えば、澱粉、α化デンプン、ショ糖、ゼラチン、アラビアゴム末、メチルセルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、プルラン、デキストリン等を用いることができる。
【0036】
滑沢剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、タルク、ステアリン酸、ショ糖脂肪酸エステル等を用いることができる。香料としては、例えば、メントール、リモネン、植物精油(ハッカ油、ミント油、ライチ油、オレンジ油、レモン油等)等が挙げられる。
【0037】
甘味料としては、例えば、サッカリンナトリウム、アスパルテーム、ステビア、グリチルリチン酸二カリウム、アセスルファムカリウム、ソーマチン、スクラロース、果糖等が挙げられる。
酸味料としては、例えば、クエン酸、酒石酸、リンゴ酸、コハク酸、フマル酸、乳酸又はそれらの塩等が挙げられる。
界面活性剤としては、ノニオン界面活性剤、アニオン界面活性剤、カチオン界面活性剤、両性界面活性剤等が挙げられる。
【0038】
固体内服医薬組成物は、粒状剤(顆粒剤、細粒剤、散剤)としたり、さらに必要に応じて打錠して錠剤、カプセル剤等の内服用固形医薬製剤にしたりすることができる。この中でも、錠剤、特に胃中で溶解する錠剤(胃中溶解錠)が好ましい。
【実施例】
【0039】
以下、実施例及び比較例を示し、本発明を具体的に説明するが、本発明は下記の実施例に制限されるものではない。なお、下記の例において特に明記のない場合は、組成の「%」は質量%、比率は質量比を示す。
【0040】
[実施例1〜15、比較例1〜5]
ビニール袋に表1の(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)結晶セルロースを添加した後、手で良く振り混ぜた。この混合粉をポリプロピレン製バットに取り出した。これを表1の各温度・時間で加熱した後、室温まで冷却した後、下記方法でバットからの剥離性を評価した。その後、(D)二酸化珪素を添加し、岩谷産業株式会社製卓上粉砕機で3分間粉砕し、固体内服医薬組成物を得た。得られた固体内服医薬組成物について、下記方法でイブプロフェンの溶出性を、粉砕性を評価した。固体内服医薬組成物の平均粒径は100〜500μmであった。なお、実施例については、DSC((株)リガク製,DSC−8230D)にて、(A)成分のピークがなくなり、非晶質化していることを確認した。
【0041】
<溶出性>
製造直後のサンプル及びアルミパウチに封入して5℃で1ヶ月間保存したサンプルについて、下記方法でイブプロフェン溶出率を測定した。
溶出試験はpH=1.2緩衝液(塩酸/塩化ナトリウム)を用い、緩衝液900mLに対しイブプロフェン130mg相当の顆粒を投入し、37℃、パドル回転数50rpmで5分間攪拌した後サンプリングし、ラボラボカンパニー製フィルター(0.45μm)でろ過した後、ろ液中のイブプロフェン濃度を液体クロマトグラフィーで測定し、式1よりイブプロフェン溶出率(%)を算出した。
溶出率(%)=顆粒を投入した5分後のイブプロフェン溶出量(mg)/イブプロフェンの仕込み量:130(mg)×100 (式1)
溶出率85%以上を○、溶出率85%未満を×とした。
【0042】
【表2】
【0043】
【表3】
【0044】
【表4】
【0045】
[実施例16]
ビニール袋に(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)二酸化珪素もしくはケイ酸カルシウムを入れ、手で良く振り混ぜた。混合粉をポリプロピレン製バットに取り出した。これを80℃で14時間加熱した後、室温まで冷却した。得られた固体内服医薬組成物について、上記実施例と同様の評価を行った。 なお、固体内服医薬組成物の平均粒径は100〜500μmであり、DSC((株)リガク製,DSC−8230D)にて、(A)成分のピークがなくなり、非晶質化していることを確認した。
【0046】
【表5】
【0047】
なお、上記実施例で得られた固体内服医薬組成物について、下記方法で粉砕性及び装置非付着性を評価したところ、いずれも全て「○」だった。
【0048】
<粉砕性評価法>
サンプルを、開きが850μmのふるいにかけ、その通過の仕方から粉砕性を下記基準に従い判断した。
[判断基準]
○:目の開き850μmのふるいを全てパスする。
×:目の開き850μmのふるいを全てはパスしない。
【0049】
<装置非付着性評価>
所定条件で加熱した後、室温まで冷却したサンプルをポリプロピレン製バットから取り出し、以下の基準で剥離性を評価した。
[判断基準]
○:手でバットから取り出すことができる。
×:強固に付着しており手ではバットから取り出すことができない。
【0050】
上記例で使用した原料を下記に示す。なお、特に明記がない限り、表中の各成分の量は純分換算量である。
【表6】
【技術分野】
【0001】
本発明は、低温保存後もイブプロフェンの溶出性が維持される、イブプロフェンを含む固体内服医薬組成物及びその製造方法に関するものである。
【背景技術】
【0002】
カルボキシル基を有する非ステロイド性抗炎症薬であるイブプロフェンは、水への溶解性が低い難溶性薬物である。この溶解性を向上させ、溶出速度を高める技術として、難溶性薬物と高分子化合物とを含有する組成物が知られている(特許文献1:特開平8−291063号公報、特許文献2:特開平3−83922号公報、特許文献3:特開2011−132219号公報)。
【0003】
例えば、特許文献1:特開平8−291063号公報には、カルボキシル基を含有する難溶性薬物、特定のアミノ基含有高分子化合物及び賦形剤を含む易吸収性製剤が提案されている。しかしながら、この易吸収性製剤は、経時的に、特に低温下での保存において難溶性薬物の結晶化が進み、溶解性の低下が生じる。このことから、プロピオン酸系又は酢酸系の非ステロイド性抗炎症薬が、低温保存時において結晶化することを抑制し、溶出性が低温保存後でも維持される低温安定化が望まれていた。
【0004】
また、特許文献3では、イブプロフェンとアミノアルキルメタクリレートE、ケイ酸カルシウムを用いてイブプロフェンの溶出性を改善することが提示されている。しかしながら、加熱温度や加熱時間の記載はなく、イブプロフェン混合物に造粒溶媒としてエタノールを添加するため、固形製剤を製造するためにはエタノールを留去する必要が生じる。また、製造後、製剤化するためには組成物を粉砕する必要があるが、イブプロフェンとアミノアルキルメタクリレートEに、ケイ酸カルシウムを添加しただけでは、粉砕機への付着が激しく粉砕することが困難であった。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平8−291063号公報
【特許文献2】特開平3−83922号公報
【特許文献3】特開2011−132219号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は上記事情に鑑みなされたもので、イブプロフェンが低温保存時において再結晶化することを抑制し、低温保存後でも上記成分の溶出性が維持された固体内服医薬組成物及び製造方法を提供することを目的とする。また、さらなる課題としては、粉砕性及び装置非付着性が良好な製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、上記目的を達成するため鋭意検討した結果、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間(hour))との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)となるように加熱し、得られた加熱混合物を粉砕する固体内服医薬組成物とすることで、上記課題を解決できることを知見し、本発明をなすに至ったものである。
【0008】
従って、本発明は、下記固体内服医薬組成物及びその製造方法を提供する。
[1].(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
[2].(B)/(A)で表される(A)成分と(B)成分との配合質量比が0.5以上である[1]記載の固体内服医薬組成物の製造方法。
[3].(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比が0.05〜3である[1]又は[2]記載の固体内服医薬組成物の製造方法。
[4].上記加熱混合物を(D)粉砕助剤と共に粉砕することを特徴とする[1]〜[3]のいずれかに記載の固体内服医薬組成物の製造方法。
[5].(C)賦型剤が、結晶セルロース、マンニトール及び二酸化ケイ素から選ばれる1種または2種以上である[1]〜[4]のいずれかに記載の固体内服医薬組成物の製造方法。
[6].(D)/[(A)+(B)+(C)]で表される、(A)〜(D)成分の配合質量比が0.05以上である[4]又は[5]記載の固体内服医薬組成物の製造方法。
[7].(D)成分が、二酸化珪素である[4]〜[6]のいずれかに記載の固体内服医薬組成物の製造方法。
[8].(C)賦型剤が、見掛比重120g/L以下である二酸化ケイ素である[1]〜[7]のいずれかに記載の固体内服医薬組成物の製造方法。
[9].(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる固体内服医薬組成物。
[10].加熱温度(X℃)と加熱時間(Y時間)との関係が、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)である[9]記載の固体内服医薬組成物。
[11].上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなる[9]又は[10]記載の固体内服医薬組成物。
【発明の効果】
【0009】
本発明によれば、低温保存後でもイブプロフェンの溶出性が維持された、固形内服医薬組成物及びその製造方法を提供することができる。さらに、特定の(C)成分の選択や、加熱混合物を(D)粉砕助剤と共に粉砕することで、粉砕性及び装置非付着性が良好な製造方法を提供することができる。
【図面の簡単な説明】
【0010】
【図1】本発明の加熱温度(X℃)と加熱時間(Y時間(h))との関係を示すグラフである。
【発明を実施するための形態】
【0011】
以下、本発明について詳細に説明する。
本発明の固体内服医薬組成物は、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる。
【0012】
[固体内服医薬組成物]
(A)イブプロフェン
イブプロフェンは解熱鎮痛薬であり、水難溶性の薬物で、溶解性の改善が望まれている。(A)成分の含有量は内服薬への配合許容範囲内(医薬品承認基準量)であれば、特に限定されない。OTC医薬品とする場合、例えば、イブプロフェン1日量として200〜600mgが好ましく、390〜450mgがより好ましい。また、固体内服医薬組成物中に1〜50質量%が好ましく、5〜50質量%がより好ましい。
【0013】
(B)アミノアルキルメタクリレートコポリマーE
アミノアルキルメタクリレートコポリマーEは、化学名:メタアクリル酸メチル・メタアクリル酸ブチル・メタアクリル酸ジメチルアミノエチルコポリマーであり、医薬品添加物規格又は日本薬局方外医薬品成分規格に記載された成分である。上記(B)成分としては、市販のものを用いることができ、例えば、エボニック社のオイドラギットE100、オイドラギットEPO(モノマーモル比;メタクリル酸メチル1:メタクリル酸ブチル1:メタクリル酸ジメチルアミノエチル2、いずれも商品名)等が挙げられ、1種単独で又は2種以上を適宜組み合わせて用いることができる。
【0014】
(A)成分は水難溶性の薬物で、溶出性の改善が課題であるが、(A)成分と(B)成分とを混合し複合化することで、(A)成分の溶出性が向上する。そのメカニズムは不明であるが、(A)イブプロフェンのカルボキシ基と、(B)成分の(メタアクリル酸ジメチルアミノエチル)のアミノ基とで、(A)成分と(B)成分とが複合化するものと予想される。複合化は(A)成分が非晶質化(非結晶化)することにより確認することができる。(A)成分の非晶質化は、例えば、DSCやXRD等での(A)成分のピークにより確認することができる。
【0015】
(B)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては60〜1800mgが好ましく、100〜1200mgがより好ましく、120〜900mgがさらに好ましい。また、(B)/(A)で表される、(A)成分に対する(B)成分の配合質量比が0.5以上が好ましく、0.5〜1.5がより好ましい。この質量比を0.5以上とすることで、溶出性がより向上する。なお、この比率が1.5を超えても、それ以上の溶出性向上がみられない。なお、本発明においては、(A)成分に対する(B)成分の含有質量比の影響が大きく、(B)成分の含有量は特に限定されないが、固体内服医薬組成物中に0.5〜60質量%が好ましく、1〜50質量%がより好ましく、2.5〜50質量%がさらに好ましく、10〜50質量%が特に好ましい。
【0016】
(C)賦型剤
(C)成分を配合することで、(A)成分と(B)成分との複合化による、前記組成物の固化を防止することができ、目的とする粉砕物からなる固体内服医薬組成物を得ることができる。
【0017】
賦型剤としては、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素、結晶セルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、クロスカルメロースナトリウム、カルボキシメチルセルロースカルシウム、低置換度ヒドロキシプロピルセルロース、クロスポピドン、乳糖、コーンスターチ、タルク、粉糖、マンニトール等が挙げられる。粉砕する工程において粉砕性が向上し、上記加熱混合物の装置非付着性が向上する点から、結晶セルロース、マンニトール及び二酸化ケイ素が好ましい。前記結晶セルロースとしては、旭化成ケミカルズのセオラスUF−711、セオラスPH−101、セオラスKG1000等が挙げられる。なお、賦型剤としてケイ酸カルシウムを用いると、粉砕性が不十分になるおそれがある。なお、「粉砕性」とは、上記加熱混合物を粉砕する際の粉砕のし易さ(装置への付着や粉砕時の凝集がなく粉砕可能)をいう。「装置非付着性」とは、上記加熱混合物が装置に付着せず、取り出しやすいことをいう。
【0018】
さらに、(C)賦型剤が、見掛比重が小さい、例えば120g/L以下、好ましくは60g/L以下、より好ましくは40g/L以下である二酸化ケイ素を用いると、(D)成分の粉砕助剤を添加しなくても、粉砕性及び装置非付着性がより向上する。前記見掛比重が小さい二酸化珪素としては、日本エアロジルのアエロジル200CF(見掛比重:30)、アエロジル300CF(1次粒子の平均粒径見掛比重 30)アエロジル200(見掛比重 50)等が挙げられる。なお、見掛比重は、ISO787/XIで測定したつき固め密度である。
【0019】
(C)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては50〜3000mgが好ましい。なお、(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比は0.05以上が好ましく、この範囲とすることで粉砕性及び装置非付着性がより向上する。上記比率は、0.1以上が好ましく、0.15以上がさらに好ましい。一方、(C)成分の量が多すぎても、一定以上の効果が得られず、錠剤にした場合に、錠剤の体積が大きくなってしまうため、(C)/[(A)+(B)]の質量比は3以下が好ましく、2以下がより好ましい。なお、本発明においては、上記配合比率の影響が大きく、(C)成分の含有量は特に限定されないが、固体内服医薬組成物中に1〜70質量%が好ましく、5〜50質量%がより好ましく、10〜50質量%がさらに好ましい。
【0020】
本発明の固体内服医薬組成物は、上記加熱温度と加熱時間とすることで、低温保存後でも、イブプロフェンの溶出性を維持することができる。
【0021】
(D)粉砕助剤
本発明の固体内服医薬組成物には、粉砕助剤を配合することが好ましく、上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなることが好ましい。このことにより、上記加熱混合物の粉砕性及び装置非付着性が向上する。粉砕助剤としては、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素、炭酸マグネシウム、炭酸カルシウム、炭酸亜鉛、石灰等のアルカリ土類金属炭酸塩、マグネシア、硫酸カルシウム、酸化亜鉛、酸化鉄、酸化チタン等の金属酸化物、珪灰石等が挙げられる。中でも、粉砕性の点から、含水二酸化ケイ素、軽質無水ケイ酸、重質無水ケイ酸等の二酸化珪素が好ましい。二酸化珪素としては、富士シリシア化学製のサイリシア、サイロスフェア等が挙げられる。
【0022】
(D)成分の含有量は内服薬として許容される範囲内(医薬品使用前例量)であれば、特に限定されないが、1日量としては1〜3000mgが好ましい。なお、(D)/[(A)+(B)+(C)]で表される含有質量比は0.05以上が好ましい。この比率を0.05以上とすることで、粉砕性がより向上する。一方、上記比率が0.5を超えると、製剤の結合力が不足して打錠できなくなったり、粉っぽくなって服用性に課題が生じたりする場合があるため、0.5以下が好ましい。なお、本発明においては、上記配合比率の影響が大きく、(D)成分の含有量は特に限定されないが、固体内服医薬組成物中に0.1〜50質量%が好ましく、1〜50質量%がより好ましい。
【0023】
[固体内服医薬組成物の製造方法]
本発明の固形内服医薬組成物は、例えば、(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)となるように加熱し、得られた加熱混合物を粉砕するものである。
【0024】
この式は、後述する実施例1〜6に記載の加熱温度(X℃)と加熱時間(Y時間(h))をグラフにプロットし、2次の多項式近似より算出して得られたものである。
Y≧0.0052X2−1.114X+60.57
また、実施例7の結果からも明らかであるように、この2次の多項式近似より算出して得られた値以上の加熱時間(Y時間、但しYは小数点第1位を四捨五入した値の範囲を含む)であれば、本発明の効果が得られるものである。加熱時間(Y)の上限は特に限定されないが、50時間、30時間、20時間としてもよい。上記範囲で、加熱温度及び加熱時間を選択することで、低温保存後でもイブプロフェンの溶出性が維持される。例えば、50℃であれば18時間以上、60℃であれば12時間以上、70℃であれば8時間以上、80℃であれば5時間以上、90℃であれば2時間以上、100℃であれば1時間以上が好ましい。なお、図1の斜線部の領域であれば特に問題はないが、加熱時間を長くしても、一定以上の溶解性向上はみられない。
【0025】
実施例のデータと上記計算式で得られた値は表1となり、実施例で得られた加熱温度(X℃)と加熱時間(Y時間)のプロットと、上記式をグラフにすると図1のようになる。この場合は、本発明の加熱温度(X℃)と加熱時間(Y時間)との関係は、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)となる。なお、図1の50℃・18時間以上、60℃・12時間以上、70℃・8時間、80℃・5時間、90℃・2時間、100℃・1時間のプロットを直線で結ぶ線の斜線部とすることもできる。
【0026】
【表1】
【0027】
なお、加熱温度(X℃)と加熱時間(Y時間)との関係は、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)であるが、Y≧0.0052X2−1.114X+(60.57−0.25)の範囲とすることもでき、Y≧0.0052X2−1.114X+(60.57−0.5)の範囲とすることもできる。
【0028】
本発明においては、上記加熱温度(X℃)及び加熱時間(Y時間)が重要であり、その混合方法は特に限定されず、(A)〜(C)成分を粉体混合すればよい。混合はビニール袋に上記成分を入れて振り混ぜてもよいし、適当な容器に混合して、モーターで撹拌してもよく、ボーレコンテナミキサー等の混合機を用いてもよい。湿式造粒や乾式造粒も選択できるが、(A)〜(C)成分をエタノール等の溶媒に溶解して造粒を行うと、エタノールを除去する工程を設ける必要がある。その際、混合物の凝集が激しく、内部にエタノールが残存すると、保存時にエタノール蒸気が発生し、製剤の安定性が保てない場合もある。
【0029】
得られた固体内服医薬組成物は、(A)成分が非晶質化したもので、(A)成分と(B)成分とが複合した複合物である。本発明の固形医薬組成物の構造は、例えば、(A)成分が非晶質化し、共溶融した(A)成分と(B)成分とからなる複合物の表面に(C)成分が付着したり、複合物中に(C)成分が混在したり、その製法によって異なる。
【0030】
加熱後は冷却するが、冷却方法は特に限定されず、室温まで冷却すればよい。
【0031】
さらに、(D)成分を配合する場合は、上記で得られた加熱混合物を、(D)粉砕助剤と共に粉砕することにより得ることができる。粉砕の条件としては特に限定されず、卓上粉砕機、フィオーレ、コーミル等の粉砕機、整粒機等の公知の方法を選択できる。
【0032】
固体内服医薬組成物の平均粒径は20〜1000μmが好ましく、50〜850μmがより好ましく、80〜600μmがさらに好ましい。なお、平均粒径はレーザー回折散乱式粒度分布測定器(乾式)による体積メジアン径(D50)である。
【0033】
本発明の固体内服医薬組成物には、本発明の効果を損なわない範囲で、1種単独で又は2種以上の各種任意成分を配合することができる。
【0034】
具体的には、生理活性成分としては、アスピリン、アセトアミノフェン、エトドラック、メフェナミック、メクロフェナミック、ピロキシカム、イソプロピルアンチピリン、トラネキサム酸等の(A)成分以外の抗炎症剤;ニトラゼパム、トリアゾラム、フェノバルビタール、アミバルビタ−ル、アリルイソプロピルアセチル尿素、ブロムワレニル尿素等の催眠・鎮静剤;フェニトイン、メタルビタール、プリミドン、クロナゼパム、カルバマゼピン、バルプロ酸等の抗てんかん剤;塩酸メクリジン、ジメンヒドリナート等の鎮うん剤;イミプラニン、ノキシプチリン、フェネルジン等の抗うつ剤;ハロペリドール、メプロバメート、クロルジアゼポキシド、ジアゼバム、オキサゼバム、スルピリド等の精神神経用剤;パパベリン、アトロピン、エトミドリン等の鎮けい剤;ジゴキシン、ジギトキシン、メチルジゴキシン、ユビデカレノン等の強心剤;ピンドロール、アジマリン、ジソピラミド等の不整脈剤;ヒドロクロロチアジド、スピロノラクトン、トリアムテレン、フロセミド、ブメタニド等の利尿剤;レセルピン、メシル酸ジヒドロエルゴトキシン、塩酸プラゾシン、メトプロロール、プロプラノロール、アテノロール等の抗高血圧剤;ニトログリセリン、硝酸イソソルビド、ジルチアゼム、ニフェジピン、ジピリダモール等の冠血管拡張剤;ノスカピン、サルブタモール、プロカテロール、ツロプテロール、トラニラスト、臭化水素酸デキストロメトルファン、リン酸ジヒドロコデイン等の鎮咳剤;ブロムヘキシン塩酸塩、アンブロキソール塩酸塩、グアイフェネシン等の去痰剤;ニカルジピン、ピンポセチン等の脳循環改善剤;塩酸メチルエフェドリン等の交感神経興奮剤;エリスロマイシン、ジョサマイシン、クロラムフェニコール、テトラサイクリン、リファンピシン、グリセオフルビン等の抗生物質;ジフェンヒドラミン、プロメタジン、メキタジン、クレマスチンフマル酸塩等の抗ヒスタミン剤;トリアムシノロン、デキサメタゾン、ベタメタゾン、プレドニソロン、ダナゾール、メチルテストステロン、酢酸クロルマジノン等のステロイド剤;ビタミンA類、ビタミンB類、ビタミンC類(アスコルビン酸等)、ビタミンD類、ビタミンE類、ビタミンK類、葉酸(ビタミンM類)等のビタミン剤;ジメチコン、ファモチジン、ラニチジン、シメチジン、ニザチジン、メトクロプラミド、ファモチジン、オメプラゾール、スルピリド、トレピブトン、スクラルファート、制酸剤(合成ヒドロタルサイト、酸化マグネシウム等)等の消化器系疾患治療剤;カフェイン、ジクマロール、シンナリジン、クロフィブラート、ゲファルナート、ブロベネシド、メルカプトプリン、メトトレキサート、ウルソデスオキシコール酸、メシル酸ジヒドロエルゴタミン、グルクロノラクトン、γ−アミノ酪酸、コンドロイチン、コンドロイチン硫酸ナトリウム、ラクトフェリン、乳性タンパク、システイン、コラーゲン等が挙げられる。
【0035】
結合剤としては、例えば、澱粉、α化デンプン、ショ糖、ゼラチン、アラビアゴム末、メチルセルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン、プルラン、デキストリン等を用いることができる。
【0036】
滑沢剤としては、例えば、ステアリン酸マグネシウム、ステアリン酸カルシウム、ポリエチレングリコール、タルク、ステアリン酸、ショ糖脂肪酸エステル等を用いることができる。香料としては、例えば、メントール、リモネン、植物精油(ハッカ油、ミント油、ライチ油、オレンジ油、レモン油等)等が挙げられる。
【0037】
甘味料としては、例えば、サッカリンナトリウム、アスパルテーム、ステビア、グリチルリチン酸二カリウム、アセスルファムカリウム、ソーマチン、スクラロース、果糖等が挙げられる。
酸味料としては、例えば、クエン酸、酒石酸、リンゴ酸、コハク酸、フマル酸、乳酸又はそれらの塩等が挙げられる。
界面活性剤としては、ノニオン界面活性剤、アニオン界面活性剤、カチオン界面活性剤、両性界面活性剤等が挙げられる。
【0038】
固体内服医薬組成物は、粒状剤(顆粒剤、細粒剤、散剤)としたり、さらに必要に応じて打錠して錠剤、カプセル剤等の内服用固形医薬製剤にしたりすることができる。この中でも、錠剤、特に胃中で溶解する錠剤(胃中溶解錠)が好ましい。
【実施例】
【0039】
以下、実施例及び比較例を示し、本発明を具体的に説明するが、本発明は下記の実施例に制限されるものではない。なお、下記の例において特に明記のない場合は、組成の「%」は質量%、比率は質量比を示す。
【0040】
[実施例1〜15、比較例1〜5]
ビニール袋に表1の(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)結晶セルロースを添加した後、手で良く振り混ぜた。この混合粉をポリプロピレン製バットに取り出した。これを表1の各温度・時間で加熱した後、室温まで冷却した後、下記方法でバットからの剥離性を評価した。その後、(D)二酸化珪素を添加し、岩谷産業株式会社製卓上粉砕機で3分間粉砕し、固体内服医薬組成物を得た。得られた固体内服医薬組成物について、下記方法でイブプロフェンの溶出性を、粉砕性を評価した。固体内服医薬組成物の平均粒径は100〜500μmであった。なお、実施例については、DSC((株)リガク製,DSC−8230D)にて、(A)成分のピークがなくなり、非晶質化していることを確認した。
【0041】
<溶出性>
製造直後のサンプル及びアルミパウチに封入して5℃で1ヶ月間保存したサンプルについて、下記方法でイブプロフェン溶出率を測定した。
溶出試験はpH=1.2緩衝液(塩酸/塩化ナトリウム)を用い、緩衝液900mLに対しイブプロフェン130mg相当の顆粒を投入し、37℃、パドル回転数50rpmで5分間攪拌した後サンプリングし、ラボラボカンパニー製フィルター(0.45μm)でろ過した後、ろ液中のイブプロフェン濃度を液体クロマトグラフィーで測定し、式1よりイブプロフェン溶出率(%)を算出した。
溶出率(%)=顆粒を投入した5分後のイブプロフェン溶出量(mg)/イブプロフェンの仕込み量:130(mg)×100 (式1)
溶出率85%以上を○、溶出率85%未満を×とした。
【0042】
【表2】
【0043】
【表3】
【0044】
【表4】
【0045】
[実施例16]
ビニール袋に(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)二酸化珪素もしくはケイ酸カルシウムを入れ、手で良く振り混ぜた。混合粉をポリプロピレン製バットに取り出した。これを80℃で14時間加熱した後、室温まで冷却した。得られた固体内服医薬組成物について、上記実施例と同様の評価を行った。 なお、固体内服医薬組成物の平均粒径は100〜500μmであり、DSC((株)リガク製,DSC−8230D)にて、(A)成分のピークがなくなり、非晶質化していることを確認した。
【0046】
【表5】
【0047】
なお、上記実施例で得られた固体内服医薬組成物について、下記方法で粉砕性及び装置非付着性を評価したところ、いずれも全て「○」だった。
【0048】
<粉砕性評価法>
サンプルを、開きが850μmのふるいにかけ、その通過の仕方から粉砕性を下記基準に従い判断した。
[判断基準]
○:目の開き850μmのふるいを全てパスする。
×:目の開き850μmのふるいを全てはパスしない。
【0049】
<装置非付着性評価>
所定条件で加熱した後、室温まで冷却したサンプルをポリプロピレン製バットから取り出し、以下の基準で剥離性を評価した。
[判断基準]
○:手でバットから取り出すことができる。
×:強固に付着しており手ではバットから取り出すことができない。
【0050】
上記例で使用した原料を下記に示す。なお、特に明記がない限り、表中の各成分の量は純分換算量である。
【表6】
【特許請求の範囲】
【請求項1】
(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
【請求項2】
(B)/(A)で表される(A)成分と(B)成分との配合質量比が0.5以上である請求項1記載の固体内服医薬組成物の製造方法。
【請求項3】
(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比が0.05〜3である請求項1又は2記載の固体内服医薬組成物の製造方法。
【請求項4】
上記加熱混合物を(D)粉砕助剤と共に粉砕することを特徴とする請求項1〜3のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項5】
(C)賦型剤が、結晶セルロース、マンニトール及び二酸化ケイ素から選ばれる1種または2種以上である請求項1〜4のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項6】
(D)/[(A)+(B)+(C)]で表される、(A)〜(D)成分の配合質量比が0.05以上である請求項4又は5記載の固体内服医薬組成物の製造方法。
【請求項7】
(D)成分が、二酸化珪素である請求項4〜6のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項8】
(C)賦型剤が、見掛比重120g/L以下である二酸化ケイ素である請求項1〜7のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項9】
(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる固体内服医薬組成物。
【請求項10】
加熱温度(X℃)と加熱時間(Y時間)との関係が、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)である請求項9記載の固体内服医薬組成物。
【請求項11】
上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなる請求項9又は10記載の固体内服医薬組成物。
【請求項1】
(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤とを混合し、得られた混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値)を満たす、加熱温度及び加熱時間で加熱し、得られた加熱混合物を粉砕する、固体内服医薬組成物の製造方法。
【請求項2】
(B)/(A)で表される(A)成分と(B)成分との配合質量比が0.5以上である請求項1記載の固体内服医薬組成物の製造方法。
【請求項3】
(C)/[(A)+(B)]で表される、(A)〜(C)成分の配合質量比が0.05〜3である請求項1又は2記載の固体内服医薬組成物の製造方法。
【請求項4】
上記加熱混合物を(D)粉砕助剤と共に粉砕することを特徴とする請求項1〜3のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項5】
(C)賦型剤が、結晶セルロース、マンニトール及び二酸化ケイ素から選ばれる1種または2種以上である請求項1〜4のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項6】
(D)/[(A)+(B)+(C)]で表される、(A)〜(D)成分の配合質量比が0.05以上である請求項4又は5記載の固体内服医薬組成物の製造方法。
【請求項7】
(D)成分が、二酸化珪素である請求項4〜6のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項8】
(C)賦型剤が、見掛比重120g/L以下である二酸化ケイ素である請求項1〜7のいずれか1項記載の固体内服医薬組成物の製造方法。
【請求項9】
(A)イブプロフェンと、(B)アミノアルキルメタクリレートコポリマーEと、(C)賦型剤との混合物を、加熱温度(X℃)と加熱時間(Y時間)との関係が、
Y≧0.0052X2−1.114X+60.57
(但し、Xは50〜100であり、Yは小数点第1位を四捨五入した値である。)を満たす、加熱温度及び加熱時間で加熱した、加熱混合物の粉砕物からなる固体内服医薬組成物。
【請求項10】
加熱温度(X℃)と加熱時間(Y時間)との関係が、図1の斜線部の領域:Y≧0.0052X2−1.114X+60.57(但し、Xは50〜100であり、斜線部にはYの小数点第1位を四捨五入した値の範囲を含む。)である請求項9記載の固体内服医薬組成物。
【請求項11】
上記加熱混合物が、さらに(D)粉砕助剤と共に粉砕されてなる請求項9又は10記載の固体内服医薬組成物。
【図1】


【公開番号】特開2013−56845(P2013−56845A)
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2011−195960(P2011−195960)
【出願日】平成23年9月8日(2011.9.8)
【出願人】(000006769)ライオン株式会社 (1,816)
【Fターム(参考)】
【公開日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願日】平成23年9月8日(2011.9.8)
【出願人】(000006769)ライオン株式会社 (1,816)
【Fターム(参考)】
[ Back to top ]