
固体酸触媒を用いたシュウ酸エステル体の製造方法
【課題】 熱分解性が高いシュウ酸エステル体を高収率で反応させることができ、更に反応終了後にシュウ酸エステル体生成物から固体酸触媒を容易に除くことのできる形状の固体酸触媒を用いたシュウ酸エステル体の製造方法を提供すること。
【解決手段】 エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法。
【解決手段】 エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、固体酸触媒であるエステル化反応触媒の存在下にシュウ酸エステル体を製造する方法、シュウ酸エステル体製造用反応装置に関する。
【背景技術】
【0002】
現在、化学工業でのエステル体の製造は、チタン系もしくは錫系の均一系触媒を用いて行っている。このような均一系触媒は生成物中に溶け込んでしまうために、単離・回収により触媒を取り除くことが難しく、残留触媒のないエステル体を合成することは非常に困難である。これらの残留触媒は、得られたエステル体中に存在する水分とエステル結合との反応を促進させ、加水分解を引き起こすため、エステル体の耐久性や保存安定性を低下させる原因となっている。
上記の問題により、均一系触媒は、通常使用可能な触媒量がごく微量に制限されるため、反応転化が遅く、結果的にエステル体の製造には長時間が必要となる。さらに触媒の単離・回収が困難であることから、エステル体の着色や物性への影響が避けられないという問題もある。
【0003】
また、本発明で目的とするシュウ酸エステル体又はシュウ酸ポリエステル体の製造を目的とした場合には、当該化合物が高い熱分解性を有しているため、従来の触媒を用いた反応系では、目的とするエステル体が生成しても反応系内で分解してしまうために、最終的に目的物が得られないという問題点があった。
【0004】
特許文献1には、固体酸触媒をエステル体重合補助触媒として使用する技術が提案されている。本文献で使用されているモリブデン酸ジルコニアはH0関数が‐12.4であり、いわゆる超強酸である。しかし、グリコールと酸との脱水縮合反応にこのような固体超強酸を用いると、グリコールの脱水反応を経たエーテル化等の副反応を起こすため、選択率に問題があり、工業的には不利である。
【0005】
特許文献2には、本発明の固体酸触媒を用いたエステル体の製造方法が記載されている。本文献で用いられる固体酸触媒の形状については、粉末状、球形粒状、不定形顆粒状等が挙げられているが、これらの形状の固体酸触媒を用いても、反応終了後の生成物中から生成物の安定性に支障のない程度まで固体酸触媒を除くのは困難である。
【0006】
特許文献3には、シュウ酸−エチレングリコールのオリゴマーを低温で製造し、オリゴマーを回収して洗浄し、高温でポリエステル化する2段階反応も記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2006‐265416号公報
【特許文献2】WO2008‐117769号公報
【特許文献3】特開平09−059359号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
これまでの固体酸触媒を用いた方法では、エステル化反応に好ましい固体酸触媒を用いたとしても、反応終了後のエステル体生成物中から、生成物の安定に支障のない程度まで固体酸触媒を除くことは困難であり、生成物の安定性を確保する上で問題となっていた。
また、これまでのシュウ酸エステル体等の熱分解性カルボン酸を原料としたエステル体の製造方法では、反応時に生成物が分解する問題点を有していた。
そこで、本発明では、熱分解性が高いシュウ酸エステル体を高収率で反応させることができ、更に反応終了後にシュウ酸エステル体生成物から固体酸触媒を容易に除くことのできる形状の固体酸触媒を用いたシュウ酸エステル体の製造方法、該固体酸触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造することを特徴とするシュウ酸エステル体製造用反応装置を提供することを課題とする。
【課題を解決するための手段】
【0009】
本発明は、エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法を提供するものである。
【発明の効果】
【0010】
本発明によれば、熱分解性が高いシュウ酸エステル体を高収率で反応させることができ、更に反応終了後にエステル体生成物から固体酸触媒を容易に除くことのできる形状の固体酸触媒を用いたシュウ酸エステル体の製造方法を提供すること、該固体酸触媒の存在下に、シュウ酸及びアルコールを反応させてシュウ酸エステル体を製造することを特徴とするシュウ酸エステル体製造用反応装置を提供することができる。
【図面の簡単な説明】
【0011】
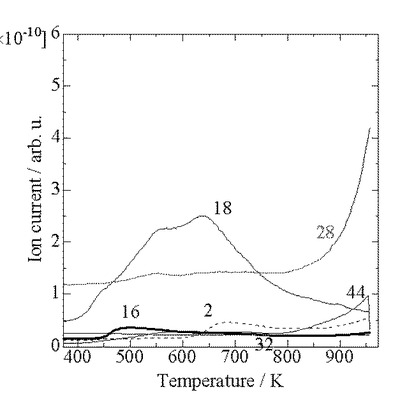
【図1】質量分析計による固体酸触媒(A1)の昇温脱離時に測定した主な質量スペクトルである。
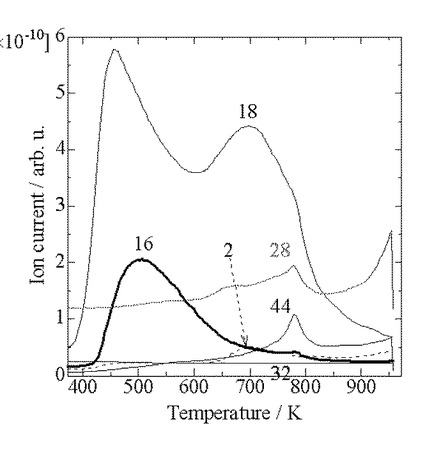
【図2】質量分析計による固体酸触媒(A2)の昇温脱離時に測定した主な質量スペクトルである。
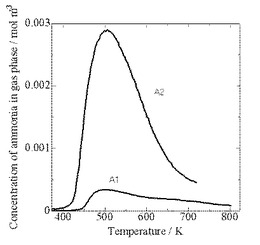
【図3】TPD‐AT‐1型昇温脱離装置による固体酸触媒(A1)及び(A2)のアンモニアTPDスペクトルである。
【発明を実施するための形態】
【0012】
即ち、本発明は、
1.エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法、
2.成型体が、バインダーを介して固体酸触媒が結合され成型されたものである1.に記載のシュウ酸エステルの製造方法、
3.成型体が、更に、滑沢剤または賦形剤を含む1.又は2.に記載のシュウ酸エステル体の製造方法、
4.1.〜3.の何れかに記載のエステル化反応触媒の存在下に、アルコールとして多価アルコールを用いてエステル化反応を行うシュウ酸ポリエステル体の製造方法、
に関する。
【0013】
本発明に用いられる固体酸触媒は、担体としてのジルコニア表面にモリブデン酸化物を担持してなる固体酸触媒である。担体としてのジルコニアは、触媒の設計・装飾の容易性、触媒能を充分に発揮すること、エステル体若しくはその原料への溶解性等を考慮して、特に好ましい。このジルコニアは、シリカ(SiO2)、アルミナ(Al2O3)、チタニア(TiO2)、酸化スズ(SnO2、SnO)、酸化ハフニウム(HfO2)、酸化鉄(Fe2O3、Fe3O4)、又はゼオライト等を併用したものであっても良い。
【0014】
これらを併用する場合、触媒中のジルコニアの含有量が、モル比で10%以上含んでいることが好ましく、さらに好ましくは30%以上含んだものである。なぜなら、これより少ないと、触媒が、エステル体生成物へ溶解することが問題となり、エステル体製造用固体酸触媒として使用し難くなるからである。
【0015】
前記担持させる金属酸化物としては、モリブデン酸化物が特に好ましい。さらに、モリブデン酸化物と共にタングステン、タンタル等他の金属元素を併用し複合化したものであっても良い。これら複合化しても良い担持する金属酸化物としては、タングステン酸化物(WO3等)、タンタル酸化物(Ta2O5等)等が挙げられる。
【0016】
本発明の触媒の金属元素であるMo/Zr(Moはモリブデン、Zrはジルコニウム)比は、質量比で0.01〜0.40が好ましい。この範囲より少ないと、反応場となる触媒の活性点としての、モリブデン酸ジルコニアが充分に形成されず、触媒能を充分に発揮しないからである。また、この範囲を超えると、担体であるジルコニア表面に比べ、担持するモリブデン酸化物が多すぎ、モリブデン酸化物はジルコニア表面に多層に担持されることになり、結果として触媒能を充分に発揮できなくなるからである。これらの観点から、さらに好ましいMo/Zrの質量比は、0.1〜0.2である。
【0017】
本発明の触媒は、例えば水酸化ジルコニウムとモリブデン酸アンモニウムとの反応生成物(モリブデン酸ジルコニア等)を溝、孔、クラック等を有するジルコニアの表面に形成することにより製造できる。その製造方法としては、水酸化ジルコニウムに、担持するモリブデン酸アンモニウムを平衡吸着法、インシピエント・ウェットネス(Incipient wetness)法、蒸発乾固法、共沈法等公知の担持方法により担持し、さらにこれら吸着混合物を焼成することにより得られる。この時の焼成温度は、好ましくは673K〜1473K、より好ましくは973K〜1273Kとするのが良い。この温度から外れた場合、例えば、焼成温度が673Kより低いと、モリブデン‐酸素‐ジルコニウム(Mo‐O‐Zr)の結合が充分に形成されず、得られた触媒の活性が不十分となる恐れがある、また1473Kより高いと、表面積が激減するために反応基質との接触面積が充分に得られず、触媒活性が激減する恐れがあるため、好ましくない。
【0018】
酸度関数とは、溶液の酸塩基の強さを定量的に表わす数値のひとつで、溶液が水素イオンを与える能力、または水素イオンを受け取る能力を示す関数であり、酸についてはルイス・ハメットによるハメットの酸度関数が一般的に用いられ、溶液が中性塩基にプロトンを移動させる傾向を表現している。
ハメットの酸度関数は、電気的に中性の塩基Bが水溶液中で下記式のように結合する。
B+H+⇔BH+
そして、BH+の酸解離定数をpKBH+とし、Bをある溶液に入れたときH+と結合する割合をCBH+、結合しない割合をCBとすると、ハメットの酸度関数(H0)は下記式で表される。
H0=‐pKBH++log(CBH+/CB)
【0019】
本発明の触媒のハメットの酸度関数(H0)は、‐3〜‐9のものである。ハメットの酸度関数(H0)は、水溶液の酸・塩基の強さがpHで表されるように、固体表面の酸・塩基点の強度を表す指標になる。この関数は、水溶液中ではpH=H0であるため、その強度が直感的に理解され、また、実験操作が簡便であるため広く受け入れられている。H0の値が小さい程強い酸性を示し、H0の値が大きい程強い塩基性を示している。
本発明におけるエステル化反応系では、本発明の固体酸触媒の酸度関数(H0)が‐3より大き過ぎると触媒活性を示さず、エステル化反応が進行しにくくなり、エステル体製造触媒として使用できない。一方、本発明の固体酸触媒の酸度関数(H0)が‐9より小さ過ぎるとグリコールの分子内脱水による炭素‐炭素二重結合の生成、さらにはこの二重結合とグリコールによるエーテル化反応等の副反応を起こすおそれがあり、エステル体製造固体酸触媒として好ましくないからである。
【0020】
<NH3‐TPD測定によるハメットの酸度関数(H0)の測定方法>
測定方法:
試料として固体酸触媒0.1gを日本ベル製TPD‐AT‐1型昇温脱離装置の石英セル(内径10mm)にセットし、ヘリウムガス(30cm3min−1,1atm)流通下で423K(150℃)まで5Kmin−1で昇温し、423Kで3時間保った。その後ヘリウムガスを流通させたまま373K(100℃)まで7.5Kmin−1で降温した後に真空脱気し、100Torr(1Torr=1/760atm=133Pa)のNH3を導入して30分間吸着させ、その後12分間脱気した後に水蒸気処理を行った。水蒸気処理としては、373Kで約25Torr(約3kPa)の蒸気圧の水蒸気を導入、そのまま30分間保ち、30分間脱気、再び30分間水蒸気導入、再び30分間脱気の順に繰り返した。その後ヘリウムガス0.041mmols−1(298K,25℃,1atmで60cm3min−1に相当する)を、減圧(100Torr)を保ちながら流通させ、373Kで30分間保った後に試料床を10Kmin−1で983K(710℃)まで昇温し、出口気体を質量分析計(ANELVA M‐QA100F)で分析した。
【0021】
測定に際しては質量数(m/e)2,4,14,15,16,17,18,26,27,28,29,30,31,32,44のマススペクトルを全て記録した。終了後に1mol%‐NH3/He標準ガスをさらにヘリウムで希釈してアンモニアガス濃度0,0.1,0.2,0.3,0.4mol%、合計流量が0.041mmols−1となるようにして検出器に流通させ、スペクトルを記録し、アンモニアの検量線を作成して検出器強度を補正した。昇温脱離時に測定した主な各質量スペクトルのアンモニア離脱TPDスペクトルから、実測に基づく1点法で、ピーク面積から酸量、ピーク位置等から平均酸強度を決定する。酸量と酸強度(ΔH)を算出し、酸度関数(H0)を計算した。
【0022】
固体酸触媒は、反応原料物に対して触媒作用を発揮してエステル化反応を進行させる。即ち、反応原料物であるアルコールとカルボン酸とは、触媒表面上の活性点に吸着、反応、脱離等のプロセスを経て反応が進行することになる。ジルコニアに担持するモリブデン酸化物とからなる活性点を固体酸触媒の表面に形成することが好ましく、特にジルコニア表面で触媒作用を発揮させることが好ましいことから、主にジルコニアの表面に担持するモリブデン酸化物を担持させることが好ましい。
【0023】
ジルコニアに、担持するモリブデン酸化物を担持する方法としては、平衡吸着法、インシピエント・ウェットネス(Incipient wetness法)、蒸発乾固法、共沈法等が挙げられる。
平衡吸着法は、ジルコニアを担持させる金属の溶液に浸して吸着させた後、過剰分の溶液を濾別する方法である。担持量は溶液濃度と細孔容積で決まる。担体を加えるにつれて溶液の組成が変化する等の問題がある。
【0024】
インシピエント・ウェットネス(Incipient Wetness)法は、ジルコニアを排気後、細孔容積分の担持させる金属の溶液を少しずつ加えてジルコニアの表面が均一に濡れた状態にする方法である。金属元素の担持量は溶液濃度で調節する。
蒸発乾固法は、ジルコニアを溶液に浸した後、溶媒を蒸発させて溶質を担持する方法である。担持量を多くできるが、担体と弱く結合した金属成分は乾燥時に濃縮されて還元処理後には大きな金属粒子になりやすい。
【0025】
共沈法は、1種類以上の触媒活性成分溶液(例えばモリブデン酸アンモニウム溶液)と、担体成分溶液(例えば硝酸ジルコニウム溶液)とを混合し、沈殿剤溶液(例えばアンモニア水)と接触させて触媒活性成分沈殿と担体沈殿を同時に作る方法、又は2種類以上の触媒活性成分溶液を混合させ沈殿させるか若しくは混合液にさらに沈殿剤を加えて沈殿を作る方法である。
【0026】
これらの中で、触媒の特性を考慮しつつ担持方法を選ぶことが好ましく、本発明の固体酸触媒では、インシピエント・ウェットネス(Incipient Wetness)法、蒸発乾固法又は共沈法が好ましく用いられる。
【0027】
本発明の固体酸触媒を製造する方法としては、例えば、モリブデン化合物及びジルコニウム化合物を上記の担持方法により共存させ、空気中もしくはHe、Ne、Ar、N2、O2等の雰囲気下で、好ましくは673K〜1473Kで焼成処理を行うことにより得られる。これらのモリブデン酸化物及びジルコニウム化合物の選定には、担体表面の等電点を考慮し、担持させる金属化合物を選定する必要がある。例えば、そのモリブデン化合物としては、モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O)が好ましく挙げられ、ジルコニウム化合物としては水酸化ジルコニウムが好ましく挙げられる。焼成温度は673K〜1473Kの範囲で行うことが好ましい。更に好ましくは773K〜1273Kの範囲である。これは、焼成温度が673Kより低いと、モリブデン‐酸素‐ジルコニウム(Mo‐O‐Zr)の結合が充分に形成されず、得られた触媒の活性が低下する恐れがあるためである。また、1473Kより高い場合、表面積が激減するために反応基質との接触面積が充分に得られないために、活性が低下する恐れがあるためである。
【0028】
本発明の固体酸触媒は、固体状の触媒であり、エステル化反応の原料であるカルボン酸、アルコールの液相に溶解しないものである。また、本発明の固体酸触媒は、必要に応じて任意の元素をさらに1種類あるいはそれ以上の種類を併用して担持させても良い。その任意の元素としてはケイ素、アルミニウム、リン、タングステン、セシウム、ニオブ、チタン、スズ、銀、銅、亜鉛、クロム、テルル、アンチモン、ビスマス、セレン、鉄、マグネシウム、カルシウム、バナジウム、セリウム、マンガン、コバルト、ヨウ素、ニッケル、ランタン、プラセオジウム、ネオジウム、プロメチウム、サマリウム、ユウロピウム、ガドリニウム、テルビウム、ジスプロシウム、ホルミウム、エルビウム、ツリウム、イッテルビウム、ルテチウム等が挙げられる。
【0029】
かかる任意に担持するモリブデン酸化物の形状としては、特に限定されるものではないが、例えば粒子状、クラスター等の形態が好ましく挙げられる。また、この担持させるモリブデン酸化物の微粒子のサイズにも限定されないが、サブミクロンからミクロン単位以下となる粒子状態等を形成する状態が好ましく、各粒子が会合・凝集等をしていても良い。
【0030】
本発明の固体酸触媒を含有する成型体は、その存在下に、本発明のエステル体製造用反応装置内で、シュウ酸及びアルコールを反応させてエステル体を製造することのできる固体酸触媒の形状であれば特に制限はないが、好ましい成型体として、例えば錠剤を挙げることができる。
これらの成型体は、公知慣用の方法によって製造することができ、例えば、錠剤を製造する場合には、常法に従って通常公知の打錠機を用いて行うことができる。また、当該錠剤には、必要に応じて滑沢剤、結着剤等の添加剤を添加しても、しなくてもよい。
【0031】
使用される滑沢剤、結着剤等の添加剤は、公知慣用の添加剤を挙げることができる。
本発明で使用する滑沢剤は、例えば、アラビアゴム末、カカオ脂、カルナウバロウ、カルメロースカルシウム、カルメロースナトリウム、カロペプタイド、含水ニ酸化ケイ素、乾燥水酸化アルミニウムゲル、グリセリン、ケイ酸マグネシウム、軽質無水ケイ酸、軽質流動パラフィン、結晶セルロース、硬化油、合成ケイ酸アルミニウム、ゴマ油、コムギデンプン、サラシミツロウ、酸化マグネシウム、ジブチルヒドロキシトルエン、2,6−ジ−t−ブチル−4−メチルフェノール、ジメチルポリシロキサン、酒石酸カリウムナトリウム、ショ糖脂肪酸エステル、シリコーン樹脂、水酸化アルミニウム・ゲル、ステアリルアルコール、ステアリン酸、ステアリン酸アルミニウム、ステアリン酸カルシウム、ステアリン酸ポリオキシル40、ステアリン酸マグネシウム、ステロテックスHM、セタノール、ゼラチン、タルク、炭酸マグネシウム、沈降炭酸カルシウム、トウモロコシデンプン、乳糖、ハードファット、白糖、バレイショデンプン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース2910、フマル酸、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール、ポリソルベート80、マクロゴール1500、マクロゴール400、マクロゴール4000、マクロゴール600、マクロゴール6000、ミツロウ、メタケイ酸アルミン酸マグネシウム、メチルセルロース、モクロウ、モノステアリン酸グリセリン、ラウリル硫酸ナトリウム、硫酸カルシウム、硫酸マグネシウム、流動パラフィン、リン酸などが挙げられる。
【0032】
本発明で行う打錠成型の条件は、φ1mm〜φ200mm、打錠圧力0.5kN/cm2〜150kN/cm2、滑沢剤の配合割合0.1%〜50.0%が好ましい。より好ましくはφ3mm〜φ15mm、打錠圧力5kN/cm2〜100kN/cm2、滑沢剤の配合割合1.0%〜10.0%である。これは、φが小さすぎると、滑沢性、及び臼杵の細さにより、打錠成型が極めて難しくなるためである。また、打錠圧力がこれより小さすぎると、錠剤に成型できたとしてもその強度が低すぎるために使い難く、一方、高すぎても得られた成型体にクラックが入るため、その部分が脆く、割れの原因が生じる。
【0033】
次に滑沢剤がこれより少ない場合、触媒の流動性が不十分であるため、打錠成型時、触媒がうまく臼杵に充填され難く、打錠成型前に造粒などを必要とすることが生じる。また、流動性が不十分であると、一旦成型体を作製することができたとしても、臼杵から排出させる際に圧力をかける必要があり、成型体が破壊される恐れもある。一方、この範囲より大きい場合、活性サイトとなる触媒成分が少なくなり、触媒性能が低下する。
【0034】
上記本発明の成型体は、固体酸触媒を加圧成型して得られるが、成型体には固体酸触媒同士の結合に与るバインダーを用いることもできる。バインダーを用いる場合には、バインダーの存在下に固体酸触媒を成型すればよい。
【0035】
バインダーとしては、例えば、無機粒子系バインダー、粘土鉱物系バインダー、又は有機粒子系バインダー等の上記のバインダーを挙げることができる。
無機粒子系バインダーとしては、シリカゾル、ジルコニアゾル、チタニアゾル、アルミナゾル、アルミナファイバー、アルミナパウダー等を挙げることができ、粘度鉱物系バインダーとしては、カオリン、ベントナイト等を挙げることができ、有機粒子系バインダーとしては、メチルセルロース、エチルセルロース、カルボキシメチルセルロース、ポリビニルアルコール等を挙げることができる。これらのバインダーは、単一で用いても、2種以上の成分を併用してもよい。
【0036】
また、本発明のエステル化反応触媒は、固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であってもよい。
【0037】
本発明に用いられるバインダーは、本発明で用いられる固体酸触媒と支持構造体を結合させるために必要である。使用されるバインダーの種類に制限はないが、具体的には、無機粒子系バインダー、粘土鉱物系バインダー、又は有機粒子系バインダー等の上記のバインダーを挙げることができる。
【0038】
本発明で用いられる支持構造体の材質は特に限定されず、たとえば、セラミックス、メタルを例示できる。セラミックスとしては、酸化物または非酸化物のものを用いることができ、具体的には、コージェライト、ムライト、アルミナ、スピネル、炭化珪素、窒化珪素、窒化アルミニウム、ジルコニア、リチウムアルミニウムシリケート、チタン酸アルミニウムなどを例示できる。支持構造体の形態は、セル壁で区画され一定方向に延びるセル通路を有するハニカムモノリス担体、粒状のペレット担体などがある。
【0039】
本発明の固体酸触媒を支持構造体に固定化するには、固体酸触媒、水及びバインダーとの混合液を調製した後に、該支持構造体に被覆し、その後、乾燥し焼成することにより得られる。
ここで、混合液は、固体酸触媒、水及びバインダーの質量比の範囲を、5〜70:30〜90:1〜30となるように調整することが好ましい。更に好ましくは、10〜25:70〜85:2〜10である。また、被覆、乾燥、及び焼成は公知慣用の方法により行うことができる。
【0040】
本発明のエステル体の製造方法は、固体酸触媒を含有する成型体の存在下に、シュウ酸及びアルコールを反応させてエステル体を製造することに特徴を有し、反応は、攪拌を行っても行わなくてもよいが、エステル体の製造途中では固体酸触媒を含有する成型体を崩壊させないようにすることが重要である。使用される反応装置は、固体酸触媒の成型体を崩壊させないようにすることができれば特に制限はないが、例えば、固体酸触媒を充填した流通式反応器又は回分式反応器に反応原料であるシュウ酸、アルコールを供給して脱水縮合反応させることが好ましい。
触媒の除去方法としては、特別な操作は特に必要無く、例えば回分式反応器を用いた場合は、簡単な濾過操作で行え、固定床流通式反応器を用いた場合は該濾過操作の必要も無く、固体酸触媒を充填したカラム内を流通して得られたエステル体中に固体酸触媒が残らない製造方法である。
【0041】
回分式反応器では、反応原料物であるシュウ酸とアルコールを反応器に仕込んで、撹拌しながら反応を行ない、一定時間後にエステル体生成物を取り出す方法で行う。非定常操作であるから、反応器内の組成は時間とともに変化することになる。遅い反応でエステル転化率を要求されるときは、回分式反応器が有利であり、小規模生産に好ましく使用できる。さらに、触媒を固定床のごとく固定化し、反応器内の原料をポンプにより触媒層に送液、流通させ、反応器に戻す固定床循環型回分式反応操作を行っても良く、触媒分離の観点から特に好ましい。
【0042】
一方、流通式反応器は、定常的な流通操作によって、物質の損失を少なくし、反応状態を安定にしてエステル体の品質を一定に保ち、生産費を低減させることが可能であり、エステル体を連続的に製造する方法としてはより有利である。これらの反応器のうち、反応終了後に触媒の回収を特殊な操作をする必要なく行える固定床流通式反応器もしくは流動床流通式反応器を用いるのが特に好ましい。
【0043】
本発明で用いられるアルコールとしては、通常エステル体の合成に用いられる脂肪族、脂環式、及び芳香族アルコールが挙げられ、主に一価、二価アルコールを使用できる。例えばエチレングリコール、1,2‐プロピレングリコール、1,3‐プロピレングリコール、1,2‐ブタンジオール、1,3‐ブタンジオール、2‐メチル‐1,3‐プロパンジオール、1,4‐ブタンジオール、ネオペンチルグリコール、1,5‐ペンタンジオール、3‐メチル‐1,5‐ペンタンジオール、1,6‐ヘキサンジオール、2‐メチル‐2‐ブチル‐1,3‐プロパンジオール、2,2,4‐トリメチル‐1,3‐ペンタンジオール、2‐エチル‐1,3‐ヘキサンジオール、2‐メチル‐1,8‐オクタンジオール、1,9‐ノナンジオール、2,4‐ジエチル‐1,5‐ペンタンジオール、2‐エチル‐2‐ブチル‐1,3‐プロパンジオール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール等の二価アルコール、グリセリン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール等が挙げられる。これらのアルコールは、二価アルコールが主に使用され、これら単独又は2種類以上組み合わせて使用することができる。
また、ヒドロキシアルキルオキセタンと1官能性エポキシ化合物とを開環反応させて得られる多分岐ポリエーテルポリオールも用いることができる。
【0044】
本発明では、カルボン酸としてシュウ酸が用いられる。本発明で得られるシュウ酸エステル体には、シュウ酸と1価アルコールとの反応により得られるシュウ酸エステル体、及びシュウ酸と多価アルコールとの反応により得られるシュウ酸ポリエステル体も含まれる。
【0045】
本発明で使用するシュウ酸とアルコールの割合は、それらの官能基数を考慮し、当量比で1:3〜3:1であることが好ましく、より好ましくは1:2〜2:1であるが、適宜当量比を選択することができる。
【0046】
本発明の固体酸触媒を用いたエステル体の製造方法は、原料であるシュウ酸、アルコールを脱水縮合させるに当り、例えば、
(1)常圧下にシュウ酸とアルコールとを縮重合させる方法、
(2)減圧下で両者を縮重合せしめる方法、
等がある。縮重合反応は、窒素等の不活性ガスの雰囲気下で行うことが、得られるエステル体の着色を防止する点で好ましい。
【0047】
従来の均一系触媒として用いられていたチタン系及び錫系の触媒は、反応温度が140℃以下ではほとんど縮重合反応が進行しないため、それ以上の温度で反応させる必要があった。しかしながら、本発明の固体酸触媒は、低温、例えば115℃でも縮重合反応を進行させることが可能であり、本発明の固体酸触媒を用いることで従来に比べ低温で、エステル化反応をすることが可能となるため、高温反応により起こりえる副生成物の抑制、省エネルギー化の観点から工業的に有利である。
また、本発明の固体酸触媒の成型体を用いた場合には、通常反応終了時に必要な反応生成物から触媒を除去する操作、例えば、水或いはアルカリ水等による洗浄が不要であり、簡便な濾過法等により容易に触媒を除去することができる。
【実施例】
【0048】
次に、実施例及び比較例により本発明を具体的に説明するが、本発明はこれに限定されるものではない。また、実施例及び比較例の部は、特記しないかぎり質量部を表す。
【0049】
(調製例1)<固体酸触媒(A1)(MoO3/ZrO2)の調製>
MoO3/ZrO2は、100℃で一晩乾燥させた水酸化ジルコニウム(Zr(OH)4、日本軽金属工業製)50gを、純水にモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O(キシダ化学製)]を必要量溶かした水溶液(0.04mol・dm−3)を用い、水酸化ジルコニウムの細孔容積分の前記モリブデン酸アンモニウム水溶液を少しずつ加えてジルコニウム担体表面が均一に濡れた状態にして得た(Incipient Wetness法)。三酸化モリブデン(MoO3)の担持量が、重量比でMo/Zr=0.1となるように溶液濃度で調節した。反応前処理として酸素雰囲気下で焼成温度1073Kで3時間焼成を行った。自然放置冷却し、常温にして、固体酸触媒(A1)を得た。
【0050】
(調整例2)<固体酸触媒(A2)(MoO3/ZrO2)の調製>
焼成温度を673Kに変えた以外は上記調整例1と同様に調製し、固体酸触媒(A2)を得た。
【0051】
<NH3−TPD測定によるH0関数の測定方法1>
測定方法:
前記固体酸触媒(A1)約0.1gを日本ベル製TPD−AT−1型昇温脱離装置の石英セル(内径10mm)にセットし、ヘリウムガス(30cm3minー1,1atm)流通下で423K(150℃)まで5Kmin−1で昇温し、423Kで3時間保った。その後ヘリウムガスを流通させたまま373K(100℃)まで7.5Kmin−1で降温した後に真空脱気し、100Torr(1Torr=1/760atm=133Pa)のNH3を導入して30分間吸着させ、その後12分間脱気した後に水蒸気処理を行った。水蒸気処理としては、373Kで約25Torr(約3kPa)の蒸気圧の水蒸気を導入、そのまま30分間保ち、30分間脱気、再び30分間水蒸気導入、再び30分間脱気の順に繰り返した。その後ヘリウムガス0.041mmols−1(298K,25℃,1atmで60cm3min−1に相当する)を減圧(100Torr)を保ちながら流通させ、373Kで30分間保った後に試料床を10Kmin−1で983K(710℃)まで昇温し、出口気体を質量分析計(ANELVAM−QA100F)で分析した。
【0052】
測定に際しては質量数(m/e)2,4,14,15,16,17,18,26,27,28,29,30,31,32,44のマススペクトルを全て記録した。終了後に1mol%−NH3/He標準ガスをさらにヘリウムで希釈してアンモニアガス濃度0,0.1,0.2,0.3,0.4mol%、合計流量が0.041mmols-1となるようにして検出器に流通させ、スペクトルを記録し、アンモニアの検量線を作成して検出器強度を補正した。
【0053】
<NH3−TPD測定によるH0関数の測定方法2>
サンプルを、固体酸触媒(A1)から固体酸触媒(A2)に変えた以外は上記NH3−TPD測定によるH0関数の測定方法1と同様に測定した。
【0054】
図1、図2に、それぞれ固体酸触媒(A1)及び(A2)の昇温脱離時に測定した主な各質量スペクトルを示した。他の質量数(m/e)の信号はほぼベースライン上にあり、ピークを示さなかった。
【0055】
どちらの試料でも、500K付近にアンモニアの脱離を示すm/e=16のピークが見られ、さらに固体酸触媒(A1)では900K以上、固体酸触媒(A2)では780K付近に小さなm/e=16のショルダーが見られる。しかし、これら高温のショルダーの出現と同時に、m/e=44の大きなピーク(CO2のフラグメント)およびm/e=28(CO2のフラグメント+N2)も見られていることから、高温のショルダーはCO2のフラグメントによるものであって、アンモニアによるものではないと考えられる。そこで、後述のアンモニアの定量ではこの部分を除いた。
図3には、m/e=16から算出した固体酸触媒(A1)及び(A2)のアンモニアTPDスペクトルを示した。これらのスペクトルから酸量と酸強度(ΔH)を算出し、表−1に示した。
【0056】
実測に基づく1点法では、ピーク面積から酸量、ピーク位置などから平均酸強度を決定できる。この方法によると質量当たりの固体酸触媒(A1)の酸量は約0.03molkg−1、固体酸触媒(A2)の酸量は約0.2molkg−1と差があるように思われるが、表面密度(酸量/表面積)は固体酸触媒(A1)及び(A2)とも0.4〜0.7nm−2程度であった。平均酸強度は固体酸触媒(A1)がΔH=133kJmol−1、H0に換算して−7.4に対して、固体酸触媒(A2)がΔH=116kJmol−1、H0に換算して−4.4とやや弱かった。
【0057】
【表1】

【0058】
(調製例3)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧11〜13kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は6〜8kgfであった。以下これを固体酸触媒(B1)と記す。
【0059】
(調製例4)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧27〜34kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は13〜16kgfであった。以下これを固体酸触媒(B2)と記す。
【0060】
(調製例5)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧50〜60kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は24〜28kgfであった。以下これを固体酸触媒(B3)と記す。
【0061】
(調製例6)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧70〜80kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は33〜40kgfであった。以下これを固体酸触媒(B4)と記す。
【0062】
(調製例7)
上記調製例1で得られた固体酸触媒(A1)にDKエステル(第一工業社製 ショ糖脂肪酸エステル)2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧40〜70kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は30〜70kgfであった。以下これを固体酸触媒(B5)と記す。
【0063】
(調製例8)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、スミ丸型φ5mmの臼杵を用い、打錠圧70〜80kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は30〜38kgfであった。以下これを固体酸触媒(B6)と記す。
【0064】
(調製例9)
上記調製例1で得られた固体酸触媒(A1)125質量部に20%アルミナゾル溶液を125質量部添加し、さらに純水750質量部加え、スラリーとし、それにアルミナからなる支持構造体を浸漬し、引上げ、大気中100℃で3時間乾燥し、さらに大気中500℃で3時間(昇温速度2℃/分)焼成して固体酸触媒(C)を得た。触媒重量から計算すると、4.94wt%の固体酸触媒(A1)がアルミナからなる支持構造体に担持されていた。
【0065】
以下、エステル化に関する実施例を用いて本発明をさらに具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、実施例および比較例の転化率(%)は下記の式により算出し、評価した。
転化率(%)=(脱水量÷理論脱水量)×100(%)
【0066】
(実施例1)
500mLの四ツ口フラスコに1、4−ブタンジオール156質量部、シュウ酸144質量部、および調製例8で調製した錠剤型固体酸触媒(B6)3質量部を、撹拌翼と接触しないように反応器の底部に仕込み、冷却管、凝集管、窒素導入管をセットし、窒素ブローしながら115℃まで昇温し、圧力雰囲気120mbarの条件下、反応を行った。反応開始から24時間後に縮合水を秤量し、転化率の計算を行った。結果、転化率は99%であった。なお、触媒(B6)は反応終了後にフラスコの底部に沈んでいるため、ピンセットで取り除いた。
【0067】
(実施例2)
原料供給槽(以後これを供給器と記す)に2‐メチル‐1,3‐プロパンジオール156質量部、シュウ酸144質量部を充填し、撹拌を行いながら約1時間で115℃まで過熱昇温を行った。115℃到達直後、調製例8で調製した固体酸触媒(B6)30質量部を充填した触媒槽(以後これを反応器と記す)に、先で115℃に過熱された原料を流速3g/minで供給器から反応器に送液し(WHSV 6h−1)、反応器内で原料の混合物と固体酸触媒(B6)とを接触させ、エステル化反応を進行させた。この時反応器内を、温度115℃、圧力雰囲気120mbarに設定し、かつ反応器内がより完全混合流れとなるように200rpmで撹拌を行い、さらに窒素40cc/minによるバブリングを反応器の底部から行った。その結果、転化率94%のポリエステルが連続的に得られた。
【0068】
(実施例3)
供給器に2‐メチル‐1,3‐プロパンジオール156質量部、シュウ酸144質量部を充填し、撹拌を行いながら約1時間で115℃まで過熱昇温を行った。115℃到達直後、調製例9で調製した固体酸触媒(C)300質量部(固体酸触媒(A1)換算で14.8質量部)を充填した反応器に、先で115℃に過熱された原料を流速3g/minで供給器から反応器に送液し(WHSV 0.6h−1、固体酸触媒(A1)換算で12.2h−1)、反応器内で原料の混合物と固体酸触媒(C)とを接触させ、エステル化反応を進行させた。この時反応器内を、温度115℃、圧力雰囲気120mbarに設定し、かつ反応器内がより完全混合流れとなるように200rpmで撹拌を行い、さらに窒素40cc/minによるバブリングを反応器の底部から行った。その結果、転化率97%のポリエステルが連続的に得られた。
【0069】
(比較例1)
500mLの四ツ口フラスコに1、4−ブタンジオール156質量部、シュウ酸144質量部、およびテトラブチルチタネート0.01質量部を仕込み、冷却管、凝集管、窒素導入管をセットし、窒素ブローしながら115℃まで昇温し、反応を行った。反応開始から24時間後に縮合水を秤量し、転化率の計算を行った。結果、反応は進行しなかった。
【0070】
<ポリエステル中に含まれる残留触媒量の定量>
実施例1〜3で合成したエステル体中の残留触媒成分の定量を、蛍光X線測定を用いて行った。結果、両エステル体とも検出限界以下だった。
【産業上の利用可能性】
【0071】
本発明の製造方法は、各種シュウ酸エステル体及びシュウ酸ポリエステル体の製造に用いることができる。本発明により得られるシュウ酸ポリエステル体は、例えば、分解することによりシュウ酸を発生することから、シュウ酸を触媒として用いる反応における潜在性触媒としての利用が可能であり、また、シュウ酸ポリエステル体は、生分解性ポリマー或いは熱分解性ポリマーとしての利用が可能である。
【技術分野】
【0001】
本発明は、固体酸触媒であるエステル化反応触媒の存在下にシュウ酸エステル体を製造する方法、シュウ酸エステル体製造用反応装置に関する。
【背景技術】
【0002】
現在、化学工業でのエステル体の製造は、チタン系もしくは錫系の均一系触媒を用いて行っている。このような均一系触媒は生成物中に溶け込んでしまうために、単離・回収により触媒を取り除くことが難しく、残留触媒のないエステル体を合成することは非常に困難である。これらの残留触媒は、得られたエステル体中に存在する水分とエステル結合との反応を促進させ、加水分解を引き起こすため、エステル体の耐久性や保存安定性を低下させる原因となっている。
上記の問題により、均一系触媒は、通常使用可能な触媒量がごく微量に制限されるため、反応転化が遅く、結果的にエステル体の製造には長時間が必要となる。さらに触媒の単離・回収が困難であることから、エステル体の着色や物性への影響が避けられないという問題もある。
【0003】
また、本発明で目的とするシュウ酸エステル体又はシュウ酸ポリエステル体の製造を目的とした場合には、当該化合物が高い熱分解性を有しているため、従来の触媒を用いた反応系では、目的とするエステル体が生成しても反応系内で分解してしまうために、最終的に目的物が得られないという問題点があった。
【0004】
特許文献1には、固体酸触媒をエステル体重合補助触媒として使用する技術が提案されている。本文献で使用されているモリブデン酸ジルコニアはH0関数が‐12.4であり、いわゆる超強酸である。しかし、グリコールと酸との脱水縮合反応にこのような固体超強酸を用いると、グリコールの脱水反応を経たエーテル化等の副反応を起こすため、選択率に問題があり、工業的には不利である。
【0005】
特許文献2には、本発明の固体酸触媒を用いたエステル体の製造方法が記載されている。本文献で用いられる固体酸触媒の形状については、粉末状、球形粒状、不定形顆粒状等が挙げられているが、これらの形状の固体酸触媒を用いても、反応終了後の生成物中から生成物の安定性に支障のない程度まで固体酸触媒を除くのは困難である。
【0006】
特許文献3には、シュウ酸−エチレングリコールのオリゴマーを低温で製造し、オリゴマーを回収して洗浄し、高温でポリエステル化する2段階反応も記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2006‐265416号公報
【特許文献2】WO2008‐117769号公報
【特許文献3】特開平09−059359号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
これまでの固体酸触媒を用いた方法では、エステル化反応に好ましい固体酸触媒を用いたとしても、反応終了後のエステル体生成物中から、生成物の安定に支障のない程度まで固体酸触媒を除くことは困難であり、生成物の安定性を確保する上で問題となっていた。
また、これまでのシュウ酸エステル体等の熱分解性カルボン酸を原料としたエステル体の製造方法では、反応時に生成物が分解する問題点を有していた。
そこで、本発明では、熱分解性が高いシュウ酸エステル体を高収率で反応させることができ、更に反応終了後にシュウ酸エステル体生成物から固体酸触媒を容易に除くことのできる形状の固体酸触媒を用いたシュウ酸エステル体の製造方法、該固体酸触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造することを特徴とするシュウ酸エステル体製造用反応装置を提供することを課題とする。
【課題を解決するための手段】
【0009】
本発明は、エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法を提供するものである。
【発明の効果】
【0010】
本発明によれば、熱分解性が高いシュウ酸エステル体を高収率で反応させることができ、更に反応終了後にエステル体生成物から固体酸触媒を容易に除くことのできる形状の固体酸触媒を用いたシュウ酸エステル体の製造方法を提供すること、該固体酸触媒の存在下に、シュウ酸及びアルコールを反応させてシュウ酸エステル体を製造することを特徴とするシュウ酸エステル体製造用反応装置を提供することができる。
【図面の簡単な説明】
【0011】
【図1】質量分析計による固体酸触媒(A1)の昇温脱離時に測定した主な質量スペクトルである。
【図2】質量分析計による固体酸触媒(A2)の昇温脱離時に測定した主な質量スペクトルである。
【図3】TPD‐AT‐1型昇温脱離装置による固体酸触媒(A1)及び(A2)のアンモニアTPDスペクトルである。
【発明を実施するための形態】
【0012】
即ち、本発明は、
1.エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法、
2.成型体が、バインダーを介して固体酸触媒が結合され成型されたものである1.に記載のシュウ酸エステルの製造方法、
3.成型体が、更に、滑沢剤または賦形剤を含む1.又は2.に記載のシュウ酸エステル体の製造方法、
4.1.〜3.の何れかに記載のエステル化反応触媒の存在下に、アルコールとして多価アルコールを用いてエステル化反応を行うシュウ酸ポリエステル体の製造方法、
に関する。
【0013】
本発明に用いられる固体酸触媒は、担体としてのジルコニア表面にモリブデン酸化物を担持してなる固体酸触媒である。担体としてのジルコニアは、触媒の設計・装飾の容易性、触媒能を充分に発揮すること、エステル体若しくはその原料への溶解性等を考慮して、特に好ましい。このジルコニアは、シリカ(SiO2)、アルミナ(Al2O3)、チタニア(TiO2)、酸化スズ(SnO2、SnO)、酸化ハフニウム(HfO2)、酸化鉄(Fe2O3、Fe3O4)、又はゼオライト等を併用したものであっても良い。
【0014】
これらを併用する場合、触媒中のジルコニアの含有量が、モル比で10%以上含んでいることが好ましく、さらに好ましくは30%以上含んだものである。なぜなら、これより少ないと、触媒が、エステル体生成物へ溶解することが問題となり、エステル体製造用固体酸触媒として使用し難くなるからである。
【0015】
前記担持させる金属酸化物としては、モリブデン酸化物が特に好ましい。さらに、モリブデン酸化物と共にタングステン、タンタル等他の金属元素を併用し複合化したものであっても良い。これら複合化しても良い担持する金属酸化物としては、タングステン酸化物(WO3等)、タンタル酸化物(Ta2O5等)等が挙げられる。
【0016】
本発明の触媒の金属元素であるMo/Zr(Moはモリブデン、Zrはジルコニウム)比は、質量比で0.01〜0.40が好ましい。この範囲より少ないと、反応場となる触媒の活性点としての、モリブデン酸ジルコニアが充分に形成されず、触媒能を充分に発揮しないからである。また、この範囲を超えると、担体であるジルコニア表面に比べ、担持するモリブデン酸化物が多すぎ、モリブデン酸化物はジルコニア表面に多層に担持されることになり、結果として触媒能を充分に発揮できなくなるからである。これらの観点から、さらに好ましいMo/Zrの質量比は、0.1〜0.2である。
【0017】
本発明の触媒は、例えば水酸化ジルコニウムとモリブデン酸アンモニウムとの反応生成物(モリブデン酸ジルコニア等)を溝、孔、クラック等を有するジルコニアの表面に形成することにより製造できる。その製造方法としては、水酸化ジルコニウムに、担持するモリブデン酸アンモニウムを平衡吸着法、インシピエント・ウェットネス(Incipient wetness)法、蒸発乾固法、共沈法等公知の担持方法により担持し、さらにこれら吸着混合物を焼成することにより得られる。この時の焼成温度は、好ましくは673K〜1473K、より好ましくは973K〜1273Kとするのが良い。この温度から外れた場合、例えば、焼成温度が673Kより低いと、モリブデン‐酸素‐ジルコニウム(Mo‐O‐Zr)の結合が充分に形成されず、得られた触媒の活性が不十分となる恐れがある、また1473Kより高いと、表面積が激減するために反応基質との接触面積が充分に得られず、触媒活性が激減する恐れがあるため、好ましくない。
【0018】
酸度関数とは、溶液の酸塩基の強さを定量的に表わす数値のひとつで、溶液が水素イオンを与える能力、または水素イオンを受け取る能力を示す関数であり、酸についてはルイス・ハメットによるハメットの酸度関数が一般的に用いられ、溶液が中性塩基にプロトンを移動させる傾向を表現している。
ハメットの酸度関数は、電気的に中性の塩基Bが水溶液中で下記式のように結合する。
B+H+⇔BH+
そして、BH+の酸解離定数をpKBH+とし、Bをある溶液に入れたときH+と結合する割合をCBH+、結合しない割合をCBとすると、ハメットの酸度関数(H0)は下記式で表される。
H0=‐pKBH++log(CBH+/CB)
【0019】
本発明の触媒のハメットの酸度関数(H0)は、‐3〜‐9のものである。ハメットの酸度関数(H0)は、水溶液の酸・塩基の強さがpHで表されるように、固体表面の酸・塩基点の強度を表す指標になる。この関数は、水溶液中ではpH=H0であるため、その強度が直感的に理解され、また、実験操作が簡便であるため広く受け入れられている。H0の値が小さい程強い酸性を示し、H0の値が大きい程強い塩基性を示している。
本発明におけるエステル化反応系では、本発明の固体酸触媒の酸度関数(H0)が‐3より大き過ぎると触媒活性を示さず、エステル化反応が進行しにくくなり、エステル体製造触媒として使用できない。一方、本発明の固体酸触媒の酸度関数(H0)が‐9より小さ過ぎるとグリコールの分子内脱水による炭素‐炭素二重結合の生成、さらにはこの二重結合とグリコールによるエーテル化反応等の副反応を起こすおそれがあり、エステル体製造固体酸触媒として好ましくないからである。
【0020】
<NH3‐TPD測定によるハメットの酸度関数(H0)の測定方法>
測定方法:
試料として固体酸触媒0.1gを日本ベル製TPD‐AT‐1型昇温脱離装置の石英セル(内径10mm)にセットし、ヘリウムガス(30cm3min−1,1atm)流通下で423K(150℃)まで5Kmin−1で昇温し、423Kで3時間保った。その後ヘリウムガスを流通させたまま373K(100℃)まで7.5Kmin−1で降温した後に真空脱気し、100Torr(1Torr=1/760atm=133Pa)のNH3を導入して30分間吸着させ、その後12分間脱気した後に水蒸気処理を行った。水蒸気処理としては、373Kで約25Torr(約3kPa)の蒸気圧の水蒸気を導入、そのまま30分間保ち、30分間脱気、再び30分間水蒸気導入、再び30分間脱気の順に繰り返した。その後ヘリウムガス0.041mmols−1(298K,25℃,1atmで60cm3min−1に相当する)を、減圧(100Torr)を保ちながら流通させ、373Kで30分間保った後に試料床を10Kmin−1で983K(710℃)まで昇温し、出口気体を質量分析計(ANELVA M‐QA100F)で分析した。
【0021】
測定に際しては質量数(m/e)2,4,14,15,16,17,18,26,27,28,29,30,31,32,44のマススペクトルを全て記録した。終了後に1mol%‐NH3/He標準ガスをさらにヘリウムで希釈してアンモニアガス濃度0,0.1,0.2,0.3,0.4mol%、合計流量が0.041mmols−1となるようにして検出器に流通させ、スペクトルを記録し、アンモニアの検量線を作成して検出器強度を補正した。昇温脱離時に測定した主な各質量スペクトルのアンモニア離脱TPDスペクトルから、実測に基づく1点法で、ピーク面積から酸量、ピーク位置等から平均酸強度を決定する。酸量と酸強度(ΔH)を算出し、酸度関数(H0)を計算した。
【0022】
固体酸触媒は、反応原料物に対して触媒作用を発揮してエステル化反応を進行させる。即ち、反応原料物であるアルコールとカルボン酸とは、触媒表面上の活性点に吸着、反応、脱離等のプロセスを経て反応が進行することになる。ジルコニアに担持するモリブデン酸化物とからなる活性点を固体酸触媒の表面に形成することが好ましく、特にジルコニア表面で触媒作用を発揮させることが好ましいことから、主にジルコニアの表面に担持するモリブデン酸化物を担持させることが好ましい。
【0023】
ジルコニアに、担持するモリブデン酸化物を担持する方法としては、平衡吸着法、インシピエント・ウェットネス(Incipient wetness法)、蒸発乾固法、共沈法等が挙げられる。
平衡吸着法は、ジルコニアを担持させる金属の溶液に浸して吸着させた後、過剰分の溶液を濾別する方法である。担持量は溶液濃度と細孔容積で決まる。担体を加えるにつれて溶液の組成が変化する等の問題がある。
【0024】
インシピエント・ウェットネス(Incipient Wetness)法は、ジルコニアを排気後、細孔容積分の担持させる金属の溶液を少しずつ加えてジルコニアの表面が均一に濡れた状態にする方法である。金属元素の担持量は溶液濃度で調節する。
蒸発乾固法は、ジルコニアを溶液に浸した後、溶媒を蒸発させて溶質を担持する方法である。担持量を多くできるが、担体と弱く結合した金属成分は乾燥時に濃縮されて還元処理後には大きな金属粒子になりやすい。
【0025】
共沈法は、1種類以上の触媒活性成分溶液(例えばモリブデン酸アンモニウム溶液)と、担体成分溶液(例えば硝酸ジルコニウム溶液)とを混合し、沈殿剤溶液(例えばアンモニア水)と接触させて触媒活性成分沈殿と担体沈殿を同時に作る方法、又は2種類以上の触媒活性成分溶液を混合させ沈殿させるか若しくは混合液にさらに沈殿剤を加えて沈殿を作る方法である。
【0026】
これらの中で、触媒の特性を考慮しつつ担持方法を選ぶことが好ましく、本発明の固体酸触媒では、インシピエント・ウェットネス(Incipient Wetness)法、蒸発乾固法又は共沈法が好ましく用いられる。
【0027】
本発明の固体酸触媒を製造する方法としては、例えば、モリブデン化合物及びジルコニウム化合物を上記の担持方法により共存させ、空気中もしくはHe、Ne、Ar、N2、O2等の雰囲気下で、好ましくは673K〜1473Kで焼成処理を行うことにより得られる。これらのモリブデン酸化物及びジルコニウム化合物の選定には、担体表面の等電点を考慮し、担持させる金属化合物を選定する必要がある。例えば、そのモリブデン化合物としては、モリブデン酸アンモニウム((NH4)6Mo7O24・4H2O)が好ましく挙げられ、ジルコニウム化合物としては水酸化ジルコニウムが好ましく挙げられる。焼成温度は673K〜1473Kの範囲で行うことが好ましい。更に好ましくは773K〜1273Kの範囲である。これは、焼成温度が673Kより低いと、モリブデン‐酸素‐ジルコニウム(Mo‐O‐Zr)の結合が充分に形成されず、得られた触媒の活性が低下する恐れがあるためである。また、1473Kより高い場合、表面積が激減するために反応基質との接触面積が充分に得られないために、活性が低下する恐れがあるためである。
【0028】
本発明の固体酸触媒は、固体状の触媒であり、エステル化反応の原料であるカルボン酸、アルコールの液相に溶解しないものである。また、本発明の固体酸触媒は、必要に応じて任意の元素をさらに1種類あるいはそれ以上の種類を併用して担持させても良い。その任意の元素としてはケイ素、アルミニウム、リン、タングステン、セシウム、ニオブ、チタン、スズ、銀、銅、亜鉛、クロム、テルル、アンチモン、ビスマス、セレン、鉄、マグネシウム、カルシウム、バナジウム、セリウム、マンガン、コバルト、ヨウ素、ニッケル、ランタン、プラセオジウム、ネオジウム、プロメチウム、サマリウム、ユウロピウム、ガドリニウム、テルビウム、ジスプロシウム、ホルミウム、エルビウム、ツリウム、イッテルビウム、ルテチウム等が挙げられる。
【0029】
かかる任意に担持するモリブデン酸化物の形状としては、特に限定されるものではないが、例えば粒子状、クラスター等の形態が好ましく挙げられる。また、この担持させるモリブデン酸化物の微粒子のサイズにも限定されないが、サブミクロンからミクロン単位以下となる粒子状態等を形成する状態が好ましく、各粒子が会合・凝集等をしていても良い。
【0030】
本発明の固体酸触媒を含有する成型体は、その存在下に、本発明のエステル体製造用反応装置内で、シュウ酸及びアルコールを反応させてエステル体を製造することのできる固体酸触媒の形状であれば特に制限はないが、好ましい成型体として、例えば錠剤を挙げることができる。
これらの成型体は、公知慣用の方法によって製造することができ、例えば、錠剤を製造する場合には、常法に従って通常公知の打錠機を用いて行うことができる。また、当該錠剤には、必要に応じて滑沢剤、結着剤等の添加剤を添加しても、しなくてもよい。
【0031】
使用される滑沢剤、結着剤等の添加剤は、公知慣用の添加剤を挙げることができる。
本発明で使用する滑沢剤は、例えば、アラビアゴム末、カカオ脂、カルナウバロウ、カルメロースカルシウム、カルメロースナトリウム、カロペプタイド、含水ニ酸化ケイ素、乾燥水酸化アルミニウムゲル、グリセリン、ケイ酸マグネシウム、軽質無水ケイ酸、軽質流動パラフィン、結晶セルロース、硬化油、合成ケイ酸アルミニウム、ゴマ油、コムギデンプン、サラシミツロウ、酸化マグネシウム、ジブチルヒドロキシトルエン、2,6−ジ−t−ブチル−4−メチルフェノール、ジメチルポリシロキサン、酒石酸カリウムナトリウム、ショ糖脂肪酸エステル、シリコーン樹脂、水酸化アルミニウム・ゲル、ステアリルアルコール、ステアリン酸、ステアリン酸アルミニウム、ステアリン酸カルシウム、ステアリン酸ポリオキシル40、ステアリン酸マグネシウム、ステロテックスHM、セタノール、ゼラチン、タルク、炭酸マグネシウム、沈降炭酸カルシウム、トウモロコシデンプン、乳糖、ハードファット、白糖、バレイショデンプン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース2910、フマル酸、ポリオキシエチレン(160)ポリオキシプロピレン(30)グリコール、ポリソルベート80、マクロゴール1500、マクロゴール400、マクロゴール4000、マクロゴール600、マクロゴール6000、ミツロウ、メタケイ酸アルミン酸マグネシウム、メチルセルロース、モクロウ、モノステアリン酸グリセリン、ラウリル硫酸ナトリウム、硫酸カルシウム、硫酸マグネシウム、流動パラフィン、リン酸などが挙げられる。
【0032】
本発明で行う打錠成型の条件は、φ1mm〜φ200mm、打錠圧力0.5kN/cm2〜150kN/cm2、滑沢剤の配合割合0.1%〜50.0%が好ましい。より好ましくはφ3mm〜φ15mm、打錠圧力5kN/cm2〜100kN/cm2、滑沢剤の配合割合1.0%〜10.0%である。これは、φが小さすぎると、滑沢性、及び臼杵の細さにより、打錠成型が極めて難しくなるためである。また、打錠圧力がこれより小さすぎると、錠剤に成型できたとしてもその強度が低すぎるために使い難く、一方、高すぎても得られた成型体にクラックが入るため、その部分が脆く、割れの原因が生じる。
【0033】
次に滑沢剤がこれより少ない場合、触媒の流動性が不十分であるため、打錠成型時、触媒がうまく臼杵に充填され難く、打錠成型前に造粒などを必要とすることが生じる。また、流動性が不十分であると、一旦成型体を作製することができたとしても、臼杵から排出させる際に圧力をかける必要があり、成型体が破壊される恐れもある。一方、この範囲より大きい場合、活性サイトとなる触媒成分が少なくなり、触媒性能が低下する。
【0034】
上記本発明の成型体は、固体酸触媒を加圧成型して得られるが、成型体には固体酸触媒同士の結合に与るバインダーを用いることもできる。バインダーを用いる場合には、バインダーの存在下に固体酸触媒を成型すればよい。
【0035】
バインダーとしては、例えば、無機粒子系バインダー、粘土鉱物系バインダー、又は有機粒子系バインダー等の上記のバインダーを挙げることができる。
無機粒子系バインダーとしては、シリカゾル、ジルコニアゾル、チタニアゾル、アルミナゾル、アルミナファイバー、アルミナパウダー等を挙げることができ、粘度鉱物系バインダーとしては、カオリン、ベントナイト等を挙げることができ、有機粒子系バインダーとしては、メチルセルロース、エチルセルロース、カルボキシメチルセルロース、ポリビニルアルコール等を挙げることができる。これらのバインダーは、単一で用いても、2種以上の成分を併用してもよい。
【0036】
また、本発明のエステル化反応触媒は、固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であってもよい。
【0037】
本発明に用いられるバインダーは、本発明で用いられる固体酸触媒と支持構造体を結合させるために必要である。使用されるバインダーの種類に制限はないが、具体的には、無機粒子系バインダー、粘土鉱物系バインダー、又は有機粒子系バインダー等の上記のバインダーを挙げることができる。
【0038】
本発明で用いられる支持構造体の材質は特に限定されず、たとえば、セラミックス、メタルを例示できる。セラミックスとしては、酸化物または非酸化物のものを用いることができ、具体的には、コージェライト、ムライト、アルミナ、スピネル、炭化珪素、窒化珪素、窒化アルミニウム、ジルコニア、リチウムアルミニウムシリケート、チタン酸アルミニウムなどを例示できる。支持構造体の形態は、セル壁で区画され一定方向に延びるセル通路を有するハニカムモノリス担体、粒状のペレット担体などがある。
【0039】
本発明の固体酸触媒を支持構造体に固定化するには、固体酸触媒、水及びバインダーとの混合液を調製した後に、該支持構造体に被覆し、その後、乾燥し焼成することにより得られる。
ここで、混合液は、固体酸触媒、水及びバインダーの質量比の範囲を、5〜70:30〜90:1〜30となるように調整することが好ましい。更に好ましくは、10〜25:70〜85:2〜10である。また、被覆、乾燥、及び焼成は公知慣用の方法により行うことができる。
【0040】
本発明のエステル体の製造方法は、固体酸触媒を含有する成型体の存在下に、シュウ酸及びアルコールを反応させてエステル体を製造することに特徴を有し、反応は、攪拌を行っても行わなくてもよいが、エステル体の製造途中では固体酸触媒を含有する成型体を崩壊させないようにすることが重要である。使用される反応装置は、固体酸触媒の成型体を崩壊させないようにすることができれば特に制限はないが、例えば、固体酸触媒を充填した流通式反応器又は回分式反応器に反応原料であるシュウ酸、アルコールを供給して脱水縮合反応させることが好ましい。
触媒の除去方法としては、特別な操作は特に必要無く、例えば回分式反応器を用いた場合は、簡単な濾過操作で行え、固定床流通式反応器を用いた場合は該濾過操作の必要も無く、固体酸触媒を充填したカラム内を流通して得られたエステル体中に固体酸触媒が残らない製造方法である。
【0041】
回分式反応器では、反応原料物であるシュウ酸とアルコールを反応器に仕込んで、撹拌しながら反応を行ない、一定時間後にエステル体生成物を取り出す方法で行う。非定常操作であるから、反応器内の組成は時間とともに変化することになる。遅い反応でエステル転化率を要求されるときは、回分式反応器が有利であり、小規模生産に好ましく使用できる。さらに、触媒を固定床のごとく固定化し、反応器内の原料をポンプにより触媒層に送液、流通させ、反応器に戻す固定床循環型回分式反応操作を行っても良く、触媒分離の観点から特に好ましい。
【0042】
一方、流通式反応器は、定常的な流通操作によって、物質の損失を少なくし、反応状態を安定にしてエステル体の品質を一定に保ち、生産費を低減させることが可能であり、エステル体を連続的に製造する方法としてはより有利である。これらの反応器のうち、反応終了後に触媒の回収を特殊な操作をする必要なく行える固定床流通式反応器もしくは流動床流通式反応器を用いるのが特に好ましい。
【0043】
本発明で用いられるアルコールとしては、通常エステル体の合成に用いられる脂肪族、脂環式、及び芳香族アルコールが挙げられ、主に一価、二価アルコールを使用できる。例えばエチレングリコール、1,2‐プロピレングリコール、1,3‐プロピレングリコール、1,2‐ブタンジオール、1,3‐ブタンジオール、2‐メチル‐1,3‐プロパンジオール、1,4‐ブタンジオール、ネオペンチルグリコール、1,5‐ペンタンジオール、3‐メチル‐1,5‐ペンタンジオール、1,6‐ヘキサンジオール、2‐メチル‐2‐ブチル‐1,3‐プロパンジオール、2,2,4‐トリメチル‐1,3‐ペンタンジオール、2‐エチル‐1,3‐ヘキサンジオール、2‐メチル‐1,8‐オクタンジオール、1,9‐ノナンジオール、2,4‐ジエチル‐1,5‐ペンタンジオール、2‐エチル‐2‐ブチル‐1,3‐プロパンジオール、ジエチレングリコール、ジプロピレングリコール、トリエチレングリコール等の二価アルコール、グリセリン、トリメチロールプロパン、ペンタエリスリトール、ソルビトール等が挙げられる。これらのアルコールは、二価アルコールが主に使用され、これら単独又は2種類以上組み合わせて使用することができる。
また、ヒドロキシアルキルオキセタンと1官能性エポキシ化合物とを開環反応させて得られる多分岐ポリエーテルポリオールも用いることができる。
【0044】
本発明では、カルボン酸としてシュウ酸が用いられる。本発明で得られるシュウ酸エステル体には、シュウ酸と1価アルコールとの反応により得られるシュウ酸エステル体、及びシュウ酸と多価アルコールとの反応により得られるシュウ酸ポリエステル体も含まれる。
【0045】
本発明で使用するシュウ酸とアルコールの割合は、それらの官能基数を考慮し、当量比で1:3〜3:1であることが好ましく、より好ましくは1:2〜2:1であるが、適宜当量比を選択することができる。
【0046】
本発明の固体酸触媒を用いたエステル体の製造方法は、原料であるシュウ酸、アルコールを脱水縮合させるに当り、例えば、
(1)常圧下にシュウ酸とアルコールとを縮重合させる方法、
(2)減圧下で両者を縮重合せしめる方法、
等がある。縮重合反応は、窒素等の不活性ガスの雰囲気下で行うことが、得られるエステル体の着色を防止する点で好ましい。
【0047】
従来の均一系触媒として用いられていたチタン系及び錫系の触媒は、反応温度が140℃以下ではほとんど縮重合反応が進行しないため、それ以上の温度で反応させる必要があった。しかしながら、本発明の固体酸触媒は、低温、例えば115℃でも縮重合反応を進行させることが可能であり、本発明の固体酸触媒を用いることで従来に比べ低温で、エステル化反応をすることが可能となるため、高温反応により起こりえる副生成物の抑制、省エネルギー化の観点から工業的に有利である。
また、本発明の固体酸触媒の成型体を用いた場合には、通常反応終了時に必要な反応生成物から触媒を除去する操作、例えば、水或いはアルカリ水等による洗浄が不要であり、簡便な濾過法等により容易に触媒を除去することができる。
【実施例】
【0048】
次に、実施例及び比較例により本発明を具体的に説明するが、本発明はこれに限定されるものではない。また、実施例及び比較例の部は、特記しないかぎり質量部を表す。
【0049】
(調製例1)<固体酸触媒(A1)(MoO3/ZrO2)の調製>
MoO3/ZrO2は、100℃で一晩乾燥させた水酸化ジルコニウム(Zr(OH)4、日本軽金属工業製)50gを、純水にモリブデン酸アンモニウム[(NH4)6Mo7O24・4H2O(キシダ化学製)]を必要量溶かした水溶液(0.04mol・dm−3)を用い、水酸化ジルコニウムの細孔容積分の前記モリブデン酸アンモニウム水溶液を少しずつ加えてジルコニウム担体表面が均一に濡れた状態にして得た(Incipient Wetness法)。三酸化モリブデン(MoO3)の担持量が、重量比でMo/Zr=0.1となるように溶液濃度で調節した。反応前処理として酸素雰囲気下で焼成温度1073Kで3時間焼成を行った。自然放置冷却し、常温にして、固体酸触媒(A1)を得た。
【0050】
(調整例2)<固体酸触媒(A2)(MoO3/ZrO2)の調製>
焼成温度を673Kに変えた以外は上記調整例1と同様に調製し、固体酸触媒(A2)を得た。
【0051】
<NH3−TPD測定によるH0関数の測定方法1>
測定方法:
前記固体酸触媒(A1)約0.1gを日本ベル製TPD−AT−1型昇温脱離装置の石英セル(内径10mm)にセットし、ヘリウムガス(30cm3minー1,1atm)流通下で423K(150℃)まで5Kmin−1で昇温し、423Kで3時間保った。その後ヘリウムガスを流通させたまま373K(100℃)まで7.5Kmin−1で降温した後に真空脱気し、100Torr(1Torr=1/760atm=133Pa)のNH3を導入して30分間吸着させ、その後12分間脱気した後に水蒸気処理を行った。水蒸気処理としては、373Kで約25Torr(約3kPa)の蒸気圧の水蒸気を導入、そのまま30分間保ち、30分間脱気、再び30分間水蒸気導入、再び30分間脱気の順に繰り返した。その後ヘリウムガス0.041mmols−1(298K,25℃,1atmで60cm3min−1に相当する)を減圧(100Torr)を保ちながら流通させ、373Kで30分間保った後に試料床を10Kmin−1で983K(710℃)まで昇温し、出口気体を質量分析計(ANELVAM−QA100F)で分析した。
【0052】
測定に際しては質量数(m/e)2,4,14,15,16,17,18,26,27,28,29,30,31,32,44のマススペクトルを全て記録した。終了後に1mol%−NH3/He標準ガスをさらにヘリウムで希釈してアンモニアガス濃度0,0.1,0.2,0.3,0.4mol%、合計流量が0.041mmols-1となるようにして検出器に流通させ、スペクトルを記録し、アンモニアの検量線を作成して検出器強度を補正した。
【0053】
<NH3−TPD測定によるH0関数の測定方法2>
サンプルを、固体酸触媒(A1)から固体酸触媒(A2)に変えた以外は上記NH3−TPD測定によるH0関数の測定方法1と同様に測定した。
【0054】
図1、図2に、それぞれ固体酸触媒(A1)及び(A2)の昇温脱離時に測定した主な各質量スペクトルを示した。他の質量数(m/e)の信号はほぼベースライン上にあり、ピークを示さなかった。
【0055】
どちらの試料でも、500K付近にアンモニアの脱離を示すm/e=16のピークが見られ、さらに固体酸触媒(A1)では900K以上、固体酸触媒(A2)では780K付近に小さなm/e=16のショルダーが見られる。しかし、これら高温のショルダーの出現と同時に、m/e=44の大きなピーク(CO2のフラグメント)およびm/e=28(CO2のフラグメント+N2)も見られていることから、高温のショルダーはCO2のフラグメントによるものであって、アンモニアによるものではないと考えられる。そこで、後述のアンモニアの定量ではこの部分を除いた。
図3には、m/e=16から算出した固体酸触媒(A1)及び(A2)のアンモニアTPDスペクトルを示した。これらのスペクトルから酸量と酸強度(ΔH)を算出し、表−1に示した。
【0056】
実測に基づく1点法では、ピーク面積から酸量、ピーク位置などから平均酸強度を決定できる。この方法によると質量当たりの固体酸触媒(A1)の酸量は約0.03molkg−1、固体酸触媒(A2)の酸量は約0.2molkg−1と差があるように思われるが、表面密度(酸量/表面積)は固体酸触媒(A1)及び(A2)とも0.4〜0.7nm−2程度であった。平均酸強度は固体酸触媒(A1)がΔH=133kJmol−1、H0に換算して−7.4に対して、固体酸触媒(A2)がΔH=116kJmol−1、H0に換算して−4.4とやや弱かった。
【0057】
【表1】
【0058】
(調製例3)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧11〜13kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は6〜8kgfであった。以下これを固体酸触媒(B1)と記す。
【0059】
(調製例4)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧27〜34kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は13〜16kgfであった。以下これを固体酸触媒(B2)と記す。
【0060】
(調製例5)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧50〜60kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は24〜28kgfであった。以下これを固体酸触媒(B3)と記す。
【0061】
(調製例6)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧70〜80kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は33〜40kgfであった。以下これを固体酸触媒(B4)と記す。
【0062】
(調製例7)
上記調製例1で得られた固体酸触媒(A1)にDKエステル(第一工業社製 ショ糖脂肪酸エステル)2wt%を添加し、フラット型φ5mmの臼杵を用い、打錠圧40〜70kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は30〜70kgfであった。以下これを固体酸触媒(B5)と記す。
【0063】
(調製例8)
上記調製例1で得られた固体酸触媒(A1)にステアリン酸カルシウム2wt%を添加し、スミ丸型φ5mmの臼杵を用い、打錠圧70〜80kN/cm2、40ストローク/minで打錠成型を行った。得られた錠剤型固体酸触媒は、硬度は30〜38kgfであった。以下これを固体酸触媒(B6)と記す。
【0064】
(調製例9)
上記調製例1で得られた固体酸触媒(A1)125質量部に20%アルミナゾル溶液を125質量部添加し、さらに純水750質量部加え、スラリーとし、それにアルミナからなる支持構造体を浸漬し、引上げ、大気中100℃で3時間乾燥し、さらに大気中500℃で3時間(昇温速度2℃/分)焼成して固体酸触媒(C)を得た。触媒重量から計算すると、4.94wt%の固体酸触媒(A1)がアルミナからなる支持構造体に担持されていた。
【0065】
以下、エステル化に関する実施例を用いて本発明をさらに具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、実施例および比較例の転化率(%)は下記の式により算出し、評価した。
転化率(%)=(脱水量÷理論脱水量)×100(%)
【0066】
(実施例1)
500mLの四ツ口フラスコに1、4−ブタンジオール156質量部、シュウ酸144質量部、および調製例8で調製した錠剤型固体酸触媒(B6)3質量部を、撹拌翼と接触しないように反応器の底部に仕込み、冷却管、凝集管、窒素導入管をセットし、窒素ブローしながら115℃まで昇温し、圧力雰囲気120mbarの条件下、反応を行った。反応開始から24時間後に縮合水を秤量し、転化率の計算を行った。結果、転化率は99%であった。なお、触媒(B6)は反応終了後にフラスコの底部に沈んでいるため、ピンセットで取り除いた。
【0067】
(実施例2)
原料供給槽(以後これを供給器と記す)に2‐メチル‐1,3‐プロパンジオール156質量部、シュウ酸144質量部を充填し、撹拌を行いながら約1時間で115℃まで過熱昇温を行った。115℃到達直後、調製例8で調製した固体酸触媒(B6)30質量部を充填した触媒槽(以後これを反応器と記す)に、先で115℃に過熱された原料を流速3g/minで供給器から反応器に送液し(WHSV 6h−1)、反応器内で原料の混合物と固体酸触媒(B6)とを接触させ、エステル化反応を進行させた。この時反応器内を、温度115℃、圧力雰囲気120mbarに設定し、かつ反応器内がより完全混合流れとなるように200rpmで撹拌を行い、さらに窒素40cc/minによるバブリングを反応器の底部から行った。その結果、転化率94%のポリエステルが連続的に得られた。
【0068】
(実施例3)
供給器に2‐メチル‐1,3‐プロパンジオール156質量部、シュウ酸144質量部を充填し、撹拌を行いながら約1時間で115℃まで過熱昇温を行った。115℃到達直後、調製例9で調製した固体酸触媒(C)300質量部(固体酸触媒(A1)換算で14.8質量部)を充填した反応器に、先で115℃に過熱された原料を流速3g/minで供給器から反応器に送液し(WHSV 0.6h−1、固体酸触媒(A1)換算で12.2h−1)、反応器内で原料の混合物と固体酸触媒(C)とを接触させ、エステル化反応を進行させた。この時反応器内を、温度115℃、圧力雰囲気120mbarに設定し、かつ反応器内がより完全混合流れとなるように200rpmで撹拌を行い、さらに窒素40cc/minによるバブリングを反応器の底部から行った。その結果、転化率97%のポリエステルが連続的に得られた。
【0069】
(比較例1)
500mLの四ツ口フラスコに1、4−ブタンジオール156質量部、シュウ酸144質量部、およびテトラブチルチタネート0.01質量部を仕込み、冷却管、凝集管、窒素導入管をセットし、窒素ブローしながら115℃まで昇温し、反応を行った。反応開始から24時間後に縮合水を秤量し、転化率の計算を行った。結果、反応は進行しなかった。
【0070】
<ポリエステル中に含まれる残留触媒量の定量>
実施例1〜3で合成したエステル体中の残留触媒成分の定量を、蛍光X線測定を用いて行った。結果、両エステル体とも検出限界以下だった。
【産業上の利用可能性】
【0071】
本発明の製造方法は、各種シュウ酸エステル体及びシュウ酸ポリエステル体の製造に用いることができる。本発明により得られるシュウ酸ポリエステル体は、例えば、分解することによりシュウ酸を発生することから、シュウ酸を触媒として用いる反応における潜在性触媒としての利用が可能であり、また、シュウ酸ポリエステル体は、生分解性ポリマー或いは熱分解性ポリマーとしての利用が可能である。
【特許請求の範囲】
【請求項1】
エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法。
【請求項2】
成型体が、バインダーを介して固体酸触媒が結合され成型されたものである請求項1に記載のシュウ酸エステルの製造方法。
【請求項3】
成型体が、更に、滑沢剤または賦形剤を含む請求項1又は2に記載のシュウ酸エステル体の製造方法。
【請求項4】
請求項1〜3の何れかに記載のエステル化反応触媒の存在下に、アルコールとして多価アルコールを用いてエステル化反応を行うシュウ酸ポリエステル体の製造方法。
【請求項1】
エステル化反応触媒の存在下にシュウ酸とアルコールを反応させてシュウ酸エステル体を製造する方法において、該エステル化反応触媒が、
1)加圧成型時の圧力が5〜100N/cm2である加圧成型法により得られる固体酸触媒を含有する成型体であるエステル化反応触媒、又は
2)固体酸触媒が支持構造体上にバインダーを介して固定化されたエステル化反応触媒であり、且つ、
前記1)及び2)における固体酸触媒が、担体としてのジルコニアにモリブデン酸化物を担持させて得られ、ハメットの酸度関数(H0)が−3〜−9であることを特徴とするシュウ酸エステル体の製造方法。
【請求項2】
成型体が、バインダーを介して固体酸触媒が結合され成型されたものである請求項1に記載のシュウ酸エステルの製造方法。
【請求項3】
成型体が、更に、滑沢剤または賦形剤を含む請求項1又は2に記載のシュウ酸エステル体の製造方法。
【請求項4】
請求項1〜3の何れかに記載のエステル化反応触媒の存在下に、アルコールとして多価アルコールを用いてエステル化反応を行うシュウ酸ポリエステル体の製造方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2011−201782(P2011−201782A)
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願番号】特願2010−67856(P2010−67856)
【出願日】平成22年3月24日(2010.3.24)
【出願人】(000002886)DIC株式会社 (2,597)
【Fターム(参考)】
【公開日】平成23年10月13日(2011.10.13)
【国際特許分類】
【出願日】平成22年3月24日(2010.3.24)
【出願人】(000002886)DIC株式会社 (2,597)
【Fターム(参考)】
[ Back to top ]