
固体NMRの測定方法
【課題】本発明の課題は、固体NMRの測定法を提供することである。
【解決手段】本発明によって、1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測または1Hの磁化を他の核種へ移動させた後に当該磁化を観測し、固体試料のNMRスペクトルを測定する。本発明においては、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱することを行う。
【解決手段】本発明によって、1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測または1Hの磁化を他の核種へ移動させた後に当該磁化を観測し、固体試料のNMRスペクトルを測定する。本発明においては、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱することを行う。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、固体NMRの測定方法に関する。
【背景技術】
【0002】
複数の化合物の混合物について、成分ごとのNMRスペクトルを選択的に取得する手法が知られている。
溶液NMRについては、例えば、DOSY法(Diffusion Ordered Spectroscopy Method)が知られている(E. O. Stejskal, J. E. Tanner, Journal of Chemical Physics, vol. 42, No.1, p.288-292 (1965))。DOSY法では、各成分が固有の自己拡散係数(分子の拡散的な運動性)を有することを利用し、成分ごとの自己拡散係数の違いを利用して成分ごとのNMRスペクトルを得る。通常、混合物のNMRスペクトルは、各成分のスペクトルの和として観測されるが、その混合物のNMRスペクトルを分子の拡散係数を利用して逆ラプラス変換(ILT:Inverse Laplace Transform)により分離すると、分子種ごとのNMRスペクトルを得ることが可能になる。通常のDOSY法は、自己拡散係数に対して化学シフトを2次元に展開する手法であるため、定量分析は困難であり、基本的に定性分析のみに適用される。また、DOSY法における測定時間の短縮を目的とした1次元のDOSY法についても報告されているが、この手法は、分子の拡散係数の測定時間の短縮化を目的としたものであるため、複数の化合物の混合物について、各成分のスペクトルを得るためものではない(N. M. Loening, J. Keeler, G. A. Morris, Journal of Magnetic Resonance, vol. 153, p.103-112 (2001))。
【0003】
一方、固体NMRについては、例えば、DECRA法(Direct Exponential Curve Resolution Algorithm)(非特許文献1)やROSY法(Relaxation Ordered Spectroscopy)(非特許文献2)が知られている。
【0004】
DECRA法は基本的に1次元NMRに適用される手法であり、分子種ごとの1Hの縦緩和時間(HT1)の違いを利用して、線形解析によって分子種ごとのNMRスペクトルを得る。DECRA法は、定性分析だけでなく、定量分析にも適用可能であるが、データ取得に莫大な時間がかかる。
【0005】
ROSY法は、DECRA法と同様に、分子種ごとの1Hの縦緩和時間(HT1)の違いを利用して、逆ラプラス変換によりシグナルを分離し、分子種ごとのNMRスペクトルを得る手法である。この手法は、1Hの縦緩和時間(HT1)に対して化学シフトを2次元に展開する手法であるため、定量分析に適用することは難しく、基本的に定性分析に用いられる。特許文献1には、混合物の固体試料についてのROSY法によるNMR測定法が記載されている。特許文献1の方法では、均一な縦磁化緩和時間を有している核に縦磁化緩和を起こさせるパルスを照射する第1の工程、所定時間tを置いて前記均一な縦磁化緩和時間を有している核のスピン拡散を切断して高分解能NMRスペクトルを測定する第2の工程、tを変えて第1の工程と第2の工程を繰り返して複数の高分解能NMRスペクトルを取得する第3の工程、第3の工程により得られた複数の高分解能NMRスペクトルを縦磁化緩和時間に依存して回復するNMR信号強度の回復速度の違いに基づいて逆ラプラス変換法により縦磁化緩和時間の値ごとに分類する第4の工程を備える。
【0006】
また、非特許文献3に記載のNMR測定方法は、複数の化合物を含む固体試料について、化合物ごとの緩和時間の違いを利用して選択的にシグナルを検出する。具体的には、混合物における1Hの縦緩和時間(HT1)について、反復回復法を利用して、消去したいシグナルのシグナル強度が励起状態から基底状態に回復する際にゼロとなるタイミングで測定を行う手法が記載されている。また、非特許文献3には、混合物における1Hの回転座標系の縦緩和時間(HT1ρ)について、スピンエコーを用いることにより、HT1ρの短い化合物のシグナル強度がゼロとなるタイミングで測定することによって、混合物中のHT1ρの長い化合物を選択的に測定する手法が記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2010−71839号公報
【非特許文献】
【0008】
【非特許文献1】B. Antalek, W. Windig, Journal of American Chemical Society, vol. 118, p.10331-10332 (1996)
【非特許文献2】Y. Nishiyama, M. H. Frey, S. Mukasa, H. Utsumi. Journal of Magnetic Resonance, vol. 202, p.135-139 (2010)
【非特許文献3】N. Zumbulyadis, Journal of Magnetic Resonance, 53, p.486-494 (1983)
【発明の概要】
【発明が解決しようとする課題】
【0009】
分子種ごとのNMRシグナルを選択的に取得するための固体NMRに関する従来の手法は、測定に莫大な時間を要したり、定量分析が困難であった。このような状況に鑑み、本発明の課題は、実用性に優れた固体NMRの測定法を提供すること、特に、混合物中の特定の分子種のシグナルを選択的に消去または減弱できる固体NMRの測定法を提供することである。
【課題を解決するための手段】
【0010】
本発明者は、固体試料のNMR測定について鋭意検討したところ、スピンロッキングを用いて1Hの回転座標系の縦緩和時間(HT1ρ)を利用することにより、固体試料のNMRスペクトルを分子種選択的に得られることを見いだし、本発明を完成させるに至った。
【0011】
すなわち、一つの態様において本発明は、複数の分子種を含む固体試料に90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、その後、各成分の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することを含む、固体NMRの測定方法である。
【0012】
本発明は以下に限定されるものではないが、以下の発明を包含する。
(1) 1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を他の核種へ移動させた後に当該磁化を観測するNMRスペクトルの測定方法であって、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱する手段を含む、固体NMRスペクトルの測定方法。
(2) 1H核の磁化を交差分極法により13C核へ移動させ、当該13C核の磁化を観測するものである、(1)に記載の方法。
(3) 1H核の磁化を13C核へ移動させ、当該13C核の磁化を観測する手法が、CP−MAS法によるものである、(2)に記載の方法。
(4) 測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、当該分子種に係る1Hの回転座標系の縦緩和時間HT1ρよりも長い時間をもってスピンロッキングを行うことにより、1Hの回転座標系の縦緩和時間HT1ρが相対的に長い分子種の磁化を観測するものである、(3)に記載の方法。
(5) 測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測する工程、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測する工程、3)1)による測定値から2)による測定値を差し引く工程、を含む、(3)に記載の方法。
(6) 1)において1H核に対して印加されるパルスが15〜60°または120〜165°である、(5)に記載の方法。
(7) 1)において1H核に対して印加されるパルスが90°である、(5)に記載の方法。
(8) 固体NMRスペクトルを測定するためのパルスプログラムであって、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させるための上記パルスプログラム。
【発明の効果】
【0013】
本発明によって、分子種の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することにより、固体試料の成分比を変えたNMRスペクトルを得ることができる。特に本発明によれば、複数の化合物または結晶形の異なる同一化合物もしくは、結晶と非晶質の混合物を含む固体試料について、特定の分子種に起因するシグナルを消去または減弱することが可能である。本発明は、一次元NMRを用いることから、定性分析のみならず定量分析にも適用可能である。また、本発明は、比較的短時間でデータを取得でき、一般的な装置およびデータ処理法で実行することができる。
【図面の簡単な説明】
【0014】
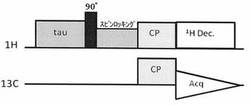
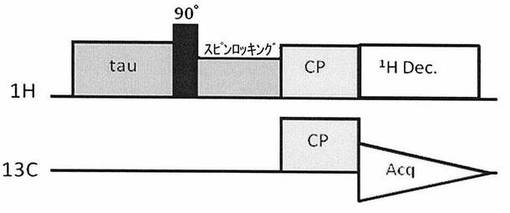
【図1】図1は、スピンロッキングの時間を長くとることにより、HT1ρの長い化合物のみを選択的に検出するためのパルスシークエンスである。
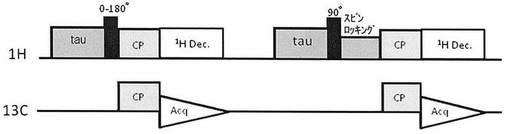
【図2】図2は、混合物中の特定成分のシグナルを選択的に消去または減弱するためのパルスシークエンスである。
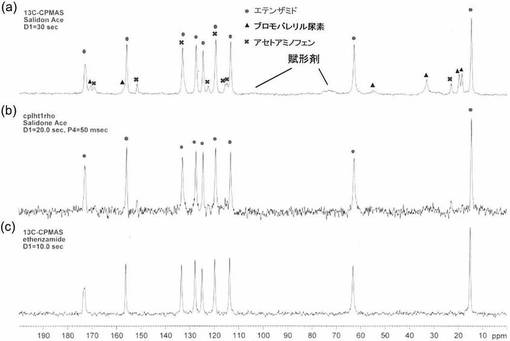
【図3】図3は、本発明の実施例1で得られたNMRチャートである。図中、○はエテンザミド、△はブロモバレリル尿素、×はアセトアミノフェンに由来するシグナルである。図3(a)は、スピンロッキング時間を0秒とした場合(=通常のCP−MAS法による測定)、図3(b)はスピンロッキング時間を50m秒とした場合であり、図3(c)はエテンザミドの標品を通常のCP−MAS法により測定した結果である。
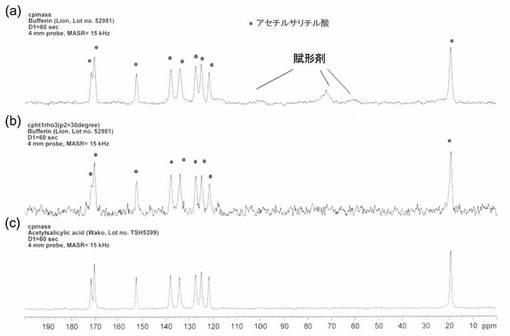
【図4】図4は、本発明の実施例2で得られたNMRチャートである。図中、○はアセチルサリチル酸に由来するシグナルである。図4(a)は、通常のCP−MAS法による測定、図4(b)は、30°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を3.44m秒としたシグナルを差し引いた場合であり、図4(c)はアセチルサリチル酸の標品を通常のCP−MAS法により測定した結果である。
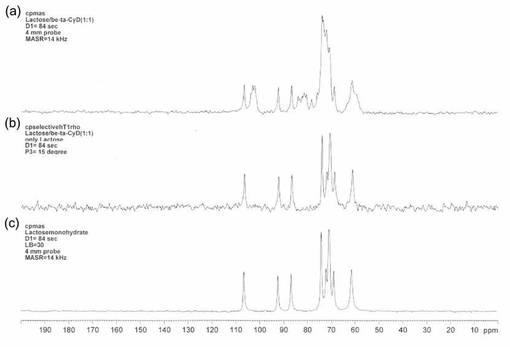
【図5】図5は、本発明の実施例3で得られたNMRチャートである。図5(a)は、ラクトース(乳糖)1水和物とβ−サイクロデキストリンの重量比1:1混合物をCP−MAS法で測定した結果、図5(b)は、同一サンプルに対し、15°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を7.84m秒としたシグナルを差し引いた場合であり、図5(c)はラクトース1水和物の標品を通常のCP−MAS法により測定した結果である。
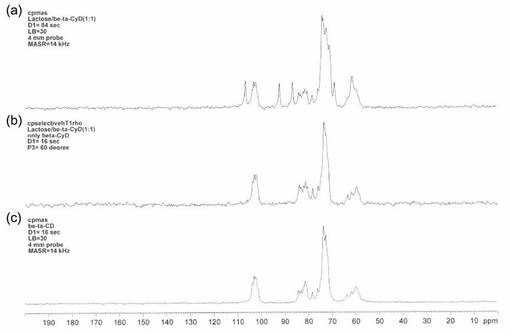
【図6】図6は、本発明の実施例4で得られたNMRチャートである。図6(a)は、ラクトース1水和物とβ−サイクロデキストリンの重量比1:1混合物を通常のCP−MAS法で測定した結果、図6(b)は、同一サンプルに対し、60°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を5.00m秒としたシグナルを差し引いた場合であり、図6(c)はβ−サイクロデキストリンの標品を通常のCP−MAS法により測定した結果である。
【発明を実施するための形態】
【0015】
本発明は、複数の分子種を含む固体試料についてNMR(核磁気共鳴)を測定する方法に関する。ここに、複数の分子種とは、構造の異なる複数の化合物のみならず、同一構造の化合物について存在する複数の結晶形または非晶質を指す。一般に固体は、溶液と比較して、分子・原子運動が遅いため、固体NMRでは、ケミカルシフト異方性や双極子相互作用などが強調された広幅なスペクトルが得られる傾向にあり、分子種に選択的な測定を行うことが難しい。
【0016】
本発明においては、複数の分子種を含む固体試料について測定を行う。本発明における固体試料とは、NMRを測定できる固体であれば特に制限はなく、広範な範囲の固体試料に本発明を適用可能である。本発明の固体試料は、複数の分子種を含んでおり、含まれる分子種の数は2以上であれば特に制限はない。本発明によれば、例えば、3以上の分子種、5以上の分子種、あるいは、10以上の分子種を含むような固体試料に利用することも可能である。複数の分子種を含む固体試料としては、例えば、製剤、または複数の結晶形あるいは結晶と非晶質の混合物を含む原薬、などを好適に挙げることができる。また、本発明に係る固体試料は、それに含まれる分子がすでに同定されていても、未同定であっても構わない。
【0017】
本発明においては、1Hの回転座標系の縦緩和時間(HT1ρ)を利用して、分子種に選択的な測定を行う。一般に、溶液NMRと比較して固体NMRでは、分子種ごと、すなわち、化合物やその固体状態(結晶多形、アモルファスなど)ごとに緩和時間が大きく異なるため、本発明では、特に各成分のHT1ρの違いを利用することにより分子種選択的な分析を行う。
【0018】
本発明においては、試料中の1Hに対して90°パルスを印加して磁化をXY平面(Z軸:静磁場方向)に倒す。
本発明では、試料中の1Hに対して90°パルスを照射した後、スピンロッキング(Spin locking)を行う。すなわち、90°パルスによってXY平面に倒された磁化に対して、倒れた方向からラジオ波を照射して、位相がバラバラにならないように保持して磁化をロックする(スピンロッキング)。これにより、スピンロッキングした状態で、1Hの回転座標系の縦緩和(HT1ρ)、つまり、1Hの回転座標系の縦緩和による磁化の減衰だけを生じさせることができ、回転座標系の縦緩和時間1HT1ρの短い信号を優先的に減衰させて消去することができる。したがって、本発明では、スピンロッキングのパルス長(スピンロッキング時間)を長く設定することにより、1HT1ρの短い信号を先に減衰させることができるため、比較的1HT1ρの長い分子種のシグナルを選択的に取得することができる。
【0019】
本発明の一つの態様において、固体試料に含まれる複数の分子種のうち、測定したくない分子種に係るHT1ρの短い成分のシグナルが、減衰して消滅するのに十分な長さでスピンロッキングを行うことによって、1Hの回転座標系の縦緩和時間HT1ρが比較的長い分子種のシグナルを選択的に取得することができる。スピンロッキングの時間としては、例えば、通常の条件では10〜50m秒が好ましい。
【0020】
本発明の一つの態様において、スピンロッキングのパワーを下げることによって長時間のスピンロッキングに伴うラジオ波の照射が可能となる。スピンロッキングを長時間行うことは、装置(パワーアンプなど)の故障を誘発するため、スピンロッキングをかける時間に制約がある場合があり、その場合、スピンロッキングの時間を短縮しつつ所望の分子種のシグナルを取得する必要が生じる。そこで、スピンロッキングで照射するラジオ波のパワーを下げることにより、長時間のスピンロッキングに伴うラジオ波の照射ができれば、例えば50m秒を超える長時間のラジオ波の照射も可能となるため、この態様によれば、通常の条件において、スピンロッキング時間を長くしないと消去できないようなHT1ρが長い分子種に起因するシグナルを消去することができる。具体的には、スピンロッキングのラジオ波のパワーを装置固有の通常の強さ(40〜90kHz)の3分の2から2分の1程度に抑えることにより装置への負荷を軽減する。この場合には、スピンロッキングの時間としては50〜200m秒の範囲も好適に用いられる。
【0021】
本発明の一つの態様において、CP−MAS法によりNMRを測定することが好ましい。CP−MAS法は、交差分極法(CP)、マジック角度回転法(MAS)、ハイパワーデカップリング(HD)を組み合わせたもので、1H核など緩和時間が比較的短い核の磁化を13C核などの緩和時間が比較的長い核に移動させることによって測定時間を短縮するとともに、大きな磁化を持つ1H核などの磁化を小さな磁化を持つ13C核などに移しているため、13C核などを観測核とした場合においてシグナル強度を大きくすることができる。すなわち、1Hの磁化を交差分極法によって13Cに移すことにより、13Cの高分解能の下で1HのT1やT1ρを評価できるため好ましい。交差分極法(CP:Cross Polarization)は、1H核と13C核に同時に共鳴周波数のラジオ波をHartmann-Hahn条件を満たすように照射後、90°位相シフトしたラジオ波を照射してスピンロッキングをかけ、1H核から13C核に磁化を移動させ、その後、FIDを取得する。この場合、緩和過程では1Hからエネルギーが散逸する。マジック角度回転法(MAS:Magic Angle Spinning)は、試料をマジック角で高速回転させてケミカルシフトの異方性を消去し、ピークをシャープにする手法である。(数kHz以上が必要)。また、ハイパワーデカップリング(HD:High Power Decoupling)は、1Hに強いRFパルスを照射して1H−13Cの双極子相互作用を除去する手法であり、例えば、CW(Continuous-Wave)デカップリングやTPPM(Two Pulse Phase Modulation)デカップリングなどが用いられる。交差分極における接触時間やラジオ波の周波数は、1HのT1ρや測定条件に応じて適宜調節することができる。通常、交差分極の時間としては1〜2m秒、1Hのラジオ波の磁場強度が40〜90kHz、13Cのラジオ波の磁場強度が40〜90kHzを使用する。
【0022】
本発明の別の態様において、複数の分子種を含む固体試料を測定する場合、ある分子種のシグナルが消滅する点(いわゆるnull点:τnull)を利用して分子種選択的な測定を行うことができる。すなわち、この態様では、第1段階として、まず(1)1Hに対して0より大きく180°未満の任意の角度のパルスを印加し、通常のCP−MAS法によりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、スピンロッキングを行う。スピンロッキングを行っている間には、1Hの回転座標系の縦緩和(HT1ρ)、つまり、1Hの回転座標系の縦緩和HT1ρによる磁化の減衰だけを生じさせることができる。さらに、(1)から(2)を差し引くことにより、ある分子種のシグナルが消滅するタイミング(τnull)が発生する。この手法によれば、複数の分子種を含む固体試料について、測定したくない分子種に起因するシグナルを消去することができ、その結果、分子種選択的な測定が可能になる。ここで、第一段階のパルス角としては、0より大きく180°未満の任意の角度を用いることができる。10〜170°の任意の角度が好ましい。混合物中の任意の分子種のシグナルを容易に消去可能という観点から、例えば、10〜80°または100〜170°パルスが好ましく、10〜70°または110〜170°パルスがより好ましく、15〜60°または120〜165°がさらに好ましい。一方、第一段階のパルス角として、90°付近のものを用いた場合、この方法により選択的に消去できる混合物中の分子種は、1Hの回転座標系の縦緩和(HT1ρ)の非常に長いものに限定されるが、シグナルの強度を上げることができる点で好適である。
【0023】
スピンロッキングの時間は、複数の分子種に関するそれぞれの1Hの回転座標系の縦緩和(HT1ρ)をスピンロッキングの時間を種々変えながら測定を行うことにより、最適な値を求める。
【0024】
さらに、本発明の一つの態様において、繰り返し時間(tau)を短くすることによって、1H核の縦緩和時間(HT1)の長いシグナルを除去することができるため、その結果、固体試料に含まれる複数の分子種のうち、ある分子種に起因するシグナルを選択的に取得することができる。例えば、HT1が60秒程度のHT1の長い化合物の場合、繰り返し時間を5秒以下に設定することによって、この化合物のシグナルを消去することが可能となる。
【0025】
本発明は、CP−MAS法に代えて、1H核の磁化を13C核、15N核、29Si核または31Pに移動させること、すなわち、観測核として13C核のみならず、15N核、29Si核、または31Pを用いる手法に適用可能である。
【0026】
また、1つの態様において、本発明は、固体NMRスペクトルを測定するためのパルスプログラムである。すなわち、本発明のプログラムは、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させる。
【0027】
一般に、固体NMRを測定する装置(ハードウェア)には、静磁場を発生させる超伝導磁石、電磁波パルスの照射とシグナルの測定を行うプローブコイル、計算機(コンピューター)などから構成される。本発明で用いるNMR装置は、上記の他にも、試料に磁場勾配をかけるためのグラジエント装置、パルスを発生させるトランスミッター、アンプ、レシーバーなどを備えていてもよい。本発明のパルスプログラムは、固体NMR測定装置に上記のシークエンスを実行させることができ、計算機を介して、パルスの照射、磁化の観測、観測データの処理を行う。
【0028】
以下の実施例により本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。なお、本明細書において特に記載しない限り、数値範囲はその端点を含み、各成分の量は重量基準で記載される。
【実施例1】
【0029】
1.試料
測定試料として、市販の固形製剤である「サリドン(登録商標)エース」(第一三共ヘルスケア社製、鎮痛・解熱薬)を用いた。1錠あたり、エテンザミド(ethenzamide)250mg、アセトアミノフェン(acetaminophen)110mg、ブロモバレリル尿素(bromovalerylurea)100mg、無水カフェイン25mgを含有し、添加物として、クロスCMC(カルボキシメチルセルロース)Na、トウモロコシデンプン、ヒドロキシプロピルセルロース、タルク、ステアリン酸Mgなどを含有する。
【0030】
【化1】

【0031】
1錠をメノウ乳鉢で磨り潰し粉末化した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρのもっとも長いエテンザミドのみを選択的に検出する条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5分21秒
・パルスシークエンス:図1に記載
・試料回転速度:14 kHz
・繰り返し時間:20 秒
・積算回数:16回
・スピンロッキング時間:50 m秒
3.測定結果
図3に、通常のCP−MAS法によるサリドンエースのNMRスペクトルと、3種類の主成分のうち、1H核の回転座標系の縦緩和時間HT1ρのもっとも長いエテンザミドのNMRスペクトルを選択的に測定した結果を示す。
【0032】
図3(a)では、試料中のエテンザミド、アセトアミノフェン、ブロモバレリル尿素、賦形剤に起因するピークを確認することができた。また、スピンロッキングを行い、緩和時間が短いものを長時間緩和により除去することによって、3種類の主成分のうち、HT1ρのもっとも長いエテンザミドのみを検出することができた(図3(b)参照)。
【実施例2】
【0033】
1.試料
測定試料として、市販の固形製剤である「バファリン(登録商標)A」(ライオン社製、鎮痛・解熱薬)を用いた。1錠あたり、アセチルサリチル酸(acetylsalicylic acid)330mg、合成ヒドロタルサイトを含有し、添加物としてトウモロコシデンプン、ヒドロキシプロピルメチルセルロース、酸化チタン、マクロゴール、青色1号などを含有する。
【0034】
【化2】
【0035】
1錠をメノウ乳鉢で磨り潰し粉末化した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρの違いを利用し、賦形剤成分を選択的に消去するための条件を用いて測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:36時間59分38秒
・パルスシークエンス:図2に記載
・試料回転速度:15 kHz
・繰り返し時間:60 秒
・積算回数:1108回
・30°パルス
・スピンロッキング時間:3.44 m秒
3.測定結果
図4に、通常のCP−MAS法によるバファリンAのNMRスペクトルと、賦形剤成分を選択的に消去し、主成分であるアセチルサリチル酸を選択的に測定した結果を示す。
【0036】
図4(a)では、試料中のアセチルサリチル酸、賦形剤に起因するピークを確認することができた。また、(1)30°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、3.44m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、アセチルサリチル酸のみを検出することができた(図4(b)参照)。
【実施例3】
【0037】
1.試料
測定試料として、ラクトース(乳糖)1水和物とβ-サイクロデキストリンを重量比1:1になるように測り取り、メノウ乳鉢で粉砕・混合した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
【0038】
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで2種類の化合物のうち、HT1ρの違いを利用し、それぞれの成分を選択的に消去するための条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:6時間9分52秒
・パルスシークエンス:図2に記載
・試料回転速度:14 kHz
・繰り返し時間:84 秒
・積算回数:132回
・第一段階のパルス角:15°
・スピンロッキング時間:7.84 m秒
3.測定結果
図5に、通常のCP−MAS法によるラクトース1水和物とβ-サイクロデキストリンの重量比1:1混合物のNMRスペクトルと、β-サイクロデキストリンを選択的に消去し、ラクトース1水和物を選択的に測定した結果を示す。
【0039】
図5(a)では、試料中のラクトース1水和物及びβ-サイクロデキストリンに起因するシグナルを確認することができた。また、(1)15°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、7.84m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、ラクトース1水和物のみを検出することができた(図5(b)参照)。
【実施例4】
【0040】
1.試料
測定試料として、ラクトース1水和物とβ-サイクロデキストリンを重量比1:1になるように測り取り、メノウ乳鉢で粉砕・混合した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
【0041】
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで2種類の化合物のうち、HT1ρの違いを利用し、それぞれの成分を選択的に消去するための条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5時間42分40秒
・パルスシークエンス:図2に記載
・試料回転速度:14 kHz
・繰り返し時間:16 秒
・積算回数:640回
・第一段階のパルス角:60°
・スピンロッキング時間:5.00 m秒
3.測定結果
図6に、通常のCP−MAS法によるラクトース1水和物とβ-サイクロデキストリンの重量比1:1混合物のNMRスペクトルと、ラクトース1水和物を選択的に消去し、β-サイクロデキストリンを選択的に測定した結果を示す。
【0042】
図6(a)では、試料中のラクトース1水和物及びβ-サイクロデキストリンに起因するシグナルを確認することができた。また、(1)60°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、5.00m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、β−サイクロデキストリンのみを検出することができた(図6(b)参照)。
【実施例5】
【0043】
1.試料
結晶化度の測定には、ニフェジピン結晶(和光純薬)、及び、非晶質ニフェジピン−ポリビニルピロリドン(PVP)固体分散体を用いた。
【0044】
検量線作成用サンプルとして、一定量のアセトアミノフェン(内部標準物質)にニフェジピン結晶を加えたものを乳鉢にて混合粉砕し、ニフェジピンの結晶成分の重量比が内部標準物質に対し、10%、50%、100%、150%及び200%となるように調製したものを準備した。
【0045】
また別途、ニフェジピン結晶の割合が50%となるように、ニフェジピン結晶と非晶質ニフェジピン−ポリビニルピロリドン固体分散体を混合したものに、一定量のアセトアミノフェン(内部標準物質)を加えたものを乳鉢にて混合・粉砕し、ニフェジピン結晶と非晶質ニフェジピンを50:50の割合で含有する試料を調製した。
【0046】
得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρの違いを利用し、非晶質成分を選択的に消去するための条件を用いて測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5時間1分12秒
・パルスシークエンス:図1に記載
・試料回転速度:14 kHz
・繰り返し時間:50 秒
・積算回数:360回
・スピンロッキング時間:150 m秒
・スピンロッキングパワー:34.7 kHz
3.測定結果
結晶化度測定用の検量線は、検量線作成用サンプル中のアセトアミノフェン(内部標準物質)とニフェジピン結晶の質量比に対して、アセトアミノフェン由来のシグナルとニフェジピン由来のシグナルの強度比をプロットして作成した。具体的には,アセトアミノフェンとニフェジピン結晶の質量比(ニフェジピン結晶/アセトアミノフェン:0.1、0.5、1.0、1.5、2.0)に対して、アセトアミノフェン由来の23.2ppmのシグナルに対するニフェジピン由来の35.2ppmのシグナルの強度比をプロットした。その相関係数は、R2=0.9974と非常に良好な相関を示した。
【0047】
この検量線を用いて、ニフェジピン結晶と非晶質ニフェジピンを50:50の割合で含有する2種類の試料(試料a:17.6%のPVPを含む、試料b:30%のPVPを含む)について、ニフェジピン結晶の比率を測定したところ、それぞれ、47.4%及び45.8%と求められた。すなわち、結晶状態の異なる化合物を複数種類含有する試料について、特定の結晶状態を有する化合物の割合を本発明によって定量することができた。
【産業上の利用可能性】
【0048】
以上、詳述したように、本発明によって、分子種の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することにより、固体試料の成分比を変えたNMRスペクトルを得ることができる。特に本発明によれば、複数の化合物または結晶形の異なる同一化合物もしくは、結晶と非晶質の混合物を含む固体試料について、特定の分子種に起因するシグナルを消去または減弱することが可能である。本発明は、一次元NMRを用いることから、定性分析のみならず定量分析にも適用可能である。また、本発明は、比較的短時間でデータを取得でき、一般的な装置およびデータ処理法で実行することができる。
【技術分野】
【0001】
本発明は、固体NMRの測定方法に関する。
【背景技術】
【0002】
複数の化合物の混合物について、成分ごとのNMRスペクトルを選択的に取得する手法が知られている。
溶液NMRについては、例えば、DOSY法(Diffusion Ordered Spectroscopy Method)が知られている(E. O. Stejskal, J. E. Tanner, Journal of Chemical Physics, vol. 42, No.1, p.288-292 (1965))。DOSY法では、各成分が固有の自己拡散係数(分子の拡散的な運動性)を有することを利用し、成分ごとの自己拡散係数の違いを利用して成分ごとのNMRスペクトルを得る。通常、混合物のNMRスペクトルは、各成分のスペクトルの和として観測されるが、その混合物のNMRスペクトルを分子の拡散係数を利用して逆ラプラス変換(ILT:Inverse Laplace Transform)により分離すると、分子種ごとのNMRスペクトルを得ることが可能になる。通常のDOSY法は、自己拡散係数に対して化学シフトを2次元に展開する手法であるため、定量分析は困難であり、基本的に定性分析のみに適用される。また、DOSY法における測定時間の短縮を目的とした1次元のDOSY法についても報告されているが、この手法は、分子の拡散係数の測定時間の短縮化を目的としたものであるため、複数の化合物の混合物について、各成分のスペクトルを得るためものではない(N. M. Loening, J. Keeler, G. A. Morris, Journal of Magnetic Resonance, vol. 153, p.103-112 (2001))。
【0003】
一方、固体NMRについては、例えば、DECRA法(Direct Exponential Curve Resolution Algorithm)(非特許文献1)やROSY法(Relaxation Ordered Spectroscopy)(非特許文献2)が知られている。
【0004】
DECRA法は基本的に1次元NMRに適用される手法であり、分子種ごとの1Hの縦緩和時間(HT1)の違いを利用して、線形解析によって分子種ごとのNMRスペクトルを得る。DECRA法は、定性分析だけでなく、定量分析にも適用可能であるが、データ取得に莫大な時間がかかる。
【0005】
ROSY法は、DECRA法と同様に、分子種ごとの1Hの縦緩和時間(HT1)の違いを利用して、逆ラプラス変換によりシグナルを分離し、分子種ごとのNMRスペクトルを得る手法である。この手法は、1Hの縦緩和時間(HT1)に対して化学シフトを2次元に展開する手法であるため、定量分析に適用することは難しく、基本的に定性分析に用いられる。特許文献1には、混合物の固体試料についてのROSY法によるNMR測定法が記載されている。特許文献1の方法では、均一な縦磁化緩和時間を有している核に縦磁化緩和を起こさせるパルスを照射する第1の工程、所定時間tを置いて前記均一な縦磁化緩和時間を有している核のスピン拡散を切断して高分解能NMRスペクトルを測定する第2の工程、tを変えて第1の工程と第2の工程を繰り返して複数の高分解能NMRスペクトルを取得する第3の工程、第3の工程により得られた複数の高分解能NMRスペクトルを縦磁化緩和時間に依存して回復するNMR信号強度の回復速度の違いに基づいて逆ラプラス変換法により縦磁化緩和時間の値ごとに分類する第4の工程を備える。
【0006】
また、非特許文献3に記載のNMR測定方法は、複数の化合物を含む固体試料について、化合物ごとの緩和時間の違いを利用して選択的にシグナルを検出する。具体的には、混合物における1Hの縦緩和時間(HT1)について、反復回復法を利用して、消去したいシグナルのシグナル強度が励起状態から基底状態に回復する際にゼロとなるタイミングで測定を行う手法が記載されている。また、非特許文献3には、混合物における1Hの回転座標系の縦緩和時間(HT1ρ)について、スピンエコーを用いることにより、HT1ρの短い化合物のシグナル強度がゼロとなるタイミングで測定することによって、混合物中のHT1ρの長い化合物を選択的に測定する手法が記載されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開2010−71839号公報
【非特許文献】
【0008】
【非特許文献1】B. Antalek, W. Windig, Journal of American Chemical Society, vol. 118, p.10331-10332 (1996)
【非特許文献2】Y. Nishiyama, M. H. Frey, S. Mukasa, H. Utsumi. Journal of Magnetic Resonance, vol. 202, p.135-139 (2010)
【非特許文献3】N. Zumbulyadis, Journal of Magnetic Resonance, 53, p.486-494 (1983)
【発明の概要】
【発明が解決しようとする課題】
【0009】
分子種ごとのNMRシグナルを選択的に取得するための固体NMRに関する従来の手法は、測定に莫大な時間を要したり、定量分析が困難であった。このような状況に鑑み、本発明の課題は、実用性に優れた固体NMRの測定法を提供すること、特に、混合物中の特定の分子種のシグナルを選択的に消去または減弱できる固体NMRの測定法を提供することである。
【課題を解決するための手段】
【0010】
本発明者は、固体試料のNMR測定について鋭意検討したところ、スピンロッキングを用いて1Hの回転座標系の縦緩和時間(HT1ρ)を利用することにより、固体試料のNMRスペクトルを分子種選択的に得られることを見いだし、本発明を完成させるに至った。
【0011】
すなわち、一つの態様において本発明は、複数の分子種を含む固体試料に90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、その後、各成分の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することを含む、固体NMRの測定方法である。
【0012】
本発明は以下に限定されるものではないが、以下の発明を包含する。
(1) 1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を他の核種へ移動させた後に当該磁化を観測するNMRスペクトルの測定方法であって、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱する手段を含む、固体NMRスペクトルの測定方法。
(2) 1H核の磁化を交差分極法により13C核へ移動させ、当該13C核の磁化を観測するものである、(1)に記載の方法。
(3) 1H核の磁化を13C核へ移動させ、当該13C核の磁化を観測する手法が、CP−MAS法によるものである、(2)に記載の方法。
(4) 測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、当該分子種に係る1Hの回転座標系の縦緩和時間HT1ρよりも長い時間をもってスピンロッキングを行うことにより、1Hの回転座標系の縦緩和時間HT1ρが相対的に長い分子種の磁化を観測するものである、(3)に記載の方法。
(5) 測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測する工程、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測する工程、3)1)による測定値から2)による測定値を差し引く工程、を含む、(3)に記載の方法。
(6) 1)において1H核に対して印加されるパルスが15〜60°または120〜165°である、(5)に記載の方法。
(7) 1)において1H核に対して印加されるパルスが90°である、(5)に記載の方法。
(8) 固体NMRスペクトルを測定するためのパルスプログラムであって、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させるための上記パルスプログラム。
【発明の効果】
【0013】
本発明によって、分子種の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することにより、固体試料の成分比を変えたNMRスペクトルを得ることができる。特に本発明によれば、複数の化合物または結晶形の異なる同一化合物もしくは、結晶と非晶質の混合物を含む固体試料について、特定の分子種に起因するシグナルを消去または減弱することが可能である。本発明は、一次元NMRを用いることから、定性分析のみならず定量分析にも適用可能である。また、本発明は、比較的短時間でデータを取得でき、一般的な装置およびデータ処理法で実行することができる。
【図面の簡単な説明】
【0014】
【図1】図1は、スピンロッキングの時間を長くとることにより、HT1ρの長い化合物のみを選択的に検出するためのパルスシークエンスである。
【図2】図2は、混合物中の特定成分のシグナルを選択的に消去または減弱するためのパルスシークエンスである。
【図3】図3は、本発明の実施例1で得られたNMRチャートである。図中、○はエテンザミド、△はブロモバレリル尿素、×はアセトアミノフェンに由来するシグナルである。図3(a)は、スピンロッキング時間を0秒とした場合(=通常のCP−MAS法による測定)、図3(b)はスピンロッキング時間を50m秒とした場合であり、図3(c)はエテンザミドの標品を通常のCP−MAS法により測定した結果である。
【図4】図4は、本発明の実施例2で得られたNMRチャートである。図中、○はアセチルサリチル酸に由来するシグナルである。図4(a)は、通常のCP−MAS法による測定、図4(b)は、30°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を3.44m秒としたシグナルを差し引いた場合であり、図4(c)はアセチルサリチル酸の標品を通常のCP−MAS法により測定した結果である。
【図5】図5は、本発明の実施例3で得られたNMRチャートである。図5(a)は、ラクトース(乳糖)1水和物とβ−サイクロデキストリンの重量比1:1混合物をCP−MAS法で測定した結果、図5(b)は、同一サンプルに対し、15°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を7.84m秒としたシグナルを差し引いた場合であり、図5(c)はラクトース1水和物の標品を通常のCP−MAS法により測定した結果である。
【図6】図6は、本発明の実施例4で得られたNMRチャートである。図6(a)は、ラクトース1水和物とβ−サイクロデキストリンの重量比1:1混合物を通常のCP−MAS法で測定した結果、図6(b)は、同一サンプルに対し、60°パルスを印加した通常のCP−MAS法による測定スペクトルから、スピンロッキング時間を5.00m秒としたシグナルを差し引いた場合であり、図6(c)はβ−サイクロデキストリンの標品を通常のCP−MAS法により測定した結果である。
【発明を実施するための形態】
【0015】
本発明は、複数の分子種を含む固体試料についてNMR(核磁気共鳴)を測定する方法に関する。ここに、複数の分子種とは、構造の異なる複数の化合物のみならず、同一構造の化合物について存在する複数の結晶形または非晶質を指す。一般に固体は、溶液と比較して、分子・原子運動が遅いため、固体NMRでは、ケミカルシフト異方性や双極子相互作用などが強調された広幅なスペクトルが得られる傾向にあり、分子種に選択的な測定を行うことが難しい。
【0016】
本発明においては、複数の分子種を含む固体試料について測定を行う。本発明における固体試料とは、NMRを測定できる固体であれば特に制限はなく、広範な範囲の固体試料に本発明を適用可能である。本発明の固体試料は、複数の分子種を含んでおり、含まれる分子種の数は2以上であれば特に制限はない。本発明によれば、例えば、3以上の分子種、5以上の分子種、あるいは、10以上の分子種を含むような固体試料に利用することも可能である。複数の分子種を含む固体試料としては、例えば、製剤、または複数の結晶形あるいは結晶と非晶質の混合物を含む原薬、などを好適に挙げることができる。また、本発明に係る固体試料は、それに含まれる分子がすでに同定されていても、未同定であっても構わない。
【0017】
本発明においては、1Hの回転座標系の縦緩和時間(HT1ρ)を利用して、分子種に選択的な測定を行う。一般に、溶液NMRと比較して固体NMRでは、分子種ごと、すなわち、化合物やその固体状態(結晶多形、アモルファスなど)ごとに緩和時間が大きく異なるため、本発明では、特に各成分のHT1ρの違いを利用することにより分子種選択的な分析を行う。
【0018】
本発明においては、試料中の1Hに対して90°パルスを印加して磁化をXY平面(Z軸:静磁場方向)に倒す。
本発明では、試料中の1Hに対して90°パルスを照射した後、スピンロッキング(Spin locking)を行う。すなわち、90°パルスによってXY平面に倒された磁化に対して、倒れた方向からラジオ波を照射して、位相がバラバラにならないように保持して磁化をロックする(スピンロッキング)。これにより、スピンロッキングした状態で、1Hの回転座標系の縦緩和(HT1ρ)、つまり、1Hの回転座標系の縦緩和による磁化の減衰だけを生じさせることができ、回転座標系の縦緩和時間1HT1ρの短い信号を優先的に減衰させて消去することができる。したがって、本発明では、スピンロッキングのパルス長(スピンロッキング時間)を長く設定することにより、1HT1ρの短い信号を先に減衰させることができるため、比較的1HT1ρの長い分子種のシグナルを選択的に取得することができる。
【0019】
本発明の一つの態様において、固体試料に含まれる複数の分子種のうち、測定したくない分子種に係るHT1ρの短い成分のシグナルが、減衰して消滅するのに十分な長さでスピンロッキングを行うことによって、1Hの回転座標系の縦緩和時間HT1ρが比較的長い分子種のシグナルを選択的に取得することができる。スピンロッキングの時間としては、例えば、通常の条件では10〜50m秒が好ましい。
【0020】
本発明の一つの態様において、スピンロッキングのパワーを下げることによって長時間のスピンロッキングに伴うラジオ波の照射が可能となる。スピンロッキングを長時間行うことは、装置(パワーアンプなど)の故障を誘発するため、スピンロッキングをかける時間に制約がある場合があり、その場合、スピンロッキングの時間を短縮しつつ所望の分子種のシグナルを取得する必要が生じる。そこで、スピンロッキングで照射するラジオ波のパワーを下げることにより、長時間のスピンロッキングに伴うラジオ波の照射ができれば、例えば50m秒を超える長時間のラジオ波の照射も可能となるため、この態様によれば、通常の条件において、スピンロッキング時間を長くしないと消去できないようなHT1ρが長い分子種に起因するシグナルを消去することができる。具体的には、スピンロッキングのラジオ波のパワーを装置固有の通常の強さ(40〜90kHz)の3分の2から2分の1程度に抑えることにより装置への負荷を軽減する。この場合には、スピンロッキングの時間としては50〜200m秒の範囲も好適に用いられる。
【0021】
本発明の一つの態様において、CP−MAS法によりNMRを測定することが好ましい。CP−MAS法は、交差分極法(CP)、マジック角度回転法(MAS)、ハイパワーデカップリング(HD)を組み合わせたもので、1H核など緩和時間が比較的短い核の磁化を13C核などの緩和時間が比較的長い核に移動させることによって測定時間を短縮するとともに、大きな磁化を持つ1H核などの磁化を小さな磁化を持つ13C核などに移しているため、13C核などを観測核とした場合においてシグナル強度を大きくすることができる。すなわち、1Hの磁化を交差分極法によって13Cに移すことにより、13Cの高分解能の下で1HのT1やT1ρを評価できるため好ましい。交差分極法(CP:Cross Polarization)は、1H核と13C核に同時に共鳴周波数のラジオ波をHartmann-Hahn条件を満たすように照射後、90°位相シフトしたラジオ波を照射してスピンロッキングをかけ、1H核から13C核に磁化を移動させ、その後、FIDを取得する。この場合、緩和過程では1Hからエネルギーが散逸する。マジック角度回転法(MAS:Magic Angle Spinning)は、試料をマジック角で高速回転させてケミカルシフトの異方性を消去し、ピークをシャープにする手法である。(数kHz以上が必要)。また、ハイパワーデカップリング(HD:High Power Decoupling)は、1Hに強いRFパルスを照射して1H−13Cの双極子相互作用を除去する手法であり、例えば、CW(Continuous-Wave)デカップリングやTPPM(Two Pulse Phase Modulation)デカップリングなどが用いられる。交差分極における接触時間やラジオ波の周波数は、1HのT1ρや測定条件に応じて適宜調節することができる。通常、交差分極の時間としては1〜2m秒、1Hのラジオ波の磁場強度が40〜90kHz、13Cのラジオ波の磁場強度が40〜90kHzを使用する。
【0022】
本発明の別の態様において、複数の分子種を含む固体試料を測定する場合、ある分子種のシグナルが消滅する点(いわゆるnull点:τnull)を利用して分子種選択的な測定を行うことができる。すなわち、この態様では、第1段階として、まず(1)1Hに対して0より大きく180°未満の任意の角度のパルスを印加し、通常のCP−MAS法によりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、スピンロッキングを行う。スピンロッキングを行っている間には、1Hの回転座標系の縦緩和(HT1ρ)、つまり、1Hの回転座標系の縦緩和HT1ρによる磁化の減衰だけを生じさせることができる。さらに、(1)から(2)を差し引くことにより、ある分子種のシグナルが消滅するタイミング(τnull)が発生する。この手法によれば、複数の分子種を含む固体試料について、測定したくない分子種に起因するシグナルを消去することができ、その結果、分子種選択的な測定が可能になる。ここで、第一段階のパルス角としては、0より大きく180°未満の任意の角度を用いることができる。10〜170°の任意の角度が好ましい。混合物中の任意の分子種のシグナルを容易に消去可能という観点から、例えば、10〜80°または100〜170°パルスが好ましく、10〜70°または110〜170°パルスがより好ましく、15〜60°または120〜165°がさらに好ましい。一方、第一段階のパルス角として、90°付近のものを用いた場合、この方法により選択的に消去できる混合物中の分子種は、1Hの回転座標系の縦緩和(HT1ρ)の非常に長いものに限定されるが、シグナルの強度を上げることができる点で好適である。
【0023】
スピンロッキングの時間は、複数の分子種に関するそれぞれの1Hの回転座標系の縦緩和(HT1ρ)をスピンロッキングの時間を種々変えながら測定を行うことにより、最適な値を求める。
【0024】
さらに、本発明の一つの態様において、繰り返し時間(tau)を短くすることによって、1H核の縦緩和時間(HT1)の長いシグナルを除去することができるため、その結果、固体試料に含まれる複数の分子種のうち、ある分子種に起因するシグナルを選択的に取得することができる。例えば、HT1が60秒程度のHT1の長い化合物の場合、繰り返し時間を5秒以下に設定することによって、この化合物のシグナルを消去することが可能となる。
【0025】
本発明は、CP−MAS法に代えて、1H核の磁化を13C核、15N核、29Si核または31Pに移動させること、すなわち、観測核として13C核のみならず、15N核、29Si核、または31Pを用いる手法に適用可能である。
【0026】
また、1つの態様において、本発明は、固体NMRスペクトルを測定するためのパルスプログラムである。すなわち、本発明のプログラムは、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させる。
【0027】
一般に、固体NMRを測定する装置(ハードウェア)には、静磁場を発生させる超伝導磁石、電磁波パルスの照射とシグナルの測定を行うプローブコイル、計算機(コンピューター)などから構成される。本発明で用いるNMR装置は、上記の他にも、試料に磁場勾配をかけるためのグラジエント装置、パルスを発生させるトランスミッター、アンプ、レシーバーなどを備えていてもよい。本発明のパルスプログラムは、固体NMR測定装置に上記のシークエンスを実行させることができ、計算機を介して、パルスの照射、磁化の観測、観測データの処理を行う。
【0028】
以下の実施例により本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。なお、本明細書において特に記載しない限り、数値範囲はその端点を含み、各成分の量は重量基準で記載される。
【実施例1】
【0029】
1.試料
測定試料として、市販の固形製剤である「サリドン(登録商標)エース」(第一三共ヘルスケア社製、鎮痛・解熱薬)を用いた。1錠あたり、エテンザミド(ethenzamide)250mg、アセトアミノフェン(acetaminophen)110mg、ブロモバレリル尿素(bromovalerylurea)100mg、無水カフェイン25mgを含有し、添加物として、クロスCMC(カルボキシメチルセルロース)Na、トウモロコシデンプン、ヒドロキシプロピルセルロース、タルク、ステアリン酸Mgなどを含有する。
【0030】
【化1】
【0031】
1錠をメノウ乳鉢で磨り潰し粉末化した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρのもっとも長いエテンザミドのみを選択的に検出する条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5分21秒
・パルスシークエンス:図1に記載
・試料回転速度:14 kHz
・繰り返し時間:20 秒
・積算回数:16回
・スピンロッキング時間:50 m秒
3.測定結果
図3に、通常のCP−MAS法によるサリドンエースのNMRスペクトルと、3種類の主成分のうち、1H核の回転座標系の縦緩和時間HT1ρのもっとも長いエテンザミドのNMRスペクトルを選択的に測定した結果を示す。
【0032】
図3(a)では、試料中のエテンザミド、アセトアミノフェン、ブロモバレリル尿素、賦形剤に起因するピークを確認することができた。また、スピンロッキングを行い、緩和時間が短いものを長時間緩和により除去することによって、3種類の主成分のうち、HT1ρのもっとも長いエテンザミドのみを検出することができた(図3(b)参照)。
【実施例2】
【0033】
1.試料
測定試料として、市販の固形製剤である「バファリン(登録商標)A」(ライオン社製、鎮痛・解熱薬)を用いた。1錠あたり、アセチルサリチル酸(acetylsalicylic acid)330mg、合成ヒドロタルサイトを含有し、添加物としてトウモロコシデンプン、ヒドロキシプロピルメチルセルロース、酸化チタン、マクロゴール、青色1号などを含有する。
【0034】
【化2】
【0035】
1錠をメノウ乳鉢で磨り潰し粉末化した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρの違いを利用し、賦形剤成分を選択的に消去するための条件を用いて測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:36時間59分38秒
・パルスシークエンス:図2に記載
・試料回転速度:15 kHz
・繰り返し時間:60 秒
・積算回数:1108回
・30°パルス
・スピンロッキング時間:3.44 m秒
3.測定結果
図4に、通常のCP−MAS法によるバファリンAのNMRスペクトルと、賦形剤成分を選択的に消去し、主成分であるアセチルサリチル酸を選択的に測定した結果を示す。
【0036】
図4(a)では、試料中のアセチルサリチル酸、賦形剤に起因するピークを確認することができた。また、(1)30°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、3.44m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、アセチルサリチル酸のみを検出することができた(図4(b)参照)。
【実施例3】
【0037】
1.試料
測定試料として、ラクトース(乳糖)1水和物とβ-サイクロデキストリンを重量比1:1になるように測り取り、メノウ乳鉢で粉砕・混合した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
【0038】
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで2種類の化合物のうち、HT1ρの違いを利用し、それぞれの成分を選択的に消去するための条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:6時間9分52秒
・パルスシークエンス:図2に記載
・試料回転速度:14 kHz
・繰り返し時間:84 秒
・積算回数:132回
・第一段階のパルス角:15°
・スピンロッキング時間:7.84 m秒
3.測定結果
図5に、通常のCP−MAS法によるラクトース1水和物とβ-サイクロデキストリンの重量比1:1混合物のNMRスペクトルと、β-サイクロデキストリンを選択的に消去し、ラクトース1水和物を選択的に測定した結果を示す。
【0039】
図5(a)では、試料中のラクトース1水和物及びβ-サイクロデキストリンに起因するシグナルを確認することができた。また、(1)15°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、7.84m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、ラクトース1水和物のみを検出することができた(図5(b)参照)。
【実施例4】
【0040】
1.試料
測定試料として、ラクトース1水和物とβ-サイクロデキストリンを重量比1:1になるように測り取り、メノウ乳鉢で粉砕・混合した後、得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
【0041】
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで2種類の化合物のうち、HT1ρの違いを利用し、それぞれの成分を選択的に消去するための条件で測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5時間42分40秒
・パルスシークエンス:図2に記載
・試料回転速度:14 kHz
・繰り返し時間:16 秒
・積算回数:640回
・第一段階のパルス角:60°
・スピンロッキング時間:5.00 m秒
3.測定結果
図6に、通常のCP−MAS法によるラクトース1水和物とβ-サイクロデキストリンの重量比1:1混合物のNMRスペクトルと、ラクトース1水和物を選択的に消去し、β-サイクロデキストリンを選択的に測定した結果を示す。
【0042】
図6(a)では、試料中のラクトース1水和物及びβ-サイクロデキストリンに起因するシグナルを確認することができた。また、(1)60°パルスを印加した通常のCP−MASによりシグナルを取り込み、引き続き、第2段階として、(2)試料中の1Hに対し90°パルスをかけた後、5.00m秒のスピンロッキングを行ってシグナルを取り込み、さらに、(1)から(2)を差し引くことにより、β−サイクロデキストリンのみを検出することができた(図6(b)参照)。
【実施例5】
【0043】
1.試料
結晶化度の測定には、ニフェジピン結晶(和光純薬)、及び、非晶質ニフェジピン−ポリビニルピロリドン(PVP)固体分散体を用いた。
【0044】
検量線作成用サンプルとして、一定量のアセトアミノフェン(内部標準物質)にニフェジピン結晶を加えたものを乳鉢にて混合粉砕し、ニフェジピンの結晶成分の重量比が内部標準物質に対し、10%、50%、100%、150%及び200%となるように調製したものを準備した。
【0045】
また別途、ニフェジピン結晶の割合が50%となるように、ニフェジピン結晶と非晶質ニフェジピン−ポリビニルピロリドン固体分散体を混合したものに、一定量のアセトアミノフェン(内部標準物質)を加えたものを乳鉢にて混合・粉砕し、ニフェジピン結晶と非晶質ニフェジピンを50:50の割合で含有する試料を調製した。
【0046】
得られた粉末を固体NMR用φ4mmのサンプルローターに詰めた。
2.NMR測定条件
上記試料を用い、CP−MAS法により固体NMRで3種類の主成分のうち、HT1ρの違いを利用し、非晶質成分を選択的に消去するための条件を用いて測定を行った。測定条件の詳細は以下のとおりである。
・装置:AVANCE 400WB(ブルカーバイオスピン製)
・測定温度:室温
・測定時間:5時間1分12秒
・パルスシークエンス:図1に記載
・試料回転速度:14 kHz
・繰り返し時間:50 秒
・積算回数:360回
・スピンロッキング時間:150 m秒
・スピンロッキングパワー:34.7 kHz
3.測定結果
結晶化度測定用の検量線は、検量線作成用サンプル中のアセトアミノフェン(内部標準物質)とニフェジピン結晶の質量比に対して、アセトアミノフェン由来のシグナルとニフェジピン由来のシグナルの強度比をプロットして作成した。具体的には,アセトアミノフェンとニフェジピン結晶の質量比(ニフェジピン結晶/アセトアミノフェン:0.1、0.5、1.0、1.5、2.0)に対して、アセトアミノフェン由来の23.2ppmのシグナルに対するニフェジピン由来の35.2ppmのシグナルの強度比をプロットした。その相関係数は、R2=0.9974と非常に良好な相関を示した。
【0047】
この検量線を用いて、ニフェジピン結晶と非晶質ニフェジピンを50:50の割合で含有する2種類の試料(試料a:17.6%のPVPを含む、試料b:30%のPVPを含む)について、ニフェジピン結晶の比率を測定したところ、それぞれ、47.4%及び45.8%と求められた。すなわち、結晶状態の異なる化合物を複数種類含有する試料について、特定の結晶状態を有する化合物の割合を本発明によって定量することができた。
【産業上の利用可能性】
【0048】
以上、詳述したように、本発明によって、分子種の1Hの回転座標系の縦緩和時間(HT1ρ)の違いを利用することにより、固体試料の成分比を変えたNMRスペクトルを得ることができる。特に本発明によれば、複数の化合物または結晶形の異なる同一化合物もしくは、結晶と非晶質の混合物を含む固体試料について、特定の分子種に起因するシグナルを消去または減弱することが可能である。本発明は、一次元NMRを用いることから、定性分析のみならず定量分析にも適用可能である。また、本発明は、比較的短時間でデータを取得でき、一般的な装置およびデータ処理法で実行することができる。
【特許請求の範囲】
【請求項1】
1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を他の核種へ移動させた後に当該磁化を観測するNMRスペクトルの測定方法であって、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱する手段を含む、固体NMRスペクトルの測定方法。
【請求項2】
1H核の磁化を交差分極法により13C核へ移動させ、当該13C核の磁化を観測するものである、請求項1に記載の方法。
【請求項3】
1H核の磁化を13C核へ移動させ、当該13C核の磁化を観測する手法が、CP−MAS法によるものである、請求項2に記載の方法。
【請求項4】
測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、当該分子種に係る1Hの回転座標系の縦緩和時間HT1ρよりも長い時間をもってスピンロッキングを行うことにより、1Hの回転座標系の縦緩和時間HT1ρが相対的に長い分子種の磁化を観測するものである、請求項3に記載の方法。
【請求項5】
測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測する工程、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測する工程、3)1)による測定値から2)による測定値を差し引く工程、を含む、請求項3に記載の方法。
【請求項6】
1)において1H核に対して印加されるパルスが15〜60°または120〜165°である、請求項5に記載の方法。
【請求項7】
1)において1H核に対して印加されるパルスが90°である、請求項5に記載の方法。
【請求項8】
固体NMRスペクトルを測定するためのパルスプログラムであって、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させるための上記パルスプログラム。
【請求項1】
1Hに対して90°パルスを試料に印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射してスピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を他の核種へ移動させた後に当該磁化を観測するNMRスペクトルの測定方法であって、複数の固体分子種を含む混合物を試料として使用し、測定を希望しない分子種に起因するシグナルを除去または減弱する手段を含む、固体NMRスペクトルの測定方法。
【請求項2】
1H核の磁化を交差分極法により13C核へ移動させ、当該13C核の磁化を観測するものである、請求項1に記載の方法。
【請求項3】
1H核の磁化を13C核へ移動させ、当該13C核の磁化を観測する手法が、CP−MAS法によるものである、請求項2に記載の方法。
【請求項4】
測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、当該分子種に係る1Hの回転座標系の縦緩和時間HT1ρよりも長い時間をもってスピンロッキングを行うことにより、1Hの回転座標系の縦緩和時間HT1ρが相対的に長い分子種の磁化を観測するものである、請求項3に記載の方法。
【請求項5】
測定を希望しない分子種に起因するシグナルを除去または減弱する手段が、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測する工程、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測する工程、3)1)による測定値から2)による測定値を差し引く工程、を含む、請求項3に記載の方法。
【請求項6】
1)において1H核に対して印加されるパルスが15〜60°または120〜165°である、請求項5に記載の方法。
【請求項7】
1)において1H核に対して印加されるパルスが90°である、請求項5に記載の方法。
【請求項8】
固体NMRスペクトルを測定するためのパルスプログラムであって、1)1H核に対して0より大きく180°未満の任意の角度のパルスを印加して磁化を倒した後、1Hの磁化を直接観測、または1Hの磁化を13C核へ移動させて当該磁化を観測させること、2)1Hに対して90°パルスを印加して磁化を倒した後、磁化の倒れている方向からラジオ波を照射して所定時間スピンロッキングを行い、1Hの磁化を直接観測、または1Hの磁化を交差分極法により13C核へ移動させて当該磁化を観測させること、3)1)による測定値から2)による測定値を差し引く処理を演算装置に行わせること、を含むシークエンスを実行させるための上記パルスプログラム。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−92436(P2013−92436A)
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2011−234407(P2011−234407)
【出願日】平成23年10月25日(2011.10.25)
【出願人】(000006677)アステラス製薬株式会社 (274)
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成23年10月25日(2011.10.25)
【出願人】(000006677)アステラス製薬株式会社 (274)
[ Back to top ]