
圧電体膜の前駆体溶液および圧電素子の製造方法
【課題】1回の成膜工程で得られる圧電体膜の膜厚を厚くすることが可能で、かつ、保存安定性のよい圧電体膜の前駆体溶液の提供。
【解決手段】酢酸及び/又はプロピオン酸からなるカルボン酸と、酢酸鉛と、Zr(OR1)4で表されるジルコニウムアルコキシドと、(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)Ti(OR2)4で表されるチタニウムアルコキシドと、(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)高分子化合物と、を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、前記ジルコニウムアルコキシドの配位子OR1または前記チタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液。
【解決手段】酢酸及び/又はプロピオン酸からなるカルボン酸と、酢酸鉛と、Zr(OR1)4で表されるジルコニウムアルコキシドと、(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)Ti(OR2)4で表されるチタニウムアルコキシドと、(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)高分子化合物と、を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、前記ジルコニウムアルコキシドの配位子OR1または前記チタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、鉛含有圧電体膜の原料である前駆体溶液に関するものであり、さらには、これを用いた圧電素子の製造方法に関するものである。
【背景技術】
【0002】
チタン酸ジルコン酸鉛(PZT)等に代表される結晶を含む圧電体膜としての強誘電体膜は、自発分極、高誘電率、電気光学効果、圧電効果、焦電効果等を有しているため、圧電素子等の広範なデバイスに応用されている。このような圧電体膜の製造方法としては、気相法と液相法の2種類に大別される。気相法としては、CVD(Chemical Vapor Deposition)法、スパッタリング法等が知られており、液相法としては、MOD(Metal Organic Decomposition)法、ゾル−ゲル法等が知られている。特に、液相法は、気相法よりも以下の点で有利である。すなわち、高価な真空装置を必要としない点、元素組成および添加物を制御しやすい点等によって、圧電体膜を低コストで、かつ簡便に成膜できる。
【0003】
ゾル−ゲル法で用いられる前駆体溶液は、金属のアルコキシド等を加水分解、重合させ、コロイド状にしたものを有機溶媒中に分散させた溶液である。MOD法で用いられる前駆体溶液は、加水分解が起きない金属の有機酸塩を有機溶媒中に溶解させた溶液である。また、ゾル−ゲル法あるいはMOD法で用いられる溶液のどちらにも区別されない(金属の有機酸塩、アルコキシドおよび錯体を併用する)前駆体溶液も広く知られている。
【0004】
ゾル−ゲル法によって成膜する場合、所望の金属酸化物の原料となるアルコキシドの混合溶液が用いられる。例えば、特許文献1には酢酸鉛3水和物を3−メトキシブタノールに溶解し、共沸脱水させることで鉛のアルコキシドを合成し、さらにチタンおよびジルコニウムそれぞれのアルコキシドを混合溶解させた金属酸化物前駆体溶液が開示されている。
MOD法によって成膜する場合、所望の金属酸化物の原料となる有機酸塩の混合溶液が用いられる。例えば、特許文献2には、金属酸化物の前駆体溶液として、鉛、チタンおよびジルコニウムそれぞれの有機酸塩の混合溶液が開示されている。
金属の有機酸塩、アルコキシドおよび錯体を混合した圧電体膜の前駆体溶液としては、例えば、特許文献3には、金属酸化物の前駆体溶液として、ストロンチウムおよびビスマスそれぞれの有機酸塩の混合溶液が開示されており、例えば、特許文献4には、金属酸化物の前駆体溶液として、アセチルアセトナート錯体の分散安定性を良好にするために、酢酸を含有した圧電体膜形成用組成物が開示されている。
【0005】
液相法による圧電体膜の製造方法としては、前駆体溶液の調製法(ゾル−ゲル法、MOD法等)に拠らず、アルコールや炭化水素等の溶媒、安定化剤および有機金属化合物を含む前駆体溶液を、シリコン基板等の被対象物上に塗布し、結晶化を目的とした加熱処理を行うことで結晶化した圧電体膜を得る。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平11−79747号公報
【特許文献2】特開平5−58636号公報
【特許文献3】特開平8−111411号公報
【特許文献4】特開2007−145657号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
前駆体溶液を塗布し、結晶化を目的とした加熱処理を行う1回の工程で成膜できる圧電体膜の膜厚が薄いと、圧電体膜に要求される性能に必要な膜厚を得るために、上述の工程を繰り返す必要がある。上述の工程で得られる圧電体膜の膜厚を厚くするには、前駆体溶液中の有機金属化合物を高濃度化する必要がある。したがって、アルコール、炭化水素等の溶媒と、アセチルアセトンやジエタノールアミン等の安定化剤を含む前駆体溶液の場合、前駆体溶液中の有機金属化合物を高濃度化するためには、溶媒の含有量を減らすか、または、安定化剤の含有量を減らす手段が講じられる。
【0008】
しかしながら、以下に述べる理由で、前駆体溶液中の有機金属化合物の濃度を濃くするのが難しい。
安定化剤の含有比率を減少させると、大気中等の水分により加水分解が進行し、前駆体溶液の保存安定性が低下する場合がある。一方、アルコールに代表される溶媒の含有比率を減少させると沈殿が発生し、均一な膜形成が困難になったり、前駆体溶液の粘度増加によって塗布膜にムラが発生したりする場合がある。
【0009】
安定化剤を必要としない前駆体溶液であっても、以下に述べる理由で、前駆体溶液中の有機金属化合物の濃度を濃くするのが難しい。
有機金属化合物として有機酸塩を用いる場合、配位子の鎖長が増すにつれて溶媒への溶解度が増大するが、含有金属濃度は低下してしまう。よって、得られる圧電体膜の膜厚と前駆体溶液との間に妥協点を見出さなければならない。特許文献3では、得られる強誘電体膜の結晶膜厚は100nm程度である。
【0010】
上述したように、従来技術では、上述の工程で得られる圧電体膜の膜厚を厚くすることが難しく、例えば、1μm以上の膜厚を得るためには上述の工程を繰り返す必要があることから、生産性を向上させることが困難であった。したがって、1回の成膜工程で得られる圧電体膜の膜厚を厚くすることが可能で、かつ、保存安定性のよい圧電体膜の前駆体溶液の開発が望まれていた。
【0011】
本発明は、上記従来技術の問題点に鑑み、課題を解決するためになされたものであり、以下様態として実現することが可能である。
【課題を解決するための手段】
【0012】
[態様1]
上記目的を達成する本発明の態様は、酢酸及び/又はプロピオン酸からなるカルボン酸と、酢酸鉛と、Zr(OR1)4で表されるジルコニウムアルコキシド(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)と、Ti(OR2)4で表されるチタニウムアルコキシド(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)と、高分子化合物と、を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、ジルコニウムアルコキシドの配位子OR1またはチタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液にある。
【0013】
本態様に用いる特定のカルボン酸は、金属アルコキシドの安定化効果を有し、さらに、前駆体溶液中の含有量が増加しても前駆体溶液としての保存安定性の低下、および粘度の増加がみられないために、溶媒として用いることができる。
上記のカルボン酸の含有量は、前駆体溶液の調製に用いる原料全量に対して20質量%以上60質量%以下とすることが好ましい。20質量%以上とすることで、大気中の水分による加水分解を防ぐことができる。一方、60質量%以下とすることで上述の工程で得られる膜の膜厚を厚くすることができる。
【0014】
従って、圧電体膜の前駆体溶液に求められる粘度、金属酸化物換算濃度、耐水性、保存安定性などの性能を満足するように、前記範囲でカルボン酸の添加量を決定することができる。
例えば、圧電体膜の前駆体溶液を用いて、スピンコート法によって薄膜を成膜する場合、圧電体膜の前駆体溶液の粘度は5〜15mPa・s、前駆体溶液に含まれる金属原料の酸化物換算濃度は12〜32%とすることができる。なお、金属原料の酸化物換算濃度は、原料組成から算出できるほか、燃焼法やICP分析などの定法によって前駆体溶液から調べることも可能である。
また、本態様では、高分子化合物を前駆体溶液に添加することによって、クラックの発生を防止することが可能となる。
【0015】
[態様2]
また、前記圧電体膜の前駆体溶液は、前記ジルコニウムアルコキシドの配位子OR1および前記チタニウムアルコキシドの配位子OR2が、ともに、分岐鎖状のアルコキシ基とすることができる。
分岐鎖状のアルコキシ基を有する金属アルコキシドを用いることで、カルボン酸エステルの生成量が抑制されるので、圧電体膜の前駆体溶液の経時変化がなく、得られる圧電体膜の膜厚の変動を抑えることが可能となる。なお、カルボン酸がカルボン酸エステルになると、圧電体膜の前駆体溶液に含有される溶媒の揮発性が変化するため、得られる圧電体膜の膜厚は厚くなる場合が多い。
【0016】
[態様3]
また、前記圧電体膜の前駆体溶液は、溶媒として、さらに、水、2級または3級のアルコール類、エーテル類、エステル類、およびケトン類から選ばれる1種または2種以上を含むことができる。
上述したカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応を抑制することが可能となる。
【0017】
[態様4]
また、前記圧電体膜の前駆体溶液は、前記高分子化合物が、ポリエチレングリコールおよびその誘導体ならびにポリプロピレングリコールおよびその誘導体から選ばれる1種または2種以上であり、さらに、前記高分子化合物の重量平均分子量が300〜800であることが好ましい。
高分子化合物を添加した圧電体膜の前駆体溶液であれば、上述の工程において、膜の収縮に起因する応力を緩和することが可能となる。一方、結晶化膜においては高分子化合物が残存しないことが圧電特性の観点からは望ましい。何故なら、高分子化合物が水や二酸化炭素等に分解・脱離せずに、残留炭素等の形態で膜中に存在すると、空隙の発生、あるいは絶縁性の低下等によって、圧電特性に悪影響を及ぼすためである。したがって、高分子化合物には良好な熱分解性が求められる。炭素−炭素結合と比べて、炭素−酸素結合は結合力が弱いために、熱分解性が良好である。ゆえに、ポリエチレングリコールおよびその誘導体、ならびにポリプロピレングリコールおよびその誘導体は、圧電体膜のクラック防止剤として好適に使用することが可能である。
【0018】
圧電体膜の前駆体溶液に含まれる高分子化合物は、熱分解性の観点に加えて、膜のクラック防止の観点から重量平均分子量を規定することが望ましい。
高分子化合物の平均分子量が小さくなると、低分子のように振舞うために圧電体膜の成膜工程における応力緩和効果が十分に得られなくなる。また、高分子化合物の平均分子量が大きくなると、成膜工程における加熱処理時に十分に熱分解しなくなるために、残留炭素等の形態として膜中に存在し、圧電体膜の絶縁性低下といったような特性の劣化につながる。
高分子化合物の重量平均分子量を300〜800にすれば、十分な応力緩和効果と熱分解性を有するため、圧電体膜の特性を劣化させることなく、圧電体膜のクラック防止を行うことが可能となる。なお、重量平均分子量は、分子量標準物質をポリエチレングリコールとしたゲル浸透クロマトグラフィー(GPC)測定により得られた分子量分布から算出される。
【0019】
[態様5]
さらに、本発明の他の態様としては、前記圧電体膜の前駆体溶液に含まれる金属元素の酸化物換算濃度が、15質量%以上27%質量%以下とすることができる。
金属元素の酸化物換算濃度とは、圧電体膜の前駆体溶液に対する、圧電体膜の前駆体溶液に含まれる各金属元素を酸化物とした質量の総和の割合である。したがって、PZT前駆体溶液の場合、PZT前駆体溶液全量に対するPbO、ZrO2およびTiO2の質量の総和の割合となる。なお、金属酸化物換算濃度は、用いる有機金属化合物から算出することができるほか、重量法やICP分析法等を用いて圧電体膜の前駆体溶液から調べることも可能である。
液相法としてスピンコート法を用いて得られる膜の膜厚は、スピンコート回転数と、金属元素の酸化物換算濃度と、に影響を受けることが知られている。金属元素の酸化物換算濃度を本態様の範囲とすることで、上述の工程で得られる膜の膜厚を厚くすることができる。
【0020】
[態様6]
本発明の他の態様としては、前記圧電体膜の前駆体溶液がさらに、少なくとも1種以上の有機金属化合物を含むことができる。
【0021】
[態様7]
本発明の他の態様は、上記圧電体膜の前駆体溶液を用いた圧電素子の製造方法にある。
本態様によれば、従来の圧電体膜の前駆体溶液を用いた場合よりも、1層あたりの圧電体膜を厚くすることが可能となるため、圧電素子の生産性を向上することが可能となる。
【図面の簡単な説明】
【0022】
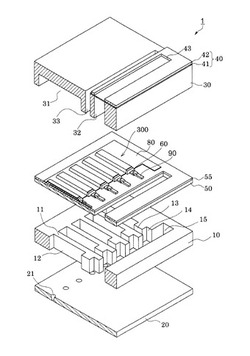
【図1】実施形態に係るインクジェット式記録ヘッドの概略構成を示す分解斜視図。
【図2】(a)はインクジェット式記録ヘッドの平面図、(b)は(a)におけるA−A断面図。
【図3】インクジェット式記録ヘッドの図2(a)におけるB−B要部拡大断面図。
【図4】インクジェット式記録ヘッドの製造方法を示す断面図。
【図5】インクジェット式記録ヘッドの製造方法を示す断面図。
【図6】インクジェット式記録ヘッドの製造方法を示す断面図。
【図7】インクジェット式記録ヘッドの製造方法を示す断面図。
【図8】実施例および実施形態の圧電体膜の前駆体溶液に用いた原料を示す図。
【図9】実施例1〜5および比較例1〜4の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図10】実施例1〜5および比較例1〜4の金属酸化物換算濃度、カルボン酸の割合、PZT膜の膜厚、耐水性および保存安定性をまとめて示す図。
【図11】実施例6〜22および比較例5〜7の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図12】実施例6〜22および比較例5〜7の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図13】実施例23〜32の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図14】実施例23〜32の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図15】実施例33〜43の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図16】実施例33〜43の金属酸化物換算濃度、カルボン酸の割合、膜厚、クラック評価、耐水性および保存安定性をまとめて示す図。
【図17】実施例44〜48の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図18】実施例44〜48および比較例8の金属酸化物換算濃度、カルボン酸の割合、スピンコート回転数、膜厚、耐水性および保存安定性をまとめて示す図。
【図19】実施例49および実施例50の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図20】実施例49および実施例50の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図21】一実施形態に係る記録装置の概略構成を示す図。
【発明を実施するための形態】
【0023】
以下、本発明の実施形態を図面に基づき詳細に説明する。
図1は、本発明の実施形態に係る液体噴射ヘッドの一例であるインクジェット式記録ヘッド1の概略構成を示す分解斜視図であり、図2(a)は、図1の平面図、図2(b)は(a)におけるA−A断面図である。
インクジェット式記録ヘッド1は、流路形成基板10、ノズルプレート20および保護基板30を備えている。
【0024】
図1および図2において、流路形成基板10は、実施形態では結晶面方位が(110)であるシリコン単結晶基板からなり、その一方面には熱酸化によって形成した酸化シリコンからなる、厚さ0.5μm〜2.0μmの弾性膜50が形成されている。流路形成基板10には、隔壁11によって区画され一方側の面が弾性膜50で構成される複数の圧力発生室12がその幅方向に並設されている。
【0025】
流路形成基板10には、圧力発生室12の長手方向一端部側に、隔壁11によって区画され各圧力発生室12に連通するインク供給路13と連通路14とが設けられている。連通路14の外側には、各連通路14と連通する連通部15が設けられている。この連通部15は、後述する保護基板30のリザーバー部32と連通して、各圧力発生室12の共通のインク室(液体室)となるリザーバー100の一部を構成する。
【0026】
ここで、インク供給路13は、圧力発生室12よりも狭い断面積となるように形成されており、連通部15から圧力発生室12に流入するインクの流路抵抗を一定に保持している。例えば、インク供給路13は、リザーバー100と各圧力発生室12との間の圧力発生室12側の流路を幅方向に絞ることで、圧力発生室12の幅より小さい幅で形成されている。なお、実施形態では、流路の幅を片側から絞ることでインク供給路を形成したが、流路の幅を両側から絞ることでインク供給路を形成してもよい。また、流路の幅を絞るのではなく、厚さ方向から絞ることでインク供給路を形成してもよい。各連通路14は、圧力発生室12の幅方向両側の隔壁11を連通部15側に延設してインク供給路13と連通部15との間の空間を区画することで形成されている。
【0027】
なお、流路形成基板10の材料として、実施形態ではシリコン単結晶基板を用いているが、勿論これに限定されず、例えば、ガラス、セラミックス、ステンレス鋼等を用いてもよい。
【0028】
流路形成基板10の開口面側には、各圧力発生室12のインク供給路13とは反対側の端部近傍に連通するノズル開口21が穿設されたノズルプレート20が、接着剤や熱溶着フィルム等によって固着されている。なお、ノズルプレート20は、例えば、ガラスセラミックス、シリコン単結晶基板、ステンレス鋼などからなる。
【0029】
一方、このような流路形成基板10の開口面とは反対側には、上述したように弾性膜50が形成され、この弾性膜50上には、厚さ0.05μm〜0.8μmの絶縁体膜55が形成されている。実施形態における絶縁体膜55は酸化ジルコニウム(ZrO2)で形成してある。さらに、この絶縁体膜55上には、厚さが例えば0.2μmの下電極60と、厚さが例えば1.3μmの圧電体層70と、厚さが例えば0.05μmの上電極80とが、後述するプロセスで積層形成されて、圧電素子300が形成されている。ここで、圧電素子300は、下電極60、圧電体層70及び上電極80を含む部分をいう。一般的には、圧電素子300の何れか一方の電極を共通電極とし、他方の電極を圧電体層70と共に圧力発生室12毎にパターニングして個別電極とする。実施形態では下電極60が個別電極、上電極80が共通電極となる、いわゆる共通上電極構造となっている。
【0030】
また、ここでは、圧電素子300と当該圧電素子300の駆動により変位が生じる振動板とを合わせて圧電アクチュエーターと称する。なお、実施形態では、弾性膜50、絶縁体膜55及び下電極60が振動板として作用する。
【0031】
ここで、実施形態に係る圧電素子300の構造について詳しく説明する。図2(a)におけるB−B要部拡大断面図である図3に示すように、圧電素子300を構成する下電極60は、各圧力発生室12に対向する領域毎に、圧力発生室12の幅よりも狭い幅で設けられて各圧電素子300の個別電極を構成している。
また、図1および図2において、下電極60は、各圧力発生室12の長手方向一端部側から周壁上まで延設されている。そして下電極60には、圧力発生室12の外側の領域で、例えば、金(Au)等からなるリード電極90がそれぞれ接続され、このリード電極90を介して各圧電素子300に選択的に電圧が印加されるようになっている。一方、圧力発生室12の長手方向のリード電極90が接続された側とは反対側の下電極60の端部は、圧力発生室12に対向する領域内に位置している。
【0032】
圧電体層70は、下電極60の幅よりも広い幅で且つ圧力発生室12の幅よりも狭い幅で設けられている。圧力発生室12の長手方向においては、圧電体層70の両端部は、圧力発生室12の端部の外側まで延設されている。すなわち、圧電体層70は、圧力発生室12に対向する領域の下電極60の上面及び端面を完全に覆うように設けられている。なお、圧力発生室12の長手方向のリード電極90が接続された側の圧電体層70の端部は、圧力発生室12の端部近傍に位置しており、その外側の領域には下電極60がさらに延設されている。
【0033】
上電極80は、複数の圧力発生室12に対向する領域に連続的に形成されるとともに、圧力発生室12の長手方向のリード電極90が接続された端部側から周壁上まで延設されている。すなわち、上電極80は、圧力発生室12に対向する領域の圧電体層70の上面及び端面のほぼ全域を覆って設けられている。
【0034】
上述の如く圧力発生室12に対向する領域の圧電体層70の上面及び端面のほぼ全域を上電極80で覆うことにより圧電体層70への大気中の水分(湿気)の浸透が実質的に防止される。したがって、水分(湿気)に起因する圧電素子300(圧電体層70)の破壊を防止することができ、圧電素子300の耐久性を著しく向上させることができる。
【0035】
また、圧力発生室12の長手方向のリード電極90が接続された側の上電極80の端部は、圧力発生室12に対向する領域内に位置している。この結果、圧電素子300の実質的な駆動部が圧力発生室12に対向する領域内に設けられている。すなわち、圧力発生室12内に位置する下電極60と上電極80との間の部分の圧電素子300が実質的な駆動部となっている。このため、圧電素子300を駆動しても、圧力発生室12の長手方向両端部近傍の振動板(弾性膜50、絶縁体膜55)には大きな変形が生じることはないため、この部分の振動板に割れが発生するのを防止することができる。なお、このような構成では、圧力発生室12に対向する領域内でも圧電体層70の表面が若干露出されることになるが、その面積は極めて狭く、また後述するように上電極80の周縁部と下電極60の間の距離が大きいため、水分に起因する圧電体層70の破壊を防止することができる。
【0036】
圧電素子300が形成された流路形成基板10上には、圧電素子300に対向する領域にその運動を阻害しない程度の空間を確保可能な圧電素子保持部31を有する保護基板30が接着剤35を介して接合されている。圧電素子300は、この圧電素子保持部31内に形成されているため、外部環境の影響を殆ど受けない状態で保護されている。また、保護基板30には、流路形成基板10の連通部15に対応する領域にリザーバー部32が設けられている。このリザーバー部32は、実施形態では、保護基板30を厚さ方向に貫通して圧力発生室12の並設方向に沿って設けられており、上述したように流路形成基板10の連通部15と連通されて各圧力発生室12の共通のインク室となるリザーバー100を構成している。
【0037】
さらに、保護基板30の圧電素子保持部31とリザーバー部32との間の領域には、保護基板30を厚さ方向に貫通する貫通孔33が設けられ、リード電極90の端部がこの貫通孔33内に露出されている。そして、図示しないが、これら下電極60及びリード電極90は、貫通孔33内に延設される接続配線によって圧電素子300を駆動するための駆動IC等に接続される。
【0038】
なお、保護基板30の材料としては、例えば、ガラス、セラミックス材料、金属、樹脂等が挙げられるが、流路形成基板10の熱膨張率と略同一の材料で形成されていることがより好ましく、実施形態では、流路形成基板10と同一材料のシリコン単結晶基板を用いて形成した。
【0039】
保護基板30上には、さらに、封止膜41及び固定板42とからなるコンプライアンス基板40が接合されている。封止膜41は、剛性が低く可撓性を有する材料からなり、この封止膜41によってリザーバー部32の一方面が封止されている。固定板42は、金属等の硬質の材料で形成される。この固定板42のリザーバー100に対向する領域は、厚さ方向に完全に除去された開口部43となっているため、リザーバー100の一方面は可撓性を有する封止膜41のみで封止されている。
【0040】
このような実施形態のインクジェット式記録ヘッド1では、図示しない外部インク供給手段からインクを取り込み、リザーバー100からノズル開口21に至るまで内部をインクで満たした後、図示しない駆動ICからの記録信号に従い、圧力発生室12に対応するそれぞれの圧電素子300に電圧を印加し、圧電素子300をたわみ変形させることにより、各圧力発生室12内の圧力が高まりノズル開口21からインク滴が吐出する。
【0041】
以下、本発明の実施形態に係る圧電素子の製造方法を、図1〜図3に示したインクジェット式記録ヘッド1の製造方法とともに、図4〜図7を参照して説明する。
【0042】
図4は、圧電素子の製造方法を表す、図2(a)におけるB−B線に沿う方向から見た断面図である。
まず、図4(a)に示すように、シリコンウェハーであり流路形成基板10が複数一体的に形成される流路形成基板用ウェハー110の表面に弾性膜50を構成する酸化膜51を形成する。
【0043】
そして、図4(b)に示すように、弾性膜50(酸化膜51)上に、絶縁体膜55を形成する。実施形態では酸化ジルコニウム(ZrO2)を形成した。
【0044】
次いで、図4(c)に示すように、絶縁体膜55上に下電極60を形成する。
この下電極60の材料は、特に限定されないが、圧電体層70としてチタン酸ジルコン酸鉛(PZT)を用いる場合には、酸化鉛の拡散による導電性の変化が少ない材料であることが望ましい。圧電体層70を液相プロセスによって形成する場合には、熱処理による下電極60の酸化が起こり難いことが望ましい。このため、下電極60の材料としては白金、イリジウム等が単体あるいは積層体として好適に用いられる。
また、下電極60は、例えば、スパッタリング法やPVD法(物理蒸着法)などにより形成することができる。なお、下電極60の成膜に先立ち、チタン、ジルコニウム又はクロムからなる、厚さが例えば0.005μm〜0.05μmである密着層(図示せず)をスパッタ法又は真空蒸着法により成膜しても良い。また、下電極60の成膜後、連続して下電極60上にチタン層(図示せず)を形成してもよい。例えばスパッタ法等により、チタン層を0.003μm〜0.02μmの厚みに成膜することで、所定の結晶配向度を備えた圧電体層70を得ることが可能である。チタン層は下電極60上に均一に成膜するが、場合によって層状ではない島状となっても構わない。
【0045】
次に、チタン酸ジルコン酸鉛(PZT)からなる圧電体としての圧電体層70を形成する。ここで、実施形態では、液相法を用いて圧電体層70を形成している。
以下に、圧電体膜を含む圧電体層70の製造方法について説明する。圧電体層70の製造方法は、調製工程と塗布工程と乾燥工程と脱脂工程と焼成工程とを含む。
【0046】
調製工程では、溶媒として特定のカルボン酸と、鉛、ジルコンおよびチタンの各有機金属化合物と、高分子化合物とを混合、撹拌することで、液相法に用いる圧電体膜の前駆体溶液を調製する。
混合は、いわゆる1ポット調製であってもよく、または複数に分けて行ってもよい。例えば、1ポット調製としては、カルボン酸に、ジルコニウムアルコキシドおよびチタニウムアルコキシドを混合し、次いで酢酸鉛を加え、最後に高分子化合物を混合することで圧電体膜の前駆体溶液を得る。複数に分ける場合は、例えば、カルボン酸と酢酸鉛を混合することで得られる第1の溶液と、カルボン酸とジルコニウムアルコキシドを混合することで得られる第2の溶液と、カルボン酸とチタニウムアルコキシドを混合することで得られる第3の溶液を、個別に調製し、3種類の溶液と高分子化合物とを混合することで最終的に圧電体膜の前駆体溶液を得る。
混合は、25℃程度の室温で行ってもよいし、加熱して行ってもよい。例えば、酢酸鉛としての酢酸鉛(II)三水和物は固体であるため、混合の際に加熱することで、溶解を促すことが可能である。また、鉛、ジルコンおよびチタン原料の混合モル比は、得られる圧電体層70の圧電特性に影響を及ぼすため、例えば、Pb:Zr:Ti=1.05〜1.25:0.46〜0.56:0.44〜0.54(モル比)となるように設定することが好ましい。
【0047】
鉛の有機金属化合物としては、一般に三水和物である酢酸鉛(II)または酢酸鉛(IV)を用いることが可能であり、取り扱いの容易性から酢酸鉛(II)を用いることが好ましい。
【0048】
ジルコンの有機金属化合物としては、アルコキシドが好ましい。ジルコニウムアルコキシドとしては、配位子として炭素数が3〜8のアルコキシ基を有するものであればよく、例えば、ジルコニウムテトラ−n−プロポキシド、ジルコニウムテトライソプロポキシド、ジルコニウムテトラ−n−ブトキシド、ジルコニウムテトラ−iso−ブトキシド、ジルコニウムテトラ−sec−ブトキシド、ジルコニウムテトラ−tert−ブトキシド、ジルコニウムテトラ−n−ペントキシド、ジルコニウムテトラ−sec−ペントキシド、ジルコニウムテトラ−tert−ペントキシド、ジルコニウムテトラ−n−ヘキソキシド、ジルコニウムテトラ−sec−ヘキソキシド、ジルコニウムテトラ−n−ヘプトキシド、ジルコニウムテトラ−sec−ヘプトキシド、ジルコニウムテトラ−n−オクトキシドおよびテトラキス(2−エチルヘキシルオキシ)オルトジルコネート等が挙げられ、単体あるいは混合体として用いることが可能である。炭素数3以上とすることで、カルボン酸エステルの生成を抑制することが可能であり、炭素数8以下とすることで、圧電体膜の前駆体溶液の金属酸化物換算濃度を高くすることが可能となる。
【0049】
チタンの有機金属化合物としては、アルコキシドが好ましい。チタニウムアルコキシドとしては、配位子として炭素数が3〜8のアルコキシ基を有するものであればよく、例えば、チタニウムテトラ−n−プロポキシド、チタニウムテトライソプロポキシド、チタニウムテトラ−n−ブトキシド、チタニウムテトラ−iso−ブトキシド、チタニウムテトラ−sec−ブトキシド、チタニウムテトラ−tert−ブトキシド、チタニウムテトラ−n−ペントキシド、チタニウムテトラ−sec−ペントキシド、チタニウムテトラ−tert−ペントキシド、チタニウムテトラ−n−ヘキソキシド、チタニウムテトラ−sec−ヘキソキシド、チタニウムテトラ−n−ヘプトキシド、チタニウムテトラ−sec−ヘプトキシド、チタニウムテトラ−n−オクトキシドおよびチタニウムテトラキス(2−エチルヘキシルオキシド)等が挙げられ、単体あるいは混合体として用いることが可能である。炭素数3以上とすることで、カルボン酸エステルの生成を抑制することが可能であり、炭素数8以下とすることで、圧電体膜の前駆体溶液の金属酸化物換算濃度を高くすることが可能となる。
【0050】
本発明の圧電体膜の前駆体溶液では、上記のジルコニウムアルコキシド又はチタニウムアルコキシドの配位子のうち、少なくとも一方の配位子が分岐鎖状であることが好ましい。これにより、圧電体膜の前駆体溶液におけるカルボン酸エステルの生成量を抑制することができるので、得られる圧電体膜の膜厚の変動を抑えることが可能となる。
【0051】
カルボン酸溶媒に金属アルコキシドを溶解させた場合、配位子交換反応が起こり、結果として、溶液中にはカルボン酸と遊離したアルコールが存在する。そして、下記式(1)で表されるカルボン酸(A−COOH)とアルコール(B−OH)とによるカルボン酸エステル(A−COO−B)の生成反応が起こる。
A−COOH+B−OH ⇔ A−COO−B+H2O ・・・(1)
カルボン酸エステルは、原料であるカルボン酸、又はアルコールと比べて、揮発性が高くなる場合が多い。したがって、圧電体膜の前駆体溶液に遊離アルコールが存在し、カルボン酸エステルの生成反応が経時的に進行する場合、成膜工程によって得られる膜厚が変動してしまう。
【0052】
カルボン酸とアルコールによるカルボン酸エステルの生成反応において、アルコールの炭素数が多くなるに従い、カルボン酸エステルの生成量が減少する。また、炭素数が同一であれば、直鎖状のアルコールのよりも、分岐鎖状のアルコールのほうがカルボン酸エステルの生成量が減少する。したがって、炭素数3〜8のアルコールを配位子として有するチタニウムアルコキシド及びジルコニウムアルコキシドを用い、かつ、チタンニウムアルコキシド又はジルコニウムアルコキシドの少なくとも一方の前記配位子を分岐鎖状とすることが好ましい。
【0053】
また、本発明の圧電体膜の前駆体溶液に含まれる溶媒として、特定のカルボン酸に加えて、さらに、水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類から選ばれる1種または2種以上を含むことができる。上述したカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応を抑制することが可能となる。
【0054】
2級または3級アルコールとしては、特に限定はされないが、イソプロピルアルコール、iso−ブチルアルコール、sec−ブチルアルコール、tert−ブチルアルコールおよびiso−ペンチルアルコール等が挙げられる。エーテル類としては、特に限定はされないが、ジイソプロピルエーテル、ジ−n−ブチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテルおよびエチレングリコールジ−n−ブチルエーテル等が挙げられる。
エステル類としては、特に限定はされないが、酢酸メチル、酢酸エチル、酢酸イソプロピル、酢酸n−ブチル、プロピオン酸エチル、n−酪酸エチルおよびエチレングリコールジアセテート等が挙げられる。さらに、エーテル類でありエステル類でもあるエチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテートおよびプロピレングリコールモノメチルエーテルアセテート等も用いることが可能である。
ケトン類としては、特に限定はされないが、例えば、ジメチルケトン(アセトン)、ジエチルケトン、ジn−プロピルケトン、ジiso−プロピルケトン、メチルエチルケトン、メチルn−プロピルケトン、メチルiso−プロピルケトン、メチルn−ブチルケトン、メチルiso−ブチルケトン、メチルtert−ブチルケトン、メチルiso−ペンチルケトン、エチルn−プロピルケトン、エチルiso−プロピルケトンおよびエチルn−ブチルケトン等が挙げられる。
【0055】
圧電体層70の具体的な作成手順を説明する。
まず、塗布工程において、図5(d)に示すように、下電極60上にPZT前駆体膜である圧電体前駆体膜71を成膜する。すなわち、下電極60が形成された流路形成基板用ウェハー110上に圧電体膜の前駆体溶液を塗布する。
圧電体膜の前駆体溶液の塗布方法としては、スピンコート法、スプレーコート法およびディップコート法等が挙げられる。実施形態では、半導体プロセスに用いられる種々の装置を利用するために、基板片面に塗布が可能で、基板面内の膜厚均一性が良好なスピンコート法を用いるのが好ましい。スピンコートの塗布条件(回転数、時間等)は、用いる圧電体膜の前駆体溶液によって適宜設定することが可能であるが、基板面内における膜厚均一性の確保や、塗布面の反対側への前駆体溶液の回りこみ防止といった観点から回転数を500〜5000rpm程度に設定するのが好ましい。また、スピンコート時にリンス液を用いて、基板外周部および塗布面の反対側を洗浄することが可能であり、これらはエッジリンスおよびバックリンスと呼称される。
【0056】
次いで、乾燥工程において、この圧電体前駆体膜71を所定温度に加熱して一定時間乾燥させる。例えば、流路形成基板用ウェハー110上に塗布されたゾルを140〜180℃で5〜30分保持することで乾燥する。乾燥温度としては、前駆体溶液の溶媒が蒸発するように設定することが好ましく、例えば、酢酸を溶媒として用いた場合には、140℃にすることが可能である。
【0057】
次に、脱脂工程において、乾燥した圧電体前駆体膜71を所定温度に加熱して一定時間保持することによって脱脂する。実施形態では、乾燥させた圧電体前駆体膜71を350〜500℃に加熱して5〜30分保持することで脱脂する。
なお、ここで言う脱脂とは、加熱することによって圧電体前駆体膜71に含まれる有機成分を、例えば、CO2、H2O、アルコール、炭化水素等として離脱させることである。したがって、脱脂温度は圧電体前駆体膜71に含まれる有機成分の状態によって設定するべきであり、脱脂温度を設定する指標としては熱分析(TG/DTA測定、DSC測定等)結果を利用することが可能である。熱分析によって、前駆体溶液の脱脂に伴う発熱反応、あるいは重量変化が起きる温度を測定することが可能である。
また、脱脂とは、PZTを圧電体膜として利用する場合、強誘電性を有さないパイロクロア相が形成されることを防ぐために、圧電体前駆体膜71が結晶化しない程度に、すなわち、非晶質の圧電体前駆体膜71を形成することを言う。圧電体前駆体膜71に含まれる有機成分の蒸発あるいは熱酸化分解による除去が目的であるため、酸素を含む雰囲気であればよく、大気雰囲気中で行うことが可能である。
【0058】
次に、焼成工程において、図5(e)に示すように、圧電体前駆体膜71を所定温度に加熱して一定時間保持することにより結晶化させ、圧電体膜72を形成する。かかる焼成工程においては、電気炉またはRTA(Rapid thermal Aannealing)装置等を利用することが可能である。PZTを圧電体膜72として利用する場合、強誘電性を有するペロブスカイト相を単相で得るために、脱脂した圧電体前駆体膜71を650〜800℃で焼成して結晶化させることが好ましい。また、PZTの酸素欠損を防ぐために、酸素雰囲気あるいは酸素フロー中で焼成することが好ましい。
【0059】
次に、図6(f)に示すように、下電極60上に1層目の圧電体膜72を形成した段階で、下電極60及び1層目の圧電体膜72をそれらの側面が傾斜するように同時にパターニングする。
【0060】
ここで、例えば、下電極60をパターニングした後、1層目の圧電体膜72を形成する場合、フォトリソグラフィー工程・エッチング工程・アッシング工程を行い、下電極60をパターニングするため、下電極60の表面や、所定の結晶配向度を有する圧電体膜を得るために下電極60の表面に設けたチタン層(図示せず)などが変質してしまう。そうすると、変質した下電極60上に圧電体膜72を形成しても当該圧電体膜72の結晶性または結晶配向度が良好なものではなくなり、2層目以降の圧電体膜72も1層目の圧電体膜72の結晶状態に影響して結晶成長するため、良好な結晶性を有する圧電体層70を形成することができない。
【0061】
これに対し実施形態の如く、1層目の圧電体膜72を形成した後に下電極60と同時に圧電体膜72をパターニングすれば、2層目以降の圧電体膜72を良好に結晶成長させることが可能である。何故なら、結晶成長を促すシード層としては、チタン等の結晶種よりも、圧電体膜72そのものを利用したほうがより好ましいからである。また、シード層としての性能が高い圧電体膜72であれば、たとえパターニングで表層に極薄い変質層が形成されていても2層目以降の圧電体膜72の結晶成長に大きな影響を与えない。
【0062】
そして、上述した前駆体膜形成工程(塗布工程、乾燥工程及び脱脂工程)と、焼成工程とを有する圧電体膜形成工程を2回以上繰り返すことで、図6(g)に示すように、2層目以降、最上層までの圧電体膜72を積層する。その後、図7(h)に示すように、2層目以降が積層された圧電体膜72をパターニングして圧電体層70を形成する。
【0063】
次いで、図7(i)に示すように、流路形成基板用ウェハー110の全面に亘って上電極80を形成し、所定形状にパターニングする。具体的には、図2に示すように、上電極80は、圧力発生室12の並設方向に亘って連続的に設け、且つ圧力発生室12の長手方向端部よりも外側まで延設するようにする。
【0064】
さらに、図示はしないが、流路形成基板用ウェハー110の全面に亘って、例えば、金(Au)等からなるリード電極90を形成後、圧電素子300毎にパターニングする。
【0065】
次いで、流路形成基板用ウェハー110の圧電素子300側に、シリコンウェハーであり複数の保護基板30となる保護基板用ウェハーを接着剤35によって接合する。なお、保護基板30には、リザーバー部32、圧電素子保持部31等が予め形成されている。また、保護基板30は、例えば、400μm程度の厚さを有するシリコン単結晶基板からなり、保護基板30を接合することで流路形成基板10の剛性は著しく向上することになる。その後、流路形成基板用ウェハー110を所定の厚さにする。
【0066】
次いで、流路形成基板用ウェハー110にマスク膜を新たに形成し、所定形状にパターニングする。そして、流路形成基板用ウェハー110を、マスク膜を介してKOH等のアルカリ溶液を用いた異方性エッチング(ウェットエッチング)することにより、圧電素子300に対応する圧力発生室12、インク供給路13、連通路14及び連通部15等を形成する。
【0067】
その後は、図2に示すように、保護基板用ウェハー上に図示しない駆動回路を実装すると共に、駆動回路とリード電極90とを接続配線によって接続する。そして、流路形成基板用ウェハー110の圧力発生室12が開口する面側のマスク膜を除去し、流路形成基板用ウェハー110及び保護基板用ウェハーの外周縁部の不要部分を、例えば、ダイシング等により切断することによって除去する。そして、流路形成基板用ウェハー110の保護基板用ウェハーとは反対側の面にノズル開口21が穿設されたノズルプレート20を接合すると共に、保護基板用ウェハーにコンプライアンス基板40を接合し、これら流路形成基板用ウェハー110等を、図1に示すような一つのチップサイズの流路形成基板10等に分割することによって上述した構造のインクジェット式記録ヘッド1を製造する。
【0068】
以下に、実施例に基づいてより詳細に説明する。実施例1〜実施例47では、圧電体膜としてPZT膜を前駆体溶液としてPZT前駆体溶液を例として説明する。
各実施例および各比較例は、圧電体膜の前駆体溶液が異なるが、塗布工程、乾燥工程、脱脂工程および焼成工程は同じ条件で行った。最初に圧電体膜の前駆体溶液の調製工程を実施例および比較例について説明し、同じ工程については、後に説明する。また、各実施例および各比較例の圧電体膜の前駆体溶液に用いた原料を図8に示した。
【0069】
(調製工程)
[実施例1]
まず、200mLのガラスフラスコに、溶媒として酢酸43.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0質量%)14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、この混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物としてポリエチレングリコール(平均分子量600)6.8gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、最終的なPZT前駆体溶液とした。
本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例のPZT前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ジルコニウムZrO2、および酸化チタンTiO2として金属酸化物換算濃度を算出すると、22.4質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、43.2質量%であった。
【0070】
[実施例2]
溶媒として酢酸50.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、46.9質量%であった。
【0071】
[実施例3]
溶媒としてプロピオン酸37.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、39.6質量%であった。
【0072】
[実施例4]
溶媒としてプロピオン酸40.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、41.4質量%であった。
【0073】
[実施例5]
溶媒として酢酸20.0gとプロピオン酸20.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、41.4質量%であった。
【0074】
[比較例1]
溶媒としてアセチルアセトン30.0gと2−n−ブトキシエタノール10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0075】
[比較例2]
溶媒として2−n−ブトキシエタノール69.0gとジエタノールアミン15.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、15.9質量%であった。
【0076】
[比較例3]
溶媒として2−n−ブトキシエタノール33.0gとジエタノールアミン7.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0077】
[比較例4]
溶媒として2−n−ブトキシエタノール25.0gとジエタノールアミン15.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0078】
(塗布工程)
塗布には、図4(c)で示される流路形成基板用ウェハー110を用いた。具体的には、直径が150mmであり、厚さが700μmであるシリコン単結晶基板上に、熱酸化によって膜厚が1.1μmのSiO2を弾性膜50として形成し、次いで、スパッタ法によって膜厚が0.4μmのZrO2を絶縁体膜55として形成し、さらに、スパッタ法により膜厚が130nmのPtと膜厚が20nmのIrとの積層体を下電極60として形成した。
また、下電極60の成膜に先立って、膜厚が10nmのチタン層を密着層として形成し、下電極60の成膜後に、膜厚が5nmであるチタン層を形成した。実施例1〜5および比較例1〜4のPZT前駆体溶液を用いて、25℃、40%RHの環境でスピンコートを行った。スピンコートの条件として、滴下するPZT前駆体溶液を4mLとし、回転数を2000rpmとし、回転時間を60秒間として、上記の基板に塗布を行った。なお、塗布は、各PZT溶液の調製からそれぞれ1日後に行った。
(乾燥工程)
塗布工程の後に、乾燥工程として、140℃のホットプレートを用いてで5分間の熱処理を行った。
(脱脂工程)
乾燥工程の後に、脱脂工程として、400℃のホットプレートで5分間の熱処理を行った。
(焼成工程)
脱脂工程の後に、焼成工程として、RTA装置を用いて酸素フローを行いながら、700℃で5分間の熱処理を行った。
【0079】
得られたPZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。各実施例および各比較例のPZT前駆体溶液の金属酸化物濃度および原料全量に対する原料として用いたカルボン酸の割合と、各実施例および各比較例のPZT前駆体溶液を用いて得られたPZT膜の膜厚とをまとめて図10に示した。
【0080】
(耐水性の評価)
実施例1〜5および比較例1〜4のPZT前駆体溶液について、耐水性試験を行った。ここで言う耐水性試験とは、各PZT前駆体溶液にイオン交換水を添加・揺動した際に、沈殿が発生するか否かで判定した。具体的には、容量が2.0mLのキャップ付ガラス瓶にPZT前駆体溶液1.0gを入れ、次いで、イオン交換水0.5gを添加・揺動して行った。沈殿の発生がなかった場合を○とし、沈殿が発生した場合を×として、各PZT前駆体溶液の耐水性を評価した。得られた評価結果をまとめて図10に示した。
【0081】
(保存安定性の評価)
実施例1〜5および比較例1〜4のPZT前駆体溶液について、保存安定性試験を行った。ここで言う保存安定性試験とは、各PZT前駆体溶液を室温にて保存した際に、沈殿が発生するか否かで判定した。具体的には、容量が100mLのキャップ付ガラス瓶にPZT前駆体溶液70gを入れ、遮光された薬品庫にて1ヶ月間、密栓保管した。なお、薬品庫内部の温度は25℃程度(室温)であった。沈殿の発生がなかった場合を○とし、沈殿が発生した場合を×として、各PZT前駆体溶液の保存安定性を評価した。得られた評価結果をまとめて図10に示した。
【0082】
図10に示した結果より、実施例1〜5のPZT前駆体溶液を用いた場合、結晶化後の膜厚が200nm以上のPZT膜を得ることが可能であり、さらに、PZT前駆体溶液の耐水性および保存安定性も良好であることが明らかである。
これに対して、比較例1のPZT前駆体溶液は、調製1日後ですでに沈殿が発生したため、塗布することができなかった。これは、アセチルアセトンの添加量が多すぎたために、溶液としての安定性の低下が起きたものである。
比較例2のPZT前駆体溶液を用いた場合では、PZT前駆体溶液の耐水性および保存安定性は良好であったが、得られたPZT膜の膜厚は90nmであった。
比較例3のPZT前駆体溶液を用いた場合では、比較的厚いPZT膜が得られたが、PZT前駆体溶液の耐水性試験結果が×となった(沈殿が発生した)。これは、ジエタノールアミンの添加量が少ないために、十分な安定化効果が得られなかったためである。
比較例4のPZT前駆体溶液を用いた場合は、PZT前駆体溶液の耐水性および保存安定性は良好であったものの、得られるPZT膜の膜ムラがひどく、評価が出来なかった。これは、ジエタノールアミンの粘度が非常に高いために、2−n−ブトキシエタノールの添加量が少なくなると、PZT前駆体溶液の粘度も高くなってしまい、基板全体に塗布膜を形成することが出来なくなったためである。
【0083】
(調製工程)
[実施例6]
まず、200mLのガラスフラスコに、溶媒として酢酸48.7gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−プロポキシド10.3gと、チタニウム原料としてチタニウムテトラ−iso−イソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物として重量平均分子量600のポリエチレングリコール6.8gと、追加溶媒としてイオン交換水5.0gを加え、25℃の室温にて1時間の撹拌を行い、これを最終的なPZT前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0084】
[実施例7]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−iso−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0085】
[実施例8]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−sec−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0086】
[実施例9]
溶媒として酢酸47.1gを用い、チタニウム原料としてチタニウムテトラ−tert−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.2質量%であった。
【0087】
[実施例10]
溶媒としてプロピオン酸41.7gを用い、チタニウム原料としてチタニウムテトラキス(2−エチルヘキシルオキシド)16.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、39.2質量%であった。
【0088】
[実施例11]
溶媒として酢酸47.1gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.2質量%であった。
【0089】
[実施例12]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0090】
[実施例13]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−sec−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0091】
[実施例14]
溶媒として酢酸46.9gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−プロポキシド8.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、49.2質量%であった。
【0092】
[実施例15]
溶媒としてプロピオン酸43.3gを用い、ジルコニウム原料としてジルコニウムテトラキス(2−エチルヘキシルオキシド)19.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、40.6質量%であった。
【0093】
[実施例16]
溶媒として酢酸48.7gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0094】
[実施例17]
溶媒としてプロピオン酸50.3gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−tert−ブトキシド10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、47.2質量%であった。
【0095】
[実施例18]
溶媒として酢酸45.2gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ペントキシド13.8gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.4質量%であった。
【0096】
[実施例19]
溶媒としてプロピオン酸44.9gを用い、ジルコニウム原料としてジルコニウムテトラキス(2−エチルヘキシルオキシド)19.1gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.2質量%であった。
【0097】
[実施例20]
溶媒としてプロピオン酸45.5gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用い、チタニウム原料としてチタニウムテトラキス(2−エチルヘキシルオキシド)16.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.7質量%であった。
【0098】
[実施例21]
溶媒として酢酸47.0gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−ブトキシド12.1gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.1質量%であった。
【0099】
[実施例22]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−iso−プロポキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0100】
[比較例5]
溶媒としてプロピオン酸60.6gを用い、ジルコニウム原料としてジルコニウムテトラメトキシド6.8gを用い、チタニウム原料としてチタニウムテトラメトキシド5.1gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、56.9質量%であった。
【0101】
[比較例6]
溶媒として酢酸52.2gを用い、ジルコニウム原料としてジルコニウムテトラエトキシド8.5gを用い、チタニウム原料としてチタニウムテトラエトキシド6.7gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、49.0質量%であった。
【0102】
[比較例7]
溶媒として酢酸48.7gを用い、チタニウム原料としてチタニウムテトラ−n−プロポキシド8.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0103】
(成膜工程)
実施例6〜22および比較例5〜7のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後および30日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0104】
(再現性評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液を用いて得られた各PZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後のPZT前駆体溶液を用いたPZT膜の膜厚を100とし、調製して30日後のPZT前駆体溶液を用いたPZT膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例6〜22および比較例5〜7の評価結果を図12に示した。
A:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が100以上110未満。
B:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が110以上120未満。
C:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が120以上。
【0105】
(耐水性の評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図12に示した。
【0106】
(保存安定性の評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図12に示した。
【0107】
図12に示した結果から明らかなように、ジルコニウムアルコキシドの配位子およびチタニウムアルコキシドの配位子が、ともに、炭素数が1〜3であり、かつ、直鎖である場合は、膜厚の再現性評価結果がいずれもCであった(比較例5〜7)。
比較例5〜7に対し、実施例6〜15では、膜厚の再現性評価結果はいずれもBであった。これは、ジルコニウムアルコキシドの配位子またはチタニウムアルコキシドの配位子のいずれかを分岐鎖としたことによって、カルボン酸エステルの生成が抑制されたためである。実施例16〜22では、膜厚の再現性評価結果はいずれもAであった。これは、ジルコニウムアルコキシドの配位子およびチタニウムアルコキシドの配位子をともに分岐鎖としたことによって、実施例6〜15よりもさらに、カルボン酸エステルの生成が抑制されたためである。
【0108】
なお、PZT前駆体溶液中に存在するアルコールおよびカルボン酸エステルの簡易的な定量手段として、NMR測定(1Hおよび13C)を行った。実施例6〜22および比較例5〜7の各PZT前駆体溶液のいずれも、調製1日後から調製30日後にかけて、カルボン酸エステルが経時的に生成していることが分かっており、さらに、膜厚の再現性はエステル変化率と相関があることが分かっている。
例えば、比較例5において、PZT前駆体溶液中の酢酸メチルとメタノールのピークからエステル変化率を算出した結果、1日後では36mol%、30日後では81mol%であった。ここで、エステル変化率は、100×[カルボン酸エステル]/([カルボン酸エステル]+[アルコール])として算出した。
実施例として、例えば、実施例16において、エステル変化率を算出すると、1日後では6mol%、30日後では10mol%であった。実施例6において、エステル変化率を算出すると、1日後では27mol%、30日後では48mol%であった。
【0109】
(調製工程)
[実施例23]
溶媒として酢酸23.5gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水23.5gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0110】
[実施例24]
溶媒としてプロピオン酸23.5gとイソプロピルアルコール23.5gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0111】
[実施例25]
溶媒として酢酸28.2gとtert−ブタノール18.8gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、26.4質量%であった。
【0112】
[実施例26]
溶媒としてプロピオン酸37.6gとジ−n−ブチルエーテル9.4gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0113】
[実施例27]
溶媒として酢酸32.9gと酢酸n−ブチル14.1gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.8質量%であった。
【0114】
[実施例28]
溶媒としてプロピオン酸32.9gとエチレングリコールモノメチルエーテルアセタート14.1gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.8質量%であった。
【0115】
[実施例29]
溶媒として酢酸23.5gとイソプロピルアルコール9.4gとを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水14.1gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0116】
[実施例30]
溶媒として酢酸28.2gと酢酸n−ブチル9.4gとを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水9.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、26.4質量%であった。
【0117】
[実施例31]
溶媒としてプロピオン酸37.6gとtert−ブタノール4.7gとエチレングリコールモノメチルエーテルアセタート4.7gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0118】
[実施例32]
溶媒としてプロピオン酸37.6gとメチルエチルケトン9.4gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0119】
(成膜工程)
実施例23〜32のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後および30日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0120】
(再現性評価)
実施例23〜32のPZT前駆体溶液を用いて得られた各PZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後のPZT前駆体溶液を用いたPZT膜の膜厚を100とし、調製して30日後のPZT前駆体溶液を用いたPZT膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例23〜32の評価結果を図14に示した。
A:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が100以上110未満。
B:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が110以上120未満。
C:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が120以上。
【0121】
(耐水性の評価)
実施例23〜32のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図14に示した。
【0122】
(保存安定性の評価)
実施例23〜32のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図14に示した。
【0123】
図14に示した結果から明らかなように、実施例23〜32のPZT前駆体溶液は、膜厚の再現性評価結果がいずれもAであった。これはカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応が抑制されたためである。
【0124】
(調製工程)
[実施例33]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物として重量平均分子量300であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的なPZT前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0125】
[実施例34]
高分子化合物として重量平均分子量300であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0126】
[実施例35]
高分子化合物として重量平均分子量400であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0127】
[実施例36]
高分子化合物として重量平均分子量400であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0128】
[実施例37]
高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0129】
[実施例38]
高分子化合物として重量平均分子量600であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0130】
[実施例39]
高分子化合物として重量平均分子量550であるポリエチレングリコールモノメチルエーテル6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0131】
[実施例40]
高分子化合物として重量平均分子量750であるポリエチレングリコールモノメチルエーテル6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0132】
[実施例41]
高分子化合物として重量平均分子量400であるポリプロピレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0133】
[実施例42]
高分子化合物として重量平均分子量700であるポリプロピレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0134】
[実施例43]
高分子化合物として重量平均分子量が400であるポリエチレングリコール1.7gと重量平均分子量が600であるポリプロピレングリコール1.7gと重量平均分子量が550であるポリエチレングリコールモノメチルエーテル1.7gと重量平均分子量が700であるポリプロピレングリコールとを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0135】
(成膜工程)
実施例33〜43のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を6回繰り返し行い、6層のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0136】
(クラック評価)
実施例33〜43のPZT前駆体溶液を用いて得られた各PZT膜について、金属顕微鏡にてクラック評価を行った。クラックが発生していない場合を○とし、クラックが発生したものを×として判定した。また、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。得られた結果をまとめて図16に示した。
【0137】
(耐水性の評価)
上述の実施例33〜43のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図16に示した。
【0138】
(保存安定性の評価)
実施例33〜43のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図16に示した。
【0139】
図16に示した結果から明らかなように、実施例33〜43の全てのPZT膜について、クラックが発生することなく、1μmを超えるPZT膜を得ることが可能であった。さらに、耐水性および保存安定性の評価結果についても良好な結果を示した。
【0140】
(調製工程)
[実施例44]
溶媒として酢酸50.0gを用い、追加溶媒としてイオン交換水23.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、17.2質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、38.6質量%であった。
【0141】
[実施例45]
溶媒として酢酸32.6gとイソプロピルアルコール14.4gとを用い、鉛原料として酢酸鉛(II)三水和物21.8gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド8.2gを用い、チタニウム原料としてチタニウムテトライソプロポキシド6.7gを用い、高分子化合物として重量平均分子量が400であるポリエチレングリコール16.4gを用い、追加溶媒としてイオン交換水14.4gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、15.6質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、28.4質量%であった。
【0142】
[実施例46]
溶媒として酢酸56.5gを用い、鉛原料として酢酸鉛(II)三水和物21.8gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液11.2gを用い、チタニウム原料としてチタニウムテトライソプロポキシド6.7gを用い、高分子化合物として重量平均分子量が600であるポリエチレングリコール5.4gを用い、追加溶媒としてイオン交換水6.3gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、16.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、52.4質量%であった。
【0143】
[実施例47]
溶媒としてプロピオン酸30.0gと酢酸n−ブチル10.0gとメチルエチルケトン6.0gを用い、高分子化合物として重量平均分子量が400であるポリエチレングリコール13.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.4質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、27.4質量%であった。
【0144】
[実施例48]
溶媒としてプロピオン酸25.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、26.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.7質量%であった。
【0145】
(成膜工程)
実施例44〜48および比較例2のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後の各PZT前駆体溶液を用いて、スピンコートの回転数を変えたほかは実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、PZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0146】
得られたPZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。各実施例および各比較例のPZT前駆体溶液を用いて得られたPZT膜の膜厚をまとめて図18に示した。
【0147】
(耐水性の評価)
上述の実施例44〜48のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図18に示した。
【0148】
(保存安定性の評価)
上述の実施例44〜48のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図18に示した。
【0149】
図18から明らかなように、実施例44〜48を用いたPZT膜は、スピンコート回転数を変えることによって、いずれも200nmを超える膜厚が得られている。これに対して、比較例2と同様のPZT前駆体溶液を用いた比較例8ではスピンコート回転数を700rpmとしても、162nmの膜厚であった。
【0150】
(調製工程)
[実施例49]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0%)14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の攪拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物25.4gと、ランタン原料として酢酸ランタン1.5水和物2.1gと、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的な鉛系圧電体膜の前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図19に示した。本実施例の鉛系圧電体膜の前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ランタンLa2O3、酸化ジルコニウムZrO2、および酸化チタンTiO2として金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0151】
[実施例50]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0%)9.8gと、チタニウム原料としてチタニウムテトライソプロポキシド5.9gと、ニオブ原料としてペンタ−n−ブトキシニオブ5.6gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の攪拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、マグネシウム原料として酢酸マグネシウム四水和物1.3gと、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的な鉛系圧電体膜の前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図19に示した。本実施例の鉛系圧電体膜の前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ジルコニウムZrO2、酸化チタンTiO2、酸化マグネシウムMgOおよび酸化ニオブNb2O5として金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0152】
(成膜工程)
実施例49および50の鉛系圧電体膜の前駆体溶液を用いて、鉛系圧電体膜の成膜を行った。調製して1日後および30日後の各前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類の鉛系圧電体膜を得た。なお、各前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0153】
(再現性評価)
上述の実施例48および49の鉛系圧電体膜の前駆体溶液を用いて得られた各鉛系圧電体膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後の前駆体溶液を用いた鉛系圧電体膜の膜厚を100とし、調製して30日後の前駆体溶液を用いた鉛系圧電体膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例49および50の評価結果を図20に示した。
A:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が100以上110未満。
B:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が110以上120未満。
C:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が120以上。
【0154】
(耐水性の評価)
上述の実施例49および50の鉛系圧電体膜の前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図20に示した。
【0155】
(保存安定性の評価)
上述の実施例49および50の鉛系圧電体膜の前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図20に示した。
【0156】
(インクジェット式記録ヘッドの製造)
実施例1〜47および比較例2のPZT前駆体溶液を用いて、上述のインクジェット式記録ヘッドの一連の製造工程に基づき、インクジェット式記録ヘッドを製造した。
なお、実施例1〜47のPZT前駆体溶液を用いる場合は、調製して1日後の各PZT前駆体溶液を用い、一連の成膜工程(塗布工程、乾燥工程、脱脂工程、焼成工程)を6回繰り返すことで、膜厚が1.2μmの圧電体層70を成膜した(なお、1層あたりの膜厚が200nmとなるように、各PZT前駆体溶液によってスピンコート回転数は適宜調整を行った)。
比較例2のPZT前駆体溶液を用いる場合は、調製して1日後の各PZT前駆体溶液を用い、一連の成膜工程を12回繰り返すことで、膜厚が1.2μmの圧電体層70を成膜した(なお、スピンコート塗布における回転数は1500rpmとした)。したがって同じ膜厚の圧電体層70を得たい場合、実施形態であれば比較例2の半分の回数の成膜工程でよい。
【0157】
得られた圧電体層70の結晶性評価(X線回折装置によるθ/2θ測定)、バーチャルグラウンド方式でのヒステリシス特性評価、レーザードップラー式振動計による圧電特性評価(変位量測定)、パルス耐久試験(初期値を基準として、200億のパルスを加えた後の変位低下率を評価)等を行った。実施例1〜46で得られた圧電体層70は、比較例2で得られた圧電体層70と同等の結果を示した。
なお、同等とは比較例2の前駆体溶液を用いて得られる圧電体を100として規格化した場合に、95〜105であることを示す。結晶性評価の場合は、(100)ピーク強度および(100)配向率であり、ヒステリシス評価の場合は、最大分極値、残留分極値および抗電圧であり、圧電特性の場合は変位量であり、パルス耐久試験では変位低下率である。
【0158】
上記実施形態で製造されたインクジェット式記録ヘッド1は、インクカートリッジ等と連通するインク流路を具備する記録ヘッドユニットの一部を構成して、インクジェット式記録装置1000に搭載される。図21は、そのインクジェット式記録装置の一例を示す概略図である。
【0159】
図21に示すインクジェット式記録装置1000において、図1に示したインクジェット式記録ヘッド1を有するインクジェット式記録ヘッドユニット1A及び1Bは、インク供給手段を構成するカートリッジ2A及び2Bが着脱可能に設けられ、このインクジェット式記録ヘッドユニット1A及び1Bを搭載したキャリッジ3は、装置本体4に取り付けられたキャリッジ軸5に軸方向移動自在に設けられている。このインクジェット式記録ヘッドユニット1A及び1Bは、例えば、それぞれブラックインク組成物及びカラーインク組成物を吐出するものとしている。
【0160】
そして、駆動モーター6の駆動力が図示しない複数の歯車およびタイミングベルト7を介してキャリッジ3に伝達されることで、インクジェット式記録ヘッドユニット1A及び1Bを搭載したキャリッジ3はキャリッジ軸5に沿って移動される。一方、装置本体4にはキャリッジ軸5に沿ってプラテン8が設けられており、図示しない給紙ローラーなどにより給紙された紙等の記録媒体である記録シートSがプラテン8に巻き掛けられて搬送されるようになっている。
【0161】
なお、図21に示す例では、インクジェット式記録ヘッドユニット1A、1Bは、それぞれ1つのインクジェット式記録ヘッド1を有するものとしたが、特にこれに限定されず、例えば、1つのインクジェット式記録ヘッドユニット1A又は1Bが2以上のインクジェット式記録ヘッド1を有するようにしてもよい。もちろん、インクジェット式記録ヘッドユニット1A、1Bという形式を取らずに、インクジェット式記録ヘッド1を直接インクジェット式記録装置1000に搭載してもよい。
【0162】
また、上述したインクジェット式記録装置1000では、インクジェット式記録ヘッド1を有するインクジェット式記録ヘッドユニット1A、1Bがキャリッジ3に搭載されて主走査方向に移動するものを例示したが、特にこれに限定されず、例えば、インクジェット式記録ヘッド1が固定されて、紙等の記録シートSを副走査方向に移動させるだけで印刷を行う、所謂ライン式記録装置にも本発明を適用することができる。
【0163】
なお、上述した実施形態では、液体噴射ヘッドの一例としてインクジェット式記録ヘッド1を挙げて説明したが、本発明は広く液体噴射ヘッド全般を対象としたものであり、インク以外の液体を噴射する液体噴射ヘッドにも勿論適用することができる。その他の液体噴射ヘッドとしては、例えば、プリンター等の画像記録装置に用いられる各種の記録ヘッド、液晶ディスプレイ等のカラーフィルターの製造に用いられる色材噴射ヘッド、有機ELディスプレイ、FED(電界放出ディスプレイ)等の電極形成に用いられる電極材料噴射ヘッド、バイオchip製造に用いられる生体有機物噴射ヘッド等が挙げられる。
【符号の説明】
【0164】
1A,1B…インクジェット式記録ヘッドユニット、2A,2B…カートリッジ、4…装置本体、6…駆動モーター、10…流路形成基板、12…圧力発生室、14…連通路、15…連通部、20…ノズルプレート、21…ノズル開口、30…保護基板、31…圧電素子保持部、32…リザーバー部、33…貫通孔、35…接着剤、40…コンプライアンス基板、41…封止膜、42…固定板、43…開口部、50…弾性膜、51…酸化膜、55…絶縁体膜、60…下電極、70…圧電体層、71…圧電体前駆体膜、72…圧電体膜、80…上電極、90…リード電極、100…リザーバー、300…圧電素子、1000…インクジェット式記録装置。
【技術分野】
【0001】
本発明は、鉛含有圧電体膜の原料である前駆体溶液に関するものであり、さらには、これを用いた圧電素子の製造方法に関するものである。
【背景技術】
【0002】
チタン酸ジルコン酸鉛(PZT)等に代表される結晶を含む圧電体膜としての強誘電体膜は、自発分極、高誘電率、電気光学効果、圧電効果、焦電効果等を有しているため、圧電素子等の広範なデバイスに応用されている。このような圧電体膜の製造方法としては、気相法と液相法の2種類に大別される。気相法としては、CVD(Chemical Vapor Deposition)法、スパッタリング法等が知られており、液相法としては、MOD(Metal Organic Decomposition)法、ゾル−ゲル法等が知られている。特に、液相法は、気相法よりも以下の点で有利である。すなわち、高価な真空装置を必要としない点、元素組成および添加物を制御しやすい点等によって、圧電体膜を低コストで、かつ簡便に成膜できる。
【0003】
ゾル−ゲル法で用いられる前駆体溶液は、金属のアルコキシド等を加水分解、重合させ、コロイド状にしたものを有機溶媒中に分散させた溶液である。MOD法で用いられる前駆体溶液は、加水分解が起きない金属の有機酸塩を有機溶媒中に溶解させた溶液である。また、ゾル−ゲル法あるいはMOD法で用いられる溶液のどちらにも区別されない(金属の有機酸塩、アルコキシドおよび錯体を併用する)前駆体溶液も広く知られている。
【0004】
ゾル−ゲル法によって成膜する場合、所望の金属酸化物の原料となるアルコキシドの混合溶液が用いられる。例えば、特許文献1には酢酸鉛3水和物を3−メトキシブタノールに溶解し、共沸脱水させることで鉛のアルコキシドを合成し、さらにチタンおよびジルコニウムそれぞれのアルコキシドを混合溶解させた金属酸化物前駆体溶液が開示されている。
MOD法によって成膜する場合、所望の金属酸化物の原料となる有機酸塩の混合溶液が用いられる。例えば、特許文献2には、金属酸化物の前駆体溶液として、鉛、チタンおよびジルコニウムそれぞれの有機酸塩の混合溶液が開示されている。
金属の有機酸塩、アルコキシドおよび錯体を混合した圧電体膜の前駆体溶液としては、例えば、特許文献3には、金属酸化物の前駆体溶液として、ストロンチウムおよびビスマスそれぞれの有機酸塩の混合溶液が開示されており、例えば、特許文献4には、金属酸化物の前駆体溶液として、アセチルアセトナート錯体の分散安定性を良好にするために、酢酸を含有した圧電体膜形成用組成物が開示されている。
【0005】
液相法による圧電体膜の製造方法としては、前駆体溶液の調製法(ゾル−ゲル法、MOD法等)に拠らず、アルコールや炭化水素等の溶媒、安定化剤および有機金属化合物を含む前駆体溶液を、シリコン基板等の被対象物上に塗布し、結晶化を目的とした加熱処理を行うことで結晶化した圧電体膜を得る。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開平11−79747号公報
【特許文献2】特開平5−58636号公報
【特許文献3】特開平8−111411号公報
【特許文献4】特開2007−145657号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
前駆体溶液を塗布し、結晶化を目的とした加熱処理を行う1回の工程で成膜できる圧電体膜の膜厚が薄いと、圧電体膜に要求される性能に必要な膜厚を得るために、上述の工程を繰り返す必要がある。上述の工程で得られる圧電体膜の膜厚を厚くするには、前駆体溶液中の有機金属化合物を高濃度化する必要がある。したがって、アルコール、炭化水素等の溶媒と、アセチルアセトンやジエタノールアミン等の安定化剤を含む前駆体溶液の場合、前駆体溶液中の有機金属化合物を高濃度化するためには、溶媒の含有量を減らすか、または、安定化剤の含有量を減らす手段が講じられる。
【0008】
しかしながら、以下に述べる理由で、前駆体溶液中の有機金属化合物の濃度を濃くするのが難しい。
安定化剤の含有比率を減少させると、大気中等の水分により加水分解が進行し、前駆体溶液の保存安定性が低下する場合がある。一方、アルコールに代表される溶媒の含有比率を減少させると沈殿が発生し、均一な膜形成が困難になったり、前駆体溶液の粘度増加によって塗布膜にムラが発生したりする場合がある。
【0009】
安定化剤を必要としない前駆体溶液であっても、以下に述べる理由で、前駆体溶液中の有機金属化合物の濃度を濃くするのが難しい。
有機金属化合物として有機酸塩を用いる場合、配位子の鎖長が増すにつれて溶媒への溶解度が増大するが、含有金属濃度は低下してしまう。よって、得られる圧電体膜の膜厚と前駆体溶液との間に妥協点を見出さなければならない。特許文献3では、得られる強誘電体膜の結晶膜厚は100nm程度である。
【0010】
上述したように、従来技術では、上述の工程で得られる圧電体膜の膜厚を厚くすることが難しく、例えば、1μm以上の膜厚を得るためには上述の工程を繰り返す必要があることから、生産性を向上させることが困難であった。したがって、1回の成膜工程で得られる圧電体膜の膜厚を厚くすることが可能で、かつ、保存安定性のよい圧電体膜の前駆体溶液の開発が望まれていた。
【0011】
本発明は、上記従来技術の問題点に鑑み、課題を解決するためになされたものであり、以下様態として実現することが可能である。
【課題を解決するための手段】
【0012】
[態様1]
上記目的を達成する本発明の態様は、酢酸及び/又はプロピオン酸からなるカルボン酸と、酢酸鉛と、Zr(OR1)4で表されるジルコニウムアルコキシド(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)と、Ti(OR2)4で表されるチタニウムアルコキシド(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)と、高分子化合物と、を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、ジルコニウムアルコキシドの配位子OR1またはチタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液にある。
【0013】
本態様に用いる特定のカルボン酸は、金属アルコキシドの安定化効果を有し、さらに、前駆体溶液中の含有量が増加しても前駆体溶液としての保存安定性の低下、および粘度の増加がみられないために、溶媒として用いることができる。
上記のカルボン酸の含有量は、前駆体溶液の調製に用いる原料全量に対して20質量%以上60質量%以下とすることが好ましい。20質量%以上とすることで、大気中の水分による加水分解を防ぐことができる。一方、60質量%以下とすることで上述の工程で得られる膜の膜厚を厚くすることができる。
【0014】
従って、圧電体膜の前駆体溶液に求められる粘度、金属酸化物換算濃度、耐水性、保存安定性などの性能を満足するように、前記範囲でカルボン酸の添加量を決定することができる。
例えば、圧電体膜の前駆体溶液を用いて、スピンコート法によって薄膜を成膜する場合、圧電体膜の前駆体溶液の粘度は5〜15mPa・s、前駆体溶液に含まれる金属原料の酸化物換算濃度は12〜32%とすることができる。なお、金属原料の酸化物換算濃度は、原料組成から算出できるほか、燃焼法やICP分析などの定法によって前駆体溶液から調べることも可能である。
また、本態様では、高分子化合物を前駆体溶液に添加することによって、クラックの発生を防止することが可能となる。
【0015】
[態様2]
また、前記圧電体膜の前駆体溶液は、前記ジルコニウムアルコキシドの配位子OR1および前記チタニウムアルコキシドの配位子OR2が、ともに、分岐鎖状のアルコキシ基とすることができる。
分岐鎖状のアルコキシ基を有する金属アルコキシドを用いることで、カルボン酸エステルの生成量が抑制されるので、圧電体膜の前駆体溶液の経時変化がなく、得られる圧電体膜の膜厚の変動を抑えることが可能となる。なお、カルボン酸がカルボン酸エステルになると、圧電体膜の前駆体溶液に含有される溶媒の揮発性が変化するため、得られる圧電体膜の膜厚は厚くなる場合が多い。
【0016】
[態様3]
また、前記圧電体膜の前駆体溶液は、溶媒として、さらに、水、2級または3級のアルコール類、エーテル類、エステル類、およびケトン類から選ばれる1種または2種以上を含むことができる。
上述したカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応を抑制することが可能となる。
【0017】
[態様4]
また、前記圧電体膜の前駆体溶液は、前記高分子化合物が、ポリエチレングリコールおよびその誘導体ならびにポリプロピレングリコールおよびその誘導体から選ばれる1種または2種以上であり、さらに、前記高分子化合物の重量平均分子量が300〜800であることが好ましい。
高分子化合物を添加した圧電体膜の前駆体溶液であれば、上述の工程において、膜の収縮に起因する応力を緩和することが可能となる。一方、結晶化膜においては高分子化合物が残存しないことが圧電特性の観点からは望ましい。何故なら、高分子化合物が水や二酸化炭素等に分解・脱離せずに、残留炭素等の形態で膜中に存在すると、空隙の発生、あるいは絶縁性の低下等によって、圧電特性に悪影響を及ぼすためである。したがって、高分子化合物には良好な熱分解性が求められる。炭素−炭素結合と比べて、炭素−酸素結合は結合力が弱いために、熱分解性が良好である。ゆえに、ポリエチレングリコールおよびその誘導体、ならびにポリプロピレングリコールおよびその誘導体は、圧電体膜のクラック防止剤として好適に使用することが可能である。
【0018】
圧電体膜の前駆体溶液に含まれる高分子化合物は、熱分解性の観点に加えて、膜のクラック防止の観点から重量平均分子量を規定することが望ましい。
高分子化合物の平均分子量が小さくなると、低分子のように振舞うために圧電体膜の成膜工程における応力緩和効果が十分に得られなくなる。また、高分子化合物の平均分子量が大きくなると、成膜工程における加熱処理時に十分に熱分解しなくなるために、残留炭素等の形態として膜中に存在し、圧電体膜の絶縁性低下といったような特性の劣化につながる。
高分子化合物の重量平均分子量を300〜800にすれば、十分な応力緩和効果と熱分解性を有するため、圧電体膜の特性を劣化させることなく、圧電体膜のクラック防止を行うことが可能となる。なお、重量平均分子量は、分子量標準物質をポリエチレングリコールとしたゲル浸透クロマトグラフィー(GPC)測定により得られた分子量分布から算出される。
【0019】
[態様5]
さらに、本発明の他の態様としては、前記圧電体膜の前駆体溶液に含まれる金属元素の酸化物換算濃度が、15質量%以上27%質量%以下とすることができる。
金属元素の酸化物換算濃度とは、圧電体膜の前駆体溶液に対する、圧電体膜の前駆体溶液に含まれる各金属元素を酸化物とした質量の総和の割合である。したがって、PZT前駆体溶液の場合、PZT前駆体溶液全量に対するPbO、ZrO2およびTiO2の質量の総和の割合となる。なお、金属酸化物換算濃度は、用いる有機金属化合物から算出することができるほか、重量法やICP分析法等を用いて圧電体膜の前駆体溶液から調べることも可能である。
液相法としてスピンコート法を用いて得られる膜の膜厚は、スピンコート回転数と、金属元素の酸化物換算濃度と、に影響を受けることが知られている。金属元素の酸化物換算濃度を本態様の範囲とすることで、上述の工程で得られる膜の膜厚を厚くすることができる。
【0020】
[態様6]
本発明の他の態様としては、前記圧電体膜の前駆体溶液がさらに、少なくとも1種以上の有機金属化合物を含むことができる。
【0021】
[態様7]
本発明の他の態様は、上記圧電体膜の前駆体溶液を用いた圧電素子の製造方法にある。
本態様によれば、従来の圧電体膜の前駆体溶液を用いた場合よりも、1層あたりの圧電体膜を厚くすることが可能となるため、圧電素子の生産性を向上することが可能となる。
【図面の簡単な説明】
【0022】
【図1】実施形態に係るインクジェット式記録ヘッドの概略構成を示す分解斜視図。
【図2】(a)はインクジェット式記録ヘッドの平面図、(b)は(a)におけるA−A断面図。
【図3】インクジェット式記録ヘッドの図2(a)におけるB−B要部拡大断面図。
【図4】インクジェット式記録ヘッドの製造方法を示す断面図。
【図5】インクジェット式記録ヘッドの製造方法を示す断面図。
【図6】インクジェット式記録ヘッドの製造方法を示す断面図。
【図7】インクジェット式記録ヘッドの製造方法を示す断面図。
【図8】実施例および実施形態の圧電体膜の前駆体溶液に用いた原料を示す図。
【図9】実施例1〜5および比較例1〜4の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図10】実施例1〜5および比較例1〜4の金属酸化物換算濃度、カルボン酸の割合、PZT膜の膜厚、耐水性および保存安定性をまとめて示す図。
【図11】実施例6〜22および比較例5〜7の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図12】実施例6〜22および比較例5〜7の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図13】実施例23〜32の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図14】実施例23〜32の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図15】実施例33〜43の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図16】実施例33〜43の金属酸化物換算濃度、カルボン酸の割合、膜厚、クラック評価、耐水性および保存安定性をまとめて示す図。
【図17】実施例44〜48の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図18】実施例44〜48および比較例8の金属酸化物換算濃度、カルボン酸の割合、スピンコート回転数、膜厚、耐水性および保存安定性をまとめて示す図。
【図19】実施例49および実施例50の溶媒、各金属原料および高分子化合物の秤量結果を示す図。
【図20】実施例49および実施例50の金属酸化物換算濃度、カルボン酸の割合、膜厚の再現性評価(膜厚変動)、耐水性および保存安定性をまとめて示す図。
【図21】一実施形態に係る記録装置の概略構成を示す図。
【発明を実施するための形態】
【0023】
以下、本発明の実施形態を図面に基づき詳細に説明する。
図1は、本発明の実施形態に係る液体噴射ヘッドの一例であるインクジェット式記録ヘッド1の概略構成を示す分解斜視図であり、図2(a)は、図1の平面図、図2(b)は(a)におけるA−A断面図である。
インクジェット式記録ヘッド1は、流路形成基板10、ノズルプレート20および保護基板30を備えている。
【0024】
図1および図2において、流路形成基板10は、実施形態では結晶面方位が(110)であるシリコン単結晶基板からなり、その一方面には熱酸化によって形成した酸化シリコンからなる、厚さ0.5μm〜2.0μmの弾性膜50が形成されている。流路形成基板10には、隔壁11によって区画され一方側の面が弾性膜50で構成される複数の圧力発生室12がその幅方向に並設されている。
【0025】
流路形成基板10には、圧力発生室12の長手方向一端部側に、隔壁11によって区画され各圧力発生室12に連通するインク供給路13と連通路14とが設けられている。連通路14の外側には、各連通路14と連通する連通部15が設けられている。この連通部15は、後述する保護基板30のリザーバー部32と連通して、各圧力発生室12の共通のインク室(液体室)となるリザーバー100の一部を構成する。
【0026】
ここで、インク供給路13は、圧力発生室12よりも狭い断面積となるように形成されており、連通部15から圧力発生室12に流入するインクの流路抵抗を一定に保持している。例えば、インク供給路13は、リザーバー100と各圧力発生室12との間の圧力発生室12側の流路を幅方向に絞ることで、圧力発生室12の幅より小さい幅で形成されている。なお、実施形態では、流路の幅を片側から絞ることでインク供給路を形成したが、流路の幅を両側から絞ることでインク供給路を形成してもよい。また、流路の幅を絞るのではなく、厚さ方向から絞ることでインク供給路を形成してもよい。各連通路14は、圧力発生室12の幅方向両側の隔壁11を連通部15側に延設してインク供給路13と連通部15との間の空間を区画することで形成されている。
【0027】
なお、流路形成基板10の材料として、実施形態ではシリコン単結晶基板を用いているが、勿論これに限定されず、例えば、ガラス、セラミックス、ステンレス鋼等を用いてもよい。
【0028】
流路形成基板10の開口面側には、各圧力発生室12のインク供給路13とは反対側の端部近傍に連通するノズル開口21が穿設されたノズルプレート20が、接着剤や熱溶着フィルム等によって固着されている。なお、ノズルプレート20は、例えば、ガラスセラミックス、シリコン単結晶基板、ステンレス鋼などからなる。
【0029】
一方、このような流路形成基板10の開口面とは反対側には、上述したように弾性膜50が形成され、この弾性膜50上には、厚さ0.05μm〜0.8μmの絶縁体膜55が形成されている。実施形態における絶縁体膜55は酸化ジルコニウム(ZrO2)で形成してある。さらに、この絶縁体膜55上には、厚さが例えば0.2μmの下電極60と、厚さが例えば1.3μmの圧電体層70と、厚さが例えば0.05μmの上電極80とが、後述するプロセスで積層形成されて、圧電素子300が形成されている。ここで、圧電素子300は、下電極60、圧電体層70及び上電極80を含む部分をいう。一般的には、圧電素子300の何れか一方の電極を共通電極とし、他方の電極を圧電体層70と共に圧力発生室12毎にパターニングして個別電極とする。実施形態では下電極60が個別電極、上電極80が共通電極となる、いわゆる共通上電極構造となっている。
【0030】
また、ここでは、圧電素子300と当該圧電素子300の駆動により変位が生じる振動板とを合わせて圧電アクチュエーターと称する。なお、実施形態では、弾性膜50、絶縁体膜55及び下電極60が振動板として作用する。
【0031】
ここで、実施形態に係る圧電素子300の構造について詳しく説明する。図2(a)におけるB−B要部拡大断面図である図3に示すように、圧電素子300を構成する下電極60は、各圧力発生室12に対向する領域毎に、圧力発生室12の幅よりも狭い幅で設けられて各圧電素子300の個別電極を構成している。
また、図1および図2において、下電極60は、各圧力発生室12の長手方向一端部側から周壁上まで延設されている。そして下電極60には、圧力発生室12の外側の領域で、例えば、金(Au)等からなるリード電極90がそれぞれ接続され、このリード電極90を介して各圧電素子300に選択的に電圧が印加されるようになっている。一方、圧力発生室12の長手方向のリード電極90が接続された側とは反対側の下電極60の端部は、圧力発生室12に対向する領域内に位置している。
【0032】
圧電体層70は、下電極60の幅よりも広い幅で且つ圧力発生室12の幅よりも狭い幅で設けられている。圧力発生室12の長手方向においては、圧電体層70の両端部は、圧力発生室12の端部の外側まで延設されている。すなわち、圧電体層70は、圧力発生室12に対向する領域の下電極60の上面及び端面を完全に覆うように設けられている。なお、圧力発生室12の長手方向のリード電極90が接続された側の圧電体層70の端部は、圧力発生室12の端部近傍に位置しており、その外側の領域には下電極60がさらに延設されている。
【0033】
上電極80は、複数の圧力発生室12に対向する領域に連続的に形成されるとともに、圧力発生室12の長手方向のリード電極90が接続された端部側から周壁上まで延設されている。すなわち、上電極80は、圧力発生室12に対向する領域の圧電体層70の上面及び端面のほぼ全域を覆って設けられている。
【0034】
上述の如く圧力発生室12に対向する領域の圧電体層70の上面及び端面のほぼ全域を上電極80で覆うことにより圧電体層70への大気中の水分(湿気)の浸透が実質的に防止される。したがって、水分(湿気)に起因する圧電素子300(圧電体層70)の破壊を防止することができ、圧電素子300の耐久性を著しく向上させることができる。
【0035】
また、圧力発生室12の長手方向のリード電極90が接続された側の上電極80の端部は、圧力発生室12に対向する領域内に位置している。この結果、圧電素子300の実質的な駆動部が圧力発生室12に対向する領域内に設けられている。すなわち、圧力発生室12内に位置する下電極60と上電極80との間の部分の圧電素子300が実質的な駆動部となっている。このため、圧電素子300を駆動しても、圧力発生室12の長手方向両端部近傍の振動板(弾性膜50、絶縁体膜55)には大きな変形が生じることはないため、この部分の振動板に割れが発生するのを防止することができる。なお、このような構成では、圧力発生室12に対向する領域内でも圧電体層70の表面が若干露出されることになるが、その面積は極めて狭く、また後述するように上電極80の周縁部と下電極60の間の距離が大きいため、水分に起因する圧電体層70の破壊を防止することができる。
【0036】
圧電素子300が形成された流路形成基板10上には、圧電素子300に対向する領域にその運動を阻害しない程度の空間を確保可能な圧電素子保持部31を有する保護基板30が接着剤35を介して接合されている。圧電素子300は、この圧電素子保持部31内に形成されているため、外部環境の影響を殆ど受けない状態で保護されている。また、保護基板30には、流路形成基板10の連通部15に対応する領域にリザーバー部32が設けられている。このリザーバー部32は、実施形態では、保護基板30を厚さ方向に貫通して圧力発生室12の並設方向に沿って設けられており、上述したように流路形成基板10の連通部15と連通されて各圧力発生室12の共通のインク室となるリザーバー100を構成している。
【0037】
さらに、保護基板30の圧電素子保持部31とリザーバー部32との間の領域には、保護基板30を厚さ方向に貫通する貫通孔33が設けられ、リード電極90の端部がこの貫通孔33内に露出されている。そして、図示しないが、これら下電極60及びリード電極90は、貫通孔33内に延設される接続配線によって圧電素子300を駆動するための駆動IC等に接続される。
【0038】
なお、保護基板30の材料としては、例えば、ガラス、セラミックス材料、金属、樹脂等が挙げられるが、流路形成基板10の熱膨張率と略同一の材料で形成されていることがより好ましく、実施形態では、流路形成基板10と同一材料のシリコン単結晶基板を用いて形成した。
【0039】
保護基板30上には、さらに、封止膜41及び固定板42とからなるコンプライアンス基板40が接合されている。封止膜41は、剛性が低く可撓性を有する材料からなり、この封止膜41によってリザーバー部32の一方面が封止されている。固定板42は、金属等の硬質の材料で形成される。この固定板42のリザーバー100に対向する領域は、厚さ方向に完全に除去された開口部43となっているため、リザーバー100の一方面は可撓性を有する封止膜41のみで封止されている。
【0040】
このような実施形態のインクジェット式記録ヘッド1では、図示しない外部インク供給手段からインクを取り込み、リザーバー100からノズル開口21に至るまで内部をインクで満たした後、図示しない駆動ICからの記録信号に従い、圧力発生室12に対応するそれぞれの圧電素子300に電圧を印加し、圧電素子300をたわみ変形させることにより、各圧力発生室12内の圧力が高まりノズル開口21からインク滴が吐出する。
【0041】
以下、本発明の実施形態に係る圧電素子の製造方法を、図1〜図3に示したインクジェット式記録ヘッド1の製造方法とともに、図4〜図7を参照して説明する。
【0042】
図4は、圧電素子の製造方法を表す、図2(a)におけるB−B線に沿う方向から見た断面図である。
まず、図4(a)に示すように、シリコンウェハーであり流路形成基板10が複数一体的に形成される流路形成基板用ウェハー110の表面に弾性膜50を構成する酸化膜51を形成する。
【0043】
そして、図4(b)に示すように、弾性膜50(酸化膜51)上に、絶縁体膜55を形成する。実施形態では酸化ジルコニウム(ZrO2)を形成した。
【0044】
次いで、図4(c)に示すように、絶縁体膜55上に下電極60を形成する。
この下電極60の材料は、特に限定されないが、圧電体層70としてチタン酸ジルコン酸鉛(PZT)を用いる場合には、酸化鉛の拡散による導電性の変化が少ない材料であることが望ましい。圧電体層70を液相プロセスによって形成する場合には、熱処理による下電極60の酸化が起こり難いことが望ましい。このため、下電極60の材料としては白金、イリジウム等が単体あるいは積層体として好適に用いられる。
また、下電極60は、例えば、スパッタリング法やPVD法(物理蒸着法)などにより形成することができる。なお、下電極60の成膜に先立ち、チタン、ジルコニウム又はクロムからなる、厚さが例えば0.005μm〜0.05μmである密着層(図示せず)をスパッタ法又は真空蒸着法により成膜しても良い。また、下電極60の成膜後、連続して下電極60上にチタン層(図示せず)を形成してもよい。例えばスパッタ法等により、チタン層を0.003μm〜0.02μmの厚みに成膜することで、所定の結晶配向度を備えた圧電体層70を得ることが可能である。チタン層は下電極60上に均一に成膜するが、場合によって層状ではない島状となっても構わない。
【0045】
次に、チタン酸ジルコン酸鉛(PZT)からなる圧電体としての圧電体層70を形成する。ここで、実施形態では、液相法を用いて圧電体層70を形成している。
以下に、圧電体膜を含む圧電体層70の製造方法について説明する。圧電体層70の製造方法は、調製工程と塗布工程と乾燥工程と脱脂工程と焼成工程とを含む。
【0046】
調製工程では、溶媒として特定のカルボン酸と、鉛、ジルコンおよびチタンの各有機金属化合物と、高分子化合物とを混合、撹拌することで、液相法に用いる圧電体膜の前駆体溶液を調製する。
混合は、いわゆる1ポット調製であってもよく、または複数に分けて行ってもよい。例えば、1ポット調製としては、カルボン酸に、ジルコニウムアルコキシドおよびチタニウムアルコキシドを混合し、次いで酢酸鉛を加え、最後に高分子化合物を混合することで圧電体膜の前駆体溶液を得る。複数に分ける場合は、例えば、カルボン酸と酢酸鉛を混合することで得られる第1の溶液と、カルボン酸とジルコニウムアルコキシドを混合することで得られる第2の溶液と、カルボン酸とチタニウムアルコキシドを混合することで得られる第3の溶液を、個別に調製し、3種類の溶液と高分子化合物とを混合することで最終的に圧電体膜の前駆体溶液を得る。
混合は、25℃程度の室温で行ってもよいし、加熱して行ってもよい。例えば、酢酸鉛としての酢酸鉛(II)三水和物は固体であるため、混合の際に加熱することで、溶解を促すことが可能である。また、鉛、ジルコンおよびチタン原料の混合モル比は、得られる圧電体層70の圧電特性に影響を及ぼすため、例えば、Pb:Zr:Ti=1.05〜1.25:0.46〜0.56:0.44〜0.54(モル比)となるように設定することが好ましい。
【0047】
鉛の有機金属化合物としては、一般に三水和物である酢酸鉛(II)または酢酸鉛(IV)を用いることが可能であり、取り扱いの容易性から酢酸鉛(II)を用いることが好ましい。
【0048】
ジルコンの有機金属化合物としては、アルコキシドが好ましい。ジルコニウムアルコキシドとしては、配位子として炭素数が3〜8のアルコキシ基を有するものであればよく、例えば、ジルコニウムテトラ−n−プロポキシド、ジルコニウムテトライソプロポキシド、ジルコニウムテトラ−n−ブトキシド、ジルコニウムテトラ−iso−ブトキシド、ジルコニウムテトラ−sec−ブトキシド、ジルコニウムテトラ−tert−ブトキシド、ジルコニウムテトラ−n−ペントキシド、ジルコニウムテトラ−sec−ペントキシド、ジルコニウムテトラ−tert−ペントキシド、ジルコニウムテトラ−n−ヘキソキシド、ジルコニウムテトラ−sec−ヘキソキシド、ジルコニウムテトラ−n−ヘプトキシド、ジルコニウムテトラ−sec−ヘプトキシド、ジルコニウムテトラ−n−オクトキシドおよびテトラキス(2−エチルヘキシルオキシ)オルトジルコネート等が挙げられ、単体あるいは混合体として用いることが可能である。炭素数3以上とすることで、カルボン酸エステルの生成を抑制することが可能であり、炭素数8以下とすることで、圧電体膜の前駆体溶液の金属酸化物換算濃度を高くすることが可能となる。
【0049】
チタンの有機金属化合物としては、アルコキシドが好ましい。チタニウムアルコキシドとしては、配位子として炭素数が3〜8のアルコキシ基を有するものであればよく、例えば、チタニウムテトラ−n−プロポキシド、チタニウムテトライソプロポキシド、チタニウムテトラ−n−ブトキシド、チタニウムテトラ−iso−ブトキシド、チタニウムテトラ−sec−ブトキシド、チタニウムテトラ−tert−ブトキシド、チタニウムテトラ−n−ペントキシド、チタニウムテトラ−sec−ペントキシド、チタニウムテトラ−tert−ペントキシド、チタニウムテトラ−n−ヘキソキシド、チタニウムテトラ−sec−ヘキソキシド、チタニウムテトラ−n−ヘプトキシド、チタニウムテトラ−sec−ヘプトキシド、チタニウムテトラ−n−オクトキシドおよびチタニウムテトラキス(2−エチルヘキシルオキシド)等が挙げられ、単体あるいは混合体として用いることが可能である。炭素数3以上とすることで、カルボン酸エステルの生成を抑制することが可能であり、炭素数8以下とすることで、圧電体膜の前駆体溶液の金属酸化物換算濃度を高くすることが可能となる。
【0050】
本発明の圧電体膜の前駆体溶液では、上記のジルコニウムアルコキシド又はチタニウムアルコキシドの配位子のうち、少なくとも一方の配位子が分岐鎖状であることが好ましい。これにより、圧電体膜の前駆体溶液におけるカルボン酸エステルの生成量を抑制することができるので、得られる圧電体膜の膜厚の変動を抑えることが可能となる。
【0051】
カルボン酸溶媒に金属アルコキシドを溶解させた場合、配位子交換反応が起こり、結果として、溶液中にはカルボン酸と遊離したアルコールが存在する。そして、下記式(1)で表されるカルボン酸(A−COOH)とアルコール(B−OH)とによるカルボン酸エステル(A−COO−B)の生成反応が起こる。
A−COOH+B−OH ⇔ A−COO−B+H2O ・・・(1)
カルボン酸エステルは、原料であるカルボン酸、又はアルコールと比べて、揮発性が高くなる場合が多い。したがって、圧電体膜の前駆体溶液に遊離アルコールが存在し、カルボン酸エステルの生成反応が経時的に進行する場合、成膜工程によって得られる膜厚が変動してしまう。
【0052】
カルボン酸とアルコールによるカルボン酸エステルの生成反応において、アルコールの炭素数が多くなるに従い、カルボン酸エステルの生成量が減少する。また、炭素数が同一であれば、直鎖状のアルコールのよりも、分岐鎖状のアルコールのほうがカルボン酸エステルの生成量が減少する。したがって、炭素数3〜8のアルコールを配位子として有するチタニウムアルコキシド及びジルコニウムアルコキシドを用い、かつ、チタンニウムアルコキシド又はジルコニウムアルコキシドの少なくとも一方の前記配位子を分岐鎖状とすることが好ましい。
【0053】
また、本発明の圧電体膜の前駆体溶液に含まれる溶媒として、特定のカルボン酸に加えて、さらに、水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類から選ばれる1種または2種以上を含むことができる。上述したカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応を抑制することが可能となる。
【0054】
2級または3級アルコールとしては、特に限定はされないが、イソプロピルアルコール、iso−ブチルアルコール、sec−ブチルアルコール、tert−ブチルアルコールおよびiso−ペンチルアルコール等が挙げられる。エーテル類としては、特に限定はされないが、ジイソプロピルエーテル、ジ−n−ブチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテルおよびエチレングリコールジ−n−ブチルエーテル等が挙げられる。
エステル類としては、特に限定はされないが、酢酸メチル、酢酸エチル、酢酸イソプロピル、酢酸n−ブチル、プロピオン酸エチル、n−酪酸エチルおよびエチレングリコールジアセテート等が挙げられる。さらに、エーテル類でありエステル類でもあるエチレングリコールモノメチルエーテルアセテート、エチレングリコールモノエチルエーテルアセテートおよびプロピレングリコールモノメチルエーテルアセテート等も用いることが可能である。
ケトン類としては、特に限定はされないが、例えば、ジメチルケトン(アセトン)、ジエチルケトン、ジn−プロピルケトン、ジiso−プロピルケトン、メチルエチルケトン、メチルn−プロピルケトン、メチルiso−プロピルケトン、メチルn−ブチルケトン、メチルiso−ブチルケトン、メチルtert−ブチルケトン、メチルiso−ペンチルケトン、エチルn−プロピルケトン、エチルiso−プロピルケトンおよびエチルn−ブチルケトン等が挙げられる。
【0055】
圧電体層70の具体的な作成手順を説明する。
まず、塗布工程において、図5(d)に示すように、下電極60上にPZT前駆体膜である圧電体前駆体膜71を成膜する。すなわち、下電極60が形成された流路形成基板用ウェハー110上に圧電体膜の前駆体溶液を塗布する。
圧電体膜の前駆体溶液の塗布方法としては、スピンコート法、スプレーコート法およびディップコート法等が挙げられる。実施形態では、半導体プロセスに用いられる種々の装置を利用するために、基板片面に塗布が可能で、基板面内の膜厚均一性が良好なスピンコート法を用いるのが好ましい。スピンコートの塗布条件(回転数、時間等)は、用いる圧電体膜の前駆体溶液によって適宜設定することが可能であるが、基板面内における膜厚均一性の確保や、塗布面の反対側への前駆体溶液の回りこみ防止といった観点から回転数を500〜5000rpm程度に設定するのが好ましい。また、スピンコート時にリンス液を用いて、基板外周部および塗布面の反対側を洗浄することが可能であり、これらはエッジリンスおよびバックリンスと呼称される。
【0056】
次いで、乾燥工程において、この圧電体前駆体膜71を所定温度に加熱して一定時間乾燥させる。例えば、流路形成基板用ウェハー110上に塗布されたゾルを140〜180℃で5〜30分保持することで乾燥する。乾燥温度としては、前駆体溶液の溶媒が蒸発するように設定することが好ましく、例えば、酢酸を溶媒として用いた場合には、140℃にすることが可能である。
【0057】
次に、脱脂工程において、乾燥した圧電体前駆体膜71を所定温度に加熱して一定時間保持することによって脱脂する。実施形態では、乾燥させた圧電体前駆体膜71を350〜500℃に加熱して5〜30分保持することで脱脂する。
なお、ここで言う脱脂とは、加熱することによって圧電体前駆体膜71に含まれる有機成分を、例えば、CO2、H2O、アルコール、炭化水素等として離脱させることである。したがって、脱脂温度は圧電体前駆体膜71に含まれる有機成分の状態によって設定するべきであり、脱脂温度を設定する指標としては熱分析(TG/DTA測定、DSC測定等)結果を利用することが可能である。熱分析によって、前駆体溶液の脱脂に伴う発熱反応、あるいは重量変化が起きる温度を測定することが可能である。
また、脱脂とは、PZTを圧電体膜として利用する場合、強誘電性を有さないパイロクロア相が形成されることを防ぐために、圧電体前駆体膜71が結晶化しない程度に、すなわち、非晶質の圧電体前駆体膜71を形成することを言う。圧電体前駆体膜71に含まれる有機成分の蒸発あるいは熱酸化分解による除去が目的であるため、酸素を含む雰囲気であればよく、大気雰囲気中で行うことが可能である。
【0058】
次に、焼成工程において、図5(e)に示すように、圧電体前駆体膜71を所定温度に加熱して一定時間保持することにより結晶化させ、圧電体膜72を形成する。かかる焼成工程においては、電気炉またはRTA(Rapid thermal Aannealing)装置等を利用することが可能である。PZTを圧電体膜72として利用する場合、強誘電性を有するペロブスカイト相を単相で得るために、脱脂した圧電体前駆体膜71を650〜800℃で焼成して結晶化させることが好ましい。また、PZTの酸素欠損を防ぐために、酸素雰囲気あるいは酸素フロー中で焼成することが好ましい。
【0059】
次に、図6(f)に示すように、下電極60上に1層目の圧電体膜72を形成した段階で、下電極60及び1層目の圧電体膜72をそれらの側面が傾斜するように同時にパターニングする。
【0060】
ここで、例えば、下電極60をパターニングした後、1層目の圧電体膜72を形成する場合、フォトリソグラフィー工程・エッチング工程・アッシング工程を行い、下電極60をパターニングするため、下電極60の表面や、所定の結晶配向度を有する圧電体膜を得るために下電極60の表面に設けたチタン層(図示せず)などが変質してしまう。そうすると、変質した下電極60上に圧電体膜72を形成しても当該圧電体膜72の結晶性または結晶配向度が良好なものではなくなり、2層目以降の圧電体膜72も1層目の圧電体膜72の結晶状態に影響して結晶成長するため、良好な結晶性を有する圧電体層70を形成することができない。
【0061】
これに対し実施形態の如く、1層目の圧電体膜72を形成した後に下電極60と同時に圧電体膜72をパターニングすれば、2層目以降の圧電体膜72を良好に結晶成長させることが可能である。何故なら、結晶成長を促すシード層としては、チタン等の結晶種よりも、圧電体膜72そのものを利用したほうがより好ましいからである。また、シード層としての性能が高い圧電体膜72であれば、たとえパターニングで表層に極薄い変質層が形成されていても2層目以降の圧電体膜72の結晶成長に大きな影響を与えない。
【0062】
そして、上述した前駆体膜形成工程(塗布工程、乾燥工程及び脱脂工程)と、焼成工程とを有する圧電体膜形成工程を2回以上繰り返すことで、図6(g)に示すように、2層目以降、最上層までの圧電体膜72を積層する。その後、図7(h)に示すように、2層目以降が積層された圧電体膜72をパターニングして圧電体層70を形成する。
【0063】
次いで、図7(i)に示すように、流路形成基板用ウェハー110の全面に亘って上電極80を形成し、所定形状にパターニングする。具体的には、図2に示すように、上電極80は、圧力発生室12の並設方向に亘って連続的に設け、且つ圧力発生室12の長手方向端部よりも外側まで延設するようにする。
【0064】
さらに、図示はしないが、流路形成基板用ウェハー110の全面に亘って、例えば、金(Au)等からなるリード電極90を形成後、圧電素子300毎にパターニングする。
【0065】
次いで、流路形成基板用ウェハー110の圧電素子300側に、シリコンウェハーであり複数の保護基板30となる保護基板用ウェハーを接着剤35によって接合する。なお、保護基板30には、リザーバー部32、圧電素子保持部31等が予め形成されている。また、保護基板30は、例えば、400μm程度の厚さを有するシリコン単結晶基板からなり、保護基板30を接合することで流路形成基板10の剛性は著しく向上することになる。その後、流路形成基板用ウェハー110を所定の厚さにする。
【0066】
次いで、流路形成基板用ウェハー110にマスク膜を新たに形成し、所定形状にパターニングする。そして、流路形成基板用ウェハー110を、マスク膜を介してKOH等のアルカリ溶液を用いた異方性エッチング(ウェットエッチング)することにより、圧電素子300に対応する圧力発生室12、インク供給路13、連通路14及び連通部15等を形成する。
【0067】
その後は、図2に示すように、保護基板用ウェハー上に図示しない駆動回路を実装すると共に、駆動回路とリード電極90とを接続配線によって接続する。そして、流路形成基板用ウェハー110の圧力発生室12が開口する面側のマスク膜を除去し、流路形成基板用ウェハー110及び保護基板用ウェハーの外周縁部の不要部分を、例えば、ダイシング等により切断することによって除去する。そして、流路形成基板用ウェハー110の保護基板用ウェハーとは反対側の面にノズル開口21が穿設されたノズルプレート20を接合すると共に、保護基板用ウェハーにコンプライアンス基板40を接合し、これら流路形成基板用ウェハー110等を、図1に示すような一つのチップサイズの流路形成基板10等に分割することによって上述した構造のインクジェット式記録ヘッド1を製造する。
【0068】
以下に、実施例に基づいてより詳細に説明する。実施例1〜実施例47では、圧電体膜としてPZT膜を前駆体溶液としてPZT前駆体溶液を例として説明する。
各実施例および各比較例は、圧電体膜の前駆体溶液が異なるが、塗布工程、乾燥工程、脱脂工程および焼成工程は同じ条件で行った。最初に圧電体膜の前駆体溶液の調製工程を実施例および比較例について説明し、同じ工程については、後に説明する。また、各実施例および各比較例の圧電体膜の前駆体溶液に用いた原料を図8に示した。
【0069】
(調製工程)
[実施例1]
まず、200mLのガラスフラスコに、溶媒として酢酸43.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0質量%)14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、この混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物としてポリエチレングリコール(平均分子量600)6.8gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、最終的なPZT前駆体溶液とした。
本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例のPZT前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ジルコニウムZrO2、および酸化チタンTiO2として金属酸化物換算濃度を算出すると、22.4質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、43.2質量%であった。
【0070】
[実施例2]
溶媒として酢酸50.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、46.9質量%であった。
【0071】
[実施例3]
溶媒としてプロピオン酸37.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、39.6質量%であった。
【0072】
[実施例4]
溶媒としてプロピオン酸40.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、41.4質量%であった。
【0073】
[実施例5]
溶媒として酢酸20.0gとプロピオン酸20.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、41.4質量%であった。
【0074】
[比較例1]
溶媒としてアセチルアセトン30.0gと2−n−ブトキシエタノール10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0075】
[比較例2]
溶媒として2−n−ブトキシエタノール69.0gとジエタノールアミン15.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、15.9質量%であった。
【0076】
[比較例3]
溶媒として2−n−ブトキシエタノール33.0gとジエタノールアミン7.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0077】
[比較例4]
溶媒として2−n−ブトキシエタノール25.0gとジエタノールアミン15.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図9に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、23.1質量%であった。
【0078】
(塗布工程)
塗布には、図4(c)で示される流路形成基板用ウェハー110を用いた。具体的には、直径が150mmであり、厚さが700μmであるシリコン単結晶基板上に、熱酸化によって膜厚が1.1μmのSiO2を弾性膜50として形成し、次いで、スパッタ法によって膜厚が0.4μmのZrO2を絶縁体膜55として形成し、さらに、スパッタ法により膜厚が130nmのPtと膜厚が20nmのIrとの積層体を下電極60として形成した。
また、下電極60の成膜に先立って、膜厚が10nmのチタン層を密着層として形成し、下電極60の成膜後に、膜厚が5nmであるチタン層を形成した。実施例1〜5および比較例1〜4のPZT前駆体溶液を用いて、25℃、40%RHの環境でスピンコートを行った。スピンコートの条件として、滴下するPZT前駆体溶液を4mLとし、回転数を2000rpmとし、回転時間を60秒間として、上記の基板に塗布を行った。なお、塗布は、各PZT溶液の調製からそれぞれ1日後に行った。
(乾燥工程)
塗布工程の後に、乾燥工程として、140℃のホットプレートを用いてで5分間の熱処理を行った。
(脱脂工程)
乾燥工程の後に、脱脂工程として、400℃のホットプレートで5分間の熱処理を行った。
(焼成工程)
脱脂工程の後に、焼成工程として、RTA装置を用いて酸素フローを行いながら、700℃で5分間の熱処理を行った。
【0079】
得られたPZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。各実施例および各比較例のPZT前駆体溶液の金属酸化物濃度および原料全量に対する原料として用いたカルボン酸の割合と、各実施例および各比較例のPZT前駆体溶液を用いて得られたPZT膜の膜厚とをまとめて図10に示した。
【0080】
(耐水性の評価)
実施例1〜5および比較例1〜4のPZT前駆体溶液について、耐水性試験を行った。ここで言う耐水性試験とは、各PZT前駆体溶液にイオン交換水を添加・揺動した際に、沈殿が発生するか否かで判定した。具体的には、容量が2.0mLのキャップ付ガラス瓶にPZT前駆体溶液1.0gを入れ、次いで、イオン交換水0.5gを添加・揺動して行った。沈殿の発生がなかった場合を○とし、沈殿が発生した場合を×として、各PZT前駆体溶液の耐水性を評価した。得られた評価結果をまとめて図10に示した。
【0081】
(保存安定性の評価)
実施例1〜5および比較例1〜4のPZT前駆体溶液について、保存安定性試験を行った。ここで言う保存安定性試験とは、各PZT前駆体溶液を室温にて保存した際に、沈殿が発生するか否かで判定した。具体的には、容量が100mLのキャップ付ガラス瓶にPZT前駆体溶液70gを入れ、遮光された薬品庫にて1ヶ月間、密栓保管した。なお、薬品庫内部の温度は25℃程度(室温)であった。沈殿の発生がなかった場合を○とし、沈殿が発生した場合を×として、各PZT前駆体溶液の保存安定性を評価した。得られた評価結果をまとめて図10に示した。
【0082】
図10に示した結果より、実施例1〜5のPZT前駆体溶液を用いた場合、結晶化後の膜厚が200nm以上のPZT膜を得ることが可能であり、さらに、PZT前駆体溶液の耐水性および保存安定性も良好であることが明らかである。
これに対して、比較例1のPZT前駆体溶液は、調製1日後ですでに沈殿が発生したため、塗布することができなかった。これは、アセチルアセトンの添加量が多すぎたために、溶液としての安定性の低下が起きたものである。
比較例2のPZT前駆体溶液を用いた場合では、PZT前駆体溶液の耐水性および保存安定性は良好であったが、得られたPZT膜の膜厚は90nmであった。
比較例3のPZT前駆体溶液を用いた場合では、比較的厚いPZT膜が得られたが、PZT前駆体溶液の耐水性試験結果が×となった(沈殿が発生した)。これは、ジエタノールアミンの添加量が少ないために、十分な安定化効果が得られなかったためである。
比較例4のPZT前駆体溶液を用いた場合は、PZT前駆体溶液の耐水性および保存安定性は良好であったものの、得られるPZT膜の膜ムラがひどく、評価が出来なかった。これは、ジエタノールアミンの粘度が非常に高いために、2−n−ブトキシエタノールの添加量が少なくなると、PZT前駆体溶液の粘度も高くなってしまい、基板全体に塗布膜を形成することが出来なくなったためである。
【0083】
(調製工程)
[実施例6]
まず、200mLのガラスフラスコに、溶媒として酢酸48.7gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−プロポキシド10.3gと、チタニウム原料としてチタニウムテトラ−iso−イソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物として重量平均分子量600のポリエチレングリコール6.8gと、追加溶媒としてイオン交換水5.0gを加え、25℃の室温にて1時間の撹拌を行い、これを最終的なPZT前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0084】
[実施例7]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−iso−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0085】
[実施例8]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−sec−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0086】
[実施例9]
溶媒として酢酸47.1gを用い、チタニウム原料としてチタニウムテトラ−tert−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.2質量%であった。
【0087】
[実施例10]
溶媒としてプロピオン酸41.7gを用い、チタニウム原料としてチタニウムテトラキス(2−エチルヘキシルオキシド)16.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、39.2質量%であった。
【0088】
[実施例11]
溶媒として酢酸47.1gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.2質量%であった。
【0089】
[実施例12]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0090】
[実施例13]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−sec−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0091】
[実施例14]
溶媒として酢酸46.9gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−n−プロポキシド8.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、49.2質量%であった。
【0092】
[実施例15]
溶媒としてプロピオン酸43.3gを用い、ジルコニウム原料としてジルコニウムテトラキス(2−エチルヘキシルオキシド)19.1gを用い、チタニウム原料としてチタニウムテトラ−n−ブトキシド10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、40.6質量%であった。
【0093】
[実施例16]
溶媒として酢酸48.7gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0094】
[実施例17]
溶媒としてプロピオン酸50.3gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−tert−ブトキシド10.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、47.2質量%であった。
【0095】
[実施例18]
溶媒として酢酸45.2gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ペントキシド13.8gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.4質量%であった。
【0096】
[実施例19]
溶媒としてプロピオン酸44.9gを用い、ジルコニウム原料としてジルコニウムテトラキス(2−エチルヘキシルオキシド)19.1gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.2質量%であった。
【0097】
[実施例20]
溶媒としてプロピオン酸45.5gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド10.3gを用い、チタニウム原料としてチタニウムテトラキス(2−エチルヘキシルオキシド)16.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.7質量%であった。
【0098】
[実施例21]
溶媒として酢酸47.0gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−ブトキシド12.1gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、44.1質量%であった。
【0099】
[実施例22]
溶媒として酢酸45.3gを用い、ジルコニウム原料としてジルコニウムテトラ−tert−ブトキシド12.1gを用い、チタニウム原料としてチタニウムテトラ−iso−プロポキシド10.0gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、42.5質量%であった。
【0100】
[比較例5]
溶媒としてプロピオン酸60.6gを用い、ジルコニウム原料としてジルコニウムテトラメトキシド6.8gを用い、チタニウム原料としてチタニウムテトラメトキシド5.1gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、56.9質量%であった。
【0101】
[比較例6]
溶媒として酢酸52.2gを用い、ジルコニウム原料としてジルコニウムテトラエトキシド8.5gを用い、チタニウム原料としてチタニウムテトラエトキシド6.7gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、49.0質量%であった。
【0102】
[比較例7]
溶媒として酢酸48.7gを用い、チタニウム原料としてチタニウムテトラ−n−プロポキシド8.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本比較例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図11に示した。本比較例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、45.8質量%であった。
【0103】
(成膜工程)
実施例6〜22および比較例5〜7のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後および30日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0104】
(再現性評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液を用いて得られた各PZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後のPZT前駆体溶液を用いたPZT膜の膜厚を100とし、調製して30日後のPZT前駆体溶液を用いたPZT膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例6〜22および比較例5〜7の評価結果を図12に示した。
A:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が100以上110未満。
B:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が110以上120未満。
C:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が120以上。
【0105】
(耐水性の評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図12に示した。
【0106】
(保存安定性の評価)
実施例6〜22および比較例5〜7のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図12に示した。
【0107】
図12に示した結果から明らかなように、ジルコニウムアルコキシドの配位子およびチタニウムアルコキシドの配位子が、ともに、炭素数が1〜3であり、かつ、直鎖である場合は、膜厚の再現性評価結果がいずれもCであった(比較例5〜7)。
比較例5〜7に対し、実施例6〜15では、膜厚の再現性評価結果はいずれもBであった。これは、ジルコニウムアルコキシドの配位子またはチタニウムアルコキシドの配位子のいずれかを分岐鎖としたことによって、カルボン酸エステルの生成が抑制されたためである。実施例16〜22では、膜厚の再現性評価結果はいずれもAであった。これは、ジルコニウムアルコキシドの配位子およびチタニウムアルコキシドの配位子をともに分岐鎖としたことによって、実施例6〜15よりもさらに、カルボン酸エステルの生成が抑制されたためである。
【0108】
なお、PZT前駆体溶液中に存在するアルコールおよびカルボン酸エステルの簡易的な定量手段として、NMR測定(1Hおよび13C)を行った。実施例6〜22および比較例5〜7の各PZT前駆体溶液のいずれも、調製1日後から調製30日後にかけて、カルボン酸エステルが経時的に生成していることが分かっており、さらに、膜厚の再現性はエステル変化率と相関があることが分かっている。
例えば、比較例5において、PZT前駆体溶液中の酢酸メチルとメタノールのピークからエステル変化率を算出した結果、1日後では36mol%、30日後では81mol%であった。ここで、エステル変化率は、100×[カルボン酸エステル]/([カルボン酸エステル]+[アルコール])として算出した。
実施例として、例えば、実施例16において、エステル変化率を算出すると、1日後では6mol%、30日後では10mol%であった。実施例6において、エステル変化率を算出すると、1日後では27mol%、30日後では48mol%であった。
【0109】
(調製工程)
[実施例23]
溶媒として酢酸23.5gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水23.5gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0110】
[実施例24]
溶媒としてプロピオン酸23.5gとイソプロピルアルコール23.5gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0111】
[実施例25]
溶媒として酢酸28.2gとtert−ブタノール18.8gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、26.4質量%であった。
【0112】
[実施例26]
溶媒としてプロピオン酸37.6gとジ−n−ブチルエーテル9.4gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0113】
[実施例27]
溶媒として酢酸32.9gと酢酸n−ブチル14.1gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.8質量%であった。
【0114】
[実施例28]
溶媒としてプロピオン酸32.9gとエチレングリコールモノメチルエーテルアセタート14.1gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.8質量%であった。
【0115】
[実施例29]
溶媒として酢酸23.5gとイソプロピルアルコール9.4gとを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水14.1gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、22.0質量%であった。
【0116】
[実施例30]
溶媒として酢酸28.2gと酢酸n−ブチル9.4gとを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用い、追加溶媒としてイオン交換水9.4gを用いたほかは、実施例6と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、26.4質量%であった。
【0117】
[実施例31]
溶媒としてプロピオン酸37.6gとtert−ブタノール4.7gとエチレングリコールモノメチルエーテルアセタート4.7gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0118】
[実施例32]
溶媒としてプロピオン酸37.6gとメチルエチルケトン9.4gとを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール10.3gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図13に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、35.2質量%であった。
【0119】
(成膜工程)
実施例23〜32のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後および30日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0120】
(再現性評価)
実施例23〜32のPZT前駆体溶液を用いて得られた各PZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後のPZT前駆体溶液を用いたPZT膜の膜厚を100とし、調製して30日後のPZT前駆体溶液を用いたPZT膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例23〜32の評価結果を図14に示した。
A:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が100以上110未満。
B:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が110以上120未満。
C:調製して30日後のPZT前駆体溶液を用いて得られたPZT膜の膜厚が120以上。
【0121】
(耐水性の評価)
実施例23〜32のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図14に示した。
【0122】
(保存安定性の評価)
実施例23〜32のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図14に示した。
【0123】
図14に示した結果から明らかなように、実施例23〜32のPZT前駆体溶液は、膜厚の再現性評価結果がいずれもAであった。これはカルボン酸エステルの原料となるカルボン酸を水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類に置き換えることで、カルボン酸エステルの生成反応が抑制されたためである。
【0124】
(調製工程)
[実施例33]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の撹拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、高分子化合物として重量平均分子量300であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的なPZT前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0125】
[実施例34]
高分子化合物として重量平均分子量300であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0126】
[実施例35]
高分子化合物として重量平均分子量400であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0127】
[実施例36]
高分子化合物として重量平均分子量400であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0128】
[実施例37]
高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0129】
[実施例38]
高分子化合物として重量平均分子量600であるポリエチレングリコール20.5gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、18.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、33.3質量%であった。
【0130】
[実施例39]
高分子化合物として重量平均分子量550であるポリエチレングリコールモノメチルエーテル6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0131】
[実施例40]
高分子化合物として重量平均分子量750であるポリエチレングリコールモノメチルエーテル6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0132】
[実施例41]
高分子化合物として重量平均分子量400であるポリプロピレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0133】
[実施例42]
高分子化合物として重量平均分子量700であるポリプロピレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0134】
[実施例43]
高分子化合物として重量平均分子量が400であるポリエチレングリコール1.7gと重量平均分子量が600であるポリプロピレングリコール1.7gと重量平均分子量が550であるポリエチレングリコールモノメチルエーテル1.7gと重量平均分子量が700であるポリプロピレングリコールとを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図15に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0135】
(成膜工程)
実施例33〜43のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後の各PZT前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を6回繰り返し行い、6層のPZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0136】
(クラック評価)
実施例33〜43のPZT前駆体溶液を用いて得られた各PZT膜について、金属顕微鏡にてクラック評価を行った。クラックが発生していない場合を○とし、クラックが発生したものを×として判定した。また、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。得られた結果をまとめて図16に示した。
【0137】
(耐水性の評価)
上述の実施例33〜43のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図16に示した。
【0138】
(保存安定性の評価)
実施例33〜43のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図16に示した。
【0139】
図16に示した結果から明らかなように、実施例33〜43の全てのPZT膜について、クラックが発生することなく、1μmを超えるPZT膜を得ることが可能であった。さらに、耐水性および保存安定性の評価結果についても良好な結果を示した。
【0140】
(調製工程)
[実施例44]
溶媒として酢酸50.0gを用い、追加溶媒としてイオン交換水23.0gを用い、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、17.2質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、38.6質量%であった。
【0141】
[実施例45]
溶媒として酢酸32.6gとイソプロピルアルコール14.4gとを用い、鉛原料として酢酸鉛(II)三水和物21.8gを用い、ジルコニウム原料としてジルコニウムテトラ−iso−プロポキシド8.2gを用い、チタニウム原料としてチタニウムテトライソプロポキシド6.7gを用い、高分子化合物として重量平均分子量が400であるポリエチレングリコール16.4gを用い、追加溶媒としてイオン交換水14.4gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、15.6質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、28.4質量%であった。
【0142】
[実施例46]
溶媒として酢酸56.5gを用い、鉛原料として酢酸鉛(II)三水和物21.8gを用い、ジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液11.2gを用い、チタニウム原料としてチタニウムテトライソプロポキシド6.7gを用い、高分子化合物として重量平均分子量が600であるポリエチレングリコール5.4gを用い、追加溶媒としてイオン交換水6.3gを用いたほかは、実施例33と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、16.5質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、52.4質量%であった。
【0143】
[実施例47]
溶媒としてプロピオン酸30.0gと酢酸n−ブチル10.0gとメチルエチルケトン6.0gを用い、高分子化合物として重量平均分子量が400であるポリエチレングリコール13.7gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、20.4質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、27.4質量%であった。
【0144】
[実施例48]
溶媒としてプロピオン酸25.0gを用いたほかは、実施例1と同様にしてPZT前駆体溶液を得た。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図17に示した。本実施例について、実施例1と同様にして金属酸化物換算濃度を算出すると、26.1質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、30.7質量%であった。
【0145】
(成膜工程)
実施例44〜48および比較例2のPZT前駆体溶液を用いて、PZT膜の成膜を行った。調製して1日後の各PZT前駆体溶液を用いて、スピンコートの回転数を変えたほかは実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、PZT膜を得た。なお、各PZT前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0146】
得られたPZT膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。各実施例および各比較例のPZT前駆体溶液を用いて得られたPZT膜の膜厚をまとめて図18に示した。
【0147】
(耐水性の評価)
上述の実施例44〜48のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図18に示した。
【0148】
(保存安定性の評価)
上述の実施例44〜48のPZT前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図18に示した。
【0149】
図18から明らかなように、実施例44〜48を用いたPZT膜は、スピンコート回転数を変えることによって、いずれも200nmを超える膜厚が得られている。これに対して、比較例2と同様のPZT前駆体溶液を用いた比較例8ではスピンコート回転数を700rpmとしても、162nmの膜厚であった。
【0150】
(調製工程)
[実施例49]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0%)14.0gと、チタニウム原料としてチタニウムテトライソプロポキシド8.4gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の攪拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物25.4gと、ランタン原料として酢酸ランタン1.5水和物2.1gと、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的な鉛系圧電体膜の前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図19に示した。本実施例の鉛系圧電体膜の前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ランタンLa2O3、酸化ジルコニウムZrO2、および酸化チタンTiO2として金属酸化物換算濃度を算出すると、20.9質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0151】
[実施例50]
まず、200mLのガラスフラスコに、溶媒として酢酸40.0gを秤量し、これにジルコニウム原料としてジルコニウムテトラ−n−ブトキシドのn−ブタノール溶液(濃度:86.0%)9.8gと、チタニウム原料としてチタニウムテトライソプロポキシド5.9gと、ニオブ原料としてペンタ−n−ブトキシニオブ5.6gを加えて、マグネチックスターラーを用いて、25℃の室温にて30分間の攪拌を行い、混合溶液を得た。次いで、混合溶液に、さらに鉛原料として酢酸鉛(II)三水和物27.3gと、マグネシウム原料として酢酸マグネシウム四水和物1.3gと、高分子化合物として重量平均分子量600であるポリエチレングリコール6.8gと、追加溶媒としてイオン交換水10.0gとを加え、80℃のオイルバスで1時間の加熱撹拌を行い、これを最終的な鉛系圧電体膜の前駆体溶液とした。本実施例に用いた溶媒、各金属原料および高分子化合物の秤量結果は図19に示した。本実施例の鉛系圧電体膜の前駆体溶液に含まれる金属成分を酸化物として、すなわち酸化鉛PbO、酸化ジルコニウムZrO2、酸化チタンTiO2、酸化マグネシウムMgOおよび酸化ニオブNb2O5として金属酸化物換算濃度を算出すると、20.8質量%であった。また、原料全量に対する原料として用いたカルボン酸の割合を算出すると、37.5質量%であった。
【0152】
(成膜工程)
実施例49および50の鉛系圧電体膜の前駆体溶液を用いて、鉛系圧電体膜の成膜を行った。調製して1日後および30日後の各前駆体溶液を用いて、実施例1と同様の成膜工程(塗布工程、乾燥工程、脱脂工程および焼成工程)を行い、各実施例につき2種類の鉛系圧電体膜を得た。なお、各前駆体溶液は、キャップ付ガラス瓶に入れ、25℃程度の室温で遮光保管していたものを使用した。
【0153】
(再現性評価)
上述の実施例48および49の鉛系圧電体膜の前駆体溶液を用いて得られた各鉛系圧電体膜について、走査型電子顕微鏡(日立:S−4700)を用いて、断面観察により膜厚を測定した。調製して1日後の前駆体溶液を用いた鉛系圧電体膜の膜厚を100とし、調製して30日後の前駆体溶液を用いた鉛系圧電体膜の膜厚を規格化することで、膜厚の再現性評価を行った。以下に示す評価基準を基にした実施例49および50の評価結果を図20に示した。
A:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が100以上110未満。
B:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が110以上120未満。
C:調製して30日後の鉛系圧電体膜の前駆体溶液を用いて得られた鉛系圧電体膜の膜厚が120以上。
【0154】
(耐水性の評価)
上述の実施例49および50の鉛系圧電体膜の前駆体溶液について、実施例1と同様の試験方法および評価基準で耐水性試験を行った。得られた評価結果をまとめて図20に示した。
【0155】
(保存安定性の評価)
上述の実施例49および50の鉛系圧電体膜の前駆体溶液について、実施例1と同様の試験方法および評価基準で保存安定性試験を行った。得られた評価結果をまとめて図20に示した。
【0156】
(インクジェット式記録ヘッドの製造)
実施例1〜47および比較例2のPZT前駆体溶液を用いて、上述のインクジェット式記録ヘッドの一連の製造工程に基づき、インクジェット式記録ヘッドを製造した。
なお、実施例1〜47のPZT前駆体溶液を用いる場合は、調製して1日後の各PZT前駆体溶液を用い、一連の成膜工程(塗布工程、乾燥工程、脱脂工程、焼成工程)を6回繰り返すことで、膜厚が1.2μmの圧電体層70を成膜した(なお、1層あたりの膜厚が200nmとなるように、各PZT前駆体溶液によってスピンコート回転数は適宜調整を行った)。
比較例2のPZT前駆体溶液を用いる場合は、調製して1日後の各PZT前駆体溶液を用い、一連の成膜工程を12回繰り返すことで、膜厚が1.2μmの圧電体層70を成膜した(なお、スピンコート塗布における回転数は1500rpmとした)。したがって同じ膜厚の圧電体層70を得たい場合、実施形態であれば比較例2の半分の回数の成膜工程でよい。
【0157】
得られた圧電体層70の結晶性評価(X線回折装置によるθ/2θ測定)、バーチャルグラウンド方式でのヒステリシス特性評価、レーザードップラー式振動計による圧電特性評価(変位量測定)、パルス耐久試験(初期値を基準として、200億のパルスを加えた後の変位低下率を評価)等を行った。実施例1〜46で得られた圧電体層70は、比較例2で得られた圧電体層70と同等の結果を示した。
なお、同等とは比較例2の前駆体溶液を用いて得られる圧電体を100として規格化した場合に、95〜105であることを示す。結晶性評価の場合は、(100)ピーク強度および(100)配向率であり、ヒステリシス評価の場合は、最大分極値、残留分極値および抗電圧であり、圧電特性の場合は変位量であり、パルス耐久試験では変位低下率である。
【0158】
上記実施形態で製造されたインクジェット式記録ヘッド1は、インクカートリッジ等と連通するインク流路を具備する記録ヘッドユニットの一部を構成して、インクジェット式記録装置1000に搭載される。図21は、そのインクジェット式記録装置の一例を示す概略図である。
【0159】
図21に示すインクジェット式記録装置1000において、図1に示したインクジェット式記録ヘッド1を有するインクジェット式記録ヘッドユニット1A及び1Bは、インク供給手段を構成するカートリッジ2A及び2Bが着脱可能に設けられ、このインクジェット式記録ヘッドユニット1A及び1Bを搭載したキャリッジ3は、装置本体4に取り付けられたキャリッジ軸5に軸方向移動自在に設けられている。このインクジェット式記録ヘッドユニット1A及び1Bは、例えば、それぞれブラックインク組成物及びカラーインク組成物を吐出するものとしている。
【0160】
そして、駆動モーター6の駆動力が図示しない複数の歯車およびタイミングベルト7を介してキャリッジ3に伝達されることで、インクジェット式記録ヘッドユニット1A及び1Bを搭載したキャリッジ3はキャリッジ軸5に沿って移動される。一方、装置本体4にはキャリッジ軸5に沿ってプラテン8が設けられており、図示しない給紙ローラーなどにより給紙された紙等の記録媒体である記録シートSがプラテン8に巻き掛けられて搬送されるようになっている。
【0161】
なお、図21に示す例では、インクジェット式記録ヘッドユニット1A、1Bは、それぞれ1つのインクジェット式記録ヘッド1を有するものとしたが、特にこれに限定されず、例えば、1つのインクジェット式記録ヘッドユニット1A又は1Bが2以上のインクジェット式記録ヘッド1を有するようにしてもよい。もちろん、インクジェット式記録ヘッドユニット1A、1Bという形式を取らずに、インクジェット式記録ヘッド1を直接インクジェット式記録装置1000に搭載してもよい。
【0162】
また、上述したインクジェット式記録装置1000では、インクジェット式記録ヘッド1を有するインクジェット式記録ヘッドユニット1A、1Bがキャリッジ3に搭載されて主走査方向に移動するものを例示したが、特にこれに限定されず、例えば、インクジェット式記録ヘッド1が固定されて、紙等の記録シートSを副走査方向に移動させるだけで印刷を行う、所謂ライン式記録装置にも本発明を適用することができる。
【0163】
なお、上述した実施形態では、液体噴射ヘッドの一例としてインクジェット式記録ヘッド1を挙げて説明したが、本発明は広く液体噴射ヘッド全般を対象としたものであり、インク以外の液体を噴射する液体噴射ヘッドにも勿論適用することができる。その他の液体噴射ヘッドとしては、例えば、プリンター等の画像記録装置に用いられる各種の記録ヘッド、液晶ディスプレイ等のカラーフィルターの製造に用いられる色材噴射ヘッド、有機ELディスプレイ、FED(電界放出ディスプレイ)等の電極形成に用いられる電極材料噴射ヘッド、バイオchip製造に用いられる生体有機物噴射ヘッド等が挙げられる。
【符号の説明】
【0164】
1A,1B…インクジェット式記録ヘッドユニット、2A,2B…カートリッジ、4…装置本体、6…駆動モーター、10…流路形成基板、12…圧力発生室、14…連通路、15…連通部、20…ノズルプレート、21…ノズル開口、30…保護基板、31…圧電素子保持部、32…リザーバー部、33…貫通孔、35…接着剤、40…コンプライアンス基板、41…封止膜、42…固定板、43…開口部、50…弾性膜、51…酸化膜、55…絶縁体膜、60…下電極、70…圧電体層、71…圧電体前駆体膜、72…圧電体膜、80…上電極、90…リード電極、100…リザーバー、300…圧電素子、1000…インクジェット式記録装置。
【特許請求の範囲】
【請求項1】
酢酸及び/又はプロピオン酸からなるカルボン酸と、
酢酸鉛と、
Zr(OR1)4で表されるジルコニウムアルコキシドと、
(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)
Ti(OR2)4で表されるチタニウムアルコキシドと、
(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)
高分子化合物と、
を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、
前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、
前記ジルコニウムアルコキシドの配位子OR1または前記チタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液。
【請求項2】
前記ジルコニウムアルコキシドの配位子OR1および前記チタニウムアルコキシドの配位子OR2が、ともに、分岐鎖状のアルコキシ基であることを特徴とする請求項1に記載の圧電体膜の前駆体溶液。
【請求項3】
前記前駆体溶液の溶媒として、さらに、水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類から選ばれる1種または2種以上を含むことを特徴とする請求項1または2に記載の圧電体膜の前駆体溶液。
【請求項4】
前記高分子化合物がポリエチレングリコールおよびその誘導体ならびにポリプロピレングリコールおよびその誘導体から選ばれる1種または2種以上であり、さらに、前記高分子化合物の重量平均分子量が300〜800であることを特徴とする請求項1〜3のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項5】
前記圧電体膜の前駆体溶液に含まれる金属元素の酸化物換算濃度が、15質量%以上27質量%以下であることを特徴とする請求項1〜4のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項6】
前記圧電体膜の前駆体溶液がさらに、少なくとも1種以上の有機金属化合物を含むことを特徴とする請求項1〜5のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項7】
請求項1〜6のいずれか一項に記載の圧電体膜の前駆体溶液を用いたことを特徴とする圧電素子の製造方法。
【請求項1】
酢酸及び/又はプロピオン酸からなるカルボン酸と、
酢酸鉛と、
Zr(OR1)4で表されるジルコニウムアルコキシドと、
(OR1は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)
Ti(OR2)4で表されるチタニウムアルコキシドと、
(OR2は炭素数が3〜8の直鎖状または分岐鎖状のアルコキシ基である)
高分子化合物と、
を含む原料を混合することで得られる圧電体膜の前駆体溶液であって、
前記原料全量に対する前記カルボン酸の割合が20質量%以上60質量%以下であり、かつ、
前記ジルコニウムアルコキシドの配位子OR1または前記チタニウムアルコキシドの配位子OR2の少なくとも一方の配位子が分岐鎖状のアルコキシ基であることを特徴とする圧電体膜の前駆体溶液。
【請求項2】
前記ジルコニウムアルコキシドの配位子OR1および前記チタニウムアルコキシドの配位子OR2が、ともに、分岐鎖状のアルコキシ基であることを特徴とする請求項1に記載の圧電体膜の前駆体溶液。
【請求項3】
前記前駆体溶液の溶媒として、さらに、水、2級または3級のアルコール類、エーテル類、エステル類およびケトン類から選ばれる1種または2種以上を含むことを特徴とする請求項1または2に記載の圧電体膜の前駆体溶液。
【請求項4】
前記高分子化合物がポリエチレングリコールおよびその誘導体ならびにポリプロピレングリコールおよびその誘導体から選ばれる1種または2種以上であり、さらに、前記高分子化合物の重量平均分子量が300〜800であることを特徴とする請求項1〜3のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項5】
前記圧電体膜の前駆体溶液に含まれる金属元素の酸化物換算濃度が、15質量%以上27質量%以下であることを特徴とする請求項1〜4のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項6】
前記圧電体膜の前駆体溶液がさらに、少なくとも1種以上の有機金属化合物を含むことを特徴とする請求項1〜5のいずれか一項に記載の圧電体膜の前駆体溶液。
【請求項7】
請求項1〜6のいずれか一項に記載の圧電体膜の前駆体溶液を用いたことを特徴とする圧電素子の製造方法。
【図1】


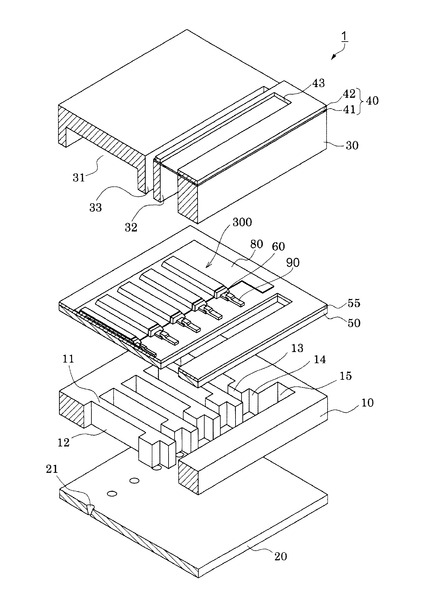
【図2】
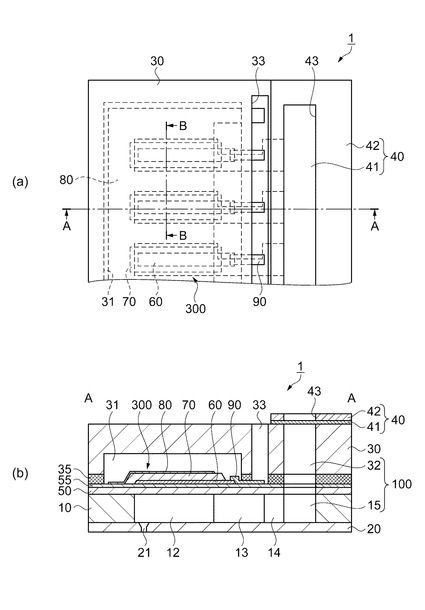
【図3】
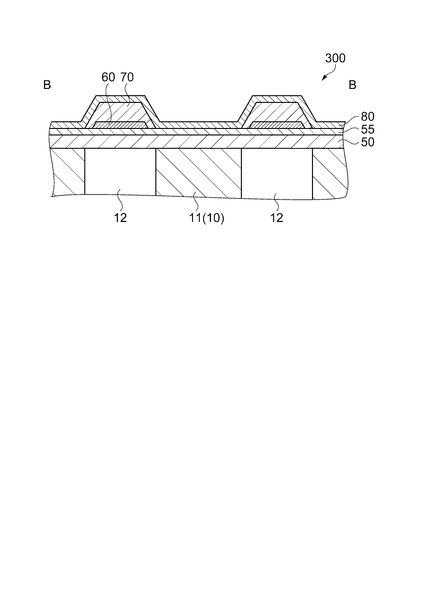
【図4】
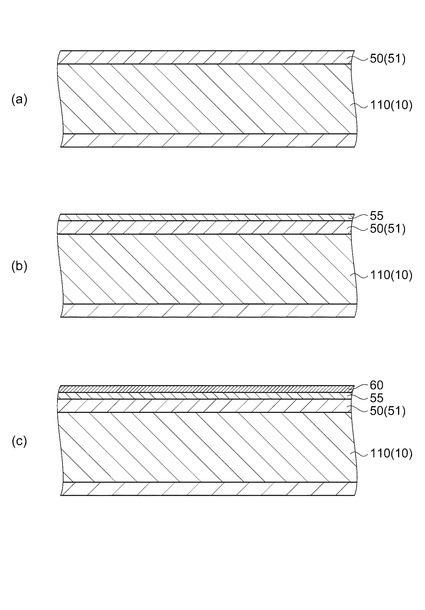
【図5】
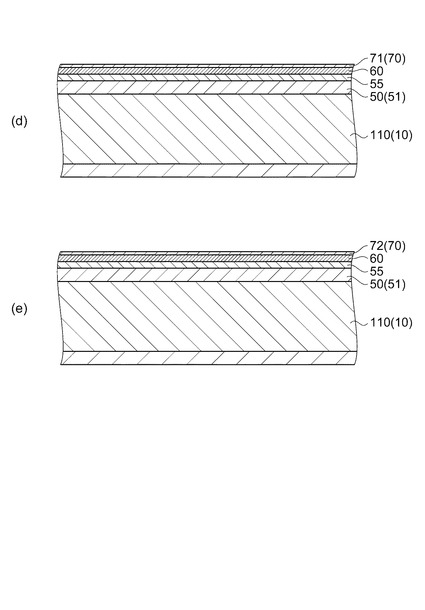
【図6】
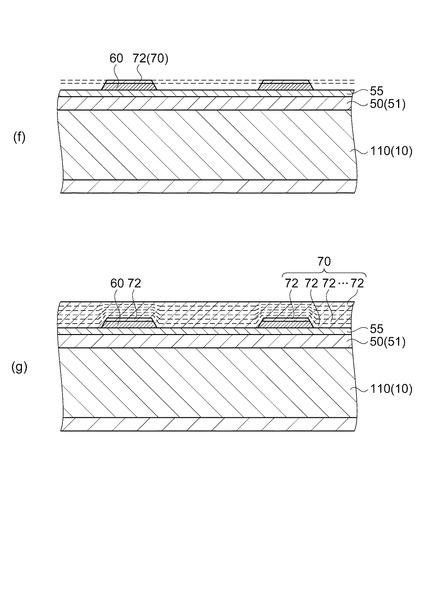
【図7】
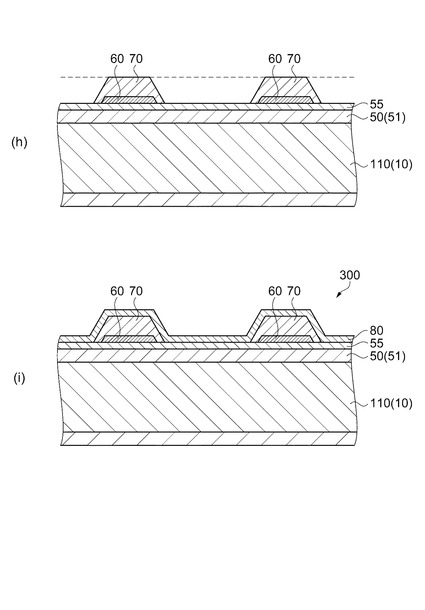
【図8】

【図9】
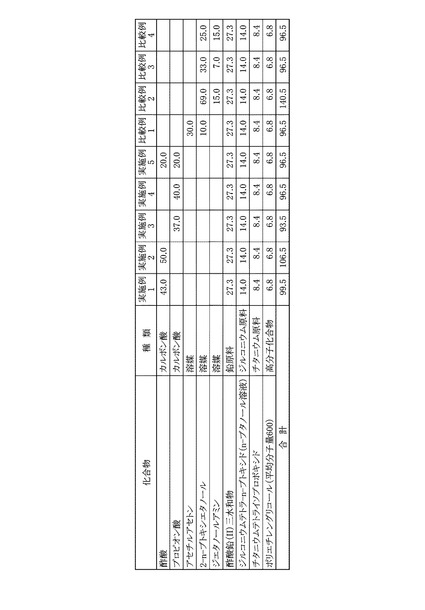
【図10】

【図11】

【図12】
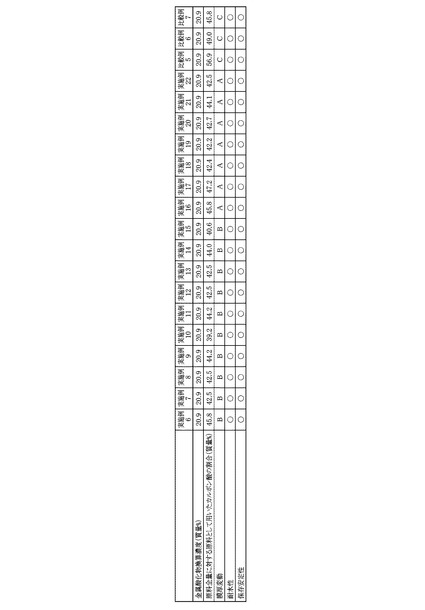
【図13】
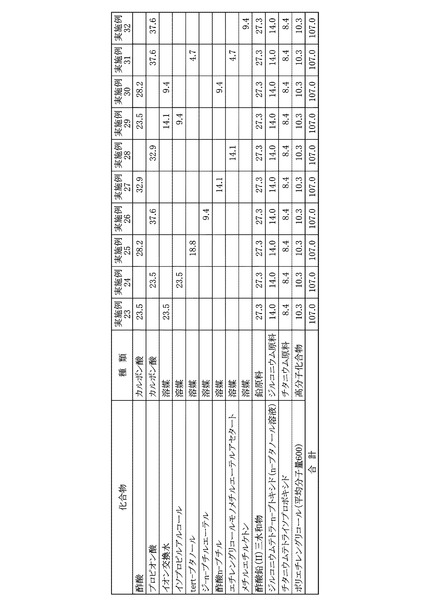
【図14】
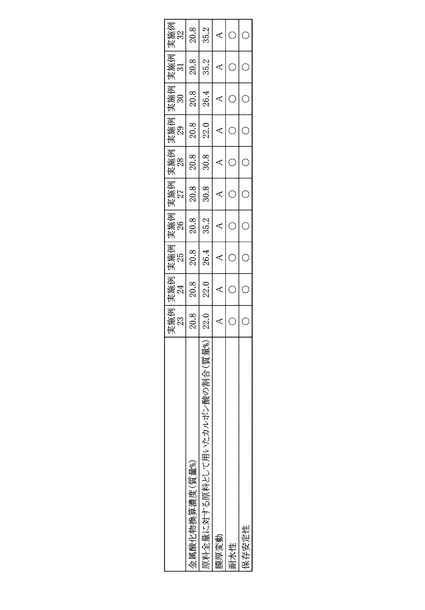
【図15】
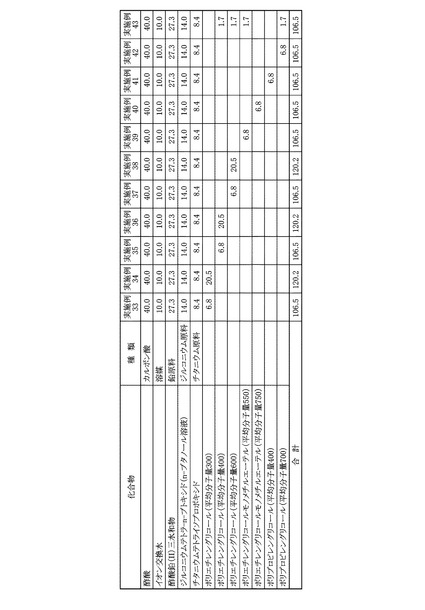
【図16】
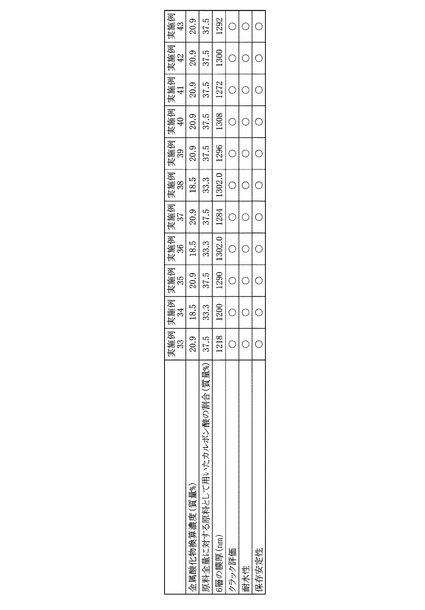
【図17】
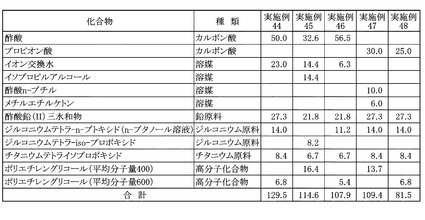
【図18】
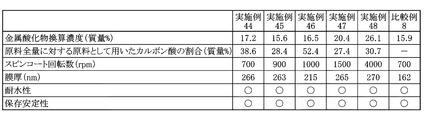
【図19】
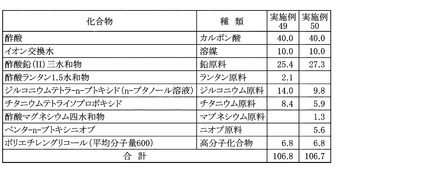
【図20】

【図21】
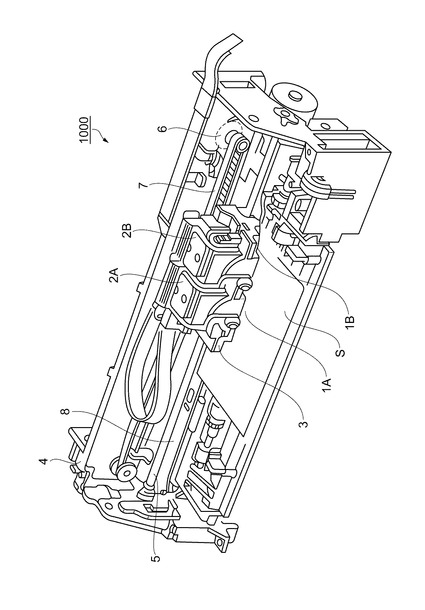
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公開番号】特開2012−235004(P2012−235004A)
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願番号】特願2011−103450(P2011−103450)
【出願日】平成23年5月6日(2011.5.6)
【出願人】(000002369)セイコーエプソン株式会社 (51,324)
【Fターム(参考)】
【公開日】平成24年11月29日(2012.11.29)
【国際特許分類】
【出願日】平成23年5月6日(2011.5.6)
【出願人】(000002369)セイコーエプソン株式会社 (51,324)
【Fターム(参考)】
[ Back to top ]