
培養細胞を用いたタンパク質発現のリアルタイム測定法
【課題】本発明は、培養細胞による目的タンパク質の発現量を、通常の細胞培養条件において細胞を回収及び破壊することなくリアルタイムで、簡易且つ正確に測定する方法を提供することをその目的とする。
【解決手段】以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【解決手段】以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、タンパク質発現の測定法に関する。より詳細には、本発明は、培養細胞において発現された目的タンパク質の量を、測定された蛍光強度を指標にして定量する方法に関する。
【背景技術】
【0002】
タンパク質の生化学・物理化学・構造生物学研究などを行うためには、研究対象とするタンパク質を多量に必要とするため、種々の細胞を用いて大量生産させる(発現させる)条件の確立が必須である。特にヒトを含む真核生物由来のタンパク質は、昆虫培養細胞や哺乳類の培養細胞など動物培養細胞を用いて生産させる例が多く見られており、なかでも膜タンパク質については動物培養細胞にて発現させることが不可欠である。ところが、それらの細胞で発現している目的タンパク質の量を調べるにあたっては、培養した細胞を回収し破砕して種々の実験手法を用いて定量する必要があり、最適な発現時間を調べるなどその条件の至適化には多大な試料量と労力が必要であった。
【0003】
近年研究対象とする目的タンパク質を緑色蛍光タンパク質(GFP)融合タンパク質として発現し、細胞の蛍光測定(これまでの報告では大腸菌や酵母)や、その後細胞を破砕し電気泳動やゲルろ過クロマトグラフィーをおこなって蛍光測定を行うことにより、目的タンパク質の発現量を見積もる技術の報告がされている(非特許文献1〜3参照)。しかしながら、従来法では細胞を培養後回収してから蛍光測定を行う必要があり、タンパク質を生産している過程の細胞培養中のタンパク質発現量をリアルタイムで測定することは不可能であった。このため、タンパク質発現の経時変化を測定するには、複数サンプルを準備しそれぞれを異なる時間培養した後回収するという労力が必要であり、さらにサンプル間の細胞増殖のばらつきによる誤差がうまれる危険性があった。さらに、これまで細胞からの蛍光強度の直接測定が報告されている大腸菌や酵母などの微生物は、細胞が不透明であり蛍光強度測定のための励起光や蛍光が細胞によって吸収されてしまうため測定が不正確であり、正確な測定をするためには細胞を破砕し電気泳動やゲルろ過クロマトグラフィーを行って蛍光観測するなどの多大な労力が必要であった。
【非特許文献1】Drew et al., 2006, Nat. Methods 3, 303-313.
【非特許文献2】Kawate et al., 2006, Structure 14, 673-681.
【非特許文献3】Newstead et al., 2007, Proc. Natl Acad. Sci. USA 104, 13936-13941.
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明は、培養細胞による目的タンパク質の発現量を、通常の細胞培養条件において細胞を回収及び破壊することなくリアルタイムで、簡易且つ正確に測定する方法を提供することをその目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、上記課題を解決するために、目的タンパク質をGFP融合タンパク質として発現し、その蛍光をプレートリーダーを用いて測定することにより当該タンパク質の発現量を測定する方法に着目し、鋭意検討を重ねた。その結果、発現量を調べたい目的タンパク質をGFP融合タンパク質として発現させる発現ベクターを構築し、動物培養細胞に遺伝子導入したものを、通常の細胞培養で行う場合と同じようにマルチウェルプレートを用いて付着培養させ、その培養しているマルチウェルプレートをそのままプレートリーダーを用いて下方測定にて蛍光測定を行うことにより、培養中の目的タンパク質の発現量を見積もることが可能であることを見出し、本発明を完成するに至った。即ち、本発明は以下の通りである。
【0006】
[1]以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
[2]蛍光強度の測定が下方測定である、上記[1]記載の方法。
[3]蛍光強度の測定が培養容器のリッドを外さないで行われる、上記[1]記載の方法。
[4]培養容器の1ウェルあたりの底面積が0.32cm2を上回るものである、上記[1]記載の方法。
[5]蛍光強度の測定が多点測定で行われる、上記[1]記載の方法。
[6]培養容器の容器辺縁部を含まない領域の底面積の70%を超える領域が多点測定に付される、上記[5]記載の方法。
[7]蛍光強度の正規化のために、培養培地のみを含み細胞が含まれていない培養容器における蛍光強度が併せて測定される、上記[1]記載の方法。
[8]細胞が動物培養細胞である、上記[1]記載の方法。
[9]蛍光強度の測定が同一細胞培養槽について継続的に行われる、上記[1]記載の方法。
【発明の効果】
【0007】
本発明の方法では、目的タンパク質の発現を測定するために細胞を回収したり培養プレートのリッド(ふた)を開閉する必要がないため、培養細胞をコンタミネーションのリスクにさらすことなくそのままに保ったまま目的タンパク質の発現をリアルタイムで評価することができ、タンパク質発現の経時測定を1回の培養で同じサンプルから行うことができる。このことによりタンパク質発現条件の確立には重要な要因である発現時間を簡便に決定することが可能である。また、これまで報告されている微生物細胞からの直接蛍光測定と比べ、透明な動物培養細胞を用いることにより、励起光や蛍光の吸収が非常に低くおさえられ、蛍光強度がタンパク質の発現量をより正確に反映していると考えられる。このため、細胞を回収し破砕して電気泳動やゲルろ過クロマトグラフィーなどを用いて行っていた定量を行うことなく、細胞の直接測定による発現量の見積もりが可能である。これらの簡便さから、多種類の目的タンパク質の発現量を一度に比較するハイスループット解析にも適した方法である。
【発明を実施するための最良の形態】
【0008】
本発明は、培養細胞において発現された目的タンパク質の量を定量する方法を提供する。当該方法は、以下の工程を含む:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【0009】
本発明の方法によって定量される前記目的タンパク質とは、培養細胞におけるその発現量を調べたい任意のタンパク質である。原核生物由来又は真核生物由来など、その由来はいかなるものであっても良く、また、分泌タンパク質、膜タンパク質、細胞質若しくは核内に局在するタンパク質など、タンパク質の種類についても限定されない。
【0010】
前記蛍光タンパク質は、融合タンパク質発現に適したものである限り、当該技術分野で使用される任意のものを好適に使用することができ、例えば、GFP、YFP、CFP、BFP、Venusなどのオワンクラゲ由来蛍光タンパク質及びそれらの類似体、ウミシイタケ由来蛍光タンパク質及びその類似体、DsRed、HcRed、AsRed、ZsGreen、ZsYellow、AmCyan、AcGFP、Kaedeなどのサンゴ由来蛍光タンパク質及びそれらの類似体などが挙げられる。使用する蛍光タンパク質の決定は、使用する発現ベクター及び培養細胞、又は価格など様々な要素を考慮してなされ得るが、これは当業者の通常の技術的知識の範囲内である。通常、オワンクラゲ由来のGFP若しくはその類似体(例、EGFP)が好適に使用される。後述するように、本発明の方法においては、目的タンパク質と蛍光タンパク質との融合タンパク質をコードする核酸を適切な発現ベクターに組み込むため、蛍光タンパク質をコードする核酸が必要となるが、それについては例えばクロンテック社などから市販されているベクターを使用すれば良い。
【0011】
目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を作製するためには、該融合タンパク質の適切な発現ベクターを構築し、それを適切な細胞に導入すれば良い。前記細胞としては、タンパク質の製造に通常使用されるあらゆる細胞を限定なく使用することができる。本発明の方法においては、細胞を破砕することなく細胞からの蛍光強度を測定するため、蛍光強度のより正確な測定を可能とするという観点から、大腸菌や酵母などの微生物細胞よりも、透明であり接着培養する性質を持つ動物培養細胞を使用することが好ましい。動物培養細胞の由来としては、哺乳類(例、ハムスター、マウス、サル、ヒトなど)、鳥類(例、ニワトリ、シチメンチョウなど)、爬虫類(例、ヘビなど)、両生類(例、カエルなど)、魚類、昆虫(例、蚊、ハエ、蛾など)などが例示されるが、通常、哺乳類細胞又は昆虫細胞が好ましい。哺乳類由来の培養細胞の例としては、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞、COS細胞、HEK293細胞などが挙げられる。一方、昆虫由来の培養細胞の例としては、Sf9細胞、Sf21細胞、High Five細胞などが挙げられる。
【0012】
融合タンパク質の発現ベクターは、目的タンパク質と蛍光タンパク質との融合タンパク質をコードする塩基配列を含み、且つ、導入された細胞内で融合タンパク質を発現させることができるものでなければならない。
融合タンパク質の発現ベクターは、例えば、目的タンパク質をコードするDNA断片、及び蛍光タンパク質をコードするDNA断片をそれぞれ切り出し、それらを適当な発現ベクター中のプロモーターの下流に連結することによって製造することができる。そのとき、目的タンパク質及び蛍光タンパク質が、融合した一つのタンパク質として合成される必要がある。その方法は、市販の蛍光タンパク質をコードする塩基配列を使用する場合は製造者による説明書を参照すれば良く、或いは多くの標準的ラボラトリーマニュアルに記載の常法に従って行えば良い。
融合タンパク質の遺伝子が連結される発現ベクターとしては、大腸菌由来のプラスミド(例、pBR322,pBR325,pUC12,pUC13)、枯草菌由来のプラスミド(例、pUB110,pTP5,pC194)、酵母由来プラスミド(例、pSH19,pSH15)、λファージなどのバクテリオファージ、レトロウイルス,ワクシニアウイルス,バキュロウイルスなどの動物ウイルスなどの他、pA1−11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neoなどが用いられる。
前記プロモーターとしては、遺伝子の発現に用いる培養細胞に対応して適切なプロモーターであればいかなるものでもよい。例えば、動物細胞を用いる場合は、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMVプロモーター、HSV-TKプロモーターなどが挙げられる。これらのうち、CMV(サイトメガロウイルス)プロモーター、SRαプロモーターなどを用いるのが好ましい。昆虫細胞を用いる場合は、IE1プロモーター、ポリヘドリンプロモーター、P10プロモーターなどが挙げられる。これらのうち、IE1プロモーターを用いるのが好ましい。
発現ベクターには、以上の他に、所望によりエンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、SV40複製オリジンなどを含有しているものを用いることができる。選択マーカーとしては、例えば、ジヒドロ葉酸還元酵素遺伝子〔メソトレキセート(MTX)耐性〕、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子〔G418耐性〕などが挙げられる。また、必要に応じて、使用する培養細胞に合ったシグナル配列を、目的タンパク質のN端末側に付加しても良い。
このようにして構築された前記融合タンパク質をコードするDNAを含有する発現ベクターを用いて、形質転換体を製造することができる。
【0013】
発現ベクターの培養細胞への導入は、細胞染色体への安定な組込みを目的とするものであっても、一過性の導入であってもよい。導入は、自体公知の方法で行えばよく、例えば、エレクトロポレーション、リポフェクション、リン酸カルシウム法(CaPi)、ポリフェクション法(PEI)、塩化カルシウム法(カルフェクション)などのトランスフェクションが挙げられる。かかる方法は、多くの標準的ラボラトリーマニュアルに記載されている。
【0014】
細胞を培養する方法は当該分野において公知である(例えば、Tissue Engineering Methods and Protocols,Morgan and Yarmush(eds.),Humana Press,Inc.,Totowa,NJ,1999参照)。当業者なら認識しているように、細胞が培養される条件は、細胞のタイプによって変わる。該条件には、環境の温度、細胞を含む培養容器、細胞培養雰囲気又は環境を構成する、様々なガス(例えば、CO2)の組成、細胞が維持される培地、培地の成分及びpH、細胞が維持される密度、培地を新しい培地に交換することを必要とするスケジュールなどが含まれる。これらのパラメータは当該分野においてはしばしば知られているものであるか、経験的に決定されうる。
培養細胞が動物細胞である場合、培地としては、例えば、約5〜20%の胎児牛血清を含むMEM培地〔Science,122巻,501(1952)〕,DMEM培地〔Virology,8巻,396(1959)〕,RPMI 1640培地〔The Journal of the American Medical Association,199巻,519(1967)〕,199培地〔Proceeding of the Society for the Biological Medicine,73巻,1(1950)〕などが用いられる。pHは約6〜8であるのが好ましい。培養は通常約30〜40℃で約15〜60時間行い、必要に応じて通気や撹拌を加える。また、培養細胞が昆虫細胞である場合、培地としては、Grace's Insect Medium(Nature,195,788(1962))に非動化した10%ウシ血清等の添加物を適宜加えたものなどが用いられる。培地のpHは約6.2〜6.4に調整するのが好ましい。培養は通常約27℃で約3〜5日間行い、必要に応じて通気や撹拌を加える。
【0015】
前記細胞培養を行うための培養容器は、細胞を培養するための培地を保持するものであって、且つ、後述するように、培養細胞を培養容器外へ移さずに蛍光強度を測定することを可能とするものであれば、特に限定されない。上記条件を満たす限りにおいて、培養容器として、例えば、マイクロプレート、チャンバ又は複数チャンバを備えた培養スライド、カバーガラス、カップ、フラスコ、チューブ、ローラーボトル、スピナーボトルなどを使用することが可能であるが、本発明の方法における蛍光強度測定は、通常マイクロプレートリーダーを使用して行われるため、好ましくはマイクロプレートが使用される。
上記マイクロプレートとしては、その各ウェルの底面積があまりに小さいと、再現性のある細胞増殖を維持することが出来ないこと、培養培地が干上がってしまう可能性があること、及び蛍光強度の測定において多点測定を許容しないことなどの欠点が生じ、そのため再現性のある結果が得られない傾向があるため、充分な大きさの底面積を持つものである必要がある。具体的には、例えば、通常市販されている96ウェルプレートの底面積である0.32cm2を上回る底面積を持つものが好ましく使用される。底面積の上限としては、蛍光強度の測定を許容する限り特に制限はないが、通常500cm2以下である。ここで、底面積は0.5〜20.0cm2がより好ましく、1.8〜9.6cm2がさらに好ましい。また、マイクロプレート以外の培養容器についても、同様に、充分な底面積を持つものである必要がある。
マイクロプレートのウェルの形状としては、平底、U底、及びV底のものなどがあるが、本発明の方法のためには、マイクロプレートリーダーでの測定焦点高さが通常固定されているという理由から、接着培養を行う際細胞が同一高さに分布する平底が好ましい。
接着培養の際細胞が付着するマイクロプレートのウェル底面の材質としては、ポリスチレン、白色ポリスチレン、ポリエチレン、ナチュラルポリプロピレン、ガラス入りポリプロピレン、又はバレックスなどのプラスチック、或いは石英ガラス又は硼珪酸ガラスなどのガラスが挙げられ、培養の際の細胞の接着性が優れており、低価格であるという理由から、プラスチック製、特にポリスチレン製のマイクロプレートが本発明の方法において好適に使用され得る。さらに蛍光観測する要請から、その底面は透明である必要があり、ポリスチレン製のものはこの用件も満たす。
上記マイクロプレートは、コーニング社製マルチプルウェルプレートなど市販のものが通常使用され、上記底面積に関する条件を満足するために、24穴以下のウェル(例、6穴、12穴、24穴など)を持つマイクロプレートが好ましく使用され得る。
また、マイクロプレート本体部分には、透明、白色、及び黒色のものなどがあるが、白色又は黒色のマイクロプレートは、透明のマイクロプレートと比較して一般的に高価であり、また市販のこれらのプレートの底面材質が必ずしも細胞接着培養に好適でないことを考慮すると、透明のものが好ましい。
以上のことから、本発明の方法において、特に好ましく使用されるマイクロプレートの例として、6穴又は24穴のポリスチレン製の平底透明マイクロプレートが挙げられる。
【0016】
本発明の方法においては、前記融合タンパク質の蛍光強度の測定は、培養細胞を破壊せず、且つ培養容器外へ移さずに行う必要がある。係る条件を満足する限り、当該分野で使用される任意の蛍光強度測定法が使用され得るが、感度、精度及び簡便性の観点から、マイクロプレートリーダーによる蛍光強度測定が好ましい。マイクロプレートリーダーとしては、例えば、サーモフィッシャーサイエンティフィック社製ヴァリオスキャンフラッシュなどが挙げられる。また、蛍光強度の測定は、使用する測定機器に付属のマニュアルを参照して、当業者は実施することが可能である。
【0017】
マイクロプレートリーダーによる蛍光強度の測定は、培養容器中の複数の点で測定された蛍光強度の平均値を容器全体における蛍光強度として得るものである多点測定により行うことが、信号対雑音比を最小限に抑える観点から好ましい。その場合、培養容器の容器辺縁部を含まない領域(以下、「測定領域」ともいう)の底面積の70%を超える領域が多点測定に付されることが好ましい。多点測定に付すべき領域の底面積の上限は特にないが、測定に要する時間を考慮すると、測定領域の95%が好ましく、90%がより好ましく、85%がさらに好ましく、80%が特に好ましい。ここで、容器辺縁部とは、容器縁部からの距離が、3mmもしくは測定装置の測定光のビーム幅の2倍の距離のいずれか大きい方の値をもつ距離の範囲内にある容器内の部分を意味する。また、上述したように本発明における蛍光強度の測定は、培養細胞を培養容器外へ移さずに測定するため、培養容器中への雑菌混入を防ぐという観点から、培養容器のリッドを外すことなく蛍光強度を測定することが好ましい。リッド付きの培養容器から蛍光強度を測定するとき、上方測定、即ちリッドのある方向からの蛍光強度測定は、励起光及び蛍光のリッドによる不規則反射などが理由で、蛍光物質なしのコントロールにおいても高い蛍光強度値を示し、そのため信号対雑音比が低くなるという問題があるため、本発明における蛍光強度の測定においては、下方測定、即ち、培養容器の底面側からの蛍光強度を測定することが好ましい。
【0018】
上述したように本発明における蛍光強度の測定は、細胞を破砕せず、且つ培養容器外へ移さずに測定するものであるため、蛍光強度の測定を同一細胞培養槽について継続的に行うことが可能である。従って、タンパク質発現の経時測定を1回の培養で同じサンプルから行うことができき、タンパク質発現条件の確立には重要な要因である発現時間を簡便に決定することが可能である。
【0019】
上述したように、本発明の方法は、測定された蛍光強度を指標に、目的タンパク質の発現量を算出することを含む。目的タンパク質の発現量の算出のために、測定された蛍光強度は、適当なコントロール由来の蛍光強度によって正規化されることが好ましい。係るコントロールとして、培養細胞が含まれておらず、細胞培養に使用したものと同種の培養培地のみを含む培養容器を利用するのが、再現性の高い結果を得られるという観点から好ましい。特にマルチウェルマイクロプレートを使用する場合、蛍光強度測定サンプルの同じ培養プレートにおける、サンプルの培養に用いたものと同種および同量の培養培地で充填されたウェルからの蛍光強度測定値を使用することが好ましい。その場合、マイクロプレートの各ウェルでの蛍光強度Fは;
相対蛍光強度=(F−F0)/F0
(ここで、F0は、測定サンプルと同じ培養プレート中の、培養培地で充填されたウェルからの蛍光強度の測定値である);
という方程式で正規化される。こうして得られる相対蛍光強度を指標にして、目的タンパク質の発現量を定量することができる。
【0020】
以下、実施例などにより本発明を更に詳しく説明するが、本発明は下記実施例などに何ら制限されるものではない。
【実施例】
【0021】
実施例1
(1)発現ベクターの構築
下記に記載の通り、昆虫培養細胞において細胞質発現及び分泌発現という二つの種類のタンパク質発現を試験するベクターを構築した(図1)。細胞質発現のための試験ベクターpCGFP_SFは、マルチクローニングサイト、トロンビン切断部位及びヘキサ・ヒスチジンタグを持つ改変GFPuv(UV領域における励起極大を持つGFPの変異体)の遺伝子を、ベクターpCGFP−BC(Eric Gouaux教授の好意により提供された。非特許文献2)を鋳型にして従来のPCR法で増幅し、当該フラグメントを制限酵素DraIIIで処理したものをpIEx4ベクター(Novagen)のNcoI部位(平滑末端化した)とDraIII部位との間に挿入した。分泌発現のための試験ベクターであるベクターpCGFP_SF+AKHは、昆虫由来の分泌シグナル配列であるadipokinetic hormone(AKH)シグナル配列をpcGFP_SFベクターのマルチクローニングサイトの上流側に連結できる様にオリゴヌクレオチドプライマーを設計し(AKHシグナル配列はpIEx-5ベクター(novagen)中に含まれる配列を参考にして設計)、通常のPCR法を用いてpcGFP_SFベクターと同様に構築した(図1)。下記実施例1における試験タンパク質のための発現ベクターの構築のために、分泌シグナルを除去したヒト由来味覚受容体リガンド結合領域T1r1(456残基、タンパク質A)、T1r2(454残基、タンパク質B)、T1r3(458残基、タンパク質C)及び分泌シグナルを含むラット由来代謝型グルタミン酸受容体1リガンド結合領域(522残基、タンパク質D)の遺伝子をpCGFP_SFベクターにサブクローニングして、C末端GFP融合とした。
【0022】
(2)発現系
本発明における組換えタンパク質発現のリアルタイム検出の評価のために、一過性タンパク質発現系でSf9細胞を使用した。Sf9発現系は、構造生物学研究分野において最も一般的に使用される組換えタンパク質発現のための系の一つである。大腸菌及び酵母細胞といった微生物での発現系とは対照的に、Sf9細胞は、他の動物培養細胞と同様、透明であり、生体細胞における発現GFPの蛍光強度のより正確な測定を可能とする。通常の動物細胞について最も標準的且つ広く適用される培養方法である単層培養(細胞は、培養プレートの底に接着する)に細胞を付した。
組換えタンパク質の発現は、具体的には次のようにして行った。Insect Genejuice(Novagen)を製造者のプロトコールに従って使用して、発現ベクターをSf9細胞(GIBCO)にトランスフェクションした。トランスフェクションした細胞を、様々の種類の組織培養プレートにおいてSf900III SFM(GIBCO)中で48〜120時間、27℃でインキュベーションし、一過的な組換えタンパク質発現をさせておいた。典型的に、融合性単層培養からのSf9細胞を6ウェル組織培養プレート(6 Well Clear TC-Treated microplates,Costar)中の各ウェルに1.0〜1.6×106細胞/ウェルの密度で添加し、その後ウェルを、10μlのInsect Genejuice及び2μgのプラスミドを含有する1mlのSf900III SFMで充填した。培養プレート中のウェルの一つを等量のSf900III SFMで充填し、コントロール(バックグラウンド)として使用した。
【0023】
(3)培養プレートの種類
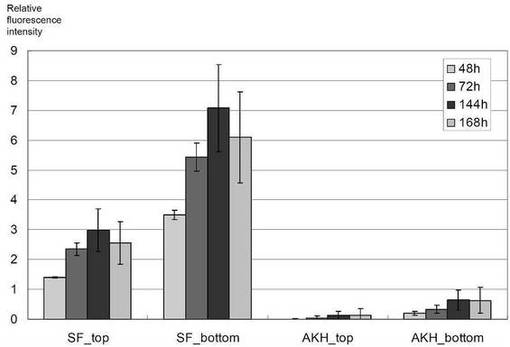
様々な細胞培養プレートを試験するために、細胞を、トランスフェクション後、6、24及び96ウェルプレートという3つの異なる種類の培養プレート中でインキュベーションした。これらのプレートのうちで、96ウェルプレートは再現性のある結果を殆ど与えなかった。96ウェルプレートのウェルは、再現性のある細胞増殖を維持するためには小さ過ぎ、しばしば培養培地の干上がりに直面した。更に、96ウェルプレートのシングルウェルは、そのサイズが小さいため一点測定のみ許容し、それが更に検出中の測定エラーを増している(データ示さず)。一方、6ウェル及び24ウェルプレートで培養された細胞は、安定した細胞増殖を示し、また、これらのウェルのサイズは多点測定を可能とし、単一ウェルの様々な領域での値を平均化することが出来、従ってウェルの細胞数の状態をより正確に反映することが出来る。次に、従来のポリスチレンの透明培養プレート(6 Well Clear TC-Treated Microplates,Costar)を、蛍光測定に好適であることが示唆されているガラス底の黒色培養プレート(EZview glass bottom culture plate LB,6ウェル,Iwaki)と比較した。しかしながら、これら2つのプレート間での検出したS/N比(相対蛍光強度の値)は、細胞質及び分泌タンパク質発現の両方について、2つの異なる時点においてほぼ同じであった(図5)。なお、図5において、SF_top及びSF_bottomは、pCGFP_SFでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ表しており、また、AKH_top及びAKH_bottomは、pCGFP_SF+AKHでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ示している。黒色培養プレートは高価であり、また必ずしも細胞培養に好適でないことを考慮すると、従来の6ウェル又は24ウェルのポリスチレンの透明組織培養プレートの使用が好適であり、リアルタイム蛍光検出のための充分な正確性を与えると考えられる。
【0024】
(4)マイクロプレートリーダーを使用する蛍光検出のための測定パラメータの決定
生体Sf9細胞におけるGFPuvの蛍光強度の正確な測定のために、マイクロプレートリーダーVarioskan Flash(Thermo Fisher Scientific)での検出のための測定パラメータを決定した。最初に、測定した蛍光強度の正規化のための適切な方法を見出すことを試みた。トランスフェクションしていないSf9細胞(プラスミドを用いないでリポフェクション試薬で処理)、及び培養培地という二つの条件を比較し、どちらが測定のための適切なコントロールとして機能するものであるのか評価した。これらの条件での蛍光強度値は、ほぼ同様の範囲であったが、コントロールとして培養培地を使用する方が、トランスフェクションしていないSf9細胞を使用するより再現性の高い値という結果であった(データ示さず)。トランスフェクションしていないSf9細胞は、トランスフェクションした細胞よりも、非常に高い細胞増殖及び蛍光強度を時に示し、その結果、正規化に使用するのに不適当なほど小さいサンプル相対蛍光強度値となる。従って、培養培地をコントロールとして採用した。
以下の実施例1及び比較例1においては、組織培養プレート中の細胞増殖において発現したGFPuvに由来する蛍光強度を、励起波長395nm、蛍光波長507nmを使用してマイクロプレートリーダーVarioskan Flash(Thermo Fisher Scientific)を用いて検出した。典型的な測定条件は、測定時間500ms及びバンド幅12nmでの多点測定(ウェルの中心からの円形領域であって測定領域の80%)にセットした。なお、今回の実験では、ウェルの測定領域の80%を超える領域の多点測定の試験は行わなかった。培養プレートの各ウェルにおいて測定された蛍光強度Fを:
相対蛍光強度=(F−F0)/F0
(ここで、F0は、測定サンプルの同じ培養プレート中のウェルに充填された培養培地の蛍光強度を表す)
という方程式で正規化した。
【0025】
次に、マイクロプレートリーダーでの蛍光検出のための様々な測定パラメータを試験した。信号対雑音比(S/N比)(相対蛍光強度の値)の観点から、以下の条件が最良の結果を与えた;測定方式は多点にセットし、測定時間は500ms、バンド幅は12nmであった。多点測定で試験される領域のうち、ウェルの測定領域の底面積の約70〜80%をカバーするウェルの中心からの円形領域が最良の結果を与えた(データ示さず)。また、上方測定及び下方測定という二つの蛍光検出法を比較した。培養プレートの上部カバーを外すプレートの下方測定及び上方測定は、同様の値を与えることをその結果は示していた(図3)。しかしながら、リッド付きのプレートの読み取りの場合、上方測定は、高いバックグラウンド(コントロールにおける高い蛍光強度値)を示し、その結果低いS/N比であったが(図4)、これは恐らく、励起光及び蛍光のリッドによる不規則反射のためである。対照的に、下方測定は、リッド無しでのプレートの測定と比較して同様の値を示した(図3及び4)。なお、図3及び4において、SF_top及びSF_bottomは、pCGFP_SFでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ表しており、また、AKH_top及びAKH_bottomは、pCGFP_SF+AKHでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ示している。細胞培養中にプレートのリッドを外すことは、コンタミネーションの危険性を大きく増加させる。それゆえ、測定のための最良の方法は、下方測定を使用し、且つ上部カバーを外すことなく培養プレートを測定することである。この方法は、培養物をインタクトに保つことを可能とし、また、単回試験の培養物を使用して細胞培養中のタンパク質産生の時間的経過を追跡することを可能とする。これらの測定パラメータは、細胞質発現及び分泌発現の両方による組換えタンパク質の量の検出で良く機能する(図3及び4)。ベクターpCGFP_SFを使用するGFPの細胞質発現について、タンパク質産生の観察された経時変化は、Sf9発現系を使用して通常みられるものと良く一致した。ベクターpCGFP_SF+AKHを使用するGFPの分泌発現については、検出値は細胞質発現のものよりも小さいとはいえ、インキュベーションの時間による観察された蛍光強度の増加は、培養培地における産生されたタンパク質の蓄積に対応しているようである。
【0026】
(5)結果
上記のプロトコールを4つの異なる試験タンパク質A、B、C、及びDに適用した。これらの試験タンパク質のうち、A、B、及びCでの結果を図7に示す。この結果は、細胞質発現および分泌発現両者において、GFP単独のみならずGFP融合で発現したこれらのタンパク質についてもこの系が良く機能することを示すものであった。観察されたGFP融合試験タンパク質の蛍光強度はGFP単体のものとは異なっており、さらに発現条件が同じである試験タンパク質A、B、及びCの間でも差異が観察された。このことから、各サンプルからの蛍光強度(従って、組換えGFP融合タンパク質発現の量)が、以前の研究で提案されたように(非特許文献1〜3参照)、試験タンパク質がSf9細胞でどれだけの量発現しうるかを反映しており、レポータータンパク質であるGFP単体の発現性には依存しないと考えられることが示唆された。この結果は、本発明の方法が、通常のタンパク質に適用可能であり、また、ハイスループットのサンプルスクリーニングに有用であることを示唆している。
【0027】
比較例1:本発明の方法の定量性の検証
本発明におけるリアルタイム蛍光検出法の定量性の検証のために、結果を、報告されている定量的方法である蛍光検出ゲルろ過クロマトグラフィー(FSEC)(非特許文献2参照)と比較した。FSEC法においては、GFP融合タンパク質の量は、蛍光によって検出されるゲルろ過クロマトグラフィーからの溶出ピーク面積を用いて評価され得る。プレートリーダーでのリアルタイム検出に使用された同一サンプルからの細胞破砕物の上清(細胞質タンパク質検出の場合)及び培養培地(分泌タンパク質検出の場合)をFSECによって分析した。
上記比較の具体的な方法は次の通りである。適当な時間でのタンパク質発現及びマイクロプレートリーダーでの蛍光検出の後、Sf9細胞及び培養培地を別々に採取した。細胞を懸濁バッファー(20 mM Tris-HCl pH 7.5, 150 mM NaCl,及び5 mM KCl)で2度洗浄し、300μlの同じバッファーで懸濁した。その後、細胞をHandy Sonic(TOMY SEIKO)を出力5で2〜3分間使用して氷上で超音波破砕した。サンプルを15k rpmで30分間4℃で遠心分離し、上清をUltrafree-MC(Millipore)でろ過した。培養培地をVivaspin(10k MWCO)(VIVASCIENCE)で濃縮し、懸濁バッファーで300μlまで希釈し、Ultrafree-MCでろ過した。
AKTA explorer instrument(GE Healthcare)に連結した Superose 6 10/300 GL column(GE Healthcare)に、ランニングバッファー(20 mM Tris-HCl pH 7.5, 200 mM NaCl)を用いて流速0.5〜1.0ml/分で200μlのサンプルをロードすることにより、FSEC分析を行った。ゲルろ過クロマトグラフィーの溶出を、励起波長395nm、蛍光波長597nmを使用して、蛍光光度計RF-10AXL(Shimadzu)を用いて検出した。発現したGFPuvタンパク質の量を、GFPuvに由来する溶出ピークの範囲を積分することにより見積もった。
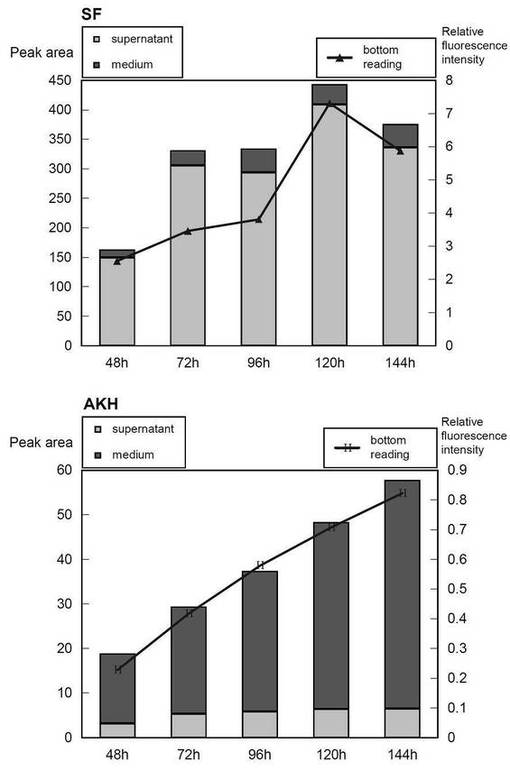
【0028】
上記比較の結果を図6に示す。FSECによる結果は、本発明におけるリアルタイム蛍光検出によって決定されたものとほぼ一致した。顕著なことに、マイクロプレートリーダーの検出範囲は、培養プレートの底に接着した細胞、及び細胞の上の培養培地の両方を含むにも関わらず、pCGFP_SFでトランスフェクションした細胞及びpCGFP_SF+AKHでトランスフェクションした細胞での結果は、これらが対象とするサンプルである、それぞれ、細胞質タンパク質及び分泌タンパク質の量を良く反映していた。
【0029】
[謝辞]
実施例での試験用発現ベクター作成のために用いたpCGFP-BCベクターを提供頂いたコロンビア大学/ハワードヒューズ医学研究所Eric Gouaux教授(現オレゴン健康科学大学ヴォーラム研究所)およびコロンビア大学に感謝します。本研究の一部は文部科学省、ターゲットタンパク研究プログラム「膜タンパク質結晶化の革新的支援法の開発」による援助を受けている。
【図面の簡単な説明】
【0030】
【図1】実施例において使用したSf9細胞用の発現プラスミドの設計を示す図である。
【図2】蛍光顕微鏡によって観察された、Sf9細胞において一過的に発現したGFPuvの蛍光を示す図である。
【図3】マイクロプレートリーダーによって検出された、リッド無しの6ウェル培養プレート中のトランスフェクションされたSf9細胞からのGFPuvの蛍光を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、培養開始から48時間、72時間、96時間、120時間、及び144時間の時点での相対蛍光強度にそれぞれ対応している。
【図4】マイクロプレートリーダーによって検出された、リッド有りの6ウェル培養プレート中のトランスフェクションされたSf9細胞からのGFPuvの蛍光を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、培養開始から48時間、72時間、144時間、及び168時間の時点での相対蛍光強度にそれぞれ対応している。
【図5】リッド無しの従来の透明の培養プレート及び黒色プレートにおけるトランスフェクションされたSf9細胞からのGFPuvの蛍光検出を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、透明プレートでの培養開始から72時間、黒色プレートでの培養開始から72時間、透明プレートでの培養開始から96時間、及び黒色プレートでの培養開始から96時間の時点での相対蛍光強度にそれぞれ対応している。
【図6】FSECを使用したタンパク質産生の見積もり(棒グラフで示した)と本発明の方法(線グラフで示した)との比較を示す図である。縦軸は、FSECからのデータについては溶出ピークの範囲の積分値、本発明の方法からのデータについては相対蛍光強度値を表している。
【図7】本発明の方法を使用した試験タンパク質の発現の評価を示す図である。SF、AKH、タンパク質A、B、C、及びDは、それぞれ、ベクターpCGFP_SF、pCGFP_SF+AKH、タンパク質A、B、C、及びDの発現ベクターでトランスフェクションした細胞での結果を表している。
【技術分野】
【0001】
本発明は、タンパク質発現の測定法に関する。より詳細には、本発明は、培養細胞において発現された目的タンパク質の量を、測定された蛍光強度を指標にして定量する方法に関する。
【背景技術】
【0002】
タンパク質の生化学・物理化学・構造生物学研究などを行うためには、研究対象とするタンパク質を多量に必要とするため、種々の細胞を用いて大量生産させる(発現させる)条件の確立が必須である。特にヒトを含む真核生物由来のタンパク質は、昆虫培養細胞や哺乳類の培養細胞など動物培養細胞を用いて生産させる例が多く見られており、なかでも膜タンパク質については動物培養細胞にて発現させることが不可欠である。ところが、それらの細胞で発現している目的タンパク質の量を調べるにあたっては、培養した細胞を回収し破砕して種々の実験手法を用いて定量する必要があり、最適な発現時間を調べるなどその条件の至適化には多大な試料量と労力が必要であった。
【0003】
近年研究対象とする目的タンパク質を緑色蛍光タンパク質(GFP)融合タンパク質として発現し、細胞の蛍光測定(これまでの報告では大腸菌や酵母)や、その後細胞を破砕し電気泳動やゲルろ過クロマトグラフィーをおこなって蛍光測定を行うことにより、目的タンパク質の発現量を見積もる技術の報告がされている(非特許文献1〜3参照)。しかしながら、従来法では細胞を培養後回収してから蛍光測定を行う必要があり、タンパク質を生産している過程の細胞培養中のタンパク質発現量をリアルタイムで測定することは不可能であった。このため、タンパク質発現の経時変化を測定するには、複数サンプルを準備しそれぞれを異なる時間培養した後回収するという労力が必要であり、さらにサンプル間の細胞増殖のばらつきによる誤差がうまれる危険性があった。さらに、これまで細胞からの蛍光強度の直接測定が報告されている大腸菌や酵母などの微生物は、細胞が不透明であり蛍光強度測定のための励起光や蛍光が細胞によって吸収されてしまうため測定が不正確であり、正確な測定をするためには細胞を破砕し電気泳動やゲルろ過クロマトグラフィーを行って蛍光観測するなどの多大な労力が必要であった。
【非特許文献1】Drew et al., 2006, Nat. Methods 3, 303-313.
【非特許文献2】Kawate et al., 2006, Structure 14, 673-681.
【非特許文献3】Newstead et al., 2007, Proc. Natl Acad. Sci. USA 104, 13936-13941.
【発明の開示】
【発明が解決しようとする課題】
【0004】
本発明は、培養細胞による目的タンパク質の発現量を、通常の細胞培養条件において細胞を回収及び破壊することなくリアルタイムで、簡易且つ正確に測定する方法を提供することをその目的とする。
【課題を解決するための手段】
【0005】
本発明者らは、上記課題を解決するために、目的タンパク質をGFP融合タンパク質として発現し、その蛍光をプレートリーダーを用いて測定することにより当該タンパク質の発現量を測定する方法に着目し、鋭意検討を重ねた。その結果、発現量を調べたい目的タンパク質をGFP融合タンパク質として発現させる発現ベクターを構築し、動物培養細胞に遺伝子導入したものを、通常の細胞培養で行う場合と同じようにマルチウェルプレートを用いて付着培養させ、その培養しているマルチウェルプレートをそのままプレートリーダーを用いて下方測定にて蛍光測定を行うことにより、培養中の目的タンパク質の発現量を見積もることが可能であることを見出し、本発明を完成するに至った。即ち、本発明は以下の通りである。
【0006】
[1]以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
[2]蛍光強度の測定が下方測定である、上記[1]記載の方法。
[3]蛍光強度の測定が培養容器のリッドを外さないで行われる、上記[1]記載の方法。
[4]培養容器の1ウェルあたりの底面積が0.32cm2を上回るものである、上記[1]記載の方法。
[5]蛍光強度の測定が多点測定で行われる、上記[1]記載の方法。
[6]培養容器の容器辺縁部を含まない領域の底面積の70%を超える領域が多点測定に付される、上記[5]記載の方法。
[7]蛍光強度の正規化のために、培養培地のみを含み細胞が含まれていない培養容器における蛍光強度が併せて測定される、上記[1]記載の方法。
[8]細胞が動物培養細胞である、上記[1]記載の方法。
[9]蛍光強度の測定が同一細胞培養槽について継続的に行われる、上記[1]記載の方法。
【発明の効果】
【0007】
本発明の方法では、目的タンパク質の発現を測定するために細胞を回収したり培養プレートのリッド(ふた)を開閉する必要がないため、培養細胞をコンタミネーションのリスクにさらすことなくそのままに保ったまま目的タンパク質の発現をリアルタイムで評価することができ、タンパク質発現の経時測定を1回の培養で同じサンプルから行うことができる。このことによりタンパク質発現条件の確立には重要な要因である発現時間を簡便に決定することが可能である。また、これまで報告されている微生物細胞からの直接蛍光測定と比べ、透明な動物培養細胞を用いることにより、励起光や蛍光の吸収が非常に低くおさえられ、蛍光強度がタンパク質の発現量をより正確に反映していると考えられる。このため、細胞を回収し破砕して電気泳動やゲルろ過クロマトグラフィーなどを用いて行っていた定量を行うことなく、細胞の直接測定による発現量の見積もりが可能である。これらの簡便さから、多種類の目的タンパク質の発現量を一度に比較するハイスループット解析にも適した方法である。
【発明を実施するための最良の形態】
【0008】
本発明は、培養細胞において発現された目的タンパク質の量を定量する方法を提供する。当該方法は、以下の工程を含む:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【0009】
本発明の方法によって定量される前記目的タンパク質とは、培養細胞におけるその発現量を調べたい任意のタンパク質である。原核生物由来又は真核生物由来など、その由来はいかなるものであっても良く、また、分泌タンパク質、膜タンパク質、細胞質若しくは核内に局在するタンパク質など、タンパク質の種類についても限定されない。
【0010】
前記蛍光タンパク質は、融合タンパク質発現に適したものである限り、当該技術分野で使用される任意のものを好適に使用することができ、例えば、GFP、YFP、CFP、BFP、Venusなどのオワンクラゲ由来蛍光タンパク質及びそれらの類似体、ウミシイタケ由来蛍光タンパク質及びその類似体、DsRed、HcRed、AsRed、ZsGreen、ZsYellow、AmCyan、AcGFP、Kaedeなどのサンゴ由来蛍光タンパク質及びそれらの類似体などが挙げられる。使用する蛍光タンパク質の決定は、使用する発現ベクター及び培養細胞、又は価格など様々な要素を考慮してなされ得るが、これは当業者の通常の技術的知識の範囲内である。通常、オワンクラゲ由来のGFP若しくはその類似体(例、EGFP)が好適に使用される。後述するように、本発明の方法においては、目的タンパク質と蛍光タンパク質との融合タンパク質をコードする核酸を適切な発現ベクターに組み込むため、蛍光タンパク質をコードする核酸が必要となるが、それについては例えばクロンテック社などから市販されているベクターを使用すれば良い。
【0011】
目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を作製するためには、該融合タンパク質の適切な発現ベクターを構築し、それを適切な細胞に導入すれば良い。前記細胞としては、タンパク質の製造に通常使用されるあらゆる細胞を限定なく使用することができる。本発明の方法においては、細胞を破砕することなく細胞からの蛍光強度を測定するため、蛍光強度のより正確な測定を可能とするという観点から、大腸菌や酵母などの微生物細胞よりも、透明であり接着培養する性質を持つ動物培養細胞を使用することが好ましい。動物培養細胞の由来としては、哺乳類(例、ハムスター、マウス、サル、ヒトなど)、鳥類(例、ニワトリ、シチメンチョウなど)、爬虫類(例、ヘビなど)、両生類(例、カエルなど)、魚類、昆虫(例、蚊、ハエ、蛾など)などが例示されるが、通常、哺乳類細胞又は昆虫細胞が好ましい。哺乳類由来の培養細胞の例としては、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞、COS細胞、HEK293細胞などが挙げられる。一方、昆虫由来の培養細胞の例としては、Sf9細胞、Sf21細胞、High Five細胞などが挙げられる。
【0012】
融合タンパク質の発現ベクターは、目的タンパク質と蛍光タンパク質との融合タンパク質をコードする塩基配列を含み、且つ、導入された細胞内で融合タンパク質を発現させることができるものでなければならない。
融合タンパク質の発現ベクターは、例えば、目的タンパク質をコードするDNA断片、及び蛍光タンパク質をコードするDNA断片をそれぞれ切り出し、それらを適当な発現ベクター中のプロモーターの下流に連結することによって製造することができる。そのとき、目的タンパク質及び蛍光タンパク質が、融合した一つのタンパク質として合成される必要がある。その方法は、市販の蛍光タンパク質をコードする塩基配列を使用する場合は製造者による説明書を参照すれば良く、或いは多くの標準的ラボラトリーマニュアルに記載の常法に従って行えば良い。
融合タンパク質の遺伝子が連結される発現ベクターとしては、大腸菌由来のプラスミド(例、pBR322,pBR325,pUC12,pUC13)、枯草菌由来のプラスミド(例、pUB110,pTP5,pC194)、酵母由来プラスミド(例、pSH19,pSH15)、λファージなどのバクテリオファージ、レトロウイルス,ワクシニアウイルス,バキュロウイルスなどの動物ウイルスなどの他、pA1−11、pXT1、pRc/CMV、pRc/RSV、pcDNAI/Neoなどが用いられる。
前記プロモーターとしては、遺伝子の発現に用いる培養細胞に対応して適切なプロモーターであればいかなるものでもよい。例えば、動物細胞を用いる場合は、SRαプロモーター、SV40プロモーター、LTRプロモーター、CMVプロモーター、HSV-TKプロモーターなどが挙げられる。これらのうち、CMV(サイトメガロウイルス)プロモーター、SRαプロモーターなどを用いるのが好ましい。昆虫細胞を用いる場合は、IE1プロモーター、ポリヘドリンプロモーター、P10プロモーターなどが挙げられる。これらのうち、IE1プロモーターを用いるのが好ましい。
発現ベクターには、以上の他に、所望によりエンハンサー、スプライシングシグナル、ポリA付加シグナル、選択マーカー、SV40複製オリジンなどを含有しているものを用いることができる。選択マーカーとしては、例えば、ジヒドロ葉酸還元酵素遺伝子〔メソトレキセート(MTX)耐性〕、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子〔G418耐性〕などが挙げられる。また、必要に応じて、使用する培養細胞に合ったシグナル配列を、目的タンパク質のN端末側に付加しても良い。
このようにして構築された前記融合タンパク質をコードするDNAを含有する発現ベクターを用いて、形質転換体を製造することができる。
【0013】
発現ベクターの培養細胞への導入は、細胞染色体への安定な組込みを目的とするものであっても、一過性の導入であってもよい。導入は、自体公知の方法で行えばよく、例えば、エレクトロポレーション、リポフェクション、リン酸カルシウム法(CaPi)、ポリフェクション法(PEI)、塩化カルシウム法(カルフェクション)などのトランスフェクションが挙げられる。かかる方法は、多くの標準的ラボラトリーマニュアルに記載されている。
【0014】
細胞を培養する方法は当該分野において公知である(例えば、Tissue Engineering Methods and Protocols,Morgan and Yarmush(eds.),Humana Press,Inc.,Totowa,NJ,1999参照)。当業者なら認識しているように、細胞が培養される条件は、細胞のタイプによって変わる。該条件には、環境の温度、細胞を含む培養容器、細胞培養雰囲気又は環境を構成する、様々なガス(例えば、CO2)の組成、細胞が維持される培地、培地の成分及びpH、細胞が維持される密度、培地を新しい培地に交換することを必要とするスケジュールなどが含まれる。これらのパラメータは当該分野においてはしばしば知られているものであるか、経験的に決定されうる。
培養細胞が動物細胞である場合、培地としては、例えば、約5〜20%の胎児牛血清を含むMEM培地〔Science,122巻,501(1952)〕,DMEM培地〔Virology,8巻,396(1959)〕,RPMI 1640培地〔The Journal of the American Medical Association,199巻,519(1967)〕,199培地〔Proceeding of the Society for the Biological Medicine,73巻,1(1950)〕などが用いられる。pHは約6〜8であるのが好ましい。培養は通常約30〜40℃で約15〜60時間行い、必要に応じて通気や撹拌を加える。また、培養細胞が昆虫細胞である場合、培地としては、Grace's Insect Medium(Nature,195,788(1962))に非動化した10%ウシ血清等の添加物を適宜加えたものなどが用いられる。培地のpHは約6.2〜6.4に調整するのが好ましい。培養は通常約27℃で約3〜5日間行い、必要に応じて通気や撹拌を加える。
【0015】
前記細胞培養を行うための培養容器は、細胞を培養するための培地を保持するものであって、且つ、後述するように、培養細胞を培養容器外へ移さずに蛍光強度を測定することを可能とするものであれば、特に限定されない。上記条件を満たす限りにおいて、培養容器として、例えば、マイクロプレート、チャンバ又は複数チャンバを備えた培養スライド、カバーガラス、カップ、フラスコ、チューブ、ローラーボトル、スピナーボトルなどを使用することが可能であるが、本発明の方法における蛍光強度測定は、通常マイクロプレートリーダーを使用して行われるため、好ましくはマイクロプレートが使用される。
上記マイクロプレートとしては、その各ウェルの底面積があまりに小さいと、再現性のある細胞増殖を維持することが出来ないこと、培養培地が干上がってしまう可能性があること、及び蛍光強度の測定において多点測定を許容しないことなどの欠点が生じ、そのため再現性のある結果が得られない傾向があるため、充分な大きさの底面積を持つものである必要がある。具体的には、例えば、通常市販されている96ウェルプレートの底面積である0.32cm2を上回る底面積を持つものが好ましく使用される。底面積の上限としては、蛍光強度の測定を許容する限り特に制限はないが、通常500cm2以下である。ここで、底面積は0.5〜20.0cm2がより好ましく、1.8〜9.6cm2がさらに好ましい。また、マイクロプレート以外の培養容器についても、同様に、充分な底面積を持つものである必要がある。
マイクロプレートのウェルの形状としては、平底、U底、及びV底のものなどがあるが、本発明の方法のためには、マイクロプレートリーダーでの測定焦点高さが通常固定されているという理由から、接着培養を行う際細胞が同一高さに分布する平底が好ましい。
接着培養の際細胞が付着するマイクロプレートのウェル底面の材質としては、ポリスチレン、白色ポリスチレン、ポリエチレン、ナチュラルポリプロピレン、ガラス入りポリプロピレン、又はバレックスなどのプラスチック、或いは石英ガラス又は硼珪酸ガラスなどのガラスが挙げられ、培養の際の細胞の接着性が優れており、低価格であるという理由から、プラスチック製、特にポリスチレン製のマイクロプレートが本発明の方法において好適に使用され得る。さらに蛍光観測する要請から、その底面は透明である必要があり、ポリスチレン製のものはこの用件も満たす。
上記マイクロプレートは、コーニング社製マルチプルウェルプレートなど市販のものが通常使用され、上記底面積に関する条件を満足するために、24穴以下のウェル(例、6穴、12穴、24穴など)を持つマイクロプレートが好ましく使用され得る。
また、マイクロプレート本体部分には、透明、白色、及び黒色のものなどがあるが、白色又は黒色のマイクロプレートは、透明のマイクロプレートと比較して一般的に高価であり、また市販のこれらのプレートの底面材質が必ずしも細胞接着培養に好適でないことを考慮すると、透明のものが好ましい。
以上のことから、本発明の方法において、特に好ましく使用されるマイクロプレートの例として、6穴又は24穴のポリスチレン製の平底透明マイクロプレートが挙げられる。
【0016】
本発明の方法においては、前記融合タンパク質の蛍光強度の測定は、培養細胞を破壊せず、且つ培養容器外へ移さずに行う必要がある。係る条件を満足する限り、当該分野で使用される任意の蛍光強度測定法が使用され得るが、感度、精度及び簡便性の観点から、マイクロプレートリーダーによる蛍光強度測定が好ましい。マイクロプレートリーダーとしては、例えば、サーモフィッシャーサイエンティフィック社製ヴァリオスキャンフラッシュなどが挙げられる。また、蛍光強度の測定は、使用する測定機器に付属のマニュアルを参照して、当業者は実施することが可能である。
【0017】
マイクロプレートリーダーによる蛍光強度の測定は、培養容器中の複数の点で測定された蛍光強度の平均値を容器全体における蛍光強度として得るものである多点測定により行うことが、信号対雑音比を最小限に抑える観点から好ましい。その場合、培養容器の容器辺縁部を含まない領域(以下、「測定領域」ともいう)の底面積の70%を超える領域が多点測定に付されることが好ましい。多点測定に付すべき領域の底面積の上限は特にないが、測定に要する時間を考慮すると、測定領域の95%が好ましく、90%がより好ましく、85%がさらに好ましく、80%が特に好ましい。ここで、容器辺縁部とは、容器縁部からの距離が、3mmもしくは測定装置の測定光のビーム幅の2倍の距離のいずれか大きい方の値をもつ距離の範囲内にある容器内の部分を意味する。また、上述したように本発明における蛍光強度の測定は、培養細胞を培養容器外へ移さずに測定するため、培養容器中への雑菌混入を防ぐという観点から、培養容器のリッドを外すことなく蛍光強度を測定することが好ましい。リッド付きの培養容器から蛍光強度を測定するとき、上方測定、即ちリッドのある方向からの蛍光強度測定は、励起光及び蛍光のリッドによる不規則反射などが理由で、蛍光物質なしのコントロールにおいても高い蛍光強度値を示し、そのため信号対雑音比が低くなるという問題があるため、本発明における蛍光強度の測定においては、下方測定、即ち、培養容器の底面側からの蛍光強度を測定することが好ましい。
【0018】
上述したように本発明における蛍光強度の測定は、細胞を破砕せず、且つ培養容器外へ移さずに測定するものであるため、蛍光強度の測定を同一細胞培養槽について継続的に行うことが可能である。従って、タンパク質発現の経時測定を1回の培養で同じサンプルから行うことができき、タンパク質発現条件の確立には重要な要因である発現時間を簡便に決定することが可能である。
【0019】
上述したように、本発明の方法は、測定された蛍光強度を指標に、目的タンパク質の発現量を算出することを含む。目的タンパク質の発現量の算出のために、測定された蛍光強度は、適当なコントロール由来の蛍光強度によって正規化されることが好ましい。係るコントロールとして、培養細胞が含まれておらず、細胞培養に使用したものと同種の培養培地のみを含む培養容器を利用するのが、再現性の高い結果を得られるという観点から好ましい。特にマルチウェルマイクロプレートを使用する場合、蛍光強度測定サンプルの同じ培養プレートにおける、サンプルの培養に用いたものと同種および同量の培養培地で充填されたウェルからの蛍光強度測定値を使用することが好ましい。その場合、マイクロプレートの各ウェルでの蛍光強度Fは;
相対蛍光強度=(F−F0)/F0
(ここで、F0は、測定サンプルと同じ培養プレート中の、培養培地で充填されたウェルからの蛍光強度の測定値である);
という方程式で正規化される。こうして得られる相対蛍光強度を指標にして、目的タンパク質の発現量を定量することができる。
【0020】
以下、実施例などにより本発明を更に詳しく説明するが、本発明は下記実施例などに何ら制限されるものではない。
【実施例】
【0021】
実施例1
(1)発現ベクターの構築
下記に記載の通り、昆虫培養細胞において細胞質発現及び分泌発現という二つの種類のタンパク質発現を試験するベクターを構築した(図1)。細胞質発現のための試験ベクターpCGFP_SFは、マルチクローニングサイト、トロンビン切断部位及びヘキサ・ヒスチジンタグを持つ改変GFPuv(UV領域における励起極大を持つGFPの変異体)の遺伝子を、ベクターpCGFP−BC(Eric Gouaux教授の好意により提供された。非特許文献2)を鋳型にして従来のPCR法で増幅し、当該フラグメントを制限酵素DraIIIで処理したものをpIEx4ベクター(Novagen)のNcoI部位(平滑末端化した)とDraIII部位との間に挿入した。分泌発現のための試験ベクターであるベクターpCGFP_SF+AKHは、昆虫由来の分泌シグナル配列であるadipokinetic hormone(AKH)シグナル配列をpcGFP_SFベクターのマルチクローニングサイトの上流側に連結できる様にオリゴヌクレオチドプライマーを設計し(AKHシグナル配列はpIEx-5ベクター(novagen)中に含まれる配列を参考にして設計)、通常のPCR法を用いてpcGFP_SFベクターと同様に構築した(図1)。下記実施例1における試験タンパク質のための発現ベクターの構築のために、分泌シグナルを除去したヒト由来味覚受容体リガンド結合領域T1r1(456残基、タンパク質A)、T1r2(454残基、タンパク質B)、T1r3(458残基、タンパク質C)及び分泌シグナルを含むラット由来代謝型グルタミン酸受容体1リガンド結合領域(522残基、タンパク質D)の遺伝子をpCGFP_SFベクターにサブクローニングして、C末端GFP融合とした。
【0022】
(2)発現系
本発明における組換えタンパク質発現のリアルタイム検出の評価のために、一過性タンパク質発現系でSf9細胞を使用した。Sf9発現系は、構造生物学研究分野において最も一般的に使用される組換えタンパク質発現のための系の一つである。大腸菌及び酵母細胞といった微生物での発現系とは対照的に、Sf9細胞は、他の動物培養細胞と同様、透明であり、生体細胞における発現GFPの蛍光強度のより正確な測定を可能とする。通常の動物細胞について最も標準的且つ広く適用される培養方法である単層培養(細胞は、培養プレートの底に接着する)に細胞を付した。
組換えタンパク質の発現は、具体的には次のようにして行った。Insect Genejuice(Novagen)を製造者のプロトコールに従って使用して、発現ベクターをSf9細胞(GIBCO)にトランスフェクションした。トランスフェクションした細胞を、様々の種類の組織培養プレートにおいてSf900III SFM(GIBCO)中で48〜120時間、27℃でインキュベーションし、一過的な組換えタンパク質発現をさせておいた。典型的に、融合性単層培養からのSf9細胞を6ウェル組織培養プレート(6 Well Clear TC-Treated microplates,Costar)中の各ウェルに1.0〜1.6×106細胞/ウェルの密度で添加し、その後ウェルを、10μlのInsect Genejuice及び2μgのプラスミドを含有する1mlのSf900III SFMで充填した。培養プレート中のウェルの一つを等量のSf900III SFMで充填し、コントロール(バックグラウンド)として使用した。
【0023】
(3)培養プレートの種類
様々な細胞培養プレートを試験するために、細胞を、トランスフェクション後、6、24及び96ウェルプレートという3つの異なる種類の培養プレート中でインキュベーションした。これらのプレートのうちで、96ウェルプレートは再現性のある結果を殆ど与えなかった。96ウェルプレートのウェルは、再現性のある細胞増殖を維持するためには小さ過ぎ、しばしば培養培地の干上がりに直面した。更に、96ウェルプレートのシングルウェルは、そのサイズが小さいため一点測定のみ許容し、それが更に検出中の測定エラーを増している(データ示さず)。一方、6ウェル及び24ウェルプレートで培養された細胞は、安定した細胞増殖を示し、また、これらのウェルのサイズは多点測定を可能とし、単一ウェルの様々な領域での値を平均化することが出来、従ってウェルの細胞数の状態をより正確に反映することが出来る。次に、従来のポリスチレンの透明培養プレート(6 Well Clear TC-Treated Microplates,Costar)を、蛍光測定に好適であることが示唆されているガラス底の黒色培養プレート(EZview glass bottom culture plate LB,6ウェル,Iwaki)と比較した。しかしながら、これら2つのプレート間での検出したS/N比(相対蛍光強度の値)は、細胞質及び分泌タンパク質発現の両方について、2つの異なる時点においてほぼ同じであった(図5)。なお、図5において、SF_top及びSF_bottomは、pCGFP_SFでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ表しており、また、AKH_top及びAKH_bottomは、pCGFP_SF+AKHでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ示している。黒色培養プレートは高価であり、また必ずしも細胞培養に好適でないことを考慮すると、従来の6ウェル又は24ウェルのポリスチレンの透明組織培養プレートの使用が好適であり、リアルタイム蛍光検出のための充分な正確性を与えると考えられる。
【0024】
(4)マイクロプレートリーダーを使用する蛍光検出のための測定パラメータの決定
生体Sf9細胞におけるGFPuvの蛍光強度の正確な測定のために、マイクロプレートリーダーVarioskan Flash(Thermo Fisher Scientific)での検出のための測定パラメータを決定した。最初に、測定した蛍光強度の正規化のための適切な方法を見出すことを試みた。トランスフェクションしていないSf9細胞(プラスミドを用いないでリポフェクション試薬で処理)、及び培養培地という二つの条件を比較し、どちらが測定のための適切なコントロールとして機能するものであるのか評価した。これらの条件での蛍光強度値は、ほぼ同様の範囲であったが、コントロールとして培養培地を使用する方が、トランスフェクションしていないSf9細胞を使用するより再現性の高い値という結果であった(データ示さず)。トランスフェクションしていないSf9細胞は、トランスフェクションした細胞よりも、非常に高い細胞増殖及び蛍光強度を時に示し、その結果、正規化に使用するのに不適当なほど小さいサンプル相対蛍光強度値となる。従って、培養培地をコントロールとして採用した。
以下の実施例1及び比較例1においては、組織培養プレート中の細胞増殖において発現したGFPuvに由来する蛍光強度を、励起波長395nm、蛍光波長507nmを使用してマイクロプレートリーダーVarioskan Flash(Thermo Fisher Scientific)を用いて検出した。典型的な測定条件は、測定時間500ms及びバンド幅12nmでの多点測定(ウェルの中心からの円形領域であって測定領域の80%)にセットした。なお、今回の実験では、ウェルの測定領域の80%を超える領域の多点測定の試験は行わなかった。培養プレートの各ウェルにおいて測定された蛍光強度Fを:
相対蛍光強度=(F−F0)/F0
(ここで、F0は、測定サンプルの同じ培養プレート中のウェルに充填された培養培地の蛍光強度を表す)
という方程式で正規化した。
【0025】
次に、マイクロプレートリーダーでの蛍光検出のための様々な測定パラメータを試験した。信号対雑音比(S/N比)(相対蛍光強度の値)の観点から、以下の条件が最良の結果を与えた;測定方式は多点にセットし、測定時間は500ms、バンド幅は12nmであった。多点測定で試験される領域のうち、ウェルの測定領域の底面積の約70〜80%をカバーするウェルの中心からの円形領域が最良の結果を与えた(データ示さず)。また、上方測定及び下方測定という二つの蛍光検出法を比較した。培養プレートの上部カバーを外すプレートの下方測定及び上方測定は、同様の値を与えることをその結果は示していた(図3)。しかしながら、リッド付きのプレートの読み取りの場合、上方測定は、高いバックグラウンド(コントロールにおける高い蛍光強度値)を示し、その結果低いS/N比であったが(図4)、これは恐らく、励起光及び蛍光のリッドによる不規則反射のためである。対照的に、下方測定は、リッド無しでのプレートの測定と比較して同様の値を示した(図3及び4)。なお、図3及び4において、SF_top及びSF_bottomは、pCGFP_SFでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ表しており、また、AKH_top及びAKH_bottomは、pCGFP_SF+AKHでトランスフェクションした細胞の上方測定又は下方測定の結果をそれぞれ示している。細胞培養中にプレートのリッドを外すことは、コンタミネーションの危険性を大きく増加させる。それゆえ、測定のための最良の方法は、下方測定を使用し、且つ上部カバーを外すことなく培養プレートを測定することである。この方法は、培養物をインタクトに保つことを可能とし、また、単回試験の培養物を使用して細胞培養中のタンパク質産生の時間的経過を追跡することを可能とする。これらの測定パラメータは、細胞質発現及び分泌発現の両方による組換えタンパク質の量の検出で良く機能する(図3及び4)。ベクターpCGFP_SFを使用するGFPの細胞質発現について、タンパク質産生の観察された経時変化は、Sf9発現系を使用して通常みられるものと良く一致した。ベクターpCGFP_SF+AKHを使用するGFPの分泌発現については、検出値は細胞質発現のものよりも小さいとはいえ、インキュベーションの時間による観察された蛍光強度の増加は、培養培地における産生されたタンパク質の蓄積に対応しているようである。
【0026】
(5)結果
上記のプロトコールを4つの異なる試験タンパク質A、B、C、及びDに適用した。これらの試験タンパク質のうち、A、B、及びCでの結果を図7に示す。この結果は、細胞質発現および分泌発現両者において、GFP単独のみならずGFP融合で発現したこれらのタンパク質についてもこの系が良く機能することを示すものであった。観察されたGFP融合試験タンパク質の蛍光強度はGFP単体のものとは異なっており、さらに発現条件が同じである試験タンパク質A、B、及びCの間でも差異が観察された。このことから、各サンプルからの蛍光強度(従って、組換えGFP融合タンパク質発現の量)が、以前の研究で提案されたように(非特許文献1〜3参照)、試験タンパク質がSf9細胞でどれだけの量発現しうるかを反映しており、レポータータンパク質であるGFP単体の発現性には依存しないと考えられることが示唆された。この結果は、本発明の方法が、通常のタンパク質に適用可能であり、また、ハイスループットのサンプルスクリーニングに有用であることを示唆している。
【0027】
比較例1:本発明の方法の定量性の検証
本発明におけるリアルタイム蛍光検出法の定量性の検証のために、結果を、報告されている定量的方法である蛍光検出ゲルろ過クロマトグラフィー(FSEC)(非特許文献2参照)と比較した。FSEC法においては、GFP融合タンパク質の量は、蛍光によって検出されるゲルろ過クロマトグラフィーからの溶出ピーク面積を用いて評価され得る。プレートリーダーでのリアルタイム検出に使用された同一サンプルからの細胞破砕物の上清(細胞質タンパク質検出の場合)及び培養培地(分泌タンパク質検出の場合)をFSECによって分析した。
上記比較の具体的な方法は次の通りである。適当な時間でのタンパク質発現及びマイクロプレートリーダーでの蛍光検出の後、Sf9細胞及び培養培地を別々に採取した。細胞を懸濁バッファー(20 mM Tris-HCl pH 7.5, 150 mM NaCl,及び5 mM KCl)で2度洗浄し、300μlの同じバッファーで懸濁した。その後、細胞をHandy Sonic(TOMY SEIKO)を出力5で2〜3分間使用して氷上で超音波破砕した。サンプルを15k rpmで30分間4℃で遠心分離し、上清をUltrafree-MC(Millipore)でろ過した。培養培地をVivaspin(10k MWCO)(VIVASCIENCE)で濃縮し、懸濁バッファーで300μlまで希釈し、Ultrafree-MCでろ過した。
AKTA explorer instrument(GE Healthcare)に連結した Superose 6 10/300 GL column(GE Healthcare)に、ランニングバッファー(20 mM Tris-HCl pH 7.5, 200 mM NaCl)を用いて流速0.5〜1.0ml/分で200μlのサンプルをロードすることにより、FSEC分析を行った。ゲルろ過クロマトグラフィーの溶出を、励起波長395nm、蛍光波長597nmを使用して、蛍光光度計RF-10AXL(Shimadzu)を用いて検出した。発現したGFPuvタンパク質の量を、GFPuvに由来する溶出ピークの範囲を積分することにより見積もった。
【0028】
上記比較の結果を図6に示す。FSECによる結果は、本発明におけるリアルタイム蛍光検出によって決定されたものとほぼ一致した。顕著なことに、マイクロプレートリーダーの検出範囲は、培養プレートの底に接着した細胞、及び細胞の上の培養培地の両方を含むにも関わらず、pCGFP_SFでトランスフェクションした細胞及びpCGFP_SF+AKHでトランスフェクションした細胞での結果は、これらが対象とするサンプルである、それぞれ、細胞質タンパク質及び分泌タンパク質の量を良く反映していた。
【0029】
[謝辞]
実施例での試験用発現ベクター作成のために用いたpCGFP-BCベクターを提供頂いたコロンビア大学/ハワードヒューズ医学研究所Eric Gouaux教授(現オレゴン健康科学大学ヴォーラム研究所)およびコロンビア大学に感謝します。本研究の一部は文部科学省、ターゲットタンパク研究プログラム「膜タンパク質結晶化の革新的支援法の開発」による援助を受けている。
【図面の簡単な説明】
【0030】
【図1】実施例において使用したSf9細胞用の発現プラスミドの設計を示す図である。
【図2】蛍光顕微鏡によって観察された、Sf9細胞において一過的に発現したGFPuvの蛍光を示す図である。
【図3】マイクロプレートリーダーによって検出された、リッド無しの6ウェル培養プレート中のトランスフェクションされたSf9細胞からのGFPuvの蛍光を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、培養開始から48時間、72時間、96時間、120時間、及び144時間の時点での相対蛍光強度にそれぞれ対応している。
【図4】マイクロプレートリーダーによって検出された、リッド有りの6ウェル培養プレート中のトランスフェクションされたSf9細胞からのGFPuvの蛍光を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、培養開始から48時間、72時間、144時間、及び168時間の時点での相対蛍光強度にそれぞれ対応している。
【図5】リッド無しの従来の透明の培養プレート及び黒色プレートにおけるトランスフェクションされたSf9細胞からのGFPuvの蛍光検出を示す図である。縦軸は相対蛍光強度値を表している。各コラム左より、透明プレートでの培養開始から72時間、黒色プレートでの培養開始から72時間、透明プレートでの培養開始から96時間、及び黒色プレートでの培養開始から96時間の時点での相対蛍光強度にそれぞれ対応している。
【図6】FSECを使用したタンパク質産生の見積もり(棒グラフで示した)と本発明の方法(線グラフで示した)との比較を示す図である。縦軸は、FSECからのデータについては溶出ピークの範囲の積分値、本発明の方法からのデータについては相対蛍光強度値を表している。
【図7】本発明の方法を使用した試験タンパク質の発現の評価を示す図である。SF、AKH、タンパク質A、B、C、及びDは、それぞれ、ベクターpCGFP_SF、pCGFP_SF+AKH、タンパク質A、B、C、及びDの発現ベクターでトランスフェクションした細胞での結果を表している。
【特許請求の範囲】
【請求項1】
以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【請求項2】
蛍光強度の測定が下方測定である、請求項1記載の方法。
【請求項3】
蛍光強度の測定が培養容器のリッドを外さないで行われる、請求項1記載の方法。
【請求項4】
培養容器の1ウェルあたりの底面積が0.32cm2を上回るものである、請求項1記載の方法。
【請求項5】
蛍光強度の測定が多点測定で行われる、請求項1記載の方法。
【請求項6】
培養容器の容器辺縁部を含まない領域の底面積の70%を超える領域が多点測定に付される、請求項5記載の方法。
【請求項7】
蛍光強度の正規化のために、培養培地のみを含み細胞が含まれていない培養容器における蛍光強度が併せて測定される、請求項1記載の方法。
【請求項8】
細胞が動物培養細胞である、請求項1記載の方法。
【請求項9】
蛍光強度の測定が同一細胞培養槽について継続的に行われる、請求項1記載の方法。
【請求項1】
以下の工程を含む、培養細胞において発現された目的タンパク質の量を定量する方法:
(1)目的タンパク質と蛍光タンパク質との融合タンパク質を発現し得る細胞を培養容器中で培養すること;
(2)(1)において培養された細胞において発現した融合タンパク質の蛍光強度を、該細胞を破砕せず、且つ培養容器外へ移さずに測定すること;及び
(3)測定された蛍光強度を指標に、目的タンパク質の発現量を算出すること。
【請求項2】
蛍光強度の測定が下方測定である、請求項1記載の方法。
【請求項3】
蛍光強度の測定が培養容器のリッドを外さないで行われる、請求項1記載の方法。
【請求項4】
培養容器の1ウェルあたりの底面積が0.32cm2を上回るものである、請求項1記載の方法。
【請求項5】
蛍光強度の測定が多点測定で行われる、請求項1記載の方法。
【請求項6】
培養容器の容器辺縁部を含まない領域の底面積の70%を超える領域が多点測定に付される、請求項5記載の方法。
【請求項7】
蛍光強度の正規化のために、培養培地のみを含み細胞が含まれていない培養容器における蛍光強度が併せて測定される、請求項1記載の方法。
【請求項8】
細胞が動物培養細胞である、請求項1記載の方法。
【請求項9】
蛍光強度の測定が同一細胞培養槽について継続的に行われる、請求項1記載の方法。
【図1】



【図2】

【図3】

【図4】
【図5】

【図6】
【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2009−183256(P2009−183256A)
【公開日】平成21年8月20日(2009.8.20)
【国際特許分類】
【出願番号】特願2008−29624(P2008−29624)
【出願日】平成20年2月8日(2008.2.8)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度、文部科学省、ターゲットタンパク研究プログラム「膜タンパク質結晶化の革新的支援法の開発」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
【公開日】平成21年8月20日(2009.8.20)
【国際特許分類】
【出願日】平成20年2月8日(2008.2.8)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成19年度、文部科学省、ターゲットタンパク研究プログラム「膜タンパク質結晶化の革新的支援法の開発」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(503359821)独立行政法人理化学研究所 (1,056)
【Fターム(参考)】
[ Back to top ]