
基質結合環状有機化合物の組み合わせライブラリー
【課題】固相出発物質上で合成された複素環式有機化合物の組み合わせライブラリーの合成、およびそのようなライブラリーを生物活性についてアッセイする方法の提供。
【解決手段】環状有機化合物のライブラリー、およびそのようなライブラリーを作製し、アッセイする方法であって、それぞれの環状有機化合物は、出発樹脂を用いて誘導体化された固体表面の形態の出発物質から構成される。化合物は、この樹脂と反応し、環状基を付加するか、またはそれを形成し、これらの反応は、ライブラリーのサイズを大きくするように、それぞれ異なる化合物が、複数のサブアマウントと反応可能であるように、分割樹脂手順を用いて行われるもの。
【解決手段】環状有機化合物のライブラリー、およびそのようなライブラリーを作製し、アッセイする方法であって、それぞれの環状有機化合物は、出発樹脂を用いて誘導体化された固体表面の形態の出発物質から構成される。化合物は、この樹脂と反応し、環状基を付加するか、またはそれを形成し、これらの反応は、ライブラリーのサイズを大きくするように、それぞれ異なる化合物が、複数のサブアマウントと反応可能であるように、分割樹脂手順を用いて行われるもの。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、広くは、化学合成技術に関する。具体的には、本発明は、固相出発物質上で合成された複素環式有機化合物の組み合わせライブラリーの合成、およびそのようなライブラリーを生物活性についてアッセイする方法に関する。
【背景技術】
【0002】
発明の背景
ペプチド合成のための伝統的な固相方法に類似する標準的な方法が、ペプチドおよびペプチド様化合物のライブラリーを合成するのに適用され得る。このような方法によれば、N,α-Fmoc-保護(および側鎖保護)アミノ酸のカルボキシレートが、活性化され得、その後、樹脂結合アミノ基に結合され得る。Fmoc基は、その後、次のモノマーの付加に続いて除去される。このようなアプローチは、保護されたN-置換アミノ酸モノマーの多様なセットを適切な量だけ調製するのに要する時間およびコストのため、望ましくない。Fmocまたはその他の保護基を付加し、除去することは、時間のかかることであり、非効率的である。
【0003】
新しい薬学的に活性な有機薬剤(すなわち、結合に必要な3-D構造をもつ化合物)発見のためのあるアプローチは、主として、精製されたレセプターのX線結晶学に依存している。すなわち、いったん結合部位が同定されると、有機分子は、利用可能な立体空間および電荷分布に適合するように設計される。しかし、精製されたレセプターを得るのは、しばしば困難であり、X線結晶学が適用され得るようにレセプターを結晶化させるのは、さらに困難である。また、結合部位が正しく同定された後でも、適切なリガンドを考案することは、取るに足らないことではない。全体として、有用で薬学的に活性な化合物を設計することは、例えば、レセプターを同定し、それらのレセプターに結合する化合物の構造を精製・同定し、その後それらの化合物を合成することに伴う困難のようないくつかの要因のため、極めて困難である。
【0004】
新しい薬剤発見のための別のアプローチとしては、既知の生物学的に活性な化合物を模倣する化合物を合成することがある。しかし、活性化合物の活性部分または活性構造成分は通常未知であるので、新しい化合物を合成するプロセスは、主として、それぞれの化合物を個別に試行錯誤の末、合成し、スクリーニングすることに依存している。この方法は、時間がかかり、高価である。なぜなら、どの1つの化合物についても、成功の可能性は比較的低いからである。
【0005】
結晶学を用いたり、既知の生物学的に活性なペプチドを模倣する特定のペプチドを合成しようと試みたりすることにより、タンパク質の特定の3次元構造を決定しようとすることよりもむしろ、組み合わせライブラリーを作製することに関する技術が発展してきている。具体的には、生物学的に活性なペプチドを単離しようと試みるものは、同一の反応容器内で極めて多数の異なるペプチドを同時に生成する。合成された組み合わせライブラリーは、その後、アッセイされ、活性分子が単離され、分析される。組み合わせライブラリー自体は、米国特許第5,266,684号に開示されている。米国特許第'684号は、ほぼ完全に、ライブラリー中の反応生成物のそれぞれが、20個の天然に存在するアミノ酸からなるペプチドである、ライブラリーの合成に関している。
【0006】
薬学的に活性な化合物は、しばしば高度に置換された複素環であるので、本発明者らは、多数の関連する置換された複素環式化合物を迅速に、かつ比較的安価に、速やかに合成する方法の必要性を見出した。このアプローチによれば、生物活性を呈する構造成分が未知である、候補化合物群のそれぞれのメンバーについて個別に合成をおこなう問題を克服し得る。
【発明の開示】
【課題を解決するための手段】
【0007】
本発明によって以下が提供される:
(1)ライブラリーを合成する方法であって、
(a)複数の固体支持体表面を提供する工程と、
(b)XHを用いて該表面のそれぞれを誘導体化する工程であって、Xが、以下のような複数の該誘導体化された支持体表面を提供するOおよびNHからなる群から選択される、工程と、
P - XH
ここで、「P」は、該支持体表面の1つであり、
(c)該誘導体化された支持体表面を、複数のサブアマウントに分割する工程と、
(d)以下の一般構造式の異なるサブモノマーと、該誘導体化された支持体表面の該サブアマウントの該-XH基とを反応させる工程と、
【0008】
【化1−1】

【0009】
ここで、R1およびR2は、独立して、炭素原子に共有結合可能な任意の部分であり、Zは、ハロゲンであり、各反応を完了させて、
【0010】
【化1−2】
【0011】
を得る工程と、
(e)該各サブアマウントを再び組み合わせて混合し、複数の異なる化合物を有する固体支持体の混合物を得る工程と、
(f)該混合物を複数のサブアマウントに分割する工程と、
(g)以下の一般構造式の異なる環状化合物と、該各サブアマウントの該-Z部分とを反応させ、
【0012】
【化1−3】
【0013】
ここで、Yは、-O-、-S-、および-NHからなる群から選択され、A1、A2、A3、A4およびA5はそれぞれ、独立して、H、ヒドロカルビル、ケトン、アルデヒド、カルボン酸、エステル、アミド、アミン、ニトリル、およびエーテルからなる群から選択される部分であり、以下の構造の化合物を得る工程と、
【0014】
【化1−4】
【0015】
(h)該サブアマウントを再び組み合わせて、該複数の固体支持体表面上に化合物のライブラリーを提供する工程と、
を包含する方法。
(2)XがNHであり、ZがBrであり、R1およびR2が、それぞれ独立して、Hまたはヒドロカルビルであり、A1、A2、A3、A4およびA5のそれぞれが独立してHまたはヒドロカルビルである、項目1に記載の方法。
(3)R1、R2、A1、A2、A3、A4、およびA5のそれぞれが独立して、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目1に記載の方法。
(4)R1、R2、A1、A2、A3、A4、およびA5のそれぞれが独立して、H、1から6個の炭酸原子を含むアルキル、および20個の天然に存在するアミノ酸の1つからなる群から選択され、各分割工程において、分割が3またはそれ以上のサブアマウントへの分割である、項目1に記載の方法。
(5) 固体支持体に共有結合した10個またはそれ以上の異なる化合物によって特徴づけられる有機化合物のライブラリーであって、該化合物が、電子供与出発物質を結合した固体支持体から得られる環状有機化合物であり、
該10個またはそれ以上の化合物のそれぞれが、回収可能かつ分析可能な量で存在する、ライブラリー。
(6)少なくとも1000個の異なる化合物を含み、該生成化合物の少なくとも1つが生物学的に活性である、項目11に記載のライブラリー。
(7)前記電子供与出発物質が、アミドを介して固体支持体に結合したアルデヒドであり、前記環状有機化合物のそれぞれが、100ピコモルまたはそれ以上の量で存在し、さらに、該環状有機化合物の環状部分が、該環状部分に共有結合した側鎖を有する、項目6に記載のライブラリー。
(8)前記側鎖が、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目7に記載のライブラリー。
(9)10個またはそれ以上の環状有機化合物のライブラリーを固体支持体上で合成する方法であって、
該化合物の官能部分が、アクセス可能かつ反応可能であり、電子供与基を提供するように、固体支持体上に化合物を誘導体化する工程と、
該誘導体化された化合物を、環化反応に供する工程であって、複数の反応物が該化合物の官能部分と反応し、複数の有機環状化合物を形成する工程と、
を包含する方法。
(10) 前記化合物が、前記固体支持体に直接結合したアミドによって該支持体に結合される、項目9に記載の方法。
(11) 複数の異なるアルデヒド化合物が前記固体支持体に結合し、それぞれが、異なる官能基を有し、
前記環状有機化合物を複数のサブアマウントに分割する工程と、
異なる反応物を該各サブアマウントとカップリングし、各カップリング反応を該各サブアマウントを用いて実質的に完了させる工程と、
既知の量の該各サブアマウントを共に組み合わせ、化合物のライブラリーを得る工程と、
をさらに包含する項目9に記載の方法。
(12) 環状有機化合物に結合した第2級アミンを-NH2による求核置換を可能にする脱離基を含む第1のサブモノマーアシル化剤でアシル化し、アシル化アミンを得る工程と、
該アシル化アミンと、-NH2基を含む十分な量の第2のサブモノマー置換剤とを反応させ、該脱離基の求核置換を実施する工程と、
該アシル化および反応工程を、所望の長さを有し、所望の置換基を含むポリマーを形成するのに適切なように連続して繰り返す工程と、
該ポリマーを環化し、共有結合したペプトイドバックボーンを有する環状生成化合物を生成する工程と、
をさらに包含する、項目9に記載の方法。
(13) それぞれが回収可能かつ分析可能な量で存在する10個またはそれ以上の異なる化合物を含む化合物のライブラリーであって、該化合物のそれぞれが、以下の一般構造式を有し、
【0016】
【化1−5】
【0017】
ここで、「a」および「b」が、それぞれ独立して、0から100までの整数であり、但し、該ライブラリーが、1またはそれ以上である「a」および「b」を有する化合物を含み、各Yが、独立して、-O-、-S-および-NHからなる群から選択され、各R1およびR2が、独立して、炭素原子に共有結合可能な任意の部分であり、各A1、A2、A3、A4およびA5が、独立して、H、ヒドロカルビル、ケトン、アルデヒド、カルボン酸、エステル、アミン、アミド、エーテル、およびニトリルからなる群から選択される、ライブラリー。
(14) A1、A2、A3、A4およびA5の少なくとも1つが、ケトン、アルデヒド、カルボン酸、エステル、アミン、アミド、エーテル、およびニトリルからなる群から選択され、
「a」および「b」が、独立して、1〜10の範囲であり、さらに、前記ライブラリーが1000個またはそれ以上の異なる化合物を含む、項目13に記載のライブラリー。
(15) R1、R2、A1、A2、A3、A4、およびA5のそれぞれが、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-
NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目13に記載のライブラリー。
(16) A1、A2、A3、A4、およびA5の1つまたはそれ以上が、前記一般構造の環構造に縮合した1つまたはそれ以上の付加環構造を提供するように定義される、項目13に記載のライブラリー。
【0018】
発明の要旨
本発明は、環状有機化合物のライブラリー、およびそのようなライブラリーを作製し、アッセイする方法に関する。本発明によれば、それぞれの環状有機化合物は、出発樹脂を用いて誘導体化された固体表面の形態の出発物質から構成される。化合物は、この樹脂と反応し、環状基を付加するか、またはそれを形成する。好ましくは、これらの反応は、ライブラリーのサイズを大きくするように、それぞれ異なる化合物が、複数のサブアマウント(subamount)と反応可能であるように、分割樹脂手順を用いて行われる。例えば、化合物は、固体支持体に結合された出発樹脂と反応し、アルデヒド官能基を有する化合物が得られる。ここで、出発樹脂と反応した(1つ以上の)アルデヒド化合物は、生成物の混合物が得られるように変えられるさまざまな置換基を有している。また、本発明は、合成が完了した後に化合物を支持体から切断することによって、環状有機化合物の組み合わせライブラリーを基質に結合された化合物から作製し、そのような化合物のライブラリーをアッセイする方法にも関している。
【0019】
本発明によるライブラリーは、すべて、出発樹脂を用いて誘導体化された固体支持体を用いて作製される。その後、出発樹脂は、順次共に結合される反応物に供され、結果として環状化合物を形成する。反応物の順次結合は、(1)分割樹脂方法論および(2)サブモノマー方法論を用いて行われ得、かつそうするのが好ましい。これらの方法論については、共に後で詳細に説明する。
【0020】
本発明の第1の目的は、固体基質に共有結合されているが、その固体支持体から切断され得る、基質に結合された樹脂から誘導された多数の環状有機化合物を含有する混合物(ライブラリー)を提供することにある。これらのライブラリーは、好ましくは、少なくとも1つの生物学的に活性な環状有機化合物を含む。
【0021】
重要な目的は、分割樹脂方法論をサブモノマー方法論と組み合わせて用いることにより、固体支持体上で誘導体化された出発樹脂から複雑な組み合わせライブラリーを迅速に合成し、ライブラリーの複雑さおよび多様性を顕著に増加させるように置換される環状化合物を、結果として得られるライブラリーに得るための効率のよい方法を提供することにある。
【0022】
本発明の別の目的は、少なくとも1つの生物学的に活性な環状有機化合物を含む、基質に結合された樹脂出発物質から誘導された環状有機化合物のライブラリーを得る方法を提供することにある。
【0023】
本発明は、高度に置換された環状構造を好ましくは有する、環状有機化合物のライブラリーを包含する。
【0024】
本発明の別の目的は、天然タンパク質またはその他の生物学的に活性な化合物の活性をある程度模倣する化合物を得るために、このような環状有機化合物ライブラリーをスクリーニングする方法論を提供することにある。
【0025】
本発明の別の目的は、薬学的に活性な薬剤のような生物活性化合物にさらに結合された、本発明の環状有機化合物である新規な化合物を生成することにより、本発明により合成された環状有機化合物の強化された結合親和力を介して、薬剤に対する生化学標的化を提供することにある。
【0026】
本発明の利点は、この方法論が、最も強力なレセプター結合親和力あるいはその他の最適化された標的生物活性を有する、固体支持体に結合された環状有機化合物を合成し、単離するのに用いられ得ることである。
【0027】
本発明の別の利点は、本発明による環状有機化合物およびライブラリーが、レセプターの相互作用、すなわちそのような化合物と天然レセプター部位との間の相互作用を探求するのに用いられ得ることである。
【0028】
本発明の別の目的は、基質に結合されたアルデヒド出発物質から誘導された環状有機化合物であって、同一のレセプター部位に結合する生物活性タンパク質あるいはその他の生物活性分子と同じか、またはより強力な、天然レセプター部位に対する親和力を有する有機化合物を設計する薬剤設計方法論を提供することにある。
【0029】
本発明の別の特徴は、この化学合成方法論が固相反応技術と関連させて用いられることにより、規定されたライブラリーの作製が可能になることであり、また、この固相反応技術が、実用的な量の環状有機化合物および/またはライブラリーを生成するように自動化され得ることである。
【0030】
本発明のさらに別の特徴は、本発明による基質に結合された環状有機化合物が、天然ペプチドあるいはその他の生物活性分子と比べて、それらの有する結合に対して異なる構造を有するのみならず、天然ペプチドあるいはその他の生物活性分子では可能ではないことがある、それぞれ異なる3次元構造をも有することである。
【0031】
本発明のこれらの目的、利点および特徴、ならびに、その他の目的、利点および特徴は、以下により詳しく説明する構造、合成および使用法の詳細を読めば、当業者には明らかになるであろう。なお、以下の説明では、その一部をなす添付の一般構造式および合成スキームを参照するが、これらの式およびスキームの全体を通して、同一の記号は、同一の分子部分を指す。
【発明を実施するための最良の形態】
【0032】
詳細な説明
本発明による固相、樹脂誘導環状有機化合物、ライブラリーおよび結合体、ならびに、それらを製造する方法を説明する前に、本発明は、以下で説明される特定の環式および複素環式化合物、ならびに、それらの置換基に限定されるわけではないことは、理解されたい。なぜなら、そのような化合物およびそれらの置換基、ならびに、方法は、もちろん改変可能であるからである。また、以下で用いられる用語法は、特定の実施形態のみの説明を目的とするものであり、限定を意図しているわけではないことも理解されたい。なぜなら、本発明の範囲は、添付の請求の範囲によってのみ限定されるからである。
【0033】
なお、この説明および添付の請求の範囲の全体を通して、単数形の「a」、「an」および「the」は、文脈により明らかにそうではないことが示されない限り、複数の指示物を含むことには留意されたい。よって、例えば、「a cyclic organic compound(ある環状有機化合物)」というときには、複数のそのような環状有機化合物および1分子の1つよりも多くのコピーの混合物が含意されており、「reactive starting compound(反応性出発化合物)」というときには、複数のそのような反応性出発化合物および/または複数のそのような出発化合物の多数のコピーの混合物への言及が含まれており、「the method of synthesis(合成方法)」というときには、以下の開示を読むことにより当業者が発想するであろう、複数のそのような方法が含意されている。
【0034】
本発明は、新規な環式または複素環式有機化合物および結合体、環状化合物のライブラリー、そのような環式または複素環式有機化合物、ライブラリーおよび結合体を合成するプロセス、ならびに、そのようなライブラリーから所望の生物活性を有する環状化合物を単離するプロセスを包含する、異なる様々な局面を含む。また、本発明のこれらの局面のそれぞれにおいて、本発明は、多数の具体的な実施形態を含んでいる。本発明の本質は、当業者が、本明細書中に開示され、記載される情報を用いて、天然に存在する分子または生物学的に活性な合成分子の生物活性を模倣する分子を生成し、単離することを可能とする、処理技術を提供することを伴うが、本発明のそのような化合物は、天然分子または合成分子と比べて異なる化学構造を有している。用語「模倣する」は、広い意味で用いられている。すなわち、生成された分子は、同じ活性を有していても、より大きな活性を有していても、より小さな活性を有していてもよく、および/または天然に存在する生物学的に活性な分子または生物学的に活性な合成分子の効果を阻害してもよい。
【0035】
本明細書中において言及される文献はすべて、本発明の特徴を開示し、記載することを目的として、参考として援用される。
【0036】
そうでないと規定しない限り、本明細書中において用いられるすべての技術的、科学的用語は、本発明の属する技術分野の当業者により通常理解されている意味と同じ意味をもっている。本明細書中において記載される方法および物質に類似する、または等価であるあらゆる方法および物質が、本発明の実施または試験において用いられ得るが、以下には、好ましい方法および物質を説明する。
【0037】
以下に便宜のために述べる定義をもつ多数の用語が、本明細書の全体を通して規定され、用いられる。
【0038】
定義
用語「ライブラリー」、「組み合わせライブラリー」、「樹脂誘導ライブラリー」などは、本明細書中においては、1つ以上の固相に結合された樹脂出発物質から固定支持体上に合成された環状有機化合物の混合物を意味する、相互に置き換え可能な用語として用いられている。このライブラリーは、互いに異なる環状有機分子を、10個以上(すなわち、10個の異なる分子であって、同一分子の10個のコピーではない)、好ましくは100個以上、より好ましくは1,000個以上、さらに好ましくは10,000個以上含む。これらの異なる分子(異なる基本構造および/または異なる置換基)のそれぞれは、その存在が何らかの手段により判定され得る(例えば、レセプターまたは適切なプローブを用いて、単離され得、分析され得、検出され得る)ような量だけ存在することになる。その存在を判定可能とするのに必要な異なるそれぞれの分子の実際の量は、用いられる実際の手順によって変化し、単離、検出および分析のための技術が進歩するにつれて変化し得る。これらの分子が実質的に等モル量だけ存在するとき、100ピコモル以上の量が検出され得る。好ましいライブラリーは、所望の反応生成物それぞれを実質的に等モル量だけ含んでおり、所与の分子の存在が優勢になったり、何らかのアッセイにおいて完全に抑圧されるように、所与の分子を比較的大量に含んだり、比較的少量に含んだりすることはない。
【0039】
用語「官能基」、「官能部分」などは、本明細書中では、炭素、酸素、水素および窒素からなる分子の有機基を表すために用いられる。典型的な官能基は、本明細書中において規定される「環状化合物」の成分に結合されるものであって、アルデヒド、ケトン、カルボン酸、エステル、アミド、アミン、エーテルおよびニトリルを含む。「環状化合物」に結合される好ましい官能基は、アルデヒドおよびケトンである。これらの基それぞれの一般構造は、よく知られている。しかし、この応用を目的として、これらの基は、「アルデヒド」の以下の定義により規定される。ここで、それぞれの基について適切な変更が施される。
【0040】
本明細書中において用いられている用語「アルデヒド」は、以下の一般構造式を有する化合物を意味するものとする。
【0041】
【化1−6】
【0042】
ここで、Rは、炭素原子および適切な固体基質または樹脂に共有結合可能であり、共有結合、または炭素原子および適切な固体基質または樹脂に共有結合可能な1個の原子または複数の原子の群であり得る。好ましくは、「R」は、1個から12個の炭素原子を含むアルキルまたは置換されたアルキルである。しかし、「R」は、任意の炭化水素ベース部分、または炭化水素ベースの置換部分でもあり得る。「R」は、本明細書中において規定される「ヒドロカルビル」または「側鎖」であり得る。
【0043】
用語「オリゴマー」は、例えば、本発明のプロセスにより生成されたポリマーを意味しており、任意の長さのホモポリマー、コポリマーおよびインターポリマーを含む。具体的には、オリゴマーは、単一の繰り返しモノマー、2つの交互モノマー単位、互いにランダムに、および/または慎重に間隔の設けられた2つまたはそれ以上のモノマー単位から構成され得る。このオリゴマーは、好ましくは、2〜100個のモノマーであり、より好ましくは2〜50個または2〜20個のモノマーであり、最も好ましくは2〜6個のモノマーである。
【0044】
用語「アシルサブモノマー」は、本発明において用いられるアシル化剤を指す。アシルサブモノマーは、反応性カルボニルあるいはカルボニル等価体、およびアミンによる求核置換により置換され得る脱離基を含む。「カルボニルあるいはカルボニル等価体」は、(本発明によるポリカルバメートの合成では)カルボン酸、エステル、アミド、無水物、アシルハライドおよびイソシアナートを含むが、これらに限定されるわけではない。用いられるエステルおよびアミドは、一般に「反応性」の形態(例えば、DIC付加物など)である。アシルサブモノマーは、さらに、側鎖を含み得る。適切なアシルサブモノマーは、ブロモ酢酸、3−ブロモプロピオン酸、2−ブロモプロピオン酸、2−ブロモエチルイソシアナート、2−ブロモエチルクロロホルメート、6−フェニル−3−ブロモヘキサン酸、4−ブロモメチル−安息香酸、4−ブロモメチル−2−メトキシ安息香酸、5−ブロモメチル−ピリジン−2−カルボン酸などを含むが、これらに限定されるわけではない。
【0045】
用語「アミノサブモノマー」は、アシルサブモノマーにおける脱離基の求核置換を行い得るアミノ基を含有する化合物を指す。このアミノ基は、第1級でも、第2級でも、第3級でもよい。第3級アミンの添加により、第4級アンモニウム塩が生じ、好ましくは、連鎖ターミネーターとして用いられる(すなわち、このオリゴマーのそれ以上のアシル化は不可能になる)。本発明による好ましいアミノサブモノマーは、第1級アミンおよびヒドラジドである。ただし、アミド、カルバメート、尿素、カルバジド、カルバゼート(carbazate)、セミカルバジドなどもまた適している。
【0046】
用語「サブモノマー」は、本発明のある工程において基質に結合された物質に付加される、本発明の方法において用いられる有機反応物を指す。本発明の「アシルサブモノマー」(スキームIAの第1のサブモノマー)は、任意のアミノ基(例えば、-NH2、-NRHまたは-NR2)により求核置換され得る脱離基を有するアシル化剤である。「アミノサブモノマー」(例えば、スキームIAの第2のサブモノマー)は、-NH2基を含む置換剤反応物である。
【0047】
本発明の別の局面においては、サブモノマーが、固体支持体またはアルデヒドで誘導体化された固体支持体樹脂に順次付加され、バックボーンを形成し、これは引き続いて環化される。環化のためのペプトイドバックボーンの調製時に、サブモノマーを段階的に付加することにより、側鎖および環置換基を最終生成物へと導入する。
【0048】
サブモノマー合成の詳細は、1994年7月18日に出願された米国特許出願第08/126,539号およびR. Zuckermannら、J. Am. Chem. Soc. (1992) 114:10646-7に記載されており、これらは本明細書中で参考として援用される。
【0049】
用語「側鎖」は、炭素原子または窒素原子を介して有機化合物に結合された基を指す。この結合は、環状基の環構造上にあってもよいし、窒素原子または炭素原子のいずれかにおける化合物のポリアミドバックボーン上にあってもよい。側鎖は、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb-、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdであり得る。ここで、Ra、Rb、RcおよびRdはそれぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
また、Ra、Rb、RcおよびRdはそれぞれ、0〜6個のハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル(alkylsufinyl)、あるいは低級アルキルスルホニルで置換される。ここで、「低級」とは、1〜6個の炭素原子を示す。
【0050】
用語「炭化水素ベースの」、「炭化水素ベースの置換基」などは、分子の残りの部分に直接結合された炭素を有し、本発明の文脈では炭化水素ベースの特性が優勢である部分を表す。
【0051】
置換基は、例えば、以下のものを含む。
【0052】
(1)炭化水素置換基、すなわち、脂肪族(例えば、アルキルまたはアルケニル)置換基、脂環族(例えば、シクロアルキルまたはシクロアルケニル)置換基、芳香族、脂肪族および脂環族置換芳香族核など、ならびに、環状置換基である。ここで、環は、分子の別の部分を介して完成される(すなわち、例えば、任意の2つの指示された置換基が、一緒になって1個の脂環基を形成する)。
【0053】
(2)置換された炭化水素置換基、すなわち、本発明の文脈では、優勢な炭化水素置換基を改変しない非炭化水素基を含む置換基である。当業者は、そのような基(例えば、ハロ(特にクロロおよびフルオロ)、アルコキシ、メルカプト、アルキルメルカプト、ニトロ、ニトロソ、スルホキシなど)を認識するであろう。
【0054】
(3)ヘテロ置換基、すなわち、本発明の文脈での優勢なヒドロカルビル特性を有しながら、他の部分は炭素原子で構成されている環または鎖に存在する炭素以外の原子を含む置換基。適切なヘテロ原子は、当業者には自明であろうが、例えば、硫黄、酸素、窒素などがあり、例えばピリジル、フラニル、チオフェニル、イミダゾリルのような置換基が、これらのヘテロ置換基の例である。ヘテロ原子および好ましくはただ1個の原子が、炭化水素ベースの置換基内のそれぞれの炭素原子について存在する。炭化水素ベースの置換基には、そのような基またはヘテロ原子が存在しないこともあるので、炭化水素ベースの置換基は、純粋な炭化水素であることもある。
【0055】
「炭化水素」は、化合物を述べているのに対して、「ヒドロカルビル」および「ヒドロカルビレン」は、それぞれ1つまたは2つの水素が除去された基を述べている。それぞれは、大まかにいうと、水素原子および炭素原子のみから構成されるが、ヘテロ原子を含んでいてもよいし、飽和であっても、不飽和であってもよく、脂肪族でも、脂環族でも、芳香族でもよい。環が含まれるとき、構造は、通常、1個、2個、3個またはそれ以上の環を含む。これらの環は、縮合されても、架橋されても、スピロ縮合されてもよい。
【0056】
用語「環状化合物」および「環状部分」は、飽和であっても、不飽和であってもよい1つ以上の閉環を特徴とする構造を有する、上記で規定した炭化水素ベースの有機化合物または部分を述べるのに用いられる。環状化合物または部分は、存在している環の個数によって、単環式でも、二環式でも、三環式でも、多環式でもよい。これらの用語は、環状化合物の主要な3つの群を包含する。すなわち、(1)脂環族、(2)芳香族(「アセン(asene)」とも呼ばれる)、および(3)複素環族の3つである。
【0057】
用語「分子部分」は、オリゴマー主鎖の窒素原子または炭素原子に結合可能であり、それにより、例えば、CH3(R1)NC(O)CH(R2)CH3(ここで、R1は、オリゴマー主鎖の窒素原子に結合可能な分子部分であり、それにより窒素原子に結合される側鎖を形成し、R2は、オリゴマー主鎖の炭素原子に結合可能な分子部分であり、それにより炭素原子に結合される側鎖を形成する)であるオリゴマーのような、形成される任意の化合物の主鎖から分離して側鎖を形成する、1個の原子または原子群を包含している。よって、ポリペプチドまたはポリアミド合成の当業者には、水素部分および、例えば、アルキル、アリール、およびアリールアルキル部分のようなヒドロカルビル部分を含むが、それらには限定されない広範で多種多様な分子部分が用いられ得ることは、容易に明らかになるであろう。
【0058】
「有機化合物」は、炭素、水素、窒素、酸素、硫黄、およびリン原子からなる分子を意味する。本明細書中において用いられる有機化合物は、そのすべてが炭素および水素から構成される環式または非環式化合物でもよいし、1つ以上のヘテロ原子(酸素、窒素、硫黄、およびリン原子を含む)を含んでいてもよい。
【0059】
「環状有機化合物」は、ペプトイドバックボーンの環化から誘導された少なくとも1つの環状構造を含む有機化合物を意味する。この環状構造は、炭素および水素から構成される炭化水素であってもよいし、脂肪族または芳香族であってもよい。この環状構造は、環状バックボーンに少なくとも1つのヘテロ原子を有する複素環であってもよい。この複素環式構造は、飽和であっても、不飽和であってもよい。環状構造は、縮合されてもよいし、環状化合物内で分離されてもよい。
【0060】
置換基は、第1分子の一部である原子または基のことをいう。なぜなら、それは第1分子の別の原子または基を置換するからである。分子が置換される場合、分子は、1つ以上の置換基を有する分子の誘導体である。本発明によるサブモノマーのいずれかにおいて有用な置換基は、ハロ、アルキル、アルコキシ、アルキルチオ、ハロアルキル、ハロアルコキシ、ハロチオ、二置換アミノなどを含んでおり、窒素または炭素に結合された水素のような原子を置換する。置換可能な位置は、第1分子の置換された原子または基の結合部位である。
【0061】
「プリンまたはピリミジン塩基」は、A、T、G、CまたはUのような天然ヌクレオシド塩基と、1以上のアルキル、カルボキシアルキル、アミノ、ヒドロキシル、ハロゲン(すなわち、フルオロ、クロロ、ブロモ、またはヨード)、チオール、またはアルキルチオール(ここで、アルキル基は、1個から6個程度の炭素原子を含む)により置換されたこれらのプリンおよびピリミジンを含むそれらの誘導体と、を含んでいる。プリンおよびピリミジンとしては、例えば、2、6−ジアミノプリン、5−フルオロウラシル、キサンチン、ヒポキサチン、8−ブロモグアニン、8−クロログアニン、8−アミノグアニン、8−ヒドロキシグアニン、8−メチルグアニン、8−チオグアニン、2−アミノプリン、5−エチルシトシン、5−メチルシトシン(5-methylcyosine)、5−ブロモウラシル、5−エチルウラシル、5−ヨードウラシル、5−プロピルウラシル、2−メチルアデニン、メチルチオアデニン、N、N−ジメチルアデニン、8−ブロモアデニン、8−ヒドロキシアデニン、6−ヒドロキシアミノプリン、6−チオプリン、4−(6−アミノヘキシル/シトシン)などがあるが、これらに限定されるわけではない。
【0062】
「脱離基」は、例えば、NH2のようなアミンにより求核置換され得る部分を意味する。求核置換により容易に除去される限り、本明細書中では、どのような脱離基が用いられてもよい。本発明において有用な脱離基としては、例えば、ブロモ、クロロ、ヨードなどのハロ、O−トシル、O−トリフリル、O−メシルなどがあるが、これらに限定されるわけではない。
【0063】
用語「基質」、「固体支持体」などは、本明細書中においては、出発樹脂材料(反応性基)が(室温で)結合され得る任意の固体材料を規定するものとして、相互に置き換え可能に用いられている。好ましい固体支持体材料は、ポリエチレン化合物およびポリスチレン化合物のようなポリマー化合物、および関連する不活性ポリマー化合物を含んでいる。よって、本明細書中では、それらのすべてを文字「P」で表す。基質は、シート、円筒形容器の内側、またはピンを含むどのような形状であってもよいが、好ましくは、直径1.0 cm未満の球状ビーズの形状であり、より好ましくは、直径1.0 mm未満である。「基質」または固体支持体は、ペプチド合成で用いられる従来の固体支持体材料である。このような基質または支持体としては、例えば、多種多様な支持体樹脂、ならびに光切断性DKP形成リンカー(DKPとはジケトピペラジンである。例えば、本明細書中でも参考として援用されるWO90/09395を参照のこと)、TFA切断性(cleavable)、HF切断性、フッ化物イオン切断性、還元的切断性および塩基活性(base-labile)リンカーのような支持体樹脂へのコネクタがあるが、これらに限定されるわけではない。固体支持体樹脂は、別々の反応用にいくつかの部分に分割され得、所望であれば再び組み合わされ得る、複数の固体支持体粒子を含んでいる。よって、用語「樹脂」が固体支持体(例えば、固体支持体樹脂)と共に用いられるとき、この用語は、-NH2基のような反応性基、またはヒドロキシル基のようなその他の電子供与基を用いて誘導体化されたポリマー材料を記載する。
【0064】
「保護基」は、その基が結合される原子(通常は、酸素または窒素)が、望ましくない反応または結合(通常は、合成反応)に関与するのを妨げ得る、任意の基を意味している。また、保護基は、カルボン酸、チオールなどの反応または結合を妨げることも知られている。このような基、ならびに、それらの調製および導入は、当該分野において従来的であり、塩、エステルなどを含む。
【0065】
「電子供与基(EDG)」は、反応物のその他の部分において電子密度を増加させ得る、反応物に共有結合された部分を意味する。本発明において有用な電子供与基としては、例えば、アルキル、アミン、ヒドロキシル、アルコキシなどがあるが、これらに限定されるわけではない。
【0066】
「電子吸引基(EWG)」とは、反応物のポリアミドバックボーン部分の求核付加を活性化し得る、反応物に共有結合された部分を意味する。本発明において有用な脱離基としては、例えば、ニトロ、カルボニル、シアノ、スルホンなどがあるが、これらに限定されるわけではない。
【0067】
「反応を完了させる」または「実質的に完了」とは、十分な反応物が(例えば、時間および温度などの十分な条件の下に)添加され、固体支持体に結合された中間化合物のすべて、または実質的にすべてが、反応物により誘導体化され得ることを意味する。「反応を実質的に完了させる」とは、反応物の濃度、触媒、温度およびその他の条件が適切である条件の下に反応を行うことにより、固体支持体に結合された中間化合物(例えば、樹脂)の80%よりも多く、好ましくは90%よりも多く、より好ましくは95%よりも多く、さらに好ましくは99%以上を、添加された反応物と反応させ、それにより検出可能なすべての中間樹脂が反応するようにすることを意味する。
【0068】
「回収可能な量」とは、回収可能な量が、分離時において、当該分野で利用可能な技術により混合物のその他の成分から分離可能であるような濃度で化合物が存在する、混合物内の化合物の量を意味する。混合物中の成分がおよそ等モル量で存在している時、好ましくは、少なくとも50 pmol、より好ましくは100 pmolの化合物が混合物中に存在している。
【0069】
「分析可能な量」とは、混合物中で検出され得、同定され得る、混合物中に存在する化合物の量を意味する。混合物中の成分がおよそ等モル量で存在している時、好ましくは、少なくとも約10 pmol、より好ましくは50 pmolの化合物が混合物中に存在している。
【0070】
用語「プール」および「プールされた量」とは、誘導体化された、または誘導体化されていない固体支持体粒子を組み合わせて、混合物を形成することを意味する。プールされた材料は、ライブラリーの調製時の中間生成物または最終生成物を含む。
用語「サブアマウント」とは、プールから分割されたプールされた量の一部を意味する。それぞれのサブアマウントは、好ましくは、その他すべてのサブアマウントとサイズが同じである。サブアマウントは、図1に示されており、本明細書中で説明されている分割樹脂方法と関連させて用いられる。プールされた量は、図1の量9、31および40を含んでおり、サブアマウントは、2〜8、11〜24などを含んでいる。
【0071】
一般的方法論
この方法論は、電子供与化合物を固相支持体上へと誘導体化することにより始まる。一般に、固体支持体は、結合された、市販の樹脂(例えば、結合されたRinkまたはMerrifield樹脂)を有している。Pが支持体を表す場合、樹脂がその上にある支持体は、P-XH(ここで、Xは、OまたはNHである)で表される。ここでAは、例えば、出発樹脂上に誘導体化されたアルデヒド基のような任意の官能基であり、P-XH-Aにより表される。アルデヒドまたはケトンのような官能基は、官能部分と反応させることにより環化され、それにより、化合物の多様なライブラリーを提供する置換された環および置換されていない環からなる環状化合物を形成する。例えば、芳香族アルデヒドAは、P-NH-Aについては、RINKアミドを介して支持体に結合される。このアルデヒドは、サルコシンのようなアミドおよびジメチルムコネートのようなエステルと同時に反応する。これらの化合物は、このアルデヒド基と反応し、環化する。得られる生成物上の置換基は、反応物上の置換基が異なる反応物の混合物を提供することにより変えることができる。例えば、サルコシン上のメチル基およびジメチルムコネート上の2つのメチル基は、独立して他のアルキル部分に変えることができる。
これらの化合物が反応する順番、その構造は、反応条件とならんで、生成物の構造を決定する。しかし、上述した合成スキームは、小さく、置換された分子から比較的複雑な分子を順番どおりの段階的手順で合成するために、いくつか共通の特徴を有していることが容易に分かり得る。それぞれの場合において、直線状の置換されたバックボーンが形成された後、環化反応により、高度に置換された環状生成物を生成する。
本明細書中において記載されている合成方法の共通の特徴について、まず説明する。
この方法論は、まず、複数の(例えば、3つ以上、10以上、1,000以上、10,000以上など)固体支持体表面ポリマービーズを提供することにより始まる。多数の支持体表面の使用は、特にサブモノマー方法論と組み合わされた時に本発明の効力を大幅に増大させる、本明細書中で説明されている「分割樹脂」方法論の適用のために重要である。これらの支持体表面は、それらを電子供与基と反応させることにより誘導体化される。この基または樹脂(例えば、-NH2)は、分子の残りの部分を構築する出発基として作用する。この出発樹脂は、多種多様な異なるタイプの反応に供され得、それにより、最終的には、例えば、アルデヒドまたはケトンのような官能基で置換される環状化合物の形成へと導かれる。例えば、出発樹脂は、本明細書中で規定されているサブモノマー反応物と反応させられ得る。その後、サブモノマー単位を以前の基のそれぞれと順次反応させることができ、それにより、任意の所望の長さをもつ連鎖を構築する。例えば、「P」が固体表面であり、「EDG」が電子供与基であり、「SM」が本明細書中で規定されているサブモノマーである場合、反応は、以下のように進行し得る。
1)P-EDG
2)P-EDG-SM
3)P-EDG-SM-SM
出発樹脂をサブモノマーと反応させる代わりに、出発樹脂は、環状化合物の電子吸引基と反応させてもよい。このような環状化合物が「CC」で表される場合、反応は、以下のようにも進行し得る。
1)P-EDG
2)P-EDG-CC
出発樹脂(EDG)を環状化合物と反応させる代わりに、出発樹脂は、環を形成する化合物、すなわち1つ以上の追加化合物と反応して環を形成する化合物と反応させてもよい。「RFC」が環を形成する化合物である場合、反応は、以下のようにも進行し得る。
1)P-EDG
2)P-EDG-RFC+RFC
3)P-EDG-環状化合物
上記一般反応のすべてにおいて、第1の反応物は、固体支持体上の反応性部位の実質的にすべてが、共有結合された反応物により占められるように、調製された(すなわち、XH基がその上にある)固体支持体と反応させられる。第1の反応物は、一般に、支持体に結合されたNH2基と反応させられる。(その後、出発樹脂として作用する)第1の反応物と反応可能な化合物は、その後、第1の反応物上の反応性基と反応させられる。
【0072】
反応した固体支持体を分割し(サブアマウントを生成し)、再び組み合わせる方法は、高度に置換された環状構造の混合物生成を可能にする、環状ペプトイド合成のサブモノマー方法の特徴である。生成物の混合物は、本発明の2つの特徴から結果として得られる。すなわち、1)サブモノマー化合物上の可変置換基の組み合わせおよびそれらの相対的位置、ならびに、2)選択されたサブモノマー添加時に、反応した固体支持体粒子を分割し、再び組み合わせることにより、環化前に前駆体直線形ペプトイドの混合物を生成することである。混合物中のそれぞれ異なる生成物化合物の個数は、1)固体樹脂に結合された、それぞれ異なる第1の反応物化合物の個数、2)第1の反応物化合物と反応した、それぞれ異なる第2の化合物の個数、ならびに、3)第1および第2の化合物それぞれの上の可変置換基の個数と共に増加する。
【0073】
本発明の1つの実施形態では、サブモノマー成分は、支持体上に誘導体化された-NH2樹脂に結合される。このサブモノマー成分は、以下の一般構造式を有する。
【0074】
【化2】
【0075】
ここで、Zはハロゲン(好ましくは、Br)であり、Rは炭素原子に結合した任意の部分であり、かつHまたはヒドロカルビル部分(例えば、-CH3)であり得る。用語「サブモノマー」は、上記で規定した通りであり、具体例も与えられている。
【0076】
このサブモノマーがP-NH2と反応すると、結果は以下のようになる。
【0077】
【化3】
【0078】
この時点において、官能部分をその上に有する環状有機化合物は、以下のようにハロゲン原子と反応する。
【0079】
【化4】
【0080】
ここで、RおよびZは上記で規定したとおりであり、Aは官能基を有する任意の部分であり、YはS、OまたはNHである。
【0081】
反応は、適切な溶媒中、適切な時間および温度条件下にて行われ、
【0082】
【化5】
【0083】
を生じる。
【0084】
大型のライブラリーを作製するためには、それぞれ異なる「複数のR」をその上に有する、異なる複数のサブモノマー基の混合物が、出発樹脂と反応させられ、その後、異なる複数の環状化合物の混合物が、ハロゲンと反応させられる。ここで、この混合物は、例えば、官能基、上記で規定した「側鎖」、または上記で規定した「ヒドロカルビル」のような置換基で置換された環状化合物を含んでいる。規定されたライブラリーは、本明細書中で説明されている「分割樹脂」方法論を用いることによって、作製され得る。
【0085】
本発明によれば、さまざまな方法論が、環状有機化合物のライブラリーの作製に適用される。具体的には、この方法は、同一の反応容器内、単一の固相支持体上で、複数の特異で、独特で、異なる環状有機化合物の混合物を調製することを伴う。すなわち、この反応容器内の環状有機生成物化合物は、互いに異なるものであり、この反応容器内の環状有機生成物化合物は、それぞれ、回収可能で、分析可能な量で存在している。サブモノマー化合物を相対量で組み合わせ、それぞれの反応が実質的に完了するように、分割樹脂方法を適用することによって、得られる環状有機化合物の混合物は、それぞれの反応生成物を予測可能で規定された量で、かつ環状有機化合物が回収し、分析し得るに十分な量で含む。これらの環状有機化合物それぞれの結果として得られる量は、予測可能である。なぜなら、それぞれの反応で用いられる、誘導体化された固体支持体の量が制御され、後続するそれぞれの反応が完了するからである。
【0086】
本発明の一般的局面によれば、反応性部分(例えば、反応性アルデヒド部分)が環状化合物またはサブモノマー化合物と反応させるのに利用可能であり、その化合物が、今度は、1つ以上の後くサブモノマーと反応した後、環化されるように、前駆体アルデヒド化合物のような前駆体化合物を固体支持体上で固定化した後、個々の環状有機化合物は、固相合成技術のような方法論を用いて生成される。環化された化合物(例えば、アルデヒド誘導体)は、固体支持体に結合されたままであり、そしてアッセイされ得るか、またはその支持体から切断され、そしてアッセイで用いられ得る。環化された化合物(例えば、アルデヒド誘導体)の切断は、必要に応じて、回収前または使用前に行われ得る。
【0087】
サブモノマー方法により調製される環化された多種多様な化合物は、サブモノマー反応の順序によって部分的に制御されるので、これらの部分が環化前に組み合わされる時、直線形アルデヒド誘導体の混合物が形成されるように、粒子状固体支持体を分割し、そして後続するサブモノマー反応のそれぞれと再び組み合わせ得ることは、容易に分かる。
【0088】
分割樹脂方法
分割樹脂方法は、合成効率を改善し、結果として得られるライブラリー反応生成物のサイズを顕著に増大させることができるように、本発明に適用され得る。分割樹脂方法は、支持体(例えば、ビーズ)上へと誘導体化された同じ出発樹脂のセット、またはそれぞれ異なる出発樹脂の混合物をそれぞれ等しいプールへと分割することを含む。これらのプールはそれぞれ、反応物に結合される。好ましくは、異なる複数の反応物がそれぞれのプールに用いられ、それぞれの反応が実質的に完了する。それぞれの反応が完了した後、これらのプールが単一のプールに再び組み合わされ、所望のライブラリーが提供される。このプロセスは、支持体上で合成されている分子のサイズを大きくし、結果として得られるライブラリー中のそれぞれ異なる分子の個数を増やすために、任意の回数だけ繰り返され得る。この分割樹脂方法論は、1993年1月26日に発行された米国特許第5,182,366号におけるペプチドの大型組み合わせライブラリーの合成に最初に適用された。この特許は、本明細書中でも、分割樹脂方法の基本的局面を開示し、説明するために参考として援用している。
【0089】
図1は、分割樹脂方法論がどのようにして行われるかを示す模式図である。工程1において、ビーズ2〜8のサブアマウントは、樹脂により占められるそれらの上の反応性部位(例えば、NH)のすべてが、反応物(例えば、本明細書中で説明されているサブモノマー)と反応するように、反応させられる。サブアマウント2〜8それぞれの反応は、完了する。反応したサブアマウントは、その後、工程9において再び組み合わされる。工程10において、9からの量が、サブアマウント11〜30に分割され、それぞれのサブアマウントが異なる反応物と反応させられて、反応はそれぞれ完了する。これらのサブアマウント11〜30は、その後、工程31において再び組み合わされ、工程32において、サブアマウント33〜39に分割される。これらのサブアマウントは工程40で組み合わされ、工程41で再びサブアマウントに分割され得る。
【0090】
これらの反応・組み合わせ・分割・反応工程は、ライブラリーのサイズおよび多様性を増すために、所望の回数だけ繰り返され得る。また、図1に示されているように、組み合わされたサブアマウントは、次の工程で所望の個数のサブアマウントに分割され得る。それぞれの反応が常に完了するので、何らかの反応生成物がその他の反応生成物を支配するということがなく、それぞれの反応生成物を等モル量だけ有する混合物を容易に得ることができる。
【0091】
この分割樹脂方法は、上述した一般的方法論に適用され得る。具体的には、P-NH2は、複数の(3以上、好ましくは5以上、より好ましくは10以上の)サブアマウントに分割される。P-NH2のそれぞれのサブアマウントは、以下の構造を有する異なるサブモノマーと反応させられる。
【0092】
【化6】
【0093】
ここで、「R」部分は可変であり、それぞれの反応は完了する。「R」は、上記で規定した「R」を含む、炭素原子に結合可能な任意の部分であり得るが、好ましくは、「R」は、H、-CH3、-CH2CH3、-CH(CH3)2、-CH2-CH2-CH3、=CH2およびフェニルからなる群から選択される。示されている構造は、単一の「R」を有している。しかし、同一の炭素原子に2つの「R」基が結合されてもよく、これらの「R」基は互いに異なっていてもよい。
【0094】
それぞれのサブアマウントで得られる反応生成物が、その後、再び組み合わされて、新しいプールを形成する。再び組み合わされた混合物すなわちプールは、その後、複数のサブアマウントに分割される。その後、それぞれのサブアマウントは、例えば、置換された環状ケトンまたはアルデヒドのような異なる置換された環状化合物と反応させられ、それぞれの反応は完了する。それぞれのサブアマウントで得られた反応生成物が、その後、再び組み合わされ、固体支持体上の環状化合物の所望のライブラリーである最終プールを提供する。レセプターは、このライブラリーに対して試験され得、または支持体から切断された後、固体支持体上のレセプターに対して試験され得る。
【0095】
サブモノマー方法
サブモノマー方法論は、アミノ酸またはアミノ酸様モノマー単位を鎖に付加するのに適用可能である。この方法は、上述した分割樹脂方法論と組み合わせて用いられる時、特に有用で効率的である。基本的方法では、それぞれのモノマーは、本明細書中では「サブモノマー」と称されている2つの反応物から、固体基質(支持体)上で直接合成される。以下の説明は、アミノ酸様化合物(ペプトイドとしても知られている)の合成について具体的に述べているが、環状化合物を形成する際に用いられる環化反応にも適用され得る。
【0096】
それぞれのモノマーは、2つの工程を有する合成サイクルにより生成される。第1の工程は、アミン(例えば、-NH2)により求核置換され得る脱離基を有する第1のサブモノマーアシル化剤(例えば、ハロ酢酸)を用いて行われる、基質に結合されたアミンのアシル化である。このモノマー合成サイクルの第2の工程は、第1級アミンのようなアミン(例えば、-NH2基)を含む第2のサブモノマー置換剤を十分な量(通常は過剰に)だけ供給することによって、例えば、ハロゲンまたはトシルのような脱離基の求核置換により側鎖を導入することを含む。この2工程プロセスは、反応スキームI. A.内に示されている。
【0097】
しかし、反応スキームI. A.は、反応スキームI. B.内に示されているように、逆の順番で行われてもよいことには、留意されたい。具体的には、反応スキームI. A.による「基質に結合されたアミン」を用いるのではなく、基質に結合されたアシル化剤サブモノマーを用いて反応を開始することが可能である。したがって、カルボン酸基は、基質の表面から延び、第1の工程においてアミンと反応する。この時点において、アミン基は、基質から外に向かって延び、上述した反応スキームI. A.の第1の工程によるサブモノマーアシル化剤を用いて、アシル化に供される。
【0098】
反応スキームI(AまたはB)の基本的な2工程プロセスは、モノマー単位を生成し、下記Iの構造をもつモノマーにより所望の長さを有するポリマー(下記の式Vによる)を生成するために繰り返され得る。これらの構造内に示されている変数は、所望の結果を得るために変えることができる。また、基本的サブモノマー構造は、構造II、IIIおよびIVにおいて異なるモノマー/ポリマー構造を得るために、以下のように変えられてもよい。
【0099】
スキームI.A
2つのサブモノマーからN-置換されたオリゴマーの固相アセンブリ
【0100】
【化7】
【0101】
スキームI.B
2つのサブモノマーからN-置換されたオリゴマーの固相アセンブリ
【0102】
【化8】
【0103】
上記のそれぞれにおいて、「P」は固相表面、R1およびR3はそれぞれ、独立的に、炭素原子に結合したあらゆる分子部分であり、R2およびR4はそれぞれ、独立的に窒素原子に結合したあらゆる分子部分であり、nは1〜10(好ましくは、1または2)の整数である。R1、R2、R3およびR4のいずれも、20の天然アミノ酸に結合した20の異なる側鎖部分(すなわち、グリシンの-H、アラニンの-CH3、バリンの-CH(CH3)2、ロイシンの-CH2CH(CH3)2、イソロイシンの-CH(CH3)CH2CH3、セリンの-CH2OH、トレオニンの-CHOHCH3、システインの-CH2SH、メチオニンの-CH2CH2SCH3、フェニルアラニンの-CH2-(フェニル)、チロシンの-CH2-(フェニル)-OH、トリプトファンの-CH2-(インドール基)、アスパラギン酸の-CH2COO−、アスパラギンの-CH2C(O)(NH2)、グルタミン酸の-CH2CH2COO-、グルタミンの-CH2CH2C(O)NH2、アルギニンの-CH2CH2CH2-N-(H)-C(NH2)+-NH2、ヒスチジンの-CH2-(イミダゾール)+基、およびリジンの-CH2(CH2)3NH3+)を含み得る。
【0104】
反応スキームI(AおよびB)には、本発明に関連して使用される試薬を指す省略形がいくつか含まれる。例えば、DMSOはジメチルスルホキシドを指し、DICはN,N-ジイソプロピルカルボジイミドを指し、DMFはN,N-ジメチルホルムアミドを指す。
【0105】
2工程法の各工程は、通常、20℃のおよそ常温にて、1気圧の圧力下で行われる。しかし、反応は、約5℃から約80℃の広い温度範囲において行われ得、使用される溶媒に依存して変わる。温度に依存して、2工程反応スキームIの時間は、約5分から約24時間の範囲内で変えられ得る。上記温度、時間、および試薬は、反応を常圧において行う際に適用される。他の圧力も採用され得る。
【0106】
サブモノマーが液体である場合、各工程は溶媒なしで行われ得る。しかし、不活性溶媒は、サブモノマーが固体である場合、または反応を容易にする場合に使用される。適切な不活性溶媒には、ジオキサンなどのエーテル、ジメチルホルムアミドなどの遮蔽アミド(blocked amides)、ジメチルスルホキシドなどのスルホキシドなどが含まれる。
【0107】
反応物の比率は変えられ得る。しかし、最高収率のためには、基質結合材料の量の約1.01から10倍、好ましくは基質結合材料の量の約1.5から5倍だけサブモノマーを多くすることが望ましい。
【0108】
スキームIに示す2工程サイクルにおいて、基質に結合した第2級アミンは、好ましくは、第1級アミンから調製されたアミンであり、基質支持体ベース面(substrate support base surface)、または固相(文字「P」で表される)に結合する(従来の方法を用いて)。
【0109】
サイクルの第1の工程は、アシル化アミンを得るための、アミン(例えば、-NH2)による求核置換が可能な脱離基を含むアシル化剤(例えば、ハロ酢酸、および特にスキームIに代表的に示されるブロモ酢酸)を含む第1のサブモノマーと、基質結合第2級アミンとを反応させることによって行われるアシル化である。
【0110】
本発明の2工程モノマー合成法の第2の工程では、モノマーユニットのバックボーン窒素、および側鎖またはR2基を付加する。第2の工程において、アシル化したアミンを、オリゴマーのモノマー位に付加されるR2基(すなわち、側鎖基)を含む第1級アミンまたは第2級アミン、アルコキシアミン、セミカルバジド、カルバゼート、またはアシルヒドラジドなどの-NH2基を含む十分な量の第2のサブモノマーと反応させる。第2のサブモノマーの反応は、脱離基の求核置換を生じる第2のサブモノマーを十分な量、通常、過剰量を添加することによって達成され、これはスキームIに臭素として代表的に示す。
【0111】
分割樹脂/サブモノマー法の適用
本発明の主要な目的は、固体支持体上に、環状有機化合物の非常に大きいライブラリ(例えば、10から10,000以上の化合物を含むライブラリ)を迅速かつ効率的に得ることである。この目的は、分割樹脂法を、それのみ、またはサブモノマー法とともに使用することによって最も容易に得られる。この方法は、まず、概して小さい重合体ビーズの形態を有する複数の固体支持体表面を得ることから始める。重合体ビーズは、文字「P」によって表され、XHで表される(ただし、Xは通常NHまたはO)電子供与基である出発樹脂の付加によって誘導体化される。電子供与基は、XH基などの電子供与基をその上に含む大きい分子であり得る。
【0112】
誘導体化されたビーズは、次いで、2以上のサブアマウントに分けられる。サブアマウントの数は、千単位であり得るが、より典型的にはおおよそ10から30である。
【0113】
サブアマウントをそれぞれ、以下の式のサブモノマーであり得る第1の反応物と反応させる:
【0114】
【化9】
【0115】
ただし、R1およびR2はそれぞれ、独立的に、炭素原子に共有結合可能なあらゆる部分であり、Zは、好ましくはBrであるハロゲン原子である。
【0116】
また代わりに、各サブアマウントの樹脂を、以下の一般構造を有する環状化合物反応物と反応させる:
【0117】
【化10】
【0118】
ただし、YはO、S、またはNHであり、A1、A2、A3、A4、およびA5のそれぞれは、独立的に、H、またはヒドロカルボニル、またはアルデヒド、カルボン酸、エステル、アミド、アミン、ニトリル、またはケトンなどの官能基部分である。好ましいライブラリには、1つ以上の官能基部分、最も好ましくはアルデヒドまたはケトンの少なくとも1つを環状構造上に有するいくらかの化合物が含まれる。
【0119】
所望であれば、サブモノマー反応物および/または環状化合物反応物を、連続的に互いに付加して、あらゆる所望の長さの鎖を作成する。しかし、通常、そのような反応化合物の1つのみを付加し、次の再結合またはプール工程までに、反応を終了させる。
【0120】
図1を参照すると、第1の工程は、複数の固体支持体ビーズ2-8を得る工程を含み得る。ビーズ2-8のプール1を、樹脂-NH2で誘導体化する。それぞれの誘導体化したビーズ2-8は、上で示し定義したように、異なるサブモノマーと個々に反応させる。それぞれの反応を終了し、以下のものを得る:
【0121】
【化11】
【0122】
ただし、Xは好ましくはNH、Zは好ましくはBr、R2およびR1は好ましくはそれぞれ独立的に、1から6の炭素原子を含むアルキルである。
【0123】
あらゆる所望の長さの鎖を提供するために、追加のサブモノマー反応物を連続的に付加し得るが、本発明の単純なバージョンにおいては、各サブアマウントとの一度の完全な反応の後に得られる各反応生成物をプールして、図1のプール9を得る。プール9を、混合し、次いで、サブアマウント11から24に分ける。
【0124】
より単純でないバージョンにおいては、各サブアマウントを、次いで、他の(かつ異なる)サブモノマー反応物と反応させ、それぞれの反応を終了させる。「アシルサブモノマー」を「アミノサブモノマー」と反応させて、アミノ酸様モノマーユニットを得る。アミノ酸様モノマーユニットのペプチド様鎖を作成するために、ビーズを所望の回数だけ、分割、反応、およびプールし得る。
【0125】
この概略化された例において、第2およびそれに続くサブモノマーとの反応は図示しておらず、サブモノマーユニットの第1の反応で得られた反応生成物をプールし、サブアマウントに分け、以下の一般構造式を有する環状化合物と反応させる:
【0126】
【化12】
【0127】
ただし、少なくとも1つの環状反応物が官能基として、A1-A5(好ましくはアルデヒドまたはケトンである)を含むという条件の下で、Yは-O、-SO、または-NHであり、A1、A2、A3、A4、およびA5のそれぞれは、独立的に、炭素原子に結合可能なあらゆる部分である。各環状化合物の各サブアマウントとの反応は終了され、以下の反応生成物を得る:
【0128】
【化13】
【0129】
各サブアマウントの反応生成物を、次いで、プールし、複数の固体支持体表面(すなわちビーズ)に結合した環状化合物のライブラリを得る。化合物を、固体支持体から開裂し得る。
【0130】
末端基は、上に示すように置換された環状化合物であり得る。しかし、ライブラリの多様性を広くするために、置換された環状部分の官能基をさらに反応させ得る。末端基がアルデヒド部分で置換された環状化合物である場合に、このことがいかにして特定の例において行われ得るかを以下で説明する。
【0131】
アルデヒドとの反応
上記説明に従って合成される本発明のライブラリは、最後に付加する反応物上にアルデヒド官能基を含む。1または全ての公知のタイプの反応を通して、アルデヒド基を反応させることによって、ライブラリの多様性を広くし得る。
【0132】
第1に、芳香族アルデヒドなどのアルデヒドを、以下のようにR-NH2と反応させ、イミン形成を得ることができる:
【0133】
【化14】
【0134】
ただし、Arはあらゆる芳香族部分、ならびにPおよびRは上記定義した通りである。
【0135】
第2に、芳香族アルデヒドなどのアルデヒドを、クネーフェナーゲルタイプの反応に供し、以下のような反応生成物を得ることができる:
【0136】
【化15】
【0137】
ただし、EWG1およびEWG2はそれぞれ、独立的に、電子吸引基であり、PおよびArは上記定義した通りである。好ましい電子吸引基には、COR、CN、CO2R、SO2R、SOR、およびSRが含まれる。
【0138】
第3に、芳香族アルデヒドなどのアルデヒドを、ウィッティヒタイプの反応に供し、以下のような反応生成物を得ることができる:
【0139】
【化16】
【0140】
ただし、P、Ar、RおよびEWGは上記定義した通りである。
【0141】
上記構造において、芳香族環は、固体支持体表面に直接結合して示している。これは、芳香族環が適切な官能基(通常、芳香族環上のNH2基)を介して支持体表面に結合、または支持体の上で誘導体化されなければならないところ、本箇所および本明細書のその他の箇所において簡潔化の目的で行っている。NH2基は、しばしば、樹脂として言及される。従って、上記構造は、以下のようにも示し得る:
【0142】
【化17】
【0143】
環状構造を形成するためのアルデヒド
その上にアルデヒド基を有する化合物を固体支持体上で合成した(上記「一般法」セクションに従って)後、アルデヒド基を種々の異なる化合物と反応させて、4、5または6員環構造を形成し得る。4員環を形成する例は以下の通りである:
【0144】
【化18】
【0145】
ArおよびPは上記定義され、R1およびR2は、独立的に、上記定義したようにあらゆるRである。
【0146】
5員環を形成する例は、以下の通りである:
【0147】
【化19】
【0148】
6員環を形成する例は、以下の通りである:
【0149】
【化20】
【0150】
ただし、R、R1、R2、R3、およびR4は、独立的に、上記Rと定義される。
【0151】
サブモノマーユニットを、互いと連続的に反応させて、ペプチドまたはペプチド様のバックボーン構造を形成し得る。反応は、分割樹脂法を用いて進行し得る。サブモノマー法を実施するために、アシルサブモノマーを、アミノサブモノマーと反応させる。アシルサブモノマーは、反応性カルボニルまたはカルボニル当量、およびアミンにより求核置換において置換され得る開裂基を含む。適切なアシルサブモノマーには、これらに限定されることなく、ブロモ酢酸、3-ブロモプロピオン酸、2-ブロモプロピオン酸、2-ブロモエチルイソシアネート、2-ブロモエチルクロロホルメート、6-フェニル-3-ブロモヘキサン酸、4-ブロモメチル-安息香酸、4-ブロモメチル-2-メトキシ安息香酸、5-ブロモメチル-ピリジン-2-カルボン酸などが含まれる。
【0152】
アシルサブモノマーを、アミノサブモノマーと反応させる。ここで好ましいアミノサブモノマーは、第1級アミンおよびヒドラジドであるが、アミド、カルバメート、尿素、カルバジド、カルバゼート、セミカルバジドなども適切である。あらゆるサブモノマーユニットも、「ヒドロカルビル」または「側鎖」と同様、置換され得るため、異なるサブモノマーを多数の異なるサブアマウントと反応させ、分割樹脂法を適用する際にそのような反応それぞれを終了させることが可能である。
【0153】
サブモノマー法を介した環状ペプトイドの調製
環状ペプトイドを、サブモノマー法によって調製した。そのための一般反応スキームを以下に示す。
【0154】
サブモノマー環化
【0155】
【化21】
【0156】
環化を生じるために要所となる反応は、N-末端ブロモアセタミドの側鎖求核剤での置換であり、樹脂上に「頭-側鎖(head-to-side-chain)」環状構造を生成する。側鎖求核剤は、標準サブモノマー状態を介してオリゴマーの所望の部分で取り入れられる。典型的な求核剤は、保護され得るチオールおよびアミンである。この目的のために好ましいサブモノマーは、Moz-NH-CH2-CH2-NH2、Alloc-NH-CH2-CH2-NH2、およびTrt-S-CH2-CH2-NH2である。オリゴマーを次いで、所望の長さが得られるまで合成し(elaborated)、ブロモアセトアミド基によって終了する。側鎖求核剤を、次いで、選択的に脱保護して環化させる。
【0157】
生成される環状ペプトイドの特定の例、および得られたパーセンテージ収率を、以下に記載する。特定の環構造を有する、ペプトイドから得られる環状化合物の例を本明細書の実施例19〜31に記載する。
【0158】
三量体
【0159】
【化22】
【0160】
固体支持体上で合成された環状有機化合物のライブラリーの有用性
高度に置換された環状構造物は、本発明のサブモノマー方法と、強力な液相化学方法とを組み合わせることによって固体支持体上で合成され得る。1個、2個、3個またはそれ以上の縮合環を含む環状化合物は、まず、線形バックボーンを合成し、次に分子内または分子間環化を行うことによってサブモノマー方法を用いて形成される。
【0161】
レセプター蛋白質または他の分子の結合領域を最も効率的にプローブするためには、種々の置換基および/または環構造物を有する環状有機化合物のライブラリーを作成することが一般に好ましい。
【0162】
種々の構造物がライブラリーに存在すると、所望の結合特性を有する化合物を単離する機会がそれだけ増加する。本願に記載する方法を、環状有機化合物の収集物を固体支持体上で合成するのに適用することによって、スクリーニング用の大きなグループの化合物が調製され得る。例えば、種々の置換基を有する環状アルデヒド類のライブラリーが、相対レセプター結合アフィニティを分析するために調製され得る。ライブラリーは、小さく(約10個の異なる構造物)ても、大きく(10,000個より多くの異なる構造物)てもよい。
【0163】
このようなライブラリーは、天然に発生する生物活性ペプチド、または必須のアフィニティによって適切なレセプターに結合する他の分子に対する環状有機アナログを同定するために有用である。例えば、仮説ペプチドが公知の細胞表面レセプターに結合する場合、細胞表面レセプターを発現する細胞の培養物を調製し、結合に良い条件下でライブラリーを適用し、ライブラリーのメンバーが細胞表面レセプターに結合する程度を決定またはレセプターの応答を顕在化し得る。
【0164】
ライブラリーの環状有機化合物とレセプターとを相互作用させた後、非結合化合物を洗浄によって除去する。多数の環状有機化合物が高結合アフィニティを示す場合、最も高いアフィニティを示す環状有機化合物のみが結合されたままになるように結合条件を変更してもよい。次に、結果として得られる選択された環状有機化合物を除去し、標準分析技術によって同定し得る。
【0165】
例えば、模倣される生物活性分子の活性部分の関連構造物が未知のものである場合、本発明の方法を用いてより大きなライブラリーを構築すればよい。ペプチドまたはエピトープの構造形態に関する手掛かりがない場合は、広範囲の置換基および/または環構造物改変体を有する「普遍的」なライブラリーが最も有用である。
【実施例】
【0166】
以下の実施例は、本発明の化合物およびライブラリーの作成方法に関する完全な開示および説明を当業者に提供するものであり、発明者が発明であると見なす範囲を越えるものではない。使用した数値(例えば、量、温度、等)に関する精度を保証するために努力をしたが、多少の実験誤差および偏差を考慮されたい。特に記載がない限り、部は重量部、分子量は平均分子量、温度は摂氏、および圧力は大気圧または大気圧付近とする。
【0167】
実施例1から11は、概してアルデヒド基を含む反応に関し、実施例12から27は、ケトン基を含む反応に関し、これらの基はすべて、概して環状有機化合物に結合される。他の官能基も、環状化合物に結合し、公知のタイプの反応にかけられ、ライブラリーの多様性を増加し得る。
【0168】
樹脂上でのピロリジン合成の実験条件
概説 Varian Unity 300分光計を用いてNMR分析を行った。以下のグラジエント:250℃で10分間、1分当たり10℃の勾配で325℃まで、および325℃で15分間を用いて、HP 5972質量選択検出器に連結されたHewlett Packard 5890ガスクロマトグラフを用いてGCMS分析を行った。以下のグラジエント:45分間にわって5%Bから95%Bを用いて、Waters 600Eシステム、Alltech Alltima C18逆相カラム(5mm粒子サイズ、4.6×250mm)、220nMでUV検出する25℃、1分間1.0mLの溶剤システムA:B(各0.1col%TFAを含む、水:アセトニトリル)を使用してHPLC分析を行った。
【0169】
実施例1
【0170】
【化23】
【0171】
15mLの無水THF中Rinkアミド樹脂(1.12mmol)上に2.0gの1を含有する20mLのガラス瓶に、517mgのLiOH・H2O(12.32mmol)および1.81mlのトリメチルホスホノアセテート(11.2mmol)を加えた。瓶に蓋をし、室温で14時間振盪させた。樹脂をCH2Cl2、H2O、MeOHおよびエーテルで連続して洗浄し、高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、純粋な化合物2が得られた。1H NMR(CDCL3、300MHz): δ3.84(s、3H)、6.51(d、J=15.6Hz、1H)、7.62(d、J=8.6Hz、2H)、7.71(d、J=15.6Hz、1H)、7.85(d、J=8.6Hz、2H)。HPLC: 1ピーク、tR=21.0分、GCMS: 1ピーク、tR=3.74分、M=205、計算値M=205)。
【0172】
実施例2
ピロリジン類をベースにしたサルコシン/サルコシン誘導体:
サルコシンと、Rinkアミド樹脂(1)に結合した芳香族アルデヒドとを結合させ、適切に置換されたアルケンを加えることによって付加環化を実施し、5を得る。
【0173】
具体的には、2.5mLのトルエンおよび110mgの樹脂上の1(0.062mmol)を有する2ドラム瓶に、220mgのサルコシン(2.46mmol)および419mgのジメチルムコネート(2.46mmol)を加え、30秒間アルゴン脱気し、しっかりと蓋をした。次に、瓶を110℃で12時間振盪させ、その後樹脂をフィルターに移し、CH2Cl2、DMF、MeOHおよびエーテルで洗浄し、次に高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、5の4個の異性体が得られた。GCMS: 4ピーク、tR=10.71分(42.9%)、11.01分(4.5%)、11.68分(40%)、11.88分(12.6%)、M=346、計算値M=346。
【0174】
【化24】
【0175】
実施例3
実施例2と同様に、サルコシンおよび芳香族アルデヒドと、2とを結合させて6を得ることができる。
【0176】
具体的には、1.5mLのトルエンおよび100mgの樹脂上の2(0.0565mmol)を有する2ドラム瓶に、220mgのサルコシン(2.24mmol)および227μLのベンズアルデヒド(2.24mmol)を加え、30秒間アルゴン脱気し、しっかりと蓋をした。瓶を110℃で12時間振盪させ、その後樹脂をフィルターに移し、CH2Cl2、DMF、MeOHおよびエーテルで洗浄し、次に高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、6の4個の異性体が得られた。GCMS: 4ピーク、tR=10.96分(9.7%)、11.78分(21.1%)、12.30分(44.6%)、13.17分(24.6%)、M=338、計算値M=338。
【0177】
【化25】
【0178】
実施例4
【0179】
【化26】
【0180】
7mlの1:1MeOH/1,4-ジオキサン中1g(0.43mmol)の樹脂に結合したアルデヒド1の懸濁液に、93μl(2.15mmol)のメチルアセトアセテート、次いで43μl(43mmol)のピペリジンを加えた。反応混合物を室温で24時間振盪させ、樹脂2を濾過し、CH2Cl2で数回洗浄した。樹脂2をCH2Cl2中10%のTFAで処理し、生成物3をGCMSによって分析したところ、所望の生成物である、m/z=227の親イオンを有する単一の生成物が示された。
【0181】
実施例5
【0182】
【化27】
【0183】
10mlのベンゼンおよびモレキュラーシーブ中1g(0.51mmol)の樹脂に結合したアルデヒド1の懸濁液に、292μl(2.55mmol)のジメチルマロネート、次いで126μl(1.28mmol)のピペリジンを加えた。次に、反応混合物を室温で一晩振盪させた。樹脂2を濾過し、CH2Cl2で数回洗浄し、CH2Cl2中10%のTFAで処理し、生成物3を定量的収率で得た。GCMSによって分析したところ、生成物に対応する、m/z=263の親イオンを有する単一の化合物の存在が示された。
【0184】
実施例6
【0185】
【化28】
【0186】
Rink樹脂に結合したブロモアセトアミド(50mg、0.024mmol、0.47mmol/g)および乾燥DMF(2mL)の混合物(23℃)に、無水K2CO3(130mg、0.94mmol)NaI(2mg、0.013mmol)およびバニリン(143mg、0.94 mmol)を加えた。75℃で16時間激しく振盪させた後、得られた混合物をH2O(10mL)、MeOH(10mL)、THF(2×10mL)、CH2Cl2(3×10mL)で希釈し、真空下(150mm)で3時間乾燥させ、54mgの淡黄色樹脂を得た。
【0187】
この樹脂(54mg、0.24mmol)に、CH2Cl2中5%のTFA溶液(2mL)を滴下した。23℃で15分間保持した後、混合物を濾過し、濾液をCH2Cl2(2×5mL)で洗浄し、溶媒を濃縮させ、3.7mg(74%)の1を淡黄色油として得た:1H NMR(300MHz、CDCl3)δ 9.86(s、1H)、7.48-7.42(m、2H)、6.97-6.94(d、1H)、4.62(s、2H)、3.96(s、3H)、3.39-3.15(br s、2H)>。
【0188】
実施例7
【0189】
【化29】
【0190】
Rink樹脂に結合した4−カルボキシベンズアミドベンジルイミン(108mg、0.085mmol)および乾燥ベンゼン(4mL)の混合物(23℃)に、i-Pr2NEt(60μL、0.34mmol)および無水1,2−フェニレンジ酢酸(59.6mg、0.34mmol)を加えた。70℃で16時間激しく振盪させた後、得られた混合物を濾過し、H2O(10mL)、MeOH(10mL)、THF(3×10mL)、CH2Cl2(3×10mL)で洗浄し、真空下(10d mm)で3時間乾燥させ、122mgの濃茶色樹脂を得た。
【0191】
この樹脂(122mg、0.085mmol)に、CH2Cl2中5%のTFA溶液(3mL)を滴下した。23℃で15分間保持した後、混合物を濾過し、濾液をCH2Cl2(2×5mL)で洗浄し、溶媒を濃縮させ、26.8mg(76%)の2を濃黄色油として得た: MS(ESP)m/e、C25H23N2O4に対する計算値415、測定値415(MH)。
【0192】
実施例8
樹脂に結合したアルデヒドの合成
4−[2',4'−ジメトキシフェニル−(4'−ホルミル−フェニルアセトアミド)]−フェノキシ樹脂{4−ベンアズアルデヒドRinkアミド樹脂}
【0193】
【化30】
【0194】
Fmoc Rinkアミド樹脂(20.0g、9.6mmol)をピペリジン(70ml)およびDMF(80ml)の混合物中で40分間振盪させた。得られた懸濁液を濾過し、DMF(4×100ml)、水(4×100ml)、THF(4×100ml)、ジクロロメタン(4×100ml)で逐次洗浄し、次に真空下で乾燥させた。
【0195】
ジクロロメタン(80ml)中の4−カルボキシベンズアルデヒド(5.0当量、7.21g)の懸濁液に、N−メチルモルホリン(5.0当量、5.3ml)およびPyBOP(5.0当量、25.0g)を加えた。得られた溶液を10分間撹拌し、乾燥樹脂に加えた。この懸濁液を一晩振盪させ、標準ニンヒドリンテストを用いて反応が完了するのをモニターした。反応物を濾過し、樹脂を上記で報告した洗浄サイクルにかけた。樹脂を完全に乾燥させた後、少量(40〜50mg)をジクロロメタン(2ml)中のTFA(40〜50μl)で15分間処理し、濾過し、濾液を真空下で濃縮させた。回収物をNMRおよびGCMSで分析し、収率および純度を決定した。
【0196】
実施例9
3−ベンズアルデヒドRinkアミド樹脂
【0197】
【化31】
【0198】
上記の方法を用いて、N−メチルモルホリン(5.0当量、5.3ml)およびPyBOP(5.0当量、25.0g)の存在下で、Rink樹脂(20.0g)と3−カルボキシベンズアルデヒド(5.0当量、7.21g)とを結合させた。
【0199】
実施例10
4−ホルミルフェノキシアセトアミドRink樹脂
【0200】
【化32】
【0201】
上記の方法を用いて、N−メチルモルホリン(5.0当量、1.32ml)およびPyBOP(5.0当量、6.24g)の存在下で、Rink樹脂(5.0g)と4−ホルミルフェノキシ酢酸(5.0当量、2.16g)とを結合させた。
【0202】
実施例11
4−ブロモメチルベンジルRinkアミド樹脂
【0203】
【化33】
【0204】
上記の方法を用いて、N−メチルモルホリン(5.0当量、1.32ml)およびPyBOP(5.0当量、6.24g)の存在下で、Rink樹脂(5.0g)と4−ブロモメチルトルイル酸(5.0当量、2.58g)とを結合させた。
【0205】
実施例12
4−アジドベンゾイル(azidobenyl)Rinkアミド樹脂
【0206】
【化34】
【0207】
上記の方法を用いて、N−メチルモルホリン(5.0当量、0.85ml)およびPyBOP(5.0当量、4.04g)の存在下でRink樹脂(5.40g)と4−アジド安息香酸(3.0当量、1.27g)とを結合させた。
【0208】
実施例13
固体支持体に結合したジケトンからの環状有機化合物の合成
固体支持体に(Rinkリンカーを介して)結合したジケトンIV(70μmol)をDMSO(1mL)中で懸濁する。シアノアセトアミド(59mg、700μmolに等しい)およびピペリジン(144μL、1400μmolに等しい)を加え、反応混合物を室温で撹拌した。反応混合物を排出し、DMSO(6回)、メタノール(3回)およびジクロロメタン(6回)で洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、次に濾過した。揮発性物質をアルゴン気流下でエバポレートし、生成物VIIを得た。
【0209】
【化35】
【0210】
実施例14
固体支持体に結合したジケトン類からのモノケトフラン類の合成
約70μmolの固体支持体に結合したIVをDMF(1mL)中で懸濁する。ヨードベンゼン(184mg、700μmolに等しい)、炭酸カリウム(193mg、1400μmolに等しい)およびテトラキス(トリフェニルホスフィン)パラジウム(O)(39mg、35μmolに等しい)を加えた。反応混合物をアルゴンで軽く分散させ、加熱(60〜100℃)した。反応混合物を冷却し、排出し、DMF(6回)、メタノール(3回)、およびジクロロメタン(6回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、生成物VおよびVIを得た。
【0211】
【化36】
【0212】
実施例15
固体支持体に結合したジケトン類からのN−置換ピラゾール類の合成
樹脂の調製:
Fmoc Rinkアミド樹脂(20.0g、9.6mmol)をピペリジン(70mL)およびDMF(80mL)の混合物中で40分間振盪させた。得られた懸濁液を濾過し、DMF(4×100mL)、水(4×100mL)、THF(4×100mL)、ジクロロメタン(4×100mL)で逐次洗浄し、次に真空下で乾燥させた。
【0213】
固体支持体に結合したモノケトンの合成:
ジクロロメタン(80mL)中の4−カルボキシアセトフェノン(5.0当量)の懸濁液に、N−メチルモルホリン(5.0当量)およびPyBOP(登録商標)(5.0当量)を加えた。得られた溶液を10分間撹拌し、乾燥樹脂に加えた。懸濁液を一晩振盪させ、標準ニンヒドリンテストを用いて反応が完了するのをモニターした。反応物を濾過し、樹脂を、DMF(4×100mL)、水(4×100mL)、THF(4×100mL)、ジクロロメタン(4×100mL)で逐次洗浄し、次に真空下で乾燥させた。樹脂を完全に乾燥させた後、少量(40〜50mg)をジクロロメタン(2mL)中のトリフルオロ酢酸(TFA、40〜50μl)で15分間処理し、濾過し、濾液を真空下で濃縮させた。回収物をNMRおよびGCMSで分析した。構造物は、固体支持体から分離した化合物1について予想されたのと一致していた。
【0214】
支持体に結合したジケトンの合成:
樹脂に結合したジケトンを調製するため、化合物2、樹脂に結合した化合物1(上記で調製した0.47mmol、1.0g)を12mLのTHF中に懸濁した。15mgのジベンゾ−18−クラウン−6および400μLの酢酸エチル(4.7mmol)をアルゴン気流下で導入した。THF(2.8mmol)中の2.8mLの1M カリウムt−ブトキシドを加える前に、懸濁液を撹拌した。反応物を1時間70℃まで加熱した。次に、冷却した樹脂を濾過し、ジクロロメタンで洗浄した。少量の樹脂をジクロロメタン中20%のTFAで処理し、濾過し、揮発物をアルゴン気流下でエバポレートした。GC-MSおよびNMR分析の結果は、化合物2への定量的変換と一致していた。
【0215】
固体支持体上で化合物4を調製するために、約100mgの固体支持体に結合したジケトン2(47μmol)を1mLの乾燥DMSOおよび50μLのフェニルヒドラジン(470μmol)中で懸濁した。反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、化合物4を得た。GC-MSによる分析の結果、所望の生成物への定量的変換(電子衝撃によりMW=277)が示された。DMSO中の生成物のNMRは、固体支持体から分離した化合物4の構造と一致していた。
【0216】
【化37】
【0217】
実施例16
N−置換ピラゾールの他の合成
化合物4および化合物9の1:1混合物の調製を以下のように行った。化合物8の合成に関して、100mgの固体支持体に結合した化合物2(47μmol)を、1mLのトルエン、147μLのDBU(940μmol)、および50μLの1−ブロモブタン(470μmol)中に懸濁した。化合物4の合成に関して、50μLのフェニルヒドラジン(470μmol)を加え、反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、GC-MS分析により化合物4および化合物9の1:1混合物を得た。
【0218】
【化38】
【0219】
実施例17
縮合ピラゾールおよびピラン環の合成
化合物IIIを以下のように調製した。固体支持体に結合したピラゾリジノンの合成を以下に示す。100mgの固体支持体に結合したβ−ケトエステル13を、1mLの乾燥DMSOおよび50μLのフェニルヒドラジン中に懸濁した。反応容器を絶えず撹拌しながら70℃まで18時間加熱し、ピラゾリジノン16(一般構造)を得た。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。70mgのピラゾリジノンIをトルエン(1mL)中に懸濁した。ピペリジン(70μL、700μmolに等しい)、ベンズアルデヒド(144μL、1400μmolに等しい)および4A0モレキュラーシーブ(1スパチュラチップ)を加え、反応混合物を室温で撹拌した。モレキュラーシーブからデカンテーションした後、固体支持体をジクロロメタンで6回洗浄した。得られた生成物である固体支持体に結合した化合物IIをDMSO(1mL)中に懸濁した。ピペリジン(70μl、700μmolに等しい)およびマロノニトリル(93mg、1400μmolに等しい)を加え、反応混合物をアルゴンで軽く分散させ、次に加熱(60〜120℃)した。冷却後、反応混合物を排出し、ジクロロメタン(6回)、メタノール(3回)およびジクロロメタン(3回)で洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、化合物IIIを得た。
【0220】
【化39】
【0221】
他のピラゾール合成:
【0222】
【化40】
【0223】
実施例18
2−アミノ−ピリミジンの合成:
化合物6の調製を以下のように行った。75mgの固体支持体に結合した化合物2(35μmol)を、67mgのグアニジン塩酸塩(700μmol)を含む750μLの乾燥DMSO中に懸濁する。次に、塩酸塩を、アルゴン気流下で、kTHF中の500μLの1M カリウムt−ブトキシド(500μmol)でインサイチュで中和した。次に、反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミド、水、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から分離した化合物6を得た。229(M+1)のエレクトロスプレーMSピークは、固体支持体から分離した6の構造と一致していた。
【0224】
【化41】
【0225】
実施例19
カルコン3の合成
50mg(0.47mmol)のベンズアルデヒドおよび100mg(0.047mmol)のRinkアミド樹脂に結合した5mLのTHF中の化合物1の混合物に12uLの2M ナトリウムエトキシドを加えた。得られた混合物を室温で2時間振盪させた。得られた樹脂に結合した化合物2を3mLの濾過カラムに収集し、水(3×2mL)、DMF(2×2mL)および塩化メチレン(3×2mL)で洗浄した。真空下で乾燥させた後、5mgの樹脂に結合した化合物2を、塩化メチレン中1.5mLの5%トリフルオロ酢酸で20分間処理した。固体を濾過し、1mLの塩化メチレンで洗浄し、合わせた塩化メチレン溶液を濃縮し、真空下で乾燥させ、0.5mgの生成物3を得た。MS m/z 252(M++1)。
【0226】
【化42】
【0227】
実施例20
ジエステル5の合成
20mg(0.009mmol)のRink樹脂に結合した化合物2および0.60mLのTHF中の25mg(0.18mmol)のマロン酸ジメチルの混合物に13uLの1,8−ジアザビシクロ[5,4,0]ウンデカ−7−エン(0.09mmol)を加えた。得られた混合物をアルゴン下、40℃で2時間振盪させた。得られたRink樹脂に結合した化合物4を3mLの濾過カラムに収集し、DMF(2×2mL)、CH2Cl2(2×2mL)で洗浄した。真空下で乾燥させた後、5mgの樹脂に結合した化合物4を、1mLのCH2Cl2で処理し、合わせたCH2Cl2溶液を真空下で濃縮し、0.8mgの生成物5を得た。MS m/z 384(M++1)。
【0228】
【化43】
【0229】
実施例21
テトラヒドロピリドン9の合成
10mg(0.0047mmol)のRinkアミド樹脂に結合した化合物4および7mg(0.047mmol)の4−メトキシベンジルアミン、ならびにTHF中の0.8mLの1M NaBH3CNの混合物に、1滴の酢酸および1滴のH2Oを加えた。この混合物をアルゴン下で密封し、75℃で24時間振盪させた。最終の樹脂に結合した化合物8をDMF(3×2mL)、H2O(2×2mL)、およびCH2Cl2(3×2mL)で洗浄した。得られた化合物8をCH2Cl2中5%のトリフルオロ酢酸で20分間処理した。固体を濾過し、1mLのCH2Cl2で洗浄し、合わせたCH2Cl2溶液を真空下で濃縮し、1.8mgの生成物9を得た。MS m/z 469(M++1)。
【0230】
【化44】
【0231】
実施例22
ジヒドロピリドン7の合成
10mg(0.0047mmol)のRinkアミド樹脂に結合した化合物4および0.8mLのトルエン中7mg(0.047mmol)の4−メトキシベンジルアミンの混合物に1滴の酢酸を加えた。この混合物をアルゴン下で密封し、80℃で15時間振盪させた。最終の樹脂に結合した化合物6をDMF(3×2mL)、H2O(2×2mL)、およびCH2Cl2(3×2mL)で洗浄した。得られた化合物6をCH2Cl2中5%のトリフルオロ酢酸で処理し、溶液を真空下で濃縮し、1.8mgの生成物7を得た。MS m/z 471(M++1)。
【0232】
【化45】
【0233】
実施例23
樹脂1(100mg、0.056mmol)を、1.5mlのEtOHおよび1,4−ジオキサンの1:1混合物と合わせ、130μl(1.12mmol)のアセトフェノンおよび87mg(1.12mmol)のNH4OAcを加えた。反応混合物を80〜85℃で一晩振盪させ、H2O、MsOHおよびCH2Cl2で洗浄した。樹脂2をCH2Cl2中10%のTFAで切断し3を得た。ESMS m/z = 351(M+1)。樹脂4を同一の反応条件で処理し、ピリジン6を得た。ESMS m/z = 315(M+1)。
【0234】
【化46】
【0235】
実施例24
イソオキサゾールの合成:
44mg(0.11mmol)のD−グルコピラノシルニトロメタンテトラ−アセテート(αアノマー)および30mg(0.016mmol)のRinkアミド樹脂に結合した化合物1を含有する0.80mLの1,2−ジクロロエタン中24uL(0.22mmol)のフェニルイソシアナートの溶液に、2mLの(0.016mmol)のトリエチルアミンをアルゴン下で加えた。この混合物を還流し、78℃で15時間振盪させた。得られた混合物を室温まで冷却した。樹脂に結合した化合物2を3mLの濾過カラム中で収集し、N,N−ジメチルホルムアミド(3×2mL)、塩化メチレン(3×2mL)で洗浄し、真空下で乾燥させた。乾燥した樹脂に結合した化合物2を塩化メチレン中2mLの20%トリフルオロ酢酸で20分間処理した。固体を濾過し、1.5mLの塩化メチレンで3回洗浄した。合わせた洗浄物を濃縮し、真空下で乾燥させ、12mgの約95%の純度の生成物3を得た。スペクトルデータは、電子スプレーMS m/z 884(M++1)からなっていた。
【0236】
【化47】
【0237】
実施例24
置換ピランの合成
化合物18、2−アミノ−3−シアノピランを以下の手法で調製した。ケトン:P-Ar-C(O)-CH2-Rからのa,b不飽和−ケトンの調製物。この時点で分割樹脂方法を行うと、次の環化工程と同様に多様性を加える。固体支持体樹脂(47μmol)に共有結合した置換α,β−不飽和ケトンを溶媒(1mL)に懸濁した。ジシアノメタンを加え、反応混合物を撹拌した。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物18を得た。
【0238】
化合物18の混合物を、まず、固体支持体樹脂上にα,β−不飽和ケトン類の混合物を調製することによって合成する。混合物の多様性を、α,β−不飽和ケトンの種々のR基によって制御する。R基のそれぞれは、独立してアルキル基またはアリール基であり得る。さらに、α,β−不飽和ケトンは、R基の任意の位置で、樹脂に共有結合され得る。次に、固体支持体に結合したケトン類の混合物を上記のようにDI−シアノメタンと反応させ、一般構造18を有する化合物の混合物を得る。
【0239】
【化48】
【0240】
α,β不飽和ケトンの一般的な合成。
【0241】
【化49】
【0242】
実施例26
ピペラジン−2−オンの合成
化合物4の一般構造を有するピペラジン−2−オンを以下の手法で調製した。化合物2などの固体支持体に結合したα,β−不飽和ケトンを溶媒(1mL)に懸濁した。yyμmolのエステルS’を加え、反応混合物を適切な温度で十分な時間撹拌し、固体支持体に結合した化合物Tを得た。誘導体化された固体支持体をジクロロメタンで(6回)洗浄し、濾過した。この時点で、固体支持体を、化合物4および5を生成する異なる反応用の部分に分割してもよい。
【0243】
化合物Uを、xxμmolの固体支持体に結合した化合物3を溶媒(1mL)に懸濁することによって生成する。第1級アミンおよびNaBH3CNを加え、反応混合物を適切な温度で適切な時間撹拌する。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物4を得た。一般構造4を有する化合物の混合物を、R基を変えることによって調製する。R1、R2、R3およびR4は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基、または水素である。Arは、任意の芳香族またはヘテロ芳香族部分である。Xは、任意の芳香族もしくはヘテロ芳香族部分、または電子吸引基である。
【0244】
【化50】
【0245】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
実施例27
テトラヒドロピラジン−2−オンの合成
化合物5を、固体支持体に結合した化合物3を酢酸を含有する溶媒(1mL)に懸濁することによって生成する。第1級アミンを加え、反応混合物を適切な温度で適切な時間撹拌する。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物5、テトラヒドロ−2−ピリドンを得た。一般構造5を有する化合物の混合物を、サブモノマー上のR基を変えることによって調製する。R1、R2、R3およびR4は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基または水素である。Arは、任意の芳香族またはヘテロ芳香族部分である。Xは、本願で定義した任意の芳香族もしくはヘテロ芳香族部分、または電子吸引基である。
【0246】
【化51】
【0247】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
実施例28
置換ピリジン類の合成
化合物8、2−アミノ−3−シアノ−ピリジンを以下のように調製した。固体支持体に結合した化合物6を酢酸アンモニウムを含む溶媒(1mL)に懸濁した。ケトン7を加え、反応混合物を適切な温度で適切な時間撹拌した。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物8、2−アミノ−3−シアノ−ピリジンを得た。一般構造8を有する化合物の混合物を、サブモノマー出発物質のR基を変えることによって調製する。R1、R2、およびR3は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基または水素である。
【0248】
【化52】
【0249】
R1、R2、R3、= 芳香族、脂肪族、ヘテロ芳香族(heteromatic)、H
ケトン類からの他の実施例
【0250】
【化53】
【0251】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
【0252】
【化54】
【0253】
【化55】
【0254】
本願は、本願と同一の譲受人に譲渡され、本願と同一日に提出された「SYNTHESIS OF N-SUBSTITUTED OLIGOMERS」という名称の代理人整理番号第06515/022001号の出願に関連する。特許出願第06515/022001号は、1994年7月18日付けで先に提出された米国特許出願第08/277,228号の一部継続出願であり、米国特許出願第08/277,228号は、1993年9月24日付けで先に提出された米国特許出願第08/126,539号の一部継続出願であり、米国特許出願第08/126,539号は、1992年9月24日付けで先に提出された米国特許出願第07/950,853号の一部継続出願であり、これらの出願はすべて同一譲受人に譲渡され、本願では参考のためにこれらの出願をそのまま援用する。
【0255】
本発明は、その具体的な実施態様に基づいて説明したが、当業者には当然のことながら、本発明の真の精神および範囲を逸脱せずに、種々の変更がなされ、等価物の代用がなされ得る。さらに、特定の反応、物質、ライブラリー、プロセス、1つのプロセス工程または複数のプロセス工程を本発明の目的、精神および範囲に適用するために、多くの改変がなされ得る。このような改変はすべて添付の請求の範囲の範囲内であるものとする。
【図面の簡単な説明】
【0256】
【図1】図1は、分割樹脂方法論を示す模式図である。
【技術分野】
【0001】
発明の分野
本発明は、広くは、化学合成技術に関する。具体的には、本発明は、固相出発物質上で合成された複素環式有機化合物の組み合わせライブラリーの合成、およびそのようなライブラリーを生物活性についてアッセイする方法に関する。
【背景技術】
【0002】
発明の背景
ペプチド合成のための伝統的な固相方法に類似する標準的な方法が、ペプチドおよびペプチド様化合物のライブラリーを合成するのに適用され得る。このような方法によれば、N,α-Fmoc-保護(および側鎖保護)アミノ酸のカルボキシレートが、活性化され得、その後、樹脂結合アミノ基に結合され得る。Fmoc基は、その後、次のモノマーの付加に続いて除去される。このようなアプローチは、保護されたN-置換アミノ酸モノマーの多様なセットを適切な量だけ調製するのに要する時間およびコストのため、望ましくない。Fmocまたはその他の保護基を付加し、除去することは、時間のかかることであり、非効率的である。
【0003】
新しい薬学的に活性な有機薬剤(すなわち、結合に必要な3-D構造をもつ化合物)発見のためのあるアプローチは、主として、精製されたレセプターのX線結晶学に依存している。すなわち、いったん結合部位が同定されると、有機分子は、利用可能な立体空間および電荷分布に適合するように設計される。しかし、精製されたレセプターを得るのは、しばしば困難であり、X線結晶学が適用され得るようにレセプターを結晶化させるのは、さらに困難である。また、結合部位が正しく同定された後でも、適切なリガンドを考案することは、取るに足らないことではない。全体として、有用で薬学的に活性な化合物を設計することは、例えば、レセプターを同定し、それらのレセプターに結合する化合物の構造を精製・同定し、その後それらの化合物を合成することに伴う困難のようないくつかの要因のため、極めて困難である。
【0004】
新しい薬剤発見のための別のアプローチとしては、既知の生物学的に活性な化合物を模倣する化合物を合成することがある。しかし、活性化合物の活性部分または活性構造成分は通常未知であるので、新しい化合物を合成するプロセスは、主として、それぞれの化合物を個別に試行錯誤の末、合成し、スクリーニングすることに依存している。この方法は、時間がかかり、高価である。なぜなら、どの1つの化合物についても、成功の可能性は比較的低いからである。
【0005】
結晶学を用いたり、既知の生物学的に活性なペプチドを模倣する特定のペプチドを合成しようと試みたりすることにより、タンパク質の特定の3次元構造を決定しようとすることよりもむしろ、組み合わせライブラリーを作製することに関する技術が発展してきている。具体的には、生物学的に活性なペプチドを単離しようと試みるものは、同一の反応容器内で極めて多数の異なるペプチドを同時に生成する。合成された組み合わせライブラリーは、その後、アッセイされ、活性分子が単離され、分析される。組み合わせライブラリー自体は、米国特許第5,266,684号に開示されている。米国特許第'684号は、ほぼ完全に、ライブラリー中の反応生成物のそれぞれが、20個の天然に存在するアミノ酸からなるペプチドである、ライブラリーの合成に関している。
【0006】
薬学的に活性な化合物は、しばしば高度に置換された複素環であるので、本発明者らは、多数の関連する置換された複素環式化合物を迅速に、かつ比較的安価に、速やかに合成する方法の必要性を見出した。このアプローチによれば、生物活性を呈する構造成分が未知である、候補化合物群のそれぞれのメンバーについて個別に合成をおこなう問題を克服し得る。
【発明の開示】
【課題を解決するための手段】
【0007】
本発明によって以下が提供される:
(1)ライブラリーを合成する方法であって、
(a)複数の固体支持体表面を提供する工程と、
(b)XHを用いて該表面のそれぞれを誘導体化する工程であって、Xが、以下のような複数の該誘導体化された支持体表面を提供するOおよびNHからなる群から選択される、工程と、
P - XH
ここで、「P」は、該支持体表面の1つであり、
(c)該誘導体化された支持体表面を、複数のサブアマウントに分割する工程と、
(d)以下の一般構造式の異なるサブモノマーと、該誘導体化された支持体表面の該サブアマウントの該-XH基とを反応させる工程と、
【0008】
【化1−1】
【0009】
ここで、R1およびR2は、独立して、炭素原子に共有結合可能な任意の部分であり、Zは、ハロゲンであり、各反応を完了させて、
【0010】
【化1−2】
【0011】
を得る工程と、
(e)該各サブアマウントを再び組み合わせて混合し、複数の異なる化合物を有する固体支持体の混合物を得る工程と、
(f)該混合物を複数のサブアマウントに分割する工程と、
(g)以下の一般構造式の異なる環状化合物と、該各サブアマウントの該-Z部分とを反応させ、
【0012】
【化1−3】
【0013】
ここで、Yは、-O-、-S-、および-NHからなる群から選択され、A1、A2、A3、A4およびA5はそれぞれ、独立して、H、ヒドロカルビル、ケトン、アルデヒド、カルボン酸、エステル、アミド、アミン、ニトリル、およびエーテルからなる群から選択される部分であり、以下の構造の化合物を得る工程と、
【0014】
【化1−4】
【0015】
(h)該サブアマウントを再び組み合わせて、該複数の固体支持体表面上に化合物のライブラリーを提供する工程と、
を包含する方法。
(2)XがNHであり、ZがBrであり、R1およびR2が、それぞれ独立して、Hまたはヒドロカルビルであり、A1、A2、A3、A4およびA5のそれぞれが独立してHまたはヒドロカルビルである、項目1に記載の方法。
(3)R1、R2、A1、A2、A3、A4、およびA5のそれぞれが独立して、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目1に記載の方法。
(4)R1、R2、A1、A2、A3、A4、およびA5のそれぞれが独立して、H、1から6個の炭酸原子を含むアルキル、および20個の天然に存在するアミノ酸の1つからなる群から選択され、各分割工程において、分割が3またはそれ以上のサブアマウントへの分割である、項目1に記載の方法。
(5) 固体支持体に共有結合した10個またはそれ以上の異なる化合物によって特徴づけられる有機化合物のライブラリーであって、該化合物が、電子供与出発物質を結合した固体支持体から得られる環状有機化合物であり、
該10個またはそれ以上の化合物のそれぞれが、回収可能かつ分析可能な量で存在する、ライブラリー。
(6)少なくとも1000個の異なる化合物を含み、該生成化合物の少なくとも1つが生物学的に活性である、項目11に記載のライブラリー。
(7)前記電子供与出発物質が、アミドを介して固体支持体に結合したアルデヒドであり、前記環状有機化合物のそれぞれが、100ピコモルまたはそれ以上の量で存在し、さらに、該環状有機化合物の環状部分が、該環状部分に共有結合した側鎖を有する、項目6に記載のライブラリー。
(8)前記側鎖が、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目7に記載のライブラリー。
(9)10個またはそれ以上の環状有機化合物のライブラリーを固体支持体上で合成する方法であって、
該化合物の官能部分が、アクセス可能かつ反応可能であり、電子供与基を提供するように、固体支持体上に化合物を誘導体化する工程と、
該誘導体化された化合物を、環化反応に供する工程であって、複数の反応物が該化合物の官能部分と反応し、複数の有機環状化合物を形成する工程と、
を包含する方法。
(10) 前記化合物が、前記固体支持体に直接結合したアミドによって該支持体に結合される、項目9に記載の方法。
(11) 複数の異なるアルデヒド化合物が前記固体支持体に結合し、それぞれが、異なる官能基を有し、
前記環状有機化合物を複数のサブアマウントに分割する工程と、
異なる反応物を該各サブアマウントとカップリングし、各カップリング反応を該各サブアマウントを用いて実質的に完了させる工程と、
既知の量の該各サブアマウントを共に組み合わせ、化合物のライブラリーを得る工程と、
をさらに包含する項目9に記載の方法。
(12) 環状有機化合物に結合した第2級アミンを-NH2による求核置換を可能にする脱離基を含む第1のサブモノマーアシル化剤でアシル化し、アシル化アミンを得る工程と、
該アシル化アミンと、-NH2基を含む十分な量の第2のサブモノマー置換剤とを反応させ、該脱離基の求核置換を実施する工程と、
該アシル化および反応工程を、所望の長さを有し、所望の置換基を含むポリマーを形成するのに適切なように連続して繰り返す工程と、
該ポリマーを環化し、共有結合したペプトイドバックボーンを有する環状生成化合物を生成する工程と、
をさらに包含する、項目9に記載の方法。
(13) それぞれが回収可能かつ分析可能な量で存在する10個またはそれ以上の異なる化合物を含む化合物のライブラリーであって、該化合物のそれぞれが、以下の一般構造式を有し、
【0016】
【化1−5】
【0017】
ここで、「a」および「b」が、それぞれ独立して、0から100までの整数であり、但し、該ライブラリーが、1またはそれ以上である「a」および「b」を有する化合物を含み、各Yが、独立して、-O-、-S-および-NHからなる群から選択され、各R1およびR2が、独立して、炭素原子に共有結合可能な任意の部分であり、各A1、A2、A3、A4およびA5が、独立して、H、ヒドロカルビル、ケトン、アルデヒド、カルボン酸、エステル、アミン、アミド、エーテル、およびニトリルからなる群から選択される、ライブラリー。
(14) A1、A2、A3、A4およびA5の少なくとも1つが、ケトン、アルデヒド、カルボン酸、エステル、アミン、アミド、エーテル、およびニトリルからなる群から選択され、
「a」および「b」が、独立して、1〜10の範囲であり、さらに、前記ライブラリーが1000個またはそれ以上の異なる化合物を含む、項目13に記載のライブラリー。
(15) R1、R2、A1、A2、A3、A4、およびA5のそれぞれが、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-
NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、項目13に記載のライブラリー。
(16) A1、A2、A3、A4、およびA5の1つまたはそれ以上が、前記一般構造の環構造に縮合した1つまたはそれ以上の付加環構造を提供するように定義される、項目13に記載のライブラリー。
【0018】
発明の要旨
本発明は、環状有機化合物のライブラリー、およびそのようなライブラリーを作製し、アッセイする方法に関する。本発明によれば、それぞれの環状有機化合物は、出発樹脂を用いて誘導体化された固体表面の形態の出発物質から構成される。化合物は、この樹脂と反応し、環状基を付加するか、またはそれを形成する。好ましくは、これらの反応は、ライブラリーのサイズを大きくするように、それぞれ異なる化合物が、複数のサブアマウント(subamount)と反応可能であるように、分割樹脂手順を用いて行われる。例えば、化合物は、固体支持体に結合された出発樹脂と反応し、アルデヒド官能基を有する化合物が得られる。ここで、出発樹脂と反応した(1つ以上の)アルデヒド化合物は、生成物の混合物が得られるように変えられるさまざまな置換基を有している。また、本発明は、合成が完了した後に化合物を支持体から切断することによって、環状有機化合物の組み合わせライブラリーを基質に結合された化合物から作製し、そのような化合物のライブラリーをアッセイする方法にも関している。
【0019】
本発明によるライブラリーは、すべて、出発樹脂を用いて誘導体化された固体支持体を用いて作製される。その後、出発樹脂は、順次共に結合される反応物に供され、結果として環状化合物を形成する。反応物の順次結合は、(1)分割樹脂方法論および(2)サブモノマー方法論を用いて行われ得、かつそうするのが好ましい。これらの方法論については、共に後で詳細に説明する。
【0020】
本発明の第1の目的は、固体基質に共有結合されているが、その固体支持体から切断され得る、基質に結合された樹脂から誘導された多数の環状有機化合物を含有する混合物(ライブラリー)を提供することにある。これらのライブラリーは、好ましくは、少なくとも1つの生物学的に活性な環状有機化合物を含む。
【0021】
重要な目的は、分割樹脂方法論をサブモノマー方法論と組み合わせて用いることにより、固体支持体上で誘導体化された出発樹脂から複雑な組み合わせライブラリーを迅速に合成し、ライブラリーの複雑さおよび多様性を顕著に増加させるように置換される環状化合物を、結果として得られるライブラリーに得るための効率のよい方法を提供することにある。
【0022】
本発明の別の目的は、少なくとも1つの生物学的に活性な環状有機化合物を含む、基質に結合された樹脂出発物質から誘導された環状有機化合物のライブラリーを得る方法を提供することにある。
【0023】
本発明は、高度に置換された環状構造を好ましくは有する、環状有機化合物のライブラリーを包含する。
【0024】
本発明の別の目的は、天然タンパク質またはその他の生物学的に活性な化合物の活性をある程度模倣する化合物を得るために、このような環状有機化合物ライブラリーをスクリーニングする方法論を提供することにある。
【0025】
本発明の別の目的は、薬学的に活性な薬剤のような生物活性化合物にさらに結合された、本発明の環状有機化合物である新規な化合物を生成することにより、本発明により合成された環状有機化合物の強化された結合親和力を介して、薬剤に対する生化学標的化を提供することにある。
【0026】
本発明の利点は、この方法論が、最も強力なレセプター結合親和力あるいはその他の最適化された標的生物活性を有する、固体支持体に結合された環状有機化合物を合成し、単離するのに用いられ得ることである。
【0027】
本発明の別の利点は、本発明による環状有機化合物およびライブラリーが、レセプターの相互作用、すなわちそのような化合物と天然レセプター部位との間の相互作用を探求するのに用いられ得ることである。
【0028】
本発明の別の目的は、基質に結合されたアルデヒド出発物質から誘導された環状有機化合物であって、同一のレセプター部位に結合する生物活性タンパク質あるいはその他の生物活性分子と同じか、またはより強力な、天然レセプター部位に対する親和力を有する有機化合物を設計する薬剤設計方法論を提供することにある。
【0029】
本発明の別の特徴は、この化学合成方法論が固相反応技術と関連させて用いられることにより、規定されたライブラリーの作製が可能になることであり、また、この固相反応技術が、実用的な量の環状有機化合物および/またはライブラリーを生成するように自動化され得ることである。
【0030】
本発明のさらに別の特徴は、本発明による基質に結合された環状有機化合物が、天然ペプチドあるいはその他の生物活性分子と比べて、それらの有する結合に対して異なる構造を有するのみならず、天然ペプチドあるいはその他の生物活性分子では可能ではないことがある、それぞれ異なる3次元構造をも有することである。
【0031】
本発明のこれらの目的、利点および特徴、ならびに、その他の目的、利点および特徴は、以下により詳しく説明する構造、合成および使用法の詳細を読めば、当業者には明らかになるであろう。なお、以下の説明では、その一部をなす添付の一般構造式および合成スキームを参照するが、これらの式およびスキームの全体を通して、同一の記号は、同一の分子部分を指す。
【発明を実施するための最良の形態】
【0032】
詳細な説明
本発明による固相、樹脂誘導環状有機化合物、ライブラリーおよび結合体、ならびに、それらを製造する方法を説明する前に、本発明は、以下で説明される特定の環式および複素環式化合物、ならびに、それらの置換基に限定されるわけではないことは、理解されたい。なぜなら、そのような化合物およびそれらの置換基、ならびに、方法は、もちろん改変可能であるからである。また、以下で用いられる用語法は、特定の実施形態のみの説明を目的とするものであり、限定を意図しているわけではないことも理解されたい。なぜなら、本発明の範囲は、添付の請求の範囲によってのみ限定されるからである。
【0033】
なお、この説明および添付の請求の範囲の全体を通して、単数形の「a」、「an」および「the」は、文脈により明らかにそうではないことが示されない限り、複数の指示物を含むことには留意されたい。よって、例えば、「a cyclic organic compound(ある環状有機化合物)」というときには、複数のそのような環状有機化合物および1分子の1つよりも多くのコピーの混合物が含意されており、「reactive starting compound(反応性出発化合物)」というときには、複数のそのような反応性出発化合物および/または複数のそのような出発化合物の多数のコピーの混合物への言及が含まれており、「the method of synthesis(合成方法)」というときには、以下の開示を読むことにより当業者が発想するであろう、複数のそのような方法が含意されている。
【0034】
本発明は、新規な環式または複素環式有機化合物および結合体、環状化合物のライブラリー、そのような環式または複素環式有機化合物、ライブラリーおよび結合体を合成するプロセス、ならびに、そのようなライブラリーから所望の生物活性を有する環状化合物を単離するプロセスを包含する、異なる様々な局面を含む。また、本発明のこれらの局面のそれぞれにおいて、本発明は、多数の具体的な実施形態を含んでいる。本発明の本質は、当業者が、本明細書中に開示され、記載される情報を用いて、天然に存在する分子または生物学的に活性な合成分子の生物活性を模倣する分子を生成し、単離することを可能とする、処理技術を提供することを伴うが、本発明のそのような化合物は、天然分子または合成分子と比べて異なる化学構造を有している。用語「模倣する」は、広い意味で用いられている。すなわち、生成された分子は、同じ活性を有していても、より大きな活性を有していても、より小さな活性を有していてもよく、および/または天然に存在する生物学的に活性な分子または生物学的に活性な合成分子の効果を阻害してもよい。
【0035】
本明細書中において言及される文献はすべて、本発明の特徴を開示し、記載することを目的として、参考として援用される。
【0036】
そうでないと規定しない限り、本明細書中において用いられるすべての技術的、科学的用語は、本発明の属する技術分野の当業者により通常理解されている意味と同じ意味をもっている。本明細書中において記載される方法および物質に類似する、または等価であるあらゆる方法および物質が、本発明の実施または試験において用いられ得るが、以下には、好ましい方法および物質を説明する。
【0037】
以下に便宜のために述べる定義をもつ多数の用語が、本明細書の全体を通して規定され、用いられる。
【0038】
定義
用語「ライブラリー」、「組み合わせライブラリー」、「樹脂誘導ライブラリー」などは、本明細書中においては、1つ以上の固相に結合された樹脂出発物質から固定支持体上に合成された環状有機化合物の混合物を意味する、相互に置き換え可能な用語として用いられている。このライブラリーは、互いに異なる環状有機分子を、10個以上(すなわち、10個の異なる分子であって、同一分子の10個のコピーではない)、好ましくは100個以上、より好ましくは1,000個以上、さらに好ましくは10,000個以上含む。これらの異なる分子(異なる基本構造および/または異なる置換基)のそれぞれは、その存在が何らかの手段により判定され得る(例えば、レセプターまたは適切なプローブを用いて、単離され得、分析され得、検出され得る)ような量だけ存在することになる。その存在を判定可能とするのに必要な異なるそれぞれの分子の実際の量は、用いられる実際の手順によって変化し、単離、検出および分析のための技術が進歩するにつれて変化し得る。これらの分子が実質的に等モル量だけ存在するとき、100ピコモル以上の量が検出され得る。好ましいライブラリーは、所望の反応生成物それぞれを実質的に等モル量だけ含んでおり、所与の分子の存在が優勢になったり、何らかのアッセイにおいて完全に抑圧されるように、所与の分子を比較的大量に含んだり、比較的少量に含んだりすることはない。
【0039】
用語「官能基」、「官能部分」などは、本明細書中では、炭素、酸素、水素および窒素からなる分子の有機基を表すために用いられる。典型的な官能基は、本明細書中において規定される「環状化合物」の成分に結合されるものであって、アルデヒド、ケトン、カルボン酸、エステル、アミド、アミン、エーテルおよびニトリルを含む。「環状化合物」に結合される好ましい官能基は、アルデヒドおよびケトンである。これらの基それぞれの一般構造は、よく知られている。しかし、この応用を目的として、これらの基は、「アルデヒド」の以下の定義により規定される。ここで、それぞれの基について適切な変更が施される。
【0040】
本明細書中において用いられている用語「アルデヒド」は、以下の一般構造式を有する化合物を意味するものとする。
【0041】
【化1−6】
【0042】
ここで、Rは、炭素原子および適切な固体基質または樹脂に共有結合可能であり、共有結合、または炭素原子および適切な固体基質または樹脂に共有結合可能な1個の原子または複数の原子の群であり得る。好ましくは、「R」は、1個から12個の炭素原子を含むアルキルまたは置換されたアルキルである。しかし、「R」は、任意の炭化水素ベース部分、または炭化水素ベースの置換部分でもあり得る。「R」は、本明細書中において規定される「ヒドロカルビル」または「側鎖」であり得る。
【0043】
用語「オリゴマー」は、例えば、本発明のプロセスにより生成されたポリマーを意味しており、任意の長さのホモポリマー、コポリマーおよびインターポリマーを含む。具体的には、オリゴマーは、単一の繰り返しモノマー、2つの交互モノマー単位、互いにランダムに、および/または慎重に間隔の設けられた2つまたはそれ以上のモノマー単位から構成され得る。このオリゴマーは、好ましくは、2〜100個のモノマーであり、より好ましくは2〜50個または2〜20個のモノマーであり、最も好ましくは2〜6個のモノマーである。
【0044】
用語「アシルサブモノマー」は、本発明において用いられるアシル化剤を指す。アシルサブモノマーは、反応性カルボニルあるいはカルボニル等価体、およびアミンによる求核置換により置換され得る脱離基を含む。「カルボニルあるいはカルボニル等価体」は、(本発明によるポリカルバメートの合成では)カルボン酸、エステル、アミド、無水物、アシルハライドおよびイソシアナートを含むが、これらに限定されるわけではない。用いられるエステルおよびアミドは、一般に「反応性」の形態(例えば、DIC付加物など)である。アシルサブモノマーは、さらに、側鎖を含み得る。適切なアシルサブモノマーは、ブロモ酢酸、3−ブロモプロピオン酸、2−ブロモプロピオン酸、2−ブロモエチルイソシアナート、2−ブロモエチルクロロホルメート、6−フェニル−3−ブロモヘキサン酸、4−ブロモメチル−安息香酸、4−ブロモメチル−2−メトキシ安息香酸、5−ブロモメチル−ピリジン−2−カルボン酸などを含むが、これらに限定されるわけではない。
【0045】
用語「アミノサブモノマー」は、アシルサブモノマーにおける脱離基の求核置換を行い得るアミノ基を含有する化合物を指す。このアミノ基は、第1級でも、第2級でも、第3級でもよい。第3級アミンの添加により、第4級アンモニウム塩が生じ、好ましくは、連鎖ターミネーターとして用いられる(すなわち、このオリゴマーのそれ以上のアシル化は不可能になる)。本発明による好ましいアミノサブモノマーは、第1級アミンおよびヒドラジドである。ただし、アミド、カルバメート、尿素、カルバジド、カルバゼート(carbazate)、セミカルバジドなどもまた適している。
【0046】
用語「サブモノマー」は、本発明のある工程において基質に結合された物質に付加される、本発明の方法において用いられる有機反応物を指す。本発明の「アシルサブモノマー」(スキームIAの第1のサブモノマー)は、任意のアミノ基(例えば、-NH2、-NRHまたは-NR2)により求核置換され得る脱離基を有するアシル化剤である。「アミノサブモノマー」(例えば、スキームIAの第2のサブモノマー)は、-NH2基を含む置換剤反応物である。
【0047】
本発明の別の局面においては、サブモノマーが、固体支持体またはアルデヒドで誘導体化された固体支持体樹脂に順次付加され、バックボーンを形成し、これは引き続いて環化される。環化のためのペプトイドバックボーンの調製時に、サブモノマーを段階的に付加することにより、側鎖および環置換基を最終生成物へと導入する。
【0048】
サブモノマー合成の詳細は、1994年7月18日に出願された米国特許出願第08/126,539号およびR. Zuckermannら、J. Am. Chem. Soc. (1992) 114:10646-7に記載されており、これらは本明細書中で参考として援用される。
【0049】
用語「側鎖」は、炭素原子または窒素原子を介して有機化合物に結合された基を指す。この結合は、環状基の環構造上にあってもよいし、窒素原子または炭素原子のいずれかにおける化合物のポリアミドバックボーン上にあってもよい。側鎖は、H、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb-、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdであり得る。ここで、Ra、Rb、RcおよびRdはそれぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、
また、Ra、Rb、RcおよびRdはそれぞれ、0〜6個のハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル(alkylsufinyl)、あるいは低級アルキルスルホニルで置換される。ここで、「低級」とは、1〜6個の炭素原子を示す。
【0050】
用語「炭化水素ベースの」、「炭化水素ベースの置換基」などは、分子の残りの部分に直接結合された炭素を有し、本発明の文脈では炭化水素ベースの特性が優勢である部分を表す。
【0051】
置換基は、例えば、以下のものを含む。
【0052】
(1)炭化水素置換基、すなわち、脂肪族(例えば、アルキルまたはアルケニル)置換基、脂環族(例えば、シクロアルキルまたはシクロアルケニル)置換基、芳香族、脂肪族および脂環族置換芳香族核など、ならびに、環状置換基である。ここで、環は、分子の別の部分を介して完成される(すなわち、例えば、任意の2つの指示された置換基が、一緒になって1個の脂環基を形成する)。
【0053】
(2)置換された炭化水素置換基、すなわち、本発明の文脈では、優勢な炭化水素置換基を改変しない非炭化水素基を含む置換基である。当業者は、そのような基(例えば、ハロ(特にクロロおよびフルオロ)、アルコキシ、メルカプト、アルキルメルカプト、ニトロ、ニトロソ、スルホキシなど)を認識するであろう。
【0054】
(3)ヘテロ置換基、すなわち、本発明の文脈での優勢なヒドロカルビル特性を有しながら、他の部分は炭素原子で構成されている環または鎖に存在する炭素以外の原子を含む置換基。適切なヘテロ原子は、当業者には自明であろうが、例えば、硫黄、酸素、窒素などがあり、例えばピリジル、フラニル、チオフェニル、イミダゾリルのような置換基が、これらのヘテロ置換基の例である。ヘテロ原子および好ましくはただ1個の原子が、炭化水素ベースの置換基内のそれぞれの炭素原子について存在する。炭化水素ベースの置換基には、そのような基またはヘテロ原子が存在しないこともあるので、炭化水素ベースの置換基は、純粋な炭化水素であることもある。
【0055】
「炭化水素」は、化合物を述べているのに対して、「ヒドロカルビル」および「ヒドロカルビレン」は、それぞれ1つまたは2つの水素が除去された基を述べている。それぞれは、大まかにいうと、水素原子および炭素原子のみから構成されるが、ヘテロ原子を含んでいてもよいし、飽和であっても、不飽和であってもよく、脂肪族でも、脂環族でも、芳香族でもよい。環が含まれるとき、構造は、通常、1個、2個、3個またはそれ以上の環を含む。これらの環は、縮合されても、架橋されても、スピロ縮合されてもよい。
【0056】
用語「環状化合物」および「環状部分」は、飽和であっても、不飽和であってもよい1つ以上の閉環を特徴とする構造を有する、上記で規定した炭化水素ベースの有機化合物または部分を述べるのに用いられる。環状化合物または部分は、存在している環の個数によって、単環式でも、二環式でも、三環式でも、多環式でもよい。これらの用語は、環状化合物の主要な3つの群を包含する。すなわち、(1)脂環族、(2)芳香族(「アセン(asene)」とも呼ばれる)、および(3)複素環族の3つである。
【0057】
用語「分子部分」は、オリゴマー主鎖の窒素原子または炭素原子に結合可能であり、それにより、例えば、CH3(R1)NC(O)CH(R2)CH3(ここで、R1は、オリゴマー主鎖の窒素原子に結合可能な分子部分であり、それにより窒素原子に結合される側鎖を形成し、R2は、オリゴマー主鎖の炭素原子に結合可能な分子部分であり、それにより炭素原子に結合される側鎖を形成する)であるオリゴマーのような、形成される任意の化合物の主鎖から分離して側鎖を形成する、1個の原子または原子群を包含している。よって、ポリペプチドまたはポリアミド合成の当業者には、水素部分および、例えば、アルキル、アリール、およびアリールアルキル部分のようなヒドロカルビル部分を含むが、それらには限定されない広範で多種多様な分子部分が用いられ得ることは、容易に明らかになるであろう。
【0058】
「有機化合物」は、炭素、水素、窒素、酸素、硫黄、およびリン原子からなる分子を意味する。本明細書中において用いられる有機化合物は、そのすべてが炭素および水素から構成される環式または非環式化合物でもよいし、1つ以上のヘテロ原子(酸素、窒素、硫黄、およびリン原子を含む)を含んでいてもよい。
【0059】
「環状有機化合物」は、ペプトイドバックボーンの環化から誘導された少なくとも1つの環状構造を含む有機化合物を意味する。この環状構造は、炭素および水素から構成される炭化水素であってもよいし、脂肪族または芳香族であってもよい。この環状構造は、環状バックボーンに少なくとも1つのヘテロ原子を有する複素環であってもよい。この複素環式構造は、飽和であっても、不飽和であってもよい。環状構造は、縮合されてもよいし、環状化合物内で分離されてもよい。
【0060】
置換基は、第1分子の一部である原子または基のことをいう。なぜなら、それは第1分子の別の原子または基を置換するからである。分子が置換される場合、分子は、1つ以上の置換基を有する分子の誘導体である。本発明によるサブモノマーのいずれかにおいて有用な置換基は、ハロ、アルキル、アルコキシ、アルキルチオ、ハロアルキル、ハロアルコキシ、ハロチオ、二置換アミノなどを含んでおり、窒素または炭素に結合された水素のような原子を置換する。置換可能な位置は、第1分子の置換された原子または基の結合部位である。
【0061】
「プリンまたはピリミジン塩基」は、A、T、G、CまたはUのような天然ヌクレオシド塩基と、1以上のアルキル、カルボキシアルキル、アミノ、ヒドロキシル、ハロゲン(すなわち、フルオロ、クロロ、ブロモ、またはヨード)、チオール、またはアルキルチオール(ここで、アルキル基は、1個から6個程度の炭素原子を含む)により置換されたこれらのプリンおよびピリミジンを含むそれらの誘導体と、を含んでいる。プリンおよびピリミジンとしては、例えば、2、6−ジアミノプリン、5−フルオロウラシル、キサンチン、ヒポキサチン、8−ブロモグアニン、8−クロログアニン、8−アミノグアニン、8−ヒドロキシグアニン、8−メチルグアニン、8−チオグアニン、2−アミノプリン、5−エチルシトシン、5−メチルシトシン(5-methylcyosine)、5−ブロモウラシル、5−エチルウラシル、5−ヨードウラシル、5−プロピルウラシル、2−メチルアデニン、メチルチオアデニン、N、N−ジメチルアデニン、8−ブロモアデニン、8−ヒドロキシアデニン、6−ヒドロキシアミノプリン、6−チオプリン、4−(6−アミノヘキシル/シトシン)などがあるが、これらに限定されるわけではない。
【0062】
「脱離基」は、例えば、NH2のようなアミンにより求核置換され得る部分を意味する。求核置換により容易に除去される限り、本明細書中では、どのような脱離基が用いられてもよい。本発明において有用な脱離基としては、例えば、ブロモ、クロロ、ヨードなどのハロ、O−トシル、O−トリフリル、O−メシルなどがあるが、これらに限定されるわけではない。
【0063】
用語「基質」、「固体支持体」などは、本明細書中においては、出発樹脂材料(反応性基)が(室温で)結合され得る任意の固体材料を規定するものとして、相互に置き換え可能に用いられている。好ましい固体支持体材料は、ポリエチレン化合物およびポリスチレン化合物のようなポリマー化合物、および関連する不活性ポリマー化合物を含んでいる。よって、本明細書中では、それらのすべてを文字「P」で表す。基質は、シート、円筒形容器の内側、またはピンを含むどのような形状であってもよいが、好ましくは、直径1.0 cm未満の球状ビーズの形状であり、より好ましくは、直径1.0 mm未満である。「基質」または固体支持体は、ペプチド合成で用いられる従来の固体支持体材料である。このような基質または支持体としては、例えば、多種多様な支持体樹脂、ならびに光切断性DKP形成リンカー(DKPとはジケトピペラジンである。例えば、本明細書中でも参考として援用されるWO90/09395を参照のこと)、TFA切断性(cleavable)、HF切断性、フッ化物イオン切断性、還元的切断性および塩基活性(base-labile)リンカーのような支持体樹脂へのコネクタがあるが、これらに限定されるわけではない。固体支持体樹脂は、別々の反応用にいくつかの部分に分割され得、所望であれば再び組み合わされ得る、複数の固体支持体粒子を含んでいる。よって、用語「樹脂」が固体支持体(例えば、固体支持体樹脂)と共に用いられるとき、この用語は、-NH2基のような反応性基、またはヒドロキシル基のようなその他の電子供与基を用いて誘導体化されたポリマー材料を記載する。
【0064】
「保護基」は、その基が結合される原子(通常は、酸素または窒素)が、望ましくない反応または結合(通常は、合成反応)に関与するのを妨げ得る、任意の基を意味している。また、保護基は、カルボン酸、チオールなどの反応または結合を妨げることも知られている。このような基、ならびに、それらの調製および導入は、当該分野において従来的であり、塩、エステルなどを含む。
【0065】
「電子供与基(EDG)」は、反応物のその他の部分において電子密度を増加させ得る、反応物に共有結合された部分を意味する。本発明において有用な電子供与基としては、例えば、アルキル、アミン、ヒドロキシル、アルコキシなどがあるが、これらに限定されるわけではない。
【0066】
「電子吸引基(EWG)」とは、反応物のポリアミドバックボーン部分の求核付加を活性化し得る、反応物に共有結合された部分を意味する。本発明において有用な脱離基としては、例えば、ニトロ、カルボニル、シアノ、スルホンなどがあるが、これらに限定されるわけではない。
【0067】
「反応を完了させる」または「実質的に完了」とは、十分な反応物が(例えば、時間および温度などの十分な条件の下に)添加され、固体支持体に結合された中間化合物のすべて、または実質的にすべてが、反応物により誘導体化され得ることを意味する。「反応を実質的に完了させる」とは、反応物の濃度、触媒、温度およびその他の条件が適切である条件の下に反応を行うことにより、固体支持体に結合された中間化合物(例えば、樹脂)の80%よりも多く、好ましくは90%よりも多く、より好ましくは95%よりも多く、さらに好ましくは99%以上を、添加された反応物と反応させ、それにより検出可能なすべての中間樹脂が反応するようにすることを意味する。
【0068】
「回収可能な量」とは、回収可能な量が、分離時において、当該分野で利用可能な技術により混合物のその他の成分から分離可能であるような濃度で化合物が存在する、混合物内の化合物の量を意味する。混合物中の成分がおよそ等モル量で存在している時、好ましくは、少なくとも50 pmol、より好ましくは100 pmolの化合物が混合物中に存在している。
【0069】
「分析可能な量」とは、混合物中で検出され得、同定され得る、混合物中に存在する化合物の量を意味する。混合物中の成分がおよそ等モル量で存在している時、好ましくは、少なくとも約10 pmol、より好ましくは50 pmolの化合物が混合物中に存在している。
【0070】
用語「プール」および「プールされた量」とは、誘導体化された、または誘導体化されていない固体支持体粒子を組み合わせて、混合物を形成することを意味する。プールされた材料は、ライブラリーの調製時の中間生成物または最終生成物を含む。
用語「サブアマウント」とは、プールから分割されたプールされた量の一部を意味する。それぞれのサブアマウントは、好ましくは、その他すべてのサブアマウントとサイズが同じである。サブアマウントは、図1に示されており、本明細書中で説明されている分割樹脂方法と関連させて用いられる。プールされた量は、図1の量9、31および40を含んでおり、サブアマウントは、2〜8、11〜24などを含んでいる。
【0071】
一般的方法論
この方法論は、電子供与化合物を固相支持体上へと誘導体化することにより始まる。一般に、固体支持体は、結合された、市販の樹脂(例えば、結合されたRinkまたはMerrifield樹脂)を有している。Pが支持体を表す場合、樹脂がその上にある支持体は、P-XH(ここで、Xは、OまたはNHである)で表される。ここでAは、例えば、出発樹脂上に誘導体化されたアルデヒド基のような任意の官能基であり、P-XH-Aにより表される。アルデヒドまたはケトンのような官能基は、官能部分と反応させることにより環化され、それにより、化合物の多様なライブラリーを提供する置換された環および置換されていない環からなる環状化合物を形成する。例えば、芳香族アルデヒドAは、P-NH-Aについては、RINKアミドを介して支持体に結合される。このアルデヒドは、サルコシンのようなアミドおよびジメチルムコネートのようなエステルと同時に反応する。これらの化合物は、このアルデヒド基と反応し、環化する。得られる生成物上の置換基は、反応物上の置換基が異なる反応物の混合物を提供することにより変えることができる。例えば、サルコシン上のメチル基およびジメチルムコネート上の2つのメチル基は、独立して他のアルキル部分に変えることができる。
これらの化合物が反応する順番、その構造は、反応条件とならんで、生成物の構造を決定する。しかし、上述した合成スキームは、小さく、置換された分子から比較的複雑な分子を順番どおりの段階的手順で合成するために、いくつか共通の特徴を有していることが容易に分かり得る。それぞれの場合において、直線状の置換されたバックボーンが形成された後、環化反応により、高度に置換された環状生成物を生成する。
本明細書中において記載されている合成方法の共通の特徴について、まず説明する。
この方法論は、まず、複数の(例えば、3つ以上、10以上、1,000以上、10,000以上など)固体支持体表面ポリマービーズを提供することにより始まる。多数の支持体表面の使用は、特にサブモノマー方法論と組み合わされた時に本発明の効力を大幅に増大させる、本明細書中で説明されている「分割樹脂」方法論の適用のために重要である。これらの支持体表面は、それらを電子供与基と反応させることにより誘導体化される。この基または樹脂(例えば、-NH2)は、分子の残りの部分を構築する出発基として作用する。この出発樹脂は、多種多様な異なるタイプの反応に供され得、それにより、最終的には、例えば、アルデヒドまたはケトンのような官能基で置換される環状化合物の形成へと導かれる。例えば、出発樹脂は、本明細書中で規定されているサブモノマー反応物と反応させられ得る。その後、サブモノマー単位を以前の基のそれぞれと順次反応させることができ、それにより、任意の所望の長さをもつ連鎖を構築する。例えば、「P」が固体表面であり、「EDG」が電子供与基であり、「SM」が本明細書中で規定されているサブモノマーである場合、反応は、以下のように進行し得る。
1)P-EDG
2)P-EDG-SM
3)P-EDG-SM-SM
出発樹脂をサブモノマーと反応させる代わりに、出発樹脂は、環状化合物の電子吸引基と反応させてもよい。このような環状化合物が「CC」で表される場合、反応は、以下のようにも進行し得る。
1)P-EDG
2)P-EDG-CC
出発樹脂(EDG)を環状化合物と反応させる代わりに、出発樹脂は、環を形成する化合物、すなわち1つ以上の追加化合物と反応して環を形成する化合物と反応させてもよい。「RFC」が環を形成する化合物である場合、反応は、以下のようにも進行し得る。
1)P-EDG
2)P-EDG-RFC+RFC
3)P-EDG-環状化合物
上記一般反応のすべてにおいて、第1の反応物は、固体支持体上の反応性部位の実質的にすべてが、共有結合された反応物により占められるように、調製された(すなわち、XH基がその上にある)固体支持体と反応させられる。第1の反応物は、一般に、支持体に結合されたNH2基と反応させられる。(その後、出発樹脂として作用する)第1の反応物と反応可能な化合物は、その後、第1の反応物上の反応性基と反応させられる。
【0072】
反応した固体支持体を分割し(サブアマウントを生成し)、再び組み合わせる方法は、高度に置換された環状構造の混合物生成を可能にする、環状ペプトイド合成のサブモノマー方法の特徴である。生成物の混合物は、本発明の2つの特徴から結果として得られる。すなわち、1)サブモノマー化合物上の可変置換基の組み合わせおよびそれらの相対的位置、ならびに、2)選択されたサブモノマー添加時に、反応した固体支持体粒子を分割し、再び組み合わせることにより、環化前に前駆体直線形ペプトイドの混合物を生成することである。混合物中のそれぞれ異なる生成物化合物の個数は、1)固体樹脂に結合された、それぞれ異なる第1の反応物化合物の個数、2)第1の反応物化合物と反応した、それぞれ異なる第2の化合物の個数、ならびに、3)第1および第2の化合物それぞれの上の可変置換基の個数と共に増加する。
【0073】
本発明の1つの実施形態では、サブモノマー成分は、支持体上に誘導体化された-NH2樹脂に結合される。このサブモノマー成分は、以下の一般構造式を有する。
【0074】
【化2】
【0075】
ここで、Zはハロゲン(好ましくは、Br)であり、Rは炭素原子に結合した任意の部分であり、かつHまたはヒドロカルビル部分(例えば、-CH3)であり得る。用語「サブモノマー」は、上記で規定した通りであり、具体例も与えられている。
【0076】
このサブモノマーがP-NH2と反応すると、結果は以下のようになる。
【0077】
【化3】
【0078】
この時点において、官能部分をその上に有する環状有機化合物は、以下のようにハロゲン原子と反応する。
【0079】
【化4】
【0080】
ここで、RおよびZは上記で規定したとおりであり、Aは官能基を有する任意の部分であり、YはS、OまたはNHである。
【0081】
反応は、適切な溶媒中、適切な時間および温度条件下にて行われ、
【0082】
【化5】
【0083】
を生じる。
【0084】
大型のライブラリーを作製するためには、それぞれ異なる「複数のR」をその上に有する、異なる複数のサブモノマー基の混合物が、出発樹脂と反応させられ、その後、異なる複数の環状化合物の混合物が、ハロゲンと反応させられる。ここで、この混合物は、例えば、官能基、上記で規定した「側鎖」、または上記で規定した「ヒドロカルビル」のような置換基で置換された環状化合物を含んでいる。規定されたライブラリーは、本明細書中で説明されている「分割樹脂」方法論を用いることによって、作製され得る。
【0085】
本発明によれば、さまざまな方法論が、環状有機化合物のライブラリーの作製に適用される。具体的には、この方法は、同一の反応容器内、単一の固相支持体上で、複数の特異で、独特で、異なる環状有機化合物の混合物を調製することを伴う。すなわち、この反応容器内の環状有機生成物化合物は、互いに異なるものであり、この反応容器内の環状有機生成物化合物は、それぞれ、回収可能で、分析可能な量で存在している。サブモノマー化合物を相対量で組み合わせ、それぞれの反応が実質的に完了するように、分割樹脂方法を適用することによって、得られる環状有機化合物の混合物は、それぞれの反応生成物を予測可能で規定された量で、かつ環状有機化合物が回収し、分析し得るに十分な量で含む。これらの環状有機化合物それぞれの結果として得られる量は、予測可能である。なぜなら、それぞれの反応で用いられる、誘導体化された固体支持体の量が制御され、後続するそれぞれの反応が完了するからである。
【0086】
本発明の一般的局面によれば、反応性部分(例えば、反応性アルデヒド部分)が環状化合物またはサブモノマー化合物と反応させるのに利用可能であり、その化合物が、今度は、1つ以上の後くサブモノマーと反応した後、環化されるように、前駆体アルデヒド化合物のような前駆体化合物を固体支持体上で固定化した後、個々の環状有機化合物は、固相合成技術のような方法論を用いて生成される。環化された化合物(例えば、アルデヒド誘導体)は、固体支持体に結合されたままであり、そしてアッセイされ得るか、またはその支持体から切断され、そしてアッセイで用いられ得る。環化された化合物(例えば、アルデヒド誘導体)の切断は、必要に応じて、回収前または使用前に行われ得る。
【0087】
サブモノマー方法により調製される環化された多種多様な化合物は、サブモノマー反応の順序によって部分的に制御されるので、これらの部分が環化前に組み合わされる時、直線形アルデヒド誘導体の混合物が形成されるように、粒子状固体支持体を分割し、そして後続するサブモノマー反応のそれぞれと再び組み合わせ得ることは、容易に分かる。
【0088】
分割樹脂方法
分割樹脂方法は、合成効率を改善し、結果として得られるライブラリー反応生成物のサイズを顕著に増大させることができるように、本発明に適用され得る。分割樹脂方法は、支持体(例えば、ビーズ)上へと誘導体化された同じ出発樹脂のセット、またはそれぞれ異なる出発樹脂の混合物をそれぞれ等しいプールへと分割することを含む。これらのプールはそれぞれ、反応物に結合される。好ましくは、異なる複数の反応物がそれぞれのプールに用いられ、それぞれの反応が実質的に完了する。それぞれの反応が完了した後、これらのプールが単一のプールに再び組み合わされ、所望のライブラリーが提供される。このプロセスは、支持体上で合成されている分子のサイズを大きくし、結果として得られるライブラリー中のそれぞれ異なる分子の個数を増やすために、任意の回数だけ繰り返され得る。この分割樹脂方法論は、1993年1月26日に発行された米国特許第5,182,366号におけるペプチドの大型組み合わせライブラリーの合成に最初に適用された。この特許は、本明細書中でも、分割樹脂方法の基本的局面を開示し、説明するために参考として援用している。
【0089】
図1は、分割樹脂方法論がどのようにして行われるかを示す模式図である。工程1において、ビーズ2〜8のサブアマウントは、樹脂により占められるそれらの上の反応性部位(例えば、NH)のすべてが、反応物(例えば、本明細書中で説明されているサブモノマー)と反応するように、反応させられる。サブアマウント2〜8それぞれの反応は、完了する。反応したサブアマウントは、その後、工程9において再び組み合わされる。工程10において、9からの量が、サブアマウント11〜30に分割され、それぞれのサブアマウントが異なる反応物と反応させられて、反応はそれぞれ完了する。これらのサブアマウント11〜30は、その後、工程31において再び組み合わされ、工程32において、サブアマウント33〜39に分割される。これらのサブアマウントは工程40で組み合わされ、工程41で再びサブアマウントに分割され得る。
【0090】
これらの反応・組み合わせ・分割・反応工程は、ライブラリーのサイズおよび多様性を増すために、所望の回数だけ繰り返され得る。また、図1に示されているように、組み合わされたサブアマウントは、次の工程で所望の個数のサブアマウントに分割され得る。それぞれの反応が常に完了するので、何らかの反応生成物がその他の反応生成物を支配するということがなく、それぞれの反応生成物を等モル量だけ有する混合物を容易に得ることができる。
【0091】
この分割樹脂方法は、上述した一般的方法論に適用され得る。具体的には、P-NH2は、複数の(3以上、好ましくは5以上、より好ましくは10以上の)サブアマウントに分割される。P-NH2のそれぞれのサブアマウントは、以下の構造を有する異なるサブモノマーと反応させられる。
【0092】
【化6】
【0093】
ここで、「R」部分は可変であり、それぞれの反応は完了する。「R」は、上記で規定した「R」を含む、炭素原子に結合可能な任意の部分であり得るが、好ましくは、「R」は、H、-CH3、-CH2CH3、-CH(CH3)2、-CH2-CH2-CH3、=CH2およびフェニルからなる群から選択される。示されている構造は、単一の「R」を有している。しかし、同一の炭素原子に2つの「R」基が結合されてもよく、これらの「R」基は互いに異なっていてもよい。
【0094】
それぞれのサブアマウントで得られる反応生成物が、その後、再び組み合わされて、新しいプールを形成する。再び組み合わされた混合物すなわちプールは、その後、複数のサブアマウントに分割される。その後、それぞれのサブアマウントは、例えば、置換された環状ケトンまたはアルデヒドのような異なる置換された環状化合物と反応させられ、それぞれの反応は完了する。それぞれのサブアマウントで得られた反応生成物が、その後、再び組み合わされ、固体支持体上の環状化合物の所望のライブラリーである最終プールを提供する。レセプターは、このライブラリーに対して試験され得、または支持体から切断された後、固体支持体上のレセプターに対して試験され得る。
【0095】
サブモノマー方法
サブモノマー方法論は、アミノ酸またはアミノ酸様モノマー単位を鎖に付加するのに適用可能である。この方法は、上述した分割樹脂方法論と組み合わせて用いられる時、特に有用で効率的である。基本的方法では、それぞれのモノマーは、本明細書中では「サブモノマー」と称されている2つの反応物から、固体基質(支持体)上で直接合成される。以下の説明は、アミノ酸様化合物(ペプトイドとしても知られている)の合成について具体的に述べているが、環状化合物を形成する際に用いられる環化反応にも適用され得る。
【0096】
それぞれのモノマーは、2つの工程を有する合成サイクルにより生成される。第1の工程は、アミン(例えば、-NH2)により求核置換され得る脱離基を有する第1のサブモノマーアシル化剤(例えば、ハロ酢酸)を用いて行われる、基質に結合されたアミンのアシル化である。このモノマー合成サイクルの第2の工程は、第1級アミンのようなアミン(例えば、-NH2基)を含む第2のサブモノマー置換剤を十分な量(通常は過剰に)だけ供給することによって、例えば、ハロゲンまたはトシルのような脱離基の求核置換により側鎖を導入することを含む。この2工程プロセスは、反応スキームI. A.内に示されている。
【0097】
しかし、反応スキームI. A.は、反応スキームI. B.内に示されているように、逆の順番で行われてもよいことには、留意されたい。具体的には、反応スキームI. A.による「基質に結合されたアミン」を用いるのではなく、基質に結合されたアシル化剤サブモノマーを用いて反応を開始することが可能である。したがって、カルボン酸基は、基質の表面から延び、第1の工程においてアミンと反応する。この時点において、アミン基は、基質から外に向かって延び、上述した反応スキームI. A.の第1の工程によるサブモノマーアシル化剤を用いて、アシル化に供される。
【0098】
反応スキームI(AまたはB)の基本的な2工程プロセスは、モノマー単位を生成し、下記Iの構造をもつモノマーにより所望の長さを有するポリマー(下記の式Vによる)を生成するために繰り返され得る。これらの構造内に示されている変数は、所望の結果を得るために変えることができる。また、基本的サブモノマー構造は、構造II、IIIおよびIVにおいて異なるモノマー/ポリマー構造を得るために、以下のように変えられてもよい。
【0099】
スキームI.A
2つのサブモノマーからN-置換されたオリゴマーの固相アセンブリ
【0100】
【化7】
【0101】
スキームI.B
2つのサブモノマーからN-置換されたオリゴマーの固相アセンブリ
【0102】
【化8】
【0103】
上記のそれぞれにおいて、「P」は固相表面、R1およびR3はそれぞれ、独立的に、炭素原子に結合したあらゆる分子部分であり、R2およびR4はそれぞれ、独立的に窒素原子に結合したあらゆる分子部分であり、nは1〜10(好ましくは、1または2)の整数である。R1、R2、R3およびR4のいずれも、20の天然アミノ酸に結合した20の異なる側鎖部分(すなわち、グリシンの-H、アラニンの-CH3、バリンの-CH(CH3)2、ロイシンの-CH2CH(CH3)2、イソロイシンの-CH(CH3)CH2CH3、セリンの-CH2OH、トレオニンの-CHOHCH3、システインの-CH2SH、メチオニンの-CH2CH2SCH3、フェニルアラニンの-CH2-(フェニル)、チロシンの-CH2-(フェニル)-OH、トリプトファンの-CH2-(インドール基)、アスパラギン酸の-CH2COO−、アスパラギンの-CH2C(O)(NH2)、グルタミン酸の-CH2CH2COO-、グルタミンの-CH2CH2C(O)NH2、アルギニンの-CH2CH2CH2-N-(H)-C(NH2)+-NH2、ヒスチジンの-CH2-(イミダゾール)+基、およびリジンの-CH2(CH2)3NH3+)を含み得る。
【0104】
反応スキームI(AおよびB)には、本発明に関連して使用される試薬を指す省略形がいくつか含まれる。例えば、DMSOはジメチルスルホキシドを指し、DICはN,N-ジイソプロピルカルボジイミドを指し、DMFはN,N-ジメチルホルムアミドを指す。
【0105】
2工程法の各工程は、通常、20℃のおよそ常温にて、1気圧の圧力下で行われる。しかし、反応は、約5℃から約80℃の広い温度範囲において行われ得、使用される溶媒に依存して変わる。温度に依存して、2工程反応スキームIの時間は、約5分から約24時間の範囲内で変えられ得る。上記温度、時間、および試薬は、反応を常圧において行う際に適用される。他の圧力も採用され得る。
【0106】
サブモノマーが液体である場合、各工程は溶媒なしで行われ得る。しかし、不活性溶媒は、サブモノマーが固体である場合、または反応を容易にする場合に使用される。適切な不活性溶媒には、ジオキサンなどのエーテル、ジメチルホルムアミドなどの遮蔽アミド(blocked amides)、ジメチルスルホキシドなどのスルホキシドなどが含まれる。
【0107】
反応物の比率は変えられ得る。しかし、最高収率のためには、基質結合材料の量の約1.01から10倍、好ましくは基質結合材料の量の約1.5から5倍だけサブモノマーを多くすることが望ましい。
【0108】
スキームIに示す2工程サイクルにおいて、基質に結合した第2級アミンは、好ましくは、第1級アミンから調製されたアミンであり、基質支持体ベース面(substrate support base surface)、または固相(文字「P」で表される)に結合する(従来の方法を用いて)。
【0109】
サイクルの第1の工程は、アシル化アミンを得るための、アミン(例えば、-NH2)による求核置換が可能な脱離基を含むアシル化剤(例えば、ハロ酢酸、および特にスキームIに代表的に示されるブロモ酢酸)を含む第1のサブモノマーと、基質結合第2級アミンとを反応させることによって行われるアシル化である。
【0110】
本発明の2工程モノマー合成法の第2の工程では、モノマーユニットのバックボーン窒素、および側鎖またはR2基を付加する。第2の工程において、アシル化したアミンを、オリゴマーのモノマー位に付加されるR2基(すなわち、側鎖基)を含む第1級アミンまたは第2級アミン、アルコキシアミン、セミカルバジド、カルバゼート、またはアシルヒドラジドなどの-NH2基を含む十分な量の第2のサブモノマーと反応させる。第2のサブモノマーの反応は、脱離基の求核置換を生じる第2のサブモノマーを十分な量、通常、過剰量を添加することによって達成され、これはスキームIに臭素として代表的に示す。
【0111】
分割樹脂/サブモノマー法の適用
本発明の主要な目的は、固体支持体上に、環状有機化合物の非常に大きいライブラリ(例えば、10から10,000以上の化合物を含むライブラリ)を迅速かつ効率的に得ることである。この目的は、分割樹脂法を、それのみ、またはサブモノマー法とともに使用することによって最も容易に得られる。この方法は、まず、概して小さい重合体ビーズの形態を有する複数の固体支持体表面を得ることから始める。重合体ビーズは、文字「P」によって表され、XHで表される(ただし、Xは通常NHまたはO)電子供与基である出発樹脂の付加によって誘導体化される。電子供与基は、XH基などの電子供与基をその上に含む大きい分子であり得る。
【0112】
誘導体化されたビーズは、次いで、2以上のサブアマウントに分けられる。サブアマウントの数は、千単位であり得るが、より典型的にはおおよそ10から30である。
【0113】
サブアマウントをそれぞれ、以下の式のサブモノマーであり得る第1の反応物と反応させる:
【0114】
【化9】
【0115】
ただし、R1およびR2はそれぞれ、独立的に、炭素原子に共有結合可能なあらゆる部分であり、Zは、好ましくはBrであるハロゲン原子である。
【0116】
また代わりに、各サブアマウントの樹脂を、以下の一般構造を有する環状化合物反応物と反応させる:
【0117】
【化10】
【0118】
ただし、YはO、S、またはNHであり、A1、A2、A3、A4、およびA5のそれぞれは、独立的に、H、またはヒドロカルボニル、またはアルデヒド、カルボン酸、エステル、アミド、アミン、ニトリル、またはケトンなどの官能基部分である。好ましいライブラリには、1つ以上の官能基部分、最も好ましくはアルデヒドまたはケトンの少なくとも1つを環状構造上に有するいくらかの化合物が含まれる。
【0119】
所望であれば、サブモノマー反応物および/または環状化合物反応物を、連続的に互いに付加して、あらゆる所望の長さの鎖を作成する。しかし、通常、そのような反応化合物の1つのみを付加し、次の再結合またはプール工程までに、反応を終了させる。
【0120】
図1を参照すると、第1の工程は、複数の固体支持体ビーズ2-8を得る工程を含み得る。ビーズ2-8のプール1を、樹脂-NH2で誘導体化する。それぞれの誘導体化したビーズ2-8は、上で示し定義したように、異なるサブモノマーと個々に反応させる。それぞれの反応を終了し、以下のものを得る:
【0121】
【化11】
【0122】
ただし、Xは好ましくはNH、Zは好ましくはBr、R2およびR1は好ましくはそれぞれ独立的に、1から6の炭素原子を含むアルキルである。
【0123】
あらゆる所望の長さの鎖を提供するために、追加のサブモノマー反応物を連続的に付加し得るが、本発明の単純なバージョンにおいては、各サブアマウントとの一度の完全な反応の後に得られる各反応生成物をプールして、図1のプール9を得る。プール9を、混合し、次いで、サブアマウント11から24に分ける。
【0124】
より単純でないバージョンにおいては、各サブアマウントを、次いで、他の(かつ異なる)サブモノマー反応物と反応させ、それぞれの反応を終了させる。「アシルサブモノマー」を「アミノサブモノマー」と反応させて、アミノ酸様モノマーユニットを得る。アミノ酸様モノマーユニットのペプチド様鎖を作成するために、ビーズを所望の回数だけ、分割、反応、およびプールし得る。
【0125】
この概略化された例において、第2およびそれに続くサブモノマーとの反応は図示しておらず、サブモノマーユニットの第1の反応で得られた反応生成物をプールし、サブアマウントに分け、以下の一般構造式を有する環状化合物と反応させる:
【0126】
【化12】
【0127】
ただし、少なくとも1つの環状反応物が官能基として、A1-A5(好ましくはアルデヒドまたはケトンである)を含むという条件の下で、Yは-O、-SO、または-NHであり、A1、A2、A3、A4、およびA5のそれぞれは、独立的に、炭素原子に結合可能なあらゆる部分である。各環状化合物の各サブアマウントとの反応は終了され、以下の反応生成物を得る:
【0128】
【化13】
【0129】
各サブアマウントの反応生成物を、次いで、プールし、複数の固体支持体表面(すなわちビーズ)に結合した環状化合物のライブラリを得る。化合物を、固体支持体から開裂し得る。
【0130】
末端基は、上に示すように置換された環状化合物であり得る。しかし、ライブラリの多様性を広くするために、置換された環状部分の官能基をさらに反応させ得る。末端基がアルデヒド部分で置換された環状化合物である場合に、このことがいかにして特定の例において行われ得るかを以下で説明する。
【0131】
アルデヒドとの反応
上記説明に従って合成される本発明のライブラリは、最後に付加する反応物上にアルデヒド官能基を含む。1または全ての公知のタイプの反応を通して、アルデヒド基を反応させることによって、ライブラリの多様性を広くし得る。
【0132】
第1に、芳香族アルデヒドなどのアルデヒドを、以下のようにR-NH2と反応させ、イミン形成を得ることができる:
【0133】
【化14】
【0134】
ただし、Arはあらゆる芳香族部分、ならびにPおよびRは上記定義した通りである。
【0135】
第2に、芳香族アルデヒドなどのアルデヒドを、クネーフェナーゲルタイプの反応に供し、以下のような反応生成物を得ることができる:
【0136】
【化15】
【0137】
ただし、EWG1およびEWG2はそれぞれ、独立的に、電子吸引基であり、PおよびArは上記定義した通りである。好ましい電子吸引基には、COR、CN、CO2R、SO2R、SOR、およびSRが含まれる。
【0138】
第3に、芳香族アルデヒドなどのアルデヒドを、ウィッティヒタイプの反応に供し、以下のような反応生成物を得ることができる:
【0139】
【化16】
【0140】
ただし、P、Ar、RおよびEWGは上記定義した通りである。
【0141】
上記構造において、芳香族環は、固体支持体表面に直接結合して示している。これは、芳香族環が適切な官能基(通常、芳香族環上のNH2基)を介して支持体表面に結合、または支持体の上で誘導体化されなければならないところ、本箇所および本明細書のその他の箇所において簡潔化の目的で行っている。NH2基は、しばしば、樹脂として言及される。従って、上記構造は、以下のようにも示し得る:
【0142】
【化17】
【0143】
環状構造を形成するためのアルデヒド
その上にアルデヒド基を有する化合物を固体支持体上で合成した(上記「一般法」セクションに従って)後、アルデヒド基を種々の異なる化合物と反応させて、4、5または6員環構造を形成し得る。4員環を形成する例は以下の通りである:
【0144】
【化18】
【0145】
ArおよびPは上記定義され、R1およびR2は、独立的に、上記定義したようにあらゆるRである。
【0146】
5員環を形成する例は、以下の通りである:
【0147】
【化19】
【0148】
6員環を形成する例は、以下の通りである:
【0149】
【化20】
【0150】
ただし、R、R1、R2、R3、およびR4は、独立的に、上記Rと定義される。
【0151】
サブモノマーユニットを、互いと連続的に反応させて、ペプチドまたはペプチド様のバックボーン構造を形成し得る。反応は、分割樹脂法を用いて進行し得る。サブモノマー法を実施するために、アシルサブモノマーを、アミノサブモノマーと反応させる。アシルサブモノマーは、反応性カルボニルまたはカルボニル当量、およびアミンにより求核置換において置換され得る開裂基を含む。適切なアシルサブモノマーには、これらに限定されることなく、ブロモ酢酸、3-ブロモプロピオン酸、2-ブロモプロピオン酸、2-ブロモエチルイソシアネート、2-ブロモエチルクロロホルメート、6-フェニル-3-ブロモヘキサン酸、4-ブロモメチル-安息香酸、4-ブロモメチル-2-メトキシ安息香酸、5-ブロモメチル-ピリジン-2-カルボン酸などが含まれる。
【0152】
アシルサブモノマーを、アミノサブモノマーと反応させる。ここで好ましいアミノサブモノマーは、第1級アミンおよびヒドラジドであるが、アミド、カルバメート、尿素、カルバジド、カルバゼート、セミカルバジドなども適切である。あらゆるサブモノマーユニットも、「ヒドロカルビル」または「側鎖」と同様、置換され得るため、異なるサブモノマーを多数の異なるサブアマウントと反応させ、分割樹脂法を適用する際にそのような反応それぞれを終了させることが可能である。
【0153】
サブモノマー法を介した環状ペプトイドの調製
環状ペプトイドを、サブモノマー法によって調製した。そのための一般反応スキームを以下に示す。
【0154】
サブモノマー環化
【0155】
【化21】
【0156】
環化を生じるために要所となる反応は、N-末端ブロモアセタミドの側鎖求核剤での置換であり、樹脂上に「頭-側鎖(head-to-side-chain)」環状構造を生成する。側鎖求核剤は、標準サブモノマー状態を介してオリゴマーの所望の部分で取り入れられる。典型的な求核剤は、保護され得るチオールおよびアミンである。この目的のために好ましいサブモノマーは、Moz-NH-CH2-CH2-NH2、Alloc-NH-CH2-CH2-NH2、およびTrt-S-CH2-CH2-NH2である。オリゴマーを次いで、所望の長さが得られるまで合成し(elaborated)、ブロモアセトアミド基によって終了する。側鎖求核剤を、次いで、選択的に脱保護して環化させる。
【0157】
生成される環状ペプトイドの特定の例、および得られたパーセンテージ収率を、以下に記載する。特定の環構造を有する、ペプトイドから得られる環状化合物の例を本明細書の実施例19〜31に記載する。
【0158】
三量体
【0159】
【化22】
【0160】
固体支持体上で合成された環状有機化合物のライブラリーの有用性
高度に置換された環状構造物は、本発明のサブモノマー方法と、強力な液相化学方法とを組み合わせることによって固体支持体上で合成され得る。1個、2個、3個またはそれ以上の縮合環を含む環状化合物は、まず、線形バックボーンを合成し、次に分子内または分子間環化を行うことによってサブモノマー方法を用いて形成される。
【0161】
レセプター蛋白質または他の分子の結合領域を最も効率的にプローブするためには、種々の置換基および/または環構造物を有する環状有機化合物のライブラリーを作成することが一般に好ましい。
【0162】
種々の構造物がライブラリーに存在すると、所望の結合特性を有する化合物を単離する機会がそれだけ増加する。本願に記載する方法を、環状有機化合物の収集物を固体支持体上で合成するのに適用することによって、スクリーニング用の大きなグループの化合物が調製され得る。例えば、種々の置換基を有する環状アルデヒド類のライブラリーが、相対レセプター結合アフィニティを分析するために調製され得る。ライブラリーは、小さく(約10個の異なる構造物)ても、大きく(10,000個より多くの異なる構造物)てもよい。
【0163】
このようなライブラリーは、天然に発生する生物活性ペプチド、または必須のアフィニティによって適切なレセプターに結合する他の分子に対する環状有機アナログを同定するために有用である。例えば、仮説ペプチドが公知の細胞表面レセプターに結合する場合、細胞表面レセプターを発現する細胞の培養物を調製し、結合に良い条件下でライブラリーを適用し、ライブラリーのメンバーが細胞表面レセプターに結合する程度を決定またはレセプターの応答を顕在化し得る。
【0164】
ライブラリーの環状有機化合物とレセプターとを相互作用させた後、非結合化合物を洗浄によって除去する。多数の環状有機化合物が高結合アフィニティを示す場合、最も高いアフィニティを示す環状有機化合物のみが結合されたままになるように結合条件を変更してもよい。次に、結果として得られる選択された環状有機化合物を除去し、標準分析技術によって同定し得る。
【0165】
例えば、模倣される生物活性分子の活性部分の関連構造物が未知のものである場合、本発明の方法を用いてより大きなライブラリーを構築すればよい。ペプチドまたはエピトープの構造形態に関する手掛かりがない場合は、広範囲の置換基および/または環構造物改変体を有する「普遍的」なライブラリーが最も有用である。
【実施例】
【0166】
以下の実施例は、本発明の化合物およびライブラリーの作成方法に関する完全な開示および説明を当業者に提供するものであり、発明者が発明であると見なす範囲を越えるものではない。使用した数値(例えば、量、温度、等)に関する精度を保証するために努力をしたが、多少の実験誤差および偏差を考慮されたい。特に記載がない限り、部は重量部、分子量は平均分子量、温度は摂氏、および圧力は大気圧または大気圧付近とする。
【0167】
実施例1から11は、概してアルデヒド基を含む反応に関し、実施例12から27は、ケトン基を含む反応に関し、これらの基はすべて、概して環状有機化合物に結合される。他の官能基も、環状化合物に結合し、公知のタイプの反応にかけられ、ライブラリーの多様性を増加し得る。
【0168】
樹脂上でのピロリジン合成の実験条件
概説 Varian Unity 300分光計を用いてNMR分析を行った。以下のグラジエント:250℃で10分間、1分当たり10℃の勾配で325℃まで、および325℃で15分間を用いて、HP 5972質量選択検出器に連結されたHewlett Packard 5890ガスクロマトグラフを用いてGCMS分析を行った。以下のグラジエント:45分間にわって5%Bから95%Bを用いて、Waters 600Eシステム、Alltech Alltima C18逆相カラム(5mm粒子サイズ、4.6×250mm)、220nMでUV検出する25℃、1分間1.0mLの溶剤システムA:B(各0.1col%TFAを含む、水:アセトニトリル)を使用してHPLC分析を行った。
【0169】
実施例1
【0170】
【化23】
【0171】
15mLの無水THF中Rinkアミド樹脂(1.12mmol)上に2.0gの1を含有する20mLのガラス瓶に、517mgのLiOH・H2O(12.32mmol)および1.81mlのトリメチルホスホノアセテート(11.2mmol)を加えた。瓶に蓋をし、室温で14時間振盪させた。樹脂をCH2Cl2、H2O、MeOHおよびエーテルで連続して洗浄し、高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、純粋な化合物2が得られた。1H NMR(CDCL3、300MHz): δ3.84(s、3H)、6.51(d、J=15.6Hz、1H)、7.62(d、J=8.6Hz、2H)、7.71(d、J=15.6Hz、1H)、7.85(d、J=8.6Hz、2H)。HPLC: 1ピーク、tR=21.0分、GCMS: 1ピーク、tR=3.74分、M=205、計算値M=205)。
【0172】
実施例2
ピロリジン類をベースにしたサルコシン/サルコシン誘導体:
サルコシンと、Rinkアミド樹脂(1)に結合した芳香族アルデヒドとを結合させ、適切に置換されたアルケンを加えることによって付加環化を実施し、5を得る。
【0173】
具体的には、2.5mLのトルエンおよび110mgの樹脂上の1(0.062mmol)を有する2ドラム瓶に、220mgのサルコシン(2.46mmol)および419mgのジメチルムコネート(2.46mmol)を加え、30秒間アルゴン脱気し、しっかりと蓋をした。次に、瓶を110℃で12時間振盪させ、その後樹脂をフィルターに移し、CH2Cl2、DMF、MeOHおよびエーテルで洗浄し、次に高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、5の4個の異性体が得られた。GCMS: 4ピーク、tR=10.71分(42.9%)、11.01分(4.5%)、11.68分(40%)、11.88分(12.6%)、M=346、計算値M=346。
【0174】
【化24】
【0175】
実施例3
実施例2と同様に、サルコシンおよび芳香族アルデヒドと、2とを結合させて6を得ることができる。
【0176】
具体的には、1.5mLのトルエンおよび100mgの樹脂上の2(0.0565mmol)を有する2ドラム瓶に、220mgのサルコシン(2.24mmol)および227μLのベンズアルデヒド(2.24mmol)を加え、30秒間アルゴン脱気し、しっかりと蓋をした。瓶を110℃で12時間振盪させ、その後樹脂をフィルターに移し、CH2Cl2、DMF、MeOHおよびエーテルで洗浄し、次に高真空下で30分間乾燥させた。この樹脂をCH2Cl2中20%のTFAで30mg、20分間切断すると、6の4個の異性体が得られた。GCMS: 4ピーク、tR=10.96分(9.7%)、11.78分(21.1%)、12.30分(44.6%)、13.17分(24.6%)、M=338、計算値M=338。
【0177】
【化25】
【0178】
実施例4
【0179】
【化26】
【0180】
7mlの1:1MeOH/1,4-ジオキサン中1g(0.43mmol)の樹脂に結合したアルデヒド1の懸濁液に、93μl(2.15mmol)のメチルアセトアセテート、次いで43μl(43mmol)のピペリジンを加えた。反応混合物を室温で24時間振盪させ、樹脂2を濾過し、CH2Cl2で数回洗浄した。樹脂2をCH2Cl2中10%のTFAで処理し、生成物3をGCMSによって分析したところ、所望の生成物である、m/z=227の親イオンを有する単一の生成物が示された。
【0181】
実施例5
【0182】
【化27】
【0183】
10mlのベンゼンおよびモレキュラーシーブ中1g(0.51mmol)の樹脂に結合したアルデヒド1の懸濁液に、292μl(2.55mmol)のジメチルマロネート、次いで126μl(1.28mmol)のピペリジンを加えた。次に、反応混合物を室温で一晩振盪させた。樹脂2を濾過し、CH2Cl2で数回洗浄し、CH2Cl2中10%のTFAで処理し、生成物3を定量的収率で得た。GCMSによって分析したところ、生成物に対応する、m/z=263の親イオンを有する単一の化合物の存在が示された。
【0184】
実施例6
【0185】
【化28】
【0186】
Rink樹脂に結合したブロモアセトアミド(50mg、0.024mmol、0.47mmol/g)および乾燥DMF(2mL)の混合物(23℃)に、無水K2CO3(130mg、0.94mmol)NaI(2mg、0.013mmol)およびバニリン(143mg、0.94 mmol)を加えた。75℃で16時間激しく振盪させた後、得られた混合物をH2O(10mL)、MeOH(10mL)、THF(2×10mL)、CH2Cl2(3×10mL)で希釈し、真空下(150mm)で3時間乾燥させ、54mgの淡黄色樹脂を得た。
【0187】
この樹脂(54mg、0.24mmol)に、CH2Cl2中5%のTFA溶液(2mL)を滴下した。23℃で15分間保持した後、混合物を濾過し、濾液をCH2Cl2(2×5mL)で洗浄し、溶媒を濃縮させ、3.7mg(74%)の1を淡黄色油として得た:1H NMR(300MHz、CDCl3)δ 9.86(s、1H)、7.48-7.42(m、2H)、6.97-6.94(d、1H)、4.62(s、2H)、3.96(s、3H)、3.39-3.15(br s、2H)>。
【0188】
実施例7
【0189】
【化29】
【0190】
Rink樹脂に結合した4−カルボキシベンズアミドベンジルイミン(108mg、0.085mmol)および乾燥ベンゼン(4mL)の混合物(23℃)に、i-Pr2NEt(60μL、0.34mmol)および無水1,2−フェニレンジ酢酸(59.6mg、0.34mmol)を加えた。70℃で16時間激しく振盪させた後、得られた混合物を濾過し、H2O(10mL)、MeOH(10mL)、THF(3×10mL)、CH2Cl2(3×10mL)で洗浄し、真空下(10d mm)で3時間乾燥させ、122mgの濃茶色樹脂を得た。
【0191】
この樹脂(122mg、0.085mmol)に、CH2Cl2中5%のTFA溶液(3mL)を滴下した。23℃で15分間保持した後、混合物を濾過し、濾液をCH2Cl2(2×5mL)で洗浄し、溶媒を濃縮させ、26.8mg(76%)の2を濃黄色油として得た: MS(ESP)m/e、C25H23N2O4に対する計算値415、測定値415(MH)。
【0192】
実施例8
樹脂に結合したアルデヒドの合成
4−[2',4'−ジメトキシフェニル−(4'−ホルミル−フェニルアセトアミド)]−フェノキシ樹脂{4−ベンアズアルデヒドRinkアミド樹脂}
【0193】
【化30】
【0194】
Fmoc Rinkアミド樹脂(20.0g、9.6mmol)をピペリジン(70ml)およびDMF(80ml)の混合物中で40分間振盪させた。得られた懸濁液を濾過し、DMF(4×100ml)、水(4×100ml)、THF(4×100ml)、ジクロロメタン(4×100ml)で逐次洗浄し、次に真空下で乾燥させた。
【0195】
ジクロロメタン(80ml)中の4−カルボキシベンズアルデヒド(5.0当量、7.21g)の懸濁液に、N−メチルモルホリン(5.0当量、5.3ml)およびPyBOP(5.0当量、25.0g)を加えた。得られた溶液を10分間撹拌し、乾燥樹脂に加えた。この懸濁液を一晩振盪させ、標準ニンヒドリンテストを用いて反応が完了するのをモニターした。反応物を濾過し、樹脂を上記で報告した洗浄サイクルにかけた。樹脂を完全に乾燥させた後、少量(40〜50mg)をジクロロメタン(2ml)中のTFA(40〜50μl)で15分間処理し、濾過し、濾液を真空下で濃縮させた。回収物をNMRおよびGCMSで分析し、収率および純度を決定した。
【0196】
実施例9
3−ベンズアルデヒドRinkアミド樹脂
【0197】
【化31】
【0198】
上記の方法を用いて、N−メチルモルホリン(5.0当量、5.3ml)およびPyBOP(5.0当量、25.0g)の存在下で、Rink樹脂(20.0g)と3−カルボキシベンズアルデヒド(5.0当量、7.21g)とを結合させた。
【0199】
実施例10
4−ホルミルフェノキシアセトアミドRink樹脂
【0200】
【化32】
【0201】
上記の方法を用いて、N−メチルモルホリン(5.0当量、1.32ml)およびPyBOP(5.0当量、6.24g)の存在下で、Rink樹脂(5.0g)と4−ホルミルフェノキシ酢酸(5.0当量、2.16g)とを結合させた。
【0202】
実施例11
4−ブロモメチルベンジルRinkアミド樹脂
【0203】
【化33】
【0204】
上記の方法を用いて、N−メチルモルホリン(5.0当量、1.32ml)およびPyBOP(5.0当量、6.24g)の存在下で、Rink樹脂(5.0g)と4−ブロモメチルトルイル酸(5.0当量、2.58g)とを結合させた。
【0205】
実施例12
4−アジドベンゾイル(azidobenyl)Rinkアミド樹脂
【0206】
【化34】
【0207】
上記の方法を用いて、N−メチルモルホリン(5.0当量、0.85ml)およびPyBOP(5.0当量、4.04g)の存在下でRink樹脂(5.40g)と4−アジド安息香酸(3.0当量、1.27g)とを結合させた。
【0208】
実施例13
固体支持体に結合したジケトンからの環状有機化合物の合成
固体支持体に(Rinkリンカーを介して)結合したジケトンIV(70μmol)をDMSO(1mL)中で懸濁する。シアノアセトアミド(59mg、700μmolに等しい)およびピペリジン(144μL、1400μmolに等しい)を加え、反応混合物を室温で撹拌した。反応混合物を排出し、DMSO(6回)、メタノール(3回)およびジクロロメタン(6回)で洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、次に濾過した。揮発性物質をアルゴン気流下でエバポレートし、生成物VIIを得た。
【0209】
【化35】
【0210】
実施例14
固体支持体に結合したジケトン類からのモノケトフラン類の合成
約70μmolの固体支持体に結合したIVをDMF(1mL)中で懸濁する。ヨードベンゼン(184mg、700μmolに等しい)、炭酸カリウム(193mg、1400μmolに等しい)およびテトラキス(トリフェニルホスフィン)パラジウム(O)(39mg、35μmolに等しい)を加えた。反応混合物をアルゴンで軽く分散させ、加熱(60〜100℃)した。反応混合物を冷却し、排出し、DMF(6回)、メタノール(3回)、およびジクロロメタン(6回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、生成物VおよびVIを得た。
【0211】
【化36】
【0212】
実施例15
固体支持体に結合したジケトン類からのN−置換ピラゾール類の合成
樹脂の調製:
Fmoc Rinkアミド樹脂(20.0g、9.6mmol)をピペリジン(70mL)およびDMF(80mL)の混合物中で40分間振盪させた。得られた懸濁液を濾過し、DMF(4×100mL)、水(4×100mL)、THF(4×100mL)、ジクロロメタン(4×100mL)で逐次洗浄し、次に真空下で乾燥させた。
【0213】
固体支持体に結合したモノケトンの合成:
ジクロロメタン(80mL)中の4−カルボキシアセトフェノン(5.0当量)の懸濁液に、N−メチルモルホリン(5.0当量)およびPyBOP(登録商標)(5.0当量)を加えた。得られた溶液を10分間撹拌し、乾燥樹脂に加えた。懸濁液を一晩振盪させ、標準ニンヒドリンテストを用いて反応が完了するのをモニターした。反応物を濾過し、樹脂を、DMF(4×100mL)、水(4×100mL)、THF(4×100mL)、ジクロロメタン(4×100mL)で逐次洗浄し、次に真空下で乾燥させた。樹脂を完全に乾燥させた後、少量(40〜50mg)をジクロロメタン(2mL)中のトリフルオロ酢酸(TFA、40〜50μl)で15分間処理し、濾過し、濾液を真空下で濃縮させた。回収物をNMRおよびGCMSで分析した。構造物は、固体支持体から分離した化合物1について予想されたのと一致していた。
【0214】
支持体に結合したジケトンの合成:
樹脂に結合したジケトンを調製するため、化合物2、樹脂に結合した化合物1(上記で調製した0.47mmol、1.0g)を12mLのTHF中に懸濁した。15mgのジベンゾ−18−クラウン−6および400μLの酢酸エチル(4.7mmol)をアルゴン気流下で導入した。THF(2.8mmol)中の2.8mLの1M カリウムt−ブトキシドを加える前に、懸濁液を撹拌した。反応物を1時間70℃まで加熱した。次に、冷却した樹脂を濾過し、ジクロロメタンで洗浄した。少量の樹脂をジクロロメタン中20%のTFAで処理し、濾過し、揮発物をアルゴン気流下でエバポレートした。GC-MSおよびNMR分析の結果は、化合物2への定量的変換と一致していた。
【0215】
固体支持体上で化合物4を調製するために、約100mgの固体支持体に結合したジケトン2(47μmol)を1mLの乾燥DMSOおよび50μLのフェニルヒドラジン(470μmol)中で懸濁した。反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、化合物4を得た。GC-MSによる分析の結果、所望の生成物への定量的変換(電子衝撃によりMW=277)が示された。DMSO中の生成物のNMRは、固体支持体から分離した化合物4の構造と一致していた。
【0216】
【化37】
【0217】
実施例16
N−置換ピラゾールの他の合成
化合物4および化合物9の1:1混合物の調製を以下のように行った。化合物8の合成に関して、100mgの固体支持体に結合した化合物2(47μmol)を、1mLのトルエン、147μLのDBU(940μmol)、および50μLの1−ブロモブタン(470μmol)中に懸濁した。化合物4の合成に関して、50μLのフェニルヒドラジン(470μmol)を加え、反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、GC-MS分析により化合物4および化合物9の1:1混合物を得た。
【0218】
【化38】
【0219】
実施例17
縮合ピラゾールおよびピラン環の合成
化合物IIIを以下のように調製した。固体支持体に結合したピラゾリジノンの合成を以下に示す。100mgの固体支持体に結合したβ−ケトエステル13を、1mLの乾燥DMSOおよび50μLのフェニルヒドラジン中に懸濁した。反応容器を絶えず撹拌しながら70℃まで18時間加熱し、ピラゾリジノン16(一般構造)を得た。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミドおよびジクロロメタンで洗浄した。70mgのピラゾリジノンIをトルエン(1mL)中に懸濁した。ピペリジン(70μL、700μmolに等しい)、ベンズアルデヒド(144μL、1400μmolに等しい)および4A0モレキュラーシーブ(1スパチュラチップ)を加え、反応混合物を室温で撹拌した。モレキュラーシーブからデカンテーションした後、固体支持体をジクロロメタンで6回洗浄した。得られた生成物である固体支持体に結合した化合物IIをDMSO(1mL)中に懸濁した。ピペリジン(70μl、700μmolに等しい)およびマロノニトリル(93mg、1400μmolに等しい)を加え、反応混合物をアルゴンで軽く分散させ、次に加熱(60〜120℃)した。冷却後、反応混合物を排出し、ジクロロメタン(6回)、メタノール(3回)およびジクロロメタン(3回)で洗浄した。固体支持体を、ジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、化合物IIIを得た。
【0220】
【化39】
【0221】
他のピラゾール合成:
【0222】
【化40】
【0223】
実施例18
2−アミノ−ピリミジンの合成:
化合物6の調製を以下のように行った。75mgの固体支持体に結合した化合物2(35μmol)を、67mgのグアニジン塩酸塩(700μmol)を含む750μLの乾燥DMSO中に懸濁する。次に、塩酸塩を、アルゴン気流下で、kTHF中の500μLの1M カリウムt−ブトキシド(500μmol)でインサイチュで中和した。次に、反応容器を絶えず撹拌しながら70℃まで18時間加熱した。次に、冷却し、誘導体化された固体支持体を濾過し、ジメチルホルムアミド、水、ジメチルホルムアミドおよびジクロロメタンで洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から分離した化合物6を得た。229(M+1)のエレクトロスプレーMSピークは、固体支持体から分離した6の構造と一致していた。
【0224】
【化41】
【0225】
実施例19
カルコン3の合成
50mg(0.47mmol)のベンズアルデヒドおよび100mg(0.047mmol)のRinkアミド樹脂に結合した5mLのTHF中の化合物1の混合物に12uLの2M ナトリウムエトキシドを加えた。得られた混合物を室温で2時間振盪させた。得られた樹脂に結合した化合物2を3mLの濾過カラムに収集し、水(3×2mL)、DMF(2×2mL)および塩化メチレン(3×2mL)で洗浄した。真空下で乾燥させた後、5mgの樹脂に結合した化合物2を、塩化メチレン中1.5mLの5%トリフルオロ酢酸で20分間処理した。固体を濾過し、1mLの塩化メチレンで洗浄し、合わせた塩化メチレン溶液を濃縮し、真空下で乾燥させ、0.5mgの生成物3を得た。MS m/z 252(M++1)。
【0226】
【化42】
【0227】
実施例20
ジエステル5の合成
20mg(0.009mmol)のRink樹脂に結合した化合物2および0.60mLのTHF中の25mg(0.18mmol)のマロン酸ジメチルの混合物に13uLの1,8−ジアザビシクロ[5,4,0]ウンデカ−7−エン(0.09mmol)を加えた。得られた混合物をアルゴン下、40℃で2時間振盪させた。得られたRink樹脂に結合した化合物4を3mLの濾過カラムに収集し、DMF(2×2mL)、CH2Cl2(2×2mL)で洗浄した。真空下で乾燥させた後、5mgの樹脂に結合した化合物4を、1mLのCH2Cl2で処理し、合わせたCH2Cl2溶液を真空下で濃縮し、0.8mgの生成物5を得た。MS m/z 384(M++1)。
【0228】
【化43】
【0229】
実施例21
テトラヒドロピリドン9の合成
10mg(0.0047mmol)のRinkアミド樹脂に結合した化合物4および7mg(0.047mmol)の4−メトキシベンジルアミン、ならびにTHF中の0.8mLの1M NaBH3CNの混合物に、1滴の酢酸および1滴のH2Oを加えた。この混合物をアルゴン下で密封し、75℃で24時間振盪させた。最終の樹脂に結合した化合物8をDMF(3×2mL)、H2O(2×2mL)、およびCH2Cl2(3×2mL)で洗浄した。得られた化合物8をCH2Cl2中5%のトリフルオロ酢酸で20分間処理した。固体を濾過し、1mLのCH2Cl2で洗浄し、合わせたCH2Cl2溶液を真空下で濃縮し、1.8mgの生成物9を得た。MS m/z 469(M++1)。
【0230】
【化44】
【0231】
実施例22
ジヒドロピリドン7の合成
10mg(0.0047mmol)のRinkアミド樹脂に結合した化合物4および0.8mLのトルエン中7mg(0.047mmol)の4−メトキシベンジルアミンの混合物に1滴の酢酸を加えた。この混合物をアルゴン下で密封し、80℃で15時間振盪させた。最終の樹脂に結合した化合物6をDMF(3×2mL)、H2O(2×2mL)、およびCH2Cl2(3×2mL)で洗浄した。得られた化合物6をCH2Cl2中5%のトリフルオロ酢酸で処理し、溶液を真空下で濃縮し、1.8mgの生成物7を得た。MS m/z 471(M++1)。
【0232】
【化45】
【0233】
実施例23
樹脂1(100mg、0.056mmol)を、1.5mlのEtOHおよび1,4−ジオキサンの1:1混合物と合わせ、130μl(1.12mmol)のアセトフェノンおよび87mg(1.12mmol)のNH4OAcを加えた。反応混合物を80〜85℃で一晩振盪させ、H2O、MsOHおよびCH2Cl2で洗浄した。樹脂2をCH2Cl2中10%のTFAで切断し3を得た。ESMS m/z = 351(M+1)。樹脂4を同一の反応条件で処理し、ピリジン6を得た。ESMS m/z = 315(M+1)。
【0234】
【化46】
【0235】
実施例24
イソオキサゾールの合成:
44mg(0.11mmol)のD−グルコピラノシルニトロメタンテトラ−アセテート(αアノマー)および30mg(0.016mmol)のRinkアミド樹脂に結合した化合物1を含有する0.80mLの1,2−ジクロロエタン中24uL(0.22mmol)のフェニルイソシアナートの溶液に、2mLの(0.016mmol)のトリエチルアミンをアルゴン下で加えた。この混合物を還流し、78℃で15時間振盪させた。得られた混合物を室温まで冷却した。樹脂に結合した化合物2を3mLの濾過カラム中で収集し、N,N−ジメチルホルムアミド(3×2mL)、塩化メチレン(3×2mL)で洗浄し、真空下で乾燥させた。乾燥した樹脂に結合した化合物2を塩化メチレン中2mLの20%トリフルオロ酢酸で20分間処理した。固体を濾過し、1.5mLの塩化メチレンで3回洗浄した。合わせた洗浄物を濃縮し、真空下で乾燥させ、12mgの約95%の純度の生成物3を得た。スペクトルデータは、電子スプレーMS m/z 884(M++1)からなっていた。
【0236】
【化47】
【0237】
実施例24
置換ピランの合成
化合物18、2−アミノ−3−シアノピランを以下の手法で調製した。ケトン:P-Ar-C(O)-CH2-Rからのa,b不飽和−ケトンの調製物。この時点で分割樹脂方法を行うと、次の環化工程と同様に多様性を加える。固体支持体樹脂(47μmol)に共有結合した置換α,β−不飽和ケトンを溶媒(1mL)に懸濁した。ジシアノメタンを加え、反応混合物を撹拌した。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物18を得た。
【0238】
化合物18の混合物を、まず、固体支持体樹脂上にα,β−不飽和ケトン類の混合物を調製することによって合成する。混合物の多様性を、α,β−不飽和ケトンの種々のR基によって制御する。R基のそれぞれは、独立してアルキル基またはアリール基であり得る。さらに、α,β−不飽和ケトンは、R基の任意の位置で、樹脂に共有結合され得る。次に、固体支持体に結合したケトン類の混合物を上記のようにDI−シアノメタンと反応させ、一般構造18を有する化合物の混合物を得る。
【0239】
【化48】
【0240】
α,β不飽和ケトンの一般的な合成。
【0241】
【化49】
【0242】
実施例26
ピペラジン−2−オンの合成
化合物4の一般構造を有するピペラジン−2−オンを以下の手法で調製した。化合物2などの固体支持体に結合したα,β−不飽和ケトンを溶媒(1mL)に懸濁した。yyμmolのエステルS’を加え、反応混合物を適切な温度で十分な時間撹拌し、固体支持体に結合した化合物Tを得た。誘導体化された固体支持体をジクロロメタンで(6回)洗浄し、濾過した。この時点で、固体支持体を、化合物4および5を生成する異なる反応用の部分に分割してもよい。
【0243】
化合物Uを、xxμmolの固体支持体に結合した化合物3を溶媒(1mL)に懸濁することによって生成する。第1級アミンおよびNaBH3CNを加え、反応混合物を適切な温度で適切な時間撹拌する。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物4を得た。一般構造4を有する化合物の混合物を、R基を変えることによって調製する。R1、R2、R3およびR4は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基、または水素である。Arは、任意の芳香族またはヘテロ芳香族部分である。Xは、任意の芳香族もしくはヘテロ芳香族部分、または電子吸引基である。
【0244】
【化50】
【0245】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
実施例27
テトラヒドロピラジン−2−オンの合成
化合物5を、固体支持体に結合した化合物3を酢酸を含有する溶媒(1mL)に懸濁することによって生成する。第1級アミンを加え、反応混合物を適切な温度で適切な時間撹拌する。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物5、テトラヒドロ−2−ピリドンを得た。一般構造5を有する化合物の混合物を、サブモノマー上のR基を変えることによって調製する。R1、R2、R3およびR4は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基または水素である。Arは、任意の芳香族またはヘテロ芳香族部分である。Xは、本願で定義した任意の芳香族もしくはヘテロ芳香族部分、または電子吸引基である。
【0246】
【化51】
【0247】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
実施例28
置換ピリジン類の合成
化合物8、2−アミノ−3−シアノ−ピリジンを以下のように調製した。固体支持体に結合した化合物6を酢酸アンモニウムを含む溶媒(1mL)に懸濁した。ケトン7を加え、反応混合物を適切な温度で適切な時間撹拌した。冷却後、誘導体化された固体支持体をジクロロメタン(6回)、メタノール(3回)、およびジクロロメタン(3回)で洗浄した。固体支持体をジクロロメタン中20%のトリフルオロ酢酸で20分間処理し、濾過し、揮発物をアルゴン気流下でエバポレートし、固体支持体から切断した生成物8、2−アミノ−3−シアノ−ピリジンを得た。一般構造8を有する化合物の混合物を、サブモノマー出発物質のR基を変えることによって調製する。R1、R2、およびR3は、独立して、任意のアルキル基、芳香族、ヘテロ芳香族基または水素である。
【0248】
【化52】
【0249】
R1、R2、R3、= 芳香族、脂肪族、ヘテロ芳香族(heteromatic)、H
ケトン類からの他の実施例
【0250】
【化53】
【0251】
R1、R2、R3、R4 = H、アルキル基、芳香族、ヘテロ芳香族
Ar = 芳香族またはヘテロ芳香族
X = ArまたはEWG
【0252】
【化54】
【0253】
【化55】
【0254】
本願は、本願と同一の譲受人に譲渡され、本願と同一日に提出された「SYNTHESIS OF N-SUBSTITUTED OLIGOMERS」という名称の代理人整理番号第06515/022001号の出願に関連する。特許出願第06515/022001号は、1994年7月18日付けで先に提出された米国特許出願第08/277,228号の一部継続出願であり、米国特許出願第08/277,228号は、1993年9月24日付けで先に提出された米国特許出願第08/126,539号の一部継続出願であり、米国特許出願第08/126,539号は、1992年9月24日付けで先に提出された米国特許出願第07/950,853号の一部継続出願であり、これらの出願はすべて同一譲受人に譲渡され、本願では参考のためにこれらの出願をそのまま援用する。
【0255】
本発明は、その具体的な実施態様に基づいて説明したが、当業者には当然のことながら、本発明の真の精神および範囲を逸脱せずに、種々の変更がなされ、等価物の代用がなされ得る。さらに、特定の反応、物質、ライブラリー、プロセス、1つのプロセス工程または複数のプロセス工程を本発明の目的、精神および範囲に適用するために、多くの改変がなされ得る。このような改変はすべて添付の請求の範囲の範囲内であるものとする。
【図面の簡単な説明】
【0256】
【図1】図1は、分割樹脂方法論を示す模式図である。
【特許請求の範囲】
【請求項1】
固体支持体に共有結合した10個またはそれ以上の異なる化合物によって特徴づけられる有機化合物のライブラリーであって、該化合物が、電子供与出発物 質を結合した固体支持体から得られる環状有機化合物であり、該10個またはそれ以上の化合物のそれぞれが、回収可能かつ分析可能な量で存在する、ライブラリー。
【請求項2】
少なくとも1000個の異なる化合物を含み、該生成化合物の少なくとも1つが生物学的に活性である、請求項1に記載のライブラリー。
【請求項3】
前記電子供与出発物質が、アミドを介して固体支持体に結合したアルデヒドであり、前記環状有機化合物のそれぞれが、100ピコモルまたはそれ以上の量で存 在し、さらに、該環状有機化合物の環状部分が、該環状部分に共有結合した側鎖を有する、請求項2に記載のライブラリー。
【請求項4】
前記側鎖が、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、請求項2に記載のライブラリー。
【請求項5】
10個またはそれ以上の環状有機化合物のライブラリーを固体支持体上で合成する方法であって、該化合物の官能部分が、アクセス可能かつ反応可能であり、電子供与基を提供するように、固体支持体上に化合物を誘導体化する工程と、 該誘導体化された化合物を、環化反応に供する工程であって、複数の反応物が該化合物の官能部分と反応し、複数の有機環状化合物を形成する工程と、を包含する方法。
【請求項6】
前記化合物が、前記固体支持体に直接結合したアミドによって該支持体に結合される、請求項5に記載の方法。
【請求項7】
複数の異なるアルデヒド化合物が前記固体支持体に結合し、それぞれが、異なる官能基を有し、前記環状有機化合物を複数のサブアマウントに分割する工程と、 異なる反応物を該各サブアマウントとカップリングし、各カップリング反応を該各サブアマウントを用いて実質的に完了させる工程と、既知の量の該各サブアマウントを共に組み合わせ、化合物のライブラリーを得る工程と、 をさらに包含する請求項5に記載の方法。
【請求項8】
.環状有機化合物に結合した第2級アミンを-NH2による求核置換を可能にする脱離基を含む第1のサブモノマーアシル化剤でアシル化し、アシル化アミンを得る工程と、該アシル化アミンと、-NH2基 を含む十分な量の第2のサブモノマー置換剤とを反応させ、該脱離基の求核置換を実施する工程と、 該アシル化および反応工程を、所望の長さを有し、所望の置換基を含むポリマーを形成するのに適切なように連続して繰り返す工程と、該ポリマーを環化し、共有結合したペプトイドバックボーンを有する環状生成化合物を生成する工程と、 をさらに包含する、請求項5に記載の方法。
【請求項9】
本願明細書等に記載されるような、基質結合環状有機化合物の組み合わせライブラリー。
【請求項1】
固体支持体に共有結合した10個またはそれ以上の異なる化合物によって特徴づけられる有機化合物のライブラリーであって、該化合物が、電子供与出発物 質を結合した固体支持体から得られる環状有機化合物であり、該10個またはそれ以上の化合物のそれぞれが、回収可能かつ分析可能な量で存在する、ライブラリー。
【請求項2】
少なくとも1000個の異なる化合物を含み、該生成化合物の少なくとも1つが生物学的に活性である、請求項1に記載のライブラリー。
【請求項3】
前記電子供与出発物質が、アミドを介して固体支持体に結合したアルデヒドであり、前記環状有機化合物のそれぞれが、100ピコモルまたはそれ以上の量で存 在し、さらに、該環状有機化合物の環状部分が、該環状部分に共有結合した側鎖を有する、請求項2に記載のライブラリー。
【請求項4】
前記側鎖が、ヒドロキシ、Ra、-ORa、-NRaRb、-SO1,2,3,4Ra、-C(O)Ra、-C(O)ORa、-OC(O)Ra、-OC(O)ORa、-NRbC(O)Ra、-C(O)NRaRb、-OC(O)NRaRb、-NRcC(O)NRaRb、-NRbC(O)ORa、-Ra-O-Rb、-Ra-NRbRc、-Ra-S-Rb、-Ra-S(O)-Rb、-Ra-S(O)2-Rb、-ORa-O-Rb、-NRaRb-O-Rc、-SO1,2,3,4Ra-O-Rb、-C(O)Ra-O-Rb、-C(O)ORa-O-Rb、-OC(O)Ra-O-Rb、-OC(O)ORa-O-Rb、-NRbC(O)Ra-O-Rc、-C(O)NRaRb-O-Rc、-OC(O)NRaRb-O-Rc、-NRcC(O)NRaRb-O-Rd、-NRbC(O)ORa-O-Rc、-ORa-S-Rb、-NRaRb-S-Rc、-SO1,2,3,4Ra-S-Rb、-C(O)Ra-S-Rb、-C(O)ORa-S-Rb、-OC(O)Ra-S-Rb、-OC(O)ORa-S-Rb、-NRbC(O)Ra-S-Rc、-C(O)NRaRb-S-Rc、-OC(O)NRaRb-S-Rc、-NRcC(O)NRaRb-S-Rd、-NRbC(O)ORa-S-Rc、-ORa-NRbRd、-NRaRb-NRcRd、-SO1,2,3,4Ra-NRbRd、-C(O)Ra-NRbRd、-C(O)ORa-NRbRd、-OC(O)Ra-N-RbRd、-OC(O)ORa-NRbRd、-NRbC(O)Ra-NRcRd、-C(O)NRaRb-NRcRd、-OC(O)NRaRb-NRcRd、-NRcC(O)NRaRb-NHRd、-NRbC(O)ORa-NRcRdからなる群から選択され、ここで、Ra、Rb、RcおよびRdは、それぞれ独立して、アルキル、アルケニル、アルキニル、アリール、アラルキル、アラルケニルまたはアラルキニルであり、ここで、Ra、Rb、RcおよびRdは、それぞれ、0-6ハロ、NO2、-OH、低級アルキル、-SH、-SO3、-NH2、低級アシル、低級アシルオキシ、低級アルキルアミノ、低級ジアルキルアミノ、トリハロメチル、-CN、低級アルキルチオ、低級アルキルスルフィニル、または低級アルキルスルホニルであり、ここで「低級」は、1から6個の炭素原子を示す、請求項2に記載のライブラリー。
【請求項5】
10個またはそれ以上の環状有機化合物のライブラリーを固体支持体上で合成する方法であって、該化合物の官能部分が、アクセス可能かつ反応可能であり、電子供与基を提供するように、固体支持体上に化合物を誘導体化する工程と、 該誘導体化された化合物を、環化反応に供する工程であって、複数の反応物が該化合物の官能部分と反応し、複数の有機環状化合物を形成する工程と、を包含する方法。
【請求項6】
前記化合物が、前記固体支持体に直接結合したアミドによって該支持体に結合される、請求項5に記載の方法。
【請求項7】
複数の異なるアルデヒド化合物が前記固体支持体に結合し、それぞれが、異なる官能基を有し、前記環状有機化合物を複数のサブアマウントに分割する工程と、 異なる反応物を該各サブアマウントとカップリングし、各カップリング反応を該各サブアマウントを用いて実質的に完了させる工程と、既知の量の該各サブアマウントを共に組み合わせ、化合物のライブラリーを得る工程と、 をさらに包含する請求項5に記載の方法。
【請求項8】
.環状有機化合物に結合した第2級アミンを-NH2による求核置換を可能にする脱離基を含む第1のサブモノマーアシル化剤でアシル化し、アシル化アミンを得る工程と、該アシル化アミンと、-NH2基 を含む十分な量の第2のサブモノマー置換剤とを反応させ、該脱離基の求核置換を実施する工程と、 該アシル化および反応工程を、所望の長さを有し、所望の置換基を含むポリマーを形成するのに適切なように連続して繰り返す工程と、該ポリマーを環化し、共有結合したペプトイドバックボーンを有する環状生成化合物を生成する工程と、 をさらに包含する、請求項5に記載の方法。
【請求項9】
本願明細書等に記載されるような、基質結合環状有機化合物の組み合わせライブラリー。
【図1】

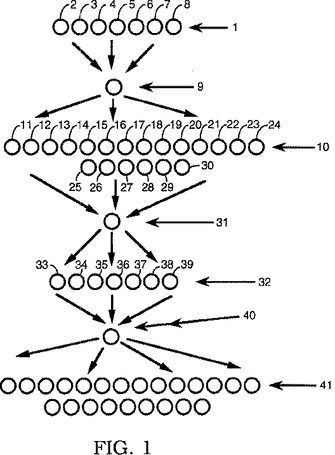
【公開番号】特開2008−50360(P2008−50360A)
【公開日】平成20年3月6日(2008.3.6)
【国際特許分類】
【出願番号】特願2007−236022(P2007−236022)
【出願日】平成19年9月11日(2007.9.11)
【分割の表示】特願平9−500720の分割
【原出願日】平成8年5月23日(1996.5.23)
【出願人】(591076811)カイロン コーポレイション (265)
【Fターム(参考)】
【公開日】平成20年3月6日(2008.3.6)
【国際特許分類】
【出願日】平成19年9月11日(2007.9.11)
【分割の表示】特願平9−500720の分割
【原出願日】平成8年5月23日(1996.5.23)
【出願人】(591076811)カイロン コーポレイション (265)
【Fターム(参考)】
[ Back to top ]