
塩の製造のための新規な方法
本発明は、医薬化合物またはそれらの中間体の塩酸塩、臭化水素酸塩またはヨウ化水素酸塩の製造および結晶化のための新規な方法であり、塩基またはその酸付加塩を溶媒中でハロゲン化トリアルキルシリルと反応させる方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬化合物またはそれらの中間体のハロゲン化水素酸塩の新規な製造および結晶化方法に関する。本発明方法によれば、確定した結晶構造から成る純粋形態のハロゲン化水素酸塩が確実にかつ良好な収率で得られる。本発明方法は工業的用途に特に好適である。
【背景技術】
【0002】
医薬化合物またはそれらの中間体の塩酸塩は一般に塩基またはその塩の溶液を塩化水素によって酸性化することによって製造され、その場合、塩化水素水溶液もしくは塩化水素ガスが使用されるかまたはHClの有機溶媒溶液が使用される。塩酸水溶液の添加による塩酸塩の製造は簡単な方法であり、塩酸を36%(w/w)水溶液として使用するのが便利である。典型的な手順は、有機塩基を溶媒に溶解し、計算量または過剰量の濃HClを添加し、結晶化が生じないならばジエチルエーテルのような有機溶媒を徐々に添加することによって結晶化を誘発できる。しかしながら、塩酸塩が水溶性のため、塩酸水溶液による塩形成には低収率という特徴がしばしば見られる。さらに、無水塩形態が望まれる場合には概して塩酸水溶液の使用は適当でない。水が固体結晶質生成物の形成および単離を妨害するならば、シリンダーから無水ガスを供給するかまたはジエチルエーテルのような無水非プロトン性溶媒中のHClガスを使用することが可能である。しかしながら、大量の塩化水素ガスを使用する代替方法は設備費の高騰および気体処理に伴う典型的な危険を招く。
【0003】
単一結晶構造から成る純粋形態の塩酸塩を製造することは非常に多くの場合に重要である。医薬基質(substance)にはこれが特に重要である。何故ならば、特に固体経口剤形配合物の場合には、結晶構造の変化が医薬製品の溶解、製造適性および安定性を左右することが有り得るからである。しかしながら所望の塩酸塩形態の優先的な形成は結晶化動力学に依存し、容易にコントロールできない。ときには塩化水素ガスを一定流量で一定時間供給することが必要であり、ガス流動中、さらに生成物の濾過中にも温度を一定に維持する必要があり、これが処理を極めて難しいものにする。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の目的は、医薬化合物またはそれらの中間体の塩酸塩、臭化水素酸塩またはヨウ化水素酸塩を確定した結晶構造から成る純粋形態で再現可能に製造するための適当な方法を提供することである。
【課題を解決するための手段】
【0005】
本発明は、医薬化合物またはそれらの中間体のハロゲン化水素酸塩の新規な製造および結晶化方法に関する。本発明の方法によれば、確定した結晶構造から成る純粋形態のハロゲン化水素酸塩を良好な収率で確実に得ることができる。本発明の方法は工業的用途に特に好適である。
【0006】
より特定的には本発明は、溶媒中で有機アミンにハロゲン化トリアルキルシリルを添加する段階を含む、有機アミンの結晶質ハロゲン化水素酸塩の製造方法に関する。有機アミンハは遊離塩基または酸付加塩の形態であり、酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である。
【0007】
本発明はさらに、塩酸モキシフロキサシンメチレンジクロリド溶媒和物、塩酸モキシフロキサシンの新規な無水IV形、塩酸モキシフロキサシンニトロメタン溶媒和物、塩酸モキシフロキサシン酢酸溶媒和物、モキシフロキサシンHClの無水IV形またはモキシフロキサシンHClの酢酸溶媒和物を有効量で含む医薬組成物、塩酸リネゾリドの新規な結晶質形、塩酸プラスグレルアセトニトリル溶媒和物および塩酸ラロキシフェンテトラヒドロフラン溶媒和物に関する。すべての新規な溶媒和物または塩が本発明の方法によって得られる。本発明はさらに、有機アミンの結晶質ハロゲン化水素酸塩の製造方法におけるハロゲン化トリアルキルシリルの使用に関する。
【図面の簡単な説明】
【0008】
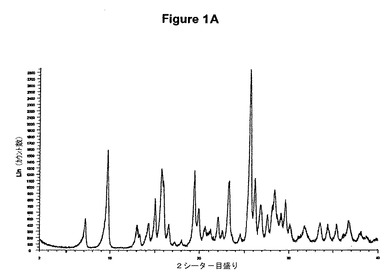
【図1A】無水塩酸ミコフェノラートモフェチルのX線粉末回折図である。
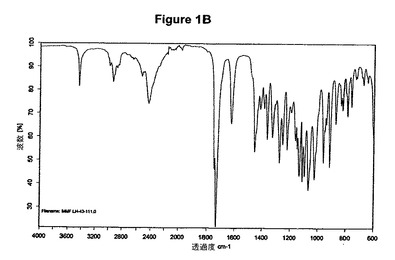
【図1B】無水塩酸ミコフェノラートモフェチルの赤外スペクトルである。
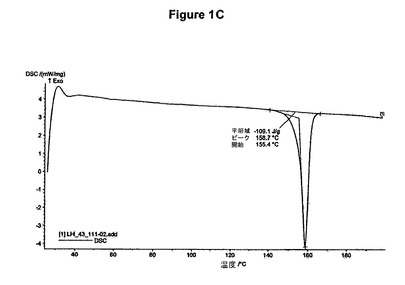
【図1C】無水塩酸ミコフェノラートモフェチルのDSC曲線である。
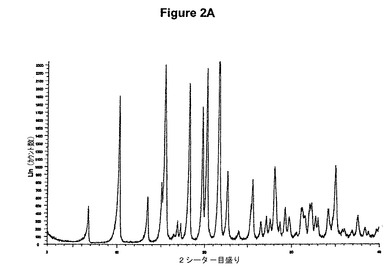
【図2A】塩酸ベンラファキシンI形のX線粉末回折図である。
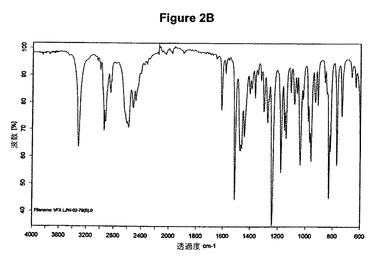
【図2B】塩酸ベンラファキシンI形の赤外スペクトルである。
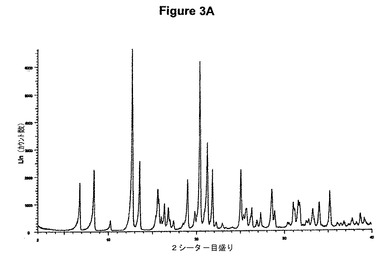
【図3A】塩酸ベンラファキシンII形のX線粉末回折図である。
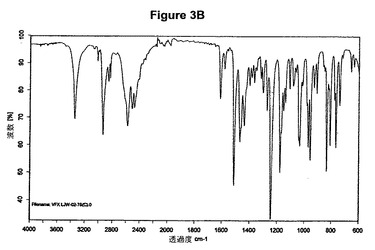
【図3B】塩酸ベンラファキシンII形の赤外スペクトルである。
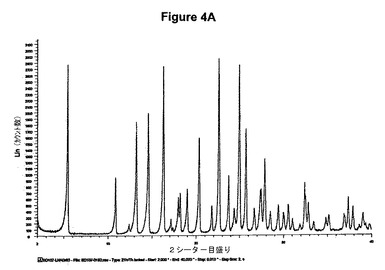
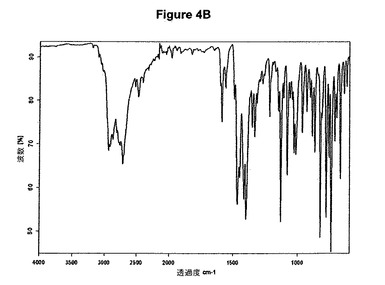
【図4A】塩酸セルトラリンII形のX線粉末回折図である。
【図4B】塩酸セルトラリンII形の赤外スペクトルである。
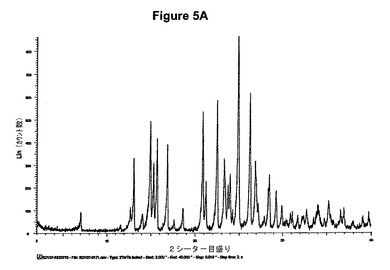
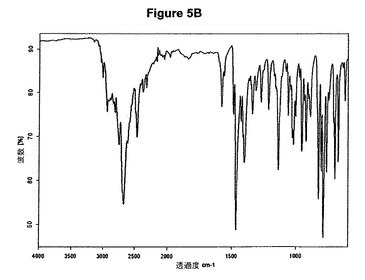
【図5A】塩酸セルトラリンI形のX線粉末回折図である。
【図5B】塩酸セルトラリンI形の赤外スペクトルである。
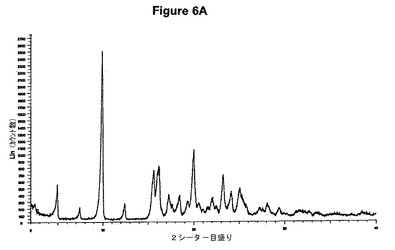
【図6A】塩酸ドネペジルII形のX線粉末回折図である。
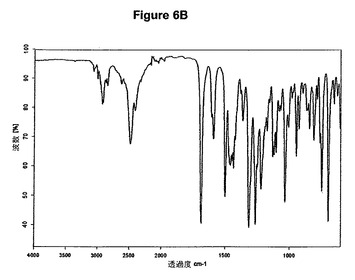
【図6B】塩酸ドネペジルII形の赤外スペクトルである。
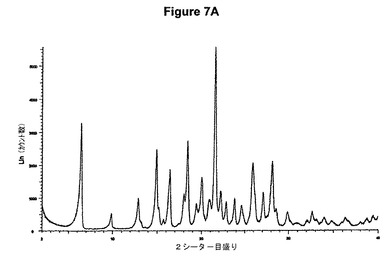
【図7A】塩酸ドネペジルIII形のX線粉末回折図である。
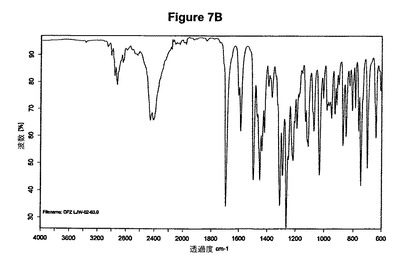
【図7B】塩酸ドネペジルIII形の赤外スペクトルである。
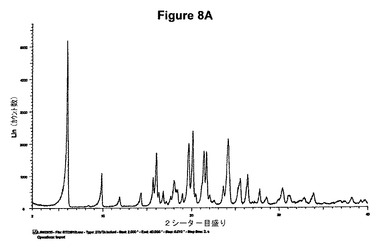
【図8A】塩酸テルビナフィンのX線粉末回折図である。
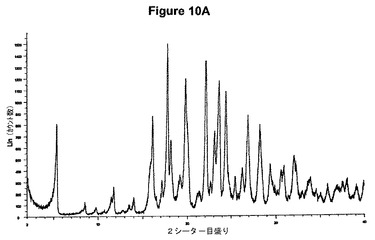
【図8B】塩酸テルビナフィンの赤外スペクトルである。
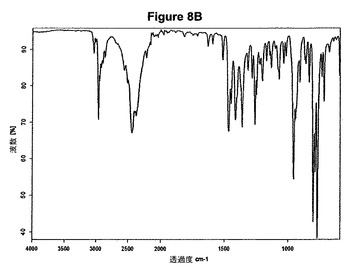
【図9A】塩酸シナカルセットのX線粉末回折図である。
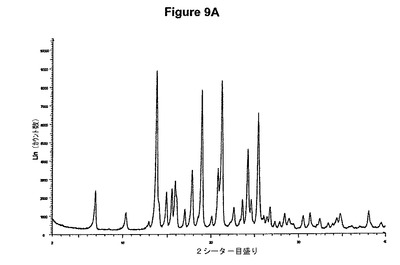
【図9B】塩酸シナカルセットの赤外スペクトルである。
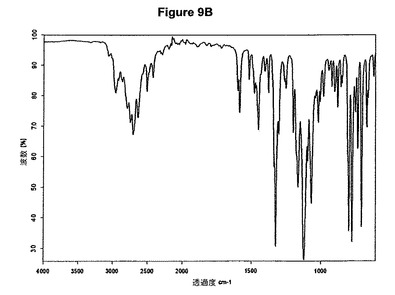
【図10A】臭化水素酸シタロプラムのX線粉末回折図である。
【図10B】臭化水素酸シタロプラムの赤外スペクトルである。
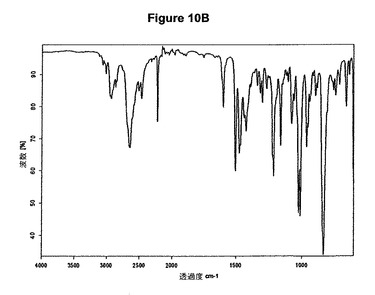
【図11A】塩酸アリピプラゾールA形のX線粉末回折図である。
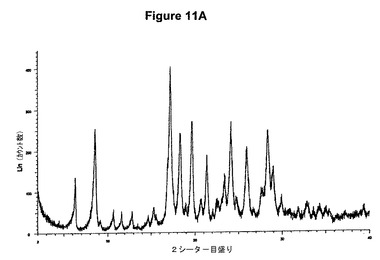
【図11B】塩酸アリピプラゾールA形の赤外スペクトルである。
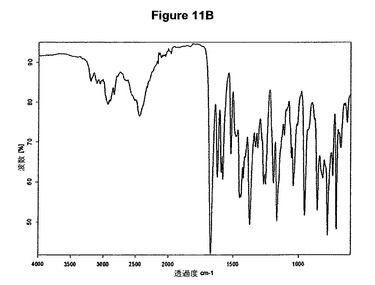
【図12A】モノ塩酸プラミペキソールのX線粉末回折図である。
【図12B】モノ塩酸プラミペキソールの赤外スペクトルである。
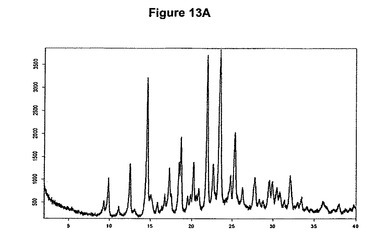
【図13A】塩酸モキシフロキサシンメチレンジクロリド溶媒和物のX線粉末回折図である。
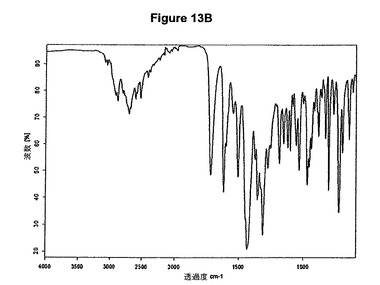
【図13B】塩酸モキシフロキサシンメチレンジクロリド溶媒和物赤外スペクトルである。
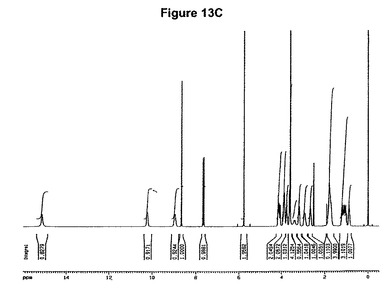
【図13C】塩酸モキシフロキサシンメチレンジクロリド溶媒和物の1H−NMRスペクトル(DMSO−d6,TMS)である。
【図14A】塩酸モキシフロキサシン無水IV形のX線粉末回折図である。
【図14B】塩酸モキシフロキサシン無水IV形の赤外スペクトルである。
【図15A】塩酸モキシフロキサシン酢酸溶媒和物のX線粉末回折図である。
【図15B】塩酸モキシフロキサシン酢酸溶媒和物の赤外スペクトルである。
【図16C】塩酸モキシフロキサシン酢酸溶媒和物の1H−NMRスペクトル(DMSO−d6,TMS)である。
【図16A】塩酸モキシフロキサシンニトロメタン溶媒和物のX線粉末回折図である。
【図16B】塩酸モキシフロキサシンニトロメタン溶媒和物の赤外スペクトルである。
【技術分野】
【0001】
本発明は、医薬化合物またはそれらの中間体のハロゲン化水素酸塩の新規な製造および結晶化方法に関する。本発明方法によれば、確定した結晶構造から成る純粋形態のハロゲン化水素酸塩が確実にかつ良好な収率で得られる。本発明方法は工業的用途に特に好適である。
【背景技術】
【0002】
医薬化合物またはそれらの中間体の塩酸塩は一般に塩基またはその塩の溶液を塩化水素によって酸性化することによって製造され、その場合、塩化水素水溶液もしくは塩化水素ガスが使用されるかまたはHClの有機溶媒溶液が使用される。塩酸水溶液の添加による塩酸塩の製造は簡単な方法であり、塩酸を36%(w/w)水溶液として使用するのが便利である。典型的な手順は、有機塩基を溶媒に溶解し、計算量または過剰量の濃HClを添加し、結晶化が生じないならばジエチルエーテルのような有機溶媒を徐々に添加することによって結晶化を誘発できる。しかしながら、塩酸塩が水溶性のため、塩酸水溶液による塩形成には低収率という特徴がしばしば見られる。さらに、無水塩形態が望まれる場合には概して塩酸水溶液の使用は適当でない。水が固体結晶質生成物の形成および単離を妨害するならば、シリンダーから無水ガスを供給するかまたはジエチルエーテルのような無水非プロトン性溶媒中のHClガスを使用することが可能である。しかしながら、大量の塩化水素ガスを使用する代替方法は設備費の高騰および気体処理に伴う典型的な危険を招く。
【0003】
単一結晶構造から成る純粋形態の塩酸塩を製造することは非常に多くの場合に重要である。医薬基質(substance)にはこれが特に重要である。何故ならば、特に固体経口剤形配合物の場合には、結晶構造の変化が医薬製品の溶解、製造適性および安定性を左右することが有り得るからである。しかしながら所望の塩酸塩形態の優先的な形成は結晶化動力学に依存し、容易にコントロールできない。ときには塩化水素ガスを一定流量で一定時間供給することが必要であり、ガス流動中、さらに生成物の濾過中にも温度を一定に維持する必要があり、これが処理を極めて難しいものにする。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の目的は、医薬化合物またはそれらの中間体の塩酸塩、臭化水素酸塩またはヨウ化水素酸塩を確定した結晶構造から成る純粋形態で再現可能に製造するための適当な方法を提供することである。
【課題を解決するための手段】
【0005】
本発明は、医薬化合物またはそれらの中間体のハロゲン化水素酸塩の新規な製造および結晶化方法に関する。本発明の方法によれば、確定した結晶構造から成る純粋形態のハロゲン化水素酸塩を良好な収率で確実に得ることができる。本発明の方法は工業的用途に特に好適である。
【0006】
より特定的には本発明は、溶媒中で有機アミンにハロゲン化トリアルキルシリルを添加する段階を含む、有機アミンの結晶質ハロゲン化水素酸塩の製造方法に関する。有機アミンハは遊離塩基または酸付加塩の形態であり、酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である。
【0007】
本発明はさらに、塩酸モキシフロキサシンメチレンジクロリド溶媒和物、塩酸モキシフロキサシンの新規な無水IV形、塩酸モキシフロキサシンニトロメタン溶媒和物、塩酸モキシフロキサシン酢酸溶媒和物、モキシフロキサシンHClの無水IV形またはモキシフロキサシンHClの酢酸溶媒和物を有効量で含む医薬組成物、塩酸リネゾリドの新規な結晶質形、塩酸プラスグレルアセトニトリル溶媒和物および塩酸ラロキシフェンテトラヒドロフラン溶媒和物に関する。すべての新規な溶媒和物または塩が本発明の方法によって得られる。本発明はさらに、有機アミンの結晶質ハロゲン化水素酸塩の製造方法におけるハロゲン化トリアルキルシリルの使用に関する。
【図面の簡単な説明】
【0008】
【図1A】無水塩酸ミコフェノラートモフェチルのX線粉末回折図である。
【図1B】無水塩酸ミコフェノラートモフェチルの赤外スペクトルである。
【図1C】無水塩酸ミコフェノラートモフェチルのDSC曲線である。
【図2A】塩酸ベンラファキシンI形のX線粉末回折図である。
【図2B】塩酸ベンラファキシンI形の赤外スペクトルである。
【図3A】塩酸ベンラファキシンII形のX線粉末回折図である。
【図3B】塩酸ベンラファキシンII形の赤外スペクトルである。
【図4A】塩酸セルトラリンII形のX線粉末回折図である。
【図4B】塩酸セルトラリンII形の赤外スペクトルである。
【図5A】塩酸セルトラリンI形のX線粉末回折図である。
【図5B】塩酸セルトラリンI形の赤外スペクトルである。
【図6A】塩酸ドネペジルII形のX線粉末回折図である。
【図6B】塩酸ドネペジルII形の赤外スペクトルである。
【図7A】塩酸ドネペジルIII形のX線粉末回折図である。
【図7B】塩酸ドネペジルIII形の赤外スペクトルである。
【図8A】塩酸テルビナフィンのX線粉末回折図である。
【図8B】塩酸テルビナフィンの赤外スペクトルである。
【図9A】塩酸シナカルセットのX線粉末回折図である。
【図9B】塩酸シナカルセットの赤外スペクトルである。
【図10A】臭化水素酸シタロプラムのX線粉末回折図である。
【図10B】臭化水素酸シタロプラムの赤外スペクトルである。
【図11A】塩酸アリピプラゾールA形のX線粉末回折図である。
【図11B】塩酸アリピプラゾールA形の赤外スペクトルである。
【図12A】モノ塩酸プラミペキソールのX線粉末回折図である。
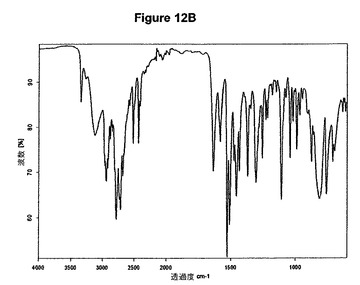
【図12B】モノ塩酸プラミペキソールの赤外スペクトルである。
【図13A】塩酸モキシフロキサシンメチレンジクロリド溶媒和物のX線粉末回折図である。
【図13B】塩酸モキシフロキサシンメチレンジクロリド溶媒和物赤外スペクトルである。
【図13C】塩酸モキシフロキサシンメチレンジクロリド溶媒和物の1H−NMRスペクトル(DMSO−d6,TMS)である。
【図14A】塩酸モキシフロキサシン無水IV形のX線粉末回折図である。
【図14B】塩酸モキシフロキサシン無水IV形の赤外スペクトルである。
【図15A】塩酸モキシフロキサシン酢酸溶媒和物のX線粉末回折図である。
【図15B】塩酸モキシフロキサシン酢酸溶媒和物の赤外スペクトルである。
【図16C】塩酸モキシフロキサシン酢酸溶媒和物の1H−NMRスペクトル(DMSO−d6,TMS)である。
【図16A】塩酸モキシフロキサシンニトロメタン溶媒和物のX線粉末回折図である。
【図16B】塩酸モキシフロキサシンニトロメタン溶媒和物の赤外スペクトルである。
【特許請求の範囲】
【請求項1】
有機アミンの結晶質ハロゲン化水素酸塩の製造方法であり、溶媒中の有機アミンにハロゲン化トリアルキルシリルが添加され、有機アミンは遊離塩基または酸付加塩の形態であり、有機アミンが酸付加塩の形態であるとき酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である方法。
【請求項2】
方法が、
(a)有機アミンをプロトン性溶媒中に溶解または懸濁させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶を形成させる段階および
(d)形成された結晶を収集する段階
を含む請求項1に記載の方法。
【請求項3】
段階(a)のプロトン性溶媒はヒドロキシル基またはカルボキシル基を含む化合物であり、特にプロトン性溶媒が芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトンまたは芳香族もしくは脂肪族カルボン酸であり、より特定的にはプロトン性溶媒がC1−C6アルキルアルコール、ギ酸または酢酸である請求項2に記載の方法。
【請求項4】
段階(b)のハロゲン化トリアルキルシリルが、トリメチルシリルクロリド、トリメチルシリルブロミドまたはトリメチルシリルヨージド、特にトリメチルシリルクロリドである請求項2または3に記載の方法。
【請求項5】
方法が、
(a)有機アミンを非プロトン性溶媒中に溶解または懸濁させる段階
(b)少なくとも1当量のプロトン性溶媒を添加する段階
(c)ハロゲン化トリアルキルシリルを添加する段階
(d)結晶を形成させる段階および
(e)形成された結晶を収集する段階
を含む請求項1の方法。
【請求項6】
段階(b)のプロトン性溶媒はヒドロキシル基またはカルボキシル基を含む化合物であり、特にプロトン性溶媒が芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトンまたは芳香族もしくは脂肪族カルボン酸であり、より特定的にはプロトン性溶媒がC1−C6アルキルアルコール、ギ酸または酢酸である請求項5の方法。
【請求項7】
段階(c)のハロゲン化トリアルキルシリルが、トリメチルシリルクロリド、トリメチルシリルブロミドまたはトリメチルシリルヨージド、特にトリメチルシリルクロリドである請求項5または6の方法。
【請求項8】
方法が、
(a)有機アミンの酸付加塩を溶媒中に溶解、懸濁または生成させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶を形成させる段階および
(d)形成された結晶を収集する段階
を含む請求項1の方法。
【請求項9】
段階(a)の有機アミンの酸付加塩は、共役酸が塩酸よりも弱い酸であり好ましくは有機酸である酸を、アミンの溶液またはスラリーに添加することによってその場に生成される請求項8の方法。
【請求項10】
段階(a)の有機酸が置換されているか置換されていないアルカン酸、芳香族カルボン酸、ジカルボン酸またはクエン酸から選択される請求項8または9の方法。
【請求項11】
有機酸が酢酸である請求項10の方法。
【請求項12】
有機アミンが医薬的に活性な化合物、特にヒト用薬剤である請求項1から10のいずれか一項の方法。
【請求項13】
薬物が一級、二級、三級または四級アミノ基を含み、特に薬物が抗欝剤、特にセロトニン再取り込みインヒビターセルトラリン、デュロキセチン、ベンラファキシンもしくはシタロプラム、精神向性薬、特にドネペジル、統合失調症治療薬、特に神経遮断薬アリピプラゾール、筋肉弛緩薬、特に鎮痙薬メマンチン、免疫抑制薬、特にミコフェノラートモフェチル、抗真菌薬、特にテルビナフィン、抗菌剤、特に例えばモキシフロキサシンもしくはオキサゾリジノンリネゾリドとしてのキノロン、カルシウム模擬薬、特にシナカルセット、ドーパミンアゴニスト、特にD2−受容体アゴニストプラミペキソール、抗肥満薬、特にリボナバント、抗血栓症薬、特にクロピドグレルおよびプラスグレル、抗骨粗鬆症薬、特にラロキシフェン、鎮痙剤、特にダリフェナシン、男性勃起機能不全治療薬、特にバルデナフィル、抗糖尿病薬、特にDPP−IVインヒビターシタグリプチンおよび抗新生物薬、特にエルロチニブからなる群から選択される、請求項12の方法。
【請求項14】
特に段階(a)においてミコフェノラートモフェチルを酢酸エチル、アセトニトリルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する結晶質無水形の塩酸ミコフェノラートモフェチルを製造するための請求項9に記載の方法。
【請求項15】
特に段階(a)においてベンラファキシンを酢酸エチルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ベンラファキシンI形を製造するための請求項9に記載の方法。
【請求項16】
特に段階(a)においてベンラファキシンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ベンラファキシンII形を製造するための請求項9に記載の方法。
【請求項17】
特に段階(a)においてセルトラリンをアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項9に記載の方法。
【請求項18】
特に段階(a)においてセルトラリンをメチルイソブチルケトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項9に記載の方法。
【請求項19】
特にセルトラリンをアセトニトリルに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項5に記載の方法。
【請求項20】
特に段階(a)においてアセトニトリル中のマンデル酸セルトラリンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項21】
特に段階(a)においてメチルエチルケトンまたはメチルイソブチルケトン中のマンデル酸セルトラリンの溶液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項22】
特に段階(a)においてアセトニトリル中のシュウ酸セルトラリンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項23】
特に段階(a)においてドネペジルをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ドネペジルIII形を製造するための請求項9に記載の方法。
【請求項24】
特に段階(a)においてテルビナフィンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸テルビナフィンを製造するための請求項9に記載の方法。
【請求項25】
特に段階(a)においてシナカルセットをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸シナカルセットを製造するための請求項9に記載の方法。
【請求項26】
特に段階(a)においてモキシフロキサシンをメチレンジクロリドに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシンメチレンジクロリド溶媒和物を製造するための請求項9に記載の方法。
【請求項27】
特に段階(a)においてモキシフロキサシンをニトロメタンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシンニトロメタン溶媒和物を製造するための請求項9に記載の方法。
【請求項28】
特に段階(a)においてモキシフロキサシンを酢酸に溶解させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシン酢酸溶媒和物を製造するための請求項2に記載の方法。
【請求項29】
特に段階(a)においてモキシフロキサシンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシン酢酸溶媒和物を製造するための請求項9に記載の方法。
【請求項30】
特に段階(a)においてシタロプラムをイソプロパノールに溶解させ、ならびに段階(b)においてトリメチルシリルブロミドを使用する臭化水素酸シタロプラムを製造するための請求項2に記載の方法。
【請求項31】
特に段階(a)においてシタロプラムを酢酸エチル、アセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルブロミドを使用する臭化水素酸シタロプラムを製造するための請求項9に記載の方法。
【請求項32】
特にアリピプラゾールをメチレンジクロリドに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルクロロシランを使用する塩酸アリピプラゾールを製造するための請求項5に記載の方法。
【請求項33】
特にプラミペキソールをアセトニトリルに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルクロロシランを使用するモノ塩酸プラミペキソールを製造するための請求項5に記載の方法。
【請求項34】
特に段階(a)においてデュロキセチンを酢酸エチルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルクロリドを使用する塩酸デュロキセチンを製造するための請求項9に記載の方法。
【請求項35】
特に段階(a)においてリネゾリドを酢酸エチルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルクロリドを使用する塩酸リネゾリドを製造するための請求項9に記載の方法。
【請求項36】
特に段階(a)においてメマンチンを酢酸エチルに溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸メマンチンを製造するための請求項5に記載の方法。
【請求項37】
有機アミンの結晶質ハロゲン化水素酸塩を製造するためのハロゲン化トリアルキルシリルの使用。
【請求項38】
有機アミンの結晶質ハロゲン化水素酸塩が前記有機アミンの複数の既存のハロゲン化水素酸塩のうちの望ましいハロゲン化水素酸塩である請求項37に記載の使用。
【請求項39】
有機アミンの無水結晶質ハロゲン化水素酸塩を製造するための請求項37または38に記載の使用。
【請求項40】
有機アミンの結晶質ハロゲン化水素酸塩溶媒和物を製造するための請求項37から39のいずれか一項に記載の使用。
【請求項41】
段階(a)においてリモナバントを酢酸エチルもしくはアセトンに溶解させるか、または、アセトニトリルに懸濁させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸リモナバントI形を製造するための請求項5に記載の方法。
【請求項42】
段階(a)においてクロピドグレルを酢酸エチル、アセトン、アセトニトリルまたはトルエンに溶解させ、ならびに段階(b)でメタノールまたは酢酸および段階(c)でトリメチルシリルクロリドを使用する塩酸クロピドグレルI形を製造するための請求項5に記載の方法。
【請求項43】
段階(a)においてクロピドグレルを酢酸エチルに溶解させ、ならびに段階(b)で酢酸および段階(c)でトリメチルシリルブロミドを使用する臭化水素酸クロピドグレルA形を製造するための請求項5に記載の方法。
【請求項44】
段階(a)においてクロピドグレルをイソプロパノールに溶解させ、ならびに段階(b)においてトリメチルシリルブロミドを使用する臭化水素酸クロピドグレルA形を製造するための請求項2に記載の方法。
【請求項45】
段階(a)においてプラスグレルをアセトンに溶解させ、ならびに段階(b)で酢酸および段階(c)でトリメチルシリルクロリドを使用する塩酸プラスグレルA形を製造するための請求項5に記載の方法。
【請求項46】
段階(a)においてプラスグレルをアセトニトリルに溶解させ、ならびに段階(b)で酢酸を使用し、ならびに段階(c)でトリメチルシリルクロリドを使用する塩酸プラスグレルアセトニトリル溶媒和物を製造するための請求項5に記載の方法。
【請求項47】
段階(a)においてラロキシフェンをエタノールに懸濁させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンA形を製造するための請求項2に記載の方法。
【請求項48】
段階(a)でアセトニトリルまたはメチルイソブチルケトン中の乳酸ラロキシフェンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンA形を製造するための請求項8に記載の方法。
【請求項49】
段階(a)においてラロキシフェンをテトラヒドロフランに懸濁させ、ならびに段階(b)でメタノールを使用し、ならびに段階(c)でトリメチルシリルクロリドを使用する塩酸ラロキシフェンテトラヒドロフラン半溶媒和物を製造するための請求項5に記載の方法。
【請求項50】
段階(a)においてテトラヒドロフラン中の乳酸ラロキシフェンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンテトラヒドロフラン半溶媒和物を製造するための請求項8に記載の方法。
【請求項51】
段階(a)においてオランザピンをアセトンまたはアセトニトリルに懸濁させ、ならびに段階(b)でメタノールを使用し、ならびに段階(c)で少なくとも2当量のトリメチルシリルクロリドを使用するジ塩酸オランザピンI形を製造するための請求項5に記載の方法。
【請求項52】
段階(a)においてダリフェナシンをメチルエチルケトンに溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルブロミドを使用する臭化水素酸ダリフェナシンを製造するための請求項5に記載の方法。
【請求項53】
段階(a)においてシタグリプチンをジエチルエーテルとメチレンジクロリドとの混合物に溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する非晶質形の塩酸シタグリプチンを製造するための請求項5に記載の方法。
【請求項54】
段階(a)においてバルデナフィルをジエチルエーテルとメチレンジクロリドとの混合物に懸濁させ、ならびに段階(b)でメタノールおよび段階(c)で少なくとも2当量のトリメチルシリルクロリドを使用するジ塩酸バルデナフィルを製造するための請求項5に記載の方法。
【請求項55】
段階(a)においてエルロチニブをイソプロパノールに懸濁させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸エルロチニブA形を製造するための請求項2に記載の方法。
【請求項1】
有機アミンの結晶質ハロゲン化水素酸塩の製造方法であり、溶媒中の有機アミンにハロゲン化トリアルキルシリルが添加され、有機アミンは遊離塩基または酸付加塩の形態であり、有機アミンが酸付加塩の形態であるとき酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である方法。
【請求項2】
方法が、
(a)有機アミンをプロトン性溶媒中に溶解または懸濁させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶を形成させる段階および
(d)形成された結晶を収集する段階
を含む請求項1に記載の方法。
【請求項3】
段階(a)のプロトン性溶媒はヒドロキシル基またはカルボキシル基を含む化合物であり、特にプロトン性溶媒が芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトンまたは芳香族もしくは脂肪族カルボン酸であり、より特定的にはプロトン性溶媒がC1−C6アルキルアルコール、ギ酸または酢酸である請求項2に記載の方法。
【請求項4】
段階(b)のハロゲン化トリアルキルシリルが、トリメチルシリルクロリド、トリメチルシリルブロミドまたはトリメチルシリルヨージド、特にトリメチルシリルクロリドである請求項2または3に記載の方法。
【請求項5】
方法が、
(a)有機アミンを非プロトン性溶媒中に溶解または懸濁させる段階
(b)少なくとも1当量のプロトン性溶媒を添加する段階
(c)ハロゲン化トリアルキルシリルを添加する段階
(d)結晶を形成させる段階および
(e)形成された結晶を収集する段階
を含む請求項1の方法。
【請求項6】
段階(b)のプロトン性溶媒はヒドロキシル基またはカルボキシル基を含む化合物であり、特にプロトン性溶媒が芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトンまたは芳香族もしくは脂肪族カルボン酸であり、より特定的にはプロトン性溶媒がC1−C6アルキルアルコール、ギ酸または酢酸である請求項5の方法。
【請求項7】
段階(c)のハロゲン化トリアルキルシリルが、トリメチルシリルクロリド、トリメチルシリルブロミドまたはトリメチルシリルヨージド、特にトリメチルシリルクロリドである請求項5または6の方法。
【請求項8】
方法が、
(a)有機アミンの酸付加塩を溶媒中に溶解、懸濁または生成させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶を形成させる段階および
(d)形成された結晶を収集する段階
を含む請求項1の方法。
【請求項9】
段階(a)の有機アミンの酸付加塩は、共役酸が塩酸よりも弱い酸であり好ましくは有機酸である酸を、アミンの溶液またはスラリーに添加することによってその場に生成される請求項8の方法。
【請求項10】
段階(a)の有機酸が置換されているか置換されていないアルカン酸、芳香族カルボン酸、ジカルボン酸またはクエン酸から選択される請求項8または9の方法。
【請求項11】
有機酸が酢酸である請求項10の方法。
【請求項12】
有機アミンが医薬的に活性な化合物、特にヒト用薬剤である請求項1から10のいずれか一項の方法。
【請求項13】
薬物が一級、二級、三級または四級アミノ基を含み、特に薬物が抗欝剤、特にセロトニン再取り込みインヒビターセルトラリン、デュロキセチン、ベンラファキシンもしくはシタロプラム、精神向性薬、特にドネペジル、統合失調症治療薬、特に神経遮断薬アリピプラゾール、筋肉弛緩薬、特に鎮痙薬メマンチン、免疫抑制薬、特にミコフェノラートモフェチル、抗真菌薬、特にテルビナフィン、抗菌剤、特に例えばモキシフロキサシンもしくはオキサゾリジノンリネゾリドとしてのキノロン、カルシウム模擬薬、特にシナカルセット、ドーパミンアゴニスト、特にD2−受容体アゴニストプラミペキソール、抗肥満薬、特にリボナバント、抗血栓症薬、特にクロピドグレルおよびプラスグレル、抗骨粗鬆症薬、特にラロキシフェン、鎮痙剤、特にダリフェナシン、男性勃起機能不全治療薬、特にバルデナフィル、抗糖尿病薬、特にDPP−IVインヒビターシタグリプチンおよび抗新生物薬、特にエルロチニブからなる群から選択される、請求項12の方法。
【請求項14】
特に段階(a)においてミコフェノラートモフェチルを酢酸エチル、アセトニトリルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する結晶質無水形の塩酸ミコフェノラートモフェチルを製造するための請求項9に記載の方法。
【請求項15】
特に段階(a)においてベンラファキシンを酢酸エチルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ベンラファキシンI形を製造するための請求項9に記載の方法。
【請求項16】
特に段階(a)においてベンラファキシンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ベンラファキシンII形を製造するための請求項9に記載の方法。
【請求項17】
特に段階(a)においてセルトラリンをアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項9に記載の方法。
【請求項18】
特に段階(a)においてセルトラリンをメチルイソブチルケトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項9に記載の方法。
【請求項19】
特にセルトラリンをアセトニトリルに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項5に記載の方法。
【請求項20】
特に段階(a)においてアセトニトリル中のマンデル酸セルトラリンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項21】
特に段階(a)においてメチルエチルケトンまたはメチルイソブチルケトン中のマンデル酸セルトラリンの溶液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項22】
特に段階(a)においてアセトニトリル中のシュウ酸セルトラリンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸セルトラリンII形を製造するための請求項8に記載の方法。
【請求項23】
特に段階(a)においてドネペジルをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ドネペジルIII形を製造するための請求項9に記載の方法。
【請求項24】
特に段階(a)においてテルビナフィンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸テルビナフィンを製造するための請求項9に記載の方法。
【請求項25】
特に段階(a)においてシナカルセットをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸シナカルセットを製造するための請求項9に記載の方法。
【請求項26】
特に段階(a)においてモキシフロキサシンをメチレンジクロリドに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシンメチレンジクロリド溶媒和物を製造するための請求項9に記載の方法。
【請求項27】
特に段階(a)においてモキシフロキサシンをニトロメタンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシンニトロメタン溶媒和物を製造するための請求項9に記載の方法。
【請求項28】
特に段階(a)においてモキシフロキサシンを酢酸に溶解させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシン酢酸溶媒和物を製造するための請求項2に記載の方法。
【請求項29】
特に段階(a)においてモキシフロキサシンをアセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸モキシフロキサシン酢酸溶媒和物を製造するための請求項9に記載の方法。
【請求項30】
特に段階(a)においてシタロプラムをイソプロパノールに溶解させ、ならびに段階(b)においてトリメチルシリルブロミドを使用する臭化水素酸シタロプラムを製造するための請求項2に記載の方法。
【請求項31】
特に段階(a)においてシタロプラムを酢酸エチル、アセトンまたはアセトニトリルに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルブロミドを使用する臭化水素酸シタロプラムを製造するための請求項9に記載の方法。
【請求項32】
特にアリピプラゾールをメチレンジクロリドに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルクロロシランを使用する塩酸アリピプラゾールを製造するための請求項5に記載の方法。
【請求項33】
特にプラミペキソールをアセトニトリルに溶解させ、ならびに段階(b)でn−ブタノールおよび段階(c)でトリメチルクロロシランを使用するモノ塩酸プラミペキソールを製造するための請求項5に記載の方法。
【請求項34】
特に段階(a)においてデュロキセチンを酢酸エチルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルクロリドを使用する塩酸デュロキセチンを製造するための請求項9に記載の方法。
【請求項35】
特に段階(a)においてリネゾリドを酢酸エチルまたはアセトンに溶解させ、および酢酸を有機酸として使用し、ならびに段階(c)においてトリメチルシリルクロリドを使用する塩酸リネゾリドを製造するための請求項9に記載の方法。
【請求項36】
特に段階(a)においてメマンチンを酢酸エチルに溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸メマンチンを製造するための請求項5に記載の方法。
【請求項37】
有機アミンの結晶質ハロゲン化水素酸塩を製造するためのハロゲン化トリアルキルシリルの使用。
【請求項38】
有機アミンの結晶質ハロゲン化水素酸塩が前記有機アミンの複数の既存のハロゲン化水素酸塩のうちの望ましいハロゲン化水素酸塩である請求項37に記載の使用。
【請求項39】
有機アミンの無水結晶質ハロゲン化水素酸塩を製造するための請求項37または38に記載の使用。
【請求項40】
有機アミンの結晶質ハロゲン化水素酸塩溶媒和物を製造するための請求項37から39のいずれか一項に記載の使用。
【請求項41】
段階(a)においてリモナバントを酢酸エチルもしくはアセトンに溶解させるか、または、アセトニトリルに懸濁させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する塩酸リモナバントI形を製造するための請求項5に記載の方法。
【請求項42】
段階(a)においてクロピドグレルを酢酸エチル、アセトン、アセトニトリルまたはトルエンに溶解させ、ならびに段階(b)でメタノールまたは酢酸および段階(c)でトリメチルシリルクロリドを使用する塩酸クロピドグレルI形を製造するための請求項5に記載の方法。
【請求項43】
段階(a)においてクロピドグレルを酢酸エチルに溶解させ、ならびに段階(b)で酢酸および段階(c)でトリメチルシリルブロミドを使用する臭化水素酸クロピドグレルA形を製造するための請求項5に記載の方法。
【請求項44】
段階(a)においてクロピドグレルをイソプロパノールに溶解させ、ならびに段階(b)においてトリメチルシリルブロミドを使用する臭化水素酸クロピドグレルA形を製造するための請求項2に記載の方法。
【請求項45】
段階(a)においてプラスグレルをアセトンに溶解させ、ならびに段階(b)で酢酸および段階(c)でトリメチルシリルクロリドを使用する塩酸プラスグレルA形を製造するための請求項5に記載の方法。
【請求項46】
段階(a)においてプラスグレルをアセトニトリルに溶解させ、ならびに段階(b)で酢酸を使用し、ならびに段階(c)でトリメチルシリルクロリドを使用する塩酸プラスグレルアセトニトリル溶媒和物を製造するための請求項5に記載の方法。
【請求項47】
段階(a)においてラロキシフェンをエタノールに懸濁させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンA形を製造するための請求項2に記載の方法。
【請求項48】
段階(a)でアセトニトリルまたはメチルイソブチルケトン中の乳酸ラロキシフェンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンA形を製造するための請求項8に記載の方法。
【請求項49】
段階(a)においてラロキシフェンをテトラヒドロフランに懸濁させ、ならびに段階(b)でメタノールを使用し、ならびに段階(c)でトリメチルシリルクロリドを使用する塩酸ラロキシフェンテトラヒドロフラン半溶媒和物を製造するための請求項5に記載の方法。
【請求項50】
段階(a)においてテトラヒドロフラン中の乳酸ラロキシフェンの懸濁液を使用し、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸ラロキシフェンテトラヒドロフラン半溶媒和物を製造するための請求項8に記載の方法。
【請求項51】
段階(a)においてオランザピンをアセトンまたはアセトニトリルに懸濁させ、ならびに段階(b)でメタノールを使用し、ならびに段階(c)で少なくとも2当量のトリメチルシリルクロリドを使用するジ塩酸オランザピンI形を製造するための請求項5に記載の方法。
【請求項52】
段階(a)においてダリフェナシンをメチルエチルケトンに溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルブロミドを使用する臭化水素酸ダリフェナシンを製造するための請求項5に記載の方法。
【請求項53】
段階(a)においてシタグリプチンをジエチルエーテルとメチレンジクロリドとの混合物に溶解させ、ならびに段階(b)でメタノールおよび段階(c)でトリメチルシリルクロリドを使用する非晶質形の塩酸シタグリプチンを製造するための請求項5に記載の方法。
【請求項54】
段階(a)においてバルデナフィルをジエチルエーテルとメチレンジクロリドとの混合物に懸濁させ、ならびに段階(b)でメタノールおよび段階(c)で少なくとも2当量のトリメチルシリルクロリドを使用するジ塩酸バルデナフィルを製造するための請求項5に記載の方法。
【請求項55】
段階(a)においてエルロチニブをイソプロパノールに懸濁させ、ならびに段階(b)においてトリメチルシリルクロリドを使用する塩酸エルロチニブA形を製造するための請求項2に記載の方法。
【図16C】塩酸モキシフロキサシンニトロメタン溶媒和物の1H−NMRスペクトル(DMSO−d6,TMS)である。
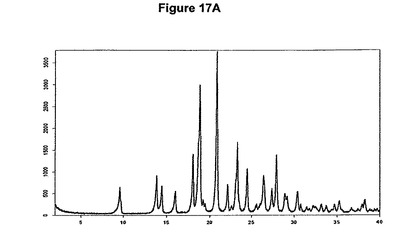
【図17A】塩酸デュロキセチンのX線粉末回折図である。
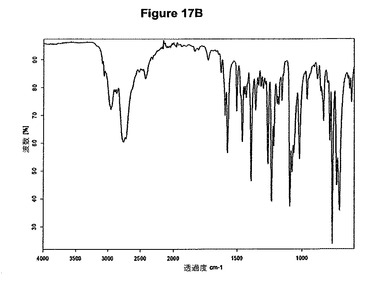
【図17B】塩酸デュロキセチンの赤外スペクトルである。
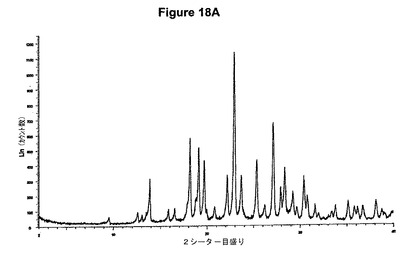
【図18A】塩酸リネゾリドのX線粉末回折図である。
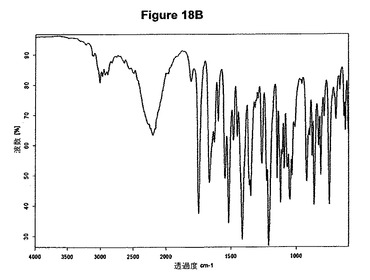
【図18B】塩酸リネゾリドの赤外スペクトルである。
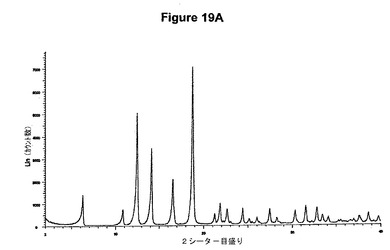
【図19A】塩酸メマンチンのX線粉末回折図である。
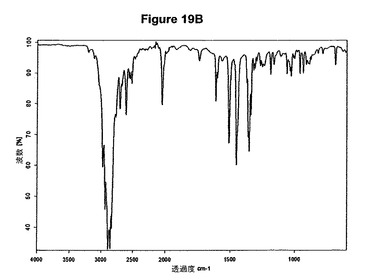
【図19B】塩酸メマンチンの赤外スペクトルである。
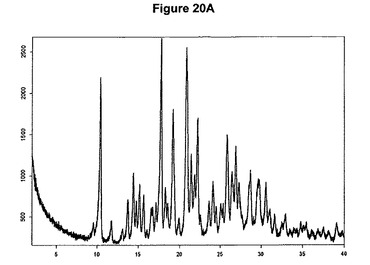
【図20A】塩酸リモナバントI形のX線粉末回折図である。
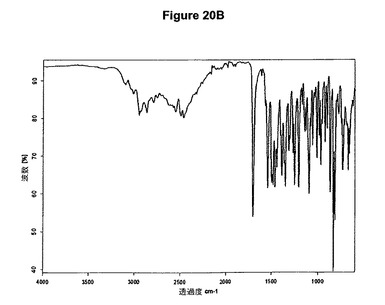
【図20B】塩酸リモナバントI形の赤外スペクトルである。
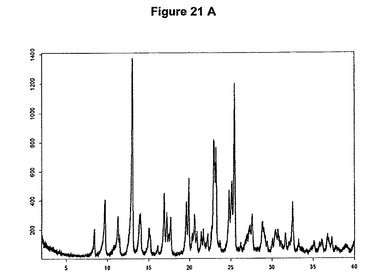
【図21A】塩酸クロピドグレルI形のX線粉末回折図である。
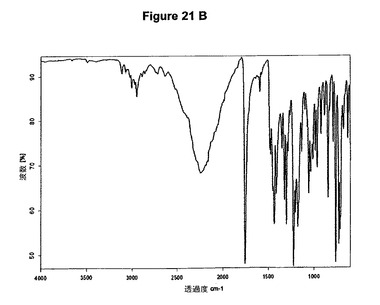
【図21B】塩酸クロピドグレルI形の赤外スペクトルである。
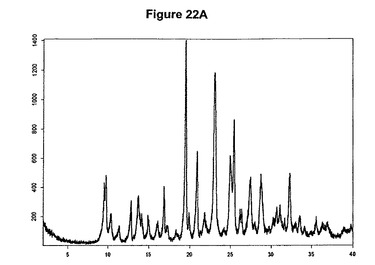
【図22A】臭化水素酸クロピドグレルA形のX線粉末回折図である。
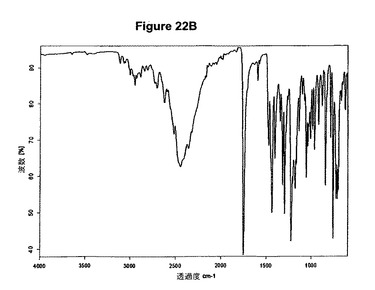
【図22B】臭化水素酸クロピドグレルA形の赤外スペクトルである。
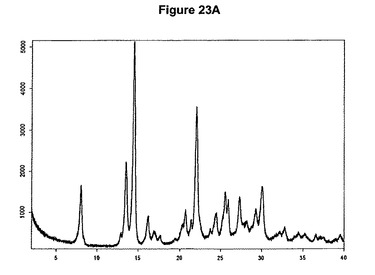
【図23A】塩酸プラスグレルB形のX線粉末回折図である。
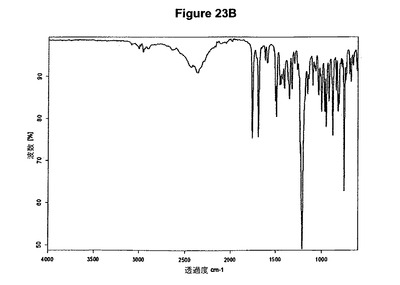
【図23B】塩酸プラスグレルB形の赤外スペクトルである。
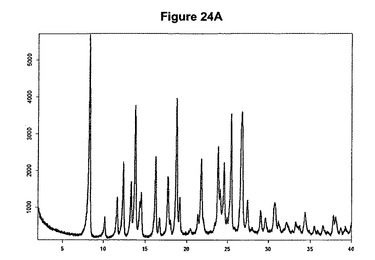
【図24A】塩酸プラスグレルアセトニトリル溶媒和物のX線粉末回折図である。
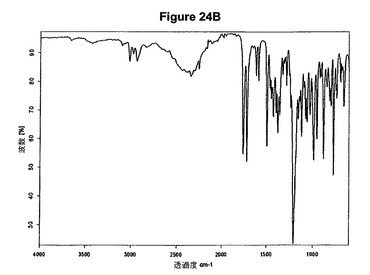
【図24B】塩酸プラスグレルアセトニトリル溶媒和物の赤外スペクトルである。
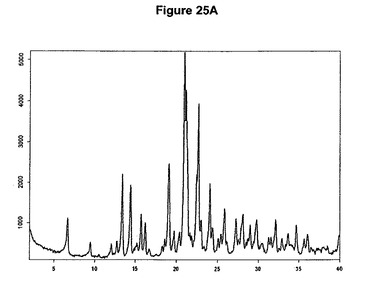
【図25A】塩酸ラロキシフェンA形のX線粉末回折図である。
【図25B】塩酸ラロキシフェンA形の赤外スペクトルである。
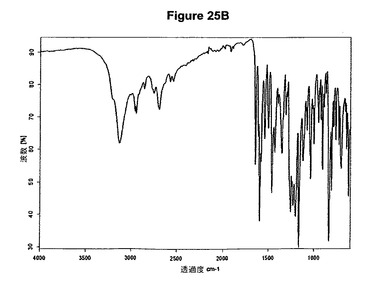
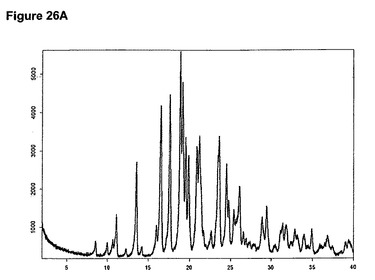
【図26A】塩酸ラロキシフェンテトラヒドロフラン溶媒和物のX線粉末回折図である。
【図26B】塩酸ラロキシフェンテトラヒドロフラン溶媒和物の赤外スペクトルである。
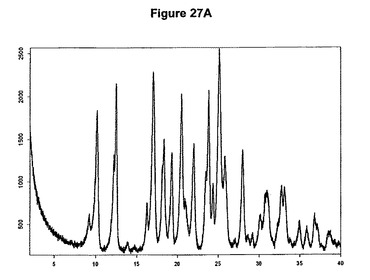
【図27A】ジ塩酸オランザピンI形のX線粉末回折図である。
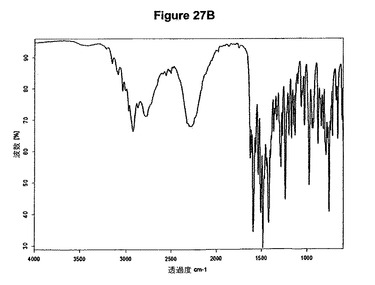
【図27B】ジ塩酸オランザピンI形の赤外スペクトルである。
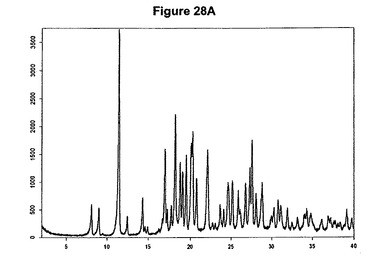
【図28A】臭化水素酸ダリフェナシンのX線粉末回折図である。
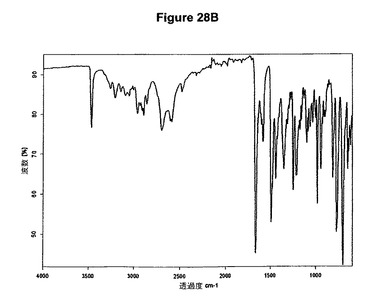
【図28B】臭化水素酸ダリフェナシンの赤外スペクトルである。
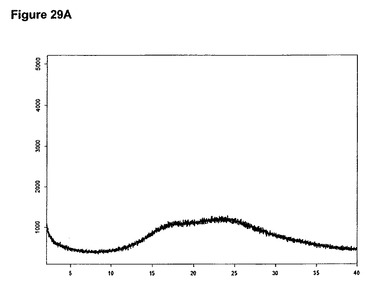
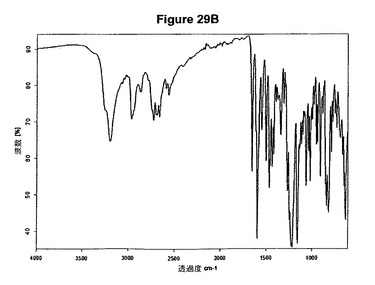
【図29A】塩酸シタグリプチン無水形のX線粉末回折図である。
【図29B】塩酸シタグリプチン無水形の赤外スペクトルである。
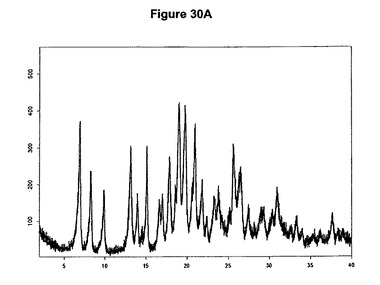
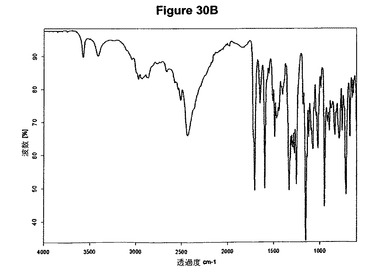
【図30A】ジ塩酸バルデナフィルのX線粉末回折図である。
【図30B】ジ塩酸バルデナフィルの赤外スペクトルである。
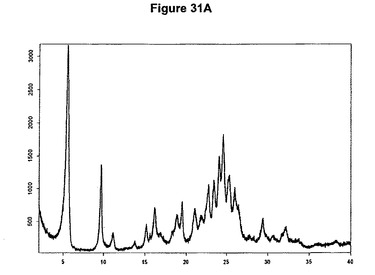
【図31A】塩酸エルロチニブA形のX線粉末回折図である。
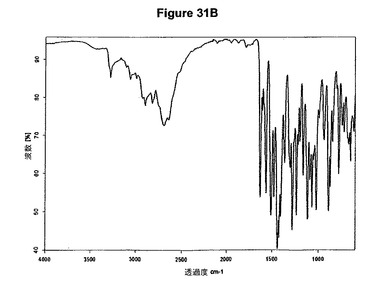
【図31B】塩酸エルロチニブA形の赤外スペクトルである。
【発明を実施するための形態】
【0009】
本発明は、ハロゲン化トリアルキルシリルが塩基性薬剤基質のハロゲン化水素酸塩または塩基性中間体のハロゲン化水素酸塩の製造に、特に無水条件が必要な場合および/または純粋形態の塩の明確に確定した結晶構造を製造しなければならない場合に、極めて好適であるという知見に関する。
【0010】
従って本発明は、ハロゲン化トリアルキルシリルを溶媒中で有機アミンに添加する段階を含む、有機アミンの結晶質ハロゲン化物の製造方法に関する。該有機アミンは遊離塩基または酸付加塩の形態であり、酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である。
【0011】
本発明方法に適した溶媒は、プロトン性溶媒、または、有機アミンに比べて少なくとも1当量のプロトン性溶媒に組合せた非プロトン性溶媒である。プロトン性溶媒は、シリル化可能溶媒、すなわち、ハロゲン化トリアルキルシリルに添加されたときにハロゲン化水素酸塩をその場に生成する能力を有している溶媒である。適当なプロトン性溶媒は例えば、脂肪族アルコールもしくは芳香族アルコール、シラノール、エノール化可能なケトン、または、脂肪族もしくは芳香族カルボン酸(carbonic acid)である。非プロトン性溶媒は、アミンを溶解もしくは懸濁させることができるか、または、形成されるハロゲン化水素酸塩を確定した結晶構造に導くことができるシラン化に対して不活性の溶媒である。
【0012】
適当な非プロトン性溶媒は例えば、エステル特に酢酸エチル、ニトリル特にアセトニトリル、ケトン特にアセトン、エーテル特に第三級ブチルメチルエーテル、ハロゲン化溶媒特にジクロロメタン、芳香族溶媒特にトルエン、アルカン特にヘキサン、および、ニトロアルケン特にニトロメタンである。
【0013】
本発明に含意される有機アミンは、第一級、第二級、第三級または第四級アミノ基と3よりも多い数の炭素−炭素結合とを含み、80Daを上回る分子量好ましくは100Daから5000Daの範囲の分子量を有している有機化合物である。
【0014】
1つの実施態様において本発明の方法は、
(a)プロトン性溶媒中に有機アミンの遊離塩基を溶解させるかまたは有機アミンの遊離塩基を好ましくは撹拌を伴って懸濁させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶(有機アミンのハロゲン化水素酸塩の結晶)を形成させる段階
(d)形成された結晶を収集する段階
を含む。
【0015】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸モキシフロキサシン酢酸溶媒和物、臭化水素酸シタロプラム、臭化水素酸クロピドグレル、塩酸ラロキシフェンA形または塩酸エルロチニブA形の製造に使用される。
【0016】
段階(b)中にハロゲンシランはシリル化剤としておそらくはプロトン性溶媒と直ちに反応しこれによりハロゲン化水素酸をその場に生成し、生成したHCl、HBrまたはHIが遊離塩基をハロゲン化水素酸塩に変換する。
【0017】
段階(d)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0018】
好ましいハロゲン化トリアルキルシリルは、3つのアルキル残基が同一であり、特にそれらがメチル、エチル、プロピル、ブチルまたはイソプロピルを表すものである。最も好ましいハロゲン化トリアルキルは、トリメチルクロロシラン、トリメチルブロモシランおよびトリメチルヨードシランである。
【0019】
本発明において使用した“プロトン性溶媒”という用語は、添加すべき有機アミンに比べてプロトン性溶媒の合計量が1分子当量を上回るような、特にプロトン性溶媒の合計量が添加すべき有機アミンの約1分子当量であるような溶媒混合物から成る溶媒系を包含する。
【0020】
段階(a)で使用される好ましいプロトン性溶媒は、ヒドロキシル基またはカルボキシル基を含む溶媒である。特に好ましいプロトン性溶媒は、芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトン、または、芳香族もしくは脂肪族カルボン酸(carbonic acid)、または、それらの混合物から成る溶媒系である。より好ましいプロトン性溶媒はメタノール、エタノール、2−プロパノール(イソプロパノール)、ブタノール(例えばn−ブタノールおよびイソブタノール)のようなC1−C6アルキルアルコール、または、C1−C6アルキルカルボン酸特にギ酸もしくは酢酸である。ハロゲン化水素酸塩の乾燥を含む最終処理(workup)および単離は当業界で公知の方法を使用して行うとよい。
【0021】
別の実施態様において、有機化合物のハロゲン化水素酸塩の製造方法は、
(a)有機アミンの遊離塩基を非プロトン性溶媒に溶解させて溶液を形成するかまたは非プロトン性溶媒中の遊離塩基の懸濁液を撹拌する段階
(b)段階(a)で形成された溶液または懸濁液に少なくとも1当量特に約1当量のプロトン性溶媒、特に上記に定義したような好ましいプロトン性溶媒を添加する段階
(c)段階(b)の溶液または懸濁液を少なくとも1当量特に約1当量のハロゲン化トリアルキルシリル特に上記に定義したような好ましいハロゲン化トリアルキルシリルで処理して対応するハロゲン化水素酸塩を形成する段階
(d)有機アミンのハロゲン化水素酸塩の結晶を形成させる段階
(e)形成された結晶を収集する段階
を含む。
【0022】
段階(e)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0023】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸セルトラリンII形、塩酸アリピプラゾール、モノ塩酸プラミペキソール、塩酸メマンチン、塩酸リモナバントI形、塩酸クロピドグレルI形、臭化水素酸クロピドグレルA形、塩酸プラスグレルA形、塩酸プラスグレルアセトニトリル溶媒和物、塩酸ラロキシフェンテトラヒドロフラン半溶媒和物、ジ塩酸オランザピンI形、臭化水素酸ダリフェナシン、塩酸シタグリプチンまたはジ塩酸バルデナフィルの製造に使用される。
【0024】
非プロトン性溶媒が使用されるとき、有機アミンの遊離塩基自体がハロゲン化トリアルキルと反応してシリル化ハロゲン化水素を形成し、シリル化生成物がその後1当量以上のアルコールのようなプロトン性溶媒の添加によって加水分解できる。従って段階(b)と(c)との順序を入れ替えてもよい。また、最初にハロゲン化トリアルキルをプロトン性溶媒と混合してハロゲン化水素酸を生成し、次にこの混合物を溶解または懸濁した有機アミンに添加してもよい。
【0025】
別の実施態様において、有機化合物のハロゲン化水素酸塩の製造方法は、
(a)溶媒中で有機アミンの酸付加塩を溶解、懸濁または生成させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)有機アミンのハロゲン化水素酸塩の結晶を形成させる段階
(d)形成された結晶を収集する段階
を含む。
【0026】
段階(d)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0027】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸セルトラリンII形、塩酸ラロキシフェンA形または塩酸ラロキシフェンテトラヒドロフラン半溶媒和物の製造などに使用される。
【0028】
段階(a)において、有機アミンの酸付加塩は溶媒中の塩基の溶液または懸濁液に有機酸を添加することによってその場に生成できる。アミンの有機酸塩の生成に使用される有機酸は塩酸よりも弱い酸でなければならないので、好ましくは置換または未置換のアルカン酸、芳香族カルボン酸、ジカルボン酸またはクエン酸から成るグループから選択され、酢酸が特に好ましい。
【0029】
段階(a)において好ましくは、有機アミンの酸付加塩は有機アミンの溶液またはスラリーに酸を添加することによってその場に生成され、共役酸は塩酸よりも弱い酸であり、好ましくは有機酸である。
【0030】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、結晶質無水形の塩酸ミコフェノラートモフェチル、塩酸ベンラファキシンI形、塩酸ベンラファキシンII形、塩酸セルトラリンII形、塩酸ドネペジルIII形、塩酸テルビナフィン、塩酸シナカルセット、塩酸モキシフロキサシンメチレンジクロリド溶媒和物、塩酸モキシフロキサシンニトロメタン溶媒和物、塩酸モキシフロキサシン酢酸溶媒和物、臭化水素酸シタロプラム、塩酸デュロキセチンまたは塩酸リネゾリドの製造などに使用される。
【0031】
塩酸塩は入手容易性および生理的寛容性があるので塩基性薬剤の塩としてこれまで最も頻繁に選択されており、すべての薬剤の塩のほぼ半数が塩酸塩であり、以後の文節中ではすべてのハロゲン化水素酸塩の代表例の役割を果たす。
【0032】
ハロゲン化水素酸塩の形成はときには厳密な無水条件を必要とし、その場合には特に上述のような塩酸のその場に生成および塩形成を誘発するために塩化トリアルキルシリルを使用するのが有利である。またあるときは、塩酸を形成するために有機アミンに対して限定された化学論的量のHClが必要であり、この要件は上述のような塩化トリアルキルシリルの使用によって容易に充足できる。溶媒中に正確な計算量のハロゲン化水素酸が必要とされるときであっても、ハロゲン化トリアルキルシリルを使用するほうがシリンダーから供給される無水ガスを用いて溶液を製造するよりも容易なかつ優れた方法である。
【0033】
言い換えると、本発明の方法においてハロゲン化水素酸をその場に生成するためのハロゲン化トリアルキルシリルの使用は、塩形成中のハロゲン化水素酸と有機アミンとの化学量論比を極めて良好にコントロールできる。このことは、有機アミンの2つ以上のハロゲン化水素酸塩例えばハロゲン化モノ水素酸塩とハロゲン化ジ水素酸塩とが存在するときに特に有利である。何故ならこのような場合、生成すべきハロゲン化水素酸の量を選択することによって所望のハロゲン化水素酸塩が得られる方向に方法を誘導できるからである。例えば、モノハロゲン化モノ水素酸塩を得ることを望むが競合するジハロゲン化水素酸塩も存在する場合、1当量のハロゲン化水素酸だけがその場に生成されるようにハロゲン化トリアルキルシリルの添加量を限定し、モノハロゲン化水素酸塩が生成する方向に結晶化を誘導できる。
【0034】
本発明の方法の別の利点は、水の実質的な非存在下で処理することもでき、その結果として有機アミンのハロゲン化水素酸塩の無水形を得るのが可能なことである。さらに、ハロゲン化トリアルキルシリルの添加は極めて安定な段階であり、広い温度範囲内で行うことができ、多様な溶媒系に適合性なので、ハロゲン化トリアルキルシリルの添加は大抵の場合、所望の結晶形を得るために最適であることが判っているまさにその条件下で行うことができる。例えば、薬剤Xのモノ塩酸塩の多形相Aは溶媒Y中で低温で得られるが同じモノ塩酸塩の多形相Bは溶媒Z中で周囲温度で得られることが判っている場合、所望の形AまたはBを形成させるように条件を選択するだけでよい。この結晶化条件の選択を可能にする理由はまさに、ハロゲン化トリアルキルシリル添加段階の安定性にある。
【0035】
別の実施態様において、本発明の方法は第一級、第二級、第三級または第四級アミノ基をもつ薬剤のハロゲン化水素酸塩の生成に使用され、薬剤は特に、セロトニン再吸収インヒビターであるセルトラリン、デュロキセチン、ベンラファキシンおよびシタロプラムなどの抗鬱薬、特にドネペジルのような精神向性薬、特に神経遮断薬アリピプラゾールおよびセロトニン/ドパミンアンタゴニストであるオランザピンのような統合失調症治療薬、特に鎮痙薬メマンチンのような筋弛緩薬、特にミコフェノラートモフェチルのような免疫抑制薬、特にテルビナフィンのような抗真菌薬、特にキノロン例えばモキシフロキサシンまたはオキサゾリジノンリネゾリドのような抗菌薬、特にシナカルセットのようなカルシウム模擬薬、特にD2受容体アゴニストであるプラミペキソールのようなドパミンアゴニスト、リモナバントのような抗肥満薬、クロピドグレルおよびプラスグレルのような抗血栓症薬、特にラロキシフェンのような抗骨粗鬆症薬、ダリフェナシンのような鎮痙薬、バルデナフィルのような男性勃起機能不全治療薬、特にDPP−IVインヒビターであるシタグリプチンのような抗糖尿病薬、エルロチニブのような抗新生物薬から選択される。
【0036】
別の実施態様において、本発明は、本発明の方法による結晶質無水塩酸ミコフェノラートモフェチルの製造に関する。より詳細には方法は、(a)ミコフェノラートモフェチル塩基を酢酸エチル、アセトニトリルまたはアセトン、特に酢酸エチルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液をトリシリルアルキルクロロシラン、特に約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む。
【0037】
この方法によって、無水塩酸ミコフェノラートモフェチルが沈殿し、97%を上回る収率で単離できる。結晶質生成物のFT−IR、DSCおよびXRPDデータはWO95/07902に示されているIR、DSCおよびX線結晶学データに一致する。WO95/07902は、無水塩酸ミコフェノラートモフェチルが一水和物塩形のほぼ2倍増の溶解度を有しながらも一水和物塩形に特有の安定性を維持していると開示している。本発明の方法は塩酸一水和物の形成を阻止するので、無水形を製造するために塩酸ミコフェノラートモフェチル一水和物を加熱する必要がない。
【0038】
別の実施態様において、本発明は、方法に塩化水素ガスを使用しない本発明の方法による塩酸ベンラファキシンの製造、特に、純粋形態の塩酸ベンラファキシンI形またはII形の製造方法に関する。好ましい実施態様において、本発明は、塩酸ベンラファキシンI形の製造方法に関する。方法は、(a)ベンラファキシン塩基を酢酸エチルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液を約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む。この方法によって無水塩酸ベンラファキシンI形が沈殿し、89%を上回る収率で単離できる。結晶質生成物のXRPDデータはWO02/45658(Teva)に示されているI形およびWO02/36542(Ciba)に示されているB形のX線結晶学データに対応する。
【0039】
別の好ましい実施態様において、本発明は、(a)ベンラファキシン塩基を溶媒となるアセトンまたはアセトニトリルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液を約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む塩酸ベンラファキシンII形の製造方法に関する。この方法によって無水塩酸ベンラファキシンII形が得られる。結晶質生成物のXRPDデータはWO02/45658(Teva)に示されているII形およびWO02/36542(Ciba)に示されているC形のX線結晶学データに対応する。
【0040】
別の実施態様において、本発明は、本発明の方法による塩酸セルトラリンII形の製造に関する。塩酸セルトラリンII形は準安定形であり、通常は有機溶媒からの塩酸セルトラリンの急速結晶化によって製造される。しかしながら、II形の優先的な形成は、容易にコントロールできない結晶化の速さに左右される。従って、塩酸セルトラリンII形の製造が必要である。本発明の方法は、この準安定II形を純粋形態でかつ簡単なやり方で工業生産できる。従って本発明はまた、I形だけの特有なXRPDピークの非存在、例えば14.9および26.3度の2シータにピークが存在しないことによって判定されるようなセルトラリンI形を検出可能なレベルで含まない、すなわち、1.0%未満、特に0.5%未満のI形を含む塩酸セルトラリンII形に関する。
【0041】
従って1つの実施態様において、本発明は、
(a)非プロトン性溶媒例えばアセトン、メチルエチルケトン、メチルイソブチルケトンまたはアセトニトリルにセルトラリン遊離塩基を溶解させる段階
(b)段階(a)の溶液にn−ブタノールまたは酢酸のようなプロトン性溶媒を1.0から約1.5当量に等しい量で添加する段階
(c)段階(b)の溶液をトリアルキルシリルクロリド例えばトリメチルクロロシランで処理する段階を含む塩酸セルトラリンII形の製造方法に関する。
【0042】
トリメチルクロロシランは全量を一度に添加してもよくまたは2回以上の分量として添加してもよくまたは漸増的に添加してもよい。トリメチルクロロシランとの反応はセルトラリンが可溶性であるいかなる温度で行わせてもよい。溶媒がアセトニトリルのとき、反応は典型的には約20から80℃の範囲の温度、より典型的には約20から50℃の範囲の温度で行う。溶媒がメチルイソブチルケトンのとき、反応は典型的には約50から100℃の範囲の温度、より典型的には約80℃の温度で行う。トリメチルクロロシランは典型的にはセルトラリン1当量あたり約1から約2当量のトリメチルクロロシランとなる量で添加する。段階(b)のプロトン性溶媒は典型的にはトリメチルクロロシランの使用量に等価の量で添加する。トリメチルクロロシランの添加後、均質混合物が得られる時間まで反応混合物を熟成させる。
【0043】
別の好ましい実施態様において、本発明は、
(a)非プロトン性溶媒例えばメチルエチルケトン、メチルイソブチルケトンまたはアセトニトリル中のセルトラリンの有機塩例えばマンデル酸塩またはシュウ酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリアルキルシリルクロリド例えばトリメチルクロロシランで処理する段階
を含む、セルトラリンの有機塩を使用する塩酸セルトラリンII形の製造方法に関する。
【0044】
典型的には、溶媒がメチルエチルケトンのときトリメチルクロロシランとの反応は約50から80℃の範囲の温度で行い、溶媒がメチルイソブチルケトンのとき反応は約50から100℃の範囲およびそれ以上の温度、典型的には約80℃で行う。溶媒がアセトニトリルのときはトリメチルクロロシランとの反応を周囲温度でも行うことができる。トリメチルクロロシランは典型的には有機塩1当量あたり約1から約2当量のトリメチルクロロシラン、より典型的には有機塩1当量あたり1.1当量のトリメチルクロロシランとなる量で添加される。
【0045】
本発明はさらに、塩酸セルトラリンI形の製造方法に関する。方法は、(a)セルトラリン遊離塩基を室温でイソプロパノールに溶解させる段階(b)溶液をトリメチルクロロシランで処理する段階とを含む。
【0046】
本発明はさらに、無水塩酸ドネペジルを製造する本発明の方法に関する。溶媒の選択次第で塩酸ドネペジルII形またはIII形を製造できる。
【0047】
第一の実施態様において、これは、
(a)ドネペジル遊離塩基を酢酸エチル、ジメトキシエタンまたはメチルイソブチルケトンに溶解させる段階
(b)少なくとも1当量のプロトン性溶媒例えばn−ブタノールまたは酢酸を添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
(d)塩酸ドネペジルII形を単離する段階
を含む塩酸ドネペジルII形の製造方法である。
【0048】
第二の実施態様において、これは、無水塩酸ドネペジルIII形の製造方法であり、この方法ではアセトンまたはアセトニトリルが溶媒として使用される。結晶質生成物である塩酸ドネペジルII形および塩酸ドネペジルIII形のXRPDデータは、WO97/46527(Eisai)に示されているII形およびIII形のX線結晶学データに一致する。
【0049】
本発明はさらに、本発明の方法による塩酸テルビナフィン、塩酸シナカルセット、塩酸デュロキセチン、塩酸メマンチンおよびモノ塩酸プラミペキソールの製造方法に関する。方法は好ましくは、
(a)テルビナフィン、シナカルセット、デュロキセチン、メマンチンまたはプラミペキソールの遊離塩基を非プロトン性溶媒に溶解させる段階
(b)少なくとも1当量のプロトン性溶媒例えば酢酸またはメタノールもしくはn−ブタノールのようなアルコールを添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
を含む。
【0050】
上記の方法において、テルビナフィンは、例えばアセトン、アセトニトリルまたはtert−ブチルメチルエーテルのような非プロトン性溶媒に溶解させることができ、シナカルセットは、例えばアセトニトリルまたは酢酸エチルのような非プロトン性溶媒に溶解させることができでき、プラミペキソールは、例えばアセトニトリルのような非プロトン性溶媒に溶解させることができる。
【0051】
結晶質生成物である塩酸テルビナフィンのXRPDデータは、E.Tedescoら,CrystEngComm,2002,4(67),393−400によって公表されたX線結晶学データに一致する。シナカルセットの結晶質塩酸塩の特性X線粉末回折図を図9Aに示し、プラミペキソールの結晶質塩酸塩の特性X線粉末回折図を図12Aに示す。さらに、シナカルセットの結晶質塩酸塩はまた図9Bに示すような典型的赤外スペクトルによって特性決定され、結晶質モノ塩酸プラミペキソールは図12Bに示すような典型的赤外スペクトルによって特性決定される。デュロキセチンの結晶質塩酸塩の特性X線粉末回折図を図18Aに示し、塩酸メマンチンの特性X線粉末回折図を図20Aに示す。さらに、デュロキセチンの結晶質塩酸塩はまた図18Bに示すような典型的赤外スペクトルによって特性決定され、結晶質塩酸メマンチンは図19Bに示すような典型的赤外スペクトルによって特性決定される。
【0052】
本発明はまた、上記に定義の一般方法による臭化水素酸シタロプラムを製造するための本発明の方法に関する。1つの実施態様において本発明は、シタロプラム遊離塩基をアルコールすなわちメタノールまたはイソプロパノールのようなプロトン性溶媒に溶解させ、1.0から約1.5当量に等しいトリメチルブロモシランを添加する段階を含む臭化水素酸シタロプラムの製造方法に関する。
【0053】
別の実施態様において、本発明は、
(a)シタロプラムの遊離塩基を酢酸エチル、アセトンまたはアセトニトリルのような非プロトン性溶媒に溶解させる段階
(b)少なくとも1、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1、特に約1当量のトリメチルブロモシランで処理する段階
(d)臭化水素酸シタロプラムを単離する段階
を含む臭化水素酸シタロプラムの製造方法に関する。結晶質臭化水素酸シタロプラムが得られる。結晶質臭化水素酸シタロプラムの特性X線粉末回折図を図10Aに示し、典型的赤外スペクトルを図10Bに示す。
【0054】
本発明はまた、塩酸アリピプラゾールA形を製造するための発明の方法に関する。方法は、
(a)アリピプラゾールの遊離塩基を非プロトン性溶媒例えばメチレンジクロリドに溶解させる段階
(b)少なくとも1、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1、特に約1当量のトリメチルクロロシランで処理する段階
(d)塩酸アリピプラゾールA形を単離する段階
を含む。結晶質生成物である塩酸アリピプラゾールのXRPDデータはWO2004/083183(Hetero Drugs Ltd.)に示されているようなX線結晶学データA形に対応する。
【0055】
リモナバントを例えばアセトニトリルに懸濁させるかまたは酢酸エチルに溶解させ、少なくとも1当量のメタノールおよびトリメチルクロロシランを添加すると、結晶形Iのリモナバントの塩酸塩が得られる。塩酸リモナバントI形のX線粉末回折図を図20Aに示す。この図は、10.4、14.4、17.8、19.2、20.8、21.9、22.2、26.4、26.9、28.7および28.5度の2シータに主要ピークを示す。塩酸リモナバントI形の赤外スペクトルを図20Bに示す。塩酸リモナバントI形はEP0656354(Sanofi)の実施例3に従って製造された生成物に一致する。該実施例では、リモナバントをエーテルに溶解させ、HClガスの飽和エーテル溶液を部分量ずつ添加する。
【0056】
I形の塩酸クロピドグレルは例えば、少なくとも1当量の酢酸およびトリメチルクロロシランを酢酸エーテル中のクロピドグレル塩基の溶液に添加することによって得ることができる。アセトンまたはアセトニトリルを溶媒として使用するときは、塩酸クロピドグレルI形を沈殿させるためにジイソプロピルエーテルのような反溶媒を添加する。1つの好ましい実施態様においては、クロピドグレル塩基をトルエンに溶解させ、1当量のメタノールを添加し、溶液をトリメチルクロロシランで処理する。トリメチルクロロシランの添加後、綿毛状の沈殿物を室温で暫時撹拌して塩酸クロピドグレルI形に変化させる。
【0057】
塩酸プラスグレルB形は例えば、少なくとも1当量の酢酸およびトリメチルクロロシランをアセトン中のプラスグレル塩基の溶液に添加することによって得ることができる。塩酸プラスグレルB形のXRPD図を図23Aに示し、赤外スペクトルを図23Bに示す。US6693115の実施例3、4および6に報告されているように特性赤外吸収バンドは1758および1690cm−1に存在する。
【0058】
本発明はさらに、新規な塩酸プラスグレルアセトニトリル溶媒和物に関する。これは、8.3、13.8、16.2、18.8、23.8、25.4および26.8度の2シータにピークを含むX線粉末回折図によって特性決定される。塩酸プラスグレルアセトニトリル溶媒和物のX線粉末回折図の一例を図24Aに示す。塩酸プラスグレルアセトニトリル溶媒和物はまた、図24Bに示すような典型的赤外スペクトルによって特性決定され得る。特性バンドは1760、1720、1499、1210および775cm−1に存在する。塩酸プラスグレルアセトニトリル溶媒和物は効率的な精製段階を可能にする。
【0059】
塩酸プラスグレルアセトニトリル溶媒和物は好ましくは、
(a)プラスグレルをアセトニトリルに溶解させる段階
(b)少なくとも1当量の酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
(d)塩酸プラスグレルアセトニトリル溶媒和物を単離する段階
を含む本発明の方法によって製造できる。
【0060】
風乾条件下で塩酸プラスグレルアセトニトリル溶媒和物は安定であり脱溶媒和しない。これは、1H−NMRスペクトル(DMSO−d6,TMS)に実質的に同一の1H−NMRスペクトルを有している。より詳細にはこれは、1モルの基質あたり約1モルのアセトニトリルに対応する2.ppm(s,3H)に特性ピークを有している。
【0061】
非晶質形の塩酸シタグリプチンは例えば、少なくとも1当量のメタノールおよびトリメチルクロロシランをメチレンクロリドおよびジエチルエーテルの混合物中のシタグリプチン塩基の溶液に添加することによって得ることができる。非晶質塩酸シタグリプチンのXRPD図を図29Aに示し、赤外スペクトルを図29Bに示す。特性赤外吸収バンドはcm−1に存在する。
【0062】
本発明はまた、アルコールすなわちメタノールまたはイソプロパノールのようなプロトン性溶媒にクロピドグレル遊離塩基を溶解させ、1.0から約1.5当量に等しいトリメチルブロモシランを添加する段階を含む、上記の定義のような一般方法による臭化水素酸クロピドグレルA形の製造方法に関する。
【0063】
臭化水素酸クロピドグレルA形の代替的製造方法は、
(a)クロピドグレルの遊離塩基を酢酸エチルのような非プロトン性溶媒に溶解させる段階
(b)少なくとも1当量の酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量のトリメチルブロモシランで処理する段階
(d)臭化水素酸クロピドグレルA形を単離する段階
を含む本発明の方法である。
【0064】
結晶質臭化水素酸クロピドグレルA形に特有なX線粉末回折図を図22Aに示し、代表的な赤外吸収スペクトルを図22Bに示す。
【0065】
本発明はさらに、塩酸ラロキシフェンA形を製造するための本発明の方法に関する。好ましい実施態様において、塩酸ラロキシフェンA形の製造方法は、
(a)エタノールのようなプロトン性溶媒中のラロキシフェン遊離塩基の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
を含む。
【0066】
塩酸ラロキシフェンA形の代替的製造方法は、
(a)エタノールのようなプロトン性溶媒またはメチルイソブチルケトンもしくはアセトニトリルのような非プロトン性溶媒中のラロキシフェンの有機塩例えば乳酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
を含む。溶媒がメチルエチルケトンのときはトリメチルクロロシランとの反応を典型的には約100℃の温度で行わせ、溶媒がアセトニトリルのときはトリメチルクロロシランとの反応を典型的には周囲温度で行わせる。塩酸ラロキシフェンA形の特性X線粉末回折図を図25Aに示し、典型的赤外スペクトルを図25Bに示す。
【0067】
本発明はまた、新規な塩酸ラロキシフェンとテトラヒドロフランとの半水和物に関する。この物質は、13.6、16.6、17.7、18.9、19.2、19.5、21.2および23.6度の2シータにピークを含むX線粉末回折図によって特性決定される。塩酸ラロキシフェンとテトラヒドロフランとの半水和物のX線粉末回折図の一例を図26Aに示す。塩酸ラロキシフェンとテトラヒドロフランとの半水和物は、図26Bに示す典型的赤外スペクトルによって特性決定される。特性バンドは1651、1595、1226、1165、838および815cm−1に存在する。塩酸ラロキシフェンとテトラヒドロフランとの半水和物は、ラロキシフェン生産のより効率的および/またはより効果的な精製段階を可能にする。
【0068】
本発明はまた、
(a)テトラヒドロフラン中のラロキシフェン遊離塩基の懸濁液を撹拌する段階
(b)1.0から約1.5当量に等しいメタノールのような極性溶媒を段階(a)の懸濁液に添加する段階
(c)段階(b)の懸濁液をトリメチルクロロシランで処理する段階
(d)塩酸ラロキシフェンのテトラヒドロフラン半溶媒和物を単離する段階
を含む塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物を製造するための本発明の方法に関する。
【0069】
塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物の製造方法の代替実施態様は、
(a)テトラヒドロフラン中のラロキシフェンの有機塩例えば乳酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
(c)塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物を単離する段階
を含むラロキシフェンの有機塩を使用する方法である。風乾条件下で塩酸ラロキシフェンTHF半溶媒和物は安定であり脱溶媒和しない。これは、1H−NMRスペクトル(DMSO−d6)に実質的に同一の1H−NMRスペクトルを有している。
【0070】
本発明はさらに、無水ジ塩酸バルデナフィルを製造するための本発明の方法に関する。方法は、
(a)非プロトン性溶媒、または、例えばEP1049695の実施例10に記載されているようなジエチルエーテルとメチレンジクロリドとの溶媒混合物にバルデナフィル塩基を溶解させる段階
(b)少なくとも2当量のメタノールのようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも2当量のトリメチルクロロシランで処理する段階
(d)無水ジ塩酸バルデナフィルを単離する段階
を含む。無水ジ塩酸バルデナフィルの特性X線粉末回折図を図30Aに示し、典型的赤外スペクトルを図30Bに示す。
【0071】
本発明はさらに、塩酸エルロチニブA形を製造するための本発明の方法に関する。方法は、エルロチニブ塩基をイソプロパノールに懸濁させ、懸濁液を少なくとも1当量のトリメチルクロロシランによって室温で処理する段階を含む。塩酸エルロチニブA形のX線粉末回折図を図31Aに示し、典型的赤外スペクトルを図31Bに示す。結晶質生成物である塩酸エルロチニブA形のXRPDデータはUS2004/0162300(Hofmann−La Roche)に示されているB形のX線結晶学データに一致する。
【0072】
本発明の方法を使用することによってわれわれはさらに、約14.6、18.7、21.9、23.5および25.3度の2シータにピークをもつX線粉末回折図によって特性決定される塩酸モキシフロキサシンメチレンジクロリド溶媒和物を発見した。従って本発明はまた、特に塩酸モキシフロキサシンメチレンジクロリド溶媒和物、特に塩酸モキシフロキサシン対メチレンジクロリドのモル比が約1:1であるこのような溶媒和物に関する。特にこの溶媒和物は、X線粉末回折図で約14.6、18.7、21.9、23.5および25.3度の2シータにピークを示す。塩酸モキシフロキサシンメチレンジクロリド溶媒和物の特性X線粉末回折図を図13Aに示す。塩酸モキシフロキサシンメチレンジクロリド溶媒和物は後述する塩酸モキシフロキサシンIV形の製造を初めて可能にする。
【0073】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物はまた、赤外スペクトルの約2704、1720、1434、1311、1272、730および702cm−1に存在する特性バンド、特に図13Bに示すような典型的赤外スペクトルによって特性決定される。
【0074】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物は、
(a)モキシフロキサシンをメチレンジクロリドに溶解させる段階
(b)少なくとも1当量、特に約1当量のプロトン性溶媒例えばn−ブタノールまたは酢酸を添加する段階
(c)溶液を少なくとも1当量、特に約1当量のトリアルキルクロロシラン例えばトリメチルクロロシランで処理する段階
(d)塩酸モキシフロキサシンメチレンジクロリド溶媒和物を単離する段階
を含む本発明の方法によって製造され得る。
【0075】
風乾条件下で塩酸モキシフロキサシンメチレンジクロリド溶媒和物は安定であり、脱溶媒和しない。塩酸モキシフロキサシンメチレンジクロリド溶媒和物のTGAは80から180℃の範囲で16%の減量を示し、これは基質1モルあたり1モルのジクロロメタンに対応する。
【0076】
従って本発明は、80から180℃の範囲で10%から20%、特に約16%の減量を示す塩酸モキシフロキサシンメチレンジクロリド溶媒和物に関する。これは基質1モルあたり約1モルのジクロロメタンに対応する。
【0077】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物は、図13Cに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細にはそれは、基質1モルあたり1モルのジクロロメタンに対応する5.77ppm(s,2H)に特性ピークを有している。
【0078】
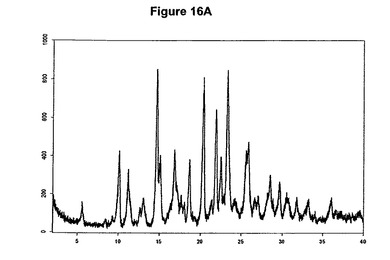
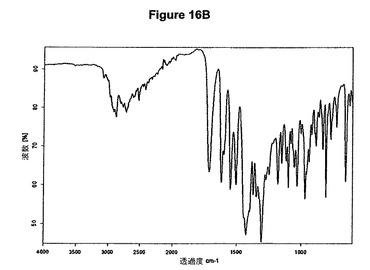
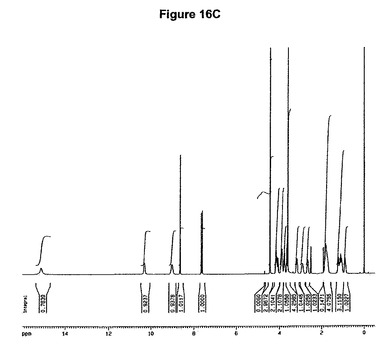
本発明の方法を使用することによってわれわれはさらに、塩酸モキシフロキサシンニトロメタン溶媒和物を発見した。従って本発明は、このような溶媒和物、特に塩酸モキシフロキサシン対ニトロメタンのモル比が約1:1であるこのような溶媒和物に関する。溶媒和物はさらに、約11.2、14.7、16.9、20.4、22.0、22.6および23.4度の2シータにピークをもつX線粉末回折図、特に図16Aに示すような塩酸モキシフロキサシンニトロメタン溶媒和物の特性X線粉末回折図によって特性決定される。塩酸モキシフロキサシンニトロメタン溶媒和物はまた、赤外スペクトルの約2879、1715、1550、1310および1032cm−1に存在する特性バンド、特に図16Bに示すような典型的赤外スペクトルによって特性決定される。塩酸モキシフロキサシンニトロメタン溶媒和物はモキシフロキサシン生産のより効率的および/または効果的な精製段階を可能にする。
【0079】
より詳細には塩酸モキシフロキサシンニトロメタン溶媒和物は、
(a)モキシフロキサシンをニトロメタンに溶解させる段階
(b)少なくとも1当量、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量、特に約1当量のトリメチルクロロシランで処理する段階
(d)塩酸モキシフロキサシンニトロメタン溶媒和物を単離する段階
を含む本発明の方法によって製造され得る。
【0080】
塩酸モキシフロキサシンニトロメタン溶媒和物は、図16Dに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細には、それは基質1モルあたり1モルのニトロメタンに対応する4.4ppm(s,3H)に特性ピークを有している。
【0081】
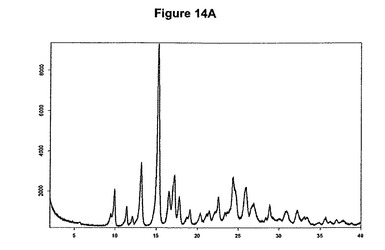
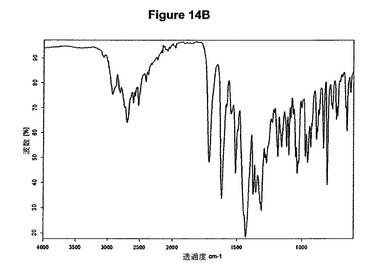
本発明はまた、この文中でIV形と呼ぶモキシフロキサシンの新規な無水塩酸塩に関する。より詳細には、IV形は、湿度レベル33%の乾燥機に48時間維持されても塩酸モキシフロキサシンの水和形に変換されない。より特定的には本発明は、約10.0、13.2、15.3、17.2および24.4度の2シータにピークをもつX線粉末回折図によって特性決定されるIV形に関する。塩酸モキシフロキサシンの無水IV形の特性X線粉末回折図を図14Aに示す。本発明はまた、約2704、1720、1434、1312および1273cm−1に存在する赤外吸収バンドによって特性決定されるIV形に関する。塩酸モキシフロキサシンIV形はさらに図14Bに示すような典型的赤外スペクトルによって特性決定される。
【0082】
新規な無水IV形は好ましくは、塩酸モキシフロキサシンメチレンジクロリド溶媒和物を真空下に約100℃で乾燥して脱溶媒和することによって製造され得る。無水IV形は、US5849752(Bayer)に記載されている無水B形よりも低吸湿性であり、また、Chemi Spaの特許出願WO2005/054240に記載されている無水B形よりも低吸湿性であるため、取扱いおよび/または保管が容易である。塩酸モキシフロキサシン無水IV形は、湿度レベル33%の乾燥機に48時間維持しても水和形の塩酸モキシフロキサシンに変換されない。これは、変質を全く示さないXRPD図および0.5%から0.6%までしか増加しない含水量によって証明される。本発明はまた、IV形を含む医薬組成物に関する。
【0083】
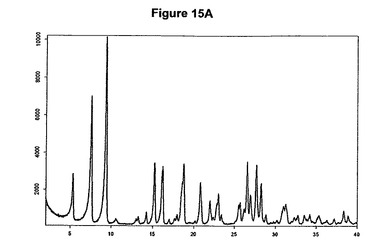
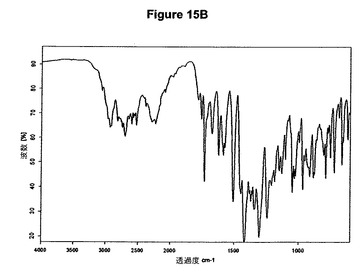
本発明の方法を使用することによってわれわれはさらに、塩酸モキシフロキサシン酢酸溶媒和物を発見した。従って本発明はこのような溶媒和物、特に塩酸モキシフロキサシン対酢酸のモル比が約1:1であるこのような溶媒和物に関する。この溶媒和物はさらに、約5.3、7.6、9.3、15.2、16.2、18.8、20.8、26.6および27.7度の2シータにピークをもつX線粉末回折図によって特性決定される。好ましい実施態様において、塩酸モキシフロキサシン酢酸溶媒和物は図15Aに示すパターンに実質的に一致するX線粉末回折図を示す。塩酸モキシフロキサシン酢酸溶媒和物はまた、図15Bに示す典型的赤外スペクトルによって特性決定される。塩酸モキシフロキサシン酢酸溶媒和物の代表的赤外吸収バンドは、2707、2289、1736、1421、1308、1246、917および757cm−1に観察される。塩酸モキシフロキサシン酢酸溶媒和物は良好な溶解度および/または安定度を示す。
【0084】
塩酸モキシフロキサシン酢酸溶媒和物は上記に定義および記載したような方法に従って製造され得る。方法は、
(a)モキシフロキサシンを酢酸または酢酸とアセトンもしくはアセトニトリルのような有機溶媒との混合物に溶解させる段階
(b)トリメチルクロロシランを添加する段階
(c)塩酸モキシフロキサシン酢酸溶媒和物を単離する段階
を含む。
【0085】
塩酸モキシフロキサシン酢酸溶媒和物は、図15Cに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細には、これは基質1モルあたり1モルの酢酸に対応する1.9ppm(s,3H)に特性ピークを有している。本発明はまた、本発明の塩酸モキシフロキサシン酢酸溶媒和物を含む医薬組成物に関する。
【0086】
本発明はまた、塩酸リネゾリドを製造するための本発明の方法に関する。方法は好ましくは、アセトンまたはアセトニトリルのような有機溶媒中のリネゾリドの溶液にn−ブタノールまたは酢酸のようなプロトン性溶媒を添加し、混合物をトリメチルクロロシランで処理する段階を含む。
【0087】
本発明はまた、結晶質塩酸リネゾリドに関する。塩酸リネゾリドは、例えばXRPD図の約13.9、18.2、19.1、23.0および27.2度の2シータ値に存在する特性ピークによって特性決定される。結晶質塩酸リネゾリドの特性X線粉末回折図を図19Aに示し、典型的赤外スペクトルを図19Bに示す。図19Aに示すように、塩酸リネゾリドは、XRPD図の約13.9、18.2、19.1、23.0および27.2度の2シータ値に特性ピークを示す。リネゾリドの化学構造に基づいて予想することはできなかったが意外にもこの塩酸塩は安定であることが知見された。塩酸リネゾリドは医薬組成物の製造を可能にするであろう。
【0088】
上記に説明したように、本発明は、結晶化溶液中に生成されるハロゲン化水素酸の量を、該ハロゲン化水素酸の生成速度を(例えばハロゲン化トリアルキルシリルの添加速度をコントロールすることによって)、および、ハロゲン化水素酸の生成条件を(例えば温度、溶媒組成または有機アミンの所望の塩(モノもしくはジハロゲン化水素酸塩など)もしくは有機アミンのハロゲン化水素酸塩の所望の多形を得るために必要な別のパラメーターを調節することによって)微調整できる。従って本発明は、有機アミンの結晶質ハロゲン化水素酸塩を製造するためのハロゲン化トリアルキルシリルの使用に関する。この使用は、有機アミンの結晶質ハロゲン化水素酸塩が該有機アミンのいくつかの既知のハロゲン化水素酸塩のうちの望ましいハロゲン化水素酸塩であるとき、例えば薬剤のジ−またはトリ−塩酸塩が存在するけれども薬剤の結晶質モノ塩酸塩が望ましいような場合に特に有利である。結晶化混合物にHClガスを添加するという慣用技術の使用は、HClの添加量に関するコントロールは難しくないかもしれないが、結晶化条件の選択に関する適応性が劣るので、操作が極めて難しいものになるであろう。
【0089】
別の利点は、ハロゲン化水素酸塩が本質的に水の非存在下または少なくとも極めて少量の水の存在下で生成できることである。従って、有機アミンの結晶質ハロゲン化水素酸塩の無水形を得ることが可能である。従って本発明はまた、有機アミンの無水結晶質ハロゲン化水素酸塩の製造におけるハロゲン化トリアルキルの使用に関する。さらに、本質的に水の非存在下でコントロール量のハロゲン化水素酸塩を製造するので、水の存在下では形成されない有機アミンの溶媒和物、例えば、対応する水和物よりも熱力学的に不安定な溶媒和物を得ることが可能である。従って、本発明はまた、有機アミンの結晶質ハロゲン化水素酸塩の溶媒和物の製造、特に無水溶媒和物の製造におけるハロゲン化トリアルキルの使用に関する。好ましいハロゲン化トリアルキルシリル、好ましい有機アミンおよびこれらの好ましい使用条件が本発明の方法に関する上記の記載によって定義されていることは当業者に明らかであろう。
【0090】
以下の実施例は本発明を例示するもので本発明の範囲を限定するものではない。使用した室温という用語は常に20から30℃の範囲の温度を表す。
【実施例】
【0091】
赤外スペクトルは、ダイアモンドATR−セルを備えたBRUKER Tensor 27 FTIR−分光計を使用して記録した。
【0092】
XRPDはAXS−BRUKER X線粉末回折計D−8で以下の取得条件を使用して記録した:チューブアノード:Cu;発電機電圧:40kV;発電機電流:40mA;出発角度:2.0度?;到着角度:40.0度?;一目盛り:0.01度?;時間/目盛り:2秒。
【0093】
示差走査熱量測定(DSC)は、DSC7(Perkin−Elmer,Norwalk,Ct.,USA)でPyris 2.0ソフトウェアを使用して行った。サンプルを25μlのAlパンで計量した。パージガスとして乾性窒素を使用した(パージ速度:20ml分−1)。
【0094】
1H−NMRスペクトルはBrucker AM−300分光計で記録した。
【実施例1】
【0095】
結晶質無水形の塩酸ミコフェノラートモフェチルの製造
2g(4.61mmol)のミコフェノラートモフェチル塩基を50mlの酢酸エチルに室温で溶解させた。この溶液に0.3ml(1.2当量)の酢酸および0.7ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、2.11g(97.6%)の塩酸ミコフェノラートモフェチルが得られた。mp.=157.2℃。
【0096】
塩酸ミコフェノラートモフェチルのXRD図を図1Aに示す。これはWO95/07902に示されているX線結晶学データをもつ結晶質無水形に一致する。得られた赤外スペクトルを図1Bに示す。塩酸ミコフェノラートモフェチルのDSCは約159℃に吸熱ピークを示す(開始温度約155℃、図1C参照)。
【実施例2】
【0097】
塩酸ベンラファキシンの製造
実施例2.a 塩酸ベンラファキシンI形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.41g(89.1%)の塩酸ベンラファキシンが得られた。mp.=208℃。
【0098】
塩酸ベンラファキシンのXRD図を図2Aに示す。これはUS03/0114536に示されているX線結晶学データをもつI形に一致する。得られた赤外スペクトルを図2Bに示す。
【0099】
実施例2.b 塩酸ベンラファキシンII形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.38g(82.6%)の塩酸ベンラファキシンII形が得られた。
【0100】
塩酸ベンラファキシンII形のXRPD図を図3Aに示す。これはWO02/45658に示されているX線結晶学データをもつII形に一致する。得られた赤外スペクトルを図3Bに示す。
【0101】
実施例2.c 塩酸ベンラファキシンII形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.23g(51.1%)の塩酸ベンラファキシンII形が得られた。
【実施例3】
【0102】
セルトラリン塩基を使用する塩酸セルトラリンII形の製造
実施例3.a
3g(9.8mmol)のセルトラリン塩基を60mlのアセトニトリルに室温で溶解させた。この溶液に0.6ml(1当量)の酢酸および1.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。添加中に塩酸セルトラリンがまさしく結晶質II形で沈殿した。懸濁液を1時間撹拌後、生成物を濾別し、50℃で3時間乾燥すると3.2g(95.3%)の塩酸セルトラリンII形が得られた。mp.:252℃。
【0103】
得られたXRD図を図4Aに示す。これは純粋なII形に一致する。得られた赤外スペクトルを図4Bに示す。
【0104】
実施例3.b
3g(9.8mmol)のセルトラリン塩基を60mlのアセトニトリルと1mlのn−ブタノールとの混合物に溶解させた。溶液を50℃に加熱し、1.4ml(1.1当量の)のトリメチルクロロシランを撹拌下で添加した。添加中に直ちに塩酸セルトラリンが結晶質II形で沈殿した。50℃で30分間撹拌後、懸濁液を室温に冷却し、約1時間撹拌した。生成物を濾別し、50℃で3時間乾燥すると、3.1g(94%)の塩酸セルトラリンII形が得られた。
【0105】
実施例3.c
200mlのメチルイソブチルケトン(MIBK)中の10g(32.7mmol)のセルトラリン塩基を約80℃に加熱した。溶液に2.4ml(1.1当量)の酢酸、4.5ml(1.1当量)のトリメチルクロロシランを撹拌下で順次に添加した。最初に得られたゼリー状の塊は80℃で1時間撹拌後に結晶質になった。反応混合物を室温に冷却し、再度約1.5時間撹拌した。生成物を濾別し、50℃で4時間乾燥すると、10.93g(97.7%)の塩酸セルトラリンII形が得られた。
【実施例4】
【0106】
マンデル酸セルトラリンを使用する塩酸セルトラリンII形の製造
実施例4.a
60mlのアセトニトリル中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を室温で撹拌し、1.4ml(1.7当量)のトリメチルクロロシランを添加した。粘性懸濁液は先ず希薄懸濁液に変化し、その後15分以内に塩酸セルトラリンII形の結晶の希薄懸濁液が得られた。約1時間撹拌後、生成物を濾別し、真空下に50℃で乾燥すると、2.09g(96.3%)の塩酸セルトラリンII形が得られた。
【0107】
実施例4.b
60mlのメチルエチルケトン中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を約80℃に加熱し、0.9ml(1.1当量)のトリメチルクロロシランを添加した。透明溶液が得られた後まもなく結晶質II形の塩酸セルトラリンが沈殿し始めた。反応混合物を撹拌下に1時間以内で室温に冷却し、次いで生成物を濾別し、真空下に50℃で乾燥すると、1.97g(87.8%)の塩酸セルトラリンII形が得られた。
【0108】
実施例4.c
60mlのメチルイソブチルケトン中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を約80℃に加熱し、0.9ml(1.1当量)のトリメチルクロロシランを添加する。懸濁液はゼリー状の塊に変化した。約5分後、結晶質II形の塩酸セルトラリンが形成され始めた。反応混合物を80℃で約20分間撹拌し、次いで撹拌下に1時間以内で室温に冷却した。生成物を濾別し、真空下に50℃で3時間乾燥すると、2.11g(94.0%)の塩酸セルトラリンII形が得られた。
【0109】
実施例4.d
300mlのメチルイソブチルケトン中の15g(32.7mmol)のマンデル酸セルトラリンの懸濁液を約100℃に加熱し、4.6ml(1.1当量)のトリメチルクロロシランを添加した。懸濁液はゼリー状の塊に変化した。約5分後、結晶質II形の塩酸セルトラリンが形成され始めた。反応混合物を100℃で約15分間撹拌し、次いで撹拌下に1.5時間以内で室温に冷却した。生成物を濾別し、10mlのアセトンで2回洗浄し、真空下に50℃で4時間乾燥すると、10.64g(94.9%)の塩酸セルトラリンII形が得られた。
【実施例5】
【0110】
シュウ酸セルトラリンを使用する塩酸セルトラリンII形の製造
実施例5.a
シュウ酸セルトラリンの製造
50mlの酢酸エチル中の3g(9.8mmol)のセルトラリン遊離塩基の溶液に50mlのメタノール中の0.97g(1.1当量)のシュウ酸の溶液を添加した。15分間撹拌後、結晶質沈殿物を濾別し、真空下に50℃で乾燥すると3.37g(86.8%)のシュウ酸セルトラリンが得られた。
【0111】
実施例5.b
シュウ酸セルトラリンからの塩酸セルトラリンII形の製造
15mlのアセトニトリル中の0.7g(1.77mmol)のシュウ酸セルトラリンの懸濁液を室温で撹拌し、250μl(1.1当量)のトリメチルクロロシランを添加した。添加後に懸濁液はほぼ溶液に変化したが同時に結晶質II形の塩酸セルトラリンが沈殿し始めた。約1時間撹拌後、生成物を濾別し、真空下50℃で乾燥すると、0.47g(77.8%)の塩酸セルトラリンII形が得られた。
【実施例6】
【0112】
塩酸セルトラリンI形の製造
60mlの2−プロパノール中の3g(9.8mmol)のセルトラリン塩基の溶液を室温で撹拌し、0,6mlの酢酸および1.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を60分間撹拌し、沈殿物を濾別した。固体を2−プロパノールで洗浄し、真空下に50℃で3時間乾燥すると、2.95g(87.8%)の塩酸セルトラリンI形が得られた。得られたXRD図を図5Aに示す。これはI形に対応する。得られた赤外スペクトルを図5Bに示す。
【実施例7】
【0113】
塩酸ドネペジルの製造
実施例7.a
塩酸ドネペジルII形の製造
0.5g(1.32mmol)のドネペジル塩基を30mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を2時間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.55g(100%)の塩酸ドネペジルII形が得られた。得られたXRD図を図6Aに示す。これはII形に対応する。得られた赤外スペクトルを図6Bに示す。
【0114】
実施例7.b
塩酸ドネペジルIII形の製造
0.5g(1.32mmol)のドネペジル塩基を30mlのアセトンに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.54g(98.5%)の塩酸ドネペジルが得られた。mp.=211℃。得られたXRD図を図7Aに示す。これはIII形に対応する。得られた赤外スペクトルを図7Bに示す。
【0115】
実施例7.c
塩酸ドネペジルIII形の製造
アセトンに代えてアセトニトリルを使用して実施例7.bを繰り返した。塩酸ドネペジルIII形の収量(収率):0.47g(85.6%)。
【実施例8】
【0116】
塩酸テルビナフィンの製造
実施例8.a
0.4g(1.37mmol)のテルビナフィン塩基を5mlのアセトンに室温で溶解させた。この溶液に86μl(1.1当量)の酢酸および191μl(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトンで洗浄し、真空下に室温で乾燥すると、0.3g(66.7%)の塩酸テルビナフィンが得られた。mp.=185℃。
【0117】
塩酸テルビナフィンのXRP図を図8Aに示す。これは文献データ(Cryst.Eng.Comm.,2002,4(67),393−400)に一致する。得られた赤外スペクトルを図8Bに示す。
【0118】
実施例8.b
アセトンに代えてアセトニトリルを使用して実施例8.aを繰り返した。塩酸テルビナフィンの収量(収率):0.19g(42.2%)。
【0119】
実施例8.c
アセトンに代えてtert.−ブチル−メチル−エーテルを使用して実施例8.aを繰り返した。塩酸テルビナフィンの収量(収率):0.41g(91,1%)。
【実施例9】
【0120】
塩酸シナカルセットの製造
実施例9.a
1.0g(2.80mmol)のシナカルセット塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.19ml(1.2当量)の酢酸および0.42ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を2時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.45g(40.8%)の塩酸シナカルセットが得られた。mp.=173℃。得られたXRD図を図9Aに示し、得られた赤外スペクトルを図9Bに示す。
【0121】
実施例9.b
アセトニトリルに代えて酢酸エチルを使用して実施例9.aを繰り返した。塩酸シナカルセットの収量(収率):0.41g(37.2%)。
【実施例10】
【0122】
臭化水素酸シタロプラムの製造
実施例10.a
0.72g(22.2mmol)のシタロプラム塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.14ml(1.1当量)の酢酸および0.32ml(1.1当量)のトリメチルブロモシランを撹拌下で添加した。室温で30分後に15mlのジエチエーテルを添加すると結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.67g(74,5%)の臭化水素酸シタロプラムが得られた。mp.=182℃。得られたXRD図を図10Aに示し、得られた赤外スペクトルを図10Bに示す。
【0123】
実施例10.b
アセトニトリルに代えて酢酸エチルを溶媒として使用し、0.5g(15.4mmol)のシタロプラム塩基で実施例10.aを繰り返した。臭化水素酸シタロプラムの収量(収率):0.53g(84.8%)。
【0124】
実施例10.c
アセトニトリルに代えてアセトンを溶媒として使用し、0.59g(18.2mmol)のシタロプラム塩基で実施例10.aを繰り返した。臭化水素酸シタロプラムの収量(収率):0.67g(90.9%)。
【0125】
実施例10.d
0.27g(0.8mmol)のシタロプラム塩基を3mlのイソプロパノールに室温で溶解させ、145μl(1.1当量)のトリメチルブロモシランを溶液に添加した。冷蔵庫で一夜静置後に結晶質沈殿物を濾別し、真空下で乾燥すると、0.25g(74.1%)の臭化水素酸シタロプラムが得られた。
【実施例11】
【0126】
塩酸アリピプラゾールA形の製造
2.0g(4.46mmol)のアリピプラゾールを20mlの1,2−ジクロロメタンに室温で溶解させた。この溶液に0.45ml(1.1当量)のn−ブタノールおよび0.63ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を15分間撹拌し、沈殿物を濾別した。固体を1,2−ジクロロメタンで洗浄し、真空下に室温で乾燥すると、2.05g(94.0%)の塩酸アリピプラゾールが得られた。mp.=210℃。
【0127】
生成物のXRD図を図11Aに示す。これはWO2004/083183(Hetero Drugs Ltd.)に示されている塩酸アリピプラゾールA形のXRPDデータに一致する。得られた赤外スペクトルを図11Bに示す。
【実施例12】
【0128】
モノ塩酸プラミペキソールの製造
0.5g(2.37mmol)のプラミペキソール塩基を20mlのアセトニトリルに室温で溶解させた。この溶液に0.24mlのn−ブタノール(2.6mmol,1.1当量)および0.33mlのトリメチルクロロシラン(2.6mmol,1.1当量)を撹拌下で添加した。室温で1分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.56g(95.5%)のモノ塩酸プラミペキソールが得られた。mp.=264℃。得られたXRD図を図12Aに示し、得られた赤外スペクトルを図12Bに示す。
【実施例13】
【0129】
無水塩酸モキシフロキサシンIV形の製造
実施例13.a
塩酸モキシフロキサシンメチレンジクロリド溶媒和物
5g(12.5mmol)のモキシフロキサシンをEP550903に従って製造し、50mlのメチレンジクロリドに室温で溶解させた。この溶液に0.9ml(1.2当量)の酢酸および2ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後直ちに結晶化が始まった。懸濁液を約30分間撹拌し、沈殿物を濾別し、真空下に室温で乾燥すると、6.3g(94.2%)の塩酸モキシフロキサシンとメチレンジクロリドとの1:1溶媒和物が得られた。
【0130】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1.9(m,4H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.5−3.7(m with Singlet at 3.6 ppm,4H),3.7−3.8(m,1H),3.8−4.0(m,2H),4.0−4.2(m,2H),5.77(s,2H),7.65(d,J=14Hz,1H),8.66(s,1H),8.95(s,broad,1H),10.2(s,broad,1H),15.1(s,broad,1H)。
【0131】
5.77ppmの一重項は基質1モルあたり約1モルのメチレンジクロリドに対応する(図13C参照)。生成物のXRD図を図13Aに示し、得られた赤外スペクトルを図13Bに示す。
【0132】
塩酸モキシフロキサシンとメチレンジクロリドとの溶媒和物は吸湿性でない(33%相対湿度に1日維持後にも全く吸水していない)。
【0133】
実施例13.b
塩酸モキシフロキサシン無水IV形
2gの塩酸モキシフロキサシンメチレンジクロリド溶媒和物を真空下に100℃で約6時間乾燥すると、1.75gの塩酸モキシフロキサシン無水IV形が脱溶媒和した。生成物のXRD図を図14Aに示し、得られた赤外スペクトルを図14Bに示す。
【実施例14】
【0134】
塩酸モキシフロキサシンと酢酸との溶媒和物の製造
実施例14.a
3.0g(7.47mmol)のモキシフロキサシンをEP550903に従って製造し、30mlの酢酸に室温で溶解させた。この溶液に2.0ml(2.1当量の)トリメチルクロロシランを撹拌下で添加した。クロロシラン添加の30分後に結晶化が始まった。懸濁液を約3.5時間撹拌し、沈殿物を濾別し、アセトニトリルで洗浄し、真空下に室温で乾燥すると、3.21g(86.3%)の塩酸モキシフロキサシンと酢酸との1:1溶媒和物が得られた。
【0135】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1. (m,4H),1.9(s,3H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.1−3.25(m,1H),3.5−3.7(m with Singlet at 3.6ppm,4H),3.7−3.8(m,1H),3.8−4.0(m,2H),4.0−4.2(m,2H),7.63(d,J=14Hz,1H),8.65(s,1H),9.1(s,broad,1H),10.4(s,broad,1H),12.7(s,broad,1H)。
【0136】
1.9ppmの一重項は基質1モルあたり約1モルの酢酸に対応する(図15C参照)。生成物のXRD図を図15Aに示し、得られた赤外スペクトルを図15Bに示す。
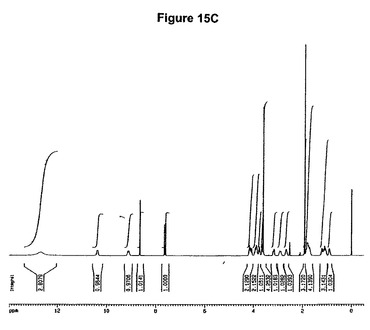
【0137】
実施例14.b
1.0g(2.49mmol)モキシフロキサシンをEP550903に従って製造し、5mlの酢酸および5mlのアセトニトリルに室温で溶解させた。溶液に0.38ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシラン添加の2時間後に結晶化は始まっていなかった。この溶液に0.38mlのトリメチルクロロシランを撹拌下でもう一度添加した。懸濁液を4℃で約17時間保存し、沈殿物を濾別し、アセトニトリルで洗浄し、真空下に室温で乾燥すると、0.93g(75.0%)の塩酸モキシフロキサシンと酢酸との1:1溶媒和物が得られた。
【実施例15】
【0138】
塩酸モキシフロキサシンとニトロメタンとの溶媒和物の製造
0.5g(1.25mmol)のモキシフロキサシンをEP550903に従って製造し、15mlのニトロメタンに約60℃で溶解させた。この溶液に0.14ml(2当量)の酢酸および(2当量)mlのトリメチルクロロシランを撹拌下で添加した。懸濁液を約17時間保存し、沈殿物を濾別し、アセトンで洗浄し、真空下に室温で乾燥すると、0.55g(88.2%)の塩酸モキシフロキサシンとニトロメタンとの1:1溶媒和物が得られた。
【0139】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1.9(m,4H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.1−3.25(m,1H),3.5−3.7(m with Singlet at 3.6ppm,4H),3.7−3.8(m,1H),3.8−3.95(m,2H),4.0−4.2(m,2H),4.44(s,3H),7.63(d,J=14Hz,1H),8.65(s,1H),9.0(s,broad,1H),10.3(s broad,1H),15.1(s,broad,1H)。
【0140】
4.44ppmの一重項は基質1モルあたり約1モルのニトロメタンに対応する。(図16C参照)。生成物のXRD図を図16Aに示し、得られた赤外スペクトルを図16Bに示す。
【実施例16】
【0141】
塩酸デュロキセチンの製造
実施例16.a
0.3g(1.0mmol)のデュロキセチン塩基を5mlの酢酸エチルに室温で溶解させた。この溶液に65μlの酢酸および0.14mlのトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、懸濁液を室温で約2時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、0.21g(62.9%)の塩酸デュロキセチンが得られた。mp.=161℃。生成物のXRD図を図17Aに示し、得られた赤外スペクトルを図17Bに示す。
【0142】
実施例16.b
酢酸エチルに代えてアセトンを使用して実施例16.aを繰り返した。塩酸デュロキセチンの収量(収率):0.26g(77.9%)。
【実施例17】
【0143】
塩酸リネゾリドの製造
実施例17.a
5g(14.8mmol)のリネゾリドを200mlのアセトニトリルに室温で溶解させた。この溶液に1.0ml(1.1当量)の酢酸および2.1ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランを添加し1時間撹拌後に沈殿物が形成され、懸濁液を室温で約3.5時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、1.43g(25.8%)の塩酸リネゾリドが得られた。mp.=163℃。生成物のXRD図を図18Aに示し、得られた赤外スペクトルを図18Bに示す。
【0144】
実施例17.b
アセトニトリルに代えてアセトンを溶媒として使用し0.5g(1.5mmol)のリネゾリドで実施例17.aを繰り返した。塩酸リネゾリドの収量(収率):0.20g(36.4%)。
【実施例18】
【0145】
塩酸メマンチンの製造
0.5g(2.8mmol)のメマンチン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)のメタノールおよび0.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、懸濁液を室温で約2時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、0.59g(98.0%)の塩酸メマンチンが得られた。m.p.=293から296℃。生成物のXRD図を図19Aに示し、得られた赤外スペクトルを図19Bに示す。
【実施例19】
【0146】
塩酸リモナバントI形の製造
実施例19a
1g(2.16mmol)のリモナバントを20mlのアセトニトリルに室温で懸濁させた。懸濁液に0.105ml(1.2当量)のメタノールおよび0.33ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。透明溶液が得られた後まもなく結晶質I形の塩酸リモナバントが沈殿し始めた。生成物を濾別し、真空下に室温で一夜乾燥させると、0.9g(83.4%)の塩酸リモナバントI形が得られた。生成物のXRD図を図20Aに示し、得られた赤外スペクトルを図20Bに示す。
【0147】
実施例19b
1g(2.16mmol)のリモナバントを10mlの酢酸エチルに溶解させた。この溶液に0.105ml(1.2当量)のメタノールおよび0.33ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、白色結晶質固体を濾別し、真空下に室温で乾燥させると、0.95g(88.1%)の塩酸リモナバントI形が得られた。
【0148】
実施例19c
酢酸エチルに代えてアセトンを溶媒として使用し実施例19bを繰り返した。塩酸リモナバントI形の収量(収率):0.89g(82.5%)。
【実施例20】
【0149】
塩酸クロピドグレルI形の製造
実施例20a
1.2g(3.73mmol)のクロピドグレルを10mlのアセトンに室温で溶解させた。溶液に255μl(1.2当量)の酢酸および565μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。6mlのジイソプロピルエーテルの添加後に塩酸クロピドグレルが沈殿し始めた。1時間撹拌後、結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.85g(63.8%)の塩酸クロピドグレルI形が得られた。生成物のXRD図を図21Aに示し、得られた赤外スペクトルを図21Bに示す。
【0150】
実施例20b
1g(3.11mmol)のクロピドグレルを10mlのアセトニトリルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および475μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。混合物を約5℃に冷却し、40mlのジイソプロピルエーテルを撹拌下で添加した。1時間撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.50g(45.0%)の塩酸クロピドグレルI形が得られた。
【0151】
実施例20c
1g(3.11mmol)のクロピドグレルを10mlの酢酸エチルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および475μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に粘性固体が沈殿し、これは約5分以内に結晶質生成物に変化した。混合物を室温で1時間撹拌した。次に生成物を濾別し、真空下に室温で一夜乾燥すると、0.88g(79.3%)の塩酸クロピドグレルI形が得られた。
【0152】
実施例20d
機械的撹拌器と滴下漏斗とを備えた1L容の反応フラスコ中で30g(93.22mmol)のクロピドグレル遊離塩基を1000mlのトルエンに溶解させた。溶液に4.5ml(1.2当量)のメタノールを添加し、撹拌によって混合した後、14.1ml(1.2当量)のトリメチルクロロシランを室温でゆっくりと撹拌しながら45分以内に滴下した。添加後、反応混合物を室温で約3時間撹拌するうちに綿毛状の沈殿物が白色結晶質固体に変化した。塩を濾別し、20mlのトルエンで洗浄した。生成物を真空下に室温で一夜乾燥すると、29.1g(87.1%)の塩酸クロピドグレルI形が得られた。
【実施例21】
【0153】
臭化水素酸クロピドグレルA形の製造
実施例21a
1g(3.11mmol)のクロピドグレルを10mlの酢酸エチルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および482μl(1,2当量)のトリメチルブロモシランを撹拌下で添加した。ブロモシランの添加後に粘性固体が沈殿し、約30分以内に結晶質生成物に変化した。混合物を室温で1時間撹拌した。次に生成物を濾別し、真空下に室温で一夜乾燥すると、1.03g(82.3%)の臭化水素酸クロピドグレルA形が得られた。生成物のXRD図を図22Aに示し、得られた赤外スペクトルを図22Bに示す。
【0154】
実施例21b
1g(3.11mmol)のクロピドグレルを10mlのイソプロパノールに溶解させた。溶液に482μl(1.2当量)のトリメチルブロモシランを添加した。90分間撹拌後、ガラス棒で引掻いた後に生成物が結晶化した。臭化水素酸塩を濾別し、真空下に室温で一夜乾燥すると、0.86g(68.7%)の臭化水素酸クロピドグレルA形が得られた。
【実施例22】
【0155】
塩酸プラスグレルB形の製造
5.0g(13.4mmol)のプラスグレル塩基を75mlのアセトンに室温で溶解させた。溶液に919μl(1.2当量)の酢酸および2053μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。添加後直ちに白色沈殿物が得られた。塩酸塩を濾別し、真空下に室温で6時間乾燥すると、4.41g(80.4%)の塩酸プラスグレルB形が得られた。生成物のXRD図を図23Aに示し、得られた赤外スペクトルを図23Bに示す。
【実施例23】
【0156】
塩酸プラスグレルアセトニトリル溶媒和物の製造
0.5g(1.34mmol)のプラスグレル塩基を7.5mlのアセトニトリルに溶解させた。非溶解分を濾別した。溶液に82μl(1.2当量)の酢酸および205μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。混合物を室温で17時間撹拌した。懸濁液を濾別し、真空下に室温で一夜乾燥させると、0.43g(78%)の塩酸プラスグレルアセトニトリル溶媒和物が得られた。
【0157】
1H−NMR(DMSO−d6):0.84−0.97(m,2H),0.98−1.16(m,2H),1.87−2.0(m,1H),2.08(s,2H),2.29(s,3H),2.96−3.15(m,2H),3.34−3.6(m,1H),3.7−4.2(m,3H),6.14(s,broad,1H),6.58(s,1H),7.3−7.55(m,2H),7.55−7.8(m,2H)。
【0158】
2.08ppmの一重項は基質1.5モルあたり約1モルのアセトニトリルに対応する。生成物のXRD図を図24Aに示し、赤外スペクトルを図24Bに示す。
【実施例24】
【0159】
塩酸ラロキシフェンA形の製造
実施例24a
1.0g(1.77mmol)の乳酸ラロキシフェンを10mlのアセトニトリルに室温で懸濁させた。懸濁液に272μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.80g(82.5%)の塩酸ラロキシフェンA形が得られた。生成物のXRD図を図25Aに示し、得られた赤外スペクトルを図25Bに示す。
【0160】
実施例24b
1.5g(2.66mmol)の乳酸ラロキシフェンを30mlのメチルイソプロピルケトン(MIBK)に室温で懸濁させた。懸濁液を100℃に加熱した。懸濁液に408μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。懸濁液を室温に冷却した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.42g(97.6%)の塩酸ラロキシフェンA形が得られた。
【0161】
実施例24c
0.51g(0.90mmol)のラロキシフェン塩基を5mlのエタノールに室温で懸濁させた。懸濁液に162μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。透明溶液が形成された後まもなく結晶質形態の塩酸ラロキシフェンが沈殿し始めた。3時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.41g(74.6%)の塩酸ラロキシフェンA形が得られた。
【実施例25】
【0162】
塩酸ラロキシフェンTHF半溶媒和物の製造
実施例25a
0.50g(0.88mmol)のラロキシフェン塩基を5mlのテトラヒドロフランに室温で懸濁させた。懸濁液に51μl(1.2当量)のメタノールおよび162μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で3時間撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.45g(97.6%)の塩酸ラロキシフェンTHF半溶媒和物が得られた。
【0163】
1H−NMR(DMSO−d6):1.25−1.4(m,1H),1.6−1.9(m,7H),2.8−3.1(m,2H),3.3−3.55(m,4H),3.55−3.65(m,2.6H),4.4(t,broad,2H),6.7−6.8(m,2H),6.9(dd,J=2.26 and 8.8Hz,1H),6.98(d,J=9Hz,1H),7.15−7.22(m,2H),7.3(d,J=8.7Hz,1H),7.4(d,J=2.3Hz.1H),7.71(d,J=9Hz,1H),9.95(d,J=9Hz,1H),10.8(s,braod,1H)。
【0164】
プロトンNMR分光法によって測定すると結晶質材料中に存在するテトラヒドロフランの量は約0.6モル当量であった。生成物のXRD図を図26Aに示し、得られた赤外スペクトルを図26Bに示す。
【0165】
実施例25b
0.30g(0.53mmol)の乳酸ラロキシフェンを3mlのテトラヒドロフランに室温で懸濁させた。懸濁液に82μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.28g(102.6%)の塩酸ラロキシフェンTHF半溶媒和物が得られた。
【実施例26】
【0166】
ジ塩酸オランザピンの製造
実施例26a
1.0g(3.20mmol)のオランザピン塩基を10mlのアセトニトリルに室温で懸濁させた。懸濁液に312μl(2.4当量)のメタノールおよび980μl(2.4当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.24g(100.54%)のジ塩酸オランザピン1形が得られた。生成物のXRD図を図27Aに示し、得られた赤外スペクトルを図27Bに示す。
【0167】
実施例26b
1.0g(3.20mmol)のオランザピン塩基を10mlのアセトンに室温で懸濁させた。懸濁液に312μl(2.4当量)のメタノールおよび980μl(2.4当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.35g(107.31%)のジ塩酸オランザピン1形が得られた。
【実施例27】
【0168】
臭化水素酸ダリフェナシンの製造
0.9g(2.11mmol)のダリフェナシン塩基を9mlのメチルエチルケトンに室温で溶解させた。溶液に103μl(1.2当量)のメタノールおよび329μl(1.2当量)のトリメチルブロモシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.88mg(82.13%)の臭化水素酸ダリフェナシンが得られた。生成物のXRD図を図28Aに示し、得られた赤外スペクトルを図28Bに示す。
【実施例28】
【0169】
非晶質形態の無水塩酸シタグリプチンの製造
0.085g(0.21mmol)のシタグリプチン塩基を2mlのジエチルエーテルおよび3mlのメチレンクロリドに室温で溶解させた。溶液に10μl(1.2当量)のメタノールおよび32μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。添加後直ちに沈殿物が得られた。懸濁液を1時間撹拌し、濾別し、真空下に室温で一夜乾燥すると、0.089g(96.8%)の非晶質形態の塩酸シタグリプチンが得られた。生成物のXRD図を図29Aに示し、得られた赤外スペクトルを図29Bに示す。
【実施例29】
【0170】
無水ジ塩酸バルデナフィルの製造
0.2g(0.359mmol)のバルデナフィル塩基を4mlのジエチルエーテルに室温で懸濁させた。懸濁液に5mlのメチレンジクロリドを添加すると溶液が得られた。35μl(2.4当量)のメタノールおよび110μl(2.4当量)のトリメチルクロロシランを溶液に添加すると、添加後直ちに白色沈殿物が得られた。懸濁液を20分間撹拌し、濾別し、真空下に室温で一夜乾燥すると、0.2g(95,.4%)のジ塩酸バルデナフィルが得られた。生成物のXRD図を図30Aに示し、得られた赤外スペクトルを図30Bに示す。
【実施例30】
【0171】
塩酸エルロチニブA形の製造
0.3g(0.82mmol)のエルロチニブ塩基を3mlのイソプロパノールに懸濁させた。懸濁液に126μl(1.2当量)のトリメチルクロロシランを添加した。白色沈殿物が得られた。1時間の撹拌後に沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.26g(80.8%)の塩酸エルロチニブA形が得られた。生成物のXRD図を図31Aに示し、得られた赤外スペクトルを図31Bに示す。
【図1A】


【図1B】
【図1C】
【図2A】
【図2B】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図10A】
【図10B】
【図11A】
【図11B】
【図12A】
【図12B】
【図13A】
【図13B】
【図13C】
【図14A】
【図14B】
【図15A】
【図15B】
【図15C】
【図16A】
【図16B】
【図16C】
【図17A】
【図17B】
【図18A】
【図18B】
【図19A】
【図19B】
【図20A】
【図20B】
【図21A】
【図21B】
【図22A】
【図22B】
【図23A】
【図23B】
【図24A】
【図24B】
【図25A】
【図25B】
【図26A】
【図26B】
【図27A】
【図27B】
【図28A】
【図28B】
【図29A】
【図29B】
【図30A】
【図30B】
【図31A】
【図31B】
【図17A】塩酸デュロキセチンのX線粉末回折図である。
【図17B】塩酸デュロキセチンの赤外スペクトルである。
【図18A】塩酸リネゾリドのX線粉末回折図である。
【図18B】塩酸リネゾリドの赤外スペクトルである。
【図19A】塩酸メマンチンのX線粉末回折図である。
【図19B】塩酸メマンチンの赤外スペクトルである。
【図20A】塩酸リモナバントI形のX線粉末回折図である。
【図20B】塩酸リモナバントI形の赤外スペクトルである。
【図21A】塩酸クロピドグレルI形のX線粉末回折図である。
【図21B】塩酸クロピドグレルI形の赤外スペクトルである。
【図22A】臭化水素酸クロピドグレルA形のX線粉末回折図である。
【図22B】臭化水素酸クロピドグレルA形の赤外スペクトルである。
【図23A】塩酸プラスグレルB形のX線粉末回折図である。
【図23B】塩酸プラスグレルB形の赤外スペクトルである。
【図24A】塩酸プラスグレルアセトニトリル溶媒和物のX線粉末回折図である。
【図24B】塩酸プラスグレルアセトニトリル溶媒和物の赤外スペクトルである。
【図25A】塩酸ラロキシフェンA形のX線粉末回折図である。
【図25B】塩酸ラロキシフェンA形の赤外スペクトルである。
【図26A】塩酸ラロキシフェンテトラヒドロフラン溶媒和物のX線粉末回折図である。
【図26B】塩酸ラロキシフェンテトラヒドロフラン溶媒和物の赤外スペクトルである。
【図27A】ジ塩酸オランザピンI形のX線粉末回折図である。
【図27B】ジ塩酸オランザピンI形の赤外スペクトルである。
【図28A】臭化水素酸ダリフェナシンのX線粉末回折図である。
【図28B】臭化水素酸ダリフェナシンの赤外スペクトルである。
【図29A】塩酸シタグリプチン無水形のX線粉末回折図である。
【図29B】塩酸シタグリプチン無水形の赤外スペクトルである。
【図30A】ジ塩酸バルデナフィルのX線粉末回折図である。
【図30B】ジ塩酸バルデナフィルの赤外スペクトルである。
【図31A】塩酸エルロチニブA形のX線粉末回折図である。
【図31B】塩酸エルロチニブA形の赤外スペクトルである。
【発明を実施するための形態】
【0009】
本発明は、ハロゲン化トリアルキルシリルが塩基性薬剤基質のハロゲン化水素酸塩または塩基性中間体のハロゲン化水素酸塩の製造に、特に無水条件が必要な場合および/または純粋形態の塩の明確に確定した結晶構造を製造しなければならない場合に、極めて好適であるという知見に関する。
【0010】
従って本発明は、ハロゲン化トリアルキルシリルを溶媒中で有機アミンに添加する段階を含む、有機アミンの結晶質ハロゲン化物の製造方法に関する。該有機アミンは遊離塩基または酸付加塩の形態であり、酸付加塩の共役酸はハロゲン化水素酸よりも弱い酸である。
【0011】
本発明方法に適した溶媒は、プロトン性溶媒、または、有機アミンに比べて少なくとも1当量のプロトン性溶媒に組合せた非プロトン性溶媒である。プロトン性溶媒は、シリル化可能溶媒、すなわち、ハロゲン化トリアルキルシリルに添加されたときにハロゲン化水素酸塩をその場に生成する能力を有している溶媒である。適当なプロトン性溶媒は例えば、脂肪族アルコールもしくは芳香族アルコール、シラノール、エノール化可能なケトン、または、脂肪族もしくは芳香族カルボン酸(carbonic acid)である。非プロトン性溶媒は、アミンを溶解もしくは懸濁させることができるか、または、形成されるハロゲン化水素酸塩を確定した結晶構造に導くことができるシラン化に対して不活性の溶媒である。
【0012】
適当な非プロトン性溶媒は例えば、エステル特に酢酸エチル、ニトリル特にアセトニトリル、ケトン特にアセトン、エーテル特に第三級ブチルメチルエーテル、ハロゲン化溶媒特にジクロロメタン、芳香族溶媒特にトルエン、アルカン特にヘキサン、および、ニトロアルケン特にニトロメタンである。
【0013】
本発明に含意される有機アミンは、第一級、第二級、第三級または第四級アミノ基と3よりも多い数の炭素−炭素結合とを含み、80Daを上回る分子量好ましくは100Daから5000Daの範囲の分子量を有している有機化合物である。
【0014】
1つの実施態様において本発明の方法は、
(a)プロトン性溶媒中に有機アミンの遊離塩基を溶解させるかまたは有機アミンの遊離塩基を好ましくは撹拌を伴って懸濁させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)結晶(有機アミンのハロゲン化水素酸塩の結晶)を形成させる段階
(d)形成された結晶を収集する段階
を含む。
【0015】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸モキシフロキサシン酢酸溶媒和物、臭化水素酸シタロプラム、臭化水素酸クロピドグレル、塩酸ラロキシフェンA形または塩酸エルロチニブA形の製造に使用される。
【0016】
段階(b)中にハロゲンシランはシリル化剤としておそらくはプロトン性溶媒と直ちに反応しこれによりハロゲン化水素酸をその場に生成し、生成したHCl、HBrまたはHIが遊離塩基をハロゲン化水素酸塩に変換する。
【0017】
段階(d)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0018】
好ましいハロゲン化トリアルキルシリルは、3つのアルキル残基が同一であり、特にそれらがメチル、エチル、プロピル、ブチルまたはイソプロピルを表すものである。最も好ましいハロゲン化トリアルキルは、トリメチルクロロシラン、トリメチルブロモシランおよびトリメチルヨードシランである。
【0019】
本発明において使用した“プロトン性溶媒”という用語は、添加すべき有機アミンに比べてプロトン性溶媒の合計量が1分子当量を上回るような、特にプロトン性溶媒の合計量が添加すべき有機アミンの約1分子当量であるような溶媒混合物から成る溶媒系を包含する。
【0020】
段階(a)で使用される好ましいプロトン性溶媒は、ヒドロキシル基またはカルボキシル基を含む溶媒である。特に好ましいプロトン性溶媒は、芳香族もしくは脂肪族アルコール、シラノール、エノール化可能なケトン、または、芳香族もしくは脂肪族カルボン酸(carbonic acid)、または、それらの混合物から成る溶媒系である。より好ましいプロトン性溶媒はメタノール、エタノール、2−プロパノール(イソプロパノール)、ブタノール(例えばn−ブタノールおよびイソブタノール)のようなC1−C6アルキルアルコール、または、C1−C6アルキルカルボン酸特にギ酸もしくは酢酸である。ハロゲン化水素酸塩の乾燥を含む最終処理(workup)および単離は当業界で公知の方法を使用して行うとよい。
【0021】
別の実施態様において、有機化合物のハロゲン化水素酸塩の製造方法は、
(a)有機アミンの遊離塩基を非プロトン性溶媒に溶解させて溶液を形成するかまたは非プロトン性溶媒中の遊離塩基の懸濁液を撹拌する段階
(b)段階(a)で形成された溶液または懸濁液に少なくとも1当量特に約1当量のプロトン性溶媒、特に上記に定義したような好ましいプロトン性溶媒を添加する段階
(c)段階(b)の溶液または懸濁液を少なくとも1当量特に約1当量のハロゲン化トリアルキルシリル特に上記に定義したような好ましいハロゲン化トリアルキルシリルで処理して対応するハロゲン化水素酸塩を形成する段階
(d)有機アミンのハロゲン化水素酸塩の結晶を形成させる段階
(e)形成された結晶を収集する段階
を含む。
【0022】
段階(e)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0023】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸セルトラリンII形、塩酸アリピプラゾール、モノ塩酸プラミペキソール、塩酸メマンチン、塩酸リモナバントI形、塩酸クロピドグレルI形、臭化水素酸クロピドグレルA形、塩酸プラスグレルA形、塩酸プラスグレルアセトニトリル溶媒和物、塩酸ラロキシフェンテトラヒドロフラン半溶媒和物、ジ塩酸オランザピンI形、臭化水素酸ダリフェナシン、塩酸シタグリプチンまたはジ塩酸バルデナフィルの製造に使用される。
【0024】
非プロトン性溶媒が使用されるとき、有機アミンの遊離塩基自体がハロゲン化トリアルキルと反応してシリル化ハロゲン化水素を形成し、シリル化生成物がその後1当量以上のアルコールのようなプロトン性溶媒の添加によって加水分解できる。従って段階(b)と(c)との順序を入れ替えてもよい。また、最初にハロゲン化トリアルキルをプロトン性溶媒と混合してハロゲン化水素酸を生成し、次にこの混合物を溶解または懸濁した有機アミンに添加してもよい。
【0025】
別の実施態様において、有機化合物のハロゲン化水素酸塩の製造方法は、
(a)溶媒中で有機アミンの酸付加塩を溶解、懸濁または生成させる段階
(b)ハロゲン化トリアルキルシリルを添加する段階
(c)有機アミンのハロゲン化水素酸塩の結晶を形成させる段階
(d)形成された結晶を収集する段階
を含む。
【0026】
段階(d)は当業界の適当な方法のいずれか例えば濾過によって行うことができる。
【0027】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、塩酸セルトラリンII形、塩酸ラロキシフェンA形または塩酸ラロキシフェンテトラヒドロフラン半溶媒和物の製造などに使用される。
【0028】
段階(a)において、有機アミンの酸付加塩は溶媒中の塩基の溶液または懸濁液に有機酸を添加することによってその場に生成できる。アミンの有機酸塩の生成に使用される有機酸は塩酸よりも弱い酸でなければならないので、好ましくは置換または未置換のアルカン酸、芳香族カルボン酸、ジカルボン酸またはクエン酸から成るグループから選択され、酢酸が特に好ましい。
【0029】
段階(a)において好ましくは、有機アミンの酸付加塩は有機アミンの溶液またはスラリーに酸を添加することによってその場に生成され、共役酸は塩酸よりも弱い酸であり、好ましくは有機酸である。
【0030】
本発明の好ましい実施態様において、この方法は、例えば後でさらに詳述するように、結晶質無水形の塩酸ミコフェノラートモフェチル、塩酸ベンラファキシンI形、塩酸ベンラファキシンII形、塩酸セルトラリンII形、塩酸ドネペジルIII形、塩酸テルビナフィン、塩酸シナカルセット、塩酸モキシフロキサシンメチレンジクロリド溶媒和物、塩酸モキシフロキサシンニトロメタン溶媒和物、塩酸モキシフロキサシン酢酸溶媒和物、臭化水素酸シタロプラム、塩酸デュロキセチンまたは塩酸リネゾリドの製造などに使用される。
【0031】
塩酸塩は入手容易性および生理的寛容性があるので塩基性薬剤の塩としてこれまで最も頻繁に選択されており、すべての薬剤の塩のほぼ半数が塩酸塩であり、以後の文節中ではすべてのハロゲン化水素酸塩の代表例の役割を果たす。
【0032】
ハロゲン化水素酸塩の形成はときには厳密な無水条件を必要とし、その場合には特に上述のような塩酸のその場に生成および塩形成を誘発するために塩化トリアルキルシリルを使用するのが有利である。またあるときは、塩酸を形成するために有機アミンに対して限定された化学論的量のHClが必要であり、この要件は上述のような塩化トリアルキルシリルの使用によって容易に充足できる。溶媒中に正確な計算量のハロゲン化水素酸が必要とされるときであっても、ハロゲン化トリアルキルシリルを使用するほうがシリンダーから供給される無水ガスを用いて溶液を製造するよりも容易なかつ優れた方法である。
【0033】
言い換えると、本発明の方法においてハロゲン化水素酸をその場に生成するためのハロゲン化トリアルキルシリルの使用は、塩形成中のハロゲン化水素酸と有機アミンとの化学量論比を極めて良好にコントロールできる。このことは、有機アミンの2つ以上のハロゲン化水素酸塩例えばハロゲン化モノ水素酸塩とハロゲン化ジ水素酸塩とが存在するときに特に有利である。何故ならこのような場合、生成すべきハロゲン化水素酸の量を選択することによって所望のハロゲン化水素酸塩が得られる方向に方法を誘導できるからである。例えば、モノハロゲン化モノ水素酸塩を得ることを望むが競合するジハロゲン化水素酸塩も存在する場合、1当量のハロゲン化水素酸だけがその場に生成されるようにハロゲン化トリアルキルシリルの添加量を限定し、モノハロゲン化水素酸塩が生成する方向に結晶化を誘導できる。
【0034】
本発明の方法の別の利点は、水の実質的な非存在下で処理することもでき、その結果として有機アミンのハロゲン化水素酸塩の無水形を得るのが可能なことである。さらに、ハロゲン化トリアルキルシリルの添加は極めて安定な段階であり、広い温度範囲内で行うことができ、多様な溶媒系に適合性なので、ハロゲン化トリアルキルシリルの添加は大抵の場合、所望の結晶形を得るために最適であることが判っているまさにその条件下で行うことができる。例えば、薬剤Xのモノ塩酸塩の多形相Aは溶媒Y中で低温で得られるが同じモノ塩酸塩の多形相Bは溶媒Z中で周囲温度で得られることが判っている場合、所望の形AまたはBを形成させるように条件を選択するだけでよい。この結晶化条件の選択を可能にする理由はまさに、ハロゲン化トリアルキルシリル添加段階の安定性にある。
【0035】
別の実施態様において、本発明の方法は第一級、第二級、第三級または第四級アミノ基をもつ薬剤のハロゲン化水素酸塩の生成に使用され、薬剤は特に、セロトニン再吸収インヒビターであるセルトラリン、デュロキセチン、ベンラファキシンおよびシタロプラムなどの抗鬱薬、特にドネペジルのような精神向性薬、特に神経遮断薬アリピプラゾールおよびセロトニン/ドパミンアンタゴニストであるオランザピンのような統合失調症治療薬、特に鎮痙薬メマンチンのような筋弛緩薬、特にミコフェノラートモフェチルのような免疫抑制薬、特にテルビナフィンのような抗真菌薬、特にキノロン例えばモキシフロキサシンまたはオキサゾリジノンリネゾリドのような抗菌薬、特にシナカルセットのようなカルシウム模擬薬、特にD2受容体アゴニストであるプラミペキソールのようなドパミンアゴニスト、リモナバントのような抗肥満薬、クロピドグレルおよびプラスグレルのような抗血栓症薬、特にラロキシフェンのような抗骨粗鬆症薬、ダリフェナシンのような鎮痙薬、バルデナフィルのような男性勃起機能不全治療薬、特にDPP−IVインヒビターであるシタグリプチンのような抗糖尿病薬、エルロチニブのような抗新生物薬から選択される。
【0036】
別の実施態様において、本発明は、本発明の方法による結晶質無水塩酸ミコフェノラートモフェチルの製造に関する。より詳細には方法は、(a)ミコフェノラートモフェチル塩基を酢酸エチル、アセトニトリルまたはアセトン、特に酢酸エチルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液をトリシリルアルキルクロロシラン、特に約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む。
【0037】
この方法によって、無水塩酸ミコフェノラートモフェチルが沈殿し、97%を上回る収率で単離できる。結晶質生成物のFT−IR、DSCおよびXRPDデータはWO95/07902に示されているIR、DSCおよびX線結晶学データに一致する。WO95/07902は、無水塩酸ミコフェノラートモフェチルが一水和物塩形のほぼ2倍増の溶解度を有しながらも一水和物塩形に特有の安定性を維持していると開示している。本発明の方法は塩酸一水和物の形成を阻止するので、無水形を製造するために塩酸ミコフェノラートモフェチル一水和物を加熱する必要がない。
【0038】
別の実施態様において、本発明は、方法に塩化水素ガスを使用しない本発明の方法による塩酸ベンラファキシンの製造、特に、純粋形態の塩酸ベンラファキシンI形またはII形の製造方法に関する。好ましい実施態様において、本発明は、塩酸ベンラファキシンI形の製造方法に関する。方法は、(a)ベンラファキシン塩基を酢酸エチルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液を約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む。この方法によって無水塩酸ベンラファキシンI形が沈殿し、89%を上回る収率で単離できる。結晶質生成物のXRPDデータはWO02/45658(Teva)に示されているI形およびWO02/36542(Ciba)に示されているB形のX線結晶学データに対応する。
【0039】
別の好ましい実施態様において、本発明は、(a)ベンラファキシン塩基を溶媒となるアセトンまたはアセトニトリルに溶解させる段階(b)酢酸、特に1.0から約1.5当量に等しい酢酸を添加する段階(c)溶液を約1.0から約1.5当量のトリメチルクロロシランで処理する段階を含む塩酸ベンラファキシンII形の製造方法に関する。この方法によって無水塩酸ベンラファキシンII形が得られる。結晶質生成物のXRPDデータはWO02/45658(Teva)に示されているII形およびWO02/36542(Ciba)に示されているC形のX線結晶学データに対応する。
【0040】
別の実施態様において、本発明は、本発明の方法による塩酸セルトラリンII形の製造に関する。塩酸セルトラリンII形は準安定形であり、通常は有機溶媒からの塩酸セルトラリンの急速結晶化によって製造される。しかしながら、II形の優先的な形成は、容易にコントロールできない結晶化の速さに左右される。従って、塩酸セルトラリンII形の製造が必要である。本発明の方法は、この準安定II形を純粋形態でかつ簡単なやり方で工業生産できる。従って本発明はまた、I形だけの特有なXRPDピークの非存在、例えば14.9および26.3度の2シータにピークが存在しないことによって判定されるようなセルトラリンI形を検出可能なレベルで含まない、すなわち、1.0%未満、特に0.5%未満のI形を含む塩酸セルトラリンII形に関する。
【0041】
従って1つの実施態様において、本発明は、
(a)非プロトン性溶媒例えばアセトン、メチルエチルケトン、メチルイソブチルケトンまたはアセトニトリルにセルトラリン遊離塩基を溶解させる段階
(b)段階(a)の溶液にn−ブタノールまたは酢酸のようなプロトン性溶媒を1.0から約1.5当量に等しい量で添加する段階
(c)段階(b)の溶液をトリアルキルシリルクロリド例えばトリメチルクロロシランで処理する段階を含む塩酸セルトラリンII形の製造方法に関する。
【0042】
トリメチルクロロシランは全量を一度に添加してもよくまたは2回以上の分量として添加してもよくまたは漸増的に添加してもよい。トリメチルクロロシランとの反応はセルトラリンが可溶性であるいかなる温度で行わせてもよい。溶媒がアセトニトリルのとき、反応は典型的には約20から80℃の範囲の温度、より典型的には約20から50℃の範囲の温度で行う。溶媒がメチルイソブチルケトンのとき、反応は典型的には約50から100℃の範囲の温度、より典型的には約80℃の温度で行う。トリメチルクロロシランは典型的にはセルトラリン1当量あたり約1から約2当量のトリメチルクロロシランとなる量で添加する。段階(b)のプロトン性溶媒は典型的にはトリメチルクロロシランの使用量に等価の量で添加する。トリメチルクロロシランの添加後、均質混合物が得られる時間まで反応混合物を熟成させる。
【0043】
別の好ましい実施態様において、本発明は、
(a)非プロトン性溶媒例えばメチルエチルケトン、メチルイソブチルケトンまたはアセトニトリル中のセルトラリンの有機塩例えばマンデル酸塩またはシュウ酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリアルキルシリルクロリド例えばトリメチルクロロシランで処理する段階
を含む、セルトラリンの有機塩を使用する塩酸セルトラリンII形の製造方法に関する。
【0044】
典型的には、溶媒がメチルエチルケトンのときトリメチルクロロシランとの反応は約50から80℃の範囲の温度で行い、溶媒がメチルイソブチルケトンのとき反応は約50から100℃の範囲およびそれ以上の温度、典型的には約80℃で行う。溶媒がアセトニトリルのときはトリメチルクロロシランとの反応を周囲温度でも行うことができる。トリメチルクロロシランは典型的には有機塩1当量あたり約1から約2当量のトリメチルクロロシラン、より典型的には有機塩1当量あたり1.1当量のトリメチルクロロシランとなる量で添加される。
【0045】
本発明はさらに、塩酸セルトラリンI形の製造方法に関する。方法は、(a)セルトラリン遊離塩基を室温でイソプロパノールに溶解させる段階(b)溶液をトリメチルクロロシランで処理する段階とを含む。
【0046】
本発明はさらに、無水塩酸ドネペジルを製造する本発明の方法に関する。溶媒の選択次第で塩酸ドネペジルII形またはIII形を製造できる。
【0047】
第一の実施態様において、これは、
(a)ドネペジル遊離塩基を酢酸エチル、ジメトキシエタンまたはメチルイソブチルケトンに溶解させる段階
(b)少なくとも1当量のプロトン性溶媒例えばn−ブタノールまたは酢酸を添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
(d)塩酸ドネペジルII形を単離する段階
を含む塩酸ドネペジルII形の製造方法である。
【0048】
第二の実施態様において、これは、無水塩酸ドネペジルIII形の製造方法であり、この方法ではアセトンまたはアセトニトリルが溶媒として使用される。結晶質生成物である塩酸ドネペジルII形および塩酸ドネペジルIII形のXRPDデータは、WO97/46527(Eisai)に示されているII形およびIII形のX線結晶学データに一致する。
【0049】
本発明はさらに、本発明の方法による塩酸テルビナフィン、塩酸シナカルセット、塩酸デュロキセチン、塩酸メマンチンおよびモノ塩酸プラミペキソールの製造方法に関する。方法は好ましくは、
(a)テルビナフィン、シナカルセット、デュロキセチン、メマンチンまたはプラミペキソールの遊離塩基を非プロトン性溶媒に溶解させる段階
(b)少なくとも1当量のプロトン性溶媒例えば酢酸またはメタノールもしくはn−ブタノールのようなアルコールを添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
を含む。
【0050】
上記の方法において、テルビナフィンは、例えばアセトン、アセトニトリルまたはtert−ブチルメチルエーテルのような非プロトン性溶媒に溶解させることができ、シナカルセットは、例えばアセトニトリルまたは酢酸エチルのような非プロトン性溶媒に溶解させることができでき、プラミペキソールは、例えばアセトニトリルのような非プロトン性溶媒に溶解させることができる。
【0051】
結晶質生成物である塩酸テルビナフィンのXRPDデータは、E.Tedescoら,CrystEngComm,2002,4(67),393−400によって公表されたX線結晶学データに一致する。シナカルセットの結晶質塩酸塩の特性X線粉末回折図を図9Aに示し、プラミペキソールの結晶質塩酸塩の特性X線粉末回折図を図12Aに示す。さらに、シナカルセットの結晶質塩酸塩はまた図9Bに示すような典型的赤外スペクトルによって特性決定され、結晶質モノ塩酸プラミペキソールは図12Bに示すような典型的赤外スペクトルによって特性決定される。デュロキセチンの結晶質塩酸塩の特性X線粉末回折図を図18Aに示し、塩酸メマンチンの特性X線粉末回折図を図20Aに示す。さらに、デュロキセチンの結晶質塩酸塩はまた図18Bに示すような典型的赤外スペクトルによって特性決定され、結晶質塩酸メマンチンは図19Bに示すような典型的赤外スペクトルによって特性決定される。
【0052】
本発明はまた、上記に定義の一般方法による臭化水素酸シタロプラムを製造するための本発明の方法に関する。1つの実施態様において本発明は、シタロプラム遊離塩基をアルコールすなわちメタノールまたはイソプロパノールのようなプロトン性溶媒に溶解させ、1.0から約1.5当量に等しいトリメチルブロモシランを添加する段階を含む臭化水素酸シタロプラムの製造方法に関する。
【0053】
別の実施態様において、本発明は、
(a)シタロプラムの遊離塩基を酢酸エチル、アセトンまたはアセトニトリルのような非プロトン性溶媒に溶解させる段階
(b)少なくとも1、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1、特に約1当量のトリメチルブロモシランで処理する段階
(d)臭化水素酸シタロプラムを単離する段階
を含む臭化水素酸シタロプラムの製造方法に関する。結晶質臭化水素酸シタロプラムが得られる。結晶質臭化水素酸シタロプラムの特性X線粉末回折図を図10Aに示し、典型的赤外スペクトルを図10Bに示す。
【0054】
本発明はまた、塩酸アリピプラゾールA形を製造するための発明の方法に関する。方法は、
(a)アリピプラゾールの遊離塩基を非プロトン性溶媒例えばメチレンジクロリドに溶解させる段階
(b)少なくとも1、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1、特に約1当量のトリメチルクロロシランで処理する段階
(d)塩酸アリピプラゾールA形を単離する段階
を含む。結晶質生成物である塩酸アリピプラゾールのXRPDデータはWO2004/083183(Hetero Drugs Ltd.)に示されているようなX線結晶学データA形に対応する。
【0055】
リモナバントを例えばアセトニトリルに懸濁させるかまたは酢酸エチルに溶解させ、少なくとも1当量のメタノールおよびトリメチルクロロシランを添加すると、結晶形Iのリモナバントの塩酸塩が得られる。塩酸リモナバントI形のX線粉末回折図を図20Aに示す。この図は、10.4、14.4、17.8、19.2、20.8、21.9、22.2、26.4、26.9、28.7および28.5度の2シータに主要ピークを示す。塩酸リモナバントI形の赤外スペクトルを図20Bに示す。塩酸リモナバントI形はEP0656354(Sanofi)の実施例3に従って製造された生成物に一致する。該実施例では、リモナバントをエーテルに溶解させ、HClガスの飽和エーテル溶液を部分量ずつ添加する。
【0056】
I形の塩酸クロピドグレルは例えば、少なくとも1当量の酢酸およびトリメチルクロロシランを酢酸エーテル中のクロピドグレル塩基の溶液に添加することによって得ることができる。アセトンまたはアセトニトリルを溶媒として使用するときは、塩酸クロピドグレルI形を沈殿させるためにジイソプロピルエーテルのような反溶媒を添加する。1つの好ましい実施態様においては、クロピドグレル塩基をトルエンに溶解させ、1当量のメタノールを添加し、溶液をトリメチルクロロシランで処理する。トリメチルクロロシランの添加後、綿毛状の沈殿物を室温で暫時撹拌して塩酸クロピドグレルI形に変化させる。
【0057】
塩酸プラスグレルB形は例えば、少なくとも1当量の酢酸およびトリメチルクロロシランをアセトン中のプラスグレル塩基の溶液に添加することによって得ることができる。塩酸プラスグレルB形のXRPD図を図23Aに示し、赤外スペクトルを図23Bに示す。US6693115の実施例3、4および6に報告されているように特性赤外吸収バンドは1758および1690cm−1に存在する。
【0058】
本発明はさらに、新規な塩酸プラスグレルアセトニトリル溶媒和物に関する。これは、8.3、13.8、16.2、18.8、23.8、25.4および26.8度の2シータにピークを含むX線粉末回折図によって特性決定される。塩酸プラスグレルアセトニトリル溶媒和物のX線粉末回折図の一例を図24Aに示す。塩酸プラスグレルアセトニトリル溶媒和物はまた、図24Bに示すような典型的赤外スペクトルによって特性決定され得る。特性バンドは1760、1720、1499、1210および775cm−1に存在する。塩酸プラスグレルアセトニトリル溶媒和物は効率的な精製段階を可能にする。
【0059】
塩酸プラスグレルアセトニトリル溶媒和物は好ましくは、
(a)プラスグレルをアセトニトリルに溶解させる段階
(b)少なくとも1当量の酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量のトリメチルクロロシランで処理する段階
(d)塩酸プラスグレルアセトニトリル溶媒和物を単離する段階
を含む本発明の方法によって製造できる。
【0060】
風乾条件下で塩酸プラスグレルアセトニトリル溶媒和物は安定であり脱溶媒和しない。これは、1H−NMRスペクトル(DMSO−d6,TMS)に実質的に同一の1H−NMRスペクトルを有している。より詳細にはこれは、1モルの基質あたり約1モルのアセトニトリルに対応する2.ppm(s,3H)に特性ピークを有している。
【0061】
非晶質形の塩酸シタグリプチンは例えば、少なくとも1当量のメタノールおよびトリメチルクロロシランをメチレンクロリドおよびジエチルエーテルの混合物中のシタグリプチン塩基の溶液に添加することによって得ることができる。非晶質塩酸シタグリプチンのXRPD図を図29Aに示し、赤外スペクトルを図29Bに示す。特性赤外吸収バンドはcm−1に存在する。
【0062】
本発明はまた、アルコールすなわちメタノールまたはイソプロパノールのようなプロトン性溶媒にクロピドグレル遊離塩基を溶解させ、1.0から約1.5当量に等しいトリメチルブロモシランを添加する段階を含む、上記の定義のような一般方法による臭化水素酸クロピドグレルA形の製造方法に関する。
【0063】
臭化水素酸クロピドグレルA形の代替的製造方法は、
(a)クロピドグレルの遊離塩基を酢酸エチルのような非プロトン性溶媒に溶解させる段階
(b)少なくとも1当量の酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量のトリメチルブロモシランで処理する段階
(d)臭化水素酸クロピドグレルA形を単離する段階
を含む本発明の方法である。
【0064】
結晶質臭化水素酸クロピドグレルA形に特有なX線粉末回折図を図22Aに示し、代表的な赤外吸収スペクトルを図22Bに示す。
【0065】
本発明はさらに、塩酸ラロキシフェンA形を製造するための本発明の方法に関する。好ましい実施態様において、塩酸ラロキシフェンA形の製造方法は、
(a)エタノールのようなプロトン性溶媒中のラロキシフェン遊離塩基の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
を含む。
【0066】
塩酸ラロキシフェンA形の代替的製造方法は、
(a)エタノールのようなプロトン性溶媒またはメチルイソブチルケトンもしくはアセトニトリルのような非プロトン性溶媒中のラロキシフェンの有機塩例えば乳酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
を含む。溶媒がメチルエチルケトンのときはトリメチルクロロシランとの反応を典型的には約100℃の温度で行わせ、溶媒がアセトニトリルのときはトリメチルクロロシランとの反応を典型的には周囲温度で行わせる。塩酸ラロキシフェンA形の特性X線粉末回折図を図25Aに示し、典型的赤外スペクトルを図25Bに示す。
【0067】
本発明はまた、新規な塩酸ラロキシフェンとテトラヒドロフランとの半水和物に関する。この物質は、13.6、16.6、17.7、18.9、19.2、19.5、21.2および23.6度の2シータにピークを含むX線粉末回折図によって特性決定される。塩酸ラロキシフェンとテトラヒドロフランとの半水和物のX線粉末回折図の一例を図26Aに示す。塩酸ラロキシフェンとテトラヒドロフランとの半水和物は、図26Bに示す典型的赤外スペクトルによって特性決定される。特性バンドは1651、1595、1226、1165、838および815cm−1に存在する。塩酸ラロキシフェンとテトラヒドロフランとの半水和物は、ラロキシフェン生産のより効率的および/またはより効果的な精製段階を可能にする。
【0068】
本発明はまた、
(a)テトラヒドロフラン中のラロキシフェン遊離塩基の懸濁液を撹拌する段階
(b)1.0から約1.5当量に等しいメタノールのような極性溶媒を段階(a)の懸濁液に添加する段階
(c)段階(b)の懸濁液をトリメチルクロロシランで処理する段階
(d)塩酸ラロキシフェンのテトラヒドロフラン半溶媒和物を単離する段階
を含む塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物を製造するための本発明の方法に関する。
【0069】
塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物の製造方法の代替実施態様は、
(a)テトラヒドロフラン中のラロキシフェンの有機塩例えば乳酸塩の懸濁液を撹拌する段階
(b)懸濁液をトリメチルクロロシランで処理する段階
(c)塩酸ラロキシフェンとテトラヒドロフランとの半溶媒和物を単離する段階
を含むラロキシフェンの有機塩を使用する方法である。風乾条件下で塩酸ラロキシフェンTHF半溶媒和物は安定であり脱溶媒和しない。これは、1H−NMRスペクトル(DMSO−d6)に実質的に同一の1H−NMRスペクトルを有している。
【0070】
本発明はさらに、無水ジ塩酸バルデナフィルを製造するための本発明の方法に関する。方法は、
(a)非プロトン性溶媒、または、例えばEP1049695の実施例10に記載されているようなジエチルエーテルとメチレンジクロリドとの溶媒混合物にバルデナフィル塩基を溶解させる段階
(b)少なくとも2当量のメタノールのようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも2当量のトリメチルクロロシランで処理する段階
(d)無水ジ塩酸バルデナフィルを単離する段階
を含む。無水ジ塩酸バルデナフィルの特性X線粉末回折図を図30Aに示し、典型的赤外スペクトルを図30Bに示す。
【0071】
本発明はさらに、塩酸エルロチニブA形を製造するための本発明の方法に関する。方法は、エルロチニブ塩基をイソプロパノールに懸濁させ、懸濁液を少なくとも1当量のトリメチルクロロシランによって室温で処理する段階を含む。塩酸エルロチニブA形のX線粉末回折図を図31Aに示し、典型的赤外スペクトルを図31Bに示す。結晶質生成物である塩酸エルロチニブA形のXRPDデータはUS2004/0162300(Hofmann−La Roche)に示されているB形のX線結晶学データに一致する。
【0072】
本発明の方法を使用することによってわれわれはさらに、約14.6、18.7、21.9、23.5および25.3度の2シータにピークをもつX線粉末回折図によって特性決定される塩酸モキシフロキサシンメチレンジクロリド溶媒和物を発見した。従って本発明はまた、特に塩酸モキシフロキサシンメチレンジクロリド溶媒和物、特に塩酸モキシフロキサシン対メチレンジクロリドのモル比が約1:1であるこのような溶媒和物に関する。特にこの溶媒和物は、X線粉末回折図で約14.6、18.7、21.9、23.5および25.3度の2シータにピークを示す。塩酸モキシフロキサシンメチレンジクロリド溶媒和物の特性X線粉末回折図を図13Aに示す。塩酸モキシフロキサシンメチレンジクロリド溶媒和物は後述する塩酸モキシフロキサシンIV形の製造を初めて可能にする。
【0073】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物はまた、赤外スペクトルの約2704、1720、1434、1311、1272、730および702cm−1に存在する特性バンド、特に図13Bに示すような典型的赤外スペクトルによって特性決定される。
【0074】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物は、
(a)モキシフロキサシンをメチレンジクロリドに溶解させる段階
(b)少なくとも1当量、特に約1当量のプロトン性溶媒例えばn−ブタノールまたは酢酸を添加する段階
(c)溶液を少なくとも1当量、特に約1当量のトリアルキルクロロシラン例えばトリメチルクロロシランで処理する段階
(d)塩酸モキシフロキサシンメチレンジクロリド溶媒和物を単離する段階
を含む本発明の方法によって製造され得る。
【0075】
風乾条件下で塩酸モキシフロキサシンメチレンジクロリド溶媒和物は安定であり、脱溶媒和しない。塩酸モキシフロキサシンメチレンジクロリド溶媒和物のTGAは80から180℃の範囲で16%の減量を示し、これは基質1モルあたり1モルのジクロロメタンに対応する。
【0076】
従って本発明は、80から180℃の範囲で10%から20%、特に約16%の減量を示す塩酸モキシフロキサシンメチレンジクロリド溶媒和物に関する。これは基質1モルあたり約1モルのジクロロメタンに対応する。
【0077】
塩酸モキシフロキサシンメチレンジクロリド溶媒和物は、図13Cに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細にはそれは、基質1モルあたり1モルのジクロロメタンに対応する5.77ppm(s,2H)に特性ピークを有している。
【0078】
本発明の方法を使用することによってわれわれはさらに、塩酸モキシフロキサシンニトロメタン溶媒和物を発見した。従って本発明は、このような溶媒和物、特に塩酸モキシフロキサシン対ニトロメタンのモル比が約1:1であるこのような溶媒和物に関する。溶媒和物はさらに、約11.2、14.7、16.9、20.4、22.0、22.6および23.4度の2シータにピークをもつX線粉末回折図、特に図16Aに示すような塩酸モキシフロキサシンニトロメタン溶媒和物の特性X線粉末回折図によって特性決定される。塩酸モキシフロキサシンニトロメタン溶媒和物はまた、赤外スペクトルの約2879、1715、1550、1310および1032cm−1に存在する特性バンド、特に図16Bに示すような典型的赤外スペクトルによって特性決定される。塩酸モキシフロキサシンニトロメタン溶媒和物はモキシフロキサシン生産のより効率的および/または効果的な精製段階を可能にする。
【0079】
より詳細には塩酸モキシフロキサシンニトロメタン溶媒和物は、
(a)モキシフロキサシンをニトロメタンに溶解させる段階
(b)少なくとも1当量、特に約1当量のn−ブタノールまたは酢酸のようなプロトン性溶媒を添加する段階
(c)溶液を少なくとも1当量、特に約1当量のトリメチルクロロシランで処理する段階
(d)塩酸モキシフロキサシンニトロメタン溶媒和物を単離する段階
を含む本発明の方法によって製造され得る。
【0080】
塩酸モキシフロキサシンニトロメタン溶媒和物は、図16Dに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細には、それは基質1モルあたり1モルのニトロメタンに対応する4.4ppm(s,3H)に特性ピークを有している。
【0081】
本発明はまた、この文中でIV形と呼ぶモキシフロキサシンの新規な無水塩酸塩に関する。より詳細には、IV形は、湿度レベル33%の乾燥機に48時間維持されても塩酸モキシフロキサシンの水和形に変換されない。より特定的には本発明は、約10.0、13.2、15.3、17.2および24.4度の2シータにピークをもつX線粉末回折図によって特性決定されるIV形に関する。塩酸モキシフロキサシンの無水IV形の特性X線粉末回折図を図14Aに示す。本発明はまた、約2704、1720、1434、1312および1273cm−1に存在する赤外吸収バンドによって特性決定されるIV形に関する。塩酸モキシフロキサシンIV形はさらに図14Bに示すような典型的赤外スペクトルによって特性決定される。
【0082】
新規な無水IV形は好ましくは、塩酸モキシフロキサシンメチレンジクロリド溶媒和物を真空下に約100℃で乾燥して脱溶媒和することによって製造され得る。無水IV形は、US5849752(Bayer)に記載されている無水B形よりも低吸湿性であり、また、Chemi Spaの特許出願WO2005/054240に記載されている無水B形よりも低吸湿性であるため、取扱いおよび/または保管が容易である。塩酸モキシフロキサシン無水IV形は、湿度レベル33%の乾燥機に48時間維持しても水和形の塩酸モキシフロキサシンに変換されない。これは、変質を全く示さないXRPD図および0.5%から0.6%までしか増加しない含水量によって証明される。本発明はまた、IV形を含む医薬組成物に関する。
【0083】
本発明の方法を使用することによってわれわれはさらに、塩酸モキシフロキサシン酢酸溶媒和物を発見した。従って本発明はこのような溶媒和物、特に塩酸モキシフロキサシン対酢酸のモル比が約1:1であるこのような溶媒和物に関する。この溶媒和物はさらに、約5.3、7.6、9.3、15.2、16.2、18.8、20.8、26.6および27.7度の2シータにピークをもつX線粉末回折図によって特性決定される。好ましい実施態様において、塩酸モキシフロキサシン酢酸溶媒和物は図15Aに示すパターンに実質的に一致するX線粉末回折図を示す。塩酸モキシフロキサシン酢酸溶媒和物はまた、図15Bに示す典型的赤外スペクトルによって特性決定される。塩酸モキシフロキサシン酢酸溶媒和物の代表的赤外吸収バンドは、2707、2289、1736、1421、1308、1246、917および757cm−1に観察される。塩酸モキシフロキサシン酢酸溶媒和物は良好な溶解度および/または安定度を示す。
【0084】
塩酸モキシフロキサシン酢酸溶媒和物は上記に定義および記載したような方法に従って製造され得る。方法は、
(a)モキシフロキサシンを酢酸または酢酸とアセトンもしくはアセトニトリルのような有機溶媒との混合物に溶解させる段階
(b)トリメチルクロロシランを添加する段階
(c)塩酸モキシフロキサシン酢酸溶媒和物を単離する段階
を含む。
【0085】
塩酸モキシフロキサシン酢酸溶媒和物は、図15Cに示す1H−NMRスペクトル(DMSO−d6,TMS)に実質的に一致する1H−NMRスペクトルを有している。より詳細には、これは基質1モルあたり1モルの酢酸に対応する1.9ppm(s,3H)に特性ピークを有している。本発明はまた、本発明の塩酸モキシフロキサシン酢酸溶媒和物を含む医薬組成物に関する。
【0086】
本発明はまた、塩酸リネゾリドを製造するための本発明の方法に関する。方法は好ましくは、アセトンまたはアセトニトリルのような有機溶媒中のリネゾリドの溶液にn−ブタノールまたは酢酸のようなプロトン性溶媒を添加し、混合物をトリメチルクロロシランで処理する段階を含む。
【0087】
本発明はまた、結晶質塩酸リネゾリドに関する。塩酸リネゾリドは、例えばXRPD図の約13.9、18.2、19.1、23.0および27.2度の2シータ値に存在する特性ピークによって特性決定される。結晶質塩酸リネゾリドの特性X線粉末回折図を図19Aに示し、典型的赤外スペクトルを図19Bに示す。図19Aに示すように、塩酸リネゾリドは、XRPD図の約13.9、18.2、19.1、23.0および27.2度の2シータ値に特性ピークを示す。リネゾリドの化学構造に基づいて予想することはできなかったが意外にもこの塩酸塩は安定であることが知見された。塩酸リネゾリドは医薬組成物の製造を可能にするであろう。
【0088】
上記に説明したように、本発明は、結晶化溶液中に生成されるハロゲン化水素酸の量を、該ハロゲン化水素酸の生成速度を(例えばハロゲン化トリアルキルシリルの添加速度をコントロールすることによって)、および、ハロゲン化水素酸の生成条件を(例えば温度、溶媒組成または有機アミンの所望の塩(モノもしくはジハロゲン化水素酸塩など)もしくは有機アミンのハロゲン化水素酸塩の所望の多形を得るために必要な別のパラメーターを調節することによって)微調整できる。従って本発明は、有機アミンの結晶質ハロゲン化水素酸塩を製造するためのハロゲン化トリアルキルシリルの使用に関する。この使用は、有機アミンの結晶質ハロゲン化水素酸塩が該有機アミンのいくつかの既知のハロゲン化水素酸塩のうちの望ましいハロゲン化水素酸塩であるとき、例えば薬剤のジ−またはトリ−塩酸塩が存在するけれども薬剤の結晶質モノ塩酸塩が望ましいような場合に特に有利である。結晶化混合物にHClガスを添加するという慣用技術の使用は、HClの添加量に関するコントロールは難しくないかもしれないが、結晶化条件の選択に関する適応性が劣るので、操作が極めて難しいものになるであろう。
【0089】
別の利点は、ハロゲン化水素酸塩が本質的に水の非存在下または少なくとも極めて少量の水の存在下で生成できることである。従って、有機アミンの結晶質ハロゲン化水素酸塩の無水形を得ることが可能である。従って本発明はまた、有機アミンの無水結晶質ハロゲン化水素酸塩の製造におけるハロゲン化トリアルキルの使用に関する。さらに、本質的に水の非存在下でコントロール量のハロゲン化水素酸塩を製造するので、水の存在下では形成されない有機アミンの溶媒和物、例えば、対応する水和物よりも熱力学的に不安定な溶媒和物を得ることが可能である。従って、本発明はまた、有機アミンの結晶質ハロゲン化水素酸塩の溶媒和物の製造、特に無水溶媒和物の製造におけるハロゲン化トリアルキルの使用に関する。好ましいハロゲン化トリアルキルシリル、好ましい有機アミンおよびこれらの好ましい使用条件が本発明の方法に関する上記の記載によって定義されていることは当業者に明らかであろう。
【0090】
以下の実施例は本発明を例示するもので本発明の範囲を限定するものではない。使用した室温という用語は常に20から30℃の範囲の温度を表す。
【実施例】
【0091】
赤外スペクトルは、ダイアモンドATR−セルを備えたBRUKER Tensor 27 FTIR−分光計を使用して記録した。
【0092】
XRPDはAXS−BRUKER X線粉末回折計D−8で以下の取得条件を使用して記録した:チューブアノード:Cu;発電機電圧:40kV;発電機電流:40mA;出発角度:2.0度?;到着角度:40.0度?;一目盛り:0.01度?;時間/目盛り:2秒。
【0093】
示差走査熱量測定(DSC)は、DSC7(Perkin−Elmer,Norwalk,Ct.,USA)でPyris 2.0ソフトウェアを使用して行った。サンプルを25μlのAlパンで計量した。パージガスとして乾性窒素を使用した(パージ速度:20ml分−1)。
【0094】
1H−NMRスペクトルはBrucker AM−300分光計で記録した。
【実施例1】
【0095】
結晶質無水形の塩酸ミコフェノラートモフェチルの製造
2g(4.61mmol)のミコフェノラートモフェチル塩基を50mlの酢酸エチルに室温で溶解させた。この溶液に0.3ml(1.2当量)の酢酸および0.7ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、2.11g(97.6%)の塩酸ミコフェノラートモフェチルが得られた。mp.=157.2℃。
【0096】
塩酸ミコフェノラートモフェチルのXRD図を図1Aに示す。これはWO95/07902に示されているX線結晶学データをもつ結晶質無水形に一致する。得られた赤外スペクトルを図1Bに示す。塩酸ミコフェノラートモフェチルのDSCは約159℃に吸熱ピークを示す(開始温度約155℃、図1C参照)。
【実施例2】
【0097】
塩酸ベンラファキシンの製造
実施例2.a 塩酸ベンラファキシンI形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.41g(89.1%)の塩酸ベンラファキシンが得られた。mp.=208℃。
【0098】
塩酸ベンラファキシンのXRD図を図2Aに示す。これはUS03/0114536に示されているX線結晶学データをもつI形に一致する。得られた赤外スペクトルを図2Bに示す。
【0099】
実施例2.b 塩酸ベンラファキシンII形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.38g(82.6%)の塩酸ベンラファキシンII形が得られた。
【0100】
塩酸ベンラファキシンII形のXRPD図を図3Aに示す。これはWO02/45658に示されているX線結晶学データをもつII形に一致する。得られた赤外スペクトルを図3Bに示す。
【0101】
実施例2.c 塩酸ベンラファキシンII形の製造
0.4g(1.44mmol)のベンラファキシン塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.23g(51.1%)の塩酸ベンラファキシンII形が得られた。
【実施例3】
【0102】
セルトラリン塩基を使用する塩酸セルトラリンII形の製造
実施例3.a
3g(9.8mmol)のセルトラリン塩基を60mlのアセトニトリルに室温で溶解させた。この溶液に0.6ml(1当量)の酢酸および1.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。添加中に塩酸セルトラリンがまさしく結晶質II形で沈殿した。懸濁液を1時間撹拌後、生成物を濾別し、50℃で3時間乾燥すると3.2g(95.3%)の塩酸セルトラリンII形が得られた。mp.:252℃。
【0103】
得られたXRD図を図4Aに示す。これは純粋なII形に一致する。得られた赤外スペクトルを図4Bに示す。
【0104】
実施例3.b
3g(9.8mmol)のセルトラリン塩基を60mlのアセトニトリルと1mlのn−ブタノールとの混合物に溶解させた。溶液を50℃に加熱し、1.4ml(1.1当量の)のトリメチルクロロシランを撹拌下で添加した。添加中に直ちに塩酸セルトラリンが結晶質II形で沈殿した。50℃で30分間撹拌後、懸濁液を室温に冷却し、約1時間撹拌した。生成物を濾別し、50℃で3時間乾燥すると、3.1g(94%)の塩酸セルトラリンII形が得られた。
【0105】
実施例3.c
200mlのメチルイソブチルケトン(MIBK)中の10g(32.7mmol)のセルトラリン塩基を約80℃に加熱した。溶液に2.4ml(1.1当量)の酢酸、4.5ml(1.1当量)のトリメチルクロロシランを撹拌下で順次に添加した。最初に得られたゼリー状の塊は80℃で1時間撹拌後に結晶質になった。反応混合物を室温に冷却し、再度約1.5時間撹拌した。生成物を濾別し、50℃で4時間乾燥すると、10.93g(97.7%)の塩酸セルトラリンII形が得られた。
【実施例4】
【0106】
マンデル酸セルトラリンを使用する塩酸セルトラリンII形の製造
実施例4.a
60mlのアセトニトリル中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を室温で撹拌し、1.4ml(1.7当量)のトリメチルクロロシランを添加した。粘性懸濁液は先ず希薄懸濁液に変化し、その後15分以内に塩酸セルトラリンII形の結晶の希薄懸濁液が得られた。約1時間撹拌後、生成物を濾別し、真空下に50℃で乾燥すると、2.09g(96.3%)の塩酸セルトラリンII形が得られた。
【0107】
実施例4.b
60mlのメチルエチルケトン中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を約80℃に加熱し、0.9ml(1.1当量)のトリメチルクロロシランを添加した。透明溶液が得られた後まもなく結晶質II形の塩酸セルトラリンが沈殿し始めた。反応混合物を撹拌下に1時間以内で室温に冷却し、次いで生成物を濾別し、真空下に50℃で乾燥すると、1.97g(87.8%)の塩酸セルトラリンII形が得られた。
【0108】
実施例4.c
60mlのメチルイソブチルケトン中の3g(6.5mmol)のマンデル酸セルトラリンの懸濁液を約80℃に加熱し、0.9ml(1.1当量)のトリメチルクロロシランを添加する。懸濁液はゼリー状の塊に変化した。約5分後、結晶質II形の塩酸セルトラリンが形成され始めた。反応混合物を80℃で約20分間撹拌し、次いで撹拌下に1時間以内で室温に冷却した。生成物を濾別し、真空下に50℃で3時間乾燥すると、2.11g(94.0%)の塩酸セルトラリンII形が得られた。
【0109】
実施例4.d
300mlのメチルイソブチルケトン中の15g(32.7mmol)のマンデル酸セルトラリンの懸濁液を約100℃に加熱し、4.6ml(1.1当量)のトリメチルクロロシランを添加した。懸濁液はゼリー状の塊に変化した。約5分後、結晶質II形の塩酸セルトラリンが形成され始めた。反応混合物を100℃で約15分間撹拌し、次いで撹拌下に1.5時間以内で室温に冷却した。生成物を濾別し、10mlのアセトンで2回洗浄し、真空下に50℃で4時間乾燥すると、10.64g(94.9%)の塩酸セルトラリンII形が得られた。
【実施例5】
【0110】
シュウ酸セルトラリンを使用する塩酸セルトラリンII形の製造
実施例5.a
シュウ酸セルトラリンの製造
50mlの酢酸エチル中の3g(9.8mmol)のセルトラリン遊離塩基の溶液に50mlのメタノール中の0.97g(1.1当量)のシュウ酸の溶液を添加した。15分間撹拌後、結晶質沈殿物を濾別し、真空下に50℃で乾燥すると3.37g(86.8%)のシュウ酸セルトラリンが得られた。
【0111】
実施例5.b
シュウ酸セルトラリンからの塩酸セルトラリンII形の製造
15mlのアセトニトリル中の0.7g(1.77mmol)のシュウ酸セルトラリンの懸濁液を室温で撹拌し、250μl(1.1当量)のトリメチルクロロシランを添加した。添加後に懸濁液はほぼ溶液に変化したが同時に結晶質II形の塩酸セルトラリンが沈殿し始めた。約1時間撹拌後、生成物を濾別し、真空下50℃で乾燥すると、0.47g(77.8%)の塩酸セルトラリンII形が得られた。
【実施例6】
【0112】
塩酸セルトラリンI形の製造
60mlの2−プロパノール中の3g(9.8mmol)のセルトラリン塩基の溶液を室温で撹拌し、0,6mlの酢酸および1.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を60分間撹拌し、沈殿物を濾別した。固体を2−プロパノールで洗浄し、真空下に50℃で3時間乾燥すると、2.95g(87.8%)の塩酸セルトラリンI形が得られた。得られたXRD図を図5Aに示す。これはI形に対応する。得られた赤外スペクトルを図5Bに示す。
【実施例7】
【0113】
塩酸ドネペジルの製造
実施例7.a
塩酸ドネペジルII形の製造
0.5g(1.32mmol)のドネペジル塩基を30mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を2時間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.55g(100%)の塩酸ドネペジルII形が得られた。得られたXRD図を図6Aに示す。これはII形に対応する。得られた赤外スペクトルを図6Bに示す。
【0114】
実施例7.b
塩酸ドネペジルIII形の製造
0.5g(1.32mmol)のドネペジル塩基を30mlのアセトンに室温で溶解させた。この溶液に0.1ml(1.1当量)の酢酸および0.2ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を30分間撹拌し、沈殿物を濾別した。固体を酢酸エチルで洗浄し、真空下に室温で乾燥すると、0.54g(98.5%)の塩酸ドネペジルが得られた。mp.=211℃。得られたXRD図を図7Aに示す。これはIII形に対応する。得られた赤外スペクトルを図7Bに示す。
【0115】
実施例7.c
塩酸ドネペジルIII形の製造
アセトンに代えてアセトニトリルを使用して実施例7.bを繰り返した。塩酸ドネペジルIII形の収量(収率):0.47g(85.6%)。
【実施例8】
【0116】
塩酸テルビナフィンの製造
実施例8.a
0.4g(1.37mmol)のテルビナフィン塩基を5mlのアセトンに室温で溶解させた。この溶液に86μl(1.1当量)の酢酸および191μl(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトンで洗浄し、真空下に室温で乾燥すると、0.3g(66.7%)の塩酸テルビナフィンが得られた。mp.=185℃。
【0117】
塩酸テルビナフィンのXRP図を図8Aに示す。これは文献データ(Cryst.Eng.Comm.,2002,4(67),393−400)に一致する。得られた赤外スペクトルを図8Bに示す。
【0118】
実施例8.b
アセトンに代えてアセトニトリルを使用して実施例8.aを繰り返した。塩酸テルビナフィンの収量(収率):0.19g(42.2%)。
【0119】
実施例8.c
アセトンに代えてtert.−ブチル−メチル−エーテルを使用して実施例8.aを繰り返した。塩酸テルビナフィンの収量(収率):0.41g(91,1%)。
【実施例9】
【0120】
塩酸シナカルセットの製造
実施例9.a
1.0g(2.80mmol)のシナカルセット塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.19ml(1.2当量)の酢酸および0.42ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を2時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.45g(40.8%)の塩酸シナカルセットが得られた。mp.=173℃。得られたXRD図を図9Aに示し、得られた赤外スペクトルを図9Bに示す。
【0121】
実施例9.b
アセトニトリルに代えて酢酸エチルを使用して実施例9.aを繰り返した。塩酸シナカルセットの収量(収率):0.41g(37.2%)。
【実施例10】
【0122】
臭化水素酸シタロプラムの製造
実施例10.a
0.72g(22.2mmol)のシタロプラム塩基を10mlのアセトニトリルに室温で溶解させた。この溶液に0.14ml(1.1当量)の酢酸および0.32ml(1.1当量)のトリメチルブロモシランを撹拌下で添加した。室温で30分後に15mlのジエチエーテルを添加すると結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.67g(74,5%)の臭化水素酸シタロプラムが得られた。mp.=182℃。得られたXRD図を図10Aに示し、得られた赤外スペクトルを図10Bに示す。
【0123】
実施例10.b
アセトニトリルに代えて酢酸エチルを溶媒として使用し、0.5g(15.4mmol)のシタロプラム塩基で実施例10.aを繰り返した。臭化水素酸シタロプラムの収量(収率):0.53g(84.8%)。
【0124】
実施例10.c
アセトニトリルに代えてアセトンを溶媒として使用し、0.59g(18.2mmol)のシタロプラム塩基で実施例10.aを繰り返した。臭化水素酸シタロプラムの収量(収率):0.67g(90.9%)。
【0125】
実施例10.d
0.27g(0.8mmol)のシタロプラム塩基を3mlのイソプロパノールに室温で溶解させ、145μl(1.1当量)のトリメチルブロモシランを溶液に添加した。冷蔵庫で一夜静置後に結晶質沈殿物を濾別し、真空下で乾燥すると、0.25g(74.1%)の臭化水素酸シタロプラムが得られた。
【実施例11】
【0126】
塩酸アリピプラゾールA形の製造
2.0g(4.46mmol)のアリピプラゾールを20mlの1,2−ジクロロメタンに室温で溶解させた。この溶液に0.45ml(1.1当量)のn−ブタノールおよび0.63ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。室温で2分後に結晶化が始まった。懸濁液を15分間撹拌し、沈殿物を濾別した。固体を1,2−ジクロロメタンで洗浄し、真空下に室温で乾燥すると、2.05g(94.0%)の塩酸アリピプラゾールが得られた。mp.=210℃。
【0127】
生成物のXRD図を図11Aに示す。これはWO2004/083183(Hetero Drugs Ltd.)に示されている塩酸アリピプラゾールA形のXRPDデータに一致する。得られた赤外スペクトルを図11Bに示す。
【実施例12】
【0128】
モノ塩酸プラミペキソールの製造
0.5g(2.37mmol)のプラミペキソール塩基を20mlのアセトニトリルに室温で溶解させた。この溶液に0.24mlのn−ブタノール(2.6mmol,1.1当量)および0.33mlのトリメチルクロロシラン(2.6mmol,1.1当量)を撹拌下で添加した。室温で1分後に結晶化が始まった。懸濁液を1時間撹拌し、沈殿物を濾別した。固体をアセトニトリルで洗浄し、真空下に室温で乾燥すると、0.56g(95.5%)のモノ塩酸プラミペキソールが得られた。mp.=264℃。得られたXRD図を図12Aに示し、得られた赤外スペクトルを図12Bに示す。
【実施例13】
【0129】
無水塩酸モキシフロキサシンIV形の製造
実施例13.a
塩酸モキシフロキサシンメチレンジクロリド溶媒和物
5g(12.5mmol)のモキシフロキサシンをEP550903に従って製造し、50mlのメチレンジクロリドに室温で溶解させた。この溶液に0.9ml(1.2当量)の酢酸および2ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後直ちに結晶化が始まった。懸濁液を約30分間撹拌し、沈殿物を濾別し、真空下に室温で乾燥すると、6.3g(94.2%)の塩酸モキシフロキサシンとメチレンジクロリドとの1:1溶媒和物が得られた。
【0130】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1.9(m,4H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.5−3.7(m with Singlet at 3.6 ppm,4H),3.7−3.8(m,1H),3.8−4.0(m,2H),4.0−4.2(m,2H),5.77(s,2H),7.65(d,J=14Hz,1H),8.66(s,1H),8.95(s,broad,1H),10.2(s,broad,1H),15.1(s,broad,1H)。
【0131】
5.77ppmの一重項は基質1モルあたり約1モルのメチレンジクロリドに対応する(図13C参照)。生成物のXRD図を図13Aに示し、得られた赤外スペクトルを図13Bに示す。
【0132】
塩酸モキシフロキサシンとメチレンジクロリドとの溶媒和物は吸湿性でない(33%相対湿度に1日維持後にも全く吸水していない)。
【0133】
実施例13.b
塩酸モキシフロキサシン無水IV形
2gの塩酸モキシフロキサシンメチレンジクロリド溶媒和物を真空下に100℃で約6時間乾燥すると、1.75gの塩酸モキシフロキサシン無水IV形が脱溶媒和した。生成物のXRD図を図14Aに示し、得られた赤外スペクトルを図14Bに示す。
【実施例14】
【0134】
塩酸モキシフロキサシンと酢酸との溶媒和物の製造
実施例14.a
3.0g(7.47mmol)のモキシフロキサシンをEP550903に従って製造し、30mlの酢酸に室温で溶解させた。この溶液に2.0ml(2.1当量の)トリメチルクロロシランを撹拌下で添加した。クロロシラン添加の30分後に結晶化が始まった。懸濁液を約3.5時間撹拌し、沈殿物を濾別し、アセトニトリルで洗浄し、真空下に室温で乾燥すると、3.21g(86.3%)の塩酸モキシフロキサシンと酢酸との1:1溶媒和物が得られた。
【0135】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1. (m,4H),1.9(s,3H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.1−3.25(m,1H),3.5−3.7(m with Singlet at 3.6ppm,4H),3.7−3.8(m,1H),3.8−4.0(m,2H),4.0−4.2(m,2H),7.63(d,J=14Hz,1H),8.65(s,1H),9.1(s,broad,1H),10.4(s,broad,1H),12.7(s,broad,1H)。
【0136】
1.9ppmの一重項は基質1モルあたり約1モルの酢酸に対応する(図15C参照)。生成物のXRD図を図15Aに示し、得られた赤外スペクトルを図15Bに示す。
【0137】
実施例14.b
1.0g(2.49mmol)モキシフロキサシンをEP550903に従って製造し、5mlの酢酸および5mlのアセトニトリルに室温で溶解させた。溶液に0.38ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシラン添加の2時間後に結晶化は始まっていなかった。この溶液に0.38mlのトリメチルクロロシランを撹拌下でもう一度添加した。懸濁液を4℃で約17時間保存し、沈殿物を濾別し、アセトニトリルで洗浄し、真空下に室温で乾燥すると、0.93g(75.0%)の塩酸モキシフロキサシンと酢酸との1:1溶媒和物が得られた。
【実施例15】
【0138】
塩酸モキシフロキサシンとニトロメタンとの溶媒和物の製造
0.5g(1.25mmol)のモキシフロキサシンをEP550903に従って製造し、15mlのニトロメタンに約60℃で溶解させた。この溶液に0.14ml(2当量)の酢酸および(2当量)mlのトリメチルクロロシランを撹拌下で添加した。懸濁液を約17時間保存し、沈殿物を濾別し、アセトンで洗浄し、真空下に室温で乾燥すると、0.55g(88.2%)の塩酸モキシフロキサシンとニトロメタンとの1:1溶媒和物が得られた。
【0139】
1H−NMR(DMSO−d6):0.8−0.95(m,1H),0.95−1.3(m,3H),1.6−1.9(m,4H),2.55−2.75(m,1H),2.8−3.0(m,1H),3.1−3.25(m,1H),3.5−3.7(m with Singlet at 3.6ppm,4H),3.7−3.8(m,1H),3.8−3.95(m,2H),4.0−4.2(m,2H),4.44(s,3H),7.63(d,J=14Hz,1H),8.65(s,1H),9.0(s,broad,1H),10.3(s broad,1H),15.1(s,broad,1H)。
【0140】
4.44ppmの一重項は基質1モルあたり約1モルのニトロメタンに対応する。(図16C参照)。生成物のXRD図を図16Aに示し、得られた赤外スペクトルを図16Bに示す。
【実施例16】
【0141】
塩酸デュロキセチンの製造
実施例16.a
0.3g(1.0mmol)のデュロキセチン塩基を5mlの酢酸エチルに室温で溶解させた。この溶液に65μlの酢酸および0.14mlのトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、懸濁液を室温で約2時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、0.21g(62.9%)の塩酸デュロキセチンが得られた。mp.=161℃。生成物のXRD図を図17Aに示し、得られた赤外スペクトルを図17Bに示す。
【0142】
実施例16.b
酢酸エチルに代えてアセトンを使用して実施例16.aを繰り返した。塩酸デュロキセチンの収量(収率):0.26g(77.9%)。
【実施例17】
【0143】
塩酸リネゾリドの製造
実施例17.a
5g(14.8mmol)のリネゾリドを200mlのアセトニトリルに室温で溶解させた。この溶液に1.0ml(1.1当量)の酢酸および2.1ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランを添加し1時間撹拌後に沈殿物が形成され、懸濁液を室温で約3.5時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、1.43g(25.8%)の塩酸リネゾリドが得られた。mp.=163℃。生成物のXRD図を図18Aに示し、得られた赤外スペクトルを図18Bに示す。
【0144】
実施例17.b
アセトニトリルに代えてアセトンを溶媒として使用し0.5g(1.5mmol)のリネゾリドで実施例17.aを繰り返した。塩酸リネゾリドの収量(収率):0.20g(36.4%)。
【実施例18】
【0145】
塩酸メマンチンの製造
0.5g(2.8mmol)のメマンチン塩基を10mlの酢酸エチルに室温で溶解させた。この溶液に0.1ml(1.1当量)のメタノールおよび0.4ml(1.1当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、懸濁液を室温で約2時間撹拌した。白色結晶質固体を濾別し、真空下に室温で乾燥すると、0.59g(98.0%)の塩酸メマンチンが得られた。m.p.=293から296℃。生成物のXRD図を図19Aに示し、得られた赤外スペクトルを図19Bに示す。
【実施例19】
【0146】
塩酸リモナバントI形の製造
実施例19a
1g(2.16mmol)のリモナバントを20mlのアセトニトリルに室温で懸濁させた。懸濁液に0.105ml(1.2当量)のメタノールおよび0.33ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。透明溶液が得られた後まもなく結晶質I形の塩酸リモナバントが沈殿し始めた。生成物を濾別し、真空下に室温で一夜乾燥させると、0.9g(83.4%)の塩酸リモナバントI形が得られた。生成物のXRD図を図20Aに示し、得られた赤外スペクトルを図20Bに示す。
【0147】
実施例19b
1g(2.16mmol)のリモナバントを10mlの酢酸エチルに溶解させた。この溶液に0.105ml(1.2当量)のメタノールおよび0.33ml(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に沈殿物が形成され、白色結晶質固体を濾別し、真空下に室温で乾燥させると、0.95g(88.1%)の塩酸リモナバントI形が得られた。
【0148】
実施例19c
酢酸エチルに代えてアセトンを溶媒として使用し実施例19bを繰り返した。塩酸リモナバントI形の収量(収率):0.89g(82.5%)。
【実施例20】
【0149】
塩酸クロピドグレルI形の製造
実施例20a
1.2g(3.73mmol)のクロピドグレルを10mlのアセトンに室温で溶解させた。溶液に255μl(1.2当量)の酢酸および565μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。6mlのジイソプロピルエーテルの添加後に塩酸クロピドグレルが沈殿し始めた。1時間撹拌後、結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.85g(63.8%)の塩酸クロピドグレルI形が得られた。生成物のXRD図を図21Aに示し、得られた赤外スペクトルを図21Bに示す。
【0150】
実施例20b
1g(3.11mmol)のクロピドグレルを10mlのアセトニトリルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および475μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。混合物を約5℃に冷却し、40mlのジイソプロピルエーテルを撹拌下で添加した。1時間撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.50g(45.0%)の塩酸クロピドグレルI形が得られた。
【0151】
実施例20c
1g(3.11mmol)のクロピドグレルを10mlの酢酸エチルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および475μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。クロロシランの添加後に粘性固体が沈殿し、これは約5分以内に結晶質生成物に変化した。混合物を室温で1時間撹拌した。次に生成物を濾別し、真空下に室温で一夜乾燥すると、0.88g(79.3%)の塩酸クロピドグレルI形が得られた。
【0152】
実施例20d
機械的撹拌器と滴下漏斗とを備えた1L容の反応フラスコ中で30g(93.22mmol)のクロピドグレル遊離塩基を1000mlのトルエンに溶解させた。溶液に4.5ml(1.2当量)のメタノールを添加し、撹拌によって混合した後、14.1ml(1.2当量)のトリメチルクロロシランを室温でゆっくりと撹拌しながら45分以内に滴下した。添加後、反応混合物を室温で約3時間撹拌するうちに綿毛状の沈殿物が白色結晶質固体に変化した。塩を濾別し、20mlのトルエンで洗浄した。生成物を真空下に室温で一夜乾燥すると、29.1g(87.1%)の塩酸クロピドグレルI形が得られた。
【実施例21】
【0153】
臭化水素酸クロピドグレルA形の製造
実施例21a
1g(3.11mmol)のクロピドグレルを10mlの酢酸エチルに室温で溶解させた。溶液に213μl(1.2当量)の酢酸および482μl(1,2当量)のトリメチルブロモシランを撹拌下で添加した。ブロモシランの添加後に粘性固体が沈殿し、約30分以内に結晶質生成物に変化した。混合物を室温で1時間撹拌した。次に生成物を濾別し、真空下に室温で一夜乾燥すると、1.03g(82.3%)の臭化水素酸クロピドグレルA形が得られた。生成物のXRD図を図22Aに示し、得られた赤外スペクトルを図22Bに示す。
【0154】
実施例21b
1g(3.11mmol)のクロピドグレルを10mlのイソプロパノールに溶解させた。溶液に482μl(1.2当量)のトリメチルブロモシランを添加した。90分間撹拌後、ガラス棒で引掻いた後に生成物が結晶化した。臭化水素酸塩を濾別し、真空下に室温で一夜乾燥すると、0.86g(68.7%)の臭化水素酸クロピドグレルA形が得られた。
【実施例22】
【0155】
塩酸プラスグレルB形の製造
5.0g(13.4mmol)のプラスグレル塩基を75mlのアセトンに室温で溶解させた。溶液に919μl(1.2当量)の酢酸および2053μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。添加後直ちに白色沈殿物が得られた。塩酸塩を濾別し、真空下に室温で6時間乾燥すると、4.41g(80.4%)の塩酸プラスグレルB形が得られた。生成物のXRD図を図23Aに示し、得られた赤外スペクトルを図23Bに示す。
【実施例23】
【0156】
塩酸プラスグレルアセトニトリル溶媒和物の製造
0.5g(1.34mmol)のプラスグレル塩基を7.5mlのアセトニトリルに溶解させた。非溶解分を濾別した。溶液に82μl(1.2当量)の酢酸および205μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。混合物を室温で17時間撹拌した。懸濁液を濾別し、真空下に室温で一夜乾燥させると、0.43g(78%)の塩酸プラスグレルアセトニトリル溶媒和物が得られた。
【0157】
1H−NMR(DMSO−d6):0.84−0.97(m,2H),0.98−1.16(m,2H),1.87−2.0(m,1H),2.08(s,2H),2.29(s,3H),2.96−3.15(m,2H),3.34−3.6(m,1H),3.7−4.2(m,3H),6.14(s,broad,1H),6.58(s,1H),7.3−7.55(m,2H),7.55−7.8(m,2H)。
【0158】
2.08ppmの一重項は基質1.5モルあたり約1モルのアセトニトリルに対応する。生成物のXRD図を図24Aに示し、赤外スペクトルを図24Bに示す。
【実施例24】
【0159】
塩酸ラロキシフェンA形の製造
実施例24a
1.0g(1.77mmol)の乳酸ラロキシフェンを10mlのアセトニトリルに室温で懸濁させた。懸濁液に272μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.80g(82.5%)の塩酸ラロキシフェンA形が得られた。生成物のXRD図を図25Aに示し、得られた赤外スペクトルを図25Bに示す。
【0160】
実施例24b
1.5g(2.66mmol)の乳酸ラロキシフェンを30mlのメチルイソプロピルケトン(MIBK)に室温で懸濁させた。懸濁液を100℃に加熱した。懸濁液に408μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。懸濁液を室温に冷却した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.42g(97.6%)の塩酸ラロキシフェンA形が得られた。
【0161】
実施例24c
0.51g(0.90mmol)のラロキシフェン塩基を5mlのエタノールに室温で懸濁させた。懸濁液に162μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。透明溶液が形成された後まもなく結晶質形態の塩酸ラロキシフェンが沈殿し始めた。3時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.41g(74.6%)の塩酸ラロキシフェンA形が得られた。
【実施例25】
【0162】
塩酸ラロキシフェンTHF半溶媒和物の製造
実施例25a
0.50g(0.88mmol)のラロキシフェン塩基を5mlのテトラヒドロフランに室温で懸濁させた。懸濁液に51μl(1.2当量)のメタノールおよび162μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で3時間撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.45g(97.6%)の塩酸ラロキシフェンTHF半溶媒和物が得られた。
【0163】
1H−NMR(DMSO−d6):1.25−1.4(m,1H),1.6−1.9(m,7H),2.8−3.1(m,2H),3.3−3.55(m,4H),3.55−3.65(m,2.6H),4.4(t,broad,2H),6.7−6.8(m,2H),6.9(dd,J=2.26 and 8.8Hz,1H),6.98(d,J=9Hz,1H),7.15−7.22(m,2H),7.3(d,J=8.7Hz,1H),7.4(d,J=2.3Hz.1H),7.71(d,J=9Hz,1H),9.95(d,J=9Hz,1H),10.8(s,braod,1H)。
【0164】
プロトンNMR分光法によって測定すると結晶質材料中に存在するテトラヒドロフランの量は約0.6モル当量であった。生成物のXRD図を図26Aに示し、得られた赤外スペクトルを図26Bに示す。
【0165】
実施例25b
0.30g(0.53mmol)の乳酸ラロキシフェンを3mlのテトラヒドロフランに室温で懸濁させた。懸濁液に82μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。室温で2時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.28g(102.6%)の塩酸ラロキシフェンTHF半溶媒和物が得られた。
【実施例26】
【0166】
ジ塩酸オランザピンの製造
実施例26a
1.0g(3.20mmol)のオランザピン塩基を10mlのアセトニトリルに室温で懸濁させた。懸濁液に312μl(2.4当量)のメタノールおよび980μl(2.4当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.24g(100.54%)のジ塩酸オランザピン1形が得られた。生成物のXRD図を図27Aに示し、得られた赤外スペクトルを図27Bに示す。
【0167】
実施例26b
1.0g(3.20mmol)のオランザピン塩基を10mlのアセトンに室温で懸濁させた。懸濁液に312μl(2.4当量)のメタノールおよび980μl(2.4当量)のトリメチルクロロシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、1.35g(107.31%)のジ塩酸オランザピン1形が得られた。
【実施例27】
【0168】
臭化水素酸ダリフェナシンの製造
0.9g(2.11mmol)のダリフェナシン塩基を9mlのメチルエチルケトンに室温で溶解させた。溶液に103μl(1.2当量)のメタノールおよび329μl(1.2当量)のトリメチルブロモシランを撹拌下で添加した。1時間の撹拌後に結晶質沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.88mg(82.13%)の臭化水素酸ダリフェナシンが得られた。生成物のXRD図を図28Aに示し、得られた赤外スペクトルを図28Bに示す。
【実施例28】
【0169】
非晶質形態の無水塩酸シタグリプチンの製造
0.085g(0.21mmol)のシタグリプチン塩基を2mlのジエチルエーテルおよび3mlのメチレンクロリドに室温で溶解させた。溶液に10μl(1.2当量)のメタノールおよび32μl(1.2当量)のトリメチルクロロシランを撹拌下で添加した。添加後直ちに沈殿物が得られた。懸濁液を1時間撹拌し、濾別し、真空下に室温で一夜乾燥すると、0.089g(96.8%)の非晶質形態の塩酸シタグリプチンが得られた。生成物のXRD図を図29Aに示し、得られた赤外スペクトルを図29Bに示す。
【実施例29】
【0170】
無水ジ塩酸バルデナフィルの製造
0.2g(0.359mmol)のバルデナフィル塩基を4mlのジエチルエーテルに室温で懸濁させた。懸濁液に5mlのメチレンジクロリドを添加すると溶液が得られた。35μl(2.4当量)のメタノールおよび110μl(2.4当量)のトリメチルクロロシランを溶液に添加すると、添加後直ちに白色沈殿物が得られた。懸濁液を20分間撹拌し、濾別し、真空下に室温で一夜乾燥すると、0.2g(95,.4%)のジ塩酸バルデナフィルが得られた。生成物のXRD図を図30Aに示し、得られた赤外スペクトルを図30Bに示す。
【実施例30】
【0171】
塩酸エルロチニブA形の製造
0.3g(0.82mmol)のエルロチニブ塩基を3mlのイソプロパノールに懸濁させた。懸濁液に126μl(1.2当量)のトリメチルクロロシランを添加した。白色沈殿物が得られた。1時間の撹拌後に沈殿物を濾別し、真空下に室温で一夜乾燥すると、0.26g(80.8%)の塩酸エルロチニブA形が得られた。生成物のXRD図を図31Aに示し、得られた赤外スペクトルを図31Bに示す。
【図1A】
【図1B】
【図1C】
【図2A】
【図2B】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図6A】
【図6B】
【図7A】
【図7B】
【図8A】
【図8B】
【図9A】
【図9B】
【図10A】
【図10B】
【図11A】
【図11B】
【図12A】
【図12B】
【図13A】
【図13B】
【図13C】
【図14A】
【図14B】
【図15A】
【図15B】
【図15C】
【図16A】
【図16B】
【図16C】
【図17A】
【図17B】
【図18A】
【図18B】
【図19A】
【図19B】
【図20A】
【図20B】
【図21A】
【図21B】
【図22A】
【図22B】
【図23A】
【図23B】
【図24A】
【図24B】
【図25A】
【図25B】
【図26A】
【図26B】
【図27A】
【図27B】
【図28A】
【図28B】
【図29A】
【図29B】
【図30A】
【図30B】
【図31A】
【図31B】
【公表番号】特表2009−541385(P2009−541385A)
【公表日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願番号】特願2009−516963(P2009−516963)
【出願日】平成19年6月25日(2007.6.25)
【国際出願番号】PCT/EP2007/005596
【国際公開番号】WO2008/000418
【国際公開日】平成20年1月3日(2008.1.3)
【出願人】(305008042)サンド・アクチエンゲゼルシヤフト (54)
【Fターム(参考)】
【公表日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願日】平成19年6月25日(2007.6.25)
【国際出願番号】PCT/EP2007/005596
【国際公開番号】WO2008/000418
【国際公開日】平成20年1月3日(2008.1.3)
【出願人】(305008042)サンド・アクチエンゲゼルシヤフト (54)
【Fターム(参考)】
[ Back to top ]