
塩基配列解析方法
【課題】DNAの塩基配列、特にDNA固定化担体に固定されたDNAの塩基配列を、短時間で簡便な処理によって解析するための手段を提供することを目的とする。
【解決手段】DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とするDNAの塩基配列解析方法。
【解決手段】DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とするDNAの塩基配列解析方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、塩基配列解析方法に関し、詳しくは、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確な解析を可能にする塩基配列解析方法に関する。
【背景技術】
【0002】
DNAチップ(マイクロアレイ)やDNAビーズアレイなどのDNA固定化担体は、疾病診断、治療のほか、ゲノム解析などにも広く用いられている。このDNA固定化担体に所望のDNAが固定化されているかどうかを確認するため、種々の塩基配列解析方法が開発されている(例えば、特許文献1および非特許文献1参照)。
しかし、これらの方法では、二次構造がハイブリダイゼーションを阻害し、かつ配列の反復が正しい解析を妨げる、あるいは、3位で修飾された塩基により、ポリメラーゼの組み込み反応が妨げられるといった問題があった。
【0003】
また、DNAの塩基配列を解析するにあたり、特定の塩基配列を認識する制限酵素によりDNAを短く切断してから調べる方法もあるが、これらの酵素が認識する塩基配列は、限られた4〜6塩基のみであるなどの限界があり、正確に特定の部位でDNAを切断する方法が必要とされていた。
酵素を使わずにDNAを切断する方法としては、加水分解による方法が提案されている。例えば、特許文献2には、1本鎖DNAの塩基配列認識部位にDNA断片(末端に切断反応の触媒が結合)を結合させ、該触媒部位で1本鎖DNAを切断のセリウム(Ce,IV)と、ポリアミン−N−ポリカルボン酸あるいはその誘導体よりなる錯体を用いた加水分解による切断方法が提案されている。
また、非特許文献2には、1本鎖DNAの塩基配列認識部位にDNA断片を2つ結合させ、Ce(IV)/EDTAを作用させて該DNA断片の間部分で1本鎖DNAを切断する方法が提案されている。
しかし、これらの方法は、いずれも加水分解による切断のみに対応したものであり、他の切断方法が想定されたものではなかった。また、加水分解反応に多大な時間がかかるなどの問題点があった。
【0004】
一方、DNA固定化担体の製造は、基体に所望のDNAを固定化して行われ、従来はリガーゼなどの酵素を利用する酵素的DNA固定化法が用いられていた(特許文献3および4参照)。
酵素的DNA固定化法では、基体を活性化した後、まず2本鎖の短いオリゴヌクレオチドをチップ上に固定して、その後で所望のオリゴヌクレオチドを制限酵素末端を利用して、DNAリガーゼを利用して固定化させていた。
しかし、この方法では、DNAを基体に確実に固定化することが困難であったほか、多くの工程を経る必要があり、多くの試薬や機器が必要なほか、処理に相当の長時間を要するという問題点があった。
【0005】
このような状況下、DNAの塩基配列、特にDNA固定化担体に固定されたDNAの塩基配列をより正確、迅速、かつ簡便に解析する方法の開発が望まれていた。
【0006】
【特許文献1】特許公表2004−529650号公報
【特許文献2】特開2001−89430号公報
【特許文献3】国際特許公開WO01−68368号公報
【特許文献4】国際特許公開WO01−40173号公報
【非特許文献1】Nucleosides & Nucleotides : 1999 Feb; 18(2): 197-201
【非特許文献2】Chem.Letters:2004;33,300-301
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、上記従来の問題に鑑みて、DNAの塩基配列、特にDNA固定化担体に固定されたDNAの塩基配列を、短時間で簡便な処理によって解析するための手段を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記目的を達成すべく鋭意検討を重ねた結果、特定の構造を有するCe4+配位子を5´末端のリン酸基に結合させると、5´末端塩基と隣接する塩基との間のホスホジエステル結合の切断が容易になり、その結果、切断された塩基をひとつひとつ解析していくことによりDNAの塩基配列を解析できることを見出した。
また、本発明者らは、マグネシウムイオンを利用したDNA固定化担体の製造方法の発明を開発したが、かかる製造方法により得られる強固に固定化されたDNAであっても、ごく短時間で正確に塩基解析できることを見出し、本発明に到達した。
【0009】
本発明の請求項1に記載のDNAの塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)
【化3】

で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とする。
請求項2に記載のDNAの塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を質量分析法により解析し、続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析し、この操作を、DNAの所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることを特徴とする。
請求項3に記載の本発明のDNAの塩基配列解析方法は、上記請求項1又は2に記載の塩基配列解析方法において、DNAが、基体に固定されてなるものであることを特徴とする。
請求項4に記載の本発明のDNAの塩基配列解析方法は、上記請求項3に記載の塩基配列解析方法において、DNAが、マグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定されてなるものであることを特徴とする。
【発明の効果】
【0010】
本発明の塩基配列解析方法によれば、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確に解析することが可能となる。
また、従来日単位であったDNA塩基配列の解析を時単位にすることが可能になり、電気泳動方式に比べ測定感度も桁違いに向上し、使用するDNA量も少量ですみ、それに伴いコストも抑えることができる。
さらに、1塩基ずつ切断しては測定するということで測定可能なDNA鎖長に限界がなくなる。さらにまた、修飾塩基であっても1塩基ずつ測定しているのでどの部分が修飾されているのか判別するのも容易となる。
【発明を実施するための最良の形態】
【0011】
本発明の塩基配列解析方法は、DNAの塩基を5´末端から1つ、あるいは複数の塩基を5´末端から順次解析する方法である。すなわち、本発明の塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とする。
【0012】
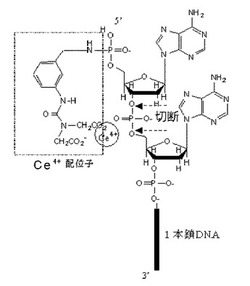
一般式(I)で表されるCe4+配位子は、図1に示すように、DNAの5´末端のリン酸基に特異的に結合する。
一般式(I)で表されるCe4+配位子の基本骨格は、後述の製造例1で記載するように、m−キシレンα,α´−ジアミンを原料とする有機合成反応を進めることにより得ることができる。
一般式(I)で表されるCe4+配位子は、Ceを保持するサイトを有しているので、機能的蛍光色素として有用である。
【0013】
本発明の塩基配列解析方法においては、まず、上記したようなDNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させる。
一般式(I)で表されるCe4+配位子の結合は、例えば、DNA断片を、dH2O等に溶かした溶液と、これに適当な緩衝液と共にCe4+配位子をDMSO等の溶媒で濃度を1〜100mMに適宜調製した後、dH2O等と混合し、20〜35℃で一晩穏やかに振盪反応後、エタノール沈殿等により精製する。
DNA断片は、予めエタノール沈殿、RPCカラム、PAGE、HPLC等により精製しておくことが好ましい。
DNAの5´末端のリン酸基と、一般式(I)で表されるCe4+配位子色素との結合率は、90%以上となることが好ましい。
【0014】
本発明の塩基配列解析方法においては、次に、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断する。切断の一例を模式図で説明すると、図2のとおりである。
切断方法については特に限定されず、加水分解やレーザーによる切断を挙げることができる。例えば、加水分解により切断する場合には、一般式(I)で表されるCe4+配位子とDNAとを反応させた後は、DNAにCe4+を導入して金属錯体化すると共に加水分解する。具体的には例えば、反応後の試料をdH2O等に溶解させた後、(NO2)2Ce(NH4)2水溶液を加え、20〜35℃で6〜24時間穏やかに振盪反応を行うことができる。振盪反応終了後、必要に応じて、エタノール沈殿等により精製させ、dH2Oに溶解させておくことができる。
一方、反応時間短縮のためレーザー照射により切断する場合には、一般式(I)で表されるCe4+配位子とDNAとを反応させた後、DNAにCe4+を導入してレーザー照射する。具体的には例えば、反応後の試料をdH2O等に溶解させた後、(NO2)2Ce(NH4)2水溶液を加え、レーザー照射後、必要に応じて、エタノール沈殿等により精製させ、dH2Oに溶解させておくことができる。ここで、レーザーとしてはYAGレーザー、色素レーザー等を用いて、照射量を1〜100mWで10〜60秒の照射条件とすることができる。
【0015】
本発明の塩基配列解析方法においては、上記切断反応により切断された末端塩基を質量分析により確認する。5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合が切断されたかどうかをも確認することができる。
質量分析は、TOF/MS、電気泳動,HPLC等質量分析に通常用いられる方法を用いることができるが、このうち正確さ、簡便さ等の点、さらには測定にかかる時間の短縮という点でTOF/MSが好ましい。TOF/MSの場合、MALDI−TOF/MS装置などを用いることができる。
TOF/MSによる測定は、例えば、以下のようにして行うことができる。マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)溶液を、切断確認の対象たるDNAとマイクロプレート上で混合し、真空中徐々に溶媒除去し、混晶を形成させる。この混晶に、レーザーを照射しマトリックス援用レーザー脱離質量分析により分子量分析し、ピーク同士の質量差から配列決定し、当初のDNAよりも5´末端の一塩基が少ないことを確認する。
【0016】
本発明の塩基配列解析方法は、上記の操作を特定の部位まで続けることにより、所望のDNA、特に固定化担体上に固定されたDNAの塩基配列を得るために用いることができる。すなわち、DNAの5´末端のリン酸基に、上記一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、この操作を、所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることにより、もとのDNAから、5´末端から何塩基かが切り離されたDNAを得ることができる。
このような本発明の切断方法は、例えば、PCRなどに用いられるプライマーやプローブなどの塩基配列を一部に含むDNAに適用することにより、その塩基配列部分のみを正確に切り出すことができる。
【0017】
このような本発明のDNAの塩基配列解析方法の対象であるDNAは、基体に固定されてなるものであること、すなわち、DNAチップ(マイクロアレイ)やDNAビーズアレイなどのDNA固定化担体が好ましい。
ここで、基体としては、素材及び形状のいずれも特に限定されない。素材としては、ガラス、ダイヤモンド、ダイヤモンドライクカーボンなどを挙げることができる。また、形状としては、チップ状、ビーズ状などのいずれであっても良い。中でも、いわゆるDNAチップとして用いられている素材及び形状であれば、特に限定されない。
【0018】
本発明のDNAの塩基配列解析方法の好ましい対象であるDNA固定化担体の製法は、特に限定されないが、DNAをマグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定させる方法が最も好ましい。マグネシウムイオンをDNA溶液中に溶解させておくことにより、DNAの立体構造を安定化し、基体との結合を効率よく媒介することができる。
【0019】
DNAをマグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定させる方法を、一例を挙げて以下に説明する。DNA溶液に含ませるDNAは、基体に固定させたいDNAであれば良く、鎖長、1本鎖か2本鎖の別、直鎖状か環状を問わないが、特に鎖長については3bp以上、中でも20bp〜100kbpであることが好ましい。また、DNAの由来や調製方法も特に限定されず、動植物、微生物、ウイルスなど天然の生物由来のものであっても良いし人工的に合成したものであっても良い。更に、DNAは、予め制限酵素処理がなされたものであっても良いし、PCR増幅産物などであっても良い。
また、DNA溶液には1種類のDNAだけでなく複数種類のDNAを含ませることもでき、この場合には、DNAライブラリーなど、塩基配列の異なる複数種類のDNAを固定化させたDNA固定化担体を得ることができる。
【0020】
DNA溶液において、DNAの濃度は、基体自身の固定化の上限によって適宜調整することができ、基本的に高い方が固定化量も増え、具体的には0.05μg/μL以上とすることが好ましい。DNAの量が0.05μg/μL未満であると、DNAを十分量基体に固定させることができないので好ましくない。一般に、用いる基体の大きさにより結合するDNA量の上限は決まるので、基本的には0.05μg/μL以上であれば問題ないが、好ましい上限は1μg/μL以下とすることができる。例えば、実施例で用いた基体のサイズの場合、もっとも好ましいDNAの濃度は、0.5〜1μg/μL、特に0.7〜1μg/μLである。
【0021】
一方、DNA溶液に含ませるマグネシウムイオンとしては、MgCl2、MgSO4、(CH3COO)2Mg等のマグネシウム塩があるが、扱いやすさ、手に入れやすさ等の理由からMgCl2を用いることが好ましい。なお、マグネシウム塩であっても水溶性でない場合は、DNA溶液中に溶解させることが困難となり、本発明の目的が十分に達成されないことから、好ましくない。
【0022】
一方、DNA溶液におけるマグネシウムイオンの濃度は、1mM以上とすることが好ましい。1mM未満であると本発明の目的が十分達成されないので好ましくない。もっとも好ましいマグネシウムイオンの濃度は、10〜20mMである。
マグネシウムイオンを含むDNA溶液は、上記マグネシウムイオン及びDNAをdH2Oなどと混合撹拌して作成することができる。DNA溶液の量は、基体のサイズなどによって適宜調製することができる。
マグネシウムイオンを含むDNA溶液中に基体を浸漬する際の浸漬時間や浸漬温度は、適宜設定することができる。一般に、浸漬時間は30分〜48時間、好ましくは30分〜24時間とすることができ、温度は20〜70℃、好ましくは30〜60℃とすることができる。
【0023】
ここで、上記したDNA固定化工程に先立って、基体を水溶性カルボジイミド及びN−ヒドロキシスクシンイミドを含む活性化反応液に浸漬する基体活性化工程を、DNA固定化工程に先立って行っておくと、より効率よくDNAの固定を進めることができるので好ましい。
活性化反応液には、水溶性カルボジイミド及びN−ヒドロキシスクシンイミドの両方を含ませておく必要がある。水溶性カルボジイミドのみとした場合には、活性基が不安定で分解しやすく、一方、N−ヒドロキシスクシンイミドのみとした場合には、活性化エステル基を生成しないので、好ましくない。
活性化反応液における水溶性カルボジイミドの濃度は、1〜50mg/mL、好ましくは5〜40mg/mL、特に好ましくは10〜30mg/mLとすることができる。一方、N−ヒドロキシスクシンイミドの濃度は、0.01〜30mg/mL、好ましくは0.1〜20mg/mL、特に好ましくは0.5〜10mg/mLとすることができる。上記数値範囲を下回ると、基体活性化工程を行う意味がなく、一方、上記数値範囲を超えると、目的の活性基が得られないとなるので好ましくない。
活性化反応液は、水溶性カルボジイミド及びN−ヒドロキシスクシンイミドを、リン酸緩衝液等の一般的な緩衝液に溶解させることにより調製される。
【0024】
基体にDNAが固定化したかどうかの確認は、PCRを利用して行うことができる。すなわち、目的のDNAを増幅させるように設計したプライマーを用いて、DNAを固定化させた基体を直接鋳型としたPCRを行い、PCR増幅産物を電気泳動等により視覚化して確認することができる。
本発明の塩基配列解析方法は、試料輸送・分析部を接続したμTAS装置のように、塩基の切断から質量分析までの過程をすべて自動で行うことができる装置を用いるとより簡易に行うことができるので、好ましい。
【実施例】
【0025】
以下、実施例によって本発明を具体的に説明する。
[製造例1](Ce4+配位子の基本骨格の合成)
出発原料として、m−キシレンα,α´−ジアミン及びジ−tert−ブチルカルボン酸を用いて、下記の図5で示す合成経路にて、以下の手順に従い、Ce4+配位子の基本骨格の合成を行った。
【0026】
(1)出発原料からの化合物2の合成
先ず、図5に示すように、m−キシレンα,α´−ジアミン(和光純薬製)0.5000mL(3.79mmol)を20mLのdryTHFに溶かし、これにジ−tert−ブチルカルボン酸(和光純薬製)0.425mL(1.85mmol)の20mL dryTHF溶液を氷水浴上で40分かけてゆっくり滴下した。室温で約15時間攪拌した後、反応液にdil NaOHaq.を加えて、酢酸エチルで抽出した。有機層は飽和食塩水で洗った後、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。
【0027】
得られた粗生成物(410mg)を5mLのdryTHFに溶かし、これに1,1´−カルボニルジイミダゾール391mg(2.47mmol)を加えた。室温で2時間攪拌した後、溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(SiO2、Merk70−230mesh、30g、酢酸エチル、Rf=0.4)で精製したところ、化合物2が317mg(52%、2steps)得られた(図5で示す2を参照)。化合物2をNMRで構造解析したところ、以下のとおりであった。
【0028】
(化合物2のNMR結果)
化合物2のNMR結果を示すと、以下のようであった。すなわち、NMR(60MHz,CDCl3)1.39(s,9H,t−Bu),4.0−4.5(two bd,4H,Ar−CH2−),5.7(br,1H,−NH−CO−),6.91,7.58,8.03(each s,3II,imidazole−H)、7.19(s,4H,Ar−H),8.71(bt,1H,−NH−CO−)
【0029】
(2)化合物3の合成
ジアミド(図5で示す2を参照)317mg(0.96mmol)を3mLのTHFに溶かし、これにdiethyliminodiacetate 0.180mL(1.00mmol)を加えた。室温で40時間(又は50−60℃で2時間)後、溶媒を減圧留去し、粗生成物550mgを得た。得られた粗生成物をシリカゲルクロマトグラフィー(SiO2、Merck70−230mesh、25g、酢酸エチル、Rf=0.6)で精製したところ、化合物3が396mg(91%)得られた(図5で示す3を参照)。化合物3をNMRで構造解析したところ、以下のとおりであった。
【0030】
(化合物3のNMR結果)
化合物3のNMR結果を示すと、以下のようであった。すなわち、NMR(60MHz,CDCl3)1.24(t,J=7 Hz,6H,−CH3),1.44(s,9H,t−Bu),4.10(s,4H,−CH2CO2−),4.0−4.4(m,4H,Ar−CH2−,−CO2−CH2−),5.3(br,1H,−NH−CO−),5.9(br,1H,−NH−CO−),7.19(s,4H,Ar−H)
【0031】
(3)化合物4の合成
前記化合物3(図5で示す3を参照)396mg(0.88mmol)にCF3COOHを0.500mL加えて、室温で2時間攪拌したところ、直ちに発泡した。過剰のCF3COOHを減圧留去し、粗生成物として化合物4を得た(図5で示す4を参照)。
【0032】
(化合物4のNMR結果)
化合物4をNMRで構造解析したところ、以下のとおりであった。すなわち、NMR(60MHz,CDCl3)1.25(t,J=7 Hz,6H,−CH3),4.0−4.4(m,10H,−CH2−),4.63(bs,2H,−CH2−),7.32(s,4H,Ar−H),7.6(m,3H,−NH3+)
【0033】
(4)化合物6の合成
前記化合物4(図5で示す4を参照)の一部を酢酸エチルに溶かして分液漏斗に移し、dilNaOHaq、次いで飽和食塩水で洗い、有機層を無水硫酸ナトリウムで乾燥し、溶媒を留去すると化合物6が得られた(図5で示す6を参照)。尚、化合物5(図5で示す5を参照)は容易に分子内環化して化合物6に変化するように思われた。
【0034】
(化合物6のNMR結果)
化合物6をNMRで構造解析したところ、以下のとおりであった。すなわち、NMR(60MHz,CDCl3)1.27(t,J=7 Hz,3H,−CH3),1.75(bs,2H,−NH2),3.84−4.41(m,8Hr,−CH2−),4.96(s,2H,−CH2−),7.30(s,4H,Ar−H)
【0035】
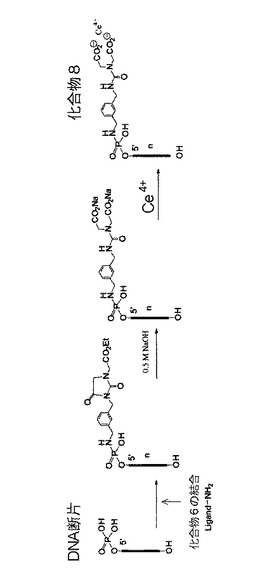
(5)化合物8の合成(図6を参照)
実際に合成する化合物としては化合物6までであるが、ここから後の工程は化合物6とDNAを結合させ環を開化させCeを配位させ、一連の流れでDNAを切断させる工程に移行する。
以下の経路にて、化合物6を環化しない方向に変換させた。即ち、下記の実施例2の(1)に従って、DNA断片(実施例2で用いたものと同じもの)の5´リン酸基に化合物6を結合させた後、0.5M NaOHを用いて加水分解を行って開環し、次いで実施例2の(2)に従ってCe錯体として、環化させない化合物を得た(これも、最終反応物のようにCeを錯化する箇所を有している蛍光色素の場合、同様に機能的蛍光色素として使える)。
【0036】
[製造例2](マグネシウムイオンを用いたDNAチップの製造)
(1)ダイヤモンドチップの活性化
まず、リン酸緩衝液(0.1M リン酸二水素カリウム及び0.01M リン酸水素二カリウム)1mL中に、水溶性カルボジイミド15.5mg及びN−ヒドロキシスクシンイミド2.3mgを含む活性化反応液を調製した。
この活性化反応液中に、ダイヤモンドチップを時々撹拌しながら30分浸した。その後、チップを取り出し、蒸留水で3〜5回洗浄して、活性化ダイヤモンドチップを得た。
【0037】
(2)DNAの固定化
λDNAをEcoRI処理して得られる21kbpのDNA断片を固定化するDNA試料として用いた。
すなわち、この21kbpのλDNA断片(1μg/μL)1μLと、25mM MgCl225μL及びdH2O74μLとを混合し、全量100μlにしてDNA固定化溶液を調製した。このDNA固定化溶液中に活性化ダイヤモンドチップを挿入し、室温で60分反応させた。その後、チップを取り出し、蒸留水で3〜5回洗浄した。
【0038】
(3)固定化の確認のためのPCR
まず、プライマー(センス:配列表の配列番号1記載の塩基配列参照)1μL、プライマー(アンチセンス:配列表の配列番号2記載の塩基配列参照)1μL、10xPCRバッファー(Applied Biosystem社製)5μL、dNTP溶液5μL、DNAポリメラーゼ0.25μL、dH2O37.75μlを加えて全量50μLとして、PCR反応液を調製した。なお、上記プライマーは、上記DNA試料である21kbpDNA断片内の500bp断片が増幅されるように設計されたものである。
このPCR反応液にDNA固定化チップを挿入し、PCRにて2サイクル増幅した。PCR条件は、(95℃30秒−65℃1分−72℃1分)×12サイクルとした。
【0039】
PCR終了後、DNA固定化チップを取り出し、蒸留水で3〜5回洗浄した。洗浄したチップを、70%エタノール中、4℃で保存した。
PCR反応液は、固定化したチップを取り出した後で残り10サイクル分PCRを行い、その後アガロースゲル電気泳動で解析した。結果を図3のレーン2に示す。
なお、対照として上記(2)においてMgCl2を添加しないDNA固定化溶液にて反応を行ったほかは、同様の処理を行った。結果を図3のレーン1に示す。
【0040】
[比較製造例1](酵素的DNA固定化法(従来法)によるDNAチップの製造)
(1)ダイヤモンドチップの活性化
実施製造例1の1.と同様の条件で活性化反応液中にダイヤモンドチップを時々攪拌しながら室温で30分間浸し、蒸留水で3〜5回洗浄して、活性化ダイヤモンドチップを得た。
【0041】
(2)オリゴヌクレオチドの固定化
5´末端に(dA)3とEcoRI部位に対応した塩基配列を含むオリゴヌクレオチド溶液を調製し、この0.5μMオリゴヌクレオチド溶液中に、上記活性化ダイヤモンドチップを室温で30〜60分浸してから(液量100μLでチップ2枚浸漬が可能である)、蒸留水で3〜5回洗浄して、オリゴヌクレオチド固定化チップを得た。
【0042】
(3)相補的オリゴヌクレオチドのハイブリダイズ
上記のオリゴヌクレオチドに相補的な0.5μMオリゴヌクレオチド溶液中に、上記のオリゴヌクレオチド固定化チップを4℃で30分間浸してから(液量100μLでチップ2枚の浸漬が可能である)、氷冷蒸留水で3〜5回洗浄して、制限酵素部位固定化チップを得た。
【0043】
(4)二本鎖DNAの固定化(酵素反応)
上記実施製造例1の(2)と同様のDNA試料(λDNAをEcoRI処理して得られる21kbpのDNA断片)を用いた。
ライゲーション反応緩衝液、T4DNAリガーゼ及び制限酵素処理された二本鎖DNA溶液とを混合してライゲーション固定化溶液を得た。ライゲーション固定化溶液は、66mM Tris−HCl(pH7.6)、6.6mM MgCl2、10mM Dithiothreitol、0.1mM ATP、300ngの二本鎖DNA、T4DNAリガーゼ 35unitsが入った計50μLの水溶液である。
ライゲーション固定化溶液中に上記の制限酵素部位固定化チップを16℃で16時間浸した。その後、蒸留水で3〜5回洗浄した。
【0044】
(5)固定化の確認のためのPCR
実施製造例1の3.と同様の条件でPCRを行い、固定化が行われたかどうか確認した。その結果を図3のレーン3に示す。
【0045】
図3から、MgCl2を利用した実施製造例1(図3のレーン2参照)は、酵素を使用した従来法による比較製造例1(図3のレーン3参照)と比較して、濃いバンドの出現が確認されたことが明らかである。
また、比較製造例1の従来法は全工程18〜20時間を要したのに対し、実施製造例1の方法では、全工程60〜90分を要したに過ぎなかった。
このことから、本発明のDNA固定法では、水溶性マグネシウム塩を利用することにより、従来法に比較してDNAを基体に効率よく、しかもごく短時間に処理を済ませることができることが明らかとなった。
【0046】
[実施例1](DNAの塩基配列解析方法)
(1)DNA試料との結合
配列表の配列番号3記載の塩基配列(ggttatcgaa atcagccaca gcgcg)からなる1本鎖DNA断片(5´リン酸化プライマー)を、dH2Oに溶かし100μMとして、DNA試料とした。このDNA試料10μLを、2×Labelling Buffer(0.24M 1−メチルイミダゾール(0.32M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを含む。)水溶液をpH9.0に調整したもの)50μL、機能的蛍光色素、すなわち、実施例2で得た化合物6をDMSOにて50mMに調製したもの(50mM配位子)20μL、及びdH2O 20μLとを混合し、室温で一晩穏やかに振盪反応後、エタノール沈殿により精製した。色素との結合率は90%以上であった。
【0047】
(2)DNA試料の金属錯体化(Ce4+配位子の導入)及び特異的部位切断
上記(1)にて機能的蛍光色素と反応し、精製処理のなされたDNA試料をdH2O 10μLに溶解させた。このDNA試料に、1mM(NO2)2Ce(NH4)2水溶液10μLを加え、室温で一晩穏やかに振盪反応後、エタノール沈殿により精製し、dH2O10μLに溶解した。
【0048】
(3)MALDI−TOF/MSによる測定
上記(2)にて切断後のDNA試料1μLについて、切断された塩基の確認を行った。
マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)30mgを水100μL、0.1M二水素クエン酸アンモニウム水溶液に溶解させた。そのうちの1μLを、DNA試料1μLと共に、マイクロプレート上で直接混合した。真空中徐々に溶媒除去し、混晶を形成させた。この混晶に、波長337nmのレーザーを照射しマトリックス援用レーザー脱離質量分析により解析した。分子量比較用の試料として、未反応のDNA試料と蛍光色素を結合させたDNA試料を同様に測定した。
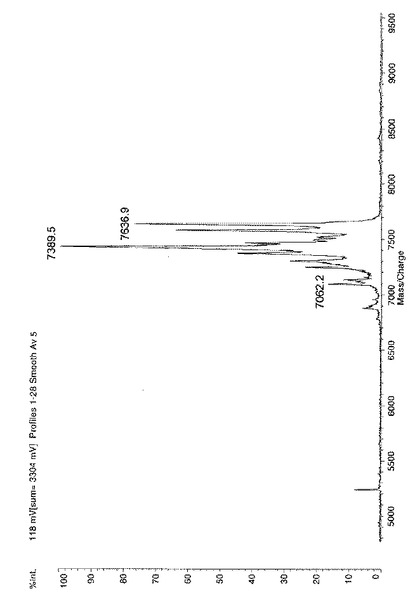
解析の結果検出されたピーク同士の質量差から配列決定した。結果を、図4に示す。
【0049】
その結果、図4から明らかなように、5´側のグアニンが切断されたと考えられるピーク(7389.5)をメインに、リン酸基のみが切断されたピーク(7636.9)、さらにはもう一つのグアニンも切断されたと考えられるピーク(7062.2)等が検出された。
このことから、本発明のCe4+配位子を用いる塩基配列解析方法により、DNAの5´末端の塩基を解析することができることが明らかとなった。
【0050】
[実施例2](DNAチップの塩基配列解析方法)
(1)DNA試料との結合
製造例2で作成したDNAチップの塩基配列の解析を行った。
すなわち、DNAチップをdH2O 100μLに浸漬し、DNAチップ試料とした。このDNAチップ試料中に、2×Labelling Buffer(0.24M 1−メチルイミダゾール(0.32M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを含む。)水溶液をpH9.0に調整したもの)500μL、機能的蛍光色素(製造例1で得た化合物6)をDMSOにて50mMに調製したもの(50mM配位子)400μLとを混合し、室温で一晩穏やかに振盪反応後、DNAチップを反応溶液中より取り出した。色素との結合率は90%以上であった。
【0051】
(2)DNA試料の金属錯体化(Ce4+ 配位子の導入)及び特異的部位切断
上記(1)にて機能的蛍光色素と反応させたDNAチップをdH2O100μLに浸漬させた。このDNA試料に、1mM(NO2)2Ce(NH4)2 水溶液100μLを加え、レーザーを照射した。
【0052】
(3)MALDI−TOF/MSによる測定
上記(2)にて切断後のDNA試料0.5μLについて、切断された塩基の確認を行った。
マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)50mgを水 200μL、アセトニトリル300μL、0.1M二水素クエン酸アンモニウム水溶液500μLに溶解させた。そのうちの0.5μLを、DNA試料0.5μLと共に、マイクロプレート上で直接混合した。真空大気圧中徐々に溶媒除去し、混晶を形成させた。この混晶に、波長337nmのレーザーを照射しマトリックス援用レーザー脱離質量分析により解析した。実施例2に従って同様に金属錯体化、レーザー照射切断を繰り返し、その都度質量分析により得られた塩基を順番に並べ、配列決定した。
【産業上の利用可能性】
【0053】
本発明の塩基配列解析方法によれば、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確に解析することが可能となる。
また、本発明により、より精密な遺伝子操作や遺伝子の解明へ応用の展開でき、迅速な遺伝子解析が可能となることで、医学分野においては、テーラーメイド医療の確立に、農水畜産分野においては、より良い形質を持ち合わせた品種を作り出すことといった遺伝子を扱う上で不可欠な配列解析のスピード化に寄与できる。
【図面の簡単な説明】
【0054】
【図1】Ce4+配位子がDNAに結合した状態の模式図である。
【図2】本発明の切断方法によるDNAの切断の模式図である。
【図3】DNAが固定化されたことを確認するための電気泳動結果である。
【図4】本発明の切断方法の結果切断されたDNAの質量分析結果の一例である。
【図5】Ce4+配位子の基本骨格の合成経路を示す概略図である。
【図6】化合物8の合成経路を示す概略図である。
【0055】
(配列表)
SEQUENCE LISTING
<110> 株式会社日本パーカーライジング広島工場
<110> 独立行政法人科学技術振興機構
<120> DNAの切断方法
<160> 3
<210> 1
<211> 25
<212> DNA
<213> Artificial Sequence
<400> gatgagttcg tgtccgtaca actgg
<210> 2
<211> 25
<212> DNA
<213> Artificial Sequence
<400> ggttatcgaa atcagccaca gcgcc
<210> 3
<211> 25
<212> DNA
<213> Artificial Sequence
<400> ggttatcgaa atcagccaca gcgcg
【技術分野】
【0001】
本発明は、塩基配列解析方法に関し、詳しくは、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確な解析を可能にする塩基配列解析方法に関する。
【背景技術】
【0002】
DNAチップ(マイクロアレイ)やDNAビーズアレイなどのDNA固定化担体は、疾病診断、治療のほか、ゲノム解析などにも広く用いられている。このDNA固定化担体に所望のDNAが固定化されているかどうかを確認するため、種々の塩基配列解析方法が開発されている(例えば、特許文献1および非特許文献1参照)。
しかし、これらの方法では、二次構造がハイブリダイゼーションを阻害し、かつ配列の反復が正しい解析を妨げる、あるいは、3位で修飾された塩基により、ポリメラーゼの組み込み反応が妨げられるといった問題があった。
【0003】
また、DNAの塩基配列を解析するにあたり、特定の塩基配列を認識する制限酵素によりDNAを短く切断してから調べる方法もあるが、これらの酵素が認識する塩基配列は、限られた4〜6塩基のみであるなどの限界があり、正確に特定の部位でDNAを切断する方法が必要とされていた。
酵素を使わずにDNAを切断する方法としては、加水分解による方法が提案されている。例えば、特許文献2には、1本鎖DNAの塩基配列認識部位にDNA断片(末端に切断反応の触媒が結合)を結合させ、該触媒部位で1本鎖DNAを切断のセリウム(Ce,IV)と、ポリアミン−N−ポリカルボン酸あるいはその誘導体よりなる錯体を用いた加水分解による切断方法が提案されている。
また、非特許文献2には、1本鎖DNAの塩基配列認識部位にDNA断片を2つ結合させ、Ce(IV)/EDTAを作用させて該DNA断片の間部分で1本鎖DNAを切断する方法が提案されている。
しかし、これらの方法は、いずれも加水分解による切断のみに対応したものであり、他の切断方法が想定されたものではなかった。また、加水分解反応に多大な時間がかかるなどの問題点があった。
【0004】
一方、DNA固定化担体の製造は、基体に所望のDNAを固定化して行われ、従来はリガーゼなどの酵素を利用する酵素的DNA固定化法が用いられていた(特許文献3および4参照)。
酵素的DNA固定化法では、基体を活性化した後、まず2本鎖の短いオリゴヌクレオチドをチップ上に固定して、その後で所望のオリゴヌクレオチドを制限酵素末端を利用して、DNAリガーゼを利用して固定化させていた。
しかし、この方法では、DNAを基体に確実に固定化することが困難であったほか、多くの工程を経る必要があり、多くの試薬や機器が必要なほか、処理に相当の長時間を要するという問題点があった。
【0005】
このような状況下、DNAの塩基配列、特にDNA固定化担体に固定されたDNAの塩基配列をより正確、迅速、かつ簡便に解析する方法の開発が望まれていた。
【0006】
【特許文献1】特許公表2004−529650号公報
【特許文献2】特開2001−89430号公報
【特許文献3】国際特許公開WO01−68368号公報
【特許文献4】国際特許公開WO01−40173号公報
【非特許文献1】Nucleosides & Nucleotides : 1999 Feb; 18(2): 197-201
【非特許文献2】Chem.Letters:2004;33,300-301
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は、上記従来の問題に鑑みて、DNAの塩基配列、特にDNA固定化担体に固定されたDNAの塩基配列を、短時間で簡便な処理によって解析するための手段を提供することを目的とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記目的を達成すべく鋭意検討を重ねた結果、特定の構造を有するCe4+配位子を5´末端のリン酸基に結合させると、5´末端塩基と隣接する塩基との間のホスホジエステル結合の切断が容易になり、その結果、切断された塩基をひとつひとつ解析していくことによりDNAの塩基配列を解析できることを見出した。
また、本発明者らは、マグネシウムイオンを利用したDNA固定化担体の製造方法の発明を開発したが、かかる製造方法により得られる強固に固定化されたDNAであっても、ごく短時間で正確に塩基解析できることを見出し、本発明に到達した。
【0009】
本発明の請求項1に記載のDNAの塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)
【化3】
で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とする。
請求項2に記載のDNAの塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を質量分析法により解析し、続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析し、この操作を、DNAの所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることを特徴とする。
請求項3に記載の本発明のDNAの塩基配列解析方法は、上記請求項1又は2に記載の塩基配列解析方法において、DNAが、基体に固定されてなるものであることを特徴とする。
請求項4に記載の本発明のDNAの塩基配列解析方法は、上記請求項3に記載の塩基配列解析方法において、DNAが、マグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定されてなるものであることを特徴とする。
【発明の効果】
【0010】
本発明の塩基配列解析方法によれば、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確に解析することが可能となる。
また、従来日単位であったDNA塩基配列の解析を時単位にすることが可能になり、電気泳動方式に比べ測定感度も桁違いに向上し、使用するDNA量も少量ですみ、それに伴いコストも抑えることができる。
さらに、1塩基ずつ切断しては測定するということで測定可能なDNA鎖長に限界がなくなる。さらにまた、修飾塩基であっても1塩基ずつ測定しているのでどの部分が修飾されているのか判別するのも容易となる。
【発明を実施するための最良の形態】
【0011】
本発明の塩基配列解析方法は、DNAの塩基を5´末端から1つ、あるいは複数の塩基を5´末端から順次解析する方法である。すなわち、本発明の塩基配列解析方法は、DNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、質量分析法により解析することを特徴とする。
【0012】
一般式(I)で表されるCe4+配位子は、図1に示すように、DNAの5´末端のリン酸基に特異的に結合する。
一般式(I)で表されるCe4+配位子の基本骨格は、後述の製造例1で記載するように、m−キシレンα,α´−ジアミンを原料とする有機合成反応を進めることにより得ることができる。
一般式(I)で表されるCe4+配位子は、Ceを保持するサイトを有しているので、機能的蛍光色素として有用である。
【0013】
本発明の塩基配列解析方法においては、まず、上記したようなDNAの5´末端のリン酸基に、一般式(I)で表されるCe4+配位子を結合させる。
一般式(I)で表されるCe4+配位子の結合は、例えば、DNA断片を、dH2O等に溶かした溶液と、これに適当な緩衝液と共にCe4+配位子をDMSO等の溶媒で濃度を1〜100mMに適宜調製した後、dH2O等と混合し、20〜35℃で一晩穏やかに振盪反応後、エタノール沈殿等により精製する。
DNA断片は、予めエタノール沈殿、RPCカラム、PAGE、HPLC等により精製しておくことが好ましい。
DNAの5´末端のリン酸基と、一般式(I)で表されるCe4+配位子色素との結合率は、90%以上となることが好ましい。
【0014】
本発明の塩基配列解析方法においては、次に、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断する。切断の一例を模式図で説明すると、図2のとおりである。
切断方法については特に限定されず、加水分解やレーザーによる切断を挙げることができる。例えば、加水分解により切断する場合には、一般式(I)で表されるCe4+配位子とDNAとを反応させた後は、DNAにCe4+を導入して金属錯体化すると共に加水分解する。具体的には例えば、反応後の試料をdH2O等に溶解させた後、(NO2)2Ce(NH4)2水溶液を加え、20〜35℃で6〜24時間穏やかに振盪反応を行うことができる。振盪反応終了後、必要に応じて、エタノール沈殿等により精製させ、dH2Oに溶解させておくことができる。
一方、反応時間短縮のためレーザー照射により切断する場合には、一般式(I)で表されるCe4+配位子とDNAとを反応させた後、DNAにCe4+を導入してレーザー照射する。具体的には例えば、反応後の試料をdH2O等に溶解させた後、(NO2)2Ce(NH4)2水溶液を加え、レーザー照射後、必要に応じて、エタノール沈殿等により精製させ、dH2Oに溶解させておくことができる。ここで、レーザーとしてはYAGレーザー、色素レーザー等を用いて、照射量を1〜100mWで10〜60秒の照射条件とすることができる。
【0015】
本発明の塩基配列解析方法においては、上記切断反応により切断された末端塩基を質量分析により確認する。5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合が切断されたかどうかをも確認することができる。
質量分析は、TOF/MS、電気泳動,HPLC等質量分析に通常用いられる方法を用いることができるが、このうち正確さ、簡便さ等の点、さらには測定にかかる時間の短縮という点でTOF/MSが好ましい。TOF/MSの場合、MALDI−TOF/MS装置などを用いることができる。
TOF/MSによる測定は、例えば、以下のようにして行うことができる。マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)溶液を、切断確認の対象たるDNAとマイクロプレート上で混合し、真空中徐々に溶媒除去し、混晶を形成させる。この混晶に、レーザーを照射しマトリックス援用レーザー脱離質量分析により分子量分析し、ピーク同士の質量差から配列決定し、当初のDNAよりも5´末端の一塩基が少ないことを確認する。
【0016】
本発明の塩基配列解析方法は、上記の操作を特定の部位まで続けることにより、所望のDNA、特に固定化担体上に固定されたDNAの塩基配列を得るために用いることができる。すなわち、DNAの5´末端のリン酸基に、上記一般式(I)で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、この操作を、所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることにより、もとのDNAから、5´末端から何塩基かが切り離されたDNAを得ることができる。
このような本発明の切断方法は、例えば、PCRなどに用いられるプライマーやプローブなどの塩基配列を一部に含むDNAに適用することにより、その塩基配列部分のみを正確に切り出すことができる。
【0017】
このような本発明のDNAの塩基配列解析方法の対象であるDNAは、基体に固定されてなるものであること、すなわち、DNAチップ(マイクロアレイ)やDNAビーズアレイなどのDNA固定化担体が好ましい。
ここで、基体としては、素材及び形状のいずれも特に限定されない。素材としては、ガラス、ダイヤモンド、ダイヤモンドライクカーボンなどを挙げることができる。また、形状としては、チップ状、ビーズ状などのいずれであっても良い。中でも、いわゆるDNAチップとして用いられている素材及び形状であれば、特に限定されない。
【0018】
本発明のDNAの塩基配列解析方法の好ましい対象であるDNA固定化担体の製法は、特に限定されないが、DNAをマグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定させる方法が最も好ましい。マグネシウムイオンをDNA溶液中に溶解させておくことにより、DNAの立体構造を安定化し、基体との結合を効率よく媒介することができる。
【0019】
DNAをマグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定させる方法を、一例を挙げて以下に説明する。DNA溶液に含ませるDNAは、基体に固定させたいDNAであれば良く、鎖長、1本鎖か2本鎖の別、直鎖状か環状を問わないが、特に鎖長については3bp以上、中でも20bp〜100kbpであることが好ましい。また、DNAの由来や調製方法も特に限定されず、動植物、微生物、ウイルスなど天然の生物由来のものであっても良いし人工的に合成したものであっても良い。更に、DNAは、予め制限酵素処理がなされたものであっても良いし、PCR増幅産物などであっても良い。
また、DNA溶液には1種類のDNAだけでなく複数種類のDNAを含ませることもでき、この場合には、DNAライブラリーなど、塩基配列の異なる複数種類のDNAを固定化させたDNA固定化担体を得ることができる。
【0020】
DNA溶液において、DNAの濃度は、基体自身の固定化の上限によって適宜調整することができ、基本的に高い方が固定化量も増え、具体的には0.05μg/μL以上とすることが好ましい。DNAの量が0.05μg/μL未満であると、DNAを十分量基体に固定させることができないので好ましくない。一般に、用いる基体の大きさにより結合するDNA量の上限は決まるので、基本的には0.05μg/μL以上であれば問題ないが、好ましい上限は1μg/μL以下とすることができる。例えば、実施例で用いた基体のサイズの場合、もっとも好ましいDNAの濃度は、0.5〜1μg/μL、特に0.7〜1μg/μLである。
【0021】
一方、DNA溶液に含ませるマグネシウムイオンとしては、MgCl2、MgSO4、(CH3COO)2Mg等のマグネシウム塩があるが、扱いやすさ、手に入れやすさ等の理由からMgCl2を用いることが好ましい。なお、マグネシウム塩であっても水溶性でない場合は、DNA溶液中に溶解させることが困難となり、本発明の目的が十分に達成されないことから、好ましくない。
【0022】
一方、DNA溶液におけるマグネシウムイオンの濃度は、1mM以上とすることが好ましい。1mM未満であると本発明の目的が十分達成されないので好ましくない。もっとも好ましいマグネシウムイオンの濃度は、10〜20mMである。
マグネシウムイオンを含むDNA溶液は、上記マグネシウムイオン及びDNAをdH2Oなどと混合撹拌して作成することができる。DNA溶液の量は、基体のサイズなどによって適宜調製することができる。
マグネシウムイオンを含むDNA溶液中に基体を浸漬する際の浸漬時間や浸漬温度は、適宜設定することができる。一般に、浸漬時間は30分〜48時間、好ましくは30分〜24時間とすることができ、温度は20〜70℃、好ましくは30〜60℃とすることができる。
【0023】
ここで、上記したDNA固定化工程に先立って、基体を水溶性カルボジイミド及びN−ヒドロキシスクシンイミドを含む活性化反応液に浸漬する基体活性化工程を、DNA固定化工程に先立って行っておくと、より効率よくDNAの固定を進めることができるので好ましい。
活性化反応液には、水溶性カルボジイミド及びN−ヒドロキシスクシンイミドの両方を含ませておく必要がある。水溶性カルボジイミドのみとした場合には、活性基が不安定で分解しやすく、一方、N−ヒドロキシスクシンイミドのみとした場合には、活性化エステル基を生成しないので、好ましくない。
活性化反応液における水溶性カルボジイミドの濃度は、1〜50mg/mL、好ましくは5〜40mg/mL、特に好ましくは10〜30mg/mLとすることができる。一方、N−ヒドロキシスクシンイミドの濃度は、0.01〜30mg/mL、好ましくは0.1〜20mg/mL、特に好ましくは0.5〜10mg/mLとすることができる。上記数値範囲を下回ると、基体活性化工程を行う意味がなく、一方、上記数値範囲を超えると、目的の活性基が得られないとなるので好ましくない。
活性化反応液は、水溶性カルボジイミド及びN−ヒドロキシスクシンイミドを、リン酸緩衝液等の一般的な緩衝液に溶解させることにより調製される。
【0024】
基体にDNAが固定化したかどうかの確認は、PCRを利用して行うことができる。すなわち、目的のDNAを増幅させるように設計したプライマーを用いて、DNAを固定化させた基体を直接鋳型としたPCRを行い、PCR増幅産物を電気泳動等により視覚化して確認することができる。
本発明の塩基配列解析方法は、試料輸送・分析部を接続したμTAS装置のように、塩基の切断から質量分析までの過程をすべて自動で行うことができる装置を用いるとより簡易に行うことができるので、好ましい。
【実施例】
【0025】
以下、実施例によって本発明を具体的に説明する。
[製造例1](Ce4+配位子の基本骨格の合成)
出発原料として、m−キシレンα,α´−ジアミン及びジ−tert−ブチルカルボン酸を用いて、下記の図5で示す合成経路にて、以下の手順に従い、Ce4+配位子の基本骨格の合成を行った。
【0026】
(1)出発原料からの化合物2の合成
先ず、図5に示すように、m−キシレンα,α´−ジアミン(和光純薬製)0.5000mL(3.79mmol)を20mLのdryTHFに溶かし、これにジ−tert−ブチルカルボン酸(和光純薬製)0.425mL(1.85mmol)の20mL dryTHF溶液を氷水浴上で40分かけてゆっくり滴下した。室温で約15時間攪拌した後、反応液にdil NaOHaq.を加えて、酢酸エチルで抽出した。有機層は飽和食塩水で洗った後、無水硫酸ナトリウムで乾燥し、溶媒を減圧留去した。
【0027】
得られた粗生成物(410mg)を5mLのdryTHFに溶かし、これに1,1´−カルボニルジイミダゾール391mg(2.47mmol)を加えた。室温で2時間攪拌した後、溶媒を減圧留去し、得られた粗生成物をシリカゲルカラムクロマトグラフィー(SiO2、Merk70−230mesh、30g、酢酸エチル、Rf=0.4)で精製したところ、化合物2が317mg(52%、2steps)得られた(図5で示す2を参照)。化合物2をNMRで構造解析したところ、以下のとおりであった。
【0028】
(化合物2のNMR結果)
化合物2のNMR結果を示すと、以下のようであった。すなわち、NMR(60MHz,CDCl3)1.39(s,9H,t−Bu),4.0−4.5(two bd,4H,Ar−CH2−),5.7(br,1H,−NH−CO−),6.91,7.58,8.03(each s,3II,imidazole−H)、7.19(s,4H,Ar−H),8.71(bt,1H,−NH−CO−)
【0029】
(2)化合物3の合成
ジアミド(図5で示す2を参照)317mg(0.96mmol)を3mLのTHFに溶かし、これにdiethyliminodiacetate 0.180mL(1.00mmol)を加えた。室温で40時間(又は50−60℃で2時間)後、溶媒を減圧留去し、粗生成物550mgを得た。得られた粗生成物をシリカゲルクロマトグラフィー(SiO2、Merck70−230mesh、25g、酢酸エチル、Rf=0.6)で精製したところ、化合物3が396mg(91%)得られた(図5で示す3を参照)。化合物3をNMRで構造解析したところ、以下のとおりであった。
【0030】
(化合物3のNMR結果)
化合物3のNMR結果を示すと、以下のようであった。すなわち、NMR(60MHz,CDCl3)1.24(t,J=7 Hz,6H,−CH3),1.44(s,9H,t−Bu),4.10(s,4H,−CH2CO2−),4.0−4.4(m,4H,Ar−CH2−,−CO2−CH2−),5.3(br,1H,−NH−CO−),5.9(br,1H,−NH−CO−),7.19(s,4H,Ar−H)
【0031】
(3)化合物4の合成
前記化合物3(図5で示す3を参照)396mg(0.88mmol)にCF3COOHを0.500mL加えて、室温で2時間攪拌したところ、直ちに発泡した。過剰のCF3COOHを減圧留去し、粗生成物として化合物4を得た(図5で示す4を参照)。
【0032】
(化合物4のNMR結果)
化合物4をNMRで構造解析したところ、以下のとおりであった。すなわち、NMR(60MHz,CDCl3)1.25(t,J=7 Hz,6H,−CH3),4.0−4.4(m,10H,−CH2−),4.63(bs,2H,−CH2−),7.32(s,4H,Ar−H),7.6(m,3H,−NH3+)
【0033】
(4)化合物6の合成
前記化合物4(図5で示す4を参照)の一部を酢酸エチルに溶かして分液漏斗に移し、dilNaOHaq、次いで飽和食塩水で洗い、有機層を無水硫酸ナトリウムで乾燥し、溶媒を留去すると化合物6が得られた(図5で示す6を参照)。尚、化合物5(図5で示す5を参照)は容易に分子内環化して化合物6に変化するように思われた。
【0034】
(化合物6のNMR結果)
化合物6をNMRで構造解析したところ、以下のとおりであった。すなわち、NMR(60MHz,CDCl3)1.27(t,J=7 Hz,3H,−CH3),1.75(bs,2H,−NH2),3.84−4.41(m,8Hr,−CH2−),4.96(s,2H,−CH2−),7.30(s,4H,Ar−H)
【0035】
(5)化合物8の合成(図6を参照)
実際に合成する化合物としては化合物6までであるが、ここから後の工程は化合物6とDNAを結合させ環を開化させCeを配位させ、一連の流れでDNAを切断させる工程に移行する。
以下の経路にて、化合物6を環化しない方向に変換させた。即ち、下記の実施例2の(1)に従って、DNA断片(実施例2で用いたものと同じもの)の5´リン酸基に化合物6を結合させた後、0.5M NaOHを用いて加水分解を行って開環し、次いで実施例2の(2)に従ってCe錯体として、環化させない化合物を得た(これも、最終反応物のようにCeを錯化する箇所を有している蛍光色素の場合、同様に機能的蛍光色素として使える)。
【0036】
[製造例2](マグネシウムイオンを用いたDNAチップの製造)
(1)ダイヤモンドチップの活性化
まず、リン酸緩衝液(0.1M リン酸二水素カリウム及び0.01M リン酸水素二カリウム)1mL中に、水溶性カルボジイミド15.5mg及びN−ヒドロキシスクシンイミド2.3mgを含む活性化反応液を調製した。
この活性化反応液中に、ダイヤモンドチップを時々撹拌しながら30分浸した。その後、チップを取り出し、蒸留水で3〜5回洗浄して、活性化ダイヤモンドチップを得た。
【0037】
(2)DNAの固定化
λDNAをEcoRI処理して得られる21kbpのDNA断片を固定化するDNA試料として用いた。
すなわち、この21kbpのλDNA断片(1μg/μL)1μLと、25mM MgCl225μL及びdH2O74μLとを混合し、全量100μlにしてDNA固定化溶液を調製した。このDNA固定化溶液中に活性化ダイヤモンドチップを挿入し、室温で60分反応させた。その後、チップを取り出し、蒸留水で3〜5回洗浄した。
【0038】
(3)固定化の確認のためのPCR
まず、プライマー(センス:配列表の配列番号1記載の塩基配列参照)1μL、プライマー(アンチセンス:配列表の配列番号2記載の塩基配列参照)1μL、10xPCRバッファー(Applied Biosystem社製)5μL、dNTP溶液5μL、DNAポリメラーゼ0.25μL、dH2O37.75μlを加えて全量50μLとして、PCR反応液を調製した。なお、上記プライマーは、上記DNA試料である21kbpDNA断片内の500bp断片が増幅されるように設計されたものである。
このPCR反応液にDNA固定化チップを挿入し、PCRにて2サイクル増幅した。PCR条件は、(95℃30秒−65℃1分−72℃1分)×12サイクルとした。
【0039】
PCR終了後、DNA固定化チップを取り出し、蒸留水で3〜5回洗浄した。洗浄したチップを、70%エタノール中、4℃で保存した。
PCR反応液は、固定化したチップを取り出した後で残り10サイクル分PCRを行い、その後アガロースゲル電気泳動で解析した。結果を図3のレーン2に示す。
なお、対照として上記(2)においてMgCl2を添加しないDNA固定化溶液にて反応を行ったほかは、同様の処理を行った。結果を図3のレーン1に示す。
【0040】
[比較製造例1](酵素的DNA固定化法(従来法)によるDNAチップの製造)
(1)ダイヤモンドチップの活性化
実施製造例1の1.と同様の条件で活性化反応液中にダイヤモンドチップを時々攪拌しながら室温で30分間浸し、蒸留水で3〜5回洗浄して、活性化ダイヤモンドチップを得た。
【0041】
(2)オリゴヌクレオチドの固定化
5´末端に(dA)3とEcoRI部位に対応した塩基配列を含むオリゴヌクレオチド溶液を調製し、この0.5μMオリゴヌクレオチド溶液中に、上記活性化ダイヤモンドチップを室温で30〜60分浸してから(液量100μLでチップ2枚浸漬が可能である)、蒸留水で3〜5回洗浄して、オリゴヌクレオチド固定化チップを得た。
【0042】
(3)相補的オリゴヌクレオチドのハイブリダイズ
上記のオリゴヌクレオチドに相補的な0.5μMオリゴヌクレオチド溶液中に、上記のオリゴヌクレオチド固定化チップを4℃で30分間浸してから(液量100μLでチップ2枚の浸漬が可能である)、氷冷蒸留水で3〜5回洗浄して、制限酵素部位固定化チップを得た。
【0043】
(4)二本鎖DNAの固定化(酵素反応)
上記実施製造例1の(2)と同様のDNA試料(λDNAをEcoRI処理して得られる21kbpのDNA断片)を用いた。
ライゲーション反応緩衝液、T4DNAリガーゼ及び制限酵素処理された二本鎖DNA溶液とを混合してライゲーション固定化溶液を得た。ライゲーション固定化溶液は、66mM Tris−HCl(pH7.6)、6.6mM MgCl2、10mM Dithiothreitol、0.1mM ATP、300ngの二本鎖DNA、T4DNAリガーゼ 35unitsが入った計50μLの水溶液である。
ライゲーション固定化溶液中に上記の制限酵素部位固定化チップを16℃で16時間浸した。その後、蒸留水で3〜5回洗浄した。
【0044】
(5)固定化の確認のためのPCR
実施製造例1の3.と同様の条件でPCRを行い、固定化が行われたかどうか確認した。その結果を図3のレーン3に示す。
【0045】
図3から、MgCl2を利用した実施製造例1(図3のレーン2参照)は、酵素を使用した従来法による比較製造例1(図3のレーン3参照)と比較して、濃いバンドの出現が確認されたことが明らかである。
また、比較製造例1の従来法は全工程18〜20時間を要したのに対し、実施製造例1の方法では、全工程60〜90分を要したに過ぎなかった。
このことから、本発明のDNA固定法では、水溶性マグネシウム塩を利用することにより、従来法に比較してDNAを基体に効率よく、しかもごく短時間に処理を済ませることができることが明らかとなった。
【0046】
[実施例1](DNAの塩基配列解析方法)
(1)DNA試料との結合
配列表の配列番号3記載の塩基配列(ggttatcgaa atcagccaca gcgcg)からなる1本鎖DNA断片(5´リン酸化プライマー)を、dH2Oに溶かし100μMとして、DNA試料とした。このDNA試料10μLを、2×Labelling Buffer(0.24M 1−メチルイミダゾール(0.32M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを含む。)水溶液をpH9.0に調整したもの)50μL、機能的蛍光色素、すなわち、実施例2で得た化合物6をDMSOにて50mMに調製したもの(50mM配位子)20μL、及びdH2O 20μLとを混合し、室温で一晩穏やかに振盪反応後、エタノール沈殿により精製した。色素との結合率は90%以上であった。
【0047】
(2)DNA試料の金属錯体化(Ce4+配位子の導入)及び特異的部位切断
上記(1)にて機能的蛍光色素と反応し、精製処理のなされたDNA試料をdH2O 10μLに溶解させた。このDNA試料に、1mM(NO2)2Ce(NH4)2水溶液10μLを加え、室温で一晩穏やかに振盪反応後、エタノール沈殿により精製し、dH2O10μLに溶解した。
【0048】
(3)MALDI−TOF/MSによる測定
上記(2)にて切断後のDNA試料1μLについて、切断された塩基の確認を行った。
マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)30mgを水100μL、0.1M二水素クエン酸アンモニウム水溶液に溶解させた。そのうちの1μLを、DNA試料1μLと共に、マイクロプレート上で直接混合した。真空中徐々に溶媒除去し、混晶を形成させた。この混晶に、波長337nmのレーザーを照射しマトリックス援用レーザー脱離質量分析により解析した。分子量比較用の試料として、未反応のDNA試料と蛍光色素を結合させたDNA試料を同様に測定した。
解析の結果検出されたピーク同士の質量差から配列決定した。結果を、図4に示す。
【0049】
その結果、図4から明らかなように、5´側のグアニンが切断されたと考えられるピーク(7389.5)をメインに、リン酸基のみが切断されたピーク(7636.9)、さらにはもう一つのグアニンも切断されたと考えられるピーク(7062.2)等が検出された。
このことから、本発明のCe4+配位子を用いる塩基配列解析方法により、DNAの5´末端の塩基を解析することができることが明らかとなった。
【0050】
[実施例2](DNAチップの塩基配列解析方法)
(1)DNA試料との結合
製造例2で作成したDNAチップの塩基配列の解析を行った。
すなわち、DNAチップをdH2O 100μLに浸漬し、DNAチップ試料とした。このDNAチップ試料中に、2×Labelling Buffer(0.24M 1−メチルイミダゾール(0.32M 1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドを含む。)水溶液をpH9.0に調整したもの)500μL、機能的蛍光色素(製造例1で得た化合物6)をDMSOにて50mMに調製したもの(50mM配位子)400μLとを混合し、室温で一晩穏やかに振盪反応後、DNAチップを反応溶液中より取り出した。色素との結合率は90%以上であった。
【0051】
(2)DNA試料の金属錯体化(Ce4+ 配位子の導入)及び特異的部位切断
上記(1)にて機能的蛍光色素と反応させたDNAチップをdH2O100μLに浸漬させた。このDNA試料に、1mM(NO2)2Ce(NH4)2 水溶液100μLを加え、レーザーを照射した。
【0052】
(3)MALDI−TOF/MSによる測定
上記(2)にて切断後のDNA試料0.5μLについて、切断された塩基の確認を行った。
マトリックスとして3−ヒドロキシピコリン酸(3−HPAと略す)50mgを水 200μL、アセトニトリル300μL、0.1M二水素クエン酸アンモニウム水溶液500μLに溶解させた。そのうちの0.5μLを、DNA試料0.5μLと共に、マイクロプレート上で直接混合した。真空大気圧中徐々に溶媒除去し、混晶を形成させた。この混晶に、波長337nmのレーザーを照射しマトリックス援用レーザー脱離質量分析により解析した。実施例2に従って同様に金属錯体化、レーザー照射切断を繰り返し、その都度質量分析により得られた塩基を順番に並べ、配列決定した。
【産業上の利用可能性】
【0053】
本発明の塩基配列解析方法によれば、短時間で簡便な処理のみで、DNA固定化担体に固定されたDNAの塩基配列を正確に解析することが可能となる。
また、本発明により、より精密な遺伝子操作や遺伝子の解明へ応用の展開でき、迅速な遺伝子解析が可能となることで、医学分野においては、テーラーメイド医療の確立に、農水畜産分野においては、より良い形質を持ち合わせた品種を作り出すことといった遺伝子を扱う上で不可欠な配列解析のスピード化に寄与できる。
【図面の簡単な説明】
【0054】
【図1】Ce4+配位子がDNAに結合した状態の模式図である。
【図2】本発明の切断方法によるDNAの切断の模式図である。
【図3】DNAが固定化されたことを確認するための電気泳動結果である。
【図4】本発明の切断方法の結果切断されたDNAの質量分析結果の一例である。
【図5】Ce4+配位子の基本骨格の合成経路を示す概略図である。
【図6】化合物8の合成経路を示す概略図である。
【0055】
(配列表)
SEQUENCE LISTING
<110> 株式会社日本パーカーライジング広島工場
<110> 独立行政法人科学技術振興機構
<120> DNAの切断方法
<160> 3
<210> 1
<211> 25
<212> DNA
<213> Artificial Sequence
<400> gatgagttcg tgtccgtaca actgg
<210> 2
<211> 25
<212> DNA
<213> Artificial Sequence
<400> ggttatcgaa atcagccaca gcgcc
<210> 3
<211> 25
<212> DNA
<213> Artificial Sequence
<400> ggttatcgaa atcagccaca gcgcg
【特許請求の範囲】
【請求項1】
DNAの5´末端のリン酸基に、一般式(I)
【化1】
で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、
質量分析法により解析することを特徴とするDNAの塩基配列解析方法。
【請求項2】
DNAの5´末端のリン酸基に、一般式(I)
【化2】
で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を質量分析法により解析し、
続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、
切断された末端塩基を、質量分析法により解析し、
この操作を、DNAの所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることを特徴とするDNAの塩基配列解析方法。
【請求項3】
DNAが、基体に固定されてなるものである請求項1又は2記載のDNAの塩基配列解析方法。
【請求項4】
DNAが、マグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定されてなるものである請求項3記載のDNAの塩基配列解析方法。
【請求項1】
DNAの5´末端のリン酸基に、一般式(I)
【化1】
で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を、
質量分析法により解析することを特徴とするDNAの塩基配列解析方法。
【請求項2】
DNAの5´末端のリン酸基に、一般式(I)
【化2】
で表されるCe4+配位子を結合させて、前記DNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、切断された末端塩基を質量分析法により解析し、
続いて切断されたDNAの5´末端に上記Ce4+配位子を結合させて、前記切断されたDNAの5´末端の塩基と該塩基に隣接する塩基との間のホスホジエステル結合を切断し、
切断された末端塩基を、質量分析法により解析し、
この操作を、DNAの所定の塩基と該塩基に隣接する3´末端の塩基との間が切断されるまで順次続けることを特徴とするDNAの塩基配列解析方法。
【請求項3】
DNAが、基体に固定されてなるものである請求項1又は2記載のDNAの塩基配列解析方法。
【請求項4】
DNAが、マグネシウムイオンを含むDNA溶液中に基体を浸漬することにより基体に固定されてなるものである請求項3記載のDNAの塩基配列解析方法。
【図1】


【図4】
【図5】

【図6】
【図2】
【図3】

【図4】
【図5】
【図6】
【図2】
【図3】
【公開番号】特開2007−60969(P2007−60969A)
【公開日】平成19年3月15日(2007.3.15)
【国際特許分類】
【出願番号】特願2005−250378(P2005−250378)
【出願日】平成17年8月30日(2005.8.30)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(591091135)株式会社日本パーカーライジング広島工場 (8)
【Fターム(参考)】
【公開日】平成19年3月15日(2007.3.15)
【国際特許分類】
【出願日】平成17年8月30日(2005.8.30)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(591091135)株式会社日本パーカーライジング広島工場 (8)
【Fターム(参考)】
[ Back to top ]