
塩酸セチリジン含有錠剤の製造方法
【課題】類縁物質を十分に抑制しながら含量均一性を高めることができる塩酸セチリジン含有錠剤の製造方法を提供する。
【解決手段】粉末状の添加剤を塩酸セチリジンの溶液(特に水溶液)と共に造粒し、この造粒物を打錠することによって塩酸セチリジン含有錠剤を製造する。前記造粒では、好ましくは、気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する。造粒では結合剤を用いる必要はない。造粒物は、例えば、崩壊剤と混合してから打錠してもよい。
【解決手段】粉末状の添加剤を塩酸セチリジンの溶液(特に水溶液)と共に造粒し、この造粒物を打錠することによって塩酸セチリジン含有錠剤を製造する。前記造粒では、好ましくは、気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する。造粒では結合剤を用いる必要はない。造粒物は、例えば、崩壊剤と混合してから打錠してもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は塩酸セチリジンの製剤化方法に関するものである。
【背景技術】
【0002】
塩酸セチリジンは、化学名が(±)−2−[4−[(4−クロロフェニル)フェニルメチル]−1−ピペラジニル]エトキシ酢酸2塩酸塩である抗アレルギー薬である。この塩酸セチリジンは、白色の結晶性の粉末であり、フィルムコート錠である市販品には、乳糖、結晶セルロース、軽質無水ケイ酸、ステアリン酸マグネシウム、ヒドロキシプロピルメチルセルロース2910、酸化チタン、マクロゴール400などの添加剤が用いられている(非特許文献1)。
【0003】
ところで塩酸セチリジンは、粒径が細かく凝集性が高いため、含量均一性を確保するのが難しい。粒子に凝集性がない場合には、粒径が細かくなるほど含量均一性が向上するが、粒子に凝集性がある場合には、粒子が細かくなるほど凝集性も増大するため、含量均一性が逆に低下する。また塩酸セチリジンは、直接打錠すると打錠障害を引き起こし易い。
【0004】
錠剤の含量均一性を高めるため、湿式造粒が推奨されている(特許文献1〜3など)。
例えば特許文献1には、活性成分と賦形剤とを混合し、次いで溶剤を添加して練り合わせて造粒すれば、含量均一性を改善できることが示されている。また特許文献2には、活性成分と賦形剤とを液体バインダーの結合力を利用しながら流動層造粒すれば、含量均一性を改善できることが示されている。特許文献3の従来技術の欄には、医薬を溶媒に溶解し、これを添加剤に加えて湿式造粒すれば含量均一性が高まる旨が記載されている。ただし医薬を溶媒に溶解すると、主薬の含量が低下する虞がある旨も記載されている。
【非特許文献1】第一製薬株式会社作製、「医薬品インタビューフォーム (ジルテック錠5 ジルテック錠10)」
【特許文献1】特開2002−338454号公報(特許請求の範囲、0006、0027)
【特許文献2】特開平9−59181号公報(特許請求の範囲、0003、0025〜0027)
【特許文献3】特開2003−119121号公報(0002)
【発明の開示】
【発明が解決しようとする課題】
【0005】
ところが本発明者らの検討によれば、塩酸セチリジンでは、特許文献1〜3の知見が必ずしも当てはまらないことが判明した。すなわち特許文献1のようにして塩酸セチリジンと賦形剤の混合粉末にバインダー液を加えて湿式攪拌造粒すると、逆に含量均一性が大きく低下した。さらには錠剤保存時に塩酸セチリジンが分解し易くなり、類縁物質が大きく増大した。特許文献2のようにして流動層造粒に変更すれば、含量均一性は改善されたが、錠剤保存時の塩酸セチリジンの分解を十分に低減することはできなかった。そして前述した様に特許文献3は、医薬を溶媒に溶解すれば、主薬の含量がさらに低下する虞があることを指摘している。
【0006】
本発明は上記の様な事情に着目してなされたものであって、その目的は、類縁物質を十分に抑制しながら含量均一性を高めることができる塩酸セチリジン含有錠剤の製造方法を提供することにある。
【0007】
本発明のより好ましい目的は、さらに主薬の溶出特性にも優れている塩酸セチリジン含有錠剤の製造方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者らが前記課題を解決するために鋭意検討を重ねた結果、塩酸セチリジンを溶解した溶液を利用して湿式造粒すると、含量均一性が高まるだけでなく、錠剤保存時の類縁物質の増大を十分に抑制できることを見出し、本発明を完成した。
【0009】
なお本発明者らは、前記湿式造粒の際に結合剤を使用しなくても優れた含量均一性を維持できること、さらには結合剤を使用することなく流動層造粒法を採用すれば、塩酸セチリジンの溶出特性を著しく高めることができることも併せて見出している。特許文献1には、結合剤を使用しない場合には溶出特性が改善されることが記載されているが(段落0008)、結合剤を使用しないだけでは塩酸セチリジンの溶出特性を高めることはできない。結合剤の不使用と流動層造粒法とを組み合わせることによって初めて塩酸セチリジンの溶出特性を改善できた。特許文献2には流動層造粒法を採用して含量均一性を高めることは記載されているが、流動層造粒法と溶出特性との関係については記載されていない。
【0010】
すなわち上記目的を達成し得た本発明の塩酸セチリジン含有錠剤の製造方法は、粉末状の添加剤を塩酸セチリジンの溶液(特に水溶液)と共に造粒し、この造粒物を打錠する点に要旨を有する。前記造粒では結合剤を用いる必要はない。好ましい造粒法は流動層造粒法である。造粒物は、例えば、崩壊剤と混合してから打錠してもよい。
【発明の効果】
【0011】
本発明によれば粉末状の添加剤を塩酸セチリジンの溶液と共に造粒(湿式造粒)している為、類縁物質を抑制しながら含量均一性を高めることができる。
【発明を実施するための最良の形態】
【0012】
本発明では、粉末状の添加剤を塩酸セチリジン溶液と共に造粒(湿式造粒)し、この造粒物を打錠することによって塩酸セチリジン含有錠剤を製造している。湿式造粒を採用することによって打錠障害(スティッキング)を防止できる。ただし湿式造粒法を採用したとしても湿式攪拌造粒では含量均一性はむしろ低下し、さらには主薬(塩酸セチリジン)の保存安定性も大きく低下し、類縁物質が大きく増大する。湿式流動層造粒法を採用することで含量均一性は向上するが、主薬(塩酸セチリジン)の保存安定性が不十分である。そこで本発明では、塩酸セチリジンを溶液にして湿式造粒することとする。塩酸セチリジン溶液を使用すると、何故か錠剤保存時の類縁物質の増大を十分に抑制することができる。また塩酸セチリジンを溶液にして使用すると、塩酸セチリジンの倍散が不要になり、錠剤の製造工程を簡略化できる。さらには塩酸セチリジンが取り扱い容器や器具に付着することも防止でき、作業性が向上する。
【0013】
塩酸セチリジン溶液に使用する溶媒としては、水が好ましい。水は塩酸セチリジンを分解する虞が少ない。また塩酸セチリジン溶液中の塩酸セチリジンの濃度は、造粒法(詳細は後述する)に応じて適宜設定できるが、例えば、5〜50質量%程度の範囲から選択できる。湿式攪拌造粒の場合、塩酸セチリジンの最も好ましい濃度は、20〜35質量%程度である。湿式流動層造粒の場合、塩酸セチリジンの最も好ましい濃度は、10〜20質量%程度である。
【0014】
塩酸セチリジン溶液と共に造粒する前記添加剤(造粒用添加剤)には、賦形剤、結合剤などが挙げられる。さらに造粒可能である限り、崩壊剤を使用してもよい。また必要に応じて、滑沢剤、着色剤、矯味・矯臭剤などを使用してもよい。好ましい造粒用添加剤は、賦形剤、結合剤である。
【0015】
賦形剤には、乳糖、ショ糖、ブドウ糖などの糖類、コムギデンプン、コメデンプン、トウモロコシデンプン、バレイショデンプンなどの水不溶性デンプン類、結晶セルロースなどの水不溶性セルロースなどが含まれる。好ましい賦形剤は、糖類、結晶セルロースなどである。
【0016】
結合剤としては、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルメロースナトリウム(カルボキシメチルセルロースナトリウム)などの水溶性又は水膨潤性のセルロース類;デキストリン、部分アルファー化デンプン、プルランなどの水溶性又は水膨潤性デンプン類;アラビアゴム、カンテン、ゼラチン、トラガント、アルギン酸ナトリウムなどの水溶性又は水膨潤性の天然高分子化合物類;ポビドン、ポリビニルアルコールなどの水溶性又は水膨潤性の合成高分子化合物類などが挙げられる。
【0017】
本発明の特に好ましい態様では、造粒用添加剤に結合剤が含まれない。本発明では、塩酸セチリジンを溶液として加えているため、結合剤を使用しなくても高い含量均一性を保ったまま造粒できる。また結合剤を省略しただけでは溶出特性を改善できないが、後述するように結合剤の省略と適切な造粒法(流動層造粒)とを組み合わせると、溶出特性を改善することもできる。
【0018】
湿式造粒では、粉末状の添加剤と塩酸セチリジン溶液とを用いて造粒できる限り種々の造粒法が採用でき、例えば、押出造粒法、転動造粒法、攪拌造粒法、流動層造粒法、噴霧乾燥造粒法などが採用できる。好ましい造粒法は、攪拌造粒法、流動層造粒法などである。
【0019】
攪拌造粒法とは、粉末状の添加剤を攪拌羽根で攪拌しながら塩酸セチリジン溶液を注液して造粒する方法のことをいう。流動層造粒法とは、気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する方法のことをいう。
【0020】
本発明では、結合剤を使用することなく前記流動層造粒を行うことが最も推奨される。湿式造粒法によって得られる塩酸セチリジン錠剤は、製造直後の溶出性に問題はないが、なぜか保管後には溶出遅延が生じる。結合剤を使用しないようにしてもこの保管後の溶出遅延を防止できず、また流動層造粒法だけを採用しても保管後の溶出遅延を防止できない。結合剤の不使用と流動層造粒法とを組み合わせて初めて保管後の溶出遅延を防止できる。
【0021】
上記のようにして得られる造粒物の大きさは、湿式造粒の方式によって異なる。例えば攪拌造粒法によって湿式造粒する場合には、850μm以上の造粒物が1%以上(特に2〜10%程度)存在する一方、75μm以下の造粒物も20%以上(特に30〜50%程度)存在するのに対し、流動層造粒法によって湿式造粒する場合には、850μm以上の造粒物が1%未満(特に0〜0.5%程度)に減少し、かつ75μm以下の造粒物も15%以下(特に5〜10%程度)に減少し、粒度分布が相対的に狭くなる。
【0022】
前記造粒物は、打錠することによって錠剤化する。この造粒物は打錠前に乾燥しておくことが望ましい。また造粒物は、必要に応じて他の医薬品用添加剤(賦形剤、結合剤、崩壊剤、滑沢剤、着色剤、矯味・矯臭剤など、好ましくは賦形剤、崩壊剤、滑沢剤など、特に崩壊剤、滑沢剤)と混合してから打錠してもよい。
【0023】
崩壊剤としては、低置換度ヒドロキシプロピルセルロース、カルメロース(カルボキシメチルセルロース)、カルメロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルエチルセルロースなどのセルロース類が挙げられる。また前記結晶セルロースやデンプン類を崩壊剤として用いてもよい。さらにはヒドロキシプロピルスターチ、カルボキシメチルスターチナトリウム(デンプングリコール酸ナトリウム)なども崩壊剤としてのデンプン類として使用してもよい。造粒物を崩壊剤と混合してから打錠することによって錠剤の溶出特性を改善することができる。
【0024】
滑沢剤としては、ステアリン酸塩、ステアリン酸、タルク、ワックス類、コムギデンプンなどが挙げられる。特に好ましい滑沢剤は、滑沢特性と抗付着特性に優れた滑沢剤(lubricants、antiadherents)であるステアリン酸塩(ステアリン酸マグネシウム、ステアリン酸カルシウムなど)である。
【0025】
各添加剤は適量使用すればよい。錠剤中の賦形剤の割合は、例えば、70〜99質量%程度、好ましくは85〜95質量%程度である。また錠剤中の崩壊剤の割合は、例えば、0〜20質量%程度、好ましくは1〜10質量%程度である。錠剤中の主薬(塩酸セチリジン)の割合は、例えば、1〜20質量%程度、好ましくは3〜15質量%程度、特に5〜10質量%程度である。
【0026】
打錠後の錠剤は、必要に応じてコーティングしてもよい。
【0027】
上記のようにして得られる塩酸セチリジン錠剤は、塩酸セチリジンを溶解した溶液を利用して湿式造粒することによって製造されているため、含量均一性が高く、しかも錠剤を保存しても類縁物質は増大しない。
【実施例】
【0028】
以下、実施例を挙げて本発明をより具体的に説明するが、本発明はもとより下記実施例によって制限を受けるものではなく、前・後記の趣旨に適合し得る範囲で適当に変更を加えて実施することも勿論可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0029】
比較例1
【0030】
【表1】

【0031】
下記の様に乾式混合粉末を直接打錠することによって、上記表1に示す処方の錠剤5000錠を製造した。
【0032】
(1)倍散工程
塩酸セチリジン25gと乳糖50gを混合し、60メッシュの篩を用いて篩過することによって倍散末を得た。
【0033】
(2)混合工程
前記倍散末と、予め一部の乳糖(20g)と共に混合して篩過(32メッシュ)しておいた軽質無水ケイ酸4.25gと、残りの乳糖171.5gと、結晶セルロース130gと、低置換度ヒドロキシプロピルセルロース20gとを混合した。さらに32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.25gを加えて混合することによって混合末を得た。
【0034】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合末を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5000錠を得た。
【0035】
比較例2
【0036】
【表2】
【0037】
下記の様に結合剤の水溶液を使用して湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表2に示す処方の錠剤5000錠を製造した。
【0038】
(1)造粒・乾燥・整粒工程
塩酸セチリジン25g、乳糖238.5g、結晶セルロース128.5g及びヒドロキシプロピルセルロース3.75gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物にヒドロキシプロピルセルロース水溶液(ヒドロキシプロピルセルロース3.75g、精製水45g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0039】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース21.25gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.25gを加えて混合することによって混合顆粒を得た。
【0040】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5000錠を得た。
【0041】
比較例3
【0042】
【表3】
【0043】
下記の様に結合剤の水溶液を使用して湿式流動層造粒することによって得られる顆粒を打錠することによって、上記表3に示す処方の錠剤50000錠を製造した。
【0044】
(1)倍散工程
塩酸セチリジン250gと乳糖500gを混合し、60メッシュの篩を用いて篩過することによって倍散末を得た。
【0045】
(2)造粒・乾燥・整粒工程
前記倍散末と乳糖2800gを流動層造粒乾燥機(フロイント産業(株)製「FLO−5A」、温度70℃)で5分間混合した。混合物にヒドロキシプロピルセルロース水溶液(ヒドロキシプロピルセルロース75g、精製水1250g)をスプレーして造粒(噴霧空気圧:2.25kg/cm2G、流速:41g/min)した後、造粒物を乾燥(70℃、30分)した。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0046】
(3)混合顆粒工程
前記整粒品と、結晶セルロース370gと、低置換度ヒドロキシプロピルセルロース212.5gとを混合機(コトブキ技研工業(株)製「ボーレコンテナーミキサー LM20−1010」)で回転数25rpmにて10分間混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム42.5gを加えて1分間混合することによって混合顆粒を得た。
【0047】
(4)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤50000錠を得た。
【0048】
実施例1
【0049】
【表4】
【0050】
下記の様に塩酸セチリジンの水溶液を使用して結合剤と共に湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表4に示す処方の錠剤4000錠を製造した。
【0051】
(1)造粒・乾燥・整粒工程
乳糖190.8g、結晶セルロース102.8g及びヒドロキシプロピルセルロース6.0gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物に塩酸セチリジン水溶液(塩酸セチリジン20g、精製水42g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0052】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース17gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム3.4gを加え混合することによって混合顆粒を得た。
【0053】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤4000錠を得た。
【0054】
実施例2
【0055】
【表5】
【0056】
下記の様に塩酸セチリジンの水溶液を使用し、結合剤なしで湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表5に示す処方の錠剤4000錠を製造した。
【0057】
(1)造粒・乾燥・整粒工程
乳糖196.8g及び結晶セルロース102.8gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物に塩酸セチリジン水溶液(塩酸セチリジン20g、精製水42g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0058】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース17gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム3.4gを加え混合することによって混合顆粒を得た。
【0059】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤4000錠を得た。
【0060】
実施例3
【0061】
【表6】
【0062】
下記の様に塩酸セチリジンの水溶液を使用し、結合剤なしで湿式流動層造粒することによって得られる顆粒を打錠することによって、上記表6に示す処方の錠剤5600錠を製造した。
【0063】
(1)造粒・乾燥・整粒工程
60メッシュの篩で篩過しておいた乳糖377.44gを流動層造粒乾燥機(フロイント産業(株)製「FRC−1」、温度60℃)に入れ、塩酸セチリジン水溶液(塩酸セチリジン28g、精製水140g)をスプレーして造粒(噴霧空気圧:0.1MPa、流速:4.7g/min)した後、造粒物を乾燥(70℃、40分)した。得られた造粒乾燥品を20メッシュの篩で整粒した。
【0064】
(2)混合顆粒工程
前記整粒品と、結晶セルロース42gと、低置換度ヒドロキシプロピルセルロース23.8gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.76gを加え混合することによって混合顆粒を得た。
【0065】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5600錠を得た。
【0066】
実施例1〜3において造粒・乾燥工程後の造粒乾燥品の粒度分布は下記表7の通りであった。この粒度分布は、受け皿の上に表7に記載の篩を目の細かい方から順に積み上げ、試料(5g)を最上段の篩から順に篩過させ、各篩に残った試料の質量を測定することによって調べたものである。
【0067】
【表7】
【0068】
上記実施例及び比較例で得られた錠剤の特性(含量均一性、類縁物質、溶出特性)を以下のようにして評価した。
【0069】
(1)含量均一性
試験製剤10錠を採取し、それぞれの塩酸セチリジンの含有量を高速液体クロマトグラフィ(HPLC)によって決定した(内部標準法)。主薬の含量(表示量(100%)に対する割合(%))とその平均値を算出し、日本薬局方に記載の計算法に従って標準偏差(下式(1)参照)及び含量均一性の判定値(下式(2)参照)を求めた。
【0070】
【数1】
【0071】
(式中、Sは標準偏差である。nは試験した試料の全個数(10)を示し、Xiは、試験した個々の試料に含まれる主薬の含量(表示量に対する割合(%))を示す。AはX1からXnまでの各含量の平均値である)
判定値=|100−A|+S×2.2 …(2)
(式中、A及びSは、前記式(1)と同じである)
【0072】
測定液調製法
試験製剤と内部標準(パラオキシ安息香酸プロピル)をそれぞれ移動相に溶解し、塩酸セチリジン濃度を約10μg/mL、内部標準濃度を約15μg/mLにした。
【0073】
HPLC条件
検出器:紫外吸光光度計(測定波長:230nm)
カラム:内径4.6mm、長さ15cmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲル((株)資生堂製「カプセルパックタイプMG 5C18」)を充填したもの
カラム温度:40℃
移動相:1−オクタンスルホン酸ナトリウム水溶液(濃度:0.01mol/L、pH:2.35)/アセトニトリル混液(前者/後者=1/1(容量比))
流量:セチリジンの保持時間が約4分になる程度
測定液の注入量:10μL
【0074】
(2)類縁物質
試験錠剤をポリ瓶に充填し、開放又は密栓状態で苛酷条件(温度55℃、湿度75%RH)で放置した。試験開始から2週間後及び4週間後の分解量をHPLCによって以下のようにして測定した。
【0075】
測定液調製方法
試験錠剤8錠を移動相50mLに溶解し、不溶成分を孔径0.45μmのフィルタでろ過した。
【0076】
HPLC条件
検出器;紫外吸光光度計(測定波長:230nm)
カラム:内径4.0mm、長さ25cmのステンレス管に粒径5μmの液体クロマトグラフ用シリカゲル(和光純薬工業(株)製「Wakosil 5SIL」)を充填したもの
カラム温度:25℃
移動相:純水で希釈した硫酸(硫酸濃度:0.04mol/L)/アセトニトリル混液(前者/後者=3/47(容量比))
流量:セチリジンの保持時間が約9分になる程度
測定液の注入量:10μL
算出法
分解量(%)=全類縁物質のピーク面積の合計/セチリジンのピーク面積×100
【0077】
(3)溶出特性
前記類縁物質試験(苛酷試験)に供した錠剤の主薬溶出性を以下のようにして調べた。
【0078】
溶出試験
日本薬局方の溶出試験第2法(パドル法)に従い、試験液:精製水(錠剤1錠当たり900mL)、回転数:50rpmの条件で錠剤から主薬(塩酸セチリジン)を溶出させた。溶出試験開始から5分後、10分後、及び15分後の主薬濃度をHPLCによって調べ(絶対検量線法)、溶出率を算出した。
【0079】
測定液調製方法
溶出試験液を一部採取し、不溶成分を孔径0.45μmのフィルタでろ過した。
【0080】
HPLC条件
検出器:紫外吸光光度計(測定波長:230nm)
カラム:内径4.6mm、長さ15cmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲル((株)資生堂製「カプセルパックタイプMG 5C18」)を充填したもの
カラム温度:40℃
移動相:1−オクタンスルホン酸ナトリウム水溶液(濃度:0.01mol/L、pH:2.35)/アセトニトリル混液(前者/後者=1/1(容量比))
流量:セチリジンの保持時間が約4分になる程度
測定液の注入量:20μL
【0081】
溶出特性を下記基準に従って評価した。
○:苛酷試験後の錠剤の溶出特性(溶出試験開始後10分時点)が、苛酷試験前の錠剤の溶出特性(溶出試験開始後10分時点)と同程度である。
△:苛酷試験後の錠剤の溶出特性(溶出試験開始後10分時点)が、苛酷試験前の錠剤の溶出特性(溶出試験開始後10分時点)に比べて劣る。
【0082】
結果を各実施例及び比較例の操作性(主薬ハンドリング性、打錠性)と共に下記表8及び図1〜6に示す。また市販されている塩酸セチリジン錠剤(「ジルテック錠5」、「ジルテック錠10」)の含量均一性も併せて示す。
【0083】
【表8】
【0084】
表8より明らかなように、市販塩酸セチリジン錠剤の含量均一性は低い。比較例1の直接打錠法によって得られる塩酸セチリジン錠剤は、市販品(ジルテック錠5)よりもスケールが小さいために含量均一性が若干高まっているが、それでも含量均一性は低い。湿式造粒法を採用しても攪拌造粒では含量均一性がさらに低下し、さらには苛酷試験下での類縁物質が大きく増大した(比較例2)。湿式流動層造粒することによって含量均一性は改善したが、類縁物質の抑制は不十分であった(比較例3)。
【0085】
塩酸セチリジンの水溶液を用いて湿式造粒することによって初めて類縁物質を十分に抑制しながら含量均一性を高めることができた(実施例1)。ただし実施例1は溶出特性に改善余地があった(図4参照)。実施例1では結合剤を使用していたため、結合剤を使用しないことにしたが、溶出特性は改善されなかった(実施例2、図5参照)。造粒法を攪拌造粒から流動層造粒に変更することによって初めて溶出特性も改善された(実施例3)。また実施例3の塩酸セチリジン錠剤では、スケールアップすることによって、含量均一性がむしろ高まった。
【図面の簡単な説明】
【0086】
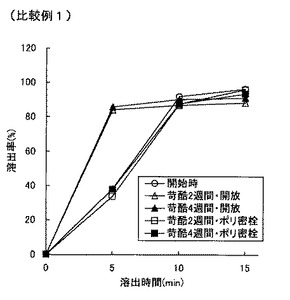
【図1】図1は比較例1の錠剤の溶出特性を示すグラフである。
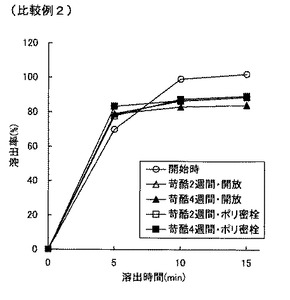
【図2】図2は比較例2の錠剤の溶出特性を示すグラフである。
【図3】図3は比較例3の錠剤の溶出特性を示すグラフである。
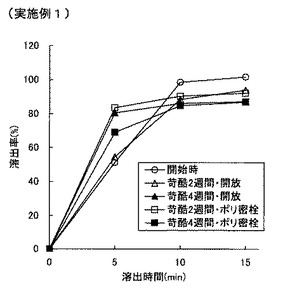
【図4】図4は実施例1の錠剤の溶出特性を示すグラフである。
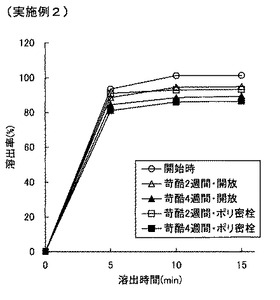
【図5】図5は実施例2の錠剤の溶出特性を示すグラフである。
【図6】図6は実施例3の錠剤の溶出特性を示すグラフである。
【技術分野】
【0001】
本発明は塩酸セチリジンの製剤化方法に関するものである。
【背景技術】
【0002】
塩酸セチリジンは、化学名が(±)−2−[4−[(4−クロロフェニル)フェニルメチル]−1−ピペラジニル]エトキシ酢酸2塩酸塩である抗アレルギー薬である。この塩酸セチリジンは、白色の結晶性の粉末であり、フィルムコート錠である市販品には、乳糖、結晶セルロース、軽質無水ケイ酸、ステアリン酸マグネシウム、ヒドロキシプロピルメチルセルロース2910、酸化チタン、マクロゴール400などの添加剤が用いられている(非特許文献1)。
【0003】
ところで塩酸セチリジンは、粒径が細かく凝集性が高いため、含量均一性を確保するのが難しい。粒子に凝集性がない場合には、粒径が細かくなるほど含量均一性が向上するが、粒子に凝集性がある場合には、粒子が細かくなるほど凝集性も増大するため、含量均一性が逆に低下する。また塩酸セチリジンは、直接打錠すると打錠障害を引き起こし易い。
【0004】
錠剤の含量均一性を高めるため、湿式造粒が推奨されている(特許文献1〜3など)。
例えば特許文献1には、活性成分と賦形剤とを混合し、次いで溶剤を添加して練り合わせて造粒すれば、含量均一性を改善できることが示されている。また特許文献2には、活性成分と賦形剤とを液体バインダーの結合力を利用しながら流動層造粒すれば、含量均一性を改善できることが示されている。特許文献3の従来技術の欄には、医薬を溶媒に溶解し、これを添加剤に加えて湿式造粒すれば含量均一性が高まる旨が記載されている。ただし医薬を溶媒に溶解すると、主薬の含量が低下する虞がある旨も記載されている。
【非特許文献1】第一製薬株式会社作製、「医薬品インタビューフォーム (ジルテック錠5 ジルテック錠10)」
【特許文献1】特開2002−338454号公報(特許請求の範囲、0006、0027)
【特許文献2】特開平9−59181号公報(特許請求の範囲、0003、0025〜0027)
【特許文献3】特開2003−119121号公報(0002)
【発明の開示】
【発明が解決しようとする課題】
【0005】
ところが本発明者らの検討によれば、塩酸セチリジンでは、特許文献1〜3の知見が必ずしも当てはまらないことが判明した。すなわち特許文献1のようにして塩酸セチリジンと賦形剤の混合粉末にバインダー液を加えて湿式攪拌造粒すると、逆に含量均一性が大きく低下した。さらには錠剤保存時に塩酸セチリジンが分解し易くなり、類縁物質が大きく増大した。特許文献2のようにして流動層造粒に変更すれば、含量均一性は改善されたが、錠剤保存時の塩酸セチリジンの分解を十分に低減することはできなかった。そして前述した様に特許文献3は、医薬を溶媒に溶解すれば、主薬の含量がさらに低下する虞があることを指摘している。
【0006】
本発明は上記の様な事情に着目してなされたものであって、その目的は、類縁物質を十分に抑制しながら含量均一性を高めることができる塩酸セチリジン含有錠剤の製造方法を提供することにある。
【0007】
本発明のより好ましい目的は、さらに主薬の溶出特性にも優れている塩酸セチリジン含有錠剤の製造方法を提供することにある。
【課題を解決するための手段】
【0008】
本発明者らが前記課題を解決するために鋭意検討を重ねた結果、塩酸セチリジンを溶解した溶液を利用して湿式造粒すると、含量均一性が高まるだけでなく、錠剤保存時の類縁物質の増大を十分に抑制できることを見出し、本発明を完成した。
【0009】
なお本発明者らは、前記湿式造粒の際に結合剤を使用しなくても優れた含量均一性を維持できること、さらには結合剤を使用することなく流動層造粒法を採用すれば、塩酸セチリジンの溶出特性を著しく高めることができることも併せて見出している。特許文献1には、結合剤を使用しない場合には溶出特性が改善されることが記載されているが(段落0008)、結合剤を使用しないだけでは塩酸セチリジンの溶出特性を高めることはできない。結合剤の不使用と流動層造粒法とを組み合わせることによって初めて塩酸セチリジンの溶出特性を改善できた。特許文献2には流動層造粒法を採用して含量均一性を高めることは記載されているが、流動層造粒法と溶出特性との関係については記載されていない。
【0010】
すなわち上記目的を達成し得た本発明の塩酸セチリジン含有錠剤の製造方法は、粉末状の添加剤を塩酸セチリジンの溶液(特に水溶液)と共に造粒し、この造粒物を打錠する点に要旨を有する。前記造粒では結合剤を用いる必要はない。好ましい造粒法は流動層造粒法である。造粒物は、例えば、崩壊剤と混合してから打錠してもよい。
【発明の効果】
【0011】
本発明によれば粉末状の添加剤を塩酸セチリジンの溶液と共に造粒(湿式造粒)している為、類縁物質を抑制しながら含量均一性を高めることができる。
【発明を実施するための最良の形態】
【0012】
本発明では、粉末状の添加剤を塩酸セチリジン溶液と共に造粒(湿式造粒)し、この造粒物を打錠することによって塩酸セチリジン含有錠剤を製造している。湿式造粒を採用することによって打錠障害(スティッキング)を防止できる。ただし湿式造粒法を採用したとしても湿式攪拌造粒では含量均一性はむしろ低下し、さらには主薬(塩酸セチリジン)の保存安定性も大きく低下し、類縁物質が大きく増大する。湿式流動層造粒法を採用することで含量均一性は向上するが、主薬(塩酸セチリジン)の保存安定性が不十分である。そこで本発明では、塩酸セチリジンを溶液にして湿式造粒することとする。塩酸セチリジン溶液を使用すると、何故か錠剤保存時の類縁物質の増大を十分に抑制することができる。また塩酸セチリジンを溶液にして使用すると、塩酸セチリジンの倍散が不要になり、錠剤の製造工程を簡略化できる。さらには塩酸セチリジンが取り扱い容器や器具に付着することも防止でき、作業性が向上する。
【0013】
塩酸セチリジン溶液に使用する溶媒としては、水が好ましい。水は塩酸セチリジンを分解する虞が少ない。また塩酸セチリジン溶液中の塩酸セチリジンの濃度は、造粒法(詳細は後述する)に応じて適宜設定できるが、例えば、5〜50質量%程度の範囲から選択できる。湿式攪拌造粒の場合、塩酸セチリジンの最も好ましい濃度は、20〜35質量%程度である。湿式流動層造粒の場合、塩酸セチリジンの最も好ましい濃度は、10〜20質量%程度である。
【0014】
塩酸セチリジン溶液と共に造粒する前記添加剤(造粒用添加剤)には、賦形剤、結合剤などが挙げられる。さらに造粒可能である限り、崩壊剤を使用してもよい。また必要に応じて、滑沢剤、着色剤、矯味・矯臭剤などを使用してもよい。好ましい造粒用添加剤は、賦形剤、結合剤である。
【0015】
賦形剤には、乳糖、ショ糖、ブドウ糖などの糖類、コムギデンプン、コメデンプン、トウモロコシデンプン、バレイショデンプンなどの水不溶性デンプン類、結晶セルロースなどの水不溶性セルロースなどが含まれる。好ましい賦形剤は、糖類、結晶セルロースなどである。
【0016】
結合剤としては、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、カルメロースナトリウム(カルボキシメチルセルロースナトリウム)などの水溶性又は水膨潤性のセルロース類;デキストリン、部分アルファー化デンプン、プルランなどの水溶性又は水膨潤性デンプン類;アラビアゴム、カンテン、ゼラチン、トラガント、アルギン酸ナトリウムなどの水溶性又は水膨潤性の天然高分子化合物類;ポビドン、ポリビニルアルコールなどの水溶性又は水膨潤性の合成高分子化合物類などが挙げられる。
【0017】
本発明の特に好ましい態様では、造粒用添加剤に結合剤が含まれない。本発明では、塩酸セチリジンを溶液として加えているため、結合剤を使用しなくても高い含量均一性を保ったまま造粒できる。また結合剤を省略しただけでは溶出特性を改善できないが、後述するように結合剤の省略と適切な造粒法(流動層造粒)とを組み合わせると、溶出特性を改善することもできる。
【0018】
湿式造粒では、粉末状の添加剤と塩酸セチリジン溶液とを用いて造粒できる限り種々の造粒法が採用でき、例えば、押出造粒法、転動造粒法、攪拌造粒法、流動層造粒法、噴霧乾燥造粒法などが採用できる。好ましい造粒法は、攪拌造粒法、流動層造粒法などである。
【0019】
攪拌造粒法とは、粉末状の添加剤を攪拌羽根で攪拌しながら塩酸セチリジン溶液を注液して造粒する方法のことをいう。流動層造粒法とは、気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する方法のことをいう。
【0020】
本発明では、結合剤を使用することなく前記流動層造粒を行うことが最も推奨される。湿式造粒法によって得られる塩酸セチリジン錠剤は、製造直後の溶出性に問題はないが、なぜか保管後には溶出遅延が生じる。結合剤を使用しないようにしてもこの保管後の溶出遅延を防止できず、また流動層造粒法だけを採用しても保管後の溶出遅延を防止できない。結合剤の不使用と流動層造粒法とを組み合わせて初めて保管後の溶出遅延を防止できる。
【0021】
上記のようにして得られる造粒物の大きさは、湿式造粒の方式によって異なる。例えば攪拌造粒法によって湿式造粒する場合には、850μm以上の造粒物が1%以上(特に2〜10%程度)存在する一方、75μm以下の造粒物も20%以上(特に30〜50%程度)存在するのに対し、流動層造粒法によって湿式造粒する場合には、850μm以上の造粒物が1%未満(特に0〜0.5%程度)に減少し、かつ75μm以下の造粒物も15%以下(特に5〜10%程度)に減少し、粒度分布が相対的に狭くなる。
【0022】
前記造粒物は、打錠することによって錠剤化する。この造粒物は打錠前に乾燥しておくことが望ましい。また造粒物は、必要に応じて他の医薬品用添加剤(賦形剤、結合剤、崩壊剤、滑沢剤、着色剤、矯味・矯臭剤など、好ましくは賦形剤、崩壊剤、滑沢剤など、特に崩壊剤、滑沢剤)と混合してから打錠してもよい。
【0023】
崩壊剤としては、低置換度ヒドロキシプロピルセルロース、カルメロース(カルボキシメチルセルロース)、カルメロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルエチルセルロースなどのセルロース類が挙げられる。また前記結晶セルロースやデンプン類を崩壊剤として用いてもよい。さらにはヒドロキシプロピルスターチ、カルボキシメチルスターチナトリウム(デンプングリコール酸ナトリウム)なども崩壊剤としてのデンプン類として使用してもよい。造粒物を崩壊剤と混合してから打錠することによって錠剤の溶出特性を改善することができる。
【0024】
滑沢剤としては、ステアリン酸塩、ステアリン酸、タルク、ワックス類、コムギデンプンなどが挙げられる。特に好ましい滑沢剤は、滑沢特性と抗付着特性に優れた滑沢剤(lubricants、antiadherents)であるステアリン酸塩(ステアリン酸マグネシウム、ステアリン酸カルシウムなど)である。
【0025】
各添加剤は適量使用すればよい。錠剤中の賦形剤の割合は、例えば、70〜99質量%程度、好ましくは85〜95質量%程度である。また錠剤中の崩壊剤の割合は、例えば、0〜20質量%程度、好ましくは1〜10質量%程度である。錠剤中の主薬(塩酸セチリジン)の割合は、例えば、1〜20質量%程度、好ましくは3〜15質量%程度、特に5〜10質量%程度である。
【0026】
打錠後の錠剤は、必要に応じてコーティングしてもよい。
【0027】
上記のようにして得られる塩酸セチリジン錠剤は、塩酸セチリジンを溶解した溶液を利用して湿式造粒することによって製造されているため、含量均一性が高く、しかも錠剤を保存しても類縁物質は増大しない。
【実施例】
【0028】
以下、実施例を挙げて本発明をより具体的に説明するが、本発明はもとより下記実施例によって制限を受けるものではなく、前・後記の趣旨に適合し得る範囲で適当に変更を加えて実施することも勿論可能であり、それらはいずれも本発明の技術的範囲に包含される。
【0029】
比較例1
【0030】
【表1】
【0031】
下記の様に乾式混合粉末を直接打錠することによって、上記表1に示す処方の錠剤5000錠を製造した。
【0032】
(1)倍散工程
塩酸セチリジン25gと乳糖50gを混合し、60メッシュの篩を用いて篩過することによって倍散末を得た。
【0033】
(2)混合工程
前記倍散末と、予め一部の乳糖(20g)と共に混合して篩過(32メッシュ)しておいた軽質無水ケイ酸4.25gと、残りの乳糖171.5gと、結晶セルロース130gと、低置換度ヒドロキシプロピルセルロース20gとを混合した。さらに32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.25gを加えて混合することによって混合末を得た。
【0034】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合末を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5000錠を得た。
【0035】
比較例2
【0036】
【表2】
【0037】
下記の様に結合剤の水溶液を使用して湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表2に示す処方の錠剤5000錠を製造した。
【0038】
(1)造粒・乾燥・整粒工程
塩酸セチリジン25g、乳糖238.5g、結晶セルロース128.5g及びヒドロキシプロピルセルロース3.75gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物にヒドロキシプロピルセルロース水溶液(ヒドロキシプロピルセルロース3.75g、精製水45g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0039】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース21.25gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.25gを加えて混合することによって混合顆粒を得た。
【0040】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5000錠を得た。
【0041】
比較例3
【0042】
【表3】
【0043】
下記の様に結合剤の水溶液を使用して湿式流動層造粒することによって得られる顆粒を打錠することによって、上記表3に示す処方の錠剤50000錠を製造した。
【0044】
(1)倍散工程
塩酸セチリジン250gと乳糖500gを混合し、60メッシュの篩を用いて篩過することによって倍散末を得た。
【0045】
(2)造粒・乾燥・整粒工程
前記倍散末と乳糖2800gを流動層造粒乾燥機(フロイント産業(株)製「FLO−5A」、温度70℃)で5分間混合した。混合物にヒドロキシプロピルセルロース水溶液(ヒドロキシプロピルセルロース75g、精製水1250g)をスプレーして造粒(噴霧空気圧:2.25kg/cm2G、流速:41g/min)した後、造粒物を乾燥(70℃、30分)した。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0046】
(3)混合顆粒工程
前記整粒品と、結晶セルロース370gと、低置換度ヒドロキシプロピルセルロース212.5gとを混合機(コトブキ技研工業(株)製「ボーレコンテナーミキサー LM20−1010」)で回転数25rpmにて10分間混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム42.5gを加えて1分間混合することによって混合顆粒を得た。
【0047】
(4)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤50000錠を得た。
【0048】
実施例1
【0049】
【表4】
【0050】
下記の様に塩酸セチリジンの水溶液を使用して結合剤と共に湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表4に示す処方の錠剤4000錠を製造した。
【0051】
(1)造粒・乾燥・整粒工程
乳糖190.8g、結晶セルロース102.8g及びヒドロキシプロピルセルロース6.0gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物に塩酸セチリジン水溶液(塩酸セチリジン20g、精製水42g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0052】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース17gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム3.4gを加え混合することによって混合顆粒を得た。
【0053】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤4000錠を得た。
【0054】
実施例2
【0055】
【表5】
【0056】
下記の様に塩酸セチリジンの水溶液を使用し、結合剤なしで湿式攪拌造粒することによって得られる顆粒を打錠することによって、上記表5に示す処方の錠剤4000錠を製造した。
【0057】
(1)造粒・乾燥・整粒工程
乳糖196.8g及び結晶セルロース102.8gを混合攪拌機(ダルトン(株)製「品川式万能混合攪拌機」)で混合(低速回転(69rpm)、10分間)した。混合物に塩酸セチリジン水溶液(塩酸セチリジン20g、精製水42g)を加えてさらに5分間、低速回転(69rpm)にて混合して造粒した後、造粒物を棚式乾燥機(ヤマト化学(株)製「送風定温恒温器DKN811」)で乾燥した(60℃、6時間)。得られた造粒乾燥品を整粒機(昭和技研(株)製「パワーミルP−04S型」、スクリーン径φ1.0mm)で整粒した。
【0058】
(2)混合顆粒工程
前記整粒品と、低置換度ヒドロキシプロピルセルロース17gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム3.4gを加え混合することによって混合顆粒を得た。
【0059】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤4000錠を得た。
【0060】
実施例3
【0061】
【表6】
【0062】
下記の様に塩酸セチリジンの水溶液を使用し、結合剤なしで湿式流動層造粒することによって得られる顆粒を打錠することによって、上記表6に示す処方の錠剤5600錠を製造した。
【0063】
(1)造粒・乾燥・整粒工程
60メッシュの篩で篩過しておいた乳糖377.44gを流動層造粒乾燥機(フロイント産業(株)製「FRC−1」、温度60℃)に入れ、塩酸セチリジン水溶液(塩酸セチリジン28g、精製水140g)をスプレーして造粒(噴霧空気圧:0.1MPa、流速:4.7g/min)した後、造粒物を乾燥(70℃、40分)した。得られた造粒乾燥品を20メッシュの篩で整粒した。
【0064】
(2)混合顆粒工程
前記整粒品と、結晶セルロース42gと、低置換度ヒドロキシプロピルセルロース23.8gとを混合した。さらに、32メッシュの篩で篩過しておいたステアリン酸マグネシウム4.76gを加え混合することによって混合顆粒を得た。
【0065】
(3)打錠工程
ロータリー式打錠機((株)菊水製作所製「VIRG 0524SS2AZ−000B00」)を用いて前記混合顆粒を打錠(打錠圧力:600kg、回転数:20rpm)することによって塩酸セチリジン含有錠剤5600錠を得た。
【0066】
実施例1〜3において造粒・乾燥工程後の造粒乾燥品の粒度分布は下記表7の通りであった。この粒度分布は、受け皿の上に表7に記載の篩を目の細かい方から順に積み上げ、試料(5g)を最上段の篩から順に篩過させ、各篩に残った試料の質量を測定することによって調べたものである。
【0067】
【表7】
【0068】
上記実施例及び比較例で得られた錠剤の特性(含量均一性、類縁物質、溶出特性)を以下のようにして評価した。
【0069】
(1)含量均一性
試験製剤10錠を採取し、それぞれの塩酸セチリジンの含有量を高速液体クロマトグラフィ(HPLC)によって決定した(内部標準法)。主薬の含量(表示量(100%)に対する割合(%))とその平均値を算出し、日本薬局方に記載の計算法に従って標準偏差(下式(1)参照)及び含量均一性の判定値(下式(2)参照)を求めた。
【0070】
【数1】
【0071】
(式中、Sは標準偏差である。nは試験した試料の全個数(10)を示し、Xiは、試験した個々の試料に含まれる主薬の含量(表示量に対する割合(%))を示す。AはX1からXnまでの各含量の平均値である)
判定値=|100−A|+S×2.2 …(2)
(式中、A及びSは、前記式(1)と同じである)
【0072】
測定液調製法
試験製剤と内部標準(パラオキシ安息香酸プロピル)をそれぞれ移動相に溶解し、塩酸セチリジン濃度を約10μg/mL、内部標準濃度を約15μg/mLにした。
【0073】
HPLC条件
検出器:紫外吸光光度計(測定波長:230nm)
カラム:内径4.6mm、長さ15cmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲル((株)資生堂製「カプセルパックタイプMG 5C18」)を充填したもの
カラム温度:40℃
移動相:1−オクタンスルホン酸ナトリウム水溶液(濃度:0.01mol/L、pH:2.35)/アセトニトリル混液(前者/後者=1/1(容量比))
流量:セチリジンの保持時間が約4分になる程度
測定液の注入量:10μL
【0074】
(2)類縁物質
試験錠剤をポリ瓶に充填し、開放又は密栓状態で苛酷条件(温度55℃、湿度75%RH)で放置した。試験開始から2週間後及び4週間後の分解量をHPLCによって以下のようにして測定した。
【0075】
測定液調製方法
試験錠剤8錠を移動相50mLに溶解し、不溶成分を孔径0.45μmのフィルタでろ過した。
【0076】
HPLC条件
検出器;紫外吸光光度計(測定波長:230nm)
カラム:内径4.0mm、長さ25cmのステンレス管に粒径5μmの液体クロマトグラフ用シリカゲル(和光純薬工業(株)製「Wakosil 5SIL」)を充填したもの
カラム温度:25℃
移動相:純水で希釈した硫酸(硫酸濃度:0.04mol/L)/アセトニトリル混液(前者/後者=3/47(容量比))
流量:セチリジンの保持時間が約9分になる程度
測定液の注入量:10μL
算出法
分解量(%)=全類縁物質のピーク面積の合計/セチリジンのピーク面積×100
【0077】
(3)溶出特性
前記類縁物質試験(苛酷試験)に供した錠剤の主薬溶出性を以下のようにして調べた。
【0078】
溶出試験
日本薬局方の溶出試験第2法(パドル法)に従い、試験液:精製水(錠剤1錠当たり900mL)、回転数:50rpmの条件で錠剤から主薬(塩酸セチリジン)を溶出させた。溶出試験開始から5分後、10分後、及び15分後の主薬濃度をHPLCによって調べ(絶対検量線法)、溶出率を算出した。
【0079】
測定液調製方法
溶出試験液を一部採取し、不溶成分を孔径0.45μmのフィルタでろ過した。
【0080】
HPLC条件
検出器:紫外吸光光度計(測定波長:230nm)
カラム:内径4.6mm、長さ15cmのステンレス管に粒径5μmの液体クロマトグラフ用オクタデシルシリル化シリカゲル((株)資生堂製「カプセルパックタイプMG 5C18」)を充填したもの
カラム温度:40℃
移動相:1−オクタンスルホン酸ナトリウム水溶液(濃度:0.01mol/L、pH:2.35)/アセトニトリル混液(前者/後者=1/1(容量比))
流量:セチリジンの保持時間が約4分になる程度
測定液の注入量:20μL
【0081】
溶出特性を下記基準に従って評価した。
○:苛酷試験後の錠剤の溶出特性(溶出試験開始後10分時点)が、苛酷試験前の錠剤の溶出特性(溶出試験開始後10分時点)と同程度である。
△:苛酷試験後の錠剤の溶出特性(溶出試験開始後10分時点)が、苛酷試験前の錠剤の溶出特性(溶出試験開始後10分時点)に比べて劣る。
【0082】
結果を各実施例及び比較例の操作性(主薬ハンドリング性、打錠性)と共に下記表8及び図1〜6に示す。また市販されている塩酸セチリジン錠剤(「ジルテック錠5」、「ジルテック錠10」)の含量均一性も併せて示す。
【0083】
【表8】
【0084】
表8より明らかなように、市販塩酸セチリジン錠剤の含量均一性は低い。比較例1の直接打錠法によって得られる塩酸セチリジン錠剤は、市販品(ジルテック錠5)よりもスケールが小さいために含量均一性が若干高まっているが、それでも含量均一性は低い。湿式造粒法を採用しても攪拌造粒では含量均一性がさらに低下し、さらには苛酷試験下での類縁物質が大きく増大した(比較例2)。湿式流動層造粒することによって含量均一性は改善したが、類縁物質の抑制は不十分であった(比較例3)。
【0085】
塩酸セチリジンの水溶液を用いて湿式造粒することによって初めて類縁物質を十分に抑制しながら含量均一性を高めることができた(実施例1)。ただし実施例1は溶出特性に改善余地があった(図4参照)。実施例1では結合剤を使用していたため、結合剤を使用しないことにしたが、溶出特性は改善されなかった(実施例2、図5参照)。造粒法を攪拌造粒から流動層造粒に変更することによって初めて溶出特性も改善された(実施例3)。また実施例3の塩酸セチリジン錠剤では、スケールアップすることによって、含量均一性がむしろ高まった。
【図面の簡単な説明】
【0086】
【図1】図1は比較例1の錠剤の溶出特性を示すグラフである。
【図2】図2は比較例2の錠剤の溶出特性を示すグラフである。
【図3】図3は比較例3の錠剤の溶出特性を示すグラフである。
【図4】図4は実施例1の錠剤の溶出特性を示すグラフである。
【図5】図5は実施例2の錠剤の溶出特性を示すグラフである。
【図6】図6は実施例3の錠剤の溶出特性を示すグラフである。
【特許請求の範囲】
【請求項1】
粉末状の添加剤を塩酸セチリジンの溶液と共に造粒し、
この造粒物を打錠することを特徴とする塩酸セチリジン含有錠剤の製造方法。
【請求項2】
結合剤を用いることなく造粒する請求項1に記載の塩酸セチリジン含有錠剤の製造方法。
【請求項3】
気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する請求項1又は2に記載の塩酸セチリジン含有錠剤の製造方法。
【請求項4】
前記塩酸セチリジン溶液の溶媒が水である請求項1〜3のいずれかに記載の塩酸セチリジン含有錠剤の製造方法。
【請求項5】
造粒物を崩壊剤と混合してから打錠する請求項1〜4のいずれかに記載の塩酸セチリジン含有錠剤の製造方法。
【請求項1】
粉末状の添加剤を塩酸セチリジンの溶液と共に造粒し、
この造粒物を打錠することを特徴とする塩酸セチリジン含有錠剤の製造方法。
【請求項2】
結合剤を用いることなく造粒する請求項1に記載の塩酸セチリジン含有錠剤の製造方法。
【請求項3】
気流によって流動化した粉末状の添加剤に、塩酸セチリジンの溶液を吹き付けて造粒する請求項1又は2に記載の塩酸セチリジン含有錠剤の製造方法。
【請求項4】
前記塩酸セチリジン溶液の溶媒が水である請求項1〜3のいずれかに記載の塩酸セチリジン含有錠剤の製造方法。
【請求項5】
造粒物を崩壊剤と混合してから打錠する請求項1〜4のいずれかに記載の塩酸セチリジン含有錠剤の製造方法。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】

【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2007−314448(P2007−314448A)
【公開日】平成19年12月6日(2007.12.6)
【国際特許分類】
【出願番号】特願2006−144574(P2006−144574)
【出願日】平成18年5月24日(2006.5.24)
【出願人】(593077308)共和薬品工業株式会社 (11)
【Fターム(参考)】
【公開日】平成19年12月6日(2007.12.6)
【国際特許分類】
【出願日】平成18年5月24日(2006.5.24)
【出願人】(593077308)共和薬品工業株式会社 (11)
【Fターム(参考)】
[ Back to top ]