
増大したバイオアベイラビリティーを有する抗真菌組成物
【課題】より安定なポサコナゾールを提供すること。
【解決手段】化学構造式(I)によって示される、抗真菌的に有効量の微粉化化合物、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む液体懸濁物が開示される。この液体懸濁物の形態は、経口投与に適切な薬学的組成物を見出し、この薬学的組成物は、従来技術を越える重要な利点を提供する。本発明の液体懸濁物の利点としては、懸濁物の改善された均質性および懸濁物の分散の容易さが挙げられる。本発明の液体懸濁物中で安定である固形物は、再分散するのが困難な固体ケーキを形成しない。
【解決手段】化学構造式(I)によって示される、抗真菌的に有効量の微粉化化合物、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む液体懸濁物が開示される。この液体懸濁物の形態は、経口投与に適切な薬学的組成物を見出し、この薬学的組成物は、従来技術を越える重要な利点を提供する。本発明の液体懸濁物の利点としては、懸濁物の改善された均質性および懸濁物の分散の容易さが挙げられる。本発明の液体懸濁物中で安定である固形物は、再分散するのが困難な固体ケーキを形成しない。
【発明の詳細な説明】
【背景技術】
【0001】
(背景技術)
本発明は、以下の化学構造式I:
【0002】
【化8】

によって示される抗真菌的(antifungally)に有効量の微粉化化合物、少なくとも1種の濃厚剤(thickening agent)、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、安定な液体懸濁物、ならびに真菌感染を処置または予防するためにこの懸濁物を使用する方法に関する。
【0003】
米国特許第5,661,151は、式Iの化合物、および広範囲の真菌(例えば、アスペルギルス属(Aspergillis)、カンジダ属、クリプトコックス属、フザリウム属および他の日和見真菌)に対するその強力な抗真菌活性を開示する。
【0004】
米国特許第5,834,472号および同第5,846,971号は、結合剤と一緒に不活性ビーズ上にコートされる構造式Iの化合物の経口の薬学的カプセル組成物を開示する。しかし、構造式Iの化合物は、高親油性であり、そして非常に低い水溶性を有する。従って、構造式Iの化合物の水性組成物は、おそらく、化合物の非常に低い水溶性に起因して、抗真菌活性および/またはバイオアベイラビリティーを減少させていることが見出された。従って、バイオアベイラビリティーを増大し、そして安定な特性を改善させる、構造式Iの化合物の経口の薬学的組成物の必要性が存在する。
【発明の開示】
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明者らは、ポサコナゾール(posaconazole)の微粉化粒子、以下の化学構造式I:
【0006】
【化9】
を有する化合物、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物の形態で、経口投与に適切な薬学的組成物を見出し、この薬学的組成物は、従来技術を越える重要な利点を提供する。
【0007】
本発明の液体懸濁物の利点としては、懸濁物の改善された均質性および懸濁物の分散の容易さが挙げられる。本発明の液体懸濁物中で安定である固形物は、再分散するのが困難な固体ケーキを形成しない。少なくとも3日間の間、本発明の再構築された液体懸濁物中に固体を実質的に沈殿しない。この驚くべき特性は、本発明の液体懸濁物を接種して、真菌感染を有する患者が、抗真菌的に有効量のポサコナゾールを接種することを確実にする。本発明の液体懸濁物は、より長い有効期間を有する。さらに、この液体懸濁物は、再構築の際に、最初に調製された懸濁物として、同じ抗真菌的に有効量のポサコナゾールを実質的に提供する。本発明の液体懸濁物のこれらの特性は、薬局、薬剤師、医師、および真菌感染を有する患者に利益を提供する。
【0008】
従って、本発明は、以下の化学構造式I:
【0009】
【化10】
を有する微粉化ポサコナゾール、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物を提供する。
【0010】
本発明はまた、以下の化学構造式I:
【0011】
【化11】
を有する、抗真菌的に有効量の微粉化ポサコナゾール、有効量の少なくとも1種の濃厚剤、約4.0〜約6.0の範囲に緩衝系のpHを維持するに有効な量の緩衝系、有効量の非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物を提供する。
【0012】
本発明はさらに、以下の化学構造式I:
【0013】
【化12】
を有する、抗真菌的に有効量の微粉化ポサコナゾール、を含む液体懸濁物を提供し、ここで、この微粉化化合物は、約1200nm〜約1600nmの範囲の平均粒径(mean particle size)を有し、有効量の、飽和または不飽和のC12〜C18酸のソルビタンエステルのポリオキシエチレン誘導体、約4.0〜約6.0の範囲にpHを維持するに十分な有効量の緩衝系、有効量の2種の濃厚剤(ここで一方は液糖である)の組み合わせ、および薬学的に受容可能な液体キャリアを含む。
したがって、本発明は、以下を提供する。
(1) 液体懸濁物であって、該液体懸濁物は、以下の化学構造式I:
【化101】
によって示される抗真菌的に有効量の微粉化化合物、少なくとも1種の濃厚剤、少なくとも1種の非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物。
(2) 前記少なくとも1種の非イオン性界面活性剤が、エチレンオキシドおよびプロピレンオキシドのブロックコポリマー、飽和または不飽和のC8〜C20酸のグリコールエステルまたはグリセリルエステル、飽和または不飽和のC8〜C20酸のポリオキシエチレンエステル、飽和または不飽和のC8〜C20酸のポリオキシエチレンエーテル、飽和または不飽和のC10〜C20酸のポリビニルアルコールまたはソルビタンエステルである、項目1に記載の液体懸濁物。
(3) 前記少なくとも1種の非イオン性界面活性剤が、飽和または不飽和のC10〜C20酸のソルビタンエステルのポリオキシエチレン誘導体である、項目1に記載の液体懸濁物。
(4) 前記少なくとも1種の濃厚剤が、キサンタンガム、液糖、澱粉、セルロースおよびそれらの混合物から選択される、項目1に記載の液体懸濁物。
(5) キサンタンガムおよび液糖の組み合わせが、前記少なくとも1種の濃厚剤として使用される、項目1に記載の液体懸濁物。
(6) 前記式Iの微粉化化合物が、約1200nm〜約1600nmの平均粒径を有する、項目1に記載の液体懸濁物。
(7) 液体懸濁物であって、該液体懸濁物が、以下:
(a)以下の化学構造式I:
【化102】
で示される、抗真菌的に有効量の微粉化化合物;
(b)少なくとも1種の有効量の濃厚剤;
(c)約4.0〜約6.0の範囲内に緩衝系pHを維持するために有効な量の緩衝系;
(d)少なくとも1種の有効量の非イオン性界面活性剤;および
(e)薬学的に受容可能な液体キャリア
を含む、液体懸濁物。
(8) 前記少なくとも1種の非イオン性界面活性剤が、飽和または非飽和のC10〜C20酸のソルビタンエステルのポリオキシエチレン誘導体である、項目7に記載の液体懸濁物。
(9) 前記ソルビタンエステルが、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンセスキオレアート、ソルビタントリオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラートおよびソルビタントリステアラート、またはそれらの混合物から選択される、ソルビタンの脂肪酸エステルである、項目7に記載の液体懸濁物。
(10) 前記少なくとも1種の濃厚剤が、ガム、液糖、澱粉、セルロースおよびそれらの混合物から選択される、項目7に記載の液体懸濁物。
(11) キサンタンガムおよび液糖の組み合わせが、前記少なくとも1種の濃厚剤として使用される、項目7に記載の液体懸濁物。
(12) キサンタンガムおよび液体グルコースの組み合わせが、前記濃厚剤として使用される、項目7に記載の液体懸濁物。
(13) 前記緩衝系が、クエン酸ナトリウムおよびクエン酸を含む、項目7に記載の液体懸濁物。
(14) 前記薬学的に受容可能な液体キャリアが、精製水、グルコース、およびグリセリンの組み合わせである、項目7に記載の液体懸濁物。
(15) 前記式Iの微粉化化合物が、約1200nm〜約1600nmの平均粒径を有する、項目7に記載の液体懸濁物。
(16) 液体懸濁物であって、該液体懸濁物が、以下:
(a)以下の化学構造式I:
【化103】
を含む、抗真菌的に有効量の微粉化化合物であって、
ここで該式Iの微粉化化合物が、約1200nm〜約1600nmの範囲内の平均粒径を有する、微粉化化合物、
(b)飽和または不飽和のC12〜C18酸のソルビタンエステルの、有効量のポリオキシエチレン誘導体;
(c)約4.0〜約6.0の範囲内にpHを維持するに十分な、有効量の緩衝系;
(d)2種の濃厚剤の、有効量の組み合わせであって、ここで一方が液糖である、組み合わせ;および
(e)薬学的に受容可能な液体キャリア
を含む、液体懸濁物。
(17)以下の化学構造式I:
【化104】
の化合物の微粉化粒子の、哺乳動物における真菌感染の処置および/または予防のための、経口懸濁物の形態における医薬の調製のための使用であって、該使用が、薬学的に受容可能なキャリア中に、抗真菌的に有効量の該式Iの化合物Iおよび少なくとも1種の濃厚剤および非イオン性界面活性剤を含む、使用。
(18) 懸濁物であって、該懸濁物が、薬学的に受容可能な液体キャリア中に、抗真菌的に有効量の、以下の化学構造式I:
【化105】
を含む化合物の微粉化化合物、少なくとも1種の濃厚剤および非イオン性界面活性剤を含む、懸濁物。
(19) 組成物であって、該組成物が、以下の構造式I:
【化106】
によって示される化合物の微粉化粒子および少なくとも1種の非イオン性界面活性剤を含
む物質の組成物。
(20)懸濁物中に、以下の化学構造式I:
【化107】
によって示される化合物の微粉化粒子を含む物質の組成物。
【0014】
(発明の詳細な説明)
本発明は、薬学的に受容可能な液体キャリア中にある抗真菌性化合物ポサコナゾール(posaconazole)の微粉化粒子の安定な懸濁液を提供する。本発明の懸濁液は、25℃で3日間よりも長い間静置して貯蔵した場合、沈澱することなく安定である。(以下の表1を参照のこと)。
【0015】
以下の表2により、懸濁液中のポサコナゾールの濃度が、12ヶ月までの期間にわたって、(HPLCによって測定した場合)初期濃度と比較して実質的に同一(±2%)である点で、本発明の液体懸濁液処方物が安定であることが示される。
【0016】
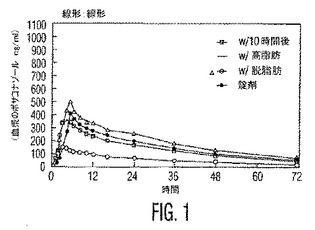
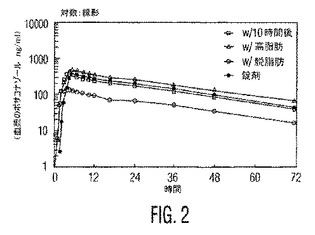
本発明者らはまた、本発明の安定な懸濁液が、各々が被験体に高脂肪の朝食と同時に投与される場合、最適化されたポサコナゾールの微粉化粒子の経口錠剤と比較して、著しくより高いバイオアベイラビリティー(23〜36%の増加)を有することを見出した。表3および表4ならびに図1および図2を参照のこと。
【0017】
本発明の1つの局面は、薬学的組成物を提供することであり、この組成物は、非イオン性界面活性剤(例えば、ソルビタンエステル)および少なくとも1つの増粘剤(好ましくは、キサンタンガムおよび液糖の組み合わせ)と組み合わせて、ポサコナゾールの微粉化粒子を含有し、これらの粒子は、精製水のような薬学的に受容可能な液体キャリア中に容易に分散可能である。この薬学的組成物は、安定した懸濁液を提供し、この懸濁液は、少なくとも3日間沈澱せず、従って患者が有効用量のポサコナゾールを得ることを確実にする。本発明の安定した懸濁液の別の特徴は、ポサコナゾールが溶液から沈澱することなしに、口腔鵞口瘡を伴うHIV−1感染を保有する患者の処置に有用であることである。本発明の別の局面は、本発明の懸濁液が、分散しにくい固体ケーキ(cake)の形成を回避することである。
【0018】
本発明の懸濁液において使用される式Iの化合物は、Schering Corporation,Kenilworth,New Jerseyから入手可能であり、そして米国特許第5,661,151号およびWO95/17407の実施例24および32に従って調製された。
【0019】
好ましいポサコナゾールのミクロンサイズの粒子は、約1000ナノメートル(nm)〜約1800nm、好ましくは、約1200nm〜約1600nm、そして最も好ましくは、約1400nmの平均粒径範囲を有する。この粒径は、式Iの抗真菌性化合物の製造の間の最終工程によってか、または慣用的な結晶化手順後の慣用的な微粉化技術の使用によってのいずれかで得られ得る。
【0020】
ポサコナゾールを望ましい平均粒径範囲に微粉化するために使用される好ましい微粉化技術は、ミクロ流動化である。ミクロ流動化は、伝統的な均質化に代わるものであり、これは、高圧での2つの物質の流れの衝突を利用して、はるかにより均一な粒径分布(Microfluidics International Co.に従う)、および約1200nm〜1600nmのより小さい平均粒径を生成する。ミクロ流動化において使用されるプロセスおよび装置は、米国特許第4,533,254号に記載されている。
【0021】
本発明の微粉化ポサコナゾールはまた、結晶形態で存在し得る。結晶形態は、好ましくは、実質的に化学的および光学的に純粋であり、そしてこれは、約10%未満のその光学異性体、エナンチオマーまたは他のジアステレオマーを含有する。結晶形態は、99%の光学純度の左旋性または右旋性の異性体であり得る。化学構造Iのこの光学的に純粋な化合物は、他の光学異性体の混合物の多くの有害な副作用を回避する。
【0022】
ポサコナゾール液体懸濁液は、目的の真菌を制御するために抗真菌的に有効な量で組成物において使用される。このような抗真菌的に有効な量は、本発明の液体懸濁液処方物の濃度が、約10mg/ml〜約100mg/ml、より好ましくは、約20mg/ml〜約60mg/ml、そして最も好ましくは、約40mg/mlの式Iの化合物の範囲であり得る。
【0023】
本発明はまた、哺乳動物における真菌感染を、処置および/または予防する方法を提供し、この方法は、このような真菌感染を処置および/または予防するために有効な量の微粉化ポサコナゾールを含む一定量の液体懸濁液を、哺乳動物に投与する工程を包含する。式Iの化合物を40mg/ml含有する、本発明の抗真菌的に有効量の液体懸濁液は、5ml(200mgの式Iの化合物を含む)で1日に3回(TID)もしくは1日に4回(QID)の用量、または10ml(400mgの式1の化合物を含む)で1日に2回(BID)の用量で経口投与される。当然、担当の臨床医は、患者の年齢、健康状態、および性別、ならびに真菌感染の重篤度を考慮して、用量および投薬レジメンを変更し得る。
【0024】
以下の用語は、本発明の薬学的組成物、その処方において使用され得る成分、およびその化合物の生物活性またはバイオアベイラビリティーを評価するための方法を記述するために使用される。
【0025】
非イオン性界面活性剤とは、正味のイオン性電荷を欠き、かつ水性媒体中で感知される程度に解離しない界面活性剤をいう。非イオン性界面活性剤の特性は、その分子における親水性基および疎水性基の割合に大きく依存する。親水性基としては、オキシエチレン基(−OCH2CH2−)およびヒドロキシ基が挙げられる。疎水性分子(例えば、脂肪酸のエステル)においてこれらの基の数を変更することによって、強力に疎水性かつ水に不溶性の化合物(例えば、モノステアリン酸グリセリン)から、強力に親水性かつ水溶性の化合物(例えば、マクロゴール)に及ぶ物質が得られる。これらの2つの極端な型の間には、親水性基および疎水性基の割合がより均等に釣り合ったもの(例えば、マクロゴールエステルおよびマクロゴールエーテルならびにソルビタン誘導体)を含む。適切な非イオン性界面活性剤は、Martindale,The Extra Pharmacopoeia,第28版,1982,The Pharmaceutical Press,London,Great Britain,pp.370〜379に見出され得る。
【0026】
このような適切な非イオン性界面活性剤としては、エチレンオキシドおよびプロピレン
オキシドのブロックコポリマー、飽和もしくは不飽和のC8〜C20酸のグリコールエステルまたはグリセリルエステル、飽和もしくは不飽和のC8〜C20酸のポリオキシエチレンエステル、飽和もしくは不飽和のC8〜C20酸のポリオキシエチレンエーテル、ならびに飽和もしくは不飽和のC10〜C20酸のポリビニルアルコールまたはソルビタンエステルが挙げられる。好ましくは、この非イオン性界面活性剤は、飽和または不飽和のC10〜C20酸のソルビタンエステルであり、そしてより好ましくは、このソルビタンエステルは、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンセスキオレアート、ソルビタントリオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラートおよびソルビタントリステアラート、またはそれらの混合物から選択されるソルビタンの脂肪酸エステルである。
【0027】
適切なソルビタンエステルとしては、例えば、ポリソルベート20、ポリソルベート40、ポリソルベート60、ポリソルベート65、ポリソルベート80、ポリソルベート85、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラート、ソルビタンセスキオレアート、ソルビタントリオレアート、およびソルビタントリステアラートが挙げられる。最も好ましい非イオン性界面活性剤は、商品名Tween80でICI Americasから入手可能なポリソルベート80であり、これは、約20モルのエチレンオキシドと縮合した主にモノエステルから構成される、ソルビトールのオレイン酸エステルおよびソルビトール無水物の混合物である。
【0028】
エチレンオキシドおよびプロピレンオキシドの適切なブロックコポリマーは、一般的に「ポロキサマー」と呼ばれ、そして以下の化学構造I:
【0029】
【化13】
によって表されるコポリマーを含み、ここで、aは、約10〜約110、好ましくは、約12〜101;より好ましくは、約12〜80にわたる整数であり;そして
bは、約20〜約60、より好ましくは、約20〜約56;または約20〜27にわたる整数である。
【0030】
脂肪酸の適切なグリコールエステルおよびグリセリルエステルとしては、モノラウリル酸グリセリンおよび同様な水溶性誘導体が挙げられる。
【0031】
脂肪酸の適切なポリオキシエチレンエステル(マクロゴールエステル)としては、ポリオキシエチレンヒマシ油および硬化ヒマシ油誘導体が挙げられる。
【0032】
脂肪酸の適切なポリオキシエチレンエーテル(マクロゴールエーテル)としては、セトマクロゲル(Cetomacrogel)1000、ラウロマクロゴール(異なる鎖長のマクロゴールの一連のラウリルエーテル)、例えば、ラウレス4、ラウレス9およびラウロマクロゴール400が挙げられる。
【0033】
組成物における非イオン性界面活性剤の有効量は、処方物の濃度が、約5mg/ml〜約50mg/ml、より好ましくは、約5mg/ml〜約25mg/ml、そして最も好ましくは、10mg/mlの範囲であり得る。
【0034】
本発明において適切と見出された増粘剤としては、このような目的のために有用な任意
の市販の薬剤が挙げられる。キサンタンガム、液糖(liquid sugar)、デンプン、セルロース、およびこれらの混合物は、好ましい増粘剤である。より好ましいのは、キサンタンガムおよび液糖の組み合わせであり、そして最も好ましくは、キサンタンガム、NFおよびグルコース、NFの組み合わせである。好ましくは、キサンタンガムが、約1mg/ml〜約5mg/mlの量で存在し、そしてより好ましくは、キサンタンガムが、約3mg/mlの量で存在する。好ましくは、グルコースNFは、約200〜約500mg/ml、そしてより好ましくは、約350mg/mlの量で存在する。本発明の増粘剤の有効量は、約1〜約500mg/ml、より好ましくは、約200〜約500mg/ml、最も好ましくは、約353mg/mlであり得る。本発明の増粘剤は、最小限の攪拌で構成した後、処方物の懸濁を容易にし、そして長時間にわたって懸濁液の迅速な沈澱およびケーキ形成を防止する。
【0035】
薬学的に受容可能なキャリアとしては、当該分野において周知のものが挙げられ、精製水USP、液体グルコース、NF、および無水グリセロールが挙げられる。最も好ましいのは、精製水USPおよび液体グルコース、NFである。薬学的に受容可能なキャリアは、約10〜約500mg/ml、より好ましくは、約200mg/ml〜約400mg/ml、最も好ましくは、約350mg/mlの量で存在し得る。
【0036】
本発明の処方物について適切な緩衝系は、液体懸濁液のpHを、約4〜約6、好ましくは、4.5〜5.0、そして最も好ましくは、約4.5の範囲に維持するものである。クエン酸ナトリウム、USPおよびクエン酸、USPの緩衝系の使用が好ましい。他の適切な緩衝系は、4〜6の望ましいpH範囲を維持するために使用され得る。緩衝化剤は、約0.4〜約1.5mg/ml、より好ましくは、約0.7〜約1.5mg/ml、最も好ましくは、約1.1mg/mlの濃度で存在し得る。
【0037】
本発明において適切と見出された消泡剤としては、このような目的のために有用な任意の市販の薬剤が挙げられ、トリメチルシロキシル単位(例えば、ジメチコーンおよびシメチコン)で末端ブロックされたメチル化直線状シロキサンポリマー、ならびに200〜250ジメチルシロキサン単位の平均鎖長を有するジメチコーンおよびシリカゲルの混合物が挙げられる。消泡剤の有効量は、約2mg/ml〜約4mg/ml、好ましくは、約3mg/mlの濃度を提供するために十分な量である。
【0038】
本発明において有用と見出された水溶性防腐剤としては、安息香酸ナトリウム、クエン酸ナトリウムおよび塩化ベンザルコニウムならびに他の薬学的に受容可能な水溶性防腐剤が挙げられる。防腐剤として安息香酸ナトリウムの使用が好ましい。水溶性防腐剤の有効量は、約0.5mg/ml〜約3mg/ml、最も好ましくは、約2mg/mlの濃度を提供するために十分な量である。
【0039】
本発明において適切と見出された乳白剤としては、薬学的に受容可能な金属酸化物、特に二酸化チタンが挙げられる。乳白剤の有効量は、約2mg/ml〜約6mg/ml、最も好ましくは、約4mg/mlの濃度を提供するために十分な量である。
【0040】
代表的な香味剤は、医薬、食品、キャンディー、飲料などを甘くする使用のためにFDAによって認可されているものであり;これらの物質は、香味(例えば、ブドウ、サクランボ、柑橘類、モモ、イチゴ、風船ガム、ペパーミントおよびその他多くのもの)を与える。香味剤の有効量は、約0.01mg/ml〜約6mg/ml、より好ましくは、約5mg/mlの濃度を提供するために十分な量である。
【0041】
以下の実施例は、ポサコナゾールを含有する本発明の組成物を記載するが、これらは、特許請求の範囲の範囲を限定するように解釈されるべきでない。
【実施例】
【0042】
【数1】
上記成分の範囲は、当業者に明らかであるように、変動され得る。本発明の範囲内の組成物の特定の実施例を、以下に示す。
【0043】
(実施例1)
【0044】
【数2】
この実施例は、以下のように調製され得る:ミキサープロペラを備えた適切な容器に20±3℃にて約5%の最終バッチ容積の精製水を充填する。工程1の精製水に40%のポリソルベート80を添加し、そして溶解するまで混合する。40%のシメチコンを添加し、そして分散するまで混合する。約5つのパスについて約30,000±5000psiで作動するマイクロフルイダイザー(microfluidizer)によって工程3の混合物を再循環する。約20±3℃にて、約7%の最終バッチ容積の精製水を添加し、そして約5分間混合する。一定に混合しながらポサコナゾールを工程5の容器に添加する。完全に分散されるまで混合し続ける。約30,000±5,000psiの圧力で作動するマイクロフルイダイザーに通して工程6からの懸濁部分を再循環する。当該分野で公知であるレーザー回折技術によって測定される場合、粒子のメジアンが約1.4±0.2μmの粒径を示すまで、濃縮物を処理する。
【0045】
この粒径が達成される場合、マイクロフルイダイザーに懸濁液を通し、そして適切な大きさの容器に収集する。約8〜10%の最終バッチ容積の精製水(20±3℃)をフィード容器に添加し、約30,000psiで作動するマイクロフルイダイザーに通す。濃縮物を含む容器中の洗浄物を収集する。濃縮物を有する容器に約22%の最終バッチ容積の精製水(20±3℃)を添加し、そして約5分間混合する。ポリソルベート80およびシメチコンの残りを添加し、そして約5分間混合する。
【0046】
安息香酸ナトリウム、クエン酸ナトリウムおよびクエン酸を添加し、そして約5分間混合する。一定に混合しながらキサンタンガムをゆっくりと添加する。キサンタンガムの添加の後、混合し続ける。混合せずに30分間、キサンタンガムを水和させる。一定に混合しながらグリセリンを添加する。一定に混合しながら液体グルコースをゆっくりと添加する。5分間または溶解するまで混合する。二酸化チタンを添加し、そして内容物が完全に分散されるまで適切なホモジナイザーを用いて混合する。人工のチェリー香料を添加し、約5分間混合する。最終容積になるまで十分な量の20±3℃の精製水を添加し、そして均一な懸濁液が達成されるまで混合する。懸濁液のpH測定および物理的観察のために約20mlのサンプルを収集する。実施例1の懸濁液のpHは、5.0であった。
【0047】
(実施例2)
実施例2は、実施例1の手順を用いて調製した本発明の範囲内での処方物の別の例であり、そしてpH4.5を有する。
【0048】
【数3】
本発明の液体懸濁液の沈降速度を、以下に示すように測定した。
【0049】
(表1:40mg/mlのポサコナゾール経口懸濁液の沈降速度)
【0050】
【表1】
本発明の懸濁液を含む2つのボトルを、振盪し、そして静置した。次いで、このボトルを、振盪の後、直ちに(時間0)、5分後、10分後、30分後、60分後および72時間後(3日間)にサンプリングした。これらのサンプル中のポサコナゾールのレベルおよび防腐剤(安息香酸ナトリウム)のレベルを、HPLCによりアッセイした。検出のHPLC方法は、当業者に周知である。
【0051】
防腐剤およびポサコナゾールのアッセイの結果は、一貫したままであり、そして変化しなかった。これらは、それぞれ、活性に対して39.8mg/ml〜40.2mg/ml(99.5%〜101%)、および保存に対して1.99mg/ml〜2.02mg/ml(99.5%〜101%)の範囲であった。この実験の結果は、上記の表1に示す。
【0052】
安息香酸ナトリウムは、沈降すると予測されなかった。驚いたことに、ポサコナゾールは、3日後でも沈降しなかった。
【0053】
従って、本発明の組成物は、表1中のサンプルの安定性によって証明されるように分散
能および均質性の両方の容易さを有する。
【0054】
次に、加速された均質性試験を、本発明の液体懸濁液に対して行った。
【0055】
(表2:40mg/mlのポサコナゾール経口懸濁液の均質性)
【0056】
【表2】
*患者の指示通りの用量を振盪しそして与える。
【0057】
これらのデータ(沈降速度試験)は、表2に示されるリアルタイムの安定性データ(40℃/75%RHで6ヶ月までおよび25℃/60%RHで24ヶ月まで)に従っていた。このアッセイの均質性結果は、驚くことに、一貫して均質なままであり、そして実質的に変化しなかった。
【0058】
40℃/75%RHで6ヶ月後、均質性結果は、それぞれ、活性に対して40.7mg/ml〜40.8mg/ml(101%)、および保存性に対して2.01mg/ml〜2.03mg/ml(101%〜102%)の範囲であった。これらの結果を、アッセイされるボトルの部分(すなわち、ボトルの上部または下部)に関係なく得た。従って、加速した安定の状態での比較的長い曝露の後でさえ、懸濁液はボトルの至るところで均質であったと結論付け得る。
【0059】
25℃/60%RHで24ヶ月後、均質性結果は、それぞれ、活性に対して41.5mg/ml〜41.6mg/ml(104%)、および保存性に対して2.01mg/ml(101%)の範囲であった。これらの結果を、アッセイされるボトルの部分(すなわち、ボトルの上部または下部)に関係なく得た。従って、25℃/60%RHでの長期(24ヶ月)の曝露の後でさえ、懸濁液はボトルの至るところで均質であったことが結論され得る。
【0060】
バイオアベイラビリティーは、活性薬物成分または治療的部分が標準またはコントロールと比較して、投与された投薬形態から体循環に吸収される速度および程度として定義される。
【0061】
Cmax値を、血清血漿中で測定された抗真菌化合物の最大濃度(すなわち「ピーク」
)として定義する。
【0062】
本発明の処方物は、増加したバイオアベイラビリティーおよび以前の処方物よりもより低い変動性を有するという利点を有する。
【0063】
ポサコナゾール経口懸濁液の相対的なバイオアベイラビリティーを、健康な被験体中で、固体投薬形態のポサコナゾールと比較した。
【0064】
第1の目的は、高脂肪朝食を与えられた場合、経口固体処方物と比較して、経口懸濁液として、与えたポサコナゾールの相対的なバイオアベイラビリティーを測定することであった。第2の目的は、経口懸濁液として与えられる場合、式1の化合物の経口バイオアベイラビリティーで絶食状態に関連して、高脂肪食物および無脂肪食物の効果を測定することであった。
【0065】
20人の健康な被検体は、この無作為化した、開放標識の、4つの方法で交差する単一投与バイオアベイラビリティーおよびポサコナゾールの食物効果研究を完了した。被験体は、少なくとも7日間の洗い出しによって分けられた以下の4つの処置のそれぞれを受けた。
【0066】
【数4】
被験体を、絶食したまま(処置A)、標準化した高脂肪朝食を受ける(処置BまたはD)または標準化した無脂肪朝食を受ける(処置C)のいずれかで無作為化した。食事を、朝の薬物投与の20分前に食べ、そして被験体は朝食を完了した5分以内に適切な処置を受けた。
【0067】
血液サンプル(6ml)を、それぞれの処置に対して、投薬の直ぐ前(0時間)に、および投薬の0.5時間後、1時間後。1.5時間後、2時間後、3時間後、4時間後、5時間後、6時間後、8時間後、10時間後、12時間後、16時間後、24時間後、36時間後、48時間後および72時間後にヘパリン処理されたチューブ内に収集した。血液を、4℃で遠心分離し、そして血漿をアッセイされるまで−20℃以下で貯蔵した。ポサコナゾールの血漿濃度を、ng/mlのLOQで有効な高速液体クロマトグラフィーアッセイを用いてアッセイした。
【0068】
個々の血漿の濃度−時間データを、モデル非依存性方法を用いて、薬物動態学的アッセイのために使用した。最大濃度(Cmax)および最大濃度の時間(Tmax)は、観察された値であった。時間0から定量化できる最終のサンプリング時間[AUC(tf)]までの血漿濃度−時間曲線の下の面積を、一次台形法(linear trapezoidal method)を使用して計算し、そして以下のように
【0069】
【数5】
無限(I)に外挿し:
ここでC(tf)は、時間(tf)で線形回帰から決定された推定の濃度である。
【0070】
全身のクリアランスを、以下によって計算した:
CL=用量/AUC(I)
分布の見掛けの容積(Vdarea/F)を、末期の速度定数(K)に対する全身クリアランスの速度から計算した。
【0071】
手短な統計学は、従来技術の錠剤処方物の濃度−時間データおよび誘導した薬物動態学的パラメーターと比較して、本発明の血漿懸濁液処方物について計算した。本来のスケールならびにlog変換CmaxおよびAUC値を、分散分析(ANOA)を用いて分析した。被験体、フェーズおよび処置に起因する効果を抽出した。
【0072】
式Iの化合物に対する血漿濃度−時間データおよび薬物動態学的パラメーターを、表3および4に表にし、そして図1および2に図示する。
【0073】
すべての被験体は、フェーズ3およびフェーズ4において被験体20を除いて以下に報告されるLOQ(5ng/ml)の1日目の0時間の濃度を有し、この被験体は、それぞれ、8.5ng/mlおよび22.5ng/mlの処置BおよびAに対して0時間でのポサコナゾールの定量化可能なレベルを有した。これらのレベルは、以前の用量からの集積の繰り越し効果におそらく起因する。
【0074】
平均aの薬物動力学パラメーターの概要を、以下の表に提供する:
【0075】
【表3】
a:バランス平均、n=20は、AUC(I)およびt1/2(n=15)を除く。
【0076】
ポサコナゾールは、ゆっくりと吸収された;平均Tmax値は、4.1〜5.5時間の範囲であった。ポスコナゾールは、処置から独立して、約22時間の平均終了t1/2でゆっくりと除去された。この研究を行って、錠剤処方物(処置D)と比較してポサコナゾール経口懸濁液(処置B)のバイオアベイラビリティーを評価した(両方ともが、高脂肪食物を与えられる)。log変換データに基づくこの結果を、以下に示す:
【0077】
【表4】
a:全ての被験体についての全ての処置について計算され得るので、AUC(tf)を、統計的な比較のために使用した。
【0078】
b:錠剤に対する懸濁液。
【0079】
平均して、本発明の懸濁液処方物は、先行技術の錠剤と比較して、Cmaxにおいて23%の増加およびAUC(tf)において36%の増加を生じた。
【0080】
研究の第2の目的は、経口懸濁液として投与されるポサコナゾールの経口アベイラビリティーで、絶食(処置A)と比較して、高脂肪食物(処置B)および無脂肪食物(処置C)の効果を評価することであった。log変換データに基づくこの結果を、以下に示す:
【0081】
【表5】
a:処置A−懸濁液/脂肪のパーセントとして表した。
【0082】
高脂肪朝食は、懸濁液中で与えられたポサコナゾールのバイオアベイラビリティーにおいて4倍の増加を生じた。食物は、錠剤処方物およびカプセル処方物の両方について3〜5倍まで、ポサコナゾ−ルのバイオアバイラビリティーを有意に増加した、以前の研究からの結果と一貫していた。絶食(処置A)と比較して無脂肪朝食(処置C)の効果は、バイオアベイラビリティーにおいて2.5〜3倍での増加であり、より少なかった。
【0083】
本発明の多くの改変および変化は、当業者に明白であるので、その精神および範囲から逸脱することなくなされ得る。本明細書中に記載される特定の実施形態は、例示のみによって提供され、そして本発明は、添付の特許請求の範囲の権利が与えられる全ての範囲の等価物と共に、このよに特許請求の範囲の用語によってのみ限定されるべきである。
【図面の簡単な説明】
【0084】
【図1】図1は、本発明の実施例1のポサコナゾール錠剤およびその液体懸濁液の平均血漿濃度の時間プロフィールを図示する。図1は、以下の4つの処置A〜Dの投与後の時間(時間)に対する式Iの化合物の血漿濃度(ng/ml)の線形:線形グラフプロフィールである:高脂肪朝食で標準化し、米国特許第5,834,472号の錠剤共沈処方物中の式Iの化合物(単一の2×100mg)(処置Dおよび記号−−);10時間絶食後、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Aおよび記号−0−);高脂肪朝食で標準化し、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Bおよび記号−−);および脱脂肪朝食で標準化し、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Cおよび記号−−)。
【図2】図2は、本発明の実施例1のポサコナゾール錠剤およびその液体懸濁液の平均血漿濃度の時間プロフィールを図示する。図2は、図1に示されるデータについての時間(時間)に対する式Iの化合物の血漿濃度(ng/ml)の対数:線形グラフプロフィールである。
【背景技術】
【0001】
(背景技術)
本発明は、以下の化学構造式I:
【0002】
【化8】
によって示される抗真菌的(antifungally)に有効量の微粉化化合物、少なくとも1種の濃厚剤(thickening agent)、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、安定な液体懸濁物、ならびに真菌感染を処置または予防するためにこの懸濁物を使用する方法に関する。
【0003】
米国特許第5,661,151は、式Iの化合物、および広範囲の真菌(例えば、アスペルギルス属(Aspergillis)、カンジダ属、クリプトコックス属、フザリウム属および他の日和見真菌)に対するその強力な抗真菌活性を開示する。
【0004】
米国特許第5,834,472号および同第5,846,971号は、結合剤と一緒に不活性ビーズ上にコートされる構造式Iの化合物の経口の薬学的カプセル組成物を開示する。しかし、構造式Iの化合物は、高親油性であり、そして非常に低い水溶性を有する。従って、構造式Iの化合物の水性組成物は、おそらく、化合物の非常に低い水溶性に起因して、抗真菌活性および/またはバイオアベイラビリティーを減少させていることが見出された。従って、バイオアベイラビリティーを増大し、そして安定な特性を改善させる、構造式Iの化合物の経口の薬学的組成物の必要性が存在する。
【発明の開示】
【課題を解決するための手段】
【0005】
(発明の要旨)
本発明者らは、ポサコナゾール(posaconazole)の微粉化粒子、以下の化学構造式I:
【0006】
【化9】
を有する化合物、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物の形態で、経口投与に適切な薬学的組成物を見出し、この薬学的組成物は、従来技術を越える重要な利点を提供する。
【0007】
本発明の液体懸濁物の利点としては、懸濁物の改善された均質性および懸濁物の分散の容易さが挙げられる。本発明の液体懸濁物中で安定である固形物は、再分散するのが困難な固体ケーキを形成しない。少なくとも3日間の間、本発明の再構築された液体懸濁物中に固体を実質的に沈殿しない。この驚くべき特性は、本発明の液体懸濁物を接種して、真菌感染を有する患者が、抗真菌的に有効量のポサコナゾールを接種することを確実にする。本発明の液体懸濁物は、より長い有効期間を有する。さらに、この液体懸濁物は、再構築の際に、最初に調製された懸濁物として、同じ抗真菌的に有効量のポサコナゾールを実質的に提供する。本発明の液体懸濁物のこれらの特性は、薬局、薬剤師、医師、および真菌感染を有する患者に利益を提供する。
【0008】
従って、本発明は、以下の化学構造式I:
【0009】
【化10】
を有する微粉化ポサコナゾール、少なくとも1種の濃厚剤、非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物を提供する。
【0010】
本発明はまた、以下の化学構造式I:
【0011】
【化11】
を有する、抗真菌的に有効量の微粉化ポサコナゾール、有効量の少なくとも1種の濃厚剤、約4.0〜約6.0の範囲に緩衝系のpHを維持するに有効な量の緩衝系、有効量の非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物を提供する。
【0012】
本発明はさらに、以下の化学構造式I:
【0013】
【化12】
を有する、抗真菌的に有効量の微粉化ポサコナゾール、を含む液体懸濁物を提供し、ここで、この微粉化化合物は、約1200nm〜約1600nmの範囲の平均粒径(mean particle size)を有し、有効量の、飽和または不飽和のC12〜C18酸のソルビタンエステルのポリオキシエチレン誘導体、約4.0〜約6.0の範囲にpHを維持するに十分な有効量の緩衝系、有効量の2種の濃厚剤(ここで一方は液糖である)の組み合わせ、および薬学的に受容可能な液体キャリアを含む。
したがって、本発明は、以下を提供する。
(1) 液体懸濁物であって、該液体懸濁物は、以下の化学構造式I:
【化101】
によって示される抗真菌的に有効量の微粉化化合物、少なくとも1種の濃厚剤、少なくとも1種の非イオン性界面活性剤、および薬学的に受容可能な液体キャリアを含む、液体懸濁物。
(2) 前記少なくとも1種の非イオン性界面活性剤が、エチレンオキシドおよびプロピレンオキシドのブロックコポリマー、飽和または不飽和のC8〜C20酸のグリコールエステルまたはグリセリルエステル、飽和または不飽和のC8〜C20酸のポリオキシエチレンエステル、飽和または不飽和のC8〜C20酸のポリオキシエチレンエーテル、飽和または不飽和のC10〜C20酸のポリビニルアルコールまたはソルビタンエステルである、項目1に記載の液体懸濁物。
(3) 前記少なくとも1種の非イオン性界面活性剤が、飽和または不飽和のC10〜C20酸のソルビタンエステルのポリオキシエチレン誘導体である、項目1に記載の液体懸濁物。
(4) 前記少なくとも1種の濃厚剤が、キサンタンガム、液糖、澱粉、セルロースおよびそれらの混合物から選択される、項目1に記載の液体懸濁物。
(5) キサンタンガムおよび液糖の組み合わせが、前記少なくとも1種の濃厚剤として使用される、項目1に記載の液体懸濁物。
(6) 前記式Iの微粉化化合物が、約1200nm〜約1600nmの平均粒径を有する、項目1に記載の液体懸濁物。
(7) 液体懸濁物であって、該液体懸濁物が、以下:
(a)以下の化学構造式I:
【化102】
で示される、抗真菌的に有効量の微粉化化合物;
(b)少なくとも1種の有効量の濃厚剤;
(c)約4.0〜約6.0の範囲内に緩衝系pHを維持するために有効な量の緩衝系;
(d)少なくとも1種の有効量の非イオン性界面活性剤;および
(e)薬学的に受容可能な液体キャリア
を含む、液体懸濁物。
(8) 前記少なくとも1種の非イオン性界面活性剤が、飽和または非飽和のC10〜C20酸のソルビタンエステルのポリオキシエチレン誘導体である、項目7に記載の液体懸濁物。
(9) 前記ソルビタンエステルが、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンセスキオレアート、ソルビタントリオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラートおよびソルビタントリステアラート、またはそれらの混合物から選択される、ソルビタンの脂肪酸エステルである、項目7に記載の液体懸濁物。
(10) 前記少なくとも1種の濃厚剤が、ガム、液糖、澱粉、セルロースおよびそれらの混合物から選択される、項目7に記載の液体懸濁物。
(11) キサンタンガムおよび液糖の組み合わせが、前記少なくとも1種の濃厚剤として使用される、項目7に記載の液体懸濁物。
(12) キサンタンガムおよび液体グルコースの組み合わせが、前記濃厚剤として使用される、項目7に記載の液体懸濁物。
(13) 前記緩衝系が、クエン酸ナトリウムおよびクエン酸を含む、項目7に記載の液体懸濁物。
(14) 前記薬学的に受容可能な液体キャリアが、精製水、グルコース、およびグリセリンの組み合わせである、項目7に記載の液体懸濁物。
(15) 前記式Iの微粉化化合物が、約1200nm〜約1600nmの平均粒径を有する、項目7に記載の液体懸濁物。
(16) 液体懸濁物であって、該液体懸濁物が、以下:
(a)以下の化学構造式I:
【化103】
を含む、抗真菌的に有効量の微粉化化合物であって、
ここで該式Iの微粉化化合物が、約1200nm〜約1600nmの範囲内の平均粒径を有する、微粉化化合物、
(b)飽和または不飽和のC12〜C18酸のソルビタンエステルの、有効量のポリオキシエチレン誘導体;
(c)約4.0〜約6.0の範囲内にpHを維持するに十分な、有効量の緩衝系;
(d)2種の濃厚剤の、有効量の組み合わせであって、ここで一方が液糖である、組み合わせ;および
(e)薬学的に受容可能な液体キャリア
を含む、液体懸濁物。
(17)以下の化学構造式I:
【化104】
の化合物の微粉化粒子の、哺乳動物における真菌感染の処置および/または予防のための、経口懸濁物の形態における医薬の調製のための使用であって、該使用が、薬学的に受容可能なキャリア中に、抗真菌的に有効量の該式Iの化合物Iおよび少なくとも1種の濃厚剤および非イオン性界面活性剤を含む、使用。
(18) 懸濁物であって、該懸濁物が、薬学的に受容可能な液体キャリア中に、抗真菌的に有効量の、以下の化学構造式I:
【化105】
を含む化合物の微粉化化合物、少なくとも1種の濃厚剤および非イオン性界面活性剤を含む、懸濁物。
(19) 組成物であって、該組成物が、以下の構造式I:
【化106】
によって示される化合物の微粉化粒子および少なくとも1種の非イオン性界面活性剤を含
む物質の組成物。
(20)懸濁物中に、以下の化学構造式I:
【化107】
によって示される化合物の微粉化粒子を含む物質の組成物。
【0014】
(発明の詳細な説明)
本発明は、薬学的に受容可能な液体キャリア中にある抗真菌性化合物ポサコナゾール(posaconazole)の微粉化粒子の安定な懸濁液を提供する。本発明の懸濁液は、25℃で3日間よりも長い間静置して貯蔵した場合、沈澱することなく安定である。(以下の表1を参照のこと)。
【0015】
以下の表2により、懸濁液中のポサコナゾールの濃度が、12ヶ月までの期間にわたって、(HPLCによって測定した場合)初期濃度と比較して実質的に同一(±2%)である点で、本発明の液体懸濁液処方物が安定であることが示される。
【0016】
本発明者らはまた、本発明の安定な懸濁液が、各々が被験体に高脂肪の朝食と同時に投与される場合、最適化されたポサコナゾールの微粉化粒子の経口錠剤と比較して、著しくより高いバイオアベイラビリティー(23〜36%の増加)を有することを見出した。表3および表4ならびに図1および図2を参照のこと。
【0017】
本発明の1つの局面は、薬学的組成物を提供することであり、この組成物は、非イオン性界面活性剤(例えば、ソルビタンエステル)および少なくとも1つの増粘剤(好ましくは、キサンタンガムおよび液糖の組み合わせ)と組み合わせて、ポサコナゾールの微粉化粒子を含有し、これらの粒子は、精製水のような薬学的に受容可能な液体キャリア中に容易に分散可能である。この薬学的組成物は、安定した懸濁液を提供し、この懸濁液は、少なくとも3日間沈澱せず、従って患者が有効用量のポサコナゾールを得ることを確実にする。本発明の安定した懸濁液の別の特徴は、ポサコナゾールが溶液から沈澱することなしに、口腔鵞口瘡を伴うHIV−1感染を保有する患者の処置に有用であることである。本発明の別の局面は、本発明の懸濁液が、分散しにくい固体ケーキ(cake)の形成を回避することである。
【0018】
本発明の懸濁液において使用される式Iの化合物は、Schering Corporation,Kenilworth,New Jerseyから入手可能であり、そして米国特許第5,661,151号およびWO95/17407の実施例24および32に従って調製された。
【0019】
好ましいポサコナゾールのミクロンサイズの粒子は、約1000ナノメートル(nm)〜約1800nm、好ましくは、約1200nm〜約1600nm、そして最も好ましくは、約1400nmの平均粒径範囲を有する。この粒径は、式Iの抗真菌性化合物の製造の間の最終工程によってか、または慣用的な結晶化手順後の慣用的な微粉化技術の使用によってのいずれかで得られ得る。
【0020】
ポサコナゾールを望ましい平均粒径範囲に微粉化するために使用される好ましい微粉化技術は、ミクロ流動化である。ミクロ流動化は、伝統的な均質化に代わるものであり、これは、高圧での2つの物質の流れの衝突を利用して、はるかにより均一な粒径分布(Microfluidics International Co.に従う)、および約1200nm〜1600nmのより小さい平均粒径を生成する。ミクロ流動化において使用されるプロセスおよび装置は、米国特許第4,533,254号に記載されている。
【0021】
本発明の微粉化ポサコナゾールはまた、結晶形態で存在し得る。結晶形態は、好ましくは、実質的に化学的および光学的に純粋であり、そしてこれは、約10%未満のその光学異性体、エナンチオマーまたは他のジアステレオマーを含有する。結晶形態は、99%の光学純度の左旋性または右旋性の異性体であり得る。化学構造Iのこの光学的に純粋な化合物は、他の光学異性体の混合物の多くの有害な副作用を回避する。
【0022】
ポサコナゾール液体懸濁液は、目的の真菌を制御するために抗真菌的に有効な量で組成物において使用される。このような抗真菌的に有効な量は、本発明の液体懸濁液処方物の濃度が、約10mg/ml〜約100mg/ml、より好ましくは、約20mg/ml〜約60mg/ml、そして最も好ましくは、約40mg/mlの式Iの化合物の範囲であり得る。
【0023】
本発明はまた、哺乳動物における真菌感染を、処置および/または予防する方法を提供し、この方法は、このような真菌感染を処置および/または予防するために有効な量の微粉化ポサコナゾールを含む一定量の液体懸濁液を、哺乳動物に投与する工程を包含する。式Iの化合物を40mg/ml含有する、本発明の抗真菌的に有効量の液体懸濁液は、5ml(200mgの式Iの化合物を含む)で1日に3回(TID)もしくは1日に4回(QID)の用量、または10ml(400mgの式1の化合物を含む)で1日に2回(BID)の用量で経口投与される。当然、担当の臨床医は、患者の年齢、健康状態、および性別、ならびに真菌感染の重篤度を考慮して、用量および投薬レジメンを変更し得る。
【0024】
以下の用語は、本発明の薬学的組成物、その処方において使用され得る成分、およびその化合物の生物活性またはバイオアベイラビリティーを評価するための方法を記述するために使用される。
【0025】
非イオン性界面活性剤とは、正味のイオン性電荷を欠き、かつ水性媒体中で感知される程度に解離しない界面活性剤をいう。非イオン性界面活性剤の特性は、その分子における親水性基および疎水性基の割合に大きく依存する。親水性基としては、オキシエチレン基(−OCH2CH2−)およびヒドロキシ基が挙げられる。疎水性分子(例えば、脂肪酸のエステル)においてこれらの基の数を変更することによって、強力に疎水性かつ水に不溶性の化合物(例えば、モノステアリン酸グリセリン)から、強力に親水性かつ水溶性の化合物(例えば、マクロゴール)に及ぶ物質が得られる。これらの2つの極端な型の間には、親水性基および疎水性基の割合がより均等に釣り合ったもの(例えば、マクロゴールエステルおよびマクロゴールエーテルならびにソルビタン誘導体)を含む。適切な非イオン性界面活性剤は、Martindale,The Extra Pharmacopoeia,第28版,1982,The Pharmaceutical Press,London,Great Britain,pp.370〜379に見出され得る。
【0026】
このような適切な非イオン性界面活性剤としては、エチレンオキシドおよびプロピレン
オキシドのブロックコポリマー、飽和もしくは不飽和のC8〜C20酸のグリコールエステルまたはグリセリルエステル、飽和もしくは不飽和のC8〜C20酸のポリオキシエチレンエステル、飽和もしくは不飽和のC8〜C20酸のポリオキシエチレンエーテル、ならびに飽和もしくは不飽和のC10〜C20酸のポリビニルアルコールまたはソルビタンエステルが挙げられる。好ましくは、この非イオン性界面活性剤は、飽和または不飽和のC10〜C20酸のソルビタンエステルであり、そしてより好ましくは、このソルビタンエステルは、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンセスキオレアート、ソルビタントリオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラートおよびソルビタントリステアラート、またはそれらの混合物から選択されるソルビタンの脂肪酸エステルである。
【0027】
適切なソルビタンエステルとしては、例えば、ポリソルベート20、ポリソルベート40、ポリソルベート60、ポリソルベート65、ポリソルベート80、ポリソルベート85、ソルビタンモノラウレート、ソルビタンモノオレアート、ソルビタンモノパルミテート、ソルビタンモノステアラート、ソルビタンセスキオレアート、ソルビタントリオレアート、およびソルビタントリステアラートが挙げられる。最も好ましい非イオン性界面活性剤は、商品名Tween80でICI Americasから入手可能なポリソルベート80であり、これは、約20モルのエチレンオキシドと縮合した主にモノエステルから構成される、ソルビトールのオレイン酸エステルおよびソルビトール無水物の混合物である。
【0028】
エチレンオキシドおよびプロピレンオキシドの適切なブロックコポリマーは、一般的に「ポロキサマー」と呼ばれ、そして以下の化学構造I:
【0029】
【化13】
によって表されるコポリマーを含み、ここで、aは、約10〜約110、好ましくは、約12〜101;より好ましくは、約12〜80にわたる整数であり;そして
bは、約20〜約60、より好ましくは、約20〜約56;または約20〜27にわたる整数である。
【0030】
脂肪酸の適切なグリコールエステルおよびグリセリルエステルとしては、モノラウリル酸グリセリンおよび同様な水溶性誘導体が挙げられる。
【0031】
脂肪酸の適切なポリオキシエチレンエステル(マクロゴールエステル)としては、ポリオキシエチレンヒマシ油および硬化ヒマシ油誘導体が挙げられる。
【0032】
脂肪酸の適切なポリオキシエチレンエーテル(マクロゴールエーテル)としては、セトマクロゲル(Cetomacrogel)1000、ラウロマクロゴール(異なる鎖長のマクロゴールの一連のラウリルエーテル)、例えば、ラウレス4、ラウレス9およびラウロマクロゴール400が挙げられる。
【0033】
組成物における非イオン性界面活性剤の有効量は、処方物の濃度が、約5mg/ml〜約50mg/ml、より好ましくは、約5mg/ml〜約25mg/ml、そして最も好ましくは、10mg/mlの範囲であり得る。
【0034】
本発明において適切と見出された増粘剤としては、このような目的のために有用な任意
の市販の薬剤が挙げられる。キサンタンガム、液糖(liquid sugar)、デンプン、セルロース、およびこれらの混合物は、好ましい増粘剤である。より好ましいのは、キサンタンガムおよび液糖の組み合わせであり、そして最も好ましくは、キサンタンガム、NFおよびグルコース、NFの組み合わせである。好ましくは、キサンタンガムが、約1mg/ml〜約5mg/mlの量で存在し、そしてより好ましくは、キサンタンガムが、約3mg/mlの量で存在する。好ましくは、グルコースNFは、約200〜約500mg/ml、そしてより好ましくは、約350mg/mlの量で存在する。本発明の増粘剤の有効量は、約1〜約500mg/ml、より好ましくは、約200〜約500mg/ml、最も好ましくは、約353mg/mlであり得る。本発明の増粘剤は、最小限の攪拌で構成した後、処方物の懸濁を容易にし、そして長時間にわたって懸濁液の迅速な沈澱およびケーキ形成を防止する。
【0035】
薬学的に受容可能なキャリアとしては、当該分野において周知のものが挙げられ、精製水USP、液体グルコース、NF、および無水グリセロールが挙げられる。最も好ましいのは、精製水USPおよび液体グルコース、NFである。薬学的に受容可能なキャリアは、約10〜約500mg/ml、より好ましくは、約200mg/ml〜約400mg/ml、最も好ましくは、約350mg/mlの量で存在し得る。
【0036】
本発明の処方物について適切な緩衝系は、液体懸濁液のpHを、約4〜約6、好ましくは、4.5〜5.0、そして最も好ましくは、約4.5の範囲に維持するものである。クエン酸ナトリウム、USPおよびクエン酸、USPの緩衝系の使用が好ましい。他の適切な緩衝系は、4〜6の望ましいpH範囲を維持するために使用され得る。緩衝化剤は、約0.4〜約1.5mg/ml、より好ましくは、約0.7〜約1.5mg/ml、最も好ましくは、約1.1mg/mlの濃度で存在し得る。
【0037】
本発明において適切と見出された消泡剤としては、このような目的のために有用な任意の市販の薬剤が挙げられ、トリメチルシロキシル単位(例えば、ジメチコーンおよびシメチコン)で末端ブロックされたメチル化直線状シロキサンポリマー、ならびに200〜250ジメチルシロキサン単位の平均鎖長を有するジメチコーンおよびシリカゲルの混合物が挙げられる。消泡剤の有効量は、約2mg/ml〜約4mg/ml、好ましくは、約3mg/mlの濃度を提供するために十分な量である。
【0038】
本発明において有用と見出された水溶性防腐剤としては、安息香酸ナトリウム、クエン酸ナトリウムおよび塩化ベンザルコニウムならびに他の薬学的に受容可能な水溶性防腐剤が挙げられる。防腐剤として安息香酸ナトリウムの使用が好ましい。水溶性防腐剤の有効量は、約0.5mg/ml〜約3mg/ml、最も好ましくは、約2mg/mlの濃度を提供するために十分な量である。
【0039】
本発明において適切と見出された乳白剤としては、薬学的に受容可能な金属酸化物、特に二酸化チタンが挙げられる。乳白剤の有効量は、約2mg/ml〜約6mg/ml、最も好ましくは、約4mg/mlの濃度を提供するために十分な量である。
【0040】
代表的な香味剤は、医薬、食品、キャンディー、飲料などを甘くする使用のためにFDAによって認可されているものであり;これらの物質は、香味(例えば、ブドウ、サクランボ、柑橘類、モモ、イチゴ、風船ガム、ペパーミントおよびその他多くのもの)を与える。香味剤の有効量は、約0.01mg/ml〜約6mg/ml、より好ましくは、約5mg/mlの濃度を提供するために十分な量である。
【0041】
以下の実施例は、ポサコナゾールを含有する本発明の組成物を記載するが、これらは、特許請求の範囲の範囲を限定するように解釈されるべきでない。
【実施例】
【0042】
【数1】
上記成分の範囲は、当業者に明らかであるように、変動され得る。本発明の範囲内の組成物の特定の実施例を、以下に示す。
【0043】
(実施例1)
【0044】
【数2】
この実施例は、以下のように調製され得る:ミキサープロペラを備えた適切な容器に20±3℃にて約5%の最終バッチ容積の精製水を充填する。工程1の精製水に40%のポリソルベート80を添加し、そして溶解するまで混合する。40%のシメチコンを添加し、そして分散するまで混合する。約5つのパスについて約30,000±5000psiで作動するマイクロフルイダイザー(microfluidizer)によって工程3の混合物を再循環する。約20±3℃にて、約7%の最終バッチ容積の精製水を添加し、そして約5分間混合する。一定に混合しながらポサコナゾールを工程5の容器に添加する。完全に分散されるまで混合し続ける。約30,000±5,000psiの圧力で作動するマイクロフルイダイザーに通して工程6からの懸濁部分を再循環する。当該分野で公知であるレーザー回折技術によって測定される場合、粒子のメジアンが約1.4±0.2μmの粒径を示すまで、濃縮物を処理する。
【0045】
この粒径が達成される場合、マイクロフルイダイザーに懸濁液を通し、そして適切な大きさの容器に収集する。約8〜10%の最終バッチ容積の精製水(20±3℃)をフィード容器に添加し、約30,000psiで作動するマイクロフルイダイザーに通す。濃縮物を含む容器中の洗浄物を収集する。濃縮物を有する容器に約22%の最終バッチ容積の精製水(20±3℃)を添加し、そして約5分間混合する。ポリソルベート80およびシメチコンの残りを添加し、そして約5分間混合する。
【0046】
安息香酸ナトリウム、クエン酸ナトリウムおよびクエン酸を添加し、そして約5分間混合する。一定に混合しながらキサンタンガムをゆっくりと添加する。キサンタンガムの添加の後、混合し続ける。混合せずに30分間、キサンタンガムを水和させる。一定に混合しながらグリセリンを添加する。一定に混合しながら液体グルコースをゆっくりと添加する。5分間または溶解するまで混合する。二酸化チタンを添加し、そして内容物が完全に分散されるまで適切なホモジナイザーを用いて混合する。人工のチェリー香料を添加し、約5分間混合する。最終容積になるまで十分な量の20±3℃の精製水を添加し、そして均一な懸濁液が達成されるまで混合する。懸濁液のpH測定および物理的観察のために約20mlのサンプルを収集する。実施例1の懸濁液のpHは、5.0であった。
【0047】
(実施例2)
実施例2は、実施例1の手順を用いて調製した本発明の範囲内での処方物の別の例であり、そしてpH4.5を有する。
【0048】
【数3】
本発明の液体懸濁液の沈降速度を、以下に示すように測定した。
【0049】
(表1:40mg/mlのポサコナゾール経口懸濁液の沈降速度)
【0050】
【表1】
本発明の懸濁液を含む2つのボトルを、振盪し、そして静置した。次いで、このボトルを、振盪の後、直ちに(時間0)、5分後、10分後、30分後、60分後および72時間後(3日間)にサンプリングした。これらのサンプル中のポサコナゾールのレベルおよび防腐剤(安息香酸ナトリウム)のレベルを、HPLCによりアッセイした。検出のHPLC方法は、当業者に周知である。
【0051】
防腐剤およびポサコナゾールのアッセイの結果は、一貫したままであり、そして変化しなかった。これらは、それぞれ、活性に対して39.8mg/ml〜40.2mg/ml(99.5%〜101%)、および保存に対して1.99mg/ml〜2.02mg/ml(99.5%〜101%)の範囲であった。この実験の結果は、上記の表1に示す。
【0052】
安息香酸ナトリウムは、沈降すると予測されなかった。驚いたことに、ポサコナゾールは、3日後でも沈降しなかった。
【0053】
従って、本発明の組成物は、表1中のサンプルの安定性によって証明されるように分散
能および均質性の両方の容易さを有する。
【0054】
次に、加速された均質性試験を、本発明の液体懸濁液に対して行った。
【0055】
(表2:40mg/mlのポサコナゾール経口懸濁液の均質性)
【0056】
【表2】
*患者の指示通りの用量を振盪しそして与える。
【0057】
これらのデータ(沈降速度試験)は、表2に示されるリアルタイムの安定性データ(40℃/75%RHで6ヶ月までおよび25℃/60%RHで24ヶ月まで)に従っていた。このアッセイの均質性結果は、驚くことに、一貫して均質なままであり、そして実質的に変化しなかった。
【0058】
40℃/75%RHで6ヶ月後、均質性結果は、それぞれ、活性に対して40.7mg/ml〜40.8mg/ml(101%)、および保存性に対して2.01mg/ml〜2.03mg/ml(101%〜102%)の範囲であった。これらの結果を、アッセイされるボトルの部分(すなわち、ボトルの上部または下部)に関係なく得た。従って、加速した安定の状態での比較的長い曝露の後でさえ、懸濁液はボトルの至るところで均質であったと結論付け得る。
【0059】
25℃/60%RHで24ヶ月後、均質性結果は、それぞれ、活性に対して41.5mg/ml〜41.6mg/ml(104%)、および保存性に対して2.01mg/ml(101%)の範囲であった。これらの結果を、アッセイされるボトルの部分(すなわち、ボトルの上部または下部)に関係なく得た。従って、25℃/60%RHでの長期(24ヶ月)の曝露の後でさえ、懸濁液はボトルの至るところで均質であったことが結論され得る。
【0060】
バイオアベイラビリティーは、活性薬物成分または治療的部分が標準またはコントロールと比較して、投与された投薬形態から体循環に吸収される速度および程度として定義される。
【0061】
Cmax値を、血清血漿中で測定された抗真菌化合物の最大濃度(すなわち「ピーク」
)として定義する。
【0062】
本発明の処方物は、増加したバイオアベイラビリティーおよび以前の処方物よりもより低い変動性を有するという利点を有する。
【0063】
ポサコナゾール経口懸濁液の相対的なバイオアベイラビリティーを、健康な被験体中で、固体投薬形態のポサコナゾールと比較した。
【0064】
第1の目的は、高脂肪朝食を与えられた場合、経口固体処方物と比較して、経口懸濁液として、与えたポサコナゾールの相対的なバイオアベイラビリティーを測定することであった。第2の目的は、経口懸濁液として与えられる場合、式1の化合物の経口バイオアベイラビリティーで絶食状態に関連して、高脂肪食物および無脂肪食物の効果を測定することであった。
【0065】
20人の健康な被検体は、この無作為化した、開放標識の、4つの方法で交差する単一投与バイオアベイラビリティーおよびポサコナゾールの食物効果研究を完了した。被験体は、少なくとも7日間の洗い出しによって分けられた以下の4つの処置のそれぞれを受けた。
【0066】
【数4】
被験体を、絶食したまま(処置A)、標準化した高脂肪朝食を受ける(処置BまたはD)または標準化した無脂肪朝食を受ける(処置C)のいずれかで無作為化した。食事を、朝の薬物投与の20分前に食べ、そして被験体は朝食を完了した5分以内に適切な処置を受けた。
【0067】
血液サンプル(6ml)を、それぞれの処置に対して、投薬の直ぐ前(0時間)に、および投薬の0.5時間後、1時間後。1.5時間後、2時間後、3時間後、4時間後、5時間後、6時間後、8時間後、10時間後、12時間後、16時間後、24時間後、36時間後、48時間後および72時間後にヘパリン処理されたチューブ内に収集した。血液を、4℃で遠心分離し、そして血漿をアッセイされるまで−20℃以下で貯蔵した。ポサコナゾールの血漿濃度を、ng/mlのLOQで有効な高速液体クロマトグラフィーアッセイを用いてアッセイした。
【0068】
個々の血漿の濃度−時間データを、モデル非依存性方法を用いて、薬物動態学的アッセイのために使用した。最大濃度(Cmax)および最大濃度の時間(Tmax)は、観察された値であった。時間0から定量化できる最終のサンプリング時間[AUC(tf)]までの血漿濃度−時間曲線の下の面積を、一次台形法(linear trapezoidal method)を使用して計算し、そして以下のように
【0069】
【数5】
無限(I)に外挿し:
ここでC(tf)は、時間(tf)で線形回帰から決定された推定の濃度である。
【0070】
全身のクリアランスを、以下によって計算した:
CL=用量/AUC(I)
分布の見掛けの容積(Vdarea/F)を、末期の速度定数(K)に対する全身クリアランスの速度から計算した。
【0071】
手短な統計学は、従来技術の錠剤処方物の濃度−時間データおよび誘導した薬物動態学的パラメーターと比較して、本発明の血漿懸濁液処方物について計算した。本来のスケールならびにlog変換CmaxおよびAUC値を、分散分析(ANOA)を用いて分析した。被験体、フェーズおよび処置に起因する効果を抽出した。
【0072】
式Iの化合物に対する血漿濃度−時間データおよび薬物動態学的パラメーターを、表3および4に表にし、そして図1および2に図示する。
【0073】
すべての被験体は、フェーズ3およびフェーズ4において被験体20を除いて以下に報告されるLOQ(5ng/ml)の1日目の0時間の濃度を有し、この被験体は、それぞれ、8.5ng/mlおよび22.5ng/mlの処置BおよびAに対して0時間でのポサコナゾールの定量化可能なレベルを有した。これらのレベルは、以前の用量からの集積の繰り越し効果におそらく起因する。
【0074】
平均aの薬物動力学パラメーターの概要を、以下の表に提供する:
【0075】
【表3】
a:バランス平均、n=20は、AUC(I)およびt1/2(n=15)を除く。
【0076】
ポサコナゾールは、ゆっくりと吸収された;平均Tmax値は、4.1〜5.5時間の範囲であった。ポスコナゾールは、処置から独立して、約22時間の平均終了t1/2でゆっくりと除去された。この研究を行って、錠剤処方物(処置D)と比較してポサコナゾール経口懸濁液(処置B)のバイオアベイラビリティーを評価した(両方ともが、高脂肪食物を与えられる)。log変換データに基づくこの結果を、以下に示す:
【0077】
【表4】
a:全ての被験体についての全ての処置について計算され得るので、AUC(tf)を、統計的な比較のために使用した。
【0078】
b:錠剤に対する懸濁液。
【0079】
平均して、本発明の懸濁液処方物は、先行技術の錠剤と比較して、Cmaxにおいて23%の増加およびAUC(tf)において36%の増加を生じた。
【0080】
研究の第2の目的は、経口懸濁液として投与されるポサコナゾールの経口アベイラビリティーで、絶食(処置A)と比較して、高脂肪食物(処置B)および無脂肪食物(処置C)の効果を評価することであった。log変換データに基づくこの結果を、以下に示す:
【0081】
【表5】
a:処置A−懸濁液/脂肪のパーセントとして表した。
【0082】
高脂肪朝食は、懸濁液中で与えられたポサコナゾールのバイオアベイラビリティーにおいて4倍の増加を生じた。食物は、錠剤処方物およびカプセル処方物の両方について3〜5倍まで、ポサコナゾ−ルのバイオアバイラビリティーを有意に増加した、以前の研究からの結果と一貫していた。絶食(処置A)と比較して無脂肪朝食(処置C)の効果は、バイオアベイラビリティーにおいて2.5〜3倍での増加であり、より少なかった。
【0083】
本発明の多くの改変および変化は、当業者に明白であるので、その精神および範囲から逸脱することなくなされ得る。本明細書中に記載される特定の実施形態は、例示のみによって提供され、そして本発明は、添付の特許請求の範囲の権利が与えられる全ての範囲の等価物と共に、このよに特許請求の範囲の用語によってのみ限定されるべきである。
【図面の簡単な説明】
【0084】
【図1】図1は、本発明の実施例1のポサコナゾール錠剤およびその液体懸濁液の平均血漿濃度の時間プロフィールを図示する。図1は、以下の4つの処置A〜Dの投与後の時間(時間)に対する式Iの化合物の血漿濃度(ng/ml)の線形:線形グラフプロフィールである:高脂肪朝食で標準化し、米国特許第5,834,472号の錠剤共沈処方物中の式Iの化合物(単一の2×100mg)(処置Dおよび記号−−);10時間絶食後、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Aおよび記号−0−);高脂肪朝食で標準化し、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Bおよび記号−−);および脱脂肪朝食で標準化し、本発明の経口懸濁液(5ml)中の式Iの化合物(200mg)(処置Cおよび記号−−)。
【図2】図2は、本発明の実施例1のポサコナゾール錠剤およびその液体懸濁液の平均血漿濃度の時間プロフィールを図示する。図2は、図1に示されるデータについての時間(時間)に対する式Iの化合物の血漿濃度(ng/ml)の対数:線形グラフプロフィールである。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

【図2】
【図2】
【公開番号】特開2008−120836(P2008−120836A)
【公開日】平成20年5月29日(2008.5.29)
【国際特許分類】
【出願番号】特願2008−36719(P2008−36719)
【出願日】平成20年2月18日(2008.2.18)
【分割の表示】特願2002−578726(P2002−578726)の分割
【原出願日】平成14年4月1日(2002.4.1)
【出願人】(596129215)シェーリング コーポレイション (785)
【氏名又は名称原語表記】Schering Corporation
【Fターム(参考)】
【公開日】平成20年5月29日(2008.5.29)
【国際特許分類】
【出願日】平成20年2月18日(2008.2.18)
【分割の表示】特願2002−578726(P2002−578726)の分割
【原出願日】平成14年4月1日(2002.4.1)
【出願人】(596129215)シェーリング コーポレイション (785)
【氏名又は名称原語表記】Schering Corporation
【Fターム(参考)】
[ Back to top ]