
変性ミクロフィブリル化植物繊維を含む樹脂組成物
【課題】オレフィン系樹脂のような疎水性の高い樹脂を用いたミクロフィブリル化植物繊維を含む樹脂組成物において、樹脂組成物内でのミクロフィブリル化植物繊維の分散性が良好で、得られる成形材料においては機械的強度を向上させることのできる、アルキル、若しくはアルケニル無水コハク酸によって変性されたミクロフィブリル化植物繊維を含む樹脂組成物及びその製造方法、並びに該樹脂組成物を用いた成形材料、及び成形体を提供する。
【解決手段】アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び熱可塑性樹脂(B)を含有する樹脂組成物に関する。
【解決手段】アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び熱可塑性樹脂(B)を含有する樹脂組成物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アルキル、若しくはアルケニル無水コハク酸によって変性されたミクロフィブリル化植物繊維を含む樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体に関する。
【背景技術】
【0002】
従来、樹脂の強度等の物性を向上させるため、充填剤を用いて樹脂を強化することが知られている。例えば、セルロース繊維をミクロフィブリル化して、繊維径がナノオーダーにまで微細化されたミクロフィブリル化植物繊維(ナノファイバー)等を充填剤とし、当該ナノファイバーを樹脂中に配合させることによって得られる成形材料は、質量が軽く、かつ高強度であるという点から、非常に有用である。
【0003】
しかしながら、ミクロフィブリル化植物繊維は非常に凝集力が強く、一般的に樹脂との相溶性が悪いため、ミクロフィブリル化繊維が均一に分散した均一な樹脂成形材料を得ることが困難である。そのため、得られる成形材料は、機械的強度が十分に発揮できないという問題があった。このような課題に対して、ミクロフィブリル化植物繊維の表面を、化学修飾剤等によって変性処理し、樹脂中での分散性を向上させようとする試みがなされている。
【0004】
例えば、特許文献1では、化学的に修飾されたセルロース繊維の集合体に、マトリクス材料を含浸させてなる複合材料について開示されている。このような化学修飾剤によって修飾されたセルロース繊維とマトリクス材料とを複合化させる方法(シート含浸法)によって、高品質の透明基板を得ることを目的としている。しかしながら、特許文献1ではシート含浸法である為に、樹脂が液状である必要があり、通常固体であるオレフィン系樹脂との複合化に用いることは難しい。また、セルロース繊維の解繊度を上げるほどち密なシートが出来やすく、内部まで樹脂を均一に含浸することが困難な為、シート内部、表面で樹脂濃度にバラツキが生じやすい。この為、機械的強度が十分に得られないという問題があった。
【0005】
特許文献2では、無水酢酸、無水酪酸、塩化アセチル、塩化ブチリル、酢酸等の有機化合物によって表面の水酸基がエステル化されたセルロースミクロフィブリルについて開示されている。また特許文献2では、該セルロースミクロフィブリルを有機溶媒に溶解した後、有機溶媒に溶解した樹脂と混合後、溶媒を除去することによって、複合材料を形成させることが記載されている。このような方法によって複合材料を得る場合、セルロースミクロフィブリルを、有機溶媒に十分に溶解させる必要がある。セルロース繊維を有機溶媒に溶解させるためには、セルロースミクロフィブリルの疎水変性を十分に行う必要がある。このような疎水変性は、セルロース繊維のI型結晶の破壊を招き、高強度の複合材料を製造することが困難であった。
【0006】
特許文献3では、イオン性液体と有機溶媒の混合溶媒中で無水酢酸、無水マレイン酸、無水コハク酸等の酸無水物によって変性されたセルロースナノファイバーとポリ乳酸をラボミルで混練後、プレス成形することによって成形材料を得る旨開示されている。このような変性セルロースナノファイバーは、ポリ乳酸のような親水性の高い樹脂では適用が可能であるが、ポリオレフィン系樹脂等の疎水性の高い樹脂の場合では、均一に混合することが困難であった。
【0007】
また特許文献4では、セルロースの水酸基の一部に無水マレイン酸や、無水コハク酸等の多塩基酸無水物によりエステル化し、カルボキシル基を導入した後、高圧ホモジナイザー処理、ニーダー、多軸押出し機等による微細繊維化処理によりナノ繊維を得る方法、及びそのナノ繊維と樹脂からなる複合材料について開示されている。該ナノ繊維は、ミクロフィブリル表面に導入された負の電荷を有するカルボキシル基の存在によりミクロフィブリル間の反発力を誘引し、分散体中での安定な分散が可能となっている。しかしながら、オレフィン系樹脂のような疎水性の高い樹脂中では十分な分散性が得られず、機械的強度において未だ改善の余地がある。
【0008】
更に特許文献5では、疎水化されたセルロース系繊維と、合成樹脂とを含有する樹脂組成物について開示されており、疎水化されたセルロース系繊維を調製するために、実際の実施例では、アルキルケテンダイマーのような疎水化剤がクラフトパルプの高圧ホモジナイザー処理物100gに対して0.01g(100ppm)用いられている。また、同実施例では濃度約0.5%のクラフトパルプ水分散物にアルキルケテンダイマーを添加しているため、添加したアルキルケテンダイマーの一部が加水分解してケトンになる副反応が起こる。そのため、実際にセルロースと共有結合しているアルキルケテンダイマーは100ppmよりかなり少ないと思われる。また、セルロース系繊維の解繊度が高い場合、この量では解繊されたセルロース表面を疎水化できるほどに覆うことが出来ず、結果としてオレフィン系樹脂のような疎水性の高い樹脂中では十分な分散性が得られず、機械的強度において未だ改善の余地がある。
【0009】
このように、ミクロフィブリル化植物繊維を含む成形材料において、樹脂としてポリエチレン、ポリプロピレン等の熱可塑性樹脂のような疎水性の高い樹脂を用いた場合、ミクロフィブリル化植物繊維の分散性が悪く、更なる機械的強度を得ることが非常に困難であった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2007−51266号公報
【特許文献2】特開平11−513425号公報
【特許文献3】特開2010−104768号公報
【特許文献4】特開2009−293167号公報
【特許文献5】特開2010−106251号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、オレフィン系樹脂のような疎水性の高い樹脂を用いたミクロフィブリル化植物繊維を含む樹脂組成物において、樹脂組成物内でのミクロフィブリル化植物繊維の分散性が良好で、得られる成形材料においては機械的強度を向上させることのできる、アルキル、若しくはアルケニル無水コハク酸によって変性されたミクロフィブリル化植物繊維を含む樹脂組成物及びその製造方法、並びに該樹脂組成物を用いた成形材料、及び成形体を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、アルキル、若しくはアルケニル無水コハク酸でエステル化することによって得られる変性ミクロフィブリル化植物繊維をオレフィン系樹脂に混合させることによって、変性ミクロフィブリル化植物繊維の分散性を向上させ、更には、得られる成形材料の機械的強度を向上することができることを見出した。
【0013】
本発明はこのような知見に基づき、更に鋭意検討を重ねて完成した発明である。すなわち、本発明は下記項に示す樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体を提供する。
【0014】
項1.アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び
熱可塑性樹脂(B)を含有する樹脂組成物であって、
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.05〜2.0である樹脂組成物。
【0015】
項2.アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、
熱可塑性樹脂(B)、及び
無機塩(C)を含有する樹脂組成物。
【0016】
項3.変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.1〜2.0である項1〜2の何れか1項に記載の樹脂組成物。
【0017】
項4.変性ミクロフィブリル化植物繊維(A)の含有量が、樹脂組成物100質量部に対して、1〜80質量部である項1〜3の何れか1項に記載の樹脂組成物。
【0018】
項5.無機塩(C)が、第2族の金属からなる塩である項2〜4の何れか1項に記載の樹脂組成物。
【0019】
項6.無機塩(C)の含有量が、樹脂組成物100質量部に対して0.1〜20質量部である項2〜5の何れか1項に記載の樹脂組成物。
【0020】
項7.(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び
(2a)工程(1a)によって得られたミクロフィブリル化植物繊維を触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維を得る工程、又は
(1b)植物繊維を、触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び
(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維を得る工程、
並びに
(3)工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程を含む樹脂組成物の製造方法。
【0021】
項8.工程(3)が、工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、及び無機塩(C)を混合する工程である項7に記載の樹脂組成物の製造方法。
【0022】
項9.工程(2a)又は工程(1b)におけるエステル化が、アルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒よりなる少なくとも1種の溶媒の存在下で行われる項7又は8に記載の樹脂組成物の製造方法。
【0023】
項10.工程(1a)又は工程(2b)における解繊が、ボールミル、ビーズミル、ブレンダー、グラインダー、リファイナー、高圧衝突分散機、ホモジナイザー、高圧ホモジナイザー、一軸又は多軸混錬機からなる群より選ばれる少なくとも1種による機械的解繊である項7〜9の何れか1項に記載の樹脂組成物の製造方法。
【0024】
項11.項7〜10の何れか1項に記載の製造方法で得られた変性ミクロフィブリル化植物繊維。
【0025】
項12.項1〜6の何れか1項に記載の樹脂組成物を用いた成形材料。
【0026】
項13.項7〜10の何れか1項で製造された樹脂組成物を用いた成形材料。
【0027】
項14.項12又は13に記載の成形材料を成形してなる成形体。
【0028】
以下、本願発明の樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体について、詳述する。
【0029】
<樹脂組成物>
本発明は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)、及び熱可塑性樹脂(B)を含有する樹脂組成物に関する。
【0030】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)の原料として用いられる植物繊維を含有する材料(植物繊維含有材料)としては、木材、竹、麻、ジュート、ケナフ、綿、ビート、農産物残廃物、布といった天然植物繊維原料から得られるパルプ、レーヨンやセロファン等の再生セルロース繊維等が挙げられる。特に、パルプが好ましい原材料として挙げられる。
【0031】
前記パルプとしては、植物原料を化学的、若しくは機械的に、又は両者を併用してパルプ化することで得られるケミカルパルプ(クラフトパルプ(KP)、亜硫酸パルプ(SP))、セミケミカルパルプ(SCP)、ケミグランドパルプ(CGP)、ケミメカニカルパルプ(CMP)、砕木パルプ(GP)、リファイナーメカニカルパルプ(RMP)、サーモメカニカルパルプ(TMP)、ケミサーモメカニカルパルプ(CTMP)、及びこれらの植物繊維を主成分とする脱墨古紙パルプ、段ボール古紙パルプ、雑誌古紙パルプが好ましいものとして挙げられる。これらの原材料は、必要に応じ、脱リグニン、又は漂白を行い、当該植物繊維中のリグニン量を調整することができる。
【0032】
これらのパルプの中でも、繊維の強度が強い針葉樹由来の各種クラフトパルプ(針葉樹未漂白クラフトパルプ(以下、NUKPということがある)、針葉樹酸素晒し未漂白クラフトパルプ(以下、NOKPということがある)、針葉樹漂白クラフトパルプ(以下、NBKPということがある))が特に好ましい。
【0033】
原料となる植物繊維は主にセルロース、ヘミセルロース、リグニンから構成される。植物繊維含有材料中のリグニン含有量は、通常0〜40質量%程度、好ましくは0〜10質量%程度である。リグニン含有量の測定は、Klason法により測定することができる。
【0034】
植物の細胞壁の中では、幅4nm程のセルロースミクロフィブリル(シングルセルロースナノファイバー)が最小単位として存在する。これが、植物の基本骨格物質(基本エレメント)である。そして、このセルロースミクロフィブリルが集まって、植物の骨格を形成している。本発明において、「ミクロフィブリル化植物繊維」とは、植物繊維を含む材料(例えば、木材パルプ等)をその繊維をナノサイズレベルまで解きほぐしたものである。
【0035】
本発明の変性ミクロフィブリル化植物繊維(A)は、ミクロフィブリル化植物繊維の水酸基とアルキル、若しくはアルケニル無水コハク酸とが反応してエステル結合している。ここで、アルキル、若しくはアルケニル無水コハク酸は水酸基との反応によりアルキル、若しくはアルケニルコハク酸のハーフエステルとなるため、変性ミクロフィブリル化植物繊維にはカルボン酸基も導入される。
【0036】
アルケニル無水コハク酸としては、より具体的には、炭素数4〜30のオレフィン由来の骨格と無水マレイン酸骨格を持つ化合物が例示される。具体的にはオクチル無水コハク酸、ドデシル無水コハク酸、ヘキサデシル無水コハク酸、オクタデシル無水コハク酸等のアルキル無水コハク酸、ペンテニル無水コハク酸、ヘキセニル無水コハク酸、オクテニル無水コハク酸、デセニル無水コハク酸、ウンデセニル無水コハク酸、ドデセニル無水コハク酸、トリデセニル無水コハク酸、ヘキサデセニルコハク酸無水物、オクタデセニルコハク酸無水物等が例示され、これらは1種類単独でも用いることが出来るし、疎水性や耐水性等の性状を制御することができるという観点から2種類以上を併用して用いても良い。
【0037】
また、アルキル無水コハク酸としては前記のアルケニル無水コハク酸の不飽和結合に水素を付加して得た水添物が例示される。
【0038】
前記アルキル、若しくはアルケニル無水コハク酸による変性ミクロフィブリル化植物繊維(A)の製造法としては、後述する<樹脂組成物の製造方法>の、<製造方法(I)>又は<製造方法(II)>によって製造されることが好ましい。
【0039】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)のエステル置換度(DS)は、親水性の高いミクロフィブリル化植物繊維を樹脂中に均一に分散させたり、ミクロフィブリル化植物繊維の耐水性を向上させる点から、0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0040】
なお、DSは、洗浄により原料として用いたアルキル、若しくはアルケニル無水コハク酸や、それらの加水分解物等の副生成物を除去した後、重量増加率、元素分析、中和滴定法、FT−IR、1H−NMR等の各種分析方法により分析することができる。
【0041】
熱可塑性樹脂(B)としては、オレフィン系樹脂等が挙げられ、オレフィン系樹脂としては、密度の異なる各種ポリエチレン系樹脂、ポリプロピレン系樹脂、ナイロン樹脂、塩化ビニル樹脂、スチレン樹脂、(メタ)アクリル樹脂、ビニルエーテル樹脂、ポリアミド樹脂、ポリカーボネート系樹脂、ポリエステル樹脂、ポリスルホン樹脂、ポリエステル樹脂、トリアセチル化セルロース、ジアセチル化セルロース等のセルロース系樹脂等が挙げられる。
【0042】
また、上記に加え、相溶化剤として上記の熱可塑性樹脂に無水マレイン酸やエポキシ等を付加し極性基を導入した樹脂、例えば無水マレイン酸変性ポリエチレン樹脂、無水マレイン酸変性ポリプロピレン樹脂、市販の各種相溶化剤を併用しても良い。
【0043】
これらの熱可塑性樹脂は、単独で使用してもよく、2種以上の混合樹脂として用いてもよい。また、2種以上の混合樹脂として用いる場合には、無水マレイン酸変性樹脂とその他のポリオレフィン系樹脂を組み合わせた混合樹脂が、高い強度の成形物を比較的得られやすい点において好ましい。
【0044】
無水マレイン酸変性樹脂とその他のポリオレフィン系樹脂を組み合わせた混合樹脂を用いる場合、無水マレイン酸変性樹脂の含有割合としては、熱可塑性樹脂(B)合計中、1〜40質量%程度が好ましく、1〜20質量%程度がより好ましい。混合樹脂として用いる場合の具体例としては、より具体的には、無水マレイン酸変性ポリプロピレン系樹脂とポリエチレン樹脂、又はポリプロピレン樹脂、無水マレイン酸変性ポリエチレン樹脂とポリエチレン樹脂、又はポリプロピレン樹脂が挙げられる。
【0045】
樹脂組成物中の変性ミクロフィブリル化植物繊維(A)の配合量は目的に応じて異なるが、例えば樹脂組成物をそのまま成形し成形材料を作る場合、樹脂組成物樹脂100質量部に対して、1〜80質量部程度が好ましく、2〜70質量部程度がより好ましく、5〜50質量部程度が更に好ましい。変性ミクロフィブリル化植物繊維(A)の配合量が増えると得られた成形物の強度・弾性率が向上するので好ましいが、一方で樹脂の成形時の流動性が落ちる為、複雑な形状に成形するのが困難になる。
【0046】
また、本発明は、前記アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)と、更に無機塩(C)を含有する樹脂組成物中にも関する。前記無機塩(C)を含有することにより、エステル化された変性ミクロフィブリル化植物繊維(A)が無機粒子と相互作用し、樹脂組成物の強度、弾性率等が向上するという効果が得られる。
【0047】
無機塩(C)としては、第1族、又は第2族の金属からなる塩が挙げられ、具体的には、第1族、又は2族の金属からなる酢酸塩、炭酸塩、硫酸塩、硝酸塩、等が挙げられる。第1族の金属としてはナトリウム、カリウムが挙げられ、第2族の金属としては、マグネシウム、カルシウム、ストロンチウム、バリウム等が挙げられ、より具体的には、硫酸マグネシウム、硫酸バリウム、炭酸バリウム、炭酸カリウム、炭酸カルシウム等が挙げられる。無機塩の粒子径は目的に応じて任意に選択することが出来るが、一般的には小さい方が好ましい。これらの中で、炭酸塩が弾性率向上効果が優れるとの点で好ましく、比較的表面積の大きな粒子径/結晶径の粉体が容易に得られることや変性ミクロフィブリル化植物繊維(A)との相互作用しやすいこと、また、得られた成形体の着色が少ないという観点から炭酸カルシウムや炭酸バリウムが更に好ましい。
【0048】
無機塩(C)の含有量は、樹脂組成物100質量部に対して、0.1〜20質量部であり、0.5〜20質量部程度が好ましく、1〜10質量部程度がより好ましく、1〜10質量部程度が更に好ましい。無機塩(C)の含有量を0.1質量部以上に設定することにより、変性ミクロフィブリル化植物繊維(A)との相互作用により、成形体の力学物性を向上させることが出来る。また、無機塩(C)の含有量を20質量部以下に設定することにより、樹脂、及び変性ミクロフィブリル化植物繊維(A)の相対量が少なくならず、強度、弾性率等の力学物性が低下や、成形性の悪化を防ぐことができる。
【0049】
樹脂組成物中に、無機塩(C)を含む場合の変性ミクロフィブリル化植物繊維(A)としては、通常、DSが0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0050】
また本発明の樹脂組成物は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、アルキル、若しくはアルケニル無水コハク酸以外にも、任意の添加剤を含有してもよい。
【0051】
例えば、相溶化剤;界面活性剤;でんぷん類、アルギン酸等の多糖類;ゼラチン、ニカワ、カゼイン等の天然たんぱく質;タンニン、ゼオライト、セラミックス、金属粉末等の無機化合物;着色剤;可塑剤;香料;顔料;流動調整剤;レベリング剤;導電剤;帯電防止剤;紫外線吸収剤;紫外線分散剤;消臭剤等の添加剤を配合してもよい。
【0052】
任意の添加剤の含有割合としては、本発明の効果が損なわれない範囲で適宜含有されてもよいが、例えば、樹脂組成物中0.01〜10質量%程度が好ましく、1〜5質量%程度がより好ましい。
【0053】
<樹脂組成物の製造方法>
樹脂組成物は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程により製造される。
【0054】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)を製造する方法としては、
(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び(2a)工程(1a)によって得られたミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維(A)を得る工程、及び(3)工程(2a)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程(以下、製造方法(I)ともいう)、又は
(1b)植物繊維を、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維(A)を得る工程、
並びに(3)工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程(以下、製造方法(II)ともいう)が挙げられる。
【0055】
<製造方法(I)>
製造方法(I)における工程(1a)に用いられる変性ミクロフィブリル化植物繊維の原料として用いられる植物繊維を含有する材料(植物繊維含有材料)としては、前記で挙げられたものを用いることが好ましい。
【0056】
植物繊維を解繊する方法としては、公知の方法が採用でき、例えば、前記セルロース繊維含有材料の水懸濁液、スラリーをリファイナー、高圧ホモジナイザー、グラインダー、一軸又は多軸混練機、ビーズミル等により機械的に摩砕、ないし叩解することにより解繊する方法が使用できる。必要に応じて、上記の解繊方法を組み合わせて処理してもよい。
【0057】
多軸混練機を用いた場合、入手のしやすさ等の観点から、二軸混練機が好ましい。
【0058】
一軸又は多軸混練機を用いる場合、スクリューの周速の下限値は、通常45m/分程度である。スクリューの周速の下限値は60m/分程度が好ましく、90m/分程度が特に好ましい。また、スクリューの周速の上限値は通常200m/分程度である。スクリューの周速の上限値は150m/分程度が好ましく、100m/分程度が特に好ましい。
【0059】
本発明において使用される混練機のL/D(スクリュー径Dと混練部の長さLの比)は、通常15〜60程度、好ましくは30〜60程度である。
【0060】
一軸又は多軸混練機による解繊時間は、セルロース繊維含有材料の種類、前記混練機のL/D等によっても異なるが、前記のL/Dの範囲内であれば、通常30〜60分程度、好ましくは30〜45分程度である。
【0061】
混練機による解繊に供する回数(パス)は、目的とするミクロフィブリル化植物繊維の繊維径、繊維長、また、前記混練機のL/D等によっても変化するが、通常1〜8回程度、好ましくは1〜4回程度である。パルプを前記混練機による解繊に供する回数(パス)があまりに多くなりすぎると、解繊はより進行するものの、同時に発熱も生じる為、セルロースが着色したり、熱ダメージ(シート強度の低下)につながる。
【0062】
混練機には、スクリューの存在する混練部は1カ所であってもよいし、2カ所以上存在してもよい。
【0063】
また、混練部が2カ所以上存在する場合、各混練部の間に1個又は2個以上のせき止め構造(返し)を有していてもよい。なお、本発明においては、スクリューの周速が45m/分以上と従来のスクリューの周速よりもかなり大きいので、混練機への負荷を軽減する為には、せき止め構造を有しない方がより好ましい。
【0064】
二軸混練機を構成する二本のスクリューの回転方向は異方向、同方向のどちらでもよい。また、二軸混練機を構成する二本のスクリューの噛み合いは、完全噛み合い型、不完全噛み合い型、非噛み合い型があるが、本発明の解繊に用いるものとしては、完全噛み合い型が好ましい。
【0065】
スクリュー長さとスクリュー直径の比(スクリュー長さ/スクリュー直径)は20〜150程度であればよい。具体的な二軸混練機としては、(株)テクノベル製「KZW」、「WDR」、「MFU」、日本製鋼所製「TEX」、東芝機械社製「TEM」、コペリオン社製「ZSK」(株)神戸製鋼所「LCM」等を用いることができる。
【0066】
一軸又は多軸混練機による解繊処理は、植物繊維と分散媒を用いて、懸濁液とし、該懸濁液を解繊することによって行われる。
【0067】
懸濁液を調製する際に用いられる分散媒としては、水を必須成分とするが、その他の任意成分を含む混合分散媒としてもよい。任意成分として含まれる水以外の分散媒としては、具体的にはメタノール、エタノール、n−プロピルアルコール、イソプロピルアルコール、n−ブタノール等の炭素数1〜4のアルコール等が挙げられる。
【0068】
一軸又は多軸混練機による解繊処理における植物繊維と分散媒の混合によって得られる懸濁液中の植物繊維の固形分濃度としては、通常10〜70質量%程度、好ましくは20〜50質量%程度である。植物繊維の固形分濃度を10質量%以上とすることにより、植物繊維を均一に解繊することができ、また、植物繊維の固形分濃度を70質量%以上とすると二軸解繊時にパルプが混練機内で詰まったり、過度なトルクが二軸にかかり二軸混練機の動作が不安定となる為、生産性、及び得られたミクロフィブリル化植物繊維の性状の両面から好ましくない。
【0069】
また、一軸又は多軸混練機による解繊時の温度には特別の制約はないが、通常0〜100℃で行うことが可能であり、特に好ましい温度は0〜50℃である。
【0070】
また、植物繊維をグラインダーにより解繊する場合には、グラインダーは通常上下2枚の砥石の間に植物繊維を含むスラリーが通過するときに発生するせん断力や衝撃力、遠心力により解繊が進行するが、植物繊維の濃度が高すぎると詰まってしまうことや薄すぎるとせん断をうけずにそのまま繊維が通ってしまう為、通常、植物繊維を分散媒で0.1〜5.0質量%、好ましくは0.1〜2%、更に好ましくは0.5〜1.5%程度へ希釈しスラリーとしてグラインダーへ投入し解繊処理を行う。解繊時の負荷によりスラリーの温度が上昇する。1パスで目的の解繊度のミクロフィブリル化植物繊維が得られない場合は繰り返してグラインダー処理を行うことにより目的の解繊度のミクロフィブリル化植物繊維を得ることが出来る。具体的には増幸産業(株)製「スーパーマスコロイダー」や(株)栗田機械製作所の「ピュアファインミル」等の市販の装置を利用することが出来る。
【0071】
植物繊維をビーズミルによって解繊する方法としては、植物繊維と分散媒を用いて懸濁液とし、該懸濁液を解繊する方法が挙げられる。使用される分散媒としては、前記一軸又は多軸混練機による解繊処理に用いられる分散媒と同様のものが用いられる。
【0072】
ビーズミルによる解繊処理において用いられる懸濁液中に含まれる植物繊維の固形分濃度としては、0.3〜2質量%程度が好ましく、0.5〜1.8質量%程度がより好ましく、0.7〜1.5質量%程度が更に好ましい。懸濁液中に含まれる植物繊維の含有割合を、0.3質量%以上に設定することで、ビーズ同士の衝突によるビーズの摩耗が抑制でき、生産性が向上させることができる。また、植物繊維の固形分濃度を2質量%以下に設定することで、粘度上昇が抑制でき、作業効率を向上させることができる。また、ビーズミルベッセル内での詰まり等を防止することができる。
【0073】
工程(1a)によって得られたミクロフィブリル化植物繊維は、そのまま、工程(2a)のアルキル、若しくはアルケニル無水コハク酸によるエステル化を行っても良いが、分散媒がアルキル、若しくはアルケニル無水コハク酸と反応しうる溶媒、例えば水、メタノール、エタノール等のアルコール系溶媒、アンモニア水、エタノールアミン等のアミン系溶媒の場合、アルキル、若しくはアルケニル無水コハク酸とミクロフィブリル化植物繊維との反応が不十分となる為、これらの分散媒を除去してから工程(2a)のアルキル、若しくはアルケニル無水コハク酸によるエステル化をするのが好ましい。
【0074】
分散媒の除去方法としてはこれらの溶媒が除去出来れば特に問わないが、ろ過、圧搾、スプレードライ、減圧留去、加熱留去、凍結乾燥等の公知の方法を用いることが出来る。
【0075】
工程(1a)によって得られたミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維(A)を得る(工程2a)。
【0076】
工程(2a)で用いられるミクロフィブリル化植物繊維は、前記工程(1a)の解繊処理で用いられた分散媒を一部含んでいてもよく、固形分濃度が、0.1〜100質量%程度、好ましくは0.5〜100質量%程度のものが用いられる。ミクロフィブリル化植物繊維の固形分濃度を高くすることでアルキル、若しくはアルケニル無水コハク酸によるミクロフィブリル化植物繊維の変性効率が高くなるが、固形分濃度が高すぎると反応が不均一となったり、反応容器内でミクロフィブリル化植物繊維の凝集物が出来る為、好ましくない。通常、0.2質量%〜50質量%、好ましくは0.4質量%〜40質量%である。
【0077】
アルキル、若しくはアルケニル無水コハク酸としては、前記で挙げられたものを用いることが好ましい。
【0078】
アルキル、若しくはアルケニル無水コハク酸によるミクロフィブリル化植物繊維をエステル化する際のアルキル、若しくはアルケニル無水コハク酸の添加量は、ミクロフィブリル化植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.1〜200モルである。ミクロフィブリル化植物繊維に対してアルキル、若しくはアルケニル無水コハク酸を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限のアルキル、若しくはアルケニル無水コハク酸を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。
【0079】
また、アルキル、若しくはアルケニル無水コハク酸は、ミクロフィブリル化植物繊維と全てエステル化させずに、一部未反応のまま残存していてもよい。
【0080】
ミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。温度が高い方が植物繊維の反応効率が高くなり好ましいが温度が高すぎると一部植物繊維の劣化が起こる為、上記の様な温度範囲とすることが好ましい。
【0081】
エステル化反応は水中で行うことができるが、反応効率が非常に低くなる為、非水系溶媒中で行った方が好ましく、非水系溶媒はアルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒であることが好ましい。具体例としては、非水系溶媒としては塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化溶媒、アセトン、メチルエチルケトン(MEK)等のケトン系溶媒;テトラヒドロフラン(THF)、エチレングリコール、プロピレングリコール、ポリエチレングリコール等のエーテル類のジメチル、ジエチル化物等のエーテル系溶媒;ジメチルホルムアミド、ジメチルアセトアミド、N−メチルピロリドン等のアミド系溶媒、ヘキサン、ヘプタン、ベンゼン、トルエン等の非極性溶媒、又はこれらの混合溶媒である。また、これらから選ばれた2種以上の混合溶媒を使用してもよい。
【0082】
なお、アルコール系溶媒、アミン系溶媒、エステル系溶媒等は、アルキル、若しくはアルケニル無水コハク酸と反応する有機溶媒である為、溶媒として含まないことが好ましい。
【0083】
植物繊維、又はミクロフィブリル化植物繊維とアルキル若しくはアルケニル無水コハク酸とのエステル化反応は、触媒を用いなくても脱水を十分に行えば加熱することによりある程度は進行させることが出来るが、触媒を用いた方がより温和な条件で、かつ高効率でエステル化反応を進行させることが出来るという点でより好ましい。
【0084】
エステル化反応において用いる触媒としては、塩酸、硫酸、酢酸等の酸類、アミン系触媒が挙げられる。酸触媒は通常、水溶液であり、酸触媒の添加によりエステル化に加え、ミクロフィブリル化植物繊維の酸加水分解が起こることがあるので、アルカリ触媒、又はアミン系触媒がより好ましい。アミン系触媒の具体例としては、ピリジン、ジメチルアミノピリジン(DMAP)等のピリジン系化合物、トリエチルアミン、トリメチルアミン、ジアザビシクロオクタン等の非環状、或いは環状三級アミン化合物、等が挙げられ、これらの中で、ピリジン、ジメチルアミノピリジン(DMAP)、ジアザビシクロオクタンが、触媒活性が優れるという観点から好ましい。必要に応じて炭酸カリウム、炭酸ナトリウム等のアルカリ化合物の粉末を触媒として使用しても良いし、アミン系化合物と併用して使用しても良い。
【0085】
アミン系触媒の配合量は、基本的には触媒量であればよいが、例えばピリジンの様に液状のアミン化合物の場合は触媒兼溶媒として多めに使用しても構わない。使用量としては例えば、ミクロフィブリル化植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.001〜10モルである。ミクロフィブリル化植物繊維に対して触媒を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限の触媒を加え、反応時間、温度等を調製することで所定のDSまで反応させることも出来る。反応後の触媒は洗浄、蒸留等により除去することが一般には好ましい。
【0086】
アルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)のDSは、前記で挙げられた範囲であることが好ましい。
【0087】
<製造方法(II)>
製造方法(II)で用いられる植物繊維としては、前記で用いられる植物繊維と同様のものが用いられる。
【0088】
工程(1b)において、植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化することによって、変性植物繊維が得られる。アルキル、若しくはアルケニル無水コハク酸としては、製造方法(I)における工程(2a)で用いられるアルキル、若しくはアルケニル無水コハク酸と同様のものが用いられる。
【0089】
アルキル、若しくはアルケニル無水コハク酸により植物繊維をエステル化する際のアルケニル無水コハク酸の添加量は、植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.1〜200モルである。植物繊維に対してアルキル、若しくはアルケニル無水コハク酸を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限のアルキル、若しくはアルケニル無水コハク酸を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。
【0090】
植物繊維をエステル化する際の溶媒、及び用いられる触媒は、前記<製造方法(I)>と同様のものが用いられる。
【0091】
アミン系触媒の配合量は、基本的には触媒量であればよいが、例えばピリジンの様に液状のアミン化合物の場合は触媒兼溶媒として多めに使用しても構わない。使用量としては例えば、植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.001〜10モルである。ミクロフィブリル化植物繊維に対して触媒を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限の触媒を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。反応後の触媒は洗浄、蒸留等により除去することが一般には好ましい。
【0092】
また、アルキル、若しくはアルケニル無水コハク酸は、植物繊維と全てエステル化させずに、一部未反応のまま残存していてもよい。
【0093】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性植物繊維のエステル置換度(DS)は、親水性の高いミクロフィブリル化植物繊維を樹脂中に均一に分散させたり、ミクロフィブリル化植物繊維の耐水性を向上させる等の観点から、0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0094】
またDSは、前記で挙げられた測定方法と同様の方法によって、測定することができる。
【0095】
植物繊維をアルキル、若しくはアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。温度が高い方が植物繊維の反応効率が高くなり好ましいが温度が高すぎると一部植物繊維の劣化が起こる為、上記の様な温度範囲とすることが好ましい。
【0096】
ミクロフィブリル化植物繊維をアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。
【0097】
前記工程(1b)によって、得られた変性植物繊維は、工程(2b)によって解繊され、変性ミクロフィブリル化植物繊維(A)を得る。
【0098】
解繊方法としては、前記製造方法(I)の工程(1a)によって行われる解繊方法と同様の方法で行われる。
【0099】
<変性ミクロフィブリル化植物繊維>
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、セルロースI型結晶を多く含む。セルロースI型結晶はX線回折における(1 −1 0)格子面(2θ=14.6°)、(2 0 0)格子面(2θ=16.5°)のピークの存在からセルロースI型結晶の存在を確認することが出来る。セルロースI型結晶の割合が高い方が繊維の強度・弾性率が高いことから樹脂の補強材として好ましい。
【0100】
セルロースI型結晶の含有割合としては、変性ミクロフィブリル化植物繊維中、20質量%程度以上が好ましく、30質量%程度以上がより好ましく、40質量%程度以上が更に好ましい。
【0101】
また、変性ミクロフィブリル化植物繊維の比表面積としては、20〜300m2/g程度が好ましく、40〜300m2/g程度がより好ましく、100〜200m2/g程度が更に好ましい。変性ミクロフィブリル化植物繊維の比表面積を高くすることで樹脂組成物の強度向上効果が向上する為、好ましい。また、比表面積が極端に高いと疎水性の樹脂中での凝集も起こりやすくなり、目的とする高強度材料が得られないことがある。この為、変性ミクロフィブリル化植物繊維の比表面積は上記の範囲とすることが好ましい。
【0102】
変性ミクロフィブリル化植物繊維の繊維径は、平均値が通常4〜800nm程度、好ましくは20〜500nm程度、特に好ましくは10〜400nm程度である。
【0103】
なお、変性ミクロフィブリル化植物繊維の繊維径の平均値は、電子顕微鏡の視野内の変性ミクロフィブリル化植物繊維の少なくとも50本以上について測定した時の平均値である。
【0104】
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、余剰な分散媒を除去し、固形分濃度を0.1〜100質量%程度としてもよく、また、完全に乾燥させ、粉末状にしてもよい。
【0105】
乾燥方法としては、凍結乾燥、減圧乾燥、加熱乾燥、静置乾燥、スプレードライ等が挙げられるが、これらの中で、得られた変性ミクロフィブリル化植物繊維を凝集させずに乾燥できるという点において良好であるという観点から、凍結乾燥が好ましい。また、二軸混練機等で加熱攪拌しながら脱水・乾燥する方法は凍結乾燥法よりも変性ミクロフィブリル化植物繊維が凝集しやすく、成型材の強度・弾性率の点で変性ミクロフィブリル化植物繊維の凍結乾燥物を用いた場合よりも劣るとの欠点がある一方で、大量にかつ効率的に変性ミクロフィブリル化植物繊維の乾燥物を得られるという点で優れている。この様に、目的に応じてこれらの方法の何れか、或いは組み合わせて用いるのが好ましい。
【0106】
乾燥方法が凍結乾燥である場合、ミクロフィブリル化植物繊維の溶媒分散物を液体窒素、ドライアイス、氷、冷蔵庫等の冷却装置を用いて冷却させた後に減圧下で溶媒を昇華させることで凍結乾燥する。溶媒としては用いる冷媒、冷却装置で固体となる溶媒であれば公知の溶媒を使用することが出来るが具体的には水、tert−ブタノール等を用いることが出来る。
【0107】
この中でもミクロフィブリル化植物繊維の凝集を防ぐことができるという観点からtert−ブタノールから凍結することが好ましい。また、用いるミクロフィブリル化植物繊維が含水物であった場合、そのまま凍結乾燥するのではなく、前処理としてエタノール、アセトン等により溶媒置換をした後にtert−ブタノールに分散させ凍結乾燥すると乾燥時に変性ミクロフィブリル化植物繊維が凝集することを防げるので好ましい。
【0108】
凍結乾燥においては溶媒が凍結し、かつ減圧にて溶媒が昇華する条件であれば、特に減圧度、温度は任意に選択することが出来る。例えば、ミクロフィブリル化植物繊維のtert−ブタノール分散物から凍結乾燥させる場合は、ミクロフィブリル化植物繊維のtert−ブタノール分散物の入った容器を液体窒素、ドライアイス/メタノール、氷浴等につけ、tert−ブタノールを十分に凍結させた後、真空乾燥機にセットし、0.1Pa以下の減圧度で乾燥させることで変性ミクロフィブリル化植物繊維の凍結乾燥物を得ることが出来る。
【0109】
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、工程(3)によって熱可塑性樹脂(B)と混合することによって、樹脂組成物が製造される。
【0110】
熱可塑性樹脂(B)としては、前記で挙げられたものを用いることが好ましい。
【0111】
また、前記、製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維、及び、工程(3)によって配合される熱可塑性樹脂(B)以外に、任意の添加剤を配合してもよい。添加剤としては、前記で挙げられたものを用いることができる。
【0112】
変性ミクロフィブリル化植物繊維と熱可塑性樹脂(B)、その他の任意の添加剤を混合する方法としては、特に限定されないが、例えば変性ミクロフィブリル化植物繊維を予め乾燥させた後、熱可塑性の粉末、或いはペレット、その他の任意の添加材をミキサー、ブレンダー二軸混練機、ニーダー、ラボプラストミル、ホモジナイザー、高速ホモジナイザー、高圧ホモジナイザー、遊星攪拌装置、3本ロール等の混合、又は攪拌出来る装置で混合、攪拌した後、二軸混練機、ニーダー固層せん断押出し機等の加熱と攪拌が出来る装置で溶融混練する方法や、水等の溶媒を含む変性ミクロフィブリル化植物繊維と熱可塑性樹脂(B)、その他の任意の添加剤を上記の装置で混合した後に脱溶媒と溶融混練を二軸混練機、ニーダー固層せん断押出し機等の加熱と攪拌が出来る装置で脱溶剤と溶融混練を同時に行う方法等を利用することが出来る。熱可塑性樹脂(B)を公知の粉砕機で粉砕した後に混合しても良い。
【0113】
溶融混練における混練温度としては、用いられる熱可塑性樹脂(B)の種類に応じて適宜設定されるが、例えば、高密度ポリエチレンの場合は160〜200℃が好ましく、ポリプロピレンの場合は160〜220℃程度が好ましく、170〜210℃程度がより好ましい。
【0114】
<成形材料及び成形体>
本発明は、前記の樹脂組成物を用いた成形材料にも関する。
【0115】
前記樹脂組成物は、所望の形状に成形され成形材料として用いることができる。成形材料の形状としては、例えば、シート、ペレット、粉末、等が挙げられる。これらの形状を有する成形材料は、例えば金型成形、射出成形、押出成形、中空成形、発泡成形等を用いて得られる。
【0116】
更に本発明は、前記成形材料を成形してなる成形体にも関する。成形の条件は樹脂の成形条件を必要に応じて適宜調整して適用すればよい。
【0117】
本発明の成形体は、ミクロフィブリル化植物繊維含有樹脂成形物が使用されていた分野に加え、より高い機械強度(引っ張り強度等)が要求される分野にも使用できる。例えば、自動車、電車、船舶、飛行機等の輸送機器の内装材、外装材、構造材等;パソコン、テレビ、電話、時計等の電化製品等の筺体、構造材、内部部品等;携帯電話等の移動通信機器等の筺体、構造材、内部部品等;携帯音楽再生機器、映像再生機器、印刷機器、複写機器、スポーツ用品等の筺体、構造材、内部部品等;建築材;文具等の事務機器等、容器、コンテナー等として有効に使用することができる。
【発明の効果】
【0118】
本発明の樹脂組成物は、ミクロフィブリル化植物繊維中のミクロフィブリル化植物繊維の表面が、アルキル、若しくはアルケニル無水コハク酸によってエステル化されている為、ミクロフィブリル化植物繊維が樹脂中で均一に分散される。よって、本発明の樹脂組成物を用いて得られる成形体は、高強度なものが得られるという効果を奏する。
【0119】
また本発明の樹脂組成物の製造方法によると、ミクロフィブリル化植物繊維の表面をアルキル、若しくはアルケニル無水コハク酸によってエステル化させることができ、樹脂中において当該変性ミクロフィブリル化植物繊維を均一に分散させることができる。
【図面の簡単な説明】
【0120】
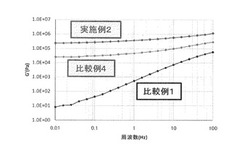
【図1】実施例2、比較例1、及び比較例4の成形物について、周波数に対する貯蔵弾性率(G’)をプロットしたグラフである(140℃)。
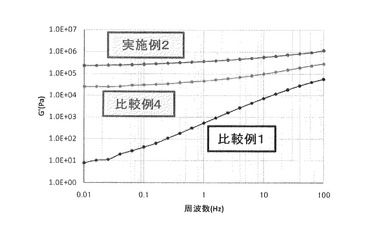
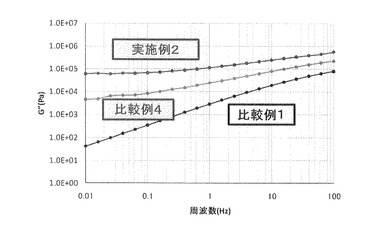
【図2】実施例2、比較例1、及び比較例4の成形物について、周波数に対する損失弾性率(G”)をプロットしたグラフである(140℃)。
【発明を実施するための形態】
【0121】
[実施例]
以下、実施例及び比較例を挙げて本発明を更に詳細に説明するが、本発明はこれらに限定されるものではない。
【0122】
・実施例1
<ビーズミルによるミクロフィブリル化植物繊維(CNF)の調製>
針葉樹漂白クラフトパルプ(NBKP)のスラリー(スラリー濃度:2質量%)をシングルディスクリファイナー(熊谷理機工業(株)製)に通液させ、カナディアンスタンダードフリーネス(CSF)が100ml以下となるまで繰り返しリファイナー処理を行った。次いで得られたスラリーを遠心分離機((株)コクサン製)を用いて20質量%まで濃縮し、NBKP(リファイナー処理)を調整した。
【0123】
次いでNBKP(リファイナー処理、濃度:20質量%)375gに水を加え、全量を10kgとした(スラリー濃度:0.75質量%)。得られたリファイナー処理NBKPスラリーをビーズミル(NVM−2、アイメックス(株)製)で以下の条件で機械的解繊処理を行った。
【0124】
[解繊条件]
ビーズ:ジルコニアビーズ(直径:1mm)
ベッセル容量:2リットル
ビーズ充填量:1216ml(4612g)
回転数:2,000rpm
ベッセル温度:20℃
吐出量:600ml/分。
【0125】
得られたCNFスラリーを吸引ろ過し、固形分濃度を12.5質量%の含水のCNFを得た。
【0126】
<比表面積測定用サンプルの調製>
上記で得られた含水のCNFを8g(固形分1g)サンプリングし、エタノールを加え0.5質量%とし、スターラーで30分間攪拌した後に遠心分離管に移し(株)コクサン製冷却高速遠心機「HR−9」を用いて遠心分離をした。遠心分離後、上澄みをデカンテーションで除いた後、残渣を再度エタノールに分散させ0.5質量%としスターラーで攪拌した後にスラリーを遠心分離した。この操作をエタノール、t-ブタノールで各3回繰り返し、溶媒置換した後、CNFのt-ブタノール分散物(濃度:0.5質量%)200gをナスフラスコに移し、このフラスコを液体窒素浴に漬け全体を凍結させた。次いで、このナスフラスコを凍結乾燥器機(FDU−1200、東京理化器械(株))にセットし凍結乾燥を行った。
【0127】
<CNFの比表面積測定>
得られた凍結乾燥後のCNFを、自動比表面積/細孔径分布測定装置「BELSORP-mini II」(日本ベル(株)製)を用いた窒素ガス吸着法によりBET比表面積を測定したところ138m2/gであった。
【0128】
<アルケニル無水コハク酸(ASA)変性ミクロフィブリル化植物繊維(ASA変性CNF)の調製>
上記の含水のCNF494g(固形分62g)にN−メチルピロリドン(NMP)を247g加え、トリミックスTX−5((株)井上製作所製)に投入した後、攪拌を開始し、40〜50℃で減圧脱水した。次いで、T−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を99.1g、ジメチルアミノピリジン(DMAP)を2.3g、炭酸カリウムを10.57g、NMPを50g加え、62℃で1.5時間反応させた。反応後、アセトン、エタノール、酢酸水、水で順次洗浄し、含水のASA変性CNFを得た。置換度を以下の方法により測定した結果、0.38であった。
【0129】
<ASA(C16)変性CNFの置換度(DS)の算出>
ASA変性の置換度(DS)は、ASA変性CNF中のASAとセルロースのエステル結合を、水酸化ナトリウム溶液中70℃で加熱攪拌することで加水分解した。その後、0.1N塩酸水溶液で逆滴定することで加水分解により生成したASA量を求めた後に算出した。なお、逆滴定の際の指示薬としては、フェノールフタレインを用いた。
【0130】
具体的には、ASA変性CNFの乾燥物を約0.5g、100mlビーカーに精秤し、エタノール15ml、蒸留水5mlを加え室温で30分攪拌した。その後、0.5N水酸化ナトリウム溶液10mlを加え、70℃で15分攪拌した後、室温まで冷却し更に1晩攪拌した。得られた混合液に85%フェノールフタレインのエタノール溶液を数滴加えた後、0.1N塩酸水溶液で逆滴定し、加水分解により生成したASA量を測定した。用いたASA変性CNF量と滴定にて測定したASA量から置換度を算出した。
【0131】
<ASA変性CNFと樹脂との複合化>
洗浄後の含水のASA変性CNF(固形分濃度:20質量%)と無水マレイン酸変性ポリプロピレン(MAPP、東洋紡績(株)製:商品名「トーヨータックPMA H1000P」、酸含有量5.7質量%、メルトフローレート:110g/10分(190℃、2.16kg))、及び高密度ポリエチレン樹脂(HDPE、住友精化(株)製:商品名「フロービーズHE3040」、融点:130℃、平均粒子径11μm)をミキサーにて1分間攪拌した。
【0132】
配合後の固形分の含有割合は下記の通りである。
【0133】
ASA変性CNF:17.6質量%(CNF由来(10質量%)+ASA由来部分(7.6質量%))
樹脂:82.4質量%(MAPP:(4.3質量%)+HDPE(78.1質量%))。
【0134】
得られた樹脂組成物を(株)テクノベル製の二軸混練機(KZW、スクリュー径:15mm、L/D:45、スクリュー回転数:200rpm、せき止め構造:0個、処理速度200g/時)にて98℃で2パスし脱水と混合を行った。次いで、上記の混合物を140℃で1パスさせ、得られた溶融混練物をペレタイザー((株)テクノベル製)を用いてペレット化した後、射出成型機(NPX7−1F、日精樹脂(株)製)に投入し、ダンベル型の試験片(厚さ1mm)を得た。なお、加熱筒(シリンダー)温度は160℃、金型温度は40℃の条件下で成形を行った。
【0135】
得られたダンベル試験片の引張り試験をインストロン3365型万能試験機(インストロンジャパンリミテッド製)を用いて測定した。測定結果を表1に示す。
【0136】
・実施例2
実施例1において、炭酸カリウムの量を2倍モル量とした以外は、実施例1と同様に行い、ASA変性CNF、及びそれを含む成形物を得た。ASA変性CNFのDSは0.44であった。成形物の各成分の配合量、及び成形物の引張り試験結果を表1に示す。
【0137】
・実施例3
実施例1において炭酸カリウムの量を2倍モル量とし、<ASA変性CNFの調製>において、71℃で1時間反応させた以外は、実施例1と同様の方法によりASA変性CNFの調製を行い、ASA変性CNF、及びそれを含む成形物を得た。ASA変性CNFのDSは0.77であった。成形物の各成分の配合量、及び成形物の引張り試験結果を表1に示す。
【0138】
・実施例4〜5
実施例1において、ASA変性CNF、及び樹脂(MAPP、HDPE)以外に、更に炭酸カルシウム(和光純薬工業(株)製、試薬1級グレード、粒子径6μm)を表1に示す含有割合で添加した。それ以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0139】
尚、炭酸カルシウムの粒子径は蒸留水で1wt%に分散させた後、レーザ回折/散乱式粒子径分布測定装置((株)堀場製作所製の「LA950V」)にて測定した。得られたデータの内、メジアン径を「粒子径」として記載した。
【0140】
・比較例1
実施例1において、樹脂(HDPE(住友精化(株)製:商品名「フロービーズHE3040」))のみを用いた以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0141】
・比較例2及び3
実施例1において、樹脂(HDPE(住友精化(株)製:商品名「フロービーズHE3040」))、及び炭酸カルシウムを表1に示す含有割合で添加した以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0142】
・比較例4
実施例1において得られたCNFを、固形分濃度20質量%まで濃縮した後、ASA変性を行わず、そのまま樹脂(MAPP及びHDPE)と複合化した以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0143】
・比較例5
実施例1と同様にしてCNFの水懸濁液(0.75質量%)を得た。
【0144】
この懸濁液5000g(固形分37.5g)にT−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を74.5g加え、1時間攪拌した後、ブフナー漏斗を用いて吸引濾過した。得られた含水のASA吸着CNFを0.5gサンプリングし、上記と同様に洗浄した後にDSを測定したところ、全く反応していなかった。
【0145】
残りの含水のASA吸着CNF(CNF:10質量%、吸着したASA:4質量%)とMAPP及びHDPEを表1に記載した配合で混合した以外は表1と同様にして成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0146】
【表1】

【0147】
<貯蔵弾性率及び損失弾性率測定>
直径3cm、厚さ1.5mmの孔のあいた金型に実施例2、比較例1、及び比較例4で得られたペレット約1gを入れ、(株)神藤金属工業製の卓上型テストプレスA YSR−5HC型で140℃/5分間加熱・プレスすることにより直径3cm、厚さ1.5mmの円盤状の成形体を得た。
【0148】
得られた円盤状の成形体をティー・エイ・インスツルメント・ジャパン(株)製の動的粘弾性測定装置 「AR−G2」にセットし円盤状の成形体の貯蔵弾性率(G’)及び損失弾性率(G”)を窒素雰囲気下で測定した。
測定条件
・測定方法:2cmパラレルプレート
・測定範囲:0.01〜100Hz
・測定ひずみ:0.1%
・測定温度:140℃
図1に周波数に対する貯蔵弾性率(G’)をプロットしたグラフを示し、図2に周波数に対する損失弾性率(G”)をプロットしたグラフを示す。
【0149】
<考察>
表1から分かるように、ASA変性CNFを樹脂組成物に配合することで得られた成形物の引張り強度は、樹脂単独、及び未変性のCNFを配合した樹脂と比較し、強度、及び弾性率のいずれも大幅に向上した。更に、図1、及び図2から分かるように、樹脂に未変性CNFを配合するだけでも、低周波域での貯蔵弾性率、損失弾性率が向上しているが、その効果はASA変性したCNFを配合することにより更に向上したことが分かる。これらのことからCNFをASA変性することにより、ASA変性CNFの樹脂中での分散性や樹脂とASA変性CNFの界面が補強されている為、成形材料の力学物性が向上したものと思われる。
【0150】
また、ASA変性CNF、樹脂(MAPP、HDPE)に、更に炭酸カルシウムを加えた成形材料も、強度、及び弾性率が向上した。
【0151】
一方、樹脂に炭酸カルシウムのみを併用した場合、樹脂単独と比較し、強度、及び弾性率が共に低下した。更に、炭酸カルシウムの配合量を2.2質量%、4.3質量%と増加しても強度、弾性率が向上していないことから、単に炭酸カルシウムを配合した効果が出ているわけではなく、ASA変性CNFと炭酸カルシウムとの間で何らかの相互作用が働いているものと推察される。
【0152】
更に比較例5より、ASAを吸着したCNFと樹脂を混合して得た成形体は強度、弾性率共に向上していない。このことからASAはCNFに単に吸着するだけでは不十分で、ASAはCNFと共有結合していることが好ましいことが分かる。
【0153】
・実施例6
実施例1で得られたNBKP(リファイナー処理、固形分濃度:20質量%)をミキサー((株)愛工舎製作所製「ケンミックスKM−800」)で粗粉砕(処理時間1時間)し、そぼろ状とした後に二軸混練機((株)テクノベル製のKZW)に入れ、解繊処理によりCNFを得た。二軸混練機による解繊条件は、以下の通りである。
【0154】
[解繊条件]
スクリュー直径:15mm
スクリュー回転数:400rpm(スクリュー周速:18.8m/分)
解繊時間:150gのパルプを500g/hr〜600g/hrの処理条件で解繊した。原料を投入してからCNFが得られる迄の時間は15分間であった。
【0155】
L/D:45
解繊処理に供した回数:1回(1パス)
せき止め構造:2個。
【0156】
得られたCNFを1gサンプリングし、実施例1と同様にして溶媒置換し、得られた凍結乾燥後のCNFを、窒素ガス吸着法(日本ベル(株)製)により比表面積を測定したところ70m2/gであった。
【0157】
残りの二軸混練機で解繊して得たCNFに、エタノールを加え1質量%とした後、吸引濾過した。これを3回繰り返した後、エタノールを含む湿潤CNF(CNF濃度:30質量%)を金属バット上に厚さ0.5〜1cm程度となるように広げ、減圧乾燥機(YAMATO社製「ADP300」)に入れ、24時間減圧乾燥(温度105℃、真空度:0.1kPa以下)し、二軸解繊CNFの乾燥物を得た。
【0158】
<ASA変性CNFの調製>
二軸解繊CNFの乾燥物5gをN−メチルピロリドンに分散させ、固形分濃度が2質量%のスラリーとした。このスラリーを攪拌装置のついた500mlフラスコに移し、T−NS136(無水コハク酸以外の炭素数が8のASA、星光PMC(株)製)19.4g、触媒としてピリジンを14.6g加え、120℃で2時間加熱攪拌した後、冷却、洗浄しASA変性CNFを得た
得られたASA変性CNFを実施例1と同様にして凍結乾燥し、ASA変性CNFの凍結乾燥物を得た。
【0159】
得られたASA変性CNFの凍結乾燥物、及び樹脂の含有割合を表2に示す割合で混合した後、小型二軸混練機((株)テクノベル製「ULTnano15TW」)で140℃、5分間混練・循環させた。混練・循環後、弁を開放し樹脂を取りだし、細かく裁断してペレット化した。
【0160】
小型二軸混練機へのサンプル投入は1回で最大20g程度であるため、この操作を3回繰り返して合計で約50gの樹脂組成物を得た。この組成物を実施例1と同様にして成形を行い、引張り試験を行った。引張り試験結果を表2に示す。
【0161】
・実施例7〜8
実施例6のT−NS136(無水コハク酸以外の炭素数が8のASA、星光PMC(株)製)をT−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)、又はT−NS146(無水コハク酸以外の炭素数が18のASA、星光PMC(株)製)とし、表2に示すDSとなるまで反応時間を延ばした以外は、実施例6と同様にしてASA変性CNFを得た。それ以降の操作は実施例6と同様に行い、鎖長の異なるASA変性CNFを配合した樹脂組成物を得た。ASA変性CNFのDS、各成分の配合量、成形体の引張り強度結果を表2に示す。
【0162】
・実施例9
実施例1で調製したNBKP(リファイナー処理、固形分:濃度20質量%)をエタノールに分散させて2質量%、1800g(固形分36g)スラリーとした後、吸引ろ過をした。これを3回、アセトンへの分散を3回繰り返し、溶媒置換をした。得られたアセトンを含有するNBKPをNMPに分散させNBKPの固形分濃度が3質量%のスラリー1200gを得た。
【0163】
次いで、T−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を215.8g、DMAPを1.4g、炭酸カリウムを30.7g加え、70℃で5時間反応させた。反応後、アセトン、エタノール、酢酸水、水で順次洗浄し、含水のASA変性CNFを得た。DSを測定した結果、0.29であった。
【0164】
得られたASA変性CNFに蒸留水を加え、総量を4,800gとした後、実施例1と同様にしてビーズミル処理を行った。
【0165】
得られたASA変性CNFを実施例6と同様に凍結乾燥、及び樹脂との複合化を行い、ASA変性CNFを配合した樹脂組成物を得た。各成分の配合量、成形体の引張り強度結果を表2に示す。
【0166】
・比較例6
実施例6で記載したASA変性CNFの凍結乾燥方法と同様にして、実施例6で調製した二軸解繊CNFの凍結乾燥物を得た。ASA変性CNFの凍結乾燥物の代わりに上記で調製した未変性の二軸解繊CNFの凍結乾燥物を用いた以外は、実施例6と同様にして樹脂組成物を得た。樹脂組成物を用いて作製した成形物の引張り強度を表2に示す。
【0167】
【表2】
【0168】
<考察>
鎖長の異なるASAを用いて調製したASA変性CNFを用いた場合でも、樹脂中での植物繊維の分散性や界面親和性が向上したため、樹脂組成物の力学物性が向上したと思われる。使用する樹脂や用途に応じて最適な置換度やASAを任意に設定することが可能である。
【0169】
・実施例10〜13
実施例4においてDSが0.56となるまで反応を進行させて得られたASA変性CNFを用いると共に、樹脂に加え、炭酸カルシウムの代わりに硫酸マグネシウム(和光純薬(株)製、試薬特級)、硫酸バリウム(和光純薬(株)製、試薬1級、粒子径:2.1μm)、炭酸バリウム(和光純薬(株)製、試薬、粒子径:0.24μm)、又は炭酸カリウム(和光純薬(株)製、試薬特級)を用いた以外は、実施例4と同様に行い成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表3に示す。尚、無機塩の粒子径は実施例4と同様にして測定した。
【0170】
なお、得られた成形物の外観は硫酸マグネシウム、硫酸バリウム、炭酸バリウムを用いた場合は無機塩を用いない場合(例えば、実施例1)や炭酸カルシウムを用いた場合(例えば実施例5)、樹脂のみ(例えば、比較例1、3)と同様に白〜淡黄色であったが、炭酸カリウムを用いた場合のみ濃褐色であった。
【0171】
・比較例7
実施例1で製造したビーズミル解繊CNFの含水物を実施例9と同様の方法で溶媒置換を行った。この溶媒置換物を無水酢酸の量を調整した以外は文献(Cellulose 9: 361-367, 2002)に記載の方法に従ってアセチル化変性CNFを得た。得られたアセチル化変性CNFのDSは0.39であった。
【0172】
ASA変性CNFの代わりに、上記で得たアセチル化変性CNFを用い、アセチル化変性CNFと樹脂を表3に記載の配合量とした以外は、実施例1と同様の方法によりアセチル化変性CNFを含む成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表3に示す。
【0173】
【表3】
【0174】
<考察>
変性CNFに硫酸マグネシウム、硫酸バリウム、炭酸バリウム、炭酸カリウムを用いた場合は無機塩として炭酸カルシウムを用いた場合と同様に高強度、高弾性率の成形体が得られた。無機塩として炭酸カルシウムを用いた場合と同様に変性CNFのカルボキシル基との相互作用がその一因であると思われる。弾性率向上効果としては炭酸カリウムが優れていたが、強度や外観の点で他の無機物よりは劣る。用途や成形条件等に応じてこれらの無機塩を使い分けることにより所望の成形材料、成形体を得る事が出来る。
【0175】
また、セルロース系繊維の疎水化方法として公知なアセチル化した場合では成形体の強度、弾性率の向上は認められていない。単に疎水化するだけでは成形体の強度、弾性率が向上しないことが分かる。このことからもCNFをASA変性することにより樹脂との親和性が向上していると推測される。
【0176】
・実施例14
実施例1においてDSが0.56となるまで反応を進行させて得られたASA変性CNFを42.4質量%用いると共に、このASA変性CNFと樹脂の配合を表4に示す含有割合とした以外は、実施例1と同様の方法により、成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表4に示す。
【0177】
なお、ASA変性CNF中のCNF由来の成分比は20質量%であり比較例8と同様である。
【0178】
・比較例8
未変性のCNF、及び樹脂の配合を表4に記載の通りとした以外は、比較例4と同様に行い成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表4に示す。
【0179】
【表4】
【0180】
<考察>
実施例14と比較例8はいずれもCNF純分は同じ20質量%であるが、実施例14は比較例8よりも強度、弾性率ともに優れていた。ミクロフィブリル化植物繊維をASA変性することにより樹脂との界面が補強されることや樹脂中での分散性が改善された為と思われる。
【技術分野】
【0001】
本発明は、アルキル、若しくはアルケニル無水コハク酸によって変性されたミクロフィブリル化植物繊維を含む樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体に関する。
【背景技術】
【0002】
従来、樹脂の強度等の物性を向上させるため、充填剤を用いて樹脂を強化することが知られている。例えば、セルロース繊維をミクロフィブリル化して、繊維径がナノオーダーにまで微細化されたミクロフィブリル化植物繊維(ナノファイバー)等を充填剤とし、当該ナノファイバーを樹脂中に配合させることによって得られる成形材料は、質量が軽く、かつ高強度であるという点から、非常に有用である。
【0003】
しかしながら、ミクロフィブリル化植物繊維は非常に凝集力が強く、一般的に樹脂との相溶性が悪いため、ミクロフィブリル化繊維が均一に分散した均一な樹脂成形材料を得ることが困難である。そのため、得られる成形材料は、機械的強度が十分に発揮できないという問題があった。このような課題に対して、ミクロフィブリル化植物繊維の表面を、化学修飾剤等によって変性処理し、樹脂中での分散性を向上させようとする試みがなされている。
【0004】
例えば、特許文献1では、化学的に修飾されたセルロース繊維の集合体に、マトリクス材料を含浸させてなる複合材料について開示されている。このような化学修飾剤によって修飾されたセルロース繊維とマトリクス材料とを複合化させる方法(シート含浸法)によって、高品質の透明基板を得ることを目的としている。しかしながら、特許文献1ではシート含浸法である為に、樹脂が液状である必要があり、通常固体であるオレフィン系樹脂との複合化に用いることは難しい。また、セルロース繊維の解繊度を上げるほどち密なシートが出来やすく、内部まで樹脂を均一に含浸することが困難な為、シート内部、表面で樹脂濃度にバラツキが生じやすい。この為、機械的強度が十分に得られないという問題があった。
【0005】
特許文献2では、無水酢酸、無水酪酸、塩化アセチル、塩化ブチリル、酢酸等の有機化合物によって表面の水酸基がエステル化されたセルロースミクロフィブリルについて開示されている。また特許文献2では、該セルロースミクロフィブリルを有機溶媒に溶解した後、有機溶媒に溶解した樹脂と混合後、溶媒を除去することによって、複合材料を形成させることが記載されている。このような方法によって複合材料を得る場合、セルロースミクロフィブリルを、有機溶媒に十分に溶解させる必要がある。セルロース繊維を有機溶媒に溶解させるためには、セルロースミクロフィブリルの疎水変性を十分に行う必要がある。このような疎水変性は、セルロース繊維のI型結晶の破壊を招き、高強度の複合材料を製造することが困難であった。
【0006】
特許文献3では、イオン性液体と有機溶媒の混合溶媒中で無水酢酸、無水マレイン酸、無水コハク酸等の酸無水物によって変性されたセルロースナノファイバーとポリ乳酸をラボミルで混練後、プレス成形することによって成形材料を得る旨開示されている。このような変性セルロースナノファイバーは、ポリ乳酸のような親水性の高い樹脂では適用が可能であるが、ポリオレフィン系樹脂等の疎水性の高い樹脂の場合では、均一に混合することが困難であった。
【0007】
また特許文献4では、セルロースの水酸基の一部に無水マレイン酸や、無水コハク酸等の多塩基酸無水物によりエステル化し、カルボキシル基を導入した後、高圧ホモジナイザー処理、ニーダー、多軸押出し機等による微細繊維化処理によりナノ繊維を得る方法、及びそのナノ繊維と樹脂からなる複合材料について開示されている。該ナノ繊維は、ミクロフィブリル表面に導入された負の電荷を有するカルボキシル基の存在によりミクロフィブリル間の反発力を誘引し、分散体中での安定な分散が可能となっている。しかしながら、オレフィン系樹脂のような疎水性の高い樹脂中では十分な分散性が得られず、機械的強度において未だ改善の余地がある。
【0008】
更に特許文献5では、疎水化されたセルロース系繊維と、合成樹脂とを含有する樹脂組成物について開示されており、疎水化されたセルロース系繊維を調製するために、実際の実施例では、アルキルケテンダイマーのような疎水化剤がクラフトパルプの高圧ホモジナイザー処理物100gに対して0.01g(100ppm)用いられている。また、同実施例では濃度約0.5%のクラフトパルプ水分散物にアルキルケテンダイマーを添加しているため、添加したアルキルケテンダイマーの一部が加水分解してケトンになる副反応が起こる。そのため、実際にセルロースと共有結合しているアルキルケテンダイマーは100ppmよりかなり少ないと思われる。また、セルロース系繊維の解繊度が高い場合、この量では解繊されたセルロース表面を疎水化できるほどに覆うことが出来ず、結果としてオレフィン系樹脂のような疎水性の高い樹脂中では十分な分散性が得られず、機械的強度において未だ改善の余地がある。
【0009】
このように、ミクロフィブリル化植物繊維を含む成形材料において、樹脂としてポリエチレン、ポリプロピレン等の熱可塑性樹脂のような疎水性の高い樹脂を用いた場合、ミクロフィブリル化植物繊維の分散性が悪く、更なる機械的強度を得ることが非常に困難であった。
【先行技術文献】
【特許文献】
【0010】
【特許文献1】特開2007−51266号公報
【特許文献2】特開平11−513425号公報
【特許文献3】特開2010−104768号公報
【特許文献4】特開2009−293167号公報
【特許文献5】特開2010−106251号公報
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明は、オレフィン系樹脂のような疎水性の高い樹脂を用いたミクロフィブリル化植物繊維を含む樹脂組成物において、樹脂組成物内でのミクロフィブリル化植物繊維の分散性が良好で、得られる成形材料においては機械的強度を向上させることのできる、アルキル、若しくはアルケニル無水コハク酸によって変性されたミクロフィブリル化植物繊維を含む樹脂組成物及びその製造方法、並びに該樹脂組成物を用いた成形材料、及び成形体を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明者らは、上記課題を解決すべく鋭意研究を重ねた結果、アルキル、若しくはアルケニル無水コハク酸でエステル化することによって得られる変性ミクロフィブリル化植物繊維をオレフィン系樹脂に混合させることによって、変性ミクロフィブリル化植物繊維の分散性を向上させ、更には、得られる成形材料の機械的強度を向上することができることを見出した。
【0013】
本発明はこのような知見に基づき、更に鋭意検討を重ねて完成した発明である。すなわち、本発明は下記項に示す樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体を提供する。
【0014】
項1.アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び
熱可塑性樹脂(B)を含有する樹脂組成物であって、
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.05〜2.0である樹脂組成物。
【0015】
項2.アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、
熱可塑性樹脂(B)、及び
無機塩(C)を含有する樹脂組成物。
【0016】
項3.変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.1〜2.0である項1〜2の何れか1項に記載の樹脂組成物。
【0017】
項4.変性ミクロフィブリル化植物繊維(A)の含有量が、樹脂組成物100質量部に対して、1〜80質量部である項1〜3の何れか1項に記載の樹脂組成物。
【0018】
項5.無機塩(C)が、第2族の金属からなる塩である項2〜4の何れか1項に記載の樹脂組成物。
【0019】
項6.無機塩(C)の含有量が、樹脂組成物100質量部に対して0.1〜20質量部である項2〜5の何れか1項に記載の樹脂組成物。
【0020】
項7.(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び
(2a)工程(1a)によって得られたミクロフィブリル化植物繊維を触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維を得る工程、又は
(1b)植物繊維を、触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び
(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維を得る工程、
並びに
(3)工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程を含む樹脂組成物の製造方法。
【0021】
項8.工程(3)が、工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、及び無機塩(C)を混合する工程である項7に記載の樹脂組成物の製造方法。
【0022】
項9.工程(2a)又は工程(1b)におけるエステル化が、アルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒よりなる少なくとも1種の溶媒の存在下で行われる項7又は8に記載の樹脂組成物の製造方法。
【0023】
項10.工程(1a)又は工程(2b)における解繊が、ボールミル、ビーズミル、ブレンダー、グラインダー、リファイナー、高圧衝突分散機、ホモジナイザー、高圧ホモジナイザー、一軸又は多軸混錬機からなる群より選ばれる少なくとも1種による機械的解繊である項7〜9の何れか1項に記載の樹脂組成物の製造方法。
【0024】
項11.項7〜10の何れか1項に記載の製造方法で得られた変性ミクロフィブリル化植物繊維。
【0025】
項12.項1〜6の何れか1項に記載の樹脂組成物を用いた成形材料。
【0026】
項13.項7〜10の何れか1項で製造された樹脂組成物を用いた成形材料。
【0027】
項14.項12又は13に記載の成形材料を成形してなる成形体。
【0028】
以下、本願発明の樹脂組成物、及びその製造方法、並びに当該樹脂組成物を用いた成形材料及び成形体について、詳述する。
【0029】
<樹脂組成物>
本発明は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)、及び熱可塑性樹脂(B)を含有する樹脂組成物に関する。
【0030】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)の原料として用いられる植物繊維を含有する材料(植物繊維含有材料)としては、木材、竹、麻、ジュート、ケナフ、綿、ビート、農産物残廃物、布といった天然植物繊維原料から得られるパルプ、レーヨンやセロファン等の再生セルロース繊維等が挙げられる。特に、パルプが好ましい原材料として挙げられる。
【0031】
前記パルプとしては、植物原料を化学的、若しくは機械的に、又は両者を併用してパルプ化することで得られるケミカルパルプ(クラフトパルプ(KP)、亜硫酸パルプ(SP))、セミケミカルパルプ(SCP)、ケミグランドパルプ(CGP)、ケミメカニカルパルプ(CMP)、砕木パルプ(GP)、リファイナーメカニカルパルプ(RMP)、サーモメカニカルパルプ(TMP)、ケミサーモメカニカルパルプ(CTMP)、及びこれらの植物繊維を主成分とする脱墨古紙パルプ、段ボール古紙パルプ、雑誌古紙パルプが好ましいものとして挙げられる。これらの原材料は、必要に応じ、脱リグニン、又は漂白を行い、当該植物繊維中のリグニン量を調整することができる。
【0032】
これらのパルプの中でも、繊維の強度が強い針葉樹由来の各種クラフトパルプ(針葉樹未漂白クラフトパルプ(以下、NUKPということがある)、針葉樹酸素晒し未漂白クラフトパルプ(以下、NOKPということがある)、針葉樹漂白クラフトパルプ(以下、NBKPということがある))が特に好ましい。
【0033】
原料となる植物繊維は主にセルロース、ヘミセルロース、リグニンから構成される。植物繊維含有材料中のリグニン含有量は、通常0〜40質量%程度、好ましくは0〜10質量%程度である。リグニン含有量の測定は、Klason法により測定することができる。
【0034】
植物の細胞壁の中では、幅4nm程のセルロースミクロフィブリル(シングルセルロースナノファイバー)が最小単位として存在する。これが、植物の基本骨格物質(基本エレメント)である。そして、このセルロースミクロフィブリルが集まって、植物の骨格を形成している。本発明において、「ミクロフィブリル化植物繊維」とは、植物繊維を含む材料(例えば、木材パルプ等)をその繊維をナノサイズレベルまで解きほぐしたものである。
【0035】
本発明の変性ミクロフィブリル化植物繊維(A)は、ミクロフィブリル化植物繊維の水酸基とアルキル、若しくはアルケニル無水コハク酸とが反応してエステル結合している。ここで、アルキル、若しくはアルケニル無水コハク酸は水酸基との反応によりアルキル、若しくはアルケニルコハク酸のハーフエステルとなるため、変性ミクロフィブリル化植物繊維にはカルボン酸基も導入される。
【0036】
アルケニル無水コハク酸としては、より具体的には、炭素数4〜30のオレフィン由来の骨格と無水マレイン酸骨格を持つ化合物が例示される。具体的にはオクチル無水コハク酸、ドデシル無水コハク酸、ヘキサデシル無水コハク酸、オクタデシル無水コハク酸等のアルキル無水コハク酸、ペンテニル無水コハク酸、ヘキセニル無水コハク酸、オクテニル無水コハク酸、デセニル無水コハク酸、ウンデセニル無水コハク酸、ドデセニル無水コハク酸、トリデセニル無水コハク酸、ヘキサデセニルコハク酸無水物、オクタデセニルコハク酸無水物等が例示され、これらは1種類単独でも用いることが出来るし、疎水性や耐水性等の性状を制御することができるという観点から2種類以上を併用して用いても良い。
【0037】
また、アルキル無水コハク酸としては前記のアルケニル無水コハク酸の不飽和結合に水素を付加して得た水添物が例示される。
【0038】
前記アルキル、若しくはアルケニル無水コハク酸による変性ミクロフィブリル化植物繊維(A)の製造法としては、後述する<樹脂組成物の製造方法>の、<製造方法(I)>又は<製造方法(II)>によって製造されることが好ましい。
【0039】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)のエステル置換度(DS)は、親水性の高いミクロフィブリル化植物繊維を樹脂中に均一に分散させたり、ミクロフィブリル化植物繊維の耐水性を向上させる点から、0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0040】
なお、DSは、洗浄により原料として用いたアルキル、若しくはアルケニル無水コハク酸や、それらの加水分解物等の副生成物を除去した後、重量増加率、元素分析、中和滴定法、FT−IR、1H−NMR等の各種分析方法により分析することができる。
【0041】
熱可塑性樹脂(B)としては、オレフィン系樹脂等が挙げられ、オレフィン系樹脂としては、密度の異なる各種ポリエチレン系樹脂、ポリプロピレン系樹脂、ナイロン樹脂、塩化ビニル樹脂、スチレン樹脂、(メタ)アクリル樹脂、ビニルエーテル樹脂、ポリアミド樹脂、ポリカーボネート系樹脂、ポリエステル樹脂、ポリスルホン樹脂、ポリエステル樹脂、トリアセチル化セルロース、ジアセチル化セルロース等のセルロース系樹脂等が挙げられる。
【0042】
また、上記に加え、相溶化剤として上記の熱可塑性樹脂に無水マレイン酸やエポキシ等を付加し極性基を導入した樹脂、例えば無水マレイン酸変性ポリエチレン樹脂、無水マレイン酸変性ポリプロピレン樹脂、市販の各種相溶化剤を併用しても良い。
【0043】
これらの熱可塑性樹脂は、単独で使用してもよく、2種以上の混合樹脂として用いてもよい。また、2種以上の混合樹脂として用いる場合には、無水マレイン酸変性樹脂とその他のポリオレフィン系樹脂を組み合わせた混合樹脂が、高い強度の成形物を比較的得られやすい点において好ましい。
【0044】
無水マレイン酸変性樹脂とその他のポリオレフィン系樹脂を組み合わせた混合樹脂を用いる場合、無水マレイン酸変性樹脂の含有割合としては、熱可塑性樹脂(B)合計中、1〜40質量%程度が好ましく、1〜20質量%程度がより好ましい。混合樹脂として用いる場合の具体例としては、より具体的には、無水マレイン酸変性ポリプロピレン系樹脂とポリエチレン樹脂、又はポリプロピレン樹脂、無水マレイン酸変性ポリエチレン樹脂とポリエチレン樹脂、又はポリプロピレン樹脂が挙げられる。
【0045】
樹脂組成物中の変性ミクロフィブリル化植物繊維(A)の配合量は目的に応じて異なるが、例えば樹脂組成物をそのまま成形し成形材料を作る場合、樹脂組成物樹脂100質量部に対して、1〜80質量部程度が好ましく、2〜70質量部程度がより好ましく、5〜50質量部程度が更に好ましい。変性ミクロフィブリル化植物繊維(A)の配合量が増えると得られた成形物の強度・弾性率が向上するので好ましいが、一方で樹脂の成形時の流動性が落ちる為、複雑な形状に成形するのが困難になる。
【0046】
また、本発明は、前記アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)と、更に無機塩(C)を含有する樹脂組成物中にも関する。前記無機塩(C)を含有することにより、エステル化された変性ミクロフィブリル化植物繊維(A)が無機粒子と相互作用し、樹脂組成物の強度、弾性率等が向上するという効果が得られる。
【0047】
無機塩(C)としては、第1族、又は第2族の金属からなる塩が挙げられ、具体的には、第1族、又は2族の金属からなる酢酸塩、炭酸塩、硫酸塩、硝酸塩、等が挙げられる。第1族の金属としてはナトリウム、カリウムが挙げられ、第2族の金属としては、マグネシウム、カルシウム、ストロンチウム、バリウム等が挙げられ、より具体的には、硫酸マグネシウム、硫酸バリウム、炭酸バリウム、炭酸カリウム、炭酸カルシウム等が挙げられる。無機塩の粒子径は目的に応じて任意に選択することが出来るが、一般的には小さい方が好ましい。これらの中で、炭酸塩が弾性率向上効果が優れるとの点で好ましく、比較的表面積の大きな粒子径/結晶径の粉体が容易に得られることや変性ミクロフィブリル化植物繊維(A)との相互作用しやすいこと、また、得られた成形体の着色が少ないという観点から炭酸カルシウムや炭酸バリウムが更に好ましい。
【0048】
無機塩(C)の含有量は、樹脂組成物100質量部に対して、0.1〜20質量部であり、0.5〜20質量部程度が好ましく、1〜10質量部程度がより好ましく、1〜10質量部程度が更に好ましい。無機塩(C)の含有量を0.1質量部以上に設定することにより、変性ミクロフィブリル化植物繊維(A)との相互作用により、成形体の力学物性を向上させることが出来る。また、無機塩(C)の含有量を20質量部以下に設定することにより、樹脂、及び変性ミクロフィブリル化植物繊維(A)の相対量が少なくならず、強度、弾性率等の力学物性が低下や、成形性の悪化を防ぐことができる。
【0049】
樹脂組成物中に、無機塩(C)を含む場合の変性ミクロフィブリル化植物繊維(A)としては、通常、DSが0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0050】
また本発明の樹脂組成物は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、アルキル、若しくはアルケニル無水コハク酸以外にも、任意の添加剤を含有してもよい。
【0051】
例えば、相溶化剤;界面活性剤;でんぷん類、アルギン酸等の多糖類;ゼラチン、ニカワ、カゼイン等の天然たんぱく質;タンニン、ゼオライト、セラミックス、金属粉末等の無機化合物;着色剤;可塑剤;香料;顔料;流動調整剤;レベリング剤;導電剤;帯電防止剤;紫外線吸収剤;紫外線分散剤;消臭剤等の添加剤を配合してもよい。
【0052】
任意の添加剤の含有割合としては、本発明の効果が損なわれない範囲で適宜含有されてもよいが、例えば、樹脂組成物中0.01〜10質量%程度が好ましく、1〜5質量%程度がより好ましい。
【0053】
<樹脂組成物の製造方法>
樹脂組成物は、アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程により製造される。
【0054】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)を製造する方法としては、
(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び(2a)工程(1a)によって得られたミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維(A)を得る工程、及び(3)工程(2a)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程(以下、製造方法(I)ともいう)、又は
(1b)植物繊維を、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維(A)を得る工程、
並びに(3)工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程(以下、製造方法(II)ともいう)が挙げられる。
【0055】
<製造方法(I)>
製造方法(I)における工程(1a)に用いられる変性ミクロフィブリル化植物繊維の原料として用いられる植物繊維を含有する材料(植物繊維含有材料)としては、前記で挙げられたものを用いることが好ましい。
【0056】
植物繊維を解繊する方法としては、公知の方法が採用でき、例えば、前記セルロース繊維含有材料の水懸濁液、スラリーをリファイナー、高圧ホモジナイザー、グラインダー、一軸又は多軸混練機、ビーズミル等により機械的に摩砕、ないし叩解することにより解繊する方法が使用できる。必要に応じて、上記の解繊方法を組み合わせて処理してもよい。
【0057】
多軸混練機を用いた場合、入手のしやすさ等の観点から、二軸混練機が好ましい。
【0058】
一軸又は多軸混練機を用いる場合、スクリューの周速の下限値は、通常45m/分程度である。スクリューの周速の下限値は60m/分程度が好ましく、90m/分程度が特に好ましい。また、スクリューの周速の上限値は通常200m/分程度である。スクリューの周速の上限値は150m/分程度が好ましく、100m/分程度が特に好ましい。
【0059】
本発明において使用される混練機のL/D(スクリュー径Dと混練部の長さLの比)は、通常15〜60程度、好ましくは30〜60程度である。
【0060】
一軸又は多軸混練機による解繊時間は、セルロース繊維含有材料の種類、前記混練機のL/D等によっても異なるが、前記のL/Dの範囲内であれば、通常30〜60分程度、好ましくは30〜45分程度である。
【0061】
混練機による解繊に供する回数(パス)は、目的とするミクロフィブリル化植物繊維の繊維径、繊維長、また、前記混練機のL/D等によっても変化するが、通常1〜8回程度、好ましくは1〜4回程度である。パルプを前記混練機による解繊に供する回数(パス)があまりに多くなりすぎると、解繊はより進行するものの、同時に発熱も生じる為、セルロースが着色したり、熱ダメージ(シート強度の低下)につながる。
【0062】
混練機には、スクリューの存在する混練部は1カ所であってもよいし、2カ所以上存在してもよい。
【0063】
また、混練部が2カ所以上存在する場合、各混練部の間に1個又は2個以上のせき止め構造(返し)を有していてもよい。なお、本発明においては、スクリューの周速が45m/分以上と従来のスクリューの周速よりもかなり大きいので、混練機への負荷を軽減する為には、せき止め構造を有しない方がより好ましい。
【0064】
二軸混練機を構成する二本のスクリューの回転方向は異方向、同方向のどちらでもよい。また、二軸混練機を構成する二本のスクリューの噛み合いは、完全噛み合い型、不完全噛み合い型、非噛み合い型があるが、本発明の解繊に用いるものとしては、完全噛み合い型が好ましい。
【0065】
スクリュー長さとスクリュー直径の比(スクリュー長さ/スクリュー直径)は20〜150程度であればよい。具体的な二軸混練機としては、(株)テクノベル製「KZW」、「WDR」、「MFU」、日本製鋼所製「TEX」、東芝機械社製「TEM」、コペリオン社製「ZSK」(株)神戸製鋼所「LCM」等を用いることができる。
【0066】
一軸又は多軸混練機による解繊処理は、植物繊維と分散媒を用いて、懸濁液とし、該懸濁液を解繊することによって行われる。
【0067】
懸濁液を調製する際に用いられる分散媒としては、水を必須成分とするが、その他の任意成分を含む混合分散媒としてもよい。任意成分として含まれる水以外の分散媒としては、具体的にはメタノール、エタノール、n−プロピルアルコール、イソプロピルアルコール、n−ブタノール等の炭素数1〜4のアルコール等が挙げられる。
【0068】
一軸又は多軸混練機による解繊処理における植物繊維と分散媒の混合によって得られる懸濁液中の植物繊維の固形分濃度としては、通常10〜70質量%程度、好ましくは20〜50質量%程度である。植物繊維の固形分濃度を10質量%以上とすることにより、植物繊維を均一に解繊することができ、また、植物繊維の固形分濃度を70質量%以上とすると二軸解繊時にパルプが混練機内で詰まったり、過度なトルクが二軸にかかり二軸混練機の動作が不安定となる為、生産性、及び得られたミクロフィブリル化植物繊維の性状の両面から好ましくない。
【0069】
また、一軸又は多軸混練機による解繊時の温度には特別の制約はないが、通常0〜100℃で行うことが可能であり、特に好ましい温度は0〜50℃である。
【0070】
また、植物繊維をグラインダーにより解繊する場合には、グラインダーは通常上下2枚の砥石の間に植物繊維を含むスラリーが通過するときに発生するせん断力や衝撃力、遠心力により解繊が進行するが、植物繊維の濃度が高すぎると詰まってしまうことや薄すぎるとせん断をうけずにそのまま繊維が通ってしまう為、通常、植物繊維を分散媒で0.1〜5.0質量%、好ましくは0.1〜2%、更に好ましくは0.5〜1.5%程度へ希釈しスラリーとしてグラインダーへ投入し解繊処理を行う。解繊時の負荷によりスラリーの温度が上昇する。1パスで目的の解繊度のミクロフィブリル化植物繊維が得られない場合は繰り返してグラインダー処理を行うことにより目的の解繊度のミクロフィブリル化植物繊維を得ることが出来る。具体的には増幸産業(株)製「スーパーマスコロイダー」や(株)栗田機械製作所の「ピュアファインミル」等の市販の装置を利用することが出来る。
【0071】
植物繊維をビーズミルによって解繊する方法としては、植物繊維と分散媒を用いて懸濁液とし、該懸濁液を解繊する方法が挙げられる。使用される分散媒としては、前記一軸又は多軸混練機による解繊処理に用いられる分散媒と同様のものが用いられる。
【0072】
ビーズミルによる解繊処理において用いられる懸濁液中に含まれる植物繊維の固形分濃度としては、0.3〜2質量%程度が好ましく、0.5〜1.8質量%程度がより好ましく、0.7〜1.5質量%程度が更に好ましい。懸濁液中に含まれる植物繊維の含有割合を、0.3質量%以上に設定することで、ビーズ同士の衝突によるビーズの摩耗が抑制でき、生産性が向上させることができる。また、植物繊維の固形分濃度を2質量%以下に設定することで、粘度上昇が抑制でき、作業効率を向上させることができる。また、ビーズミルベッセル内での詰まり等を防止することができる。
【0073】
工程(1a)によって得られたミクロフィブリル化植物繊維は、そのまま、工程(2a)のアルキル、若しくはアルケニル無水コハク酸によるエステル化を行っても良いが、分散媒がアルキル、若しくはアルケニル無水コハク酸と反応しうる溶媒、例えば水、メタノール、エタノール等のアルコール系溶媒、アンモニア水、エタノールアミン等のアミン系溶媒の場合、アルキル、若しくはアルケニル無水コハク酸とミクロフィブリル化植物繊維との反応が不十分となる為、これらの分散媒を除去してから工程(2a)のアルキル、若しくはアルケニル無水コハク酸によるエステル化をするのが好ましい。
【0074】
分散媒の除去方法としてはこれらの溶媒が除去出来れば特に問わないが、ろ過、圧搾、スプレードライ、減圧留去、加熱留去、凍結乾燥等の公知の方法を用いることが出来る。
【0075】
工程(1a)によって得られたミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維(A)を得る(工程2a)。
【0076】
工程(2a)で用いられるミクロフィブリル化植物繊維は、前記工程(1a)の解繊処理で用いられた分散媒を一部含んでいてもよく、固形分濃度が、0.1〜100質量%程度、好ましくは0.5〜100質量%程度のものが用いられる。ミクロフィブリル化植物繊維の固形分濃度を高くすることでアルキル、若しくはアルケニル無水コハク酸によるミクロフィブリル化植物繊維の変性効率が高くなるが、固形分濃度が高すぎると反応が不均一となったり、反応容器内でミクロフィブリル化植物繊維の凝集物が出来る為、好ましくない。通常、0.2質量%〜50質量%、好ましくは0.4質量%〜40質量%である。
【0077】
アルキル、若しくはアルケニル無水コハク酸としては、前記で挙げられたものを用いることが好ましい。
【0078】
アルキル、若しくはアルケニル無水コハク酸によるミクロフィブリル化植物繊維をエステル化する際のアルキル、若しくはアルケニル無水コハク酸の添加量は、ミクロフィブリル化植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.1〜200モルである。ミクロフィブリル化植物繊維に対してアルキル、若しくはアルケニル無水コハク酸を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限のアルキル、若しくはアルケニル無水コハク酸を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。
【0079】
また、アルキル、若しくはアルケニル無水コハク酸は、ミクロフィブリル化植物繊維と全てエステル化させずに、一部未反応のまま残存していてもよい。
【0080】
ミクロフィブリル化植物繊維をアルキル、若しくはアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。温度が高い方が植物繊維の反応効率が高くなり好ましいが温度が高すぎると一部植物繊維の劣化が起こる為、上記の様な温度範囲とすることが好ましい。
【0081】
エステル化反応は水中で行うことができるが、反応効率が非常に低くなる為、非水系溶媒中で行った方が好ましく、非水系溶媒はアルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒であることが好ましい。具体例としては、非水系溶媒としては塩化メチレン、クロロホルム、四塩化炭素等のハロゲン化溶媒、アセトン、メチルエチルケトン(MEK)等のケトン系溶媒;テトラヒドロフラン(THF)、エチレングリコール、プロピレングリコール、ポリエチレングリコール等のエーテル類のジメチル、ジエチル化物等のエーテル系溶媒;ジメチルホルムアミド、ジメチルアセトアミド、N−メチルピロリドン等のアミド系溶媒、ヘキサン、ヘプタン、ベンゼン、トルエン等の非極性溶媒、又はこれらの混合溶媒である。また、これらから選ばれた2種以上の混合溶媒を使用してもよい。
【0082】
なお、アルコール系溶媒、アミン系溶媒、エステル系溶媒等は、アルキル、若しくはアルケニル無水コハク酸と反応する有機溶媒である為、溶媒として含まないことが好ましい。
【0083】
植物繊維、又はミクロフィブリル化植物繊維とアルキル若しくはアルケニル無水コハク酸とのエステル化反応は、触媒を用いなくても脱水を十分に行えば加熱することによりある程度は進行させることが出来るが、触媒を用いた方がより温和な条件で、かつ高効率でエステル化反応を進行させることが出来るという点でより好ましい。
【0084】
エステル化反応において用いる触媒としては、塩酸、硫酸、酢酸等の酸類、アミン系触媒が挙げられる。酸触媒は通常、水溶液であり、酸触媒の添加によりエステル化に加え、ミクロフィブリル化植物繊維の酸加水分解が起こることがあるので、アルカリ触媒、又はアミン系触媒がより好ましい。アミン系触媒の具体例としては、ピリジン、ジメチルアミノピリジン(DMAP)等のピリジン系化合物、トリエチルアミン、トリメチルアミン、ジアザビシクロオクタン等の非環状、或いは環状三級アミン化合物、等が挙げられ、これらの中で、ピリジン、ジメチルアミノピリジン(DMAP)、ジアザビシクロオクタンが、触媒活性が優れるという観点から好ましい。必要に応じて炭酸カリウム、炭酸ナトリウム等のアルカリ化合物の粉末を触媒として使用しても良いし、アミン系化合物と併用して使用しても良い。
【0085】
アミン系触媒の配合量は、基本的には触媒量であればよいが、例えばピリジンの様に液状のアミン化合物の場合は触媒兼溶媒として多めに使用しても構わない。使用量としては例えば、ミクロフィブリル化植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.001〜10モルである。ミクロフィブリル化植物繊維に対して触媒を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限の触媒を加え、反応時間、温度等を調製することで所定のDSまで反応させることも出来る。反応後の触媒は洗浄、蒸留等により除去することが一般には好ましい。
【0086】
アルケニル無水コハク酸によって変性された変性ミクロフィブリル化植物繊維(A)のDSは、前記で挙げられた範囲であることが好ましい。
【0087】
<製造方法(II)>
製造方法(II)で用いられる植物繊維としては、前記で用いられる植物繊維と同様のものが用いられる。
【0088】
工程(1b)において、植物繊維をアルキル、若しくはアルケニル無水コハク酸でエステル化することによって、変性植物繊維が得られる。アルキル、若しくはアルケニル無水コハク酸としては、製造方法(I)における工程(2a)で用いられるアルキル、若しくはアルケニル無水コハク酸と同様のものが用いられる。
【0089】
アルキル、若しくはアルケニル無水コハク酸により植物繊維をエステル化する際のアルケニル無水コハク酸の添加量は、植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.1〜200モルである。植物繊維に対してアルキル、若しくはアルケニル無水コハク酸を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限のアルキル、若しくはアルケニル無水コハク酸を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。
【0090】
植物繊維をエステル化する際の溶媒、及び用いられる触媒は、前記<製造方法(I)>と同様のものが用いられる。
【0091】
アミン系触媒の配合量は、基本的には触媒量であればよいが、例えばピリジンの様に液状のアミン化合物の場合は触媒兼溶媒として多めに使用しても構わない。使用量としては例えば、植物繊維を構成するセルロースのグルコース単位1モルに対して通常、0.001〜10モルである。ミクロフィブリル化植物繊維に対して触媒を過剰に加えた後、所定のDSまで反応させた後、反応を停止させることも出来るし、必要最小限の触媒を加え、反応時間、温度、触媒量等を調製することで所定のDSまで反応させることも出来る。反応後の触媒は洗浄、蒸留等により除去することが一般には好ましい。
【0092】
また、アルキル、若しくはアルケニル無水コハク酸は、植物繊維と全てエステル化させずに、一部未反応のまま残存していてもよい。
【0093】
アルキル、若しくはアルケニル無水コハク酸によって変性された変性植物繊維のエステル置換度(DS)は、親水性の高いミクロフィブリル化植物繊維を樹脂中に均一に分散させたり、ミクロフィブリル化植物繊維の耐水性を向上させる等の観点から、0.05〜2.0程度が好ましく、0.1〜2.0程度がより好ましく、0.1〜0.8程度が更に好ましい。
【0094】
またDSは、前記で挙げられた測定方法と同様の方法によって、測定することができる。
【0095】
植物繊維をアルキル、若しくはアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。温度が高い方が植物繊維の反応効率が高くなり好ましいが温度が高すぎると一部植物繊維の劣化が起こる為、上記の様な温度範囲とすることが好ましい。
【0096】
ミクロフィブリル化植物繊維をアルケニル無水コハク酸によりエステル化する際の反応温度としては、20〜160℃程度が好ましく、40〜120℃程度がより好ましく、60〜100℃程度が更に好ましい。
【0097】
前記工程(1b)によって、得られた変性植物繊維は、工程(2b)によって解繊され、変性ミクロフィブリル化植物繊維(A)を得る。
【0098】
解繊方法としては、前記製造方法(I)の工程(1a)によって行われる解繊方法と同様の方法で行われる。
【0099】
<変性ミクロフィブリル化植物繊維>
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、セルロースI型結晶を多く含む。セルロースI型結晶はX線回折における(1 −1 0)格子面(2θ=14.6°)、(2 0 0)格子面(2θ=16.5°)のピークの存在からセルロースI型結晶の存在を確認することが出来る。セルロースI型結晶の割合が高い方が繊維の強度・弾性率が高いことから樹脂の補強材として好ましい。
【0100】
セルロースI型結晶の含有割合としては、変性ミクロフィブリル化植物繊維中、20質量%程度以上が好ましく、30質量%程度以上がより好ましく、40質量%程度以上が更に好ましい。
【0101】
また、変性ミクロフィブリル化植物繊維の比表面積としては、20〜300m2/g程度が好ましく、40〜300m2/g程度がより好ましく、100〜200m2/g程度が更に好ましい。変性ミクロフィブリル化植物繊維の比表面積を高くすることで樹脂組成物の強度向上効果が向上する為、好ましい。また、比表面積が極端に高いと疎水性の樹脂中での凝集も起こりやすくなり、目的とする高強度材料が得られないことがある。この為、変性ミクロフィブリル化植物繊維の比表面積は上記の範囲とすることが好ましい。
【0102】
変性ミクロフィブリル化植物繊維の繊維径は、平均値が通常4〜800nm程度、好ましくは20〜500nm程度、特に好ましくは10〜400nm程度である。
【0103】
なお、変性ミクロフィブリル化植物繊維の繊維径の平均値は、電子顕微鏡の視野内の変性ミクロフィブリル化植物繊維の少なくとも50本以上について測定した時の平均値である。
【0104】
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、余剰な分散媒を除去し、固形分濃度を0.1〜100質量%程度としてもよく、また、完全に乾燥させ、粉末状にしてもよい。
【0105】
乾燥方法としては、凍結乾燥、減圧乾燥、加熱乾燥、静置乾燥、スプレードライ等が挙げられるが、これらの中で、得られた変性ミクロフィブリル化植物繊維を凝集させずに乾燥できるという点において良好であるという観点から、凍結乾燥が好ましい。また、二軸混練機等で加熱攪拌しながら脱水・乾燥する方法は凍結乾燥法よりも変性ミクロフィブリル化植物繊維が凝集しやすく、成型材の強度・弾性率の点で変性ミクロフィブリル化植物繊維の凍結乾燥物を用いた場合よりも劣るとの欠点がある一方で、大量にかつ効率的に変性ミクロフィブリル化植物繊維の乾燥物を得られるという点で優れている。この様に、目的に応じてこれらの方法の何れか、或いは組み合わせて用いるのが好ましい。
【0106】
乾燥方法が凍結乾燥である場合、ミクロフィブリル化植物繊維の溶媒分散物を液体窒素、ドライアイス、氷、冷蔵庫等の冷却装置を用いて冷却させた後に減圧下で溶媒を昇華させることで凍結乾燥する。溶媒としては用いる冷媒、冷却装置で固体となる溶媒であれば公知の溶媒を使用することが出来るが具体的には水、tert−ブタノール等を用いることが出来る。
【0107】
この中でもミクロフィブリル化植物繊維の凝集を防ぐことができるという観点からtert−ブタノールから凍結することが好ましい。また、用いるミクロフィブリル化植物繊維が含水物であった場合、そのまま凍結乾燥するのではなく、前処理としてエタノール、アセトン等により溶媒置換をした後にtert−ブタノールに分散させ凍結乾燥すると乾燥時に変性ミクロフィブリル化植物繊維が凝集することを防げるので好ましい。
【0108】
凍結乾燥においては溶媒が凍結し、かつ減圧にて溶媒が昇華する条件であれば、特に減圧度、温度は任意に選択することが出来る。例えば、ミクロフィブリル化植物繊維のtert−ブタノール分散物から凍結乾燥させる場合は、ミクロフィブリル化植物繊維のtert−ブタノール分散物の入った容器を液体窒素、ドライアイス/メタノール、氷浴等につけ、tert−ブタノールを十分に凍結させた後、真空乾燥機にセットし、0.1Pa以下の減圧度で乾燥させることで変性ミクロフィブリル化植物繊維の凍結乾燥物を得ることが出来る。
【0109】
前記製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維は、工程(3)によって熱可塑性樹脂(B)と混合することによって、樹脂組成物が製造される。
【0110】
熱可塑性樹脂(B)としては、前記で挙げられたものを用いることが好ましい。
【0111】
また、前記、製造方法(I)又は(II)によって得られた変性ミクロフィブリル化植物繊維、及び、工程(3)によって配合される熱可塑性樹脂(B)以外に、任意の添加剤を配合してもよい。添加剤としては、前記で挙げられたものを用いることができる。
【0112】
変性ミクロフィブリル化植物繊維と熱可塑性樹脂(B)、その他の任意の添加剤を混合する方法としては、特に限定されないが、例えば変性ミクロフィブリル化植物繊維を予め乾燥させた後、熱可塑性の粉末、或いはペレット、その他の任意の添加材をミキサー、ブレンダー二軸混練機、ニーダー、ラボプラストミル、ホモジナイザー、高速ホモジナイザー、高圧ホモジナイザー、遊星攪拌装置、3本ロール等の混合、又は攪拌出来る装置で混合、攪拌した後、二軸混練機、ニーダー固層せん断押出し機等の加熱と攪拌が出来る装置で溶融混練する方法や、水等の溶媒を含む変性ミクロフィブリル化植物繊維と熱可塑性樹脂(B)、その他の任意の添加剤を上記の装置で混合した後に脱溶媒と溶融混練を二軸混練機、ニーダー固層せん断押出し機等の加熱と攪拌が出来る装置で脱溶剤と溶融混練を同時に行う方法等を利用することが出来る。熱可塑性樹脂(B)を公知の粉砕機で粉砕した後に混合しても良い。
【0113】
溶融混練における混練温度としては、用いられる熱可塑性樹脂(B)の種類に応じて適宜設定されるが、例えば、高密度ポリエチレンの場合は160〜200℃が好ましく、ポリプロピレンの場合は160〜220℃程度が好ましく、170〜210℃程度がより好ましい。
【0114】
<成形材料及び成形体>
本発明は、前記の樹脂組成物を用いた成形材料にも関する。
【0115】
前記樹脂組成物は、所望の形状に成形され成形材料として用いることができる。成形材料の形状としては、例えば、シート、ペレット、粉末、等が挙げられる。これらの形状を有する成形材料は、例えば金型成形、射出成形、押出成形、中空成形、発泡成形等を用いて得られる。
【0116】
更に本発明は、前記成形材料を成形してなる成形体にも関する。成形の条件は樹脂の成形条件を必要に応じて適宜調整して適用すればよい。
【0117】
本発明の成形体は、ミクロフィブリル化植物繊維含有樹脂成形物が使用されていた分野に加え、より高い機械強度(引っ張り強度等)が要求される分野にも使用できる。例えば、自動車、電車、船舶、飛行機等の輸送機器の内装材、外装材、構造材等;パソコン、テレビ、電話、時計等の電化製品等の筺体、構造材、内部部品等;携帯電話等の移動通信機器等の筺体、構造材、内部部品等;携帯音楽再生機器、映像再生機器、印刷機器、複写機器、スポーツ用品等の筺体、構造材、内部部品等;建築材;文具等の事務機器等、容器、コンテナー等として有効に使用することができる。
【発明の効果】
【0118】
本発明の樹脂組成物は、ミクロフィブリル化植物繊維中のミクロフィブリル化植物繊維の表面が、アルキル、若しくはアルケニル無水コハク酸によってエステル化されている為、ミクロフィブリル化植物繊維が樹脂中で均一に分散される。よって、本発明の樹脂組成物を用いて得られる成形体は、高強度なものが得られるという効果を奏する。
【0119】
また本発明の樹脂組成物の製造方法によると、ミクロフィブリル化植物繊維の表面をアルキル、若しくはアルケニル無水コハク酸によってエステル化させることができ、樹脂中において当該変性ミクロフィブリル化植物繊維を均一に分散させることができる。
【図面の簡単な説明】
【0120】
【図1】実施例2、比較例1、及び比較例4の成形物について、周波数に対する貯蔵弾性率(G’)をプロットしたグラフである(140℃)。
【図2】実施例2、比較例1、及び比較例4の成形物について、周波数に対する損失弾性率(G”)をプロットしたグラフである(140℃)。
【発明を実施するための形態】
【0121】
[実施例]
以下、実施例及び比較例を挙げて本発明を更に詳細に説明するが、本発明はこれらに限定されるものではない。
【0122】
・実施例1
<ビーズミルによるミクロフィブリル化植物繊維(CNF)の調製>
針葉樹漂白クラフトパルプ(NBKP)のスラリー(スラリー濃度:2質量%)をシングルディスクリファイナー(熊谷理機工業(株)製)に通液させ、カナディアンスタンダードフリーネス(CSF)が100ml以下となるまで繰り返しリファイナー処理を行った。次いで得られたスラリーを遠心分離機((株)コクサン製)を用いて20質量%まで濃縮し、NBKP(リファイナー処理)を調整した。
【0123】
次いでNBKP(リファイナー処理、濃度:20質量%)375gに水を加え、全量を10kgとした(スラリー濃度:0.75質量%)。得られたリファイナー処理NBKPスラリーをビーズミル(NVM−2、アイメックス(株)製)で以下の条件で機械的解繊処理を行った。
【0124】
[解繊条件]
ビーズ:ジルコニアビーズ(直径:1mm)
ベッセル容量:2リットル
ビーズ充填量:1216ml(4612g)
回転数:2,000rpm
ベッセル温度:20℃
吐出量:600ml/分。
【0125】
得られたCNFスラリーを吸引ろ過し、固形分濃度を12.5質量%の含水のCNFを得た。
【0126】
<比表面積測定用サンプルの調製>
上記で得られた含水のCNFを8g(固形分1g)サンプリングし、エタノールを加え0.5質量%とし、スターラーで30分間攪拌した後に遠心分離管に移し(株)コクサン製冷却高速遠心機「HR−9」を用いて遠心分離をした。遠心分離後、上澄みをデカンテーションで除いた後、残渣を再度エタノールに分散させ0.5質量%としスターラーで攪拌した後にスラリーを遠心分離した。この操作をエタノール、t-ブタノールで各3回繰り返し、溶媒置換した後、CNFのt-ブタノール分散物(濃度:0.5質量%)200gをナスフラスコに移し、このフラスコを液体窒素浴に漬け全体を凍結させた。次いで、このナスフラスコを凍結乾燥器機(FDU−1200、東京理化器械(株))にセットし凍結乾燥を行った。
【0127】
<CNFの比表面積測定>
得られた凍結乾燥後のCNFを、自動比表面積/細孔径分布測定装置「BELSORP-mini II」(日本ベル(株)製)を用いた窒素ガス吸着法によりBET比表面積を測定したところ138m2/gであった。
【0128】
<アルケニル無水コハク酸(ASA)変性ミクロフィブリル化植物繊維(ASA変性CNF)の調製>
上記の含水のCNF494g(固形分62g)にN−メチルピロリドン(NMP)を247g加え、トリミックスTX−5((株)井上製作所製)に投入した後、攪拌を開始し、40〜50℃で減圧脱水した。次いで、T−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を99.1g、ジメチルアミノピリジン(DMAP)を2.3g、炭酸カリウムを10.57g、NMPを50g加え、62℃で1.5時間反応させた。反応後、アセトン、エタノール、酢酸水、水で順次洗浄し、含水のASA変性CNFを得た。置換度を以下の方法により測定した結果、0.38であった。
【0129】
<ASA(C16)変性CNFの置換度(DS)の算出>
ASA変性の置換度(DS)は、ASA変性CNF中のASAとセルロースのエステル結合を、水酸化ナトリウム溶液中70℃で加熱攪拌することで加水分解した。その後、0.1N塩酸水溶液で逆滴定することで加水分解により生成したASA量を求めた後に算出した。なお、逆滴定の際の指示薬としては、フェノールフタレインを用いた。
【0130】
具体的には、ASA変性CNFの乾燥物を約0.5g、100mlビーカーに精秤し、エタノール15ml、蒸留水5mlを加え室温で30分攪拌した。その後、0.5N水酸化ナトリウム溶液10mlを加え、70℃で15分攪拌した後、室温まで冷却し更に1晩攪拌した。得られた混合液に85%フェノールフタレインのエタノール溶液を数滴加えた後、0.1N塩酸水溶液で逆滴定し、加水分解により生成したASA量を測定した。用いたASA変性CNF量と滴定にて測定したASA量から置換度を算出した。
【0131】
<ASA変性CNFと樹脂との複合化>
洗浄後の含水のASA変性CNF(固形分濃度:20質量%)と無水マレイン酸変性ポリプロピレン(MAPP、東洋紡績(株)製:商品名「トーヨータックPMA H1000P」、酸含有量5.7質量%、メルトフローレート:110g/10分(190℃、2.16kg))、及び高密度ポリエチレン樹脂(HDPE、住友精化(株)製:商品名「フロービーズHE3040」、融点:130℃、平均粒子径11μm)をミキサーにて1分間攪拌した。
【0132】
配合後の固形分の含有割合は下記の通りである。
【0133】
ASA変性CNF:17.6質量%(CNF由来(10質量%)+ASA由来部分(7.6質量%))
樹脂:82.4質量%(MAPP:(4.3質量%)+HDPE(78.1質量%))。
【0134】
得られた樹脂組成物を(株)テクノベル製の二軸混練機(KZW、スクリュー径:15mm、L/D:45、スクリュー回転数:200rpm、せき止め構造:0個、処理速度200g/時)にて98℃で2パスし脱水と混合を行った。次いで、上記の混合物を140℃で1パスさせ、得られた溶融混練物をペレタイザー((株)テクノベル製)を用いてペレット化した後、射出成型機(NPX7−1F、日精樹脂(株)製)に投入し、ダンベル型の試験片(厚さ1mm)を得た。なお、加熱筒(シリンダー)温度は160℃、金型温度は40℃の条件下で成形を行った。
【0135】
得られたダンベル試験片の引張り試験をインストロン3365型万能試験機(インストロンジャパンリミテッド製)を用いて測定した。測定結果を表1に示す。
【0136】
・実施例2
実施例1において、炭酸カリウムの量を2倍モル量とした以外は、実施例1と同様に行い、ASA変性CNF、及びそれを含む成形物を得た。ASA変性CNFのDSは0.44であった。成形物の各成分の配合量、及び成形物の引張り試験結果を表1に示す。
【0137】
・実施例3
実施例1において炭酸カリウムの量を2倍モル量とし、<ASA変性CNFの調製>において、71℃で1時間反応させた以外は、実施例1と同様の方法によりASA変性CNFの調製を行い、ASA変性CNF、及びそれを含む成形物を得た。ASA変性CNFのDSは0.77であった。成形物の各成分の配合量、及び成形物の引張り試験結果を表1に示す。
【0138】
・実施例4〜5
実施例1において、ASA変性CNF、及び樹脂(MAPP、HDPE)以外に、更に炭酸カルシウム(和光純薬工業(株)製、試薬1級グレード、粒子径6μm)を表1に示す含有割合で添加した。それ以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0139】
尚、炭酸カルシウムの粒子径は蒸留水で1wt%に分散させた後、レーザ回折/散乱式粒子径分布測定装置((株)堀場製作所製の「LA950V」)にて測定した。得られたデータの内、メジアン径を「粒子径」として記載した。
【0140】
・比較例1
実施例1において、樹脂(HDPE(住友精化(株)製:商品名「フロービーズHE3040」))のみを用いた以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0141】
・比較例2及び3
実施例1において、樹脂(HDPE(住友精化(株)製:商品名「フロービーズHE3040」))、及び炭酸カルシウムを表1に示す含有割合で添加した以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0142】
・比較例4
実施例1において得られたCNFを、固形分濃度20質量%まで濃縮した後、ASA変性を行わず、そのまま樹脂(MAPP及びHDPE)と複合化した以外は、実施例1と同様の方法により成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0143】
・比較例5
実施例1と同様にしてCNFの水懸濁液(0.75質量%)を得た。
【0144】
この懸濁液5000g(固形分37.5g)にT−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を74.5g加え、1時間攪拌した後、ブフナー漏斗を用いて吸引濾過した。得られた含水のASA吸着CNFを0.5gサンプリングし、上記と同様に洗浄した後にDSを測定したところ、全く反応していなかった。
【0145】
残りの含水のASA吸着CNF(CNF:10質量%、吸着したASA:4質量%)とMAPP及びHDPEを表1に記載した配合で混合した以外は表1と同様にして成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表1に示す。
【0146】
【表1】
【0147】
<貯蔵弾性率及び損失弾性率測定>
直径3cm、厚さ1.5mmの孔のあいた金型に実施例2、比較例1、及び比較例4で得られたペレット約1gを入れ、(株)神藤金属工業製の卓上型テストプレスA YSR−5HC型で140℃/5分間加熱・プレスすることにより直径3cm、厚さ1.5mmの円盤状の成形体を得た。
【0148】
得られた円盤状の成形体をティー・エイ・インスツルメント・ジャパン(株)製の動的粘弾性測定装置 「AR−G2」にセットし円盤状の成形体の貯蔵弾性率(G’)及び損失弾性率(G”)を窒素雰囲気下で測定した。
測定条件
・測定方法:2cmパラレルプレート
・測定範囲:0.01〜100Hz
・測定ひずみ:0.1%
・測定温度:140℃
図1に周波数に対する貯蔵弾性率(G’)をプロットしたグラフを示し、図2に周波数に対する損失弾性率(G”)をプロットしたグラフを示す。
【0149】
<考察>
表1から分かるように、ASA変性CNFを樹脂組成物に配合することで得られた成形物の引張り強度は、樹脂単独、及び未変性のCNFを配合した樹脂と比較し、強度、及び弾性率のいずれも大幅に向上した。更に、図1、及び図2から分かるように、樹脂に未変性CNFを配合するだけでも、低周波域での貯蔵弾性率、損失弾性率が向上しているが、その効果はASA変性したCNFを配合することにより更に向上したことが分かる。これらのことからCNFをASA変性することにより、ASA変性CNFの樹脂中での分散性や樹脂とASA変性CNFの界面が補強されている為、成形材料の力学物性が向上したものと思われる。
【0150】
また、ASA変性CNF、樹脂(MAPP、HDPE)に、更に炭酸カルシウムを加えた成形材料も、強度、及び弾性率が向上した。
【0151】
一方、樹脂に炭酸カルシウムのみを併用した場合、樹脂単独と比較し、強度、及び弾性率が共に低下した。更に、炭酸カルシウムの配合量を2.2質量%、4.3質量%と増加しても強度、弾性率が向上していないことから、単に炭酸カルシウムを配合した効果が出ているわけではなく、ASA変性CNFと炭酸カルシウムとの間で何らかの相互作用が働いているものと推察される。
【0152】
更に比較例5より、ASAを吸着したCNFと樹脂を混合して得た成形体は強度、弾性率共に向上していない。このことからASAはCNFに単に吸着するだけでは不十分で、ASAはCNFと共有結合していることが好ましいことが分かる。
【0153】
・実施例6
実施例1で得られたNBKP(リファイナー処理、固形分濃度:20質量%)をミキサー((株)愛工舎製作所製「ケンミックスKM−800」)で粗粉砕(処理時間1時間)し、そぼろ状とした後に二軸混練機((株)テクノベル製のKZW)に入れ、解繊処理によりCNFを得た。二軸混練機による解繊条件は、以下の通りである。
【0154】
[解繊条件]
スクリュー直径:15mm
スクリュー回転数:400rpm(スクリュー周速:18.8m/分)
解繊時間:150gのパルプを500g/hr〜600g/hrの処理条件で解繊した。原料を投入してからCNFが得られる迄の時間は15分間であった。
【0155】
L/D:45
解繊処理に供した回数:1回(1パス)
せき止め構造:2個。
【0156】
得られたCNFを1gサンプリングし、実施例1と同様にして溶媒置換し、得られた凍結乾燥後のCNFを、窒素ガス吸着法(日本ベル(株)製)により比表面積を測定したところ70m2/gであった。
【0157】
残りの二軸混練機で解繊して得たCNFに、エタノールを加え1質量%とした後、吸引濾過した。これを3回繰り返した後、エタノールを含む湿潤CNF(CNF濃度:30質量%)を金属バット上に厚さ0.5〜1cm程度となるように広げ、減圧乾燥機(YAMATO社製「ADP300」)に入れ、24時間減圧乾燥(温度105℃、真空度:0.1kPa以下)し、二軸解繊CNFの乾燥物を得た。
【0158】
<ASA変性CNFの調製>
二軸解繊CNFの乾燥物5gをN−メチルピロリドンに分散させ、固形分濃度が2質量%のスラリーとした。このスラリーを攪拌装置のついた500mlフラスコに移し、T−NS136(無水コハク酸以外の炭素数が8のASA、星光PMC(株)製)19.4g、触媒としてピリジンを14.6g加え、120℃で2時間加熱攪拌した後、冷却、洗浄しASA変性CNFを得た
得られたASA変性CNFを実施例1と同様にして凍結乾燥し、ASA変性CNFの凍結乾燥物を得た。
【0159】
得られたASA変性CNFの凍結乾燥物、及び樹脂の含有割合を表2に示す割合で混合した後、小型二軸混練機((株)テクノベル製「ULTnano15TW」)で140℃、5分間混練・循環させた。混練・循環後、弁を開放し樹脂を取りだし、細かく裁断してペレット化した。
【0160】
小型二軸混練機へのサンプル投入は1回で最大20g程度であるため、この操作を3回繰り返して合計で約50gの樹脂組成物を得た。この組成物を実施例1と同様にして成形を行い、引張り試験を行った。引張り試験結果を表2に示す。
【0161】
・実施例7〜8
実施例6のT−NS136(無水コハク酸以外の炭素数が8のASA、星光PMC(株)製)をT−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)、又はT−NS146(無水コハク酸以外の炭素数が18のASA、星光PMC(株)製)とし、表2に示すDSとなるまで反応時間を延ばした以外は、実施例6と同様にしてASA変性CNFを得た。それ以降の操作は実施例6と同様に行い、鎖長の異なるASA変性CNFを配合した樹脂組成物を得た。ASA変性CNFのDS、各成分の配合量、成形体の引張り強度結果を表2に示す。
【0162】
・実施例9
実施例1で調製したNBKP(リファイナー処理、固形分:濃度20質量%)をエタノールに分散させて2質量%、1800g(固形分36g)スラリーとした後、吸引ろ過をした。これを3回、アセトンへの分散を3回繰り返し、溶媒置換をした。得られたアセトンを含有するNBKPをNMPに分散させNBKPの固形分濃度が3質量%のスラリー1200gを得た。
【0163】
次いで、T−NS135(無水コハク酸以外の炭素数が16のASA、星光PMC(株)製)を215.8g、DMAPを1.4g、炭酸カリウムを30.7g加え、70℃で5時間反応させた。反応後、アセトン、エタノール、酢酸水、水で順次洗浄し、含水のASA変性CNFを得た。DSを測定した結果、0.29であった。
【0164】
得られたASA変性CNFに蒸留水を加え、総量を4,800gとした後、実施例1と同様にしてビーズミル処理を行った。
【0165】
得られたASA変性CNFを実施例6と同様に凍結乾燥、及び樹脂との複合化を行い、ASA変性CNFを配合した樹脂組成物を得た。各成分の配合量、成形体の引張り強度結果を表2に示す。
【0166】
・比較例6
実施例6で記載したASA変性CNFの凍結乾燥方法と同様にして、実施例6で調製した二軸解繊CNFの凍結乾燥物を得た。ASA変性CNFの凍結乾燥物の代わりに上記で調製した未変性の二軸解繊CNFの凍結乾燥物を用いた以外は、実施例6と同様にして樹脂組成物を得た。樹脂組成物を用いて作製した成形物の引張り強度を表2に示す。
【0167】
【表2】
【0168】
<考察>
鎖長の異なるASAを用いて調製したASA変性CNFを用いた場合でも、樹脂中での植物繊維の分散性や界面親和性が向上したため、樹脂組成物の力学物性が向上したと思われる。使用する樹脂や用途に応じて最適な置換度やASAを任意に設定することが可能である。
【0169】
・実施例10〜13
実施例4においてDSが0.56となるまで反応を進行させて得られたASA変性CNFを用いると共に、樹脂に加え、炭酸カルシウムの代わりに硫酸マグネシウム(和光純薬(株)製、試薬特級)、硫酸バリウム(和光純薬(株)製、試薬1級、粒子径:2.1μm)、炭酸バリウム(和光純薬(株)製、試薬、粒子径:0.24μm)、又は炭酸カリウム(和光純薬(株)製、試薬特級)を用いた以外は、実施例4と同様に行い成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表3に示す。尚、無機塩の粒子径は実施例4と同様にして測定した。
【0170】
なお、得られた成形物の外観は硫酸マグネシウム、硫酸バリウム、炭酸バリウムを用いた場合は無機塩を用いない場合(例えば、実施例1)や炭酸カルシウムを用いた場合(例えば実施例5)、樹脂のみ(例えば、比較例1、3)と同様に白〜淡黄色であったが、炭酸カリウムを用いた場合のみ濃褐色であった。
【0171】
・比較例7
実施例1で製造したビーズミル解繊CNFの含水物を実施例9と同様の方法で溶媒置換を行った。この溶媒置換物を無水酢酸の量を調整した以外は文献(Cellulose 9: 361-367, 2002)に記載の方法に従ってアセチル化変性CNFを得た。得られたアセチル化変性CNFのDSは0.39であった。
【0172】
ASA変性CNFの代わりに、上記で得たアセチル化変性CNFを用い、アセチル化変性CNFと樹脂を表3に記載の配合量とした以外は、実施例1と同様の方法によりアセチル化変性CNFを含む成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表3に示す。
【0173】
【表3】
【0174】
<考察>
変性CNFに硫酸マグネシウム、硫酸バリウム、炭酸バリウム、炭酸カリウムを用いた場合は無機塩として炭酸カルシウムを用いた場合と同様に高強度、高弾性率の成形体が得られた。無機塩として炭酸カルシウムを用いた場合と同様に変性CNFのカルボキシル基との相互作用がその一因であると思われる。弾性率向上効果としては炭酸カリウムが優れていたが、強度や外観の点で他の無機物よりは劣る。用途や成形条件等に応じてこれらの無機塩を使い分けることにより所望の成形材料、成形体を得る事が出来る。
【0175】
また、セルロース系繊維の疎水化方法として公知なアセチル化した場合では成形体の強度、弾性率の向上は認められていない。単に疎水化するだけでは成形体の強度、弾性率が向上しないことが分かる。このことからもCNFをASA変性することにより樹脂との親和性が向上していると推測される。
【0176】
・実施例14
実施例1においてDSが0.56となるまで反応を進行させて得られたASA変性CNFを42.4質量%用いると共に、このASA変性CNFと樹脂の配合を表4に示す含有割合とした以外は、実施例1と同様の方法により、成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表4に示す。
【0177】
なお、ASA変性CNF中のCNF由来の成分比は20質量%であり比較例8と同様である。
【0178】
・比較例8
未変性のCNF、及び樹脂の配合を表4に記載の通りとした以外は、比較例4と同様に行い成形物を得た。成形物の各成分の配合、及び成形物の引張り試験結果を表4に示す。
【0179】
【表4】
【0180】
<考察>
実施例14と比較例8はいずれもCNF純分は同じ20質量%であるが、実施例14は比較例8よりも強度、弾性率ともに優れていた。ミクロフィブリル化植物繊維をASA変性することにより樹脂との界面が補強されることや樹脂中での分散性が改善された為と思われる。
【特許請求の範囲】
【請求項1】
アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び
熱可塑性樹脂(B)を含有する樹脂組成物であって、
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.05〜2.0である樹脂組成物。
【請求項2】
アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、
熱可塑性樹脂(B)、及び
無機塩(C)を含有する樹脂組成物。
【請求項3】
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.1〜2.0である請求項1〜2の何れか1項に記載の樹脂組成物。
【請求項4】
変性ミクロフィブリル化植物繊維(A)の含有量が、樹脂組成物100質量部に対して、1〜80質量部である請求項1〜3の何れか1項に記載の樹脂組成物。
【請求項5】
無機塩(C)が、第2族の金属からなる塩である請求項2〜4の何れか1項に記載の樹脂組成物。
【請求項6】
無機塩(C)の含有量が、樹脂組成物100質量部に対して0.1〜20質量部である請求項2〜5の何れか1項に記載の樹脂組成物。
【請求項7】
(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び
(2a)工程(1a)によって得られたミクロフィブリル化植物繊維を触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維を得る工程、又は
(1b)植物繊維を、触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び
(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維を得る工程、
並びに
(3)工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程を含む樹脂組成物の製造方法。
【請求項8】
工程(3)が、工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、及び無機塩(C)を混合する工程である請求項7に記載の樹脂組成物の製造方法。
【請求項9】
工程(2a)又は工程(1b)におけるエステル化が、アルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒よりなる少なくとも1種の溶媒の存在下で行われる請求項7又は8に記載の樹脂組成物の製造方法。
【請求項10】
工程(1a)又は工程(2b)における解繊が、ボールミル、ビーズミル、ブレンダー、グラインダー、リファイナー、高圧衝突分散機、ホモジナイザー、高圧ホモジナイザー、一軸又は多軸混錬機からなる群より選ばれる少なくとも1種による機械的解繊である請求項7〜9の何れか1項に記載の樹脂組成物の製造方法。
【請求項11】
請求項7〜10の何れか1項に記載の製造方法で得られた変性ミクロフィブリル化植物繊維。
【請求項12】
請求項1〜6の何れか1項に記載の樹脂組成物を用いた成形材料。
【請求項13】
請求項7〜10の何れか1項で製造された樹脂組成物を用いた成形材料。
【請求項14】
請求項12又は13に記載の成形材料を成形してなる成形体。
【請求項1】
アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、及び
熱可塑性樹脂(B)を含有する樹脂組成物であって、
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.05〜2.0である樹脂組成物。
【請求項2】
アルキル、若しくはアルケニル無水コハク酸でエステル化された変性ミクロフィブリル化植物繊維(A)、
熱可塑性樹脂(B)、及び
無機塩(C)を含有する樹脂組成物。
【請求項3】
変性ミクロフィブリル化植物繊維(A)のエステル置換度が0.1〜2.0である請求項1〜2の何れか1項に記載の樹脂組成物。
【請求項4】
変性ミクロフィブリル化植物繊維(A)の含有量が、樹脂組成物100質量部に対して、1〜80質量部である請求項1〜3の何れか1項に記載の樹脂組成物。
【請求項5】
無機塩(C)が、第2族の金属からなる塩である請求項2〜4の何れか1項に記載の樹脂組成物。
【請求項6】
無機塩(C)の含有量が、樹脂組成物100質量部に対して0.1〜20質量部である請求項2〜5の何れか1項に記載の樹脂組成物。
【請求項7】
(1a)植物繊維を解繊し、ミクロフィブリル化植物繊維を得る工程、及び
(2a)工程(1a)によって得られたミクロフィブリル化植物繊維を触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性ミクロフィブリル化植物繊維を得る工程、又は
(1b)植物繊維を、触媒の存在下で、アルキル、若しくはアルケニル無水コハク酸でエステル化し、変性植物繊維を得る工程、及び
(2b)工程(1b)によって得られた変性植物繊維を解繊し、変性ミクロフィブリル化植物繊維を得る工程、
並びに
(3)工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)と熱可塑性樹脂(B)を混合する工程を含む樹脂組成物の製造方法。
【請求項8】
工程(3)が、工程(2a)又は工程(2b)によって得られた変性ミクロフィブリル化植物繊維(A)、熱可塑性樹脂(B)、及び無機塩(C)を混合する工程である請求項7に記載の樹脂組成物の製造方法。
【請求項9】
工程(2a)又は工程(1b)におけるエステル化が、アルキル、若しくはアルケニル無水コハク酸と反応しない有機溶媒よりなる少なくとも1種の溶媒の存在下で行われる請求項7又は8に記載の樹脂組成物の製造方法。
【請求項10】
工程(1a)又は工程(2b)における解繊が、ボールミル、ビーズミル、ブレンダー、グラインダー、リファイナー、高圧衝突分散機、ホモジナイザー、高圧ホモジナイザー、一軸又は多軸混錬機からなる群より選ばれる少なくとも1種による機械的解繊である請求項7〜9の何れか1項に記載の樹脂組成物の製造方法。
【請求項11】
請求項7〜10の何れか1項に記載の製造方法で得られた変性ミクロフィブリル化植物繊維。
【請求項12】
請求項1〜6の何れか1項に記載の樹脂組成物を用いた成形材料。
【請求項13】
請求項7〜10の何れか1項で製造された樹脂組成物を用いた成形材料。
【請求項14】
請求項12又は13に記載の成形材料を成形してなる成形体。
【図1】

【図2】
【図2】
【公開番号】特開2012−214563(P2012−214563A)
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2011−79440(P2011−79440)
【出願日】平成23年3月31日(2011.3.31)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成22年度独立行政法人新エネルギー・産業技術総合開発機構「グリーン・サステイナブルケミカルプロセス基盤技術開発/研究開発項目(4)化学品原料の転換・多様化を可能とする革新グリーン技術の開発/セルロースナノファイバー強化による自動車用高機能化グリーン部材の研究開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願日】平成23年3月31日(2011.3.31)
【国等の委託研究の成果に係る記載事項】(出願人による申告)国等の委託研究の成果に係る特許出願(平成22年度独立行政法人新エネルギー・産業技術総合開発機構「グリーン・サステイナブルケミカルプロセス基盤技術開発/研究開発項目(4)化学品原料の転換・多様化を可能とする革新グリーン技術の開発/セルロースナノファイバー強化による自動車用高機能化グリーン部材の研究開発」委託研究、産業技術力強化法第19条の適用を受ける特許出願)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]