
変性疾患及び炎症性疾患の治療に有用な新規化合物
下記の(I)により表される式を有する化合物が開示される。この化合物は医薬組成物として製造でき、非限定的な例として、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患を含む、ヒトを含む哺乳動物における種々の病態の予防及び治療に使用できる。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患に関与するチロシンキナーゼのファミリーであるJAKの阻害剤である化合物に関する。本発明は、この化合物の製造方法、この化合物を含む医薬組成物、並びに炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の、本発明の化合物を投与することによる予防及び/又は治療の方法も提供する。
【0002】
ヤヌスキナーゼ(JAK)は、膜受容体からSTAT転写因子へサイトカインシグナリングを伝達する細胞質チロシンキナーゼである。4種のJAKファミリーメンバー、JAK1、JAK2、JAK3、及びTYK2が記載されている。サイトカインがその受容体に結合すると、JAKファミリーメンバーは、自己リン酸化及び/又は互いにトランスリン酸化し、続いてSTATがリン酸化されて核へ移動し、転写が調節される。JAK-STAT細胞内シグナル伝達は、インターフェロン、ほとんどのインターロイキン、並びにEPO、TPO、GH、OSM、LIF、CNTF、GM-CSF、及びPRLなどの種々のサイトカイン及び内分泌因子の役に立つ(Vainchenker W.らの文献(2008))。
【0003】
遺伝モデルと小分子JAK阻害剤探索との組合せは、いくつかのJAKの治療上での可能性を明らかにした。JAK3は、免疫抑制の標的としてマウス及びヒト遺伝学で検証されている(O'Shea J.らの文献(2004))。JAK3阻害剤は、当初、臓器移植拒絶反応への臨床開発の取り組みに成功したが、後に、関節リウマチ(RA)、乾癬、及びクローン病などの他の免疫炎症性の徴候においても臨床開発の取り組みに成功した(http://clinicaltrials.gov/)。
【0004】
TYK2は、免疫炎症性疾患の潜在的な標的であり、ヒト遺伝学及びマウスノックアウト研究によって検証されている(Levy D.及びLoomis C.の文献(2007))。
【0005】
JAK1は、免疫炎症性疾患分野における新規の標的である。JAK1は、他のJAKとヘテロ二量体化して、サイトカイン駆動性の炎症促進性シグナルを伝達する。したがって、JAK1及び/又は他のJAKを阻害することで、様々な炎症病態、並びにJAK媒介性のシグナル伝達によって駆動される他の疾患に対する治療上の利益が期待される。
【背景技術】
【0006】
(発明の背景)
軟骨の変性は、様々な疾患の特徴となっており、その中でも、関節リウマチ及び変形性関節症が最も顕著なものである。関節リウマチ(RA)は、慢性の関節変性疾患であり、関節構造の炎症及び破壊によって特徴付けられる。該疾患を野放しにすると、関節機能性の喪失により相当な身体障害及び疼痛、並びに早死すら招く。したがって、RA治療の目的は、該関節破壊を阻止するために、該疾患の進行を減速するだけでなく、寛解も達成することである。該疾患の予後の厳しさに加えて、RAの高い罹患率(世界中で成人の約0.8%が罹患している)も、社会経済的影響が大きいことを意味している(RAの概説については、Smolen及びSteinerの文献(2003);Lee及びWeinblattの文献(2001);Choy及びPanayiの文献(2001);O'Dellの文献(2004)、及びFiresteinの文献(2003)を参照されたい。)。
【0007】
変形性関節症(OA、又は磨耗性関節炎とも称される)は、最も一般的な形態の関節炎であり、関節軟骨の喪失により特徴付けられ、多くの場合、骨の肥大化及び疼痛と関連する。変形性関節症の広範な概説については、Wielandらの文献(2005)を参照されたい。
【0008】
変形性関節症は治療が困難である。現在のところ、治癒を得ることはできず、治療は疼痛を緩和すること及び罹患した関節が変形するのを防ぐことに重点をおく。一般的な治療には、非ステロイド性抗炎症薬(NSAID)の使用が含まれる。コンドロイチン及びグルコサミン硫酸塩などの栄養補助食品が、変形性関節症の治療の安全でかつ有効な選択肢として推奨されているが、最近の臨床治験から、両治療が変形性関節症に関連する疼痛を低減させないことが明らかとなった。(Cleggらの文献、2006)。まとめると、疾患修飾性の変形性関節症の薬剤は全く利用できない。
【0009】
同化プロセスの刺激、異化プロセスの阻止、又はこれら2つを組み合わせると、軟骨が安定化し、障害の回復さえももたらされる可能性があり、したがって、疾患のさらなる進行を防ぐことができる。種々のトリガーが軟骨細胞の同化刺激を刺激することができる。インスリン様成長因子-I (IGF-I)は、滑液における主要な同化成長因子であり、プロテオグリカン及びコラーゲンの両方の合成を刺激する。骨形成タンパク質(BMP)ファミリーのメンバー、特にBMP2、BMP4、BMP6、及びBMP7、並びにヒト形質転換成長因子-β(TGF-β)ファミリーのメンバーが軟骨細胞同化刺激を誘発できることも示された(Chubinskaya及びKuettnerの文献、2003)。軟骨細胞の同化刺激を誘発する化合物が最近特定された(米国特許第6,500,854号;欧州特許第1391211号)。しかし、これらの化合物のほとんどは重度の副作用を示し、そのため、軟骨細胞分化を刺激する、これらの副作用のない化合物が強く必要とされている。
【0010】
Vandeghinsteらは、その阻害がOAを含む様々な疾患について治療上の関連性を有する可能性がある標的としてJAK1を発見した(WO2005/124342)。JAK1は、サイトカイン受容体媒介性細胞内シグナル伝達に関与する、細胞質チロシンキナーゼのヤヌスキナーゼ(JAK)ファミリーに属する。JAKファミリーは4種のメンバー、JAK1、JAK2、JAK3、及びTYK2からなる。サイトカインの結合と同時に、サイトカイン受容体にJAKがリクルートされ、次いで、サイトカイン受容体及び共有された受容体サブユニット(共通γ-c鎖、gp130)のヘテロ二量体化が起こる。次いで、JAKは自己リン酸化及び/又は他のJAKによりトランスリン酸化により活性化され、受容体のリン酸化並びにシグナル伝達性転写因子(STAT)のメンバーのリクルートメント及びリン酸化をもたらす。リン酸化されたSTATは二量体化し、核に移動してサイトカイン反応性遺伝子のエンハンサー領域に結合する。マウスのJAK1遺伝子のノックアウトから、JAK1は、発生の間に必須かつ非重複の役割を果たすことが示された:JAK1-/-マウスは生後24時間以内に死亡し、かつリンパ球の発生は著しく損なわれた。さらに、JAK1-/-細胞は、クラスIIサイトカイン受容体を使用するサイトカイン、シグナル伝達にγ-cサブユニットを使用するサイトカイン受容体、及びシグナル伝達にgp130サブユニットを使用するサイトカイン受容体ファミリーに反応しないか、又は反応性が低かった(Rodigらの文献、1998)。
【0011】
様々な群が、軟骨細胞生物学におけるJAK-STATシグナル伝達に関係している。Liら(2001)は、JAK/STAT及びMAPKシグナル伝達経路の活性化によって、オンコスタチンMが初代軟骨細胞におけるMMP及びTIMP3遺伝子の発現を誘導することを示した。Osakiら(2003)は、軟骨細胞におけるインターフェロン-γ媒介性のII型コラーゲンの阻害がJAK-STATシグナル伝達を含むことを示した。基質構成成分の発現を低下させることによって、並びにコラゲナーゼ及び一酸化窒素(NO)の産生を媒介する誘導型一酸化窒素合成酵素(NOS2)の発現を誘導することによって、IL1-βは軟骨異化反応を誘導する。Oteroら(2005)は、レプチン及びIL1-βが軟骨細胞におけるNO産生又はNOS2 mRNAの発現を相乗的に誘導し、しかも、それがJAK阻害剤によって阻止されることを示した。Legendreら(2003)は、ウシ関節の軟骨細胞において、IL6/IL6受容体が軟骨-特異的基質遺伝子II型コラーゲン、アグリカンコア及びリンクタンパク質の発現低下を誘導し、しかも、これがJAK/STATシグナル伝達によって媒介されることを示した。したがって、これらの観察は、軟骨恒常性維持におけるJAKキナーゼ活性の役割、及びJAKキナーゼ阻害剤に対する治療機会を示唆している。
【0012】
JAKファミリーメンバーは、骨髄増殖性疾患を含むさらなる病態に関与しており(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506)、JAK2の突然変異が同定されている。これは、JAK、特にJAK2の阻害剤が骨髄増殖性疾患の治療に有用となり得ることも示している。加えて、該JAKファミリー、特にJAK1、JAK2及びJAK3は、ガン、特に白血病、例えば、急性骨髄性白血病(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506;Xiangらの文献、2008、「急性骨髄性白血病患者における体細胞JAK1突然変異の同定(Identification of somatic JAK1 mutations in patients with acute myeloid leukemia)」Blood First Edition Paper, prepublished online 12月26日、2007;DOI 10.1182/blood-2007-05-090308)及び急性リンパ芽球性白血病(Mullighanらの文献、2009)、又は固形腫瘍、例えば、子宮平滑筋肉腫(Constantinescuらの文献、2007、Trends in Biochemical Sciences 33(3): 122-131)、前立腺ガン(Tamらの文献、2007、British Journal of Cancer, 97, 378-383)に関係している。これらの結果は、JAK、特にJAK1及び/又はJAK2の阻害剤が、ガン(白血病及び固形腫瘍、例えば、子宮平滑筋肉腫、前立腺ガン)の治療にも利用できることを示している。
【0013】
加えて、キャッスルマン病、多発性骨髄腫、メサンギウム増殖性糸球体腎炎、乾癬及びカポジ肉腫は、サイトカインIL-6の分泌過多に起因する可能性があり、その生物学的作用は細胞内JAK-STATシグナル伝達により媒介される(Tetsuji Naka、Norihiro Nishimoto及びTadamitsu Kishimotoの文献、Arthritis Res 2002, 4 (suppl 3):S233-S242)。この結果は、前記疾患の治療においてJAKの阻害剤の有用性が見出され得ることを示している。
【0014】
JAK3及びTYK2では自己免疫疾患との関連が確立された。JAK3の突然変異だけでなく、上流シグナル伝達成分γ-c受容体鎖及びIL7受容体の突然変異も、総計で、ヒトの重症複合型免疫不全症の症例の約70%の原因である('OSheaらの文献、2004)。γ-c受容体鎖からのシグナル伝達において、JAK1はJAK3と協同することに留意されたい。Tyk2の多形が全身性エリテマトーデス(SLE)に見られる(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506)。したがって、JAKファミリーの標的化は、免疫炎症分野における治療機会を与え得る。
【0015】
現在の治療は十分なものではなく、したがって、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療に有用となり得るさらなる化合物同定の必要性が残されている。したがって、本発明は、化合物、その製造方法、及び本発明の化合物を好適な医薬担体とともに含む医薬組成物を提供する。特に、該化合物は、385キナーゼ及び非キナーゼ標的に加え、他のJAKファミリーメンバーよりもJAK1への高い効力及び選択性を示す。さらに、データは、該化合物は良好な安全域を有することを示す。したがって、本発明がJAK1媒介性の疾患、特にSLE(全身性エリテマトーデス)及びRAなどの炎症性疾患の治療に新規な機会を与えることが結論付けられる。
【0016】
本発明は、これらの疾患及び病態を治療するための医薬の製造における本発明の化合物の使用も提供する。
【発明の概要】
【0017】
本発明は、JAK、特にJAK1の阻害剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療に有用であるという発見に基づいている。本発明は、これらの化合物の製造方法、これらの化合物を含む医薬組成物、並びに炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患を、本発明の化合物を投与することにより治療する方法も提供する。
【0018】
したがって、本発明の第一の態様において、式Iを有する本発明の化合物が開示される。
【化1】
【0019】
本発明の化合物は、385キナーゼ及び非キナーゼ標的、及び他のJAKファミリーメンバーよりもJAK1への高い効力及び選択性を示すと同時に、良好な安全域も示す、JAKの新規な阻害剤である。このプロファイルを有する化合物を使用すると、オフターゲット効果の低い発生率により、炎症性疾患、特にSLE及びRAの治療において、利点が生まれることがある。
【0020】
さらなる態様において、本発明は、本発明の化合物、及び医薬用担体、賦形剤又は希釈剤を含む医薬組成物を提供する。さらに、本発明の化合物は、本明細書に開示される医薬組成物及び治療方法に有用であり、製造及び使用されるときに医薬として許容し得るものである。本発明のこの態様において、該医薬組成物は、本発明の化合物と組み合わせた使用に好適なさらなる有効成分をさらに含むことがある。
【0021】
本発明のさらなる態様において、本発明は、本明細書に列記されるものから選ばれる病態、特に異常なJAK活性に関連し得る病態であって、例えば、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患に罹患し易い又は苦しんでいる哺乳動物の治療方法であって、治療上有効な量の本明細書に記載される本発明の化合物又は1種以上の医薬組成物を投与することを含む方法を提供する。具体的な態様において、本発明は、異常なJAK1活性に関連し得る病態、特に、炎症性疾患、増殖性疾患、及び軟骨代謝回転の異常を含む疾患に罹患し易い又は苦しんでいる哺乳動物を治療する方法を提供する。
【0022】
さらなる態様において、本発明は、本明細書に列記されるものから選ばれる病態、特に、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患などの異常なJAK活性に関連し得る病態の治療又は予防に使用するための本発明の化合物を提供する。具体的な態様において、本発明は、異常なJAK1活性に関連する病態、特に、炎症性疾患、増殖性疾患、及び軟骨代謝回転の異常を含む疾患の治療又は予防に使用するための本発明の化合物を提供する。
【0023】
治療態様のさらに他の方法において、本発明は、原因として本明細書に記載される異常なJAK活性に関連する病態に罹患し易い又は苦しんでいる哺乳動物の治療方法であって、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む方法を提供する。具体的な実施態様において、該異常なJAK活性は異常なJAK1活性である。
【0024】
さらなる態様において、本発明は、原因として異常なJAK活性に関連する病態の治療又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該異常なJAK活性は異常なJAK1活性である。
【0025】
追加の態様において、本発明は、本明細書の下記に開示される代表的な合成手順及び経路とともに本発明の化合物の合成方法を提供する。
【0026】
したがって、本発明の主な目的は、JAKの活性を修正できる化合物を提供し、ひいては原因としてそれに関連し得る任意の病態を予防又は治療することである。特に、原因としてJAK1の活性に関連し得る任意の病態。
【0027】
本発明のさらなる目的は、原因としてJAKの活性に関連し得る、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患などの病態若しくは疾病又はその症状を治療又は軽減することができる化合物を提供することである。特に、原因としてJAK1の活性に関連し得る疾病又は病態。
【0028】
本発明のなおさらなる目的は、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患などのJAK活性に関連する疾患を含む、種々の疾病状態の治療又は予防に使用できる医薬組成物を提供することである。特に、JAK1活性に関連する病態。
【0029】
他の目的及び利点は、次の詳細な説明を考慮することにより、当業者に明らかとなろう。
【発明を実施するための形態】
【0030】
(発明の詳細な説明)
(定義)
以下の用語は、それとともに以下に提示される意味を有するものとし、本発明の記載及び意図される範囲を理解するのに有用である。
【0031】
本発明を記載する場合、化合物、該化合物を含む医薬組成物、並びに該化合物及び組成物を使用する方法を含み得るが、以下の用語は、存在する場合、他に示さない限り、以下の意味を有する。本明細書に記載される場合、以下に定義される部分はいずれも多様な置換基で置換できること、及びそれぞれの定義が以下に記載されるそれらの範囲内でこのような置換部分を含むものとすることも理解されたい。他に明記しない限り、用語「置換」とは、以下に記載されるように定義できよう。本明細書に使用される場合、用語「基」及び「ラジカル」が交換可能であるとみなし得ることをさらに理解されたい。
【0032】
冠詞「a」及び「an」は、本明細書において、1又は2以上(すなわち、少なくとも1)の該冠詞の文法上の対象を言及するのに使用できる。例として、「アナログ(an analogue)」は、1のアナログ又は2以上のアナログを意味する。
【0033】
「医薬として許容し得る」とは、連邦政府若しくは州政府の監督機関又は米国以外の国の対応機関により承認されたか又は承認され得ることを意味するか、あるいは、動物、より具体的にはヒトにおける使用のための米国薬局方又は他の公認された薬局方に収載されていることを意味する。
【0034】
「医薬として許容し得る塩」とは、医薬として許容することができ、かつ親化合物の所望の薬理活性を有する、本発明の化合物の塩をいう。特に、そのような塩は無毒性であり、無機又は有機の酸付加塩及び塩基付加塩とすることができる。具体的には、そのような塩には下記がある:(1)塩化水素酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸で形成された酸付加塩;酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、3-(4-ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、1,2-エタン-ジスルホン酸、2-ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、4-クロロベンゼンスルホン酸、2-ナフタレンスルホン酸、4-トルエンスルホン酸、カンファスルホン酸、4-メチルビシクロ[2.2.2]-オクト-2-エン-1-カルボン酸、グルコヘプトン酸、3-フェニルプロピオン酸、トリメチル酢酸、三級ブチル酢酸、ラウリル硫酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸などの有機酸で形成された酸付加塩;又は、(2)親化合物に存在する酸性プロトンが、金属イオン、例えば、アルカリ金属イオン、アルカリ土類イオン、又はアルミニウムイオンにより置換されるか、;又はエタノールアミン、ジエタノールアミン、トリエタノールアミン、N-メチルグルカミンなどの有機塩基と配位する場合に形成された塩。塩は、単なる例として、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウム、テトラアルキルアンモニウムなどをさらに含み、化合物が塩基性官能性を含む場合、塩酸塩、臭化水素酸塩、酒石酸塩、メシル酸塩、酢酸塩、マレイン酸塩、シュウ酸塩などの無毒の有機又は無機酸の塩をさらに含む。用語「医薬として許容し得るカチオン」とは、酸性官能基の許容し得るカチオン性対イオンをいう。そのようなカチオンは、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウム、テトラアルキルアンモニウムカチオンなどにより例示される。
【0035】
「医薬として許容し得るビヒクル」とは、本発明の化合物とともに投与される希釈剤、補助剤、賦形剤、又は担体をいう。
【0036】
「プロドラッグ」は、切断可能な基を有し、加溶媒分解により、又は生理的な条件下で、インビボで医薬として活性な本発明の化合物になる、本発明の化合物の誘導体を含む化合物を意味する。そのような例には、コリンエステル誘導体など、N-アルキルモルホリンエステルなどがあるが、これらに限定されない。
【0037】
「溶媒和物」とは、通常、加溶媒分解反応によって溶媒と会合する化合物の形態をいう。この物理的会合には水素結合がある。従来型の溶媒には、水、エタノール、酢酸などがある。本発明の化合物は、例えば結晶形で調製でき、溶媒和できるか又は水和できる。適切な溶媒和物には、水和物などの医薬として許容し得る溶媒和物があり、化学量論的溶媒和物及び非化学量論的溶媒和物の両方もさらに含まれる。特定の例において、溶媒和物は、例えば1以上の溶媒分子が結晶質固体の結晶格子に取り込まれている場合に単離できる。「溶媒和物」は、液相及び単離可能な溶媒和物の両方を包含する。代表的な溶媒和物には、水和物、エタノラート及びメタノラートがある。
【0038】
「対象」はヒトを含む。「ヒト」、「患者」及び「対象」という用語は、本明細書において交換可能に使用される。
【0039】
「治療上有効な量」とは、疾患を治療するために対象へ投与される場合に、該疾患のそのような治療を発効させるのに十分である化合物の量を意味する。「治療上有効な量」は、化合物、疾患及びその重症性、並びに治療される対象の年齢、体重などに応じて変化し得る。
【0040】
「予防する(preventing)」又は「予防(prevention)」とは、疾患又は障害を得るか又はかかるリスクの低減を意味する(すなわち、該疾患の臨床症状のうちの少なくとも1つを、疾患原因物質に曝露されている可能性がある対象、又は疾患の発症に先立って該疾患に罹りやすい対象において起こさせない)。
【0041】
用語「予防(prophylaxis)」は「予防(prevention)」に関連し、その目的が疾患を治療又は治癒することよりも予防することにある処置又は手段をいう。予防処置の非限定的な例には、ワクチンの投与;例えば不動化に起因する血栓症のリスクのある入院患者に対する低分子量ヘパリンの投与;及び、マラリアが風土性である、又はマラリアになるリスクが高い地理的領域への訪問に先立つクロロキンなどの抗マラリア薬の投与があり得る。
【0042】
任意の疾患又は障害の「治療する(treating)」又は「治療(treatment)」とは、一実施態様において、疾患又は障害を回復させること(すなわち、該疾患を抑止するか又はその臨床症状のうちの少なくとも1つの徴候、程度、又は重症度を減少させる)をいう。別の実施態様において、「治療する」又は「治療」とは、少なくとも1つの肉体的パラメーターを回復させることをいい、これは対象によって認識できない場合もある。さらに別の実施態様において、「治療する」又は「治療」とは、肉体的に、(例えば、認識可能な症状の安定化)、生理的に、(例えば、肉体的パラメーターの安定化)、又はその両方のいずれかで、疾患又は障害を調節することをいう。さらなる実施態様において、「治療する」又は「治療」とは、疾患の進行を減速させることに関する。
【0043】
本明細書では、「炎症性病態(複数可)」という用語は、関節リウマチ、変形性関節症、若年性特発性関節炎、乾癬、アレルギー性気道疾患(例えば、喘息、鼻炎)、炎症性腸疾患(例えば、クローン病、大腸炎)、エンドトキシン駆動性疾病状態(例えば、バイパス手術後の合併症又は例えば、慢性心不全の原因となる慢性的なエンドトキシン状態)、及び関節の軟骨などの軟骨に関する関連疾患を含む病態の群をいう。特に該用語は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患をいう。
【0044】
本明細書では、「自己免疫疾患(複数可)」は、COPDなどの病態を含む閉塞性気道疾患、喘息(例えば、内因性喘息、外因性喘息、塵埃性喘息(dust asthma)、小児喘息)、特に慢性又は難治性喘息(例えば、遅発型喘息及び気道過敏)、気管支喘息を含む気管支炎、全身性エリテマトーデス(SLE)、多発性硬化症、I型糖尿病及びそれと関連する合併症、アトピー性湿疹(アトピー性皮膚炎)、接触性皮膚炎及びさらなる湿疹性皮膚炎、炎症性腸疾患(例えば、クローン病及び潰瘍性大腸炎)、アテローム硬化症、並びに筋萎縮性側索硬化症を含む疾患の群をいう。特に、該用語は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患を意味する。
【0045】
本明細書では、「増殖性疾患(複数可)」という用語は、ガン(例えば、子宮平滑筋肉腫又は前立腺ガン)、骨髄増殖性疾患(例えば、真性多血症、本態性血小板血症、及び骨髄線維症)、白血病(例えば、急性骨髄性白血病及び急性リンパ芽球性白血病)、多発性骨髄腫、乾癬、再狭窄、硬化性皮膚炎、又は線維症などの病態をいう。特に、該用語は、ガン、白血病、多発性骨髄腫、及び乾癬をいう。
【0046】
本明細書では、「ガン」という用語は、皮膚又は体内臓器、例えば、限定はされないが、乳房、前立腺、肺、腎臓、膵臓、胃又は腸の細胞の悪性又は良性の増殖をいう。ガンは、隣接する組織に浸潤し、例えば、骨、肝臓、肺又は脳など遠位の臓器に広がる(転移する)傾向がある。本明細書では、ガンという用語は、限定はされないが、メラノーマ、リンパ腫、白血病、線維肉腫、横紋筋肉腫、及びマスト細胞腫などの転移性腫瘍細胞型、並びに限定はされないが、結腸直腸ガン、前立腺ガン、小細胞肺ガン及び非小細胞肺ガン、乳ガン、膵ガン、膀胱ガン、腎ガン、胃ガン、神経膠芽腫、原発性肝ガン、卵巣ガン、前立腺ガン、及び子宮平滑筋肉腫などの組織細胞腫(tissue carcinoma)型の両方を含む。
【0047】
本明細書では、「白血病」という用語は、血液及び造血器官の腫瘍性疾患をいう。そのような疾患は、宿主を感染及び出血しやすい状態にさせる、骨髄及び免疫系の機能障害を引き起こし得る。特に、白血病という用語は、急性骨髄性白血病(AML)及び急性リンパ芽球性白血病(ALL)をいう。
【0048】
本明細書では、「移植拒絶」という用語は、例えば、膵島、幹細胞、骨髄、皮膚、筋肉、角膜組織、神経細胞組織、心臓、肺、心肺の組み合わせ、腎臓、肝臓、腸、膵臓、気管、又は食道など、細胞、組織又は固形臓器の同種又は異種移植片の急性又は慢性拒絶反応、又は移植片対宿主病をいう。
【0049】
本明細書では、「軟骨代謝回転の障害を含む疾患」という用語は、変形性関節症、乾癬性関節炎、若年性関節リウマチ、痛風関節炎、化膿性又は感染性関節炎、反応性関節炎、反射性交感神経性ジストロフィー、疼痛性ジストロフィー、ティーツェ症候群又は肋骨軟骨炎、線維筋痛症、骨軟骨炎、神経性又はニューロパチー性関節炎、関節症、地方性変形性骨軟骨関節症(osteoarthritis deformans endemica)などの風土病型関節炎、ムセレニ病(Mseleni disease)及びハンディゴデュ病(Handigodu disease);線維筋痛症に起因する変性、全身性エリテマトーデス、強皮症及び強直性脊椎炎などの病態を含む。
【0050】
本明細書では、「先天性軟骨形成異常(複数可)」という用語は、遺伝性軟骨溶解、軟骨異形成症、及び偽性軟骨異形成症などの病態、特に、限定はされないが、小耳症、無耳症、骨幹端軟骨異形成症、及び関連の疾患を含む。
【0051】
本明細書では、「IL6の分泌過多と関連する疾患(複数可)」という用語は、キャッスルマン病、多発性骨髄腫、乾癬、カポジ肉腫、及び/又はメサンギウム増殖性糸球体腎炎などの病態を含む。
【0052】
本明細書では、「JAK」という用語は、膜受容体からのサイトカインシグナル伝達をSTAT転写因子に伝達する細胞質チロシンキナーゼであるヤヌスキナーゼ(JAK)のファミリーに関連する。4種のJAKファミリーメンバー、JAK1、JAK2、JAK3、及びTYK2が記載され、JAKという用語は、文脈が示すとおり、JAKファミリーメンバー全てをまとめて意味することも、1つ以上のJAKファミリーメンバーを意味することもある。
【0053】
「本発明の化合物(複数可)」及び等価な表現は、本明細書に記載された式の化合物を包含することを意味し、文脈が許す場合、その表現は、医薬として許容し得る塩及びその溶媒和物、例えば、水和物、並びに該医薬として許容し得る塩の溶媒和物を含む。同様に、中間体への言及は、それら自体が特許請求されているか否かに関わらず、文脈が許す場合、それらの塩及び溶媒和物を包含することを意味する。
【0054】
例えば、限定はされないがC1-C8アルキルなど、本明細書において範囲が言及される場合、範囲の引用は、前記範囲の各要素の説明であるとみなされたい。
【0055】
本発明の化合物の他の誘導体は、それらの酸形態及び酸誘導体形態の両方で活性を有するが、該酸感受性形態では多くの場合、哺乳動物において溶解性、組織適合性又は遅延放出の利点を付与する(Bundgard, H.の文献、「プロドラッグの設計(Design of Prodrugs)」, pp. 7-9, 21-24, Elsevier, Amsterdam 1985参照)。プロドラッグには、当業者に周知である酸誘導体があり、例えば、親の酸と好適なアルコールとの反応により製造されるエステル、又は親の酸化合物と置換若しくは非置換のアミンとの反応により製造されるアミド、又は酸無水物、又は混合無水物がある。本発明の化合物の懸垂した酸性基から誘導される単純な脂肪族又は芳香族のエステル、アミド、及び酸無水物は特に有用なプロドラッグである。いくつかの場合で、(アシルオキシ)アルキルエステル又は((アルコキシカルボニル)オキシ)アルキルエステルなどの二重エステルタイプのプロドラッグを製造するのが望ましい。そのような特定のプロドラッグは、本発明の化合物のC1からC8アルキル、C2-C8アルケニル、アリール、C7-C12置換アリール、及びC7-C12アリールアルキルエステルである。
【0056】
本明細書では、「同位体異形」という用語は、そのような化合物を構成する1以上の原子で非天然の同位体比率を含む化合物をいう。例えば、化合物の「同位体異形」は、例えば、重水素(2H又はD)、炭素-13(13C)、窒素-15(15N)などの1以上の非放射性同位体を含むことができる。そのような同位体的置換がなされる化合物において、以下の原子は存在する場合、例えば、任意の水素が2H/Dとなり、任意の炭素が13Cとなり、又は任意の窒素が15Nとなり、かつそのような原子の存在及び配置が当分野の技術内で決定できるように変更し得ることが理解されよう。同様に、本発明は、放射性同位体を有する同位体異形の製造を含むことができ、例えば、得られた化合物を、薬剤及び/又は基質組織分布研究に使用する場合がある。放射性同位体トリチウム、すなわち3H、及び炭素-14、すなわち14Cは、それらの取込みが容易であり、かつ検出手段が整っているという観点から本目的に特に有用である。さらに、化合物は11C、18F、15O、及び13Nなどの陽電子放出同位体で置換されて調整することができ、基質受容体占有率を検討するため陽電子放射型断層撮影法(PET)研究において有用であり得るだろう。
【0057】
本明細書に提供される化合物の全ての同位体異形は、放射性であるか否かに関わらず、本発明の範囲内に包含されるものとする。
【0058】
互いの鏡像ではない立体異性体は「ジアステレオマー」と称され、互いの重ねあわすことのできない鏡像であるものは「エナンチオマー」と称される。化合物が不斉中心を有する場合、例えば4つの異なる基に結合している場合、一組のエナンチオマーが存在し得る。エナンチオマーは、その不斉中心の絶対配置により特徴づけることができ、Cahn及びPrelogのR-及びS-順序付け規則か、又は分子が偏光面を回転させる様態により説明され、右旋性又は左旋性(すなわち、それぞれ(+)又は(-)-異性体)であると呼ばれる。キラルな化合物は、個別なエナンチオマーとして、又はその混合物として存在し得る。等しい比率のエナンチオマーを含む混合物は「ラセミ混合物」と呼ばれる。
【0059】
「互変異性体」とは、特定の化合物構造の交換可能な形態であり、かつ水素原子及び電子の配置において異なる化合物をいう。したがって、2つの構造は、π電子及び原子(通常、H)の動きを介して平衡となり得る。例えば、エノール及びケトンは、酸又は塩基のいずれかを用いた処理により迅速に相互変換されるので、互変異性体である。互変異性の別の例には、フェニルニトロメタンのアシ-及びニトロ-形態があり、これらは酸又は塩基の処理により同様に形成される。
【0060】
互変異性形態は、目的とする化合物の最適な化学反応性及び生物活性の実現に関連し得る。
【0061】
本発明の化合物は、1つ以上の不斉中心を持つことがある。したがって、そのような化合物は、個別な(R)-若しくは(S)-立体異性体として、又はその混合物として製造することができる。
【0062】
特記されない限り、本明細書及び請求項中の特定の化合物の記載又は呼称は、その個別のエナンチオマー、並びにラセミ体及びその他の混合物の両方を含むものとする。立体化学の決定及び立体異性体の分離の方法は当分野に周知である。
【0063】
(化合物)
本発明は、JAKの阻害剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患の治療に有用であるという発見に基づいている。本発明は、該化合物の製造方法、該化合物を含む医薬組成物、並びに軟骨の破壊、骨及び/又は関節の破壊、及び/又は炎症を含む疾患を、本発明の化合物を投与することにより治療する方法も提供する。本化合物は、JAKファミリーのメンバーの阻害剤である。具体的には、それは、JAK1、JAK2、JAK3、及びTYK2の活性を阻害する。特に、それはJAK1の活性を阻害する。
【0064】
したがって、本発明の第一の態様において、式Iを有する本発明の化合物が開示される。
【化2】
【0065】
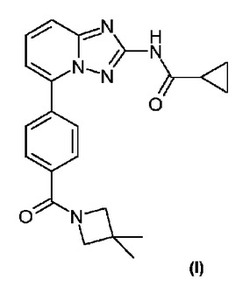
本発明の化合物は、シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミドである。
【0066】
一実施態様において、本発明の化合物は、同位体異形ではない。
【0067】
一態様において、本発明の化合物は、遊離の塩基として存在する。
【0068】
一態様において、本発明の化合物は、医薬として許容し得る塩である。
【0069】
一態様において、本発明の化合物は、該化合物の溶媒和物である。
【0070】
一態様において、本発明の化合物は、該化合物の医薬として許容し得る塩の溶媒和物である。
【0071】
特定の態様において、本発明は、上記の式の化合物のプロドラッグ及び誘導体を提供する。プロドラッグは、代謝により切断可能な基を有し、加溶媒分解により、又は生理的な条件下で、インビボで医薬として活性な本発明の化合物になる、本発明の化合物の誘導体である。そのような例には、コリンエステル誘導体など、N-アルキルモルホリンエステルなどがあるが、これらに限定されない。
【0072】
本発明の化合物の他の誘導体は、それらの酸形態及び酸誘導体形態の両方で活性を有するが、該酸感受性形態では多くの場合、哺乳動物において溶解性、組織適合性又は遅延放出の利点を付与する(Bundgard, H.の文献、「プロドラッグの設計(Design of Prodrugs)」, pp. 7-9, 21-24, Elsevier, Amsterdam 1985参照)。プロドラッグには当業者に周知である酸誘導体があり、例えば、親の酸と好適なアルコールとの反応により製造されるエステル、又は親の酸化合物と置換若しくは非置換のアミンとの反応により製造されるアミド、又は酸無水物、又は混合無水物がある。本発明の化合物の垂れている酸性基から誘導される単純な脂肪族又は芳香族のエステル、アミド、及び酸無水物は好ましいプロドラッグである。いくつかの場合で、(アシルオキシ)アルキルエステル又は((アルコキシカルボニル)オキシ)アルキルエステルなどの二重エステルタイプのプロドラッグを製造するのが望ましい。本発明の化合物のC1からC8アルキル、C2-C8アルケニル、アリール、C7-C12置換アリール、及びC7-C12アリールアルキルエステルが特に有用である。
【0073】
(医薬組成物)
医薬として使用される場合、本発明の化合物は、典型的には医薬組成物の形態で投与される。そのような組成物は、医薬分野において周知の様式で製造でき、少なくとも1つの活性化合物を含むことができる。通常、本発明の化合物は、医薬として有効な量で投与される。実際に投与される化合物の量は、典型的には、治療される状態、選択した投与経路、実際に投与される化合物、個々の患者の年齢、体重及び反応、患者の症状の重症度などを含む関連した状況に照らして、医師により決定される。
【0074】
本発明の医薬組成物は、経口経路、直腸経路、経皮経路、皮下経路、関節内経路、静脈経路、筋肉内経路、及び鼻腔内経路を含む様々な経路により投与できる。意図された送達経路に応じて、本発明の化合物は、好ましくは、注射可能若しくは経口組成物として、又は全て経皮投与のための塗剤として、ローション剤として、若しくはパッチとして、製剤化される。
【0075】
経口投与用組成物は、バルク溶液若しくは懸濁液、又はバルク粉末の形態をとり得る。しかしながら、より一般的には、本組成物は、正確な投与を容易にする単位投与形態で提供される。「単位投与形態」という用語は、ヒトの対象及び他の哺乳動物への単位投与量として適切な物理的に個別の単位をいい、各単位は適当な医薬用賦形剤、ビヒクル、又は担体とともに、所望の治療効果を提供するように算出された既定量の活性物質を含む。固体組成物の場合、典型的な単位投与形態には、液体組成物が予め充填され予め計量されたアンプル若しくはシリンジ又は丸剤、錠剤、カプセル等などを含む。そのような組成物において、本発明の化合物は、通常、微量成分(約0.1〜約50重量%又は好ましくは約1〜約40重量%)であり、その残りは様々なビヒクル又は担体及び所望の投与形態を形成するのを補助する加工助剤である。
【0076】
経口投与に適した液体形態には、緩衝液、懸濁剤及び分配剤、着色剤、香料などとともに好適な水性又は非水性の溶媒を含むことができる。固体形態は、例えば、以下の成分のいずれか、又は類似の性質の化合物を含むことができる:微結晶性セルロース、トラガカントガム又はゼラチンなどの結合剤;デンプン又はラクトースなどの賦形剤、アルギン酸、プリモゲル、又はコーンスターチなどの崩壊剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド状二酸化ケイ素などの流動促進剤;スクロース又はサッカリンなどの甘味剤;又は、ハッカ、サリチル酸メチル、又はオレンジ香料などの香料。
【0077】
注射可能組成物は、典型的には、当該技術分野において公知の注射可能な無菌の食塩水若しくはリン酸緩衝食塩水又は他の注射可能な担体に基づく。前記のように、そのような組成物における活性化合物は、典型的には微量成分であり、多くの場合、約0.05〜10重量%であり、その残りは注射可能な担体などである。
【0078】
経皮組成物は、典型的には、一般に約0.01〜約20重量%、好ましくは約0.1〜約20重量%、好ましくは約0.1〜約10重量%、より好ましくは約0.5〜約15重量%にわたる量で有効成分(複数可)を含む、局所用軟膏又はクリームとして製剤化される。軟膏として製剤化される場合、有効成分は、典型的には、パラフィン性又は水混和性の軟膏基剤のいずれかと混合される。あるいは、有効成分は、例えば水中油クリーム基剤とともにクリームに配合できる。そのような経皮製剤は当該技術分野において周知であり、有効成分又は製剤の真皮浸透の安定性を促進するために一般的には添加成分を含む。そのような公知の経皮製剤及び成分の全ては、本発明の範囲内に含まれる。
【0079】
本発明の化合物は、経皮装置により投与することもできる。したがって、経皮投与は、リザーバー型若しくは多孔膜型いずれかのパッチ又は固体マトリクス多様体を使用して達成できる。
【0080】
経口投与可能な、注射可能な、又は局所的に投与可能な組成物のための上記成分は、代表的なものにすぎない。他の材料並びに加工技術などは、「レミントンの薬学(Remington's Pharmaceutical Sciences)」、第17版, 1985, Mack Publishing Company, Easton, Pennsylvaniaのパート8に記載され、これは引用により本明細書に組み込まれる。
【0081】
本発明の化合物は、徐放性形態で、又は徐放性製剤送達系から投与することもできる。代表的な徐放物質の説明は、レミントンの薬学に見出すことができる。
【0082】
以下の製剤例は、本発明に従って製造できる代表的な医薬組成物を表す。しかし、本発明は、以下の医薬組成物に限定されない。
【0083】
(製剤1-錠剤)
本発明の化合物は乾燥粉末として、乾燥ゼラチン結合剤とおよそ1:2の重量比で混合できる。微量のステアリン酸マグネシウムを滑沢剤として加えてよい。該混合物は、錠剤成形機で240〜270mgの錠剤(錠剤当たり80〜90mgの活性アミド化合物)に成形することができる。
【0084】
(製剤2-カプセル剤)
本発明の化合物は乾燥粉末として、デンプン希釈剤とおよそ1:1の重量比で混合できる。該混合物は、250mgのカプセル(カプセル当たり125mgの活性アミド化合物)に充填できる。
【0085】
(製剤3-液剤)
本発明の化合物(125mg)は、スクロース(1.75g)及びキサンタンガム(4mg)と混合でき、その結果として生じる混合物は混合され、No.10メッシュU.S.シーブに通され、その後、微結晶性セルロース及びカルボキシメチルセルロースナトリウム(11:89、50mg)の予め製造した水溶液と混合できる。安息香酸ナトリウム(10mg)、香料及び着色料を水で希釈して、撹拌しながら加えてよい。次いで、十分な水を撹拌しながら、加えることができる。次いで、十分な水を添加し、5mLの総容積にすることができる。
【0086】
(製剤4-錠剤)
本発明の化合物は乾燥粉末として、乾燥ゼラチン結合剤とおよそ1:2の重量比で混合できる。微量のステアリン酸マグネシウムを、滑沢剤として加えてよい。該混合物は、錠剤成形機で450〜900mgの錠剤(150〜300mgの活性アミド化合物)に成形される。
【0087】
(製剤5-注射製剤)
本発明の化合物は、緩衝化無菌食塩水の注射可能水性媒体中、およそ5mg/mLの濃度に、溶解又は懸濁できる。
【0088】
(製剤6-局所製剤)
ステアリルアルコール(250g)及び白色ワセリン(250g)を約75℃で溶かすことができ、その後、水(約370g)に溶かした本発明の化合物(50g)、メチルパラベン(0.25g)、プロピルパラベン(0.15g)、ラウリル硫酸ナトリウム(10g)、及びプロピレングリコール(120g)の混合物を添加でき、その結果生じた混合物を凝結するまで撹拌してよい。
【0089】
(治療の方法)
本発明の化合物は、JAKの異常な活性に原因として関連しているか、又はそれに起因している、哺乳動物の病態の治療のための治療剤として使用できる。特に、JAK1の異常な活性に関連した病態。したがって、本発明の化合物及び医薬組成物は、ヒトを含む哺乳動物の炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患の予防及び/又は治療に治療法として使用される。
【0090】
治療態様の追加の方法において、本発明は、炎症性病態に罹患しやすい又は苦しんでいる哺乳動物を治療する方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、該炎症性病態は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患から選択される。
【0091】
他の態様において、本発明は、炎症性病態の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該炎症性病態は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患から選択される。
【0092】
治療態様のさらなる方法において、本発明は、自己免疫疾患に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、該自己免疫疾患は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患から選択される。
【0093】
他の態様において、本発明は、自己免疫疾患の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該自己免疫疾患は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患から選択される。
【0094】
治療態様のさらなる方法において、本発明は、増殖性疾患、特にガン(例えば、子宮平滑筋肉腫又は前立腺ガンなどの固形腫瘍)、白血病(例えば、AML又はALL)、多発性骨髄腫及び/又は乾癬に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0095】
他の態様において、本発明は、増殖性疾患、特にガン(例えば、子宮平滑筋肉腫又は前立腺ガンなどの固形腫瘍)、白血病(例えば、AML又はALL)、多発性骨髄腫及び/又は乾癬の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0096】
治療態様のさらなる方法において、本発明は、移植拒絶に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、本発明は、臓器移植拒絶を治療する方法を提供する。
【0097】
他の態様において、本発明は、移植拒絶の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、本発明は臓器移植拒絶を治療する方法を提供する。
【0098】
治療態様の方法において、本発明は、軟骨代謝回転の異常を含む疾患に罹患しやすい又は苦しんでいる哺乳動物の治療、予防、又は予防の方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0099】
他の態様において、本発明は、軟骨代謝回転の異常を含む疾患の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0100】
本発明は、先天性軟骨形成異常を治療する方法も提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0101】
他の態様において、本発明は、先天性軟骨形成異常の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0102】
治療態様のさらなる方法において、本発明は、IL6の分泌過多に関連する疾患、特にキャッスルマン病又はメサンギウム増殖性糸球体腎炎に罹患し易い又は苦しんでいる哺乳動物を治療する方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0103】
他の態様において、本発明は、IL6の分泌過多に関連する疾患、特にキャッスルマン病又はメサンギウム増殖性糸球体腎炎の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0104】
本発明のさらなる態様として、特に上述の病態及び疾患の治療又は予防における医薬品として使用するための本発明の化合物が提供される。また、上述の病態及び疾患の1つを治療又は予防するための医薬の製造における該化合物の使用も本明細書に提供される。
【0105】
本方法の特定の療法は、炎症性病態に苦しんでいる対象に、該対象における炎症のレベルを低下させ、かつ好ましくは、前記炎症の原因であるプロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。本方法の特別な実施態様は、関節リウマチの進行に苦しんでいる又は罹患し易い対象患者に、前記患者の関節における炎症をそれぞれ低下させ若しくは予防し、かつ好ましくは、前記炎症の原因であるプロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。
【0106】
本方法のさらに特別な療法は、軟骨代謝回転の異常を含む疾患(例えば、変形性関節症)に苦しんでいる対象に、前記破壊の原因である永続的なプロセスを低下させ、かつ好ましくは終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。該方法の特別な実施態様は、変形性関節症の進行に苦しんでいる又は罹患し易い対象患者に、前記患者の関節における軟骨破壊をそれぞれ低下させるか又は予防し、かつ好ましくは前記破壊の原因である永続的プロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。特別な実施態様において、前記化合物は、軟骨同化特性及び/又は抗異化特性を示すことがある。
【0107】
注入用量レベルは、約1〜約120時間及び特に24〜96時間の全てについて、約0.1mg/kg/時〜少なくとも10mg/kg/時にわたる。また、約0.1mg/kg〜約10mg/kg又はこれを超える予充填ボーラスを、妥当な定常状態レベルを達成するために投与することもできる。最大の全用量は、40〜80kgのヒト患者について約2g/日を超えるとは予想されない。
【0108】
長期の変性状態などの状態の予防及び/又は治療について、治療のための療法は、通常、数ヶ月又は数年にわたるため、患者の便宜及び許容度のために経口投与が好ましい。経口投与では、1日あたり1〜5回、特に2〜4回、典型的には3回の経口投与が代表的な療法である。これらの投与様式を利用して、各投与量は、個別の投与量をそれぞれ、約0.1〜約10mg/kg、特に約1〜約5mg/kgで提供することによって、約0.01〜約20mg/kgの本発明の化合物を提供する。
【0109】
経皮用量は、一般に、注入用量を使用して達成されるのと同等又はそれよりも低い血中濃度を提供するように選択される。
【0110】
炎症性病態の発症を予防するために使用する場合、本発明の化合物は、典型的には医師の助言に従い、かつその管理下で、上記の投与量レベルにて、該病態にかかる危険性のある患者に投与される。特定の病態にかかる危険性のある患者は、一般に、該病態の家族歴を有する者、又は遺伝的試験若しくはスクリーニングにより、特に該病態にかかりやすいと同定された者を含む。
【0111】
本発明の化合物は、単一の活性薬剤として投与することができるか、又は同じ若しくは類似の治療活性を示し、かつ併用投与に安全かつ有効であることが判断された他の化合物を含む他の治療剤との組合せで投与することもできる。具体的な実施態様において、2種の(又はそれより多い)薬剤の同時投与は、使用されるそれぞれの用量を顕著に減少させることができ、それによって、見られる副作用を低減させる。
【0112】
一実施態様において、本発明の化合物は、炎症性病態の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、免疫調節性薬剤、例えば、アザチオプリン、コルチコステロイド(例えば、プレドニゾロン又はデキサメタゾン)、シクロホスファミド、シクロスポリンA、タクロリムス、ミコフェノール酸モフェチル、ムロモナブ-CD3(OKT-3、例えば、Orthocolone(登録商標))、ATG、アスピリン、アセトアミノフェン、イブプロフェン、ナプロキセン、及びピロキシカムを含むが、これらに限定されない。
【0113】
一実施態様において、本発明の化合物は、関節炎(例えば、関節リウマチ)の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、鎮痛薬、非ステロイド性抗炎症薬(NSAID)、ステロイド、合成DMARD(例えば、限定はされないが、メトトレキサート、レフルノミド、スルファサラジン、オーラノフィン、金チオリンゴ酸ナトリウム、ペニシラミン、クロロキン、ヒドロキシクロロキン、アザチオプリン、及びシクロスポリン)、及び生物学的DMARD(例えば、限定はされないが、インフリキシマブ、エタネルセプト、アダリムマブ、リツキシマブ、及びアバタセプト)を含むが、これらに限定されない。
【0114】
一実施態様において、本発明の化合物は、増殖性疾患の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、メトトレキサート、ロイコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロホスファミド、5-フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗-HER2モノクローナル抗体(例えば、Herceptin(商標))、カペシタビン、塩酸ラロキシフェン、EGFR阻害剤(例えば、lressa(登録商標)、Tarceva(商標)、Erbitux(商標))、VEGF阻害剤(例えば、Avastin(商標))、プロテアソーム阻害剤(例えば、Velcade(商標))、Glivec(登録商標)及びhsp90阻害剤(例えば、17-AAG)を含むが、これらに限定されない。加えて、本発明の化合物は、限定はされないが、放射線療法又は外科手術を含む他の治療法と組合せて投与することができる。具体的な実施態様において、該増殖性疾患は、ガン、骨髄増殖性疾患、又は白血病から選択される。
【0115】
一実施態様において、本発明の化合物は、自己免疫疾患の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、グルココルチコイド、細胞分裂阻害剤(例えば、プリンアナログ)、アルキル化剤(例えば、ナイトロジェンマスタード(シクロホスファミド)、ニトロソウレア、白金化合物、その他)、代謝拮抗薬(例えば、メトトレキサート、アザチオプリン及びメルカプトプリン)、細胞毒性抗生物質(例えば、ダクチノマイシンアントラサイクリン系薬剤、マイトマイシンC、ブレオマイシン及びミトラマイシン)、抗体(例えば、抗-CD20、抗-CD25、又は抗-CD3(OTK3)モノクローナル抗体、Atgam(登録商標)及びThymoglobuline(登録商標))、シクロスポリン、タクロリムス、ラパマイシン(シロリムス)、インターフェロン(例えば、IFN-β)、TNF結合タンパク質(例えば、インフリキシマブ(Remicade(商標))、エタネルセプト(Enbrel(商標))、又はアダリムマブ(Humira(商標)))、ミコフェノレート、フィンゴリモド、及びミリオシンを含むが、これらに限定されない。
【0116】
一実施態様において、本発明の化合物は、移植拒絶反応の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、カルシニューリン阻害剤(例えば、シクロスポリン又はタクロリムス(FK506))、mTOR阻害剤(例えば、シロリムス、エベロリムス)、抗増殖剤(例えば、アザチオプリン、ミコフェノール酸)、副腎皮質ステロイド(例えば、プレドニゾロン、ヒドロコルチゾン)、抗体(例えば、モノクローナル抗-IL-2Rα受容体抗体、バシリキシマブ、ダクリズマブ)、ポリクローナル抗T細胞抗体(例えば、抗胸腺細胞グロブリン(ATG)、抗リンパ球グロブリン(ALG))を含むが、これらに限定されない。
【0117】
一実施態様において、本発明の化合物は、喘息及び/又は鼻炎及び/又はCOPDの治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、β2-アドレナリン受容体刺激薬(例えば、サルブタモール、レバルブテロール、テルブタリン、及びビトルテロール)、エピネフリン(吸入又は錠剤)、抗コリン薬(例えば、臭化イプラトロピウム)、グルココルチコイド(経口又は吸入)、長時間作用性β2-刺激薬(例えば、サルメテロール、ホルモテロール、バンブテロール、及び徐放性経口アルブテロール)、吸入ステロイド薬と長時間作用性気管支拡張薬との組合せ(例えば、フルチカゾン/サルメテロール、ブデソニド/ホルモテロール)、ロイコトリエン拮抗薬及び合成阻害剤(例えば、モンテルカスト、ザフィルルカスト、及びジロートン)、メディエーター放出の阻害剤(例えば、クロモグリケート及びケトチフェン)、IgE応答の生体調節因子(例えば、オマリズマブ)、抗ヒスタミン剤(例えば、セテリジン、シンナリジン、フェキソフェナジン)、及び血管収縮薬(例えば、オキシメタゾリン、キシロメタゾリン、ナファゾリン、及びトラマゾリン)を含むが、これらに限定されない。
【0118】
加えて、本発明の化合物は、喘息及び/又はCOPDの緊急治療と組合せて投与することができ、そのような治療には、酸素又はヘリオックス投与、サルブタモール又はテルブタリン噴霧(抗コリン薬(例えば、イプラトロピウム)と任意に併用)、全身性ステロイド薬(経口又は静脈内、例えば、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、デキサメタゾン、又はヒドロコルチゾン)、静脈内サルブタモール、非特異的β-刺激薬、注射又は吸入(例えば、エピネフリン、イソエタリン、イソプロテレノール、メタプロテレノール)、抗コリン薬(静脈内又は噴霧、例えば、グリコピロレート、アトロピン、イプラトロピウム)、メチルキサンチン(テオフィリン、アミノフィリン、バミフィリン)、気管支拡張性効果を有する吸入麻酔(例えば、イソフルラン、ハロタン、エンフルラン)、ケタミン、及び静脈内硫酸マグネシウムを含む。
【0119】
一実施態様において、本発明の化合物は、過敏性腸疾患(IBD)の治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、グルココルチコイド(例えば、プレドニゾン、ブデソニド)、合成疾患修飾性免疫調節薬(例えば、メトトレキサート、レフルノミド、スルファサラジン、メサラジン、アザチオプリン、6-メルカプトプリン、及びシクロスポリン)及び生物学的疾患修飾性免疫調節薬(インフリキシマブ、アダリムマブ、リツキシマブ及びアバタセプト)を含むが、これらに限定されない。
【0120】
一実施態様において、本発明の化合物は、全身性エリテマトーデスの治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、疾患修飾性抗リウマチ薬(DMARD)、例えば、抗マラリア薬(例えば、プラキニル、ヒドロキシクロロキン)、免疫抑制剤(例えば、メトトレキサート及びアザチオプリン)、シクロホスファミド及びミコフェノール酸;免疫抑制薬及び鎮痛薬、例えば、非ステロイド性抗炎症薬、アヘン剤(例えば、デキストロプロポキシフェン及びココダモール)、オピオイド(例えば、ヒドロコドン、オキシコドン、MSコンチン又はメサドン)及びフェンタニルデュラゲシク経皮吸収パッチを含むが、これらに限定されない。
【0121】
一実施態様において、本発明の化合物は、乾癬の治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、コールタール、ジトラノール(アントラリン)、デスオキシメタゾン(Topicort(商標))などの副腎皮質ステロイド、フルオシノニド、ビタミンD3アナログ(例えば、カルシポトリオール)、アルガン油、及びレチノイド(エトレチナート、アシトレチン、タザロテン)を含有する浴溶液、保湿剤、薬用クリーム、及び軟膏剤などの局所的治療薬、メトトレキサート、シクロスポリン、レチノイド、チオグアニン、ヒドロキシウレア、スルファサラジン、ミコフェノール酸モフェチル、アザチオプリン、タクロリムス、フマル酸エステルなどの全身治療薬、又はAmevive(商標)、Enbrel(商標)、Humira(商標)、Remicade(商標)、Raptiva(商標)、及びウステキヌマブ(IL-12及びIL-23遮断薬)などの生物製剤を含むが、これらに限定されない。加えて、本発明の化合物は、限定はされないが、光線療法又は光化学療法(例えば、ソラレン長波長紫外線治療(PUVA))を含む他の治療法と組合せて投与することができる。
【0122】
当業者に明らかであるように、同時投与とは、同じ治療計画の一部として2種以上の治療薬を患者に送達する任意の手段を含むものである。2種以上の薬剤は単一製剤で同時に投与できるが、これは必須ではない。該薬剤は、異なる製剤で、かつ異なる時間に、投与してもよい。
【0123】
(全般的合成手順)
(全般)
本発明の化合物は、以下の全般的方法及び手順を利用して容易に利用できる出発物質から製造できる。典型的又は好ましいプロセス条件(すなわち、反応温度、時間、反応物のモル比、溶媒、圧力など)が与えられる場合、他のプロセス条件も他に明記しない限り利用できることは理解されるであろう。最適な反応条件は、使用する特定の反応物又は溶媒により変わり得るが、そのような条件は、通例の最適化手順によって当業者により決定できる。
【0124】
さらに、当業者には明らかであるように、特定の官能基が望ましくない反応を受けるのを防ぐために、従来型の保護基が必要となることがある。特定の官能基のための好適な保護基、並びに保護及び脱保護のための適当な条件の選択は、当該分野において周知である。例えば、多数の保護基並びにそれらの導入及び除去は、T. W. Greene及びP. G. M. Wutsの文献、「有機合成における保護基(Protecting Groups in Organic Synthesis)」、第2版、Wiley, New York, 1991及びその中で引用された文献に記載されている。
【0125】
以下の方法は、先に列記された代表的なビシクロヘテロアリールの製造に関して詳細に提示されている。本発明の化合物は、公知又は市販の出発物質及び試薬から、有機合成の分野の当業者により製造できる。
【0126】
全ての試薬は市販の等級であり、他に明記しない限り、さらに精製せずに入手したままで使用した。市販の無水溶媒を、不活性雰囲気下で実施される反応に使用した。他に特に明記しない限り、試薬等級溶媒を他の全ての場合において使用した。カラムクロマトグラフィーをシリカゲル60(35〜70μm)で実施した。薄層クロマトグラフィーは、予め被覆してあるシリカゲルF-254プレート(0.25mm厚)を使用して実施した。1H NMRスペクトルは、Bruker社製DPX 400核磁気共鳴分光計(400MHz)で記録した。1H NMRスペクトルの化学シフト(δ)は、内部標準としてのテトラメチルシラン(δ0.00)又は適切な残留溶媒ピーク、すなわちCHCl3(δ7.27)に対して百万分率(ppm)で報告される。多重度は、シングレット(s)、ダブレット(d)、トリプレット(t)、カルテット(q)、マルチプレット(m)及びブロード(br)として与えられる。結合定数(J)は、Hzで与えられる。エレクトロスプレーMSスペクトルは、MicromassプラットフォームLC/MSスペクトロメータで得られた。全てのLCMS分析に使用したカラムは以下のものである:Waters社製Acquity UPLC BEH C18 1.7μm、2.1mm ID×50mm L(Part No.186002350)。分取HPLC:Waters社製XBridge Prep C18 5μm ODB 19mm ID×100mm L(Part No.186002978)。全ての方法は、MeCN/H2O勾配を使用している。H2Oは、0.1%TFA又は0.1%NH3のいずれかを含有する。
【0127】
下記は、実験セクションにおいて使用する省略形のリストである。
【表1】
【0128】
(本発明の化合物の合成製造)
本発明の化合物は、以下のスキームに従って製造できる。
【化3】
式中、Arはフェニル-L1-ヘテロシクロアルキルを表し、ここで、式中、L1は-CO-であり、ヘテロシクロアルキル基は任意に置換されている。
【0129】
(全般)
(1.1.1 1-(6-ブロモ-ピリジン-2-イル)-3-カルボエトキシ-チオ尿素(2))
【化4】
5℃に冷却した2-アミノ-6-ブロモピリジン(1)(253.8g、1.467mol)のDCM(2.5L)溶液に、イソチオシアン酸エトキシカルボニル(173.0mL、1.467mol)を15分にわたって滴加する。次いで、該反応混合物を室温(20℃)に温め、16時間撹拌する。真空での蒸発により固形物を得て、これを濾過により回収し、石油(3×600mL)で完全に洗浄し、風乾させて(2)を得る。該チオ尿素は、精製することなくそのまま次工程に使用することができる。
【化5】
【0130】
(1.1.2 5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イルアミン(3))
【化6】
EtOH/MeOH(1:1、900mL)中の塩酸ヒドロキシルアミン(101.8g、1.465mol)の懸濁液に、N,N-ジイソプロピルエチルアミン(145.3mL、0.879mol)を添加し、該混合物を室温(20℃)で1時間撹拌する。次に、1-(6-ブロモ-ピリジン-2-イル)-3-カルボエトキシ-チオ尿素(2)(89.0g、0.293mol)を添加し、該混合物を還流までゆっくりと加熱する(注意:発生するH2Sをクエンチするのにブリーチスクラバーが必要である)。3時間の還流後、該混合物を放冷し、濾過して沈殿した固形物を回収する。該濾液を真空中で蒸発させ、H2O(250mL)を添加し、濾過することによって、さらに生成物を回収する。合わせた固形物をH2O(250mL)、EtOH/MeOH(1:1、250mL)及びEt2O(250mL)で連続的に洗浄し、次いで、真空中で乾燥させて、トリアゾロピリジン誘導体(3)を固形物として得る。該化合物は、精製することなくそのまま次工程に使用することができる。
【化7】
【0131】
(1.1.3 シクロプロパンカルボン酸(5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル)-アミド(4))
【化8】
5℃の2-アミノ-トリアゾロピリジン(3)(7.10g、33.3mmol)の無水CH3CN(150mL)溶液に、Et3N(11.6mL、83.3mmol)を添加し、続いてシクロプロパンカルボニルクロリド(83.3mmol)を添加する。次いで、該反応混合物を周囲温度まで温め、すべての出発物質(3)が消費されるまで撹拌する。必要であれば、Et3N(4.64mL、33.3mmol)及びシクロプロパンカルボニルクロリド(33.3mmol)をさらに添加して、完全な反応を確実にする。真空中で溶媒を蒸発させた後、生じた残渣を7Nのメタノール性アンモニア溶液(50mL)で処理し、周囲温度で撹拌して(1〜16時間)、ビス-アシル化生成物を加水分解する。生成物の単離は、真空中で揮発性物質を除去し、続いて、Et2O(50mL)でトリチュレイトすることによってなされる。該固形物を濾過によって回収し、H2O(2×50mL)、アセトン(50mL)、及びEt2O(50mL)で洗浄し、次いで、真空中で乾燥させて、必要なブロモ中間体(4)を得る。
【0132】
(本発明の化合物の製造のための合成手順)
(化合物1)
(工程1:スズキカップリング)
【化9】
4-カルボキシフェニルボロン酸(3.5 g, 0.021 mol.)を、シクロプロパンカルボン酸(5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル)-アミド(中間体4、5 g, 0.018 mol)の1,4-ジオキサン/水(5:1)溶液に加えた。K2CO3 (5.0 g, 0.036 mol)及びPdCl2dppf (5%)を該溶液に加えた。次いで、生じた混合物を、90℃の油浴中で16時間従来の加熱により加熱した。1M HCl溶液を加えると、酸性溶液中に沈殿物が生じた。沈殿物を濾過し、真空下で乾燥させると、標記化合物を与え、さらに精製することなく次の工程に使用した。
【0133】
(工程2:シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミド(化合物1))
【化10】
EDCI (3.59 g, 0.019 mol)、HOBt (2.53 g, 0.019 mol)、及びDIPEA (4.48 mL)を、4-[2-(シクロプロパンカルボニル-アミノ)-[1,2,4]トリアゾロ[1,5-a]ピリジン-5-イル]-安息香酸(4g, 0.012 mol)の室温のDCM (150 mL)溶液に加えた。生じた混合物を室温で10分間撹拌した。ジメチルアゼチジン塩酸塩(1.64 g, 0.013 mol)を該溶液に加え、反応物を16時間撹拌する。水を該反応混合物に加えた。有機層を分離し、2N NaOH溶液、2N HCl溶液、及び水で洗浄した。有機相をMgSO4で乾燥させ、濾過し、真空下で蒸発させた。フラッシュクロマトグラフィー(溶離液: 1:1 ぺトロール/EtOAcからニートEtOAc)により精製すると、シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミドを与えた。
【0134】
本明細書に記載された合成方法により製造された、又は製造可能な本発明の化合物を、以下の表Iに列記する。本発明の化合物のNMRスペクトルデータを表IIに与える。
【0135】
【表2】
【0136】
【表3】
【0137】
(生物学的実施例)
(実施例1−インビトロアッセイ)
(実施例1.1 JAK1阻害アッセイ)
組換えヒトJAK1(触媒ドメイン、アミノ酸866-1154;カタログ番号PV4774)をInvitrogen社から購入した。1ngのJAK1を、白色384 Luminotrac 200プレート(Greiner社製、カタログ番号781075)中で、被験化合物又はビヒクル(DMSO、最終濃度1%)を含む4μLとともに、又はなしで、総量20μLのキナーゼ反応緩衝液(25mM MOPS pH6.8, 0.016% Brij-35, 8.33mM MgCl2, 3.33mM DTT, 7μM ATP)中で、20 nMのUlight-JAK1(tyr1023)ペプチド(Perkin Elmer社製、カタログ番号TRF0121)とともにインキュベートした。室温で60分後、20μL/ウェルの検出混合液(1×検出緩衝液 (Perkin Elmer社製、カタログ番号CR97-100C)、0.5nMのユーロピウム-抗-ホスホチロシン(PT66) (Perkin Elmer社製、カタログ番号AD0068)、10 mM EDTA)の添加により反応を停止させた。Envisionを使用し、320nmでの励起及び615nmでの発光測定により読み取りを行う(Perkin Elmer)。キナーゼ活性は、ビヒクルの存在下で得られた相対蛍光単位(RFU)からポジティブコントロール阻害剤(10μMスタウロスポリン)の存在下で得られたRFUを引くことにより計算した。この活性を阻害する被験化合物の能力を以下のとおり決定した。
【0138】
阻害率=((被験化合物を含む試料で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU)/(ビヒクルの存在下で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU))×100。
【0139】
JAK1アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/5の段階希釈で、1%DMSOの最終濃度で8点(20μM-4μM-800nM-160nM-32nM-6.4nM-1.28nM-0.26nM)で試験する。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させる(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0140】
【表4】
【0141】
(実施例1.2 JAK1 Ki決定アッセイ)
Kiの決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。JAK1(Invitrogen社製、PV4774)を、最終濃度500ng/mlで使用した。基質は、ポリ(Glu, Tyr)ナトリウム塩(4:1)、MW 20000-50000(Sigma社製, P0275)であった。反応は、25mM Hepes pH 7.5; 0,01% Tween20, 10 mM MgCl2中で、ATP及び化合物の濃度を変えながら実施し、150mMのリン酸を添加して停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0142】
化合物1をこのアッセイで試験すると、6.9nMのKi値を測定した。
【0143】
別法としては、Ki決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。1 ngのJAK1(Invitrogen社製、PV4774)をアッセイに使用した。基質は、50nM Ulight-JAK-1(Tyr1023) Peptide(Perkin Elmer社製、TRF0121)であった。反応は、25mM MOPS pH 6.8, 0.01%, 2 mM DTT, 5 mM MgCl2 Brij-35中で、ATP及び化合物の濃度を変えながら実施した。リン酸化された基質を、Eu標識抗ホスホチロシン抗体PT66(Perkin Elmer社製、AD0068)を使用して測定した。読み取りは、envision(Perkin Elmer社製)で、320nmでの励起及び615nm及び665nmでの発光で実施した。
【0144】
化合物1をこのアッセイで試験すると、5.6nMのKi値を測定した。
【0145】
(実施例1.3 JAK2阻害アッセイ)
組換えヒトJAK2(触媒ドメイン、アミノ酸866-1154;カタログ番号PV4210)をInvitrogen社から購入した。0.0125mUのJAK2を、白色384 Luminotrac 200プレート(Greiner社製、カタログ番号781075)中で、被験化合物又はビヒクル(DMSO、最終濃度1%)を含む4μLとともに、又はなしで、総量20μLのキナーゼ反応緩衝液(41.66mM HEPES pH7.0, 0.016% Triton X-100, 12.5mM MgCl2, 3.33mM DTT, 7.5μM ATP)中で、25 nMのUlight-JAK1(tyr1023)ペプチド(Perkin Elmer社製、カタログ番号TRF0121)とともにインキュベートした。室温で60分後、20μL/ウェルの検出混合液(1×検出緩衝液 (Perkin Elmer社製、カタログ番号CR97-100C)、0.5nMのユーロピウム-抗-ホスホチロシン(PT66) (Perkin Elmer社製、カタログ番号AD0068)、10 mM EDTA)の添加により反応を停止させた。Envision(Perkin Elmer社製)を使用し、320nmでの励起及び615nmでの発光測定により読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られた相対蛍光単位(RFU)からポジティブコントロール阻害剤(10μMスタウロスポリン)の存在下で得られたRFUを引くことにより計算した。この活性を阻害する被験化合物の能力を以下のとおり決定した。
【0146】
阻害率=((被験化合物を含む試料で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU)/(ビヒクルの存在下で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU))×100。
【0147】
JAK2アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製する。各化合物を通常20μMの濃度で試験し、続いて、1/5の段階希釈で、1%DMSOの最終濃度で8点(20μM-4μM-800nM-160nM-32nM-6.4nM-1.28nM-0.26nM)で試験する。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させる(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0148】
【表5】
【0149】
(実施例1.4 JAK2 Ki決定アッセイ)
Kiの決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。0.025mUのJAK2(Invitrogen社製、PV4210)を、アッセイに使用した。基質は、ポリ(Glu, Tyr)ナトリウム塩(4:1)、MW 20000-50000(Sigma社製, P0275)であった。反応は、10 mM MOPS pH 7.5, 0.5 mM EDTA, 0.01% Brij-35, 1 mM DTT, 15 mM MgAc中で、ATP及び化合物の濃度を変えながら実施し、150mMのリン酸を添加して停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0150】
化合物1をこのアッセイで試験すると、126nMのKi値を測定した。
【0151】
別法としては、Ki測定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。0.0125mUのJAK2(Invitrogen社製、PV4210)をアッセイに使用した。基質は、50nM Ulight-JAK-1(Tyr1023) Peptide(Perkin Elmer社製、TRF0121)であった。反応は、25 mM HEPES pH 7.0, 0.01% Triton X-100, 2 mM DTT, 7.5 mM MgCl2中で、ATP及び化合物の濃度を変えながら実施した。リン酸化された基質を、Eu標識抗ホスホチロシン抗体PT66(Perkin Elmer社製、AD0068)を使用して測定した。読み取りは、envision(Perkin Elmer社製)で、320 nmでの励起及び615 nm及び665 nmでの発光で実施した。
【0152】
化合物1をこのアッセイで試験すると、35nMのKi値を測定した。
【0153】
(実施例1.5 JAK3阻害アッセイ)
組換えヒトJAK3触媒ドメイン(アミノ酸781-1124;カタログ番号PV3855)をInvitrogen社から購入した。ポリプロピレン製96ウェルプレート(Greiner社製、V字底)において、0.025mUのJAK3を2.5μgのポリGT基質(Sigma社製、カタログ番号P0275)とともに、被験化合物又はビヒクル(DMSO、最終濃度1%)を含有する5μLとともに、又はなしで、総量25μLのキナーゼ反応緩衝液(25mMのTris pH7.5、0.5mMのEGTA、0.5mMのNa3VO4、5mMのb-グリセロールホスフェート、0.01%のTritonX-100、1μMの非放射性ATP、0.25μCiの33P-γ-ATP(GE Healthcare社製、カタログ番号AH9968)最終濃度)中でインキュベートした。30℃で105分後、25μL/ウェルの150mMリン酸を添加して反応を停止させた。該終結したキナーゼ反応物の全てを、セルハーベスター(Perkin Elmer社製)を使用して、予洗い(75mMのリン酸)した96ウェルフィルタープレート(Perkin Elmer社製、カタログ番号6005177)に移した。プレートを1ウェルあたり300μLの75mMリン酸溶液で6回洗浄し、該プレートの底を密閉した。40μL/ウェルのMicroscint-20を添加し、該プレートの上部を密閉し、Topcount(Perkin Elmer社製)を使用して読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られたカウント毎分(cpm)から、ポジティブコントロール阻害剤(10μMのスタウロスポリン)の存在下で得られたcpmを引くことによって算出した。この活性を阻害する被験化合物の能力は、以下のように決定した。
阻害率=((被験化合物が存在する試料について測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm)/(ビヒクルの存在下で測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm))×100。
【0154】
JAK3アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/3の段階希釈で、1%DMSOの最終濃度で8点(20μM-6.67μM-2.22μM-740nM-247nM-82nM-27nM-9nM)で試験した。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させた(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0155】
【表6】
【0156】
(実施例1.6 JAK3 Ki測定アッセイ)
Kiの測定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。JAK3 (Carna Biosciences社製、09CBS-0625B)を、最終濃度10 ng/mlで使用した。基質はポリ(Glu,Tyr)ナトリウム塩(4:1)、MW 20000-50000 (Sigma社製、P0275)であった。反応を、25mM Tris pH 7.5, 0.01% Triton X-100, 0.5mM EGTA, 2.5mM DTT, 0.5mM Na3VO4, 5mM b-グリセロールホスフェート, 10mM MgCl2中で、ATP及び化合物の濃度を変えながら実施し、150mMリン酸の添加により停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0157】
化合物1をこのアッセイで試験すると、188nMのKi値を測定した。
【0158】
(実施例1.7 TYK2阻害アッセイ)
組換えヒトTYK2触媒ドメイン(アミノ酸871-1187;カタログ番号08-147)をCarna biosciences社から購入した。ポリプロピレン製96ウェルプレート(Greiner社製、V字底)において、5ngのTYK2を12.5μgのポリGT基質(Sigma社製、カタログ番号P0275)とともに、被験化合物又はビヒクル(DMSO、最終濃度1%)を含有する5μLとともに、又はなしで、総量25μLのキナーゼ反応緩衝液(25mMのHepes pH7.5、100mMのNaCl、0.2mMのNa3VO4、0.1%のNP-40、0.1μMの非放射性ATP、0.125μCiの33P-γ-ATP (GE Healthcare社製、カタログ番号AH9968)最終濃度)中でインキュベートした。30℃で90分後、25μL/ウェルの150mMリン酸を添加して反応を停止させた。該終結したキナーゼ反応物の全てを、セルハーベスター(Perkin Elmer社製)を使用して、予洗い(75mMのリン酸)した96ウェルフィルタープレート(Perkin Elmer社製、カタログ番号6005177)に移した。プレートを1ウェルあたり300μLの75mM リン酸溶液で6回洗浄し、該プレートの底を密閉した。40μL/ウェルのMicroscint-20を添加し、該プレートの上部を密閉し、Topcount(Perkin Elmer社製)を使用して読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られたカウント毎分(cpm)から、ポジティブコントロール阻害剤(10μMのスタウロスポリン)の存在下で得られたcpmを引くことによって算出した。この活性を阻害する被験化合物の能力は以下のように決定した。
阻害率=((被験化合物が存在する試料について測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm)/(ビヒクルの存在下で測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm))×100。
【0159】
TYK2アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/3の段階希釈で、1%DMSOの最終濃度で8点(20μM-6.67μM-2.22μM-740nM-247nM-82nM-27nM-9nM)で試験した。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させた(例えば、5μM、1μM)。
【0160】
【表7】
【0161】
(実施例2 細胞アッセイ)
(実施例2.1 JAK-STATシグナル伝達アッセイ)
HeLa細胞を、10%の熱失活したウシ胎児血清、100U/mLのペニシリン、及び100μg/mLのストレプトマイシンを含有するダルベッコ変法イーグル培地(DMEM)中に維持した。トランスフェクションのために70%コンフルエンスでHeLa細胞を使用した。87μLの細胞培地中20,000個の細胞を、96ウェルプレートフォーマットにおいて、1ウェルあたり0.32μLのJet-PEI(Polyplus社製)をトランスフェクション試薬として使用して、40ngのpSTAT1(2)-ルシフェラーゼレポーター(Panomics社製)、内部標準レポーターとして8ngのLacZレポーター、及び52ngのpBSKで一過性にトランスフェクションさせた。37℃、10%CO2で一晩インキュベートした後、トランスフェクション培地を除去した。75μLのDMEM+1.5%の熱失活したウシ胎児血清を添加した。6.7倍濃縮した15μLの化合物を60分間添加し、次いで、最終濃度33ng/mLの10μLのヒトOSM(Peprotech社製)を添加した。
【0162】
全ての化合物を20μMから開始して、1/3段階希釈し、0.2%DMSOの最終濃度で全8用量(20μM-6.6μM-2.2μM-740nM-250nM-82nM-27nM-9nM)で二連で試験した。
【0163】
37℃、10%CO2で一晩インキュベートした後、細胞を100μLの溶解緩衝液/ウェル(PBS、0.9mMのCaCl2、0.5mMのMgCl2、5%のトレハロース、0.025%のTergitol NP9、0.15%のBSA)で溶解した。
【0164】
40μLの細胞溶解物を使用して、180μLのβGal溶液(30μlのONPG 4mg/mL+150μLのβ-ガラクトシダーゼ緩衝液(0.06MのNa2HPO4、0.04MのNaH2PO4、1mMのMgCl2))を20分間添加することによりβ-ガラクトシダーゼ活性を読み取った。50μLのNa2CO3 1Mの添加によって該反応を停止させた。405nmで吸光度を読み取った。
【0165】
ルシフェラーゼ活性を、製造元(Perkin Elmer社)による説明どおりに、40μlの細胞溶解物+40μlのSteadylite(登録商標)を使用してEnvison(Perkin Elmer社製)で測定した。
【0166】
10μMの汎JAK阻害剤をポジティブコントロール(100%阻害)として使用した。ネガティブコントロールとして、0.5%のDMSO(0%阻害)を使用した。ポジティブ及びネガティブコントロールを使用して、z'及び「阻害率」(PIN)値を算出した。
【0167】
阻害率=((ビヒクル存在下で測定された蛍光−被験化合物が存在する試料について測定された蛍光)/(ビヒクルの存在下で測定された蛍光−トリガーなしの試料について測定された蛍光))×100である。
【0168】
用量−反応において、試験した化合物についてPIN値をプロットし、EC50値を算出した。
【0169】
【表8】
【0170】
(実施例2.2 OSM/IL-1βシグナル伝達アッセイ)
OSM及びIL-1βは、ヒト軟骨肉腫株化細胞系SW1353において、MMP13レベルを相乗的に上方制御することが示された。該細胞を96ウェルプレートにおいて、37℃、5%CO2でインキュベートした10%(v/v)のFBS及び1%のペニシリン/ストレプトマイシン(InVitrogen社製)を含有するDMEM(Invitrogen社製)の容量120μL中、15,000細胞/ウェルで播種した。細胞を、25ng/mLのOSM及び1ng/mLのIL-1βに達するまで、15μLのOSM及びIL-1βでトリガーする1時間前に、2%のDMSOを有するM199培地中で15μLの化合物とともにプレインキュベートし、トリガーした48時間後に条件培地でMMP13レベルを測定した。抗体捕獲活性アッセイを利用してMMP13活性を測定した。この目的のために、384ウェルプレート(NUNC社製、460518、MaxiSorb black)を35μLの1.5μg/mL抗ヒトMMP13抗体(R&D Systems社製、MAB511)溶液で24時間4℃で被覆した。該ウェルをPBS+0.05%Tweenで2回洗浄した後、残りの結合部位を100μLのPBS中5%の脱脂粉乳(Santa Cruz社製、sc-2325、Blotto)で24時間4℃でブロックした。次に、該ウェルをPBS+0.05%Tweenで2回洗浄し、100倍希釈したブロッキング緩衝液中の、MMP13を含む培養上清の1/10希釈を35μL添加し、室温で4時間インキュベートした。次に、該ウェルをPBS+0.05% Tweenで2回洗浄し、続いて、35μLの1.5mM 4-アミノフェニル水銀アセタート(APMA)(Sigma社製、A9563)溶液を添加し、37℃で1時間インキュベートすることによってMMP13を活性化した。該ウェルをPBS+0.05%Tweenで再度洗浄し、35μLのMMP13基質(Biomol社製、P-126、OmniMMP蛍光発生基質)を添加した。37℃で24時間のインキュベーションの後、転化した基質の蛍光をPerkin Elmer社製Wallac EnVision2102マルチラベルリーダー(励起波長:320nm、発光波長:405nm)で測定した。
【0171】
阻害率=((ビヒクル存在下で測定された蛍光−被験化合物が存在する試料について測定された蛍光)/(ビヒクルの存在下で測定された蛍光−トリガーなしの試料について測定された蛍光))×100である。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0172】
【表9】
【0173】
(実施例2.3 PBL増殖アッセイ)
ヒト末梢血リンパ球(PBL)をIL-2で刺激し、BrdU取り込みアッセイを使用して増殖を測定する。まず、PHAを用いてPBLを72時間刺激してIL-2受容体を誘発し、24時間絶食させて細胞増殖を停止させ、続いて、さらに72時間(24時間のBrdU標識化を含む)IL-2刺激を行う。細胞を被験化合物とともにIL-2添加の1時間前にプレインキュベートする。10%(v/v)FBSを含有するRPMI1640で細胞を培養する。
【0174】
(実施例2.4 全血アッセイ(WBA))
(2.4.1 IFNα刺激プロトコール)
被験化合物がインビボでJAK2依存性シグナル伝達経路よりもJAK1シグナル伝達経路を阻害する選択性を予測するため、生理学的に関連あるインビトロモデルを、ヒトの全血を使用して開発した。WBAアッセイにおいて、インフォームドコンセントを与えたヒトの志願者から採取された血液を化合物によりエクスビボで処理し(1時間)、次いで、インターフェロンα(IFNα、JAK1依存性経路)により30分間、又は顆粒球マクロファージコロニー刺激因子(GM-CSF、JAK2依存性経路)により2時間のいずれかで刺激した。
【0175】
(2.4.1.1 ホスホ-STAT1アッセイ)
IFNα刺激に関して、白血球抽出物中でのIFNαによるシグナル伝達性転写因子1のリン酸化(pSTAT1)の増加をpSTAT1 ELISAアッセイを利用して測定した。インターフェロンα(IFNα)トリガリングの後のシグナル伝達性転写因子1(STAT1)のリン酸化はJAK1-媒介性事象である。ホスホ-STAT1アッセイは、細胞抽出物におけるホスホ-STAT1レベルの測定に利用したが、化合物がJAK1-依存性シグナル伝達経路を阻害する能力を評価するために開発された。
【0176】
インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血を、化合物によりエクスビボで処理し(1時間)、次いで、IFNαにより30分間刺激した。白血球抽出物におけるIFNαによるSTAT1のリン酸化の増加を、ホスホ-STAT1 ELISAを利用して測定した。
【0177】
ACK溶解緩衝液は、0.15 M NH4Cl、10 mM KHCO3、0.1 mM EDTAからなっていた。該緩衝液のpHは7.3であった。
【0178】
10×細胞溶解緩衝濃縮液(Cell Signaling社製、PathScan Phospho-STAT1 (Tyr701) sandwich ELISAキットの一部)を、水で10倍に希釈した。使用前にプロテイナーゼ阻害剤を緩衝液に加えた。
【0179】
20μgのIFNαを40μLのH2Oに溶かして、500μg/mLのストック溶液を得る。ストック溶液を-20℃で保存した。
【0180】
化合物の3倍の希釈系列をDMSOで調製した(最高濃度:10 mM)。その後、化合物をさらに培地で希釈した(希釈係数は、所望の最終化合物濃度による)。
【0181】
(2.4.1.1.1 化合物と血液とのインキュベーション及びIFNαによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を392μLのアリコートに分けた。その後、4μLの化合物希釈液を各アリコートに加え、血液試料を37℃で1時間インキュベートした。IFNαストック溶液をRPMI培地で1000倍に希釈し、500 ng/mLの使用液を得た。4μLの500 ng/mL使用液を血液試料に加えた(最終濃度IFNα:5ng/ml)。該試料を37℃で30分間インキュベートした。
【0182】
(2.4.1.1.2 細胞抽出物の調製)
刺激期間の最後に、7.6 mLのACK緩衝液を血液試料に加え、赤血球を溶解させた。チューブを5回反転させて試料を混合し、反応物を氷上で5分間インキュベートした。RBCの溶解は、このインキュベーションの間に明らかであるはずである。300g、4℃で7分間の遠心分離により細胞をペレットにし、上清を除いた。10 mLの1×PBSを各チューブに加え、細胞ペレットを再懸濁化した。試料を300g、4℃で7分間再び遠心分離した。上清を除き、ペレットを500μLの1×PBSで再懸濁化した。次いで、細胞懸濁液をきれいな1.5 mL微量遠心チューブに移した。700g、4℃で5分間での遠心分離により細胞をペレットにした。上清を除き、ペレットを150μLの細胞溶解緩衝液に溶かした。試料を氷上で15分間インキュベートした。その後、試料を、さらに処理するまで-80℃で保存した。
【0183】
(2.4.1.1.3 ELISAによるSTAT1リン酸化の測定)
Cell Signaling社製のPathscan Phospho-STAT1 (Tyr701) Sandwich ELISAキット(カタログ番号7234)を使用して、ホスホ-STAT1レベルを測定した。
【0184】
細胞抽出物を氷上で解凍した。チューブを16,000g、4℃で5分間遠心分離し、清澄化された溶解物を採取した。その間に、キットのマイクロウェルストリップを室温に平衡化し、20×洗浄緩衝液を水で希釈して洗浄緩衝液を調製した。試料を、試料希釈液で2倍に希釈し、100μLをマイクロウェルストリップに加えた。ストリップを4℃で一晩インキュベートした。
【0185】
翌日、ウェルを洗浄緩衝液により3回洗浄した。100μLの検出抗体をウェルに加えた。ストリップを37℃で1時間インキュベートした。次いで、ウェルを洗浄緩衝液で再び3回洗浄した。100μLのHRP結合二次抗体を各ウェルに加え、試料を37℃でインキュベートした。30分後、ウェルを再び3回洗浄し、100μLのTMB基質を全てのウェルに加えた。試料が青くなった場合、100μLのSTOP溶液を加えて反応を停止させた。吸光度を450 nmで測定した。
【0186】
(2.4.1.2 IL-8 ELISA)
GM-CSF刺激に関して、血漿中のインターロイキン-8(IL-8)レベルの増加をIL-8 ELISA アッセイを利用して測定した。顆粒球マクロファージコロニー刺激因子(GM-CSF)-誘導インターロイキン8(IL-8)発現は、JAK2-媒介性事象である。IL-8 ELISAは、血漿試料中のIL-8レベルの測定に使用できるが、化合物がJAK2-依存性シグナル伝達経路を阻害する能力を評価するために開発された。
【0187】
インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血を化合物によりエクスビボで処理し(1時間)、次いでGM-CSFで2時間刺激した。血漿中のIL-8レベルの増加を、IL-8 ELISAアッセイを利用して測定した。
【0188】
10μgのGM-CSFを100μLのH2Oに溶かし、100μg/mLのストック溶液を得た。ストック溶液を-20℃で保存した。
【0189】
被験化合物の3倍の希釈系列をDMSO中に調製した(最高濃度:10 mM)。次いで、化合物を培地でさらに希釈した(希釈率は、所望の最終化合物濃度による)。
【0190】
(2.4.1.2.1 血液の化合物とのインキュベーション及びGM-CSFによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を245μLのアリコートに分けた。その後、2.5μLの被験化合物希釈液を各アリコートに加え、血液試料を37℃で1時間インキュベートした。GM-CSFストック溶液をRPMI培地で100倍に希釈し、1μg/mLの使用液を得た。2.5μLの1μg/mL使用液を血液試料に加えた(最終濃度GM-CSF:10 ng/ml)。該試料を37℃で2時間インキュベートした。
【0191】
(2.4.1.2.2 血漿試料の調製)
試料を1,000g、4℃で15分間遠心分離した。100μLの血漿を採取し、さらに使用するまで-80℃で保存した。
【0192】
(2.4.1.2.3 ELISAによるIL-8レベルの測定)
R&D Systems社製のHuman IL-8 Chemiluminescent Immunoassayキット(カタログ番号:Q8000B)を使用してIL-8レベルを測定した。
【0193】
洗浄緩衝液は、10×洗浄緩衝液をH2Oに希釈して調製した。使用グロ試薬は、1部のGlo Reagent 1を2部のGlo Reagent Bに、使用の15分から4時間前に添加して調製した。
【0194】
100μLのアッセイ希釈液RD1-86を各ウェルに加えた。その後、50μLの試料(血漿)を加えた。ELISAプレートを室温で2時間500rpmでインキュベートした。全てのウェルを洗浄緩衝液で4回洗浄し、200μLのIL-8コンジュゲートを各ウェルに加えた。室温で3時間インキュベートした後、ウェルを洗浄緩衝液で4回洗浄し、100μLの使用グロ試薬を各ウェルに加えた。ELISAプレートを室温で5分間インキュベートした(遮光する)。発光を測定した(0.5秒/ウェル読み取り時間)。
【0195】
(2.4.1.3 結果)
INFαに誘導されるpSTAT1レベル増加を阻害する化合物1のpIC50は、6.32±0.07(SEM)であり、一方GM-CSFに誘導されたIL-8の増加の阻害に対するpIC50は、4.87±0.13(SEM)であった(6人の血液提供者を利用して得られた結果)。これは、化合物1が、JAK2経路よりもJAK1経路に対して28倍選択性があることを示す。したがって、細胞環境では、化合物1がJAK2よりもJAK1に対して選択性を示すことは明らかである。化合物1は、構造的に関連する公知の化合物又は他の公知のJAK阻害剤に比べて、JAK2よりもJAK1に対して高い選択性を示すようである。
【0196】
(2.4.1.3 データキュレーション処理及び結果)
結果をさらに精密にするため、細胞抽出物中のIFNαによるホスホSTAT1誘導の阻害又は血漿中のGM-CSFによるIL-8誘導の阻害を、化合物濃度に対してプロットし、Graphpadソフトウェアを利用してIC50値を誘導した。R2が0.8より大きく、ヒルスロープが3より小さい場合にデータを保持し、両アッセイに対して有効なデータを得られた提供者のみを保持した。
【0197】
例えば、この追加のデータ分析プロトコールに供された場合、INFα誘導性のpSTAT1レベルの増加の阻害に関する化合物1のpIC50測定値は6.21±0.09 (SEM)であり、一方GM-CSF誘導性のIL-8の増加の阻害に関するpIC50は4.97±0.14 (SEM)であった(6人の血液提供者を利用して得られた結果)。このことは、化合物1が、JAK2経路に対してJAK1経路に対する選択性が17.6倍であることを立証している。したがって、細胞環境中で、化合物1がJAK2よりもJAK1に対して選択性を示すことは明らかである。化合物1は、構造的に関連する公知の化合物又は他の公知のJAK阻害剤に比べて、JAK2よりもJAK1に対して高い選択性を示すようである。
【0198】
(2.4.2 IL-6刺激プロトコール)
フローサイトメトリー分析も実施して、ヒトの全血を利用してエクスビボで、JAK2を上回るJAK1への化合物選択性を確立した。したがって、インフォームドコンセントを与えたヒトの志願者から血液を採取した。次いで、血液を穏やかに揺らしながら37℃で30分間平衡化し、エッペンドルフチューブに小分けした。化合物を異なる濃度で加え、穏やかに揺らしながら37℃で30分間インキュベートし、次いで、穏やかに揺らしながら20分間37℃で、JAK1依存性経路刺激にはインターロイキン6 (IL-6)により、又はJAK2依存性経路刺激にはGM-CSFにより刺激した。次いで、FACS分析を利用して、ホスホ-STAT1及びホスホ-STAT5を評価した。
【0199】
(2.4.2.1 ホスホ-STAT1アッセイ)
白血球におけるシグナル伝達性転写因子1(pSTAT1)リン酸化のIL-6刺激性の増加には、インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血をエクスビボで化合物により30分間処理し、その後IL-6により20分間刺激した。リンパ球におけるIL-6によるSTAT1のリン酸化の増加を、抗ホスホ-STAT1抗体を利用して、FACSにより測定した。
【0200】
5×Lyse/Fix緩衝液(BD PhosFlow社製、カタログ番号558049)を蒸留水で5倍に希釈し、37℃に予熱した。残りの希釈したLyse/Fix緩衝液を廃棄した。
【0201】
10μgのrhIL-6(R&D Systems社製、カタログ番号206-IL)を、1mlの、PBS 0.1% BSAに溶かし、10μg/mlのストック溶液を得た。ストック溶液を小分けした状態で-80℃で保存した。
【0202】
化合物の3倍の希釈系列をDMSO中に調製した(10 mMストック溶液)。コントロール処理試料には化合物の代わりにDMSOを与えた。試料は全て1%の最終DMSO濃度でインキュベートした。
【0203】
(2.4.2.1.1 血液の化合物とのインキュベーション及びIL-6による刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を148.5μLのアリコートに分けた。その後、1.5μLの化合物希釈液を各アリコートに加え、血液試料を穏やかに揺らしながら37℃で30分間インキュベートした。IL-6ストック溶液(1.5μl)を血液試料に加え(最終濃度10ng/ml)、該試料を穏やかに揺らしながら37℃で20分間インキュベートした。
【0204】
(2.4.2.1.2 白血球調製及びCD4標識付け)
刺激期間の最後に、赤血球を溶解し白血球を固定するために、3mlの1×予熱されたLyse/Fix緩衝液を直ちに血液試料に加え、手短にボルテックスにかけ、水浴中で37℃で15分間インキュベートし、次いでさらに使用するまで-80℃で凍結した。
【0205】
以下の工程のために、チューブを37℃でおよそ20分間解凍し、400gで4℃で5分間遠心分離した。細胞ペレットを3mlの冷1×PBSで洗浄し、遠心分離後、細胞ペレットを100μlの3%BSA含有PBSに再懸濁化した。FITC-結合抗-CD4抗体又はコントロールのFITC-結合アイソタイプ抗体を加え、暗所にて室温で20分間インキュベートした。
【0206】
(2.4.2.1.3 細胞透過処理及び抗ホスホ-STAT1抗体による標識付け)
細胞を1×PBSにより洗浄した後、細胞ペレットを100μlの氷冷1×PBSに再懸濁化し、900μlの氷冷100%メタノールを加えた。次いで、細胞を、透過処理のために4℃で30分間インキュベートした。
【0207】
次いで、透過処理された細胞を1×3%BSA含有PBSにより洗浄し、最後に、80μlの1×3%BSA含有PBXに再懸濁化した。
【0208】
20μLのPEマウス抗-STAT1(pY701)又はPEマウスIgG2aκアイソタイプコントロール抗体(BD Biosciences社製、それぞれカタログ番号612564及び559319)を加え、混合し、次いで、暗所にて4℃で30分間インキュベートした。
【0209】
次いで、細胞を1×PBSで1回洗浄し、FACSCanto IIフローサイトメーター(BD Biosciences社製)で分析した。
【0210】
(2.4.2.1.4 FACSCanto IIによる蛍光分析)
50,000の総事象を計数し、ホスホ-STAT1陽性細胞を、リンパ球ゲートにおいてCD4+細胞のゲーティングの後測定した。データはFACSDivaソフトウェアを利用して分析され、CD4+細胞上のホスホ-STAT1に陽性な細胞のパーセンテージに対して計算されたIL-6刺激の阻害率に相当する。
【0211】
(2.4.2.2 ホスホ-STAT5アッセイ)
白血球におけるシグナル伝達性転写因子5(pSTAT5)リン酸化のGM-CSF-刺激性の増加には、インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血をエクスビボで化合物により30分間処理し、次いで20分間GM-CSFにより刺激した。単球におけるGM-CSFによるSTAT5のリン酸化の増加を、抗ホスホ-STAT5抗体を利用してFACSにより測定した。
【0212】
5×Lyse/Fix緩衝液(BD PhosFlow社製、カタログ番号558049)を蒸留水で5倍に希釈し、37℃に予熱した。残りの希釈したLyse/Fix緩衝液を廃棄した。
【0213】
10μgのrhGM-CSF(AbCys S.A.社製、カタログ番号P300-03)を100μlのPBS 0.1% BSAに溶かし、100μg/mlのストック溶液を得た。ストック溶液を小分けした状態で-80℃で保存した。
【0214】
化合物の3倍希釈系列をDMSO中に調製した(10 mMストック溶液)。コントロール処理試料には、全く被験化合物なしでDMSOを与えた。試料を全て、1%の最終DMSO濃度でインキュベートした。
【0215】
(2.4.2.2.1 血液の化合物とのインキュベーション及びGM-CSFによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を148.5μlのアリコートに分けた。その後、1.5μlの化合物希釈液を各アリコートに加え、血液試料を穏やかに揺らしながら37℃で30分間インキュベートした。GM-CSFストック溶液(1.5μl)を血液試料に加え(最終濃度20pg/ml)、試料を穏やかに揺らしながら37℃で20分間インキュベートした。
【0216】
(2.4.2.2.2 白血球調製及びCD14標識付け)
刺激期間の最後に、赤血球を溶解し白血球を固定するために、3mlの1×予熱されたLyse/Fix緩衝液を直ちに血液試料に加え、手短にボルテックスにかけ、水浴中で37℃で15分間インキュベートし、次いで、さらに使用するまで-80℃で凍結した。
【0217】
以下の工程のために、チューブを37℃でおよそ20分間解凍し、400gで4℃で5分間遠心分離した。細胞ペレットを3mlの冷1×PBSで洗浄し、遠心分離後、細胞ペレットを100μlの3%BSA含有PBSに再懸濁化した。FITCマウス抗-CD14抗体(BD Biosciences社製、カタログ番号345784)又はコントロールFITCマウスIgG2bκアイソタイプ抗体(BD Biosciences社製、カタログ番号555057)を加え、暗所にて室温で20分間インキュベートした。
【0218】
(2.4.2.2.3 細胞透過処理及び抗ホスホ-STAT5抗体による標識付け)
細胞を1×PBSで洗浄した後、細胞ペレットを100μlの氷冷1×PBSに再懸濁化し、900μlの氷冷100%メタノールを加えた。次いで、細胞を、透過処理のために、4℃で30分間インキュベートした。
【0219】
次いで、透過処理された細胞を1×3%BSA含有PBSで洗浄し、最後に80μlの1×3%BSA含有PBSに再懸濁化した。
【0220】
20μLのPEマウス抗-STAT5(pY694)又はPEマウスIgG1κアイソタイプコントロール抗体(BD Biosciences社製、それぞれカタログ番号612567及び554680)を加え、混合し、次いで暗所にて4℃で30分間インキュベートした。
【0221】
次いで、細胞を1×PBSで1回洗浄し、FACSCanto IIフローサイトメーター(BD Biosciences社製)で分析する。
【0222】
(2.4.2.2.4 FACSCanto IIによる蛍光分析)
50,000の総事象を計数し、ホスホ-STAT5陽性細胞を、CD14+細胞のゲーティングの後測定した。データはFACSDivaソフトウェアを利用して分析され、CD14+細胞上のホスホ-STAT5に陽性な細胞のパーセンテージに対して計算されたGM-CSF刺激の阻害率に相当する。
【0223】
(2.4.2.3 結果)
このプロトコールに供されたとき、3名の健常な志願者の平均から得られた阻害率(PIN)を、本発明の化合物に対して決定した。例えば、化合物1を試験し、STAT1リン酸化の阻害においてpIC50=6.61を、STAT5リン酸化の阻害においてpIC50=5.37を生み出した。
【0224】
STAT1(JAK1-依存性経路)及びSTAT5(JAK2-依存性経路)に対する化合物1の効果を比較すると、JAK2よりJAK1に対して17.6倍の選択性が測定された。
【0225】
(実施例3 インビボモデル)
(実施例3.1 CIAモデル)
(3.1.1 材料)
完全フロイントアジュバント(CFA)及び不完全フロイントアジュバント(IFA)をDifco社から購入した。ウシII型コラーゲン(CII)、リポ多糖(LPS)及びエンブレルをそれぞれ、Chondrex社(Isle d'Abeau、フランス)、Sigma社(P4252、L'Isle d'Abeau、フランス)、Whyett(25mg注射可能シリンジ、フランス)、Acros Organics社(カリフォルニア州パロアルト)から入手した。使用した他の全ての試薬は試薬等級のものであり、全ての溶媒は分析用のものであった。
【0226】
(3.1.2 動物)
Dark Agoutiラット(雄、7〜8週齢)をHarlan Laboratories社(Maison-Alfort、フランス)から入手する。DBA/1Jマウス(雄、7週齢)を、Centre d’Elevage et de Reproduction JANVIER (CERJ) (Laval, フランス)から得た。ラット及びマウスを12時間の明暗サイクル(0700-1900)においた。温度を22℃に維持し、餌及び水を適宜与えた。
【0227】
(3.1.3 コラーゲン誘発関節炎(CIA))
実験の前日に、CII溶液(2mg/mL)を0.05Mの酢酸を用いて調製し、4℃で保管した。免疫化の直前に、等量のアジュバント(IFA)及びCIIを氷水浴において、予冷却したガラス瓶中でホモジナイザーにより混合した。乳濁液が形成されない場合は、さらなるアジュバント及びホモジナイズの延長が必要となる場合がある。
【0228】
マウス:1日目に0.1mLの乳濁液を各マウスの尾の根元に皮内注射し、21日目に第2の追加免疫の皮内注射(CFA 0.1mLの生理食塩水中1mg/mLのCII溶液)を行った。この免疫化法は既報の方法(David D Brand Kary A Latham及びEdward F Rosloniecの文献、「コラーゲン誘発関節炎(Collagen-induced arthritis)」Nature Methods 2 (5): 1269-1275, 2007)を変更したものであった。
【0229】
ラット:1日目に0.2mLの乳濁液を各ラットの尾の根元に皮内注射し、9日目に第2の追加免疫の皮内注射(CFA 0.1mLの生理食塩水中2mg/mLのCII溶液)を行った。この免疫化法は既報の方法(Sims NAらの文献、(2004)「ゾレドロン酸による破骨細胞の標的化はコラーゲン誘導性関節炎における骨破壊を予防する(Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis)」 Arthritis Rheum. 50 2338-2346;Jouらの文献、2005)を変更したものである。
【0230】
(3.1.4 研究設計)
被験化合物の治療効果をラット又はマウスのCIAモデルで試験した。動物を無作為に、均等な群に分け、各群は10匹の動物を含んだ。全てのラットを1日目に免疫化し、9日目に追加免疫する。全てのマウスを1日目に免疫化し、21日目に追加免疫した。治療量を16日目〜30日目まで与え続けた。ネガティブコントロール群をビヒクル(MC 0.5%)で処理し、ポジティブコントロール群をエンブレル(10mg/kg、3×週、皮下注射)で処理した。対象とする化合物を通常3用量、例えば、3、10、30mg/kg、経口で試験した。
【0231】
(3.1.5 関節炎の臨床評価)
関節炎をKhachigianの文献2006、Linらの文献2007及びNishidaらの文献2004の方法に従い得点付けした。4足それぞれの腫脹を以下のように関節炎スコアによってランク付けした:0−症状なし;1−軽度だが、足首又は手首などの1種の関節の明確な紅化及び腫脹、又は罹患した指の数にかかわらず個々の指に限られた明らかな紅化及び腫脹;2−2種以上の関節の中等度の紅化及び腫脹;3−指を含む足全体の重度の紅化及び腫脹;4−多数の関節が関与する極度に炎症を起こした肢(動物あたりの最大の累積臨床関節炎スコアは16)(Nishidaらの文献2004)。
【0232】
複数の研究のメタ分析を可能にするため、臨床スコア値を以下のとおり規格化した。
【0233】
臨床スコアのAUC(AUCスコア):個々のラットに対して、1日から14日の曲線下面積(AUC)を計算した。各動物のAUCを、その動物のデータが得られた研究におけるビヒクルに対して得られた平均AUCで割り、100をかけた(すなわち、AUCを、研究あたりの平均ビヒクルAUCのパーセンテージとして表した)。
【0234】
1日から14日までの臨床スコアの増加(エンドポイントスコア):各動物の臨床スコアの差を、その動物のデータが得られた研究におけるビヒクルに対して得られた平均臨床スコアの差で割り、100をかけた(すなわち、差を、研究あたりビヒクルの平均臨床スコアの差のパーセンテージとして表した)。
【0235】
(3.1.6 関節炎発症後の体重の変化(%))
臨床的に、体重減少は関節炎と関連している(Sheltonらの文献、2005;Argilesらの文献、1998;Rallの文献、2004;Walsmithらの文献2004)。したがって、関節炎発症後の体重の変化は、ラットモデルにおける治療術の効果を評価する非特異的なエンドポイントとして利用できた。関節炎発症後の体重の変化(%)を以下のように計算した。
【数1】
【0236】
(3.1.7 ラーセンスコア)
死後に、少なくとも2人の科学者により全ラットの両後足に対してラーセンスコアを与えた。これらのスコアの平均を計算して、ラットあたりの1つのラーセンスコアを得た。ラーセンスコアの研究間の比較を可能にするため、各ラットのラーセンスコアを、そのラットが属する研究におけるビヒクルで得られた平均ラーセンスコアにより割り、100をかけた(すなわち、ラーセンスコアを、研究あたりの平均ビヒクルラーセンスコアのパーセンテージとして表した)。異なる治療群の平均ラーセンスコアを計算して比較した。
【0237】
(3.1.8 放射線学)
個々の動物の後足のX線写真を撮った。無作為のブラインド・アイデンティティ番号を、写真のそれぞれに割り当て、以下の放射線学的ラーセンのスコアシステムを用いて2人の独立したスコアラーにより骨びらんの重症度をランク付けした:0−無傷の骨の外郭及び正常な関節腔を有する正常状態;1−1又は2の外側中足骨に軽微な骨びらんが見られる軽度の異常;2−3から5の外側中足骨に骨びらんがみられる明確な初期の異常;3−全ての外側中足骨並びに1又は2の内側中足骨に明らかな骨びらんが見られる中等度破壊性異常;4−全ての中足骨に明らかな骨びらんが見られ、かつ少なくとも1の内側中足骨関節が完全に侵食され、骨の関節外郭のいくらかが部分的に残されている重度破壊性異常;5−骨の外郭を有さないムチランス型異常。このスコア付けシステムは、Salveminiらの文献、2001;Bushらの文献、2002;Simsらの文献、2004;Jouらの文献、2005の変形である。
【0238】
(3.1.9 組織学)
放射線学的分析の後、マウスの後足を10%リン酸-緩衝ホルマリン(pH7.4)に固定し、微細組織診のための急速骨脱灰剤(Laboratories Eurobio社製)によって脱灰しパラフィンに包埋した。関節炎の関節の詳細な評価を確実に行うために、少なくとも4つの連続切片(5μm厚)を切り出し、各系列の切片の間を100μmとした。該切片をヘマトキシリン及びエオシン(H&E)で染色した。滑膜の炎症、並びに骨及び軟骨損傷の組織学的実験を二重盲検で行った。各足において、4点スケールを用いて4つのパラメーターを評価した。該パラメーターは、細胞浸潤、パンヌス重症度、軟骨侵食、及び骨びらんとした。以下のようにスコア付けを行った:1−正常、2−軽度、3−中等度、4−顕著。これらの4つのスコアを総合し、さらなるスコア、つまり「RA総計スコア」として表した。組織学的読み取りの研究間比較を可能にするために、各ラットの全体的な組織学的スコアを、そのラットが属する研究におけるビヒクルで得られた平均の全体的な組織学的スコアにより割り、100をかけた(すなわち、全体的な組織学的スコアを、研究あたりの平均ビヒクル全体的組織学的スコアのパーセンテージとして表した)。異なる治療群の平均の全体的な組織学的スコアを計算して比較した。
【0239】
(3.1.10 踵骨(踵の骨)のマイクロ-コンピュータ断層撮影法(μCT)分析)
RAに見られる骨破壊は特に皮質骨で生じ、μCT分析により明らかとなり得る(Sims NAらの文献、(2004);Oste Lらの文献、ECTC Montreal 2007)。踵骨の走査及び3Dボリューム再構築後に、骨破壊を、インシリコで骨の縦軸に対して垂直に単離されたスライドあたりに存在する分離した対象の数として測定した。破壊されている骨が多いほど、測定される個別の対象が多い。踵骨に沿って均一に分布した1000の切片(約10.8μmの間隔)を分析する。
【0240】
(3.1.11 結果)
化合物1を、30 mg/kgでマウスCIA試験で、30、10、3、及び1 mg/kgでラットCIA試験で試験した。化合物1は、ラットCIA試験において実施された全ての読み取りにおいて有効であり、いくつかの読み取りにおいて統計的に有意であり、特に下記では有意な向上が見られた:臨床スコアのAUC(10mg/kgから)、エンドポイント臨床スコア(1 mg/kgから)、ラーセンスコア(30 mg/kgから)、及び足の腫脹(1 mg/kgから)。
【0241】
(実施例3.2 敗血症ショックモデル)
リポ多糖(LPS)の注射は、可溶性腫瘍壊死因子(TNF-α)の末梢への急速な放出を誘導する。このモデルを使用して、インビボでのTNF放出の有望な遮断薬を分析する。
【0242】
1群あたり6匹の雌のBALB/cJマウス(20g)を、一度の意図された投与、経口投与で処理する。30分後、LPS(15μg/kg;大腸菌血清型0111:B4)を腹腔内に注射する。90分後、マウスを安楽死させ、血液を回収する。循環しているTNFαのレベルを、市販のELISAキットを使用して測定する。デキサメタゾン(5μg/kg)を参照抗炎症化合物として使用する。選択された化合物を1回投与又は複数回投与、例えば、3及び/又は10及び/又は30mg/kg、経口投与で試験する。
【0243】
化合物1は、経口投与30 mg/kgでTNF放出の統計的に有意な減少(>50%)を示した。
【0244】
(実施例3.3 MABモデル)
MABモデルは、治療法によるRA様炎症応答の調節の迅速な評価を可能にする(Kachigian LMの文献、Nature Protocols (2006) 2512-2516:「コラーゲン抗体誘発性関節炎(Collagen antibody-induced arthritis)」)。DBA/Jマウスに、II型コラーゲンに対するmAbカクテルを静脈内注射する。1日後、化合物処理を開始する(ビヒクル:10% (v/v) HPβCD)。3日後、マウスに腹腔内LPS注射(50μg/マウス)を施し、速やかな炎症の発症を起こさせる。該mAb注射の10日後まで、化合物処理を続ける。足の腫脹の測定及び各足の臨床スコアの記録によって、炎症を読み取る。四肢の累積的臨床関節炎スコアを提示して炎症の重症度を示す。4が最も重症の炎症である0〜4のスケールを使用して、スコア付けシステムを各肢に適用する。
0 症状なし
1 軽度だが、足首又は手首などの1種の関節の明確な紅化及び腫脹、又は罹患した指の数にかかわらず個々の指に限られた明らかな紅化及び腫脹
2 2種以上の関節の中等度の紅化及び腫脹
3 指を含む足全体の重度の紅化及び腫脹
4 多数の関節が関与する極度に炎症を起こした肢。
【0245】
(実施例3.4 オンコロジーモデル)
JAK2駆動性の骨髄増殖性疾患に対する小分子の有効性を検証するためのインビボモデルは、Wernigらの文献、Cancer Cell 13, 311, 2008、及びGeronらの文献、Cancer Cell 13, 321, 2008に記載されている。
【0246】
(実施例3.5 マウスIBDモデル)
IBDに対する小分子の有効性を検証するためのインビトロ及びインビボのモデルは、Wirtzらの文献、2007に記載されている。
【0247】
(実施例3.6 マウス喘息モデル)
喘息に対する小分子の有効性を検証するためのインビトロ及びインビボのモデルは、Nialsらの文献、2008;Ipらの文献、2006;Pernisらの文献、2002;Kudlaczらの文献、2008に記載されている。
【0248】
(実施例4:毒性、DMPK、及び安全性モデル)
(実施例4.1 熱力学的溶解性)
1mg/mLの被験化合物の溶液を、ガラス小瓶において室温で、0.2Mのリン酸緩衝液pH7.4又は0.1Mのクエン酸緩衝液pH3.0に調製する。
【0249】
該試料をローテータードライブSTR4(Stuart Scientific社製、Bibby社製)において、速度3.0、室温で24時間回転させる。
【0250】
24時間後、800μLの試料をエッペンドルフチューブに移し、14000rpmで5分間遠心分離する。次いで、200μLの試料の上清をMultiscreen(登録商標)溶解度用プレート(Millipore社製、MSSLBPC50)に移し、該上清を真空マニホールドを用いて、清潔なGreiner社製ポリプロピレンV字底96ウェルプレート(カタログ番号651201)に濾過する(25.4-30.5cmHg)。5μLの濾液を、検量線を有するプレート(Greiner社製、カタログ番号651201)で、インキュベートに使用するのと同じ緩衝液95μL(20倍)に希釈する。
【0251】
該化合物の検量線をDMSO中で新たに作成する。10mMのDMSOストック溶液から開始して、DMSOに2倍に希釈し(5000μM)、次いで、19.5μMまでDMSOでさらに希釈する。次いで、5000μMからの希釈系列3μLを、97μlのアセトニトリル-緩衝液混合物(50/50)に移す。最終濃度の範囲は2.5〜150μMである。
【0252】
該プレートをシーリングマット(MA96RD-04S、www.kinesis.co.uk)で密閉し、分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定する。
【0253】
該試料を、LCMSにおいて1ml/分の流量で分析する。溶媒Aは15mMのアンモニアであり、溶媒Bはアセトニトリルである。該試料を、Waters社製のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作する。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲である。
【0254】
ピーク面積をMasslynxソフトウェアパッケージを用いて分析し、試料のピーク面積を検量線に対してプロットして、該化合物の溶解度を得る。
【0255】
溶解度値をμM又はμg/mLで報告する。
【0256】
(実施例4.2 水溶性)
10mMのDMSO中のストック溶液から開始して、化合物の段階希釈をDMSOで調製する。該希釈系列を、96NUNC Maxisorbプレート平底(カタログ番号442404)に移し、室温の0.2Mのリン酸緩衝液pH7.4又は0.1Mのクエン酸緩衝液pH3.0を添加する。
【0257】
最終濃度は、5回の同じ希釈工程で、200μM〜2.5μMの範囲にある。最終DMSO濃度は2%を超えなかった。200μMのピレンを各96ウェルプレートの角点に添加し、顕微鏡のZ軸の補正のための参照点とする。
【0258】
該アッセイプレートを密閉し、230rpmで振盪しながら37℃で1時間インキュベートする。次いで、該プレートを白色光顕微鏡下で走査し、濃度あたりの沈殿の個々の写真を得る。該沈殿を分析し、数値に変換して、これをグラフ上にプロットする。該化合物が完全に溶解したように見える最初の濃度は報告する濃度であるが、しかし、本当の濃度は、この濃度と1つ上の希釈段階との間のどこかにある。
【0259】
溶解度値をμg/mLで報告する。
【0260】
(実施例4.3 血漿タンパク質結合(平衡透析))
DMSO中の化合物の10mMストック溶液を、DMSOで5倍に希釈する。この溶液を、新たに解凍したヒト、ラット、マウス又はイヌの血漿(BioReclamation社製)で、最終濃度10μM及びDMSO最終濃度0.5%にさらに希釈する(PP-Masterblock 96ウェル(Greiner社製、カタログ番号780285)において1094.5μlの血漿中5.5μl)。
【0261】
インサートを有するPierce Red Deviceプレート(ThermoScientific社製、カタログ番号89809)を準備し、緩衝液チャンバーを750μLのPBSで満たし、かつ血漿チャンバーを500μLの添加血漿(spiked plasma)で満たす。該プレートを230rpmで振盪しながら、37℃で4時間インキュベートする。インキュベーション後、両チャンバーの120μLを、96ウェル丸底、PPディープウェルプレート(Nunc社製、カタログ番号278743)の360μLのアセトニトリルに移し、アルミ箔の蓋で密閉する。該試料を混合し、30分間氷上に置く。次いで、このプレートを1200rcf、4℃で30分遠心分離し、該上澄みをLCMSでの分析用に96V字底PPプレート(Greiner社製、651201)に移す。
【0262】
該プレートを、www.kinesis.co.ukのシーリングマット(MA96RD-04S)で密閉し、分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定する。
【0263】
該試料を、LCMSで1mL/分の流量で分析する。溶媒Aは15mMのアンモニアであり、溶媒Bはアセトニトリルである。該試料を、Waters社製のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作した。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲である。
【0264】
緩衝液チャンバー及び血漿チャンバー中の化合物のピーク面積は、100%の化合物であると考えられる。血漿への結合率を、これらの結果から導き、血漿への結合率としてLIMSに報告した。
【0265】
PBS中の最終試験濃度の化合物の溶解度を顕微鏡で調査して、沈殿が見られるか否かを示した。
【0266】
(実施例4.4 QT延長の傾向)
QT延長の可能性は、hERGパッチクランプアッセイにおいて評価する。
【0267】
(4.4.1 従来の全細胞パッチクランプ)
全細胞パッチクランプ記録は、Pulse v8.77ソフトウェア(HEKA社)により制御されたEPC10増幅器を利用して実施する。直列抵抗は典型的には10 MΩ未満であり、60%超補償されており、記録はリーク減算されない。電極は、GC150TFピペットガラス(Harvard社)から製造されている。
【0268】
外部浴溶液は、135 mM NaCl、5 mM KCl、1.8 mM CaCl2、5 mMグルコース、10 mM HEPES, pH 7.4を含む。
【0269】
内部パッチピペット溶液は、100mMグルコン酸カリウム、20 mM KCl、1mM CaCl2、1 mM MgCl2、5mM Na2ATP、2mMグルタチオン、11 mM EGTA、10 mM HEPES, pH 7.2を含む。
【0270】
Biologic MEV-9/EVH-9迅速潅流システムを利用して、薬物を潅流させる。
【0271】
記録は全てhERGチャネルを安定に発現しているHEK293細胞で行う。細胞を、2本の白金ロッド(Goodfellow社製)を用い、記録チャンバーに固定された12 mmの円形カバースリップ(German glass、Bellco社)上に培養する。hERG電流を、活性化パルスを用いて+40 mVに1000ミリ秒間惹起し、次いでテール電流パルスにより2000ミリ秒間-50 mVにし、保持電位は-80 mVであった。パルスを20秒ごとに印加し、実験は全て室温で行う。
【0272】
(4.4.2 データ分析)
IC50及びIC20値を、試験した各化合物について計算する。ラットCIAモデルから得られた結果により決定される、関連する治療投与量で得られた被験化合物のIC20と非結合のCmax濃度とのフォールド差(fold difference)を計算する。
【0273】
濃度反応曲線では、ピークテール電流振幅を、-50 mVへの電圧ステップの間に測定する。濃度反応データのカーブフィッティングを、以下の式を利用して実施する。
y = a + [( b - a)/ ( 1 + 10^ ( ( logc-x ) d )]
【0274】
式中、aは最小反応であり、bは最大反応であり、dはヒルスロープであり、この式は、IC50(式中、y = 50かつcがIC50値)とIC20(式中、y = 20かつcがIC20値)の両方の計算に利用できる。GraphPad(登録商標)Prism(登録商標)(Graphpad(登録商標)Software社製)ソフトウェアを、全てのカーブフィッティングに利用した。100倍以上の差は、QT延長の低い可能性を示す。
【0275】
(実施例4.5 ミクロソーム安定性)
DMSO中の化合物の10mMストック溶液を、96ディープウェルプレート(Greiner社製、カタログ番号780285)において182mMのリン酸緩衝液pH7.4で1000倍に希釈し、37℃でプレインキュベートした。
【0276】
40μLの脱イオン水をポリプロピレン製マトリックス2Dバーコード標識化した保存チューブ(Thermo Scientific社製)のウェルに添加し、37℃でプレインキュベートした。
【0277】
グルコース-6-リン酸脱水素酵素(G6PDH)作業溶液を182mMのリン酸緩衝液pH7.4で調製し、使用前に氷上に置いた。MgCl2、グルコース-6-リン酸及びNADP+を含む補因子を脱イオン水で調製し、使用前に氷上に置いた。
【0278】
対象とする種(ヒト、マウス、ラット、イヌ)の肝ミクロソーム(Xenotech社)、前述のG6PDH、及び補因子を含む最終作業溶液を調製し、この混合物を室温で20分以内でインキュベートした。
【0279】
30μLの予熱した化合物希釈液を、マトリックスチューブ中の予熱した水40μLに添加し、30μLのミクロソーム混合物を添加した。最終反応濃度は、化合物3μM、ミクロソーム1mg、GDPDH 0.4U/mL、MgCl2 3.3mM、グルコース-6-リン酸3.3mM及びNADP+ 1.3mMであった。
【0280】
ゼロ時間での化合物の残存率を測定するために、ミクロソーム混合物の添加前に、MeOH又はACNを該ウェルに添加した(1:1)。該プレートをMatrix Sepra seals(商標)(Matrix社、カタログ番号4464)で密閉し、数秒間振盪して、確実に全ての成分の混合が行われるようにした。
【0281】
停止ししなかった試料を、37℃、300rpmでインキュベートし、1時間のインキュベート後、該反応をMeOH又はACNで停止させた(1:1)。
【0282】
該反応の停止後、試料を混合し、氷上に30分間置いてタンパク質を沈殿させた。次いで、該プレートを1200rcf、4℃で30分間遠心分離し、LCMSでの分析用に上澄みを96V字底PPプレート(Greiner社、651201)に移した。
【0283】
これらのプレートを、www.kinesis.co.ukのシーリングマット(MA96RD-04S)で密閉し、親分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定した。
【0284】
該試料を、LCMSにおいて1ml/分の流量で分析した。溶媒Aは15mMのアンモニアであり、溶媒Bは、使用した停止溶液に応じてメタノール又はアセトニトリルであった。該試料を、Waters社のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作した。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲であった。
【0285】
0時間での親化合物のピーク面積を100%の残存とした。1時間のインキュベーション後の残存率を0時間から算出し、残存率として算出した。緩衝液中の最終試験濃度の化合物の溶解度を顕微鏡で調査し、結果を報告する。
【0286】
ミクロソーム安定性のデータを、60分後に残存している化合物の総量の割合として表す。
【0287】
【表10】
【0288】
(実施例4.6 Caco2透過性)
2方向Caco-2アッセイを下記のように行った。Caco-2細胞を欧州細胞カルチャーコレクション(ECACC、カタログ86010202)から入手し、24ウェルトランスウェルプレート(Fisher社、TKT-545-020B)での21日間の細胞培養後に使用した。
【0289】
DMEM+GlutaMAXI+1%のNEAA+10%のFBS(FetalClone II)+1%のPen/Strepからなる播種培地に2×105細胞/ウェルを播種した。培地は2〜3日毎に交換した。
【0290】
被験化合物及び参照化合物(プロプラノロール及びローダミン123又はビンブラスチン、全てSigma社から購入した)を、25mMのHEPES(pH7.4)を含有するハンクス平衡塩類溶液で調製し、0.25%の最終DMSO濃度を有する10μMの濃度で、トランスウェルプレートアセンブリの頂端側チャンバー(125μL)又は基底側チャンバー(600μL)のいずれかに加えた。
【0291】
50μMのルシファーイエロー(Sigma社製)を全ウェルのドナー緩衝液に添加し、ルシファーイエローの透過を監視することによって細胞層の一体性を評価した。ルシファーイエロー(LY)は親油性バリアを自由に透過することができないので、LY輸送の程度が高いと該細胞層の一体性が乏しいことを示唆する。
【0292】
オービタルシェーカーで150rpmで振盪しながら37℃で1時間インキュベーションした後、頂端側(A)及び基底側(B)の両チャンバーから70μLのアリコートを取り出し、96ウェルプレートにて、分析用内部標準(0.5μMのカルバマゼピン)を含有する、100μLの50:50 アセトニトリル:水の溶液に添加した。
【0293】
基底側及び頂端側から得た液体150μLを含む新たな96ウェルプレートで、Spectramax Gemini XS(励起426nm及び発光538nm)を用いてルシファーイエローを測定した。
【0294】
試料中の化合物濃度を高速液体クロマトグラフィー/質量分析計(LC-MS/MS)により測定した。
【0295】
見掛けの透過度(Papp)の値を以下の関係から算出した:
Papp=[化合物]受容体最終×V受容体/([化合物]供与体初期×V供与体)/Tinc×V供与体/表面積×60×10-6cm/秒
V=チャンバー容積
Tinc=インキュベーション時間
表面積=0.33cm2。
【0296】
Papp B>A/Papp A>Bの比を用いて、頂端細胞表面からの能動的な流出の指標としての流出率を算出した。
【0297】
下記のアッセイ許容基準を使用した:
プロプラノロール:Papp(A>B)値≧20(×10-6cm/秒)
ローダミン123又はビンブラスチン:Papp(A>B)値<5(×10-6cm/秒)流出率≧5を伴う
ルシファーイエロー透過度:≦100nm/秒。
【0298】
【表11】
(実施例4.7 薬物動態学的研究)
(4.7.1. 齧歯類における薬物動態学的研究)
化合物を、経静脈経路用にはPEG200/生理食塩水又はPEG400/DMSO/生理食塩水混合液に、経口経路用には0.5%メチルセルロース又は10〜30%ヒドロキシプロピル-β-シクロデキストリンpH3又はpH7.4に製剤する。被験化合物を、5〜10 mg/kgで単一の食道経管栄養法として経口投与し、ボーラスとして尾静脈により1 mg/kgで経静脈投与する。各群は3匹のラットからなる。下記の範囲の時点に抗凝血剤としてのヘパリンリチウムとともに血液試料を、カニューレ処置されたラットを利用して頸静脈から、又は後方眼窩洞(retro-orbital sinus)で採取する:0.05から8時間(経静脈経路)、並びに0.25から6、又は24時間(経口経路)。全血試料を5000 rpmで10分間遠心分離し、生じた血漿試料を分析未決のまま-20℃で保存する。
【0299】
(4.7.2 イヌにおける薬物動態学的研究)
化合物を、経静脈経路用にはPEG200/生理食塩水混合液に、経口経路用にはクエン酸でpH2〜3に酸性化させた0.5%メチルセルロース又はPEG400/ヒドロキシプロピル-β-シクロデキストリン混合液に製剤する。被験化合物を、5〜30 mg/kgで経管栄養法により経口投与し、ボーラスとして、又は橈側皮静脈により10分間の点滴として1 mg/kgで経静脈投与する。各群は3匹の雄又は雌のビーグルドッグからなる。投与後0.083から24時間の時点に抗凝血剤としてのヘパリンリチウムとともに、血液試料を、頸静脈から採取する。全血試料を5000 rpmで10分間遠心分離し、生じた血漿試料を分析未決のまま-20℃で保存する。
【0300】
(4.7.3 血漿中の化合物レベルの定量化)
各被験化合物の血漿濃度を、質量分析計がポジティブエレクトロスプレーモードで運転しているLC-MS/MS法により測定する。
【0301】
(4.7.4 薬物動態学的パラメーターの決定)
薬物動態学的パラメーターを、Winnonlin(登録商標)(Pharsight(登録商標)、アメリカ合衆国)を使用して計算する。
【0302】
(実施例4.8 7日間ラット毒性研究)
被験化合物を用いた7日間経口毒性研究を、Sprague-Dawley系の雄のラットで行い、それらの毒性及び毒物動態を、5mL/kg/日の一定の投与体積で、経管栄養法による100、300及び500mg/kg/日の一日量で評価する。
【0303】
被験化合物を精製水中30%(v/v)のHPβCDに製剤する。各群は、5匹の主要な雄のラット並びに3匹の毒物動態のためのサテライト動物を含む。第4群に、30%(v/v)HPβCD水溶液のみを、同じ頻度、投与体積で、かつ同じ投与経路により与え、ビヒクルコントロール群として役割を果たす。
【0304】
該研究の目標は、確認される有害事象のない最低用量(最大無毒性量-NOAEL)を決定することである。
【0305】
(実施例4.9 肝細胞安定性)
肝細胞における代謝クリアランスを評価するモデルは、McGinnityらの文献Drug Metabolism and Disposition 2008, 32, 11, 1247に記載されている。
【0306】
先の記載が、本来例示的及び説明的なものであり、示されているとおり本発明及びその好ましい実施態様を説明するように意図されていることが、当業者には理解されるであろう。日常の試験を通して、本発明の趣旨を逸脱せずになし得る明らかな改良及び変形を、当業者は認識するだろう。したがって、本発明は、上記記載によってではなく、以下の特許請求の範囲及びそれらの均等物で規定されるものとする。
【0307】
(引用文献)
【化11】
【0308】
本明細書に引用された、特許及び特許出願を含むがこれらに限定されない刊行物は全て、個々の刊行物が完全に記載されているかのように引用により本明細書に組み込まれるように具体的かつ個別的に示されているかのように、引用により本明細書に組み込まれる。
【0309】
上記記載から、本発明の組成物及び方法における様々な改良及び変更を当業者は思いつくだろう。添付の特許請求の範囲内にあるそのような改良は全て、本件出願に含まれるものとする。
【0310】
様々な化合物の示差的な細胞浸透能などの因子がインビトロ生化学アッセイにおける化合物活性と細胞アッセイにおける化合物活性との間の相違に関与し得ることを理解されたい。
【0311】
本件出願に与えられ、かつ記載される本発明の化合物の化学名の少なくともいくつかは、市販の化学命名ソフトウェアプログラムを使用して自動的に生成された場合があり、独立に検証していない場合がある。この機能を実行する代表的なプログラムには、Open Eye Software社により販売されるLexichem命名ツール及びMDL社により販売されるAutonom Softwareツールがある。示された化学名と表された構造とが異なる場合、表された構造が基準となる。
【0312】
本明細書に示された化学構造は、ChemDraw(登録商標)又はISIS(登録商標)/DRAWのいずれかを使用して作成された。本明細書中の構造における炭素、酸素又は窒素原子に現れている任意の開放原子価は、水素原子の存在を示す。構造中にキラル中心が存在するが、該キラル中心について具体的な立体化学が示されない場合、該キラル構造と関連する両方の鏡像異性体が該構造により包含される。
【技術分野】
【0001】
(発明の分野)
本発明は、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患に関与するチロシンキナーゼのファミリーであるJAKの阻害剤である化合物に関する。本発明は、この化合物の製造方法、この化合物を含む医薬組成物、並びに炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の、本発明の化合物を投与することによる予防及び/又は治療の方法も提供する。
【0002】
ヤヌスキナーゼ(JAK)は、膜受容体からSTAT転写因子へサイトカインシグナリングを伝達する細胞質チロシンキナーゼである。4種のJAKファミリーメンバー、JAK1、JAK2、JAK3、及びTYK2が記載されている。サイトカインがその受容体に結合すると、JAKファミリーメンバーは、自己リン酸化及び/又は互いにトランスリン酸化し、続いてSTATがリン酸化されて核へ移動し、転写が調節される。JAK-STAT細胞内シグナル伝達は、インターフェロン、ほとんどのインターロイキン、並びにEPO、TPO、GH、OSM、LIF、CNTF、GM-CSF、及びPRLなどの種々のサイトカイン及び内分泌因子の役に立つ(Vainchenker W.らの文献(2008))。
【0003】
遺伝モデルと小分子JAK阻害剤探索との組合せは、いくつかのJAKの治療上での可能性を明らかにした。JAK3は、免疫抑制の標的としてマウス及びヒト遺伝学で検証されている(O'Shea J.らの文献(2004))。JAK3阻害剤は、当初、臓器移植拒絶反応への臨床開発の取り組みに成功したが、後に、関節リウマチ(RA)、乾癬、及びクローン病などの他の免疫炎症性の徴候においても臨床開発の取り組みに成功した(http://clinicaltrials.gov/)。
【0004】
TYK2は、免疫炎症性疾患の潜在的な標的であり、ヒト遺伝学及びマウスノックアウト研究によって検証されている(Levy D.及びLoomis C.の文献(2007))。
【0005】
JAK1は、免疫炎症性疾患分野における新規の標的である。JAK1は、他のJAKとヘテロ二量体化して、サイトカイン駆動性の炎症促進性シグナルを伝達する。したがって、JAK1及び/又は他のJAKを阻害することで、様々な炎症病態、並びにJAK媒介性のシグナル伝達によって駆動される他の疾患に対する治療上の利益が期待される。
【背景技術】
【0006】
(発明の背景)
軟骨の変性は、様々な疾患の特徴となっており、その中でも、関節リウマチ及び変形性関節症が最も顕著なものである。関節リウマチ(RA)は、慢性の関節変性疾患であり、関節構造の炎症及び破壊によって特徴付けられる。該疾患を野放しにすると、関節機能性の喪失により相当な身体障害及び疼痛、並びに早死すら招く。したがって、RA治療の目的は、該関節破壊を阻止するために、該疾患の進行を減速するだけでなく、寛解も達成することである。該疾患の予後の厳しさに加えて、RAの高い罹患率(世界中で成人の約0.8%が罹患している)も、社会経済的影響が大きいことを意味している(RAの概説については、Smolen及びSteinerの文献(2003);Lee及びWeinblattの文献(2001);Choy及びPanayiの文献(2001);O'Dellの文献(2004)、及びFiresteinの文献(2003)を参照されたい。)。
【0007】
変形性関節症(OA、又は磨耗性関節炎とも称される)は、最も一般的な形態の関節炎であり、関節軟骨の喪失により特徴付けられ、多くの場合、骨の肥大化及び疼痛と関連する。変形性関節症の広範な概説については、Wielandらの文献(2005)を参照されたい。
【0008】
変形性関節症は治療が困難である。現在のところ、治癒を得ることはできず、治療は疼痛を緩和すること及び罹患した関節が変形するのを防ぐことに重点をおく。一般的な治療には、非ステロイド性抗炎症薬(NSAID)の使用が含まれる。コンドロイチン及びグルコサミン硫酸塩などの栄養補助食品が、変形性関節症の治療の安全でかつ有効な選択肢として推奨されているが、最近の臨床治験から、両治療が変形性関節症に関連する疼痛を低減させないことが明らかとなった。(Cleggらの文献、2006)。まとめると、疾患修飾性の変形性関節症の薬剤は全く利用できない。
【0009】
同化プロセスの刺激、異化プロセスの阻止、又はこれら2つを組み合わせると、軟骨が安定化し、障害の回復さえももたらされる可能性があり、したがって、疾患のさらなる進行を防ぐことができる。種々のトリガーが軟骨細胞の同化刺激を刺激することができる。インスリン様成長因子-I (IGF-I)は、滑液における主要な同化成長因子であり、プロテオグリカン及びコラーゲンの両方の合成を刺激する。骨形成タンパク質(BMP)ファミリーのメンバー、特にBMP2、BMP4、BMP6、及びBMP7、並びにヒト形質転換成長因子-β(TGF-β)ファミリーのメンバーが軟骨細胞同化刺激を誘発できることも示された(Chubinskaya及びKuettnerの文献、2003)。軟骨細胞の同化刺激を誘発する化合物が最近特定された(米国特許第6,500,854号;欧州特許第1391211号)。しかし、これらの化合物のほとんどは重度の副作用を示し、そのため、軟骨細胞分化を刺激する、これらの副作用のない化合物が強く必要とされている。
【0010】
Vandeghinsteらは、その阻害がOAを含む様々な疾患について治療上の関連性を有する可能性がある標的としてJAK1を発見した(WO2005/124342)。JAK1は、サイトカイン受容体媒介性細胞内シグナル伝達に関与する、細胞質チロシンキナーゼのヤヌスキナーゼ(JAK)ファミリーに属する。JAKファミリーは4種のメンバー、JAK1、JAK2、JAK3、及びTYK2からなる。サイトカインの結合と同時に、サイトカイン受容体にJAKがリクルートされ、次いで、サイトカイン受容体及び共有された受容体サブユニット(共通γ-c鎖、gp130)のヘテロ二量体化が起こる。次いで、JAKは自己リン酸化及び/又は他のJAKによりトランスリン酸化により活性化され、受容体のリン酸化並びにシグナル伝達性転写因子(STAT)のメンバーのリクルートメント及びリン酸化をもたらす。リン酸化されたSTATは二量体化し、核に移動してサイトカイン反応性遺伝子のエンハンサー領域に結合する。マウスのJAK1遺伝子のノックアウトから、JAK1は、発生の間に必須かつ非重複の役割を果たすことが示された:JAK1-/-マウスは生後24時間以内に死亡し、かつリンパ球の発生は著しく損なわれた。さらに、JAK1-/-細胞は、クラスIIサイトカイン受容体を使用するサイトカイン、シグナル伝達にγ-cサブユニットを使用するサイトカイン受容体、及びシグナル伝達にgp130サブユニットを使用するサイトカイン受容体ファミリーに反応しないか、又は反応性が低かった(Rodigらの文献、1998)。
【0011】
様々な群が、軟骨細胞生物学におけるJAK-STATシグナル伝達に関係している。Liら(2001)は、JAK/STAT及びMAPKシグナル伝達経路の活性化によって、オンコスタチンMが初代軟骨細胞におけるMMP及びTIMP3遺伝子の発現を誘導することを示した。Osakiら(2003)は、軟骨細胞におけるインターフェロン-γ媒介性のII型コラーゲンの阻害がJAK-STATシグナル伝達を含むことを示した。基質構成成分の発現を低下させることによって、並びにコラゲナーゼ及び一酸化窒素(NO)の産生を媒介する誘導型一酸化窒素合成酵素(NOS2)の発現を誘導することによって、IL1-βは軟骨異化反応を誘導する。Oteroら(2005)は、レプチン及びIL1-βが軟骨細胞におけるNO産生又はNOS2 mRNAの発現を相乗的に誘導し、しかも、それがJAK阻害剤によって阻止されることを示した。Legendreら(2003)は、ウシ関節の軟骨細胞において、IL6/IL6受容体が軟骨-特異的基質遺伝子II型コラーゲン、アグリカンコア及びリンクタンパク質の発現低下を誘導し、しかも、これがJAK/STATシグナル伝達によって媒介されることを示した。したがって、これらの観察は、軟骨恒常性維持におけるJAKキナーゼ活性の役割、及びJAKキナーゼ阻害剤に対する治療機会を示唆している。
【0012】
JAKファミリーメンバーは、骨髄増殖性疾患を含むさらなる病態に関与しており(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506)、JAK2の突然変異が同定されている。これは、JAK、特にJAK2の阻害剤が骨髄増殖性疾患の治療に有用となり得ることも示している。加えて、該JAKファミリー、特にJAK1、JAK2及びJAK3は、ガン、特に白血病、例えば、急性骨髄性白血病(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506;Xiangらの文献、2008、「急性骨髄性白血病患者における体細胞JAK1突然変異の同定(Identification of somatic JAK1 mutations in patients with acute myeloid leukemia)」Blood First Edition Paper, prepublished online 12月26日、2007;DOI 10.1182/blood-2007-05-090308)及び急性リンパ芽球性白血病(Mullighanらの文献、2009)、又は固形腫瘍、例えば、子宮平滑筋肉腫(Constantinescuらの文献、2007、Trends in Biochemical Sciences 33(3): 122-131)、前立腺ガン(Tamらの文献、2007、British Journal of Cancer, 97, 378-383)に関係している。これらの結果は、JAK、特にJAK1及び/又はJAK2の阻害剤が、ガン(白血病及び固形腫瘍、例えば、子宮平滑筋肉腫、前立腺ガン)の治療にも利用できることを示している。
【0013】
加えて、キャッスルマン病、多発性骨髄腫、メサンギウム増殖性糸球体腎炎、乾癬及びカポジ肉腫は、サイトカインIL-6の分泌過多に起因する可能性があり、その生物学的作用は細胞内JAK-STATシグナル伝達により媒介される(Tetsuji Naka、Norihiro Nishimoto及びTadamitsu Kishimotoの文献、Arthritis Res 2002, 4 (suppl 3):S233-S242)。この結果は、前記疾患の治療においてJAKの阻害剤の有用性が見出され得ることを示している。
【0014】
JAK3及びTYK2では自己免疫疾患との関連が確立された。JAK3の突然変異だけでなく、上流シグナル伝達成分γ-c受容体鎖及びIL7受容体の突然変異も、総計で、ヒトの重症複合型免疫不全症の症例の約70%の原因である('OSheaらの文献、2004)。γ-c受容体鎖からのシグナル伝達において、JAK1はJAK3と協同することに留意されたい。Tyk2の多形が全身性エリテマトーデス(SLE)に見られる(O'Sullivanらの文献、2007、Mol Immunol. 44(10):2497-506)。したがって、JAKファミリーの標的化は、免疫炎症分野における治療機会を与え得る。
【0015】
現在の治療は十分なものではなく、したがって、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療に有用となり得るさらなる化合物同定の必要性が残されている。したがって、本発明は、化合物、その製造方法、及び本発明の化合物を好適な医薬担体とともに含む医薬組成物を提供する。特に、該化合物は、385キナーゼ及び非キナーゼ標的に加え、他のJAKファミリーメンバーよりもJAK1への高い効力及び選択性を示す。さらに、データは、該化合物は良好な安全域を有することを示す。したがって、本発明がJAK1媒介性の疾患、特にSLE(全身性エリテマトーデス)及びRAなどの炎症性疾患の治療に新規な機会を与えることが結論付けられる。
【0016】
本発明は、これらの疾患及び病態を治療するための医薬の製造における本発明の化合物の使用も提供する。
【発明の概要】
【0017】
本発明は、JAK、特にJAK1の阻害剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療に有用であるという発見に基づいている。本発明は、これらの化合物の製造方法、これらの化合物を含む医薬組成物、並びに炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患を、本発明の化合物を投与することにより治療する方法も提供する。
【0018】
したがって、本発明の第一の態様において、式Iを有する本発明の化合物が開示される。
【化1】
【0019】
本発明の化合物は、385キナーゼ及び非キナーゼ標的、及び他のJAKファミリーメンバーよりもJAK1への高い効力及び選択性を示すと同時に、良好な安全域も示す、JAKの新規な阻害剤である。このプロファイルを有する化合物を使用すると、オフターゲット効果の低い発生率により、炎症性疾患、特にSLE及びRAの治療において、利点が生まれることがある。
【0020】
さらなる態様において、本発明は、本発明の化合物、及び医薬用担体、賦形剤又は希釈剤を含む医薬組成物を提供する。さらに、本発明の化合物は、本明細書に開示される医薬組成物及び治療方法に有用であり、製造及び使用されるときに医薬として許容し得るものである。本発明のこの態様において、該医薬組成物は、本発明の化合物と組み合わせた使用に好適なさらなる有効成分をさらに含むことがある。
【0021】
本発明のさらなる態様において、本発明は、本明細書に列記されるものから選ばれる病態、特に異常なJAK活性に関連し得る病態であって、例えば、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患に罹患し易い又は苦しんでいる哺乳動物の治療方法であって、治療上有効な量の本明細書に記載される本発明の化合物又は1種以上の医薬組成物を投与することを含む方法を提供する。具体的な態様において、本発明は、異常なJAK1活性に関連し得る病態、特に、炎症性疾患、増殖性疾患、及び軟骨代謝回転の異常を含む疾患に罹患し易い又は苦しんでいる哺乳動物を治療する方法を提供する。
【0022】
さらなる態様において、本発明は、本明細書に列記されるものから選ばれる病態、特に、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患などの異常なJAK活性に関連し得る病態の治療又は予防に使用するための本発明の化合物を提供する。具体的な態様において、本発明は、異常なJAK1活性に関連する病態、特に、炎症性疾患、増殖性疾患、及び軟骨代謝回転の異常を含む疾患の治療又は予防に使用するための本発明の化合物を提供する。
【0023】
治療態様のさらに他の方法において、本発明は、原因として本明細書に記載される異常なJAK活性に関連する病態に罹患し易い又は苦しんでいる哺乳動物の治療方法であって、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む方法を提供する。具体的な実施態様において、該異常なJAK活性は異常なJAK1活性である。
【0024】
さらなる態様において、本発明は、原因として異常なJAK活性に関連する病態の治療又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該異常なJAK活性は異常なJAK1活性である。
【0025】
追加の態様において、本発明は、本明細書の下記に開示される代表的な合成手順及び経路とともに本発明の化合物の合成方法を提供する。
【0026】
したがって、本発明の主な目的は、JAKの活性を修正できる化合物を提供し、ひいては原因としてそれに関連し得る任意の病態を予防又は治療することである。特に、原因としてJAK1の活性に関連し得る任意の病態。
【0027】
本発明のさらなる目的は、原因としてJAKの活性に関連し得る、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患などの病態若しくは疾病又はその症状を治療又は軽減することができる化合物を提供することである。特に、原因としてJAK1の活性に関連し得る疾病又は病態。
【0028】
本発明のなおさらなる目的は、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患などのJAK活性に関連する疾患を含む、種々の疾病状態の治療又は予防に使用できる医薬組成物を提供することである。特に、JAK1活性に関連する病態。
【0029】
他の目的及び利点は、次の詳細な説明を考慮することにより、当業者に明らかとなろう。
【発明を実施するための形態】
【0030】
(発明の詳細な説明)
(定義)
以下の用語は、それとともに以下に提示される意味を有するものとし、本発明の記載及び意図される範囲を理解するのに有用である。
【0031】
本発明を記載する場合、化合物、該化合物を含む医薬組成物、並びに該化合物及び組成物を使用する方法を含み得るが、以下の用語は、存在する場合、他に示さない限り、以下の意味を有する。本明細書に記載される場合、以下に定義される部分はいずれも多様な置換基で置換できること、及びそれぞれの定義が以下に記載されるそれらの範囲内でこのような置換部分を含むものとすることも理解されたい。他に明記しない限り、用語「置換」とは、以下に記載されるように定義できよう。本明細書に使用される場合、用語「基」及び「ラジカル」が交換可能であるとみなし得ることをさらに理解されたい。
【0032】
冠詞「a」及び「an」は、本明細書において、1又は2以上(すなわち、少なくとも1)の該冠詞の文法上の対象を言及するのに使用できる。例として、「アナログ(an analogue)」は、1のアナログ又は2以上のアナログを意味する。
【0033】
「医薬として許容し得る」とは、連邦政府若しくは州政府の監督機関又は米国以外の国の対応機関により承認されたか又は承認され得ることを意味するか、あるいは、動物、より具体的にはヒトにおける使用のための米国薬局方又は他の公認された薬局方に収載されていることを意味する。
【0034】
「医薬として許容し得る塩」とは、医薬として許容することができ、かつ親化合物の所望の薬理活性を有する、本発明の化合物の塩をいう。特に、そのような塩は無毒性であり、無機又は有機の酸付加塩及び塩基付加塩とすることができる。具体的には、そのような塩には下記がある:(1)塩化水素酸、臭化水素酸、硫酸、硝酸、リン酸などの無機酸で形成された酸付加塩;酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、3-(4-ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、1,2-エタン-ジスルホン酸、2-ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、4-クロロベンゼンスルホン酸、2-ナフタレンスルホン酸、4-トルエンスルホン酸、カンファスルホン酸、4-メチルビシクロ[2.2.2]-オクト-2-エン-1-カルボン酸、グルコヘプトン酸、3-フェニルプロピオン酸、トリメチル酢酸、三級ブチル酢酸、ラウリル硫酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸などの有機酸で形成された酸付加塩;又は、(2)親化合物に存在する酸性プロトンが、金属イオン、例えば、アルカリ金属イオン、アルカリ土類イオン、又はアルミニウムイオンにより置換されるか、;又はエタノールアミン、ジエタノールアミン、トリエタノールアミン、N-メチルグルカミンなどの有機塩基と配位する場合に形成された塩。塩は、単なる例として、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウム、テトラアルキルアンモニウムなどをさらに含み、化合物が塩基性官能性を含む場合、塩酸塩、臭化水素酸塩、酒石酸塩、メシル酸塩、酢酸塩、マレイン酸塩、シュウ酸塩などの無毒の有機又は無機酸の塩をさらに含む。用語「医薬として許容し得るカチオン」とは、酸性官能基の許容し得るカチオン性対イオンをいう。そのようなカチオンは、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウム、テトラアルキルアンモニウムカチオンなどにより例示される。
【0035】
「医薬として許容し得るビヒクル」とは、本発明の化合物とともに投与される希釈剤、補助剤、賦形剤、又は担体をいう。
【0036】
「プロドラッグ」は、切断可能な基を有し、加溶媒分解により、又は生理的な条件下で、インビボで医薬として活性な本発明の化合物になる、本発明の化合物の誘導体を含む化合物を意味する。そのような例には、コリンエステル誘導体など、N-アルキルモルホリンエステルなどがあるが、これらに限定されない。
【0037】
「溶媒和物」とは、通常、加溶媒分解反応によって溶媒と会合する化合物の形態をいう。この物理的会合には水素結合がある。従来型の溶媒には、水、エタノール、酢酸などがある。本発明の化合物は、例えば結晶形で調製でき、溶媒和できるか又は水和できる。適切な溶媒和物には、水和物などの医薬として許容し得る溶媒和物があり、化学量論的溶媒和物及び非化学量論的溶媒和物の両方もさらに含まれる。特定の例において、溶媒和物は、例えば1以上の溶媒分子が結晶質固体の結晶格子に取り込まれている場合に単離できる。「溶媒和物」は、液相及び単離可能な溶媒和物の両方を包含する。代表的な溶媒和物には、水和物、エタノラート及びメタノラートがある。
【0038】
「対象」はヒトを含む。「ヒト」、「患者」及び「対象」という用語は、本明細書において交換可能に使用される。
【0039】
「治療上有効な量」とは、疾患を治療するために対象へ投与される場合に、該疾患のそのような治療を発効させるのに十分である化合物の量を意味する。「治療上有効な量」は、化合物、疾患及びその重症性、並びに治療される対象の年齢、体重などに応じて変化し得る。
【0040】
「予防する(preventing)」又は「予防(prevention)」とは、疾患又は障害を得るか又はかかるリスクの低減を意味する(すなわち、該疾患の臨床症状のうちの少なくとも1つを、疾患原因物質に曝露されている可能性がある対象、又は疾患の発症に先立って該疾患に罹りやすい対象において起こさせない)。
【0041】
用語「予防(prophylaxis)」は「予防(prevention)」に関連し、その目的が疾患を治療又は治癒することよりも予防することにある処置又は手段をいう。予防処置の非限定的な例には、ワクチンの投与;例えば不動化に起因する血栓症のリスクのある入院患者に対する低分子量ヘパリンの投与;及び、マラリアが風土性である、又はマラリアになるリスクが高い地理的領域への訪問に先立つクロロキンなどの抗マラリア薬の投与があり得る。
【0042】
任意の疾患又は障害の「治療する(treating)」又は「治療(treatment)」とは、一実施態様において、疾患又は障害を回復させること(すなわち、該疾患を抑止するか又はその臨床症状のうちの少なくとも1つの徴候、程度、又は重症度を減少させる)をいう。別の実施態様において、「治療する」又は「治療」とは、少なくとも1つの肉体的パラメーターを回復させることをいい、これは対象によって認識できない場合もある。さらに別の実施態様において、「治療する」又は「治療」とは、肉体的に、(例えば、認識可能な症状の安定化)、生理的に、(例えば、肉体的パラメーターの安定化)、又はその両方のいずれかで、疾患又は障害を調節することをいう。さらなる実施態様において、「治療する」又は「治療」とは、疾患の進行を減速させることに関する。
【0043】
本明細書では、「炎症性病態(複数可)」という用語は、関節リウマチ、変形性関節症、若年性特発性関節炎、乾癬、アレルギー性気道疾患(例えば、喘息、鼻炎)、炎症性腸疾患(例えば、クローン病、大腸炎)、エンドトキシン駆動性疾病状態(例えば、バイパス手術後の合併症又は例えば、慢性心不全の原因となる慢性的なエンドトキシン状態)、及び関節の軟骨などの軟骨に関する関連疾患を含む病態の群をいう。特に該用語は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患をいう。
【0044】
本明細書では、「自己免疫疾患(複数可)」は、COPDなどの病態を含む閉塞性気道疾患、喘息(例えば、内因性喘息、外因性喘息、塵埃性喘息(dust asthma)、小児喘息)、特に慢性又は難治性喘息(例えば、遅発型喘息及び気道過敏)、気管支喘息を含む気管支炎、全身性エリテマトーデス(SLE)、多発性硬化症、I型糖尿病及びそれと関連する合併症、アトピー性湿疹(アトピー性皮膚炎)、接触性皮膚炎及びさらなる湿疹性皮膚炎、炎症性腸疾患(例えば、クローン病及び潰瘍性大腸炎)、アテローム硬化症、並びに筋萎縮性側索硬化症を含む疾患の群をいう。特に、該用語は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患を意味する。
【0045】
本明細書では、「増殖性疾患(複数可)」という用語は、ガン(例えば、子宮平滑筋肉腫又は前立腺ガン)、骨髄増殖性疾患(例えば、真性多血症、本態性血小板血症、及び骨髄線維症)、白血病(例えば、急性骨髄性白血病及び急性リンパ芽球性白血病)、多発性骨髄腫、乾癬、再狭窄、硬化性皮膚炎、又は線維症などの病態をいう。特に、該用語は、ガン、白血病、多発性骨髄腫、及び乾癬をいう。
【0046】
本明細書では、「ガン」という用語は、皮膚又は体内臓器、例えば、限定はされないが、乳房、前立腺、肺、腎臓、膵臓、胃又は腸の細胞の悪性又は良性の増殖をいう。ガンは、隣接する組織に浸潤し、例えば、骨、肝臓、肺又は脳など遠位の臓器に広がる(転移する)傾向がある。本明細書では、ガンという用語は、限定はされないが、メラノーマ、リンパ腫、白血病、線維肉腫、横紋筋肉腫、及びマスト細胞腫などの転移性腫瘍細胞型、並びに限定はされないが、結腸直腸ガン、前立腺ガン、小細胞肺ガン及び非小細胞肺ガン、乳ガン、膵ガン、膀胱ガン、腎ガン、胃ガン、神経膠芽腫、原発性肝ガン、卵巣ガン、前立腺ガン、及び子宮平滑筋肉腫などの組織細胞腫(tissue carcinoma)型の両方を含む。
【0047】
本明細書では、「白血病」という用語は、血液及び造血器官の腫瘍性疾患をいう。そのような疾患は、宿主を感染及び出血しやすい状態にさせる、骨髄及び免疫系の機能障害を引き起こし得る。特に、白血病という用語は、急性骨髄性白血病(AML)及び急性リンパ芽球性白血病(ALL)をいう。
【0048】
本明細書では、「移植拒絶」という用語は、例えば、膵島、幹細胞、骨髄、皮膚、筋肉、角膜組織、神経細胞組織、心臓、肺、心肺の組み合わせ、腎臓、肝臓、腸、膵臓、気管、又は食道など、細胞、組織又は固形臓器の同種又は異種移植片の急性又は慢性拒絶反応、又は移植片対宿主病をいう。
【0049】
本明細書では、「軟骨代謝回転の障害を含む疾患」という用語は、変形性関節症、乾癬性関節炎、若年性関節リウマチ、痛風関節炎、化膿性又は感染性関節炎、反応性関節炎、反射性交感神経性ジストロフィー、疼痛性ジストロフィー、ティーツェ症候群又は肋骨軟骨炎、線維筋痛症、骨軟骨炎、神経性又はニューロパチー性関節炎、関節症、地方性変形性骨軟骨関節症(osteoarthritis deformans endemica)などの風土病型関節炎、ムセレニ病(Mseleni disease)及びハンディゴデュ病(Handigodu disease);線維筋痛症に起因する変性、全身性エリテマトーデス、強皮症及び強直性脊椎炎などの病態を含む。
【0050】
本明細書では、「先天性軟骨形成異常(複数可)」という用語は、遺伝性軟骨溶解、軟骨異形成症、及び偽性軟骨異形成症などの病態、特に、限定はされないが、小耳症、無耳症、骨幹端軟骨異形成症、及び関連の疾患を含む。
【0051】
本明細書では、「IL6の分泌過多と関連する疾患(複数可)」という用語は、キャッスルマン病、多発性骨髄腫、乾癬、カポジ肉腫、及び/又はメサンギウム増殖性糸球体腎炎などの病態を含む。
【0052】
本明細書では、「JAK」という用語は、膜受容体からのサイトカインシグナル伝達をSTAT転写因子に伝達する細胞質チロシンキナーゼであるヤヌスキナーゼ(JAK)のファミリーに関連する。4種のJAKファミリーメンバー、JAK1、JAK2、JAK3、及びTYK2が記載され、JAKという用語は、文脈が示すとおり、JAKファミリーメンバー全てをまとめて意味することも、1つ以上のJAKファミリーメンバーを意味することもある。
【0053】
「本発明の化合物(複数可)」及び等価な表現は、本明細書に記載された式の化合物を包含することを意味し、文脈が許す場合、その表現は、医薬として許容し得る塩及びその溶媒和物、例えば、水和物、並びに該医薬として許容し得る塩の溶媒和物を含む。同様に、中間体への言及は、それら自体が特許請求されているか否かに関わらず、文脈が許す場合、それらの塩及び溶媒和物を包含することを意味する。
【0054】
例えば、限定はされないがC1-C8アルキルなど、本明細書において範囲が言及される場合、範囲の引用は、前記範囲の各要素の説明であるとみなされたい。
【0055】
本発明の化合物の他の誘導体は、それらの酸形態及び酸誘導体形態の両方で活性を有するが、該酸感受性形態では多くの場合、哺乳動物において溶解性、組織適合性又は遅延放出の利点を付与する(Bundgard, H.の文献、「プロドラッグの設計(Design of Prodrugs)」, pp. 7-9, 21-24, Elsevier, Amsterdam 1985参照)。プロドラッグには、当業者に周知である酸誘導体があり、例えば、親の酸と好適なアルコールとの反応により製造されるエステル、又は親の酸化合物と置換若しくは非置換のアミンとの反応により製造されるアミド、又は酸無水物、又は混合無水物がある。本発明の化合物の懸垂した酸性基から誘導される単純な脂肪族又は芳香族のエステル、アミド、及び酸無水物は特に有用なプロドラッグである。いくつかの場合で、(アシルオキシ)アルキルエステル又は((アルコキシカルボニル)オキシ)アルキルエステルなどの二重エステルタイプのプロドラッグを製造するのが望ましい。そのような特定のプロドラッグは、本発明の化合物のC1からC8アルキル、C2-C8アルケニル、アリール、C7-C12置換アリール、及びC7-C12アリールアルキルエステルである。
【0056】
本明細書では、「同位体異形」という用語は、そのような化合物を構成する1以上の原子で非天然の同位体比率を含む化合物をいう。例えば、化合物の「同位体異形」は、例えば、重水素(2H又はD)、炭素-13(13C)、窒素-15(15N)などの1以上の非放射性同位体を含むことができる。そのような同位体的置換がなされる化合物において、以下の原子は存在する場合、例えば、任意の水素が2H/Dとなり、任意の炭素が13Cとなり、又は任意の窒素が15Nとなり、かつそのような原子の存在及び配置が当分野の技術内で決定できるように変更し得ることが理解されよう。同様に、本発明は、放射性同位体を有する同位体異形の製造を含むことができ、例えば、得られた化合物を、薬剤及び/又は基質組織分布研究に使用する場合がある。放射性同位体トリチウム、すなわち3H、及び炭素-14、すなわち14Cは、それらの取込みが容易であり、かつ検出手段が整っているという観点から本目的に特に有用である。さらに、化合物は11C、18F、15O、及び13Nなどの陽電子放出同位体で置換されて調整することができ、基質受容体占有率を検討するため陽電子放射型断層撮影法(PET)研究において有用であり得るだろう。
【0057】
本明細書に提供される化合物の全ての同位体異形は、放射性であるか否かに関わらず、本発明の範囲内に包含されるものとする。
【0058】
互いの鏡像ではない立体異性体は「ジアステレオマー」と称され、互いの重ねあわすことのできない鏡像であるものは「エナンチオマー」と称される。化合物が不斉中心を有する場合、例えば4つの異なる基に結合している場合、一組のエナンチオマーが存在し得る。エナンチオマーは、その不斉中心の絶対配置により特徴づけることができ、Cahn及びPrelogのR-及びS-順序付け規則か、又は分子が偏光面を回転させる様態により説明され、右旋性又は左旋性(すなわち、それぞれ(+)又は(-)-異性体)であると呼ばれる。キラルな化合物は、個別なエナンチオマーとして、又はその混合物として存在し得る。等しい比率のエナンチオマーを含む混合物は「ラセミ混合物」と呼ばれる。
【0059】
「互変異性体」とは、特定の化合物構造の交換可能な形態であり、かつ水素原子及び電子の配置において異なる化合物をいう。したがって、2つの構造は、π電子及び原子(通常、H)の動きを介して平衡となり得る。例えば、エノール及びケトンは、酸又は塩基のいずれかを用いた処理により迅速に相互変換されるので、互変異性体である。互変異性の別の例には、フェニルニトロメタンのアシ-及びニトロ-形態があり、これらは酸又は塩基の処理により同様に形成される。
【0060】
互変異性形態は、目的とする化合物の最適な化学反応性及び生物活性の実現に関連し得る。
【0061】
本発明の化合物は、1つ以上の不斉中心を持つことがある。したがって、そのような化合物は、個別な(R)-若しくは(S)-立体異性体として、又はその混合物として製造することができる。
【0062】
特記されない限り、本明細書及び請求項中の特定の化合物の記載又は呼称は、その個別のエナンチオマー、並びにラセミ体及びその他の混合物の両方を含むものとする。立体化学の決定及び立体異性体の分離の方法は当分野に周知である。
【0063】
(化合物)
本発明は、JAKの阻害剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患の治療に有用であるという発見に基づいている。本発明は、該化合物の製造方法、該化合物を含む医薬組成物、並びに軟骨の破壊、骨及び/又は関節の破壊、及び/又は炎症を含む疾患を、本発明の化合物を投与することにより治療する方法も提供する。本化合物は、JAKファミリーのメンバーの阻害剤である。具体的には、それは、JAK1、JAK2、JAK3、及びTYK2の活性を阻害する。特に、それはJAK1の活性を阻害する。
【0064】
したがって、本発明の第一の態様において、式Iを有する本発明の化合物が開示される。
【化2】
【0065】
本発明の化合物は、シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミドである。
【0066】
一実施態様において、本発明の化合物は、同位体異形ではない。
【0067】
一態様において、本発明の化合物は、遊離の塩基として存在する。
【0068】
一態様において、本発明の化合物は、医薬として許容し得る塩である。
【0069】
一態様において、本発明の化合物は、該化合物の溶媒和物である。
【0070】
一態様において、本発明の化合物は、該化合物の医薬として許容し得る塩の溶媒和物である。
【0071】
特定の態様において、本発明は、上記の式の化合物のプロドラッグ及び誘導体を提供する。プロドラッグは、代謝により切断可能な基を有し、加溶媒分解により、又は生理的な条件下で、インビボで医薬として活性な本発明の化合物になる、本発明の化合物の誘導体である。そのような例には、コリンエステル誘導体など、N-アルキルモルホリンエステルなどがあるが、これらに限定されない。
【0072】
本発明の化合物の他の誘導体は、それらの酸形態及び酸誘導体形態の両方で活性を有するが、該酸感受性形態では多くの場合、哺乳動物において溶解性、組織適合性又は遅延放出の利点を付与する(Bundgard, H.の文献、「プロドラッグの設計(Design of Prodrugs)」, pp. 7-9, 21-24, Elsevier, Amsterdam 1985参照)。プロドラッグには当業者に周知である酸誘導体があり、例えば、親の酸と好適なアルコールとの反応により製造されるエステル、又は親の酸化合物と置換若しくは非置換のアミンとの反応により製造されるアミド、又は酸無水物、又は混合無水物がある。本発明の化合物の垂れている酸性基から誘導される単純な脂肪族又は芳香族のエステル、アミド、及び酸無水物は好ましいプロドラッグである。いくつかの場合で、(アシルオキシ)アルキルエステル又は((アルコキシカルボニル)オキシ)アルキルエステルなどの二重エステルタイプのプロドラッグを製造するのが望ましい。本発明の化合物のC1からC8アルキル、C2-C8アルケニル、アリール、C7-C12置換アリール、及びC7-C12アリールアルキルエステルが特に有用である。
【0073】
(医薬組成物)
医薬として使用される場合、本発明の化合物は、典型的には医薬組成物の形態で投与される。そのような組成物は、医薬分野において周知の様式で製造でき、少なくとも1つの活性化合物を含むことができる。通常、本発明の化合物は、医薬として有効な量で投与される。実際に投与される化合物の量は、典型的には、治療される状態、選択した投与経路、実際に投与される化合物、個々の患者の年齢、体重及び反応、患者の症状の重症度などを含む関連した状況に照らして、医師により決定される。
【0074】
本発明の医薬組成物は、経口経路、直腸経路、経皮経路、皮下経路、関節内経路、静脈経路、筋肉内経路、及び鼻腔内経路を含む様々な経路により投与できる。意図された送達経路に応じて、本発明の化合物は、好ましくは、注射可能若しくは経口組成物として、又は全て経皮投与のための塗剤として、ローション剤として、若しくはパッチとして、製剤化される。
【0075】
経口投与用組成物は、バルク溶液若しくは懸濁液、又はバルク粉末の形態をとり得る。しかしながら、より一般的には、本組成物は、正確な投与を容易にする単位投与形態で提供される。「単位投与形態」という用語は、ヒトの対象及び他の哺乳動物への単位投与量として適切な物理的に個別の単位をいい、各単位は適当な医薬用賦形剤、ビヒクル、又は担体とともに、所望の治療効果を提供するように算出された既定量の活性物質を含む。固体組成物の場合、典型的な単位投与形態には、液体組成物が予め充填され予め計量されたアンプル若しくはシリンジ又は丸剤、錠剤、カプセル等などを含む。そのような組成物において、本発明の化合物は、通常、微量成分(約0.1〜約50重量%又は好ましくは約1〜約40重量%)であり、その残りは様々なビヒクル又は担体及び所望の投与形態を形成するのを補助する加工助剤である。
【0076】
経口投与に適した液体形態には、緩衝液、懸濁剤及び分配剤、着色剤、香料などとともに好適な水性又は非水性の溶媒を含むことができる。固体形態は、例えば、以下の成分のいずれか、又は類似の性質の化合物を含むことができる:微結晶性セルロース、トラガカントガム又はゼラチンなどの結合剤;デンプン又はラクトースなどの賦形剤、アルギン酸、プリモゲル、又はコーンスターチなどの崩壊剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド状二酸化ケイ素などの流動促進剤;スクロース又はサッカリンなどの甘味剤;又は、ハッカ、サリチル酸メチル、又はオレンジ香料などの香料。
【0077】
注射可能組成物は、典型的には、当該技術分野において公知の注射可能な無菌の食塩水若しくはリン酸緩衝食塩水又は他の注射可能な担体に基づく。前記のように、そのような組成物における活性化合物は、典型的には微量成分であり、多くの場合、約0.05〜10重量%であり、その残りは注射可能な担体などである。
【0078】
経皮組成物は、典型的には、一般に約0.01〜約20重量%、好ましくは約0.1〜約20重量%、好ましくは約0.1〜約10重量%、より好ましくは約0.5〜約15重量%にわたる量で有効成分(複数可)を含む、局所用軟膏又はクリームとして製剤化される。軟膏として製剤化される場合、有効成分は、典型的には、パラフィン性又は水混和性の軟膏基剤のいずれかと混合される。あるいは、有効成分は、例えば水中油クリーム基剤とともにクリームに配合できる。そのような経皮製剤は当該技術分野において周知であり、有効成分又は製剤の真皮浸透の安定性を促進するために一般的には添加成分を含む。そのような公知の経皮製剤及び成分の全ては、本発明の範囲内に含まれる。
【0079】
本発明の化合物は、経皮装置により投与することもできる。したがって、経皮投与は、リザーバー型若しくは多孔膜型いずれかのパッチ又は固体マトリクス多様体を使用して達成できる。
【0080】
経口投与可能な、注射可能な、又は局所的に投与可能な組成物のための上記成分は、代表的なものにすぎない。他の材料並びに加工技術などは、「レミントンの薬学(Remington's Pharmaceutical Sciences)」、第17版, 1985, Mack Publishing Company, Easton, Pennsylvaniaのパート8に記載され、これは引用により本明細書に組み込まれる。
【0081】
本発明の化合物は、徐放性形態で、又は徐放性製剤送達系から投与することもできる。代表的な徐放物質の説明は、レミントンの薬学に見出すことができる。
【0082】
以下の製剤例は、本発明に従って製造できる代表的な医薬組成物を表す。しかし、本発明は、以下の医薬組成物に限定されない。
【0083】
(製剤1-錠剤)
本発明の化合物は乾燥粉末として、乾燥ゼラチン結合剤とおよそ1:2の重量比で混合できる。微量のステアリン酸マグネシウムを滑沢剤として加えてよい。該混合物は、錠剤成形機で240〜270mgの錠剤(錠剤当たり80〜90mgの活性アミド化合物)に成形することができる。
【0084】
(製剤2-カプセル剤)
本発明の化合物は乾燥粉末として、デンプン希釈剤とおよそ1:1の重量比で混合できる。該混合物は、250mgのカプセル(カプセル当たり125mgの活性アミド化合物)に充填できる。
【0085】
(製剤3-液剤)
本発明の化合物(125mg)は、スクロース(1.75g)及びキサンタンガム(4mg)と混合でき、その結果として生じる混合物は混合され、No.10メッシュU.S.シーブに通され、その後、微結晶性セルロース及びカルボキシメチルセルロースナトリウム(11:89、50mg)の予め製造した水溶液と混合できる。安息香酸ナトリウム(10mg)、香料及び着色料を水で希釈して、撹拌しながら加えてよい。次いで、十分な水を撹拌しながら、加えることができる。次いで、十分な水を添加し、5mLの総容積にすることができる。
【0086】
(製剤4-錠剤)
本発明の化合物は乾燥粉末として、乾燥ゼラチン結合剤とおよそ1:2の重量比で混合できる。微量のステアリン酸マグネシウムを、滑沢剤として加えてよい。該混合物は、錠剤成形機で450〜900mgの錠剤(150〜300mgの活性アミド化合物)に成形される。
【0087】
(製剤5-注射製剤)
本発明の化合物は、緩衝化無菌食塩水の注射可能水性媒体中、およそ5mg/mLの濃度に、溶解又は懸濁できる。
【0088】
(製剤6-局所製剤)
ステアリルアルコール(250g)及び白色ワセリン(250g)を約75℃で溶かすことができ、その後、水(約370g)に溶かした本発明の化合物(50g)、メチルパラベン(0.25g)、プロピルパラベン(0.15g)、ラウリル硫酸ナトリウム(10g)、及びプロピレングリコール(120g)の混合物を添加でき、その結果生じた混合物を凝結するまで撹拌してよい。
【0089】
(治療の方法)
本発明の化合物は、JAKの異常な活性に原因として関連しているか、又はそれに起因している、哺乳動物の病態の治療のための治療剤として使用できる。特に、JAK1の異常な活性に関連した病態。したがって、本発明の化合物及び医薬組成物は、ヒトを含む哺乳動物の炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及びIL6の分泌過多に関連する疾患の予防及び/又は治療に治療法として使用される。
【0090】
治療態様の追加の方法において、本発明は、炎症性病態に罹患しやすい又は苦しんでいる哺乳動物を治療する方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、該炎症性病態は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患から選択される。
【0091】
他の態様において、本発明は、炎症性病態の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該炎症性病態は、関節リウマチ、変形性関節症、アレルギー性気道疾患(例えば、喘息)、及び炎症性腸疾患から選択される。
【0092】
治療態様のさらなる方法において、本発明は、自己免疫疾患に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、該自己免疫疾患は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患から選択される。
【0093】
他の態様において、本発明は、自己免疫疾患の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、該自己免疫疾患は、COPD、喘息、全身性エリテマトーデス、I型糖尿病、及び炎症性腸疾患から選択される。
【0094】
治療態様のさらなる方法において、本発明は、増殖性疾患、特にガン(例えば、子宮平滑筋肉腫又は前立腺ガンなどの固形腫瘍)、白血病(例えば、AML又はALL)、多発性骨髄腫及び/又は乾癬に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0095】
他の態様において、本発明は、増殖性疾患、特にガン(例えば、子宮平滑筋肉腫又は前立腺ガンなどの固形腫瘍)、白血病(例えば、AML又はALL)、多発性骨髄腫及び/又は乾癬の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0096】
治療態様のさらなる方法において、本発明は、移植拒絶に罹患し易い又は苦しんでいる哺乳動物の治療方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。具体的な実施態様において、本発明は、臓器移植拒絶を治療する方法を提供する。
【0097】
他の態様において、本発明は、移植拒絶の治療、予防、又は予防に使用するための本発明の化合物を提供する。具体的な実施態様において、本発明は臓器移植拒絶を治療する方法を提供する。
【0098】
治療態様の方法において、本発明は、軟骨代謝回転の異常を含む疾患に罹患しやすい又は苦しんでいる哺乳動物の治療、予防、又は予防の方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を、病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0099】
他の態様において、本発明は、軟骨代謝回転の異常を含む疾患の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0100】
本発明は、先天性軟骨形成異常を治療する方法も提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0101】
他の態様において、本発明は、先天性軟骨形成異常の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0102】
治療態様のさらなる方法において、本発明は、IL6の分泌過多に関連する疾患、特にキャッスルマン病又はメサンギウム増殖性糸球体腎炎に罹患し易い又は苦しんでいる哺乳動物を治療する方法を提供する。該方法は、本明細書に記載される1種以上の医薬組成物又は本発明の化合物を病態の治療に有効な量又は病態の予防に有効な量投与することを含む。
【0103】
他の態様において、本発明は、IL6の分泌過多に関連する疾患、特にキャッスルマン病又はメサンギウム増殖性糸球体腎炎の治療、予防、又は予防に使用するための本発明の化合物を提供する。
【0104】
本発明のさらなる態様として、特に上述の病態及び疾患の治療又は予防における医薬品として使用するための本発明の化合物が提供される。また、上述の病態及び疾患の1つを治療又は予防するための医薬の製造における該化合物の使用も本明細書に提供される。
【0105】
本方法の特定の療法は、炎症性病態に苦しんでいる対象に、該対象における炎症のレベルを低下させ、かつ好ましくは、前記炎症の原因であるプロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。本方法の特別な実施態様は、関節リウマチの進行に苦しんでいる又は罹患し易い対象患者に、前記患者の関節における炎症をそれぞれ低下させ若しくは予防し、かつ好ましくは、前記炎症の原因であるプロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。
【0106】
本方法のさらに特別な療法は、軟骨代謝回転の異常を含む疾患(例えば、変形性関節症)に苦しんでいる対象に、前記破壊の原因である永続的なプロセスを低下させ、かつ好ましくは終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。該方法の特別な実施態様は、変形性関節症の進行に苦しんでいる又は罹患し易い対象患者に、前記患者の関節における軟骨破壊をそれぞれ低下させるか又は予防し、かつ好ましくは前記破壊の原因である永続的プロセスを終結させるのに十分な時間、有効量の本発明の化合物を投与することを含む。特別な実施態様において、前記化合物は、軟骨同化特性及び/又は抗異化特性を示すことがある。
【0107】
注入用量レベルは、約1〜約120時間及び特に24〜96時間の全てについて、約0.1mg/kg/時〜少なくとも10mg/kg/時にわたる。また、約0.1mg/kg〜約10mg/kg又はこれを超える予充填ボーラスを、妥当な定常状態レベルを達成するために投与することもできる。最大の全用量は、40〜80kgのヒト患者について約2g/日を超えるとは予想されない。
【0108】
長期の変性状態などの状態の予防及び/又は治療について、治療のための療法は、通常、数ヶ月又は数年にわたるため、患者の便宜及び許容度のために経口投与が好ましい。経口投与では、1日あたり1〜5回、特に2〜4回、典型的には3回の経口投与が代表的な療法である。これらの投与様式を利用して、各投与量は、個別の投与量をそれぞれ、約0.1〜約10mg/kg、特に約1〜約5mg/kgで提供することによって、約0.01〜約20mg/kgの本発明の化合物を提供する。
【0109】
経皮用量は、一般に、注入用量を使用して達成されるのと同等又はそれよりも低い血中濃度を提供するように選択される。
【0110】
炎症性病態の発症を予防するために使用する場合、本発明の化合物は、典型的には医師の助言に従い、かつその管理下で、上記の投与量レベルにて、該病態にかかる危険性のある患者に投与される。特定の病態にかかる危険性のある患者は、一般に、該病態の家族歴を有する者、又は遺伝的試験若しくはスクリーニングにより、特に該病態にかかりやすいと同定された者を含む。
【0111】
本発明の化合物は、単一の活性薬剤として投与することができるか、又は同じ若しくは類似の治療活性を示し、かつ併用投与に安全かつ有効であることが判断された他の化合物を含む他の治療剤との組合せで投与することもできる。具体的な実施態様において、2種の(又はそれより多い)薬剤の同時投与は、使用されるそれぞれの用量を顕著に減少させることができ、それによって、見られる副作用を低減させる。
【0112】
一実施態様において、本発明の化合物は、炎症性病態の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、免疫調節性薬剤、例えば、アザチオプリン、コルチコステロイド(例えば、プレドニゾロン又はデキサメタゾン)、シクロホスファミド、シクロスポリンA、タクロリムス、ミコフェノール酸モフェチル、ムロモナブ-CD3(OKT-3、例えば、Orthocolone(登録商標))、ATG、アスピリン、アセトアミノフェン、イブプロフェン、ナプロキセン、及びピロキシカムを含むが、これらに限定されない。
【0113】
一実施態様において、本発明の化合物は、関節炎(例えば、関節リウマチ)の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、鎮痛薬、非ステロイド性抗炎症薬(NSAID)、ステロイド、合成DMARD(例えば、限定はされないが、メトトレキサート、レフルノミド、スルファサラジン、オーラノフィン、金チオリンゴ酸ナトリウム、ペニシラミン、クロロキン、ヒドロキシクロロキン、アザチオプリン、及びシクロスポリン)、及び生物学的DMARD(例えば、限定はされないが、インフリキシマブ、エタネルセプト、アダリムマブ、リツキシマブ、及びアバタセプト)を含むが、これらに限定されない。
【0114】
一実施態様において、本発明の化合物は、増殖性疾患の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、メトトレキサート、ロイコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロホスファミド、5-フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗-HER2モノクローナル抗体(例えば、Herceptin(商標))、カペシタビン、塩酸ラロキシフェン、EGFR阻害剤(例えば、lressa(登録商標)、Tarceva(商標)、Erbitux(商標))、VEGF阻害剤(例えば、Avastin(商標))、プロテアソーム阻害剤(例えば、Velcade(商標))、Glivec(登録商標)及びhsp90阻害剤(例えば、17-AAG)を含むが、これらに限定されない。加えて、本発明の化合物は、限定はされないが、放射線療法又は外科手術を含む他の治療法と組合せて投与することができる。具体的な実施態様において、該増殖性疾患は、ガン、骨髄増殖性疾患、又は白血病から選択される。
【0115】
一実施態様において、本発明の化合物は、自己免疫疾患の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、グルココルチコイド、細胞分裂阻害剤(例えば、プリンアナログ)、アルキル化剤(例えば、ナイトロジェンマスタード(シクロホスファミド)、ニトロソウレア、白金化合物、その他)、代謝拮抗薬(例えば、メトトレキサート、アザチオプリン及びメルカプトプリン)、細胞毒性抗生物質(例えば、ダクチノマイシンアントラサイクリン系薬剤、マイトマイシンC、ブレオマイシン及びミトラマイシン)、抗体(例えば、抗-CD20、抗-CD25、又は抗-CD3(OTK3)モノクローナル抗体、Atgam(登録商標)及びThymoglobuline(登録商標))、シクロスポリン、タクロリムス、ラパマイシン(シロリムス)、インターフェロン(例えば、IFN-β)、TNF結合タンパク質(例えば、インフリキシマブ(Remicade(商標))、エタネルセプト(Enbrel(商標))、又はアダリムマブ(Humira(商標)))、ミコフェノレート、フィンゴリモド、及びミリオシンを含むが、これらに限定されない。
【0116】
一実施態様において、本発明の化合物は、移植拒絶反応の治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、カルシニューリン阻害剤(例えば、シクロスポリン又はタクロリムス(FK506))、mTOR阻害剤(例えば、シロリムス、エベロリムス)、抗増殖剤(例えば、アザチオプリン、ミコフェノール酸)、副腎皮質ステロイド(例えば、プレドニゾロン、ヒドロコルチゾン)、抗体(例えば、モノクローナル抗-IL-2Rα受容体抗体、バシリキシマブ、ダクリズマブ)、ポリクローナル抗T細胞抗体(例えば、抗胸腺細胞グロブリン(ATG)、抗リンパ球グロブリン(ALG))を含むが、これらに限定されない。
【0117】
一実施態様において、本発明の化合物は、喘息及び/又は鼻炎及び/又はCOPDの治療及び/又は予防のための別の治療薬と同時投与され;特定の薬剤には、β2-アドレナリン受容体刺激薬(例えば、サルブタモール、レバルブテロール、テルブタリン、及びビトルテロール)、エピネフリン(吸入又は錠剤)、抗コリン薬(例えば、臭化イプラトロピウム)、グルココルチコイド(経口又は吸入)、長時間作用性β2-刺激薬(例えば、サルメテロール、ホルモテロール、バンブテロール、及び徐放性経口アルブテロール)、吸入ステロイド薬と長時間作用性気管支拡張薬との組合せ(例えば、フルチカゾン/サルメテロール、ブデソニド/ホルモテロール)、ロイコトリエン拮抗薬及び合成阻害剤(例えば、モンテルカスト、ザフィルルカスト、及びジロートン)、メディエーター放出の阻害剤(例えば、クロモグリケート及びケトチフェン)、IgE応答の生体調節因子(例えば、オマリズマブ)、抗ヒスタミン剤(例えば、セテリジン、シンナリジン、フェキソフェナジン)、及び血管収縮薬(例えば、オキシメタゾリン、キシロメタゾリン、ナファゾリン、及びトラマゾリン)を含むが、これらに限定されない。
【0118】
加えて、本発明の化合物は、喘息及び/又はCOPDの緊急治療と組合せて投与することができ、そのような治療には、酸素又はヘリオックス投与、サルブタモール又はテルブタリン噴霧(抗コリン薬(例えば、イプラトロピウム)と任意に併用)、全身性ステロイド薬(経口又は静脈内、例えば、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、デキサメタゾン、又はヒドロコルチゾン)、静脈内サルブタモール、非特異的β-刺激薬、注射又は吸入(例えば、エピネフリン、イソエタリン、イソプロテレノール、メタプロテレノール)、抗コリン薬(静脈内又は噴霧、例えば、グリコピロレート、アトロピン、イプラトロピウム)、メチルキサンチン(テオフィリン、アミノフィリン、バミフィリン)、気管支拡張性効果を有する吸入麻酔(例えば、イソフルラン、ハロタン、エンフルラン)、ケタミン、及び静脈内硫酸マグネシウムを含む。
【0119】
一実施態様において、本発明の化合物は、過敏性腸疾患(IBD)の治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、グルココルチコイド(例えば、プレドニゾン、ブデソニド)、合成疾患修飾性免疫調節薬(例えば、メトトレキサート、レフルノミド、スルファサラジン、メサラジン、アザチオプリン、6-メルカプトプリン、及びシクロスポリン)及び生物学的疾患修飾性免疫調節薬(インフリキシマブ、アダリムマブ、リツキシマブ及びアバタセプト)を含むが、これらに限定されない。
【0120】
一実施態様において、本発明の化合物は、全身性エリテマトーデスの治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、疾患修飾性抗リウマチ薬(DMARD)、例えば、抗マラリア薬(例えば、プラキニル、ヒドロキシクロロキン)、免疫抑制剤(例えば、メトトレキサート及びアザチオプリン)、シクロホスファミド及びミコフェノール酸;免疫抑制薬及び鎮痛薬、例えば、非ステロイド性抗炎症薬、アヘン剤(例えば、デキストロプロポキシフェン及びココダモール)、オピオイド(例えば、ヒドロコドン、オキシコドン、MSコンチン又はメサドン)及びフェンタニルデュラゲシク経皮吸収パッチを含むが、これらに限定されない。
【0121】
一実施態様において、本発明の化合物は、乾癬の治療及び/又は予防のための別の治療薬と同時投与され、特定の薬剤には、コールタール、ジトラノール(アントラリン)、デスオキシメタゾン(Topicort(商標))などの副腎皮質ステロイド、フルオシノニド、ビタミンD3アナログ(例えば、カルシポトリオール)、アルガン油、及びレチノイド(エトレチナート、アシトレチン、タザロテン)を含有する浴溶液、保湿剤、薬用クリーム、及び軟膏剤などの局所的治療薬、メトトレキサート、シクロスポリン、レチノイド、チオグアニン、ヒドロキシウレア、スルファサラジン、ミコフェノール酸モフェチル、アザチオプリン、タクロリムス、フマル酸エステルなどの全身治療薬、又はAmevive(商標)、Enbrel(商標)、Humira(商標)、Remicade(商標)、Raptiva(商標)、及びウステキヌマブ(IL-12及びIL-23遮断薬)などの生物製剤を含むが、これらに限定されない。加えて、本発明の化合物は、限定はされないが、光線療法又は光化学療法(例えば、ソラレン長波長紫外線治療(PUVA))を含む他の治療法と組合せて投与することができる。
【0122】
当業者に明らかであるように、同時投与とは、同じ治療計画の一部として2種以上の治療薬を患者に送達する任意の手段を含むものである。2種以上の薬剤は単一製剤で同時に投与できるが、これは必須ではない。該薬剤は、異なる製剤で、かつ異なる時間に、投与してもよい。
【0123】
(全般的合成手順)
(全般)
本発明の化合物は、以下の全般的方法及び手順を利用して容易に利用できる出発物質から製造できる。典型的又は好ましいプロセス条件(すなわち、反応温度、時間、反応物のモル比、溶媒、圧力など)が与えられる場合、他のプロセス条件も他に明記しない限り利用できることは理解されるであろう。最適な反応条件は、使用する特定の反応物又は溶媒により変わり得るが、そのような条件は、通例の最適化手順によって当業者により決定できる。
【0124】
さらに、当業者には明らかであるように、特定の官能基が望ましくない反応を受けるのを防ぐために、従来型の保護基が必要となることがある。特定の官能基のための好適な保護基、並びに保護及び脱保護のための適当な条件の選択は、当該分野において周知である。例えば、多数の保護基並びにそれらの導入及び除去は、T. W. Greene及びP. G. M. Wutsの文献、「有機合成における保護基(Protecting Groups in Organic Synthesis)」、第2版、Wiley, New York, 1991及びその中で引用された文献に記載されている。
【0125】
以下の方法は、先に列記された代表的なビシクロヘテロアリールの製造に関して詳細に提示されている。本発明の化合物は、公知又は市販の出発物質及び試薬から、有機合成の分野の当業者により製造できる。
【0126】
全ての試薬は市販の等級であり、他に明記しない限り、さらに精製せずに入手したままで使用した。市販の無水溶媒を、不活性雰囲気下で実施される反応に使用した。他に特に明記しない限り、試薬等級溶媒を他の全ての場合において使用した。カラムクロマトグラフィーをシリカゲル60(35〜70μm)で実施した。薄層クロマトグラフィーは、予め被覆してあるシリカゲルF-254プレート(0.25mm厚)を使用して実施した。1H NMRスペクトルは、Bruker社製DPX 400核磁気共鳴分光計(400MHz)で記録した。1H NMRスペクトルの化学シフト(δ)は、内部標準としてのテトラメチルシラン(δ0.00)又は適切な残留溶媒ピーク、すなわちCHCl3(δ7.27)に対して百万分率(ppm)で報告される。多重度は、シングレット(s)、ダブレット(d)、トリプレット(t)、カルテット(q)、マルチプレット(m)及びブロード(br)として与えられる。結合定数(J)は、Hzで与えられる。エレクトロスプレーMSスペクトルは、MicromassプラットフォームLC/MSスペクトロメータで得られた。全てのLCMS分析に使用したカラムは以下のものである:Waters社製Acquity UPLC BEH C18 1.7μm、2.1mm ID×50mm L(Part No.186002350)。分取HPLC:Waters社製XBridge Prep C18 5μm ODB 19mm ID×100mm L(Part No.186002978)。全ての方法は、MeCN/H2O勾配を使用している。H2Oは、0.1%TFA又は0.1%NH3のいずれかを含有する。
【0127】
下記は、実験セクションにおいて使用する省略形のリストである。
【表1】
【0128】
(本発明の化合物の合成製造)
本発明の化合物は、以下のスキームに従って製造できる。
【化3】
式中、Arはフェニル-L1-ヘテロシクロアルキルを表し、ここで、式中、L1は-CO-であり、ヘテロシクロアルキル基は任意に置換されている。
【0129】
(全般)
(1.1.1 1-(6-ブロモ-ピリジン-2-イル)-3-カルボエトキシ-チオ尿素(2))
【化4】
5℃に冷却した2-アミノ-6-ブロモピリジン(1)(253.8g、1.467mol)のDCM(2.5L)溶液に、イソチオシアン酸エトキシカルボニル(173.0mL、1.467mol)を15分にわたって滴加する。次いで、該反応混合物を室温(20℃)に温め、16時間撹拌する。真空での蒸発により固形物を得て、これを濾過により回収し、石油(3×600mL)で完全に洗浄し、風乾させて(2)を得る。該チオ尿素は、精製することなくそのまま次工程に使用することができる。
【化5】
【0130】
(1.1.2 5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イルアミン(3))
【化6】
EtOH/MeOH(1:1、900mL)中の塩酸ヒドロキシルアミン(101.8g、1.465mol)の懸濁液に、N,N-ジイソプロピルエチルアミン(145.3mL、0.879mol)を添加し、該混合物を室温(20℃)で1時間撹拌する。次に、1-(6-ブロモ-ピリジン-2-イル)-3-カルボエトキシ-チオ尿素(2)(89.0g、0.293mol)を添加し、該混合物を還流までゆっくりと加熱する(注意:発生するH2Sをクエンチするのにブリーチスクラバーが必要である)。3時間の還流後、該混合物を放冷し、濾過して沈殿した固形物を回収する。該濾液を真空中で蒸発させ、H2O(250mL)を添加し、濾過することによって、さらに生成物を回収する。合わせた固形物をH2O(250mL)、EtOH/MeOH(1:1、250mL)及びEt2O(250mL)で連続的に洗浄し、次いで、真空中で乾燥させて、トリアゾロピリジン誘導体(3)を固形物として得る。該化合物は、精製することなくそのまま次工程に使用することができる。
【化7】
【0131】
(1.1.3 シクロプロパンカルボン酸(5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル)-アミド(4))
【化8】
5℃の2-アミノ-トリアゾロピリジン(3)(7.10g、33.3mmol)の無水CH3CN(150mL)溶液に、Et3N(11.6mL、83.3mmol)を添加し、続いてシクロプロパンカルボニルクロリド(83.3mmol)を添加する。次いで、該反応混合物を周囲温度まで温め、すべての出発物質(3)が消費されるまで撹拌する。必要であれば、Et3N(4.64mL、33.3mmol)及びシクロプロパンカルボニルクロリド(33.3mmol)をさらに添加して、完全な反応を確実にする。真空中で溶媒を蒸発させた後、生じた残渣を7Nのメタノール性アンモニア溶液(50mL)で処理し、周囲温度で撹拌して(1〜16時間)、ビス-アシル化生成物を加水分解する。生成物の単離は、真空中で揮発性物質を除去し、続いて、Et2O(50mL)でトリチュレイトすることによってなされる。該固形物を濾過によって回収し、H2O(2×50mL)、アセトン(50mL)、及びEt2O(50mL)で洗浄し、次いで、真空中で乾燥させて、必要なブロモ中間体(4)を得る。
【0132】
(本発明の化合物の製造のための合成手順)
(化合物1)
(工程1:スズキカップリング)
【化9】
4-カルボキシフェニルボロン酸(3.5 g, 0.021 mol.)を、シクロプロパンカルボン酸(5-ブロモ-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル)-アミド(中間体4、5 g, 0.018 mol)の1,4-ジオキサン/水(5:1)溶液に加えた。K2CO3 (5.0 g, 0.036 mol)及びPdCl2dppf (5%)を該溶液に加えた。次いで、生じた混合物を、90℃の油浴中で16時間従来の加熱により加熱した。1M HCl溶液を加えると、酸性溶液中に沈殿物が生じた。沈殿物を濾過し、真空下で乾燥させると、標記化合物を与え、さらに精製することなく次の工程に使用した。
【0133】
(工程2:シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミド(化合物1))
【化10】
EDCI (3.59 g, 0.019 mol)、HOBt (2.53 g, 0.019 mol)、及びDIPEA (4.48 mL)を、4-[2-(シクロプロパンカルボニル-アミノ)-[1,2,4]トリアゾロ[1,5-a]ピリジン-5-イル]-安息香酸(4g, 0.012 mol)の室温のDCM (150 mL)溶液に加えた。生じた混合物を室温で10分間撹拌した。ジメチルアゼチジン塩酸塩(1.64 g, 0.013 mol)を該溶液に加え、反応物を16時間撹拌する。水を該反応混合物に加えた。有機層を分離し、2N NaOH溶液、2N HCl溶液、及び水で洗浄した。有機相をMgSO4で乾燥させ、濾過し、真空下で蒸発させた。フラッシュクロマトグラフィー(溶離液: 1:1 ぺトロール/EtOAcからニートEtOAc)により精製すると、シクロプロパンカルボン酸{5-[4-(3,3-ジメチル-アゼチジン-1-カルボニル)-フェニル]-[1,2,4]トリアゾロ[1,5-a]ピリジン-2-イル}-アミドを与えた。
【0134】
本明細書に記載された合成方法により製造された、又は製造可能な本発明の化合物を、以下の表Iに列記する。本発明の化合物のNMRスペクトルデータを表IIに与える。
【0135】
【表2】
【0136】
【表3】
【0137】
(生物学的実施例)
(実施例1−インビトロアッセイ)
(実施例1.1 JAK1阻害アッセイ)
組換えヒトJAK1(触媒ドメイン、アミノ酸866-1154;カタログ番号PV4774)をInvitrogen社から購入した。1ngのJAK1を、白色384 Luminotrac 200プレート(Greiner社製、カタログ番号781075)中で、被験化合物又はビヒクル(DMSO、最終濃度1%)を含む4μLとともに、又はなしで、総量20μLのキナーゼ反応緩衝液(25mM MOPS pH6.8, 0.016% Brij-35, 8.33mM MgCl2, 3.33mM DTT, 7μM ATP)中で、20 nMのUlight-JAK1(tyr1023)ペプチド(Perkin Elmer社製、カタログ番号TRF0121)とともにインキュベートした。室温で60分後、20μL/ウェルの検出混合液(1×検出緩衝液 (Perkin Elmer社製、カタログ番号CR97-100C)、0.5nMのユーロピウム-抗-ホスホチロシン(PT66) (Perkin Elmer社製、カタログ番号AD0068)、10 mM EDTA)の添加により反応を停止させた。Envisionを使用し、320nmでの励起及び615nmでの発光測定により読み取りを行う(Perkin Elmer)。キナーゼ活性は、ビヒクルの存在下で得られた相対蛍光単位(RFU)からポジティブコントロール阻害剤(10μMスタウロスポリン)の存在下で得られたRFUを引くことにより計算した。この活性を阻害する被験化合物の能力を以下のとおり決定した。
【0138】
阻害率=((被験化合物を含む試料で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU)/(ビヒクルの存在下で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU))×100。
【0139】
JAK1アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/5の段階希釈で、1%DMSOの最終濃度で8点(20μM-4μM-800nM-160nM-32nM-6.4nM-1.28nM-0.26nM)で試験する。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させる(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0140】
【表4】
【0141】
(実施例1.2 JAK1 Ki決定アッセイ)
Kiの決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。JAK1(Invitrogen社製、PV4774)を、最終濃度500ng/mlで使用した。基質は、ポリ(Glu, Tyr)ナトリウム塩(4:1)、MW 20000-50000(Sigma社製, P0275)であった。反応は、25mM Hepes pH 7.5; 0,01% Tween20, 10 mM MgCl2中で、ATP及び化合物の濃度を変えながら実施し、150mMのリン酸を添加して停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0142】
化合物1をこのアッセイで試験すると、6.9nMのKi値を測定した。
【0143】
別法としては、Ki決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。1 ngのJAK1(Invitrogen社製、PV4774)をアッセイに使用した。基質は、50nM Ulight-JAK-1(Tyr1023) Peptide(Perkin Elmer社製、TRF0121)であった。反応は、25mM MOPS pH 6.8, 0.01%, 2 mM DTT, 5 mM MgCl2 Brij-35中で、ATP及び化合物の濃度を変えながら実施した。リン酸化された基質を、Eu標識抗ホスホチロシン抗体PT66(Perkin Elmer社製、AD0068)を使用して測定した。読み取りは、envision(Perkin Elmer社製)で、320nmでの励起及び615nm及び665nmでの発光で実施した。
【0144】
化合物1をこのアッセイで試験すると、5.6nMのKi値を測定した。
【0145】
(実施例1.3 JAK2阻害アッセイ)
組換えヒトJAK2(触媒ドメイン、アミノ酸866-1154;カタログ番号PV4210)をInvitrogen社から購入した。0.0125mUのJAK2を、白色384 Luminotrac 200プレート(Greiner社製、カタログ番号781075)中で、被験化合物又はビヒクル(DMSO、最終濃度1%)を含む4μLとともに、又はなしで、総量20μLのキナーゼ反応緩衝液(41.66mM HEPES pH7.0, 0.016% Triton X-100, 12.5mM MgCl2, 3.33mM DTT, 7.5μM ATP)中で、25 nMのUlight-JAK1(tyr1023)ペプチド(Perkin Elmer社製、カタログ番号TRF0121)とともにインキュベートした。室温で60分後、20μL/ウェルの検出混合液(1×検出緩衝液 (Perkin Elmer社製、カタログ番号CR97-100C)、0.5nMのユーロピウム-抗-ホスホチロシン(PT66) (Perkin Elmer社製、カタログ番号AD0068)、10 mM EDTA)の添加により反応を停止させた。Envision(Perkin Elmer社製)を使用し、320nmでの励起及び615nmでの発光測定により読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られた相対蛍光単位(RFU)からポジティブコントロール阻害剤(10μMスタウロスポリン)の存在下で得られたRFUを引くことにより計算した。この活性を阻害する被験化合物の能力を以下のとおり決定した。
【0146】
阻害率=((被験化合物を含む試料で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU)/(ビヒクルの存在下で測定されたRFU−ポジティブコントロール阻害剤を含む試料で測定されたRFU))×100。
【0147】
JAK2アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製する。各化合物を通常20μMの濃度で試験し、続いて、1/5の段階希釈で、1%DMSOの最終濃度で8点(20μM-4μM-800nM-160nM-32nM-6.4nM-1.28nM-0.26nM)で試験する。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させる(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0148】
【表5】
【0149】
(実施例1.4 JAK2 Ki決定アッセイ)
Kiの決定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。0.025mUのJAK2(Invitrogen社製、PV4210)を、アッセイに使用した。基質は、ポリ(Glu, Tyr)ナトリウム塩(4:1)、MW 20000-50000(Sigma社製, P0275)であった。反応は、10 mM MOPS pH 7.5, 0.5 mM EDTA, 0.01% Brij-35, 1 mM DTT, 15 mM MgAc中で、ATP及び化合物の濃度を変えながら実施し、150mMのリン酸を添加して停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0150】
化合物1をこのアッセイで試験すると、126nMのKi値を測定した。
【0151】
別法としては、Ki測定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。0.0125mUのJAK2(Invitrogen社製、PV4210)をアッセイに使用した。基質は、50nM Ulight-JAK-1(Tyr1023) Peptide(Perkin Elmer社製、TRF0121)であった。反応は、25 mM HEPES pH 7.0, 0.01% Triton X-100, 2 mM DTT, 7.5 mM MgCl2中で、ATP及び化合物の濃度を変えながら実施した。リン酸化された基質を、Eu標識抗ホスホチロシン抗体PT66(Perkin Elmer社製、AD0068)を使用して測定した。読み取りは、envision(Perkin Elmer社製)で、320 nmでの励起及び615 nm及び665 nmでの発光で実施した。
【0152】
化合物1をこのアッセイで試験すると、35nMのKi値を測定した。
【0153】
(実施例1.5 JAK3阻害アッセイ)
組換えヒトJAK3触媒ドメイン(アミノ酸781-1124;カタログ番号PV3855)をInvitrogen社から購入した。ポリプロピレン製96ウェルプレート(Greiner社製、V字底)において、0.025mUのJAK3を2.5μgのポリGT基質(Sigma社製、カタログ番号P0275)とともに、被験化合物又はビヒクル(DMSO、最終濃度1%)を含有する5μLとともに、又はなしで、総量25μLのキナーゼ反応緩衝液(25mMのTris pH7.5、0.5mMのEGTA、0.5mMのNa3VO4、5mMのb-グリセロールホスフェート、0.01%のTritonX-100、1μMの非放射性ATP、0.25μCiの33P-γ-ATP(GE Healthcare社製、カタログ番号AH9968)最終濃度)中でインキュベートした。30℃で105分後、25μL/ウェルの150mMリン酸を添加して反応を停止させた。該終結したキナーゼ反応物の全てを、セルハーベスター(Perkin Elmer社製)を使用して、予洗い(75mMのリン酸)した96ウェルフィルタープレート(Perkin Elmer社製、カタログ番号6005177)に移した。プレートを1ウェルあたり300μLの75mMリン酸溶液で6回洗浄し、該プレートの底を密閉した。40μL/ウェルのMicroscint-20を添加し、該プレートの上部を密閉し、Topcount(Perkin Elmer社製)を使用して読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られたカウント毎分(cpm)から、ポジティブコントロール阻害剤(10μMのスタウロスポリン)の存在下で得られたcpmを引くことによって算出した。この活性を阻害する被験化合物の能力は、以下のように決定した。
阻害率=((被験化合物が存在する試料について測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm)/(ビヒクルの存在下で測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm))×100。
【0154】
JAK3アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/3の段階希釈で、1%DMSOの最終濃度で8点(20μM-6.67μM-2.22μM-740nM-247nM-82nM-27nM-9nM)で試験した。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させた(例えば、5μM、1μM)。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0155】
【表6】
【0156】
(実施例1.6 JAK3 Ki測定アッセイ)
Kiの測定のために、異なる量の阻害剤を酵素と混合し、酵素反応をATP濃度の関数として追跡した。Km対化合物濃度の二重逆数プロット(ラインウィーバー・バークプロット)により、Kiを決定した。JAK3 (Carna Biosciences社製、09CBS-0625B)を、最終濃度10 ng/mlで使用した。基質はポリ(Glu,Tyr)ナトリウム塩(4:1)、MW 20000-50000 (Sigma社製、P0275)であった。反応を、25mM Tris pH 7.5, 0.01% Triton X-100, 0.5mM EGTA, 2.5mM DTT, 0.5mM Na3VO4, 5mM b-グリセロールホスフェート, 10mM MgCl2中で、ATP及び化合物の濃度を変えながら実施し、150mMリン酸の添加により停止させた。基質ポリGTに取り込まれたリン酸の測定は、試料をフィルタープレートに載せ(ハーベスター使用、Perkin Elmer社製)、次に洗浄することにより行った。ポリGT中に取り込まれた33Pは、フィルタープレート(Perkin Elmer社製)へのシンチレーション液体の添加後にTopcountシンチレーションカウンターで測定する。
【0157】
化合物1をこのアッセイで試験すると、188nMのKi値を測定した。
【0158】
(実施例1.7 TYK2阻害アッセイ)
組換えヒトTYK2触媒ドメイン(アミノ酸871-1187;カタログ番号08-147)をCarna biosciences社から購入した。ポリプロピレン製96ウェルプレート(Greiner社製、V字底)において、5ngのTYK2を12.5μgのポリGT基質(Sigma社製、カタログ番号P0275)とともに、被験化合物又はビヒクル(DMSO、最終濃度1%)を含有する5μLとともに、又はなしで、総量25μLのキナーゼ反応緩衝液(25mMのHepes pH7.5、100mMのNaCl、0.2mMのNa3VO4、0.1%のNP-40、0.1μMの非放射性ATP、0.125μCiの33P-γ-ATP (GE Healthcare社製、カタログ番号AH9968)最終濃度)中でインキュベートした。30℃で90分後、25μL/ウェルの150mMリン酸を添加して反応を停止させた。該終結したキナーゼ反応物の全てを、セルハーベスター(Perkin Elmer社製)を使用して、予洗い(75mMのリン酸)した96ウェルフィルタープレート(Perkin Elmer社製、カタログ番号6005177)に移した。プレートを1ウェルあたり300μLの75mM リン酸溶液で6回洗浄し、該プレートの底を密閉した。40μL/ウェルのMicroscint-20を添加し、該プレートの上部を密閉し、Topcount(Perkin Elmer社製)を使用して読み取りを行った。キナーゼ活性は、ビヒクルの存在下で得られたカウント毎分(cpm)から、ポジティブコントロール阻害剤(10μMのスタウロスポリン)の存在下で得られたcpmを引くことによって算出した。この活性を阻害する被験化合物の能力は以下のように決定した。
阻害率=((被験化合物が存在する試料について測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm)/(ビヒクルの存在下で測定されたcpm−ポジティブコントロール阻害剤を有する試料について測定されたcpm))×100。
【0159】
TYK2アッセイにおける用量−反応効果の試験、及び化合物についてのIC50の算出を可能にする、化合物についての用量希釈系列を調製した。各化合物を通常20μMの濃度で試験し、続いて、1/3の段階希釈で、1%DMSOの最終濃度で8点(20μM-6.67μM-2.22μM-740nM-247nM-82nM-27nM-9nM)で試験した。化合物系列の効力が大きい場合、より希釈したものを調製する、及び/又は最大濃度をより低下させた(例えば、5μM、1μM)。
【0160】
【表7】
【0161】
(実施例2 細胞アッセイ)
(実施例2.1 JAK-STATシグナル伝達アッセイ)
HeLa細胞を、10%の熱失活したウシ胎児血清、100U/mLのペニシリン、及び100μg/mLのストレプトマイシンを含有するダルベッコ変法イーグル培地(DMEM)中に維持した。トランスフェクションのために70%コンフルエンスでHeLa細胞を使用した。87μLの細胞培地中20,000個の細胞を、96ウェルプレートフォーマットにおいて、1ウェルあたり0.32μLのJet-PEI(Polyplus社製)をトランスフェクション試薬として使用して、40ngのpSTAT1(2)-ルシフェラーゼレポーター(Panomics社製)、内部標準レポーターとして8ngのLacZレポーター、及び52ngのpBSKで一過性にトランスフェクションさせた。37℃、10%CO2で一晩インキュベートした後、トランスフェクション培地を除去した。75μLのDMEM+1.5%の熱失活したウシ胎児血清を添加した。6.7倍濃縮した15μLの化合物を60分間添加し、次いで、最終濃度33ng/mLの10μLのヒトOSM(Peprotech社製)を添加した。
【0162】
全ての化合物を20μMから開始して、1/3段階希釈し、0.2%DMSOの最終濃度で全8用量(20μM-6.6μM-2.2μM-740nM-250nM-82nM-27nM-9nM)で二連で試験した。
【0163】
37℃、10%CO2で一晩インキュベートした後、細胞を100μLの溶解緩衝液/ウェル(PBS、0.9mMのCaCl2、0.5mMのMgCl2、5%のトレハロース、0.025%のTergitol NP9、0.15%のBSA)で溶解した。
【0164】
40μLの細胞溶解物を使用して、180μLのβGal溶液(30μlのONPG 4mg/mL+150μLのβ-ガラクトシダーゼ緩衝液(0.06MのNa2HPO4、0.04MのNaH2PO4、1mMのMgCl2))を20分間添加することによりβ-ガラクトシダーゼ活性を読み取った。50μLのNa2CO3 1Mの添加によって該反応を停止させた。405nmで吸光度を読み取った。
【0165】
ルシフェラーゼ活性を、製造元(Perkin Elmer社)による説明どおりに、40μlの細胞溶解物+40μlのSteadylite(登録商標)を使用してEnvison(Perkin Elmer社製)で測定した。
【0166】
10μMの汎JAK阻害剤をポジティブコントロール(100%阻害)として使用した。ネガティブコントロールとして、0.5%のDMSO(0%阻害)を使用した。ポジティブ及びネガティブコントロールを使用して、z'及び「阻害率」(PIN)値を算出した。
【0167】
阻害率=((ビヒクル存在下で測定された蛍光−被験化合物が存在する試料について測定された蛍光)/(ビヒクルの存在下で測定された蛍光−トリガーなしの試料について測定された蛍光))×100である。
【0168】
用量−反応において、試験した化合物についてPIN値をプロットし、EC50値を算出した。
【0169】
【表8】
【0170】
(実施例2.2 OSM/IL-1βシグナル伝達アッセイ)
OSM及びIL-1βは、ヒト軟骨肉腫株化細胞系SW1353において、MMP13レベルを相乗的に上方制御することが示された。該細胞を96ウェルプレートにおいて、37℃、5%CO2でインキュベートした10%(v/v)のFBS及び1%のペニシリン/ストレプトマイシン(InVitrogen社製)を含有するDMEM(Invitrogen社製)の容量120μL中、15,000細胞/ウェルで播種した。細胞を、25ng/mLのOSM及び1ng/mLのIL-1βに達するまで、15μLのOSM及びIL-1βでトリガーする1時間前に、2%のDMSOを有するM199培地中で15μLの化合物とともにプレインキュベートし、トリガーした48時間後に条件培地でMMP13レベルを測定した。抗体捕獲活性アッセイを利用してMMP13活性を測定した。この目的のために、384ウェルプレート(NUNC社製、460518、MaxiSorb black)を35μLの1.5μg/mL抗ヒトMMP13抗体(R&D Systems社製、MAB511)溶液で24時間4℃で被覆した。該ウェルをPBS+0.05%Tweenで2回洗浄した後、残りの結合部位を100μLのPBS中5%の脱脂粉乳(Santa Cruz社製、sc-2325、Blotto)で24時間4℃でブロックした。次に、該ウェルをPBS+0.05%Tweenで2回洗浄し、100倍希釈したブロッキング緩衝液中の、MMP13を含む培養上清の1/10希釈を35μL添加し、室温で4時間インキュベートした。次に、該ウェルをPBS+0.05% Tweenで2回洗浄し、続いて、35μLの1.5mM 4-アミノフェニル水銀アセタート(APMA)(Sigma社製、A9563)溶液を添加し、37℃で1時間インキュベートすることによってMMP13を活性化した。該ウェルをPBS+0.05%Tweenで再度洗浄し、35μLのMMP13基質(Biomol社製、P-126、OmniMMP蛍光発生基質)を添加した。37℃で24時間のインキュベーションの後、転化した基質の蛍光をPerkin Elmer社製Wallac EnVision2102マルチラベルリーダー(励起波長:320nm、発光波長:405nm)で測定した。
【0171】
阻害率=((ビヒクル存在下で測定された蛍光−被験化合物が存在する試料について測定された蛍光)/(ビヒクルの存在下で測定された蛍光−トリガーなしの試料について測定された蛍光))×100である。データを、アッセイで得た平均IC50±平均値の標準誤差として表す。
【0172】
【表9】
【0173】
(実施例2.3 PBL増殖アッセイ)
ヒト末梢血リンパ球(PBL)をIL-2で刺激し、BrdU取り込みアッセイを使用して増殖を測定する。まず、PHAを用いてPBLを72時間刺激してIL-2受容体を誘発し、24時間絶食させて細胞増殖を停止させ、続いて、さらに72時間(24時間のBrdU標識化を含む)IL-2刺激を行う。細胞を被験化合物とともにIL-2添加の1時間前にプレインキュベートする。10%(v/v)FBSを含有するRPMI1640で細胞を培養する。
【0174】
(実施例2.4 全血アッセイ(WBA))
(2.4.1 IFNα刺激プロトコール)
被験化合物がインビボでJAK2依存性シグナル伝達経路よりもJAK1シグナル伝達経路を阻害する選択性を予測するため、生理学的に関連あるインビトロモデルを、ヒトの全血を使用して開発した。WBAアッセイにおいて、インフォームドコンセントを与えたヒトの志願者から採取された血液を化合物によりエクスビボで処理し(1時間)、次いで、インターフェロンα(IFNα、JAK1依存性経路)により30分間、又は顆粒球マクロファージコロニー刺激因子(GM-CSF、JAK2依存性経路)により2時間のいずれかで刺激した。
【0175】
(2.4.1.1 ホスホ-STAT1アッセイ)
IFNα刺激に関して、白血球抽出物中でのIFNαによるシグナル伝達性転写因子1のリン酸化(pSTAT1)の増加をpSTAT1 ELISAアッセイを利用して測定した。インターフェロンα(IFNα)トリガリングの後のシグナル伝達性転写因子1(STAT1)のリン酸化はJAK1-媒介性事象である。ホスホ-STAT1アッセイは、細胞抽出物におけるホスホ-STAT1レベルの測定に利用したが、化合物がJAK1-依存性シグナル伝達経路を阻害する能力を評価するために開発された。
【0176】
インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血を、化合物によりエクスビボで処理し(1時間)、次いで、IFNαにより30分間刺激した。白血球抽出物におけるIFNαによるSTAT1のリン酸化の増加を、ホスホ-STAT1 ELISAを利用して測定した。
【0177】
ACK溶解緩衝液は、0.15 M NH4Cl、10 mM KHCO3、0.1 mM EDTAからなっていた。該緩衝液のpHは7.3であった。
【0178】
10×細胞溶解緩衝濃縮液(Cell Signaling社製、PathScan Phospho-STAT1 (Tyr701) sandwich ELISAキットの一部)を、水で10倍に希釈した。使用前にプロテイナーゼ阻害剤を緩衝液に加えた。
【0179】
20μgのIFNαを40μLのH2Oに溶かして、500μg/mLのストック溶液を得る。ストック溶液を-20℃で保存した。
【0180】
化合物の3倍の希釈系列をDMSOで調製した(最高濃度:10 mM)。その後、化合物をさらに培地で希釈した(希釈係数は、所望の最終化合物濃度による)。
【0181】
(2.4.1.1.1 化合物と血液とのインキュベーション及びIFNαによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を392μLのアリコートに分けた。その後、4μLの化合物希釈液を各アリコートに加え、血液試料を37℃で1時間インキュベートした。IFNαストック溶液をRPMI培地で1000倍に希釈し、500 ng/mLの使用液を得た。4μLの500 ng/mL使用液を血液試料に加えた(最終濃度IFNα:5ng/ml)。該試料を37℃で30分間インキュベートした。
【0182】
(2.4.1.1.2 細胞抽出物の調製)
刺激期間の最後に、7.6 mLのACK緩衝液を血液試料に加え、赤血球を溶解させた。チューブを5回反転させて試料を混合し、反応物を氷上で5分間インキュベートした。RBCの溶解は、このインキュベーションの間に明らかであるはずである。300g、4℃で7分間の遠心分離により細胞をペレットにし、上清を除いた。10 mLの1×PBSを各チューブに加え、細胞ペレットを再懸濁化した。試料を300g、4℃で7分間再び遠心分離した。上清を除き、ペレットを500μLの1×PBSで再懸濁化した。次いで、細胞懸濁液をきれいな1.5 mL微量遠心チューブに移した。700g、4℃で5分間での遠心分離により細胞をペレットにした。上清を除き、ペレットを150μLの細胞溶解緩衝液に溶かした。試料を氷上で15分間インキュベートした。その後、試料を、さらに処理するまで-80℃で保存した。
【0183】
(2.4.1.1.3 ELISAによるSTAT1リン酸化の測定)
Cell Signaling社製のPathscan Phospho-STAT1 (Tyr701) Sandwich ELISAキット(カタログ番号7234)を使用して、ホスホ-STAT1レベルを測定した。
【0184】
細胞抽出物を氷上で解凍した。チューブを16,000g、4℃で5分間遠心分離し、清澄化された溶解物を採取した。その間に、キットのマイクロウェルストリップを室温に平衡化し、20×洗浄緩衝液を水で希釈して洗浄緩衝液を調製した。試料を、試料希釈液で2倍に希釈し、100μLをマイクロウェルストリップに加えた。ストリップを4℃で一晩インキュベートした。
【0185】
翌日、ウェルを洗浄緩衝液により3回洗浄した。100μLの検出抗体をウェルに加えた。ストリップを37℃で1時間インキュベートした。次いで、ウェルを洗浄緩衝液で再び3回洗浄した。100μLのHRP結合二次抗体を各ウェルに加え、試料を37℃でインキュベートした。30分後、ウェルを再び3回洗浄し、100μLのTMB基質を全てのウェルに加えた。試料が青くなった場合、100μLのSTOP溶液を加えて反応を停止させた。吸光度を450 nmで測定した。
【0186】
(2.4.1.2 IL-8 ELISA)
GM-CSF刺激に関して、血漿中のインターロイキン-8(IL-8)レベルの増加をIL-8 ELISA アッセイを利用して測定した。顆粒球マクロファージコロニー刺激因子(GM-CSF)-誘導インターロイキン8(IL-8)発現は、JAK2-媒介性事象である。IL-8 ELISAは、血漿試料中のIL-8レベルの測定に使用できるが、化合物がJAK2-依存性シグナル伝達経路を阻害する能力を評価するために開発された。
【0187】
インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血を化合物によりエクスビボで処理し(1時間)、次いでGM-CSFで2時間刺激した。血漿中のIL-8レベルの増加を、IL-8 ELISAアッセイを利用して測定した。
【0188】
10μgのGM-CSFを100μLのH2Oに溶かし、100μg/mLのストック溶液を得た。ストック溶液を-20℃で保存した。
【0189】
被験化合物の3倍の希釈系列をDMSO中に調製した(最高濃度:10 mM)。次いで、化合物を培地でさらに希釈した(希釈率は、所望の最終化合物濃度による)。
【0190】
(2.4.1.2.1 血液の化合物とのインキュベーション及びGM-CSFによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を245μLのアリコートに分けた。その後、2.5μLの被験化合物希釈液を各アリコートに加え、血液試料を37℃で1時間インキュベートした。GM-CSFストック溶液をRPMI培地で100倍に希釈し、1μg/mLの使用液を得た。2.5μLの1μg/mL使用液を血液試料に加えた(最終濃度GM-CSF:10 ng/ml)。該試料を37℃で2時間インキュベートした。
【0191】
(2.4.1.2.2 血漿試料の調製)
試料を1,000g、4℃で15分間遠心分離した。100μLの血漿を採取し、さらに使用するまで-80℃で保存した。
【0192】
(2.4.1.2.3 ELISAによるIL-8レベルの測定)
R&D Systems社製のHuman IL-8 Chemiluminescent Immunoassayキット(カタログ番号:Q8000B)を使用してIL-8レベルを測定した。
【0193】
洗浄緩衝液は、10×洗浄緩衝液をH2Oに希釈して調製した。使用グロ試薬は、1部のGlo Reagent 1を2部のGlo Reagent Bに、使用の15分から4時間前に添加して調製した。
【0194】
100μLのアッセイ希釈液RD1-86を各ウェルに加えた。その後、50μLの試料(血漿)を加えた。ELISAプレートを室温で2時間500rpmでインキュベートした。全てのウェルを洗浄緩衝液で4回洗浄し、200μLのIL-8コンジュゲートを各ウェルに加えた。室温で3時間インキュベートした後、ウェルを洗浄緩衝液で4回洗浄し、100μLの使用グロ試薬を各ウェルに加えた。ELISAプレートを室温で5分間インキュベートした(遮光する)。発光を測定した(0.5秒/ウェル読み取り時間)。
【0195】
(2.4.1.3 結果)
INFαに誘導されるpSTAT1レベル増加を阻害する化合物1のpIC50は、6.32±0.07(SEM)であり、一方GM-CSFに誘導されたIL-8の増加の阻害に対するpIC50は、4.87±0.13(SEM)であった(6人の血液提供者を利用して得られた結果)。これは、化合物1が、JAK2経路よりもJAK1経路に対して28倍選択性があることを示す。したがって、細胞環境では、化合物1がJAK2よりもJAK1に対して選択性を示すことは明らかである。化合物1は、構造的に関連する公知の化合物又は他の公知のJAK阻害剤に比べて、JAK2よりもJAK1に対して高い選択性を示すようである。
【0196】
(2.4.1.3 データキュレーション処理及び結果)
結果をさらに精密にするため、細胞抽出物中のIFNαによるホスホSTAT1誘導の阻害又は血漿中のGM-CSFによるIL-8誘導の阻害を、化合物濃度に対してプロットし、Graphpadソフトウェアを利用してIC50値を誘導した。R2が0.8より大きく、ヒルスロープが3より小さい場合にデータを保持し、両アッセイに対して有効なデータを得られた提供者のみを保持した。
【0197】
例えば、この追加のデータ分析プロトコールに供された場合、INFα誘導性のpSTAT1レベルの増加の阻害に関する化合物1のpIC50測定値は6.21±0.09 (SEM)であり、一方GM-CSF誘導性のIL-8の増加の阻害に関するpIC50は4.97±0.14 (SEM)であった(6人の血液提供者を利用して得られた結果)。このことは、化合物1が、JAK2経路に対してJAK1経路に対する選択性が17.6倍であることを立証している。したがって、細胞環境中で、化合物1がJAK2よりもJAK1に対して選択性を示すことは明らかである。化合物1は、構造的に関連する公知の化合物又は他の公知のJAK阻害剤に比べて、JAK2よりもJAK1に対して高い選択性を示すようである。
【0198】
(2.4.2 IL-6刺激プロトコール)
フローサイトメトリー分析も実施して、ヒトの全血を利用してエクスビボで、JAK2を上回るJAK1への化合物選択性を確立した。したがって、インフォームドコンセントを与えたヒトの志願者から血液を採取した。次いで、血液を穏やかに揺らしながら37℃で30分間平衡化し、エッペンドルフチューブに小分けした。化合物を異なる濃度で加え、穏やかに揺らしながら37℃で30分間インキュベートし、次いで、穏やかに揺らしながら20分間37℃で、JAK1依存性経路刺激にはインターロイキン6 (IL-6)により、又はJAK2依存性経路刺激にはGM-CSFにより刺激した。次いで、FACS分析を利用して、ホスホ-STAT1及びホスホ-STAT5を評価した。
【0199】
(2.4.2.1 ホスホ-STAT1アッセイ)
白血球におけるシグナル伝達性転写因子1(pSTAT1)リン酸化のIL-6刺激性の増加には、インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血をエクスビボで化合物により30分間処理し、その後IL-6により20分間刺激した。リンパ球におけるIL-6によるSTAT1のリン酸化の増加を、抗ホスホ-STAT1抗体を利用して、FACSにより測定した。
【0200】
5×Lyse/Fix緩衝液(BD PhosFlow社製、カタログ番号558049)を蒸留水で5倍に希釈し、37℃に予熱した。残りの希釈したLyse/Fix緩衝液を廃棄した。
【0201】
10μgのrhIL-6(R&D Systems社製、カタログ番号206-IL)を、1mlの、PBS 0.1% BSAに溶かし、10μg/mlのストック溶液を得た。ストック溶液を小分けした状態で-80℃で保存した。
【0202】
化合物の3倍の希釈系列をDMSO中に調製した(10 mMストック溶液)。コントロール処理試料には化合物の代わりにDMSOを与えた。試料は全て1%の最終DMSO濃度でインキュベートした。
【0203】
(2.4.2.1.1 血液の化合物とのインキュベーション及びIL-6による刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を148.5μLのアリコートに分けた。その後、1.5μLの化合物希釈液を各アリコートに加え、血液試料を穏やかに揺らしながら37℃で30分間インキュベートした。IL-6ストック溶液(1.5μl)を血液試料に加え(最終濃度10ng/ml)、該試料を穏やかに揺らしながら37℃で20分間インキュベートした。
【0204】
(2.4.2.1.2 白血球調製及びCD4標識付け)
刺激期間の最後に、赤血球を溶解し白血球を固定するために、3mlの1×予熱されたLyse/Fix緩衝液を直ちに血液試料に加え、手短にボルテックスにかけ、水浴中で37℃で15分間インキュベートし、次いでさらに使用するまで-80℃で凍結した。
【0205】
以下の工程のために、チューブを37℃でおよそ20分間解凍し、400gで4℃で5分間遠心分離した。細胞ペレットを3mlの冷1×PBSで洗浄し、遠心分離後、細胞ペレットを100μlの3%BSA含有PBSに再懸濁化した。FITC-結合抗-CD4抗体又はコントロールのFITC-結合アイソタイプ抗体を加え、暗所にて室温で20分間インキュベートした。
【0206】
(2.4.2.1.3 細胞透過処理及び抗ホスホ-STAT1抗体による標識付け)
細胞を1×PBSにより洗浄した後、細胞ペレットを100μlの氷冷1×PBSに再懸濁化し、900μlの氷冷100%メタノールを加えた。次いで、細胞を、透過処理のために4℃で30分間インキュベートした。
【0207】
次いで、透過処理された細胞を1×3%BSA含有PBSにより洗浄し、最後に、80μlの1×3%BSA含有PBXに再懸濁化した。
【0208】
20μLのPEマウス抗-STAT1(pY701)又はPEマウスIgG2aκアイソタイプコントロール抗体(BD Biosciences社製、それぞれカタログ番号612564及び559319)を加え、混合し、次いで、暗所にて4℃で30分間インキュベートした。
【0209】
次いで、細胞を1×PBSで1回洗浄し、FACSCanto IIフローサイトメーター(BD Biosciences社製)で分析した。
【0210】
(2.4.2.1.4 FACSCanto IIによる蛍光分析)
50,000の総事象を計数し、ホスホ-STAT1陽性細胞を、リンパ球ゲートにおいてCD4+細胞のゲーティングの後測定した。データはFACSDivaソフトウェアを利用して分析され、CD4+細胞上のホスホ-STAT1に陽性な細胞のパーセンテージに対して計算されたIL-6刺激の阻害率に相当する。
【0211】
(2.4.2.2 ホスホ-STAT5アッセイ)
白血球におけるシグナル伝達性転写因子5(pSTAT5)リン酸化のGM-CSF-刺激性の増加には、インフォームドコンセントを与えたヒトの志願者から採取されたヒトの全血をエクスビボで化合物により30分間処理し、次いで20分間GM-CSFにより刺激した。単球におけるGM-CSFによるSTAT5のリン酸化の増加を、抗ホスホ-STAT5抗体を利用してFACSにより測定した。
【0212】
5×Lyse/Fix緩衝液(BD PhosFlow社製、カタログ番号558049)を蒸留水で5倍に希釈し、37℃に予熱した。残りの希釈したLyse/Fix緩衝液を廃棄した。
【0213】
10μgのrhGM-CSF(AbCys S.A.社製、カタログ番号P300-03)を100μlのPBS 0.1% BSAに溶かし、100μg/mlのストック溶液を得た。ストック溶液を小分けした状態で-80℃で保存した。
【0214】
化合物の3倍希釈系列をDMSO中に調製した(10 mMストック溶液)。コントロール処理試料には、全く被験化合物なしでDMSOを与えた。試料を全て、1%の最終DMSO濃度でインキュベートした。
【0215】
(2.4.2.2.1 血液の化合物とのインキュベーション及びGM-CSFによる刺激)
ヒトの血液をヘパリン処理したチューブに集めた。血液を148.5μlのアリコートに分けた。その後、1.5μlの化合物希釈液を各アリコートに加え、血液試料を穏やかに揺らしながら37℃で30分間インキュベートした。GM-CSFストック溶液(1.5μl)を血液試料に加え(最終濃度20pg/ml)、試料を穏やかに揺らしながら37℃で20分間インキュベートした。
【0216】
(2.4.2.2.2 白血球調製及びCD14標識付け)
刺激期間の最後に、赤血球を溶解し白血球を固定するために、3mlの1×予熱されたLyse/Fix緩衝液を直ちに血液試料に加え、手短にボルテックスにかけ、水浴中で37℃で15分間インキュベートし、次いで、さらに使用するまで-80℃で凍結した。
【0217】
以下の工程のために、チューブを37℃でおよそ20分間解凍し、400gで4℃で5分間遠心分離した。細胞ペレットを3mlの冷1×PBSで洗浄し、遠心分離後、細胞ペレットを100μlの3%BSA含有PBSに再懸濁化した。FITCマウス抗-CD14抗体(BD Biosciences社製、カタログ番号345784)又はコントロールFITCマウスIgG2bκアイソタイプ抗体(BD Biosciences社製、カタログ番号555057)を加え、暗所にて室温で20分間インキュベートした。
【0218】
(2.4.2.2.3 細胞透過処理及び抗ホスホ-STAT5抗体による標識付け)
細胞を1×PBSで洗浄した後、細胞ペレットを100μlの氷冷1×PBSに再懸濁化し、900μlの氷冷100%メタノールを加えた。次いで、細胞を、透過処理のために、4℃で30分間インキュベートした。
【0219】
次いで、透過処理された細胞を1×3%BSA含有PBSで洗浄し、最後に80μlの1×3%BSA含有PBSに再懸濁化した。
【0220】
20μLのPEマウス抗-STAT5(pY694)又はPEマウスIgG1κアイソタイプコントロール抗体(BD Biosciences社製、それぞれカタログ番号612567及び554680)を加え、混合し、次いで暗所にて4℃で30分間インキュベートした。
【0221】
次いで、細胞を1×PBSで1回洗浄し、FACSCanto IIフローサイトメーター(BD Biosciences社製)で分析する。
【0222】
(2.4.2.2.4 FACSCanto IIによる蛍光分析)
50,000の総事象を計数し、ホスホ-STAT5陽性細胞を、CD14+細胞のゲーティングの後測定した。データはFACSDivaソフトウェアを利用して分析され、CD14+細胞上のホスホ-STAT5に陽性な細胞のパーセンテージに対して計算されたGM-CSF刺激の阻害率に相当する。
【0223】
(2.4.2.3 結果)
このプロトコールに供されたとき、3名の健常な志願者の平均から得られた阻害率(PIN)を、本発明の化合物に対して決定した。例えば、化合物1を試験し、STAT1リン酸化の阻害においてpIC50=6.61を、STAT5リン酸化の阻害においてpIC50=5.37を生み出した。
【0224】
STAT1(JAK1-依存性経路)及びSTAT5(JAK2-依存性経路)に対する化合物1の効果を比較すると、JAK2よりJAK1に対して17.6倍の選択性が測定された。
【0225】
(実施例3 インビボモデル)
(実施例3.1 CIAモデル)
(3.1.1 材料)
完全フロイントアジュバント(CFA)及び不完全フロイントアジュバント(IFA)をDifco社から購入した。ウシII型コラーゲン(CII)、リポ多糖(LPS)及びエンブレルをそれぞれ、Chondrex社(Isle d'Abeau、フランス)、Sigma社(P4252、L'Isle d'Abeau、フランス)、Whyett(25mg注射可能シリンジ、フランス)、Acros Organics社(カリフォルニア州パロアルト)から入手した。使用した他の全ての試薬は試薬等級のものであり、全ての溶媒は分析用のものであった。
【0226】
(3.1.2 動物)
Dark Agoutiラット(雄、7〜8週齢)をHarlan Laboratories社(Maison-Alfort、フランス)から入手する。DBA/1Jマウス(雄、7週齢)を、Centre d’Elevage et de Reproduction JANVIER (CERJ) (Laval, フランス)から得た。ラット及びマウスを12時間の明暗サイクル(0700-1900)においた。温度を22℃に維持し、餌及び水を適宜与えた。
【0227】
(3.1.3 コラーゲン誘発関節炎(CIA))
実験の前日に、CII溶液(2mg/mL)を0.05Mの酢酸を用いて調製し、4℃で保管した。免疫化の直前に、等量のアジュバント(IFA)及びCIIを氷水浴において、予冷却したガラス瓶中でホモジナイザーにより混合した。乳濁液が形成されない場合は、さらなるアジュバント及びホモジナイズの延長が必要となる場合がある。
【0228】
マウス:1日目に0.1mLの乳濁液を各マウスの尾の根元に皮内注射し、21日目に第2の追加免疫の皮内注射(CFA 0.1mLの生理食塩水中1mg/mLのCII溶液)を行った。この免疫化法は既報の方法(David D Brand Kary A Latham及びEdward F Rosloniecの文献、「コラーゲン誘発関節炎(Collagen-induced arthritis)」Nature Methods 2 (5): 1269-1275, 2007)を変更したものであった。
【0229】
ラット:1日目に0.2mLの乳濁液を各ラットの尾の根元に皮内注射し、9日目に第2の追加免疫の皮内注射(CFA 0.1mLの生理食塩水中2mg/mLのCII溶液)を行った。この免疫化法は既報の方法(Sims NAらの文献、(2004)「ゾレドロン酸による破骨細胞の標的化はコラーゲン誘導性関節炎における骨破壊を予防する(Targeting osteoclasts with zoledronic acid prevents bone destruction in collagen-induced arthritis)」 Arthritis Rheum. 50 2338-2346;Jouらの文献、2005)を変更したものである。
【0230】
(3.1.4 研究設計)
被験化合物の治療効果をラット又はマウスのCIAモデルで試験した。動物を無作為に、均等な群に分け、各群は10匹の動物を含んだ。全てのラットを1日目に免疫化し、9日目に追加免疫する。全てのマウスを1日目に免疫化し、21日目に追加免疫した。治療量を16日目〜30日目まで与え続けた。ネガティブコントロール群をビヒクル(MC 0.5%)で処理し、ポジティブコントロール群をエンブレル(10mg/kg、3×週、皮下注射)で処理した。対象とする化合物を通常3用量、例えば、3、10、30mg/kg、経口で試験した。
【0231】
(3.1.5 関節炎の臨床評価)
関節炎をKhachigianの文献2006、Linらの文献2007及びNishidaらの文献2004の方法に従い得点付けした。4足それぞれの腫脹を以下のように関節炎スコアによってランク付けした:0−症状なし;1−軽度だが、足首又は手首などの1種の関節の明確な紅化及び腫脹、又は罹患した指の数にかかわらず個々の指に限られた明らかな紅化及び腫脹;2−2種以上の関節の中等度の紅化及び腫脹;3−指を含む足全体の重度の紅化及び腫脹;4−多数の関節が関与する極度に炎症を起こした肢(動物あたりの最大の累積臨床関節炎スコアは16)(Nishidaらの文献2004)。
【0232】
複数の研究のメタ分析を可能にするため、臨床スコア値を以下のとおり規格化した。
【0233】
臨床スコアのAUC(AUCスコア):個々のラットに対して、1日から14日の曲線下面積(AUC)を計算した。各動物のAUCを、その動物のデータが得られた研究におけるビヒクルに対して得られた平均AUCで割り、100をかけた(すなわち、AUCを、研究あたりの平均ビヒクルAUCのパーセンテージとして表した)。
【0234】
1日から14日までの臨床スコアの増加(エンドポイントスコア):各動物の臨床スコアの差を、その動物のデータが得られた研究におけるビヒクルに対して得られた平均臨床スコアの差で割り、100をかけた(すなわち、差を、研究あたりビヒクルの平均臨床スコアの差のパーセンテージとして表した)。
【0235】
(3.1.6 関節炎発症後の体重の変化(%))
臨床的に、体重減少は関節炎と関連している(Sheltonらの文献、2005;Argilesらの文献、1998;Rallの文献、2004;Walsmithらの文献2004)。したがって、関節炎発症後の体重の変化は、ラットモデルにおける治療術の効果を評価する非特異的なエンドポイントとして利用できた。関節炎発症後の体重の変化(%)を以下のように計算した。
【数1】
【0236】
(3.1.7 ラーセンスコア)
死後に、少なくとも2人の科学者により全ラットの両後足に対してラーセンスコアを与えた。これらのスコアの平均を計算して、ラットあたりの1つのラーセンスコアを得た。ラーセンスコアの研究間の比較を可能にするため、各ラットのラーセンスコアを、そのラットが属する研究におけるビヒクルで得られた平均ラーセンスコアにより割り、100をかけた(すなわち、ラーセンスコアを、研究あたりの平均ビヒクルラーセンスコアのパーセンテージとして表した)。異なる治療群の平均ラーセンスコアを計算して比較した。
【0237】
(3.1.8 放射線学)
個々の動物の後足のX線写真を撮った。無作為のブラインド・アイデンティティ番号を、写真のそれぞれに割り当て、以下の放射線学的ラーセンのスコアシステムを用いて2人の独立したスコアラーにより骨びらんの重症度をランク付けした:0−無傷の骨の外郭及び正常な関節腔を有する正常状態;1−1又は2の外側中足骨に軽微な骨びらんが見られる軽度の異常;2−3から5の外側中足骨に骨びらんがみられる明確な初期の異常;3−全ての外側中足骨並びに1又は2の内側中足骨に明らかな骨びらんが見られる中等度破壊性異常;4−全ての中足骨に明らかな骨びらんが見られ、かつ少なくとも1の内側中足骨関節が完全に侵食され、骨の関節外郭のいくらかが部分的に残されている重度破壊性異常;5−骨の外郭を有さないムチランス型異常。このスコア付けシステムは、Salveminiらの文献、2001;Bushらの文献、2002;Simsらの文献、2004;Jouらの文献、2005の変形である。
【0238】
(3.1.9 組織学)
放射線学的分析の後、マウスの後足を10%リン酸-緩衝ホルマリン(pH7.4)に固定し、微細組織診のための急速骨脱灰剤(Laboratories Eurobio社製)によって脱灰しパラフィンに包埋した。関節炎の関節の詳細な評価を確実に行うために、少なくとも4つの連続切片(5μm厚)を切り出し、各系列の切片の間を100μmとした。該切片をヘマトキシリン及びエオシン(H&E)で染色した。滑膜の炎症、並びに骨及び軟骨損傷の組織学的実験を二重盲検で行った。各足において、4点スケールを用いて4つのパラメーターを評価した。該パラメーターは、細胞浸潤、パンヌス重症度、軟骨侵食、及び骨びらんとした。以下のようにスコア付けを行った:1−正常、2−軽度、3−中等度、4−顕著。これらの4つのスコアを総合し、さらなるスコア、つまり「RA総計スコア」として表した。組織学的読み取りの研究間比較を可能にするために、各ラットの全体的な組織学的スコアを、そのラットが属する研究におけるビヒクルで得られた平均の全体的な組織学的スコアにより割り、100をかけた(すなわち、全体的な組織学的スコアを、研究あたりの平均ビヒクル全体的組織学的スコアのパーセンテージとして表した)。異なる治療群の平均の全体的な組織学的スコアを計算して比較した。
【0239】
(3.1.10 踵骨(踵の骨)のマイクロ-コンピュータ断層撮影法(μCT)分析)
RAに見られる骨破壊は特に皮質骨で生じ、μCT分析により明らかとなり得る(Sims NAらの文献、(2004);Oste Lらの文献、ECTC Montreal 2007)。踵骨の走査及び3Dボリューム再構築後に、骨破壊を、インシリコで骨の縦軸に対して垂直に単離されたスライドあたりに存在する分離した対象の数として測定した。破壊されている骨が多いほど、測定される個別の対象が多い。踵骨に沿って均一に分布した1000の切片(約10.8μmの間隔)を分析する。
【0240】
(3.1.11 結果)
化合物1を、30 mg/kgでマウスCIA試験で、30、10、3、及び1 mg/kgでラットCIA試験で試験した。化合物1は、ラットCIA試験において実施された全ての読み取りにおいて有効であり、いくつかの読み取りにおいて統計的に有意であり、特に下記では有意な向上が見られた:臨床スコアのAUC(10mg/kgから)、エンドポイント臨床スコア(1 mg/kgから)、ラーセンスコア(30 mg/kgから)、及び足の腫脹(1 mg/kgから)。
【0241】
(実施例3.2 敗血症ショックモデル)
リポ多糖(LPS)の注射は、可溶性腫瘍壊死因子(TNF-α)の末梢への急速な放出を誘導する。このモデルを使用して、インビボでのTNF放出の有望な遮断薬を分析する。
【0242】
1群あたり6匹の雌のBALB/cJマウス(20g)を、一度の意図された投与、経口投与で処理する。30分後、LPS(15μg/kg;大腸菌血清型0111:B4)を腹腔内に注射する。90分後、マウスを安楽死させ、血液を回収する。循環しているTNFαのレベルを、市販のELISAキットを使用して測定する。デキサメタゾン(5μg/kg)を参照抗炎症化合物として使用する。選択された化合物を1回投与又は複数回投与、例えば、3及び/又は10及び/又は30mg/kg、経口投与で試験する。
【0243】
化合物1は、経口投与30 mg/kgでTNF放出の統計的に有意な減少(>50%)を示した。
【0244】
(実施例3.3 MABモデル)
MABモデルは、治療法によるRA様炎症応答の調節の迅速な評価を可能にする(Kachigian LMの文献、Nature Protocols (2006) 2512-2516:「コラーゲン抗体誘発性関節炎(Collagen antibody-induced arthritis)」)。DBA/Jマウスに、II型コラーゲンに対するmAbカクテルを静脈内注射する。1日後、化合物処理を開始する(ビヒクル:10% (v/v) HPβCD)。3日後、マウスに腹腔内LPS注射(50μg/マウス)を施し、速やかな炎症の発症を起こさせる。該mAb注射の10日後まで、化合物処理を続ける。足の腫脹の測定及び各足の臨床スコアの記録によって、炎症を読み取る。四肢の累積的臨床関節炎スコアを提示して炎症の重症度を示す。4が最も重症の炎症である0〜4のスケールを使用して、スコア付けシステムを各肢に適用する。
0 症状なし
1 軽度だが、足首又は手首などの1種の関節の明確な紅化及び腫脹、又は罹患した指の数にかかわらず個々の指に限られた明らかな紅化及び腫脹
2 2種以上の関節の中等度の紅化及び腫脹
3 指を含む足全体の重度の紅化及び腫脹
4 多数の関節が関与する極度に炎症を起こした肢。
【0245】
(実施例3.4 オンコロジーモデル)
JAK2駆動性の骨髄増殖性疾患に対する小分子の有効性を検証するためのインビボモデルは、Wernigらの文献、Cancer Cell 13, 311, 2008、及びGeronらの文献、Cancer Cell 13, 321, 2008に記載されている。
【0246】
(実施例3.5 マウスIBDモデル)
IBDに対する小分子の有効性を検証するためのインビトロ及びインビボのモデルは、Wirtzらの文献、2007に記載されている。
【0247】
(実施例3.6 マウス喘息モデル)
喘息に対する小分子の有効性を検証するためのインビトロ及びインビボのモデルは、Nialsらの文献、2008;Ipらの文献、2006;Pernisらの文献、2002;Kudlaczらの文献、2008に記載されている。
【0248】
(実施例4:毒性、DMPK、及び安全性モデル)
(実施例4.1 熱力学的溶解性)
1mg/mLの被験化合物の溶液を、ガラス小瓶において室温で、0.2Mのリン酸緩衝液pH7.4又は0.1Mのクエン酸緩衝液pH3.0に調製する。
【0249】
該試料をローテータードライブSTR4(Stuart Scientific社製、Bibby社製)において、速度3.0、室温で24時間回転させる。
【0250】
24時間後、800μLの試料をエッペンドルフチューブに移し、14000rpmで5分間遠心分離する。次いで、200μLの試料の上清をMultiscreen(登録商標)溶解度用プレート(Millipore社製、MSSLBPC50)に移し、該上清を真空マニホールドを用いて、清潔なGreiner社製ポリプロピレンV字底96ウェルプレート(カタログ番号651201)に濾過する(25.4-30.5cmHg)。5μLの濾液を、検量線を有するプレート(Greiner社製、カタログ番号651201)で、インキュベートに使用するのと同じ緩衝液95μL(20倍)に希釈する。
【0251】
該化合物の検量線をDMSO中で新たに作成する。10mMのDMSOストック溶液から開始して、DMSOに2倍に希釈し(5000μM)、次いで、19.5μMまでDMSOでさらに希釈する。次いで、5000μMからの希釈系列3μLを、97μlのアセトニトリル-緩衝液混合物(50/50)に移す。最終濃度の範囲は2.5〜150μMである。
【0252】
該プレートをシーリングマット(MA96RD-04S、www.kinesis.co.uk)で密閉し、分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定する。
【0253】
該試料を、LCMSにおいて1ml/分の流量で分析する。溶媒Aは15mMのアンモニアであり、溶媒Bはアセトニトリルである。該試料を、Waters社製のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作する。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲である。
【0254】
ピーク面積をMasslynxソフトウェアパッケージを用いて分析し、試料のピーク面積を検量線に対してプロットして、該化合物の溶解度を得る。
【0255】
溶解度値をμM又はμg/mLで報告する。
【0256】
(実施例4.2 水溶性)
10mMのDMSO中のストック溶液から開始して、化合物の段階希釈をDMSOで調製する。該希釈系列を、96NUNC Maxisorbプレート平底(カタログ番号442404)に移し、室温の0.2Mのリン酸緩衝液pH7.4又は0.1Mのクエン酸緩衝液pH3.0を添加する。
【0257】
最終濃度は、5回の同じ希釈工程で、200μM〜2.5μMの範囲にある。最終DMSO濃度は2%を超えなかった。200μMのピレンを各96ウェルプレートの角点に添加し、顕微鏡のZ軸の補正のための参照点とする。
【0258】
該アッセイプレートを密閉し、230rpmで振盪しながら37℃で1時間インキュベートする。次いで、該プレートを白色光顕微鏡下で走査し、濃度あたりの沈殿の個々の写真を得る。該沈殿を分析し、数値に変換して、これをグラフ上にプロットする。該化合物が完全に溶解したように見える最初の濃度は報告する濃度であるが、しかし、本当の濃度は、この濃度と1つ上の希釈段階との間のどこかにある。
【0259】
溶解度値をμg/mLで報告する。
【0260】
(実施例4.3 血漿タンパク質結合(平衡透析))
DMSO中の化合物の10mMストック溶液を、DMSOで5倍に希釈する。この溶液を、新たに解凍したヒト、ラット、マウス又はイヌの血漿(BioReclamation社製)で、最終濃度10μM及びDMSO最終濃度0.5%にさらに希釈する(PP-Masterblock 96ウェル(Greiner社製、カタログ番号780285)において1094.5μlの血漿中5.5μl)。
【0261】
インサートを有するPierce Red Deviceプレート(ThermoScientific社製、カタログ番号89809)を準備し、緩衝液チャンバーを750μLのPBSで満たし、かつ血漿チャンバーを500μLの添加血漿(spiked plasma)で満たす。該プレートを230rpmで振盪しながら、37℃で4時間インキュベートする。インキュベーション後、両チャンバーの120μLを、96ウェル丸底、PPディープウェルプレート(Nunc社製、カタログ番号278743)の360μLのアセトニトリルに移し、アルミ箔の蓋で密閉する。該試料を混合し、30分間氷上に置く。次いで、このプレートを1200rcf、4℃で30分遠心分離し、該上澄みをLCMSでの分析用に96V字底PPプレート(Greiner社製、651201)に移す。
【0262】
該プレートを、www.kinesis.co.ukのシーリングマット(MA96RD-04S)で密閉し、分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定する。
【0263】
該試料を、LCMSで1mL/分の流量で分析する。溶媒Aは15mMのアンモニアであり、溶媒Bはアセトニトリルである。該試料を、Waters社製のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作した。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲である。
【0264】
緩衝液チャンバー及び血漿チャンバー中の化合物のピーク面積は、100%の化合物であると考えられる。血漿への結合率を、これらの結果から導き、血漿への結合率としてLIMSに報告した。
【0265】
PBS中の最終試験濃度の化合物の溶解度を顕微鏡で調査して、沈殿が見られるか否かを示した。
【0266】
(実施例4.4 QT延長の傾向)
QT延長の可能性は、hERGパッチクランプアッセイにおいて評価する。
【0267】
(4.4.1 従来の全細胞パッチクランプ)
全細胞パッチクランプ記録は、Pulse v8.77ソフトウェア(HEKA社)により制御されたEPC10増幅器を利用して実施する。直列抵抗は典型的には10 MΩ未満であり、60%超補償されており、記録はリーク減算されない。電極は、GC150TFピペットガラス(Harvard社)から製造されている。
【0268】
外部浴溶液は、135 mM NaCl、5 mM KCl、1.8 mM CaCl2、5 mMグルコース、10 mM HEPES, pH 7.4を含む。
【0269】
内部パッチピペット溶液は、100mMグルコン酸カリウム、20 mM KCl、1mM CaCl2、1 mM MgCl2、5mM Na2ATP、2mMグルタチオン、11 mM EGTA、10 mM HEPES, pH 7.2を含む。
【0270】
Biologic MEV-9/EVH-9迅速潅流システムを利用して、薬物を潅流させる。
【0271】
記録は全てhERGチャネルを安定に発現しているHEK293細胞で行う。細胞を、2本の白金ロッド(Goodfellow社製)を用い、記録チャンバーに固定された12 mmの円形カバースリップ(German glass、Bellco社)上に培養する。hERG電流を、活性化パルスを用いて+40 mVに1000ミリ秒間惹起し、次いでテール電流パルスにより2000ミリ秒間-50 mVにし、保持電位は-80 mVであった。パルスを20秒ごとに印加し、実験は全て室温で行う。
【0272】
(4.4.2 データ分析)
IC50及びIC20値を、試験した各化合物について計算する。ラットCIAモデルから得られた結果により決定される、関連する治療投与量で得られた被験化合物のIC20と非結合のCmax濃度とのフォールド差(fold difference)を計算する。
【0273】
濃度反応曲線では、ピークテール電流振幅を、-50 mVへの電圧ステップの間に測定する。濃度反応データのカーブフィッティングを、以下の式を利用して実施する。
y = a + [( b - a)/ ( 1 + 10^ ( ( logc-x ) d )]
【0274】
式中、aは最小反応であり、bは最大反応であり、dはヒルスロープであり、この式は、IC50(式中、y = 50かつcがIC50値)とIC20(式中、y = 20かつcがIC20値)の両方の計算に利用できる。GraphPad(登録商標)Prism(登録商標)(Graphpad(登録商標)Software社製)ソフトウェアを、全てのカーブフィッティングに利用した。100倍以上の差は、QT延長の低い可能性を示す。
【0275】
(実施例4.5 ミクロソーム安定性)
DMSO中の化合物の10mMストック溶液を、96ディープウェルプレート(Greiner社製、カタログ番号780285)において182mMのリン酸緩衝液pH7.4で1000倍に希釈し、37℃でプレインキュベートした。
【0276】
40μLの脱イオン水をポリプロピレン製マトリックス2Dバーコード標識化した保存チューブ(Thermo Scientific社製)のウェルに添加し、37℃でプレインキュベートした。
【0277】
グルコース-6-リン酸脱水素酵素(G6PDH)作業溶液を182mMのリン酸緩衝液pH7.4で調製し、使用前に氷上に置いた。MgCl2、グルコース-6-リン酸及びNADP+を含む補因子を脱イオン水で調製し、使用前に氷上に置いた。
【0278】
対象とする種(ヒト、マウス、ラット、イヌ)の肝ミクロソーム(Xenotech社)、前述のG6PDH、及び補因子を含む最終作業溶液を調製し、この混合物を室温で20分以内でインキュベートした。
【0279】
30μLの予熱した化合物希釈液を、マトリックスチューブ中の予熱した水40μLに添加し、30μLのミクロソーム混合物を添加した。最終反応濃度は、化合物3μM、ミクロソーム1mg、GDPDH 0.4U/mL、MgCl2 3.3mM、グルコース-6-リン酸3.3mM及びNADP+ 1.3mMであった。
【0280】
ゼロ時間での化合物の残存率を測定するために、ミクロソーム混合物の添加前に、MeOH又はACNを該ウェルに添加した(1:1)。該プレートをMatrix Sepra seals(商標)(Matrix社、カタログ番号4464)で密閉し、数秒間振盪して、確実に全ての成分の混合が行われるようにした。
【0281】
停止ししなかった試料を、37℃、300rpmでインキュベートし、1時間のインキュベート後、該反応をMeOH又はACNで停止させた(1:1)。
【0282】
該反応の停止後、試料を混合し、氷上に30分間置いてタンパク質を沈殿させた。次いで、該プレートを1200rcf、4℃で30分間遠心分離し、LCMSでの分析用に上澄みを96V字底PPプレート(Greiner社、651201)に移した。
【0283】
これらのプレートを、www.kinesis.co.ukのシーリングマット(MA96RD-04S)で密閉し、親分子の適切な質量を決定するためにQuanoptimizeを使用して最適条件下で、室温でLCMS(Waters社製、ZQ1525)にて試料を測定した。
【0284】
該試料を、LCMSにおいて1ml/分の流量で分析した。溶媒Aは15mMのアンモニアであり、溶媒Bは、使用した停止溶液に応じてメタノール又はアセトニトリルであった。該試料を、Waters社のXBridge C18 3.5μM(2.1×30mm)カラムを用いてポジティブイオンスプレー下で操作した。溶媒勾配は、総実行時間2分であり、5% B〜95% Bの範囲であった。
【0285】
0時間での親化合物のピーク面積を100%の残存とした。1時間のインキュベーション後の残存率を0時間から算出し、残存率として算出した。緩衝液中の最終試験濃度の化合物の溶解度を顕微鏡で調査し、結果を報告する。
【0286】
ミクロソーム安定性のデータを、60分後に残存している化合物の総量の割合として表す。
【0287】
【表10】
【0288】
(実施例4.6 Caco2透過性)
2方向Caco-2アッセイを下記のように行った。Caco-2細胞を欧州細胞カルチャーコレクション(ECACC、カタログ86010202)から入手し、24ウェルトランスウェルプレート(Fisher社、TKT-545-020B)での21日間の細胞培養後に使用した。
【0289】
DMEM+GlutaMAXI+1%のNEAA+10%のFBS(FetalClone II)+1%のPen/Strepからなる播種培地に2×105細胞/ウェルを播種した。培地は2〜3日毎に交換した。
【0290】
被験化合物及び参照化合物(プロプラノロール及びローダミン123又はビンブラスチン、全てSigma社から購入した)を、25mMのHEPES(pH7.4)を含有するハンクス平衡塩類溶液で調製し、0.25%の最終DMSO濃度を有する10μMの濃度で、トランスウェルプレートアセンブリの頂端側チャンバー(125μL)又は基底側チャンバー(600μL)のいずれかに加えた。
【0291】
50μMのルシファーイエロー(Sigma社製)を全ウェルのドナー緩衝液に添加し、ルシファーイエローの透過を監視することによって細胞層の一体性を評価した。ルシファーイエロー(LY)は親油性バリアを自由に透過することができないので、LY輸送の程度が高いと該細胞層の一体性が乏しいことを示唆する。
【0292】
オービタルシェーカーで150rpmで振盪しながら37℃で1時間インキュベーションした後、頂端側(A)及び基底側(B)の両チャンバーから70μLのアリコートを取り出し、96ウェルプレートにて、分析用内部標準(0.5μMのカルバマゼピン)を含有する、100μLの50:50 アセトニトリル:水の溶液に添加した。
【0293】
基底側及び頂端側から得た液体150μLを含む新たな96ウェルプレートで、Spectramax Gemini XS(励起426nm及び発光538nm)を用いてルシファーイエローを測定した。
【0294】
試料中の化合物濃度を高速液体クロマトグラフィー/質量分析計(LC-MS/MS)により測定した。
【0295】
見掛けの透過度(Papp)の値を以下の関係から算出した:
Papp=[化合物]受容体最終×V受容体/([化合物]供与体初期×V供与体)/Tinc×V供与体/表面積×60×10-6cm/秒
V=チャンバー容積
Tinc=インキュベーション時間
表面積=0.33cm2。
【0296】
Papp B>A/Papp A>Bの比を用いて、頂端細胞表面からの能動的な流出の指標としての流出率を算出した。
【0297】
下記のアッセイ許容基準を使用した:
プロプラノロール:Papp(A>B)値≧20(×10-6cm/秒)
ローダミン123又はビンブラスチン:Papp(A>B)値<5(×10-6cm/秒)流出率≧5を伴う
ルシファーイエロー透過度:≦100nm/秒。
【0298】
【表11】
(実施例4.7 薬物動態学的研究)
(4.7.1. 齧歯類における薬物動態学的研究)
化合物を、経静脈経路用にはPEG200/生理食塩水又はPEG400/DMSO/生理食塩水混合液に、経口経路用には0.5%メチルセルロース又は10〜30%ヒドロキシプロピル-β-シクロデキストリンpH3又はpH7.4に製剤する。被験化合物を、5〜10 mg/kgで単一の食道経管栄養法として経口投与し、ボーラスとして尾静脈により1 mg/kgで経静脈投与する。各群は3匹のラットからなる。下記の範囲の時点に抗凝血剤としてのヘパリンリチウムとともに血液試料を、カニューレ処置されたラットを利用して頸静脈から、又は後方眼窩洞(retro-orbital sinus)で採取する:0.05から8時間(経静脈経路)、並びに0.25から6、又は24時間(経口経路)。全血試料を5000 rpmで10分間遠心分離し、生じた血漿試料を分析未決のまま-20℃で保存する。
【0299】
(4.7.2 イヌにおける薬物動態学的研究)
化合物を、経静脈経路用にはPEG200/生理食塩水混合液に、経口経路用にはクエン酸でpH2〜3に酸性化させた0.5%メチルセルロース又はPEG400/ヒドロキシプロピル-β-シクロデキストリン混合液に製剤する。被験化合物を、5〜30 mg/kgで経管栄養法により経口投与し、ボーラスとして、又は橈側皮静脈により10分間の点滴として1 mg/kgで経静脈投与する。各群は3匹の雄又は雌のビーグルドッグからなる。投与後0.083から24時間の時点に抗凝血剤としてのヘパリンリチウムとともに、血液試料を、頸静脈から採取する。全血試料を5000 rpmで10分間遠心分離し、生じた血漿試料を分析未決のまま-20℃で保存する。
【0300】
(4.7.3 血漿中の化合物レベルの定量化)
各被験化合物の血漿濃度を、質量分析計がポジティブエレクトロスプレーモードで運転しているLC-MS/MS法により測定する。
【0301】
(4.7.4 薬物動態学的パラメーターの決定)
薬物動態学的パラメーターを、Winnonlin(登録商標)(Pharsight(登録商標)、アメリカ合衆国)を使用して計算する。
【0302】
(実施例4.8 7日間ラット毒性研究)
被験化合物を用いた7日間経口毒性研究を、Sprague-Dawley系の雄のラットで行い、それらの毒性及び毒物動態を、5mL/kg/日の一定の投与体積で、経管栄養法による100、300及び500mg/kg/日の一日量で評価する。
【0303】
被験化合物を精製水中30%(v/v)のHPβCDに製剤する。各群は、5匹の主要な雄のラット並びに3匹の毒物動態のためのサテライト動物を含む。第4群に、30%(v/v)HPβCD水溶液のみを、同じ頻度、投与体積で、かつ同じ投与経路により与え、ビヒクルコントロール群として役割を果たす。
【0304】
該研究の目標は、確認される有害事象のない最低用量(最大無毒性量-NOAEL)を決定することである。
【0305】
(実施例4.9 肝細胞安定性)
肝細胞における代謝クリアランスを評価するモデルは、McGinnityらの文献Drug Metabolism and Disposition 2008, 32, 11, 1247に記載されている。
【0306】
先の記載が、本来例示的及び説明的なものであり、示されているとおり本発明及びその好ましい実施態様を説明するように意図されていることが、当業者には理解されるであろう。日常の試験を通して、本発明の趣旨を逸脱せずになし得る明らかな改良及び変形を、当業者は認識するだろう。したがって、本発明は、上記記載によってではなく、以下の特許請求の範囲及びそれらの均等物で規定されるものとする。
【0307】
(引用文献)
【化11】
【0308】
本明細書に引用された、特許及び特許出願を含むがこれらに限定されない刊行物は全て、個々の刊行物が完全に記載されているかのように引用により本明細書に組み込まれるように具体的かつ個別的に示されているかのように、引用により本明細書に組み込まれる。
【0309】
上記記載から、本発明の組成物及び方法における様々な改良及び変更を当業者は思いつくだろう。添付の特許請求の範囲内にあるそのような改良は全て、本件出願に含まれるものとする。
【0310】
様々な化合物の示差的な細胞浸透能などの因子がインビトロ生化学アッセイにおける化合物活性と細胞アッセイにおける化合物活性との間の相違に関与し得ることを理解されたい。
【0311】
本件出願に与えられ、かつ記載される本発明の化合物の化学名の少なくともいくつかは、市販の化学命名ソフトウェアプログラムを使用して自動的に生成された場合があり、独立に検証していない場合がある。この機能を実行する代表的なプログラムには、Open Eye Software社により販売されるLexichem命名ツール及びMDL社により販売されるAutonom Softwareツールがある。示された化学名と表された構造とが異なる場合、表された構造が基準となる。
【0312】
本明細書に示された化学構造は、ChemDraw(登録商標)又はISIS(登録商標)/DRAWのいずれかを使用して作成された。本明細書中の構造における炭素、酸素又は窒素原子に現れている任意の開放原子価は、水素原子の存在を示す。構造中にキラル中心が存在するが、該キラル中心について具体的な立体化学が示されない場合、該キラル構造と関連する両方の鏡像異性体が該構造により包含される。
【特許請求の範囲】
【請求項1】
式Iの化合物又はその医薬として許容し得る塩:
【化1】
。
【請求項2】
医薬として許容し得る担体、及び医薬として有効な量の請求項1記載の化合物若しくはその医薬として許容し得る塩を含む、医薬組成物。
【請求項3】
さらなる治療剤を含む、請求項2記載の医薬組成物。
【請求項4】
医薬の製造における、請求項1記載の化合物又はその医薬として許容し得る塩の使用。
【請求項5】
医薬として使用するための、請求項1記載の化合物又はその医薬として許容し得る塩。
【請求項6】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防に使用するための、請求項1記載の化合物又はその医薬として許容し得る塩。
【請求項7】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防のための医薬の製造における、請求項1記載の化合物又はその医薬として許容し得る塩の使用。
【請求項8】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療又は予防の方法であって、有効量の請求項1、4、又は5のいずれか一項記載の化合物若しくはその医薬として許容し得る塩、又は請求項2記載の医薬組成物を投与することを含む、前記方法。
【請求項9】
前記炎症性病態が関節リウマチである、請求項8記載の方法。
【請求項10】
前記病態又は疾患が炎症を含む、請求項8記載の方法。
【請求項11】
請求項1記載の化合物がさらなる治療剤と組み合わされて投与される、請求項5又は8記載の化合物又は方法。
【請求項12】
前記さらなる治療剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防のための薬剤である、請求項2記載の医薬組成物又は請求項11記載の方法。
【請求項1】
式Iの化合物又はその医薬として許容し得る塩:
【化1】
。
【請求項2】
医薬として許容し得る担体、及び医薬として有効な量の請求項1記載の化合物若しくはその医薬として許容し得る塩を含む、医薬組成物。
【請求項3】
さらなる治療剤を含む、請求項2記載の医薬組成物。
【請求項4】
医薬の製造における、請求項1記載の化合物又はその医薬として許容し得る塩の使用。
【請求項5】
医薬として使用するための、請求項1記載の化合物又はその医薬として許容し得る塩。
【請求項6】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防に使用するための、請求項1記載の化合物又はその医薬として許容し得る塩。
【請求項7】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防のための医薬の製造における、請求項1記載の化合物又はその医薬として許容し得る塩の使用。
【請求項8】
炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療又は予防の方法であって、有効量の請求項1、4、又は5のいずれか一項記載の化合物若しくはその医薬として許容し得る塩、又は請求項2記載の医薬組成物を投与することを含む、前記方法。
【請求項9】
前記炎症性病態が関節リウマチである、請求項8記載の方法。
【請求項10】
前記病態又は疾患が炎症を含む、請求項8記載の方法。
【請求項11】
請求項1記載の化合物がさらなる治療剤と組み合わされて投与される、請求項5又は8記載の化合物又は方法。
【請求項12】
前記さらなる治療剤が、炎症性病態、自己免疫疾患、増殖性疾患、移植拒絶、軟骨代謝回転の異常を含む疾患、先天性軟骨形成異常、及び/又はIL6の分泌過多に関連する疾患の治療、予防、又は予防のための薬剤である、請求項2記載の医薬組成物又は請求項11記載の方法。
【公表番号】特表2012−530767(P2012−530767A)
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2012−516762(P2012−516762)
【出願日】平成22年6月25日(2010.6.25)
【国際出願番号】PCT/EP2010/059067
【国際公開番号】WO2010/149771
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(504064364)ガラパゴス・ナムローゼ・フェンノートシャップ (27)
【氏名又は名称原語表記】Galapagos N.V.
【Fターム(参考)】
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成22年6月25日(2010.6.25)
【国際出願番号】PCT/EP2010/059067
【国際公開番号】WO2010/149771
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(504064364)ガラパゴス・ナムローゼ・フェンノートシャップ (27)
【氏名又は名称原語表記】Galapagos N.V.
【Fターム(参考)】
[ Back to top ]