
変異型尿酸オキシダーゼおよびその利用
【課題】 トランケートされ、構造安定性が向上した変異型組み換えウリカーゼタンパク質の提供。
【解決手段】 本発明は、尿酸分解活性を有する遺伝的に改変したタンパク質に関する。より具体的には、本発明は切断型尿酸オキシダーゼを含むタンパク質、ならびにその製造法、および、切断型尿酸オキシダーゼを含むPEG化タンパク質を含む。
【解決手段】 本発明は、尿酸分解活性を有する遺伝的に改変したタンパク質に関する。より具体的には、本発明は切断型尿酸オキシダーゼを含むタンパク質、ならびにその製造法、および、切断型尿酸オキシダーゼを含むPEG化タンパク質を含む。
【発明の詳細な説明】
【技術分野】
【0001】
関連文献の相互参照
本出願は、参照によりここに援用されるところの、2005年4月11日出願の米国特許仮出願番号第60/670,541号の優先権並びに利益を請求するものである。
【0002】
技術分野
本発明は、尿酸分解活性を有する遺伝的に改変されたタンパク質に関する。より具体的には切断型尿酸オキシダーゼを含むタンパク質および、その製造法に関する。
【0003】
背景技術
以下、「尿酸オキシダーゼ」と「ウリカーゼ」を同義語として用いる。尿酸オキシダーゼ(ウリカーゼ、E.C.1.7.3.3)は、酸化を触媒する事によって尿酸を、より可溶性が高く、より容易に排泄されるプリン代謝物質、アラントインへ転換する酵素である。ヒトは、高等霊長類の進化の過程で蓄積したウリカーゼ遺伝子へのいくつかの変異により、酵素活性を有するウリカーゼを生産しない。Wu,X,et al.,(1992)J Mol Evol 34:78−84を参照することにより、その全文を本明細書に含む。この結果、感受性個体では、血中での過剰濃度の尿酸(高尿酸血症)が疼痛性関節炎(痛風)、関節破壊を伴う尿酸塩沈着(痛風結節)および腎不全を引き起こすことがある。アロプリノール(尿酸合成阻害剤)などの薬剤が利用可能であるが、一部の罹患個体では、治療を限定する有害作用を生じるか、またはこれらの症状を適切に軽減できない場合がある。Hande,KR,et al.,(1984)Am J Med 76:47−56、Fam,AG,(1990) Bailliere’s Clin Rheumatol 4:177−192を、それぞれ参照することによってその全文を本明細書に含む。ウリカーゼの注入は、高尿酸血症および高尿酸尿症を少なくとも一次的に低減し得る。しかしながら、ウリカーゼはヒトにおいては外来タンパク質であるため、Aspergillus flavus由来の非修飾タンパク質の一次注射でさえ、処置患者の数パーセントにおいてアナフィラキシー反応を誘導し(Pui,C−H,et al.,(1997)Leukemia 11:1813−1816を、参照することによってその全文を本明細書に含む)、免疫応答は慢性的または間欠的治療における有用性を限定する(Donadio,D,et al.,(1981) Nouv Presse Med 10:711−712、Leaustic,M,et al.,(1983) Rev Rhum Mal Osteoartic 50:553−554をそれぞれ参照することによって全文を本明細書に含む)。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、トランケートされ、構造安定性が向上した変異型組み換えウリカーゼタンパク質に関する。
【課題を解決するための手段】
【0005】
本発明は新規の組み換えウリカーゼタンパク質を提供する。本発明のウリカーゼはトランケートされており、天然のウリカーゼタンパク質に対して、変異したアミノ酸を有する。
【0006】
本発明はSEQ ID NO.8のアミノ酸配列を含む変異型組み換えウリカーゼを提供する。さらにSEQ ID NO.13のアミノ酸配列を含む変異型組み換えウリカーゼも提供する。
【0007】
本発明のさらなる目的は、尿酸分解活性を有する新規の組み換えウリカーゼタンパク質を含む、尿酸を代謝する手段を提供することである。尿酸分解活性とは、本明細書では酵素による尿酸のアラントインへの変換を指す。
【0008】
特定の一実施形態にあっては、ウリカーゼは、SEQ ID NO.7またはSEQ ID NO.12の位置8から位置287のアミノ酸配列を含む。さらにSEQ ID NO.8またはSEQ ID NO.13のアミノ酸配列を含むウリカーゼが提供される。一実施形態にあっては、ウリカーゼはアミノ末端アミノ酸を含み、該アミノ末端アミノ酸はアラニン、グリシン、プロリン、セリンまたはトレオニンである。特定の一実施形態にあっては、アミノ末端アミノ酸はメチオニンである。
【0009】
本発明は、本発明のウリカーゼをコードする核酸配列を含む、単離された核酸も提供する。一実施形態にあっては、該ウリカーゼをコードする核酸は動作可能なように異種性のプロモータ、例えばosmBプロモータに連結されている。さらに、ウリカーゼをエンコードする核酸を含む核酸ベクター、ならびにこのようなベクターを含む宿主細胞も提供される。さらに一実施形態にあっては、核酸はSEQ ID NO.7の配列を有する。
【0010】
このような宿主細胞を、核酸配列が宿主細胞によって発現されるような条件下で培養し、発現されたウリカーゼを単離する工程を含む、ウリカーゼを生成する方法も、提供される。
【図面の簡単な説明】
【0011】
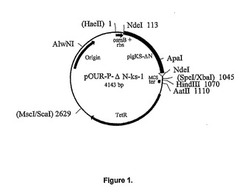
【図1】図1はプラスミドpOUR−P−ΔN−ks−1の構造を示している。制限酵素サイトの横に示された番号は、1で示したHaeIIサイトから数えた、ヌクレオチドの位置を示している。クローニングの過程で失われた制限酵素サイトは、括弧付きで示している。
【0012】
【図2】図2は、Pig−KS−ΔNウリカーゼのDNA配列、および推定されるアミノ酸配列を示したものである(それぞれSEQ ID NO.9およびSEQ ID NO.7)。図2のアミノ酸番号は、全長ブタウリカーゼに対応している。イニシエーターであるメチオニン残基に続いて、ブタウリカーゼ配列のアスパラギン酸7がトレオニンに置換されている。サブクローニングの様々な過程で使用した制限酵素サイトが、示されている。3′側の翻訳されない配列は小文字で示されている。翻訳終止コドンは、アスタリスクで示してある。
【0013】
【図3】図3は様々な組み換えブタ(SEQ ID NO.11)、PBC−ΔNC (SEQ ID NO.12)およびPig−KS−ΔN(SEQ ID NO.7)ウリカーゼ配列のアラインメントを示している。Pig−KS−ΔNで、公開されているブタウリカーゼ配列とアミノ酸の相違がある位置をアスタリスクで示している。丸印は、Pig−KS−ΔNとPBC−ΔNでアミノ酸の相違がある位置を示している。点線は、アミノ酸の欠失を示している。
【0014】
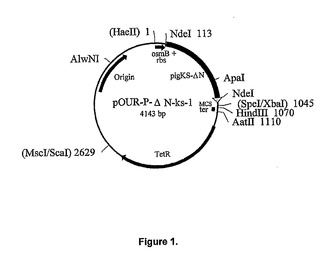
【図4】図4はブタウリカーゼおよび、高度に精製した、実施例1から3に従って作成した変異型ウリカーゼのSDS−PAGEを示している。生成日(月/年)およびそれぞれのサンプルに対応するレーン番号は以下に示してある。Y軸は分子量マーカーの分子量でラベルされており、図の上部にレーン番号を示してある。レーン番号は以下の通りである:レーン1−分子量マーカー、レーン2−PigKS−ΔN(7/98)、レーン3−Pig(9/98)、レーン4−PigKS(6/99)、レーン5−PigKS(6/99)、レーン6−Pig−ΔN(6/99)、レーン7−PigKS−ΔN(7/99)、レーン8−PigKS−ΔN(8/99)。
【0015】
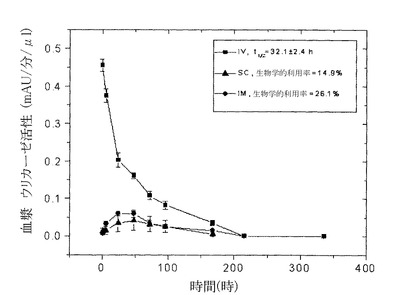
【図5】図5は血液サンプルでの酵素活性を測定することによって求めた、ラットでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与)、SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0016】
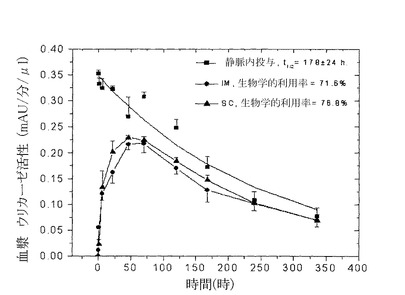
【図6】図6は血液サンプルでの酵素活性を測定することによって求めた、ウサギでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0017】
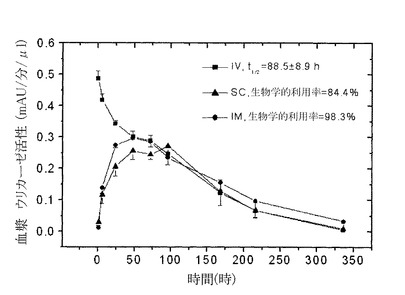
【図7】図7は血液サンプルでの酵素活性を測定することによって求めた、イヌでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0018】
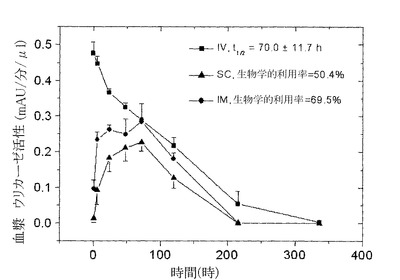
【図8】図8は血液サンプルでの酵素活性を測定することによって求めた、ブタでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【発明を実施するための形態】
【0019】
これまでの研究は、PEG化によってウリカーゼの免疫原性および/または抗原性の大幅な低減が達成された場合は、必ず尿酸分解活性も大幅に失われてしまうことを示している。生物薬剤の安全性、利便性および対費用効果は、効力が失われ、また従って投与量を上げなければならないことによって悪影響を受ける。従って、血液を含む体液において、上昇した尿酸レベルを下げる安全で効果的な代替的な方法が必要とされている。本発明はSEQ ID NO.7、SEQ ID NO.8、SEQ ID NO.12またはSEQ ID NO.13のアミノ酸配列を含む変異型組み換えウリカーゼを提供する。
【0020】
特に記載のない限り、本明細書では用語「ウリカーゼ」を、個々のサブユニットならびにその四量体両方を指すものとして用いる。
【0021】
本明細書では、用語「変異型ウリカーゼ」を、他のアミノ酸に置換されたアミノ酸を有するウリカーゼ分子を指す語として用いる。
【0022】
特定の実施態様では、本発明のウリカーゼはアミノ末端メチオニンを有する。特定の実施態様では、アミノ末端メチオニンはウリカーゼが産生された後に除去される。特定の実施態様では、該メチオニンは内在するバクテリアのアミノペプチダーゼによって除去される。N末端のメチオニンの最も完全な除去を可能にするアミノ酸は、アラニン、グリシン、プロリン、セリンおよびトレオニンである。特定の一実施形態にあっては、ウリカーゼはアミノ末端に2つのアミノ酸を有し、この2つのアミノ酸は一方がメチオニンであって、これに引き続いてアラニン、グリシン、プロリン、セリンおよびトレオニンからなる群から選んだアミノ酸1つが引き続くものである。
【0023】
本発明はウリカーゼをコードする核酸配列を提供する。
【0024】
本発明は、当該核酸配列を含むベクターを提供する。
【0025】
特定の一実施形態にあっては、ウリカーゼは単離される。特定の一実施形態にあっては、ウリカーゼは精製される。特定の実施形態にあっては、ウリカーゼは単離され精製される。
【0026】
本発明はウリカーゼをコードする核酸配列を含むベクターを含む宿主細胞を提供する。
【0027】
本発明はPCR(ポリメラーゼ連鎖反応法)技術による、ウリカーゼをコードする核酸配列の改変を含む、ウリカーゼをコードする核酸配列を作成する方法を提供する。当該業務に精通する者には目的の核酸配列がPCRによって、増幅する標的DNAの領域に相補的な合成オリゴヌクレオチドプライマー(それぞれの鎖に1つずつ)を介して、調製できることが既知である。プライマーを、過剰量のデオキシヌクレオチドおよび、耐熱性DNAポリメーラーゼであるTaqポリメラーゼの存在下で標的DNA(純粋なものでなくてよい)に加える。標的DNAは、連続する温度サイクル(典型的な場合で、30回)を経る中で反復して変性(約90℃)、プライマーとのアニーリング(典型的な場合で、50から60℃)、プライマーからの娘鎖の伸長(72℃)を繰り返す。次のサイクルからは娘鎖もテンプレートとして働くので、両方のプライマーとマッチするDNA断片は線形にではなく、指数関数的に増幅される。
【0028】
本発明は、変異型組み換えウリカーゼを生成する方法を提供するものであって、宿主細胞のベクターによる形質転換、宿主細胞によるウリカーゼの発現、変異型組み換えウリカーゼの宿主細胞からの単離、精製された変異型組み換えウリカーゼの、例えばクロマトグラフィー技術による単離、および変異型組み換えウリカーゼの精製を含む。例えば、ウリカーゼは参照することによってその全文を本明細書に含む、国際公開第00/08196号パンフレット、および米国特許出願番号60/095,489に記載の方法によって作成することができる。
【0029】
好ましい一実施形態にあっては、変異型組み換えウリカーゼの発現を引き起こすよう、宿主細胞に処置を施す。当該業務に精通する者には、細胞のベクターによる形質転換は通常カルシウムイオンと沈殿させたDNAを用いて達成されるが、他にも様々な方法(例えば電気穿孔法)を用いることができることを理解している。
【0030】
ウリカーゼは、当該業務に精通した者に既知であるあらゆる方法によって単離および/または精製されうる。一般に、本発明で発現させたポリペプチドは実質的に純粋な形で単離される。好ましくは、ポリペプチドは重量で少なくとも80%の純度で単離される。より好ましくは、重量で少なくとも95%の純度、そして最も好ましくは、少なくとも99%の純度で単離される。一般にこのような精製は、例えば硫安分画法、SDS−PAGE電気泳動法およびアフィニティークロマトグラフィーの標準的な技法を用いて達成されうる。ウリカーゼは、好ましくは陽イオン界面活性剤、例えば、参照することによってその全文を本明細書に含む、2005年4月11日出願の同時係属出願、代理人整理番号第103864.146644号、タイトルが陽イオン界面活性剤によるタンパク質の精製である、米国特許仮出願番号第60/670,520号に記載の方法に従って塩化セチルピリジニウム(CPC)によって単離される。
【0031】
本発明の一実施形態にあっては、ベクターは浸透圧感受性プロモータの制御下にある。プロモータとは、RNAポリメラーゼが、DNAのRNAへの転写プロセスを開始するに先立って結合する、DNA領域のことを言う。浸透圧感受性プロモータは、細胞が浸透圧の上昇を感知した場合に転写を開始する。
【0032】
本発明のウリカーゼはポリマーと結合したウリカーゼ、たとえばポリエチレングリコールと結合したウリカーゼ、すなわち、PEG化されたウリカーゼを含む。
【0033】
本発明の一実施形態にあっては、ウリカーゼを含む医薬組成物が提供される。一実施形態にあっては、組成物はウリカーゼ溶液である。好ましい一実施形態にあっては、溶液は滅菌済みであり、注射に適している。一実施形態にあっては、このような組成物はウリカーゼのリン酸緩衝生理食塩水溶液を含む。一実施形態にあっては、組成物はバイアルで提供され、さらに、任意にラバー注射ストッパーを備えることができる。特定の実施形態にあっては、組成物は、ウリカーゼを溶液1ミリリットルあたり2から16ミリグラムの濃度、溶液1ミリリットルあたり4から12ミリグラムの濃度、溶液1ミリリットルあたり6から10ミリグラムの濃度で含む。好ましい一実施形態にあっては、組成物はウリカーゼを1ミリリットルあたり8ミリグラムの濃度で含む。好ましくは、ウリカーゼの重量は、タンパク質重量に対して測定される。
【0034】
本発明の組成物の効果的な投与計画は、当該業務に精通した者によって決定されうる。任意の投与計画の、有効性の評価に適した指標は、当該業務に精通した者に既知である。このような指標の例としては、血漿尿酸レベル(PUA)の正常化または低下、ならびにPUAを6.8mg/dL以下、好ましくは6mg/dL以下に低下または維持させることが挙げられる。好ましい一実施形態にあっては、本発明の組成物による治療の対象者は、総治療期間の70%以上、80%以上、または90%以上の期間、PUAが6mg/ml以下である。例えば治療期間が24週間である場合、対象者のPUAは24週間の治療期間の80%以上の間、すなわち、少なくとも134.4日(24週間x7日間/週間x0.8=134.4日)に等しい時間の間、6mg/ml以下である。
【0035】
特定の実施形態にあっては、溶液としてウリカーゼ0.5から24mgが2から4週に1回投与される。ウリカーゼは、当該業務に精通した者に既知のあらゆる適切な方法、例えば静脈内投与、筋肉内投与、皮下投与によって投与することができる。好ましくは、投与が静脈内投与による場合は、ウリカーゼとして0.5mgから12mgが投与される。好ましくは、投与が皮下投与による場合は、ウリカーゼとして4mgから24mgが投与される。好ましい一実施形態にあっては、ウリカーゼは静脈点滴によって、30から240分の点滴時間で投与される。一実施形態にあっては、2週間に1回、ウリカーゼとして8mgが投与される。特定の実施形態にあっては、100から500mLの生理食塩水を用いて点滴を行うことができる。好ましい一実施形態にあっては、2週間に1回または4週間に1回、点滴時間120分で、溶液として8mgのウリカーゼが投与される。好ましくは、点滴に用いるウリカーゼは250mLの生理食塩水に溶解される。特定の実施形態にあっては、ウリカーゼの投与は3ヶ月間、6ヶ月間、8ヶ月間または12ヶ月間の治療期間にわたって行う。他の実施形態にあっては、治療期間は12週間、24週間、36週間または48週間である。特定の一実施形態にあっては、治療期間は長期間にわたるものであって、例えば2年以上、治療対象者の全生存期間にわたる。加えて、複数の治療期間を、治療休止期間ではさむことも可能である。例えば、6ヶ月間の治療に引き続いて3ヶ月間の治療休止期間をおき、その後再び6ヶ月間治療を行うことなどが、可能である。
【0036】
特定の実施形態にあっては、ウリカーゼの投与による注入反応の出現を阻止または低減するために、予防的に抗炎症性物質を投与することができる。一実施形態にあっては、少なくとも1つのコルチコステロイド、少なくとも1つの抗ヒスタミン薬、少なくとも1つのNSAID、またはこれらの組み合わせが、よって投与される。有用なコルチコステロイドには、ベータメタゾン、ブデソニド、コルチゾン、デキサメタゾン、ヒドロコルチゾン、メチルプレドニソロン、プレドニソロン、プレドニゾンおよびトリアムシノロンが含まれる。有用なNSAIDにはイブプロフェン、インドメタシン、ナプロキセン、アスピリン、アセトアミノフェン、セレコキシブおよびバルデコキシブが含まれる。有用な抗ヒスタミン薬には、アザタジン、ブロムフェニラミン、セチリジン、クロルフェニラミン、クレマスチン、シプロヘプタジン、デスロラタジン、デクスクロルフェニラミン、ジメンヒドリナート、ジフェンヒドラミン、ドキシルアミン、フェキソフェナジン、ヒドロキシジン、ロラタジンおよびフェニンダミンが含まれる。
【0037】
好ましい一実施形態にあっては、抗ヒスタミン薬はフェキソフェナジンであり、NSAIDはアセトアミノフェンであり、コルチコステロイドはヒドロコルチゾンおよび/またはプレドニゾンである。好ましくは、3つ全ての組み合わせ(必ずしも同時にではない)を、ウリカーゼ溶液の注入投与の前に投与する。好ましい一実施形態にあっては、NSAIDと抗ヒスタミン薬はウリカーゼ注入の1から4時間前に経口投与される。フェキソフェナジンの適切な用量は約30から約180mg、約40から約150mg、約50から約120mg、約60から約90mg、約60mgを含み、好ましくは60mgである。アセトアミノフェンの適切な用量は約500から約1500mg、約700から約1200mg、約800から約1100mg、約1000mgを含み、好ましくは1000mgである。ヒドロコルチゾンの適切な用量は約100から約500mg、約150から約300mg、約200mgを含み、好ましくは200mgである。一実施形態にあっては、抗ヒスタミン薬はジフェンヒドラミンではない。他の実施形態にあっては、NSAIDはアセトアミノフェンではない。好ましい一実施形態にあっては、フェキソフェナジン60mgをウリカーゼ注入の前夜に経口投与し、フェキソフェナジン60mgおよびアセトアミノフェン1000mgを次の朝に経口投与し、最後に、ウリカーゼ注入の直前にヒドロコルチゾン200mgを投与する。一実施形態にあっては、プレドニゾンをウリカーゼ投与の前日に、好ましくは夕方、投与する。プレドニゾンの適切な用量は5から50mgを含み、好ましくは20mgである。特定の実施形態にあっては、注入反応の出現を阻止または低減させるためのこのような予防措置は、PEG化ウリカーゼおよび非PEG化ウリカーゼを含むウリカーゼ投与を、受けているかまたは受ける予定の治療対象者に対して講じられる。特定の実施形態にあっては、このような予防措置はウリカーゼ以外の、PEG化治療薬ペプチドおよび非PEG化治療薬ペプチドを含む、治療薬ペプチド投与を受けているかまたは受ける予定の対象者に対して講じられる。
【0038】
本発明の一実施形態にあっては、医薬組成物はポリマーと結合されたウリカーゼを含有するものであって、修飾されたウリカーゼは尿酸分解活性を保持している。好ましい実施態様においては、ウリカーゼはPEG化されたウリカーゼである。
【0039】
本発明の一実施形態にあっては、医薬組成物はポリマーとの結合によって修飾されたウリカーゼを含有するものであって、修飾されたウリカーゼは尿酸分解活性を保持している。特定の一実施形態にあっては、ポリマー−ウリカーゼ結合体は参照によってその全文を本明細書に含める、国際公開第01/59078号パンフレットおよび米国特許出願番号第09/501730号に記載されている方法に従って調製される。
【0040】
本発明の一実施形態にあっては、ポリマーはポリエチレングリコール、デキストラン、ポリプロピレングリコール、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロース、ポリビニールピロリドンおよびポリビニールアルコールを含む群から選択される。好ましい実施態様においては、ポリマーはポリエチレングリコールであり、ウリカーゼはPEG化されたウリカーゼである。
【0041】
本発明の一実施形態にあっては、組成物は2−12のポリマー分子をそれぞれのウリカーゼサブユニットに含有し、好ましくは、3から10ポリマー分子をそれぞれのウリカーゼサブユニットに含有する。本発明の一実施形態にあっては、それぞれのポリマー分子は約1kDから約100kDの分子量を有する。
【0042】
本発明の他の実施形態にあっては、それぞれのポリマー分子は約1kDから約50kDの分子量を有する。本発明の好ましい一実施形態にあっては、それぞれのポリマー分子は約5kDから約20kD、約8kDから約15kD、約10kDから12kDの分子量を有し、好ましくは、約10kDの分子量を有する。好ましい一実施形態にあっては、それぞれのポリマー分子は約5kDまたは約20kDの分子量を有する。特に好ましい本発明の一実施形態にあっては、それぞれのポリマー分子は10kDの分子量を有する。
【0043】
本発明の実施態様においては、組成物は、組成物の反復投与に適している。
【0044】
本発明は当該ウリカーゼを用いて尿酸を代謝する方法を提供する。
【0045】
本発明は、ウリカーゼ組成物の、体液中尿酸レベルの低減への利用を提供する。
【0046】
本発明の一実施形態にあっては、ウリカーゼ組成物は血液を含む体液における尿酸の低減に用いられる。
【0047】
また、本発明のウリカーゼをコードする新規核酸分子も提供される。これらを生産する技法は、当該業務に精通した者によく知られている。例えば、ウリカーゼ核酸配列は当該業界に既知である、多くのあらゆる戦略によって改変されうる(Maniatis,T.,1990,Molecular Cloning,A Laboratory Manual,2d ed.,Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y.)。配列は、適切な場所で1つまたはそれ以上の制限エンドヌクレアーゼによって切断でき、必要であればさらに酵素による改変を施し、単離し、in vitroでライゲーションすることができる。ウリカーゼをコードする遺伝子の作成にあっては、改変された遺伝子が翻訳停止シグナルに遮られることなく、適切な翻訳読み枠を保持するよう注意すべきである。
【0048】
ウリカーゼタンパク質をコードする核酸配列は適切な発現ベクター、すなわち、挿入されたタンパク質コーディング配列の転写および翻訳に必要なエレメントを含むベクターに、挿入することができる。タンパク質コーディング配列を発現させるためには、多様な宿主ベクター系を用いることができる。これらはウィルス(例えばワクシニアウィルス、アデノウィルス等)を感染させた哺乳類細胞系、ウィルス(例えばバキュロウィルス)を感染させた昆虫細胞系、酵母ベクターを含む酵母などの微生物、バクテリオファージDNA、プラスミドDNA、コスミドDNAなどで形質転換したバクテリアを含むが、これに限定されない。これらのベクターの発現エレメントはそれらの強度と特異性において異なる。用いる宿主ベクター系に応じて、あらゆる適切な転写および翻訳エレメントを用いることができる。
【0049】
適切な転写/翻訳制御シグナルと、タンパクをコードする配列からなるキメラ遺伝子を含んだ発現ベクターの作製には、DNA断片をベクターに挿入する既知のあらゆる方法を用いることができる。このような方法には、in vitro組換えDNAおよび合成的技術およびin vivo組換え(遺伝的組換え)を含めることができる。ウリカーゼタンパク質をコードしている核酸配列の発現は、組換えDNA分子で形質転換した宿主内でウリカーゼタンパク質が発現されるよう、第二の核酸配列によって制御することができる。例えば、ウリカーゼの発現は、技術的に知られているあらゆるプロモータ/エンハンサーエレメントによって制御され得る。ウリカーゼの発現を制御するのに用いることのできるプロモータはSV40初期プロモータ領域(Bernoist and Chambon,1981,Nature 290:304−310)、ラウス肉腫ウィルスの3’末端反復配列に含まれるプロモータ(Yamamoto,et al.,1980,Cell 22:787−797)、ヘルペスチミジンキナーゼプロモータ(Wagner et al.,1981,Proc.Natl.Acad.Sci.U.S.A.78:144−1445)、メタロチオネイン遺伝子の制御配列(Brinster et al., 1982,Nature 296:39−42)、βラクタマーゼプロモータなどの原核生物発現ベクター(Villa−Kamaroff,et al.,1978,Proc.Natl.Acad.Sci.U.S.A.75:3727−3731)、tacプロモータ(DeBoer,et al.,1983,Proc.Natl.Acad.Sci.U.S.A.80:21−25)、osmBプロモータを含むが、これに限定されない。特定の実施形態にあっては、核酸は動作可能な形で異種性プロモータに結合されたウリカーゼ配列を含む。
【0050】
核酸配列がコードされている特定の組み換えDNA分子が一旦調製され単離されたならば、技術的に知られている数種の方法によって、増殖させることができる。一旦宿主系ならびに培養条件が確立されれば、組み換え発現ベクターは増殖させ、大量に調製することができる。先に説明したように、使用できる発現ベクターは数例を挙げるならば、ワクシニアウィルスまたはアデノウィルスなどのヒトまたは動物ウィルス、バキュロウィルスなどの昆虫ウィルス、酵母ベクター、バクテリオファージベクター(例:ラムダ)およびプラスミドおよびコスミドDNAベクターならびに、これらの誘導体を含むが、これに限定されない。
【0051】
加えて、挿入された配列の発現を制御する、または遺伝子産物に、目的に合致した特定の修飾ならびに処理を施す宿主細胞株を選択することが可能である。特定のプロモータによる発現は特定の誘導物質の存在下で上昇し、よって遺伝的に改変したウリカーゼタンパク質の発現を制御することができる。さらに、異なる宿主細胞はそれぞれ特徴的ならびに特異的な、タンパク質の翻訳処理および翻訳後処理および修飾(例えばグリコシル化、切断)メカニズムを有している。発現された外来タンパク質に、目的の修飾および処理が施されることが保証されるよう、適切な細胞株を選ぶことができる。異なったベクター/宿主発現系は、タンパク質分解性開裂反応などの処理反応の程度を変えるなどの影響を与えうる。
【0052】
本発明の特定の実施形態にあっては、ウリカーゼ をE.coliで発現させることが好ましく、この際osmBプロモータを含むベクターを用いる。
【0053】
実施例
実施例 1. 遺伝子の作成ならびにウリカーゼ発現のための発現プラスミド
組み換えブタウリカーゼ(尿酸酸化酵素)、Pig−KS−ΔN(アミノ末端がトランケーションされたブタウリカーゼタンパク質で、第291番目と第301番目のアミノ酸が、それぞれリシンとセリンに置換されている)をE.coli K−12株W3110F−で発現させた。pOUR−P−ΔN−ks−1に到る、一連のプラスミドを作成したが、pOUR−P−ΔN−ks−1でE.coli宿主細胞を形質転換したところ、ウリカーゼを効率的に発現する能力を獲得した。
【0054】
ウリカーゼcDNAのブタおよびヒヒ肝臓からの単離およびサブクローニング
ウリカーゼcDNAは、当該RNAのサブクローニングおよび単離によって、ブタおよびヒヒ肝臓より調製した。全細胞RNAをブタおよびヒヒ肝臓より抽出し(Erlich,H.A.(1989).PCR Technology,Principles and Application for DNA Amplification、Sambrook,J.,et al.(1989).Molecular Cloning:A Laboratory Manual,2nd edition、Ausubel,F.M. et al.(1998).Current protocols in molecular Biology)、ファースト−ストランドcDNA合成キット(Pharmacia Biotech)を用いて逆転写した。PCR増幅反応は、Taq DNAポリメラーゼ(Gibco BRL,Life Technologies)を用いて行った。
【0055】
ブタならびにヒヒ尿酸オキシダーゼ(ウリカーゼ)のPCR増幅反応に用いた合成オリゴヌクレオチドプライマーは表1に示した。
表 1. ウリカーゼcDNAのPCR増幅に用いたプライマー
【表1】

【0056】
プライマー末端に設定した制限酵素配列はセンスEcoRIおよびNcoI(ブタおよびヒヒ)およびアンチセンスNcoI、HindIIIおよびXbaI(ブタ)、XbaIおよびNcoI(ヒヒ)であり、表1に小文字で示してある。ヒヒのセンスプライマーでは、ヒヒウリカーゼに存在する3番目のコドンGAC(アスパラギン酸)を、ヒト尿酸オキシダーゼ偽遺伝子をコードする配列のこの位置に存在するCAC(ヒスチジン)で置換した。これらのプライマーを用いて作成した組み換えヒヒウリカーゼ構造体をD3Hヒヒウリカーゼとする。
【0057】
EcoRIとHindIIIでブタウリカーゼPCR産物を処理し、pUC18にクローニングしてpUC18−ブタウリカーゼを得た。D3HヒヒウリカーゼPCR産物はTA Cloning(登録商標)(Invitrogen,Carlsbad,CA)を用いて、直接pCR(登録商標)IIベクターにクローニングし、pCR(登録商標)II−D3Hヒヒウリカーゼを得た。
【0058】
ライゲーションしたcDNAでE.coli株XL1−Blue(Stratagene,LaJolla,CA)を形質転換した。クローニングしたウリカーゼcDNAを含むプラスミドDNAを調製し、公開されたウリカーゼDNAコーディング配列を含むクローン(表1に記載の、ヒヒウリカーゼでのD3Hの置換を除く)をセレクションし、単離した。選んだpCR(登録商標)II−D3HヒヒウリカーゼクローンにおいてはPCRによって配列を削除したことで、pCR(登録商標)II配列がウリカーゼのストップコドンの隣にくるようになっている。この結果として、3′側の翻訳されない領域にあったXbaIおよびNcoIの制限酵素サイトが除かれているので、PCR産物の5′末端に存在するNcoIサイトとpCR(登録商標)IIベクター由来のBamHIサイトを用いて、デクレショナルクローニングをすることができた。
【0059】
ウリカーゼcDNAの、pET発現ベクターへのサブクローニング
ヒヒウリカーゼのサブクローニング
ウリカーゼコード配列の全長を含むD3HヒヒcDNAをpET−3d発現ベクター(Novagen,Madison,WI)に導入した。pCR(登録商標)II−D3HヒヒウリカーゼをNcoIおよびBamHIで処理し、得られた960bpの断片を単離した。発現プラスミドpET−3dはNcoIおよびBamHIで処理し、得られた4600bpの断片を単離した。この2つの断片をライゲーションしてpET−3d−D3H−Baboonを得た。
【0060】
ブタ−ヒヒキメラウリカーゼのサブクローニング
ブタ−ヒヒキメラ(PBC)ウリカーゼは、発現量、安定性および活性がより大きい組み換え遺伝子を得るために作成された。PBCの作成にはまず、pET−3d−D3H−ヒヒクローンから4936bp長のNcoI−ApaI断片を単離し、これをpUC18−ブタウリカーゼから単離した624bp長のNcoI−ApaI断片とライゲーションしてpET−3d−PBCを得た。PBCウリカーゼcDNAは、ブタウリカーゼのコドン1−225と、ヒヒウリカーゼのコドン226−304をインフレームで結合したものからなる。
【0061】
Pig−KSウリカーゼのサブクローニング
Pig−KSウリカーゼは、PEG化できる部位を1つ増やすことが期待されるリシン残基を、1つ加える目的で作成した。KSとはブタウリカーゼの291番目の位置にあるアルギニンの代わりにリシンを挿入したアミノ酸置換(R291K)を指す。加えて、301番目の位置にあるトレオニンをセリンで置換した(T301S)。Pig−KSウリカーゼプラスミドは、pET−3d−D3H−Baboonから4696bp長のNcoI−NdeI断片を単離し、pUC18−ブタウリカーゼから単離した得た864bp長のNcoI−NdeI断片をライゲーションして作成し、pET−3d−Pig−KSとした。得られたPigKSウリカーゼ配列は、ブタウリカーゼコドン1−288がインフレームにヒヒウリカーゼコドン289−304と結合されたものとなっている。
【0062】
osmBプロモータの制御下にあるウリカーゼ配列のサブクローニング
ウリカーゼ遺伝子を、osmBプロモータを含む発現ベクターにサブクローニングした(参照することによってその全文を本明細書に含む、米国特許番号5,795,776号に従った)。このベクターは、高浸透圧または長時間の培養によってタンパク質発現を誘導することを可能にした。発現プラスミドpMFOA−18はosmBプロモータ、リボソーム結合部位配列(rbs)および転写ターミネーター配列(ter)を含んでいる。また、アンピシリン耐性(AmpR)を付与し、組み換えヒトアセチルコリンエステラーゼ(AChE)を発現する。
【0063】
D3H−ヒヒウリカーゼのサブクローニング
プラスミドpMFOA−18をNcoIおよびBamHIで処理し、大きな断片を単離した。pET−3d−D3H−BaboonをNcoIおよびBamHIで処理し、D3Hヒヒウリカーゼを含む960bp長の断片を単離した。この2つの断片をライゲーションしてpMFOU18を得た。
【0064】
発現プラスミドpMFXT133はosmBプロモータ、rbs(E.colideoオペロン)、ter(E.coliTrypA)、組み換え型第Xa因子阻害ポリペプチド(FxaI)を含み、テトラサイクリン耐性遺伝子(TetR)を宿主に与える。ヒヒウリカーゼ遺伝子をこのプラスミドに挿入し、抗生物質耐性遺伝子と交換した。プラスミドpMFOU18をNcoIで処理し、平滑化後、XhoIで処理し、1030bp長の断片を単離した。プラスミドpMFXT133はNdeIで処理した後、平滑化し、その後XhoIで処理し、大きな断片を単離した。この2断片をライゲーションしてヒヒウリカーゼ発現ベクターpURBA16を得た。
【0065】
ブタ−ヒヒキメラウリカーゼのサブクローニング
プラスミドpURBA16をApaIおよびAlwNIで処理し、2320bp長の断片を単離した。プラスミドpMFXT133をNdeIで処理し、平滑化し、次にAlwNIで処理し、620bp長の断片を単離した。プラスミドpET−3d−PBCをXbaIで処理し、平滑化し、次にApaIで処理し、得た710bp長の断片を単離した。この3つの断片をライゲーションし、pET−3dベクター由来の、osmBプロモータならびにrbsならびにT7rbsの制御下でPBCウリカーゼを発現するプラスミドpUR−PBを得た。
【0066】
T7rbsは追加的な工程で取り除いた。pUR−PBをNcoIで処理し、平滑化し、次にAlwNIで処理し、得た3000bp長の断片を単離した。プラスミドpMFXT133をNdeIで処理し、平滑化し、次にAlwNIで処理し、得た620bp長の断片を単離した。この2つの断片をライゲーションして、PBCをosmBプロモータの制御下で発現するpDUR−PBを得た。
【0067】
pOUR−PB−ΔNCの作成
ウリカーゼcDNAを何カ所か改変したところ、安定性が大幅に高まった組み換え体酵素を得ることができた。プラスミドpOUR−PBC−ΔNCは、invivoでペルオキシソームターゲティングシグナルとして機能する、C末端のトリペプチドとマチュレーションペプチドであるN末端の6残基とを取り除いて作成した。これは、プラスミドpDUR−PBのPBC配列と、表2に示した特異的なオリゴヌクレオチドプライマーを用いたPCR増幅反応によって行った。
表2.PBC−ΔNCウリカーゼのPCR増幅に用いたプライマー
【表2】
【0068】
プライマーの末端に導入した制限酵素切断部位は太字で、また翻訳されない領域は小文字で、それぞれ表2に示してある。NdeIはセンス方向であり、XbaIはアンチセンス方向である。アンチセンスプライマーは、アミノ酸配列に影響しない点変異(下線で示している)を導入することによって内部に存在するNdeIサイトを除去し、NdeIを用いたサブクローニングを可能にするためにも用いられた。
【0069】
pDUR−PBのPCR増幅反応で得た900塩基長の断片をNdeIおよびXbaIで切断し、単離した。得た断片を、次にE.coli由来のdeo−P1P2プロモータおよびrbsを含み、恒常的にヒト組み換え型インシュリン前駆体を発現するdeo発現プラスミドpDBAST−RAT−Nに挿入した。プラスミドをNdeIおよびXbaIで処理し、得た4035bp長の断片を単離し、PBC−ウリカーゼPCR産物とライゲーションした。よって得たpDUR−PB−ΔNCで、E.coliK−12SΦ733(F−cytRstrA)を形質転換したところ、活性を有する切断型ウリカーゼを多量に発現した。
【0070】
両端でトランケーションしたPBC−ΔNC配列は、osmBプロモータの制御下でも発現させた。プラスミドpDUR−PB−ΔNCをAlwNI−NdeIで処理し、得た3459bp長の断片を単離した。先に述べたプラスミドpMFXT133をNdeI−AlwNIで処理し、得た660bp長の断片を単離した。これらの断片をライゲーションし、pOUR−PB−ΔNCを得、これでE.coliK−12株W3110F−を形質転換したところ、活性を有する切断型ウリカーゼを多量に発現した。
【0071】
ウリカーゼ発現プラスミドpOUR−P−ΔN−ks−1の作成
このプラスミドは、組み換え型酵素の活性ならびに安定性を向上させるために作成した。Pig−KS−ΔNウリカーゼは、N末端のマチュレーションペプチド6残基が除去される形でN末端のみをトランケーションし(ΔN)、S46TならびにR291KならびにT301S変異を有するように作成した。46番目の位置には、PCR増幅反応ならびにクローニングの過程で発生した保存的変異によって、セリン残基の代わりにトレオニン残基が存在していた。291番目の位置ではアルギニンがリシンに、また301番目の位置では、トレオニンがセリンに置換されていた。この両方が、ヒヒウリカーゼ配列由来である。R291KならびにT301Sの両改変は先にKSと命名し、説明したとおりである。リシン残基を追加することで、潜在的なPEG化サイトが1つ追加されている。
【0072】
pOUR−P−ΔN−ks−1(図1)の作成には、プラスミドpOUR−PB−ΔNCをApaI−XbaIで処理し、得た3873bp長の断片を単離した。プラスミドpET−3d−PKS(構造を図4に示してある)をApaI−SpeIで処理し、得た270bp長の断片を単離した。SpeIによる5′CTAGでの切断の結果、XbaIによって生じたDNA断片と効率的にライゲーションする、5′CTAGの突出部が生じた。この2つの断片をライゲーションしてpOUR−P−ΔN−ks−1を得た。ライゲーションの結果、SpeIおよびXbaI認識サイトは失われた(図9に括弧で示してある)。プラスミドpOUR−P−ΔN−ks−1をE.coliK−12株W3110F−原栄養株ATCC#27325に導入した。osmBプロモータの制御下で発現させて得たPig−KS−ΔNウリカーゼにより、優れた活性ならびに安定性を有する組み換え型酵素を多量に得ることができた。
【0073】
図1はプラスミドpOUR−P−ΔN−ks−1の構造を示したものである。制限酵素サイトの横に示した番号は、1と設定したHaeIIサイトから数えた、ヌクレオチドの位置を示している。クローニングの過程で失われた制限酵素サイトは括弧で示してある。Pig−KS−ΔNウリカーゼをコードするプラスミドpOUR−P−ΔN−ks−1は4143塩基対(bp)長で、下記のエレメントを含む。
1. osmBプロモータおよびリボソーム結合サイト(rbs)を含む、1番目のヌクレオチドから、NdeIサイト(113番目の位置)に到る、113bp長のDNA断片。
2. Pig−KS−ΔN(アミノ末端がトランケーションされており、291番目および301番目のアミノ酸がそれぞれリシンおよびセリンで置換されているブタウリカーゼタンパク質の核酸配列)コーディング領域から900bp、ならびにpCR(登録商標)II由来の、TAクローニング部位から上流方向にSpeI/XbaI制限酵素サイトまでの同フランキング配列32bpを含む、NdeI(113番目の位置)からSpeI/XbaIジャンクション(第1045番目の位置)に到る932bp長のDNA断片。
3. SpeI/XbaIジャンクション(1045番目の位置)からHindIII(1070番目の位置)にわたる、25bpのマルチクローニングサイト(MCS)配列。
4. HindIII(1070番目の位置)およびAatII(1110番目の位置)を末端に有し、TrpA転写ターミネーター(ter)を含む40bpの合成オリゴヌクレオチド。
5. pBR322上の、AatII(1110番目の位置)からMscI/ScaI(2629番目の位置)にわたる、テトラサイクリン耐性遺伝子(TetR)を含む1519bp長のDNA断片。
6. DNA複製起点を含み、pBR322上のScaI(2629番目の位置)からHaeII(4143番目の位置)にわたる、1514bp長のDNA断片。
【0074】
図2は、Pig−KS−ΔNウリカーゼのDNA配列ならびに推定アミノ酸配列を示している。この図では、アミノ酸の位置を示す番号は全長ブタウリカーゼ配列に対応している。イニシエーターであるメチオニン残基に続いて、ブタウリカーゼ配列ではアスパラギン酸となっている位置に、代わりにトレオニンを挿入した。このトレオニン残基によって、バクテリアのアミノペプチダーゼによるメチオニンの除去が可能となっている。アミノ酸配列にある間隙は、削除されたN末端のマチュレーションペプチドを示している。異なるウリカーゼ配列のサブクローニングの様々な過程で用いた制限酵素サイト(ApaI、NdeI、BamHI、EcoRIおよびSpeI)が示してある。小文字で示した3′側の翻訳されない領域は、pCR(登録商標)IIに由来する配列である。翻訳停止コドンはアスタリスクで示してある。
【0075】
図3は様々な組み換え型ウリカーゼ配列のアミノ酸配列のアラインメントを示したものである。1行目は全長アミノ酸配列を含むブタウリカーゼの配列である。2行目は両端をトランケーションしたブタ−ヒヒキメラウリカーゼ(PBC−ΔNC)である。3行目はN末端側のみでトランケーションされており、変異S46Tおよびウリカーゼコーディング配列のカルボキシ末端のヒヒ由来物を反映するアミノ酸変異R291KおよびT301Sを有するPig−KS−ΔNウリカーゼ配列である。アスタリスクは、Pig−KS−ΔNと公開されているブタウリカーゼ配列とでアミノ酸配列に相違がある位置を示している。丸印は、Pig−KS−ΔNとブタ−ヒヒキメラPBC−ΔNとでアミノ酸配列に相違がある位置を示している。点線はアミノ酸の欠損を示している。
【0076】
Y97H変異を有するネィティブのヒヒ、ブタ、ウサギウリカーゼおよびブタ/ヒヒキメラ(PBC)のcDNAを、E.coliへのクローニングのために作成した。全クローンでosmBによって発現が制御されており、W3110F−E.coliであるように、変異型ウリカーゼを多量に発現しているクローンを作成して選択した。プラスミドDNAを単離し、DNAシーケンシングならびに制限酵素分析によって確認し、細胞を培養した。
【0077】
Pig−ΔNおよびPig−KS−ΔNを含む切断型ウリカーゼの作成は、PBC−ΔNCとPig−KSを、制限酵素ApaIおよびXbaI、ならびにApaIおよびSpeIでそれぞれ処理後、クロスライゲーションして行った。N末端の「マチュレーションペプチド」(1−2)6残基、ならびにC末端のトリペプチド「ペルオキシソームターゲティングシグナル」(3−5)は酵素活性に大きく影響する機能を有さないので、これらのトランケーションされた変異体は活性を保つものと思われ、また、これらの配列は免疫原性を有する可能性が考えられる。ウリカーゼを非常に多量に発現しているクローンを選択した。
【0078】
実施例2.発現プラスミドの、バクテリア宿主細胞への形質転換
発現プラスミドpOUR−P−ΔN−ks−1を、E.coliK−12株W3110F−に導入した。ルリア培地(LB)で対数増殖期中期にまで培養した形質転換用のバクテリア細胞を、遠心処理によって集菌し、冷水で洗浄後10%グリセロールに懸濁、さらに水に懸濁し、1mlあたり約3x1010細胞の最終濃度とした。細胞はアリコットとして−70℃で保存した。プラスミドDNAはエタノールで沈殿させ水に溶解した。
【0079】
バクテリア細胞とプラスミドDNAを混合し、BIO−RADからのGenePulser IIを用いた高電圧電気穿孔法(Trevors et al(1992)Electrotransformation of bacteria by plasmid DNA,in Guide to Electroporation and Electrofusion(D.C.Chang,B.M.Chassy,J.A.Saunders and A.E.Sowers,eds.),pp.265−290,Academic Press Inc.,San Diego、Hanahan et al(1991)Meth.Enzymol.,204,63−113)によって形質転換した。形質転換した細胞はSOC培地(2% トリプトン、0.5% 酵母抽出物、10 mM NaCl、2.5mM KCl、10mM MgCl2、10mM MgSO4、20mM グルコース)に懸濁し、37℃で1時間インキュベーションした後テトラサイクリン耐性を指標に選択した。高発現クローンを選択した。
【0080】
実施例3.組み換え型ウリカーゼの調製
形質転換したものなど(上記参照)のバクテリアは、グルコースを含有する培地で培養した。約37℃で、pHは7.2±0.2に保った。
【0081】
培養の終期5−6時間にかけて培地にKClを、最終濃度が0.3Mとなるように加えた。ウリカーゼの蓄積を生じさせるため、培養を続けた。
【0082】
組み換え型ウリカーゼはバクテリア細胞内で、封入体(IB)に類似した不溶性の沈殿物として蓄積した。細胞懸濁液を遠心処理で洗浄し、pH8.0の50mM Trisバッファーおよび10mM EDTAに懸濁し、最終体積を乾燥菌体重量の約40倍とした。
【0083】
細胞をライソザイムと高圧力によって破壊した後、遠心処理によって組み換え型ウリカーゼを含有するIBを単離した。ライソザイム処理(2000−3000units/ml)はpH8.0、7±3℃で16−20時間、混合しながら行った。ペレットを水で洗浄し、使用するまで−20℃で保存した。
【0084】
濃縮したIBは、pH10.3±0.1の50mM NaHCO3バッファーで懸濁後さらに処理した。懸濁液は一晩室温でインキュベーションし、IB由来のウリカーゼを可溶化させた後、遠心処理によって沈殿物を除いた。
【0085】
ウリカーゼはさらに、複数のクロマトグラフィー工程によって精製した。最初のクロマトグラフィーはQ−セファロースFFカラムを用いて行った。カラムにサンプルをロードした後、150mM NaClを含有する重炭酸バッファーで洗浄し、ウリカーゼの溶出は250mM NaClを含有する重炭酸バッファーによって行った。次に、キサンチン−アガロース樹脂(Sigma)によって少量の不純物をウリカーゼ調製物より除いた。Q−セファロースFF溶出物はpH10.3±0.1の50mMグリシンバッファーで希釈し、タンパク質最終濃度を約0.25mg/mlとしてロードした。カラムを100mM NaClを含有するpH10.3±0.1の重炭酸バッファーで洗浄し、同じバッファーに60μMキサンチンを加えたものでウリカーゼを溶出した。この段階で、ウリカーゼをQ−セファロースクロマトグラフィーで再精製し、凝集したものを除いた。
【0086】
それぞれのウリカーゼ調製物の純度を、サイズ排除クロマトグラフィーで測定したところ、95%以上であった。それぞれの調製物でSuperdex200カラムを用いて行った分析では、凝集体は0.5%以下しか検出されなかった。
【0087】
表3は25リットルの発酵培地から得たIB由来のPig−KSΔNウリカーゼの精製をまとめたものである。
表3.Pig−KSΔNウリカーゼの精製
【表3】
【0088】
実施例 4. 組み換え型ウリカーゼの性質
SDS−PAGE
高度に精製された変異型ウリカーゼのSDS−PAGE分析(図4)では、かなり特徴的なパターンが現れた。サンプルはpH10.3の炭酸バッファーに、4℃で最高数ヶ月保存した。ブタ、ブタ−KSならびにPBCの、全長変異型タンパク質では、分子量が20および15kDの顕著な分解産物の蓄積が見られた。これは、少なくとも1つのニックがウリカーゼサブユニット分子を開裂していることを示唆している。アミノ末端を短くしたクローンならびにウサギウリカーゼでは、これと異なった分解パターンが検出されたが、割合は小さかった。ウサギウリカーゼのアミノ末端は、短くしたクローンに類似している。精製並びに保管の過程で生じたウリカーゼ断片のアミノ末端配列を決定した。
【0089】
ペプチドシーケンシング
原体ウリカーゼ調製物のN末端を、エドマン分解法でシーケンシングした。サイクル数は10であった。組み換え型ブタウリカーゼ(全長クローン)はPig−KS−ΔNよりも多くの分解断片を産した。分解断片を生じる切断部位の予想位置は以下の通りである:
1)下記の配列を有する、168番目の位置の、メジャーサイト:
−−QSG ↓ FEGFI−−
2) 下記の配列を有する、142番目の位置の、マイナーサイト:
−−IRN ↓ GPPVI−−
【0090】
上記の配列は、既知のいかなるタンパク質分解性開裂も示唆しない。しかしながら、開裂はタンパク質分解またはなんらかの化学反応によって生じうる。アミノ末端切断型ウリカーゼは非アミノ末端切断型ウリカーゼに比べて驚くほど安定性が高い。PBC−ΔNCも、他のΔN分子と類似した、非アミノ切断型PBCよりも少ない安定性を有していた。
【0091】
効力
ウリカーゼ活性の測定はUV法によって行った。酵素反応速度は、尿酸のアラントインへの酸化による、292nm吸光度の減少を測定することによって求めた。1活性単位は、25℃、1分間で、示した特定の条件下で1μモルの尿酸を酸化するのに必要なウリカーゼの量と定義される。ウリカーゼの効力は、タンパク質1mgあたりの活性単位として表される(U/mg)。
【0092】
292nmにおける1mM尿酸の消衰係数は12.2mM−1cm−1である。ゆえに、反応溶液1mlあたりの、尿酸1μモルの酸化は、12.2mA292の吸光度の減少となる。経時的な吸光度の変化(毎分ΔA292)は、曲線の直線部より得た。
【0093】
タンパク質濃度は、改変ブラッドフォード法によって求めた(MacartandGerbaut(1982) Clin Chim Acta 122:93−101)。ウリカーゼの比活性度(効力)は、U/mlで表される活性を、mg/mlで表されるタンパク質濃度で割ることによって計算した。様々な組み換え型ウリカーゼの酵素活性結果を、表4にまとめた。市販の調製物の結果も、参考値としてこの表に示した。結果からは、ウリカーゼタンパク質のトランケーションがその酵素活性に大きな影響を与えないことが明らかである。
表4. 組み換え型およびネィティブ型のウリカーゼの、速度パラメータのまとめ
【表4】
【0094】
表4注釈:
(1) タンパク質濃度は、278nmにおける吸光度から、10mg/mlウリカーゼ溶液に対して消衰係数として11.3を用いて求めた(Mahler,1963)。
(2)1単位のウリカーゼ活性は、25℃、1分間で1μモルの尿酸をアラントインに酸化するのに必要な酵素の量と定義される。
(3)比活性度は、60μMに等しい基質濃度の下で行ったラインウィーバーバークプロットによって求めた。
(4)反応混合物は、以下のストック溶液を様々な組み合わせで混合して作成した。
pH9.2の100mMホウ酸ナトリウムバッファー
pH9.2の50mMホウ酸ナトリウムに溶解した300μM尿酸
pH9.2の50mMホウ酸ナトリウムに溶解した1mg/mlBSA
(5)Kcatは、Vmax(対応するラインウィーバーバークプロットから算出)を反応溶液のウリカーゼ濃度(リカーゼの四量体の分子量に基づいてモル量として表現したもの)で割ることで求めた。
【0095】
実施例5.ウリカーゼとm−PEGの結合(PEG化)
Pig−KS−ΔNウリカーゼは、m−PEG−NPC(モノメトキシ−ポリ(エチレングリコール)−ニトロフェニルカーボネート)と結合体を形成させた。1ウリカーゼサブユニットあたり5、10または20kDのPEG鎖2から12本が結合する条件を確立した。m−PEG−NPCは、徐々にタンパク質溶液に加えた。PEGの添加が終わった後、ウリカーゼ/m−PEG−NPC反応溶液を2から8℃で16から18時間、未結合のm−PEG鎖が最大限ウリカーゼに結合するようインキュベーションした。
【0096】
PEG−ウリカーゼモノマー1つあたりのPEG鎖の数は、PEGおよびウリカーゼ標準試料を用いて、Superose6サイズ排除クロマトグラフィー(SEC)によって同定した。サブユニット1つあたりに結合したPEGの数は、下記の式によって算出した:
【数1】
【0097】
紫外線(UV)および屈折率(RI)検出器の配列(Kunitani,et al.,1991の開発による)を用いて、サイズ排除クロマトグラフィー(SEC)によってPEG−ウリカーゼサンプルにおける、PEG並びにタンパク質成分濃度を同定した。3つの較正曲線を作成した:タンパク質曲線(220nmで測定した吸光度)、タンパク質曲線(RIの測定による)、PEG曲線(RIの測定による)。次に、PEG−ウリカーゼサンプルを同様のシステムで測定した。得られた試験サンプルのUVならびにRIピークエリア値を、較正曲線に対するPEGならびにタンパク質の濃度を計算するのに用いた。3.42は、10kD PEGに対するウリカーゼ分子モノマーの分子量(34,192ダルトン)の比である。
【0098】
結合させたPEGは、生理的pHの溶液においてウリカーゼの可溶性を向上させた。表5はPEG化Pig−KS−ΔNウリカーゼ産生物のバッチ間のばらつきを示したものである。一般に、結合したPEG鎖数と酵素が保持する比活性度(SA)との間には、反比例の関係がある。
表5.PEG化Pig−KS−ΔNウリカーゼ結合体の酵素活性
【表5】
【0099】
実施例6.1000Dならびに100,000DのPEGによるウリカーゼのPEG化
Pig−KS−ΔNウリカーゼを、実施例5に記載の通りの方法で、1000Dならびに100,000Dのm−PEG−NPCを用いてPEG化した。1ウリカーゼサブユニットあたりPEG鎖2〜11本が結合する条件を用いた。PEGの添加が終わった後、ウリカーゼ/m−PEG−NPC反応溶液を2から8℃で16から18時間、未結合のm−PEG鎖が最大限ウリカーゼに結合するようインキュベーションした。
【0100】
PEG−ウリカーゼモノマー1つあたりのPEG鎖数は、上記に記載の方法で求めた。
【0101】
結合させたPEGは、生理的pHの溶液においてウリカーゼの可溶性を向上させた。
【0102】
実施例7.PEGと結合させたPig−KS−ΔNウリカーゼの薬物動態
治療上の利益を提供するために、PEG化の最適な程度ならびにサイズを決定するために、生物学的な実験を行った。
【0103】
ラットで、体重1kgあたり0.4mg(2U)の改変されていないウリカーゼを、1日目ならびに8日目に静脈投与して行った薬物動態試験では、循環系半減期が約10分であるとの結果が得られた。しかしながら、ラットで2−11x10kDのPEG−Pig−KS−ΔNウリカーゼを、週1回、9回に渡って注射してクリアランス速度を調べた試験では、クリアランスはPEG鎖の数に(この範囲においては)依存していないことが示唆され、試験期間を通じて比較的同じ値に保たれた(表6参照、半減期が約30時間)。各週間の違いは誤差の範囲内である。10x5kD PEGならびに10x20kD PEG−ウリカーゼ結合体の9回の注射後でも、同様のパターンが観察された。結果は、この範囲においては、ウリカーゼのPEG化の程度にかかわらず、ラットモデルにおいて類似した生理的影響が観察されたことを示している。
表6.PEG化Pig−KS−ΔNウリカーゼ調製物の、ラットにおける半減期
【表6】
【0104】
表6注釈: 結果は時間±標準誤差で示している。括弧で示した数字は試験した実験動物個体数を示している。
【0105】
ラットに、1週間に1回、体重1kgあたり0.4mgの、表に示したとおりの改変を施したPig−KS−ΔNウリカーゼを静脈投与した。それぞれの個体群は元々15個体のラットから成っており、5個体のサブグループで交互に採血した。ラット数例が、麻酔によって実験中に死亡した。半減期は、注射5分後、および6、24、48時間後に採取した血漿サンプルのウリカーゼ活性(比色測定)を測定することによって同定した。
【0106】
表5に、実験に用いられたPEG化ウリカーゼを記載している。
【0107】
6x5kD PEG−Pig−KS−ΔNウリカーゼを用いた、ウサギでの生物学的利用率試験によると、最初の注射後では、循環系半減期は98.2±1.8時間(i.v.)であり、筋肉投与(i.m.)ならびに皮下投与(s.c.)では、それぞれ71%、52%であった。しかしながら、2回目のi.m.ならびにs.c.注射の後、全てのウサギで相当量の抗ウリカーゼ抗体タイターが検出され、以降の注射でクリアランスが促進された。ラットに同じ結合体を注射した実験では、26±1.6時間の半減期(i.v.)が得られ、生物学的利用率はi.m.ならびにs.c.注射についてそれぞれ33%、22%であった。
【0108】
9x10kD PEG−Pig−KS−ΔNウリカーゼを用いたラットでの試験では、1回目の注射後の循環系半減期が42.4時間(i.v.)であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ28.9%、14.5%だった。(図5ならびに表7参照)。4回目の注射後では、循環系半減期は32.1±2.4時間であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ26.1%、14.9%だった。
【0109】
9x10kD PEG−Pig−KS−ΔNウリカーゼを用いた、ウサギでの同様の薬物動態試験では、この結合体の注射に引き続くクリアランスの促進は観察されなかった(隔週で4回注射として投与した)。これらの個体では、最初の注射後の循環系半減期は88.5時間(i.v.)であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ98.3%、84.4%であった。(図6ならびに表7参照)。4回目の注射後の循環系半減期は141.1±15.4時間であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ85%、83%であった。
【0110】
9x10k DPEG−Pig−KS−ΔNを用いた同様の試験によって、ビーグル犬における生物学的利用率を測定した(それぞれの群にオス2個体、雌2個体)。1回目のi.v.注射後で、70±11.7時間の循環系半減期が記録され、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ69.5%、50.4%だった(図6ならびに表7参照)。
【0111】
9x10kD PEG−Pig−KS−ΔN調製物を用いて、ブタで試験を行った。1つの群あたり3個体にi.v.、s.c.ならびにi.m.ルートによる投与を行った。1回目のi.v.注射後で、178±24時間の循環系半減期が記録され、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ71.6%、76.8%であった(図8ならびに表7参照)。
表7.9x10kDPEG−Pig−KS−ΔNウリカーゼを用いた薬物
【表7】
【0112】
9x10kD PEG−Pig−KS−ΔNウリカーゼの、ボルトン&ハンター法による、125Iでのヨウ素化に引き続いて、吸収、分布、代謝、排泄(ADME)試験を実施した。ラベルした結合体を、それぞれ4個体のラット(雄2個体と雌2個体)からなる7群に注射した。放射線の分布を、1時間後ならびに、7日間にわたって24時間毎に(5日目を除く)測定した。それぞれの群を順番に屠殺し、異なる臓器を摘出して分析した。7番目の群は代謝ケージで飼育し、尿と便を回収した。物質の、動物体内における分布は、それぞれの臓器の総放射活性ならびに、TCAによる沈殿が可能なカウント(腎臓、肝臓、肺、脾臓)のフラクション(すなわち、臓器のサイズで標準化した結合したタンパク質)を測定することによって評価した。摘出した臓器で、他の臓器よりも比放射能が高いものはなかった。すなわち、例えば肝臓または腎臓における特筆すべき蓄積は見られなかった。放射活性の70%が、7日目までに排出された。
【0113】
実施例8.治験結果
従来の治療法に不応答性または不耐性の、ヒト高尿酸血症または重篤な痛風に罹患している患者における、PEG−ウリカーゼ(Puricase(登録商標),Savient Pharmaceuticals)の尿酸応答ならびに、薬物動態的および安全性プロフィールを評価するために無作為化非盲検多施設並行群間試を実施した。平均罹患期間は14年で、被験者集団の70%が1つまたはそれ以上の痛風結節を有していた。
【0114】
治験では、41例の患者(平均年齢58.1歳)を無作為に、次に示す4つの12週間にわたる、静脈投与によるPEG−ウリカーゼ治療投与量計画に振り分けた:2週間毎に1回4mg(7例)、2週間毎に1回8mg(8例)、4週間毎に1回8mg(13例)、4週間毎に1回12mg(13例)。血漿ウリカーゼ活性ならびに尿酸レベルを定められた時間間隔で測定した。薬物動態学的パラメータ、平均血漿尿酸濃度および血漿尿酸レベルが6mg/dL以下であった時間の百分率を、ウリカーゼ活性並びに尿酸レベルの分析によって求めた。
【0115】
PEG−ウリカーゼを2週間毎に1回8mg投与した群で、最も大きなPUAの低減が見られ、治療期間の92%で6mg/dL未満のレベルであった。(12週間で、平均血漿尿酸濃度が治療前の9.1mg/dLに対して、治療後は1.4mg/dL)。
【0116】
他の投与量を用いたPEG−ウリカーゼ治療群でも、顕著で持続的な低い血漿尿酸レベルが観察された:4週間毎に1回8mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の86%(12週間で、平均血漿尿酸濃度が治療前の9.1mg/dLに対して、治療後は2.6mg/dL)、4週間毎に1回12mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の84%(12週間で、平均血漿尿酸濃度が治療前の8.5mg/dLに対して、治療後は2.6mg/dL)、2週間毎に1回4mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の73%(12週間で、平均血漿尿酸濃度が治療前の7.6mg/dLに対して、治療後は4.2mg/dL)。
【0117】
最初のPEG−ウリカーゼ投与から24時間以内での、血漿中尿酸の基線レベルからの減少割合の最大値は4mg/2週間の群で72%(p=.0002)、8mg/2週間の群で94%(p<.0001)、8mg/4週間の群で87%(p<.0001)、12mg/4週間の群で93%(p<.0001)であった。
【0118】
12週間の治療期間における血漿中尿酸の基線レベルからの減少割合は4mg/2週間の群で38%(p=.0002)、8mg/2週間の群で86%(p<.0001)、8mg/4週間の群で58%(p<.0003)、12mg/4週間の群で67%(p<.0001)であった。
【0119】
驚くべき事に、PEG−ウリカーゼの投与を受けた患者の一部で注入に関連した有害事象、例えば注入反応が出現した。この反応はすべての注入のうち14%で出現した。
【0120】
本出願に引用される全ての文献を、各個々の刊行物、特許または特許出願を具体的かつ個々に、参照することによって援用すると示したのと同程度に、全ての目的のためにその全文を参照することによって本明細書に含める。
【0121】
当該分野に精通する者に理解されるように、本発明にあっては、その思想および範囲から逸脱することなく、様々な変更ならびに形態様が可能である。ここに記載された具体的な実施例は単に例を示すのみであって、本発明は添付した特許請求項、ならびに、このような請求に権利が与えられるあらゆる等価例によってのみ制限されるものである。
【技術分野】
【0001】
関連文献の相互参照
本出願は、参照によりここに援用されるところの、2005年4月11日出願の米国特許仮出願番号第60/670,541号の優先権並びに利益を請求するものである。
【0002】
技術分野
本発明は、尿酸分解活性を有する遺伝的に改変されたタンパク質に関する。より具体的には切断型尿酸オキシダーゼを含むタンパク質および、その製造法に関する。
【0003】
背景技術
以下、「尿酸オキシダーゼ」と「ウリカーゼ」を同義語として用いる。尿酸オキシダーゼ(ウリカーゼ、E.C.1.7.3.3)は、酸化を触媒する事によって尿酸を、より可溶性が高く、より容易に排泄されるプリン代謝物質、アラントインへ転換する酵素である。ヒトは、高等霊長類の進化の過程で蓄積したウリカーゼ遺伝子へのいくつかの変異により、酵素活性を有するウリカーゼを生産しない。Wu,X,et al.,(1992)J Mol Evol 34:78−84を参照することにより、その全文を本明細書に含む。この結果、感受性個体では、血中での過剰濃度の尿酸(高尿酸血症)が疼痛性関節炎(痛風)、関節破壊を伴う尿酸塩沈着(痛風結節)および腎不全を引き起こすことがある。アロプリノール(尿酸合成阻害剤)などの薬剤が利用可能であるが、一部の罹患個体では、治療を限定する有害作用を生じるか、またはこれらの症状を適切に軽減できない場合がある。Hande,KR,et al.,(1984)Am J Med 76:47−56、Fam,AG,(1990) Bailliere’s Clin Rheumatol 4:177−192を、それぞれ参照することによってその全文を本明細書に含む。ウリカーゼの注入は、高尿酸血症および高尿酸尿症を少なくとも一次的に低減し得る。しかしながら、ウリカーゼはヒトにおいては外来タンパク質であるため、Aspergillus flavus由来の非修飾タンパク質の一次注射でさえ、処置患者の数パーセントにおいてアナフィラキシー反応を誘導し(Pui,C−H,et al.,(1997)Leukemia 11:1813−1816を、参照することによってその全文を本明細書に含む)、免疫応答は慢性的または間欠的治療における有用性を限定する(Donadio,D,et al.,(1981) Nouv Presse Med 10:711−712、Leaustic,M,et al.,(1983) Rev Rhum Mal Osteoartic 50:553−554をそれぞれ参照することによって全文を本明細書に含む)。
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明は、トランケートされ、構造安定性が向上した変異型組み換えウリカーゼタンパク質に関する。
【課題を解決するための手段】
【0005】
本発明は新規の組み換えウリカーゼタンパク質を提供する。本発明のウリカーゼはトランケートされており、天然のウリカーゼタンパク質に対して、変異したアミノ酸を有する。
【0006】
本発明はSEQ ID NO.8のアミノ酸配列を含む変異型組み換えウリカーゼを提供する。さらにSEQ ID NO.13のアミノ酸配列を含む変異型組み換えウリカーゼも提供する。
【0007】
本発明のさらなる目的は、尿酸分解活性を有する新規の組み換えウリカーゼタンパク質を含む、尿酸を代謝する手段を提供することである。尿酸分解活性とは、本明細書では酵素による尿酸のアラントインへの変換を指す。
【0008】
特定の一実施形態にあっては、ウリカーゼは、SEQ ID NO.7またはSEQ ID NO.12の位置8から位置287のアミノ酸配列を含む。さらにSEQ ID NO.8またはSEQ ID NO.13のアミノ酸配列を含むウリカーゼが提供される。一実施形態にあっては、ウリカーゼはアミノ末端アミノ酸を含み、該アミノ末端アミノ酸はアラニン、グリシン、プロリン、セリンまたはトレオニンである。特定の一実施形態にあっては、アミノ末端アミノ酸はメチオニンである。
【0009】
本発明は、本発明のウリカーゼをコードする核酸配列を含む、単離された核酸も提供する。一実施形態にあっては、該ウリカーゼをコードする核酸は動作可能なように異種性のプロモータ、例えばosmBプロモータに連結されている。さらに、ウリカーゼをエンコードする核酸を含む核酸ベクター、ならびにこのようなベクターを含む宿主細胞も提供される。さらに一実施形態にあっては、核酸はSEQ ID NO.7の配列を有する。
【0010】
このような宿主細胞を、核酸配列が宿主細胞によって発現されるような条件下で培養し、発現されたウリカーゼを単離する工程を含む、ウリカーゼを生成する方法も、提供される。
【図面の簡単な説明】
【0011】
【図1】図1はプラスミドpOUR−P−ΔN−ks−1の構造を示している。制限酵素サイトの横に示された番号は、1で示したHaeIIサイトから数えた、ヌクレオチドの位置を示している。クローニングの過程で失われた制限酵素サイトは、括弧付きで示している。
【0012】
【図2】図2は、Pig−KS−ΔNウリカーゼのDNA配列、および推定されるアミノ酸配列を示したものである(それぞれSEQ ID NO.9およびSEQ ID NO.7)。図2のアミノ酸番号は、全長ブタウリカーゼに対応している。イニシエーターであるメチオニン残基に続いて、ブタウリカーゼ配列のアスパラギン酸7がトレオニンに置換されている。サブクローニングの様々な過程で使用した制限酵素サイトが、示されている。3′側の翻訳されない配列は小文字で示されている。翻訳終止コドンは、アスタリスクで示してある。
【0013】
【図3】図3は様々な組み換えブタ(SEQ ID NO.11)、PBC−ΔNC (SEQ ID NO.12)およびPig−KS−ΔN(SEQ ID NO.7)ウリカーゼ配列のアラインメントを示している。Pig−KS−ΔNで、公開されているブタウリカーゼ配列とアミノ酸の相違がある位置をアスタリスクで示している。丸印は、Pig−KS−ΔNとPBC−ΔNでアミノ酸の相違がある位置を示している。点線は、アミノ酸の欠失を示している。
【0014】
【図4】図4はブタウリカーゼおよび、高度に精製した、実施例1から3に従って作成した変異型ウリカーゼのSDS−PAGEを示している。生成日(月/年)およびそれぞれのサンプルに対応するレーン番号は以下に示してある。Y軸は分子量マーカーの分子量でラベルされており、図の上部にレーン番号を示してある。レーン番号は以下の通りである:レーン1−分子量マーカー、レーン2−PigKS−ΔN(7/98)、レーン3−Pig(9/98)、レーン4−PigKS(6/99)、レーン5−PigKS(6/99)、レーン6−Pig−ΔN(6/99)、レーン7−PigKS−ΔN(7/99)、レーン8−PigKS−ΔN(8/99)。
【0015】
【図5】図5は血液サンプルでの酵素活性を測定することによって求めた、ラットでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与)、SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0016】
【図6】図6は血液サンプルでの酵素活性を測定することによって求めた、ウサギでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0017】
【図7】図7は血液サンプルでの酵素活性を測定することによって求めた、イヌでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【0018】
【図8】図8は血液サンプルでの酵素活性を測定することによって求めた、ブタでのPEG化された(9x10kD)Pig−KS−ΔNウリカーゼのIM(筋肉内投与),SC(皮下投与)およびIV(静脈内投与)注射後の薬物動態プロファイルを示している。示した時間間隔で採取した血漿サンプルにおけるウリカーゼ活性は比色分析によって求めた。活性の値(mAU=ミリ吸光度単位)は、血漿サンプル1μlあたりの酵素反応速度を表している。注射されたウリカーゼの生物学的利用率(IV注射の内、循環系に到達する薬剤の量)はグラフの曲線の下のエリアから計算した。
【発明を実施するための形態】
【0019】
これまでの研究は、PEG化によってウリカーゼの免疫原性および/または抗原性の大幅な低減が達成された場合は、必ず尿酸分解活性も大幅に失われてしまうことを示している。生物薬剤の安全性、利便性および対費用効果は、効力が失われ、また従って投与量を上げなければならないことによって悪影響を受ける。従って、血液を含む体液において、上昇した尿酸レベルを下げる安全で効果的な代替的な方法が必要とされている。本発明はSEQ ID NO.7、SEQ ID NO.8、SEQ ID NO.12またはSEQ ID NO.13のアミノ酸配列を含む変異型組み換えウリカーゼを提供する。
【0020】
特に記載のない限り、本明細書では用語「ウリカーゼ」を、個々のサブユニットならびにその四量体両方を指すものとして用いる。
【0021】
本明細書では、用語「変異型ウリカーゼ」を、他のアミノ酸に置換されたアミノ酸を有するウリカーゼ分子を指す語として用いる。
【0022】
特定の実施態様では、本発明のウリカーゼはアミノ末端メチオニンを有する。特定の実施態様では、アミノ末端メチオニンはウリカーゼが産生された後に除去される。特定の実施態様では、該メチオニンは内在するバクテリアのアミノペプチダーゼによって除去される。N末端のメチオニンの最も完全な除去を可能にするアミノ酸は、アラニン、グリシン、プロリン、セリンおよびトレオニンである。特定の一実施形態にあっては、ウリカーゼはアミノ末端に2つのアミノ酸を有し、この2つのアミノ酸は一方がメチオニンであって、これに引き続いてアラニン、グリシン、プロリン、セリンおよびトレオニンからなる群から選んだアミノ酸1つが引き続くものである。
【0023】
本発明はウリカーゼをコードする核酸配列を提供する。
【0024】
本発明は、当該核酸配列を含むベクターを提供する。
【0025】
特定の一実施形態にあっては、ウリカーゼは単離される。特定の一実施形態にあっては、ウリカーゼは精製される。特定の実施形態にあっては、ウリカーゼは単離され精製される。
【0026】
本発明はウリカーゼをコードする核酸配列を含むベクターを含む宿主細胞を提供する。
【0027】
本発明はPCR(ポリメラーゼ連鎖反応法)技術による、ウリカーゼをコードする核酸配列の改変を含む、ウリカーゼをコードする核酸配列を作成する方法を提供する。当該業務に精通する者には目的の核酸配列がPCRによって、増幅する標的DNAの領域に相補的な合成オリゴヌクレオチドプライマー(それぞれの鎖に1つずつ)を介して、調製できることが既知である。プライマーを、過剰量のデオキシヌクレオチドおよび、耐熱性DNAポリメーラーゼであるTaqポリメラーゼの存在下で標的DNA(純粋なものでなくてよい)に加える。標的DNAは、連続する温度サイクル(典型的な場合で、30回)を経る中で反復して変性(約90℃)、プライマーとのアニーリング(典型的な場合で、50から60℃)、プライマーからの娘鎖の伸長(72℃)を繰り返す。次のサイクルからは娘鎖もテンプレートとして働くので、両方のプライマーとマッチするDNA断片は線形にではなく、指数関数的に増幅される。
【0028】
本発明は、変異型組み換えウリカーゼを生成する方法を提供するものであって、宿主細胞のベクターによる形質転換、宿主細胞によるウリカーゼの発現、変異型組み換えウリカーゼの宿主細胞からの単離、精製された変異型組み換えウリカーゼの、例えばクロマトグラフィー技術による単離、および変異型組み換えウリカーゼの精製を含む。例えば、ウリカーゼは参照することによってその全文を本明細書に含む、国際公開第00/08196号パンフレット、および米国特許出願番号60/095,489に記載の方法によって作成することができる。
【0029】
好ましい一実施形態にあっては、変異型組み換えウリカーゼの発現を引き起こすよう、宿主細胞に処置を施す。当該業務に精通する者には、細胞のベクターによる形質転換は通常カルシウムイオンと沈殿させたDNAを用いて達成されるが、他にも様々な方法(例えば電気穿孔法)を用いることができることを理解している。
【0030】
ウリカーゼは、当該業務に精通した者に既知であるあらゆる方法によって単離および/または精製されうる。一般に、本発明で発現させたポリペプチドは実質的に純粋な形で単離される。好ましくは、ポリペプチドは重量で少なくとも80%の純度で単離される。より好ましくは、重量で少なくとも95%の純度、そして最も好ましくは、少なくとも99%の純度で単離される。一般にこのような精製は、例えば硫安分画法、SDS−PAGE電気泳動法およびアフィニティークロマトグラフィーの標準的な技法を用いて達成されうる。ウリカーゼは、好ましくは陽イオン界面活性剤、例えば、参照することによってその全文を本明細書に含む、2005年4月11日出願の同時係属出願、代理人整理番号第103864.146644号、タイトルが陽イオン界面活性剤によるタンパク質の精製である、米国特許仮出願番号第60/670,520号に記載の方法に従って塩化セチルピリジニウム(CPC)によって単離される。
【0031】
本発明の一実施形態にあっては、ベクターは浸透圧感受性プロモータの制御下にある。プロモータとは、RNAポリメラーゼが、DNAのRNAへの転写プロセスを開始するに先立って結合する、DNA領域のことを言う。浸透圧感受性プロモータは、細胞が浸透圧の上昇を感知した場合に転写を開始する。
【0032】
本発明のウリカーゼはポリマーと結合したウリカーゼ、たとえばポリエチレングリコールと結合したウリカーゼ、すなわち、PEG化されたウリカーゼを含む。
【0033】
本発明の一実施形態にあっては、ウリカーゼを含む医薬組成物が提供される。一実施形態にあっては、組成物はウリカーゼ溶液である。好ましい一実施形態にあっては、溶液は滅菌済みであり、注射に適している。一実施形態にあっては、このような組成物はウリカーゼのリン酸緩衝生理食塩水溶液を含む。一実施形態にあっては、組成物はバイアルで提供され、さらに、任意にラバー注射ストッパーを備えることができる。特定の実施形態にあっては、組成物は、ウリカーゼを溶液1ミリリットルあたり2から16ミリグラムの濃度、溶液1ミリリットルあたり4から12ミリグラムの濃度、溶液1ミリリットルあたり6から10ミリグラムの濃度で含む。好ましい一実施形態にあっては、組成物はウリカーゼを1ミリリットルあたり8ミリグラムの濃度で含む。好ましくは、ウリカーゼの重量は、タンパク質重量に対して測定される。
【0034】
本発明の組成物の効果的な投与計画は、当該業務に精通した者によって決定されうる。任意の投与計画の、有効性の評価に適した指標は、当該業務に精通した者に既知である。このような指標の例としては、血漿尿酸レベル(PUA)の正常化または低下、ならびにPUAを6.8mg/dL以下、好ましくは6mg/dL以下に低下または維持させることが挙げられる。好ましい一実施形態にあっては、本発明の組成物による治療の対象者は、総治療期間の70%以上、80%以上、または90%以上の期間、PUAが6mg/ml以下である。例えば治療期間が24週間である場合、対象者のPUAは24週間の治療期間の80%以上の間、すなわち、少なくとも134.4日(24週間x7日間/週間x0.8=134.4日)に等しい時間の間、6mg/ml以下である。
【0035】
特定の実施形態にあっては、溶液としてウリカーゼ0.5から24mgが2から4週に1回投与される。ウリカーゼは、当該業務に精通した者に既知のあらゆる適切な方法、例えば静脈内投与、筋肉内投与、皮下投与によって投与することができる。好ましくは、投与が静脈内投与による場合は、ウリカーゼとして0.5mgから12mgが投与される。好ましくは、投与が皮下投与による場合は、ウリカーゼとして4mgから24mgが投与される。好ましい一実施形態にあっては、ウリカーゼは静脈点滴によって、30から240分の点滴時間で投与される。一実施形態にあっては、2週間に1回、ウリカーゼとして8mgが投与される。特定の実施形態にあっては、100から500mLの生理食塩水を用いて点滴を行うことができる。好ましい一実施形態にあっては、2週間に1回または4週間に1回、点滴時間120分で、溶液として8mgのウリカーゼが投与される。好ましくは、点滴に用いるウリカーゼは250mLの生理食塩水に溶解される。特定の実施形態にあっては、ウリカーゼの投与は3ヶ月間、6ヶ月間、8ヶ月間または12ヶ月間の治療期間にわたって行う。他の実施形態にあっては、治療期間は12週間、24週間、36週間または48週間である。特定の一実施形態にあっては、治療期間は長期間にわたるものであって、例えば2年以上、治療対象者の全生存期間にわたる。加えて、複数の治療期間を、治療休止期間ではさむことも可能である。例えば、6ヶ月間の治療に引き続いて3ヶ月間の治療休止期間をおき、その後再び6ヶ月間治療を行うことなどが、可能である。
【0036】
特定の実施形態にあっては、ウリカーゼの投与による注入反応の出現を阻止または低減するために、予防的に抗炎症性物質を投与することができる。一実施形態にあっては、少なくとも1つのコルチコステロイド、少なくとも1つの抗ヒスタミン薬、少なくとも1つのNSAID、またはこれらの組み合わせが、よって投与される。有用なコルチコステロイドには、ベータメタゾン、ブデソニド、コルチゾン、デキサメタゾン、ヒドロコルチゾン、メチルプレドニソロン、プレドニソロン、プレドニゾンおよびトリアムシノロンが含まれる。有用なNSAIDにはイブプロフェン、インドメタシン、ナプロキセン、アスピリン、アセトアミノフェン、セレコキシブおよびバルデコキシブが含まれる。有用な抗ヒスタミン薬には、アザタジン、ブロムフェニラミン、セチリジン、クロルフェニラミン、クレマスチン、シプロヘプタジン、デスロラタジン、デクスクロルフェニラミン、ジメンヒドリナート、ジフェンヒドラミン、ドキシルアミン、フェキソフェナジン、ヒドロキシジン、ロラタジンおよびフェニンダミンが含まれる。
【0037】
好ましい一実施形態にあっては、抗ヒスタミン薬はフェキソフェナジンであり、NSAIDはアセトアミノフェンであり、コルチコステロイドはヒドロコルチゾンおよび/またはプレドニゾンである。好ましくは、3つ全ての組み合わせ(必ずしも同時にではない)を、ウリカーゼ溶液の注入投与の前に投与する。好ましい一実施形態にあっては、NSAIDと抗ヒスタミン薬はウリカーゼ注入の1から4時間前に経口投与される。フェキソフェナジンの適切な用量は約30から約180mg、約40から約150mg、約50から約120mg、約60から約90mg、約60mgを含み、好ましくは60mgである。アセトアミノフェンの適切な用量は約500から約1500mg、約700から約1200mg、約800から約1100mg、約1000mgを含み、好ましくは1000mgである。ヒドロコルチゾンの適切な用量は約100から約500mg、約150から約300mg、約200mgを含み、好ましくは200mgである。一実施形態にあっては、抗ヒスタミン薬はジフェンヒドラミンではない。他の実施形態にあっては、NSAIDはアセトアミノフェンではない。好ましい一実施形態にあっては、フェキソフェナジン60mgをウリカーゼ注入の前夜に経口投与し、フェキソフェナジン60mgおよびアセトアミノフェン1000mgを次の朝に経口投与し、最後に、ウリカーゼ注入の直前にヒドロコルチゾン200mgを投与する。一実施形態にあっては、プレドニゾンをウリカーゼ投与の前日に、好ましくは夕方、投与する。プレドニゾンの適切な用量は5から50mgを含み、好ましくは20mgである。特定の実施形態にあっては、注入反応の出現を阻止または低減させるためのこのような予防措置は、PEG化ウリカーゼおよび非PEG化ウリカーゼを含むウリカーゼ投与を、受けているかまたは受ける予定の治療対象者に対して講じられる。特定の実施形態にあっては、このような予防措置はウリカーゼ以外の、PEG化治療薬ペプチドおよび非PEG化治療薬ペプチドを含む、治療薬ペプチド投与を受けているかまたは受ける予定の対象者に対して講じられる。
【0038】
本発明の一実施形態にあっては、医薬組成物はポリマーと結合されたウリカーゼを含有するものであって、修飾されたウリカーゼは尿酸分解活性を保持している。好ましい実施態様においては、ウリカーゼはPEG化されたウリカーゼである。
【0039】
本発明の一実施形態にあっては、医薬組成物はポリマーとの結合によって修飾されたウリカーゼを含有するものであって、修飾されたウリカーゼは尿酸分解活性を保持している。特定の一実施形態にあっては、ポリマー−ウリカーゼ結合体は参照によってその全文を本明細書に含める、国際公開第01/59078号パンフレットおよび米国特許出願番号第09/501730号に記載されている方法に従って調製される。
【0040】
本発明の一実施形態にあっては、ポリマーはポリエチレングリコール、デキストラン、ポリプロピレングリコール、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロース、ポリビニールピロリドンおよびポリビニールアルコールを含む群から選択される。好ましい実施態様においては、ポリマーはポリエチレングリコールであり、ウリカーゼはPEG化されたウリカーゼである。
【0041】
本発明の一実施形態にあっては、組成物は2−12のポリマー分子をそれぞれのウリカーゼサブユニットに含有し、好ましくは、3から10ポリマー分子をそれぞれのウリカーゼサブユニットに含有する。本発明の一実施形態にあっては、それぞれのポリマー分子は約1kDから約100kDの分子量を有する。
【0042】
本発明の他の実施形態にあっては、それぞれのポリマー分子は約1kDから約50kDの分子量を有する。本発明の好ましい一実施形態にあっては、それぞれのポリマー分子は約5kDから約20kD、約8kDから約15kD、約10kDから12kDの分子量を有し、好ましくは、約10kDの分子量を有する。好ましい一実施形態にあっては、それぞれのポリマー分子は約5kDまたは約20kDの分子量を有する。特に好ましい本発明の一実施形態にあっては、それぞれのポリマー分子は10kDの分子量を有する。
【0043】
本発明の実施態様においては、組成物は、組成物の反復投与に適している。
【0044】
本発明は当該ウリカーゼを用いて尿酸を代謝する方法を提供する。
【0045】
本発明は、ウリカーゼ組成物の、体液中尿酸レベルの低減への利用を提供する。
【0046】
本発明の一実施形態にあっては、ウリカーゼ組成物は血液を含む体液における尿酸の低減に用いられる。
【0047】
また、本発明のウリカーゼをコードする新規核酸分子も提供される。これらを生産する技法は、当該業務に精通した者によく知られている。例えば、ウリカーゼ核酸配列は当該業界に既知である、多くのあらゆる戦略によって改変されうる(Maniatis,T.,1990,Molecular Cloning,A Laboratory Manual,2d ed.,Cold Spring Harbor Laboratory,Cold Spring Harbor,N.Y.)。配列は、適切な場所で1つまたはそれ以上の制限エンドヌクレアーゼによって切断でき、必要であればさらに酵素による改変を施し、単離し、in vitroでライゲーションすることができる。ウリカーゼをコードする遺伝子の作成にあっては、改変された遺伝子が翻訳停止シグナルに遮られることなく、適切な翻訳読み枠を保持するよう注意すべきである。
【0048】
ウリカーゼタンパク質をコードする核酸配列は適切な発現ベクター、すなわち、挿入されたタンパク質コーディング配列の転写および翻訳に必要なエレメントを含むベクターに、挿入することができる。タンパク質コーディング配列を発現させるためには、多様な宿主ベクター系を用いることができる。これらはウィルス(例えばワクシニアウィルス、アデノウィルス等)を感染させた哺乳類細胞系、ウィルス(例えばバキュロウィルス)を感染させた昆虫細胞系、酵母ベクターを含む酵母などの微生物、バクテリオファージDNA、プラスミドDNA、コスミドDNAなどで形質転換したバクテリアを含むが、これに限定されない。これらのベクターの発現エレメントはそれらの強度と特異性において異なる。用いる宿主ベクター系に応じて、あらゆる適切な転写および翻訳エレメントを用いることができる。
【0049】
適切な転写/翻訳制御シグナルと、タンパクをコードする配列からなるキメラ遺伝子を含んだ発現ベクターの作製には、DNA断片をベクターに挿入する既知のあらゆる方法を用いることができる。このような方法には、in vitro組換えDNAおよび合成的技術およびin vivo組換え(遺伝的組換え)を含めることができる。ウリカーゼタンパク質をコードしている核酸配列の発現は、組換えDNA分子で形質転換した宿主内でウリカーゼタンパク質が発現されるよう、第二の核酸配列によって制御することができる。例えば、ウリカーゼの発現は、技術的に知られているあらゆるプロモータ/エンハンサーエレメントによって制御され得る。ウリカーゼの発現を制御するのに用いることのできるプロモータはSV40初期プロモータ領域(Bernoist and Chambon,1981,Nature 290:304−310)、ラウス肉腫ウィルスの3’末端反復配列に含まれるプロモータ(Yamamoto,et al.,1980,Cell 22:787−797)、ヘルペスチミジンキナーゼプロモータ(Wagner et al.,1981,Proc.Natl.Acad.Sci.U.S.A.78:144−1445)、メタロチオネイン遺伝子の制御配列(Brinster et al., 1982,Nature 296:39−42)、βラクタマーゼプロモータなどの原核生物発現ベクター(Villa−Kamaroff,et al.,1978,Proc.Natl.Acad.Sci.U.S.A.75:3727−3731)、tacプロモータ(DeBoer,et al.,1983,Proc.Natl.Acad.Sci.U.S.A.80:21−25)、osmBプロモータを含むが、これに限定されない。特定の実施形態にあっては、核酸は動作可能な形で異種性プロモータに結合されたウリカーゼ配列を含む。
【0050】
核酸配列がコードされている特定の組み換えDNA分子が一旦調製され単離されたならば、技術的に知られている数種の方法によって、増殖させることができる。一旦宿主系ならびに培養条件が確立されれば、組み換え発現ベクターは増殖させ、大量に調製することができる。先に説明したように、使用できる発現ベクターは数例を挙げるならば、ワクシニアウィルスまたはアデノウィルスなどのヒトまたは動物ウィルス、バキュロウィルスなどの昆虫ウィルス、酵母ベクター、バクテリオファージベクター(例:ラムダ)およびプラスミドおよびコスミドDNAベクターならびに、これらの誘導体を含むが、これに限定されない。
【0051】
加えて、挿入された配列の発現を制御する、または遺伝子産物に、目的に合致した特定の修飾ならびに処理を施す宿主細胞株を選択することが可能である。特定のプロモータによる発現は特定の誘導物質の存在下で上昇し、よって遺伝的に改変したウリカーゼタンパク質の発現を制御することができる。さらに、異なる宿主細胞はそれぞれ特徴的ならびに特異的な、タンパク質の翻訳処理および翻訳後処理および修飾(例えばグリコシル化、切断)メカニズムを有している。発現された外来タンパク質に、目的の修飾および処理が施されることが保証されるよう、適切な細胞株を選ぶことができる。異なったベクター/宿主発現系は、タンパク質分解性開裂反応などの処理反応の程度を変えるなどの影響を与えうる。
【0052】
本発明の特定の実施形態にあっては、ウリカーゼ をE.coliで発現させることが好ましく、この際osmBプロモータを含むベクターを用いる。
【0053】
実施例
実施例 1. 遺伝子の作成ならびにウリカーゼ発現のための発現プラスミド
組み換えブタウリカーゼ(尿酸酸化酵素)、Pig−KS−ΔN(アミノ末端がトランケーションされたブタウリカーゼタンパク質で、第291番目と第301番目のアミノ酸が、それぞれリシンとセリンに置換されている)をE.coli K−12株W3110F−で発現させた。pOUR−P−ΔN−ks−1に到る、一連のプラスミドを作成したが、pOUR−P−ΔN−ks−1でE.coli宿主細胞を形質転換したところ、ウリカーゼを効率的に発現する能力を獲得した。
【0054】
ウリカーゼcDNAのブタおよびヒヒ肝臓からの単離およびサブクローニング
ウリカーゼcDNAは、当該RNAのサブクローニングおよび単離によって、ブタおよびヒヒ肝臓より調製した。全細胞RNAをブタおよびヒヒ肝臓より抽出し(Erlich,H.A.(1989).PCR Technology,Principles and Application for DNA Amplification、Sambrook,J.,et al.(1989).Molecular Cloning:A Laboratory Manual,2nd edition、Ausubel,F.M. et al.(1998).Current protocols in molecular Biology)、ファースト−ストランドcDNA合成キット(Pharmacia Biotech)を用いて逆転写した。PCR増幅反応は、Taq DNAポリメラーゼ(Gibco BRL,Life Technologies)を用いて行った。
【0055】
ブタならびにヒヒ尿酸オキシダーゼ(ウリカーゼ)のPCR増幅反応に用いた合成オリゴヌクレオチドプライマーは表1に示した。
表 1. ウリカーゼcDNAのPCR増幅に用いたプライマー
【表1】
【0056】
プライマー末端に設定した制限酵素配列はセンスEcoRIおよびNcoI(ブタおよびヒヒ)およびアンチセンスNcoI、HindIIIおよびXbaI(ブタ)、XbaIおよびNcoI(ヒヒ)であり、表1に小文字で示してある。ヒヒのセンスプライマーでは、ヒヒウリカーゼに存在する3番目のコドンGAC(アスパラギン酸)を、ヒト尿酸オキシダーゼ偽遺伝子をコードする配列のこの位置に存在するCAC(ヒスチジン)で置換した。これらのプライマーを用いて作成した組み換えヒヒウリカーゼ構造体をD3Hヒヒウリカーゼとする。
【0057】
EcoRIとHindIIIでブタウリカーゼPCR産物を処理し、pUC18にクローニングしてpUC18−ブタウリカーゼを得た。D3HヒヒウリカーゼPCR産物はTA Cloning(登録商標)(Invitrogen,Carlsbad,CA)を用いて、直接pCR(登録商標)IIベクターにクローニングし、pCR(登録商標)II−D3Hヒヒウリカーゼを得た。
【0058】
ライゲーションしたcDNAでE.coli株XL1−Blue(Stratagene,LaJolla,CA)を形質転換した。クローニングしたウリカーゼcDNAを含むプラスミドDNAを調製し、公開されたウリカーゼDNAコーディング配列を含むクローン(表1に記載の、ヒヒウリカーゼでのD3Hの置換を除く)をセレクションし、単離した。選んだpCR(登録商標)II−D3HヒヒウリカーゼクローンにおいてはPCRによって配列を削除したことで、pCR(登録商標)II配列がウリカーゼのストップコドンの隣にくるようになっている。この結果として、3′側の翻訳されない領域にあったXbaIおよびNcoIの制限酵素サイトが除かれているので、PCR産物の5′末端に存在するNcoIサイトとpCR(登録商標)IIベクター由来のBamHIサイトを用いて、デクレショナルクローニングをすることができた。
【0059】
ウリカーゼcDNAの、pET発現ベクターへのサブクローニング
ヒヒウリカーゼのサブクローニング
ウリカーゼコード配列の全長を含むD3HヒヒcDNAをpET−3d発現ベクター(Novagen,Madison,WI)に導入した。pCR(登録商標)II−D3HヒヒウリカーゼをNcoIおよびBamHIで処理し、得られた960bpの断片を単離した。発現プラスミドpET−3dはNcoIおよびBamHIで処理し、得られた4600bpの断片を単離した。この2つの断片をライゲーションしてpET−3d−D3H−Baboonを得た。
【0060】
ブタ−ヒヒキメラウリカーゼのサブクローニング
ブタ−ヒヒキメラ(PBC)ウリカーゼは、発現量、安定性および活性がより大きい組み換え遺伝子を得るために作成された。PBCの作成にはまず、pET−3d−D3H−ヒヒクローンから4936bp長のNcoI−ApaI断片を単離し、これをpUC18−ブタウリカーゼから単離した624bp長のNcoI−ApaI断片とライゲーションしてpET−3d−PBCを得た。PBCウリカーゼcDNAは、ブタウリカーゼのコドン1−225と、ヒヒウリカーゼのコドン226−304をインフレームで結合したものからなる。
【0061】
Pig−KSウリカーゼのサブクローニング
Pig−KSウリカーゼは、PEG化できる部位を1つ増やすことが期待されるリシン残基を、1つ加える目的で作成した。KSとはブタウリカーゼの291番目の位置にあるアルギニンの代わりにリシンを挿入したアミノ酸置換(R291K)を指す。加えて、301番目の位置にあるトレオニンをセリンで置換した(T301S)。Pig−KSウリカーゼプラスミドは、pET−3d−D3H−Baboonから4696bp長のNcoI−NdeI断片を単離し、pUC18−ブタウリカーゼから単離した得た864bp長のNcoI−NdeI断片をライゲーションして作成し、pET−3d−Pig−KSとした。得られたPigKSウリカーゼ配列は、ブタウリカーゼコドン1−288がインフレームにヒヒウリカーゼコドン289−304と結合されたものとなっている。
【0062】
osmBプロモータの制御下にあるウリカーゼ配列のサブクローニング
ウリカーゼ遺伝子を、osmBプロモータを含む発現ベクターにサブクローニングした(参照することによってその全文を本明細書に含む、米国特許番号5,795,776号に従った)。このベクターは、高浸透圧または長時間の培養によってタンパク質発現を誘導することを可能にした。発現プラスミドpMFOA−18はosmBプロモータ、リボソーム結合部位配列(rbs)および転写ターミネーター配列(ter)を含んでいる。また、アンピシリン耐性(AmpR)を付与し、組み換えヒトアセチルコリンエステラーゼ(AChE)を発現する。
【0063】
D3H−ヒヒウリカーゼのサブクローニング
プラスミドpMFOA−18をNcoIおよびBamHIで処理し、大きな断片を単離した。pET−3d−D3H−BaboonをNcoIおよびBamHIで処理し、D3Hヒヒウリカーゼを含む960bp長の断片を単離した。この2つの断片をライゲーションしてpMFOU18を得た。
【0064】
発現プラスミドpMFXT133はosmBプロモータ、rbs(E.colideoオペロン)、ter(E.coliTrypA)、組み換え型第Xa因子阻害ポリペプチド(FxaI)を含み、テトラサイクリン耐性遺伝子(TetR)を宿主に与える。ヒヒウリカーゼ遺伝子をこのプラスミドに挿入し、抗生物質耐性遺伝子と交換した。プラスミドpMFOU18をNcoIで処理し、平滑化後、XhoIで処理し、1030bp長の断片を単離した。プラスミドpMFXT133はNdeIで処理した後、平滑化し、その後XhoIで処理し、大きな断片を単離した。この2断片をライゲーションしてヒヒウリカーゼ発現ベクターpURBA16を得た。
【0065】
ブタ−ヒヒキメラウリカーゼのサブクローニング
プラスミドpURBA16をApaIおよびAlwNIで処理し、2320bp長の断片を単離した。プラスミドpMFXT133をNdeIで処理し、平滑化し、次にAlwNIで処理し、620bp長の断片を単離した。プラスミドpET−3d−PBCをXbaIで処理し、平滑化し、次にApaIで処理し、得た710bp長の断片を単離した。この3つの断片をライゲーションし、pET−3dベクター由来の、osmBプロモータならびにrbsならびにT7rbsの制御下でPBCウリカーゼを発現するプラスミドpUR−PBを得た。
【0066】
T7rbsは追加的な工程で取り除いた。pUR−PBをNcoIで処理し、平滑化し、次にAlwNIで処理し、得た3000bp長の断片を単離した。プラスミドpMFXT133をNdeIで処理し、平滑化し、次にAlwNIで処理し、得た620bp長の断片を単離した。この2つの断片をライゲーションして、PBCをosmBプロモータの制御下で発現するpDUR−PBを得た。
【0067】
pOUR−PB−ΔNCの作成
ウリカーゼcDNAを何カ所か改変したところ、安定性が大幅に高まった組み換え体酵素を得ることができた。プラスミドpOUR−PBC−ΔNCは、invivoでペルオキシソームターゲティングシグナルとして機能する、C末端のトリペプチドとマチュレーションペプチドであるN末端の6残基とを取り除いて作成した。これは、プラスミドpDUR−PBのPBC配列と、表2に示した特異的なオリゴヌクレオチドプライマーを用いたPCR増幅反応によって行った。
表2.PBC−ΔNCウリカーゼのPCR増幅に用いたプライマー
【表2】
【0068】
プライマーの末端に導入した制限酵素切断部位は太字で、また翻訳されない領域は小文字で、それぞれ表2に示してある。NdeIはセンス方向であり、XbaIはアンチセンス方向である。アンチセンスプライマーは、アミノ酸配列に影響しない点変異(下線で示している)を導入することによって内部に存在するNdeIサイトを除去し、NdeIを用いたサブクローニングを可能にするためにも用いられた。
【0069】
pDUR−PBのPCR増幅反応で得た900塩基長の断片をNdeIおよびXbaIで切断し、単離した。得た断片を、次にE.coli由来のdeo−P1P2プロモータおよびrbsを含み、恒常的にヒト組み換え型インシュリン前駆体を発現するdeo発現プラスミドpDBAST−RAT−Nに挿入した。プラスミドをNdeIおよびXbaIで処理し、得た4035bp長の断片を単離し、PBC−ウリカーゼPCR産物とライゲーションした。よって得たpDUR−PB−ΔNCで、E.coliK−12SΦ733(F−cytRstrA)を形質転換したところ、活性を有する切断型ウリカーゼを多量に発現した。
【0070】
両端でトランケーションしたPBC−ΔNC配列は、osmBプロモータの制御下でも発現させた。プラスミドpDUR−PB−ΔNCをAlwNI−NdeIで処理し、得た3459bp長の断片を単離した。先に述べたプラスミドpMFXT133をNdeI−AlwNIで処理し、得た660bp長の断片を単離した。これらの断片をライゲーションし、pOUR−PB−ΔNCを得、これでE.coliK−12株W3110F−を形質転換したところ、活性を有する切断型ウリカーゼを多量に発現した。
【0071】
ウリカーゼ発現プラスミドpOUR−P−ΔN−ks−1の作成
このプラスミドは、組み換え型酵素の活性ならびに安定性を向上させるために作成した。Pig−KS−ΔNウリカーゼは、N末端のマチュレーションペプチド6残基が除去される形でN末端のみをトランケーションし(ΔN)、S46TならびにR291KならびにT301S変異を有するように作成した。46番目の位置には、PCR増幅反応ならびにクローニングの過程で発生した保存的変異によって、セリン残基の代わりにトレオニン残基が存在していた。291番目の位置ではアルギニンがリシンに、また301番目の位置では、トレオニンがセリンに置換されていた。この両方が、ヒヒウリカーゼ配列由来である。R291KならびにT301Sの両改変は先にKSと命名し、説明したとおりである。リシン残基を追加することで、潜在的なPEG化サイトが1つ追加されている。
【0072】
pOUR−P−ΔN−ks−1(図1)の作成には、プラスミドpOUR−PB−ΔNCをApaI−XbaIで処理し、得た3873bp長の断片を単離した。プラスミドpET−3d−PKS(構造を図4に示してある)をApaI−SpeIで処理し、得た270bp長の断片を単離した。SpeIによる5′CTAGでの切断の結果、XbaIによって生じたDNA断片と効率的にライゲーションする、5′CTAGの突出部が生じた。この2つの断片をライゲーションしてpOUR−P−ΔN−ks−1を得た。ライゲーションの結果、SpeIおよびXbaI認識サイトは失われた(図9に括弧で示してある)。プラスミドpOUR−P−ΔN−ks−1をE.coliK−12株W3110F−原栄養株ATCC#27325に導入した。osmBプロモータの制御下で発現させて得たPig−KS−ΔNウリカーゼにより、優れた活性ならびに安定性を有する組み換え型酵素を多量に得ることができた。
【0073】
図1はプラスミドpOUR−P−ΔN−ks−1の構造を示したものである。制限酵素サイトの横に示した番号は、1と設定したHaeIIサイトから数えた、ヌクレオチドの位置を示している。クローニングの過程で失われた制限酵素サイトは括弧で示してある。Pig−KS−ΔNウリカーゼをコードするプラスミドpOUR−P−ΔN−ks−1は4143塩基対(bp)長で、下記のエレメントを含む。
1. osmBプロモータおよびリボソーム結合サイト(rbs)を含む、1番目のヌクレオチドから、NdeIサイト(113番目の位置)に到る、113bp長のDNA断片。
2. Pig−KS−ΔN(アミノ末端がトランケーションされており、291番目および301番目のアミノ酸がそれぞれリシンおよびセリンで置換されているブタウリカーゼタンパク質の核酸配列)コーディング領域から900bp、ならびにpCR(登録商標)II由来の、TAクローニング部位から上流方向にSpeI/XbaI制限酵素サイトまでの同フランキング配列32bpを含む、NdeI(113番目の位置)からSpeI/XbaIジャンクション(第1045番目の位置)に到る932bp長のDNA断片。
3. SpeI/XbaIジャンクション(1045番目の位置)からHindIII(1070番目の位置)にわたる、25bpのマルチクローニングサイト(MCS)配列。
4. HindIII(1070番目の位置)およびAatII(1110番目の位置)を末端に有し、TrpA転写ターミネーター(ter)を含む40bpの合成オリゴヌクレオチド。
5. pBR322上の、AatII(1110番目の位置)からMscI/ScaI(2629番目の位置)にわたる、テトラサイクリン耐性遺伝子(TetR)を含む1519bp長のDNA断片。
6. DNA複製起点を含み、pBR322上のScaI(2629番目の位置)からHaeII(4143番目の位置)にわたる、1514bp長のDNA断片。
【0074】
図2は、Pig−KS−ΔNウリカーゼのDNA配列ならびに推定アミノ酸配列を示している。この図では、アミノ酸の位置を示す番号は全長ブタウリカーゼ配列に対応している。イニシエーターであるメチオニン残基に続いて、ブタウリカーゼ配列ではアスパラギン酸となっている位置に、代わりにトレオニンを挿入した。このトレオニン残基によって、バクテリアのアミノペプチダーゼによるメチオニンの除去が可能となっている。アミノ酸配列にある間隙は、削除されたN末端のマチュレーションペプチドを示している。異なるウリカーゼ配列のサブクローニングの様々な過程で用いた制限酵素サイト(ApaI、NdeI、BamHI、EcoRIおよびSpeI)が示してある。小文字で示した3′側の翻訳されない領域は、pCR(登録商標)IIに由来する配列である。翻訳停止コドンはアスタリスクで示してある。
【0075】
図3は様々な組み換え型ウリカーゼ配列のアミノ酸配列のアラインメントを示したものである。1行目は全長アミノ酸配列を含むブタウリカーゼの配列である。2行目は両端をトランケーションしたブタ−ヒヒキメラウリカーゼ(PBC−ΔNC)である。3行目はN末端側のみでトランケーションされており、変異S46Tおよびウリカーゼコーディング配列のカルボキシ末端のヒヒ由来物を反映するアミノ酸変異R291KおよびT301Sを有するPig−KS−ΔNウリカーゼ配列である。アスタリスクは、Pig−KS−ΔNと公開されているブタウリカーゼ配列とでアミノ酸配列に相違がある位置を示している。丸印は、Pig−KS−ΔNとブタ−ヒヒキメラPBC−ΔNとでアミノ酸配列に相違がある位置を示している。点線はアミノ酸の欠損を示している。
【0076】
Y97H変異を有するネィティブのヒヒ、ブタ、ウサギウリカーゼおよびブタ/ヒヒキメラ(PBC)のcDNAを、E.coliへのクローニングのために作成した。全クローンでosmBによって発現が制御されており、W3110F−E.coliであるように、変異型ウリカーゼを多量に発現しているクローンを作成して選択した。プラスミドDNAを単離し、DNAシーケンシングならびに制限酵素分析によって確認し、細胞を培養した。
【0077】
Pig−ΔNおよびPig−KS−ΔNを含む切断型ウリカーゼの作成は、PBC−ΔNCとPig−KSを、制限酵素ApaIおよびXbaI、ならびにApaIおよびSpeIでそれぞれ処理後、クロスライゲーションして行った。N末端の「マチュレーションペプチド」(1−2)6残基、ならびにC末端のトリペプチド「ペルオキシソームターゲティングシグナル」(3−5)は酵素活性に大きく影響する機能を有さないので、これらのトランケーションされた変異体は活性を保つものと思われ、また、これらの配列は免疫原性を有する可能性が考えられる。ウリカーゼを非常に多量に発現しているクローンを選択した。
【0078】
実施例2.発現プラスミドの、バクテリア宿主細胞への形質転換
発現プラスミドpOUR−P−ΔN−ks−1を、E.coliK−12株W3110F−に導入した。ルリア培地(LB)で対数増殖期中期にまで培養した形質転換用のバクテリア細胞を、遠心処理によって集菌し、冷水で洗浄後10%グリセロールに懸濁、さらに水に懸濁し、1mlあたり約3x1010細胞の最終濃度とした。細胞はアリコットとして−70℃で保存した。プラスミドDNAはエタノールで沈殿させ水に溶解した。
【0079】
バクテリア細胞とプラスミドDNAを混合し、BIO−RADからのGenePulser IIを用いた高電圧電気穿孔法(Trevors et al(1992)Electrotransformation of bacteria by plasmid DNA,in Guide to Electroporation and Electrofusion(D.C.Chang,B.M.Chassy,J.A.Saunders and A.E.Sowers,eds.),pp.265−290,Academic Press Inc.,San Diego、Hanahan et al(1991)Meth.Enzymol.,204,63−113)によって形質転換した。形質転換した細胞はSOC培地(2% トリプトン、0.5% 酵母抽出物、10 mM NaCl、2.5mM KCl、10mM MgCl2、10mM MgSO4、20mM グルコース)に懸濁し、37℃で1時間インキュベーションした後テトラサイクリン耐性を指標に選択した。高発現クローンを選択した。
【0080】
実施例3.組み換え型ウリカーゼの調製
形質転換したものなど(上記参照)のバクテリアは、グルコースを含有する培地で培養した。約37℃で、pHは7.2±0.2に保った。
【0081】
培養の終期5−6時間にかけて培地にKClを、最終濃度が0.3Mとなるように加えた。ウリカーゼの蓄積を生じさせるため、培養を続けた。
【0082】
組み換え型ウリカーゼはバクテリア細胞内で、封入体(IB)に類似した不溶性の沈殿物として蓄積した。細胞懸濁液を遠心処理で洗浄し、pH8.0の50mM Trisバッファーおよび10mM EDTAに懸濁し、最終体積を乾燥菌体重量の約40倍とした。
【0083】
細胞をライソザイムと高圧力によって破壊した後、遠心処理によって組み換え型ウリカーゼを含有するIBを単離した。ライソザイム処理(2000−3000units/ml)はpH8.0、7±3℃で16−20時間、混合しながら行った。ペレットを水で洗浄し、使用するまで−20℃で保存した。
【0084】
濃縮したIBは、pH10.3±0.1の50mM NaHCO3バッファーで懸濁後さらに処理した。懸濁液は一晩室温でインキュベーションし、IB由来のウリカーゼを可溶化させた後、遠心処理によって沈殿物を除いた。
【0085】
ウリカーゼはさらに、複数のクロマトグラフィー工程によって精製した。最初のクロマトグラフィーはQ−セファロースFFカラムを用いて行った。カラムにサンプルをロードした後、150mM NaClを含有する重炭酸バッファーで洗浄し、ウリカーゼの溶出は250mM NaClを含有する重炭酸バッファーによって行った。次に、キサンチン−アガロース樹脂(Sigma)によって少量の不純物をウリカーゼ調製物より除いた。Q−セファロースFF溶出物はpH10.3±0.1の50mMグリシンバッファーで希釈し、タンパク質最終濃度を約0.25mg/mlとしてロードした。カラムを100mM NaClを含有するpH10.3±0.1の重炭酸バッファーで洗浄し、同じバッファーに60μMキサンチンを加えたものでウリカーゼを溶出した。この段階で、ウリカーゼをQ−セファロースクロマトグラフィーで再精製し、凝集したものを除いた。
【0086】
それぞれのウリカーゼ調製物の純度を、サイズ排除クロマトグラフィーで測定したところ、95%以上であった。それぞれの調製物でSuperdex200カラムを用いて行った分析では、凝集体は0.5%以下しか検出されなかった。
【0087】
表3は25リットルの発酵培地から得たIB由来のPig−KSΔNウリカーゼの精製をまとめたものである。
表3.Pig−KSΔNウリカーゼの精製
【表3】
【0088】
実施例 4. 組み換え型ウリカーゼの性質
SDS−PAGE
高度に精製された変異型ウリカーゼのSDS−PAGE分析(図4)では、かなり特徴的なパターンが現れた。サンプルはpH10.3の炭酸バッファーに、4℃で最高数ヶ月保存した。ブタ、ブタ−KSならびにPBCの、全長変異型タンパク質では、分子量が20および15kDの顕著な分解産物の蓄積が見られた。これは、少なくとも1つのニックがウリカーゼサブユニット分子を開裂していることを示唆している。アミノ末端を短くしたクローンならびにウサギウリカーゼでは、これと異なった分解パターンが検出されたが、割合は小さかった。ウサギウリカーゼのアミノ末端は、短くしたクローンに類似している。精製並びに保管の過程で生じたウリカーゼ断片のアミノ末端配列を決定した。
【0089】
ペプチドシーケンシング
原体ウリカーゼ調製物のN末端を、エドマン分解法でシーケンシングした。サイクル数は10であった。組み換え型ブタウリカーゼ(全長クローン)はPig−KS−ΔNよりも多くの分解断片を産した。分解断片を生じる切断部位の予想位置は以下の通りである:
1)下記の配列を有する、168番目の位置の、メジャーサイト:
−−QSG ↓ FEGFI−−
2) 下記の配列を有する、142番目の位置の、マイナーサイト:
−−IRN ↓ GPPVI−−
【0090】
上記の配列は、既知のいかなるタンパク質分解性開裂も示唆しない。しかしながら、開裂はタンパク質分解またはなんらかの化学反応によって生じうる。アミノ末端切断型ウリカーゼは非アミノ末端切断型ウリカーゼに比べて驚くほど安定性が高い。PBC−ΔNCも、他のΔN分子と類似した、非アミノ切断型PBCよりも少ない安定性を有していた。
【0091】
効力
ウリカーゼ活性の測定はUV法によって行った。酵素反応速度は、尿酸のアラントインへの酸化による、292nm吸光度の減少を測定することによって求めた。1活性単位は、25℃、1分間で、示した特定の条件下で1μモルの尿酸を酸化するのに必要なウリカーゼの量と定義される。ウリカーゼの効力は、タンパク質1mgあたりの活性単位として表される(U/mg)。
【0092】
292nmにおける1mM尿酸の消衰係数は12.2mM−1cm−1である。ゆえに、反応溶液1mlあたりの、尿酸1μモルの酸化は、12.2mA292の吸光度の減少となる。経時的な吸光度の変化(毎分ΔA292)は、曲線の直線部より得た。
【0093】
タンパク質濃度は、改変ブラッドフォード法によって求めた(MacartandGerbaut(1982) Clin Chim Acta 122:93−101)。ウリカーゼの比活性度(効力)は、U/mlで表される活性を、mg/mlで表されるタンパク質濃度で割ることによって計算した。様々な組み換え型ウリカーゼの酵素活性結果を、表4にまとめた。市販の調製物の結果も、参考値としてこの表に示した。結果からは、ウリカーゼタンパク質のトランケーションがその酵素活性に大きな影響を与えないことが明らかである。
表4. 組み換え型およびネィティブ型のウリカーゼの、速度パラメータのまとめ
【表4】
【0094】
表4注釈:
(1) タンパク質濃度は、278nmにおける吸光度から、10mg/mlウリカーゼ溶液に対して消衰係数として11.3を用いて求めた(Mahler,1963)。
(2)1単位のウリカーゼ活性は、25℃、1分間で1μモルの尿酸をアラントインに酸化するのに必要な酵素の量と定義される。
(3)比活性度は、60μMに等しい基質濃度の下で行ったラインウィーバーバークプロットによって求めた。
(4)反応混合物は、以下のストック溶液を様々な組み合わせで混合して作成した。
pH9.2の100mMホウ酸ナトリウムバッファー
pH9.2の50mMホウ酸ナトリウムに溶解した300μM尿酸
pH9.2の50mMホウ酸ナトリウムに溶解した1mg/mlBSA
(5)Kcatは、Vmax(対応するラインウィーバーバークプロットから算出)を反応溶液のウリカーゼ濃度(リカーゼの四量体の分子量に基づいてモル量として表現したもの)で割ることで求めた。
【0095】
実施例5.ウリカーゼとm−PEGの結合(PEG化)
Pig−KS−ΔNウリカーゼは、m−PEG−NPC(モノメトキシ−ポリ(エチレングリコール)−ニトロフェニルカーボネート)と結合体を形成させた。1ウリカーゼサブユニットあたり5、10または20kDのPEG鎖2から12本が結合する条件を確立した。m−PEG−NPCは、徐々にタンパク質溶液に加えた。PEGの添加が終わった後、ウリカーゼ/m−PEG−NPC反応溶液を2から8℃で16から18時間、未結合のm−PEG鎖が最大限ウリカーゼに結合するようインキュベーションした。
【0096】
PEG−ウリカーゼモノマー1つあたりのPEG鎖の数は、PEGおよびウリカーゼ標準試料を用いて、Superose6サイズ排除クロマトグラフィー(SEC)によって同定した。サブユニット1つあたりに結合したPEGの数は、下記の式によって算出した:
【数1】
【0097】
紫外線(UV)および屈折率(RI)検出器の配列(Kunitani,et al.,1991の開発による)を用いて、サイズ排除クロマトグラフィー(SEC)によってPEG−ウリカーゼサンプルにおける、PEG並びにタンパク質成分濃度を同定した。3つの較正曲線を作成した:タンパク質曲線(220nmで測定した吸光度)、タンパク質曲線(RIの測定による)、PEG曲線(RIの測定による)。次に、PEG−ウリカーゼサンプルを同様のシステムで測定した。得られた試験サンプルのUVならびにRIピークエリア値を、較正曲線に対するPEGならびにタンパク質の濃度を計算するのに用いた。3.42は、10kD PEGに対するウリカーゼ分子モノマーの分子量(34,192ダルトン)の比である。
【0098】
結合させたPEGは、生理的pHの溶液においてウリカーゼの可溶性を向上させた。表5はPEG化Pig−KS−ΔNウリカーゼ産生物のバッチ間のばらつきを示したものである。一般に、結合したPEG鎖数と酵素が保持する比活性度(SA)との間には、反比例の関係がある。
表5.PEG化Pig−KS−ΔNウリカーゼ結合体の酵素活性
【表5】
【0099】
実施例6.1000Dならびに100,000DのPEGによるウリカーゼのPEG化
Pig−KS−ΔNウリカーゼを、実施例5に記載の通りの方法で、1000Dならびに100,000Dのm−PEG−NPCを用いてPEG化した。1ウリカーゼサブユニットあたりPEG鎖2〜11本が結合する条件を用いた。PEGの添加が終わった後、ウリカーゼ/m−PEG−NPC反応溶液を2から8℃で16から18時間、未結合のm−PEG鎖が最大限ウリカーゼに結合するようインキュベーションした。
【0100】
PEG−ウリカーゼモノマー1つあたりのPEG鎖数は、上記に記載の方法で求めた。
【0101】
結合させたPEGは、生理的pHの溶液においてウリカーゼの可溶性を向上させた。
【0102】
実施例7.PEGと結合させたPig−KS−ΔNウリカーゼの薬物動態
治療上の利益を提供するために、PEG化の最適な程度ならびにサイズを決定するために、生物学的な実験を行った。
【0103】
ラットで、体重1kgあたり0.4mg(2U)の改変されていないウリカーゼを、1日目ならびに8日目に静脈投与して行った薬物動態試験では、循環系半減期が約10分であるとの結果が得られた。しかしながら、ラットで2−11x10kDのPEG−Pig−KS−ΔNウリカーゼを、週1回、9回に渡って注射してクリアランス速度を調べた試験では、クリアランスはPEG鎖の数に(この範囲においては)依存していないことが示唆され、試験期間を通じて比較的同じ値に保たれた(表6参照、半減期が約30時間)。各週間の違いは誤差の範囲内である。10x5kD PEGならびに10x20kD PEG−ウリカーゼ結合体の9回の注射後でも、同様のパターンが観察された。結果は、この範囲においては、ウリカーゼのPEG化の程度にかかわらず、ラットモデルにおいて類似した生理的影響が観察されたことを示している。
表6.PEG化Pig−KS−ΔNウリカーゼ調製物の、ラットにおける半減期
【表6】
【0104】
表6注釈: 結果は時間±標準誤差で示している。括弧で示した数字は試験した実験動物個体数を示している。
【0105】
ラットに、1週間に1回、体重1kgあたり0.4mgの、表に示したとおりの改変を施したPig−KS−ΔNウリカーゼを静脈投与した。それぞれの個体群は元々15個体のラットから成っており、5個体のサブグループで交互に採血した。ラット数例が、麻酔によって実験中に死亡した。半減期は、注射5分後、および6、24、48時間後に採取した血漿サンプルのウリカーゼ活性(比色測定)を測定することによって同定した。
【0106】
表5に、実験に用いられたPEG化ウリカーゼを記載している。
【0107】
6x5kD PEG−Pig−KS−ΔNウリカーゼを用いた、ウサギでの生物学的利用率試験によると、最初の注射後では、循環系半減期は98.2±1.8時間(i.v.)であり、筋肉投与(i.m.)ならびに皮下投与(s.c.)では、それぞれ71%、52%であった。しかしながら、2回目のi.m.ならびにs.c.注射の後、全てのウサギで相当量の抗ウリカーゼ抗体タイターが検出され、以降の注射でクリアランスが促進された。ラットに同じ結合体を注射した実験では、26±1.6時間の半減期(i.v.)が得られ、生物学的利用率はi.m.ならびにs.c.注射についてそれぞれ33%、22%であった。
【0108】
9x10kD PEG−Pig−KS−ΔNウリカーゼを用いたラットでの試験では、1回目の注射後の循環系半減期が42.4時間(i.v.)であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ28.9%、14.5%だった。(図5ならびに表7参照)。4回目の注射後では、循環系半減期は32.1±2.4時間であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ26.1%、14.9%だった。
【0109】
9x10kD PEG−Pig−KS−ΔNウリカーゼを用いた、ウサギでの同様の薬物動態試験では、この結合体の注射に引き続くクリアランスの促進は観察されなかった(隔週で4回注射として投与した)。これらの個体では、最初の注射後の循環系半減期は88.5時間(i.v.)であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ98.3%、84.4%であった。(図6ならびに表7参照)。4回目の注射後の循環系半減期は141.1±15.4時間であり、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ85%、83%であった。
【0110】
9x10k DPEG−Pig−KS−ΔNを用いた同様の試験によって、ビーグル犬における生物学的利用率を測定した(それぞれの群にオス2個体、雌2個体)。1回目のi.v.注射後で、70±11.7時間の循環系半減期が記録され、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ69.5%、50.4%だった(図6ならびに表7参照)。
【0111】
9x10kD PEG−Pig−KS−ΔN調製物を用いて、ブタで試験を行った。1つの群あたり3個体にi.v.、s.c.ならびにi.m.ルートによる投与を行った。1回目のi.v.注射後で、178±24時間の循環系半減期が記録され、i.m.ならびにs.c.注射後の生物学的利用率はそれぞれ71.6%、76.8%であった(図8ならびに表7参照)。
表7.9x10kDPEG−Pig−KS−ΔNウリカーゼを用いた薬物
【表7】
【0112】
9x10kD PEG−Pig−KS−ΔNウリカーゼの、ボルトン&ハンター法による、125Iでのヨウ素化に引き続いて、吸収、分布、代謝、排泄(ADME)試験を実施した。ラベルした結合体を、それぞれ4個体のラット(雄2個体と雌2個体)からなる7群に注射した。放射線の分布を、1時間後ならびに、7日間にわたって24時間毎に(5日目を除く)測定した。それぞれの群を順番に屠殺し、異なる臓器を摘出して分析した。7番目の群は代謝ケージで飼育し、尿と便を回収した。物質の、動物体内における分布は、それぞれの臓器の総放射活性ならびに、TCAによる沈殿が可能なカウント(腎臓、肝臓、肺、脾臓)のフラクション(すなわち、臓器のサイズで標準化した結合したタンパク質)を測定することによって評価した。摘出した臓器で、他の臓器よりも比放射能が高いものはなかった。すなわち、例えば肝臓または腎臓における特筆すべき蓄積は見られなかった。放射活性の70%が、7日目までに排出された。
【0113】
実施例8.治験結果
従来の治療法に不応答性または不耐性の、ヒト高尿酸血症または重篤な痛風に罹患している患者における、PEG−ウリカーゼ(Puricase(登録商標),Savient Pharmaceuticals)の尿酸応答ならびに、薬物動態的および安全性プロフィールを評価するために無作為化非盲検多施設並行群間試を実施した。平均罹患期間は14年で、被験者集団の70%が1つまたはそれ以上の痛風結節を有していた。
【0114】
治験では、41例の患者(平均年齢58.1歳)を無作為に、次に示す4つの12週間にわたる、静脈投与によるPEG−ウリカーゼ治療投与量計画に振り分けた:2週間毎に1回4mg(7例)、2週間毎に1回8mg(8例)、4週間毎に1回8mg(13例)、4週間毎に1回12mg(13例)。血漿ウリカーゼ活性ならびに尿酸レベルを定められた時間間隔で測定した。薬物動態学的パラメータ、平均血漿尿酸濃度および血漿尿酸レベルが6mg/dL以下であった時間の百分率を、ウリカーゼ活性並びに尿酸レベルの分析によって求めた。
【0115】
PEG−ウリカーゼを2週間毎に1回8mg投与した群で、最も大きなPUAの低減が見られ、治療期間の92%で6mg/dL未満のレベルであった。(12週間で、平均血漿尿酸濃度が治療前の9.1mg/dLに対して、治療後は1.4mg/dL)。
【0116】
他の投与量を用いたPEG−ウリカーゼ治療群でも、顕著で持続的な低い血漿尿酸レベルが観察された:4週間毎に1回8mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の86%(12週間で、平均血漿尿酸濃度が治療前の9.1mg/dLに対して、治療後は2.6mg/dL)、4週間毎に1回12mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の84%(12週間で、平均血漿尿酸濃度が治療前の8.5mg/dLに対して、治療後は2.6mg/dL)、2週間毎に1回4mgを投与した群で、PUAレベルが6mg/ml未満である期間が治療期間の73%(12週間で、平均血漿尿酸濃度が治療前の7.6mg/dLに対して、治療後は4.2mg/dL)。
【0117】
最初のPEG−ウリカーゼ投与から24時間以内での、血漿中尿酸の基線レベルからの減少割合の最大値は4mg/2週間の群で72%(p=.0002)、8mg/2週間の群で94%(p<.0001)、8mg/4週間の群で87%(p<.0001)、12mg/4週間の群で93%(p<.0001)であった。
【0118】
12週間の治療期間における血漿中尿酸の基線レベルからの減少割合は4mg/2週間の群で38%(p=.0002)、8mg/2週間の群で86%(p<.0001)、8mg/4週間の群で58%(p<.0003)、12mg/4週間の群で67%(p<.0001)であった。
【0119】
驚くべき事に、PEG−ウリカーゼの投与を受けた患者の一部で注入に関連した有害事象、例えば注入反応が出現した。この反応はすべての注入のうち14%で出現した。
【0120】
本出願に引用される全ての文献を、各個々の刊行物、特許または特許出願を具体的かつ個々に、参照することによって援用すると示したのと同程度に、全ての目的のためにその全文を参照することによって本明細書に含める。
【0121】
当該分野に精通する者に理解されるように、本発明にあっては、その思想および範囲から逸脱することなく、様々な変更ならびに形態様が可能である。ここに記載された具体的な実施例は単に例を示すのみであって、本発明は添付した特許請求項、ならびに、このような請求に権利が与えられるあらゆる等価例によってのみ制限されるものである。
【特許請求の範囲】
【請求項1】
SEQ ID NO.7の位置8から位置287のアミノ酸配列を含む単離されたウリカーゼ、ここでアミノ酸番号は、SEQ ID NO.11のアミノ酸配列を有する全長ブタウリカーゼに対応している。
【請求項2】
アミノ末端アミノ酸を有し、該アミノ末端アミノ酸がアラニン、グリシン、プロリン、セリン、またはトレオニンである請求項1に記載のウリカーゼ。
【請求項3】
アミノ末端アミノ酸を有し、アミノ末端アミノ酸がメチオニンである請求項1に記載のウリカーゼ。
【請求項4】
SEQ ID NO.12の位置8から位置287のアミノ酸配列を含む単離されたウリカーゼ、ここでアミノ酸番号は、SEQ ID NO.11のアミノ酸配列を有する全長ブタウリカーゼに対応している。
【請求項5】
SEQ ID NO.13のアミノ酸配列を含む請求項4に記載のウリカーゼ。
【請求項6】
アミノ末端アミノ酸を有し、該アミノ末端アミノ酸がアラニン、グリシン、プロリン、セリン、またはトレオニンである請求項4に記載のウリカーゼ。
【請求項7】
アミノ末端アミノ酸を有し、アミノ末端アミノ酸がメチオニンである請求項5に記載のウリカーゼ。
【請求項8】
ウリカーゼがPEG化されたウリカーゼである、請求項1記載のウリカーゼ。
【請求項9】
請求項1記載のウリカーゼ、請求項2記載のウリカーゼ、請求項3記載のウリカーゼ、請求項5記載のウリカーゼ、および請求項6記載のウリカーゼからなる群から選択されたウリカーゼをコードする核酸配列を含む、単離された核酸。
【請求項10】
核酸配列が動作可能な形で異種性プロモータに結合されている、請求項9記載の単離された核酸。
【請求項11】
プロモータがosmBプロモータである、請求項9に記載の核酸。
【請求項12】
請求項10に記載の核酸を含む、核酸ベクター。
【請求項13】
請求項12に記載のベクターを含む、宿主細胞。
【請求項14】
請求項13記載の宿主細胞を、宿主細胞により当該核酸配列が発現される条件下で培養し、発現されたウリカーゼを単離する工程を含む、ウリカーゼを生成するための方法。
【請求項15】
請求項1から7のいずれか1項記載のウリカーゼを含む、医薬組成物。
【請求項16】
請求項8記載のPEG化されたウリカーゼを含む、医薬組成物。
【請求項17】
反復投与に適している、請求項15記載の医薬組成物。
【請求項18】
反復投与に適している、請求項16記載の医薬組成物。
【請求項19】
必要とされる対象の体液中の尿酸レベルを低下させる医薬組成物の製造のための、請求項1から7のいずれか1項に記載されたウリカーゼの使用。
【請求項20】
必要とされる対象の体液中の尿酸レベルを低下させる医薬組成物の製造のための、請求項8に記載されたウリカーゼの使用。
【請求項21】
該体液が血液である、請求項19記載の使用。
【請求項22】
該体液が血液である、請求項20記載の使用。
【請求項1】
SEQ ID NO.7の位置8から位置287のアミノ酸配列を含む単離されたウリカーゼ、ここでアミノ酸番号は、SEQ ID NO.11のアミノ酸配列を有する全長ブタウリカーゼに対応している。
【請求項2】
アミノ末端アミノ酸を有し、該アミノ末端アミノ酸がアラニン、グリシン、プロリン、セリン、またはトレオニンである請求項1に記載のウリカーゼ。
【請求項3】
アミノ末端アミノ酸を有し、アミノ末端アミノ酸がメチオニンである請求項1に記載のウリカーゼ。
【請求項4】
SEQ ID NO.12の位置8から位置287のアミノ酸配列を含む単離されたウリカーゼ、ここでアミノ酸番号は、SEQ ID NO.11のアミノ酸配列を有する全長ブタウリカーゼに対応している。
【請求項5】
SEQ ID NO.13のアミノ酸配列を含む請求項4に記載のウリカーゼ。
【請求項6】
アミノ末端アミノ酸を有し、該アミノ末端アミノ酸がアラニン、グリシン、プロリン、セリン、またはトレオニンである請求項4に記載のウリカーゼ。
【請求項7】
アミノ末端アミノ酸を有し、アミノ末端アミノ酸がメチオニンである請求項5に記載のウリカーゼ。
【請求項8】
ウリカーゼがPEG化されたウリカーゼである、請求項1記載のウリカーゼ。
【請求項9】
請求項1記載のウリカーゼ、請求項2記載のウリカーゼ、請求項3記載のウリカーゼ、請求項5記載のウリカーゼ、および請求項6記載のウリカーゼからなる群から選択されたウリカーゼをコードする核酸配列を含む、単離された核酸。
【請求項10】
核酸配列が動作可能な形で異種性プロモータに結合されている、請求項9記載の単離された核酸。
【請求項11】
プロモータがosmBプロモータである、請求項9に記載の核酸。
【請求項12】
請求項10に記載の核酸を含む、核酸ベクター。
【請求項13】
請求項12に記載のベクターを含む、宿主細胞。
【請求項14】
請求項13記載の宿主細胞を、宿主細胞により当該核酸配列が発現される条件下で培養し、発現されたウリカーゼを単離する工程を含む、ウリカーゼを生成するための方法。
【請求項15】
請求項1から7のいずれか1項記載のウリカーゼを含む、医薬組成物。
【請求項16】
請求項8記載のPEG化されたウリカーゼを含む、医薬組成物。
【請求項17】
反復投与に適している、請求項15記載の医薬組成物。
【請求項18】
反復投与に適している、請求項16記載の医薬組成物。
【請求項19】
必要とされる対象の体液中の尿酸レベルを低下させる医薬組成物の製造のための、請求項1から7のいずれか1項に記載されたウリカーゼの使用。
【請求項20】
必要とされる対象の体液中の尿酸レベルを低下させる医薬組成物の製造のための、請求項8に記載されたウリカーゼの使用。
【請求項21】
該体液が血液である、請求項19記載の使用。
【請求項22】
該体液が血液である、請求項20記載の使用。
【図1】

【図2】

【図3】

【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2013−90636(P2013−90636A)
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2013−9960(P2013−9960)
【出願日】平成25年1月23日(2013.1.23)
【分割の表示】特願2008−505656(P2008−505656)の分割
【原出願日】平成18年4月11日(2006.4.11)
【出願人】(507334679)サビエント ファーマセウティカルズ インク. (4)
【Fターム(参考)】
【公開日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成25年1月23日(2013.1.23)
【分割の表示】特願2008−505656(P2008−505656)の分割
【原出願日】平成18年4月11日(2006.4.11)
【出願人】(507334679)サビエント ファーマセウティカルズ インク. (4)
【Fターム(参考)】
[ Back to top ]