
変異型蛍光タンパク質及びそれを用いた高効率FRET検出
【課題】FRET効率が高く、しかも分子デザインが容易なFRETプローブを提供する。
【解決手段】自己集合して二量体を形成する蛍光タンパク質(未変異型蛍光タンパク質)に変異を導入して、自己集合性を維持しながら励起極大波長及び吸収極大波長がともに異なるように改変した蛍光タンパク質(変異型蛍光タンパク質)を作出し、変異型及び未変異型の蛍光タンパク質からなる、又は、互いに異なる変異型蛍光タンパク質からなるヘテロ二量体を形成する蛍光タンパク質のペアを用いてプローブを構成することにより、様々なタンパク質やペプチド等の生体分子の分子間相互作用や分子内構造変化を検出するための、FRET効率が高く、しかも分子デザインが容易なFRETプローブが提供される。
【解決手段】自己集合して二量体を形成する蛍光タンパク質(未変異型蛍光タンパク質)に変異を導入して、自己集合性を維持しながら励起極大波長及び吸収極大波長がともに異なるように改変した蛍光タンパク質(変異型蛍光タンパク質)を作出し、変異型及び未変異型の蛍光タンパク質からなる、又は、互いに異なる変異型蛍光タンパク質からなるヘテロ二量体を形成する蛍光タンパク質のペアを用いてプローブを構成することにより、様々なタンパク質やペプチド等の生体分子の分子間相互作用や分子内構造変化を検出するための、FRET効率が高く、しかも分子デザインが容易なFRETプローブが提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、FRET効率が高く、しかも分子デザインが容易なFRETプローブに関する。より詳細には、本発明は、自己集合性をもち、ヘテロ二量体を形成しやすい蛍光タンパク質を用いる、FRET効率が高く、しかも分子デザインが容易なFRETプローブ及びその利用に関する。
【背景技術】
【0002】
蛍光共鳴エネルギー移動(FRET)とは、2つの蛍光分子が存在するとき、一方の分子の励起エネルギーが、他方の分子にトランスファーされる現象であり、その結果、エネルギーをトランスファーされた分子から蛍光が発せられる。励起エネルギーをトランスファーする方をドナー、される方をアクセプターと呼ぶ。
【0003】
FRET技術は、生体分子を可視化・画像化するための技術の1つであり、例えば、生体分子の分子間相互作用や分子内構造変化をモニタリングするために用いられ、特に、遺伝子の機能、及び疾病の発症と深く関わるシグナル伝達や遺伝子発現を解明するために好適である。これは、生体分子が分子間相互作用又は分子内構造変化によって、その生体分子を標識する2つの蛍光分子間の距離や相対的向きが変化すると、FRETが誘起又は解消されることを利用するものである。
【0004】
FRETの誘起又は解消を検出するための蛍光センサーがFRETプローブ(単に「プローブ」ともいう。)である。FRETプローブには、二分子間相互作用をする2つの生体分子のそれぞれを蛍光分子で標識したもの(2分子プローブ)と分子内構造変化又は分子間相互作用をする生体分子部分が挿入されたリンカー分子の両端を2つの蛍光分子で標識したもの(1分子プローブ)とがある。どちらの場合も、蛍光分子は合成小分子や蛍光タンパク質を利用できるが、蛍光タンパク質を用いると、フュージョンさせた融合タンパク質を生きた細胞内で発現させることができるため、低侵襲的に細胞体タンパク質の分子動態を可視化・映像化できる。
【0005】
FRETの誘起・解消の検出は、ドナーとアクセプターの蛍光強度比から近似的に求められるのが普通である。これをレシオメトリック測定法といい、2種類の蛍光フィルターセットを用いて、FRETにより減弱したドナーの蛍光強度(Dda)とFRETにより増強したアクセプターの蛍光強度(Ada)を測定し、その比をFRET効率(Ada/Dda)とするものである。
【0006】
この2色の蛍光強度比を用いる定量方法(レシオメトリー測定法)で、シグナル/ノイズ比(S/N比)を高めるためには、2つの蛍光タンパク質が予め同じ分子数で細胞内に存在することが必要である。しかし、2分子FRETでは、2つの蛍光タンパク質の分子数を揃えることが困難あり、これまでに比較的高いFRET効率を実現するのは、1分子FRETの場合が非常に多かった。
【0007】
現在までに実用化されているプローブには、FRET効率が非常に低いものが多く、そのようなプローブを用いる場合、わずかなFRET効率変化を2色の蛍光強度比として定量化して検出することとなる。実際、よく用いられている開発済みのプローブでさえも、FRET効率が非常に低く、2色の蛍光強度の比が、1.0から1.5への変化程度であるものも多い。
【0008】
FRET効率を算出する方法としては、レシオメトリー測定法の他に、ドナー蛍光の蛍光寿命を測定する方法がある。蛍光寿命の測定は、蛍光寿命測定装置を用いて行われ、蛍光の強さが時間経過とともに減衰していく変化を測定する。
【0009】
しかしながら、もともとFRET効率が非常に低い多くのプローブでは、蛍光寿命の変化もごくわずかであるため、FRET効率を精度よく算出することが難しい。
【0010】
例えば、非特許文献2(第58頁〜第59頁、実証実験)には、CFP(シアン色蛍光タンパク質)とRas(癌遺伝子)との融合タンパク質と、YFP(黄色蛍光タンパク質)とRaf(セリン・スレオニンリン酸化酵素)との融合タンパク質とからなるプローブを293F細胞で発現させ、Flicyme(登録商標)を使用して10万個の細胞についてドナー(CFP)の蛍光寿命を測定したところ、FRET誘起時に3.78nsから0.32ns程度短くなる方向にシフトし、FRET効率が8.5%と求められたことが記載されている。
【0011】
プローブの分子デザインをするとき、通常は、そのプローブが置かれている環境やそのプローブ分子の状態変化によって、蛍光分子間の距離が変動することを狙って分子デザインを行う。しかし、FRETは双極子−双極子相互作用により起こるものであり、蛍光分子間の距離だけでなく、蛍光分子の相対的向きもまた、考慮すべき要素である。特に、巨大な蛍光タンパク質分子の場合には、合成小分子のように自由回転する双極子とみなすこともできないので、分子の向きを考慮することは、より重要である。言い換えると、2つの蛍光分子が適切な角度をもって会合するようにデザインできなければ、FRET効率は非常に低いものとなる。
【0012】
ところが、実際には、相互作用する2分子プローブどころか、1本のポリペプチド鎖として発現される1分子プローブであっても、その一次構造からタンパク質が折り畳まれた際の2つの蛍光タンパク質間の距離や角度を予測することが困難であり、蛍光タンパク質分子間の距離を至適化し、さらに角度までも最適化することは非常に困難である。
【0013】
この問題に対する解決策のひとつとして、蛍光タンパク質のN末端とC末端の位置を作り替えた円順列変異体を用いることによって、蛍光タンパク質同士の角度を変更しようとする方法が開発されている(例えば、特許文献1)。
【0014】
この方法は多種類の円順列変異体を用いたプローブを全て作成してみて、その中からFRET効率の高いものを探す方法である。
【0015】
そのため、作成した円順列変異体の中に適切なものがなかった場合には、問題の解決が困難であり、さらに、あらゆる蛍光タンパク質で円順列変異体が作成されている訳ではないため、プローブ作成が、やはり困難である。
【0016】
【特許文献1】国際公開WO2005/036178号公報
【特許文献2】特開2007−232559号公報
【非特許文献1】中田ら,三井造船技報,第190号,54〜60頁(2007年).
【非特許文献2】Bairdら,米国科学アカデミー紀要,97:11984−11989(2000年).
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明は、上記課題を解決するため、FRET効率が高く、蛍光寿命測定によってFRETの誘起・解消を検出するために好適に用いることができ、しかも分子デザインが容易なFRETプローブを提供することを目的とする。
【課題を解決するための手段】
【0018】
上記の目的を達成するため、発明者らが鋭意検討をした結果、FRETプローブに用いる蛍光タンパク質には自己集合性が低いものを用いることが技術常識であるところ、あえて自己集合性の蛍光タンパク質を用いてプローブを構成することによって、前記の目的を達成できることを知得した。以下に本発明を詳細に説明する。
【0019】
生体分子を標識するための蛍光分子としては、例えば、クマリン(黄緑色)やフルオレセイン(緑色)のような合成小分子、CFP(シアン色)やYFP(黄色)のような蛍光タンパク質が使用される。特に、蛍光タンパク質の遺伝子が同定され、遺伝子組み換え技術を用いて細胞内で生体分子との融合タンパク質として発現できるようになってからは、蛍光タンパク質の利用が進み、新しい蛍光タンパク質の分離や新しいバリアントの開発が相次いでいる。
【0020】
最初に発見された蛍光タンパク質は、オワンクラゲ(Aequorea victoria)に由来する緑色蛍光タンパク質(GFP)である。前述のCFPとYFPは、ともにGFPのバリアントであり、プローブ標識に用いられる極めて一般的な蛍光タンパク質のペアとなっている。
【0021】
また、サンゴ(Discosoma sp.)に由来する赤色蛍光タンパク質(DsRed)及びそのバリアントもまた、一般的に使用されている(例えば、非特許文献2)。さらに、バリアントには、異色変異体のみならず、培養細胞での大量発現に好適なようにコドン使用頻度が最適化された変異体や細胞内での凝集性を減少した変異体も存在する。
【0022】
例えば、サンゴ由来の野生型のDsRedに対して、DsRed1(クロンテック社)はヒト細胞でのコドン使用頻度に最適化された変異体であり、DsRed2(クロンテック社)は野生型のDsRedが四量体を形成しやすいのに対して、凝集性が減少し、成熟が早い変異体である。なお、これら市販の蛍光タンパク質は、例えば、それをコードするDNAが組み込まれた発現ベクター(発現コンストラクト)として提供される。
【0023】
従来の蛍光タンパク質を用いたプローブは、FRET効率が低いものが多いことは既に述べた通りである。しかも、従来の技術では、プローブの分子デザインは試行錯誤の要素が大きく、誰でもが容易にできるものでもない。
【0024】
FRET効率とはドナー/アクセプター間の蛍光共鳴エネルギーの移動効率、つまり、ドナーに吸収されたエネルギーのうち、アクセプターに転送されたエネルギーの比率を意味する。
【0025】
ここで、フェルスターの関係式から、FRET効率(E)は、
E={1+(r/R0)6}−1 (1)
と表すことができる。ただし、r[nm]はドナーとアクセプターのクロモフォア間距離であり、R0[nm]は、このドナーとアクセプターのペアのフェルスター距離(FRET効率が50%のクロモフォア間距離)である。
さらに、R0[nm]は、
R0=[8.8×10−23・n−4・κ2・Q・J]1/6 (2)
と表すことができる。ただし、κ=配向因子(0≦κ2≦4。自由回転している場合はκ2≒2/3。)、Q=アクセプターがないときのドナーの量子収率、n=溶媒の屈折率、J=重なり積分(ドナーの蛍光スペクトルとアクセプターの吸収スペクトルの重なり。)である。
【0026】
従って、2つの蛍光タンパク質分子間でのFRET効率は、クロモフォア間距離(r)、配向因子(κ)、及び重なり積分(J)の3つのパラメタに依存する。ところが、配向因子は自由回転する分子ではκ2≒2/3と近似できること、さらに、重なり積分はドナーの蛍光スペクトルとアクセプターの励起スペクトルから決まり、2つの蛍光タンパク質の位置関係には依存しないことから、プローブの分子デザインをする上では、クロモフォア間距離が特に重視されてきた。配向因子については分子デザイン段階ではあまり考慮せず、プローブを作製してからFRET効率が高いものを選別するというスタンスである。従って、実用性のあるプローブを作製することは、非常に手間がかかることであり、誰もが容易にできることではない。
【0027】
しかし、発明者らは、二量体を形成する蛍光タンパク質は、2分子間の相対的位置関係が実質的に固定されることに着目し、高いFRET効率を与えるために好ましいクロモフォア間距離(r)と配向因子(κ)となるように自己集合して二量体を形成する蛍光タンパク質の、片方又は両方を改変し、重なり積分が存在するようにすることで、高いFRET効率を実現できる蛍光タンパク質のペアを得られることに想到した。
【0028】
つまり、本発明の蛍光タンパク質は、二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質(以下「未変異型蛍光タンパク質」という。)のアミノ酸配列を、好ましくは、クロモフォア(発色団)形成に関わるアミノ酸を置換、挿入又は欠失等の変異を導入することによって、自己集合性を維持しながら、励起極大波長及び蛍光極大波長がともに異なる蛍光タンパク質(以下「変異型蛍光タンパク質」という。)に改変したものである。
【0029】
前記未変異型蛍光タンパク質と前記変異型蛍光タンパク質とのヘテロ二量体、又は、前記変異型蛍光タンパク質の励起極大波長及び蛍光極大波長がともに相違する2つの変異型蛍光タンパク質からなるヘテロ二量体は、FRET効率のパラメタである、クロモフォア間距離、配向因子、及び、重なり積分が、蛍光タンパク質のペアによって決まるため、プローブの分子デザインが非常に容易である。
【0030】
しかも、これらのヘテロ二量体を形成する蛍光タンパク質は、実質的に励起極大波長及び蛍光極大波長のみが異なるものであり、非常にFRET効率が高いペアが存在すれば、他のペアについては、実質的に、重なり積分のみを考慮すればよいため、高いFRET効率のプローブの分子デザインも容易となる。
【0031】
一方、FRET効率(E)は、次の(3)式で表すこともできる。
E=1−τD´/τD (3)
ここで、τD´はアクセプター存在時のドナーの蛍光寿命、τDはアクセプターがない場合のドナー蛍光寿命を、それぞれ表す。
【0032】
即ち、ドナーの蛍光寿命を測定すれば、FRET効率も容易に得ることができる。
【0033】
ところが、従来の技術水準では、プライマーのFRET効率が低く、FRETの誘起時と解消時で蛍光寿命にあまり差が出ず、10−10〜10−9秒のオーダーの蛍光寿命を精度よく蛍光寿命を測定することが困難であるため、蛍光寿命を測定してFRET効率を求めるのではなく、代わりに、レシオメトリック測定法で近似的にFRET効率が求められる。
【0034】
しかし、本発明の蛍光タンパク質を用いてFRETプローブを構成することによって、FRETの誘起・解消に伴う蛍光寿命変化が大きくなるため、蛍光寿命を測定してFRET効率を求めることが容易である。
【0035】
蛍光寿命測定法では、ドナーのみの蛍光寿命を測定すればよく、細胞内のドナー/アクセプター2種類の蛍光タンパク質の分子数が揃っている必要がない。従って、この測定法は、1分子FRETの場合だけではなく、レシオメトリック測定法ではS/N比が悪い2分子FRETの場合にも、非常に有効である。
【0036】
なお、2分子FRETは、分子間相互作用をする2つの生体分子を、それぞれ、異なる色の蛍光分子で標識し、FRETプローブを構成するものであり、標識された2分子間の相互作用によって、蛍光分子間の距離又は配行性が変化し、FRETが誘起又は解消される。また、一方、1分子FRETは、2つの蛍光分子部分がリンカー部分で連結された構造をもつFRETプローブを使用するものであり、リンカー部分に生体分子の全部又は一部が挿入され、他の生体分子との分子間相互作用又はリンカー部分に挿入された生体分子の全部又は一部の部分の間での相互作用によってリンカー部分の構造が変化し、FRETが誘起又は解消される。
【0037】
また、蛍光寿命測定とフローサイトメトリーのメリットを組み合わせ、細胞を高速で流しながら1細胞ごとに蛍光強度と蛍光寿命を同時に測定できる蛍光寿命フローサイトメーター(例えば、特許文献2)を好適に利用することができる。これにより、多数の細胞を高スループットで処理し、精度良く生体分子の分子間相互作用や分子内構造変化を検出できる。
【0038】
本発明では、前記の変異型蛍光タンパク質をコードする核酸、及び、該核酸を組み込んだ遺伝子発現ベクター(発現コンストラクト)を提供する。発現ベクター(発現コンストラクト)を培養細胞にトランスフェクトして形質転換し、細胞内で前記変異型蛍光タンパク質を発現することができる。
【0039】
本発明では、前記の変異型蛍光タンパク質を含むプローブ(蛍光タンパク質部分と生体分子部分の融合タンパク質)をコードする核酸、及び、該核酸を組み込んだ発現ベクター(発現コンストラクト)及び該ベクターを作成する方法を提供する。また、該ベクターによって形質転換された培養細胞を提供する。さらに、前記核酸、前記発現ベクター、又は前記形質転換された培養細胞を含むキットが提供される。これらにより、プローブの作成と使用が、より容易となる。
【0040】
さらに、細胞内で起こるFRET効率の変化を算出するための、好ましい方法として、ドナーの蛍光寿命を測定してFRET効率を算出する方法が提供される。好ましい一態様では特に、三井造船(株)製の蛍光寿命フローサイトメーター“Flicyme”(登録商標)を使用して、効率的に大量の細胞の蛍光寿命を測定することができる。
【0041】
即ち、本発明は以下のものである。
〔1〕二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質のアミノ酸配列に変異が導入された変異型蛍光タンパク質であって、励起極大波長及び蛍光極大波長がともに変異導入前の蛍光タンパク質(以下、未変異型蛍光タンパク質という。)からシフトし、かつ、該未変異型蛍光タンパク質若しくは励起極大波長及び蛍光極大波長がともに相違する他の変異型蛍光タンパク質とヘテロ二量体を形成できる、変異型蛍光タンパク質。
〔2〕〔1〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔3〕前記未変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができる、〔1〕に記載の変異型蛍光タンパク質。
〔4〕〔3〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔5〕前記変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができ、変異型蛍光タンパク質とは励起極大波長及び蛍光極大波長がともに相違する、〔1〕に記載の変異型蛍光タンパク質。
〔6〕〔5〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔7〕前記未変異型蛍光タンパク質が、サンゴ(Discosoma sp.)に由来する蛍光タンパク質である、〔1〕に記載の変異型蛍光タンパク質。
〔8〕前記蛍光タンパク質が、DsRed1である、〔7〕に記載の変異型蛍光タンパク質。
〔9〕配列番号1に記載のDNA塩基配列を翻訳して得られるアミノ酸配列をもつ蛍光タンパク質。
〔10〕配列番号2に記載のアミノ酸配列をもつ蛍光タンパク質。
〔11〕〔3〕又は〔5〕に記載の2つの蛍光タンパク質を含むFRETプローブ。
〔12〕〔3〕又は〔5〕に記載の2つの蛍光タンパク質がポリペプチドからなるリンカーを介して連結される構造をもつ融合タンパク質であるFRETプローブ。
〔13〕前記リンカーが生体分子の全部又は一部のアミノ酸配列を含む、〔11〕に記載のFRETプローブ。
〔14〕前記2つの蛍光タンパク質がDsG2(配列番号2)及びDsRed1である、〔11〕又は12に記載のFRETプローブ。
〔15〕前記生体分子がSUMO1である〔12〕又は〔13〕に記載のFRETプローブ。
〔16〕〔11〕〜〔14〕のいずれかに記載のFRETプローブをコードするポリヌクレオチド。
〔17〕〔11〕〜〔14〕のいずれかに記載のFRETプローブをコードするDNAを遺伝子発現ベクターに組み込んで構築した発現ベクター。
〔18〕前記遺伝子発現ベクターが、pEB6CAGmcs、pEB6SRαmcs及びpEB6CMVmcsからなる群から選ばれる、〔17〕に記載の発現ベクター。
〔19〕前記発現ベクターが、pEB6CMVmcs−SRZである、〔18〕に記載の発現ベクター。
〔20〕〔17〕〜〔19〕のいずれかに記載の発現ベクターを培養細胞に導入した形質転換細胞。
〔21〕前記培養細胞がヒト由来細胞株である、〔20〕に記載の形質転換細胞。
〔22〕前記ヒト由来細胞株がHEp−2細胞株である、〔21〕に記載の形質転換細胞。
〔23〕〔17〕〜〔19〕のいずれかに記載の発現ベクターを含んでなるキット。
〔24〕〔20〕〜〔22〕のいずれかに記載の形質転換細胞を含んでなるキット。
〔25〕〔11〕〜〔15〕のいずれかに記載のFRETプローブを用いる、ドナー蛍光タンパク質の蛍光寿命を測定して細胞内FRETを検出する方法。
〔26〕蛍光寿命フローサイトメーターを使用して蛍光寿命を測定する、〔25〕に記載の方法。
〔27〕前記蛍光寿命フローサイトメーターが、好ましくは、Flicyme(登録商標)である、〔26〕に記載の方法。
【発明の効果】
【0042】
本発明の自己集合性蛍光タンパク質のペアを用いることによって、自発的に会合して二量体が形成されるため、プローブの分子デザインをする際に分子間距離と分子の配向性を考慮する必要がなく、スペクトルの重なりを考慮すれば、FRET効率の高いプローブの分子デザインをすることが従来に比べて非常に容易となった。また、FRET効率が高く、蛍光寿命変化も大きいことから、蛍光寿命フローサイトメーターを用いて蛍光寿命を精度良く測定し、FRET効率の変化を算出する用途に好適に用いることができる。
【発明を実施するための最良の形態】
【0043】
<1>本発明の蛍光タンパク質について説明する。
本発明の蛍光タンパク質は、二量体又は三量体以上の多量体を形成しやすい、自己集合性をもつ蛍光タンパク質である。好ましくは、自己集合性をもち、二量体又は三量体以上の多量体を形成しやすい蛍光タンパク質(以下、原型蛍光タンパク質という。)の、アミノ酸配列に変異を導入することによって、自己集合性を維持しながら蛍光極大波長及び励起極大波長をシフトさせた、変異型蛍光タンパク質である。より好ましくは、原型蛍光タンパク質と変異型蛍光タンパク質とで、又は、互いに異なる変異型蛍光タンパク質同士で、ヘテロ二量体を形成する。さらに好ましくは、前記へテロ二量体形成時に、2つの蛍光タンパク質の、クロモフォア間距離、遷移双極子モーメントの相対的向き、及び、励起波長が短い方の分子(ドナー)の蛍光スペクトルと、励起波長が長い方の分子(アクセプター)の励起スペクトルとの重なり(スペクトル・オーバーラップ)が、該ドナーから該アクセプターへのFRETを起こす条件を満たす。いっそう好ましくは、前記ドナーと前記アクセプターの間の、クロモフォア間距離rとフェルスター距離(FRET効率が50%になるクロモフォア間距離)について(r/R0)6を最小にするような、r、κ及びJ(但し、R0=[8.8×10−23・n−4・κ2・Q・J]1/6、κは双極子の配向因子、Jはドナーの蛍光スペクトルとアクセプターの励起スペクトルの重なり積分、Qはアクセプターがないときのドナーの量子収率、nは葉液の屈折率である。)を与えるヘテロ二量体を構成する変異型蛍光タンパク質である。
【0044】
前記変異型蛍光タンパク質は、当業者にとって自明な、コンベンショナルな遺伝子工学的手法を用いて、発現及び精製をして、取得することができる。
より詳細には、例えば、変異型蛍光タンパク質の原型蛍光タンパク質をコードする組み換えcDNAを合成し、これを適当な遺伝子発現ベクターに組み込んで発現ベクターを構築し、適切な細胞にコンベンショナルな方法でトランスフェクトしてトランスフォーメーションし、蛍光タンパク質を細胞中に発現させる。所望により、前記発現ベクターは、トランスフェクション前に大腸菌等を用いて大量調製することもできる。さらに、前記遺伝子発現ベクターに組み込まれている選択マーカーを利用して、トランスフェクションの成否とトランスフォーメーションの成否によって細胞を選択することができる。細胞内で発現される蛍光タンパク質は、コンベンショナルな方法で分離・精製をすることができる。
【0045】
本発明の好ましい変異型蛍光タンパク質は、サンゴ(Discosoma sp.)由来の赤色蛍光タンパク質DsRedの変異体であるDsRed1に、遺伝子組み換えにより、83番目のリジンをアルギニン、120番目のチロシンをヒスチジン、197番目のセリンをスレオニンに置換する変異を導入して作出した緑色蛍光タンパク質DsG2である。DsG2はDsRed1とヘテロ二量体を形成し、そのとき、DsG2の励起エネルギーがDsRed1に移動してFRETが起こり、DsRed1の赤い蛍光が発生する。
【0046】
DsRedは、そのクロモフォア形成及び成熟の過程において、イクオレアGFPと同様に一度酸化還元反応を行って緑色の発色団を形成した後に、再度酸化還元反応を行い赤色の発色団を形成することが知られている(図1)が、緑色蛍光を発するクロモフォアが形成された段階で酸化還元反応が停止したDsRed1の緑色変異体は、DsRed1とヘテロ二量体を形成し、該DsRed1緑色変異体の蛍光スペクトルとDsRed1の励起スペクトルの重なりがFRETを誘起するのに十分であるといえるから、本発明の変異型蛍光タンパク質の範囲に含まれる。
【0047】
なお、本明細書において、DsRedは、サンゴ(学名:Discosoma sp.)の赤色蛍光タンパク質であり、DsRed1は、サンゴのDsRedのコドン使用パターンをヒト型に置換した変異型であり、DsRed2は、DsRed1に点変異を導入してタンパク質凝集の傾向が抑えられた変異型である。また、GFPは、オワンクラゲ(Aquorea victoria)の緑色蛍光タンパク質であり、EGFPはクラゲのGFPのコドン使用パターンをヒト型に置換した変異型である。
【0048】
<2>本発明のプローブについて説明する。
本発明のプローブに、技術常識に反して自己集合性を持つ蛍光タンパク質をあえて用いる理由は、アクセプターとドナーの2つの蛍光タンパク質が、必ずその分子特性によって決まった角度でしかも安定に結合することを意図したものであるが、必ずしもこの意図に限定されない。この角度がFRETを起こすため好ましい角度であるならば、2種類の蛍光タンパク質に付加するアミノ酸配列や他のタンパク質に関して、所望により、あまり詳細な検討を必要とせず、確実に高いFRET効率を実現できるように蛍光タンパク質が自発的に会合することを期待してもよい。従って、これまでに開発されてきたプローブと比較して、著しくFRET効率の高いプローブを誰もが非常に簡便に開発することが可能になることを意図するものであるが、必ずしもこの意図に限定して解釈されるべきではない。
【0049】
本発明のプローブは、1分子プローブであっても2分子プローブであってもよいが、1分子プローブのときは、2つの蛍光タンパク質部分がリンカー部分を介して連結される構造となる。ここで、2つの蛍光タンパク質部分は、一方が前記変異型蛍光タンパク質であれば、他方は原型蛍光タンパク質であっても変異型蛍光タンパク質であってもよい。リンカー部分には、好ましくは、細胞内で分子間相互作用又は分子内構造変化を受けるタンパク質又はポリペプチドのアミノ酸配列を含む。さらに、前記アミノ酸配列は、前記タンパク質又はポリペプチドの全体又は部分であってもよいし、複数の全体又は部分が含まれてもよい。細胞内での分子間相互作用を解析するためには、好ましくは、ある一態様では、リンカー部分にタンパク質又はポリペプチドの全体又は部分を1つ含み、細胞内の標識されていない分子と相互作用することでFRETが誘起又は解消することによって分子間相互作用を検出する。また、細胞内での分子間相互作用を解析するためには、好ましくは、別の一態様では、リンカー部分にタンパク質又はポリペプチドの全体又は部分を1つ又は2つリンカーに含み、リンカー部分が構造変化することでFRETが誘起又は解消することによって分子内構造変化を検出する。前記1分子プローブは、好ましくは、蛍光タンパク質−リンカー−蛍光タンパク質の三重融合タンパク質として細胞内で発現される。また、2分子プローブのときには、好ましくは、2つの分子間相互作用をするタンパク質又はポリペプチドを、それぞれ蛍光タンパク質で標識する構造となる。ここで、2つの蛍光タンパク質は、一方が前記変異型蛍光タンパク質であれば、他方は原型蛍光タンパク質であっても変異型蛍光タンパク質であってもよい。好ましい態様では、前記タンパク質又はポリペプチドが結合すると、2つの蛍光タンパク質も接近し、会合し、ヘテロダイマーを形成し、FRETが起こり、前記結合が解かれると、2つの蛍光タンパク質も解離し、FRETも解消する。前記プローブは、好ましくは、蛍光タンパク質−タンパク質若しくはペプチド、又は、タンパク質若しくはペプチド−蛍光タンパク質の構造をもつ融合タンパク質として細胞内で発現される。蛍光タンパク質は、前記タンパク質又はポリペプチドのN末端又はC末端のどちらか一方に連結されればよい。前記融合タンパク質は、プローブを構成する2つの融合タンパク質分子が、別個の発現ベクター上にコードされ、二重トランスフェクトされた細胞で発現されてもよい。
【0050】
本発明の1分子プローブは、好ましい一態様では、DsG2とDsRed1をSUMO1のアミノ酸配列を含むリンカーで連結した三重融合タンパク質である。ユビキチン様タンパク質SUMO1の前駆型タンパク質のC末端のグリシンで切断されて成熟型となることから、前記のリンカー部分にSUMO1前駆型タンパク質のアミノ酸配列を含むFRETプローブでは、リンカー部分が切断されることによってFRETが解消し、アクセプターの蛍光からドナーの蛍光に変化することによって、細胞内で起こる分子間相互作用を検出することを意図したものであるが、この意図に限定されて解釈されるものではない。
【0051】
また、前記の三重融合タンパク質は、例えば、ヒトSUMO1遺伝子のORFをはさむオリゴDNAを合成し、これを用いてヒトHEp−2細胞由来のcDNAライブラリーよりPCR反応によって野生型SUMO1のcDNAを調製し、SUMO1の終止コドンのかわりに他のアミノ酸をコードするように置換した塩基配列とその下流に制限酵素によって認識されて切断される配列を有するオリゴDNAを合成し、これを用いてPCR反応を行い、終止コドンを持たないSUMO1のcDNAを構築し、DsG2のcDNAの下流に、オリゴDNA、Link3−1SE(配列情報3)とLink3−1AS(配列情報5)をアニーリングさせた19アミノ酸からなるリンカー配列(LG3)を連結し、作成したSUMO1のcDNAを読み枠が合うように連結し、さらにその下流に再度LG3を連結し、読み枠が合うようにDsRed1のcDNAを連結して、三重融合タンパク質「DsG2−SUMO1−DsRed1」をコードするcDNAを作成することができるが、この方法に限定されるものではなく、当業者が想到し得るいかなる方法でも用いることが可能である。
前記のcDNAを細胞中で発現する好ましい態様は、前記cDNAを“pEB6CAGmcs”遺伝子発現ベクターに組み込み、「DsG2−SUMO1−DsRed1」を発現するベクターを構築し、大腸菌から大量調製し、調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)存在下で培養することによって、細胞中で発現することができるが、この手順に限定されず、当業者が想到し得るいかなる方法でも用いることが可能である。
【0052】
本発明の2分子プローブは、好ましい一態様では、DsG2とRunx2RDを連結した融合タンパク質とDsRed1とCBFβを連結した融合タンパク質の2分子からなる。Runx2RDとCBFβは1:1で結合するため、細胞中で前記の融合タンパク質が両方とも発現され、両者が結合するとFRETが起こり、分離するとFRETが解消することによって、細胞内で起こる2分子間の相互作用を検出することを意図するものであるが、この意図に限定して解釈されるものではない。
【0053】
また、前記の2種類の融合タンパク質は、例えば、マウスRUNX2遺伝子のRuntドメイン(RD)をコードするDNA断片を調製し、前記の「DsG2−SUMO1−DsRed1」を発現するベクターのSUMO1−DsRed1をコードする部分と置き換えて、「DsG2−Runx2RD」を構築することができる。同様に、CBFβをコードするDNA断片を調製し、前記の「DsG2−SUMO1−DsRed1」を発現するベクターのDsG2−SUMO1をコードする部分と置き換えて「CBFβ−DsRed1」を発現するベクターを構築することができる。この方法に限定されることなく、当業者が想到し得るいかなる方法でも用いることが可能である。
【0054】
さらに、好ましくは、このcDNAを切り出してゼオシン耐性遺伝子を持つベクター“pEB6CAGmcs−SRZ”に乗せ替えることにより、G418とゼオシンの二重選択によって、「DsG2−Runx2RD」と「CBFβ−DsRed1」を同時に細胞に発現できるベクターセットを構築することができる。
【0055】
また、所望により、FRETが起こらない場合の比較実験用に、「CBFβ−DsRed1」のDsRed1の発色団(クロモフォア)を構成するアミノ酸配列を改変し、FRETアクセプターとしてまったく機能できないようにしたものを作成して用いてもよい。好ましくは、67番目のチロシンをスレオニンに置換した変異を導入した「CBFβ−DsRed1Y67T」を構築してもよい。
【0056】
構築した発現ベクターは、好ましくは、市販のDNA精製キットを用いて大腸菌から大量調製することができる。調製した発現ベクターは、好ましくは、市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)、0.1mg/ml ゼオシン(インビトロジェン社)両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択することができる。この手順に限定されず、当業者が想到し得るいかなる方法でも用いることができる。
【0057】
<3>本発明のFRET検出法について説明する。
本発明の好ましい一態様では、フローサイトメーターによって蛍光強度を測定し、FRETの誘起・解消を検出することができる。
例えば、SUMO1を用いたプローブは細胞内で切断を受けるため、FRETが解消して緑色蛍光が多くなるが、SUMO1GAは切断を受けないためFRETが誘起されて赤色蛍光が多くなる。ある態様では、SUMO1を用いたプローブでは、赤い蛍光と緑の蛍光の比は1.1と最も低くなるのに対して、SUMO1GAを用いたプローブでは切断を受けないため、高い効率のFRETが起こり、緑の蛍光は減弱し赤い蛍光が強くなるために蛍光強度比が19.7にまで上がる。
【0058】
本発明の別の好ましい一態様では、蛍光顕微鏡による観察によってFRETを検出することができる。例えば、DsG2−SUMO1−DsRed1又は変異RUBYG2−SUMO1GA−DsRed1を発現させた細胞を倒立型顕微鏡で撮影すると、DsG2−SUMO1−DsRed1は緑も赤も核内にドット状に観察されるが、変異RUBYG2−SUMO1GA−DsRed1は赤のみが細胞質に観察され、これは、SUMO1が活性化されるとFRETが解消され緑の蛍光が観察されるようになるが、SUMO1の活性化がおこらない場合にはFRETが誘起されDsG2の蛍光エネルギーがDsRed1に移動していると解釈できる。
【0059】
本発明のさらに別の好ましい一態様では、フローサイトメーターによって蛍光緩和時定数(蛍光寿命)を測定してFRETを検出することができる。
【0060】
例えば、フローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて、td−DsG2、td−DsRed1及びDsG2−LG3−DsRed1の蛍光緩和時定数を測定すると、td−DsG2の緑色の緩和時定数は2.1nsであり、td−DsG2とtd−DsRed1を同一細胞に共発現させると緑も赤もそれぞれ蛍光を発するため緑色の緩和時定数はtd−DsG2単独発現時とほとんど同じ2.0nsであるが、DsG2−LG3−DsRed1の緑色の緩和時定数は1.3nsに減少することから、DsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移していることが確認される。また別の例では、細胞内でのSUMO1の活性化をFRETにより検出することを試みるとすると、DsG2−SUMO1−LG3−DsRed1の緑色緩和時定数は2.1ns(td−DsG2と同じ)であるものの、切断されないSUMO1GA変異を用いたDsG2−SUMO1GA−LG3−DsRed1の緑色緩和時定数は1.5nsに短縮することから、蛍光強度の著しい変動は、FRETによるものであり、しかも蛍光寿命の測定によってタンパク質の切断を検出可能であることが示される。
【0061】
また、例えば、フローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて、HEp−2細胞で発現させたDsG2−Runx2RDとCBFβ−野生型DsRed1とからなる2分子プローブの蛍光緩和時定数(蛍光寿命)を測定すると、ドナー(DsG2)の緑の蛍光の緩和時定数は1.52nsであるのに対し、DsG2−Runx2RDとCBFβ−DsRed1Y67T変異(DsRed1Y67T変異体:発色団となる67番目のチロシンがスレオニンに置換され、アクセプターにならない。)とからなる2分子プローブの蛍光緩和時定数(蛍光寿命)を測定すると、ドナーの蛍光緩和時定数は2.23nsであり、前者では高い効率でFRETが起こっていることが確認できる。
【0062】
よって、本発明のFRETプローブを用いる場合、FRETの検出は、好ましくは、蛍光顕微鏡法、レシオメトリー測定法又は蛍光寿命測定法によって行うことができる。より好ましくは、蛍光寿命測定法によって行うことができる。また、フローサイトメトリと組み合わせてFRETの検出を行ってもよい。フローサイトメトリと組み合わせるのは、単位時間あたりにより多くの細胞を観察することを意図したものであるが、その意図に限定されて解釈されるものではない。
【0063】
以下に、実施例により本発明を説明するが、本発明の範囲はこれらの実施例によって限定されない。
【実施例】
【0064】
1:変異型DsRedの調製及び蛍光特性の検証
[1]材料及び方法
A.cDNAの調製
サンゴ(Discosoma sp.)由来の赤色蛍光タンパク質DsRedのバリアントであるDsRed1に、遺伝子組み換えにより、83番目のリジンをアルギニン、120番目のチロシンをヒスチジン、197番目のセリンをスレオニンに置換する変異を導入し、変異型DsRed1である緑色蛍光タンパク質を作出した。この緑色蛍光タンパク質をDsG2と命名した(配列番号2)。手順は、以下の通りである。
(1)導入しようとするアミノ酸置換を含むアミノ酸配列をコードするオリゴヌクレオチドを合成し、これを用いてPCR反応によって、変異を含むDNA断片を調製した。
(2)これを野生型DsRed1タンパク質をコードするDNAの当該部分と入れ替えることにより、上記変異を導入したcDNAを調製した。
【0065】
次に、前記cDNAをタンデムに融合させた。手順は、以下の通りである。
(1)DsRed1をコードするDNA及びDsG2をコードするDNA(配列番号1)の終止コドンを他のアミノ酸に対応するコドンに置換した塩基配列をもち、その下流に制限酵素によって認識されて切断される配列を含むオリゴDNAを合成した。
(2)これを用いてPCR反応を行い、終止コドンを持たない野生型又は変異導入DsRed1をコードするDNA断片を調製した。
(3)同一のタンパク質同士を合成の翻訳の読み枠が合うように連結した(タンデムダイマーDsRed1;td−DsRed1)。
【0066】
B.発現ベクターの作製
上記のように作製したcDNAを、本発明者らが開発した、ヒト細胞中で安定に複製・維持される遺伝子発現ベクター“pEB6CAGmcs”、“pEB6SRαmcs”又は“pEB6CMVmcs”に組み込んだ(これらの発現ベクターは本発明者から入手可能である)。構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0067】
C.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間1.5mg/ml G418存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(FACSCalibur(登録商標),ベクトン・ディッキンソン社)で蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0068】
D.蛍光顕微鏡による観察
形質転換細胞の蛍光特性及び細胞内局在を、蛍光顕微鏡(IX70,オリンパス社)及びそれに接続したCCDカメラ(SenSys(登録商標),フォトメトリクス社)で観察した。緑色蛍光の観察は495±10nmの励起フィルター及び535±15nmの蛍光フィルターで、赤色蛍光の観察は555±12nmの励起フィルター及び620±30nmの蛍光フィルターで観察した。
【0069】
[2]結果
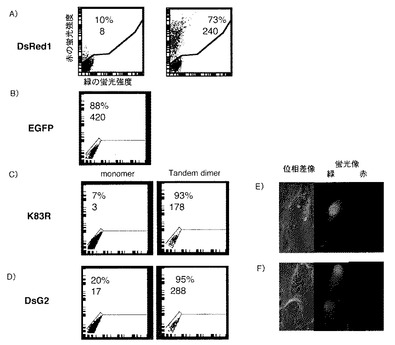
フローサイトメーター及び蛍光顕微鏡による蛍光強度の測定
図2A〜Dは、緑色蛍光タンパク質EGFP、野生型DsRed1及び変異DsRed1の蛍光特性を、フローサイトメーターを用いて試験した結果を示すプロットである。
図2Aに示すように、野生型td−DsRed1を発現させた細胞では赤色蛍光が認められた。
野生型DsRed1の83番目のリシンをアルギニンに置換した変異体K83Rでは、確かに赤から緑に蛍光特性が変化しているものの(図2C)、まだ波長の変化が不十分であり赤い波長成分がかなり残っているため、一般的な蛍光顕微鏡において赤い蛍光を観察するためのフィルターセットで観察されてしまう(図2E)。
一方、本発明でさらにY120H及びS197Tの変異を2つ追加して開発したDsG2では赤い蛍光成分が軽減しているため、フローサイトメーターの結果において、より下方に細胞のプロットが現れていた(図2D)。
図2FはDsG2の蛍光特性を蛍光顕微鏡を用いて試験した結果を示す図である。図2Eと同様のフィルターセットにおいて、赤い蛍光がほとんど消失していた。このことから、本発明で開発したDsG2は、緑の蛍光タンパク質として用いることができる。
【0070】
2:DsG2−DsRed1間のFRET効率の評価
[1]材料及び方法
A.cDNAの作製
上記のように作製したtd−DsG2とtd−DsRed1のcDNAを途中のリンカー部分で組み換えてタンデムに融合させ、DsG2−DsRed1を作成した。
【0071】
B.遺伝子発現ベクターの作製
上記のように作製したcDNAを、本発明者らが開発した、ヒト細胞中で安定に複製・維持される遺伝子発現ベクター“pEB6CAGmcs”に組み込んだ。またtd−DsRed1のcDNAを異なる薬剤耐性遺伝子をもつ同等のベクター構築した“pEB6CAGmcs−SRZ”に組み込んで二重トランスフェクションができるようにした。発現ベクターDNAは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0072】
C.形質転換細胞の作製及び選択
調製した遺伝子発現ベクターDNAを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し4日間、1.5mg/ml G418(ロシュ社)存在下、0.1mg/ml ゼオシン(インビトロジェン社)存在下、又は両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(FACSCalibur(登録商標),ベクトン・ディッキンソン社)で蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0073】
[2]結果
フローサイトメーターによる蛍光強度の測定
図3Aに示すように、DsG2単独の細胞は水平方向に、図3Bに示すように、DsRed1単独の細胞は垂直方向にプロットされるように、装置の設定を調整した。この条件下で、DsG2とDsRed1を二重導入した細胞を測定したところ、緑の平均蛍光強度はやや弱くなり、赤の平均蛍光強度はやや強くなっており、赤い蛍光と緑の蛍光の強度比は2.1であった。
一方、DsG2とDsRed1がタンデムに連結された融合タンパク質を発現する細胞を解析したところ、赤い蛍光は図3B及び図3Cと比較して著しく増強されていたのに対し、緑の蛍光強度は、細胞内で緑色蛍光タンパク質がかなり発現されているにもかかわらず、図3A及び図3Cと比較して著しく減弱していた。赤い蛍光と緑の蛍光の強度比は16.4と別々に発現させた場合の8倍の比を示し、明らかにDsG2の励起エネルギーがDsRed1に移動するFRETが高い効率で起こっていることが示唆された。
【0074】
3:FRET検出システムの応用と性能の評価
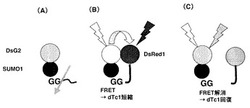
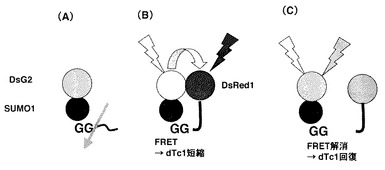
生きた細胞内でのFRET効率の変化と生命現象との関連を調べるため、ユビキチン様(UBL)タンパク質のひとつであるSUMO1について応用することを試みた。
SUMO1は、SUMOタンパク質のひとつである。SUMOタンパク質は、細胞内の他のタンパク質に一時的に共有結合してその機能を助ける小さなタンパク質であり、タンパク質のSUMO化は、細胞核−細胞質の輸送、転写制御、アポトーシス、タンパク質の安定化、ストレス応答、細胞周期の進行など様々な細胞内のプロセスに関係する翻訳後修飾の1つである。
図4はプローブのデザインと、働く機構の概念図である。SUMO1は、前駆型タンパク質のC末端付近のグリシンの後ろでプロペプチドが切断されて成熟型となり、活性化して、基質に結合することが知られている(図4A)。そこで、DsG2の下流にSUMO1を融合し、さらにその下流にDsRed1を融合した、「DsG2−SUMO1−DsRed1」三重融合タンパク質を細胞中に発現させると、初めはSUMO1が切断による活性化を受けていない(図4B)ため、特別に工夫されたリンカー配列を使っていなくても、DsG2とDsRed1が分子内で自発的に会合し、極めて高い効率でFRETが誘起されるために、緑の蛍光は著しく減弱し、赤い蛍光が高くなる。これに伴って、ドナーの蛍光寿命dTc1が短縮する。一方、SUMO1が活性化され切断が起こると、DsG2とDsRed1が解離し(図4C)、FRETが解消されて緑の蛍光が強く検出されるようになり、蛍光寿命dTc1がもとの長さに回復する。
【0075】
[1]材料及び方法
以上のモデルを検証するために、以下の手順で実験を行った。
A.cDNAの調製
ヒトSUMO1遺伝子のORFをはさむオリゴDNAを合成し、これを用いてヒトHEp−2細胞由来のcDNAライブラリーよりPCR反応によって野生型SUMO1のcDNAを調製した。SUMO1の終止コドンのかわりに他のアミノ酸をコードするように置換した塩基配列とその下流に制限酵素によって認識されて切断される配列を有するオリゴDNAを合成し、これを用いてPCR反応を行い、終止コドンを持たないSUMO1のcDNAを構築した。実施例1で作製したDsG2のcDNAの下流に、19アミノ酸からなるリンカー配列(LG3)を連結し、作成したSUMO1のcDNAを読み枠が合うように連結した。さらにその下流に再度LG3を連結した後、読み枠が合うようにDsRed1のcDNAを連結しDsG2−LG3−SUMO1−LG3−DsRed1を作成した。
また、切断を受けない比較実験用に、SUMO1のC末近くのグリシンをアラニンに置換した変異体SUMO1GAも作成し、同様のプローブDsG2−LG3−SUMO1GA−LG3−DsRed1を作成した。
【0076】
B.発現ベクターの作製
上記のように作製したcDNAを、“pEB6CAGmcs”ベクターに組み込んだ。構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0077】
C.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(Flicyme(登録商標),三井造船(株))で蛍光緩和時定数(蛍光寿命)、蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0078】
D.蛍光顕微鏡による観察
形質転換細胞を用い、形質転換細胞の蛍光特性及び細胞内局在を、蛍光顕微鏡(IX70,オリンパス社)及びそれに接続したCCDカメラ(SenSys(登録商標),フォトメトリクス社)で観察した。緑色蛍光の観察は495±10nmの励起フィルター及び535±15nmの蛍光フィルターで、赤色蛍光の観察は555±12nmの励起フィルター及び620±30nmの蛍光フィルターで観察した。
【0079】
[2]結果
A.フローサイトメーターによる蛍光強度の測定
実施例2と同様にフローサイトメーターのパラメーターを調整して測定を行った。図5Eに示すように、赤い蛍光が非常に弱くなり検出される蛍光は緑が多くなったため、赤い蛍光と緑の蛍光の比は1.1と最も低くなった。一方、切断を受けないSUMO1GAを用いたプローブでは図5Dと同様に高い効率のFRETが起こり、緑の蛍光は減弱し赤い蛍光が強くなったために蛍光強度比が19.7にまで上がった。以上のことから、2つの蛍光タンパク質の間にSUMO1を挿入した場合にも、非常に簡単に高いFRET効率を実現することができ、しかもSUMO1の切断によって、このFRETが解消することから、タンパク質の切断を極めて高感度にFRETを用いて解析できることが明らかとなった。
【0080】
B.蛍光顕微鏡による観察
図5FとHは、DsG2−SUMO1−DsRed1あるいはDsG2−SUMO1GA−DsRed1を発現させた細胞を倒立型顕微鏡で撮影した図である。示されるように、DsG2−SUMO1−DsRed1は緑も赤も核内にドット状に観察された。一方、変異RUBYG2−SUMO1GA−DsRed1は赤のみが細胞質に観察された。このことは図4の概念図に示した通り、SUMO1が活性化されるとFRETが解消され緑の蛍光が観察されるようになるが、SUMO1の活性化がおこらない場合にはFRETが誘起されDsG2の蛍光エネルギーがDsRed1に移動していることを示唆する。また、SUMO1の場合には、切断による活性化に伴う細胞内局在の変化(細胞質から核への移行)もイメージングできることが示された。
【0081】
C.フローサイトメーターによる蛍光緩和時定数の測定
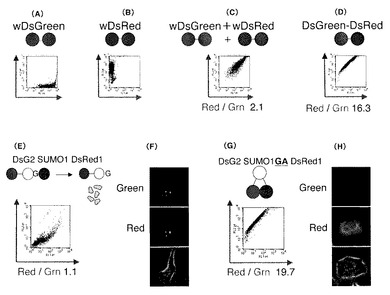
図6は、td−DsG2、td−野生型DsRed1及びDsG2−LG3−DsRed1の蛍光緩和時定数をフローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて試験した結果を示すヒストグラムである。
td−DsG2の緑色の緩和時定数は2.1nsだった(図6A)。
td−DsRed1の赤色の蛍光は緑のチャンネルにはほとんど入らないため細胞の自家蛍光成分と考えられる弱い蛍光のみが検出され、緑色の緩和時定数は1.3nsだったが細胞間のばらつきが非常に大きく、蛍光タンパク質由来の蛍光寿命とは明らかに異なった。(図6B)。
td−DsG2とtd−DsRed1を同一細胞に共発現させると緑も赤もそれぞれ蛍光を発するため緑色の緩和時定数はtd−DsG2単独発現細胞の図7Aとほとんど同じ2.0nsだった(図6C)。
しかし、DsG2−LG3−DsRed1の緑色の緩和時定数は1.3nsに減少した。また赤い蛍光においてはわずかだが蛍光寿命の延長も見られた(図6D)。
このことからDsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移していることが確認された。
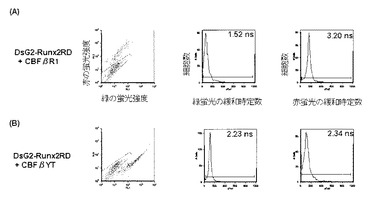
そこで、細胞内でのSUMO1の活性化をFRETにより検出することを試みた。DsG2−SUMO1−LG3−DsRed1の緑色緩和時定数は2.1nsで、td−DsG2と同じだった(図7A)。
しかしながら、切断されないSUMO1GA変異を用いたDsG2−SUMO1GA−LG3−DsRed1の緑色緩和時定数は1.5nsに短縮した(図7B)。
このことから、蛍光強度の著しい変動は、FRETによるものであり、しかも蛍光寿命の測定によってタンパク質の切断を検出可能であることが示された。
【0082】
次に、この蛍光寿命の変化が、DsG2がSUMO1と結合したことによる何らかの影響によるものかどうかを確認する実験を行った。
図8はEGFPあるいはDsG2にSUMO1を融合させたときの蛍光寿命dTc1を示した図である。EGFP、DsG2ともにSUMO1あるいはSUMO1GAを融合させてもdTc1はtd−DSG2とほとんどかわらなかった。これによりSUMO1との融合で蛍光寿命が減少した訳ではないことが確認された。
従って、DsG2−SUMO1GA−DsRed1での蛍光寿命の短縮は、DsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移したことがわかった。
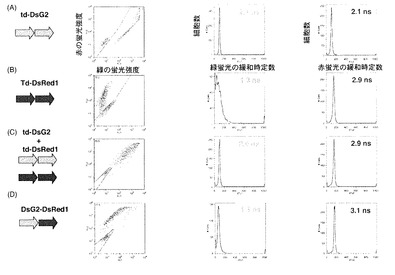
図9はDsG2の蛍光強度が蛍光寿命dTc1に及ぼす影響を検討した結果を示す図である。蛍光強度を変えるために用いたプロモーターは、本発明者らがプロモーター強度を確認済みのCAGプロモーター、SRαプロモーター。CMVプロモーターである(プロモーター強度:CAGプロモーター>SRαプロモーター>CMVプロモーター)。
図9Aに示すように、いずれのプロモーターでもほとんどすべての細胞で蛍光が観察され、その蛍光強度はCAGプロモーター>SRαプロモーター>CMVプロモーターだった。
一方、図9Bに示すように、dTc1にはほとんど違いはなかった。
このことからDsG2の蛍光強度はdTc1にほとんど影響しておらず、dTc1の短縮は蛍光強度の違いによるものではなく、FRETによるものであることが確認された。
【0083】
4:2分子間FRET検出システムへの応用と性能の評価
生きた細胞内でのタンパク質間相互作用を本発明を応用した2分子間FRETによって検出できる可能性を調べるため、転写因子Runx2とCBFβ間での結合について応用することを試みた。
転写因子Runx2は、骨芽細胞の分化を制御することで知られる転写因子であり、その結合パートナーであるCBFβと1:1で結合する。
【0084】
[1]材料及び方法
以上のモデルを検証するために、以下の手順で実験を行った。
A.発現ベクターの作製
マウスRUNX2遺伝子のRuntドメイン(RD)をコードするDNA断片を調製し、上記の「DsG2−SUMO1−DsRed1」を発現するベクターのSUMO1−DsRed1をコードする部分と置き換えて、「DsG2−Runx2RD」を構築した。同様にCBFβをコードするDNA断片を、「DsG2−SUMO1−DsRed1」を発現するベクターのDsG2−SUMO1をコードする部分と置き換えて「CBFβ−DsRed1」を発現するベクターを構築した後、このcDNAを切り出してゼオシン耐性遺伝子を持つベクター“pEB6CAGmcs−SRZ”に乗せ替えた。これにより、G418とゼオシンの二重選択によって、「DsG2−Runx2RD」と「CBFβ−DsRed1」を同時に細胞に発現できるベクターセットを構築した。
また、FRETが起こらない場合の比較実験用に、「CBFβ−DsRed1」のDsRed1の67番目のチロシンをスレオニンに置換した変異を導入した「CBFβ−DsRed1Y67T」も構築し、FRETアクセプターとしてまったく機能できないようにした。
構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0085】
B.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)、0.1mg/ml ゼオシン(インビトロジェン社)両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(Flicyme(登録商標),三井造船(株))で蛍光緩和時定数、蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0086】
[2]結果
フローサイトメーターによる蛍光緩和時定数の測定
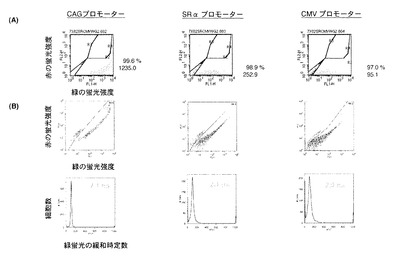
図10は、DsG2−Runx2RDと組み合わせて、CBFβ−野生型DsRed1又はCBFβ−DsRed1Y67T変異を細胞に発現させての蛍光緩和時定数をフローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて試験した結果を示すヒストグラムである。
DsG2−Runx2RDとCBFβ−野生型DsRed1(野生型のtd−DsRed1)との組み合わせを用いた場合には、ドナー(DsG2)の緑の蛍光の緩和時定数は1.52nsであった(図10A)。
一方、DsG2−Runx2RDとCBFβ−DsRed1Y67T変異(67番目のチロシンがスレオニンに置換されている)との組合せを用いた場合には、ドナーの蛍光緩和時定数は2.23nsであり、DsG2のままであった(図10B)。CBFβ−DsRed1Y67T変異はタンパク質としては生合成されるが、発色団(クロモフォア)のY67がTに置換されているため、アクセプターとして機能できないので蛍光寿命の短縮は起こらないためである。
以上から、DsG2−Runx2RDとCBFβ−野生型DsRed1との組み合わせを用いた場合に、ドナーの蛍光緩和時定数が1.52nsまで短縮し、高い効率でFRETが起こっていることが確認できた。
このように本発明を用いると、2分子間での相互作用もFRETによって簡便に検出できることが明らかとなった。
【産業上の利用可能性】
【0087】
本発明のFRETプローブは、分子デザインが容易で、様々な生体分子の解析に利用することができ、生体分子が生きた細胞中の、いつ、どこで、分子間相互作用や分子内構造変化を受けているのかを可視化・画像化することができる。それによって、細胞内での機能分子の動態を明らかにすることができるため、創薬スクリーニングのためのツールとして利用が可能である。
【図面の簡単な説明】
【0088】
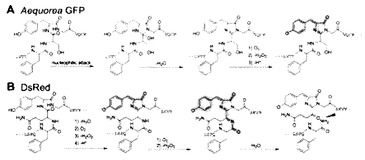
【図1】(A)AequoreaGFPと(B)DsRedについて、蛍光タンパク質の発色団(クロモフォア)の自己触媒的な形成と成熟の過程を比較する図である。切断部位が矢尻形で示される。可視光吸収のためのπ共役系が緑又は赤で示される。
【図2】(A−D):DsRed1、EGFP及びDsG2を細胞に発現させ、蛍光強度をFACSCaliburで測定した結果を示す図である。図中数値は上段が蛍光陽性細胞割合、下段が平均蛍光強度を示す(但しA:赤色蛍光について、B:緑色蛍光について)。(E,F):変異型DsRed1を細胞に発現させ、倒立顕微鏡を用いて観察した結果を示す図である。
【図3】DsG2からDsRed1へのFRET効率をフローサイトメーターを用いて検出した図である。
【図4】SUMO1のタンパク質切断をFRETを用いて検出するしくみの模式図である。
【図5】SUMO1活性化検出ベクターを細胞に発現させ、フローサイトメーター及び倒立顕微鏡を用いて観察した結果を示す図である。
【図6】DsG2、DsRed1を細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。A:td−DsG2、B:td−DsRed1、C:td−DsG2及びtd−DsRed1、D:DsG2とDsRed1を融合
【図7】SUMO1とその不活性化型変異体SUMO1GAの切断を検出できるプローブをを細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。A;野生型SUMO1、B:不活性化型変異体SUMO1GA
【図8】EGFPあるいはDsG2にSUMO1及びその不活性化型SUMO1GAを融合させ、細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。
【図9】強度の異なるプロモーターを用いてtd−DsG2を発現させ、蛍光強度及びdTc1を測定した結果を示す図である。 A:FACSCaliburによる測定、B:Flicymeによる測定
【図10】Runx2とCBFβの間の相互作用を、2分子間FRETで検出した結果を示す図である。 A:FACSCaliburによる測定、B:Flicymeによる測定
【配列表フリーテキスト】
【0089】
配列番号1−DsG2のcDNA塩基配列
配列番号2−DsG2のアミノ酸配列
配列番号3−オリゴDNA(Link3−1SE)の塩基配列
配列番号4−LG3のアミノ酸配列(Link3−1SE)
配列番号5−オリゴDNA(Link3−1AS)の塩基配列
【技術分野】
【0001】
本発明は、FRET効率が高く、しかも分子デザインが容易なFRETプローブに関する。より詳細には、本発明は、自己集合性をもち、ヘテロ二量体を形成しやすい蛍光タンパク質を用いる、FRET効率が高く、しかも分子デザインが容易なFRETプローブ及びその利用に関する。
【背景技術】
【0002】
蛍光共鳴エネルギー移動(FRET)とは、2つの蛍光分子が存在するとき、一方の分子の励起エネルギーが、他方の分子にトランスファーされる現象であり、その結果、エネルギーをトランスファーされた分子から蛍光が発せられる。励起エネルギーをトランスファーする方をドナー、される方をアクセプターと呼ぶ。
【0003】
FRET技術は、生体分子を可視化・画像化するための技術の1つであり、例えば、生体分子の分子間相互作用や分子内構造変化をモニタリングするために用いられ、特に、遺伝子の機能、及び疾病の発症と深く関わるシグナル伝達や遺伝子発現を解明するために好適である。これは、生体分子が分子間相互作用又は分子内構造変化によって、その生体分子を標識する2つの蛍光分子間の距離や相対的向きが変化すると、FRETが誘起又は解消されることを利用するものである。
【0004】
FRETの誘起又は解消を検出するための蛍光センサーがFRETプローブ(単に「プローブ」ともいう。)である。FRETプローブには、二分子間相互作用をする2つの生体分子のそれぞれを蛍光分子で標識したもの(2分子プローブ)と分子内構造変化又は分子間相互作用をする生体分子部分が挿入されたリンカー分子の両端を2つの蛍光分子で標識したもの(1分子プローブ)とがある。どちらの場合も、蛍光分子は合成小分子や蛍光タンパク質を利用できるが、蛍光タンパク質を用いると、フュージョンさせた融合タンパク質を生きた細胞内で発現させることができるため、低侵襲的に細胞体タンパク質の分子動態を可視化・映像化できる。
【0005】
FRETの誘起・解消の検出は、ドナーとアクセプターの蛍光強度比から近似的に求められるのが普通である。これをレシオメトリック測定法といい、2種類の蛍光フィルターセットを用いて、FRETにより減弱したドナーの蛍光強度(Dda)とFRETにより増強したアクセプターの蛍光強度(Ada)を測定し、その比をFRET効率(Ada/Dda)とするものである。
【0006】
この2色の蛍光強度比を用いる定量方法(レシオメトリー測定法)で、シグナル/ノイズ比(S/N比)を高めるためには、2つの蛍光タンパク質が予め同じ分子数で細胞内に存在することが必要である。しかし、2分子FRETでは、2つの蛍光タンパク質の分子数を揃えることが困難あり、これまでに比較的高いFRET効率を実現するのは、1分子FRETの場合が非常に多かった。
【0007】
現在までに実用化されているプローブには、FRET効率が非常に低いものが多く、そのようなプローブを用いる場合、わずかなFRET効率変化を2色の蛍光強度比として定量化して検出することとなる。実際、よく用いられている開発済みのプローブでさえも、FRET効率が非常に低く、2色の蛍光強度の比が、1.0から1.5への変化程度であるものも多い。
【0008】
FRET効率を算出する方法としては、レシオメトリー測定法の他に、ドナー蛍光の蛍光寿命を測定する方法がある。蛍光寿命の測定は、蛍光寿命測定装置を用いて行われ、蛍光の強さが時間経過とともに減衰していく変化を測定する。
【0009】
しかしながら、もともとFRET効率が非常に低い多くのプローブでは、蛍光寿命の変化もごくわずかであるため、FRET効率を精度よく算出することが難しい。
【0010】
例えば、非特許文献2(第58頁〜第59頁、実証実験)には、CFP(シアン色蛍光タンパク質)とRas(癌遺伝子)との融合タンパク質と、YFP(黄色蛍光タンパク質)とRaf(セリン・スレオニンリン酸化酵素)との融合タンパク質とからなるプローブを293F細胞で発現させ、Flicyme(登録商標)を使用して10万個の細胞についてドナー(CFP)の蛍光寿命を測定したところ、FRET誘起時に3.78nsから0.32ns程度短くなる方向にシフトし、FRET効率が8.5%と求められたことが記載されている。
【0011】
プローブの分子デザインをするとき、通常は、そのプローブが置かれている環境やそのプローブ分子の状態変化によって、蛍光分子間の距離が変動することを狙って分子デザインを行う。しかし、FRETは双極子−双極子相互作用により起こるものであり、蛍光分子間の距離だけでなく、蛍光分子の相対的向きもまた、考慮すべき要素である。特に、巨大な蛍光タンパク質分子の場合には、合成小分子のように自由回転する双極子とみなすこともできないので、分子の向きを考慮することは、より重要である。言い換えると、2つの蛍光分子が適切な角度をもって会合するようにデザインできなければ、FRET効率は非常に低いものとなる。
【0012】
ところが、実際には、相互作用する2分子プローブどころか、1本のポリペプチド鎖として発現される1分子プローブであっても、その一次構造からタンパク質が折り畳まれた際の2つの蛍光タンパク質間の距離や角度を予測することが困難であり、蛍光タンパク質分子間の距離を至適化し、さらに角度までも最適化することは非常に困難である。
【0013】
この問題に対する解決策のひとつとして、蛍光タンパク質のN末端とC末端の位置を作り替えた円順列変異体を用いることによって、蛍光タンパク質同士の角度を変更しようとする方法が開発されている(例えば、特許文献1)。
【0014】
この方法は多種類の円順列変異体を用いたプローブを全て作成してみて、その中からFRET効率の高いものを探す方法である。
【0015】
そのため、作成した円順列変異体の中に適切なものがなかった場合には、問題の解決が困難であり、さらに、あらゆる蛍光タンパク質で円順列変異体が作成されている訳ではないため、プローブ作成が、やはり困難である。
【0016】
【特許文献1】国際公開WO2005/036178号公報
【特許文献2】特開2007−232559号公報
【非特許文献1】中田ら,三井造船技報,第190号,54〜60頁(2007年).
【非特許文献2】Bairdら,米国科学アカデミー紀要,97:11984−11989(2000年).
【発明の開示】
【発明が解決しようとする課題】
【0017】
従って、本発明は、上記課題を解決するため、FRET効率が高く、蛍光寿命測定によってFRETの誘起・解消を検出するために好適に用いることができ、しかも分子デザインが容易なFRETプローブを提供することを目的とする。
【課題を解決するための手段】
【0018】
上記の目的を達成するため、発明者らが鋭意検討をした結果、FRETプローブに用いる蛍光タンパク質には自己集合性が低いものを用いることが技術常識であるところ、あえて自己集合性の蛍光タンパク質を用いてプローブを構成することによって、前記の目的を達成できることを知得した。以下に本発明を詳細に説明する。
【0019】
生体分子を標識するための蛍光分子としては、例えば、クマリン(黄緑色)やフルオレセイン(緑色)のような合成小分子、CFP(シアン色)やYFP(黄色)のような蛍光タンパク質が使用される。特に、蛍光タンパク質の遺伝子が同定され、遺伝子組み換え技術を用いて細胞内で生体分子との融合タンパク質として発現できるようになってからは、蛍光タンパク質の利用が進み、新しい蛍光タンパク質の分離や新しいバリアントの開発が相次いでいる。
【0020】
最初に発見された蛍光タンパク質は、オワンクラゲ(Aequorea victoria)に由来する緑色蛍光タンパク質(GFP)である。前述のCFPとYFPは、ともにGFPのバリアントであり、プローブ標識に用いられる極めて一般的な蛍光タンパク質のペアとなっている。
【0021】
また、サンゴ(Discosoma sp.)に由来する赤色蛍光タンパク質(DsRed)及びそのバリアントもまた、一般的に使用されている(例えば、非特許文献2)。さらに、バリアントには、異色変異体のみならず、培養細胞での大量発現に好適なようにコドン使用頻度が最適化された変異体や細胞内での凝集性を減少した変異体も存在する。
【0022】
例えば、サンゴ由来の野生型のDsRedに対して、DsRed1(クロンテック社)はヒト細胞でのコドン使用頻度に最適化された変異体であり、DsRed2(クロンテック社)は野生型のDsRedが四量体を形成しやすいのに対して、凝集性が減少し、成熟が早い変異体である。なお、これら市販の蛍光タンパク質は、例えば、それをコードするDNAが組み込まれた発現ベクター(発現コンストラクト)として提供される。
【0023】
従来の蛍光タンパク質を用いたプローブは、FRET効率が低いものが多いことは既に述べた通りである。しかも、従来の技術では、プローブの分子デザインは試行錯誤の要素が大きく、誰でもが容易にできるものでもない。
【0024】
FRET効率とはドナー/アクセプター間の蛍光共鳴エネルギーの移動効率、つまり、ドナーに吸収されたエネルギーのうち、アクセプターに転送されたエネルギーの比率を意味する。
【0025】
ここで、フェルスターの関係式から、FRET効率(E)は、
E={1+(r/R0)6}−1 (1)
と表すことができる。ただし、r[nm]はドナーとアクセプターのクロモフォア間距離であり、R0[nm]は、このドナーとアクセプターのペアのフェルスター距離(FRET効率が50%のクロモフォア間距離)である。
さらに、R0[nm]は、
R0=[8.8×10−23・n−4・κ2・Q・J]1/6 (2)
と表すことができる。ただし、κ=配向因子(0≦κ2≦4。自由回転している場合はκ2≒2/3。)、Q=アクセプターがないときのドナーの量子収率、n=溶媒の屈折率、J=重なり積分(ドナーの蛍光スペクトルとアクセプターの吸収スペクトルの重なり。)である。
【0026】
従って、2つの蛍光タンパク質分子間でのFRET効率は、クロモフォア間距離(r)、配向因子(κ)、及び重なり積分(J)の3つのパラメタに依存する。ところが、配向因子は自由回転する分子ではκ2≒2/3と近似できること、さらに、重なり積分はドナーの蛍光スペクトルとアクセプターの励起スペクトルから決まり、2つの蛍光タンパク質の位置関係には依存しないことから、プローブの分子デザインをする上では、クロモフォア間距離が特に重視されてきた。配向因子については分子デザイン段階ではあまり考慮せず、プローブを作製してからFRET効率が高いものを選別するというスタンスである。従って、実用性のあるプローブを作製することは、非常に手間がかかることであり、誰もが容易にできることではない。
【0027】
しかし、発明者らは、二量体を形成する蛍光タンパク質は、2分子間の相対的位置関係が実質的に固定されることに着目し、高いFRET効率を与えるために好ましいクロモフォア間距離(r)と配向因子(κ)となるように自己集合して二量体を形成する蛍光タンパク質の、片方又は両方を改変し、重なり積分が存在するようにすることで、高いFRET効率を実現できる蛍光タンパク質のペアを得られることに想到した。
【0028】
つまり、本発明の蛍光タンパク質は、二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質(以下「未変異型蛍光タンパク質」という。)のアミノ酸配列を、好ましくは、クロモフォア(発色団)形成に関わるアミノ酸を置換、挿入又は欠失等の変異を導入することによって、自己集合性を維持しながら、励起極大波長及び蛍光極大波長がともに異なる蛍光タンパク質(以下「変異型蛍光タンパク質」という。)に改変したものである。
【0029】
前記未変異型蛍光タンパク質と前記変異型蛍光タンパク質とのヘテロ二量体、又は、前記変異型蛍光タンパク質の励起極大波長及び蛍光極大波長がともに相違する2つの変異型蛍光タンパク質からなるヘテロ二量体は、FRET効率のパラメタである、クロモフォア間距離、配向因子、及び、重なり積分が、蛍光タンパク質のペアによって決まるため、プローブの分子デザインが非常に容易である。
【0030】
しかも、これらのヘテロ二量体を形成する蛍光タンパク質は、実質的に励起極大波長及び蛍光極大波長のみが異なるものであり、非常にFRET効率が高いペアが存在すれば、他のペアについては、実質的に、重なり積分のみを考慮すればよいため、高いFRET効率のプローブの分子デザインも容易となる。
【0031】
一方、FRET効率(E)は、次の(3)式で表すこともできる。
E=1−τD´/τD (3)
ここで、τD´はアクセプター存在時のドナーの蛍光寿命、τDはアクセプターがない場合のドナー蛍光寿命を、それぞれ表す。
【0032】
即ち、ドナーの蛍光寿命を測定すれば、FRET効率も容易に得ることができる。
【0033】
ところが、従来の技術水準では、プライマーのFRET効率が低く、FRETの誘起時と解消時で蛍光寿命にあまり差が出ず、10−10〜10−9秒のオーダーの蛍光寿命を精度よく蛍光寿命を測定することが困難であるため、蛍光寿命を測定してFRET効率を求めるのではなく、代わりに、レシオメトリック測定法で近似的にFRET効率が求められる。
【0034】
しかし、本発明の蛍光タンパク質を用いてFRETプローブを構成することによって、FRETの誘起・解消に伴う蛍光寿命変化が大きくなるため、蛍光寿命を測定してFRET効率を求めることが容易である。
【0035】
蛍光寿命測定法では、ドナーのみの蛍光寿命を測定すればよく、細胞内のドナー/アクセプター2種類の蛍光タンパク質の分子数が揃っている必要がない。従って、この測定法は、1分子FRETの場合だけではなく、レシオメトリック測定法ではS/N比が悪い2分子FRETの場合にも、非常に有効である。
【0036】
なお、2分子FRETは、分子間相互作用をする2つの生体分子を、それぞれ、異なる色の蛍光分子で標識し、FRETプローブを構成するものであり、標識された2分子間の相互作用によって、蛍光分子間の距離又は配行性が変化し、FRETが誘起又は解消される。また、一方、1分子FRETは、2つの蛍光分子部分がリンカー部分で連結された構造をもつFRETプローブを使用するものであり、リンカー部分に生体分子の全部又は一部が挿入され、他の生体分子との分子間相互作用又はリンカー部分に挿入された生体分子の全部又は一部の部分の間での相互作用によってリンカー部分の構造が変化し、FRETが誘起又は解消される。
【0037】
また、蛍光寿命測定とフローサイトメトリーのメリットを組み合わせ、細胞を高速で流しながら1細胞ごとに蛍光強度と蛍光寿命を同時に測定できる蛍光寿命フローサイトメーター(例えば、特許文献2)を好適に利用することができる。これにより、多数の細胞を高スループットで処理し、精度良く生体分子の分子間相互作用や分子内構造変化を検出できる。
【0038】
本発明では、前記の変異型蛍光タンパク質をコードする核酸、及び、該核酸を組み込んだ遺伝子発現ベクター(発現コンストラクト)を提供する。発現ベクター(発現コンストラクト)を培養細胞にトランスフェクトして形質転換し、細胞内で前記変異型蛍光タンパク質を発現することができる。
【0039】
本発明では、前記の変異型蛍光タンパク質を含むプローブ(蛍光タンパク質部分と生体分子部分の融合タンパク質)をコードする核酸、及び、該核酸を組み込んだ発現ベクター(発現コンストラクト)及び該ベクターを作成する方法を提供する。また、該ベクターによって形質転換された培養細胞を提供する。さらに、前記核酸、前記発現ベクター、又は前記形質転換された培養細胞を含むキットが提供される。これらにより、プローブの作成と使用が、より容易となる。
【0040】
さらに、細胞内で起こるFRET効率の変化を算出するための、好ましい方法として、ドナーの蛍光寿命を測定してFRET効率を算出する方法が提供される。好ましい一態様では特に、三井造船(株)製の蛍光寿命フローサイトメーター“Flicyme”(登録商標)を使用して、効率的に大量の細胞の蛍光寿命を測定することができる。
【0041】
即ち、本発明は以下のものである。
〔1〕二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質のアミノ酸配列に変異が導入された変異型蛍光タンパク質であって、励起極大波長及び蛍光極大波長がともに変異導入前の蛍光タンパク質(以下、未変異型蛍光タンパク質という。)からシフトし、かつ、該未変異型蛍光タンパク質若しくは励起極大波長及び蛍光極大波長がともに相違する他の変異型蛍光タンパク質とヘテロ二量体を形成できる、変異型蛍光タンパク質。
〔2〕〔1〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔3〕前記未変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができる、〔1〕に記載の変異型蛍光タンパク質。
〔4〕〔3〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔5〕前記変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができ、変異型蛍光タンパク質とは励起極大波長及び蛍光極大波長がともに相違する、〔1〕に記載の変異型蛍光タンパク質。
〔6〕〔5〕に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
〔7〕前記未変異型蛍光タンパク質が、サンゴ(Discosoma sp.)に由来する蛍光タンパク質である、〔1〕に記載の変異型蛍光タンパク質。
〔8〕前記蛍光タンパク質が、DsRed1である、〔7〕に記載の変異型蛍光タンパク質。
〔9〕配列番号1に記載のDNA塩基配列を翻訳して得られるアミノ酸配列をもつ蛍光タンパク質。
〔10〕配列番号2に記載のアミノ酸配列をもつ蛍光タンパク質。
〔11〕〔3〕又は〔5〕に記載の2つの蛍光タンパク質を含むFRETプローブ。
〔12〕〔3〕又は〔5〕に記載の2つの蛍光タンパク質がポリペプチドからなるリンカーを介して連結される構造をもつ融合タンパク質であるFRETプローブ。
〔13〕前記リンカーが生体分子の全部又は一部のアミノ酸配列を含む、〔11〕に記載のFRETプローブ。
〔14〕前記2つの蛍光タンパク質がDsG2(配列番号2)及びDsRed1である、〔11〕又は12に記載のFRETプローブ。
〔15〕前記生体分子がSUMO1である〔12〕又は〔13〕に記載のFRETプローブ。
〔16〕〔11〕〜〔14〕のいずれかに記載のFRETプローブをコードするポリヌクレオチド。
〔17〕〔11〕〜〔14〕のいずれかに記載のFRETプローブをコードするDNAを遺伝子発現ベクターに組み込んで構築した発現ベクター。
〔18〕前記遺伝子発現ベクターが、pEB6CAGmcs、pEB6SRαmcs及びpEB6CMVmcsからなる群から選ばれる、〔17〕に記載の発現ベクター。
〔19〕前記発現ベクターが、pEB6CMVmcs−SRZである、〔18〕に記載の発現ベクター。
〔20〕〔17〕〜〔19〕のいずれかに記載の発現ベクターを培養細胞に導入した形質転換細胞。
〔21〕前記培養細胞がヒト由来細胞株である、〔20〕に記載の形質転換細胞。
〔22〕前記ヒト由来細胞株がHEp−2細胞株である、〔21〕に記載の形質転換細胞。
〔23〕〔17〕〜〔19〕のいずれかに記載の発現ベクターを含んでなるキット。
〔24〕〔20〕〜〔22〕のいずれかに記載の形質転換細胞を含んでなるキット。
〔25〕〔11〕〜〔15〕のいずれかに記載のFRETプローブを用いる、ドナー蛍光タンパク質の蛍光寿命を測定して細胞内FRETを検出する方法。
〔26〕蛍光寿命フローサイトメーターを使用して蛍光寿命を測定する、〔25〕に記載の方法。
〔27〕前記蛍光寿命フローサイトメーターが、好ましくは、Flicyme(登録商標)である、〔26〕に記載の方法。
【発明の効果】
【0042】
本発明の自己集合性蛍光タンパク質のペアを用いることによって、自発的に会合して二量体が形成されるため、プローブの分子デザインをする際に分子間距離と分子の配向性を考慮する必要がなく、スペクトルの重なりを考慮すれば、FRET効率の高いプローブの分子デザインをすることが従来に比べて非常に容易となった。また、FRET効率が高く、蛍光寿命変化も大きいことから、蛍光寿命フローサイトメーターを用いて蛍光寿命を精度良く測定し、FRET効率の変化を算出する用途に好適に用いることができる。
【発明を実施するための最良の形態】
【0043】
<1>本発明の蛍光タンパク質について説明する。
本発明の蛍光タンパク質は、二量体又は三量体以上の多量体を形成しやすい、自己集合性をもつ蛍光タンパク質である。好ましくは、自己集合性をもち、二量体又は三量体以上の多量体を形成しやすい蛍光タンパク質(以下、原型蛍光タンパク質という。)の、アミノ酸配列に変異を導入することによって、自己集合性を維持しながら蛍光極大波長及び励起極大波長をシフトさせた、変異型蛍光タンパク質である。より好ましくは、原型蛍光タンパク質と変異型蛍光タンパク質とで、又は、互いに異なる変異型蛍光タンパク質同士で、ヘテロ二量体を形成する。さらに好ましくは、前記へテロ二量体形成時に、2つの蛍光タンパク質の、クロモフォア間距離、遷移双極子モーメントの相対的向き、及び、励起波長が短い方の分子(ドナー)の蛍光スペクトルと、励起波長が長い方の分子(アクセプター)の励起スペクトルとの重なり(スペクトル・オーバーラップ)が、該ドナーから該アクセプターへのFRETを起こす条件を満たす。いっそう好ましくは、前記ドナーと前記アクセプターの間の、クロモフォア間距離rとフェルスター距離(FRET効率が50%になるクロモフォア間距離)について(r/R0)6を最小にするような、r、κ及びJ(但し、R0=[8.8×10−23・n−4・κ2・Q・J]1/6、κは双極子の配向因子、Jはドナーの蛍光スペクトルとアクセプターの励起スペクトルの重なり積分、Qはアクセプターがないときのドナーの量子収率、nは葉液の屈折率である。)を与えるヘテロ二量体を構成する変異型蛍光タンパク質である。
【0044】
前記変異型蛍光タンパク質は、当業者にとって自明な、コンベンショナルな遺伝子工学的手法を用いて、発現及び精製をして、取得することができる。
より詳細には、例えば、変異型蛍光タンパク質の原型蛍光タンパク質をコードする組み換えcDNAを合成し、これを適当な遺伝子発現ベクターに組み込んで発現ベクターを構築し、適切な細胞にコンベンショナルな方法でトランスフェクトしてトランスフォーメーションし、蛍光タンパク質を細胞中に発現させる。所望により、前記発現ベクターは、トランスフェクション前に大腸菌等を用いて大量調製することもできる。さらに、前記遺伝子発現ベクターに組み込まれている選択マーカーを利用して、トランスフェクションの成否とトランスフォーメーションの成否によって細胞を選択することができる。細胞内で発現される蛍光タンパク質は、コンベンショナルな方法で分離・精製をすることができる。
【0045】
本発明の好ましい変異型蛍光タンパク質は、サンゴ(Discosoma sp.)由来の赤色蛍光タンパク質DsRedの変異体であるDsRed1に、遺伝子組み換えにより、83番目のリジンをアルギニン、120番目のチロシンをヒスチジン、197番目のセリンをスレオニンに置換する変異を導入して作出した緑色蛍光タンパク質DsG2である。DsG2はDsRed1とヘテロ二量体を形成し、そのとき、DsG2の励起エネルギーがDsRed1に移動してFRETが起こり、DsRed1の赤い蛍光が発生する。
【0046】
DsRedは、そのクロモフォア形成及び成熟の過程において、イクオレアGFPと同様に一度酸化還元反応を行って緑色の発色団を形成した後に、再度酸化還元反応を行い赤色の発色団を形成することが知られている(図1)が、緑色蛍光を発するクロモフォアが形成された段階で酸化還元反応が停止したDsRed1の緑色変異体は、DsRed1とヘテロ二量体を形成し、該DsRed1緑色変異体の蛍光スペクトルとDsRed1の励起スペクトルの重なりがFRETを誘起するのに十分であるといえるから、本発明の変異型蛍光タンパク質の範囲に含まれる。
【0047】
なお、本明細書において、DsRedは、サンゴ(学名:Discosoma sp.)の赤色蛍光タンパク質であり、DsRed1は、サンゴのDsRedのコドン使用パターンをヒト型に置換した変異型であり、DsRed2は、DsRed1に点変異を導入してタンパク質凝集の傾向が抑えられた変異型である。また、GFPは、オワンクラゲ(Aquorea victoria)の緑色蛍光タンパク質であり、EGFPはクラゲのGFPのコドン使用パターンをヒト型に置換した変異型である。
【0048】
<2>本発明のプローブについて説明する。
本発明のプローブに、技術常識に反して自己集合性を持つ蛍光タンパク質をあえて用いる理由は、アクセプターとドナーの2つの蛍光タンパク質が、必ずその分子特性によって決まった角度でしかも安定に結合することを意図したものであるが、必ずしもこの意図に限定されない。この角度がFRETを起こすため好ましい角度であるならば、2種類の蛍光タンパク質に付加するアミノ酸配列や他のタンパク質に関して、所望により、あまり詳細な検討を必要とせず、確実に高いFRET効率を実現できるように蛍光タンパク質が自発的に会合することを期待してもよい。従って、これまでに開発されてきたプローブと比較して、著しくFRET効率の高いプローブを誰もが非常に簡便に開発することが可能になることを意図するものであるが、必ずしもこの意図に限定して解釈されるべきではない。
【0049】
本発明のプローブは、1分子プローブであっても2分子プローブであってもよいが、1分子プローブのときは、2つの蛍光タンパク質部分がリンカー部分を介して連結される構造となる。ここで、2つの蛍光タンパク質部分は、一方が前記変異型蛍光タンパク質であれば、他方は原型蛍光タンパク質であっても変異型蛍光タンパク質であってもよい。リンカー部分には、好ましくは、細胞内で分子間相互作用又は分子内構造変化を受けるタンパク質又はポリペプチドのアミノ酸配列を含む。さらに、前記アミノ酸配列は、前記タンパク質又はポリペプチドの全体又は部分であってもよいし、複数の全体又は部分が含まれてもよい。細胞内での分子間相互作用を解析するためには、好ましくは、ある一態様では、リンカー部分にタンパク質又はポリペプチドの全体又は部分を1つ含み、細胞内の標識されていない分子と相互作用することでFRETが誘起又は解消することによって分子間相互作用を検出する。また、細胞内での分子間相互作用を解析するためには、好ましくは、別の一態様では、リンカー部分にタンパク質又はポリペプチドの全体又は部分を1つ又は2つリンカーに含み、リンカー部分が構造変化することでFRETが誘起又は解消することによって分子内構造変化を検出する。前記1分子プローブは、好ましくは、蛍光タンパク質−リンカー−蛍光タンパク質の三重融合タンパク質として細胞内で発現される。また、2分子プローブのときには、好ましくは、2つの分子間相互作用をするタンパク質又はポリペプチドを、それぞれ蛍光タンパク質で標識する構造となる。ここで、2つの蛍光タンパク質は、一方が前記変異型蛍光タンパク質であれば、他方は原型蛍光タンパク質であっても変異型蛍光タンパク質であってもよい。好ましい態様では、前記タンパク質又はポリペプチドが結合すると、2つの蛍光タンパク質も接近し、会合し、ヘテロダイマーを形成し、FRETが起こり、前記結合が解かれると、2つの蛍光タンパク質も解離し、FRETも解消する。前記プローブは、好ましくは、蛍光タンパク質−タンパク質若しくはペプチド、又は、タンパク質若しくはペプチド−蛍光タンパク質の構造をもつ融合タンパク質として細胞内で発現される。蛍光タンパク質は、前記タンパク質又はポリペプチドのN末端又はC末端のどちらか一方に連結されればよい。前記融合タンパク質は、プローブを構成する2つの融合タンパク質分子が、別個の発現ベクター上にコードされ、二重トランスフェクトされた細胞で発現されてもよい。
【0050】
本発明の1分子プローブは、好ましい一態様では、DsG2とDsRed1をSUMO1のアミノ酸配列を含むリンカーで連結した三重融合タンパク質である。ユビキチン様タンパク質SUMO1の前駆型タンパク質のC末端のグリシンで切断されて成熟型となることから、前記のリンカー部分にSUMO1前駆型タンパク質のアミノ酸配列を含むFRETプローブでは、リンカー部分が切断されることによってFRETが解消し、アクセプターの蛍光からドナーの蛍光に変化することによって、細胞内で起こる分子間相互作用を検出することを意図したものであるが、この意図に限定されて解釈されるものではない。
【0051】
また、前記の三重融合タンパク質は、例えば、ヒトSUMO1遺伝子のORFをはさむオリゴDNAを合成し、これを用いてヒトHEp−2細胞由来のcDNAライブラリーよりPCR反応によって野生型SUMO1のcDNAを調製し、SUMO1の終止コドンのかわりに他のアミノ酸をコードするように置換した塩基配列とその下流に制限酵素によって認識されて切断される配列を有するオリゴDNAを合成し、これを用いてPCR反応を行い、終止コドンを持たないSUMO1のcDNAを構築し、DsG2のcDNAの下流に、オリゴDNA、Link3−1SE(配列情報3)とLink3−1AS(配列情報5)をアニーリングさせた19アミノ酸からなるリンカー配列(LG3)を連結し、作成したSUMO1のcDNAを読み枠が合うように連結し、さらにその下流に再度LG3を連結し、読み枠が合うようにDsRed1のcDNAを連結して、三重融合タンパク質「DsG2−SUMO1−DsRed1」をコードするcDNAを作成することができるが、この方法に限定されるものではなく、当業者が想到し得るいかなる方法でも用いることが可能である。
前記のcDNAを細胞中で発現する好ましい態様は、前記cDNAを“pEB6CAGmcs”遺伝子発現ベクターに組み込み、「DsG2−SUMO1−DsRed1」を発現するベクターを構築し、大腸菌から大量調製し、調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)存在下で培養することによって、細胞中で発現することができるが、この手順に限定されず、当業者が想到し得るいかなる方法でも用いることが可能である。
【0052】
本発明の2分子プローブは、好ましい一態様では、DsG2とRunx2RDを連結した融合タンパク質とDsRed1とCBFβを連結した融合タンパク質の2分子からなる。Runx2RDとCBFβは1:1で結合するため、細胞中で前記の融合タンパク質が両方とも発現され、両者が結合するとFRETが起こり、分離するとFRETが解消することによって、細胞内で起こる2分子間の相互作用を検出することを意図するものであるが、この意図に限定して解釈されるものではない。
【0053】
また、前記の2種類の融合タンパク質は、例えば、マウスRUNX2遺伝子のRuntドメイン(RD)をコードするDNA断片を調製し、前記の「DsG2−SUMO1−DsRed1」を発現するベクターのSUMO1−DsRed1をコードする部分と置き換えて、「DsG2−Runx2RD」を構築することができる。同様に、CBFβをコードするDNA断片を調製し、前記の「DsG2−SUMO1−DsRed1」を発現するベクターのDsG2−SUMO1をコードする部分と置き換えて「CBFβ−DsRed1」を発現するベクターを構築することができる。この方法に限定されることなく、当業者が想到し得るいかなる方法でも用いることが可能である。
【0054】
さらに、好ましくは、このcDNAを切り出してゼオシン耐性遺伝子を持つベクター“pEB6CAGmcs−SRZ”に乗せ替えることにより、G418とゼオシンの二重選択によって、「DsG2−Runx2RD」と「CBFβ−DsRed1」を同時に細胞に発現できるベクターセットを構築することができる。
【0055】
また、所望により、FRETが起こらない場合の比較実験用に、「CBFβ−DsRed1」のDsRed1の発色団(クロモフォア)を構成するアミノ酸配列を改変し、FRETアクセプターとしてまったく機能できないようにしたものを作成して用いてもよい。好ましくは、67番目のチロシンをスレオニンに置換した変異を導入した「CBFβ−DsRed1Y67T」を構築してもよい。
【0056】
構築した発現ベクターは、好ましくは、市販のDNA精製キットを用いて大腸菌から大量調製することができる。調製した発現ベクターは、好ましくは、市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)、0.1mg/ml ゼオシン(インビトロジェン社)両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択することができる。この手順に限定されず、当業者が想到し得るいかなる方法でも用いることができる。
【0057】
<3>本発明のFRET検出法について説明する。
本発明の好ましい一態様では、フローサイトメーターによって蛍光強度を測定し、FRETの誘起・解消を検出することができる。
例えば、SUMO1を用いたプローブは細胞内で切断を受けるため、FRETが解消して緑色蛍光が多くなるが、SUMO1GAは切断を受けないためFRETが誘起されて赤色蛍光が多くなる。ある態様では、SUMO1を用いたプローブでは、赤い蛍光と緑の蛍光の比は1.1と最も低くなるのに対して、SUMO1GAを用いたプローブでは切断を受けないため、高い効率のFRETが起こり、緑の蛍光は減弱し赤い蛍光が強くなるために蛍光強度比が19.7にまで上がる。
【0058】
本発明の別の好ましい一態様では、蛍光顕微鏡による観察によってFRETを検出することができる。例えば、DsG2−SUMO1−DsRed1又は変異RUBYG2−SUMO1GA−DsRed1を発現させた細胞を倒立型顕微鏡で撮影すると、DsG2−SUMO1−DsRed1は緑も赤も核内にドット状に観察されるが、変異RUBYG2−SUMO1GA−DsRed1は赤のみが細胞質に観察され、これは、SUMO1が活性化されるとFRETが解消され緑の蛍光が観察されるようになるが、SUMO1の活性化がおこらない場合にはFRETが誘起されDsG2の蛍光エネルギーがDsRed1に移動していると解釈できる。
【0059】
本発明のさらに別の好ましい一態様では、フローサイトメーターによって蛍光緩和時定数(蛍光寿命)を測定してFRETを検出することができる。
【0060】
例えば、フローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて、td−DsG2、td−DsRed1及びDsG2−LG3−DsRed1の蛍光緩和時定数を測定すると、td−DsG2の緑色の緩和時定数は2.1nsであり、td−DsG2とtd−DsRed1を同一細胞に共発現させると緑も赤もそれぞれ蛍光を発するため緑色の緩和時定数はtd−DsG2単独発現時とほとんど同じ2.0nsであるが、DsG2−LG3−DsRed1の緑色の緩和時定数は1.3nsに減少することから、DsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移していることが確認される。また別の例では、細胞内でのSUMO1の活性化をFRETにより検出することを試みるとすると、DsG2−SUMO1−LG3−DsRed1の緑色緩和時定数は2.1ns(td−DsG2と同じ)であるものの、切断されないSUMO1GA変異を用いたDsG2−SUMO1GA−LG3−DsRed1の緑色緩和時定数は1.5nsに短縮することから、蛍光強度の著しい変動は、FRETによるものであり、しかも蛍光寿命の測定によってタンパク質の切断を検出可能であることが示される。
【0061】
また、例えば、フローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて、HEp−2細胞で発現させたDsG2−Runx2RDとCBFβ−野生型DsRed1とからなる2分子プローブの蛍光緩和時定数(蛍光寿命)を測定すると、ドナー(DsG2)の緑の蛍光の緩和時定数は1.52nsであるのに対し、DsG2−Runx2RDとCBFβ−DsRed1Y67T変異(DsRed1Y67T変異体:発色団となる67番目のチロシンがスレオニンに置換され、アクセプターにならない。)とからなる2分子プローブの蛍光緩和時定数(蛍光寿命)を測定すると、ドナーの蛍光緩和時定数は2.23nsであり、前者では高い効率でFRETが起こっていることが確認できる。
【0062】
よって、本発明のFRETプローブを用いる場合、FRETの検出は、好ましくは、蛍光顕微鏡法、レシオメトリー測定法又は蛍光寿命測定法によって行うことができる。より好ましくは、蛍光寿命測定法によって行うことができる。また、フローサイトメトリと組み合わせてFRETの検出を行ってもよい。フローサイトメトリと組み合わせるのは、単位時間あたりにより多くの細胞を観察することを意図したものであるが、その意図に限定されて解釈されるものではない。
【0063】
以下に、実施例により本発明を説明するが、本発明の範囲はこれらの実施例によって限定されない。
【実施例】
【0064】
1:変異型DsRedの調製及び蛍光特性の検証
[1]材料及び方法
A.cDNAの調製
サンゴ(Discosoma sp.)由来の赤色蛍光タンパク質DsRedのバリアントであるDsRed1に、遺伝子組み換えにより、83番目のリジンをアルギニン、120番目のチロシンをヒスチジン、197番目のセリンをスレオニンに置換する変異を導入し、変異型DsRed1である緑色蛍光タンパク質を作出した。この緑色蛍光タンパク質をDsG2と命名した(配列番号2)。手順は、以下の通りである。
(1)導入しようとするアミノ酸置換を含むアミノ酸配列をコードするオリゴヌクレオチドを合成し、これを用いてPCR反応によって、変異を含むDNA断片を調製した。
(2)これを野生型DsRed1タンパク質をコードするDNAの当該部分と入れ替えることにより、上記変異を導入したcDNAを調製した。
【0065】
次に、前記cDNAをタンデムに融合させた。手順は、以下の通りである。
(1)DsRed1をコードするDNA及びDsG2をコードするDNA(配列番号1)の終止コドンを他のアミノ酸に対応するコドンに置換した塩基配列をもち、その下流に制限酵素によって認識されて切断される配列を含むオリゴDNAを合成した。
(2)これを用いてPCR反応を行い、終止コドンを持たない野生型又は変異導入DsRed1をコードするDNA断片を調製した。
(3)同一のタンパク質同士を合成の翻訳の読み枠が合うように連結した(タンデムダイマーDsRed1;td−DsRed1)。
【0066】
B.発現ベクターの作製
上記のように作製したcDNAを、本発明者らが開発した、ヒト細胞中で安定に複製・維持される遺伝子発現ベクター“pEB6CAGmcs”、“pEB6SRαmcs”又は“pEB6CMVmcs”に組み込んだ(これらの発現ベクターは本発明者から入手可能である)。構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0067】
C.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間1.5mg/ml G418存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(FACSCalibur(登録商標),ベクトン・ディッキンソン社)で蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0068】
D.蛍光顕微鏡による観察
形質転換細胞の蛍光特性及び細胞内局在を、蛍光顕微鏡(IX70,オリンパス社)及びそれに接続したCCDカメラ(SenSys(登録商標),フォトメトリクス社)で観察した。緑色蛍光の観察は495±10nmの励起フィルター及び535±15nmの蛍光フィルターで、赤色蛍光の観察は555±12nmの励起フィルター及び620±30nmの蛍光フィルターで観察した。
【0069】
[2]結果
フローサイトメーター及び蛍光顕微鏡による蛍光強度の測定
図2A〜Dは、緑色蛍光タンパク質EGFP、野生型DsRed1及び変異DsRed1の蛍光特性を、フローサイトメーターを用いて試験した結果を示すプロットである。
図2Aに示すように、野生型td−DsRed1を発現させた細胞では赤色蛍光が認められた。
野生型DsRed1の83番目のリシンをアルギニンに置換した変異体K83Rでは、確かに赤から緑に蛍光特性が変化しているものの(図2C)、まだ波長の変化が不十分であり赤い波長成分がかなり残っているため、一般的な蛍光顕微鏡において赤い蛍光を観察するためのフィルターセットで観察されてしまう(図2E)。
一方、本発明でさらにY120H及びS197Tの変異を2つ追加して開発したDsG2では赤い蛍光成分が軽減しているため、フローサイトメーターの結果において、より下方に細胞のプロットが現れていた(図2D)。
図2FはDsG2の蛍光特性を蛍光顕微鏡を用いて試験した結果を示す図である。図2Eと同様のフィルターセットにおいて、赤い蛍光がほとんど消失していた。このことから、本発明で開発したDsG2は、緑の蛍光タンパク質として用いることができる。
【0070】
2:DsG2−DsRed1間のFRET効率の評価
[1]材料及び方法
A.cDNAの作製
上記のように作製したtd−DsG2とtd−DsRed1のcDNAを途中のリンカー部分で組み換えてタンデムに融合させ、DsG2−DsRed1を作成した。
【0071】
B.遺伝子発現ベクターの作製
上記のように作製したcDNAを、本発明者らが開発した、ヒト細胞中で安定に複製・維持される遺伝子発現ベクター“pEB6CAGmcs”に組み込んだ。またtd−DsRed1のcDNAを異なる薬剤耐性遺伝子をもつ同等のベクター構築した“pEB6CAGmcs−SRZ”に組み込んで二重トランスフェクションができるようにした。発現ベクターDNAは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0072】
C.形質転換細胞の作製及び選択
調製した遺伝子発現ベクターDNAを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し4日間、1.5mg/ml G418(ロシュ社)存在下、0.1mg/ml ゼオシン(インビトロジェン社)存在下、又は両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(FACSCalibur(登録商標),ベクトン・ディッキンソン社)で蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0073】
[2]結果
フローサイトメーターによる蛍光強度の測定
図3Aに示すように、DsG2単独の細胞は水平方向に、図3Bに示すように、DsRed1単独の細胞は垂直方向にプロットされるように、装置の設定を調整した。この条件下で、DsG2とDsRed1を二重導入した細胞を測定したところ、緑の平均蛍光強度はやや弱くなり、赤の平均蛍光強度はやや強くなっており、赤い蛍光と緑の蛍光の強度比は2.1であった。
一方、DsG2とDsRed1がタンデムに連結された融合タンパク質を発現する細胞を解析したところ、赤い蛍光は図3B及び図3Cと比較して著しく増強されていたのに対し、緑の蛍光強度は、細胞内で緑色蛍光タンパク質がかなり発現されているにもかかわらず、図3A及び図3Cと比較して著しく減弱していた。赤い蛍光と緑の蛍光の強度比は16.4と別々に発現させた場合の8倍の比を示し、明らかにDsG2の励起エネルギーがDsRed1に移動するFRETが高い効率で起こっていることが示唆された。
【0074】
3:FRET検出システムの応用と性能の評価
生きた細胞内でのFRET効率の変化と生命現象との関連を調べるため、ユビキチン様(UBL)タンパク質のひとつであるSUMO1について応用することを試みた。
SUMO1は、SUMOタンパク質のひとつである。SUMOタンパク質は、細胞内の他のタンパク質に一時的に共有結合してその機能を助ける小さなタンパク質であり、タンパク質のSUMO化は、細胞核−細胞質の輸送、転写制御、アポトーシス、タンパク質の安定化、ストレス応答、細胞周期の進行など様々な細胞内のプロセスに関係する翻訳後修飾の1つである。
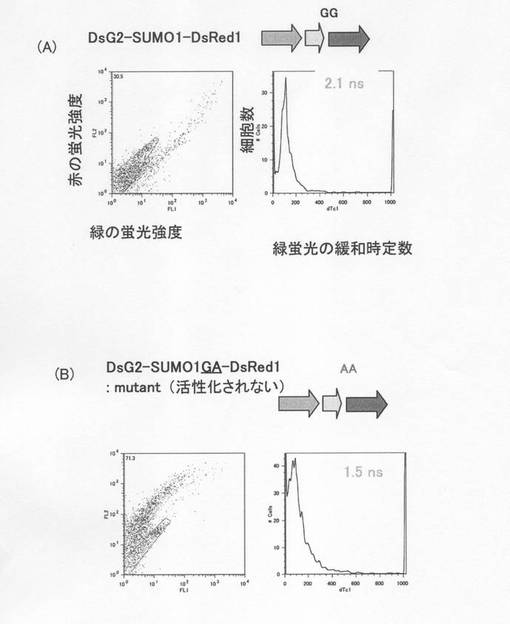
図4はプローブのデザインと、働く機構の概念図である。SUMO1は、前駆型タンパク質のC末端付近のグリシンの後ろでプロペプチドが切断されて成熟型となり、活性化して、基質に結合することが知られている(図4A)。そこで、DsG2の下流にSUMO1を融合し、さらにその下流にDsRed1を融合した、「DsG2−SUMO1−DsRed1」三重融合タンパク質を細胞中に発現させると、初めはSUMO1が切断による活性化を受けていない(図4B)ため、特別に工夫されたリンカー配列を使っていなくても、DsG2とDsRed1が分子内で自発的に会合し、極めて高い効率でFRETが誘起されるために、緑の蛍光は著しく減弱し、赤い蛍光が高くなる。これに伴って、ドナーの蛍光寿命dTc1が短縮する。一方、SUMO1が活性化され切断が起こると、DsG2とDsRed1が解離し(図4C)、FRETが解消されて緑の蛍光が強く検出されるようになり、蛍光寿命dTc1がもとの長さに回復する。
【0075】
[1]材料及び方法
以上のモデルを検証するために、以下の手順で実験を行った。
A.cDNAの調製
ヒトSUMO1遺伝子のORFをはさむオリゴDNAを合成し、これを用いてヒトHEp−2細胞由来のcDNAライブラリーよりPCR反応によって野生型SUMO1のcDNAを調製した。SUMO1の終止コドンのかわりに他のアミノ酸をコードするように置換した塩基配列とその下流に制限酵素によって認識されて切断される配列を有するオリゴDNAを合成し、これを用いてPCR反応を行い、終止コドンを持たないSUMO1のcDNAを構築した。実施例1で作製したDsG2のcDNAの下流に、19アミノ酸からなるリンカー配列(LG3)を連結し、作成したSUMO1のcDNAを読み枠が合うように連結した。さらにその下流に再度LG3を連結した後、読み枠が合うようにDsRed1のcDNAを連結しDsG2−LG3−SUMO1−LG3−DsRed1を作成した。
また、切断を受けない比較実験用に、SUMO1のC末近くのグリシンをアラニンに置換した変異体SUMO1GAも作成し、同様のプローブDsG2−LG3−SUMO1GA−LG3−DsRed1を作成した。
【0076】
B.発現ベクターの作製
上記のように作製したcDNAを、“pEB6CAGmcs”ベクターに組み込んだ。構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0077】
C.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(Flicyme(登録商標),三井造船(株))で蛍光緩和時定数(蛍光寿命)、蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0078】
D.蛍光顕微鏡による観察
形質転換細胞を用い、形質転換細胞の蛍光特性及び細胞内局在を、蛍光顕微鏡(IX70,オリンパス社)及びそれに接続したCCDカメラ(SenSys(登録商標),フォトメトリクス社)で観察した。緑色蛍光の観察は495±10nmの励起フィルター及び535±15nmの蛍光フィルターで、赤色蛍光の観察は555±12nmの励起フィルター及び620±30nmの蛍光フィルターで観察した。
【0079】
[2]結果
A.フローサイトメーターによる蛍光強度の測定
実施例2と同様にフローサイトメーターのパラメーターを調整して測定を行った。図5Eに示すように、赤い蛍光が非常に弱くなり検出される蛍光は緑が多くなったため、赤い蛍光と緑の蛍光の比は1.1と最も低くなった。一方、切断を受けないSUMO1GAを用いたプローブでは図5Dと同様に高い効率のFRETが起こり、緑の蛍光は減弱し赤い蛍光が強くなったために蛍光強度比が19.7にまで上がった。以上のことから、2つの蛍光タンパク質の間にSUMO1を挿入した場合にも、非常に簡単に高いFRET効率を実現することができ、しかもSUMO1の切断によって、このFRETが解消することから、タンパク質の切断を極めて高感度にFRETを用いて解析できることが明らかとなった。
【0080】
B.蛍光顕微鏡による観察
図5FとHは、DsG2−SUMO1−DsRed1あるいはDsG2−SUMO1GA−DsRed1を発現させた細胞を倒立型顕微鏡で撮影した図である。示されるように、DsG2−SUMO1−DsRed1は緑も赤も核内にドット状に観察された。一方、変異RUBYG2−SUMO1GA−DsRed1は赤のみが細胞質に観察された。このことは図4の概念図に示した通り、SUMO1が活性化されるとFRETが解消され緑の蛍光が観察されるようになるが、SUMO1の活性化がおこらない場合にはFRETが誘起されDsG2の蛍光エネルギーがDsRed1に移動していることを示唆する。また、SUMO1の場合には、切断による活性化に伴う細胞内局在の変化(細胞質から核への移行)もイメージングできることが示された。
【0081】
C.フローサイトメーターによる蛍光緩和時定数の測定
図6は、td−DsG2、td−野生型DsRed1及びDsG2−LG3−DsRed1の蛍光緩和時定数をフローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて試験した結果を示すヒストグラムである。
td−DsG2の緑色の緩和時定数は2.1nsだった(図6A)。
td−DsRed1の赤色の蛍光は緑のチャンネルにはほとんど入らないため細胞の自家蛍光成分と考えられる弱い蛍光のみが検出され、緑色の緩和時定数は1.3nsだったが細胞間のばらつきが非常に大きく、蛍光タンパク質由来の蛍光寿命とは明らかに異なった。(図6B)。
td−DsG2とtd−DsRed1を同一細胞に共発現させると緑も赤もそれぞれ蛍光を発するため緑色の緩和時定数はtd−DsG2単独発現細胞の図7Aとほとんど同じ2.0nsだった(図6C)。
しかし、DsG2−LG3−DsRed1の緑色の緩和時定数は1.3nsに減少した。また赤い蛍光においてはわずかだが蛍光寿命の延長も見られた(図6D)。
このことからDsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移していることが確認された。
そこで、細胞内でのSUMO1の活性化をFRETにより検出することを試みた。DsG2−SUMO1−LG3−DsRed1の緑色緩和時定数は2.1nsで、td−DsG2と同じだった(図7A)。
しかしながら、切断されないSUMO1GA変異を用いたDsG2−SUMO1GA−LG3−DsRed1の緑色緩和時定数は1.5nsに短縮した(図7B)。
このことから、蛍光強度の著しい変動は、FRETによるものであり、しかも蛍光寿命の測定によってタンパク質の切断を検出可能であることが示された。
【0082】
次に、この蛍光寿命の変化が、DsG2がSUMO1と結合したことによる何らかの影響によるものかどうかを確認する実験を行った。
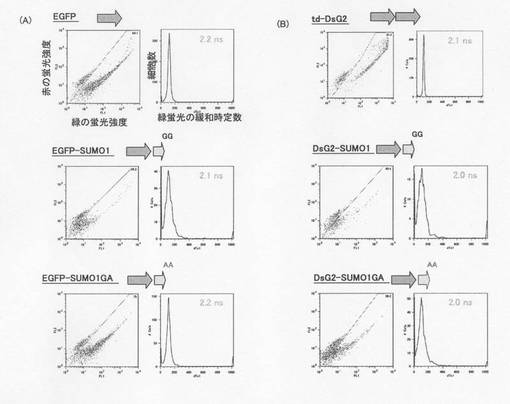
図8はEGFPあるいはDsG2にSUMO1を融合させたときの蛍光寿命dTc1を示した図である。EGFP、DsG2ともにSUMO1あるいはSUMO1GAを融合させてもdTc1はtd−DSG2とほとんどかわらなかった。これによりSUMO1との融合で蛍光寿命が減少した訳ではないことが確認された。
従って、DsG2−SUMO1GA−DsRed1での蛍光寿命の短縮は、DsG2の蛍光エネルギーがFRETによってDsRed1の励起エネルギーに転移したことがわかった。
図9はDsG2の蛍光強度が蛍光寿命dTc1に及ぼす影響を検討した結果を示す図である。蛍光強度を変えるために用いたプロモーターは、本発明者らがプロモーター強度を確認済みのCAGプロモーター、SRαプロモーター。CMVプロモーターである(プロモーター強度:CAGプロモーター>SRαプロモーター>CMVプロモーター)。
図9Aに示すように、いずれのプロモーターでもほとんどすべての細胞で蛍光が観察され、その蛍光強度はCAGプロモーター>SRαプロモーター>CMVプロモーターだった。
一方、図9Bに示すように、dTc1にはほとんど違いはなかった。
このことからDsG2の蛍光強度はdTc1にほとんど影響しておらず、dTc1の短縮は蛍光強度の違いによるものではなく、FRETによるものであることが確認された。
【0083】
4:2分子間FRET検出システムへの応用と性能の評価
生きた細胞内でのタンパク質間相互作用を本発明を応用した2分子間FRETによって検出できる可能性を調べるため、転写因子Runx2とCBFβ間での結合について応用することを試みた。
転写因子Runx2は、骨芽細胞の分化を制御することで知られる転写因子であり、その結合パートナーであるCBFβと1:1で結合する。
【0084】
[1]材料及び方法
以上のモデルを検証するために、以下の手順で実験を行った。
A.発現ベクターの作製
マウスRUNX2遺伝子のRuntドメイン(RD)をコードするDNA断片を調製し、上記の「DsG2−SUMO1−DsRed1」を発現するベクターのSUMO1−DsRed1をコードする部分と置き換えて、「DsG2−Runx2RD」を構築した。同様にCBFβをコードするDNA断片を、「DsG2−SUMO1−DsRed1」を発現するベクターのDsG2−SUMO1をコードする部分と置き換えて「CBFβ−DsRed1」を発現するベクターを構築した後、このcDNAを切り出してゼオシン耐性遺伝子を持つベクター“pEB6CAGmcs−SRZ”に乗せ替えた。これにより、G418とゼオシンの二重選択によって、「DsG2−Runx2RD」と「CBFβ−DsRed1」を同時に細胞に発現できるベクターセットを構築した。
また、FRETが起こらない場合の比較実験用に、「CBFβ−DsRed1」のDsRed1の67番目のチロシンをスレオニンに置換した変異を導入した「CBFβ−DsRed1Y67T」も構築し、FRETアクセプターとしてまったく機能できないようにした。
構築した発現ベクターは、市販のDNA精製キットを用いて大腸菌から大量調製した。
【0085】
B.形質転換細胞の作製及び選択
調製した発現ベクターを市販のリポフェクション試薬を用いてヒト細胞株HEp−2細胞に導入し、4日間、1.5mg/ml G418(ロシュ社)、0.1mg/ml ゼオシン(インビトロジェン社)両抗生物質の存在下で培養し、DNAが導入された細胞だけを選択した。細胞をトリプシン処理して回収し、フローサイトメーター(Flicyme(登録商標),三井造船(株))で蛍光緩和時定数、蛍光陽性細胞率及び個々の細胞の蛍光強度を解析した。
【0086】
[2]結果
フローサイトメーターによる蛍光緩和時定数の測定
図10は、DsG2−Runx2RDと組み合わせて、CBFβ−野生型DsRed1又はCBFβ−DsRed1Y67T変異を細胞に発現させての蛍光緩和時定数をフローサイトメーター(Flicyme(登録商標),三井造船(株))を用いて試験した結果を示すヒストグラムである。
DsG2−Runx2RDとCBFβ−野生型DsRed1(野生型のtd−DsRed1)との組み合わせを用いた場合には、ドナー(DsG2)の緑の蛍光の緩和時定数は1.52nsであった(図10A)。
一方、DsG2−Runx2RDとCBFβ−DsRed1Y67T変異(67番目のチロシンがスレオニンに置換されている)との組合せを用いた場合には、ドナーの蛍光緩和時定数は2.23nsであり、DsG2のままであった(図10B)。CBFβ−DsRed1Y67T変異はタンパク質としては生合成されるが、発色団(クロモフォア)のY67がTに置換されているため、アクセプターとして機能できないので蛍光寿命の短縮は起こらないためである。
以上から、DsG2−Runx2RDとCBFβ−野生型DsRed1との組み合わせを用いた場合に、ドナーの蛍光緩和時定数が1.52nsまで短縮し、高い効率でFRETが起こっていることが確認できた。
このように本発明を用いると、2分子間での相互作用もFRETによって簡便に検出できることが明らかとなった。
【産業上の利用可能性】
【0087】
本発明のFRETプローブは、分子デザインが容易で、様々な生体分子の解析に利用することができ、生体分子が生きた細胞中の、いつ、どこで、分子間相互作用や分子内構造変化を受けているのかを可視化・画像化することができる。それによって、細胞内での機能分子の動態を明らかにすることができるため、創薬スクリーニングのためのツールとして利用が可能である。
【図面の簡単な説明】
【0088】
【図1】(A)AequoreaGFPと(B)DsRedについて、蛍光タンパク質の発色団(クロモフォア)の自己触媒的な形成と成熟の過程を比較する図である。切断部位が矢尻形で示される。可視光吸収のためのπ共役系が緑又は赤で示される。
【図2】(A−D):DsRed1、EGFP及びDsG2を細胞に発現させ、蛍光強度をFACSCaliburで測定した結果を示す図である。図中数値は上段が蛍光陽性細胞割合、下段が平均蛍光強度を示す(但しA:赤色蛍光について、B:緑色蛍光について)。(E,F):変異型DsRed1を細胞に発現させ、倒立顕微鏡を用いて観察した結果を示す図である。
【図3】DsG2からDsRed1へのFRET効率をフローサイトメーターを用いて検出した図である。
【図4】SUMO1のタンパク質切断をFRETを用いて検出するしくみの模式図である。
【図5】SUMO1活性化検出ベクターを細胞に発現させ、フローサイトメーター及び倒立顕微鏡を用いて観察した結果を示す図である。
【図6】DsG2、DsRed1を細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。A:td−DsG2、B:td−DsRed1、C:td−DsG2及びtd−DsRed1、D:DsG2とDsRed1を融合
【図7】SUMO1とその不活性化型変異体SUMO1GAの切断を検出できるプローブをを細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。A;野生型SUMO1、B:不活性化型変異体SUMO1GA
【図8】EGFPあるいはDsG2にSUMO1及びその不活性化型SUMO1GAを融合させ、細胞に発現させ、FlicymeによってdTc1を測定した結果を示す図である。
【図9】強度の異なるプロモーターを用いてtd−DsG2を発現させ、蛍光強度及びdTc1を測定した結果を示す図である。 A:FACSCaliburによる測定、B:Flicymeによる測定
【図10】Runx2とCBFβの間の相互作用を、2分子間FRETで検出した結果を示す図である。 A:FACSCaliburによる測定、B:Flicymeによる測定
【配列表フリーテキスト】
【0089】
配列番号1−DsG2のcDNA塩基配列
配列番号2−DsG2のアミノ酸配列
配列番号3−オリゴDNA(Link3−1SE)の塩基配列
配列番号4−LG3のアミノ酸配列(Link3−1SE)
配列番号5−オリゴDNA(Link3−1AS)の塩基配列
【特許請求の範囲】
【請求項1】
二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質のアミノ酸配列に変異が導入された変異型蛍光タンパク質であって、励起極大波長及び蛍光極大波長がともに変異導入前の蛍光タンパク質(以下、未変異型蛍光タンパク質という。)からシフトし、かつ、該未変異型蛍光タンパク質若しくは励起極大波長及び蛍光極大波長がともに相違する他の変異型蛍光タンパク質とヘテロ二量体を形成できる、変異型蛍光タンパク質。
【請求項2】
請求項1に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項3】
前記未変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができる、請求項1に記載の変異型蛍光タンパク質。
【請求項4】
請求項3に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項5】
前記変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができ、変異型蛍光タンパク質とは励起極大波長及び蛍光極大波長がともに相違する、請求項1に記載の変異型蛍光タンパク質。
【請求項6】
請求項5に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項7】
前記未変異型蛍光タンパク質が、サンゴ(学名:Discosoma sp.)に由来する蛍光タンパク質である、請求項1に記載の変異型蛍光タンパク質。
【請求項8】
前記サンゴに由来する蛍光タンパク質が、DsRed1である、請求項7に記載の変異型蛍光タンパク質。
【請求項9】
配列番号1に記載のDNA塩基配列を翻訳して得られるアミノ酸配列をもつ蛍光タンパク質。
【請求項10】
配列番号2に記載のアミノ酸配列をもつ蛍光タンパク質。
【請求項11】
請求項3又は5に記載の2つの蛍光タンパク質を含むFRETプローブ。
【請求項12】
請求項3又は5に記載の2つの蛍光タンパク質がポリペプチドからなるリンカーを介して連結される構造をもつ融合タンパク質であるFRETプローブ。
【請求項13】
前記リンカーが生体分子の全部又は一部のアミノ酸配列を含む、請求項11に記載のFRETプローブ。
【請求項14】
前記2つの蛍光タンパク質がDsG2及びDsRed1である、請求項11又は12に記載のFRETプローブ。
【請求項15】
前記生体分子がSUMO1である請求項12又は13に記載のFRETプローブ。
【請求項16】
請求項11〜14のいずれかに記載のFRETプローブをコードするポリヌクレオチド。
【請求項17】
請求項11〜14のいずれかに記載のFRETプローブをコードするDNAを遺伝子発現ベクターに組み込んで構築した発現ベクター。
【請求項18】
前記遺伝子発現ベクターが、pEB6CAGmcs、pEB6SRαmcs及びpEB6CMVmcsからなる群から選ばれる、請求項17に記載の発現ベクター。
【請求項19】
前記発現ベクターが、pEB6CMVmcs−SRZである、請求項18に記載の発現ベクター。
【請求項20】
請求項17〜19のいずれかに記載の発現ベクターを培養細胞に導入した形質転換細胞。
【請求項21】
前記培養細胞がヒト由来細胞株である、請求項20に記載の形質転換細胞。
【請求項22】
前記ヒト由来細胞株がHEp−2細胞株である、請求項21に記載の形質転換細胞。
【請求項23】
請求項17〜19のいずれかに記載の発現ベクターを含んでなるキット。
【請求項24】
請求項20〜22のいずれかに記載の形質転換細胞を含んでなるキット。
【請求項25】
請求項11〜15のいずれかに記載のFRETプローブを用いる、ドナー蛍光タンパク質の蛍光寿命を測定して細胞内FRETを検出する方法。
【請求項26】
蛍光寿命フローサイトメーターを使用して蛍光寿命を測定する、請求項25に記載の方法。
【請求項1】
二量体又は三量体以上の多量体を形成する自己集合性をもつ蛍光タンパク質のアミノ酸配列に変異が導入された変異型蛍光タンパク質であって、励起極大波長及び蛍光極大波長がともに変異導入前の蛍光タンパク質(以下、未変異型蛍光タンパク質という。)からシフトし、かつ、該未変異型蛍光タンパク質若しくは励起極大波長及び蛍光極大波長がともに相違する他の変異型蛍光タンパク質とヘテロ二量体を形成できる、変異型蛍光タンパク質。
【請求項2】
請求項1に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項3】
前記未変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができる、請求項1に記載の変異型蛍光タンパク質。
【請求項4】
請求項3に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項5】
前記変異型蛍光タンパク質とヘテロ二量体を形成し、両蛍光タンパク質間でFRETを起こすことができ、変異型蛍光タンパク質とは励起極大波長及び蛍光極大波長がともに相違する、請求項1に記載の変異型蛍光タンパク質。
【請求項6】
請求項5に記載の変異型蛍光タンパク質をコードするポリヌクレオチド。
【請求項7】
前記未変異型蛍光タンパク質が、サンゴ(学名:Discosoma sp.)に由来する蛍光タンパク質である、請求項1に記載の変異型蛍光タンパク質。
【請求項8】
前記サンゴに由来する蛍光タンパク質が、DsRed1である、請求項7に記載の変異型蛍光タンパク質。
【請求項9】
配列番号1に記載のDNA塩基配列を翻訳して得られるアミノ酸配列をもつ蛍光タンパク質。
【請求項10】
配列番号2に記載のアミノ酸配列をもつ蛍光タンパク質。
【請求項11】
請求項3又は5に記載の2つの蛍光タンパク質を含むFRETプローブ。
【請求項12】
請求項3又は5に記載の2つの蛍光タンパク質がポリペプチドからなるリンカーを介して連結される構造をもつ融合タンパク質であるFRETプローブ。
【請求項13】
前記リンカーが生体分子の全部又は一部のアミノ酸配列を含む、請求項11に記載のFRETプローブ。
【請求項14】
前記2つの蛍光タンパク質がDsG2及びDsRed1である、請求項11又は12に記載のFRETプローブ。
【請求項15】
前記生体分子がSUMO1である請求項12又は13に記載のFRETプローブ。
【請求項16】
請求項11〜14のいずれかに記載のFRETプローブをコードするポリヌクレオチド。
【請求項17】
請求項11〜14のいずれかに記載のFRETプローブをコードするDNAを遺伝子発現ベクターに組み込んで構築した発現ベクター。
【請求項18】
前記遺伝子発現ベクターが、pEB6CAGmcs、pEB6SRαmcs及びpEB6CMVmcsからなる群から選ばれる、請求項17に記載の発現ベクター。
【請求項19】
前記発現ベクターが、pEB6CMVmcs−SRZである、請求項18に記載の発現ベクター。
【請求項20】
請求項17〜19のいずれかに記載の発現ベクターを培養細胞に導入した形質転換細胞。
【請求項21】
前記培養細胞がヒト由来細胞株である、請求項20に記載の形質転換細胞。
【請求項22】
前記ヒト由来細胞株がHEp−2細胞株である、請求項21に記載の形質転換細胞。
【請求項23】
請求項17〜19のいずれかに記載の発現ベクターを含んでなるキット。
【請求項24】
請求項20〜22のいずれかに記載の形質転換細胞を含んでなるキット。
【請求項25】
請求項11〜15のいずれかに記載のFRETプローブを用いる、ドナー蛍光タンパク質の蛍光寿命を測定して細胞内FRETを検出する方法。
【請求項26】
蛍光寿命フローサイトメーターを使用して蛍光寿命を測定する、請求項25に記載の方法。
【図1】


【図2】
【図3】

【図4】
【図5】
【図6】
【図9】
【図10】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図9】
【図10】
【図7】
【図8】
【公開番号】特開2009−261259(P2009−261259A)
【公開日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願番号】特願2008−111636(P2008−111636)
【出願日】平成20年4月22日(2008.4.22)
【出願人】(000005902)三井造船株式会社 (1,723)
【出願人】(504171134)国立大学法人 筑波大学 (510)
【Fターム(参考)】
【公開日】平成21年11月12日(2009.11.12)
【国際特許分類】
【出願日】平成20年4月22日(2008.4.22)
【出願人】(000005902)三井造船株式会社 (1,723)
【出願人】(504171134)国立大学法人 筑波大学 (510)
【Fターム(参考)】
[ Back to top ]