
外来性タンパク質の高生産形質転換体及びその利用
【課題】本発明は、外来性タンパク質を安定的に高生産可能なより実用的な酵母形質転換体を提供する。
【解決手段】1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23等からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体が提供される。
【解決手段】1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23等からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、外来性タンパク質の高生産形質転換体及びその利用に関する。
【背景技術】
【0002】
近年、有限である石油資源を代替するものとして、植物の光合成作用に由来するバイオマスへの期待が高まってきており、バイオマスをエネルギーや各種材料に利用するための各種の試みがなされている。バイオマスを、エネルギー源やその他の原料として有効利用するためには、バイオマスを動物や微生物が容易に利用可能な炭素源に糖化することが必要である。
【0003】
典型的なバイオマスであるセルロースやヘミセルロースを利用するには、大量のセルラーゼが必要である。こうしたバイオマスをグルコースやキシロース等に糖化するために用いられる酵素製剤費のコスト比率は相当高いものとなっている。セルロースを低コストで糖化し、有用物質に変換するには、セルラーゼの使用をどれだけ低減できるかに大きく依存している。
【0004】
1つの解決策としては、グルコースからエタノールや有機酸などの有用物質を発酵生産する酵母などの微生物に直接セルラーゼを生産させ、糖化させることにより添加すべきセルラーゼ量を抑制することが考えられる。酵母はセルラーゼ遺伝子を有しておらず、外来遺伝子としてセルラーゼ遺伝子を導入し形質転換体を得る必要がある。セルラーゼを高生産する変異株を古典的な変異誘発剤による変異導入法により取得されたことが報告されている(非特許文献1)。しかしながらこうした変異体の取得には大変な時間と労力を要する。
【0005】
一方、酵母での異種タンパク質の高生産変異株及びそれに関与する遺伝子が開示されている(特許文献1)。この文献には、サッカロマイセス セレビジエ(Saccharomyces cerevisiae)の遺伝子破壊株ライブラリーに異種遺伝子であるラッカーゼ遺伝子を導入して、ラッカーゼを高発現する遺伝子を網羅的に探索して、合計18株の変異株を選抜し、18種のラッカーゼの分泌生産を促進する遺伝子が記載されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2005−58143号公報
【非特許文献】
【0007】
【非特許文献1】Aho S., Arffman A., Korhola M., Saccharomyces cerevisiae mutants selected for increased production of Trichoderma reesei cellulases., Appl. Microbiol. Biotechnol., 1996, 46 (1), 36-45.
【発明の概要】
【発明が解決しようとする課題】
【0008】
上記特許文献1に記載の方法では、増殖に影響を与えないタンパク質を高生産させる遺伝子破壊株を調べるために、外来遺伝子を導入した遺伝子破壊株を固体培地上で培養しつつ培地内の基質分解量を指標として高生産変異株を取得し、タンパク質の分泌生産を促進する遺伝子を特定している。こうした遺伝子としては、BUB2、BUB3、BUL1、END3.FYV10、IKI3、MAD1、MAD2、MAD3、NCL1、PER1、PFK26、RPS24A、VHS2、VIK1、VRP1、YKL053W、YML094C-A等が挙げられている。こうした遺伝子の破壊が、酵母を液体培地で培養したときにおいても異種タンパク質の分泌を促進する方向に作用する可能性もあった。一方、タンパク質の分泌に関し固体培養と液体培養とにおける環境は全く相違するものとも考えられ、液体培養では全く異なる遺伝子の破壊が分泌促進に関与する可能性があると考えられた。
【0009】
上記先行技術文献はいずれも、染色体外プラスミドにおいて外来タンパク質を生産するものである。プラスミドは、培養条件によっては脱落する場合があり、プラスミド上に外来遺伝子を保持する場合には、安定して当該遺伝子を発現できない。
【0010】
そこで、本発明は、外来性タンパク質を安定的に高生産可能なより実用的な酵母形質転換体及び当該高生産に関与する遺伝子並びにこれらの利用を提供することを一つの目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記した課題を解決するべく、S. cerevisiaeの遺伝子破壊株に外来性遺伝子であって分泌性タンパク質であるセルラーゼの遺伝子の染色体外及び染色体上への導入を試み、これらの形質転換体を液体培養して高いセルラーゼ活性を示す破壊株のスクリーニングを行った。この結果、染色体外に外来遺伝子を導入した場合に高いセルラーゼ活性を示す破壊株のうちから、染色体上に外来遺伝子を導入した場合に高いセルラーゼ活性を示す破壊株を見出した。すなわち、染色体上に外来遺伝子を導入した場合において、当該外来遺伝子がコードするタンパク質の分泌を促進する遺伝子破壊株を見出した。これらの知見に基づき、以下の手段が提供される。
【0012】
本発明によれば、1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体が提供される。
【0013】
本発明の酵母形質転換体においては、前記外来性タンパク質は分泌性タンパク質であることが好ましい。また、前記外来性タンパク質を液体培養で生産するのに用いる酵母形質転換体であることも好ましい。さらに、前記外来性タンパク質はセルラーゼから選択される1種又は2種以上であることが好ましい。
【0014】
本発明の酵母形質転換体においては、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、 VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることが好ましい。また、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることも好ましい。より好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であり、さらに、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることも好ましい。最も好ましくは、GCN5、RPL19B及びVPS3から選択される1種又は2種以上である。
【0015】
本発明の酵母形質転換体において、不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上を含むことが好ましく、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45及びYPT7からなる群から選択される2種以上を含むことがより好ましい。さらに好ましくは、不活性化されている前記遺伝子は、FAR8, RAV1, RRD1及びVPS3からなる群から選択される2種以上を含むことが好ましい。さらにまた、不活性化されている前記遺伝子は、以下の(a)〜(c)から選択されるいずれかの組み合わせを含むことが好ましい。
(a)RAV1とFAR8
(b)RAV1とRRD1
(c)RAV1とVPS3
【0016】
本発明の酵母形質転換体においては、前記外来性タンパク質は、β−グルコシダーゼ、セロビオヒドロラーゼ及びエンドグルカナーゼから選択される1種又は2種以上であることが好ましい。
【0017】
さらに、本発明の酵母形質転換体においては、前記宿主はサッカロマイセス属酵母であることが好ましく、より好ましくは、前記宿主はサッカロマイセス・セレビジエである。
【0018】
本発明によれば、酵母において外来性タンパク質を発現させるためのベクターであって、
以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上を不活性化するためのベクターが提供される。
【0019】
本発明によれば、外来性のタンパク質を発現させるための宿主酵母であって、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上が不活性化されている、宿主酵母が提供される。
【0020】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、 YPT7及びRRD1からなる群から選択される2種以上である、請求項16に記載の宿主酵母。
【0021】
本発明によれば、セルロース含有材料の分解産物の製造方法であって、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、を備える、製造方法が提供される。
【0022】
本発明によれば、セルロース含有材料から有用物質を製造する方法であって、セルラーゼを外来性タンパク質として分泌生産する上記いずれかに記載の酵母形質転換体を準備する工程と、前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、を備える、製造方法が提供される。
【0023】
本発明によれば、タンパク質の製造方法であって、上記いずれかに記載の酵母形質転換体を準備する工程と、液体培地で前記酵母形質転換体を培養する工程と、を備える、製造方法が提供される。
【図面の簡単な説明】
【0024】
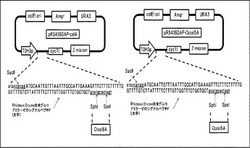
【図1】セルラーゼを酵母で発現させるためのプラスミドの構造を示す図である。
【図2】各種培地の培養上清についてのCtcel8Aのエンドグルカナーゼ活性の評価結果を示す図である。
【図3】Ctcel8A発現プラスミドの酵母遺伝子破壊ライブラリーへの形質転換を示す図である。
【図4】Ctcel8A形質転換体の高活性株のスクリーニング結果を示す図である。
【図5】Ctcel8Aの高生産酵母変異株(活性程度+++)を示す図である。
【図6】Ctcel8Aの高生産酵母変異株(活性程度++)を示す図である。
【図7】Ctcel8Aの高生産酵母変異株(活性程度+)を示す図である。
【図8】富栄養培地(YPD)でのCtcel8Aの活性を示す図である。
【図9】富栄養培地(YPD)でのCccel5Aの活性を示す図である。
【図10】Ctcel8A及びCccel5Aの染色体導入用プラスミドの構造を示す図である。
【図11】富栄養培地で培養したセルラーゼ染色体導入株でのvps3破壊株の活性の評価結果を示す図である。
【図12】貧栄養培地で培養したセルラーゼ染色体導入株でのvps3破壊株の活性の評価結果を示す図である。
【図13】Ctcel8Aの染色体導入用プラスミドpAL-HOR7p-Ctcel8Aの構造を示す図である。
【図14】Ctcel8A染色体導入株のCMCプレートによるセルラーゼ活性の評価結果の一例を示す図である。
【図15】基準株の分解活性を1としたときのCtcel8Aを染色体導入した各種遺伝子破壊株の培養上清のエンドグルカナーゼによるCMC分解活性を示す図である。
【図16】Ctcel8A染色体導入株のcel8A高生産株の培養上清を用いたCMCプレートアッセイの結果を示す図である。
【図17】AaBGLを染色体に導入するためのベクターpDU-HOR7p-AaBGLsecの構造を示す図である。
【図18】DYN3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図19】GCN5破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図20】OPI3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図21】PER1破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図22】RPL13B破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図23】RPL19B破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図24】RP136A破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図25】SNF7破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図26】VMA7破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図27】VPS3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図28】VPS4破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図29】VPS16破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図30】VPS27破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図31】YKL118W破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図32】YOL138C破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図33】BGL導入株の細胞表面(白抜き)及び培地上清(グレー)におけるBGLの相対比活性を示す図である。
【図34】rav1×far8、rav1×rrd1の各二重破壊株のCMC分解プレートアッセイの結果を示す図である。
【図35】rav1×far8、rav1×rrd1の各二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図36】rav1×vps3の二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図37】rpl19bについての二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図38】55種類の遺伝子の細胞内の局在等をSaccharomycees cerevisiae genome database (http://www.yeastgenome.org/) で調べた結果を示す図である。
【発明を実施するための形態】
【0025】
本発明は、外来性タンパク質を安定して高生産可能な酵母形質転換体、そのための宿主酵母、ベクター及び酵母形質転換体を利用した製造方法に関する。本発明は、酵母において以下の55種の遺伝子から選択される1種又は2種以上が不活性化されることにより、外来性タンパク質の分泌生産が安定して促進されることに基づいている。
【0026】
本発明者らは、酵母の遺伝子破壊株ライブラリーを利用して、不活性化されることにより液体培養において外来性の分泌性タンパク質の分泌生産を促進する遺伝子を探索したところ、従来、酵母について固体培地でのスクリーニングにより得られていたこの種の遺伝子とは全く相違する多数の遺伝子を見出した。液体培養でのスクリーニングにより見出される遺伝子は、固体培養でのスクリーニングにより見出される遺伝子とはある程度は一致するものと考えられていたところ、本発明者らが分泌性タンパク質の分泌促進を再確認できた96種の遺伝子のうち、2遺伝子(PER1,VRP1のみ)が固体培養でのスクリーニングにより得られた遺伝子と重複しているに過ぎなかった。このような遺伝子の相違は予想を超えるものであった。
【0027】
また、染色体上に外来タンパク質をコードする遺伝子を保持する場合には、これら96種の遺伝子のうち55種の遺伝子の破壊のみが外来タンパク質の分泌生産に有効であることがわかった。すなわち、外来遺伝子がコードするタンパク質の分泌生産に関し、染色体外のプラスミド上にある外来遺伝子がコードするタンパク質の分泌生産を促進する遺伝子破壊が、必ずしも染色体上にある外来遺伝子がコードするタンパク質の分泌生産を促進するわけではないことがわかった。
【0028】
本発明者らが見出した外来性タンパク質高生産株は、使用した遺伝子破壊株において特定の遺伝子が破壊されたことにより染色体上に導入された外来遺伝子がコードする異種分泌性タンパク質が高生産されたと考えられる。特定遺伝子が破壊される前の正常な状態では、その遺伝子によって染色体上に導入された外来遺伝子がコードする外来性タンパク質の細胞外への生産量が直接または間接的に抑えられていたと考えられる。外来性タンパク質高生産株の中には、vacuolar protein mis-sorting(vps)遺伝子破壊株の一部が含まれた。これらの遺伝子破壊株は、通常細胞内の液胞へソーティングされる内在性タンパク質が細胞外へミスソーティングされ細胞外へ分泌される株である。内在性のタンパク質でも本来の機能を果たせない異常なタンパク質は、液胞内で分解される。本発明を拘束するものではないが、外来性タンパク質が異常なタンパク質と認識され、液胞で処理されるところが、遺伝子が破壊されたために細胞外へ分泌されているのではないかと考えられる。
【0029】
本発明によれば、本発明者らが見出した、液体培養において、不活性化されることにより外来性タンパク質の分泌生産を促進する遺伝子を利用することで、異種分泌性タンパク質を安定して高生産する酵母形質転換体のほか、このような形質転換体に利用する宿主、ベクターも提供される。さらに、こうした酵母形質転換体を利用することでタンパク質を高効率で生産することができるほか、セルロース含有材料の分解、利用も効率的に実施することができる。
【0030】
以下、本発明の各種実施形態について詳細に説明する。
【0031】
(形質転換体)
本発明の酵母形質転換体は、1種又は2種以上の外来性タンパク質をコードするDNAを保持し、宿主の第1の遺伝子群から選択される1種又は2種以上が不活性化されている。以下、宿主、異種分泌性タンパク質及び不活性化対象である遺伝子群につき、順次説明する。
【0032】
(宿主)
本発明の形質転換体の宿主酵母は、特に限定されないで公知の各種酵母を利用できる。後述するエタノール発酵等を考慮すると、サッカロマイセス・セレビジエ(Saccharomyces cerevisiae)等のサッカロマイセス属の酵母、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)等のシゾサッカロマイセス属の酵母、キャンディダ・シェハーテ(Candida shehatae)等のキャンディダ属の酵母、ピヒア・スティピティス(Pichia stipitis)等のピヒア属の酵母、ハンセヌラ(Hansenula)属の酵母、トリコスポロン(Trichosporon)属の酵母、ブレタノマイセス(Brettanomyces)属の酵母、パチソレン(Pachysolen)属の酵母、ヤマダジマ(Yamadazyma)属の酵母、クルイベロマイセス・マーキシアヌス(Kluyveromyces marxianus)、クルイベロマイセス・ラクティス(Kluveromyces lactis)等のクルイベロマイセス属の酵母が挙げられる。なかでも、工業的利用性等の観点からサッカロマイセス属酵母が好ましい。なかでも、サッカロマイセス・セレビジエが好ましい。
【0033】
(外来性のタンパク質をコードするDNA)
外来性のタンパク質とは、宿主酵母にとって外来性であればよい。したがって、他の酵母に由来するタンパク質であっても外来性タンパク質に包含される。外来性タンパク質とは、好ましくは、酵母以外の微生物を含む生物に由来するタンパク質である。
【0034】
外来性タンパク質は、酵母において分泌性を有していることが好ましい。本明細書においてタンパク質の分泌性とは、宿主酵母において膜タンパク質(細胞表層提示タンパク質を含む)又は分泌タンパク質として生産されるタンパク質であることを意味する。分泌性を有する外来タンパク質としては、それ自体が酵母においても膜タンパク質又は分泌タンパク質となりうるものを用いることができるほか、そのような分泌性を有していなくとも、公知の分泌シグナルを連結した融合タンパク質であってもよい。また、それ自体分泌性を有していても、宿主酵母に合わせて分泌シグナルが改変されていてもよい。分泌シグナル等については後段にて説明する。なお、本明細書において、細胞表層提示タンパク質における表層提示形態は特に限定されない。酵母の細胞表層に直接的であっても間接的であっても保持され提示されていればよい。
【0035】
このような外来性の分泌性タンパク質としては、適宜選択できるが、例えば、セルロース含有材料を分解する観点からは、エンドグルカナーゼ、セロビオヒドロラーゼ、β−グルコシダーゼなどの各種セルラーゼのほか、リグニンペルオキシダーゼ、マンガンペルオキシダーゼ及びラッカーゼなどのリグニン分解酵素が挙げられる。また、例えば、セルロース緩和タンパク質であるスウォレニンやエクスパンシン、セルロソームやセルラーゼの構成部分であるセルロース結合ドメイン(タンパク質)が挙げられる。また、キシラナーゼやヘミセルラーゼ等のその他のバイオマス分解酵素も挙げられる。これらのタンパク質は、いずれもセルロースへのセルラーゼのアクセシビリティを向上させることができる。
【0036】
外来性の分泌性タンパク質としては好ましくは、エンドグルカナーゼ(EC 3.2.1.74)、セロビオヒドロラーゼ(EC 3.2.1.91)及びβ−グルコシダーゼ(EC23.2.4.1、EC 3.2.1.21)が挙げられる。これらを酵母に大量に分泌生産させることで、セルロース含有材料を効率的に分解できるようになる。なかでも、結晶性セルロースを分解するほか、セルロースを効率的に低分子化するためにエンドグルカナーゼを発現させることが好ましい。なお、セルラーゼは、そのアミノ酸配列の類似性に基づきGHF(Glycoside Hydrolase family)(http://www.cazy.org/fam/acc.gh.html)の13(5,6,7,8,9,10,12,44,45,48,51,61,74)のファミリーに分類されている。異なるファミリーに分類される同種又は異種のセルラーゼを組み合わせてもよい。
【0037】
セルラーゼとしては、特に限定しないが、それ自体活性の高いセルラーゼであることが好ましい。このようなセルラーゼとしては、例えば、ファネロケーテ(Phanerochaete)属菌、Trichoderma reeseiなどのトリコデルマ属(Trichoderma)菌、フザリウム属(Fusarium)菌、トレメテス属(Tremetes)菌、ペニシリウム属(Penicillium)菌、フミコーラ属(Humicola)菌、アクレモニウム属(Acremonium)菌、アスペルギルス属(Aspergillus)菌等の糸状菌の他に、クロストリジウム属(Clostridium)菌、シュードモナス属(Pseudomonas)菌、セルロモナス属(Cellulomonas)菌、ルミノコッカス属(Ruminococcus)菌、バチルス属(Bacillus)菌等の細菌、スルフォロバス属(Sulfolobus)菌等の始原菌、さらにストレプトマイセス属(Streptomyces)菌、サーモアクチノマイセス属(Thermoactinomyces)菌などの放射菌由来のセルラーゼが挙げられる。なお、こうしたセルラーゼは、人工的に改変されていてもよい。
【0038】
本発明の形質転換体は、外来性のタンパク質をコードするDNAの少なくとも一つを宿主染色体上に保持している。また、このコード化DNAは、宿主酵母内においてタンパク質として発現可能に保持されている。具体的には、宿主酵母で作動可能なプロモーター(典型的には酵母プロモーター)の制御下に連結されるとともに適切なターミネーターをその下流に有した状態で保持されている。プロモーターは、構成的プロモーターであっても誘導的プロモーターであってもよい。一般には、こうした外来DNAの導入に伴って、宿主において利用可能な選択マーカー遺伝子も同時に宿主染色体上に保持されている。
【0039】
(不活性化対象となる遺伝子)
染色体上に分泌性タンパク質をコードするDNAを保持する本発明の形質転換体において不活性化されていることが好ましい遺伝子、すなわち、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性を有する遺伝子を、以下の表に示す。なお、表1に示す55種類の遺伝子は、クロストリジウム・サーモセラム(Clostridium thermocellum)のGHF8に属するセルラーゼ(エンドグルカナーゼ)であるCel8A(以下、Ctcel8Aという。)をコードするDNAを、特定遺伝子が不活性化された酵母の染色体外に導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングし、さらに選抜された遺伝子不活性化株の酵母の染色体上に前記エンドグルカナーゼをコードするDNAを導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングされたものである。
【0040】
【表1】

【0041】
これらの遺伝子は、外来性タンパク質を分泌生産するのに不活性化されていることが好ましい。これらの遺伝子から選択される1種又は2種以上が不活性化され、かつ、外来タンパク質が分泌される形態でそのコード化DNAが染色体上に保持される酵母は、当該遺伝子が不活性化されていない酵母であって同じコード化DNAを染色体上に保持する酵母よりも外来タンパク質の分泌生産が促進される。外来タンパク質は、エンドグルカナーゼなどセルラーゼから選択される1種又は2種以上であることが好ましい。
【0042】
不活性化対象となる遺伝子は、PER1及びVRP1以外の53種類の遺伝子から選択される1種又は2種以上である。これらの遺伝子が不活性化されていると、液体培地で外来性タンパク質を分泌生産させやすくなる。さらに好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。これらの遺伝子が不活性化されていると、エンドグルカナーゼを分泌生産させるのに都合がよい。なお、PER1が除かれていてもよい。
【0043】
また、不活性化対象となる遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であってもよい。これらの遺伝子の不活性化は、β−グルコシダーゼなどのセルラーゼの分泌生産に有利である。より好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。これらの群から、PER1が除かれていてもよい。一層好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。最も好ましくは、GCN5、RPL19B及びVPS3から選択される1種又は2種以上である。
【0044】
なお、表2には、染色体外に分泌性タンパク質をコードするDNAを保持する本発明の形質転換体において不活性化されていることが好ましい遺伝子、すなわち、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性を有する遺伝子を示す。
【0045】
【表2】
【0046】
表2中A欄の計96種の遺伝子は、表1の遺伝子と同様、クロストリジウム・サーモセラム(Clostridium thermocellum)のGHF8に属するセルラーゼ(エンドグルカナーゼ)であるCel8A(以下、Ctcel8Aという。)を外来性の分泌性タンパク質として特定遺伝子が不活性化された酵母に導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングされたものである。
【0047】
これら96種の遺伝子は、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性の程度に応じて3群(表2B欄〜D欄)に分けることができる。表2B欄に記載の遺伝子は、96種のうち不活性化による分泌生産促進活性が最も高いものであり、表1C欄に記載の遺伝子は、次に分泌生産促進活性が高いものであり、表2D欄に記載の遺伝子は、次いで分泌生産促進活性が高いものである。
【0048】
また、表2A欄に示す96種の遺伝子のうち、不活性化によりGHF8に属するセルラーゼの富栄養培地での分泌生産を促進する遺伝子を一つのサブグループ(表2E欄)に分類できる。さらに、不活性化によりGHF5に属するクロストリジウム・セルロヴォランス(Clostridium cellulovorans)の富栄養培地でのセルラーゼ(エンドグルカナーゼ)(Cccel5A)の分泌生産を促進する遺伝子を一つのサブグループ(表2F欄)に分類できる。GHF8及び15に共通する遺伝子は、表2のE欄及びF欄において下線を施した遺伝子である。これらの遺伝子は、双方のファミリーのセルラーゼを活性化できるため、これら2種類のセルラーゼを同時に分泌生産させるのに好ましく用いることできる。
【0049】
表2の遺伝子群は、GHF8に属するエンドグルカナーゼを外来性分泌性タンパク質として分泌生産させたときの活性で評価して得られたものであるが、表2E欄及びF欄に示すように、異なる培養条件及び異なるGHFのセルラーゼについても表2A欄記載の遺伝子の不活性化が有効であることがわかる。このことは、表2A欄に記載の遺伝子の不活性化が、アミノ酸類似性を超えて外来性分泌性タンパク質の分泌生産に広く有用であることを支持している。
【0050】
表2A欄に示す遺伝子群から選択される2種類以上が不活性化されていてもよい。2種類以上の遺伝子が不活性化されていることにより、外来性タンパク質の分泌生産がさらに促進される場合があるからである。2種類以上の遺伝子を不活性化する場合、例えば、以下の表3から選択される2種類以上とすることができる。
【0051】
【表3】
【0052】
表3において、表3A欄には、2種類以上組み合わせて不活性化するのに好ましい遺伝子を記載している。表3A欄には、表3B欄及び表3C欄の遺伝子とRRD1を含んでいる。図38に示すように、表3B欄及びC欄に示す遺伝子は、いずれもタンパク質の輸送経路に関するタンパク質をコードする遺伝子である。表3B欄に示す遺伝子は、粗面小胞体、ゴルジ体に局在するタンパク質をコードする遺伝子(ARV1, CHO2, CSG2, ERG6, FAR8, OPI3, PER1, SAC1)であり、表3C欄に示す、エンドソーム等に局在し、主としてゴルジ体からエンドソーム、液胞への輸送に関わるタンパク質をコードする遺伝子(ERG3, MON2, RAV1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45, YPT7)である(Saccharomycees cerevisiae genome database (http://www.yeastgenome.org/による)。これらタンパク質の輸送経路に関わる遺伝子の破壊、より好ましくは二重破壊は、高生産効果につき相乗効果を有している。図38に示すように、タンパク質が細胞外へ分泌されるときは、粗面小胞体、ゴルジ体を通る。また、正しくフォールディングされなかったタンパク質などの異常なたんぱく質は、ゴルジ体からエンドソームへタンパク質の輸送を経て液胞にて分解される。本発明者らによれば、RAV1などエンドソームに局在するタンパク質をコードする遺伝子と、FAR8等の粗面小胞体に局在するタンパク質をコードする遺伝子又はVPS3等の液胞たんぱく質の輸送に関わるタンパク質をコードする遺伝子の2重破壊により、外来性タンパク質の分泌生産活性が向上したことがわかっている。
【0053】
表3A欄は、表3B欄記載の遺伝子と表3C欄記載の遺伝子に加えて、RRD1を含んでいる。本発明において組み合わされて不活性化される遺伝子は、表3A欄に記載の遺伝子から選択される2種類以上を含むことができる。RRD1は、核、細胞質に局在するタンパク質をコードするが、タンパク質輸送に関するタンパク質をコードする遺伝子とともに不活性化されることにより、相乗的なタンパク質高生産効果をもたらすことができる。好ましくは、表3B欄及び表3C欄に記載の遺伝子群から選択される2種類以上である。2種類以上の不活性化遺伝子は、表3B欄の遺伝子群及び表3C欄の遺伝子群の双方を含んでいてもよいし、いずれか一方であってもよい。
【0054】
不活性化される遺伝子は表3B欄中からはFAR8が選択されることが好ましい。また、不活性化される遺伝子は表3C欄中からはRAV1及びVPS3から選択される1種又2種であることが好ましい。特に、RAV1が選択されることが好ましい。また、不活性化される遺伝子は、FAR8、RAV1、VPS3及びRRD1からなる群から選択される2種類以上を含んでいてもよいし、FAR8、RAV1及びVPS3から選択される2種類以上を含んでいてもよい。こうした不活性化遺伝子の組み合わせとしては、例えば、FAR8とRAV1、RAV1とVPS3、RAV1とRRD1等が挙げられる。
【0055】
なお、Rav1タンパク質は、エンドソームに局在し、Rav1タンパク質、Rav2タンパク質、Skp1タンパク質で複合体を形成することが知られており、初期エンドソームの液胞への輸送にとって必要であることが分かっている(Soi3p/Rav1p functions at the early endosome to regulate endocytic trafficking to the vacuole and localization of trans-Golgi network transmembrane proteins., Mol Biol Cell. 2004 Jul;15(7):3196-209、Seol J. H., Shevchenko A., Shevchenko A., Deshaies R. J., Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat Cell Biol. 2001,Apr;3(4):384-91)。VPS3遺伝子は、液胞タンパク質のソーティングやプロセシングに必要なタンパク質で、H+−液胞ATPaseタンパク質複合体の集合に必要である(Rothman J. H., Yamashiro C. T., Raymond C. K., Kane P. M., Stevens T. H.., Acidification of the lysosome-like vacuole and the vacuolar H+-ATPase are deficient in two yeast mutants that fail to sort vacuolar proteins., J Cell Biol. 1989 109(1):93-100)。Far8タンパク質は、粗面小胞体に局在し、細胞周期のG1期の停止に関わることが分かっている。RRD1タンパク質は、核、細胞質に局在し、タンパク質脱リン酸化酵素PP2Aの活性化するタンパク質であることがわかっている(Kemp H. A., Sprague G. F. Jr., Far3 and five interacting proteins prevent premature recovery from pheromone arrest in the budding yeast Saccharomyces cerevisiae., Mol Cell Biol. 2003 Mar;23(5):1750-63))。
【0056】
表1、表2及び表3に示す酵母の各遺伝子の塩基配列等については、特にS. cerevisiaeなどのサッカロマイセス属に関しては、サッカロマイセスゲノムデータベース(http://www.yeastgenome.org/)から取得することができる。また、サッカロマイセス属を含め、これらの遺伝子については、米国国立衛生研究所のホームページ(http://www.ncbi.nlm.nih.gov/)のブラストサーチサイト(http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)等において取得できる。
【0057】
本発明の形質転換体においては、表1に示す遺伝子又は表2に示す遺伝子から選択される1種又は2種以上、又は表3に示す遺伝子から選択されるは2種類以上が不活性化されている。ここで、遺伝子が不活性化されているとは、少なくとも、宿主酵母内で、(1)当該遺伝子の転写又は翻訳が完全に抑制又は部分的に抑制されている状態、(2)当該遺伝子に変異が導入されており、本来の機能を全く発揮できない程度に不完全なタンパク質を発現する状態が含まれる。こうした不活性化状態は、典型的には、当該遺伝子のプロモーターなどの調節領域又はコード領域に変異が導入されることにより実現される。具体的には、相同組換えによりこうした不活性化体を得ることができる。表1〜表3において示す各酵母遺伝子が破壊された株は、Open Biosystems社(米国)より入手することができる。また、2種類以上の酵母遺伝子が破壊された株は、これらの株に対してさらに遺伝子破壊を施すことなどにより得ることができる。
【0058】
本発明の形質転換体は、液体培養における外来性タンパク質の分泌生産が、表1に示す遺伝子から選択される1種又は2種以上の遺伝子が不活性化されていないときよりも促進されているという特徴を有することができる。すなわち、宿主が野生型(表1に示す遺伝子が不活性化されていない)である以外は、同様に外来性の分泌性タンパク質をコードするDNAを導入して得られた形質転換体よりも外来性の分泌性タンパク質の分泌生産が促進されている。
【0059】
液体培養の条件は特に限定しないで、例えば、実施しようとしている液体培養プロセスを考慮して適宜設定できる。したがって、富栄養培地であっても貧栄養培地であってもよいし、使用する酵母に合わせてpHも適宜設定することができるほか、培地にpH緩衝能を付与してもしなくてもよい。通常、酵母の一般的な培地であるYPD培地やSD培地及びこれらに対して適宜改変した培地を用いることができる。
【0060】
外来性の分泌性タンパク質の分泌生産が促進されているかどうかは、当該外来性の分泌性タンパク質の分泌生産量又は当該分泌性タンパク質の活性により評価することができる。好ましくは、本発明の形質転換体と対比すべき野生型形質転換体とを一定期間液体培養して得られた培養上清につき、当該タンパク質量等を評価する。
【0061】
タンパク質量及びタンパク質の活性測定方法は、タンパク質の種類に応じて適宜選択することができる。当該タンパク質が酵素等であれば、公知の手法で酵素活性を測定してもよい。例えば、外来性分泌性タンパク質がエンドグルカナーゼなどのセルラーゼである場合には、採取した培養上清につき、適当なセルラーゼ基質(カルボキシメチルセルロース、リン酸セルロース、結晶性セルロース等)と反応させて反応生成物量や基質量等を測定することで酵素活性を評価できる。反応温度、pH及び時間は、酵素の種類等において適宜設定することができる。なお、酵素反応の結果生じる還元糖量の定量法としてはSomogyi法、Tauber-Kleiner法、Hanes法(滴定法)、Park-Johnson法、3,5-ジニトロサリチル酸(DNS)法、TZ法(Journal of Biochemical Methods, 11(1985)109-115)等の公知の方法を適宜採用すればよい。
【0062】
また、エンドグルカナーゼなどのセルラーゼ活性は、カルボキシルメチルセルロースなどのセルロースを含有する固相体からなる評価領域に本発明としての可能性のある被験タンパク質を供給し、当該領域の固相体中のセルロースを分解させて、固相体中でセルロースが分解されて消失した領域(ハロー:固相体においてバイオマスの分解により淡色化又は無色化する領域)の大きさでエンドグルカナーゼ活性を評価することもできる。固相体におけるセルロース消失に基づくハローは、目視等により視認することができるほか、ハロー検出時にコンゴーレッドなどで、セルロースを染色することで明瞭にハローを検出することができる。また、バイオマスとして色素結合セルロース(例えば、Cellulose Azure、Sigma社製など)を用いた場合、セルロースの分解により色素が固相体中に拡散しハローを形成するため、容易にセルロース分解活性を検出することができる。同様に蛍光色素を結合したセルロースもバイオマスとして利用することでハローを容易に検出することができる。さらに、酸処理セルロースなどをバイオマスとして用いた場合にもセルロースの分解により透明なハローを形成するため、セルロース分解活性を容易に検出することができる。
【0063】
ハロー形成用の固相体は、例えば、バイオマスを担持するゲルやフィルムが挙げられる。ゲルやフィルムの構成材料は特に限定しないで、天然又は人工の高分子材料を好ましく用いることができる。このような高分子材料としては、アガロース(寒天)を好ましく用いることができる。
【0064】
なお、本発明の形質転換体は、表1、表2又は表3に示す遺伝子の1種類以上又は2種類以上が不活性化され、かつセルラーゼなどの外来タンパク質をコードするDNAの一つが少なくとも染色体上に導入されているとともに、さらに他の外来性タンパク質、例えば、後述する有用物質を生産するような改変がなされていてもよい。
【0065】
(酵母形質転換体の作製)
以上説明した本発明の形質転換体は、モレキュラークローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載されている方法に準じて作製することができる。表1、表2又は表3に示す遺伝子の1種又は2種以上が予め不活性化された宿主酵母に対して、外来性の分泌性タンパク質をコードするDNAを有する発現ベクター等を導入し、宿主酵母を形質転換することにより得ることもできる。
【0066】
宿主酵母としては、Open Biosystems社(米国)より入手することができるほか、破壊しようとする遺伝子のプロモーターなどの調節領域又はコード領域に変異を導入することで取得できる。特定遺伝子の不活性化は、例えば、相同組換え法を利用して、特定の遺伝子に変異を導入したり、別の遺伝子を導入したりすること等により可能である。2種以上の特定遺伝子が不活性化された宿主酵母も、公知の各種方法による遺伝子導入や入手可能な破壊株を利用することによって取得することができる。
【0067】
本発明の好ましい宿主酵母は、表1〜表3に示す遺伝子の1種又は2種以上が不活性化されている酵母である。本発明の宿主酵母は、好適な不活性化遺伝子として既に説明した遺伝子又はその組み合わせが不活性化されていることが好ましい。
【0068】
本発明の酵母において外来性タンパク質を発現させるためのベクターの一つの形態は、表1〜表3に記載の遺伝子から選択される1種又は2種以上を不活性化するためのベクターが挙げられる。このベクターは、不活性化する遺伝子又はその近傍と相同領域を有しており、この相同領域を介して当該ベクターが保持するDNA領域で当該遺伝子又はその近傍を置換することで当該遺伝子を不活性化するものである。このような相同組換えのためのベクター及びその作製は、当業者において周知であって、モレキュラークローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に開示されている。
【0069】
また、酵母において外来性の分泌性タンパク質を発現させるためのベクターの他の一つの形態は、所望の外来性分泌性タンパク質のコード化領域を有している。発現させようとするタンパク質は、酵母において細胞外への分泌性を備えていることが好ましい。こうした分泌性は意図するタンパク質が本来的に有する場合のほか、分泌シグナル等を用いて人工的に付与することができる。細胞外に分泌発現する形態としては、特に限定されない。分泌発現の形態としては、例えば、細胞表層に拘束することなく分泌する形態とタンパク質を細胞表層に提示する形態とが挙げられる。例えば、前者の形態は、酵母において機能するシグナルペプチドを付与するなどすればよく、例えば、酵母におけるタンパク質の分泌のためには、天然シグナル配列は、例えば酵母インベルターゼリーダー、α因子リーダー、またはRhizopus oryzae やC. albicansグルコアミラーゼリーダーなどが挙げられる。また、後者の形態は公知の酵母の細胞表層提示システムを利用することができる。このような提示システムとしては、凝集性タンパク質又はその一部を用いることでタンパク質を酵母の表層に提示した状態に分泌させることができる。例えば、分泌シグナルに加えて凝集性タンパク質であるα−アグルチニンC末端側の320アミノ酸残基からなるペプチドが利用される。所望のタンパク質を細胞表層に提示するためのポリペプチドや手法は、WO01/79483号公報や、特開2003−235579号公報、WO2002/042483号パンフレット、WO2003/016525号パンフレット、特開2006−136223号公報、藤田らの文献(藤田ら,2004. Appl Environ Microbiol 70:1207-1212および藤田ら, 2002. Appl Environ Microbiol 68:5136-5141.)、村井ら, 1998. Appl Environ Microbiol 64:4857-4861.に開示されている。例えば、シグナル配列は、ベクターに組み込まれる要素であってもよいが、セルラーゼ遺伝子の一部であってもよい。
【0070】
本発明のベクターは、上記したタンパク質のコード化DNAとともに調節領域(プロモーター及びターミネーター等)を備えるほか、適宜選択マーカー遺伝子の発現カセットを備えることができる。ベクターは、形質転換の手法や宿主細胞における当該コード化DNAの保持形態(染色体に導入する形態や染色体外に保持する形態等)に応じて、上記コード領域以外の構成要素(相同組換えにより染色体への導入を意図する場合には、酵母染色体DNAとの相同性領域が必要となる。)が適宜決定される。また、ベクターの形態は、使用形態に応じて様々な形態を採ることができる。例えば、DNA断片の形態を採ることができるほか、2マイクロプラスミドなどの適当な酵母用ベクターの形態を採ることもできる。
【0071】
このようなベクターで適当な酵母宿主を適宜形質転換することによって本発明の酵母形質転換体を得ることができる。形質転換にあたり、従来公知の各種方法、例えば、トランスフォーメーション法や、トランスフェクション法、接合法、プロトプラスト法、エレクトロポレーション法、リポフェクション法、酢酸リチウム法等を用いることができる。
【0072】
(セルロース含有材料の分解産物の製造方法)
本発明のセルロース含有材料の分解産物の製造方法は、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、を備えることができる。本発明の製造方法によると、セルロース含有材料を糖化するためのセルラーゼの添加をすることなく又はその添加量を抑制してセルロース含有材料を酵母により直接利用できる。このため、効率的にセルロース含有材料を利用できる。
【0073】
(形質転換体の準備)
酵母形質転換体が分泌生産するセルラーゼとしては、既に説明した各種セルラーゼのうち1種又は2種以上組み合わせて用いることができる。好ましくは2種類以上のセルラーゼ(例えば、エンドグルカナーゼとセロビオヒドロラーゼ等)とすることができる。なお、2種類以上のセルラーゼは、1種の形質転換体が分泌生産するものであってもよいが、それぞれのセルラーゼを分泌生産する2種類以上の形質転換体であってもよい。そのほか、本発明の製造方法で用いる形質転換体については、既に説明した本発明の形質転換体についての各種態様を適用することができる。
【0074】
(液体培養)
上記のような酵母形質転換体を液体培地で液体培養するには、セルロース含有材料を炭素源の少なくとも1種類として用いる以外は、公知の液体培地を利用しあるいは適宜改変して用いることができる。培地組成、温度、pH、通気条件等は必要に応じて適宜設定することができる。
【0075】
セルロース含有材料は、グルコースがβ-1,4-グルコシド結合により重合した重合体及びその誘導体を含んでいる。グルコースの重合度は特に限定しない。また、誘導体としては、カルボキシメチル化、アルデヒド化、若しくはエステル化などの誘導体が挙げられる。セルロースは、結晶性セルロースであってもよいし、非結晶性セルロースであってもよい。また、セルロースは、その部分分解物である、セロオリゴ糖、セロビオースであってもよい。セルロース含有材料は、配糖体であるβグルコシド、リグニン及び/又はヘミセルロースとの複合体であるリグノセルロース、さらにペクチンなどとの複合体であってもよい。セルロース含有材料はその由来も特に限定しない。植物由来のものでも、真菌由来のものでも、細菌由来のものであってもよい。セルロース含有材料は、綿や麻などの天然繊維品、レーヨン、キュプラ、アセテート、リヨセルなどの再生繊維品、稲ワラ、籾殻、木材チップなどの農産廃棄物などが挙げられる。
【0076】
本発明方法で得られるセルロースの分解産物は、グルコースのほか、セロビオース及びセロオリゴ糖が挙げられる。こうして得られるセルロースの分解産物は、通常のグルコースと同様、有用物質の発酵原料等に利用できる。なお、本発明の製造方法で用いる形質転換体としては、酵母本来のアルコール発酵性を有している場合には、後述するように、培養工程を実施することでアルコール(エタノール)などの有用物質を製造することができる。また、使用する形質転換体が遺伝子工学的に乳酸などの有機酸を製造するよう改変されている場合には、当該有機酸を製造することができる。
【0077】
(セルロース含有材料から有用物質を製造する方法)
本発明の有用物質の製造方法は、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、を備えることができる。本発明の製造方法によると、セルロース含有材料を糖化するためのセルラーゼを添加をすることなく又はその添加量を抑制してセルロース含有材料を酵母により直接利用して有用物質を製造できる。このため、従来に比してセルラーゼコストを低減して有用物質を製造することができる。
【0078】
有用物質としては特に限定しないが、グルコースを利用して酵母が生産可能なものであればよい。例えば、酵母におけるグルコースからの代謝系の1種又は2種以上の酵素を遺伝子組換えにより置換、追加等して本来の代謝物でない化合物であってもよい。具体的には、エタノールなどの低級アルコール、乳酸などの有機酸の他、イソプレノド合成経路の追加によるファインケミカル(コエンザイムQ10、ビタミン及びその原料等)、解糖系の改変によるグリセリン、プラスチック・化成品原料など、バイオリファイナリー技術が対象とする材料が挙げられる。酵母は、アルコール発酵能が高いため、本形質転換酵母は、セルロース含有材料からエタノールを効率的に生産することができる。
【0079】
形質転換酵母の発酵には、セルロース含有材料を含む炭素源を用いる。培養開始時には、グルコースやスクロースなど酵母が利用しやすい形態の炭素源を利用することもできる。また、培地中にセルラーゼを添加してもよい。添加するセルラーゼは、特に限定しないが、本発明の形質転換酵母を培養して生産したものであってもよい。
【0080】
そのほか、形質転換酵母の発酵には、酵母に一般的に適用される培養条件を適宜選択して用いることができる。典型的には、発酵のための培養は、静置培養、振とう培養または通気攪拌培養等を用いることができる。通気条件は、嫌気条件下、微好気条件下及び好気条件等、適宜選択することができる。培養温度も、特に限定しないが、25℃〜55℃等の範囲とすることができる。また、培養時間も必要に応じて設定されるが、数時間〜150時間程度とすることができる。また、pHの調整は、無機あるいは有機酸、アルカリ溶液等を用いて行うことができる。培養中は、必要に応じてアンピシリン、テトラサイクリンなどの抗生物質を培地に添加することができる。
【0081】
発酵終了後、培養液から有用物質含有画分を回収する工程、さらにこれを精製又は濃縮する工程を実施することもできる。回収工程や精製等の工程は有用物質の種類等に応じて適宜選択される。
【0082】
本発明のエタノール製造方法は、本発明のセルロース含有材料の分解産物の製造方法において、酵母形質転換体としてアルコール発酵性を有しているものであればよい。
【0083】
(タンパク質の製造方法)
本発明のタンパク質の製造方法は、本発明の形質転換体を準備する工程と、前記形質転換体を液体培地で培養する工程と、を備えることができる。本発明の酵母形質転換体によれば、所望の外来性タンパク質を効率的に分泌生産させることができるため、タンパク質の製造にも有利である。培養工程において用いる液体培地の組成、温度、pH等の条件は、適宜設定することができる。液体培地は、前記酵母形質転換体が利用可能な炭素源を有していればよい。したがって、前記酵母形質転換体に外来タンパク質としてセルラーゼを分泌生産させる場合には、液体培地は、セルロース含有材料を炭素源として含んでいてもよい。培養工程で得られる培養上清には、本発明の形質転換体によって分泌生産されたタンパク質が含まれている。当該タンパク質が酵素等の場合には、これを精製してもよいが、培養上清をそのまま酵素液として用いてもよい。
【0084】
以下、本発明を、実施例を挙げて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、以下に述べる遺伝子組換え操作はMolecular Cloning: A Laboratory Manual (T. Maniatis, et al., Cold Spring Harbor Laboratory) に従い行った。
【実施例1】
【0085】
セルラーゼ発現プラスミドの構築
TDH3プロモーター、CYC1ターミネーター、マーカーURA3を持つ酵母発現用2ミクロンベクターのSacII-SpeI間にRhizopus Oryzae由来グルコアミラーゼのシグナルペプチドを導入した。本シグナルペプチドの下流SphI-SpeI間にClostridium thermocellum(以下C.thermocellum)のエンドグルカナーゼCel8A(以下Ctcel8A)、またはClostridium cellulovorans (以下C.cellulovorans)のエンドグルカナーゼcel5A(以下Cccel5A)の導入を行った(図1)。作製したプラスミドDNAはCtcel8AはpRSA436GAPcelA, Cccel5AはpRS436GAPCccel5Aと名付けた。
【実施例2】
【0086】
酵母菌でのエンドグルカナーゼ活性の確認
Saccharomyces cerevisiaeのS288C株由来の2倍体酵母菌BY4743(遺伝型:MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/LYS2 MET15/met15Δ0 ura3Δ0/ura3Δ0)へpRS436GAPcelAを酢酸リチウム法によって形質転換した。YPDプレートに酵母をストリークし、30℃で1日培養後、形質転換液(35μlの60%ポリエチレングリコール平均分子量3350+2.5μlの4M酢酸リチウム+5μlの1M ジチオスレイトール+5μlの10mg/ml 鮭精子DNAにpRS436GAPcelAを1μlを混ぜ、全量50μlになるように滅菌水を加える)に酵母を培養したYPDプレートからかきとり混ぜた。形質転換液を42℃で2時間熱を加え、SD-uracil(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(-uracil))培地にまいた。アミノ酸ミックス(-uracil)は、アデニン0.5g、トリプトファン2g、ヒスチジン塩酸塩2g、メチオニン2g、ロイシン4g、リシン塩酸塩2gを1リットル当たり0.6g加えた。培地のpHを制御した場合にエンドグルカナーゼ活性に変化が起こるかどうかを調べた。YPD培地(1% yeast extract, 2% peptone+2% glucose)、SD-uracil培地、SD-uracilに0.1 M KPO4を加えpH緩衝した培地で培養した培養上清を、エンドグルカナーゼ活性を検出することができる0.1% カルボキシメチルセルロース入りの2% 寒天培地 (以下、CMC培地)にスポットし、40℃で1日反応させた。0.1% CongoRed /1 M Tris (pH9)(以下、コンゴーレッド液)でプレートを染色し、1 M NaClで脱色後、エンドグルカナーゼ活性がある染色されないハロ(円)を確認した(図2)。培養pHの低下によってcelAのエンドグルカナーゼ活性は大きく低下した。以上のことから、以後のエンドグルカナーゼ活性の評価にあたっては、pH低下を抑制した状態を確保することとした。
【実施例3】
【0087】
酵母遺伝子破壊株ライブラリーへの形質転換
「実施例1」で構築したプラスミドpRS436GAPcelAを2倍体酵母遺伝子破壊株ライブラリーへ形質転換した。方法は、YPDプレートに保存した破壊株ライブラリーをYPD 25μlを入れた96ウェルマイクロプレートに植菌し、30℃で1日静地培養後、培養液を混ぜ、文献(kitagawa et al.,Biosci Biotechnol Biochem. 2007 Mar;71(3):772-82. Epub 2007 Mar 7.Screening of drugs that suppress Ste11 MAPKKK activation in yeast identified a c-Abl tyrosine kinase inhibitor.)に記載の方法に鮭精子DNAを加えた形質転換液(3500μlの60% ポリエチレングリコール平均分子量3350+250μlの1 M ジチオスレイトール+ 250μlの4 M 酢酸ナトリウム+250μlの10 mg/ml鮭精子DNA+250μlのpRS436GAPcelAのDNA溶液)と混ぜた後、42℃で2時間熱を加え、SD-uracil培地へスポットした(図3)。
【実施例4】
【0088】
Ctcel8A高生産酵母変異株のスクリーニング
Ctcel8A高生産する酵母変異株をスクリーニングするために、Ctcel8Aの形質転換したプレートから各株を採取し、SD-uracilが50μl入った96ウェルマイクロプレートで30℃で1日振とう培養した。培養上清5μlを8レーンピペットでCMC培地にスポットし、40℃で1日反応させ、コンゴーレッド液で染色し、1 M NaClで脱色した。エンドグルカナーゼ活性のあるハロが他の遺伝子破壊株よりも大きい株を226株取得した(図4)。
【実施例5】
【0089】
Ctcel8A高生産酵母変異株の再確認
スクリーニングで得られたCtcel8A高活性株226株の再現性を得るために、取得した高活性株をひと株ずつ試験管でSD-uracil培地で振とう培養し、実施例2と同様の操作で野生型のBY4743よりも高活性な株として147株の再現性を得た。Ctcel8A高生産株147株の栄養要求性を確認するためにSD-U-M-K (0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(-U-M-K))培地へストリークし、増殖できない株を高生産株から除き、117株になった。SD-U-M-K培地のアミノ酸ミックス(-U-M-K)は、アデニン0.5g、トリプトファン2g、ヒスチジン塩酸塩2g、ロイシン4gを1リットル当たり0.4gをSD培地に混ぜた。使用した破壊株ライブラリーは2倍体なので、取得した高活性株に1倍体が混ざっていると1倍体とmatingするはずである。一倍体を除くために、N435-1A (遺伝型:MATa his7 lys7 met6 arg1 gal4 MAL2 SUC)、N435-2A (遺伝型:MATα his7 lys7 met6 arg1 gal4 MAL2 SUC)と高生産株をYPDプレート上で混ざるようにストリークし、30℃で1日培養した。培養したプレートを最小培地(0.67% YNB w/o a. a. +2% グルコース)へレプリカした。7株が最小培地で増殖したので除き、110株となった。もう一度野生型(BY4743株)と活性の差がほとんどない株を除くために、再度試験管でSD-uracil培地で培養し、実施例2と同様の方法で再実験を行い96株の再現性が確認できた。96株をCtcel8A高活性株とし、活性の強さを+++、++、+の3段階に分類した(図5〜図7)。
【0090】
これら96株については、外来性タンパク質を分泌生産するのに好ましい変異を有しているといえる。すなわち、これらの株において破壊されている遺伝子は、外来性タンパク質を分泌生産させるのに不活性化することが好ましい遺伝子であることがわかった。
【実施例6】
【0091】
Ctcel8A高活性株の富栄養培地での活性
スクリーニングで得られた96株のCtcel8A高活性株を富栄養培地であるYPD培地で培養し、100倍希釈した培養上清の活性を実施例2と同様の方法(CMC培地にスポットし、40℃で1日反応させた後、0.1% CongoRed /1 M Tris (pH9)(以下、コンゴーレッド液)でプレートを染色し、1 M NaClで脱色した。)で調べたところ、25株が野生型のBY4743よりも高活性であった。これらの遺伝子破壊株に対してCtcel8Aを再形質転換し、増殖の悪かった2株は除き、21株が野生型のBY4743よりも高活性であった(図8)。2株は再現性が得られなかった。以上のことから、この実施例で取得できた株は、培地の組成に関わらず、高活性を示すことがわかった。
【実施例7】
【0092】
Cccel5Aでの富栄養培地での活性
実施例6で再形質転換して得られた23株に対して、Clostridium cellulolyticum,のエンドグルカナーゼCccel5Aを発現する実施例1記載のプラスミドpRS436GAPCccel5A(図1)を形質転換した。YPD培地で培養し、50倍希釈した培養上清の活性を実施例2と同様の方法で調べたところ、14株が野生型のBY4743よりも高活性であった(図9)。残り9株は、BY4743と活性は変わらなかった 。以上のことから、本実施例で高活性の確認された変異株は、GHF5及び8の双方を分泌生産するのに有用であること、すなわち、これらの変異株において破壊されている遺伝子の不活性化が外来性タンパク質の分泌生産に有効であることがわかった。
【実施例8】
【0093】
Ctcel8A及びCccel5Aの染色代導入用ベクターの作製
セルラーゼを染色体に導入するためのベクターpAH-HORp-Ctcel8A, pAH-HOR7p-Cccel5Aを作製した(図10)。AAP1遺伝子の上流-3000bpから-2000bpまでの配列(以下AAP1Uと呼ぶ)をS288CのゲノムDNAからPCR反応により、クローニングした。AAP1遺伝子の上流-2000bpから-1000bpまでの配列(以下AAP1D)をS288CのゲノムDNAからPCR反応により、クローニングした。遺伝子導入の確認用マーカーとしてハイグロマイシン耐性遺伝子を連結した。C.thermocellum (ATCC27405), C.cellulolyticum (ATCC35319)の培養菌体からゲノムを抽出後、分泌シグナル配列を除くセルラーゼ遺伝子C.thermocellum由来Cel8A (ABN51508)約1.3kb及びC.cellulolyticum由来Cel5A(AAA51444)約1.3kbをPCR法により取得した。取得した遺伝子断片をCel8Aは制限酵素KpnI-BglII、Cel5AはKpnI-BamHI部位にて染色体導入用ベクター内Rhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【実施例9】
【0094】
vps3遺伝子破壊株でのCtcel8A及びCccel5A染色体導入株の効果
実施例8で作製したプラスミドpAH-HOR7p-Ctcel8A、pAH-HOR7p-Cccel5Aを形質転換した。形質転換方法はBY4743及び、vps3破壊株をYPD培地で30℃、1日培養し、YPD培地で再度5時間30℃で培養した。培養液は遠心して回収し、再度滅菌水で洗浄後、遠心して回収した。60%ポリエチレングリコール平均分子量3350を120μl、4M 酢酸リチウムを5μl、1M ジチオスレイトールを10μl、制限酵素Sse8387IでプラスミドDNAの染色体導入部を線上DNAにした後、DNA溶液を5μl加え、細胞懸濁液を50μl加え、懸濁後、42℃で1時間反応させた。反応液を遠心後、YPD培地を加え、30℃で一晩回復培養を行った。培養液はYPDに最終濃度が150μg/mlのハイグロマイシンBが入ったプレートにまき、30℃で増殖させた。 ハイグロマイシン耐性になったコロニーを再びシングルコロニー化した。コロニーをYPDプレートで再度増殖させた。YPD培地で30℃1日培養後、培養液を遠心し、培養上清をx1, x2, x4, x8, x16, x32倍希釈した。上清8μlをCMC培地へスポットし、40℃で1日反応させた。コンゴーレッド液で染色し1M NaClで脱色し、活性を確認した(図11)。BY4743と比べて、vps3破壊株はCtcel8Aでは8倍以上、Cccel5Aでは32倍以上の活性上昇が認められた。
【0095】
上記のCtcel8A及びCccel5A染色体導入株をSD+complement培地(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(complement)、 pHをNaOHで6に調整した)培地とSD+complemnent+0.1M NaAc培地(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(complement)+0.1M 酢酸ナトリウムを加えpHをHClで6に調整)培地で培養した。アミノ酸ミックス(complement)は、アデニン0.5g、ウラシル2g、トリプトファン2g、ヒスチジン塩酸塩2g、メチオニン2g、ロイシン4g、リシン塩酸塩2gを混ぜ、1リットル当たり0.6gを加えた。培養後、培養上清をCMC培地へ8μlスポットした。プレートを40℃で1日反応後、コンゴーレッド液で染色し1M NaClで脱色した(図12)。酢酸ナトリウムで培地のpHを下げないように緩衝した場合(SD+complement+0.1M NaAc培地)では、野生型(BY4743株)よりもvps3破壊株のほうが活性が約2倍程度上昇した。一方pHを緩衝しない培地(SD+complement)では、培地pHの低下によりBY4743は活性が確認できなかったが、vps3破壊株では活性が確認できた。よって、YPD培地と同様に、SD培地のような貧栄養培地でもvps3破壊株はセルラーゼを高生産できることが確認できた。
【0096】
以上のことから、vps3遺伝子を不活性化した酵母においては、外来性タンパク質のコード化DNAが染色体導入形態であっても外来性タンパク質の高度な分泌生産活性が得られることがわかった。すなわち、安定して外来タンパク質を高活性で分泌生産できることがわかった。また、貧栄養及び低pHに対する耐性も野生型よりも向上していることがわかった。
【実施例10】
【0097】
遺伝子破壊株へのcel8A遺伝子の染色体導入
−80℃で保存された1倍体の遺伝子破壊株ライブラリー(インビトロジェン株式会社)から表1に記載の遺伝子に相当する1倍体の株を96株選び、遺伝子の破壊をコロニーPCRにより確認した。それぞれの株を、20mMのNaOH5μlに少量加え、100℃で10分ボイルした。ボイルしたサンプルに15μlの超純水を加えた。使用したプライマーは表3に示すとおりであった。Reverse側のプライマーはKANMX+600R(ATCAAGTGAGAAATCACCATGAGTGACGAC)(配列番号1)を共通に使用し、Forward側のプライマーは、それぞれKANMX+600R以外の遺伝子固有のプライマーを使用した。これらのPCR反応は、Extaq DNAポリメラーゼ(タカラバイオ株式会社)を用いた。なお、PCRは以下のようにして行った。すなわち、PCR反応液6.5μlに1.5μlの100℃で10分ボイルして破砕した細胞懸濁液を加えた。反応条件は、94℃、2分ボイルし、94℃、15秒、50℃、30秒、72℃、2分を40サイクル繰り返した。この結果、96株中、13株(vma4、kap120、mtq2、ydr433W、ygr102C、yvh1、adh1、ado1、pat1、mdm34、rpl1B、vps33、ynl296W破壊株)は遺伝子破壊が確認できなかった。
【0098】
【表4A】
【表4B】
【0099】
遺伝子破壊が確認できなかった13株を除いた83株の遺伝子破壊株及び基準株(BY4741株、遺伝型:MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0)に対してCtcel8Aの染色体導入を行った。染色体導入用ベクターとして図13に示すpAL-HOR7p-Ctcel8Aを構築した。このベクターは、以下のようにして作製した。AAP1遺伝子の上流-3000bpから-2000bpまでの配列(以下AAP1Uと呼ぶ)をS288CのゲノムDNAからPCR反応により、クローニングした。AAP1遺伝子の上流-2000bpから-1000bpまでの配列(以下AAP1D)をS288CのゲノムDNAからPCR反応により、クローニングした。遺伝子導入の確認用マーカーとしてLEU2遺伝子を連結した。C.thermocellum (ATCC27405)の培養菌体からゲノムを抽出後、分泌シグナル配列を除くセルラーゼ遺伝子C.thermocellum由来Cel8A (ABN51508)約1.3kbをPCR法により取得した。取得した遺伝子断片をCel8Aは制限酵素KpnI-BglII部位にて染色体導入用ベクター内Rhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【0100】
それぞれの株をYPD培地で30℃、24時間培養した。培養液1mlを10mlのYPD培地に植菌し、再び5時間培養を行った。培養液を遠心し、超純水で洗浄後再び遠心して細胞をペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに1μlのSse8387Iで処理したpAL-HOR7p-Ctcel8Aと細胞懸濁液を54μl加え、42℃で1時間熱ショックを与えた。この細胞懸濁液をSD-leucine培地へ接種し、30℃で数日培養した。生えたコロニーをYPD+0.1%CMC培地に塗抹し、30℃、1日培養し、CMC分解活性があるかどうかを確認した。
【0101】
CMC含有培地上のコロニーにおいてCMC分解活性が認められた株をそれぞれYPD液体培地に植菌し、30℃で24時間培養した。培養液を遠心し、培養上清を回収した。培養上清をx1、x2、x4、x8、x16、x32倍に希釈し、0.1%カルボキシメチルセルロース、50mMの酢酸ナトリウム入り(pH6)の2%寒天(以下CMCプレート)に8μlずつスポットし、40℃で1日反応させた。反応後、0.1%コンゴーレッド、1M Tris-Base (pH9)(以下コンゴーレッド液)と30分室温で反応させ、1MのNaClで脱色した。結果を図14及び図15に示す。
【0102】
図14に示すようにして、Ctcel8Aの染色体導入株を2クローンずつ調べたところ、基準株よりも活性の強い株と基準株と活性が変わらない株に分かれた。2クローンとも基準株よりも2倍以上高活性であった株を染色体導入でも高生産できる株とした。その結果、図15に示すように、83株中、55株が基準株と比べて、2倍から4倍高活性であることがわかった。
【実施例11】
【0103】
Ctcel8A染色体導入株のcel8A高生産の再確認
再現性をとるために、実施例10で選抜した55株を再度YPD液体培地で、30℃で24時間培養し、培養上清を8倍希釈し、CMCプレートへスポットし、40℃で1日反応させ、コンゴーレッド液で染色し、1M NaClで脱色した。結果を図16に示す。選抜された55株の遺伝子破壊株はいずれも8倍希釈したハローは基準株(BY4741株)よりも高活性であることが確認できた。実施例10及び11の結果から、55株のなかでも、dyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138cの各遺伝子破壊株がセルラーゼの分泌生産が高いことがわかった。
【実施例12】
【0104】
BGL染色体導入用ベクターの作製
エンドグルカナーゼ以外のセルラーゼにおいて高生産効果があるかどうかを調べるために、Aspergillus aculeatusのβ-グルコシダーゼBGL1(以下AaBGLと称する。)を酵母染色体に導入するベクターを作製した。
【0105】
AaBGLを染色体に導入するためのベクターpDU-HOR7p-AaBGLsecを作製した(図17)。染色体導入のための相同配列として、ADH3遺伝子の上流-3000bpから-2000bpまでの配列(以下ADH3Uと称する。)をS288Cのゲノムからクローニングした。同様に、ADH3遺伝子の上流-2000bpから-1000bpまでの配列(以下ADH3Dと呼ぶ)をS288Cのゲノムからクローニングした。遺伝子導入の確認用マーカーとしてURA3遺伝子を連結した。Aspergillus aculeatusのβ-グルコシダーゼ遺伝子(Genbank Accesion No.D64088)の分泌配列を除く配列をクローニングした。染色体導入用ベクター内のRhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【実施例13】
【0106】
BGL染色体導入株のBGL活性とセロビオース培地でのエタノール発酵
実施例12で作製したプラスミドDNA、pDU-HOR7p-AaBGLsecをSse8387Iで処理したDNA溶液を実施例10と同様の方法で基準株であるBY4741及び遺伝子破壊株の染色体へ導入した。遺伝子破壊株は、dyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138c破壊株(合計15種類)を使用した。AaBGLを染色体へ導入した株3クローンをYPD培地で30℃、120rpm, 24時間培養した。培養液の培養上清及び細胞表面のBGL活性をp-Nitrophenyl-α-glucopyranoside (pNPG)の分解によって測定した。培養上清のBGL活性の測定方法は、695μlのMQ(ミリポア社)水、200μlの250mM NaAc(pH5)、100μlの20mM pNPGに細胞を除いた培養上清を5μl加え、30℃で振とうしながら60分反応させた。反応液100μlに50μlの1N Na2CO3を加え、反応を停止させた。反応停止液をA400の吸光高度計で測定した。また、細胞表面でのBGL活性は、細胞を超純水で2回洗浄し、OD600を測定し、650μlの超純水、200μlの250mM NaAc(pH5)、100μlの20mM pNPGに、50μlのOD600を1に合わせた細胞懸濁液を加え(最終OD600=0.5)、30℃で振とうしながら、60分反応させた。培養上清と同様に反応液を測定した。これらの結果を図33に示す。
【0107】
図33に示すように、細胞表面ではsnf7破壊株以外のdyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138c破壊株は基準株(BY4741株)よりも活性が向上した。培養上清は、dyn3, rpl13b, rpl36b, snf7, vps4, vps27, ykl118w破壊株以外のgcn5, opi3, per1, rpl19b, vps3, vps16, yol138c破壊株は基準株(BY4741株)よりも活性が向上した。snf7破壊株以外は、培養上清及び細胞表面のいずれか又は双方で分解活性が高まり、分泌生産が向上したことがわかった。snf7破壊株は細胞表面、培養上清両方において活性が低下した。
【0108】
さらに、AaBGL導入株3クローンのセロビオースを炭素源としたエタノール発酵を行った。2mlのYPD培地で、30℃、120rpm、24時間培養後、再度20mlのYPD培地で30℃、80rpm、24時間培養した。OD600を測定し、OD600=10に相当する培養液を遠心し、超純水10mlで細胞を洗浄後、5%セロビオース入りのYPcellobiose培地10mlでエタノール生産を行った(図18−図32)。培養開始時並びに4,6,8,10及び12時間後の発酵液をサンプリングした。発酵時の細胞濃度(OD600)は吸光光度計で測定した。残存グルコース、残存セロビオース、エタノールはバイオセンサー(王子計測機器株式会社、BF-4)にて測定した。残存セロビオースは、培養サンプルを20倍希釈したものに0.25%になるようにNovozyme188(インビトロジェン株式会社)を加え、45℃、1時間振とうしながら反応させ、セロビオースを完全糖化させたものを測定し、残存グルコースの測定値を引いた。発酵時の増vps16, vps27, snf7は基準株よりも低下した。snf7, vps16以外のdyn3, gcn5, opi3, per1,殖速度はrpl13b, rpl19b, rpl36a, vma7, vps3, vps4, vps27, ykl118w, yol138c破壊株はエタノール生産速度が向上した。
【実施例14】
【0109】
MET17遺伝子のクローニング
形質転換用のマーカー遺伝子をクローニングするために、MET17遺伝子をクローニングした。すなわち、MET17遺伝子を、S288CのゲノムDNAを鋳型として、KOD Plus(東洋紡株式会社)酵素及び以下のプライマーを用いて、94℃で15秒、55℃で30秒、68℃で2分を40サイクル繰り返して合成した。
attB2-MET17-500F (ggggacagctttcttgtacaaagtggATTTAATGCTATAATAGACATTTAAATCCA)(配列番号98)
attB3-MET17+1632R (ggggacaactttgtataataaagttgACCTCCATCATCCTCTTTTGTAACTTGGTC)(配列番号99)
【0110】
PCR産物をGFX PCR DNA and gel band purification kit (GE)で精製した。精製したPCR産物2μlに、pDONR P2R-P3 (インビトロジェン株式会社)プラスミド1μl、TE buffer 5μl、BPクロナーゼenzyme mix(インビトロジェン株式会社)2μlを加え、25℃で1時間反応させた。その後、proteinase Kを1μl加え、37℃で10分反応後、大腸菌DH5αへ形質転換した。形質転換した大腸菌につき、Extaq (タカラバイオ株式会社)及び以下のプライマーを用いてインサートを確認した。また、確認したプラスミドDNAをシークエンスし、変異がないことを確認した。得られたプラスミドを、pE3-MET17と命名した。
M13−F (GTAAAACGACGGCCAG)(配列番号100)
M13-R (GGAAACAGCTATGACCATG)(配列番号101)
【実施例15】
【0111】
RAV1遺伝子とRRD1遺伝子またはFAR8遺伝子との2重破壊によるセルラーゼ高生産の効果
Rav1タンパク質、RRD1タンパク質、Far8タンパク質は、細胞内の局在または機能が異なっていると考えられる。こうした機能が異なる遺伝子の2重破壊でタンパク質高生産の更なる効果が認められるのかどうかを調べるために、一倍体のrrd1またはfar8破壊株に対して、RAV1遺伝子の破壊を行った。
【0112】
pE3-MET17を鋳型DNAとして、以下のプライマーを用いて、PrimeSTAR MAX DNA polymerase(タカラバイオ株式会社)でrav1::MET17破壊のDNA断片を98℃で2分ボイル後、98℃で10秒、55℃で15秒、72℃で1分を30サイクル繰り返して合成した。
RAV1-MET17F: (TTAAAGGGATCTAAGAACACCGCAAACAATTGAGAAAGTAAACAAACGTGCACACGCAAAGGTAGTTCTCCACGTGAAGCTGTCGATATTGGGGAACTGT)(配列番号102)
RAV1-MET17R:(AAAACACTACATTAATAAGATGTAATTATTTTTATTTATAAATCCATTTATTTGATAAAAAATATGATATTACGAGTCACGACATGTAACAAGGGAAAAA)(配列番号103)
【0113】
1倍体の酵母遺伝子破壊株ライブラリーから出したrrd1破壊株及びfar8破壊株をYPD培地(1% yeast extract, 2% peptone, 2% glucose)で30℃、120rpm、24時間培養した。10mlのYPD培地に1mlのこの培養液を加え、さらに30℃で5時間培養した。得られた培養液を遠心し、ペレットの細胞を超純水5mlで洗浄し、再び遠心してペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに10μlのrav1::MET17破壊のDNA断片と細胞懸濁液を45μl加え、42℃で1時間熱ショックを与えた。サンプルはSD-methionine培地へ播種し、30℃で数日培養した。
【0114】
生えたコロニーをExtaq DNAポリメラーゼ(タカラバイオ株式会社)を使い、以下のプライマーを用いて、94℃で2分ボイル後、94℃で15秒、55℃で30秒、72℃で2分を40サイクル行ってPCR反応を行い、インサートを確認した。far8とrav1の2重破壊株をTTK175、rrd1とrav1の2重破壊株をTTK176と命名した。
RAV1-500F:
(AACTTTCCTTTGCACAAGTTTTCGTACTGT)(配列番号104)
RAV1+4280R :
(AGTTATAGAACAAAGAATAGAATATGTCAA)(配列番号105)
【0115】
次に、作製した2重破壊株TTK175およびTTK176へCtcel8Aの染色体への導入を次のような操作で行った。TTK175 (rav1 x far8 2重破壊株)及びTTK176 (rav1 x rrd1 2重破壊株)をYPD培地で30℃、120rpm、24時間培養した。10mlのYPD培地に1mlの培養液を加えさらに30℃で5時間培養した。培養液を遠心し、ペレットの細胞を超純水5mlで洗浄し、再び遠心してペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに1μlのSse8387I制限酵素処理したpAL-HOR7p-Ctcel8Aと細胞懸濁液を54μl加え、42℃で1時間熱ショックを与えた。サンプルはSD-leucine培地へ播種し、30℃で数日培養した。生えたコロニーをYPD+0.1%CMC培地にぬり、30℃、1日培養し、CMC分解活性があるかどうかを確認した。
【0116】
確認できた株をYPD培地で30℃、1日培養した。培養液を遠心し、培養上清と細胞に分けた。培養上清を8倍希釈し、0.1%CMC/50mMNaAc(pH6)2% agarに8μlずつスポットした。プレートを40℃で1日反応させ、コンゴレッド液で30分染色し、1MのNaClで脱色させた。結果を図34に示す。プレートレベルでは、2重破壊株のほうがわずかにCMCの分解されたハローが大きかった。
【0117】
次に、培養上清の活性の定量を次の方法で測定した。Ctcel8A遺伝子を導入していない酵母BY4741(基準株)、rav1破壊株、rrd1破壊株、far8破壊株、TTK175、TTK176に対して、Ctcel8Aを導入した株と導入していない株をYPD培地で培養した。培養液を遠心し培養上清と細胞にわけた。250μlの1% CMC、100μlの250mM NaAc buffer(pH6)、140μlの超純水に培養上清を10μl加え、40℃で60分反応させた。反応液5μlを100μlのTZ bufferへ入れ、100℃で3分反応させた。反応液を分光光度計の吸光度A660を測定した。活性の数値は、Ctcel8Aを導入した酵母の数値からCtcel8Aを導入していない酵母の培養上清の数値を引いて算出した。結果を図35に示す。
【0118】
図35に示すように、基準株と比べ、far8破壊株、rav1破壊株、rrd1破壊株の活性は、2.4、2.1、1.4倍活性が上昇した。rav1 x far8 2重破壊株(TTK175)は3.9倍活性が上昇した。また、rav1 x rrd1 2重破壊株(TTK176)は、活性が2.6倍上昇した。それぞれの2重破壊株は単独の破壊株と比べて活性が上昇することが分かった。以上のことから、細胞内の局在や機能が異なるタンパク質をコードする遺伝子を組み合わせて破壊することが外来タンパク質の高生産に寄与することがわかった。
【実施例16】
【0119】
RAV1遺伝子とVPS3との2重破壊によるセルラーゼ高生産の効果
VPS3遺伝子及びRAV1遺伝子は、似た作用部位で働くタンパク質をコードする遺伝子であるが、こうした遺伝子を2重に破壊した場合でも更なる高生産効果が得られるのかどうかを調べるために、vps3破壊株に対してrav1遺伝子の破壊を行い、2重破壊株を作製した。2重破壊株の作製方法は実施例15の方法に従って行った。rav1とvps3の2重破壊株をTTK179と名付けた。作製した2重破壊株に対してCtcel8Aを実施例15と同様の方法で導入した。染色体導入の確認は、実施例15と同様の方法で行った。Ctcel8Aを導入したBY4741(基準株)、rav1破壊株、vps3破壊株、rav1 x vps3 2重破壊株、Ctcel8Aを導入していない同様の株をそれぞれYPD培地で30℃、24時間培養した。培養液を遠心し培養上清と細胞に分けた。CMC分解による活性の定量は実施例15に記載のTZ法を用いて行った。結果を図36に示す。
【0120】
図36に示すように、rav1及びvps3破壊株はBY4741(基準株)と比べて、活性が2.5倍、3.5倍活性が上昇したが、rav1 x vps3 2重破壊株はBY4741(基準株)と比べて4.9倍活性が上昇した。以上の結果から、細胞内の近接ないし類似する作用部位で働くrav1 タンパク質及びvps3タンパク質をそれぞれコードする遺伝子を組み合わせて破壊することが、外来タンパク質の高生産に寄与することがわかった。
【実施例17】
【0121】
RPL19B遺伝子との2重破壊によるセルラーゼ高生産の効果
RPL19B遺伝子はタンパク質の翻訳に必要なリボソームの構成成分のひとつである(Song J. M., Cheung E., Rabinowitz J. C., Organization and characterization of the two yeast ribosomal protein YL19 genes., Curr Genet. 1996 Sep;30(4):273-8.
)。RPL19B遺伝子の破壊により、Ctcel8Aを高生産できたが、リボソーム構成要素と他の機能との2重破壊による高生産効果を調べるために、4つのCtcel8Aを高生産できる遺伝子(dyn3, gcn5, opi3, ykl118w)を選択した。DYN3遺伝子は、核の分配に関わる遺伝子(Lee WL, Kaiser MA, Cooper JA., The offloading model for dynein function: differential function of motor subunits., J Cell Biol. 2005 Jan 17;168(2):201-7)、GCN5遺伝子は、ヒストンアセチルトランスフェラーゼ(Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD., Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation., Cell. 1996 Mar 22;84(6):843-51.)、OPI3遺伝子は、リン脂質メチル転移酵素であり(McGraw P, Henry SA., Mutations in the Saccharomyces cerevisiae opi3 gene: effects on phospholipid methylation, growth and cross-pathway regulation of inositol synthesis., Genetics. 1989 Jun;122(2):317-30)、YKL118W遺伝子は、機能未知で遺伝子の一部が液胞のvacuolar H+-ATPase (V-ATPase) であるVPH2遺伝子と重複している。6種の遺伝子はRPL19Bの翻訳機能とは全く異なった作用で機能しているが、これらの遺伝子との2重破壊株を以下の通りに作製した。
【0122】
rpl18b::MET17破壊のDNA断片を、実施例14に記載のpEG-MET17プラスミドDNAを鋳型として、以下のプライマー及びKOD-Plusで98℃で30秒ボイル後、98℃で10秒、60℃で2分、72℃で2分を35サイクル繰り返してPCR反応を行い合成した。
RPL19B-MET17F:(ATACTTTCAATTCTTGAGTTGTAAGGCTGCATCCCTCCGTAGAACAAACCAGAGAGCCGAGCTTAGTAATCACGTGAAGCTGTCGATATTGGGGAACTGT)(配列番号106)
RPL19B-MET17R:(AGAAATTCAGTACAAAAAATATCGTTACATCTTATTTCCCCCTATAGCAGCAAATCAATTCACTCCTGGGTACGAGTCACGACATGTAACAAGGGAAAAA)(配列番号107)
【0123】
実施例15と同様の操作で、BY4741株にPCR合成したrpl19b::MET17破壊のDNA断片を形質転換して、コロニーを得た。遺伝子破壊できたかどうかの確認のために、以下のプライマーを用いて生えたコロニー実施例15と同様の方法でPCR反応を行い、目的のDNA断片が得られることを確認した。それぞれの株をTTK309 (dyn3Δ rpl19bΔ::MET17)、TTK310 (ykl118wΔ rpl19bΔ::MET17)、TTK311 (opi3Δ rpl19bΔ::MET17)、TTK312 (gcn5Δ rpl19bΔ:MET17)と命名した。それぞれの株にCtcel8Aを染色体へ導入するために、実施例15と同様の操作でCtcel8Aを染色体へ挿入した。
MET17+1F:
(ATGCCATCTCATTTCGATACTGTTCAACTA)(配列番号108)
RPL19B+1530R:
(ATGTAGATAAGCTATCATAAACTGGTAGTG)(配列番号109)
【0124】
それぞれの株をYPD培地で24時間培養し、細胞と培養上清に分けた。細胞上清の活性を実施例15に記載のTZ法を用いてCMCの分解活性を測定した。結果を図37に示す。図37に示すように、rpl19bとの2重破壊は、rpl19b x opi3 2重破壊株は、opi3破壊株よりも活性が上昇したが、rpl19b破壊株とほとんど同程度の活性であった。rpl19b x dyn3の2重破壊株、rpl19b x ykl118wの2重破壊株はdyn3及びykl118w破壊株単独と同程度の活性であった。また、rpl19b x gcn5の二重破壊株はgcn5単独破壊株よりも活性が低下した。
【0125】
以上説明したように、遺伝子を二重破壊しても活性が変わらない株、活性が低下する株も見られた。取得した55種類の遺伝子の細胞内の局在をSaccharomycees cerevisiae genome database (http://www.yeastgenome.org/) で調べた(図38)。タンパク質が細胞外へ分泌されるときは、粗面小胞体、ゴルジ体を通る。また、正しくフォールディングされなかったタンパク質などの異常なたんぱく質は、ゴルジ体からエンドソームへタンパク質の輸送を経て液胞にて分解される。エンドソームに局在するRAV1と粗面小胞体に局在するFAR8遺伝子また、液胞たんぱく質の輸送に関わるVPS3の2重破壊はCMC分解活性が上昇した。このことから、タンパク質の輸送経路に関わる遺伝子の2重破壊がより高生産効果があることがわかった。特に粗面小胞体、ゴルジ体に局在する遺伝子群(ARV1, CHO2, CSG2, ERG6, FAR8, OPI3, PER1, SAC1)、エンドソーム等に局在し、主として、ゴルジ体からエンドソーム、液胞への輸送に関連する遺伝子群(ERG3, MON2, RAV1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45, YPT7)が最も効果があることが考えられた。
【配列表フリーテキスト】
【0126】
配列番号1〜109:プライマー
【技術分野】
【0001】
本発明は、外来性タンパク質の高生産形質転換体及びその利用に関する。
【背景技術】
【0002】
近年、有限である石油資源を代替するものとして、植物の光合成作用に由来するバイオマスへの期待が高まってきており、バイオマスをエネルギーや各種材料に利用するための各種の試みがなされている。バイオマスを、エネルギー源やその他の原料として有効利用するためには、バイオマスを動物や微生物が容易に利用可能な炭素源に糖化することが必要である。
【0003】
典型的なバイオマスであるセルロースやヘミセルロースを利用するには、大量のセルラーゼが必要である。こうしたバイオマスをグルコースやキシロース等に糖化するために用いられる酵素製剤費のコスト比率は相当高いものとなっている。セルロースを低コストで糖化し、有用物質に変換するには、セルラーゼの使用をどれだけ低減できるかに大きく依存している。
【0004】
1つの解決策としては、グルコースからエタノールや有機酸などの有用物質を発酵生産する酵母などの微生物に直接セルラーゼを生産させ、糖化させることにより添加すべきセルラーゼ量を抑制することが考えられる。酵母はセルラーゼ遺伝子を有しておらず、外来遺伝子としてセルラーゼ遺伝子を導入し形質転換体を得る必要がある。セルラーゼを高生産する変異株を古典的な変異誘発剤による変異導入法により取得されたことが報告されている(非特許文献1)。しかしながらこうした変異体の取得には大変な時間と労力を要する。
【0005】
一方、酵母での異種タンパク質の高生産変異株及びそれに関与する遺伝子が開示されている(特許文献1)。この文献には、サッカロマイセス セレビジエ(Saccharomyces cerevisiae)の遺伝子破壊株ライブラリーに異種遺伝子であるラッカーゼ遺伝子を導入して、ラッカーゼを高発現する遺伝子を網羅的に探索して、合計18株の変異株を選抜し、18種のラッカーゼの分泌生産を促進する遺伝子が記載されている。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2005−58143号公報
【非特許文献】
【0007】
【非特許文献1】Aho S., Arffman A., Korhola M., Saccharomyces cerevisiae mutants selected for increased production of Trichoderma reesei cellulases., Appl. Microbiol. Biotechnol., 1996, 46 (1), 36-45.
【発明の概要】
【発明が解決しようとする課題】
【0008】
上記特許文献1に記載の方法では、増殖に影響を与えないタンパク質を高生産させる遺伝子破壊株を調べるために、外来遺伝子を導入した遺伝子破壊株を固体培地上で培養しつつ培地内の基質分解量を指標として高生産変異株を取得し、タンパク質の分泌生産を促進する遺伝子を特定している。こうした遺伝子としては、BUB2、BUB3、BUL1、END3.FYV10、IKI3、MAD1、MAD2、MAD3、NCL1、PER1、PFK26、RPS24A、VHS2、VIK1、VRP1、YKL053W、YML094C-A等が挙げられている。こうした遺伝子の破壊が、酵母を液体培地で培養したときにおいても異種タンパク質の分泌を促進する方向に作用する可能性もあった。一方、タンパク質の分泌に関し固体培養と液体培養とにおける環境は全く相違するものとも考えられ、液体培養では全く異なる遺伝子の破壊が分泌促進に関与する可能性があると考えられた。
【0009】
上記先行技術文献はいずれも、染色体外プラスミドにおいて外来タンパク質を生産するものである。プラスミドは、培養条件によっては脱落する場合があり、プラスミド上に外来遺伝子を保持する場合には、安定して当該遺伝子を発現できない。
【0010】
そこで、本発明は、外来性タンパク質を安定的に高生産可能なより実用的な酵母形質転換体及び当該高生産に関与する遺伝子並びにこれらの利用を提供することを一つの目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記した課題を解決するべく、S. cerevisiaeの遺伝子破壊株に外来性遺伝子であって分泌性タンパク質であるセルラーゼの遺伝子の染色体外及び染色体上への導入を試み、これらの形質転換体を液体培養して高いセルラーゼ活性を示す破壊株のスクリーニングを行った。この結果、染色体外に外来遺伝子を導入した場合に高いセルラーゼ活性を示す破壊株のうちから、染色体上に外来遺伝子を導入した場合に高いセルラーゼ活性を示す破壊株を見出した。すなわち、染色体上に外来遺伝子を導入した場合において、当該外来遺伝子がコードするタンパク質の分泌を促進する遺伝子破壊株を見出した。これらの知見に基づき、以下の手段が提供される。
【0012】
本発明によれば、1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体が提供される。
【0013】
本発明の酵母形質転換体においては、前記外来性タンパク質は分泌性タンパク質であることが好ましい。また、前記外来性タンパク質を液体培養で生産するのに用いる酵母形質転換体であることも好ましい。さらに、前記外来性タンパク質はセルラーゼから選択される1種又は2種以上であることが好ましい。
【0014】
本発明の酵母形質転換体においては、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、 VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることが好ましい。また、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることも好ましい。より好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であり、さらに、前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であることも好ましい。最も好ましくは、GCN5、RPL19B及びVPS3から選択される1種又は2種以上である。
【0015】
本発明の酵母形質転換体において、不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上を含むことが好ましく、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45及びYPT7からなる群から選択される2種以上を含むことがより好ましい。さらに好ましくは、不活性化されている前記遺伝子は、FAR8, RAV1, RRD1及びVPS3からなる群から選択される2種以上を含むことが好ましい。さらにまた、不活性化されている前記遺伝子は、以下の(a)〜(c)から選択されるいずれかの組み合わせを含むことが好ましい。
(a)RAV1とFAR8
(b)RAV1とRRD1
(c)RAV1とVPS3
【0016】
本発明の酵母形質転換体においては、前記外来性タンパク質は、β−グルコシダーゼ、セロビオヒドロラーゼ及びエンドグルカナーゼから選択される1種又は2種以上であることが好ましい。
【0017】
さらに、本発明の酵母形質転換体においては、前記宿主はサッカロマイセス属酵母であることが好ましく、より好ましくは、前記宿主はサッカロマイセス・セレビジエである。
【0018】
本発明によれば、酵母において外来性タンパク質を発現させるためのベクターであって、
以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上を不活性化するためのベクターが提供される。
【0019】
本発明によれば、外来性のタンパク質を発現させるための宿主酵母であって、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上が不活性化されている、宿主酵母が提供される。
【0020】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、 YPT7及びRRD1からなる群から選択される2種以上である、請求項16に記載の宿主酵母。
【0021】
本発明によれば、セルロース含有材料の分解産物の製造方法であって、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、を備える、製造方法が提供される。
【0022】
本発明によれば、セルロース含有材料から有用物質を製造する方法であって、セルラーゼを外来性タンパク質として分泌生産する上記いずれかに記載の酵母形質転換体を準備する工程と、前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、を備える、製造方法が提供される。
【0023】
本発明によれば、タンパク質の製造方法であって、上記いずれかに記載の酵母形質転換体を準備する工程と、液体培地で前記酵母形質転換体を培養する工程と、を備える、製造方法が提供される。
【図面の簡単な説明】
【0024】
【図1】セルラーゼを酵母で発現させるためのプラスミドの構造を示す図である。
【図2】各種培地の培養上清についてのCtcel8Aのエンドグルカナーゼ活性の評価結果を示す図である。
【図3】Ctcel8A発現プラスミドの酵母遺伝子破壊ライブラリーへの形質転換を示す図である。
【図4】Ctcel8A形質転換体の高活性株のスクリーニング結果を示す図である。
【図5】Ctcel8Aの高生産酵母変異株(活性程度+++)を示す図である。
【図6】Ctcel8Aの高生産酵母変異株(活性程度++)を示す図である。
【図7】Ctcel8Aの高生産酵母変異株(活性程度+)を示す図である。
【図8】富栄養培地(YPD)でのCtcel8Aの活性を示す図である。
【図9】富栄養培地(YPD)でのCccel5Aの活性を示す図である。
【図10】Ctcel8A及びCccel5Aの染色体導入用プラスミドの構造を示す図である。
【図11】富栄養培地で培養したセルラーゼ染色体導入株でのvps3破壊株の活性の評価結果を示す図である。
【図12】貧栄養培地で培養したセルラーゼ染色体導入株でのvps3破壊株の活性の評価結果を示す図である。
【図13】Ctcel8Aの染色体導入用プラスミドpAL-HOR7p-Ctcel8Aの構造を示す図である。
【図14】Ctcel8A染色体導入株のCMCプレートによるセルラーゼ活性の評価結果の一例を示す図である。
【図15】基準株の分解活性を1としたときのCtcel8Aを染色体導入した各種遺伝子破壊株の培養上清のエンドグルカナーゼによるCMC分解活性を示す図である。
【図16】Ctcel8A染色体導入株のcel8A高生産株の培養上清を用いたCMCプレートアッセイの結果を示す図である。
【図17】AaBGLを染色体に導入するためのベクターpDU-HOR7p-AaBGLsecの構造を示す図である。
【図18】DYN3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図19】GCN5破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図20】OPI3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図21】PER1破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図22】RPL13B破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図23】RPL19B破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図24】RP136A破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図25】SNF7破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図26】VMA7破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図27】VPS3破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図28】VPS4破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化について示す図である。
【図29】VPS16破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図30】VPS27破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図31】YKL118W破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図32】YOL138C破壊株のYPcellobiose(5%)培地におけるセロビオースの発酵試験結果を示す図である。上段は、増殖速度を示すグラフであり、下段は、エタノール濃度、セロビオース濃度及びグルコース濃度の変化を示すグラフである。
【図33】BGL導入株の細胞表面(白抜き)及び培地上清(グレー)におけるBGLの相対比活性を示す図である。
【図34】rav1×far8、rav1×rrd1の各二重破壊株のCMC分解プレートアッセイの結果を示す図である。
【図35】rav1×far8、rav1×rrd1の各二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図36】rav1×vps3の二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図37】rpl19bについての二重破壊株のTZ法によるCMC分解活性の測定結果を示す図である。
【図38】55種類の遺伝子の細胞内の局在等をSaccharomycees cerevisiae genome database (http://www.yeastgenome.org/) で調べた結果を示す図である。
【発明を実施するための形態】
【0025】
本発明は、外来性タンパク質を安定して高生産可能な酵母形質転換体、そのための宿主酵母、ベクター及び酵母形質転換体を利用した製造方法に関する。本発明は、酵母において以下の55種の遺伝子から選択される1種又は2種以上が不活性化されることにより、外来性タンパク質の分泌生産が安定して促進されることに基づいている。
【0026】
本発明者らは、酵母の遺伝子破壊株ライブラリーを利用して、不活性化されることにより液体培養において外来性の分泌性タンパク質の分泌生産を促進する遺伝子を探索したところ、従来、酵母について固体培地でのスクリーニングにより得られていたこの種の遺伝子とは全く相違する多数の遺伝子を見出した。液体培養でのスクリーニングにより見出される遺伝子は、固体培養でのスクリーニングにより見出される遺伝子とはある程度は一致するものと考えられていたところ、本発明者らが分泌性タンパク質の分泌促進を再確認できた96種の遺伝子のうち、2遺伝子(PER1,VRP1のみ)が固体培養でのスクリーニングにより得られた遺伝子と重複しているに過ぎなかった。このような遺伝子の相違は予想を超えるものであった。
【0027】
また、染色体上に外来タンパク質をコードする遺伝子を保持する場合には、これら96種の遺伝子のうち55種の遺伝子の破壊のみが外来タンパク質の分泌生産に有効であることがわかった。すなわち、外来遺伝子がコードするタンパク質の分泌生産に関し、染色体外のプラスミド上にある外来遺伝子がコードするタンパク質の分泌生産を促進する遺伝子破壊が、必ずしも染色体上にある外来遺伝子がコードするタンパク質の分泌生産を促進するわけではないことがわかった。
【0028】
本発明者らが見出した外来性タンパク質高生産株は、使用した遺伝子破壊株において特定の遺伝子が破壊されたことにより染色体上に導入された外来遺伝子がコードする異種分泌性タンパク質が高生産されたと考えられる。特定遺伝子が破壊される前の正常な状態では、その遺伝子によって染色体上に導入された外来遺伝子がコードする外来性タンパク質の細胞外への生産量が直接または間接的に抑えられていたと考えられる。外来性タンパク質高生産株の中には、vacuolar protein mis-sorting(vps)遺伝子破壊株の一部が含まれた。これらの遺伝子破壊株は、通常細胞内の液胞へソーティングされる内在性タンパク質が細胞外へミスソーティングされ細胞外へ分泌される株である。内在性のタンパク質でも本来の機能を果たせない異常なタンパク質は、液胞内で分解される。本発明を拘束するものではないが、外来性タンパク質が異常なタンパク質と認識され、液胞で処理されるところが、遺伝子が破壊されたために細胞外へ分泌されているのではないかと考えられる。
【0029】
本発明によれば、本発明者らが見出した、液体培養において、不活性化されることにより外来性タンパク質の分泌生産を促進する遺伝子を利用することで、異種分泌性タンパク質を安定して高生産する酵母形質転換体のほか、このような形質転換体に利用する宿主、ベクターも提供される。さらに、こうした酵母形質転換体を利用することでタンパク質を高効率で生産することができるほか、セルロース含有材料の分解、利用も効率的に実施することができる。
【0030】
以下、本発明の各種実施形態について詳細に説明する。
【0031】
(形質転換体)
本発明の酵母形質転換体は、1種又は2種以上の外来性タンパク質をコードするDNAを保持し、宿主の第1の遺伝子群から選択される1種又は2種以上が不活性化されている。以下、宿主、異種分泌性タンパク質及び不活性化対象である遺伝子群につき、順次説明する。
【0032】
(宿主)
本発明の形質転換体の宿主酵母は、特に限定されないで公知の各種酵母を利用できる。後述するエタノール発酵等を考慮すると、サッカロマイセス・セレビジエ(Saccharomyces cerevisiae)等のサッカロマイセス属の酵母、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)等のシゾサッカロマイセス属の酵母、キャンディダ・シェハーテ(Candida shehatae)等のキャンディダ属の酵母、ピヒア・スティピティス(Pichia stipitis)等のピヒア属の酵母、ハンセヌラ(Hansenula)属の酵母、トリコスポロン(Trichosporon)属の酵母、ブレタノマイセス(Brettanomyces)属の酵母、パチソレン(Pachysolen)属の酵母、ヤマダジマ(Yamadazyma)属の酵母、クルイベロマイセス・マーキシアヌス(Kluyveromyces marxianus)、クルイベロマイセス・ラクティス(Kluveromyces lactis)等のクルイベロマイセス属の酵母が挙げられる。なかでも、工業的利用性等の観点からサッカロマイセス属酵母が好ましい。なかでも、サッカロマイセス・セレビジエが好ましい。
【0033】
(外来性のタンパク質をコードするDNA)
外来性のタンパク質とは、宿主酵母にとって外来性であればよい。したがって、他の酵母に由来するタンパク質であっても外来性タンパク質に包含される。外来性タンパク質とは、好ましくは、酵母以外の微生物を含む生物に由来するタンパク質である。
【0034】
外来性タンパク質は、酵母において分泌性を有していることが好ましい。本明細書においてタンパク質の分泌性とは、宿主酵母において膜タンパク質(細胞表層提示タンパク質を含む)又は分泌タンパク質として生産されるタンパク質であることを意味する。分泌性を有する外来タンパク質としては、それ自体が酵母においても膜タンパク質又は分泌タンパク質となりうるものを用いることができるほか、そのような分泌性を有していなくとも、公知の分泌シグナルを連結した融合タンパク質であってもよい。また、それ自体分泌性を有していても、宿主酵母に合わせて分泌シグナルが改変されていてもよい。分泌シグナル等については後段にて説明する。なお、本明細書において、細胞表層提示タンパク質における表層提示形態は特に限定されない。酵母の細胞表層に直接的であっても間接的であっても保持され提示されていればよい。
【0035】
このような外来性の分泌性タンパク質としては、適宜選択できるが、例えば、セルロース含有材料を分解する観点からは、エンドグルカナーゼ、セロビオヒドロラーゼ、β−グルコシダーゼなどの各種セルラーゼのほか、リグニンペルオキシダーゼ、マンガンペルオキシダーゼ及びラッカーゼなどのリグニン分解酵素が挙げられる。また、例えば、セルロース緩和タンパク質であるスウォレニンやエクスパンシン、セルロソームやセルラーゼの構成部分であるセルロース結合ドメイン(タンパク質)が挙げられる。また、キシラナーゼやヘミセルラーゼ等のその他のバイオマス分解酵素も挙げられる。これらのタンパク質は、いずれもセルロースへのセルラーゼのアクセシビリティを向上させることができる。
【0036】
外来性の分泌性タンパク質としては好ましくは、エンドグルカナーゼ(EC 3.2.1.74)、セロビオヒドロラーゼ(EC 3.2.1.91)及びβ−グルコシダーゼ(EC23.2.4.1、EC 3.2.1.21)が挙げられる。これらを酵母に大量に分泌生産させることで、セルロース含有材料を効率的に分解できるようになる。なかでも、結晶性セルロースを分解するほか、セルロースを効率的に低分子化するためにエンドグルカナーゼを発現させることが好ましい。なお、セルラーゼは、そのアミノ酸配列の類似性に基づきGHF(Glycoside Hydrolase family)(http://www.cazy.org/fam/acc.gh.html)の13(5,6,7,8,9,10,12,44,45,48,51,61,74)のファミリーに分類されている。異なるファミリーに分類される同種又は異種のセルラーゼを組み合わせてもよい。
【0037】
セルラーゼとしては、特に限定しないが、それ自体活性の高いセルラーゼであることが好ましい。このようなセルラーゼとしては、例えば、ファネロケーテ(Phanerochaete)属菌、Trichoderma reeseiなどのトリコデルマ属(Trichoderma)菌、フザリウム属(Fusarium)菌、トレメテス属(Tremetes)菌、ペニシリウム属(Penicillium)菌、フミコーラ属(Humicola)菌、アクレモニウム属(Acremonium)菌、アスペルギルス属(Aspergillus)菌等の糸状菌の他に、クロストリジウム属(Clostridium)菌、シュードモナス属(Pseudomonas)菌、セルロモナス属(Cellulomonas)菌、ルミノコッカス属(Ruminococcus)菌、バチルス属(Bacillus)菌等の細菌、スルフォロバス属(Sulfolobus)菌等の始原菌、さらにストレプトマイセス属(Streptomyces)菌、サーモアクチノマイセス属(Thermoactinomyces)菌などの放射菌由来のセルラーゼが挙げられる。なお、こうしたセルラーゼは、人工的に改変されていてもよい。
【0038】
本発明の形質転換体は、外来性のタンパク質をコードするDNAの少なくとも一つを宿主染色体上に保持している。また、このコード化DNAは、宿主酵母内においてタンパク質として発現可能に保持されている。具体的には、宿主酵母で作動可能なプロモーター(典型的には酵母プロモーター)の制御下に連結されるとともに適切なターミネーターをその下流に有した状態で保持されている。プロモーターは、構成的プロモーターであっても誘導的プロモーターであってもよい。一般には、こうした外来DNAの導入に伴って、宿主において利用可能な選択マーカー遺伝子も同時に宿主染色体上に保持されている。
【0039】
(不活性化対象となる遺伝子)
染色体上に分泌性タンパク質をコードするDNAを保持する本発明の形質転換体において不活性化されていることが好ましい遺伝子、すなわち、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性を有する遺伝子を、以下の表に示す。なお、表1に示す55種類の遺伝子は、クロストリジウム・サーモセラム(Clostridium thermocellum)のGHF8に属するセルラーゼ(エンドグルカナーゼ)であるCel8A(以下、Ctcel8Aという。)をコードするDNAを、特定遺伝子が不活性化された酵母の染色体外に導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングし、さらに選抜された遺伝子不活性化株の酵母の染色体上に前記エンドグルカナーゼをコードするDNAを導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングされたものである。
【0040】
【表1】
【0041】
これらの遺伝子は、外来性タンパク質を分泌生産するのに不活性化されていることが好ましい。これらの遺伝子から選択される1種又は2種以上が不活性化され、かつ、外来タンパク質が分泌される形態でそのコード化DNAが染色体上に保持される酵母は、当該遺伝子が不活性化されていない酵母であって同じコード化DNAを染色体上に保持する酵母よりも外来タンパク質の分泌生産が促進される。外来タンパク質は、エンドグルカナーゼなどセルラーゼから選択される1種又は2種以上であることが好ましい。
【0042】
不活性化対象となる遺伝子は、PER1及びVRP1以外の53種類の遺伝子から選択される1種又は2種以上である。これらの遺伝子が不活性化されていると、液体培地で外来性タンパク質を分泌生産させやすくなる。さらに好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。これらの遺伝子が不活性化されていると、エンドグルカナーゼを分泌生産させるのに都合がよい。なお、PER1が除かれていてもよい。
【0043】
また、不活性化対象となる遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上であってもよい。これらの遺伝子の不活性化は、β−グルコシダーゼなどのセルラーゼの分泌生産に有利である。より好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。これらの群から、PER1が除かれていてもよい。一層好ましくは、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上である。最も好ましくは、GCN5、RPL19B及びVPS3から選択される1種又は2種以上である。
【0044】
なお、表2には、染色体外に分泌性タンパク質をコードするDNAを保持する本発明の形質転換体において不活性化されていることが好ましい遺伝子、すなわち、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性を有する遺伝子を示す。
【0045】
【表2】
【0046】
表2中A欄の計96種の遺伝子は、表1の遺伝子と同様、クロストリジウム・サーモセラム(Clostridium thermocellum)のGHF8に属するセルラーゼ(エンドグルカナーゼ)であるCel8A(以下、Ctcel8Aという。)を外来性の分泌性タンパク質として特定遺伝子が不活性化された酵母に導入し、液体培養して得られた培養上清のエンドグルカナーゼ活性に基づいてスクリーニングされたものである。
【0047】
これら96種の遺伝子は、不活性化により外来性の分泌性タンパク質の分泌生産を促進する活性の程度に応じて3群(表2B欄〜D欄)に分けることができる。表2B欄に記載の遺伝子は、96種のうち不活性化による分泌生産促進活性が最も高いものであり、表1C欄に記載の遺伝子は、次に分泌生産促進活性が高いものであり、表2D欄に記載の遺伝子は、次いで分泌生産促進活性が高いものである。
【0048】
また、表2A欄に示す96種の遺伝子のうち、不活性化によりGHF8に属するセルラーゼの富栄養培地での分泌生産を促進する遺伝子を一つのサブグループ(表2E欄)に分類できる。さらに、不活性化によりGHF5に属するクロストリジウム・セルロヴォランス(Clostridium cellulovorans)の富栄養培地でのセルラーゼ(エンドグルカナーゼ)(Cccel5A)の分泌生産を促進する遺伝子を一つのサブグループ(表2F欄)に分類できる。GHF8及び15に共通する遺伝子は、表2のE欄及びF欄において下線を施した遺伝子である。これらの遺伝子は、双方のファミリーのセルラーゼを活性化できるため、これら2種類のセルラーゼを同時に分泌生産させるのに好ましく用いることできる。
【0049】
表2の遺伝子群は、GHF8に属するエンドグルカナーゼを外来性分泌性タンパク質として分泌生産させたときの活性で評価して得られたものであるが、表2E欄及びF欄に示すように、異なる培養条件及び異なるGHFのセルラーゼについても表2A欄記載の遺伝子の不活性化が有効であることがわかる。このことは、表2A欄に記載の遺伝子の不活性化が、アミノ酸類似性を超えて外来性分泌性タンパク質の分泌生産に広く有用であることを支持している。
【0050】
表2A欄に示す遺伝子群から選択される2種類以上が不活性化されていてもよい。2種類以上の遺伝子が不活性化されていることにより、外来性タンパク質の分泌生産がさらに促進される場合があるからである。2種類以上の遺伝子を不活性化する場合、例えば、以下の表3から選択される2種類以上とすることができる。
【0051】
【表3】
【0052】
表3において、表3A欄には、2種類以上組み合わせて不活性化するのに好ましい遺伝子を記載している。表3A欄には、表3B欄及び表3C欄の遺伝子とRRD1を含んでいる。図38に示すように、表3B欄及びC欄に示す遺伝子は、いずれもタンパク質の輸送経路に関するタンパク質をコードする遺伝子である。表3B欄に示す遺伝子は、粗面小胞体、ゴルジ体に局在するタンパク質をコードする遺伝子(ARV1, CHO2, CSG2, ERG6, FAR8, OPI3, PER1, SAC1)であり、表3C欄に示す、エンドソーム等に局在し、主としてゴルジ体からエンドソーム、液胞への輸送に関わるタンパク質をコードする遺伝子(ERG3, MON2, RAV1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45, YPT7)である(Saccharomycees cerevisiae genome database (http://www.yeastgenome.org/による)。これらタンパク質の輸送経路に関わる遺伝子の破壊、より好ましくは二重破壊は、高生産効果につき相乗効果を有している。図38に示すように、タンパク質が細胞外へ分泌されるときは、粗面小胞体、ゴルジ体を通る。また、正しくフォールディングされなかったタンパク質などの異常なたんぱく質は、ゴルジ体からエンドソームへタンパク質の輸送を経て液胞にて分解される。本発明者らによれば、RAV1などエンドソームに局在するタンパク質をコードする遺伝子と、FAR8等の粗面小胞体に局在するタンパク質をコードする遺伝子又はVPS3等の液胞たんぱく質の輸送に関わるタンパク質をコードする遺伝子の2重破壊により、外来性タンパク質の分泌生産活性が向上したことがわかっている。
【0053】
表3A欄は、表3B欄記載の遺伝子と表3C欄記載の遺伝子に加えて、RRD1を含んでいる。本発明において組み合わされて不活性化される遺伝子は、表3A欄に記載の遺伝子から選択される2種類以上を含むことができる。RRD1は、核、細胞質に局在するタンパク質をコードするが、タンパク質輸送に関するタンパク質をコードする遺伝子とともに不活性化されることにより、相乗的なタンパク質高生産効果をもたらすことができる。好ましくは、表3B欄及び表3C欄に記載の遺伝子群から選択される2種類以上である。2種類以上の不活性化遺伝子は、表3B欄の遺伝子群及び表3C欄の遺伝子群の双方を含んでいてもよいし、いずれか一方であってもよい。
【0054】
不活性化される遺伝子は表3B欄中からはFAR8が選択されることが好ましい。また、不活性化される遺伝子は表3C欄中からはRAV1及びVPS3から選択される1種又2種であることが好ましい。特に、RAV1が選択されることが好ましい。また、不活性化される遺伝子は、FAR8、RAV1、VPS3及びRRD1からなる群から選択される2種類以上を含んでいてもよいし、FAR8、RAV1及びVPS3から選択される2種類以上を含んでいてもよい。こうした不活性化遺伝子の組み合わせとしては、例えば、FAR8とRAV1、RAV1とVPS3、RAV1とRRD1等が挙げられる。
【0055】
なお、Rav1タンパク質は、エンドソームに局在し、Rav1タンパク質、Rav2タンパク質、Skp1タンパク質で複合体を形成することが知られており、初期エンドソームの液胞への輸送にとって必要であることが分かっている(Soi3p/Rav1p functions at the early endosome to regulate endocytic trafficking to the vacuole and localization of trans-Golgi network transmembrane proteins., Mol Biol Cell. 2004 Jul;15(7):3196-209、Seol J. H., Shevchenko A., Shevchenko A., Deshaies R. J., Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat Cell Biol. 2001,Apr;3(4):384-91)。VPS3遺伝子は、液胞タンパク質のソーティングやプロセシングに必要なタンパク質で、H+−液胞ATPaseタンパク質複合体の集合に必要である(Rothman J. H., Yamashiro C. T., Raymond C. K., Kane P. M., Stevens T. H.., Acidification of the lysosome-like vacuole and the vacuolar H+-ATPase are deficient in two yeast mutants that fail to sort vacuolar proteins., J Cell Biol. 1989 109(1):93-100)。Far8タンパク質は、粗面小胞体に局在し、細胞周期のG1期の停止に関わることが分かっている。RRD1タンパク質は、核、細胞質に局在し、タンパク質脱リン酸化酵素PP2Aの活性化するタンパク質であることがわかっている(Kemp H. A., Sprague G. F. Jr., Far3 and five interacting proteins prevent premature recovery from pheromone arrest in the budding yeast Saccharomyces cerevisiae., Mol Cell Biol. 2003 Mar;23(5):1750-63))。
【0056】
表1、表2及び表3に示す酵母の各遺伝子の塩基配列等については、特にS. cerevisiaeなどのサッカロマイセス属に関しては、サッカロマイセスゲノムデータベース(http://www.yeastgenome.org/)から取得することができる。また、サッカロマイセス属を含め、これらの遺伝子については、米国国立衛生研究所のホームページ(http://www.ncbi.nlm.nih.gov/)のブラストサーチサイト(http://www.ncbi.nlm.nih.gov/blast/Blast.cgi)等において取得できる。
【0057】
本発明の形質転換体においては、表1に示す遺伝子又は表2に示す遺伝子から選択される1種又は2種以上、又は表3に示す遺伝子から選択されるは2種類以上が不活性化されている。ここで、遺伝子が不活性化されているとは、少なくとも、宿主酵母内で、(1)当該遺伝子の転写又は翻訳が完全に抑制又は部分的に抑制されている状態、(2)当該遺伝子に変異が導入されており、本来の機能を全く発揮できない程度に不完全なタンパク質を発現する状態が含まれる。こうした不活性化状態は、典型的には、当該遺伝子のプロモーターなどの調節領域又はコード領域に変異が導入されることにより実現される。具体的には、相同組換えによりこうした不活性化体を得ることができる。表1〜表3において示す各酵母遺伝子が破壊された株は、Open Biosystems社(米国)より入手することができる。また、2種類以上の酵母遺伝子が破壊された株は、これらの株に対してさらに遺伝子破壊を施すことなどにより得ることができる。
【0058】
本発明の形質転換体は、液体培養における外来性タンパク質の分泌生産が、表1に示す遺伝子から選択される1種又は2種以上の遺伝子が不活性化されていないときよりも促進されているという特徴を有することができる。すなわち、宿主が野生型(表1に示す遺伝子が不活性化されていない)である以外は、同様に外来性の分泌性タンパク質をコードするDNAを導入して得られた形質転換体よりも外来性の分泌性タンパク質の分泌生産が促進されている。
【0059】
液体培養の条件は特に限定しないで、例えば、実施しようとしている液体培養プロセスを考慮して適宜設定できる。したがって、富栄養培地であっても貧栄養培地であってもよいし、使用する酵母に合わせてpHも適宜設定することができるほか、培地にpH緩衝能を付与してもしなくてもよい。通常、酵母の一般的な培地であるYPD培地やSD培地及びこれらに対して適宜改変した培地を用いることができる。
【0060】
外来性の分泌性タンパク質の分泌生産が促進されているかどうかは、当該外来性の分泌性タンパク質の分泌生産量又は当該分泌性タンパク質の活性により評価することができる。好ましくは、本発明の形質転換体と対比すべき野生型形質転換体とを一定期間液体培養して得られた培養上清につき、当該タンパク質量等を評価する。
【0061】
タンパク質量及びタンパク質の活性測定方法は、タンパク質の種類に応じて適宜選択することができる。当該タンパク質が酵素等であれば、公知の手法で酵素活性を測定してもよい。例えば、外来性分泌性タンパク質がエンドグルカナーゼなどのセルラーゼである場合には、採取した培養上清につき、適当なセルラーゼ基質(カルボキシメチルセルロース、リン酸セルロース、結晶性セルロース等)と反応させて反応生成物量や基質量等を測定することで酵素活性を評価できる。反応温度、pH及び時間は、酵素の種類等において適宜設定することができる。なお、酵素反応の結果生じる還元糖量の定量法としてはSomogyi法、Tauber-Kleiner法、Hanes法(滴定法)、Park-Johnson法、3,5-ジニトロサリチル酸(DNS)法、TZ法(Journal of Biochemical Methods, 11(1985)109-115)等の公知の方法を適宜採用すればよい。
【0062】
また、エンドグルカナーゼなどのセルラーゼ活性は、カルボキシルメチルセルロースなどのセルロースを含有する固相体からなる評価領域に本発明としての可能性のある被験タンパク質を供給し、当該領域の固相体中のセルロースを分解させて、固相体中でセルロースが分解されて消失した領域(ハロー:固相体においてバイオマスの分解により淡色化又は無色化する領域)の大きさでエンドグルカナーゼ活性を評価することもできる。固相体におけるセルロース消失に基づくハローは、目視等により視認することができるほか、ハロー検出時にコンゴーレッドなどで、セルロースを染色することで明瞭にハローを検出することができる。また、バイオマスとして色素結合セルロース(例えば、Cellulose Azure、Sigma社製など)を用いた場合、セルロースの分解により色素が固相体中に拡散しハローを形成するため、容易にセルロース分解活性を検出することができる。同様に蛍光色素を結合したセルロースもバイオマスとして利用することでハローを容易に検出することができる。さらに、酸処理セルロースなどをバイオマスとして用いた場合にもセルロースの分解により透明なハローを形成するため、セルロース分解活性を容易に検出することができる。
【0063】
ハロー形成用の固相体は、例えば、バイオマスを担持するゲルやフィルムが挙げられる。ゲルやフィルムの構成材料は特に限定しないで、天然又は人工の高分子材料を好ましく用いることができる。このような高分子材料としては、アガロース(寒天)を好ましく用いることができる。
【0064】
なお、本発明の形質転換体は、表1、表2又は表3に示す遺伝子の1種類以上又は2種類以上が不活性化され、かつセルラーゼなどの外来タンパク質をコードするDNAの一つが少なくとも染色体上に導入されているとともに、さらに他の外来性タンパク質、例えば、後述する有用物質を生産するような改変がなされていてもよい。
【0065】
(酵母形質転換体の作製)
以上説明した本発明の形質転換体は、モレキュラークローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に記載されている方法に準じて作製することができる。表1、表2又は表3に示す遺伝子の1種又は2種以上が予め不活性化された宿主酵母に対して、外来性の分泌性タンパク質をコードするDNAを有する発現ベクター等を導入し、宿主酵母を形質転換することにより得ることもできる。
【0066】
宿主酵母としては、Open Biosystems社(米国)より入手することができるほか、破壊しようとする遺伝子のプロモーターなどの調節領域又はコード領域に変異を導入することで取得できる。特定遺伝子の不活性化は、例えば、相同組換え法を利用して、特定の遺伝子に変異を導入したり、別の遺伝子を導入したりすること等により可能である。2種以上の特定遺伝子が不活性化された宿主酵母も、公知の各種方法による遺伝子導入や入手可能な破壊株を利用することによって取得することができる。
【0067】
本発明の好ましい宿主酵母は、表1〜表3に示す遺伝子の1種又は2種以上が不活性化されている酵母である。本発明の宿主酵母は、好適な不活性化遺伝子として既に説明した遺伝子又はその組み合わせが不活性化されていることが好ましい。
【0068】
本発明の酵母において外来性タンパク質を発現させるためのベクターの一つの形態は、表1〜表3に記載の遺伝子から選択される1種又は2種以上を不活性化するためのベクターが挙げられる。このベクターは、不活性化する遺伝子又はその近傍と相同領域を有しており、この相同領域を介して当該ベクターが保持するDNA領域で当該遺伝子又はその近傍を置換することで当該遺伝子を不活性化するものである。このような相同組換えのためのベクター及びその作製は、当業者において周知であって、モレキュラークローニング第3版、カレント・プロトコールズ・イン・モレキュラー・バイオロジー等に開示されている。
【0069】
また、酵母において外来性の分泌性タンパク質を発現させるためのベクターの他の一つの形態は、所望の外来性分泌性タンパク質のコード化領域を有している。発現させようとするタンパク質は、酵母において細胞外への分泌性を備えていることが好ましい。こうした分泌性は意図するタンパク質が本来的に有する場合のほか、分泌シグナル等を用いて人工的に付与することができる。細胞外に分泌発現する形態としては、特に限定されない。分泌発現の形態としては、例えば、細胞表層に拘束することなく分泌する形態とタンパク質を細胞表層に提示する形態とが挙げられる。例えば、前者の形態は、酵母において機能するシグナルペプチドを付与するなどすればよく、例えば、酵母におけるタンパク質の分泌のためには、天然シグナル配列は、例えば酵母インベルターゼリーダー、α因子リーダー、またはRhizopus oryzae やC. albicansグルコアミラーゼリーダーなどが挙げられる。また、後者の形態は公知の酵母の細胞表層提示システムを利用することができる。このような提示システムとしては、凝集性タンパク質又はその一部を用いることでタンパク質を酵母の表層に提示した状態に分泌させることができる。例えば、分泌シグナルに加えて凝集性タンパク質であるα−アグルチニンC末端側の320アミノ酸残基からなるペプチドが利用される。所望のタンパク質を細胞表層に提示するためのポリペプチドや手法は、WO01/79483号公報や、特開2003−235579号公報、WO2002/042483号パンフレット、WO2003/016525号パンフレット、特開2006−136223号公報、藤田らの文献(藤田ら,2004. Appl Environ Microbiol 70:1207-1212および藤田ら, 2002. Appl Environ Microbiol 68:5136-5141.)、村井ら, 1998. Appl Environ Microbiol 64:4857-4861.に開示されている。例えば、シグナル配列は、ベクターに組み込まれる要素であってもよいが、セルラーゼ遺伝子の一部であってもよい。
【0070】
本発明のベクターは、上記したタンパク質のコード化DNAとともに調節領域(プロモーター及びターミネーター等)を備えるほか、適宜選択マーカー遺伝子の発現カセットを備えることができる。ベクターは、形質転換の手法や宿主細胞における当該コード化DNAの保持形態(染色体に導入する形態や染色体外に保持する形態等)に応じて、上記コード領域以外の構成要素(相同組換えにより染色体への導入を意図する場合には、酵母染色体DNAとの相同性領域が必要となる。)が適宜決定される。また、ベクターの形態は、使用形態に応じて様々な形態を採ることができる。例えば、DNA断片の形態を採ることができるほか、2マイクロプラスミドなどの適当な酵母用ベクターの形態を採ることもできる。
【0071】
このようなベクターで適当な酵母宿主を適宜形質転換することによって本発明の酵母形質転換体を得ることができる。形質転換にあたり、従来公知の各種方法、例えば、トランスフォーメーション法や、トランスフェクション法、接合法、プロトプラスト法、エレクトロポレーション法、リポフェクション法、酢酸リチウム法等を用いることができる。
【0072】
(セルロース含有材料の分解産物の製造方法)
本発明のセルロース含有材料の分解産物の製造方法は、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、を備えることができる。本発明の製造方法によると、セルロース含有材料を糖化するためのセルラーゼの添加をすることなく又はその添加量を抑制してセルロース含有材料を酵母により直接利用できる。このため、効率的にセルロース含有材料を利用できる。
【0073】
(形質転換体の準備)
酵母形質転換体が分泌生産するセルラーゼとしては、既に説明した各種セルラーゼのうち1種又は2種以上組み合わせて用いることができる。好ましくは2種類以上のセルラーゼ(例えば、エンドグルカナーゼとセロビオヒドロラーゼ等)とすることができる。なお、2種類以上のセルラーゼは、1種の形質転換体が分泌生産するものであってもよいが、それぞれのセルラーゼを分泌生産する2種類以上の形質転換体であってもよい。そのほか、本発明の製造方法で用いる形質転換体については、既に説明した本発明の形質転換体についての各種態様を適用することができる。
【0074】
(液体培養)
上記のような酵母形質転換体を液体培地で液体培養するには、セルロース含有材料を炭素源の少なくとも1種類として用いる以外は、公知の液体培地を利用しあるいは適宜改変して用いることができる。培地組成、温度、pH、通気条件等は必要に応じて適宜設定することができる。
【0075】
セルロース含有材料は、グルコースがβ-1,4-グルコシド結合により重合した重合体及びその誘導体を含んでいる。グルコースの重合度は特に限定しない。また、誘導体としては、カルボキシメチル化、アルデヒド化、若しくはエステル化などの誘導体が挙げられる。セルロースは、結晶性セルロースであってもよいし、非結晶性セルロースであってもよい。また、セルロースは、その部分分解物である、セロオリゴ糖、セロビオースであってもよい。セルロース含有材料は、配糖体であるβグルコシド、リグニン及び/又はヘミセルロースとの複合体であるリグノセルロース、さらにペクチンなどとの複合体であってもよい。セルロース含有材料はその由来も特に限定しない。植物由来のものでも、真菌由来のものでも、細菌由来のものであってもよい。セルロース含有材料は、綿や麻などの天然繊維品、レーヨン、キュプラ、アセテート、リヨセルなどの再生繊維品、稲ワラ、籾殻、木材チップなどの農産廃棄物などが挙げられる。
【0076】
本発明方法で得られるセルロースの分解産物は、グルコースのほか、セロビオース及びセロオリゴ糖が挙げられる。こうして得られるセルロースの分解産物は、通常のグルコースと同様、有用物質の発酵原料等に利用できる。なお、本発明の製造方法で用いる形質転換体としては、酵母本来のアルコール発酵性を有している場合には、後述するように、培養工程を実施することでアルコール(エタノール)などの有用物質を製造することができる。また、使用する形質転換体が遺伝子工学的に乳酸などの有機酸を製造するよう改変されている場合には、当該有機酸を製造することができる。
【0077】
(セルロース含有材料から有用物質を製造する方法)
本発明の有用物質の製造方法は、セルラーゼを外来性タンパク質として分泌生産する本発明の酵母形質転換体を準備する工程と、セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、を備えることができる。本発明の製造方法によると、セルロース含有材料を糖化するためのセルラーゼを添加をすることなく又はその添加量を抑制してセルロース含有材料を酵母により直接利用して有用物質を製造できる。このため、従来に比してセルラーゼコストを低減して有用物質を製造することができる。
【0078】
有用物質としては特に限定しないが、グルコースを利用して酵母が生産可能なものであればよい。例えば、酵母におけるグルコースからの代謝系の1種又は2種以上の酵素を遺伝子組換えにより置換、追加等して本来の代謝物でない化合物であってもよい。具体的には、エタノールなどの低級アルコール、乳酸などの有機酸の他、イソプレノド合成経路の追加によるファインケミカル(コエンザイムQ10、ビタミン及びその原料等)、解糖系の改変によるグリセリン、プラスチック・化成品原料など、バイオリファイナリー技術が対象とする材料が挙げられる。酵母は、アルコール発酵能が高いため、本形質転換酵母は、セルロース含有材料からエタノールを効率的に生産することができる。
【0079】
形質転換酵母の発酵には、セルロース含有材料を含む炭素源を用いる。培養開始時には、グルコースやスクロースなど酵母が利用しやすい形態の炭素源を利用することもできる。また、培地中にセルラーゼを添加してもよい。添加するセルラーゼは、特に限定しないが、本発明の形質転換酵母を培養して生産したものであってもよい。
【0080】
そのほか、形質転換酵母の発酵には、酵母に一般的に適用される培養条件を適宜選択して用いることができる。典型的には、発酵のための培養は、静置培養、振とう培養または通気攪拌培養等を用いることができる。通気条件は、嫌気条件下、微好気条件下及び好気条件等、適宜選択することができる。培養温度も、特に限定しないが、25℃〜55℃等の範囲とすることができる。また、培養時間も必要に応じて設定されるが、数時間〜150時間程度とすることができる。また、pHの調整は、無機あるいは有機酸、アルカリ溶液等を用いて行うことができる。培養中は、必要に応じてアンピシリン、テトラサイクリンなどの抗生物質を培地に添加することができる。
【0081】
発酵終了後、培養液から有用物質含有画分を回収する工程、さらにこれを精製又は濃縮する工程を実施することもできる。回収工程や精製等の工程は有用物質の種類等に応じて適宜選択される。
【0082】
本発明のエタノール製造方法は、本発明のセルロース含有材料の分解産物の製造方法において、酵母形質転換体としてアルコール発酵性を有しているものであればよい。
【0083】
(タンパク質の製造方法)
本発明のタンパク質の製造方法は、本発明の形質転換体を準備する工程と、前記形質転換体を液体培地で培養する工程と、を備えることができる。本発明の酵母形質転換体によれば、所望の外来性タンパク質を効率的に分泌生産させることができるため、タンパク質の製造にも有利である。培養工程において用いる液体培地の組成、温度、pH等の条件は、適宜設定することができる。液体培地は、前記酵母形質転換体が利用可能な炭素源を有していればよい。したがって、前記酵母形質転換体に外来タンパク質としてセルラーゼを分泌生産させる場合には、液体培地は、セルロース含有材料を炭素源として含んでいてもよい。培養工程で得られる培養上清には、本発明の形質転換体によって分泌生産されたタンパク質が含まれている。当該タンパク質が酵素等の場合には、これを精製してもよいが、培養上清をそのまま酵素液として用いてもよい。
【0084】
以下、本発明を、実施例を挙げて具体的に説明するが、本発明はこれらの実施例に限定されるものではない。なお、以下に述べる遺伝子組換え操作はMolecular Cloning: A Laboratory Manual (T. Maniatis, et al., Cold Spring Harbor Laboratory) に従い行った。
【実施例1】
【0085】
セルラーゼ発現プラスミドの構築
TDH3プロモーター、CYC1ターミネーター、マーカーURA3を持つ酵母発現用2ミクロンベクターのSacII-SpeI間にRhizopus Oryzae由来グルコアミラーゼのシグナルペプチドを導入した。本シグナルペプチドの下流SphI-SpeI間にClostridium thermocellum(以下C.thermocellum)のエンドグルカナーゼCel8A(以下Ctcel8A)、またはClostridium cellulovorans (以下C.cellulovorans)のエンドグルカナーゼcel5A(以下Cccel5A)の導入を行った(図1)。作製したプラスミドDNAはCtcel8AはpRSA436GAPcelA, Cccel5AはpRS436GAPCccel5Aと名付けた。
【実施例2】
【0086】
酵母菌でのエンドグルカナーゼ活性の確認
Saccharomyces cerevisiaeのS288C株由来の2倍体酵母菌BY4743(遺伝型:MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/LYS2 MET15/met15Δ0 ura3Δ0/ura3Δ0)へpRS436GAPcelAを酢酸リチウム法によって形質転換した。YPDプレートに酵母をストリークし、30℃で1日培養後、形質転換液(35μlの60%ポリエチレングリコール平均分子量3350+2.5μlの4M酢酸リチウム+5μlの1M ジチオスレイトール+5μlの10mg/ml 鮭精子DNAにpRS436GAPcelAを1μlを混ぜ、全量50μlになるように滅菌水を加える)に酵母を培養したYPDプレートからかきとり混ぜた。形質転換液を42℃で2時間熱を加え、SD-uracil(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(-uracil))培地にまいた。アミノ酸ミックス(-uracil)は、アデニン0.5g、トリプトファン2g、ヒスチジン塩酸塩2g、メチオニン2g、ロイシン4g、リシン塩酸塩2gを1リットル当たり0.6g加えた。培地のpHを制御した場合にエンドグルカナーゼ活性に変化が起こるかどうかを調べた。YPD培地(1% yeast extract, 2% peptone+2% glucose)、SD-uracil培地、SD-uracilに0.1 M KPO4を加えpH緩衝した培地で培養した培養上清を、エンドグルカナーゼ活性を検出することができる0.1% カルボキシメチルセルロース入りの2% 寒天培地 (以下、CMC培地)にスポットし、40℃で1日反応させた。0.1% CongoRed /1 M Tris (pH9)(以下、コンゴーレッド液)でプレートを染色し、1 M NaClで脱色後、エンドグルカナーゼ活性がある染色されないハロ(円)を確認した(図2)。培養pHの低下によってcelAのエンドグルカナーゼ活性は大きく低下した。以上のことから、以後のエンドグルカナーゼ活性の評価にあたっては、pH低下を抑制した状態を確保することとした。
【実施例3】
【0087】
酵母遺伝子破壊株ライブラリーへの形質転換
「実施例1」で構築したプラスミドpRS436GAPcelAを2倍体酵母遺伝子破壊株ライブラリーへ形質転換した。方法は、YPDプレートに保存した破壊株ライブラリーをYPD 25μlを入れた96ウェルマイクロプレートに植菌し、30℃で1日静地培養後、培養液を混ぜ、文献(kitagawa et al.,Biosci Biotechnol Biochem. 2007 Mar;71(3):772-82. Epub 2007 Mar 7.Screening of drugs that suppress Ste11 MAPKKK activation in yeast identified a c-Abl tyrosine kinase inhibitor.)に記載の方法に鮭精子DNAを加えた形質転換液(3500μlの60% ポリエチレングリコール平均分子量3350+250μlの1 M ジチオスレイトール+ 250μlの4 M 酢酸ナトリウム+250μlの10 mg/ml鮭精子DNA+250μlのpRS436GAPcelAのDNA溶液)と混ぜた後、42℃で2時間熱を加え、SD-uracil培地へスポットした(図3)。
【実施例4】
【0088】
Ctcel8A高生産酵母変異株のスクリーニング
Ctcel8A高生産する酵母変異株をスクリーニングするために、Ctcel8Aの形質転換したプレートから各株を採取し、SD-uracilが50μl入った96ウェルマイクロプレートで30℃で1日振とう培養した。培養上清5μlを8レーンピペットでCMC培地にスポットし、40℃で1日反応させ、コンゴーレッド液で染色し、1 M NaClで脱色した。エンドグルカナーゼ活性のあるハロが他の遺伝子破壊株よりも大きい株を226株取得した(図4)。
【実施例5】
【0089】
Ctcel8A高生産酵母変異株の再確認
スクリーニングで得られたCtcel8A高活性株226株の再現性を得るために、取得した高活性株をひと株ずつ試験管でSD-uracil培地で振とう培養し、実施例2と同様の操作で野生型のBY4743よりも高活性な株として147株の再現性を得た。Ctcel8A高生産株147株の栄養要求性を確認するためにSD-U-M-K (0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(-U-M-K))培地へストリークし、増殖できない株を高生産株から除き、117株になった。SD-U-M-K培地のアミノ酸ミックス(-U-M-K)は、アデニン0.5g、トリプトファン2g、ヒスチジン塩酸塩2g、ロイシン4gを1リットル当たり0.4gをSD培地に混ぜた。使用した破壊株ライブラリーは2倍体なので、取得した高活性株に1倍体が混ざっていると1倍体とmatingするはずである。一倍体を除くために、N435-1A (遺伝型:MATa his7 lys7 met6 arg1 gal4 MAL2 SUC)、N435-2A (遺伝型:MATα his7 lys7 met6 arg1 gal4 MAL2 SUC)と高生産株をYPDプレート上で混ざるようにストリークし、30℃で1日培養した。培養したプレートを最小培地(0.67% YNB w/o a. a. +2% グルコース)へレプリカした。7株が最小培地で増殖したので除き、110株となった。もう一度野生型(BY4743株)と活性の差がほとんどない株を除くために、再度試験管でSD-uracil培地で培養し、実施例2と同様の方法で再実験を行い96株の再現性が確認できた。96株をCtcel8A高活性株とし、活性の強さを+++、++、+の3段階に分類した(図5〜図7)。
【0090】
これら96株については、外来性タンパク質を分泌生産するのに好ましい変異を有しているといえる。すなわち、これらの株において破壊されている遺伝子は、外来性タンパク質を分泌生産させるのに不活性化することが好ましい遺伝子であることがわかった。
【実施例6】
【0091】
Ctcel8A高活性株の富栄養培地での活性
スクリーニングで得られた96株のCtcel8A高活性株を富栄養培地であるYPD培地で培養し、100倍希釈した培養上清の活性を実施例2と同様の方法(CMC培地にスポットし、40℃で1日反応させた後、0.1% CongoRed /1 M Tris (pH9)(以下、コンゴーレッド液)でプレートを染色し、1 M NaClで脱色した。)で調べたところ、25株が野生型のBY4743よりも高活性であった。これらの遺伝子破壊株に対してCtcel8Aを再形質転換し、増殖の悪かった2株は除き、21株が野生型のBY4743よりも高活性であった(図8)。2株は再現性が得られなかった。以上のことから、この実施例で取得できた株は、培地の組成に関わらず、高活性を示すことがわかった。
【実施例7】
【0092】
Cccel5Aでの富栄養培地での活性
実施例6で再形質転換して得られた23株に対して、Clostridium cellulolyticum,のエンドグルカナーゼCccel5Aを発現する実施例1記載のプラスミドpRS436GAPCccel5A(図1)を形質転換した。YPD培地で培養し、50倍希釈した培養上清の活性を実施例2と同様の方法で調べたところ、14株が野生型のBY4743よりも高活性であった(図9)。残り9株は、BY4743と活性は変わらなかった 。以上のことから、本実施例で高活性の確認された変異株は、GHF5及び8の双方を分泌生産するのに有用であること、すなわち、これらの変異株において破壊されている遺伝子の不活性化が外来性タンパク質の分泌生産に有効であることがわかった。
【実施例8】
【0093】
Ctcel8A及びCccel5Aの染色代導入用ベクターの作製
セルラーゼを染色体に導入するためのベクターpAH-HORp-Ctcel8A, pAH-HOR7p-Cccel5Aを作製した(図10)。AAP1遺伝子の上流-3000bpから-2000bpまでの配列(以下AAP1Uと呼ぶ)をS288CのゲノムDNAからPCR反応により、クローニングした。AAP1遺伝子の上流-2000bpから-1000bpまでの配列(以下AAP1D)をS288CのゲノムDNAからPCR反応により、クローニングした。遺伝子導入の確認用マーカーとしてハイグロマイシン耐性遺伝子を連結した。C.thermocellum (ATCC27405), C.cellulolyticum (ATCC35319)の培養菌体からゲノムを抽出後、分泌シグナル配列を除くセルラーゼ遺伝子C.thermocellum由来Cel8A (ABN51508)約1.3kb及びC.cellulolyticum由来Cel5A(AAA51444)約1.3kbをPCR法により取得した。取得した遺伝子断片をCel8Aは制限酵素KpnI-BglII、Cel5AはKpnI-BamHI部位にて染色体導入用ベクター内Rhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【実施例9】
【0094】
vps3遺伝子破壊株でのCtcel8A及びCccel5A染色体導入株の効果
実施例8で作製したプラスミドpAH-HOR7p-Ctcel8A、pAH-HOR7p-Cccel5Aを形質転換した。形質転換方法はBY4743及び、vps3破壊株をYPD培地で30℃、1日培養し、YPD培地で再度5時間30℃で培養した。培養液は遠心して回収し、再度滅菌水で洗浄後、遠心して回収した。60%ポリエチレングリコール平均分子量3350を120μl、4M 酢酸リチウムを5μl、1M ジチオスレイトールを10μl、制限酵素Sse8387IでプラスミドDNAの染色体導入部を線上DNAにした後、DNA溶液を5μl加え、細胞懸濁液を50μl加え、懸濁後、42℃で1時間反応させた。反応液を遠心後、YPD培地を加え、30℃で一晩回復培養を行った。培養液はYPDに最終濃度が150μg/mlのハイグロマイシンBが入ったプレートにまき、30℃で増殖させた。 ハイグロマイシン耐性になったコロニーを再びシングルコロニー化した。コロニーをYPDプレートで再度増殖させた。YPD培地で30℃1日培養後、培養液を遠心し、培養上清をx1, x2, x4, x8, x16, x32倍希釈した。上清8μlをCMC培地へスポットし、40℃で1日反応させた。コンゴーレッド液で染色し1M NaClで脱色し、活性を確認した(図11)。BY4743と比べて、vps3破壊株はCtcel8Aでは8倍以上、Cccel5Aでは32倍以上の活性上昇が認められた。
【0095】
上記のCtcel8A及びCccel5A染色体導入株をSD+complement培地(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(complement)、 pHをNaOHで6に調整した)培地とSD+complemnent+0.1M NaAc培地(0.67% YNB w/o a. a. +2% グルコース+アミノ酸ミックス(complement)+0.1M 酢酸ナトリウムを加えpHをHClで6に調整)培地で培養した。アミノ酸ミックス(complement)は、アデニン0.5g、ウラシル2g、トリプトファン2g、ヒスチジン塩酸塩2g、メチオニン2g、ロイシン4g、リシン塩酸塩2gを混ぜ、1リットル当たり0.6gを加えた。培養後、培養上清をCMC培地へ8μlスポットした。プレートを40℃で1日反応後、コンゴーレッド液で染色し1M NaClで脱色した(図12)。酢酸ナトリウムで培地のpHを下げないように緩衝した場合(SD+complement+0.1M NaAc培地)では、野生型(BY4743株)よりもvps3破壊株のほうが活性が約2倍程度上昇した。一方pHを緩衝しない培地(SD+complement)では、培地pHの低下によりBY4743は活性が確認できなかったが、vps3破壊株では活性が確認できた。よって、YPD培地と同様に、SD培地のような貧栄養培地でもvps3破壊株はセルラーゼを高生産できることが確認できた。
【0096】
以上のことから、vps3遺伝子を不活性化した酵母においては、外来性タンパク質のコード化DNAが染色体導入形態であっても外来性タンパク質の高度な分泌生産活性が得られることがわかった。すなわち、安定して外来タンパク質を高活性で分泌生産できることがわかった。また、貧栄養及び低pHに対する耐性も野生型よりも向上していることがわかった。
【実施例10】
【0097】
遺伝子破壊株へのcel8A遺伝子の染色体導入
−80℃で保存された1倍体の遺伝子破壊株ライブラリー(インビトロジェン株式会社)から表1に記載の遺伝子に相当する1倍体の株を96株選び、遺伝子の破壊をコロニーPCRにより確認した。それぞれの株を、20mMのNaOH5μlに少量加え、100℃で10分ボイルした。ボイルしたサンプルに15μlの超純水を加えた。使用したプライマーは表3に示すとおりであった。Reverse側のプライマーはKANMX+600R(ATCAAGTGAGAAATCACCATGAGTGACGAC)(配列番号1)を共通に使用し、Forward側のプライマーは、それぞれKANMX+600R以外の遺伝子固有のプライマーを使用した。これらのPCR反応は、Extaq DNAポリメラーゼ(タカラバイオ株式会社)を用いた。なお、PCRは以下のようにして行った。すなわち、PCR反応液6.5μlに1.5μlの100℃で10分ボイルして破砕した細胞懸濁液を加えた。反応条件は、94℃、2分ボイルし、94℃、15秒、50℃、30秒、72℃、2分を40サイクル繰り返した。この結果、96株中、13株(vma4、kap120、mtq2、ydr433W、ygr102C、yvh1、adh1、ado1、pat1、mdm34、rpl1B、vps33、ynl296W破壊株)は遺伝子破壊が確認できなかった。
【0098】
【表4A】
【表4B】
【0099】
遺伝子破壊が確認できなかった13株を除いた83株の遺伝子破壊株及び基準株(BY4741株、遺伝型:MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0)に対してCtcel8Aの染色体導入を行った。染色体導入用ベクターとして図13に示すpAL-HOR7p-Ctcel8Aを構築した。このベクターは、以下のようにして作製した。AAP1遺伝子の上流-3000bpから-2000bpまでの配列(以下AAP1Uと呼ぶ)をS288CのゲノムDNAからPCR反応により、クローニングした。AAP1遺伝子の上流-2000bpから-1000bpまでの配列(以下AAP1D)をS288CのゲノムDNAからPCR反応により、クローニングした。遺伝子導入の確認用マーカーとしてLEU2遺伝子を連結した。C.thermocellum (ATCC27405)の培養菌体からゲノムを抽出後、分泌シグナル配列を除くセルラーゼ遺伝子C.thermocellum由来Cel8A (ABN51508)約1.3kbをPCR法により取得した。取得した遺伝子断片をCel8Aは制限酵素KpnI-BglII部位にて染色体導入用ベクター内Rhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【0100】
それぞれの株をYPD培地で30℃、24時間培養した。培養液1mlを10mlのYPD培地に植菌し、再び5時間培養を行った。培養液を遠心し、超純水で洗浄後再び遠心して細胞をペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに1μlのSse8387Iで処理したpAL-HOR7p-Ctcel8Aと細胞懸濁液を54μl加え、42℃で1時間熱ショックを与えた。この細胞懸濁液をSD-leucine培地へ接種し、30℃で数日培養した。生えたコロニーをYPD+0.1%CMC培地に塗抹し、30℃、1日培養し、CMC分解活性があるかどうかを確認した。
【0101】
CMC含有培地上のコロニーにおいてCMC分解活性が認められた株をそれぞれYPD液体培地に植菌し、30℃で24時間培養した。培養液を遠心し、培養上清を回収した。培養上清をx1、x2、x4、x8、x16、x32倍に希釈し、0.1%カルボキシメチルセルロース、50mMの酢酸ナトリウム入り(pH6)の2%寒天(以下CMCプレート)に8μlずつスポットし、40℃で1日反応させた。反応後、0.1%コンゴーレッド、1M Tris-Base (pH9)(以下コンゴーレッド液)と30分室温で反応させ、1MのNaClで脱色した。結果を図14及び図15に示す。
【0102】
図14に示すようにして、Ctcel8Aの染色体導入株を2クローンずつ調べたところ、基準株よりも活性の強い株と基準株と活性が変わらない株に分かれた。2クローンとも基準株よりも2倍以上高活性であった株を染色体導入でも高生産できる株とした。その結果、図15に示すように、83株中、55株が基準株と比べて、2倍から4倍高活性であることがわかった。
【実施例11】
【0103】
Ctcel8A染色体導入株のcel8A高生産の再確認
再現性をとるために、実施例10で選抜した55株を再度YPD液体培地で、30℃で24時間培養し、培養上清を8倍希釈し、CMCプレートへスポットし、40℃で1日反応させ、コンゴーレッド液で染色し、1M NaClで脱色した。結果を図16に示す。選抜された55株の遺伝子破壊株はいずれも8倍希釈したハローは基準株(BY4741株)よりも高活性であることが確認できた。実施例10及び11の結果から、55株のなかでも、dyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138cの各遺伝子破壊株がセルラーゼの分泌生産が高いことがわかった。
【実施例12】
【0104】
BGL染色体導入用ベクターの作製
エンドグルカナーゼ以外のセルラーゼにおいて高生産効果があるかどうかを調べるために、Aspergillus aculeatusのβ-グルコシダーゼBGL1(以下AaBGLと称する。)を酵母染色体に導入するベクターを作製した。
【0105】
AaBGLを染色体に導入するためのベクターpDU-HOR7p-AaBGLsecを作製した(図17)。染色体導入のための相同配列として、ADH3遺伝子の上流-3000bpから-2000bpまでの配列(以下ADH3Uと称する。)をS288Cのゲノムからクローニングした。同様に、ADH3遺伝子の上流-2000bpから-1000bpまでの配列(以下ADH3Dと呼ぶ)をS288Cのゲノムからクローニングした。遺伝子導入の確認用マーカーとしてURA3遺伝子を連結した。Aspergillus aculeatusのβ-グルコシダーゼ遺伝子(Genbank Accesion No.D64088)の分泌配列を除く配列をクローニングした。染色体導入用ベクター内のRhizopus Oryzae由来グルコアミラーゼのシグナルペプチド下流に挿入した。
【実施例13】
【0106】
BGL染色体導入株のBGL活性とセロビオース培地でのエタノール発酵
実施例12で作製したプラスミドDNA、pDU-HOR7p-AaBGLsecをSse8387Iで処理したDNA溶液を実施例10と同様の方法で基準株であるBY4741及び遺伝子破壊株の染色体へ導入した。遺伝子破壊株は、dyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138c破壊株(合計15種類)を使用した。AaBGLを染色体へ導入した株3クローンをYPD培地で30℃、120rpm, 24時間培養した。培養液の培養上清及び細胞表面のBGL活性をp-Nitrophenyl-α-glucopyranoside (pNPG)の分解によって測定した。培養上清のBGL活性の測定方法は、695μlのMQ(ミリポア社)水、200μlの250mM NaAc(pH5)、100μlの20mM pNPGに細胞を除いた培養上清を5μl加え、30℃で振とうしながら60分反応させた。反応液100μlに50μlの1N Na2CO3を加え、反応を停止させた。反応停止液をA400の吸光高度計で測定した。また、細胞表面でのBGL活性は、細胞を超純水で2回洗浄し、OD600を測定し、650μlの超純水、200μlの250mM NaAc(pH5)、100μlの20mM pNPGに、50μlのOD600を1に合わせた細胞懸濁液を加え(最終OD600=0.5)、30℃で振とうしながら、60分反応させた。培養上清と同様に反応液を測定した。これらの結果を図33に示す。
【0107】
図33に示すように、細胞表面ではsnf7破壊株以外のdyn3, gcn5, opi3, per1, rpl13b, rpl19b, rpl36a, snf7, vma7, vps3, vps4, vps16, vps27, ykl118w, yol138c破壊株は基準株(BY4741株)よりも活性が向上した。培養上清は、dyn3, rpl13b, rpl36b, snf7, vps4, vps27, ykl118w破壊株以外のgcn5, opi3, per1, rpl19b, vps3, vps16, yol138c破壊株は基準株(BY4741株)よりも活性が向上した。snf7破壊株以外は、培養上清及び細胞表面のいずれか又は双方で分解活性が高まり、分泌生産が向上したことがわかった。snf7破壊株は細胞表面、培養上清両方において活性が低下した。
【0108】
さらに、AaBGL導入株3クローンのセロビオースを炭素源としたエタノール発酵を行った。2mlのYPD培地で、30℃、120rpm、24時間培養後、再度20mlのYPD培地で30℃、80rpm、24時間培養した。OD600を測定し、OD600=10に相当する培養液を遠心し、超純水10mlで細胞を洗浄後、5%セロビオース入りのYPcellobiose培地10mlでエタノール生産を行った(図18−図32)。培養開始時並びに4,6,8,10及び12時間後の発酵液をサンプリングした。発酵時の細胞濃度(OD600)は吸光光度計で測定した。残存グルコース、残存セロビオース、エタノールはバイオセンサー(王子計測機器株式会社、BF-4)にて測定した。残存セロビオースは、培養サンプルを20倍希釈したものに0.25%になるようにNovozyme188(インビトロジェン株式会社)を加え、45℃、1時間振とうしながら反応させ、セロビオースを完全糖化させたものを測定し、残存グルコースの測定値を引いた。発酵時の増vps16, vps27, snf7は基準株よりも低下した。snf7, vps16以外のdyn3, gcn5, opi3, per1,殖速度はrpl13b, rpl19b, rpl36a, vma7, vps3, vps4, vps27, ykl118w, yol138c破壊株はエタノール生産速度が向上した。
【実施例14】
【0109】
MET17遺伝子のクローニング
形質転換用のマーカー遺伝子をクローニングするために、MET17遺伝子をクローニングした。すなわち、MET17遺伝子を、S288CのゲノムDNAを鋳型として、KOD Plus(東洋紡株式会社)酵素及び以下のプライマーを用いて、94℃で15秒、55℃で30秒、68℃で2分を40サイクル繰り返して合成した。
attB2-MET17-500F (ggggacagctttcttgtacaaagtggATTTAATGCTATAATAGACATTTAAATCCA)(配列番号98)
attB3-MET17+1632R (ggggacaactttgtataataaagttgACCTCCATCATCCTCTTTTGTAACTTGGTC)(配列番号99)
【0110】
PCR産物をGFX PCR DNA and gel band purification kit (GE)で精製した。精製したPCR産物2μlに、pDONR P2R-P3 (インビトロジェン株式会社)プラスミド1μl、TE buffer 5μl、BPクロナーゼenzyme mix(インビトロジェン株式会社)2μlを加え、25℃で1時間反応させた。その後、proteinase Kを1μl加え、37℃で10分反応後、大腸菌DH5αへ形質転換した。形質転換した大腸菌につき、Extaq (タカラバイオ株式会社)及び以下のプライマーを用いてインサートを確認した。また、確認したプラスミドDNAをシークエンスし、変異がないことを確認した。得られたプラスミドを、pE3-MET17と命名した。
M13−F (GTAAAACGACGGCCAG)(配列番号100)
M13-R (GGAAACAGCTATGACCATG)(配列番号101)
【実施例15】
【0111】
RAV1遺伝子とRRD1遺伝子またはFAR8遺伝子との2重破壊によるセルラーゼ高生産の効果
Rav1タンパク質、RRD1タンパク質、Far8タンパク質は、細胞内の局在または機能が異なっていると考えられる。こうした機能が異なる遺伝子の2重破壊でタンパク質高生産の更なる効果が認められるのかどうかを調べるために、一倍体のrrd1またはfar8破壊株に対して、RAV1遺伝子の破壊を行った。
【0112】
pE3-MET17を鋳型DNAとして、以下のプライマーを用いて、PrimeSTAR MAX DNA polymerase(タカラバイオ株式会社)でrav1::MET17破壊のDNA断片を98℃で2分ボイル後、98℃で10秒、55℃で15秒、72℃で1分を30サイクル繰り返して合成した。
RAV1-MET17F: (TTAAAGGGATCTAAGAACACCGCAAACAATTGAGAAAGTAAACAAACGTGCACACGCAAAGGTAGTTCTCCACGTGAAGCTGTCGATATTGGGGAACTGT)(配列番号102)
RAV1-MET17R:(AAAACACTACATTAATAAGATGTAATTATTTTTATTTATAAATCCATTTATTTGATAAAAAATATGATATTACGAGTCACGACATGTAACAAGGGAAAAA)(配列番号103)
【0113】
1倍体の酵母遺伝子破壊株ライブラリーから出したrrd1破壊株及びfar8破壊株をYPD培地(1% yeast extract, 2% peptone, 2% glucose)で30℃、120rpm、24時間培養した。10mlのYPD培地に1mlのこの培養液を加え、さらに30℃で5時間培養した。得られた培養液を遠心し、ペレットの細胞を超純水5mlで洗浄し、再び遠心してペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに10μlのrav1::MET17破壊のDNA断片と細胞懸濁液を45μl加え、42℃で1時間熱ショックを与えた。サンプルはSD-methionine培地へ播種し、30℃で数日培養した。
【0114】
生えたコロニーをExtaq DNAポリメラーゼ(タカラバイオ株式会社)を使い、以下のプライマーを用いて、94℃で2分ボイル後、94℃で15秒、55℃で30秒、72℃で2分を40サイクル行ってPCR反応を行い、インサートを確認した。far8とrav1の2重破壊株をTTK175、rrd1とrav1の2重破壊株をTTK176と命名した。
RAV1-500F:
(AACTTTCCTTTGCACAAGTTTTCGTACTGT)(配列番号104)
RAV1+4280R :
(AGTTATAGAACAAAGAATAGAATATGTCAA)(配列番号105)
【0115】
次に、作製した2重破壊株TTK175およびTTK176へCtcel8Aの染色体への導入を次のような操作で行った。TTK175 (rav1 x far8 2重破壊株)及びTTK176 (rav1 x rrd1 2重破壊株)をYPD培地で30℃、120rpm、24時間培養した。10mlのYPD培地に1mlの培養液を加えさらに30℃で5時間培養した。培養液を遠心し、ペレットの細胞を超純水5mlで洗浄し、再び遠心してペレット状にした。120μlの60%ポリエチレングリコール平均分子量3350、5μlの4M 酢酸ナトリウム、10μlの1M ジチオスレイトール、10μlの鮭精子DNAに1μlのSse8387I制限酵素処理したpAL-HOR7p-Ctcel8Aと細胞懸濁液を54μl加え、42℃で1時間熱ショックを与えた。サンプルはSD-leucine培地へ播種し、30℃で数日培養した。生えたコロニーをYPD+0.1%CMC培地にぬり、30℃、1日培養し、CMC分解活性があるかどうかを確認した。
【0116】
確認できた株をYPD培地で30℃、1日培養した。培養液を遠心し、培養上清と細胞に分けた。培養上清を8倍希釈し、0.1%CMC/50mMNaAc(pH6)2% agarに8μlずつスポットした。プレートを40℃で1日反応させ、コンゴレッド液で30分染色し、1MのNaClで脱色させた。結果を図34に示す。プレートレベルでは、2重破壊株のほうがわずかにCMCの分解されたハローが大きかった。
【0117】
次に、培養上清の活性の定量を次の方法で測定した。Ctcel8A遺伝子を導入していない酵母BY4741(基準株)、rav1破壊株、rrd1破壊株、far8破壊株、TTK175、TTK176に対して、Ctcel8Aを導入した株と導入していない株をYPD培地で培養した。培養液を遠心し培養上清と細胞にわけた。250μlの1% CMC、100μlの250mM NaAc buffer(pH6)、140μlの超純水に培養上清を10μl加え、40℃で60分反応させた。反応液5μlを100μlのTZ bufferへ入れ、100℃で3分反応させた。反応液を分光光度計の吸光度A660を測定した。活性の数値は、Ctcel8Aを導入した酵母の数値からCtcel8Aを導入していない酵母の培養上清の数値を引いて算出した。結果を図35に示す。
【0118】
図35に示すように、基準株と比べ、far8破壊株、rav1破壊株、rrd1破壊株の活性は、2.4、2.1、1.4倍活性が上昇した。rav1 x far8 2重破壊株(TTK175)は3.9倍活性が上昇した。また、rav1 x rrd1 2重破壊株(TTK176)は、活性が2.6倍上昇した。それぞれの2重破壊株は単独の破壊株と比べて活性が上昇することが分かった。以上のことから、細胞内の局在や機能が異なるタンパク質をコードする遺伝子を組み合わせて破壊することが外来タンパク質の高生産に寄与することがわかった。
【実施例16】
【0119】
RAV1遺伝子とVPS3との2重破壊によるセルラーゼ高生産の効果
VPS3遺伝子及びRAV1遺伝子は、似た作用部位で働くタンパク質をコードする遺伝子であるが、こうした遺伝子を2重に破壊した場合でも更なる高生産効果が得られるのかどうかを調べるために、vps3破壊株に対してrav1遺伝子の破壊を行い、2重破壊株を作製した。2重破壊株の作製方法は実施例15の方法に従って行った。rav1とvps3の2重破壊株をTTK179と名付けた。作製した2重破壊株に対してCtcel8Aを実施例15と同様の方法で導入した。染色体導入の確認は、実施例15と同様の方法で行った。Ctcel8Aを導入したBY4741(基準株)、rav1破壊株、vps3破壊株、rav1 x vps3 2重破壊株、Ctcel8Aを導入していない同様の株をそれぞれYPD培地で30℃、24時間培養した。培養液を遠心し培養上清と細胞に分けた。CMC分解による活性の定量は実施例15に記載のTZ法を用いて行った。結果を図36に示す。
【0120】
図36に示すように、rav1及びvps3破壊株はBY4741(基準株)と比べて、活性が2.5倍、3.5倍活性が上昇したが、rav1 x vps3 2重破壊株はBY4741(基準株)と比べて4.9倍活性が上昇した。以上の結果から、細胞内の近接ないし類似する作用部位で働くrav1 タンパク質及びvps3タンパク質をそれぞれコードする遺伝子を組み合わせて破壊することが、外来タンパク質の高生産に寄与することがわかった。
【実施例17】
【0121】
RPL19B遺伝子との2重破壊によるセルラーゼ高生産の効果
RPL19B遺伝子はタンパク質の翻訳に必要なリボソームの構成成分のひとつである(Song J. M., Cheung E., Rabinowitz J. C., Organization and characterization of the two yeast ribosomal protein YL19 genes., Curr Genet. 1996 Sep;30(4):273-8.
)。RPL19B遺伝子の破壊により、Ctcel8Aを高生産できたが、リボソーム構成要素と他の機能との2重破壊による高生産効果を調べるために、4つのCtcel8Aを高生産できる遺伝子(dyn3, gcn5, opi3, ykl118w)を選択した。DYN3遺伝子は、核の分配に関わる遺伝子(Lee WL, Kaiser MA, Cooper JA., The offloading model for dynein function: differential function of motor subunits., J Cell Biol. 2005 Jan 17;168(2):201-7)、GCN5遺伝子は、ヒストンアセチルトランスフェラーゼ(Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, Roth SY, Allis CD., Tetrahymena histone acetyltransferase A: a homolog to yeast Gcn5p linking histone acetylation to gene activation., Cell. 1996 Mar 22;84(6):843-51.)、OPI3遺伝子は、リン脂質メチル転移酵素であり(McGraw P, Henry SA., Mutations in the Saccharomyces cerevisiae opi3 gene: effects on phospholipid methylation, growth and cross-pathway regulation of inositol synthesis., Genetics. 1989 Jun;122(2):317-30)、YKL118W遺伝子は、機能未知で遺伝子の一部が液胞のvacuolar H+-ATPase (V-ATPase) であるVPH2遺伝子と重複している。6種の遺伝子はRPL19Bの翻訳機能とは全く異なった作用で機能しているが、これらの遺伝子との2重破壊株を以下の通りに作製した。
【0122】
rpl18b::MET17破壊のDNA断片を、実施例14に記載のpEG-MET17プラスミドDNAを鋳型として、以下のプライマー及びKOD-Plusで98℃で30秒ボイル後、98℃で10秒、60℃で2分、72℃で2分を35サイクル繰り返してPCR反応を行い合成した。
RPL19B-MET17F:(ATACTTTCAATTCTTGAGTTGTAAGGCTGCATCCCTCCGTAGAACAAACCAGAGAGCCGAGCTTAGTAATCACGTGAAGCTGTCGATATTGGGGAACTGT)(配列番号106)
RPL19B-MET17R:(AGAAATTCAGTACAAAAAATATCGTTACATCTTATTTCCCCCTATAGCAGCAAATCAATTCACTCCTGGGTACGAGTCACGACATGTAACAAGGGAAAAA)(配列番号107)
【0123】
実施例15と同様の操作で、BY4741株にPCR合成したrpl19b::MET17破壊のDNA断片を形質転換して、コロニーを得た。遺伝子破壊できたかどうかの確認のために、以下のプライマーを用いて生えたコロニー実施例15と同様の方法でPCR反応を行い、目的のDNA断片が得られることを確認した。それぞれの株をTTK309 (dyn3Δ rpl19bΔ::MET17)、TTK310 (ykl118wΔ rpl19bΔ::MET17)、TTK311 (opi3Δ rpl19bΔ::MET17)、TTK312 (gcn5Δ rpl19bΔ:MET17)と命名した。それぞれの株にCtcel8Aを染色体へ導入するために、実施例15と同様の操作でCtcel8Aを染色体へ挿入した。
MET17+1F:
(ATGCCATCTCATTTCGATACTGTTCAACTA)(配列番号108)
RPL19B+1530R:
(ATGTAGATAAGCTATCATAAACTGGTAGTG)(配列番号109)
【0124】
それぞれの株をYPD培地で24時間培養し、細胞と培養上清に分けた。細胞上清の活性を実施例15に記載のTZ法を用いてCMCの分解活性を測定した。結果を図37に示す。図37に示すように、rpl19bとの2重破壊は、rpl19b x opi3 2重破壊株は、opi3破壊株よりも活性が上昇したが、rpl19b破壊株とほとんど同程度の活性であった。rpl19b x dyn3の2重破壊株、rpl19b x ykl118wの2重破壊株はdyn3及びykl118w破壊株単独と同程度の活性であった。また、rpl19b x gcn5の二重破壊株はgcn5単独破壊株よりも活性が低下した。
【0125】
以上説明したように、遺伝子を二重破壊しても活性が変わらない株、活性が低下する株も見られた。取得した55種類の遺伝子の細胞内の局在をSaccharomycees cerevisiae genome database (http://www.yeastgenome.org/) で調べた(図38)。タンパク質が細胞外へ分泌されるときは、粗面小胞体、ゴルジ体を通る。また、正しくフォールディングされなかったタンパク質などの異常なたんぱく質は、ゴルジ体からエンドソームへタンパク質の輸送を経て液胞にて分解される。エンドソームに局在するRAV1と粗面小胞体に局在するFAR8遺伝子また、液胞たんぱく質の輸送に関わるVPS3の2重破壊はCMC分解活性が上昇した。このことから、タンパク質の輸送経路に関わる遺伝子の2重破壊がより高生産効果があることがわかった。特に粗面小胞体、ゴルジ体に局在する遺伝子群(ARV1, CHO2, CSG2, ERG6, FAR8, OPI3, PER1, SAC1)、エンドソーム等に局在し、主として、ゴルジ体からエンドソーム、液胞への輸送に関連する遺伝子群(ERG3, MON2, RAV1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45, YPT7)が最も効果があることが考えられた。
【配列表フリーテキスト】
【0126】
配列番号1〜109:プライマー
【特許請求の範囲】
【請求項1】
1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体。
【請求項2】
前記外来性タンパク質は分泌性タンパク質である、請求項1に記載の酵母形質転換体。
【請求項3】
前記外来性タンパク質を液体培養で生産するのに用いる、請求項1又は2に記載の酵母形質転換体。
【請求項4】
前記外来性タンパク質はセルラーゼから選択される1種又は2種以上である、請求項1〜3のいずれかに記載の酵母形質転換体。
【請求項5】
不活性化されている遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、 VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜4のいずれかに記載の酵母形質転換体。
【請求項6】
不活性化されている前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜5のいずれかに記載の酵母形質転換体。
【請求項7】
不活性化されている前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜6のいずれかに記載の酵母形質転換体。
【請求項8】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上を含む、請求項1〜7のいずれかにに記載の酵母形質転換体。
【請求項9】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45及びYPT7からなる群から選択される2種以上を含む、請求項8に記載の酵母形質転換体。
【請求項10】
不活性化されている前記遺伝子は、FAR8, RAV1及びVPS3からなる群から選択される1種又は2種以上を含む、請求項8又は9に記載の酵母形質転換体。
【請求項11】
不活性化されている前記遺伝子は、以下の(a)〜(c)から選択されるいずれかの組み合わせを含む、請求項8〜10のいずれかに記載の酵母形質転換体。
(a)RAV1とFAR8
(b)RAV1とRRD1
(c)RAV1とVPS3
【請求項12】
前記外来性タンパク質は、β−グルコシダーゼ、セロビオヒドロラーゼ及びエンドグルカナーゼから選択される1種又は2種以上である、請求項1〜11のいずれかに記載の酵母形質転換体。
【請求項13】
前記宿主は、サッカロマイセス属酵母である、請求項1〜12のいずれかに記載の酵母形質転換体。
【請求項14】
前記宿主は、サッカロマイセス・セレビジエである、請求項1〜13のいずれかに記載の酵母形質転換体。
【請求項15】
酵母において外来性タンパク質を発現させるためのベクターであって、
以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上を不活性化するためのベクター。
【請求項16】
外来性のタンパク質を発現させるための宿主酵母であって、
宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上が不活性化されている、宿主酵母。
【請求項17】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上である、請求項16に記載の宿主酵母。
【請求項18】
セルロース含有材料の分解産物の製造方法であって、
セルラーゼを外来性タンパク質として分泌生産する請求項1〜9のいずれかに記載の酵母形質転換体を準備する工程と、
前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、
を備える、製造方法。
【請求項19】
セルロース含有材料から有用物質を製造する方法であって、
セルラーゼを外来性タンパク質として分泌生産する請求項1〜14のいずれかに記載の酵母形質転換体を準備する工程と、
前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、
を備える、製造方法。
【請求項20】
タンパク質の製造方法であって、
請求項1〜14のいずれかに記載の酵母形質転換体を準備する工程と、
セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を用いて培養する工程と、
を備える、製造方法。
【請求項1】
1種又は2種以上の外来性タンパク質をコードするDNAを染色体上に保持し、宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上の遺伝子が不活性化されている、酵母形質転換体。
【請求項2】
前記外来性タンパク質は分泌性タンパク質である、請求項1に記載の酵母形質転換体。
【請求項3】
前記外来性タンパク質を液体培養で生産するのに用いる、請求項1又は2に記載の酵母形質転換体。
【請求項4】
前記外来性タンパク質はセルラーゼから選択される1種又は2種以上である、請求項1〜3のいずれかに記載の酵母形質転換体。
【請求項5】
不活性化されている遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、SNF7、VMA7、VPS3、 VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜4のいずれかに記載の酵母形質転換体。
【請求項6】
不活性化されている前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、RPL36A、VMA7、VPS3、VPS4、VPS16、VPS27、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜5のいずれかに記載の酵母形質転換体。
【請求項7】
不活性化されている前記遺伝子は、DYN3、GCN5、OPI3、PER1、RPL13B、RPL19B、VPS3、YKL118W及びYOL138Cからなる群から選択される1種又は2種以上を含む、請求項1〜6のいずれかに記載の酵母形質転換体。
【請求項8】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上を含む、請求項1〜7のいずれかにに記載の酵母形質転換体。
【請求項9】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45及びYPT7からなる群から選択される2種以上を含む、請求項8に記載の酵母形質転換体。
【請求項10】
不活性化されている前記遺伝子は、FAR8, RAV1及びVPS3からなる群から選択される1種又は2種以上を含む、請求項8又は9に記載の酵母形質転換体。
【請求項11】
不活性化されている前記遺伝子は、以下の(a)〜(c)から選択されるいずれかの組み合わせを含む、請求項8〜10のいずれかに記載の酵母形質転換体。
(a)RAV1とFAR8
(b)RAV1とRRD1
(c)RAV1とVPS3
【請求項12】
前記外来性タンパク質は、β−グルコシダーゼ、セロビオヒドロラーゼ及びエンドグルカナーゼから選択される1種又は2種以上である、請求項1〜11のいずれかに記載の酵母形質転換体。
【請求項13】
前記宿主は、サッカロマイセス属酵母である、請求項1〜12のいずれかに記載の酵母形質転換体。
【請求項14】
前記宿主は、サッカロマイセス・セレビジエである、請求項1〜13のいずれかに記載の酵母形質転換体。
【請求項15】
酵母において外来性タンパク質を発現させるためのベクターであって、
以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上を不活性化するためのベクター。
【請求項16】
外来性のタンパク質を発現させるための宿主酵母であって、
宿主の以下の遺伝子;ADA2、ADE5,7、ARV1、BUD23、BUR2、CHO2、CSG2、DYN3、ERG3、ERG6、FAR8、FYV6、GCN5、GDH1、GND1、HMO1、MMM1、MON2、NBP2、NPR2、OPI3、OPI9、PER1、RAV1、RMD11、RPL13B、RPL19B、RPL36A、RPS6A、RRD1、RSA1、SAC1、SFP1、SNF7、SNF8、URM1、VMA7、VPS16、VPS25、VPS27、VPS28、VPS3、VPS36、VPS4、VPS41、VPS45、VRP1、YDR065W、YKL118W、YNL120C、YNL228W、YOL138C、YOR331C、YPT7及びZUO1からなる群から選択される1種又は2種以上が不活性化されている、宿主酵母。
【請求項17】
不活性化されている前記遺伝子は、ARV1, CHO2, CSG2, ERG3, ERG6, FAR8, MON2, OPI3, PER1, RAV1, SAC1, SNF7, SNF8, VPS3, VPS4, VPS16, VPS25, VPS27, VPS28, VPS36, VPS45、YPT7及びRRD1からなる群から選択される2種以上である、請求項16に記載の宿主酵母。
【請求項18】
セルロース含有材料の分解産物の製造方法であって、
セルラーゼを外来性タンパク質として分泌生産する請求項1〜9のいずれかに記載の酵母形質転換体を準備する工程と、
前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養する工程と、
を備える、製造方法。
【請求項19】
セルロース含有材料から有用物質を製造する方法であって、
セルラーゼを外来性タンパク質として分泌生産する請求項1〜14のいずれかに記載の酵母形質転換体を準備する工程と、
前記セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を培養して有用物質を発酵生産させる工程と、
を備える、製造方法。
【請求項20】
タンパク質の製造方法であって、
請求項1〜14のいずれかに記載の酵母形質転換体を準備する工程と、
セルロース含有材料を炭素源として含有する液体培地で前記酵母形質転換体を用いて培養する工程と、
を備える、製造方法。
【図1】

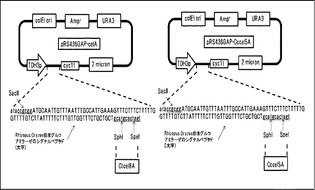
【図10】
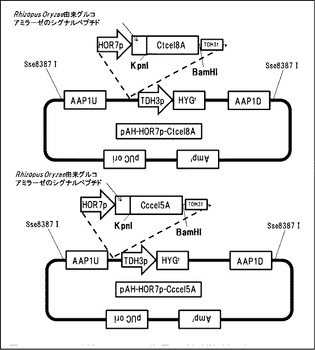
【図13】
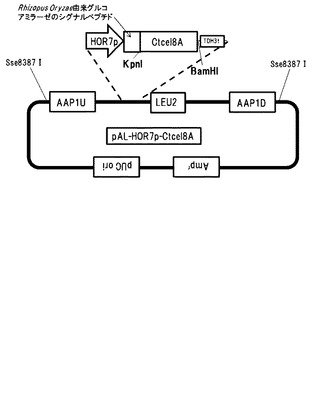
【図17】
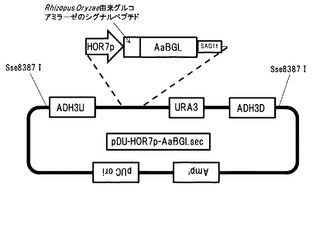
【図2】
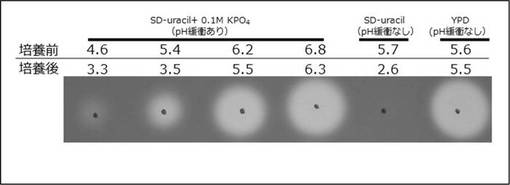
【図3】
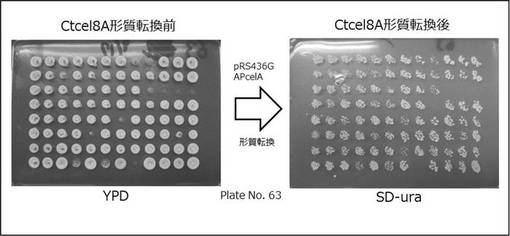
【図4】
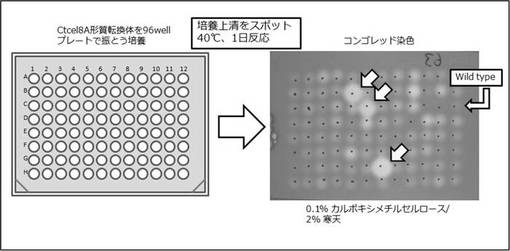
【図5】

【図6】

【図7】

【図8】

【図9】
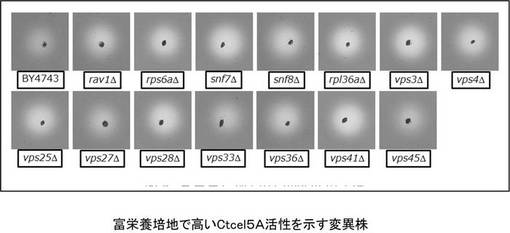
【図11】

【図12】

【図14】

【図15】

【図16】

【図18】

【図19】
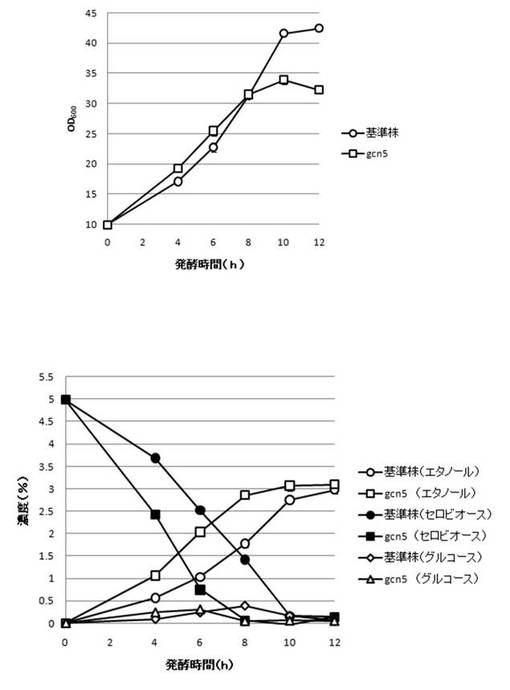
【図20】
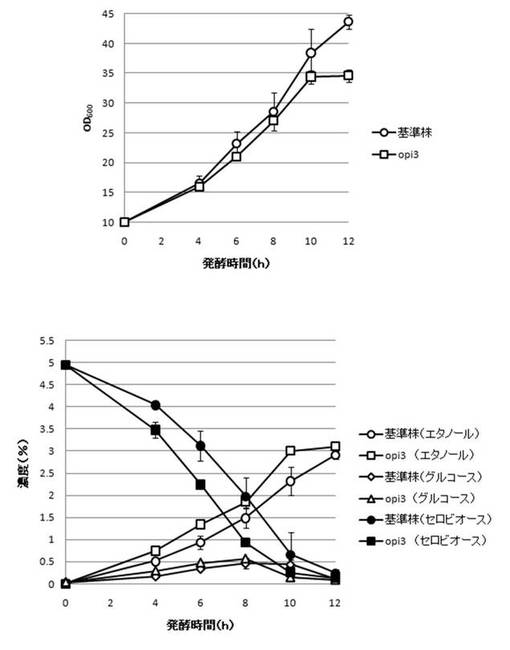
【図21】
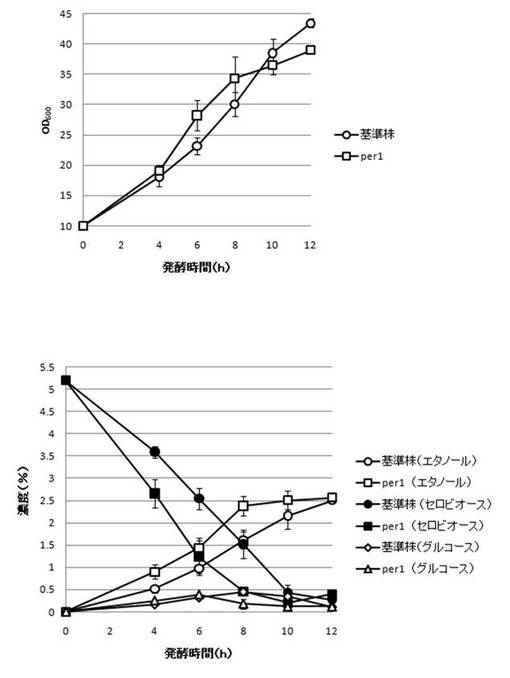
【図22】
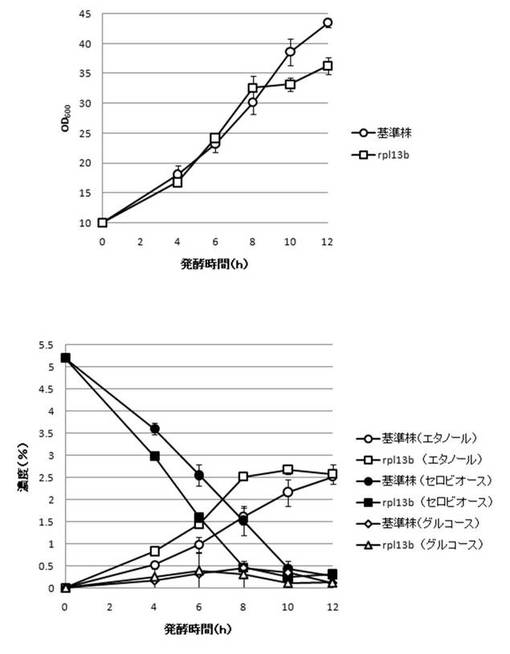
【図23】

【図24】
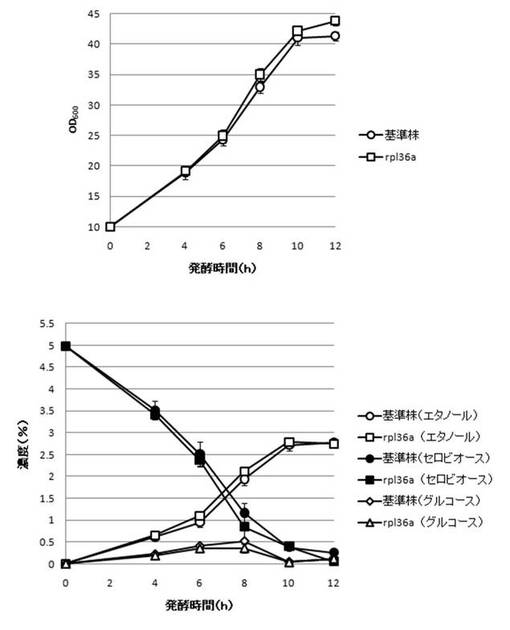
【図25】
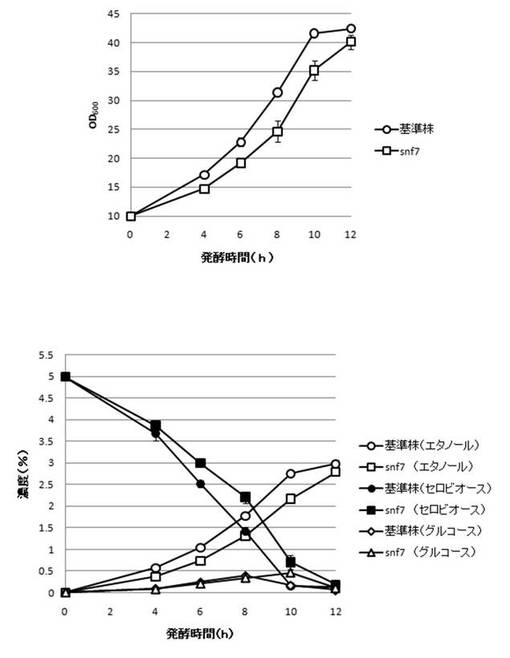
【図26】

【図27】
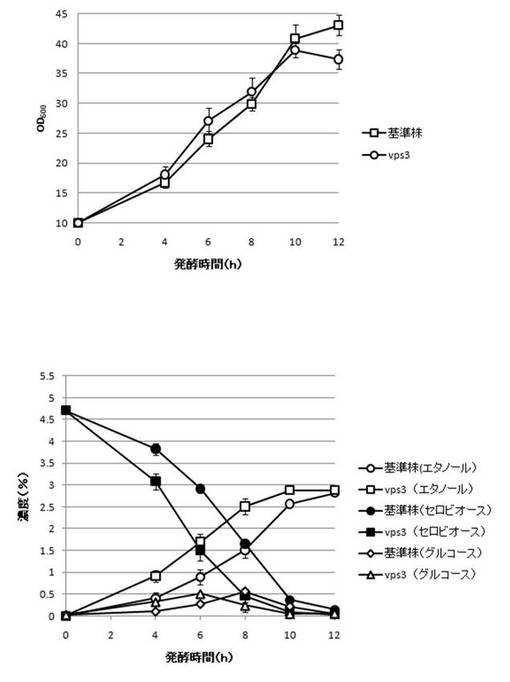
【図28】
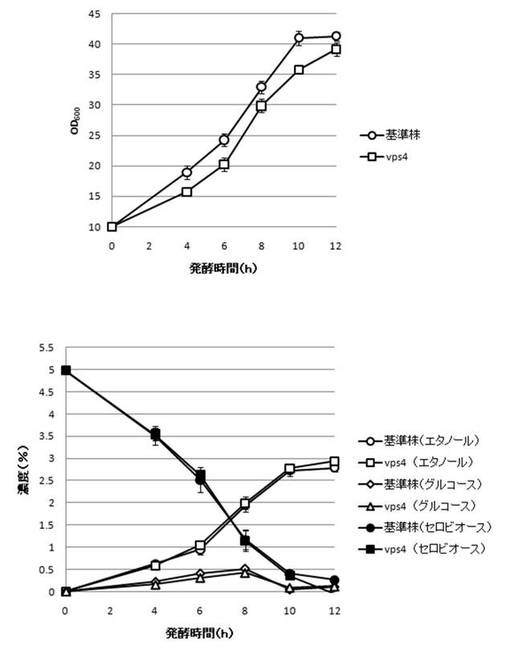
【図29】
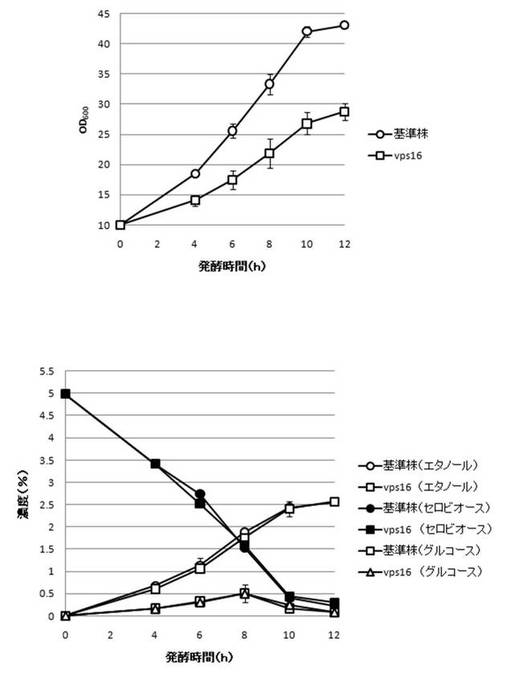
【図30】
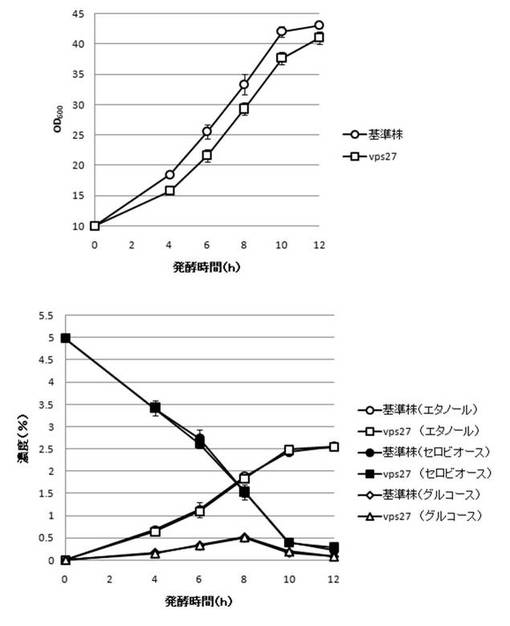
【図31】
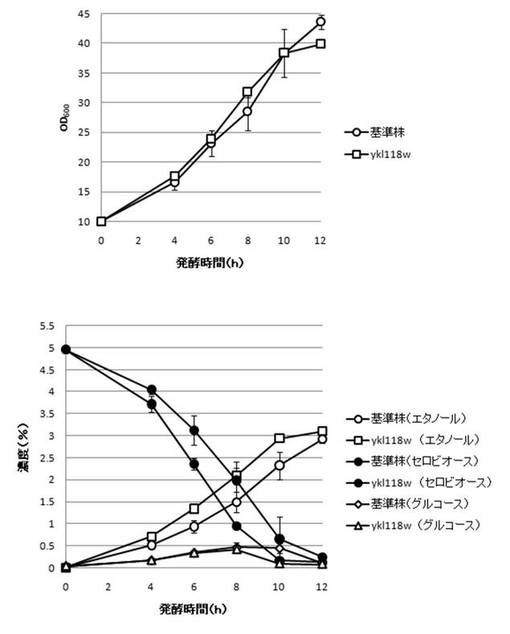
【図32】

【図33】
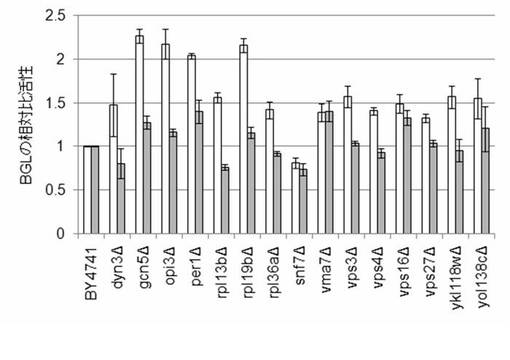
【図34】
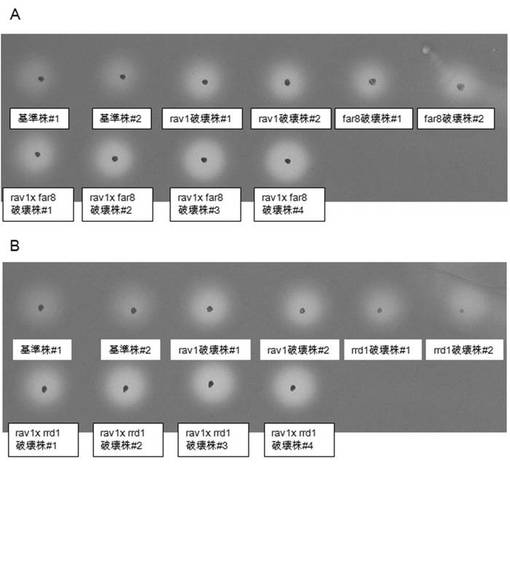
【図35】
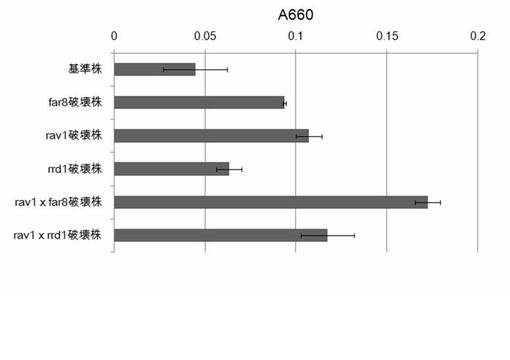
【図36】

【図37】
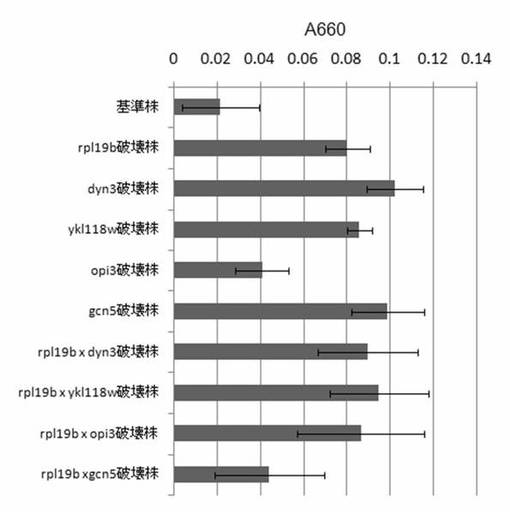
【図38】
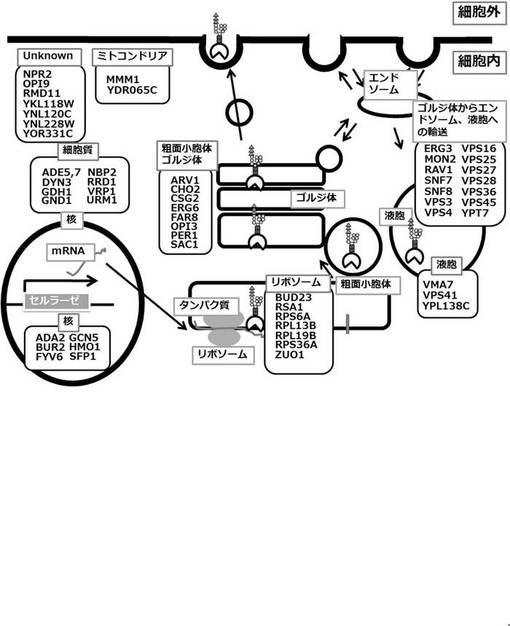
【図10】
【図13】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図11】
【図12】
【図14】
【図15】
【図16】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【公開番号】特開2010−193869(P2010−193869A)
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願番号】特願2009−51248(P2009−51248)
【出願日】平成21年3月4日(2009.3.4)
【出願人】(000003609)株式会社豊田中央研究所 (4,200)
【Fターム(参考)】
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願日】平成21年3月4日(2009.3.4)
【出願人】(000003609)株式会社豊田中央研究所 (4,200)
【Fターム(参考)】
[ Back to top ]