
外来性DNAを含む組換えイヌアデノウィルス(CAV)
【課題】ゲノムの本質的でないまたはその一部が欠失された、外来性DNAを含む組換え型アデノウィルス、好ましくは組換え型イヌアデノウィルスのタイプ2(CAV2)を提供する。
【解決手段】CAV2遺伝子のE3領域に欠失部分を含み、且つ(i)CAV2遺伝子のE3領域、(ii)CAV2遺伝子のE4領域と右ITR領域の間の領域、(iii)上記(i)および(ii)の両方の領域、に外来DNAが挿入されている組換えCAV2および上記組換えCAV2を含むベクター。
【解決手段】CAV2遺伝子のE3領域に欠失部分を含み、且つ(i)CAV2遺伝子のE3領域、(ii)CAV2遺伝子のE4領域と右ITR領域の間の領域、(iii)上記(i)および(ii)の両方の領域、に外来DNAが挿入されている組換えCAV2および上記組換えCAV2を含むベクター。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は組換え型アデノウィルス(adenoviruses)と、その製造方法と、その使用(複製DNAのためのベクターとしての使用を含む)と、その発現産物と、発現産物の使用とに関するものである。
本発明はさらに、プロモーターと形質発現カセットに関するものであり、特に切出したプロモーターとそれを含む形質発現カセットに関するものである。
本発明は特に、組換え型イヌアデノウィルス(canine adenoviruses, CAV)と、その製造方法と、その使用(DNAを複製するためのベクターとしての使用を含む)と、その発現産物と、発現産物の使用とに関するものである。
本発明は特に、組換え型CAV2ウィルス、外来性DNAがCAV2 E3および/または右ITRとE4転写単位との間のゲノムの右端に挿入されたものと、その製造方法と、その使用(免疫学、免疫療法での使用、ワクチン治療用組成物またはクローン化、複製、DNA発現用のためのベクターとしての使用、組成物またはベクターの使用方法を含む)と、その発現産物および発現産物の使用は関するものである。
【0002】
より一般的には、本発明はCAVゲノムの本質的でない領域またはそれの一部が欠失された外来DNAを含むように合成で修正されたCAVに関するものである。このCAVは、CAVが自然に複製される細胞に対して感染力をもつCAVとして入れられるのが好ましい。CAVゲノムの重要でない領域(またはその一部がCAVから欠失された領域)はE3領域またはそれの一部であるのが好ましい。外来性DNAはE3領域、El領域、E4領域または右ITRとE4領域との間に位置する領域に存在するのが好ましい。このCAVはCAV2であることができる。
【0003】
この組換え型CAVはヘテロジニアス(heterologous)なDNAの発現またはクローン化のためのベクターであることができる。このヘテロジニアスなデオキシリボ核酸は任意の所望の発現産物をコードできる。好ましい発現産物は以下を含む:そのエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療用遺伝子または融合蛋白。すなわち、このヘテロジニアスなデオキシリボ核酸はこれらの任意または全ての産物をコード化することができる。従って、ヘテロジニアスなDNAはトランスジェーンであることができる。
上記エピトープは抗原または免疫原であることができ、人間または家畜の病原またはトキシンのエピトープであることができる。従って、本発明は免疫学、抗原またはワクチンとしての組成物と、その発現産物にも関するものである。このCAVベクターは場合によっては宿主に直接投与できるので、本発明はCAVベクターを含む組成物にも関するものである。本発明組成物は免疫学的組成物、抗原性組成物、ワクチン組成物あるいは治療用組成物(例えば、局所または全身的な免疫応答(保護応答または遺伝子治療を含む)を刺激する組成)であることができる。従って、本発明は適切な脊椎動物宿主(動物または人間)に上記が組成を投与して免疫応答を誘発する遺伝情報を転写方法(例えば遺伝子治療)に関するものである。
【0004】
本発明の発現産物はインヴィトロでCAVベクターから単離でき、また、CAVベクターでインヴィトロに感染またはトランスフェクションされた細胞から単離できるので、本発明は生産物を発現するための方法、例えば細胞から遺伝子組換え、分割(cleaving)または連鎖反応(ligating)を用いて外来性DNAをCAVへ挿入し、得られた組換え型CAVをインヴィトロで適切な細胞へ感染またはトランスフェクションし、必要に応じてその発現産物を抽出または分離することから成る方法に関するものである。
発現産物は抗原性、免疫性または保護(ワクチン)応答物を提供するので、本発明はその生産物すなわち抗体およびそれの使用にも関するものである。特に、発現産物は誘導(elicit)抗体にすることができる。抗体はモノクローナル抗体の形に作ることができる。抗体または発現産物はキット、アッセイ、バインディングを含むテストなどで使うことができる。従って、本発明はこれらの使用にも関するものである。
【0005】
本発明の組換え型はDNAの複製に使えるので、本発明は、細胞を組換え物で感染させ、そこからDNAを収穫する、組換え型CAVをベクターとして用いたDNAの複製方法にも関するものである。得られたDNAはプローブまたはプライマとして使用でき、また、増殖(amplification)のためにも使える。
本発明はさらに、、組換え型ウィルスまたはプラスミドのためのプロモータおよびそのプロモータを含む発現カセットにも関するものである。
この観点は、本発明はウィルスまたはプラスミドが挿入される系で与えられたトランスアクチベイティング蛋白がトアンスアクチベイトされた(transactivated)領域と、プロモータの最小のプロモーター領域とを有する組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータに関するものである。本発明はプロモータから成る形質発現カセットと、プロモータまたは形質発現カセットを含むウィルスまたはプラスミドにも関するものである。この形質発現カセットは切り出した(truncated)機能性ポリアデニル化信号を含むことができる。
以下、明細書では多数の刊行物が引用されるが、これらの刊行物の情報は「参考文献」の項に記載されおり、引用時点でも説明してある。本明細書で引用される刊行物およびそれら刊行物で引用される文献の内容は本願明細書の一部をなすものとする。
【背景技術】
【0006】
各種ウイルス性ベクター系と、その使用、この種の系を用いた蛋白の形質発現のための外来性DNAと、この種の蛋白質類の使用およびこの種の蛋白からの産物の使用は多数の特許および科学刊行物に記載されている。
例えば以下のものを挙げることができる:ウイルス性ベクター系の発現のための組換え型ポックスウィルス(例えばワクシニア、アビポック(avipox)ウィルス)および外来性DNA(例えば、組換え型アビポック(avipox)ウィルス(ワクシニアウィルス)(特許文献1および特許文献2に記載されている);狂犬病グリコプロテイン(G)、遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、ウシ属の白血病ウィルスのgp5l,30エンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAV‐1 env遺伝子、NP(Chicken/ペンシルバニア/1/83インフルエンザウィルスのヌーデオプロテイン(nudeoprotein)遺伝子)、マトリックス、そして、伝染性の気管支炎ウィルス;HSV gDのプレポリマー遺伝子;エントモポックス(entomopox)プロモータ(特に特許文献3)、例えば、組換え型ワクシニアウィルス(アビポック(avipox)ウィルス);特にヘルペスウイルス・グリコプロテインをコード化しているDNA;特許文献4(例えば、組換え型ワクシニア(avipox);狂犬病、ヘパチチス(Hepatitis)B、JEV、YF、Dengue、はしか、仮性狂犬病、エピスタイン(Epstein)から抗原をコード化している外来性DNA‐バー、HSV、HIV、SIV、EHV、BHV、HCMV、犬パルボウィルス、馬の風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、特にお多福風邪(NDV));特許文献5(例えば組換え型ワクシニア、アビポック(avipox)、モリビリウイルス(例えば、はしかF、ヘマグルチニン);特許文献6(例えば、組換え型ワクシニアウィルス; HSV tk、グリコプロテイン(例えばgB、gD)、(風邪HA)ヘパチチス(Hepatitis)B(例えばHBsAg);特許文献7、特許文献8(組換え型ポックスウィルス、フラビウイルス構造蛋白質); 特許文献9(例えば、組換え型ポックスウィルス、免疫不全ウイルス);特許文献10(例えば、組換え型ポックスウィルス、特にIBDV);特許文献11および特許文献12(例えば、組換え型ポックスウィルス;特にサイトカインおよび/または腫瘍関連抗原); PCT/US94/06652(Plasmodiumライフサイクルの各段階からのプラズモディウム抗体)。
【特許文献1】米国特許第5,174,993号明細書
【特許文献2】米国特許第5,505,941号明細書
【特許文献3】米国特許第5,338,683号明細書
【特許文献4】米国特許第5,494,807号明細書
【特許文献5】米国特許第5,503,834号明細書
【特許文献6】米国特許第4,722,848号明細書
【特許文献7】英国特許GB 2 269 820 B明細書
【特許文献8】米国特許第5,514,375号明細書
【特許文献9】WO 92/22641
【特許文献10】WO 93/03145
【特許文献11】WO 94/16716号
【特許文献12】米国特許出願第08/184009号明細書
【0007】
バクロウイルス発現系と、発現のための外来性DNAおよび組換え型蛋白の精製法は非特許文献1(例えば風邪HA発現のためのCh.18、組換え型蛋白精製技術のためのCh.19参照)、非特許文献2(皮膚テストと、AIDSのためのテストキット、HIV1 env遺伝子の一部を含むバクロウイルス発現系を考察、特許文献13および特許文献14を引用)に記載されている。
特許文献15はヘルペスウィルスをベクターに関するものである。
ポリオウィルスおよびアデノウィルス・ベクター系もある(例えば非特許文献3〜6参照)。
特許文献16はSmaI部位から早い領域4(E4)のためのプロモータまでの逆終末反複領域近くの終わりまでの領域内にプロモータ-遺伝子配列を含む変性されたCAV2に関するものである。
【非特許文献1】リチャードソン、C.D.(編)、「Methods in Molecular Bioloav 39, "Baculovirus Expression Protocols」 (1995 Humana Press Inc.)
【非特許文献2】スミス達、「Production of Huma Beta Interferon in Insect Cells Infected with a Baculovirus Expression Vector」Molecular and Cellular Biology, Dec., 1983, Vol. 3, No. 12, p. 2156-2165; EPA 0 370 573
【非特許文献3】Kitson達、J. Virol. 65, 3068、 3075, 1991)
【非特許文献4】Grunhaus 達, 1992, "Adenovirus as cloning vectors," Seminars in Virology (Vol. 3) p. 237‐52, 1993
【非特許文献5】Ballay達EMBO Journal, vol. 4, p. 3861‐65
【非特許文献6】グレアム(Tibtech 8, 85-87, April, 1990;Prevec達)J. Gen Virol. 70, 429‐434
【特許文献13】第1986年10月16日に出願の米国出願第920,197号明細書
【特許文献14】欧州特許公報No.265785
【特許文献15】米国特許第4,769,331号明細書
【特許文献16】PCT WO91/11525
【0008】
CAV、特にCAV2には多くの問題がある。これらの問題のいくつかを以下説明する。重大な問題はCAVゲノムは外来性DNAの限られた量しか受けないということである。すなわち、外来性DNAの限られた量しかCAVゲノムに挿入できない。従って、CAVは「挿入できる大きさが制限され」。これはCAVをクローン化および発現で使うベクターの場合には重大な問題である。
口腔鼻腔径路での多くのウィルス感染の伝染効率は同じ径路でのウィルスベクターをベースにしたワクチン候補の効果で与えられている。しかし、ワクチン接種を受けた人でワクチンは大抵複製し、一般の生育環境と接触し、展開することが分かっている(例えばシュワルツ達、1974、Mueller達、1989、Oualikene達、1994を参照)ので、適切なウイルス性ベクターを採択したか否かは明らかでない。
完全な安全性を確保するためには、ベクターの選抜で明らかな安全性が確立されるような生きた弱毒ワクチンを特徴付けるのが好ましい。人間のワクチン接種のために生きた弱毒ウィルスを複製する各種ベクターが考えられている。現在公知の文献には以下のアプローチが記載されている:ヒトアデノウイルス(HAVs)セロタイプ4および7(Lubeck達、1989、チャンダ達、1990、Chengalvala達、1991、1994、Hsu達、1994)(インフルエンザウィルス(Garcia-Sastre and Palese, 1995のレビュー)およびポリオウィルスと関連ウィルス(Girard et al., 1995のレビュー)。
【0009】
獣医学の分野では、非経口または自然径路の感染(局所回復を刺激)によるワクチンを目的とした組換え型ベクターとして生きた弱毒ウィルスを複製したいくつかのベクターが現在分析されている。ポクスビリダ(poxviridae)科がこの点で最も特徴的なものである[例えば鶏痘ベースのベクター(Edbauer達、1990、テーラー達、1995とその参照文献参照)、herpesviridae科(例えば、仮性狂犬病ウイルスベースのベクター(Sedegah達、1992、Mettenleiter達、1994、Hooft van Iddekinge達、1996とその参照文献参照)、シチメンチョウ‐ヘルペスウィルスベースのベクター(ロス達、1995、Darteil達、1993 とその参照文献参照)、ネコ科のヘルペスウィルスべースのベクター(コール達、1990、Wardley達、1992、Willense達、1996)、伝染性のウイルス性呼吸器疾患ウィルスをベースにするベクター(Guo達、1994)、ウシ属のヘルペスウィルスをベースにするベクター(キット達、1991)]およびAdenoviridae科のより小さい構成メンバ(ウシ属のアデノウィルス3をベースにしたベクター(Mittal達、1995)]。
【0010】
犬種は口腔鼻腔免疫化に適したモデルを提供する。すなわち、弱毒化されたワクチン株が存在する犬アデノウィルス セロタイプ2(CAV2)は非経口経路または口腔鼻腔径路で安全に投与することができる犬のワクチン接種用の生きた免疫化媒体である。イヌのジステンパーウィルス(CDV)感染はこの標的種の呼吸感染のよい例を提供する。更により直接的な実験的CDV抗原投与系を用いることによって、CAV2 をベースにしたワクチン候補と以前に開発された古典的なCDVワクチンとを直接対比することができる。
CAV2はディチフィールド(Ditchfield)達(1962)によって、イヌの上気道感染の大発生から最初に単離された。その後、このウィルスは米国および欧州で呼吸疾患を有するイヌの呼吸管から単離された(Binn 達、 1967, Appel and Percy, 1970, Assaf 達、 1978, Danskin 1973)。実験研究によってCAV2のエーロゾルを植菌すると穏やかな呼吸疾患になるようになった(Swango達、1970、Appel、1970)。いくつかのCAV2 ベースのワクチンが開発され、広く子犬および成犬のワクチン接種で世界的に用いられている。CAV2の免疫化によってCAV1の血清学的株(予防注射されないイヌでは致命的な株)に実験的に抗原暴露した時に保護されることが証明された(フェアチャイルドとコーエン(1969)、Appel達、1973、バス達、1980)。ワクチンとしてのCAV2の明白な安全性はイヌワクチンによって誘発される疾患およびワクチンに付随する合併症が人類を含む他の動物種で過去30年間無いことでよく証拠される。更に、血清学的調査の結果から、多くの野生動物(キツネ、アライグマ、ゲジゲジおよびマングース)がCAV2またはそれに抗体的に関連したウイルス感染(Summer達(1988))症候に曝されていることが示されている。従って、犬アデノウィルス セロタイプ2(CAV2)のワクチン株はイヌの安全な複製コンピテント(ワクチン接種のための効果的なベクターをベースとするワクチン候補として考慮できる宿主制限ウィルス)の特異な例を提供する。
【0011】
HAVsは価値ある哺乳類の細胞発現ベクトル(グレアム達1988を参照)であり、組換えウイルス型ワクチン候補(Randrianarison-Jewtoukoff and Perricaudet 1995, Imler 1995を参照)であり、現在、遺伝子治療(Perricaudet and Perricaudet 1995を参照)のベクターとして評価されている。2つの主要なグループのHAVsがあり、組換え型HAVsは第3のグループである。
【0012】
これらのアデノウィルスベクターの最初のグループは、El領域を欠失したウィルスをベースにした複製不適格な組換え型アデノウィルスに対応する。El領域は組織培養のウイルス複製にとって重要な蛋白質類をコード化する。しかし、無能力の組換え型アデノウィルスは、このエル領域(Haj Ahmad達(1986)を本質的に発現する293の細胞系(グレアム達、1977)でエル領域の欠失の複製を伝播することが証明された。
El領域の欠失はHAVsに挿入ができる外来性DNAの量を増やすだけでなく、ヒト細胞の複製を制限し、人間での組換え型HAVsの形質転換の安全性を改善する。家畜および人間の病原を防ぐための現在の大部分のHAVベースのワクチン候補はEl欠失したベクターをベースにしている。その転写能力は限られているが、抗原投与実験での保護データは報告されている(Prevec達、1989、McDermott達、1989、Lubeck達、1989、Eloit達、1990、Ragot達、1993、Wesseling達、1993、Both達、1993、Gallichan達、1993、Hsu達、1994、Breker-Kiasser達、1995)。ベクター複製がない場合でも、防御免疫応答誘発特性は他の宿主制限ウイルス性ベクター与えられる。その最も有望なものはカナリポックス(canarypox)ウイルスベースのベクターALVACである(テーラー達(1991)、Perkus達(1995)を参照)。
【0013】
最終ゴールが複製コンピテント・アデノウィルス・ベクターである場合には挿入部位としてのEl領域を使用することは望ましくないので、El領域は今まで欠点であり、問題が有った。人間に対して安全な複製コンピテント・アデノウィルスが要求されるときにはこの欠点と上記問題が複合する。特に、HAVsのEl領域欠失がヒト細胞の複製を制限し、人間に関する安全性を改善するが、以下の文献に記載されているように、El形質転換細胞系とEl欠失した組換え型アデノウィルスとの間の遺伝子組換えの可能性、従って、El形質転換細胞系の安全性プロファイルは疑わしく見えるので、欠失したEl領域を用いることの利点は単なる可能性で、El領域を欠失したアデノウィルスを用いることは上記の欠点および問題点の解決にはならないと思える(El領域を欠失したアデノウィルスの増殖がEl領域を表現する細胞であるため)。
アデノウィルス・ベクターの第2のグループはヒト細胞の複製コンピテントで、人間以外の大部分の動物細胞では複製である組換え型アデノウィルスに対応する。これらのウィルスは異種遺伝子発現カセットを有するE3領域の一部の置換で特徴づけられる。E3領域がインビトロおよびインビボの両方で本質的でないことは伝染性ウィルス発生で示された(ケリーとルイス、1973、Kapoor達、1981、Morin達、1987、Lubeck達、1989)。従って、E3領域の一部の置換で多くの組換え型HAVsが作り出された(モリン達、1987、Chengalvala達、1991、1994、Prevec達、1989、ジョンソン達、1988、Lubeck達、1989、Dewar達、1989、Natuk達、1993、Hsu達、1994)。
【0014】
しかし、E3領域を用いてコードされる蛋白は宿主免疫応答(WoldとGooding 1991を参照)を種種の観点で変えるので、E3欠失は対応する組換え型ウィルスの病原プロファイルに何らかの影響を与えると思える。事実、HAVセロタイプ5からのE3領域の欠失がウィルス肺の病原性(ギンズバーグ達、1989)を増やすことはコトンラット・モデルで示された。しかし、E3領域内で部分的に欠失した組換え型ウシ属のAd3が親のwtウシ属のAd3で観測されることと類似したコトンラットの疾患を生産することも示されたので、ウシ属の動物の安全性がベクターをAd3をベースにすることはウシ用の生きた組換え型ウィルス・ワクチンでは充分であることを示唆している(Mittal達、1996)。この結果も、アデノウィルスものE3領域内の欠失の影響はケースバイケースであることを示している。
【0015】
CAV2 E3領域は既に同定され、特徴付けられている(Linne、1992)。しかし、利用できる発表されたデータ(Linne 1992)からは、CAV2 E3領域の挿入部位の明確な定義は明らかではない。DNAの塩基配列解析から、CAV2 E3領域の組織はHAVsのものとは大幅に異なることが明らかになっている。CAV2 E3領域は長さが1.5kbpであって、3つの転写解読枠(orf)のみを含むのに対して、ヒトアデノウイルスE3領域は少なくとも8つの転写解読枠(orf)を含んだ、少なくとも3kbpの範囲に延びている。これらのorfsはどれもHAV E3 orfsと有意レベルの相同性を有していない。この予備的な比較解析から、人間と犬のアデノウィルス・ゲノムは違って発展したと考えるのが合理的に見える。
アデノウィルス科(Imperiale達(1995)参照)を特徴づける錯体スプライシングおよびポリアデニル化パターンを用いたCAV2 E3領域内の挿入部位の定義はさらに複雑である。RNAスプライシング・ドナーおよびアクセプター部位は、たとえそれらの暗号配列がE3領域の外側に局所化されているとしても、E3領域内に局所化されたいくつかの重要なメッセンジャーRNAの成熟にとって重要である。
【0016】
更に、E3領域は高転写活性条件のゲノム領域内に位置する(シャープ達(1984)参照)ので、この部位での外来性DNAの組込みは組換え型ウィルスの生態学に潜在的に有害な影響を与える。さらに、E3領域は主要な後期プロモータ(MLP)の下流に位置するので、組換遺伝子の転写とMLPで始まる転写との間の干渉が示された(Zu達、1995)。
従来法の課題は組換え型ウィルスの表現型改変個所および転写活性でない領域の挿入部位の定義を最小にすることにある。一般的には、従来技術の問題点はE3領域にあるといえる。
【0017】
第3のグループの組換え型HAVsは右の反転終末反複(ITR)とE4プロモータとの間の組換えDNAの組込みをベースにしている。このITRはウイルス性ゲノムDNAのウィルスDNA反復および効率的パッケージングにとって重要な配列を含む。右の反転終末反複(ITR)とE4プロモータとの間の領域が外来性DNA配列(斎藤達、1985、チャンダ達、1990)を受け入れが、アデノウィルスをベースにしたベクターは組換え型hAd5のパッケージング能力が野生型ゲノム(Bett達、1993)の約105%のゲノムに限られるので、それらが運ぶことができる外来性DNAの量には厳しい限界があり、従来法と同じ問題点がある。
右のITRとE4領域との間の領域はCAV2組換え型ウィルス生成のための追加の挿入部位の候補になる。PCT WO 91/11525はSmaI部位がITRの左端部の近くの潜在的な挿入部位に関するものである。W091/11525の教えとは逆に、本発明者は組込み部位の上限があると考えられる。それは400のbp DNAフラグメントが挿入でき、約1 kbpののような大きいフラグメントの組込みは繰り返し失敗したものである。従来法の課題はこの部位の利用にある。
すなわち、これまではE4プロモーター領域が欠失物であり、それが問題になっていた。
【特許文献17】WO 91/11525
【0018】
分子レベルでのCAV2ゲノムの初期の性格付けは文献に記載されている。CAV2およびCAViの菌株(Jouvenne達、1987、Macartney達、1988、SpibeyとCavanagh 、1989)およびEl、E3およびITRs領域に対応する配列解析の制限解析が報告されている。イヌアデノウィルスのゲノムの全体機構は他のAdenoviridae科構成メンバに類似しているが、CAV2 の遺伝子的なE3領域の明確な組織はユニークである。
従って、Adenoviridaeの他の構成メンバから単に1つの構成メンバを推論することはできないので、従来法にはさらに他の問題がある。
【0019】
さらに、前記の欠失または問題を解決するときには、E3のような外来プロモータまたはMLPプロモータに対する依存を避けるのを好ましい。しかし、組換遺伝子の発現のパターンは全体的な発現、従ってワクチン組成または免疫学組成(Darteil達、1995、Xu達、1995、Hooft Van Iddekinge達、1996)の組換え効能の重大なパラメータである。
いくつかの細胞およびウィルスのプロモータは組換え型HAVsの誘導に関係する。中でも、最も特徴づけるものはb-actin、SV40 early、SV40 late、hAD MLPおよびhCMV-IE(Zu達,1995)である。hCMV―IEプロモータは組織培養で組換え型蛋白発現の最も高レベル且つ最も長い存在と関連するので、これが上流域制御領域になることは確かである。このプロモータもここまでにテストされたほとんどの細胞系で作動するように思える。細胞型にインデペンデントなプロモータ活性の可能性は明らかに強みである。
【0020】
hCMV-IEプロモータはHAV感染(ゴーマン達、1989)で転写活性化できることは証明されている。このプロモータ(約850bp)の大きさは組換え型CAVベクターの寸法限界で問題である。従って、このプロモータを有する過去の成功から推論することができるのは組換え型CAVベクターだけである。
アデノウィルスがウィルスDNA反復の開始期の後に、強く細胞の蛋白合成を抑制することは公知である(チャンとシュナイダー(1993)参照)。従って、複製コンピテントな組換え型アデノウィルスはDNA合成の開始期の後は組換え型蛋白発現で限界されてきた。
【0021】
同様に、斎藤達(1985)は組換え型ヒトアデノウイルス・セロタイプ5ではほとんど組換え型蛋白が得られられないことを組換え型メッセンジャーRNAの高生産で証明している。
最近のアデノウィルス・メッセンジャーRNAはその5'の翻訳されていない領域(5'UTR)でのトリパr−テーリーダー(TPL)配列の存在で特徴づけられる。このTPLの存在は最近のアデノウィルス・メッセンジャーRNAの翻訳可能性の重要な成分である。更に、hAd5バックグラウンドにおいて、TPLの存在がアデノウィルス後期プロモータ(Thummel達1983)から発現される組換え型SV40 T抗原の翻訳調節の特色であることも証明されている。
【0022】
発現カセットのデザインでの他の重要な問題はポリアデニル化信号の大きさである。従来法のさらに他の問題は、CAV2 DNAを単層にトランスフェクションするために条件を確立することにある。精製された裸のアデノウィルスDNAの感染性は低い。リン酸カルシウムを用いた手順で、グレアムとVan derバーグ(1973)は1 pfu/mgの精製されたDNAの収率を報告しているが、換え型ウィルスを単離させるための効率的なプロセスではない。他にいくつかのアプローチは本問題を解決するために提案されたが、従来法の問題は充分に解決されず、特に安全性の問題は言及されていない。
【0023】
例えば、DNA蛋白複合体が精製され、裸のDNAで感染性が向上する(5x103 pfu/mg)と報告されている(Sharp 達、1976)。同様に、アデノウィルスDNAを共有結合で閉環したものが感染力をもつことが示された(グレアム、1984)。
組換え型HAVsを誘導するのに広く使われている手順はHAV El領域で変形した293細胞系を用いたものである(グレアム達、1977)。ウシ属のおよび犬アデノウィルス組換え型の誘導が対応するアデノウィルスEl領域(PCT WO 91/11525、Mittal達、1995a)で変形する細胞系の利用に依存することが報告されている。いくらかのアデノウィルスのEl領域を用いてコード化される遺伝子が齧歯動物細胞(Grand、1987)を用いてチェックされ、トランスフォーメーションに貢献することは示されたが、El形質転換細胞系の安全性プロファイルは疑わしい。また、アデノウィルスEl領域内の有力なtransactivatorsの存在はよく確認され、El形質転換細胞系に関する安全性に関心が延びている(ネヴィンズ(1993)参照)。
従って、El形質転換細胞系の使用から独立した、好収率を有するトランスフェクション条件は技術の重要な前進になる。
【特許文献18】WO 91/11525
【0024】
以上のとおり、組換え型CAV、好ましくは外来性DNAと本質的でない領域またはそれの一部が欠失された領域息とを有する組換え型CAV2を挿入し、特にCAVが自然に模写される細胞に対して感染力をもつCAVのパックとして挿入し、または、E3内および/または右のITRとE4転写単位との間の右の端を含むゲノム内に外来性DNAを含むCAVを挿入することと、この種の組換え体の製造方法およびその使用は上記および下記の文献には提案されなかった。また、ウィルスまたはプラスミドが挿入される系を用いてトアンスアクチベイトされた領域と、このプロモータの最小のプロモーター領域とを有する組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータや、このプロモータから成る発現カセットや、プロモータまたは発現カセットを含んでいるウィルスまたはプラスミドは提案されなかった。この組換え型CAV、プロモータ、この組換え型CAVおよびプロモータの製造方法、発現カセット、プロモータまたは発現カセットを含むウィルスまたはプラスミドは、人間に関してはCAVが非複製ベクターであり、プロモータおよび発現カセットは組換え型ウィルスの挿入寸法限度にしてあるので、従来の組換え体より優れたものである。
【発明の開示】
【発明が解決しようとする課題】
【0025】
本発明の目的は、組換え型アデノウィルス、好ましくは組換え型イヌアデノウィルス2(CAV2)のような組換え型イヌアデノウィルス(CAV)を提供することにある。
【0026】
本発明のさらに他の目的は、外来性DNAを含む上記組換え体、好ましくは本質的でない領域に外来性DNAを含み、CAVゲノムの本質的でない領域を有し、またはその一部を削除したものを提供することにある。そして、好ましくは、CAVを自然に複製する細胞に対してこの組換え体を感染性CAVとしてパックした組換え体を提供することにある。
本発明のさらに他の目的は、外来性DNAがE3またはE3と右のITRとE4形質移入単位との間の領域の両方に挿入される外来性DNAを含んだ上記組換え型CAVを提供することにある。
【0027】
本発明のさらに他の目的は、複写活性のある切り詰めたプロモータと、このプロモータを含む発現カセットと、プロモータまたは発現カセットを含むウィルスまたはプラスミドを提供することにある。また、ポリアデニル化信号を含む切り詰めた発現カセットを提供する。
本発明のさらに他の目的は、上記組換え体、この組換え体からの生産物を発現させる方法、組換え体または発現産物を含む組成、発現産物の使用方法、組成の使用方法、組換え体からのDNA、組換え体からDNAを複製する方法のいずれか一つまたは全てを提供することにある。
本発明の別の目的は、アデノウィルスベース、例えばCAVベース、好ましくはCAV2ベースのベクターまたはこのベクターを含む組成または上記の任意または全ての欠点および/または従来法の問題に関連したベクターの製造方法または使用方法を提供することにある。
【課題を解決するための手段】
【0028】
驚くべきことに、本発明はCAVゲノムの本質的でない領域またはその一部がCAVから欠失された、外来性DNAを含む合成で変性されたCAVを提供する。CAVはCAVを自然に複製する細胞に対して感染性CAVとしたパックされるのが好ましい。本質的でない領域またはその一部はCAVゲノムから欠失できる。欠失した領域またはその一部が本当に本質的でないかどうかは欠失に起因する組換え型CAVの生育性および安定度から確かめる。CAVゲノムまたはその一部の領域がCAVから欠失する必須でない物はE3領域またはそれの一部であるのが好ましい。外来性DNAは本質的でない領域にも存在する。外来性DNAが挿入された領域またはその一部が本質的でないかどうかは外来性DNAの組込みに起因する組換え型CAVの生育性および安定度から確かめる。CAVゲノムに外来性DNAの組込みのための本質的でない領域としてはE3領域、El領域、E4領域または右のITRとE4領域との間の領域が好ましい。
【0029】
本発明は、驚くことに、ヘテロジニアスなDNAがE3にあるか、E3および右のITRとE4形質移入単位との間の領域にあるヘテロジニアスなDNAをCAVゲノムの本質的でない領域に含む組換え型CAVを提供する。
これら具体例のCAVは好ましくはCAV2である。
【0030】
本発明はさらに、組換え型CAVから成るヘテロジニアスなDNAの発現またはクローン化ベクターを提供する。
ヘテロジニアスなDNAは重要なエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療用遺伝子または融合蛋白から成る発現産物をコードする。
このエピトープは抗原または免疫原であるか、家畜または人間の病原またはトキシンからの免疫学的に活性なフラグメントである。
【0031】
このエピトープは家畜の病原またはトキシンまたは家畜の病原またはトキシンの抗原または他の抗原からの抗原であることができ、または、例えば病原に対して応答を導き出す他の抗原またはトキシンからの、病原に関して応答を導き出すトキシン、例えばモリビリウイルス抗原イヌジステンパーウィルスまたははしかまたは牛疫抗原HAまたはF;狂犬病グリコプロテイン(例えば狂犬病グリコプロテインG);鳥風邪抗原(例えばシチメンチョウ風邪HA)Chicken/ペンシルバニア/1/83風邪抗原、例えばヌーデオプロテイン(nudeoprotein)(NP);ウシ属の白血病ウィルス抗原(例えばgp5l,30エンベロープ);ニューカッスル病ウイルス(NDV)抗原(例えばHNまたはF);ネコ白血病ウィルス抗原(猫白血病ウイルス)(例えば猫白血病ウイルス外膜蛋白);RAV-1 env; RAV-1 env;伝染性の気管支炎ウィルスのマトリックスおよび/またはプレポリマー;ヘルペスウイルス・グリコプロテイン(例えばねこヘルペスウィルスからのグリコプロテイン、馬のヘルペスウィルス、ウシ属のヘルペスウィルス、仮性狂犬病ウイルス、犬ヘルペスウィルスまたはサイトメガロウィルス);フラビウイルス抗原(例えば日本脳炎ウィルス(JEV)抗原);免疫不全ウイルス抗原(例えばネコ科の免疫不全ウイルス(FIV)抗原またはサル免疫不全ウイルス(SIV)抗原);パルボウィルス抗原(例えば犬パルボウィルス);馬の風邪抗原;マレック病ウィルス抗原;ポックスウィルス抗原(例えば先天性四股欠損症抗原、カナリポックス(canarypox)ウィルス抗原または鶏痘ウィルス抗原)または伝染性包性(bursal)疾患ウィルス抗原(例えばVP2、VP3、VP4)にすることができる。
【0032】
このエピトープは人間の病原またはトキシンまたは人間の病原またはトキシンの抗原または他の抗原からの抗原であることができ、または、病原に対してたとえば応答を導き出す他の抗原またはトキシンから応答を導き出すトキシン:モリビリウイルス抗原(例えばはしかウィルス抗原(例えばHAまたはF));狂犬病グリコプロテイン(例えば狂犬病ウィルス・グリコプロテインG);風邪抗原(例えばインフルエンザウィルスHAまたはN);ヘルペスウイルス抗原(例えば単純ヘルペスウィルス(HSV)のグリコプロテイン、ヒトサイトメガロウイルス(HCMV)、エピスタイン(Epstein)-バー);フラビウイルス抗原、JEV、Yellow FeverウィルスまたはDengueウィルス抗原;Hepatitisウィルス抗原(例えばHBsAg);免疫不全ウイルス抗原(例えばgp120のようなHIV抗原、gpl6O);ハンタン(Hantaan)ウィルス抗原;C.テアニ(tetani)抗原;お多福風邪抗原;pneumococcalな抗原(例えばPspA);Borrelia抗原(例えばOspA、OspB、Borrelia burgdorferiのようなライム病と関連があるBorreliaのOspC、Borrelia afzelliおよびBorrelia garinii);水ぼうそう(水痘帯状ヘルペス)抗原またはPlasmodium抗原にすることができる。
もちろん、上記リストは典型的なもので、このエピトープは任意の家畜または人間の病原または家畜または人間の病原の抗原からの抗原であることができる。
【0033】
ヘテロジニアスなDNAは成育因子または治療の遺伝子であることができるので、本発明の組換え型CAVは遺伝子治療で使うことができる。遺伝子治療は遺伝情報を積み換えることを含む。遺伝子治療および免疫療法に関しては米国特許第5,252,479号、さらにはWO 94/16716および第1994年1月19日に出願の米国特許願第08/184009号を参照。これらの内容は本願明細書の一部をなす。成育因子または治療遺伝子は疾患ファイティング蛋白、癌を処理する分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードすることができる;成育因子はさらに例えば治療用遺伝子がαグロビンをコードする遺伝子から成るグループ、β‐グロビン、γ‐グロビン、顆粒球マクロファージ-コロニー刺激因子、腫瘍壊死因子、インターロイキン、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、EPO、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随する抗原またはB7から選ばれる。
【0034】
本発明はさらに、組換え型CAVウィルスまたはベクターと、薬学的に許容可能なキャリアまたは希釈剤を含む免疫組成物、免疫学組成物またはワクチン組成物を提供する、組換え型CAVウィルスまたはベクター(またはそれの発現産物)を含む免疫学の組成は局所的または全身的な免疫応答を導き出す。応答は保護であってもよいが、そうでなくてもよい。組換え型CAVウィルスを含む免疫的組成またはベクター(またはそれの発現産物)も同様に局所または全身的な免疫応答を導く。ワクチン組成は局所的または全身的な保護応答を導き出す。従って、「免疫学組成物」および「免疫組成物」は「ワクチン組成物」を含む(前者2つの用語は保護組成物でもよい)。
【0035】
従って、本発明は、組換え型CAVウィルスまたはベクターと、薬学的に許容可能な担体または希釈剤から成る免疫組成物、免疫学組成物またはワクチン組成物を投与して宿主脊椎動物の免疫応答を誘発する方法を提供する。本明細書では人間を除いて「動物」は全ての脊椎動物種を含む;「脊椎動物」は動物および人間を含む全ての脊椎動物を含む。もちろん、「動物」のサブセットが「哺乳類」である。本明細書では人間以外の全ての哺乳類を含む。
人間の投与では、本発明組換え型CAV(特にCAV2)は生産的複製のない発現をするという利点がある。従って、免疫的にバランスした(immunocompromised)個体で本発明組換え体を使用することができ、本発明の組換え型と接触する研究者の安全性のレベルを提供する。従って、本発明は組換え型CAVウィルスまたはベクター(例えばCAV2)を投与するか宿主に接種して増殖または蛋白を発現する方法と理解できる。それによって宿主が犬でなく、組換え型ウィルスまたはベクターの自然の宿主でない場合にも生産的複製のない発現ができる。
【0036】
さらに、CAV、特にCAV2がイヌのワクチン用株として使われるので、本発明はワクチン用CAVに、犬病原またはトキシンの抗原の関心の追加のエピトープ、例えばCAV2を導入して、それらの追加のエピトープを発現する組換え型CAVとするための手段を提供し、イヌに予防接種をすることでこれらエピトープおよび犬アデノウィルスの応答を導き出す手段を提供する。
追加のエピトープは犬病原(アデノウィルス以外の)またはトキシン、犬の病原性抗原(アデノウィルス以外の)またはトキシンに対してイヌまたは子犬で応答を導き出す他の抗原または犬病原(アデノウィルス以外)またはトキシン(後者の例、イヌまたは子犬のイヌジステンパーウィルスに対して保護応答を導き出すはしかHAおよびFおよびそのエピトープ、米国特許第5,503,834号参照)にすることができる。
【0037】
従って、本発明は組換え型ワクチン用CAV、たとえば、犬病原またはトキシンの任意の抗原:狂犬病、犬ヘルペスウィルス、イヌジステンパーウィルス、犬パルボウィルスなどからのエピトープをコード化するヘテロジニアスなDNAを含むことができる。この点については、1995年6月7日に出願された米国特許第5,529,780号(狂犬病化合組成)および第1995年3月29日に出願のアメリカ出願第08/413118号(犬ヘルペスウィルスDNA)、1994年4月6日に出願の第08/224657号(イヌジステンパー)、1995年4月5日に出願の第08/416646号(イヌジステンパー)および08/486,969(犬ヘルペスウィルスDNA)を参照。ここで引用される文献の内容は本発明の一部をなす。従って、本発明は複数の蛋白、例えば犬病原の抗原のような少なくとも2つのエピトープをコードする外来性DNAを含むCAV組換え体に関するものである。
【0038】
本発明はさらに、他の抗原をさらに含むCAV組換え体を含む組成物に関するものでもある。
本発明が、組換え型CAVウィルスまたはベクターおよび薬学的に許容範囲内である担体または希釈剤を含む治療用組成物を提供する。この治療用組成物は遺伝子治療および本発明の免疫療法の具体化、すなわち、遺伝情報をそれを必要とする動物および人間に移入する方法で用いられる。従って、本発明は遺伝情報を移入する方法を含む。
【0039】
他の具体例として、本発明は蛋白または遺伝子生産物を発現する方法を提供する。この方法では蛋白、遺伝子生産物または発現産物を感染させるか、本発明の組換え型CAVウィルスまたはベクターを有する細胞をインビトロでトランスフェクションし、必要に応じて抽出し、細胞から遺伝子産物、発現産物またはDNAを精製または単離する。本発明はさらにヘテロジニアスなDNAの塩基配列のクローン化または複製方法を提供する。この方法では本発明の組換え型CAVウィルスまたはベクターを細胞にインビトロまたはインビボで感染またはトランスフェクションし、必要に応じて抽出し、細胞またはウィルスからDNAを精製または分離する。
【0040】
別の態様において、本発明はCAVゲノムの本質的でない領域に外来性DNAを本発明のCAVウィルスまたはベクターに挿入することから成る組換え型の調製方法を提供する。
この方法では、好ましくは外来性DNAを挿入する前に、必須でない領域をCAVゲノムから欠失することができる。
この方法はインビボ遺伝子組換え(CAV DNAが感染力をもつものでもよい)から成ることができる。本発明方法はCAVゲノムの一部と相同なDNAの塩基配列でフランクした外来性DNAから成るドナーDNAの存在下で細胞と相溶性のある培地中でCAV DNAで細胞をトランスフェクションする。それによって外来性DNAをCAVのゲノム中に導入し、必要な場合には生体内の遺伝子組換えで修正されたCAVを回収する。
【0041】
本発明はさらに、CAV DNAを切断して切断されたCAV DNAを得、切断されたCAV DNAを外来性DNAと連鎖反応させてハイブリッドCAV−外来DNAを得、この複合型CAV-外来DNAで細胞を形質移入し、必要な場合にはそれから外来性DNAの存在下で、変性されたCAVを回収することから成る方法を提供する。
インビボ遺伝子組換えが理解されたので、本発明はCAVに対して外来のポリペチドをコードするCAVでは自然には起らないドナーDNAから成るプラスミドを提供する。このドナーDNAはCAVの本質的でない領域と同一直線上にあるCAV DNAのセグメント内にあり、CAVゲノムの本質的でない領域からのDNAがドナーDNAが側面に位置する。
【0042】
外来性DNAはDNAおよび必要な場合にはその発現物の安定した組込みを生ずる任意の向きでCAVに挿入して組換え型CAVを作ることができる。本発明の組換え型CAVウィルスの外来性DNAまたはベクターはプロモータを含むことができる。このプロモータはヘルペスウィルスからのものにすることができる。例えば、このプロモータはサイトメガロウィルス(CMV)プロモータ(例えば人間のCNV(HCMV)または鼠CMVプロモータ)であることができる。
このプロモータは好ましくは切り詰めた転写活性プロモータであるのが好ましい。この転写活性プロモータはウィルスからのトランスアクチベイティング蛋白でトアンスアクチベイトされた領域および完全長プロモータ(これから切り詰めた転写活性プロモータが誘導される)の最小プロモーター領域とから成る。
【0043】
本明細書では「プロモータ」は最小のプロモータと上流域調節配列に対応するのDNA塩基配列の会合から成る。「最小のプロモータ」はCAP部位+TATAボックスから成る(転写の基礎的レベルの最小配列;転写の無秩序なレベル)から成る。「上流域調節配列」は上流要素とエンハンサ配列とから成る。「切り詰められた」とは完全長プロモータが完全に存在しないこと、すなわち、完全長プロモータの一部が除去されたことを示す。この切り詰めたプロモータはMCMVまたはHCMV、例えばHCMV-IEまたはNCMV-IEのようなヘルペスウィルスに由来することができる。
プロモータは、例えば鼠CMV-IEプロモータおよびHCMV-IEプロモータで、塩基対をベースにして完全長プロモータからの寸法が40%、さらには90%まで切り詰めることができる。本発明の切り詰めたプロモータは、切り詰めたプロモータが挿入されるウィルスまたは系で与えられるトランスアクチベイティング蛋白および最小プロモータを用いて転写活性化されるエンハンサ領域から成ることができる。従って、本発明の切り詰めたプロモータ中に存在するプロモータは完全長の当初の塩基対の60%、さらには10%にすることができる。
プロモータはより多くの塩基対を必要とするので、本発明の切り詰めたプロモータを含む発現カセット、ウィルスおよびプラスミドは驚くべきである。本発明のプロモータは完全長プロモータに比較して優れた性能を有する。いかなる特定の理論に縛られるものではないが、この優れた性能は切り詰めた(トランケーション)に起因すると思われる。更に、プロモータの切り詰めた(トランケーション)は組換え型ウィルスおよびプラスミド(特にCAV)の挿入寸法制限の問題を解決する。
【0044】
従って、本発明はさらに、プラスミドが挿入されるウィルスまたは系によって与えられるトランスアクチベイティング蛋白によってトアンスアクチベイトした領域および切り詰めた転写活性プロモータが得られる完全長のプロモータの最小のプロモーター領域とから成る組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータに関するものである。
前記のプロモータと同様に、本発明のプロモータはヘルペスウィルス、例えばMCMVまたはHCMV(例えばHCMV-IEまたはHCMV-IEプロモータ)であるのが好ましく、塩基対をベースにして完全長プロモータの寸法の40%まで、さらには90%まで小さくすることができる。
【0045】
従って、本発明はさらに、切り詰めた転写活性プロモータから成る組換え型ウィルスまたはプラスミドへ組込むための発現カセットを提供する。この発現カセットは機能を有する切り詰めたポリアデニル化信号、例えば機能を保持した切り詰めたSV4Oポリアデニル化信号を含むことができる。大きい信号をだすことを考えると、切り詰めたポリアデニル化信号が機能することは驚くべきことである。そして、切り詰めたポリアデニル化信号はCAVのような組換え型ウィルスの挿入寸法限界問題を解決する。この発現カセットはさらに、外来性またはヘテロジニアスなDNAをそれが挿入されるウィルスまたは系に関して含むことができる。これらの外来性またはヘテロジニアスなDNAは上記と同じのDNAにすることができる。
【0046】
さらに更に驚くことは、本発明は組換え型を生成するために少なくとも一つの本質的でない遺伝子座、例えばE3領域を用いて組換え型CAV、好ましくはCAV2を提供する。HAVおよびウシのAd3から得られたデータから、この領域の一部はインビトロおよびインビボで伝染性ウィルスの形成には本質的でなく、従って、組込み領域とみなすことができる。従って、本発明の一つの観点はCAV E3欠失または部分的欠失の突然変異体(例えばE3 ORF1および/またはORF2)の生成物を提供することにある。この突然変異体は全CAV E3領域が組織培養に必要でなく、従って、組換え型CAV形成の挿入部位として使用できることを証明した。従って、本発明は外来性DNAが欠失したおよび/またはE3領域中、好ましくはE3領域の少なくとも1つの本質的でない領域、例えばORF2中に外来性DNAを挿入した組換え型CAVに関するものである。
E3領域の欠失はCAVゲノムへヘテロジニアス配列を組込む能力をさらに追加する。例えばこの欠失はゲノムの右端に導入した大きな発現カセットを補償することができる。当業者は過度の実験なしに本明細書に記載の方法を用いることで追加の本質的でない領域、好ましくはE3領域および追加の本質的でない区域を容易に確認できる。
【0047】
別の態様において、本発明はヘテロジニアスなDNAの挿入に対して必須でない領域中で欠失をした組換え型CAV、好ましくはCAV2を提供する。例えば、この必須でない領域内の欠失は他の領域、例えばE4/右ITR領域へのヘテロジニアスDNAの挿入と実質的に同じにする(例えば、補償する)ことができる。
多様なCAV2株からのITRa間のヌクレオチド配列の比較で右ITRの直ぐ上流に多様性があることが分っている(Cavanagh達、1991、Spibey、1991)。本発明者はこの領域内に新規で自明でない挿入部位を設計した。従って、更なる観点から、本発明は外来性DNAを挿入したるCAV組換え型にある。更に、驚くことに、E4/右ITR領域は上記SmaIより大きなヘテロジニアスDNAの断片を挿入でき、挿入寸法制限の問題が解決する。
E4/右ITR部位が転写活性が低いCAVゲノムの領域に局所化している(シャープ達(1984)を参照)ので、そこへの挿入はCAV組換え型ウィルスの生態学に大きな影響を与えない。
【0048】
既に示したように、本発明の一つの実施例では、本発明はCAVに外来性DNAを挿入するための新規で非自明な発現カセットを提供する。このカセットはヘテロジニアスな成熟核調節配列から成る。好ましい実施例では、本発明はウィルスおよびプラスミド、例えばアデノウィルスベースのベクターと組合されたパッケージング限界およびと生物学的特性を考慮に入れて合理的に設計された発現カセットを提供する。
【0049】
MCMVおよびHCMVプロモータをプロモータが挿入されるウィルスまたは系が与えるトランスアクチベイティング蛋白で転写活性化されるエンハンサ領域として小さく切り詰める能力および最小のプロモータから、本明細書の開示および従来の知識からの過度の実験なしで、他の成熟核ウィルス、特に他のヘルペスウィルスから同じように切り詰めできることが示される。本発明はそうした他のウィルスから切り詰めたプロモータを含む。
【0050】
より具体的な観点から、本発明はHCMV-IEまたはHCMV-IEプロモータ、好ましくはその切り詰めたプロモータから成るCAVまたは組換え体、好ましくはCAV2または組換え体を含む。これらのHCMVN−IEまたはHCMVN−IEプロモータまたはその切り詰めたプロモータはCAV-誘導遺伝子生産物でトアンスアクチベイトされるのが好ましい。
切り詰めた転写活性(またはコンピテント)プロモータ(好ましくはヘルペスウィルス・プロモータのような切り詰めた転写活性成熟核ウィルス・プロモータ、例えばHCMVまたはNOWプロモータ)を含む本発明の態様では、「活性」(または「コンピテント」)によって切り詰めた転写活性プロモータは元の完全長のプロモータの転写活性の少なくとも80%、好ましくは少なくとも85%、より好ましくは少なくとも90%および最も好ましくは少なくとも95%を示さなければならない。
【0051】
ヌクレオチドまたは完全長のプロモータの領域の一部の欠失は過度の実験なしででき、例示したもの以外の活性フラグメントも生成できる。切り詰めたプロモータが「活性」または「コンピテント」である限り、トランケーションの程度、すなわち欠失される塩基対の量は、完全長または元のプロモータの90%までの任意の量にすることができる。すなわち、切り詰めた転写活性プロモータは完全長または元のプロモータに対して塩基対で完全長または元のプロモータの約5%〜約95%、好ましくは約10%〜約90%、より好ましくは約10%〜約60%、最も好ましくは約10%〜約40%であり、具体例では、完全長または元のプロモータの約10%〜約40%である(完全長または元のプロモータの塩基対で約95%〜約5%の欠失、好ましくは約90%〜約10%の欠失、さらに好ましくは約90%〜約60%の欠失、最も好ましくは完全長または元のプロモータの塩基対の約90%〜約60%の欠失)。完全長または元のプロモータで最低限度保持する必要のあるものは、プロモータが挿入されるウィルスまたは系で与えられるトランスアクチベイティング蛋白で転写活性化される最小のプロモータおよび領域である。
【0052】
プロモータの一部の欠失、例えばHCMV-IEの欠失で寸法が小さくなり、HCMV-IEプロモータのようなプロモータの不足および/または寸法の問題とアデノウィルスのパッキング限界寸法の問題を解決することができる。
【0053】
特定の観点から、本発明は91bpの大きさを有するHCMV-IEの活性フラグメントまたは466bpの大きさを有するHCMV-IEの活性フラグメント、すなわち、切り詰めた約91bpの転写活性HCMV-IEまたは切り詰めた約466bpの転写活性HCMV-IEを提供する(本発明は相当塩基対寸法および/または91bpまたは466bpフラグメントに対して相同を有するHCMV-IEまたはHCMV-IEフラグメントを含む。例えば塩基対寸法および/または相同性は91bpまたは466bpフラグメントの少なくとも80%、好ましくは少なくとも85%、さらに好ましくは少なくとも90%、最も好ましくは少なくとも95%である)。このフラグメントはCAV2のようなCAVに挿入できる。従って、本発明はHCMV-IEまたはNCMV-IEの活性断片、例えばHCMV-IEまたはHCMV-IEから誘導される切り詰めた転写活性プロモータ、好ましくは91bpまたは466bpの断片または91のbpまたは466bp断片の実質的な塩基対寸法および/または相同性を有する活性断片から成るCAV2のような組換え型CAVを含む。
【0054】
91bpまたは466bpの断片およびその他のHCMV-IEまたはNCMV-IEプロモータの任意の活性断片を製造するための寸法は上記のHCMV-IEまたはHCMVN−IEプロモータの公知の分子構成(Boshart、1985)から減少させることができる。
例えば91bpまたは466bp断片のような完全長または元のプロモータの上記のような小さい断片が「活性」(上記定義の用語)を有し、それぞれHCMVN−IEの850bp体およびMCMV-IEの766bp体としてCAV、特にCAV2感染細胞で同様な高い転写活性レベルを導くということは驚くべきである。
【0055】
91bpフラグメントすなわち実質的な塩基対の寸法が91bp断片の活性フラグメントアデノウイルス−ベースの組換え型ウィルスにおいて使われていた最小プロモータ要素であると思われるのは特に驚くべきことである。
「活性」フラグメントの生成のためのHCMV-IEおよびMCMV-IEプロモータに適用される以下の説明から、他の成熟核ウィルス、例えばアデノウィルスに外来性である他のヘルペスウィルス、例えばCAV2からHCMVN−IEまたはMCMVN−IE以外のプロモータの「活性」フラグメントをつくることは過度の実験なしで行なうことができる。従って、本発明はアデノウィルスに外来性のプロモータのフラグメント、例えば、アデノウィルスに導入したときにアデノウィルスの完全長プロモータのように活性な切り詰めた転写活性プロモータを提供する。アデノウィルスはCAV2のようなCAVであるのが好ましい。
【0056】
従って、本発明の別の態様で、本発明は鼠CMVN−IE(MCMV-IE)のフラグメント(Dorsh-Hasier達(1985))、例えばアデノウィルス(例えばCAV2)で活性なMCMV-IEに由来するプロモータの切り詰めた転写活性プロモータを提供する。CAV2感染細胞のようなアデノウィルスでは、466bp MCMV-IEプロモータ要素は、91bp のHCMV−IEプロモータ要素のような活性を示す。
【0057】
本発明のさらに他の観点で、本発明はウイルスサイクルの後期に組換え型メッセンジャーRNAの翻訳をするアデノウィルス(例えばCAV2)の活性なプロモータと、このプロモータを含む組換え体と、この組換え体から成る組成物と、プロモータの製造方法および使用方法と、組換え体および組成物とを提供する。そうしたプロモータは、HCMV-IEプロモータまたはその活性フラグメントから成り、5'UTRはヒトのAd2 TPLに置換された。
【0058】
本発明のさらに他の観点で、本発明は組換え型アデノウィルス(例えばCAV2)を製造するためのカセットと、このカセットを含む組換え体と、このカセット製造方法および使用方法と、組換え体と、組成物とを提供する。このカセットは最小化されたポリアデニル化配列(「最小化したポリA」)、例えばSV4Oからの最小化されたポリアデニル化配列(「最小化したSV4OポリA」)から成るのが好ましい。最小化されたSV4OポリAは完全長または元のSV4OポリAより短く、約153bp(±10%)の大きさにすることができる。
【0059】
この種の最小化されたSV4OポリAはアデノウィルス(例えばCAV2)感染細胞で野生型と同じ高条件レベルの安定した安定したメッセンジャーRNAと組合せることができることがわかっている。最小化されたSV4OポリAカセットはアデノウィルスに挿入されるDNAを最小にするために用いることができ、それによって能力不足およびアデノウィルスの諸問題を解決することができる。更に、SV4Oポリアデニル化信号の最小化によって過度の実験無しに他の供与源から他の類似した配列を誘くことができる。
組換え型蛋白の発現のためにアデノウィルスをベースにしたベクターを用いて最適化された寸法および成分を有する発現カセットは今までの文献には記載がないと思われる。
【0060】
本発明のさらに他の観点で、本発明は精製されたアデノウィルス、例えばCAV、好ましくはCAV2 DNAを犬単層中へトランスフェクションするための条件および方法を提供する。
【0061】
本発明の好ましい実施例では、トランスフェクション条件は、El変性した犬細胞系の利用とは独立している。この手順で好ましい収率、精製されたCAV DNAを約5x103pfu/ugの収率で得られる。この手順を用いるとCAV組換え型ウィルスの使用および増殖のためのEl変性細胞の使用を避けることができ、それによってEl変性細胞をに関する安全問題を避けることができる。
従って、本発明は組換え型アデノウィルス、好ましくはCAV、より好ましくは、CAV2と、その製造方法および使用方法と、それらおよびその発現産物を含む組成物を提供する。ゲノムへの組込みまたはゲノムからの欠失に任意の適切な本質的でない領域を使用することができる。そうした部位はE4、ElおよびE3を含む。2つの挿入部位が存在するのが好ましい。その第1のものはE3領域にあり、第2のものは右ITRとE4転写単位(好ましくはSmaI部位)との間に位置し、前者の部位または両方の部位(組合せ)が好ましい。CAV E3 0RF2、例えばCAV2 E2 0RF2が現在のところ最も好ましい。
【0062】
本発明の結果はCAV E3は組織培養の複製に必須ではないことをが証明している。これが組換え型CAVウィルスを誘導する最初の成功した試み表し、従って、組換え型CAV、例えばCAV2をベースとする生産物、例えば、組換え型CAVまたは発現産物を含む免疫学組成物、抗原性組成物またはワクチン組成物のベースが構成される。
従って、本発明はCAV2ゲノムの本質的でない領域に外来性DNA(CAVでは自然には起らないか、挿入部位で自然にCAVでは起らないDNA)を含む合成で修正されたCAV2のようなCAVであると理解できる。
本質的でない領域すなわち必須でない領域はCAV E3またはCAV E3およびSmaI部位のようなゲノムの右終端の両方であるのが好ましい。
【0063】
本発明はさらに、本発明組成物および/または組換え体を用いて得られる抗体とこの抗体の使用と理解することができる。抗体またはその生産物(エピトープ)、抗体から導かれるモノクローナル抗体は抗原または抗体の有無を決定する結合アッセイ、テストまたはキットで使用できる。
本発明で使われるフランキングDNAは挿入部位またはそれに隣接するゲノムの一部にすることができる(ここで、「隣接」とは同じ隣接する配列(例えばコードンまたはコードン群)および多数の配列、例えばコードンまたはコードン群を含み、その前に挿入部位がある)。
【0064】
外来性またはヘテロジニアスなDNA(またはCAVに異質なDNAまたは自然にCAVでは起らないDNA)は上記エピトープのいずれかをコードするDNAであることができる。この点については、Borrelia DNAに関して米国特許第5,523,089号、W093/08306、PCT/U892/08697、Molecular Microbiology(1989)、2(4)、479N486およびPCT公報WO 93/04175および、WO 96/06165を参照。肺炎球菌(pneumococcal)エピトープに関してはBriles達のWO 92/14488を参照。腫瘍ウィルス参照に関してはMolecular Biology of Tumor Viruses, RNA TUMOR VIRUSES(Edition、ワイス達d、Spring Laboratory 1982)(例えば44ページRetroviruses)を参照。他のエピトープをコードするDNAに関しては例えば「発明の背景」で引用した文献:米国特許第5,174,993号、第5,505,941号を参照(例えば、組換え型アビポック(avipox)ウィルス、ワクシニアウィルス;狂犬病グリコプロテイン(G)、遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、ウシ白血病ウィルス、gp5l、30エンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAVN1 env遺伝子、NP(Chicken/ペンシルバニア/1/83インフルエンザウィルスのヌーデオプロテイン(nudeoprotein)遺伝子)、伝染性の気管支炎ウィルスのマトリックスおよびプレポリマー遺伝子;HSV gD; HSV gD;昆虫ポックスウイルス(entomopox) プロモータ、inter alia)、米国特許第5,338,683号, 例えば組換えワクチンウイルス、組換え型アビポック;エントモポックス(entomopox)プロモータ、グリコプロテインをコード化しているDNA、米国特許第5,494,807号、例えば組換え型ワクシニアウィルス、アビポック(avipox)ウィルス;ウサギからの抗原をコードする外来DNA、狂犬病、ヘパチチス(Hepatitis)B、JEV、YF、Dengue、はしか、仮性狂犬病、エピスタインバー(Epstein-Barr)、HSV、HIV、SIV、EHV、BHV、HCMV、犬パルボウィルス、馬風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、特にお多福風邪(NDV):米国特許第5,503,834号、例えば、組換え型ワクシニア、アビポック(avipox)、モリビリウイルス(例えばはしかF、ヘマグルチニン);米国特許第4,722,848号、例えば、組換え型ワクシニアウィルス;HSV tK、グリコプロテイン(例えばgB、gD、風邪HA、ヘパチチス(Hepatitis)B(例えばHBsAg);英国特許第GB 2 269 820 Bおよび米国特許第5,514,375号(組換え型ポックスウィルス;フラビウイルス構造蛋白質);WO92/22641、例えば組換え型ポックスウィルス; WO 92/22641、例えば、組換え型ポックスウィルス; 免疫不全ウィルス;WO 93/03145、例えば組換え型ポックスウィルス; WO 93/03145、例えば、組換え型ポックスウィルス;IBDV; WO 94/16716および1994年1月19日出願の米国特許出願第08/184009号、例えば組換え型ポックスウィルス;サイトカインおよび/または腫瘍関連抗原);PCT/U594/06652(プラスモジウム(Plasmodium)の各ライフサイクル段階からのプラズモディウム)。
【0065】
実施例に記載の組換え体のように、タグおよび他の外来性DNAがCAV2に組み込まれるので、他の外来性DNAをCAV2に組み込むことができる。vCAl、vCA2、vCA3、vCA4、vCA5、vCA6、vCA7、vCA8およびvCA-CDVF1-Ql2bp-up-Smalを作るのに用いる外来性DNAの代わりに、上記文献および/またはここで引用した文献に記載の外来性DNAを使用して追加のCAV2組換え体を作ることができる。すなわち、vCA2のように、vCA8およびvCA-CDVF1-Ql2bp-up-SmaIを介した挿入、vCA2 のように、多重抗原をコードする組換え体を含むvCA8およびvCA-CDVFlQl2bp-up-SmaIを介した欠失(例えばE3中または右ITRとE4転写単位との間の領域または両方の領域への挿入およびE3領域の欠失)(サブフラグメントプロモータ、縮小または変性されたポリアデニル化カセットおよび5'UTR置換プロモータを含む)。分析で発現が示された。脊椎動物(動物または人間)宿主に投与して抗体応答を含む応答を導くための本発明組成物は担体または希釈剤と混合して調製される。
外来性DNAはマーカー(例えば色または光顕マーカー)を含むことができる。外来性DNAはさらに昆虫宿主に有害な生産物をコードすることができ、発現産物が殺虫剤または殺虫剤にすることができる。外来性DNAは抗菌ポリペチド、そのポリペチドおよびDNA上の情報をコードできる(この点に関しては米国特許第5,421,839号を参照)。
【0066】
本発明はさらに、例えばアデノウィルスゲノム、好ましくはCAV、例えばCAV2の必須でない領域の地図を作る方法を提供する。この方法はインビボ遺伝子組換えでドナーDNAをCAVゲノムの一つの領域に挿入できるように、CAV DNAのセグメント内にCAVゲノムの一部と同一直線上にCAV中では自然に生じないDNAから成るドナーDNAを調製し、このドナーDNAをインビボ遺伝子組換えでCAVゲノム中に導入し、組換え体を回収し、安定度およびその生死およびCAV中の自然には起り得ないDNA発現および存在および/または組換え体中の外来性CAV DNAの発現および非存在を決定し、この安定度およびその生死およびCAV中の自然には起り得ないDNA発現および存在および/または組換え体中の外来性CAV DNAの発現および非存在によってDNAが挿入された上記領域が非必須領域であることを示す。この方法は下記実施例で使われる。ドナーDNAをマーカーDNAにして、例えばマーカーDNAのハイブリッド化できるか、マーカーと置換した外来DNAとのハイブリド化ができないかで、ハイブリダイゼーションを用いてそれがゲノムに組み込まれたかどうかを決定することができる。
【0067】
本発明内の上記およびその他の目的および具体化は下記の詳細な説明から明らかになろう。
【発明を実施するための最良の形態】
【0068】
既に述べたように、本発明は組換え型アデノウィルス、CAV、好ましくはCAV2、その製造方法、使用、その発現産物およびその発現カセットに関するものである。本発明は特に、組換え型CAV(例えばCAV2)に関するものである。特に外来性DNAが本質的でない領域および/または本質的でない領域領域に挿入された欠失したものと、その製造方法と、使用または(ベクターまたは複写DNAとしてを含む)それの発現産物および発現産物の使用にある。組込みおよび/または欠失のためにはCAV E3領域、特に0RF2が好ましい。
【0069】
組換え型ウィルスおよびその生産物の使用は、過度の実験なしで発明の背景および討論に発明の要約の下に記載される文献から決定できる。
本発明の組換え体のヘテロジニアスな外来性DNAは、好ましくは下記を含む発現産物をコードする:エピトープ、生化学反応モジュレータ、成育因子、認識配列、治療の遺伝子または融合蛋白。これらの用語に関しては下記を参照:Kendrew、THE ENCYCLOPEDIA OF MOLECULAR BIOLOGY(Blackwell Science Ltd 1995)およびSambrook, Fritsch, Haniatis, Molecular Cloning, A LABORATORY MANUAL(2版 EditioNcoId spring Harbor Laboratory Press、1989)。
【0070】
ワクチンまたは免疫学の組成に用いられる抗原に関しては発明の背景に記載のStedmans Medical Dictionaryを参照(例えば第24版(1982)。例えば、ワクチンの定義(ワクチン製剤において使われる抗原のリスト;この種の抗原または抗原のエピトープが本発明で使用できる。本発明の組換え型ウィルスの発現産物または本発明の組換え型ウィルスまたは組換え型ウィルスを含む多価組成物)。
エピトープに関しては、当業者は過度の実験なしでアミノ酸および対応するペプチドまたはポリペチドのDNAの塩基配列の知識および特定のアミノ酸の特性(例えば大きさ、電荷、その他)およびコードン辞書からエピトープすなわちペプチドまたはポリペチドの免疫領域およびそのコード付けDNAを決定できる。
【0071】
免疫学組成物で蛋白のどの部分を使用するかに関する一般的な方法で所望抗原の寸法と配列を決定する。一般に、大きな蛋白はより多くのポテンシャルを有するので、小さいものよりよい抗原である。より多く外来抗原(寛容性を誘発する自己配位とは違う)は免疫応答を引き起こすのにより効果的である(Ivan Roitt, Essential Innunology, 1988)。
大きさに関しては:当業者はウイルス性ベクターに挿入されるDNAの塩基配列でコードされる蛋白の大きさをベクターのパッケージング限界を考えて最大にすることができる。挿入されるDNAを最小に且つ発現する蛋白の大きさを最大にするために、DNAの塩基配列はイントロン(転写されるが一次RNA転写産物から削除した遺伝子領域)を無しにすることができる。
【0072】
DNAの塩基配列は少なくともペプチド長が少なくとも8または9のアミノ酸をコードできるものである。このペプチドはCD4+T細胞(これがウィルス感染細胞または癌細胞を認識する)を刺激するのに必要な最小長さであるである。CD8+ T細胞応答(病原を飲み込んだ特別な抗原提示細胞を認識する)を刺激するための最小限のペプチド長さは13〜25のアミノ酸である(上記、Kendrewを参照)。これらが極小長さであるが、これらのペプチドは免疫応答すなわち抗体またはT細胞応答をし難いので、保護応答(ワクチン組成物)のためにはそれより長いペプチドにするのが好ましい。
【0073】
配列に関しては、DNA塩基配列は少なくとも抗体応答またはT細胞応答を作り出すペプチド領域をコードするのが好ましい。TおよびB細胞エピトープを決定する1つの方法はエピトープマッピングである。問題の蛋白を蛋白分解酵素で重複ペプチドに断片化し、次に、各ペプチドを検査して、天然蛋白を用いて導いた抗体に結合するか否か、T細胞またはB細胞活性化を誘発するか否かを調べる。T細胞はMHC分子と複合した短い線形のペプチドを認識するので、このアプローチは特にマッピングT細胞エピトープに役立った。B細胞エピトープは線形のアミノ酸配列でなく、むしろ折れた3次元の蛋白の三次構造からできるので、上記方法はB細胞エピトープを決定するのには効果的でない(Janis Kuby、Immunology;(1992)pp. 79N80)。
【0074】
問題のエピトープを決定するための他の方法は親水性である蛋白の領域を選ぶことである。親水性残基は蛋白の外表面上にあることが多いので、抗体にアクセスできる蛋白領域にある(Janis Kuby, Immunology, (1992) p. 81)。
エピトープを決定するさらに他の方法は抗原(全長)抗体錯体のX線結晶学的解析を実行することである(Janis Kuby, Immunology (1992)80ページ)。
T細胞応答を作り出すことができるエピトープを選ぶためのさらに別の方法は、MHC分子が結合し易いことが知られているペプチド配列である蛋白配列潜在HLAアンカー結合単位から同定する方法である。
T細胞応答を作り出すための推定のエピトープであるペプチドはMHC錯体に提示されなければならない。このペプチドは好ましくはMHC分子に接合する適当なアンカー単位を含み、免疫応答を作り出す十分に高い親和性で結合しなければならない。考慮すべきファクタは以下の通りである:免疫化する患者(脊椎動物、動物または人間)のHLA型、蛋白配列、適当なアンカー単位の存在および他の生きた細胞中のペプチド配列。
【0075】
一般に、免疫応答は次のように作り出される:蛋白がより小さいペプチドに切断され、他の細胞の界面に「主要組織適合遺伝子複合体MHC」と呼ばれる錯体が提示された場合にだけT細胞は蛋白を認識する。MHC複合体は2つのクラスIおよびIIがあり、各クラスは多くの異なるアレルから成る。各患者はそれぞれ異なる種類のMHC錯体アレレを有し、「異なる HLAタイプ」とよばれる。
クラスIのMHC複合体はほぼ全ての細胞で見られ、細胞内部で生産された蛋白からのペプチドを有する。すなわち、クラスIのMHC複合体は、ウィルスに感染するか、オンコジーンの発現の結果としガン化した時に細胞を殺す役目をする。表面上にCD4とよばれる蛋白を有するT細胞はこのクラスIのMHC細胞と結合し、リンフォカインを分泌する。リンフォカインは応答を刺激し、細胞はウイルス性感染細胞を殺す。
クラスIIのMHC複合体は抗体提示細胞上にだけ見つかっており、抗体提示細胞細胞でエンドサイトーシスされた循環病原からペプチドを提示するのに用いられる。CD8とよばれる蛋白を有するT細胞はクラスIIのMHC細胞と結合し、溶菌顆粒のエキソサイトーシスで細胞を殺す。
【0076】
蛋白がT細胞応答を刺激するエピトープであるかどうか決定する際のいくつかのガイドラインは以下を含む:ペプチドの長さ:ペプチドはクラスIのMHCに合う少なくとも8または9つのアミノ酸長と、クラスIIのMCH錯体に合った少なくとも13〜25のアミノ酸長でなければならない。この長さはMHC錯体に結合するペプチドの最小長さである。細胞は発現されたペプチドを切るので、これらの長さより長いペプチドであるのが好ましい。ペプチドが免疫応答を作り出す十分に高い特異性で各クラスIおよびIIと結合できるようにする適当なアンカー単位を含まなければならない(Bocchia、H.ほか、Soecific Bindina of Leukemia Oncoacrie Fusion Protein Peptides to HLA Class I Molecules, Blood 85:2680N2684; Englehard、VH, クラスIおよびクラスIIのMHC分子と関連があるペプチドの構造 Ann. Rev. liumunol. 12:181 (1994)を参照)。これは過度の実験なしで MHC分子と関連するペプチドの発表された構造と問題の蛋白の配列とを比較することで行なうことができる。T細胞受容体ペプチドで認識される蛋白エピトープは蛋白質分子の酵素分解で作られ、クラスIまたはクラスIIのMHC分子と一緒に細胞表面上に提示されるペプチドでである。
【0077】
更に、当業者は蛋白データベースにリストアップされた配列と蛋白配列とを比較することで問題のエピトープを確認できる。相同性がほとんどないか、ない蛋白領域はその蛋白のエピトープとして選択でき、ワクチンまたは免疫組成物として有用である。生きた細胞中に広く存在する配列と相同が大きい領域は避けなければならない。
【0078】
さらに他の方法は問題の蛋白の一部を単に作り、発現し、問題の蛋白の一部に対するモノクローナル抗体を作り、それらの抗体が蛋白を誘導した病原のインビトロ成長を抑制するか否かを確認することである。当業者は本明細書に開示および抗体がインビトロ成長を抑制するか否かに関する分析するために問題の蛋白の一部を作り、発現する公知の他のガイドラインを使用できる。例えば、当業者は問題の蛋白の一部を作るのに、8〜9または13〜25のアミノ酸長のその蛋白の一部を選択し、親水性領域を選択し、抗原(全長)−抗体錯体のX線データから結合する部分を選択し、他の蛋白とは違う配列の領域を選択し、潜在的HLAアンカー結合単位を選択する。また、これらの方法またはその他の公知方法を任意に組合せることができる。
抗体が認識するエピトープは蛋白の表面に発現する。抗体応答を刺激しそうな蛋白の領域を決定する最も好ましい方法として当業者は、上記の一般的方法またはその他公知技術のマッピング法を使用してエピトープ地図を作ることができる。
【0079】
上記のように、本明細書の開示および従来法の知識から、過度の実験なしで、当業者はT細胞、B細胞および/または抗体応答を得るためのエピトープのアミノ酸および対応するDNAの塩基配列を確認できる。Gefter達の米国特許第5,019,384号およびそれが引用した文献も参照(注、特に上記特許の「関連文献」部分を参照。この特許の第13欄には多くの重要な生物の多数のエピトープが開示されている。特に、中和抗体が目指すエピトープは重要である。そうしたエピトープは「関連文献」に記載の文献に開示されている)。
生化学反応モジュレータの発現に関しては1993年9月30日公開のWohlstadterのWO 93/19170「選択方法」とそこで引用されている文献を参照。
たとえば、生化学反応モジュレータは生物学的活性度を調整し、例えば、生化学反応モジュレータは非-NMDA興奮性アミノ酸受容体と関連した高分子量蛋白のような調節的な成分であり、アロステリックにAMPA結合の親和性を調整する。本発明の組換え型はそうした高分子量蛋白を発現する(上記Kendrewを参照)。
【0080】
より一般には自然は多数の生化学反応モジュレータを提供している。活性の調節は存在/非共存系、例えば発現/劣化系へのアロステリックに誘発された単純な四元変化として複雑な機構によって実行できる。多くの生体分子の発現の抑制/活性化はそれ自体活性が種々の機構によって調整される分子を用いて調整される。
バクテリア蛋白の化学修飾のリストはNcidhardt達のPhysiology Bacterial Cell(Sinauer Associates社、Publishers、1990)の73ページの表2に示されている。この表が示すようにある変異には適当な会合体が関係し、他の変異では関与がないが、いずれの場合もこの種の変異は機能の変位の原因となる。表から、過度の実験なしで、他の細胞の蛋白の類似した化学的変位を決定できる。
【0081】
生物学的機能の変位は単に分子の適当/不適当な局在化によって調節できる。分子はそれが特定の位置を目標とした場合にだけ、成長に利点または欠点を与えるように機能する。例えば、分子は一般にその分子の機能としてそれを劣化させる酵素の分泌で最初に劣化されると細胞に取りこまれたり、利用されない。従って、組換え型による酵素の生産では細胞による分子の利用または取込みを調節できる。同様に、組換え型は分子の取込みまたは使用に必要な酵素に結合した分子を発現でき、従って、その取込みまたは使用を調節できる。
シグナルペプチドの開裂によって実行される蛋白の局在化のターゲットは別の種類の変更または調節である。この場合は特定のエンドプロテアーゼ触媒活性が組換え型によって発現できる。
【0082】
機能の変化が起こる機構の別の例はRNAウィルスポリ蛋白、アロステリック効果および一般的な共有結合性および非共役な立体障害である。HIVは非機能的ポリ蛋白質構造物を発現するRNAウィルスの典型的な例である。HIV ではgag, pol, env-ポリ蛋白を加工してウィルス構造蛋白p17、p24およびp15−逆転トランスクリプターゼおよびインテグラーゼ−および2つのエンベロップ蛋白gp4lおよびgpl20O(コール達(PNAS USA 85:4686N 90(1988))になる。ポリ蛋白質の適当な開裂はウィルスの複製に重要であり、不活発な突然変異体HIVプロテアーゼを運んでいるウィリオンは非感染性である(同上)。これは活性を低下調節する蛋白フュージョンの他の例である。従って、ある種の蛋白の自然の発現を阻止または増進するために(開裂を阻止するか、増進させて)エンドプロテアーゼと干渉する分子を発現するか、エンドプロテアーゼを出す組換え型ウィルスを造ることが可能である。
【0083】
酵素機能の有用性はその反応に対する触媒作用の能力を変えることで調整することができる。調整された分子の例はチモーゲン、多重サブユニット機能複合体の形成/解離、RNAウィルスポリ蛋白鎖、アロステリック相互作用、一般立体障害(共有結合性および非共有結合性)およびリン酸化、メチル化、アセチル化、アデニル化およびユリデニル化等の各種化学修飾である(上記Neidhardtの表1、315ページおよび表2、73ページを参照)。
【0084】
チモーゲンは酵素活性の変位を生じさせる自然に起こる蛋白融合の例である。チモーゲンは一定の蛋白加水分解により活性状態に変換される蛋白の1つのクラスある。Proteases、Biological Control(第2巻)(1975)、ライヒ、表3、54ページを参照)。自然は一定の酵素、例えばトリプシンの活性をこれらの酵素でアミノ末端で追加の「リーダー」ペプチド配列を発現することで低下調節する機構を開発している。余分なペプチド配列で酵素は不活性チモーゲン状態になる。この配列の開裂でチモーゲンは酵素活性状態に変わる。チモーゲンの全反応速度は対応する酵素の活性の約l05〜106倍である(上記ライヒの54ページの表3を参照)。
従って、ある酵素のその末端の1つに単にペプチド配列を加えるだけで酵素の機能を低下調節することができる。例としてはこの性質の知識を用いて、組換え型が追加のアミノ酸の一旦または両端に含むペプチド配列を発現することができる。
【0085】
多重サブユニット酵素の形成または脱会合は変移を起こさせる他の方法である。多重サブユニット酵素の形成または脱会合に管用する機構は種々ある。
従って、適当な特定のサブユニットの相互作用を立体障害することで触媒活性は低下変移する。従って、本発明の組換え型は生物学的機能を調整するために自然に起る酵素または酵素複合体を立体障害する分子を発現する。
【0086】
いくつかの酵素阻害剤は共有結合性立体障害または変異による機能の低下変異のよい例である。活性サイト中の触媒的に重要なアミノ酸の所で酵素の活性サイトに不可逆的に結合した自殺基質は酵素活性サイトを立体的にブロックする共有結合の例である。酵素自殺基質の例はキモトリプシンのためのTPCKである(Enzyme Structure、Mechanism、2d ed Fritach;フリーマン社(1984))。この種類の変異は適切な酵素自殺基質を発現している組換え型を用いて可能であり、それによって生化学反応を(例えば、酵素活性を制限することで)調整することができる。
【0087】
多くのリプレッサ分子を含む非共有性立体障害のさらに他の例がある。組換え型が立体障害可能なリプレッサ分子を発現でき、従って、特定のDNA-RNAポリメラーゼ干渉を防ぐことによってDNAの塩基配列の機能を低下調整する。
アロステリック効果はある種の生物系において実行される他の変異方法である。アスパラギン酸トランスカルバモイラーゼは良く特徴づけられたアロステリック酵素である。触媒サブユニットとの相互作用が規制ドメインである。CTPまたはUTPに結合で制御サブユニットはホロエンザイムの四次構造変化を誘発し、触媒活性の低下変異が起こる。これとは対照的に、制御サブユニットにATPが結合すると触媒活性が増加変異する(上記、Fritach)。本発明方法を使用すると分子は結合および調節可能な第四または第三の変化を引き起こす発現をする。
【0088】
さらに、各種の化学的修飾、例えばリン酸化、メチル化、アセチル化、アデニル化およびウリデニル化を機能の調整のために行うこともできる。これらの変異は多くの重要な細胞成分の調節で重要な役割をすることは公知である。上記Neidhardtの表2、73ページにはこの種の変異を経た各種の細菌酵素の一覧が示されている。このリストから当業者は過度の実験なしで同じか類似の変異を経て他の系の他の酵素を確認することができる。さらに、人間の疾患に関係する多くの蛋白はこの種の化学修飾を経ることができる。例えば、多くのオンコジーンはリン酸化を用いて変性されるか、リン酸化または脱リン酸化により他の蛋白を修正する。従って、機能、例えばリン酸化を修正または変えることができるモジュレータを発現する本発明によって与えられる能力は重要である。
上記から、当業者は過度の実験なしで本発明を使用して生化学反応モジュレータを発現することができる。
【0089】
本発明の組換え体を用いた融合蛋白の発現に関してはSambrook、Fritsch、Maniatis、Molecular Cloning、A LABORATORY MANUAL(2d EditioNcoIdSpring Harbor LaboratoryPress(1989)(特に第3巻)および上記Kendrewが参照できる。Sambrook達の教えは過度の実験なしで適当に変更でき、当業者は融合蛋白を発現する組換え体を作ることができる。
遺伝子治療および免疫療法に関しては米国特許第4,690,915号および第5,252,479号が参照できる。これらの内容および組成物の引用文献は本願明細書の一部を成す。また、WO 94/16716および1994年1月19日に出願の米国特許出願第第08/184009号とその引用文献は本願明細書の一部を成す。
【0090】
成育因子は組織および機能の個体発生および維持を制御する多機能で局部的に働く細胞間信号ペプチドと定義できる(Kendrew、et seq.、特に455ページを参照)。
成育因子または治療遺伝子は、例えば疾患攻撃蛋白、癌治療分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードできる。さらに、成育因子または治療遺伝子は、例えばαグロビン、β‐グロビン、γ-グロビン、顆粒球マクロファージ-コロニー刺激因子、腫瘍壊死因子、インターロイキン(例えば、インターロイキン1〜14または1〜11またはこれらの任意の組合せの中から選択されるインターロイキン)、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、EPO、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随抗原またはB7から成る群の中から選択できる。米国特許第5,252,479号には遺伝子治療のためにアデノウィルス系で発現できる蛋白のリストが記載されている。当業者はこの開示を見ることができる。WO 94/16716および1994年1月19日に出願の米国特許出願第08/184009号にはサイトカインおよび腫瘍関連抗原のための遺伝子および免疫療法方法が記載されている。当業者はこれらの開示を見ることができる。
【0091】
従って、当業者は本発明の開示および従来法の知識から、過度の実験なしで、成育因子または治療遺伝子を発現する組換え体を作ることができ、組換え体を使用することができる。
上記および従来法の知識から、過度の実験無しに、当業者は生化学反応モジュレータ、成育因子、認識配列、治療遺伝子または融合蛋白のエピトープを発現する本発明お組換え型を作ることができ、また、その組換え型を使用することができる。
【0092】
外来性またはヘテロジニアスなDNAはそれ自体が組換え型CAVでの発現に導くプロモータを含む点に注意されたい。すなわち、外来性DNAは単にコード化DNAであることができ、発現を導く内因性プロモータから適当な下流を配置できる。更に、発現を増幅するかまたは増やすために、コード化DNAの多重コピーまたは強いか早期プロモータまたは早期と後期のプロモータまたはその任意の組合せを使用することができる。従って、外来性またはヘテロジニアスなDNAはE3のような内因性プロモータまたはMLPプロモータに対して最適に配置でき、また、これらのプロモータの位置を変えて外来性またはヘテロジニアスなDNAと一緒に他の位置に挿入できる。コード化DNAは組換え型CAVから複数の生産物を発現するための複数の蛋白のためのコード化DNAであることができる。
発現産物は抗原、免疫源またはエピトープであることができる。従って、本発明は発現産物を含む免疫学的組成物、抗原性組成物またはワクチン組成物に関するものでもある。ある例ではCAVベクターは直接適切な宿主へ投与できるので、更に、本発明はCAV、好ましくはCAV2、ベクターを含む組成物に関するものである。さらに、発現産物はインビトロでCAV、好ましくはCAV2ベクターから単離でき、また、インヴィトロでCAVベクターに感染したまたはトランスフェクションされた細胞から単離できるので、本発明は製品の発現方法にも関するものである。この方法は例えばベクターとしてのCAV中へ外来性DNAを挿入する、例えば、制限/リゲーション組換えを用い、適当な細胞をインヴィトロでCAVで感染またはトランスフェクションし、必要な場合には発現産物をさらに抽出し、精製し、分離することから成る。任意の抽出、精製または単離技術が使用でき、この点に関しては「発明の背景」および「発明の要約」に記載の文献が参照できる。
【0093】
組換え型CAVで感染後、外来性DNAの発現からの蛋白はクロマトグラフィのような公知の技術を用いて回収される(ロビンス、EPA 0162738A1;Panicali、EPA 0261940A2;リチャードソン;スミス達、Pennock達;欧州特許第0265785)回収した蛋白は抗原組成物、免疫組成物または適当な担体を含むワクチンにすることができる。
組換え型CAVは例えば抗原、免疫源、エピトープ、その他の蛋白の製造に使用でき、それから抗原性組成物、免疫学組成物、ワクチン組成物を調製することができる。昆虫の成長または発達に有害な生産物を発現する組換え型CAVは殺虫剤の調製に使用でき、また、植物の成長または発達に有害な生産物を発現する組換え型CAVは除草剤の調製に使用でき(発現産物を単離し、殺虫剤または除草剤としての許容範囲内で担体または希釈剤と混合して)、抗菌類ポリペチドを発現する組換え型CAVは抗菌剤の製造に使用できる(発現産物を単離し、適切な担体または希釈剤と混合して)点に注意されたい。
【0094】
発現産物が抗原性、免疫学的または保護(ワクチン)応答を示すので、本発明はさらに、その生産物すなわち抗体とその使用にも関するものである。発現産物は抗体を導き出すことができる。この抗体はモノクローナル抗体を作ることができ、抗体または発現産物はキット、アッセイ、テスト等で使用できる。従って、本発明はこれらの使用にも関するものである。さらに、本発明の組換え型はDNAの複製に使用できるので、本発明はベクターとしての組換え型CAVと、細胞を組換え型に感染またはトランスフェクションさせ、DNAを回収する方法でもある。得られたDNAはプローブまたはプライマーとして使用できる。
本発明の組換え型CAV、または、その発現産物、例えば免疫学組成物、抗原性組成物またはワクチン組成物または治療組成物は非経口径路(皮内、筋内、皮下)で投与できる。この種の投与は全身的な免疫応答を可能にする。投与は粘膜径路、例えば経口、経鼻、経生殖器、その他)を通して行なうこともできる。そうした投与では局所免疫応答ができる。
【0095】
一般に、本発明の抗原性組成物、免疫学組成物、ワクチン組成物または治療組成物(CAV、好ましくはCAV2、本発明の組換え体または発現産物を含む組成物)は周知の標準的技術によって医薬または獣医技術に熟練した当業者が調製できる。この種の組成物は医学技術に熟練した当業者が周知技術を用いて各種ファクタ、例えば品種、性差、年齢、体重、投与対象疾患の状態および投与径路を考慮した供与量で投与できる。本発明の組成物はそれ単独で投与でき、また、他の抗原性組成物、免疫組成物、ワクチン組成物または治療組成物と一緒または一定順番で投与できる。この種の他の組成物は精製した生来抗原またはエピトープまたは組換え型CAVまたはその他のベクター系を用いて発現した抗原またはエピトープをを含むことができ、上記の点を考慮して投与できる。
本発明の組成物の実施例は体腔、例えば鼻、肛門、口、生殖器、膣、その他用の液状製剤にすることができ、懸濁液、シロップまたはエリキシルで投与でき、非経口、経皮、経皮内、経筋内または経静脈での投与、例えば懸濁液またはエマルションの注射投与用に製造することができる。これら組成物では本発明の組換え型を適切な担体、希釈剤または賦形剤(例えば無菌水、生理食塩液、グルコース、その他)との混合製剤にすることができる。
【0096】
抗原性組成物、免疫学組成物またはワクチン組成物はアジュバントと、発現産物の応答を導き出すための所望量の組換え型CAVとを含むことができる。人間の用途では明礬(リン酸アルミニウムまたは水酸化アルミニウム)が典型的なアジュバントである。家畜の用途において研究、使用されているサポニンおよびその精製された成分Quil A、フロインドの完全アジュバント、その他のアジュバントは毒性を有し、人間のワクチンでの使用には制限がある。化学的に定義された製剤、例えばムラミルジペプチド、モノホスホリルリピドA、ホスホリピド共役物(Goodman-Snitkoff達147:410N415(1991)の記載を参照)、プロテオリポソーム内の蛋白のカプセル化法(ミラー達、メリーランド、176:1739N1744、1992)、そして、ノバソウム(Novasome、登録商標)脂肪小胞(Micro Vescular Systems社、Nashua、NH)のような脂肪小胞蛋白のカプセル化法に使用できる。
【0097】
本発明組成物は、非経口投与(すなわち筋内、皮内、皮下)投与または経体腔投与(例えば経舌)、胃内投与、経口、経肛門、径膣を含む経粘膜で投与ができる。有効投与量は発全製品の種類、組換え型CAV2を用いる場合には発現レベル、その他の公知ファクタ、例えば品種、性差、年齢、ホストの状態および種類、さらにはLD50や公知のスクリーニング法を用いて過度の実験を必要せずに決めることができる。発現産物の有効投与量は数マイクログラムから数百マイクログラム、例えば5〜500μgにすることができる。本発明の発現産物は発現が実現できる任意の投与量で投与できる。すなわち、本発明の組換え型は少なくともこの量で投与され、ワクチンCAV2は約、l03.5 pfuの量、好ましくはl04 pfu〜約106pfuの量を投与できる。他の適切な担体または希釈剤は水または緩衝食塩水で、保存剤を含んでもよい。本発明の発現産物または組換え型CAVは凍結乾燥でき、投与の時点で再懸濁または溶液にすることができる。
【0098】
担体は重合体遅延系にすることができる。組成物を放出制御するには合成ポリマーが有効である。この例は1ミクロンのナノ粒子の球状メタクリル酸メチル重合体である(Kreuter、J.、Microcapsules and Nanoparticles in Medicine and Pharmacology, M. Doribrow(Ed),CRC Press、p. 125N148)。
マイクロカプセル化してマイクロカプセル化された医薬品を注射し、放出制御することができる。マイクロカプセル化のポリマーの選択には多数のファクタがある。ポリマー合成およびマイクロカプセル化プロセスの再現性、マイクロカプセル化材料およびプロセスのコスト、毒物学プロファイル、ポリマーの放出速度、およびポリマーと抗原の物理化学的適合性等の全てのファクタを考慮する必要がある。有用なポリマーの例はポリカーボネート、ポリエステル、ポリウレタン、ポリオルトエステルおよびポリアミド、特に(生物分解可能なものである。
【0099】
医薬品用および抗原用に最近選択される担体はポリアクリル酸エステル(d,1-ラクチド-co-グリコリド)(PLGA)である。これは侵食性縫合、硬骨プレート、その他の一次的人工器官の医療で長い歴史を有する生物分解可能なポリエステルで、毒性はない。各種医薬品はPLGAマイクロカプセル化されたペプチドおよび抗原を含む。抗原の制御放出用のPLGAのデータは例えばEldridge, J.H. et al、Current Topics in Microbiology and Immunology 1989、146:59-66に記載されている。直径1〜10ミクロンのPLGAのミクロスフィアの抗原の包括体が経口投与時に特に有効なアジュバント効果を示す。PLGAマイクロカプセル化プロセスには油中水エマルションの相分離を使用する。問題の化合物は水溶液として調製され、PLGAは適当な有機溶媒、例えばジクロロメタンおよび酢酸エチル中に溶かされる。これらの2つの非相溶性の溶液は高速攪拌機を用いてエマルジョン化される。次いでポリマーの非溶剤を加え、水溶液滴のまわりにポリマーを沈降させて胎生期マイクロカプセルを形成する。マイクロカプセルを回収し、ポリビニールアルコール(PVA)、ゼラチン、アルギナート、ポリビニルピロリドン(PVP)、メチル-フルロースで安定させ、真空乾燥または液液抽出法で溶剤を除去する。
【0100】
固形物を含む液体、液体およびゲル(gel caps)を含む固形物組成物が得られる。また、本発明の組換え型CAV2およびその発現産物は動物において免疫性すなわち抗体応答を刺激できる。これら抗体から周知の技術を用いてモノクローナル抗体が調製でき、そのモノクローナル抗体は周知の抗体結合アッセイ、診断キットまたは抗原の有無およびそこから抗原の自然に起因する因子の有無を決定し、免疫応答がその抗原または因子に単に刺激されたかどうか否かのテストで使用できる。
モノクローナル抗体はハイブリドーマ細胞を用いて得られる免疫グロブリンである。モノクローナル抗体は単一の抗原決定基と化学反応し、従来の血漿誘導抗体よりも大きい特異性を与える。さらに、多数のモノクローナル抗体を使用することによって所望の特異性、結合活性およびイソタイプを有する個体抗体を選択できる。ハイブリドーマ細胞株は化学的に全く同じ一定の抗体の安い供与源を提供し、この種の抗体の製造は簡単に標準化できる。モノクローナル抗体を生産する法は当業者に周知で、例えばKoprowski、H.達の1989年4月1日発行の米国特許第4,196,265号に記載されている。その内容は本明細書に引用したものとする。
モノクローナル抗体の使用は公知で、その使用の例は1983年3月8日発行のH.デイビッド、G.グリーンの米国特許第4,376,110号に記載されている。
モノクローナル抗体は免疫吸着クロマトグラフィによる材料の回収にも費用できる。例えばミルシテイン、C.、1980、Scientific American 243:66、70を参照。
【0101】
本発明の組換え型CAVまたはその発現産物は、インビトロでまたはインビボでの細胞の刺激と、その患者への自己輸血で使用できる。患者が血清陰性な場合には自己輸血は免疫応答を刺激し、例えば能動免疫のような免疫応答または抗原性応答をする。血清陽性な個体では自己輸血は病原に対して免疫系を刺激またはブーストする。
本発明の組換え型CAVはさらに、プローブまたはPCRプライマのためのDNAを作るのに有用であり、ハイブリット可能なDNAの有無の検出、DNAの増幅でき、例えば、サンプル中の病原を検出でき、また、DNAを増幅できる。
【0102】
本発明は、アデノウィルス系およびトランスアクチベイティング蛋白を与える任意のウイルス性または細胞系に有用なプロモータおよび発現カセットをに関するものである。このプロモータは切り詰めた転写活性プロモータまたは組換え型で、プラスミドが挿入されたウィルスまたは系によって与えられたトアンスアクチベイト蛋白でトアンスアクチベイトされた領域と切り詰めた転写活性プロモータが誘導される完全長プロモータの最小プロモーター領域とから成るのが好ましい。
【0103】
本発明のプロモータはヘルペスウィルス、例えばMCMV-IEまたはMCMVN−IEプロモータのような、MCMVまたはMCMVのような完全成熟ウイルスから誘導し、塩基対にベースで完全長プロモータの40%、さらには90%まで短くしたものであるのが好ましい。
【0104】
本発明の発現カセットは機能的な切り詰めたポリアデニル化信号、例えば切り詰めても機能するSV4Oポリアデニル化信号含むことができる。この発現カセットは(プロモータまたは発現カセットが挿入されているウィルスまたは系に対して)外来性またはヘテロジニアスなDNA、例えば上記の外来性またはヘテロジニアスなコード化DNAを含むことができる。このDNAは最適に配置され、リンクされて発現用プロモータとして使用することができる。発現カセットは任意の位置、好ましくは発現カセットが挿入された系またはウィルスから最高の発現が得られる位置に挿入できる。
【0105】
プロモータおよび発現カセットを特にアデノウィルスに関して説明したが、当業者は単にウィルス、プラスミド、細胞または系がトランスアクチベイティング蛋白を提供するか否かを確認するだけで、過度の実験のなしに、本発明を細胞、例えば真核細胞に対する他のウィルスおよびプラスミドに適応できる。
HCNVプロモータに関しては米国特許第5,168,062および5,385,839を参照(本願明細書に引用したもの)。発現のためのプラスミドDNAを有する細胞をそこからトランスフェクションすることに関してはFelgner達J. Biol.chem. 269, 1994, 2550-2561を参照。いろいろな感染症を防ぐためのワクチン接種の単純かつ効果的な方法、プラスミドDNAの直接注射に関してはScience、259:1745-49、1993を参照。本発明プロモータおよび発現カセットはアデノウィルス以外の系;例えばプラスミドDNAの直接注射で使われるプラスミドも本発明の範囲内である。
【0106】
本発明の具体化のためにの他の利用も存在する。
以下の本発明の実施例を説明するが、本発明が下記実施例に限定されるものではない。
【実施例】
【0107】
実施例1
ウイルスおよび細胞系の同定
イヌアデノウィルスタイプ2(CAV2)株は、ローヌメリュー(Rhone Merieux)社(アテネ(Ga))から参照番号CAV2ロット# 0830プールN 033093生産されたもので、タイターは107.4 TCID50/ml。また、Hadinおよびダービー犬腎臓(MDCK)細胞系はRhone Herieux Incから提供された。このCAV2は犬ワクチンとしてローヌメリュー社から市販されている。
【0108】
実施例2
ウィルス培養とコロニー化
MDCK細胞懸濁液をMEM(Gibco、グランドアイランド、NY)に種付けし、7.5%ウシ胎児血清(シグマ、Stルイ、HO)、ナトリウム・ピルベート(Gibco(1mM最終))、グルタミン(Gibco(2mM最終))、ペニシリン(Gibco(50のU/ml))、ストレプトマイシン(50mg/ml、Gibco)および非必須アミノ酸(NRA)(Gibco、0.1 mM最終)を供給し、5% CO2中で37itchで培養した。合体したMDCK細胞をCAV2の希釈液で感染させ、0.6%アガロース・オーバーレ下で、5% CO2中で37itchで培養した。CAV2はいくつかのラウンドでプラーク精製した。プラーク精製されたCAV2はT25 MDCKフラスコで培養した。培養CPEが完了したときに感染細胞を集め、アガロース下のMDCK細胞単層上でCAV2含量を定量した。ウィルス株はT175 MDCKフラスコを0.1の感染多重度(HOI)に感染させることで更に増殖させた。T-175 MDCKフラスコ増殖させたウィルスの力価は108p.f.u/mlであった。
【0109】
実施例3
ウィルスDNA精製
合体したHDCK細胞単層を含んでいるローラーボトル(108細胞/ボトル)を0.1pfu/細胞のプラーク精製されたCAV2ウィルスで感染させた。3日後に感染した単層は取り入まれ、低速遠心(1Kg、15分、l5℃)した。細胞ペレットは‐7O℃で保存された。次いで、凍ったペレットを37℃で解凍し、慎重に10mMトリスHCl pH 8.0と10mM EDTA緩衝液に懸濁し、細胞のDNAシアリングを制限した(35ml/108細胞)。再懸濁したペレットにSDSを加え、1%の最終濃度にした。室温で15分間培養した後、NaClを加えて1.25Mno濃度にした。4℃で3時間培養後、4℃、25Kgで20分間遠心分離した。塩を含んでいる濃い白いペレットとび細胞のDNAは捨て、上澄を42℃で4時間、次いで、60℃で30分間のProteinase K(300μg/ml最終濃度)で消化した。ウィルスDNAの回収の前に石炭酸-クロロホルムおよびクロロホルム抽出を二回行い、0.3M酢酸ナトリウムpH 6.0の存在で70%エタノールで沈殿させた。ウィルスDNAペレットを1時間風乾し、次いで2mlの水に再懸濁した。この手順で典型的には約4mgの精製されたCAV2 DNAを生ずる。精製されたウィルスDNAは利用まで-2O℃で保存した。
【0110】
実施例4
ウィルスDNA制限解析
精製されたCAV2 DNAのアリクォットをBoehringer Manriheim社(インディアナポリス(IN))の一組の限定酵素でメーカー明細に従って消化した。制限DNAサンプルは1%アガロースゲル上で電気泳動を用いて分別し、対応する制限フラグメントはUV光下でエチジウムブロマイド(4つの~g/ml)でゲル染色して視覚化した。表1に各種制限フラグメントの大きさを要約した。
【0111】
実施例5
E3を含む制限フラグメントの同定と特徴付け
1. CAV2 DNAのエンドヌクレアーゼのサザンブロット分析
それぞれ精製されたCAV2 DNAの4μgアリクォットをBamHI、Bg1I、HindII、HindIIIおよびPastIで消化し、1%アガロースゲルによる電気泳動で分画した。ゲルは、0.25MのHClに30分漬けた後、H20 で5分間洗った。次いで、ウィルスDNAを0.5M NaOHと0.9M NaCl溶液で変性した。H20 で5分間洗った後、DNAは3M NaClを含んだ0.5MトリスHCl( pH 7.5)の2つのバスで再度変性した。
DNAはナイロンメンブレン(Hybond N、Amersham Life Sciences、クリーブランド、OH)上でlOxSSC(1.5MのNaCl、0.15MのNa Citrate pH 7.4)緩衝中で一晩おいた。ナイロンメンブレンは1時間風乾し、3分間UV架橋した。65℃で6時間4xSSC、25% Denhardt's溶液(v/v)、0.1% SDS(v/v)、0.1% Naピロ燐酸塩および変性heringDNA溶液(500のmg/ml)で予備ハイブリッド化した。
【0112】
2. CAV2 PVIIIおよびFiber遺伝子に特有のプローブの製造
大部分のアデノウィルスのE3領域は2つの構造遺伝子、PVIIIおよび繊維の間にあるので、本出願人は各遺伝子のための2つのプライマ対を設計する(ライム、1992)ためにCAV2(マンハッタン株)の既に発表されている部分鎖のゲノムを利用した。オリゴヌクレオチドLF189(5'-TCAGTCATAGCCATCGACAGA-3')(配列番号:26)およびLFl9O(5'-GTGCTGGCTGGCACGGGCATT-3')(配列番号:27)およびオリゴヌクレオチドLFl91(5'-ATGTCCACCAAAGTCCCCTCTN‐3')(配列番号:28)はCAV2 PVIII遺伝子の3'端内の配列に対応するように設計され、LF192(5'-CCCGGGGCGTCGTATGGATAT‐3')(配列番号:29)はCAV2繊維遺伝子の5'端内の配列に対応するように設計された。
302bp のDNA PVIIIプローブは、lOX PCR緩衝、2mM dNTPsの3.75μl、26μlのH20、Taqポリメラーゼ(5.0u/μl)の0.25μl、5μM5'端HプライマLF189および5μlの5μM 3'端のプライマLF190と混合して作った。増殖は90℃で1分、55℃で1分、72℃で1分のプロフィルで40μlの鉱油を含む0.5mlの間で30回行った。190bpのDNA 繊維プローブはプライマLFl91でプライマLF189を交換することで上記プロトコールでVPCRで作られた。両方のPCR反応は1%アガロースゲルで電気透析され、対応するPCR生産物はメーカー指定の手順を使用して単離した(Bio 101, Inc., La Jolla, CA)。各プローブの100ngのアリクォットをlμgのランダムな六量体(Pharmacia, Piscataway, NJ)(全体積13μl)と混合し、3分間沸騰し、2.5μ1のdCTP 、dTTPおよびdGTP混合液(各0.5M濃度)、2.3μlのクレノーlOX緩衝液、1.5μlのクレノウ酵素(2u/μl)および5μlの32P−32P‐a‐ dATP(300℃i/mmol、10mCi/ml、NEN、ボストン、MA)で4時間培養した。反応はStop溶液(IBI Prime Timeキット)の100μlを加えることで停止した。各プローブの25μlを3分間50mlのprehybridization溶液の全体積で上記ナイロンメンブレンで65℃で一晩培養した後、変性した(100℃)。このナイロンメンブレンを6xSSC、0.1% SDSおよび2時間の50mM Na Pyrophosphate溶液の65℃で、次いで、蒸留水に通した。放射性同位元素でラベル化されたDNAプローブに対するウィルスDNA制限フラグメント相補性はオートラジオグラフィを用いて同定した。
【0113】
3. E3領域を含んだ制限フラグメントの同定およびクローン化
最も短いものは両方のPVIIIおよびFiberプローブで認識される制限フラグメントをよく単離するので、HindIllフラグメントA(4.0のKbp)が同定され、全CAV2 E3領域を含むことができることを示している。このフラグメントは上記遺伝子Clean手順を用して単離され、次いで、プラスミドpLFO27生成ベクターpBluescript SK+のHinndIII部位にサブクローン化される(Strantagene, La Jolla, CA)。
【0114】
4. CAV2 E3領域の特徴付け
CAV2 E3領域は、pLF027の規制変異消化作用と、配列2.0のキット指示(US Bi℃hemical、クリーブランド、OH)に従うシークエンシングpLF027を用いて分析された。配列解析はMacVectorソフトウェア(イーストマン・コダック、ロチェスター、NY)を使用して実行された。pLF027制限地図は図 2で示される。CAV2 E3領域を含むpLF027の対応配列(PVIIIストップコードン(pLFO27の#1413)および繊維ATG開始コードン間のDNA範囲として定義する(pLFO27)の#2945は図1に表される。シークエンシング・データの解析は、CAV2 E3が1,533 bps 100%以前に同定されたCAV2(マンハッタン株)E3領域(ライム(1992))については相応することを明らかにした。ヌクレオチド配列から推論されるアミノ酸配列の解析はCAV2 E3領域の右へのコード付けストランドが2つの潜在的なポリペチド(ORF1および0RF2)をコード化することを明らかにし、左へのコード付けストランドは単極電位ポリペチド(ORF3)をコード化する。これらのORFsの形質は表2で示される。
【0115】
実施例6
ドナー・プラスミドnLFOS6の生成
1. CAV2 E3配列の中央へのBglIIおよびMluI制限サイトの導入
取扱いを容易にするためにユニークなBglIIおよびMluI制限サイトを含む24 bp DNAリンカ(5'-GATACGCGTTCCATTAGCAGATCT-3')(配列番号:30)を、CAV2 E3領域(図1にて説明したように)のヌクレオチド#1487とび#1966との間にダブルラウンドPCR増殖手順を用いて導いた。最初のPCR増殖は鋳型としてpLF027 DNAを使用し、以下のプライマ一対を使用して実行した(LF327(5'-GGACACCTTTCTGATCAGTTCATT-3')/ LF3 24(5'‐GATACGCGTTCCATTAGCAGATCTTTGAGGGGC CTGGAAATAGGC-3')(配列番号:31、32)]、(LF326(5'−GGTTGTGTGGAAGACCCGGG GGCG-3')/LF325(5'-AGATCTGCTAATGGAA CGCGTATCGTGCCCCCACAGTACAGCAA-3')(配列番号:33、34)。それぞれ838bpおよび956bpの2つの部分的に重なり合うDNAフラグメントを作る。PCR増殖の第2ラウンドは部分的に重なり合う精製されたDNAフラグメントおよび両方の外部のプライマLF327およびLF326の存在下で実行された。得られた1,794bp DNAフラグメントはPstIおよびAatIで消化された。その結果生じる890bp の PstI/AatI フラグメントを精製し、6,069bp のPstI/ AatIl DNAフラグメントで連鎖反応させてpLFO27(pLF047A(図3および4)を作る。全てのPCR増殖は、上記条件を使用して実行された。次いで、6,944bp のMluI/BglII pLF047AをprearinealedしたオリゴヌクレオチドLF328(5'-GATCTGTTAACCCTAAGGCCATGGCATATGTCGCGAGGCCATCGTGGCCGCGGCCGC A-3')(配列番号:35)およびLF329(5'-CGCGTGCGGCCGCGGCCACGATGGCCTCGCGACATATGCCATGGCCTTAGGGTTA ACA-3')(配列番号:36)と連鎖反応させてpLF049A(図5および6)を作る。この結果、60bpのCAV2 E3領域は60bp BglII/HluIポリリンカDNAフラグメントと交換する。E3領域の大きさは修正されず、E3 ORF1はごく自然なままだった。しかし、E3 ORF2に対応する配列は分裂させられ、 E3 ORF3は完全に除去された。
【0116】
2. ドナー・プラスミドpLFO86の生成
CAV2 E3領域の一部を欠失するために、428bpの欠失がpLF049A MluI部位の3'を設計した。537bp DNAフラグメントをpLF027鋳型およびプライマ対LF361(5'-CTAGTCATCTTAACGCGTGTCCTCAACATCACCCGCGA-3')/LF334(5'-CTT GCTTGTTATTAAAAAAAG-3')(配列番号:37、38)を上記のように用いてPCRを用いて作った。この551bpフラグメントはMluIおよびAatIIで消化されて、精製され、pLFO49A の6,284bp のMluI/AatII DNAフラグメントと連鎖反応させてpLFO86(図7および8)を作る。この処理でE3領域の27%(428bp)が欠失し、更にその3'端の方のE3 ORF2にの欠失が広がるが、E3 ORF1暗号配列の邪魔はしない。
【0117】
実施例7
ウィルスゲノムの右の端を含む制限フラグメントのクローン化および特徴付け
1. ウィルスゲノムの右端む制限フラグメントのクローン化
CAV2(グラスゴー株)ゲノムの以前に発表された制限地図は84.0のマップ単位(SpibeyおよびCavanagh 1989)に位置するユニークなSalI制限サイトの存在を示した。CAV2 DNA(30μg)のSalI消化作用は、予測された3.2kbpおよび29kbp DNAフラグメントを作った。CAV2 DNA SalI Bフラグメント(3.2kbp)は、前述したように、ジーンClean手順を使用して精製されるゲルであって、20μ1のH20に予備懸濁した。約、3μgの精製されたSalI Bフラグメントが、22μ1 の1N NaOH (全体積22μ1)に90分で添加され、室温で5'-アデノウィルス・ゲノムの末端)に共有結合した蛋白部分(ロビンソン達(1973))を除去するために変成した。DNAは、2MのトリスHCl pH 7.5の1.3μ1を添加し、次いで、変形されて65℃で1時間培養され、pBluescript SK+の2.919bp SalI/SmaIフラグメント連鎖反応されて、pLFOS6を作った。
【0118】
2. ウィルスゲノムの右の端を含む制限フラグメントの性格付け
CAV2ゲノムのbp右の3.2K端は、pLF056の規制変異消化作用を用いて、そして、Sequenase 2.0のキット規定に従う同じプラスミドのシークエンシングを用いて分析された。配列解析は、MacVectorソフトウェアを使用して実行された。
pLF056制限地図は図10に示される、そして、図9はDNAの塩基配列を示す。
シークエンシング・データは、CAV2 DNA SalI Bフラグメントが長さの3,274bpであることを明らかにした。CAV2ゲノム内の2つのユニークな制限サイトは、CAV2 DNA SalI Bフラグメント内に局所化された:分類学的位置#587でのIglLtおよび分類学的位置#2133でのSalI。CAV2のヌクレオチド配列が右の末端で位置させた196のbp ITR(図9)が、100%、CAV2 Vaxitasおよびグラスゴー株のためにCAV2左右のITR配列については相応する(Cavanagh達1991)。残りのCAV2のDNAの塩基配列対CAV2 SalINBフラグメントDNAの解析から45%相同だけが示された。
【0119】
実施例8
PLF06lの生成
NruI/EcoRV 312のbpタグDNAフラグメント(図11)は、SmaI線形にされたpLF056についてはpLF06l(図11;図12に示される制限地図)を作り出すために連鎖反応させられた。
【0120】
実施例9
MDCIへの精製されたウィルスDNAのトランスフェクション
溶液Aは5μgの精製されたCAV2 DNAと血清遊離HEM(前述したように最終体積が300μlになるように補う)を混合して調製した。
溶液Bは、40μlのLipofectamine試薬(Gibco)に260μlの血清を含まないHEM培地を補って調製した。溶液AおよびBを一緒に混ぜ、30分間の室温で培養した。CAV2 DNA/リポソーム複合体に2.4mlのHEM培地(血清遊離)を混合して補った後、75%のHDCK細胞単層を合せた。5% C02の存在下、37℃で24時間インキュベーション後、血清を含まない培地を除去し、5% CO2を含む3mlのHEM培地と交換した。培養は5% CO2の存在下で37℃で8日間行ない、3日目に2mlのHEM培地を補った。このインキュベーションの間、CPEなしを証拠としてもよい。8日目にトランスフェクトされたMDCK細胞を掻き取り、全体積5mlにした。氷上で2分間超音波処理を2回した後、2mlのトランスフェクトされた培養を用いて、5% CO2の存在下で37℃で1時間、150mm直径の組織培養皿で100% MDCK単層を感染させた。その後、培養を0.6%アガロースを含む培地で行った。5日以後、5%CO2の存在下で37℃でプラークが現れる。観測される典型的な収率は10μgの精製されたDNA当り少なくとも2,000pfuである。
【0121】
実施例10
組換え型CAV2ウィルスの生成
1. 組換え型CAV2ゲノムのインビトロ生成
精製された20μg のCAV2 DNAを30U のSalIで一晩37℃で消化した。消化されたDNAは石炭酸クロロホルムで抽出て、H20中でエチルアルコールに再懸濁し、370ng/μl濃度にした。5μg のSalIで消化されたCAV2 DNAを5μgの3,557bp SalI/SacI pLF06l DNAフラグメントで400UのDNAリガーゼ(NEB、ベヴァリー、MA)の存在下でl5℃で一晩全体積50μ1にして結合反応した。
【0122】
2. CAV2組換え型ウィルスvCA1の単離
次いで、全部の結合反応を用いて75% MDCK単層をトランスフェクションした。収穫したトランスフェクトされた4mlの培養物を用いて2つの150のmm直径組織培養皿を感染させた。10日インキュベーションした後に合計8つのプラークが現れた。全てのプラークは回収し、1mlの追加のHEM培地に再兼濁し、氷上で2x 2'間、超音波処理した。浄化した培地を希釈し、連続的に60-mm直径組織培養皿で100% MDCK細胞単層を感染させた。6日の培養後、アガロースオーバーレを捨て、感染した単層をPerkus達1993に記載の手順でニトロセルロースフィルタ上へに塗布した。濾過処理し、ラベル化したNruI/EcorV 312bpのタグDNAフラグメントと上記の通常の方法でハイブリッドした。オートラジオグラフィ実験で初めに検出された8つのプラークの中の5つが組換え型CAV2ウィルスを含むことがわかった。プラークハイブリダイゼーションを用いて同定された1つのプラークを単離し、MDCK細胞上でプラーク精製を繰り返した。プローブとのハイブリダイゼーションを各精製後に確認した。プラーク精製した組換え型CAV2ウィルスにはvCAlという名前をつけた。
【0123】
3. vCAlの性格付け
vCAlを特徴づけるために小規模なDNA精製を行った。手短に言うと、精製されたvCAl組換え型ウィルスを100% MDCK単層(106の細胞)を感染させるために用いた。5日以後、CPEが完了後、感染した培養物を掻き取り、収穫した。超音波処理および浄化した培地を42℃で2時間、proteiriase K(500μg/mi終濃度)で処理した。この酵素を65℃で20分間加熱して機能をなくし、次いで、全DNAを石炭酸クロロホルムで抽出し、H20でエチルアルコールに再懸濁した。精製された全DNAをRNアーゼT1で処理し、石炭酸クロロホルムで抽出し、エチルアルコールで沈殿させ、H20中に再懸濁し、最終濃度を1.2μg/mlとした。5μgの精製されたvCAlのアリクォットをそれぞれBglII/SpeIで消化した。これらの2つの部位はCAV2ゲノム中でにユニークであるので、29kbpおよび3kbpのフラグメントはBg BglIIでの消化が予想される、30.5kbpおよび1.5kbpフラグメントはSpeIの消化が予想される。これら制限フラグメントは、vCA1がそのゲノムの右端内に300bpのヘテロジニアスなDNAを取り入れた組換え型CAV2ウィルスであることを証明している。
vCAlが本当に予想されたタグDNAフラグメントを取り入れたことを更に証明するために、VCAl DNAをサザンブロッティングを用いて分析した。そして、vCAlがタグDNAフラグメントを取り入れたことを確認した。
CAV2 Smalが挿入部位として使われことを確認するために、精製したvCA1 DNAからの1.9kbpDNAフラグメントをプライマLF379(5'-TCACGCCCTGGTCAGGGTGTT-3')(配列番号:39)およびLF407(5'-GCCATCGCGTCAACCTGA-3')(配列番号:40)の対で増殖した。
プライマLF63(5'-ATGATGTCTGGGGACATGN3')(配列番号:41)、LF379(5'-TCACGCCCTGGTCAGGGTGTT-3')(配列番号:42)およびLF384(5'-ACCACGCGCCCACATTTT-3')(配列番号:43)を使用した1.940bp DNAフラグメントの部分鎖解析からヘテロジニアスなタグDNAがCAV2 SmaI部位に本当に挿入されvCAlになったことが確認された。
【0124】
実施例11
組換え型CAV2ウィルスの生成
10μgのpLFO86をHindIIIで消化し、得られた3.6kbp DNAフラグメントを上記ジーンClean手順を使用して単離し、H20中に再懸濁し、最終濃度を100ng/μlにした。MDCK細胞を上記Lipofectamineをベースにした手順を使用してトランスフェクションした。溶液Aは3μgの精製したCAV2 DNAと0.5μgの3.600bp HindIIIDNAフラグメントとを混合して調製した。血清を含まないHEM培地を補って溶液Aの全容積を300μlにした。Tトランスフェクションされた細胞を8日目に収穫し、150mm直径組織培養皿上の付けた。上記能方法でプラークを5'端をラベル化したオリゴヌクレオチドLF328とハイブリッドした。プローブと交差反応させた5つのウイルス性プラークを掻き取り、プラーク精製を4回行った。プラーク精製した組換え型CAV2ウィルスにvCA2という名前を付けた(プラーク精製はDNAまたはウィルスの複製での組換え型の使用、すなわち組換え型のベクターの使用で、外来性DNAの制限または限界がないことを示す点に注意)。
【0125】
2. vCA2の性格付け
vCA2を特徴づけるために上記方法でvCAlの小規模DNA精製を実行した。精製したvCA2 DNAおよび野生型CAV2 DNAはHindIIIを用いてそれぞれに消化し、制限されたDNAを1%アガロースゲルで電気泳動した。VCA2サンプル中の3.6kbp HindIIIフラグメントが視覚化された。野性-型CAV2サンプル中には4.0kbpフラグメントが存在するので、vCA2ゲノム中で428bpのE3領域が欠失したことが証明された。
予想されたタグ(オリゴヌクレオチドLF328/LF329)が本当にvCA2 E3領域に組み込まれたことを更に証明するために、Southernブロットが実行した。その結果、タグの移入が確認された。
この結果は完全なCAV2 E3 ORF2が組織培養に必要でないことを示している。さらに、CAV2 E3 ORF2配列の一部がヘテロジニアスなDNAに置換でき、従って、CAV2ゲノム中にに第2の挿入部位がることを証明している。この結果はさらにCAV2 E3領域の一部が欠失でき、vCA1の所で述べたSmal部位に外来性DNAを導入して補償できることを証明している。
【0126】
実施例 12
再断片化プロモータ、短く変性されたポリアデニル化カセット、5'UTR置換プロモータおよびプラスミドおよびそれを含む組換え体の生成
1.1 pLFO22(CATリポータ遺伝子がHCMV-IEプロモータの再断片化(145bp)の制御下に置かれた発現ベクトル)の生成:
ヒトサイトメガロウイルス(hCMV)(タウン株)からのDNAをLafemina達(1989)に記載の方法で調製された。プライマ組LFl7 2(5'(ATCGTAAAGCTTAATGTCGTAATAACCCCGC-3')/LF159(5'-TCTACTGCAGCCGGTGTCTTCTATGGAG GTCA-3')およびhCMV DNA(lOng)を鋳型として使用して、ヒトサイトメガロウイルス即時早期プロモータ(hCMVNIE)の3'端の増殖をPCRを用いて上記方法で実行した。166bp DNAフラグメントをジーンClean手順を使用して精製し、PstI/HindIIIで消化し、pCAT-Basicベクター(Promega、マディソン、WI)の4,348bp PstI/HindIIIDNAフラグメントで直接連鎖反応させ、pLF022(図13、14(配列番号:7))を作った。pLF022発現カセット中に存在する調節配列はhCMV IEプロモータおよびSV4O小さいt抗原およびポリアデニル化信号を含んでいる856bpカセットの145bpフラグメントである。
【0127】
1.2 pLFO62(SV4Oポリアデニル化カセットが241bpに縮小されたpLF022の誘導体)の生成:
SV4O小さいt抗原の寸法およびpLFO22のポリアデニル化信号カセット(856bp)を減らすために、以下の処理を実行した。170bp DNAフラグメントはプライマLF377(5'-TCTTCGCCCCCGTTTTCACCATGG-3')およびLF378(5'-ATCACGCCGCGGCTTAAAAAAA TTACGCCCCGCCCT-3')およびpLF022 DNA(長く続く)を鋳型としてPCRを用いて増殖した。精製された増殖されたフラグメントを18mlのH20に再懸濁し、室温でまたは30分間、800μMのdNTPs存在下で1lUのクレノウ酵素(Boehringer Manriheim、インディアナポリス、IN)で培養した。変性されたDNAフラグメントを石炭酸-クロロホルムで抽出し、エタノール沈殿し、NcoIで消化し、回収した。得られた136bpフラグメントを3,655bp NcoI/BsaBI DNAフラグメントと連鎖反応してpLFO22(pLF062(図15、16(配列番号:8))を作った。pLF062はCAT遺伝子の下流に2つのコンセンサスポリアデニル化信号AATAAAの反複を含む。pLF062のCAT発現カセットの寸法はpLFO22の1,804bpと比較して1,119bpである。pLF062カセット発現カセット中の調節配列は、hCMV-IEプロモータの145bpフラグメントおよび241bp のSV4Oポリアデニル化信号を含んでいる。
【0128】
1.3 Ad2 TPLがHCMV-IEプロモータのクローン化した下流であるpLF062誘導体 pLFO66の生成:
CAV2複製の開始後にリポータ遺伝子の発現を許すためにhCMV-IEプロモータ転写開始部位の下流のヒトAd2三分節リーダー(Ad2 TPL)をクローン化して変性されたpLF062 CAT発現カセット
オリゴヌクレオチドSPH6ETr1(5'-AATTCGGTACCAAGCTTCTTTATTCTATACTTAAAAAGTGA AAATAAATACAAAGGTTCTTGACTCTCTTC-3O、SPH6ETr2(5'-CGCATCGCTGTCTGCGAGGGCCAGCTGT TGGGCTCGCGGTTGAGGACAAACTCTTCGCGGTCTTTCCAGT-3')、SPH6ETr3(5'-ACTCTTGGATCGGA AACCCGTCGGCCTCCGAACGTACTCCGCCACCGAGGGACCTGAGCGAGTCCGCATC-3')、SPH6ETr4(5'-GACCGGATCGGAAAACCTTCGAGAAAGGCGTCTAACC AGTcACAGTCGCAAGCCCGGGT-3')、SPH6ETrS(5'-CTTTGTATTTATTTTCAC TTTTTAAGTATAGAATAAAGAAGCTTGGTACCG-3')、SPH6 ETr6(5'-GAAGAGTTTGTCCTCAACCGCGAGCCCAACAGCTGGCCCTCGCAGACAGCGATGCGG~GAGAG TCAAGAAC-3')、SPH6ETr7(5'-GCTCAGGTCCCTCGGTGGCGGAGTACGTTCGGAGGCCGACGGGTTTCCGATCCGAGTACTGG AAAGACCGC-3')およびSPH6ETr8(5'-CTAGACCCGGGCTTGCGACTGTGACTGGTTAGACGCCTTT CTCGAGAGGTTTTCCGATCCGGTCGATGCGGACTC-3')はをキナーゼ処理し、アニールし、271bpの生産物をゲル精製した。
次に、完全なAd2 TPLはプライマLF394(5'ATCGTCCTGCAGACTCTCTTCCGCATCGCT GTCTGC-3')およびLF3 95(5'-GCTCTAGACTTGCGACTGTGACTGGTTAG-3')を用いてPCRで培養した。精製ゲルは鋳型としてオリゴヌクレオチドをアニールした。
得られた220bp DNAフラグメントをPstI/XkaIで消化し、上記ジーンClean手順を使用して精製し、3,800bp PstI/XbaI pLF062フラグメントで連鎖反応させてpLF066(図17、18(配列番号:9))を作る。pLF066発現カセットの調節配列はhCMVNIEプロモータの145bpフラグメントで、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号を含む241bpカセットと置換している。
【0129】
1.4 pLFO69(HCMVNIE 5'UTRがAd2 TPLで置換されたpLF066の誘導体)の生成:
pLF062中に存在するHCMV-IEプロモータ5'UTR(54bp)を以下の手順を使用して欠失した。アニールしたオリゴヌクレオチドLF397(5'-CGTTTAGTGA.ACCGTCTGCA-3')およびLF398(5'-GACGGTTCACTAAACGAGCT-3')をpLFO62の3,936bp DNAフラグメントで連鎖反応してpLF069(図19、20(配列番号:10))を作った。pLF069発現カセットの調節配列は、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号を含む241bpカセットと置換したHCMV-IEプロモータの91bpフラグメントである。
【0130】
1.5 pLFO77(SV4Oポリアデニル化カセットが153bpに縮小されたpLF069の誘導体)の生成:
SV4Oポリアデニル化配列の160bpのサブフラグメントをオリゴヌクレオチドHX3R(5'-GTAAAACGACGGCCAGT-3')およびLF409(5'-ATCGTCCCGCGGAATTGTTGTTGTTAACTTGTT-3')を使用し、pCAT Basic DNA(長く続く)を鋳型として用いてPCRで増殖した。得られた145bp DNAフラグメントをKspl/BamIIを用いて消化し、上記ジーンClean手順を使用して精製し、pLFO69の3,716 bp Kspl/BamII DNAフラグメントで直接連鎖反応してpLF077 (図21, 22, 配列番号: 11)を作る。pLF077のCAT発現カセットのサイズは1,161bpである。一方、pLF022では1,804bpである(36%縮小)。pLF069発現カセットの調節配列は、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号の一部を含む153bpカセットと交換したHCMV-IEプロモータの91bpフラグメントである。
【0131】
1.6 pLFO9l(ポリアデニル化信号の3O端が修正されたpLF077の誘導体)の生成:
CAV2ゲノムの右ITR配列の5'端のSmaI部位の上流域を局所化した12 bp(5'-TTTTTGGGCGTT-3')を以下の手順を使用してpLF077ポリアデニル化カセットの下流に導入した。1,000bp DNAフラグメントをオリゴヌクレオチドLF423(5'-ACGACCCGTAGAGGGCGTTGGACAGCAACTTGGCCTCGCGGTTGAGGACAAACTCTT-3')およびLF432(5'-ATCGTCCCCGGGTTTTTGGGCGTTATCCAGACATGAT~GATACA-3')を使用しpLF077 DNA(10人の黒ん坊)を鋳型として用いてPCRで増殖した。1,000bp PCR DNAフラグメントをジーンCleanで精製し、クレノー処置で修正し、NcoIを用いて消化した。PCR反応では1.2%アガロースゲルで電気透析し、295bpフラグメントをジーンClean手順を使用し単離した。
pLF077をBamHIを用いて消化し、クレノー酵素の作用で修正し、次いでNcoIを用いて消化した。消化反応では1%アガロースゲルで電気透析し、ジーンClean手順を使用して3,567bp制限フラグメントを単離し、上記295bp DNAフラグメントで連鎖反応してpLF09l(図23、24(配列番号:12))を得た。
【0132】
1.7 pLFO92(CAT発現カセットドナープラスミド)の生成:
pLFO9lの1,180bp HindIII/SmaI DNAフラグメント(全CAT発現カセットを含む)をKlenov酵素の作用で変性し、6.2kbp SmaIで線形にした pLF056で連鎖反応してpLF092(図25、26(配列番号:13))を作った。このプラスミドは、CAV2ゲノム5'端のSmaI部位の12bp上流域の挿入部位にCAT発現カセットを組込むためのドナー・プラスミドに対応する。
【0133】
1.8 pLFlO5(CAV2ゲノムの右のITR配列の5'端のSmaI部位の12bp上流域に外来性DNAを組込むドナープラスミド)の生成
予備アニールされたオリゴヌクレオチドLF446(5'-GGGTTTTTGGGCGTTTCGCGA ACCGGTGTTCACGCGTGTCGACCCC-3')およびLF447(5'−CCCAAAAACCCGCALGCGCTTGGCCA CTTGTGCGCACAGCTGGGG-3')から成るポリリンカ(NruI-AgeI-EcoRI-HluI-SalI-smaI]をSmaIで線形にした6.2kbp pLF056と連鎖反応させてpLF105(図27、28(配列番号:14)を作った。
【0134】
1.9 CAV2ゲノムの右終末端に挿入されるCAT発現カセットを含む組換え型CAV2ウィルスvCA3の生成:
l0μgのpLF092をHindIIIおよびBamHIで消化し、得られた4.3kbp DNAフラグメントをジーンClean手順を使用して単離し、H20中に再懸濁し、100ng/μlの濃度にした。MDCK細胞はLipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.4μgの4.3kbp HindIII/BamHI pLF092フラグメントと4.4μgの精製されたCAV2 DNAとを混合して調製した。溶液Aの全容積を血清無しのHEM培地を補って300μlにした。8日後にトランスフェクション細胞を収穫し、前述したように150mm直径組織培養皿上に塗布した。CATリポータ遺伝子用プローブはpCAT Basic DNA(10ng)を鋳型とし、プライマ対のLF218(5'-ATCGTACATATGGAGAAAAAAATCACTGGATAT-3')/LF2 31(5'-ATCGTAGATATCCTCGAGTTACGCCC CGCCCTGCCACTC-3')を用いたPCRで作った。結果として得られた660bp DNAフラグメントを上記方法でランダムプライミングでラベル化し、ニトロセルロース膜で上記手順を使用してリフト・ウイルス性プラークにハイブリッド化した。プローブに対応するプラークを掻き取り、上記方法でプラーク精製を4回行った。プラーク精製した組換え型CAV2ウィルスにvCA3という名前を付けた。
【0135】
2. vCA3の性格付け。
2.1 組換え型ウィルスvCA3を用いたCAT遺伝子形質発現の解析
2.1.1 vCA3感染MDCK細胞溶菌液中のCAT 酵素活性の検出
精製したvCA3組換え型ウィルスと野生型CAV2をそれぞれ別々にM.O.Iが10で、100%-MDCK単層(106の細胞)で感染させた。5% C02の存在下で37℃で24時間、感染した倍養液を掻き取り、収穫した。細胞ペレットを予熱された(37℃)PBS(Ca2+ およびMg2+遊離)で3回洗浄し、40mMトリス-HCl、pH 7.5、1mM EDTA、および150mM NaClのlml中に、室温で5分間培養した。細胞を4℃では30秒間、12Kgで遠心分離した。得られたペレットを100mlの0.25Hトリス-HCl pH 8.0に再懸濁し、激しく攪拌して急速凍結/融解サイクルを3回行う。溶菌液を65℃で10分間培養して端部遺伝子デアセチル活性の機能をなくされた。室温で12Kgで2分間遠心分離し、上澄をクロラムフェニコールアセチルトランスフェラーゼ(CAT)アッセイで次のように分析した。25mlの細胞溶菌液を37℃での2時間、3mlの(14Cクロラムフェニコール(0.005mCi/ml)(NEN、ボストン、MA)、n-Butyryl Coenzyme A(5mg/ml)の5mlおよび92mlの0.25のM TrisN HCl(pH 8.0)で培養した。反応は各チューブに500mlの酢酸エチル(シグマ、Stルイ、MO)を加えて終了させた。反応物を30秒間、キシレンで攪拌し、1分間の12Kgで遠心分離した。上側有機相は新しいチューブに入れ換え、乾燥蒸発した。残部は25mlのn-Butyryl Coerizyme A(5mg/ml)に再懸濁し、再懸濁された材料の10mlをシリカゲル薄層クロマトグラフィ(TCL)シリカプレート(ベーカー、Philisburg、NY)上に点状に付けた。シリカプレートクロマトグラフィは閉じたチャンバー内で溶剤がプレートの中間にくるまで約1時間運転した。シリカプレートを乾燥し、放射化した。ブチリル化されたクロラムフェニコールがvCA2サンプル中に明らかに検出され、変性クロラムフェニコールは対照の野生型CAV2サンプル中には見られなかった。この結果は組換え型ウィルスvCA3が機能的なCAT活性を発現し、従って、我々が設計した発現カセットおよび我々が選んだ挿入部位を確認した。
【0136】
2.1.2 vCA3感染MDCK細胞溶菌液からの放射化免疫沈殿によるCAT蛋白の検出
放射化免疫沈殿分析をvCA3-感染させたMDCK細胞およびCATシャトルポリクローナル血清(5'3'Inc、ボールダー社)に由来する[35S]メチオニン(1000Ci/nmol、NEN)ラベル化溶菌液を用いて上記(Pincus達(1992)方法で行う。放射化免疫沈殿したCATポリペチドをSDS−ページに溶かし、サリチル酸ナトリウムを用いたフルオログラフィで視覚化した。
制限酵素活性を用いたvCA3遺伝子組織を解析:
vCA3 DNAを上記方法で精製した。精製した全DNAをH20中に再懸濁して1.3μg/mlの最終濃度にした。精製し2μgのvCA3のアリクォットをBgllIおよびSalIで消化した。これら2つの部位はCAV2ゲノム中でユニークであるので、28.2kbpと3.8kbpのフラグメントがBgllIの消化から予想され、27.8kbpおよび4.2kbpフラグメントがSalI消化作用から予想される。これらの制限フラグメント・が確認され、VCA3がそのゲノムの右端内にCAT発現カセットの1,000bpを取り入れた組換え型CAV2ウィルスであることが証明された。
【0137】
実施例13
ドナープラスミド pLFl02の生成
E3 ORFlを修正せずにE3 ORF2の3'端を欠失するために、以下の手順を開発した。
pLF027 DNAを鋳型とし、プライマ対LF437(5'ATCTTAACGCGTCCCTcAGCTTCTAATGGGAC−3')およびLF334(5'−CTTGCTTGTTATTAAAAAAAG−3')を用いて上記方法でPCR増殖した。増殖された329bp DNAフラグメントをMlulおよびSmalを用いて消化し、上記ジーンClean手順を使用して精製した。得られた287bpのMiul/Snal DNAフラグメントをゲル精製し、pLFO86の6,079bp Miul/Smal DNAフラグメントと連鎖反応してpLFO95を作った。pLFO95の63bp BglIl/Miulリンカーを以下の手順を使用して無関係な外来性DNAの305bpのBglII/MiuIリンカーと交換した。305 bp DNAフラグメント(図29および30に記載のヌクレオチド配列、下記参照]は MiulおよびBglIIで無関係なプラスミドを消化して得た。MiulおよびBglII消化したDNAフラグメントをゲル精製し、pLFO9Sの6,315bp MiuI/BglII DNAフラグメントと連鎖反応させてpLFl02(図29、配列番号:15)を作った。
pLFl02のこの処理で305bpの外来性DNAとCAV2 E3の688bpフラグメント(全E3の寸法の45%)とが交換され、CAV2 E3領域内に必須でないサブドメインの制限を定義できる。
【0138】
実施例14
ドナープラスミド DLFl16Aの生成
2つのSphI制限サイト(位置#3770と#3870)を含むpLF027 EcoRV/AatIIの1.8kbp DNAフラグメントを欠失するために、pLF027 EcoRV/AatIIの5,163bpフラグメントをゲル精製し、クレノウ酵素で処理し、再リゲートしてpLFO94を作った。
ユニークなBglIIおよびMiul制限サイトを含む24bp DNAリンカ(5'-GATACGCGTTCCATTAGCAGATCT-3')を2回のPCR増殖手順を用いてE3 ORF1とE3 ORF2と間のpLFO94の遺伝子間配列に挿入した。最初のPCR増殖はpLF027 DNAを鋳型として使用し、下記プライマ一対を用いて実行した:LF243 (5'-CGCGCACAAACTGGTAGGTGC-3')/LF436(5-'AGATCTGCTA~TGGAACGCGTATC~GmAATAATATTATC-3')およびLF4 35 (5'-GATACGCGTTCCATTAGCAGATCTGTTTTACAGCTACCA-3')/LF277(5'-GTACAGTTATGTTGAAGG -3')。487bpおよび698bpの2つの部分的に重なり合うDNAフラグメントが作られた。第2回目のPCR増殖は部分的に重なり合う精製されたDNAフラグメントおよび両方の外部のプライマLF243およびLF277の存在下で、実行した。増殖された1,185bp DNAフラグメントをSphIおよびPatIで消化し、得られた566bp PatI/SphIフラグメントを精製し、pLFO94の4,653bp SphI/PatI部分消化で連鎖反応させてpLFO93を作った。全てのPCR増殖は上記条件を使用して実行された。
E3 ORFlを修正しないE3 ORF2の5'端の欠失を以下の手順を用いて行った。pLFO93 XhoI/MluI 1,062bpフラグメントをゲルは精製し、pLFO86の5,081bp XhoI/MluIフラグメントと連鎖反応させてpLFll5を作った。Miulで線形化したpLF115 DNAを無関係な外来性DNAの311bp Mlul/Mlulフラグメントと連鎖反応させてpLFll6AおよびBを作った。無関係な311のbp Mlulの配列/外来性DNAのMlulフラグメントを含むpLFll6Aの完全なDNAの塩基配列は図31(配列番号:16)に示され、制限地図は図32に示されている。
このpLFll6Aの遺伝子操作の結果、CAV2 E3876bpフラグメント(全E3寸法の57%)が311bpの外来性DNAと交換され、CAV2 E3領域内に必須でないサブドメインの制限を定義することができる。
【0139】
実施例14
ドナープラスミドpLFl00の生成
E3 0RF2、E3 ORFlの3'端および完全なE3 ORF3の5'端を同時に欠失するために、pLFOB6(図7、図8)のMluI(#1529)とDraIII(#889)制限サイトの間の634bpフラグメントを欠失し、以下の手順を使用して無関係な外来性DNAの302bpフラグメントとは交換した。
MiulおよびDraIIIで無関係なプラスミドを消化して302bpのDNAフラグメントを得た。MlulとDraIIIで消化したDNAフラグメントをゲルは精製し、pLFO86の5,946bp MluI/DraIII DNAフラグメントと連鎖反応させてpLFlOO(図33、34の配列番号:17)を作った。302bpフラグメントのヌクレオチド配列は図33に示され、制限地図は図34に示してある。
このpLFlOOの遺伝子操作でCAV2 E3の1,060bpフラグメント(全E3の寸法の69%)を302bpの外来性DNAと交換され、CAV2 E3領域内に更に必須でないサブドメインの制限を定義することができる。
【0140】
実施例15
ドナープラスミドpLFI20の生成
E3 ORF1、ほとんど完全なE3 ORF2および完全なE3 ORF3の3'端を同時に欠失するために、pLF1O2のMluI(#1771)とDraIII(#889)制限サイトの間で882bpフラグメントを欠失し、以下の手順を使用して無関係な外来性DNAの311bpフラグメントと交換した。
pLF1O2 DNAをMlulを用いて線形にし、DraIIIで部分的に消化した。得られた5,733bp MluI/DraIIIを無関係な外来性DNAの311bp MluI/DraIIIフラグメントと連鎖反応させてpLFl2O(図35、36(配列番号:18))を作った。この無関係な外来性DNAの311bp MluI/DraIIIフラグメントのヌクレオチド配列は図35に示され、制限地図は図36に示してある。
このpLFl2Oの遺伝子操作の結果で、CAV2 E3の1,261bpフラグメント(全E3寸法の82%)が311bpの外来性DNAと交換され、CAV2 E3領域内に更に必須でないサブドメインの制限を定義できる。
この最も大きい欠失でE3領域の約80%〜約100%、例えばE3領域の約80%〜95%、または約80%〜90%または80%〜約85%の全てを欠失できることを示す。
【0141】
実施例16
イヌジステンパーウィルス(CDV)ヘマグルチニン(HA)暗号配列を含むpLF043pBSSK+ の生成
1. プラスミドpSDCDVHAの生成
イヌジステンパーウィルス(CDV)のOnderstepoort株はH. Appel博士(コーネル大学、イサカ、NY)から得られた。RNAはCDV感染Vero細胞から収穫し、相補DNAは以下の方法で調整した。
CDV感染Vero細胞からのRNAはChirgwin達のグアニジウムイソチオシアネート-セシウム・クロライド法(1979)を用いて単離した。最初のストランド相補DNAはAMVリバーストランスクリプターゼ(Bio Sciences、セントピーターズバーグ、FL)、オリゴヌクレオチドプライマCDVFSP(配列番号:44)(5'-CCAGGACATAGCAAGCCAACAGGTC-3')およびリボゾームリボ核酸を用いてCDV感染細胞から合成した。CDVFSP(配列番号:44)はCDV融合(F)開始コードンの80bp上流域に入り、上記とヘマグルチニン(HA)暗号配列を含むポジティブなセンス信号ストランド状相補DNA生産物を生じる。
【0142】
HA−特異的転写解読枠(ORF)は最初のストランド相補DNA生産物からPCR法(PCR)を用いて上記方法で増殖した。オリゴヌクレオチドプライマCDVHA1(配列番号:45)(5'-CGATATCCGTTAAGTTTGTATCGTAATGCTCCCCTACCAAGAC-3')およびCDVHA2(配列番号:46)(5'-GGGATAAAAATTAACGGTTACATGAGAATCTTATACGGAC-3')をCDVFSP誘導の最初のストランド相補DNAとのPCRで鋳型として使用した。CDVHA1はワクシニアウィルスH6プロモータの3'末端領域を有し、翻訳開始コドンからCDVにHA ORYに入る配列が続く(Perkus達、1989)。CDVHA2(配列番号:46)はHA ORFのストップコードンからCDV HA 5'端に入る。その結果得られる1.8kbp PCR生産物は20mM dNTPsの存在下で大腸菌DNAポリメラーゼからのクレノー断片化処理でフラグメントをブラントエンドにする。1.8kbpのブラントエンドフラグメントがH6プロモータ内のNruI部位とpSD554(下記参照)のH6プロモータのSinaI部位3'との間に挿入される。得られたプラスミドpCDVHAはH6プロモートされたCDV HA ORFを含まなければならないが、予想外に、CDV HA 5'が欠失した。この欠失を下記方法で回復する。
【0143】
プラスミドpSD554はワクシニアフランキングアーム中にワクシニアK1Lホスト範囲遺伝子(Gillard達、1986)と、挿入部位が続くワクシニアH6プロモータとを含む。ワクシニア・フランキングアームはATI領域:転写解読枠A25LおよびA26Lを置換する(1990a,b、ゲーベル達)。pSD554は以下の方法で調製された。
左右のワクシニアフランキングアームはワクシニアSalI B(1990a,b、ゲーベル達)を含むpSD4l4を鋳型として使用してPCRで造られる。左アームは鋳型pSD4l4でオリゴヌクレオチドプライマMPSYN267(配列番号:47)(5'-GGGCTGAAGCTTGCTGGCCGCTCATTAGACAAGCGAATGAGGGAC-3')およびMP5YN268(配列番号:48)(5'-AGATCTCCCGGGCTCGAGTAATTAATTAATTTTTATTACACCAGAAAAGACGGCTTGAGATC-3')を使用してPCRで合成した。右アームは鋳型pSD4l4でオリゴヌクレオチドプライマMPSYN269(配列番号:49)(5'-TAATTACTCGAGCCCGGGAGATCTAATTTAATT TAATTTATATAACTCATTTTTTGAATATACT-3')およびMPSYN27O(配列番号:50)(5'-TATCTCGAATTCCCGCGGCTTTAAA TGGACGGAACTCTTTTCCCC-3')を使用してPCRで合成した。左右のアームを含む2つのPCRからのフラグメントはPCR中に結合された。結果として得られたPCR生産物をEcoRIおよびHindIIIで消化し、O.9 kbpフラグメントを単離した。この0.9kbフラグメントをpUC8 EcoRIとHindIII部位との間に挿入した。得られたプラスミドpSD541は以下の方法でKlL遺伝子および追加の挿入部位を受けた。
【0144】
プラスミドpSDS4lをBglIlおよびXhoIで消化し、アニールされた相補オリゴヌクレオチドMPSYN333(配列番号:5l)(5'-GATCTTTTGTTAACAAAAACTAATCAGC TATCGCGAATCGATTCCCGGGGGATCCGGTACCC-3')およびMPSYN334(配列番号:52)(5'-TCGAGGGTACCGGATCCCCCGGGAATCGATTCGCGATAGCTGATTAGTTTTTGTTAACAAAA-3')と連鎖反応させてプラスミドpSD552を作った。pSD452(Perkus達、1990)はKlL遺伝子を含む。pSD452をHpal で消化し、BglIIで部分的に消化した。得られたKlL遺伝子を含むlk bpのフラグメントをpSD5S2 HglIIとHpal部位との間に挿入した。得られたプラスミドpSD553をNroIで消化し、ワクシニアH6プロモータ(Perkus達、1989)を含むSmaI/NroIフラグメントを挿入した。合成プラスミド(pMPSS3H6)はA26L組込み遺伝子座内にKlL遺伝子から下流にワクシニアH6プロモータを含む。
プラスミドpMP5S3H6をNruIおよびBamHIで消化し、アニールされた合成オリゴヌクレオチドMPSYN347(配列番号:53)(5'-CGATATCCGTTAAGTTTGTATCGTAAT CTGCAGCCCGGGGGGG-3')およびMPSYN348(配列番号:54)(5'-GATCCCCCGGGCTGCAGA TTACGATACAAACTTAACGGATATCG-3')と連鎖反応させた。得られたプラスミドpSD554はATI領域と交換したフランキングワクシニア配列の範囲内の挿入部位にKlL遺伝子およびH6プロモータを含む。
PCR誘導フラグメントとして、ワクシニアウィルスH6プロモータおよびCDV HA ORFの5'端がpCDVHAに加えた。制御領域H6のATGはPCR誘導フラグメントのCDV HA翻訳開始コドンと重なる。ワクシニアウィルスH6プロモータはPerkus達、1989に記載されている。
【0145】
pEIVC5Lは変性されたH6プロモータおよび無関係な遺伝子を含む。pEIVC5LはオリゴヌクレオチドプライマH65PH(配列番号:55)(5'-ATCATCAAGCTTGA TTCTTTATTCTATAC-3')およびCDVHAH6(配列番号:56)(5'-GTCTTGGTAGGGGAGCAT TACGATACAAACTTAACG-3')とのPCR法で156bpフラグメントを作るのに用いた。CDVHAH6はH6プロモータの5'端へ向かう翻訳開始コドンから始まるCDV HAの5'端の18塩基対を含む。H6SPH(配列番号:55)はHindIII部位を含み、5'から3'へ向かH6プロモータからの配列が続く。156塩基対のPCR誘導H6SPH/CDVHAH6(配列番号:56)はCDV HA暗号化配列の5'端の18塩基対を含む。
【0146】
CDVFSP(配列番号:44)の最初のストランド相補DNA生産物はオリゴヌクレオチドプライマCDVHAATG(配列番号:57)(5'-ATGCTCCCCTACCAAGAC-3')およびCDVHAECO(配列番号:58)(5'-GTAATTAGTAAAATTCACCTTG-3')でPCRされて459塩基対のフラグメントを作る。CDVHAATG(配列番号:57)は翻訳開始コドンからCDV HA 3'端に入る。CDVHAECO(配列番号:58)プライムスはCDV HA 5'端に向かうH6プロモータ化されたCDV HA配列の位置583から始まる。 156塩基対および459塩基対のPCR誘導フラグメントをプールし、H6SPH(配列番号:55)およびCDVHAECOとPCRで597塩基対フラグメントを作る(配列番号:58)。PCR誘導生産物をHindIIIおよびEcoRIで消化してH6プロモータおよび5'端のCDV HA暗号配列の387塩基対を含む520塩基対フラグメントを作る。520塩基対のHindIll/EcoRI消化PCRフラグメントをpBSSK+のHindIIIおよびEcoRI部位との間に挿入してpBSCDVHA5Sを作る。プラスミドpBSCDVHASSはpBSSK+のCDV HA ORFのH6プロモートされた5'端を含む。CDV HA ORFの3'端は以下の方法で加えた。
プラスミドpCDVHAをSinaIで消化し、次にEcoRIで部分消化して3'がCDV HA ORYで終わるl.4kbpフラグメントを作る。この1.4kbpのpCDVHA EcoRI/SmalフラグメントをpBSCDVHA5SのEcoRIとSinaI部位との間に挿入した。得られたプラスミドpBSCDVHAをBamHIで消化し、XhoIで部分的に消化して、H6プロモートされたCDV HA転写解読枠を含む1.9kbpフラグメントを作った。この1.9kbp HamHI/XhoI pBSCDVHAフラグメントをpSD553(上記参照)のBamHIとXhoI部位との間に挿入した。得られたプラスミドpSDCDVHAは、ATI挿入部位のH6プロモートされたCDV HA遺伝子を含む。
【0147】
2. pLFO43の生成
CDV HA暗号配列とワクシニアウィルスH6プロモータの3'端領域を含むpSDCDVHA 1,975 bpのBainHIおよびHindIllをゲル精製し、pBSSK+の対応する制限部位の間に挿入してpLFO43(図37および38)(配列番号:19)を作る。
【0148】
実施例17
完全なCDV HA発現カセットを含むpLFO9Sの生成
XhaI制限サイトを以下の方法でCDV HA開始コードン(ATG)の直ぐ上流に設計した。409bp DNAフラグメントをプライマ対LF412(5'-CTGATCTCTAG AATGCTCCCCTACCAAGACAAG 3')(配列番号:59)およびLF413(5'-TGGAGATCGCGGAAGTCG 3')(配列番号:60)を用い、pLFO43 DNAを鋳型として用いてPCRで増殖した。PCR増殖されたフラグメントを上記ジーンClean手順を使用して単離し、20mM dNTPsの存在下で大腸菌DNAポリメラーゼからクレノーフラグメント化し、SpalおよびEcoRIを用いて消化した。得られたブラントエンドな/Spalの192bp DNAフラグメントをpLFO43の4,891bp Nrul/SpaIフラグメントで連鎖反応してpLF096を作った。
KspI制限サイトはpLFO96 CDV HAストップコードン(TAA)の直ぐ下流に以下の方法で設計された。204bp DNAフラグメントをpLFO43を鋳型として使用し、プライマLF438(5'-TGTTTATGACCCAATCG-3')(配列番号:61)およびLF4 39(5'ATGC TCCCGCGGTTAAC GGTTACATGAGAATCT 3O)(配列番号:62)として上記方法でPCRで増殖した。PCR増殖されたフラグメントをKspIおよびAcoIで消化し、上記ジーンClean手順を使用して単離した。得られた143bp DNAフラグメントをゲル精製し、pLFO96の4,594bp KspI/AcoIフラグメントと連鎖反応させてpLF097を作った。
CDV HA暗号配列を含む1,821bp pLF097 KspI/XhaIフラグメントをpLFO69の3,246bp HpI/XhaIフラグメントと連鎖反応させてpLFO98(図39、40)(配列番号:20)を作った。
【0149】
実施例18
pLFO99A(CAV2ゲノム右端のSmaI部位のCDVヘクタール発現カセット12bp上流域の組込みのためのドナープラスミド)の生成
pLF069に定義された調節配列と連結されたCDV HA暗号配列を含む2,372bp BamHI/HindIII pLFO98フラグメントを大腸菌DNAポリメラーゼからのクレノーフラグメントで処理し、6,243bp NruIで線形化したpLF1OS、と連鎖反応させてpLFO99AおよびpLFO99Bを作った。pLFO99Aは発現カセット(図41、42)(配列番号:21)の右側に対応する。
【0150】
実施例 19
組換え型CAV2ウィルスvCA4 1の生成および性格付け
1. 組換え型CAV2ウィルスvCA4の生成
10μgのpLF1O2をHindIIIで消化し、得られた3,652bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oで100ng/mlの濃度に再沈殿した。MDK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.6kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEM培地を補って溶液Aの全容積を300mlにした。トランスフェクション細胞を150 mmの直径の組織培養皿上に塗布し、8日後に収穫した。pLF1O2 DNA(10ng)を鋳型に使用し、プライマ対LF4 40(5'-ATCAGTACGCGTATGGGCCACACACGGAGG-3')/LF44 1(5'-ATCAGTAGATCTGT TATTAGTGATATCAAA-3')でPCRして、pLF1O2に挿入される外来性DNAの305bpフラグメントに特有のプローブを作った。得られた305bp DNAフラグメントは上記ハイブリッド手順を使用してランダムプライミングでラベル化し、ウイルス性プラークを持ち上げるためにニトロセルロース膜を用いてハイブリッド化した。プローブに対応する5つのウイルス性プラークを取り、上記方法でプラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスはvCA4という名前をつけた。
【0151】
2. vCA4の特徴付け
vCA4 DNAは上記方法で精製した。精製された全DNAをH2Oに再沈殿して最終濃度を1.9μg/mlにした。精製されたvCA4の2μgアリクォットをHindlIlで消化した。予想された3667bp HindIIIフラグメントがvCA4サンプル中に視覚化され、4.0kbpフラグメントは野性型CAV2サンプル中に存在していたので、vCA4ゲノムDNAがpLF102に記載の部分的に欠失したE3領域を含むということが証明された。サウザンブロットを用いてVCA4 DNAを分析し、VCA4が371bpは野性型E3領域より短いE3領域を有することを示した。
この結果はCAV2 E3領域の必須でないサブドメインを示す。より詳しくは、vCA4の誘導体は、pLFO27(図1(配列番号:1)参照)に記載の位置#1470と位置#2157との間[すなわちE3領域の45%]から成るCAV2 E3配列がヘテロジニアスなDNAに置換できることを証明している。これはさらに、CAV2 E3がCAV2ゲノム内の挿入部位であることを確認するものである。この結果はさらに、CAV2 E3領域の一部を欠失でき、CAV2ゲノムの右端にvCA3の所で説明したように外来性DNAを導入して保証できることを証明している。
【0152】
実施例20
組換え型CAV2ウィルスvCA5の生成および特徴付け
1. 組換え型CAV2ウィルスvCA5の生成
10μgのpLFll6AをHindIIIandで消化し、得られた3,487bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して濃度を100ng/mlにした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.5kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞を150mm直径の組織培養皿上にプレートアウトし、8日後に上記方法で収穫した。
pLFll6Aに挿入される外来性DNAの311bpフラグメントに特有のプローブはpLFll6A DNA(10ng)を鋳型として使用し、プライマ対LF453(5'-ATCGTCATTGCCACGCGTATGGC AGAAGGATTTGCAGCCAAT-3')/LF4 54(5'-ATCGTCATTGCCA CGCGTAACCA GGGACAATACTTGTTCATC-3')でPCRを用いて作った。得られた311bp DNAフラグメントは上記手順を用いてランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜を用いてハイブリッド化した。プローブに対応する5つのウイルス性プラークを取り、プラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスにはvCA5という名前をつけた。
【0153】
2. vCA5の特徴付け
vCA5 DNAを上記方法で精製した。精製された全DNAをH2Oに再沈殿して、最終濃度を1.9μg/mlにした。精製されたvCA5の2μgアリクォットをHindlIlで消化した。予想された3,487bp HindIIIフラグメントはvCA5サンプル中に視覚化され、4.0kbpフラグメントは野生型CAV2サンプル中に存在するので、vCA5ゲノムDNAがpLFl16Aに記載された部分的に欠失したE3領域を含むということが証明された。この結果は、CAV2 E3領域が必須でないサブドメインであることを示す。特に、このVCA5の誘導体はpLFO27(図1(配列番号:1)を参照)で説明したように、位置#1088と位置#1964 [E3領域の57%]との間から成るCAV2 E3配列はヘテロジニアスなDNAと交換できということを証明している。これはさらに、CAV2 E3がCAV2ゲノム内の挿入部位であることを確認するものである。この結果もCAV2 E3の一部を欠失でき、vCA3で述べたように、CAV2ゲノムの右端に外来性DNAを導入して補償できるということを証明している。
【0154】
実施例21
組換え型CAV2ウィルスvCA6の生成および性格付け
1. 組換え型CAV2ウィルスvCA6の生成
10μgのpLFlOOをHindIIIで消化し、得られた3,284bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿し、100ng/ml濃度にした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.3kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞を150mm直径の組織培養皿上にプレートアウトし、8日の後に収穫した。
pLFlOODNA(10ng)を鋳型としては使用し、プライマ対LF442(5'-ATCAGTCACGGTGTGTAAATGG GCCACACACGGAGG-3')/LF44 3(5'-ATCAGTACGCGTGTTATTAGTG ATATCAAA-3')でPCRでpLFlOOに挿入するたプローブ用に外来性DNAの311bpフラグメントを作った。得られた302bp DNAフラグメントは上記ランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜とハイブリッドした。プローブと交差した5つのウイルス性プラークを掻き取り、プラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスはvCA6という名前をつけた。
【0155】
2. vCA6の特徴付け
vCA6 DNAは上記のようにして精製した。精製された全DNAをH2Oに再沈殿して最終濃度を1.9μg/mlにした。精製されたvCA6の2μgアリクォットをHindlIlで消化した。予想された3,284のbp HindIIIフラグメントはvCA6サンプル中に視覚化され、4.0kbpフラグメントは野生型CAV2サンプル中に存在するので、vCA6ゲノムDNAがpLFlOOに記載の部分的に欠失したE3領域を含むということが証明された。この結果はCAV2 E3領域が必須でないサブドメインであることを示す。より詳しくは、このvCA6の誘導体は、pLFO27(図1(配列番号:1)を参照)で説明したように、位置#898と位置#1949の間 [E3領域の69%] から成るCAV2 E3配列がヘテロジニアスなDNAと交換できることを証明している。これはまたCAV2ゲノム内に挿入部位としてCAV2 E3があることを確認している。この結果もCAV2 E3領域の一部が欠失でき、、vCA3の誘導体の所で説明したように、CAV2ゲノムの右端に外来性DNAの導入することで補償できることを証明している。
【0156】
実施例22
組換え型CAV2ウィルスvCA7の生成および特徴付け
1. 組換え型CAV2ウィルスvCA7の生成
10μgのpLFl2OをHindIIIで消化し、得られた3,085bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して100ng/mlの濃度にした。MDCK細胞は5μg の3kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞は150mm直径組織培養皿上にプレートアウトし、8日後に収穫した。pLFlOOに挿入される外来性DNAの311bpフラグメントに特有のプローブはpLFlOO DNA(10ng)を鋳型に使用し、プライマ対LF458(5'-ATCCGTACGCGTTAGAGGGCAAAGCCC GTGCAGCAGCGC-3')/LF4 59(5'-ATCCGTCACGGTGTGTAGAT GGGTTGTTTTGTG GAGAAT-3')でPCRで作った。得られた311bp DNAフラグメントを上記方法でランダムべヌライミングし、ウイルス性プラークを持ち上げるためにニトロセルロース膜を用いてハイブリッド化した。プローブとウィルスDNAとの間の交差反応が確認された。
この結果はpLFO27(図1(配列番号:1)参照)で説明したように、位置#898と位置#2157との間の1,259bpの欠失が組織培養でのウイルス性複製と互換性を持ち、実質的にE3領域の全てを欠失できることを示している。
【0157】
実施例23
vCA5の生成
10μgのpLFO99AをHglIIおよびNotIで消化し、得られた5,131bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿し、100ng/mlの濃度にした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションした。溶液Aは0.5μgの5.1kbp HglII/NotI DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞は150mm直径の組織培養皿上にプレートアウトし、8日後に収穫した。pSDCDVHAの440bp EcoRIフラグメントは上記ランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜とハイブリッドした。プローブと交差した2つのウイルス性プラークを掻き取り、プラーク精製を行った。プラーク精製した組換え型CAV2ウィルスはvCA8という名前を付けた。
【0158】
2. vCA8の特徴付け
vCA8 DNAの精製、制限消化、サウザンブロット、放射化免疫沈殿、CDV HA発現の解析によって組込みおよび発現を確認した。
【0159】
実施例24
pLF1OS (イヌジステンパーウィルス(CDV)融合(Fl)をコードするプラスミドから誘導されたpBSSK+)の生成
1. pATICDVF1の生成
CDV融合(F)特異性転写解読枠(ORF)をCDVFSP(配列番号:44)を最初のストランド相補DNAから誘導したCDVFSP(配列番号:44)を鋳型として相補DNAからオリゴヌクレオチドプライマCDVATGF1(配列番号:63)(5'-CATAAATTATTTCATTATCGCGATATCCGTTAAGTTTGTA TCGTAATGCACAAGGGAATCCCCAAAAGC-3')およびCDVFT(配列番号:64)(5'-ATCATCGGATCCATAAAAATCAGTGTGATCTCACATAGGATTTCGAAG-3')を用いてPCRで増殖した。CDVATGF1(配列番号:63)はワクシニアウィルスH6プロモータの3'端領域(Perkus、達、1989)を含み、CDV F翻訳開始コドンからCDV F ORFまでの配列が続く。CDVFT(配列番号:64)はBamHI部位を含み、CDV FストップコードンからCDV Fの 5'端の方への配列が続く。得られたPCR生産物をNruIおよびBamHIで消化し、NruIとBamHI部位との間のpSD554に2kbpフラグメントを挿入した。得られたプラスミドpATICDVYlはワクシニアウィルスATI組込み遺伝子座にH6プロモーテッドCDV F ORFを含む。
【0160】
2. HC5LSP28の生成
CS ORFを除去するためにCSベクタープラスミドHCSLSP28を以下の方法で作った。遺伝子カナリポックス(canarypox)DNAを鋳型としてPCRでオリゴヌクレオチドプライマC5A(配列番号:65)(5'-ATCATCGAATTCTGAATGTTAAATGTTATACTTTG-3')およびCSB(配列番号:66)(5'-GGGGGTACCTTTGAGAGTACCACTTCAG-3')を用いた。得られた1.5kbpフラグメントのC5A端をEc1QRIで消化し、他端はpUC8のEcoR1とSmaI部位との間に組込みためにブラントにしたままにしたプラスミドC5LABを作った。遺伝子カナリポックス(canarypox)DNAを鋳型としてPCRでオリゴヌクレオチドプライマC5C(配列番号:67)(5'-GGGTCTAGAGCGGCCGCTTATAAAGATCTAAAATGCATAATTTC-3')およびC5DA(配列番号:68)(5'-ATCATCCTGCAGGTATTCTAAACTAGGAATAGATG-3')を使った。得られた400塩基対フラグメントのC5DA端をPatIで消化し、他端はのSmaIとPatI部位との間の組込みのためにブラントのままにしてプラスミドpCSLを作った。アニールされた相補なオリゴヌクレオチドCP26(配列番号:69)(5'-GTACGTGACTAATTAGCTATAAAAAGGATCCGGTACCCTCGAGTCTAGAATCGATCCCGGGTTTTTATGACTAGTTAATCAC-3')およびCP27(配列番号:70)(5'-GGCCGTGATTAACTAGTCATAAAAACCCGGGATCG ATTCTAGACTCGAGGGTACCGGATCCTTTTTATAGCTAATTAGTCAC-3')をpCSL p7l8とNotI部位との間に挿入した。得られたプラスミドHCSLSP28は遺伝子座CSベクタープラスミドである。
【0161】
3. pBSCDVHAVQの生成
オリゴヌクレオチドRW132(配列番号:71)(5'-AGCTTCCCGGGTTAA TTAATTAGTCATCAGGCAGGGCGAGAACGAGACTATCTGCTC GTTAATTA ATTAG-3')およびRW133(配列番号:72)(5'-AGCTCTAATTAATTAACGAGCAGATAGTCTCGTTCTCGCCCTGCCTGATGACTAATTAATTAACC CGGGA-3')をアニールしてはダブルリンカ配列を形成した。RW132/RW133(配列番号:71/配列番号:72)ダブルストランド配列をpBSCDVHA5SのH6プロモートされたCDV HA ORFのHindIII部位5'に挿入してプラスミドpBSCDVHAVQを作った。
【0162】
4. pCSCDVHAYlの生成
H6プロモートされたCDV HA ORFを含む2kbp pBSCDVHAVQ SmaIフラグメントをHC5LSP28 SmaI部位に挿入してプラスミドpCSLCDVHAを作った。CS遺伝子座において、お互いから離れて目指すそれらの転写産物で 、H6プロモートされたCDV Fを含む2.1kbp pATICDVF1 HpaI/BamHIフラグメントをpCSLCDVHA Smal/ BamHIの6.5kbp DNAフラグメントで連鎖反応してH6プロモートされたCDV F およびCDV HA ORFと含むプラスミドpC5LCDVHAY1を作る。
【0163】
6. ベクタープラスミドpC6Lの生成
C6 ORFを除去するためにC6ベクターpC6Lを以下の方法で作った。オリゴヌクレオチドプライマC6Al(配列番号:73)(5'-ATCATCGAGCTCGCGGCCGCCTATCAAAAG TCTTAATGAGTT-3')、C6B1(配列番号:74)(5'-GAATTCCTCGAGCTGCAGCCCGGGTTTTTATAGC TAATTAGTCATTTTTTCGTAAGTAAGTATTTTTATTTAA-3')、C6Cl(配列番号:75)(5'-CCCGGGCTGCAGCTCGAGGAATTCTTTTTATTGATTAACTAGTCAAATGAGTATATATAATTGAA AAAGTAA-3O)およびC6D1(配列番号:76)(5'-GATGATGGTACCTTCATAAA TACAAGTTTGATTAAACTTAAGTTG-3')を用いてpC6Lを作った。オリゴヌクレオチドプライマC6Al(配列番号:73)およびC6B1(配列番号:74)はカナリポックス(canarypox)DNAを鋳型にしてPCRで用い、380塩基対フラグメントを作った。カナリポックス(canarypox)DNAを鋳型にした第2回目のPCR反応はC6Cl(配列番号:75)およびC6D1(配列番号:76)は、オリゴヌクレオチドプライマを用い、1,155塩基対フラグメントを作った。2回のPCR反応生成物を合せ、C6Al(配列番号:73)およびC6D1(配列番号:76)で最後のPCRを行って1,613塩基対フラグメントを作った。最終PCR生産物をSacIおよびKonIで消化し、pBSSK+のSacIとKonIとの間に挿入した。得られたC6組込みプラスミドはpC6Lと名付けた。
【0164】
7. pNl03の生成
pC5LCDVHAY1をBamHIで消化し、20μM dNTPSの存在下で大腸菌DNAポリメラーゼからのクレノーフラグメントで処理し、BamHI部位をブラントエンドし、続けてSmalで消化した。H6プロモートされたCDV FとH6プロモートされたCDV HA ORFsとを含む4.2kbp BamHIからSmaI部位へのブラントエンドをpC6LのSmaIフラグメントに挿入てプラスミドpMMl03を作った。
【0165】
8. pLFl08の生成
CDV Fl暗号配列とワクシニアウィルスH6プロモータの3'端領域とを含むpMM103 HindlIl/ BamHI 1,961bp DNAフラグメントをゲル精製し、pBSSK+の対応する制限部位の間に挿入してpLFl08(図43、44(配列番号:22))を作った。
【0166】
実施例25
完全なCDV F1発現カセットを含むpLFlllの生成
pLFl08 XbaI制限サイトをCDV F1開始コードン(ATG)の直ぐ上流に以下の方法で設計した。473bp DNAフラグメントをpLFl08 DNAを鋳型とし、LF44 8A(5'-ACTGTACTCGAGTCTAGAATGCACAAGGGAATCCCCAAAAGC-3')(配列番号:77)およびRW830(5'-ATTCCAATGTATCTGAGC-3')(配列番号:78)をプライマとして使用してPCRで増殖した。PCR増殖されたフラグメントをXhoIおよびCo1IIで消化し、上記ジーンClean手順を使用して単離し、XhoI/Co1IIで消化した。得られたXhoI/ColII 136bpDNAフラグメントをpLF1O8の4,783bpのXhoI/ColIIフラグメントで連鎖反応してpLF109を作った。
XhaI(#2035)を欠失され、以下の方法でHpI制限サイトをpLFl08 CDV F1ストップコードン(TGA#2016)の直ぐ下流に設計した。431bp DNAフラグメントをpLF109を鋳型として使用しLF449(5'ACTGTACCGCGGTCAGTGTGATCTCACATAGGATTTCGA 3')(配列番号:79)およびCDV-FG(5'-GGTTGAAATAGATGGTG 3')(配列番号:80)をプライマとしてPCRで増殖した。PCR増殖されたフラグメントを上記ジーンClean手順を使用して単離し、KspIおよびBfrIで消化した。得られた255bp DNAフラグメントをゲル精製し、pLFl09の4,631bp KspI/BfrIフラグメントで連鎖反応させてpLYllOを作った。
CDV F1暗号配列を含む1,997bp pLYllO KspI/XbaIフラグメントをpLFO69の3,244bp KspI/XbaIフラグメントで連鎖反応してpLFlll(図45、46(配列番号:23))を作った。
【0167】
実施例26
変性された完全なCDV F1発現カセットを含むpLFl28の生成
CDV F1発現カセットのポリアデニル化カセットの大きさを241bpから153bpまで減らすために以下方法を実行した。pLYO77 KspI/BamHI 146bpフラグメントは上記のようにゲル精製し、pLFlll KanI/BamHI 5,002bpフラグメントで連鎖反応してpLFl28(図47、48(配列番号:24))を作った。
【0168】
実施例27
pLFL3OA(CAV2ゲノム右端のSmaI部位のCDV F1発現カセット12bp上流域に組込むためのドナー・プラスミド)の生成
プラスミドpLFl28をBamHIで消化し、次にHindlIlを用いて部分的に消化した。pLFO77中に制限配列と組合されたCDV F1コード配列を含む2,451bpのBamHI/HindIIIフラグメントをE. coli DNA ポリメラーゼからのクレノーフラグメントで処理し、6,243 bpの NruI 直線化 pLF105で連鎖反応してpLYl30A と PLY130Bとを作った。pLYl30Aは発現カセットの右側に対応している(図49, 50, 配列番号: 29).
【0169】
実施例28
vCA-CDVFl-Ql2bl-up-SmaIの生成
10μgのpLYl30AをNotIおよびHglIIで消化し、得られた5,305bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して100ng/mlの濃度にした。MDCK細胞は上記Lipofectamineベースの手順を使用してトランスフェクションした。溶液Aは0.9μgの9.3kbp HglII/NotI DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。8日後にトランスフェクションされた細胞を収穫し、上記150mm直径組織培養皿上に塗布した。pATICDVF1の1.4kbp EccRI/BamHI DNAフラグメントを上記のランダムプライミングでラベル化し、リフトウイルスプラークを得るためにニトロセルロース膜とハイブリッドした。プローブと交差した2つのウイルス性プラークを取り、上記プラーク精製法を行い、vCA-CDVFl-@l2bp-up-SmaIを得た。このウィルスを制限消化(DNA解析)およびサウザンブロット放射化免疫沈殿(発現解析)を用いて特徴付けた。
【0170】
実施例29
追加の組換え体
タグおよびその他の外来性DNAがCAV2に組み込まれたので、他の外来性DNAもCAV2に組み込むことができる。従ってvCAl、vCA2、vCA3、vCA4、vCA5を作りたものである。vCA6、vCA7、vCA8およびvCA-CDVY1-@l2bp-up-SmaIを作るために使用した外来性DNAの代わりに、米国特許第5,174,993および第9,509,941号に記載のものを使用できる。例えば、組換え型アビポック(avipox)ウィルス、ワクシニアウィルス;狂犬病グリコプロテイン(G)遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、gp 5l,30ウシ属の白血病ウィルスのエンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAV-l env遺伝子、NP(Chicken/ペンシルバニア/1/83風邪(ウィルス)のヌーデオプロテイン(nudeoprotein)遺伝子)、マトリックス、プレポリマー伝染性気管支炎ウィルス;HSV gD;特に、エントモポックス(entomopox)プロモータの遺伝子、特に、米国特許第5,338,683号に記載の外来性DNA、例えば組換え型ワクシニアウィルス(アビポック(avipox)ウィルス;ヘルペスウイルス・グリコプロテインをコードするDNA;米国特許第5,494,807号に記載の抗原をコードする外来DNA、例えば組換え型ワクシニア、アビポック(avipox);特に、狂犬病、ヘパチチス(Hepatitis)B、JEV、YY、Dengue、はしか、仮性狂犬病、エピスタイン(Epstein)-バー、HSV、HIV、SIV、EHV、BHV、HcMV、犬パルボウィルス、馬の風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、お多福風邪、NDV;米国特許第9,903,834号に記載のもの、例えば、組換え型ワクシニア、アビポック(avipox)、Norbillivirus、例えばはしかF、ヘマグルチニン;米国特許第4,722,848号に記載のもの、例えば組換え型ワクシニアウィルス;HSV tk、グリコプロテイン(例えばgB、gD)(風邪HA)ヘパチチス(Hepatitis)B(例えば、特に、HBsAgJ);英国特許GB 2 269 820 Bおよび米国特許第9,514,375号に記載のもの(組換え型ポックスウィルス;フラビウイルス構造蛋白質);WO 92/22641に記載のもの(例えば、組換え型ポックスウィルス;特に、免疫不全ウイルス);WO 93/03149に記載のもの(例えば、組換え型ポックスウィルス;特に、IBDV);WO 94/16716および1994年1月19日出願の米国特許出願第08/184009に記載のもの(例えば、組換え型ポックスウィルス;特に、cytoicineおよび/または腫瘍関連された抗原);PCT/US94/066521994年1月19日(Plasraodiumライフサイクルの各段階からのようなプラズモディウム抗原);米国特許第5,523,089号(W093/08306)PCT/US92/08697、Molecular 5 Microbiology(1989)、3(4)、479-486、PCT公報WO 93/04175、WO 96/06165に記載のもの(Borrelia抗原とめそのDNA);Briles達のWO 92/14488(pneumococcalDNA)。これらはvCA2〜vCA8およびvCA-CDVF1-@12bp-up-SmaI中のような外来性DNAで追加のCAV2組換え体およびvCA2〜vCA8およびおよびvCA-CDVF1-@12bp-up-SmaI中のような欠失を作るのに用いることができる。例えば多重抗原のコードを含む組換え体を含む(サブフラグメントプロモータ、減少または変性したポリアデニル化カセットおよび交換した5'UTRを有するプロモータを含む)E3または右ITRとE4転写単位との間の領域またはその両方への挿入およびE3領域での欠失で使用できる。発現は解析で示される。本発明組成物は投与のための担体または希釈剤との混合製剤で宿主(抗体応答を含む応答を生じる脊椎動物、動物または人間)に与える。
【0171】

【0172】
【0173】
【図面の簡単な説明】
【0174】
【図1a】pLF027の完全なDNAの塩基配列を示す(6,995bp、配列番号:1)。CAV2 HindIll Aフラグメントはヌクレオチド#4725で始まり、ヌクレオチド#689で終わる。CAV2 E3領域はヌクレオチド#1414で始まり、ヌクレオチド#2945で終わる。CAV2 E3 ORF1はヌクレオチド#8で始まり、ヌクレオチド#346で終わる。CAV2 E3 ORF2はヌクレオチド#384で始まり、ヌクレオチド#1478で終わる。CAV2 E3 ORF3はヌクレオチド#1019で始まり、ヌクレオチド#483で終わる。残りのヌクレオチドとpBSSK+に対応する);
【図1b】図1aの続きである。
【図1c】図1bの続きである。
【図2】pLF027の制限地図を示す。
【図3a】pLF047Aの完全なDNAの塩基配列を示す(6,959bp、配列番号:2)。23 bp BlgII/MluIリンカーはヌクレオチド#1485で始まり、ヌクレオチド#1508で終わる。残りの配列はpLF027に対応する。
【図3b】図3aの続きである。
【図3c】図3bの続きである。
【図4】pLF047Aの制限地図を示す。
【図5a】pLF049Aの完全なDNAの塩基配列を示す(7,002bp、配列番号:3)。63 bp BlgII/MluIリンカーはヌクレオチド#2138で始まり、ヌクレオチド#2201で終わる。残りの配列はpLF047Aに対応する。
【図5b】図5aの続きである。
【図5c】図5bの続きである。
【図6】pLFO49Aの制限地図を示す。
【図7a】pLFO86の完全なDNAの塩基配列を示す(6,581bp、配列番号:4)。63 bp BlgII/MluIリンカーはヌクレオチド#2295で始まり、ヌクレオチド#2358で終わる。残りの配列はpLF047Aに対応する。
【図7b】図7aの続きである。
【図7c】図7bの続きである。
【図8】pLF086の制限地図を示す。
【図9a】pLF056の完全なDNAの塩基配列を示す(6,196bp、配列番号:5)。CAV2 SalI Bフラグメントはヌクレオチド#1で始まり、ヌクレオチド#3274で終わる。右ITR(196bp)はヌクレオチド#3078で始まり、ヌクレオチド#3274で終わる。SmaI部位は位置#3088に局所化している。残りのヌクレオチドはpBSSK+に対応する。
【図9b】図9aの続きである。
【図9c】図9bの続きである。
【図10】pLF056の制限地図を示す;
【図11a】pLF06lの完全なDNAの塩基配列を示す(6,503bp、配列番号:6)。306bpヘテロジニアスDNAタグはヌクレオチド#3091で始まり、ヌクレオチド#3397で終わる。残留りのヌクレオチドはpLF056に対応する。
【図11b】図11aの続きである。
【図11c】図11bの続きである。
【図12】pLF061の制限地図を示す;
【図13a】pLF022の完全なDNAの塩基配列を示す(4,504bp、配列番号:7)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。全ての他のヌクレオチドはpCATの基礎的配列に対応し、以下を含む:ヌクレオチド#209で始まり、ヌクレオチド#868で終わるCATリポータ遺伝子、ヌクレオチド#958で始まり、ヌクレオチド#1814で終わるSV4O小t抗原およびポリアデニル化信号(856bp)、ヌクレオチド#2467で始まり、ヌクレオチド#3327で終わるアンピシリン耐性遺伝子。
【図13b】図13aの続きである。
【図13c】図13bの続きである。
【図14】pLF022の制限地図を示す;
【図15a】pLF062のDNAの完全な塩基配列を示す(3,812bp、配列番号:8)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。CATリポータ遺伝子はヌクレオチド#209で始まり、ヌクレオチド#868で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#881で始まり、ヌクレオチド#1122で終わる。アンピシリン耐性遺伝子はヌクレオチド#1775で始まり、ヌクレオチド#2635で終わる。
【図15b】図15aの続きである。
【図16】pLF062の制限地図を示す。
【図17a】pLF066の完全なDNAの塩基配列を示す(4,009bp、配列番号:9)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。Ad2 TPL(202bp)はヌクレオチド#154で始まり、ヌクレオチド#356で終わる。CATリポータ遺伝子はヌクレオチド#406で始まり、ヌクレオチド#1065で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#1077で始まり、ヌクレオチド#1319で終わる。アンピシリン耐性遺伝子はヌクレオチド#1972で始まり、ヌクレオチド#2832で終わる。
【図17b】図17aの続きである。
【図18】pLFO66の制限地図を示す。
【図19a】pLF06の完全なDNAの塩基配列を示す(3,955bp、配列番号:10)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#1024で始まり、ヌクレオチド#1265で終わる。アンピシリン耐性遺伝子はヌクレオチド#191.8で始まり、ヌクレオチド#2778で終わる。
【図19b】図19aの続きである。
【図20】pLF069の制限地図を示す。
【図21a】pLF077の完全なDNAの塩基配列を示す(3,861bp、配列番号:11)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド# 93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(153bp)はヌクレオチド#1018で始まり、ヌクレオチド#1171で終わる。アンピシリン耐性遺伝子はヌクレオチド#1824で始まり、ヌクレオチド#2684で終わる。
【図21b】図21aの続きである。
【図22】pLFO77の制限地図を示す;
【図23a】pLF09lの完全なDNAの塩基配列を示す(3,888bp、配列番号:12)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(153bp)はヌクレオチド#1018で始まり、ヌクレオチド#1164で終わる。SV4Oポリアデニル化信号の3'端に挿入されるCAV2 12のヌクレオチドはヌクレオチド#1165で始まり、ヌクレオチド#1176で終わる。アンピシリン耐性遺伝子はヌクレオチド#1851で始まり、ヌクレオチド#2711で終わる。
【図23b】図23aの続きである。
【図24】pLF09lの制限地図を示す。
【図25a】pLF092の完全なDNAの塩基配列を示す(7,379bp、配列番号:13)。CAT発現カセット(pLF091で定義)はヌクレオチド#1で始まり、ヌクレオチド#1179で終わる。CAV2の左フランキングアーム(182bp)はヌクレオチド#1180で始まり、ヌクレオチド#1362で終わる。CAV2右フランキングアーム(3,090bp)はヌクレオチド#4285で始まり、ヌクレオチドヌクレオチド#7375終わる。残りの配列はpBSSK+に対応する。
【図25b】図25aの続きである。
【図25c】図25bの続きである。
【図25d】図25cの続きである。
【図26】pLF092の制限地図を示す。
【図27a】pLF105の完全なDNAの塩基配列を示す(6,243bp、配列番号:14)。ポリリンカはヌクレオチド#3092で始まり、ヌクレオチド#3123で終わる。CAV2左フランキングアーム(182bp)はヌクレオチド#3123で始まり、ヌクレオチド#3321で終わる。CAV2右フランキングアーム(3,090bp)はヌクレオチド#1で始まり、ヌクレオチド#3091で終わる。残りのヌクレオチドはpBSSK+に対応する。
【図27b】図27aの続きである。
【図27c】図27bの続きである。
【図28】pLF105の制限地図を示す。
【図29a】pLFl02の完全なDNAの塩基配列を示す(6,615bp、配列番号:15)。305 bp BigII/MiuIリンカーはヌクレオチド#1471で始まり、ヌクレオチド#1776で終わる。残りの配列はpLF086に対応する。
【図29b】図29aの続きである。
【図29c】図29bの続きである。
【図30】pLFl02の制限地図を示す。
【図31a】pLFll16Aの完全なDNAの塩基配列を示す(6,450bp、配列番号:16)。311bp Miul/Miulリンカーはヌクレオチド#1092で始まり、ヌクレオチド#1403で終わる。残りの配列はpLF086対応する。
【図31b】図31aの続きである。
【図31c】図31bの続きである。
【図32】pLF1116Aの制限地図を示す。
【図33a】pLFl00の完全なDNAの塩基配列を示す(6,247bp、配列番号:17)。302 bp DraIII/MiuIリンカーはヌクレオチド#898で始まり、ヌクレオチド#1200で終わる。残りの配列はpLF086に対応する。
【図33b】図33aの続きである。
【図33c】図33bの続きである。
【図34】pLFl00の制限地図を示す。
【図35a】pLFl20の完全なDNAの塩基配列を示す(6,048bp、配列番号:18)。311 bp DraIII/MiuIリンカーはヌクレオチド#898で始まり、ヌクレオチド#1209で終わる。残りの配列はpLF086に対応する。
【図35b】図35aの続きである。
【図35c】図35bの続きである。
【図36】pLF120の制限地図を示す。
【図37a】pLF043の完全なDNAの塩基配列を示す(5,109bp、配列番号:19)。CDV HAコード配列はヌクレオチド#35で始まり、ヌクレオチド#2175で終わる。CDV HA ORFストップコードンは#1847である。部分的ワクシニアH6プロモータはヌクレオチド#7で始まり、ヌクレオチド#35で終わる。残りの配列はpBSSK+に対応する。
【図37b】図37aの続きである。
【図37c】図37bの続きである。
【図38】pLFO43の制限地図を示す。
【図39a】pLFO98の完全なDNAの塩基配列を示す(5,070bp、配列番号:20)。CDV HA発現カセットはヌクレオチド#1で始まり、ヌクレオチド#2372で終わる。残りの配列はpLF069に対応する。
【図39b】図39aの続きである。
【図39c】図39bの続きである。
【図40】pLF098の制限地図を示す。
【図41a】pLF099Aの完全なDNAの塩基配列を示す(8,618bp、配列番号:21)。CDV HA発現カセットはヌクレオチド#3120で始まり、ヌクレオチド#5494で終わる。残りの配列はpLFI05に対応する。
【図41b】図41aの続きである。
【図41c】図41bの続きである。
【図41d】図41cの続きである。
【図42】pLF099Aの制限地図を示す。
【図43a】pLFl08の完全なDNAの塩基配列を示す(4,965bp、配列番号:22)。ワクシニアウィルスH6プロモータの3'端領域は位置#1と#29との間に位置する。CDV Flコード配列は位置#30から始まって、位置#2018で終わる。残りの配列はpBSSK+に対応する。
【図43b】図43aの続きである。
【図43c】図43bの続きである。
【図44】pLFl08の制限地図を示す。
【図45a】pLF111の完全なDNAの塩基配列を示す(5,241bp、配列番号:23)。CDV Fl発現カセットは位置#1から始まって位置#2(556)で終わる。残りの配列はpLF069に対応する。
【図45b】図45aの続きである。
【図45c】図45bの続きである。
【図46】pLF11lの制限地図を示す;
【図47a】pLF128の完全なDNAの塩基配列を示す(5,147bp、配列番号:24)。CDV Fl発現カセットは位置#1から始まって位置#2452で終わる。残りの配列はpLF077に対応する。
【図47b】図47aの続きである。
【図47c】図47bの続きである。
【図48】pLFl28の制限地図を示す。
【図49a】pLF130Aの完全なDNAの塩基配列を示す(8,792bp、配列番号:25)。CDV Fl発現カセットは位置#3126から始まってヌクレオチド#5669で終わる。CAV2 SalI.B左フランキングアーム(3,091bp)は位置#1と3,091との間に位置する。CAV2 SalI.B右フランキングアーム(182bp)は位置#5688と5,870との間に位置する。残りの配列はpLF105に対応する。
【図49b】図49aの続きである。
【図49c】図49bの続きである。
【図49d】図49cの続きである。
【図50】pLFl30Aの制限地図を示す。
【技術分野】
【0001】
本発明は組換え型アデノウィルス(adenoviruses)と、その製造方法と、その使用(複製DNAのためのベクターとしての使用を含む)と、その発現産物と、発現産物の使用とに関するものである。
本発明はさらに、プロモーターと形質発現カセットに関するものであり、特に切出したプロモーターとそれを含む形質発現カセットに関するものである。
本発明は特に、組換え型イヌアデノウィルス(canine adenoviruses, CAV)と、その製造方法と、その使用(DNAを複製するためのベクターとしての使用を含む)と、その発現産物と、発現産物の使用とに関するものである。
本発明は特に、組換え型CAV2ウィルス、外来性DNAがCAV2 E3および/または右ITRとE4転写単位との間のゲノムの右端に挿入されたものと、その製造方法と、その使用(免疫学、免疫療法での使用、ワクチン治療用組成物またはクローン化、複製、DNA発現用のためのベクターとしての使用、組成物またはベクターの使用方法を含む)と、その発現産物および発現産物の使用は関するものである。
【0002】
より一般的には、本発明はCAVゲノムの本質的でない領域またはそれの一部が欠失された外来DNAを含むように合成で修正されたCAVに関するものである。このCAVは、CAVが自然に複製される細胞に対して感染力をもつCAVとして入れられるのが好ましい。CAVゲノムの重要でない領域(またはその一部がCAVから欠失された領域)はE3領域またはそれの一部であるのが好ましい。外来性DNAはE3領域、El領域、E4領域または右ITRとE4領域との間に位置する領域に存在するのが好ましい。このCAVはCAV2であることができる。
【0003】
この組換え型CAVはヘテロジニアス(heterologous)なDNAの発現またはクローン化のためのベクターであることができる。このヘテロジニアスなデオキシリボ核酸は任意の所望の発現産物をコードできる。好ましい発現産物は以下を含む:そのエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療用遺伝子または融合蛋白。すなわち、このヘテロジニアスなデオキシリボ核酸はこれらの任意または全ての産物をコード化することができる。従って、ヘテロジニアスなDNAはトランスジェーンであることができる。
上記エピトープは抗原または免疫原であることができ、人間または家畜の病原またはトキシンのエピトープであることができる。従って、本発明は免疫学、抗原またはワクチンとしての組成物と、その発現産物にも関するものである。このCAVベクターは場合によっては宿主に直接投与できるので、本発明はCAVベクターを含む組成物にも関するものである。本発明組成物は免疫学的組成物、抗原性組成物、ワクチン組成物あるいは治療用組成物(例えば、局所または全身的な免疫応答(保護応答または遺伝子治療を含む)を刺激する組成)であることができる。従って、本発明は適切な脊椎動物宿主(動物または人間)に上記が組成を投与して免疫応答を誘発する遺伝情報を転写方法(例えば遺伝子治療)に関するものである。
【0004】
本発明の発現産物はインヴィトロでCAVベクターから単離でき、また、CAVベクターでインヴィトロに感染またはトランスフェクションされた細胞から単離できるので、本発明は生産物を発現するための方法、例えば細胞から遺伝子組換え、分割(cleaving)または連鎖反応(ligating)を用いて外来性DNAをCAVへ挿入し、得られた組換え型CAVをインヴィトロで適切な細胞へ感染またはトランスフェクションし、必要に応じてその発現産物を抽出または分離することから成る方法に関するものである。
発現産物は抗原性、免疫性または保護(ワクチン)応答物を提供するので、本発明はその生産物すなわち抗体およびそれの使用にも関するものである。特に、発現産物は誘導(elicit)抗体にすることができる。抗体はモノクローナル抗体の形に作ることができる。抗体または発現産物はキット、アッセイ、バインディングを含むテストなどで使うことができる。従って、本発明はこれらの使用にも関するものである。
【0005】
本発明の組換え型はDNAの複製に使えるので、本発明は、細胞を組換え物で感染させ、そこからDNAを収穫する、組換え型CAVをベクターとして用いたDNAの複製方法にも関するものである。得られたDNAはプローブまたはプライマとして使用でき、また、増殖(amplification)のためにも使える。
本発明はさらに、、組換え型ウィルスまたはプラスミドのためのプロモータおよびそのプロモータを含む発現カセットにも関するものである。
この観点は、本発明はウィルスまたはプラスミドが挿入される系で与えられたトランスアクチベイティング蛋白がトアンスアクチベイトされた(transactivated)領域と、プロモータの最小のプロモーター領域とを有する組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータに関するものである。本発明はプロモータから成る形質発現カセットと、プロモータまたは形質発現カセットを含むウィルスまたはプラスミドにも関するものである。この形質発現カセットは切り出した(truncated)機能性ポリアデニル化信号を含むことができる。
以下、明細書では多数の刊行物が引用されるが、これらの刊行物の情報は「参考文献」の項に記載されおり、引用時点でも説明してある。本明細書で引用される刊行物およびそれら刊行物で引用される文献の内容は本願明細書の一部をなすものとする。
【背景技術】
【0006】
各種ウイルス性ベクター系と、その使用、この種の系を用いた蛋白の形質発現のための外来性DNAと、この種の蛋白質類の使用およびこの種の蛋白からの産物の使用は多数の特許および科学刊行物に記載されている。
例えば以下のものを挙げることができる:ウイルス性ベクター系の発現のための組換え型ポックスウィルス(例えばワクシニア、アビポック(avipox)ウィルス)および外来性DNA(例えば、組換え型アビポック(avipox)ウィルス(ワクシニアウィルス)(特許文献1および特許文献2に記載されている);狂犬病グリコプロテイン(G)、遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、ウシ属の白血病ウィルスのgp5l,30エンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAV‐1 env遺伝子、NP(Chicken/ペンシルバニア/1/83インフルエンザウィルスのヌーデオプロテイン(nudeoprotein)遺伝子)、マトリックス、そして、伝染性の気管支炎ウィルス;HSV gDのプレポリマー遺伝子;エントモポックス(entomopox)プロモータ(特に特許文献3)、例えば、組換え型ワクシニアウィルス(アビポック(avipox)ウィルス);特にヘルペスウイルス・グリコプロテインをコード化しているDNA;特許文献4(例えば、組換え型ワクシニア(avipox);狂犬病、ヘパチチス(Hepatitis)B、JEV、YF、Dengue、はしか、仮性狂犬病、エピスタイン(Epstein)から抗原をコード化している外来性DNA‐バー、HSV、HIV、SIV、EHV、BHV、HCMV、犬パルボウィルス、馬の風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、特にお多福風邪(NDV));特許文献5(例えば組換え型ワクシニア、アビポック(avipox)、モリビリウイルス(例えば、はしかF、ヘマグルチニン);特許文献6(例えば、組換え型ワクシニアウィルス; HSV tk、グリコプロテイン(例えばgB、gD)、(風邪HA)ヘパチチス(Hepatitis)B(例えばHBsAg);特許文献7、特許文献8(組換え型ポックスウィルス、フラビウイルス構造蛋白質); 特許文献9(例えば、組換え型ポックスウィルス、免疫不全ウイルス);特許文献10(例えば、組換え型ポックスウィルス、特にIBDV);特許文献11および特許文献12(例えば、組換え型ポックスウィルス;特にサイトカインおよび/または腫瘍関連抗原); PCT/US94/06652(Plasmodiumライフサイクルの各段階からのプラズモディウム抗体)。
【特許文献1】米国特許第5,174,993号明細書
【特許文献2】米国特許第5,505,941号明細書
【特許文献3】米国特許第5,338,683号明細書
【特許文献4】米国特許第5,494,807号明細書
【特許文献5】米国特許第5,503,834号明細書
【特許文献6】米国特許第4,722,848号明細書
【特許文献7】英国特許GB 2 269 820 B明細書
【特許文献8】米国特許第5,514,375号明細書
【特許文献9】WO 92/22641
【特許文献10】WO 93/03145
【特許文献11】WO 94/16716号
【特許文献12】米国特許出願第08/184009号明細書
【0007】
バクロウイルス発現系と、発現のための外来性DNAおよび組換え型蛋白の精製法は非特許文献1(例えば風邪HA発現のためのCh.18、組換え型蛋白精製技術のためのCh.19参照)、非特許文献2(皮膚テストと、AIDSのためのテストキット、HIV1 env遺伝子の一部を含むバクロウイルス発現系を考察、特許文献13および特許文献14を引用)に記載されている。
特許文献15はヘルペスウィルスをベクターに関するものである。
ポリオウィルスおよびアデノウィルス・ベクター系もある(例えば非特許文献3〜6参照)。
特許文献16はSmaI部位から早い領域4(E4)のためのプロモータまでの逆終末反複領域近くの終わりまでの領域内にプロモータ-遺伝子配列を含む変性されたCAV2に関するものである。
【非特許文献1】リチャードソン、C.D.(編)、「Methods in Molecular Bioloav 39, "Baculovirus Expression Protocols」 (1995 Humana Press Inc.)
【非特許文献2】スミス達、「Production of Huma Beta Interferon in Insect Cells Infected with a Baculovirus Expression Vector」Molecular and Cellular Biology, Dec., 1983, Vol. 3, No. 12, p. 2156-2165; EPA 0 370 573
【非特許文献3】Kitson達、J. Virol. 65, 3068、 3075, 1991)
【非特許文献4】Grunhaus 達, 1992, "Adenovirus as cloning vectors," Seminars in Virology (Vol. 3) p. 237‐52, 1993
【非特許文献5】Ballay達EMBO Journal, vol. 4, p. 3861‐65
【非特許文献6】グレアム(Tibtech 8, 85-87, April, 1990;Prevec達)J. Gen Virol. 70, 429‐434
【特許文献13】第1986年10月16日に出願の米国出願第920,197号明細書
【特許文献14】欧州特許公報No.265785
【特許文献15】米国特許第4,769,331号明細書
【特許文献16】PCT WO91/11525
【0008】
CAV、特にCAV2には多くの問題がある。これらの問題のいくつかを以下説明する。重大な問題はCAVゲノムは外来性DNAの限られた量しか受けないということである。すなわち、外来性DNAの限られた量しかCAVゲノムに挿入できない。従って、CAVは「挿入できる大きさが制限され」。これはCAVをクローン化および発現で使うベクターの場合には重大な問題である。
口腔鼻腔径路での多くのウィルス感染の伝染効率は同じ径路でのウィルスベクターをベースにしたワクチン候補の効果で与えられている。しかし、ワクチン接種を受けた人でワクチンは大抵複製し、一般の生育環境と接触し、展開することが分かっている(例えばシュワルツ達、1974、Mueller達、1989、Oualikene達、1994を参照)ので、適切なウイルス性ベクターを採択したか否かは明らかでない。
完全な安全性を確保するためには、ベクターの選抜で明らかな安全性が確立されるような生きた弱毒ワクチンを特徴付けるのが好ましい。人間のワクチン接種のために生きた弱毒ウィルスを複製する各種ベクターが考えられている。現在公知の文献には以下のアプローチが記載されている:ヒトアデノウイルス(HAVs)セロタイプ4および7(Lubeck達、1989、チャンダ達、1990、Chengalvala達、1991、1994、Hsu達、1994)(インフルエンザウィルス(Garcia-Sastre and Palese, 1995のレビュー)およびポリオウィルスと関連ウィルス(Girard et al., 1995のレビュー)。
【0009】
獣医学の分野では、非経口または自然径路の感染(局所回復を刺激)によるワクチンを目的とした組換え型ベクターとして生きた弱毒ウィルスを複製したいくつかのベクターが現在分析されている。ポクスビリダ(poxviridae)科がこの点で最も特徴的なものである[例えば鶏痘ベースのベクター(Edbauer達、1990、テーラー達、1995とその参照文献参照)、herpesviridae科(例えば、仮性狂犬病ウイルスベースのベクター(Sedegah達、1992、Mettenleiter達、1994、Hooft van Iddekinge達、1996とその参照文献参照)、シチメンチョウ‐ヘルペスウィルスベースのベクター(ロス達、1995、Darteil達、1993 とその参照文献参照)、ネコ科のヘルペスウィルスべースのベクター(コール達、1990、Wardley達、1992、Willense達、1996)、伝染性のウイルス性呼吸器疾患ウィルスをベースにするベクター(Guo達、1994)、ウシ属のヘルペスウィルスをベースにするベクター(キット達、1991)]およびAdenoviridae科のより小さい構成メンバ(ウシ属のアデノウィルス3をベースにしたベクター(Mittal達、1995)]。
【0010】
犬種は口腔鼻腔免疫化に適したモデルを提供する。すなわち、弱毒化されたワクチン株が存在する犬アデノウィルス セロタイプ2(CAV2)は非経口経路または口腔鼻腔径路で安全に投与することができる犬のワクチン接種用の生きた免疫化媒体である。イヌのジステンパーウィルス(CDV)感染はこの標的種の呼吸感染のよい例を提供する。更により直接的な実験的CDV抗原投与系を用いることによって、CAV2 をベースにしたワクチン候補と以前に開発された古典的なCDVワクチンとを直接対比することができる。
CAV2はディチフィールド(Ditchfield)達(1962)によって、イヌの上気道感染の大発生から最初に単離された。その後、このウィルスは米国および欧州で呼吸疾患を有するイヌの呼吸管から単離された(Binn 達、 1967, Appel and Percy, 1970, Assaf 達、 1978, Danskin 1973)。実験研究によってCAV2のエーロゾルを植菌すると穏やかな呼吸疾患になるようになった(Swango達、1970、Appel、1970)。いくつかのCAV2 ベースのワクチンが開発され、広く子犬および成犬のワクチン接種で世界的に用いられている。CAV2の免疫化によってCAV1の血清学的株(予防注射されないイヌでは致命的な株)に実験的に抗原暴露した時に保護されることが証明された(フェアチャイルドとコーエン(1969)、Appel達、1973、バス達、1980)。ワクチンとしてのCAV2の明白な安全性はイヌワクチンによって誘発される疾患およびワクチンに付随する合併症が人類を含む他の動物種で過去30年間無いことでよく証拠される。更に、血清学的調査の結果から、多くの野生動物(キツネ、アライグマ、ゲジゲジおよびマングース)がCAV2またはそれに抗体的に関連したウイルス感染(Summer達(1988))症候に曝されていることが示されている。従って、犬アデノウィルス セロタイプ2(CAV2)のワクチン株はイヌの安全な複製コンピテント(ワクチン接種のための効果的なベクターをベースとするワクチン候補として考慮できる宿主制限ウィルス)の特異な例を提供する。
【0011】
HAVsは価値ある哺乳類の細胞発現ベクトル(グレアム達1988を参照)であり、組換えウイルス型ワクチン候補(Randrianarison-Jewtoukoff and Perricaudet 1995, Imler 1995を参照)であり、現在、遺伝子治療(Perricaudet and Perricaudet 1995を参照)のベクターとして評価されている。2つの主要なグループのHAVsがあり、組換え型HAVsは第3のグループである。
【0012】
これらのアデノウィルスベクターの最初のグループは、El領域を欠失したウィルスをベースにした複製不適格な組換え型アデノウィルスに対応する。El領域は組織培養のウイルス複製にとって重要な蛋白質類をコード化する。しかし、無能力の組換え型アデノウィルスは、このエル領域(Haj Ahmad達(1986)を本質的に発現する293の細胞系(グレアム達、1977)でエル領域の欠失の複製を伝播することが証明された。
El領域の欠失はHAVsに挿入ができる外来性DNAの量を増やすだけでなく、ヒト細胞の複製を制限し、人間での組換え型HAVsの形質転換の安全性を改善する。家畜および人間の病原を防ぐための現在の大部分のHAVベースのワクチン候補はEl欠失したベクターをベースにしている。その転写能力は限られているが、抗原投与実験での保護データは報告されている(Prevec達、1989、McDermott達、1989、Lubeck達、1989、Eloit達、1990、Ragot達、1993、Wesseling達、1993、Both達、1993、Gallichan達、1993、Hsu達、1994、Breker-Kiasser達、1995)。ベクター複製がない場合でも、防御免疫応答誘発特性は他の宿主制限ウイルス性ベクター与えられる。その最も有望なものはカナリポックス(canarypox)ウイルスベースのベクターALVACである(テーラー達(1991)、Perkus達(1995)を参照)。
【0013】
最終ゴールが複製コンピテント・アデノウィルス・ベクターである場合には挿入部位としてのEl領域を使用することは望ましくないので、El領域は今まで欠点であり、問題が有った。人間に対して安全な複製コンピテント・アデノウィルスが要求されるときにはこの欠点と上記問題が複合する。特に、HAVsのEl領域欠失がヒト細胞の複製を制限し、人間に関する安全性を改善するが、以下の文献に記載されているように、El形質転換細胞系とEl欠失した組換え型アデノウィルスとの間の遺伝子組換えの可能性、従って、El形質転換細胞系の安全性プロファイルは疑わしく見えるので、欠失したEl領域を用いることの利点は単なる可能性で、El領域を欠失したアデノウィルスを用いることは上記の欠点および問題点の解決にはならないと思える(El領域を欠失したアデノウィルスの増殖がEl領域を表現する細胞であるため)。
アデノウィルス・ベクターの第2のグループはヒト細胞の複製コンピテントで、人間以外の大部分の動物細胞では複製である組換え型アデノウィルスに対応する。これらのウィルスは異種遺伝子発現カセットを有するE3領域の一部の置換で特徴づけられる。E3領域がインビトロおよびインビボの両方で本質的でないことは伝染性ウィルス発生で示された(ケリーとルイス、1973、Kapoor達、1981、Morin達、1987、Lubeck達、1989)。従って、E3領域の一部の置換で多くの組換え型HAVsが作り出された(モリン達、1987、Chengalvala達、1991、1994、Prevec達、1989、ジョンソン達、1988、Lubeck達、1989、Dewar達、1989、Natuk達、1993、Hsu達、1994)。
【0014】
しかし、E3領域を用いてコードされる蛋白は宿主免疫応答(WoldとGooding 1991を参照)を種種の観点で変えるので、E3欠失は対応する組換え型ウィルスの病原プロファイルに何らかの影響を与えると思える。事実、HAVセロタイプ5からのE3領域の欠失がウィルス肺の病原性(ギンズバーグ達、1989)を増やすことはコトンラット・モデルで示された。しかし、E3領域内で部分的に欠失した組換え型ウシ属のAd3が親のwtウシ属のAd3で観測されることと類似したコトンラットの疾患を生産することも示されたので、ウシ属の動物の安全性がベクターをAd3をベースにすることはウシ用の生きた組換え型ウィルス・ワクチンでは充分であることを示唆している(Mittal達、1996)。この結果も、アデノウィルスものE3領域内の欠失の影響はケースバイケースであることを示している。
【0015】
CAV2 E3領域は既に同定され、特徴付けられている(Linne、1992)。しかし、利用できる発表されたデータ(Linne 1992)からは、CAV2 E3領域の挿入部位の明確な定義は明らかではない。DNAの塩基配列解析から、CAV2 E3領域の組織はHAVsのものとは大幅に異なることが明らかになっている。CAV2 E3領域は長さが1.5kbpであって、3つの転写解読枠(orf)のみを含むのに対して、ヒトアデノウイルスE3領域は少なくとも8つの転写解読枠(orf)を含んだ、少なくとも3kbpの範囲に延びている。これらのorfsはどれもHAV E3 orfsと有意レベルの相同性を有していない。この予備的な比較解析から、人間と犬のアデノウィルス・ゲノムは違って発展したと考えるのが合理的に見える。
アデノウィルス科(Imperiale達(1995)参照)を特徴づける錯体スプライシングおよびポリアデニル化パターンを用いたCAV2 E3領域内の挿入部位の定義はさらに複雑である。RNAスプライシング・ドナーおよびアクセプター部位は、たとえそれらの暗号配列がE3領域の外側に局所化されているとしても、E3領域内に局所化されたいくつかの重要なメッセンジャーRNAの成熟にとって重要である。
【0016】
更に、E3領域は高転写活性条件のゲノム領域内に位置する(シャープ達(1984)参照)ので、この部位での外来性DNAの組込みは組換え型ウィルスの生態学に潜在的に有害な影響を与える。さらに、E3領域は主要な後期プロモータ(MLP)の下流に位置するので、組換遺伝子の転写とMLPで始まる転写との間の干渉が示された(Zu達、1995)。
従来法の課題は組換え型ウィルスの表現型改変個所および転写活性でない領域の挿入部位の定義を最小にすることにある。一般的には、従来技術の問題点はE3領域にあるといえる。
【0017】
第3のグループの組換え型HAVsは右の反転終末反複(ITR)とE4プロモータとの間の組換えDNAの組込みをベースにしている。このITRはウイルス性ゲノムDNAのウィルスDNA反復および効率的パッケージングにとって重要な配列を含む。右の反転終末反複(ITR)とE4プロモータとの間の領域が外来性DNA配列(斎藤達、1985、チャンダ達、1990)を受け入れが、アデノウィルスをベースにしたベクターは組換え型hAd5のパッケージング能力が野生型ゲノム(Bett達、1993)の約105%のゲノムに限られるので、それらが運ぶことができる外来性DNAの量には厳しい限界があり、従来法と同じ問題点がある。
右のITRとE4領域との間の領域はCAV2組換え型ウィルス生成のための追加の挿入部位の候補になる。PCT WO 91/11525はSmaI部位がITRの左端部の近くの潜在的な挿入部位に関するものである。W091/11525の教えとは逆に、本発明者は組込み部位の上限があると考えられる。それは400のbp DNAフラグメントが挿入でき、約1 kbpののような大きいフラグメントの組込みは繰り返し失敗したものである。従来法の課題はこの部位の利用にある。
すなわち、これまではE4プロモーター領域が欠失物であり、それが問題になっていた。
【特許文献17】WO 91/11525
【0018】
分子レベルでのCAV2ゲノムの初期の性格付けは文献に記載されている。CAV2およびCAViの菌株(Jouvenne達、1987、Macartney達、1988、SpibeyとCavanagh 、1989)およびEl、E3およびITRs領域に対応する配列解析の制限解析が報告されている。イヌアデノウィルスのゲノムの全体機構は他のAdenoviridae科構成メンバに類似しているが、CAV2 の遺伝子的なE3領域の明確な組織はユニークである。
従って、Adenoviridaeの他の構成メンバから単に1つの構成メンバを推論することはできないので、従来法にはさらに他の問題がある。
【0019】
さらに、前記の欠失または問題を解決するときには、E3のような外来プロモータまたはMLPプロモータに対する依存を避けるのを好ましい。しかし、組換遺伝子の発現のパターンは全体的な発現、従ってワクチン組成または免疫学組成(Darteil達、1995、Xu達、1995、Hooft Van Iddekinge達、1996)の組換え効能の重大なパラメータである。
いくつかの細胞およびウィルスのプロモータは組換え型HAVsの誘導に関係する。中でも、最も特徴づけるものはb-actin、SV40 early、SV40 late、hAD MLPおよびhCMV-IE(Zu達,1995)である。hCMV―IEプロモータは組織培養で組換え型蛋白発現の最も高レベル且つ最も長い存在と関連するので、これが上流域制御領域になることは確かである。このプロモータもここまでにテストされたほとんどの細胞系で作動するように思える。細胞型にインデペンデントなプロモータ活性の可能性は明らかに強みである。
【0020】
hCMV-IEプロモータはHAV感染(ゴーマン達、1989)で転写活性化できることは証明されている。このプロモータ(約850bp)の大きさは組換え型CAVベクターの寸法限界で問題である。従って、このプロモータを有する過去の成功から推論することができるのは組換え型CAVベクターだけである。
アデノウィルスがウィルスDNA反復の開始期の後に、強く細胞の蛋白合成を抑制することは公知である(チャンとシュナイダー(1993)参照)。従って、複製コンピテントな組換え型アデノウィルスはDNA合成の開始期の後は組換え型蛋白発現で限界されてきた。
【0021】
同様に、斎藤達(1985)は組換え型ヒトアデノウイルス・セロタイプ5ではほとんど組換え型蛋白が得られられないことを組換え型メッセンジャーRNAの高生産で証明している。
最近のアデノウィルス・メッセンジャーRNAはその5'の翻訳されていない領域(5'UTR)でのトリパr−テーリーダー(TPL)配列の存在で特徴づけられる。このTPLの存在は最近のアデノウィルス・メッセンジャーRNAの翻訳可能性の重要な成分である。更に、hAd5バックグラウンドにおいて、TPLの存在がアデノウィルス後期プロモータ(Thummel達1983)から発現される組換え型SV40 T抗原の翻訳調節の特色であることも証明されている。
【0022】
発現カセットのデザインでの他の重要な問題はポリアデニル化信号の大きさである。従来法のさらに他の問題は、CAV2 DNAを単層にトランスフェクションするために条件を確立することにある。精製された裸のアデノウィルスDNAの感染性は低い。リン酸カルシウムを用いた手順で、グレアムとVan derバーグ(1973)は1 pfu/mgの精製されたDNAの収率を報告しているが、換え型ウィルスを単離させるための効率的なプロセスではない。他にいくつかのアプローチは本問題を解決するために提案されたが、従来法の問題は充分に解決されず、特に安全性の問題は言及されていない。
【0023】
例えば、DNA蛋白複合体が精製され、裸のDNAで感染性が向上する(5x103 pfu/mg)と報告されている(Sharp 達、1976)。同様に、アデノウィルスDNAを共有結合で閉環したものが感染力をもつことが示された(グレアム、1984)。
組換え型HAVsを誘導するのに広く使われている手順はHAV El領域で変形した293細胞系を用いたものである(グレアム達、1977)。ウシ属のおよび犬アデノウィルス組換え型の誘導が対応するアデノウィルスEl領域(PCT WO 91/11525、Mittal達、1995a)で変形する細胞系の利用に依存することが報告されている。いくらかのアデノウィルスのEl領域を用いてコード化される遺伝子が齧歯動物細胞(Grand、1987)を用いてチェックされ、トランスフォーメーションに貢献することは示されたが、El形質転換細胞系の安全性プロファイルは疑わしい。また、アデノウィルスEl領域内の有力なtransactivatorsの存在はよく確認され、El形質転換細胞系に関する安全性に関心が延びている(ネヴィンズ(1993)参照)。
従って、El形質転換細胞系の使用から独立した、好収率を有するトランスフェクション条件は技術の重要な前進になる。
【特許文献18】WO 91/11525
【0024】
以上のとおり、組換え型CAV、好ましくは外来性DNAと本質的でない領域またはそれの一部が欠失された領域息とを有する組換え型CAV2を挿入し、特にCAVが自然に模写される細胞に対して感染力をもつCAVのパックとして挿入し、または、E3内および/または右のITRとE4転写単位との間の右の端を含むゲノム内に外来性DNAを含むCAVを挿入することと、この種の組換え体の製造方法およびその使用は上記および下記の文献には提案されなかった。また、ウィルスまたはプラスミドが挿入される系を用いてトアンスアクチベイトされた領域と、このプロモータの最小のプロモーター領域とを有する組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータや、このプロモータから成る発現カセットや、プロモータまたは発現カセットを含んでいるウィルスまたはプラスミドは提案されなかった。この組換え型CAV、プロモータ、この組換え型CAVおよびプロモータの製造方法、発現カセット、プロモータまたは発現カセットを含むウィルスまたはプラスミドは、人間に関してはCAVが非複製ベクターであり、プロモータおよび発現カセットは組換え型ウィルスの挿入寸法限度にしてあるので、従来の組換え体より優れたものである。
【発明の開示】
【発明が解決しようとする課題】
【0025】
本発明の目的は、組換え型アデノウィルス、好ましくは組換え型イヌアデノウィルス2(CAV2)のような組換え型イヌアデノウィルス(CAV)を提供することにある。
【0026】
本発明のさらに他の目的は、外来性DNAを含む上記組換え体、好ましくは本質的でない領域に外来性DNAを含み、CAVゲノムの本質的でない領域を有し、またはその一部を削除したものを提供することにある。そして、好ましくは、CAVを自然に複製する細胞に対してこの組換え体を感染性CAVとしてパックした組換え体を提供することにある。
本発明のさらに他の目的は、外来性DNAがE3またはE3と右のITRとE4形質移入単位との間の領域の両方に挿入される外来性DNAを含んだ上記組換え型CAVを提供することにある。
【0027】
本発明のさらに他の目的は、複写活性のある切り詰めたプロモータと、このプロモータを含む発現カセットと、プロモータまたは発現カセットを含むウィルスまたはプラスミドを提供することにある。また、ポリアデニル化信号を含む切り詰めた発現カセットを提供する。
本発明のさらに他の目的は、上記組換え体、この組換え体からの生産物を発現させる方法、組換え体または発現産物を含む組成、発現産物の使用方法、組成の使用方法、組換え体からのDNA、組換え体からDNAを複製する方法のいずれか一つまたは全てを提供することにある。
本発明の別の目的は、アデノウィルスベース、例えばCAVベース、好ましくはCAV2ベースのベクターまたはこのベクターを含む組成または上記の任意または全ての欠点および/または従来法の問題に関連したベクターの製造方法または使用方法を提供することにある。
【課題を解決するための手段】
【0028】
驚くべきことに、本発明はCAVゲノムの本質的でない領域またはその一部がCAVから欠失された、外来性DNAを含む合成で変性されたCAVを提供する。CAVはCAVを自然に複製する細胞に対して感染性CAVとしたパックされるのが好ましい。本質的でない領域またはその一部はCAVゲノムから欠失できる。欠失した領域またはその一部が本当に本質的でないかどうかは欠失に起因する組換え型CAVの生育性および安定度から確かめる。CAVゲノムまたはその一部の領域がCAVから欠失する必須でない物はE3領域またはそれの一部であるのが好ましい。外来性DNAは本質的でない領域にも存在する。外来性DNAが挿入された領域またはその一部が本質的でないかどうかは外来性DNAの組込みに起因する組換え型CAVの生育性および安定度から確かめる。CAVゲノムに外来性DNAの組込みのための本質的でない領域としてはE3領域、El領域、E4領域または右のITRとE4領域との間の領域が好ましい。
【0029】
本発明は、驚くことに、ヘテロジニアスなDNAがE3にあるか、E3および右のITRとE4形質移入単位との間の領域にあるヘテロジニアスなDNAをCAVゲノムの本質的でない領域に含む組換え型CAVを提供する。
これら具体例のCAVは好ましくはCAV2である。
【0030】
本発明はさらに、組換え型CAVから成るヘテロジニアスなDNAの発現またはクローン化ベクターを提供する。
ヘテロジニアスなDNAは重要なエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療用遺伝子または融合蛋白から成る発現産物をコードする。
このエピトープは抗原または免疫原であるか、家畜または人間の病原またはトキシンからの免疫学的に活性なフラグメントである。
【0031】
このエピトープは家畜の病原またはトキシンまたは家畜の病原またはトキシンの抗原または他の抗原からの抗原であることができ、または、例えば病原に対して応答を導き出す他の抗原またはトキシンからの、病原に関して応答を導き出すトキシン、例えばモリビリウイルス抗原イヌジステンパーウィルスまたははしかまたは牛疫抗原HAまたはF;狂犬病グリコプロテイン(例えば狂犬病グリコプロテインG);鳥風邪抗原(例えばシチメンチョウ風邪HA)Chicken/ペンシルバニア/1/83風邪抗原、例えばヌーデオプロテイン(nudeoprotein)(NP);ウシ属の白血病ウィルス抗原(例えばgp5l,30エンベロープ);ニューカッスル病ウイルス(NDV)抗原(例えばHNまたはF);ネコ白血病ウィルス抗原(猫白血病ウイルス)(例えば猫白血病ウイルス外膜蛋白);RAV-1 env; RAV-1 env;伝染性の気管支炎ウィルスのマトリックスおよび/またはプレポリマー;ヘルペスウイルス・グリコプロテイン(例えばねこヘルペスウィルスからのグリコプロテイン、馬のヘルペスウィルス、ウシ属のヘルペスウィルス、仮性狂犬病ウイルス、犬ヘルペスウィルスまたはサイトメガロウィルス);フラビウイルス抗原(例えば日本脳炎ウィルス(JEV)抗原);免疫不全ウイルス抗原(例えばネコ科の免疫不全ウイルス(FIV)抗原またはサル免疫不全ウイルス(SIV)抗原);パルボウィルス抗原(例えば犬パルボウィルス);馬の風邪抗原;マレック病ウィルス抗原;ポックスウィルス抗原(例えば先天性四股欠損症抗原、カナリポックス(canarypox)ウィルス抗原または鶏痘ウィルス抗原)または伝染性包性(bursal)疾患ウィルス抗原(例えばVP2、VP3、VP4)にすることができる。
【0032】
このエピトープは人間の病原またはトキシンまたは人間の病原またはトキシンの抗原または他の抗原からの抗原であることができ、または、病原に対してたとえば応答を導き出す他の抗原またはトキシンから応答を導き出すトキシン:モリビリウイルス抗原(例えばはしかウィルス抗原(例えばHAまたはF));狂犬病グリコプロテイン(例えば狂犬病ウィルス・グリコプロテインG);風邪抗原(例えばインフルエンザウィルスHAまたはN);ヘルペスウイルス抗原(例えば単純ヘルペスウィルス(HSV)のグリコプロテイン、ヒトサイトメガロウイルス(HCMV)、エピスタイン(Epstein)-バー);フラビウイルス抗原、JEV、Yellow FeverウィルスまたはDengueウィルス抗原;Hepatitisウィルス抗原(例えばHBsAg);免疫不全ウイルス抗原(例えばgp120のようなHIV抗原、gpl6O);ハンタン(Hantaan)ウィルス抗原;C.テアニ(tetani)抗原;お多福風邪抗原;pneumococcalな抗原(例えばPspA);Borrelia抗原(例えばOspA、OspB、Borrelia burgdorferiのようなライム病と関連があるBorreliaのOspC、Borrelia afzelliおよびBorrelia garinii);水ぼうそう(水痘帯状ヘルペス)抗原またはPlasmodium抗原にすることができる。
もちろん、上記リストは典型的なもので、このエピトープは任意の家畜または人間の病原または家畜または人間の病原の抗原からの抗原であることができる。
【0033】
ヘテロジニアスなDNAは成育因子または治療の遺伝子であることができるので、本発明の組換え型CAVは遺伝子治療で使うことができる。遺伝子治療は遺伝情報を積み換えることを含む。遺伝子治療および免疫療法に関しては米国特許第5,252,479号、さらにはWO 94/16716および第1994年1月19日に出願の米国特許願第08/184009号を参照。これらの内容は本願明細書の一部をなす。成育因子または治療遺伝子は疾患ファイティング蛋白、癌を処理する分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードすることができる;成育因子はさらに例えば治療用遺伝子がαグロビンをコードする遺伝子から成るグループ、β‐グロビン、γ‐グロビン、顆粒球マクロファージ-コロニー刺激因子、腫瘍壊死因子、インターロイキン、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、EPO、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随する抗原またはB7から選ばれる。
【0034】
本発明はさらに、組換え型CAVウィルスまたはベクターと、薬学的に許容可能なキャリアまたは希釈剤を含む免疫組成物、免疫学組成物またはワクチン組成物を提供する、組換え型CAVウィルスまたはベクター(またはそれの発現産物)を含む免疫学の組成は局所的または全身的な免疫応答を導き出す。応答は保護であってもよいが、そうでなくてもよい。組換え型CAVウィルスを含む免疫的組成またはベクター(またはそれの発現産物)も同様に局所または全身的な免疫応答を導く。ワクチン組成は局所的または全身的な保護応答を導き出す。従って、「免疫学組成物」および「免疫組成物」は「ワクチン組成物」を含む(前者2つの用語は保護組成物でもよい)。
【0035】
従って、本発明は、組換え型CAVウィルスまたはベクターと、薬学的に許容可能な担体または希釈剤から成る免疫組成物、免疫学組成物またはワクチン組成物を投与して宿主脊椎動物の免疫応答を誘発する方法を提供する。本明細書では人間を除いて「動物」は全ての脊椎動物種を含む;「脊椎動物」は動物および人間を含む全ての脊椎動物を含む。もちろん、「動物」のサブセットが「哺乳類」である。本明細書では人間以外の全ての哺乳類を含む。
人間の投与では、本発明組換え型CAV(特にCAV2)は生産的複製のない発現をするという利点がある。従って、免疫的にバランスした(immunocompromised)個体で本発明組換え体を使用することができ、本発明の組換え型と接触する研究者の安全性のレベルを提供する。従って、本発明は組換え型CAVウィルスまたはベクター(例えばCAV2)を投与するか宿主に接種して増殖または蛋白を発現する方法と理解できる。それによって宿主が犬でなく、組換え型ウィルスまたはベクターの自然の宿主でない場合にも生産的複製のない発現ができる。
【0036】
さらに、CAV、特にCAV2がイヌのワクチン用株として使われるので、本発明はワクチン用CAVに、犬病原またはトキシンの抗原の関心の追加のエピトープ、例えばCAV2を導入して、それらの追加のエピトープを発現する組換え型CAVとするための手段を提供し、イヌに予防接種をすることでこれらエピトープおよび犬アデノウィルスの応答を導き出す手段を提供する。
追加のエピトープは犬病原(アデノウィルス以外の)またはトキシン、犬の病原性抗原(アデノウィルス以外の)またはトキシンに対してイヌまたは子犬で応答を導き出す他の抗原または犬病原(アデノウィルス以外)またはトキシン(後者の例、イヌまたは子犬のイヌジステンパーウィルスに対して保護応答を導き出すはしかHAおよびFおよびそのエピトープ、米国特許第5,503,834号参照)にすることができる。
【0037】
従って、本発明は組換え型ワクチン用CAV、たとえば、犬病原またはトキシンの任意の抗原:狂犬病、犬ヘルペスウィルス、イヌジステンパーウィルス、犬パルボウィルスなどからのエピトープをコード化するヘテロジニアスなDNAを含むことができる。この点については、1995年6月7日に出願された米国特許第5,529,780号(狂犬病化合組成)および第1995年3月29日に出願のアメリカ出願第08/413118号(犬ヘルペスウィルスDNA)、1994年4月6日に出願の第08/224657号(イヌジステンパー)、1995年4月5日に出願の第08/416646号(イヌジステンパー)および08/486,969(犬ヘルペスウィルスDNA)を参照。ここで引用される文献の内容は本発明の一部をなす。従って、本発明は複数の蛋白、例えば犬病原の抗原のような少なくとも2つのエピトープをコードする外来性DNAを含むCAV組換え体に関するものである。
【0038】
本発明はさらに、他の抗原をさらに含むCAV組換え体を含む組成物に関するものでもある。
本発明が、組換え型CAVウィルスまたはベクターおよび薬学的に許容範囲内である担体または希釈剤を含む治療用組成物を提供する。この治療用組成物は遺伝子治療および本発明の免疫療法の具体化、すなわち、遺伝情報をそれを必要とする動物および人間に移入する方法で用いられる。従って、本発明は遺伝情報を移入する方法を含む。
【0039】
他の具体例として、本発明は蛋白または遺伝子生産物を発現する方法を提供する。この方法では蛋白、遺伝子生産物または発現産物を感染させるか、本発明の組換え型CAVウィルスまたはベクターを有する細胞をインビトロでトランスフェクションし、必要に応じて抽出し、細胞から遺伝子産物、発現産物またはDNAを精製または単離する。本発明はさらにヘテロジニアスなDNAの塩基配列のクローン化または複製方法を提供する。この方法では本発明の組換え型CAVウィルスまたはベクターを細胞にインビトロまたはインビボで感染またはトランスフェクションし、必要に応じて抽出し、細胞またはウィルスからDNAを精製または分離する。
【0040】
別の態様において、本発明はCAVゲノムの本質的でない領域に外来性DNAを本発明のCAVウィルスまたはベクターに挿入することから成る組換え型の調製方法を提供する。
この方法では、好ましくは外来性DNAを挿入する前に、必須でない領域をCAVゲノムから欠失することができる。
この方法はインビボ遺伝子組換え(CAV DNAが感染力をもつものでもよい)から成ることができる。本発明方法はCAVゲノムの一部と相同なDNAの塩基配列でフランクした外来性DNAから成るドナーDNAの存在下で細胞と相溶性のある培地中でCAV DNAで細胞をトランスフェクションする。それによって外来性DNAをCAVのゲノム中に導入し、必要な場合には生体内の遺伝子組換えで修正されたCAVを回収する。
【0041】
本発明はさらに、CAV DNAを切断して切断されたCAV DNAを得、切断されたCAV DNAを外来性DNAと連鎖反応させてハイブリッドCAV−外来DNAを得、この複合型CAV-外来DNAで細胞を形質移入し、必要な場合にはそれから外来性DNAの存在下で、変性されたCAVを回収することから成る方法を提供する。
インビボ遺伝子組換えが理解されたので、本発明はCAVに対して外来のポリペチドをコードするCAVでは自然には起らないドナーDNAから成るプラスミドを提供する。このドナーDNAはCAVの本質的でない領域と同一直線上にあるCAV DNAのセグメント内にあり、CAVゲノムの本質的でない領域からのDNAがドナーDNAが側面に位置する。
【0042】
外来性DNAはDNAおよび必要な場合にはその発現物の安定した組込みを生ずる任意の向きでCAVに挿入して組換え型CAVを作ることができる。本発明の組換え型CAVウィルスの外来性DNAまたはベクターはプロモータを含むことができる。このプロモータはヘルペスウィルスからのものにすることができる。例えば、このプロモータはサイトメガロウィルス(CMV)プロモータ(例えば人間のCNV(HCMV)または鼠CMVプロモータ)であることができる。
このプロモータは好ましくは切り詰めた転写活性プロモータであるのが好ましい。この転写活性プロモータはウィルスからのトランスアクチベイティング蛋白でトアンスアクチベイトされた領域および完全長プロモータ(これから切り詰めた転写活性プロモータが誘導される)の最小プロモーター領域とから成る。
【0043】
本明細書では「プロモータ」は最小のプロモータと上流域調節配列に対応するのDNA塩基配列の会合から成る。「最小のプロモータ」はCAP部位+TATAボックスから成る(転写の基礎的レベルの最小配列;転写の無秩序なレベル)から成る。「上流域調節配列」は上流要素とエンハンサ配列とから成る。「切り詰められた」とは完全長プロモータが完全に存在しないこと、すなわち、完全長プロモータの一部が除去されたことを示す。この切り詰めたプロモータはMCMVまたはHCMV、例えばHCMV-IEまたはNCMV-IEのようなヘルペスウィルスに由来することができる。
プロモータは、例えば鼠CMV-IEプロモータおよびHCMV-IEプロモータで、塩基対をベースにして完全長プロモータからの寸法が40%、さらには90%まで切り詰めることができる。本発明の切り詰めたプロモータは、切り詰めたプロモータが挿入されるウィルスまたは系で与えられるトランスアクチベイティング蛋白および最小プロモータを用いて転写活性化されるエンハンサ領域から成ることができる。従って、本発明の切り詰めたプロモータ中に存在するプロモータは完全長の当初の塩基対の60%、さらには10%にすることができる。
プロモータはより多くの塩基対を必要とするので、本発明の切り詰めたプロモータを含む発現カセット、ウィルスおよびプラスミドは驚くべきである。本発明のプロモータは完全長プロモータに比較して優れた性能を有する。いかなる特定の理論に縛られるものではないが、この優れた性能は切り詰めた(トランケーション)に起因すると思われる。更に、プロモータの切り詰めた(トランケーション)は組換え型ウィルスおよびプラスミド(特にCAV)の挿入寸法制限の問題を解決する。
【0044】
従って、本発明はさらに、プラスミドが挿入されるウィルスまたは系によって与えられるトランスアクチベイティング蛋白によってトアンスアクチベイトした領域および切り詰めた転写活性プロモータが得られる完全長のプロモータの最小のプロモーター領域とから成る組換え型ウィルスまたはプラスミドのための切り詰めた転写活性プロモータに関するものである。
前記のプロモータと同様に、本発明のプロモータはヘルペスウィルス、例えばMCMVまたはHCMV(例えばHCMV-IEまたはHCMV-IEプロモータ)であるのが好ましく、塩基対をベースにして完全長プロモータの寸法の40%まで、さらには90%まで小さくすることができる。
【0045】
従って、本発明はさらに、切り詰めた転写活性プロモータから成る組換え型ウィルスまたはプラスミドへ組込むための発現カセットを提供する。この発現カセットは機能を有する切り詰めたポリアデニル化信号、例えば機能を保持した切り詰めたSV4Oポリアデニル化信号を含むことができる。大きい信号をだすことを考えると、切り詰めたポリアデニル化信号が機能することは驚くべきことである。そして、切り詰めたポリアデニル化信号はCAVのような組換え型ウィルスの挿入寸法限界問題を解決する。この発現カセットはさらに、外来性またはヘテロジニアスなDNAをそれが挿入されるウィルスまたは系に関して含むことができる。これらの外来性またはヘテロジニアスなDNAは上記と同じのDNAにすることができる。
【0046】
さらに更に驚くことは、本発明は組換え型を生成するために少なくとも一つの本質的でない遺伝子座、例えばE3領域を用いて組換え型CAV、好ましくはCAV2を提供する。HAVおよびウシのAd3から得られたデータから、この領域の一部はインビトロおよびインビボで伝染性ウィルスの形成には本質的でなく、従って、組込み領域とみなすことができる。従って、本発明の一つの観点はCAV E3欠失または部分的欠失の突然変異体(例えばE3 ORF1および/またはORF2)の生成物を提供することにある。この突然変異体は全CAV E3領域が組織培養に必要でなく、従って、組換え型CAV形成の挿入部位として使用できることを証明した。従って、本発明は外来性DNAが欠失したおよび/またはE3領域中、好ましくはE3領域の少なくとも1つの本質的でない領域、例えばORF2中に外来性DNAを挿入した組換え型CAVに関するものである。
E3領域の欠失はCAVゲノムへヘテロジニアス配列を組込む能力をさらに追加する。例えばこの欠失はゲノムの右端に導入した大きな発現カセットを補償することができる。当業者は過度の実験なしに本明細書に記載の方法を用いることで追加の本質的でない領域、好ましくはE3領域および追加の本質的でない区域を容易に確認できる。
【0047】
別の態様において、本発明はヘテロジニアスなDNAの挿入に対して必須でない領域中で欠失をした組換え型CAV、好ましくはCAV2を提供する。例えば、この必須でない領域内の欠失は他の領域、例えばE4/右ITR領域へのヘテロジニアスDNAの挿入と実質的に同じにする(例えば、補償する)ことができる。
多様なCAV2株からのITRa間のヌクレオチド配列の比較で右ITRの直ぐ上流に多様性があることが分っている(Cavanagh達、1991、Spibey、1991)。本発明者はこの領域内に新規で自明でない挿入部位を設計した。従って、更なる観点から、本発明は外来性DNAを挿入したるCAV組換え型にある。更に、驚くことに、E4/右ITR領域は上記SmaIより大きなヘテロジニアスDNAの断片を挿入でき、挿入寸法制限の問題が解決する。
E4/右ITR部位が転写活性が低いCAVゲノムの領域に局所化している(シャープ達(1984)を参照)ので、そこへの挿入はCAV組換え型ウィルスの生態学に大きな影響を与えない。
【0048】
既に示したように、本発明の一つの実施例では、本発明はCAVに外来性DNAを挿入するための新規で非自明な発現カセットを提供する。このカセットはヘテロジニアスな成熟核調節配列から成る。好ましい実施例では、本発明はウィルスおよびプラスミド、例えばアデノウィルスベースのベクターと組合されたパッケージング限界およびと生物学的特性を考慮に入れて合理的に設計された発現カセットを提供する。
【0049】
MCMVおよびHCMVプロモータをプロモータが挿入されるウィルスまたは系が与えるトランスアクチベイティング蛋白で転写活性化されるエンハンサ領域として小さく切り詰める能力および最小のプロモータから、本明細書の開示および従来の知識からの過度の実験なしで、他の成熟核ウィルス、特に他のヘルペスウィルスから同じように切り詰めできることが示される。本発明はそうした他のウィルスから切り詰めたプロモータを含む。
【0050】
より具体的な観点から、本発明はHCMV-IEまたはHCMV-IEプロモータ、好ましくはその切り詰めたプロモータから成るCAVまたは組換え体、好ましくはCAV2または組換え体を含む。これらのHCMVN−IEまたはHCMVN−IEプロモータまたはその切り詰めたプロモータはCAV-誘導遺伝子生産物でトアンスアクチベイトされるのが好ましい。
切り詰めた転写活性(またはコンピテント)プロモータ(好ましくはヘルペスウィルス・プロモータのような切り詰めた転写活性成熟核ウィルス・プロモータ、例えばHCMVまたはNOWプロモータ)を含む本発明の態様では、「活性」(または「コンピテント」)によって切り詰めた転写活性プロモータは元の完全長のプロモータの転写活性の少なくとも80%、好ましくは少なくとも85%、より好ましくは少なくとも90%および最も好ましくは少なくとも95%を示さなければならない。
【0051】
ヌクレオチドまたは完全長のプロモータの領域の一部の欠失は過度の実験なしででき、例示したもの以外の活性フラグメントも生成できる。切り詰めたプロモータが「活性」または「コンピテント」である限り、トランケーションの程度、すなわち欠失される塩基対の量は、完全長または元のプロモータの90%までの任意の量にすることができる。すなわち、切り詰めた転写活性プロモータは完全長または元のプロモータに対して塩基対で完全長または元のプロモータの約5%〜約95%、好ましくは約10%〜約90%、より好ましくは約10%〜約60%、最も好ましくは約10%〜約40%であり、具体例では、完全長または元のプロモータの約10%〜約40%である(完全長または元のプロモータの塩基対で約95%〜約5%の欠失、好ましくは約90%〜約10%の欠失、さらに好ましくは約90%〜約60%の欠失、最も好ましくは完全長または元のプロモータの塩基対の約90%〜約60%の欠失)。完全長または元のプロモータで最低限度保持する必要のあるものは、プロモータが挿入されるウィルスまたは系で与えられるトランスアクチベイティング蛋白で転写活性化される最小のプロモータおよび領域である。
【0052】
プロモータの一部の欠失、例えばHCMV-IEの欠失で寸法が小さくなり、HCMV-IEプロモータのようなプロモータの不足および/または寸法の問題とアデノウィルスのパッキング限界寸法の問題を解決することができる。
【0053】
特定の観点から、本発明は91bpの大きさを有するHCMV-IEの活性フラグメントまたは466bpの大きさを有するHCMV-IEの活性フラグメント、すなわち、切り詰めた約91bpの転写活性HCMV-IEまたは切り詰めた約466bpの転写活性HCMV-IEを提供する(本発明は相当塩基対寸法および/または91bpまたは466bpフラグメントに対して相同を有するHCMV-IEまたはHCMV-IEフラグメントを含む。例えば塩基対寸法および/または相同性は91bpまたは466bpフラグメントの少なくとも80%、好ましくは少なくとも85%、さらに好ましくは少なくとも90%、最も好ましくは少なくとも95%である)。このフラグメントはCAV2のようなCAVに挿入できる。従って、本発明はHCMV-IEまたはNCMV-IEの活性断片、例えばHCMV-IEまたはHCMV-IEから誘導される切り詰めた転写活性プロモータ、好ましくは91bpまたは466bpの断片または91のbpまたは466bp断片の実質的な塩基対寸法および/または相同性を有する活性断片から成るCAV2のような組換え型CAVを含む。
【0054】
91bpまたは466bpの断片およびその他のHCMV-IEまたはNCMV-IEプロモータの任意の活性断片を製造するための寸法は上記のHCMV-IEまたはHCMVN−IEプロモータの公知の分子構成(Boshart、1985)から減少させることができる。
例えば91bpまたは466bp断片のような完全長または元のプロモータの上記のような小さい断片が「活性」(上記定義の用語)を有し、それぞれHCMVN−IEの850bp体およびMCMV-IEの766bp体としてCAV、特にCAV2感染細胞で同様な高い転写活性レベルを導くということは驚くべきである。
【0055】
91bpフラグメントすなわち実質的な塩基対の寸法が91bp断片の活性フラグメントアデノウイルス−ベースの組換え型ウィルスにおいて使われていた最小プロモータ要素であると思われるのは特に驚くべきことである。
「活性」フラグメントの生成のためのHCMV-IEおよびMCMV-IEプロモータに適用される以下の説明から、他の成熟核ウィルス、例えばアデノウィルスに外来性である他のヘルペスウィルス、例えばCAV2からHCMVN−IEまたはMCMVN−IE以外のプロモータの「活性」フラグメントをつくることは過度の実験なしで行なうことができる。従って、本発明はアデノウィルスに外来性のプロモータのフラグメント、例えば、アデノウィルスに導入したときにアデノウィルスの完全長プロモータのように活性な切り詰めた転写活性プロモータを提供する。アデノウィルスはCAV2のようなCAVであるのが好ましい。
【0056】
従って、本発明の別の態様で、本発明は鼠CMVN−IE(MCMV-IE)のフラグメント(Dorsh-Hasier達(1985))、例えばアデノウィルス(例えばCAV2)で活性なMCMV-IEに由来するプロモータの切り詰めた転写活性プロモータを提供する。CAV2感染細胞のようなアデノウィルスでは、466bp MCMV-IEプロモータ要素は、91bp のHCMV−IEプロモータ要素のような活性を示す。
【0057】
本発明のさらに他の観点で、本発明はウイルスサイクルの後期に組換え型メッセンジャーRNAの翻訳をするアデノウィルス(例えばCAV2)の活性なプロモータと、このプロモータを含む組換え体と、この組換え体から成る組成物と、プロモータの製造方法および使用方法と、組換え体および組成物とを提供する。そうしたプロモータは、HCMV-IEプロモータまたはその活性フラグメントから成り、5'UTRはヒトのAd2 TPLに置換された。
【0058】
本発明のさらに他の観点で、本発明は組換え型アデノウィルス(例えばCAV2)を製造するためのカセットと、このカセットを含む組換え体と、このカセット製造方法および使用方法と、組換え体と、組成物とを提供する。このカセットは最小化されたポリアデニル化配列(「最小化したポリA」)、例えばSV4Oからの最小化されたポリアデニル化配列(「最小化したSV4OポリA」)から成るのが好ましい。最小化されたSV4OポリAは完全長または元のSV4OポリAより短く、約153bp(±10%)の大きさにすることができる。
【0059】
この種の最小化されたSV4OポリAはアデノウィルス(例えばCAV2)感染細胞で野生型と同じ高条件レベルの安定した安定したメッセンジャーRNAと組合せることができることがわかっている。最小化されたSV4OポリAカセットはアデノウィルスに挿入されるDNAを最小にするために用いることができ、それによって能力不足およびアデノウィルスの諸問題を解決することができる。更に、SV4Oポリアデニル化信号の最小化によって過度の実験無しに他の供与源から他の類似した配列を誘くことができる。
組換え型蛋白の発現のためにアデノウィルスをベースにしたベクターを用いて最適化された寸法および成分を有する発現カセットは今までの文献には記載がないと思われる。
【0060】
本発明のさらに他の観点で、本発明は精製されたアデノウィルス、例えばCAV、好ましくはCAV2 DNAを犬単層中へトランスフェクションするための条件および方法を提供する。
【0061】
本発明の好ましい実施例では、トランスフェクション条件は、El変性した犬細胞系の利用とは独立している。この手順で好ましい収率、精製されたCAV DNAを約5x103pfu/ugの収率で得られる。この手順を用いるとCAV組換え型ウィルスの使用および増殖のためのEl変性細胞の使用を避けることができ、それによってEl変性細胞をに関する安全問題を避けることができる。
従って、本発明は組換え型アデノウィルス、好ましくはCAV、より好ましくは、CAV2と、その製造方法および使用方法と、それらおよびその発現産物を含む組成物を提供する。ゲノムへの組込みまたはゲノムからの欠失に任意の適切な本質的でない領域を使用することができる。そうした部位はE4、ElおよびE3を含む。2つの挿入部位が存在するのが好ましい。その第1のものはE3領域にあり、第2のものは右ITRとE4転写単位(好ましくはSmaI部位)との間に位置し、前者の部位または両方の部位(組合せ)が好ましい。CAV E3 0RF2、例えばCAV2 E2 0RF2が現在のところ最も好ましい。
【0062】
本発明の結果はCAV E3は組織培養の複製に必須ではないことをが証明している。これが組換え型CAVウィルスを誘導する最初の成功した試み表し、従って、組換え型CAV、例えばCAV2をベースとする生産物、例えば、組換え型CAVまたは発現産物を含む免疫学組成物、抗原性組成物またはワクチン組成物のベースが構成される。
従って、本発明はCAV2ゲノムの本質的でない領域に外来性DNA(CAVでは自然には起らないか、挿入部位で自然にCAVでは起らないDNA)を含む合成で修正されたCAV2のようなCAVであると理解できる。
本質的でない領域すなわち必須でない領域はCAV E3またはCAV E3およびSmaI部位のようなゲノムの右終端の両方であるのが好ましい。
【0063】
本発明はさらに、本発明組成物および/または組換え体を用いて得られる抗体とこの抗体の使用と理解することができる。抗体またはその生産物(エピトープ)、抗体から導かれるモノクローナル抗体は抗原または抗体の有無を決定する結合アッセイ、テストまたはキットで使用できる。
本発明で使われるフランキングDNAは挿入部位またはそれに隣接するゲノムの一部にすることができる(ここで、「隣接」とは同じ隣接する配列(例えばコードンまたはコードン群)および多数の配列、例えばコードンまたはコードン群を含み、その前に挿入部位がある)。
【0064】
外来性またはヘテロジニアスなDNA(またはCAVに異質なDNAまたは自然にCAVでは起らないDNA)は上記エピトープのいずれかをコードするDNAであることができる。この点については、Borrelia DNAに関して米国特許第5,523,089号、W093/08306、PCT/U892/08697、Molecular Microbiology(1989)、2(4)、479N486およびPCT公報WO 93/04175および、WO 96/06165を参照。肺炎球菌(pneumococcal)エピトープに関してはBriles達のWO 92/14488を参照。腫瘍ウィルス参照に関してはMolecular Biology of Tumor Viruses, RNA TUMOR VIRUSES(Edition、ワイス達d、Spring Laboratory 1982)(例えば44ページRetroviruses)を参照。他のエピトープをコードするDNAに関しては例えば「発明の背景」で引用した文献:米国特許第5,174,993号、第5,505,941号を参照(例えば、組換え型アビポック(avipox)ウィルス、ワクシニアウィルス;狂犬病グリコプロテイン(G)、遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、ウシ白血病ウィルス、gp5l、30エンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAVN1 env遺伝子、NP(Chicken/ペンシルバニア/1/83インフルエンザウィルスのヌーデオプロテイン(nudeoprotein)遺伝子)、伝染性の気管支炎ウィルスのマトリックスおよびプレポリマー遺伝子;HSV gD; HSV gD;昆虫ポックスウイルス(entomopox) プロモータ、inter alia)、米国特許第5,338,683号, 例えば組換えワクチンウイルス、組換え型アビポック;エントモポックス(entomopox)プロモータ、グリコプロテインをコード化しているDNA、米国特許第5,494,807号、例えば組換え型ワクシニアウィルス、アビポック(avipox)ウィルス;ウサギからの抗原をコードする外来DNA、狂犬病、ヘパチチス(Hepatitis)B、JEV、YF、Dengue、はしか、仮性狂犬病、エピスタインバー(Epstein-Barr)、HSV、HIV、SIV、EHV、BHV、HCMV、犬パルボウィルス、馬風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、特にお多福風邪(NDV):米国特許第5,503,834号、例えば、組換え型ワクシニア、アビポック(avipox)、モリビリウイルス(例えばはしかF、ヘマグルチニン);米国特許第4,722,848号、例えば、組換え型ワクシニアウィルス;HSV tK、グリコプロテイン(例えばgB、gD、風邪HA、ヘパチチス(Hepatitis)B(例えばHBsAg);英国特許第GB 2 269 820 Bおよび米国特許第5,514,375号(組換え型ポックスウィルス;フラビウイルス構造蛋白質);WO92/22641、例えば組換え型ポックスウィルス; WO 92/22641、例えば、組換え型ポックスウィルス; 免疫不全ウィルス;WO 93/03145、例えば組換え型ポックスウィルス; WO 93/03145、例えば、組換え型ポックスウィルス;IBDV; WO 94/16716および1994年1月19日出願の米国特許出願第08/184009号、例えば組換え型ポックスウィルス;サイトカインおよび/または腫瘍関連抗原);PCT/U594/06652(プラスモジウム(Plasmodium)の各ライフサイクル段階からのプラズモディウム)。
【0065】
実施例に記載の組換え体のように、タグおよび他の外来性DNAがCAV2に組み込まれるので、他の外来性DNAをCAV2に組み込むことができる。vCAl、vCA2、vCA3、vCA4、vCA5、vCA6、vCA7、vCA8およびvCA-CDVF1-Ql2bp-up-Smalを作るのに用いる外来性DNAの代わりに、上記文献および/またはここで引用した文献に記載の外来性DNAを使用して追加のCAV2組換え体を作ることができる。すなわち、vCA2のように、vCA8およびvCA-CDVF1-Ql2bp-up-SmaIを介した挿入、vCA2 のように、多重抗原をコードする組換え体を含むvCA8およびvCA-CDVFlQl2bp-up-SmaIを介した欠失(例えばE3中または右ITRとE4転写単位との間の領域または両方の領域への挿入およびE3領域の欠失)(サブフラグメントプロモータ、縮小または変性されたポリアデニル化カセットおよび5'UTR置換プロモータを含む)。分析で発現が示された。脊椎動物(動物または人間)宿主に投与して抗体応答を含む応答を導くための本発明組成物は担体または希釈剤と混合して調製される。
外来性DNAはマーカー(例えば色または光顕マーカー)を含むことができる。外来性DNAはさらに昆虫宿主に有害な生産物をコードすることができ、発現産物が殺虫剤または殺虫剤にすることができる。外来性DNAは抗菌ポリペチド、そのポリペチドおよびDNA上の情報をコードできる(この点に関しては米国特許第5,421,839号を参照)。
【0066】
本発明はさらに、例えばアデノウィルスゲノム、好ましくはCAV、例えばCAV2の必須でない領域の地図を作る方法を提供する。この方法はインビボ遺伝子組換えでドナーDNAをCAVゲノムの一つの領域に挿入できるように、CAV DNAのセグメント内にCAVゲノムの一部と同一直線上にCAV中では自然に生じないDNAから成るドナーDNAを調製し、このドナーDNAをインビボ遺伝子組換えでCAVゲノム中に導入し、組換え体を回収し、安定度およびその生死およびCAV中の自然には起り得ないDNA発現および存在および/または組換え体中の外来性CAV DNAの発現および非存在を決定し、この安定度およびその生死およびCAV中の自然には起り得ないDNA発現および存在および/または組換え体中の外来性CAV DNAの発現および非存在によってDNAが挿入された上記領域が非必須領域であることを示す。この方法は下記実施例で使われる。ドナーDNAをマーカーDNAにして、例えばマーカーDNAのハイブリッド化できるか、マーカーと置換した外来DNAとのハイブリド化ができないかで、ハイブリダイゼーションを用いてそれがゲノムに組み込まれたかどうかを決定することができる。
【0067】
本発明内の上記およびその他の目的および具体化は下記の詳細な説明から明らかになろう。
【発明を実施するための最良の形態】
【0068】
既に述べたように、本発明は組換え型アデノウィルス、CAV、好ましくはCAV2、その製造方法、使用、その発現産物およびその発現カセットに関するものである。本発明は特に、組換え型CAV(例えばCAV2)に関するものである。特に外来性DNAが本質的でない領域および/または本質的でない領域領域に挿入された欠失したものと、その製造方法と、使用または(ベクターまたは複写DNAとしてを含む)それの発現産物および発現産物の使用にある。組込みおよび/または欠失のためにはCAV E3領域、特に0RF2が好ましい。
【0069】
組換え型ウィルスおよびその生産物の使用は、過度の実験なしで発明の背景および討論に発明の要約の下に記載される文献から決定できる。
本発明の組換え体のヘテロジニアスな外来性DNAは、好ましくは下記を含む発現産物をコードする:エピトープ、生化学反応モジュレータ、成育因子、認識配列、治療の遺伝子または融合蛋白。これらの用語に関しては下記を参照:Kendrew、THE ENCYCLOPEDIA OF MOLECULAR BIOLOGY(Blackwell Science Ltd 1995)およびSambrook, Fritsch, Haniatis, Molecular Cloning, A LABORATORY MANUAL(2版 EditioNcoId spring Harbor Laboratory Press、1989)。
【0070】
ワクチンまたは免疫学の組成に用いられる抗原に関しては発明の背景に記載のStedmans Medical Dictionaryを参照(例えば第24版(1982)。例えば、ワクチンの定義(ワクチン製剤において使われる抗原のリスト;この種の抗原または抗原のエピトープが本発明で使用できる。本発明の組換え型ウィルスの発現産物または本発明の組換え型ウィルスまたは組換え型ウィルスを含む多価組成物)。
エピトープに関しては、当業者は過度の実験なしでアミノ酸および対応するペプチドまたはポリペチドのDNAの塩基配列の知識および特定のアミノ酸の特性(例えば大きさ、電荷、その他)およびコードン辞書からエピトープすなわちペプチドまたはポリペチドの免疫領域およびそのコード付けDNAを決定できる。
【0071】
免疫学組成物で蛋白のどの部分を使用するかに関する一般的な方法で所望抗原の寸法と配列を決定する。一般に、大きな蛋白はより多くのポテンシャルを有するので、小さいものよりよい抗原である。より多く外来抗原(寛容性を誘発する自己配位とは違う)は免疫応答を引き起こすのにより効果的である(Ivan Roitt, Essential Innunology, 1988)。
大きさに関しては:当業者はウイルス性ベクターに挿入されるDNAの塩基配列でコードされる蛋白の大きさをベクターのパッケージング限界を考えて最大にすることができる。挿入されるDNAを最小に且つ発現する蛋白の大きさを最大にするために、DNAの塩基配列はイントロン(転写されるが一次RNA転写産物から削除した遺伝子領域)を無しにすることができる。
【0072】
DNAの塩基配列は少なくともペプチド長が少なくとも8または9のアミノ酸をコードできるものである。このペプチドはCD4+T細胞(これがウィルス感染細胞または癌細胞を認識する)を刺激するのに必要な最小長さであるである。CD8+ T細胞応答(病原を飲み込んだ特別な抗原提示細胞を認識する)を刺激するための最小限のペプチド長さは13〜25のアミノ酸である(上記、Kendrewを参照)。これらが極小長さであるが、これらのペプチドは免疫応答すなわち抗体またはT細胞応答をし難いので、保護応答(ワクチン組成物)のためにはそれより長いペプチドにするのが好ましい。
【0073】
配列に関しては、DNA塩基配列は少なくとも抗体応答またはT細胞応答を作り出すペプチド領域をコードするのが好ましい。TおよびB細胞エピトープを決定する1つの方法はエピトープマッピングである。問題の蛋白を蛋白分解酵素で重複ペプチドに断片化し、次に、各ペプチドを検査して、天然蛋白を用いて導いた抗体に結合するか否か、T細胞またはB細胞活性化を誘発するか否かを調べる。T細胞はMHC分子と複合した短い線形のペプチドを認識するので、このアプローチは特にマッピングT細胞エピトープに役立った。B細胞エピトープは線形のアミノ酸配列でなく、むしろ折れた3次元の蛋白の三次構造からできるので、上記方法はB細胞エピトープを決定するのには効果的でない(Janis Kuby、Immunology;(1992)pp. 79N80)。
【0074】
問題のエピトープを決定するための他の方法は親水性である蛋白の領域を選ぶことである。親水性残基は蛋白の外表面上にあることが多いので、抗体にアクセスできる蛋白領域にある(Janis Kuby, Immunology, (1992) p. 81)。
エピトープを決定するさらに他の方法は抗原(全長)抗体錯体のX線結晶学的解析を実行することである(Janis Kuby, Immunology (1992)80ページ)。
T細胞応答を作り出すことができるエピトープを選ぶためのさらに別の方法は、MHC分子が結合し易いことが知られているペプチド配列である蛋白配列潜在HLAアンカー結合単位から同定する方法である。
T細胞応答を作り出すための推定のエピトープであるペプチドはMHC錯体に提示されなければならない。このペプチドは好ましくはMHC分子に接合する適当なアンカー単位を含み、免疫応答を作り出す十分に高い親和性で結合しなければならない。考慮すべきファクタは以下の通りである:免疫化する患者(脊椎動物、動物または人間)のHLA型、蛋白配列、適当なアンカー単位の存在および他の生きた細胞中のペプチド配列。
【0075】
一般に、免疫応答は次のように作り出される:蛋白がより小さいペプチドに切断され、他の細胞の界面に「主要組織適合遺伝子複合体MHC」と呼ばれる錯体が提示された場合にだけT細胞は蛋白を認識する。MHC複合体は2つのクラスIおよびIIがあり、各クラスは多くの異なるアレルから成る。各患者はそれぞれ異なる種類のMHC錯体アレレを有し、「異なる HLAタイプ」とよばれる。
クラスIのMHC複合体はほぼ全ての細胞で見られ、細胞内部で生産された蛋白からのペプチドを有する。すなわち、クラスIのMHC複合体は、ウィルスに感染するか、オンコジーンの発現の結果としガン化した時に細胞を殺す役目をする。表面上にCD4とよばれる蛋白を有するT細胞はこのクラスIのMHC細胞と結合し、リンフォカインを分泌する。リンフォカインは応答を刺激し、細胞はウイルス性感染細胞を殺す。
クラスIIのMHC複合体は抗体提示細胞上にだけ見つかっており、抗体提示細胞細胞でエンドサイトーシスされた循環病原からペプチドを提示するのに用いられる。CD8とよばれる蛋白を有するT細胞はクラスIIのMHC細胞と結合し、溶菌顆粒のエキソサイトーシスで細胞を殺す。
【0076】
蛋白がT細胞応答を刺激するエピトープであるかどうか決定する際のいくつかのガイドラインは以下を含む:ペプチドの長さ:ペプチドはクラスIのMHCに合う少なくとも8または9つのアミノ酸長と、クラスIIのMCH錯体に合った少なくとも13〜25のアミノ酸長でなければならない。この長さはMHC錯体に結合するペプチドの最小長さである。細胞は発現されたペプチドを切るので、これらの長さより長いペプチドであるのが好ましい。ペプチドが免疫応答を作り出す十分に高い特異性で各クラスIおよびIIと結合できるようにする適当なアンカー単位を含まなければならない(Bocchia、H.ほか、Soecific Bindina of Leukemia Oncoacrie Fusion Protein Peptides to HLA Class I Molecules, Blood 85:2680N2684; Englehard、VH, クラスIおよびクラスIIのMHC分子と関連があるペプチドの構造 Ann. Rev. liumunol. 12:181 (1994)を参照)。これは過度の実験なしで MHC分子と関連するペプチドの発表された構造と問題の蛋白の配列とを比較することで行なうことができる。T細胞受容体ペプチドで認識される蛋白エピトープは蛋白質分子の酵素分解で作られ、クラスIまたはクラスIIのMHC分子と一緒に細胞表面上に提示されるペプチドでである。
【0077】
更に、当業者は蛋白データベースにリストアップされた配列と蛋白配列とを比較することで問題のエピトープを確認できる。相同性がほとんどないか、ない蛋白領域はその蛋白のエピトープとして選択でき、ワクチンまたは免疫組成物として有用である。生きた細胞中に広く存在する配列と相同が大きい領域は避けなければならない。
【0078】
さらに他の方法は問題の蛋白の一部を単に作り、発現し、問題の蛋白の一部に対するモノクローナル抗体を作り、それらの抗体が蛋白を誘導した病原のインビトロ成長を抑制するか否かを確認することである。当業者は本明細書に開示および抗体がインビトロ成長を抑制するか否かに関する分析するために問題の蛋白の一部を作り、発現する公知の他のガイドラインを使用できる。例えば、当業者は問題の蛋白の一部を作るのに、8〜9または13〜25のアミノ酸長のその蛋白の一部を選択し、親水性領域を選択し、抗原(全長)−抗体錯体のX線データから結合する部分を選択し、他の蛋白とは違う配列の領域を選択し、潜在的HLAアンカー結合単位を選択する。また、これらの方法またはその他の公知方法を任意に組合せることができる。
抗体が認識するエピトープは蛋白の表面に発現する。抗体応答を刺激しそうな蛋白の領域を決定する最も好ましい方法として当業者は、上記の一般的方法またはその他公知技術のマッピング法を使用してエピトープ地図を作ることができる。
【0079】
上記のように、本明細書の開示および従来法の知識から、過度の実験なしで、当業者はT細胞、B細胞および/または抗体応答を得るためのエピトープのアミノ酸および対応するDNAの塩基配列を確認できる。Gefter達の米国特許第5,019,384号およびそれが引用した文献も参照(注、特に上記特許の「関連文献」部分を参照。この特許の第13欄には多くの重要な生物の多数のエピトープが開示されている。特に、中和抗体が目指すエピトープは重要である。そうしたエピトープは「関連文献」に記載の文献に開示されている)。
生化学反応モジュレータの発現に関しては1993年9月30日公開のWohlstadterのWO 93/19170「選択方法」とそこで引用されている文献を参照。
たとえば、生化学反応モジュレータは生物学的活性度を調整し、例えば、生化学反応モジュレータは非-NMDA興奮性アミノ酸受容体と関連した高分子量蛋白のような調節的な成分であり、アロステリックにAMPA結合の親和性を調整する。本発明の組換え型はそうした高分子量蛋白を発現する(上記Kendrewを参照)。
【0080】
より一般には自然は多数の生化学反応モジュレータを提供している。活性の調節は存在/非共存系、例えば発現/劣化系へのアロステリックに誘発された単純な四元変化として複雑な機構によって実行できる。多くの生体分子の発現の抑制/活性化はそれ自体活性が種々の機構によって調整される分子を用いて調整される。
バクテリア蛋白の化学修飾のリストはNcidhardt達のPhysiology Bacterial Cell(Sinauer Associates社、Publishers、1990)の73ページの表2に示されている。この表が示すようにある変異には適当な会合体が関係し、他の変異では関与がないが、いずれの場合もこの種の変異は機能の変位の原因となる。表から、過度の実験なしで、他の細胞の蛋白の類似した化学的変位を決定できる。
【0081】
生物学的機能の変位は単に分子の適当/不適当な局在化によって調節できる。分子はそれが特定の位置を目標とした場合にだけ、成長に利点または欠点を与えるように機能する。例えば、分子は一般にその分子の機能としてそれを劣化させる酵素の分泌で最初に劣化されると細胞に取りこまれたり、利用されない。従って、組換え型による酵素の生産では細胞による分子の利用または取込みを調節できる。同様に、組換え型は分子の取込みまたは使用に必要な酵素に結合した分子を発現でき、従って、その取込みまたは使用を調節できる。
シグナルペプチドの開裂によって実行される蛋白の局在化のターゲットは別の種類の変更または調節である。この場合は特定のエンドプロテアーゼ触媒活性が組換え型によって発現できる。
【0082】
機能の変化が起こる機構の別の例はRNAウィルスポリ蛋白、アロステリック効果および一般的な共有結合性および非共役な立体障害である。HIVは非機能的ポリ蛋白質構造物を発現するRNAウィルスの典型的な例である。HIV ではgag, pol, env-ポリ蛋白を加工してウィルス構造蛋白p17、p24およびp15−逆転トランスクリプターゼおよびインテグラーゼ−および2つのエンベロップ蛋白gp4lおよびgpl20O(コール達(PNAS USA 85:4686N 90(1988))になる。ポリ蛋白質の適当な開裂はウィルスの複製に重要であり、不活発な突然変異体HIVプロテアーゼを運んでいるウィリオンは非感染性である(同上)。これは活性を低下調節する蛋白フュージョンの他の例である。従って、ある種の蛋白の自然の発現を阻止または増進するために(開裂を阻止するか、増進させて)エンドプロテアーゼと干渉する分子を発現するか、エンドプロテアーゼを出す組換え型ウィルスを造ることが可能である。
【0083】
酵素機能の有用性はその反応に対する触媒作用の能力を変えることで調整することができる。調整された分子の例はチモーゲン、多重サブユニット機能複合体の形成/解離、RNAウィルスポリ蛋白鎖、アロステリック相互作用、一般立体障害(共有結合性および非共有結合性)およびリン酸化、メチル化、アセチル化、アデニル化およびユリデニル化等の各種化学修飾である(上記Neidhardtの表1、315ページおよび表2、73ページを参照)。
【0084】
チモーゲンは酵素活性の変位を生じさせる自然に起こる蛋白融合の例である。チモーゲンは一定の蛋白加水分解により活性状態に変換される蛋白の1つのクラスある。Proteases、Biological Control(第2巻)(1975)、ライヒ、表3、54ページを参照)。自然は一定の酵素、例えばトリプシンの活性をこれらの酵素でアミノ末端で追加の「リーダー」ペプチド配列を発現することで低下調節する機構を開発している。余分なペプチド配列で酵素は不活性チモーゲン状態になる。この配列の開裂でチモーゲンは酵素活性状態に変わる。チモーゲンの全反応速度は対応する酵素の活性の約l05〜106倍である(上記ライヒの54ページの表3を参照)。
従って、ある酵素のその末端の1つに単にペプチド配列を加えるだけで酵素の機能を低下調節することができる。例としてはこの性質の知識を用いて、組換え型が追加のアミノ酸の一旦または両端に含むペプチド配列を発現することができる。
【0085】
多重サブユニット酵素の形成または脱会合は変移を起こさせる他の方法である。多重サブユニット酵素の形成または脱会合に管用する機構は種々ある。
従って、適当な特定のサブユニットの相互作用を立体障害することで触媒活性は低下変移する。従って、本発明の組換え型は生物学的機能を調整するために自然に起る酵素または酵素複合体を立体障害する分子を発現する。
【0086】
いくつかの酵素阻害剤は共有結合性立体障害または変異による機能の低下変異のよい例である。活性サイト中の触媒的に重要なアミノ酸の所で酵素の活性サイトに不可逆的に結合した自殺基質は酵素活性サイトを立体的にブロックする共有結合の例である。酵素自殺基質の例はキモトリプシンのためのTPCKである(Enzyme Structure、Mechanism、2d ed Fritach;フリーマン社(1984))。この種類の変異は適切な酵素自殺基質を発現している組換え型を用いて可能であり、それによって生化学反応を(例えば、酵素活性を制限することで)調整することができる。
【0087】
多くのリプレッサ分子を含む非共有性立体障害のさらに他の例がある。組換え型が立体障害可能なリプレッサ分子を発現でき、従って、特定のDNA-RNAポリメラーゼ干渉を防ぐことによってDNAの塩基配列の機能を低下調整する。
アロステリック効果はある種の生物系において実行される他の変異方法である。アスパラギン酸トランスカルバモイラーゼは良く特徴づけられたアロステリック酵素である。触媒サブユニットとの相互作用が規制ドメインである。CTPまたはUTPに結合で制御サブユニットはホロエンザイムの四次構造変化を誘発し、触媒活性の低下変異が起こる。これとは対照的に、制御サブユニットにATPが結合すると触媒活性が増加変異する(上記、Fritach)。本発明方法を使用すると分子は結合および調節可能な第四または第三の変化を引き起こす発現をする。
【0088】
さらに、各種の化学的修飾、例えばリン酸化、メチル化、アセチル化、アデニル化およびウリデニル化を機能の調整のために行うこともできる。これらの変異は多くの重要な細胞成分の調節で重要な役割をすることは公知である。上記Neidhardtの表2、73ページにはこの種の変異を経た各種の細菌酵素の一覧が示されている。このリストから当業者は過度の実験なしで同じか類似の変異を経て他の系の他の酵素を確認することができる。さらに、人間の疾患に関係する多くの蛋白はこの種の化学修飾を経ることができる。例えば、多くのオンコジーンはリン酸化を用いて変性されるか、リン酸化または脱リン酸化により他の蛋白を修正する。従って、機能、例えばリン酸化を修正または変えることができるモジュレータを発現する本発明によって与えられる能力は重要である。
上記から、当業者は過度の実験なしで本発明を使用して生化学反応モジュレータを発現することができる。
【0089】
本発明の組換え体を用いた融合蛋白の発現に関してはSambrook、Fritsch、Maniatis、Molecular Cloning、A LABORATORY MANUAL(2d EditioNcoIdSpring Harbor LaboratoryPress(1989)(特に第3巻)および上記Kendrewが参照できる。Sambrook達の教えは過度の実験なしで適当に変更でき、当業者は融合蛋白を発現する組換え体を作ることができる。
遺伝子治療および免疫療法に関しては米国特許第4,690,915号および第5,252,479号が参照できる。これらの内容および組成物の引用文献は本願明細書の一部を成す。また、WO 94/16716および1994年1月19日に出願の米国特許出願第第08/184009号とその引用文献は本願明細書の一部を成す。
【0090】
成育因子は組織および機能の個体発生および維持を制御する多機能で局部的に働く細胞間信号ペプチドと定義できる(Kendrew、et seq.、特に455ページを参照)。
成育因子または治療遺伝子は、例えば疾患攻撃蛋白、癌治療分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードできる。さらに、成育因子または治療遺伝子は、例えばαグロビン、β‐グロビン、γ-グロビン、顆粒球マクロファージ-コロニー刺激因子、腫瘍壊死因子、インターロイキン(例えば、インターロイキン1〜14または1〜11またはこれらの任意の組合せの中から選択されるインターロイキン)、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、EPO、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随抗原またはB7から成る群の中から選択できる。米国特許第5,252,479号には遺伝子治療のためにアデノウィルス系で発現できる蛋白のリストが記載されている。当業者はこの開示を見ることができる。WO 94/16716および1994年1月19日に出願の米国特許出願第08/184009号にはサイトカインおよび腫瘍関連抗原のための遺伝子および免疫療法方法が記載されている。当業者はこれらの開示を見ることができる。
【0091】
従って、当業者は本発明の開示および従来法の知識から、過度の実験なしで、成育因子または治療遺伝子を発現する組換え体を作ることができ、組換え体を使用することができる。
上記および従来法の知識から、過度の実験無しに、当業者は生化学反応モジュレータ、成育因子、認識配列、治療遺伝子または融合蛋白のエピトープを発現する本発明お組換え型を作ることができ、また、その組換え型を使用することができる。
【0092】
外来性またはヘテロジニアスなDNAはそれ自体が組換え型CAVでの発現に導くプロモータを含む点に注意されたい。すなわち、外来性DNAは単にコード化DNAであることができ、発現を導く内因性プロモータから適当な下流を配置できる。更に、発現を増幅するかまたは増やすために、コード化DNAの多重コピーまたは強いか早期プロモータまたは早期と後期のプロモータまたはその任意の組合せを使用することができる。従って、外来性またはヘテロジニアスなDNAはE3のような内因性プロモータまたはMLPプロモータに対して最適に配置でき、また、これらのプロモータの位置を変えて外来性またはヘテロジニアスなDNAと一緒に他の位置に挿入できる。コード化DNAは組換え型CAVから複数の生産物を発現するための複数の蛋白のためのコード化DNAであることができる。
発現産物は抗原、免疫源またはエピトープであることができる。従って、本発明は発現産物を含む免疫学的組成物、抗原性組成物またはワクチン組成物に関するものでもある。ある例ではCAVベクターは直接適切な宿主へ投与できるので、更に、本発明はCAV、好ましくはCAV2、ベクターを含む組成物に関するものである。さらに、発現産物はインビトロでCAV、好ましくはCAV2ベクターから単離でき、また、インヴィトロでCAVベクターに感染したまたはトランスフェクションされた細胞から単離できるので、本発明は製品の発現方法にも関するものである。この方法は例えばベクターとしてのCAV中へ外来性DNAを挿入する、例えば、制限/リゲーション組換えを用い、適当な細胞をインヴィトロでCAVで感染またはトランスフェクションし、必要な場合には発現産物をさらに抽出し、精製し、分離することから成る。任意の抽出、精製または単離技術が使用でき、この点に関しては「発明の背景」および「発明の要約」に記載の文献が参照できる。
【0093】
組換え型CAVで感染後、外来性DNAの発現からの蛋白はクロマトグラフィのような公知の技術を用いて回収される(ロビンス、EPA 0162738A1;Panicali、EPA 0261940A2;リチャードソン;スミス達、Pennock達;欧州特許第0265785)回収した蛋白は抗原組成物、免疫組成物または適当な担体を含むワクチンにすることができる。
組換え型CAVは例えば抗原、免疫源、エピトープ、その他の蛋白の製造に使用でき、それから抗原性組成物、免疫学組成物、ワクチン組成物を調製することができる。昆虫の成長または発達に有害な生産物を発現する組換え型CAVは殺虫剤の調製に使用でき、また、植物の成長または発達に有害な生産物を発現する組換え型CAVは除草剤の調製に使用でき(発現産物を単離し、殺虫剤または除草剤としての許容範囲内で担体または希釈剤と混合して)、抗菌類ポリペチドを発現する組換え型CAVは抗菌剤の製造に使用できる(発現産物を単離し、適切な担体または希釈剤と混合して)点に注意されたい。
【0094】
発現産物が抗原性、免疫学的または保護(ワクチン)応答を示すので、本発明はさらに、その生産物すなわち抗体とその使用にも関するものである。発現産物は抗体を導き出すことができる。この抗体はモノクローナル抗体を作ることができ、抗体または発現産物はキット、アッセイ、テスト等で使用できる。従って、本発明はこれらの使用にも関するものである。さらに、本発明の組換え型はDNAの複製に使用できるので、本発明はベクターとしての組換え型CAVと、細胞を組換え型に感染またはトランスフェクションさせ、DNAを回収する方法でもある。得られたDNAはプローブまたはプライマーとして使用できる。
本発明の組換え型CAV、または、その発現産物、例えば免疫学組成物、抗原性組成物またはワクチン組成物または治療組成物は非経口径路(皮内、筋内、皮下)で投与できる。この種の投与は全身的な免疫応答を可能にする。投与は粘膜径路、例えば経口、経鼻、経生殖器、その他)を通して行なうこともできる。そうした投与では局所免疫応答ができる。
【0095】
一般に、本発明の抗原性組成物、免疫学組成物、ワクチン組成物または治療組成物(CAV、好ましくはCAV2、本発明の組換え体または発現産物を含む組成物)は周知の標準的技術によって医薬または獣医技術に熟練した当業者が調製できる。この種の組成物は医学技術に熟練した当業者が周知技術を用いて各種ファクタ、例えば品種、性差、年齢、体重、投与対象疾患の状態および投与径路を考慮した供与量で投与できる。本発明の組成物はそれ単独で投与でき、また、他の抗原性組成物、免疫組成物、ワクチン組成物または治療組成物と一緒または一定順番で投与できる。この種の他の組成物は精製した生来抗原またはエピトープまたは組換え型CAVまたはその他のベクター系を用いて発現した抗原またはエピトープをを含むことができ、上記の点を考慮して投与できる。
本発明の組成物の実施例は体腔、例えば鼻、肛門、口、生殖器、膣、その他用の液状製剤にすることができ、懸濁液、シロップまたはエリキシルで投与でき、非経口、経皮、経皮内、経筋内または経静脈での投与、例えば懸濁液またはエマルションの注射投与用に製造することができる。これら組成物では本発明の組換え型を適切な担体、希釈剤または賦形剤(例えば無菌水、生理食塩液、グルコース、その他)との混合製剤にすることができる。
【0096】
抗原性組成物、免疫学組成物またはワクチン組成物はアジュバントと、発現産物の応答を導き出すための所望量の組換え型CAVとを含むことができる。人間の用途では明礬(リン酸アルミニウムまたは水酸化アルミニウム)が典型的なアジュバントである。家畜の用途において研究、使用されているサポニンおよびその精製された成分Quil A、フロインドの完全アジュバント、その他のアジュバントは毒性を有し、人間のワクチンでの使用には制限がある。化学的に定義された製剤、例えばムラミルジペプチド、モノホスホリルリピドA、ホスホリピド共役物(Goodman-Snitkoff達147:410N415(1991)の記載を参照)、プロテオリポソーム内の蛋白のカプセル化法(ミラー達、メリーランド、176:1739N1744、1992)、そして、ノバソウム(Novasome、登録商標)脂肪小胞(Micro Vescular Systems社、Nashua、NH)のような脂肪小胞蛋白のカプセル化法に使用できる。
【0097】
本発明組成物は、非経口投与(すなわち筋内、皮内、皮下)投与または経体腔投与(例えば経舌)、胃内投与、経口、経肛門、径膣を含む経粘膜で投与ができる。有効投与量は発全製品の種類、組換え型CAV2を用いる場合には発現レベル、その他の公知ファクタ、例えば品種、性差、年齢、ホストの状態および種類、さらにはLD50や公知のスクリーニング法を用いて過度の実験を必要せずに決めることができる。発現産物の有効投与量は数マイクログラムから数百マイクログラム、例えば5〜500μgにすることができる。本発明の発現産物は発現が実現できる任意の投与量で投与できる。すなわち、本発明の組換え型は少なくともこの量で投与され、ワクチンCAV2は約、l03.5 pfuの量、好ましくはl04 pfu〜約106pfuの量を投与できる。他の適切な担体または希釈剤は水または緩衝食塩水で、保存剤を含んでもよい。本発明の発現産物または組換え型CAVは凍結乾燥でき、投与の時点で再懸濁または溶液にすることができる。
【0098】
担体は重合体遅延系にすることができる。組成物を放出制御するには合成ポリマーが有効である。この例は1ミクロンのナノ粒子の球状メタクリル酸メチル重合体である(Kreuter、J.、Microcapsules and Nanoparticles in Medicine and Pharmacology, M. Doribrow(Ed),CRC Press、p. 125N148)。
マイクロカプセル化してマイクロカプセル化された医薬品を注射し、放出制御することができる。マイクロカプセル化のポリマーの選択には多数のファクタがある。ポリマー合成およびマイクロカプセル化プロセスの再現性、マイクロカプセル化材料およびプロセスのコスト、毒物学プロファイル、ポリマーの放出速度、およびポリマーと抗原の物理化学的適合性等の全てのファクタを考慮する必要がある。有用なポリマーの例はポリカーボネート、ポリエステル、ポリウレタン、ポリオルトエステルおよびポリアミド、特に(生物分解可能なものである。
【0099】
医薬品用および抗原用に最近選択される担体はポリアクリル酸エステル(d,1-ラクチド-co-グリコリド)(PLGA)である。これは侵食性縫合、硬骨プレート、その他の一次的人工器官の医療で長い歴史を有する生物分解可能なポリエステルで、毒性はない。各種医薬品はPLGAマイクロカプセル化されたペプチドおよび抗原を含む。抗原の制御放出用のPLGAのデータは例えばEldridge, J.H. et al、Current Topics in Microbiology and Immunology 1989、146:59-66に記載されている。直径1〜10ミクロンのPLGAのミクロスフィアの抗原の包括体が経口投与時に特に有効なアジュバント効果を示す。PLGAマイクロカプセル化プロセスには油中水エマルションの相分離を使用する。問題の化合物は水溶液として調製され、PLGAは適当な有機溶媒、例えばジクロロメタンおよび酢酸エチル中に溶かされる。これらの2つの非相溶性の溶液は高速攪拌機を用いてエマルジョン化される。次いでポリマーの非溶剤を加え、水溶液滴のまわりにポリマーを沈降させて胎生期マイクロカプセルを形成する。マイクロカプセルを回収し、ポリビニールアルコール(PVA)、ゼラチン、アルギナート、ポリビニルピロリドン(PVP)、メチル-フルロースで安定させ、真空乾燥または液液抽出法で溶剤を除去する。
【0100】
固形物を含む液体、液体およびゲル(gel caps)を含む固形物組成物が得られる。また、本発明の組換え型CAV2およびその発現産物は動物において免疫性すなわち抗体応答を刺激できる。これら抗体から周知の技術を用いてモノクローナル抗体が調製でき、そのモノクローナル抗体は周知の抗体結合アッセイ、診断キットまたは抗原の有無およびそこから抗原の自然に起因する因子の有無を決定し、免疫応答がその抗原または因子に単に刺激されたかどうか否かのテストで使用できる。
モノクローナル抗体はハイブリドーマ細胞を用いて得られる免疫グロブリンである。モノクローナル抗体は単一の抗原決定基と化学反応し、従来の血漿誘導抗体よりも大きい特異性を与える。さらに、多数のモノクローナル抗体を使用することによって所望の特異性、結合活性およびイソタイプを有する個体抗体を選択できる。ハイブリドーマ細胞株は化学的に全く同じ一定の抗体の安い供与源を提供し、この種の抗体の製造は簡単に標準化できる。モノクローナル抗体を生産する法は当業者に周知で、例えばKoprowski、H.達の1989年4月1日発行の米国特許第4,196,265号に記載されている。その内容は本明細書に引用したものとする。
モノクローナル抗体の使用は公知で、その使用の例は1983年3月8日発行のH.デイビッド、G.グリーンの米国特許第4,376,110号に記載されている。
モノクローナル抗体は免疫吸着クロマトグラフィによる材料の回収にも費用できる。例えばミルシテイン、C.、1980、Scientific American 243:66、70を参照。
【0101】
本発明の組換え型CAVまたはその発現産物は、インビトロでまたはインビボでの細胞の刺激と、その患者への自己輸血で使用できる。患者が血清陰性な場合には自己輸血は免疫応答を刺激し、例えば能動免疫のような免疫応答または抗原性応答をする。血清陽性な個体では自己輸血は病原に対して免疫系を刺激またはブーストする。
本発明の組換え型CAVはさらに、プローブまたはPCRプライマのためのDNAを作るのに有用であり、ハイブリット可能なDNAの有無の検出、DNAの増幅でき、例えば、サンプル中の病原を検出でき、また、DNAを増幅できる。
【0102】
本発明は、アデノウィルス系およびトランスアクチベイティング蛋白を与える任意のウイルス性または細胞系に有用なプロモータおよび発現カセットをに関するものである。このプロモータは切り詰めた転写活性プロモータまたは組換え型で、プラスミドが挿入されたウィルスまたは系によって与えられたトアンスアクチベイト蛋白でトアンスアクチベイトされた領域と切り詰めた転写活性プロモータが誘導される完全長プロモータの最小プロモーター領域とから成るのが好ましい。
【0103】
本発明のプロモータはヘルペスウィルス、例えばMCMV-IEまたはMCMVN−IEプロモータのような、MCMVまたはMCMVのような完全成熟ウイルスから誘導し、塩基対にベースで完全長プロモータの40%、さらには90%まで短くしたものであるのが好ましい。
【0104】
本発明の発現カセットは機能的な切り詰めたポリアデニル化信号、例えば切り詰めても機能するSV4Oポリアデニル化信号含むことができる。この発現カセットは(プロモータまたは発現カセットが挿入されているウィルスまたは系に対して)外来性またはヘテロジニアスなDNA、例えば上記の外来性またはヘテロジニアスなコード化DNAを含むことができる。このDNAは最適に配置され、リンクされて発現用プロモータとして使用することができる。発現カセットは任意の位置、好ましくは発現カセットが挿入された系またはウィルスから最高の発現が得られる位置に挿入できる。
【0105】
プロモータおよび発現カセットを特にアデノウィルスに関して説明したが、当業者は単にウィルス、プラスミド、細胞または系がトランスアクチベイティング蛋白を提供するか否かを確認するだけで、過度の実験のなしに、本発明を細胞、例えば真核細胞に対する他のウィルスおよびプラスミドに適応できる。
HCNVプロモータに関しては米国特許第5,168,062および5,385,839を参照(本願明細書に引用したもの)。発現のためのプラスミドDNAを有する細胞をそこからトランスフェクションすることに関してはFelgner達J. Biol.chem. 269, 1994, 2550-2561を参照。いろいろな感染症を防ぐためのワクチン接種の単純かつ効果的な方法、プラスミドDNAの直接注射に関してはScience、259:1745-49、1993を参照。本発明プロモータおよび発現カセットはアデノウィルス以外の系;例えばプラスミドDNAの直接注射で使われるプラスミドも本発明の範囲内である。
【0106】
本発明の具体化のためにの他の利用も存在する。
以下の本発明の実施例を説明するが、本発明が下記実施例に限定されるものではない。
【実施例】
【0107】
実施例1
ウイルスおよび細胞系の同定
イヌアデノウィルスタイプ2(CAV2)株は、ローヌメリュー(Rhone Merieux)社(アテネ(Ga))から参照番号CAV2ロット# 0830プールN 033093生産されたもので、タイターは107.4 TCID50/ml。また、Hadinおよびダービー犬腎臓(MDCK)細胞系はRhone Herieux Incから提供された。このCAV2は犬ワクチンとしてローヌメリュー社から市販されている。
【0108】
実施例2
ウィルス培養とコロニー化
MDCK細胞懸濁液をMEM(Gibco、グランドアイランド、NY)に種付けし、7.5%ウシ胎児血清(シグマ、Stルイ、HO)、ナトリウム・ピルベート(Gibco(1mM最終))、グルタミン(Gibco(2mM最終))、ペニシリン(Gibco(50のU/ml))、ストレプトマイシン(50mg/ml、Gibco)および非必須アミノ酸(NRA)(Gibco、0.1 mM最終)を供給し、5% CO2中で37itchで培養した。合体したMDCK細胞をCAV2の希釈液で感染させ、0.6%アガロース・オーバーレ下で、5% CO2中で37itchで培養した。CAV2はいくつかのラウンドでプラーク精製した。プラーク精製されたCAV2はT25 MDCKフラスコで培養した。培養CPEが完了したときに感染細胞を集め、アガロース下のMDCK細胞単層上でCAV2含量を定量した。ウィルス株はT175 MDCKフラスコを0.1の感染多重度(HOI)に感染させることで更に増殖させた。T-175 MDCKフラスコ増殖させたウィルスの力価は108p.f.u/mlであった。
【0109】
実施例3
ウィルスDNA精製
合体したHDCK細胞単層を含んでいるローラーボトル(108細胞/ボトル)を0.1pfu/細胞のプラーク精製されたCAV2ウィルスで感染させた。3日後に感染した単層は取り入まれ、低速遠心(1Kg、15分、l5℃)した。細胞ペレットは‐7O℃で保存された。次いで、凍ったペレットを37℃で解凍し、慎重に10mMトリスHCl pH 8.0と10mM EDTA緩衝液に懸濁し、細胞のDNAシアリングを制限した(35ml/108細胞)。再懸濁したペレットにSDSを加え、1%の最終濃度にした。室温で15分間培養した後、NaClを加えて1.25Mno濃度にした。4℃で3時間培養後、4℃、25Kgで20分間遠心分離した。塩を含んでいる濃い白いペレットとび細胞のDNAは捨て、上澄を42℃で4時間、次いで、60℃で30分間のProteinase K(300μg/ml最終濃度)で消化した。ウィルスDNAの回収の前に石炭酸-クロロホルムおよびクロロホルム抽出を二回行い、0.3M酢酸ナトリウムpH 6.0の存在で70%エタノールで沈殿させた。ウィルスDNAペレットを1時間風乾し、次いで2mlの水に再懸濁した。この手順で典型的には約4mgの精製されたCAV2 DNAを生ずる。精製されたウィルスDNAは利用まで-2O℃で保存した。
【0110】
実施例4
ウィルスDNA制限解析
精製されたCAV2 DNAのアリクォットをBoehringer Manriheim社(インディアナポリス(IN))の一組の限定酵素でメーカー明細に従って消化した。制限DNAサンプルは1%アガロースゲル上で電気泳動を用いて分別し、対応する制限フラグメントはUV光下でエチジウムブロマイド(4つの~g/ml)でゲル染色して視覚化した。表1に各種制限フラグメントの大きさを要約した。
【0111】
実施例5
E3を含む制限フラグメントの同定と特徴付け
1. CAV2 DNAのエンドヌクレアーゼのサザンブロット分析
それぞれ精製されたCAV2 DNAの4μgアリクォットをBamHI、Bg1I、HindII、HindIIIおよびPastIで消化し、1%アガロースゲルによる電気泳動で分画した。ゲルは、0.25MのHClに30分漬けた後、H20 で5分間洗った。次いで、ウィルスDNAを0.5M NaOHと0.9M NaCl溶液で変性した。H20 で5分間洗った後、DNAは3M NaClを含んだ0.5MトリスHCl( pH 7.5)の2つのバスで再度変性した。
DNAはナイロンメンブレン(Hybond N、Amersham Life Sciences、クリーブランド、OH)上でlOxSSC(1.5MのNaCl、0.15MのNa Citrate pH 7.4)緩衝中で一晩おいた。ナイロンメンブレンは1時間風乾し、3分間UV架橋した。65℃で6時間4xSSC、25% Denhardt's溶液(v/v)、0.1% SDS(v/v)、0.1% Naピロ燐酸塩および変性heringDNA溶液(500のmg/ml)で予備ハイブリッド化した。
【0112】
2. CAV2 PVIIIおよびFiber遺伝子に特有のプローブの製造
大部分のアデノウィルスのE3領域は2つの構造遺伝子、PVIIIおよび繊維の間にあるので、本出願人は各遺伝子のための2つのプライマ対を設計する(ライム、1992)ためにCAV2(マンハッタン株)の既に発表されている部分鎖のゲノムを利用した。オリゴヌクレオチドLF189(5'-TCAGTCATAGCCATCGACAGA-3')(配列番号:26)およびLFl9O(5'-GTGCTGGCTGGCACGGGCATT-3')(配列番号:27)およびオリゴヌクレオチドLFl91(5'-ATGTCCACCAAAGTCCCCTCTN‐3')(配列番号:28)はCAV2 PVIII遺伝子の3'端内の配列に対応するように設計され、LF192(5'-CCCGGGGCGTCGTATGGATAT‐3')(配列番号:29)はCAV2繊維遺伝子の5'端内の配列に対応するように設計された。
302bp のDNA PVIIIプローブは、lOX PCR緩衝、2mM dNTPsの3.75μl、26μlのH20、Taqポリメラーゼ(5.0u/μl)の0.25μl、5μM5'端HプライマLF189および5μlの5μM 3'端のプライマLF190と混合して作った。増殖は90℃で1分、55℃で1分、72℃で1分のプロフィルで40μlの鉱油を含む0.5mlの間で30回行った。190bpのDNA 繊維プローブはプライマLFl91でプライマLF189を交換することで上記プロトコールでVPCRで作られた。両方のPCR反応は1%アガロースゲルで電気透析され、対応するPCR生産物はメーカー指定の手順を使用して単離した(Bio 101, Inc., La Jolla, CA)。各プローブの100ngのアリクォットをlμgのランダムな六量体(Pharmacia, Piscataway, NJ)(全体積13μl)と混合し、3分間沸騰し、2.5μ1のdCTP 、dTTPおよびdGTP混合液(各0.5M濃度)、2.3μlのクレノーlOX緩衝液、1.5μlのクレノウ酵素(2u/μl)および5μlの32P−32P‐a‐ dATP(300℃i/mmol、10mCi/ml、NEN、ボストン、MA)で4時間培養した。反応はStop溶液(IBI Prime Timeキット)の100μlを加えることで停止した。各プローブの25μlを3分間50mlのprehybridization溶液の全体積で上記ナイロンメンブレンで65℃で一晩培養した後、変性した(100℃)。このナイロンメンブレンを6xSSC、0.1% SDSおよび2時間の50mM Na Pyrophosphate溶液の65℃で、次いで、蒸留水に通した。放射性同位元素でラベル化されたDNAプローブに対するウィルスDNA制限フラグメント相補性はオートラジオグラフィを用いて同定した。
【0113】
3. E3領域を含んだ制限フラグメントの同定およびクローン化
最も短いものは両方のPVIIIおよびFiberプローブで認識される制限フラグメントをよく単離するので、HindIllフラグメントA(4.0のKbp)が同定され、全CAV2 E3領域を含むことができることを示している。このフラグメントは上記遺伝子Clean手順を用して単離され、次いで、プラスミドpLFO27生成ベクターpBluescript SK+のHinndIII部位にサブクローン化される(Strantagene, La Jolla, CA)。
【0114】
4. CAV2 E3領域の特徴付け
CAV2 E3領域は、pLF027の規制変異消化作用と、配列2.0のキット指示(US Bi℃hemical、クリーブランド、OH)に従うシークエンシングpLF027を用いて分析された。配列解析はMacVectorソフトウェア(イーストマン・コダック、ロチェスター、NY)を使用して実行された。pLF027制限地図は図 2で示される。CAV2 E3領域を含むpLF027の対応配列(PVIIIストップコードン(pLFO27の#1413)および繊維ATG開始コードン間のDNA範囲として定義する(pLFO27)の#2945は図1に表される。シークエンシング・データの解析は、CAV2 E3が1,533 bps 100%以前に同定されたCAV2(マンハッタン株)E3領域(ライム(1992))については相応することを明らかにした。ヌクレオチド配列から推論されるアミノ酸配列の解析はCAV2 E3領域の右へのコード付けストランドが2つの潜在的なポリペチド(ORF1および0RF2)をコード化することを明らかにし、左へのコード付けストランドは単極電位ポリペチド(ORF3)をコード化する。これらのORFsの形質は表2で示される。
【0115】
実施例6
ドナー・プラスミドnLFOS6の生成
1. CAV2 E3配列の中央へのBglIIおよびMluI制限サイトの導入
取扱いを容易にするためにユニークなBglIIおよびMluI制限サイトを含む24 bp DNAリンカ(5'-GATACGCGTTCCATTAGCAGATCT-3')(配列番号:30)を、CAV2 E3領域(図1にて説明したように)のヌクレオチド#1487とび#1966との間にダブルラウンドPCR増殖手順を用いて導いた。最初のPCR増殖は鋳型としてpLF027 DNAを使用し、以下のプライマ一対を使用して実行した(LF327(5'-GGACACCTTTCTGATCAGTTCATT-3')/ LF3 24(5'‐GATACGCGTTCCATTAGCAGATCTTTGAGGGGC CTGGAAATAGGC-3')(配列番号:31、32)]、(LF326(5'−GGTTGTGTGGAAGACCCGGG GGCG-3')/LF325(5'-AGATCTGCTAATGGAA CGCGTATCGTGCCCCCACAGTACAGCAA-3')(配列番号:33、34)。それぞれ838bpおよび956bpの2つの部分的に重なり合うDNAフラグメントを作る。PCR増殖の第2ラウンドは部分的に重なり合う精製されたDNAフラグメントおよび両方の外部のプライマLF327およびLF326の存在下で実行された。得られた1,794bp DNAフラグメントはPstIおよびAatIで消化された。その結果生じる890bp の PstI/AatI フラグメントを精製し、6,069bp のPstI/ AatIl DNAフラグメントで連鎖反応させてpLFO27(pLF047A(図3および4)を作る。全てのPCR増殖は、上記条件を使用して実行された。次いで、6,944bp のMluI/BglII pLF047AをprearinealedしたオリゴヌクレオチドLF328(5'-GATCTGTTAACCCTAAGGCCATGGCATATGTCGCGAGGCCATCGTGGCCGCGGCCGC A-3')(配列番号:35)およびLF329(5'-CGCGTGCGGCCGCGGCCACGATGGCCTCGCGACATATGCCATGGCCTTAGGGTTA ACA-3')(配列番号:36)と連鎖反応させてpLF049A(図5および6)を作る。この結果、60bpのCAV2 E3領域は60bp BglII/HluIポリリンカDNAフラグメントと交換する。E3領域の大きさは修正されず、E3 ORF1はごく自然なままだった。しかし、E3 ORF2に対応する配列は分裂させられ、 E3 ORF3は完全に除去された。
【0116】
2. ドナー・プラスミドpLFO86の生成
CAV2 E3領域の一部を欠失するために、428bpの欠失がpLF049A MluI部位の3'を設計した。537bp DNAフラグメントをpLF027鋳型およびプライマ対LF361(5'-CTAGTCATCTTAACGCGTGTCCTCAACATCACCCGCGA-3')/LF334(5'-CTT GCTTGTTATTAAAAAAAG-3')(配列番号:37、38)を上記のように用いてPCRを用いて作った。この551bpフラグメントはMluIおよびAatIIで消化されて、精製され、pLFO49A の6,284bp のMluI/AatII DNAフラグメントと連鎖反応させてpLFO86(図7および8)を作る。この処理でE3領域の27%(428bp)が欠失し、更にその3'端の方のE3 ORF2にの欠失が広がるが、E3 ORF1暗号配列の邪魔はしない。
【0117】
実施例7
ウィルスゲノムの右の端を含む制限フラグメントのクローン化および特徴付け
1. ウィルスゲノムの右端む制限フラグメントのクローン化
CAV2(グラスゴー株)ゲノムの以前に発表された制限地図は84.0のマップ単位(SpibeyおよびCavanagh 1989)に位置するユニークなSalI制限サイトの存在を示した。CAV2 DNA(30μg)のSalI消化作用は、予測された3.2kbpおよび29kbp DNAフラグメントを作った。CAV2 DNA SalI Bフラグメント(3.2kbp)は、前述したように、ジーンClean手順を使用して精製されるゲルであって、20μ1のH20に予備懸濁した。約、3μgの精製されたSalI Bフラグメントが、22μ1 の1N NaOH (全体積22μ1)に90分で添加され、室温で5'-アデノウィルス・ゲノムの末端)に共有結合した蛋白部分(ロビンソン達(1973))を除去するために変成した。DNAは、2MのトリスHCl pH 7.5の1.3μ1を添加し、次いで、変形されて65℃で1時間培養され、pBluescript SK+の2.919bp SalI/SmaIフラグメント連鎖反応されて、pLFOS6を作った。
【0118】
2. ウィルスゲノムの右の端を含む制限フラグメントの性格付け
CAV2ゲノムのbp右の3.2K端は、pLF056の規制変異消化作用を用いて、そして、Sequenase 2.0のキット規定に従う同じプラスミドのシークエンシングを用いて分析された。配列解析は、MacVectorソフトウェアを使用して実行された。
pLF056制限地図は図10に示される、そして、図9はDNAの塩基配列を示す。
シークエンシング・データは、CAV2 DNA SalI Bフラグメントが長さの3,274bpであることを明らかにした。CAV2ゲノム内の2つのユニークな制限サイトは、CAV2 DNA SalI Bフラグメント内に局所化された:分類学的位置#587でのIglLtおよび分類学的位置#2133でのSalI。CAV2のヌクレオチド配列が右の末端で位置させた196のbp ITR(図9)が、100%、CAV2 Vaxitasおよびグラスゴー株のためにCAV2左右のITR配列については相応する(Cavanagh達1991)。残りのCAV2のDNAの塩基配列対CAV2 SalINBフラグメントDNAの解析から45%相同だけが示された。
【0119】
実施例8
PLF06lの生成
NruI/EcoRV 312のbpタグDNAフラグメント(図11)は、SmaI線形にされたpLF056についてはpLF06l(図11;図12に示される制限地図)を作り出すために連鎖反応させられた。
【0120】
実施例9
MDCIへの精製されたウィルスDNAのトランスフェクション
溶液Aは5μgの精製されたCAV2 DNAと血清遊離HEM(前述したように最終体積が300μlになるように補う)を混合して調製した。
溶液Bは、40μlのLipofectamine試薬(Gibco)に260μlの血清を含まないHEM培地を補って調製した。溶液AおよびBを一緒に混ぜ、30分間の室温で培養した。CAV2 DNA/リポソーム複合体に2.4mlのHEM培地(血清遊離)を混合して補った後、75%のHDCK細胞単層を合せた。5% C02の存在下、37℃で24時間インキュベーション後、血清を含まない培地を除去し、5% CO2を含む3mlのHEM培地と交換した。培養は5% CO2の存在下で37℃で8日間行ない、3日目に2mlのHEM培地を補った。このインキュベーションの間、CPEなしを証拠としてもよい。8日目にトランスフェクトされたMDCK細胞を掻き取り、全体積5mlにした。氷上で2分間超音波処理を2回した後、2mlのトランスフェクトされた培養を用いて、5% CO2の存在下で37℃で1時間、150mm直径の組織培養皿で100% MDCK単層を感染させた。その後、培養を0.6%アガロースを含む培地で行った。5日以後、5%CO2の存在下で37℃でプラークが現れる。観測される典型的な収率は10μgの精製されたDNA当り少なくとも2,000pfuである。
【0121】
実施例10
組換え型CAV2ウィルスの生成
1. 組換え型CAV2ゲノムのインビトロ生成
精製された20μg のCAV2 DNAを30U のSalIで一晩37℃で消化した。消化されたDNAは石炭酸クロロホルムで抽出て、H20中でエチルアルコールに再懸濁し、370ng/μl濃度にした。5μg のSalIで消化されたCAV2 DNAを5μgの3,557bp SalI/SacI pLF06l DNAフラグメントで400UのDNAリガーゼ(NEB、ベヴァリー、MA)の存在下でl5℃で一晩全体積50μ1にして結合反応した。
【0122】
2. CAV2組換え型ウィルスvCA1の単離
次いで、全部の結合反応を用いて75% MDCK単層をトランスフェクションした。収穫したトランスフェクトされた4mlの培養物を用いて2つの150のmm直径組織培養皿を感染させた。10日インキュベーションした後に合計8つのプラークが現れた。全てのプラークは回収し、1mlの追加のHEM培地に再兼濁し、氷上で2x 2'間、超音波処理した。浄化した培地を希釈し、連続的に60-mm直径組織培養皿で100% MDCK細胞単層を感染させた。6日の培養後、アガロースオーバーレを捨て、感染した単層をPerkus達1993に記載の手順でニトロセルロースフィルタ上へに塗布した。濾過処理し、ラベル化したNruI/EcorV 312bpのタグDNAフラグメントと上記の通常の方法でハイブリッドした。オートラジオグラフィ実験で初めに検出された8つのプラークの中の5つが組換え型CAV2ウィルスを含むことがわかった。プラークハイブリダイゼーションを用いて同定された1つのプラークを単離し、MDCK細胞上でプラーク精製を繰り返した。プローブとのハイブリダイゼーションを各精製後に確認した。プラーク精製した組換え型CAV2ウィルスにはvCAlという名前をつけた。
【0123】
3. vCAlの性格付け
vCAlを特徴づけるために小規模なDNA精製を行った。手短に言うと、精製されたvCAl組換え型ウィルスを100% MDCK単層(106の細胞)を感染させるために用いた。5日以後、CPEが完了後、感染した培養物を掻き取り、収穫した。超音波処理および浄化した培地を42℃で2時間、proteiriase K(500μg/mi終濃度)で処理した。この酵素を65℃で20分間加熱して機能をなくし、次いで、全DNAを石炭酸クロロホルムで抽出し、H20でエチルアルコールに再懸濁した。精製された全DNAをRNアーゼT1で処理し、石炭酸クロロホルムで抽出し、エチルアルコールで沈殿させ、H20中に再懸濁し、最終濃度を1.2μg/mlとした。5μgの精製されたvCAlのアリクォットをそれぞれBglII/SpeIで消化した。これらの2つの部位はCAV2ゲノム中でにユニークであるので、29kbpおよび3kbpのフラグメントはBg BglIIでの消化が予想される、30.5kbpおよび1.5kbpフラグメントはSpeIの消化が予想される。これら制限フラグメントは、vCA1がそのゲノムの右端内に300bpのヘテロジニアスなDNAを取り入れた組換え型CAV2ウィルスであることを証明している。
vCAlが本当に予想されたタグDNAフラグメントを取り入れたことを更に証明するために、VCAl DNAをサザンブロッティングを用いて分析した。そして、vCAlがタグDNAフラグメントを取り入れたことを確認した。
CAV2 Smalが挿入部位として使われことを確認するために、精製したvCA1 DNAからの1.9kbpDNAフラグメントをプライマLF379(5'-TCACGCCCTGGTCAGGGTGTT-3')(配列番号:39)およびLF407(5'-GCCATCGCGTCAACCTGA-3')(配列番号:40)の対で増殖した。
プライマLF63(5'-ATGATGTCTGGGGACATGN3')(配列番号:41)、LF379(5'-TCACGCCCTGGTCAGGGTGTT-3')(配列番号:42)およびLF384(5'-ACCACGCGCCCACATTTT-3')(配列番号:43)を使用した1.940bp DNAフラグメントの部分鎖解析からヘテロジニアスなタグDNAがCAV2 SmaI部位に本当に挿入されvCAlになったことが確認された。
【0124】
実施例11
組換え型CAV2ウィルスの生成
10μgのpLFO86をHindIIIで消化し、得られた3.6kbp DNAフラグメントを上記ジーンClean手順を使用して単離し、H20中に再懸濁し、最終濃度を100ng/μlにした。MDCK細胞を上記Lipofectamineをベースにした手順を使用してトランスフェクションした。溶液Aは3μgの精製したCAV2 DNAと0.5μgの3.600bp HindIIIDNAフラグメントとを混合して調製した。血清を含まないHEM培地を補って溶液Aの全容積を300μlにした。Tトランスフェクションされた細胞を8日目に収穫し、150mm直径組織培養皿上の付けた。上記能方法でプラークを5'端をラベル化したオリゴヌクレオチドLF328とハイブリッドした。プローブと交差反応させた5つのウイルス性プラークを掻き取り、プラーク精製を4回行った。プラーク精製した組換え型CAV2ウィルスにvCA2という名前を付けた(プラーク精製はDNAまたはウィルスの複製での組換え型の使用、すなわち組換え型のベクターの使用で、外来性DNAの制限または限界がないことを示す点に注意)。
【0125】
2. vCA2の性格付け
vCA2を特徴づけるために上記方法でvCAlの小規模DNA精製を実行した。精製したvCA2 DNAおよび野生型CAV2 DNAはHindIIIを用いてそれぞれに消化し、制限されたDNAを1%アガロースゲルで電気泳動した。VCA2サンプル中の3.6kbp HindIIIフラグメントが視覚化された。野性-型CAV2サンプル中には4.0kbpフラグメントが存在するので、vCA2ゲノム中で428bpのE3領域が欠失したことが証明された。
予想されたタグ(オリゴヌクレオチドLF328/LF329)が本当にvCA2 E3領域に組み込まれたことを更に証明するために、Southernブロットが実行した。その結果、タグの移入が確認された。
この結果は完全なCAV2 E3 ORF2が組織培養に必要でないことを示している。さらに、CAV2 E3 ORF2配列の一部がヘテロジニアスなDNAに置換でき、従って、CAV2ゲノム中にに第2の挿入部位がることを証明している。この結果はさらにCAV2 E3領域の一部が欠失でき、vCA1の所で述べたSmal部位に外来性DNAを導入して補償できることを証明している。
【0126】
実施例 12
再断片化プロモータ、短く変性されたポリアデニル化カセット、5'UTR置換プロモータおよびプラスミドおよびそれを含む組換え体の生成
1.1 pLFO22(CATリポータ遺伝子がHCMV-IEプロモータの再断片化(145bp)の制御下に置かれた発現ベクトル)の生成:
ヒトサイトメガロウイルス(hCMV)(タウン株)からのDNAをLafemina達(1989)に記載の方法で調製された。プライマ組LFl7 2(5'(ATCGTAAAGCTTAATGTCGTAATAACCCCGC-3')/LF159(5'-TCTACTGCAGCCGGTGTCTTCTATGGAG GTCA-3')およびhCMV DNA(lOng)を鋳型として使用して、ヒトサイトメガロウイルス即時早期プロモータ(hCMVNIE)の3'端の増殖をPCRを用いて上記方法で実行した。166bp DNAフラグメントをジーンClean手順を使用して精製し、PstI/HindIIIで消化し、pCAT-Basicベクター(Promega、マディソン、WI)の4,348bp PstI/HindIIIDNAフラグメントで直接連鎖反応させ、pLF022(図13、14(配列番号:7))を作った。pLF022発現カセット中に存在する調節配列はhCMV IEプロモータおよびSV4O小さいt抗原およびポリアデニル化信号を含んでいる856bpカセットの145bpフラグメントである。
【0127】
1.2 pLFO62(SV4Oポリアデニル化カセットが241bpに縮小されたpLF022の誘導体)の生成:
SV4O小さいt抗原の寸法およびpLFO22のポリアデニル化信号カセット(856bp)を減らすために、以下の処理を実行した。170bp DNAフラグメントはプライマLF377(5'-TCTTCGCCCCCGTTTTCACCATGG-3')およびLF378(5'-ATCACGCCGCGGCTTAAAAAAA TTACGCCCCGCCCT-3')およびpLF022 DNA(長く続く)を鋳型としてPCRを用いて増殖した。精製された増殖されたフラグメントを18mlのH20に再懸濁し、室温でまたは30分間、800μMのdNTPs存在下で1lUのクレノウ酵素(Boehringer Manriheim、インディアナポリス、IN)で培養した。変性されたDNAフラグメントを石炭酸-クロロホルムで抽出し、エタノール沈殿し、NcoIで消化し、回収した。得られた136bpフラグメントを3,655bp NcoI/BsaBI DNAフラグメントと連鎖反応してpLFO22(pLF062(図15、16(配列番号:8))を作った。pLF062はCAT遺伝子の下流に2つのコンセンサスポリアデニル化信号AATAAAの反複を含む。pLF062のCAT発現カセットの寸法はpLFO22の1,804bpと比較して1,119bpである。pLF062カセット発現カセット中の調節配列は、hCMV-IEプロモータの145bpフラグメントおよび241bp のSV4Oポリアデニル化信号を含んでいる。
【0128】
1.3 Ad2 TPLがHCMV-IEプロモータのクローン化した下流であるpLF062誘導体 pLFO66の生成:
CAV2複製の開始後にリポータ遺伝子の発現を許すためにhCMV-IEプロモータ転写開始部位の下流のヒトAd2三分節リーダー(Ad2 TPL)をクローン化して変性されたpLF062 CAT発現カセット
オリゴヌクレオチドSPH6ETr1(5'-AATTCGGTACCAAGCTTCTTTATTCTATACTTAAAAAGTGA AAATAAATACAAAGGTTCTTGACTCTCTTC-3O、SPH6ETr2(5'-CGCATCGCTGTCTGCGAGGGCCAGCTGT TGGGCTCGCGGTTGAGGACAAACTCTTCGCGGTCTTTCCAGT-3')、SPH6ETr3(5'-ACTCTTGGATCGGA AACCCGTCGGCCTCCGAACGTACTCCGCCACCGAGGGACCTGAGCGAGTCCGCATC-3')、SPH6ETr4(5'-GACCGGATCGGAAAACCTTCGAGAAAGGCGTCTAACC AGTcACAGTCGCAAGCCCGGGT-3')、SPH6ETrS(5'-CTTTGTATTTATTTTCAC TTTTTAAGTATAGAATAAAGAAGCTTGGTACCG-3')、SPH6 ETr6(5'-GAAGAGTTTGTCCTCAACCGCGAGCCCAACAGCTGGCCCTCGCAGACAGCGATGCGG~GAGAG TCAAGAAC-3')、SPH6ETr7(5'-GCTCAGGTCCCTCGGTGGCGGAGTACGTTCGGAGGCCGACGGGTTTCCGATCCGAGTACTGG AAAGACCGC-3')およびSPH6ETr8(5'-CTAGACCCGGGCTTGCGACTGTGACTGGTTAGACGCCTTT CTCGAGAGGTTTTCCGATCCGGTCGATGCGGACTC-3')はをキナーゼ処理し、アニールし、271bpの生産物をゲル精製した。
次に、完全なAd2 TPLはプライマLF394(5'ATCGTCCTGCAGACTCTCTTCCGCATCGCT GTCTGC-3')およびLF3 95(5'-GCTCTAGACTTGCGACTGTGACTGGTTAG-3')を用いてPCRで培養した。精製ゲルは鋳型としてオリゴヌクレオチドをアニールした。
得られた220bp DNAフラグメントをPstI/XkaIで消化し、上記ジーンClean手順を使用して精製し、3,800bp PstI/XbaI pLF062フラグメントで連鎖反応させてpLF066(図17、18(配列番号:9))を作る。pLF066発現カセットの調節配列はhCMVNIEプロモータの145bpフラグメントで、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号を含む241bpカセットと置換している。
【0129】
1.4 pLFO69(HCMVNIE 5'UTRがAd2 TPLで置換されたpLF066の誘導体)の生成:
pLF062中に存在するHCMV-IEプロモータ5'UTR(54bp)を以下の手順を使用して欠失した。アニールしたオリゴヌクレオチドLF397(5'-CGTTTAGTGA.ACCGTCTGCA-3')およびLF398(5'-GACGGTTCACTAAACGAGCT-3')をpLFO62の3,936bp DNAフラグメントで連鎖反応してpLF069(図19、20(配列番号:10))を作った。pLF069発現カセットの調節配列は、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号を含む241bpカセットと置換したHCMV-IEプロモータの91bpフラグメントである。
【0130】
1.5 pLFO77(SV4Oポリアデニル化カセットが153bpに縮小されたpLF069の誘導体)の生成:
SV4Oポリアデニル化配列の160bpのサブフラグメントをオリゴヌクレオチドHX3R(5'-GTAAAACGACGGCCAGT-3')およびLF409(5'-ATCGTCCCGCGGAATTGTTGTTGTTAACTTGTT-3')を使用し、pCAT Basic DNA(長く続く)を鋳型として用いてPCRで増殖した。得られた145bp DNAフラグメントをKspl/BamIIを用いて消化し、上記ジーンClean手順を使用して精製し、pLFO69の3,716 bp Kspl/BamII DNAフラグメントで直接連鎖反応してpLF077 (図21, 22, 配列番号: 11)を作る。pLF077のCAT発現カセットのサイズは1,161bpである。一方、pLF022では1,804bpである(36%縮小)。pLF069発現カセットの調節配列は、5'UTRが202bp Ad2 TPLおよびSV4Oポリアデニル化信号の一部を含む153bpカセットと交換したHCMV-IEプロモータの91bpフラグメントである。
【0131】
1.6 pLFO9l(ポリアデニル化信号の3O端が修正されたpLF077の誘導体)の生成:
CAV2ゲノムの右ITR配列の5'端のSmaI部位の上流域を局所化した12 bp(5'-TTTTTGGGCGTT-3')を以下の手順を使用してpLF077ポリアデニル化カセットの下流に導入した。1,000bp DNAフラグメントをオリゴヌクレオチドLF423(5'-ACGACCCGTAGAGGGCGTTGGACAGCAACTTGGCCTCGCGGTTGAGGACAAACTCTT-3')およびLF432(5'-ATCGTCCCCGGGTTTTTGGGCGTTATCCAGACATGAT~GATACA-3')を使用しpLF077 DNA(10人の黒ん坊)を鋳型として用いてPCRで増殖した。1,000bp PCR DNAフラグメントをジーンCleanで精製し、クレノー処置で修正し、NcoIを用いて消化した。PCR反応では1.2%アガロースゲルで電気透析し、295bpフラグメントをジーンClean手順を使用し単離した。
pLF077をBamHIを用いて消化し、クレノー酵素の作用で修正し、次いでNcoIを用いて消化した。消化反応では1%アガロースゲルで電気透析し、ジーンClean手順を使用して3,567bp制限フラグメントを単離し、上記295bp DNAフラグメントで連鎖反応してpLF09l(図23、24(配列番号:12))を得た。
【0132】
1.7 pLFO92(CAT発現カセットドナープラスミド)の生成:
pLFO9lの1,180bp HindIII/SmaI DNAフラグメント(全CAT発現カセットを含む)をKlenov酵素の作用で変性し、6.2kbp SmaIで線形にした pLF056で連鎖反応してpLF092(図25、26(配列番号:13))を作った。このプラスミドは、CAV2ゲノム5'端のSmaI部位の12bp上流域の挿入部位にCAT発現カセットを組込むためのドナー・プラスミドに対応する。
【0133】
1.8 pLFlO5(CAV2ゲノムの右のITR配列の5'端のSmaI部位の12bp上流域に外来性DNAを組込むドナープラスミド)の生成
予備アニールされたオリゴヌクレオチドLF446(5'-GGGTTTTTGGGCGTTTCGCGA ACCGGTGTTCACGCGTGTCGACCCC-3')およびLF447(5'−CCCAAAAACCCGCALGCGCTTGGCCA CTTGTGCGCACAGCTGGGG-3')から成るポリリンカ(NruI-AgeI-EcoRI-HluI-SalI-smaI]をSmaIで線形にした6.2kbp pLF056と連鎖反応させてpLF105(図27、28(配列番号:14)を作った。
【0134】
1.9 CAV2ゲノムの右終末端に挿入されるCAT発現カセットを含む組換え型CAV2ウィルスvCA3の生成:
l0μgのpLF092をHindIIIおよびBamHIで消化し、得られた4.3kbp DNAフラグメントをジーンClean手順を使用して単離し、H20中に再懸濁し、100ng/μlの濃度にした。MDCK細胞はLipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.4μgの4.3kbp HindIII/BamHI pLF092フラグメントと4.4μgの精製されたCAV2 DNAとを混合して調製した。溶液Aの全容積を血清無しのHEM培地を補って300μlにした。8日後にトランスフェクション細胞を収穫し、前述したように150mm直径組織培養皿上に塗布した。CATリポータ遺伝子用プローブはpCAT Basic DNA(10ng)を鋳型とし、プライマ対のLF218(5'-ATCGTACATATGGAGAAAAAAATCACTGGATAT-3')/LF2 31(5'-ATCGTAGATATCCTCGAGTTACGCCC CGCCCTGCCACTC-3')を用いたPCRで作った。結果として得られた660bp DNAフラグメントを上記方法でランダムプライミングでラベル化し、ニトロセルロース膜で上記手順を使用してリフト・ウイルス性プラークにハイブリッド化した。プローブに対応するプラークを掻き取り、上記方法でプラーク精製を4回行った。プラーク精製した組換え型CAV2ウィルスにvCA3という名前を付けた。
【0135】
2. vCA3の性格付け。
2.1 組換え型ウィルスvCA3を用いたCAT遺伝子形質発現の解析
2.1.1 vCA3感染MDCK細胞溶菌液中のCAT 酵素活性の検出
精製したvCA3組換え型ウィルスと野生型CAV2をそれぞれ別々にM.O.Iが10で、100%-MDCK単層(106の細胞)で感染させた。5% C02の存在下で37℃で24時間、感染した倍養液を掻き取り、収穫した。細胞ペレットを予熱された(37℃)PBS(Ca2+ およびMg2+遊離)で3回洗浄し、40mMトリス-HCl、pH 7.5、1mM EDTA、および150mM NaClのlml中に、室温で5分間培養した。細胞を4℃では30秒間、12Kgで遠心分離した。得られたペレットを100mlの0.25Hトリス-HCl pH 8.0に再懸濁し、激しく攪拌して急速凍結/融解サイクルを3回行う。溶菌液を65℃で10分間培養して端部遺伝子デアセチル活性の機能をなくされた。室温で12Kgで2分間遠心分離し、上澄をクロラムフェニコールアセチルトランスフェラーゼ(CAT)アッセイで次のように分析した。25mlの細胞溶菌液を37℃での2時間、3mlの(14Cクロラムフェニコール(0.005mCi/ml)(NEN、ボストン、MA)、n-Butyryl Coenzyme A(5mg/ml)の5mlおよび92mlの0.25のM TrisN HCl(pH 8.0)で培養した。反応は各チューブに500mlの酢酸エチル(シグマ、Stルイ、MO)を加えて終了させた。反応物を30秒間、キシレンで攪拌し、1分間の12Kgで遠心分離した。上側有機相は新しいチューブに入れ換え、乾燥蒸発した。残部は25mlのn-Butyryl Coerizyme A(5mg/ml)に再懸濁し、再懸濁された材料の10mlをシリカゲル薄層クロマトグラフィ(TCL)シリカプレート(ベーカー、Philisburg、NY)上に点状に付けた。シリカプレートクロマトグラフィは閉じたチャンバー内で溶剤がプレートの中間にくるまで約1時間運転した。シリカプレートを乾燥し、放射化した。ブチリル化されたクロラムフェニコールがvCA2サンプル中に明らかに検出され、変性クロラムフェニコールは対照の野生型CAV2サンプル中には見られなかった。この結果は組換え型ウィルスvCA3が機能的なCAT活性を発現し、従って、我々が設計した発現カセットおよび我々が選んだ挿入部位を確認した。
【0136】
2.1.2 vCA3感染MDCK細胞溶菌液からの放射化免疫沈殿によるCAT蛋白の検出
放射化免疫沈殿分析をvCA3-感染させたMDCK細胞およびCATシャトルポリクローナル血清(5'3'Inc、ボールダー社)に由来する[35S]メチオニン(1000Ci/nmol、NEN)ラベル化溶菌液を用いて上記(Pincus達(1992)方法で行う。放射化免疫沈殿したCATポリペチドをSDS−ページに溶かし、サリチル酸ナトリウムを用いたフルオログラフィで視覚化した。
制限酵素活性を用いたvCA3遺伝子組織を解析:
vCA3 DNAを上記方法で精製した。精製した全DNAをH20中に再懸濁して1.3μg/mlの最終濃度にした。精製し2μgのvCA3のアリクォットをBgllIおよびSalIで消化した。これら2つの部位はCAV2ゲノム中でユニークであるので、28.2kbpと3.8kbpのフラグメントがBgllIの消化から予想され、27.8kbpおよび4.2kbpフラグメントがSalI消化作用から予想される。これらの制限フラグメント・が確認され、VCA3がそのゲノムの右端内にCAT発現カセットの1,000bpを取り入れた組換え型CAV2ウィルスであることが証明された。
【0137】
実施例13
ドナープラスミド pLFl02の生成
E3 ORFlを修正せずにE3 ORF2の3'端を欠失するために、以下の手順を開発した。
pLF027 DNAを鋳型とし、プライマ対LF437(5'ATCTTAACGCGTCCCTcAGCTTCTAATGGGAC−3')およびLF334(5'−CTTGCTTGTTATTAAAAAAAG−3')を用いて上記方法でPCR増殖した。増殖された329bp DNAフラグメントをMlulおよびSmalを用いて消化し、上記ジーンClean手順を使用して精製した。得られた287bpのMiul/Snal DNAフラグメントをゲル精製し、pLFO86の6,079bp Miul/Smal DNAフラグメントと連鎖反応してpLFO95を作った。pLFO95の63bp BglIl/Miulリンカーを以下の手順を使用して無関係な外来性DNAの305bpのBglII/MiuIリンカーと交換した。305 bp DNAフラグメント(図29および30に記載のヌクレオチド配列、下記参照]は MiulおよびBglIIで無関係なプラスミドを消化して得た。MiulおよびBglII消化したDNAフラグメントをゲル精製し、pLFO9Sの6,315bp MiuI/BglII DNAフラグメントと連鎖反応させてpLFl02(図29、配列番号:15)を作った。
pLFl02のこの処理で305bpの外来性DNAとCAV2 E3の688bpフラグメント(全E3の寸法の45%)とが交換され、CAV2 E3領域内に必須でないサブドメインの制限を定義できる。
【0138】
実施例14
ドナープラスミド DLFl16Aの生成
2つのSphI制限サイト(位置#3770と#3870)を含むpLF027 EcoRV/AatIIの1.8kbp DNAフラグメントを欠失するために、pLF027 EcoRV/AatIIの5,163bpフラグメントをゲル精製し、クレノウ酵素で処理し、再リゲートしてpLFO94を作った。
ユニークなBglIIおよびMiul制限サイトを含む24bp DNAリンカ(5'-GATACGCGTTCCATTAGCAGATCT-3')を2回のPCR増殖手順を用いてE3 ORF1とE3 ORF2と間のpLFO94の遺伝子間配列に挿入した。最初のPCR増殖はpLF027 DNAを鋳型として使用し、下記プライマ一対を用いて実行した:LF243 (5'-CGCGCACAAACTGGTAGGTGC-3')/LF436(5-'AGATCTGCTA~TGGAACGCGTATC~GmAATAATATTATC-3')およびLF4 35 (5'-GATACGCGTTCCATTAGCAGATCTGTTTTACAGCTACCA-3')/LF277(5'-GTACAGTTATGTTGAAGG -3')。487bpおよび698bpの2つの部分的に重なり合うDNAフラグメントが作られた。第2回目のPCR増殖は部分的に重なり合う精製されたDNAフラグメントおよび両方の外部のプライマLF243およびLF277の存在下で、実行した。増殖された1,185bp DNAフラグメントをSphIおよびPatIで消化し、得られた566bp PatI/SphIフラグメントを精製し、pLFO94の4,653bp SphI/PatI部分消化で連鎖反応させてpLFO93を作った。全てのPCR増殖は上記条件を使用して実行された。
E3 ORFlを修正しないE3 ORF2の5'端の欠失を以下の手順を用いて行った。pLFO93 XhoI/MluI 1,062bpフラグメントをゲルは精製し、pLFO86の5,081bp XhoI/MluIフラグメントと連鎖反応させてpLFll5を作った。Miulで線形化したpLF115 DNAを無関係な外来性DNAの311bp Mlul/Mlulフラグメントと連鎖反応させてpLFll6AおよびBを作った。無関係な311のbp Mlulの配列/外来性DNAのMlulフラグメントを含むpLFll6Aの完全なDNAの塩基配列は図31(配列番号:16)に示され、制限地図は図32に示されている。
このpLFll6Aの遺伝子操作の結果、CAV2 E3876bpフラグメント(全E3寸法の57%)が311bpの外来性DNAと交換され、CAV2 E3領域内に必須でないサブドメインの制限を定義することができる。
【0139】
実施例14
ドナープラスミドpLFl00の生成
E3 0RF2、E3 ORFlの3'端および完全なE3 ORF3の5'端を同時に欠失するために、pLFOB6(図7、図8)のMluI(#1529)とDraIII(#889)制限サイトの間の634bpフラグメントを欠失し、以下の手順を使用して無関係な外来性DNAの302bpフラグメントとは交換した。
MiulおよびDraIIIで無関係なプラスミドを消化して302bpのDNAフラグメントを得た。MlulとDraIIIで消化したDNAフラグメントをゲルは精製し、pLFO86の5,946bp MluI/DraIII DNAフラグメントと連鎖反応させてpLFlOO(図33、34の配列番号:17)を作った。302bpフラグメントのヌクレオチド配列は図33に示され、制限地図は図34に示してある。
このpLFlOOの遺伝子操作でCAV2 E3の1,060bpフラグメント(全E3の寸法の69%)を302bpの外来性DNAと交換され、CAV2 E3領域内に更に必須でないサブドメインの制限を定義することができる。
【0140】
実施例15
ドナープラスミドpLFI20の生成
E3 ORF1、ほとんど完全なE3 ORF2および完全なE3 ORF3の3'端を同時に欠失するために、pLF1O2のMluI(#1771)とDraIII(#889)制限サイトの間で882bpフラグメントを欠失し、以下の手順を使用して無関係な外来性DNAの311bpフラグメントと交換した。
pLF1O2 DNAをMlulを用いて線形にし、DraIIIで部分的に消化した。得られた5,733bp MluI/DraIIIを無関係な外来性DNAの311bp MluI/DraIIIフラグメントと連鎖反応させてpLFl2O(図35、36(配列番号:18))を作った。この無関係な外来性DNAの311bp MluI/DraIIIフラグメントのヌクレオチド配列は図35に示され、制限地図は図36に示してある。
このpLFl2Oの遺伝子操作の結果で、CAV2 E3の1,261bpフラグメント(全E3寸法の82%)が311bpの外来性DNAと交換され、CAV2 E3領域内に更に必須でないサブドメインの制限を定義できる。
この最も大きい欠失でE3領域の約80%〜約100%、例えばE3領域の約80%〜95%、または約80%〜90%または80%〜約85%の全てを欠失できることを示す。
【0141】
実施例16
イヌジステンパーウィルス(CDV)ヘマグルチニン(HA)暗号配列を含むpLF043pBSSK+ の生成
1. プラスミドpSDCDVHAの生成
イヌジステンパーウィルス(CDV)のOnderstepoort株はH. Appel博士(コーネル大学、イサカ、NY)から得られた。RNAはCDV感染Vero細胞から収穫し、相補DNAは以下の方法で調整した。
CDV感染Vero細胞からのRNAはChirgwin達のグアニジウムイソチオシアネート-セシウム・クロライド法(1979)を用いて単離した。最初のストランド相補DNAはAMVリバーストランスクリプターゼ(Bio Sciences、セントピーターズバーグ、FL)、オリゴヌクレオチドプライマCDVFSP(配列番号:44)(5'-CCAGGACATAGCAAGCCAACAGGTC-3')およびリボゾームリボ核酸を用いてCDV感染細胞から合成した。CDVFSP(配列番号:44)はCDV融合(F)開始コードンの80bp上流域に入り、上記とヘマグルチニン(HA)暗号配列を含むポジティブなセンス信号ストランド状相補DNA生産物を生じる。
【0142】
HA−特異的転写解読枠(ORF)は最初のストランド相補DNA生産物からPCR法(PCR)を用いて上記方法で増殖した。オリゴヌクレオチドプライマCDVHA1(配列番号:45)(5'-CGATATCCGTTAAGTTTGTATCGTAATGCTCCCCTACCAAGAC-3')およびCDVHA2(配列番号:46)(5'-GGGATAAAAATTAACGGTTACATGAGAATCTTATACGGAC-3')をCDVFSP誘導の最初のストランド相補DNAとのPCRで鋳型として使用した。CDVHA1はワクシニアウィルスH6プロモータの3'末端領域を有し、翻訳開始コドンからCDVにHA ORYに入る配列が続く(Perkus達、1989)。CDVHA2(配列番号:46)はHA ORFのストップコードンからCDV HA 5'端に入る。その結果得られる1.8kbp PCR生産物は20mM dNTPsの存在下で大腸菌DNAポリメラーゼからのクレノー断片化処理でフラグメントをブラントエンドにする。1.8kbpのブラントエンドフラグメントがH6プロモータ内のNruI部位とpSD554(下記参照)のH6プロモータのSinaI部位3'との間に挿入される。得られたプラスミドpCDVHAはH6プロモートされたCDV HA ORFを含まなければならないが、予想外に、CDV HA 5'が欠失した。この欠失を下記方法で回復する。
【0143】
プラスミドpSD554はワクシニアフランキングアーム中にワクシニアK1Lホスト範囲遺伝子(Gillard達、1986)と、挿入部位が続くワクシニアH6プロモータとを含む。ワクシニア・フランキングアームはATI領域:転写解読枠A25LおよびA26Lを置換する(1990a,b、ゲーベル達)。pSD554は以下の方法で調製された。
左右のワクシニアフランキングアームはワクシニアSalI B(1990a,b、ゲーベル達)を含むpSD4l4を鋳型として使用してPCRで造られる。左アームは鋳型pSD4l4でオリゴヌクレオチドプライマMPSYN267(配列番号:47)(5'-GGGCTGAAGCTTGCTGGCCGCTCATTAGACAAGCGAATGAGGGAC-3')およびMP5YN268(配列番号:48)(5'-AGATCTCCCGGGCTCGAGTAATTAATTAATTTTTATTACACCAGAAAAGACGGCTTGAGATC-3')を使用してPCRで合成した。右アームは鋳型pSD4l4でオリゴヌクレオチドプライマMPSYN269(配列番号:49)(5'-TAATTACTCGAGCCCGGGAGATCTAATTTAATT TAATTTATATAACTCATTTTTTGAATATACT-3')およびMPSYN27O(配列番号:50)(5'-TATCTCGAATTCCCGCGGCTTTAAA TGGACGGAACTCTTTTCCCC-3')を使用してPCRで合成した。左右のアームを含む2つのPCRからのフラグメントはPCR中に結合された。結果として得られたPCR生産物をEcoRIおよびHindIIIで消化し、O.9 kbpフラグメントを単離した。この0.9kbフラグメントをpUC8 EcoRIとHindIII部位との間に挿入した。得られたプラスミドpSD541は以下の方法でKlL遺伝子および追加の挿入部位を受けた。
【0144】
プラスミドpSDS4lをBglIlおよびXhoIで消化し、アニールされた相補オリゴヌクレオチドMPSYN333(配列番号:5l)(5'-GATCTTTTGTTAACAAAAACTAATCAGC TATCGCGAATCGATTCCCGGGGGATCCGGTACCC-3')およびMPSYN334(配列番号:52)(5'-TCGAGGGTACCGGATCCCCCGGGAATCGATTCGCGATAGCTGATTAGTTTTTGTTAACAAAA-3')と連鎖反応させてプラスミドpSD552を作った。pSD452(Perkus達、1990)はKlL遺伝子を含む。pSD452をHpal で消化し、BglIIで部分的に消化した。得られたKlL遺伝子を含むlk bpのフラグメントをpSD5S2 HglIIとHpal部位との間に挿入した。得られたプラスミドpSD553をNroIで消化し、ワクシニアH6プロモータ(Perkus達、1989)を含むSmaI/NroIフラグメントを挿入した。合成プラスミド(pMPSS3H6)はA26L組込み遺伝子座内にKlL遺伝子から下流にワクシニアH6プロモータを含む。
プラスミドpMP5S3H6をNruIおよびBamHIで消化し、アニールされた合成オリゴヌクレオチドMPSYN347(配列番号:53)(5'-CGATATCCGTTAAGTTTGTATCGTAAT CTGCAGCCCGGGGGGG-3')およびMPSYN348(配列番号:54)(5'-GATCCCCCGGGCTGCAGA TTACGATACAAACTTAACGGATATCG-3')と連鎖反応させた。得られたプラスミドpSD554はATI領域と交換したフランキングワクシニア配列の範囲内の挿入部位にKlL遺伝子およびH6プロモータを含む。
PCR誘導フラグメントとして、ワクシニアウィルスH6プロモータおよびCDV HA ORFの5'端がpCDVHAに加えた。制御領域H6のATGはPCR誘導フラグメントのCDV HA翻訳開始コドンと重なる。ワクシニアウィルスH6プロモータはPerkus達、1989に記載されている。
【0145】
pEIVC5Lは変性されたH6プロモータおよび無関係な遺伝子を含む。pEIVC5LはオリゴヌクレオチドプライマH65PH(配列番号:55)(5'-ATCATCAAGCTTGA TTCTTTATTCTATAC-3')およびCDVHAH6(配列番号:56)(5'-GTCTTGGTAGGGGAGCAT TACGATACAAACTTAACG-3')とのPCR法で156bpフラグメントを作るのに用いた。CDVHAH6はH6プロモータの5'端へ向かう翻訳開始コドンから始まるCDV HAの5'端の18塩基対を含む。H6SPH(配列番号:55)はHindIII部位を含み、5'から3'へ向かH6プロモータからの配列が続く。156塩基対のPCR誘導H6SPH/CDVHAH6(配列番号:56)はCDV HA暗号化配列の5'端の18塩基対を含む。
【0146】
CDVFSP(配列番号:44)の最初のストランド相補DNA生産物はオリゴヌクレオチドプライマCDVHAATG(配列番号:57)(5'-ATGCTCCCCTACCAAGAC-3')およびCDVHAECO(配列番号:58)(5'-GTAATTAGTAAAATTCACCTTG-3')でPCRされて459塩基対のフラグメントを作る。CDVHAATG(配列番号:57)は翻訳開始コドンからCDV HA 3'端に入る。CDVHAECO(配列番号:58)プライムスはCDV HA 5'端に向かうH6プロモータ化されたCDV HA配列の位置583から始まる。 156塩基対および459塩基対のPCR誘導フラグメントをプールし、H6SPH(配列番号:55)およびCDVHAECOとPCRで597塩基対フラグメントを作る(配列番号:58)。PCR誘導生産物をHindIIIおよびEcoRIで消化してH6プロモータおよび5'端のCDV HA暗号配列の387塩基対を含む520塩基対フラグメントを作る。520塩基対のHindIll/EcoRI消化PCRフラグメントをpBSSK+のHindIIIおよびEcoRI部位との間に挿入してpBSCDVHA5Sを作る。プラスミドpBSCDVHASSはpBSSK+のCDV HA ORFのH6プロモートされた5'端を含む。CDV HA ORFの3'端は以下の方法で加えた。
プラスミドpCDVHAをSinaIで消化し、次にEcoRIで部分消化して3'がCDV HA ORYで終わるl.4kbpフラグメントを作る。この1.4kbpのpCDVHA EcoRI/SmalフラグメントをpBSCDVHA5SのEcoRIとSinaI部位との間に挿入した。得られたプラスミドpBSCDVHAをBamHIで消化し、XhoIで部分的に消化して、H6プロモートされたCDV HA転写解読枠を含む1.9kbpフラグメントを作った。この1.9kbp HamHI/XhoI pBSCDVHAフラグメントをpSD553(上記参照)のBamHIとXhoI部位との間に挿入した。得られたプラスミドpSDCDVHAは、ATI挿入部位のH6プロモートされたCDV HA遺伝子を含む。
【0147】
2. pLFO43の生成
CDV HA暗号配列とワクシニアウィルスH6プロモータの3'端領域を含むpSDCDVHA 1,975 bpのBainHIおよびHindIllをゲル精製し、pBSSK+の対応する制限部位の間に挿入してpLFO43(図37および38)(配列番号:19)を作る。
【0148】
実施例17
完全なCDV HA発現カセットを含むpLFO9Sの生成
XhaI制限サイトを以下の方法でCDV HA開始コードン(ATG)の直ぐ上流に設計した。409bp DNAフラグメントをプライマ対LF412(5'-CTGATCTCTAG AATGCTCCCCTACCAAGACAAG 3')(配列番号:59)およびLF413(5'-TGGAGATCGCGGAAGTCG 3')(配列番号:60)を用い、pLFO43 DNAを鋳型として用いてPCRで増殖した。PCR増殖されたフラグメントを上記ジーンClean手順を使用して単離し、20mM dNTPsの存在下で大腸菌DNAポリメラーゼからクレノーフラグメント化し、SpalおよびEcoRIを用いて消化した。得られたブラントエンドな/Spalの192bp DNAフラグメントをpLFO43の4,891bp Nrul/SpaIフラグメントで連鎖反応してpLF096を作った。
KspI制限サイトはpLFO96 CDV HAストップコードン(TAA)の直ぐ下流に以下の方法で設計された。204bp DNAフラグメントをpLFO43を鋳型として使用し、プライマLF438(5'-TGTTTATGACCCAATCG-3')(配列番号:61)およびLF4 39(5'ATGC TCCCGCGGTTAAC GGTTACATGAGAATCT 3O)(配列番号:62)として上記方法でPCRで増殖した。PCR増殖されたフラグメントをKspIおよびAcoIで消化し、上記ジーンClean手順を使用して単離した。得られた143bp DNAフラグメントをゲル精製し、pLFO96の4,594bp KspI/AcoIフラグメントと連鎖反応させてpLF097を作った。
CDV HA暗号配列を含む1,821bp pLF097 KspI/XhaIフラグメントをpLFO69の3,246bp HpI/XhaIフラグメントと連鎖反応させてpLFO98(図39、40)(配列番号:20)を作った。
【0149】
実施例18
pLFO99A(CAV2ゲノム右端のSmaI部位のCDVヘクタール発現カセット12bp上流域の組込みのためのドナープラスミド)の生成
pLF069に定義された調節配列と連結されたCDV HA暗号配列を含む2,372bp BamHI/HindIII pLFO98フラグメントを大腸菌DNAポリメラーゼからのクレノーフラグメントで処理し、6,243bp NruIで線形化したpLF1OS、と連鎖反応させてpLFO99AおよびpLFO99Bを作った。pLFO99Aは発現カセット(図41、42)(配列番号:21)の右側に対応する。
【0150】
実施例 19
組換え型CAV2ウィルスvCA4 1の生成および性格付け
1. 組換え型CAV2ウィルスvCA4の生成
10μgのpLF1O2をHindIIIで消化し、得られた3,652bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oで100ng/mlの濃度に再沈殿した。MDK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.6kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEM培地を補って溶液Aの全容積を300mlにした。トランスフェクション細胞を150 mmの直径の組織培養皿上に塗布し、8日後に収穫した。pLF1O2 DNA(10ng)を鋳型に使用し、プライマ対LF4 40(5'-ATCAGTACGCGTATGGGCCACACACGGAGG-3')/LF44 1(5'-ATCAGTAGATCTGT TATTAGTGATATCAAA-3')でPCRして、pLF1O2に挿入される外来性DNAの305bpフラグメントに特有のプローブを作った。得られた305bp DNAフラグメントは上記ハイブリッド手順を使用してランダムプライミングでラベル化し、ウイルス性プラークを持ち上げるためにニトロセルロース膜を用いてハイブリッド化した。プローブに対応する5つのウイルス性プラークを取り、上記方法でプラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスはvCA4という名前をつけた。
【0151】
2. vCA4の特徴付け
vCA4 DNAは上記方法で精製した。精製された全DNAをH2Oに再沈殿して最終濃度を1.9μg/mlにした。精製されたvCA4の2μgアリクォットをHindlIlで消化した。予想された3667bp HindIIIフラグメントがvCA4サンプル中に視覚化され、4.0kbpフラグメントは野性型CAV2サンプル中に存在していたので、vCA4ゲノムDNAがpLF102に記載の部分的に欠失したE3領域を含むということが証明された。サウザンブロットを用いてVCA4 DNAを分析し、VCA4が371bpは野性型E3領域より短いE3領域を有することを示した。
この結果はCAV2 E3領域の必須でないサブドメインを示す。より詳しくは、vCA4の誘導体は、pLFO27(図1(配列番号:1)参照)に記載の位置#1470と位置#2157との間[すなわちE3領域の45%]から成るCAV2 E3配列がヘテロジニアスなDNAに置換できることを証明している。これはさらに、CAV2 E3がCAV2ゲノム内の挿入部位であることを確認するものである。この結果はさらに、CAV2 E3領域の一部を欠失でき、CAV2ゲノムの右端にvCA3の所で説明したように外来性DNAを導入して保証できることを証明している。
【0152】
実施例20
組換え型CAV2ウィルスvCA5の生成および特徴付け
1. 組換え型CAV2ウィルスvCA5の生成
10μgのpLFll6AをHindIIIandで消化し、得られた3,487bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して濃度を100ng/mlにした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.5kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞を150mm直径の組織培養皿上にプレートアウトし、8日後に上記方法で収穫した。
pLFll6Aに挿入される外来性DNAの311bpフラグメントに特有のプローブはpLFll6A DNA(10ng)を鋳型として使用し、プライマ対LF453(5'-ATCGTCATTGCCACGCGTATGGC AGAAGGATTTGCAGCCAAT-3')/LF4 54(5'-ATCGTCATTGCCA CGCGTAACCA GGGACAATACTTGTTCATC-3')でPCRを用いて作った。得られた311bp DNAフラグメントは上記手順を用いてランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜を用いてハイブリッド化した。プローブに対応する5つのウイルス性プラークを取り、プラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスにはvCA5という名前をつけた。
【0153】
2. vCA5の特徴付け
vCA5 DNAを上記方法で精製した。精製された全DNAをH2Oに再沈殿して、最終濃度を1.9μg/mlにした。精製されたvCA5の2μgアリクォットをHindlIlで消化した。予想された3,487bp HindIIIフラグメントはvCA5サンプル中に視覚化され、4.0kbpフラグメントは野生型CAV2サンプル中に存在するので、vCA5ゲノムDNAがpLFl16Aに記載された部分的に欠失したE3領域を含むということが証明された。この結果は、CAV2 E3領域が必須でないサブドメインであることを示す。特に、このVCA5の誘導体はpLFO27(図1(配列番号:1)を参照)で説明したように、位置#1088と位置#1964 [E3領域の57%]との間から成るCAV2 E3配列はヘテロジニアスなDNAと交換できということを証明している。これはさらに、CAV2 E3がCAV2ゲノム内の挿入部位であることを確認するものである。この結果もCAV2 E3の一部を欠失でき、vCA3で述べたように、CAV2ゲノムの右端に外来性DNAを導入して補償できるということを証明している。
【0154】
実施例21
組換え型CAV2ウィルスvCA6の生成および性格付け
1. 組換え型CAV2ウィルスvCA6の生成
10μgのpLFlOOをHindIIIで消化し、得られた3,284bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿し、100ng/ml濃度にした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションされた。溶液Aは0.5μgの3.3kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞を150mm直径の組織培養皿上にプレートアウトし、8日の後に収穫した。
pLFlOODNA(10ng)を鋳型としては使用し、プライマ対LF442(5'-ATCAGTCACGGTGTGTAAATGG GCCACACACGGAGG-3')/LF44 3(5'-ATCAGTACGCGTGTTATTAGTG ATATCAAA-3')でPCRでpLFlOOに挿入するたプローブ用に外来性DNAの311bpフラグメントを作った。得られた302bp DNAフラグメントは上記ランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜とハイブリッドした。プローブと交差した5つのウイルス性プラークを掻き取り、プラーク精製を4回行った。プラーク精製された組換え型CAV2ウィルスはvCA6という名前をつけた。
【0155】
2. vCA6の特徴付け
vCA6 DNAは上記のようにして精製した。精製された全DNAをH2Oに再沈殿して最終濃度を1.9μg/mlにした。精製されたvCA6の2μgアリクォットをHindlIlで消化した。予想された3,284のbp HindIIIフラグメントはvCA6サンプル中に視覚化され、4.0kbpフラグメントは野生型CAV2サンプル中に存在するので、vCA6ゲノムDNAがpLFlOOに記載の部分的に欠失したE3領域を含むということが証明された。この結果はCAV2 E3領域が必須でないサブドメインであることを示す。より詳しくは、このvCA6の誘導体は、pLFO27(図1(配列番号:1)を参照)で説明したように、位置#898と位置#1949の間 [E3領域の69%] から成るCAV2 E3配列がヘテロジニアスなDNAと交換できることを証明している。これはまたCAV2ゲノム内に挿入部位としてCAV2 E3があることを確認している。この結果もCAV2 E3領域の一部が欠失でき、、vCA3の誘導体の所で説明したように、CAV2ゲノムの右端に外来性DNAの導入することで補償できることを証明している。
【0156】
実施例22
組換え型CAV2ウィルスvCA7の生成および特徴付け
1. 組換え型CAV2ウィルスvCA7の生成
10μgのpLFl2OをHindIIIで消化し、得られた3,085bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して100ng/mlの濃度にした。MDCK細胞は5μg の3kbp HindIII DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞は150mm直径組織培養皿上にプレートアウトし、8日後に収穫した。pLFlOOに挿入される外来性DNAの311bpフラグメントに特有のプローブはpLFlOO DNA(10ng)を鋳型に使用し、プライマ対LF458(5'-ATCCGTACGCGTTAGAGGGCAAAGCCC GTGCAGCAGCGC-3')/LF4 59(5'-ATCCGTCACGGTGTGTAGAT GGGTTGTTTTGTG GAGAAT-3')でPCRで作った。得られた311bp DNAフラグメントを上記方法でランダムべヌライミングし、ウイルス性プラークを持ち上げるためにニトロセルロース膜を用いてハイブリッド化した。プローブとウィルスDNAとの間の交差反応が確認された。
この結果はpLFO27(図1(配列番号:1)参照)で説明したように、位置#898と位置#2157との間の1,259bpの欠失が組織培養でのウイルス性複製と互換性を持ち、実質的にE3領域の全てを欠失できることを示している。
【0157】
実施例23
vCA5の生成
10μgのpLFO99AをHglIIおよびNotIで消化し、得られた5,131bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿し、100ng/mlの濃度にした。MDCK細胞は上記Lipofectamineベースとする手順を使用してトランスフェクションした。溶液Aは0.5μgの5.1kbp HglII/NotI DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。トランスフェクション細胞は150mm直径の組織培養皿上にプレートアウトし、8日後に収穫した。pSDCDVHAの440bp EcoRIフラグメントは上記ランダムプライミングし、ウイルス性プラークを取るためにニトロセルロース膜とハイブリッドした。プローブと交差した2つのウイルス性プラークを掻き取り、プラーク精製を行った。プラーク精製した組換え型CAV2ウィルスはvCA8という名前を付けた。
【0158】
2. vCA8の特徴付け
vCA8 DNAの精製、制限消化、サウザンブロット、放射化免疫沈殿、CDV HA発現の解析によって組込みおよび発現を確認した。
【0159】
実施例24
pLF1OS (イヌジステンパーウィルス(CDV)融合(Fl)をコードするプラスミドから誘導されたpBSSK+)の生成
1. pATICDVF1の生成
CDV融合(F)特異性転写解読枠(ORF)をCDVFSP(配列番号:44)を最初のストランド相補DNAから誘導したCDVFSP(配列番号:44)を鋳型として相補DNAからオリゴヌクレオチドプライマCDVATGF1(配列番号:63)(5'-CATAAATTATTTCATTATCGCGATATCCGTTAAGTTTGTA TCGTAATGCACAAGGGAATCCCCAAAAGC-3')およびCDVFT(配列番号:64)(5'-ATCATCGGATCCATAAAAATCAGTGTGATCTCACATAGGATTTCGAAG-3')を用いてPCRで増殖した。CDVATGF1(配列番号:63)はワクシニアウィルスH6プロモータの3'端領域(Perkus、達、1989)を含み、CDV F翻訳開始コドンからCDV F ORFまでの配列が続く。CDVFT(配列番号:64)はBamHI部位を含み、CDV FストップコードンからCDV Fの 5'端の方への配列が続く。得られたPCR生産物をNruIおよびBamHIで消化し、NruIとBamHI部位との間のpSD554に2kbpフラグメントを挿入した。得られたプラスミドpATICDVYlはワクシニアウィルスATI組込み遺伝子座にH6プロモーテッドCDV F ORFを含む。
【0160】
2. HC5LSP28の生成
CS ORFを除去するためにCSベクタープラスミドHCSLSP28を以下の方法で作った。遺伝子カナリポックス(canarypox)DNAを鋳型としてPCRでオリゴヌクレオチドプライマC5A(配列番号:65)(5'-ATCATCGAATTCTGAATGTTAAATGTTATACTTTG-3')およびCSB(配列番号:66)(5'-GGGGGTACCTTTGAGAGTACCACTTCAG-3')を用いた。得られた1.5kbpフラグメントのC5A端をEc1QRIで消化し、他端はpUC8のEcoR1とSmaI部位との間に組込みためにブラントにしたままにしたプラスミドC5LABを作った。遺伝子カナリポックス(canarypox)DNAを鋳型としてPCRでオリゴヌクレオチドプライマC5C(配列番号:67)(5'-GGGTCTAGAGCGGCCGCTTATAAAGATCTAAAATGCATAATTTC-3')およびC5DA(配列番号:68)(5'-ATCATCCTGCAGGTATTCTAAACTAGGAATAGATG-3')を使った。得られた400塩基対フラグメントのC5DA端をPatIで消化し、他端はのSmaIとPatI部位との間の組込みのためにブラントのままにしてプラスミドpCSLを作った。アニールされた相補なオリゴヌクレオチドCP26(配列番号:69)(5'-GTACGTGACTAATTAGCTATAAAAAGGATCCGGTACCCTCGAGTCTAGAATCGATCCCGGGTTTTTATGACTAGTTAATCAC-3')およびCP27(配列番号:70)(5'-GGCCGTGATTAACTAGTCATAAAAACCCGGGATCG ATTCTAGACTCGAGGGTACCGGATCCTTTTTATAGCTAATTAGTCAC-3')をpCSL p7l8とNotI部位との間に挿入した。得られたプラスミドHCSLSP28は遺伝子座CSベクタープラスミドである。
【0161】
3. pBSCDVHAVQの生成
オリゴヌクレオチドRW132(配列番号:71)(5'-AGCTTCCCGGGTTAA TTAATTAGTCATCAGGCAGGGCGAGAACGAGACTATCTGCTC GTTAATTA ATTAG-3')およびRW133(配列番号:72)(5'-AGCTCTAATTAATTAACGAGCAGATAGTCTCGTTCTCGCCCTGCCTGATGACTAATTAATTAACC CGGGA-3')をアニールしてはダブルリンカ配列を形成した。RW132/RW133(配列番号:71/配列番号:72)ダブルストランド配列をpBSCDVHA5SのH6プロモートされたCDV HA ORFのHindIII部位5'に挿入してプラスミドpBSCDVHAVQを作った。
【0162】
4. pCSCDVHAYlの生成
H6プロモートされたCDV HA ORFを含む2kbp pBSCDVHAVQ SmaIフラグメントをHC5LSP28 SmaI部位に挿入してプラスミドpCSLCDVHAを作った。CS遺伝子座において、お互いから離れて目指すそれらの転写産物で 、H6プロモートされたCDV Fを含む2.1kbp pATICDVF1 HpaI/BamHIフラグメントをpCSLCDVHA Smal/ BamHIの6.5kbp DNAフラグメントで連鎖反応してH6プロモートされたCDV F およびCDV HA ORFと含むプラスミドpC5LCDVHAY1を作る。
【0163】
6. ベクタープラスミドpC6Lの生成
C6 ORFを除去するためにC6ベクターpC6Lを以下の方法で作った。オリゴヌクレオチドプライマC6Al(配列番号:73)(5'-ATCATCGAGCTCGCGGCCGCCTATCAAAAG TCTTAATGAGTT-3')、C6B1(配列番号:74)(5'-GAATTCCTCGAGCTGCAGCCCGGGTTTTTATAGC TAATTAGTCATTTTTTCGTAAGTAAGTATTTTTATTTAA-3')、C6Cl(配列番号:75)(5'-CCCGGGCTGCAGCTCGAGGAATTCTTTTTATTGATTAACTAGTCAAATGAGTATATATAATTGAA AAAGTAA-3O)およびC6D1(配列番号:76)(5'-GATGATGGTACCTTCATAAA TACAAGTTTGATTAAACTTAAGTTG-3')を用いてpC6Lを作った。オリゴヌクレオチドプライマC6Al(配列番号:73)およびC6B1(配列番号:74)はカナリポックス(canarypox)DNAを鋳型にしてPCRで用い、380塩基対フラグメントを作った。カナリポックス(canarypox)DNAを鋳型にした第2回目のPCR反応はC6Cl(配列番号:75)およびC6D1(配列番号:76)は、オリゴヌクレオチドプライマを用い、1,155塩基対フラグメントを作った。2回のPCR反応生成物を合せ、C6Al(配列番号:73)およびC6D1(配列番号:76)で最後のPCRを行って1,613塩基対フラグメントを作った。最終PCR生産物をSacIおよびKonIで消化し、pBSSK+のSacIとKonIとの間に挿入した。得られたC6組込みプラスミドはpC6Lと名付けた。
【0164】
7. pNl03の生成
pC5LCDVHAY1をBamHIで消化し、20μM dNTPSの存在下で大腸菌DNAポリメラーゼからのクレノーフラグメントで処理し、BamHI部位をブラントエンドし、続けてSmalで消化した。H6プロモートされたCDV FとH6プロモートされたCDV HA ORFsとを含む4.2kbp BamHIからSmaI部位へのブラントエンドをpC6LのSmaIフラグメントに挿入てプラスミドpMMl03を作った。
【0165】
8. pLFl08の生成
CDV Fl暗号配列とワクシニアウィルスH6プロモータの3'端領域とを含むpMM103 HindlIl/ BamHI 1,961bp DNAフラグメントをゲル精製し、pBSSK+の対応する制限部位の間に挿入してpLFl08(図43、44(配列番号:22))を作った。
【0166】
実施例25
完全なCDV F1発現カセットを含むpLFlllの生成
pLFl08 XbaI制限サイトをCDV F1開始コードン(ATG)の直ぐ上流に以下の方法で設計した。473bp DNAフラグメントをpLFl08 DNAを鋳型とし、LF44 8A(5'-ACTGTACTCGAGTCTAGAATGCACAAGGGAATCCCCAAAAGC-3')(配列番号:77)およびRW830(5'-ATTCCAATGTATCTGAGC-3')(配列番号:78)をプライマとして使用してPCRで増殖した。PCR増殖されたフラグメントをXhoIおよびCo1IIで消化し、上記ジーンClean手順を使用して単離し、XhoI/Co1IIで消化した。得られたXhoI/ColII 136bpDNAフラグメントをpLF1O8の4,783bpのXhoI/ColIIフラグメントで連鎖反応してpLF109を作った。
XhaI(#2035)を欠失され、以下の方法でHpI制限サイトをpLFl08 CDV F1ストップコードン(TGA#2016)の直ぐ下流に設計した。431bp DNAフラグメントをpLF109を鋳型として使用しLF449(5'ACTGTACCGCGGTCAGTGTGATCTCACATAGGATTTCGA 3')(配列番号:79)およびCDV-FG(5'-GGTTGAAATAGATGGTG 3')(配列番号:80)をプライマとしてPCRで増殖した。PCR増殖されたフラグメントを上記ジーンClean手順を使用して単離し、KspIおよびBfrIで消化した。得られた255bp DNAフラグメントをゲル精製し、pLFl09の4,631bp KspI/BfrIフラグメントで連鎖反応させてpLYllOを作った。
CDV F1暗号配列を含む1,997bp pLYllO KspI/XbaIフラグメントをpLFO69の3,244bp KspI/XbaIフラグメントで連鎖反応してpLFlll(図45、46(配列番号:23))を作った。
【0167】
実施例26
変性された完全なCDV F1発現カセットを含むpLFl28の生成
CDV F1発現カセットのポリアデニル化カセットの大きさを241bpから153bpまで減らすために以下方法を実行した。pLYO77 KspI/BamHI 146bpフラグメントは上記のようにゲル精製し、pLFlll KanI/BamHI 5,002bpフラグメントで連鎖反応してpLFl28(図47、48(配列番号:24))を作った。
【0168】
実施例27
pLFL3OA(CAV2ゲノム右端のSmaI部位のCDV F1発現カセット12bp上流域に組込むためのドナー・プラスミド)の生成
プラスミドpLFl28をBamHIで消化し、次にHindlIlを用いて部分的に消化した。pLFO77中に制限配列と組合されたCDV F1コード配列を含む2,451bpのBamHI/HindIIIフラグメントをE. coli DNA ポリメラーゼからのクレノーフラグメントで処理し、6,243 bpの NruI 直線化 pLF105で連鎖反応してpLYl30A と PLY130Bとを作った。pLYl30Aは発現カセットの右側に対応している(図49, 50, 配列番号: 29).
【0169】
実施例28
vCA-CDVFl-Ql2bl-up-SmaIの生成
10μgのpLYl30AをNotIおよびHglIIで消化し、得られた5,305bp DNAフラグメントを上記ジーンClean手順を使用して単離し、H2Oに再沈殿して100ng/mlの濃度にした。MDCK細胞は上記Lipofectamineベースの手順を使用してトランスフェクションした。溶液Aは0.9μgの9.3kbp HglII/NotI DNAフラグメントと3μgの精製されたvCA2 DNAとを混合して調製した。血清なしのMEN培地を補って溶液Aの全容積を300m1にした。8日後にトランスフェクションされた細胞を収穫し、上記150mm直径組織培養皿上に塗布した。pATICDVF1の1.4kbp EccRI/BamHI DNAフラグメントを上記のランダムプライミングでラベル化し、リフトウイルスプラークを得るためにニトロセルロース膜とハイブリッドした。プローブと交差した2つのウイルス性プラークを取り、上記プラーク精製法を行い、vCA-CDVFl-@l2bp-up-SmaIを得た。このウィルスを制限消化(DNA解析)およびサウザンブロット放射化免疫沈殿(発現解析)を用いて特徴付けた。
【0170】
実施例29
追加の組換え体
タグおよびその他の外来性DNAがCAV2に組み込まれたので、他の外来性DNAもCAV2に組み込むことができる。従ってvCAl、vCA2、vCA3、vCA4、vCA5を作りたものである。vCA6、vCA7、vCA8およびvCA-CDVY1-@l2bp-up-SmaIを作るために使用した外来性DNAの代わりに、米国特許第5,174,993および第9,509,941号に記載のものを使用できる。例えば、組換え型アビポック(avipox)ウィルス、ワクシニアウィルス;狂犬病グリコプロテイン(G)遺伝子、シチメンチョウ風邪ヘマグルチニン遺伝子、gp 5l,30ウシ属の白血病ウィルスのエンベロープ遺伝子、ニューカッスル病ウイルス(NDV)抗原、FelVエンベロープ遺伝子、RAV-l env遺伝子、NP(Chicken/ペンシルバニア/1/83風邪(ウィルス)のヌーデオプロテイン(nudeoprotein)遺伝子)、マトリックス、プレポリマー伝染性気管支炎ウィルス;HSV gD;特に、エントモポックス(entomopox)プロモータの遺伝子、特に、米国特許第5,338,683号に記載の外来性DNA、例えば組換え型ワクシニアウィルス(アビポック(avipox)ウィルス;ヘルペスウイルス・グリコプロテインをコードするDNA;米国特許第5,494,807号に記載の抗原をコードする外来DNA、例えば組換え型ワクシニア、アビポック(avipox);特に、狂犬病、ヘパチチス(Hepatitis)B、JEV、YY、Dengue、はしか、仮性狂犬病、エピスタイン(Epstein)-バー、HSV、HIV、SIV、EHV、BHV、HcMV、犬パルボウィルス、馬の風邪、猫白血病ウイルス、FHV、ハンタン(Hantaan)、C.テアニ(tetani)、鳥風邪、お多福風邪、NDV;米国特許第9,903,834号に記載のもの、例えば、組換え型ワクシニア、アビポック(avipox)、Norbillivirus、例えばはしかF、ヘマグルチニン;米国特許第4,722,848号に記載のもの、例えば組換え型ワクシニアウィルス;HSV tk、グリコプロテイン(例えばgB、gD)(風邪HA)ヘパチチス(Hepatitis)B(例えば、特に、HBsAgJ);英国特許GB 2 269 820 Bおよび米国特許第9,514,375号に記載のもの(組換え型ポックスウィルス;フラビウイルス構造蛋白質);WO 92/22641に記載のもの(例えば、組換え型ポックスウィルス;特に、免疫不全ウイルス);WO 93/03149に記載のもの(例えば、組換え型ポックスウィルス;特に、IBDV);WO 94/16716および1994年1月19日出願の米国特許出願第08/184009に記載のもの(例えば、組換え型ポックスウィルス;特に、cytoicineおよび/または腫瘍関連された抗原);PCT/US94/066521994年1月19日(Plasraodiumライフサイクルの各段階からのようなプラズモディウム抗原);米国特許第5,523,089号(W093/08306)PCT/US92/08697、Molecular 5 Microbiology(1989)、3(4)、479-486、PCT公報WO 93/04175、WO 96/06165に記載のもの(Borrelia抗原とめそのDNA);Briles達のWO 92/14488(pneumococcalDNA)。これらはvCA2〜vCA8およびvCA-CDVF1-@12bp-up-SmaI中のような外来性DNAで追加のCAV2組換え体およびvCA2〜vCA8およびおよびvCA-CDVF1-@12bp-up-SmaI中のような欠失を作るのに用いることができる。例えば多重抗原のコードを含む組換え体を含む(サブフラグメントプロモータ、減少または変性したポリアデニル化カセットおよび交換した5'UTRを有するプロモータを含む)E3または右ITRとE4転写単位との間の領域またはその両方への挿入およびE3領域での欠失で使用できる。発現は解析で示される。本発明組成物は投与のための担体または希釈剤との混合製剤で宿主(抗体応答を含む応答を生じる脊椎動物、動物または人間)に与える。
【0171】
【0172】
【0173】
【図面の簡単な説明】
【0174】
【図1a】pLF027の完全なDNAの塩基配列を示す(6,995bp、配列番号:1)。CAV2 HindIll Aフラグメントはヌクレオチド#4725で始まり、ヌクレオチド#689で終わる。CAV2 E3領域はヌクレオチド#1414で始まり、ヌクレオチド#2945で終わる。CAV2 E3 ORF1はヌクレオチド#8で始まり、ヌクレオチド#346で終わる。CAV2 E3 ORF2はヌクレオチド#384で始まり、ヌクレオチド#1478で終わる。CAV2 E3 ORF3はヌクレオチド#1019で始まり、ヌクレオチド#483で終わる。残りのヌクレオチドとpBSSK+に対応する);
【図1b】図1aの続きである。
【図1c】図1bの続きである。
【図2】pLF027の制限地図を示す。
【図3a】pLF047Aの完全なDNAの塩基配列を示す(6,959bp、配列番号:2)。23 bp BlgII/MluIリンカーはヌクレオチド#1485で始まり、ヌクレオチド#1508で終わる。残りの配列はpLF027に対応する。
【図3b】図3aの続きである。
【図3c】図3bの続きである。
【図4】pLF047Aの制限地図を示す。
【図5a】pLF049Aの完全なDNAの塩基配列を示す(7,002bp、配列番号:3)。63 bp BlgII/MluIリンカーはヌクレオチド#2138で始まり、ヌクレオチド#2201で終わる。残りの配列はpLF047Aに対応する。
【図5b】図5aの続きである。
【図5c】図5bの続きである。
【図6】pLFO49Aの制限地図を示す。
【図7a】pLFO86の完全なDNAの塩基配列を示す(6,581bp、配列番号:4)。63 bp BlgII/MluIリンカーはヌクレオチド#2295で始まり、ヌクレオチド#2358で終わる。残りの配列はpLF047Aに対応する。
【図7b】図7aの続きである。
【図7c】図7bの続きである。
【図8】pLF086の制限地図を示す。
【図9a】pLF056の完全なDNAの塩基配列を示す(6,196bp、配列番号:5)。CAV2 SalI Bフラグメントはヌクレオチド#1で始まり、ヌクレオチド#3274で終わる。右ITR(196bp)はヌクレオチド#3078で始まり、ヌクレオチド#3274で終わる。SmaI部位は位置#3088に局所化している。残りのヌクレオチドはpBSSK+に対応する。
【図9b】図9aの続きである。
【図9c】図9bの続きである。
【図10】pLF056の制限地図を示す;
【図11a】pLF06lの完全なDNAの塩基配列を示す(6,503bp、配列番号:6)。306bpヘテロジニアスDNAタグはヌクレオチド#3091で始まり、ヌクレオチド#3397で終わる。残留りのヌクレオチドはpLF056に対応する。
【図11b】図11aの続きである。
【図11c】図11bの続きである。
【図12】pLF061の制限地図を示す;
【図13a】pLF022の完全なDNAの塩基配列を示す(4,504bp、配列番号:7)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。全ての他のヌクレオチドはpCATの基礎的配列に対応し、以下を含む:ヌクレオチド#209で始まり、ヌクレオチド#868で終わるCATリポータ遺伝子、ヌクレオチド#958で始まり、ヌクレオチド#1814で終わるSV4O小t抗原およびポリアデニル化信号(856bp)、ヌクレオチド#2467で始まり、ヌクレオチド#3327で終わるアンピシリン耐性遺伝子。
【図13b】図13aの続きである。
【図13c】図13bの続きである。
【図14】pLF022の制限地図を示す;
【図15a】pLF062のDNAの完全な塩基配列を示す(3,812bp、配列番号:8)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。CATリポータ遺伝子はヌクレオチド#209で始まり、ヌクレオチド#868で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#881で始まり、ヌクレオチド#1122で終わる。アンピシリン耐性遺伝子はヌクレオチド#1775で始まり、ヌクレオチド#2635で終わる。
【図15b】図15aの続きである。
【図16】pLF062の制限地図を示す。
【図17a】pLF066の完全なDNAの塩基配列を示す(4,009bp、配列番号:9)。hCMV−IE(145bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#147で終わる。Ad2 TPL(202bp)はヌクレオチド#154で始まり、ヌクレオチド#356で終わる。CATリポータ遺伝子はヌクレオチド#406で始まり、ヌクレオチド#1065で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#1077で始まり、ヌクレオチド#1319で終わる。アンピシリン耐性遺伝子はヌクレオチド#1972で始まり、ヌクレオチド#2832で終わる。
【図17b】図17aの続きである。
【図18】pLFO66の制限地図を示す。
【図19a】pLF06の完全なDNAの塩基配列を示す(3,955bp、配列番号:10)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(241bp)はヌクレオチド#1024で始まり、ヌクレオチド#1265で終わる。アンピシリン耐性遺伝子はヌクレオチド#191.8で始まり、ヌクレオチド#2778で終わる。
【図19b】図19aの続きである。
【図20】pLF069の制限地図を示す。
【図21a】pLF077の完全なDNAの塩基配列を示す(3,861bp、配列番号:11)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド# 93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(153bp)はヌクレオチド#1018で始まり、ヌクレオチド#1171で終わる。アンピシリン耐性遺伝子はヌクレオチド#1824で始まり、ヌクレオチド#2684で終わる。
【図21b】図21aの続きである。
【図22】pLFO77の制限地図を示す;
【図23a】pLF09lの完全なDNAの塩基配列を示す(3,888bp、配列番号:12)。hCMV−IE(91bp)プロモータはヌクレオチド#2で始まり、ヌクレオチド#93で終わる。Ad2 TPL(202bp)はヌクレオチド#100で始まり、ヌクレオチド#302で終わる。CATリポータ遺伝子はヌクレオチド#352で始まり、ヌクレオチド#1011で終わる。SV4Oポリアデニル化信号(153bp)はヌクレオチド#1018で始まり、ヌクレオチド#1164で終わる。SV4Oポリアデニル化信号の3'端に挿入されるCAV2 12のヌクレオチドはヌクレオチド#1165で始まり、ヌクレオチド#1176で終わる。アンピシリン耐性遺伝子はヌクレオチド#1851で始まり、ヌクレオチド#2711で終わる。
【図23b】図23aの続きである。
【図24】pLF09lの制限地図を示す。
【図25a】pLF092の完全なDNAの塩基配列を示す(7,379bp、配列番号:13)。CAT発現カセット(pLF091で定義)はヌクレオチド#1で始まり、ヌクレオチド#1179で終わる。CAV2の左フランキングアーム(182bp)はヌクレオチド#1180で始まり、ヌクレオチド#1362で終わる。CAV2右フランキングアーム(3,090bp)はヌクレオチド#4285で始まり、ヌクレオチドヌクレオチド#7375終わる。残りの配列はpBSSK+に対応する。
【図25b】図25aの続きである。
【図25c】図25bの続きである。
【図25d】図25cの続きである。
【図26】pLF092の制限地図を示す。
【図27a】pLF105の完全なDNAの塩基配列を示す(6,243bp、配列番号:14)。ポリリンカはヌクレオチド#3092で始まり、ヌクレオチド#3123で終わる。CAV2左フランキングアーム(182bp)はヌクレオチド#3123で始まり、ヌクレオチド#3321で終わる。CAV2右フランキングアーム(3,090bp)はヌクレオチド#1で始まり、ヌクレオチド#3091で終わる。残りのヌクレオチドはpBSSK+に対応する。
【図27b】図27aの続きである。
【図27c】図27bの続きである。
【図28】pLF105の制限地図を示す。
【図29a】pLFl02の完全なDNAの塩基配列を示す(6,615bp、配列番号:15)。305 bp BigII/MiuIリンカーはヌクレオチド#1471で始まり、ヌクレオチド#1776で終わる。残りの配列はpLF086に対応する。
【図29b】図29aの続きである。
【図29c】図29bの続きである。
【図30】pLFl02の制限地図を示す。
【図31a】pLFll16Aの完全なDNAの塩基配列を示す(6,450bp、配列番号:16)。311bp Miul/Miulリンカーはヌクレオチド#1092で始まり、ヌクレオチド#1403で終わる。残りの配列はpLF086対応する。
【図31b】図31aの続きである。
【図31c】図31bの続きである。
【図32】pLF1116Aの制限地図を示す。
【図33a】pLFl00の完全なDNAの塩基配列を示す(6,247bp、配列番号:17)。302 bp DraIII/MiuIリンカーはヌクレオチド#898で始まり、ヌクレオチド#1200で終わる。残りの配列はpLF086に対応する。
【図33b】図33aの続きである。
【図33c】図33bの続きである。
【図34】pLFl00の制限地図を示す。
【図35a】pLFl20の完全なDNAの塩基配列を示す(6,048bp、配列番号:18)。311 bp DraIII/MiuIリンカーはヌクレオチド#898で始まり、ヌクレオチド#1209で終わる。残りの配列はpLF086に対応する。
【図35b】図35aの続きである。
【図35c】図35bの続きである。
【図36】pLF120の制限地図を示す。
【図37a】pLF043の完全なDNAの塩基配列を示す(5,109bp、配列番号:19)。CDV HAコード配列はヌクレオチド#35で始まり、ヌクレオチド#2175で終わる。CDV HA ORFストップコードンは#1847である。部分的ワクシニアH6プロモータはヌクレオチド#7で始まり、ヌクレオチド#35で終わる。残りの配列はpBSSK+に対応する。
【図37b】図37aの続きである。
【図37c】図37bの続きである。
【図38】pLFO43の制限地図を示す。
【図39a】pLFO98の完全なDNAの塩基配列を示す(5,070bp、配列番号:20)。CDV HA発現カセットはヌクレオチド#1で始まり、ヌクレオチド#2372で終わる。残りの配列はpLF069に対応する。
【図39b】図39aの続きである。
【図39c】図39bの続きである。
【図40】pLF098の制限地図を示す。
【図41a】pLF099Aの完全なDNAの塩基配列を示す(8,618bp、配列番号:21)。CDV HA発現カセットはヌクレオチド#3120で始まり、ヌクレオチド#5494で終わる。残りの配列はpLFI05に対応する。
【図41b】図41aの続きである。
【図41c】図41bの続きである。
【図41d】図41cの続きである。
【図42】pLF099Aの制限地図を示す。
【図43a】pLFl08の完全なDNAの塩基配列を示す(4,965bp、配列番号:22)。ワクシニアウィルスH6プロモータの3'端領域は位置#1と#29との間に位置する。CDV Flコード配列は位置#30から始まって、位置#2018で終わる。残りの配列はpBSSK+に対応する。
【図43b】図43aの続きである。
【図43c】図43bの続きである。
【図44】pLFl08の制限地図を示す。
【図45a】pLF111の完全なDNAの塩基配列を示す(5,241bp、配列番号:23)。CDV Fl発現カセットは位置#1から始まって位置#2(556)で終わる。残りの配列はpLF069に対応する。
【図45b】図45aの続きである。
【図45c】図45bの続きである。
【図46】pLF11lの制限地図を示す;
【図47a】pLF128の完全なDNAの塩基配列を示す(5,147bp、配列番号:24)。CDV Fl発現カセットは位置#1から始まって位置#2452で終わる。残りの配列はpLF077に対応する。
【図47b】図47aの続きである。
【図47c】図47bの続きである。
【図48】pLFl28の制限地図を示す。
【図49a】pLF130Aの完全なDNAの塩基配列を示す(8,792bp、配列番号:25)。CDV Fl発現カセットは位置#3126から始まってヌクレオチド#5669で終わる。CAV2 SalI.B左フランキングアーム(3,091bp)は位置#1と3,091との間に位置する。CAV2 SalI.B右フランキングアーム(182bp)は位置#5688と5,870との間に位置する。残りの配列はpLF105に対応する。
【図49b】図49aの続きである。
【図49c】図49bの続きである。
【図49d】図49cの続きである。
【図50】pLFl30Aの制限地図を示す。
【特許請求の範囲】
【請求項1】
イヌアデノウィルスのタイプ2(CAV2)遺伝子のE3領域に欠失部分を含み且つ(i)CAV2遺伝子のE3領域、(ii) CAV2遺伝子のE4領域と右ITR領域の間の領域、または(iii)CAV2遺伝子の上記E3領域およびCAV2遺伝子のE4領域と右ITR領域の間の領域の両方の領域に外来DNAが挿入されていることを特徴とする組み換えイヌアデノウィルスタイプ2(CAV2)。
【請求項2】
CAVが自然に複製する細胞に対して感染性CAV2としてパックされた請求項1に記載のCAV2。
【請求項3】
請求項1または2に記載のCAV2を含む外来DNAの発現ベクター。
【請求項4】
請求項1または2に記載のCAV2を含む外来DNAのクローン化用ベクター。
【請求項5】
外来DNAがエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療遺伝子または融合蛋白から成る群の中から選択される発現産物をコード化する請求項3に記載のベクター。
【請求項6】
外来DNAがエピトープをコードする請求項5のベクター。
【請求項7】
上記エピトープが家畜の病原またはトキシンの抗原性エピトープである請求項6に記載のベクター。
【請求項8】
上記抗原性エピトープが麻疹ウイルス(Morbillivirus)抗原、狂犬病グリコプロテイン、鳥風邪抗原、ウシ属白血病ウィルス抗原、ニューカッスル病ウイルス(NDV)抗原、猫白血病ウイルス外膜蛋白(FeLV)、ラウス関連ウイルス(RAV-1 env)、伝染性気管支炎ウィルスマトリックスおよび/またはプレポリマー、ヘルペスウイルスグリコプロテイン、フラビウイルス抗原、免疫不全ウィルス抗原、パルボウィルス抗原のプレポリマー、馬の風邪抗原、マレック病(Marek's Disease)ウィルス抗原、ポックスウィルス抗原または伝染性滑液嚢膿瘍(bursal)ウィルス抗原から成る請求項7に記載のベクター。
【請求項9】
麻疹ウイルス(Morbillivirus)抗原がイヌジステンパーウィルス血球凝集素プロテイン(HA)または融合プロテイン(F)から成る請求項8に記載のベクター
【請求項10】
上記エピトープが人間の病原またはトキシンの抗原性エピトープから成る請求項6に記載のベクター。
【請求項11】
上記抗原性エピトープが麻疹ウイルス抗原、狂犬病グリコプロテイン、風邪抗原、ヘルペスウイルス抗原、フラビウイルス抗原、ヘパチチス(Hepatitis)ウィルス抗原、免疫不全ウイルス抗原、ハンタン(Hantaan)ウィルス抗原、C.テアニ(tetani)抗原、お多福風邪抗原、肺炎球菌(pneumococcal)抗原、ボレリア(Borrella)抗原、マラリア病原虫(Plasmodium)抗原または水疱瘡抗原から成る請求項10に記載のベクター。
【請求項12】
外来DNAが成育因子または治療遺伝子をコードする請求項5に記載のベクター。
【請求項13】
上記の成長因子または治療遺伝子が疾患ファイティング蛋白、癌治療分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードする請求項12に記載のベクター。
【請求項14】
上記の成長因子または治療遺伝子がα―グロビン、β‐グロビン、γ-グロビン、顆粒球マクロファージ・コロニー刺激因子、腫瘍壊死因子、インターロイキン、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、エリスロポイエチン(EPO)、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随抗原またはメラノーマ付随抗原(B7)をコードする遺伝子から成るグループから選択される請求項13に記載のベクター。
【請求項15】
請求項1または2に記載の組み換えイヌアデノウィルスタイプ2(CAV2)と、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物。
【請求項16】
請求項3および5〜11のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物。
【請求項17】
請求項12〜14のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む治療用組成物。
【請求項18】
請求項6〜8のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物をヒトを除く宿主に投与することから成るヒトを除く宿主の脊椎動物の免疫応答を誘発する方法。
【請求項19】
請求項9に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物を宿主のネコに投与することから成る宿主のネコの免疫応答を誘発する方法。
【請求項20】
請求項17に記載の治療用組成物をヒトを除く宿主に投与することから成るヒトを除く動物へ遺伝情報を移入する方法。
【請求項21】
請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)をインヴィトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞から蛋白、遺伝子産物または発現産物を抽出、精製または分離することから成る、蛋白、遺伝子産物または発現産物の発現方法。
【請求項22】
請求項3、5〜7および10のいずれか一項に記載のベクターをインヴィトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞から蛋白、遺伝子産物または発現産物を抽出、精製または分離することから成る、蛋白、遺伝子産物または発現産物の発現方法。
【請求項23】
請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)をインビボまたはインビトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞からDNAまたは子ウィルスを抽出、精製または分離することから成る外来性DNAの塩基配列のクローン化方法。
【請求項24】
CAVゲノムの本質的でない領域中に外来性DNAを挿入することから成る請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)の製造方法。
【請求項25】
外来性DNAがプロモータを含むことを特徴とする請求項1または2に記載の組み換えイヌアデノウィルスタイプ2(CAV2)。
【請求項26】
上記プロモータがヘルペスウィルスである請求項25に記載のCAV2。
【請求項27】
上記プロモータがサイトメガロウイルス(CMV)プロモータである請求項26に記載のCAV2。
【請求項28】
上記プロモータが鼠CMV−IEプロモータである請求項27に記載のCAV2。
【請求項29】
上記プロモータがHCMV−IEプロモータである請求項27に記載のCAV2。
【請求項30】
上記プロモータが切り詰めた転写活性プロモーターであり、この切り詰めた転写活性プロモーターはウィルスに起因する転写活性化タンパクによって転写活性化された領域とこの切り詰めた転写活性プロモータの元になる完全長プロモータの最小のプロモーター領域とから成る請求項25に記載のCAV2。
【請求項31】
上記プロモータがヘルペスウィルスである請求項30に記載のCAV2。
【請求項32】
上記プロモータがCMVからのプロモータである請求項31に記載のCAV2。
【請求項33】
上記プロモータが鼠CMV−IEからのプロモータである請求項32に記載のCAV2。
【請求項34】
上記プロモータがHCMV−IEからのプロモータである請求項32に記載のCAV2。
【請求項35】
上記プロモータが、塩基対基準で完全長の鼠CMV−IEプロモータの10〜40%の寸法にまで切り詰められたものである請求項33に記載のCAV2。
【請求項36】
上記プロモータが、塩基対基準で完全長のHCMV‐IEプロモータの5〜90%の寸法にまで切り詰められたものである請求項34に記載のCAV2。
【請求項37】
組み換えウィルスまたはプラスミド用の切り詰めた転写活性プロモータであって、ウィルスまたは上記プラスミドが挿入された系によって与えられた転写活性化タンパクによって転写活性化された領域と、上記の切り詰めた転写活性プロモータの元になる完全長プロモータの最小のプロモーター領域とから成ることを特徴とする、切り詰めた転写活性プロモータ。
【請求項38】
上記プロモータがヘルペスウイルスである請求項37に記載の切り詰めた転写活性プロモータ。
【請求項39】
上記プロモータがCMVからのプロモータである請求項38に記載の切り詰めた転写活性プロモータ。
【請求項40】
上記プロモータが鼠CMV−IEからのプロモータである請求項39に記載の切り詰めた転写活性プロモータ。
【請求項41】
上記プロモータがHCMV−IEプロモータである請求項39に記載の切り詰めた転写活性プロモータ。
【請求項42】
上記プロモータが、塩基対基準で完全長の鼠CMV−IEプロモータの10〜40%の寸法にまで切り詰められている請求項請求項40に記載の切り詰めた転写活性プロモータ。
【請求項43】
上記プロモータが、塩基対基準で完全長のHCMV‐IEプロモータの5〜90%の寸法にまで切り詰められている請求項40に記載の切り詰めた転写活性プロモータ。
【請求項44】
(i)図21に記載の91塩基対のDNAおよび(ii) 図13に記載の145塩基対のDNAの中から選択されるDNA配列によって基本的に構成されることを特徴とする、切り詰めた転写活性なサイトメガロウイルスの前初期プロモータ。
【請求項45】
DNA配列が図21に記載の91塩基対のDNA配列である請求項44に記載の切り詰めたプロモータ。
【請求項46】
DNA配列が図13に記載の145塩基対のDNA配列である請求項44に記載の切り詰めたプロモータ。
【請求項47】
請求項37〜46のいずれか一項に記載のプロモータからなる、組換えウィルスへの組込み用発現カセット。
【請求項48】
切り詰めた機能性ポリアデニル化信号を含む請求項47に記載の発現カセット。
【請求項49】
請求項37または44のプロモータを含む組換え型CAV。
【請求項50】
請求項47の発現カセットを含む組換え型CAV。
【請求項51】
請求項48の発現カセットを含む組換え型CAV。
【請求項52】
CAV2である請求項49〜51のいずれか一項に記載の組換え型CAV。
【請求項53】
CAV2に対して外来のポリペチドをコードするCAV2中では自然には生じないドナーDNAから成り、このドナーDNAはCAV2の本質的でない領域と同一直線上になるCAV2 DNAのセグメント内にあって、CAV2の本質的でない領域からのDNAがドナーDNAをフランクリングしているプラスミド。
【請求項54】
請求項15または16に記載の免疫組成物またはワクチン組成物のワクチン接種(vaccination)での使用。
【請求項1】
イヌアデノウィルスのタイプ2(CAV2)遺伝子のE3領域に欠失部分を含み且つ(i)CAV2遺伝子のE3領域、(ii) CAV2遺伝子のE4領域と右ITR領域の間の領域、または(iii)CAV2遺伝子の上記E3領域およびCAV2遺伝子のE4領域と右ITR領域の間の領域の両方の領域に外来DNAが挿入されていることを特徴とする組み換えイヌアデノウィルスタイプ2(CAV2)。
【請求項2】
CAVが自然に複製する細胞に対して感染性CAV2としてパックされた請求項1に記載のCAV2。
【請求項3】
請求項1または2に記載のCAV2を含む外来DNAの発現ベクター。
【請求項4】
請求項1または2に記載のCAV2を含む外来DNAのクローン化用ベクター。
【請求項5】
外来DNAがエピトープ、生化学反応モジュレータ、成育因子、認識配列、治療遺伝子または融合蛋白から成る群の中から選択される発現産物をコード化する請求項3に記載のベクター。
【請求項6】
外来DNAがエピトープをコードする請求項5のベクター。
【請求項7】
上記エピトープが家畜の病原またはトキシンの抗原性エピトープである請求項6に記載のベクター。
【請求項8】
上記抗原性エピトープが麻疹ウイルス(Morbillivirus)抗原、狂犬病グリコプロテイン、鳥風邪抗原、ウシ属白血病ウィルス抗原、ニューカッスル病ウイルス(NDV)抗原、猫白血病ウイルス外膜蛋白(FeLV)、ラウス関連ウイルス(RAV-1 env)、伝染性気管支炎ウィルスマトリックスおよび/またはプレポリマー、ヘルペスウイルスグリコプロテイン、フラビウイルス抗原、免疫不全ウィルス抗原、パルボウィルス抗原のプレポリマー、馬の風邪抗原、マレック病(Marek's Disease)ウィルス抗原、ポックスウィルス抗原または伝染性滑液嚢膿瘍(bursal)ウィルス抗原から成る請求項7に記載のベクター。
【請求項9】
麻疹ウイルス(Morbillivirus)抗原がイヌジステンパーウィルス血球凝集素プロテイン(HA)または融合プロテイン(F)から成る請求項8に記載のベクター
【請求項10】
上記エピトープが人間の病原またはトキシンの抗原性エピトープから成る請求項6に記載のベクター。
【請求項11】
上記抗原性エピトープが麻疹ウイルス抗原、狂犬病グリコプロテイン、風邪抗原、ヘルペスウイルス抗原、フラビウイルス抗原、ヘパチチス(Hepatitis)ウィルス抗原、免疫不全ウイルス抗原、ハンタン(Hantaan)ウィルス抗原、C.テアニ(tetani)抗原、お多福風邪抗原、肺炎球菌(pneumococcal)抗原、ボレリア(Borrella)抗原、マラリア病原虫(Plasmodium)抗原または水疱瘡抗原から成る請求項10に記載のベクター。
【請求項12】
外来DNAが成育因子または治療遺伝子をコードする請求項5に記載のベクター。
【請求項13】
上記の成長因子または治療遺伝子が疾患ファイティング蛋白、癌治療分子、腫瘍サプレッサ、サイトカイン、腫瘍関連抗原またはインターフェロンをコードする請求項12に記載のベクター。
【請求項14】
上記の成長因子または治療遺伝子がα―グロビン、β‐グロビン、γ-グロビン、顆粒球マクロファージ・コロニー刺激因子、腫瘍壊死因子、インターロイキン、マクロファージ・コロニー刺激因子、顆粒球コロニー刺激因子、エリスロポイエチン(EPO)、マストセル成長因子、腫瘍サプレッサp53、網膜芽細胞腫、インターフェロン、メラノーマ付随抗原またはメラノーマ付随抗原(B7)をコードする遺伝子から成るグループから選択される請求項13に記載のベクター。
【請求項15】
請求項1または2に記載の組み換えイヌアデノウィルスタイプ2(CAV2)と、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物。
【請求項16】
請求項3および5〜11のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物。
【請求項17】
請求項12〜14のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む治療用組成物。
【請求項18】
請求項6〜8のいずれか一項に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物をヒトを除く宿主に投与することから成るヒトを除く宿主の脊椎動物の免疫応答を誘発する方法。
【請求項19】
請求項9に記載のベクターと、薬学的に許容される担体または希釈剤とを含む免疫組成物またはワクチン組成物を宿主のネコに投与することから成る宿主のネコの免疫応答を誘発する方法。
【請求項20】
請求項17に記載の治療用組成物をヒトを除く宿主に投与することから成るヒトを除く動物へ遺伝情報を移入する方法。
【請求項21】
請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)をインヴィトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞から蛋白、遺伝子産物または発現産物を抽出、精製または分離することから成る、蛋白、遺伝子産物または発現産物の発現方法。
【請求項22】
請求項3、5〜7および10のいずれか一項に記載のベクターをインヴィトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞から蛋白、遺伝子産物または発現産物を抽出、精製または分離することから成る、蛋白、遺伝子産物または発現産物の発現方法。
【請求項23】
請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)をインビボまたはインビトロで細胞に感染またはトランスフェクションし、必要な場合にはさらに上記細胞からDNAまたは子ウィルスを抽出、精製または分離することから成る外来性DNAの塩基配列のクローン化方法。
【請求項24】
CAVゲノムの本質的でない領域中に外来性DNAを挿入することから成る請求項1または2に記載のイヌアデノウィルスのタイプ2(CAV2)の製造方法。
【請求項25】
外来性DNAがプロモータを含むことを特徴とする請求項1または2に記載の組み換えイヌアデノウィルスタイプ2(CAV2)。
【請求項26】
上記プロモータがヘルペスウィルスである請求項25に記載のCAV2。
【請求項27】
上記プロモータがサイトメガロウイルス(CMV)プロモータである請求項26に記載のCAV2。
【請求項28】
上記プロモータが鼠CMV−IEプロモータである請求項27に記載のCAV2。
【請求項29】
上記プロモータがHCMV−IEプロモータである請求項27に記載のCAV2。
【請求項30】
上記プロモータが切り詰めた転写活性プロモーターであり、この切り詰めた転写活性プロモーターはウィルスに起因する転写活性化タンパクによって転写活性化された領域とこの切り詰めた転写活性プロモータの元になる完全長プロモータの最小のプロモーター領域とから成る請求項25に記載のCAV2。
【請求項31】
上記プロモータがヘルペスウィルスである請求項30に記載のCAV2。
【請求項32】
上記プロモータがCMVからのプロモータである請求項31に記載のCAV2。
【請求項33】
上記プロモータが鼠CMV−IEからのプロモータである請求項32に記載のCAV2。
【請求項34】
上記プロモータがHCMV−IEからのプロモータである請求項32に記載のCAV2。
【請求項35】
上記プロモータが、塩基対基準で完全長の鼠CMV−IEプロモータの10〜40%の寸法にまで切り詰められたものである請求項33に記載のCAV2。
【請求項36】
上記プロモータが、塩基対基準で完全長のHCMV‐IEプロモータの5〜90%の寸法にまで切り詰められたものである請求項34に記載のCAV2。
【請求項37】
組み換えウィルスまたはプラスミド用の切り詰めた転写活性プロモータであって、ウィルスまたは上記プラスミドが挿入された系によって与えられた転写活性化タンパクによって転写活性化された領域と、上記の切り詰めた転写活性プロモータの元になる完全長プロモータの最小のプロモーター領域とから成ることを特徴とする、切り詰めた転写活性プロモータ。
【請求項38】
上記プロモータがヘルペスウイルスである請求項37に記載の切り詰めた転写活性プロモータ。
【請求項39】
上記プロモータがCMVからのプロモータである請求項38に記載の切り詰めた転写活性プロモータ。
【請求項40】
上記プロモータが鼠CMV−IEからのプロモータである請求項39に記載の切り詰めた転写活性プロモータ。
【請求項41】
上記プロモータがHCMV−IEプロモータである請求項39に記載の切り詰めた転写活性プロモータ。
【請求項42】
上記プロモータが、塩基対基準で完全長の鼠CMV−IEプロモータの10〜40%の寸法にまで切り詰められている請求項請求項40に記載の切り詰めた転写活性プロモータ。
【請求項43】
上記プロモータが、塩基対基準で完全長のHCMV‐IEプロモータの5〜90%の寸法にまで切り詰められている請求項40に記載の切り詰めた転写活性プロモータ。
【請求項44】
(i)図21に記載の91塩基対のDNAおよび(ii) 図13に記載の145塩基対のDNAの中から選択されるDNA配列によって基本的に構成されることを特徴とする、切り詰めた転写活性なサイトメガロウイルスの前初期プロモータ。
【請求項45】
DNA配列が図21に記載の91塩基対のDNA配列である請求項44に記載の切り詰めたプロモータ。
【請求項46】
DNA配列が図13に記載の145塩基対のDNA配列である請求項44に記載の切り詰めたプロモータ。
【請求項47】
請求項37〜46のいずれか一項に記載のプロモータからなる、組換えウィルスへの組込み用発現カセット。
【請求項48】
切り詰めた機能性ポリアデニル化信号を含む請求項47に記載の発現カセット。
【請求項49】
請求項37または44のプロモータを含む組換え型CAV。
【請求項50】
請求項47の発現カセットを含む組換え型CAV。
【請求項51】
請求項48の発現カセットを含む組換え型CAV。
【請求項52】
CAV2である請求項49〜51のいずれか一項に記載の組換え型CAV。
【請求項53】
CAV2に対して外来のポリペチドをコードするCAV2中では自然には生じないドナーDNAから成り、このドナーDNAはCAV2の本質的でない領域と同一直線上になるCAV2 DNAのセグメント内にあって、CAV2の本質的でない領域からのDNAがドナーDNAをフランクリングしているプラスミド。
【請求項54】
請求項15または16に記載の免疫組成物またはワクチン組成物のワクチン接種(vaccination)での使用。
【図1a】


【図1b】
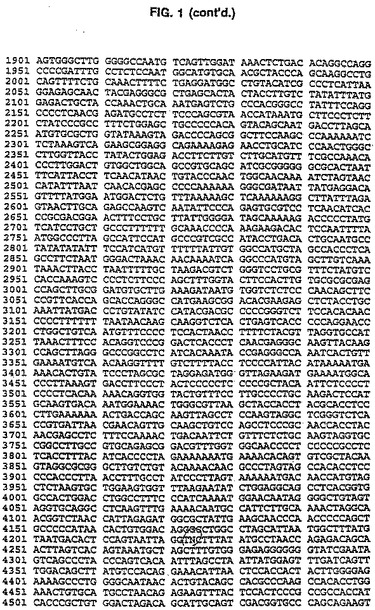
【図1c】

【図2】
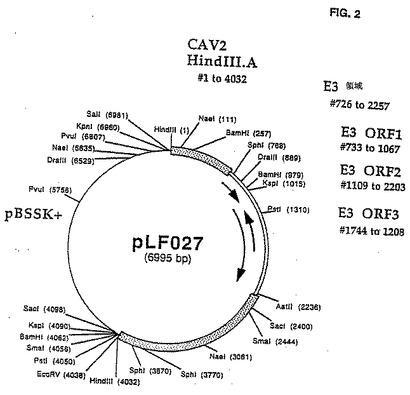
【図3a】

【図3b】
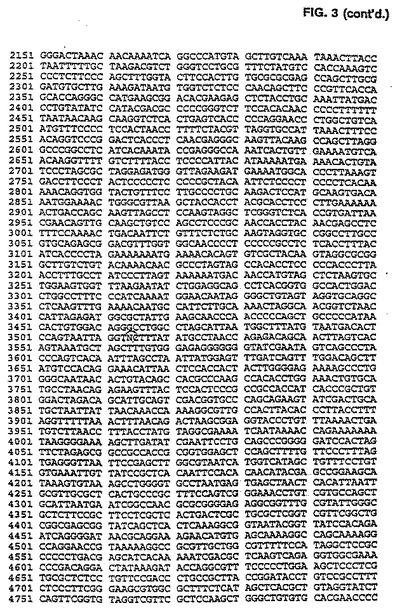
【図3c】

【図4】
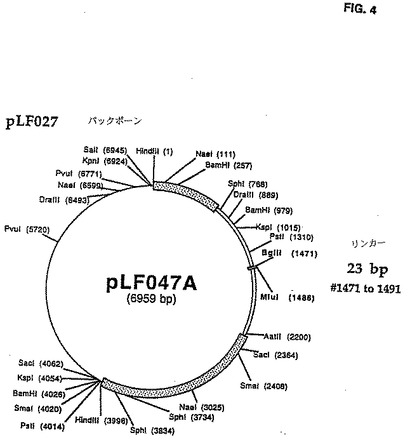
【図5a】

【図5b】

【図5c】

【図6】
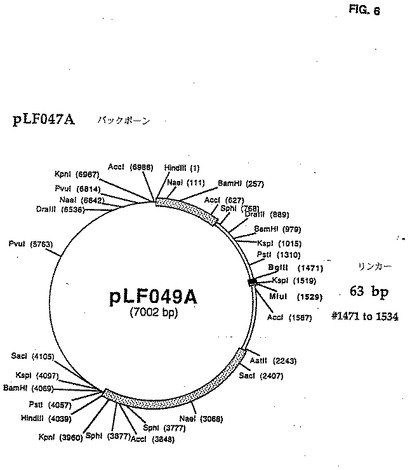
【図7a】

【図7b】
【図7c】

【図8】

【図9a】

【図9b】

【図9c】

【図10】
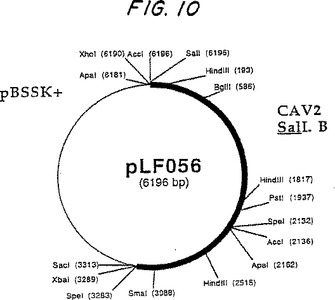
【図11a】

【図11b】

【図11c】

【図12】
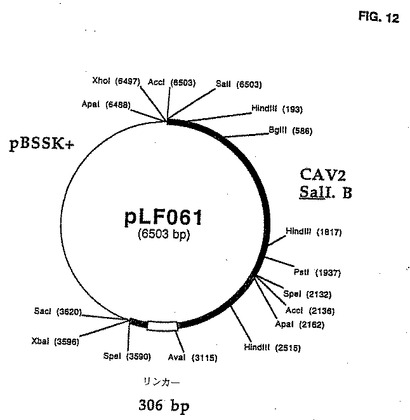
【図13a】

【図13b】

【図13c】

【図14】
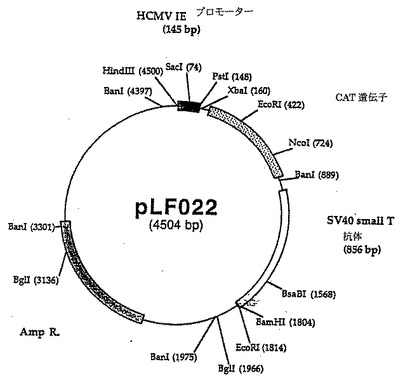
【図15a】

【図15b】

【図16】
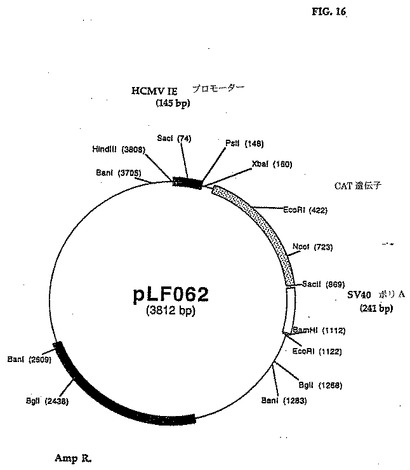
【図17a】

【図17b】
【図18】

【図19a】

【図19b】
【図20】

【図21a】

【図21b】
【図22】

【図23a】

【図23b】

【図24】
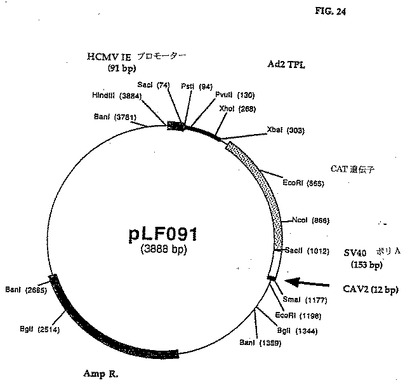
【図25a】

【図25b】
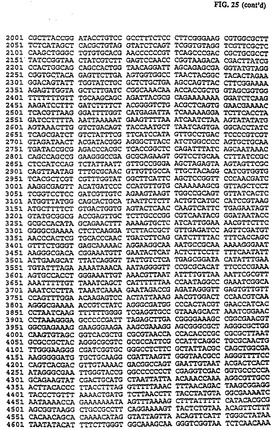
【図25c】

【図25d】

【図26】
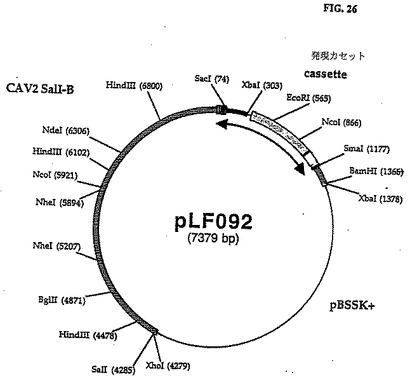
【図27a】

【図27b】

【図27c】

【図28】

【図29a】

【図29b】
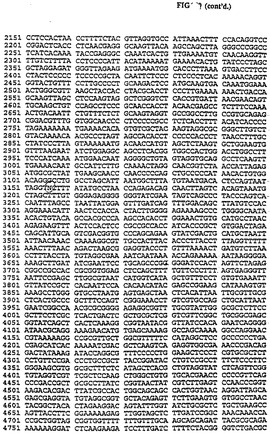
【図29c】

【図30】

【図31a】

【図31b】

【図31c】

【図32】
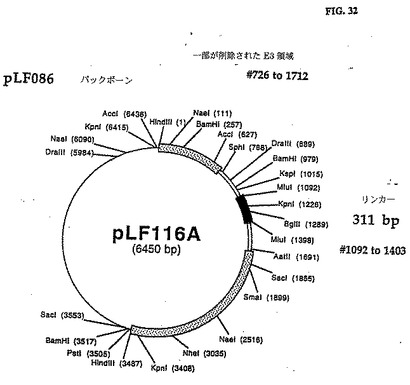
【図33a】

【図33b】
【図33c】

【図34】
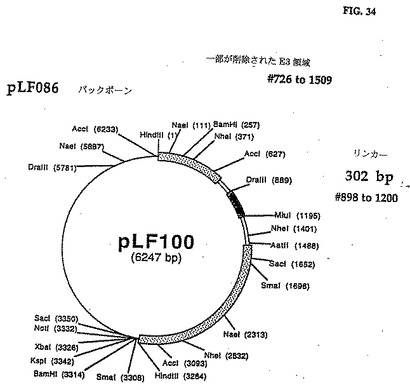
【図35a】

【図35b】

【図35c】

【図36】
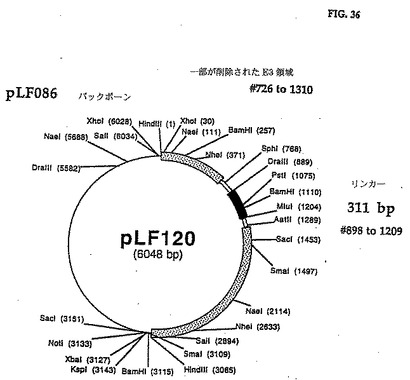
【図37a】

【図37b】

【図37c】

【図38】
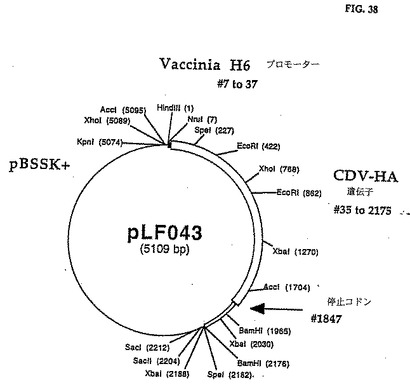
【図39a】

【図39b】

【図39c】

【図40】
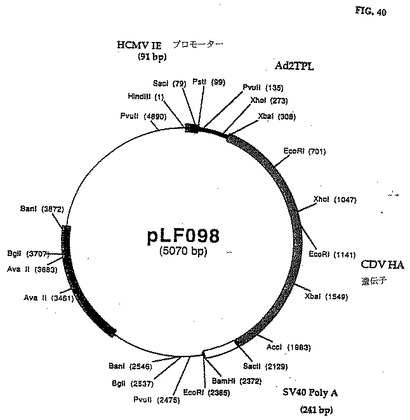
【図41a】

【図41b】

【図41c】
【図41d】

【図42】

【図43a】

【図43b】

【図43c】

【図44】
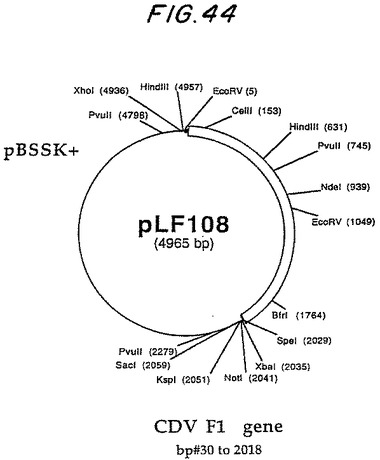
【図45a】

【図45b】
【図45c】

【図46】
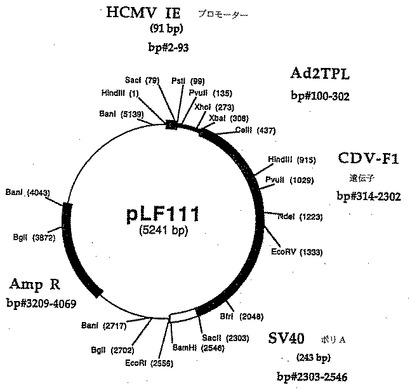
【図47a】
【図47b】

【図47c】

【図48】
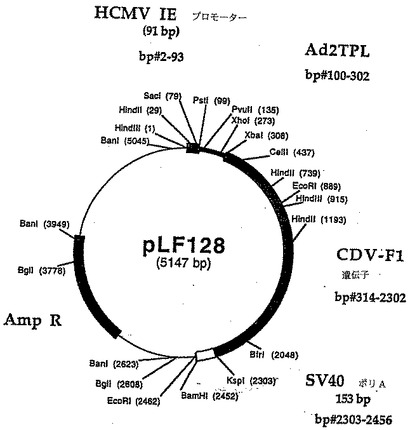
【図49a】

【図49b】
【図49c】

【図49d】

【図50】
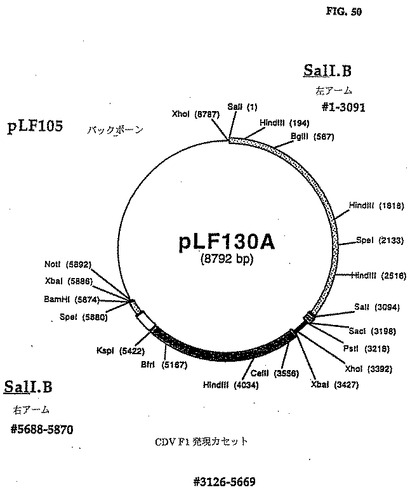
【図1b】
【図1c】
【図2】
【図3a】
【図3b】
【図3c】
【図4】
【図5a】
【図5b】
【図5c】
【図6】
【図7a】
【図7b】
【図7c】
【図8】
【図9a】
【図9b】
【図9c】
【図10】
【図11a】
【図11b】
【図11c】
【図12】
【図13a】
【図13b】
【図13c】
【図14】
【図15a】
【図15b】
【図16】
【図17a】
【図17b】
【図18】
【図19a】
【図19b】
【図20】
【図21a】
【図21b】
【図22】
【図23a】
【図23b】
【図24】
【図25a】
【図25b】
【図25c】
【図25d】
【図26】
【図27a】
【図27b】
【図27c】
【図28】
【図29a】
【図29b】
【図29c】
【図30】
【図31a】
【図31b】
【図31c】
【図32】
【図33a】
【図33b】
【図33c】
【図34】
【図35a】
【図35b】
【図35c】
【図36】
【図37a】
【図37b】
【図37c】
【図38】
【図39a】
【図39b】
【図39c】
【図40】
【図41a】
【図41b】
【図41c】
【図41d】
【図42】
【図43a】
【図43b】
【図43c】
【図44】
【図45a】
【図45b】
【図45c】
【図46】
【図47a】
【図47b】
【図47c】
【図48】
【図49a】
【図49b】
【図49c】
【図49d】
【図50】
【公開番号】特開2008−283969(P2008−283969A)
【公開日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願番号】特願2008−121726(P2008−121726)
【出願日】平成20年5月7日(2008.5.7)
【分割の表示】特願平10−504421の分割
【原出願日】平成9年6月30日(1997.6.30)
【出願人】(399013029)メリアル,インコーポレイテッド (1)
【氏名又は名称原語表記】MERIAL,Inc.
【Fターム(参考)】
【公開日】平成20年11月27日(2008.11.27)
【国際特許分類】
【出願日】平成20年5月7日(2008.5.7)
【分割の表示】特願平10−504421の分割
【原出願日】平成9年6月30日(1997.6.30)
【出願人】(399013029)メリアル,インコーポレイテッド (1)
【氏名又は名称原語表記】MERIAL,Inc.
【Fターム(参考)】
[ Back to top ]