
外来抗原に対する宿主の寛容を壊すためのキメラ抗原
抗原に対して免疫応答を引き出すための組成物および方法が、本明細書において開示される。具体的には、本化合物および方法は、本来は宿主により「自己」抗原として認識される外来性の抗原に対して免疫応答を引き出し、従って、これらの抗原に対する宿主の寛容を破壊する。免疫応答ドメインと、抗体フラグメントを含む標的結合ドメインを含むキメラ抗原を宿主免疫系に提示することは、外来性または寛容化された抗原に対する免疫応答を増強する。抗原提示細胞は、キメラ抗原を取り込み、処理し、提示して、所望の抗原に対して体液性免疫応答と細胞性免疫応答の両方を引き出す。
【発明の詳細な説明】
【技術分野】
【0001】
(導入)
(A.関連出願)
本出願は、米国仮出願第60/493,449号(2003年8月8日出願)の優先権を主張し、米国仮出願第60/493,449号は、本明細書中に参考として援用される。
【0002】
(B.技術分野)
本発明は、免疫応答を引き出すか、または増強させるための方法および組成物、ならびに外来抗原に対して宿主寛容を壊すための方法および組成物に関する。
【背景技術】
【0003】
(C.背景)
健康な宿主(ヒトまたは動物)が、細菌、ウイルスおよび/または寄生生物由来のタンパク質等の外来抗原に出会うと、宿主は通常は免疫応答を開始する。この免疫応答は体液性応答および/または細胞性免疫応答でありうる。体液性応答において、抗体は、B細胞により作られ、抗原刺激に対して血液および/またはリンパに分泌される。次に、抗体は、抗原、例えばウイルスを、その表面上の抗原に特異的に結合して、食作用細胞および/もしくは補体仲介機構による破壊のためにそれをマークすることによって、または結合をブロックするか、もしくは循環からの遊離抗原の排除を増強することによって、中和する。細胞性応答は、抗原を含有する細胞を直接または間接的に除去することのできる特定のヘルパーT細胞および細胞障害性Tリンパ球の選択と増殖により特性付けられる。
【0004】
一部の個体において、免疫系はある種の外来抗原に応答できない。抗原が特定の抗体および/またはキラーT細胞の産生を刺激しない場合、免疫系は生じる病気を防止できない。よって、感染因子、例えば、ウイルスは、慢性感染を確立させることができ、宿主免疫系は感染因子により作られる抗原に対して寛容があるものとなる。
【0005】
感染因子が宿主免疫機構を侵す機構は明確には確立されていないが、宿主免疫系への適当な外来抗原の提示の欠如が慢性的な感染の進展に寄与する因子であろう。抗原提示細胞(APC)は、抗原の局在性に基づきながら出会った抗原を種々に処理する。外因性の抗原はエンドサイトーシスを受け、引き続いて抗原提示細胞のエンドソーム内で処理される。外因性の抗原から生じたペプチドフラグメントは主要組織適合複合体(MHC)クラスIIと複合した細胞表面に提示される。この複合体のCD4+T細胞への提示は、CD4+Tヘルパー細胞を刺激して、外因性の抗原に対して抗体を産生するB細胞を刺激するサイトカインを分泌させる(体液性応答)。一方、細胞内抗原は処理され、抗原提示細胞の表面上のMHCクラスIとの複合体として提示される。CD8+T細胞への抗原提示は、抗原を有する宿主細胞に対する細胞障害性T細胞(CTL)の免疫応答をもたらす。
【0006】
ウイルスまたは寄生生物の慢性的な感染を有する被験体において(この生物は生活環中のある時点で宿主細胞内に住む)、抗原は宿主細胞により作られ、宿主細胞中に発現され、そして分泌された抗原は、循環中に存在する。例として、慢性的なヒトB型肝炎ウイルス(HBV)キャリアの場合、ビリオンおよびHBV表面抗原および(e抗原の形態の)コア抗原の代用物が、血液中で検出され得る。
【0007】
慢性的な感染の効果的な治療は、感染因子に関連した抗原に対する強いCTL応答を必要とする。これは、抗原を宿主細胞内に作ることによるか、または抗原を適切な細胞区分に送達して、細胞応答を引き出すように抗原が処理および提示されることによって達成することができる。抗原を細胞内に運ぶために、いくつかのアプローチが文献化されている。これらのうち、ウイルスベクター(非特許文献1)、cDNAトランスフェクト細胞の使用(非特許文献2)、および注入cDNAベクターによる抗原の発現(非特許文献3);ならびに特許文献1)が文献化されている。さらに、樹状細胞を標的とする抗原を発現させるDNAワクチンが記載されている(非特許文献4)。
【0008】
MHCクラスI経路プロセシングのために、細胞の細胞質ゾル区画まで抗原を運ぶことができる輸送ビヒクルもまた、利用されてきた。Hilgersら(非特許文献5)は、同目標を達成するアジュバントの使用を詳細に説明している。別のアプローチは、オボアルブミンペプチドに対するTh1免疫応答の発生により例示される、抗原の細胞質輸送のための生分解性ミクロスフェアの使用である(非特許文献6;および非特許文献7)。さらに、抗原提示細胞、例えば樹状細胞はPLGAナノスフェアを取り込む(非特許文献8)。
【0009】
抗原を捕捉し、処理し、そして提示し、かつナイーブT細胞を刺激する樹状細胞の能力から、樹状細胞は治療用ワクチンの開発のために非常に重要な手段となっている(非特許文献9)。抗原を樹状細胞の標的とすることは、抗原提示において重大な工程であり、抗体のFc領域に関して樹状細胞上のいくつかのレセプターの存在が、この目的のために利用されている(非特許文献10)。このアプローチのさらなる例として、卵巣癌Mab−B43.13、抗PSA抗体ならびに抗HBV抗体抗原の複合体が挙げられる(非特許文献11)。腫瘍関連抗原を負荷した樹状細胞を用いる癌免疫治療は、腫瘍に特異的な免疫応答と抗腫瘍活性とを作ることが示されている(非特許文献12;および非特許文献13)。腫瘍抗原のパルスを受けた樹状細胞を用いたインビボ臨床試験で有望な結果が得られた(非特許文献14)。これらの研究は、癌抗原に対して免疫応答を作るために樹状細胞を用いる効能を明らかに示している。
【0010】
抗原提示はまた、抗原提示細胞上のFcレセプターの利用の代わり、またはそれに加えてマンノースレセプターによって影響を受けることもできる。CD206としても公知のマクロファージマンノースレセプター(MMR)は、樹状細胞(DC)等の抗原提示細胞上に発現する。この分子は、エンドサイトーシスレセプターのC型レクチンファミリーのメンバーである。マンノシル化抗原は、CD206によって、結合し得、そして内在化され得る。一般的に、外因性の抗原は主にMHCクラスII経路により処理、提示されると考えられる。しかし、CD206を経た標的化の場合、MHCクラスIとクラスIIとの両方の経路が関わっているとの証拠が存在する(非特許文献15;非特許文献16;非特許文献17)。
【特許文献1】米国特許第5,589,466号明細書
【非特許文献1】Lorenzら、Hum.Gen.Ther.10:623−631(1999)
【非特許文献2】Donnellyら、Ann.Rev.Immunol.15:617(1997)
【非特許文献3】Laiら、Crit.Rev.Immunol.18:449−484(1988)
【非特許文献4】Youら、Cancer Res 61:3704−3711(2001)
【非特許文献5】Hilgersら、Vaccine 17:219−228(1999)
【非特許文献6】Newmanら、J.Control Release 54:49−59(1998)
【非特許文献7】Newmanら、J Biomed Mater Res 50:591−597(2000)
【非特許文献8】Newmanら、J.Biomed Mater Res 60:480−486(2002)
【非特許文献9】Laupezeら、Hum Immunol 60:591−597(1999)
【非特許文献10】Regnaultら、J Exp Med 189:371−380(1999)
【非特許文献11】Wenら、Int Rev Immunol 18:251−258(1999)
【非特許文献12】FongおよびEngleman、Ann Rev Immunol 96:1865−1972(2000)
【非特許文献13】Camptonら、J Invest Dermatol 115:57−61(2000)
【非特許文献14】TarteおよびKlein、Leukemia 13:653−663(1999)
【非特許文献15】Apostolopoulosら、Eur.J.Immunol.30:1714(2000)
【非特許文献16】ApostolopoulosおよびMcKenzie、Curr.Mol.Med.1:469(2001)
【非特許文献17】Ramakrishnaら、J.Immunol.172:2845−2852(2004)
【発明の開示】
【発明が解決しようとする課題】
【0011】
感染性の病気および癌は、大きな社会的健康問題である。例えば、世界保健機関の統計は、20億を超える人々がHBVに感染していることを示す。これらのうち、3億7千万人が慢性感染であり、その結果、肝臓の肝硬変および肝臓細胞の癌腫の発症の高い確率を有する。世界中の約1億7千万の人々がHCVの慢性キャリアであり、それに対する効果的な予防ワクチンまたは治療ワクチンは存在しない。世界保健機関は、毎年1千万の人々が癌であると診断されていると報告している。癌は毎年、世界中の死の12%にあたる6百万人の死を引き起こす。よって、感染および癌に対して免疫応答を引き出すための治療的に有効な新しい組成物および方法、ならびにこのような組成物を生成するための方法に対する必要性が存在する。
【課題を解決するための手段】
【0012】
(発明の要旨)
本発明は、免疫応答を引き出すためのキメラ抗原を提供し、このキメラ抗原は、免疫応答ドメインおよび標的結合ドメインを含み、上記標的結合ドメインは、抗体フラグメントを含む。
【0013】
本発明のもう一つの特徴は、抗原提示細胞において抗原提示を増強する方法であって、上記抗原提示細胞を本発明のキメラ抗原を含む組成物と接触させる工程を包含する方法を提供する。
【0014】
本発明のさらにもう一つの特徴は、抗原提示細胞を活性化する方法であって、上記抗原提示細胞を本発明のキメラ抗原に接触させる工程を包含する方法を提供する。
【0015】
本発明の一つの特徴は、免疫応答を引き出す方法であって、本発明のキメラ抗原を含有する組成物を被験体に投与する工程を包含する方法を提供する。
【0016】
本発明のもう一つの特徴は、寛容を壊す方法であって、本発明のキメラ抗原を被験体に投与する工程を包含する方法を提供する。好ましい実施態様において、上記被験体は、ウイルスまたは偏性細胞内寄生生物に慢性感染している。
【0017】
本発明の一つの特徴は、免疫治療が可能な症状を治療する方法であって、治療的有効量の本発明のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する方法を提供する。好ましい実施態様において、免疫治療が可能な症状は感染、特に慢性感染または癌である。
【0018】
本発明のさらにもう一つの特徴は、感染に対して被験体をワクチン接種する方法であって、本発明のキメラ抗原を被験体に投与する工程を包含する方法を提供する。上記被験体は、予防的または治療的にワクチン接種され得る。好ましい実施態様において、上記被験体は、キメラ抗原の二つ以上のエピトープ、そしてより好ましくは免疫応答ドメインの二つ以上のエピトープに対する免疫応答を発達させる。好ましくは、上記感染は、ウイルス感染または偏性細胞内寄生生物の感染である。
【0019】
本発明のもう一つの特徴は、本発明のキメラ抗原および薬学的に受容可能な賦形剤を含む薬学的組成物を提供する。
【0020】
本発明の一つの特徴は、本発明のキメラ抗原と、治療が必要な被験体に対するキメラ抗原を投与するための説明書とを含む製品を提供する。
【0021】
本発明のもう一つの特徴は、キメラ抗原をコードするポリヌクレオチドを提供し、このポリヌクレオチドは免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二ポリヌクレオチド部分とを含み、この標的結合ドメインが抗体フラグメントを含む。また、本発明は、このようなポリヌクレオチドを含む微生物および細胞株を提供する。
【0022】
本発明のさらにもう一つの特徴は、本発明のキメラ抗原を作製する方法を提供し、この方法は、本発明のキメラ抗原をコードするポリヌクレオチドを含む微生物または細胞株を提供する工程、および上記キメラ抗原が発現される条件下で上記微生物または細胞株を培養する工程を包含する。
【0023】
本発明の一つ以上の実施態様の詳細を、添付の図面および下記の説明に示す。本発明の他の特徴、目的および利点は、下記の説明および図面から、および請求の範囲から明らかであろう。
【発明を実施するための最良の形態】
【0024】
(IV.詳細な説明)
(A.概説)
抗原に対する免疫応答を引き出すための組成物および方法を、本明細書において開示する。具体的には、この化合物および方法は、本来は宿主により「自己」抗原として認識される外来性の抗原に対して免疫応答を引き出すので、これらの抗原に対する宿主の寛容を壊す。免疫応答ドメインおよび標的結合ドメイン(この標的結合ドメインは、抗体フラグメントを含む)を含むキメラ抗原を、宿主の免疫系に提示することは、外来性または寛容化された抗原に対する免疫応答を増強する。抗原提示細胞は、上記キメラ抗原を取り込み、処理し、そして提示して、所望の抗原に対して体液性免疫応答と細胞性免疫応答の両方を引き出す。
【0025】
(B.定義)
本発明をさらに詳しく説明する前に、本出願で用いられる用語は、特に示さないかぎり下記のように定義される。
【0026】
「抗体」とは、抗原(免疫原)によりヒトまたは他の動物に引き起こされた特定のアミノ酸配列を有する、Bリンパ系細胞によって産生された免疫グロブリン分子をいう。これらの分子は他の用語で各々定義される抗原と特異的に反応することにより特徴付けられる。
【0027】
「抗体応答」または「体液性応答」とは、抗体がBリンパ系細胞により作られ、抗原の刺激により血液および/またはリンパに分泌される、ある型の免疫応答をいう。適切に機能する免疫応答において、上記抗体は、細胞(例えば、病原体)表面上の抗原に特異的に結合し、食細胞および/または補体仲介機構による破壊のために上記細胞にマークする。また、抗体は全身を循環し、遊離ビリオンに結合することができる。この抗体結合は、ビリオンを中和してビリオンが細胞に感染することを妨げ得、そして食作用や腎臓でのろ過によって循環から除去するために、ビリオンをマークする。
【0028】
「抗原」とは、適当な細胞との接触の結果、感受性および/または免疫応答性の状態を誘導し、インビボまたはエキソビボで感作被験体の抗体および/または免疫細胞に明白な方法で反応する任意の物質をいう。
【0029】
「抗原提示細胞」とは、主に抗原を扱って、これをリンパ球に提示することにより機能する抗原誘導事象の補助細胞をいう。抗原提示細胞の抗原との相互作用は、リンパ球を抗原分子に会わせて認識させ、リンパ球を活性化させることができるので、免疫誘導において必須の工程である。例示的な抗原提示細胞としては、マクロファージ、ランゲルハンス樹状細胞、濾胞樹状細胞およびB細胞が挙げられる。
【0030】
「B細胞」とは、抗原と相互作用する免疫グロブリンまたは抗体を産生する、ある型のリンパ球をいう。
【0031】
「CH1領域」とは、抗体の抗原結合フラグメント上の重鎖定常ドメインの領域をいう。
【0032】
「細胞性応答」とは、ウイルス感染細胞または癌細胞を直接に除去することができる特定のヘルパーT細胞およびキラーT細胞により仲介される、ある型の免疫応答をいう。
【0033】
本明細書中で使用される場合、「キメラ抗原」との用語とは、免疫応答ドメインおよび標的結合ドメインを含むポリペプチドをいう。免疫応答ドメインおよび標的結合ドメインは、共有的手段または非共有的手段により直接的に結合してもよいし、間接的に結合してもよい。
【0034】
「細胞障害性Tリンパ球」とは、外来細胞、およびウイルス性抗原を作る感染因子に感染した宿主細胞を破壊することのできる、特化された型のリンパ球である。
【0035】
「エピトープ」とは、複合抗原分子上の最も単純な形の抗原決定基をいう。これは免疫グロブリンまたはT細胞レセプターにより認識される抗原の特定部分である。
【0036】
「融合タンパク質」とは、二つ以上の遺伝子配列を組み合わせることにより作られたハイブリッド遺伝子の発現により形成されるタンパク質をいう。
【0037】
「ヒンジ領域」とは、FabフラグメントをFcフラグメントに結合させる抗体部分をいう。このヒンジ領域は、二つの重鎖を共有結合させるジスルヒド結合を含有する。
【0038】
「相同」との用語は、例えば、対応する位置で同じまたは類似である化学残基の配列を有することによって、別の分子と相同性を示す分子をいう。「%相同の」または「%相同性」の語句は、同一または類似である相同性ポリヌクレオチドまたはポリペプチドの同一位置におけるヌクレオチドまたはアミノ酸のパーセントをいう。例えば、二つのタンパク質中で80残基のうち75残基が同一である場合、それら二つのタンパク質は93.75%相同性である。%相同性は当業者に公知の多様なソフトウエアプログラムを用いて決めることができる。
【0039】
「宿主」とは、感染または癌等の免疫治療が可能な症状に苦しむ温血動物(ヒトを含む)をいう。ここで用いる「宿主」はまた、キメラ抗原が投与される温血動物(ヒトを含む)も言及する。
【0040】
本発明の文脈において、「ハイブリダイゼーション」とは、オリゴマー化合物の相補鎖が対となることを意味する。本発明において、対となる好ましい機構は、水素結合を伴うもので、これは、オリゴマー化合物の鎖の相補的なヌクレオシド塩基またはヌクレオチド塩基(ヌクレオ塩基)間でのWatson−Crick、Hoogsteenまたは逆向きHoogsteen水素結合であってよい。例えば、アデニンとチミンとは、水素結合の形成により対となる相補的なヌクレオ塩基である。ハイブリダイゼーションは、多様な状況下で起こりうる。ポリヌクレオチドの文脈において用いられる場合、「ハイブリダイズする」、「ハイブリダイズしている」等の用語は、慣用的なハイブリダイゼーション条件、好ましくは、ハイブリダイゼーション温度が37℃超で、0.1×SSC/0.1%SDSでの洗浄温度が55℃超である、50%ホルムアルデヒド/6×SSC/0.1%SDS/100μg/ml mDNAでのハイブリダイゼーション等の条件をいう。
【0041】
「免疫」または「免疫応答」は、抗原に対する体の応答をいう。特定の実施態様において、体が感染疾患に対してそれ自体を抵抗するか、または保護する能力をいう。
【0042】
「免疫応答ドメイン(IRD)」とは、二官能性キメラ抗原分子の多様な構造の抗原性部分をいう。この免疫応答ドメインは、一つ以上の抗原および/または一つ以上の組換え抗原を含む。
【0043】
本明細書において使用される場合、「免疫治療が可能な症状」との語句は、被験体の免疫応答を引き出すか、または調節することにより防止、抑制あるいは緩和することのできる症状または病気をいう。
【0044】
「リンパ球」とは、特定の免疫応答を仲介する血液中に見られる有核細胞の部分集合をいう。
【0045】
「モノクローナル抗体」または「mAb」とは、融合ハイブリッド細胞、すなわちハイブリドーマ細胞のクローンまたは遺伝的に均一な集団から作られた抗体をいう。ハイブッド細胞は、クローニングされて、化学的および免疫学的に均一な(すなわち一つの型のみの抗原を認識する)特定のモノクローナル抗体を産生する細胞株を確立する。
【0046】
「ペプチド結合」は、1つのアミノ酸のαアミノ基と別のアミノ基のαカルボキシル基との間の置換アミド結合により共有結合した二つ以上のアミノ基をいう。
【0047】
「薬学的に受容可能な」とは、ヒトまたは他の動物に生理学的に適合する毒性のない組成物をいう。
【0048】
「薬学的に受容可能な賦形剤」は、毒性がなく、ヒトまたは他の動物に生理学的に適合性のあるアジュバント、キャリア、pH調節剤および緩衝剤、浸透圧調節剤、湿潤剤、保存剤等の物質を含む。
【0049】
本明細書において使用される場合、「ポリヌクレオチド」との用語は、リボヌクレオチドかデオキシリボヌクレオチドのいずれかの長さのヌクレオチドの多量形をいう。この用語は、分子の一次構造のみをいう。よって、この用語は、二本鎖または一本鎖のDNAまたはRNAを含む。また、公知の型の改変、例えば、当該分野で公知の標識、メチル化、「キャップ」、一つ以上の天然に存在するヌクレオチドの類似体による置換、ヌクレオチド間の改変(例えば、未荷電結合(例えば、メチルホスホネート、ホスホトリエステル、ホスホアミデート、カルバメート等)および荷電結合(例えば、ホスホロチオエート、ホスホロジチオエート等)による修飾、ペンダント部分(例えばタンパク質(例えば、ヌクレアーゼ、トキシン、抗体、シグナルペプチド、ポリ−L−リジン等)を含む)を含む改変、インターカレーター(例えば、アクリジン、ソラレン等)による改変)、キレーター(例えば、金属、放射性金属、ホウ素、酸化金属等)を含む改変、アルキル化剤を含む改変、改変結合(例えば、αアノマー核酸等)による改変、ならびにポリヌクレオチドの非改変形態など、公知の種類の改変も挙げられる。
【0050】
「プロテアーゼ切断部位」とは、タンパク質分解酵素がポリペプチド鎖を加水分解する(壊す)部位をいう。
【0051】
本発明において、「ストリンジェントなハイブリダイゼーション条件」または「ストリンジェント条件」との語句は、本発明の化合物がその標的配列とハイブリダイズするが、他の配列とのハイブリダイズは最小限とする条件をいう。
【0052】
「被験体」との用語は、温血動物、好ましくはヒトをいう。
【0053】
「タグ」とは、タグを含む分子を単離または精製するために用いられる、マーカーまたはマーカー配列をいう。例示的なタグとしては6×Hisタグが挙げられる。
【0054】
「T細胞」とは、抗原特異的な細胞相互作用に関与し、体液性免疫応答および細胞性免疫応答を仲介する型のリンパ球をいう。
【0055】
「標的結合ドメイン(TBD)」とは、抗原提示細胞上、特に樹状細胞上のレセプターに結合することができ、引き続いてレセプター仲介取り込みにより抗原提示細胞内に輸送される、抗体フラグメントを含むタンパク質をいう。
【0056】
「治療的有効量」とは、抗原に対して効果的なB細胞、細胞障害性Tリンパ球(CTL)および/またはヘルパーTリンパ球(Th)の応答を引き出して、病気または疾患の症状および/または合併症を阻止するか、または治療するか、もしくは少なくとも部分的に抑制するか、もしくは遅らせるのに十分なキメラ抗原の量またはキメラ抗原をコードするポリヌクレオチドの量をいう。
【0057】
本明細書中で使用される場合、「治療する」および「治療」との用語は、動物(特に、ヒト)においてキメラ抗原により治療可能な症状の任意の治療を包含し、(i)症状が起こりやすいが、その症状があるとはまだ診断されていない被験体にこの症状が起こることを妨ぐ、(ii)症状の抑制(例えば、その進展を停止または遅らせる);または(iii)症状の緩和(例えば、症状またはその徴候の低減を引き起こすこと)を含む。
【0058】
本明細書において使用される場合、「異種型」とは、宿主以外の異なる種に由来することをいう。例えば、マウスのゲノムからクローニングされた組換え発現の抗体は、組換え発現の抗体が細菌、昆虫またはマウスの細胞のいずれで作られるかにかかわらず、ヒトに対して異種型であるが、マウスに対しては異種型ではないということである。
【0059】
(C.新規なキメラ抗原)
本発明は、免疫応答ドメインおよび標的結合ドメインを含む免疫応答を引き出すためのキメラ抗原において、この標的結合ドメインが抗体フラグメントを含むキメラ抗原を提供する。本発明に従って、キメラ抗原は好ましくはFcレセプターおよび/またはマクロファージマンノースレセプターに結合できる。この抗体フラグメントは宿主に対して異種型であっても、宿主に対して異種型でなくてもよい。
【0060】
本発明の好ましい実施態様において、キメラ抗原は体液性免疫応答および/または細胞性免疫応答を誘導することができる。細胞性免疫応答として、Th1応答、Th2応答および/または細胞障害性Tリンパ球(CTL)応答を挙げることができる。さらに別の好ましい実施態様において、キメラ抗原は多エピトープ性免疫応答を引き出す。多エピトープ性免疫応答としては、免疫応答ドメインの少なくとも一つのエピトープに対する応答および/または標的結合ドメインの少なくとも一つのエピトープに対する応答を挙げることができる。あるいは、多エピトープ性応答は免疫応答ドメインの二つ以上のエピトープに対する応答に限定してもよい。
【0061】
本発明のキメラ抗原は、二つの部分、すなわち、抗原性配列(ウイルス抗原等)を含む免疫応答ドメインと、抗体フラグメントを含む標的結合ドメインとを含む(図1)。好ましい実施態様において、上記免疫応答ドメインは、当業者に公知の任意の方法によって標的結合ドメインに結合させることができる。免疫応答ドメインを標的結合ドメインに結合させるためのリンカーとしては、共有ペプチド結合、化学接合、ロイシンジッパーおよびビオチン/アビジンが挙げられるが、これらに限定されない。好ましい実施態様において、上記免疫応答ドメインおよび標的結合ドメインは、単一の融合タンパク質としてクローニングすることができる。融合タンパク質の共有ペプチド結合は、付加的なペプチド配列、例えばSRPQGGGSまたはVRPQGGGS(配列番号1)を含んでよい。さらに別の好ましい実施態様において、多様な免疫応答ドメインがビオチン化され、そして標的結合ドメインは、ストレプトアビジンを有する融合タンパク質として作られて、キメラ抗原の広い類別の産生に役立つ。あるいは、免疫応答ドメインと標的結合応答領域はそれぞれ、ロイシンジッパー部分との融合物として発現させることができ、キメラ抗原の二つの部分は混合の際に会合させられる。最後に、免疫応答ドメインと標的結合ドメインを別個に発現させて、次に当業者に公知の方法を用いて化学的に接合させることができる。例示的な方法は、それら2つのドメインを共有結合させるスベルイミノ酸ジメチル等のタンパク質架橋剤の使用を含む。
【0062】
免疫応答ドメインは主にキメラ抗原の抗原性の部分を提供する。この免疫応答ドメインは、免疫応答が望まれるものの全体の少なくとも一つの抗原性部分を含有する。このキメラ抗原は、必要に応じて二つ以上の免疫応答ドメインを含有してもよい。好ましい実施態様において、上記免疫応答ドメインは、ウイルスまたは偏性細胞内寄生生物等の感染因子、あるいは癌抗原の少なくとも一つの抗原性部分を含有する。より好ましくは、上記免疫応答ドメインは、感染性ウイルスの少なくとも一つの抗原性部分を含む。
【0063】
好ましい感染性ウイルスの例としては、レトロウイルス科(例えば、ヒト免疫不全ウイルス、例えばHTLV−III、LAVまたはHTLV−III/LAVまたはHIV−IIIとも称されるヒト免疫不全ウイルス1(HIV−1);および他の単離物、例えばHIV−LP;ピコルナウイルス科(例えば、ポリオウイルス、A型肝炎ウイルス;エンテロウイルス、ヒトコクサッキーウイルス、ライノウイルス、エコーウイルス);カルシウイルス科(Calciviridae)(例えば、胃腸炎を引き起こす株);トガウイルス科(例えば、ウマ脳炎ウイルス、風疹ウイルス);フラビウイルス科(例えば、C型肝炎ウイルス、デング熱ウイルス、脳炎ウイルス、黄熱病ウイルス);コロナウイルス科(例えば、コロナウイルス);ラブドウイルス科(例えば、水ほう性口内炎ウイルス、狂犬病ウイルス);フィロウイルス科(例えば、エボラウイルス);パラミクソウイルス科(例えば、パラインフルエンザウイルス、流行性耳下腺炎ウイルス、麻疹ウイルス、RSウイルス);オルトミクソウイルス科(例えば、インフルエンザウイルス);ブンガウイルス科(Bungaviridae)(例えば、ハンターンウイルス、ブンガウイルス、フレボウイルスおよびナイロウイルス);アレナウイルス科(出血熱ウイルス);レオウイルス科(例えば、レオウイルス、オルビウイルスおよびロタウイルス);ビルナウイルス科;ヘパドナウイルス科(ヒトB型肝炎ウイルス(HBV)、アヒルB型肝炎ウイルス(DHBV));パルボウイルス科(パルボウイルス);パポバウイルス科(パピローマウイルス、ポリオーマウイルス);アデノウイルス科(ほとんどのアデノウイルス);ヘルペルウイルス科(単純ヘルペスウイルス(HSV)1および2、水痘帯状疱疹ウイルス、サイトメガロウイルス(CMV)、エプスタイン−バーウイルス、ヘルペスウイルス);ポックスウイルス科(痘そうウイルス、ワクシニアウイルス、ポックスウイルス)およびイリドウイルス科(例えば、アフリカブタ熱ウイルス(African swine fever virus));および分類されていないウイルス(例えば、δ肝炎(hepatitides)因子、非A型、非B型肝炎因子(クラス1、内部伝染);クラス2、非経口伝染;ノルウォークウイルスと関連ウイルス、ならびにアストロウイルス)が挙げられる。本発明の一部実施態様において、キメラ抗原の免疫応答ドメインは、HBVタンパク質、DHBVタンパク質およびHCVタンパク質からなる群から選択される一つ以上のタンパク質の少なくとも一つの抗原性部分を含む。本発明に使用される特に好ましいHBVタンパク質として、HBV S1/S2、HBV S1/S2/S、HBV Core、HBV Core ctm(C末端改変)、HBV e抗原およびHBVポリメラーゼが挙げられるが、これらに限定されない。本発明に使用される特に好ましいDHBVタンパク質としては、DHBV PreS/S、DHBV PreS、DHBV CoreおよびDHBVポリメラーゼが挙げられるが、これらに限定されない。本発明に使用される特に好ましいHCVタンパク質としては、HCV Core(1−191)、HCV Core(1−177)、HCV E1−E2、HCV E1、HCV E2、HCV NS3、HCV NS5AおよびHCV NS4Aが挙げられるが、これらに限定されない。本発明に用いられる他の好ましいウイルス抗原としては、HIV gp120、HSVアルカリヌクレアーゼおよびヒトパピローマウイルス(HPV)キャプシドタンパク質L1およびL2、および初期領域タンパク質HPV E1、HPV E2、HPV E4、HPV E5、HPV E6およびHPV E7が挙げられる。
【0064】
好ましい偏性細胞内寄生生物の例として、Tetrahymena sp.(例えば、T.pyriformis)、Plasmodium sp.(例えば、P.falciparum)、Cryptospiridium sp.、Spraguea sp.(例えば、S.lophii)、Giardia sp.,Toxoplasma sp.(例えば、T.gondii、T.cruzi)、Leishmania sp.、Rickettsia sp.(例えば、R.prowazekii)、Chlamydia sp.、Mycobacterium sp.(例えば、M.tuberculosis)、Legionella sp.、Listeria sp.(例えば、L.monocytogenes)、Coxiella sp.(例えば、C.brunette)、Shigella sp.、Erlichia sp.およびBartonelia sp.が挙げられる。好ましい癌抗原として、前立腺特異的抗原(PSA)、前立腺特異的膜抗原(PSMA)、MUC1、CA 125、WT1、Her−2/neu、癌胎児性抗原(CEA)、MAGE−3、MART−1、gp100、NY−ESSO−1、CA19.9、TAG72、CA 15.3、CA 27.9、gp120、前立腺酸性ホスファターゼ(PAP)、ヒートショックタンパク質、αフェトタンパク質(AFP)、テロメラーゼおよびrasが挙げられる。
【0065】
本発明のもう一つの実施態様において、キメラ抗原の免疫応答ドメインは、一つ以上の抗原性部分に融合させた6×Hisタグを包含する。
【0066】
本発明にしたがえば、上記キメラ抗原は、抗原提示細胞上、特に樹状細胞上のFcレセプターおよび/またはCD206に結合することのできるタンパク質であり、このキメラ抗原は、次いでレセプター仲介取り込みにより抗原提示細胞中に輸送される。本発明にしたがえば、抗体フラグメントの存在は、抗原提示細胞上、具体的には樹状細胞上のFcレセプターによるキメラ抗原の取り込みを増強する。ウイルスのこの特異的な結合と細胞内への取り込みによって、ウイルス抗原が処理されて、外来物として提示される。よって、免疫応答は、前もって寛容化された抗原に対して効果的に引き出され得る。標的結合ドメインは宿主に対して異種型であっても、異種型でなくてもよい抗体フラグメントを含んでいる。本発明の好ましい実施態様において、上記抗体フラグメントはマウスのFcフラグメントを含む。本発明のさらに好ましい実施態様において、標的結合ドメインは、Fcフラグメント、ヒンジ領域、およびCH1領域の一部を含み、上記キメラ抗原は、標的結合ドメインを免疫応答ドメインに結合させるのに適当なペプチドリンカーを含む。もう一つの好ましい実施態様において、標的結合ドメインは免疫グロブリン重鎖フラグメントを含み、必要に応じてさらにヒンジ領域を含む。特に好ましい実施態様において、重鎖フラグメントは、CH1ドメインのアミノ酸VDKKI(配列番号2)および/またはCH2ドメインおよびCH3ドメインの一部またはすべてを含む。
【0067】
上記のように、CD206により結合され、細胞内に取り込まれた抗原は、MHCクラスIとクラスIIの両方によって提示されて、細胞性と体液性の両方の免疫応答を引き出すことができる。したがって、好ましい実施態様では、キメラ抗原がグリコシル化される。免疫応答ドメインおよび/または標的結合ドメインは、グリコシル化することができる。特に好ましい実施態様において、キメラ抗原は高マンノースグリコシル化(high mannose glycosylation)または低マンノースグリコシル化(pauci mannose glycosylation)のいずれかによりマンノースグリコシル化されている(Jarvis, Virology 310:1−7(2003))。
【0068】
(D.キメラ抗原を利用する新規な方法)
本発明は、免疫応答を引き出す方法を包含し、この方法は、本発明のキメラ抗原を含有する組成物を、被験体に投与する工程を包含する。
【0069】
抗原の十分な提示を提供するために、本発明者らは、免疫応答ドメインと標的結合ドメインとを含む新規のキメラ抗原を開発している。この標的結合ドメインは、抗体フラグメントを含む。発明の特別な理論に限定されるわけではないが、この分子は、その抗体フラグメントのおかげで、抗原提示細胞上の特定のレセプターに結合し、ウイルス抗原が主要組織適合複合体(MHC)クラスIおよびクラスIIと複合体化するように処理、提示される。MHCクラスIによるそのような処理と抗原提示は、細胞障害性Tリンパ球による増強応答を引き出して、結果として免疫応答ドメインの抗原に会合する任意の感染因子の除去をもたらす。さらに、MHCクラスII分子による抗原提示は、感染細胞および/または循環から抗原のクリアランスをも助ける体液性応答を引き出す。
【0070】
また、本発明は、寛容を壊す方法を包含し、この方法は、本発明のキメラ抗原を被験体に投与する工程を包含する。キメラ抗原を用いることによって細胞性免疫応答および/または体液性免疫応答を引き出すための抗原の提示において、慢性的な感染の間に「自己」として扱われた抗原は、「外来性」として認識される。したがって、宿主の免疫系はCTL応答を開始させて感染細胞を除去する。同時に、キメラ抗原に対する応答において誘発された抗体は、感染因子に結合し、感染因子を循環から除去するか、または宿主細胞に対する感染因子の結合を阻止する。したがって、本来は宿主免疫系により寛容化される慢性的な感染を有する被験体において、広い免疫応答を誘導しうるキメラ抗原を作るように、本発明は設計される。好ましい実施態様において、上記キメラ抗原は、ウイルスまたは寄生生物等の感染因子により慢性的に感染しているか、または癌を有する被験体において、抗原に対する寛容を破壊する。さらに好ましくは、感染因子はその生活環中のある時点で宿主細胞に内在する。
【0071】
好ましい実施態様において、宿主免疫系により認識されないか、寛容化される前選択された抗原の免疫原性は、抗原フラグメントの存在に起因して、ならびに真核細胞(例えば昆虫細胞)発現系に導入されたグリコシル化の存在により増強される。そのようなキメラ抗原は、抗体成分およびグリコシル化の存在に起因して、樹状細胞、マクロファージ、B細胞および顆粒球等の多様な免疫細胞型に存在する特定のレセプターに結合する。
【0072】
本発明のさらに別の特徴は、抗原提示細胞を活性化する方法を提供し、この方法は、上記抗原提示細胞を本発明のキメラ抗原に接触させる工程を包含する。また、本発明は、抗原提示細胞において抗原提示を増強する方法を提供し、この方法は、本発明のキメラ抗原を含有する組成物に抗原提示細胞を接触させる工程を包含する。上記キメラ抗原は抗原提示細胞、好ましくは樹状細胞にインビボまたはエキソビボで接触させることができる。好ましい実施態様において、キメラ抗原を抗原提示細胞に接触させることは、抗原提示細胞を活性化し、二つ以上のエピトープの抗原提示を促進する。この多エピトープ性応答としては、免疫応答ドメインの一つ以上のエピトープの提示および/または標的結合ドメインの一つ以上のエピトープの提示を挙げることができる。
【0073】
また、本発明は、免疫治療が可能な症状を治療する方法を提供し、この方法は、治療的有効量の本発明のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する。好ましい実施態様において、免疫治療が可能な症状は感染または癌である。上記感染は、ウイルス感染、寄生生物感染または細菌感染であってよい。好ましくは、この感染は、感染因子が宿主細胞内に見られる段階を有する。さらに好ましくは、免疫治療が可能な症状は慢性のウイルス感染である。最も好ましくは、免疫治療が可能な症状は慢性B型肝炎ウイルス(HBV)感染または慢性C型肝炎ウイルス(HCV)感染である。HBVの治療のために、上記免疫応答ドメインは、好ましくは、HBV Coreタンパク質、HBV Sタンパク質、HBV S1タンパク質、HBV S2タンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む。HCVの治療のために、上記免疫応答ドメインは好ましくはHCV Core(1−191)タンパク質、HCV Core(1−177)タンパク質、HCV E1タンパク質、HCV E2タンパク質、HCV E1−E2タンパク質、HCV NS3Aタンパク質、HCV NS5Aタンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む。
【0074】
好ましい実施態様において、キメラ抗原の投与は免疫応答ドメインのみの投与よりもさらに高い免疫応答を引き出す。免疫応答の大きさは、例えば、(i)被験体に存在する抗原特異的な抗体の量により、(ii)キメラ抗原または免疫応答ドメインのみを負荷した抗原提示細胞に対して曝されることに応答してT細胞により分泌されたインターフェロンγの量により、または(iii)キメラ抗原または免疫応答ドメインのみを負荷した抗原提示細胞に対して曝されることに応答してもたらされた抗原特異的なCD8+T細胞の量により、測定され得る。
【0075】
キメラ抗原は、免疫応答を生じるその効能について、該キメラ抗原を樹状細胞にエキソビボまたはインビボで提示することによって評価することができる。上記樹状細胞は、T細胞応答のマーカーとしてT細胞の増殖およびインターフェロンγの産生に関して評価されるTリンパ球に対して、キメラ抗原を処理し、提示する。具体的に、エキソビボ状況で、ナイーブ樹状細胞は、末梢血液から単離される。上記樹状細胞によるT細胞の活性化は、マーカー、例えばインターフェロンγレベルを公知の操作で測定することによって評価される。例えば、Berlynら、Clin.Immunol 101(3):276−283(2001)を参照のこと。インターフェロンγを分泌するT細胞の比率の少なくとも50%までの増加は、インビボでの効能を予想させる。インビボ状況の場合、上記キメラ抗原は、利用可能な樹状細胞および他の抗原処理細胞が抗原と相互作用でき、よってそれらを処理する能力を有する宿主に直接に非経口で導入される。
【0076】
さらに、本発明は、感染に対して被験体をワクチン接種する方法を包含し、本方法は、本発明のキメラ抗原を被験体に投与する工程を包含する。上記被験体は、予防的または治療的にワクチン接種され得る。好ましくは、上記感染はウイルス感染である。分子の二官能性の性質は、抗原提示細胞、例えば樹状細胞を抗原の標的とするのに役立ち、抗体に対して抗原の最も効果的な化学量にて特異的に抗原提示細胞を標的とすることによって、慢性的な感染疾病の治療において独自のアプローチとなる。これは、B型肝炎、C型肝炎、ヒト免疫不全ウイルス、ヒトパピローマウイルスおよび単純ヘルペスウイルス等の慢性ウイルス感染、偏性細胞内寄生生物感染を治療する治療ワクチンの開発に有用であり、癌および自己免疫疾患等の病気におけるすべての自己抗原に適用可能でもあり得る。これらの融合タンパク質の投与は、細胞性および体液性の両方の応答を含め、宿主から広い免疫応答を引き出すことができる。よって、これら融合タンパク質は、特別な感染を発症させる危険のある被験体を免疫するための予防ワクチンとして有用であることに加えて、既存の感染に対して免疫寛容性である被験体を治療する治療ワクチンとして使用することができる。
【0077】
(E.キメラ抗原の製造方法)
本発明の一つの特徴は、キメラ抗原を作製するための方法を提供し、この方法は、以下、(a)キメラ抗原をコードするポリヌクレオチドを含む微生物または細胞株を提供する工程であって、この微生物または細胞株は、好ましくは真核細胞であって、より好ましくは非哺乳動物の微生物または細胞株である、工程;および(b)上記微生物または細胞株を、キメラ抗原が発現される条件下で培養する工程、を包含する方法である。好ましくは、上記微生物または細胞株は酵母、植物細胞株または昆虫細胞株である。さらに好ましくは、上記細胞株は、Sf9、Sf21、Drosophila S2およびHigh Five(登録商標)からなる群から選択される昆虫細胞株である。
【0078】
本発明の一実施態様は、本発明を実施するために必要な選択された抗原と標的結合ドメインとの融合タンパク質を作るために、確立された組換えDNA技術を使用する。融合タンパク質構築物は、所望のDNAフラグメントを発現ベクターに導入するために利用される特定の制限酵素部位を導入して、DNAレベルで作られ、これを用いて異種発現系において所望の融合タンパク質を発現させる。ここで用いる「ベクター」との用語は、所望のタンパク質をコードするDNAを運ぶことのできるプラスミドをいう。本発明に使用される好ましいプラスミドベクターとしては、pFastBac HTaおよびDH10Bac(登録商標)E.coli(Invitrogen)で作られたその対応する組換え「バクミド(bacmid)」が挙げられるが、これらに限定されない。
【0079】
標的結合ドメインをコードする遺伝子は、抗体産生細胞、例えばモノクローナル抗体を産生するハイブリドーマから、ポリメラーゼ連鎖反応(PCR)により得ることができる。後者のクローニング工程を容易にするために、独自の制限酵素認識部位を加えるためのオリゴヌクレオチドプライマーを設計することが望ましい。同様に、免疫応答ドメインの抗原性部分は、所望の標的の抗原性部分をコードする遺伝子を含む任意の細胞またはウイルスRNAもしくはウイルスDNAから得ることができる。好ましくは、PCRを用いて、免疫応答ドメインの抗原性部分をコードするDNAを得て、PCRプライマーを設計して、クローニングを容易にする独自の制限酵素認識部位を付加する。しかし、任意の組換えDNA法を用いて、免疫応答ドメインの抗原性部分をコードするDNAが得られ得る。次に、標準的なクローニング技術を用いて、標的結合ドメインをコードするポリヌクレオチドと免疫応答ドメインをコードするポリヌクレオチドとを単一の構築物に組み合わせることができる。もしくは、それら別々のドメインは、ロイシンジッパー、ストレプトアビジンまたはビオチニル化シグナル等のDNAコードリンカーとともにクローニングされ得る。
【0080】
大量の異種タンパク質が作られるという理由のみならず、真核生物のタンパク質のリン酸化やグリコシル化等の翻訳後改変が感染昆虫細胞内で起こるという理由から、バキュロウイルス系を用いて本発明のキメラ抗原を発現させるのが好ましい。昆虫細胞への直接のクローニングは困難でありうるために、キメラ抗原をコードするポリヌクレオチドを細菌系において作成し、その最終構築物をバキュロウイルス/昆虫細胞発現系に転移させることが好ましい。転移システム、例えば、Bac−To−Bac(登録商標)系(Invitrogen)は当業者に公知である。このBac−to−Bac(登録商標)系は、細菌トランスポゾンTn7による部位特異的転位を利用して、所定の遺伝子をE.coli−昆虫細胞シャトルベクター(バクミド)に転移させる。得られる組換えバクミドは、昆虫細胞にトランスフェクションされ、組換えタンパク質を発現するバキュロウイルスを作る。
【0081】
バキュロウイルスを作るために、上記バクミドを、Sf9細胞等の昆虫細胞にトランスフェクションする。トランスフェクション後、この細胞を、バキュロウイルス集団を増殖させるのに十分な時間インキュベートする。バキュロウイルスを含む培地を集めて、4℃、暗所で保存する。このトランスフェクションは、所望のDNA挿入物に対して特異的なプライマーを利用するPCRにウイルス培養物を供して、バキュロウイルスDNAの生産を調べることにより実証され得る。上記細胞中の異種タンパク質の発現は、任意の公知の方法、例えば、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)またはウエスタンブロットにより実証され得る。
【0082】
標準的な感染効率(MOI)の組換えバクミドを用いて、昆虫細胞を感染させる。細胞を約3×105細胞/mLの密度で撒き、細胞密度が約2〜3×106細胞/mLに達するまで懸濁培養(27.5℃、攪拌下)にてインキュベートする。次に、標準化量の各組換えバキュロウイルスを細胞に加える。インキュベーション温度は27.5℃で、適切な感染時間は個々のタンパク質発現のについて標準化される。細胞を遠心分離により集め、組換えタンパク質の精製に用いる。細胞の使用しない部分は、液体窒素中で瞬時に凍らせて、−70℃で保存され得る。
【0083】
キメラ抗原は、好ましくは変性条件下で精製される。キメラ抗原を発現する細胞は変性緩衝液、例えば6Mグアニジン−HClを含有する緩衝液に可溶化される。可溶化は超音波処理等の機械的手段により高めることができる。溶解物を遠心して、未破壊細胞と細胞破片を除去する。次に、上清を、前もって可溶化緩衝液で平衡化したNi−NTA Super Flow(Qiagen)ビーズカラムにロードする。ロードした後に、カラムを緩衝性変性溶液、好ましくは、約pH8の6MグアニジンHClを含有する変性溶液で洗浄する。この時点で、この変性剤を例えば緩衝溶液中の8M尿素に交換することができる。可溶化緩衝液、ローディング緩衝液および洗浄緩衝液は好ましくは低濃度、例えば1〜40mMのイミダゾールを含有する。緩衝液の交換後、カラムをOD280が例えば<0.1に低下するまで緩衝液で洗浄しなければならない。結合タンパク質は、8M尿素と250mMイミダゾール(pH8)を含有する緩衝液(溶出緩衝液)で溶出させ得る。タンパク質を含有する画分をプールして、低濃度(例えば、100mM)塩の(好ましくは8M尿素を含有する)変性透析緩衝液に対して緩衝液を数回交換しながら4℃で透析する。次に、透析タンパク質を、DEAE(ジエチルアミノエチル)等のイオン交換カラムにかける。好ましい実施態様において、イオン交換カラムにかける前に、ジチオスレイトール(DTT)または他の還元剤をタンパク質に加える。キメラ抗原はDEAEカラムを通過する。したがって、このDEAE通過分を集めて、低減した濃度の変性剤を含有する緩衝液に対して段階的な様式で透析する。例示的な方法において、次に上記タンパク質を緩衝性4M尿素に対して少なくとも12時間以上、次に緩衝性2M尿素に対して少なくとも12時間以上、次に緩衝性1M尿素に対して少なくとも12時間以上、次に緩衝性0.5M尿素に対して少なくとも12時間以上、そして最後に変性剤を含まない緩衝液に対して少なくとも12時間以上透析した後に、好ましくは変性剤を含まない新しい緩衝液に対して12時間の透析をさらに二回行う。精製され再び折りたたまれたタンパク質を濃縮して、例えば、SDSゲル電気泳動、等電点電気泳動(isoelectric focusing)または発現タンパク質の異なるドメインに対する抗体を用いるウエスタンブロット分析等の標準的な生化学的手法を用いて特性付けることができる。
【0084】
本発明の実施は、特に他に示さなければ、当業者の技量の範囲内にある分子生物学、微生物学、組換えDNAおよび免疫学の慣用の技術を用いる。そのような技法は、文献により十分に説明されている。例えば、Sambrookら、Molecular Cloning:A Laboratory Manual,第2版,New York:Cold Spring Harbor Press,1989;およびAusubelら、Current Protocols in Molecular Biology,Wiley Interscience Publishers((著作権)1995,2004年4月、補足66で補足されたもの)を参照のこと。
【0085】
(F.新規なポリヌクレオチド)
本発明のもう一つの特徴は、免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二のポリヌクレオチド部分を含むキメラ抗原をコードするポリヌクレオチドを提供する。第一のポリヌクレオチド部分と第二のポリヌクレオチド部分は同一または異なるヌクレオチド鎖に配置され得る。
【0086】
本発明は、キメラ抗原をコードする遺伝子、mRNAおよび/またはコード配列に対応するか、またはこれらに相補的なポリヌクレオチドを、好ましくは単離された形態で提供する。これらとしては、キメラ抗原変異タンパク質をコードするポリヌクレオチド、キメラ抗原をコードする遺伝子もしくはmRNA配列もしくはその一部に対して相補的であるか、または少なくとも90%の相同性を有するDNA、RNA、DNA/RNAハイブリッドおよびその関連する分子、ポリヌクレオチドもしくはオリゴヌクレオチド、ならびにキメラ抗原をコードする遺伝子、mRNA、またはキメラ抗原をコードするポリヌクレオチドに対してハイブリダイズするポリヌクレオチドもしくはオリゴヌクレオチドが挙げられる。
【0087】
さらに、本発明はここに具体的に開示されたキメラ抗原をコードする遺伝子の類似体を包含する。類似体としては、例えば、免疫応答を引き出す能力を保持する変異体であって、キメラ抗原をコードするポリヌクレオチドの、好ましくは少なくとも80%、より好ましくは90%、そして最も好ましくは95%の相同性を有する変異体が挙げられる。これらは具体的には、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号44に記載される。典型的には、そのような類似体はわずかに1〜15コドン変化のみが異なるだけである。例としては、ウイルス抗原または抗体フラグメントの天然アミノ酸配列由来の小さなアミノ酸変化(特に、保存的なアミノ酸の置換)を有するポリペプチドが挙げられる。保存的置換は、側鎖に関連性のあるアミノ酸ファミリー内で起こる置換である。遺伝的にコードされるアミノ酸は、一般的に下記の4ファミリーに分けられる:(1)酸性=アスパラギン酸、グルタミン酸;(2)塩基性=リジン、アルギニン、ヒスチジン;(3)非極性=アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン;および(4)非電荷極性=グリシン、アスパラギン、グルタミン、シスチン、セリン、スレオニン、チロシン。フェニルアラニン、トリプトファンおよびチロシンは、ときどき一緒にされて芳香族アミノ酸に分類される。例えば、ロイシンのイソロイシンまたはバリンへの単離された置換、アスパラギン酸のグルタミン酸への単離された置換、スレオニンのセリンへの単離された置換、またはあるアミノ酸の構造的に関連するアミノ酸への類似の保存的置換は、生物学的活性に対して大きな影響を及ぼさないと予想することは合理的である。配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49のいずれかに開示されたポリペプチドのいずれかと実質的に同じアミノ酸配列を有するが、免疫応答を引き出すキメラ抗原の能力に実質的に影響を与えない重要でないアミノ酸置換を有するポリペプチド分子は、配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49にそれぞれ示された配列を有するキメラ抗原の定義内である。誘導体としては、他のキメラ抗原分子との集合複合体(aggregative conjugate)および関連しない化学部分との共有結合複合体が挙げられる。共有結合誘導体は、キメラ抗原のアミノ酸鎖において、またはN末端残基もしくはC末端残基に見られる基に対して、公知の手段によって官能基を結合させることにより調製される。
【0088】
アミノ酸の略語を表1に示す。
【0089】
【表1】

タンパク質のコンフォメーションまたは機能のいずれも変更することなしに、保存的アミノ酸置換をタンパク質中で行うことができる。本発明のタンパク質は、1個〜15個の保存的置換を含み得る。そのような変化としては、イソロイシン(I)、バリン(V)およびロイシン(L)のいずれかの、これらの疎水性アミノ酸のいずれか他のものへの置換;アスパラギン酸(D)のグルタミン酸(E)による置換およびその逆の置換;グルタミン(Q)のアスパラギン(N)による置換およびその逆の置換;ならびにセリン(S)のスレオニン(T)による置換およびその逆の置換が挙げられる。他の置換も、特定のアミノ酸の環境およびタンパク質の三次元構造におけるその役割に依存して、保存的であると考えられ得る。例えば、グリシン(G)とアラニン(A)とは、アラニン(A)とバリン(V)とがそうであり得るように、しばしば相互交換できる。比較的疎水性であるメチオニン(M)は、ロイシンおよびイソロイシンとしばしば相互交換可能であり得、バリンと置換可能である場合もある。リジン(K)とアルギニン(R)とは、アミノ酸残基の顕著な特徴がその電荷であり、これら二つのアミノ酸残基の異なるpKが顕著ではないような位置において、しばしば相互交換可能である。さらに他の変化は特定の環境において「保存的」であると考えられ得る(例えば、Biochemistry 第4版,Lubert Stryer(編)(W.H.Freeman and Co.),18−23頁;HenikoffおよびHenikoff,Proc Nat’l Acad Sci USA 89:10915−10919(1992);Leiら、J Biol Chem 270(20):11882−6(1995)を参照のこと)。
【0090】
また、本発明は、キメラ抗原をコードするポリヌクレオチドに対して、好ましくはストリンジェント条件下にハイブリダイズする、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号44に具体的に記されたポリヌクレオチドを提供する。ハイブリダイゼーション反応のストリンジェンシーは、当業者により容易に決定可能であり、通常、プローブの長さ、洗浄温度および塩濃度に依存する経験的な計算である。一般的に、より長いプローブは適切なアニーリングのためにより高い温度を必要とする一方で、より短いプローブはより低い温度を必要とする。相補鎖がそれらの溶融温度を下回る環境に存在する場合、ハイブリダイゼーションは、通常、変性核酸配列が再アニーリングする能力に依存する。プローブとハイブリダイズ可能な配列との間の所望の相同性の度合いが高ければ高いほど、使用できる相対温度は高くなる。結果として、より高い相対的温度は反応条件をよりストリンジェントとする傾向があるが、より低い温度はそうではない。ハイブリダイゼーション反応のストリンジェンシーのさらなる詳細と説明に関しては、Ausubelら、前出、2.9.1〜2.10.8および4.9.1〜4.9.13を参照されたい。
【0091】
本明細書において定義される「ストリンジェント条件」または「高ストリンジェント条件」とは、(1)低イオン強度および高洗浄温度(例えば、0.015M塩化ナトリウム/0.0015Mクエン酸ナトリウム/0.1%ドデシル硫酸ナトリウム、50℃)を用いる条件;(2)ハイブリダイゼーション中、ホルムアミド等の変性剤を用いる条件(例えば、50%(v/v)ホルムアミド+0.1%ウシ血清アルブミン/0.1% Ficoll/0.1%ポリビニルピロリドン/50mMリン酸ナトリウム緩衝液(pH6.5)+750mM塩化ナトリウム、75mMクエン酸ナトリウム、42℃;または(3)50%ホルムアミド、5×SSC(0.75M NaCl、0.075Mクエン酸ナトリウム)、50mMリン酸ナトリウム(pH6.8)、0.1%ピロリン酸ナトリウム、5×Denhardtの溶液、超音波処理済サケ精子DNA(50μg/ml)、0.1%SDSおよび10%硫酸デキストラン、42℃を用いる条件であって、42℃での0.2×SSC(塩化ナトリウム/クエン酸ナトリウム)での洗浄と、55℃での50%ホルムアミドでの洗浄と、それに引き続く55℃でのEDTAを含む0.1×SSCを含む高ストリンジェント洗浄とを用いる条件が挙げられるが、これらに限定されない。「適度なストリンジェント条件」とは、Sambrookら(前出)において記載された条件であるが、これに限定されず、そして上記に記載された条件よりも低いストリンジェントな洗浄溶液およびハイブリダイゼーション条件(例えば、温度、イオン強度およびSDS%)の使用を包含する。適度なストリンジェント条件の例は、20%ホルムアミド、5×SSC(150mM NaCl、15mMクエン酸三ナトリウム)、50mMリン酸ナトリウム(pH7.6)、5×Denhardt溶液、10%硫酸デキストランおよび20mg/mL変性せん断サケ精子DNAを含む溶液中での、37℃における一晩のインキュベーション、そしてそれに引き続く約37〜50℃における1×SSC中でのフィルター洗浄である。当業者らは、必要な場合、プローブの長さ等の因子に適合させるための温度、イオン強度等を調節する方法を認識する。
【0092】
キメラ抗原をコードするポリヌクレオチドの実施態様としては、配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49のいずれかから選択された配列を有するキメラ抗原をコードするポリヌクレオチド、または配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号48のいずれかから選択されたキメラ抗原のヌクレオチド配列(ここで、Tは必要に応じてUであり得る)が挙げられる。例えば、キメラ抗原ヌクレオチドの実施態様は、限定するものではないが、以下を含む:
(a)配列番号26のヌクレオチド1〜1326、配列番号28のヌクレオチド1〜2004、配列番号30のヌクレオチド1〜1350、配列番号32のヌクレオチド1〜1293、配列番号34のヌクレオチド1〜1794、配列番号36のヌクレオチド1〜1581、配列番号38のヌクレオチド1〜1389、配列番号40のヌクレオチド1〜1347、配列番号42のヌクレオチド1〜2157、配列番号44のヌクレオチド1〜1395、配列番号46のヌクレオチド1〜1905、または配列番号48のヌクレオチド1〜2484(ここで、Tはまた、Uでもあり得る)の配列番号に記載された配列を含むか、あるいはこれらの配列からなるポリヌクレオチド;
(b)配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46または配列番号48に記載された配列に対して、少なくとも80%相同性である配列を有するポリヌクレオチド;
(c)登録番号080504−03、登録番号080504−04、登録番号080504−05、登録番号080504−02および登録番号080504−01としてそれぞれカナダ国際寄託当局(Bureau of Microbiology at Health Canada)に寄託された、pFastBacHTa HBV S1/S2−TBD、pFastBacHTa HBV core−TBD、pFastBacHTa HCV core(1−177)−TBD、pFastBacHTa HCV NS5A−TBDおよびpFastBacHTa HCV E2−TBDと命名されたプラスミドの一つに含まれるDNAによってコードされる配列を有するキメラ抗原をコードするポリヌクレオチド;
(d)キメラ抗原の配列が、配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635または配列番号49のアミノ酸1〜828の配列である、キメラ抗原をコードするポリヌクレオチド;
(e)配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635または配列番号49のアミノ酸1〜828により記載される全アミノ酸配列に対して、少なくとも90%同一性であるキメラ抗原関連タンパク質をコードするポリヌクレオチド;
(f)(а)〜(d)のいずれか一つのポリヌクレオチドに完全に相補的であるポリヌクレオチド;ならびに
(g)(а)〜(f)のポリヌクレオチドに対してストリンジェント条件下で選択的にハイブリダイズするポリヌクレオチド。
【0093】
また、本発明は、キメラ抗原ポリヌクレオチド、その類似体または相同体を含む組換えDNA分子またはRNA分子を提供する。これらとしては、ファージ、プラスミド、ファージミド、コスミド、YAC(酵母の人工的染色体)、BAC(細菌の人工的染色体)、ならびに当該分野で周知の種々のウイルスベクターおよび非ウイルスベクター、およびこのような組換えのDNA分子もしくはRNA分子で形質転換されたか、またはトランスフェクションされた細胞が挙げられるが、これらに限定されない。このような分子を作製する方法は、周知である(例えば、Sambrookら(前出)(1989)を参照のこと)。
【0094】
さらに、本発明は、キメラ抗原ポリヌクレオチド、その類似体または相同体を含む組換えDNA分子を、適切な原核細胞または真核細胞の宿主細胞内に含む宿主−ベクター系を提供する。適切な真核宿主細胞の例としては、酵母細胞、植物細胞または、哺乳動物細胞もしくは昆虫細胞等の動物細胞(例えば、Sf9、Sf21、Drosophila S2またはHigh Five(登録商標)細胞のようなバキュロウイルス感染細胞)が挙げられる。適切な哺乳動物細胞の例としては、DU145およびTsuPr1等の多様な前立腺癌細胞株、他のトランスフェクション可能または形質導入可能な前立腺癌細胞株、初代細胞(PrEC)、ならびに組換えタンパク質の発現に日常的に用いられる多くの哺乳動物細胞(例えば、COS、CHO、293、293T細胞)が挙げられる。より具体的には、キメラ抗原またはフラグメント、その類似体または相同体のコード配列を含むポリヌクレオチドは、当該分野で日常的に用いられ、広く知られている多くの宿主−ベクター系を用いてそのキメラ抗原を作製するために用いられ得る。
【0095】
キメラ抗原の発現に適する広い範囲の宿主−ベクター系が利用できる(例えば、Sambrookら(1989)(前出);Ausubelら、1.0.1〜1.16.16、9.01〜9.17.3および13.4.1〜13.6.5を参照のこと)。昆虫細胞発現用の好ましいベクターとしては、pFastBac HTa(Invitrogen)が挙げられるが、これに限定されない。そのような発現ベクターを用いて、キメラ抗原を、例えばSf9、Sf21、Drosophila S2およびHigh Fiveを含むいくつかの昆虫細胞株において発現させることができる。もしくは、好ましい酵母発現系としては、Saccharomyces cerevisiae、Schizosaccharomyces pombe、Pichia pastorisおよびPichia augustが挙げられる。本発明の宿主−ベクター系は、キメラ抗原の生産に有用である。
【0096】
キメラ抗原またはその類似体または相同体は、キメラ抗原をコードする構築物をトランスフェクトした細胞により作ることができる。例えば、Sf9細胞は、キメラ抗原またはその類似体または相同体をコードする発現プラスミドでトランスフェクションされ得、そのキメラ抗原は、Sf9細胞において発現され、そしてそのキメラ抗原は、標準的な精製法を用いて単離される。当該分野で周知の多様な他の発現系もまた、用いられ得る。キメラ抗原をコードする配列にフレーム内で連結させたリーダーペプチドをコードする構築物の発現は、分泌型キメラ抗原を作製するために使用され得る。
【0097】
ここで論じるように、遺伝子コードの重複性が、キメラ抗原遺伝子配列中の変異を可能にする。特に、特定の宿主種はしばしば特定のコドン選択性を有することが当該分野で公知であり、所望の宿主について好ましいように、開示された配列を適合し得る。例えば、望ましい類似体コドン配列は、典型的にはより高い頻度コドンにより置換された稀なコドン(すなわち、所望の宿主の公知配列において、約20%未満の利用頻度を有するコドン)を有する。特定の種についてのコドン選択性は、例えば、世界的なウェブのURL www.kazusa.or.jp/codon等のインターネット上で利用できるコドン利用表を用いて計算される。
【0098】
さらなる配列の改変が、細胞宿主中のタンパク質発現を増強することは公知である。これら改変としては、擬似ポリアデニル化シグナル、エキソン/イントロンスプライス部位シグナル、トランスポゾン様リピートをコードする配列、および/または遺伝子発現に有害な他のよく特性付けられた配列を排除することを包含する。上記配列のGC含量は、宿主細胞において発現された公知の遺伝子を参照することによって計算された、所与の細胞宿主に対しての平均的なレベルに調節される。可能な場合、上記配列は、予想されるヘアピンの二次mRNA構造を避けるように改変される。他の有用な改変としては、Kozak,Mol.Cell Biol.,9:5073−5080(1989)に記載のような、オープンリーディングフレームの開始点における翻訳開始共通配列の付加を包含する。真核生物のリボゾームが5’付近のAUGコドンにおいてのみ翻訳を開始するという一般的な規則は、稀な条件下においてのみ無効とされることを当業者らは理解する(例えば、Kozak Proc Nat’l Acad Sci USA 92(7):2662−2666(1995)およびKozak Nucl Acids Res 15(20):8125−8148(1987)を参照のこと)。
【0099】
(G.本発明の薬学的組成物)
本発明の一つの特徴は、免疫応答ドメインと、抗体フラグメントを含む標的結合ドメインとを含むキメラ抗原および薬学的に受容可能な賦形剤を含む薬学的組成物に関する。治療利用において、上記薬学的組成物は、抗原に対する効果的なB細胞、細胞障害性Tリンパ球(CTL)および/またはヘルパーTリンパ球(Th)の応答を引き起こし、かつ感染を遮断するか、あるいは症状および/もしくは合併症もしくは病気もしくは疾患を治療するか、または少なくとも部分的に抑制または遅らせるのに十分な量で、被験体に投与され得る。この使用に効果的な量は、例えば、投与される特定の組成物、投与方法、処置される病気の段階と重症度、その被験体の体重および健康の一般的な状態、ならびに処方する医師の判断に依存する。
【0100】
(キメラ抗原による)最初の治療的免疫化のための投薬量は、一般的には、低い値が約1ng、5ng、50ng、500ngまたは1,000ngであり、高い値が約10,000μg、20,000μg、30,000μgまたは50,000μgであるような単位投薬量範囲に存在する。ヒト用の投薬量値は、被験体70キログラムあたり典型的には約500ng〜約50,000μgの範囲にある。数週間〜数ヶ月にわたる追加免疫レジメンに準じる約1.0ng〜約50,000μgの間のキメラ抗原の追加免疫投薬量が、被験体の応答と状態に基づきながら投与され得る。少なくとも、臨床的な症状または実験室試験から、症状が防止されるか、抑止されるか、遅らされるか、またはなくなることが示されるまで、そしてその後のある期間にわたって、投与は継続されなければならない。投薬量、投与経路および投与計画は、当該分野で公知の方法にしたがって調節される。
【0101】
キメラ抗原のヒト単位用量形態は、典型的には、ヒト単位用量の受容可能なキャリア(1つの実施態様においては、水性キャリア)を含有する薬学的組成物中に含まれ、そしてこのようなポリペプチドのヒトへの投与に有用であることが当業者に公知である体積/量で投与される(例えば、Remington:The Science and Practice of Pharmacy,第20版,A.Gennaro,Editor,Lippincott Williams & Wilkins,Baltimore,Md.,2000を参照)。当業者により理解されるように、多様な因子が、特定の場合における理想的な用量に影響を及ぼしうる。そのような因子としては、例えば、キメラ抗原の半減期、キメラ抗原の結合親和性、組成物の免疫原性、所望の定常状態の濃度レベル、投与経路、治療頻度、および本発明の治療方法と組み合わせて使用される他の物質の影響、ならびに特定の被験体の健康状態が挙げられる。
【0102】
ある実施態様において、本発明の組成物は、深刻な病状(すなわち、生命を脅かすか、または潜在的に生命を脅かす状態)に使用される。そのような場合、本発明の好ましい組成物中のキメラ抗原が比較的毒性のない性質であるという結果として、これら述べられた投薬量に対して実質的に過剰なこれらキメラ抗原を投与することが可能であり、治療を行う医師には望ましいと感じられ得る。
【0103】
薬学的処方物における本発明のキメラ抗原の濃度は、広く変動し得る。すなわち、約0.1重量%未満、通常は約2重量%、または少なくとも約2重量%から20〜50重量%またはそれ以上まで変動し得、選択された投与の具体的な態様にしたがって主に液体容量、粘度等によって選択される。
【0104】
この薬学的組成物は、非経口、髄腔内、血管内、静脈内、筋肉内、経皮、皮内、皮下、鼻内、局所、経口、直腸、膣、肺または腹腔内等の当該分野で公知の任意の経路を介して送達され得る。好ましくは、上記組成物は、皮下投与または皮膚内投与等の非経口経路により送達される。
【0105】
上記薬学的組成物は、所望のキメラ抗原を、意図される投与経路に適する適切なビヒクルと混合することによって調製され得る。本発明の薬学的組成物の作製において、このキメラ抗原は、通常賦形剤と混合されるか、賦形剤で希釈されるか、またはカプセル、匂い袋、紙または他の容器の形態であり得るキャリア内に封入される。薬学的に受容可能な賦形剤が希釈剤として役立つ場合、この賦形剤は治療剤用のビヒクル、キャリアまたは媒体として作用する固体、半固体または液体の物質であり得る。よって、上記組成物は、錠剤、丸剤、散薬、ロゼンジ、匂い袋、カシェ剤、エリキシール、懸濁液、乳化液、溶液、シロップ、(固体としてまたは液体媒体中の)エーロゾル、例えば10重量%までのキメラ抗原を含有する軟膏、軟性または硬性のゼラチンカプセル、座薬、滅菌注射溶液および滅菌包装散薬の形態であり得る。
【0106】
適当な賦形剤のいくつかの例としては、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アカシアゴム、リン酸カルシウム、アルギン酸塩、トラガカント、ゼラチン、ケイ酸カルシウム、微結晶セルロース、ポリビニルピロリドン、セルロース、滅菌水、シロップおよびメチルセルロースが挙げられるが、これらに限定されない。これらの処方物はさらに、タルク、ステアリン酸マグネシウムおよび鉱油等の潤滑剤、湿潤剤、乳化懸濁剤、メチルベンゾエートおよびプロピルヒドロキシベンゾエート等の保存剤、甘味剤、ならびに調味剤が挙げられ得る。本発明の組成物は、当該分野で公知の操作を用いることにより、被験体に投与した後にキメラ抗原の即効性放出、徐放性放出または遅延性放出を提供するように処方され得る。例えば、Remington(前出)の903〜92ページおよび1015〜1050ページを参照のこと。
【0107】
錠剤等の固体組成物を調製するために、上記キメラ抗原は、薬学的賦形剤と混合されて本発明のキメラ抗原の均一混合物を含む固体の予備処方組成物を形成する。これらの予備処方組成物を均一であると称する場合、上記キメラ抗原が、その組成物が錠剤、丸剤およびカプセル等の効果の等しい単位投薬形態に容易に再分割され得るように、組成物中に均一に分散していることを意味する。
【0108】
本発明の錠剤または丸剤は、被覆され得るか、または長期間の作用という利点を与える投薬形態を提供するように配合され得る。例えば、上記錠剤または丸剤は、内投薬成分と外投薬成分とを含み得、後者は前者を被う封筒のような形である。これらの二成分は、腸溶性層によって分離され得る。この層は、胃内の分解に対して抵抗性があり、内成分をそのまま十二指腸に通過させ得るか、または放出を遅らせ得る。多様な物質がそのような腸溶性層または被覆に用いられ得、そのような物質として、多くのポリマー酸およびポリマー酸とセラック、セチルアルコールおよび酢酸セルロースのような物質との混合物が挙げられる。
【0109】
本発明の新規組成物が経口または注射による投与で取り込まれ得るような液体形態としては、水性溶液、適当に味付けしたシロップ、水性または油性の懸濁液、およびトウモロコシ油、綿実油、ゴマ油、ココナッツ油またはピーナッツ油等の食用油を有する味付けした乳化物ならびにエリキシールおよび類似の薬学的ビヒクルが挙げられる。
【0110】
非経口投与用組成物の調製において、刺激を減少させる浸透圧調節に対して、厳密な注意を払う必要がある。再構成可能な組成物は、乾燥形態の包装された滅菌固体である。再構成可能な組成物が望ましいのは、乾燥固体として保存した場合、即座に投与するために溶液を準備するよりもさらに安定なためである。通常、乾燥固体は、その固体が最適な湿度範囲に確実に維持されるようにブチルゴムの蓋を有する滅菌容器に包装される。再構成可能な乾燥固体は、乾燥充填法、噴霧乾燥法または凍結乾燥法により形成される。これら方法の説明は、例えばRemington(前出)、681〜685ページと802〜803ページに見出され得る。
【0111】
非経口注射用の組成物は、一般的に希釈されており、高い比率で存在する成分はビヒクルである。通常、このビヒクルは治療活性を持たず、非毒性であるが、吸収に適する形態で、上記キメラ抗原を体組織に提示する。通常、吸収は、キメラ抗原が水溶液として提示される場合に、最も容易かつ完全に起こる。しかし、液体混和性液体によるビヒクルの改変または水非混和性液体による置換は、吸収速度に影響を与えうる。好ましくは、本組成物について最も価値のあるビヒクルは、等張食塩水である。注射に適する組成物の調製において、水性ビヒクル、水混和性ビヒクルおよび非水性ビヒクルを使用し得る。
【0112】
さらなる物質が、上記組成物の品質を向上または保護するために、本発明の注射可能な組成物中に含まれ得る。したがって、加えられた物質は、溶解性に影響を与え得、被験体の快適さを提供し得、化学安定性を増強し得るか、または微生物の成長に対して調製物を保護し得る。よって、この組成物は、適当な可溶化剤、抗酸化剤として働く物質および微生物の成長を妨げる保存剤として働く物質を含有し得る。これらの物質は、それらの機能に適切であるが、組成物の作用に悪影響を与えないような量で存在する。適当な抗微生物剤の例としては、チメロサール、塩化ベンゼトニウム、塩化ベンズアルコニウム、フェノール、p−ヒドロキシ安息香酸メチルおよびp−ヒドロキシ安息香酸プロピルが挙げられる。適当な抗酸化剤は、Remington(前出)、1015〜1017ページに見出され得る。
【0113】
ある実施態様において、リポソーム、ナノカプセル、マイクロカプセル、液体粒子、ベシクル等が、本発明のキメラ抗原の投与ために用いられる。特に、本発明の組成物は、送達のために、液体粒子、リポソーム、ベシクル、ナノスフェアまたはナノ粒子等にカプセル化されて処方され得る。あるいは、本発明の組成物は、共有結合または非共有結合のいずれかにより、このようなキャリアビヒクルの表面に結合され得る。
【0114】
リポソームを介して投与される組成物はまた、以下のように役立ち得る:1)リンパ系組織等の特別な組織に対してキメラ抗原を標的化する;2)抗原提示細胞に対して選択的に標的化する;または3)ペプチド組成物の半減期を増加させる。リポソームとしては、乳化物、泡、ミセル、不溶性単層、液晶、リン脂質分散物、薄層等が挙げられる。これらの調製物において、送達されるキメラ抗原は、リポソームの一部として、単独またはCD45抗原に結合するモノクローナル抗体等のリンパ系細胞間に広く認められるレセプターに結合する分子と組み合わせて取り込まれるか、あるいは他の治療組成物または免疫原組成物とともに取り込まれる。よって、本発明の所望のキメラ抗原により充填または装飾されたリポソームは、そのリポソームがキメラ抗原を送達するリンパ系細胞の部位に向かい得る。本発明に従って用いられるリポソームは、中性および負電荷のリン脂質およびコレステロール等のステロールを一般的に包含する、標準的ベシクル形成脂質から形成される。脂質の選択は、例えば、血流中のリポソームの大きさ、酸不安定性と安定性を考慮することにより通常行われる。Szokaら、Ann.Rev.Biophys.Bioeng.9:467−508(1980)および米国特許4,235,871号、米国特許4,501,728号、米国特許4,837,028号および米国特許5,019,369号に記載されるように、多様な方法がリポソームを調製するために利用できる。キメラ抗原を含むリポソーム懸濁液は、とりわけ投与法、送達されるキメラ抗原および処置される病気の段階にしたがって変動する用量において静脈内、局所的、局部等に投与され得る。
【0115】
吸入用または吹入れ用の組成物としては、薬学的に受容可能な水性溶媒もしくは有機溶媒、またはそれらの混合物中の溶液または懸濁液、ならびに粉末が挙げられる。液体または固体の組成物は、本明細書中に記載されるような適当な薬学的に受容可能な賦形剤を含有し得る。この組成物は、局所性効果または全身性効果のために、口または鼻の呼吸経路により投与され得る。薬学的に受容可能な溶媒中の組成物は、不活性気体を用いることにより霧状化され得る。霧状化された溶液は、噴霧用装置から直接的に吸入され得るか、またはこの噴霧用装置は顔マスクテント(facemask tent)または間欠的陽圧呼吸装置に装着され得る。溶液、懸濁または粉末組成物は、適切な方法で処方物を送達する装置から、好ましくは経口または経鼻にて投与され得る。
【0116】
本発明の方法に用いられるもう一つの処方は、経皮送達装置(「パッチ」)を用いる。そのような経皮パッチを用いて、本発明のキメラ抗原の連続的または非連続的な注入を制御された量で提供し得る。薬学的物質の送達のための経皮パッチの構築と使用は、当分野で周知である。例えば、本明細書において参考として援用される米国特許5,023,252号を参照のこと。そのようなパッチは、薬学的物質の連続的送達、拍動性送達またはオンデマンド送達のために構築され得る。
【0117】
さらに、キメラ抗原および薬学的賦形剤に加えて、少なくとも一つの抗ウイルス治療剤または化学療法剤を含むことが有利であり得る。抗ウイルス治療剤としては、以下が挙げられるが、これらに限定されない:擬似ペプチド(例えば、アンプレナビル、インジナビル、ロピナビル、ネルフィナビル、リトナビルおよびサキナビル)、ポリヌクレオチド(例えば、アンプリゲンおよびホミビルセン)、プリン/ピリミジノン類(例えば、アバカビル、アシクロビル、アデフォビル、シドフォビル、シタラビン、ジダノシン、ジデオキシアデノシン、ジピボキシル、エドクスウジン、エムトリシタビン、エンテコビル、ファムシクロビル、ガンシクロビル、イドクスウリジン、イノシン・プラノベクス、ラミブジン、MADU、ペンシクロビル、ソリブジン、スタブジン、テノフォビル、トリフルリジン、バラシクロビル、バルガンシクロビル、ビダラビン、ザルシタビンおよびジドブジン)、シアル酸類似体(例えば、オセルタミビルおよびザナミビル)、アセマンナン、アセチルロイシンモノエタノールアミン、アマンタジン、アミジノマイシン、アテビリジン、カプラビリン、デラビルジン、n−ドコサノール、エファビレンズ、ナトリウムフォスカーネット、インターフェロン−α、インターフェロン−β、インターフェロン−γ、ケトキサール、リゾチーム、メチサゾン、モロキシジン、ネビラピン、ペンタフシド、プレコナリル、ポドフィルロトキシン、リバビリン、リマンチジン、スタリマイシン、スタトロン、ターマカムラおよびトラオマンタジン。他の適当な抗ウイルス剤は、Remington(前出)、87章、抗感染剤、1507〜1561ページ、特に1555〜1560ページに説明されている。本発明の薬学的組成物に包含される好ましい抗ウイルス治療剤として、アデフォビル、ジピボキシル、エンテコビル、ラミブジンおよびリバビリンが挙げられる。
【0118】
いくつかの実施態様において、Bリンパ球またはTリンパ球を初回刺激する(prime)少なくとも一つの成分を本発明の薬学的組成物に含むことが望ましい。脂質はCTLをインビボで初回刺激し得る物質として同定されている。例えば、パルミチン酸残基を、リジン残基のεアミノ基とαアミノ基に結合させ、次にこれをGly、Gly−Gly−、Ser、Ser−Ser等の一つ以上の連結残基を介して免疫原性ペプチドに連結し得る。次に、この脂質化ペプチドは、ミセルまたは粒子のいずれかで直接投与され得るか、リポソームに取り込まれ得るか、またはアジュバント、例えば不完全フロイントアジュバント中に乳化させ得る。好ましい実施態様において、特に効果的な免疫原性組成物は、免疫原性ペプチドのアミノ末端に対して例えばSer−Serの連結により結合させたLysのεアミノ基とαアミノ基とに結合させたパルミチン酸を含む。
【0119】
CTL応答の脂質初回刺激のもう一つの例として、トリパルミトイル−S−グリセリルシステインリセリル−セリン(P3CSS)等のE.coliリポタンパク質を用いて、適当なペプチドに共有結合させた場合のウイルス特異的CTLを初回刺激し得る(例えば、Deresら、Nature 342:561(1989))。本発明のキメラ抗原は、例えばP3CSSに結合させ得、リポペプチドを個体に投与して、標的抗原に対する免疫応答を特異的に初回刺激し得る。
【0120】
本発明の組成物はアジュバントの使用を必要とすべきではないが、アジュバントを用いることはできる。宿主の種に基づいて、免疫学的応答を高める多様なアジュバントが用いられ得、そのようなアジュバントとしては、フロイント(完全および不完全)、水酸化アルミニウム等の無機ゲル、リソレシチン等の表面活性剤、洗剤、プルロニックポリオール、ポリアニオン、ペプチド、油性乳化物、キーホールリンペットヘモシアニン、ジニトロフェノール、免疫促進ポリ塩基配列およびBCG(カルメット‐ゲラン杆菌)およびCorynebacterium parvum等の潜在的に有用なヒトアジュバントが挙げられるが、これらに限定されない。さらなるアジュバントもまた、当該分野において周知である。
【0121】
(H.製品)
本発明のもう一つの特徴は、キメラ抗原を含有する組成物を保持する容器を含む製品を提供する。この製品は、上記組成物を非経口(例えば、皮下、筋内、皮内、鼻内または血管内)で、どのように投与するかを説明する印刷されたラベル説明と組み合わせて、注射に適切であるか、または注射用に再構築するためのものである。上記組成物は、この組成物と顕著に相互作用せず、かつそれが非経口使用であることを示す適当なラベルで標識された任意の適当な容器に含まれる。容器に組み合わされるものは、上記の治療方法に合わせたラベル説明である。本発明の組成物を保持する容器は、患者、医師、看護婦またはそれ以外の専門家がキメラ抗原を投与し得るように、注射用の適当な針とシリンジとを有する注射に適する液体組成物を有する容器であり得る。あるいは、上記組成物は、この組成物を溶解または懸濁させる水性ビヒクルまたは非水性ビヒクルと組み合わされるか、またはそれらのビヒクルで希釈されるキメラ抗原の可溶性の種類を含有する乾燥組成物あるいは濃縮組成物であり得る。あるいは、上記容器は、液体中に懸濁剤を有するか、塩の不溶性の種類が懸濁されるビヒクルと組み合わせるための塩の不可溶性の種類であり得る。適当な容器は、Remington(前出)の788〜789ページ、805ページ、850〜851ページおよび1005〜1014ページに説明されている。
【0122】
本発明のキットは、典型的には、上記の容器と、緩衝液、希釈液、フィルター、針、シリンジおよび使用説明書を備える包装挿入物を含む商品と使用者の観点から望ましい材料を含む一つ以上の他の容器を含む。組成物が特定の治療または非治療的利用のために用いられことを示すラベルが容器上に存在してよく、かつそのラベルはインビボまたはエキソビボのいずれかでの使用についての説明、例えば上記のような説明を示すこともできる。説明および/または他の情報は、キットに含まれる挿入物に含ませることもできる。
【実施例】
【0123】
(V.実施例)
以下の非限定的な実施例は、本発明のさらなる説明を提供する。
【0124】
(A.実施例1:TBD発現ベクターの構築)
CH1−ヒンジ−CH2−CH3領域の一部のアミノ酸をコードするマウスIgG1 DNA配列を、HBV表面抗原(sAg)に対してmAbを産生するハイブリドーマ(2C12)から単離されたmRNAから作った。全mRNAを、Trizol(登録商標)試薬(Gibco BRLカタログ番号15596−026)を用いて単離し、標的結合ドメイン(TBD;マウス免疫グロブリンフラグメント)のcDNAを、Superscript First−strand Synthesis(Invitrogenカタログ番号11904−018)を用いたRT−PCRにより作った。このPCRプライマーは、リンカーペプチド(5’末端の−SRPQGGGS−(配列番号1))をコードするリンカー配列、5’末端の独自のNotI部位および3’末端の独自のHindIII制限部位を含んだ。得られたcDNAは(5’NotI)−リンカー配列−CH1(VDKKI)(配列番号2)、−ヒンジ領域−CH2−CH3−(3’HindIII)を含む。各酵素による消化後に、同じ制限酵素部位を用いて、フラグメントをpFastBac HTa発現ベクタープラスミド(Invitrogen)に連結する。PCR増幅に用いられる5’プライマーは(センス)5’TGTCATTCTGCGGCCGCAAGGCGGCGGATCCGTGGACAAGAAAATTGTGCCCAGG(配列番号3)であり、そして3’プライマーは(アンチセンス)5’ACGAATCAAGCTTTGCAGCCCAGGAGAGTGGGAGAG(配列番号4)であり、それぞれNotIとHindIII部位を有した。以下は、方向性クローニング(directional cloning)に用いられるプロトコールである。生成したフラグメントを各酵素で消化し、アガロースゲルで精製し、ベクタープラスミドにクローニングした。このDNA配列とORFの正しさを標準的な配列決定方法を用いて実証した。
【0125】
標的結合ドメインをコードするDNAのpFastBac HTaドナープラスミドへのクローニング後に、組換えタンパク質をBac−to−BacTMバキュロウイルス発現系(Invitrogen)を用いて発現させた。クローニングした遺伝子を部位特異的転位によりE.coli(DH10Bac)株中のバキュロウイルスシャトルベクターに移した。DH10Bac細胞は、カナマイシン耐性を付与するシャトルベクターと、トランスポザーゼをコードし、かつテトラサイクリンに対する耐性を付与するヘルパープラスミドとを含む。コンピテントなDH10Bac細胞の100μlのアリコートを氷上で解凍し、pFastBac HTaベースのプラスミドを加え、その混合物を氷上で30分間インキュベートした。この混合物を45秒間42℃で熱ショックを与え、次に氷上で2分間冷やした。次に、この混合物を900μLのLB培地に加え、4時間、37℃でインキュベートした。この形質転換細胞を、LBで、10−1〜10−2まで連続的に希釈し、100μlの各希釈物を、50μl/mlカナマイシン、7μg/mlゲンタマイシン、10μg/mlテトラサイクリン、100μg/ml X−galおよび40μg/ml IPTGを補充したLBアガープレートにプレートし、少なくとも36時間、37℃でインキュベートした。ゲンタマイシン耐性をpFastBac HTaにより賦与し、X−galとIPTG(イソプロピルチオ−β−D−ガラクトシド)を用いて、白色コロニー(組換えプラスミド)と青色コロニー(非組換え体)との間を区別した。白色コロニーを取り、50μg/mlカナマイシン、7μg/mlゲンタマイシンおよび10μg/mlテトラサイクリンを補充した2mlのLBに接種し、攪拌下に37℃で一晩インキュベートした。滅菌ループを用いて少量の一晩培養物を採取し、得られた試料を、50μg/mlカナマイシン、7μg/mlゲンタマイシン、10μg/mlテトラサイクリン、100μg/ml X−galおよび40μg/ml IPTGを補充した新鮮なLB寒天プレートに縞状に塗布し、少なくとも36時間、37℃でインキュベートして白色表現型を確認した。組換えバクミドを標準的なプロトコール(Sambrook(前出))により単離し、そのDNA試料を40μlのTE(10mMトリス塩酸、pH8、1mM EDTA)に溶解し、トランスフェクションに用いた。
【0126】
バキュロウイルスを作るために、バクミドをSf9昆虫細胞にトランスフェクションした。Sf9細胞(9×105)を、2mlのESF921(Expression Systems)中の6ウエル細胞培養皿(35mmウエル)の各ウエルに撒き、少なくとも1時間、27℃で付着させた。Sf9細胞の供給者により提供されたプロトコールにしたがってCellfection(登録商標)試薬(Invitrogen、カタログ番号10362−010)を用いて、トランススフェクションを行った。トランスフェクション後、その細胞を27℃で72時間インキュベートした。バキュロウイルスを含有する培地を集めて、4℃、暗所で保存した。
【0127】
トランスフェクションの効率はバキュロウイルスDNAの生産について検査することによって実証した。単離されたバキュロウイルスDNAをPCRに供して、TBDをコードする挿入遺伝子をスクリーニングした。使用されたプライマーは(センス)5’TATTCCGGATTATTCATACCG(配列番号5)および3’(アンチセンス)5’CTCTACAAATGTGGTATGGC(配列番号6)であった。増幅生成物をアガロースゲル(0.8%)で泳動した。細胞中の異種タンパク質の発現を、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)および6×Hisタグモノクローナル抗体(Clonetech)をプローブとして用いたウエスタンブロットにより実証した。
【0128】
バキュロウイルスの生産とタンパク質の発現を一旦確認したら、ウイルス生産を増幅して、標的結合ドメインをコードする遺伝子を有するバキュロウイルスの濃縮原液を作製した。バキュロウイルスを少なくとも二回増幅することは当分野で標準的な慣行であり、ここに記載のすべてのプロトコールにおいて、この標準的な慣行を遵守した。二回目の増幅の後、作製したバキュロウイルスの濃度を、キットの製造者(Invitrogen)により記載されたプロトコールによるプラークアッセイを用いて定量した。High Five細胞を感染させるためのウイルスの最も適当な濃度と、所望のタンパク質生産の最適な時間とを設定した。一般的に、TBDの発現のために、1のMOIと48時間を用いた。
【0129】
(B.実施例2:キメラ抗原発現ベクターの構築)
表2に示した5’センスプライマーと3’アンチセンスプライマーとを用いたPCR法を使用して、所望のウイルス抗原をコードするDNAを鋳型から作った。得られた増幅フラグメントの5’末端は、独自の制限部位「5’酵素(enzy)」を含み、そして3’末端は独自の制限部位「3’酵素(enzy)」を含んでおり、それら各々をライゲーションのために用いた。
【0130】
【表2】
増幅DNAを適当な5’制限酵素および3’制限酵素で消化し、pFastBac HTa発現ベクターに連結して、ウイルス抗原のみの発現プラスミドを作った。各酵素による消化後に、実施例1に記載のように、DNAの同フラグメントをプラスミドpFastBac HTa−TBDに連結させて、標的結合ドメインに融合させたウイルス抗原の発現プラスミドを作った。得られたプラスミドを用いて、実施例1に記載のように、組換えバキュロウイルスを作り、続いてキメラ抗原の発現に用いた。このキメラ抗原のDNA配列とアミノ酸配列とを表3に示す。
【0131】
【表3】
(C.実施例3:TBD、ウイルス抗原およびキメラ抗原の発現および精製)
標準化感染多重度(MOI)の組換えバクミドを用いて、High Five(登録商標)昆虫細胞を感染させた。懸濁培養のために、細胞を3×105細胞/mLの密度で撒き、細胞密度が2〜3×106細胞/mLに達するまで27.5℃、138rpmでの攪拌下にインキュベートした。標準化量の各組換えバキュロウイルスを細胞に加えた。インキュベーション温度は27.5℃で、適当な感染時間を個々のタンパク質発現について標準化した。これらの細胞を10分間、4℃で2,500rpmにおける遠心分離により集め、組換えタンパク質の精製に用いた。細胞の使用しない部分は液体窒素中に瞬時に凍らせて、−70℃で保存した。
【0132】
組換えタンパク質を変性条件下で精製した。この細胞を、100mMのNaH2PO4、10mM Tris、300mM NaCl、10mMイミダゾール、pH8.0に6MグアニジウムHClを含有する緩衝液(可溶化緩衝液)中に可溶化した。その懸濁物を60ワットの出力設定で1分/パルスの5パルスにて氷上で超音波処理し、室温で1時間混合した。この溶解物を10,000×gで10分間遠心分離して、未破壊細胞と細胞破片を除去した。この上清を、前もって可溶化緩衝液で平衡化させておいたNi−NTAアガロース(Qiagen)ビーズカラム(1×5cm/100mL細胞溶解物)にロードした。ローディングの後に、カラムを100mMのNaH2PO4、10mM Tris、300mM NaCl、40mMイミダゾール、pH8.0中の6MグアニウムHCl(洗浄緩衝液1)の20カラム体積分で洗浄し、引き続いて100mMのNaH2PO4、10mM Tris、300mM NaCl、40mMイミダゾール、pH8.0中の8M尿素(洗浄緩衝液2)の20カラム体積分で洗浄した。結合タンパク質を、8M尿素、100mMのNaH2PO4、10mM Tris、300mM NaCl、250mMイミダゾール、pH8.0を含有する緩衝液(溶出緩衝液)で溶出した。タンパク質を含有する画分を集めて、4℃で透析緩衝液(10mM NaH2PO4、300mM NaCl)を複数回変えながら透析した。精製タンパク質を、SDSゲル電気泳動、等電点電気泳動および発現タンパク質の異なるドメインに対する抗体を用いたウエスタンブロット分析等の標準的な生化学技術を用いて特性付けた。
【0133】
(D.実施例4:キメラ抗原融合タンパク質を用いた「自己」タンパク質に対する寛容の破壊)
キメラ抗原融合タンパク質に対する免疫応答を評価するために、精製したHBV S1/S2、TBDまたはS1/S2−TBDタンパク質でマウスを免疫し、個々のタンパク質に対して作られた抗体を定量した。免疫されたマウスから集めた脾臓T細胞の増殖を、各タンパク質による誘発(challenge)後に評価した。
【0134】
15週齢のBALB/cマウスを免疫に用いた。マウスにS1/S2−TBD(4.15μg)、S1/S2(4.15μg)またはTBD(4.15μg)を二週間の間隔で4回皮下注射した。免疫の開始前と各免疫の一週間後に、血液試料を集めた。血清を凝固血液試料から調製し、注射した各抗原に対して宿主動物により作られた抗体量の予測に用いた。
【0135】
(1.HBV S1/S2、TBDまたはS1/S2−TBDに対する抗体の検出のためのELISA)
96ウエルプレートに、抗原HBV S1/S2、TBDまたはS1/S2−TBDを1.0μg/mLの濃度にて一晩、4℃で被覆した。このプレートを2%BSAを含有するPBSで洗浄した。各動物からの希釈血清を各ウエルに多様な希釈率(1:10〜1:500)で加え、37℃で1時間インキュベートした。このプレートを0.05%のTween 20を含有するPBS(洗浄緩衝液)で洗浄した。ヤギ抗マウスIgG Fab西洋ワサビペルオキシダーゼ(HRP)(1:5000)希釈物をウエルに加えて、37℃で1時間インキュベートした。プレートを洗浄緩衝液で洗浄し、2−2’アジノ−ジ−(3−エチルベンジルチアゾリン−6−スルフォネート)(KPL、Guildford、UK)を用いて発色させた。試料中の発色の光学密度をELISAプレートリーダー(Molecular Devices、USA)を用いて405nmの波長で測定した。実験の陰性対照は、同じ動物の免疫前の血清であり、その値をすべての実験値から引いた。HBV S1/S2−TBDで免疫したマウスに関する結果を表4に示す。このキメラ抗原は、キメラ抗原(S1/S2−TBD)に対する強い抗体応答を引き出す。
【0136】
【表4】
ここでの抗体応答は、多価(または多エピトープ)性のものである。表4に示す結果は、HBV S1/S2−TBDで免疫したマウスによって作られた抗体はキメラ抗原に結合し、かつプレート上に被覆されたS1/S2タンパク質標的に結合することを示す。したがって、抗体はキメラ抗原のS1/S2成分に対して作られる。同じように、HBV S1/S2−TBDで免疫したマウスにより作られた抗体は標的結合ドメインタンパク質に結合する(表4)。マウス起源のタンパク質を含有するキメラ抗体は、マウスに体液性免疫応答を生じさせ得、このキメラ抗原が「自己」抗原を「外来性」に変換し得るという証拠を示す。したがって、本来は「自己」タンパク質として処理されるタンパク質に対する寛容を壊すことが可能である。
【0137】
(2.T細胞増殖アッセイ)
四回目の免疫の一週間後に動物を屠殺し、脾臓を切除して単一の細胞懸濁液を作った。細胞を三重で、96ウエルプレートに4×105細胞/ウエルの細胞密度で撒いた。それらに、HBV S1/S2、TBDまたはS1/S2−TBDの各抗原を0.1μg/mL、1.0μg/mLおよび10μg/mLの濃度で加えた。陰性対照の細胞は、培地のみであり、T細胞増殖についての陽性対照は1.0〜5.0μg/mLのフィトヘマグルチニン(PHA)であった。細胞培養物を4日間37℃で、7%CO2の雰囲気下でインキュベートした。細胞の各ウエルに1.0mCiの3[H]チミジンを与え、さらに18時間インキュベートした。細胞を、TOMTECMACH3細胞ハーベスター(Hamden、CT、USA)を用いて採取し、ガラス繊維フィルター(Wallac Oy、Turku、フィンランド)に結合した放射活性を、Wallac Trilux 1450マイクロベータ液体シンチレーションおよび発光計数器(Wallac、USA)を用いて定量した。その結果を表5に示す。
【0138】
【表5】
T細胞増殖は、HBV S1/S2−TBD、S1/S2またはTBDで誘発されたときに見られた。このキメラ抗原による免疫は、多価のT細胞応答、すなわち同一タンパク質の異なる部分に対する応答を誘導した。マウス起源のタンパク質を含有するキメラ抗原は、マウスにおいて細胞性免疫応答を生じ得、キメラ抗原が「自己」抗原を「外来性」に変換し得るということを証拠付ける。したがって、本来は「自己」タンパク質として処置されるタンパク質に対する寛容を壊すことが可能である。
【0139】
(E.実施例5:抗原提示アッセイ)
HBV S1/S2−TBDの免疫応答を引き出す能力を、エキソビボ抗原提示アッセイを用いて測定した。樹状細胞(DC)等の抗原を負荷された抗原提示細胞(APC)によってナイーブT細胞の多刺激後に効果的なT細胞応答が発生することを、抗原特異的T細胞の数の増加ならびにTh1サイトカインIFN−γを産生するT細胞の能力を定量化することにより評価した。
【0140】
(1.接着による単球の選択)
末梢血液単核細胞(PBMC)にAIM−Vを加えて解凍した(1mlの凍結細胞に対して9mlの添加AIM−Vの比率)。次に、この細胞を200×gで5分間遠心分離し、上清を除去し、細胞を、AIM−V/1%調和血清に再懸濁し、100mm培養皿またはT−25培養フラスコに加えた。このPBMCを、7%CO2下の加湿インキュベーターで1時間、37℃でインキュベートした。非接着細胞を除去するために、培養物を数回粉砕し、上清を捨て、細胞をAIM−V培地で一回洗浄した。単球を細胞スクレーパーで採取し、300×gで5分間遠心分離した。細胞ペレットをAIM−V/2.5%調和血清に2×106細胞/mlで再懸濁し、24ウエルプレートに撒いた。サイトカインIL−4とGM−CSF(それぞれ1000IU/ml)を加えて、単球の未成熟DCへの分化を誘導した。
【0141】
(2.迅速または遅延抗原提示アッセイ)
迅速抗原提示アッセイ(APA)のために、抗原を、未成熟DCに単離の4〜24時間内に加えた。24時間後に、抗原を負荷した未成熟単球を、PGE2(1μM)、IL−1β(10ng/ml)およびTNF−α(10ng/ml)とともに24時間培養することにより誘導して成熟させた。次に、成熟DCを自己のT細胞とともに共培養した(一回目の刺激)。このT細胞は、磁気T細胞単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択を用いて、DCとして同一のPBMCからT細胞を作製された。
【0142】
次に、T細胞を、7日後にIL−2(20IU/ml)、IL−7(10ng/ml)およびIL−15(5ng/ml)の存在下で抗原を負荷した成熟DCにより再刺激した。7日間のインキュベーション後に、T細胞を抗原を負荷した成熟DCにより三回目の再刺激を行った。この三回目の刺激は6時間続け、この際にT細胞を採取し、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0143】
遅延APAのために、単球を、抗原の添加前の5〜6日間、GM−CSFおよびIL−4の存在下において未成熟DCに分化させた。抗原添加の二時間後に、未成熟DCをTNF−α(10ng/ml)およびIFN−α(50IU/ml)により成熟させた。単離の7日後に、成熟DCを(上記の)自己T細胞とともに共培養した(一回目の刺激)。
【0144】
次に、T細胞を、IL−2、IL−7およびIL−15の存在下において抗原を負荷した成熟DCによって7日後に再刺激した。7日間のインキュベーションの後、細胞を抗原を負荷した成熟DCにより三回目の再刺激を行った。18時間のインキュベーション後に、T細胞を集めて、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0145】
(2.PBMC抗原提示アッセイ)
このアッセイにおいて、最初の培養物は、抗原とIL−2とともにインキュベートされる全PBMC(すなわち、リンパ球および単球)からなり、ここではすべての細胞タイプが存在して関与するのでこの機構はインビボ免疫応答に似ていることが想定されている(Maini,M.K.ら、J.Exp.Med.191:1269−1280,2000)。PBMCを解凍し、洗浄し、すぐに抗原とともにインキュベートした。抗原の取り込みと提示とを可能にする4日間の培養後に、IL−2(20IU/ml)を加えて、さらに8日間放置した(すなわち実験の12日目)。二回目の刺激の2日前(すなわち実験の10日目)、DCを上記のように接着により単離し、すぐにGM−CSF、IL−4および抗原とともに24時間インキュベートする。Fast APAと同じように、未成熟DCを、PGE2、IL−1βおよびTNF−αの添加した後の24時間分化させる。次に、負荷した成熟DCを、IL−2、IL−7およびIL−15の存在下においてPBMC培養物に加える(二回目の刺激、実験の12日目)。三回目の刺激は、上記のように二日前に調製された抗原を負荷した成熟DCを用いて、実験の21日目に行った。6時間のインキュベーション後に、T細胞を集め、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0146】
上記のすべての抗原提示アッセイのために、アッセイの最後のT細胞の一部をさらに3〜5日間インキュベートし、テトラマー分析(下記を参照のこと)により特異的T細胞について調べた。
【0147】
(4.HBV S1/S2はHBV S1およびHBV S2のペプチドに対してT細胞応答を引き出す)
PBMC APAを用いてT細胞を作り、次にこれを抗原特異性に関して検定した。よって、健康なHLA−A2個体からのPBMCを、96ウエルプレートにて2.5%調和血清を含有するAIM−V中、5×105細胞/mlで培養した。抗原(すなわち10μg/ml S1/S2−TBD)を加え、その細胞を4日間、37℃で培養した。次に、IL−2を20IU/mlで加え、細胞を、2〜3日毎に培地(AIM−V/2.5%調和血清および20IU/ml IL−2)を交換しながらさらに8日間培養した。12日間の培養の終わりに残る細胞のほとんどはT細胞であり、これらのT細胞を、IL−2(20IU/ml)、IL−7(10ng/ml)およびIL−15(5ng/ml)の存在下において自己抗原を負荷した成熟DCにより再刺激した。
【0148】
APAにおいてT細胞の二回目と三回目の刺激のために、抗原を負荷した成熟DCを、下記の操作を用いて48時間にわたって作った。単球を、プラスチック組織培養皿上への接着により全PBMCから単離した。その細胞(そのうち、約85%がFACS分析により決定された単球である(CD11c+、CD14+、CD19−およびCD3−))を、96ウエルプレートで、1000IU/mlのサイトカインIL−4とGM−CSFとを含む100μlのAIM−V/2.5%調和血清を含む1×105細胞/ウエルで培養し、4時間後、S1/S2−TBD等の抗原を加えた。20時間のインキュベーション後に、作られた未成熟DCを、PGE2(1×10−6M)、IL−1β(10ng/ml)およびTNF−α(10ng/ml)の存在下でさらに24時間培養することにより成熟DCに分化させた。
【0149】
T細胞を、1〜2日毎に培地(2.5%調和血清と20IU/ml IL−2を有するAIM V)を交換しながら、二回目の刺激後の7日間培養した。次に、T細胞(培養の19日目)を(上記の)IL−2、IL−7およびIL−15の存在下において(上記のように2日間の操作にわたって作られた)抗原を負荷した成熟DCによる三回目の刺激を行い、そして6時間培養後のIFN−γ産生を検定するか、または(1〜2日毎にAIM−V/2.5%調和血清および20IU/ml IL−2の培地を変更しながら)5日間培養し、次にHBV preSテトラマーを用いてHBV preS抗原に対するT細胞特異性に関する検定のいずれかを行った(培養の24日目)。
【0150】
カスタム合成iTag MHCクラスIテトラマー(Beckman Coulter)を製造者のプロトコールに従って用いてテトラマー分析を行った。すなわち、細胞を採取し、洗浄し、96ウエルv底プレートに、20μl中約2×105細胞/ウエルで移した。2μlのPE複合HLA−A*0201 preS1テトラマー(GMLTPVSTI、配列番号50)またはpreS2テトラマー(NIASHISSI、配列番号51)のどちらかとともに、CD3(抗CD3−FITC)およびCD8(抗CD8−Cy−Chrome)に特異的なmAbによって、その細胞を20℃で30分間標識した。次に、細胞を洗浄し、PBS中の2%パラホルムアルデヒドで固定し、5mlのFACSチューブに移した。この細胞を、一試料あたり80,000〜100,000個の細胞でFACSCalibur(BD Biosciences)上に得た。生存(FSC/SSCプロフィールに基づく)CD3+集団上のゲートにより、CellQuestソフトウエア(BD Biosciences)を用いて分析を行い、テトラマーによるCD8+細胞標識の比率を決定した。PBMCを、10μg/mlのHBV S1/S2−TBDとともに培養し、HBV S1/S2−TBDを負荷した成熟DCにより二回再刺激した場合に、顕著な比率の細胞をS1テトラマー(図2)およびS2テトラマー(図3)によって陽性標識した。これは、テトラマー陽性細胞の数が顕著でない場合、抗原を負荷していない成熟DCとともに培養されたT細胞とは対照的である。よって、S1/S2−TBDで負荷したDCは、HBV S1およびHBV S2抗原の決定基に特異性を有する顕著な数のT細胞の生成を誘導することができた。
【0151】
(F.実施例6:キメラ抗原融合タンパク質を用いるDHBV抗原およびDHBV抗原に対する寛容の破壊)
DHBVは、HBVに対する抗ウイルス療法を開発する際の強力な動物模型として役立っている。ウイルスの複製機構の研究を行い、抗ウイルス化合物のスクリーニングのために、DHBVに先天的に感染させた北京ダック(アヒル)を用いた。二種類のアヒル模型を本発明に用いた。最初のものは先天的にDHBVに感染させたアヒルである。これはヒトにおけるHBV感染の垂直感染に似ている。第二の模型は、新しく孵化したアヒルの子がDHBVに感染し、これらが感染を有するという持続的な感染模型である。この第二の模型はヒトにおけるHBV感染の水平感染に似ている。
【0152】
(1.先天的にDHBVに感染させたアヒル)
四週齢の先天的にDHBVに感染させたアヒルを二つの集団に分けた。血液試料(1.0mL)を免疫前抗体量の参照のために集めて、ワクチン接種の前に、血液試料を毎週集めた。実験集団は、DHBV Core TBDキメラ抗原融合タンパク質を19.95mg/用量で5週目まで毎週同じ曜日に皮下注射した。6週目は、用量を二倍として、ワクチン接種を26週目に停止するまで、四週毎に一回注射した。プラセーボ集団には等量の緩衝液(20mMリン酸ナトリウム、pH8.0、300mM NaCl)を投与した。
【0153】
DHBV Core、TBDまたはDHBV Core TBDの各抗原を1.0μg/mlの濃度で一晩4℃にて96ウエルプレートに被覆した。このプレートを、2%BSAを含有するリン酸緩衝性食塩水(PBS)で洗浄した。各動物からの希釈血清を各ウエルに多様な希釈度(1:10〜1:500)で加え、37℃で1時間インキュベートした。このプレートを、0.05%のTween20を含有するPBS(洗浄緩衝液)で洗浄した。ヤギ抗アヒルIgG−HRP(1:5000)希釈物をウエルに加え、37℃で1時間インキュベートした。プレートを洗浄緩衝液で洗浄し、2,2’アジノ−ジ−(3−エチルベンジルチアゾリン−6−スルフォネート)(KPL、Guildford、UK)を用いて発色させた。試料中の発色の光学密度をELISAプレートリーダー(Molecular Devices、USA)を用いて測定した。抗体力価は同一動物からの免疫前血清に対して計算した。
【0154】
0週目、3週目および6週目のアヒルの対照集団と実験集団とにおける、先天的にDHBVに感染させたアヒル由来の血清中の抗コア抗体レベルを表6に示す。アヒルは慢性DHBV感染を有するが、感染の慢性的性質と抗原を外来分子として認識しない免疫系に起因して、抗体レベルは低い。DHBV Core−TBDキメラ抗原による免疫を行うと、宿主の免疫系はウイルス抗原を認識し、宿主に既に存在するコア抗原に対して体液性応答を開始し、ウイルス抗原に対する宿主寛容を壊した。
【0155】
【表6】
同様に、アヒルの免疫系は、キメラ抗原のTBD成分を外来抗原として認識し、融合タンパク質のこの部分に対する免疫応答も同じく生じさせた。プレートをTBDで被覆し、個々のアヒル由来の血清をELISAによる抗体レベルについて評価した。この研究結果を表6に示す。
【0156】
(2.孵化後にDHBV感染したアヒル)
正常な子アヒルに、子アヒルが孵化した後の日に、DHBV含有アヒル血清を感染させた。これはDHBV研究の分野での標準的な操作である。確立された技術を用いて、持続的なウイルス血症の存在を免疫開始の四週前に確認した。DHBV感染アヒルを二つの集団に分けた。血液(1.0mL)の試料を免疫前抗体レベルの参照のために各アヒルより集めて、ワクチン接種の前に、血液試料を毎週集めた。実験集団はDHBV Core−TBDキメラ抗原融合タンパク質を19.95μg/用量で5週目まで毎週同じ曜日に皮下注射した。6週目、用量を二倍として、ワクチン接種を30週目に停止するまで、四週毎に一回注射した。血液試料は、等量の緩衝液(20mMリン酸ナトリウム、pH8.0、300mM NaCl)を投与したプラセーボ集団から集めた。
【0157】
0週目、3週目および6週目にアヒルから集めた血清の抗体レベルを示す。アヒルの対照集団と実験集団とのにおける、孵化後DHBV感染アヒル由来の血清中の抗コア抗体レベルを表7に示す。DHBVは持続的感染を確立しているために、免疫系はウイルス抗原を外来分子として認識せず抗体レベルは低い。DHBV Core−TBDキメラ抗原による免疫を行うと、宿主の免疫系はウイルス抗原を認識し、宿主に既に存在するコア抗原に対して体液性応答を開始し、ウイルス抗原に対する宿主寛容を破壊した。TBDに対する抗体レベルも増加した(表7)。したがって、同一のキメラ抗原の異なる部分に対する多価(または多エピトープ性)免疫応答が存在する。
【0158】
【表7】
(G.実施例7:化学架橋HBV sAg−Fc(マウス))
100μgのsAg(US Biologicals;カタログ番号H 1910−27)および100μgのマウスポリクローナルIgG Fcフラグメント(Harlan Sera−Lab Ltd.、カタログ番号PP−19−01)の溶液を、100mMのHEPES、pH8.7に対して一晩4℃で透析した。タンパク質溶液を混合して一緒にして、すぐにジメチルスベリミデート(DMS;Pierceカタログ番号20700)を10mMの最終濃度にて加え、この混合物を室温で1時間インキュベートした。この反応を、0.1MトリスHCl、pH7.8を加えることで停止させた。反応混合物をSephadex G 75カラム(0.7×12cm)にかけて、画分をリン酸緩衝性食塩水を用いて溶出させた。0.5ml画分を集め、各抗体を用いたELISAにより予測してsAg/Fcを1:1のモル比で含有する画分をプールした。
【0159】
プールした画分を抗原提示アッセイに用いた(Berlynら、Clin.Immunol.101:276−283(2001))。未成熟樹状細胞を、GM−CSF/IL4とともに4日間培養し、sAg−Fc複合体とともにインキュベートし、TNFαとインターフェロンαとの存在下において成熟させた。自己CD3+T細胞を成熟樹状細胞に加えた。成熟樹状細胞に対して3回曝した後に、T細胞刺激を、フローサイトメトリーを用いて細胞内インターフェロンγの産生を測定することにより定量した。複合体の存在下においてT細胞中に産生された細胞内インターフェロンγのレベルは、sAgまたはFcフラグメントのみの存在下におけるよりも実質的に高かった(表8)。
【0160】
【表8】
(H.実施例8:抗原提示アッセイ)
ヒトPBMC由来の樹状細胞を確立されたプロトコール(Berlynら(前出)(2001))に従って用いることで抗原提示アッセイを実施した。T細胞刺激アッセイのプロトコールの要約をスキームの形で示す。
【0161】
【化1】
(1.成熟した負荷樹状細胞の調製)
健康なドナーからの白血球搬出の試料から単球を作り、抗CD2抗体、抗CD7抗体、抗CD16抗体、抗CD19抗体および抗CD56抗体とともにインキュベートすることによりリンパ球と顆粒球とを枯渇させた。これに続いて、磁気ビーズ複合抗マウスIgGとのインキュベーションと磁石(Dynal)による分離を行った。ネガティブ選択細胞は、広いCDマーカーパネル(CD14+、CD11c+、CD19−、CD3−、CD4−、CD64+、CD32+、CD86+、CD16−)を用いたフローサイトメトリーにより特性付けられたように95%を超える純度の単球であった。次に、単球を、IL−4とGM−CSF(R&D Systems)とともに、2.5%調和ヒト血清を加えたAIM V中で4日間インキュベートして、未成熟樹状細胞を作った。再び、細胞のアリコートを、細胞の純度と同一性を保証する広いCDマーカーパネルにより染色した。次に、細胞に、HBV S1/S2−TBD(5.0μg/ml)、HBV S1/S2(2.5μg/ml)またはTBD(2.5μg/ml)を2〜4時間37℃で負荷し、インターフェロンαとTNF−αとにより3日間成熟させた。細胞が適切な成熟を受けたことを保証するために、再びフローサイトメトリーを用いてCDマーカーのアレイに対する樹状細胞の確認を行った。得られた成熟した負荷樹状細胞をT細胞刺激アッセイに用いた。
【0162】
(2.T細胞刺激アッセイ:サイトカイン分析)
樹状細胞としてPMBCの同一の試料から、磁気T細胞単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択により、T細胞を生じさせた。成熟した負荷樹状細胞(DC−1)を完全に洗浄し、T細胞に加えた(0日目)。T細胞および樹状細胞を7日間インキュベートした。7日目に、このT細胞を、成熟した負荷樹状細胞(DC−2)により再刺激した。細胞のアリコートを二時間後に採取した。この細胞のアリコートを、ブレフェルジンA(GolgiPlug(登録商標)、R&D Systems)とともに18時間インキュベートし、次に下記の細胞内サイトカイン染色についてアッセイした。
【0163】
残っている細胞をさらに7日間インキュベートした。14日目に、残っている細胞を、第三群の成熟した負荷樹状細胞(DC−3)で刺激した。細胞のアリコートを二時間後に採取した。この細胞のアリコートをブレフェルジンA(GolgiPlug(登録商標)、R&D Systems)とともに18時間インキュベートし、次に下記の細胞内サイトカイン染色についてアッセイした。
【0164】
細胞内サイトカイン染色のために、細胞を抗CD3−FITCと抗CD8−Cy−Chromeとで30分間染色し、洗浄し、固定し、浸透化処理し、次に抗インターフェロン−γ−PEで30分間氷上にて染色した。細胞を洗浄し、フローサイトメトリー(FACScan、BD Biosciences)で分析した。この結果を表9に示す。
【0165】
【表9】
14日目にアリコートを除去した後、残っているT細胞をさらに3日間インキュベートし、次にその上清を用いてELISA(Opt E1A ELISAキット、BD Biosciences)により分泌インターフェロンγのレベルを測定した。T細胞刺激を、細胞内インターフェロンγおよび分泌インターフェロンγのレベルを測定することにより評価した。この結果を表10に示す。キメラ抗原S1/S2−TBDは、単独で試験した場合、分子の免疫応答ドメインまたはTBDドメインのいずれかと比較して、等濃度においてより高い量のインターフェロンγの産生を引き起こした。S1/S2−TBDの5μgの用量は、おおよそ上記成分のそれぞれを2.5μg含むことを指摘しておかなければならない。
【0166】
【表10】
T細胞応答に関して、多様な濃度のS1/S2−TBDを調べた。S1/S2−TBDの効果は、同様な濃度において破傷風トキソイド治療よりも高かった。5μg/mLよりも低い濃度において、キメラ抗原はインターフェロンγの産生および分泌の濃度依存的増加を引き出した。CD3+T細胞によるインターフェロンγの産生および分泌は、表11に示されるように、樹状細胞によるS1/S2−TBD抗原の提示後に、濃度依存的様式で増加した。低濃度での陽性応答は、ワクチン接種に必要な用量およびワクチンの製造費用に関して有利である。
【0167】
【表11】
(I.実施例9:キメラ抗原の結合および取り込み)
(1.成熟した負荷樹状細胞の調製)
末梢血液単核細胞(PBMC)を、白血球搬出細胞調製物のFicoll/Histopaque(Sigma)処理から得た(Berlynら(前出)(2001))。単球単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択により、単球をPBMC集団から分離した。単球は、抗体分析およびフローサイトメトリー(CD14+、CD11c+、CD19−、CD3−、CD4−、CD64+、CD32+、CD86+、CD16−)による検定で95%を超える純度であった。単球を、1%のドナー調和血清(Berlynら(前出)(2001)による記載のように単離されたもの)を含む、L−グルタミン、硫酸ストレプトマイシン(50μg/mL)および硫酸ゲンタマイシン(10μg/mL)を含むAIM−V(Invitrogen)培地で二回洗浄した。次に、この単球を、2.5%ドナー調和血清とサイトカインGM−CSFとIL−4とを含むAIM−V培地中で培養し、細胞を樹状細胞(DC)系統へと分化させた。この細胞を12ウエル組織培養皿中、37℃、7%CO2雰囲気でインキュベートした。
【0168】
単球由来の樹状細胞を1日目〜4日目に集めた。次に細胞を、0.1%BSA(Sigma)を含むAIM−V培地で一回洗浄し、0.1%(w/v)BSA(PBSB)を含むダルベッコリン酸緩衝性食塩水(Invitrogen)で二回洗浄した。単球由来の樹状細胞を4℃の標識または結合アッセイまたは37℃の結合/取り込みアッセイに用いた。
【0169】
(2.キメラ抗原の成熟樹状細胞への結合)
マウスIgG1およびIgG2aに対してS1/S2−TBDの成熟樹状細胞への結合の程度を比較した。樹状細胞を、エキソビボ培養(0日目〜4日目)の種々の日に単離し、S1/S2−TBD(10μg/mL)、またはマウスIgG1(2C12、TBDが生産された親mAb)もしくはIgG2a(G155−178、90μg/mL)で、樹状細胞を1時間、4℃で処理した。この細胞を、PBSB中のF(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)で20分間処理した。細胞をPBSBで二回洗浄し、2%パラホルムアルデヒド(PF)を含むPBSBに再懸濁し、CellQuest獲得分析(acquisition and analysis)ソフトウエア(BD)に適合させたBecton Dickinson(BD)FACScanにより得た。FSCとSSC分散プロフィールにより決定された生存細胞集団上にゲートを作り、≧10,000細胞を得た。陽性細胞の比率を決定するために、ゲートを、陰性対照処置細胞(F(ab’)2ヤギ抗マウスAlexa−488のみで標識したアイソタイプ対照または細胞)に基づいて設定した。特異的な陽性細胞の比率は下記のように計算した:
【0170】
【化2】
相対平均蛍光強度(MFI)を、試験試料のMFIから対照試料のMFIを引いたものとして決定した。
【0171】
培養1日〜4日後のDC上のIgG1およびIgG2aに比較したS1/S2−TBDの結合を表12に示す。
【0172】
【表12】
S1/S2−TBD結合は、IgG1またはIgG2aのいずれか結合よりも明らかにかなり高く、さらにS1/S2−TBD結合は4日目よりも1日目で明らかであった。これらの実験は、S1/S2−TBDは成熟樹状細胞に対して高い効率で結合したことを示した。
【0173】
(3.成熟樹状細胞へのキメラ抗原の取り込み)
IgG1およびIgG2aに比較した場合のキメラ抗原(例えば、HBV S1/S2−TBD)の取り込みの程度を決定するために、0.1%BSAを含むAIM V培地中で1時間、37℃で多様な濃度の抗原IgG1(2C12、TBDが生産された親mAb)またはIgG2a(G155−178)と共に細胞をインキュベートした。細胞をPBSBで二回洗浄し、2%PFを含むPBSで一晩4℃にて固定した。続いて、この細胞をPBSBで二回洗浄し、0.1%(w/v)サポニン(Sigma)を含有するPBSで40分間、20℃にて浸透化処理した。
【0174】
細胞をPBSBで二回洗浄し、0.1%(w/v)サポニンを含有するPBSB中のF(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)と共に20分間、4℃にてインキュベートした。PBSBで二回洗浄した後、細胞をPBSBに再懸濁した。このアッセイの変法では、細胞を、キメラ抗原、IgG1またはIgG2aにより10分間上記のように処理した後に、50分間F(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)を加えた。続いて、細胞を洗浄し、2%PFを含むPBSに再懸濁した。
【0175】
Cellquest獲得分析ソフトウエア(BD)で適合されたBecton Dickinson(BD)FACScanにより細胞を得た。FSCおよびSSCの分散プロフィールにより決定された生存細胞集団上にゲートを作り、≧10,000細胞を得た。陽性細胞の比率を決定するために、ゲートを陰性対照処置細胞(F(ab’)2ヤギ抗マウスAlexa−488のみで標識されたアイソタイプ対照または細胞)に基づいて設定した。特異的な陽性細胞の比率は下記のように計算した:
【0176】
【化3】
相対平均蛍光強度(MFI)は、試験試料のMFIから対照試料のMFIを引いたものとして決定した。
【0177】
マウスIgG1およびIgG2aと比較したS1/S2−TBDの取り込みを、樹状細胞の成熟の4日目の濃度の関数と予測した。取り込みを37℃で1時間定量し、その結果を図4に示す。濃度とともにS1/S2−TBDの取り込みは直線的に増加した。IgG1はかなり低いレベルで取り込まれ、IgG2aの取り込みはほとんどなかった。したがって、キメラ抗原S1/S2−TBDは、免疫グロブリンよりもさらに効率的に樹状細胞に取り込まれる。
【0178】
(J.実施例10:成熟DC上でのFc−γレセプターおよびCD206の発現)
抗原に結合し、これを取り込む抗原提示細胞上にいくつかのレセプターが存在する。成熟樹状細胞上のこれらレセプターの量を、蛍光標識レセプターに特異的な抗体を用いて評価した。FACS分析を用いて、樹状細胞の全集団における、特異的レセプターに陽性な細胞の比率を予測した。レセプター発現の程度は、相対的平均蛍光強度の測定により、そして相対的蛍光強度の関数として評価した(表13)。
【0179】
【表13】
CD64(FcγレセプターI)の発現は培養時間とともに減少し、4日目にほとんど無視できた。対照的に、CD32(FcγレセプターII)、およびさらに低い程度でCD16(FcγレセプターIII)は、DC培養の4日後まで発現し続けた。培養の0日目では、CD206(マンノースマクロファージレセプター)発現は基本的に存在しなかった。しかし、発現はIL−4とGM−CSFとの培養により誘導され、4日目までに、CD206は非常に高いレベルで発現した。よって、4日目に抗原を抗原提示アッセイにかけたときに、樹状細胞は、キメラ抗原の結合のための少なくとも二つの潜在的なレセプター(CD32およびCD206)を有した。さらに、それらのレセプターは共刺激分子の完全な補体を有した(データは示さず)。HLA−DR(クラスII)およびHLA−ABC(クラスI)の発現も培養時間とともに増加した。共刺激分子のCD86(B7.2)およびCD80(B7.1)がアッセイの間中発現した。これらの結果は、単球由来の樹状細胞が成熟樹状細胞に分化し、これらが抗原処理とT細胞への提示をし得ることを示す。
【0180】
(K.実施例11:CD32/CD206発現と成熟DCに対するS1/S2−TBD結合との相関)
CD32/CD206レセプターの発現と成熟樹状細胞へのS1/S2−TBD結合との間に直接の相関が存在する。マウスIgG1は、ヒトCD32へ結合することが公知であるので、IgG1のマウスFc成分を含むS1/S2−TBDはCD32にも結合することが予測された。さらに、その高いマンノースグリコシル化のために、S1/S2−TBDが、CD206レセプターを経て樹状細胞に結合することもまた予測される。
【0181】
図5のドットプロットは、S1/S2−TBD結合(10μg/mL)およびCD32発現、ならびにS1/S2−TBD結合およびCD206発現を示す。S1/S2−TBD結合の程度とCD32発現の程度(この発現は比較的不均一であり、すなわち広い程度の発現が存在した)との間には直接の相関があった。これらの結果は、S1/S2−TBDはCD32に結合することを示し、CD32の発現が高いほど、キメラ抗原S1/S2−TBDの結合の程度が高いことを示す。S1/S2−TBD結合とCD206発現とのドットプロットは、CD206を発現するほとんど大部分の細胞が、S1/S2−TBDにも結合したことを示す。この細胞集団の低い割合でCD206陰性であり、したがってS1/S2−TBD結合に対して陰性であった。したがって、CD32とCD206との両方のレセプターがS1/S2−TBDの結合に相関する。
【0182】
(L.実施例12:S1/S2−TBDの結合および取り込みは、主にCD32によるものであり、CD206はそれよりも低い程度で関与する)
マウスIgG1およびIgG2aと比較したS1/S2−TBDの取り込みは、DC成熟の4日目の濃度の関数と予測した。この取り込みを、37℃で1時間、培地、マンナン(2mg/ml、Sigma)および/またはマウスFcγ(2mg/ml、Jackson ImmunoResearch Laboratories)の存在下において定量した。マンナンはCD206結合の競合的インヒビターであり、よって樹状細胞上のCD206による抗原の取り込みのインヒビターである。FcγはCD32結合の競合的インヒビターであり、よってCD32が媒介する抗原取り込みのインヒビターである。この結果を表14に示す。
【0183】
【表14】
濃度とともにキメラ抗原の結合の漸進的な増加が存在した。高い濃度のマウスFcγフラグメントとの細胞のインキュベーションは、この結合を無効にしたが、CD206レセプター結合のインヒビターであるマンナンは些細な効果を有するのみであった。したがって、CD32は、キメラ抗原の結合と取り込みとに関わる主要なレセプターであり得る。
【0184】
(M.実施例13:HBV S1/S2抗原のグリコシル化は免疫原性を付与する)
昆虫細胞で合成されたタンパク質が、分泌タンパク質において高マンノース含量および末端シアル酸残基の欠如をもたらすグリコシル化を受ける点で、タンパク質グリコシル化の昆虫細胞経路は哺乳動物細胞のものとは異なる(Altmanら、Glycoconjug 16:109−123(1999))。キメラ抗原の抗原成分であるHBV S1/S2は、E.coli(グリコシル化なし)とHigh Five(登録商標)昆虫細胞(マンノールグリコシル化)との両方で発現させた。
【0185】
(1.抗原結合に対するグリコシル化の効果)
樹状細胞に対する結合に関して、これらの抗原を実施例9に記載のように比較した。成熟樹状細胞に、昆虫細胞またはE.coli.で発現した10μg/mlのHBV S1/S2を負荷した。グリコシル化タンパク質は樹状細胞による良好な結合を示した(表15)。
【0186】
【表15】
(2.免疫応答を引き出すことに対するグリコシル化の効果)
HBV S1/S2のグリコシル化は、増強された免疫原性とT細胞応答とを引き出す。樹状細胞により提示された場合のT細胞応答について、E.coliとHigh Five(登録商標)昆虫細胞との両方で発現させたHBV S1/S2を比較した。実施例8に記載されたように(2.5μg/ml HBV S1/S2タンパク質を用いて)細胞内インターフェロンγと分泌インターフェロンγのレベルを測定し、その結果を表16に示す。
【0187】
【表16】
昆虫細胞で発現したHBV S1/S2は、E.coli.で発現したグリコシル化されていないタンパク質と比較して、細胞内インターフェロンと分泌インターフェロンとの両方をより高いレベルで生じた。
【0188】
上記明細書に示したすべての公刊物および特許は、本明細書において参考として援用される。本発明の記載された方法と系の多様な改良および変化物は、当業者にとって本発明の精神と範囲から離れることなく明らかであろう。本発明は特定の実施態様との関連において記載されているが、本発明の特許請求の範囲は、そのような特定の実施態様に過度に限定すべきではないことが理解されるべきである。実際、本発明を実施するための記載された態様の多様な改変は、当業者に明らかであり、それらの改変は、以下の請求項の範囲の内にあることが意図される。
【0189】
【化4】
【0190】
【化5】
【0191】
【化6】
【0192】
【化7】
【0193】
【化8】
【0194】
【化9】
【0195】
【化10】
【0196】
【化11】
【0197】
【化12】
【0198】
【化13】
【図面の簡単な説明】
【0199】
【図1A】図1Aは、本発明のキメラ抗原の単量体としての構造を示す略図であり、このキメラ抗原は二つの部分(すなわち、免疫応答ドメインおよび標的結合ドメイン)を有する。この略図はまた、ヒンジ領域が存在する好ましい実施態様も示している。
【図1B】図1Bは、二量体として組み立てられた正常な状態での本発明のキメラ抗原の構造を示す略図である。この略図は、キメラ抗原が免疫応答ドメインと標的結合とに加えて6×Hisタグおよびペプチドリンカーを含む特に好ましい実施態様を示す。
【図2】図2は、HBV S1/S2−TBDによるT細胞の刺激が、HBV S1タンパク質由来のエピトープに対する細胞障害性T細胞(CTL)応答を特異的に発生させることを示す。
【図3】図3は、HBV S1/S2−TBDによるT細胞の刺激が、HBV S2タンパク質由来のエピトープに対する細胞障害性T細胞(CTL)応答を特異的に発生させることを示す。
【図4】図4は、樹状細胞を成熟させることによるHBV S1/S2−TBD、IgG1およびIgG2の取り込みの比較を濃度の関数として示す。
【図5】図5は、CD32へのHBV S1/S2−TBDの結合と樹状細胞上のCD206発現との相関を示す。
【技術分野】
【0001】
(導入)
(A.関連出願)
本出願は、米国仮出願第60/493,449号(2003年8月8日出願)の優先権を主張し、米国仮出願第60/493,449号は、本明細書中に参考として援用される。
【0002】
(B.技術分野)
本発明は、免疫応答を引き出すか、または増強させるための方法および組成物、ならびに外来抗原に対して宿主寛容を壊すための方法および組成物に関する。
【背景技術】
【0003】
(C.背景)
健康な宿主(ヒトまたは動物)が、細菌、ウイルスおよび/または寄生生物由来のタンパク質等の外来抗原に出会うと、宿主は通常は免疫応答を開始する。この免疫応答は体液性応答および/または細胞性免疫応答でありうる。体液性応答において、抗体は、B細胞により作られ、抗原刺激に対して血液および/またはリンパに分泌される。次に、抗体は、抗原、例えばウイルスを、その表面上の抗原に特異的に結合して、食作用細胞および/もしくは補体仲介機構による破壊のためにそれをマークすることによって、または結合をブロックするか、もしくは循環からの遊離抗原の排除を増強することによって、中和する。細胞性応答は、抗原を含有する細胞を直接または間接的に除去することのできる特定のヘルパーT細胞および細胞障害性Tリンパ球の選択と増殖により特性付けられる。
【0004】
一部の個体において、免疫系はある種の外来抗原に応答できない。抗原が特定の抗体および/またはキラーT細胞の産生を刺激しない場合、免疫系は生じる病気を防止できない。よって、感染因子、例えば、ウイルスは、慢性感染を確立させることができ、宿主免疫系は感染因子により作られる抗原に対して寛容があるものとなる。
【0005】
感染因子が宿主免疫機構を侵す機構は明確には確立されていないが、宿主免疫系への適当な外来抗原の提示の欠如が慢性的な感染の進展に寄与する因子であろう。抗原提示細胞(APC)は、抗原の局在性に基づきながら出会った抗原を種々に処理する。外因性の抗原はエンドサイトーシスを受け、引き続いて抗原提示細胞のエンドソーム内で処理される。外因性の抗原から生じたペプチドフラグメントは主要組織適合複合体(MHC)クラスIIと複合した細胞表面に提示される。この複合体のCD4+T細胞への提示は、CD4+Tヘルパー細胞を刺激して、外因性の抗原に対して抗体を産生するB細胞を刺激するサイトカインを分泌させる(体液性応答)。一方、細胞内抗原は処理され、抗原提示細胞の表面上のMHCクラスIとの複合体として提示される。CD8+T細胞への抗原提示は、抗原を有する宿主細胞に対する細胞障害性T細胞(CTL)の免疫応答をもたらす。
【0006】
ウイルスまたは寄生生物の慢性的な感染を有する被験体において(この生物は生活環中のある時点で宿主細胞内に住む)、抗原は宿主細胞により作られ、宿主細胞中に発現され、そして分泌された抗原は、循環中に存在する。例として、慢性的なヒトB型肝炎ウイルス(HBV)キャリアの場合、ビリオンおよびHBV表面抗原および(e抗原の形態の)コア抗原の代用物が、血液中で検出され得る。
【0007】
慢性的な感染の効果的な治療は、感染因子に関連した抗原に対する強いCTL応答を必要とする。これは、抗原を宿主細胞内に作ることによるか、または抗原を適切な細胞区分に送達して、細胞応答を引き出すように抗原が処理および提示されることによって達成することができる。抗原を細胞内に運ぶために、いくつかのアプローチが文献化されている。これらのうち、ウイルスベクター(非特許文献1)、cDNAトランスフェクト細胞の使用(非特許文献2)、および注入cDNAベクターによる抗原の発現(非特許文献3);ならびに特許文献1)が文献化されている。さらに、樹状細胞を標的とする抗原を発現させるDNAワクチンが記載されている(非特許文献4)。
【0008】
MHCクラスI経路プロセシングのために、細胞の細胞質ゾル区画まで抗原を運ぶことができる輸送ビヒクルもまた、利用されてきた。Hilgersら(非特許文献5)は、同目標を達成するアジュバントの使用を詳細に説明している。別のアプローチは、オボアルブミンペプチドに対するTh1免疫応答の発生により例示される、抗原の細胞質輸送のための生分解性ミクロスフェアの使用である(非特許文献6;および非特許文献7)。さらに、抗原提示細胞、例えば樹状細胞はPLGAナノスフェアを取り込む(非特許文献8)。
【0009】
抗原を捕捉し、処理し、そして提示し、かつナイーブT細胞を刺激する樹状細胞の能力から、樹状細胞は治療用ワクチンの開発のために非常に重要な手段となっている(非特許文献9)。抗原を樹状細胞の標的とすることは、抗原提示において重大な工程であり、抗体のFc領域に関して樹状細胞上のいくつかのレセプターの存在が、この目的のために利用されている(非特許文献10)。このアプローチのさらなる例として、卵巣癌Mab−B43.13、抗PSA抗体ならびに抗HBV抗体抗原の複合体が挙げられる(非特許文献11)。腫瘍関連抗原を負荷した樹状細胞を用いる癌免疫治療は、腫瘍に特異的な免疫応答と抗腫瘍活性とを作ることが示されている(非特許文献12;および非特許文献13)。腫瘍抗原のパルスを受けた樹状細胞を用いたインビボ臨床試験で有望な結果が得られた(非特許文献14)。これらの研究は、癌抗原に対して免疫応答を作るために樹状細胞を用いる効能を明らかに示している。
【0010】
抗原提示はまた、抗原提示細胞上のFcレセプターの利用の代わり、またはそれに加えてマンノースレセプターによって影響を受けることもできる。CD206としても公知のマクロファージマンノースレセプター(MMR)は、樹状細胞(DC)等の抗原提示細胞上に発現する。この分子は、エンドサイトーシスレセプターのC型レクチンファミリーのメンバーである。マンノシル化抗原は、CD206によって、結合し得、そして内在化され得る。一般的に、外因性の抗原は主にMHCクラスII経路により処理、提示されると考えられる。しかし、CD206を経た標的化の場合、MHCクラスIとクラスIIとの両方の経路が関わっているとの証拠が存在する(非特許文献15;非特許文献16;非特許文献17)。
【特許文献1】米国特許第5,589,466号明細書
【非特許文献1】Lorenzら、Hum.Gen.Ther.10:623−631(1999)
【非特許文献2】Donnellyら、Ann.Rev.Immunol.15:617(1997)
【非特許文献3】Laiら、Crit.Rev.Immunol.18:449−484(1988)
【非特許文献4】Youら、Cancer Res 61:3704−3711(2001)
【非特許文献5】Hilgersら、Vaccine 17:219−228(1999)
【非特許文献6】Newmanら、J.Control Release 54:49−59(1998)
【非特許文献7】Newmanら、J Biomed Mater Res 50:591−597(2000)
【非特許文献8】Newmanら、J.Biomed Mater Res 60:480−486(2002)
【非特許文献9】Laupezeら、Hum Immunol 60:591−597(1999)
【非特許文献10】Regnaultら、J Exp Med 189:371−380(1999)
【非特許文献11】Wenら、Int Rev Immunol 18:251−258(1999)
【非特許文献12】FongおよびEngleman、Ann Rev Immunol 96:1865−1972(2000)
【非特許文献13】Camptonら、J Invest Dermatol 115:57−61(2000)
【非特許文献14】TarteおよびKlein、Leukemia 13:653−663(1999)
【非特許文献15】Apostolopoulosら、Eur.J.Immunol.30:1714(2000)
【非特許文献16】ApostolopoulosおよびMcKenzie、Curr.Mol.Med.1:469(2001)
【非特許文献17】Ramakrishnaら、J.Immunol.172:2845−2852(2004)
【発明の開示】
【発明が解決しようとする課題】
【0011】
感染性の病気および癌は、大きな社会的健康問題である。例えば、世界保健機関の統計は、20億を超える人々がHBVに感染していることを示す。これらのうち、3億7千万人が慢性感染であり、その結果、肝臓の肝硬変および肝臓細胞の癌腫の発症の高い確率を有する。世界中の約1億7千万の人々がHCVの慢性キャリアであり、それに対する効果的な予防ワクチンまたは治療ワクチンは存在しない。世界保健機関は、毎年1千万の人々が癌であると診断されていると報告している。癌は毎年、世界中の死の12%にあたる6百万人の死を引き起こす。よって、感染および癌に対して免疫応答を引き出すための治療的に有効な新しい組成物および方法、ならびにこのような組成物を生成するための方法に対する必要性が存在する。
【課題を解決するための手段】
【0012】
(発明の要旨)
本発明は、免疫応答を引き出すためのキメラ抗原を提供し、このキメラ抗原は、免疫応答ドメインおよび標的結合ドメインを含み、上記標的結合ドメインは、抗体フラグメントを含む。
【0013】
本発明のもう一つの特徴は、抗原提示細胞において抗原提示を増強する方法であって、上記抗原提示細胞を本発明のキメラ抗原を含む組成物と接触させる工程を包含する方法を提供する。
【0014】
本発明のさらにもう一つの特徴は、抗原提示細胞を活性化する方法であって、上記抗原提示細胞を本発明のキメラ抗原に接触させる工程を包含する方法を提供する。
【0015】
本発明の一つの特徴は、免疫応答を引き出す方法であって、本発明のキメラ抗原を含有する組成物を被験体に投与する工程を包含する方法を提供する。
【0016】
本発明のもう一つの特徴は、寛容を壊す方法であって、本発明のキメラ抗原を被験体に投与する工程を包含する方法を提供する。好ましい実施態様において、上記被験体は、ウイルスまたは偏性細胞内寄生生物に慢性感染している。
【0017】
本発明の一つの特徴は、免疫治療が可能な症状を治療する方法であって、治療的有効量の本発明のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する方法を提供する。好ましい実施態様において、免疫治療が可能な症状は感染、特に慢性感染または癌である。
【0018】
本発明のさらにもう一つの特徴は、感染に対して被験体をワクチン接種する方法であって、本発明のキメラ抗原を被験体に投与する工程を包含する方法を提供する。上記被験体は、予防的または治療的にワクチン接種され得る。好ましい実施態様において、上記被験体は、キメラ抗原の二つ以上のエピトープ、そしてより好ましくは免疫応答ドメインの二つ以上のエピトープに対する免疫応答を発達させる。好ましくは、上記感染は、ウイルス感染または偏性細胞内寄生生物の感染である。
【0019】
本発明のもう一つの特徴は、本発明のキメラ抗原および薬学的に受容可能な賦形剤を含む薬学的組成物を提供する。
【0020】
本発明の一つの特徴は、本発明のキメラ抗原と、治療が必要な被験体に対するキメラ抗原を投与するための説明書とを含む製品を提供する。
【0021】
本発明のもう一つの特徴は、キメラ抗原をコードするポリヌクレオチドを提供し、このポリヌクレオチドは免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二ポリヌクレオチド部分とを含み、この標的結合ドメインが抗体フラグメントを含む。また、本発明は、このようなポリヌクレオチドを含む微生物および細胞株を提供する。
【0022】
本発明のさらにもう一つの特徴は、本発明のキメラ抗原を作製する方法を提供し、この方法は、本発明のキメラ抗原をコードするポリヌクレオチドを含む微生物または細胞株を提供する工程、および上記キメラ抗原が発現される条件下で上記微生物または細胞株を培養する工程を包含する。
【0023】
本発明の一つ以上の実施態様の詳細を、添付の図面および下記の説明に示す。本発明の他の特徴、目的および利点は、下記の説明および図面から、および請求の範囲から明らかであろう。
【発明を実施するための最良の形態】
【0024】
(IV.詳細な説明)
(A.概説)
抗原に対する免疫応答を引き出すための組成物および方法を、本明細書において開示する。具体的には、この化合物および方法は、本来は宿主により「自己」抗原として認識される外来性の抗原に対して免疫応答を引き出すので、これらの抗原に対する宿主の寛容を壊す。免疫応答ドメインおよび標的結合ドメイン(この標的結合ドメインは、抗体フラグメントを含む)を含むキメラ抗原を、宿主の免疫系に提示することは、外来性または寛容化された抗原に対する免疫応答を増強する。抗原提示細胞は、上記キメラ抗原を取り込み、処理し、そして提示して、所望の抗原に対して体液性免疫応答と細胞性免疫応答の両方を引き出す。
【0025】
(B.定義)
本発明をさらに詳しく説明する前に、本出願で用いられる用語は、特に示さないかぎり下記のように定義される。
【0026】
「抗体」とは、抗原(免疫原)によりヒトまたは他の動物に引き起こされた特定のアミノ酸配列を有する、Bリンパ系細胞によって産生された免疫グロブリン分子をいう。これらの分子は他の用語で各々定義される抗原と特異的に反応することにより特徴付けられる。
【0027】
「抗体応答」または「体液性応答」とは、抗体がBリンパ系細胞により作られ、抗原の刺激により血液および/またはリンパに分泌される、ある型の免疫応答をいう。適切に機能する免疫応答において、上記抗体は、細胞(例えば、病原体)表面上の抗原に特異的に結合し、食細胞および/または補体仲介機構による破壊のために上記細胞にマークする。また、抗体は全身を循環し、遊離ビリオンに結合することができる。この抗体結合は、ビリオンを中和してビリオンが細胞に感染することを妨げ得、そして食作用や腎臓でのろ過によって循環から除去するために、ビリオンをマークする。
【0028】
「抗原」とは、適当な細胞との接触の結果、感受性および/または免疫応答性の状態を誘導し、インビボまたはエキソビボで感作被験体の抗体および/または免疫細胞に明白な方法で反応する任意の物質をいう。
【0029】
「抗原提示細胞」とは、主に抗原を扱って、これをリンパ球に提示することにより機能する抗原誘導事象の補助細胞をいう。抗原提示細胞の抗原との相互作用は、リンパ球を抗原分子に会わせて認識させ、リンパ球を活性化させることができるので、免疫誘導において必須の工程である。例示的な抗原提示細胞としては、マクロファージ、ランゲルハンス樹状細胞、濾胞樹状細胞およびB細胞が挙げられる。
【0030】
「B細胞」とは、抗原と相互作用する免疫グロブリンまたは抗体を産生する、ある型のリンパ球をいう。
【0031】
「CH1領域」とは、抗体の抗原結合フラグメント上の重鎖定常ドメインの領域をいう。
【0032】
「細胞性応答」とは、ウイルス感染細胞または癌細胞を直接に除去することができる特定のヘルパーT細胞およびキラーT細胞により仲介される、ある型の免疫応答をいう。
【0033】
本明細書中で使用される場合、「キメラ抗原」との用語とは、免疫応答ドメインおよび標的結合ドメインを含むポリペプチドをいう。免疫応答ドメインおよび標的結合ドメインは、共有的手段または非共有的手段により直接的に結合してもよいし、間接的に結合してもよい。
【0034】
「細胞障害性Tリンパ球」とは、外来細胞、およびウイルス性抗原を作る感染因子に感染した宿主細胞を破壊することのできる、特化された型のリンパ球である。
【0035】
「エピトープ」とは、複合抗原分子上の最も単純な形の抗原決定基をいう。これは免疫グロブリンまたはT細胞レセプターにより認識される抗原の特定部分である。
【0036】
「融合タンパク質」とは、二つ以上の遺伝子配列を組み合わせることにより作られたハイブリッド遺伝子の発現により形成されるタンパク質をいう。
【0037】
「ヒンジ領域」とは、FabフラグメントをFcフラグメントに結合させる抗体部分をいう。このヒンジ領域は、二つの重鎖を共有結合させるジスルヒド結合を含有する。
【0038】
「相同」との用語は、例えば、対応する位置で同じまたは類似である化学残基の配列を有することによって、別の分子と相同性を示す分子をいう。「%相同の」または「%相同性」の語句は、同一または類似である相同性ポリヌクレオチドまたはポリペプチドの同一位置におけるヌクレオチドまたはアミノ酸のパーセントをいう。例えば、二つのタンパク質中で80残基のうち75残基が同一である場合、それら二つのタンパク質は93.75%相同性である。%相同性は当業者に公知の多様なソフトウエアプログラムを用いて決めることができる。
【0039】
「宿主」とは、感染または癌等の免疫治療が可能な症状に苦しむ温血動物(ヒトを含む)をいう。ここで用いる「宿主」はまた、キメラ抗原が投与される温血動物(ヒトを含む)も言及する。
【0040】
本発明の文脈において、「ハイブリダイゼーション」とは、オリゴマー化合物の相補鎖が対となることを意味する。本発明において、対となる好ましい機構は、水素結合を伴うもので、これは、オリゴマー化合物の鎖の相補的なヌクレオシド塩基またはヌクレオチド塩基(ヌクレオ塩基)間でのWatson−Crick、Hoogsteenまたは逆向きHoogsteen水素結合であってよい。例えば、アデニンとチミンとは、水素結合の形成により対となる相補的なヌクレオ塩基である。ハイブリダイゼーションは、多様な状況下で起こりうる。ポリヌクレオチドの文脈において用いられる場合、「ハイブリダイズする」、「ハイブリダイズしている」等の用語は、慣用的なハイブリダイゼーション条件、好ましくは、ハイブリダイゼーション温度が37℃超で、0.1×SSC/0.1%SDSでの洗浄温度が55℃超である、50%ホルムアルデヒド/6×SSC/0.1%SDS/100μg/ml mDNAでのハイブリダイゼーション等の条件をいう。
【0041】
「免疫」または「免疫応答」は、抗原に対する体の応答をいう。特定の実施態様において、体が感染疾患に対してそれ自体を抵抗するか、または保護する能力をいう。
【0042】
「免疫応答ドメイン(IRD)」とは、二官能性キメラ抗原分子の多様な構造の抗原性部分をいう。この免疫応答ドメインは、一つ以上の抗原および/または一つ以上の組換え抗原を含む。
【0043】
本明細書において使用される場合、「免疫治療が可能な症状」との語句は、被験体の免疫応答を引き出すか、または調節することにより防止、抑制あるいは緩和することのできる症状または病気をいう。
【0044】
「リンパ球」とは、特定の免疫応答を仲介する血液中に見られる有核細胞の部分集合をいう。
【0045】
「モノクローナル抗体」または「mAb」とは、融合ハイブリッド細胞、すなわちハイブリドーマ細胞のクローンまたは遺伝的に均一な集団から作られた抗体をいう。ハイブッド細胞は、クローニングされて、化学的および免疫学的に均一な(すなわち一つの型のみの抗原を認識する)特定のモノクローナル抗体を産生する細胞株を確立する。
【0046】
「ペプチド結合」は、1つのアミノ酸のαアミノ基と別のアミノ基のαカルボキシル基との間の置換アミド結合により共有結合した二つ以上のアミノ基をいう。
【0047】
「薬学的に受容可能な」とは、ヒトまたは他の動物に生理学的に適合する毒性のない組成物をいう。
【0048】
「薬学的に受容可能な賦形剤」は、毒性がなく、ヒトまたは他の動物に生理学的に適合性のあるアジュバント、キャリア、pH調節剤および緩衝剤、浸透圧調節剤、湿潤剤、保存剤等の物質を含む。
【0049】
本明細書において使用される場合、「ポリヌクレオチド」との用語は、リボヌクレオチドかデオキシリボヌクレオチドのいずれかの長さのヌクレオチドの多量形をいう。この用語は、分子の一次構造のみをいう。よって、この用語は、二本鎖または一本鎖のDNAまたはRNAを含む。また、公知の型の改変、例えば、当該分野で公知の標識、メチル化、「キャップ」、一つ以上の天然に存在するヌクレオチドの類似体による置換、ヌクレオチド間の改変(例えば、未荷電結合(例えば、メチルホスホネート、ホスホトリエステル、ホスホアミデート、カルバメート等)および荷電結合(例えば、ホスホロチオエート、ホスホロジチオエート等)による修飾、ペンダント部分(例えばタンパク質(例えば、ヌクレアーゼ、トキシン、抗体、シグナルペプチド、ポリ−L−リジン等)を含む)を含む改変、インターカレーター(例えば、アクリジン、ソラレン等)による改変)、キレーター(例えば、金属、放射性金属、ホウ素、酸化金属等)を含む改変、アルキル化剤を含む改変、改変結合(例えば、αアノマー核酸等)による改変、ならびにポリヌクレオチドの非改変形態など、公知の種類の改変も挙げられる。
【0050】
「プロテアーゼ切断部位」とは、タンパク質分解酵素がポリペプチド鎖を加水分解する(壊す)部位をいう。
【0051】
本発明において、「ストリンジェントなハイブリダイゼーション条件」または「ストリンジェント条件」との語句は、本発明の化合物がその標的配列とハイブリダイズするが、他の配列とのハイブリダイズは最小限とする条件をいう。
【0052】
「被験体」との用語は、温血動物、好ましくはヒトをいう。
【0053】
「タグ」とは、タグを含む分子を単離または精製するために用いられる、マーカーまたはマーカー配列をいう。例示的なタグとしては6×Hisタグが挙げられる。
【0054】
「T細胞」とは、抗原特異的な細胞相互作用に関与し、体液性免疫応答および細胞性免疫応答を仲介する型のリンパ球をいう。
【0055】
「標的結合ドメイン(TBD)」とは、抗原提示細胞上、特に樹状細胞上のレセプターに結合することができ、引き続いてレセプター仲介取り込みにより抗原提示細胞内に輸送される、抗体フラグメントを含むタンパク質をいう。
【0056】
「治療的有効量」とは、抗原に対して効果的なB細胞、細胞障害性Tリンパ球(CTL)および/またはヘルパーTリンパ球(Th)の応答を引き出して、病気または疾患の症状および/または合併症を阻止するか、または治療するか、もしくは少なくとも部分的に抑制するか、もしくは遅らせるのに十分なキメラ抗原の量またはキメラ抗原をコードするポリヌクレオチドの量をいう。
【0057】
本明細書中で使用される場合、「治療する」および「治療」との用語は、動物(特に、ヒト)においてキメラ抗原により治療可能な症状の任意の治療を包含し、(i)症状が起こりやすいが、その症状があるとはまだ診断されていない被験体にこの症状が起こることを妨ぐ、(ii)症状の抑制(例えば、その進展を停止または遅らせる);または(iii)症状の緩和(例えば、症状またはその徴候の低減を引き起こすこと)を含む。
【0058】
本明細書において使用される場合、「異種型」とは、宿主以外の異なる種に由来することをいう。例えば、マウスのゲノムからクローニングされた組換え発現の抗体は、組換え発現の抗体が細菌、昆虫またはマウスの細胞のいずれで作られるかにかかわらず、ヒトに対して異種型であるが、マウスに対しては異種型ではないということである。
【0059】
(C.新規なキメラ抗原)
本発明は、免疫応答ドメインおよび標的結合ドメインを含む免疫応答を引き出すためのキメラ抗原において、この標的結合ドメインが抗体フラグメントを含むキメラ抗原を提供する。本発明に従って、キメラ抗原は好ましくはFcレセプターおよび/またはマクロファージマンノースレセプターに結合できる。この抗体フラグメントは宿主に対して異種型であっても、宿主に対して異種型でなくてもよい。
【0060】
本発明の好ましい実施態様において、キメラ抗原は体液性免疫応答および/または細胞性免疫応答を誘導することができる。細胞性免疫応答として、Th1応答、Th2応答および/または細胞障害性Tリンパ球(CTL)応答を挙げることができる。さらに別の好ましい実施態様において、キメラ抗原は多エピトープ性免疫応答を引き出す。多エピトープ性免疫応答としては、免疫応答ドメインの少なくとも一つのエピトープに対する応答および/または標的結合ドメインの少なくとも一つのエピトープに対する応答を挙げることができる。あるいは、多エピトープ性応答は免疫応答ドメインの二つ以上のエピトープに対する応答に限定してもよい。
【0061】
本発明のキメラ抗原は、二つの部分、すなわち、抗原性配列(ウイルス抗原等)を含む免疫応答ドメインと、抗体フラグメントを含む標的結合ドメインとを含む(図1)。好ましい実施態様において、上記免疫応答ドメインは、当業者に公知の任意の方法によって標的結合ドメインに結合させることができる。免疫応答ドメインを標的結合ドメインに結合させるためのリンカーとしては、共有ペプチド結合、化学接合、ロイシンジッパーおよびビオチン/アビジンが挙げられるが、これらに限定されない。好ましい実施態様において、上記免疫応答ドメインおよび標的結合ドメインは、単一の融合タンパク質としてクローニングすることができる。融合タンパク質の共有ペプチド結合は、付加的なペプチド配列、例えばSRPQGGGSまたはVRPQGGGS(配列番号1)を含んでよい。さらに別の好ましい実施態様において、多様な免疫応答ドメインがビオチン化され、そして標的結合ドメインは、ストレプトアビジンを有する融合タンパク質として作られて、キメラ抗原の広い類別の産生に役立つ。あるいは、免疫応答ドメインと標的結合応答領域はそれぞれ、ロイシンジッパー部分との融合物として発現させることができ、キメラ抗原の二つの部分は混合の際に会合させられる。最後に、免疫応答ドメインと標的結合ドメインを別個に発現させて、次に当業者に公知の方法を用いて化学的に接合させることができる。例示的な方法は、それら2つのドメインを共有結合させるスベルイミノ酸ジメチル等のタンパク質架橋剤の使用を含む。
【0062】
免疫応答ドメインは主にキメラ抗原の抗原性の部分を提供する。この免疫応答ドメインは、免疫応答が望まれるものの全体の少なくとも一つの抗原性部分を含有する。このキメラ抗原は、必要に応じて二つ以上の免疫応答ドメインを含有してもよい。好ましい実施態様において、上記免疫応答ドメインは、ウイルスまたは偏性細胞内寄生生物等の感染因子、あるいは癌抗原の少なくとも一つの抗原性部分を含有する。より好ましくは、上記免疫応答ドメインは、感染性ウイルスの少なくとも一つの抗原性部分を含む。
【0063】
好ましい感染性ウイルスの例としては、レトロウイルス科(例えば、ヒト免疫不全ウイルス、例えばHTLV−III、LAVまたはHTLV−III/LAVまたはHIV−IIIとも称されるヒト免疫不全ウイルス1(HIV−1);および他の単離物、例えばHIV−LP;ピコルナウイルス科(例えば、ポリオウイルス、A型肝炎ウイルス;エンテロウイルス、ヒトコクサッキーウイルス、ライノウイルス、エコーウイルス);カルシウイルス科(Calciviridae)(例えば、胃腸炎を引き起こす株);トガウイルス科(例えば、ウマ脳炎ウイルス、風疹ウイルス);フラビウイルス科(例えば、C型肝炎ウイルス、デング熱ウイルス、脳炎ウイルス、黄熱病ウイルス);コロナウイルス科(例えば、コロナウイルス);ラブドウイルス科(例えば、水ほう性口内炎ウイルス、狂犬病ウイルス);フィロウイルス科(例えば、エボラウイルス);パラミクソウイルス科(例えば、パラインフルエンザウイルス、流行性耳下腺炎ウイルス、麻疹ウイルス、RSウイルス);オルトミクソウイルス科(例えば、インフルエンザウイルス);ブンガウイルス科(Bungaviridae)(例えば、ハンターンウイルス、ブンガウイルス、フレボウイルスおよびナイロウイルス);アレナウイルス科(出血熱ウイルス);レオウイルス科(例えば、レオウイルス、オルビウイルスおよびロタウイルス);ビルナウイルス科;ヘパドナウイルス科(ヒトB型肝炎ウイルス(HBV)、アヒルB型肝炎ウイルス(DHBV));パルボウイルス科(パルボウイルス);パポバウイルス科(パピローマウイルス、ポリオーマウイルス);アデノウイルス科(ほとんどのアデノウイルス);ヘルペルウイルス科(単純ヘルペスウイルス(HSV)1および2、水痘帯状疱疹ウイルス、サイトメガロウイルス(CMV)、エプスタイン−バーウイルス、ヘルペスウイルス);ポックスウイルス科(痘そうウイルス、ワクシニアウイルス、ポックスウイルス)およびイリドウイルス科(例えば、アフリカブタ熱ウイルス(African swine fever virus));および分類されていないウイルス(例えば、δ肝炎(hepatitides)因子、非A型、非B型肝炎因子(クラス1、内部伝染);クラス2、非経口伝染;ノルウォークウイルスと関連ウイルス、ならびにアストロウイルス)が挙げられる。本発明の一部実施態様において、キメラ抗原の免疫応答ドメインは、HBVタンパク質、DHBVタンパク質およびHCVタンパク質からなる群から選択される一つ以上のタンパク質の少なくとも一つの抗原性部分を含む。本発明に使用される特に好ましいHBVタンパク質として、HBV S1/S2、HBV S1/S2/S、HBV Core、HBV Core ctm(C末端改変)、HBV e抗原およびHBVポリメラーゼが挙げられるが、これらに限定されない。本発明に使用される特に好ましいDHBVタンパク質としては、DHBV PreS/S、DHBV PreS、DHBV CoreおよびDHBVポリメラーゼが挙げられるが、これらに限定されない。本発明に使用される特に好ましいHCVタンパク質としては、HCV Core(1−191)、HCV Core(1−177)、HCV E1−E2、HCV E1、HCV E2、HCV NS3、HCV NS5AおよびHCV NS4Aが挙げられるが、これらに限定されない。本発明に用いられる他の好ましいウイルス抗原としては、HIV gp120、HSVアルカリヌクレアーゼおよびヒトパピローマウイルス(HPV)キャプシドタンパク質L1およびL2、および初期領域タンパク質HPV E1、HPV E2、HPV E4、HPV E5、HPV E6およびHPV E7が挙げられる。
【0064】
好ましい偏性細胞内寄生生物の例として、Tetrahymena sp.(例えば、T.pyriformis)、Plasmodium sp.(例えば、P.falciparum)、Cryptospiridium sp.、Spraguea sp.(例えば、S.lophii)、Giardia sp.,Toxoplasma sp.(例えば、T.gondii、T.cruzi)、Leishmania sp.、Rickettsia sp.(例えば、R.prowazekii)、Chlamydia sp.、Mycobacterium sp.(例えば、M.tuberculosis)、Legionella sp.、Listeria sp.(例えば、L.monocytogenes)、Coxiella sp.(例えば、C.brunette)、Shigella sp.、Erlichia sp.およびBartonelia sp.が挙げられる。好ましい癌抗原として、前立腺特異的抗原(PSA)、前立腺特異的膜抗原(PSMA)、MUC1、CA 125、WT1、Her−2/neu、癌胎児性抗原(CEA)、MAGE−3、MART−1、gp100、NY−ESSO−1、CA19.9、TAG72、CA 15.3、CA 27.9、gp120、前立腺酸性ホスファターゼ(PAP)、ヒートショックタンパク質、αフェトタンパク質(AFP)、テロメラーゼおよびrasが挙げられる。
【0065】
本発明のもう一つの実施態様において、キメラ抗原の免疫応答ドメインは、一つ以上の抗原性部分に融合させた6×Hisタグを包含する。
【0066】
本発明にしたがえば、上記キメラ抗原は、抗原提示細胞上、特に樹状細胞上のFcレセプターおよび/またはCD206に結合することのできるタンパク質であり、このキメラ抗原は、次いでレセプター仲介取り込みにより抗原提示細胞中に輸送される。本発明にしたがえば、抗体フラグメントの存在は、抗原提示細胞上、具体的には樹状細胞上のFcレセプターによるキメラ抗原の取り込みを増強する。ウイルスのこの特異的な結合と細胞内への取り込みによって、ウイルス抗原が処理されて、外来物として提示される。よって、免疫応答は、前もって寛容化された抗原に対して効果的に引き出され得る。標的結合ドメインは宿主に対して異種型であっても、異種型でなくてもよい抗体フラグメントを含んでいる。本発明の好ましい実施態様において、上記抗体フラグメントはマウスのFcフラグメントを含む。本発明のさらに好ましい実施態様において、標的結合ドメインは、Fcフラグメント、ヒンジ領域、およびCH1領域の一部を含み、上記キメラ抗原は、標的結合ドメインを免疫応答ドメインに結合させるのに適当なペプチドリンカーを含む。もう一つの好ましい実施態様において、標的結合ドメインは免疫グロブリン重鎖フラグメントを含み、必要に応じてさらにヒンジ領域を含む。特に好ましい実施態様において、重鎖フラグメントは、CH1ドメインのアミノ酸VDKKI(配列番号2)および/またはCH2ドメインおよびCH3ドメインの一部またはすべてを含む。
【0067】
上記のように、CD206により結合され、細胞内に取り込まれた抗原は、MHCクラスIとクラスIIの両方によって提示されて、細胞性と体液性の両方の免疫応答を引き出すことができる。したがって、好ましい実施態様では、キメラ抗原がグリコシル化される。免疫応答ドメインおよび/または標的結合ドメインは、グリコシル化することができる。特に好ましい実施態様において、キメラ抗原は高マンノースグリコシル化(high mannose glycosylation)または低マンノースグリコシル化(pauci mannose glycosylation)のいずれかによりマンノースグリコシル化されている(Jarvis, Virology 310:1−7(2003))。
【0068】
(D.キメラ抗原を利用する新規な方法)
本発明は、免疫応答を引き出す方法を包含し、この方法は、本発明のキメラ抗原を含有する組成物を、被験体に投与する工程を包含する。
【0069】
抗原の十分な提示を提供するために、本発明者らは、免疫応答ドメインと標的結合ドメインとを含む新規のキメラ抗原を開発している。この標的結合ドメインは、抗体フラグメントを含む。発明の特別な理論に限定されるわけではないが、この分子は、その抗体フラグメントのおかげで、抗原提示細胞上の特定のレセプターに結合し、ウイルス抗原が主要組織適合複合体(MHC)クラスIおよびクラスIIと複合体化するように処理、提示される。MHCクラスIによるそのような処理と抗原提示は、細胞障害性Tリンパ球による増強応答を引き出して、結果として免疫応答ドメインの抗原に会合する任意の感染因子の除去をもたらす。さらに、MHCクラスII分子による抗原提示は、感染細胞および/または循環から抗原のクリアランスをも助ける体液性応答を引き出す。
【0070】
また、本発明は、寛容を壊す方法を包含し、この方法は、本発明のキメラ抗原を被験体に投与する工程を包含する。キメラ抗原を用いることによって細胞性免疫応答および/または体液性免疫応答を引き出すための抗原の提示において、慢性的な感染の間に「自己」として扱われた抗原は、「外来性」として認識される。したがって、宿主の免疫系はCTL応答を開始させて感染細胞を除去する。同時に、キメラ抗原に対する応答において誘発された抗体は、感染因子に結合し、感染因子を循環から除去するか、または宿主細胞に対する感染因子の結合を阻止する。したがって、本来は宿主免疫系により寛容化される慢性的な感染を有する被験体において、広い免疫応答を誘導しうるキメラ抗原を作るように、本発明は設計される。好ましい実施態様において、上記キメラ抗原は、ウイルスまたは寄生生物等の感染因子により慢性的に感染しているか、または癌を有する被験体において、抗原に対する寛容を破壊する。さらに好ましくは、感染因子はその生活環中のある時点で宿主細胞に内在する。
【0071】
好ましい実施態様において、宿主免疫系により認識されないか、寛容化される前選択された抗原の免疫原性は、抗原フラグメントの存在に起因して、ならびに真核細胞(例えば昆虫細胞)発現系に導入されたグリコシル化の存在により増強される。そのようなキメラ抗原は、抗体成分およびグリコシル化の存在に起因して、樹状細胞、マクロファージ、B細胞および顆粒球等の多様な免疫細胞型に存在する特定のレセプターに結合する。
【0072】
本発明のさらに別の特徴は、抗原提示細胞を活性化する方法を提供し、この方法は、上記抗原提示細胞を本発明のキメラ抗原に接触させる工程を包含する。また、本発明は、抗原提示細胞において抗原提示を増強する方法を提供し、この方法は、本発明のキメラ抗原を含有する組成物に抗原提示細胞を接触させる工程を包含する。上記キメラ抗原は抗原提示細胞、好ましくは樹状細胞にインビボまたはエキソビボで接触させることができる。好ましい実施態様において、キメラ抗原を抗原提示細胞に接触させることは、抗原提示細胞を活性化し、二つ以上のエピトープの抗原提示を促進する。この多エピトープ性応答としては、免疫応答ドメインの一つ以上のエピトープの提示および/または標的結合ドメインの一つ以上のエピトープの提示を挙げることができる。
【0073】
また、本発明は、免疫治療が可能な症状を治療する方法を提供し、この方法は、治療的有効量の本発明のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する。好ましい実施態様において、免疫治療が可能な症状は感染または癌である。上記感染は、ウイルス感染、寄生生物感染または細菌感染であってよい。好ましくは、この感染は、感染因子が宿主細胞内に見られる段階を有する。さらに好ましくは、免疫治療が可能な症状は慢性のウイルス感染である。最も好ましくは、免疫治療が可能な症状は慢性B型肝炎ウイルス(HBV)感染または慢性C型肝炎ウイルス(HCV)感染である。HBVの治療のために、上記免疫応答ドメインは、好ましくは、HBV Coreタンパク質、HBV Sタンパク質、HBV S1タンパク質、HBV S2タンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む。HCVの治療のために、上記免疫応答ドメインは好ましくはHCV Core(1−191)タンパク質、HCV Core(1−177)タンパク質、HCV E1タンパク質、HCV E2タンパク質、HCV E1−E2タンパク質、HCV NS3Aタンパク質、HCV NS5Aタンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む。
【0074】
好ましい実施態様において、キメラ抗原の投与は免疫応答ドメインのみの投与よりもさらに高い免疫応答を引き出す。免疫応答の大きさは、例えば、(i)被験体に存在する抗原特異的な抗体の量により、(ii)キメラ抗原または免疫応答ドメインのみを負荷した抗原提示細胞に対して曝されることに応答してT細胞により分泌されたインターフェロンγの量により、または(iii)キメラ抗原または免疫応答ドメインのみを負荷した抗原提示細胞に対して曝されることに応答してもたらされた抗原特異的なCD8+T細胞の量により、測定され得る。
【0075】
キメラ抗原は、免疫応答を生じるその効能について、該キメラ抗原を樹状細胞にエキソビボまたはインビボで提示することによって評価することができる。上記樹状細胞は、T細胞応答のマーカーとしてT細胞の増殖およびインターフェロンγの産生に関して評価されるTリンパ球に対して、キメラ抗原を処理し、提示する。具体的に、エキソビボ状況で、ナイーブ樹状細胞は、末梢血液から単離される。上記樹状細胞によるT細胞の活性化は、マーカー、例えばインターフェロンγレベルを公知の操作で測定することによって評価される。例えば、Berlynら、Clin.Immunol 101(3):276−283(2001)を参照のこと。インターフェロンγを分泌するT細胞の比率の少なくとも50%までの増加は、インビボでの効能を予想させる。インビボ状況の場合、上記キメラ抗原は、利用可能な樹状細胞および他の抗原処理細胞が抗原と相互作用でき、よってそれらを処理する能力を有する宿主に直接に非経口で導入される。
【0076】
さらに、本発明は、感染に対して被験体をワクチン接種する方法を包含し、本方法は、本発明のキメラ抗原を被験体に投与する工程を包含する。上記被験体は、予防的または治療的にワクチン接種され得る。好ましくは、上記感染はウイルス感染である。分子の二官能性の性質は、抗原提示細胞、例えば樹状細胞を抗原の標的とするのに役立ち、抗体に対して抗原の最も効果的な化学量にて特異的に抗原提示細胞を標的とすることによって、慢性的な感染疾病の治療において独自のアプローチとなる。これは、B型肝炎、C型肝炎、ヒト免疫不全ウイルス、ヒトパピローマウイルスおよび単純ヘルペスウイルス等の慢性ウイルス感染、偏性細胞内寄生生物感染を治療する治療ワクチンの開発に有用であり、癌および自己免疫疾患等の病気におけるすべての自己抗原に適用可能でもあり得る。これらの融合タンパク質の投与は、細胞性および体液性の両方の応答を含め、宿主から広い免疫応答を引き出すことができる。よって、これら融合タンパク質は、特別な感染を発症させる危険のある被験体を免疫するための予防ワクチンとして有用であることに加えて、既存の感染に対して免疫寛容性である被験体を治療する治療ワクチンとして使用することができる。
【0077】
(E.キメラ抗原の製造方法)
本発明の一つの特徴は、キメラ抗原を作製するための方法を提供し、この方法は、以下、(a)キメラ抗原をコードするポリヌクレオチドを含む微生物または細胞株を提供する工程であって、この微生物または細胞株は、好ましくは真核細胞であって、より好ましくは非哺乳動物の微生物または細胞株である、工程;および(b)上記微生物または細胞株を、キメラ抗原が発現される条件下で培養する工程、を包含する方法である。好ましくは、上記微生物または細胞株は酵母、植物細胞株または昆虫細胞株である。さらに好ましくは、上記細胞株は、Sf9、Sf21、Drosophila S2およびHigh Five(登録商標)からなる群から選択される昆虫細胞株である。
【0078】
本発明の一実施態様は、本発明を実施するために必要な選択された抗原と標的結合ドメインとの融合タンパク質を作るために、確立された組換えDNA技術を使用する。融合タンパク質構築物は、所望のDNAフラグメントを発現ベクターに導入するために利用される特定の制限酵素部位を導入して、DNAレベルで作られ、これを用いて異種発現系において所望の融合タンパク質を発現させる。ここで用いる「ベクター」との用語は、所望のタンパク質をコードするDNAを運ぶことのできるプラスミドをいう。本発明に使用される好ましいプラスミドベクターとしては、pFastBac HTaおよびDH10Bac(登録商標)E.coli(Invitrogen)で作られたその対応する組換え「バクミド(bacmid)」が挙げられるが、これらに限定されない。
【0079】
標的結合ドメインをコードする遺伝子は、抗体産生細胞、例えばモノクローナル抗体を産生するハイブリドーマから、ポリメラーゼ連鎖反応(PCR)により得ることができる。後者のクローニング工程を容易にするために、独自の制限酵素認識部位を加えるためのオリゴヌクレオチドプライマーを設計することが望ましい。同様に、免疫応答ドメインの抗原性部分は、所望の標的の抗原性部分をコードする遺伝子を含む任意の細胞またはウイルスRNAもしくはウイルスDNAから得ることができる。好ましくは、PCRを用いて、免疫応答ドメインの抗原性部分をコードするDNAを得て、PCRプライマーを設計して、クローニングを容易にする独自の制限酵素認識部位を付加する。しかし、任意の組換えDNA法を用いて、免疫応答ドメインの抗原性部分をコードするDNAが得られ得る。次に、標準的なクローニング技術を用いて、標的結合ドメインをコードするポリヌクレオチドと免疫応答ドメインをコードするポリヌクレオチドとを単一の構築物に組み合わせることができる。もしくは、それら別々のドメインは、ロイシンジッパー、ストレプトアビジンまたはビオチニル化シグナル等のDNAコードリンカーとともにクローニングされ得る。
【0080】
大量の異種タンパク質が作られるという理由のみならず、真核生物のタンパク質のリン酸化やグリコシル化等の翻訳後改変が感染昆虫細胞内で起こるという理由から、バキュロウイルス系を用いて本発明のキメラ抗原を発現させるのが好ましい。昆虫細胞への直接のクローニングは困難でありうるために、キメラ抗原をコードするポリヌクレオチドを細菌系において作成し、その最終構築物をバキュロウイルス/昆虫細胞発現系に転移させることが好ましい。転移システム、例えば、Bac−To−Bac(登録商標)系(Invitrogen)は当業者に公知である。このBac−to−Bac(登録商標)系は、細菌トランスポゾンTn7による部位特異的転位を利用して、所定の遺伝子をE.coli−昆虫細胞シャトルベクター(バクミド)に転移させる。得られる組換えバクミドは、昆虫細胞にトランスフェクションされ、組換えタンパク質を発現するバキュロウイルスを作る。
【0081】
バキュロウイルスを作るために、上記バクミドを、Sf9細胞等の昆虫細胞にトランスフェクションする。トランスフェクション後、この細胞を、バキュロウイルス集団を増殖させるのに十分な時間インキュベートする。バキュロウイルスを含む培地を集めて、4℃、暗所で保存する。このトランスフェクションは、所望のDNA挿入物に対して特異的なプライマーを利用するPCRにウイルス培養物を供して、バキュロウイルスDNAの生産を調べることにより実証され得る。上記細胞中の異種タンパク質の発現は、任意の公知の方法、例えば、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)またはウエスタンブロットにより実証され得る。
【0082】
標準的な感染効率(MOI)の組換えバクミドを用いて、昆虫細胞を感染させる。細胞を約3×105細胞/mLの密度で撒き、細胞密度が約2〜3×106細胞/mLに達するまで懸濁培養(27.5℃、攪拌下)にてインキュベートする。次に、標準化量の各組換えバキュロウイルスを細胞に加える。インキュベーション温度は27.5℃で、適切な感染時間は個々のタンパク質発現のについて標準化される。細胞を遠心分離により集め、組換えタンパク質の精製に用いる。細胞の使用しない部分は、液体窒素中で瞬時に凍らせて、−70℃で保存され得る。
【0083】
キメラ抗原は、好ましくは変性条件下で精製される。キメラ抗原を発現する細胞は変性緩衝液、例えば6Mグアニジン−HClを含有する緩衝液に可溶化される。可溶化は超音波処理等の機械的手段により高めることができる。溶解物を遠心して、未破壊細胞と細胞破片を除去する。次に、上清を、前もって可溶化緩衝液で平衡化したNi−NTA Super Flow(Qiagen)ビーズカラムにロードする。ロードした後に、カラムを緩衝性変性溶液、好ましくは、約pH8の6MグアニジンHClを含有する変性溶液で洗浄する。この時点で、この変性剤を例えば緩衝溶液中の8M尿素に交換することができる。可溶化緩衝液、ローディング緩衝液および洗浄緩衝液は好ましくは低濃度、例えば1〜40mMのイミダゾールを含有する。緩衝液の交換後、カラムをOD280が例えば<0.1に低下するまで緩衝液で洗浄しなければならない。結合タンパク質は、8M尿素と250mMイミダゾール(pH8)を含有する緩衝液(溶出緩衝液)で溶出させ得る。タンパク質を含有する画分をプールして、低濃度(例えば、100mM)塩の(好ましくは8M尿素を含有する)変性透析緩衝液に対して緩衝液を数回交換しながら4℃で透析する。次に、透析タンパク質を、DEAE(ジエチルアミノエチル)等のイオン交換カラムにかける。好ましい実施態様において、イオン交換カラムにかける前に、ジチオスレイトール(DTT)または他の還元剤をタンパク質に加える。キメラ抗原はDEAEカラムを通過する。したがって、このDEAE通過分を集めて、低減した濃度の変性剤を含有する緩衝液に対して段階的な様式で透析する。例示的な方法において、次に上記タンパク質を緩衝性4M尿素に対して少なくとも12時間以上、次に緩衝性2M尿素に対して少なくとも12時間以上、次に緩衝性1M尿素に対して少なくとも12時間以上、次に緩衝性0.5M尿素に対して少なくとも12時間以上、そして最後に変性剤を含まない緩衝液に対して少なくとも12時間以上透析した後に、好ましくは変性剤を含まない新しい緩衝液に対して12時間の透析をさらに二回行う。精製され再び折りたたまれたタンパク質を濃縮して、例えば、SDSゲル電気泳動、等電点電気泳動(isoelectric focusing)または発現タンパク質の異なるドメインに対する抗体を用いるウエスタンブロット分析等の標準的な生化学的手法を用いて特性付けることができる。
【0084】
本発明の実施は、特に他に示さなければ、当業者の技量の範囲内にある分子生物学、微生物学、組換えDNAおよび免疫学の慣用の技術を用いる。そのような技法は、文献により十分に説明されている。例えば、Sambrookら、Molecular Cloning:A Laboratory Manual,第2版,New York:Cold Spring Harbor Press,1989;およびAusubelら、Current Protocols in Molecular Biology,Wiley Interscience Publishers((著作権)1995,2004年4月、補足66で補足されたもの)を参照のこと。
【0085】
(F.新規なポリヌクレオチド)
本発明のもう一つの特徴は、免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二のポリヌクレオチド部分を含むキメラ抗原をコードするポリヌクレオチドを提供する。第一のポリヌクレオチド部分と第二のポリヌクレオチド部分は同一または異なるヌクレオチド鎖に配置され得る。
【0086】
本発明は、キメラ抗原をコードする遺伝子、mRNAおよび/またはコード配列に対応するか、またはこれらに相補的なポリヌクレオチドを、好ましくは単離された形態で提供する。これらとしては、キメラ抗原変異タンパク質をコードするポリヌクレオチド、キメラ抗原をコードする遺伝子もしくはmRNA配列もしくはその一部に対して相補的であるか、または少なくとも90%の相同性を有するDNA、RNA、DNA/RNAハイブリッドおよびその関連する分子、ポリヌクレオチドもしくはオリゴヌクレオチド、ならびにキメラ抗原をコードする遺伝子、mRNA、またはキメラ抗原をコードするポリヌクレオチドに対してハイブリダイズするポリヌクレオチドもしくはオリゴヌクレオチドが挙げられる。
【0087】
さらに、本発明はここに具体的に開示されたキメラ抗原をコードする遺伝子の類似体を包含する。類似体としては、例えば、免疫応答を引き出す能力を保持する変異体であって、キメラ抗原をコードするポリヌクレオチドの、好ましくは少なくとも80%、より好ましくは90%、そして最も好ましくは95%の相同性を有する変異体が挙げられる。これらは具体的には、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号44に記載される。典型的には、そのような類似体はわずかに1〜15コドン変化のみが異なるだけである。例としては、ウイルス抗原または抗体フラグメントの天然アミノ酸配列由来の小さなアミノ酸変化(特に、保存的なアミノ酸の置換)を有するポリペプチドが挙げられる。保存的置換は、側鎖に関連性のあるアミノ酸ファミリー内で起こる置換である。遺伝的にコードされるアミノ酸は、一般的に下記の4ファミリーに分けられる:(1)酸性=アスパラギン酸、グルタミン酸;(2)塩基性=リジン、アルギニン、ヒスチジン;(3)非極性=アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン;および(4)非電荷極性=グリシン、アスパラギン、グルタミン、シスチン、セリン、スレオニン、チロシン。フェニルアラニン、トリプトファンおよびチロシンは、ときどき一緒にされて芳香族アミノ酸に分類される。例えば、ロイシンのイソロイシンまたはバリンへの単離された置換、アスパラギン酸のグルタミン酸への単離された置換、スレオニンのセリンへの単離された置換、またはあるアミノ酸の構造的に関連するアミノ酸への類似の保存的置換は、生物学的活性に対して大きな影響を及ぼさないと予想することは合理的である。配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49のいずれかに開示されたポリペプチドのいずれかと実質的に同じアミノ酸配列を有するが、免疫応答を引き出すキメラ抗原の能力に実質的に影響を与えない重要でないアミノ酸置換を有するポリペプチド分子は、配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49にそれぞれ示された配列を有するキメラ抗原の定義内である。誘導体としては、他のキメラ抗原分子との集合複合体(aggregative conjugate)および関連しない化学部分との共有結合複合体が挙げられる。共有結合誘導体は、キメラ抗原のアミノ酸鎖において、またはN末端残基もしくはC末端残基に見られる基に対して、公知の手段によって官能基を結合させることにより調製される。
【0088】
アミノ酸の略語を表1に示す。
【0089】
【表1】
タンパク質のコンフォメーションまたは機能のいずれも変更することなしに、保存的アミノ酸置換をタンパク質中で行うことができる。本発明のタンパク質は、1個〜15個の保存的置換を含み得る。そのような変化としては、イソロイシン(I)、バリン(V)およびロイシン(L)のいずれかの、これらの疎水性アミノ酸のいずれか他のものへの置換;アスパラギン酸(D)のグルタミン酸(E)による置換およびその逆の置換;グルタミン(Q)のアスパラギン(N)による置換およびその逆の置換;ならびにセリン(S)のスレオニン(T)による置換およびその逆の置換が挙げられる。他の置換も、特定のアミノ酸の環境およびタンパク質の三次元構造におけるその役割に依存して、保存的であると考えられ得る。例えば、グリシン(G)とアラニン(A)とは、アラニン(A)とバリン(V)とがそうであり得るように、しばしば相互交換できる。比較的疎水性であるメチオニン(M)は、ロイシンおよびイソロイシンとしばしば相互交換可能であり得、バリンと置換可能である場合もある。リジン(K)とアルギニン(R)とは、アミノ酸残基の顕著な特徴がその電荷であり、これら二つのアミノ酸残基の異なるpKが顕著ではないような位置において、しばしば相互交換可能である。さらに他の変化は特定の環境において「保存的」であると考えられ得る(例えば、Biochemistry 第4版,Lubert Stryer(編)(W.H.Freeman and Co.),18−23頁;HenikoffおよびHenikoff,Proc Nat’l Acad Sci USA 89:10915−10919(1992);Leiら、J Biol Chem 270(20):11882−6(1995)を参照のこと)。
【0090】
また、本発明は、キメラ抗原をコードするポリヌクレオチドに対して、好ましくはストリンジェント条件下にハイブリダイズする、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号44に具体的に記されたポリヌクレオチドを提供する。ハイブリダイゼーション反応のストリンジェンシーは、当業者により容易に決定可能であり、通常、プローブの長さ、洗浄温度および塩濃度に依存する経験的な計算である。一般的に、より長いプローブは適切なアニーリングのためにより高い温度を必要とする一方で、より短いプローブはより低い温度を必要とする。相補鎖がそれらの溶融温度を下回る環境に存在する場合、ハイブリダイゼーションは、通常、変性核酸配列が再アニーリングする能力に依存する。プローブとハイブリダイズ可能な配列との間の所望の相同性の度合いが高ければ高いほど、使用できる相対温度は高くなる。結果として、より高い相対的温度は反応条件をよりストリンジェントとする傾向があるが、より低い温度はそうではない。ハイブリダイゼーション反応のストリンジェンシーのさらなる詳細と説明に関しては、Ausubelら、前出、2.9.1〜2.10.8および4.9.1〜4.9.13を参照されたい。
【0091】
本明細書において定義される「ストリンジェント条件」または「高ストリンジェント条件」とは、(1)低イオン強度および高洗浄温度(例えば、0.015M塩化ナトリウム/0.0015Mクエン酸ナトリウム/0.1%ドデシル硫酸ナトリウム、50℃)を用いる条件;(2)ハイブリダイゼーション中、ホルムアミド等の変性剤を用いる条件(例えば、50%(v/v)ホルムアミド+0.1%ウシ血清アルブミン/0.1% Ficoll/0.1%ポリビニルピロリドン/50mMリン酸ナトリウム緩衝液(pH6.5)+750mM塩化ナトリウム、75mMクエン酸ナトリウム、42℃;または(3)50%ホルムアミド、5×SSC(0.75M NaCl、0.075Mクエン酸ナトリウム)、50mMリン酸ナトリウム(pH6.8)、0.1%ピロリン酸ナトリウム、5×Denhardtの溶液、超音波処理済サケ精子DNA(50μg/ml)、0.1%SDSおよび10%硫酸デキストラン、42℃を用いる条件であって、42℃での0.2×SSC(塩化ナトリウム/クエン酸ナトリウム)での洗浄と、55℃での50%ホルムアミドでの洗浄と、それに引き続く55℃でのEDTAを含む0.1×SSCを含む高ストリンジェント洗浄とを用いる条件が挙げられるが、これらに限定されない。「適度なストリンジェント条件」とは、Sambrookら(前出)において記載された条件であるが、これに限定されず、そして上記に記載された条件よりも低いストリンジェントな洗浄溶液およびハイブリダイゼーション条件(例えば、温度、イオン強度およびSDS%)の使用を包含する。適度なストリンジェント条件の例は、20%ホルムアミド、5×SSC(150mM NaCl、15mMクエン酸三ナトリウム)、50mMリン酸ナトリウム(pH7.6)、5×Denhardt溶液、10%硫酸デキストランおよび20mg/mL変性せん断サケ精子DNAを含む溶液中での、37℃における一晩のインキュベーション、そしてそれに引き続く約37〜50℃における1×SSC中でのフィルター洗浄である。当業者らは、必要な場合、プローブの長さ等の因子に適合させるための温度、イオン強度等を調節する方法を認識する。
【0092】
キメラ抗原をコードするポリヌクレオチドの実施態様としては、配列番号27、配列番号29、配列番号31、配列番号33、配列番号35、配列番号37、配列番号39、配列番号41、配列番号43、配列番号45、配列番号47および配列番号49のいずれかから選択された配列を有するキメラ抗原をコードするポリヌクレオチド、または配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および配列番号48のいずれかから選択されたキメラ抗原のヌクレオチド配列(ここで、Tは必要に応じてUであり得る)が挙げられる。例えば、キメラ抗原ヌクレオチドの実施態様は、限定するものではないが、以下を含む:
(a)配列番号26のヌクレオチド1〜1326、配列番号28のヌクレオチド1〜2004、配列番号30のヌクレオチド1〜1350、配列番号32のヌクレオチド1〜1293、配列番号34のヌクレオチド1〜1794、配列番号36のヌクレオチド1〜1581、配列番号38のヌクレオチド1〜1389、配列番号40のヌクレオチド1〜1347、配列番号42のヌクレオチド1〜2157、配列番号44のヌクレオチド1〜1395、配列番号46のヌクレオチド1〜1905、または配列番号48のヌクレオチド1〜2484(ここで、Tはまた、Uでもあり得る)の配列番号に記載された配列を含むか、あるいはこれらの配列からなるポリヌクレオチド;
(b)配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46または配列番号48に記載された配列に対して、少なくとも80%相同性である配列を有するポリヌクレオチド;
(c)登録番号080504−03、登録番号080504−04、登録番号080504−05、登録番号080504−02および登録番号080504−01としてそれぞれカナダ国際寄託当局(Bureau of Microbiology at Health Canada)に寄託された、pFastBacHTa HBV S1/S2−TBD、pFastBacHTa HBV core−TBD、pFastBacHTa HCV core(1−177)−TBD、pFastBacHTa HCV NS5A−TBDおよびpFastBacHTa HCV E2−TBDと命名されたプラスミドの一つに含まれるDNAによってコードされる配列を有するキメラ抗原をコードするポリヌクレオチド;
(d)キメラ抗原の配列が、配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635または配列番号49のアミノ酸1〜828の配列である、キメラ抗原をコードするポリヌクレオチド;
(e)配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635または配列番号49のアミノ酸1〜828により記載される全アミノ酸配列に対して、少なくとも90%同一性であるキメラ抗原関連タンパク質をコードするポリヌクレオチド;
(f)(а)〜(d)のいずれか一つのポリヌクレオチドに完全に相補的であるポリヌクレオチド;ならびに
(g)(а)〜(f)のポリヌクレオチドに対してストリンジェント条件下で選択的にハイブリダイズするポリヌクレオチド。
【0093】
また、本発明は、キメラ抗原ポリヌクレオチド、その類似体または相同体を含む組換えDNA分子またはRNA分子を提供する。これらとしては、ファージ、プラスミド、ファージミド、コスミド、YAC(酵母の人工的染色体)、BAC(細菌の人工的染色体)、ならびに当該分野で周知の種々のウイルスベクターおよび非ウイルスベクター、およびこのような組換えのDNA分子もしくはRNA分子で形質転換されたか、またはトランスフェクションされた細胞が挙げられるが、これらに限定されない。このような分子を作製する方法は、周知である(例えば、Sambrookら(前出)(1989)を参照のこと)。
【0094】
さらに、本発明は、キメラ抗原ポリヌクレオチド、その類似体または相同体を含む組換えDNA分子を、適切な原核細胞または真核細胞の宿主細胞内に含む宿主−ベクター系を提供する。適切な真核宿主細胞の例としては、酵母細胞、植物細胞または、哺乳動物細胞もしくは昆虫細胞等の動物細胞(例えば、Sf9、Sf21、Drosophila S2またはHigh Five(登録商標)細胞のようなバキュロウイルス感染細胞)が挙げられる。適切な哺乳動物細胞の例としては、DU145およびTsuPr1等の多様な前立腺癌細胞株、他のトランスフェクション可能または形質導入可能な前立腺癌細胞株、初代細胞(PrEC)、ならびに組換えタンパク質の発現に日常的に用いられる多くの哺乳動物細胞(例えば、COS、CHO、293、293T細胞)が挙げられる。より具体的には、キメラ抗原またはフラグメント、その類似体または相同体のコード配列を含むポリヌクレオチドは、当該分野で日常的に用いられ、広く知られている多くの宿主−ベクター系を用いてそのキメラ抗原を作製するために用いられ得る。
【0095】
キメラ抗原の発現に適する広い範囲の宿主−ベクター系が利用できる(例えば、Sambrookら(1989)(前出);Ausubelら、1.0.1〜1.16.16、9.01〜9.17.3および13.4.1〜13.6.5を参照のこと)。昆虫細胞発現用の好ましいベクターとしては、pFastBac HTa(Invitrogen)が挙げられるが、これに限定されない。そのような発現ベクターを用いて、キメラ抗原を、例えばSf9、Sf21、Drosophila S2およびHigh Fiveを含むいくつかの昆虫細胞株において発現させることができる。もしくは、好ましい酵母発現系としては、Saccharomyces cerevisiae、Schizosaccharomyces pombe、Pichia pastorisおよびPichia augustが挙げられる。本発明の宿主−ベクター系は、キメラ抗原の生産に有用である。
【0096】
キメラ抗原またはその類似体または相同体は、キメラ抗原をコードする構築物をトランスフェクトした細胞により作ることができる。例えば、Sf9細胞は、キメラ抗原またはその類似体または相同体をコードする発現プラスミドでトランスフェクションされ得、そのキメラ抗原は、Sf9細胞において発現され、そしてそのキメラ抗原は、標準的な精製法を用いて単離される。当該分野で周知の多様な他の発現系もまた、用いられ得る。キメラ抗原をコードする配列にフレーム内で連結させたリーダーペプチドをコードする構築物の発現は、分泌型キメラ抗原を作製するために使用され得る。
【0097】
ここで論じるように、遺伝子コードの重複性が、キメラ抗原遺伝子配列中の変異を可能にする。特に、特定の宿主種はしばしば特定のコドン選択性を有することが当該分野で公知であり、所望の宿主について好ましいように、開示された配列を適合し得る。例えば、望ましい類似体コドン配列は、典型的にはより高い頻度コドンにより置換された稀なコドン(すなわち、所望の宿主の公知配列において、約20%未満の利用頻度を有するコドン)を有する。特定の種についてのコドン選択性は、例えば、世界的なウェブのURL www.kazusa.or.jp/codon等のインターネット上で利用できるコドン利用表を用いて計算される。
【0098】
さらなる配列の改変が、細胞宿主中のタンパク質発現を増強することは公知である。これら改変としては、擬似ポリアデニル化シグナル、エキソン/イントロンスプライス部位シグナル、トランスポゾン様リピートをコードする配列、および/または遺伝子発現に有害な他のよく特性付けられた配列を排除することを包含する。上記配列のGC含量は、宿主細胞において発現された公知の遺伝子を参照することによって計算された、所与の細胞宿主に対しての平均的なレベルに調節される。可能な場合、上記配列は、予想されるヘアピンの二次mRNA構造を避けるように改変される。他の有用な改変としては、Kozak,Mol.Cell Biol.,9:5073−5080(1989)に記載のような、オープンリーディングフレームの開始点における翻訳開始共通配列の付加を包含する。真核生物のリボゾームが5’付近のAUGコドンにおいてのみ翻訳を開始するという一般的な規則は、稀な条件下においてのみ無効とされることを当業者らは理解する(例えば、Kozak Proc Nat’l Acad Sci USA 92(7):2662−2666(1995)およびKozak Nucl Acids Res 15(20):8125−8148(1987)を参照のこと)。
【0099】
(G.本発明の薬学的組成物)
本発明の一つの特徴は、免疫応答ドメインと、抗体フラグメントを含む標的結合ドメインとを含むキメラ抗原および薬学的に受容可能な賦形剤を含む薬学的組成物に関する。治療利用において、上記薬学的組成物は、抗原に対する効果的なB細胞、細胞障害性Tリンパ球(CTL)および/またはヘルパーTリンパ球(Th)の応答を引き起こし、かつ感染を遮断するか、あるいは症状および/もしくは合併症もしくは病気もしくは疾患を治療するか、または少なくとも部分的に抑制または遅らせるのに十分な量で、被験体に投与され得る。この使用に効果的な量は、例えば、投与される特定の組成物、投与方法、処置される病気の段階と重症度、その被験体の体重および健康の一般的な状態、ならびに処方する医師の判断に依存する。
【0100】
(キメラ抗原による)最初の治療的免疫化のための投薬量は、一般的には、低い値が約1ng、5ng、50ng、500ngまたは1,000ngであり、高い値が約10,000μg、20,000μg、30,000μgまたは50,000μgであるような単位投薬量範囲に存在する。ヒト用の投薬量値は、被験体70キログラムあたり典型的には約500ng〜約50,000μgの範囲にある。数週間〜数ヶ月にわたる追加免疫レジメンに準じる約1.0ng〜約50,000μgの間のキメラ抗原の追加免疫投薬量が、被験体の応答と状態に基づきながら投与され得る。少なくとも、臨床的な症状または実験室試験から、症状が防止されるか、抑止されるか、遅らされるか、またはなくなることが示されるまで、そしてその後のある期間にわたって、投与は継続されなければならない。投薬量、投与経路および投与計画は、当該分野で公知の方法にしたがって調節される。
【0101】
キメラ抗原のヒト単位用量形態は、典型的には、ヒト単位用量の受容可能なキャリア(1つの実施態様においては、水性キャリア)を含有する薬学的組成物中に含まれ、そしてこのようなポリペプチドのヒトへの投与に有用であることが当業者に公知である体積/量で投与される(例えば、Remington:The Science and Practice of Pharmacy,第20版,A.Gennaro,Editor,Lippincott Williams & Wilkins,Baltimore,Md.,2000を参照)。当業者により理解されるように、多様な因子が、特定の場合における理想的な用量に影響を及ぼしうる。そのような因子としては、例えば、キメラ抗原の半減期、キメラ抗原の結合親和性、組成物の免疫原性、所望の定常状態の濃度レベル、投与経路、治療頻度、および本発明の治療方法と組み合わせて使用される他の物質の影響、ならびに特定の被験体の健康状態が挙げられる。
【0102】
ある実施態様において、本発明の組成物は、深刻な病状(すなわち、生命を脅かすか、または潜在的に生命を脅かす状態)に使用される。そのような場合、本発明の好ましい組成物中のキメラ抗原が比較的毒性のない性質であるという結果として、これら述べられた投薬量に対して実質的に過剰なこれらキメラ抗原を投与することが可能であり、治療を行う医師には望ましいと感じられ得る。
【0103】
薬学的処方物における本発明のキメラ抗原の濃度は、広く変動し得る。すなわち、約0.1重量%未満、通常は約2重量%、または少なくとも約2重量%から20〜50重量%またはそれ以上まで変動し得、選択された投与の具体的な態様にしたがって主に液体容量、粘度等によって選択される。
【0104】
この薬学的組成物は、非経口、髄腔内、血管内、静脈内、筋肉内、経皮、皮内、皮下、鼻内、局所、経口、直腸、膣、肺または腹腔内等の当該分野で公知の任意の経路を介して送達され得る。好ましくは、上記組成物は、皮下投与または皮膚内投与等の非経口経路により送達される。
【0105】
上記薬学的組成物は、所望のキメラ抗原を、意図される投与経路に適する適切なビヒクルと混合することによって調製され得る。本発明の薬学的組成物の作製において、このキメラ抗原は、通常賦形剤と混合されるか、賦形剤で希釈されるか、またはカプセル、匂い袋、紙または他の容器の形態であり得るキャリア内に封入される。薬学的に受容可能な賦形剤が希釈剤として役立つ場合、この賦形剤は治療剤用のビヒクル、キャリアまたは媒体として作用する固体、半固体または液体の物質であり得る。よって、上記組成物は、錠剤、丸剤、散薬、ロゼンジ、匂い袋、カシェ剤、エリキシール、懸濁液、乳化液、溶液、シロップ、(固体としてまたは液体媒体中の)エーロゾル、例えば10重量%までのキメラ抗原を含有する軟膏、軟性または硬性のゼラチンカプセル、座薬、滅菌注射溶液および滅菌包装散薬の形態であり得る。
【0106】
適当な賦形剤のいくつかの例としては、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アカシアゴム、リン酸カルシウム、アルギン酸塩、トラガカント、ゼラチン、ケイ酸カルシウム、微結晶セルロース、ポリビニルピロリドン、セルロース、滅菌水、シロップおよびメチルセルロースが挙げられるが、これらに限定されない。これらの処方物はさらに、タルク、ステアリン酸マグネシウムおよび鉱油等の潤滑剤、湿潤剤、乳化懸濁剤、メチルベンゾエートおよびプロピルヒドロキシベンゾエート等の保存剤、甘味剤、ならびに調味剤が挙げられ得る。本発明の組成物は、当該分野で公知の操作を用いることにより、被験体に投与した後にキメラ抗原の即効性放出、徐放性放出または遅延性放出を提供するように処方され得る。例えば、Remington(前出)の903〜92ページおよび1015〜1050ページを参照のこと。
【0107】
錠剤等の固体組成物を調製するために、上記キメラ抗原は、薬学的賦形剤と混合されて本発明のキメラ抗原の均一混合物を含む固体の予備処方組成物を形成する。これらの予備処方組成物を均一であると称する場合、上記キメラ抗原が、その組成物が錠剤、丸剤およびカプセル等の効果の等しい単位投薬形態に容易に再分割され得るように、組成物中に均一に分散していることを意味する。
【0108】
本発明の錠剤または丸剤は、被覆され得るか、または長期間の作用という利点を与える投薬形態を提供するように配合され得る。例えば、上記錠剤または丸剤は、内投薬成分と外投薬成分とを含み得、後者は前者を被う封筒のような形である。これらの二成分は、腸溶性層によって分離され得る。この層は、胃内の分解に対して抵抗性があり、内成分をそのまま十二指腸に通過させ得るか、または放出を遅らせ得る。多様な物質がそのような腸溶性層または被覆に用いられ得、そのような物質として、多くのポリマー酸およびポリマー酸とセラック、セチルアルコールおよび酢酸セルロースのような物質との混合物が挙げられる。
【0109】
本発明の新規組成物が経口または注射による投与で取り込まれ得るような液体形態としては、水性溶液、適当に味付けしたシロップ、水性または油性の懸濁液、およびトウモロコシ油、綿実油、ゴマ油、ココナッツ油またはピーナッツ油等の食用油を有する味付けした乳化物ならびにエリキシールおよび類似の薬学的ビヒクルが挙げられる。
【0110】
非経口投与用組成物の調製において、刺激を減少させる浸透圧調節に対して、厳密な注意を払う必要がある。再構成可能な組成物は、乾燥形態の包装された滅菌固体である。再構成可能な組成物が望ましいのは、乾燥固体として保存した場合、即座に投与するために溶液を準備するよりもさらに安定なためである。通常、乾燥固体は、その固体が最適な湿度範囲に確実に維持されるようにブチルゴムの蓋を有する滅菌容器に包装される。再構成可能な乾燥固体は、乾燥充填法、噴霧乾燥法または凍結乾燥法により形成される。これら方法の説明は、例えばRemington(前出)、681〜685ページと802〜803ページに見出され得る。
【0111】
非経口注射用の組成物は、一般的に希釈されており、高い比率で存在する成分はビヒクルである。通常、このビヒクルは治療活性を持たず、非毒性であるが、吸収に適する形態で、上記キメラ抗原を体組織に提示する。通常、吸収は、キメラ抗原が水溶液として提示される場合に、最も容易かつ完全に起こる。しかし、液体混和性液体によるビヒクルの改変または水非混和性液体による置換は、吸収速度に影響を与えうる。好ましくは、本組成物について最も価値のあるビヒクルは、等張食塩水である。注射に適する組成物の調製において、水性ビヒクル、水混和性ビヒクルおよび非水性ビヒクルを使用し得る。
【0112】
さらなる物質が、上記組成物の品質を向上または保護するために、本発明の注射可能な組成物中に含まれ得る。したがって、加えられた物質は、溶解性に影響を与え得、被験体の快適さを提供し得、化学安定性を増強し得るか、または微生物の成長に対して調製物を保護し得る。よって、この組成物は、適当な可溶化剤、抗酸化剤として働く物質および微生物の成長を妨げる保存剤として働く物質を含有し得る。これらの物質は、それらの機能に適切であるが、組成物の作用に悪影響を与えないような量で存在する。適当な抗微生物剤の例としては、チメロサール、塩化ベンゼトニウム、塩化ベンズアルコニウム、フェノール、p−ヒドロキシ安息香酸メチルおよびp−ヒドロキシ安息香酸プロピルが挙げられる。適当な抗酸化剤は、Remington(前出)、1015〜1017ページに見出され得る。
【0113】
ある実施態様において、リポソーム、ナノカプセル、マイクロカプセル、液体粒子、ベシクル等が、本発明のキメラ抗原の投与ために用いられる。特に、本発明の組成物は、送達のために、液体粒子、リポソーム、ベシクル、ナノスフェアまたはナノ粒子等にカプセル化されて処方され得る。あるいは、本発明の組成物は、共有結合または非共有結合のいずれかにより、このようなキャリアビヒクルの表面に結合され得る。
【0114】
リポソームを介して投与される組成物はまた、以下のように役立ち得る:1)リンパ系組織等の特別な組織に対してキメラ抗原を標的化する;2)抗原提示細胞に対して選択的に標的化する;または3)ペプチド組成物の半減期を増加させる。リポソームとしては、乳化物、泡、ミセル、不溶性単層、液晶、リン脂質分散物、薄層等が挙げられる。これらの調製物において、送達されるキメラ抗原は、リポソームの一部として、単独またはCD45抗原に結合するモノクローナル抗体等のリンパ系細胞間に広く認められるレセプターに結合する分子と組み合わせて取り込まれるか、あるいは他の治療組成物または免疫原組成物とともに取り込まれる。よって、本発明の所望のキメラ抗原により充填または装飾されたリポソームは、そのリポソームがキメラ抗原を送達するリンパ系細胞の部位に向かい得る。本発明に従って用いられるリポソームは、中性および負電荷のリン脂質およびコレステロール等のステロールを一般的に包含する、標準的ベシクル形成脂質から形成される。脂質の選択は、例えば、血流中のリポソームの大きさ、酸不安定性と安定性を考慮することにより通常行われる。Szokaら、Ann.Rev.Biophys.Bioeng.9:467−508(1980)および米国特許4,235,871号、米国特許4,501,728号、米国特許4,837,028号および米国特許5,019,369号に記載されるように、多様な方法がリポソームを調製するために利用できる。キメラ抗原を含むリポソーム懸濁液は、とりわけ投与法、送達されるキメラ抗原および処置される病気の段階にしたがって変動する用量において静脈内、局所的、局部等に投与され得る。
【0115】
吸入用または吹入れ用の組成物としては、薬学的に受容可能な水性溶媒もしくは有機溶媒、またはそれらの混合物中の溶液または懸濁液、ならびに粉末が挙げられる。液体または固体の組成物は、本明細書中に記載されるような適当な薬学的に受容可能な賦形剤を含有し得る。この組成物は、局所性効果または全身性効果のために、口または鼻の呼吸経路により投与され得る。薬学的に受容可能な溶媒中の組成物は、不活性気体を用いることにより霧状化され得る。霧状化された溶液は、噴霧用装置から直接的に吸入され得るか、またはこの噴霧用装置は顔マスクテント(facemask tent)または間欠的陽圧呼吸装置に装着され得る。溶液、懸濁または粉末組成物は、適切な方法で処方物を送達する装置から、好ましくは経口または経鼻にて投与され得る。
【0116】
本発明の方法に用いられるもう一つの処方は、経皮送達装置(「パッチ」)を用いる。そのような経皮パッチを用いて、本発明のキメラ抗原の連続的または非連続的な注入を制御された量で提供し得る。薬学的物質の送達のための経皮パッチの構築と使用は、当分野で周知である。例えば、本明細書において参考として援用される米国特許5,023,252号を参照のこと。そのようなパッチは、薬学的物質の連続的送達、拍動性送達またはオンデマンド送達のために構築され得る。
【0117】
さらに、キメラ抗原および薬学的賦形剤に加えて、少なくとも一つの抗ウイルス治療剤または化学療法剤を含むことが有利であり得る。抗ウイルス治療剤としては、以下が挙げられるが、これらに限定されない:擬似ペプチド(例えば、アンプレナビル、インジナビル、ロピナビル、ネルフィナビル、リトナビルおよびサキナビル)、ポリヌクレオチド(例えば、アンプリゲンおよびホミビルセン)、プリン/ピリミジノン類(例えば、アバカビル、アシクロビル、アデフォビル、シドフォビル、シタラビン、ジダノシン、ジデオキシアデノシン、ジピボキシル、エドクスウジン、エムトリシタビン、エンテコビル、ファムシクロビル、ガンシクロビル、イドクスウリジン、イノシン・プラノベクス、ラミブジン、MADU、ペンシクロビル、ソリブジン、スタブジン、テノフォビル、トリフルリジン、バラシクロビル、バルガンシクロビル、ビダラビン、ザルシタビンおよびジドブジン)、シアル酸類似体(例えば、オセルタミビルおよびザナミビル)、アセマンナン、アセチルロイシンモノエタノールアミン、アマンタジン、アミジノマイシン、アテビリジン、カプラビリン、デラビルジン、n−ドコサノール、エファビレンズ、ナトリウムフォスカーネット、インターフェロン−α、インターフェロン−β、インターフェロン−γ、ケトキサール、リゾチーム、メチサゾン、モロキシジン、ネビラピン、ペンタフシド、プレコナリル、ポドフィルロトキシン、リバビリン、リマンチジン、スタリマイシン、スタトロン、ターマカムラおよびトラオマンタジン。他の適当な抗ウイルス剤は、Remington(前出)、87章、抗感染剤、1507〜1561ページ、特に1555〜1560ページに説明されている。本発明の薬学的組成物に包含される好ましい抗ウイルス治療剤として、アデフォビル、ジピボキシル、エンテコビル、ラミブジンおよびリバビリンが挙げられる。
【0118】
いくつかの実施態様において、Bリンパ球またはTリンパ球を初回刺激する(prime)少なくとも一つの成分を本発明の薬学的組成物に含むことが望ましい。脂質はCTLをインビボで初回刺激し得る物質として同定されている。例えば、パルミチン酸残基を、リジン残基のεアミノ基とαアミノ基に結合させ、次にこれをGly、Gly−Gly−、Ser、Ser−Ser等の一つ以上の連結残基を介して免疫原性ペプチドに連結し得る。次に、この脂質化ペプチドは、ミセルまたは粒子のいずれかで直接投与され得るか、リポソームに取り込まれ得るか、またはアジュバント、例えば不完全フロイントアジュバント中に乳化させ得る。好ましい実施態様において、特に効果的な免疫原性組成物は、免疫原性ペプチドのアミノ末端に対して例えばSer−Serの連結により結合させたLysのεアミノ基とαアミノ基とに結合させたパルミチン酸を含む。
【0119】
CTL応答の脂質初回刺激のもう一つの例として、トリパルミトイル−S−グリセリルシステインリセリル−セリン(P3CSS)等のE.coliリポタンパク質を用いて、適当なペプチドに共有結合させた場合のウイルス特異的CTLを初回刺激し得る(例えば、Deresら、Nature 342:561(1989))。本発明のキメラ抗原は、例えばP3CSSに結合させ得、リポペプチドを個体に投与して、標的抗原に対する免疫応答を特異的に初回刺激し得る。
【0120】
本発明の組成物はアジュバントの使用を必要とすべきではないが、アジュバントを用いることはできる。宿主の種に基づいて、免疫学的応答を高める多様なアジュバントが用いられ得、そのようなアジュバントとしては、フロイント(完全および不完全)、水酸化アルミニウム等の無機ゲル、リソレシチン等の表面活性剤、洗剤、プルロニックポリオール、ポリアニオン、ペプチド、油性乳化物、キーホールリンペットヘモシアニン、ジニトロフェノール、免疫促進ポリ塩基配列およびBCG(カルメット‐ゲラン杆菌)およびCorynebacterium parvum等の潜在的に有用なヒトアジュバントが挙げられるが、これらに限定されない。さらなるアジュバントもまた、当該分野において周知である。
【0121】
(H.製品)
本発明のもう一つの特徴は、キメラ抗原を含有する組成物を保持する容器を含む製品を提供する。この製品は、上記組成物を非経口(例えば、皮下、筋内、皮内、鼻内または血管内)で、どのように投与するかを説明する印刷されたラベル説明と組み合わせて、注射に適切であるか、または注射用に再構築するためのものである。上記組成物は、この組成物と顕著に相互作用せず、かつそれが非経口使用であることを示す適当なラベルで標識された任意の適当な容器に含まれる。容器に組み合わされるものは、上記の治療方法に合わせたラベル説明である。本発明の組成物を保持する容器は、患者、医師、看護婦またはそれ以外の専門家がキメラ抗原を投与し得るように、注射用の適当な針とシリンジとを有する注射に適する液体組成物を有する容器であり得る。あるいは、上記組成物は、この組成物を溶解または懸濁させる水性ビヒクルまたは非水性ビヒクルと組み合わされるか、またはそれらのビヒクルで希釈されるキメラ抗原の可溶性の種類を含有する乾燥組成物あるいは濃縮組成物であり得る。あるいは、上記容器は、液体中に懸濁剤を有するか、塩の不溶性の種類が懸濁されるビヒクルと組み合わせるための塩の不可溶性の種類であり得る。適当な容器は、Remington(前出)の788〜789ページ、805ページ、850〜851ページおよび1005〜1014ページに説明されている。
【0122】
本発明のキットは、典型的には、上記の容器と、緩衝液、希釈液、フィルター、針、シリンジおよび使用説明書を備える包装挿入物を含む商品と使用者の観点から望ましい材料を含む一つ以上の他の容器を含む。組成物が特定の治療または非治療的利用のために用いられことを示すラベルが容器上に存在してよく、かつそのラベルはインビボまたはエキソビボのいずれかでの使用についての説明、例えば上記のような説明を示すこともできる。説明および/または他の情報は、キットに含まれる挿入物に含ませることもできる。
【実施例】
【0123】
(V.実施例)
以下の非限定的な実施例は、本発明のさらなる説明を提供する。
【0124】
(A.実施例1:TBD発現ベクターの構築)
CH1−ヒンジ−CH2−CH3領域の一部のアミノ酸をコードするマウスIgG1 DNA配列を、HBV表面抗原(sAg)に対してmAbを産生するハイブリドーマ(2C12)から単離されたmRNAから作った。全mRNAを、Trizol(登録商標)試薬(Gibco BRLカタログ番号15596−026)を用いて単離し、標的結合ドメイン(TBD;マウス免疫グロブリンフラグメント)のcDNAを、Superscript First−strand Synthesis(Invitrogenカタログ番号11904−018)を用いたRT−PCRにより作った。このPCRプライマーは、リンカーペプチド(5’末端の−SRPQGGGS−(配列番号1))をコードするリンカー配列、5’末端の独自のNotI部位および3’末端の独自のHindIII制限部位を含んだ。得られたcDNAは(5’NotI)−リンカー配列−CH1(VDKKI)(配列番号2)、−ヒンジ領域−CH2−CH3−(3’HindIII)を含む。各酵素による消化後に、同じ制限酵素部位を用いて、フラグメントをpFastBac HTa発現ベクタープラスミド(Invitrogen)に連結する。PCR増幅に用いられる5’プライマーは(センス)5’TGTCATTCTGCGGCCGCAAGGCGGCGGATCCGTGGACAAGAAAATTGTGCCCAGG(配列番号3)であり、そして3’プライマーは(アンチセンス)5’ACGAATCAAGCTTTGCAGCCCAGGAGAGTGGGAGAG(配列番号4)であり、それぞれNotIとHindIII部位を有した。以下は、方向性クローニング(directional cloning)に用いられるプロトコールである。生成したフラグメントを各酵素で消化し、アガロースゲルで精製し、ベクタープラスミドにクローニングした。このDNA配列とORFの正しさを標準的な配列決定方法を用いて実証した。
【0125】
標的結合ドメインをコードするDNAのpFastBac HTaドナープラスミドへのクローニング後に、組換えタンパク質をBac−to−BacTMバキュロウイルス発現系(Invitrogen)を用いて発現させた。クローニングした遺伝子を部位特異的転位によりE.coli(DH10Bac)株中のバキュロウイルスシャトルベクターに移した。DH10Bac細胞は、カナマイシン耐性を付与するシャトルベクターと、トランスポザーゼをコードし、かつテトラサイクリンに対する耐性を付与するヘルパープラスミドとを含む。コンピテントなDH10Bac細胞の100μlのアリコートを氷上で解凍し、pFastBac HTaベースのプラスミドを加え、その混合物を氷上で30分間インキュベートした。この混合物を45秒間42℃で熱ショックを与え、次に氷上で2分間冷やした。次に、この混合物を900μLのLB培地に加え、4時間、37℃でインキュベートした。この形質転換細胞を、LBで、10−1〜10−2まで連続的に希釈し、100μlの各希釈物を、50μl/mlカナマイシン、7μg/mlゲンタマイシン、10μg/mlテトラサイクリン、100μg/ml X−galおよび40μg/ml IPTGを補充したLBアガープレートにプレートし、少なくとも36時間、37℃でインキュベートした。ゲンタマイシン耐性をpFastBac HTaにより賦与し、X−galとIPTG(イソプロピルチオ−β−D−ガラクトシド)を用いて、白色コロニー(組換えプラスミド)と青色コロニー(非組換え体)との間を区別した。白色コロニーを取り、50μg/mlカナマイシン、7μg/mlゲンタマイシンおよび10μg/mlテトラサイクリンを補充した2mlのLBに接種し、攪拌下に37℃で一晩インキュベートした。滅菌ループを用いて少量の一晩培養物を採取し、得られた試料を、50μg/mlカナマイシン、7μg/mlゲンタマイシン、10μg/mlテトラサイクリン、100μg/ml X−galおよび40μg/ml IPTGを補充した新鮮なLB寒天プレートに縞状に塗布し、少なくとも36時間、37℃でインキュベートして白色表現型を確認した。組換えバクミドを標準的なプロトコール(Sambrook(前出))により単離し、そのDNA試料を40μlのTE(10mMトリス塩酸、pH8、1mM EDTA)に溶解し、トランスフェクションに用いた。
【0126】
バキュロウイルスを作るために、バクミドをSf9昆虫細胞にトランスフェクションした。Sf9細胞(9×105)を、2mlのESF921(Expression Systems)中の6ウエル細胞培養皿(35mmウエル)の各ウエルに撒き、少なくとも1時間、27℃で付着させた。Sf9細胞の供給者により提供されたプロトコールにしたがってCellfection(登録商標)試薬(Invitrogen、カタログ番号10362−010)を用いて、トランススフェクションを行った。トランスフェクション後、その細胞を27℃で72時間インキュベートした。バキュロウイルスを含有する培地を集めて、4℃、暗所で保存した。
【0127】
トランスフェクションの効率はバキュロウイルスDNAの生産について検査することによって実証した。単離されたバキュロウイルスDNAをPCRに供して、TBDをコードする挿入遺伝子をスクリーニングした。使用されたプライマーは(センス)5’TATTCCGGATTATTCATACCG(配列番号5)および3’(アンチセンス)5’CTCTACAAATGTGGTATGGC(配列番号6)であった。増幅生成物をアガロースゲル(0.8%)で泳動した。細胞中の異種タンパク質の発現を、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)および6×Hisタグモノクローナル抗体(Clonetech)をプローブとして用いたウエスタンブロットにより実証した。
【0128】
バキュロウイルスの生産とタンパク質の発現を一旦確認したら、ウイルス生産を増幅して、標的結合ドメインをコードする遺伝子を有するバキュロウイルスの濃縮原液を作製した。バキュロウイルスを少なくとも二回増幅することは当分野で標準的な慣行であり、ここに記載のすべてのプロトコールにおいて、この標準的な慣行を遵守した。二回目の増幅の後、作製したバキュロウイルスの濃度を、キットの製造者(Invitrogen)により記載されたプロトコールによるプラークアッセイを用いて定量した。High Five細胞を感染させるためのウイルスの最も適当な濃度と、所望のタンパク質生産の最適な時間とを設定した。一般的に、TBDの発現のために、1のMOIと48時間を用いた。
【0129】
(B.実施例2:キメラ抗原発現ベクターの構築)
表2に示した5’センスプライマーと3’アンチセンスプライマーとを用いたPCR法を使用して、所望のウイルス抗原をコードするDNAを鋳型から作った。得られた増幅フラグメントの5’末端は、独自の制限部位「5’酵素(enzy)」を含み、そして3’末端は独自の制限部位「3’酵素(enzy)」を含んでおり、それら各々をライゲーションのために用いた。
【0130】
【表2】
増幅DNAを適当な5’制限酵素および3’制限酵素で消化し、pFastBac HTa発現ベクターに連結して、ウイルス抗原のみの発現プラスミドを作った。各酵素による消化後に、実施例1に記載のように、DNAの同フラグメントをプラスミドpFastBac HTa−TBDに連結させて、標的結合ドメインに融合させたウイルス抗原の発現プラスミドを作った。得られたプラスミドを用いて、実施例1に記載のように、組換えバキュロウイルスを作り、続いてキメラ抗原の発現に用いた。このキメラ抗原のDNA配列とアミノ酸配列とを表3に示す。
【0131】
【表3】
(C.実施例3:TBD、ウイルス抗原およびキメラ抗原の発現および精製)
標準化感染多重度(MOI)の組換えバクミドを用いて、High Five(登録商標)昆虫細胞を感染させた。懸濁培養のために、細胞を3×105細胞/mLの密度で撒き、細胞密度が2〜3×106細胞/mLに達するまで27.5℃、138rpmでの攪拌下にインキュベートした。標準化量の各組換えバキュロウイルスを細胞に加えた。インキュベーション温度は27.5℃で、適当な感染時間を個々のタンパク質発現について標準化した。これらの細胞を10分間、4℃で2,500rpmにおける遠心分離により集め、組換えタンパク質の精製に用いた。細胞の使用しない部分は液体窒素中に瞬時に凍らせて、−70℃で保存した。
【0132】
組換えタンパク質を変性条件下で精製した。この細胞を、100mMのNaH2PO4、10mM Tris、300mM NaCl、10mMイミダゾール、pH8.0に6MグアニジウムHClを含有する緩衝液(可溶化緩衝液)中に可溶化した。その懸濁物を60ワットの出力設定で1分/パルスの5パルスにて氷上で超音波処理し、室温で1時間混合した。この溶解物を10,000×gで10分間遠心分離して、未破壊細胞と細胞破片を除去した。この上清を、前もって可溶化緩衝液で平衡化させておいたNi−NTAアガロース(Qiagen)ビーズカラム(1×5cm/100mL細胞溶解物)にロードした。ローディングの後に、カラムを100mMのNaH2PO4、10mM Tris、300mM NaCl、40mMイミダゾール、pH8.0中の6MグアニウムHCl(洗浄緩衝液1)の20カラム体積分で洗浄し、引き続いて100mMのNaH2PO4、10mM Tris、300mM NaCl、40mMイミダゾール、pH8.0中の8M尿素(洗浄緩衝液2)の20カラム体積分で洗浄した。結合タンパク質を、8M尿素、100mMのNaH2PO4、10mM Tris、300mM NaCl、250mMイミダゾール、pH8.0を含有する緩衝液(溶出緩衝液)で溶出した。タンパク質を含有する画分を集めて、4℃で透析緩衝液(10mM NaH2PO4、300mM NaCl)を複数回変えながら透析した。精製タンパク質を、SDSゲル電気泳動、等電点電気泳動および発現タンパク質の異なるドメインに対する抗体を用いたウエスタンブロット分析等の標準的な生化学技術を用いて特性付けた。
【0133】
(D.実施例4:キメラ抗原融合タンパク質を用いた「自己」タンパク質に対する寛容の破壊)
キメラ抗原融合タンパク質に対する免疫応答を評価するために、精製したHBV S1/S2、TBDまたはS1/S2−TBDタンパク質でマウスを免疫し、個々のタンパク質に対して作られた抗体を定量した。免疫されたマウスから集めた脾臓T細胞の増殖を、各タンパク質による誘発(challenge)後に評価した。
【0134】
15週齢のBALB/cマウスを免疫に用いた。マウスにS1/S2−TBD(4.15μg)、S1/S2(4.15μg)またはTBD(4.15μg)を二週間の間隔で4回皮下注射した。免疫の開始前と各免疫の一週間後に、血液試料を集めた。血清を凝固血液試料から調製し、注射した各抗原に対して宿主動物により作られた抗体量の予測に用いた。
【0135】
(1.HBV S1/S2、TBDまたはS1/S2−TBDに対する抗体の検出のためのELISA)
96ウエルプレートに、抗原HBV S1/S2、TBDまたはS1/S2−TBDを1.0μg/mLの濃度にて一晩、4℃で被覆した。このプレートを2%BSAを含有するPBSで洗浄した。各動物からの希釈血清を各ウエルに多様な希釈率(1:10〜1:500)で加え、37℃で1時間インキュベートした。このプレートを0.05%のTween 20を含有するPBS(洗浄緩衝液)で洗浄した。ヤギ抗マウスIgG Fab西洋ワサビペルオキシダーゼ(HRP)(1:5000)希釈物をウエルに加えて、37℃で1時間インキュベートした。プレートを洗浄緩衝液で洗浄し、2−2’アジノ−ジ−(3−エチルベンジルチアゾリン−6−スルフォネート)(KPL、Guildford、UK)を用いて発色させた。試料中の発色の光学密度をELISAプレートリーダー(Molecular Devices、USA)を用いて405nmの波長で測定した。実験の陰性対照は、同じ動物の免疫前の血清であり、その値をすべての実験値から引いた。HBV S1/S2−TBDで免疫したマウスに関する結果を表4に示す。このキメラ抗原は、キメラ抗原(S1/S2−TBD)に対する強い抗体応答を引き出す。
【0136】
【表4】
ここでの抗体応答は、多価(または多エピトープ)性のものである。表4に示す結果は、HBV S1/S2−TBDで免疫したマウスによって作られた抗体はキメラ抗原に結合し、かつプレート上に被覆されたS1/S2タンパク質標的に結合することを示す。したがって、抗体はキメラ抗原のS1/S2成分に対して作られる。同じように、HBV S1/S2−TBDで免疫したマウスにより作られた抗体は標的結合ドメインタンパク質に結合する(表4)。マウス起源のタンパク質を含有するキメラ抗体は、マウスに体液性免疫応答を生じさせ得、このキメラ抗原が「自己」抗原を「外来性」に変換し得るという証拠を示す。したがって、本来は「自己」タンパク質として処理されるタンパク質に対する寛容を壊すことが可能である。
【0137】
(2.T細胞増殖アッセイ)
四回目の免疫の一週間後に動物を屠殺し、脾臓を切除して単一の細胞懸濁液を作った。細胞を三重で、96ウエルプレートに4×105細胞/ウエルの細胞密度で撒いた。それらに、HBV S1/S2、TBDまたはS1/S2−TBDの各抗原を0.1μg/mL、1.0μg/mLおよび10μg/mLの濃度で加えた。陰性対照の細胞は、培地のみであり、T細胞増殖についての陽性対照は1.0〜5.0μg/mLのフィトヘマグルチニン(PHA)であった。細胞培養物を4日間37℃で、7%CO2の雰囲気下でインキュベートした。細胞の各ウエルに1.0mCiの3[H]チミジンを与え、さらに18時間インキュベートした。細胞を、TOMTECMACH3細胞ハーベスター(Hamden、CT、USA)を用いて採取し、ガラス繊維フィルター(Wallac Oy、Turku、フィンランド)に結合した放射活性を、Wallac Trilux 1450マイクロベータ液体シンチレーションおよび発光計数器(Wallac、USA)を用いて定量した。その結果を表5に示す。
【0138】
【表5】
T細胞増殖は、HBV S1/S2−TBD、S1/S2またはTBDで誘発されたときに見られた。このキメラ抗原による免疫は、多価のT細胞応答、すなわち同一タンパク質の異なる部分に対する応答を誘導した。マウス起源のタンパク質を含有するキメラ抗原は、マウスにおいて細胞性免疫応答を生じ得、キメラ抗原が「自己」抗原を「外来性」に変換し得るということを証拠付ける。したがって、本来は「自己」タンパク質として処置されるタンパク質に対する寛容を壊すことが可能である。
【0139】
(E.実施例5:抗原提示アッセイ)
HBV S1/S2−TBDの免疫応答を引き出す能力を、エキソビボ抗原提示アッセイを用いて測定した。樹状細胞(DC)等の抗原を負荷された抗原提示細胞(APC)によってナイーブT細胞の多刺激後に効果的なT細胞応答が発生することを、抗原特異的T細胞の数の増加ならびにTh1サイトカインIFN−γを産生するT細胞の能力を定量化することにより評価した。
【0140】
(1.接着による単球の選択)
末梢血液単核細胞(PBMC)にAIM−Vを加えて解凍した(1mlの凍結細胞に対して9mlの添加AIM−Vの比率)。次に、この細胞を200×gで5分間遠心分離し、上清を除去し、細胞を、AIM−V/1%調和血清に再懸濁し、100mm培養皿またはT−25培養フラスコに加えた。このPBMCを、7%CO2下の加湿インキュベーターで1時間、37℃でインキュベートした。非接着細胞を除去するために、培養物を数回粉砕し、上清を捨て、細胞をAIM−V培地で一回洗浄した。単球を細胞スクレーパーで採取し、300×gで5分間遠心分離した。細胞ペレットをAIM−V/2.5%調和血清に2×106細胞/mlで再懸濁し、24ウエルプレートに撒いた。サイトカインIL−4とGM−CSF(それぞれ1000IU/ml)を加えて、単球の未成熟DCへの分化を誘導した。
【0141】
(2.迅速または遅延抗原提示アッセイ)
迅速抗原提示アッセイ(APA)のために、抗原を、未成熟DCに単離の4〜24時間内に加えた。24時間後に、抗原を負荷した未成熟単球を、PGE2(1μM)、IL−1β(10ng/ml)およびTNF−α(10ng/ml)とともに24時間培養することにより誘導して成熟させた。次に、成熟DCを自己のT細胞とともに共培養した(一回目の刺激)。このT細胞は、磁気T細胞単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択を用いて、DCとして同一のPBMCからT細胞を作製された。
【0142】
次に、T細胞を、7日後にIL−2(20IU/ml)、IL−7(10ng/ml)およびIL−15(5ng/ml)の存在下で抗原を負荷した成熟DCにより再刺激した。7日間のインキュベーション後に、T細胞を抗原を負荷した成熟DCにより三回目の再刺激を行った。この三回目の刺激は6時間続け、この際にT細胞を採取し、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0143】
遅延APAのために、単球を、抗原の添加前の5〜6日間、GM−CSFおよびIL−4の存在下において未成熟DCに分化させた。抗原添加の二時間後に、未成熟DCをTNF−α(10ng/ml)およびIFN−α(50IU/ml)により成熟させた。単離の7日後に、成熟DCを(上記の)自己T細胞とともに共培養した(一回目の刺激)。
【0144】
次に、T細胞を、IL−2、IL−7およびIL−15の存在下において抗原を負荷した成熟DCによって7日後に再刺激した。7日間のインキュベーションの後、細胞を抗原を負荷した成熟DCにより三回目の再刺激を行った。18時間のインキュベーション後に、T細胞を集めて、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0145】
(2.PBMC抗原提示アッセイ)
このアッセイにおいて、最初の培養物は、抗原とIL−2とともにインキュベートされる全PBMC(すなわち、リンパ球および単球)からなり、ここではすべての細胞タイプが存在して関与するのでこの機構はインビボ免疫応答に似ていることが想定されている(Maini,M.K.ら、J.Exp.Med.191:1269−1280,2000)。PBMCを解凍し、洗浄し、すぐに抗原とともにインキュベートした。抗原の取り込みと提示とを可能にする4日間の培養後に、IL−2(20IU/ml)を加えて、さらに8日間放置した(すなわち実験の12日目)。二回目の刺激の2日前(すなわち実験の10日目)、DCを上記のように接着により単離し、すぐにGM−CSF、IL−4および抗原とともに24時間インキュベートする。Fast APAと同じように、未成熟DCを、PGE2、IL−1βおよびTNF−αの添加した後の24時間分化させる。次に、負荷した成熟DCを、IL−2、IL−7およびIL−15の存在下においてPBMC培養物に加える(二回目の刺激、実験の12日目)。三回目の刺激は、上記のように二日前に調製された抗原を負荷した成熟DCを用いて、実験の21日目に行った。6時間のインキュベーション後に、T細胞を集め、CD3、CD8およびIFN−γ発現について免疫染色し、フローサイトメトリーにより分析した。
【0146】
上記のすべての抗原提示アッセイのために、アッセイの最後のT細胞の一部をさらに3〜5日間インキュベートし、テトラマー分析(下記を参照のこと)により特異的T細胞について調べた。
【0147】
(4.HBV S1/S2はHBV S1およびHBV S2のペプチドに対してT細胞応答を引き出す)
PBMC APAを用いてT細胞を作り、次にこれを抗原特異性に関して検定した。よって、健康なHLA−A2個体からのPBMCを、96ウエルプレートにて2.5%調和血清を含有するAIM−V中、5×105細胞/mlで培養した。抗原(すなわち10μg/ml S1/S2−TBD)を加え、その細胞を4日間、37℃で培養した。次に、IL−2を20IU/mlで加え、細胞を、2〜3日毎に培地(AIM−V/2.5%調和血清および20IU/ml IL−2)を交換しながらさらに8日間培養した。12日間の培養の終わりに残る細胞のほとんどはT細胞であり、これらのT細胞を、IL−2(20IU/ml)、IL−7(10ng/ml)およびIL−15(5ng/ml)の存在下において自己抗原を負荷した成熟DCにより再刺激した。
【0148】
APAにおいてT細胞の二回目と三回目の刺激のために、抗原を負荷した成熟DCを、下記の操作を用いて48時間にわたって作った。単球を、プラスチック組織培養皿上への接着により全PBMCから単離した。その細胞(そのうち、約85%がFACS分析により決定された単球である(CD11c+、CD14+、CD19−およびCD3−))を、96ウエルプレートで、1000IU/mlのサイトカインIL−4とGM−CSFとを含む100μlのAIM−V/2.5%調和血清を含む1×105細胞/ウエルで培養し、4時間後、S1/S2−TBD等の抗原を加えた。20時間のインキュベーション後に、作られた未成熟DCを、PGE2(1×10−6M)、IL−1β(10ng/ml)およびTNF−α(10ng/ml)の存在下でさらに24時間培養することにより成熟DCに分化させた。
【0149】
T細胞を、1〜2日毎に培地(2.5%調和血清と20IU/ml IL−2を有するAIM V)を交換しながら、二回目の刺激後の7日間培養した。次に、T細胞(培養の19日目)を(上記の)IL−2、IL−7およびIL−15の存在下において(上記のように2日間の操作にわたって作られた)抗原を負荷した成熟DCによる三回目の刺激を行い、そして6時間培養後のIFN−γ産生を検定するか、または(1〜2日毎にAIM−V/2.5%調和血清および20IU/ml IL−2の培地を変更しながら)5日間培養し、次にHBV preSテトラマーを用いてHBV preS抗原に対するT細胞特異性に関する検定のいずれかを行った(培養の24日目)。
【0150】
カスタム合成iTag MHCクラスIテトラマー(Beckman Coulter)を製造者のプロトコールに従って用いてテトラマー分析を行った。すなわち、細胞を採取し、洗浄し、96ウエルv底プレートに、20μl中約2×105細胞/ウエルで移した。2μlのPE複合HLA−A*0201 preS1テトラマー(GMLTPVSTI、配列番号50)またはpreS2テトラマー(NIASHISSI、配列番号51)のどちらかとともに、CD3(抗CD3−FITC)およびCD8(抗CD8−Cy−Chrome)に特異的なmAbによって、その細胞を20℃で30分間標識した。次に、細胞を洗浄し、PBS中の2%パラホルムアルデヒドで固定し、5mlのFACSチューブに移した。この細胞を、一試料あたり80,000〜100,000個の細胞でFACSCalibur(BD Biosciences)上に得た。生存(FSC/SSCプロフィールに基づく)CD3+集団上のゲートにより、CellQuestソフトウエア(BD Biosciences)を用いて分析を行い、テトラマーによるCD8+細胞標識の比率を決定した。PBMCを、10μg/mlのHBV S1/S2−TBDとともに培養し、HBV S1/S2−TBDを負荷した成熟DCにより二回再刺激した場合に、顕著な比率の細胞をS1テトラマー(図2)およびS2テトラマー(図3)によって陽性標識した。これは、テトラマー陽性細胞の数が顕著でない場合、抗原を負荷していない成熟DCとともに培養されたT細胞とは対照的である。よって、S1/S2−TBDで負荷したDCは、HBV S1およびHBV S2抗原の決定基に特異性を有する顕著な数のT細胞の生成を誘導することができた。
【0151】
(F.実施例6:キメラ抗原融合タンパク質を用いるDHBV抗原およびDHBV抗原に対する寛容の破壊)
DHBVは、HBVに対する抗ウイルス療法を開発する際の強力な動物模型として役立っている。ウイルスの複製機構の研究を行い、抗ウイルス化合物のスクリーニングのために、DHBVに先天的に感染させた北京ダック(アヒル)を用いた。二種類のアヒル模型を本発明に用いた。最初のものは先天的にDHBVに感染させたアヒルである。これはヒトにおけるHBV感染の垂直感染に似ている。第二の模型は、新しく孵化したアヒルの子がDHBVに感染し、これらが感染を有するという持続的な感染模型である。この第二の模型はヒトにおけるHBV感染の水平感染に似ている。
【0152】
(1.先天的にDHBVに感染させたアヒル)
四週齢の先天的にDHBVに感染させたアヒルを二つの集団に分けた。血液試料(1.0mL)を免疫前抗体量の参照のために集めて、ワクチン接種の前に、血液試料を毎週集めた。実験集団は、DHBV Core TBDキメラ抗原融合タンパク質を19.95mg/用量で5週目まで毎週同じ曜日に皮下注射した。6週目は、用量を二倍として、ワクチン接種を26週目に停止するまで、四週毎に一回注射した。プラセーボ集団には等量の緩衝液(20mMリン酸ナトリウム、pH8.0、300mM NaCl)を投与した。
【0153】
DHBV Core、TBDまたはDHBV Core TBDの各抗原を1.0μg/mlの濃度で一晩4℃にて96ウエルプレートに被覆した。このプレートを、2%BSAを含有するリン酸緩衝性食塩水(PBS)で洗浄した。各動物からの希釈血清を各ウエルに多様な希釈度(1:10〜1:500)で加え、37℃で1時間インキュベートした。このプレートを、0.05%のTween20を含有するPBS(洗浄緩衝液)で洗浄した。ヤギ抗アヒルIgG−HRP(1:5000)希釈物をウエルに加え、37℃で1時間インキュベートした。プレートを洗浄緩衝液で洗浄し、2,2’アジノ−ジ−(3−エチルベンジルチアゾリン−6−スルフォネート)(KPL、Guildford、UK)を用いて発色させた。試料中の発色の光学密度をELISAプレートリーダー(Molecular Devices、USA)を用いて測定した。抗体力価は同一動物からの免疫前血清に対して計算した。
【0154】
0週目、3週目および6週目のアヒルの対照集団と実験集団とにおける、先天的にDHBVに感染させたアヒル由来の血清中の抗コア抗体レベルを表6に示す。アヒルは慢性DHBV感染を有するが、感染の慢性的性質と抗原を外来分子として認識しない免疫系に起因して、抗体レベルは低い。DHBV Core−TBDキメラ抗原による免疫を行うと、宿主の免疫系はウイルス抗原を認識し、宿主に既に存在するコア抗原に対して体液性応答を開始し、ウイルス抗原に対する宿主寛容を壊した。
【0155】
【表6】
同様に、アヒルの免疫系は、キメラ抗原のTBD成分を外来抗原として認識し、融合タンパク質のこの部分に対する免疫応答も同じく生じさせた。プレートをTBDで被覆し、個々のアヒル由来の血清をELISAによる抗体レベルについて評価した。この研究結果を表6に示す。
【0156】
(2.孵化後にDHBV感染したアヒル)
正常な子アヒルに、子アヒルが孵化した後の日に、DHBV含有アヒル血清を感染させた。これはDHBV研究の分野での標準的な操作である。確立された技術を用いて、持続的なウイルス血症の存在を免疫開始の四週前に確認した。DHBV感染アヒルを二つの集団に分けた。血液(1.0mL)の試料を免疫前抗体レベルの参照のために各アヒルより集めて、ワクチン接種の前に、血液試料を毎週集めた。実験集団はDHBV Core−TBDキメラ抗原融合タンパク質を19.95μg/用量で5週目まで毎週同じ曜日に皮下注射した。6週目、用量を二倍として、ワクチン接種を30週目に停止するまで、四週毎に一回注射した。血液試料は、等量の緩衝液(20mMリン酸ナトリウム、pH8.0、300mM NaCl)を投与したプラセーボ集団から集めた。
【0157】
0週目、3週目および6週目にアヒルから集めた血清の抗体レベルを示す。アヒルの対照集団と実験集団とのにおける、孵化後DHBV感染アヒル由来の血清中の抗コア抗体レベルを表7に示す。DHBVは持続的感染を確立しているために、免疫系はウイルス抗原を外来分子として認識せず抗体レベルは低い。DHBV Core−TBDキメラ抗原による免疫を行うと、宿主の免疫系はウイルス抗原を認識し、宿主に既に存在するコア抗原に対して体液性応答を開始し、ウイルス抗原に対する宿主寛容を破壊した。TBDに対する抗体レベルも増加した(表7)。したがって、同一のキメラ抗原の異なる部分に対する多価(または多エピトープ性)免疫応答が存在する。
【0158】
【表7】
(G.実施例7:化学架橋HBV sAg−Fc(マウス))
100μgのsAg(US Biologicals;カタログ番号H 1910−27)および100μgのマウスポリクローナルIgG Fcフラグメント(Harlan Sera−Lab Ltd.、カタログ番号PP−19−01)の溶液を、100mMのHEPES、pH8.7に対して一晩4℃で透析した。タンパク質溶液を混合して一緒にして、すぐにジメチルスベリミデート(DMS;Pierceカタログ番号20700)を10mMの最終濃度にて加え、この混合物を室温で1時間インキュベートした。この反応を、0.1MトリスHCl、pH7.8を加えることで停止させた。反応混合物をSephadex G 75カラム(0.7×12cm)にかけて、画分をリン酸緩衝性食塩水を用いて溶出させた。0.5ml画分を集め、各抗体を用いたELISAにより予測してsAg/Fcを1:1のモル比で含有する画分をプールした。
【0159】
プールした画分を抗原提示アッセイに用いた(Berlynら、Clin.Immunol.101:276−283(2001))。未成熟樹状細胞を、GM−CSF/IL4とともに4日間培養し、sAg−Fc複合体とともにインキュベートし、TNFαとインターフェロンαとの存在下において成熟させた。自己CD3+T細胞を成熟樹状細胞に加えた。成熟樹状細胞に対して3回曝した後に、T細胞刺激を、フローサイトメトリーを用いて細胞内インターフェロンγの産生を測定することにより定量した。複合体の存在下においてT細胞中に産生された細胞内インターフェロンγのレベルは、sAgまたはFcフラグメントのみの存在下におけるよりも実質的に高かった(表8)。
【0160】
【表8】
(H.実施例8:抗原提示アッセイ)
ヒトPBMC由来の樹状細胞を確立されたプロトコール(Berlynら(前出)(2001))に従って用いることで抗原提示アッセイを実施した。T細胞刺激アッセイのプロトコールの要約をスキームの形で示す。
【0161】
【化1】
(1.成熟した負荷樹状細胞の調製)
健康なドナーからの白血球搬出の試料から単球を作り、抗CD2抗体、抗CD7抗体、抗CD16抗体、抗CD19抗体および抗CD56抗体とともにインキュベートすることによりリンパ球と顆粒球とを枯渇させた。これに続いて、磁気ビーズ複合抗マウスIgGとのインキュベーションと磁石(Dynal)による分離を行った。ネガティブ選択細胞は、広いCDマーカーパネル(CD14+、CD11c+、CD19−、CD3−、CD4−、CD64+、CD32+、CD86+、CD16−)を用いたフローサイトメトリーにより特性付けられたように95%を超える純度の単球であった。次に、単球を、IL−4とGM−CSF(R&D Systems)とともに、2.5%調和ヒト血清を加えたAIM V中で4日間インキュベートして、未成熟樹状細胞を作った。再び、細胞のアリコートを、細胞の純度と同一性を保証する広いCDマーカーパネルにより染色した。次に、細胞に、HBV S1/S2−TBD(5.0μg/ml)、HBV S1/S2(2.5μg/ml)またはTBD(2.5μg/ml)を2〜4時間37℃で負荷し、インターフェロンαとTNF−αとにより3日間成熟させた。細胞が適切な成熟を受けたことを保証するために、再びフローサイトメトリーを用いてCDマーカーのアレイに対する樹状細胞の確認を行った。得られた成熟した負荷樹状細胞をT細胞刺激アッセイに用いた。
【0162】
(2.T細胞刺激アッセイ:サイトカイン分析)
樹状細胞としてPMBCの同一の試料から、磁気T細胞単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択により、T細胞を生じさせた。成熟した負荷樹状細胞(DC−1)を完全に洗浄し、T細胞に加えた(0日目)。T細胞および樹状細胞を7日間インキュベートした。7日目に、このT細胞を、成熟した負荷樹状細胞(DC−2)により再刺激した。細胞のアリコートを二時間後に採取した。この細胞のアリコートを、ブレフェルジンA(GolgiPlug(登録商標)、R&D Systems)とともに18時間インキュベートし、次に下記の細胞内サイトカイン染色についてアッセイした。
【0163】
残っている細胞をさらに7日間インキュベートした。14日目に、残っている細胞を、第三群の成熟した負荷樹状細胞(DC−3)で刺激した。細胞のアリコートを二時間後に採取した。この細胞のアリコートをブレフェルジンA(GolgiPlug(登録商標)、R&D Systems)とともに18時間インキュベートし、次に下記の細胞内サイトカイン染色についてアッセイした。
【0164】
細胞内サイトカイン染色のために、細胞を抗CD3−FITCと抗CD8−Cy−Chromeとで30分間染色し、洗浄し、固定し、浸透化処理し、次に抗インターフェロン−γ−PEで30分間氷上にて染色した。細胞を洗浄し、フローサイトメトリー(FACScan、BD Biosciences)で分析した。この結果を表9に示す。
【0165】
【表9】
14日目にアリコートを除去した後、残っているT細胞をさらに3日間インキュベートし、次にその上清を用いてELISA(Opt E1A ELISAキット、BD Biosciences)により分泌インターフェロンγのレベルを測定した。T細胞刺激を、細胞内インターフェロンγおよび分泌インターフェロンγのレベルを測定することにより評価した。この結果を表10に示す。キメラ抗原S1/S2−TBDは、単独で試験した場合、分子の免疫応答ドメインまたはTBDドメインのいずれかと比較して、等濃度においてより高い量のインターフェロンγの産生を引き起こした。S1/S2−TBDの5μgの用量は、おおよそ上記成分のそれぞれを2.5μg含むことを指摘しておかなければならない。
【0166】
【表10】
T細胞応答に関して、多様な濃度のS1/S2−TBDを調べた。S1/S2−TBDの効果は、同様な濃度において破傷風トキソイド治療よりも高かった。5μg/mLよりも低い濃度において、キメラ抗原はインターフェロンγの産生および分泌の濃度依存的増加を引き出した。CD3+T細胞によるインターフェロンγの産生および分泌は、表11に示されるように、樹状細胞によるS1/S2−TBD抗原の提示後に、濃度依存的様式で増加した。低濃度での陽性応答は、ワクチン接種に必要な用量およびワクチンの製造費用に関して有利である。
【0167】
【表11】
(I.実施例9:キメラ抗原の結合および取り込み)
(1.成熟した負荷樹状細胞の調製)
末梢血液単核細胞(PBMC)を、白血球搬出細胞調製物のFicoll/Histopaque(Sigma)処理から得た(Berlynら(前出)(2001))。単球単離キット(Dynal)を製造者の指示に従って用いるネガティブ選択により、単球をPBMC集団から分離した。単球は、抗体分析およびフローサイトメトリー(CD14+、CD11c+、CD19−、CD3−、CD4−、CD64+、CD32+、CD86+、CD16−)による検定で95%を超える純度であった。単球を、1%のドナー調和血清(Berlynら(前出)(2001)による記載のように単離されたもの)を含む、L−グルタミン、硫酸ストレプトマイシン(50μg/mL)および硫酸ゲンタマイシン(10μg/mL)を含むAIM−V(Invitrogen)培地で二回洗浄した。次に、この単球を、2.5%ドナー調和血清とサイトカインGM−CSFとIL−4とを含むAIM−V培地中で培養し、細胞を樹状細胞(DC)系統へと分化させた。この細胞を12ウエル組織培養皿中、37℃、7%CO2雰囲気でインキュベートした。
【0168】
単球由来の樹状細胞を1日目〜4日目に集めた。次に細胞を、0.1%BSA(Sigma)を含むAIM−V培地で一回洗浄し、0.1%(w/v)BSA(PBSB)を含むダルベッコリン酸緩衝性食塩水(Invitrogen)で二回洗浄した。単球由来の樹状細胞を4℃の標識または結合アッセイまたは37℃の結合/取り込みアッセイに用いた。
【0169】
(2.キメラ抗原の成熟樹状細胞への結合)
マウスIgG1およびIgG2aに対してS1/S2−TBDの成熟樹状細胞への結合の程度を比較した。樹状細胞を、エキソビボ培養(0日目〜4日目)の種々の日に単離し、S1/S2−TBD(10μg/mL)、またはマウスIgG1(2C12、TBDが生産された親mAb)もしくはIgG2a(G155−178、90μg/mL)で、樹状細胞を1時間、4℃で処理した。この細胞を、PBSB中のF(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)で20分間処理した。細胞をPBSBで二回洗浄し、2%パラホルムアルデヒド(PF)を含むPBSBに再懸濁し、CellQuest獲得分析(acquisition and analysis)ソフトウエア(BD)に適合させたBecton Dickinson(BD)FACScanにより得た。FSCとSSC分散プロフィールにより決定された生存細胞集団上にゲートを作り、≧10,000細胞を得た。陽性細胞の比率を決定するために、ゲートを、陰性対照処置細胞(F(ab’)2ヤギ抗マウスAlexa−488のみで標識したアイソタイプ対照または細胞)に基づいて設定した。特異的な陽性細胞の比率は下記のように計算した:
【0170】
【化2】
相対平均蛍光強度(MFI)を、試験試料のMFIから対照試料のMFIを引いたものとして決定した。
【0171】
培養1日〜4日後のDC上のIgG1およびIgG2aに比較したS1/S2−TBDの結合を表12に示す。
【0172】
【表12】
S1/S2−TBD結合は、IgG1またはIgG2aのいずれか結合よりも明らかにかなり高く、さらにS1/S2−TBD結合は4日目よりも1日目で明らかであった。これらの実験は、S1/S2−TBDは成熟樹状細胞に対して高い効率で結合したことを示した。
【0173】
(3.成熟樹状細胞へのキメラ抗原の取り込み)
IgG1およびIgG2aに比較した場合のキメラ抗原(例えば、HBV S1/S2−TBD)の取り込みの程度を決定するために、0.1%BSAを含むAIM V培地中で1時間、37℃で多様な濃度の抗原IgG1(2C12、TBDが生産された親mAb)またはIgG2a(G155−178)と共に細胞をインキュベートした。細胞をPBSBで二回洗浄し、2%PFを含むPBSで一晩4℃にて固定した。続いて、この細胞をPBSBで二回洗浄し、0.1%(w/v)サポニン(Sigma)を含有するPBSで40分間、20℃にて浸透化処理した。
【0174】
細胞をPBSBで二回洗浄し、0.1%(w/v)サポニンを含有するPBSB中のF(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)と共に20分間、4℃にてインキュベートした。PBSBで二回洗浄した後、細胞をPBSBに再懸濁した。このアッセイの変法では、細胞を、キメラ抗原、IgG1またはIgG2aにより10分間上記のように処理した後に、50分間F(ab’)2ヤギ抗マウスAlexa−488(10μg/mL)を加えた。続いて、細胞を洗浄し、2%PFを含むPBSに再懸濁した。
【0175】
Cellquest獲得分析ソフトウエア(BD)で適合されたBecton Dickinson(BD)FACScanにより細胞を得た。FSCおよびSSCの分散プロフィールにより決定された生存細胞集団上にゲートを作り、≧10,000細胞を得た。陽性細胞の比率を決定するために、ゲートを陰性対照処置細胞(F(ab’)2ヤギ抗マウスAlexa−488のみで標識されたアイソタイプ対照または細胞)に基づいて設定した。特異的な陽性細胞の比率は下記のように計算した:
【0176】
【化3】
相対平均蛍光強度(MFI)は、試験試料のMFIから対照試料のMFIを引いたものとして決定した。
【0177】
マウスIgG1およびIgG2aと比較したS1/S2−TBDの取り込みを、樹状細胞の成熟の4日目の濃度の関数と予測した。取り込みを37℃で1時間定量し、その結果を図4に示す。濃度とともにS1/S2−TBDの取り込みは直線的に増加した。IgG1はかなり低いレベルで取り込まれ、IgG2aの取り込みはほとんどなかった。したがって、キメラ抗原S1/S2−TBDは、免疫グロブリンよりもさらに効率的に樹状細胞に取り込まれる。
【0178】
(J.実施例10:成熟DC上でのFc−γレセプターおよびCD206の発現)
抗原に結合し、これを取り込む抗原提示細胞上にいくつかのレセプターが存在する。成熟樹状細胞上のこれらレセプターの量を、蛍光標識レセプターに特異的な抗体を用いて評価した。FACS分析を用いて、樹状細胞の全集団における、特異的レセプターに陽性な細胞の比率を予測した。レセプター発現の程度は、相対的平均蛍光強度の測定により、そして相対的蛍光強度の関数として評価した(表13)。
【0179】
【表13】
CD64(FcγレセプターI)の発現は培養時間とともに減少し、4日目にほとんど無視できた。対照的に、CD32(FcγレセプターII)、およびさらに低い程度でCD16(FcγレセプターIII)は、DC培養の4日後まで発現し続けた。培養の0日目では、CD206(マンノースマクロファージレセプター)発現は基本的に存在しなかった。しかし、発現はIL−4とGM−CSFとの培養により誘導され、4日目までに、CD206は非常に高いレベルで発現した。よって、4日目に抗原を抗原提示アッセイにかけたときに、樹状細胞は、キメラ抗原の結合のための少なくとも二つの潜在的なレセプター(CD32およびCD206)を有した。さらに、それらのレセプターは共刺激分子の完全な補体を有した(データは示さず)。HLA−DR(クラスII)およびHLA−ABC(クラスI)の発現も培養時間とともに増加した。共刺激分子のCD86(B7.2)およびCD80(B7.1)がアッセイの間中発現した。これらの結果は、単球由来の樹状細胞が成熟樹状細胞に分化し、これらが抗原処理とT細胞への提示をし得ることを示す。
【0180】
(K.実施例11:CD32/CD206発現と成熟DCに対するS1/S2−TBD結合との相関)
CD32/CD206レセプターの発現と成熟樹状細胞へのS1/S2−TBD結合との間に直接の相関が存在する。マウスIgG1は、ヒトCD32へ結合することが公知であるので、IgG1のマウスFc成分を含むS1/S2−TBDはCD32にも結合することが予測された。さらに、その高いマンノースグリコシル化のために、S1/S2−TBDが、CD206レセプターを経て樹状細胞に結合することもまた予測される。
【0181】
図5のドットプロットは、S1/S2−TBD結合(10μg/mL)およびCD32発現、ならびにS1/S2−TBD結合およびCD206発現を示す。S1/S2−TBD結合の程度とCD32発現の程度(この発現は比較的不均一であり、すなわち広い程度の発現が存在した)との間には直接の相関があった。これらの結果は、S1/S2−TBDはCD32に結合することを示し、CD32の発現が高いほど、キメラ抗原S1/S2−TBDの結合の程度が高いことを示す。S1/S2−TBD結合とCD206発現とのドットプロットは、CD206を発現するほとんど大部分の細胞が、S1/S2−TBDにも結合したことを示す。この細胞集団の低い割合でCD206陰性であり、したがってS1/S2−TBD結合に対して陰性であった。したがって、CD32とCD206との両方のレセプターがS1/S2−TBDの結合に相関する。
【0182】
(L.実施例12:S1/S2−TBDの結合および取り込みは、主にCD32によるものであり、CD206はそれよりも低い程度で関与する)
マウスIgG1およびIgG2aと比較したS1/S2−TBDの取り込みは、DC成熟の4日目の濃度の関数と予測した。この取り込みを、37℃で1時間、培地、マンナン(2mg/ml、Sigma)および/またはマウスFcγ(2mg/ml、Jackson ImmunoResearch Laboratories)の存在下において定量した。マンナンはCD206結合の競合的インヒビターであり、よって樹状細胞上のCD206による抗原の取り込みのインヒビターである。FcγはCD32結合の競合的インヒビターであり、よってCD32が媒介する抗原取り込みのインヒビターである。この結果を表14に示す。
【0183】
【表14】
濃度とともにキメラ抗原の結合の漸進的な増加が存在した。高い濃度のマウスFcγフラグメントとの細胞のインキュベーションは、この結合を無効にしたが、CD206レセプター結合のインヒビターであるマンナンは些細な効果を有するのみであった。したがって、CD32は、キメラ抗原の結合と取り込みとに関わる主要なレセプターであり得る。
【0184】
(M.実施例13:HBV S1/S2抗原のグリコシル化は免疫原性を付与する)
昆虫細胞で合成されたタンパク質が、分泌タンパク質において高マンノース含量および末端シアル酸残基の欠如をもたらすグリコシル化を受ける点で、タンパク質グリコシル化の昆虫細胞経路は哺乳動物細胞のものとは異なる(Altmanら、Glycoconjug 16:109−123(1999))。キメラ抗原の抗原成分であるHBV S1/S2は、E.coli(グリコシル化なし)とHigh Five(登録商標)昆虫細胞(マンノールグリコシル化)との両方で発現させた。
【0185】
(1.抗原結合に対するグリコシル化の効果)
樹状細胞に対する結合に関して、これらの抗原を実施例9に記載のように比較した。成熟樹状細胞に、昆虫細胞またはE.coli.で発現した10μg/mlのHBV S1/S2を負荷した。グリコシル化タンパク質は樹状細胞による良好な結合を示した(表15)。
【0186】
【表15】
(2.免疫応答を引き出すことに対するグリコシル化の効果)
HBV S1/S2のグリコシル化は、増強された免疫原性とT細胞応答とを引き出す。樹状細胞により提示された場合のT細胞応答について、E.coliとHigh Five(登録商標)昆虫細胞との両方で発現させたHBV S1/S2を比較した。実施例8に記載されたように(2.5μg/ml HBV S1/S2タンパク質を用いて)細胞内インターフェロンγと分泌インターフェロンγのレベルを測定し、その結果を表16に示す。
【0187】
【表16】
昆虫細胞で発現したHBV S1/S2は、E.coli.で発現したグリコシル化されていないタンパク質と比較して、細胞内インターフェロンと分泌インターフェロンとの両方をより高いレベルで生じた。
【0188】
上記明細書に示したすべての公刊物および特許は、本明細書において参考として援用される。本発明の記載された方法と系の多様な改良および変化物は、当業者にとって本発明の精神と範囲から離れることなく明らかであろう。本発明は特定の実施態様との関連において記載されているが、本発明の特許請求の範囲は、そのような特定の実施態様に過度に限定すべきではないことが理解されるべきである。実際、本発明を実施するための記載された態様の多様な改変は、当業者に明らかであり、それらの改変は、以下の請求項の範囲の内にあることが意図される。
【0189】
【化4】
【0190】
【化5】
【0191】
【化6】
【0192】
【化7】
【0193】
【化8】
【0194】
【化9】
【0195】
【化10】
【0196】
【化11】
【0197】
【化12】
【0198】
【化13】
【図面の簡単な説明】
【0199】
【図1A】図1Aは、本発明のキメラ抗原の単量体としての構造を示す略図であり、このキメラ抗原は二つの部分(すなわち、免疫応答ドメインおよび標的結合ドメイン)を有する。この略図はまた、ヒンジ領域が存在する好ましい実施態様も示している。
【図1B】図1Bは、二量体として組み立てられた正常な状態での本発明のキメラ抗原の構造を示す略図である。この略図は、キメラ抗原が免疫応答ドメインと標的結合とに加えて6×Hisタグおよびペプチドリンカーを含む特に好ましい実施態様を示す。
【図2】図2は、HBV S1/S2−TBDによるT細胞の刺激が、HBV S1タンパク質由来のエピトープに対する細胞障害性T細胞(CTL)応答を特異的に発生させることを示す。
【図3】図3は、HBV S1/S2−TBDによるT細胞の刺激が、HBV S2タンパク質由来のエピトープに対する細胞障害性T細胞(CTL)応答を特異的に発生させることを示す。
【図4】図4は、樹状細胞を成熟させることによるHBV S1/S2−TBD、IgG1およびIgG2の取り込みの比較を濃度の関数として示す。
【図5】図5は、CD32へのHBV S1/S2−TBDの結合と樹状細胞上のCD206発現との相関を示す。
【特許請求の範囲】
【請求項1】
免疫応答ドメインおよび標的結合ドメインを含む免疫応答を引き出すためのキメラ抗原であって、該標的結合ドメインが抗体フラグメントを含む、キメラ抗原。
【請求項2】
前記抗体フラグメントが、異種型抗体フラグメントである、請求項1に記載のキメラ抗原。
【請求項3】
前記抗体フラグメントが、異種型抗体フラグメントでない、請求項1に記載のキメラ抗原。
【請求項4】
前記キメラ抗原が、二つ以上の免疫応答ドメインを含む、請求項1に記載のキメラ抗原。
【請求項5】
前記免疫応答ドメインが、HBVタンパク質、DHBVタンパク質およびHCVタンパク質を含む群から選択されるタンパク質の少なくとも一つの抗原性部分を含む、請求項1に記載のキメラ抗原。
【請求項6】
前記免疫応答ドメインが、HBV S1/S2、HBV S1/S2/S、HBV Core、HBV Core ctmおよびHBVポリメラーゼからなる群から選択されるHBVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項7】
前記免疫応答ドメインが、DHBV PreS/S、DHBV PreS、DHBV CoreおよびDHBVポリメラーゼからなる群から選択されるDHBVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項8】
前記免疫応答ドメインが、HCV Core(1−191)、HCV Core(1−177)、HCV E1−E2、HCV E1、HCV E2、HCV NS3、HCV NS5AおよびHCV NS4Aからなる群から選択されるHCVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項9】
前記免疫応答ドメインが、前記タンパク質に融合した6×Hisタグをさらに含む、請求項1〜8のいずれかに記載のキメラ抗原。
【請求項10】
前記免疫応答ドメインが、HPV、HIV、HSV、偏性細胞内寄生生物または癌抗原由来のタンパク質の一つ以上の抗原性部分を含む、請求項1〜9のいずれかに記載のキメラ抗原。
【請求項11】
前記標的結合ドメインが、抗原提示細胞に結合し得る、請求項1〜10のいずれかに記載のキメラ抗原。
【請求項12】
前記抗原フラグメントが、免疫グロブリン重鎖フラグメントを含む、請求項1〜11のいずれかに記載のキメラ抗原。
【請求項13】
前記免疫グロブリン重鎖フラグメントが、ヒンジ領域を含む、請求項12に記載のキメラ抗原。
【請求項14】
前記キメラ抗原が、アミノ酸VDKKI(配列番号2)をさらに含む、請求項12に記載のキメラ抗原。
【請求項15】
前記免疫グロブリン重鎖フラグメントが、CH2ドメインおよびCH3ドメインを含む、請求項12に記載のキメラ抗原。
【請求項16】
前記抗体フラグメントが、Fcフラグメントである、請求項1〜15のいずれかに記載のキメラ抗原。
【請求項17】
前記抗体フラグメントが、CH1領域の一部および抗体ヒンジ領域を含む、請求項1〜15のいずれかに記載のキメラ抗原。
【請求項18】
前記免疫応答ドメインおよび前記標的結合ドメインを結合するためのリンカーをさらに含む、請求項1〜17のいずれかに記載のキメラ抗原。
【請求項19】
前記リンカーが、共有ペプチド結合である、請求項18に記載のキメラ抗原。
【請求項20】
前記ペプチド結合が、配列SRPQGGGSまたは配列VRPQGGGS(配列番号1)を含む、請求項19に記載のキメラ抗原。
【請求項21】
前記リンカーが、ロイシンジッパーおよびビオチン/アビジンからなる群から選択される、請求項18に記載のキメラ抗原。
【請求項22】
前記キメラ抗原が、マンノースグリコシル化されている、請求項1〜21のいずれかに記載のキメラ抗原。
【請求項23】
前記免疫応答ドメインが、マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項24】
前記標的結合ドメインが、マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項25】
前記キメラ抗原が、低マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項26】
前記キメラ抗原が、高マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項27】
前記標的結合ドメインが、アミノ酸配列SRPQGGGS(配列番号1)、VDKKI(配列番号2)、ならびにマウス免疫グロブリン重鎖定常領域のCH2ドメインおよびCH3ドメインを含む、請求項1に記載のキメラ抗原。
【請求項28】
多エピトープ性応答を引き出す請求項1〜27のいずれかに記載のキメラ抗原。
【請求項29】
前記免疫応答ドメインの少なくとも一つのエピトープに対する免疫応答を引き出す、請求項1に記載のキメラ抗原。
【請求項30】
体液性免疫応答を引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項31】
細胞性免疫応答を引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項32】
Th1免疫応答、Th2免疫応答、CTL応答またはそれらの組合せを引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項33】
抗原提示細胞において抗原提示を増強する方法であって、該方法は、該抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原を含む組成物と接触させる工程を包含する、方法。
【請求項34】
前記抗原提示細胞が、樹状細胞である、請求項33に記載の方法。
【請求項35】
前記方法が、二つ以上のエピトープの抗原提示を増強する、請求項34に記載の方法。
【請求項36】
前記方法が、前記免疫応答ドメインの少なくとも一つのエピトープの抗原提示を増強する、請求項34に記載の方法。
【請求項37】
抗原提示細胞を活性化する方法であって、該方法は、該抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原と接触させる工程を包含する、方法。
【請求項38】
前記接触させる工程が、エキソビボで行われる、請求項33または37に記載の方法。
【請求項39】
前記接触させる工程が、インビボで行われる、請求項33または37に記載の方法。
【請求項40】
前記接触させる工程が、ヒトにおいて行われる、請求項39に記載の方法。
【請求項41】
免疫応答を引き出す方法であって、該方法は、請求項1〜32のいずれかに記載のキメラ抗原を含む組成物を被験体に投与する工程を包含する、方法。
【請求項42】
寛容を壊す方法であって、該方法は、抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原に接触させる工程を包含する、方法。
【請求項43】
免疫治療可能な症状を治療するための方法であって、該方法は、請求項1〜32いずれかに記載のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する、方法。
【請求項44】
前記免疫応答ドメイン単独の投与が、免疫応答を引き出さない、請求項41〜43のいずれかに記載の方法。
【請求項45】
前記キメラ抗原の投与が、免疫応答ドメイン単独の投与よりも高い免疫応答を引き出す請求項41〜43のいずれかに記載の方法。
【請求項46】
前記免疫応答が、前記被験体に存在する抗原特異的な抗体の量によって測定される、請求項45に記載の方法。
【請求項47】
前記免疫応答が、前記キメラ抗原または免疫応答ドメインを単独で負荷した抗原提示細胞に曝された応答において、T細胞により分泌されたインターフェロンγの量によって測定される、請求項45に記載の方法。
【請求項48】
前記免疫応答が、前記キメラ抗原または免疫応答ドメインを単独で負荷した抗原提示細胞に曝された応答において引き出された抗原特異的CD8+T細胞の量によって測定される、請求項45に記載の方法。
【請求項49】
前記被験体が、感染または癌を有する請求項41〜43のいずれかに記載の方法。
【請求項50】
前記感染が、ウイルス感染または寄生生物感染である、請求項49に記載の方法。
【請求項51】
前記感染が、慢性のウイルス感染である、請求項50に記載の方法。
【請求項52】
前記慢性のウイルス感染が、慢性B型肝炎ウイルス感染もしくは慢性C型肝炎ウイルス感染、慢性ヒトパピローマウイルス感染、慢性ヒト免疫不全ウイルス感染、または慢性単純ヘルペスウイルス感染である、請求項51に記載の方法。
【請求項53】
前記感染が、B型肝炎ウイルス感染であり、そして前記免疫応答ドメインが、HBV Coreタンパク質、HBV Sタンパク質、HBV S1タンパク質、およびHBV S2タンパク質、ならびにそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む、請求項52に記載の方法。
【請求項54】
前記感染が、C型肝炎ウイルス感染であり、そして前記免疫応答ドメインが、HCV core(1−191)タンパク質、HCV Core(1−177)タンパク質、HCV E1タンパク質、HCV E2タンパク質、HCV E1−E2タンパク質、HCV NS3タンパク質、HCV NS5Aタンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む請求項52に記載の方法。
【請求項55】
ウイルス感染に対して被験体にワクチン接種する方法であって、該方法は、請求項1〜32のいずれかに記載のキメラ抗原を該被験体に投与する工程を包含する、方法。
【請求項56】
前記被験体が、既存のウイルス感染に対して治療的にワクチン接種される、請求項55に記載の方法。
【請求項57】
前記被験体が、ウイルス感染に対して予防的にワクチン接種される、請求項55に記載の方法。
【請求項58】
前記被験体が、前記キメラ抗原の二つ以上のエピトープに対する免疫応答を発達させる、請求項55に記載の方法。
【請求項59】
前記被験体が、前記免疫応答ドメインの二つ以上のエピトープに対する免疫応答を発達させる、請求項58に記載の方法。
【請求項60】
薬学的に受容可能な賦形剤と請求項1〜32のいずれかに記載のキメラ抗原とを含む、薬学的組成物。
【請求項61】
前記薬学的組成物が、非経口投与のために処方されている、請求項60に記載の薬学的組成物。
【請求項62】
前記薬学的組成物が、経皮投与、皮内投与、静脈内投与、皮下投与、筋肉内投与、鼻投与、肺投与または経口投与のために処方されている、請求項60に記載の薬学的組成物。
【請求項63】
請求項1〜32のいずれかに記載のキメラ抗原と、該キメラ抗原の投与が必要な被験体に対して該キメラ抗原を投与するための説明書とを備える、製品。
【請求項64】
キメラ抗原を作製する方法であって、該方法は、以下:
(a)微生物または細胞株を提供する工程であって、該微生物または細胞株は、請求項1〜32のいずれかに記載のキメラ抗原をコードするポリヌクレオチドを含む、工程;および
(b)該微生物または細胞株を、該キメラ抗原が発現される条件下で培養する工程、
を包含する、方法。
【請求項65】
前記微生物または細胞株が、真核細胞の微生物または細胞株である、請求項64に記載の方法。
【請求項66】
前記微生物または細胞株が、酵母、植物細胞株または昆虫細胞株である、請求項65に記載の方法。
【請求項67】
前記キメラ抗原が、マンノースグリコシル化を含むように翻訳後修飾されている、請求項64に記載の方法。
【請求項68】
キメラ抗原をコードするポリヌクレオチドであって、該ポリヌクレオチドは、免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二のポリヌクレオチド部分とを含み、該標的結合ドメインが抗体フラグメントを含む、ポリヌクレオチド。
【請求項69】
前記免疫応答ドメインが、ウイルスタンパク質の少なくとも一つの抗原性部分を含む、請求項68に記載のポリヌクレオチド。
【請求項70】
前記ウイルスタンパク質が、HBVタンパク質またはHCVタンパク質である、請求項69に記載のポリヌクレオチド。
【請求項71】
前記抗体フラグメントが、異種型フラグメントである、請求項68に記載のポリヌクレオチド。
【請求項72】
前記抗体フラグメントが、異種型フラグメントではない、請求項68に記載のポリヌクレオチド。
【請求項73】
前記ポリヌクレオチドが、配列番号26のヌクレオチド1〜1326、配列番号28のヌクレオチド1〜2004、配列番号30のヌクレオチド1〜1350、配列番号32のヌクレオチド1〜1293、配列番号34のヌクレオチド1〜1794、配列番号36のヌクレオチド1〜1581、配列番号38のヌクレオチド1〜1389、配列番号40のヌクレオチド1〜1347、配列番号42のヌクレオチド1〜2157、配列番号44のヌクレオチド1〜1395、配列番号46のヌクレオチド1〜1905および配列番号48のヌクレオチド1〜2484からなる群から選択されるヌクレオチド配列を含む、請求項68に記載のポリヌクレオチド。
【請求項74】
前記ポリヌクレオチドが、配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635および配列番号49のアミノ酸1〜828からなる群から選択される、全アミノ酸配列に対して少なくとも90%同一性であるキメラ抗原をコードする、請求項68に記載のポリヌクレオチド。
【請求項75】
前記ポリヌクレオチドが、ストリンジェントな条件下で、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および請求項48からなる群から選択されるヌクレオチド配列を有するポリヌクレオチドに選択的にハイブリダイズする、請求項68に記載のポリヌクレオチド。
【請求項76】
請求項68に記載のポリヌクレオチドを含む、微生物または細胞株。
【請求項77】
請求項68に記載のポリヌクレオチドを含む、ベクター。
【請求項1】
免疫応答ドメインおよび標的結合ドメインを含む免疫応答を引き出すためのキメラ抗原であって、該標的結合ドメインが抗体フラグメントを含む、キメラ抗原。
【請求項2】
前記抗体フラグメントが、異種型抗体フラグメントである、請求項1に記載のキメラ抗原。
【請求項3】
前記抗体フラグメントが、異種型抗体フラグメントでない、請求項1に記載のキメラ抗原。
【請求項4】
前記キメラ抗原が、二つ以上の免疫応答ドメインを含む、請求項1に記載のキメラ抗原。
【請求項5】
前記免疫応答ドメインが、HBVタンパク質、DHBVタンパク質およびHCVタンパク質を含む群から選択されるタンパク質の少なくとも一つの抗原性部分を含む、請求項1に記載のキメラ抗原。
【請求項6】
前記免疫応答ドメインが、HBV S1/S2、HBV S1/S2/S、HBV Core、HBV Core ctmおよびHBVポリメラーゼからなる群から選択されるHBVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項7】
前記免疫応答ドメインが、DHBV PreS/S、DHBV PreS、DHBV CoreおよびDHBVポリメラーゼからなる群から選択されるDHBVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項8】
前記免疫応答ドメインが、HCV Core(1−191)、HCV Core(1−177)、HCV E1−E2、HCV E1、HCV E2、HCV NS3、HCV NS5AおよびHCV NS4Aからなる群から選択されるHCVタンパク質の少なくとも一つの抗原性部分を含む、請求項5に記載のキメラ抗原。
【請求項9】
前記免疫応答ドメインが、前記タンパク質に融合した6×Hisタグをさらに含む、請求項1〜8のいずれかに記載のキメラ抗原。
【請求項10】
前記免疫応答ドメインが、HPV、HIV、HSV、偏性細胞内寄生生物または癌抗原由来のタンパク質の一つ以上の抗原性部分を含む、請求項1〜9のいずれかに記載のキメラ抗原。
【請求項11】
前記標的結合ドメインが、抗原提示細胞に結合し得る、請求項1〜10のいずれかに記載のキメラ抗原。
【請求項12】
前記抗原フラグメントが、免疫グロブリン重鎖フラグメントを含む、請求項1〜11のいずれかに記載のキメラ抗原。
【請求項13】
前記免疫グロブリン重鎖フラグメントが、ヒンジ領域を含む、請求項12に記載のキメラ抗原。
【請求項14】
前記キメラ抗原が、アミノ酸VDKKI(配列番号2)をさらに含む、請求項12に記載のキメラ抗原。
【請求項15】
前記免疫グロブリン重鎖フラグメントが、CH2ドメインおよびCH3ドメインを含む、請求項12に記載のキメラ抗原。
【請求項16】
前記抗体フラグメントが、Fcフラグメントである、請求項1〜15のいずれかに記載のキメラ抗原。
【請求項17】
前記抗体フラグメントが、CH1領域の一部および抗体ヒンジ領域を含む、請求項1〜15のいずれかに記載のキメラ抗原。
【請求項18】
前記免疫応答ドメインおよび前記標的結合ドメインを結合するためのリンカーをさらに含む、請求項1〜17のいずれかに記載のキメラ抗原。
【請求項19】
前記リンカーが、共有ペプチド結合である、請求項18に記載のキメラ抗原。
【請求項20】
前記ペプチド結合が、配列SRPQGGGSまたは配列VRPQGGGS(配列番号1)を含む、請求項19に記載のキメラ抗原。
【請求項21】
前記リンカーが、ロイシンジッパーおよびビオチン/アビジンからなる群から選択される、請求項18に記載のキメラ抗原。
【請求項22】
前記キメラ抗原が、マンノースグリコシル化されている、請求項1〜21のいずれかに記載のキメラ抗原。
【請求項23】
前記免疫応答ドメインが、マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項24】
前記標的結合ドメインが、マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項25】
前記キメラ抗原が、低マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項26】
前記キメラ抗原が、高マンノースグリコシル化されている、請求項22に記載のキメラ抗原。
【請求項27】
前記標的結合ドメインが、アミノ酸配列SRPQGGGS(配列番号1)、VDKKI(配列番号2)、ならびにマウス免疫グロブリン重鎖定常領域のCH2ドメインおよびCH3ドメインを含む、請求項1に記載のキメラ抗原。
【請求項28】
多エピトープ性応答を引き出す請求項1〜27のいずれかに記載のキメラ抗原。
【請求項29】
前記免疫応答ドメインの少なくとも一つのエピトープに対する免疫応答を引き出す、請求項1に記載のキメラ抗原。
【請求項30】
体液性免疫応答を引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項31】
細胞性免疫応答を引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項32】
Th1免疫応答、Th2免疫応答、CTL応答またはそれらの組合せを引き出す、請求項1〜29のいずれかに記載のキメラ抗原。
【請求項33】
抗原提示細胞において抗原提示を増強する方法であって、該方法は、該抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原を含む組成物と接触させる工程を包含する、方法。
【請求項34】
前記抗原提示細胞が、樹状細胞である、請求項33に記載の方法。
【請求項35】
前記方法が、二つ以上のエピトープの抗原提示を増強する、請求項34に記載の方法。
【請求項36】
前記方法が、前記免疫応答ドメインの少なくとも一つのエピトープの抗原提示を増強する、請求項34に記載の方法。
【請求項37】
抗原提示細胞を活性化する方法であって、該方法は、該抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原と接触させる工程を包含する、方法。
【請求項38】
前記接触させる工程が、エキソビボで行われる、請求項33または37に記載の方法。
【請求項39】
前記接触させる工程が、インビボで行われる、請求項33または37に記載の方法。
【請求項40】
前記接触させる工程が、ヒトにおいて行われる、請求項39に記載の方法。
【請求項41】
免疫応答を引き出す方法であって、該方法は、請求項1〜32のいずれかに記載のキメラ抗原を含む組成物を被験体に投与する工程を包含する、方法。
【請求項42】
寛容を壊す方法であって、該方法は、抗原提示細胞を請求項1〜32のいずれかに記載のキメラ抗原に接触させる工程を包含する、方法。
【請求項43】
免疫治療可能な症状を治療するための方法であって、該方法は、請求項1〜32いずれかに記載のキメラ抗原を、治療を必要とする被験体に投与する工程を包含する、方法。
【請求項44】
前記免疫応答ドメイン単独の投与が、免疫応答を引き出さない、請求項41〜43のいずれかに記載の方法。
【請求項45】
前記キメラ抗原の投与が、免疫応答ドメイン単独の投与よりも高い免疫応答を引き出す請求項41〜43のいずれかに記載の方法。
【請求項46】
前記免疫応答が、前記被験体に存在する抗原特異的な抗体の量によって測定される、請求項45に記載の方法。
【請求項47】
前記免疫応答が、前記キメラ抗原または免疫応答ドメインを単独で負荷した抗原提示細胞に曝された応答において、T細胞により分泌されたインターフェロンγの量によって測定される、請求項45に記載の方法。
【請求項48】
前記免疫応答が、前記キメラ抗原または免疫応答ドメインを単独で負荷した抗原提示細胞に曝された応答において引き出された抗原特異的CD8+T細胞の量によって測定される、請求項45に記載の方法。
【請求項49】
前記被験体が、感染または癌を有する請求項41〜43のいずれかに記載の方法。
【請求項50】
前記感染が、ウイルス感染または寄生生物感染である、請求項49に記載の方法。
【請求項51】
前記感染が、慢性のウイルス感染である、請求項50に記載の方法。
【請求項52】
前記慢性のウイルス感染が、慢性B型肝炎ウイルス感染もしくは慢性C型肝炎ウイルス感染、慢性ヒトパピローマウイルス感染、慢性ヒト免疫不全ウイルス感染、または慢性単純ヘルペスウイルス感染である、請求項51に記載の方法。
【請求項53】
前記感染が、B型肝炎ウイルス感染であり、そして前記免疫応答ドメインが、HBV Coreタンパク質、HBV Sタンパク質、HBV S1タンパク質、およびHBV S2タンパク質、ならびにそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む、請求項52に記載の方法。
【請求項54】
前記感染が、C型肝炎ウイルス感染であり、そして前記免疫応答ドメインが、HCV core(1−191)タンパク質、HCV Core(1−177)タンパク質、HCV E1タンパク質、HCV E2タンパク質、HCV E1−E2タンパク質、HCV NS3タンパク質、HCV NS5Aタンパク質およびそれらの組合せからなる群から選択されるタンパク質の少なくとも一つの抗原性部分を含む請求項52に記載の方法。
【請求項55】
ウイルス感染に対して被験体にワクチン接種する方法であって、該方法は、請求項1〜32のいずれかに記載のキメラ抗原を該被験体に投与する工程を包含する、方法。
【請求項56】
前記被験体が、既存のウイルス感染に対して治療的にワクチン接種される、請求項55に記載の方法。
【請求項57】
前記被験体が、ウイルス感染に対して予防的にワクチン接種される、請求項55に記載の方法。
【請求項58】
前記被験体が、前記キメラ抗原の二つ以上のエピトープに対する免疫応答を発達させる、請求項55に記載の方法。
【請求項59】
前記被験体が、前記免疫応答ドメインの二つ以上のエピトープに対する免疫応答を発達させる、請求項58に記載の方法。
【請求項60】
薬学的に受容可能な賦形剤と請求項1〜32のいずれかに記載のキメラ抗原とを含む、薬学的組成物。
【請求項61】
前記薬学的組成物が、非経口投与のために処方されている、請求項60に記載の薬学的組成物。
【請求項62】
前記薬学的組成物が、経皮投与、皮内投与、静脈内投与、皮下投与、筋肉内投与、鼻投与、肺投与または経口投与のために処方されている、請求項60に記載の薬学的組成物。
【請求項63】
請求項1〜32のいずれかに記載のキメラ抗原と、該キメラ抗原の投与が必要な被験体に対して該キメラ抗原を投与するための説明書とを備える、製品。
【請求項64】
キメラ抗原を作製する方法であって、該方法は、以下:
(a)微生物または細胞株を提供する工程であって、該微生物または細胞株は、請求項1〜32のいずれかに記載のキメラ抗原をコードするポリヌクレオチドを含む、工程;および
(b)該微生物または細胞株を、該キメラ抗原が発現される条件下で培養する工程、
を包含する、方法。
【請求項65】
前記微生物または細胞株が、真核細胞の微生物または細胞株である、請求項64に記載の方法。
【請求項66】
前記微生物または細胞株が、酵母、植物細胞株または昆虫細胞株である、請求項65に記載の方法。
【請求項67】
前記キメラ抗原が、マンノースグリコシル化を含むように翻訳後修飾されている、請求項64に記載の方法。
【請求項68】
キメラ抗原をコードするポリヌクレオチドであって、該ポリヌクレオチドは、免疫応答ドメインをコードする第一のポリヌクレオチド部分と標的結合ドメインをコードする第二のポリヌクレオチド部分とを含み、該標的結合ドメインが抗体フラグメントを含む、ポリヌクレオチド。
【請求項69】
前記免疫応答ドメインが、ウイルスタンパク質の少なくとも一つの抗原性部分を含む、請求項68に記載のポリヌクレオチド。
【請求項70】
前記ウイルスタンパク質が、HBVタンパク質またはHCVタンパク質である、請求項69に記載のポリヌクレオチド。
【請求項71】
前記抗体フラグメントが、異種型フラグメントである、請求項68に記載のポリヌクレオチド。
【請求項72】
前記抗体フラグメントが、異種型フラグメントではない、請求項68に記載のポリヌクレオチド。
【請求項73】
前記ポリヌクレオチドが、配列番号26のヌクレオチド1〜1326、配列番号28のヌクレオチド1〜2004、配列番号30のヌクレオチド1〜1350、配列番号32のヌクレオチド1〜1293、配列番号34のヌクレオチド1〜1794、配列番号36のヌクレオチド1〜1581、配列番号38のヌクレオチド1〜1389、配列番号40のヌクレオチド1〜1347、配列番号42のヌクレオチド1〜2157、配列番号44のヌクレオチド1〜1395、配列番号46のヌクレオチド1〜1905および配列番号48のヌクレオチド1〜2484からなる群から選択されるヌクレオチド配列を含む、請求項68に記載のポリヌクレオチド。
【請求項74】
前記ポリヌクレオチドが、配列番号27のアミノ酸1〜442、配列番号29のアミノ酸1〜668、配列番号31のアミノ酸1〜450、配列番号33のアミノ酸1〜431、配列番号35のアミノ酸1〜598、配列番号37のアミノ酸1〜527、配列番号39のアミノ酸1〜463、配列番号41のアミノ酸1〜449、配列番号43のアミノ酸1〜719、配列番号45のアミノ酸1〜465、配列番号47のアミノ酸1〜635および配列番号49のアミノ酸1〜828からなる群から選択される、全アミノ酸配列に対して少なくとも90%同一性であるキメラ抗原をコードする、請求項68に記載のポリヌクレオチド。
【請求項75】
前記ポリヌクレオチドが、ストリンジェントな条件下で、配列番号26、配列番号28、配列番号30、配列番号32、配列番号34、配列番号36、配列番号38、配列番号40、配列番号42、配列番号44、配列番号46および請求項48からなる群から選択されるヌクレオチド配列を有するポリヌクレオチドに選択的にハイブリダイズする、請求項68に記載のポリヌクレオチド。
【請求項76】
請求項68に記載のポリヌクレオチドを含む、微生物または細胞株。
【請求項77】
請求項68に記載のポリヌクレオチドを含む、ベクター。
【図1A】

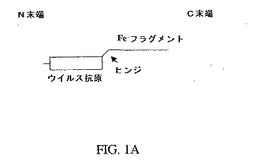
【図1B】
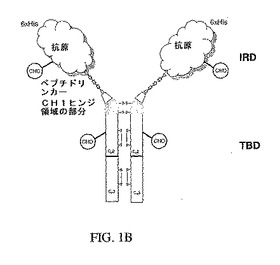
【図2】
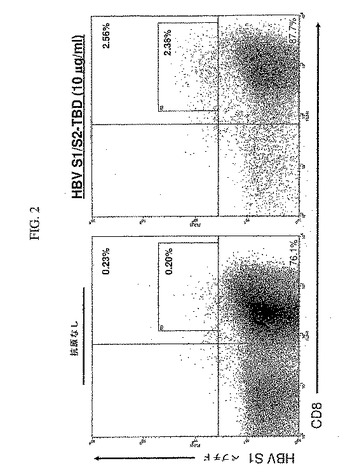
【図3】
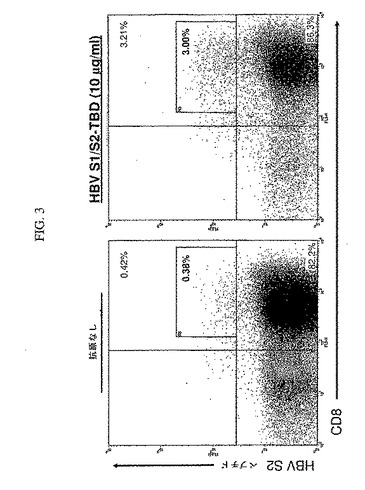
【図4】
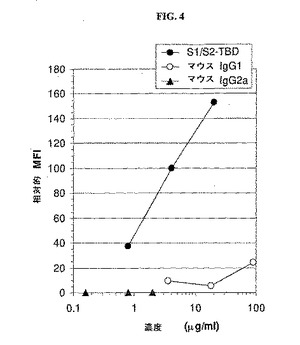
【図5】
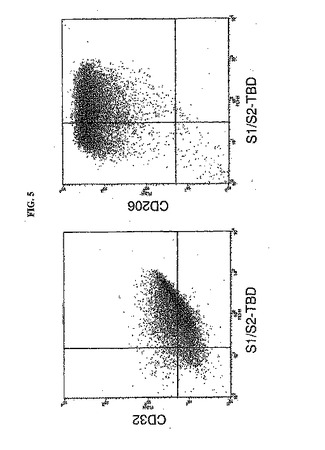
【図1B】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2007−501602(P2007−501602A)
【公表日】平成19年2月1日(2007.2.1)
【国際特許分類】
【出願番号】特願2006−522192(P2006−522192)
【出願日】平成16年8月6日(2004.8.6)
【国際出願番号】PCT/CA2004/001469
【国際公開番号】WO2005/014838
【国際公開日】平成17年2月17日(2005.2.17)
【出願人】(306013902)ヴィレックス メディカル コーポレイション (3)
【Fターム(参考)】
【公表日】平成19年2月1日(2007.2.1)
【国際特許分類】
【出願日】平成16年8月6日(2004.8.6)
【国際出願番号】PCT/CA2004/001469
【国際公開番号】WO2005/014838
【国際公開日】平成17年2月17日(2005.2.17)
【出願人】(306013902)ヴィレックス メディカル コーポレイション (3)
【Fターム(参考)】
[ Back to top ]