
多孔質シリカ内包粒子の製造方法および多孔質シリカ、多孔質シリカ内包粒子
【課題】良好な多孔質シリカ内包粒子の製造方法を提供する。
【解決手段】(a)多孔質シリカを準備する工程と、(b)多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有する。また、無溶媒系でアルコキシシランの加水分解することにより多孔質シリカを合成すれば、微細な孔径を有する多孔質シリカの合成が可能であり、この多孔質シリカを鋳型とすることで、バルクの状態では見られない特異な性質を示す粒子(例えば、W、Cu、Cr、Mn、Fe、CoまたはNiやこれらの金属酸化物)も容易に形成することができる。
【解決手段】(a)多孔質シリカを準備する工程と、(b)多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有する。また、無溶媒系でアルコキシシランの加水分解することにより多孔質シリカを合成すれば、微細な孔径を有する多孔質シリカの合成が可能であり、この多孔質シリカを鋳型とすることで、バルクの状態では見られない特異な性質を示す粒子(例えば、W、Cu、Cr、Mn、Fe、CoまたはNiやこれらの金属酸化物)も容易に形成することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、多孔質シリカ内包粒子の製造方法および多孔質シリカ、多孔質シリカ内包粒子に関し、特に、微細な孔を有する多孔質シリカを利用し、その孔の内部に微細な粒子を内包させる技術に関する。
【背景技術】
【0002】
金属、セラミックス、炭素などの材料はその粒子径を減少させることでバルクの状態では見られない特異な性質を示す(下記非特許文献1参照)。また、多くの場合それらの特異な性質はシングルナノメートルのサイズで発現し始め、サブナノメートルのサイズ領域において最大になることが、一部の実験と理論予測から報告されている(下記非特許文献2参照)。
【0003】
例えば、金属サブナノ粒子の場合は、粒子サイズの減少に伴い金属的な性質を失い、離散的なバンド構造を構築することが知られている(下記非特許文献3参照)。そのため、バンド構造由来の光吸収が起こり、半導体や色素分子のような性質を示すことから次世代の色素増感太陽電池などへの応用が検討されている(下記特許文献4参照)。さらに、バルクでは見られない発光や磁化などの特異な現象も発現することから広い分野への応用が期待されている(下記非特許文献5参照)。
【0004】
また、金属酸化物や硫化物などに代表される半導体セラミックス材料の場合、サブナノ粒子にすることで、量子サイズ効果によりバンドギャップエネルギーの大幅な増大が見られる(下記特許文献1、2参照)。可変なバンドギャップエネルギーは光触媒などの分野において有用であり、触媒活性の飛躍的な向上、反応選択制の向上、さらにはバルクでは起こらない反応を起こさせることも期待されている。
【0005】
また、ナノ炭素材料は近年多くの注目を集める材料である。炭素材料もサブナノ粒子化することで特異な性質を示す。特に、紫外光照射下での白色発光やアップコンバージョン発光などの性質はバルクの炭素では起こらない現象であり、近年でも研究の対象となっている(下記非特許文献6参照)。
【0006】
また、下記非特許文献7〜12には、後述するように、金属などの微細粒子の合成についての開示がある。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Y. Wang, N. Herron, J. Phys. Chem., 1991, 95, 525.
【非特許文献2】L. Brus, J. Phys. Chem., 1986, 90, 2555.
【非特許文献3】J. N. Solanki, Z.V.P. Murthy, Colloids and Surfaces A: Physicochem. Eng. Aspects, 2010, 359, 31.
【非特許文献4】A. Kogo, N. Sakai, T. Tatsuma, Electrochemistry Communications, 2010, 12, 996.
【非特許文献5】X. Liu, M. Bauer, H. Bertagnolli, E. Roduner, J. V. Slageren, F. Phillip, Phys. Rev. Lett., 2006, 97, 253401.
【非特許文献6】J. Zong, Y. Zhu, X. Yang, J. Shen, C. Li, Chem. Commun., 2011, 47, 764.
【非特許文献7】S.L.Hu,K.Y.Niu,J.Sun,J.Yang,N.Q.ZhaoandX.W.Du, J. Mater. Chem., 2009, 19, 484.
【非特許文献8】Y. Negishi, K. Nobusada, T. Tsukuda, J. AM. CHEM. SOC., 2005, 127, 5261.
【非特許文献9】N. Satoh, T. Nakashima, K. Kamikura, K. Yamamoto, Nature Nanotech., 2008, 3, 106.
【非特許文献10】G. A. Ozin, S. Ozkar, R. A. Prokopowicz, Acc. Chem. Res., 1992, 25, 553.
【非特許文献11】D. Tanaka, Y. Oaki, H. Imai, Chem. Commun., 2010, 46, 5286.
【非特許文献12】J. S. Beck, J. C. Vartuli, G. J. Kennedy, C.T.Kresge, W.J.Roth, S. E. Schramm, Chem. Mater., 1994, 6, 1816.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明者は、多孔質シリカ、多孔質シリカ内包粒子および孔中に粒子を内包した多孔質シリカについての研究・開発に従事し、各種の検討を重ねている。この多孔質シリカ内包粒子は、多孔質シリカを利用して孔の内部に内包させた微細粒子を意味する。多孔質シリカの孔中に内包された状態のもの、また、多孔質シリカの孔中に内包された状態から粒子だけを取り出した状態のものを意味する。
【0009】
このように、多孔質シリカを利用して孔の内部に微細粒子を形成することで、微細粒子自身若しくは微細粒子を内包する多孔質シリカ全体として種々の機能を持たせることができ、その機能の向上を図るため、本発明者は、各種の検討を重ねている。
【0010】
微細粒子、例えば、サブナノ粒子の合成は、古くはレーザーアブレーション(上記非特許文献7参照)などの方法で行われていたが、生産性やサイズの制御の点で問題がある。
【0011】
また、金属粒子に限定すれば、金属表面とチオール基などの特定の官能基の相互作用を利用した合成例が存在する(上記非特許文献8参照)。しかし、この方法も生産性とサイズ制御性にやや問題があり、また、合成対象が金属粒子に限定されることが問題であり幅広い物質群に適応することはできない。
【0012】
さらに、骨格に金属イオンの配位子となる官能基を有する樹枝状高分子(デンドリマー)を用いた合成例が存在する(上記非特許文献9参照)。このデンドリマーの性質を制御することで粒子サイズのそろったナノ〜サブナノメートル粒子を合成可能だが、サブナノメートル領域での制御性の悪さや、デンドリマーの合成の煩雑さ等で問題がある。
【0013】
また、ゼオライトに代表される多孔質材料を粒子合成の鋳型にする手法は、合成の簡便さと基質の一般性において長所がある。ゼオライトを鋳型としたサブナノ粒子の合成はいくつかの報告例が存在する(下記非特許文献10参照)。しかし、ゼオライトの細孔は一般的に制御性が悪く、また、スーパーケージと呼ばれる内部細孔において大きな粒子が成長してしまうことが問題点としてあげられる。
【0014】
これに対し、メソポーラスシリカは細孔径の制御性が高いことから、ナノ粒子の合成には適当な鋳型材料である(下記非特許文献11参照)。また、生成した粒子が優秀な吸着材であるメソポーラスシリカ内に均一に分散することも利点の一つである。しかし、従来のメソポーラスシリカにおいては細孔径を1.5nm以下に制御することが困難であるため、サブナノ粒子の合成には用いることができないという問題がある(下記非特許文献12参照)。
【0015】
以上のように、微細粒子の合成方法には各種方法が提案されているが、一長一短があり、微細な粒子を制御性良く合成することは困難であった。
【0016】
そこで、本発明の目的は、多孔質シリカ内包粒子の特性を向上させることができる技術を提供することにある。特に、粒径分布特性が良好で、特性の良い多孔質シリカ内包粒子を提供することにある。
【0017】
また、本発明の他の目的は、良好な多孔質シリカ内包粒子の製造方法を提供することにある。また、内包させる粒子の材料について、幅広い材料を内包可能な多孔質シリカ内包粒子の製造方法を提供することにある。
【0018】
本発明の上記目的およびその他の目的と新規な特徴は、本願明細書の記載および添付図面から明らかになるであろう。
【課題を解決するための手段】
【0019】
本願において開示される発明のうち、代表的なものの概要を簡単に説明すれば、次のとおりである。
【0020】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に上記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0021】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に上記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0022】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと、ビスマスまたはビスマスを元素組成として有する化合物を含有する第1液と、バナジウムまたはバナジウムを元素組成として有する化合物を含有する第2液との混合液とを接触させ、前記多孔質シリカの孔内に上記混合液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内においてバナジン酸ビスマスを含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0023】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する気体とを接触させ、前記多孔質シリカの孔内に上記気体を導入し、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0024】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)アルコキシシランの加水分解による多孔質シリカの製造工程であって、(a1)界面活性剤およびアルコキシシランを混合し、混合液を形成する工程と、(a2)前記混合液に水を添加することにより、前記アルコキシシランの加水分解反応を行わせる工程と、(a3)前記(a2)工程の後、熱処理を施すことにより前記界面活性剤を除去する工程と、を有する多孔質シリカの製造工程と、(b)前記(a)工程により製造された多孔質シリカの孔内において粒子を合成する工程であって、前記多孔質シリカと粒子材料または前記粒子材料を元素組成として有する液または気体とを接触させ、前記多孔質シリカの孔内に上記液または気体を導入させ、前記多孔質シリカの前記孔内において前記粒子材料または前記粒子材料化合物を含有する粒子を形成する工程と、を有する。
【0025】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカは、平均孔直径が0.5以上1.5nm以下である孔を有し、金属、金属化合物および炭素のうちのいずれかを含有する粒子を、前記孔中に内包している。
【0026】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有する。
【0027】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属酸化物を含有する微細粒子を形成する工程と、(d)前記(c)工程の後、前記金属酸化物を含有する微細粒子を還元することにより、前記金属の微細粒子を形成する工程と、を有する。
【0028】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と(c)前記(b)工程の後、前記多孔質シリカの孔内の前記液を還元することにより、前記金属の微細粒子を形成する工程と、を有する。
【0029】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径を制御することにより、前記微細粒子の発光波長を調整する。
【発明の効果】
【0030】
本願において開示される発明のうち、以下に示す代表的な実施の形態に示される多孔質シリカ内包粒子によれば、多孔質シリカ内包粒子の特性を向上させることができる。
【0031】
また、本願において開示される発明のうち、以下に示す代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法によれば、良好な多孔質シリカ内包粒子を製造することができる。
【図面の簡単な説明】
【0032】
【図1】実施例1で得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)および内包させた酸化タングステンの粒子径の分布(グラフ)を示す図である。
【図2】実施例1で得られた酸化タングステン内包多孔質シリカのTaucプロットを示す図である。
【図3】実施例1で得られた酸化タングステン内包多孔質シリカの粒子径とバンドギャップエネルギーとの関係を示す図である。
【図4】実施例1で得られた酸化タングステン内包多孔質シリカのESRスペクトルを示す図である。
【図5】実施例2で得られた酸化タングステン内包多孔質シリカのTaucプロットを示す図である。
【図6】図6(a)は、実施例3で得られた金内包多孔質シリカを示す図(写真)であり、図6(b)は、紫外線照射下の金内包多孔質シリカを示す図(写真)である。
【図7】実施例3で得られた金内包多孔質シリカの発光現象時の発光スペクトルを示す図である。
【図8】図8(a)は、実施例4で得られた炭素内包多孔質シリカを示す図(写真)であり、図8(b)は、紫外線照射下の炭素内包多孔質シリカを示す図(写真)である。
【図9】実施例4で得られた炭素内包多孔質シリカの発光現象時の発光スペクトルを示す図である。
【図10】実施例5で得られたバナジン酸ビスマス内包多孔質シリカの透過型電子顕微鏡像(TEM像)を示す図である。
【図11】実施例5で得られたバナジン酸ビスマス内包多孔質シリカ(実線)およびバルクのバナジン酸ビスマス(破線)のTaucプロットを示す図である。
【図12】実施例6においてC16MPSを鋳型にして得られたCuO粒子の高角度散乱暗視野走査型透過電子顕微鏡像(HAADF-STEM像)である。
【図13】実施例6においてC6SMPSを鋳型にして得られたCuO粒子のHAADF-STEM像である。
【図14】実施例6においてC16MPSを鋳型にして得られたCuO粒子の35℃〜600℃でのTaucプロットを示す図である。
【図15】実施例6においてC6SMPSを鋳型にして得られたCuO粒子の35℃〜600℃でのTaucプロットを示す図である。
【図16】実施例6においてC6SMPSを鋳型にして得られたCuO粒子およびC16MPSを鋳型にして得られたCuO粒子のバンドギャップの温度依存性を示す図である。
【図17】細孔径の異なる多孔質シリカを鋳型に用いて合成した酸化銅の微細粒子のUV−Visスペクトルである。
【図18】酸化銅の微細粒子(量子ドット)のXPS(X線光電子分光)スペクトルである。
【図19】還元反応により生成した銅の微細粒子(量子ドット)のXPSスペクトルである。
【図20】酸化時と還元時における紫外・可視吸収(UV−Vis)スペクトルである。
【図21】合成した金属銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図22】合成した酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図23】実施の形態4〜実施の形態8(実施例7〜実施例11)の合成ルートの概念図である。
【図24】合成した酸化クロムの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図25】合成した酸化マンガンの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図26】合成した酸化鉄の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図27】合成した酸化コバルトの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図28】合成した酸化ニッケルの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図29】クロムの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図30】コバルトの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図31】得られた炭素の微細粒子の発光スペクトル(Ex.340nm)である。
【図32】得られた炭素の微細粒子の発光スペクトルである。
【発明を実施するための形態】
【0033】
以下、本発明の実施の形態を図面を参照しながら詳細に説明する。なお、同一の機能を有するものには同一もしくは関連の符号を付し、その繰り返しの説明を省略する。
【0034】
(実施の形態1)
<1>多孔質シリカの合成
界面活性剤を鋳型としたゾルゲル法により、多孔質シリカを合成する。このような合成法を分子鋳型法(テンプレート法)ということがある。
【0035】
一般的には、溶液中に界面活性剤を溶解させると、界面活性剤の種類と濃度に応じて例えば筒状のミセル粒子が形成される。ここで溶液中にシリカ源となるテトラエトキシシランなどを加えると、ミセル粒子の隙間でシリケートイオンの吸着および成長反応が進行し、シリカゲル骨格が形成される。このシリケートイオンの吸着および成長反応により、ゾル状態からゲル状態へ変化するためゾルゲル反応と呼ばれる。この後、焼成(熱処理)すると、鋳型とした界面活性剤が分解・除去されて多孔質シリカが得られる。言い換えれば、複数の孔(細孔、微細孔)を有するシリカ骨格が得られる。
【0036】
本実施の形態においては、無溶媒系で上記ゾルゲル反応を進行させることにより制御性良く、多孔質シリカを合成する。ここで言う“無溶媒系”とは、アルコキシシランと界面活性剤との混合の際の溶媒を言い、これらを混合しやすくするため、予めアルコキシシランを溶解し、また、界面活性剤を溶解するための溶媒、または、アルコキシシランと界面活性剤との混合液に添加する多量の溶媒(50等量以上の溶媒)をいう。このような溶媒を用いず、アルコキシシランと界面活性剤とを直接混合し、反応剤として必要な量の水(H2O)を添加し多孔質シリカを合成することで、制御性良く、多孔質シリカを形成することができる。
【0037】
以下に、本実施の形態の多孔質シリカの合成方法について詳細に説明する。
【0038】
アルコキシシランとカチオン性界面活性剤とをそのまま混合し、攪拌する。この混合液中に、水を添加し攪拌することで前駆体溶液を形成する。この前駆体溶液は、攪拌とともにゲル化する。
【0039】
添加する水(H2O)は、アルコキシシランの加水分解のための反応剤として寄与する。また、添加する水のpHは、アルコキシシランの等電点であるpH2程度に調整することが望ましい。等電点においては、アルコキシシランの加水分解、およびシリケートイオンのゲル化速度が最も遅いため、界面活性剤のミセル形成のための時間を十分に確保できるからである。
【0040】
また、水のpH0〜1程度においては加水分解の加速が起こるが、シリケートイオンのゲル化速度が十分に遅いため同様の効果が得られる。そのため、添加する水のpHは、0〜2の範囲に調整することが好ましい。pH3以上では加水分解およびゲル化速度が速すぎるため、界面活性剤の溶解およびミセル形成のための時間が十分に確保できない恐れがある。
【0041】
pH調整用の酸としては、塩酸、硫酸、硝酸等の無機酸、または酢酸などの有機酸を使用することができる。
【0042】
多孔質シリカの成型性の向上のためには、できるだけ少ない溶媒で加水分解することが好ましい。そのため、アルコキシシランに対する水の添加量は、反応に最低限必要な2等量(eq)以上20等量以下の範囲、より好ましくは、2等量以上10等量以下の範囲とする。このように、少溶媒の系とすることにより、反応系をほぼ純粋なシリケートイオンと界面活性剤との混合物に維持することができ、鋳型となる界面活性剤のミセルの安定性を確保しつつ、ゾルゲル反応を促進することができる。例えば、後述する炭素数が8未満の界面活性剤を用いても多孔質シリカを合成することができる。よって、多孔質シリカの平均細孔径(平均孔径、平均孔直径、D)を小さくすることができる。別の捉え方をすれば、界面活性剤の炭素数を調整することで、容易に多孔質シリカの孔径を調整することが可能となる。
【0043】
カチオン性界面活性剤としては、一般式R1R2R3R4N+X−で示される界面活性剤を用いることができる。R1は、例えば、炭素数1〜24のアルキル基、ベンジル基、フェニル基であり、R2R3R4は、例えば、メチル基、エチル基、プロピル基、ブチル基である。また、Xは、例えば、F、Cl、Br、Iなどのハロゲンイオンである。カチオン性界面活性剤としては、4級のカチオン性界面活性剤を用いることが好ましい。また、R1のアルキル基は直鎖型でも分岐型でもよい。
【0044】
以下、実施例に基づき本実施の形態をさらに詳細に説明する。なお、本発明は、これらの実施例によって限定されるものではない。
【0045】
(実施例A)…多孔質シリカ(ポーラスシリカ)の合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いてカチオン性界面活性剤を、0.2〜1.2eq(0.038mol×0.2〜0.038mol×1.2)を分散させ、撹拌した。この時点で、TEOSと界面活性剤とは混じり合わない。即ち、均一な溶液とならない。カチオン性界面活性剤としては、オクタデシルトリメチルアンモニウムクロライド(C18TAC)ヘキサデシルトリメチルアンモニウムクロライド(C16TAC)、テトラデシルトリメチルアンモニウムブロミド(C14TAB)、ドデシルトリメチルアンモニウムブロミド(C12TAB)、デシルトリメチルアンモニウムブロミド(C10TAB)、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)の8種類を用いて、それぞれポーラスシリカを合成した。
【0046】
次いで、上記混合液に、塩酸を用いてpHを0〜2程度に調整した水を2〜4eq(0.038mol×2〜0.038mol×4)程度、添加し、室温で撹拌した。一時間程度の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。この溶液(前駆体溶液)を室温または60℃に保持し、継続して撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤を除去した。これにより、無色透明のモノリス状のポーラスシリカが得られた。
【0047】
このように、反応系において、高濃度なシリケートイオン溶液を前躯体溶液として用いることで、界面活性剤のミセル形成を溶媒分子などにより阻害させることなく、シリカの合成を促進させることができる。特に、従来困難であった炭素数が小さい(例えば、7以下)の界面活性剤を用いてもミセル形成が可能となり微細な細孔を有するポーラスシリカを形成することが可能となる。
【0048】
得られたポーラスシリカの細孔を解析した。比表面積(SSA)、細孔容積(TPV)、平均細孔径(D)を測定した。比表面積(SSA)は、BET法により測定した。平均細孔径は、BJH法、HK法、GCMC法を用いて測定した。平均細孔径については、BJH法よりHK法において、より微細な細孔径の算出(解析)が可能である。また、HK法よりGCMC法において、より微細な細孔径の算出(解析)が可能である。
【0049】
C18TACを用いたポーラスシリカ(C18)の、BET比表面積は1361m2/g、細孔容積は0.96cm3/gであった。平均細孔径は、BJH法では、3.00nm、HK法では、3.36nm、GCMC法では、3.27nmであった。
【0050】
C16TACを用いたポーラスシリカ(C16)の、BET比表面積は1452m2/g、細孔容積は0.79cm3/gであった。平均細孔径は、BJH法では、2.70nm、HK法では、2.86nm、GCMC法では、2.82nmであった。
【0051】
C14TABを用いたポーラスシリカ(C14)の、BET比表面積は1234m2/g、細孔容積は0.60cm3/gであった。平均細孔径は、HK法では2.40nm、GCMC法では、2.26nmであった。
【0052】
C12TABを用いたポーラスシリカ(C12)の、BET比表面積は1056m2/g、細孔容積は0.53cm3/gであった。平均細孔径は、HK法では、2.00nm、GCMC法では、1.82nmであった。
【0053】
C10TABを用いたポーラスシリカ(C10)の、BET比表面積は916m2/g、細孔容積は0.45cm3/gであった。平均細孔径は、HK法では、1.60nm、GCMC法では、1.58nmであった。
【0054】
C8TABを用いたポーラスシリカ(C8)の、BET比表面積は810m2/g、細孔容積は0.41cm3/gであった。平均細孔径は、GCMC法では、1.28nmであった。
【0055】
C6TABを用いたポーラスシリカ(C6)の、BET比表面積は632m2/g、細孔容積は0.32cm3/gであった。平均細孔径は、GCMC法では、1.12nmであった。
【0056】
C4TACを用いたポーラスシリカ(C4)の、BET比表面積は586m2/g、細孔容積は0.29cm3/gであった。平均細孔径は、GCMC法では、0.92nmであった。
【0057】
このように、鎖長に対応した細孔を有するポーラスシリカが得られた。即ち、炭素数が18から4まで低下するにしたがって、平均細孔径(D)が小さくなることが判明した。特に、炭素数が12以下の界面活性剤を用いたポーラスシリカの平均細孔径は、2nm以下となり、ミクロ孔が確認された。また、従来法では、合成が困難であった、炭素数が7以下の界面活性剤を用いたポーラスシリカの合成が可能となり、炭素数が6のC6TABを用いたポーラスシリカの平均細孔径は、GCMC法で、1.12nmであり、また、炭素数が4のC4TABを用いたポーラスシリカの平均細孔径は、GCMC法で、0.92nmであった。このように、炭素数が8未満の界面活性剤を用い、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカの形成が可能であることが判明した。また、細孔容積も大きく、0.25cm3/g以上のポーラスシリカの形成が可能であることが判明した。
【0058】
なお、上記メソポーラスシリカの合成においては、2〜4eqの水を用いたが、8eqの水を用いても良好に加水分解が進むことが確認された。このように、成型性の向上のためには、溶媒が存在しない(無溶媒である)、言い換えれば、溶媒としての水を含有しないことが要求される。溶媒としての水とは、例えば、アルコキシシランやカチオン性界面活性剤などの溶解や分散に必要な、これらの材料の数十倍等量(例えば、50倍等量以上)の水(溶媒)をいう。これに対し、本発明でいう無溶媒とは、アルコキシシランに対する水の添加量で言えば、反応に最低限必要な2等量(eq)から、その10倍程度、即ち、2等量以上20等量以下の範囲である。また、より好ましくは、2等量以上10等量以下の範囲である。この条件を用いることで、系を高濃度なシリケートイオンと界面活性剤との混合物にすることができ、成型性と界面活性剤ミセルの安定性を確保することができる。
【0059】
(実施例B)…有機シラン添加による細孔径制御
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)とトリエトキシビニルシラン(TEVS)を8g×5%(0.038mol×5%)とを混合し、続いて界面活性剤を0.2〜1.2等量を添加し、撹拌した。この混合物に、塩酸を用いてpH0〜2に調整した水を2〜4等量の範囲で添加し、室温で撹拌した。1時間程度の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。さらに、この溶液(前駆体溶液)を室温または60℃に保持し、継続して撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤を除去した。界面活性剤としては、カチオン性界面活性剤である、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)の3種類を用いて、それぞれについてポーラスシリカを形成した。
【0060】
得られたポーラスシリカの細孔を解析した。比表面積(SSA)、細孔容積(TPV)、平均細孔径(D)および細孔壁厚さ(Dwall)を測定した。比表面積(SSA)は、BET法により測定した。平均細孔径は、GCMC法を用いて測定した。細孔壁厚さ、即ち、筒を構成する壁の厚さは、X線回折結果などにより算出することができる。
【0061】
C8TABを用いたポーラスシリカ(C8V)の、BET比表面積は519m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.99nmであった。細孔壁厚さは、2.37nmであった。
【0062】
C6TABを用いたポーラスシリカ(C6V)の、BET比表面積は582m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.82nmであった。細孔壁厚さは、2.00nmであった。
【0063】
C4TACを用いたポーラスシリカ(C4V)の、BET比表面積は355m2/g、細孔容積は0.16cm3/gであった。平均細孔径は、0.77nmであった。細孔壁厚さは、1.98nmであった。
【0064】
これに対し、上記実施例Aにおいては、C8TABを用いた場合、平均細孔径は、1.28nmであるため、有機シラン化合物の添加により、平均細孔径の、1.28nmから0.99nmへの縮小効果が確認された。細孔径の差は、0.29nmである。
【0065】
同様に、上記実施例Aとの比較において、C6TABを用いた場合、有機シラン化合物の添加により、平均細孔径の、1.12nmから0.82nmへの縮小効果が確認された。細孔径の差は、0.30nmである。
【0066】
また、C4TACを用いた場合、有機シラン化合物の添加により、平均細孔径の、0.92nmから0.77nmへの縮小効果が確認された。細孔径の差は、0.15nmである。
【0067】
このように、有機シラン化合物の添加により、ポーラスシリカの細孔径の微調整が可能であることが分かった。特に、炭素数が8の界面活性剤を用いても、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカの形成が可能であることが判明した。また、上記縮小効果から推測すれば、炭素数10や12の界面活性剤を用いても、有機シラン化合物の添加により、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカが形成される。また、さらに炭素数の少ない界面活性剤の使用、界面活性剤や有機シラン添加量の増減、反応温度の低下によっても平均細孔径を0.5nm程度まで低下させることが可能である。
【0068】
さらに、吸着質(例えば、分子径など)に応じて必要とされる平均細孔径を算出し、当該平均細孔径に合うよう界面活性剤の炭素数を選択する、または、有機シラン化合物を添加してポーラスシリカの形成を行うことで、細孔径の微細な調整(例えば、細孔径の約0.1nm刻みの調整)を行うことができる。例えば、サブナノメートルのオーダー、言い換えれば、m×10−10の単位で細孔径の微細な制御を行うことができる。
【0069】
(実施例C)…ポーラスシリカナノ粒子の合成
ポリプロピレン製容器にシリカ源としてTEOS8g(0.038mol;1eq)を入れ、界面活性剤を0.2〜1.2等量を添加した後、さらに、平均分子量1000のポリエチレングリコール(PEG)を7.5g添加し、撹拌した。この混合物に、塩酸を用いてpH0〜2に調整した水を2〜4等量の範囲で添加し、室温で撹拌した。1時間の撹拌でTEOSの加水分解が進行し、界面活性剤およびポリエチレングリコールが溶解したほぼ均一な溶液が得られた。この溶液(前躯体溶液)を室温あるいは60℃に保持し、撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。
【0070】
カチオン性界面活性剤としては、オクタデシルトリメチルアンモニウムクロライド(C18TAC)、ヘキサデシルトリメチルアンモニウムクロライド(C16TAC)、テトラデシルトリメチルアンモニウムブロミド(C14TAB)、ドデシルトリメチルアンモニウムブロミド(C12TAB)、デシルトリメチルアンモニウムブロミド(C10TAB)、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)のいずれかを用いることができる。
【0071】
ここで、上記工程のように、単にゲル化、焼成を行っただけでは、炭素数16以上の界面活性剤を用いた場合は、ナノ粒子の集合体よりなるモノリス状のポーラスシリカが得られるのに対し、炭素数16未満や臭化物塩の界面活性剤においては、細孔径分布が広い、アモルファス状のポーラスシリカが得られるにすぎなかった。
【0072】
そこで、上記前躯体溶液のうち、C6TABを用いて形成した前躯体溶液を、塩基性水溶液に滴下した。塩基性水溶液としては、28%のアンモニア水溶液を用いた。pHは約13である。滴下された略粒状の前躯体溶液は、ゲル状となり、アンモニア水溶液中に沈殿した。得られたゲルを、60℃で乾燥、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。得られたポーラスシリカは、白色のビーズ状で得られた。ビーズ状は、上記前躯体溶液の滴下形状に対応する。
【0073】
塩基性水溶液としては、上記アンモニア水溶液の他、アミン類の水溶液などを用いることができる。これらの塩基は、乾燥、焼成過程での除去が容易で、塩基性水溶液として用いて好適である。また、シリカは、pH14以上の高pH領域で、溶解が始まるため、高pH領域の塩基性水溶液を用いる場合には、反応後(ゲル化後、重合後)速やかに溶液外に取り出すことが好ましい。また、反応系中に、アルカリ金属やアルカリ土類金属のイオンが共存するとシリカの溶解速度が高まるため、水酸化ナトリウムなどの水溶液よりも上記アンモニアやアミン類を用いた塩基性水溶液を用いることがより好ましい。
【0074】
C16TACを用いて合成した多孔質シリカの細孔径を測定したところ、当該ポーラスシリカにおいては、2つの細孔径が確認された。即ち、約2nm程度の界面活性剤に由来するメソ孔と、約20〜50nm程度の粒子間隙に対応するメソ孔との2つの細孔を有するポーラスシリカの構成が確認できた。
【0075】
C6TABを用いて合成したポーラスシリカについても、約1nm程度の界面活性剤に由来するミクロ孔と、約5〜10nm程度の粒子間隙に対応するメソ孔との2つの細孔が確認できた。
【0076】
以上詳細に説明したように、単にゲル化、焼成を行っただけでは、炭素数16以上の界面活性剤を用いた場合は、ナノ粒子の集合体よりなるモノリス状のポーラスシリカが得られるのが、炭素数16未満の界面活性剤においては、アモルファス状のポーラスシリカが得られるにすぎない。これに対し、塩基性水溶液に前躯体溶液を接触させた場合には、炭素数16未満の界面活性剤を用いた場合でも、ナノ粒子化が可能となる。
【0077】
上記現象は次のように考察できる。pH0〜2の前駆体溶液中では、シリケートイオンは中性、あるいは正に帯電している。よって、シリケートイオンは、ポリエチレングリコールとは水素結合で相互作用し、界面活性剤とはカウンターアニオンを介して静電相互作用している。炭素鎖の短い、即ち、炭素数の少ない界面活性剤の場合、ミセル形成能が低いため、シリカの重合に伴う界面活性剤の集合、およびそれに伴うポリエチレングリコールの系外への相分離を十分に行うことができない。そのため、アモルファス状のシリカのみが得られる。一方、塩基性水溶液への滴下により系のpHを急激に上昇させた場合には、シリケートイオンは負に帯電し、カチオン性界面活性剤との間には、カウンターアニオンを介さない、より強固な静電相互作用が生じる。また、ポリエチレングリコールとの間の水素結合は解消され、逆に静電反発により相分離を誘導する。この二つの現象は界面活性剤のミセル形成とポリエチレングリコールの相分離を誘導し、界面活性剤の鎖長に対応した細孔を有したポーラスシリカのナノ粒子化を可能とすると考えられる。
【0078】
<2>微細粒子の合成方法
次いで、上記<1>で合成された多孔質シリカを使用し、その孔(細孔、微細孔)に金属などの材料を内包させることにより微細粒子(粒子、ナノ粒子、サブナノ粒子)を合成する。
【0079】
多孔質シリカを金属化合物水溶液と接触させ、多孔質シリカの孔内に金属化合物水溶液を浸透させる。次いで、多孔質シリカを乾燥した後、焼成し、多孔質シリカの孔内に金属を内包させる。
【0080】
焼成により金属または金属化合物が析出(残存)する金属化合物水溶液であれば、用いる金属化合物水溶液に制限はないが、例えば、当該水溶液として、過酸化タングステン酸水溶液などの、粒子材料を元素組成として有する化合物をを用いることができる。
【0081】
上記過酸化タングステン水溶液を用いた場合には、多孔質シリカの孔内に酸化タングステン(WO3)を内包させることができる。また、酢酸銅や硝酸銅などの銅塩を用いた場合には、多孔質シリカの孔内に酸化銅を内包させることができる。また、塩化鉄や硝酸鉄などの鉄塩を用いた場合には、多孔質シリカの孔内に酸化鉄を内包させることができる。また、硝酸マンガンなどのマンガン塩を用いた場合には、多孔質シリカの孔内に酸化マンガンを内包させることができる。また、硫酸チタニルなどの塩を用いた場合には、多孔質シリカの孔内に酸化チタンを内包させることができる。
【0082】
用いることの出来る金属塩は任意の溶媒に溶解し、溶液にすることが可能な化合物であれば種類は問わない。酸化物を得るためには焼成により容易に酸化することが望ましく、そのためには硝酸塩や酢酸塩などの熱分解しやすい塩を用いることが望ましい。用いる溶媒は水であっても有機溶媒であっても良く、金属塩の溶解度を制御するために硝酸や塩酸、アンモニアなどによりpHをコントロールした溶媒であっても良い。
【0083】
多孔質シリカは吸着機能を有するため、液体や気体と接触させるだけで容易に多孔質シリカの孔中に液体や気体を導入させることができる。また、微細な孔を有する多孔質シリカを用いれば、内包させる粒子材料(金属や金属化合物等)を容易に微細化することができる。
【0084】
このように、本実施の形態の微細粒子の合成方法によれば、簡易な処理により、多孔質シリカ内包粒子を形成することができる。また、上記<1>においては、0.5以上1.5nm以下の微細な孔径を有する多孔質シリカも合成可能であるため、このような多孔質シリカを鋳型に用いることで、内包させる粒子の粒径(粒子の直径)を容易に小さくすることができる。例えば、粒径(粒子の直径)を1.5nm以下と、より好ましくは、1nm以下(サブナノオーダー)とすることができる。もちろん、上記<1>の合成方法によれば、炭素数の大きな界面活性剤を用いることで、シングルナノメートルのオーダー(10nm以下、より好ましくは、5nm以下)の粒子も容易に調整することができる。このように、内包させる粒子を微細化することで、バルクの状態では見られない特異な性質を有する多孔質シリカ内包粒子(機能性複合体)を形成することができる。
【0085】
また、上記<1>においては、孔径の分布を小さくでき、言い換えれば、孔径のばらつきを小さくでき、狭い粒径分布を有する微細粒子(ここでは、サブナノ粒子)の合成が可能となる。別の言い方をすれば、バンドギャップなどの物理・化学的特性のばらつきの少ない量子ドットの合成が可能となる。
【0086】
また、孔内への粒子材料(粒子を構成する元素、または、その元素を元素組成として有する化合物)の導入も、上記液体を用いれば容易に行うことができる。このような液体プロセス(液相プロセス)では、導入する液体(前駆体)も、塩、錯体等、種類を選ばない。例えば、金属または金属化合物を孔内で合成するには、金属イオンを有する塩、金属錯体等を用いることができる。
【0087】
また、粒子材料を気体で導入しても良い。例えば、粒子材料を合成し得る原料ガスと接触させることにより、気相成長(気相での化学反応)により孔内において微細粒子を形成してもよい。また、粒子材料を気化(蒸発)させた気体を用いてもよい。
【0088】
特に、多孔質シリカ自身の特性として吸着性を有するため、粒子材料を容易に孔内に取り込むことができ、均一性の高い微細粒子を形成することができる。
【0089】
なお、多孔質シリカ(固体)と固体の原材料とを乳鉢等で混合し、乳棒等により圧力を加えることにより孔内に固体の粒子材料を導入することも可能である。
【0090】
また、多孔質シリカ中の微細粒子の形成割合は、細孔容積と液体プロセスに用いられる液体(前駆体)の濃度、浸漬回数で制御が可能である。また、気体プロセスにおいては、原料ガスの流量などで制御可能である。
【0091】
また、多孔質シリカ自身は、高い熱的・化学的安定性を有する。例えば、耐熱性や耐薬剤性を有するため、微細粒子を内包させた状態で、酸化雰囲気での熱処理(焼成)により微細粒子を酸化させ、また、薬液処理により所望の処理(例えば、炭化)を行うことが可能である。また、酸化状態の微細粒子を還元雰囲気(例えば、水素雰囲気)で熱処理することにより還元してもよい。また、光照射による還元を行ってもよい。また、硫化水素雰囲気下で熱処理し、硫化物を得ることができる。
【0092】
さらに、内包した粒子と、化学反応を用いて発生させたガス状の化合物をシリカ細孔内で反応させ、目的の化合物を得ることもできる。この方法はテルル化カドミウムのサブナノ粒子を得る場合に特に有効である。その場合には、塩化カドミウムの粒子を細孔内で生成した後、テルル化水素ガスなどと反応させることにより、テルル化カドミウムを得ることができる。テルル化カドミウムは太陽電池や蛍光体に応用した場合に、高い変換効率や発光効率を示すことから注目される物質であるが、その毒性から使用が懸念されている。本発明の多孔質シリカ内での合成法を用いれば、テルル化カドミウム自体がシリカ細孔内に内包された形で得られるため、毒性によるリスクを低減できると共に、サブナノ粒子の環境中への排出リスクを低減することが可能である。
【0093】
このように、内包させる微細粒子の材料としては、金属および金属化合物の他、炭素や炭素化合物(例えば、SiCなど)など多種多様の材料に対応することができる。また、金属化合物(半導体、セラミックスなどを含む)としては、金属酸化物、金属窒化物、金属硫化物などが例示でき、金属酸化物としては、酸化タングステンや酸化チタンなど、金属窒化物としては、窒化タングステン(WN)など、金属硫化物としては、硫化カドミウム(CdS)などを例示することができる。
【0094】
また、上記微細粒子の機能性材料としての利用は、多孔質シリカ内包させた状態で(微細粒子とシリカの複合体として)行ってもよい。特に、多孔質シリカは、前述したように、熱的・化学的安定性を有し、また、光透過性にも優れるため、光触媒や光学素子等への応用の場合には、有効な担持体としてそのまま用いることが可能である。
【0095】
また、多孔質シリカの鋳型を分解することにより、微細粒子のみを取り出し、この粒子を他の材料(担体)に固定(担持)させ、機能性材料として使用してもよい。多孔質シリカから微細粒子を取りだすには、例えば、水酸化ナトリウム水溶液やフッ酸等によりシリカ骨格を溶解させることで微細粒子を取り出すことができる。
【0096】
また、内包させる粒子を量子ドット化することで、多孔質シリカ内包粒子に各種機能を持たせることができる。
【0097】
例えば、量子サイズ効果によるバンドギャップの調整により、特定波長の光の吸収や発光が生じ、発光材料として用いることができる。発光波長はバンドギャップと相関するため、粒子サイズの制御により発光波長を制御可能になる。また、内包させる化合物を適宜選択(例えば、炭素や硫化カドミウム、テルル化カドミウム等)することにより、アップコンバージョン蛍光特性を持たせることも可能である。これにより、吸収光と発光光の周波数変換が可能となり、例えば、近赤外光を可視光に、あるいは可視光を紫外光に周波数変換する素子(光学素子)などとして利用することができる。
【0098】
また、光触媒材料として用いることができる。例えば、二酸化チタンに光(紫外線)が当たると、価電子帯の電子が伝導帯へと励起され、電子(e-)とホール(h+)が生成する。このホールは強い酸化力をもち、周囲の分子から電子を奪い、酸化させる。水分子を酸化した場合、OHラジカルを生成する。また、伝導帯に励起された電子は電荷移動により、周囲の分子に電子を与え、還元させる。酸素を還元した場合にはスーパーオキシドラジカルアニオン(O2-・)を生成する。このようなラジカルは高い反応性を持つために空気中や水中の有機物(有害物質)を二酸化炭素まで分解する。これにより有害物質の除去が可能となる。このような酸化還元反応は、光触媒の価電子帯や伝導帯の準位と反応基質の酸化還元準位との差が、それぞれの電子移動の方向に対してエネルギー的に安定化に向かう方向でなければ進行しない。すなわち、光触媒の伝導帯準位は目的の還元反応の準位よりも負である必要があり、価電子帯の準位は目的の酸化反応準位よりも正である必要がある。そのため、光触媒として有効に活用できる化合物はこの制限のために限定される。例えば酸化タングステンの価電子帯は他の半導体と比較して低い準位に存在するため、強い酸化力を有する。しかし、伝導帯は二酸化チタンなどと比較して低く、一般的に還元力が低いと言われている。事実、酸化タングステンの伝導帯準位は酸素の還元準位よりも低く、酸素の還元反応を起こすことができない。そのため、酸化タングステンを水中の有機物の分解反応に用いようとした場合、ホールによる直接酸化やOHラジカルの生成により有機物を分解することが可能だが、伝導帯の電子による反応を起こすことができないため、すぐに電子過剰の状態になる。この電子過剰な状態は励起子の再結合を促進するため、光触媒効率の著しい低下を引き起こす。そのため、酸化タングステンを光触媒として用いるためには伝導帯の電子を効率よく使用する必要がある。また、価電子帯と伝導帯の準位が目的の反応を起こす準位を満足していたとしても、活性化エネルギーの障壁を超えるためには過剰のエネルギーが必要になる。この過剰のエネルギーは目的の反応準位と、価電子帯と伝導帯準位との差分のエネルギーで供給することができるため、準位差が大きければ、目的の反応の効率を向上させることができる。このように、光触媒の反応性や反応効率には価電子帯と伝導帯の準位が大きく関わっている。そのため、価電子帯と伝導帯の準位の制御が可能になれば、同じ物質を用いたまま、任意に反応性や反応効率をコントロールすることが可能になる。粒子サイズの制御で価電子帯と伝導帯の準位をコントロール可能な量子サイズ効果を用いる方法はこの目的において非常に有効な手段である。量子サイズ効果が発現する粒径領域は、一般的に直径がシングルナノメートルの領域であり、サブナノメートルの領域でより顕著になる。従って、大きな量子サイズ効果を発現しつつ制御するためには、サブナノメートル領域での粒径制御が必須である。これまでに提案されてきたナノ粒子合成法はサブナノメートル領域での制御性が悪く、また、適応可能な化合物群も限定されている。本発明の方法では、通常の化合物合成プロセスをそのまま用いることが可能なため、現在までに化学合成可能な化合物群全てに適応可能である。また、シリンダー状の細孔を鋳型とするため、細孔径よりも大きな粒子が複生成することが無く、非常にシャープな粒度分布で生成物を得ることが可能である。さらに、ゼオライトのような三次元的な細孔構造を有さないため、内包粒子同士が合一化して量子サイズ効果の低い大粒子を生成することも無く、安定に使用することができる。このように、量子サイズ効果により、有効な光触媒として利用可能である。
【0099】
また、色素増感型太陽電池の色素材料として、また、量子ドット型太陽電池の量子ドットとして用いることができる。
【0100】
具体的には、増感剤から電子を受け取る半導体粒子や増感剤そのものとして用いることができる。色素増感型太陽電池の構成に制限はないが、例えば、酸化チタンのナノ粒子の集合体を備える透明電極と、他の電極と、これらの電極間に封止された電解液とを有するように構成する。酸化チタンは、紫外線しか吸収しないが、酸化チタンの表面に増感剤が吸着することで、可視光にも感度を有するようになる(色素増感)。太陽電池に光が当たると酸化チタンに吸着した色素が励起状態となり、電子を放出する。この電子は酸化チタンを経由して透明電極に達し外部に流れる。一方、電子を放出して陽イオンになった増感剤は、もう片方の電極から供給される電子を、電解液中の陰イオンを経由して受け取り、還元され、もとに戻る。負極上に存在する半導体粒子のフェルミ準位と対極にある金属のフェルミ準位差が太陽電池の起電力(開放電圧)を決定する。そのため、半導体粒子の伝導帯準位を量子サイズ効果によって制御すれば、太陽電池の起電力をコントロールすることが可能になる。また、金サブナノ粒子や炭素サブナノ粒子は、増感剤として用いることも可能であり、有機色素と比較して劣化の少ない増感剤として機能する。
【0101】
このように、本実施の形態の多孔質シリカ内包粒子は、光触媒材料や色素増感型太陽電池の色素材料として用いることができる。
【0102】
以下に効果をまとめて説明する。上記において詳細に説明したように、本実施の形態においては、サイズ制御性が高く、粒度分布の狭いサブナノ粒子の製造が可能となる。
【0103】
また、内包させる粒子材料を金属や半導体などとすれば、粒径の制御によりバンドギャップエネルギーを制御することができ、発光スペクトルや触媒活性の精密な制御が可能になる。これらは耐久性の高い発光素子やバイオイメージング、触媒として利用できる。
【0104】
また、内包させる粒子材料を光触媒機能を有する半導体などとすれば、量子サイズ効果によるバンドギャップの増大に伴う機能制御が可能になる。後述する実施例では、酸化タングステンのサブナノ粒子が酸素の単電子還元を可能にすることを示した。これはバルクの酸化タングステンでは起こらない現象である。これらは、高性能な触媒、特に水の分解による水素の製造や二酸化炭素固定化反応において有効な光触媒として期待される材料となりうる。さらに、半導体粒子をサブナノ粒子化することで、後述する酸化タングステンではフォトクロミズムを、酸化銅ではサーモクロミズムを示し、これらはセンサなどへの応用が可能である。
【0105】
また、内包させる粒子材料を炭素などとすれば、後述する実施例において説明するように、紫外線照射下での発光が見られる。これらも同様に発光素子やバイオイメージング等への応用が可能である。
【0106】
このように、各種粒子材料の微細化(特に、サブナノ粒子化)の効果は、粒子サイズの制御でその性質を制御することが可能であり、触媒活性、反応選択制、発光スペクトル分布、等をコントロールすることができる。特に、本実施の形態においては、0.5〜1.5nmの孔径を有する多孔質シリカを鋳型とすることで、非常に高いサイズ選択的合成が可能になっており、また、狭い粒度分布を有するサブナノ粒子が製造可能である。
【0107】
さらに、生成した粒子は多孔質シリカ内に分散性よく内包されている。また、多孔質シリカ自体が優秀な吸着材として働き、また、光透過性に優れるため、光触媒や光学素子等への応用の場合には、有効な担持・複合体としてそのまま用いることが可能である。また、多孔質シリカ自体をナノ粒子化すれば、より効率の良い触媒の構築や、ナノ蛍光体としての応用が可能である。
【実施例】
【0108】
以下、実施例に基づき本実施の形態をさらに詳細に説明する。なお、本発明は、これらの実施例によって限定されるものではない。
【0109】
(実施例1)
<1−1>多孔質シリカの合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いて界面活性剤を0.0075〜0.038molの範囲で添加した混合液を用いた。この混合液を攪拌しつつ、TEOSに界面活性剤を分散させた。
【0110】
界面活性剤としては、ヘキシルトリメチルアンモニウムブロミド(C6TAB)およびブチルトリメチルアンモニウムクロライド(C4TAC)のいずれかを用いた。また、界面活性剤に加え、トリメトキシビニルシラン(TEVS)をTEOSに対して5mol%添加した混合液を用いた。
【0111】
上記4種類の混合液に、それぞれ塩酸を用いてpH2に調整した水を2.74g(0.152mol;4eq)入れ、室温に保持し、継続して撹拌した。いずれの混合液を用いた場合も、12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し(熱処理を施し)、界面活性剤を除去することにより4種類の多孔質シリカ(Samples)を得た。
【0112】
得られた多孔質シリカについて、窒素吸着機(Tristar3000、マイクロメリティクス社製)を用い、試料の細孔構造と、比表面積、細孔容積および平均細孔径とを調べた。試料は、直前にVacPrep061(マイクロメリティクス社製)にて、160℃で3時間脱気したものを用いて測定した。窒素吸着機(BELSORP−max、日本ベル社製)を用い、試料の孔径分布についてGCMC法にて解析した。
【0113】
表1に、得られた多孔質シリカの孔の解析結果を示す。即ち、各試料(Sample)について、比表面積(SSA、[m2/g])、細孔容積(TPV、[cm3/g])、平均細孔径(Dpore、[nm]、直径)を測定した。なお、比表面積(SSA)は、BET法により測定した。細孔容積(TPV)は、総細孔容積である。平均細孔径(D)は、GCMC法を用いて測定した。
【0114】
【表1】

C6TABを用いたポーラスシリカ(C6SMPS)の、BET比表面積は632m2/g、細孔容積は0.32cm3/gであった。平均細孔径は、1.12nmであった。
【0115】
C4TACを用いたポーラスシリカ(C4SMPS)の、BET比表面積は586m2/g、細孔容積は0.29cm3/gであった。平均細孔径は、0.92nmであった。
【0116】
C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)の、BET比表面積は582m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.82nmであった。
【0117】
C4TACおよびTEVSを用いたポーラスシリカ(C4VSMPS)の、BET比表面積は355m2/g、細孔容積は0.16cm3/gであった。平均細孔径は、0.77nmであった。
【0118】
このように、無溶媒系でのゾルゲル反応により、制御性良く、具体的には、孔径の制御性や反応性が良い状態で、多孔質シリカを合成することができた。特に、炭素数が8未満の界面活性剤を用い、孔径が小さい、例えば、孔径が0.5nm以上1.5nm以下の多孔質シリカの合成が可能となった。このような孔径の多孔質シリカをスーパーミクロポーラスシリカ(SMPS)と呼ぶことがある。
【0119】
また、TEVSのような有機シラン化合物の添加により、C6TABを用いたポーラスシリカについては、平均細孔径が、1.12nmから0.82nmへ縮小し、また、C4TACを用いたポーラスシリカについては、平均細孔径が、0.92nmから0.77nmへ縮小した。このように有機シラン化合物の添加により、孔径のさらなる微細化が可能となることが判明した。
【0120】
<1−2>微細粒子の合成
次いで、上記多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成した。上記4種類の多孔質シリカのそれぞれ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、真空ポンプで減圧し、水等の吸着物質を除去した(減圧乾燥工程)。その後、減圧を一時停止し、各多孔質シリカに0.2M(mol)の過酸化タングステン酸水溶液を添加した。この時にフラスコ内は陰圧を維持している。水溶液と粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における過酸化タングステン酸水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、その孔内に酸化タングステンの微細粒子を内包した多孔質シリカを得た。
【0121】
本実施例においては前駆体水溶液の導入に減圧下での浸透を行っているが、これは、内包粒子の重量を一定にしつつ、分析の妨げになる細孔外で生成した粒子を最小限にするためである。触媒等に用いる場合など細孔外に多少の粒子が生成してもかまわない場合には常圧での浸透や、水溶液への浸漬等によって前駆体溶液を浸透させてもかまわない。また、粒子外部に付着した余剰の前駆体水溶液を水洗等により除去すれば、減圧下での浸透と同じようにシリカ細孔内にのみ粒子を生成することが可能である。
【0122】
(ア)得られた酸化タングステン内包多孔質シリカについて、透過型電子顕微鏡(TEM)による観察を行った。具体的には、FE−TEM(TECNAI F20:FEI)を用いて、試料の形状、粒子サイズを測定した。観察試料は、粉砕した試料をコロジオン膜付銅メッシュに分散させることで作製した。
【0123】
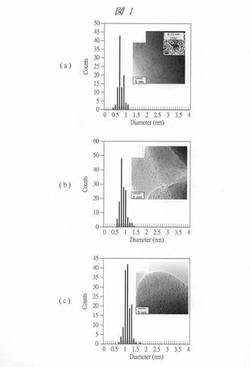
図1は、得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)および内包させた酸化タングステンの粒子径の分布(グラフ)を示す図である。グラフの横軸は、粒子径(Diameter、[nm])であり、縦軸は、個数(Counts、[個])である。
【0124】
図1中の(a)は、酸化タングステン内包させたC4VSMPS(C4VW)の画像である。(b)は、酸化タングステン内包させたC4SMPS(C4W)の画像である。(c)は、酸化タングステン内包させたC6SMPS(C6W)の画像である。いずれの画像においても、内包させた酸化タングステンの粒子径が、1nm以下であることが確認された。また、内包箇所に偏りがなく、ほぼ均一に高濃度に内包している、言い換えれば、高分散であることが確認された。
【0125】
また、内包させた酸化タングステンの粒子径については、図1(a)に示すように、酸化タングステン内包させたC4VSMPS(C4VW)については、0.8nm程度の粒子径の割合が高く、平均粒径は、0.77nmであった。また、図1(b)に示すように、酸化タングステン内包させたC4SMPS(C4W)については、0.8nm程度の粒子径の割合が高く、平均粒径は、0.89nmであった。また、図1(c)に示すように、酸化タングステン内包させたC6SMPS(C6W)については、0.9nm程度の粒子径の割合が高く、平均粒径は、1.11nmであった。
【0126】
このように、多孔質シリカの孔内に酸化タングステンなどの金属化合物を内包させることができた。特に、孔径が0.5nm以上1.5nm以下の微細な孔を有する多孔質シリカにも酸化タングステンなどの金属化合物を内包させることができた。また、孔径のより小さい多孔質シリカに内包させた場合、酸化タングステンの粒子径は小さくなり、多孔質シリカの孔径に対応した大きさの微細粒子を形成できることが判明した。また、粒径分布が狭く、例えば、粒径分布が、平均粒径から±0.3nm程度の範囲となる、良好な微細粒子を形成できることが判明した。
【0127】
(イ)得られた酸化タングステン内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて紫外・可視吸収スペクトルを求め、Taucプロットにより、酸化タングステンの微細粒子のバンドギャップエネルギーを求めた。ここでTaucプロットとは、半導体に対して紫外・可視吸収スペクトルの吸収端からバンドギャップを求める方法である。半導体のバンド間の光学的遷移による光吸収においては、吸光度と光子エネルギーの関係において「α=k(E−Eg)2/E(kは定数)」の関係が成り立つ。よって、横軸に光子エネルギーE(Energy、hν[eV])を、縦軸に吸光度(α)と光子エネルギー(hν[eV])の積のn乗((αhν)n)をとり、接線を引く。ここでnの値は直接遷移型の場合n=2となり、間接遷移型の場合n=1/2となる。この接線とベースラインとの交点がバンドギャップエネルギー(Eg)となる。
【0128】
図2に、得られた酸化タングステン内包多孔質シリカのTaucプロットを示す。グラフ(a)は、酸化タングステン内包させたC4VSMPS(C4VW)のプロットである。グラフ(b)は、酸化タングステン内包させたC4SMPS(C4W)のプロットである。グラフ(c)は、酸化タングステン内包させたC6SMPS(C6W)のプロットである。(d)は、バルクの酸化タングステン、即ち、微粒子化していない酸化タングステン、言い換えれば、酸化タングステンの塊のプロットである。
【0129】
各グラフ(a)〜(c)について、バンドギャップエネルギー(Eg)を算出したところ、(a)の酸化タングステン内包させたC4VSMPS(C4VW)のEgは、3.69eV、(b)の酸化タングステン内包させたC4SMPS(C4W)のEgは、3.43、(c)の酸化タングステン内包させたC6SMPS(C6W)のEgは、3.32となった。
【0130】
表2に、得られた酸化タングステン内包多孔質シリカについて、鋳型となった多孔質シリカの孔径(Pore diameter of SMPS、[nm])、内包させた酸化タングステンの粒子径(Particle size)、上記バンドギャップエネルギー(Eg)から算出された粒径(Caluculated particle size、[nm])および上記バンドギャップエネルギー(Eg、[eV])を示す。なお、バンドギャップエネルギー(Eg)から算出された粒径については、いわゆるBrusの式を用いて算出した。Brusの式は、「Eg=Egb+h2π2/2R2・(1/Me+1/Mh)−1.8e2/εR」で示され、Egは、上記バンドギャップエネルギー[eV]、Egbは、バルクのバンドギャップエネルギー[eV]、hは、プランク定数(4.136×10−15eV・s)である。Rは、粒子半径で、MeとMhは、それぞれ電子と正孔の有効質量、εは比誘電率である。
【0131】
【表2】
表2に示すように、酸化タングステン内包させたC6SMPS(C6W)について、孔径が1.12nmの多孔質シリカ中に、直径1.11nmの粒子が確認でき、その粒子のEgは、3.32eVであった。このEgから算出された粒径は0.97nmであった。
【0132】
また、酸化タングステン内包させたC4SMPS(C4W)について、孔径が0.92nmの多孔質シリカ中に、直径0.89nmの粒子が確認でき、その粒子のEgは、3.43eVであった。このEgから算出された粒径は0.87nmであった。
【0133】
また、酸化タングステン内包させたC4VSMPS(C4VW)について、孔径が0.77nmの多孔質シリカ中に、直径0.70nmの粒子が確認でき、その粒子のEgは、3.69eVであった。このEgから算出された粒径は0.70nmであった。
【0134】
このように、孔径と粒径とは、良い相関性を示している。また、粒径が小さくなるにしたがってEgが大きくなっており、量子サイズ効果を確認することができた。また、このEgから算出した粒径も、TEMによる測定結果(粒径)と良好な相関関係を有していることから、微細粒子(2nm以下、より好ましくは、サブナノオーダー(1.0nm以下)の微細粒子)が形成されていることが確認できた。
【0135】
図3に、得られた酸化タングステン内包多孔質シリカの粒子径(Particle size)とバンドギャップエネルギーEgとの関係を示す。点(a)は、酸化タングステン内包させたC4VSMPS(C4VW)、点(b)は、酸化タングステン内包させたC4SMPS(C4W)、点(c)は、酸化タングステン内包させたC6SMPS(C6W)のプロットである。また、ここでは、炭素数が8以上の界面活性剤を用いて形成した孔径の大きい多孔質シリカに酸化タングステンを内包させた試料((d)、(e))についても粒子径とEgとの関係を示してある。(d)の試料の粒子径は1.4nmであり、(e)の試料の粒子径は1.8nmである。また、点(f)は、バルクの酸化タングステン(粒子径が1000nm)のプロットである。
【0136】
この図3からも、粒子径がサブナノ領域になるにしたがってEgが急激に大きくなっており、顕著な量子サイズ効果を確認することができる。
【0137】
(ウ)得られた酸化タングステン内包多孔質シリカについて、酸素の一電子還元能の評価を行った。得られた試料を紫外線照射下に置き、電子スピン共鳴E−500(Bulker社製)を用いて、電子スピン共鳴(ESR:Electron Spin Resonance)を利用したスピントラップ法により評価を行った。スピントラップ剤としては、ジメチルピロリン−N−オキシド(DMPO)を用いた。
【0138】
即ち、酸素の一電子還元反応の生成物であるスーパーオキシドラジカルアニオン(O2-・)をDMPOで補足し、生成するDMPO-OOHラジカルのシグナルをESRで観測した。具体的には次のような実験を行った。
【0139】
酸化タングステン内包させたC4SMPS(C4W)を40mgと、タングステン酸を600℃で3時間焼成して得られたバルクの酸化タングステンを4mg計測し、それぞれを40mMのDMPOのエタノール溶液5ml中に添加し、分散させた。試料の量が異なるのは、それぞれの試料中の酸化タングステンの質量が同量になるように調整したためである。
【0140】
上記分散液をESRのフラットセルに導入し、市販のブラックライトを光源として90分の紫外線照射を行った。この紫外線(UV)照射前後のESRスペクトルから酸素−電子還元能の評価を行った。なお、全てのスペクトルの強度(シグナル強度)は、既知濃度のテトラメチルピロリジノキシフリーラジカルを標準試料として用い校正してある。
【0141】
図4に、ESRスペクトルを示す。横軸は、磁場の強さ(Magnetic Field、[G])を示し、縦軸は、シグナル強度(Intensity)を示す。このシグナル強度は、フリーラジカルの個数、言い換えれば生成したO2-・量に対応している。
【0142】
図4(a)は、酸化タングステン内包させたC4SMPS(C4W)のESRスペクトルであり、破線(点線)は、UV照射前のESRスペクトルを示し、実線は、UV照射後のESRスペクトルを示す。図示するように、酸化タングステン内包させたC4SMPS(C4W)においては、UV照射によりシグナル強度の増大が確認できた。即ち、DMPO−OOHラジカルの生成が観測された。これに対し、図4(b)に示すバルクの酸化タングステンの場合は、UV照射の前後でシグナル強度の変化はほとんどなかった。
【0143】
これにより、酸化タングステン内包させたC4SMPS(C4W)においては、紫外線照射による酸素の一電子還元反応が進行していることが確認できた。これにより、生成した電子とホールの両方を速やかに消費する反応経路が確保される。そのため、電子とホールの再結合が抑制され、反応効率が著しく増大する。たとえば、環境浄化の用途においては、紫外線照射により、非常に酸化作用の強いスーパーオキシドラジカルアニオン(O2-・)を生成し、これによって、悪臭成分等を分解するなど、光触媒作用を有することが確認される。上記悪臭成分の他、水質浄化、シックハウスガス分解、排ガス中有害物質分解、土壌汚染物質の除去、環境ホルモンやダイオキシン分解、抗菌など、の機能も備え得る。
【0144】
(エ)得られた酸化タングステン粒子のフォトクロミック挙動について観測した。酸化タングステンは薄膜状にすることでフォトクロミズムを示すことが知られている。本発明で得られた酸化タングステン粒子も紫外線照射下で可逆的なフォトクロミズムを示すことが観測された。得られた酸化タングステン-シリカ複合体を時計皿に入れ、365nmの紫外光を照射した。試料にエタノールのようなアルコールを滴下すると、より顕著な色の変化が観測された。これらの現象は可逆的であり、目視で観測することができる。エタノールの滴下によるフォトクロミック挙動の向上はその着色メカニズムに起因する。酸化タングステンの場合、光照射により還元されたW5+が生成し、W5+のd−d遷移が青色の着色に起因する。一方でエタノールなどのαプロトンを有するアルコールが共存した場合、その分解過程でホールには一電子余剰に注入される。そのため、通常よりもW5+の生成効率が向上し、より顕著なフォトクロミズムを示すと考えられる。これらの性質を応用すればセンサやディスプレイ材料等への応用が可能である。さらに通常の酸化タングステン薄膜では青から白への脱色速度が遅く、加速するには加熱が必要であるとされている。しかし、本発明で得られた酸化タングステンサブナノ粒子の場合、脱色に要する時間は室温で10分未満である。この高い可逆性はセンサ等に用いた場合に有効である。
【0145】
(実施例2)
本実施例においては、多孔質シリカを粒子化(ナノ粒子化)することで、多孔質シリカ単体が有する第1孔(微細孔)と、複数の多孔質シリカの集合体である各粒子の粒子間の隙間よりなる第2孔を有する多孔質シリカを合成し、これら2種の孔を有する多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成する。このように、ナノ粒子化した多孔質シリカを用いることで、含浸させる溶液の物質拡散効率を向上させることができ、効率的に微細粒子を内包させることができる。また、触媒として用いる際には、分子の拡散効率と触媒界面が増加し、触媒活性などの機能が向上する。発光体として用いる際には媒質中への分散性が向上することで機能が向上する。
【0146】
<2−1>多孔質シリカの合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いて界面活性剤としてヘキシルトリメチルアンモニウムブロミド(C6TAB)を0.0075〜0.038molの範囲で添加した混合液を用いた。この混合液を攪拌しつつ、TEOSに界面活性剤を分散させた。次いで、混合液中に平均分子量1000のポリエチレングリコール(PEG)を0.0075mol添加し、撹拌した。
【0147】
この後、混合液に、それぞれ塩酸を用いてpH2に調整した水を2.74g(0.152mol;4eq)入れ、室温に保持し、継続して撹拌したところ、1時間の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。この溶液を室温で静置し、数時間から数日間熟成させ、前駆体液とした。
【0148】
次いで、上記前駆体液を、シリンジで28%のアンモニア水溶液に滴下した。滴下した前駆体液は、アンモニア水溶液に入った瞬間に球形を保ったままゲル化した。沈降した白色の球状ゲルを回収し、乾燥させた後、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。これにより白色で、直径約2〜3mmの球形の多孔質シリカを得ることができた。
【0149】
得られた多孔質シリカについて、BJH法を用いて平均細孔径を測定したところ、2つの孔径が確認された。第1孔種として1.1nmの平均孔径を有するミクロ孔と、第2孔種として20nmの平均孔径を有するメソ孔とを確認することができた。
【0150】
<2−2>微細粒子の合成
次いで、上記2種の孔を有する多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。その後、各多孔質シリカに0.2Mの過酸化タングステン酸水溶液を添加し、減圧下において、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における過酸化タングステン酸水溶液の添加量は、第1孔(ミクロ孔)の細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、第1孔のみに酸化タングステンの微細粒子を内包した多孔質シリカを得た。第1孔のみに粒子が生成する理由は以下の通りである。細孔径の大きな違いにより、ポアフィリングによる凝縮相の安定性が、第1孔では第2孔と比較して非常に高く、第1孔の容積に相当する体積の溶液を含侵させた場合に、優先的に第1孔への浸透が起こるためである。これにより、第1孔には量子ドットが内包されつつ、第2孔は物質拡散のための空間として確保することが可能になる。
【0151】
得られた酸化タングステン内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて電子スペクトル(可視・紫外吸収スペクトル)を求め、Taucプロットにより、酸化タングステンの微細粒子のバンドギャップエネルギーを求めた。
【0152】
図5に、得られた酸化タングステン内包多孔質シリカのTaucプロットを示す。横軸は、光子エネルギーE(Energy、hν[eV])であり、縦軸は、吸光度(α)と光子エネルギー(hν[eV])の積の平方根((αhν)1/2)である。図示するグラフから、バンドギャップエネルギー(Eg)は、3.3eV程度であることが分かる。前述の表2において、酸化タングステンを内包させたC6SMPS(C6W)の中の粒子のEgは、3.32eVであり、当初の多孔質シリカの孔径が1.12nmであることから、第1孔(ミクロ孔)にのみ酸化タングステンが形成したことが分かる。
【0153】
(実施例3)
本実施例においては、多孔質シリカを鋳型として金(Au)の微細粒子を合成する。
【0154】
<3−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C4TACおよびTEVSを用いたポーラスシリカ(C4VSMPS)を形成した。
【0155】
<3−2>微細粒子の合成
次いで、上記多孔質シリカ(C4VSMPS)を鋳型として金(Au)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。その後、多孔質シリカに0.2Mの塩化金酸水溶液を添加し、減圧下において、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における塩化金酸水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、回収した試料を水洗し、さらに、減圧乾燥を行った。その後、4%程度のH2(水素)を含有するAr雰囲気中で、350℃で、2時間程度焼成し、塩化金酸を還元し、金の微細粒子を内包した多孔質シリカを得た。
【0156】
得られた金内包多孔質シリカに、360nm〜370nmの波長の紫外線を照射したところ、目視において発光現象を確認することができた。図6(A)に、得られた金内包多孔質シリカの写真を、図6(B)に、紫外線照射下の金内包多孔質シリカの写真を示す。このように、発光(蛍光)材料としての機能を有することが判明した。
【0157】
また、上記発光現象時の発光スペクトル(Ex.360nm)を図7に示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、PL強度(PL Intensity、PL;フォトルミネッセンス(Photoluminescence)、[a.u.])である。発光スペクトルは、分光蛍光光度計(PL)FP−6500(日本分光社製)を用いて測定した。
【0158】
(実施例4)
本実施例においては、多孔質シリカを鋳型として炭素(C)の微細粒子を合成する。
【0159】
<4−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)を形成した。
【0160】
<4−2>微細粒子の合成
次いで、上記多孔質シリカ(C6VSMPS)を鋳型として炭素(C)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、0.26Mのブドウ糖(グルコース;C6H12O6)の水溶液を添加し、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。その後、回収した試料を水洗し、300℃で2時間程度、空気中で焼成し、ブドウ糖を炭化することにより、炭素の微細粒子を内包した多孔質シリカを得た。なお、上記焼成工程に代えて、濃硫酸に数時間、浸漬することでブドウ糖を炭化してもよい。また、本実施例では、ブドウ糖を用いたが、この他、適当な分子サイズを有する有機化合物の溶液を用い、焼成により炭化してもよい。また、有機化合物として、糖類を用いた場合には硫酸を用いた脱水反応で炭化させることができる。
【0161】
得られた炭素内包多孔質シリカに、360nm〜370nmの波長の紫外線を照射したところ、目視において発光現象を確認することができた。図8(A)に、得られた炭素内包多孔質シリカの写真を、図8(B)に、紫外線照射下の炭素内包多孔質シリカの写真を示す。このように、発光(蛍光)材料としての機能を有することが判明した。
【0162】
また、上記発光現象時の発光スペクトル(Ex.320nm)を図9に示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、PL強度(PL Intensity、PL;フォトルミネッセンス(Photoluminescence)、[a.u.])である。発光スペクトルは、分光蛍光光度計(PL)FP−6500(日本分光社製)を用いて測定した。
【0163】
(実施の形態2)
実施の形態1においては、多孔質シリカに金属化合物(WO3)、金属(Au)または炭素(C)の微細粒子を内包させたが、本実施の形態においては、バナジン酸ビスマス(BiVO4)の微細粒子を内包させる。
【0164】
バナジン酸ビスマスは、有害視されているカドミウムイエローやクロムイエローの代替材料として黄色の顔料として用いられているが、光触媒として水の分解(水素製造)や有機物の分解などの機能を有する。また、光触媒として二酸化炭素(CO2)をエタノール(C2H5OH)に変換する機能を有する。しかし、その触媒活性は低く、高効率化が望まれる。
【0165】
一般的に光触媒を用いた上記二酸化炭素の固定化反応は、炭素が1個であるメタン、メタノール、ホルムアルデヒドまたは蟻酸などが生成されることが多い。これに対し、上記バナジン酸ビスマスの場合は、二酸化炭素(CO2)を炭素が2個のエタノール(C2H5OH)に変換することが可能である。このエタノールの生成メカニズムは、二酸化炭素の還元によって生じた炭素ラジカル(C・)中間体の二量化反応が触媒表面で優先的に進行するためでる。このような優先的な反応が生じるのは、バナジン酸ビスマスの結晶表面の特殊性にあると考えられている。
【0166】
この特性を維持したまま、活性のみを向上させるには、結晶構造を維持したまま、還元準位を上昇させる必要がある。これを実現するためには、バナジン酸ビスマスをサブナノ粒子化し、量子サイズ効果によりバンドギャップエネルギーを増大させることが有効である。
【0167】
上記バナジン酸ビスマスは、硝酸ビスマス(Bi(NO3)3)などのビスマス塩とアンモニウムオルトバナジン酸(NH4VO3)などのバナジン酸塩(バナジウム酸塩)を水溶液中で反応させ合成することができる。すなわち、バナジン酸ビスマスを合成するためにはビスマスカチオンとバナジン酸アニオンを水中で反応させる。
【0168】
(実施例5)
本実施例においては、多孔質シリカを鋳型としてバナジン酸ビスマスの微細粒子を合成する。
【0169】
<5−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)を形成した。
【0170】
<5−2>微細粒子の合成
次いで、上記多孔質シリカ(C6VSMPS)を鋳型としてバナジン酸ビスマスの微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。また、0.6MのBi(NO3)3・5H2Oと0.6MのNH4VO3をそれぞれ2Mの硝酸に溶解させ、0.2Mの溶液を得た。この二つの溶液を混合し、それぞれのイオンが0.1Mの濃度で溶解した混合溶液を得た。この溶液を上記多孔質シリカの粉末に添加し、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回(1回〜4回程度)繰り返し行った後、乾燥工程後の試料、450℃で2時間程度、空気中で焼成し、バナジン酸ビスマスの微細粒子を内包した多孔質シリカを得た。
【0171】
得られたバナジン酸ビスマス内包多孔質シリカについて、透過型電子顕微鏡(TEM)による観察を行った。具体的には、FE−TEM(TECNAI F20:FEI)を用いて、試料の形状、粒子サイズを測定した。観察試料は、粉砕した試料をマイクログリッド膜付銅メッシュに分散させることで作製した。
【0172】
図10は、得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)である。多孔質シリカの孔内にバナジン酸ビスマスを内包している構造が確認できた。
【0173】
また、得られたバナジン酸ビスマス内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて電子スペクトル(可視・紫外吸収スペクトル)を求め、Taucプロットにより、バンドギャップエネルギーを求めた。
【0174】
図11に、得られたバナジン酸ビスマス内包多孔質シリカ(実線)およびバルクのバナジン酸ビスマス(破線)のTaucプロットを示す。横軸は、光子エネルギーE(Energy、hν[eV])であり、縦軸は、吸光度(α)と光子エネルギー(hν[eV])の積の平方根((αhν)1/2)である。図11の実線のグラフからバンドギャップエネルギー(Eg)を算出したところ、得られたバナジン酸ビスマス内包多孔質シリカのEgは、3.0eVであり、バルクのバナジン酸ビスマス(破線)のEgは、2.2eVとなった。このように、2,2eVから3.0eVまでバンドギャップエネルギーが増大し、顕著な量子サイズ効果を有することが確認できた。
【0175】
よって、バナジン酸ビスマス内包多孔質シリカを光触媒として利用することで、触媒活性を向上することができ、高効率な光触媒作用を実現することができる。特に、二酸化炭素(CO2)をエタノール(C2H5OH)に変換する光触媒作用の触媒活性を向上することができ、高効率な変換を実現することができる。
【0176】
以上詳細に説明したように、本実施の形態のバナジン酸ビスマス内包多孔質シリカは、地球温暖化の要因である二酸化炭素(CO2)を低減するとともに、エネルギー源であるエタノール(C2H5OH)を生成することができ、地球温暖化対策および再生可能エネルギーとして広く有用な技術に応用可能である。
【0177】
一方で、微細粒子の形成方法としては、デンドリマーを鋳型に用いた合成方法が考えられる。しかしながら、この方法は、含窒素有機化合物からなるデンドリマーの窒素部へ金属カチオンを配位させ、熱処理や還元処理を行うことで金属酸化物や金属の微細粒子を合成するものである。よって、バナジン酸ビスマスのようなカチオンとアニオンを反応させて生成するような物質を合成するためには、両方のイオンをデンドリマー内に配位させる必要があり、このためには特殊な構造を有するデンドリマーを合成する必要があり、その合成は困難を極めるものである。
【0178】
これに対し、本実施の形態においては、ビスマスカチオンとバナジン酸アニオンを容易に孔中で溶液反応させることができ、簡易な製造工程で、効率的に、バナジン酸ビスマス内包多孔質シリカを得ることができる。
【0179】
(実施の形態3)
実施の形態1においては、多孔質シリカに金属化合物(WO3)、金属(Au)または炭素(C)の微細粒子を、実施の形態2においては、バナジン酸ビスマス(BiVO4)を内包させたが、本実施の形態においては、酸化銅(CuO)の微細粒子を内包させる。
【0180】
CuOは約1.3 eVにバンドギャップを有する半導体である。他の半導体と比較してバンドギャップエネルギーが小さいが、チタニア(TiO2)等に比べて高い伝導帯準位を有する。この性質を利用してTiO2のホールとCuOの電子を利用するTiO2-CuO複合光触媒系等が開発されている。バルクの状態でのバンドギャップエネルギーが小さいため、量子サイズ効果を発現させ、伝導帯準位を上昇させても可視光応答性を維持できる可能性がある。これらの性質を用いれば、伝導帯準位が非常に高くかつ可視光応答性を有する高機能性光触媒材料を設計できる可能性がある。
【0181】
(実施例6)
本実施例においては、多孔質シリカを鋳型として酸化銅(CuO)の微細粒子を合成する。
【0182】
<6−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよび比較のためにC16TACを用いたポーラスシリカ(C6SMPSおよびC16MPS)を形成した。
【0183】
<6−2>微細粒子の合成
次いで、上記多孔質シリカを鋳型として酸化銅(CuO)の微細粒子を合成した。上記2種類の多孔質シリカのそれぞれ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、真空ポンプで減圧し、水等の吸着物質を除去した(減圧乾燥工程)。その後、減圧を一時停止し、各多孔質シリカに0.2M(mol)の酢酸銅(Cu(AcO)2)水溶液を添加した。この時にフラスコ内は陰圧を維持している。水溶液と粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における酢酸銅水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、その孔内にCuOの微細粒子を内包した多孔質シリカを得た。
【0184】
図12は、C16MPSを鋳型にして得られたCuO粒子の高角度散乱暗視野走査型透過電子顕微鏡(HAADF−STEM)像である。白色に見える点が内包された粒子を示しており、2nm以上のCuO粒子が均一に分散している様子が分かる。図13は、C6SMPSを鋳型にして得られたCuO粒子のHAADF−STEM像である。1nm以下のCuO粒子がシリカ中に高分散状態で存在していることが分かる。
【0185】
図14は、C16MPSを鋳型にして得られたCuO粒子の紫外・可視吸収スペクトルから求めたTaucプロットである。なお、ここでは試料の温度変化(35℃〜600℃)に伴う、各Taucプロットの変化も示してある(図15についても同じ)。例えば、35℃のグラフから、バルクでは1.3 eV程度の位置に存在するバンドギャップが、約2.3eVまで増大していることが確認できる。一方で、図15に示すようにC6SMPSを鋳型にして得られたCuO粒子の場合には、バンドギャップエネルギーはさらに増加し、約3.2eVまで増加した(35℃のグラフ参照)。これは1nm以下で量子サイズ効果が顕著になることに起因する。また、800nm付近に存在するブロードなピークはCu2+のd−d遷移に起因する。この吸収は通常のCuOでは間接遷移の吸収に埋もれて観測できないが、量子サイズ効果による吸収端の顕著なブルーシフトより初めて観測されるようになった。その結果、バルクのCuOの色が黒であるのに対し、得られた粒子はC16MPSを鋳型にして得られたCuO粒子の場合、緑色であり、C6SMPSを鋳型にして得られたCuO粒子の場合、青色であった。
【0186】
得られた粒子は顕著なサーモクロミズム(温度変化による色変化)を示した。図14および15には、前述したとおり、得られた粒子の35℃〜600℃での紫外・可視吸収スペクトルから求めたTaucプロットも示してある。各図から分かるように吸収端は温度の上昇とともに長波長側にシフトした。
【0187】
図16に、それぞれの試料のバンドギャップエネルギーの温度依存性を示した。縦軸がバンドギャップエネルギー(Eg;eV)で、横軸が温度(Temperature;K)である。C16MPSを鋳型にして得られたCuO粒子の場合(CuO@C16MPS)、バンドギャップの温度依存性は約0.8meV/Kであり、バルクで報告されている値とほぼ同程度であった。一方、C6SMPSを鋳型にして得られたCuO粒子の場合(CuO@C6SMPS)には、約2.3meV/Kと大きく、サブナノ粒子化により著しく温度依存性が高まっていることが観測された。また、これらのサーモクロミズムは可逆的であり、室温まで冷却するともとのスペクトルに戻ることも確認された。また、同温度領域における熱重量測定の結果から、温度上昇中も降下中も酸化や水酸化などの化学反応や相転移等が起こっていないことも確認された。
【0188】
この吸収スペクトルの変化に伴い、目視では緑ないしは青から黄色への顕著なサーモクロミズムが観測された。このサーモクロミズムのメカニズムは格子振動が電子軌道に影響を及ぼす、エレクトロン-フォノンカップリングによるものであると考えられる。代表的なサーモクロミズムを示す無機物である酸化亜鉛では、温度上昇に伴う白色から黄色への色変化が観測される。この要因もエレクトロン-フォノンカップリングことにあると考えられている。バルクのCuOでもこの挙動は観測されるが、バルク体は黒色であるため、目視で色の変化は観測されない。本実施の形態のCuOサブナノ粒子において顕著なサーモクロミズムが観測された要因は間接遷移の吸収端が量子サイズ効果によって紫外領域までシフトしたことと、温度変化に対する吸収端のレッドシフト幅が非常に大きくなったことに起因する。これらは通常のナノ粒子では観測されない現象であり、本実施の形態においてサブナノ粒子が高温でも焼結すること無くシリカ中に単分散で存在できる状態にあることにより初めて実現可能になったものである。
【0189】
このようにサブナノ粒子化することでバンドギャップエネルギーの温度依存性が著しく向上することが明らかになった。このような材料はシリカの熱安定性と相まって、温度インジケータとしての応用用途が期待できる。さらに、半導体発光材料をサブナノ粒子化した場合には温度によって発光波長を大きく制御可能になり、新規な機能性を有する材料を合成することが可能である。
【0190】
(実施の形態4)
本実施の形態においては、浸漬法と焼成による金属酸化物の微細粒子(量子ドット)の合成について説明する。
【0191】
(実施例7)
本実施例においては、多孔質シリカを鋳型として酸化銅(CuO)の微細粒子(量子ドット)を合成する。
【0192】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。濃度の低下に伴い粒径が減少する。そのため、金属塩の水溶液(前駆体液、前駆体溶液)の濃度を低下させることで、細孔径より小さい粒子を合成することができる。このように、実施の形態1の実施例A等で説明した多孔質シリカの細孔径の調整に加え、さらに、金属塩の水溶液(前駆体液)の濃度を低下させることで、多孔質シリカ内包粒子(ここでは、酸化銅の微細粒子)の粒径を微調整することができる。
【0193】
本実施例では、金属塩の水溶液(前駆体液)として、0.6M(mol/L)の硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径3.0nm〜0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0194】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面を洗浄する。洗浄にはエタノールなどのアルコール類を用いるのが好ましい。アルコール類に代えて、水を用いることも可能である。但し、水を用いた場合には溶質(金属塩)の溶出が起こる場合があるため、アルコール類を用いるのが好ましい。
【0195】
洗浄後、多孔質シリカを乾燥させ、細孔内に金属塩の微粒子を析出させる。乾燥方法としては真空乾燥や凍結乾燥を用いることが好ましい。自然乾燥や温風乾燥を用いることも可能であるが、真空乾燥や凍結乾燥の方が、迅速な乾燥が可能であり、より均一な粒子を得ることができる。
【0196】
本実施例においては、真空乾燥を用いて、多孔質シリカを乾燥し、細孔内に金属塩の微粒子を析出させた。
【0197】
この後、細孔内に金属塩の微粒子が析出した多孔質シリカを、空気中で焼成することで、金属酸化物の微細粒子(ここでは、酸化銅の微細粒子)を合成することができる。
【0198】
本実施例においては、空気中において450℃で1時間の焼成を行い、酸化銅の微細粒子を得た。図17に細孔径の異なる多孔質シリカを鋳型に用いて合成した酸化銅の微細粒子のUV−Visスペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。各グラフの添え字は、細孔径(nm)を示す。図17に示すように、各グラフとも、250nm〜450nmの間に、酸化銅のスペクトルピークを確認できる。さらに、細孔径(nm)が右側から3.0nm、2.8nm、1.8nm、1.3nm、0.8nm、および0.7nmの順に、スペクトルピークが左にシフトすることが判明した。また、細孔径(nm)が同じ0.7nmであっても、硝酸銅水溶液の濃度を0.2Mと低くしたグラフ(0.7(0.2M))については、硝酸銅水溶液の濃度を0.6Mとしたグラフ(0.7)より、スペクトルピークが左にシフトしている。このように、細孔径に応じた酸化銅の微細粒子が合成されていることが判明し、また、硝酸銅水溶液の濃度を低くすることで、微細粒子の粒径が小さくなることが判明した。
【0199】
また、バルクの酸化銅のバンドギャップエネルギーは約1.3eV程度であり、目視では黒色である。これに対し、上記酸化銅の微細粒子(金属酸化物量子ドット)においては、量子サイズ効果によってバンドギャップエネルギーが顕著に増大し、目視では緑色から青色となることが確認できた。表3に、上記酸化銅の微細粒子(金属酸化物量子ドット)を内包する多孔質シリカの細孔径(Dpore;nm)と酸化銅の微細粒子のバンドギャップエネルギー(Eg;eV)の関係を示す。バンドギャップエネルギーはTaucプロットにより求めた。
【0200】
【表3】
このように、本実施例においては、多孔質シリカを粉砕(微細化)することなく、モノリス状(例えば、粒子の直径が1mm以上の塊状態)のまま前駆体液に浸漬し、焼成することで、容易に金属酸化物の微細粒子(量子ドット)を合成することができることが判明した。また、内包させる粒子材料の粒径の制御が可能であり、これによりバンドギャップエネルギーを制御することができることが判明した。これは、発光スペクトルや触媒活性の精密な制御が可能になることを示唆しており、例えば、伝導帯準位が非常に高くかつ可視光応答性を有する高機能性光触媒材料などに有用である。
【0201】
なお、酸化銅のみならず、クロムやコバルトの酸化物についても、硝酸クロムや硝酸コバルトを用い、同様にして、金属酸化物の微細粒子(量子ドット)を合成することが可能である(実施例12参照)。
【0202】
(実施の形態5)
本実施の形態においては、浸漬法と焼成による金属の微細粒子(量子ドット)の合成について説明する。
【0203】
(実施例8)
本実施例においては、多孔質シリカを鋳型として銀(Ag)の微細粒子(量子ドット)を合成する。
【0204】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、硝酸銀の水溶液(前駆体液)に浸漬した。硝酸銀の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。ここでも、金属塩の水溶液(前駆体液、前駆体溶液)の濃度を低下させることで、細孔径より小さい粒子を合成することができる。
【0205】
本実施例では、0.6Mの硝酸銀水溶液を用い、モノリス状の多孔質シリカ(細孔径3.0nm〜0.7nm)を浸漬させた。浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを真空乾燥させ、細孔内に硝酸銀の微粒子を析出させた。
【0206】
この後、多孔質シリカを空気中において600℃で3時間の焼成を行い、銀の微細粒子を得た。紫外線照射により蛍光を示すことより、内包物が銀であることを確認した。上記焼成温度においては、酸化銀よりも金属銀の方が安定的に存在するため、銀の微細粒子(銀量子ドット)を合成することが可能となる。得られた銀の微細粒子(銀量子ドット)は、365nmの紫外線照射下で黄色の発光を示した。
【0207】
このように、前駆体液を構成する金属(金属化合物)によっては、浸漬後に焼成を行っても金属酸化物とならず、金属(金属の微細粒子)として合成できる場合もある。また、得られた金属が大気中の酸素などにより僅かに酸化された場合であっても、再焼成を行うことで金属の微細粒子に戻すことが可能である。
【0208】
また、酸素と反応させることにより、酸化銀の微細粒子を合成することも可能である。酸化物になると金属微粒子特有の蛍光がなくなるため、酸化を確認できる。
【0209】
このように、上記銀の微細粒子(銀量子ドット)は、紫外線照射下での発光が見られ、発光素子やバイオイメージング等への応用が可能である。
【0210】
(実施の形態6)
本実施の形態においては、浸漬法と焼成により得られた金属酸化物の微細粒子(量子ドット)を還元することにより金属の微細粒子(量子ドット)を合成する方法について説明する。
【0211】
(実施例9)
本実施例においては、実施例7で合成した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。還元剤としては、例えば、過酸化水素水溶液を用いることができる。
【0212】
ここでは、実施例7で合成した、銅、クロムおよびコバルトの金属酸化物の微細粒子を前駆体として、15%の過酸化水素水溶液に浸漬することにより金属の微細粒子(量子ドット)を得た。
【0213】
図18に、酸化銅の微細粒子(量子ドット)のXPS(X線光電子分光)スペクトルを示し、図19に、還元反応により生成した銅の微細粒子(量子ドット)のXPSスペクトルを示す。横軸は、結合エネルギー(Binding Energy、[eV])であり、縦軸は、強度([a.u.]、CPS)である。
【0214】
図18のスペクトルにおいては、933.7eVに銅(Cu)の2p3/2軌道のピークが観測され、また、サテライトピークも確認されることから、微細粒子が酸化銅(CuO)であることが確認できる。これに対し、図19のスペクトルにおいては、932.8eVにピークが観測され、金属(0価)の銅の微細粒子が生成していることが確認できる。
【0215】
図20に、酸化時と還元時における紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。グラフaのスペクトルを示す酸化銅の微細粒子を、過酸化水素により還元し、銅の微細粒子としたところ、グラフbのスペクトルを示した。さらに、グラフbのスペクトルを示す銅の微細粒子を空気中の酸素により再酸化し、酸化銅の微細粒子としたところ、グラフcのスペクトルを示した。さらに、グラフcのスペクトルを示す酸化銅の微細粒子を、過酸化水素により再還元し、銅の微細粒子としたところ、グラフdのスペクトルを示した。このように、酸化還元反応が可逆的に進行することが確認できる。
【0216】
このように、本実施例によれば、簡易な酸化還元処理により、多孔質シリカに内包される金属の酸化または金属酸化物の還元が行えることが判明した。また、これらが、可逆的に進行することから、目的物(例えば、金属または金属酸化物)の再生が容易に行えることが判明した。
【0217】
(実施の形態7)
本実施の形態においては、浸漬法と還元剤による還元による金属の微細粒子(量子ドット)の合成について説明する。
【0218】
(実施例10)
本実施例においては、多孔質シリカを鋳型として銅(Cu)の微細粒子(量子ドット)を合成する。
【0219】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。本実施例では、金属塩の水溶液(前駆体液)として、0.6Mの硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0220】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを乾燥させ、細孔内に金属塩の微粒子を析出させた。乾燥方法は真空乾燥や凍結乾燥を用いることが好ましい。本実施例においては、真空乾燥を用いて、多孔質シリカを乾燥し、細孔内に金属塩(ここでは、硝酸銅)の微粒子を析出させた。
【0221】
この後、細孔内に金属塩の微粒子が析出した多孔質シリカを還元剤を用いて直接還元することにより、金属銅の微細粒子を合成した。本実施例では、15%の過酸化水素水溶液に、金属塩の微粒子が析出した多孔質シリカを浸漬することにより、金属銅の微細粒子を合成した。
【0222】
図21に、合成した金属銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。酸化銅(CuO)に特有の750nm近傍のスペクトルピークが確認されず、銅の微細粒子の合成を確認することができる。
【0223】
このように、本実施例によれば、浸漬法と還元剤による還元による金属の微細粒子(量子ドット)の合成が可能であることが判明した。
【0224】
また、得られた銅の微細粒子は、空気中の酸素によって緩やかに酸化され、酸化銅の微細粒子となる。このため、上記銅の微細粒子を空気中などの酸素雰囲気へ暴露するだけで、酸化銅の微細粒子を合成することが可能である。このような工程によれば、熱処理(焼成)を行うことなく、室温程度の温度雰囲気において、酸化銅の微細粒子の合成が可能である。
【0225】
本実施の形態の直接還元は、銅の微細粒子の合成のみならず、後述の実施例13等を参酌することにより、クロム(Cr)、コバルトなどの金属微細粒子(量子ドット)の合成にも適用可能である。
【0226】
また、銅の微細粒子としてより安定的に多孔質シリカの内部に存在させるためには、多孔質シリカの細孔内に過酸化水素などの還元剤を残存させて置くことが有用である。例えば、過酸化水素水に浸漬したのち、乾燥させずにおく、または、乾燥時間を短くするなどの乾燥条件を調整することにより、還元剤を残存させて置くことができる。細孔内に過酸化水素などの還元剤が残っている状態では、酸化反応が抑制されるため銅の微細粒子を安定的に存在させることができる。
【0227】
(実施の形態8)
本実施の形態においては、マイクロ波加熱によるの微細粒子(量子ドット)の合成について説明する。
【0228】
(実施例11)
本実施例においては、多孔質シリカを鋳型としてマイクロ波加熱を利用して酸化銅(CuO)の微細粒子(量子ドット)を合成する。なお、ここで言うマイクロ波(Microwave)とは、電波の周波数による分類の一つであり、電波の中で比較的短い波長域であることを意味する。一般的には波長1mから100μm、周波数300MHzから3THzの電波(電磁波)を指す。代表的なマイクロ波の照射手段としては、いわゆる電子レンジ(周波数2.45GHz)が挙げられる。
【0229】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上とすることが好ましい。本実施例では、金属塩の水溶液(前駆体液)として、0.6Mおよび3.0Mの硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0230】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、電子レンジを用いて700Wで15分の条件でマイクロ波照射を行った。マイクロ波照射により多孔質シリカに内包された前駆体液が加熱され、酸化銅の微細粒子が得られた。比較のため、洗浄後の前駆体液を内包した多孔質シリカを電気炉を用いて450℃で1時間の焼成を行った試料も作成した。
【0231】
合成した各酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを測定した。図22に、合成した酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。図22に示すように、金属塩の水溶液(前駆体液)として、0.6Mまたは3.0Mの硝酸銅水溶液を用い、マイクロ波加熱を行った酸化銅の微細粒子(マイクロ波加熱(3.0M)、マイクロ波加熱(0.6M))のスペクトルについては、250nm〜450nmの間のスペクトルがほぼ一致している。これに対し、3.0Mの硝酸銅水溶液を用い、電気炉加熱を行った酸化銅の微細粒子(電気炉450℃(3.0M))のスペクトルは、250nm〜450nmの間にスペクトルのピークを有するものの、ピークの位置が長波長側にずれ、“吸収端のレッドシフト”が観察された。
【0232】
これは、高濃度の前駆体液を用いた電気炉加熱においては、多孔質シリカの各細孔内における微細粒子の合一化(凝集)、焼結が起こり、多孔質シリカの細孔径に対応した球状粒子を得ることができないことに起因するものと考えられる。一方、マイクロ波加熱においては、急速加熱が可能であり、高濃度の前駆体液を用いた場合でも多孔質シリカの細孔径に対応した均一かつ高分散な球状粒子を得ることが可能である。
【0233】
このように、マイクロ波加熱のような簡易な加熱手段を用いても、多孔質シリカに内包された金属酸化物の微細粒子の合成が可能であることが判明した。また、加熱手法によって用いる金属塩の水溶液(前駆体液)の濃度を調整することが好ましいことが判明した。
【0234】
(まとめ)
ここで、上記実施の形態4〜実施の形態8(実施例7〜実施例11)で説明した合成ルートをまとめて説明する。図23は、実施の形態4〜実施の形態8(実施例7〜実施例11)の合成ルートの概念図である。
【0235】
図23に示すように、多孔質シリカの細孔内に前駆体液(金属塩)を注入し、加熱することで、細孔内に金属酸化物の微細粒子を生成することができる(ルート1;加熱ルート)。また、多孔質シリカの細孔内に前駆体液を注入し、これを乾燥し、還元することで、細孔内に金属の微細粒子を生成することができる(ルート2;直接還元ルート)。また、細孔内の金属酸化物の微細粒子を還元することにより、細孔内に金属の微細粒子を生成することができる(ルート3)。また、細孔内の金属の微細粒子を酸化することにより、細孔内に金属酸化物の微細粒子を生成することができる(ルート3)。このように、ルート3の酸化体/還元体は相互変換可能である。また、ルート2によれば、室温での処理が可能である。
【0236】
また、多孔質シリカの細孔内への前駆体液の注入は、浸漬法でも、細孔容積に対応した量の溶液を浸透させる方法でも構わない。また、加熱(焼成)は、電気炉加熱やマイクロ波加熱が有効である。特に、高濃度の前駆体液を用い、均一な微細粒子を得るためにはマイクロ波加熱が有効である。還元反応に要する還元剤の種類は問わないが、金属酸化物を金属化することが可能であること、微細粒子の溶出を促進させないことなどが条件となる。上記実施例では過酸化水素が良好な結果を示した。酸化工程には、酸素などの酸化剤を用いることができる。また、ルート2(直接還元ルート)においては、各工程が室温下で行えるため、エネルギーコストの低減が可能である。
【0237】
(実施の形態9)
上記実施の形態4〜実施の形態8(実施例7〜実施例11)においては、銅や銀の微細粒子や銅の酸化物の微細粒子の合成について説明したが、本実施の形態においては、その他の金属(特に、遷移金属)の酸化物の微細粒子の合成について説明する。
【0238】
(実施例12)
本実施例においては、多孔質シリカを鋳型として、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の金属酸化物の微細粒子(量子ドット)を合成する。
【0239】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。本実施例では、金属塩の水溶液(前駆体液)として、それぞれ、クロム、マンガン、鉄、コバルトおよびニッケルの硝酸塩水溶液を用いた。硝酸塩水溶液の濃度は、それぞれ0.5Mとした。
【0240】
各硝酸塩水溶液に、モノリス状の多孔質シリカ(細孔径0.8nm〜3.0nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0241】
浸漬の完了後、多孔質シリカを硝酸塩水溶液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、電子レンジを用いて700Wで15分の条件でマイクロ波照射を行った。マイクロ波照射により多孔質シリカに内包された硝酸塩水溶液が加熱され、対応する金属酸化物の微細粒子、即ち、クロム、マンガン、鉄、コバルトおよびニッケルの酸化物の微細粒子が得られた。
【0242】
各金属酸化物の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを測定した(図24〜図28)。図24は、合成した酸化クロムの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図25は、合成した酸化マンガンの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図26は、合成した酸化鉄の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図27は、合成した酸化コバルトの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図28は、合成した酸化ニッケルの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。それぞれ、グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。また、各グラフの添え字は、細孔径(nm)を示す。
【0243】
図24〜図28に示すように、いずれの金属酸化物においても細孔径の低下に伴う量子サイズ効果が観測された。特に、サブナノ領域(細孔径1nm未満)において、顕著な量子サイズ効果が観測された。
【0244】
表4に、得られた金属酸化物の目視における色を示す。なお、表4においては、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の酸化物の他、実施の形態4(実施例7)で説明した銅(Cu)の酸化物の色についても表示してある。
【0245】
表4に示すように、いずれのいずれの金属酸化物においても鋳型となるシリカの細孔径の低下に伴い、色の変化が観測された。
【0246】
【表4】
(実施の形態10)
上記実施の形態4〜実施の形態8(実施例7〜実施例11)においては、銅や銀の微細粒子や銅の酸化物の微細粒子の合成について説明したが、本実施の形態においては、その他の金属(特に、遷移金属)の微細粒子の合成について説明する。具体的には、実施の形態9(実施例12)で説明した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。
【0247】
(実施例13)
本実施例においては、実施例12で合成した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。還元剤としては、例えば、過酸化水素水溶液を用いることができる。
【0248】
ここでは、実施例12で合成した、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の酸化物の微細粒子を前駆体として、15%の過酸化水素水溶液に浸漬することにより対応する金属の微細粒子、即ち、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の微細粒子(量子ドット)を得た。
【0249】
図29に、クロムの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。図30に、コバルトの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。図29および図30のいずれも、鋳型として、細孔径が0.8nmの多孔質シリカを用いた。図29および図30に示すように、酸化物と金属との間で大きなスペクトルの変化が確認できた。
【0250】
金属の種類によって再酸化されやすいものとそうでないものが存在する。コバルトは再酸化されにくく、クロムは再酸化されやすい傾向がある。いずれの場合も多孔質シリカの細孔内に還元剤を残存させることで、金属の状態を安定的に維持することが可能である。
【0251】
(実施の形態11)
本実施の形態においては、多孔質シリカを鋳型として炭素の微細粒子(量子ドット)の合成について説明する。実施の形態1の実施例4においては、多孔質シリカを乳鉢で粉砕し粉末状とした後、ブドウ糖の水溶液を添加したが、以下の実施例14においては、モノリス状の多孔質シリカをクエン酸水溶液に浸漬する。
【0252】
(実施例14)
本実施例においては、多孔質シリカを鋳型として炭素の微細粒子(量子ドット)を合成する。
【0253】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(細孔径1.1nm、2.8nm)をそのまま、0.6Mのクエン酸水溶液に浸漬した。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0254】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを乾燥させ、焼成することで、炭素の微細粒子を合成した。本実施例においては、電気炉を用い、空気中において300℃で1時間の焼成を行い、多孔質シリカの細孔中のクエン酸を炭化させ、炭素の微細粒子を得た。
【0255】
図31に、得られた炭素の微細粒子の発光スペクトル(Ex.340nm)を示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、強度(Intensity、[a.u.])である。各グラフの添え字は、細孔径(nm)を示す。図32に、得られた炭素の微細粒子の発光スペクトルを示す。横軸は、励起波長(Em、[nm])であり、縦軸は、発光波長(Ex、[nm])である。図32の上部の発光スペクトルは、細孔径1.1nmのものであり、下部の発光スペクトルは、細孔径2.8nmのものである。この、図32中の破線で囲んだ領域は、強発光部を示し、その中心(励起波長)をラインC1、C2で示してある。
【0256】
図31に示すように、細孔径を変化(2.8nm→1.1nm)させることで、強度のピークが変化することが判明した。また、図32に示すように、細孔径を変化(2.8nm→1.1nm)させることで、強発光部を示し、その中心がC1からC2へ変化することが判明した。
【0257】
このように、細孔径を変化させることで、発光波長が変化することが判明した。即ち、細孔径を変化させることで、発光波長をコントロールすることが可能である。言い換えれば、多孔質シリカの細孔径(平均孔直径)を制御することにより、微細粒子の発光波長を調整することができる。細孔径を低下させることで、より小さな炭素の微細粒子を合成することが可能であり、発光波長は短波長側にシフトする。目視においては、細孔径1.1nmの多孔質シリカを鋳型とした炭素の微細粒子は、青色の発光が観測され、細孔径2.8nmの多孔質シリカを鋳型とした炭素の微細粒子は、黄緑の発光が観測された。
【0258】
以上、本発明者によってなされた発明をその実施の形態に基づき具体的に説明したが、本発明は前記実施の形態に限定されるものではなく、その要旨を逸脱しない範囲で種々変更可能である。
【産業上の利用可能性】
【0259】
本発明は、多孔質シリカ内包粒子の製造方法および多孔質シリカ、多孔質シリカ内包粒子に関し、特に、微細な孔を有する多孔質シリカを利用し、その孔の内部に微細な粒子を内包させる技術に適用して有効である。
【技術分野】
【0001】
本発明は、多孔質シリカ内包粒子の製造方法および多孔質シリカ、多孔質シリカ内包粒子に関し、特に、微細な孔を有する多孔質シリカを利用し、その孔の内部に微細な粒子を内包させる技術に関する。
【背景技術】
【0002】
金属、セラミックス、炭素などの材料はその粒子径を減少させることでバルクの状態では見られない特異な性質を示す(下記非特許文献1参照)。また、多くの場合それらの特異な性質はシングルナノメートルのサイズで発現し始め、サブナノメートルのサイズ領域において最大になることが、一部の実験と理論予測から報告されている(下記非特許文献2参照)。
【0003】
例えば、金属サブナノ粒子の場合は、粒子サイズの減少に伴い金属的な性質を失い、離散的なバンド構造を構築することが知られている(下記非特許文献3参照)。そのため、バンド構造由来の光吸収が起こり、半導体や色素分子のような性質を示すことから次世代の色素増感太陽電池などへの応用が検討されている(下記特許文献4参照)。さらに、バルクでは見られない発光や磁化などの特異な現象も発現することから広い分野への応用が期待されている(下記非特許文献5参照)。
【0004】
また、金属酸化物や硫化物などに代表される半導体セラミックス材料の場合、サブナノ粒子にすることで、量子サイズ効果によりバンドギャップエネルギーの大幅な増大が見られる(下記特許文献1、2参照)。可変なバンドギャップエネルギーは光触媒などの分野において有用であり、触媒活性の飛躍的な向上、反応選択制の向上、さらにはバルクでは起こらない反応を起こさせることも期待されている。
【0005】
また、ナノ炭素材料は近年多くの注目を集める材料である。炭素材料もサブナノ粒子化することで特異な性質を示す。特に、紫外光照射下での白色発光やアップコンバージョン発光などの性質はバルクの炭素では起こらない現象であり、近年でも研究の対象となっている(下記非特許文献6参照)。
【0006】
また、下記非特許文献7〜12には、後述するように、金属などの微細粒子の合成についての開示がある。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Y. Wang, N. Herron, J. Phys. Chem., 1991, 95, 525.
【非特許文献2】L. Brus, J. Phys. Chem., 1986, 90, 2555.
【非特許文献3】J. N. Solanki, Z.V.P. Murthy, Colloids and Surfaces A: Physicochem. Eng. Aspects, 2010, 359, 31.
【非特許文献4】A. Kogo, N. Sakai, T. Tatsuma, Electrochemistry Communications, 2010, 12, 996.
【非特許文献5】X. Liu, M. Bauer, H. Bertagnolli, E. Roduner, J. V. Slageren, F. Phillip, Phys. Rev. Lett., 2006, 97, 253401.
【非特許文献6】J. Zong, Y. Zhu, X. Yang, J. Shen, C. Li, Chem. Commun., 2011, 47, 764.
【非特許文献7】S.L.Hu,K.Y.Niu,J.Sun,J.Yang,N.Q.ZhaoandX.W.Du, J. Mater. Chem., 2009, 19, 484.
【非特許文献8】Y. Negishi, K. Nobusada, T. Tsukuda, J. AM. CHEM. SOC., 2005, 127, 5261.
【非特許文献9】N. Satoh, T. Nakashima, K. Kamikura, K. Yamamoto, Nature Nanotech., 2008, 3, 106.
【非特許文献10】G. A. Ozin, S. Ozkar, R. A. Prokopowicz, Acc. Chem. Res., 1992, 25, 553.
【非特許文献11】D. Tanaka, Y. Oaki, H. Imai, Chem. Commun., 2010, 46, 5286.
【非特許文献12】J. S. Beck, J. C. Vartuli, G. J. Kennedy, C.T.Kresge, W.J.Roth, S. E. Schramm, Chem. Mater., 1994, 6, 1816.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明者は、多孔質シリカ、多孔質シリカ内包粒子および孔中に粒子を内包した多孔質シリカについての研究・開発に従事し、各種の検討を重ねている。この多孔質シリカ内包粒子は、多孔質シリカを利用して孔の内部に内包させた微細粒子を意味する。多孔質シリカの孔中に内包された状態のもの、また、多孔質シリカの孔中に内包された状態から粒子だけを取り出した状態のものを意味する。
【0009】
このように、多孔質シリカを利用して孔の内部に微細粒子を形成することで、微細粒子自身若しくは微細粒子を内包する多孔質シリカ全体として種々の機能を持たせることができ、その機能の向上を図るため、本発明者は、各種の検討を重ねている。
【0010】
微細粒子、例えば、サブナノ粒子の合成は、古くはレーザーアブレーション(上記非特許文献7参照)などの方法で行われていたが、生産性やサイズの制御の点で問題がある。
【0011】
また、金属粒子に限定すれば、金属表面とチオール基などの特定の官能基の相互作用を利用した合成例が存在する(上記非特許文献8参照)。しかし、この方法も生産性とサイズ制御性にやや問題があり、また、合成対象が金属粒子に限定されることが問題であり幅広い物質群に適応することはできない。
【0012】
さらに、骨格に金属イオンの配位子となる官能基を有する樹枝状高分子(デンドリマー)を用いた合成例が存在する(上記非特許文献9参照)。このデンドリマーの性質を制御することで粒子サイズのそろったナノ〜サブナノメートル粒子を合成可能だが、サブナノメートル領域での制御性の悪さや、デンドリマーの合成の煩雑さ等で問題がある。
【0013】
また、ゼオライトに代表される多孔質材料を粒子合成の鋳型にする手法は、合成の簡便さと基質の一般性において長所がある。ゼオライトを鋳型としたサブナノ粒子の合成はいくつかの報告例が存在する(下記非特許文献10参照)。しかし、ゼオライトの細孔は一般的に制御性が悪く、また、スーパーケージと呼ばれる内部細孔において大きな粒子が成長してしまうことが問題点としてあげられる。
【0014】
これに対し、メソポーラスシリカは細孔径の制御性が高いことから、ナノ粒子の合成には適当な鋳型材料である(下記非特許文献11参照)。また、生成した粒子が優秀な吸着材であるメソポーラスシリカ内に均一に分散することも利点の一つである。しかし、従来のメソポーラスシリカにおいては細孔径を1.5nm以下に制御することが困難であるため、サブナノ粒子の合成には用いることができないという問題がある(下記非特許文献12参照)。
【0015】
以上のように、微細粒子の合成方法には各種方法が提案されているが、一長一短があり、微細な粒子を制御性良く合成することは困難であった。
【0016】
そこで、本発明の目的は、多孔質シリカ内包粒子の特性を向上させることができる技術を提供することにある。特に、粒径分布特性が良好で、特性の良い多孔質シリカ内包粒子を提供することにある。
【0017】
また、本発明の他の目的は、良好な多孔質シリカ内包粒子の製造方法を提供することにある。また、内包させる粒子の材料について、幅広い材料を内包可能な多孔質シリカ内包粒子の製造方法を提供することにある。
【0018】
本発明の上記目的およびその他の目的と新規な特徴は、本願明細書の記載および添付図面から明らかになるであろう。
【課題を解決するための手段】
【0019】
本願において開示される発明のうち、代表的なものの概要を簡単に説明すれば、次のとおりである。
【0020】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に上記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0021】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に上記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0022】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと、ビスマスまたはビスマスを元素組成として有する化合物を含有する第1液と、バナジウムまたはバナジウムを元素組成として有する化合物を含有する第2液との混合液とを接触させ、前記多孔質シリカの孔内に上記混合液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内においてバナジン酸ビスマスを含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0023】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する気体とを接触させ、前記多孔質シリカの孔内に上記気体を導入し、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下である。
【0024】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)アルコキシシランの加水分解による多孔質シリカの製造工程であって、(a1)界面活性剤およびアルコキシシランを混合し、混合液を形成する工程と、(a2)前記混合液に水を添加することにより、前記アルコキシシランの加水分解反応を行わせる工程と、(a3)前記(a2)工程の後、熱処理を施すことにより前記界面活性剤を除去する工程と、を有する多孔質シリカの製造工程と、(b)前記(a)工程により製造された多孔質シリカの孔内において粒子を合成する工程であって、前記多孔質シリカと粒子材料または前記粒子材料を元素組成として有する液または気体とを接触させ、前記多孔質シリカの孔内に上記液または気体を導入させ、前記多孔質シリカの前記孔内において前記粒子材料または前記粒子材料化合物を含有する粒子を形成する工程と、を有する。
【0025】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカは、平均孔直径が0.5以上1.5nm以下である孔を有し、金属、金属化合物および炭素のうちのいずれかを含有する粒子を、前記孔中に内包している。
【0026】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、を有する。
【0027】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属酸化物を含有する微細粒子を形成する工程と、(d)前記(c)工程の後、前記金属酸化物を含有する微細粒子を還元することにより、前記金属の微細粒子を形成する工程と、を有する。
【0028】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と(c)前記(b)工程の後、前記多孔質シリカの孔内の前記液を還元することにより、前記金属の微細粒子を形成する工程と、を有する。
【0029】
本願において開示される発明のうち、代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法は、(a)多孔質シリカを準備する工程と、(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、を有し、前記多孔質シリカの平均孔直径を制御することにより、前記微細粒子の発光波長を調整する。
【発明の効果】
【0030】
本願において開示される発明のうち、以下に示す代表的な実施の形態に示される多孔質シリカ内包粒子によれば、多孔質シリカ内包粒子の特性を向上させることができる。
【0031】
また、本願において開示される発明のうち、以下に示す代表的な実施の形態に示される多孔質シリカ内包粒子の製造方法によれば、良好な多孔質シリカ内包粒子を製造することができる。
【図面の簡単な説明】
【0032】
【図1】実施例1で得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)および内包させた酸化タングステンの粒子径の分布(グラフ)を示す図である。
【図2】実施例1で得られた酸化タングステン内包多孔質シリカのTaucプロットを示す図である。
【図3】実施例1で得られた酸化タングステン内包多孔質シリカの粒子径とバンドギャップエネルギーとの関係を示す図である。
【図4】実施例1で得られた酸化タングステン内包多孔質シリカのESRスペクトルを示す図である。
【図5】実施例2で得られた酸化タングステン内包多孔質シリカのTaucプロットを示す図である。
【図6】図6(a)は、実施例3で得られた金内包多孔質シリカを示す図(写真)であり、図6(b)は、紫外線照射下の金内包多孔質シリカを示す図(写真)である。
【図7】実施例3で得られた金内包多孔質シリカの発光現象時の発光スペクトルを示す図である。
【図8】図8(a)は、実施例4で得られた炭素内包多孔質シリカを示す図(写真)であり、図8(b)は、紫外線照射下の炭素内包多孔質シリカを示す図(写真)である。
【図9】実施例4で得られた炭素内包多孔質シリカの発光現象時の発光スペクトルを示す図である。
【図10】実施例5で得られたバナジン酸ビスマス内包多孔質シリカの透過型電子顕微鏡像(TEM像)を示す図である。
【図11】実施例5で得られたバナジン酸ビスマス内包多孔質シリカ(実線)およびバルクのバナジン酸ビスマス(破線)のTaucプロットを示す図である。
【図12】実施例6においてC16MPSを鋳型にして得られたCuO粒子の高角度散乱暗視野走査型透過電子顕微鏡像(HAADF-STEM像)である。
【図13】実施例6においてC6SMPSを鋳型にして得られたCuO粒子のHAADF-STEM像である。
【図14】実施例6においてC16MPSを鋳型にして得られたCuO粒子の35℃〜600℃でのTaucプロットを示す図である。
【図15】実施例6においてC6SMPSを鋳型にして得られたCuO粒子の35℃〜600℃でのTaucプロットを示す図である。
【図16】実施例6においてC6SMPSを鋳型にして得られたCuO粒子およびC16MPSを鋳型にして得られたCuO粒子のバンドギャップの温度依存性を示す図である。
【図17】細孔径の異なる多孔質シリカを鋳型に用いて合成した酸化銅の微細粒子のUV−Visスペクトルである。
【図18】酸化銅の微細粒子(量子ドット)のXPS(X線光電子分光)スペクトルである。
【図19】還元反応により生成した銅の微細粒子(量子ドット)のXPSスペクトルである。
【図20】酸化時と還元時における紫外・可視吸収(UV−Vis)スペクトルである。
【図21】合成した金属銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図22】合成した酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図23】実施の形態4〜実施の形態8(実施例7〜実施例11)の合成ルートの概念図である。
【図24】合成した酸化クロムの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図25】合成した酸化マンガンの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図26】合成した酸化鉄の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図27】合成した酸化コバルトの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図28】合成した酸化ニッケルの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図29】クロムの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図30】コバルトの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。
【図31】得られた炭素の微細粒子の発光スペクトル(Ex.340nm)である。
【図32】得られた炭素の微細粒子の発光スペクトルである。
【発明を実施するための形態】
【0033】
以下、本発明の実施の形態を図面を参照しながら詳細に説明する。なお、同一の機能を有するものには同一もしくは関連の符号を付し、その繰り返しの説明を省略する。
【0034】
(実施の形態1)
<1>多孔質シリカの合成
界面活性剤を鋳型としたゾルゲル法により、多孔質シリカを合成する。このような合成法を分子鋳型法(テンプレート法)ということがある。
【0035】
一般的には、溶液中に界面活性剤を溶解させると、界面活性剤の種類と濃度に応じて例えば筒状のミセル粒子が形成される。ここで溶液中にシリカ源となるテトラエトキシシランなどを加えると、ミセル粒子の隙間でシリケートイオンの吸着および成長反応が進行し、シリカゲル骨格が形成される。このシリケートイオンの吸着および成長反応により、ゾル状態からゲル状態へ変化するためゾルゲル反応と呼ばれる。この後、焼成(熱処理)すると、鋳型とした界面活性剤が分解・除去されて多孔質シリカが得られる。言い換えれば、複数の孔(細孔、微細孔)を有するシリカ骨格が得られる。
【0036】
本実施の形態においては、無溶媒系で上記ゾルゲル反応を進行させることにより制御性良く、多孔質シリカを合成する。ここで言う“無溶媒系”とは、アルコキシシランと界面活性剤との混合の際の溶媒を言い、これらを混合しやすくするため、予めアルコキシシランを溶解し、また、界面活性剤を溶解するための溶媒、または、アルコキシシランと界面活性剤との混合液に添加する多量の溶媒(50等量以上の溶媒)をいう。このような溶媒を用いず、アルコキシシランと界面活性剤とを直接混合し、反応剤として必要な量の水(H2O)を添加し多孔質シリカを合成することで、制御性良く、多孔質シリカを形成することができる。
【0037】
以下に、本実施の形態の多孔質シリカの合成方法について詳細に説明する。
【0038】
アルコキシシランとカチオン性界面活性剤とをそのまま混合し、攪拌する。この混合液中に、水を添加し攪拌することで前駆体溶液を形成する。この前駆体溶液は、攪拌とともにゲル化する。
【0039】
添加する水(H2O)は、アルコキシシランの加水分解のための反応剤として寄与する。また、添加する水のpHは、アルコキシシランの等電点であるpH2程度に調整することが望ましい。等電点においては、アルコキシシランの加水分解、およびシリケートイオンのゲル化速度が最も遅いため、界面活性剤のミセル形成のための時間を十分に確保できるからである。
【0040】
また、水のpH0〜1程度においては加水分解の加速が起こるが、シリケートイオンのゲル化速度が十分に遅いため同様の効果が得られる。そのため、添加する水のpHは、0〜2の範囲に調整することが好ましい。pH3以上では加水分解およびゲル化速度が速すぎるため、界面活性剤の溶解およびミセル形成のための時間が十分に確保できない恐れがある。
【0041】
pH調整用の酸としては、塩酸、硫酸、硝酸等の無機酸、または酢酸などの有機酸を使用することができる。
【0042】
多孔質シリカの成型性の向上のためには、できるだけ少ない溶媒で加水分解することが好ましい。そのため、アルコキシシランに対する水の添加量は、反応に最低限必要な2等量(eq)以上20等量以下の範囲、より好ましくは、2等量以上10等量以下の範囲とする。このように、少溶媒の系とすることにより、反応系をほぼ純粋なシリケートイオンと界面活性剤との混合物に維持することができ、鋳型となる界面活性剤のミセルの安定性を確保しつつ、ゾルゲル反応を促進することができる。例えば、後述する炭素数が8未満の界面活性剤を用いても多孔質シリカを合成することができる。よって、多孔質シリカの平均細孔径(平均孔径、平均孔直径、D)を小さくすることができる。別の捉え方をすれば、界面活性剤の炭素数を調整することで、容易に多孔質シリカの孔径を調整することが可能となる。
【0043】
カチオン性界面活性剤としては、一般式R1R2R3R4N+X−で示される界面活性剤を用いることができる。R1は、例えば、炭素数1〜24のアルキル基、ベンジル基、フェニル基であり、R2R3R4は、例えば、メチル基、エチル基、プロピル基、ブチル基である。また、Xは、例えば、F、Cl、Br、Iなどのハロゲンイオンである。カチオン性界面活性剤としては、4級のカチオン性界面活性剤を用いることが好ましい。また、R1のアルキル基は直鎖型でも分岐型でもよい。
【0044】
以下、実施例に基づき本実施の形態をさらに詳細に説明する。なお、本発明は、これらの実施例によって限定されるものではない。
【0045】
(実施例A)…多孔質シリカ(ポーラスシリカ)の合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いてカチオン性界面活性剤を、0.2〜1.2eq(0.038mol×0.2〜0.038mol×1.2)を分散させ、撹拌した。この時点で、TEOSと界面活性剤とは混じり合わない。即ち、均一な溶液とならない。カチオン性界面活性剤としては、オクタデシルトリメチルアンモニウムクロライド(C18TAC)ヘキサデシルトリメチルアンモニウムクロライド(C16TAC)、テトラデシルトリメチルアンモニウムブロミド(C14TAB)、ドデシルトリメチルアンモニウムブロミド(C12TAB)、デシルトリメチルアンモニウムブロミド(C10TAB)、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)の8種類を用いて、それぞれポーラスシリカを合成した。
【0046】
次いで、上記混合液に、塩酸を用いてpHを0〜2程度に調整した水を2〜4eq(0.038mol×2〜0.038mol×4)程度、添加し、室温で撹拌した。一時間程度の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。この溶液(前駆体溶液)を室温または60℃に保持し、継続して撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤を除去した。これにより、無色透明のモノリス状のポーラスシリカが得られた。
【0047】
このように、反応系において、高濃度なシリケートイオン溶液を前躯体溶液として用いることで、界面活性剤のミセル形成を溶媒分子などにより阻害させることなく、シリカの合成を促進させることができる。特に、従来困難であった炭素数が小さい(例えば、7以下)の界面活性剤を用いてもミセル形成が可能となり微細な細孔を有するポーラスシリカを形成することが可能となる。
【0048】
得られたポーラスシリカの細孔を解析した。比表面積(SSA)、細孔容積(TPV)、平均細孔径(D)を測定した。比表面積(SSA)は、BET法により測定した。平均細孔径は、BJH法、HK法、GCMC法を用いて測定した。平均細孔径については、BJH法よりHK法において、より微細な細孔径の算出(解析)が可能である。また、HK法よりGCMC法において、より微細な細孔径の算出(解析)が可能である。
【0049】
C18TACを用いたポーラスシリカ(C18)の、BET比表面積は1361m2/g、細孔容積は0.96cm3/gであった。平均細孔径は、BJH法では、3.00nm、HK法では、3.36nm、GCMC法では、3.27nmであった。
【0050】
C16TACを用いたポーラスシリカ(C16)の、BET比表面積は1452m2/g、細孔容積は0.79cm3/gであった。平均細孔径は、BJH法では、2.70nm、HK法では、2.86nm、GCMC法では、2.82nmであった。
【0051】
C14TABを用いたポーラスシリカ(C14)の、BET比表面積は1234m2/g、細孔容積は0.60cm3/gであった。平均細孔径は、HK法では2.40nm、GCMC法では、2.26nmであった。
【0052】
C12TABを用いたポーラスシリカ(C12)の、BET比表面積は1056m2/g、細孔容積は0.53cm3/gであった。平均細孔径は、HK法では、2.00nm、GCMC法では、1.82nmであった。
【0053】
C10TABを用いたポーラスシリカ(C10)の、BET比表面積は916m2/g、細孔容積は0.45cm3/gであった。平均細孔径は、HK法では、1.60nm、GCMC法では、1.58nmであった。
【0054】
C8TABを用いたポーラスシリカ(C8)の、BET比表面積は810m2/g、細孔容積は0.41cm3/gであった。平均細孔径は、GCMC法では、1.28nmであった。
【0055】
C6TABを用いたポーラスシリカ(C6)の、BET比表面積は632m2/g、細孔容積は0.32cm3/gであった。平均細孔径は、GCMC法では、1.12nmであった。
【0056】
C4TACを用いたポーラスシリカ(C4)の、BET比表面積は586m2/g、細孔容積は0.29cm3/gであった。平均細孔径は、GCMC法では、0.92nmであった。
【0057】
このように、鎖長に対応した細孔を有するポーラスシリカが得られた。即ち、炭素数が18から4まで低下するにしたがって、平均細孔径(D)が小さくなることが判明した。特に、炭素数が12以下の界面活性剤を用いたポーラスシリカの平均細孔径は、2nm以下となり、ミクロ孔が確認された。また、従来法では、合成が困難であった、炭素数が7以下の界面活性剤を用いたポーラスシリカの合成が可能となり、炭素数が6のC6TABを用いたポーラスシリカの平均細孔径は、GCMC法で、1.12nmであり、また、炭素数が4のC4TABを用いたポーラスシリカの平均細孔径は、GCMC法で、0.92nmであった。このように、炭素数が8未満の界面活性剤を用い、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカの形成が可能であることが判明した。また、細孔容積も大きく、0.25cm3/g以上のポーラスシリカの形成が可能であることが判明した。
【0058】
なお、上記メソポーラスシリカの合成においては、2〜4eqの水を用いたが、8eqの水を用いても良好に加水分解が進むことが確認された。このように、成型性の向上のためには、溶媒が存在しない(無溶媒である)、言い換えれば、溶媒としての水を含有しないことが要求される。溶媒としての水とは、例えば、アルコキシシランやカチオン性界面活性剤などの溶解や分散に必要な、これらの材料の数十倍等量(例えば、50倍等量以上)の水(溶媒)をいう。これに対し、本発明でいう無溶媒とは、アルコキシシランに対する水の添加量で言えば、反応に最低限必要な2等量(eq)から、その10倍程度、即ち、2等量以上20等量以下の範囲である。また、より好ましくは、2等量以上10等量以下の範囲である。この条件を用いることで、系を高濃度なシリケートイオンと界面活性剤との混合物にすることができ、成型性と界面活性剤ミセルの安定性を確保することができる。
【0059】
(実施例B)…有機シラン添加による細孔径制御
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)とトリエトキシビニルシラン(TEVS)を8g×5%(0.038mol×5%)とを混合し、続いて界面活性剤を0.2〜1.2等量を添加し、撹拌した。この混合物に、塩酸を用いてpH0〜2に調整した水を2〜4等量の範囲で添加し、室温で撹拌した。1時間程度の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。さらに、この溶液(前駆体溶液)を室温または60℃に保持し、継続して撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤を除去した。界面活性剤としては、カチオン性界面活性剤である、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)の3種類を用いて、それぞれについてポーラスシリカを形成した。
【0060】
得られたポーラスシリカの細孔を解析した。比表面積(SSA)、細孔容積(TPV)、平均細孔径(D)および細孔壁厚さ(Dwall)を測定した。比表面積(SSA)は、BET法により測定した。平均細孔径は、GCMC法を用いて測定した。細孔壁厚さ、即ち、筒を構成する壁の厚さは、X線回折結果などにより算出することができる。
【0061】
C8TABを用いたポーラスシリカ(C8V)の、BET比表面積は519m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.99nmであった。細孔壁厚さは、2.37nmであった。
【0062】
C6TABを用いたポーラスシリカ(C6V)の、BET比表面積は582m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.82nmであった。細孔壁厚さは、2.00nmであった。
【0063】
C4TACを用いたポーラスシリカ(C4V)の、BET比表面積は355m2/g、細孔容積は0.16cm3/gであった。平均細孔径は、0.77nmであった。細孔壁厚さは、1.98nmであった。
【0064】
これに対し、上記実施例Aにおいては、C8TABを用いた場合、平均細孔径は、1.28nmであるため、有機シラン化合物の添加により、平均細孔径の、1.28nmから0.99nmへの縮小効果が確認された。細孔径の差は、0.29nmである。
【0065】
同様に、上記実施例Aとの比較において、C6TABを用いた場合、有機シラン化合物の添加により、平均細孔径の、1.12nmから0.82nmへの縮小効果が確認された。細孔径の差は、0.30nmである。
【0066】
また、C4TACを用いた場合、有機シラン化合物の添加により、平均細孔径の、0.92nmから0.77nmへの縮小効果が確認された。細孔径の差は、0.15nmである。
【0067】
このように、有機シラン化合物の添加により、ポーラスシリカの細孔径の微調整が可能であることが分かった。特に、炭素数が8の界面活性剤を用いても、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカの形成が可能であることが判明した。また、上記縮小効果から推測すれば、炭素数10や12の界面活性剤を用いても、有機シラン化合物の添加により、平均細孔径が0.7nm以上1.5nm以下のスーパーミクロ孔を有するポーラスシリカが形成される。また、さらに炭素数の少ない界面活性剤の使用、界面活性剤や有機シラン添加量の増減、反応温度の低下によっても平均細孔径を0.5nm程度まで低下させることが可能である。
【0068】
さらに、吸着質(例えば、分子径など)に応じて必要とされる平均細孔径を算出し、当該平均細孔径に合うよう界面活性剤の炭素数を選択する、または、有機シラン化合物を添加してポーラスシリカの形成を行うことで、細孔径の微細な調整(例えば、細孔径の約0.1nm刻みの調整)を行うことができる。例えば、サブナノメートルのオーダー、言い換えれば、m×10−10の単位で細孔径の微細な制御を行うことができる。
【0069】
(実施例C)…ポーラスシリカナノ粒子の合成
ポリプロピレン製容器にシリカ源としてTEOS8g(0.038mol;1eq)を入れ、界面活性剤を0.2〜1.2等量を添加した後、さらに、平均分子量1000のポリエチレングリコール(PEG)を7.5g添加し、撹拌した。この混合物に、塩酸を用いてpH0〜2に調整した水を2〜4等量の範囲で添加し、室温で撹拌した。1時間の撹拌でTEOSの加水分解が進行し、界面活性剤およびポリエチレングリコールが溶解したほぼ均一な溶液が得られた。この溶液(前躯体溶液)を室温あるいは60℃に保持し、撹拌あるいは静置した。12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。
【0070】
カチオン性界面活性剤としては、オクタデシルトリメチルアンモニウムクロライド(C18TAC)、ヘキサデシルトリメチルアンモニウムクロライド(C16TAC)、テトラデシルトリメチルアンモニウムブロミド(C14TAB)、ドデシルトリメチルアンモニウムブロミド(C12TAB)、デシルトリメチルアンモニウムブロミド(C10TAB)、オクチルトリメチルアンモニウムブロミド(C8TAB)、ヘキシルトリメチルアンモニウムブロミド(C6TAB)、ブチルトリメチルアンモニウムクロライド(C4TAC)のいずれかを用いることができる。
【0071】
ここで、上記工程のように、単にゲル化、焼成を行っただけでは、炭素数16以上の界面活性剤を用いた場合は、ナノ粒子の集合体よりなるモノリス状のポーラスシリカが得られるのに対し、炭素数16未満や臭化物塩の界面活性剤においては、細孔径分布が広い、アモルファス状のポーラスシリカが得られるにすぎなかった。
【0072】
そこで、上記前躯体溶液のうち、C6TABを用いて形成した前躯体溶液を、塩基性水溶液に滴下した。塩基性水溶液としては、28%のアンモニア水溶液を用いた。pHは約13である。滴下された略粒状の前躯体溶液は、ゲル状となり、アンモニア水溶液中に沈殿した。得られたゲルを、60℃で乾燥、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。得られたポーラスシリカは、白色のビーズ状で得られた。ビーズ状は、上記前躯体溶液の滴下形状に対応する。
【0073】
塩基性水溶液としては、上記アンモニア水溶液の他、アミン類の水溶液などを用いることができる。これらの塩基は、乾燥、焼成過程での除去が容易で、塩基性水溶液として用いて好適である。また、シリカは、pH14以上の高pH領域で、溶解が始まるため、高pH領域の塩基性水溶液を用いる場合には、反応後(ゲル化後、重合後)速やかに溶液外に取り出すことが好ましい。また、反応系中に、アルカリ金属やアルカリ土類金属のイオンが共存するとシリカの溶解速度が高まるため、水酸化ナトリウムなどの水溶液よりも上記アンモニアやアミン類を用いた塩基性水溶液を用いることがより好ましい。
【0074】
C16TACを用いて合成した多孔質シリカの細孔径を測定したところ、当該ポーラスシリカにおいては、2つの細孔径が確認された。即ち、約2nm程度の界面活性剤に由来するメソ孔と、約20〜50nm程度の粒子間隙に対応するメソ孔との2つの細孔を有するポーラスシリカの構成が確認できた。
【0075】
C6TABを用いて合成したポーラスシリカについても、約1nm程度の界面活性剤に由来するミクロ孔と、約5〜10nm程度の粒子間隙に対応するメソ孔との2つの細孔が確認できた。
【0076】
以上詳細に説明したように、単にゲル化、焼成を行っただけでは、炭素数16以上の界面活性剤を用いた場合は、ナノ粒子の集合体よりなるモノリス状のポーラスシリカが得られるのが、炭素数16未満の界面活性剤においては、アモルファス状のポーラスシリカが得られるにすぎない。これに対し、塩基性水溶液に前躯体溶液を接触させた場合には、炭素数16未満の界面活性剤を用いた場合でも、ナノ粒子化が可能となる。
【0077】
上記現象は次のように考察できる。pH0〜2の前駆体溶液中では、シリケートイオンは中性、あるいは正に帯電している。よって、シリケートイオンは、ポリエチレングリコールとは水素結合で相互作用し、界面活性剤とはカウンターアニオンを介して静電相互作用している。炭素鎖の短い、即ち、炭素数の少ない界面活性剤の場合、ミセル形成能が低いため、シリカの重合に伴う界面活性剤の集合、およびそれに伴うポリエチレングリコールの系外への相分離を十分に行うことができない。そのため、アモルファス状のシリカのみが得られる。一方、塩基性水溶液への滴下により系のpHを急激に上昇させた場合には、シリケートイオンは負に帯電し、カチオン性界面活性剤との間には、カウンターアニオンを介さない、より強固な静電相互作用が生じる。また、ポリエチレングリコールとの間の水素結合は解消され、逆に静電反発により相分離を誘導する。この二つの現象は界面活性剤のミセル形成とポリエチレングリコールの相分離を誘導し、界面活性剤の鎖長に対応した細孔を有したポーラスシリカのナノ粒子化を可能とすると考えられる。
【0078】
<2>微細粒子の合成方法
次いで、上記<1>で合成された多孔質シリカを使用し、その孔(細孔、微細孔)に金属などの材料を内包させることにより微細粒子(粒子、ナノ粒子、サブナノ粒子)を合成する。
【0079】
多孔質シリカを金属化合物水溶液と接触させ、多孔質シリカの孔内に金属化合物水溶液を浸透させる。次いで、多孔質シリカを乾燥した後、焼成し、多孔質シリカの孔内に金属を内包させる。
【0080】
焼成により金属または金属化合物が析出(残存)する金属化合物水溶液であれば、用いる金属化合物水溶液に制限はないが、例えば、当該水溶液として、過酸化タングステン酸水溶液などの、粒子材料を元素組成として有する化合物をを用いることができる。
【0081】
上記過酸化タングステン水溶液を用いた場合には、多孔質シリカの孔内に酸化タングステン(WO3)を内包させることができる。また、酢酸銅や硝酸銅などの銅塩を用いた場合には、多孔質シリカの孔内に酸化銅を内包させることができる。また、塩化鉄や硝酸鉄などの鉄塩を用いた場合には、多孔質シリカの孔内に酸化鉄を内包させることができる。また、硝酸マンガンなどのマンガン塩を用いた場合には、多孔質シリカの孔内に酸化マンガンを内包させることができる。また、硫酸チタニルなどの塩を用いた場合には、多孔質シリカの孔内に酸化チタンを内包させることができる。
【0082】
用いることの出来る金属塩は任意の溶媒に溶解し、溶液にすることが可能な化合物であれば種類は問わない。酸化物を得るためには焼成により容易に酸化することが望ましく、そのためには硝酸塩や酢酸塩などの熱分解しやすい塩を用いることが望ましい。用いる溶媒は水であっても有機溶媒であっても良く、金属塩の溶解度を制御するために硝酸や塩酸、アンモニアなどによりpHをコントロールした溶媒であっても良い。
【0083】
多孔質シリカは吸着機能を有するため、液体や気体と接触させるだけで容易に多孔質シリカの孔中に液体や気体を導入させることができる。また、微細な孔を有する多孔質シリカを用いれば、内包させる粒子材料(金属や金属化合物等)を容易に微細化することができる。
【0084】
このように、本実施の形態の微細粒子の合成方法によれば、簡易な処理により、多孔質シリカ内包粒子を形成することができる。また、上記<1>においては、0.5以上1.5nm以下の微細な孔径を有する多孔質シリカも合成可能であるため、このような多孔質シリカを鋳型に用いることで、内包させる粒子の粒径(粒子の直径)を容易に小さくすることができる。例えば、粒径(粒子の直径)を1.5nm以下と、より好ましくは、1nm以下(サブナノオーダー)とすることができる。もちろん、上記<1>の合成方法によれば、炭素数の大きな界面活性剤を用いることで、シングルナノメートルのオーダー(10nm以下、より好ましくは、5nm以下)の粒子も容易に調整することができる。このように、内包させる粒子を微細化することで、バルクの状態では見られない特異な性質を有する多孔質シリカ内包粒子(機能性複合体)を形成することができる。
【0085】
また、上記<1>においては、孔径の分布を小さくでき、言い換えれば、孔径のばらつきを小さくでき、狭い粒径分布を有する微細粒子(ここでは、サブナノ粒子)の合成が可能となる。別の言い方をすれば、バンドギャップなどの物理・化学的特性のばらつきの少ない量子ドットの合成が可能となる。
【0086】
また、孔内への粒子材料(粒子を構成する元素、または、その元素を元素組成として有する化合物)の導入も、上記液体を用いれば容易に行うことができる。このような液体プロセス(液相プロセス)では、導入する液体(前駆体)も、塩、錯体等、種類を選ばない。例えば、金属または金属化合物を孔内で合成するには、金属イオンを有する塩、金属錯体等を用いることができる。
【0087】
また、粒子材料を気体で導入しても良い。例えば、粒子材料を合成し得る原料ガスと接触させることにより、気相成長(気相での化学反応)により孔内において微細粒子を形成してもよい。また、粒子材料を気化(蒸発)させた気体を用いてもよい。
【0088】
特に、多孔質シリカ自身の特性として吸着性を有するため、粒子材料を容易に孔内に取り込むことができ、均一性の高い微細粒子を形成することができる。
【0089】
なお、多孔質シリカ(固体)と固体の原材料とを乳鉢等で混合し、乳棒等により圧力を加えることにより孔内に固体の粒子材料を導入することも可能である。
【0090】
また、多孔質シリカ中の微細粒子の形成割合は、細孔容積と液体プロセスに用いられる液体(前駆体)の濃度、浸漬回数で制御が可能である。また、気体プロセスにおいては、原料ガスの流量などで制御可能である。
【0091】
また、多孔質シリカ自身は、高い熱的・化学的安定性を有する。例えば、耐熱性や耐薬剤性を有するため、微細粒子を内包させた状態で、酸化雰囲気での熱処理(焼成)により微細粒子を酸化させ、また、薬液処理により所望の処理(例えば、炭化)を行うことが可能である。また、酸化状態の微細粒子を還元雰囲気(例えば、水素雰囲気)で熱処理することにより還元してもよい。また、光照射による還元を行ってもよい。また、硫化水素雰囲気下で熱処理し、硫化物を得ることができる。
【0092】
さらに、内包した粒子と、化学反応を用いて発生させたガス状の化合物をシリカ細孔内で反応させ、目的の化合物を得ることもできる。この方法はテルル化カドミウムのサブナノ粒子を得る場合に特に有効である。その場合には、塩化カドミウムの粒子を細孔内で生成した後、テルル化水素ガスなどと反応させることにより、テルル化カドミウムを得ることができる。テルル化カドミウムは太陽電池や蛍光体に応用した場合に、高い変換効率や発光効率を示すことから注目される物質であるが、その毒性から使用が懸念されている。本発明の多孔質シリカ内での合成法を用いれば、テルル化カドミウム自体がシリカ細孔内に内包された形で得られるため、毒性によるリスクを低減できると共に、サブナノ粒子の環境中への排出リスクを低減することが可能である。
【0093】
このように、内包させる微細粒子の材料としては、金属および金属化合物の他、炭素や炭素化合物(例えば、SiCなど)など多種多様の材料に対応することができる。また、金属化合物(半導体、セラミックスなどを含む)としては、金属酸化物、金属窒化物、金属硫化物などが例示でき、金属酸化物としては、酸化タングステンや酸化チタンなど、金属窒化物としては、窒化タングステン(WN)など、金属硫化物としては、硫化カドミウム(CdS)などを例示することができる。
【0094】
また、上記微細粒子の機能性材料としての利用は、多孔質シリカ内包させた状態で(微細粒子とシリカの複合体として)行ってもよい。特に、多孔質シリカは、前述したように、熱的・化学的安定性を有し、また、光透過性にも優れるため、光触媒や光学素子等への応用の場合には、有効な担持体としてそのまま用いることが可能である。
【0095】
また、多孔質シリカの鋳型を分解することにより、微細粒子のみを取り出し、この粒子を他の材料(担体)に固定(担持)させ、機能性材料として使用してもよい。多孔質シリカから微細粒子を取りだすには、例えば、水酸化ナトリウム水溶液やフッ酸等によりシリカ骨格を溶解させることで微細粒子を取り出すことができる。
【0096】
また、内包させる粒子を量子ドット化することで、多孔質シリカ内包粒子に各種機能を持たせることができる。
【0097】
例えば、量子サイズ効果によるバンドギャップの調整により、特定波長の光の吸収や発光が生じ、発光材料として用いることができる。発光波長はバンドギャップと相関するため、粒子サイズの制御により発光波長を制御可能になる。また、内包させる化合物を適宜選択(例えば、炭素や硫化カドミウム、テルル化カドミウム等)することにより、アップコンバージョン蛍光特性を持たせることも可能である。これにより、吸収光と発光光の周波数変換が可能となり、例えば、近赤外光を可視光に、あるいは可視光を紫外光に周波数変換する素子(光学素子)などとして利用することができる。
【0098】
また、光触媒材料として用いることができる。例えば、二酸化チタンに光(紫外線)が当たると、価電子帯の電子が伝導帯へと励起され、電子(e-)とホール(h+)が生成する。このホールは強い酸化力をもち、周囲の分子から電子を奪い、酸化させる。水分子を酸化した場合、OHラジカルを生成する。また、伝導帯に励起された電子は電荷移動により、周囲の分子に電子を与え、還元させる。酸素を還元した場合にはスーパーオキシドラジカルアニオン(O2-・)を生成する。このようなラジカルは高い反応性を持つために空気中や水中の有機物(有害物質)を二酸化炭素まで分解する。これにより有害物質の除去が可能となる。このような酸化還元反応は、光触媒の価電子帯や伝導帯の準位と反応基質の酸化還元準位との差が、それぞれの電子移動の方向に対してエネルギー的に安定化に向かう方向でなければ進行しない。すなわち、光触媒の伝導帯準位は目的の還元反応の準位よりも負である必要があり、価電子帯の準位は目的の酸化反応準位よりも正である必要がある。そのため、光触媒として有効に活用できる化合物はこの制限のために限定される。例えば酸化タングステンの価電子帯は他の半導体と比較して低い準位に存在するため、強い酸化力を有する。しかし、伝導帯は二酸化チタンなどと比較して低く、一般的に還元力が低いと言われている。事実、酸化タングステンの伝導帯準位は酸素の還元準位よりも低く、酸素の還元反応を起こすことができない。そのため、酸化タングステンを水中の有機物の分解反応に用いようとした場合、ホールによる直接酸化やOHラジカルの生成により有機物を分解することが可能だが、伝導帯の電子による反応を起こすことができないため、すぐに電子過剰の状態になる。この電子過剰な状態は励起子の再結合を促進するため、光触媒効率の著しい低下を引き起こす。そのため、酸化タングステンを光触媒として用いるためには伝導帯の電子を効率よく使用する必要がある。また、価電子帯と伝導帯の準位が目的の反応を起こす準位を満足していたとしても、活性化エネルギーの障壁を超えるためには過剰のエネルギーが必要になる。この過剰のエネルギーは目的の反応準位と、価電子帯と伝導帯準位との差分のエネルギーで供給することができるため、準位差が大きければ、目的の反応の効率を向上させることができる。このように、光触媒の反応性や反応効率には価電子帯と伝導帯の準位が大きく関わっている。そのため、価電子帯と伝導帯の準位の制御が可能になれば、同じ物質を用いたまま、任意に反応性や反応効率をコントロールすることが可能になる。粒子サイズの制御で価電子帯と伝導帯の準位をコントロール可能な量子サイズ効果を用いる方法はこの目的において非常に有効な手段である。量子サイズ効果が発現する粒径領域は、一般的に直径がシングルナノメートルの領域であり、サブナノメートルの領域でより顕著になる。従って、大きな量子サイズ効果を発現しつつ制御するためには、サブナノメートル領域での粒径制御が必須である。これまでに提案されてきたナノ粒子合成法はサブナノメートル領域での制御性が悪く、また、適応可能な化合物群も限定されている。本発明の方法では、通常の化合物合成プロセスをそのまま用いることが可能なため、現在までに化学合成可能な化合物群全てに適応可能である。また、シリンダー状の細孔を鋳型とするため、細孔径よりも大きな粒子が複生成することが無く、非常にシャープな粒度分布で生成物を得ることが可能である。さらに、ゼオライトのような三次元的な細孔構造を有さないため、内包粒子同士が合一化して量子サイズ効果の低い大粒子を生成することも無く、安定に使用することができる。このように、量子サイズ効果により、有効な光触媒として利用可能である。
【0099】
また、色素増感型太陽電池の色素材料として、また、量子ドット型太陽電池の量子ドットとして用いることができる。
【0100】
具体的には、増感剤から電子を受け取る半導体粒子や増感剤そのものとして用いることができる。色素増感型太陽電池の構成に制限はないが、例えば、酸化チタンのナノ粒子の集合体を備える透明電極と、他の電極と、これらの電極間に封止された電解液とを有するように構成する。酸化チタンは、紫外線しか吸収しないが、酸化チタンの表面に増感剤が吸着することで、可視光にも感度を有するようになる(色素増感)。太陽電池に光が当たると酸化チタンに吸着した色素が励起状態となり、電子を放出する。この電子は酸化チタンを経由して透明電極に達し外部に流れる。一方、電子を放出して陽イオンになった増感剤は、もう片方の電極から供給される電子を、電解液中の陰イオンを経由して受け取り、還元され、もとに戻る。負極上に存在する半導体粒子のフェルミ準位と対極にある金属のフェルミ準位差が太陽電池の起電力(開放電圧)を決定する。そのため、半導体粒子の伝導帯準位を量子サイズ効果によって制御すれば、太陽電池の起電力をコントロールすることが可能になる。また、金サブナノ粒子や炭素サブナノ粒子は、増感剤として用いることも可能であり、有機色素と比較して劣化の少ない増感剤として機能する。
【0101】
このように、本実施の形態の多孔質シリカ内包粒子は、光触媒材料や色素増感型太陽電池の色素材料として用いることができる。
【0102】
以下に効果をまとめて説明する。上記において詳細に説明したように、本実施の形態においては、サイズ制御性が高く、粒度分布の狭いサブナノ粒子の製造が可能となる。
【0103】
また、内包させる粒子材料を金属や半導体などとすれば、粒径の制御によりバンドギャップエネルギーを制御することができ、発光スペクトルや触媒活性の精密な制御が可能になる。これらは耐久性の高い発光素子やバイオイメージング、触媒として利用できる。
【0104】
また、内包させる粒子材料を光触媒機能を有する半導体などとすれば、量子サイズ効果によるバンドギャップの増大に伴う機能制御が可能になる。後述する実施例では、酸化タングステンのサブナノ粒子が酸素の単電子還元を可能にすることを示した。これはバルクの酸化タングステンでは起こらない現象である。これらは、高性能な触媒、特に水の分解による水素の製造や二酸化炭素固定化反応において有効な光触媒として期待される材料となりうる。さらに、半導体粒子をサブナノ粒子化することで、後述する酸化タングステンではフォトクロミズムを、酸化銅ではサーモクロミズムを示し、これらはセンサなどへの応用が可能である。
【0105】
また、内包させる粒子材料を炭素などとすれば、後述する実施例において説明するように、紫外線照射下での発光が見られる。これらも同様に発光素子やバイオイメージング等への応用が可能である。
【0106】
このように、各種粒子材料の微細化(特に、サブナノ粒子化)の効果は、粒子サイズの制御でその性質を制御することが可能であり、触媒活性、反応選択制、発光スペクトル分布、等をコントロールすることができる。特に、本実施の形態においては、0.5〜1.5nmの孔径を有する多孔質シリカを鋳型とすることで、非常に高いサイズ選択的合成が可能になっており、また、狭い粒度分布を有するサブナノ粒子が製造可能である。
【0107】
さらに、生成した粒子は多孔質シリカ内に分散性よく内包されている。また、多孔質シリカ自体が優秀な吸着材として働き、また、光透過性に優れるため、光触媒や光学素子等への応用の場合には、有効な担持・複合体としてそのまま用いることが可能である。また、多孔質シリカ自体をナノ粒子化すれば、より効率の良い触媒の構築や、ナノ蛍光体としての応用が可能である。
【実施例】
【0108】
以下、実施例に基づき本実施の形態をさらに詳細に説明する。なお、本発明は、これらの実施例によって限定されるものではない。
【0109】
(実施例1)
<1−1>多孔質シリカの合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いて界面活性剤を0.0075〜0.038molの範囲で添加した混合液を用いた。この混合液を攪拌しつつ、TEOSに界面活性剤を分散させた。
【0110】
界面活性剤としては、ヘキシルトリメチルアンモニウムブロミド(C6TAB)およびブチルトリメチルアンモニウムクロライド(C4TAC)のいずれかを用いた。また、界面活性剤に加え、トリメトキシビニルシラン(TEVS)をTEOSに対して5mol%添加した混合液を用いた。
【0111】
上記4種類の混合液に、それぞれ塩酸を用いてpH2に調整した水を2.74g(0.152mol;4eq)入れ、室温に保持し、継続して撹拌した。いずれの混合液を用いた場合も、12時間から数日でゲル化が完了し、溶液全体が目視で無色透明のゲル状となった。このゲルを60℃で乾燥、600℃で3時間焼成し(熱処理を施し)、界面活性剤を除去することにより4種類の多孔質シリカ(Samples)を得た。
【0112】
得られた多孔質シリカについて、窒素吸着機(Tristar3000、マイクロメリティクス社製)を用い、試料の細孔構造と、比表面積、細孔容積および平均細孔径とを調べた。試料は、直前にVacPrep061(マイクロメリティクス社製)にて、160℃で3時間脱気したものを用いて測定した。窒素吸着機(BELSORP−max、日本ベル社製)を用い、試料の孔径分布についてGCMC法にて解析した。
【0113】
表1に、得られた多孔質シリカの孔の解析結果を示す。即ち、各試料(Sample)について、比表面積(SSA、[m2/g])、細孔容積(TPV、[cm3/g])、平均細孔径(Dpore、[nm]、直径)を測定した。なお、比表面積(SSA)は、BET法により測定した。細孔容積(TPV)は、総細孔容積である。平均細孔径(D)は、GCMC法を用いて測定した。
【0114】
【表1】
C6TABを用いたポーラスシリカ(C6SMPS)の、BET比表面積は632m2/g、細孔容積は0.32cm3/gであった。平均細孔径は、1.12nmであった。
【0115】
C4TACを用いたポーラスシリカ(C4SMPS)の、BET比表面積は586m2/g、細孔容積は0.29cm3/gであった。平均細孔径は、0.92nmであった。
【0116】
C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)の、BET比表面積は582m2/g、細孔容積は0.25cm3/gであった。平均細孔径は、0.82nmであった。
【0117】
C4TACおよびTEVSを用いたポーラスシリカ(C4VSMPS)の、BET比表面積は355m2/g、細孔容積は0.16cm3/gであった。平均細孔径は、0.77nmであった。
【0118】
このように、無溶媒系でのゾルゲル反応により、制御性良く、具体的には、孔径の制御性や反応性が良い状態で、多孔質シリカを合成することができた。特に、炭素数が8未満の界面活性剤を用い、孔径が小さい、例えば、孔径が0.5nm以上1.5nm以下の多孔質シリカの合成が可能となった。このような孔径の多孔質シリカをスーパーミクロポーラスシリカ(SMPS)と呼ぶことがある。
【0119】
また、TEVSのような有機シラン化合物の添加により、C6TABを用いたポーラスシリカについては、平均細孔径が、1.12nmから0.82nmへ縮小し、また、C4TACを用いたポーラスシリカについては、平均細孔径が、0.92nmから0.77nmへ縮小した。このように有機シラン化合物の添加により、孔径のさらなる微細化が可能となることが判明した。
【0120】
<1−2>微細粒子の合成
次いで、上記多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成した。上記4種類の多孔質シリカのそれぞれ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、真空ポンプで減圧し、水等の吸着物質を除去した(減圧乾燥工程)。その後、減圧を一時停止し、各多孔質シリカに0.2M(mol)の過酸化タングステン酸水溶液を添加した。この時にフラスコ内は陰圧を維持している。水溶液と粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における過酸化タングステン酸水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、その孔内に酸化タングステンの微細粒子を内包した多孔質シリカを得た。
【0121】
本実施例においては前駆体水溶液の導入に減圧下での浸透を行っているが、これは、内包粒子の重量を一定にしつつ、分析の妨げになる細孔外で生成した粒子を最小限にするためである。触媒等に用いる場合など細孔外に多少の粒子が生成してもかまわない場合には常圧での浸透や、水溶液への浸漬等によって前駆体溶液を浸透させてもかまわない。また、粒子外部に付着した余剰の前駆体水溶液を水洗等により除去すれば、減圧下での浸透と同じようにシリカ細孔内にのみ粒子を生成することが可能である。
【0122】
(ア)得られた酸化タングステン内包多孔質シリカについて、透過型電子顕微鏡(TEM)による観察を行った。具体的には、FE−TEM(TECNAI F20:FEI)を用いて、試料の形状、粒子サイズを測定した。観察試料は、粉砕した試料をコロジオン膜付銅メッシュに分散させることで作製した。
【0123】
図1は、得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)および内包させた酸化タングステンの粒子径の分布(グラフ)を示す図である。グラフの横軸は、粒子径(Diameter、[nm])であり、縦軸は、個数(Counts、[個])である。
【0124】
図1中の(a)は、酸化タングステン内包させたC4VSMPS(C4VW)の画像である。(b)は、酸化タングステン内包させたC4SMPS(C4W)の画像である。(c)は、酸化タングステン内包させたC6SMPS(C6W)の画像である。いずれの画像においても、内包させた酸化タングステンの粒子径が、1nm以下であることが確認された。また、内包箇所に偏りがなく、ほぼ均一に高濃度に内包している、言い換えれば、高分散であることが確認された。
【0125】
また、内包させた酸化タングステンの粒子径については、図1(a)に示すように、酸化タングステン内包させたC4VSMPS(C4VW)については、0.8nm程度の粒子径の割合が高く、平均粒径は、0.77nmであった。また、図1(b)に示すように、酸化タングステン内包させたC4SMPS(C4W)については、0.8nm程度の粒子径の割合が高く、平均粒径は、0.89nmであった。また、図1(c)に示すように、酸化タングステン内包させたC6SMPS(C6W)については、0.9nm程度の粒子径の割合が高く、平均粒径は、1.11nmであった。
【0126】
このように、多孔質シリカの孔内に酸化タングステンなどの金属化合物を内包させることができた。特に、孔径が0.5nm以上1.5nm以下の微細な孔を有する多孔質シリカにも酸化タングステンなどの金属化合物を内包させることができた。また、孔径のより小さい多孔質シリカに内包させた場合、酸化タングステンの粒子径は小さくなり、多孔質シリカの孔径に対応した大きさの微細粒子を形成できることが判明した。また、粒径分布が狭く、例えば、粒径分布が、平均粒径から±0.3nm程度の範囲となる、良好な微細粒子を形成できることが判明した。
【0127】
(イ)得られた酸化タングステン内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて紫外・可視吸収スペクトルを求め、Taucプロットにより、酸化タングステンの微細粒子のバンドギャップエネルギーを求めた。ここでTaucプロットとは、半導体に対して紫外・可視吸収スペクトルの吸収端からバンドギャップを求める方法である。半導体のバンド間の光学的遷移による光吸収においては、吸光度と光子エネルギーの関係において「α=k(E−Eg)2/E(kは定数)」の関係が成り立つ。よって、横軸に光子エネルギーE(Energy、hν[eV])を、縦軸に吸光度(α)と光子エネルギー(hν[eV])の積のn乗((αhν)n)をとり、接線を引く。ここでnの値は直接遷移型の場合n=2となり、間接遷移型の場合n=1/2となる。この接線とベースラインとの交点がバンドギャップエネルギー(Eg)となる。
【0128】
図2に、得られた酸化タングステン内包多孔質シリカのTaucプロットを示す。グラフ(a)は、酸化タングステン内包させたC4VSMPS(C4VW)のプロットである。グラフ(b)は、酸化タングステン内包させたC4SMPS(C4W)のプロットである。グラフ(c)は、酸化タングステン内包させたC6SMPS(C6W)のプロットである。(d)は、バルクの酸化タングステン、即ち、微粒子化していない酸化タングステン、言い換えれば、酸化タングステンの塊のプロットである。
【0129】
各グラフ(a)〜(c)について、バンドギャップエネルギー(Eg)を算出したところ、(a)の酸化タングステン内包させたC4VSMPS(C4VW)のEgは、3.69eV、(b)の酸化タングステン内包させたC4SMPS(C4W)のEgは、3.43、(c)の酸化タングステン内包させたC6SMPS(C6W)のEgは、3.32となった。
【0130】
表2に、得られた酸化タングステン内包多孔質シリカについて、鋳型となった多孔質シリカの孔径(Pore diameter of SMPS、[nm])、内包させた酸化タングステンの粒子径(Particle size)、上記バンドギャップエネルギー(Eg)から算出された粒径(Caluculated particle size、[nm])および上記バンドギャップエネルギー(Eg、[eV])を示す。なお、バンドギャップエネルギー(Eg)から算出された粒径については、いわゆるBrusの式を用いて算出した。Brusの式は、「Eg=Egb+h2π2/2R2・(1/Me+1/Mh)−1.8e2/εR」で示され、Egは、上記バンドギャップエネルギー[eV]、Egbは、バルクのバンドギャップエネルギー[eV]、hは、プランク定数(4.136×10−15eV・s)である。Rは、粒子半径で、MeとMhは、それぞれ電子と正孔の有効質量、εは比誘電率である。
【0131】
【表2】
表2に示すように、酸化タングステン内包させたC6SMPS(C6W)について、孔径が1.12nmの多孔質シリカ中に、直径1.11nmの粒子が確認でき、その粒子のEgは、3.32eVであった。このEgから算出された粒径は0.97nmであった。
【0132】
また、酸化タングステン内包させたC4SMPS(C4W)について、孔径が0.92nmの多孔質シリカ中に、直径0.89nmの粒子が確認でき、その粒子のEgは、3.43eVであった。このEgから算出された粒径は0.87nmであった。
【0133】
また、酸化タングステン内包させたC4VSMPS(C4VW)について、孔径が0.77nmの多孔質シリカ中に、直径0.70nmの粒子が確認でき、その粒子のEgは、3.69eVであった。このEgから算出された粒径は0.70nmであった。
【0134】
このように、孔径と粒径とは、良い相関性を示している。また、粒径が小さくなるにしたがってEgが大きくなっており、量子サイズ効果を確認することができた。また、このEgから算出した粒径も、TEMによる測定結果(粒径)と良好な相関関係を有していることから、微細粒子(2nm以下、より好ましくは、サブナノオーダー(1.0nm以下)の微細粒子)が形成されていることが確認できた。
【0135】
図3に、得られた酸化タングステン内包多孔質シリカの粒子径(Particle size)とバンドギャップエネルギーEgとの関係を示す。点(a)は、酸化タングステン内包させたC4VSMPS(C4VW)、点(b)は、酸化タングステン内包させたC4SMPS(C4W)、点(c)は、酸化タングステン内包させたC6SMPS(C6W)のプロットである。また、ここでは、炭素数が8以上の界面活性剤を用いて形成した孔径の大きい多孔質シリカに酸化タングステンを内包させた試料((d)、(e))についても粒子径とEgとの関係を示してある。(d)の試料の粒子径は1.4nmであり、(e)の試料の粒子径は1.8nmである。また、点(f)は、バルクの酸化タングステン(粒子径が1000nm)のプロットである。
【0136】
この図3からも、粒子径がサブナノ領域になるにしたがってEgが急激に大きくなっており、顕著な量子サイズ効果を確認することができる。
【0137】
(ウ)得られた酸化タングステン内包多孔質シリカについて、酸素の一電子還元能の評価を行った。得られた試料を紫外線照射下に置き、電子スピン共鳴E−500(Bulker社製)を用いて、電子スピン共鳴(ESR:Electron Spin Resonance)を利用したスピントラップ法により評価を行った。スピントラップ剤としては、ジメチルピロリン−N−オキシド(DMPO)を用いた。
【0138】
即ち、酸素の一電子還元反応の生成物であるスーパーオキシドラジカルアニオン(O2-・)をDMPOで補足し、生成するDMPO-OOHラジカルのシグナルをESRで観測した。具体的には次のような実験を行った。
【0139】
酸化タングステン内包させたC4SMPS(C4W)を40mgと、タングステン酸を600℃で3時間焼成して得られたバルクの酸化タングステンを4mg計測し、それぞれを40mMのDMPOのエタノール溶液5ml中に添加し、分散させた。試料の量が異なるのは、それぞれの試料中の酸化タングステンの質量が同量になるように調整したためである。
【0140】
上記分散液をESRのフラットセルに導入し、市販のブラックライトを光源として90分の紫外線照射を行った。この紫外線(UV)照射前後のESRスペクトルから酸素−電子還元能の評価を行った。なお、全てのスペクトルの強度(シグナル強度)は、既知濃度のテトラメチルピロリジノキシフリーラジカルを標準試料として用い校正してある。
【0141】
図4に、ESRスペクトルを示す。横軸は、磁場の強さ(Magnetic Field、[G])を示し、縦軸は、シグナル強度(Intensity)を示す。このシグナル強度は、フリーラジカルの個数、言い換えれば生成したO2-・量に対応している。
【0142】
図4(a)は、酸化タングステン内包させたC4SMPS(C4W)のESRスペクトルであり、破線(点線)は、UV照射前のESRスペクトルを示し、実線は、UV照射後のESRスペクトルを示す。図示するように、酸化タングステン内包させたC4SMPS(C4W)においては、UV照射によりシグナル強度の増大が確認できた。即ち、DMPO−OOHラジカルの生成が観測された。これに対し、図4(b)に示すバルクの酸化タングステンの場合は、UV照射の前後でシグナル強度の変化はほとんどなかった。
【0143】
これにより、酸化タングステン内包させたC4SMPS(C4W)においては、紫外線照射による酸素の一電子還元反応が進行していることが確認できた。これにより、生成した電子とホールの両方を速やかに消費する反応経路が確保される。そのため、電子とホールの再結合が抑制され、反応効率が著しく増大する。たとえば、環境浄化の用途においては、紫外線照射により、非常に酸化作用の強いスーパーオキシドラジカルアニオン(O2-・)を生成し、これによって、悪臭成分等を分解するなど、光触媒作用を有することが確認される。上記悪臭成分の他、水質浄化、シックハウスガス分解、排ガス中有害物質分解、土壌汚染物質の除去、環境ホルモンやダイオキシン分解、抗菌など、の機能も備え得る。
【0144】
(エ)得られた酸化タングステン粒子のフォトクロミック挙動について観測した。酸化タングステンは薄膜状にすることでフォトクロミズムを示すことが知られている。本発明で得られた酸化タングステン粒子も紫外線照射下で可逆的なフォトクロミズムを示すことが観測された。得られた酸化タングステン-シリカ複合体を時計皿に入れ、365nmの紫外光を照射した。試料にエタノールのようなアルコールを滴下すると、より顕著な色の変化が観測された。これらの現象は可逆的であり、目視で観測することができる。エタノールの滴下によるフォトクロミック挙動の向上はその着色メカニズムに起因する。酸化タングステンの場合、光照射により還元されたW5+が生成し、W5+のd−d遷移が青色の着色に起因する。一方でエタノールなどのαプロトンを有するアルコールが共存した場合、その分解過程でホールには一電子余剰に注入される。そのため、通常よりもW5+の生成効率が向上し、より顕著なフォトクロミズムを示すと考えられる。これらの性質を応用すればセンサやディスプレイ材料等への応用が可能である。さらに通常の酸化タングステン薄膜では青から白への脱色速度が遅く、加速するには加熱が必要であるとされている。しかし、本発明で得られた酸化タングステンサブナノ粒子の場合、脱色に要する時間は室温で10分未満である。この高い可逆性はセンサ等に用いた場合に有効である。
【0145】
(実施例2)
本実施例においては、多孔質シリカを粒子化(ナノ粒子化)することで、多孔質シリカ単体が有する第1孔(微細孔)と、複数の多孔質シリカの集合体である各粒子の粒子間の隙間よりなる第2孔を有する多孔質シリカを合成し、これら2種の孔を有する多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成する。このように、ナノ粒子化した多孔質シリカを用いることで、含浸させる溶液の物質拡散効率を向上させることができ、効率的に微細粒子を内包させることができる。また、触媒として用いる際には、分子の拡散効率と触媒界面が増加し、触媒活性などの機能が向上する。発光体として用いる際には媒質中への分散性が向上することで機能が向上する。
【0146】
<2−1>多孔質シリカの合成
ポリプロピレン製容器にシリカ源としてテトラエトキシシラン(TEOS)8g(0.038mol;1eq)を入れ、続いて界面活性剤としてヘキシルトリメチルアンモニウムブロミド(C6TAB)を0.0075〜0.038molの範囲で添加した混合液を用いた。この混合液を攪拌しつつ、TEOSに界面活性剤を分散させた。次いで、混合液中に平均分子量1000のポリエチレングリコール(PEG)を0.0075mol添加し、撹拌した。
【0147】
この後、混合液に、それぞれ塩酸を用いてpH2に調整した水を2.74g(0.152mol;4eq)入れ、室温に保持し、継続して撹拌したところ、1時間の撹拌でTEOSの加水分解が進行し、ほぼ均一な溶液が得られた。この溶液を室温で静置し、数時間から数日間熟成させ、前駆体液とした。
【0148】
次いで、上記前駆体液を、シリンジで28%のアンモニア水溶液に滴下した。滴下した前駆体液は、アンモニア水溶液に入った瞬間に球形を保ったままゲル化した。沈降した白色の球状ゲルを回収し、乾燥させた後、600℃で3時間焼成し、界面活性剤およびポリエチレングリコールを除去した。これにより白色で、直径約2〜3mmの球形の多孔質シリカを得ることができた。
【0149】
得られた多孔質シリカについて、BJH法を用いて平均細孔径を測定したところ、2つの孔径が確認された。第1孔種として1.1nmの平均孔径を有するミクロ孔と、第2孔種として20nmの平均孔径を有するメソ孔とを確認することができた。
【0150】
<2−2>微細粒子の合成
次いで、上記2種の孔を有する多孔質シリカを鋳型として酸化タングステン(WO3)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。その後、各多孔質シリカに0.2Mの過酸化タングステン酸水溶液を添加し、減圧下において、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における過酸化タングステン酸水溶液の添加量は、第1孔(ミクロ孔)の細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、第1孔のみに酸化タングステンの微細粒子を内包した多孔質シリカを得た。第1孔のみに粒子が生成する理由は以下の通りである。細孔径の大きな違いにより、ポアフィリングによる凝縮相の安定性が、第1孔では第2孔と比較して非常に高く、第1孔の容積に相当する体積の溶液を含侵させた場合に、優先的に第1孔への浸透が起こるためである。これにより、第1孔には量子ドットが内包されつつ、第2孔は物質拡散のための空間として確保することが可能になる。
【0151】
得られた酸化タングステン内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて電子スペクトル(可視・紫外吸収スペクトル)を求め、Taucプロットにより、酸化タングステンの微細粒子のバンドギャップエネルギーを求めた。
【0152】
図5に、得られた酸化タングステン内包多孔質シリカのTaucプロットを示す。横軸は、光子エネルギーE(Energy、hν[eV])であり、縦軸は、吸光度(α)と光子エネルギー(hν[eV])の積の平方根((αhν)1/2)である。図示するグラフから、バンドギャップエネルギー(Eg)は、3.3eV程度であることが分かる。前述の表2において、酸化タングステンを内包させたC6SMPS(C6W)の中の粒子のEgは、3.32eVであり、当初の多孔質シリカの孔径が1.12nmであることから、第1孔(ミクロ孔)にのみ酸化タングステンが形成したことが分かる。
【0153】
(実施例3)
本実施例においては、多孔質シリカを鋳型として金(Au)の微細粒子を合成する。
【0154】
<3−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C4TACおよびTEVSを用いたポーラスシリカ(C4VSMPS)を形成した。
【0155】
<3−2>微細粒子の合成
次いで、上記多孔質シリカ(C4VSMPS)を鋳型として金(Au)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。その後、多孔質シリカに0.2Mの塩化金酸水溶液を添加し、減圧下において、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における塩化金酸水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、回収した試料を水洗し、さらに、減圧乾燥を行った。その後、4%程度のH2(水素)を含有するAr雰囲気中で、350℃で、2時間程度焼成し、塩化金酸を還元し、金の微細粒子を内包した多孔質シリカを得た。
【0156】
得られた金内包多孔質シリカに、360nm〜370nmの波長の紫外線を照射したところ、目視において発光現象を確認することができた。図6(A)に、得られた金内包多孔質シリカの写真を、図6(B)に、紫外線照射下の金内包多孔質シリカの写真を示す。このように、発光(蛍光)材料としての機能を有することが判明した。
【0157】
また、上記発光現象時の発光スペクトル(Ex.360nm)を図7に示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、PL強度(PL Intensity、PL;フォトルミネッセンス(Photoluminescence)、[a.u.])である。発光スペクトルは、分光蛍光光度計(PL)FP−6500(日本分光社製)を用いて測定した。
【0158】
(実施例4)
本実施例においては、多孔質シリカを鋳型として炭素(C)の微細粒子を合成する。
【0159】
<4−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)を形成した。
【0160】
<4−2>微細粒子の合成
次いで、上記多孔質シリカ(C6VSMPS)を鋳型として炭素(C)の微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、0.26Mのブドウ糖(グルコース;C6H12O6)の水溶液を添加し、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。その後、回収した試料を水洗し、300℃で2時間程度、空気中で焼成し、ブドウ糖を炭化することにより、炭素の微細粒子を内包した多孔質シリカを得た。なお、上記焼成工程に代えて、濃硫酸に数時間、浸漬することでブドウ糖を炭化してもよい。また、本実施例では、ブドウ糖を用いたが、この他、適当な分子サイズを有する有機化合物の溶液を用い、焼成により炭化してもよい。また、有機化合物として、糖類を用いた場合には硫酸を用いた脱水反応で炭化させることができる。
【0161】
得られた炭素内包多孔質シリカに、360nm〜370nmの波長の紫外線を照射したところ、目視において発光現象を確認することができた。図8(A)に、得られた炭素内包多孔質シリカの写真を、図8(B)に、紫外線照射下の炭素内包多孔質シリカの写真を示す。このように、発光(蛍光)材料としての機能を有することが判明した。
【0162】
また、上記発光現象時の発光スペクトル(Ex.320nm)を図9に示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、PL強度(PL Intensity、PL;フォトルミネッセンス(Photoluminescence)、[a.u.])である。発光スペクトルは、分光蛍光光度計(PL)FP−6500(日本分光社製)を用いて測定した。
【0163】
(実施の形態2)
実施の形態1においては、多孔質シリカに金属化合物(WO3)、金属(Au)または炭素(C)の微細粒子を内包させたが、本実施の形態においては、バナジン酸ビスマス(BiVO4)の微細粒子を内包させる。
【0164】
バナジン酸ビスマスは、有害視されているカドミウムイエローやクロムイエローの代替材料として黄色の顔料として用いられているが、光触媒として水の分解(水素製造)や有機物の分解などの機能を有する。また、光触媒として二酸化炭素(CO2)をエタノール(C2H5OH)に変換する機能を有する。しかし、その触媒活性は低く、高効率化が望まれる。
【0165】
一般的に光触媒を用いた上記二酸化炭素の固定化反応は、炭素が1個であるメタン、メタノール、ホルムアルデヒドまたは蟻酸などが生成されることが多い。これに対し、上記バナジン酸ビスマスの場合は、二酸化炭素(CO2)を炭素が2個のエタノール(C2H5OH)に変換することが可能である。このエタノールの生成メカニズムは、二酸化炭素の還元によって生じた炭素ラジカル(C・)中間体の二量化反応が触媒表面で優先的に進行するためでる。このような優先的な反応が生じるのは、バナジン酸ビスマスの結晶表面の特殊性にあると考えられている。
【0166】
この特性を維持したまま、活性のみを向上させるには、結晶構造を維持したまま、還元準位を上昇させる必要がある。これを実現するためには、バナジン酸ビスマスをサブナノ粒子化し、量子サイズ効果によりバンドギャップエネルギーを増大させることが有効である。
【0167】
上記バナジン酸ビスマスは、硝酸ビスマス(Bi(NO3)3)などのビスマス塩とアンモニウムオルトバナジン酸(NH4VO3)などのバナジン酸塩(バナジウム酸塩)を水溶液中で反応させ合成することができる。すなわち、バナジン酸ビスマスを合成するためにはビスマスカチオンとバナジン酸アニオンを水中で反応させる。
【0168】
(実施例5)
本実施例においては、多孔質シリカを鋳型としてバナジン酸ビスマスの微細粒子を合成する。
【0169】
<5−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよびTEVSを用いたポーラスシリカ(C6VSMPS)を形成した。
【0170】
<5−2>微細粒子の合成
次いで、上記多孔質シリカ(C6VSMPS)を鋳型としてバナジン酸ビスマスの微細粒子を合成した。上記多孔質シリカ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、減圧状態下に維持し、水等の吸着物質を除去した(減圧乾燥工程)。また、0.6MのBi(NO3)3・5H2Oと0.6MのNH4VO3をそれぞれ2Mの硝酸に溶解させ、0.2Mの溶液を得た。この二つの溶液を混合し、それぞれのイオンが0.1Mの濃度で溶解した混合溶液を得た。この溶液を上記多孔質シリカの粉末に添加し、水溶液中の粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回(1回〜4回程度)繰り返し行った後、乾燥工程後の試料、450℃で2時間程度、空気中で焼成し、バナジン酸ビスマスの微細粒子を内包した多孔質シリカを得た。
【0171】
得られたバナジン酸ビスマス内包多孔質シリカについて、透過型電子顕微鏡(TEM)による観察を行った。具体的には、FE−TEM(TECNAI F20:FEI)を用いて、試料の形状、粒子サイズを測定した。観察試料は、粉砕した試料をマイクログリッド膜付銅メッシュに分散させることで作製した。
【0172】
図10は、得られた酸化タングステン内包多孔質シリカ透過型電子顕微鏡像(TEM像)である。多孔質シリカの孔内にバナジン酸ビスマスを内包している構造が確認できた。
【0173】
また、得られたバナジン酸ビスマス内包多孔質シリカについて、紫外・可視吸光光度計(UV−Vis) V−550(日本分光社製)を用いて電子スペクトル(可視・紫外吸収スペクトル)を求め、Taucプロットにより、バンドギャップエネルギーを求めた。
【0174】
図11に、得られたバナジン酸ビスマス内包多孔質シリカ(実線)およびバルクのバナジン酸ビスマス(破線)のTaucプロットを示す。横軸は、光子エネルギーE(Energy、hν[eV])であり、縦軸は、吸光度(α)と光子エネルギー(hν[eV])の積の平方根((αhν)1/2)である。図11の実線のグラフからバンドギャップエネルギー(Eg)を算出したところ、得られたバナジン酸ビスマス内包多孔質シリカのEgは、3.0eVであり、バルクのバナジン酸ビスマス(破線)のEgは、2.2eVとなった。このように、2,2eVから3.0eVまでバンドギャップエネルギーが増大し、顕著な量子サイズ効果を有することが確認できた。
【0175】
よって、バナジン酸ビスマス内包多孔質シリカを光触媒として利用することで、触媒活性を向上することができ、高効率な光触媒作用を実現することができる。特に、二酸化炭素(CO2)をエタノール(C2H5OH)に変換する光触媒作用の触媒活性を向上することができ、高効率な変換を実現することができる。
【0176】
以上詳細に説明したように、本実施の形態のバナジン酸ビスマス内包多孔質シリカは、地球温暖化の要因である二酸化炭素(CO2)を低減するとともに、エネルギー源であるエタノール(C2H5OH)を生成することができ、地球温暖化対策および再生可能エネルギーとして広く有用な技術に応用可能である。
【0177】
一方で、微細粒子の形成方法としては、デンドリマーを鋳型に用いた合成方法が考えられる。しかしながら、この方法は、含窒素有機化合物からなるデンドリマーの窒素部へ金属カチオンを配位させ、熱処理や還元処理を行うことで金属酸化物や金属の微細粒子を合成するものである。よって、バナジン酸ビスマスのようなカチオンとアニオンを反応させて生成するような物質を合成するためには、両方のイオンをデンドリマー内に配位させる必要があり、このためには特殊な構造を有するデンドリマーを合成する必要があり、その合成は困難を極めるものである。
【0178】
これに対し、本実施の形態においては、ビスマスカチオンとバナジン酸アニオンを容易に孔中で溶液反応させることができ、簡易な製造工程で、効率的に、バナジン酸ビスマス内包多孔質シリカを得ることができる。
【0179】
(実施の形態3)
実施の形態1においては、多孔質シリカに金属化合物(WO3)、金属(Au)または炭素(C)の微細粒子を、実施の形態2においては、バナジン酸ビスマス(BiVO4)を内包させたが、本実施の形態においては、酸化銅(CuO)の微細粒子を内包させる。
【0180】
CuOは約1.3 eVにバンドギャップを有する半導体である。他の半導体と比較してバンドギャップエネルギーが小さいが、チタニア(TiO2)等に比べて高い伝導帯準位を有する。この性質を利用してTiO2のホールとCuOの電子を利用するTiO2-CuO複合光触媒系等が開発されている。バルクの状態でのバンドギャップエネルギーが小さいため、量子サイズ効果を発現させ、伝導帯準位を上昇させても可視光応答性を維持できる可能性がある。これらの性質を用いれば、伝導帯準位が非常に高くかつ可視光応答性を有する高機能性光触媒材料を設計できる可能性がある。
【0181】
(実施例6)
本実施例においては、多孔質シリカを鋳型として酸化銅(CuO)の微細粒子を合成する。
【0182】
<6−1>多孔質シリカの合成
(実施例1)の<1−1>で説明した多孔質シリカの合成方法と同様の方法により、C6TABおよび比較のためにC16TACを用いたポーラスシリカ(C6SMPSおよびC16MPS)を形成した。
【0183】
<6−2>微細粒子の合成
次いで、上記多孔質シリカを鋳型として酸化銅(CuO)の微細粒子を合成した。上記2種類の多孔質シリカのそれぞれ2gを乳鉢で粉砕し粉末状とした後、二口フラスコに入れ、数時間〜一昼夜、真空ポンプで減圧し、水等の吸着物質を除去した(減圧乾燥工程)。その後、減圧を一時停止し、各多孔質シリカに0.2M(mol)の酢酸銅(Cu(AcO)2)水溶液を添加した。この時にフラスコ内は陰圧を維持している。水溶液と粉末を撹拌、震盪しながら水溶液を均一に細孔内に浸透させた(含浸工程)。1回の含浸工程における酢酸銅水溶液の添加量は、細孔容積(TPV、[cm3/g])×2gと同程度あるいはそれ以下とする。この後、再度、減圧乾燥を行った。この含浸工程および減圧乾燥工程を数回繰り返し行った後、乾燥した試料を400℃〜600℃で3時間程度、空気中で焼成し、その孔内にCuOの微細粒子を内包した多孔質シリカを得た。
【0184】
図12は、C16MPSを鋳型にして得られたCuO粒子の高角度散乱暗視野走査型透過電子顕微鏡(HAADF−STEM)像である。白色に見える点が内包された粒子を示しており、2nm以上のCuO粒子が均一に分散している様子が分かる。図13は、C6SMPSを鋳型にして得られたCuO粒子のHAADF−STEM像である。1nm以下のCuO粒子がシリカ中に高分散状態で存在していることが分かる。
【0185】
図14は、C16MPSを鋳型にして得られたCuO粒子の紫外・可視吸収スペクトルから求めたTaucプロットである。なお、ここでは試料の温度変化(35℃〜600℃)に伴う、各Taucプロットの変化も示してある(図15についても同じ)。例えば、35℃のグラフから、バルクでは1.3 eV程度の位置に存在するバンドギャップが、約2.3eVまで増大していることが確認できる。一方で、図15に示すようにC6SMPSを鋳型にして得られたCuO粒子の場合には、バンドギャップエネルギーはさらに増加し、約3.2eVまで増加した(35℃のグラフ参照)。これは1nm以下で量子サイズ効果が顕著になることに起因する。また、800nm付近に存在するブロードなピークはCu2+のd−d遷移に起因する。この吸収は通常のCuOでは間接遷移の吸収に埋もれて観測できないが、量子サイズ効果による吸収端の顕著なブルーシフトより初めて観測されるようになった。その結果、バルクのCuOの色が黒であるのに対し、得られた粒子はC16MPSを鋳型にして得られたCuO粒子の場合、緑色であり、C6SMPSを鋳型にして得られたCuO粒子の場合、青色であった。
【0186】
得られた粒子は顕著なサーモクロミズム(温度変化による色変化)を示した。図14および15には、前述したとおり、得られた粒子の35℃〜600℃での紫外・可視吸収スペクトルから求めたTaucプロットも示してある。各図から分かるように吸収端は温度の上昇とともに長波長側にシフトした。
【0187】
図16に、それぞれの試料のバンドギャップエネルギーの温度依存性を示した。縦軸がバンドギャップエネルギー(Eg;eV)で、横軸が温度(Temperature;K)である。C16MPSを鋳型にして得られたCuO粒子の場合(CuO@C16MPS)、バンドギャップの温度依存性は約0.8meV/Kであり、バルクで報告されている値とほぼ同程度であった。一方、C6SMPSを鋳型にして得られたCuO粒子の場合(CuO@C6SMPS)には、約2.3meV/Kと大きく、サブナノ粒子化により著しく温度依存性が高まっていることが観測された。また、これらのサーモクロミズムは可逆的であり、室温まで冷却するともとのスペクトルに戻ることも確認された。また、同温度領域における熱重量測定の結果から、温度上昇中も降下中も酸化や水酸化などの化学反応や相転移等が起こっていないことも確認された。
【0188】
この吸収スペクトルの変化に伴い、目視では緑ないしは青から黄色への顕著なサーモクロミズムが観測された。このサーモクロミズムのメカニズムは格子振動が電子軌道に影響を及ぼす、エレクトロン-フォノンカップリングによるものであると考えられる。代表的なサーモクロミズムを示す無機物である酸化亜鉛では、温度上昇に伴う白色から黄色への色変化が観測される。この要因もエレクトロン-フォノンカップリングことにあると考えられている。バルクのCuOでもこの挙動は観測されるが、バルク体は黒色であるため、目視で色の変化は観測されない。本実施の形態のCuOサブナノ粒子において顕著なサーモクロミズムが観測された要因は間接遷移の吸収端が量子サイズ効果によって紫外領域までシフトしたことと、温度変化に対する吸収端のレッドシフト幅が非常に大きくなったことに起因する。これらは通常のナノ粒子では観測されない現象であり、本実施の形態においてサブナノ粒子が高温でも焼結すること無くシリカ中に単分散で存在できる状態にあることにより初めて実現可能になったものである。
【0189】
このようにサブナノ粒子化することでバンドギャップエネルギーの温度依存性が著しく向上することが明らかになった。このような材料はシリカの熱安定性と相まって、温度インジケータとしての応用用途が期待できる。さらに、半導体発光材料をサブナノ粒子化した場合には温度によって発光波長を大きく制御可能になり、新規な機能性を有する材料を合成することが可能である。
【0190】
(実施の形態4)
本実施の形態においては、浸漬法と焼成による金属酸化物の微細粒子(量子ドット)の合成について説明する。
【0191】
(実施例7)
本実施例においては、多孔質シリカを鋳型として酸化銅(CuO)の微細粒子(量子ドット)を合成する。
【0192】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。濃度の低下に伴い粒径が減少する。そのため、金属塩の水溶液(前駆体液、前駆体溶液)の濃度を低下させることで、細孔径より小さい粒子を合成することができる。このように、実施の形態1の実施例A等で説明した多孔質シリカの細孔径の調整に加え、さらに、金属塩の水溶液(前駆体液)の濃度を低下させることで、多孔質シリカ内包粒子(ここでは、酸化銅の微細粒子)の粒径を微調整することができる。
【0193】
本実施例では、金属塩の水溶液(前駆体液)として、0.6M(mol/L)の硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径3.0nm〜0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0194】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面を洗浄する。洗浄にはエタノールなどのアルコール類を用いるのが好ましい。アルコール類に代えて、水を用いることも可能である。但し、水を用いた場合には溶質(金属塩)の溶出が起こる場合があるため、アルコール類を用いるのが好ましい。
【0195】
洗浄後、多孔質シリカを乾燥させ、細孔内に金属塩の微粒子を析出させる。乾燥方法としては真空乾燥や凍結乾燥を用いることが好ましい。自然乾燥や温風乾燥を用いることも可能であるが、真空乾燥や凍結乾燥の方が、迅速な乾燥が可能であり、より均一な粒子を得ることができる。
【0196】
本実施例においては、真空乾燥を用いて、多孔質シリカを乾燥し、細孔内に金属塩の微粒子を析出させた。
【0197】
この後、細孔内に金属塩の微粒子が析出した多孔質シリカを、空気中で焼成することで、金属酸化物の微細粒子(ここでは、酸化銅の微細粒子)を合成することができる。
【0198】
本実施例においては、空気中において450℃で1時間の焼成を行い、酸化銅の微細粒子を得た。図17に細孔径の異なる多孔質シリカを鋳型に用いて合成した酸化銅の微細粒子のUV−Visスペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。各グラフの添え字は、細孔径(nm)を示す。図17に示すように、各グラフとも、250nm〜450nmの間に、酸化銅のスペクトルピークを確認できる。さらに、細孔径(nm)が右側から3.0nm、2.8nm、1.8nm、1.3nm、0.8nm、および0.7nmの順に、スペクトルピークが左にシフトすることが判明した。また、細孔径(nm)が同じ0.7nmであっても、硝酸銅水溶液の濃度を0.2Mと低くしたグラフ(0.7(0.2M))については、硝酸銅水溶液の濃度を0.6Mとしたグラフ(0.7)より、スペクトルピークが左にシフトしている。このように、細孔径に応じた酸化銅の微細粒子が合成されていることが判明し、また、硝酸銅水溶液の濃度を低くすることで、微細粒子の粒径が小さくなることが判明した。
【0199】
また、バルクの酸化銅のバンドギャップエネルギーは約1.3eV程度であり、目視では黒色である。これに対し、上記酸化銅の微細粒子(金属酸化物量子ドット)においては、量子サイズ効果によってバンドギャップエネルギーが顕著に増大し、目視では緑色から青色となることが確認できた。表3に、上記酸化銅の微細粒子(金属酸化物量子ドット)を内包する多孔質シリカの細孔径(Dpore;nm)と酸化銅の微細粒子のバンドギャップエネルギー(Eg;eV)の関係を示す。バンドギャップエネルギーはTaucプロットにより求めた。
【0200】
【表3】
このように、本実施例においては、多孔質シリカを粉砕(微細化)することなく、モノリス状(例えば、粒子の直径が1mm以上の塊状態)のまま前駆体液に浸漬し、焼成することで、容易に金属酸化物の微細粒子(量子ドット)を合成することができることが判明した。また、内包させる粒子材料の粒径の制御が可能であり、これによりバンドギャップエネルギーを制御することができることが判明した。これは、発光スペクトルや触媒活性の精密な制御が可能になることを示唆しており、例えば、伝導帯準位が非常に高くかつ可視光応答性を有する高機能性光触媒材料などに有用である。
【0201】
なお、酸化銅のみならず、クロムやコバルトの酸化物についても、硝酸クロムや硝酸コバルトを用い、同様にして、金属酸化物の微細粒子(量子ドット)を合成することが可能である(実施例12参照)。
【0202】
(実施の形態5)
本実施の形態においては、浸漬法と焼成による金属の微細粒子(量子ドット)の合成について説明する。
【0203】
(実施例8)
本実施例においては、多孔質シリカを鋳型として銀(Ag)の微細粒子(量子ドット)を合成する。
【0204】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、硝酸銀の水溶液(前駆体液)に浸漬した。硝酸銀の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。ここでも、金属塩の水溶液(前駆体液、前駆体溶液)の濃度を低下させることで、細孔径より小さい粒子を合成することができる。
【0205】
本実施例では、0.6Mの硝酸銀水溶液を用い、モノリス状の多孔質シリカ(細孔径3.0nm〜0.7nm)を浸漬させた。浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを真空乾燥させ、細孔内に硝酸銀の微粒子を析出させた。
【0206】
この後、多孔質シリカを空気中において600℃で3時間の焼成を行い、銀の微細粒子を得た。紫外線照射により蛍光を示すことより、内包物が銀であることを確認した。上記焼成温度においては、酸化銀よりも金属銀の方が安定的に存在するため、銀の微細粒子(銀量子ドット)を合成することが可能となる。得られた銀の微細粒子(銀量子ドット)は、365nmの紫外線照射下で黄色の発光を示した。
【0207】
このように、前駆体液を構成する金属(金属化合物)によっては、浸漬後に焼成を行っても金属酸化物とならず、金属(金属の微細粒子)として合成できる場合もある。また、得られた金属が大気中の酸素などにより僅かに酸化された場合であっても、再焼成を行うことで金属の微細粒子に戻すことが可能である。
【0208】
また、酸素と反応させることにより、酸化銀の微細粒子を合成することも可能である。酸化物になると金属微粒子特有の蛍光がなくなるため、酸化を確認できる。
【0209】
このように、上記銀の微細粒子(銀量子ドット)は、紫外線照射下での発光が見られ、発光素子やバイオイメージング等への応用が可能である。
【0210】
(実施の形態6)
本実施の形態においては、浸漬法と焼成により得られた金属酸化物の微細粒子(量子ドット)を還元することにより金属の微細粒子(量子ドット)を合成する方法について説明する。
【0211】
(実施例9)
本実施例においては、実施例7で合成した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。還元剤としては、例えば、過酸化水素水溶液を用いることができる。
【0212】
ここでは、実施例7で合成した、銅、クロムおよびコバルトの金属酸化物の微細粒子を前駆体として、15%の過酸化水素水溶液に浸漬することにより金属の微細粒子(量子ドット)を得た。
【0213】
図18に、酸化銅の微細粒子(量子ドット)のXPS(X線光電子分光)スペクトルを示し、図19に、還元反応により生成した銅の微細粒子(量子ドット)のXPSスペクトルを示す。横軸は、結合エネルギー(Binding Energy、[eV])であり、縦軸は、強度([a.u.]、CPS)である。
【0214】
図18のスペクトルにおいては、933.7eVに銅(Cu)の2p3/2軌道のピークが観測され、また、サテライトピークも確認されることから、微細粒子が酸化銅(CuO)であることが確認できる。これに対し、図19のスペクトルにおいては、932.8eVにピークが観測され、金属(0価)の銅の微細粒子が生成していることが確認できる。
【0215】
図20に、酸化時と還元時における紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。グラフaのスペクトルを示す酸化銅の微細粒子を、過酸化水素により還元し、銅の微細粒子としたところ、グラフbのスペクトルを示した。さらに、グラフbのスペクトルを示す銅の微細粒子を空気中の酸素により再酸化し、酸化銅の微細粒子としたところ、グラフcのスペクトルを示した。さらに、グラフcのスペクトルを示す酸化銅の微細粒子を、過酸化水素により再還元し、銅の微細粒子としたところ、グラフdのスペクトルを示した。このように、酸化還元反応が可逆的に進行することが確認できる。
【0216】
このように、本実施例によれば、簡易な酸化還元処理により、多孔質シリカに内包される金属の酸化または金属酸化物の還元が行えることが判明した。また、これらが、可逆的に進行することから、目的物(例えば、金属または金属酸化物)の再生が容易に行えることが判明した。
【0217】
(実施の形態7)
本実施の形態においては、浸漬法と還元剤による還元による金属の微細粒子(量子ドット)の合成について説明する。
【0218】
(実施例10)
本実施例においては、多孔質シリカを鋳型として銅(Cu)の微細粒子(量子ドット)を合成する。
【0219】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上0.8M以下の範囲が好ましい。本実施例では、金属塩の水溶液(前駆体液)として、0.6Mの硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0220】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを乾燥させ、細孔内に金属塩の微粒子を析出させた。乾燥方法は真空乾燥や凍結乾燥を用いることが好ましい。本実施例においては、真空乾燥を用いて、多孔質シリカを乾燥し、細孔内に金属塩(ここでは、硝酸銅)の微粒子を析出させた。
【0221】
この後、細孔内に金属塩の微粒子が析出した多孔質シリカを還元剤を用いて直接還元することにより、金属銅の微細粒子を合成した。本実施例では、15%の過酸化水素水溶液に、金属塩の微粒子が析出した多孔質シリカを浸漬することにより、金属銅の微細粒子を合成した。
【0222】
図21に、合成した金属銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、吸光度[Absorbance;a.u.]である。酸化銅(CuO)に特有の750nm近傍のスペクトルピークが確認されず、銅の微細粒子の合成を確認することができる。
【0223】
このように、本実施例によれば、浸漬法と還元剤による還元による金属の微細粒子(量子ドット)の合成が可能であることが判明した。
【0224】
また、得られた銅の微細粒子は、空気中の酸素によって緩やかに酸化され、酸化銅の微細粒子となる。このため、上記銅の微細粒子を空気中などの酸素雰囲気へ暴露するだけで、酸化銅の微細粒子を合成することが可能である。このような工程によれば、熱処理(焼成)を行うことなく、室温程度の温度雰囲気において、酸化銅の微細粒子の合成が可能である。
【0225】
本実施の形態の直接還元は、銅の微細粒子の合成のみならず、後述の実施例13等を参酌することにより、クロム(Cr)、コバルトなどの金属微細粒子(量子ドット)の合成にも適用可能である。
【0226】
また、銅の微細粒子としてより安定的に多孔質シリカの内部に存在させるためには、多孔質シリカの細孔内に過酸化水素などの還元剤を残存させて置くことが有用である。例えば、過酸化水素水に浸漬したのち、乾燥させずにおく、または、乾燥時間を短くするなどの乾燥条件を調整することにより、還元剤を残存させて置くことができる。細孔内に過酸化水素などの還元剤が残っている状態では、酸化反応が抑制されるため銅の微細粒子を安定的に存在させることができる。
【0227】
(実施の形態8)
本実施の形態においては、マイクロ波加熱によるの微細粒子(量子ドット)の合成について説明する。
【0228】
(実施例11)
本実施例においては、多孔質シリカを鋳型としてマイクロ波加熱を利用して酸化銅(CuO)の微細粒子(量子ドット)を合成する。なお、ここで言うマイクロ波(Microwave)とは、電波の周波数による分類の一つであり、電波の中で比較的短い波長域であることを意味する。一般的には波長1mから100μm、周波数300MHzから3THzの電波(電磁波)を指す。代表的なマイクロ波の照射手段としては、いわゆる電子レンジ(周波数2.45GHz)が挙げられる。
【0229】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。金属塩の水溶液の濃度は、0.5M以上とすることが好ましい。本実施例では、金属塩の水溶液(前駆体液)として、0.6Mおよび3.0Mの硝酸銅水溶液を用い、モノリス状の多孔質シリカ(細孔径0.7nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0230】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、電子レンジを用いて700Wで15分の条件でマイクロ波照射を行った。マイクロ波照射により多孔質シリカに内包された前駆体液が加熱され、酸化銅の微細粒子が得られた。比較のため、洗浄後の前駆体液を内包した多孔質シリカを電気炉を用いて450℃で1時間の焼成を行った試料も作成した。
【0231】
合成した各酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを測定した。図22に、合成した酸化銅の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。図22に示すように、金属塩の水溶液(前駆体液)として、0.6Mまたは3.0Mの硝酸銅水溶液を用い、マイクロ波加熱を行った酸化銅の微細粒子(マイクロ波加熱(3.0M)、マイクロ波加熱(0.6M))のスペクトルについては、250nm〜450nmの間のスペクトルがほぼ一致している。これに対し、3.0Mの硝酸銅水溶液を用い、電気炉加熱を行った酸化銅の微細粒子(電気炉450℃(3.0M))のスペクトルは、250nm〜450nmの間にスペクトルのピークを有するものの、ピークの位置が長波長側にずれ、“吸収端のレッドシフト”が観察された。
【0232】
これは、高濃度の前駆体液を用いた電気炉加熱においては、多孔質シリカの各細孔内における微細粒子の合一化(凝集)、焼結が起こり、多孔質シリカの細孔径に対応した球状粒子を得ることができないことに起因するものと考えられる。一方、マイクロ波加熱においては、急速加熱が可能であり、高濃度の前駆体液を用いた場合でも多孔質シリカの細孔径に対応した均一かつ高分散な球状粒子を得ることが可能である。
【0233】
このように、マイクロ波加熱のような簡易な加熱手段を用いても、多孔質シリカに内包された金属酸化物の微細粒子の合成が可能であることが判明した。また、加熱手法によって用いる金属塩の水溶液(前駆体液)の濃度を調整することが好ましいことが判明した。
【0234】
(まとめ)
ここで、上記実施の形態4〜実施の形態8(実施例7〜実施例11)で説明した合成ルートをまとめて説明する。図23は、実施の形態4〜実施の形態8(実施例7〜実施例11)の合成ルートの概念図である。
【0235】
図23に示すように、多孔質シリカの細孔内に前駆体液(金属塩)を注入し、加熱することで、細孔内に金属酸化物の微細粒子を生成することができる(ルート1;加熱ルート)。また、多孔質シリカの細孔内に前駆体液を注入し、これを乾燥し、還元することで、細孔内に金属の微細粒子を生成することができる(ルート2;直接還元ルート)。また、細孔内の金属酸化物の微細粒子を還元することにより、細孔内に金属の微細粒子を生成することができる(ルート3)。また、細孔内の金属の微細粒子を酸化することにより、細孔内に金属酸化物の微細粒子を生成することができる(ルート3)。このように、ルート3の酸化体/還元体は相互変換可能である。また、ルート2によれば、室温での処理が可能である。
【0236】
また、多孔質シリカの細孔内への前駆体液の注入は、浸漬法でも、細孔容積に対応した量の溶液を浸透させる方法でも構わない。また、加熱(焼成)は、電気炉加熱やマイクロ波加熱が有効である。特に、高濃度の前駆体液を用い、均一な微細粒子を得るためにはマイクロ波加熱が有効である。還元反応に要する還元剤の種類は問わないが、金属酸化物を金属化することが可能であること、微細粒子の溶出を促進させないことなどが条件となる。上記実施例では過酸化水素が良好な結果を示した。酸化工程には、酸素などの酸化剤を用いることができる。また、ルート2(直接還元ルート)においては、各工程が室温下で行えるため、エネルギーコストの低減が可能である。
【0237】
(実施の形態9)
上記実施の形態4〜実施の形態8(実施例7〜実施例11)においては、銅や銀の微細粒子や銅の酸化物の微細粒子の合成について説明したが、本実施の形態においては、その他の金属(特に、遷移金属)の酸化物の微細粒子の合成について説明する。
【0238】
(実施例12)
本実施例においては、多孔質シリカを鋳型として、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の金属酸化物の微細粒子(量子ドット)を合成する。
【0239】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(ポーラスシリカ)をそのまま、金属塩の水溶液(前駆体液)に浸漬した。本実施例では、金属塩の水溶液(前駆体液)として、それぞれ、クロム、マンガン、鉄、コバルトおよびニッケルの硝酸塩水溶液を用いた。硝酸塩水溶液の濃度は、それぞれ0.5Mとした。
【0240】
各硝酸塩水溶液に、モノリス状の多孔質シリカ(細孔径0.8nm〜3.0nm)を浸漬させた。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0241】
浸漬の完了後、多孔質シリカを硝酸塩水溶液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、電子レンジを用いて700Wで15分の条件でマイクロ波照射を行った。マイクロ波照射により多孔質シリカに内包された硝酸塩水溶液が加熱され、対応する金属酸化物の微細粒子、即ち、クロム、マンガン、鉄、コバルトおよびニッケルの酸化物の微細粒子が得られた。
【0242】
各金属酸化物の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを測定した(図24〜図28)。図24は、合成した酸化クロムの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図25は、合成した酸化マンガンの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図26は、合成した酸化鉄の微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図27は、合成した酸化コバルトの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。図28は、合成した酸化ニッケルの微細粒子の紫外・可視吸収(UV−Vis)スペクトルである。それぞれ、グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。また、各グラフの添え字は、細孔径(nm)を示す。
【0243】
図24〜図28に示すように、いずれの金属酸化物においても細孔径の低下に伴う量子サイズ効果が観測された。特に、サブナノ領域(細孔径1nm未満)において、顕著な量子サイズ効果が観測された。
【0244】
表4に、得られた金属酸化物の目視における色を示す。なお、表4においては、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の酸化物の他、実施の形態4(実施例7)で説明した銅(Cu)の酸化物の色についても表示してある。
【0245】
表4に示すように、いずれのいずれの金属酸化物においても鋳型となるシリカの細孔径の低下に伴い、色の変化が観測された。
【0246】
【表4】
(実施の形態10)
上記実施の形態4〜実施の形態8(実施例7〜実施例11)においては、銅や銀の微細粒子や銅の酸化物の微細粒子の合成について説明したが、本実施の形態においては、その他の金属(特に、遷移金属)の微細粒子の合成について説明する。具体的には、実施の形態9(実施例12)で説明した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。
【0247】
(実施例13)
本実施例においては、実施例12で合成した多孔質シリカに内包される金属酸化物の微細粒子(量子ドット)を還元することにより、金属の微細粒子を合成する。還元剤としては、例えば、過酸化水素水溶液を用いることができる。
【0248】
ここでは、実施例12で合成した、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の酸化物の微細粒子を前駆体として、15%の過酸化水素水溶液に浸漬することにより対応する金属の微細粒子、即ち、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)およびニッケル(Ni)の微細粒子(量子ドット)を得た。
【0249】
図29に、クロムの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。図30に、コバルトの酸化物の微細粒子と金属の微細粒子の紫外・可視吸収(UV−Vis)スペクトルを示す。グラフの横軸は、波長[Wavelength;nm]であり、縦軸は、強度[Intensity;a.u.]である。図29および図30のいずれも、鋳型として、細孔径が0.8nmの多孔質シリカを用いた。図29および図30に示すように、酸化物と金属との間で大きなスペクトルの変化が確認できた。
【0250】
金属の種類によって再酸化されやすいものとそうでないものが存在する。コバルトは再酸化されにくく、クロムは再酸化されやすい傾向がある。いずれの場合も多孔質シリカの細孔内に還元剤を残存させることで、金属の状態を安定的に維持することが可能である。
【0251】
(実施の形態11)
本実施の形態においては、多孔質シリカを鋳型として炭素の微細粒子(量子ドット)の合成について説明する。実施の形態1の実施例4においては、多孔質シリカを乳鉢で粉砕し粉末状とした後、ブドウ糖の水溶液を添加したが、以下の実施例14においては、モノリス状の多孔質シリカをクエン酸水溶液に浸漬する。
【0252】
(実施例14)
本実施例においては、多孔質シリカを鋳型として炭素の微細粒子(量子ドット)を合成する。
【0253】
ここでは、例えば、実施の形態1の実施例A等で説明したモノリス状の多孔質シリカ(細孔径1.1nm、2.8nm)をそのまま、0.6Mのクエン酸水溶液に浸漬した。浸漬に伴い、多孔質シリカの細孔内の空気が放出され、前駆体液が充填される。空気の放出がなくなった段階を浸漬の完了時とする。おおよそ半日〜2日の浸漬で、空気と前駆体液との置換が完了する。
【0254】
浸漬の完了後、多孔質シリカを前駆体液から取り出し、外表面をエタノールなどのアルコール類を用いて洗浄した。洗浄後、多孔質シリカを乾燥させ、焼成することで、炭素の微細粒子を合成した。本実施例においては、電気炉を用い、空気中において300℃で1時間の焼成を行い、多孔質シリカの細孔中のクエン酸を炭化させ、炭素の微細粒子を得た。
【0255】
図31に、得られた炭素の微細粒子の発光スペクトル(Ex.340nm)を示す。横軸は、波長(Wavelength、[nm])であり、縦軸は、強度(Intensity、[a.u.])である。各グラフの添え字は、細孔径(nm)を示す。図32に、得られた炭素の微細粒子の発光スペクトルを示す。横軸は、励起波長(Em、[nm])であり、縦軸は、発光波長(Ex、[nm])である。図32の上部の発光スペクトルは、細孔径1.1nmのものであり、下部の発光スペクトルは、細孔径2.8nmのものである。この、図32中の破線で囲んだ領域は、強発光部を示し、その中心(励起波長)をラインC1、C2で示してある。
【0256】
図31に示すように、細孔径を変化(2.8nm→1.1nm)させることで、強度のピークが変化することが判明した。また、図32に示すように、細孔径を変化(2.8nm→1.1nm)させることで、強発光部を示し、その中心がC1からC2へ変化することが判明した。
【0257】
このように、細孔径を変化させることで、発光波長が変化することが判明した。即ち、細孔径を変化させることで、発光波長をコントロールすることが可能である。言い換えれば、多孔質シリカの細孔径(平均孔直径)を制御することにより、微細粒子の発光波長を調整することができる。細孔径を低下させることで、より小さな炭素の微細粒子を合成することが可能であり、発光波長は短波長側にシフトする。目視においては、細孔径1.1nmの多孔質シリカを鋳型とした炭素の微細粒子は、青色の発光が観測され、細孔径2.8nmの多孔質シリカを鋳型とした炭素の微細粒子は、黄緑の発光が観測された。
【0258】
以上、本発明者によってなされた発明をその実施の形態に基づき具体的に説明したが、本発明は前記実施の形態に限定されるものではなく、その要旨を逸脱しない範囲で種々変更可能である。
【産業上の利用可能性】
【0259】
本発明は、多孔質シリカ内包粒子の製造方法および多孔質シリカ、多孔質シリカ内包粒子に関し、特に、微細な孔を有する多孔質シリカを利用し、その孔の内部に微細な粒子を内包させる技術に適用して有効である。
【特許請求の範囲】
【請求項1】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項2】
前記金属化合物は、金属酸化物、金属硫化物または金属窒化物であることを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項3】
前記微細粒子の直径は、0.5nm以上1.0nm以下であることを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項4】
前記微細粒子は、金を含有し、前記液は、金化合物を含有することを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項5】
前記液は、塩化金酸水溶液を含有することを特徴とする請求項4記載の多孔質シリカ内包粒子の製造方法。
【請求項6】
前記微細粒子は、酸化タングステンを含有し、前記液は、タングステン化合物を含有することを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項7】
前記液は、過酸化タングステン酸水溶液を含有することを特徴とする請求項6記載の多孔質シリカ内包粒子の製造方法。
【請求項8】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項9】
前記液は、有機化合物を含有することを特徴とする請求項8記載の多孔質シリカ内包粒子の製造方法。
【請求項10】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと、ビスマスまたはビスマスを元素組成として有する化合物を含有する第1液と、バナジウムまたはバナジウムを元素組成として有する化合物を含有する第2液との混合液とを接触させ、前記多孔質シリカの孔内に前記混合液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内においてバナジン酸ビスマスを含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項11】
前記第1液は、Bi(NO3)3を含有し、前記第2液は、NH4VO3を含有することを特徴とする請求項10記載の多孔質シリカ内包粒子の製造方法。
【請求項12】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する気体とを接触させ、前記多孔質シリカの孔内に前記気体を導入し、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項13】
前記(b)工程は、前記気体の気相での化学反応により前記微細粒子を形成する工程であることを特徴とする請求項12記載の多孔質シリカ内包粒子の製造方法。
【請求項14】
(a)アルコキシシランの加水分解による多孔質シリカの製造工程であって、
(a1)界面活性剤およびアルコキシシランを混合し、混合液を形成する工程と、
(a2)前記混合液に水を添加することにより、前記アルコキシシランの加水分解反応を行わせる工程と、
(a3)前記(a2)工程の後、熱処理を施すことにより前記界面活性剤を除去する工程と、を有する多孔質シリカの製造工程と、
(b)前記(a)工程により製造された多孔質シリカの孔内において粒子を合成する工程であって、
前記多孔質シリカと粒子材料または前記粒子材料を元素組成として有する化合物を含有する液または気体とを接触させ、前記多孔質シリカの孔内に前記液または前記気体を導入させ、前記多孔質シリカの前記孔内において前記粒子材料または前記粒子材料化合物を含有する粒子を形成する工程と、
を有することを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項15】
(c)前記(b)工程の後、前記多孔質シリカを除去し、前記微細粒子を取り出す工程を有することを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項16】
前記(a2)工程は、
前記水の量が、前記アルコキシシランと前記水との化学量論比をアルコキシシラン:水=1:nとした場合、nを20以下とした量であり、
前記水のpHが、0〜2である
条件下において、前記加水分解反応により、前記界面活性剤のミセルを鋳型として、シリカを形成するものであることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項17】
前記界面活性剤は、疎水部の炭素数が2〜7であるカチオン性界面活性剤であることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項18】
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項19】
平均孔直径が0.5以上1.5nm以下である孔を有し、
金属、金属化合物および炭素のうちのいずれかを含有する粒子を、前記孔中に内包していることを特徴とする多孔質シリカ。
【請求項20】
前記粒子は、平均直径が、0.5以上1.0nm以下であることを特徴とする請求項19記載の多孔質シリカ。
【請求項21】
前記粒子は、酸化タングステンを含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項22】
前記粒子は、金を含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項23】
前記粒子は、バナジン酸ビスマスを含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項24】
前記粒子は、酸化銅を含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項25】
平均孔直径が0.5以上1.5nm以下である孔を有する多孔質シリカの前記孔中に内包された粒子であって、金属、金属化合物および炭素のうちのいずれかを含有することを特徴とする多孔質シリカ内包粒子。
【請求項26】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項27】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項28】
前記多孔質シリカはモノリス状であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項29】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項30】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項29記載の多孔質シリカ内包粒子の製造方法。
【請求項31】
前記液は、硝酸塩であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項32】
前記微細粒子は、銀(Au)の微細粒子を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項33】
前記微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の金属酸化物の微細粒子を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項34】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項35】
前記液の濃度は、0.5M以上であることを特徴とする請求項34記載の多孔質シリカ内包粒子の製造方法。
【請求項36】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属酸化物を含有する微細粒子を形成する工程と、
(d)前記(c)工程の後、前記金属酸化物を含有する微細粒子を還元することにより、前記金属の微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項37】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項38】
前記多孔質シリカはモノリス状であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項39】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項40】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項39記載の多孔質シリカ内包粒子の製造方法。
【請求項41】
前記(d)工程は、前記金属酸化物を含有する微細粒子と還元剤を接触させる工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項42】
前記液は、硝酸塩であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項43】
前記(c)工程の前記金属酸化物を含有する微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の金属酸化物の微細粒子を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項44】
前記(d)工程の前記金属の微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の微細粒子を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項45】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項46】
前記液の濃度は、0.5M以上であることを特徴とする請求項45記載の多孔質シリカ内包粒子の製造方法。
【請求項47】
前記(d)工程の後、前記金属の微細粒子を再酸化することにより、前記金属酸化物の微細粒子を形成する工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項48】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、前記多孔質シリカの孔内の前記液を還元することにより、前記金属の微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項49】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項50】
前記多孔質シリカはモノリス状であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項51】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項52】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項51記載の多孔質シリカ内包粒子の製造方法。
【請求項53】
前記(c)工程は、前記金属酸化物を含有する微細粒子と還元剤を接触させる工程を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項54】
前記液は、硝酸塩であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項55】
前記(c)工程の前記金属の微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の微細粒子を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項56】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径を制御することにより、前記微細粒子の発光波長を調整することを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項57】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項56記載の多孔質シリカ内包粒子の製造方法。
【請求項1】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項2】
前記金属化合物は、金属酸化物、金属硫化物または金属窒化物であることを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項3】
前記微細粒子の直径は、0.5nm以上1.0nm以下であることを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項4】
前記微細粒子は、金を含有し、前記液は、金化合物を含有することを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項5】
前記液は、塩化金酸水溶液を含有することを特徴とする請求項4記載の多孔質シリカ内包粒子の製造方法。
【請求項6】
前記微細粒子は、酸化タングステンを含有し、前記液は、タングステン化合物を含有することを特徴とする請求項1記載の多孔質シリカ内包粒子の製造方法。
【請求項7】
前記液は、過酸化タングステン酸水溶液を含有することを特徴とする請求項6記載の多孔質シリカ内包粒子の製造方法。
【請求項8】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項9】
前記液は、有機化合物を含有することを特徴とする請求項8記載の多孔質シリカ内包粒子の製造方法。
【請求項10】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと、ビスマスまたはビスマスを元素組成として有する化合物を含有する第1液と、バナジウムまたはバナジウムを元素組成として有する化合物を含有する第2液との混合液とを接触させ、前記多孔質シリカの孔内に前記混合液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内においてバナジン酸ビスマスを含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項11】
前記第1液は、Bi(NO3)3を含有し、前記第2液は、NH4VO3を含有することを特徴とする請求項10記載の多孔質シリカ内包粒子の製造方法。
【請求項12】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する気体とを接触させ、前記多孔質シリカの孔内に前記気体を導入し、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項13】
前記(b)工程は、前記気体の気相での化学反応により前記微細粒子を形成する工程であることを特徴とする請求項12記載の多孔質シリカ内包粒子の製造方法。
【請求項14】
(a)アルコキシシランの加水分解による多孔質シリカの製造工程であって、
(a1)界面活性剤およびアルコキシシランを混合し、混合液を形成する工程と、
(a2)前記混合液に水を添加することにより、前記アルコキシシランの加水分解反応を行わせる工程と、
(a3)前記(a2)工程の後、熱処理を施すことにより前記界面活性剤を除去する工程と、を有する多孔質シリカの製造工程と、
(b)前記(a)工程により製造された多孔質シリカの孔内において粒子を合成する工程であって、
前記多孔質シリカと粒子材料または前記粒子材料を元素組成として有する化合物を含有する液または気体とを接触させ、前記多孔質シリカの孔内に前記液または前記気体を導入させ、前記多孔質シリカの前記孔内において前記粒子材料または前記粒子材料化合物を含有する粒子を形成する工程と、
を有することを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項15】
(c)前記(b)工程の後、前記多孔質シリカを除去し、前記微細粒子を取り出す工程を有することを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項16】
前記(a2)工程は、
前記水の量が、前記アルコキシシランと前記水との化学量論比をアルコキシシラン:水=1:nとした場合、nを20以下とした量であり、
前記水のpHが、0〜2である
条件下において、前記加水分解反応により、前記界面活性剤のミセルを鋳型として、シリカを形成するものであることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項17】
前記界面活性剤は、疎水部の炭素数が2〜7であるカチオン性界面活性剤であることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項18】
前記多孔質シリカの平均孔直径は、0.5以上1.5nm以下であることを特徴とする請求項14記載の多孔質シリカ内包粒子の製造方法。
【請求項19】
平均孔直径が0.5以上1.5nm以下である孔を有し、
金属、金属化合物および炭素のうちのいずれかを含有する粒子を、前記孔中に内包していることを特徴とする多孔質シリカ。
【請求項20】
前記粒子は、平均直径が、0.5以上1.0nm以下であることを特徴とする請求項19記載の多孔質シリカ。
【請求項21】
前記粒子は、酸化タングステンを含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項22】
前記粒子は、金を含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項23】
前記粒子は、バナジン酸ビスマスを含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項24】
前記粒子は、酸化銅を含有することを特徴とする請求項19記載の多孔質シリカ。
【請求項25】
平均孔直径が0.5以上1.5nm以下である孔を有する多孔質シリカの前記孔中に内包された粒子であって、金属、金属化合物および炭素のうちのいずれかを含有することを特徴とする多孔質シリカ内包粒子。
【請求項26】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または前記金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属または前記金属化合物を含有する微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項27】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項28】
前記多孔質シリカはモノリス状であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項29】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項30】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項29記載の多孔質シリカ内包粒子の製造方法。
【請求項31】
前記液は、硝酸塩であることを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項32】
前記微細粒子は、銀(Au)の微細粒子を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項33】
前記微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の金属酸化物の微細粒子を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項34】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項26記載の多孔質シリカ内包粒子の製造方法。
【請求項35】
前記液の濃度は、0.5M以上であることを特徴とする請求項34記載の多孔質シリカ内包粒子の製造方法。
【請求項36】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において前記金属酸化物を含有する微細粒子を形成する工程と、
(d)前記(c)工程の後、前記金属酸化物を含有する微細粒子を還元することにより、前記金属の微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項37】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項38】
前記多孔質シリカはモノリス状であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項39】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項40】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項39記載の多孔質シリカ内包粒子の製造方法。
【請求項41】
前記(d)工程は、前記金属酸化物を含有する微細粒子と還元剤を接触させる工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項42】
前記液は、硝酸塩であることを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項43】
前記(c)工程の前記金属酸化物を含有する微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の金属酸化物の微細粒子を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項44】
前記(d)工程の前記金属の微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の微細粒子を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項45】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項46】
前記液の濃度は、0.5M以上であることを特徴とする請求項45記載の多孔質シリカ内包粒子の製造方法。
【請求項47】
前記(d)工程の後、前記金属の微細粒子を再酸化することにより、前記金属酸化物の微細粒子を形成する工程を有することを特徴とする請求項36記載の多孔質シリカ内包粒子の製造方法。
【請求項48】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと金属または金属を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、前記多孔質シリカの孔内の前記液を還元することにより、前記金属の微細粒子を形成する工程と、
を有する多孔質シリカ内包粒子の製造方法。
【請求項49】
前記(b)工程は、前記多孔質シリカを前記液に浸漬する工程であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項50】
前記多孔質シリカはモノリス状であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項51】
前記(b)工程の後、前記(c)工程の前に、
前記多孔質シリカを洗浄する工程を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項52】
前記洗浄は、アルコールを用いた洗浄であることを特徴とする請求項51記載の多孔質シリカ内包粒子の製造方法。
【請求項53】
前記(c)工程は、前記金属酸化物を含有する微細粒子と還元剤を接触させる工程を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項54】
前記液は、硝酸塩であることを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項55】
前記(c)工程の前記金属の微細粒子は、銅(Cu)、クロム(Cr)、マンガン(Mn)、鉄(Fe)、コバルト(Co)またはニッケル(Ni)の微細粒子を有することを特徴とする請求項48記載の多孔質シリカ内包粒子の製造方法。
【請求項56】
(a)多孔質シリカを準備する工程と、
(b)前記多孔質シリカと炭素または炭素を元素組成として有する化合物を含有する液とを接触させ、前記多孔質シリカの孔内に前記液を含浸させる工程と、
(c)前記(b)工程の後、熱処理を施すことにより、前記多孔質シリカの孔内において炭素または炭素化合物を含有する微細粒子を形成する工程と、
を有し、
前記多孔質シリカの平均孔直径を制御することにより、前記微細粒子の発光波長を調整することを特徴とする多孔質シリカ内包粒子の製造方法。
【請求項57】
前記(c)工程の熱処理は、マイクロ波照射工程を有することを特徴とする請求項56記載の多孔質シリカ内包粒子の製造方法。
【図1】

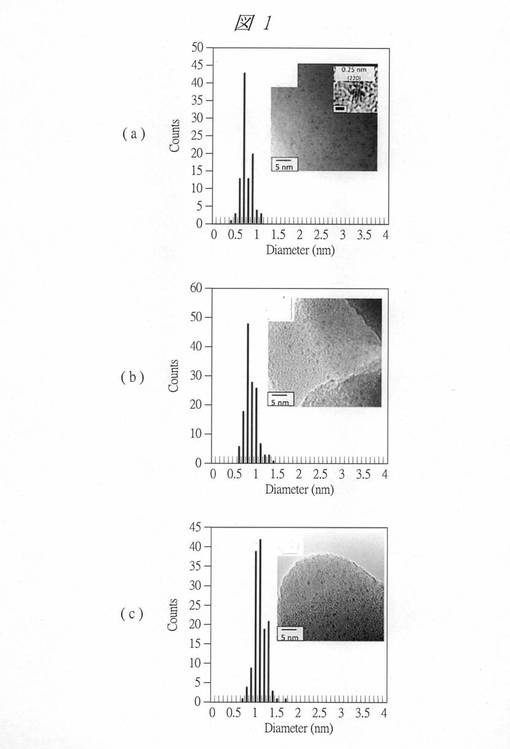
【図2】
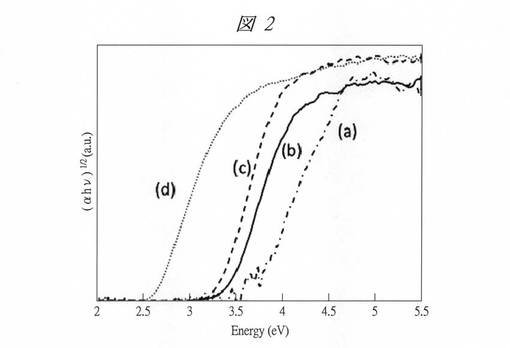
【図3】
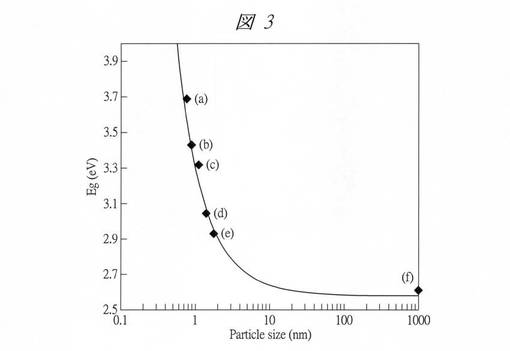
【図4】
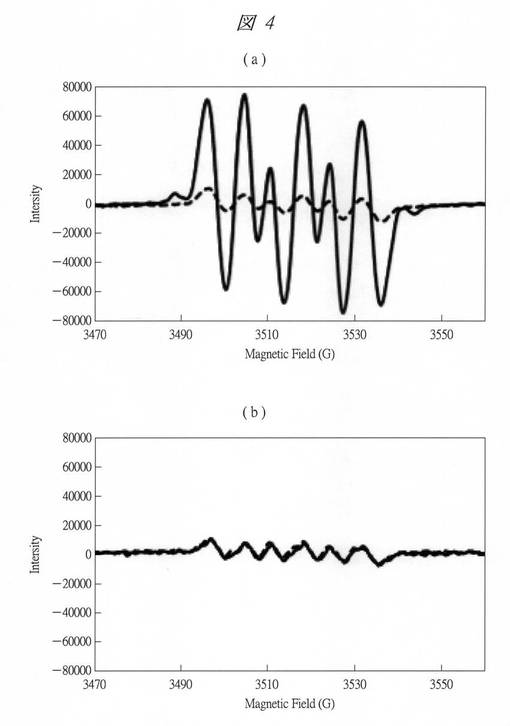
【図5】
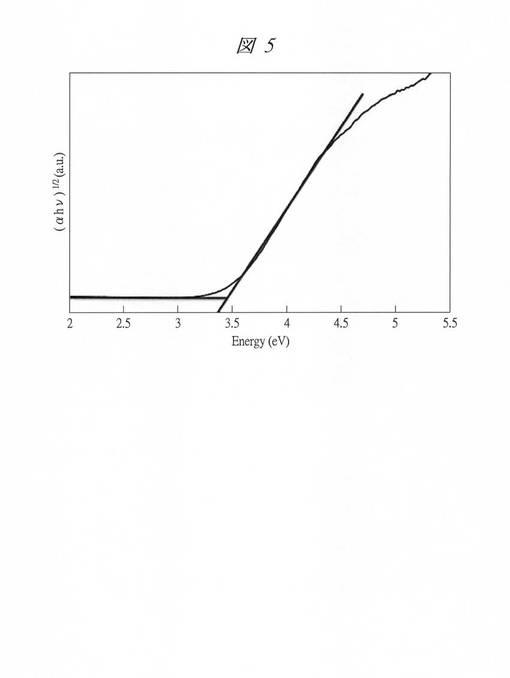
【図6】

【図7】
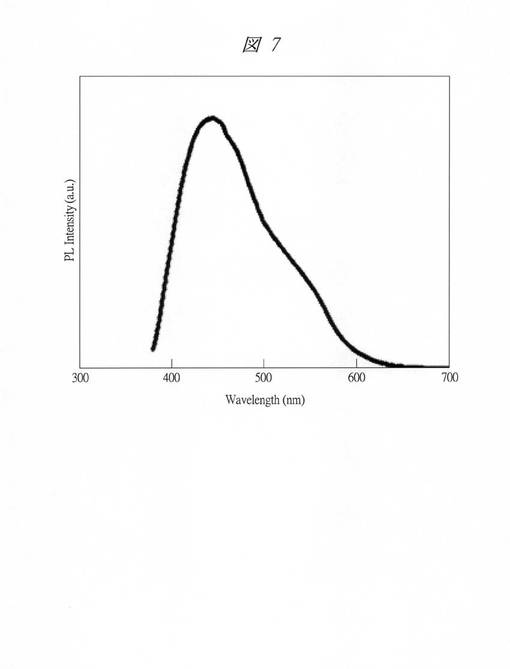
【図8】

【図9】
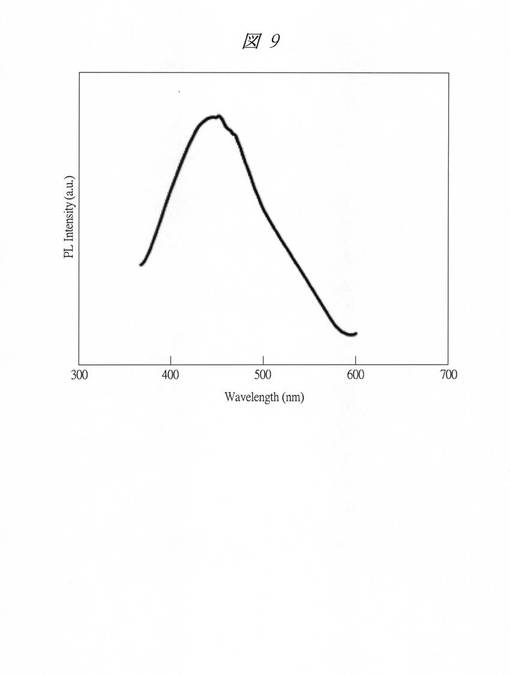
【図10】

【図11】
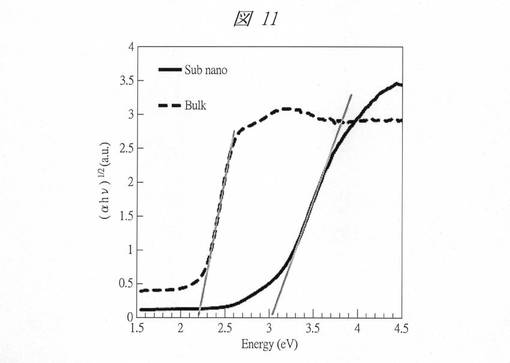
【図12】
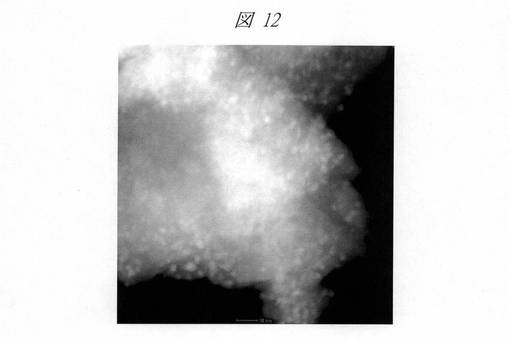
【図13】
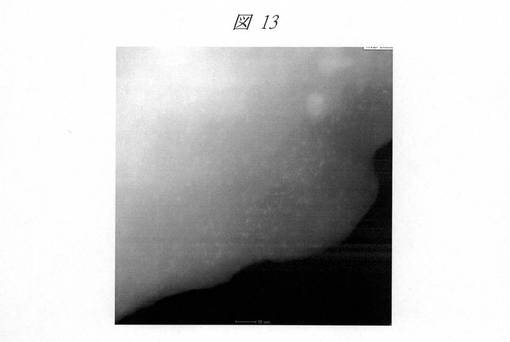
【図14】
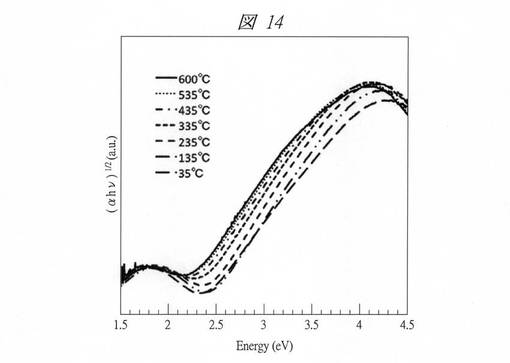
【図15】
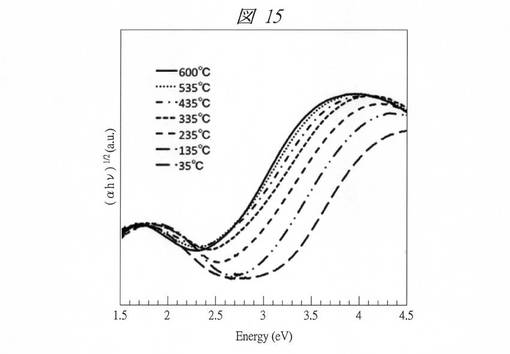
【図16】
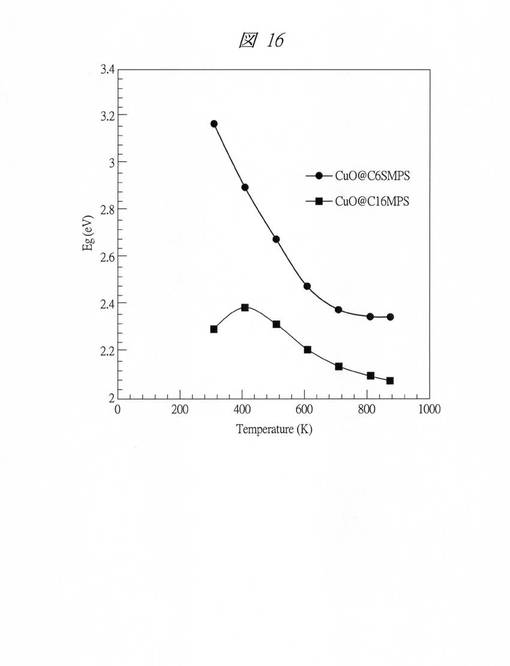
【図17】
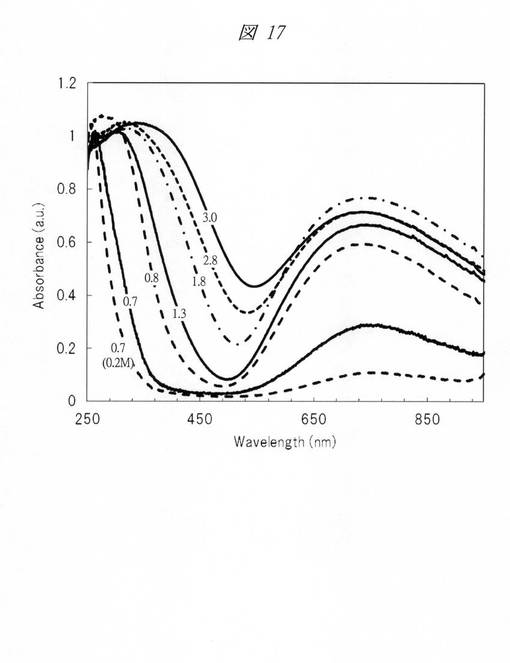
【図18】
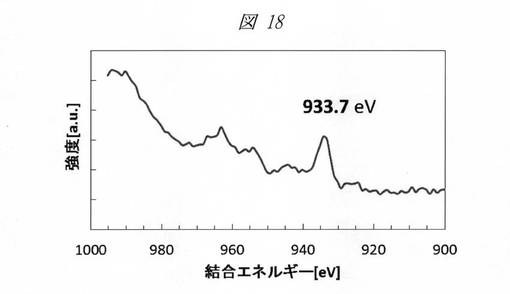
【図19】
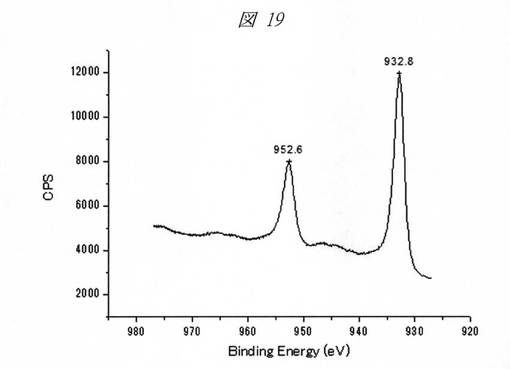
【図20】
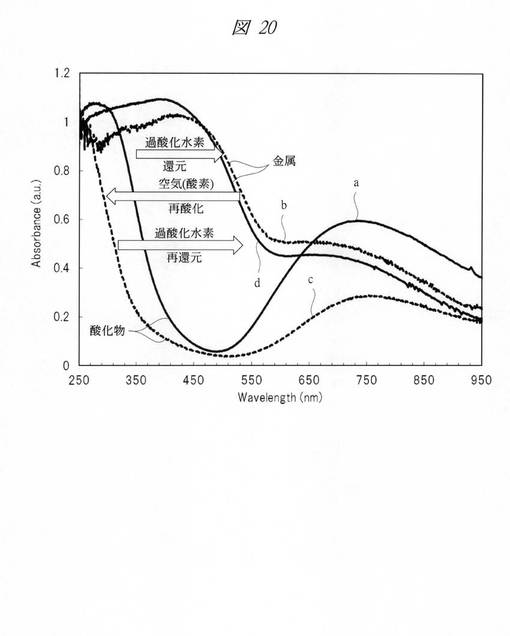
【図21】
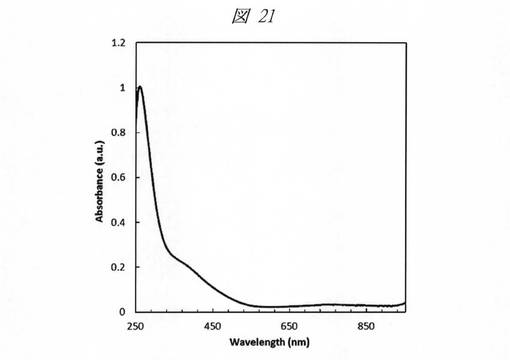
【図22】
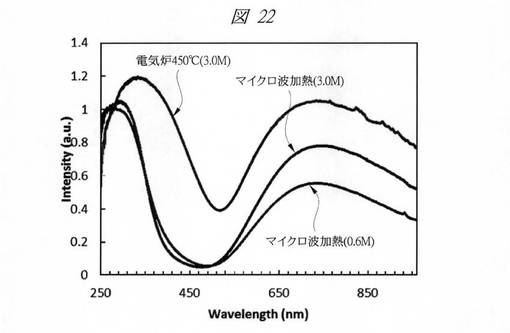
【図23】
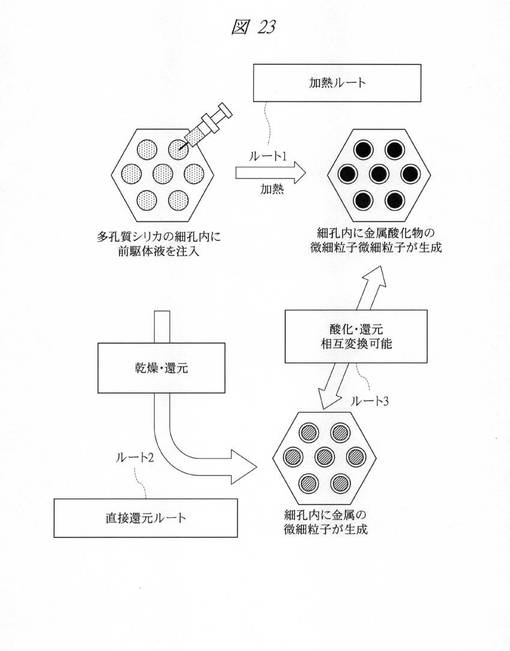
【図24】
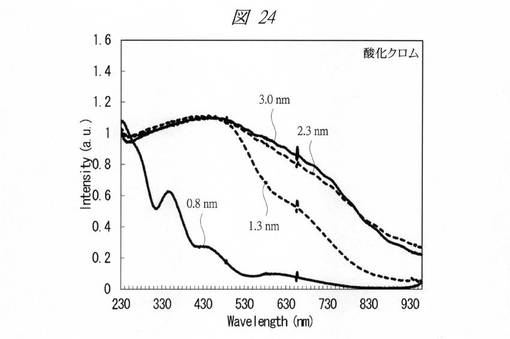
【図25】
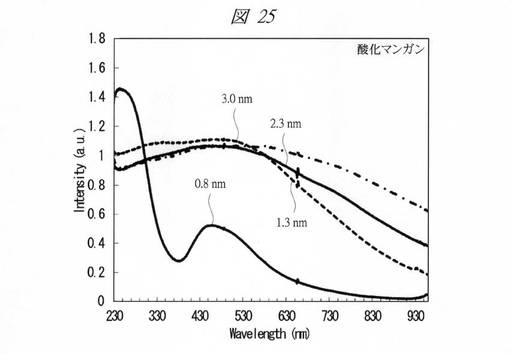
【図26】
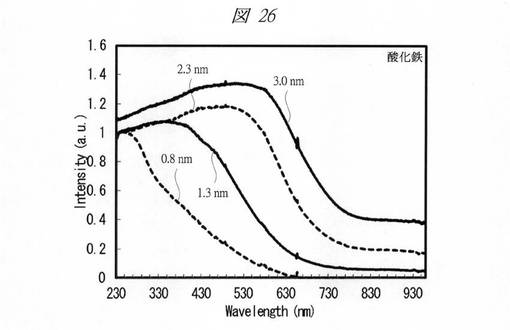
【図27】
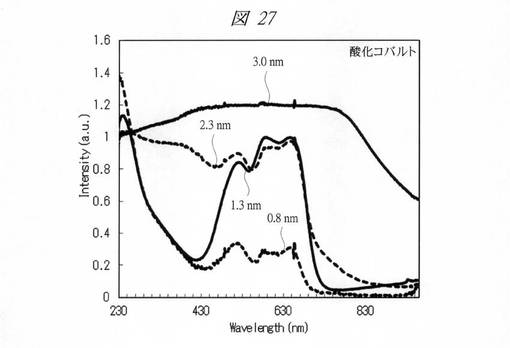
【図28】
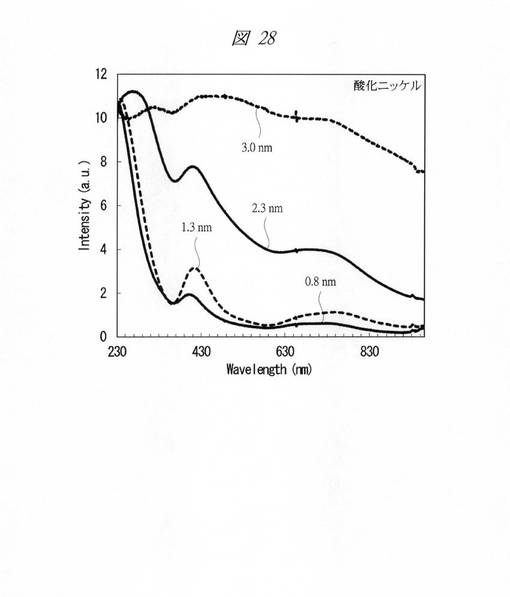
【図29】
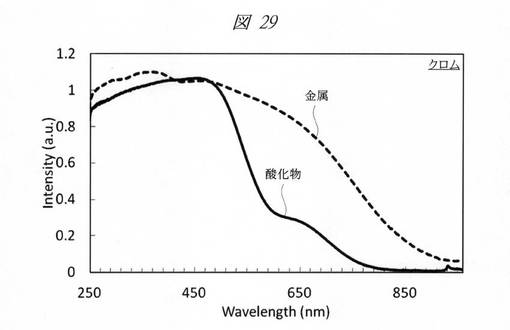
【図30】
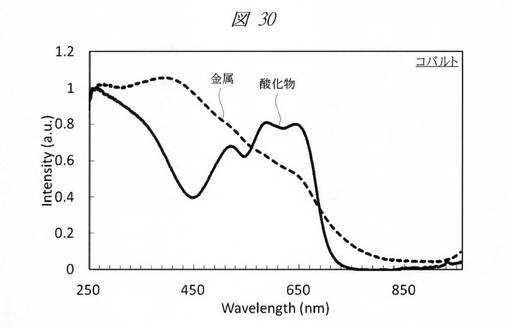
【図31】
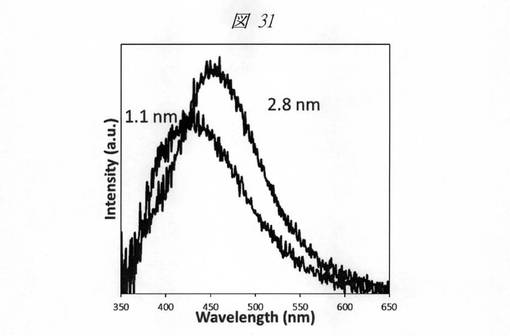
【図32】
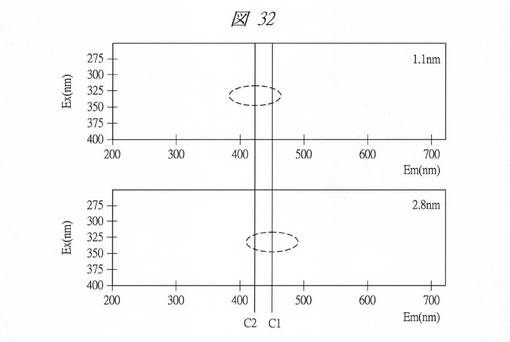
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【公開番号】特開2013−63895(P2013−63895A)
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願番号】特願2012−186879(P2012−186879)
【出願日】平成24年8月27日(2012.8.27)
【出願人】(506209422)地方独立行政法人 東京都立産業技術研究センター (134)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願日】平成24年8月27日(2012.8.27)
【出願人】(506209422)地方独立行政法人 東京都立産業技術研究センター (134)
【出願人】(899000079)学校法人慶應義塾 (742)
【Fターム(参考)】
[ Back to top ]