
多次元分析の計算方法およびシステム
少なくとも2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム(10、50、60)内の少なくとも1つの試料から得られたデータを分析する方法であって、2つの変数の関数として表される、少なくとも1つの試料を表すデータをそのシステムから得ること、連続するレベルを有するデータ・スタック(70、74、78、82、84)を形成することであって、各レベルが、少なくとも1つの試料を表す連続データを含むこと、データ・スタック内のすべてのデータのコンパイルを表すデータ配列(R)を形成すること、および、データ配列を一連の行列に分離することを含む方法。この方法に従って動作する化学分析システム、およびシステムにこの方法を実行させるコンピュータ可読プログラム・コードを有する媒体。
【発明の詳細な説明】
【技術分野】
【0001】
本願は、2003年4月28日出願の米国仮出願第60/466010号、米国仮出願第60/466011号、および米国仮出願第60/466012号からの優先権を主張するものであり、すべてその全体を本明細書に組み込む。本願は、その内容全体を参照により本明細書に組み込む、2003年10月20日出願の米国仮出願第10/689313号からの優先権も主張するものである。
【0002】
本発明は、化学分析システムに関する。より詳細には、本発明は、タンパク質、環境汚染物、石油化学化合物などの大きい有機分子を含む分子の複雑な混合物の分析に有用なシステム、それに使用される分析の方法、およびコンピュータまたはコンピュータと質量分析計の組合せにそのような分析を行わせるためにコンピュータ・コードが組み入れられたコンピュータ・プログラム製品に関する。さらに具体的には、本発明は、質量分析計部分を有するシステムに関する。
【背景技術】
【0003】
過去数年間のヒト・ゲノム解析競争は、ゲノミクスという名前の新しい化学の分野および産業を生み、ゲノミクスは、DNA配列を研究して、メッセンジャRNA(mRNA)での発現およびタンパク質のもとであるペプチドの後続のコーディングを介して遺伝病の原因である遺伝子および遺伝子突然変異を探す。この分野では、遺伝子が、多数の形の癌を含む多数の病気の根底にあるが、これらの遺伝子から翻訳されたタンパク質が、実際の生物学的機能を実行するものであることが明確に立証されている。したがって、これらのタンパク質およびその相互作用の同定および定量化は、病状の理解および新しい治療学の開発の鍵として働く。したがって、2000年夏にヒト・ゲノム・プロジェクトが成功裡に完了し約35,000種のヒト遺伝子が同定された後で、商業投資と学術研究のどちらにおいても遺伝子(ゲノミクス)からタンパク質(プロテオミクス)に急速にシフトしたことは、驚くに値しない。種ごとにより限定できる終点を有するゲノミクスとは異なって、プロテオミクスは、遺伝子発現レベル、環境要因、およびタンパク質間相互作用の変化がタンパク質変動に寄与する可能性があるので、はるかに制約がない。さらに、個人の遺伝子構造は、比較的安定しているが、タンパク質の発現は、さまざまな病状および他の要因に応じて、はるかに動的なものになる可能性がある。この「ポスト・ゲノミクス時代」における課題は、さまざまな生理学的条件の下での複雑な細胞経路、ネットワーク、および「モジュール」を理解する助けとなるように、組織、細胞、または他の生物試料内の有機体によって発現される複雑なタンパク質(すなわちプロテオーム)を分析することである。正常な状態および病気の状態の両方で発現するタンパク質の同定および定量化は、バイオマーカまたは標的タンパク質の発見でクリティカルな役割を演じる。
【0004】
プロテオミクスという急速に進展する分野が提示する課題は、試料調製、試料分離、イメージング、安定同位体標識(isotope labeling)から、質量スペクトル検出(mass spectral detection)まで、所持する非常に高機能の科学機器をもたらした。ますます高次元になる大量のデータ配列が、全世界の産業界と学究的世界の両方において、ゲノミクスおよびプロテオミクスの成果を獲得しようと競って日常的に生成されている。プロテオミクス研究に通常用いられるタンパク質の複雑さおよび(容易に数千種に達する)数の多さに起因して、複雑で時間がかかり骨の折れる物理的分離が、複合試料内の個々のタンパク質を同定し、時として定量化するために実行される。これらの物理的分離は、単一の試料の内容を完全に解明するのに通常要する日、週、さらには月は言うまでもなく、試料処理および情報追跡に対して大量の課題を生み出す。
【0005】
ヒト・ゲノムには約35,000種の遺伝子しかないが、一般集団について及び治療中
または他の病態にある個人についても研究できるヒト・プロテオームには、推定500,
000種から2,000,000種のタンパク質がある。たとえば細胞、血液、または尿から取られる通常の試料には、通常、大量の数千種に及ぶ異なるタンパク質が含まれる。過去10年間に、産業界は、試料に存在する多数のタンパク質を分析するために、複数のステージを含むプロセスを普及させてきた。このプロセスを、特筆すべき特徴と共に表1に要約する。
【0006】
【表1】

【0007】
a.単一の試料の分析を完了するのに、数日から数週間または数ヶ月を要する可能性がある。
b.巨大なハードウェア・システムは、600,000ドルから100万ドルのコスト
がかかり、かなりの運営コスト(人件費および消耗品)、保守コスト、およびそれと関連した研究室スペース・コストを伴う。
【0008】
c.これは、本質的に、それぞれが固体−液体−固体化学処理の別のサイクルを介して1つずつ分析される必要がある数百個から数千個の個々の固体スポットに1つの液体試料を分離する、複数の異なるロボットおよび少数の異なるタイプの機器を含む極端に時間のかかる複雑なプロセスである。
d.急速に変化する産業界のためにこれらの部分/ステップを一緒に統合することは、小さい課題ではなく、その結果として、これらのステップのすべてを完全に統合し、自動化する市販システムはまだない。このため、このプロセスは、人間の誤りならびに機械の誤差を伴う。
【0009】
e.このプロセスは、途中のすべてのステップからの試料およびデータの追跡も要求するが、これは、現在の情報科学にとってさえ小さい課題ではない。
f.完全な試料およびデータを追跡する情報科学システムを有する完全に自動化されたプロセスについても、これらのデータをどのように管理し、ナビゲートし、そして最も重要なこととして、分析しなければならないかは、明らかでない。
【0010】
g.プロテオミクスのこの早期の段階では、多数の研究者が、タンパク質の定性的同定に満足している。しかし、プロテオミクスの究極の目的は、同定と定量化の両方であり、これによって、卵巣癌診断の血液試料からのタンパク質プロフィールの使用に関する最近の刊行物(Pertricoin,E.F.III他、Lancet、Vol.359、573〜77ページ、2002年)から生じた強い関心によって明示されているように、創薬のためのバイオマーカ同定の領域だけではなく、臨床診断に関する刺激的な応用例の門戸が開かれることになる。現在のプロセスは、タンパク質消失、試料汚染、またはゲル溶解性の欠如に起因して、定量分析に簡単に適合させることができないが、ICAT(同位体コードアフィニティータグ)すなわち、2つの異なる試料供給源からのタンパク質またはタンパク質消化物が同位体原子の対によってラベル付けされ、その後、1つの質量分析法分析で混合される、定量化に対する一般的な手法(Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年)などの複雑な化学プロセスの使用による定量的プロテオミクスの試みが行われてきた。
【0011】
同位体コードアフィニティータグ(ICAT)は、米国カリフォルニア州フォスタ・シティのApplied Biosystems社が最近導入した手法の市販版である。この技法では、2つの異なる細胞プールからのタンパク質が、通常の試薬(軽)および重水素置換された試薬(重)を用いてラベル付けされ、1つの混合物に組み合わされる。トリプシン消化の後に、組み合わされた消化混合物が、ビオチン−アフィニティ・クロマトグラフィによる分離を受けて、システインを含むペプチド混合物がもたらされる。この混合物が、さらに、逆相HPLCによって分離され、データ依存質量分析法およびそれに続くデータベース検索によって分析される。
【0012】
この方法では、複雑なペプチド混合物がシステインを含むペプチド混合物に大幅に単純化され、SEQUESTデータベース検索によるタンパク質同定と重ペプチドに対する軽ペプチドの比率による定量化が同時に可能になる。LC/LC/MS/MSと同様、ICATにより、不溶性問題も回避される。というのは、両方の技法が、分離および分析の前に、タンパク質混合物全体をペプチド断片(peptide fragment)に消化するからである。
【0013】
ICAT技法は、非常に強力であるが、ラベル付けおよび事前分離処理に複数ステップ・プロセスを必要とし、少量タンパク質の消失と、追加の試薬コストをもたらし、さらに、既に低速のプロテオーム解析のスループットを下げる。システインを含むペプチドだけが分析されるので、配列カバレッジ(sequence coverage)は、通常、ICATでは非常に低い。通常のLC/MS/MS実験でそうであるように、タンパク質同定は、うまくいけばシグネチャ・ペプチド(signature peptide)の限られた回数のMS/MS分析を介して達成され、比率定量化に関する唯一の、多くとも少数の標識ペプチドをもたらす。
【0014】
タンデム質量分析と適合された液体クロマトグラフィ(LC/MS/MS)が、プロテイン・シーケンシングに関するえりぬきの方法になってきた(Yates Jr.他、Anal.Chem.67、1426〜1436ページ、1995年)。この方法には、タンパク質の消化、タンパク質消化物から生成されたペプチド混合物のLC分離、得られたペプチドのMS/MS分析、およびタンパク質同定のためのデータベース検索を含む少しのプロセスが含まれる。LC/MS/MSを用いてタンパク質を効果的に同定するための鍵は、データベース検索中の信頼性のあるマッチングを可能にするために、できる限り多くの良い質のMS/MSスペクトルを生じさせることである。これは、四重極機器またはイオン・トラップ機器でのデータ依存走査技法によって達成される。この技法を用いると、質量分析計が、全走査MSスペクトル内で最多のイオンの強度および信号対雑音比を検査し、最多イオンの強度および信号対雑音比が事前に設定された閾値を超えるときMS/MS実験を実行する。通常、3つの最も豊富なイオンが、配列情報を最大にし、必要な時間を最小にするためにプロダクト・イオン走査について選択される。というのも、MS/MS実験に関する3つを超えるイオンの選択は、ことによるとLCから質量分析計に現在溶出している他の資格のあるペプチドの消失をもたらすからである。
【0015】
タンパク質の同定に関するLC/MS/MSの成功は、主に、その多数の顕著な分析特性に起因する。第1に、これは、優れた再現性を有する非常に堅牢な技法である。これは、タンパク質同定に関する高スループットLC/MS/MS分析について信頼性があることが実証されている。第2に、ナノスプレイ・イオン化を使用するとき、この技法は、サブフェンタモル(sub−fentamole)レベルでペプチドの高品質MS/MSスペクトルを生む。第3に、MS/MSスペクトルは、C末端イオンとN末端イオンの両方の配列情報を担持する。この貴重な情報は、タンパク質の同定だけではなく、どの翻訳後修飾(PTM)がタンパク質に生じたかと、どのアミノ酸残基でPTMが起こったかをピンポイントで指摘するのに使用することができる。
【0016】
有機体、細胞系、または組織タイプからの総タンパク質消化物について、LC/MS/MSだけでは、タンパク質の同定に十分な数の良い質のMS/MSスペクトルを生成するのは不十分である。したがって、LC/MS/MSは、通常、2次元電気泳動(2DE)によって分離されたタンパク質などの単一のタンパク質またはタンパク質の単純な混合物の消化を分析するのに使用され、総分析時間に最少の数日を追加し、機器コストと、試料処理の複雑さと、試料追跡のための情報科学の必要を増大させる。全MS走査には試料に関する豊富な情報が含まれる可能性があり、通常は含んでいるが、現在のLC/MS/MS法は、全MS走査内のごく少数のイオンだけについて提供できるMS/MS分析に依拠する。さらに、LC/MS/MSで使用されるエレクトロスプレイ・イオン化(ESI)は、試料からの塩濃度に対して許容範囲が少なく、厳重な試料クリーン・アップ・ステップを必要とする。
【0017】
有機体、細胞系、および組織タイプでのタンパク質の同定は、これらの系の非常に多くのタンパク質(数千種から数万種と推定される)に起因して、極端にむずかしい作業である。LC/LC/MS/MS技術の開発(Link,A.J.他、Nat.Biotechnol.17、676〜682ページ、1999年、およびWashburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年)は、LC分離の1つの特別な次元を追い求めることによってこの課題を処理する試みの1つである。この手法は、タンパク質混合物全体の消化から始まり、強陽イオン交換(SCX)LCを使用して、塩濃度の階段状の勾配によってタンパク質消化物を分離する。この分離は、極端に複雑なタンパク質混合物を比較的単純化された混合物に変えるのに、通常は10〜20ステップを要する。SCXカラムから溶出した混合物は、さらに、逆相LCに導入され、その後、質量分析法によって分析される。この方法は、イーストおよびヒト骨髄性白血病細胞のミクロゾームからの大量のタンパク質を同定することが実証されている。
【0018】
この技法の明白な利益の1つは、2DEでの不溶性問題が回避されることである。というのは、すべてのタンパク質が、ペプチド断片に消化され、このペプチド断片は、通常、タンパク質よりはるかに溶けやすいからである。その結果、LC/LC/MS/MSを用いると、より多くのタンパク質を検出でき、より広いダイナミック・レンジを達成することができる。もう1つの利益は、クロマトグラフ分離が、広範囲の2D LC分離を介してはなはだしく増え、その結果、より完全で信頼性のあるタンパク質同定のために、より高品質のペプチドのMS/MSスペクトルを生成できることである。第3の利益は、この手法が、潜在的に高いスループットのプロテオーム解析のために現在のLC/MSシステムの枠組みの中でたやすく自動化されることである。
【0019】
しかし、LC/LC/MS/MSでの広範囲の2D LC分離は、完了に1〜2日を要する可能性がある。さらに、この技法だけでは、同定されるタンパク質の定量的情報を提供することができず、ICATなどの定量的方式は、試料損失および余分の複雑さと共に余分の時間および労力を必要とする。広範囲の2D LC分離にかかわらず、まだ、MS/MSデータ収集と連続LC溶出の間の時間の制約に起因してMS/MS実験について選択されないかなりの量のペプチド・イオンがあり、配列カバレッジが低くなる(25%カバレッジが、既に非常に良いと考えられる)。LCトレースを後のMS/MS分析用の固体サポートに堆積させる最近の開発は、潜在的に、限られたMS/MSカバレッジの問題に対処できるが、かなり多くの試料処理およびタンパク質損失をもたらし、さらに、試料追跡作業および情報管理作業が複雑になる。
【0020】
マトリックス介助レーザーデソープションイオン化法(MALDI)は、集束レーザー・ビームを使用して、伝導性試料プレート上のマトリックス化合物と共結晶化されているターゲット試料を照射する。イオン化された分子は、パルス化技法としてそれらが共有する特性に起因して、通常、飛行時間型(TOF)質量分析計によって検出される。
【0021】
MALDI/TOFは、その優れた速度、高い感度、広い質量範囲、高い分解能、および汚染物への寛大さのゆえに、2DE分離された無傷のタンパク質を検出するのに一般的に使用されている。遅延抽出および反射型イオン光学系の機能を有するMALDI/TOFは、1〜10ppmの印象的な質量精度と、ペプチドの正確な分析に関して10000〜15000のm/Δmを有する質量分解能を達成することができる。しかし、MALDI/TOFにMS/MS機能がないことが、プロテオミクス応用例での使用に関する主要な制限の1つである。MALDI/TOFでのポストソース分解(PSD)は、ペプチドに関する配列様のMS/MS情報を生成するが、PSDの動作は、しばしば、三連四重極質量分析計またはイオン・トラップ質量分析計の動作ほど堅牢でない。さらに、PSDデータ収集は、ペプチド依存になる可能性があるので、自動化が困難である。
【0022】
新たに開発されたMALDI TOF/TOFシステム(Rejtar,T.他、J.Proteomr.Res.1(2)、171〜179ページ、2002年)は、多くの魅力的な特徴を与える。このシステムは、2つのTOFおよび1つの衝突セルからなり、タンデム四重極システムの構成に似ている。第1TOFは、断片イオンを生成するためにセル内で衝突誘起解離(CID)を受ける前駆イオンを選択するのに使用される。その後、断片イオンが、第2TOFによって検出される。魅力的な特徴の1つは、TOF/TOFが、必要なだけ何度でもデータ依存MS/MS実験を実行できることであり、これに対して、通常のLC/MS/MSシステムは、実験のためにほんの僅かの豊富なイオンだけを選択する。この独自の開発によって、TOF/TOFで産業スケールのプロテオーム解析を実行することが可能になった。提案された解決策は、2D LC実験から分画(fraction)を収集し、MS/MS用のMALDIプレート上に分画をスポッティングすることである。その結果、より多くのMS/MSスペクトルを、データベース検索によるより信頼できるタンパク質同定のために獲得することができる。というのは、TOF/TOF内の高エネルギCIDによって生成されるMS/MSスペクトルの質が、PSDスペクトルよりはるかに良いからである。
【0023】
この手法の主要な短所は、機器の高いコスト(750,000ドル)、長い2D分離、LC分画に関する試料処理の複雑さ、MALDIのための面倒な試料調製プロセス、MALDIを用いる定量化に固有のむずかしさ、ならびにデータおよび試料追跡に関する膨大な情報科学の課題である。LC分離および必要な試料調製時間に起因して、1試料内の数百種のタンパク質の分析は、少なくとも2日を要する。
【0024】
フーリエ変換イオンサイクロトロン共鳴型(FTICR)MSは、高い感度、高い質量分解能、広い質量範囲、および高い質量精度を提供できる強力な技法である。最近、LCと結合されたFTICR/MSが、精密質量タグ(AMT)を介するプロテオーム解析に関する印象的な能力を示した(Smith,R.D.他、Proteomics、2、513〜523ページ、2002年)。AMTは、タンパク質を排他的に同定するのに使用できる、ペプチドの正確なm/z値である。AMT手法を使用することによって、単一LC/FTICR−MS分析が、潜在的に、1ppmより良い質量精度で105種を超えるタンパク質を同定できることが実証された。それでも、AMT単独では、ペプチドのアミノ酸残基特異的翻訳後修飾を正確に指摘するのに不十分である可能性がある。さらに、この機器は、75万ドル以上のコストと高い保守要件を有して非常に高価である。
【0025】
プロテイン・アレイおよびプロテイン・チップは、設計概念において遺伝子発現プロファイリングに使用されるオリゴヌクレオチドチップに似た、新生の技術である(Issaq,H.J.他、Biochem Biophys Res Commun.292(3)、587〜592ページ、2002年)。プロテイン・アレイは、注目のタンパク質との特定の相互作用のために化学的に(陽イオン、陰イオン、疎水性、親水性など)または生化学的に(抗体、受容体、DNAなど)処理された表面を含むプロテイン・チップからなる。これらの技術は、親和性化学作用によって提供される特異性およびMALDI/TOFの高い感度を利用し、タンパク質の高スループット検出を提供する。通常のプロテイン・アレイ実験では、多数のタンパク質試料を、特定の表面化学作用を用いて処理されたチップのアレイに同時に適用することができる。望ましくない化学的バックグラウンドおよび生物分子バックグラウンドを洗い落とすことによって、注目のタンパク質が、アフィニティ・キャプチャリング(affinity capturing)に起因してチップにドッキングし、したがって「純化」される。MALDI−TOFによる個々のチップのさらなる分析が、試料内のタンパク質プロフィールをもたらす。これらの技術は、タンパク質間相互作用の調査に理想的である。というのは、タンパク質を親和性試薬として使用して表面を処理して、他の特定のタンパク質との相互作用を監視できるからである。この技術のもう1つの有用な応用例は、病気診断の潜在的ツールとして、正常な組織の試料と病気の組織の試料の間の比較パターンを生成することである。
【0026】
関係する複雑な表面化学作用と、変性、折り畳み、および溶解性の問題など、タンパク質または他のタンパク質様結合剤によって追加される複雑化とに起因して、プロテイン・アレイおよびプロテイン・チップは、遺伝子チップまたは遺伝子発現アレイほど広い応用分野を有しないと予想される。
【0027】
したがって、過去100年間に、MS計測器に関する長足の進歩が見られ、高スループット、高分解能、および高感度動作のために多数の異なるタイプの機器が設計され、作られてきた。計測器は、ほとんどの市販MSシステムで単一イオン検出をおおむね達成できる段階まで開発され、単位質量分解能は、異なる同位元素から得られるイオン・フラグメントの観察を可能にしている。ハードウェアの高度化と全く対照的に、現代のMS計測器によって生成された大量のMSデータを組織的かつ効果的に分析することに、ほとんど何も行われてこなかった。
【0028】
通常の質量分析計では、ユーザは、通常、注目の質量スペクトルm/z範囲をカバーする複数の断片イオンを有する標準材料を要求されるか、これを供給される。ベースライン効果、同位元素干渉、質量分解能、および質量に対する分解能依存を受けて、少数のイオン・フラグメントのピーク位置は、ピーク頂部での低次多項式あてはめを介して重心またはピーク最大値のいずれかに関して決定される。次に、このピーク位置が、質量(m/z)軸を較正するための1次または高次のいずれかの多項式フィットを介して、これらのイオンの既知のピーク位置にあてはめられる。
【0029】
質量軸較正の後に、通常の質量スペクトル・データ・トレースがピーク分析を受け、ピーク(イオン)が同定されることになる。このピーク検出ルーチンは、ピークの肩、データ・トレース中の雑音、化学的バックグラウンドまたは汚染に起因するベースライン、同位元素ピーク干渉などが考慮される、非常に経験的で複雑な処理である。
【0030】
同定されたピークについて、通常、セントロイディング(centroiding)と称する処理を適用して、積分されたピーク面積およびピーク位置を計算する。上記で概要を示した多数の干渉する要因ならびに他のピークおよび/またはベースラインが存在する中でのピーク面積の決定に特有のむずかしさに起因して、これは、セントロイディング品質の客観的尺度なしで同位元素ピークを出現させるか消滅させる可能性がある多数の調整可能なパラメータに悩まされる処理である。
【0031】
したがって、そのみかけの高度化にかかわりなく、現在の手法は、複数の著しい短所を有する。これには、次が含まれる。
【0032】
質量精度の欠如。現在使用されている質量較正は、通常、単位質量分解能(重要な同位元素ピークの存在または不在を視覚化する能力)を有する従来のMSシステムでの質量決定精度において、0.1amu(m/z単位)より良いものを提供しない。より高い質量精度を達成し、タンパク質同定用のペプチド・マッチングなどの分子フィンガープリント法での曖昧さを減らすためには、かなりコストが高い四重極TOF(qTOF)またはFT ICR MSなどのより高分解能のMSシステムに切り替えなければならない。
【0033】
大きいピーク積分誤差。質量スペクトル・ピーク形状、その可変性、同位元素ピーク、ベースラインおよび他のバックグラウンド信号、ならびにランダム・ノイズの寄与に起因して、現在のピーク面積積分は、強い質量スペクトル・ピークおよび弱いスペクトル・ピークの両方に関して大きい誤差(系統誤差およびランダム誤差の両方)を有する。
【0034】
同位元素ピークに関する問題。現在の手法は、単位質量分解能を有する従来のMSシステムで通常は部分的にオーバーラップした質量スペクトル・ピークを発するさまざまな同位元素からの寄与を分離する良い方法を有しない。経験的手法では、隣接同位元素ピークからの寄与の無視またはその過大評価のいずれかが使用され、優位を占める同位元素ピークに関する誤差と、より弱い同位元素ピークに関する大きい偏りまたはより弱いピークの完全な無視とがもたらされた。多重荷電のイオンがかかわるとき、隣接同位元素ピーク間の質量単位の減らされた分離に起因して、状況はさらに悪くなる。
【0035】
非線形動作。現在の手法では、各ステージ中に多数の経験的に調整可能なパラメータを有する、複数ステージのばらばらな処理が使用される。系統誤差(偏り)は、各ステージで生成され、制御されない予測不能で非線形な形で後のステージに伝搬され、アルゴリズムがデータ処理品質および信頼性の尺度として意味のある統計を報告することを不可能にする。
【0036】
支配的な系統誤差。産業プロセス制御および環境監視からタンパク質同定またはバイオマーカ発見に及ぶほとんどのMS応用例で、機器の感度または検出限度は、常に焦点であり、多くの機器システムで、測定誤差または信号への雑音寄与を最小にするために大いに努力がされてきた。残念ながら、現在使用されているピーク処理手法は、生データのランダム・ノイズよりずっと大きい系統誤差の供給源を作り、したがって、機器の感度または信頼性の制限要因になっている。
【0037】
数学的矛盾および統計的矛盾。現在使用されている多くの経験的手法は、質量スペクトル・ピーク処理全体を、数学的にも統計的にも矛盾したものにしている。ピーク処理結果は、ランダム・ノイズがないわずかに異なるデータまたはわずかに異なる雑音を伴う同一の合成データに対して、劇的に変化する可能性がある。言い換えると、ピーク処理の結果は、堅牢でなく、特定の実験またはデータ収集に依存して不安定になる可能性がある。
【0038】
機器間変動。通常、機械的許容範囲、電磁許容範囲、または環境の許容範囲の変動に起因して、異なるMS機器からの生の質量スペクトル・データを直接に比較することは困難であった。生データに対して適用される現在のアド・ホックなピーク処理を用いると、異なるMS機器からの結果を定量的に比較することのむずかしさが増すだけである。その一方で、異なる機器または異なるタイプの機器からの生質量スペクトル・データを直接に比較するかピーク処理結果を比較する必要が、不純物検出または確立されたMSライブラリの検索を介するタンパク質同定のためにますます高まってきた。
【0039】
2次機器は、各試料のデータの行列を生成し、データ行列が正しく構成されているならば、1次機器より高い分析能力を有することができる。最も広範囲で使用されているプロテオミクス機器であるLC/MSは、通常、現在達成されているものより潜在的にはるかに高い分析能力が可能な2次機器の典型例である。他の2次プロテオミクス機器に、単一UV波長検出を用いるLC/LC、MALDI−TOF MS検出を用いる1Dゲル、MALDI MS検出を用いる1Dプロテイン・アレイなどが含まれる。
【0040】
2次元ゲル電気泳動(2Dゲル)は、細胞または尿などの複雑な生物試料でのタンパク質の分離に広く使用されてきた。通常、タンパク質によって形成されるスポットは、可視イメージング・システムを用いる簡単な同定のために、銀を用いて着色される。これらのスポットは、その後、除去され、酵素を用いて溶解/消化され、MALDIターゲットに移され、乾燥させてMALDI飛行時間型質量分析計を使用してペプチド・シグネチャについて分析される。
【0041】
このプロセスから、次の複数の複雑化が生じる。
1.タンパク質スポットは、特に分離パラメータ(電荷の場合にpI、分子量の場合にMW)の両端で、単一のタンパク質だけを含むことが保証されない。これによって、通常、ペプチド検索が、不可能ではないにしてもむずかしくなる。除去されたスポットごとに追加の液体クロマトグラフィ分離が必要になる可能性があり、分析がさらに低速になる。
【0042】
2.液相から固相(ゲル上の)へ、液相へ戻し(消化のため)、そして最後に再び固相へ(MALDI TOF分析のため)移す生物試料の変換は、誤り、キャリーオーバー、および汚染を受けやすい非常に面倒なプロセスである。
【0043】
3.用いられる試料変換プロセスと、サンプリングおよびイオン化でのMALDI−TOF再生不可能性の事実に起因して、この分析は、定性的であるのみで定量的でないと広く認められてきた。
【0044】
したがって、1次分析および0次分析に対するはなはだしい潜在的な利益および明瞭な利益にもかかわらず、2次機器および2次分析は、これまでは、商品化のきざしがない、試料が少数の合成検体からなる学術研究に制限されてきた。この手法がその巨大な可能性に達するために超えなければならない複数の障壁がある。これには、次が含まれる。
【0045】
a.2次タンパク質分析では、現在事実上すべてのMS応用例で使用されている重心データではなく、生プロフィールMS走査を使用することがさらに重要である。双一次データ構造を維持するために、LCから溶出した特定のイオンの連続MS走査が、同一の質量スペクトル・ピーク形状(明らかに、異なるピーク高さでの)すなわち、セントロイディングおよびデアイソトーピング(de−isotoping)(すべての同位元素ピークを1つの積分された面積に合計すること)によって破壊されるクリティカルな2次構造を有する必要がある。データのセントロイディングからのスティック(stick)は、同一イオンの連続MS走査からの異なる質量位置(0.5amu誤差まで)に現れる。
【0046】
b.より高次の機器および分析分析は、より堅牢な機器および測定プロセスを必要とし、次元のうちの1または2におけるシフトなどのアーチファクトは、学究的世界での最近の進歩(Bro,R.他、J.Chemometrics 13、295ページ、1999年)にもかかわらず、分析の定量的結果および定性的結果さえ深刻に損なう可能性がある(Wang,Y.他、Anal.Chem.63、2750ページ、1991年、Wang,Y.他、Anal.Chem.,65、1174ページ、1993年、およびKiers,H.A.L.他、J.Chemometrics 13、275ページ、1999年)。非線形性または非双一次性などの他のアーチファクトも、複雑化につながる可能性がある(Wang,Y.他、J.Chemometrics、7、439ページ、1993年)。2次プロテオミクス・データの双一次性を維持するために、標準化およびアルゴリズムの訂正を開発する必要がある。
【0047】
c.四重極MSなどの多数のMS機器で、質量スペクトル走査時間は、タンパク質またはペプチドの溶出時間と比較して無視できない。したがって、GC/MSについて報告されたものに似て(Stein,S.E.他、J.Am.Soc.Mass Spectrom.5、859ページ、1994年)、ある質量スペクトル走査で測定されたイオンが、LC溶出中の異なる時点から得られるという重要なスキューが存在する。
【0048】
したがって、プロテオミクス研究がなるであろうものと現在の姿の間には大きいギャップが存在する。
【特許文献1】米国仮出願第60/466010号
【特許文献2】米国仮出願第60/466011号
【特許文献3】米国仮出願第60/466012号
【特許文献4】米国仮出願第10/689313号
【非特許文献1】Pertricoin,E.F.III他、Lancet、Vol.359、573〜77ページ、2002年
【非特許文献2】Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年
【非特許文献3】Yates Jr.他、Anal.Chem.67、1426〜1436ページ、1995年
【非特許文献4】Link,A.J.他、Nat.Biotechnol.17、676〜682ページ、1999年
【非特許文献5】Washburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年
【非特許文献6】Rejtar,T.他、J.Proteomr.Res.1(2)、171〜179ページ、2002年
【非特許文献7】Smith,R.D.他、Proteomics、2、513〜523ページ、2002年
【非特許文献8】Issaq,H.J.他、Biochem Biophys Res Commun.292(3)、587〜592ページ、2002年
【非特許文献9】Bro,R.他、J.Chemometrics 13、295ページ、1999年
【非特許文献10】Wang,Y.他、Anal.Chem.63、2750ページ、1991年
【非特許文献11】Wang,Y.他、Anal.Chem.,65、1174ページ1993年
【非特許文献12】Kiers,H.A.L.他、J.Chemometrics 13、275ページ、1999年
【非特許文献13】Wang,Y.他、J.Chemometrics、7、439ページ、1993年
【非特許文献14】Stein,S.E.他、J.Am.Soc.Mass Spectrom.5、859ページ、1994年
【非特許文献15】Hannesh,S.M.、Electrophoresis 21、1202〜1209ページ、2000年
【非特許文献16】Sanchez,E.他、J.Chemometrics 4、29ページ、1990年
【非特許文献17】Carroll,J.他、Psychometrika 3、45ページ、1980年
【非特許文献18】Bezemer,E.他、Anal.Chem.73、4403ページ、2001年
【非特許文献19】http://www.matrixscience.com
【非特許文献20】http://us.expasy.org/sprot/
【発明の開示】
【発明が解決しようとする課題】
【0049】
本発明の目的は、上述の欠点を克服する、質量分析計を含めることができる化学分析システムと、化学分析システムを動作させる方法を提供することである。
【0050】
本発明のもう1つの目的は、質量分析計を有する化学分析システムを含む化学分析システムに、本発明による方法を実行させるコンピュータ可読プログラム・コードを有する記憶媒体を提供することである。
【課題を解決するための手段】
【0051】
上記および他の目的は、本発明の第1の態様によれば、タンパク質スポット・オーバーラップの存在する状態で質量分析計を使用することなく、無傷のタンパク質から獲得された2Dゲル・イメージング・データを使用して、定性分析および定量分析の両方を実行することによって達成される。さらに、本発明は、より広い母集団範囲にわたって(病気と健康)、同一の母集団のある時間期間にわたって(病気の進行)、および異なる治療方法にまたがって(潜在的な治療に応答して)などのいずれかで収集された多数の異なる試料の間の直接の定量的比較を容易にする。ゲル・スポット割り当ておよびマッチングは、最良の総合結果を作るために、データ分析に自動的に組み込まれる。本発明による手法は、タンパク質同定とタンパク質発現分析の両方の、高速、安価、定量的、かつ定性的なツールを表す。
【0052】
一般に、本発明は、少なくとも2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の少なくとも1つの試料から得られたデータを分析する方法であって、前記2つの変数の関数として表される、少なくとも1つの試料を表すデータを前記システムから得ること、連続するレベルを有するデータ・スタックを形成することであって、各レベルが、少なくとも1つの試料を表す連続データを含むこと、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、変数のうちの第1変数の関数としての成分の第1プロフィール、および変数のうちの第2変数の関数としての成分の第2プロフィールであることを含む方法を対象とする。唯一または単一の試料があることができ、連続データは、時間の関数として試料を表す。連続データは、その成分の質量の関数として単一の試料を表すことができる。代替では、複数の試料があることができ、連続データは、連続試料を表す。
【0053】
本発明は、より具体的には、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の複数の試料から得られたデータを分析する方法であって、2つの変数の関数として表される、複数の試料を表すデータをシステムから得ること、各レベルがデータ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を対象とする。第1プロフィールおよび第2プロフィールは、実質的に純粋な成分のプロフィールを表す。この方法は、第1プロフィールおよび第2プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む。
【0054】
この方法は、標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、データ自体、および第2標準化行列の積の形にすることによってデータを標準化することをさらに含む。第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、2つの変数に対する位置でデータを表現させる値を有することができる。第1標準化行列は、第1変数に対してデータをシフトし、第2標準化行列は、第2変数に対してデータをシフトする。第1標準化行列および第2標準化行列の項は、それぞれ第1変数および第2変数に関してデータの分布形状を標準化するように働く値を有する。第1標準化行列および第2標準化行列の項は、既知の成分を有する試料を装置に適用すること、および、既知の成分によって生成されたデータを第1変数および第2変数に対して正しく位置決めさせる第1標準化行列および第2標準化行列の項を選択することによって決定することができる。項は、標準化されたデータ行列内の第1変数および第2変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定することができる。第1標準化行列および第2標準化行列の項は、試料ごとに計算されることが好ましく、その結果全ての試料に対して最少の誤差を生じる。第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる。第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づくものとすることができる。
【0055】
第1標準化行列および第2標準化行列の項の値は、データ配列R、すなわち
【数1】
を解くことによって決定され、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、p個の試料のすべてのこれらの成分の濃度を含み、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である。
【0056】
分離装置は、2次元電気泳動分離システムとすることができる。第1変数は、等電点であり、第2変数は、分子量である。
【0057】
変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果とすることができる。
【0058】
2つの変数は、質量分析計の質量軸に関連する質量とすることができる。
【0059】
装置は、質量分析計に試料を供給するクロマトグラフィ・システムをさらに含むことができ、保持時間は、2つの変数のうちの他方である。
【0060】
装置は、質量分析計に試料を供給する電気泳動分離システムをさらに含むことができ、試料の移動特性は、2つの変数のうちの他方である。
【0061】
この方法では、データは、連続体質量スペクトル・データであることが好ましい。データは、セントロイディングなしで使用されることが好ましい。時間スキューに関してデータを補正することができる。質量および質量スペクトル・ピーク形状に関してデータの較正を実行することが好ましい。
【0062】
第1変数および第2変数のうちの1つは、複数のタンパク質親和性領域を有するプロテイン・チップ上の領域の変数とすることができる。
【0063】
この方法は、単一チャネル・アナライザを使用することによって、また試料を連続して分析することによって前記データ配列のデータを得ることをさらに含むことができる。単一チャネル・検出器は、光吸収、光放出、光反射、光透過、光散乱、屈折率、化学作用、導電性、放射能、またこれらの任意の組合せのうちの1つに基づくものとすることができる。試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合することができる。
【0064】
本発明は、複数の試料から得られたデータを分析するデータ分析部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、化学分析システムは、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、コンピュータ可読コードは、コンピュータに、2つの変数の関数として表される、複数の試料を表すデータをシステムから得ること、各レベルがデータ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を実行させるコンピュータ可読媒体も対象とする。このコンピュータ可読媒体は、上述の用法のいずれか1つのステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含むことができる。
【0065】
本発明は、さらに、複数の試料から得られたデータを分析する化学分析システムであって、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、2つの変数の関数として表される、複数の試料を表すデータを化学分析システムから得ること、各レベルがデータ試料の1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を実行する装置を有するシステムを対象とする。この化学分析システムは、上述の方法のいずれかのステップを実行するための機構を有することができる。
【0066】
本発明は、さらに、複数の成分を含む試料の成分を分離する能力を有する分離システム内の試料から得られたデータを分析する方法であって、複数の成分を含む試料を少なくとも第1変数に関して分離し、その分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を含む。第1プロフィール、第2プロフィール、および第3プロフィールは、実質的に純粋な成分のプロフィールを表す。この方法は、さらに、第1プロフィール、第2プロフィール、および第3プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む。
【0067】
この方法は、標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、データ自体、および第2標準化行列の積の形にすることによってデータを標準化することをさらに含む。第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、3つの変数のうちの2つに対する位置でデータを表現させる値を有する。第1標準化行列は、2つの変数のうちの一方に対してデータをシフトし、第2標準化行列は、2つの変数のうちの他方に対してデータをシフトする。第1標準化行列および第2標準化行列の項は、それぞれ2つの変数に関してデータの分布形状を標準化するように働く値を有することができる。第1標準化行列および第2標準化行列の項は、既知の成分を有する試料を装置に適用すること、および、既知の成分によって生成されたデータを2つの変数に対して正しく位置決めさせる第1標準化行列および第2標準化行列の項を選択することによって決定される。
【0068】
項は、標準化されたデータ行列内の2つの変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される。第1標準化行列および第2標準化行列の項は、単一チャネルについて計算することができる。第1標準化行列および第2標準化行列の項は、チャネルについて最小の誤差を生じるように計算される。
【0069】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる。第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づくことが好ましい。
【0070】
本発明によれば、第1標準化行列および第2標準化行列の項の値は、データ配列R、すなわち
【数2】
を解くことによって決定され、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、マルチチャネル・検出器または第3変数に関するこれらの成分の純粋なプロフィールを含み、I(k×k×k)は、k個の成分のすべての濃度を表す唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である。
【0071】
使用される分離装置は、1次元電気泳動分離システムとすることができ、変数は、等電点および分子量のうちの1つである。
【0072】
2つの分離変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果とすることができる。
【0073】
3つの変数のうちの1つは、質量分析計の質量軸に関連する質量とすることができる。
【0074】
使用される装置は、質量分析計に分離された試料を供給する少なくとも1つのクロマトグラフィ・システムをさらに含むことができ、保持時間は、変数のうちの少なくとも1つである。装置は、質量分析計に分離された試料を供給する少なくとも1つの電気泳動分離システムをさらに含むこともでき、試料の移動特性は、変数のうちの少なくとも1つである。データは、連続体質量スペクトル・データであることが好ましい。データは、セントロイディングなしで使用されることが好ましい。
【0075】
この方法は、時間スキューに関して前記データを補正することをさらに含む。この方法は、質量およびスペクトル・ピーク形状に関して前記データの較正を実行することをさらに含む。
【0076】
使用される装置は、複数のタンパク質親和性領域を有するプロテイン・チップを含み、領域の位置は、3つの変数のうちの1つである。
【0077】
使用されるマルチチャネル・アナライザは、光吸収、光放出、光反射、光透過、光散乱、屈折率、化学作用、導電性、放射能、またこれらの任意の組合せのうちの1つに基づくものとすることができる。試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合することができる。
【0078】
使用される装置は、2次元電気泳動分離システムを含むことができ、少なくとも1つの変数のうちの第1は、等電点であり、少なくとも1つの変数のうちの第2は、分子量である。
【0079】
本発明は、試料から得られたデータを分析するデータ分析部分を有し、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、コンピュータ可読コードは、コンピュータに、複数の成分を含む試料を少なくとも第1変数に関して分離し、その分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を実行させるコンピュータ可読媒体も対象とする。このコンピュータ可読媒体は、上記で示した方法のいずれかのステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含むことができる。
【0080】
本発明は、試料から得られたデータを分析する化学分析システムであって、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を実行する装置を有するシステムも対象とする。この化学分析システムは、上に記載の方法のステップを実行するための機構をさらに含むことができる。
【0081】
本発明の前述の態様および他の特徴を、添付図面と共に次の説明で説明するが、同一の符号は、同一の構成要素を表す。
【発明を実施するための最良の形態】
【0082】
図1を参照すると、上記で示したようにタンパク質または他の分子の分析に使用できる、本発明の特徴を組み込んだ分析システム10のブロック図が示されている。本発明を、図面に示された単一の実施形態に関して説明するが、本発明は、実施形態の多数の代替形態で実施できることを理解されたい。さらに、すべての適切なタイプの構成要素を使用することができる。
【0083】
分析システム10は、試料調製部分12、質量分析計部分14、データ分析システム16、およびコンピュータ・システム18を有する。試料調製部分12は、米国マサチューセッツ州ウォルサムのThermo Electron Corporation社が製造するFinnegan LCQ Deca XP Maxなど、注目の分子を含む試料をシステム10に導入するタイプの試料導入ユニット20を含むことができる。試料調製部分12は、システム10によって分析されるタンパク質などの検体の予備分離を実行するのに使用される検体分離ユニット22をも含むことができる。検体分離ユニット22は、クロマトグラフィ・カラム、米国カリフォルニア州ハーキュリーズのBio−Rad Laboratories,Inc.社が製造するものなどのゲル分離ユニットのいずれかとすることができ、当技術分野で周知である。一般に、電圧またはPH勾配をゲルに印加して、1次元分離の場合に毛細管を介する移動速度(分子量、MW)および等電点電気泳動点(Hannesh,S.M.、Electrophoresis 21、1202〜1209ページ、2000年)などの1変数の関数として、または等電点電気泳動およびMW(2次元分離)によるなど、これらの変数のうちの複数によって、タンパク質などの分子を分離させる。後者の例を、SDS−PAGEと称する。
【0084】
質量分離部分14は、従来の質量分析計とすることができ、使用可能な任意のものとすることができるが、MALDI−TOF、四重極MS、イオン・トラップMS、またはFTICR−MSのうちの1つであることが好ましい。MALDIまたはエレクトロスプレイ・イオン化イオン源を有する場合に、そのようなイオン源は、質量分析計部分14への試料入力を提供することができる。一般に、質量分析計部分14は、イオン源24、質量対荷電比(または単に質量と呼ばれる)によって、イオン源24によって生成されたイオンを分離する質量スペクトルアナライザ26、質量スペクトルアナライザ26からのイオンを検出するイオン検出器部分28、および質量分析計部分14が能率的に動作するのに十分な真空を維持する真空装置30を含むことができる。質量分析計部分14が、イオン移動度スペクトロメータである場合には、一般に、真空装置は不要である。
【0085】
データ分析システム16には、イオン検出器部分28からの信号をディジタル・データに変換する1つまたは一連のアナログ・ディジタル変換器(図示せず)を含むことができるデータ収集部分32が含まれる。このディジタル・データは、リアル・タイム・データ処理部分34に供給され、リアル・タイム・データ処理部分34は、合計および/または平均などの演算を介してディジタル・データを処理する。後処理部分36は、ライブラリ検索、データ保管、およびデータ報告を含む、リアル・タイム・データ処理部分34からのデータの追加処理を行うのに使用することができる。
【0086】
コンピュータ・システム18は、下記で説明する形で、試料調製部分12、質量分析計部分14、およびデータ分析システム16を制御する。コンピュータ・システム18は、適切なスクリーン・ディスプレイでのデータの入力および実行された分析の結果の表示を可能にするために、従来のコンピュータ・モニタ40を有することができる。コンピュータ・システム18は、たとえばWindows(登録商標)またはUNIX(登録商標)オペレーティング・システムあるいは他の適切なオペレーティング・システムを用いて動作する、適切なパーソナル・コンピュータに基づくものとすることができる。コンピュータ・システム18は、通常はハード・ドライブ42を有し、このハード・ドライブ42に、オペレーティング・システムと、下記で説明するデータ分析を実行するプログラムとが保管される。CDまたはフロッピ・ディスクを受け入れるドライブ44が、本発明によるプログラムをコンピュータ・システム18にロードするのに使用される。試料調製部分12および質量分析計部分14を制御するプログラムは、通常、システム10のこれらの部分のファームウェアとしてダウンロードされる。データ分析システム16は、下記で述べる処理ステップを実施するために、C++、JAVA、またはVisual Basicなどの複数のプログラミング言語のいずれかで記述されたプログラムとすることができる。
【0087】
図2は、試料調製部分12に試料導入ユニット20および1次元試料分離装置52が含まれる分析システム50のブロック図である。たとえば、装置52は、1次元電気泳動装置とすることができる。分離された試料成分が、たとえば一連の紫外線センサまたは質量分析計などのマルチチャネル検出装置54によって分析される。データ分析を行える形を、下記で述べる。
【0088】
図3は、試料調製部分12に、試料導入ユニット20、第1次元試料分離装置62、および第2次元試料分離装置64が含まれる分析システム60のブロック図である。たとえば、第1次元試料分離装置62および第2次元試料分離装置64は、2つの連続する異なる液体クロマトグラフィ・ユニットとすることができ、あるいは、2次元電気泳動装置として合体させることができる。分離された試料成分は、たとえば245nm帯域フィルタを有する紫外線センサ、またはグレイ・スケール・ゲル・イメージャ(gel imager)など、単一チャネル検出装置66によって分析される。また、データ分析を行える形を、下記で述べる。
【0089】
図4Aに、分析される成分の混合物の連続試料を表す、一連の2次元配列72Aから72Nよりコンパイルされた3次元データ配列70を示す。2次元データ配列72Aから72Nは、たとえば、図3に関して上述したように2次元ゲル電気泳動または連続クロマトグラフ分離、あるいは他の分離技法の組合せによって作ることができる。
【0090】
図4Bに、分析される成分の混合物の連続試料を表す、一連の2次元配列76Aから76Nよりコンパイルされた3次元データ配列74を示す。2次元データ配列76Aから76Nは、たとえば、図2に関して上述したように1次元ゲル電気泳動または液体クロマトグラフィとその後のマルチチャネル分析、あるいはガス・クロマトグラフィ/赤外線スペクトル法(GC/IR)またはLC/蛍光などの他の技法によって作ることができる。
【0091】
図4Cに、分析される成分の混合物の連続試料を表す、一連の2次元配列80Aから80Nからコンパイルされた3次元データ配列78を示す。2次元データ配列80Aから80Nは、たとえば、米国カリフォルニア州フレモントのCiphergen Biosystems,Inc.社が販売するタイプの、表面で画定された領域(スポット)にタンパク質を選択的に結合することのできるタンパク質親和性チップと、その後の、図2に関して説明したものなどのシステムのうちの1つとすることができる、表面増強レーザー脱離/イオン化(SELDI)飛行時間型質量分析法などのマルチチャネル分析によって作られる。使用できる他の技法は、マルチチャネル蛍光検出と組み合わされた1Dプロテイン・アレイである。
【0092】
図5に、分析されるべき成分の混合物の単一の試料を表す、一連の2次元配列84Aから84Nからコンパイルされた3次元データ配列82を示す。2次元データ配列84Aから84Nは、たとえば、図1に関して上述したように、2次元ゲル電気泳動または連続液体クロマトグラフィによって作ることができる。たとえば、図1に関して上述したものなどの質量分析法によるマルチチャネル検出は、第3次元のデータを作る。他の適切な技法は、マルチチャネルUV検出またはマルチチャネル蛍光検出を用いる2D LC、IR検出を用いる2D LC、質量分析法を用いる2Dプロテイン・アレイである。
【0093】
図6に、2次元液相分離(たとえば、強陽イオン交換クロマトグラフィとその後の逆相クロマトグラフィ)によって得られたデータ配列84を示す。第3次元は、質量スペクトル検出からの質量軸86に沿ったデータによって表されている。
【0094】
図4A、図4B、図4C、図5、および図6のデータ配列には、場合によっては、すべての試料または単一の試料のすべての成分を表す項が含まれる(較正標準の成分を含む)。
【0095】
図7に、所定の走査時間中のある質量範囲を掃引する質量分析計にLCが接続されるLC/MSの場合にそうであるように、時間ベースの分離に接続された走査型マルチチャネル検出器の時間スキューの補正を示す。このタイプの時間スキューは、すべての質量のイオンを同時に検出する磁気セクタ・システム(magnetic sector system)などの同時システムを除く質量分析計のほとんどに存在する。他の例には、揮発化合物が、IRスペクトルが走査モノクロメータまたは干渉計のいずれかを介して獲得されている間にカラムを通過した後の保持時間に関して分離されるGC/IRが含まれる。分離または反応などの時間依存事象が、複数のチャネルを順次走査する検出システムに接続されているとき、前に走査されたチャネルが事象の以前の時点に対応し、後に走査されたチャネルが事象の後の時点に対応する時間スキューが生成される。この時間スキューは、すべてのチャネルについて同一の時点に対応するマルチチャネル・データを生成するすなわち、図7の傾いた実線から対応する破線の水平線へ各チャネルを補間するために、チャネルごとの基礎での補間によって補正することができる。
【0096】
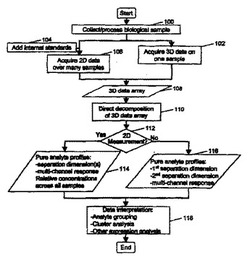
図8は、本発明に従って試料データを獲得し、処理する方法の一般的な流れ図である。生物試料などの試料の収集および処理が、100で行われる。単一の試料を処理する場合には、102で3次元データを獲得する。106で複数の試料を用いて2次元データを獲得する場合には、任意選択で、104で内部標準を試料に追加する。上記の技法およびシステムのいずれかに関して説明したように、108で、3次元データ配列を形成する。110で、3次元データ配列が、直接分解を受ける。2次元測定を行ったか否かに基づいて、112で異なる経路を選択する。2次元測定が行われた場合には、114で、各次元の純粋な検体プロフィールを、すべての試料にまたがるそれぞれの濃度と共に得る。3次元測定が単一の試料に対して行われた場合には、116で、3つのすべての次元に沿った試料のすべての検体の純粋な検体プロフィールを得る。どの場合でも、検体グループ化、クラスタ分析、ならびに他のタイプの発現および分析を含むデータ解釈を、118で行い、その結果を、図1、図2、または図3のうちの一つのシステムに関連するコンピュータ・システム18のモニタ40で報告する。
【0097】
データの分析のモードを、本発明の範囲の制限ではなく理解を容易にするために提供される特定の例に関して、下記で説明する。
【0098】
通常の試料の応答行列Rj(m×n)を次の双一次形で表現できる場合:
【数3】
ここで、ciは、第i検体の濃度であり、xi(m×1)は、行軸に沿ったこの検体の応答(たとえば、LC/MSでのこの検体のLC溶出プロフィールまたはクロマトグラム)であり、yi(n×1)は、列軸に沿ったこの検体の応答(たとえば、LC/MSでのこの
検体のMSスペクトル)であり、kは、試料内の検体の数である。複数の試料(j=1,2,…,p)の応答行列がコンパイルされるとき、3Dデータ配列R(m×n×p)を形成することができる。
【0099】
したがって、2Dゲル・ランの終りに、グレイスケール・イメージを生成し、2D行列Rj(m×nの次元を有し、試料jの、行にディジタル化されたm個の異なるpI値および列にディジタル化されたn個の異なるMW値に対応する)で表すことができる。この生イメージ・データは、標準化されたイメージRjを作るために、pI軸とMW軸の両方で較正される必要がある。
Rj=AjRjBj
ここで、Ajは、m×mの次元を有し、主対角に沿っておよびその周囲に非0要素を有する正方行列(帯対角行列)であり、Bjは、主対角に沿っておよびその周囲に非0要素を有するもう1つの正方行列(n×n)(もう1つの帯対角行列)である。行列AjおよびBjは、対角行列(単純な線形スケーリングを表す)のように単純なものまたは主対角に沿って増加するもしくは減少する帯域幅を有するもの(バンド・シフト、拡幅、およびひずみのうちの少なくとも1つまたは他のタイプの非線形に補正する)のように複雑なものになる可能性がある。上式の一般形でのグラフィカル表現を、図9に示されているように与えることができる。
【0100】
【数4】
複数の試料からの2−Dゲル・データが収集されるとき、Rjの集合を配置して、3Dデータ配列Rを
【数5】
として形成することができ、ここで、pは、生物試料の数であり、Rは、m×n×pの次元を有する。このデータ配列(立方体または直方体)は、GRAM(Generalized Rank Annihilation Method、反復なしの行列演算を介する直接分解、Sanchez,E.他、J.Chemometrics 4、29ページ、1990年)またはPARAFAC(PARAllel FACtor analysis、交互最小二乗法を用いる反復分解、Carroll,J.他、Psychometrika 3、45ページ、1980年およびBezemer,E.他、Anal.Chem.73、4403ページ、2001年)に基づくトリリニア分解法を用いて、4つの異なる配列および残余データ配列Eに分解することができる。
【0101】
【数6】
ここで、Cは、p個の試料のすべてでのすべての同定可能タンパク質(そのうちのk個、k≦min(m,n))の相対濃度を表し、Qは、各タンパク質(そのうちのk個)のm個のpI値でディジタル化されたpIプロフィールを表し、Wは、各タンパク質のn個の値でディジタル化された分子量プロフィール(理想的には、各タンパク質に対応する単一のピークが観察される)であり、Iは、唯一の非0要素としてそのスーパーダイアゴナル(super−diagonal)にスカラを有する新しいデータ・キューブである。
【0102】
すべてのタンパク質が別個であり(異なるpI値および異なるMWを有する)、発現レベルが、試料ごとに線形独立の形で変動するとき、結果の次の直接解釈を予想することができる。
1.上記の分解からのk値は、自動的に、タンパク質の数と等しくなる。
2.行列Cの各行の値は、Iのスーパーダイアゴナル要素によるスケーリングの後に、特定の試料内のこれらのタンパク質の相対濃度を表す。
3.行列Qの各列は、特定のタンパク質の逆畳み込みされたpIプロフィールを表す。
4.行列Wの各列は、特定のタンパク質の逆畳み込みされたMWプロフィールを表す。
【0103】
これらのタンパク質が、別個であるが、試料ごとに相関する発現レベルを有する(行列Cが線形依存列を有する)場合に、解釈は、各個々のタンパク質ではなく、相関する発現レベルを有するタンパク質のグループに対してのみ実行することができ、プロテオミクス研究の重要性の発見である。
【0104】
上記で提示した分解に基づいて、そのような多次元のシステムおよび分析の威力を、すぐに見ることができる。
【0105】
a.成分応答を各次元の個々のタンパク質応答の線形組合せに分離するこの分解の結果として、他のすべてのタンパク質の存在下で、各タンパク質について量的情報を得ることができる。
【0106】
b.この分解は、各個々のタンパク質のプロフィールを各次元で分離し、両方の次元(2DEでpIおよびMW、LC/MSでクロマトグラフ次元および質量スペクトル次元)でのこれらのタンパク質の同定に関する質的情報も提供する。
【0107】
c.3Dデータ配列Rの各試料に、タンパク質の異なる集合が含まれる可能性があり、注目のタンパク質を、未知のタンパク質の存在下で同定でき、定量化でき、データ配列のすべての試料が共有する共通タンパク質だけが、分解された行列Cですべて非0濃度を有することが暗示される。
【0108】
d.この分析には、最低限で別個の試料が2つ必要とされるだけであり、他の関心のないタンパク質の存在下で注目のタンパク質を素早く信頼できる形でとりあげるために、ICATのようなラベルなしで異なるプロテオーム解析を実行するはるかに良い方法を提供する。
【0109】
e.分析できる検体の数は、応答行列Rjごとの最大の許容可能な擬似ランクによって制限され、これは、簡単に数千(イオン・トラップMS)または数十万(TOFまたはFTICR−MS)に達することができ、複雑な生物試料に対する大規模なプロテオーム解析が可能になる。
【0110】
f.通常のLC/MSランは、関係する他の化学プロセスまたは試料調製ステップを用いることなく2時間未満で完了することができ、スループットにおける少なくとも10倍の向上および情報科学における著しい単純化を示す。
【0111】
g.完全なLC/MSデータが分析に使用されるので、MS/MS実験なしでほぼ100%の配列カバレッジを達成することができる。
【0112】
2−Dゲル分離のイメージに基づく上記の分析の重要な利益は、これが非破壊的であり、たとえばMALDI TOFの使用を介してさらなる確認を引き続き行えることである。上記の分析は、同一のタンパク質からのすべてのペプチドを分析および解釈に関して別個のグループとして扱うことができるタンパク質消化物にも適用することができる。pIプロフィールおよびMWプロフィールの個々のタンパク質への分離は、個々のペプチドへの分離が実現可能でない時であっても実行することができる。
【0113】
左右の変換行列AjおよびBjを、好ましくは各試料に追加される内部標準を使用して決定することができる。これらの内部標準は、すべてのpI範囲およびMW範囲をカバーするように選択され、たとえば、2Dゲル・イメージの各角に1つと中央の右1つの5つの内部標準が選択される。これらの内部標準の濃度は、試料ごとに変化し、その結果、上記の分解の対応する行列Cを、
C=[Cs|Cunk]
として区分することができ、ここで、Csのすべての列が独立すなわち、Csが最大階数であるか、よりよくは、最大の特異値と最小の特異値の間の比が最小にされる。上記の分解において既知の行列Cの一部を用いて、各試料の変換行列AjおよびBj(j=1,2,…,p)を同一の分解処理で決定して総合的な残余Eを最小にすることができるように分解を実行することができる。問題のスケールは、AjおよびBjの非0対角帯をパラメータ化することによって、たとえば、Ajの各行およびBjの各列についてガウス形状の帯を広げるフィルタを指定し、Ajの行を下げおよびBjの列を横切る(across)ガウス・パラメータの滑らかな変動を可能にすることによって、劇的に減らすことができる。行列AjおよびBjが正しくパラメータ化され、パラメータに関する導関数の分析方法を導出したならば、効率的なガウス−ニュートン反復手法をトリリニア分解またはPARAFACアルゴリズムに適用して、各試料の所望の分解および正しい変換行列AjおよびBjの両方に達することができる。
【0114】
ICAT(同位体コードアフィニティータグ、Gygi,S.P.他、Nature Biotech.1999年、17、994ページ)と比較して、この手法は、2つの試料だけの分析に制限されず、タンパク質同定にペプチド・シーケンシングを必要としない。定量化できる試料の数は、数百から数千、あるいは数万にさえなる可能性があり、タンパク質同定は、すべてのこれらのタンパク質が数学的に解かれ、分離されたならば、質量スペクトル・データだけを介して達成することができる。さらに、同位元素ラベルを用いる追加の化学作用がなく、これによって、ICATに必要な面倒な試料調製ステージ中に多数の重要なタンパク質を失う危険性が減る。
【0115】
短く言うと、本発明は、上述の分析の方法を使用して、次の特徴を有する2Dデータを使用するタンパク質同定およびタンパク質発現分析の技法を提供する。
−複数の試料からの2Dゲル・データが、3Dデータ配列を形成するのに使用される。
−次のシナリオのそれぞれについて、適用可能な解釈の異なる組がある。
a)すべてのタンパク質が別個であり、発現レベルが試料の間で独立に変化する場合
b)すべてのタンパク質が別個であり、試料の間に相関する発現レベルがある場合
−質量スペクトル連続体データに対するセントロイディングが回避される。
−生の質量スペクトル・データだけを、直接に使用することができ、データ配列分解への入力としてこれだけで十分である。
−たとえば米国仮出願第10/689313号で実行されたもののような、完全な質量スペクトル較正を、生連続体データに対して任意選択で実行して、分析への入力としての完全に較正された連続データを得ることができ、逆畳み込み質量スペクトルが配列分解後に個々のタンパク質について使用可能になったならば、タンパク質同定のためにさらに正確な質量決定およびライブラリ検索が可能になる。
−この手法は、タンパク質を分解し、分離するのに物理的シーケンシングではなく数学に基づき、タンパク質同定にペプチド・シーケンシングを必要としない。
−結果は、定性的であると同時に定量的である。
−ゲル・スポットの整列およびマッチングは、データ分析に自動的に組み込まれる。
【0116】
さらに、その短い要約を下記で示す同時係属の米国仮出願第10/689313号に記載のように、質量整列およびスペクトル・ピーク形状一貫性をさらに改善するために、本発明で完全に較正された連続質量スペクトル・データを有することが好ましい。
【0117】
完全に較正された連続質量スペクトル・データの作成
m=f(m0) (式A)
の形の較正関係を、測定された重心と、質量範囲にまたがる質量スペクトル標準で使用可能なすべての明瞭に同定可能な同位元素クラスタを使用して計算された重心との間での最小二乗多項式フィットを介して確立することができる。
【0118】
この単純な質量較正に加えて、追加の完全なスペクトル較正フィルタが、同時に2つの目的すなわち質量スペクトル・ピーク形状および質量スペクトル・ピーク位置の較正に役立つために計算される。質量軸は、事前に較正することができるので、フィルタ機能の質量較正部分は、この場合に、質量較正でのさらなる洗練を達成するためすなわち式Aによって与えられる多項式フィットの後の任意の残留質量誤差を考慮に入れるために縮小される。
【0119】
この較正処理全体は、質量スペクトル・ピーク幅(半値全幅またはFWHM)が一般に動作質量範囲内でほぼ一定である、イオン・トラップを含む四重極タイプのMSに簡単に適用される。磁気セクタ、TOF、またはFTMSなどの他のタイプの質量分析計システムについて、質量スペクトル・ピーク形状は、動作原理および/または特定の機器設計によって指定される関係で、質量に伴って変化すると予想される。同一の質量依存較正手順が、それでも適用可能ではあるが、ピーク幅/位置と質量の間の所与の関係と一貫する変換されたデータ空間で較正全体を実行することが好ましい可能性がある。
【0120】
TOFの場合に、質量スペクトル・ピーク幅(FWHM)Δmが、次の関係で質量(m)に関係することが既知である:
【数7】
ここで、aは、既知の較正係数である。言い換えると、質量範囲にわたって測定されたピーク幅は、質量の平方根に伴って増加する。平方根を用いると、質量軸から新しい関数に変換する変換は、次のようになる:
【数8】
ここで、変換後の質量軸で測定されたピーク幅(FWHM)は、
【数9】
によって与えられ、これは、スペクトル範囲全体にわたって変化しない。
【0121】
その一方で、FT MS機器について、ピーク幅(FWHM)Δmは、質量mに正比例し、したがって、対数変換が必要である:
m’=ln(m)
ここで、変換後の対数空間で測定されたピーク幅(FWHM)は、
【数10】
によって与えられ、これは、質量と独立に固定されている。通常、FTMSでは、Δm/mを10-5程度にすなわち、分解能m/Δmに関して105に管理することができる。
【0122】
磁気セクタ機器について、特定の設計に依存して、スペクトル・ピーク幅および質量サンプリング間隔は、質量との既知の数学的関係に従い、これはそれ自体、TOFおよびFTMSに関して平方根変換および対数変換が行うのによく似た形で、期待される質量スペクトル・ピーク幅がそれを介して質量と独立になる特定の形の変換に役立つ可能性がある。
【0123】
FTMSでの対数変換およびTOF−MSでの平方根変換などの適切な変換または十分に設計され正しく調整された四重極MSまたはイオン・トラップMSなどの特定の機器に固有の性質のいずれかに起因して、予想される質量スペクトル・ピーク幅が、質量と独立になるとき、計算時間の大幅な節約が、質量スペクトル範囲全体に適用可能な単一の較正フィルタを用いて達成される。これによって、質量スペクトル較正標準に対する要件も単純化される。すなわち、単一の質量スペクトル・ピークが較正に必要とされ、(存在する場合には)追加ピークが検査または確認としてだけ働き、測定される各試料に追加された内部標準に基づくすべてのMSの完全な質量スペクトル較正のために道を開く。
【0124】
通常、質量スペクトル較正全体を達成する上で2つのステップがある。第1ステップは、実際の質量スペクトル・ピーク形状関数を導出することであり、第2ステップは、導出された実際のピーク形状関数を、正しい質量位置を中心とする指定された目標ピーク形状関数に変換することである。測定された生連続質量スペクトルy0を有する内部標準または外部標準は、標準イオンまたは標準イオン・フラグメントの同位元素分布yに、
【数11】
によって関係する。ここで、pは、計算された実際のピーク形状関数である。この実際のピーク形状関数は、その後、
【数12】
によって与えられる1つまたはそれ以上の較正フィルタを介して、指定された目標ピーク形状関数t(たとえば、あるFWHMのガウシアン)に変換される。
【0125】
上記で計算された較正フィルタを、次の帯対角フィルタ行列に配置することができる:
【数13】
ここで、対角線上の各短い列ベクトルfiは、対応する中心質量に関する上記で計算された畳み込みフィルタからとられる。fiの要素は、逆の順序の畳み込みフィルタの要素から取られる、すなわち
【数14】
である。
【0126】
例として、この較正行列は、1/8amuデータ間隔で1000amuまでの質量カバレッジを有する四重極MSで8000×8000の次元を有する。しかし、その疎な性質に起因して、通常のストレージ要件は、5amu質量範囲をカバーする40要素の有効フィルタ長で約40×8000だけである。
【0127】
本発明に戻ると、さらなる多変量統計分析を行列Cに適用して、異なる試料と異なるタンパク質の間の関係を研究し、理解することができる。試料およびタンパク質をグループ化するかクラスタ分析して、どのタンパク質がどの試料グループ内により多く発現するかを調べることができる。たとえば、C行列の主成分分析からのスコアまたはローディングを使用して、デンドログラムを作成することができる。典型的な結論は、健康な個人からの細胞試料はお互いの周囲にクラスタ化するが、病気の個人からの細胞試料は異なるグループでクラスタ化することを含む。ある治療の後のある時間期間にわたって収集された試料について、その試料が、タンパク質レベルに関して治療に対する生物反応を示す、あるタンパク質の発現レベルの継続的変化を示す場合がある。一連の投薬量に対して収集された試料について、関連するタンパク質の変化が、タンパク質のこの集合に対する投薬量の影響およびその潜在的調整を示す可能性がある。
【0128】
タンパク質が分析の前にペプチドに事前消化される場合に、行列Cの各列は、同一のタンパク質から得られるペプチドのグループまたは試料の間で類似する発現パターンを示すタンパク質のグループの線形組合せを表す。図11に示されたものなどの行列Cの列を分類するために実行されたデンドログラムでは、個々のペプチドがそれぞれのタンパク質にグループ化され、したがって、プロテオーム・レベルでの分析が達成される。
【0129】
同定されたタンパク質の定性的(またはシグナトリ(signatory))情報を、pIプロフィール行列QおよびMW行列Wで見つけることができる。定性的情報は、特に分子量情報が十分な精度で決定される場合に、タンパク質同定およびライブラリ検索のために働くことができる。要約すると、3つの行列C、Q、およびWは、組み合わされたとき、AjおよびBjによって表される変換行列の決定からの自動ゲル・マッチングおよびスポット整列と共に、タンパク質の定量化と同定の両方を可能にする。
【0130】
上記の2−Dデータは、異なる形式および形状になる可能性がある。2−Dゲル・スポットの除去/消化の後のMALDI−TOFに対する代替は、これらの試料を従来のLC/MS、たとえばThermal Finnigan LCQシステムにかけて、MS分析の前に各ゲル・スポットからタンパク質をさらに分離することである。この手法の非常に重要な応用は、2−Dゲル(2DE)分離を完全に回避することによる素早く直接のタンパク質の同定および定量化を可能にし、したがって、スループットを数桁高める。これは、次のステップを介して達成することができる。
【0131】
1.分離なしで数十万種のタンパク質を含む試料を直接に消化する
2.消化された試薬を従来のLC/MS機器にかけて、2次元配列を得る。MS/MS機能はこの場合に不要であるが、試料をLC/MS/MSシステムにかけることを選択することができ、これによって追加のシーケンシング情報が生成されることに留意されたい。
【0132】
3.複数の試料について1および2を繰り返して、3次元データ配列を生成する。
4.上記で概要を示した手法を使用してデータ配列を分解する。
【0133】
5.タンパク質同定のための解釈および質量スペクトル検索で、pI軸をLC保持時間に置換し、MW軸を質量軸に置換する。数学的に分離された質量スペクトルを、セントロイディングおよびデアイソトーピングを介してさらに処理して、http://www.matrixscience.comまたはhttp://us.expasy.org/sprot/からオンラインで使用可能なMascotまたはSwissProtなどの従来のデータベースおよび検索エンジンと互換のスティック・スペクトルを作ることができる。しかし、データ配列分解の前に、生の質量スペクトル連続体データを較正済み連続体データに完全に較正し、各逆畳み込みされるタンパク質またはペプチドについて完全に較正された連続体質量スペクトル・データを生み出すことが好ましい。この連続体質量スペクトル・データは、その後、セントロイディングなしのその高い質量精度と共に、同時係属の特許の新規のデータベース検索を介するタンパク質同定に使用される。
【0134】
LCカラムの性質に応じて、LCは、2−DゲルのpI軸に似た別の形の電荷分離として働くことができる。この場合の質量分析計は、2−Dゲル分析のWM軸に似た、分子量測定の正確な手段として働く。質量分析計で使用可能な高い質量精度に起因して、変換行列Bjを対角行列に縮小して、質量依存のイオン化効率変化を訂正するか、または特に上述の完全な質量スペクトル較正の後に、単位行列を式から消すことができる。大きいタンパク質分子を扱うために、タンパク質試料は、通常、酵素または化学反応、たとえばトリプシンの使用を介してペプチドに事前消化される。したがって、通常、注目のタンパク質ごとに、複数のLCピークならびに複数の質量が見られる。これによって、試料処理が複雑になる可能性があるが、ライブラリ検索およびタンパク質同定の選択性が大幅に強化される。複数の消化を使用して、選択性をさらに強化することができる。極端に言うと、各タンパク質を事前にさまざまな長さのペプチドに消化して(Erdman degradation)、行列Wから完全なタンパク質配列情報を作ることができる。これは、LCタンデム質量分析に対する代替としての、物理シーケンシングではなく数学に基づくタンパク質シーケンシングの新しい技法である。MSを含む応用例で、この手法は、多数の非系統誤差を受けやすい市販計測器で通常行われているセントロイディングおよびデアイソトーピングなどの質量走査からの連続データに対するデータ処理を必要としない。生のカウント・データをデータ配列分解への入力として供給し、直接に使用することができる。
【0135】
同一の手法を用いて類似する結果を作ることができる他の2−Dデータに、単一点検出を用いる2−D分離、または複数チャネル検出もしくは2−Dマルチチャネル検出を用いる1−D分離を有する以下のが含まれるが、これに制限はされない。
【0136】
1.1−Dまたは2−Dのゲル・スポットのそれぞれを、後続のLC/MS分析に関して独立の試料として扱って、スポットごとに1つのLC/MS 2−Dデータ配列と、すべてのゲル・スポットおよびそのLC/MSデータ配列を含むデータ配列とを生成することができる。ゲルおよびLC分離の両方から得られる追加の解像度に起因して、より多くのタンパク質をより正確に同定することができる。
【0137】
2.pI/疎水性、MW/疎水性などの他のタイプの2−D分離、またはpI、MW、もしくは疎水性のいずれかを使用する1−D分離と、on−the−gel MALDI TOFと組み合わされた1−Dゲル、またはLC/TOF、LC/UV、LC/蛍光などのマルチチャネル電磁検出または質量スペクトル検出のある形を使用することができる。
【0138】
3.単一チャネル検出(245nmのUV、または1波長で測定されるようにタグ付けされた蛍光)を用いる、2−D液体クロマトグラフィなどの他のタイプの2−D分離を使用することができる。
【0139】
4.アレイの各要素がLCカラムと異ならない形でタンパク質の特定の組合せを取り込む、質量スペクトル検出または他のマルチチャネル検出と結合された1Dまたは2Dのプロテイン・アレイを使用することができる。これらの1Dまたは2Dのスポットを、2−D配列の1次元に配置し、他方の次元を質量分析法とすることができる。これらのタンパク質スポットは、表面弾性波センサ(SAW、あるクラスの化合物に選択的に結合するためにGCカラム材料をコーティングされている)などのセンサ・アレイ、またはその上での結合事象が別個の電気信号を生成する導電性重合体アレイなどの電子鼻に似ている。
【0140】
5.差別的にまたはタンパク質配列のセグメントに特異的にタグ付けされた異なるタンパク質を有する単一の試料に対する複数波長EEM(emission and excitation fluorescence)を使用することができる。
【0141】
2次プロテオミクス分析では、データ配列が、複数の試料からの2D応答行列によって形成される。データ配列を作成するもう1つの効果的な形は、3次機器と呼ばれるもので単一の試料からデータ配列を生成できるように、測定自体にもう1つの次元を含めることである。プロテオミクスで広く注目を集め始めているそのような機器の1つが、試料に存在する各タンパク質のLC次元とMSスペクトル応答の両方で数学的に分離された溶出プロフィールを作るために同一の分解に従うLC/LC/MSである。
【0142】
したがって、上記で概要を示した2次元手法は、当技術分野における主要な改善であるが、3次元手法は、試料がプロセス全体を通じて液相のままであるという事実から生じる、より高速な、より再現可能な、単純なものであるという利益を有する。しかし、多数のタンパク質は、従来の質量分析計には大きすぎ、試料内のすべてのタンパク質が、LC分離および質量スペクトル検出の前にペプチド断片に消化される可能性があるので、ペプチドの数およびシステムの複雑さが、少なくとも1桁増える。これは、データ・ハンドリングおよびデータ解釈に関する解決しにくい問題と思われるものをもたらす。さらに、使用可能な手法は、イーストなどの非常に限られた複雑さの試料の定性的タンパク質同定のレベルで止まる(Washburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年)。下記に提示する手法は、単一の2次元液体クロマトグラフィ−質量分析法(LC/LC/MSまたは2D−LC/MS)ランで数百種から数万種のタンパク質の同定と定量化の両方を達成する。
【0143】
たとえば、サイズ排除逆相液体クロマトグラフィ(SEC−RPLC)または強陽イオン交換逆相液体クロマトグラフィ(SCX−RPLC)のいずれかを、最初の分離に使用することができる。これに、エレクトロスプレイ・イオン化(ESI)質量分析法または飛行時間型質量分析法のいずれかの形の質量分析検出(MS)が続く。生成されたデータの集合は、3次元データ配列Rに配置され、このRには、保持時間(t1およびt2、m個およびn個の異なる時点でディジタル化された、各LC次元での保持時間、たとえばSEC保持時間およびRPHL保持時間に対応する)と質量(注目の質量範囲をカバーするp個の異なる値でディジタル化された)の異なる組合せでの質量強度(カウント)データが含まれる。このデータ配列のグラフ表示を、図6に示す。
【0144】
質量スペクトル・データを、セントロイディングおよびデアイソトーピングを介してスティック・スペクトルの形に再処理することができるが、これは、この手法が働くために望ましくないことに留意することが重要である。生の質量スペクトル連続体データは、分析全体を通じてのスペクトル・ピーク形状情報の保存および上述のすべてのタイプのセントロイディング誤差およびデアイソトーピング誤差の除去に起因して、よりよく働くことができる。好ましい手法は、連続生質量スペクトル・データを較正された連続データに完全に較正して、高い質量精度を達成し、より正確なライブラリ検索を可能にすることである。
【0145】
データ配列R(m×n×pの次元を有する)の保持時間t1およびt2の各組合せで、質量分析計に注入される試料の分画は、オリジナル試料内のペプチドの部分集合のある線形組合せからなる。試料のこの分画は、少数のペプチドと数万種のペプチドの間のどれかを含む可能性が高い。そのような試料分画に対応する質量スペクトルは、非常に複雑である可能性が高く、上記で指摘したように、タンパク質同定および特に定量化のためにそのような混合物を個々のタンパク質に分解するという課題は、解決しがたいと思われる。
【0146】
しかし、この3次元データ配列は、2次元分析に関して上記で指摘したように、GRAM(Generalized Rank Annihilation Method、反復なしの行列演算を介する直接分解)またはPARAFAC(PARAllel FACtor analysis、交互最小二乗法を用いる反復分解)に基づくトリリニア分解法を用いて、上記で指摘したように4つの異なる行列および残余データ・キューブEに分解することができる。
【0147】
この3次元分析で、Cは、すべての同定可能ペプチド(そのうちのk種、k≦min(m,n))のt1に関するクロマトグラムを表し、Qは、すべての同定可能ペプチド(そのうちのk種)のt2に関するクロマトグラムを表し、Wは、すべてのペプチド(そのうちのk種)の逆畳み込みされた連続質量スペクトルを表し、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列である。言い換えると、このデータ配列の分解を介して、2つの保持時間(t1およびt2)が、Wに含まれる各ペプチドの質量スペクトル連続体の正確な決定と共に、試料に存在するそれぞれすべてのペプチドについて同定されている。
【0148】
前述の分析は、無傷のタンパク質が、消化なしで、より大きい質量を処理できる質量分析計を用いて直接に分析されない限り、ペプチド・レベルでの情報を作る。しかし、タンパク質レベルの情報は、次の追加ステップを介して複数の試料から得ることができる。
【0149】
1.同一の治療に関する時間期間にわたって、または治療のうち異なる投薬量を用いる固定された時間に、または異なる病状の複数の個人から収集された複数の試料(そのうちのl個)について、上述の2D−LC/MSランを実行する。
【0150】
2.上述の試料ごとにデータ分解を実行し、各試料を用いてすべてのペプチドを完全に同定する。
【0151】
3.各試料内のすべてのペプチドの相対濃度を、Iのスーパーダイアゴナル要素から直接に読み取ることができる。すべての試料にまたがるこれらの濃度からなる新しい行列Sを、l個の試料×全試料中のq個の別個のペプチド(q□max(k1,k2,…,kp)
、ただし、kiは、試料i(i=1,2,…,p)のペプチドの数)という次元を用いて形成することができる。他の試料に存在するペプチドの一部を含まない試料について、これらのペプチド(列に配置される)に対応する行の項目は、0になる。
【0152】
4.行列Sの統計的検討によって、試料ごとに互いに比例して変化するペプチドの調査が可能になる。これらのペプチドは、同一のタンパク質からのすべてのペプチドに潜在的に対応する可能性がある。S行列の特異値分解(SVD)または主成分分析(PCA)から計算されたマハラノビス距離に基づくデンドログラムによって、これらのペプチドの相互関連性を示すことができる。しかし、試料ごとに協調して変化するタンパク質のグループがあり、したがって、それに対応するペプチドのすべてが同一のクラスタにグループ化されることを指摘しなければならない。このプロセスのグラフ表示を、図11に示す。
【0153】
5.上記のグループ化に従って区分された行列Sは、多数の試料にまたがる異なるタンパク質の発現レベルを示すディファレンシャル・プロテオミクス分析の結果を表す。
【0154】
6.真上のステップ5で同定された各グループのすべてのペプチドについて、Wに含まれる分解された質量スペクトル応答を組み合わせて、その発現レベルにおいて協調して変化する各タンパク質またはタンパク質のグループに含まれるすべてのペプチドの複合質量スペクトル(composite mass spectrum)シグネチャを形成する。そのような複合質量スペクトルは、スティック/重心スペクトルにさらに処理する(まだそのように処理されていない場合)か、好ましくは、同時係属の出願で開示されている連続質量スペクトル・データを使用するタンパク質同定のためにMascotおよびSwissProtなどの標準タンパク質データベースに対して直接に検索することができる。
【0155】
ICAT(Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年)と比較して、本明細書で提案される定量化は、追加の試料調製を必要とせず、数千個の試料を処理する能力を有し、相対的なタンパク質発現レベルに達するために全体的な最小二乗フィットにすべての使用可能なペプチド(同位元素タグ付けに使用可能な少数ではなく)を使用する。すべてのペプチドの数学的分離およびその後のタンパク質へのグループ化にも起因して、このタンパク質同定は、ICATの場合のようなペプチド・シーケンシングなしで達成することができる。無傷のタンパク質2D−LC/MS分析の場合に、すべてのタンパク質濃度を、さらなる再グループ化なしで、Iのスーパーダイアゴナルから直接に読み取ることができる。しかし、上記で示したようにS行列を形成し、ディファレンシャル・プロテオミクス分析またはタンパク質発現分析のためにこの行列に対して統計分析を実行することが望ましい場合がある。
【0156】
短く言うと、本発明は、次の特徴を有する3次元データを使用するタンパク質同定およびタンパク質発現分析の方法を提供する。
【0157】
−次の方法のいずれかから生成されるデータの集合を、3Dデータ配列に配置する
i)タンパク質消化後のペプチドに関するエレクトロスプレイ・イオン化(ESI)質量分析法または
ii)ペプチドまたは無傷のタンパク質に関する飛行時間(TOF)型質量分析法と組み合わされた、
a)サイズ排除逆相液体クロマトグラフィ(SEC−RPLC)または
b)強酸性陽イオン交換逆相液体クロマトグラフィ(SCX−RPLC)
【0158】
−ここで、生質量スペクトル連続体を完全に較正することが望ましいが、質量スペクトル・データは、セントロイディングおよび/またはデアイソトーピングを介して前処理される必要がない。
【0159】
−質量スペクトル連続体データは、直接に使用することができ、実際に好ましく、したがって、分析全体を通じてスペクトル・ピーク形状情報が保存される。
【0160】
−この手法は、すべてのペプチドを数学的に分離し、その後、タンパク質にグループ化する方法であり、したがって、タンパク質同定を、ペプチド・シーケンシングなしで行うことができる。
【0161】
−本発明は、追加試料調製を必要とせず、数千個の試料を処理する能力を有し、相対タンパク質発現レベルに達するために全体的な最小二乗フィットにすべての使用可能なペプチドを使用する定量的ツールを提供する。
【0162】
上記の3−Dデータは、異なる形式および形状で得られる可能性がある。2D−LC/MSに対する代替は、エレクトロスプレイ・イオン化(ESI)質量分析法(従来のイオン・トラップか四重極MSかTOF−MS)と結合された2D電気泳動分離を実行することである。分析の手法および処理は、上述のものと同一である。この手法に従う他のタイプの3Dデータに、次が含まれるが、これに制限はされない。
【0163】
UV、蛍光(その蛍光がタンパク質配列のセグメントによって影響される配列特異的な1つまたは複数のタグを用いる)によって検出される他のマルチチャネル・スペクトル検出を用いる2D−LCなど。
【0164】
単一チャネル検出(たとえば、245nmのUV)を用いる3D電気泳動または3D LC。3D分離は、たとえばpI、MW、および疎水性において分離するために無傷のタンパク質に適用することができる。
【0165】
1D電気泳動とその後の消化されたタンパク質または無傷のタンパク質のいずれかに対する1D−LC/MS。
【0166】
2Dゲル分離とその後のMSマルチチャネル検出。消化が必要な場合には、on the gel TOF分析用の適当なMALDIマトリックスを用いてゲルに対して達成することができる。
【0167】
マルチチャネル検出と結合された分離の他の2D手段。
【0168】
2Dスペクトル検出と結合された1D分離、LC/MS/MS。
【0169】
2Dスペクトル検出、たとえば、発光励起2D蛍光(EEM)と結合された1D LCまたは1Dゲル電気泳動。
【0170】
本発明の分析の方法は、ハードウェア、ソフトウェア、またはハードウェアとソフトウェアの組合せで実現することができる。どの種類のコンピュータ・システムでも、あるいは本明細書に記載の方法および/または機能を実行するように適合された他の装置も好適である。ハードウェアとソフトウェアの通常の組合せは、ロードされ実行されたときコンピュータ・システムを制御するコンピュータ・プログラムを有する汎用コンピュータ・システムとすることができ、このコンピュータ・システムは、分析システムが本明細書に記載の方法を実行するように、その分析システムを制御する。本発明は、本明細書に記載の方法の実装を可能にするすべての特徴を含み、コンピュータ・システム(次に分析システムを制御する)にロードされたときこれらの方法を実行することができる、コンピュータ・プログラム製品で実施することもできる。
【0171】
この文脈でのコンピュータ・プログラム手段またはコンピュータ・プログラムは、別の言語、コード、もしくは表記への変換および/または異なる材料形態での再生の後にあるいは直接に、情報処理機能を有するシステムに特定の機能を実行させることを意図された命令の組の任意の表現を、任意の言語、コード、または表記で含む。
【0172】
したがって、本発明は、上述の機能を引き起こすコンピュータ可読プログラム・コード手段が組み込まれたコンピュータ使用可能媒体を含む製造品を含む。この製造品内のコンピュータ可読プログラム・コード手段は、コンピュータに本発明の方法のステップを行わせるコンピュータ可読プログラム・コード手段を含む。同様に、本発明は、上述の機能を引き起こすコンピュータ可読プログラム・コード手段が組み込まれたコンピュータ使用可能媒体を含むコンピュータ・プログラム製品として実施することができる。このコンピュータ・プログラム製品内のコンピュータ可読プログラム・コード手段は、コンピュータに本発明の1つまたはそれ以上の機能を行わせるコンピュータ可読プログラム・コード手段を含む。さらに、本発明は、本発明の1つまたはそれ以上の機能を行うための方法ステップを実行するために機械によって実行可能な命令のプログラムを明らかに実施する、機械によって可読のプログラム・ストレージ・デバイスとして実施することができる。
【0173】
前述したものは、本発明のより関連する目的および実施形態の一部の概要を示したものであることに留意されたい。本発明の概念は、多数の応用分野に使用することができる。したがって、当該説明は特定の装置および方法に関して述べられているが、本発明の意図および概念は、他の装置および応用例に適し、適用可能である。開示された実施形態に対する他の変更を、本発明の趣旨および範囲から逸脱せずにおこなえることが、当業者には明白であろう。説明された実施形態は、本発明のより顕著な特徴および応用例の一部を例示するに過ぎないと解釈すべきである。したがって、前述の説明が、本発明の例示にすぎないことを理解されたい。当業者は、本発明から逸脱せずに、さまざまな代替形態および修正形態を考案することができる。開示された発明を異なる形で適用することまたは本発明を当業者に既知の形で変更することによって、他の有益な結果を実現することができる。したがって、この実施形態が、制限ではなく例として提供されたことを理解されたい。したがって、本発明は、請求項の範囲に含まれるすべての代替形態、修正形態、および変形形態を含むことが意図されている。
【図面の簡単な説明】
【0174】
【図1】質量分析計を含む、本発明による分析システムを示すブロック図である。
【図2】1次元試料分離およびマルチチャネル検出器を有するシステムを示すブロック図である。
【図3】2次元試料分離および単一チャネル検出器を有するシステムを示すブロック図である。
【図4A】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図4B】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図4C】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図5】1試料に関する単一の3次元測定に基づく3次元データ配列を示す図である。
【図6】2次元液相分離とその後の質量スペクトル検出に基づく3次元データ配列を示す図である。
【図7】順次走査を用いるマルチチャネル検出の時間スキュー補正を示す図である。
【図8】本発明による分析の方法を示す流れ図である。
【図9】本発明による、分離軸および対応するプロフィールの自動整列の変換を示す図である。
【図10】3次元データ配列の直接分解を示す図である。
【図11】本発明による、タンパク質への酵素消化から生じたペプチドのクラスタ分析を介するグループ化(デンドログラム)を示す図である。
【技術分野】
【0001】
本願は、2003年4月28日出願の米国仮出願第60/466010号、米国仮出願第60/466011号、および米国仮出願第60/466012号からの優先権を主張するものであり、すべてその全体を本明細書に組み込む。本願は、その内容全体を参照により本明細書に組み込む、2003年10月20日出願の米国仮出願第10/689313号からの優先権も主張するものである。
【0002】
本発明は、化学分析システムに関する。より詳細には、本発明は、タンパク質、環境汚染物、石油化学化合物などの大きい有機分子を含む分子の複雑な混合物の分析に有用なシステム、それに使用される分析の方法、およびコンピュータまたはコンピュータと質量分析計の組合せにそのような分析を行わせるためにコンピュータ・コードが組み入れられたコンピュータ・プログラム製品に関する。さらに具体的には、本発明は、質量分析計部分を有するシステムに関する。
【背景技術】
【0003】
過去数年間のヒト・ゲノム解析競争は、ゲノミクスという名前の新しい化学の分野および産業を生み、ゲノミクスは、DNA配列を研究して、メッセンジャRNA(mRNA)での発現およびタンパク質のもとであるペプチドの後続のコーディングを介して遺伝病の原因である遺伝子および遺伝子突然変異を探す。この分野では、遺伝子が、多数の形の癌を含む多数の病気の根底にあるが、これらの遺伝子から翻訳されたタンパク質が、実際の生物学的機能を実行するものであることが明確に立証されている。したがって、これらのタンパク質およびその相互作用の同定および定量化は、病状の理解および新しい治療学の開発の鍵として働く。したがって、2000年夏にヒト・ゲノム・プロジェクトが成功裡に完了し約35,000種のヒト遺伝子が同定された後で、商業投資と学術研究のどちらにおいても遺伝子(ゲノミクス)からタンパク質(プロテオミクス)に急速にシフトしたことは、驚くに値しない。種ごとにより限定できる終点を有するゲノミクスとは異なって、プロテオミクスは、遺伝子発現レベル、環境要因、およびタンパク質間相互作用の変化がタンパク質変動に寄与する可能性があるので、はるかに制約がない。さらに、個人の遺伝子構造は、比較的安定しているが、タンパク質の発現は、さまざまな病状および他の要因に応じて、はるかに動的なものになる可能性がある。この「ポスト・ゲノミクス時代」における課題は、さまざまな生理学的条件の下での複雑な細胞経路、ネットワーク、および「モジュール」を理解する助けとなるように、組織、細胞、または他の生物試料内の有機体によって発現される複雑なタンパク質(すなわちプロテオーム)を分析することである。正常な状態および病気の状態の両方で発現するタンパク質の同定および定量化は、バイオマーカまたは標的タンパク質の発見でクリティカルな役割を演じる。
【0004】
プロテオミクスという急速に進展する分野が提示する課題は、試料調製、試料分離、イメージング、安定同位体標識(isotope labeling)から、質量スペクトル検出(mass spectral detection)まで、所持する非常に高機能の科学機器をもたらした。ますます高次元になる大量のデータ配列が、全世界の産業界と学究的世界の両方において、ゲノミクスおよびプロテオミクスの成果を獲得しようと競って日常的に生成されている。プロテオミクス研究に通常用いられるタンパク質の複雑さおよび(容易に数千種に達する)数の多さに起因して、複雑で時間がかかり骨の折れる物理的分離が、複合試料内の個々のタンパク質を同定し、時として定量化するために実行される。これらの物理的分離は、単一の試料の内容を完全に解明するのに通常要する日、週、さらには月は言うまでもなく、試料処理および情報追跡に対して大量の課題を生み出す。
【0005】
ヒト・ゲノムには約35,000種の遺伝子しかないが、一般集団について及び治療中
または他の病態にある個人についても研究できるヒト・プロテオームには、推定500,
000種から2,000,000種のタンパク質がある。たとえば細胞、血液、または尿から取られる通常の試料には、通常、大量の数千種に及ぶ異なるタンパク質が含まれる。過去10年間に、産業界は、試料に存在する多数のタンパク質を分析するために、複数のステージを含むプロセスを普及させてきた。このプロセスを、特筆すべき特徴と共に表1に要約する。
【0006】
【表1】
【0007】
a.単一の試料の分析を完了するのに、数日から数週間または数ヶ月を要する可能性がある。
b.巨大なハードウェア・システムは、600,000ドルから100万ドルのコスト
がかかり、かなりの運営コスト(人件費および消耗品)、保守コスト、およびそれと関連した研究室スペース・コストを伴う。
【0008】
c.これは、本質的に、それぞれが固体−液体−固体化学処理の別のサイクルを介して1つずつ分析される必要がある数百個から数千個の個々の固体スポットに1つの液体試料を分離する、複数の異なるロボットおよび少数の異なるタイプの機器を含む極端に時間のかかる複雑なプロセスである。
d.急速に変化する産業界のためにこれらの部分/ステップを一緒に統合することは、小さい課題ではなく、その結果として、これらのステップのすべてを完全に統合し、自動化する市販システムはまだない。このため、このプロセスは、人間の誤りならびに機械の誤差を伴う。
【0009】
e.このプロセスは、途中のすべてのステップからの試料およびデータの追跡も要求するが、これは、現在の情報科学にとってさえ小さい課題ではない。
f.完全な試料およびデータを追跡する情報科学システムを有する完全に自動化されたプロセスについても、これらのデータをどのように管理し、ナビゲートし、そして最も重要なこととして、分析しなければならないかは、明らかでない。
【0010】
g.プロテオミクスのこの早期の段階では、多数の研究者が、タンパク質の定性的同定に満足している。しかし、プロテオミクスの究極の目的は、同定と定量化の両方であり、これによって、卵巣癌診断の血液試料からのタンパク質プロフィールの使用に関する最近の刊行物(Pertricoin,E.F.III他、Lancet、Vol.359、573〜77ページ、2002年)から生じた強い関心によって明示されているように、創薬のためのバイオマーカ同定の領域だけではなく、臨床診断に関する刺激的な応用例の門戸が開かれることになる。現在のプロセスは、タンパク質消失、試料汚染、またはゲル溶解性の欠如に起因して、定量分析に簡単に適合させることができないが、ICAT(同位体コードアフィニティータグ)すなわち、2つの異なる試料供給源からのタンパク質またはタンパク質消化物が同位体原子の対によってラベル付けされ、その後、1つの質量分析法分析で混合される、定量化に対する一般的な手法(Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年)などの複雑な化学プロセスの使用による定量的プロテオミクスの試みが行われてきた。
【0011】
同位体コードアフィニティータグ(ICAT)は、米国カリフォルニア州フォスタ・シティのApplied Biosystems社が最近導入した手法の市販版である。この技法では、2つの異なる細胞プールからのタンパク質が、通常の試薬(軽)および重水素置換された試薬(重)を用いてラベル付けされ、1つの混合物に組み合わされる。トリプシン消化の後に、組み合わされた消化混合物が、ビオチン−アフィニティ・クロマトグラフィによる分離を受けて、システインを含むペプチド混合物がもたらされる。この混合物が、さらに、逆相HPLCによって分離され、データ依存質量分析法およびそれに続くデータベース検索によって分析される。
【0012】
この方法では、複雑なペプチド混合物がシステインを含むペプチド混合物に大幅に単純化され、SEQUESTデータベース検索によるタンパク質同定と重ペプチドに対する軽ペプチドの比率による定量化が同時に可能になる。LC/LC/MS/MSと同様、ICATにより、不溶性問題も回避される。というのは、両方の技法が、分離および分析の前に、タンパク質混合物全体をペプチド断片(peptide fragment)に消化するからである。
【0013】
ICAT技法は、非常に強力であるが、ラベル付けおよび事前分離処理に複数ステップ・プロセスを必要とし、少量タンパク質の消失と、追加の試薬コストをもたらし、さらに、既に低速のプロテオーム解析のスループットを下げる。システインを含むペプチドだけが分析されるので、配列カバレッジ(sequence coverage)は、通常、ICATでは非常に低い。通常のLC/MS/MS実験でそうであるように、タンパク質同定は、うまくいけばシグネチャ・ペプチド(signature peptide)の限られた回数のMS/MS分析を介して達成され、比率定量化に関する唯一の、多くとも少数の標識ペプチドをもたらす。
【0014】
タンデム質量分析と適合された液体クロマトグラフィ(LC/MS/MS)が、プロテイン・シーケンシングに関するえりぬきの方法になってきた(Yates Jr.他、Anal.Chem.67、1426〜1436ページ、1995年)。この方法には、タンパク質の消化、タンパク質消化物から生成されたペプチド混合物のLC分離、得られたペプチドのMS/MS分析、およびタンパク質同定のためのデータベース検索を含む少しのプロセスが含まれる。LC/MS/MSを用いてタンパク質を効果的に同定するための鍵は、データベース検索中の信頼性のあるマッチングを可能にするために、できる限り多くの良い質のMS/MSスペクトルを生じさせることである。これは、四重極機器またはイオン・トラップ機器でのデータ依存走査技法によって達成される。この技法を用いると、質量分析計が、全走査MSスペクトル内で最多のイオンの強度および信号対雑音比を検査し、最多イオンの強度および信号対雑音比が事前に設定された閾値を超えるときMS/MS実験を実行する。通常、3つの最も豊富なイオンが、配列情報を最大にし、必要な時間を最小にするためにプロダクト・イオン走査について選択される。というのも、MS/MS実験に関する3つを超えるイオンの選択は、ことによるとLCから質量分析計に現在溶出している他の資格のあるペプチドの消失をもたらすからである。
【0015】
タンパク質の同定に関するLC/MS/MSの成功は、主に、その多数の顕著な分析特性に起因する。第1に、これは、優れた再現性を有する非常に堅牢な技法である。これは、タンパク質同定に関する高スループットLC/MS/MS分析について信頼性があることが実証されている。第2に、ナノスプレイ・イオン化を使用するとき、この技法は、サブフェンタモル(sub−fentamole)レベルでペプチドの高品質MS/MSスペクトルを生む。第3に、MS/MSスペクトルは、C末端イオンとN末端イオンの両方の配列情報を担持する。この貴重な情報は、タンパク質の同定だけではなく、どの翻訳後修飾(PTM)がタンパク質に生じたかと、どのアミノ酸残基でPTMが起こったかをピンポイントで指摘するのに使用することができる。
【0016】
有機体、細胞系、または組織タイプからの総タンパク質消化物について、LC/MS/MSだけでは、タンパク質の同定に十分な数の良い質のMS/MSスペクトルを生成するのは不十分である。したがって、LC/MS/MSは、通常、2次元電気泳動(2DE)によって分離されたタンパク質などの単一のタンパク質またはタンパク質の単純な混合物の消化を分析するのに使用され、総分析時間に最少の数日を追加し、機器コストと、試料処理の複雑さと、試料追跡のための情報科学の必要を増大させる。全MS走査には試料に関する豊富な情報が含まれる可能性があり、通常は含んでいるが、現在のLC/MS/MS法は、全MS走査内のごく少数のイオンだけについて提供できるMS/MS分析に依拠する。さらに、LC/MS/MSで使用されるエレクトロスプレイ・イオン化(ESI)は、試料からの塩濃度に対して許容範囲が少なく、厳重な試料クリーン・アップ・ステップを必要とする。
【0017】
有機体、細胞系、および組織タイプでのタンパク質の同定は、これらの系の非常に多くのタンパク質(数千種から数万種と推定される)に起因して、極端にむずかしい作業である。LC/LC/MS/MS技術の開発(Link,A.J.他、Nat.Biotechnol.17、676〜682ページ、1999年、およびWashburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年)は、LC分離の1つの特別な次元を追い求めることによってこの課題を処理する試みの1つである。この手法は、タンパク質混合物全体の消化から始まり、強陽イオン交換(SCX)LCを使用して、塩濃度の階段状の勾配によってタンパク質消化物を分離する。この分離は、極端に複雑なタンパク質混合物を比較的単純化された混合物に変えるのに、通常は10〜20ステップを要する。SCXカラムから溶出した混合物は、さらに、逆相LCに導入され、その後、質量分析法によって分析される。この方法は、イーストおよびヒト骨髄性白血病細胞のミクロゾームからの大量のタンパク質を同定することが実証されている。
【0018】
この技法の明白な利益の1つは、2DEでの不溶性問題が回避されることである。というのは、すべてのタンパク質が、ペプチド断片に消化され、このペプチド断片は、通常、タンパク質よりはるかに溶けやすいからである。その結果、LC/LC/MS/MSを用いると、より多くのタンパク質を検出でき、より広いダイナミック・レンジを達成することができる。もう1つの利益は、クロマトグラフ分離が、広範囲の2D LC分離を介してはなはだしく増え、その結果、より完全で信頼性のあるタンパク質同定のために、より高品質のペプチドのMS/MSスペクトルを生成できることである。第3の利益は、この手法が、潜在的に高いスループットのプロテオーム解析のために現在のLC/MSシステムの枠組みの中でたやすく自動化されることである。
【0019】
しかし、LC/LC/MS/MSでの広範囲の2D LC分離は、完了に1〜2日を要する可能性がある。さらに、この技法だけでは、同定されるタンパク質の定量的情報を提供することができず、ICATなどの定量的方式は、試料損失および余分の複雑さと共に余分の時間および労力を必要とする。広範囲の2D LC分離にかかわらず、まだ、MS/MSデータ収集と連続LC溶出の間の時間の制約に起因してMS/MS実験について選択されないかなりの量のペプチド・イオンがあり、配列カバレッジが低くなる(25%カバレッジが、既に非常に良いと考えられる)。LCトレースを後のMS/MS分析用の固体サポートに堆積させる最近の開発は、潜在的に、限られたMS/MSカバレッジの問題に対処できるが、かなり多くの試料処理およびタンパク質損失をもたらし、さらに、試料追跡作業および情報管理作業が複雑になる。
【0020】
マトリックス介助レーザーデソープションイオン化法(MALDI)は、集束レーザー・ビームを使用して、伝導性試料プレート上のマトリックス化合物と共結晶化されているターゲット試料を照射する。イオン化された分子は、パルス化技法としてそれらが共有する特性に起因して、通常、飛行時間型(TOF)質量分析計によって検出される。
【0021】
MALDI/TOFは、その優れた速度、高い感度、広い質量範囲、高い分解能、および汚染物への寛大さのゆえに、2DE分離された無傷のタンパク質を検出するのに一般的に使用されている。遅延抽出および反射型イオン光学系の機能を有するMALDI/TOFは、1〜10ppmの印象的な質量精度と、ペプチドの正確な分析に関して10000〜15000のm/Δmを有する質量分解能を達成することができる。しかし、MALDI/TOFにMS/MS機能がないことが、プロテオミクス応用例での使用に関する主要な制限の1つである。MALDI/TOFでのポストソース分解(PSD)は、ペプチドに関する配列様のMS/MS情報を生成するが、PSDの動作は、しばしば、三連四重極質量分析計またはイオン・トラップ質量分析計の動作ほど堅牢でない。さらに、PSDデータ収集は、ペプチド依存になる可能性があるので、自動化が困難である。
【0022】
新たに開発されたMALDI TOF/TOFシステム(Rejtar,T.他、J.Proteomr.Res.1(2)、171〜179ページ、2002年)は、多くの魅力的な特徴を与える。このシステムは、2つのTOFおよび1つの衝突セルからなり、タンデム四重極システムの構成に似ている。第1TOFは、断片イオンを生成するためにセル内で衝突誘起解離(CID)を受ける前駆イオンを選択するのに使用される。その後、断片イオンが、第2TOFによって検出される。魅力的な特徴の1つは、TOF/TOFが、必要なだけ何度でもデータ依存MS/MS実験を実行できることであり、これに対して、通常のLC/MS/MSシステムは、実験のためにほんの僅かの豊富なイオンだけを選択する。この独自の開発によって、TOF/TOFで産業スケールのプロテオーム解析を実行することが可能になった。提案された解決策は、2D LC実験から分画(fraction)を収集し、MS/MS用のMALDIプレート上に分画をスポッティングすることである。その結果、より多くのMS/MSスペクトルを、データベース検索によるより信頼できるタンパク質同定のために獲得することができる。というのは、TOF/TOF内の高エネルギCIDによって生成されるMS/MSスペクトルの質が、PSDスペクトルよりはるかに良いからである。
【0023】
この手法の主要な短所は、機器の高いコスト(750,000ドル)、長い2D分離、LC分画に関する試料処理の複雑さ、MALDIのための面倒な試料調製プロセス、MALDIを用いる定量化に固有のむずかしさ、ならびにデータおよび試料追跡に関する膨大な情報科学の課題である。LC分離および必要な試料調製時間に起因して、1試料内の数百種のタンパク質の分析は、少なくとも2日を要する。
【0024】
フーリエ変換イオンサイクロトロン共鳴型(FTICR)MSは、高い感度、高い質量分解能、広い質量範囲、および高い質量精度を提供できる強力な技法である。最近、LCと結合されたFTICR/MSが、精密質量タグ(AMT)を介するプロテオーム解析に関する印象的な能力を示した(Smith,R.D.他、Proteomics、2、513〜523ページ、2002年)。AMTは、タンパク質を排他的に同定するのに使用できる、ペプチドの正確なm/z値である。AMT手法を使用することによって、単一LC/FTICR−MS分析が、潜在的に、1ppmより良い質量精度で105種を超えるタンパク質を同定できることが実証された。それでも、AMT単独では、ペプチドのアミノ酸残基特異的翻訳後修飾を正確に指摘するのに不十分である可能性がある。さらに、この機器は、75万ドル以上のコストと高い保守要件を有して非常に高価である。
【0025】
プロテイン・アレイおよびプロテイン・チップは、設計概念において遺伝子発現プロファイリングに使用されるオリゴヌクレオチドチップに似た、新生の技術である(Issaq,H.J.他、Biochem Biophys Res Commun.292(3)、587〜592ページ、2002年)。プロテイン・アレイは、注目のタンパク質との特定の相互作用のために化学的に(陽イオン、陰イオン、疎水性、親水性など)または生化学的に(抗体、受容体、DNAなど)処理された表面を含むプロテイン・チップからなる。これらの技術は、親和性化学作用によって提供される特異性およびMALDI/TOFの高い感度を利用し、タンパク質の高スループット検出を提供する。通常のプロテイン・アレイ実験では、多数のタンパク質試料を、特定の表面化学作用を用いて処理されたチップのアレイに同時に適用することができる。望ましくない化学的バックグラウンドおよび生物分子バックグラウンドを洗い落とすことによって、注目のタンパク質が、アフィニティ・キャプチャリング(affinity capturing)に起因してチップにドッキングし、したがって「純化」される。MALDI−TOFによる個々のチップのさらなる分析が、試料内のタンパク質プロフィールをもたらす。これらの技術は、タンパク質間相互作用の調査に理想的である。というのは、タンパク質を親和性試薬として使用して表面を処理して、他の特定のタンパク質との相互作用を監視できるからである。この技術のもう1つの有用な応用例は、病気診断の潜在的ツールとして、正常な組織の試料と病気の組織の試料の間の比較パターンを生成することである。
【0026】
関係する複雑な表面化学作用と、変性、折り畳み、および溶解性の問題など、タンパク質または他のタンパク質様結合剤によって追加される複雑化とに起因して、プロテイン・アレイおよびプロテイン・チップは、遺伝子チップまたは遺伝子発現アレイほど広い応用分野を有しないと予想される。
【0027】
したがって、過去100年間に、MS計測器に関する長足の進歩が見られ、高スループット、高分解能、および高感度動作のために多数の異なるタイプの機器が設計され、作られてきた。計測器は、ほとんどの市販MSシステムで単一イオン検出をおおむね達成できる段階まで開発され、単位質量分解能は、異なる同位元素から得られるイオン・フラグメントの観察を可能にしている。ハードウェアの高度化と全く対照的に、現代のMS計測器によって生成された大量のMSデータを組織的かつ効果的に分析することに、ほとんど何も行われてこなかった。
【0028】
通常の質量分析計では、ユーザは、通常、注目の質量スペクトルm/z範囲をカバーする複数の断片イオンを有する標準材料を要求されるか、これを供給される。ベースライン効果、同位元素干渉、質量分解能、および質量に対する分解能依存を受けて、少数のイオン・フラグメントのピーク位置は、ピーク頂部での低次多項式あてはめを介して重心またはピーク最大値のいずれかに関して決定される。次に、このピーク位置が、質量(m/z)軸を較正するための1次または高次のいずれかの多項式フィットを介して、これらのイオンの既知のピーク位置にあてはめられる。
【0029】
質量軸較正の後に、通常の質量スペクトル・データ・トレースがピーク分析を受け、ピーク(イオン)が同定されることになる。このピーク検出ルーチンは、ピークの肩、データ・トレース中の雑音、化学的バックグラウンドまたは汚染に起因するベースライン、同位元素ピーク干渉などが考慮される、非常に経験的で複雑な処理である。
【0030】
同定されたピークについて、通常、セントロイディング(centroiding)と称する処理を適用して、積分されたピーク面積およびピーク位置を計算する。上記で概要を示した多数の干渉する要因ならびに他のピークおよび/またはベースラインが存在する中でのピーク面積の決定に特有のむずかしさに起因して、これは、セントロイディング品質の客観的尺度なしで同位元素ピークを出現させるか消滅させる可能性がある多数の調整可能なパラメータに悩まされる処理である。
【0031】
したがって、そのみかけの高度化にかかわりなく、現在の手法は、複数の著しい短所を有する。これには、次が含まれる。
【0032】
質量精度の欠如。現在使用されている質量較正は、通常、単位質量分解能(重要な同位元素ピークの存在または不在を視覚化する能力)を有する従来のMSシステムでの質量決定精度において、0.1amu(m/z単位)より良いものを提供しない。より高い質量精度を達成し、タンパク質同定用のペプチド・マッチングなどの分子フィンガープリント法での曖昧さを減らすためには、かなりコストが高い四重極TOF(qTOF)またはFT ICR MSなどのより高分解能のMSシステムに切り替えなければならない。
【0033】
大きいピーク積分誤差。質量スペクトル・ピーク形状、その可変性、同位元素ピーク、ベースラインおよび他のバックグラウンド信号、ならびにランダム・ノイズの寄与に起因して、現在のピーク面積積分は、強い質量スペクトル・ピークおよび弱いスペクトル・ピークの両方に関して大きい誤差(系統誤差およびランダム誤差の両方)を有する。
【0034】
同位元素ピークに関する問題。現在の手法は、単位質量分解能を有する従来のMSシステムで通常は部分的にオーバーラップした質量スペクトル・ピークを発するさまざまな同位元素からの寄与を分離する良い方法を有しない。経験的手法では、隣接同位元素ピークからの寄与の無視またはその過大評価のいずれかが使用され、優位を占める同位元素ピークに関する誤差と、より弱い同位元素ピークに関する大きい偏りまたはより弱いピークの完全な無視とがもたらされた。多重荷電のイオンがかかわるとき、隣接同位元素ピーク間の質量単位の減らされた分離に起因して、状況はさらに悪くなる。
【0035】
非線形動作。現在の手法では、各ステージ中に多数の経験的に調整可能なパラメータを有する、複数ステージのばらばらな処理が使用される。系統誤差(偏り)は、各ステージで生成され、制御されない予測不能で非線形な形で後のステージに伝搬され、アルゴリズムがデータ処理品質および信頼性の尺度として意味のある統計を報告することを不可能にする。
【0036】
支配的な系統誤差。産業プロセス制御および環境監視からタンパク質同定またはバイオマーカ発見に及ぶほとんどのMS応用例で、機器の感度または検出限度は、常に焦点であり、多くの機器システムで、測定誤差または信号への雑音寄与を最小にするために大いに努力がされてきた。残念ながら、現在使用されているピーク処理手法は、生データのランダム・ノイズよりずっと大きい系統誤差の供給源を作り、したがって、機器の感度または信頼性の制限要因になっている。
【0037】
数学的矛盾および統計的矛盾。現在使用されている多くの経験的手法は、質量スペクトル・ピーク処理全体を、数学的にも統計的にも矛盾したものにしている。ピーク処理結果は、ランダム・ノイズがないわずかに異なるデータまたはわずかに異なる雑音を伴う同一の合成データに対して、劇的に変化する可能性がある。言い換えると、ピーク処理の結果は、堅牢でなく、特定の実験またはデータ収集に依存して不安定になる可能性がある。
【0038】
機器間変動。通常、機械的許容範囲、電磁許容範囲、または環境の許容範囲の変動に起因して、異なるMS機器からの生の質量スペクトル・データを直接に比較することは困難であった。生データに対して適用される現在のアド・ホックなピーク処理を用いると、異なるMS機器からの結果を定量的に比較することのむずかしさが増すだけである。その一方で、異なる機器または異なるタイプの機器からの生質量スペクトル・データを直接に比較するかピーク処理結果を比較する必要が、不純物検出または確立されたMSライブラリの検索を介するタンパク質同定のためにますます高まってきた。
【0039】
2次機器は、各試料のデータの行列を生成し、データ行列が正しく構成されているならば、1次機器より高い分析能力を有することができる。最も広範囲で使用されているプロテオミクス機器であるLC/MSは、通常、現在達成されているものより潜在的にはるかに高い分析能力が可能な2次機器の典型例である。他の2次プロテオミクス機器に、単一UV波長検出を用いるLC/LC、MALDI−TOF MS検出を用いる1Dゲル、MALDI MS検出を用いる1Dプロテイン・アレイなどが含まれる。
【0040】
2次元ゲル電気泳動(2Dゲル)は、細胞または尿などの複雑な生物試料でのタンパク質の分離に広く使用されてきた。通常、タンパク質によって形成されるスポットは、可視イメージング・システムを用いる簡単な同定のために、銀を用いて着色される。これらのスポットは、その後、除去され、酵素を用いて溶解/消化され、MALDIターゲットに移され、乾燥させてMALDI飛行時間型質量分析計を使用してペプチド・シグネチャについて分析される。
【0041】
このプロセスから、次の複数の複雑化が生じる。
1.タンパク質スポットは、特に分離パラメータ(電荷の場合にpI、分子量の場合にMW)の両端で、単一のタンパク質だけを含むことが保証されない。これによって、通常、ペプチド検索が、不可能ではないにしてもむずかしくなる。除去されたスポットごとに追加の液体クロマトグラフィ分離が必要になる可能性があり、分析がさらに低速になる。
【0042】
2.液相から固相(ゲル上の)へ、液相へ戻し(消化のため)、そして最後に再び固相へ(MALDI TOF分析のため)移す生物試料の変換は、誤り、キャリーオーバー、および汚染を受けやすい非常に面倒なプロセスである。
【0043】
3.用いられる試料変換プロセスと、サンプリングおよびイオン化でのMALDI−TOF再生不可能性の事実に起因して、この分析は、定性的であるのみで定量的でないと広く認められてきた。
【0044】
したがって、1次分析および0次分析に対するはなはだしい潜在的な利益および明瞭な利益にもかかわらず、2次機器および2次分析は、これまでは、商品化のきざしがない、試料が少数の合成検体からなる学術研究に制限されてきた。この手法がその巨大な可能性に達するために超えなければならない複数の障壁がある。これには、次が含まれる。
【0045】
a.2次タンパク質分析では、現在事実上すべてのMS応用例で使用されている重心データではなく、生プロフィールMS走査を使用することがさらに重要である。双一次データ構造を維持するために、LCから溶出した特定のイオンの連続MS走査が、同一の質量スペクトル・ピーク形状(明らかに、異なるピーク高さでの)すなわち、セントロイディングおよびデアイソトーピング(de−isotoping)(すべての同位元素ピークを1つの積分された面積に合計すること)によって破壊されるクリティカルな2次構造を有する必要がある。データのセントロイディングからのスティック(stick)は、同一イオンの連続MS走査からの異なる質量位置(0.5amu誤差まで)に現れる。
【0046】
b.より高次の機器および分析分析は、より堅牢な機器および測定プロセスを必要とし、次元のうちの1または2におけるシフトなどのアーチファクトは、学究的世界での最近の進歩(Bro,R.他、J.Chemometrics 13、295ページ、1999年)にもかかわらず、分析の定量的結果および定性的結果さえ深刻に損なう可能性がある(Wang,Y.他、Anal.Chem.63、2750ページ、1991年、Wang,Y.他、Anal.Chem.,65、1174ページ、1993年、およびKiers,H.A.L.他、J.Chemometrics 13、275ページ、1999年)。非線形性または非双一次性などの他のアーチファクトも、複雑化につながる可能性がある(Wang,Y.他、J.Chemometrics、7、439ページ、1993年)。2次プロテオミクス・データの双一次性を維持するために、標準化およびアルゴリズムの訂正を開発する必要がある。
【0047】
c.四重極MSなどの多数のMS機器で、質量スペクトル走査時間は、タンパク質またはペプチドの溶出時間と比較して無視できない。したがって、GC/MSについて報告されたものに似て(Stein,S.E.他、J.Am.Soc.Mass Spectrom.5、859ページ、1994年)、ある質量スペクトル走査で測定されたイオンが、LC溶出中の異なる時点から得られるという重要なスキューが存在する。
【0048】
したがって、プロテオミクス研究がなるであろうものと現在の姿の間には大きいギャップが存在する。
【特許文献1】米国仮出願第60/466010号
【特許文献2】米国仮出願第60/466011号
【特許文献3】米国仮出願第60/466012号
【特許文献4】米国仮出願第10/689313号
【非特許文献1】Pertricoin,E.F.III他、Lancet、Vol.359、573〜77ページ、2002年
【非特許文献2】Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年
【非特許文献3】Yates Jr.他、Anal.Chem.67、1426〜1436ページ、1995年
【非特許文献4】Link,A.J.他、Nat.Biotechnol.17、676〜682ページ、1999年
【非特許文献5】Washburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年
【非特許文献6】Rejtar,T.他、J.Proteomr.Res.1(2)、171〜179ページ、2002年
【非特許文献7】Smith,R.D.他、Proteomics、2、513〜523ページ、2002年
【非特許文献8】Issaq,H.J.他、Biochem Biophys Res Commun.292(3)、587〜592ページ、2002年
【非特許文献9】Bro,R.他、J.Chemometrics 13、295ページ、1999年
【非特許文献10】Wang,Y.他、Anal.Chem.63、2750ページ、1991年
【非特許文献11】Wang,Y.他、Anal.Chem.,65、1174ページ1993年
【非特許文献12】Kiers,H.A.L.他、J.Chemometrics 13、275ページ、1999年
【非特許文献13】Wang,Y.他、J.Chemometrics、7、439ページ、1993年
【非特許文献14】Stein,S.E.他、J.Am.Soc.Mass Spectrom.5、859ページ、1994年
【非特許文献15】Hannesh,S.M.、Electrophoresis 21、1202〜1209ページ、2000年
【非特許文献16】Sanchez,E.他、J.Chemometrics 4、29ページ、1990年
【非特許文献17】Carroll,J.他、Psychometrika 3、45ページ、1980年
【非特許文献18】Bezemer,E.他、Anal.Chem.73、4403ページ、2001年
【非特許文献19】http://www.matrixscience.com
【非特許文献20】http://us.expasy.org/sprot/
【発明の開示】
【発明が解決しようとする課題】
【0049】
本発明の目的は、上述の欠点を克服する、質量分析計を含めることができる化学分析システムと、化学分析システムを動作させる方法を提供することである。
【0050】
本発明のもう1つの目的は、質量分析計を有する化学分析システムを含む化学分析システムに、本発明による方法を実行させるコンピュータ可読プログラム・コードを有する記憶媒体を提供することである。
【課題を解決するための手段】
【0051】
上記および他の目的は、本発明の第1の態様によれば、タンパク質スポット・オーバーラップの存在する状態で質量分析計を使用することなく、無傷のタンパク質から獲得された2Dゲル・イメージング・データを使用して、定性分析および定量分析の両方を実行することによって達成される。さらに、本発明は、より広い母集団範囲にわたって(病気と健康)、同一の母集団のある時間期間にわたって(病気の進行)、および異なる治療方法にまたがって(潜在的な治療に応答して)などのいずれかで収集された多数の異なる試料の間の直接の定量的比較を容易にする。ゲル・スポット割り当ておよびマッチングは、最良の総合結果を作るために、データ分析に自動的に組み込まれる。本発明による手法は、タンパク質同定とタンパク質発現分析の両方の、高速、安価、定量的、かつ定性的なツールを表す。
【0052】
一般に、本発明は、少なくとも2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の少なくとも1つの試料から得られたデータを分析する方法であって、前記2つの変数の関数として表される、少なくとも1つの試料を表すデータを前記システムから得ること、連続するレベルを有するデータ・スタックを形成することであって、各レベルが、少なくとも1つの試料を表す連続データを含むこと、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、変数のうちの第1変数の関数としての成分の第1プロフィール、および変数のうちの第2変数の関数としての成分の第2プロフィールであることを含む方法を対象とする。唯一または単一の試料があることができ、連続データは、時間の関数として試料を表す。連続データは、その成分の質量の関数として単一の試料を表すことができる。代替では、複数の試料があることができ、連続データは、連続試料を表す。
【0053】
本発明は、より具体的には、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の複数の試料から得られたデータを分析する方法であって、2つの変数の関数として表される、複数の試料を表すデータをシステムから得ること、各レベルがデータ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を対象とする。第1プロフィールおよび第2プロフィールは、実質的に純粋な成分のプロフィールを表す。この方法は、第1プロフィールおよび第2プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む。
【0054】
この方法は、標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、データ自体、および第2標準化行列の積の形にすることによってデータを標準化することをさらに含む。第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、2つの変数に対する位置でデータを表現させる値を有することができる。第1標準化行列は、第1変数に対してデータをシフトし、第2標準化行列は、第2変数に対してデータをシフトする。第1標準化行列および第2標準化行列の項は、それぞれ第1変数および第2変数に関してデータの分布形状を標準化するように働く値を有する。第1標準化行列および第2標準化行列の項は、既知の成分を有する試料を装置に適用すること、および、既知の成分によって生成されたデータを第1変数および第2変数に対して正しく位置決めさせる第1標準化行列および第2標準化行列の項を選択することによって決定することができる。項は、標準化されたデータ行列内の第1変数および第2変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定することができる。第1標準化行列および第2標準化行列の項は、試料ごとに計算されることが好ましく、その結果全ての試料に対して最少の誤差を生じる。第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる。第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づくものとすることができる。
【0055】
第1標準化行列および第2標準化行列の項の値は、データ配列R、すなわち
【数1】
を解くことによって決定され、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、p個の試料のすべてのこれらの成分の濃度を含み、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である。
【0056】
分離装置は、2次元電気泳動分離システムとすることができる。第1変数は、等電点であり、第2変数は、分子量である。
【0057】
変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果とすることができる。
【0058】
2つの変数は、質量分析計の質量軸に関連する質量とすることができる。
【0059】
装置は、質量分析計に試料を供給するクロマトグラフィ・システムをさらに含むことができ、保持時間は、2つの変数のうちの他方である。
【0060】
装置は、質量分析計に試料を供給する電気泳動分離システムをさらに含むことができ、試料の移動特性は、2つの変数のうちの他方である。
【0061】
この方法では、データは、連続体質量スペクトル・データであることが好ましい。データは、セントロイディングなしで使用されることが好ましい。時間スキューに関してデータを補正することができる。質量および質量スペクトル・ピーク形状に関してデータの較正を実行することが好ましい。
【0062】
第1変数および第2変数のうちの1つは、複数のタンパク質親和性領域を有するプロテイン・チップ上の領域の変数とすることができる。
【0063】
この方法は、単一チャネル・アナライザを使用することによって、また試料を連続して分析することによって前記データ配列のデータを得ることをさらに含むことができる。単一チャネル・検出器は、光吸収、光放出、光反射、光透過、光散乱、屈折率、化学作用、導電性、放射能、またこれらの任意の組合せのうちの1つに基づくものとすることができる。試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合することができる。
【0064】
本発明は、複数の試料から得られたデータを分析するデータ分析部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、化学分析システムは、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、コンピュータ可読コードは、コンピュータに、2つの変数の関数として表される、複数の試料を表すデータをシステムから得ること、各レベルがデータ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を実行させるコンピュータ可読媒体も対象とする。このコンピュータ可読媒体は、上述の用法のいずれか1つのステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含むことができる。
【0065】
本発明は、さらに、複数の試料から得られたデータを分析する化学分析システムであって、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、2つの変数の関数として表される、複数の試料を表すデータを化学分析システムから得ること、各レベルがデータ試料の1つを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列に分離し、行列は、試料内の各成分の濃度を表す濃度行列、第1変数の関数としての成分の第1プロフィール、および第2変数の関数としての成分の第2プロフィールであることを含む方法を実行する装置を有するシステムを対象とする。この化学分析システムは、上述の方法のいずれかのステップを実行するための機構を有することができる。
【0066】
本発明は、さらに、複数の成分を含む試料の成分を分離する能力を有する分離システム内の試料から得られたデータを分析する方法であって、複数の成分を含む試料を少なくとも第1変数に関して分離し、その分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を含む。第1プロフィール、第2プロフィール、および第3プロフィールは、実質的に純粋な成分のプロフィールを表す。この方法は、さらに、第1プロフィール、第2プロフィール、および第3プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む。
【0067】
この方法は、標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、データ自体、および第2標準化行列の積の形にすることによってデータを標準化することをさらに含む。第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、3つの変数のうちの2つに対する位置でデータを表現させる値を有する。第1標準化行列は、2つの変数のうちの一方に対してデータをシフトし、第2標準化行列は、2つの変数のうちの他方に対してデータをシフトする。第1標準化行列および第2標準化行列の項は、それぞれ2つの変数に関してデータの分布形状を標準化するように働く値を有することができる。第1標準化行列および第2標準化行列の項は、既知の成分を有する試料を装置に適用すること、および、既知の成分によって生成されたデータを2つの変数に対して正しく位置決めさせる第1標準化行列および第2標準化行列の項を選択することによって決定される。
【0068】
項は、標準化されたデータ行列内の2つの変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される。第1標準化行列および第2標準化行列の項は、単一チャネルについて計算することができる。第1標準化行列および第2標準化行列の項は、チャネルについて最小の誤差を生じるように計算される。
【0069】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる。第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づくことが好ましい。
【0070】
本発明によれば、第1標準化行列および第2標準化行列の項の値は、データ配列R、すなわち
【数2】
を解くことによって決定され、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、マルチチャネル・検出器または第3変数に関するこれらの成分の純粋なプロフィールを含み、I(k×k×k)は、k個の成分のすべての濃度を表す唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である。
【0071】
使用される分離装置は、1次元電気泳動分離システムとすることができ、変数は、等電点および分子量のうちの1つである。
【0072】
2つの分離変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果とすることができる。
【0073】
3つの変数のうちの1つは、質量分析計の質量軸に関連する質量とすることができる。
【0074】
使用される装置は、質量分析計に分離された試料を供給する少なくとも1つのクロマトグラフィ・システムをさらに含むことができ、保持時間は、変数のうちの少なくとも1つである。装置は、質量分析計に分離された試料を供給する少なくとも1つの電気泳動分離システムをさらに含むこともでき、試料の移動特性は、変数のうちの少なくとも1つである。データは、連続体質量スペクトル・データであることが好ましい。データは、セントロイディングなしで使用されることが好ましい。
【0075】
この方法は、時間スキューに関して前記データを補正することをさらに含む。この方法は、質量およびスペクトル・ピーク形状に関して前記データの較正を実行することをさらに含む。
【0076】
使用される装置は、複数のタンパク質親和性領域を有するプロテイン・チップを含み、領域の位置は、3つの変数のうちの1つである。
【0077】
使用されるマルチチャネル・アナライザは、光吸収、光放出、光反射、光透過、光散乱、屈折率、化学作用、導電性、放射能、またこれらの任意の組合せのうちの1つに基づくものとすることができる。試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合することができる。
【0078】
使用される装置は、2次元電気泳動分離システムを含むことができ、少なくとも1つの変数のうちの第1は、等電点であり、少なくとも1つの変数のうちの第2は、分子量である。
【0079】
本発明は、試料から得られたデータを分析するデータ分析部分を有し、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、コンピュータ可読コードは、コンピュータに、複数の成分を含む試料を少なくとも第1変数に関して分離し、その分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を実行させるコンピュータ可読媒体も対象とする。このコンピュータ可読媒体は、上記で示した方法のいずれかのステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含むことができる。
【0080】
本発明は、試料から得られたデータを分析する化学分析システムであって、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、その分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、3つの変数の関数として表される、さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、各レベルがマルチチャネル・アナライザの1チャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、データ配列を一連の行列または配列に分離し、行列または配列は、そのスーパーダイアゴナル上の、試料内の各成分の濃度を表す濃度データ配列、第1変数の関数としての各成分の第1プロフィール、第2変数の関数としての各成分の第2プロフィール、および第3変数の関数としての各成分の第3プロフィールであることを含む方法を実行する装置を有するシステムも対象とする。この化学分析システムは、上に記載の方法のステップを実行するための機構をさらに含むことができる。
【0081】
本発明の前述の態様および他の特徴を、添付図面と共に次の説明で説明するが、同一の符号は、同一の構成要素を表す。
【発明を実施するための最良の形態】
【0082】
図1を参照すると、上記で示したようにタンパク質または他の分子の分析に使用できる、本発明の特徴を組み込んだ分析システム10のブロック図が示されている。本発明を、図面に示された単一の実施形態に関して説明するが、本発明は、実施形態の多数の代替形態で実施できることを理解されたい。さらに、すべての適切なタイプの構成要素を使用することができる。
【0083】
分析システム10は、試料調製部分12、質量分析計部分14、データ分析システム16、およびコンピュータ・システム18を有する。試料調製部分12は、米国マサチューセッツ州ウォルサムのThermo Electron Corporation社が製造するFinnegan LCQ Deca XP Maxなど、注目の分子を含む試料をシステム10に導入するタイプの試料導入ユニット20を含むことができる。試料調製部分12は、システム10によって分析されるタンパク質などの検体の予備分離を実行するのに使用される検体分離ユニット22をも含むことができる。検体分離ユニット22は、クロマトグラフィ・カラム、米国カリフォルニア州ハーキュリーズのBio−Rad Laboratories,Inc.社が製造するものなどのゲル分離ユニットのいずれかとすることができ、当技術分野で周知である。一般に、電圧またはPH勾配をゲルに印加して、1次元分離の場合に毛細管を介する移動速度(分子量、MW)および等電点電気泳動点(Hannesh,S.M.、Electrophoresis 21、1202〜1209ページ、2000年)などの1変数の関数として、または等電点電気泳動およびMW(2次元分離)によるなど、これらの変数のうちの複数によって、タンパク質などの分子を分離させる。後者の例を、SDS−PAGEと称する。
【0084】
質量分離部分14は、従来の質量分析計とすることができ、使用可能な任意のものとすることができるが、MALDI−TOF、四重極MS、イオン・トラップMS、またはFTICR−MSのうちの1つであることが好ましい。MALDIまたはエレクトロスプレイ・イオン化イオン源を有する場合に、そのようなイオン源は、質量分析計部分14への試料入力を提供することができる。一般に、質量分析計部分14は、イオン源24、質量対荷電比(または単に質量と呼ばれる)によって、イオン源24によって生成されたイオンを分離する質量スペクトルアナライザ26、質量スペクトルアナライザ26からのイオンを検出するイオン検出器部分28、および質量分析計部分14が能率的に動作するのに十分な真空を維持する真空装置30を含むことができる。質量分析計部分14が、イオン移動度スペクトロメータである場合には、一般に、真空装置は不要である。
【0085】
データ分析システム16には、イオン検出器部分28からの信号をディジタル・データに変換する1つまたは一連のアナログ・ディジタル変換器(図示せず)を含むことができるデータ収集部分32が含まれる。このディジタル・データは、リアル・タイム・データ処理部分34に供給され、リアル・タイム・データ処理部分34は、合計および/または平均などの演算を介してディジタル・データを処理する。後処理部分36は、ライブラリ検索、データ保管、およびデータ報告を含む、リアル・タイム・データ処理部分34からのデータの追加処理を行うのに使用することができる。
【0086】
コンピュータ・システム18は、下記で説明する形で、試料調製部分12、質量分析計部分14、およびデータ分析システム16を制御する。コンピュータ・システム18は、適切なスクリーン・ディスプレイでのデータの入力および実行された分析の結果の表示を可能にするために、従来のコンピュータ・モニタ40を有することができる。コンピュータ・システム18は、たとえばWindows(登録商標)またはUNIX(登録商標)オペレーティング・システムあるいは他の適切なオペレーティング・システムを用いて動作する、適切なパーソナル・コンピュータに基づくものとすることができる。コンピュータ・システム18は、通常はハード・ドライブ42を有し、このハード・ドライブ42に、オペレーティング・システムと、下記で説明するデータ分析を実行するプログラムとが保管される。CDまたはフロッピ・ディスクを受け入れるドライブ44が、本発明によるプログラムをコンピュータ・システム18にロードするのに使用される。試料調製部分12および質量分析計部分14を制御するプログラムは、通常、システム10のこれらの部分のファームウェアとしてダウンロードされる。データ分析システム16は、下記で述べる処理ステップを実施するために、C++、JAVA、またはVisual Basicなどの複数のプログラミング言語のいずれかで記述されたプログラムとすることができる。
【0087】
図2は、試料調製部分12に試料導入ユニット20および1次元試料分離装置52が含まれる分析システム50のブロック図である。たとえば、装置52は、1次元電気泳動装置とすることができる。分離された試料成分が、たとえば一連の紫外線センサまたは質量分析計などのマルチチャネル検出装置54によって分析される。データ分析を行える形を、下記で述べる。
【0088】
図3は、試料調製部分12に、試料導入ユニット20、第1次元試料分離装置62、および第2次元試料分離装置64が含まれる分析システム60のブロック図である。たとえば、第1次元試料分離装置62および第2次元試料分離装置64は、2つの連続する異なる液体クロマトグラフィ・ユニットとすることができ、あるいは、2次元電気泳動装置として合体させることができる。分離された試料成分は、たとえば245nm帯域フィルタを有する紫外線センサ、またはグレイ・スケール・ゲル・イメージャ(gel imager)など、単一チャネル検出装置66によって分析される。また、データ分析を行える形を、下記で述べる。
【0089】
図4Aに、分析される成分の混合物の連続試料を表す、一連の2次元配列72Aから72Nよりコンパイルされた3次元データ配列70を示す。2次元データ配列72Aから72Nは、たとえば、図3に関して上述したように2次元ゲル電気泳動または連続クロマトグラフ分離、あるいは他の分離技法の組合せによって作ることができる。
【0090】
図4Bに、分析される成分の混合物の連続試料を表す、一連の2次元配列76Aから76Nよりコンパイルされた3次元データ配列74を示す。2次元データ配列76Aから76Nは、たとえば、図2に関して上述したように1次元ゲル電気泳動または液体クロマトグラフィとその後のマルチチャネル分析、あるいはガス・クロマトグラフィ/赤外線スペクトル法(GC/IR)またはLC/蛍光などの他の技法によって作ることができる。
【0091】
図4Cに、分析される成分の混合物の連続試料を表す、一連の2次元配列80Aから80Nからコンパイルされた3次元データ配列78を示す。2次元データ配列80Aから80Nは、たとえば、米国カリフォルニア州フレモントのCiphergen Biosystems,Inc.社が販売するタイプの、表面で画定された領域(スポット)にタンパク質を選択的に結合することのできるタンパク質親和性チップと、その後の、図2に関して説明したものなどのシステムのうちの1つとすることができる、表面増強レーザー脱離/イオン化(SELDI)飛行時間型質量分析法などのマルチチャネル分析によって作られる。使用できる他の技法は、マルチチャネル蛍光検出と組み合わされた1Dプロテイン・アレイである。
【0092】
図5に、分析されるべき成分の混合物の単一の試料を表す、一連の2次元配列84Aから84Nからコンパイルされた3次元データ配列82を示す。2次元データ配列84Aから84Nは、たとえば、図1に関して上述したように、2次元ゲル電気泳動または連続液体クロマトグラフィによって作ることができる。たとえば、図1に関して上述したものなどの質量分析法によるマルチチャネル検出は、第3次元のデータを作る。他の適切な技法は、マルチチャネルUV検出またはマルチチャネル蛍光検出を用いる2D LC、IR検出を用いる2D LC、質量分析法を用いる2Dプロテイン・アレイである。
【0093】
図6に、2次元液相分離(たとえば、強陽イオン交換クロマトグラフィとその後の逆相クロマトグラフィ)によって得られたデータ配列84を示す。第3次元は、質量スペクトル検出からの質量軸86に沿ったデータによって表されている。
【0094】
図4A、図4B、図4C、図5、および図6のデータ配列には、場合によっては、すべての試料または単一の試料のすべての成分を表す項が含まれる(較正標準の成分を含む)。
【0095】
図7に、所定の走査時間中のある質量範囲を掃引する質量分析計にLCが接続されるLC/MSの場合にそうであるように、時間ベースの分離に接続された走査型マルチチャネル検出器の時間スキューの補正を示す。このタイプの時間スキューは、すべての質量のイオンを同時に検出する磁気セクタ・システム(magnetic sector system)などの同時システムを除く質量分析計のほとんどに存在する。他の例には、揮発化合物が、IRスペクトルが走査モノクロメータまたは干渉計のいずれかを介して獲得されている間にカラムを通過した後の保持時間に関して分離されるGC/IRが含まれる。分離または反応などの時間依存事象が、複数のチャネルを順次走査する検出システムに接続されているとき、前に走査されたチャネルが事象の以前の時点に対応し、後に走査されたチャネルが事象の後の時点に対応する時間スキューが生成される。この時間スキューは、すべてのチャネルについて同一の時点に対応するマルチチャネル・データを生成するすなわち、図7の傾いた実線から対応する破線の水平線へ各チャネルを補間するために、チャネルごとの基礎での補間によって補正することができる。
【0096】
図8は、本発明に従って試料データを獲得し、処理する方法の一般的な流れ図である。生物試料などの試料の収集および処理が、100で行われる。単一の試料を処理する場合には、102で3次元データを獲得する。106で複数の試料を用いて2次元データを獲得する場合には、任意選択で、104で内部標準を試料に追加する。上記の技法およびシステムのいずれかに関して説明したように、108で、3次元データ配列を形成する。110で、3次元データ配列が、直接分解を受ける。2次元測定を行ったか否かに基づいて、112で異なる経路を選択する。2次元測定が行われた場合には、114で、各次元の純粋な検体プロフィールを、すべての試料にまたがるそれぞれの濃度と共に得る。3次元測定が単一の試料に対して行われた場合には、116で、3つのすべての次元に沿った試料のすべての検体の純粋な検体プロフィールを得る。どの場合でも、検体グループ化、クラスタ分析、ならびに他のタイプの発現および分析を含むデータ解釈を、118で行い、その結果を、図1、図2、または図3のうちの一つのシステムに関連するコンピュータ・システム18のモニタ40で報告する。
【0097】
データの分析のモードを、本発明の範囲の制限ではなく理解を容易にするために提供される特定の例に関して、下記で説明する。
【0098】
通常の試料の応答行列Rj(m×n)を次の双一次形で表現できる場合:
【数3】
ここで、ciは、第i検体の濃度であり、xi(m×1)は、行軸に沿ったこの検体の応答(たとえば、LC/MSでのこの検体のLC溶出プロフィールまたはクロマトグラム)であり、yi(n×1)は、列軸に沿ったこの検体の応答(たとえば、LC/MSでのこの
検体のMSスペクトル)であり、kは、試料内の検体の数である。複数の試料(j=1,2,…,p)の応答行列がコンパイルされるとき、3Dデータ配列R(m×n×p)を形成することができる。
【0099】
したがって、2Dゲル・ランの終りに、グレイスケール・イメージを生成し、2D行列Rj(m×nの次元を有し、試料jの、行にディジタル化されたm個の異なるpI値および列にディジタル化されたn個の異なるMW値に対応する)で表すことができる。この生イメージ・データは、標準化されたイメージRjを作るために、pI軸とMW軸の両方で較正される必要がある。
Rj=AjRjBj
ここで、Ajは、m×mの次元を有し、主対角に沿っておよびその周囲に非0要素を有する正方行列(帯対角行列)であり、Bjは、主対角に沿っておよびその周囲に非0要素を有するもう1つの正方行列(n×n)(もう1つの帯対角行列)である。行列AjおよびBjは、対角行列(単純な線形スケーリングを表す)のように単純なものまたは主対角に沿って増加するもしくは減少する帯域幅を有するもの(バンド・シフト、拡幅、およびひずみのうちの少なくとも1つまたは他のタイプの非線形に補正する)のように複雑なものになる可能性がある。上式の一般形でのグラフィカル表現を、図9に示されているように与えることができる。
【0100】
【数4】
複数の試料からの2−Dゲル・データが収集されるとき、Rjの集合を配置して、3Dデータ配列Rを
【数5】
として形成することができ、ここで、pは、生物試料の数であり、Rは、m×n×pの次元を有する。このデータ配列(立方体または直方体)は、GRAM(Generalized Rank Annihilation Method、反復なしの行列演算を介する直接分解、Sanchez,E.他、J.Chemometrics 4、29ページ、1990年)またはPARAFAC(PARAllel FACtor analysis、交互最小二乗法を用いる反復分解、Carroll,J.他、Psychometrika 3、45ページ、1980年およびBezemer,E.他、Anal.Chem.73、4403ページ、2001年)に基づくトリリニア分解法を用いて、4つの異なる配列および残余データ配列Eに分解することができる。
【0101】
【数6】
ここで、Cは、p個の試料のすべてでのすべての同定可能タンパク質(そのうちのk個、k≦min(m,n))の相対濃度を表し、Qは、各タンパク質(そのうちのk個)のm個のpI値でディジタル化されたpIプロフィールを表し、Wは、各タンパク質のn個の値でディジタル化された分子量プロフィール(理想的には、各タンパク質に対応する単一のピークが観察される)であり、Iは、唯一の非0要素としてそのスーパーダイアゴナル(super−diagonal)にスカラを有する新しいデータ・キューブである。
【0102】
すべてのタンパク質が別個であり(異なるpI値および異なるMWを有する)、発現レベルが、試料ごとに線形独立の形で変動するとき、結果の次の直接解釈を予想することができる。
1.上記の分解からのk値は、自動的に、タンパク質の数と等しくなる。
2.行列Cの各行の値は、Iのスーパーダイアゴナル要素によるスケーリングの後に、特定の試料内のこれらのタンパク質の相対濃度を表す。
3.行列Qの各列は、特定のタンパク質の逆畳み込みされたpIプロフィールを表す。
4.行列Wの各列は、特定のタンパク質の逆畳み込みされたMWプロフィールを表す。
【0103】
これらのタンパク質が、別個であるが、試料ごとに相関する発現レベルを有する(行列Cが線形依存列を有する)場合に、解釈は、各個々のタンパク質ではなく、相関する発現レベルを有するタンパク質のグループに対してのみ実行することができ、プロテオミクス研究の重要性の発見である。
【0104】
上記で提示した分解に基づいて、そのような多次元のシステムおよび分析の威力を、すぐに見ることができる。
【0105】
a.成分応答を各次元の個々のタンパク質応答の線形組合せに分離するこの分解の結果として、他のすべてのタンパク質の存在下で、各タンパク質について量的情報を得ることができる。
【0106】
b.この分解は、各個々のタンパク質のプロフィールを各次元で分離し、両方の次元(2DEでpIおよびMW、LC/MSでクロマトグラフ次元および質量スペクトル次元)でのこれらのタンパク質の同定に関する質的情報も提供する。
【0107】
c.3Dデータ配列Rの各試料に、タンパク質の異なる集合が含まれる可能性があり、注目のタンパク質を、未知のタンパク質の存在下で同定でき、定量化でき、データ配列のすべての試料が共有する共通タンパク質だけが、分解された行列Cですべて非0濃度を有することが暗示される。
【0108】
d.この分析には、最低限で別個の試料が2つ必要とされるだけであり、他の関心のないタンパク質の存在下で注目のタンパク質を素早く信頼できる形でとりあげるために、ICATのようなラベルなしで異なるプロテオーム解析を実行するはるかに良い方法を提供する。
【0109】
e.分析できる検体の数は、応答行列Rjごとの最大の許容可能な擬似ランクによって制限され、これは、簡単に数千(イオン・トラップMS)または数十万(TOFまたはFTICR−MS)に達することができ、複雑な生物試料に対する大規模なプロテオーム解析が可能になる。
【0110】
f.通常のLC/MSランは、関係する他の化学プロセスまたは試料調製ステップを用いることなく2時間未満で完了することができ、スループットにおける少なくとも10倍の向上および情報科学における著しい単純化を示す。
【0111】
g.完全なLC/MSデータが分析に使用されるので、MS/MS実験なしでほぼ100%の配列カバレッジを達成することができる。
【0112】
2−Dゲル分離のイメージに基づく上記の分析の重要な利益は、これが非破壊的であり、たとえばMALDI TOFの使用を介してさらなる確認を引き続き行えることである。上記の分析は、同一のタンパク質からのすべてのペプチドを分析および解釈に関して別個のグループとして扱うことができるタンパク質消化物にも適用することができる。pIプロフィールおよびMWプロフィールの個々のタンパク質への分離は、個々のペプチドへの分離が実現可能でない時であっても実行することができる。
【0113】
左右の変換行列AjおよびBjを、好ましくは各試料に追加される内部標準を使用して決定することができる。これらの内部標準は、すべてのpI範囲およびMW範囲をカバーするように選択され、たとえば、2Dゲル・イメージの各角に1つと中央の右1つの5つの内部標準が選択される。これらの内部標準の濃度は、試料ごとに変化し、その結果、上記の分解の対応する行列Cを、
C=[Cs|Cunk]
として区分することができ、ここで、Csのすべての列が独立すなわち、Csが最大階数であるか、よりよくは、最大の特異値と最小の特異値の間の比が最小にされる。上記の分解において既知の行列Cの一部を用いて、各試料の変換行列AjおよびBj(j=1,2,…,p)を同一の分解処理で決定して総合的な残余Eを最小にすることができるように分解を実行することができる。問題のスケールは、AjおよびBjの非0対角帯をパラメータ化することによって、たとえば、Ajの各行およびBjの各列についてガウス形状の帯を広げるフィルタを指定し、Ajの行を下げおよびBjの列を横切る(across)ガウス・パラメータの滑らかな変動を可能にすることによって、劇的に減らすことができる。行列AjおよびBjが正しくパラメータ化され、パラメータに関する導関数の分析方法を導出したならば、効率的なガウス−ニュートン反復手法をトリリニア分解またはPARAFACアルゴリズムに適用して、各試料の所望の分解および正しい変換行列AjおよびBjの両方に達することができる。
【0114】
ICAT(同位体コードアフィニティータグ、Gygi,S.P.他、Nature Biotech.1999年、17、994ページ)と比較して、この手法は、2つの試料だけの分析に制限されず、タンパク質同定にペプチド・シーケンシングを必要としない。定量化できる試料の数は、数百から数千、あるいは数万にさえなる可能性があり、タンパク質同定は、すべてのこれらのタンパク質が数学的に解かれ、分離されたならば、質量スペクトル・データだけを介して達成することができる。さらに、同位元素ラベルを用いる追加の化学作用がなく、これによって、ICATに必要な面倒な試料調製ステージ中に多数の重要なタンパク質を失う危険性が減る。
【0115】
短く言うと、本発明は、上述の分析の方法を使用して、次の特徴を有する2Dデータを使用するタンパク質同定およびタンパク質発現分析の技法を提供する。
−複数の試料からの2Dゲル・データが、3Dデータ配列を形成するのに使用される。
−次のシナリオのそれぞれについて、適用可能な解釈の異なる組がある。
a)すべてのタンパク質が別個であり、発現レベルが試料の間で独立に変化する場合
b)すべてのタンパク質が別個であり、試料の間に相関する発現レベルがある場合
−質量スペクトル連続体データに対するセントロイディングが回避される。
−生の質量スペクトル・データだけを、直接に使用することができ、データ配列分解への入力としてこれだけで十分である。
−たとえば米国仮出願第10/689313号で実行されたもののような、完全な質量スペクトル較正を、生連続体データに対して任意選択で実行して、分析への入力としての完全に較正された連続データを得ることができ、逆畳み込み質量スペクトルが配列分解後に個々のタンパク質について使用可能になったならば、タンパク質同定のためにさらに正確な質量決定およびライブラリ検索が可能になる。
−この手法は、タンパク質を分解し、分離するのに物理的シーケンシングではなく数学に基づき、タンパク質同定にペプチド・シーケンシングを必要としない。
−結果は、定性的であると同時に定量的である。
−ゲル・スポットの整列およびマッチングは、データ分析に自動的に組み込まれる。
【0116】
さらに、その短い要約を下記で示す同時係属の米国仮出願第10/689313号に記載のように、質量整列およびスペクトル・ピーク形状一貫性をさらに改善するために、本発明で完全に較正された連続質量スペクトル・データを有することが好ましい。
【0117】
完全に較正された連続質量スペクトル・データの作成
m=f(m0) (式A)
の形の較正関係を、測定された重心と、質量範囲にまたがる質量スペクトル標準で使用可能なすべての明瞭に同定可能な同位元素クラスタを使用して計算された重心との間での最小二乗多項式フィットを介して確立することができる。
【0118】
この単純な質量較正に加えて、追加の完全なスペクトル較正フィルタが、同時に2つの目的すなわち質量スペクトル・ピーク形状および質量スペクトル・ピーク位置の較正に役立つために計算される。質量軸は、事前に較正することができるので、フィルタ機能の質量較正部分は、この場合に、質量較正でのさらなる洗練を達成するためすなわち式Aによって与えられる多項式フィットの後の任意の残留質量誤差を考慮に入れるために縮小される。
【0119】
この較正処理全体は、質量スペクトル・ピーク幅(半値全幅またはFWHM)が一般に動作質量範囲内でほぼ一定である、イオン・トラップを含む四重極タイプのMSに簡単に適用される。磁気セクタ、TOF、またはFTMSなどの他のタイプの質量分析計システムについて、質量スペクトル・ピーク形状は、動作原理および/または特定の機器設計によって指定される関係で、質量に伴って変化すると予想される。同一の質量依存較正手順が、それでも適用可能ではあるが、ピーク幅/位置と質量の間の所与の関係と一貫する変換されたデータ空間で較正全体を実行することが好ましい可能性がある。
【0120】
TOFの場合に、質量スペクトル・ピーク幅(FWHM)Δmが、次の関係で質量(m)に関係することが既知である:
【数7】
ここで、aは、既知の較正係数である。言い換えると、質量範囲にわたって測定されたピーク幅は、質量の平方根に伴って増加する。平方根を用いると、質量軸から新しい関数に変換する変換は、次のようになる:
【数8】
ここで、変換後の質量軸で測定されたピーク幅(FWHM)は、
【数9】
によって与えられ、これは、スペクトル範囲全体にわたって変化しない。
【0121】
その一方で、FT MS機器について、ピーク幅(FWHM)Δmは、質量mに正比例し、したがって、対数変換が必要である:
m’=ln(m)
ここで、変換後の対数空間で測定されたピーク幅(FWHM)は、
【数10】
によって与えられ、これは、質量と独立に固定されている。通常、FTMSでは、Δm/mを10-5程度にすなわち、分解能m/Δmに関して105に管理することができる。
【0122】
磁気セクタ機器について、特定の設計に依存して、スペクトル・ピーク幅および質量サンプリング間隔は、質量との既知の数学的関係に従い、これはそれ自体、TOFおよびFTMSに関して平方根変換および対数変換が行うのによく似た形で、期待される質量スペクトル・ピーク幅がそれを介して質量と独立になる特定の形の変換に役立つ可能性がある。
【0123】
FTMSでの対数変換およびTOF−MSでの平方根変換などの適切な変換または十分に設計され正しく調整された四重極MSまたはイオン・トラップMSなどの特定の機器に固有の性質のいずれかに起因して、予想される質量スペクトル・ピーク幅が、質量と独立になるとき、計算時間の大幅な節約が、質量スペクトル範囲全体に適用可能な単一の較正フィルタを用いて達成される。これによって、質量スペクトル較正標準に対する要件も単純化される。すなわち、単一の質量スペクトル・ピークが較正に必要とされ、(存在する場合には)追加ピークが検査または確認としてだけ働き、測定される各試料に追加された内部標準に基づくすべてのMSの完全な質量スペクトル較正のために道を開く。
【0124】
通常、質量スペクトル較正全体を達成する上で2つのステップがある。第1ステップは、実際の質量スペクトル・ピーク形状関数を導出することであり、第2ステップは、導出された実際のピーク形状関数を、正しい質量位置を中心とする指定された目標ピーク形状関数に変換することである。測定された生連続質量スペクトルy0を有する内部標準または外部標準は、標準イオンまたは標準イオン・フラグメントの同位元素分布yに、
【数11】
によって関係する。ここで、pは、計算された実際のピーク形状関数である。この実際のピーク形状関数は、その後、
【数12】
によって与えられる1つまたはそれ以上の較正フィルタを介して、指定された目標ピーク形状関数t(たとえば、あるFWHMのガウシアン)に変換される。
【0125】
上記で計算された較正フィルタを、次の帯対角フィルタ行列に配置することができる:
【数13】
ここで、対角線上の各短い列ベクトルfiは、対応する中心質量に関する上記で計算された畳み込みフィルタからとられる。fiの要素は、逆の順序の畳み込みフィルタの要素から取られる、すなわち
【数14】
である。
【0126】
例として、この較正行列は、1/8amuデータ間隔で1000amuまでの質量カバレッジを有する四重極MSで8000×8000の次元を有する。しかし、その疎な性質に起因して、通常のストレージ要件は、5amu質量範囲をカバーする40要素の有効フィルタ長で約40×8000だけである。
【0127】
本発明に戻ると、さらなる多変量統計分析を行列Cに適用して、異なる試料と異なるタンパク質の間の関係を研究し、理解することができる。試料およびタンパク質をグループ化するかクラスタ分析して、どのタンパク質がどの試料グループ内により多く発現するかを調べることができる。たとえば、C行列の主成分分析からのスコアまたはローディングを使用して、デンドログラムを作成することができる。典型的な結論は、健康な個人からの細胞試料はお互いの周囲にクラスタ化するが、病気の個人からの細胞試料は異なるグループでクラスタ化することを含む。ある治療の後のある時間期間にわたって収集された試料について、その試料が、タンパク質レベルに関して治療に対する生物反応を示す、あるタンパク質の発現レベルの継続的変化を示す場合がある。一連の投薬量に対して収集された試料について、関連するタンパク質の変化が、タンパク質のこの集合に対する投薬量の影響およびその潜在的調整を示す可能性がある。
【0128】
タンパク質が分析の前にペプチドに事前消化される場合に、行列Cの各列は、同一のタンパク質から得られるペプチドのグループまたは試料の間で類似する発現パターンを示すタンパク質のグループの線形組合せを表す。図11に示されたものなどの行列Cの列を分類するために実行されたデンドログラムでは、個々のペプチドがそれぞれのタンパク質にグループ化され、したがって、プロテオーム・レベルでの分析が達成される。
【0129】
同定されたタンパク質の定性的(またはシグナトリ(signatory))情報を、pIプロフィール行列QおよびMW行列Wで見つけることができる。定性的情報は、特に分子量情報が十分な精度で決定される場合に、タンパク質同定およびライブラリ検索のために働くことができる。要約すると、3つの行列C、Q、およびWは、組み合わされたとき、AjおよびBjによって表される変換行列の決定からの自動ゲル・マッチングおよびスポット整列と共に、タンパク質の定量化と同定の両方を可能にする。
【0130】
上記の2−Dデータは、異なる形式および形状になる可能性がある。2−Dゲル・スポットの除去/消化の後のMALDI−TOFに対する代替は、これらの試料を従来のLC/MS、たとえばThermal Finnigan LCQシステムにかけて、MS分析の前に各ゲル・スポットからタンパク質をさらに分離することである。この手法の非常に重要な応用は、2−Dゲル(2DE)分離を完全に回避することによる素早く直接のタンパク質の同定および定量化を可能にし、したがって、スループットを数桁高める。これは、次のステップを介して達成することができる。
【0131】
1.分離なしで数十万種のタンパク質を含む試料を直接に消化する
2.消化された試薬を従来のLC/MS機器にかけて、2次元配列を得る。MS/MS機能はこの場合に不要であるが、試料をLC/MS/MSシステムにかけることを選択することができ、これによって追加のシーケンシング情報が生成されることに留意されたい。
【0132】
3.複数の試料について1および2を繰り返して、3次元データ配列を生成する。
4.上記で概要を示した手法を使用してデータ配列を分解する。
【0133】
5.タンパク質同定のための解釈および質量スペクトル検索で、pI軸をLC保持時間に置換し、MW軸を質量軸に置換する。数学的に分離された質量スペクトルを、セントロイディングおよびデアイソトーピングを介してさらに処理して、http://www.matrixscience.comまたはhttp://us.expasy.org/sprot/からオンラインで使用可能なMascotまたはSwissProtなどの従来のデータベースおよび検索エンジンと互換のスティック・スペクトルを作ることができる。しかし、データ配列分解の前に、生の質量スペクトル連続体データを較正済み連続体データに完全に較正し、各逆畳み込みされるタンパク質またはペプチドについて完全に較正された連続体質量スペクトル・データを生み出すことが好ましい。この連続体質量スペクトル・データは、その後、セントロイディングなしのその高い質量精度と共に、同時係属の特許の新規のデータベース検索を介するタンパク質同定に使用される。
【0134】
LCカラムの性質に応じて、LCは、2−DゲルのpI軸に似た別の形の電荷分離として働くことができる。この場合の質量分析計は、2−Dゲル分析のWM軸に似た、分子量測定の正確な手段として働く。質量分析計で使用可能な高い質量精度に起因して、変換行列Bjを対角行列に縮小して、質量依存のイオン化効率変化を訂正するか、または特に上述の完全な質量スペクトル較正の後に、単位行列を式から消すことができる。大きいタンパク質分子を扱うために、タンパク質試料は、通常、酵素または化学反応、たとえばトリプシンの使用を介してペプチドに事前消化される。したがって、通常、注目のタンパク質ごとに、複数のLCピークならびに複数の質量が見られる。これによって、試料処理が複雑になる可能性があるが、ライブラリ検索およびタンパク質同定の選択性が大幅に強化される。複数の消化を使用して、選択性をさらに強化することができる。極端に言うと、各タンパク質を事前にさまざまな長さのペプチドに消化して(Erdman degradation)、行列Wから完全なタンパク質配列情報を作ることができる。これは、LCタンデム質量分析に対する代替としての、物理シーケンシングではなく数学に基づくタンパク質シーケンシングの新しい技法である。MSを含む応用例で、この手法は、多数の非系統誤差を受けやすい市販計測器で通常行われているセントロイディングおよびデアイソトーピングなどの質量走査からの連続データに対するデータ処理を必要としない。生のカウント・データをデータ配列分解への入力として供給し、直接に使用することができる。
【0135】
同一の手法を用いて類似する結果を作ることができる他の2−Dデータに、単一点検出を用いる2−D分離、または複数チャネル検出もしくは2−Dマルチチャネル検出を用いる1−D分離を有する以下のが含まれるが、これに制限はされない。
【0136】
1.1−Dまたは2−Dのゲル・スポットのそれぞれを、後続のLC/MS分析に関して独立の試料として扱って、スポットごとに1つのLC/MS 2−Dデータ配列と、すべてのゲル・スポットおよびそのLC/MSデータ配列を含むデータ配列とを生成することができる。ゲルおよびLC分離の両方から得られる追加の解像度に起因して、より多くのタンパク質をより正確に同定することができる。
【0137】
2.pI/疎水性、MW/疎水性などの他のタイプの2−D分離、またはpI、MW、もしくは疎水性のいずれかを使用する1−D分離と、on−the−gel MALDI TOFと組み合わされた1−Dゲル、またはLC/TOF、LC/UV、LC/蛍光などのマルチチャネル電磁検出または質量スペクトル検出のある形を使用することができる。
【0138】
3.単一チャネル検出(245nmのUV、または1波長で測定されるようにタグ付けされた蛍光)を用いる、2−D液体クロマトグラフィなどの他のタイプの2−D分離を使用することができる。
【0139】
4.アレイの各要素がLCカラムと異ならない形でタンパク質の特定の組合せを取り込む、質量スペクトル検出または他のマルチチャネル検出と結合された1Dまたは2Dのプロテイン・アレイを使用することができる。これらの1Dまたは2Dのスポットを、2−D配列の1次元に配置し、他方の次元を質量分析法とすることができる。これらのタンパク質スポットは、表面弾性波センサ(SAW、あるクラスの化合物に選択的に結合するためにGCカラム材料をコーティングされている)などのセンサ・アレイ、またはその上での結合事象が別個の電気信号を生成する導電性重合体アレイなどの電子鼻に似ている。
【0140】
5.差別的にまたはタンパク質配列のセグメントに特異的にタグ付けされた異なるタンパク質を有する単一の試料に対する複数波長EEM(emission and excitation fluorescence)を使用することができる。
【0141】
2次プロテオミクス分析では、データ配列が、複数の試料からの2D応答行列によって形成される。データ配列を作成するもう1つの効果的な形は、3次機器と呼ばれるもので単一の試料からデータ配列を生成できるように、測定自体にもう1つの次元を含めることである。プロテオミクスで広く注目を集め始めているそのような機器の1つが、試料に存在する各タンパク質のLC次元とMSスペクトル応答の両方で数学的に分離された溶出プロフィールを作るために同一の分解に従うLC/LC/MSである。
【0142】
したがって、上記で概要を示した2次元手法は、当技術分野における主要な改善であるが、3次元手法は、試料がプロセス全体を通じて液相のままであるという事実から生じる、より高速な、より再現可能な、単純なものであるという利益を有する。しかし、多数のタンパク質は、従来の質量分析計には大きすぎ、試料内のすべてのタンパク質が、LC分離および質量スペクトル検出の前にペプチド断片に消化される可能性があるので、ペプチドの数およびシステムの複雑さが、少なくとも1桁増える。これは、データ・ハンドリングおよびデータ解釈に関する解決しにくい問題と思われるものをもたらす。さらに、使用可能な手法は、イーストなどの非常に限られた複雑さの試料の定性的タンパク質同定のレベルで止まる(Washburn,M.P.他、Nat.Biotechnol.19、242〜247ページ、2001年)。下記に提示する手法は、単一の2次元液体クロマトグラフィ−質量分析法(LC/LC/MSまたは2D−LC/MS)ランで数百種から数万種のタンパク質の同定と定量化の両方を達成する。
【0143】
たとえば、サイズ排除逆相液体クロマトグラフィ(SEC−RPLC)または強陽イオン交換逆相液体クロマトグラフィ(SCX−RPLC)のいずれかを、最初の分離に使用することができる。これに、エレクトロスプレイ・イオン化(ESI)質量分析法または飛行時間型質量分析法のいずれかの形の質量分析検出(MS)が続く。生成されたデータの集合は、3次元データ配列Rに配置され、このRには、保持時間(t1およびt2、m個およびn個の異なる時点でディジタル化された、各LC次元での保持時間、たとえばSEC保持時間およびRPHL保持時間に対応する)と質量(注目の質量範囲をカバーするp個の異なる値でディジタル化された)の異なる組合せでの質量強度(カウント)データが含まれる。このデータ配列のグラフ表示を、図6に示す。
【0144】
質量スペクトル・データを、セントロイディングおよびデアイソトーピングを介してスティック・スペクトルの形に再処理することができるが、これは、この手法が働くために望ましくないことに留意することが重要である。生の質量スペクトル連続体データは、分析全体を通じてのスペクトル・ピーク形状情報の保存および上述のすべてのタイプのセントロイディング誤差およびデアイソトーピング誤差の除去に起因して、よりよく働くことができる。好ましい手法は、連続生質量スペクトル・データを較正された連続データに完全に較正して、高い質量精度を達成し、より正確なライブラリ検索を可能にすることである。
【0145】
データ配列R(m×n×pの次元を有する)の保持時間t1およびt2の各組合せで、質量分析計に注入される試料の分画は、オリジナル試料内のペプチドの部分集合のある線形組合せからなる。試料のこの分画は、少数のペプチドと数万種のペプチドの間のどれかを含む可能性が高い。そのような試料分画に対応する質量スペクトルは、非常に複雑である可能性が高く、上記で指摘したように、タンパク質同定および特に定量化のためにそのような混合物を個々のタンパク質に分解するという課題は、解決しがたいと思われる。
【0146】
しかし、この3次元データ配列は、2次元分析に関して上記で指摘したように、GRAM(Generalized Rank Annihilation Method、反復なしの行列演算を介する直接分解)またはPARAFAC(PARAllel FACtor analysis、交互最小二乗法を用いる反復分解)に基づくトリリニア分解法を用いて、上記で指摘したように4つの異なる行列および残余データ・キューブEに分解することができる。
【0147】
この3次元分析で、Cは、すべての同定可能ペプチド(そのうちのk種、k≦min(m,n))のt1に関するクロマトグラムを表し、Qは、すべての同定可能ペプチド(そのうちのk種)のt2に関するクロマトグラムを表し、Wは、すべてのペプチド(そのうちのk種)の逆畳み込みされた連続質量スペクトルを表し、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列である。言い換えると、このデータ配列の分解を介して、2つの保持時間(t1およびt2)が、Wに含まれる各ペプチドの質量スペクトル連続体の正確な決定と共に、試料に存在するそれぞれすべてのペプチドについて同定されている。
【0148】
前述の分析は、無傷のタンパク質が、消化なしで、より大きい質量を処理できる質量分析計を用いて直接に分析されない限り、ペプチド・レベルでの情報を作る。しかし、タンパク質レベルの情報は、次の追加ステップを介して複数の試料から得ることができる。
【0149】
1.同一の治療に関する時間期間にわたって、または治療のうち異なる投薬量を用いる固定された時間に、または異なる病状の複数の個人から収集された複数の試料(そのうちのl個)について、上述の2D−LC/MSランを実行する。
【0150】
2.上述の試料ごとにデータ分解を実行し、各試料を用いてすべてのペプチドを完全に同定する。
【0151】
3.各試料内のすべてのペプチドの相対濃度を、Iのスーパーダイアゴナル要素から直接に読み取ることができる。すべての試料にまたがるこれらの濃度からなる新しい行列Sを、l個の試料×全試料中のq個の別個のペプチド(q□max(k1,k2,…,kp)
、ただし、kiは、試料i(i=1,2,…,p)のペプチドの数)という次元を用いて形成することができる。他の試料に存在するペプチドの一部を含まない試料について、これらのペプチド(列に配置される)に対応する行の項目は、0になる。
【0152】
4.行列Sの統計的検討によって、試料ごとに互いに比例して変化するペプチドの調査が可能になる。これらのペプチドは、同一のタンパク質からのすべてのペプチドに潜在的に対応する可能性がある。S行列の特異値分解(SVD)または主成分分析(PCA)から計算されたマハラノビス距離に基づくデンドログラムによって、これらのペプチドの相互関連性を示すことができる。しかし、試料ごとに協調して変化するタンパク質のグループがあり、したがって、それに対応するペプチドのすべてが同一のクラスタにグループ化されることを指摘しなければならない。このプロセスのグラフ表示を、図11に示す。
【0153】
5.上記のグループ化に従って区分された行列Sは、多数の試料にまたがる異なるタンパク質の発現レベルを示すディファレンシャル・プロテオミクス分析の結果を表す。
【0154】
6.真上のステップ5で同定された各グループのすべてのペプチドについて、Wに含まれる分解された質量スペクトル応答を組み合わせて、その発現レベルにおいて協調して変化する各タンパク質またはタンパク質のグループに含まれるすべてのペプチドの複合質量スペクトル(composite mass spectrum)シグネチャを形成する。そのような複合質量スペクトルは、スティック/重心スペクトルにさらに処理する(まだそのように処理されていない場合)か、好ましくは、同時係属の出願で開示されている連続質量スペクトル・データを使用するタンパク質同定のためにMascotおよびSwissProtなどの標準タンパク質データベースに対して直接に検索することができる。
【0155】
ICAT(Gygi,S.P.他、Nat.Biotechnol.17、994〜999ページ、1999年)と比較して、本明細書で提案される定量化は、追加の試料調製を必要とせず、数千個の試料を処理する能力を有し、相対的なタンパク質発現レベルに達するために全体的な最小二乗フィットにすべての使用可能なペプチド(同位元素タグ付けに使用可能な少数ではなく)を使用する。すべてのペプチドの数学的分離およびその後のタンパク質へのグループ化にも起因して、このタンパク質同定は、ICATの場合のようなペプチド・シーケンシングなしで達成することができる。無傷のタンパク質2D−LC/MS分析の場合に、すべてのタンパク質濃度を、さらなる再グループ化なしで、Iのスーパーダイアゴナルから直接に読み取ることができる。しかし、上記で示したようにS行列を形成し、ディファレンシャル・プロテオミクス分析またはタンパク質発現分析のためにこの行列に対して統計分析を実行することが望ましい場合がある。
【0156】
短く言うと、本発明は、次の特徴を有する3次元データを使用するタンパク質同定およびタンパク質発現分析の方法を提供する。
【0157】
−次の方法のいずれかから生成されるデータの集合を、3Dデータ配列に配置する
i)タンパク質消化後のペプチドに関するエレクトロスプレイ・イオン化(ESI)質量分析法または
ii)ペプチドまたは無傷のタンパク質に関する飛行時間(TOF)型質量分析法と組み合わされた、
a)サイズ排除逆相液体クロマトグラフィ(SEC−RPLC)または
b)強酸性陽イオン交換逆相液体クロマトグラフィ(SCX−RPLC)
【0158】
−ここで、生質量スペクトル連続体を完全に較正することが望ましいが、質量スペクトル・データは、セントロイディングおよび/またはデアイソトーピングを介して前処理される必要がない。
【0159】
−質量スペクトル連続体データは、直接に使用することができ、実際に好ましく、したがって、分析全体を通じてスペクトル・ピーク形状情報が保存される。
【0160】
−この手法は、すべてのペプチドを数学的に分離し、その後、タンパク質にグループ化する方法であり、したがって、タンパク質同定を、ペプチド・シーケンシングなしで行うことができる。
【0161】
−本発明は、追加試料調製を必要とせず、数千個の試料を処理する能力を有し、相対タンパク質発現レベルに達するために全体的な最小二乗フィットにすべての使用可能なペプチドを使用する定量的ツールを提供する。
【0162】
上記の3−Dデータは、異なる形式および形状で得られる可能性がある。2D−LC/MSに対する代替は、エレクトロスプレイ・イオン化(ESI)質量分析法(従来のイオン・トラップか四重極MSかTOF−MS)と結合された2D電気泳動分離を実行することである。分析の手法および処理は、上述のものと同一である。この手法に従う他のタイプの3Dデータに、次が含まれるが、これに制限はされない。
【0163】
UV、蛍光(その蛍光がタンパク質配列のセグメントによって影響される配列特異的な1つまたは複数のタグを用いる)によって検出される他のマルチチャネル・スペクトル検出を用いる2D−LCなど。
【0164】
単一チャネル検出(たとえば、245nmのUV)を用いる3D電気泳動または3D LC。3D分離は、たとえばpI、MW、および疎水性において分離するために無傷のタンパク質に適用することができる。
【0165】
1D電気泳動とその後の消化されたタンパク質または無傷のタンパク質のいずれかに対する1D−LC/MS。
【0166】
2Dゲル分離とその後のMSマルチチャネル検出。消化が必要な場合には、on the gel TOF分析用の適当なMALDIマトリックスを用いてゲルに対して達成することができる。
【0167】
マルチチャネル検出と結合された分離の他の2D手段。
【0168】
2Dスペクトル検出と結合された1D分離、LC/MS/MS。
【0169】
2Dスペクトル検出、たとえば、発光励起2D蛍光(EEM)と結合された1D LCまたは1Dゲル電気泳動。
【0170】
本発明の分析の方法は、ハードウェア、ソフトウェア、またはハードウェアとソフトウェアの組合せで実現することができる。どの種類のコンピュータ・システムでも、あるいは本明細書に記載の方法および/または機能を実行するように適合された他の装置も好適である。ハードウェアとソフトウェアの通常の組合せは、ロードされ実行されたときコンピュータ・システムを制御するコンピュータ・プログラムを有する汎用コンピュータ・システムとすることができ、このコンピュータ・システムは、分析システムが本明細書に記載の方法を実行するように、その分析システムを制御する。本発明は、本明細書に記載の方法の実装を可能にするすべての特徴を含み、コンピュータ・システム(次に分析システムを制御する)にロードされたときこれらの方法を実行することができる、コンピュータ・プログラム製品で実施することもできる。
【0171】
この文脈でのコンピュータ・プログラム手段またはコンピュータ・プログラムは、別の言語、コード、もしくは表記への変換および/または異なる材料形態での再生の後にあるいは直接に、情報処理機能を有するシステムに特定の機能を実行させることを意図された命令の組の任意の表現を、任意の言語、コード、または表記で含む。
【0172】
したがって、本発明は、上述の機能を引き起こすコンピュータ可読プログラム・コード手段が組み込まれたコンピュータ使用可能媒体を含む製造品を含む。この製造品内のコンピュータ可読プログラム・コード手段は、コンピュータに本発明の方法のステップを行わせるコンピュータ可読プログラム・コード手段を含む。同様に、本発明は、上述の機能を引き起こすコンピュータ可読プログラム・コード手段が組み込まれたコンピュータ使用可能媒体を含むコンピュータ・プログラム製品として実施することができる。このコンピュータ・プログラム製品内のコンピュータ可読プログラム・コード手段は、コンピュータに本発明の1つまたはそれ以上の機能を行わせるコンピュータ可読プログラム・コード手段を含む。さらに、本発明は、本発明の1つまたはそれ以上の機能を行うための方法ステップを実行するために機械によって実行可能な命令のプログラムを明らかに実施する、機械によって可読のプログラム・ストレージ・デバイスとして実施することができる。
【0173】
前述したものは、本発明のより関連する目的および実施形態の一部の概要を示したものであることに留意されたい。本発明の概念は、多数の応用分野に使用することができる。したがって、当該説明は特定の装置および方法に関して述べられているが、本発明の意図および概念は、他の装置および応用例に適し、適用可能である。開示された実施形態に対する他の変更を、本発明の趣旨および範囲から逸脱せずにおこなえることが、当業者には明白であろう。説明された実施形態は、本発明のより顕著な特徴および応用例の一部を例示するに過ぎないと解釈すべきである。したがって、前述の説明が、本発明の例示にすぎないことを理解されたい。当業者は、本発明から逸脱せずに、さまざまな代替形態および修正形態を考案することができる。開示された発明を異なる形で適用することまたは本発明を当業者に既知の形で変更することによって、他の有益な結果を実現することができる。したがって、この実施形態が、制限ではなく例として提供されたことを理解されたい。したがって、本発明は、請求項の範囲に含まれるすべての代替形態、修正形態、および変形形態を含むことが意図されている。
【図面の簡単な説明】
【0174】
【図1】質量分析計を含む、本発明による分析システムを示すブロック図である。
【図2】1次元試料分離およびマルチチャネル検出器を有するシステムを示すブロック図である。
【図3】2次元試料分離および単一チャネル検出器を有するシステムを示すブロック図である。
【図4A】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図4B】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図4C】本発明による、2次元測定に基づく3次元データ配列のコンパイルを示す図である。
【図5】1試料に関する単一の3次元測定に基づく3次元データ配列を示す図である。
【図6】2次元液相分離とその後の質量スペクトル検出に基づく3次元データ配列を示す図である。
【図7】順次走査を用いるマルチチャネル検出の時間スキュー補正を示す図である。
【図8】本発明による分析の方法を示す流れ図である。
【図9】本発明による、分離軸および対応するプロフィールの自動整列の変換を示す図である。
【図10】3次元データ配列の直接分解を示す図である。
【図11】本発明による、タンパク質への酵素消化から生じたペプチドのクラスタ分析を介するグループ化(デンドログラム)を示す図である。
【特許請求の範囲】
【請求項1】
2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の複数の試料から得られたデータを分析する方法であって、
前記2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む上記の方法。
【請求項2】
第1プロフィールおよび第2プロフィールは、実質的に純粋な成分のプロフィールを表す請求項1に記載の方法。
【請求項3】
第1プロフィールおよび第2プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む請求項1に記載の方法。
【請求項4】
標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、前記データ自体、および第2標準化行列の積の形にすることによって前記データを標準化することをさらに含む請求項1に記載の方法。
【請求項5】
第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、2つの変数に対する位置でデータを表現させる値を有する請求項4に記載の方法。
【請求項6】
第1標準化行列は、第1変数に対してデータをシフトし、第2標準化行列は、第2変数に対してデータをシフトする請求項5に記載の方法。
【請求項7】
第1標準化行列および第2標準化行列の項は、それぞれ第1変数および第2変数に関してデータの分布形状を標準化するように働く値を有する請求項5に記載の方法。
【請求項8】
第1標準化行列および第2標準化行列の項は、
既知の成分を有する試料を装置に適用すること、および、
前記既知の成分によって生成されたデータを第1変数および第2変数に対して正しく位置決めさせる前記第1標準化行列および前記第2標準化行列の項を選択すること
によって決定される請求項4に記載の方法。
【請求項9】
項は、標準化されたデータ行列内の第1変数および第2変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される請求項8に記載の方法。
【請求項10】
第1標準化行列および第2標準化行列の項は、試料ごとに計算される請求項9に記載の方法。
【請求項11】
第1標準化行列および第2標準化行列の項は、すべての試料にわたって最小の誤差を生じるように計算される請求項10に記載の方法。
【請求項12】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる請求項4に記載の方法。
【請求項13】
第1標準化行列および第2標準化行列の項は、変数に対する前記項のパラメータ化された既知の機能的依存性に基づく請求項4に記載の方法。
【請求項14】
第1標準化行列および第2標準化行列の項は、データ配列R:
【数1】
[ここで、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、前記成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、p個の試料のすべてのこれらの成分の濃度を含み、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である]を解くことによって決定される、請求項8に記載の方法。
【請求項15】
装置は、2次元電気泳動分離システムである請求項1に記載の方法。
【請求項16】
第1変数は、等電点であり、第2変数は、分子量である請求項15に記載の方法。
【請求項17】
変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果である請求項1に記載の方法。
【請求項18】
2つの変数のうちの1つは、質量分析計の質量軸に関連する質量である請求項1に記載の方法。
【請求項19】
装置は、質量分析計に試料を供給するクロマトグラフィ・システムをさらに含み、保持時間は、2つの変数のうちの他方である請求項18に記載の方法。
【請求項20】
装置は、質量分析計に試料を供給する電気泳動分離システムをさらに含み、前記試料の移動特性は、2つの変数のうちの他方である請求項18に記載の方法。
【請求項21】
データは、連続質量スペクトル・データである請求項18に記載の方法。
【請求項22】
データは、セントロイディングなしで使用される請求項18に記載の方法。
【請求項23】
時間スキューに関してデータを補正することをさらに含む請求項18に記載の方法。
【請求項24】
質量および質量スペクトル・ピーク形状に関してデータの較正を実行することをさらに含む請求項18に記載の方法。
【請求項25】
第1変数および第2変数のうちの他方は、複数のタンパク質親和性領域を有するプロテイン・チップ上の領域の変数である請求項18に記載の方法。
【請求項26】
単一チャネル・アナライザを使用することによって、また試料を連続して分析することによってデータ配列のデータを得ることをさらに含む請求項1に記載の方法。
【請求項27】
単一チャネル検出器は、光吸収、光放出、光反射、光透過、光散乱、屈折率、電気化学、導電性、放射能、またこれらの任意の組合せのうちの1つに基づく請求項26に記載の方法。
【請求項28】
試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合される請求項27に記載の方法。
【請求項29】
複数の試料から得られたデータを分析するデータ分析部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、前記化学分析システムは、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、前記コンピュータ可読コードは、コンピュータに、
2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む方法を実行させるコンピュータ可読媒体。
【請求項30】
請求項2〜28のいずれか一項に記載のステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含む請求項29に記載のコンピュータ可読媒体。
【請求項31】
複数の試料から得られたデータを分析する化学分析システムであって、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、
2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む方法を実行する装置を有するシステム。
【請求項32】
方法は、請求項2〜28のいずれか一項に記載のステップをさらに含む請求項31に記載の化学分析システム。
【請求項33】
複数の成分を含む試料の成分を分離する能力を有する分離システム内の試料から得られたデータを分析する方法であって、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む上記の方法。
【請求項34】
第1プロフィール、第2プロフィール、および第3プロフィールは、実質的に純粋な成分のプロフィールを表す請求項33に記載の方法。
【請求項35】
第1プロフィール、第2プロフィール、および第3プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む請求項33に記載の方法。
【請求項36】
標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、前記データ自体、および第2標準化行列の積の形にすることによって前記データを標準化することをさらに含む請求項33に記載の方法。
【請求項37】
第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、3つの変数のうちの2つに対する位置でデータを表現させる値を有する請求項36に記載の方法。
【請求項38】
第1標準化行列は、2つの変数のうちの一方に対してデータをシフトし、第2標準化行列は、前記2つの変数のうちの他方に対してデータをシフトする請求項37に記載の方法。
【請求項39】
第1標準化行列および第2標準化行列の項は、それぞれ2つの変数に関してデータの分布形状を標準化するように働く値を有する請求項37に記載の方法。
【請求項40】
第1標準化行列および第2標準化行列の項は、
既知の成分を有する試料を装置に適用すること、および、
既知の成分によって生成されたデータを2つの変数に対して正しく位置決めさせる前記第1標準化行列および前記第2標準化行列の項を選択すること
によって決定される請求項36に記載の方法。
【請求項41】
項は、標準化されたデータ行列内の2つの変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される請求項40に記載の方法。
【請求項42】
第1標準化行列および第2標準化行列の項は、単一チャネルについて計算される請求項41に記載の方法。
【請求項43】
第1標準化行列および第2標準化行列の項は、チャネルについて最小の誤差を生じるように計算される請求項42に記載の方法。
【請求項44】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる請求項36に記載の方法。
【請求項45】
第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づく請求項36に記載の方法。
【請求項46】
第1標準化行列および第2標準化行列の項の値は、データ配列R:
【数2】
[ここで、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、前記成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、マルチチャネル・アナライザまたは第3変数に関するこれらの成分の純粋なプロフィールを含み、I(k×k×k)は、前記k個の成分のすべての濃度を表す唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、そしてE(m×n×p)は、残余データ配列である]を解くことによって決定される、請求項41に記載の方法。
【請求項47】
分離装置のうちの1つは、1次元電気泳動分離システムである請求項33に記載の方法。
【請求項48】
変数は、等電点および分子量のうちの1つである請求項47に記載の方法。
【請求項49】
2つの分離変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果である請求項33に記載の方法。
【請求項50】
3つの変数のうちの1つは、質量分析計の質量軸に関連する質量である請求項33に記載の方法。
【請求項51】
装置は、質量分析計に分離された試料を供給する少なくとも1つのクロマトグラフィ・システムをさらに含み、保持時間は、変数のうちの少なくとも1つである請求項50に記載の方法。
【請求項52】
装置は、質量分析計に分離された試料を供給する少なくとも1つの電気泳動分離システムをさらに含み、前記試料の移動特性は、変数のうちの少なくとも1つである請求項50に記載の方法。
【請求項53】
データは、連続質量スペクトル・データである請求項50に記載の方法。
【請求項54】
データは、セントロイディングなしで使用される請求項50に記載の方法。
【請求項55】
時間スキューに関してデータを補正することをさらに含む請求項50に記載の方法。
【請求項56】
質量およびスペクトル・ピーク形状に関してデータの較正を実行することをさらに含む請求項50に記載の方法。
【請求項57】
装置は、複数のタンパク質親和性領域を有するプロテイン・チップを含み、領域の位置は、3つの変数のうちの1つである請求項50に記載の方法。
【請求項58】
マルチチャネル・アナライザは、光吸収、光放出、光反射、光透過、光散乱、屈折率、電気化学、導電性、放射能、またこれらの任意の組合せのうちの1つに基づく請求項33に記載の方法。
【請求項59】
試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合される請求項58に記載の方法。
【請求項60】
装置は、2次元電気泳動分離システムを含む請求項33に記載の方法。
【請求項61】
少なくとも1つの変数のうちの第1は、等電点であり、前記少なくとも1つの変数のうちの第2は、分子量である請求項60に記載の方法。
【請求項62】
試料から得られたデータを分析するデータ分析部分を有し、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、前記コンピュータ可読コードは、コンピュータに、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む方法を実行させるためのものである上記コンピュータ可読媒体。
【請求項63】
請求項34から61のいずれか一項に記載のステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含む請求項62に記載のコンピュータ可読媒体。
【請求項64】
試料から得られたデータを分析する化学分析システムであって、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む方法を実行する装置を有する上記システム。
【請求項65】
方法は、請求項34〜61のいずれか一項に記載のステップをさらに含む請求項64に記載の化学分析システム。
【請求項66】
少なくとも2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の少なくとも1つの試料から得られたデータを分析する方法であって、
前記2つの変数の関数として表される、少なくとも1つの試料を表すデータを前記システムから得ること、
連続するレベルを有するデータ・スタックを形成することであって、各レベルが、前記少なくとも1つの試料を表す連続データを含むこと、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記変数のうちの第1変数の関数としての前記成分の第1プロフィール、および
前記変数のうちの第2変数の関数としての前記成分の第2プロフィールである)
を含む上記の方法。
【請求項67】
少なくとも1つの試料は、単一の試料を含み、連続データは、時間の関数として前記試料を表す請求項66に記載の方法。
【請求項68】
少なくとも1つの試料は、単一の試料を含み、連続データは、その成分の質量の関数として前記試料を表す請求項66に記載の方法。
【請求項69】
少なくとも1つの試料は、複数の試料を含み、連続データは、連続試料を表す請求項68に記載の方法。
【請求項1】
2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の複数の試料から得られたデータを分析する方法であって、
前記2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む上記の方法。
【請求項2】
第1プロフィールおよび第2プロフィールは、実質的に純粋な成分のプロフィールを表す請求項1に記載の方法。
【請求項3】
第1プロフィールおよび第2プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む請求項1に記載の方法。
【請求項4】
標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、前記データ自体、および第2標準化行列の積の形にすることによって前記データを標準化することをさらに含む請求項1に記載の方法。
【請求項5】
第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、2つの変数に対する位置でデータを表現させる値を有する請求項4に記載の方法。
【請求項6】
第1標準化行列は、第1変数に対してデータをシフトし、第2標準化行列は、第2変数に対してデータをシフトする請求項5に記載の方法。
【請求項7】
第1標準化行列および第2標準化行列の項は、それぞれ第1変数および第2変数に関してデータの分布形状を標準化するように働く値を有する請求項5に記載の方法。
【請求項8】
第1標準化行列および第2標準化行列の項は、
既知の成分を有する試料を装置に適用すること、および、
前記既知の成分によって生成されたデータを第1変数および第2変数に対して正しく位置決めさせる前記第1標準化行列および前記第2標準化行列の項を選択すること
によって決定される請求項4に記載の方法。
【請求項9】
項は、標準化されたデータ行列内の第1変数および第2変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される請求項8に記載の方法。
【請求項10】
第1標準化行列および第2標準化行列の項は、試料ごとに計算される請求項9に記載の方法。
【請求項11】
第1標準化行列および第2標準化行列の項は、すべての試料にわたって最小の誤差を生じるように計算される請求項10に記載の方法。
【請求項12】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる請求項4に記載の方法。
【請求項13】
第1標準化行列および第2標準化行列の項は、変数に対する前記項のパラメータ化された既知の機能的依存性に基づく請求項4に記載の方法。
【請求項14】
第1標準化行列および第2標準化行列の項は、データ配列R:
【数1】
[ここで、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、前記成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、p個の試料のすべてのこれらの成分の濃度を含み、Iは、唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、E(m×n×p)は、残余データ配列である]を解くことによって決定される、請求項8に記載の方法。
【請求項15】
装置は、2次元電気泳動分離システムである請求項1に記載の方法。
【請求項16】
第1変数は、等電点であり、第2変数は、分子量である請求項15に記載の方法。
【請求項17】
変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果である請求項1に記載の方法。
【請求項18】
2つの変数のうちの1つは、質量分析計の質量軸に関連する質量である請求項1に記載の方法。
【請求項19】
装置は、質量分析計に試料を供給するクロマトグラフィ・システムをさらに含み、保持時間は、2つの変数のうちの他方である請求項18に記載の方法。
【請求項20】
装置は、質量分析計に試料を供給する電気泳動分離システムをさらに含み、前記試料の移動特性は、2つの変数のうちの他方である請求項18に記載の方法。
【請求項21】
データは、連続質量スペクトル・データである請求項18に記載の方法。
【請求項22】
データは、セントロイディングなしで使用される請求項18に記載の方法。
【請求項23】
時間スキューに関してデータを補正することをさらに含む請求項18に記載の方法。
【請求項24】
質量および質量スペクトル・ピーク形状に関してデータの較正を実行することをさらに含む請求項18に記載の方法。
【請求項25】
第1変数および第2変数のうちの他方は、複数のタンパク質親和性領域を有するプロテイン・チップ上の領域の変数である請求項18に記載の方法。
【請求項26】
単一チャネル・アナライザを使用することによって、また試料を連続して分析することによってデータ配列のデータを得ることをさらに含む請求項1に記載の方法。
【請求項27】
単一チャネル検出器は、光吸収、光放出、光反射、光透過、光散乱、屈折率、電気化学、導電性、放射能、またこれらの任意の組合せのうちの1つに基づく請求項26に記載の方法。
【請求項28】
試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合される請求項27に記載の方法。
【請求項29】
複数の試料から得られたデータを分析するデータ分析部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、前記化学分析システムは、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有し、前記コンピュータ可読コードは、コンピュータに、
2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む方法を実行させるコンピュータ可読媒体。
【請求項30】
請求項2〜28のいずれか一項に記載のステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含む請求項29に記載のコンピュータ可読媒体。
【請求項31】
複数の試料から得られたデータを分析する化学分析システムであって、2つの異なる変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、
2つの変数の関数として表される、複数の試料を表すデータを前記システムから得ること、
各レベルが前記データ試料のうちの1つを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記第1変数の関数としての前記成分の第1プロフィール、および
前記第2変数の関数としての前記成分の第2プロフィールである)
を含む方法を実行する装置を有するシステム。
【請求項32】
方法は、請求項2〜28のいずれか一項に記載のステップをさらに含む請求項31に記載の化学分析システム。
【請求項33】
複数の成分を含む試料の成分を分離する能力を有する分離システム内の試料から得られたデータを分析する方法であって、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む上記の方法。
【請求項34】
第1プロフィール、第2プロフィール、および第3プロフィールは、実質的に純粋な成分のプロフィールを表す請求項33に記載の方法。
【請求項35】
第1プロフィール、第2プロフィール、および第3プロフィールのうちの少なくとも1つを使用して定性分析を実行することをさらに含む請求項33に記載の方法。
【請求項36】
標準化されたデータ行列を形成するために、試料を表すデータのデータ行列乗算を実行して、第1標準化行列、前記データ自体、および第2標準化行列の積の形にすることによって前記データを標準化することをさらに含む請求項33に記載の方法。
【請求項37】
第1標準化行列および第2標準化行列の項は、データ配列内のものと標準化されたデータ行列内で異なる、3つの変数のうちの2つに対する位置でデータを表現させる値を有する請求項36に記載の方法。
【請求項38】
第1標準化行列は、2つの変数のうちの一方に対してデータをシフトし、第2標準化行列は、前記2つの変数のうちの他方に対してデータをシフトする請求項37に記載の方法。
【請求項39】
第1標準化行列および第2標準化行列の項は、それぞれ2つの変数に関してデータの分布形状を標準化するように働く値を有する請求項37に記載の方法。
【請求項40】
第1標準化行列および第2標準化行列の項は、
既知の成分を有する試料を装置に適用すること、および、
既知の成分によって生成されたデータを2つの変数に対して正しく位置決めさせる前記第1標準化行列および前記第2標準化行列の項を選択すること
によって決定される請求項36に記載の方法。
【請求項41】
項は、標準化されたデータ行列内の2つの変数に対するデータの位置の最小の誤差を生じる項を選択することによって決定される請求項40に記載の方法。
【請求項42】
第1標準化行列および第2標準化行列の項は、単一チャネルについて計算される請求項41に記載の方法。
【請求項43】
第1標準化行列および第2標準化行列の項は、チャネルについて最小の誤差を生じるように計算される請求項42に記載の方法。
【請求項44】
第1標準化行列および第2標準化行列のうちの少なくとも1つを、対角行列または単位行列のいずれかに単純化することができる請求項36に記載の方法。
【請求項45】
第1標準化行列および第2標準化行列の項は、変数に対する項のパラメータ化された既知の機能的依存性に基づく請求項36に記載の方法。
【請求項46】
第1標準化行列および第2標準化行列の項の値は、データ配列R:
【数2】
[ここで、Q(m×k)は、第1変数に関してk個の成分のすべての純粋なプロフィールを含み、W(n×k)は、前記成分の第2変数に関して純粋なプロフィールを含み、C(p×k)は、マルチチャネル・アナライザまたは第3変数に関するこれらの成分の純粋なプロフィールを含み、I(k×k×k)は、前記k個の成分のすべての濃度を表す唯一の非0要素としてそのスーパーダイアゴナルにスカラを有する新しいデータ配列であり、そしてE(m×n×p)は、残余データ配列である]を解くことによって決定される、請求項41に記載の方法。
【請求項47】
分離装置のうちの1つは、1次元電気泳動分離システムである請求項33に記載の方法。
【請求項48】
変数は、等電点および分子量のうちの1つである請求項47に記載の方法。
【請求項49】
2つの分離変数は、クロマトグラフ分離、キャピラリ電気泳動分離、ゲルベース分離、親和性分離、および抗体分離の、それ自体の組合せを含めて特定の順序でない任意の組合せの結果である請求項33に記載の方法。
【請求項50】
3つの変数のうちの1つは、質量分析計の質量軸に関連する質量である請求項33に記載の方法。
【請求項51】
装置は、質量分析計に分離された試料を供給する少なくとも1つのクロマトグラフィ・システムをさらに含み、保持時間は、変数のうちの少なくとも1つである請求項50に記載の方法。
【請求項52】
装置は、質量分析計に分離された試料を供給する少なくとも1つの電気泳動分離システムをさらに含み、前記試料の移動特性は、変数のうちの少なくとも1つである請求項50に記載の方法。
【請求項53】
データは、連続質量スペクトル・データである請求項50に記載の方法。
【請求項54】
データは、セントロイディングなしで使用される請求項50に記載の方法。
【請求項55】
時間スキューに関してデータを補正することをさらに含む請求項50に記載の方法。
【請求項56】
質量およびスペクトル・ピーク形状に関してデータの較正を実行することをさらに含む請求項50に記載の方法。
【請求項57】
装置は、複数のタンパク質親和性領域を有するプロテイン・チップを含み、領域の位置は、3つの変数のうちの1つである請求項50に記載の方法。
【請求項58】
マルチチャネル・アナライザは、光吸収、光放出、光反射、光透過、光散乱、屈折率、電気化学、導電性、放射能、またこれらの任意の組合せのうちの1つに基づく請求項33に記載の方法。
【請求項59】
試料内の成分は、蛍光タグ、同位元素タグ、染色剤、親和性タグ、または抗体タグのうちの少なくとも1つに結合される請求項58に記載の方法。
【請求項60】
装置は、2次元電気泳動分離システムを含む請求項33に記載の方法。
【請求項61】
少なくとも1つの変数のうちの第1は、等電点であり、前記少なくとも1つの変数のうちの第2は、分子量である請求項60に記載の方法。
【請求項62】
試料から得られたデータを分析するデータ分析部分を有し、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離部分を有する化学分析システムと共に使用される、コンピュータ可読コードを有するコンピュータ可読媒体であって、前記コンピュータ可読コードは、コンピュータに、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む方法を実行させるためのものである上記コンピュータ可読媒体。
【請求項63】
請求項34から61のいずれか一項に記載のステップを実行することによってコンピュータにデータを分析させるコンピュータ可読コードをさらに含む請求項62に記載のコンピュータ可読媒体。
【請求項64】
試料から得られたデータを分析する化学分析システムであって、少なくとも1つの変数の関数として複数の成分を含む試料の成分を分離する機能を有する分離システムを有し、
複数の成分を含む試料を少なくとも第1変数に関して分離し、分離された試料を形成すること、
前記分離された試料を少なくとも第2変数に関して分離し、さらに分離された試料を形成すること、
3つの変数の関数として表される、前記さらに分離された試料を表すデータをマルチチャネル・アナライザから得ること、
各レベルが前記マルチチャネル・アナライザの1つのチャネルからのデータを含む連続するレベルを有するデータ・スタックを形成すること、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列または配列に分離すること(該行列または配列は、
そのスーパーダイアゴナル上の、前記試料内の各成分の濃度を表す濃度データ配列、
第1変数の関数としての各成分の第1プロフィール、
第2変数の関数としての各成分の第2プロフィール、および
第3変数の関数としての各成分の第3プロフィールである)
を含む方法を実行する装置を有する上記システム。
【請求項65】
方法は、請求項34〜61のいずれか一項に記載のステップをさらに含む請求項64に記載の化学分析システム。
【請求項66】
少なくとも2つの異なる変数の関数として複数の成分を含む試料の成分を分離する能力を有する分離システム内の少なくとも1つの試料から得られたデータを分析する方法であって、
前記2つの変数の関数として表される、少なくとも1つの試料を表すデータを前記システムから得ること、
連続するレベルを有するデータ・スタックを形成することであって、各レベルが、前記少なくとも1つの試料を表す連続データを含むこと、
前記データ・スタック内のすべてのデータのコンパイルを表すデータ配列を形成すること、および、
前記データ配列を一連の行列に分離すること(該行列は、
前記試料内の各成分の濃度を表す濃度行列、
前記変数のうちの第1変数の関数としての前記成分の第1プロフィール、および
前記変数のうちの第2変数の関数としての前記成分の第2プロフィールである)
を含む上記の方法。
【請求項67】
少なくとも1つの試料は、単一の試料を含み、連続データは、時間の関数として前記試料を表す請求項66に記載の方法。
【請求項68】
少なくとも1つの試料は、単一の試料を含み、連続データは、その成分の質量の関数として前記試料を表す請求項66に記載の方法。
【請求項69】
少なくとも1つの試料は、複数の試料を含み、連続データは、連続試料を表す請求項68に記載の方法。
【図1】

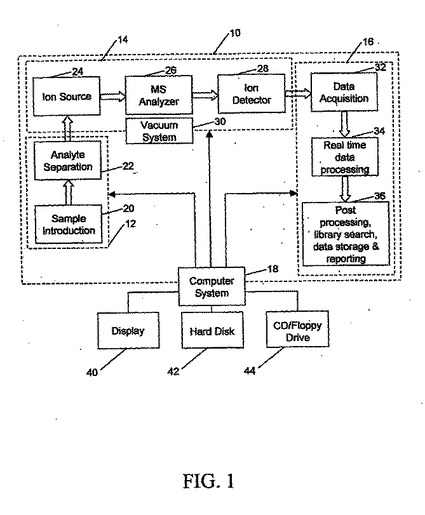
【図2】
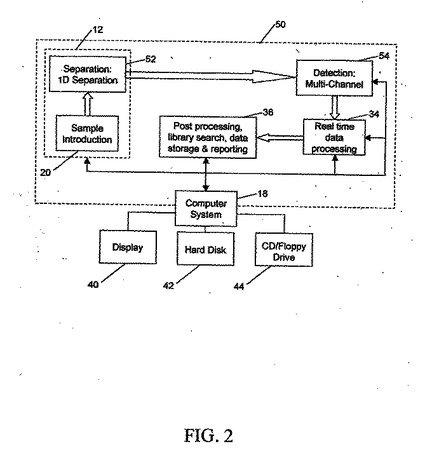
【図3】
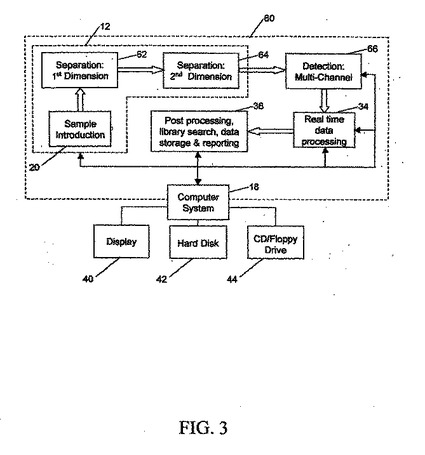
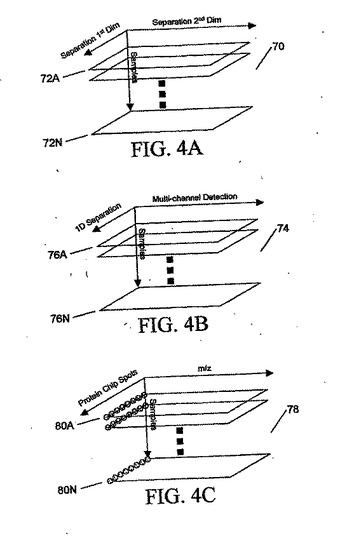
【図5】
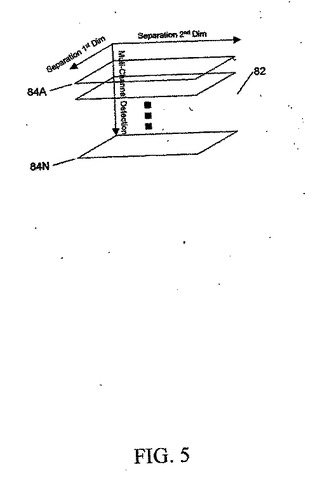
【図6】
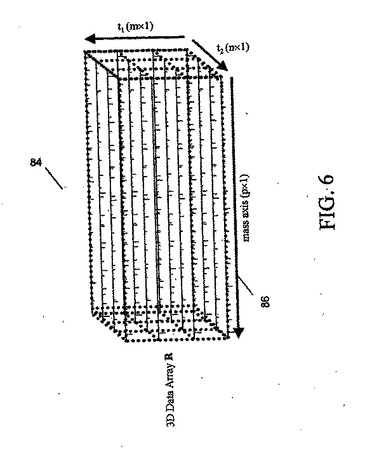
【図7】
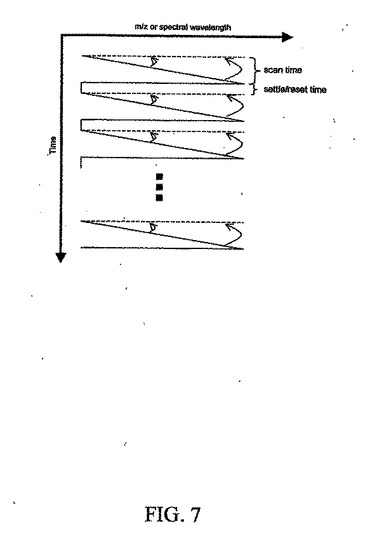
【図8】
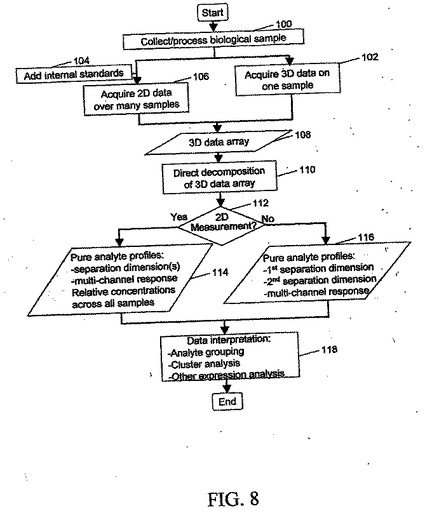
【図9】
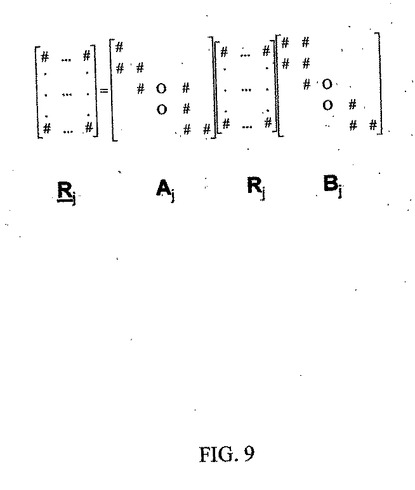
【図10】
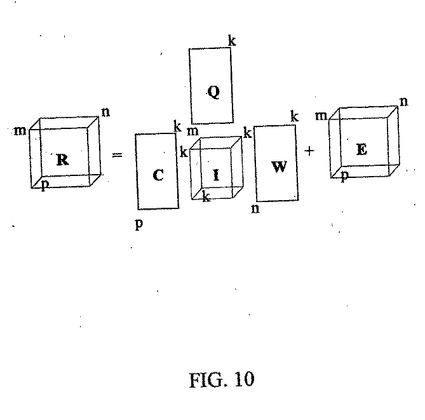
【図11】
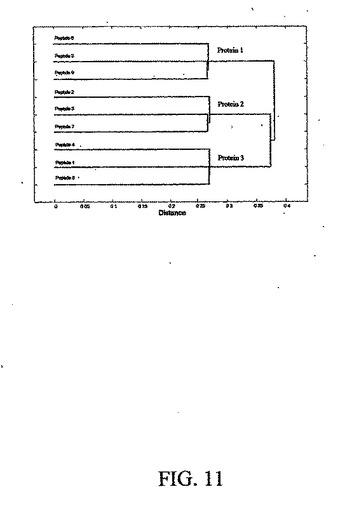
【図2】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2007−525645(P2007−525645A)
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願番号】特願2006−513397(P2006−513397)
【出願日】平成16年4月28日(2004.4.28)
【国際出願番号】PCT/US2004/013097
【国際公開番号】WO2004/097582
【国際公開日】平成16年11月11日(2004.11.11)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.JAVA
【出願人】(505401366)セルノ・バイオサイエンス・エルエルシー (7)
【Fターム(参考)】
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願日】平成16年4月28日(2004.4.28)
【国際出願番号】PCT/US2004/013097
【国際公開番号】WO2004/097582
【国際公開日】平成16年11月11日(2004.11.11)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.JAVA
【出願人】(505401366)セルノ・バイオサイエンス・エルエルシー (7)
【Fターム(参考)】
[ Back to top ]