
多環系化合物の新規形態
【解決手段】 化合物Iの代替形態、それらの再現性のある方法、及びそれらを使って患者を治療する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、多環系化合物の新規形態(以後化合物Iとする)を含有する組成物、それらの再現性のある方法、及び化合物Iを有する薬学的組成物に関するものである。
【背景技術】
【0002】
医薬品原料(Active pharmaceutical ingredients;APIs)は、例えば、化学誘導体、溶媒和化合物、水和物、共結晶或いは塩類などの様々な形態で調合され得る。APIsはさらに、無定型でもあり、異なる結晶性多形(polymorphs)を持つ、若しくは異なる溶媒和或いは水和状態で存在する。APIsの形態を変えることによって、それらの物理学的特性を変えることが可能となる。例えば、より熱力学的に安定でない多形と比較して、より熱力学的に安定な多形がより低い溶解性を持つように、結晶性多形は一般的に異なる溶解度を持つ。多形はさらに、安定性、生物学的利用度、形態、蒸気圧、密度、色及び圧縮率などの特性も異なる。従って、APIの結晶性状態のバリエーションは、それらの物理学的及び薬理学的特性を調節するたくさんの方法の1つである。
【0003】
ポリ(ADP−リボース)ポリメラーゼ(PARP、ポリ(ADP−リボース)合成酵素或いはPARSとも呼ばれる)は、DNA修復過程の一部としての一本鎖DNA切断に反応したNAD+からのポリ(ADP−リボース)鎖の合成を触媒する核内酵素である(de Murcia, G; de Murcia, J.M. Poly(ADP−ribose) polymerase: a molecular nick−sensor. Trends Biochem. Sci. 1994, 19,172−176; Alvarez−Gonzalez, R.; Pacheco−Rodriguez, G.; Mendoza−Alvarez, H.Enzymology of ADP−ribose polymer synthesis. Mol. Cell. Biochem. 1994, 138, 33)。PARPの低分子阻害剤は、神経変性疾患、癌、及び他のPARP及びキナーゼ−関連疾患の治療において潜在的な役割を担うと仮定されていた。
【0004】
特異的PARP阻害化合物は、化学名4,5,6,7−テトラヒドロ−11−メトキシ−2−[(4−メチル−1−ピペラジニル)メチル]−1H−シクロペンタ [a]ピロロ[3,4−c]カルバゾール−1,3(2H)−ジオンであり、乳癌及び卵巣がんの治療に有用であり、他の薬剤耐性癌の治療に対する化学療法或いは放射線療法と組み合わされる。この化合物は以下の化学式(I):
【化1】

【0005】
によって表され、これ以後「化合物I」として参照される。米国特許第7,122,679号及び米国出願第2006/0276497号には、化合物I及びその有用性が記載されている。
【0006】
化合物Iの異なる形態は、異なる融解点、可溶性或いは溶解率を持つものである;これらの物理的特性は、単独或いは組み合わせで、例えば生物学的利用のうなどに影響を与えるものである。APIsの代替形態の潜在的利益の点において、化合物Iの代替形態を同定し調合する必要性が存在するものである。
【発明の概要】
【課題を解決するための手段】
【0007】
化合物Iの様々な形態は、それらを調合する方法と共に記載されるものである。特に、無水結晶形態の2つの多形(形態A0及びB0)、結晶性一水和物形態の3つの多形(HA0、HC0及びHD0)、及び9つの溶媒和化合物(S20、S30、S40、S50、S60、S70、S90、S100及びS120)は、本明細書において記載されるものである。これらの形態の1若しくはそれ以上を有する薬学的組成物、さらには化合物Iの非結晶形態(As)をさらに有する薬学的組成物も記載されるものである。これらの形態の1若しくはそれ以上を有する薬学的組成物、さらにそのような組成物を利用して治療する方法も記載されるものである。
【0008】
本発明の薬学的組成物は、これに限定されるものではないが、放射線の抗腫瘍活性或いはDNA障害化学療法剤の増強を含む、様々な方法において使用される(Griffin, R.J.; Curtin, N.J.; Newell, D.R.; Golding, B.T.; Durkacz. B.W.; Calvert, A.H. The role of inhibitors of poly(ADP−ribose) polymerase as resistance−modifying agents in cancer therapy. Biochemie 1995, 77, 408)。
【図面の簡単な説明】
【0009】
【図1】図1は、形態A0のX線粉末回折(X−ray Powder Diffractogram;XRPD)を図示したものである。
【図2】図2は、形態B0のX線粉末回折(XRPD)を図示したものである。
【図3】図3は、形態HA0のX線粉末回折(XRPD)を図示したものである。
【図4】図4は、形態HC0のX線粉末回折(XRPD)を図示したものである。
【図5】図5は、形態HD0のX線粉末回折(XRPD)を図示したものである。
【図6】図6は、形態S20のX線粉末回折(XRPD)を図示したものである。
【図7】図7は、形態S30のX線粉末回折(XRPD)を図示したものである。
【図8】図8は、形態S40のX線粉末回折(XRPD)を図示したものである。
【図9】図9は、形態S50のX線粉末回折(XRPD)を図示したものである。
【図10】図10は、形態S60のX線粉末回折(XRPD)を図示したものである。
【図11】図11は、形態S70のX線粉末回折(XRPD)を図示したものである。
【図12】図12は、形態S90のX線粉末回折(XRPD)を図示したものである。
【図13】図13は、形態S100のX線粉末回折(XRPD)を図示したものである。
【図14】図14は、形態S120のX線粉末回折(XRPD)を図示したものである。
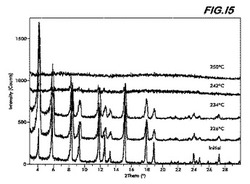
【図15】図15は、形態A0の可変温度X線粉末解析(VT−XRPD)を図示したものである。
【図16】図16は、形態A0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図17】図17は、動的水蒸気吸収装置(DVS)規則等温プロットを図示したものである。
【図18】図18は、動的水蒸気吸収装置(DVS)分析前後の形態A0のX線粉末回折(XRPD)を図示したものである。
【図19】図19は、形態A0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図20】図20は、形態A0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図21】図21は、形態A0のラマンスペクトルを図示したものである。
【図22】図22は、形態B0の可変温度X線粉末回折(VT−XRPD)を図示したものである。
【図23】図23は、形態B0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図24】図24は、形態HA0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図25】図25は、形態HA0の動的水蒸気吸収装置(DVS)等温プロットを図示したものである。
【図26】図26は、形態HA0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図27】図27は、形態HA0のラマンスペクトルを図示したものである。
【図28】図28は、形態HC0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図29】図29は、形態HC0の動的水蒸気吸収装置(DVS)等温プロットを図示したものである。
【図30】図30は、動的水蒸気吸収装置(DVS)分析前後の形態HC0のX線粉末回折(XRPD)を図示したものである。
【図31】図31は、形態HC0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図32】図32は、形態HC0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図33】図33は、形態HC0のラマンスペクトルを図示したものである。
【図34】図34は、形態HD0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図35】図35は、形態HD0の動的水蒸気吸収装置(DVS)規則等温プロットを図示したものである。
【図36】図36は、動的水蒸気吸収装置(DVS)分析前後の形態HD0のX線粉末回折(XRPD)を図示したものである。
【図37】図37は、形態HD0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図38】図38は、形態HD0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図39】図39は、形態HD0のラマンスペクトルを図示したものである。
【図40】図40は、形態S20の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図41】図41は、形態S30の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図42】図42は、形態S40の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図43】図43は、形態S50の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図44】図44は、形態S60の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図45】図45は、形態S70の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図46】図46は、形態S90の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図47】図47は、形態S100の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図48】図48は、形態S120の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図49】図49は、粉砕後の形態A0のX線粉末回折(XRPD)パターンのオーバーレイを図示したものである。
【図50】図50は、15分間の粉砕後の形態HC0 及びHD0のX線粉末回折(XRPD)パターンのオーバーレイを図示したものである。
【図51】図51は、15分間の粉砕後の形態HC0 及びHD0の示差走査熱量測定(DSC)サーモグラムを図示したものである。
【発明を実施するための形態】
【0010】
多数の化合物Iの形態の存在が現在見つかっている。これらの形態の調合及び記述は、本明細書に記載した。これらの形態に関連したスペクトルデータは、図1〜51に示した。
【0011】
より特異的に、多数の化合物Iの異なる物理的形態の存在が見つかっている。化合物Iの無水結晶性形態の2つの多形(形態A0及びB0)、及び結晶性一水和物形態の3つの多形(HA0、HC0及びHD0)が発見されていた。A及びBという文字は、これら無水形態及び水和物を指定し、前にあるHは特に水和物形態を意味するものである。下付き文字「0」はさらに、遊離塩基形態を同定するために指定するものである。加えて、化合物Iの9つの溶媒(S20、S30、S40、S50、S60、S70、S90、S100及びS120)は、本明細書で記載されている。これら形態の1若しくはそれ以上を有する薬学的組成物、さらには化合物Iの非結晶形態(As)をさらに有する薬学的組成物も記載されている。
【0012】
形態A0に対する代表的なXRPDピークは、以下の表1に記載した。形態A0のX線回折パターン特徴は、図1に示した。
【0013】
【表1】
【0014】
形態B0に対する代表的なXRPDピークは、以下の表2に記載した。形態B0のX線回折パターン特徴は、図2に示した。
【0015】
【表2】
【0016】
形態HA0に対する代表的なXRPDピークは、以下の表3に記載した。形態HA0のX線回折パターン特徴は、図3に示した。
【0017】
【表3】
【0018】
形態HC0に対する代表的なXRPDピークは、以下の表4に記載した。形態HC0のX線回折パターン特徴は、図4に示した。
【0019】
【表4】
【0020】
形態HD0に対する代表的なXRPDピークは、以下の表5に記載した。形態HD0のX線回折パターン特徴は、図5に示した。
【0021】
【表5】
【0022】
形態S20に対する代表的なXRPDピークは、以下の表6に記載した。形態S20のX線回折パターン特徴は、図6に示した。
【0023】
【表6】
【0024】
形態S30に対する代表的なXRPDピークは、以下の表7に記載した。形態S30のX線回折パターン特徴は、図7に示した。
【0025】
【表7】
【0026】
形態S40に対する代表的なXRPDピークは、以下の表8に記載した。形態S40のX線回折パターン特徴は、図8に示した。
【0027】
【表8】
【0028】
形態S50に対する代表的なXRPDピークは、以下の表9に記載した。形態S50のX線回折パターン特徴は、図9に示した。
【0029】
【表9】
【0030】
形態S60に対する代表的なXRPDピークは、以下の表10に記載した。形態S60のX線回折パターン特徴は、図10に示した。
【0031】
【表10】
【0032】
形態S70に対する代表的なXRPDピークは、以下の表11に記載した。形態S70のX線回折パターン特徴は、図11に示した。
【0033】
【表11】
【0034】
形態S90に対する代表的なXRPDピークは、以下の表12に記載した。形態S90のX線回折パターン特徴は、図12に示した。
【0035】
【表12】
【0036】
形態S100に対する代表的なXRPDピークは、以下の表13に記載した。形態S100のX線回折パターン特徴は、図13に示した。
【0037】
【表13】
【0038】
形態S120に対する代表的なXRPDピークは、以下の表14に記載した。形態S120のX線回折パターン特徴は、図7に示した。
【0039】
【表14】
【0040】
従って、1つの観点において、本発明は、形態A0、形態B0、或いはそれらの混合物である化合物Iの結晶形態に関連するものである。さらなる観点において、前記結晶形態は形態A0である。別の観点において、前記結晶形態は形態B0である。さらなる観点において、前記結晶形態は、以下のピーク:4.32、6.07、8.55、12.07及び/若しくは15.37±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図1に記載されたようなX線粉末回折パターンを持つものである。付加的な観点において、前記結晶形態は、以下のピーク:7.16、7.89、10.77、16.54及び/若しくは21.20±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図2に記載されたようなX線粉末回折パターンを持つものである。
【0041】
本発明のさらなる観点は、形態HA0、形態HC0、形態HD0或いはそれらの混合物である化合物Iの結晶形態に関連するものである。別の観点において、前記結晶形態は、形態HA0である。さらなる観点において、前記結晶形態は、形態HC0である。付加的な観点において、前記結晶形態は、形態HD0である。さらなる別の観点において、前記結晶形態は、以下のピーク:7.59、15.12、16.06、17.94及び/若しくは23.89±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる観点において、前記結晶形態は、実質的に図3に記載されたようなX線粉末回折パターンを持つものである。付加的な観点において、前記結晶形態は、以下のピーク:8.36、8.71、16.69、17.39及び/若しくは24.59±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図4に記載されたようなX線粉末回折パターンを持つものである。別の観点において、前記結晶形態は、以下のピーク:7.60、8.99及び/若しくは15.16±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる観点において、前記結晶形態は、実質的に図5に記載されたようなX線粉末回折パターンを持つものである。
【0042】
本発明のさらなる別の観点は、形態S20、形態S30、形態S40、形態S50、形態S60、形態S70、形態S90、形態S100、形態S120或いはそれらの混合物である化合物Iの結晶形態に関するものである。さらなる観点において、前記結晶形態は、形態S20である。さらなる別の観点において、前記結晶形態は、形態S30である。付加的な観点において、前記結晶形態は、形態S40である。さらなる観点において、前記結晶形態は、形態S50である。さらなる付加的な観点において、前記結晶形態は、形態S60である。別の観点において、前記結晶形態は、形態S70である。さらなる観点において、前記結晶形態は、形態S90である。さらなる別の観点において、前記結晶形態は、形態S100である。さらなる観点において、前記結晶形態は、形態S120である。さらなる観点において、前記結晶形態は、以下のピーク:8.56、14.64、16.07、22.24及び/若しくは23.02±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:6.07、8.67、13.36、16.80及び/若しくは16.85±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:8.42、8.60、13.92、17.20及び/若しくは24.46±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:4.46、7.67、8.86及び/若しくは11.71±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:8.68、11.10、16.94、17.39及び/若しくは23.31±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:4.50、7.70、8.90及び/若しくは11.76±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、以下のピーク:8.34、8.67、16.68、17.33及び/若しくは24.57±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:4.45、7.62、8.79、11.62及び/若しくは17.67±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:7.63、7.67、9.00、17.99及び/若しくは24.46±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。
【0043】
本発明の付加的な観点は、形態A0である化合物Iの結晶形態を調合する方法に関するものであり、この方法は:(a)炭化水素(ヘプタン或いはトルエンなど)において化合物Iをスラリー化する工程と;(b)結果生じたスラリーを冷却する工程と;(c)前記結果生じたスラリーを濾過する工程と;(d)濾過ケーキを乾燥させる工程とを有するものである。1つの観点において、化合物Iは、26〜45容積のヘプタンにおいてスラリー化させるものである。別の観点において、化合物Iは、45容積のヘプタンにおいてスラリー化させるものである。付加的な観点において、工程(a)は、79〜83℃で実行されるものである。さらなる別の観点において、工程(a)は、85℃で実行されるものである。さらなる別の観点において、工程(a)は、24〜48時間実行されるものである。さらなる観点において、工程(a)は、45時間実行されるものである。別の観点において、工程(b)は、30〜65℃の温度で起こるものである。さらなる別の観点において、工程(b)は、65℃で実行されるものである。別の観点において、工程(d)は、0.33〜3時間、室温で実行されるものである。さらなる別の観点において、工程(d)は、3時間室温で実行されるものである。
【0044】
本発明のさらなる観点は、形態A0である化合物Iの結晶形態を調合する方法に関するものであり、この方法は:(a)溶媒に化合物Iを溶解させる工程と;(b)結果生じた溶液を濾過する工程と;(c)化合物Iを沈殿させるために抗溶媒を添加する間に前記溶媒を部分的に蒸留する工程と;(d)工程(a)で使用した前記溶媒の容量を減らすために付加的な抗溶媒を添加する間に結果生じたスラリーをさらに蒸留する工程と;(e)形態A0への完全な変換を達成するように前記スラリーを加熱する工程と;(f)冷却する工程と;(g)濾過を介して産物を回収する工程と;(h)乾燥させる工程とを有するものである。さらなる観点において、工程(a)は、27〜35容積のTHFを用いて実行されるものである。別の観点において、工程(a)は、30容積のTHFを用いて実行されるものである。さらなる観点において、工程(a)を介して産生された前記溶液は、任意に金属スカベンジャー或いは炭素で処理されるものである。さらなる観点において、前記濾過する工程(b)は、以下の工程:(i)前記金属スカベンジャーを除去するために濾過する工程;及び(ii)1ミクロンインライン(inline)カートリッジフィルターを通じて研磨濾過(polish filtering)する工程の1若しくは両者を有するものである。さらなる観点において、工程(c)で存在する前記溶媒は、そのオリジナル容量の60〜90%へ蒸留されるものである。付加的な観点において、工程(c)は、抗溶媒として炭化水素(ヘプタンなど)を用いて実行されるものである。別の観点において、工程(d)は、容量で5%以下のTHFが残るまで実行されるものである。さらなる別の観点において、工程(e)は、約90〜96℃の温度で実行されるものである。付加的な観点において、工程(e)は、任意に省略されるものである。別の観点において、前記スラリーは、約3〜5時間撹拌させるものである。さらなる観点において、工程(f)は、外界温度(25±5℃)で実行されるものである。付加的な観点において、工程(g)の濾過は、乾燥した不活性ガスを用いて実行されるものである。別の観点において、工程(h)は、80℃までの温度で実行されるものである。さらなる別の観点において、残留水及び/若しくは溶媒和化合物(群)は、共沸的に除去されるものである。
【0045】
本発明の別の観点は、形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物に関するものである。さらなる観点は、癌を治療する方法に関するものであり、この方法は、形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物の治療上有効な量を必要とする患者へ投与する工程を有するものである。付加的な観点において、本発明は、癌を治療する方法に関するものであり、この方法は、形態A0を有する薬学的組成物の治療上有効な量を必要な患者へ投与する工程を有するものである。
【0046】
専門用語
本明細書で用いられたように「非結晶」という用語は、特徴的な結晶形状或いは結晶構造を欠いていることを意味するものである。
【0047】
本明細書で用いられたように「抗溶媒」という用語は、そこにおいては化合物が実質的に不溶性であることを意味するものである。
【0048】
本明細書で用いられたように「結晶」という用語は、分子或いは外表面(external face planes)の規則的に繰り返される配列を持つことを意味するものである。
【0049】
本明細書で用いられたように「結晶形態」という用語は、X線粉末回折によって分析した場合、ピークの特徴的なパターンを提供する、固形化学化合物或いは化合物の混合物を意味するものであり;これには、限定するものではないが、多形(polymorphs)、溶媒、水和物、共結晶及び脱溶媒された(de−solvated)溶媒が含まれるものである。
【0050】
「多形」或いは「多型(polymorphism)」という用語は、同じ化学分子に対して少なくとも2つの異なる結晶配列の可能性として定義されるものである。
【0051】
本明細書で用いられたように「溶質」という用語は、通常溶液の構成成分はより少ない量で存在するものである、別の物質に溶解された物質を意味するものである。
【0052】
本明細書で用いられたように「溶液」という用語は、少なくとも1つの溶媒、及び前記溶媒に少なくとも部分的に溶解した少なくとも1つの化合物を含有する混合物を意味するものである。
【0053】
本明細書で用いられたように「溶媒和化合物」という用語は、前記結晶構造内に溶媒分子を含有する結晶物質を意味するものである。
【0054】
本明細書で用いられたように「溶媒」という用語は、別の物質、一般的には固形を完全に或いは部分的に溶解することができる物質、一般的には液体を意味するものである。本発明の実行に対する溶媒には、これに限定されるものではないが、水、酢酸、アセトン、アセトニトリル(ACN)、ベンジルアルコール、1−ブタノール、2−ブタノール、2−ブタノン、ブチロニトリル、三級(tert)−ブタノール、N−ブチル酢酸、クロロベンゼン、クロロホルム、シクロヘキサン、1−2ジクロロエタン(DCE)、ジクロロメタン(DCM)、ジエチレングリコールジブチルエーテル(DGDE)、ジイソプロピルアミン(DIPA)、ジイソプロピルエーテル(DIPE)、1,2−ジメトキシエタン(DE)、N,N−ジメチルアセトアミド(DMA)、4−ジメチルアミノピリジン(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド、1,4−ジオキサン、エチレングリコール、ジメチルエーテル、エタノール、酢酸エチル、エチルジイソプロピルアミン、エチレングリコール、ギ酸エチル、ギ酸、ヘプタン、イソブチルアルコール、酢酸イソプロピル(IPAC)、イソプロピルアルコール(IPA)、イソプロピルアミン、リチウムジイソプロピルアミド(LDA)、メタノール、メトキシベンゼン(MTB)、酢酸メチル、メチルエチルケトン(MEK)、メチルイソブチルケトン(MIK)、2−メチルテトラヒドロフラン、メチル三級(tert)−ブチルエーテル(MTBE)、1:1のホルムアミド:水、1:1のN−メチルピロリジノン(NMP):水、2−ペンタノン、3−ペンタノン、1−ペンタノール、1,2−プロパンジオール、2−プロパノール(IPA)、1−プロパノール、プロパノニトリル、プロプレンカーボネート、1,2−プロピレングリコール(PG)、ピリジン、テトラヒドロフラン(THF)、テトラヒドロピラン(THP)、トルエン、トリエチルアミン、キシレン、それらの混合物、及びそれらと同等物が含まれる。これらの溶媒は、それらの機能基に従って5つのクラスへ分類される:クラス1:「プロトン性」或いは水素結合供与溶媒(ルイス酸)、これにはベンジルアルコール、エタノール、IPA、メタノール及び水が含まれる;クラス2:水素結合受容溶媒(ルイス塩基)、これにはアセトン、1,4−ジオキサン、DMF、酢酸エチル、MEK、MTBE、THF及び水が含まれる;クラス3:極性非プロトン性溶媒、「非ヒドロキシル性溶媒」とも呼ばれる、これにはアセトニトリル、DMA、DMF及びDMSOが含まれる;クラス4:クロロカーボン溶媒、これにはクロロホルムが含まれる;クラス5:ヒドロカーボン溶媒、飽和及び不飽和の両者を含み、これらにはn−ヘプタン、トルエン、p−キシレン及びキシレンが含まれる。
【0055】
本明細書で用いられたように「治療上有効な量」という用語は、所定の投与経路で、意図され、確立された薬物動態法及び技術に従って測定されたような所定の薬剤と関連する生理学的効果を得るのに必要であると決定された量を意味するものである。適切で特異的な治療上有効な量は、従来の技術の使用によって、本分野の当業者である専門診断医によって容易に決定され得る。有効な量は、疾患或いは疾病の進行のタイプ及び程度、特定の患者の全体の健康状態、選択された化合物の関連する生物学的有効性、適切な賦形剤での有効薬剤の処方及び投与経路を含む、多数の因子に依存して変えられるであろう。一般的に、結晶形態は、目的の効果が達成されるまで徐々に増加させながら、より低い投与量レベルで投与される。
【0056】
他に記述がない限り、本明細書中で記載されるパーセンテージは、重量/重量(w/w)パーセンテージである。
【0057】
本明細書で用いられたように「薬学的に許容可能な賦形剤」という用語は、あらゆる及び全ての溶媒、分散媒体、コーティング剤、抗菌剤及び抗真菌剤、等張性吸収遅延剤及びそれらと同等物を含む。薬学的に有効な物質に対するそのような媒体及び薬剤の使用は、本分野ではよく知られており、例えば、Remington:The Science and Practice of Pharmacy,20th ed.;Gennaro,A.R.,Ed.;Lippincott Williams&Wilkins:Philadelphia,PA,2000などを参照のこと。あらゆる従来の媒体或いは薬剤が前記有効成分と不適合である限りを除いて、治療用組成物におけるその使用は考慮される。
【0058】
治療目的で、本発明の結晶形態は、対象の体内における薬剤の作用部位との有効薬剤の接触を生じるあらゆる手段で投与され得る。前記結晶形態は、個々の薬剤として、或いは例えば鎮痛剤などの他の薬剤との組み合わせで、医薬品との併用で利用可能なあらゆる従来の手段によって投与される。本発明の結晶形態は、好ましくは、それが必要な対象へ、本明細書で記載された疾病及び疾患の治療に対して治療上有効な量で投与される。
【0059】
治療用或いは予防用使用において、本発明の結晶形態は、薬剤が従来投与されるあらゆる経路によって投与される。そのような投与経路は、腹腔内、静脈内、筋肉内、皮下、くも膜下腔内、気管内、脳室内、経口、頬側、直腸、非経口、鼻腔内、経皮及び皮内を含む。投与は、全身的或いは局所的である。
【0060】
本明細書に記載された結晶形態は、純粋な形態で、他の有効成分との組み合わせで、或いは薬学的に許容可能な無毒賦形剤或いは担体との組み合わせで投与される。経口組成物には、一般的に不活性希釈剤担体或いは食用担体が含まれる。薬学的に適合性な結合剤及び/若しくはアジュバント物質は、前記組成物の一部として含まれ得る。錠剤、丸剤、カプセル、トローチ及びそれらと同等物は、あらゆる以下の成分、或いは同様な性質の化合物:結晶セルロース、ゴムトラガント或いはゼラチンなどの結合剤;スターチ或いはラクトースなどの賦形剤、アルギン酸、プリモゲル(Primogel)或いはコーンスターチなどの分布剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド性二酸化ケイ素などの流動促進剤;スクロース或いはサッカリンなどの甘味料;或いはペパーミント、サリチル酸メチル或いはオレンジ香料などの香味料、を含有することができる。投薬量ユニット形態がカプセルである場合、上記タイプの物質に加えて、それは脂肪油などの液体担体を含有することができる。加えて、投薬量ユニット形態は、例えば、砂糖のコーティング剤、セラック或いは腸溶性剤などの前記投薬量ユニットの物理的形態を修飾する様々な他の物質を含有することができる。さらに、前記有効化合物に加えて、シロップは甘味料としてのスクロース、及び特定の保存料、色素、着色料及び香料を含有する。
【0061】
投与に対する代替調合には、蒸留水或いは非水溶液、懸濁液及び乳化剤が含まれる。非水溶液の例としては、ジメチルスルホキシド、アルコール、プロピレン、グリコール、ポリエチレングリコール、オリーブオイルなどの植物油、及びオレイン酸エチルなどの注入可能な有機エステルである。水溶性担体には、アルコールと水の混合物、緩衝媒体及び生理食塩水が含まれる。静脈内媒体には、液体及び栄養補充薬、リンガーブドウ糖に基づいたものなどの電解質補充剤、及びそれらと同等物が含まれる。例えば抗菌剤、抗酸化剤、キレート剤、不活性ガス及びそれらと同等物などの保存料及び他の添加物も存在する。
【0062】
ほ乳類への結晶形態の投与の好ましい方法には、腹腔内注射、筋肉内注射、及び静脈内注入が含まれる。生理食塩水、アルコール、DMSO及び水ベース溶液を含む様々な液体処方は、これらの運搬方法に対して可能である。濃度は、運搬される投与量及び容量に従って変えられ、約1〜約1000mg/mLの範囲であり得る。前記液体処方の他の成分には、保存料、無機塩類、酸、塩基、緩衝液、栄養剤、ビタミン群、或いは鎮痛薬或いは付加的PARP及びキナーゼ阻害剤などの他の薬剤が含まれる。
【0063】
計測手段
X線粉末回折(XRPD)
XRPDパターンは、40kVおよび40mAで、CuKα放射線を使用してPANalytical X’ Pert Pro回折装置で記録した。シリコン標準は、X線管のアラインメントをチェックするために試験した。試料は、アルミホルダー中のゼロ−バックグラウンド石英プレート上に押しつけた。一般的なX線粉末パターンのスキャンは、約2〜40°2θ間を刻み幅0.0080°および計数時間96.06秒で収集し、約0.5°/分のスキャン速度で行った。
【0064】
単結晶の研究では、選択した結晶にパラトン油(paratone oil)を塗布し、Oxford回折CCD回折計(サファイア検出器搭載Xcalibur S)上で急速冷凍した。データは、標準領域検出法で収集した。構造はSHELXTLパッケージで解析し,精密化した。測定したXRPDパターンに対する単結晶パラメータのデフォルトのリートベルト(Reitveld)法精密化により、良い適合が得られ、説明のつかないピークは無かった。
【0065】
温度可変X線粉末回折(VT−XRPD)
温度可変の研究は、Anton Paar TTK450温度槽を用い、Anton Paar TCU100温度制御ユニットによる、コンピュータ制御下、窒素雰囲気で行った。限定および連続測定方式の2つを使用した。限定モードにおいては、前記TTK450チャンバーが所定温度に到達した後でのみ、測定を実施した。連続モードでは、試料を10℃/分で加熱し、温度が変化する間、高速スキャンを測定した。所定温度に到達後、試料を30または35℃/分で冷却し、より低速なスキャンを25℃で測定した。前記選択した温度は、DSCの結果に基づいたものである。
【0066】
示差走査熱量測定(DSC)
熱曲線は、Pyrisソフトウェア・バージョン6.0実行下の自動試料採取装置搭載し、分析前にインジウムで較正したPerkin−Elmer Sapphire DSCユニットを使用して取得した。固体試料1〜11mgを蓋無しの、20μLアルミ製試料パンに秤量した。次いで、DSCセルは、窒素でパージし、温度0℃から275℃まで10℃/分の速度で加熱した。
【0067】
熱重量分析(TGA)
熱曲線は、Pyrisソフトウェア−バージョン6.0を実行し、シュウ酸カルシウム1水和物で較正したPerkin−Elmer Pyris 1 TGA装置を使用して取得した。TGA試料1〜15mgを約50mL/分のヘリウムでパージした炉内において10℃/分で25℃から400℃に加熱した時の%重量減を測定した。
【0068】
動的水蒸気吸着(DVS)
重量による水蒸気吸着実験は、DVS−HT装置(英国ロンドン、Surface Measurement Systems,London,UK)を用いて行った。この装置は、重量測定±0.1μgの質量分解能を有する記録式超微量天秤を用いて水蒸気の吸収および減量を重量で測定する。試料周辺水蒸気分圧(±1.0%)は、電子質量流量コントローラを使用して飽和および乾燥キャリアガス流を混合することによって制御した。所望の温度を±0.1℃に維持した。
【0069】
試料(10〜25mg)を所定温度でDVS−HT装置内に配置した。2種の動的蒸気吸着実験を実施した。
【0070】
1.試料は、最初20時間乾燥空気流中(<0.1%未満の相対湿度(RH))で乾燥して乾燥質量を決定し、次いで2つの0〜90%RHサイクル(RHを10%単位で変化)にさらした。
【0071】
2.試料は20時間90%RHにさらし、次いで2つの90〜0%RHサイクル(RHを10%単位で変化)にさらした。
【0072】
赤外分光(FTIR)
スペクトルは、ダイヤモンド結晶ウィンドーを含むSmart Orbit ATR付属装置付きThermo Electron−Nicolet Avatar 370 DTGSを用いて取得した。Thermo Electron Omnic(商標)ソフトウェア(version 3.1) を使用して、初期干渉写真(interferogram)から4000〜400cm−1のスペクトルを計算した。各試料のスペクトルを取得する以前にバックグラウンドスキャンを収集した。各試料について、4cm−1のスペクトル分解能でスキャンを32回行い、平均値を得た。
【0073】
ラマン分光測定
前記試料のラマンスペクトルは、FT−ラマンモジュールを用い、Vertex70 FTIR分光器(Bruker RAM II,Bruker OPtics,ドイツ)で記録した。 YAGゲルマニウムフォトダイオードを使用して、ND : Yag レーザー(蛍光の抑制)で励起したFT−ラマンスペクトルを記録した。試料の分析前にポリスチレン標準を試験した。各スペクトルの取得時間は4cm−1の分解能で1分であり、1064nm レーザーパワーは、試料部で50mWであった。
【0074】
独自性(identity)、アッセイ(assay)および純度
通常、試料溶液分量10μLをアセトニトリルで1mLに希釈し、アッセイ濃度は、以下のHPLC法を用いて二回注入物の平均値から決定した。純度および不純物の分析は、従来のHPLCを使用して実施する。
【0075】
【表15】
【0076】
実施例
化合物I調製プロセス
化合物Iは、図式Iに従って調製することが出来る。
【0077】
【化2】
【0078】
図式Iにおいて、前記合成は、市販の出発原料4−メトキシインドールを用いて開始する。ジtert−ブチルジカーボネート((Boc)2O)でインドールの窒素を保護(マスキング)した後、当該インドール誘導体を、リチウムジイソプロピルアミド(LDA)で活性化して、当該インドールの2位置にカルバニオンを生成し、それはその場で(in−situ)ホウ酸トリイソプロピルと反応する。酸性処理でボロン酸エステル中間体を加水分解して、対応するインドールボロン酸化合物Aにする。次いで、化合物Aを鈴木法条件下、触媒量の酢酸パラジウムとトリフェニルフォスフィンの存在下で、1−シクロペンテニルトリフルオロメタンスルホネート(また、本明細書ではエノールトリフレートとも呼ぶ)と結合させ、重要なジエン中間体化合物Bを得る。ナトリウムメトキシドで前記Boc保護基を除去した後、ジエン化合物Cを、酢酸中、ディールス・アルダー反応によってマレイミドと結合させて、5環性中間体化合物Dを生成する。クロルアニル酸化による芳香族化で、化合物Dを化合物Eに転換し、それをマンニッヒ反応条件下1−メチルピペラジンと結合させて、目的とする分子の化合物Iを生成する。この合成の詳細な内容を以下に記載する。
【0079】
N−Boc−4−メトキシインドールの合成
100ガロン・ガラス内張反応装置に4−メトキシインドール(20.0kg、136モル、Yixing Zhongyu Medicine Technology Co.,Ltd.)を仕込み、次いでDMAP(0.50kg、4.1モル、Aldrich)およびトルエン(92kg、CORCO試薬等級)を加えた。得られた混合物を攪拌し、約40℃に加熱した。別に、ジ−tert−ブチルジカーボネート(31.8kg、146モル、Lacamas Laboratories、Inc.)のトルエン(60kg、Corco試薬等級)溶液を第2反応装置で調製した。この溶液を、約1.75時間かけて前記インドール溶液に添加した。わずかに発熱した反応(最高温度約41℃)は、ガスの発生を伴った。40℃でさらに1時間撹拌後、前記反応液を20±3℃に冷却した。工程内試験(in−process test)で、4−メトキシインドールが完全に反応したことが判明した。脱イオン水(15ガロン)を加えて過剰の(Boc)2O(注意:ガス発生)を分解した。次いで、得られた混合物を0.5時間激しく攪拌し,次いで一晩放置した。下側の水層を除去後、当該有機層を部分的に減圧下(60℃のジャケットで最高60mmHg)で濃縮し、留出液約145Lを除去した。この時点で、追加のトルエン(30kg、Corco試薬等級)を仕込み、合計約200Lの留出物が収集されるまで蒸留を続けた。このバッチは、その後、室温に冷却し、ポリ・ドラムに空けて、N−Boc−4−メトキシインドール(理論収率と想定)33.6kgを含む濃色琥珀色の溶液62.3kg得た。これをさらに精製することなく次の工程で使用した。
【0080】
化合物A
2−ボロノ−4−メトキシ−1H−インドール−1−カルボン酸−1−(1,1−ジメチルエチル)エステル)の合成
上記溶液の約半分を、100ガロン・ガラス内張反応装置に仕込み、次いでトルエン、(3.0kgで仕込み量を50重量%に希釈)、ホウ酸トリイソプロピル(19.9kg、105.9モル、Anderson Development Co.)、およびTHF(91kg、CORCO試薬等級)を添加した。得られた溶液を攪拌し、−2℃に冷却した。この時点で、リチウムジイソプロピルアミド(37.3kg、91.8モル、エチルベンゼン/テトラヒドロフラン/ヘプタン27%溶液、FMC Lithium社)を、当該バッチの温度を3℃未満(−10℃のジャケット)に維持しながら、一時間かけて添加した。LDAの添加後、HPLC(LDAの添加後30分で、N−Boc−4−メトキシインドールは、0.6面積%残存)で反応完了が認められまで、得られた反応混合物を0±3℃で攪拌した。別に,第2反応装置で、濃塩酸27kgを脱イオン水16.3ガロンで希釈して3N塩酸溶液を調製し、約5℃に冷却した。この希塩酸を、バッチ温度を15℃未満に維持するよう1時間かけて当該バッチに添加した(バッチ温度は添加完了時に8℃に到達した)。次いで、ジャケット温度を20℃に設定した。反応装置および添加管路を脱イオン水(6ガロン)で水洗し、この水洗液をバッチと合わせた。次いでMTBE(27kg、Pride)を添加した。得られた混合物を0.5時間攪拌し、次いで停止して相分離させた。水層を分離し、第2反応装置でMTBE(14kg、Pride)を用いて逆抽出した。合併した有機層を5%NaCl(34L)、5%NaHCO3(34 L)、10%NaCl(19L)で順次洗浄した。当該有機相をドラム缶に投下し、検量後(172.2kg)、反応装置に戻し、減圧下(反応装置のジャケット設定温度:30℃)で濃縮し、3時間かけて留出物116kgを除去した。得られたスラリーをn−ヘプタン(75kg、CORCO試薬等級)で希釈し、次いで蒸留し、さらに留出液75Lを除去した。室温で一晩撹拌後、当該スラリーを1時間約5℃に冷却した。生成物はAuroraフィルター上に収集し、n−ヘプタン33kgで洗浄した。濾過ケーキを窒素気流中加熱せずに備え付きの真空下トレイ上で一晩乾燥させた。灰色がかった固体として化合物A17.8kgを得た(補正収率88.8%)。HPLC純度:100LC面積%(LCAP)、95.8LC重量%(LCWP)。
【0081】
1,1,1−トリフルオロメタンスルホン酸1−シクロペンテン−1−イルエステルの合成
室温で100ガロン・ガラス内張反応装置にシクロペンタノン(8.95kg、106.5モル)を仕込み、次いで、トルエン(116.40kg、CORCO試薬等級)及びエチルジイソプロピルアミン(16.6kg、128.7モル)加えた。得られた溶液を攪拌し、45±5℃に加熱した。この時点で、トリフルオロメタンスルホン酸無水物(36.2kg、128.4モル)を30−L添加フラスコから約1時間かけて添加した。トリフルオロメタンスルホン酸無水物の添加は非常に発熱した。ジャケット冷却(10℃に設定)を実施して、バッチ温度を45±5℃に維持した。当該バッチは44分の添加時間中、7分間40℃未満に降下した。トリフルオロメタンスルホン酸無水物の添加後、20分間39〜45℃で攪拌を続けた。この20分後の工程内試験では、シクロペンタノンが全量反応したことが判った。19.6℃に冷却後、当該バッチをフィルター中のセライト(Celite)のパッド(18.0kg)を通して濾過した。当該濾液は清潔なポリ内張スチールドラム缶に収集された。前記セライトパッドは、トルエン(37.0kg、CORCO試薬等級)でリンスした。概リンス液は、同じポリ内張スチールドラム缶のバッチと合併した。濾液(159.85kg)は、参照標準に対して分析し、エノールトリフレート(triflate)を19.50kg(83.3%収率)含むことを示した。このエノールトリフレートのトルエン溶液を一晩低温室に貯蔵し、これ以上精製することなく、次の鈴木カップリング法に使用した。
【0082】
化合物B
2−(1−シクロペンテン−1−イル)−4−メトキシ−1H−インドール−1−カルボン酸1,1−ジメチルエチルエステルの合成
室温で100ガロン・ガラス内張反応装置に化合物A(18.00kg、61.8モル)、トリフェニルホスフィン(648.8グラム、2.47モル)および酢酸パラジウム(277.0グラム、1.23モル)を仕込んだ。反応装置は、その後3回脱気・窒素充填を行った。トルエン(78.3kg、CORCO試薬等級)を反応装置内に噴射注入し、次いでジシクロヘキシルアミン(44.5kg、245.4モル)を注入した。この添加に4分かかった。得られたスラリーを21分間室温で(125rpm)を激しく攪拌し、次いで43分かかってエノールトリフレートのトルエン溶液ストリーム(131.7kg、エノールトリフレートを16.07kg、74.3モル含有)をゆっくり注入添加した。エノールトリフレートの添加により発熱した。ジャケット冷却を実施し、バッチ温度を18.8〜27.5℃に保持した。得られた不均一混合物は、前記反応の完了がHPLCで認められるまで、18.4〜22.3℃で攪拌した(注、当該反応は、1時間未満で完了したが、当該処理を続ける前に、猶一晩室温で攪拌した。これは厳密には便宜上の為であった。前記バッチは、生成物に悪影響を与えることなく、100時間までは室温に放置できる)。当該バッチを10分間室温で撹拌し、次いでフィルター中のセライト(Celite)のパッド(2.70kg)を通して濾過した。濾液は清潔なポリ内張スチールドラム缶2個に収集した。濾過ケーキをトルエン(47.8kg、CORCO試薬等級)でリンスした。リンス液は、同じポリ内張スチールドラム缶のバッチと混合した。濾液(260.45kg)を参照標準に対して分析し、化合物Bを20.59kg(106.4%収率)含有することが示された。この検定(assay)に基づいて本反応は100%収率に達したと見なされ、次の工程の仕込みは、反応が100%収率で進行したとして実施した。化合物Bのトルエン溶液は、室温でパイロットプラント内に貯蔵し、これ以上精製することなく、その後の脱保護手法で使用した。
【0083】
化合物C
2−(シクロペンテン−1−イル)−4−メトキシ−1H−インドールの合成
室温で100ガロン・ガラス内張反応装置に、化合物Bのトルエン溶液流(化合物B12.82kg、40.96モル)を仕込み、次いでナトリウムメトキシド(44.0kg、25〜30重量%MeOH溶液、203.7モル)を加えた。得られた溶液を攪拌し、45±5℃に加熱した。反応の完了がHPLCで認められるまで45±5℃で攪拌を続けた(約4時間で反応が完了、HPLCデータは約8時間目に報告された)。前記バッチは、その後26分かかって23.5℃に冷却した。当該バッチを22±2℃で一晩攪拌した。22℃で約17時間後、バッチの約1/2(111.15kg)を第2反応装置に移して、別々に処理した。第1反応装置には脱イオン水(21ガロン)を仕込んだ。得られた混合物を16分間攪拌し、次いで停止した。バッチを46分間室温に静置させた後、底水層を除去した。次いで、ラグ層の小部分を清潔なカーボイ(大型瓶)に排出した。残りの有機層は、フィルター中のセライト(Celite)のパッド(3.84kg)を通して濾過した。濾液は清潔なポリ内張 スチールドラム缶に収集した。前記ラグ層は、同じセライトパッドを通して濾過し、濾液を新規のカーボイに回収した。前記セライトパッドは、トルエン(6.20kg、CORCO試薬等級)で洗浄し、この洗浄液は濾過したラグ層と合併した。濾過ラグ層は、ガラス製添加容器に移し、底水層を除去し、ラグ由来有機層を元の有機層と合併した。上記の処理手法をバッチの第2半量部に繰り返し、化合物Cの第2トルエン溶液を生成した。入るだけの第2溶液を第1有機層(164.70kg、化合物C8.14kg含む)の入ったポリ内張スチールドラム缶に入れた。残りの第2有機層は、小さなポリドラム(19.05kg、化合物Cを0.49kg含有)に収納した。これら2つの溶液は、それ以上精製せずに次の工程でさらなる処理用に、パイロットプラントに貯蔵した。化合物Cは、合計8.63kg(99.2%収率)生成された。
【0084】
化合物D
3a、4、5、6、6a、7、11c−オクタヒドロ−11−メトキシ−1H−シクロペンタ[a]ピロロ[3,4−c〕カルバゾール−1,3(2H)−ジオンの合成
室温で100ガロン・ガラス内張反応装置に、化合物Cのトルエン溶液流(化合物C、、12.58kg、59.1モル)を仕込んだ。この溶液を備え付き真空全開で40℃未満の内部温度下、残渣が化合物Cの重量の約6倍(目標体積約75.5 L)に達するまで、約7時間かけて、濃縮した。この残渣は、清潔なポリエチレンドラムに排出させ、これ以上精製せずに次のディールス・アルダー反応に使用した。第2・100ガロン・ガラス内張反応装置に,マレイミド(7.45kg、76.8モル、Carbosynth Limited)を仕込み、次いで氷酢酸(145.70kg)を加えた。得られた混合物を攪拌して溶液を得た。この時点で、上方から化合物Cの濃縮溶液(84.95kg)を、バッチ温度20±10℃(ジャケット温度を15℃に設定した)に制御するよう、約20分かけて仕込んだ。得られた混合物は、反応の完了が、HPLC(反応は約15.5時間で完了、HPLCデータは約17.5時間目に受領する)で認められるまで30℃±3℃で攪拌した。当該バッチは、約20分かけて23.2℃に冷却した。母液を重量基準HPLCアッセイにより分析し、化合物Dが10%未満(検出:5.88%)含まれていることを確認後、当該バッチをAuroraフィルタで濾過した(23.2℃に達してから濾過時まで2.5時間)。当該濾過ケーキを氷酢酸(39.65kg)でリンスし、化合物Dの純度が、HPLCの重量基準アッセイで(3晩乾燥させ、3晩後の純度は99.5重量%であった)所定の仕様(90重量%超過)に到達するまで、窒素気流、真空下当該フィルタで乾燥した。次いで生成物を2重ポリエチレン袋内張ファイバードラムに取り出し、所望化合物D13.43kg(73.3%収率)を褐色固体として得た。この物質は、これ以上精製せずに、次のクロラニルによる酸化に使用した。
【0085】
化合物E
4、5、6、7−テトラヒドロ−11−メトキシ−1H−シクロペンタ[a]ピロロ[3、4−c〕カルバゾール−1、3(2H)−ジオンの合成
室温で100ガロン・ガラス内張反応装置に化合物D(28.66kg、92.45モル)を仕込み、次いでテトラクロロ−p−ベンゾキノン(45.50kg、185.0モル、99%、ACROS)およびTHF(253.1kg、CORCO試薬等級)を加えた。得られた不均一混合物を65±5℃に加熱し、反応の完了がHPLC(反応は約22時間で完了、HPLCデータ約23時間目に受理)で認められまで、この温度で攪拌した。次に当該バッチを35分間かけて22±5℃に冷却し、溶液中に化合物Eが消失したか分析し(仕様では10%未満、検出:1.9%)てから、フィルターで濾過した。反応装置、管路、および濾過ケーキをTHF−EtOH−H2Oの混合物(第2反応装置でTHF62.0kgをEtOH41.25kgおよび脱イオン水4.64ガロンと混合して調製)でリンスした。生成物が所定の仕様(仕様では80重量%超過、5日後化合物Eを80.8重量%検出)に到達するまで湿潤ケーキを窒素気流、真空下、フィルターで乾燥した。生成物は、その後、2つの二重ポリエチレン袋内張プラスチック缶に取り出し、化合物E23.84kg(86.7%収率)を暗緑黄色の固体として得た。$$$
化合物Iの合成
100ガロン・ガラス内張反応装置に化合物E(15.20kg、40.1モル)を仕込み、次いでパラホルムアルデヒド(2.51kg、80.9モル、97%、ACROS)および変性エタノール(223.45kg、試薬等級)を加えた。得られた混合物を撹拌(121rpm)し、その間添加フラスコから約10分かけて、1−メチルピペラジン(6.65kg、65.77モル、ACROS、99%)を添加した。得られた反応混合物を70℃で加熱、攪拌した。反応の進行は、HPLC(約5時間後、化合物E1.35面積%残存)により観察した。 合計9時間70℃で攪拌後、当該バッチを20±3℃に冷却し、この温度で一晩攪拌した。生成物はフィルターで濾過した。濾過ケーキをエタノール(43.9kg、試薬等級)でリンスし、残留エタノールが1HNMRで12重量%(化合物Iに対して8.4重量%)未満になるまで窒素気流下フィルター上で減圧乾燥した。生成物は、その後、ポリエチレン袋内張ファイバー・ドラム缶に取り出し、粗化合物I18.05kg(95.8パーセント収率)を黄色固体として取得した:98.6LCAP(LC面積%)、89.2LCWP(LC重量%)。この物質は、これ以上精製せずに、下流プロセスに使用した。
【0086】
多形態のスクリーニング研究
結晶化の研究を実施し、48種の異なる溶媒における多形態を調べた。溶媒は、許容性に基づいて、(ICHクラス2および3)、一連の誘電率、双極子モーメントおよび官能基の範囲を網羅するよう選択した。出発材料2種:形態A0およびロット7(形態A0、形態HC0および形態HD0の混合物)を選択した。可能な場合には、化合物Iの多形態のスクリーニング中に発生した新しい形態に就いて、完全な特性解析を行った。この特性解析は、XRPD、熱解析、DVS、40℃/75%RHでの保存、および純度から構成されたものである。化合物(I)の異なる多形態を得るために、冷却、蒸発、貧溶媒添加を含む4つの結晶化手法を利用した。それぞれ結晶化手法の詳細は以下に示す。これらの手法で各溶媒から得た固体形態を表15に要約する。
【0087】
結晶化の手法:
1.迅速結晶化スクリーン法
2種の小規模スクリーニング手法を使用した。
【0088】
A.化合物I約1mgを0.5mLポリプロピレン製遠心管と溶媒0.5 mL中に秤量した。当該遠心管は室温で18時間撹乱せずに放置させて、変化があるか観察した。次いで、前記管を52.5℃で2.5時間攪拌し、各管を変化に関して観察した。当該加熱遠心管を2〜8℃で20時間攪拌し、初期室温状態から結晶化度の変化(もしあれば)に就いての観測を記録した。
【0089】
B.10倍容積の化合物I(400μL中40 mgのロット7)を含むプレートを20℃から初期温度80℃に4.8℃/分の速度で加熱し、30分後に緩やかな(0.28℃/分)または早い(10℃/分)速度で最終温度5℃に冷却し、18時間その温度に保持した。結晶化実験は、ガラスバイアル(4mL)ウエル・プレート中で実施し、濾過によって固体物質を単離した。当該固形物を10時間57℃で乾燥した。
【0090】
2.迅速冷却結晶化
試料は、沸点において飽和状態を確保するために、溶剤容量に化合物I固体物質40 mg(±2)を添加して調製した。当該混合物を冷却し、0.2μナイロン膜フィルターで濾過し、暖めたガラスバイアルまたは三角フラスコに入れた。この溶液を室温に冷却し、目視検査による決定で結晶形成が終結したと見なされるまで、冷蔵庫(約4℃)に置いた。各冷蔵庫の試料をデカントし、結晶を秤量紙に移し、常温の実験室条件下で恒量になるまで乾燥した。デカントし難い試料は、4分間12000rpmで遠心分離した。迅速冷却手法で固体物質が生成しなかった場合は、前記溶媒を約半分量蒸発させてこれらの試料を濃縮した。当該溶液は、再び冷蔵庫に入れ、固体材料はすべて、デカントまたは遠心分離により単離した。
【0091】
3.ロット7および形態A0での熟成による結晶化
2つのタイプの熟成研究を実施した。
【0092】
A.試料は、ロット7または形態A0を約10 mgねじ蓋バイアル(容積約4.0 mL)中の各溶媒1.0mLに添加して調製した。次いで、これらを振盪しつつ64℃に加熱した。64℃で40分間保持後、試料を5℃(−0.25℃/分の速度で)に冷却した。試料を合計18時間、5℃に保持し、ピペットで1.5mLポリプロピレン製遠心管に移し、1分間12000rpmで回転させた。上清液をデカントした。遠心管またはガラスバイアル中の残渣は、その後、110℃で18時間真空乾燥器で乾燥し、XRPDで分析した。
【0093】
B.形態A0約40mgを異なる溶媒10容量(400μL中に40mg)中でスラリー化した。当該スラリーを50℃(0.5℃/分)および5℃(−0.5℃/分)で4時間づつ交代に48時間振盪した。次いで、固体物質をすべて濾過により単離し、XRPDおよび熱分析で分析した。
【0094】
4.形態A0含有スラリーによる結晶化
スラリー(各溶媒500μL中の形態A020mg)を25℃で時間を変えて振盪した。固体を濾過により単離し、2時間57℃で乾燥し、XRPDで分析した。
【0095】
4つの結晶化方法で単離した固体から得たXRPDの結果を以下表15に記録する。
【0096】
【表16】
【0097】
化合物Iの多形態のスクリーニングで14種の形態および120℃を超えて水和物を加熱することでのみ得られた新しい形態1種(形態B0)が得られた。単離した形態の結果の概要は以下の表16に示す。
【0098】
【表17】
【0099】
安定した固体状形態の説明
無水形態A0の調製
化合物I約200mgを、85℃で45時間、ヘプタン45容量中でスラリー化し、65℃に冷却し、高真空下、3時間室温でフィルター乾燥した。形態A0の回収率は97%であった。
【0100】
別の手法では、次のプロセスに従って化合物Iを形態A0に転換した:
1)化合物Iを30容量のTHFに溶解した。この時点で必要に応じて、この溶液は、金属消去剤(スカベンジャー)または炭素で処理してもよい。
【0101】
2)前記金属スカベンジャーまたは炭素を除去するために、得られた溶液を濾過し、次いですべての外部微粒子を除去する為に、1ミクロンのインライン・カートリッジフィルターを通して仕上げ濾過した。
【0102】
3)前記溶媒(THF)の一部を真空下常温で蒸留して元の容積の約60%にし、続いて同等容積の貧溶媒(ヘプタン)を緩やかに添加して化合物Iを沈殿させた。
【0103】
4)前記溶媒が容積でTHFを5%未満含むようになるまで真空蒸留とさらなるヘプタンの添加を続けた。
【0104】
5)得られたスラリーを90〜96℃に加熱し、この温度で3〜5時間攪拌し、完全なA0への転換を達成した。
【0105】
6)当該スラリーを常温(25±5℃)に冷却した。
【0106】
7)前記生成物/化合物Iは、生成物を通して湿気が吸い込まれるのを避けるために、乾燥不活性ガス下濾過して収集した。
【0107】
8)生成物中の残留溶媒が仕様に適合するまで、湿潤ケーキを最高80℃で乾燥した。乾燥は大気圧または真空下で実施してよい。
【0108】
温度可変粉末X線回折(VT−XRPD)を用いた形態A0の特性評価
形態A0に関して固体−固体変換は20℃から250℃までの温度範囲では起こらない。周囲環境条件に暴露した後、220℃まで加熱することにより得たサンプルのXRPDパターンにおいて有意な変化は認められない(図15を参照)。
【0109】
熱重量分析(TGA)による形態A0の特性評価
形態A0は84.4J/gの融解エンタルピー(ΔHFUS)で、約239℃において単一のピークを示す。TGAにより質量の減少は認められない。TGAにより重量の減少が認められなかったため、脱溶媒和過程の存在は考慮しない(図16を参照)。
【0110】
水分収着(DVS)による形態A0の特性評価
規則的DVS(0〜90%RH)
75%RHで吸着される水分量は0.08%、および90%RHでは約0.1%未満であった。この吸着および脱離曲線の重複は形態A0が吸湿性でないことを示唆する(図17および表25を参照)。DVS後のXRPD再分析により有意な変化は認められなかった(図18)。
【0111】
【表18】
【0112】
変則的DVS(0〜90%RH)
試料質量は90%RHで0.5%のみ増加する。ヒステリシスギャップは表面水吸着のみが生じていることを示唆する。等温線は、全質量増加の0.6%以下で可逆的である(図19および表26)。DVS後のXRPD再分析により有意な変化は認められなかった(図18)。
【0113】
【表19】
【0114】
フーリエ変換赤外線分光(FTIR)およびラマン分光法による形態A0の特性評価
結晶形態A0のFTIRおよびラマンスペクトルはそれぞれ図20および図21に示す。
【0115】
無水形態B0の調製
窒素フロー下で20mgの化合物Iを125℃まで加熱することにより、形態B0を得た。
【0116】
温度可変X線粉末回折(VT−XRPD)による形態B0の特性評価
脱水の後、形態B0に関して固体−固体変換は150℃から200℃までの温度範囲では起こらない(図22を参照)。
【0117】
熱分析による形態B0の特性評価
形態B0の示差走査熱量測定(DSC)図は、68.2J/gの融解エンタルピー(ΔHfus)で、約197℃での溶解を示す(図23)。固体−固体転移が、形態化合物I−B0の融点の前に起こる。形態B0は脱溶媒和のみにより得られた。本形態の相対的な熱力学安定性は、120℃〜199℃の間で示されるDSCデータに反映される。
【0118】
水和物形態HA0の調製
THF/ヘプタンからの結晶化
200mgの化合物Iを室温で143倍容量のヘプタンで70倍容量のTHFから沈殿させ形態HA0を得た。固体は濾過により単離した。物質は18時間57℃で乾燥させた。
【0119】
固体−固体転移による調製
20mgの化合物Iを125℃まで加熱し、窒素フローなしで室温に冷却し、形態HA0を得た。
【0120】
熱分析による形態HA0の特性評価
形態HA0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を表す(図24および表27)。オープンパンにおいて水和物は約60〜120℃の間で幅の広い吸熱ピークを示し、結晶から抜け出る総水量に相当する。吸熱反応の事象は格子からの水分流出を伴う脱水プロセスと一致する。脱溶媒和は、吸熱ピークで固体の状態で起こる。この吸熱ピークの位置およびエネルギーは製剤原料および溶媒の2つの化学成分の状態図に依存し、当該化学成分の安定性が生じる。脱溶媒和のプロセスに割当てることが可能なそれらのそれぞれの溶媒の沸点近くの温度で幅の広い吸熱ピークを示す溶媒和物のDSCサーモグラムはTGAにより確認する。TGAにより解析した場合、一水和物形態HA0は50〜120℃の間で平均4.0%の重量損失を示した。これは1モルの化合物Iの1モルの水への取り込みの理論値である4.1%と一致する。
【0121】
【表20】
【0122】
水吸収による形態HA0の特性評価
図25は形態HA0で集積した動的蒸気収着データを示す。乾燥すると即座に、湿気への暴露による即時の取り込みが起こる。形態HA0の等温線は、20〜30%RHの間で、1.25%の重量の減少を示す。30〜90%RHから、取り込みは平衡に達し始める。最初の脱離段階の間、表面の吸着のみを示すわずかなヒステリシスがある。第2の脱離段階の間ではほとんど脱離は起こらないが、サンプルは〜0.4%の2回目の変化をする。吸収はこの形態がチャンネル水和物であるという証拠を示す。非化学量論的水和は、格子のチャンネルの不完全な水和に起因する。DVS後のXRPD再分析において有意な変化は認められなかった。
【0123】
FTIRおよびラマン分光法による特性評価
結晶形態HA0のFTIRおよびラマンスペクトルはそれぞれ図26および図27に示す。
【0124】
水和物形態HC0の調製
エタノール/水からの再結晶化
40mgのLot7を400μLのエタノールと100μLの水に加え、形態HC0を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0125】
エタノール溶媒和物の40℃/75%RHでの保存
化合物Iのエタノール溶媒和物20mgを9日間40℃/75%RHで保存し、形態HC0を得た。
【0126】
結晶構造の調製
一水和物HC0が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をテトラヒドロフランに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0127】
単結晶X線回折による結晶構造の同定
HC0に関する単結晶X線データを得た。データから得られた格子パラメータは表28に示す。
【0128】
データは、ω−2θスキャンテクニックを用い103Kの温度で集積した。大きさが約0.30×0.16×0.11mmのC24H28N4O4の無色の金属板をランダムな方向にガラス繊維上にのせた。三斜晶系格子パラメータ(P−1,Z=2)および算出したボリュームは、以下の通りである。
a=7.6128(10) α=65.839(18)°
b=11.5697(15) β=79.137(16)°
c=13.193(4)Å γ=86.800(10)°
V=1040.9(3)Å3
【0129】
【表21】
【0130】
【表22】
【0131】
熱分析による形態HC0の特性評価
形態HC0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を示す(図28および表29)。TGAを行う場合、一水和物形態HC0は50〜120℃の間で平均3.9%の重量損失を示した。これは1モルの化合物Iの1モルの水への取り込みの理論値である4.1%と一致する。
【0132】
【表23】
【0133】
水分収着(DVS)による形態HC0の特性評価
規則的DVS(0〜90%RH)
ヒステリシスギャップは全取り込みの0.4%で表面水吸着のみが生じていることを示唆する(図29および表30)。DVS後のXRPD再分析により有意な変化は認められなかった(図30)。
【0134】
【表24】
【0135】
変則的DVS(90〜0%RH)
ヒステリシスギャップは0〜40%RHで表面水吸着のみが生じていることを示唆する。40〜90%RHでバルク吸収が生じているものと思われる(図31および表31)。DVS後のXRPD再分析により有意な変化は認められなかった(図30)。
【0136】
【表25】
【0137】
FTIRおよびラマン分光法による特性評価
結晶形態HC0のFTIRおよびラマンスペクトルはそれぞれ図32および図33に示す。
【0138】
水和物形態HD0の調製
アセトン/水からの再結晶化
40mgのLot7を400μLのアセトンと100μLの水に加え、形態HD0を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0139】
2−メチル−2−プロパノールからの再結晶化
55mLの2−メチル−2−プロパノールに0.54gの化合物Iを沸点まで加熱することによりほぼ完全に溶解させ、形態HD0を得た。透明な溶液を得るため濁った溶液を5μナイロンメンブレンシリンジフィルターを用いシリンジ濾過した(約15%流出し、失った)。溶液を25〜30mLに濃縮し、固体を与えるため2〜8℃で4.5〜5時間冷却した。固体を50℃のオーブン中で溶かし、不溶性物質をt− ブチルアルコールの凍結を妨ぐため暖ためた器具で吸引濾過により単離した。50℃のオーブンで2時間乾燥させ、結果的に0.42g の固体を得た(収率75%)。
【0140】
酢酸イソプロピルからの再結晶
7.5mLの酢酸イソプロピルにおいて0.45gの化合物Iを軽く蓋を閉めたガラス製の20mLのシンチレーション小びん内でマグネティック撹拌子を用い室温で20時間撹拌し、形態HD0を得た。懸濁物を吸引濾過し、固体をドラフト内で110時間以上空気にさらし乾燥させた。乾燥させた物質の重さは380mgであった(収率84%)。
【0141】
結晶構造の調製
一水和物HC0が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をテトラヒドロフランに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0142】
単結晶X線回折による結晶構造の同定
HD0に関する単結晶X線データを得た。データから得た格子パラメータを下記の表32に示す。データは、ω−2θスキャンテクニックを用い103Kの温度で集積した。大きさが約0.40×0.25×0.08mmのC24H28N4O4の無色の金属板をランダムな方向にガラス繊維上にのせた。三斜晶系格子パラメータ(P−1,Z=2)および 算出したボリュームは、以下の通りである。
a=8.171(2) α=111.173(18)°
b=11.419(3) β=92.863(17)°
c=12.7305(19)Å γ=102.07(2)°
V=1072.8(4)Å3
【0143】
【表26】
【0144】
熱分析による形態HD0の特性評価
形態HD0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を示す(図34および表33)。TGAを行う場合、一水和物形態HD0は50〜120℃の間で平均4.0%の重量損失を示した。1モルの化合物Iの1モルの水への取り込みの理論値は4.1%である。
【0145】
【表27】
【0146】
水分収着による形態HD0の特性評価
規則的DVS(0〜90%RH)
試料質量は90%RHで0.6%のみ増加する。ヒステリシスギャップは表面水吸着およびバルク吸収が生じていることを示唆する(図35および表34)。DVS後のXRPD再分析により有意な変化は認められなかった(図36)。
【0147】
【表28】
【0148】
変則的DVS(0〜90%RH)
試料質量は90%RHで0.8%のみ増加する。ヒステリシスギャップは表面水吸着および限られたバルク吸収が生じていることを示唆する(図37および表26)。DVS後のXRPD再分析により有意な変化は認められなかった(図27)。
【0149】
【表29】
【0150】
FTIRおよびラマン分光法による特性評価
結晶形態HD0のFTIRおよびラマンスペクトルはそれぞれ図38および図39に示す。
【0151】
化合物Iの溶解和物形態
メタノールからの再結晶化
40mgのLot7を400μLのメタノールに加え、形態S20を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0152】
2−プロパノールからの再結晶化
40mgのLot7を400μLの2−プロパノールに加え、形態S30を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0153】
エタノールからの再結晶化
40mgのLot7を400μLのエタノールに加え、形態S30を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0154】
結晶構造の調製
エタノール和物が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をエタノールに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0155】
N−N−ジメチルホルムアミドからの再結晶化
40mgのLot7を400μLのN−N−ジメチルホルムアミド(DMF)に添加し、形態S50を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体を濾過により単離した。物質は10時間57℃で乾燥させた。
【0156】
エチレングリコールからの再結晶化
40mgのLot7を400μLのエチレングリコールに加え、形態S60を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0157】
ピリジンからの再結晶化
40mgのLot7を400μLのピリジンに加え、形態S70を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0158】
1−プロパノールからの再結晶化
沸点で飽和状況を確保するため1−プロパノールに40mgの化合物Iを添加して、形態S90を得た。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。結晶形成が視覚的検査により完了したように見えるまで溶液を室温まで冷却し、冷蔵庫(約4℃)に静置した。上澄みの液体を別の容器へ静かかに移すのが困難なサンプルは4分間12000rpmで遠心分離した。
【0159】
N−N−ジメチルアセトアミドからの再結晶化
40mgのLot7を400μLのN−N−ジメチルアセトアミド(DMA)に加え、形態S100を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0160】
イソブタノールからの再結晶化
沸点で飽和状況を確保するためイソブタノールに40mgの化合物Iを添加して、形態S120を得た。混合物を冷却し、暖めたガラス製の小型薬瓶に5μナイロンメンブレンフィルターで濾過した。結晶形成が視覚的検査により完了したように見えるまで溶液を室温まで冷却し、冷蔵庫(約4℃)に静置した。上澄みの液体を別の容器へ静かかに移すのが困難なサンプルは4分間12000rpmで遠心分離した。
【0161】
単結晶X線回折による結晶構造の同定
形態S40に関する単結晶X線データを得た。データから得られた格子パラメータは表28に示す。
【0162】
【表30】
【0163】
化合物Iの溶媒和物形態の熱分析
DSC熱曲線は、全ての溶媒和化合物に関し化合物Iの融点の前に、大きく幅の広い吸熱の存在を示した。TGA分析によりこれらの吸熱が形態S20(図40)、S30(図41)、S40(図42)、S50(図43)、S60(図44)、S70(図45)、S90(図46)、S100(図47)およびS120(図48)に関する脱溶媒和過程に起因していることが示された。これらの溶媒において導かれる溶媒の重量減少の計算を表29に提示する。
【0164】
【表31】
【0165】
化合物I一水和物の結晶構造の同定
水和物形態HC0およびHD0は同形であり、これは個々の「同形の溶媒和物」が、単位格子次元がわずかに歪み、ホスト分子の分子ネットワークが同じタイプである同一の空間群で結晶化することを意味する(Reutzel−Edens S.M.,Newman A W.,Polymorphism in the Pharmaceutical Industry,Edited by Rolf Hilfiker,2006,Wiley−VCH Verlag GmbH&Co.KGaA ISBN:9783527311460)。形態HC0および形態HD0はテトラヒドロピラジン環の立体配座において異なる。2つの水和物形態のX線粉末回折パターンは、開始点として単結晶パラメータを用い、リートベルトテクニック(Rietveld H.M.「A profile refinement method for nuclear and magnetic structures」、Journal of Applied Crystallography 2:65〜71(1969))を使用する事で精密化に成功した。セルデータ、データ集積および精密化の詳細は表28および表32に要約する。
【0166】
識別分析のためのFTIRおよびFT−ラマン法
図20および図21にある形態A0および水和物((図26、図32、図38)および(図27、図33、図39))に関するFTIRおよびラマンスペクトルの比較はカルボニル伸縮領域を除きほとんど違いが認められない。
【0167】
形態A0において、FTIRに関しては中程度の強度である1765cm−1、およびラマンに関しては1770 cm−1でピークを生じた。水和物形態において、FTIRに関しては中程度の強度である1742cm−1、およびラマンに関しては1754〜1695cm−1で本領域においてピークを生じた。この吸収ピークは化合物I構造の五員環の中に含まれるイミド・カルボニル官能性に当てはまる。この違いはほぼ純粋な固体形態の識別に使用するのに十分大きいものである。水和物形態およびA0に関するIRスペクトルはいくらかの違いを示すが、 水酸基における−OH結合の伸縮のために、水和物形態に存在する最も重要な広帯域(3800〜2800)に懸念を残す。
【0168】
【表32】
【0169】
【表33】
【0170】
固体形態間の関係
水中における化合物Iの懸濁物の相対的な安定性
水和物HA0およびA0を水媒体から結晶化する場合、形態HC0+HD0の混合物が産生される(表32)。
【0171】
【表34】
【0172】
2mLの水を数ミリグラムの化合物I形態に添加した。試料を一晩スラリーにした。小さな固体試料を除去し、XRPDにより分析した。スラリーにした後、 形態HA0およびA0が分析を行った全ての状況下において水和物形態(形態HC0およびHD0の混合物)に変換した事を見いだした(表33、表34、表35、表36および表37)。水和物形態は5〜45℃の間の形態A0より熱力学的に安定であるように見える。
【0173】
【表35】
【0174】
【表36】
【0175】
【表37】
【0176】
【表38】
【0177】
【表39】
【0178】
一水和物の相対的な安定性
実際的な温度範囲で2つの多形体(形態HC0および形態HD0)の熱力学溶解度を計測する事により、どちらがより安定でそれらの関係が単変性または互変性であるかどうか決定することが可能である。室温および55℃における これらの一水和物形態の酢酸エチル、MTBEおよび1−ペンタノールへの熱力学溶解度を計測するために実験を行った。化合物Iが多形体スクリーンの間、これらの溶媒で溶媒和化合物を形成しなかったため、これらの溶媒を選択した。
【0179】
【表40】
【0180】
表38にまとめた結果は使用した溶媒および調査を行った温度範囲を示し、2つの水和多形の可溶度値が非常に近似するが、常に形態HD0がより高い値であることを示す。室温〜55℃の間で、溶液形態HD0が形態HC0および形態HA0よりもより熱力学的に安定なことを示す。
【0181】
固体応力安定性
形態安定性における温度と湿気の影響について適切な影響を得るため応力安定性解析を行った。安定性を示すHPLC分析法は、化合物Iおよびその主要な分解産物である、これまで「化合物E」と呼んだ7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザベンゾ[a]トリンデン−4,6−ジオンの定量を行うため開発された。開発した方法は特異的であり、正確で、緻密で、強固である。この手法により化合物Iおよび化合物Eの正確で量的な同定が可能となった。 強制的分解実験の間、形成された全ての分解産物が主要ピークと十分に分離し、開発された方法が特異的であり、かつ安定性を示すことが証明された。
【0182】
形態A0
固体の状態では、無水形態A0は周囲環境から水を吸収する傾向を示し、3ヵ月後に形態HC0およびHD0を水和させる40℃および75%RHの標準的なICH応力状況下で上昇する傾向を示した。化学分解はこれらの応力状況下において化合物Iサンプルで認められなかった。化合物Iを110℃に暴露した場合のみ、化学分解を観察した(表39、表40および表41)。
【0183】
【表41】
【0184】
【表42】
【0185】
【表43】
【0186】
一水和物形態
固体の状態において、表42、表43および表44は40℃で75%の相対湿度で保存する場合、すべての結晶性一水和物が28日間安定であったことを示す。
【0187】
【表44】
【0188】
【表45】
【0189】
【表46】
【0190】
形態S40
形態S40(エタノール溶媒和物)は40℃/75%RHにおいて9日後に一水和物形態HC0に変わり、62日間、この状態のままであった(表45)。
【0191】
【表47】
【0192】
機械的応力による形態の転換
乳鉢および乳棒による粉砕
約100mgの化合物Iをメノウ乳鉢で5〜27分にわたる異なる時間、粉砕した。試料をXRPDと熱分析のため移動させた。確実に均一な破砕を行うため、かき集め、乳鉢の湾曲端部にこびりついた粉末を再度混ぜ直すため、破砕プロセスを5分おきに中断した。
【0193】
Wig−L−Bug(登録商標)によるフライス加工
Wig−L−Bug(登録商標)(米国、Piketch)を化合物I形態A0、HA0およびHB0 (形態HC0およびHD0の混合物)の粉砕に用いた。5から10分間、または変化が観察されなくなるまで、各々の試料(50mg)を粉砕した。2.82cm3の容器で0.9gのステンレス製のボール(直径0.6mm)を用い各々のフライス加工を行った。小びんは3200rpm、円弧6.5°で振とうさせ、ボールを100Hz以上で小びんの端に打ちつけさせた。
【0194】
形態A0の安定性
20分(すり鉢およびすりこぎ)および5分( Wig−L−Bug(登録商標))の処理後、XRPDパターンは結晶化度が有意に低下した事を示した。 残りのピークが出発物質と同じ位置にあった為、試料は完全な非結晶質にはならなかった(図49)。
【0195】
形態A0、HC0およびHD0の安定性
5分間の破砕を3回行った後、破砕形態HB0(形態HC0およびHD0の混合物)のXRPDパターン(図50)は 破砕形態HA0 のパターンと類似する。7.6°(2θ)におけるXRPDピークは、およそ30倍で強度が低下する。
【0196】
DSC曲線は、水の放出に起因すると考えられる50から100℃までの幅の広い吸熱を示す。サーモグラムは最初 、約113℃に位置するガラス転移温度Tgを示す(図51)。このDSCは観測されたひとつの発熱が136℃で準安定性形態B0へ向かう再結晶化の第一段階に相当する事を示す。幅の広い吸熱事象 は形態B0の融解と一致し、231℃(形態A0)で最終溶解となる。形態A0およびB0が単変性とし考えるならばこれらの事象の説明が可能であり、形態A0はより安定した形態である。
【0197】
XRPD、VT−XRPDおよび単結晶回折パターン実験の結果として得たピークの高さが異なる可能性があり、温度、結晶のサイズまたは形態、試料調製またはPANalytical X Pert Pro回折計またはオックスフォード回折CCD回折計の分析ウェルにおける試料の高さのような変化しやすいものに依存する可能性があることは理解されるはずである。
【0198】
異なる放射線源で測定する場合、ピークの位置が変化する可能性があることもまた理解されるはずである。たとえば、それぞれ1.54060Å、0.7107Å、および1.9373Åの波長を持つCu−Kα1、Mo−Kα、Co−KαおよびFe−Kα放射線がCu−Kα放射線で測定したそれらのピークと異なるピークの位置を提供する可能性がある。
【0199】
さらに、一連のピークの位置の後に続く「±0.2度2−シータ」という単語が±0.2度2−シータの変動性で角度位置に関して報告されるそれに続く群のピークの全てを意味することは理解されるはずである。たとえば、「6.81、8.52、9.73、12.04、および/または13.25±0.2度2−シータ」は「6.81±0.2度2−シータ、8.52±0.2度2−シータ、9.73±0.2度2−シータ、12.04±0.2度2−シータ、および/または13.25±0.2度2−シータ」を意味する。
【0200】
当業者が正しく理解するならば本発明における多数の修正および変更は上記の内容を考慮した上で可能である。したがって、添付の特許請求の範囲内で、特にここに解説したこと以上に、本発明が熟練されることが可能であると理解され、本発明の範囲はそのようなすべての変化を含むことを意図するものである。
【技術分野】
【0001】
本発明は、多環系化合物の新規形態(以後化合物Iとする)を含有する組成物、それらの再現性のある方法、及び化合物Iを有する薬学的組成物に関するものである。
【背景技術】
【0002】
医薬品原料(Active pharmaceutical ingredients;APIs)は、例えば、化学誘導体、溶媒和化合物、水和物、共結晶或いは塩類などの様々な形態で調合され得る。APIsはさらに、無定型でもあり、異なる結晶性多形(polymorphs)を持つ、若しくは異なる溶媒和或いは水和状態で存在する。APIsの形態を変えることによって、それらの物理学的特性を変えることが可能となる。例えば、より熱力学的に安定でない多形と比較して、より熱力学的に安定な多形がより低い溶解性を持つように、結晶性多形は一般的に異なる溶解度を持つ。多形はさらに、安定性、生物学的利用度、形態、蒸気圧、密度、色及び圧縮率などの特性も異なる。従って、APIの結晶性状態のバリエーションは、それらの物理学的及び薬理学的特性を調節するたくさんの方法の1つである。
【0003】
ポリ(ADP−リボース)ポリメラーゼ(PARP、ポリ(ADP−リボース)合成酵素或いはPARSとも呼ばれる)は、DNA修復過程の一部としての一本鎖DNA切断に反応したNAD+からのポリ(ADP−リボース)鎖の合成を触媒する核内酵素である(de Murcia, G; de Murcia, J.M. Poly(ADP−ribose) polymerase: a molecular nick−sensor. Trends Biochem. Sci. 1994, 19,172−176; Alvarez−Gonzalez, R.; Pacheco−Rodriguez, G.; Mendoza−Alvarez, H.Enzymology of ADP−ribose polymer synthesis. Mol. Cell. Biochem. 1994, 138, 33)。PARPの低分子阻害剤は、神経変性疾患、癌、及び他のPARP及びキナーゼ−関連疾患の治療において潜在的な役割を担うと仮定されていた。
【0004】
特異的PARP阻害化合物は、化学名4,5,6,7−テトラヒドロ−11−メトキシ−2−[(4−メチル−1−ピペラジニル)メチル]−1H−シクロペンタ [a]ピロロ[3,4−c]カルバゾール−1,3(2H)−ジオンであり、乳癌及び卵巣がんの治療に有用であり、他の薬剤耐性癌の治療に対する化学療法或いは放射線療法と組み合わされる。この化合物は以下の化学式(I):
【化1】
【0005】
によって表され、これ以後「化合物I」として参照される。米国特許第7,122,679号及び米国出願第2006/0276497号には、化合物I及びその有用性が記載されている。
【0006】
化合物Iの異なる形態は、異なる融解点、可溶性或いは溶解率を持つものである;これらの物理的特性は、単独或いは組み合わせで、例えば生物学的利用のうなどに影響を与えるものである。APIsの代替形態の潜在的利益の点において、化合物Iの代替形態を同定し調合する必要性が存在するものである。
【発明の概要】
【課題を解決するための手段】
【0007】
化合物Iの様々な形態は、それらを調合する方法と共に記載されるものである。特に、無水結晶形態の2つの多形(形態A0及びB0)、結晶性一水和物形態の3つの多形(HA0、HC0及びHD0)、及び9つの溶媒和化合物(S20、S30、S40、S50、S60、S70、S90、S100及びS120)は、本明細書において記載されるものである。これらの形態の1若しくはそれ以上を有する薬学的組成物、さらには化合物Iの非結晶形態(As)をさらに有する薬学的組成物も記載されるものである。これらの形態の1若しくはそれ以上を有する薬学的組成物、さらにそのような組成物を利用して治療する方法も記載されるものである。
【0008】
本発明の薬学的組成物は、これに限定されるものではないが、放射線の抗腫瘍活性或いはDNA障害化学療法剤の増強を含む、様々な方法において使用される(Griffin, R.J.; Curtin, N.J.; Newell, D.R.; Golding, B.T.; Durkacz. B.W.; Calvert, A.H. The role of inhibitors of poly(ADP−ribose) polymerase as resistance−modifying agents in cancer therapy. Biochemie 1995, 77, 408)。
【図面の簡単な説明】
【0009】
【図1】図1は、形態A0のX線粉末回折(X−ray Powder Diffractogram;XRPD)を図示したものである。
【図2】図2は、形態B0のX線粉末回折(XRPD)を図示したものである。
【図3】図3は、形態HA0のX線粉末回折(XRPD)を図示したものである。
【図4】図4は、形態HC0のX線粉末回折(XRPD)を図示したものである。
【図5】図5は、形態HD0のX線粉末回折(XRPD)を図示したものである。
【図6】図6は、形態S20のX線粉末回折(XRPD)を図示したものである。
【図7】図7は、形態S30のX線粉末回折(XRPD)を図示したものである。
【図8】図8は、形態S40のX線粉末回折(XRPD)を図示したものである。
【図9】図9は、形態S50のX線粉末回折(XRPD)を図示したものである。
【図10】図10は、形態S60のX線粉末回折(XRPD)を図示したものである。
【図11】図11は、形態S70のX線粉末回折(XRPD)を図示したものである。
【図12】図12は、形態S90のX線粉末回折(XRPD)を図示したものである。
【図13】図13は、形態S100のX線粉末回折(XRPD)を図示したものである。
【図14】図14は、形態S120のX線粉末回折(XRPD)を図示したものである。
【図15】図15は、形態A0の可変温度X線粉末解析(VT−XRPD)を図示したものである。
【図16】図16は、形態A0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図17】図17は、動的水蒸気吸収装置(DVS)規則等温プロットを図示したものである。
【図18】図18は、動的水蒸気吸収装置(DVS)分析前後の形態A0のX線粉末回折(XRPD)を図示したものである。
【図19】図19は、形態A0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図20】図20は、形態A0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図21】図21は、形態A0のラマンスペクトルを図示したものである。
【図22】図22は、形態B0の可変温度X線粉末回折(VT−XRPD)を図示したものである。
【図23】図23は、形態B0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図24】図24は、形態HA0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図25】図25は、形態HA0の動的水蒸気吸収装置(DVS)等温プロットを図示したものである。
【図26】図26は、形態HA0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図27】図27は、形態HA0のラマンスペクトルを図示したものである。
【図28】図28は、形態HC0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図29】図29は、形態HC0の動的水蒸気吸収装置(DVS)等温プロットを図示したものである。
【図30】図30は、動的水蒸気吸収装置(DVS)分析前後の形態HC0のX線粉末回折(XRPD)を図示したものである。
【図31】図31は、形態HC0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図32】図32は、形態HC0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図33】図33は、形態HC0のラマンスペクトルを図示したものである。
【図34】図34は、形態HD0の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図35】図35は、形態HD0の動的水蒸気吸収装置(DVS)規則等温プロットを図示したものである。
【図36】図36は、動的水蒸気吸収装置(DVS)分析前後の形態HD0のX線粉末回折(XRPD)を図示したものである。
【図37】図37は、形態HD0の動的水蒸気吸収装置(DVS)不規則等温プロットを図示したものである。
【図38】図38は、形態HD0のフーリエ変換赤外(FTIR)スペクトルを図示したものである。
【図39】図39は、形態HD0のラマンスペクトルを図示したものである。
【図40】図40は、形態S20の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図41】図41は、形態S30の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図42】図42は、形態S40の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図43】図43は、形態S50の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図44】図44は、形態S60の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図45】図45は、形態S70の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図46】図46は、形態S90の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図47】図47は、形態S100の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図48】図48は、形態S120の示差走査熱量測定(DSC)サーモグラム及び熱重量分析(TGA)サーモグラムオーバーレイを図示したものである。
【図49】図49は、粉砕後の形態A0のX線粉末回折(XRPD)パターンのオーバーレイを図示したものである。
【図50】図50は、15分間の粉砕後の形態HC0 及びHD0のX線粉末回折(XRPD)パターンのオーバーレイを図示したものである。
【図51】図51は、15分間の粉砕後の形態HC0 及びHD0の示差走査熱量測定(DSC)サーモグラムを図示したものである。
【発明を実施するための形態】
【0010】
多数の化合物Iの形態の存在が現在見つかっている。これらの形態の調合及び記述は、本明細書に記載した。これらの形態に関連したスペクトルデータは、図1〜51に示した。
【0011】
より特異的に、多数の化合物Iの異なる物理的形態の存在が見つかっている。化合物Iの無水結晶性形態の2つの多形(形態A0及びB0)、及び結晶性一水和物形態の3つの多形(HA0、HC0及びHD0)が発見されていた。A及びBという文字は、これら無水形態及び水和物を指定し、前にあるHは特に水和物形態を意味するものである。下付き文字「0」はさらに、遊離塩基形態を同定するために指定するものである。加えて、化合物Iの9つの溶媒(S20、S30、S40、S50、S60、S70、S90、S100及びS120)は、本明細書で記載されている。これら形態の1若しくはそれ以上を有する薬学的組成物、さらには化合物Iの非結晶形態(As)をさらに有する薬学的組成物も記載されている。
【0012】
形態A0に対する代表的なXRPDピークは、以下の表1に記載した。形態A0のX線回折パターン特徴は、図1に示した。
【0013】
【表1】
【0014】
形態B0に対する代表的なXRPDピークは、以下の表2に記載した。形態B0のX線回折パターン特徴は、図2に示した。
【0015】
【表2】
【0016】
形態HA0に対する代表的なXRPDピークは、以下の表3に記載した。形態HA0のX線回折パターン特徴は、図3に示した。
【0017】
【表3】
【0018】
形態HC0に対する代表的なXRPDピークは、以下の表4に記載した。形態HC0のX線回折パターン特徴は、図4に示した。
【0019】
【表4】
【0020】
形態HD0に対する代表的なXRPDピークは、以下の表5に記載した。形態HD0のX線回折パターン特徴は、図5に示した。
【0021】
【表5】
【0022】
形態S20に対する代表的なXRPDピークは、以下の表6に記載した。形態S20のX線回折パターン特徴は、図6に示した。
【0023】
【表6】
【0024】
形態S30に対する代表的なXRPDピークは、以下の表7に記載した。形態S30のX線回折パターン特徴は、図7に示した。
【0025】
【表7】
【0026】
形態S40に対する代表的なXRPDピークは、以下の表8に記載した。形態S40のX線回折パターン特徴は、図8に示した。
【0027】
【表8】
【0028】
形態S50に対する代表的なXRPDピークは、以下の表9に記載した。形態S50のX線回折パターン特徴は、図9に示した。
【0029】
【表9】
【0030】
形態S60に対する代表的なXRPDピークは、以下の表10に記載した。形態S60のX線回折パターン特徴は、図10に示した。
【0031】
【表10】
【0032】
形態S70に対する代表的なXRPDピークは、以下の表11に記載した。形態S70のX線回折パターン特徴は、図11に示した。
【0033】
【表11】
【0034】
形態S90に対する代表的なXRPDピークは、以下の表12に記載した。形態S90のX線回折パターン特徴は、図12に示した。
【0035】
【表12】
【0036】
形態S100に対する代表的なXRPDピークは、以下の表13に記載した。形態S100のX線回折パターン特徴は、図13に示した。
【0037】
【表13】
【0038】
形態S120に対する代表的なXRPDピークは、以下の表14に記載した。形態S120のX線回折パターン特徴は、図7に示した。
【0039】
【表14】
【0040】
従って、1つの観点において、本発明は、形態A0、形態B0、或いはそれらの混合物である化合物Iの結晶形態に関連するものである。さらなる観点において、前記結晶形態は形態A0である。別の観点において、前記結晶形態は形態B0である。さらなる観点において、前記結晶形態は、以下のピーク:4.32、6.07、8.55、12.07及び/若しくは15.37±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図1に記載されたようなX線粉末回折パターンを持つものである。付加的な観点において、前記結晶形態は、以下のピーク:7.16、7.89、10.77、16.54及び/若しくは21.20±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図2に記載されたようなX線粉末回折パターンを持つものである。
【0041】
本発明のさらなる観点は、形態HA0、形態HC0、形態HD0或いはそれらの混合物である化合物Iの結晶形態に関連するものである。別の観点において、前記結晶形態は、形態HA0である。さらなる観点において、前記結晶形態は、形態HC0である。付加的な観点において、前記結晶形態は、形態HD0である。さらなる別の観点において、前記結晶形態は、以下のピーク:7.59、15.12、16.06、17.94及び/若しくは23.89±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる観点において、前記結晶形態は、実質的に図3に記載されたようなX線粉末回折パターンを持つものである。付加的な観点において、前記結晶形態は、以下のピーク:8.36、8.71、16.69、17.39及び/若しくは24.59±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、実質的に図4に記載されたようなX線粉末回折パターンを持つものである。別の観点において、前記結晶形態は、以下のピーク:7.60、8.99及び/若しくは15.16±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる観点において、前記結晶形態は、実質的に図5に記載されたようなX線粉末回折パターンを持つものである。
【0042】
本発明のさらなる別の観点は、形態S20、形態S30、形態S40、形態S50、形態S60、形態S70、形態S90、形態S100、形態S120或いはそれらの混合物である化合物Iの結晶形態に関するものである。さらなる観点において、前記結晶形態は、形態S20である。さらなる別の観点において、前記結晶形態は、形態S30である。付加的な観点において、前記結晶形態は、形態S40である。さらなる観点において、前記結晶形態は、形態S50である。さらなる付加的な観点において、前記結晶形態は、形態S60である。別の観点において、前記結晶形態は、形態S70である。さらなる観点において、前記結晶形態は、形態S90である。さらなる別の観点において、前記結晶形態は、形態S100である。さらなる観点において、前記結晶形態は、形態S120である。さらなる観点において、前記結晶形態は、以下のピーク:8.56、14.64、16.07、22.24及び/若しくは23.02±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:6.07、8.67、13.36、16.80及び/若しくは16.85±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:8.42、8.60、13.92、17.20及び/若しくは24.46±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:4.46、7.67、8.86及び/若しくは11.71±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:8.68、11.10、16.94、17.39及び/若しくは23.31±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:4.50、7.70、8.90及び/若しくは11.76±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。さらなる別の観点において、前記結晶形態は、以下のピーク:8.34、8.67、16.68、17.33及び/若しくは24.57±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。付加的な観点において、前記結晶形態は、以下のピーク:4.45、7.62、8.79、11.62及び/若しくは17.67±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。別の観点において、前記結晶形態は、以下のピーク:7.63、7.67、9.00、17.99及び/若しくは24.46±0.2度2シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである。
【0043】
本発明の付加的な観点は、形態A0である化合物Iの結晶形態を調合する方法に関するものであり、この方法は:(a)炭化水素(ヘプタン或いはトルエンなど)において化合物Iをスラリー化する工程と;(b)結果生じたスラリーを冷却する工程と;(c)前記結果生じたスラリーを濾過する工程と;(d)濾過ケーキを乾燥させる工程とを有するものである。1つの観点において、化合物Iは、26〜45容積のヘプタンにおいてスラリー化させるものである。別の観点において、化合物Iは、45容積のヘプタンにおいてスラリー化させるものである。付加的な観点において、工程(a)は、79〜83℃で実行されるものである。さらなる別の観点において、工程(a)は、85℃で実行されるものである。さらなる別の観点において、工程(a)は、24〜48時間実行されるものである。さらなる観点において、工程(a)は、45時間実行されるものである。別の観点において、工程(b)は、30〜65℃の温度で起こるものである。さらなる別の観点において、工程(b)は、65℃で実行されるものである。別の観点において、工程(d)は、0.33〜3時間、室温で実行されるものである。さらなる別の観点において、工程(d)は、3時間室温で実行されるものである。
【0044】
本発明のさらなる観点は、形態A0である化合物Iの結晶形態を調合する方法に関するものであり、この方法は:(a)溶媒に化合物Iを溶解させる工程と;(b)結果生じた溶液を濾過する工程と;(c)化合物Iを沈殿させるために抗溶媒を添加する間に前記溶媒を部分的に蒸留する工程と;(d)工程(a)で使用した前記溶媒の容量を減らすために付加的な抗溶媒を添加する間に結果生じたスラリーをさらに蒸留する工程と;(e)形態A0への完全な変換を達成するように前記スラリーを加熱する工程と;(f)冷却する工程と;(g)濾過を介して産物を回収する工程と;(h)乾燥させる工程とを有するものである。さらなる観点において、工程(a)は、27〜35容積のTHFを用いて実行されるものである。別の観点において、工程(a)は、30容積のTHFを用いて実行されるものである。さらなる観点において、工程(a)を介して産生された前記溶液は、任意に金属スカベンジャー或いは炭素で処理されるものである。さらなる観点において、前記濾過する工程(b)は、以下の工程:(i)前記金属スカベンジャーを除去するために濾過する工程;及び(ii)1ミクロンインライン(inline)カートリッジフィルターを通じて研磨濾過(polish filtering)する工程の1若しくは両者を有するものである。さらなる観点において、工程(c)で存在する前記溶媒は、そのオリジナル容量の60〜90%へ蒸留されるものである。付加的な観点において、工程(c)は、抗溶媒として炭化水素(ヘプタンなど)を用いて実行されるものである。別の観点において、工程(d)は、容量で5%以下のTHFが残るまで実行されるものである。さらなる別の観点において、工程(e)は、約90〜96℃の温度で実行されるものである。付加的な観点において、工程(e)は、任意に省略されるものである。別の観点において、前記スラリーは、約3〜5時間撹拌させるものである。さらなる観点において、工程(f)は、外界温度(25±5℃)で実行されるものである。付加的な観点において、工程(g)の濾過は、乾燥した不活性ガスを用いて実行されるものである。別の観点において、工程(h)は、80℃までの温度で実行されるものである。さらなる別の観点において、残留水及び/若しくは溶媒和化合物(群)は、共沸的に除去されるものである。
【0045】
本発明の別の観点は、形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物に関するものである。さらなる観点は、癌を治療する方法に関するものであり、この方法は、形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物の治療上有効な量を必要とする患者へ投与する工程を有するものである。付加的な観点において、本発明は、癌を治療する方法に関するものであり、この方法は、形態A0を有する薬学的組成物の治療上有効な量を必要な患者へ投与する工程を有するものである。
【0046】
専門用語
本明細書で用いられたように「非結晶」という用語は、特徴的な結晶形状或いは結晶構造を欠いていることを意味するものである。
【0047】
本明細書で用いられたように「抗溶媒」という用語は、そこにおいては化合物が実質的に不溶性であることを意味するものである。
【0048】
本明細書で用いられたように「結晶」という用語は、分子或いは外表面(external face planes)の規則的に繰り返される配列を持つことを意味するものである。
【0049】
本明細書で用いられたように「結晶形態」という用語は、X線粉末回折によって分析した場合、ピークの特徴的なパターンを提供する、固形化学化合物或いは化合物の混合物を意味するものであり;これには、限定するものではないが、多形(polymorphs)、溶媒、水和物、共結晶及び脱溶媒された(de−solvated)溶媒が含まれるものである。
【0050】
「多形」或いは「多型(polymorphism)」という用語は、同じ化学分子に対して少なくとも2つの異なる結晶配列の可能性として定義されるものである。
【0051】
本明細書で用いられたように「溶質」という用語は、通常溶液の構成成分はより少ない量で存在するものである、別の物質に溶解された物質を意味するものである。
【0052】
本明細書で用いられたように「溶液」という用語は、少なくとも1つの溶媒、及び前記溶媒に少なくとも部分的に溶解した少なくとも1つの化合物を含有する混合物を意味するものである。
【0053】
本明細書で用いられたように「溶媒和化合物」という用語は、前記結晶構造内に溶媒分子を含有する結晶物質を意味するものである。
【0054】
本明細書で用いられたように「溶媒」という用語は、別の物質、一般的には固形を完全に或いは部分的に溶解することができる物質、一般的には液体を意味するものである。本発明の実行に対する溶媒には、これに限定されるものではないが、水、酢酸、アセトン、アセトニトリル(ACN)、ベンジルアルコール、1−ブタノール、2−ブタノール、2−ブタノン、ブチロニトリル、三級(tert)−ブタノール、N−ブチル酢酸、クロロベンゼン、クロロホルム、シクロヘキサン、1−2ジクロロエタン(DCE)、ジクロロメタン(DCM)、ジエチレングリコールジブチルエーテル(DGDE)、ジイソプロピルアミン(DIPA)、ジイソプロピルエーテル(DIPE)、1,2−ジメトキシエタン(DE)、N,N−ジメチルアセトアミド(DMA)、4−ジメチルアミノピリジン(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド(DMAP)、N,N−ジメチルホルムアミド(DMF)、ジメチルスルホキシド、1,4−ジオキサン、エチレングリコール、ジメチルエーテル、エタノール、酢酸エチル、エチルジイソプロピルアミン、エチレングリコール、ギ酸エチル、ギ酸、ヘプタン、イソブチルアルコール、酢酸イソプロピル(IPAC)、イソプロピルアルコール(IPA)、イソプロピルアミン、リチウムジイソプロピルアミド(LDA)、メタノール、メトキシベンゼン(MTB)、酢酸メチル、メチルエチルケトン(MEK)、メチルイソブチルケトン(MIK)、2−メチルテトラヒドロフラン、メチル三級(tert)−ブチルエーテル(MTBE)、1:1のホルムアミド:水、1:1のN−メチルピロリジノン(NMP):水、2−ペンタノン、3−ペンタノン、1−ペンタノール、1,2−プロパンジオール、2−プロパノール(IPA)、1−プロパノール、プロパノニトリル、プロプレンカーボネート、1,2−プロピレングリコール(PG)、ピリジン、テトラヒドロフラン(THF)、テトラヒドロピラン(THP)、トルエン、トリエチルアミン、キシレン、それらの混合物、及びそれらと同等物が含まれる。これらの溶媒は、それらの機能基に従って5つのクラスへ分類される:クラス1:「プロトン性」或いは水素結合供与溶媒(ルイス酸)、これにはベンジルアルコール、エタノール、IPA、メタノール及び水が含まれる;クラス2:水素結合受容溶媒(ルイス塩基)、これにはアセトン、1,4−ジオキサン、DMF、酢酸エチル、MEK、MTBE、THF及び水が含まれる;クラス3:極性非プロトン性溶媒、「非ヒドロキシル性溶媒」とも呼ばれる、これにはアセトニトリル、DMA、DMF及びDMSOが含まれる;クラス4:クロロカーボン溶媒、これにはクロロホルムが含まれる;クラス5:ヒドロカーボン溶媒、飽和及び不飽和の両者を含み、これらにはn−ヘプタン、トルエン、p−キシレン及びキシレンが含まれる。
【0055】
本明細書で用いられたように「治療上有効な量」という用語は、所定の投与経路で、意図され、確立された薬物動態法及び技術に従って測定されたような所定の薬剤と関連する生理学的効果を得るのに必要であると決定された量を意味するものである。適切で特異的な治療上有効な量は、従来の技術の使用によって、本分野の当業者である専門診断医によって容易に決定され得る。有効な量は、疾患或いは疾病の進行のタイプ及び程度、特定の患者の全体の健康状態、選択された化合物の関連する生物学的有効性、適切な賦形剤での有効薬剤の処方及び投与経路を含む、多数の因子に依存して変えられるであろう。一般的に、結晶形態は、目的の効果が達成されるまで徐々に増加させながら、より低い投与量レベルで投与される。
【0056】
他に記述がない限り、本明細書中で記載されるパーセンテージは、重量/重量(w/w)パーセンテージである。
【0057】
本明細書で用いられたように「薬学的に許容可能な賦形剤」という用語は、あらゆる及び全ての溶媒、分散媒体、コーティング剤、抗菌剤及び抗真菌剤、等張性吸収遅延剤及びそれらと同等物を含む。薬学的に有効な物質に対するそのような媒体及び薬剤の使用は、本分野ではよく知られており、例えば、Remington:The Science and Practice of Pharmacy,20th ed.;Gennaro,A.R.,Ed.;Lippincott Williams&Wilkins:Philadelphia,PA,2000などを参照のこと。あらゆる従来の媒体或いは薬剤が前記有効成分と不適合である限りを除いて、治療用組成物におけるその使用は考慮される。
【0058】
治療目的で、本発明の結晶形態は、対象の体内における薬剤の作用部位との有効薬剤の接触を生じるあらゆる手段で投与され得る。前記結晶形態は、個々の薬剤として、或いは例えば鎮痛剤などの他の薬剤との組み合わせで、医薬品との併用で利用可能なあらゆる従来の手段によって投与される。本発明の結晶形態は、好ましくは、それが必要な対象へ、本明細書で記載された疾病及び疾患の治療に対して治療上有効な量で投与される。
【0059】
治療用或いは予防用使用において、本発明の結晶形態は、薬剤が従来投与されるあらゆる経路によって投与される。そのような投与経路は、腹腔内、静脈内、筋肉内、皮下、くも膜下腔内、気管内、脳室内、経口、頬側、直腸、非経口、鼻腔内、経皮及び皮内を含む。投与は、全身的或いは局所的である。
【0060】
本明細書に記載された結晶形態は、純粋な形態で、他の有効成分との組み合わせで、或いは薬学的に許容可能な無毒賦形剤或いは担体との組み合わせで投与される。経口組成物には、一般的に不活性希釈剤担体或いは食用担体が含まれる。薬学的に適合性な結合剤及び/若しくはアジュバント物質は、前記組成物の一部として含まれ得る。錠剤、丸剤、カプセル、トローチ及びそれらと同等物は、あらゆる以下の成分、或いは同様な性質の化合物:結晶セルロース、ゴムトラガント或いはゼラチンなどの結合剤;スターチ或いはラクトースなどの賦形剤、アルギン酸、プリモゲル(Primogel)或いはコーンスターチなどの分布剤;ステアリン酸マグネシウムなどの潤滑剤;コロイド性二酸化ケイ素などの流動促進剤;スクロース或いはサッカリンなどの甘味料;或いはペパーミント、サリチル酸メチル或いはオレンジ香料などの香味料、を含有することができる。投薬量ユニット形態がカプセルである場合、上記タイプの物質に加えて、それは脂肪油などの液体担体を含有することができる。加えて、投薬量ユニット形態は、例えば、砂糖のコーティング剤、セラック或いは腸溶性剤などの前記投薬量ユニットの物理的形態を修飾する様々な他の物質を含有することができる。さらに、前記有効化合物に加えて、シロップは甘味料としてのスクロース、及び特定の保存料、色素、着色料及び香料を含有する。
【0061】
投与に対する代替調合には、蒸留水或いは非水溶液、懸濁液及び乳化剤が含まれる。非水溶液の例としては、ジメチルスルホキシド、アルコール、プロピレン、グリコール、ポリエチレングリコール、オリーブオイルなどの植物油、及びオレイン酸エチルなどの注入可能な有機エステルである。水溶性担体には、アルコールと水の混合物、緩衝媒体及び生理食塩水が含まれる。静脈内媒体には、液体及び栄養補充薬、リンガーブドウ糖に基づいたものなどの電解質補充剤、及びそれらと同等物が含まれる。例えば抗菌剤、抗酸化剤、キレート剤、不活性ガス及びそれらと同等物などの保存料及び他の添加物も存在する。
【0062】
ほ乳類への結晶形態の投与の好ましい方法には、腹腔内注射、筋肉内注射、及び静脈内注入が含まれる。生理食塩水、アルコール、DMSO及び水ベース溶液を含む様々な液体処方は、これらの運搬方法に対して可能である。濃度は、運搬される投与量及び容量に従って変えられ、約1〜約1000mg/mLの範囲であり得る。前記液体処方の他の成分には、保存料、無機塩類、酸、塩基、緩衝液、栄養剤、ビタミン群、或いは鎮痛薬或いは付加的PARP及びキナーゼ阻害剤などの他の薬剤が含まれる。
【0063】
計測手段
X線粉末回折(XRPD)
XRPDパターンは、40kVおよび40mAで、CuKα放射線を使用してPANalytical X’ Pert Pro回折装置で記録した。シリコン標準は、X線管のアラインメントをチェックするために試験した。試料は、アルミホルダー中のゼロ−バックグラウンド石英プレート上に押しつけた。一般的なX線粉末パターンのスキャンは、約2〜40°2θ間を刻み幅0.0080°および計数時間96.06秒で収集し、約0.5°/分のスキャン速度で行った。
【0064】
単結晶の研究では、選択した結晶にパラトン油(paratone oil)を塗布し、Oxford回折CCD回折計(サファイア検出器搭載Xcalibur S)上で急速冷凍した。データは、標準領域検出法で収集した。構造はSHELXTLパッケージで解析し,精密化した。測定したXRPDパターンに対する単結晶パラメータのデフォルトのリートベルト(Reitveld)法精密化により、良い適合が得られ、説明のつかないピークは無かった。
【0065】
温度可変X線粉末回折(VT−XRPD)
温度可変の研究は、Anton Paar TTK450温度槽を用い、Anton Paar TCU100温度制御ユニットによる、コンピュータ制御下、窒素雰囲気で行った。限定および連続測定方式の2つを使用した。限定モードにおいては、前記TTK450チャンバーが所定温度に到達した後でのみ、測定を実施した。連続モードでは、試料を10℃/分で加熱し、温度が変化する間、高速スキャンを測定した。所定温度に到達後、試料を30または35℃/分で冷却し、より低速なスキャンを25℃で測定した。前記選択した温度は、DSCの結果に基づいたものである。
【0066】
示差走査熱量測定(DSC)
熱曲線は、Pyrisソフトウェア・バージョン6.0実行下の自動試料採取装置搭載し、分析前にインジウムで較正したPerkin−Elmer Sapphire DSCユニットを使用して取得した。固体試料1〜11mgを蓋無しの、20μLアルミ製試料パンに秤量した。次いで、DSCセルは、窒素でパージし、温度0℃から275℃まで10℃/分の速度で加熱した。
【0067】
熱重量分析(TGA)
熱曲線は、Pyrisソフトウェア−バージョン6.0を実行し、シュウ酸カルシウム1水和物で較正したPerkin−Elmer Pyris 1 TGA装置を使用して取得した。TGA試料1〜15mgを約50mL/分のヘリウムでパージした炉内において10℃/分で25℃から400℃に加熱した時の%重量減を測定した。
【0068】
動的水蒸気吸着(DVS)
重量による水蒸気吸着実験は、DVS−HT装置(英国ロンドン、Surface Measurement Systems,London,UK)を用いて行った。この装置は、重量測定±0.1μgの質量分解能を有する記録式超微量天秤を用いて水蒸気の吸収および減量を重量で測定する。試料周辺水蒸気分圧(±1.0%)は、電子質量流量コントローラを使用して飽和および乾燥キャリアガス流を混合することによって制御した。所望の温度を±0.1℃に維持した。
【0069】
試料(10〜25mg)を所定温度でDVS−HT装置内に配置した。2種の動的蒸気吸着実験を実施した。
【0070】
1.試料は、最初20時間乾燥空気流中(<0.1%未満の相対湿度(RH))で乾燥して乾燥質量を決定し、次いで2つの0〜90%RHサイクル(RHを10%単位で変化)にさらした。
【0071】
2.試料は20時間90%RHにさらし、次いで2つの90〜0%RHサイクル(RHを10%単位で変化)にさらした。
【0072】
赤外分光(FTIR)
スペクトルは、ダイヤモンド結晶ウィンドーを含むSmart Orbit ATR付属装置付きThermo Electron−Nicolet Avatar 370 DTGSを用いて取得した。Thermo Electron Omnic(商標)ソフトウェア(version 3.1) を使用して、初期干渉写真(interferogram)から4000〜400cm−1のスペクトルを計算した。各試料のスペクトルを取得する以前にバックグラウンドスキャンを収集した。各試料について、4cm−1のスペクトル分解能でスキャンを32回行い、平均値を得た。
【0073】
ラマン分光測定
前記試料のラマンスペクトルは、FT−ラマンモジュールを用い、Vertex70 FTIR分光器(Bruker RAM II,Bruker OPtics,ドイツ)で記録した。 YAGゲルマニウムフォトダイオードを使用して、ND : Yag レーザー(蛍光の抑制)で励起したFT−ラマンスペクトルを記録した。試料の分析前にポリスチレン標準を試験した。各スペクトルの取得時間は4cm−1の分解能で1分であり、1064nm レーザーパワーは、試料部で50mWであった。
【0074】
独自性(identity)、アッセイ(assay)および純度
通常、試料溶液分量10μLをアセトニトリルで1mLに希釈し、アッセイ濃度は、以下のHPLC法を用いて二回注入物の平均値から決定した。純度および不純物の分析は、従来のHPLCを使用して実施する。
【0075】
【表15】
【0076】
実施例
化合物I調製プロセス
化合物Iは、図式Iに従って調製することが出来る。
【0077】
【化2】
【0078】
図式Iにおいて、前記合成は、市販の出発原料4−メトキシインドールを用いて開始する。ジtert−ブチルジカーボネート((Boc)2O)でインドールの窒素を保護(マスキング)した後、当該インドール誘導体を、リチウムジイソプロピルアミド(LDA)で活性化して、当該インドールの2位置にカルバニオンを生成し、それはその場で(in−situ)ホウ酸トリイソプロピルと反応する。酸性処理でボロン酸エステル中間体を加水分解して、対応するインドールボロン酸化合物Aにする。次いで、化合物Aを鈴木法条件下、触媒量の酢酸パラジウムとトリフェニルフォスフィンの存在下で、1−シクロペンテニルトリフルオロメタンスルホネート(また、本明細書ではエノールトリフレートとも呼ぶ)と結合させ、重要なジエン中間体化合物Bを得る。ナトリウムメトキシドで前記Boc保護基を除去した後、ジエン化合物Cを、酢酸中、ディールス・アルダー反応によってマレイミドと結合させて、5環性中間体化合物Dを生成する。クロルアニル酸化による芳香族化で、化合物Dを化合物Eに転換し、それをマンニッヒ反応条件下1−メチルピペラジンと結合させて、目的とする分子の化合物Iを生成する。この合成の詳細な内容を以下に記載する。
【0079】
N−Boc−4−メトキシインドールの合成
100ガロン・ガラス内張反応装置に4−メトキシインドール(20.0kg、136モル、Yixing Zhongyu Medicine Technology Co.,Ltd.)を仕込み、次いでDMAP(0.50kg、4.1モル、Aldrich)およびトルエン(92kg、CORCO試薬等級)を加えた。得られた混合物を攪拌し、約40℃に加熱した。別に、ジ−tert−ブチルジカーボネート(31.8kg、146モル、Lacamas Laboratories、Inc.)のトルエン(60kg、Corco試薬等級)溶液を第2反応装置で調製した。この溶液を、約1.75時間かけて前記インドール溶液に添加した。わずかに発熱した反応(最高温度約41℃)は、ガスの発生を伴った。40℃でさらに1時間撹拌後、前記反応液を20±3℃に冷却した。工程内試験(in−process test)で、4−メトキシインドールが完全に反応したことが判明した。脱イオン水(15ガロン)を加えて過剰の(Boc)2O(注意:ガス発生)を分解した。次いで、得られた混合物を0.5時間激しく攪拌し,次いで一晩放置した。下側の水層を除去後、当該有機層を部分的に減圧下(60℃のジャケットで最高60mmHg)で濃縮し、留出液約145Lを除去した。この時点で、追加のトルエン(30kg、Corco試薬等級)を仕込み、合計約200Lの留出物が収集されるまで蒸留を続けた。このバッチは、その後、室温に冷却し、ポリ・ドラムに空けて、N−Boc−4−メトキシインドール(理論収率と想定)33.6kgを含む濃色琥珀色の溶液62.3kg得た。これをさらに精製することなく次の工程で使用した。
【0080】
化合物A
2−ボロノ−4−メトキシ−1H−インドール−1−カルボン酸−1−(1,1−ジメチルエチル)エステル)の合成
上記溶液の約半分を、100ガロン・ガラス内張反応装置に仕込み、次いでトルエン、(3.0kgで仕込み量を50重量%に希釈)、ホウ酸トリイソプロピル(19.9kg、105.9モル、Anderson Development Co.)、およびTHF(91kg、CORCO試薬等級)を添加した。得られた溶液を攪拌し、−2℃に冷却した。この時点で、リチウムジイソプロピルアミド(37.3kg、91.8モル、エチルベンゼン/テトラヒドロフラン/ヘプタン27%溶液、FMC Lithium社)を、当該バッチの温度を3℃未満(−10℃のジャケット)に維持しながら、一時間かけて添加した。LDAの添加後、HPLC(LDAの添加後30分で、N−Boc−4−メトキシインドールは、0.6面積%残存)で反応完了が認められまで、得られた反応混合物を0±3℃で攪拌した。別に,第2反応装置で、濃塩酸27kgを脱イオン水16.3ガロンで希釈して3N塩酸溶液を調製し、約5℃に冷却した。この希塩酸を、バッチ温度を15℃未満に維持するよう1時間かけて当該バッチに添加した(バッチ温度は添加完了時に8℃に到達した)。次いで、ジャケット温度を20℃に設定した。反応装置および添加管路を脱イオン水(6ガロン)で水洗し、この水洗液をバッチと合わせた。次いでMTBE(27kg、Pride)を添加した。得られた混合物を0.5時間攪拌し、次いで停止して相分離させた。水層を分離し、第2反応装置でMTBE(14kg、Pride)を用いて逆抽出した。合併した有機層を5%NaCl(34L)、5%NaHCO3(34 L)、10%NaCl(19L)で順次洗浄した。当該有機相をドラム缶に投下し、検量後(172.2kg)、反応装置に戻し、減圧下(反応装置のジャケット設定温度:30℃)で濃縮し、3時間かけて留出物116kgを除去した。得られたスラリーをn−ヘプタン(75kg、CORCO試薬等級)で希釈し、次いで蒸留し、さらに留出液75Lを除去した。室温で一晩撹拌後、当該スラリーを1時間約5℃に冷却した。生成物はAuroraフィルター上に収集し、n−ヘプタン33kgで洗浄した。濾過ケーキを窒素気流中加熱せずに備え付きの真空下トレイ上で一晩乾燥させた。灰色がかった固体として化合物A17.8kgを得た(補正収率88.8%)。HPLC純度:100LC面積%(LCAP)、95.8LC重量%(LCWP)。
【0081】
1,1,1−トリフルオロメタンスルホン酸1−シクロペンテン−1−イルエステルの合成
室温で100ガロン・ガラス内張反応装置にシクロペンタノン(8.95kg、106.5モル)を仕込み、次いで、トルエン(116.40kg、CORCO試薬等級)及びエチルジイソプロピルアミン(16.6kg、128.7モル)加えた。得られた溶液を攪拌し、45±5℃に加熱した。この時点で、トリフルオロメタンスルホン酸無水物(36.2kg、128.4モル)を30−L添加フラスコから約1時間かけて添加した。トリフルオロメタンスルホン酸無水物の添加は非常に発熱した。ジャケット冷却(10℃に設定)を実施して、バッチ温度を45±5℃に維持した。当該バッチは44分の添加時間中、7分間40℃未満に降下した。トリフルオロメタンスルホン酸無水物の添加後、20分間39〜45℃で攪拌を続けた。この20分後の工程内試験では、シクロペンタノンが全量反応したことが判った。19.6℃に冷却後、当該バッチをフィルター中のセライト(Celite)のパッド(18.0kg)を通して濾過した。当該濾液は清潔なポリ内張スチールドラム缶に収集された。前記セライトパッドは、トルエン(37.0kg、CORCO試薬等級)でリンスした。概リンス液は、同じポリ内張スチールドラム缶のバッチと合併した。濾液(159.85kg)は、参照標準に対して分析し、エノールトリフレート(triflate)を19.50kg(83.3%収率)含むことを示した。このエノールトリフレートのトルエン溶液を一晩低温室に貯蔵し、これ以上精製することなく、次の鈴木カップリング法に使用した。
【0082】
化合物B
2−(1−シクロペンテン−1−イル)−4−メトキシ−1H−インドール−1−カルボン酸1,1−ジメチルエチルエステルの合成
室温で100ガロン・ガラス内張反応装置に化合物A(18.00kg、61.8モル)、トリフェニルホスフィン(648.8グラム、2.47モル)および酢酸パラジウム(277.0グラム、1.23モル)を仕込んだ。反応装置は、その後3回脱気・窒素充填を行った。トルエン(78.3kg、CORCO試薬等級)を反応装置内に噴射注入し、次いでジシクロヘキシルアミン(44.5kg、245.4モル)を注入した。この添加に4分かかった。得られたスラリーを21分間室温で(125rpm)を激しく攪拌し、次いで43分かかってエノールトリフレートのトルエン溶液ストリーム(131.7kg、エノールトリフレートを16.07kg、74.3モル含有)をゆっくり注入添加した。エノールトリフレートの添加により発熱した。ジャケット冷却を実施し、バッチ温度を18.8〜27.5℃に保持した。得られた不均一混合物は、前記反応の完了がHPLCで認められるまで、18.4〜22.3℃で攪拌した(注、当該反応は、1時間未満で完了したが、当該処理を続ける前に、猶一晩室温で攪拌した。これは厳密には便宜上の為であった。前記バッチは、生成物に悪影響を与えることなく、100時間までは室温に放置できる)。当該バッチを10分間室温で撹拌し、次いでフィルター中のセライト(Celite)のパッド(2.70kg)を通して濾過した。濾液は清潔なポリ内張スチールドラム缶2個に収集した。濾過ケーキをトルエン(47.8kg、CORCO試薬等級)でリンスした。リンス液は、同じポリ内張スチールドラム缶のバッチと混合した。濾液(260.45kg)を参照標準に対して分析し、化合物Bを20.59kg(106.4%収率)含有することが示された。この検定(assay)に基づいて本反応は100%収率に達したと見なされ、次の工程の仕込みは、反応が100%収率で進行したとして実施した。化合物Bのトルエン溶液は、室温でパイロットプラント内に貯蔵し、これ以上精製することなく、その後の脱保護手法で使用した。
【0083】
化合物C
2−(シクロペンテン−1−イル)−4−メトキシ−1H−インドールの合成
室温で100ガロン・ガラス内張反応装置に、化合物Bのトルエン溶液流(化合物B12.82kg、40.96モル)を仕込み、次いでナトリウムメトキシド(44.0kg、25〜30重量%MeOH溶液、203.7モル)を加えた。得られた溶液を攪拌し、45±5℃に加熱した。反応の完了がHPLCで認められるまで45±5℃で攪拌を続けた(約4時間で反応が完了、HPLCデータは約8時間目に報告された)。前記バッチは、その後26分かかって23.5℃に冷却した。当該バッチを22±2℃で一晩攪拌した。22℃で約17時間後、バッチの約1/2(111.15kg)を第2反応装置に移して、別々に処理した。第1反応装置には脱イオン水(21ガロン)を仕込んだ。得られた混合物を16分間攪拌し、次いで停止した。バッチを46分間室温に静置させた後、底水層を除去した。次いで、ラグ層の小部分を清潔なカーボイ(大型瓶)に排出した。残りの有機層は、フィルター中のセライト(Celite)のパッド(3.84kg)を通して濾過した。濾液は清潔なポリ内張 スチールドラム缶に収集した。前記ラグ層は、同じセライトパッドを通して濾過し、濾液を新規のカーボイに回収した。前記セライトパッドは、トルエン(6.20kg、CORCO試薬等級)で洗浄し、この洗浄液は濾過したラグ層と合併した。濾過ラグ層は、ガラス製添加容器に移し、底水層を除去し、ラグ由来有機層を元の有機層と合併した。上記の処理手法をバッチの第2半量部に繰り返し、化合物Cの第2トルエン溶液を生成した。入るだけの第2溶液を第1有機層(164.70kg、化合物C8.14kg含む)の入ったポリ内張スチールドラム缶に入れた。残りの第2有機層は、小さなポリドラム(19.05kg、化合物Cを0.49kg含有)に収納した。これら2つの溶液は、それ以上精製せずに次の工程でさらなる処理用に、パイロットプラントに貯蔵した。化合物Cは、合計8.63kg(99.2%収率)生成された。
【0084】
化合物D
3a、4、5、6、6a、7、11c−オクタヒドロ−11−メトキシ−1H−シクロペンタ[a]ピロロ[3,4−c〕カルバゾール−1,3(2H)−ジオンの合成
室温で100ガロン・ガラス内張反応装置に、化合物Cのトルエン溶液流(化合物C、、12.58kg、59.1モル)を仕込んだ。この溶液を備え付き真空全開で40℃未満の内部温度下、残渣が化合物Cの重量の約6倍(目標体積約75.5 L)に達するまで、約7時間かけて、濃縮した。この残渣は、清潔なポリエチレンドラムに排出させ、これ以上精製せずに次のディールス・アルダー反応に使用した。第2・100ガロン・ガラス内張反応装置に,マレイミド(7.45kg、76.8モル、Carbosynth Limited)を仕込み、次いで氷酢酸(145.70kg)を加えた。得られた混合物を攪拌して溶液を得た。この時点で、上方から化合物Cの濃縮溶液(84.95kg)を、バッチ温度20±10℃(ジャケット温度を15℃に設定した)に制御するよう、約20分かけて仕込んだ。得られた混合物は、反応の完了が、HPLC(反応は約15.5時間で完了、HPLCデータは約17.5時間目に受領する)で認められるまで30℃±3℃で攪拌した。当該バッチは、約20分かけて23.2℃に冷却した。母液を重量基準HPLCアッセイにより分析し、化合物Dが10%未満(検出:5.88%)含まれていることを確認後、当該バッチをAuroraフィルタで濾過した(23.2℃に達してから濾過時まで2.5時間)。当該濾過ケーキを氷酢酸(39.65kg)でリンスし、化合物Dの純度が、HPLCの重量基準アッセイで(3晩乾燥させ、3晩後の純度は99.5重量%であった)所定の仕様(90重量%超過)に到達するまで、窒素気流、真空下当該フィルタで乾燥した。次いで生成物を2重ポリエチレン袋内張ファイバードラムに取り出し、所望化合物D13.43kg(73.3%収率)を褐色固体として得た。この物質は、これ以上精製せずに、次のクロラニルによる酸化に使用した。
【0085】
化合物E
4、5、6、7−テトラヒドロ−11−メトキシ−1H−シクロペンタ[a]ピロロ[3、4−c〕カルバゾール−1、3(2H)−ジオンの合成
室温で100ガロン・ガラス内張反応装置に化合物D(28.66kg、92.45モル)を仕込み、次いでテトラクロロ−p−ベンゾキノン(45.50kg、185.0モル、99%、ACROS)およびTHF(253.1kg、CORCO試薬等級)を加えた。得られた不均一混合物を65±5℃に加熱し、反応の完了がHPLC(反応は約22時間で完了、HPLCデータ約23時間目に受理)で認められまで、この温度で攪拌した。次に当該バッチを35分間かけて22±5℃に冷却し、溶液中に化合物Eが消失したか分析し(仕様では10%未満、検出:1.9%)てから、フィルターで濾過した。反応装置、管路、および濾過ケーキをTHF−EtOH−H2Oの混合物(第2反応装置でTHF62.0kgをEtOH41.25kgおよび脱イオン水4.64ガロンと混合して調製)でリンスした。生成物が所定の仕様(仕様では80重量%超過、5日後化合物Eを80.8重量%検出)に到達するまで湿潤ケーキを窒素気流、真空下、フィルターで乾燥した。生成物は、その後、2つの二重ポリエチレン袋内張プラスチック缶に取り出し、化合物E23.84kg(86.7%収率)を暗緑黄色の固体として得た。$$$
化合物Iの合成
100ガロン・ガラス内張反応装置に化合物E(15.20kg、40.1モル)を仕込み、次いでパラホルムアルデヒド(2.51kg、80.9モル、97%、ACROS)および変性エタノール(223.45kg、試薬等級)を加えた。得られた混合物を撹拌(121rpm)し、その間添加フラスコから約10分かけて、1−メチルピペラジン(6.65kg、65.77モル、ACROS、99%)を添加した。得られた反応混合物を70℃で加熱、攪拌した。反応の進行は、HPLC(約5時間後、化合物E1.35面積%残存)により観察した。 合計9時間70℃で攪拌後、当該バッチを20±3℃に冷却し、この温度で一晩攪拌した。生成物はフィルターで濾過した。濾過ケーキをエタノール(43.9kg、試薬等級)でリンスし、残留エタノールが1HNMRで12重量%(化合物Iに対して8.4重量%)未満になるまで窒素気流下フィルター上で減圧乾燥した。生成物は、その後、ポリエチレン袋内張ファイバー・ドラム缶に取り出し、粗化合物I18.05kg(95.8パーセント収率)を黄色固体として取得した:98.6LCAP(LC面積%)、89.2LCWP(LC重量%)。この物質は、これ以上精製せずに、下流プロセスに使用した。
【0086】
多形態のスクリーニング研究
結晶化の研究を実施し、48種の異なる溶媒における多形態を調べた。溶媒は、許容性に基づいて、(ICHクラス2および3)、一連の誘電率、双極子モーメントおよび官能基の範囲を網羅するよう選択した。出発材料2種:形態A0およびロット7(形態A0、形態HC0および形態HD0の混合物)を選択した。可能な場合には、化合物Iの多形態のスクリーニング中に発生した新しい形態に就いて、完全な特性解析を行った。この特性解析は、XRPD、熱解析、DVS、40℃/75%RHでの保存、および純度から構成されたものである。化合物(I)の異なる多形態を得るために、冷却、蒸発、貧溶媒添加を含む4つの結晶化手法を利用した。それぞれ結晶化手法の詳細は以下に示す。これらの手法で各溶媒から得た固体形態を表15に要約する。
【0087】
結晶化の手法:
1.迅速結晶化スクリーン法
2種の小規模スクリーニング手法を使用した。
【0088】
A.化合物I約1mgを0.5mLポリプロピレン製遠心管と溶媒0.5 mL中に秤量した。当該遠心管は室温で18時間撹乱せずに放置させて、変化があるか観察した。次いで、前記管を52.5℃で2.5時間攪拌し、各管を変化に関して観察した。当該加熱遠心管を2〜8℃で20時間攪拌し、初期室温状態から結晶化度の変化(もしあれば)に就いての観測を記録した。
【0089】
B.10倍容積の化合物I(400μL中40 mgのロット7)を含むプレートを20℃から初期温度80℃に4.8℃/分の速度で加熱し、30分後に緩やかな(0.28℃/分)または早い(10℃/分)速度で最終温度5℃に冷却し、18時間その温度に保持した。結晶化実験は、ガラスバイアル(4mL)ウエル・プレート中で実施し、濾過によって固体物質を単離した。当該固形物を10時間57℃で乾燥した。
【0090】
2.迅速冷却結晶化
試料は、沸点において飽和状態を確保するために、溶剤容量に化合物I固体物質40 mg(±2)を添加して調製した。当該混合物を冷却し、0.2μナイロン膜フィルターで濾過し、暖めたガラスバイアルまたは三角フラスコに入れた。この溶液を室温に冷却し、目視検査による決定で結晶形成が終結したと見なされるまで、冷蔵庫(約4℃)に置いた。各冷蔵庫の試料をデカントし、結晶を秤量紙に移し、常温の実験室条件下で恒量になるまで乾燥した。デカントし難い試料は、4分間12000rpmで遠心分離した。迅速冷却手法で固体物質が生成しなかった場合は、前記溶媒を約半分量蒸発させてこれらの試料を濃縮した。当該溶液は、再び冷蔵庫に入れ、固体材料はすべて、デカントまたは遠心分離により単離した。
【0091】
3.ロット7および形態A0での熟成による結晶化
2つのタイプの熟成研究を実施した。
【0092】
A.試料は、ロット7または形態A0を約10 mgねじ蓋バイアル(容積約4.0 mL)中の各溶媒1.0mLに添加して調製した。次いで、これらを振盪しつつ64℃に加熱した。64℃で40分間保持後、試料を5℃(−0.25℃/分の速度で)に冷却した。試料を合計18時間、5℃に保持し、ピペットで1.5mLポリプロピレン製遠心管に移し、1分間12000rpmで回転させた。上清液をデカントした。遠心管またはガラスバイアル中の残渣は、その後、110℃で18時間真空乾燥器で乾燥し、XRPDで分析した。
【0093】
B.形態A0約40mgを異なる溶媒10容量(400μL中に40mg)中でスラリー化した。当該スラリーを50℃(0.5℃/分)および5℃(−0.5℃/分)で4時間づつ交代に48時間振盪した。次いで、固体物質をすべて濾過により単離し、XRPDおよび熱分析で分析した。
【0094】
4.形態A0含有スラリーによる結晶化
スラリー(各溶媒500μL中の形態A020mg)を25℃で時間を変えて振盪した。固体を濾過により単離し、2時間57℃で乾燥し、XRPDで分析した。
【0095】
4つの結晶化方法で単離した固体から得たXRPDの結果を以下表15に記録する。
【0096】
【表16】
【0097】
化合物Iの多形態のスクリーニングで14種の形態および120℃を超えて水和物を加熱することでのみ得られた新しい形態1種(形態B0)が得られた。単離した形態の結果の概要は以下の表16に示す。
【0098】
【表17】
【0099】
安定した固体状形態の説明
無水形態A0の調製
化合物I約200mgを、85℃で45時間、ヘプタン45容量中でスラリー化し、65℃に冷却し、高真空下、3時間室温でフィルター乾燥した。形態A0の回収率は97%であった。
【0100】
別の手法では、次のプロセスに従って化合物Iを形態A0に転換した:
1)化合物Iを30容量のTHFに溶解した。この時点で必要に応じて、この溶液は、金属消去剤(スカベンジャー)または炭素で処理してもよい。
【0101】
2)前記金属スカベンジャーまたは炭素を除去するために、得られた溶液を濾過し、次いですべての外部微粒子を除去する為に、1ミクロンのインライン・カートリッジフィルターを通して仕上げ濾過した。
【0102】
3)前記溶媒(THF)の一部を真空下常温で蒸留して元の容積の約60%にし、続いて同等容積の貧溶媒(ヘプタン)を緩やかに添加して化合物Iを沈殿させた。
【0103】
4)前記溶媒が容積でTHFを5%未満含むようになるまで真空蒸留とさらなるヘプタンの添加を続けた。
【0104】
5)得られたスラリーを90〜96℃に加熱し、この温度で3〜5時間攪拌し、完全なA0への転換を達成した。
【0105】
6)当該スラリーを常温(25±5℃)に冷却した。
【0106】
7)前記生成物/化合物Iは、生成物を通して湿気が吸い込まれるのを避けるために、乾燥不活性ガス下濾過して収集した。
【0107】
8)生成物中の残留溶媒が仕様に適合するまで、湿潤ケーキを最高80℃で乾燥した。乾燥は大気圧または真空下で実施してよい。
【0108】
温度可変粉末X線回折(VT−XRPD)を用いた形態A0の特性評価
形態A0に関して固体−固体変換は20℃から250℃までの温度範囲では起こらない。周囲環境条件に暴露した後、220℃まで加熱することにより得たサンプルのXRPDパターンにおいて有意な変化は認められない(図15を参照)。
【0109】
熱重量分析(TGA)による形態A0の特性評価
形態A0は84.4J/gの融解エンタルピー(ΔHFUS)で、約239℃において単一のピークを示す。TGAにより質量の減少は認められない。TGAにより重量の減少が認められなかったため、脱溶媒和過程の存在は考慮しない(図16を参照)。
【0110】
水分収着(DVS)による形態A0の特性評価
規則的DVS(0〜90%RH)
75%RHで吸着される水分量は0.08%、および90%RHでは約0.1%未満であった。この吸着および脱離曲線の重複は形態A0が吸湿性でないことを示唆する(図17および表25を参照)。DVS後のXRPD再分析により有意な変化は認められなかった(図18)。
【0111】
【表18】
【0112】
変則的DVS(0〜90%RH)
試料質量は90%RHで0.5%のみ増加する。ヒステリシスギャップは表面水吸着のみが生じていることを示唆する。等温線は、全質量増加の0.6%以下で可逆的である(図19および表26)。DVS後のXRPD再分析により有意な変化は認められなかった(図18)。
【0113】
【表19】
【0114】
フーリエ変換赤外線分光(FTIR)およびラマン分光法による形態A0の特性評価
結晶形態A0のFTIRおよびラマンスペクトルはそれぞれ図20および図21に示す。
【0115】
無水形態B0の調製
窒素フロー下で20mgの化合物Iを125℃まで加熱することにより、形態B0を得た。
【0116】
温度可変X線粉末回折(VT−XRPD)による形態B0の特性評価
脱水の後、形態B0に関して固体−固体変換は150℃から200℃までの温度範囲では起こらない(図22を参照)。
【0117】
熱分析による形態B0の特性評価
形態B0の示差走査熱量測定(DSC)図は、68.2J/gの融解エンタルピー(ΔHfus)で、約197℃での溶解を示す(図23)。固体−固体転移が、形態化合物I−B0の融点の前に起こる。形態B0は脱溶媒和のみにより得られた。本形態の相対的な熱力学安定性は、120℃〜199℃の間で示されるDSCデータに反映される。
【0118】
水和物形態HA0の調製
THF/ヘプタンからの結晶化
200mgの化合物Iを室温で143倍容量のヘプタンで70倍容量のTHFから沈殿させ形態HA0を得た。固体は濾過により単離した。物質は18時間57℃で乾燥させた。
【0119】
固体−固体転移による調製
20mgの化合物Iを125℃まで加熱し、窒素フローなしで室温に冷却し、形態HA0を得た。
【0120】
熱分析による形態HA0の特性評価
形態HA0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を表す(図24および表27)。オープンパンにおいて水和物は約60〜120℃の間で幅の広い吸熱ピークを示し、結晶から抜け出る総水量に相当する。吸熱反応の事象は格子からの水分流出を伴う脱水プロセスと一致する。脱溶媒和は、吸熱ピークで固体の状態で起こる。この吸熱ピークの位置およびエネルギーは製剤原料および溶媒の2つの化学成分の状態図に依存し、当該化学成分の安定性が生じる。脱溶媒和のプロセスに割当てることが可能なそれらのそれぞれの溶媒の沸点近くの温度で幅の広い吸熱ピークを示す溶媒和物のDSCサーモグラムはTGAにより確認する。TGAにより解析した場合、一水和物形態HA0は50〜120℃の間で平均4.0%の重量損失を示した。これは1モルの化合物Iの1モルの水への取り込みの理論値である4.1%と一致する。
【0121】
【表20】
【0122】
水吸収による形態HA0の特性評価
図25は形態HA0で集積した動的蒸気収着データを示す。乾燥すると即座に、湿気への暴露による即時の取り込みが起こる。形態HA0の等温線は、20〜30%RHの間で、1.25%の重量の減少を示す。30〜90%RHから、取り込みは平衡に達し始める。最初の脱離段階の間、表面の吸着のみを示すわずかなヒステリシスがある。第2の脱離段階の間ではほとんど脱離は起こらないが、サンプルは〜0.4%の2回目の変化をする。吸収はこの形態がチャンネル水和物であるという証拠を示す。非化学量論的水和は、格子のチャンネルの不完全な水和に起因する。DVS後のXRPD再分析において有意な変化は認められなかった。
【0123】
FTIRおよびラマン分光法による特性評価
結晶形態HA0のFTIRおよびラマンスペクトルはそれぞれ図26および図27に示す。
【0124】
水和物形態HC0の調製
エタノール/水からの再結晶化
40mgのLot7を400μLのエタノールと100μLの水に加え、形態HC0を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0125】
エタノール溶媒和物の40℃/75%RHでの保存
化合物Iのエタノール溶媒和物20mgを9日間40℃/75%RHで保存し、形態HC0を得た。
【0126】
結晶構造の調製
一水和物HC0が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をテトラヒドロフランに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0127】
単結晶X線回折による結晶構造の同定
HC0に関する単結晶X線データを得た。データから得られた格子パラメータは表28に示す。
【0128】
データは、ω−2θスキャンテクニックを用い103Kの温度で集積した。大きさが約0.30×0.16×0.11mmのC24H28N4O4の無色の金属板をランダムな方向にガラス繊維上にのせた。三斜晶系格子パラメータ(P−1,Z=2)および算出したボリュームは、以下の通りである。
a=7.6128(10) α=65.839(18)°
b=11.5697(15) β=79.137(16)°
c=13.193(4)Å γ=86.800(10)°
V=1040.9(3)Å3
【0129】
【表21】
【0130】
【表22】
【0131】
熱分析による形態HC0の特性評価
形態HC0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を示す(図28および表29)。TGAを行う場合、一水和物形態HC0は50〜120℃の間で平均3.9%の重量損失を示した。これは1モルの化合物Iの1モルの水への取り込みの理論値である4.1%と一致する。
【0132】
【表23】
【0133】
水分収着(DVS)による形態HC0の特性評価
規則的DVS(0〜90%RH)
ヒステリシスギャップは全取り込みの0.4%で表面水吸着のみが生じていることを示唆する(図29および表30)。DVS後のXRPD再分析により有意な変化は認められなかった(図30)。
【0134】
【表24】
【0135】
変則的DVS(90〜0%RH)
ヒステリシスギャップは0〜40%RHで表面水吸着のみが生じていることを示唆する。40〜90%RHでバルク吸収が生じているものと思われる(図31および表31)。DVS後のXRPD再分析により有意な変化は認められなかった(図30)。
【0136】
【表25】
【0137】
FTIRおよびラマン分光法による特性評価
結晶形態HC0のFTIRおよびラマンスペクトルはそれぞれ図32および図33に示す。
【0138】
水和物形態HD0の調製
アセトン/水からの再結晶化
40mgのLot7を400μLのアセトンと100μLの水に加え、形態HD0を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0139】
2−メチル−2−プロパノールからの再結晶化
55mLの2−メチル−2−プロパノールに0.54gの化合物Iを沸点まで加熱することによりほぼ完全に溶解させ、形態HD0を得た。透明な溶液を得るため濁った溶液を5μナイロンメンブレンシリンジフィルターを用いシリンジ濾過した(約15%流出し、失った)。溶液を25〜30mLに濃縮し、固体を与えるため2〜8℃で4.5〜5時間冷却した。固体を50℃のオーブン中で溶かし、不溶性物質をt− ブチルアルコールの凍結を妨ぐため暖ためた器具で吸引濾過により単離した。50℃のオーブンで2時間乾燥させ、結果的に0.42g の固体を得た(収率75%)。
【0140】
酢酸イソプロピルからの再結晶
7.5mLの酢酸イソプロピルにおいて0.45gの化合物Iを軽く蓋を閉めたガラス製の20mLのシンチレーション小びん内でマグネティック撹拌子を用い室温で20時間撹拌し、形態HD0を得た。懸濁物を吸引濾過し、固体をドラフト内で110時間以上空気にさらし乾燥させた。乾燥させた物質の重さは380mgであった(収率84%)。
【0141】
結晶構造の調製
一水和物HC0が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をテトラヒドロフランに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0142】
単結晶X線回折による結晶構造の同定
HD0に関する単結晶X線データを得た。データから得た格子パラメータを下記の表32に示す。データは、ω−2θスキャンテクニックを用い103Kの温度で集積した。大きさが約0.40×0.25×0.08mmのC24H28N4O4の無色の金属板をランダムな方向にガラス繊維上にのせた。三斜晶系格子パラメータ(P−1,Z=2)および 算出したボリュームは、以下の通りである。
a=8.171(2) α=111.173(18)°
b=11.419(3) β=92.863(17)°
c=12.7305(19)Å γ=102.07(2)°
V=1072.8(4)Å3
【0143】
【表26】
【0144】
熱分析による形態HD0の特性評価
形態HD0のDSCサーモグラムは、2つの異なる吸熱ピークの存在を示す(図34および表33)。TGAを行う場合、一水和物形態HD0は50〜120℃の間で平均4.0%の重量損失を示した。1モルの化合物Iの1モルの水への取り込みの理論値は4.1%である。
【0145】
【表27】
【0146】
水分収着による形態HD0の特性評価
規則的DVS(0〜90%RH)
試料質量は90%RHで0.6%のみ増加する。ヒステリシスギャップは表面水吸着およびバルク吸収が生じていることを示唆する(図35および表34)。DVS後のXRPD再分析により有意な変化は認められなかった(図36)。
【0147】
【表28】
【0148】
変則的DVS(0〜90%RH)
試料質量は90%RHで0.8%のみ増加する。ヒステリシスギャップは表面水吸着および限られたバルク吸収が生じていることを示唆する(図37および表26)。DVS後のXRPD再分析により有意な変化は認められなかった(図27)。
【0149】
【表29】
【0150】
FTIRおよびラマン分光法による特性評価
結晶形態HD0のFTIRおよびラマンスペクトルはそれぞれ図38および図39に示す。
【0151】
化合物Iの溶解和物形態
メタノールからの再結晶化
40mgのLot7を400μLのメタノールに加え、形態S20を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0152】
2−プロパノールからの再結晶化
40mgのLot7を400μLの2−プロパノールに加え、形態S30を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0153】
エタノールからの再結晶化
40mgのLot7を400μLのエタノールに加え、形態S30を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0154】
結晶構造の調製
エタノール和物が沸点において飽和状態にあることを確実にするため200mgのLot7固体物質をエタノールに添加することにより単結晶を調製した。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。溶液は過飽和値を増加させるために20℃±0.2℃に冷却させ、均一な溶液はそのまま数日間おいた。
【0155】
N−N−ジメチルホルムアミドからの再結晶化
40mgのLot7を400μLのN−N−ジメチルホルムアミド(DMF)に添加し、形態S50を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体を濾過により単離した。物質は10時間57℃で乾燥させた。
【0156】
エチレングリコールからの再結晶化
40mgのLot7を400μLのエチレングリコールに加え、形態S60を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0157】
ピリジンからの再結晶化
40mgのLot7を400μLのピリジンに加え、形態S70を得た。サンプルを4.8℃/分の速度で80℃の初期温度まで加熱し、30分後に、5℃の最終温度まで0.28℃/分の速度で冷却し、18時間その温度で保温した。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0158】
1−プロパノールからの再結晶化
沸点で飽和状況を確保するため1−プロパノールに40mgの化合物Iを添加して、形態S90を得た。混合物を冷却し、暖めたガラス製の小型薬瓶に0.22μナイロンメンブレンフィルターで濾過した。結晶形成が視覚的検査により完了したように見えるまで溶液を室温まで冷却し、冷蔵庫(約4℃)に静置した。上澄みの液体を別の容器へ静かかに移すのが困難なサンプルは4分間12000rpmで遠心分離した。
【0159】
N−N−ジメチルアセトアミドからの再結晶化
40mgのLot7を400μLのN−N−ジメチルアセトアミド(DMA)に加え、形態S100を得た。サンプルを3日間、20℃±0.2でスラリーにした。固体は濾過により単離した。物質は10時間57℃で乾燥させた。
【0160】
イソブタノールからの再結晶化
沸点で飽和状況を確保するためイソブタノールに40mgの化合物Iを添加して、形態S120を得た。混合物を冷却し、暖めたガラス製の小型薬瓶に5μナイロンメンブレンフィルターで濾過した。結晶形成が視覚的検査により完了したように見えるまで溶液を室温まで冷却し、冷蔵庫(約4℃)に静置した。上澄みの液体を別の容器へ静かかに移すのが困難なサンプルは4分間12000rpmで遠心分離した。
【0161】
単結晶X線回折による結晶構造の同定
形態S40に関する単結晶X線データを得た。データから得られた格子パラメータは表28に示す。
【0162】
【表30】
【0163】
化合物Iの溶媒和物形態の熱分析
DSC熱曲線は、全ての溶媒和化合物に関し化合物Iの融点の前に、大きく幅の広い吸熱の存在を示した。TGA分析によりこれらの吸熱が形態S20(図40)、S30(図41)、S40(図42)、S50(図43)、S60(図44)、S70(図45)、S90(図46)、S100(図47)およびS120(図48)に関する脱溶媒和過程に起因していることが示された。これらの溶媒において導かれる溶媒の重量減少の計算を表29に提示する。
【0164】
【表31】
【0165】
化合物I一水和物の結晶構造の同定
水和物形態HC0およびHD0は同形であり、これは個々の「同形の溶媒和物」が、単位格子次元がわずかに歪み、ホスト分子の分子ネットワークが同じタイプである同一の空間群で結晶化することを意味する(Reutzel−Edens S.M.,Newman A W.,Polymorphism in the Pharmaceutical Industry,Edited by Rolf Hilfiker,2006,Wiley−VCH Verlag GmbH&Co.KGaA ISBN:9783527311460)。形態HC0および形態HD0はテトラヒドロピラジン環の立体配座において異なる。2つの水和物形態のX線粉末回折パターンは、開始点として単結晶パラメータを用い、リートベルトテクニック(Rietveld H.M.「A profile refinement method for nuclear and magnetic structures」、Journal of Applied Crystallography 2:65〜71(1969))を使用する事で精密化に成功した。セルデータ、データ集積および精密化の詳細は表28および表32に要約する。
【0166】
識別分析のためのFTIRおよびFT−ラマン法
図20および図21にある形態A0および水和物((図26、図32、図38)および(図27、図33、図39))に関するFTIRおよびラマンスペクトルの比較はカルボニル伸縮領域を除きほとんど違いが認められない。
【0167】
形態A0において、FTIRに関しては中程度の強度である1765cm−1、およびラマンに関しては1770 cm−1でピークを生じた。水和物形態において、FTIRに関しては中程度の強度である1742cm−1、およびラマンに関しては1754〜1695cm−1で本領域においてピークを生じた。この吸収ピークは化合物I構造の五員環の中に含まれるイミド・カルボニル官能性に当てはまる。この違いはほぼ純粋な固体形態の識別に使用するのに十分大きいものである。水和物形態およびA0に関するIRスペクトルはいくらかの違いを示すが、 水酸基における−OH結合の伸縮のために、水和物形態に存在する最も重要な広帯域(3800〜2800)に懸念を残す。
【0168】
【表32】
【0169】
【表33】
【0170】
固体形態間の関係
水中における化合物Iの懸濁物の相対的な安定性
水和物HA0およびA0を水媒体から結晶化する場合、形態HC0+HD0の混合物が産生される(表32)。
【0171】
【表34】
【0172】
2mLの水を数ミリグラムの化合物I形態に添加した。試料を一晩スラリーにした。小さな固体試料を除去し、XRPDにより分析した。スラリーにした後、 形態HA0およびA0が分析を行った全ての状況下において水和物形態(形態HC0およびHD0の混合物)に変換した事を見いだした(表33、表34、表35、表36および表37)。水和物形態は5〜45℃の間の形態A0より熱力学的に安定であるように見える。
【0173】
【表35】
【0174】
【表36】
【0175】
【表37】
【0176】
【表38】
【0177】
【表39】
【0178】
一水和物の相対的な安定性
実際的な温度範囲で2つの多形体(形態HC0および形態HD0)の熱力学溶解度を計測する事により、どちらがより安定でそれらの関係が単変性または互変性であるかどうか決定することが可能である。室温および55℃における これらの一水和物形態の酢酸エチル、MTBEおよび1−ペンタノールへの熱力学溶解度を計測するために実験を行った。化合物Iが多形体スクリーンの間、これらの溶媒で溶媒和化合物を形成しなかったため、これらの溶媒を選択した。
【0179】
【表40】
【0180】
表38にまとめた結果は使用した溶媒および調査を行った温度範囲を示し、2つの水和多形の可溶度値が非常に近似するが、常に形態HD0がより高い値であることを示す。室温〜55℃の間で、溶液形態HD0が形態HC0および形態HA0よりもより熱力学的に安定なことを示す。
【0181】
固体応力安定性
形態安定性における温度と湿気の影響について適切な影響を得るため応力安定性解析を行った。安定性を示すHPLC分析法は、化合物Iおよびその主要な分解産物である、これまで「化合物E」と呼んだ7−メトキシ−1,2,3,11−テトラヒドロ−5,11−ジアザベンゾ[a]トリンデン−4,6−ジオンの定量を行うため開発された。開発した方法は特異的であり、正確で、緻密で、強固である。この手法により化合物Iおよび化合物Eの正確で量的な同定が可能となった。 強制的分解実験の間、形成された全ての分解産物が主要ピークと十分に分離し、開発された方法が特異的であり、かつ安定性を示すことが証明された。
【0182】
形態A0
固体の状態では、無水形態A0は周囲環境から水を吸収する傾向を示し、3ヵ月後に形態HC0およびHD0を水和させる40℃および75%RHの標準的なICH応力状況下で上昇する傾向を示した。化学分解はこれらの応力状況下において化合物Iサンプルで認められなかった。化合物Iを110℃に暴露した場合のみ、化学分解を観察した(表39、表40および表41)。
【0183】
【表41】
【0184】
【表42】
【0185】
【表43】
【0186】
一水和物形態
固体の状態において、表42、表43および表44は40℃で75%の相対湿度で保存する場合、すべての結晶性一水和物が28日間安定であったことを示す。
【0187】
【表44】
【0188】
【表45】
【0189】
【表46】
【0190】
形態S40
形態S40(エタノール溶媒和物)は40℃/75%RHにおいて9日後に一水和物形態HC0に変わり、62日間、この状態のままであった(表45)。
【0191】
【表47】
【0192】
機械的応力による形態の転換
乳鉢および乳棒による粉砕
約100mgの化合物Iをメノウ乳鉢で5〜27分にわたる異なる時間、粉砕した。試料をXRPDと熱分析のため移動させた。確実に均一な破砕を行うため、かき集め、乳鉢の湾曲端部にこびりついた粉末を再度混ぜ直すため、破砕プロセスを5分おきに中断した。
【0193】
Wig−L−Bug(登録商標)によるフライス加工
Wig−L−Bug(登録商標)(米国、Piketch)を化合物I形態A0、HA0およびHB0 (形態HC0およびHD0の混合物)の粉砕に用いた。5から10分間、または変化が観察されなくなるまで、各々の試料(50mg)を粉砕した。2.82cm3の容器で0.9gのステンレス製のボール(直径0.6mm)を用い各々のフライス加工を行った。小びんは3200rpm、円弧6.5°で振とうさせ、ボールを100Hz以上で小びんの端に打ちつけさせた。
【0194】
形態A0の安定性
20分(すり鉢およびすりこぎ)および5分( Wig−L−Bug(登録商標))の処理後、XRPDパターンは結晶化度が有意に低下した事を示した。 残りのピークが出発物質と同じ位置にあった為、試料は完全な非結晶質にはならなかった(図49)。
【0195】
形態A0、HC0およびHD0の安定性
5分間の破砕を3回行った後、破砕形態HB0(形態HC0およびHD0の混合物)のXRPDパターン(図50)は 破砕形態HA0 のパターンと類似する。7.6°(2θ)におけるXRPDピークは、およそ30倍で強度が低下する。
【0196】
DSC曲線は、水の放出に起因すると考えられる50から100℃までの幅の広い吸熱を示す。サーモグラムは最初 、約113℃に位置するガラス転移温度Tgを示す(図51)。このDSCは観測されたひとつの発熱が136℃で準安定性形態B0へ向かう再結晶化の第一段階に相当する事を示す。幅の広い吸熱事象 は形態B0の融解と一致し、231℃(形態A0)で最終溶解となる。形態A0およびB0が単変性とし考えるならばこれらの事象の説明が可能であり、形態A0はより安定した形態である。
【0197】
XRPD、VT−XRPDおよび単結晶回折パターン実験の結果として得たピークの高さが異なる可能性があり、温度、結晶のサイズまたは形態、試料調製またはPANalytical X Pert Pro回折計またはオックスフォード回折CCD回折計の分析ウェルにおける試料の高さのような変化しやすいものに依存する可能性があることは理解されるはずである。
【0198】
異なる放射線源で測定する場合、ピークの位置が変化する可能性があることもまた理解されるはずである。たとえば、それぞれ1.54060Å、0.7107Å、および1.9373Åの波長を持つCu−Kα1、Mo−Kα、Co−KαおよびFe−Kα放射線がCu−Kα放射線で測定したそれらのピークと異なるピークの位置を提供する可能性がある。
【0199】
さらに、一連のピークの位置の後に続く「±0.2度2−シータ」という単語が±0.2度2−シータの変動性で角度位置に関して報告されるそれに続く群のピークの全てを意味することは理解されるはずである。たとえば、「6.81、8.52、9.73、12.04、および/または13.25±0.2度2−シータ」は「6.81±0.2度2−シータ、8.52±0.2度2−シータ、9.73±0.2度2−シータ、12.04±0.2度2−シータ、および/または13.25±0.2度2−シータ」を意味する。
【0200】
当業者が正しく理解するならば本発明における多数の修正および変更は上記の内容を考慮した上で可能である。したがって、添付の特許請求の範囲内で、特にここに解説したこと以上に、本発明が熟練されることが可能であると理解され、本発明の範囲はそのようなすべての変化を含むことを意図するものである。
【特許請求の範囲】
【請求項1】
形態A0或いは形態B0、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項2】
請求項1の結晶形態において、前記結晶形態は形態A0である、結晶形態。
【請求項3】
請求項1の結晶形態において、前記結晶形態は形態B0である、結晶形態。
【請求項4】
請求項2の結晶形態であって、これは以下のピーク:4.32、6.07、8.55、12.07及び15.37±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項5】
請求項2の結晶形態であって、これは実質的に図1に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項6】
請求項3の結晶形態であって、これは以下のピーク:7.16、7.89、10.77、16.54及び21.20±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項7】
請求項3の結晶形態であって、これは実質的に図2に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項8】
形態HA0、形態HC0或いは形態HD0、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項9】
請求項8の結晶形態において、前記結晶形態は形態HA0である、結晶形態。
【請求項10】
請求項8の結晶形態において、前記結晶形態は形態HC0である、結晶形態。
【請求項11】
請求項8の結晶形態において、前記結晶形態は形態HD0である、結晶形態。
【請求項12】
請求項9の結晶形態であって、これは以下のピーク:7.59、15.12、16.06、17.94及び23.89±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項13】
請求項9の結晶形態であって、これは実質的に図3に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項14】
請求項10の結晶形態であって、これは以下のピーク:8.36、8.71、16.69、17.39及び24.59±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項15】
請求項10の結晶形態であって、これは実質的に図4に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項16】
請求項11の結晶形態であって、これは以下のピーク:7.60、8.99及び15.16±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項17】
請求項11の結晶形態であって、これは実質的に図5に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項18】
形態S20、形態S30、形態S40、形態S50、形態S60、形態S70、形態S90、形態S100或いは形態S120、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項19】
請求項18の結晶形態において、前記結晶形態は形態S20である、結晶形態。
【請求項20】
請求項18の結晶形態において、前記結晶形態は形態S30である、結晶形態。
【請求項21】
請求項18の結晶形態において、前記結晶形態は形態S40である、結晶形態。
【請求項22】
請求項18の結晶形態において、前記結晶形態は形態S50である、結晶形態。
【請求項23】
請求項18の結晶形態において、前記結晶形態は形態S60である、結晶形態。
【請求項24】
請求項18の結晶形態において、前記結晶形態は形態S70である、結晶形態。
【請求項25】
請求項18の結晶形態において、前記結晶形態は形態S90である、結晶形態。
【請求項26】
請求項18の結晶形態において、前記結晶形態は形態S100である、結晶形態。
【請求項27】
請求項18の結晶形態において、前記結晶形態は形態S120である、結晶形態。
【請求項28】
請求項19の結晶形態であって、これは以下のピーク:8.56、14.64、16.07、22.24及び23.02±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項29】
請求項20の結晶形態であって、これは以下のピーク:6.70、8.67、13.36、16.80及び16/85±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項30】
請求項21の結晶形態であって、これは以下のピーク:8.42、8.60、13.92、17.20及び24.46±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項31】
請求項22の結晶形態であって、これは以下のピーク:4.46、7.67、8.86及び11.71±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項32】
請求項23の結晶形態であって、これは以下のピーク:8.68、11.10、16.94、17.39及び23.31±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項33】
請求項24の結晶形態であって、これは以下のピーク:4.50、7.70、8.90及び11.76±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項34】
請求項25の結晶形態であって、これは以下のピーク:8.34、8.67、16.68、17.33及び24.57±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項35】
請求項26の結晶形態であって、これは以下のピーク:4.45、7.62、8.79、11.62及び/若しくは17.67±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項36】
請求項27の結晶形態であって、これは以下のピーク:7.63、7.67、9.00、17.99及び24.46±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項37】
形態A0である化合物Iの結晶形態を調合するための方法であって、この方法は:
a.炭化水素(群)(ヘプタン或いはトルエンなど)において化合物Iをスラリー化する工程と;
b.結果生じたスラリーを冷却する工程と;
c.前記結果生じたスラリーを濾過する工程と;
d.濾過ケーキを乾燥させる工程とを有するものである、方法。
【請求項38】
請求項37の方法において、前記化合物Iは、26〜45容積のヘプタンにおいてスラリー化させるものである、方法。
【請求項39】
請求項38の方法において、前記化合物Iは、45容積のヘプタンにおいてスラリー化させるものである、方法。
【請求項40】
請求項37の方法において、前記工程(a)は79〜83℃で実行されるものである、方法。
【請求項41】
請求項40の方法において、前記工程(a)は85℃で実行されるものである、方法。
【請求項42】
請求項37の方法において、前記工程(a)は24〜48時間実行されるものである、方法。
【請求項43】
請求項42の方法において、前記工程(a)は45時間実行されるものである、方法。
【請求項44】
請求項37の方法において、前記工程(b)は30〜65℃の温度で生じるものである、方法。
【請求項45】
請求項44の方法において、前記工程(b)は65℃の温度で生じるものである、方法。
【請求項46】
請求項37の方法において、前記工程(c)は0.33〜3時間、室温で実行されるものである、方法。
【請求項47】
請求項46の方法において、前記工程(c)は3時間、室温で実行されるものである、方法。
【請求項48】
形態A0である化合物Iの結晶形態を調合する方法であって、この方法は:
a.溶媒に化合物Iを溶解させる工程と;
b.結果生じた溶液を濾過する工程と;
c.化合物Iを沈殿させるために抗溶媒を添加する間に前記溶媒を部分的に蒸留する工程と;
d.工程(a)で使用した前記溶媒の容量を減らすために付加的な抗溶媒を添加する間に結果生じたスラリーをさらに蒸留する工程と;
e.形態A0への完全な変換を達成するように前記スラリーを加熱する工程と;
f.冷却する工程と;
g.濾過を介して産物を回収する工程と;
h.乾燥させる工程とを有するものである、方法。
【請求項49】
請求項48の方法において、前記工程(a)は27〜35容積のTHFを用いて実行されるものである、方法。
【請求項50】
請求項49の方法において、前記工程(a)は30容積のTHFを用いて実行されるものである、方法。
【請求項51】
請求項48の方法において、前記工程(a)を介して産生された前記溶液は任意に金属スカベンジャー或いは炭素で処理されるものである、方法。
【請求項52】
請求項48の方法において、前記濾過する工程(b)は以下の工程:(i)前記金属スカベンジャーを除去するために濾過する工程;及び(ii)1ミクロンインライン(inline)カートリッジフィルターを通じて研磨濾過(polish filtering)する工程の1若しくは両者を有するものである、方法。
【請求項53】
請求項48の方法において、前記工程(c)で存在する前記溶媒はそのオリジナル容量の60〜90%へ蒸留されるものである、方法。
【請求項54】
請求項48の方法において、前記工程(c)は抗溶媒としてヘプタンを用いて実行されるものである、方法。
【請求項55】
請求項48の方法において、前記工程(d)は容量で5%以下のTHFが残るまで実行されるものである、方法。
【請求項56】
請求項48の方法において、前記工程(e)は約90〜96℃の温度で実行されるものである、方法。
【請求項57】
請求項48の方法において、前記工程(e)は任意に省略されるものである、方法。
【請求項58】
請求項56の方法において、前記スラリーは約3〜5時間撹拌させるものである、方法。
【請求項59】
請求項48の方法において、前記工程(f)は外界温度(25±5℃)で実行されるものである、方法。
【請求項60】
請求項48の方法において、前記工程(g)の濾過は乾燥した不活性ガスを用いて実行されるものである、方法。
【請求項61】
請求項48の方法において、前記工程(h)は80℃までの温度で実行されるものである、方法。
【請求項62】
請求項48の方法において、残留水及び/若しくは溶媒和化合物(群)は共沸的に除去されるものである、方法。
【請求項63】
形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物。
【請求項64】
化合物Iを調合する方法であって、この方法は、
【化3】
化合物Aを
【化4】
1,1,1−トリフルオロメタンスルホン酸1−シクロペンテン−1−イルエステルと反応させ、化合物Bを
【化5】
産生する工程を有するものである、方法。
【請求項65】
化合物Iを調合する方法であって、この方法は、
【化6】
化合物Cを
【化7】
マレイミドと反応させ、化合物Dを
【化8】
産生する工程を有するものである、方法。
【請求項1】
形態A0或いは形態B0、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項2】
請求項1の結晶形態において、前記結晶形態は形態A0である、結晶形態。
【請求項3】
請求項1の結晶形態において、前記結晶形態は形態B0である、結晶形態。
【請求項4】
請求項2の結晶形態であって、これは以下のピーク:4.32、6.07、8.55、12.07及び15.37±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項5】
請求項2の結晶形態であって、これは実質的に図1に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項6】
請求項3の結晶形態であって、これは以下のピーク:7.16、7.89、10.77、16.54及び21.20±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項7】
請求項3の結晶形態であって、これは実質的に図2に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項8】
形態HA0、形態HC0或いは形態HD0、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項9】
請求項8の結晶形態において、前記結晶形態は形態HA0である、結晶形態。
【請求項10】
請求項8の結晶形態において、前記結晶形態は形態HC0である、結晶形態。
【請求項11】
請求項8の結晶形態において、前記結晶形態は形態HD0である、結晶形態。
【請求項12】
請求項9の結晶形態であって、これは以下のピーク:7.59、15.12、16.06、17.94及び23.89±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項13】
請求項9の結晶形態であって、これは実質的に図3に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項14】
請求項10の結晶形態であって、これは以下のピーク:8.36、8.71、16.69、17.39及び24.59±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項15】
請求項10の結晶形態であって、これは実質的に図4に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項16】
請求項11の結晶形態であって、これは以下のピーク:7.60、8.99及び15.16±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項17】
請求項11の結晶形態であって、これは実質的に図5に記載されたようなX線粉末回折パターンを持つものである、結晶形態。
【請求項18】
形態S20、形態S30、形態S40、形態S50、形態S60、形態S70、形態S90、形態S100或いは形態S120、若しくはそれらの混合物である、化合物Iの結晶形態。
【請求項19】
請求項18の結晶形態において、前記結晶形態は形態S20である、結晶形態。
【請求項20】
請求項18の結晶形態において、前記結晶形態は形態S30である、結晶形態。
【請求項21】
請求項18の結晶形態において、前記結晶形態は形態S40である、結晶形態。
【請求項22】
請求項18の結晶形態において、前記結晶形態は形態S50である、結晶形態。
【請求項23】
請求項18の結晶形態において、前記結晶形態は形態S60である、結晶形態。
【請求項24】
請求項18の結晶形態において、前記結晶形態は形態S70である、結晶形態。
【請求項25】
請求項18の結晶形態において、前記結晶形態は形態S90である、結晶形態。
【請求項26】
請求項18の結晶形態において、前記結晶形態は形態S100である、結晶形態。
【請求項27】
請求項18の結晶形態において、前記結晶形態は形態S120である、結晶形態。
【請求項28】
請求項19の結晶形態であって、これは以下のピーク:8.56、14.64、16.07、22.24及び23.02±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項29】
請求項20の結晶形態であって、これは以下のピーク:6.70、8.67、13.36、16.80及び16/85±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項30】
請求項21の結晶形態であって、これは以下のピーク:8.42、8.60、13.92、17.20及び24.46±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項31】
請求項22の結晶形態であって、これは以下のピーク:4.46、7.67、8.86及び11.71±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項32】
請求項23の結晶形態であって、これは以下のピーク:8.68、11.10、16.94、17.39及び23.31±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項33】
請求項24の結晶形態であって、これは以下のピーク:4.50、7.70、8.90及び11.76±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項34】
請求項25の結晶形態であって、これは以下のピーク:8.34、8.67、16.68、17.33及び24.57±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項35】
請求項26の結晶形態であって、これは以下のピーク:4.45、7.62、8.79、11.62及び/若しくは17.67±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項36】
請求項27の結晶形態であって、これは以下のピーク:7.63、7.67、9.00、17.99及び24.46±0.2度2−シータの1若しくはそれ以上を有するX線粉末回折パターンによって特徴付けされるものである、結晶形態。
【請求項37】
形態A0である化合物Iの結晶形態を調合するための方法であって、この方法は:
a.炭化水素(群)(ヘプタン或いはトルエンなど)において化合物Iをスラリー化する工程と;
b.結果生じたスラリーを冷却する工程と;
c.前記結果生じたスラリーを濾過する工程と;
d.濾過ケーキを乾燥させる工程とを有するものである、方法。
【請求項38】
請求項37の方法において、前記化合物Iは、26〜45容積のヘプタンにおいてスラリー化させるものである、方法。
【請求項39】
請求項38の方法において、前記化合物Iは、45容積のヘプタンにおいてスラリー化させるものである、方法。
【請求項40】
請求項37の方法において、前記工程(a)は79〜83℃で実行されるものである、方法。
【請求項41】
請求項40の方法において、前記工程(a)は85℃で実行されるものである、方法。
【請求項42】
請求項37の方法において、前記工程(a)は24〜48時間実行されるものである、方法。
【請求項43】
請求項42の方法において、前記工程(a)は45時間実行されるものである、方法。
【請求項44】
請求項37の方法において、前記工程(b)は30〜65℃の温度で生じるものである、方法。
【請求項45】
請求項44の方法において、前記工程(b)は65℃の温度で生じるものである、方法。
【請求項46】
請求項37の方法において、前記工程(c)は0.33〜3時間、室温で実行されるものである、方法。
【請求項47】
請求項46の方法において、前記工程(c)は3時間、室温で実行されるものである、方法。
【請求項48】
形態A0である化合物Iの結晶形態を調合する方法であって、この方法は:
a.溶媒に化合物Iを溶解させる工程と;
b.結果生じた溶液を濾過する工程と;
c.化合物Iを沈殿させるために抗溶媒を添加する間に前記溶媒を部分的に蒸留する工程と;
d.工程(a)で使用した前記溶媒の容量を減らすために付加的な抗溶媒を添加する間に結果生じたスラリーをさらに蒸留する工程と;
e.形態A0への完全な変換を達成するように前記スラリーを加熱する工程と;
f.冷却する工程と;
g.濾過を介して産物を回収する工程と;
h.乾燥させる工程とを有するものである、方法。
【請求項49】
請求項48の方法において、前記工程(a)は27〜35容積のTHFを用いて実行されるものである、方法。
【請求項50】
請求項49の方法において、前記工程(a)は30容積のTHFを用いて実行されるものである、方法。
【請求項51】
請求項48の方法において、前記工程(a)を介して産生された前記溶液は任意に金属スカベンジャー或いは炭素で処理されるものである、方法。
【請求項52】
請求項48の方法において、前記濾過する工程(b)は以下の工程:(i)前記金属スカベンジャーを除去するために濾過する工程;及び(ii)1ミクロンインライン(inline)カートリッジフィルターを通じて研磨濾過(polish filtering)する工程の1若しくは両者を有するものである、方法。
【請求項53】
請求項48の方法において、前記工程(c)で存在する前記溶媒はそのオリジナル容量の60〜90%へ蒸留されるものである、方法。
【請求項54】
請求項48の方法において、前記工程(c)は抗溶媒としてヘプタンを用いて実行されるものである、方法。
【請求項55】
請求項48の方法において、前記工程(d)は容量で5%以下のTHFが残るまで実行されるものである、方法。
【請求項56】
請求項48の方法において、前記工程(e)は約90〜96℃の温度で実行されるものである、方法。
【請求項57】
請求項48の方法において、前記工程(e)は任意に省略されるものである、方法。
【請求項58】
請求項56の方法において、前記スラリーは約3〜5時間撹拌させるものである、方法。
【請求項59】
請求項48の方法において、前記工程(f)は外界温度(25±5℃)で実行されるものである、方法。
【請求項60】
請求項48の方法において、前記工程(g)の濾過は乾燥した不活性ガスを用いて実行されるものである、方法。
【請求項61】
請求項48の方法において、前記工程(h)は80℃までの温度で実行されるものである、方法。
【請求項62】
請求項48の方法において、残留水及び/若しくは溶媒和化合物(群)は共沸的に除去されるものである、方法。
【請求項63】
形態A0、形態B0、形態HA0、形態HC0或いは形態HD0若しくはそれらの混合物を有する薬学的組成物。
【請求項64】
化合物Iを調合する方法であって、この方法は、
【化3】
化合物Aを
【化4】
1,1,1−トリフルオロメタンスルホン酸1−シクロペンテン−1−イルエステルと反応させ、化合物Bを
【化5】
産生する工程を有するものである、方法。
【請求項65】
化合物Iを調合する方法であって、この方法は、
【化6】
化合物Cを
【化7】
マレイミドと反応させ、化合物Dを
【化8】
産生する工程を有するものである、方法。
【図1】

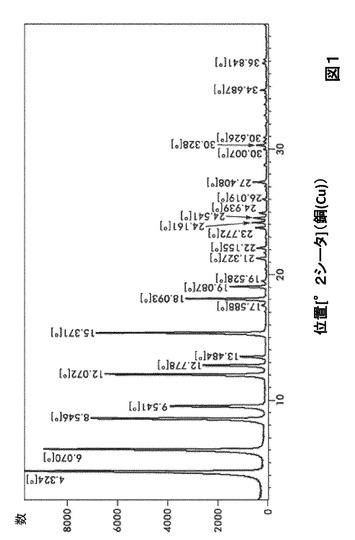
【図2】
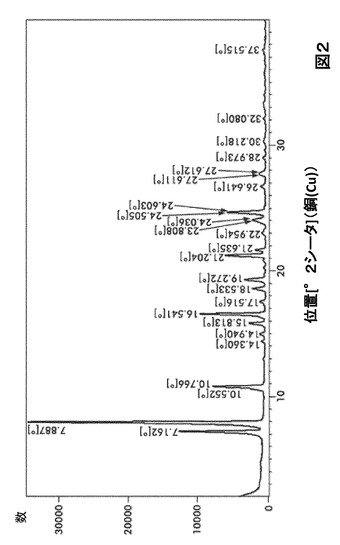
【図3】

【図4】
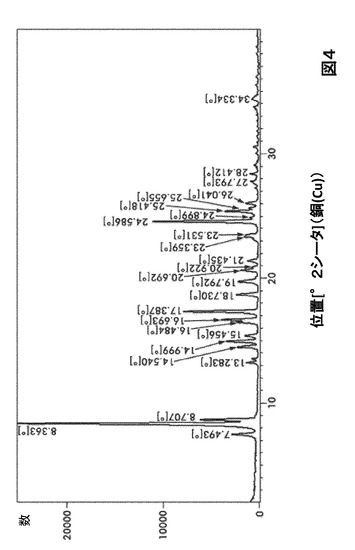
【図5】
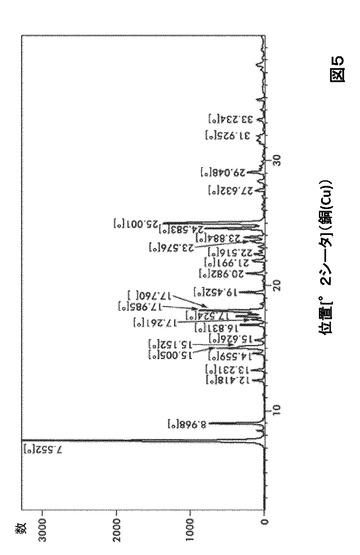
【図6】
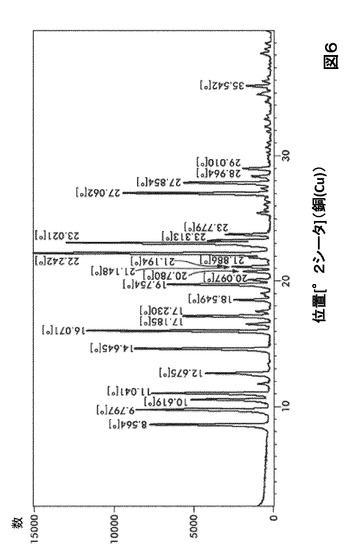
【図7】

【図8】

【図9】

【図10】
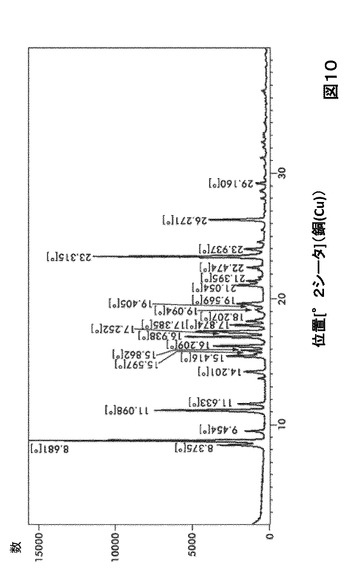
【図11】
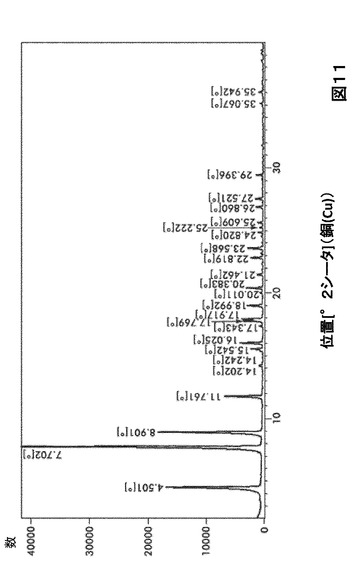
【図12】

【図13】
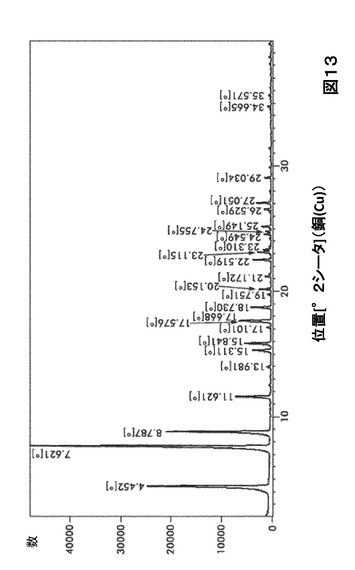
【図14】
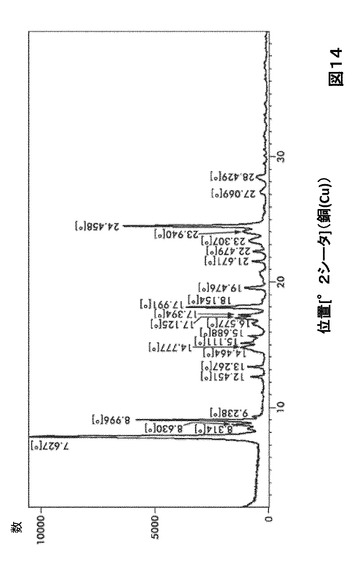
【図15】
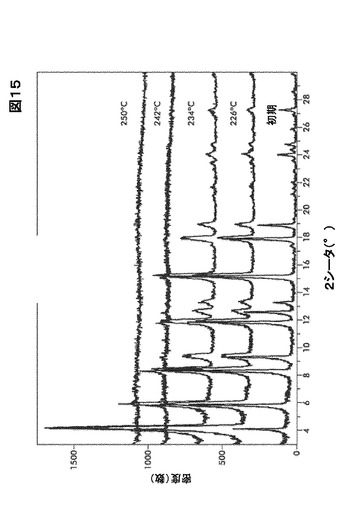
【図16】
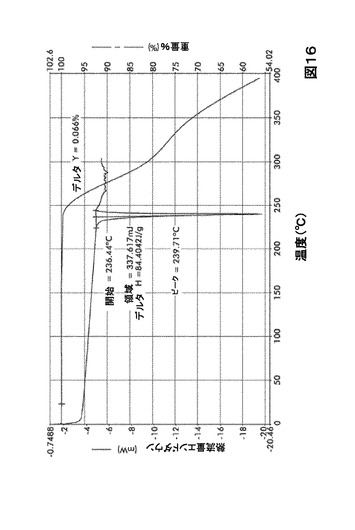
【図17】
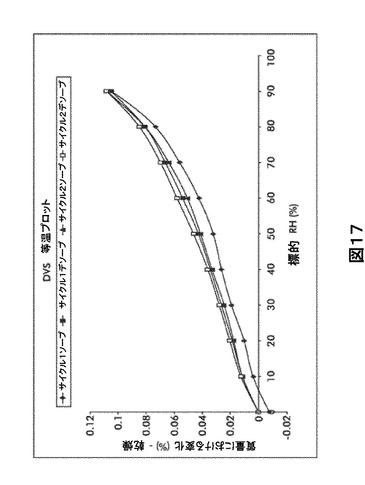
【図18】

【図19】
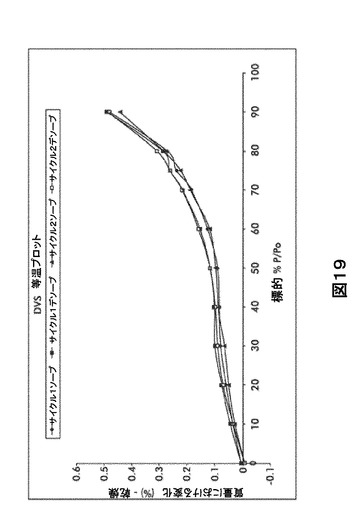
【図20】
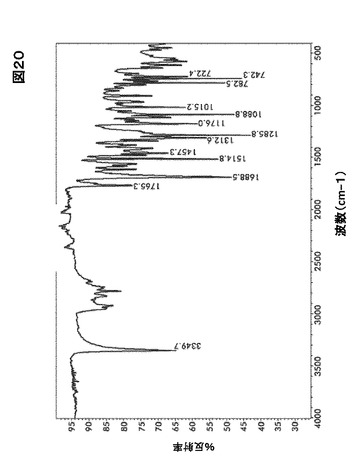
【図21】

【図22】

【図23】

【図24】

【図25】
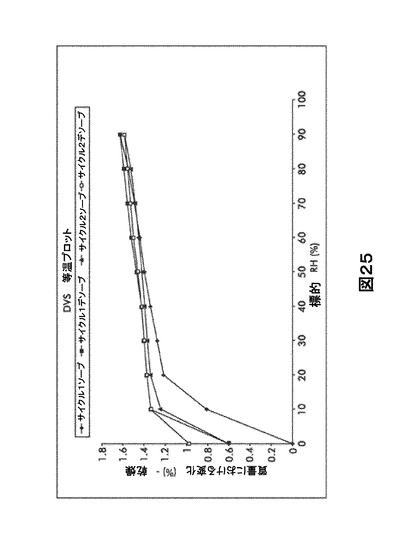
【図26】
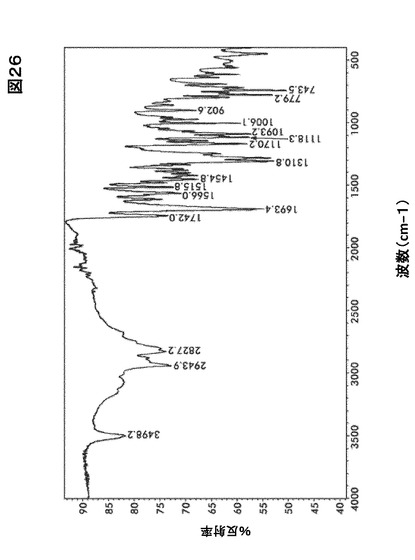
【図27】
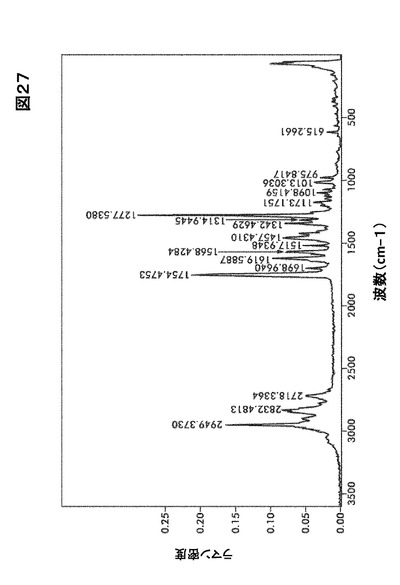
【図28】
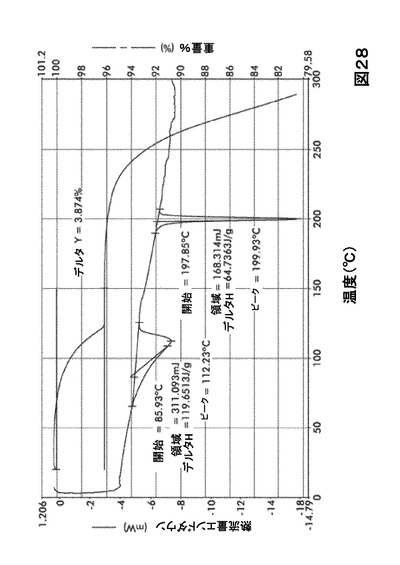
【図29】

【図30】

【図31】

【図32】
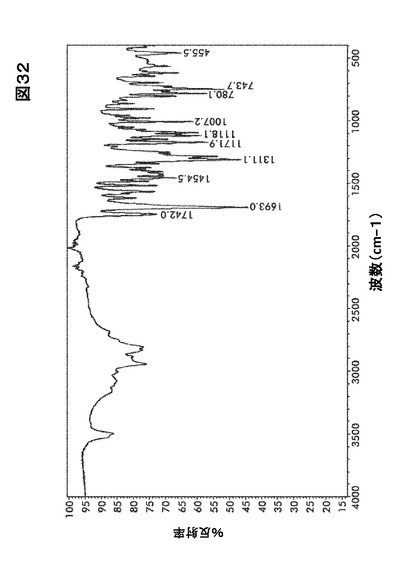
【図33】

【図34】

【図35】
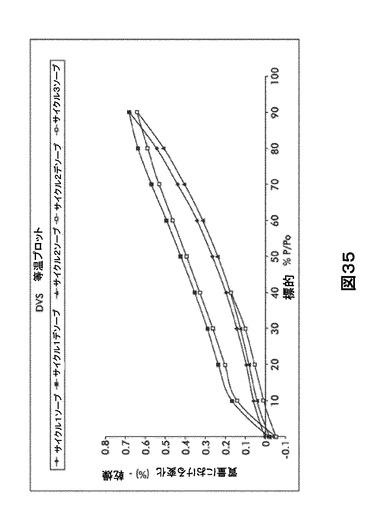
【図36】
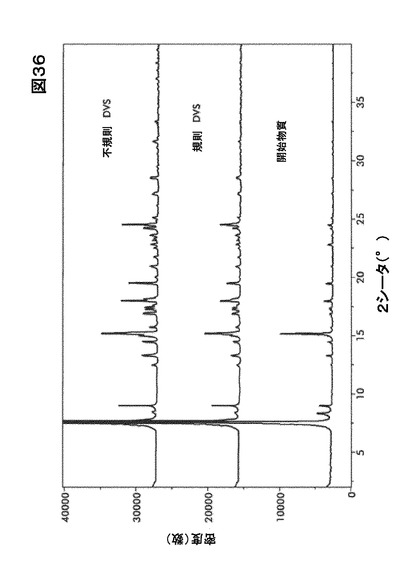
【図37】
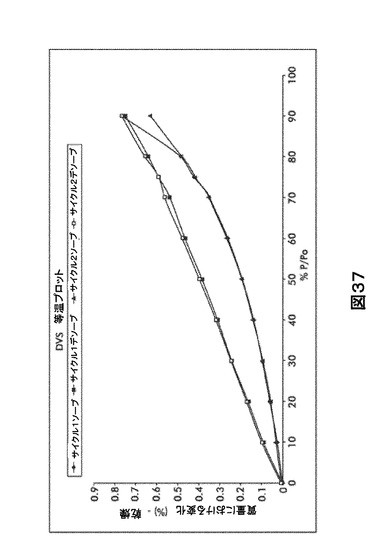
【図38】
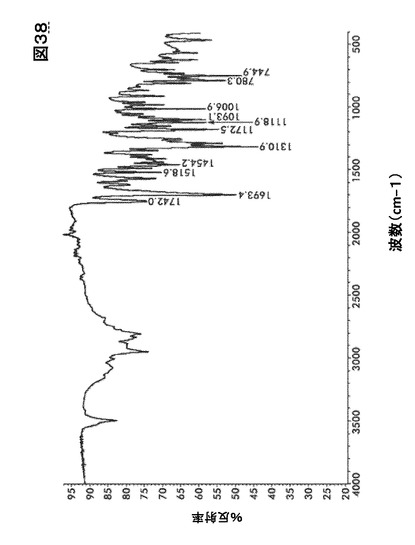
【図39】
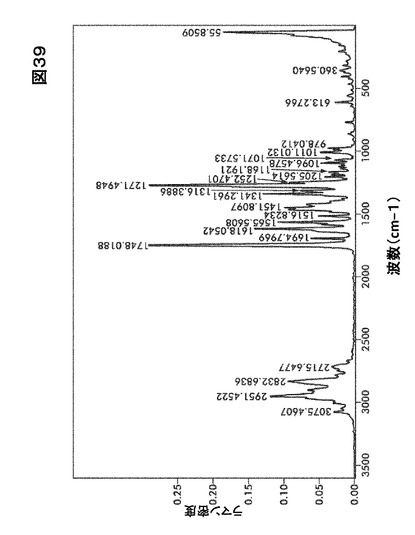
【図40】

【図41】
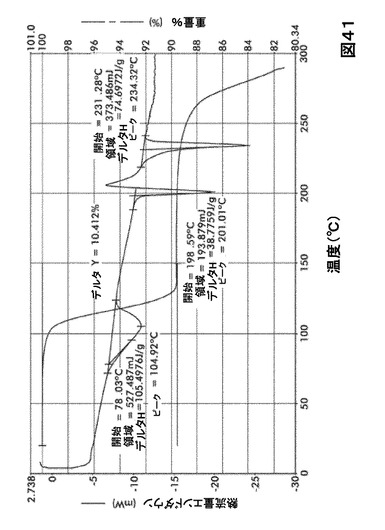
【図42】
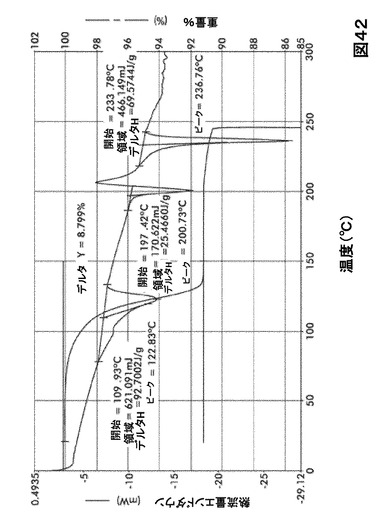
【図43】
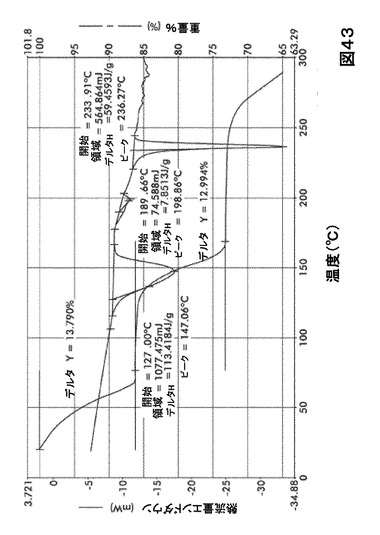
【図44】
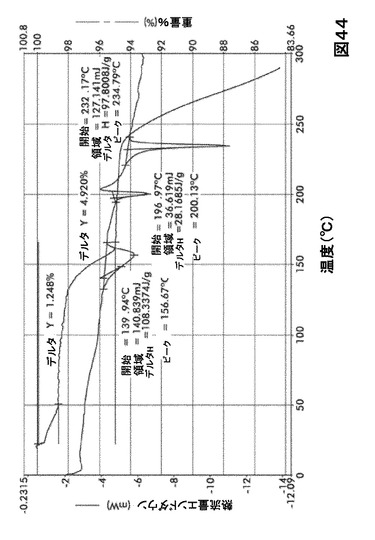
【図45】
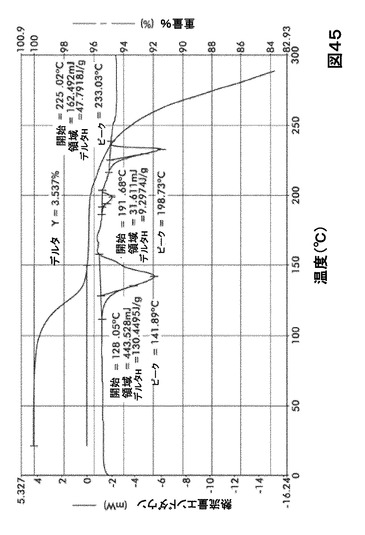
【図46】
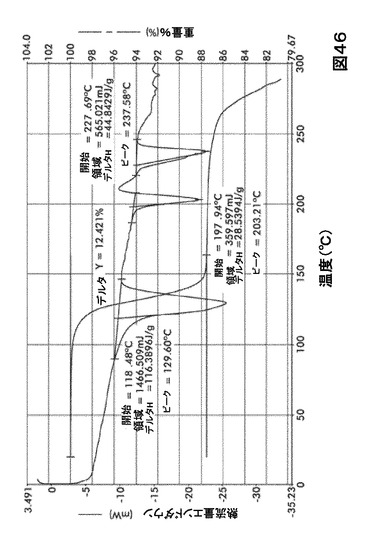
【図47】
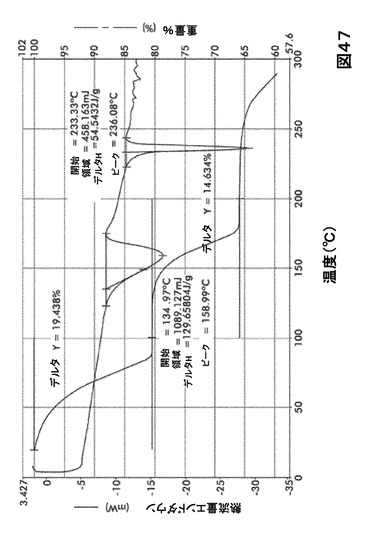
【図48】

【図49】
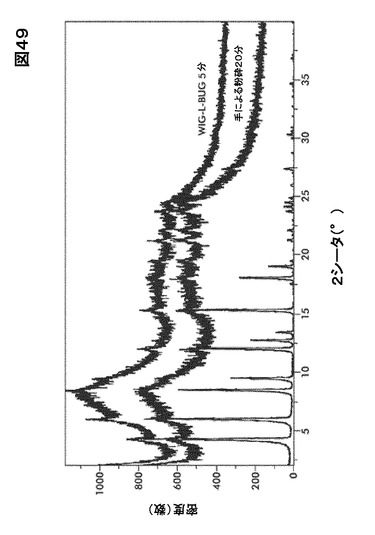
【図50】

【図51】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【公表番号】特表2013−503173(P2013−503173A)
【公表日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−526945(P2012−526945)
【出願日】平成22年8月25日(2010.8.25)
【国際出願番号】PCT/US2010/046671
【国際公開番号】WO2011/028580
【国際公開日】平成23年3月10日(2011.3.10)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
【公表日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成22年8月25日(2010.8.25)
【国際出願番号】PCT/US2010/046671
【国際公開番号】WO2011/028580
【国際公開日】平成23年3月10日(2011.3.10)
【出願人】(509021085)セファロン、インク. (24)
【Fターム(参考)】
[ Back to top ]