
多発性骨髄腫細胞を死滅させるためのeIF−5Aの使用
本発明は、真核生物翻訳開始因子5A、および、癌細胞の成長を阻害し、転移を阻害するための、該因子をコード化するポリヌクレオチドの使用に関する。好ましい実施形態において、eIF−5A1は多発性骨髄腫細胞を死滅させるために使用される。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、2005年12月13日に出願された米国仮特許出願第60/749,604号、および、2006年4月27日に出願された米国仮特許出願第60/795,168号の利益を主張するものであり、参照することにより、そのすべてを本明細書に援用する。
【0002】
発明の分野
本発明は、アポトーシスに特異的な真核生物開始因子(「eIF−5A」)、および該因子をコード化するポリヌクレオチドの多発性骨髄腫細胞やその他の癌細胞を死滅させるための使用に関する。本発明は、多発性骨髄腫を阻害するため、多発性骨髄腫細胞を死滅させるため、そして、その他の癌細胞の成長を阻害するおよび/または死滅させるための、アポトーシスに特異的なeIF−5A、または「アポトーシス特異的eIF−5A」、または「eIF−5A1」の使用と、eIF−5A2アイソフォームの使用に関する。
【背景技術】
【0003】
発明の背景
アポトーシスは、細胞の縮小、クロマチンの濃縮、核断片化、および膜泡形成といった明確な形態学的特性によって特徴付けられる、遺伝子的にプログラムされた細胞事象である。Kerr et al(1972)Br.J.Cancer,26,239−257;Wyllie et al(1980)Int.Rev.Cytol.,68,251−306。アポトーシスは、正常な組織発達およびホメオスタシスにおいて重要な役割を果たし、アポトーシスのプログラムにおける欠陥は、神経変性疾患および自己免疫疾患から腫瘍まで、種々のヒト疾患の原因になると考えられている。Thompson(1995)Science,267,1456−1462;Mullauer et al(2001)Mutat.Res,488,211−231。アポトーシス細胞の形態学的特徴については明らかになっているものの、その過程を制御する分子経路が解明されるようになってからまだ間もない。
【0004】
アポトーシスに関与する別の主要なタンパク質は、腫瘍抑制遺伝子p53によりコード化されるタンパク質である。このタンパク質は、おそらくはBaxの上方制御によって、細胞の成長を制御し、損傷を受けて遺伝的に不安定な細胞にアポトーシスを誘導する、転写因子である。Bold et al(1997)Surgical Oncology,6,133−142;Ronen et al.,1996;Schuler & Green(2001)Biochem.Soc.Trans.,29,684−688;Ryan et al(2001)Curr.Opin.Cell Biol.,13,332−337;Zornig et al(2001)Biochem.Biophys.Acta,1551,F1−F37。
【0005】
アポトーシス経路における改変は、癌など多数の疾患過程において重要な役割を果たすと考えられる。Wyllie et al(1980)Int.Rev.Cytol.,68,251−306;Thompson(1995)Science,267,1456−1462、Sen & D’Incalci(1992)FEBS Letters,307,122−127;McDonnell et al(1995)Seminars in Cancer and Biology,6,53−60。従来、癌の発生および進行についての調査は、細胞増殖に焦点を当ててきた。しかし、腫瘍形成においてアポトーシスが果たす重要な役割が最近明らかになった。実際、アポトーシスの制御には、腫瘍細胞において必ず何らかの改変が生じるため、アポトーシスに関して現在分かっていることのほとんどが、腫瘍モデルを用いて分かってきたことである。Bold et al.(1997)Surgical Oncology,6,133−142。
【0006】
サイトカインもアポトーシス経路に関与してきた。生態系は、自らを制御するために細胞間相互作用を必要とし、細胞間のクロストークには概して多種類のサイトカインが必要となる。サイトカインは、数多くの異なる種類の細胞の型による、種々の刺激に反応して産生されるメディエータである。サイトカインは、数多くの異なる種類の細胞に、数多くの異なる効果を及ぼすことができる多面的分子であるが、特に、免疫応答の制御と、造血細胞の増殖および分化において重要である。標的細胞に対するサイトカインの作用は、特定のサイトカイン、相対濃度、その他のメディエータの存在に依存して、細胞の生存、増殖、活性化、分化、またはアポトーシスを促進することが可能である。
【0007】
デオキシハイプシン合成酵素(DHS)およびハイプシン含有の真核生物翻訳開始因子5A(eIF−5A)は、細胞の成長や分化など多くの細胞過程において重要な役割を果たすことで知られている。独特のアミノ酸であるハイプシンは、検査するすべての真核生物および古細菌中に見られるが真正細菌中には見られず、eIF−5Aが唯一知られるハイプシン含有タンパク質である。Park(1988)J.Biol.Chem.,263,7447−7449;Schumann & Klink(1989)System.Appl.Microbiol.,11,103−107、Bartig et al.(1990)System.Appl.Microbiol.,13,112−116;Gordon et al.(1987a)J.Biol.Chem.,262,16585−16589。活性eIF−5Aは、翻訳後2つのステップにおいて形成される。第1のステップは、デオキシハイプシン合成酵素により触媒される、前駆物質eIF−5Aの特定のリジンのα‐アミノ基へ、スペルミジンの4‐アミノブチル部分を転移することによる、デオキシハイプシン残基の形成であり、第2のステップは、この4‐アミノブチル部分をデオキシハイプシン水酸化酵素によって水酸化してハイプシンを形成することを含む。
【0008】
eIF−5Aのアミノ酸配列は種間でよく保存されており、eIF−5A内のハイプシン残基を取り囲むアミノ酸配列には徹底した保存が見られ、この修飾が生存のために重要である可能性を示している。Park et al.(1993)Biofactors,4,95−104。今まで酵母中に発見されたeIF−5Aの両アイソフォームの不活性化、またはアイソフォーム活性化の第1のステップを触媒するDHS遺伝子の不活性化が、細胞分裂を阻止するという観察は、この仮説をさらに支持するものである。Schnier et al.(1991)Mol.Cell.Biol.,11,3105−3114;Sasaki et al.(1996)FEBS Lett.,384,151−154,Park et al.(1998)J.Biol.Chem.,273,1677−1683。しかし、酵母におけるeIF−5Aタンパク質の枯渇は、総タンパク質合成をわずかに減少させたに過ぎず、タンパク質全体の合成にではなく、mRNAの特定のサブセットの翻訳に、eIF−5Aが必要である可能性を示している。Kang et al. (1993)「Effect of initiation factor eIF−5A depletion on cell proliferation and protein synthesis,」in Tuite,M.(ed.),Protein Synthesis and Targeting in Yeast,NATO Series H。eIF−5Aを結合するリガンドが、非常によく保存された部分を共有するという最近の発見もまた、eIF−5Aの重要性を支持するものである。Xu & Chen(2001)J.Biol.Chem.,276,2555−2561。また、修飾されたeIF−5Aのハイプシン残基が、RNAへの配列特異的結合に必要不可欠であることが分かり、結合はリボヌクレアーゼからの保護を提供しなかった。
【0009】
さらに、細胞内におけるeIF−5Aの枯渇が、核内に特定のmRNAの有意な堆積をもたらし、eIF−5Aが、特定のクラスのmRNAを核から細胞質へと輸送する役割を担っている可能性があることを示唆している。Liu & Tartakoff(1997)Supplement to Molecular Biology of the Cell,8,426a.Abstract No.2476,37th American Society for Cell Biology Annual Meeting。核孔に関連する核内繊維にeIF−5Aが堆積すること、そして一般の核外輸送受容体と相互作用することは、eIF−5Aが、ポリソームの一構成要素であるよりも、むしろ核細胞質間輸送タンパク質である可能性をさらに示すものである。Rosorius et al.(1999)J.Cell Science,112,2369−2380。
【0010】
1989年に、初めてSmit−McBrideらによってヒトからeIF−5AのcDNAがクローン化されて以来、酵母、ラット、ニワトリ胚、アルファルファ、トマトなど、種々の真核生物からeIF−5AのcDNAまたは遺伝子がクローン化されてきた。Smit−McBride et al.(1989)J.Biol.Chem.,264,1578−1583;Schnier et al.(1991)(酵母);Sano,A.(1995)in Imahori,M.et al.(eds),Polyamines,Basic and Clinical Aspects,VNU Science Press,The Netherlands,81−88(ラット);Rinaudo & Park(1992)FASEB J.,6,A453(ニワトリ胚);Pay et al.(1991)Plant Mol.Biol.,17,927−929(アルファルファ);Wang et al.(2001)J.Biol.Chem.,276,17541−17549(トマト)。
【0011】
多発性骨髄腫は、骨髄中における悪性形質細胞の増大、および溶骨性病変の存在を特徴とする、進行性および致命的な疾患である。多発性骨髄腫は、不治ではあるが治療可能な形質細胞の癌である。形質細胞は免疫系の重要な部分であり、感染や疾患との闘いを助ける免疫グロブリン(抗体)を産生する。多発性骨髄腫は、骨髄における過剰な数の異常な形質細胞、そして、インタクトなモノクローナル免疫グロブリン(IgG、IgA、IgD、またはIgE:「Mタンパク質」)、またはベンス・ジョーンズタンパク質(遊離のモノクローナルなL鎖)の、過剰産生によって特徴付けられる。低カルシウム血症、貧血、腎損傷、細菌感染に対する感受性の増加、正常な免疫グロブリンの産生低下が、多発性骨髄腫に共通する臨床症状である。また、多発性骨髄腫は、骨盤、脊椎、肋骨、および頭蓋骨におけるびまん性の骨粗しょう症を特徴とすることも多い。
【0012】
多発性骨髄腫に対する従来の治療法には、化学療法、幹細胞移植、幹細胞移植を併用した大量化学療法、およびサルベージ療法がある。化学療法には、Thalomid(登録商標)(サリドマイド)、ボルテゾミブ、Aredia(登録商標)(パミドロネート)、ステロイド、およびZometa(登録商標)(ゾレドロン酸)を用いる治療などがある。しかし、多くの化学療法薬は、骨髄、胃および腸の粘膜、毛包など、活発に分裂する非癌性細胞に毒性を有する。従って、化学療法は、血球数の減少、悪心、嘔吐、下痢、脱毛を引き起こす場合がある。
【0013】
従来の化学療法、すなわち標準用量の化学療法は、多発性骨髄腫を罹患する患者に対して1次的にまたは初期的に通常行われる治療である。また、大量化学療法や幹細胞移植の準備として、患者が化学療法を受ける場合もある。移植前に腫瘍量を減少させるために、導入療法(幹細胞移植の前に行う従来の化学療法)が使用されることもある。特定の化学療法薬は、骨髄細胞に対する毒性がより低く、骨髄からより多くの幹細胞を得ることができるため、他の薬よりも導入療法に適している。導入療法に適した化学療法薬の例には、デキサメタゾン、サリドマイド/デキサメタゾン、VAD(ビンクリスチン、Adriamicyn(登録商標)(ドキソルビシン)、およびデキサメタゾンの併用)、およびDVd(ペグ化リポソームドキソルビシン(Doxil(登録商標)、Caelyx(登録商標))、ビンクリスチン、および低用量デキサメサゾンの併用)が挙げられる。
【0014】
多発性骨髄腫の標準的な治療は、メルファランをプレドニゾン(副腎皮質ステロイド剤)と併用することであり、50%の奏功率を示している。残念ながら、メルファランはアルキル化剤であるため、導入治療には不向きである。副腎皮質ステロイド(特にデキサメサゾン)は、多発性骨髄腫の治療、特に化学療法に耐えられない高齢患者に対して、単独で使用される場合がある。デキサメサゾンも、単独あるいは他の薬剤と併用して、導入療法に使用される場合がある。VADは最も広く使用される導入療法だが、DVdが導入療法において有効であることは最近明らかになった。ボルテゾミブは、多発性骨髄腫の治療に最近承認されたが、毒性が非常に高い。しかし、現存するいかなる治療法も、治癒に対する顕著な可能性を提供することはできない。つまり、多発性骨髄腫細胞を死滅させるための適切な治療法が、依然として必要である。本発明はその必要とされているものを提供する。
【発明の開示】
【0015】
本発明は、癌細胞の成長を阻害する、および/または、癌細胞を死滅させる方法を提供する。また本発明は、癌細胞が転移する能力を阻害する、または遅らせる方法も提供する。癌の成長を阻害することは、腫瘍の縮小、腫瘍成長の低下を含み、また、腫瘍の完全な寛解を含む場合もある。癌はいかなる癌または腫瘍でもあり得るとし、大腸癌、結腸直腸腺癌、膀胱癌、子宮頸部腺癌、および肺癌を含むが、これらに限定されない。本発明の方法は、eIF−5Aの投与、好ましくはヒトeIF−5A1を、該癌を罹患する患者(哺乳類、好ましくはヒト)に投与することに関連する。eIF−5A1が好ましいが、eIF−5A2アイソフォームを使用してもよい。eIF−5Aは、当該技術分野で既知である任意の適切な方法によって、それを必要とする患者に送達され得る。eIF−5Aは、生物学的に適切な媒体中のDNAのような裸のDNAとして、IVや皮下注射を介して、あるいはその他の任意の生物学的に適切な送達機構を介して送達され得る。あるいは、eIF−5Aはアデノウイルスベクターのようなベクターの中で送達され得る。あるいは、DNAは、標的癌細胞へのDNAの送達を提供する、リポソームやその他の任意の適切な「担体」の中で送達され得る。また、eIF−5Aは腫瘍の部位に直接送達され得る。当業者は、eIF−5Aの送達について、用量および治療投与計画の期間を決定することが可能であろう。
【0016】
eIF−5A1およびeIF−5A2は、09/909,796(U.S.6,867,237);10/141,647(許可);10/200,148;10/277,969;10/383,614;10/792,893;11/287,460;10/861,980;11/134,445;11/184,982;11/293,391;60/749,604;60/795,168などの先の同時係属出願において既知、かつ説明されており、参照することにより、それらのすべてを本明細書に援用するものとする。eIF−5Aはヒト、ラット、マウス、イヌ等の種間でよく保存されているため、任意のeIF−5Aが本発明において使用され得る。好ましくは、ヒトeIF−5Aがヒト等の治療に使用される。突然変異したeIF−5Asが、eIF−5Aの発現を上方制御する、または増加することが可能であり、それにより癌の成長を阻害する、または癌細胞を死滅させることができる限り、eIF−5AはeIF−5Aの突然変異体も含む。
【0017】
本発明は、細胞にeIF−5A1をコード化するヌクレオチドを提供することにより、上記細胞内のMAPK/SAPKシグナル経路を活性化する方法も提供する。該eIF−5A1ポリヌクレオチドおよびeIF−5A1タンパク質は上述の通りである。
【0018】
本発明は、eIF−5Aをコード化するポリヌクレオチドを含む、骨髄腫細胞を死滅させるために有用な医薬組成物も提供する。該eIF5Aは、eIF5A1、eIF5A2、またはeIF5A1の突然変異体であってもよい。好ましくは、該eIF5AはeIF5A1である。該組成物は、送達ビヒクルをさらに含んでもよい。該送達ビヒクルは、ベクター、プラスミド、リポソーム、またはデンドリマーであってもよいが、これらに限定されない。
【0019】
本発明は、多発性骨髄腫を罹患する対象において多発性骨髄腫細胞を死滅させるための薬物を作るためのeIF5A(好ましくはeIF5A1)の使用も提供する。
【0020】
本発明は、eIF5A1をコード化するポリヌクレオチドを含む組成物を骨髄腫細胞に投与することを含む、多発性骨髄腫細胞を死滅させる方法をさらに提供し、該組成物は多発性骨髄腫細胞を死滅させる。該eIF5A1は突然変異体であってもよく、該突然変異体は別のアミノ酸に変化した保存リジンを有し、上記突然変異体はハイプシン化されることができない。治療の方法において有用な組成物は本明細書に記載する通りである。
【0021】
本発明は、多発性骨髄腫細胞を死滅させる方法をさらに提供し、eIF−5A1をコード化するポリヌクレオチドを含む組成物のほかに、eIF−5A1に対するsiRNAを含む組成物が提供される。該siRNAはeIF−5A1の内因性発現を下方制御し、それによってIL−6の発現を下方制御し、ひいては骨髄腫細胞内のアポトーシスを引き起こす。eIF−5A1 siRNAを含む組成物は、静脈内投与されてもよく、または、プラスミド、ベクター、リポソーム、またはデンドリマーのような送達ビヒクルの中で投与されてもよい。
【0022】
発明の詳細な説明
真核生物翻訳開始因子5A1(eIF5A1)は、細胞増殖に関与するmRNAのサブセットの翻訳促進に関与する、核細胞質間輸送シャトルタンパク質として機能すると仮説を立てられてきた。しかし、eIF5A1は、アポトーシスの制御因子(Taylor et al.,Invest Ophthalmol Vis Sci.;45(10):3568−76(2004))として、また、p53の発現を制御することのできるアポトーシス促進性のタンパク質としても認識されている(Li et al.(2004)J.Biol.Chem.;279:49251−49258;および本明細書の図23)。腫瘍抑制タンパク質p53は、細胞周期停止を仲介する上で、またストレスやDNA損傷に応答するアポトーシスにおいて、中心的な役割を担っている。eIF5A1の過剰発現は、癌細胞株のp53発現を上方制御することが可能であるが、(Li et al.(2004)J.Biol.Chem.;279:49251−49258;および本明細書の図23)、eIF5A1の過剰発現は、p53欠損細胞株にアポトーシスを誘導することも可能であり(Taylor et al.(2006)Journal of Molecular and Cellular Biology,Feb.9,2006(決定保留))、eIF5A1の過剰発現が複数の機構によってアポトーシスを誘導することを示唆している。
【0023】
eIF5A1の発現はアポトーシスと相関性を有する。トポイソメラーゼ阻害剤であるアクチノマイシンDで処理した、正常な大腸線維芽細胞のウエスタンブロット(図1A)は、アクチノマイシンD処理を開始してから24時間後に、p53と並行してeIF5A1タンパク質発現が上方制御されたことを実証している。一酸化窒素(NO)供与体であるSNPへの暴露に起因するアポトーシス中のRKO細胞も、eIF5A1タンパク質発現を上方制御する(図1B)。eIF5A1のRNA転写レベルは、これらの条件下では増加せず、これは、eIF5A1タンパク質が、mRNAの転写後制御の結果として堆積していることを示唆している(図1Aおよび1B)。これらの結果は、eIF5A1が、遺伝毒性ストレスや一酸化窒素に起因するアポトーシスに関与する可能性を示唆するものである。
【0024】
細胞周期停止やアポトーシスを誘導するDHSおよびDHH阻害剤に関する報告や、細胞の成長を維持するにはハイプシン化されたeIF5A1が必要であるに違いないという結論に一部基づいて、細胞増殖におけるeIF5A1の必要性が長い間提案されてきた。しかし、siRNAの使用によるeIF5A1発現の特異的抑制は、細胞の成長に何の影響も与えなかったことが判明した。HT−29細胞のeIF5A1発現を90%超抑制したことは、5日間にわたる細胞増殖に何の影響も与えなかった(図2A〜E)。しかし、DHS阻害剤であるGC7は、これらの細胞の成長に絶大な影響を与えた(図2C)。これらの結果は、eIF5A1が細胞の成長に必要ではない可能性があることを示唆している。また、eIF5A1の抑制が、アクチノマイシンD誘導性の細胞毒性からHT‐29細胞を一部保護できたことは興味深く(図2B)、これは、遺伝毒性ストレスに起因するアポトーシスにeIF5A1が関与することを、さらに支持するものである。
【0025】
eIF5A1 siRNAが、アポトーシスから細胞を保護する能力は、eIF5A1タンパク質抑制がp53発現に与えた影響についての試験を促すことになった。RKO細胞は、ストレス条件下以外では堆積しない機能的p53タンパク質を有する。siRNAによるeIF5A1の抑制は、アクチノマイシンD処理から24時間後にp53タンパク質の堆積を69%阻害することが可能であり(図3Aおよび3B)、これは遺伝毒性ストレス下でのp53の適切な発現に、eIF5A1が必要であることを示唆している。異なる配列を有するeIF5A1に対する第2のsiRNAも、アクチノマイシンDに反応してp53の上方制御を防止することができ(図8B)、この効果がsiRNAの非特異的な効果ではないことを示唆している。しかし、eIF5A1は、RKO細胞(機能的p53を含む)およびRKO−E6(機能的p53を含まない、図8A)の両方でアポトーシスを誘導することが可能であったため(図4Aおよび4B)、eIF5A1がp53から独立した機構によってもアポトーシスを誘導することができることを示唆している。
【0026】
eIF5A1またはハイプシン修飾に必要な保存リジン(K)(50位)において点突然変異を含むeIF5A1{eIF5A1(K50A)}のいずれかを発現しているアデノウイルスコンストラクトを構築した。点突然変異はリジンがアラニン(A)となる原因となった。HT‐29細胞がいずれかのコンストラクトに感染することで、それらの細胞にアポトーシスを誘導し(図5Cおよび5Dおよび図6)、細胞のバイアビリティーを大きく減少した(図5E)。Ad−eIF5A1またはAd−eIF5A1(K50A)による感染の結果として、どのような形態のeIF5A1が堆積していたかを判定するために、2次元ゲル電気泳動を使用した(図5B)。予想したとおり、Ad−eIF5A1(K50A)感染後に、非修飾eIF5A1が堆積した。Ad−eIF5A1による感染は、非修飾およびデオキシハイプシン修飾eIF5A1の両方の堆積を著しく増加させる結果となり、ウイルスによって産生されたeIF5A1タンパク質の大部分をハイプシン化するためには、DHSおよびDHHの活性が不十分であることを示した(図5B)。これらの結果は、非修飾eIF5A1、そしておそらくはデオキシハイプシン修飾eIF5A1が、細胞のアポトーシスを引き起こす形態であることを強く示唆するものである。ハイプシン化eIF5A1もこの能力を有するか否かはまだ明らかではない。
【0027】
eIF5A1の核細胞質間輸送機能が示唆されてきたが、eIF5A1は主として細胞質の中で発現すると報告されている。(Shi et al.,Exp Cell Res.;225:348−356(1996b))。核細胞質間輸送シャトルタンパク質は、核局在化も行うことが予想される。アポトーシス中のeIF5A1の機能が発見されたため、アポトーシス中のeIF5A1の局在変化についての研究を行った。IFN−γおよびTNF−αを用いた処理による細胞死受容体の活性化と、アクチノマイシンDを用いた処理による遺伝毒性ストレスという、2つの異なる機構によってアポトーシスをHT‐29細胞に誘導した。間接免疫蛍光法によって、eIF5A1は未処理の成長中の細胞の細胞質内に局在することが明らかになった(図7)。しかし、アポトーシス刺激剤を用いた処理は、eIF5A1の動きを非常に迅速に刺激し、主に細胞質内に局在していたeIF5A1が、主に核に局在するようになった(図7Aおよび7B)。このeIF5A1の局在における変化は、IFNγ/TNF−α処理細胞では10分以内(図7A)、アクチノマイシンD処理細胞では90分以内(図7B)と、非常に迅速に発生した。これらの結果は、細胞死受容体の活性化および遺伝毒性ストレスにより誘導されたアポトーシスの間、eIF5A1が核機能を有することを示唆している。
【0028】
アポトーシスに関与しているのが、非修飾eIF5A1なのか、デオキシハイプシン修飾型eIF5A1なのか、あるいはハイプシン修飾型eIF5A1なのかを明確にするため、アポトーシス中に堆積するeIF5A1の形態を2Dゲル電気泳動を用いて調査した。アクチノマイシンD(遺伝毒性ストレス)を用いた刺激によっても、Fas(細胞死受容体経路)に対するアゴニスト抗体と一緒にインキューベーションすることによっても、HT−29細胞においてアポトーシスの誘発が起こった。1〜24時間までの異なる時点で細胞ライセートを収集し、2Dゲル電気泳動、およびeIF5A抗体を用いたウエスタンブロットで分析した(図9)。文献に報告されるとおり、未処理の細胞においてeIF5A1タンパク質は主にハイプシン化型で存在した。両方のアポトーシス誘発剤が、存在するデオキシハイプシン化型eIF5A1の増加を刺激した。処理から1時間後に増加し始めたが、処理から24時間に停止した。非修飾eIF5A1の堆積も、処理から2時間後に観察された。これらの結果は、アポトーシスの制御に関与するデオキシハイプシン化および/または非修飾eIF5A1と一致していた。
【0029】
eIF5A1、eIF5A1(K50A)(eIF5A1の突然変異体)、およびeIF5A2が、アポトーシスを誘導する能力、および増殖を阻害する能力を、種々の癌細胞株において調査した。eIF5A1およびeIF5A1(K50A)は両方とも、大腸癌細胞株HT−29(図5、6および10)と、膀胱癌細胞株HTB−9(図11)およびHTB−4(図12)に、アポトーシスを誘導することができた。また、eIF5A2を発現するアデノウイルスによる感染は、膀胱癌細胞株HTB−9(図11)およびHTB−4(図12)にアポトーシスを誘導することができた。Ad−eIF5A1、Ad−eIF5A1(K50A)、およびAd−eIF5A2は、膀胱癌細胞株HTB−4(図13)、HTB−9(図14)、J82HTB−1(図15)およびUMUC−3(図16)の成長を阻害することができた。これらウイルス性のコンストラクトは、HeLa子宮頚部腺癌細胞(図17)の成長を阻害することもできた。Ad−eIF5A1、Ad−eIF5A1(K50A)、およびAd−eIF5A2の感染に反応したPARP切断により、HTB−4細胞におけるアポトーシスも観察された(図18)。PARPは、普通はDNAの修復、DNAの安定化、およびその他の細胞事象に関与しているが、アポトーシス初期にカスパーゼファミリーのメンバーによって切断される。PARPのカスパーゼによる切断の検出は、アポトーシスを代表する特徴として示されてきた。Lazebnik Y et al.,Nature 371,346−347(1994)。これらの結果は、eIF5A1とeIF5A2が、実際には重複する機能を有している可能性があることを示唆するものである。さらに、eIF5A1の突然変異型は、種々の細胞株において成長を停止し、アポトーシスを誘導することができた。野生型eIF5A1(およびeIF5A2)のアポトーシス誘導能は、DHSおよびDHHの制限的な活性のためにAd−eIF5A1を細胞に導入した時に生じる(図5B)、デオキシハイプシン化および非修飾eIF5A1の堆積に関係している可能性がある。最近の報告は、外因性eIF5A1がハイプシン化型として細胞内で過剰発現するには、DHSおよびDHHの両方が同一細胞内で過剰発現する必要があるとしている(Park et al.2006,PNAS;103(1):51−56)。これらの結果は、DHSおよびDHH阻害剤が細胞成長を阻害する能力は、細胞増殖に必要とされるハイプシン化eIF5A1の減少によるものではなく、細胞内で細胞周期停止および/またはアポトーシスをトリガーする、非修飾(DHS阻害剤)あるいはデオキシハイプシン修飾型(DHH阻害剤)eIF5A1の堆積によるものである可能性を示唆している。DHSは特定の細胞株において過剰発現されることが判明したこと(Clement et al.2006,FEBS J;273(6):1102−14)、およびDHSは腫瘍転移における重要な増幅された遺伝子の組として認識されてきたこと(Ramaswamy et al.2003,Nat Genet;33,49−54.)は興味深い。この情報の解釈として、非修飾あるいはデオキシハイプシン修飾型eIF5A1の堆積(アポトーシスを起こすように細胞がトリガーされた時に堆積する。図9参照。)によってトリガーされるアポトーシスを避けるために、特定の癌細胞がDHSを過剰発現するということが考えられる。DHSを過剰発現することにより、eIF5A1をハイプシン化した状態、つまり「安全な」形態を維持することによって、癌細胞は遺伝毒性ストレスやその他のストレスによるアポトーシスを減少できる可能性がある。
【0030】
eIF5A1の過剰発現によって、どのシグナル経路が影響を受けているのかを解明するために、A549肺癌細胞におけるAd−eIF5A1またはAd−eIF5A1(K50A)感染に反応した、マイトジェン活性化タンパク質キナーゼ(MAPK)/ストレス活性化タンパク質キナーゼ(SAPK)[MAPK/SAPK]経路の活性化を調査した。3つの主要なMAPK経路は、ERK MAPK経路、p38 MAPK経路、およびJNK SAPK経路である。ERK MAPK経路は、主に上皮増殖因子(EGF)などの成長ホルモンなどのマイトジェン刺激に反応してトリガーされ、種々の腫瘍の成長および生存を支持する。p38 MAPK経路は、細胞ストレス、UV光線、成長因子の使用中止、および炎症促進性サイトカインに反応して活性化される。リン酸化によりp38を活性化することは、p53などの転写因子のリン酸化をもたらし、ひいてはp53の活性または安定性を増加させる。p38の活性化は、アポトーシス促進性と抗アポトーシスの2つの経路、および炎症に関与している。JNK/SAPK経路は、UV光線、DNA損傷、炎症促進性サイトカインなどの細胞ストレスに対する反応を仲介し、c−junのような転写因子のリン酸化および活性増加を引き起こす。JNK経路の活性化は、成長、形質転換、アポトーシスなど、数多くの細胞応答をもたらすことができる。JNK経路は、A549細胞におけるEGF誘導性の成長の1次的なエフェクター経路であると思われる(Bost et al.1997,JBC;272:33422−33429)。A549細胞をAd−eIF5A1に感染させることは、これら3つの経路すべての活性化を誘発する。EGFを用いて30分間刺激したA549細胞を、Ad−eIF5A1を増量しながら感染させることは、ERKおよびその下流標的であるp90RSKのリン酸化/活性化の増加を引き起こしたが、非リン酸化ERKの量には変化は見られなかった(図20)。Ad−eIF5A1感染に反応して、リン酸化/活性化p38およびリン酸化/活性化JNKの、用量反応的な強い上方制御も観察された(図20)。Ad−eIF5A1感染の時間の経過とともに、ERK、p38およびJNK経路の活性化が、感染後24〜72時間持続したことが明らかになった(図21)。非ハイプシン化eIF5A1突然変異体であるAd−eIF5A1(K50A)による感染に反応して、これらの経路の活性化において用量反応的な増加が見られた(図22)。MEK1阻害剤U1026を用いてERKの活性化を阻害することが、JNKの活性化をも阻害した(図22)ということは興味深く、これは、ERK活性化に反応してこれらの細胞内でJNKが活性化されたことを裏付けるものである(Bost et al.1997,JBC;272:33422−33429)。反対に、MEK1阻害剤は、Ad−eIF5A1またはAd−eIF5A1(K50A)による感染に反応して一貫してp38の活性化を増加し、ERK経路の活性化がeIF5A1に反応して、p38経路の活性化を負に制御することを示唆している(図22)。
【0031】
図38は、eIF−5A1がMAPK/SAPK経路およびA549肺癌細胞のアポトーシスに与える影響を示す仮説モデルを提供する。eIF5A1(野生型あるいはハイプシン化されることのできない突然変異体)(K50A)の過剰発現は、A549細胞にアポトーシスを誘導する。eIF5A1の過剰発現は、p38、JNK、およびERKなどのMAPK/SAPK経路の増加も伴う。eIF5A1の過剰発現は、p53の安定性および活性の増加に関連するリン酸化型p53の増加など、p53タンパク質レベルの増加を誘発する。このp53の増加は、MEK/ERK経路の活性およびp53転写活性に依存する。しかし、MEK/ERK経路の阻害、そしてそれほど多くはないがJNK経路の阻害は、eIF5A1によって誘導されたアポトーシスを増強する。これらの結果は、癌細胞にアポトーシスを誘導するため、そして、抗癌剤のMEK阻害剤クラスの癌細胞死滅能を増加するために、eIF5A1を治療薬として使用できる可能性を示唆している。
【0032】
Ad−eIF5A1およびAd−eIF5A1(K50A)が、p53の発現およびリン酸化に与える影響は、図23で見ることができる。野生型および突然変異体eIF5A1の過剰発現は、用量反応的にp53タンパク質の全体的な発現増加をもたらす(図23)。セリン15がリン酸化されたp53の増加も、eIF5A1の両形態について観察された(図23)。セリン46がリン酸化されたp53の増加はAd−eIF5A1(K50A)について用量反応的に観察された。p53は、セリン15においてATM、ATRおよびp38など幾つかのキナーゼによってリン酸化することが可能であり、この修飾は、MDM2のp53に対する結合能を減少してユビキネーションを標的とし、それによってp53の堆積および機能活性を促進する。p53はセリン46において、PKCdeltaおよびp38など種々のキナーゼによってリン酸化され、この修飾はp53のアポトーシス促進プロモータに対する親和性を増加し、これはp53誘導性アポトーシスにとって重要であると考えられる。
【0033】
腫瘍の浸潤および転移は、複雑なプロセスである。そのためには腫瘍細胞が、付着する能力を備え、周辺の細胞外基質を分解し、2次的部位に移動して増殖し、そして最後には、成長の増加を維持するために血管新生を促進することが要求される。基底膜は、細胞外基質(ECM)に近接するシートであり、すべての臓器を覆って、巨大分子および細胞に対するバリアの役目を果たしている。再構成ECM(Matrigel(商標))を含む8μm膜でトランスウェルを被膜し、走化性刺激に反応して、この膜を貫通して向こう側まで到達することのできる細胞を染色することにより、腫瘍細胞の侵襲性を測定できる。A549肺癌細胞は侵襲性が高く、ECMの成分を消化することのできるゼラチナーゼであるマトリックスメタロプロテアーゼなどのタンパク質を分泌することができる。eIF5A1の過剰発現がA549細胞の侵襲性に与える影響を調査した。Ad−eIF5A1またはAd−eIF5A1(K50A)による感染が、Matrigel(商標)で被膜したトランスウェルを通って浸潤した細胞数を有意に減少させた(図24)。
【0034】
こうして得られた結果は、腫瘍をeIF5A1で処理することは治療効果がある可能性を示していた。そこで、マウスにおける実験的転移のモデルを使用した。実験IIにおいて、侵襲性の高いマウスメラノーマ細胞株B16F10をマウスに注射することにより、実験的転移を開始した(第0日目)。LacZ遺伝子(DNA対照として)またはeIF5A1をコード化するプラスミドDNAを、2、4、7、11、16、21、26および31日目に尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した(図25)。eIF5A1をコード化するプラスミドDNAでマウスを処理した時、腫瘍量における用量反応性の減少が観察された(図25および26)。1×の用量よりも2×の用量の方が効果が少ないように見えたが、0.1×用量と1×用量の間には、腫瘍量に有意な減少が観察された。腫瘍が巨視的に成長する能力は、新しい血管の形成(血管新生)に依存する。血管内皮成長因子(VEGF)は、多くの腫瘍細胞によって分泌されるサイトカインであり、腫瘍の血管新生の促進における重要な因子である。実験IIで単離された、B16F10を有する肺からの細胞ライセートを、VEGF発現について、ELISA法を用いて分析した(図27)。eIF5AプラスミドDNAを与えたマウスのVEGFレベルにおいて、有意な用量反応性の減少が見られ、eIF5A1がVEGFを制御する可能性があることを示唆している。
【0035】
実験IIIでは、尾静脈注射したB16F10細胞を用いた実験的転移のモデルを再び使用した。今回は、血清中DNAの半減期を延長し、プラスミドの肺腫瘍への取り込みを増加するために、プラスミドDNAをDOTAPと複合化した。7、14および21日目に、DNA/DOTAP複合体を注射した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した(図28)。LacZプラスミドDNA/DOTAJP複合体を与えたマウスと比較すると、eIF5A1プラスミドDNA/DOTAP複合体で処理したマウスの肺重量に、平均60%の減少が見られた(図29)。
【0036】
eIF5A1処理がメラノーマ腫瘍にアポトーシスを誘導するかどうかを判定するために、B16F0またはB16F10皮下腫瘍を有するマウスに、Ad−eIF5A1を腫瘍内注射した(実験IV)。48時間後、腫瘍を切除し、パラフィンで包埋してから薄切した。薄切した組織をTUNEL染色することにより、Ad−eIF5A1が腫瘍細胞のアポトーシスを誘導したことが明らかになった(図30)。実験Vは、アポトーシスを誘導するために腫瘍内に注射したAd−eIF5A1の能力が、B16F10皮下腫瘍の成長を減少し、生存期間を延長するかどうかを判定するためにデザインされた。B16F10細胞をC57BL/6マウスの側腹部に皮下注射した。腫瘍が直径約8mmに到達した時、Ad−LacZ、Ad−eIF5A1または緩衝液を腫瘍内に注射した。最初の3日間は毎日、それ以降は腫瘍の大きさが直径15〜16mmを超えるまで1日置きにマウスに注射をし、その時点でマウスを屠殺した。キャリパーを用いて毎日腫瘍の大きさを計測し、腫瘍体積を推測するために使用した。マウスをAd−eIF5A1で処理した時は、腫瘍成長の遅延が明らかに観察された(図31)。緩衝液のみを注射したマウスは、治療開始から最長4〜6日間生存し、Ad−LacZのみを注射したマウスは、治療開始から4日間のみ生存した(図32)。Ad−eIF5A1を注射したマウスは、治療開始から少なくとも8日間、最長で25日間生存し、Ad−eIF5A1を用いた治療が、腫瘍を有するマウスの生存期間を著しく改善することが可能であることを実証した。(図32)。
【0037】
従って、本発明は癌の成長を阻害する方法を提供する。また本発明は、癌細胞が転移する能力を阻害する、あるいは遅らせる方法も提供する。癌の成長を阻害することは、腫瘍の縮小、腫瘍成長の低下を含み、また、腫瘍の完全な寛解をも含む場合もある。癌の成長を阻害することは、癌細胞を死滅させることも含む。癌はいかなる癌または腫瘍でもあり得るとし、大腸癌、結腸直腸腺癌、膀胱癌、子宮頸部腺癌、および肺癌を含むが、これらに限定されない。
【0038】
本発明の方法は、eIF−5Aをコード化するポリヌクレオチドの投与、好ましくはヒトeIF−5A1の該癌を有する患者(哺乳類、好ましくはヒト)への投与、好ましくはeIF−5A1受入番号NM001970(図44参照)の投与を含む。eIF−5A2アイソフォーム(受入番号NM020390。ただしeIF−5A1が好ましい。)を使用してもよい。eIF−5Aは、当該技術分野で既知の任意の適切な方法で送達することができる。eIF−5Aは、生物学的に適切な媒体中のDNAのような裸のDNAとして、IVや皮下注射を介して、あるいはその他の任意の生物学的に適切な送達機構を介して送達され得る。あるいは、eIF−5Aは、プラスミド、アデノウイルスベクターのようなベクター、または任意の適切な発現ベクターの中で送達され得る。
【0039】
あるいは、上記DNAは、標的とする腫瘍または癌細胞へDNA(またはプラスミドまたは発現ベクター)の送達を提供する、リポソームやその他任意の適切な「担体」又は「ビヒクル」中で送達され得る。例えば合成DNA送達システムの考察について、Luo,Dan,et al.,Nature Biotechnology,Vol.18,January 2000,pp.33−37を参照のこと。本発明の発明者は、eIF−5A1は正常組織にとって有毒ではないと以前にも述べているが(参照することにより、そのすべてを本明細書に援用した、2005年11月28日出願の、係属出願第11/293,391号を参照のこと)、(eIF5Aポリヌクレオチド/プラスミド/発現ベクターの直接投与と比較して)送達システムが好ましい。好ましい送達システムは、有効量のeIF−5Aを腫瘍または一群の癌細胞に提供し、また好ましくは標的送達を腫瘍または一群の癌細胞に提供する。よって、eIF−5Aヌクレオチド/プラスミド/発現ベクターを、リポソーム、デンドリマー、類似する無毒性のナノ粒子のような、ナノメートルサイズのビヒクルを介して送達することが好ましい。さらに、該ビヒクルは、有効量のeIF−5Aヌクレオチド/プラスミド/発現ベクターを腫瘍または一群の癌細胞に送達する間に、好ましくはeIF−5Aヌクレオチド/プラスミド/発現ベクターを、早急過ぎるクリアランスや免疫応答を引き起こすことから保護する。例示的なビヒクルは、eIF−5Aヌクレオチド/プラスミド/発現ベクターと会合した単純なナノ粒子から、特異的細胞受容体を標的とするために表面にリガンドを結合させたペグ化リポソームのような、より複雑なペグ化ビヒクルまで様々であってもよい。
【0040】
リポソームおよびペグ化リポソームは当該技術分野において既知である。従来のリポソームにおいて、送達される分子(すなわち低分子量の薬剤、タンパク質、ヌクレオチドまたはプラスミド)は、リポソームの中心腔内に含まれる。当業者は、分子送達に有用な「ステルス」、標的化、および陽イオン性リポソームもあることを理解するであろう。例えば、Hortobagyi,Gabriel N.,et al.,J.Clinical Oncology,Vol.19,Issue 14(July)2001:3422−3433、およびYu,Wei,et al.,Nucleic Acids Research.2004,32(5);e48を参照のこと。リポソームは、静脈内注射することができ、また、血流における循環時間が長くなるように、その表面をより親水性にするため(2重層にポリエチレングリコールを付加(「ペグ化」)することにより)に修飾することができる。これらは「ステルス」リポソームとして知られており、ドキソルビシンやミトキサントロンなど親水性(水溶性)の抗癌剤の担体として特に有用である。薬剤運搬リポソームの特異的結合特性を腫瘍細胞のような標的細胞にまで広げるために、抗体、タンパク質、ペプチド等の特異的分子がリポソームの表面に結合していてもよい。例えば、癌細胞に存在する受容体に対する抗体は、リポソームに癌細胞を標的とさせるために使用され得る。多発性骨髄腫を標的とする場合には、例えば葉酸、IL−6、またはトランスフェリンを、リポソームに多発性骨髄腫細胞を標的とさせるために使用してもよい。
【0041】
デンドリマーもまた、当該技術分野において既知であり、好ましい送達ビヒクルを提供する。デンドリマーの考察について、例えば、Marjoros,Istvan,J.,et al,「PAMAM Dendrimer−Based Multifunctional Conjugate for Cancer Therapy:Synthesis,Characterization,and Functionality,」Biomacromolecules,Vol.7,No.2,2006;572−579、およびMajoros,IstvanJ.,et al.,J.Med.Chem,2005.48,5892−5899を参照のこと。
【0042】
またeIF−5Aは、腫瘍の部位に直接送達されてもよい。当業者は、eIF−5Aの送達に関して、用量および治療投与計画の期間を決定することが可能であろう。
【0043】
本発明の別の実施態様は、多発性骨髄腫細胞において細胞死を誘導する方法を提供する。多発性骨髄腫は、骨病変および腫瘍の悪化を引き起こす、高レベルの炎症性サイトカインを産生する骨髄癌の一種である。サイトカインIL−1BおよびIL−6は骨髄腫細胞の成長因子の役割を果たす。
【0044】
eIF−5A1をコード化するポリヌクレオチド(完全長コード領域)を含むアデノウイルスベクターコンストラクトを、多発性骨髄腫細胞株KAS6/1細胞に投与した。KAS6/1細胞株は、Mayo Clinicで作成され、Westendorf,JJ.,et al.,Leukemia(1996)10,866−876において報告されている。細胞株は、侵襲性多発性骨髄腫を罹患する患者からの分離株から直接作製した。本発明の発明者は、eIF−5Aを含むアデノウイルスコンストラクトをKAS6/1細胞に投与すると、対照ベクターのみを用いて処理された細胞と比較して(図1で「CTL」と表示)、死滅したあるいは死滅する細胞数が増加すること(より少ない生きた癌細胞を残す)(図41で「WT」と表示)を示した。図41参照。未処理の細胞の約25%に比べて、Factor5A1で処理した癌細胞は約90%が死滅した。従って、本発明の一実施形態は、eIF−5A1の発現を上方制御し、多発性骨髄腫細胞を死滅させるために、eIF−5A1をコード化するポリヌクレオチドを提供することにより骨髄腫細胞を死滅させる方法を提供する。
【0045】
また、対照(「CTL+IL−6」と表示)およびeIF−5Aコンストラクト(図41において「WT+IL−6」と表示)とともに、IL−6も投与した。IL−6は、骨髄腫細胞の成長因子の役割を果たすサイトカインである。結果は、たとえIL−6をeIF−5Aコンストラクトと同時投与しても、アポトーシスの増加を達成することができたことを示している。図41参照。これは、Factor5A1が骨髄腫細胞を死滅させるだけでなく、IL−6の存在下において骨髄腫細胞を除去することも可能であることを示している。IL−6の存在下で、デキサメタゾンのような標準的治療薬を用いて骨髄腫細胞にアポトーシスを誘導することは、非常に難しいことが証明されているため、この発見は興味深い。
【0046】
さらに、突然変異体eIF−5A(K50A)(50位の保存リジンを別のアミノ酸に変更するため、ハイプシン化されることができない)を含むアデノウイルスコンストラクトも、単独投与(「MUT」)またはIL−6と併用投与した(「MUT+IL−6」)。結果は、IL−6の存在下においても、対照細胞と比較して、eIF−5A1の突然変異体もアポトーシスを増加させることができたことを示す。図41を参照。従って、本発明の一実施形態は、eIF−5A1の突然変異体を投与することにより骨髄腫細胞を死滅させる方法を提供し、該突然変異体はハイプシン化されることができない。eIF−5A1の突然変異体は、骨髄腫細胞における細胞死を増加させる非ハイプシン化eIF−5A1の発現増加を引き起こす。
【0047】
IL−6は骨髄腫細胞の成長因子としての役割を果たすため、IL−6の発現を下方制御することも、骨髄腫細胞を死滅させる方法を提供する。本発明の発明者は、eIF−5A1に対するsiRNA(siRNAコンストラクトについては図43、46Aおよび46B参照)が、内因性eIF−5A1の発現を下方制御するだけでなく、種々の炎症促進性サイトカインの発現も下方制御すると示した。よって、本発明の一実施形態は、IL−1Bの内因性発現を下方制御するためにアポトーシス特異的eIF−5Aに対するsiRNAを投与することにより、骨髄腫細胞を死滅させる方法を提供し、それによってIL−6の発現を下方制御し、ひいては骨髄腫細胞の成長因子としての役割を果たすIL−6を減少させる。IL−6の発現を下方制御することにより、骨髄腫細胞の成長および生存を存続するために必要な、利用可能なIL−6はより少なくなる。
【0048】
別の実施形態において、eIF−5Aをコード化するポリヌクレオチドを、骨髄腫細胞のアポトーシス増加を提供するために、eIF−5Aに対するsiRNAと併せて投与する。eIF−5A1をコード化するポリヌクレオチドは、siRNAが外因性eIF−5A1の発現を阻害しないように、好ましくはアデノウイルスベクターなどのベクター中で投与する。例えば、siRNAは3’UTRを標的にするが、外因性eIF5A1をコード化するポリヌクレオチドは、好ましくは読み取り枠(ORF)全体を含み、したがってsiRNAによって標的とされる3’UTRを有しない。適切なsiRNAコンストラクトは、11/287,460;11/134,445;11/184,982;および11/293,391などの同時係属出願において先に説明されており、参照することにより、そのすべてを本明細書に援用する。図43、46Aおよび46Bも参照のこと。eIF−5A1は骨髄腫細胞において発現し、細胞死を引き起こす。さらに、eIF−5A1 siRNAは、eIF−5Aの内因性発現を減少させ、それによってIL−6の発現を減少させ、ひいては骨髄腫細胞における細胞死を増加させる。本方法において、上述したeIF−5Aの突然変異体もまたベクターコンストラクトに用いてもよい。
【0049】
また、本発明は、多発性骨髄腫細胞を死滅させる併用療法をも提供する。eIF5A、好ましくはeIF5A1をコード化するポリヌクレオチドを含む組成物は、標準的治療法と併せて投与されてもよい。eIF5A組成物は、標準的療法の前、途中、後に投与してもよい。eIF5Aは、医薬組成物として投与しても、または上述したように、送達ビヒクル内で投与してもよい。
【実施例】
【0050】
実施例1:In vitro実験
薬品
DHSの阻害剤であるN1−グアニル−1,7−ジアミノヘプタン(GC7、Biosearch Technologies)を濃度50μMで使用した。アクチノマイシンD(Calbiochem)を0.5または1.0μg/mlで使用した。ニトロプルシドナトリウムおよびデスフェリオキサミンをSigma社より購入し、それぞれ濃度3mMおよび500μMで使用した。ブレフェルジンAもSigma社から調達し、濃度4nMで使用した。
【0051】
細胞培養および処理
Anita Antes氏(ニュージャージー医科歯科大学)のご好意により頂いたヒト大腸腺癌細胞株HT−29を、細胞増殖およびeIF5A局在化研究に使用した。HT‐29細胞は、1mMのピルビン酸ナトリウム、10mMのHEPES、および10%のウシ胎仔血清(FBS)を添加したRPMI1640に維持した。その他の細胞株はすべてAmerican Type Culture Collectionより入手した。CCD112Coは、正常な大腸の繊維芽細胞株である。RKOは、野生型p53を含むヒト結腸直腸癌細胞株(CRL−2577)である。RKO−E6細胞株(CRL−2578)はRKO細胞株に由来する。安定的に組み込まれたヒトパピローマウイルスE6癌遺伝子を含み、従ってかなりの機能性p53腫瘍抑制タンパク質が欠損している。RKO、RKO−E6、A549および細胞株CCD112Coは、2mMのL−グルタミンと、1.5g/Lの炭酸水素ナトリウム、0.1mMの非必須アミノ酸、および1mMのピルビン酸ナトリウムを含むように調製したEarle’s Balanced Salt Solutionを含み、10%のFBSを添加したModified Eagle Minimum Essential Mediumで成長させた。細胞はCO2を5%含む加湿環境において37℃に維持した。
【0052】
プラスミドのクローニングおよび構築
RKO細胞から単離した総RNAから、製造者の付着細胞向けプロトコルに従ってGenElute Mammalian RNA Mini−Prep Kit(Sigma)を用いて、ヒトeIF5AをRT−PCR法によりクローン化した。使用したプライマーは、フォワードプライマー5’−CGAGTTGGAATCGAAGCCTC−3’、リバースプライマー5’−GGTTCAGAGGATCACTGCTG−3’であった。得られた532塩基対の生成物を、pGEM−T Easy (Promega)にサブクローニングして、配列決定した。得られたプラスミドを、フォワードプライマー5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー5’−CCTGAATTCCAGTTATTTTGCCATGG−3’を用いてPCRのテンプレートとして使用し、PCR生成物をpHM6(赤血球凝集素[HA]タグ付き、Roche Molecular Biochemicals)のHindIII及びEcoRI部位にサブクローンし、pHM6−eIF5Aベクターを生成した。eIF5AのC末端切断コンストラクト(pHM6−eIF5AA37)をPCR法により以下のプライマーを用いて生成した。フォワードプライマー:5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー:5’−GCCGAATTCTCCCTCAGGCAGAGAAG−3’。得られたPCR生成物をpHM6ベクターにサブクローンした。トランスフェクションを最適化するため、およびトランスフェクションがアポトーシスに与える影響を知るための対照として、pHM6−LacZベクター(Roche Molecular Biochemicals)を使用した。
【0053】
ノーザンブロット
RKO細胞を6ウェルプレート上にコンフルエントになるよう成長させ、アクチノマイシンD1.0μg/mlを用いて0、1、4、または8時間処理した。GenElute Mammalian RNA Mini−Prep Kit(Sigma)を用いて総RNAを細胞から単離し、5μgのRNAを1.2%のアガロース/ホルムアルデヒドゲル上で分画した。確立された方法に従い、eIF5Aの3’非翻訳領域(3’−UTR)と相同である32P標識cDNAで膜をプローブした。ノーザンブロットに用いたeIF5Aの3’−UTR cDNAは、次のプライマーを用いてRT−PCR法によりRKO細胞からクローン化した。フォワードプライマー:5’−GAGGAATTCGCTGTTGCAATCAAGGC−3’、リバースプライマー:5’−TTTAAGCTTTGTGTCGGGGAGAGAGC−3’。ノーザンブロットの負荷対照として使用したβ−アクチンcDNAを、次のプライマーを用いてRT−PCR法によりクローン化した。フォワードプライマー:5’−GATGATATCGCCGCGCTCGT−3’、リバースプライマー:5’−GTAGATGGGCACAGTGTGGGTG−3’。
【0054】
プラスミドのトランスフェクションおよびアポトーシスの検出
製造者の推奨プロトコルに従ってLipofectamine 2000(Invitrogen)を用いて、RKOおよびRKO−E6細胞をプラスミドDNAで一時的にトランスフェクトした。トランスフェクションから48時間後、製造者のプロトコルに従ってDNA Fragmentation Detection Kit(Oncogene Research Products)を用いて、末端デオキシリボヌクレオチジルトランスフェラーゼ仲介dUTP−ジゴキシゲニンニック末端標識法(TUNEL)により、断片化DNAを含むアポトーシス細胞を検出した。蛍光顕微鏡による分析のため、細胞を8ウェルのカルチャースライドグラス上でトランスフェクトし、4%のホルムアルデヒドで固定した後に、TUNEL法を用いて標識化し、Taylorら(2004)によって記述された方法に従って、Hoescht33258で染色した。
【0055】
siRNAのトランスフェクション
siRNAはすべてDharmacon社より入手した。eIF5A mRNAの3’UTR(受入番号BC085015)を標的にするeIF5A siRNAは次の配列を有していた。センス鎖:5’−GCUGGACUCCUCCUACACAdTdT−3’、アンチセンス鎖:3’−dTdTCGACCUGAGGAGGAUGUGU−5’。eIF5Aに対する第2のsiRNA(5A−2)は次の配列を有していた。センス鎖:5’−AGGAAUGACUUCCAGCUGAdTdT−3’、アンチセンス鎖:3’−dTdTUCCUUACUGAAGGUCGACU−5’。使用した対照siRNAは、eIF5A特異的siRNAの逆の配列を有しており、いかなる既知であるヒト遺伝子生成物に対する同一性も示さなかった。対照siRNAは次の配列を有していた。センス鎖:5’−ACACAUCCUCCUCAGGUCGdTdT−3’、アンチセンス鎖:3’−dTdTUGUGUAGGAGGAGUCCAGC−5’。Lipofectamine 2000を用いて細胞をsiRNA12でトランスフェクトし、増殖研究またはウエスタンブロットに使用した。
【0056】
ウエスタンブロット
ウエスタンブロット用のタンパク質は、沸騰している溶解緩衝液[SDS2%、50mMのTris−HCl(pH7.4)]を用いて単離した。Bicinchoninic Acid Kit(Sigma)を用いてタンパク質濃度を判定した。ウエスタンブロットするために、5μgの総タンパク質を12%のSDS−ポリアクリルアミドゲル上で分画し、二フッ化ポリビニリデン膜に移した。使用した1次抗体は、抗eIF5A(BD Transduction Laboratories、マウスIgG)および、抗β−アクチン(Oncogene、マウスIgM)で、両方とも5%乳汁中1:20,000の希釈度であった。2次抗体は、西洋わさびペルオキシダーゼ(HRP、Sigma)に結合した抗マウスIgGおよび抗マウスIgM−HRP(Oncogene)であった。抗体‐タンパク質複合体は、増感化学発光法(ECL、Amersham Biosciences)を用いて可視化した。eIF5A検出後、ECL Plus Western blotting検出システムの提供するプロトコルに従ってブロットをストリップに切断し、同等負荷を確認するために、抗β−アクチン抗体でプローブした。
【0057】
MAPK/SAPK経路分析に使用したA549細胞のライセートのウエスタンブロットは、MAPK溶解緩衝液(10mM Tris−pH7.4、2% SDS、10%グリセロール)中で収集したライセートを用いて行った。ライセート10μgを10%のSDS−PAGEゲル上で分離し、PVDF膜に移した。PBS中の無脂肪5%脱脂乳により膜をブロックし、PBSで洗浄し、1:1000の1次抗体を用いて、5%BSA/PBS−T中で振動を与えながら4℃で一晩インキュベートした。MAPK/SAPK抗体(P−p38、p38、P−JNK、JNK、P−ERK、ERK、p90RSK)は、Cell Signalingより購入した。A549の研究に使用したp53抗体もCell Signalingより入手し、同様の方法で用いた。
【0058】
2Dゲル電気泳動
2Dゲル電気泳動のために、低温の溶解緩衝液(7M尿素、2Mチオ尿素、30mMトリス、4%CHAPS、プロテアーゼ阻害剤カクテル)中でHT−29細胞ライセートを採取して、超音波処理し、遠心分離によりデブリを除去した。Bradford法を用いてタンパク質濃度を判定した。製造者の指示に従ってEttan IPGphor Isoelectric Focusing System(Amersham Biosciences)を用いて、1次元等電点電気泳動を行った。再水和緩衝液(8Mの尿素、2%のCHAPS、0.2%のDTT,0.5%のpH4〜7のIPG緩衝液、0.002%のブロモフェノールブルー)の中で、Immobiline DryStrips(7cm pH 4〜7、Amersham Biosciences)を細胞ライセートとともに室温で12時間再水和した。500Vで30分、1000Vで30分、および5000Vで1時間40分、等電点電気泳動を行った。IPGストリップゲル上のタンパク質をSDS−PAGEで分離し、PVDF膜(Amersham Biosciences)に移した。eIF5A抗体(BD Biosciences)を用いてウエスタンブロットを行った。
【0059】
アデノウイルスの生成
ヒトeIF5A、または、ハイプシン化を防ぐ単一の点突然変異(K50→A50)を含むeIF5A[eIF5A(K50A)]を発現するアデノウイルス(アデノウイルス5血清型、E1、E3−欠失)を、AdMax(商標) Hi−IQシステム(カナダ、トロントのMicrobix Biosystems Inc)を用いて構築した。eIF5A cDNA内に、PCR法を用いて部位特異的突然変異を作製した。プラスミドDNAをテンプレートとして用いて、PCR法でeIF5A cDNAを増幅し、アデノウイルスシャトルベクターpDC516(io)のSmaI部位に連結した。PCRプライマーの配列は、フォワードプライマー5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー5’−CCTGAATTCCAGTTATTTTGCCATGG−3’であった。アデノウイルスゲノムのプラスミドベクターpBHGfrt(del)E1,3FLPおよびシャトルベクターをE.coli DH5a中で増殖させ、Qiagen EndoFree Plasmid Mega Kitを用いて精製した。それぞれ5μgのアデノウイルスゲノムプラスミド pBHGfrt(del)E1、3FLP、シャトルベクターpDC516(io)−eIF5A、またはpDC516(io)−eIF5A(K50A)を、Microbix Biosystems Inc.推奨のCaCl2法を用いて、60mm培養皿内の60〜80%コンフルエントな293−IQ細胞(Microbix Biosystems)にトランスフェクトした。37°Cで7〜10日間培養した後プラークが現れ、得られたアデノウイルス粒子[Ad−eIF5A、およびAd−eIF5A(K50A)]を293−IQ細胞内で増殖した。純粋な、力値の高いアデノウイルス株を、Microbix Biosystems Inc.提供の製造者プロトコルに従って、CsCl勾配超遠心法を用いて調製した。LacZを発現するアデノウイルスベクター(Ad−LacZ;血清型5、E1,E3−欠失)を、Qbiogene(米国カリフォルニア州)より購入し、これらの実験における対照およびレポーターとして使用した。Ad−LacZアデノウイルスをAd−eIF5AおよびAd−eIF5A(K50A)ウイルスと同様の方法で増幅および精製した。
【0060】
アデノウイルス感染およびアネキシンV標識
100mm組織培養プレート内に、HT−29、HTB−9、またはHTB−4細胞を、プレートあたり1×106細胞で播種し、翌日、5mlのRPMI1640+2%FBS中において、細胞あたり3000感染単位でアデノウイルスに感染させた。4時間後、細胞にさらなる培地を追加し、FBS濃度を10%になるよう調製した。感染から24、48、または72時間後、細胞をトリプシン処理により剥離し、洗浄してから製造者のプロトコル(BD Biosciences)に従ってAnnexin V−FITCで染色した。フルオレセイン検出のための488nmアルゴンレーザー光源及びフィルターを用いたフローサイメトリー(Coulter Epics XL−MCL)により細胞をソートし、WinMDI2.8でデータを分析した。
【0061】
HT‐29細胞を、細胞あたり3000感染単位を用いて感染させ、細胞あたり1500感染単位でA549細胞を用いた実験を行った。
【0062】
増殖アッセイ
Lipofectamine 2000(Invitrogen)を用いて、siRNAでHT‐29細胞を96ウェルプレート上でトランスフェクトした。XTT Cell Proliferation Kit(Roche Applied Science)を用いて、増殖する細胞の代謝活性を測定した。DNA合成を測定するためには、製造者のプロトコルに従ってBrdU Cell Proliferation Kit(Roche Applied Science)を用いた。アデノウイルス感染後に実行したXTTアッセイのために、96ウェルプレート内に、ウェルあたり5000個の細胞を播種した。翌日、Ad−lacZ、Ad−eIF5A1、およびAd−eIF5A1(K50A)(Ad−eIF5A1M)をそれぞれ用いて細胞を感染させ、未処理の細胞を負の対照とし、アクチノマイシンDで処理した細胞を正の対照とした。XTT基質を追加し、A690nmを基準としてA475nmを測定した。
【0063】
間接免疫蛍光法
ポリ−L−リジンを被膜したカバーガラス上でHT‐29細胞を培養した。サブコンフルエントな状態の細胞を、200 単位のインターフェロンガンマ(IFN−γ、Roche Applied Science)を用いて16時間インキュベートした後、TNF−α(100ng/ml、Leinco Technologies)で10分〜8時間の異なる時間インキュベートした。あるいは、30分から16時間まで時間を増加させながら、1.0μg/mlのアクチノマイシンDを用いて細胞を処理した。処理済の細胞を3%のホルムアルデヒド(メタノールを含まない;Polysciences Inc.)で20分間固定し、PBSで5分間、2回洗浄し、100mMのグリシンを含むPBSで5分間1回洗浄し、PBS中の0.2%のTriton X−100で4分間透過処理した。その後、標準プロトコルを用いて免疫蛍光検査のために細胞を標識化した。1次抗体は、抗−eIF5A(BD Transduction Laboratories、マウスIgG)を用いて、1:250の希釈度で1時間インキュベートした。2次抗体は、抗マウスIgG−AlexaFluor 488(Molecular Probes)で、希釈度1:200で1時間使用した。抗体標識に続いて、Hoescht33258で核を染色し、標識化した細胞を蛍光顕微鏡で観察した。
【0064】
Matrigel(商標)細胞浸潤アッセイ
A549細胞を、アデノウイルスを用いて細胞あたり1500感染単位で感染させ、24時間インキュベートした。トリプシンで細胞を剥離して、無血清培地で洗浄し、Matrigel(商標) Basement Membrane Matrix (BD Biosciences)で事前に被膜しておいたトランスウェル(Falcon 8.0μm セルカルチャーインサート)上に、ウェルあたり30,000個の細胞を無血清培地中に播種した。10%のFBSを含む培地を下のウェル(トランスウェルが乗っている24ウェルプレートのウェル)に播種し、さらに細胞を24時間インキュベートした。インキュベーション後、トランスウェルの上部チャンバから培地を除去し、トランスウェルを外して500マイクロリットルのクリスタルバイオレットを含む24ウェルプレートのウェル内に置いた。トランスウェルを色素の中で20分間インキュベートしてから、ビーカーに入れた水にトランスウェルを何回も浸漬することにより洗浄した。あらかじめ濡らしておいた綿棒を用いてトランスウェルの上面から細胞をこすり取った。トランスウェルの底面まで移動した細胞を、光学顕微鏡を用いて観察し、写真を撮影し、領域ごとに移動した細胞数を数えた。図24参照。
【0065】
統計
統計分析にはスチューデントのt‐検定を用いた。信頼度95%超(P<0.05)で有意性を判定した。
【0066】
実施例2:In vivo実験
マウスおよび腫瘍の確立
カナダのケベック州にあるCharles Riverより、5〜7週齢のC57BL/6マウスを購入した。実験開始前の1週間、マウスを馴化させた。B16F10マウスメラノーマ細胞をATCCより購入し、10%FBS含有DMEM内で培養した。細胞の単層をトリプシン処理し、10%FBS含有MEMで中性化した。細胞をPBSで2回洗浄し、細胞の生存率をトリパンブルー染色によって判定した。実験的転移の実験(実験IIおよびIII)のため、B16F10細胞を6週齢マウスに尾静脈注射することにより、メラノーマ腫瘍を肺の中に確立した。PBS中で、B16F10細胞を1×106生細胞/mlまで希釈した。200ulの細胞を各マウスの尾静脈から注射した。皮下腫瘍実験(実験IVおよびV)のために、500,000個のB16F10細胞を10〜14週齢のマウスの右側腹部に皮下注射してメラノーマ腫瘍を確立した。実験がすべて終了した時(マウスが瀕死状態となった時、または腫瘍が所定の大きさを超えた時)に、CO2吸入によりマウスを安楽死させた。
【0067】
実施例3:実験II
プラスミドDNAの構築および精製
CpGジヌクレオチドを欠損するpCpG−lacZ発現ベクターを米国サンディエゴのInvivoGenより購入した。まずNcoIおよびNheIでプラスミドを消化させ、3.1kbのpCpGベクターバックボーンを単離し(LacZコード化配列を除去)、HAタグを含むeIF5A1のPCRで増幅したcDNAと結合させて(pCpG−HA5A1)、HAタグ付きeIF5A1 cDNAを、pCpG−lacZベクターにサブクローンした。PCRプライマーは次の通りであった。eIF5A1フォワード:HA−5A1フォワード:5’−GCTCCATGGATGTACCCATACGACGTCCC−3’、eIF5A1リバース:5’−CGCGCTAGCCAGTTATTTTGCCATCGCC−3’。25μg/mlのゼオシンを含むLB培地または2XYT培地で培養したE.coli GT115中で、pCpG−lacZおよびpCpG−HA5A1を増幅した。プラスミドを抽出し、QIAGEN Endofree Plasmid Giga kitを用いて精製した。波長260nmのUV吸収法およびアガロースゲル電気泳動法を用いてDNA濃度を測定した。
【0068】
プラスミドDNAの尾静脈注射(実験II)
1×PBS(体重に基づいて約200μl)に溶解したプラスミドDNAを、2、4、7、11、16、21、26、31日目に、マウスの尾静脈に注射した。プラスミドDNAの濃度は、2×(6.6mg/kg)に対して660ng/μl、1×(3.3mg/kg)に対して330ng/μl、0.1×(0.33mg/kg)に対して33ng/μlであった。
【0069】
体重および肺重量
尾静脈注射の前、または毎週月曜および金曜日に体重を測定した。マウスが瀕死状態(嗜眠性、呼吸困難)になった時にCO2を用いて安楽死させ、肺を摘出して重量を測り、写真を撮ってから冷凍して−70℃で保管した。図25〜27参照。
【0070】
VEGF ELISA
採取した肺組織をPBSで洗浄し、ドライアイスの中で冷凍し、−70℃で保管した。粉砕した肺組織からタンパク質ライセートを単離した。50μgの肺組織タンパク質を使用して、Mouse VEGF Immunoassay kit(米国ミネアポリス州、R&D Systems,Inc.)を用いて、マウス肺中のVEGF濃度を判定した。
【0071】
実施例4:実験III
プラスミドDNA/DOTAP複合体の注射
6週齢のマウスの尾静脈に、B16F10メラノーマ細胞(マウス1匹あたりPBS200μl中に50,000個の細胞)、プラスミドDNA、および50μgのエンドトキシンフリーキットで精製したプラスミドDNAを含むDNA担体複合体を注射し、80μgのDOTAPを7、14、および21日目にマウスの尾静脈に注射した。25日目にマウスを屠殺した。肺を摘出し、重量を測定して写真を撮影した。図28〜29参照。
【0072】
実証例5:実験IV
アデノウイルスコンストラクトの注射およびTUNEL法
500,000個のB16F0またはB16F10細胞を、10週齢のC57BL/6マウスの右側腹部に皮下注射した。腫瘍の大きさが直径約4mmに到達した時(B16細胞注射から10〜12日後)、PBS50ul中の1×109pfu Ad−5A1を腫瘍内に注射した。48時間後にマウスを屠殺し、腫瘍を摘出して固定し、パラフィンで包埋した。製造者のプロトコルに従ってTUNEL(Promega)を用いて、各細胞の腫瘍の種類ごとに(Ad−5A1−1およびAd−5A1−2)2ヶ所染色した。細胞の種類ごとに、TUNEL反応の後TdT酵素が残った、負の対照スライド(Ad−5A1陰性)を示した。図30参照。
【0073】
実施例6:実験V
腫瘍の確立
14週齢のC57BL/6マウスの右側腹部に、(PBS100ul中の)500,000個のB16F10細胞を皮下注射した。腫瘍の大きさが直径約8mmに到達するまで、毎日腫瘍形成の経過を調べた。
【0074】
アデノウイルスを用いた注射
腫瘍の直径が8mmに到達した時に治療を開始した。Ad−eIF5A1またはAd−LacZの1×109プラーク形成単位(pfu)をマウスに注射した。注射は腫瘍の3つの部位に分散させた。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。緩衝液のみ与えた緩衝液マウスには、アデノウイルスを懸濁させた緩衝液のみを与えた(10mM Tris−HCl pH 7.4、10 mMのMgCl2、10%グリセロール)。キャリパーを用いて毎日腫瘍の大きさを計測し、次の方程式を用いて腫瘍体積を算出した。
腫瘍体積(mm3)=L*W2*0.52
L=長さ、W=幅(常に短いほうの寸法)
図31および32参照。
【0075】
実施例7
A549肺癌細胞を、LacZ(Lac)またはeIF5A1(5A)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのp38阻害剤SB203580(Calbiochem)、10μMのJNK阻害剤II(Calbiochem)、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、EGFで30分間細胞を処理し、細胞ライセートを採取した。総p53(p53)、セリン15位におけるリン酸化p53[P−p53(ser15)]、またはセリン37位におけるリン酸化p53[P−p53(ser37)]に対する抗体のいずれかを用いて、ライセートにウエスタンブロットを行った。図33参照。
【0076】
図33に示す結果は、感染から48時間後までにAd−eIF5A1がp53の堆積を誘発することを表している。また、図は、Ad−eIF5A1が感染から48時間後までにp53のリン酸化を誘発すること、p53の堆積およびリン酸化がMEKの阻害剤によって阻害されること、p53の堆積およびリン酸化がp53活性の阻害剤によって阻害されること、eIF5A1は、p53のMEK依存リン酸化を刺激すること、そしてeIF5A1が、p53のp53依存性の堆積を刺激することを示している。
【0077】
実施例8
A549肺癌細胞を、LacZ(Ad−LacZ)、またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。48時間後、総RNAを細胞から単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53のレベルおよびbax mRNAの転写産物レベルを判定した。p53プライマーは、5’−CGCTGCTCAGATAGCGATGGTC−3’(5’−プライマー)、および5’−CTTCTTTGGCTGGGGAGAGGAG−3’(3’−プライマー)[これらのp53プライマー配列は、Li et al.(2004).A Novel eIF5A/complex Functions as a Regulator of p53 and p53−dependent Apoptosis,J Biol.Chem.279 49251−49258より入手した]であった。eIF5A1の過剰発現がmRNAレベルでp53の堆積を誘発するが、baxへの影響は見られなかったことが分かる、図34を参照のこと。
【0078】
実施例9
A549肺癌細胞を、LacZ(Ad−LacZ)またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞から総RNAを単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53転写産物レベルを判定した。p53プライマーは、5’−CGCTGCTCAGATAGCGATGGTC−3’ (5’−プライマー)、および5’−CTTCTTTGGCTGGGGAGAGGAG−3’(3’−プライマー)[これらのp53プライマー配列は、Li et al.(2004).A Novel eIF5A/complex Functions as a Regulator of p53 and p53−dependent Apoptosis,J Biol.Chem.279 49251−49258より入手した]であった。p53のeIF5A1依存性の堆積がp53の転写活性に依存しており、eIF5A1に反応して生じるp53タンパク質の上方制御が、p53 mRNAの転写を増加させていることが分かる、図35を参照のこと。
【0079】
実施例10
A549肺癌細胞を、LacZ(Ad−LacZ)またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地を、DMSO、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞から総RNAを単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53転写産物レベル濃度を判定した。TNFR1プライマーは、TNFR1−F 5’ATCTCTTCTTGCACAGTGG 3’、およびTNFR1−R 5’CAATGGAGTAGAGCTTGGAC 3’であった。TNFR1 mRNAレベルがAd−eIF5A1の感染によって上方制御されること、また、このTNFR1 mRNAの堆積が一部MEKに依存することを示す、図36を参照のこと。このTNFR1 mRNAの堆積はp53の転写活性に依存するものである。
【0080】
実施例11
A549肺癌細胞を、LacZ(Lac)またはeIF5A1(5A)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのp38阻害剤SB203580(Calbiochem)、10μMのJNK阻害剤II(Calbiochem)、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞を採取し、Annexin/PI染色(BD Bioscience)を用いてアポトーシス中の細胞の割合を判定し、フローサイメトリーによる分析を行った。図37参照。
【0081】
図37は、感染から48時間後までにAd−eIF5A1がアポトーシスを誘発すること、JNKの阻害がeIF5A1によって誘導されたアポトーシスを増加させること、MEKの阻害がeIF5A1によって誘導されたアポトーシスを増加させること、そして、p53の阻害がeIF5A1によって誘導されたアポトーシスを減少させることを示している。
【0082】
実施例12
50,000個のB16−F0メラノーマ細胞をマウスに皮下注射した。腫瘍の大きさが約5×5mm(65mm3)に到達した時、腫瘍内注射を開始した。50〜100μlのPBS/10%グリセロール緩衝液で希釈した、1×109pfuのAd−lacZ(群2)、Ad−eIF5A1(群3)、またはAd−eIF5A2(群4)あるいは緩衝液のみ(群1)を腫瘍内の3つの部位に1日置きに注射した。一日置きに腫瘍の大きさを測定し、腫瘍の大きさが体重の10%に到達した時、マウスを屠殺した。図39参照。
【0083】
実施例13
50,000個のB16−F0メラノーマ細胞をマウスに皮下注射した。腫瘍の大きさが約5×5mm(65mm3)に到達した時、腫瘍内注射を開始した。50〜100μlのPBS/10%グリセロール緩衝液で希釈した、1×109pfuのAd−lacZ(群2)、Ad−eIF5A1(群3)、またはAd−eIF5A2(群4)あるいは緩衝液のみ(群1)のいずれかを、腫瘍内の3つの部位に、1日おきに注射した。腫瘍の大きさが体重の10%に到達した時、マウスを屠殺した。図40参照。
【0084】
実施例14:多発性骨髄腫
第0日目、KAS6/1多発性骨髄腫細胞を、3000IFU/細胞の野生型または突然変異体のeIF5a(保存リジンが突然変異しているため、ハイプシン化されることができない)アデノウイルスベクターコンストラクトに4時間感染させた。感染後の培養培地に存在するIL−6を含む、あるいは含まない、3つの複写物を設定した。また、対照(感染していない)のためにKAS細胞を播種した。
【0085】
第2日目および4日目に、MTTおよびアネキシン/PIアッセイを行った。上清を採取した。結果を図41に示す。
【図面の簡単な説明】
【0086】
【図1】図1は、eIF5A1の発現が、遺伝毒性ストレスおよび一酸化窒素によって増加することを示す。A)アクチノマイシンD0.5μg/mlで0、1、4、8および24時間処理した、正常な大腸繊維芽細胞における、eIF5Aの発現のノーザンブロット(上のパネル)およびウエスタンブロット(下のパネル)分析。ウエスタンブロットは、eIF5A、p53、およびβ−アクチンに対する抗体でプローブした。B)3mMのニトロプルシドナトリウムで、0、2、4、8および24時間処理したRKO細胞における、eIF5A1の発現のノーザンブロット(上のパネル)およびウエスタンブロット(下のパネル)分析。ウエスタンブロットは、eIF5Aおよびβ‐アクチンに対する抗体でプローブした。RKO細胞は有意なレベルでeIF5A2を発現しない。また、eIF−5A2のサイズは、eIF−5A1よりもアミノ酸1個長いだけであるが、SDS PAGEゲル上で高い位置を流れるため、eIF5A2が発現した場合、eIF−5A1とは別のバンドとして見えることになる。
【図2】図2は、eIF5A1発現の抑制が、細胞増殖に対して何の影響も与えないことを示す。A)eIF5A1 siRNAまたは対照siRNAを用いたトランスフェクションから72時間後にHT−29細胞から単離した細胞ライセートのウエスタンブロット。2つの独立した実験からのウエスタンブロットを示す。ブロットは、eIF5Aおよびβ−アクチンに対する抗体でプローブした。B)eIF5A1 siRNAを用いてトランスフェクトした細胞の代謝活性を、XTT細胞増殖アッセイを用いて測定した。対照siRNAまたはeIF5A1 siRNAを用いたトランスフェクションの24時間前に、HT−29細胞を96ウェルプレートに播種した。トランスフェクションから24時間後、細胞を未処理のまま放置するか、または、代謝活性測定の前に、アクチノマイシンD(1.0μg/ml)を用いて48時間処理した。値は、4通り行われた2つの実験からの平均値であり、1に設定された0時間対照のために得られた値まで正規化した。C)対照またはeIF5A1 siRNAを用いてトランスフェクトしたHT−29細胞の増殖能を、50μMのGC7とともに72時間インキュベートした細胞の増殖能と比較した。細胞増殖はBrdU取り込みによって測定した。値は、n=4に対して平均値±SEで、1に設定したGC7(+)血清サンプルの値に合わせて正規化した。アスタリスク(*)は、対応のあるスチューデントのt検定(p<0.01)により、有意に差があると思われた値を示す。対照siRNAまたはeIF5A siRNAのどちらかを用いてトランスフェクトしたHT−29細胞のトランスフェクションの日(第0日目)からトランスフェクション後5日目までのD)XTTおよびE)BrdU取り込み細胞増殖アッセイ。第0日目の値は1に設定した。
【図3】図3は、eIF5A1が、アクチノマイシンDに反応してp53の発現を制御することを示す。RKO細胞を、対照siRNA(C)またはeIF5A siRNA(5A−1)のいずれかを用いてトランスフェクトした。トランスフェクションから72時間後、アクチノマイシンD0.5μg/mlを用いて、0、4、8、または24時間細胞を処理した。A)eIF5A、p53、またはβ−アクチンに対する抗体を用いてブロッティングした細胞ライセートのウエスタンブロット。結果は3つの独立した実験を代表するものである。B)対応するアクチンバンドまで正規化したウエスタンブロットにおけるp53の相対強度のプロット。値は最小値n=3に対して平均値±SE。
【図4】図4は、ヒトeIF5A1の過剰発現が、p53から独立してアポトーシスを誘導することを示す。RKO細胞(A)またはRKO−E6細胞(B)を、pHM6−LacZ、pHM6 eIF5A、またはpHM6−eIF5A△37(C末端側で37位のアミノ酸を切断)を用いてトランスフェクトした。トランスフェクションから48時間後、TUNEL法を用いて細胞を固定し、標識化した。核は、Hoescht33258で染色し、標識化した細胞を蛍光顕微鏡を用いて観察した。明るい緑色に染色された細胞を、アポトーシス性であるとスコア化した。Hoeschtで染色した核を総細胞数を決定するのに使用した。値はn=4(A)またはn=3(B)についての平均値±SE。アスタリスク(*)は、対応のあるスチューデントのt検定(p<0.02)による、対照(pHM6−LacZ)からの有意差を示す。C)アクチノマイシンD0.5μg/mlで処理してから0、4、8、および24時間後に採取したRKOおよびRKO−E6細胞ライセートのウエスタンブロット。ブロットは、p53およびβ−アクチンに対する抗体を用いてプローブした。
【図5】図5は、eIF5A1アデノウイルスコンストラクトが大腸癌細胞にアポトーシスを誘導することを示す。A)Ad−LacZ(L)、AdeIF5A1(5A)、またはAd−eIF5A(K50A)(K50A)で感染させてから72時間後にHT−29細胞から単離した、細胞ライセートのウエスタンブロット。B)GC7またはDFOで72時間処理した後、またはアデノウイルスコンストラクトに感染させてから72時間後、eIF5Aに対する抗体を用いてウエスタンブロットを行った後、HT−29細胞から単離した、細胞ライセートの2Dゲル電気泳動。eIF5Aを過剰発現するアデノウイルス感染細胞からのライセートから0.3μgのタンパク質が分離されたことを除いては、7μgのタンパク質が分離された。C)上のパネル:アデノウイルスコンストラクト感染から48時間後のHT−29細胞のTUNEL染色。下のパネル:Hoechst染色した同じ領域内の細胞の核。すべての写真は400倍の倍率で撮影した。結果は3つの独立した実験を代表するものである。D)アデノウイルスコンストラクト感染から48時間後のHT−29細胞のアポトーシスの割合。値は、n=3に対して平均値±SE。E)アデノウイルスコンストラクト感染から7日後の、HT−29細胞のXTT細胞増殖アッセイ。値は、n=4に対して平均値±SE。
【図6】図6は、eIFA1アデノウイルスコンストラクトに感染したHT‐29細胞のアネキシンVおよびPI染色を示す。A)アデノウイルスコンストラクト感染から48時間後のHT‐29細胞のアネキシン−FITCおよびヨウ化プロピジウム(PI)標識のヒストグラム。B)アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染後24、48、および72時間後、またはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンAで処理した後の、HT−29細胞アポトーシスの割合。値はn=2に対して平均値±SE(事象数>5000)。
【図7】図7は、免疫蛍光法によるeIF5A1の局在化を示す。IFN−γおよびTNF−α(A)またはアクチノマイシンD(B)で刺激を与えたHT‐29細胞内におけるeIF5Aタンパク質の細胞内局在を、間接免疫蛍光法によって判定した。Hoechst33258を核の染色に使用した。A)TNF−αで0分[(−)TNF−α]、10分、または30分間刺激する前に、HT‐29細胞を未処理のまま放置するか、あるいはIFN−γで16時間予備刺激した。上のパネル:eIF5A1の免疫蛍光検出。中央のパネル:Hoechst染色した同領域内にある細胞の核。下のパネル:合併したイメージ。B)HT‐29細胞を未処理のまま放置するか、あるいはアクチノマイシンDで0.5時間、1.5時間、または4時間処理した。上のパネル:eIF5A1の免疫蛍光検出。中央のパネル:Hoechst染色した同領域内にある細胞の核、下のパネル:合併したイメージ。すべての写真は400倍の倍率で撮影した。結果は3つの独立した実験を代表するものである。
【図8】図8は、eIF5A1が、アクチノマイシンDに反応してp53の発現を制御することを示す。RKO−E6細胞はp53を発現しない。A)RKOまたはRKO−E6細胞を、アクチノマイシンD0.5μg/mlで1、4、8、または24時間処理した。細胞ライセートを採取し、ウエスタンブロットを用いてp53の発現について分析した。B)対照siRNA(C)、eIF5A siRNA(5A−1)、またはeIF5Aの異なる領域を標的とする第2のeIF5A siRNA(5A−2)のいずれかを用いて、RKO細胞をトランスフェクトした。トランスフェクトから72時間後、アクチノマイシンD0.5μg/mlで0、4、8、または24時間細胞を処理した。A)eIF5A、p53、またはβ−アクチンに対する抗体でブロットした細胞ライセートのウエスタンブロット。結果は3つの独立した実験を代表するものである。
【図9】図9は、アポトーシスの間、デオキシハイプシン化したeIF5A1が堆積することを示す。この図は、アクチノマイシンD(A)または作動薬Fas抗体(B)で、1〜24時間処理してから、eIF5Aに対する抗体でウエスタンブロットした後、HT‐29細胞から単離した細胞ライセートの2Dゲル電気泳動を提供する。
【図10】図10は、Ad−eIF5A1またはAd−eIF5A1(K50A){Ad−eIF5A1M)による感染が、HT−29結腸直腸腺癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HT−29細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図11】図11は、Ad−eIF5A1またはAd−eIF5A2による感染が、HTB−9膀胱癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HTB−9細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図12】図12は、Ad−eIF5A1またはAd−eIF5A2による感染が、HTB−4膀胱癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HTB−4細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図13】図13は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HTB−4膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−4細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図14】図14は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染がHTB−9膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−9細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図15】図15は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HTB−1膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−1細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図16】図16は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、UMUC−3膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、UMUC−3細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図17】図17は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HeLa子宮頚部腺癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HeLa細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図18】図18は、Ad−eIF5A1、Ad−eIF5A1(K50A)(Ad−eIF5A1M)、またはAd−eIF5A2による感染が、HTB−4細胞にPARP切断を誘発することを示す。この図は、アデノウイルスコンストラクト感染から48時間または72時間後に、HTB−4細胞から単離された細胞ライセートを、PARP、eIF5A、およびβ−アクチンに対する抗体を用いてウエスタンブロットした結果を示す。
【図19】図19は、Ad−eIF5A1、Ad−eIF5A1(K50A)(Ad−eIF5A1M)、またはAd−eIF5A2による感染が、HTB−1細胞にPARP切断を誘発しないことを示す。この図は、アデノウイルスコンストラクト感染から48時間または72時間後に、HTB−1細胞から単離した細胞ライセートを、PARP、eIF5A、およびβ−アクチンに対する抗体を用いてウエスタンブロットした結果を示す。
【図20】図20は、A549肺癌細胞におけるeIF5A1の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、感染効率(MOI)を次第に高くしながらAd−eIF5A1(5A)に感染させた。比較のために、Ad−LacZ(Lac)を用いて細胞を感染させた。感染から48時間後、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図21】図21は、A549肺癌細胞におけるeIF5A1の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、Ad−eIF5A1(5A)またはAd−LacZ(L)を用いて50MOIで感染させた。感染から6、24、48、または72時間後に、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図22】図22は、ハイプシン化されることのできないeIF5A1の突然変異体の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、感染効率(MOI)を次第に高めながらAd−eIF5A1(K50A)(M)に感染させた。比較のために、Ad−eIF5A1(5A)およびAd−LacZ(Lac)を用いて細胞を感染させた。MEK阻害剤U1026を用いて、あるいは用いずに細胞をインキュベートした。感染から48時間後、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図23】図23は、A549肺癌細胞におけるeIF5A1または突然変異体eIF5A1(K50A)の過剰発現が、p53の発現を増加させることを示す。A549細胞は感染効率(MOI)を次第に高くしながらAd−eIF5A1(5A)またはAd−eIF5A1(K50A)(M)を用いて感染させた。比較のために、Ad−LacZ(Lac)に細胞を感染させた。MEK阻害剤U1026を用いて、あるいは用いずに細胞をインキュベートした。感染から48時間後、EGFで30分間細胞を処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図24】図24は、eIF5A1または突然変異体eIF5A1(K50A)の過剰発現は、A549肺癌細胞がMatrigel(商標)細胞外基質に浸潤する能力を減少させることを示す。アデノウイルスコンストラクト感染から24時間後、A549細胞をMatrigel(商標)を塗布した無血清培地のトランスウェルに播種した。血清含有培地は、化学誘引物質として作用するように下のウェルに配置した。播種から24時間後、トランスウェルの下部表面まで浸潤した細胞を、クリスタルバイオレットで染色し、光学顕微鏡下で細胞を数えることにより、領域あたりの平均細胞数を判定した。サンプルあたり最低6個の領域が数えられた。
【図25】図25は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、eIF5A1によって肺腫瘍量が有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した。
【図26】図26は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、eIF5A1によって肺重量が有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して重量を測定した。
【図27】図27は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、VEGF発現がeIF5A1によって有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して重量を測定した。肺重量が判明してからその肺を冷凍し、後にVEGF ELISAに使用した。肺組織は溶解緩衝液中で粉砕し、ライセート中に存在するVEGFの量をELISA法によって測定した。
【図28】図28は、実験IIIの結果を示す。DNAをDOTAPと複合化することにより、eIF5A1遺伝子療法の抗腫瘍効果が向上した。6週齢のC57BL/6マウスの尾静脈に、50,000個のB16F10細胞を注射した(第0日目)。7,14、および21日目に尾静脈注射を行う前に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAをDOTAPと複合化した。プラスミドDNAを含まないDOTAPを注射した腫瘍の無いマウスを、DOTAPの効果の対照として使用した。マウスは25日目に屠殺し、肺を摘出して写真を撮影した。
【図29】図29は、実験IIIの結果を示す。DNAをDOTAPと複合化することにより、eIF5A1遺伝子療法の抗腫瘍効果が向上した。6週齢のC57BL/6マウスの尾静脈に、50,000個のB16F10細胞を注射した(第0日目)。7,14、および21日目に尾静脈注射を行う前に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAをDOTAPと複合化した。プラスミドDNAを含まないDOTAPを注射した腫瘍の無いマウスを、DOTAPの効果の対照として使用した。マウスは25日目に屠殺し、肺を摘出して重量を測定した。
【図30】図30は、実験IVの結果を示す。Ad−eIF5A1の注射はB16F0およびB16F10腫瘍にアポトーシスを誘導する。500,000個のB16F0またはB16F10細胞を、C57BL/6マウスの右側腹部に皮下注射した。腫瘍の直径が約4mmに到達した時、50μlのPBS中の1×109pfu Ad−5A1を腫瘍に注射した(B16細胞注射から10〜12日後)。マウスは48時間後に屠殺し、腫瘍を切除して、固定し、パラフィンに包埋した。各細胞腫瘍の種類(Ad−5A1−1およびAd−5A1−2)に対して2ヶ所を、製造者のプロトコルに従ってTUNEL(Promega)を用いて染色した。TUNEL反応からTdT酵素が残った陰性対照のスライド(Ad−5A1陰性)を、各細胞の種類ごとに含ませた。
【図31】図31は、実験Vの結果を示す。Ad−eIF5A1の注射により腫瘍成長が有意に遅延した。皮下にB16−F10を有するC57BL/6に、1×109pfuのAd−eIF5A1またはAd−LacZまたは等体積の緩衝液を注射した。腫瘍の直径が約8mmに到達した時に治療を開始し、その日を「第0日目」に指定した。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。各群につきマウス3匹とした。群1のマウス(1−1、1−2、1−3)はAd−eIF5A1で処理した。群2のマウス(2−1、2−2、2−3)にはAd−LacZを注射し、群3(3−1、3−2、3−3)のマウスには緩衝液のみを与えた。キャリパーを用いて毎日腫瘍の大きさを計測し、腫瘍体積を推測するために使用した。
【図32】図32は、実験Vの結果を示す。Ad−eIF5A1の注射により生存期間が有意に伸びた。皮下にB16−F10を有するC57BL/6に、1×109pfuのAd−eIF5A1またはAd−LacZまたは等体積の緩衝液を注射した。腫瘍の直径が約8mmに到達した時に治療を開始し、その日を「第0日目」に指定した。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。各群につきマウス3匹とした。群1のマウス(1−1、1−2、1−3)はAd−eIF5A1で処理した。群2のマウス(2−1、2−2、2−3)にはAd−LacZを注射し、群3(3−1、3−2、3−3)のマウスには緩衝液のみを与えた。
【図33】図33は、eIF−5A1が、A549肺癌細胞におけるp53腫瘍抑制タンパク質の堆積およびリン酸化を増加することを示す。
【図34】図34は、eIF−5A1が、A549肺癌細胞におけるp53 mRNAレベルを上昇させることを示す。
【図35】図35は、p53レベルの上昇は、A549肺癌細胞におけるp53の転写活性に依存することを示す。
【図36】図36は、TNFR1 mRNAレベルは、A549肺癌細胞においてeIF−5A1に感染することにより上方制御されることを示す。
【図37】図37は、eIF−5A1が、A549肺癌細胞におけるアポトーシスを誘導し、MEK阻害剤の使用が、eIF−5A1によって誘導されたアポトーシス量を増加することを示す。
【図38】図38は、eIF−5A1の仮説モデルが、MAPK/SAPK経路およびA549肺癌細胞におけるアポトーシスに影響することを示す。
【図39】図39は、腫瘍成長の抑制において、eIF−5A1がeIF−5A2より優れていることを示す(マウス異種移植モデル(B16−FOメラノーマ細胞))。
【図40】図40は、マウスの生存期間延長において、eIF−5A1がeIF−5A2より優れていることを示す(マウス異種移植モデル(B16−FOメラノーマ細胞))。
【図41】図41は、IL−6を含む、あるいは含まない対照を用いて処理された細胞と比較すると、eIF−5Aが、死滅したあるいは死滅する細胞数を増加することを示す。
【図42】図42は、eIF−5A2の配列を提供する。
【図43】図43は、eIF−5A1 siRNAの位置および配列を提供する。
【図44】図44は、ヒトeIF−5A1およびヒトeIF−5A2のヌクレオチドアラインメントを提供する。
【図45】図45は、ヒトeIF−5A1およびヒトeIF−5A2のアミノ酸アラインメントを提供する。
【図46A】図46Aは、eIF−5A1 siRNAの位置および配列を提供する。
【図46B】図46Bは、eIF−5A1 siRNAの位置および配列を提供する。
【技術分野】
【0001】
関連出願
本出願は、2005年12月13日に出願された米国仮特許出願第60/749,604号、および、2006年4月27日に出願された米国仮特許出願第60/795,168号の利益を主張するものであり、参照することにより、そのすべてを本明細書に援用する。
【0002】
発明の分野
本発明は、アポトーシスに特異的な真核生物開始因子(「eIF−5A」)、および該因子をコード化するポリヌクレオチドの多発性骨髄腫細胞やその他の癌細胞を死滅させるための使用に関する。本発明は、多発性骨髄腫を阻害するため、多発性骨髄腫細胞を死滅させるため、そして、その他の癌細胞の成長を阻害するおよび/または死滅させるための、アポトーシスに特異的なeIF−5A、または「アポトーシス特異的eIF−5A」、または「eIF−5A1」の使用と、eIF−5A2アイソフォームの使用に関する。
【背景技術】
【0003】
発明の背景
アポトーシスは、細胞の縮小、クロマチンの濃縮、核断片化、および膜泡形成といった明確な形態学的特性によって特徴付けられる、遺伝子的にプログラムされた細胞事象である。Kerr et al(1972)Br.J.Cancer,26,239−257;Wyllie et al(1980)Int.Rev.Cytol.,68,251−306。アポトーシスは、正常な組織発達およびホメオスタシスにおいて重要な役割を果たし、アポトーシスのプログラムにおける欠陥は、神経変性疾患および自己免疫疾患から腫瘍まで、種々のヒト疾患の原因になると考えられている。Thompson(1995)Science,267,1456−1462;Mullauer et al(2001)Mutat.Res,488,211−231。アポトーシス細胞の形態学的特徴については明らかになっているものの、その過程を制御する分子経路が解明されるようになってからまだ間もない。
【0004】
アポトーシスに関与する別の主要なタンパク質は、腫瘍抑制遺伝子p53によりコード化されるタンパク質である。このタンパク質は、おそらくはBaxの上方制御によって、細胞の成長を制御し、損傷を受けて遺伝的に不安定な細胞にアポトーシスを誘導する、転写因子である。Bold et al(1997)Surgical Oncology,6,133−142;Ronen et al.,1996;Schuler & Green(2001)Biochem.Soc.Trans.,29,684−688;Ryan et al(2001)Curr.Opin.Cell Biol.,13,332−337;Zornig et al(2001)Biochem.Biophys.Acta,1551,F1−F37。
【0005】
アポトーシス経路における改変は、癌など多数の疾患過程において重要な役割を果たすと考えられる。Wyllie et al(1980)Int.Rev.Cytol.,68,251−306;Thompson(1995)Science,267,1456−1462、Sen & D’Incalci(1992)FEBS Letters,307,122−127;McDonnell et al(1995)Seminars in Cancer and Biology,6,53−60。従来、癌の発生および進行についての調査は、細胞増殖に焦点を当ててきた。しかし、腫瘍形成においてアポトーシスが果たす重要な役割が最近明らかになった。実際、アポトーシスの制御には、腫瘍細胞において必ず何らかの改変が生じるため、アポトーシスに関して現在分かっていることのほとんどが、腫瘍モデルを用いて分かってきたことである。Bold et al.(1997)Surgical Oncology,6,133−142。
【0006】
サイトカインもアポトーシス経路に関与してきた。生態系は、自らを制御するために細胞間相互作用を必要とし、細胞間のクロストークには概して多種類のサイトカインが必要となる。サイトカインは、数多くの異なる種類の細胞の型による、種々の刺激に反応して産生されるメディエータである。サイトカインは、数多くの異なる種類の細胞に、数多くの異なる効果を及ぼすことができる多面的分子であるが、特に、免疫応答の制御と、造血細胞の増殖および分化において重要である。標的細胞に対するサイトカインの作用は、特定のサイトカイン、相対濃度、その他のメディエータの存在に依存して、細胞の生存、増殖、活性化、分化、またはアポトーシスを促進することが可能である。
【0007】
デオキシハイプシン合成酵素(DHS)およびハイプシン含有の真核生物翻訳開始因子5A(eIF−5A)は、細胞の成長や分化など多くの細胞過程において重要な役割を果たすことで知られている。独特のアミノ酸であるハイプシンは、検査するすべての真核生物および古細菌中に見られるが真正細菌中には見られず、eIF−5Aが唯一知られるハイプシン含有タンパク質である。Park(1988)J.Biol.Chem.,263,7447−7449;Schumann & Klink(1989)System.Appl.Microbiol.,11,103−107、Bartig et al.(1990)System.Appl.Microbiol.,13,112−116;Gordon et al.(1987a)J.Biol.Chem.,262,16585−16589。活性eIF−5Aは、翻訳後2つのステップにおいて形成される。第1のステップは、デオキシハイプシン合成酵素により触媒される、前駆物質eIF−5Aの特定のリジンのα‐アミノ基へ、スペルミジンの4‐アミノブチル部分を転移することによる、デオキシハイプシン残基の形成であり、第2のステップは、この4‐アミノブチル部分をデオキシハイプシン水酸化酵素によって水酸化してハイプシンを形成することを含む。
【0008】
eIF−5Aのアミノ酸配列は種間でよく保存されており、eIF−5A内のハイプシン残基を取り囲むアミノ酸配列には徹底した保存が見られ、この修飾が生存のために重要である可能性を示している。Park et al.(1993)Biofactors,4,95−104。今まで酵母中に発見されたeIF−5Aの両アイソフォームの不活性化、またはアイソフォーム活性化の第1のステップを触媒するDHS遺伝子の不活性化が、細胞分裂を阻止するという観察は、この仮説をさらに支持するものである。Schnier et al.(1991)Mol.Cell.Biol.,11,3105−3114;Sasaki et al.(1996)FEBS Lett.,384,151−154,Park et al.(1998)J.Biol.Chem.,273,1677−1683。しかし、酵母におけるeIF−5Aタンパク質の枯渇は、総タンパク質合成をわずかに減少させたに過ぎず、タンパク質全体の合成にではなく、mRNAの特定のサブセットの翻訳に、eIF−5Aが必要である可能性を示している。Kang et al. (1993)「Effect of initiation factor eIF−5A depletion on cell proliferation and protein synthesis,」in Tuite,M.(ed.),Protein Synthesis and Targeting in Yeast,NATO Series H。eIF−5Aを結合するリガンドが、非常によく保存された部分を共有するという最近の発見もまた、eIF−5Aの重要性を支持するものである。Xu & Chen(2001)J.Biol.Chem.,276,2555−2561。また、修飾されたeIF−5Aのハイプシン残基が、RNAへの配列特異的結合に必要不可欠であることが分かり、結合はリボヌクレアーゼからの保護を提供しなかった。
【0009】
さらに、細胞内におけるeIF−5Aの枯渇が、核内に特定のmRNAの有意な堆積をもたらし、eIF−5Aが、特定のクラスのmRNAを核から細胞質へと輸送する役割を担っている可能性があることを示唆している。Liu & Tartakoff(1997)Supplement to Molecular Biology of the Cell,8,426a.Abstract No.2476,37th American Society for Cell Biology Annual Meeting。核孔に関連する核内繊維にeIF−5Aが堆積すること、そして一般の核外輸送受容体と相互作用することは、eIF−5Aが、ポリソームの一構成要素であるよりも、むしろ核細胞質間輸送タンパク質である可能性をさらに示すものである。Rosorius et al.(1999)J.Cell Science,112,2369−2380。
【0010】
1989年に、初めてSmit−McBrideらによってヒトからeIF−5AのcDNAがクローン化されて以来、酵母、ラット、ニワトリ胚、アルファルファ、トマトなど、種々の真核生物からeIF−5AのcDNAまたは遺伝子がクローン化されてきた。Smit−McBride et al.(1989)J.Biol.Chem.,264,1578−1583;Schnier et al.(1991)(酵母);Sano,A.(1995)in Imahori,M.et al.(eds),Polyamines,Basic and Clinical Aspects,VNU Science Press,The Netherlands,81−88(ラット);Rinaudo & Park(1992)FASEB J.,6,A453(ニワトリ胚);Pay et al.(1991)Plant Mol.Biol.,17,927−929(アルファルファ);Wang et al.(2001)J.Biol.Chem.,276,17541−17549(トマト)。
【0011】
多発性骨髄腫は、骨髄中における悪性形質細胞の増大、および溶骨性病変の存在を特徴とする、進行性および致命的な疾患である。多発性骨髄腫は、不治ではあるが治療可能な形質細胞の癌である。形質細胞は免疫系の重要な部分であり、感染や疾患との闘いを助ける免疫グロブリン(抗体)を産生する。多発性骨髄腫は、骨髄における過剰な数の異常な形質細胞、そして、インタクトなモノクローナル免疫グロブリン(IgG、IgA、IgD、またはIgE:「Mタンパク質」)、またはベンス・ジョーンズタンパク質(遊離のモノクローナルなL鎖)の、過剰産生によって特徴付けられる。低カルシウム血症、貧血、腎損傷、細菌感染に対する感受性の増加、正常な免疫グロブリンの産生低下が、多発性骨髄腫に共通する臨床症状である。また、多発性骨髄腫は、骨盤、脊椎、肋骨、および頭蓋骨におけるびまん性の骨粗しょう症を特徴とすることも多い。
【0012】
多発性骨髄腫に対する従来の治療法には、化学療法、幹細胞移植、幹細胞移植を併用した大量化学療法、およびサルベージ療法がある。化学療法には、Thalomid(登録商標)(サリドマイド)、ボルテゾミブ、Aredia(登録商標)(パミドロネート)、ステロイド、およびZometa(登録商標)(ゾレドロン酸)を用いる治療などがある。しかし、多くの化学療法薬は、骨髄、胃および腸の粘膜、毛包など、活発に分裂する非癌性細胞に毒性を有する。従って、化学療法は、血球数の減少、悪心、嘔吐、下痢、脱毛を引き起こす場合がある。
【0013】
従来の化学療法、すなわち標準用量の化学療法は、多発性骨髄腫を罹患する患者に対して1次的にまたは初期的に通常行われる治療である。また、大量化学療法や幹細胞移植の準備として、患者が化学療法を受ける場合もある。移植前に腫瘍量を減少させるために、導入療法(幹細胞移植の前に行う従来の化学療法)が使用されることもある。特定の化学療法薬は、骨髄細胞に対する毒性がより低く、骨髄からより多くの幹細胞を得ることができるため、他の薬よりも導入療法に適している。導入療法に適した化学療法薬の例には、デキサメタゾン、サリドマイド/デキサメタゾン、VAD(ビンクリスチン、Adriamicyn(登録商標)(ドキソルビシン)、およびデキサメタゾンの併用)、およびDVd(ペグ化リポソームドキソルビシン(Doxil(登録商標)、Caelyx(登録商標))、ビンクリスチン、および低用量デキサメサゾンの併用)が挙げられる。
【0014】
多発性骨髄腫の標準的な治療は、メルファランをプレドニゾン(副腎皮質ステロイド剤)と併用することであり、50%の奏功率を示している。残念ながら、メルファランはアルキル化剤であるため、導入治療には不向きである。副腎皮質ステロイド(特にデキサメサゾン)は、多発性骨髄腫の治療、特に化学療法に耐えられない高齢患者に対して、単独で使用される場合がある。デキサメサゾンも、単独あるいは他の薬剤と併用して、導入療法に使用される場合がある。VADは最も広く使用される導入療法だが、DVdが導入療法において有効であることは最近明らかになった。ボルテゾミブは、多発性骨髄腫の治療に最近承認されたが、毒性が非常に高い。しかし、現存するいかなる治療法も、治癒に対する顕著な可能性を提供することはできない。つまり、多発性骨髄腫細胞を死滅させるための適切な治療法が、依然として必要である。本発明はその必要とされているものを提供する。
【発明の開示】
【0015】
本発明は、癌細胞の成長を阻害する、および/または、癌細胞を死滅させる方法を提供する。また本発明は、癌細胞が転移する能力を阻害する、または遅らせる方法も提供する。癌の成長を阻害することは、腫瘍の縮小、腫瘍成長の低下を含み、また、腫瘍の完全な寛解を含む場合もある。癌はいかなる癌または腫瘍でもあり得るとし、大腸癌、結腸直腸腺癌、膀胱癌、子宮頸部腺癌、および肺癌を含むが、これらに限定されない。本発明の方法は、eIF−5Aの投与、好ましくはヒトeIF−5A1を、該癌を罹患する患者(哺乳類、好ましくはヒト)に投与することに関連する。eIF−5A1が好ましいが、eIF−5A2アイソフォームを使用してもよい。eIF−5Aは、当該技術分野で既知である任意の適切な方法によって、それを必要とする患者に送達され得る。eIF−5Aは、生物学的に適切な媒体中のDNAのような裸のDNAとして、IVや皮下注射を介して、あるいはその他の任意の生物学的に適切な送達機構を介して送達され得る。あるいは、eIF−5Aはアデノウイルスベクターのようなベクターの中で送達され得る。あるいは、DNAは、標的癌細胞へのDNAの送達を提供する、リポソームやその他の任意の適切な「担体」の中で送達され得る。また、eIF−5Aは腫瘍の部位に直接送達され得る。当業者は、eIF−5Aの送達について、用量および治療投与計画の期間を決定することが可能であろう。
【0016】
eIF−5A1およびeIF−5A2は、09/909,796(U.S.6,867,237);10/141,647(許可);10/200,148;10/277,969;10/383,614;10/792,893;11/287,460;10/861,980;11/134,445;11/184,982;11/293,391;60/749,604;60/795,168などの先の同時係属出願において既知、かつ説明されており、参照することにより、それらのすべてを本明細書に援用するものとする。eIF−5Aはヒト、ラット、マウス、イヌ等の種間でよく保存されているため、任意のeIF−5Aが本発明において使用され得る。好ましくは、ヒトeIF−5Aがヒト等の治療に使用される。突然変異したeIF−5Asが、eIF−5Aの発現を上方制御する、または増加することが可能であり、それにより癌の成長を阻害する、または癌細胞を死滅させることができる限り、eIF−5AはeIF−5Aの突然変異体も含む。
【0017】
本発明は、細胞にeIF−5A1をコード化するヌクレオチドを提供することにより、上記細胞内のMAPK/SAPKシグナル経路を活性化する方法も提供する。該eIF−5A1ポリヌクレオチドおよびeIF−5A1タンパク質は上述の通りである。
【0018】
本発明は、eIF−5Aをコード化するポリヌクレオチドを含む、骨髄腫細胞を死滅させるために有用な医薬組成物も提供する。該eIF5Aは、eIF5A1、eIF5A2、またはeIF5A1の突然変異体であってもよい。好ましくは、該eIF5AはeIF5A1である。該組成物は、送達ビヒクルをさらに含んでもよい。該送達ビヒクルは、ベクター、プラスミド、リポソーム、またはデンドリマーであってもよいが、これらに限定されない。
【0019】
本発明は、多発性骨髄腫を罹患する対象において多発性骨髄腫細胞を死滅させるための薬物を作るためのeIF5A(好ましくはeIF5A1)の使用も提供する。
【0020】
本発明は、eIF5A1をコード化するポリヌクレオチドを含む組成物を骨髄腫細胞に投与することを含む、多発性骨髄腫細胞を死滅させる方法をさらに提供し、該組成物は多発性骨髄腫細胞を死滅させる。該eIF5A1は突然変異体であってもよく、該突然変異体は別のアミノ酸に変化した保存リジンを有し、上記突然変異体はハイプシン化されることができない。治療の方法において有用な組成物は本明細書に記載する通りである。
【0021】
本発明は、多発性骨髄腫細胞を死滅させる方法をさらに提供し、eIF−5A1をコード化するポリヌクレオチドを含む組成物のほかに、eIF−5A1に対するsiRNAを含む組成物が提供される。該siRNAはeIF−5A1の内因性発現を下方制御し、それによってIL−6の発現を下方制御し、ひいては骨髄腫細胞内のアポトーシスを引き起こす。eIF−5A1 siRNAを含む組成物は、静脈内投与されてもよく、または、プラスミド、ベクター、リポソーム、またはデンドリマーのような送達ビヒクルの中で投与されてもよい。
【0022】
発明の詳細な説明
真核生物翻訳開始因子5A1(eIF5A1)は、細胞増殖に関与するmRNAのサブセットの翻訳促進に関与する、核細胞質間輸送シャトルタンパク質として機能すると仮説を立てられてきた。しかし、eIF5A1は、アポトーシスの制御因子(Taylor et al.,Invest Ophthalmol Vis Sci.;45(10):3568−76(2004))として、また、p53の発現を制御することのできるアポトーシス促進性のタンパク質としても認識されている(Li et al.(2004)J.Biol.Chem.;279:49251−49258;および本明細書の図23)。腫瘍抑制タンパク質p53は、細胞周期停止を仲介する上で、またストレスやDNA損傷に応答するアポトーシスにおいて、中心的な役割を担っている。eIF5A1の過剰発現は、癌細胞株のp53発現を上方制御することが可能であるが、(Li et al.(2004)J.Biol.Chem.;279:49251−49258;および本明細書の図23)、eIF5A1の過剰発現は、p53欠損細胞株にアポトーシスを誘導することも可能であり(Taylor et al.(2006)Journal of Molecular and Cellular Biology,Feb.9,2006(決定保留))、eIF5A1の過剰発現が複数の機構によってアポトーシスを誘導することを示唆している。
【0023】
eIF5A1の発現はアポトーシスと相関性を有する。トポイソメラーゼ阻害剤であるアクチノマイシンDで処理した、正常な大腸線維芽細胞のウエスタンブロット(図1A)は、アクチノマイシンD処理を開始してから24時間後に、p53と並行してeIF5A1タンパク質発現が上方制御されたことを実証している。一酸化窒素(NO)供与体であるSNPへの暴露に起因するアポトーシス中のRKO細胞も、eIF5A1タンパク質発現を上方制御する(図1B)。eIF5A1のRNA転写レベルは、これらの条件下では増加せず、これは、eIF5A1タンパク質が、mRNAの転写後制御の結果として堆積していることを示唆している(図1Aおよび1B)。これらの結果は、eIF5A1が、遺伝毒性ストレスや一酸化窒素に起因するアポトーシスに関与する可能性を示唆するものである。
【0024】
細胞周期停止やアポトーシスを誘導するDHSおよびDHH阻害剤に関する報告や、細胞の成長を維持するにはハイプシン化されたeIF5A1が必要であるに違いないという結論に一部基づいて、細胞増殖におけるeIF5A1の必要性が長い間提案されてきた。しかし、siRNAの使用によるeIF5A1発現の特異的抑制は、細胞の成長に何の影響も与えなかったことが判明した。HT−29細胞のeIF5A1発現を90%超抑制したことは、5日間にわたる細胞増殖に何の影響も与えなかった(図2A〜E)。しかし、DHS阻害剤であるGC7は、これらの細胞の成長に絶大な影響を与えた(図2C)。これらの結果は、eIF5A1が細胞の成長に必要ではない可能性があることを示唆している。また、eIF5A1の抑制が、アクチノマイシンD誘導性の細胞毒性からHT‐29細胞を一部保護できたことは興味深く(図2B)、これは、遺伝毒性ストレスに起因するアポトーシスにeIF5A1が関与することを、さらに支持するものである。
【0025】
eIF5A1 siRNAが、アポトーシスから細胞を保護する能力は、eIF5A1タンパク質抑制がp53発現に与えた影響についての試験を促すことになった。RKO細胞は、ストレス条件下以外では堆積しない機能的p53タンパク質を有する。siRNAによるeIF5A1の抑制は、アクチノマイシンD処理から24時間後にp53タンパク質の堆積を69%阻害することが可能であり(図3Aおよび3B)、これは遺伝毒性ストレス下でのp53の適切な発現に、eIF5A1が必要であることを示唆している。異なる配列を有するeIF5A1に対する第2のsiRNAも、アクチノマイシンDに反応してp53の上方制御を防止することができ(図8B)、この効果がsiRNAの非特異的な効果ではないことを示唆している。しかし、eIF5A1は、RKO細胞(機能的p53を含む)およびRKO−E6(機能的p53を含まない、図8A)の両方でアポトーシスを誘導することが可能であったため(図4Aおよび4B)、eIF5A1がp53から独立した機構によってもアポトーシスを誘導することができることを示唆している。
【0026】
eIF5A1またはハイプシン修飾に必要な保存リジン(K)(50位)において点突然変異を含むeIF5A1{eIF5A1(K50A)}のいずれかを発現しているアデノウイルスコンストラクトを構築した。点突然変異はリジンがアラニン(A)となる原因となった。HT‐29細胞がいずれかのコンストラクトに感染することで、それらの細胞にアポトーシスを誘導し(図5Cおよび5Dおよび図6)、細胞のバイアビリティーを大きく減少した(図5E)。Ad−eIF5A1またはAd−eIF5A1(K50A)による感染の結果として、どのような形態のeIF5A1が堆積していたかを判定するために、2次元ゲル電気泳動を使用した(図5B)。予想したとおり、Ad−eIF5A1(K50A)感染後に、非修飾eIF5A1が堆積した。Ad−eIF5A1による感染は、非修飾およびデオキシハイプシン修飾eIF5A1の両方の堆積を著しく増加させる結果となり、ウイルスによって産生されたeIF5A1タンパク質の大部分をハイプシン化するためには、DHSおよびDHHの活性が不十分であることを示した(図5B)。これらの結果は、非修飾eIF5A1、そしておそらくはデオキシハイプシン修飾eIF5A1が、細胞のアポトーシスを引き起こす形態であることを強く示唆するものである。ハイプシン化eIF5A1もこの能力を有するか否かはまだ明らかではない。
【0027】
eIF5A1の核細胞質間輸送機能が示唆されてきたが、eIF5A1は主として細胞質の中で発現すると報告されている。(Shi et al.,Exp Cell Res.;225:348−356(1996b))。核細胞質間輸送シャトルタンパク質は、核局在化も行うことが予想される。アポトーシス中のeIF5A1の機能が発見されたため、アポトーシス中のeIF5A1の局在変化についての研究を行った。IFN−γおよびTNF−αを用いた処理による細胞死受容体の活性化と、アクチノマイシンDを用いた処理による遺伝毒性ストレスという、2つの異なる機構によってアポトーシスをHT‐29細胞に誘導した。間接免疫蛍光法によって、eIF5A1は未処理の成長中の細胞の細胞質内に局在することが明らかになった(図7)。しかし、アポトーシス刺激剤を用いた処理は、eIF5A1の動きを非常に迅速に刺激し、主に細胞質内に局在していたeIF5A1が、主に核に局在するようになった(図7Aおよび7B)。このeIF5A1の局在における変化は、IFNγ/TNF−α処理細胞では10分以内(図7A)、アクチノマイシンD処理細胞では90分以内(図7B)と、非常に迅速に発生した。これらの結果は、細胞死受容体の活性化および遺伝毒性ストレスにより誘導されたアポトーシスの間、eIF5A1が核機能を有することを示唆している。
【0028】
アポトーシスに関与しているのが、非修飾eIF5A1なのか、デオキシハイプシン修飾型eIF5A1なのか、あるいはハイプシン修飾型eIF5A1なのかを明確にするため、アポトーシス中に堆積するeIF5A1の形態を2Dゲル電気泳動を用いて調査した。アクチノマイシンD(遺伝毒性ストレス)を用いた刺激によっても、Fas(細胞死受容体経路)に対するアゴニスト抗体と一緒にインキューベーションすることによっても、HT−29細胞においてアポトーシスの誘発が起こった。1〜24時間までの異なる時点で細胞ライセートを収集し、2Dゲル電気泳動、およびeIF5A抗体を用いたウエスタンブロットで分析した(図9)。文献に報告されるとおり、未処理の細胞においてeIF5A1タンパク質は主にハイプシン化型で存在した。両方のアポトーシス誘発剤が、存在するデオキシハイプシン化型eIF5A1の増加を刺激した。処理から1時間後に増加し始めたが、処理から24時間に停止した。非修飾eIF5A1の堆積も、処理から2時間後に観察された。これらの結果は、アポトーシスの制御に関与するデオキシハイプシン化および/または非修飾eIF5A1と一致していた。
【0029】
eIF5A1、eIF5A1(K50A)(eIF5A1の突然変異体)、およびeIF5A2が、アポトーシスを誘導する能力、および増殖を阻害する能力を、種々の癌細胞株において調査した。eIF5A1およびeIF5A1(K50A)は両方とも、大腸癌細胞株HT−29(図5、6および10)と、膀胱癌細胞株HTB−9(図11)およびHTB−4(図12)に、アポトーシスを誘導することができた。また、eIF5A2を発現するアデノウイルスによる感染は、膀胱癌細胞株HTB−9(図11)およびHTB−4(図12)にアポトーシスを誘導することができた。Ad−eIF5A1、Ad−eIF5A1(K50A)、およびAd−eIF5A2は、膀胱癌細胞株HTB−4(図13)、HTB−9(図14)、J82HTB−1(図15)およびUMUC−3(図16)の成長を阻害することができた。これらウイルス性のコンストラクトは、HeLa子宮頚部腺癌細胞(図17)の成長を阻害することもできた。Ad−eIF5A1、Ad−eIF5A1(K50A)、およびAd−eIF5A2の感染に反応したPARP切断により、HTB−4細胞におけるアポトーシスも観察された(図18)。PARPは、普通はDNAの修復、DNAの安定化、およびその他の細胞事象に関与しているが、アポトーシス初期にカスパーゼファミリーのメンバーによって切断される。PARPのカスパーゼによる切断の検出は、アポトーシスを代表する特徴として示されてきた。Lazebnik Y et al.,Nature 371,346−347(1994)。これらの結果は、eIF5A1とeIF5A2が、実際には重複する機能を有している可能性があることを示唆するものである。さらに、eIF5A1の突然変異型は、種々の細胞株において成長を停止し、アポトーシスを誘導することができた。野生型eIF5A1(およびeIF5A2)のアポトーシス誘導能は、DHSおよびDHHの制限的な活性のためにAd−eIF5A1を細胞に導入した時に生じる(図5B)、デオキシハイプシン化および非修飾eIF5A1の堆積に関係している可能性がある。最近の報告は、外因性eIF5A1がハイプシン化型として細胞内で過剰発現するには、DHSおよびDHHの両方が同一細胞内で過剰発現する必要があるとしている(Park et al.2006,PNAS;103(1):51−56)。これらの結果は、DHSおよびDHH阻害剤が細胞成長を阻害する能力は、細胞増殖に必要とされるハイプシン化eIF5A1の減少によるものではなく、細胞内で細胞周期停止および/またはアポトーシスをトリガーする、非修飾(DHS阻害剤)あるいはデオキシハイプシン修飾型(DHH阻害剤)eIF5A1の堆積によるものである可能性を示唆している。DHSは特定の細胞株において過剰発現されることが判明したこと(Clement et al.2006,FEBS J;273(6):1102−14)、およびDHSは腫瘍転移における重要な増幅された遺伝子の組として認識されてきたこと(Ramaswamy et al.2003,Nat Genet;33,49−54.)は興味深い。この情報の解釈として、非修飾あるいはデオキシハイプシン修飾型eIF5A1の堆積(アポトーシスを起こすように細胞がトリガーされた時に堆積する。図9参照。)によってトリガーされるアポトーシスを避けるために、特定の癌細胞がDHSを過剰発現するということが考えられる。DHSを過剰発現することにより、eIF5A1をハイプシン化した状態、つまり「安全な」形態を維持することによって、癌細胞は遺伝毒性ストレスやその他のストレスによるアポトーシスを減少できる可能性がある。
【0030】
eIF5A1の過剰発現によって、どのシグナル経路が影響を受けているのかを解明するために、A549肺癌細胞におけるAd−eIF5A1またはAd−eIF5A1(K50A)感染に反応した、マイトジェン活性化タンパク質キナーゼ(MAPK)/ストレス活性化タンパク質キナーゼ(SAPK)[MAPK/SAPK]経路の活性化を調査した。3つの主要なMAPK経路は、ERK MAPK経路、p38 MAPK経路、およびJNK SAPK経路である。ERK MAPK経路は、主に上皮増殖因子(EGF)などの成長ホルモンなどのマイトジェン刺激に反応してトリガーされ、種々の腫瘍の成長および生存を支持する。p38 MAPK経路は、細胞ストレス、UV光線、成長因子の使用中止、および炎症促進性サイトカインに反応して活性化される。リン酸化によりp38を活性化することは、p53などの転写因子のリン酸化をもたらし、ひいてはp53の活性または安定性を増加させる。p38の活性化は、アポトーシス促進性と抗アポトーシスの2つの経路、および炎症に関与している。JNK/SAPK経路は、UV光線、DNA損傷、炎症促進性サイトカインなどの細胞ストレスに対する反応を仲介し、c−junのような転写因子のリン酸化および活性増加を引き起こす。JNK経路の活性化は、成長、形質転換、アポトーシスなど、数多くの細胞応答をもたらすことができる。JNK経路は、A549細胞におけるEGF誘導性の成長の1次的なエフェクター経路であると思われる(Bost et al.1997,JBC;272:33422−33429)。A549細胞をAd−eIF5A1に感染させることは、これら3つの経路すべての活性化を誘発する。EGFを用いて30分間刺激したA549細胞を、Ad−eIF5A1を増量しながら感染させることは、ERKおよびその下流標的であるp90RSKのリン酸化/活性化の増加を引き起こしたが、非リン酸化ERKの量には変化は見られなかった(図20)。Ad−eIF5A1感染に反応して、リン酸化/活性化p38およびリン酸化/活性化JNKの、用量反応的な強い上方制御も観察された(図20)。Ad−eIF5A1感染の時間の経過とともに、ERK、p38およびJNK経路の活性化が、感染後24〜72時間持続したことが明らかになった(図21)。非ハイプシン化eIF5A1突然変異体であるAd−eIF5A1(K50A)による感染に反応して、これらの経路の活性化において用量反応的な増加が見られた(図22)。MEK1阻害剤U1026を用いてERKの活性化を阻害することが、JNKの活性化をも阻害した(図22)ということは興味深く、これは、ERK活性化に反応してこれらの細胞内でJNKが活性化されたことを裏付けるものである(Bost et al.1997,JBC;272:33422−33429)。反対に、MEK1阻害剤は、Ad−eIF5A1またはAd−eIF5A1(K50A)による感染に反応して一貫してp38の活性化を増加し、ERK経路の活性化がeIF5A1に反応して、p38経路の活性化を負に制御することを示唆している(図22)。
【0031】
図38は、eIF−5A1がMAPK/SAPK経路およびA549肺癌細胞のアポトーシスに与える影響を示す仮説モデルを提供する。eIF5A1(野生型あるいはハイプシン化されることのできない突然変異体)(K50A)の過剰発現は、A549細胞にアポトーシスを誘導する。eIF5A1の過剰発現は、p38、JNK、およびERKなどのMAPK/SAPK経路の増加も伴う。eIF5A1の過剰発現は、p53の安定性および活性の増加に関連するリン酸化型p53の増加など、p53タンパク質レベルの増加を誘発する。このp53の増加は、MEK/ERK経路の活性およびp53転写活性に依存する。しかし、MEK/ERK経路の阻害、そしてそれほど多くはないがJNK経路の阻害は、eIF5A1によって誘導されたアポトーシスを増強する。これらの結果は、癌細胞にアポトーシスを誘導するため、そして、抗癌剤のMEK阻害剤クラスの癌細胞死滅能を増加するために、eIF5A1を治療薬として使用できる可能性を示唆している。
【0032】
Ad−eIF5A1およびAd−eIF5A1(K50A)が、p53の発現およびリン酸化に与える影響は、図23で見ることができる。野生型および突然変異体eIF5A1の過剰発現は、用量反応的にp53タンパク質の全体的な発現増加をもたらす(図23)。セリン15がリン酸化されたp53の増加も、eIF5A1の両形態について観察された(図23)。セリン46がリン酸化されたp53の増加はAd−eIF5A1(K50A)について用量反応的に観察された。p53は、セリン15においてATM、ATRおよびp38など幾つかのキナーゼによってリン酸化することが可能であり、この修飾は、MDM2のp53に対する結合能を減少してユビキネーションを標的とし、それによってp53の堆積および機能活性を促進する。p53はセリン46において、PKCdeltaおよびp38など種々のキナーゼによってリン酸化され、この修飾はp53のアポトーシス促進プロモータに対する親和性を増加し、これはp53誘導性アポトーシスにとって重要であると考えられる。
【0033】
腫瘍の浸潤および転移は、複雑なプロセスである。そのためには腫瘍細胞が、付着する能力を備え、周辺の細胞外基質を分解し、2次的部位に移動して増殖し、そして最後には、成長の増加を維持するために血管新生を促進することが要求される。基底膜は、細胞外基質(ECM)に近接するシートであり、すべての臓器を覆って、巨大分子および細胞に対するバリアの役目を果たしている。再構成ECM(Matrigel(商標))を含む8μm膜でトランスウェルを被膜し、走化性刺激に反応して、この膜を貫通して向こう側まで到達することのできる細胞を染色することにより、腫瘍細胞の侵襲性を測定できる。A549肺癌細胞は侵襲性が高く、ECMの成分を消化することのできるゼラチナーゼであるマトリックスメタロプロテアーゼなどのタンパク質を分泌することができる。eIF5A1の過剰発現がA549細胞の侵襲性に与える影響を調査した。Ad−eIF5A1またはAd−eIF5A1(K50A)による感染が、Matrigel(商標)で被膜したトランスウェルを通って浸潤した細胞数を有意に減少させた(図24)。
【0034】
こうして得られた結果は、腫瘍をeIF5A1で処理することは治療効果がある可能性を示していた。そこで、マウスにおける実験的転移のモデルを使用した。実験IIにおいて、侵襲性の高いマウスメラノーマ細胞株B16F10をマウスに注射することにより、実験的転移を開始した(第0日目)。LacZ遺伝子(DNA対照として)またはeIF5A1をコード化するプラスミドDNAを、2、4、7、11、16、21、26および31日目に尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した(図25)。eIF5A1をコード化するプラスミドDNAでマウスを処理した時、腫瘍量における用量反応性の減少が観察された(図25および26)。1×の用量よりも2×の用量の方が効果が少ないように見えたが、0.1×用量と1×用量の間には、腫瘍量に有意な減少が観察された。腫瘍が巨視的に成長する能力は、新しい血管の形成(血管新生)に依存する。血管内皮成長因子(VEGF)は、多くの腫瘍細胞によって分泌されるサイトカインであり、腫瘍の血管新生の促進における重要な因子である。実験IIで単離された、B16F10を有する肺からの細胞ライセートを、VEGF発現について、ELISA法を用いて分析した(図27)。eIF5AプラスミドDNAを与えたマウスのVEGFレベルにおいて、有意な用量反応性の減少が見られ、eIF5A1がVEGFを制御する可能性があることを示唆している。
【0035】
実験IIIでは、尾静脈注射したB16F10細胞を用いた実験的転移のモデルを再び使用した。今回は、血清中DNAの半減期を延長し、プラスミドの肺腫瘍への取り込みを増加するために、プラスミドDNAをDOTAPと複合化した。7、14および21日目に、DNA/DOTAP複合体を注射した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した(図28)。LacZプラスミドDNA/DOTAJP複合体を与えたマウスと比較すると、eIF5A1プラスミドDNA/DOTAP複合体で処理したマウスの肺重量に、平均60%の減少が見られた(図29)。
【0036】
eIF5A1処理がメラノーマ腫瘍にアポトーシスを誘導するかどうかを判定するために、B16F0またはB16F10皮下腫瘍を有するマウスに、Ad−eIF5A1を腫瘍内注射した(実験IV)。48時間後、腫瘍を切除し、パラフィンで包埋してから薄切した。薄切した組織をTUNEL染色することにより、Ad−eIF5A1が腫瘍細胞のアポトーシスを誘導したことが明らかになった(図30)。実験Vは、アポトーシスを誘導するために腫瘍内に注射したAd−eIF5A1の能力が、B16F10皮下腫瘍の成長を減少し、生存期間を延長するかどうかを判定するためにデザインされた。B16F10細胞をC57BL/6マウスの側腹部に皮下注射した。腫瘍が直径約8mmに到達した時、Ad−LacZ、Ad−eIF5A1または緩衝液を腫瘍内に注射した。最初の3日間は毎日、それ以降は腫瘍の大きさが直径15〜16mmを超えるまで1日置きにマウスに注射をし、その時点でマウスを屠殺した。キャリパーを用いて毎日腫瘍の大きさを計測し、腫瘍体積を推測するために使用した。マウスをAd−eIF5A1で処理した時は、腫瘍成長の遅延が明らかに観察された(図31)。緩衝液のみを注射したマウスは、治療開始から最長4〜6日間生存し、Ad−LacZのみを注射したマウスは、治療開始から4日間のみ生存した(図32)。Ad−eIF5A1を注射したマウスは、治療開始から少なくとも8日間、最長で25日間生存し、Ad−eIF5A1を用いた治療が、腫瘍を有するマウスの生存期間を著しく改善することが可能であることを実証した。(図32)。
【0037】
従って、本発明は癌の成長を阻害する方法を提供する。また本発明は、癌細胞が転移する能力を阻害する、あるいは遅らせる方法も提供する。癌の成長を阻害することは、腫瘍の縮小、腫瘍成長の低下を含み、また、腫瘍の完全な寛解をも含む場合もある。癌の成長を阻害することは、癌細胞を死滅させることも含む。癌はいかなる癌または腫瘍でもあり得るとし、大腸癌、結腸直腸腺癌、膀胱癌、子宮頸部腺癌、および肺癌を含むが、これらに限定されない。
【0038】
本発明の方法は、eIF−5Aをコード化するポリヌクレオチドの投与、好ましくはヒトeIF−5A1の該癌を有する患者(哺乳類、好ましくはヒト)への投与、好ましくはeIF−5A1受入番号NM001970(図44参照)の投与を含む。eIF−5A2アイソフォーム(受入番号NM020390。ただしeIF−5A1が好ましい。)を使用してもよい。eIF−5Aは、当該技術分野で既知の任意の適切な方法で送達することができる。eIF−5Aは、生物学的に適切な媒体中のDNAのような裸のDNAとして、IVや皮下注射を介して、あるいはその他の任意の生物学的に適切な送達機構を介して送達され得る。あるいは、eIF−5Aは、プラスミド、アデノウイルスベクターのようなベクター、または任意の適切な発現ベクターの中で送達され得る。
【0039】
あるいは、上記DNAは、標的とする腫瘍または癌細胞へDNA(またはプラスミドまたは発現ベクター)の送達を提供する、リポソームやその他任意の適切な「担体」又は「ビヒクル」中で送達され得る。例えば合成DNA送達システムの考察について、Luo,Dan,et al.,Nature Biotechnology,Vol.18,January 2000,pp.33−37を参照のこと。本発明の発明者は、eIF−5A1は正常組織にとって有毒ではないと以前にも述べているが(参照することにより、そのすべてを本明細書に援用した、2005年11月28日出願の、係属出願第11/293,391号を参照のこと)、(eIF5Aポリヌクレオチド/プラスミド/発現ベクターの直接投与と比較して)送達システムが好ましい。好ましい送達システムは、有効量のeIF−5Aを腫瘍または一群の癌細胞に提供し、また好ましくは標的送達を腫瘍または一群の癌細胞に提供する。よって、eIF−5Aヌクレオチド/プラスミド/発現ベクターを、リポソーム、デンドリマー、類似する無毒性のナノ粒子のような、ナノメートルサイズのビヒクルを介して送達することが好ましい。さらに、該ビヒクルは、有効量のeIF−5Aヌクレオチド/プラスミド/発現ベクターを腫瘍または一群の癌細胞に送達する間に、好ましくはeIF−5Aヌクレオチド/プラスミド/発現ベクターを、早急過ぎるクリアランスや免疫応答を引き起こすことから保護する。例示的なビヒクルは、eIF−5Aヌクレオチド/プラスミド/発現ベクターと会合した単純なナノ粒子から、特異的細胞受容体を標的とするために表面にリガンドを結合させたペグ化リポソームのような、より複雑なペグ化ビヒクルまで様々であってもよい。
【0040】
リポソームおよびペグ化リポソームは当該技術分野において既知である。従来のリポソームにおいて、送達される分子(すなわち低分子量の薬剤、タンパク質、ヌクレオチドまたはプラスミド)は、リポソームの中心腔内に含まれる。当業者は、分子送達に有用な「ステルス」、標的化、および陽イオン性リポソームもあることを理解するであろう。例えば、Hortobagyi,Gabriel N.,et al.,J.Clinical Oncology,Vol.19,Issue 14(July)2001:3422−3433、およびYu,Wei,et al.,Nucleic Acids Research.2004,32(5);e48を参照のこと。リポソームは、静脈内注射することができ、また、血流における循環時間が長くなるように、その表面をより親水性にするため(2重層にポリエチレングリコールを付加(「ペグ化」)することにより)に修飾することができる。これらは「ステルス」リポソームとして知られており、ドキソルビシンやミトキサントロンなど親水性(水溶性)の抗癌剤の担体として特に有用である。薬剤運搬リポソームの特異的結合特性を腫瘍細胞のような標的細胞にまで広げるために、抗体、タンパク質、ペプチド等の特異的分子がリポソームの表面に結合していてもよい。例えば、癌細胞に存在する受容体に対する抗体は、リポソームに癌細胞を標的とさせるために使用され得る。多発性骨髄腫を標的とする場合には、例えば葉酸、IL−6、またはトランスフェリンを、リポソームに多発性骨髄腫細胞を標的とさせるために使用してもよい。
【0041】
デンドリマーもまた、当該技術分野において既知であり、好ましい送達ビヒクルを提供する。デンドリマーの考察について、例えば、Marjoros,Istvan,J.,et al,「PAMAM Dendrimer−Based Multifunctional Conjugate for Cancer Therapy:Synthesis,Characterization,and Functionality,」Biomacromolecules,Vol.7,No.2,2006;572−579、およびMajoros,IstvanJ.,et al.,J.Med.Chem,2005.48,5892−5899を参照のこと。
【0042】
またeIF−5Aは、腫瘍の部位に直接送達されてもよい。当業者は、eIF−5Aの送達に関して、用量および治療投与計画の期間を決定することが可能であろう。
【0043】
本発明の別の実施態様は、多発性骨髄腫細胞において細胞死を誘導する方法を提供する。多発性骨髄腫は、骨病変および腫瘍の悪化を引き起こす、高レベルの炎症性サイトカインを産生する骨髄癌の一種である。サイトカインIL−1BおよびIL−6は骨髄腫細胞の成長因子の役割を果たす。
【0044】
eIF−5A1をコード化するポリヌクレオチド(完全長コード領域)を含むアデノウイルスベクターコンストラクトを、多発性骨髄腫細胞株KAS6/1細胞に投与した。KAS6/1細胞株は、Mayo Clinicで作成され、Westendorf,JJ.,et al.,Leukemia(1996)10,866−876において報告されている。細胞株は、侵襲性多発性骨髄腫を罹患する患者からの分離株から直接作製した。本発明の発明者は、eIF−5Aを含むアデノウイルスコンストラクトをKAS6/1細胞に投与すると、対照ベクターのみを用いて処理された細胞と比較して(図1で「CTL」と表示)、死滅したあるいは死滅する細胞数が増加すること(より少ない生きた癌細胞を残す)(図41で「WT」と表示)を示した。図41参照。未処理の細胞の約25%に比べて、Factor5A1で処理した癌細胞は約90%が死滅した。従って、本発明の一実施形態は、eIF−5A1の発現を上方制御し、多発性骨髄腫細胞を死滅させるために、eIF−5A1をコード化するポリヌクレオチドを提供することにより骨髄腫細胞を死滅させる方法を提供する。
【0045】
また、対照(「CTL+IL−6」と表示)およびeIF−5Aコンストラクト(図41において「WT+IL−6」と表示)とともに、IL−6も投与した。IL−6は、骨髄腫細胞の成長因子の役割を果たすサイトカインである。結果は、たとえIL−6をeIF−5Aコンストラクトと同時投与しても、アポトーシスの増加を達成することができたことを示している。図41参照。これは、Factor5A1が骨髄腫細胞を死滅させるだけでなく、IL−6の存在下において骨髄腫細胞を除去することも可能であることを示している。IL−6の存在下で、デキサメタゾンのような標準的治療薬を用いて骨髄腫細胞にアポトーシスを誘導することは、非常に難しいことが証明されているため、この発見は興味深い。
【0046】
さらに、突然変異体eIF−5A(K50A)(50位の保存リジンを別のアミノ酸に変更するため、ハイプシン化されることができない)を含むアデノウイルスコンストラクトも、単独投与(「MUT」)またはIL−6と併用投与した(「MUT+IL−6」)。結果は、IL−6の存在下においても、対照細胞と比較して、eIF−5A1の突然変異体もアポトーシスを増加させることができたことを示す。図41を参照。従って、本発明の一実施形態は、eIF−5A1の突然変異体を投与することにより骨髄腫細胞を死滅させる方法を提供し、該突然変異体はハイプシン化されることができない。eIF−5A1の突然変異体は、骨髄腫細胞における細胞死を増加させる非ハイプシン化eIF−5A1の発現増加を引き起こす。
【0047】
IL−6は骨髄腫細胞の成長因子としての役割を果たすため、IL−6の発現を下方制御することも、骨髄腫細胞を死滅させる方法を提供する。本発明の発明者は、eIF−5A1に対するsiRNA(siRNAコンストラクトについては図43、46Aおよび46B参照)が、内因性eIF−5A1の発現を下方制御するだけでなく、種々の炎症促進性サイトカインの発現も下方制御すると示した。よって、本発明の一実施形態は、IL−1Bの内因性発現を下方制御するためにアポトーシス特異的eIF−5Aに対するsiRNAを投与することにより、骨髄腫細胞を死滅させる方法を提供し、それによってIL−6の発現を下方制御し、ひいては骨髄腫細胞の成長因子としての役割を果たすIL−6を減少させる。IL−6の発現を下方制御することにより、骨髄腫細胞の成長および生存を存続するために必要な、利用可能なIL−6はより少なくなる。
【0048】
別の実施形態において、eIF−5Aをコード化するポリヌクレオチドを、骨髄腫細胞のアポトーシス増加を提供するために、eIF−5Aに対するsiRNAと併せて投与する。eIF−5A1をコード化するポリヌクレオチドは、siRNAが外因性eIF−5A1の発現を阻害しないように、好ましくはアデノウイルスベクターなどのベクター中で投与する。例えば、siRNAは3’UTRを標的にするが、外因性eIF5A1をコード化するポリヌクレオチドは、好ましくは読み取り枠(ORF)全体を含み、したがってsiRNAによって標的とされる3’UTRを有しない。適切なsiRNAコンストラクトは、11/287,460;11/134,445;11/184,982;および11/293,391などの同時係属出願において先に説明されており、参照することにより、そのすべてを本明細書に援用する。図43、46Aおよび46Bも参照のこと。eIF−5A1は骨髄腫細胞において発現し、細胞死を引き起こす。さらに、eIF−5A1 siRNAは、eIF−5Aの内因性発現を減少させ、それによってIL−6の発現を減少させ、ひいては骨髄腫細胞における細胞死を増加させる。本方法において、上述したeIF−5Aの突然変異体もまたベクターコンストラクトに用いてもよい。
【0049】
また、本発明は、多発性骨髄腫細胞を死滅させる併用療法をも提供する。eIF5A、好ましくはeIF5A1をコード化するポリヌクレオチドを含む組成物は、標準的治療法と併せて投与されてもよい。eIF5A組成物は、標準的療法の前、途中、後に投与してもよい。eIF5Aは、医薬組成物として投与しても、または上述したように、送達ビヒクル内で投与してもよい。
【実施例】
【0050】
実施例1:In vitro実験
薬品
DHSの阻害剤であるN1−グアニル−1,7−ジアミノヘプタン(GC7、Biosearch Technologies)を濃度50μMで使用した。アクチノマイシンD(Calbiochem)を0.5または1.0μg/mlで使用した。ニトロプルシドナトリウムおよびデスフェリオキサミンをSigma社より購入し、それぞれ濃度3mMおよび500μMで使用した。ブレフェルジンAもSigma社から調達し、濃度4nMで使用した。
【0051】
細胞培養および処理
Anita Antes氏(ニュージャージー医科歯科大学)のご好意により頂いたヒト大腸腺癌細胞株HT−29を、細胞増殖およびeIF5A局在化研究に使用した。HT‐29細胞は、1mMのピルビン酸ナトリウム、10mMのHEPES、および10%のウシ胎仔血清(FBS)を添加したRPMI1640に維持した。その他の細胞株はすべてAmerican Type Culture Collectionより入手した。CCD112Coは、正常な大腸の繊維芽細胞株である。RKOは、野生型p53を含むヒト結腸直腸癌細胞株(CRL−2577)である。RKO−E6細胞株(CRL−2578)はRKO細胞株に由来する。安定的に組み込まれたヒトパピローマウイルスE6癌遺伝子を含み、従ってかなりの機能性p53腫瘍抑制タンパク質が欠損している。RKO、RKO−E6、A549および細胞株CCD112Coは、2mMのL−グルタミンと、1.5g/Lの炭酸水素ナトリウム、0.1mMの非必須アミノ酸、および1mMのピルビン酸ナトリウムを含むように調製したEarle’s Balanced Salt Solutionを含み、10%のFBSを添加したModified Eagle Minimum Essential Mediumで成長させた。細胞はCO2を5%含む加湿環境において37℃に維持した。
【0052】
プラスミドのクローニングおよび構築
RKO細胞から単離した総RNAから、製造者の付着細胞向けプロトコルに従ってGenElute Mammalian RNA Mini−Prep Kit(Sigma)を用いて、ヒトeIF5AをRT−PCR法によりクローン化した。使用したプライマーは、フォワードプライマー5’−CGAGTTGGAATCGAAGCCTC−3’、リバースプライマー5’−GGTTCAGAGGATCACTGCTG−3’であった。得られた532塩基対の生成物を、pGEM−T Easy (Promega)にサブクローニングして、配列決定した。得られたプラスミドを、フォワードプライマー5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー5’−CCTGAATTCCAGTTATTTTGCCATGG−3’を用いてPCRのテンプレートとして使用し、PCR生成物をpHM6(赤血球凝集素[HA]タグ付き、Roche Molecular Biochemicals)のHindIII及びEcoRI部位にサブクローンし、pHM6−eIF5Aベクターを生成した。eIF5AのC末端切断コンストラクト(pHM6−eIF5AA37)をPCR法により以下のプライマーを用いて生成した。フォワードプライマー:5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー:5’−GCCGAATTCTCCCTCAGGCAGAGAAG−3’。得られたPCR生成物をpHM6ベクターにサブクローンした。トランスフェクションを最適化するため、およびトランスフェクションがアポトーシスに与える影響を知るための対照として、pHM6−LacZベクター(Roche Molecular Biochemicals)を使用した。
【0053】
ノーザンブロット
RKO細胞を6ウェルプレート上にコンフルエントになるよう成長させ、アクチノマイシンD1.0μg/mlを用いて0、1、4、または8時間処理した。GenElute Mammalian RNA Mini−Prep Kit(Sigma)を用いて総RNAを細胞から単離し、5μgのRNAを1.2%のアガロース/ホルムアルデヒドゲル上で分画した。確立された方法に従い、eIF5Aの3’非翻訳領域(3’−UTR)と相同である32P標識cDNAで膜をプローブした。ノーザンブロットに用いたeIF5Aの3’−UTR cDNAは、次のプライマーを用いてRT−PCR法によりRKO細胞からクローン化した。フォワードプライマー:5’−GAGGAATTCGCTGTTGCAATCAAGGC−3’、リバースプライマー:5’−TTTAAGCTTTGTGTCGGGGAGAGAGC−3’。ノーザンブロットの負荷対照として使用したβ−アクチンcDNAを、次のプライマーを用いてRT−PCR法によりクローン化した。フォワードプライマー:5’−GATGATATCGCCGCGCTCGT−3’、リバースプライマー:5’−GTAGATGGGCACAGTGTGGGTG−3’。
【0054】
プラスミドのトランスフェクションおよびアポトーシスの検出
製造者の推奨プロトコルに従ってLipofectamine 2000(Invitrogen)を用いて、RKOおよびRKO−E6細胞をプラスミドDNAで一時的にトランスフェクトした。トランスフェクションから48時間後、製造者のプロトコルに従ってDNA Fragmentation Detection Kit(Oncogene Research Products)を用いて、末端デオキシリボヌクレオチジルトランスフェラーゼ仲介dUTP−ジゴキシゲニンニック末端標識法(TUNEL)により、断片化DNAを含むアポトーシス細胞を検出した。蛍光顕微鏡による分析のため、細胞を8ウェルのカルチャースライドグラス上でトランスフェクトし、4%のホルムアルデヒドで固定した後に、TUNEL法を用いて標識化し、Taylorら(2004)によって記述された方法に従って、Hoescht33258で染色した。
【0055】
siRNAのトランスフェクション
siRNAはすべてDharmacon社より入手した。eIF5A mRNAの3’UTR(受入番号BC085015)を標的にするeIF5A siRNAは次の配列を有していた。センス鎖:5’−GCUGGACUCCUCCUACACAdTdT−3’、アンチセンス鎖:3’−dTdTCGACCUGAGGAGGAUGUGU−5’。eIF5Aに対する第2のsiRNA(5A−2)は次の配列を有していた。センス鎖:5’−AGGAAUGACUUCCAGCUGAdTdT−3’、アンチセンス鎖:3’−dTdTUCCUUACUGAAGGUCGACU−5’。使用した対照siRNAは、eIF5A特異的siRNAの逆の配列を有しており、いかなる既知であるヒト遺伝子生成物に対する同一性も示さなかった。対照siRNAは次の配列を有していた。センス鎖:5’−ACACAUCCUCCUCAGGUCGdTdT−3’、アンチセンス鎖:3’−dTdTUGUGUAGGAGGAGUCCAGC−5’。Lipofectamine 2000を用いて細胞をsiRNA12でトランスフェクトし、増殖研究またはウエスタンブロットに使用した。
【0056】
ウエスタンブロット
ウエスタンブロット用のタンパク質は、沸騰している溶解緩衝液[SDS2%、50mMのTris−HCl(pH7.4)]を用いて単離した。Bicinchoninic Acid Kit(Sigma)を用いてタンパク質濃度を判定した。ウエスタンブロットするために、5μgの総タンパク質を12%のSDS−ポリアクリルアミドゲル上で分画し、二フッ化ポリビニリデン膜に移した。使用した1次抗体は、抗eIF5A(BD Transduction Laboratories、マウスIgG)および、抗β−アクチン(Oncogene、マウスIgM)で、両方とも5%乳汁中1:20,000の希釈度であった。2次抗体は、西洋わさびペルオキシダーゼ(HRP、Sigma)に結合した抗マウスIgGおよび抗マウスIgM−HRP(Oncogene)であった。抗体‐タンパク質複合体は、増感化学発光法(ECL、Amersham Biosciences)を用いて可視化した。eIF5A検出後、ECL Plus Western blotting検出システムの提供するプロトコルに従ってブロットをストリップに切断し、同等負荷を確認するために、抗β−アクチン抗体でプローブした。
【0057】
MAPK/SAPK経路分析に使用したA549細胞のライセートのウエスタンブロットは、MAPK溶解緩衝液(10mM Tris−pH7.4、2% SDS、10%グリセロール)中で収集したライセートを用いて行った。ライセート10μgを10%のSDS−PAGEゲル上で分離し、PVDF膜に移した。PBS中の無脂肪5%脱脂乳により膜をブロックし、PBSで洗浄し、1:1000の1次抗体を用いて、5%BSA/PBS−T中で振動を与えながら4℃で一晩インキュベートした。MAPK/SAPK抗体(P−p38、p38、P−JNK、JNK、P−ERK、ERK、p90RSK)は、Cell Signalingより購入した。A549の研究に使用したp53抗体もCell Signalingより入手し、同様の方法で用いた。
【0058】
2Dゲル電気泳動
2Dゲル電気泳動のために、低温の溶解緩衝液(7M尿素、2Mチオ尿素、30mMトリス、4%CHAPS、プロテアーゼ阻害剤カクテル)中でHT−29細胞ライセートを採取して、超音波処理し、遠心分離によりデブリを除去した。Bradford法を用いてタンパク質濃度を判定した。製造者の指示に従ってEttan IPGphor Isoelectric Focusing System(Amersham Biosciences)を用いて、1次元等電点電気泳動を行った。再水和緩衝液(8Mの尿素、2%のCHAPS、0.2%のDTT,0.5%のpH4〜7のIPG緩衝液、0.002%のブロモフェノールブルー)の中で、Immobiline DryStrips(7cm pH 4〜7、Amersham Biosciences)を細胞ライセートとともに室温で12時間再水和した。500Vで30分、1000Vで30分、および5000Vで1時間40分、等電点電気泳動を行った。IPGストリップゲル上のタンパク質をSDS−PAGEで分離し、PVDF膜(Amersham Biosciences)に移した。eIF5A抗体(BD Biosciences)を用いてウエスタンブロットを行った。
【0059】
アデノウイルスの生成
ヒトeIF5A、または、ハイプシン化を防ぐ単一の点突然変異(K50→A50)を含むeIF5A[eIF5A(K50A)]を発現するアデノウイルス(アデノウイルス5血清型、E1、E3−欠失)を、AdMax(商標) Hi−IQシステム(カナダ、トロントのMicrobix Biosystems Inc)を用いて構築した。eIF5A cDNA内に、PCR法を用いて部位特異的突然変異を作製した。プラスミドDNAをテンプレートとして用いて、PCR法でeIF5A cDNAを増幅し、アデノウイルスシャトルベクターpDC516(io)のSmaI部位に連結した。PCRプライマーの配列は、フォワードプライマー5’−GCCAAGCTTAATGGCAGATGATTTGG−3’、リバースプライマー5’−CCTGAATTCCAGTTATTTTGCCATGG−3’であった。アデノウイルスゲノムのプラスミドベクターpBHGfrt(del)E1,3FLPおよびシャトルベクターをE.coli DH5a中で増殖させ、Qiagen EndoFree Plasmid Mega Kitを用いて精製した。それぞれ5μgのアデノウイルスゲノムプラスミド pBHGfrt(del)E1、3FLP、シャトルベクターpDC516(io)−eIF5A、またはpDC516(io)−eIF5A(K50A)を、Microbix Biosystems Inc.推奨のCaCl2法を用いて、60mm培養皿内の60〜80%コンフルエントな293−IQ細胞(Microbix Biosystems)にトランスフェクトした。37°Cで7〜10日間培養した後プラークが現れ、得られたアデノウイルス粒子[Ad−eIF5A、およびAd−eIF5A(K50A)]を293−IQ細胞内で増殖した。純粋な、力値の高いアデノウイルス株を、Microbix Biosystems Inc.提供の製造者プロトコルに従って、CsCl勾配超遠心法を用いて調製した。LacZを発現するアデノウイルスベクター(Ad−LacZ;血清型5、E1,E3−欠失)を、Qbiogene(米国カリフォルニア州)より購入し、これらの実験における対照およびレポーターとして使用した。Ad−LacZアデノウイルスをAd−eIF5AおよびAd−eIF5A(K50A)ウイルスと同様の方法で増幅および精製した。
【0060】
アデノウイルス感染およびアネキシンV標識
100mm組織培養プレート内に、HT−29、HTB−9、またはHTB−4細胞を、プレートあたり1×106細胞で播種し、翌日、5mlのRPMI1640+2%FBS中において、細胞あたり3000感染単位でアデノウイルスに感染させた。4時間後、細胞にさらなる培地を追加し、FBS濃度を10%になるよう調製した。感染から24、48、または72時間後、細胞をトリプシン処理により剥離し、洗浄してから製造者のプロトコル(BD Biosciences)に従ってAnnexin V−FITCで染色した。フルオレセイン検出のための488nmアルゴンレーザー光源及びフィルターを用いたフローサイメトリー(Coulter Epics XL−MCL)により細胞をソートし、WinMDI2.8でデータを分析した。
【0061】
HT‐29細胞を、細胞あたり3000感染単位を用いて感染させ、細胞あたり1500感染単位でA549細胞を用いた実験を行った。
【0062】
増殖アッセイ
Lipofectamine 2000(Invitrogen)を用いて、siRNAでHT‐29細胞を96ウェルプレート上でトランスフェクトした。XTT Cell Proliferation Kit(Roche Applied Science)を用いて、増殖する細胞の代謝活性を測定した。DNA合成を測定するためには、製造者のプロトコルに従ってBrdU Cell Proliferation Kit(Roche Applied Science)を用いた。アデノウイルス感染後に実行したXTTアッセイのために、96ウェルプレート内に、ウェルあたり5000個の細胞を播種した。翌日、Ad−lacZ、Ad−eIF5A1、およびAd−eIF5A1(K50A)(Ad−eIF5A1M)をそれぞれ用いて細胞を感染させ、未処理の細胞を負の対照とし、アクチノマイシンDで処理した細胞を正の対照とした。XTT基質を追加し、A690nmを基準としてA475nmを測定した。
【0063】
間接免疫蛍光法
ポリ−L−リジンを被膜したカバーガラス上でHT‐29細胞を培養した。サブコンフルエントな状態の細胞を、200 単位のインターフェロンガンマ(IFN−γ、Roche Applied Science)を用いて16時間インキュベートした後、TNF−α(100ng/ml、Leinco Technologies)で10分〜8時間の異なる時間インキュベートした。あるいは、30分から16時間まで時間を増加させながら、1.0μg/mlのアクチノマイシンDを用いて細胞を処理した。処理済の細胞を3%のホルムアルデヒド(メタノールを含まない;Polysciences Inc.)で20分間固定し、PBSで5分間、2回洗浄し、100mMのグリシンを含むPBSで5分間1回洗浄し、PBS中の0.2%のTriton X−100で4分間透過処理した。その後、標準プロトコルを用いて免疫蛍光検査のために細胞を標識化した。1次抗体は、抗−eIF5A(BD Transduction Laboratories、マウスIgG)を用いて、1:250の希釈度で1時間インキュベートした。2次抗体は、抗マウスIgG−AlexaFluor 488(Molecular Probes)で、希釈度1:200で1時間使用した。抗体標識に続いて、Hoescht33258で核を染色し、標識化した細胞を蛍光顕微鏡で観察した。
【0064】
Matrigel(商標)細胞浸潤アッセイ
A549細胞を、アデノウイルスを用いて細胞あたり1500感染単位で感染させ、24時間インキュベートした。トリプシンで細胞を剥離して、無血清培地で洗浄し、Matrigel(商標) Basement Membrane Matrix (BD Biosciences)で事前に被膜しておいたトランスウェル(Falcon 8.0μm セルカルチャーインサート)上に、ウェルあたり30,000個の細胞を無血清培地中に播種した。10%のFBSを含む培地を下のウェル(トランスウェルが乗っている24ウェルプレートのウェル)に播種し、さらに細胞を24時間インキュベートした。インキュベーション後、トランスウェルの上部チャンバから培地を除去し、トランスウェルを外して500マイクロリットルのクリスタルバイオレットを含む24ウェルプレートのウェル内に置いた。トランスウェルを色素の中で20分間インキュベートしてから、ビーカーに入れた水にトランスウェルを何回も浸漬することにより洗浄した。あらかじめ濡らしておいた綿棒を用いてトランスウェルの上面から細胞をこすり取った。トランスウェルの底面まで移動した細胞を、光学顕微鏡を用いて観察し、写真を撮影し、領域ごとに移動した細胞数を数えた。図24参照。
【0065】
統計
統計分析にはスチューデントのt‐検定を用いた。信頼度95%超(P<0.05)で有意性を判定した。
【0066】
実施例2:In vivo実験
マウスおよび腫瘍の確立
カナダのケベック州にあるCharles Riverより、5〜7週齢のC57BL/6マウスを購入した。実験開始前の1週間、マウスを馴化させた。B16F10マウスメラノーマ細胞をATCCより購入し、10%FBS含有DMEM内で培養した。細胞の単層をトリプシン処理し、10%FBS含有MEMで中性化した。細胞をPBSで2回洗浄し、細胞の生存率をトリパンブルー染色によって判定した。実験的転移の実験(実験IIおよびIII)のため、B16F10細胞を6週齢マウスに尾静脈注射することにより、メラノーマ腫瘍を肺の中に確立した。PBS中で、B16F10細胞を1×106生細胞/mlまで希釈した。200ulの細胞を各マウスの尾静脈から注射した。皮下腫瘍実験(実験IVおよびV)のために、500,000個のB16F10細胞を10〜14週齢のマウスの右側腹部に皮下注射してメラノーマ腫瘍を確立した。実験がすべて終了した時(マウスが瀕死状態となった時、または腫瘍が所定の大きさを超えた時)に、CO2吸入によりマウスを安楽死させた。
【0067】
実施例3:実験II
プラスミドDNAの構築および精製
CpGジヌクレオチドを欠損するpCpG−lacZ発現ベクターを米国サンディエゴのInvivoGenより購入した。まずNcoIおよびNheIでプラスミドを消化させ、3.1kbのpCpGベクターバックボーンを単離し(LacZコード化配列を除去)、HAタグを含むeIF5A1のPCRで増幅したcDNAと結合させて(pCpG−HA5A1)、HAタグ付きeIF5A1 cDNAを、pCpG−lacZベクターにサブクローンした。PCRプライマーは次の通りであった。eIF5A1フォワード:HA−5A1フォワード:5’−GCTCCATGGATGTACCCATACGACGTCCC−3’、eIF5A1リバース:5’−CGCGCTAGCCAGTTATTTTGCCATCGCC−3’。25μg/mlのゼオシンを含むLB培地または2XYT培地で培養したE.coli GT115中で、pCpG−lacZおよびpCpG−HA5A1を増幅した。プラスミドを抽出し、QIAGEN Endofree Plasmid Giga kitを用いて精製した。波長260nmのUV吸収法およびアガロースゲル電気泳動法を用いてDNA濃度を測定した。
【0068】
プラスミドDNAの尾静脈注射(実験II)
1×PBS(体重に基づいて約200μl)に溶解したプラスミドDNAを、2、4、7、11、16、21、26、31日目に、マウスの尾静脈に注射した。プラスミドDNAの濃度は、2×(6.6mg/kg)に対して660ng/μl、1×(3.3mg/kg)に対して330ng/μl、0.1×(0.33mg/kg)に対して33ng/μlであった。
【0069】
体重および肺重量
尾静脈注射の前、または毎週月曜および金曜日に体重を測定した。マウスが瀕死状態(嗜眠性、呼吸困難)になった時にCO2を用いて安楽死させ、肺を摘出して重量を測り、写真を撮ってから冷凍して−70℃で保管した。図25〜27参照。
【0070】
VEGF ELISA
採取した肺組織をPBSで洗浄し、ドライアイスの中で冷凍し、−70℃で保管した。粉砕した肺組織からタンパク質ライセートを単離した。50μgの肺組織タンパク質を使用して、Mouse VEGF Immunoassay kit(米国ミネアポリス州、R&D Systems,Inc.)を用いて、マウス肺中のVEGF濃度を判定した。
【0071】
実施例4:実験III
プラスミドDNA/DOTAP複合体の注射
6週齢のマウスの尾静脈に、B16F10メラノーマ細胞(マウス1匹あたりPBS200μl中に50,000個の細胞)、プラスミドDNA、および50μgのエンドトキシンフリーキットで精製したプラスミドDNAを含むDNA担体複合体を注射し、80μgのDOTAPを7、14、および21日目にマウスの尾静脈に注射した。25日目にマウスを屠殺した。肺を摘出し、重量を測定して写真を撮影した。図28〜29参照。
【0072】
実証例5:実験IV
アデノウイルスコンストラクトの注射およびTUNEL法
500,000個のB16F0またはB16F10細胞を、10週齢のC57BL/6マウスの右側腹部に皮下注射した。腫瘍の大きさが直径約4mmに到達した時(B16細胞注射から10〜12日後)、PBS50ul中の1×109pfu Ad−5A1を腫瘍内に注射した。48時間後にマウスを屠殺し、腫瘍を摘出して固定し、パラフィンで包埋した。製造者のプロトコルに従ってTUNEL(Promega)を用いて、各細胞の腫瘍の種類ごとに(Ad−5A1−1およびAd−5A1−2)2ヶ所染色した。細胞の種類ごとに、TUNEL反応の後TdT酵素が残った、負の対照スライド(Ad−5A1陰性)を示した。図30参照。
【0073】
実施例6:実験V
腫瘍の確立
14週齢のC57BL/6マウスの右側腹部に、(PBS100ul中の)500,000個のB16F10細胞を皮下注射した。腫瘍の大きさが直径約8mmに到達するまで、毎日腫瘍形成の経過を調べた。
【0074】
アデノウイルスを用いた注射
腫瘍の直径が8mmに到達した時に治療を開始した。Ad−eIF5A1またはAd−LacZの1×109プラーク形成単位(pfu)をマウスに注射した。注射は腫瘍の3つの部位に分散させた。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。緩衝液のみ与えた緩衝液マウスには、アデノウイルスを懸濁させた緩衝液のみを与えた(10mM Tris−HCl pH 7.4、10 mMのMgCl2、10%グリセロール)。キャリパーを用いて毎日腫瘍の大きさを計測し、次の方程式を用いて腫瘍体積を算出した。
腫瘍体積(mm3)=L*W2*0.52
L=長さ、W=幅(常に短いほうの寸法)
図31および32参照。
【0075】
実施例7
A549肺癌細胞を、LacZ(Lac)またはeIF5A1(5A)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのp38阻害剤SB203580(Calbiochem)、10μMのJNK阻害剤II(Calbiochem)、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、EGFで30分間細胞を処理し、細胞ライセートを採取した。総p53(p53)、セリン15位におけるリン酸化p53[P−p53(ser15)]、またはセリン37位におけるリン酸化p53[P−p53(ser37)]に対する抗体のいずれかを用いて、ライセートにウエスタンブロットを行った。図33参照。
【0076】
図33に示す結果は、感染から48時間後までにAd−eIF5A1がp53の堆積を誘発することを表している。また、図は、Ad−eIF5A1が感染から48時間後までにp53のリン酸化を誘発すること、p53の堆積およびリン酸化がMEKの阻害剤によって阻害されること、p53の堆積およびリン酸化がp53活性の阻害剤によって阻害されること、eIF5A1は、p53のMEK依存リン酸化を刺激すること、そしてeIF5A1が、p53のp53依存性の堆積を刺激することを示している。
【0077】
実施例8
A549肺癌細胞を、LacZ(Ad−LacZ)、またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。48時間後、総RNAを細胞から単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53のレベルおよびbax mRNAの転写産物レベルを判定した。p53プライマーは、5’−CGCTGCTCAGATAGCGATGGTC−3’(5’−プライマー)、および5’−CTTCTTTGGCTGGGGAGAGGAG−3’(3’−プライマー)[これらのp53プライマー配列は、Li et al.(2004).A Novel eIF5A/complex Functions as a Regulator of p53 and p53−dependent Apoptosis,J Biol.Chem.279 49251−49258より入手した]であった。eIF5A1の過剰発現がmRNAレベルでp53の堆積を誘発するが、baxへの影響は見られなかったことが分かる、図34を参照のこと。
【0078】
実施例9
A549肺癌細胞を、LacZ(Ad−LacZ)またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞から総RNAを単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53転写産物レベルを判定した。p53プライマーは、5’−CGCTGCTCAGATAGCGATGGTC−3’ (5’−プライマー)、および5’−CTTCTTTGGCTGGGGAGAGGAG−3’(3’−プライマー)[これらのp53プライマー配列は、Li et al.(2004).A Novel eIF5A/complex Functions as a Regulator of p53 and p53−dependent Apoptosis,J Biol.Chem.279 49251−49258より入手した]であった。p53のeIF5A1依存性の堆積がp53の転写活性に依存しており、eIF5A1に反応して生じるp53タンパク質の上方制御が、p53 mRNAの転写を増加させていることが分かる、図35を参照のこと。
【0079】
実施例10
A549肺癌細胞を、LacZ(Ad−LacZ)またはeIF5A1(Ad−eIF5A1)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地を、DMSO、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞から総RNAを単離した。Real Time PCR法を用いて、GAPDHを参照の遺伝子としてp53転写産物レベル濃度を判定した。TNFR1プライマーは、TNFR1−F 5’ATCTCTTCTTGCACAGTGG 3’、およびTNFR1−R 5’CAATGGAGTAGAGCTTGGAC 3’であった。TNFR1 mRNAレベルがAd−eIF5A1の感染によって上方制御されること、また、このTNFR1 mRNAの堆積が一部MEKに依存することを示す、図36を参照のこと。このTNFR1 mRNAの堆積はp53の転写活性に依存するものである。
【0080】
実施例11
A549肺癌細胞を、LacZ(Lac)またはeIF5A1(5A)のいずれかを発現するアデノウイルスに感染させた。感染から4時間後、培地をDMSO、10μMのp38阻害剤SB203580(Calbiochem)、10μMのJNK阻害剤II(Calbiochem)、10μMのMEK阻害剤U1026(Calbiochem)、または30μMのp53阻害剤Pifithrin−a(Calbiochem)のいずれかを含む培地と交換した。48時間後、細胞を採取し、Annexin/PI染色(BD Bioscience)を用いてアポトーシス中の細胞の割合を判定し、フローサイメトリーによる分析を行った。図37参照。
【0081】
図37は、感染から48時間後までにAd−eIF5A1がアポトーシスを誘発すること、JNKの阻害がeIF5A1によって誘導されたアポトーシスを増加させること、MEKの阻害がeIF5A1によって誘導されたアポトーシスを増加させること、そして、p53の阻害がeIF5A1によって誘導されたアポトーシスを減少させることを示している。
【0082】
実施例12
50,000個のB16−F0メラノーマ細胞をマウスに皮下注射した。腫瘍の大きさが約5×5mm(65mm3)に到達した時、腫瘍内注射を開始した。50〜100μlのPBS/10%グリセロール緩衝液で希釈した、1×109pfuのAd−lacZ(群2)、Ad−eIF5A1(群3)、またはAd−eIF5A2(群4)あるいは緩衝液のみ(群1)を腫瘍内の3つの部位に1日置きに注射した。一日置きに腫瘍の大きさを測定し、腫瘍の大きさが体重の10%に到達した時、マウスを屠殺した。図39参照。
【0083】
実施例13
50,000個のB16−F0メラノーマ細胞をマウスに皮下注射した。腫瘍の大きさが約5×5mm(65mm3)に到達した時、腫瘍内注射を開始した。50〜100μlのPBS/10%グリセロール緩衝液で希釈した、1×109pfuのAd−lacZ(群2)、Ad−eIF5A1(群3)、またはAd−eIF5A2(群4)あるいは緩衝液のみ(群1)のいずれかを、腫瘍内の3つの部位に、1日おきに注射した。腫瘍の大きさが体重の10%に到達した時、マウスを屠殺した。図40参照。
【0084】
実施例14:多発性骨髄腫
第0日目、KAS6/1多発性骨髄腫細胞を、3000IFU/細胞の野生型または突然変異体のeIF5a(保存リジンが突然変異しているため、ハイプシン化されることができない)アデノウイルスベクターコンストラクトに4時間感染させた。感染後の培養培地に存在するIL−6を含む、あるいは含まない、3つの複写物を設定した。また、対照(感染していない)のためにKAS細胞を播種した。
【0085】
第2日目および4日目に、MTTおよびアネキシン/PIアッセイを行った。上清を採取した。結果を図41に示す。
【図面の簡単な説明】
【0086】
【図1】図1は、eIF5A1の発現が、遺伝毒性ストレスおよび一酸化窒素によって増加することを示す。A)アクチノマイシンD0.5μg/mlで0、1、4、8および24時間処理した、正常な大腸繊維芽細胞における、eIF5Aの発現のノーザンブロット(上のパネル)およびウエスタンブロット(下のパネル)分析。ウエスタンブロットは、eIF5A、p53、およびβ−アクチンに対する抗体でプローブした。B)3mMのニトロプルシドナトリウムで、0、2、4、8および24時間処理したRKO細胞における、eIF5A1の発現のノーザンブロット(上のパネル)およびウエスタンブロット(下のパネル)分析。ウエスタンブロットは、eIF5Aおよびβ‐アクチンに対する抗体でプローブした。RKO細胞は有意なレベルでeIF5A2を発現しない。また、eIF−5A2のサイズは、eIF−5A1よりもアミノ酸1個長いだけであるが、SDS PAGEゲル上で高い位置を流れるため、eIF5A2が発現した場合、eIF−5A1とは別のバンドとして見えることになる。
【図2】図2は、eIF5A1発現の抑制が、細胞増殖に対して何の影響も与えないことを示す。A)eIF5A1 siRNAまたは対照siRNAを用いたトランスフェクションから72時間後にHT−29細胞から単離した細胞ライセートのウエスタンブロット。2つの独立した実験からのウエスタンブロットを示す。ブロットは、eIF5Aおよびβ−アクチンに対する抗体でプローブした。B)eIF5A1 siRNAを用いてトランスフェクトした細胞の代謝活性を、XTT細胞増殖アッセイを用いて測定した。対照siRNAまたはeIF5A1 siRNAを用いたトランスフェクションの24時間前に、HT−29細胞を96ウェルプレートに播種した。トランスフェクションから24時間後、細胞を未処理のまま放置するか、または、代謝活性測定の前に、アクチノマイシンD(1.0μg/ml)を用いて48時間処理した。値は、4通り行われた2つの実験からの平均値であり、1に設定された0時間対照のために得られた値まで正規化した。C)対照またはeIF5A1 siRNAを用いてトランスフェクトしたHT−29細胞の増殖能を、50μMのGC7とともに72時間インキュベートした細胞の増殖能と比較した。細胞増殖はBrdU取り込みによって測定した。値は、n=4に対して平均値±SEで、1に設定したGC7(+)血清サンプルの値に合わせて正規化した。アスタリスク(*)は、対応のあるスチューデントのt検定(p<0.01)により、有意に差があると思われた値を示す。対照siRNAまたはeIF5A siRNAのどちらかを用いてトランスフェクトしたHT−29細胞のトランスフェクションの日(第0日目)からトランスフェクション後5日目までのD)XTTおよびE)BrdU取り込み細胞増殖アッセイ。第0日目の値は1に設定した。
【図3】図3は、eIF5A1が、アクチノマイシンDに反応してp53の発現を制御することを示す。RKO細胞を、対照siRNA(C)またはeIF5A siRNA(5A−1)のいずれかを用いてトランスフェクトした。トランスフェクションから72時間後、アクチノマイシンD0.5μg/mlを用いて、0、4、8、または24時間細胞を処理した。A)eIF5A、p53、またはβ−アクチンに対する抗体を用いてブロッティングした細胞ライセートのウエスタンブロット。結果は3つの独立した実験を代表するものである。B)対応するアクチンバンドまで正規化したウエスタンブロットにおけるp53の相対強度のプロット。値は最小値n=3に対して平均値±SE。
【図4】図4は、ヒトeIF5A1の過剰発現が、p53から独立してアポトーシスを誘導することを示す。RKO細胞(A)またはRKO−E6細胞(B)を、pHM6−LacZ、pHM6 eIF5A、またはpHM6−eIF5A△37(C末端側で37位のアミノ酸を切断)を用いてトランスフェクトした。トランスフェクションから48時間後、TUNEL法を用いて細胞を固定し、標識化した。核は、Hoescht33258で染色し、標識化した細胞を蛍光顕微鏡を用いて観察した。明るい緑色に染色された細胞を、アポトーシス性であるとスコア化した。Hoeschtで染色した核を総細胞数を決定するのに使用した。値はn=4(A)またはn=3(B)についての平均値±SE。アスタリスク(*)は、対応のあるスチューデントのt検定(p<0.02)による、対照(pHM6−LacZ)からの有意差を示す。C)アクチノマイシンD0.5μg/mlで処理してから0、4、8、および24時間後に採取したRKOおよびRKO−E6細胞ライセートのウエスタンブロット。ブロットは、p53およびβ−アクチンに対する抗体を用いてプローブした。
【図5】図5は、eIF5A1アデノウイルスコンストラクトが大腸癌細胞にアポトーシスを誘導することを示す。A)Ad−LacZ(L)、AdeIF5A1(5A)、またはAd−eIF5A(K50A)(K50A)で感染させてから72時間後にHT−29細胞から単離した、細胞ライセートのウエスタンブロット。B)GC7またはDFOで72時間処理した後、またはアデノウイルスコンストラクトに感染させてから72時間後、eIF5Aに対する抗体を用いてウエスタンブロットを行った後、HT−29細胞から単離した、細胞ライセートの2Dゲル電気泳動。eIF5Aを過剰発現するアデノウイルス感染細胞からのライセートから0.3μgのタンパク質が分離されたことを除いては、7μgのタンパク質が分離された。C)上のパネル:アデノウイルスコンストラクト感染から48時間後のHT−29細胞のTUNEL染色。下のパネル:Hoechst染色した同じ領域内の細胞の核。すべての写真は400倍の倍率で撮影した。結果は3つの独立した実験を代表するものである。D)アデノウイルスコンストラクト感染から48時間後のHT−29細胞のアポトーシスの割合。値は、n=3に対して平均値±SE。E)アデノウイルスコンストラクト感染から7日後の、HT−29細胞のXTT細胞増殖アッセイ。値は、n=4に対して平均値±SE。
【図6】図6は、eIFA1アデノウイルスコンストラクトに感染したHT‐29細胞のアネキシンVおよびPI染色を示す。A)アデノウイルスコンストラクト感染から48時間後のHT‐29細胞のアネキシン−FITCおよびヨウ化プロピジウム(PI)標識のヒストグラム。B)アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染後24、48、および72時間後、またはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンAで処理した後の、HT−29細胞アポトーシスの割合。値はn=2に対して平均値±SE(事象数>5000)。
【図7】図7は、免疫蛍光法によるeIF5A1の局在化を示す。IFN−γおよびTNF−α(A)またはアクチノマイシンD(B)で刺激を与えたHT‐29細胞内におけるeIF5Aタンパク質の細胞内局在を、間接免疫蛍光法によって判定した。Hoechst33258を核の染色に使用した。A)TNF−αで0分[(−)TNF−α]、10分、または30分間刺激する前に、HT‐29細胞を未処理のまま放置するか、あるいはIFN−γで16時間予備刺激した。上のパネル:eIF5A1の免疫蛍光検出。中央のパネル:Hoechst染色した同領域内にある細胞の核。下のパネル:合併したイメージ。B)HT‐29細胞を未処理のまま放置するか、あるいはアクチノマイシンDで0.5時間、1.5時間、または4時間処理した。上のパネル:eIF5A1の免疫蛍光検出。中央のパネル:Hoechst染色した同領域内にある細胞の核、下のパネル:合併したイメージ。すべての写真は400倍の倍率で撮影した。結果は3つの独立した実験を代表するものである。
【図8】図8は、eIF5A1が、アクチノマイシンDに反応してp53の発現を制御することを示す。RKO−E6細胞はp53を発現しない。A)RKOまたはRKO−E6細胞を、アクチノマイシンD0.5μg/mlで1、4、8、または24時間処理した。細胞ライセートを採取し、ウエスタンブロットを用いてp53の発現について分析した。B)対照siRNA(C)、eIF5A siRNA(5A−1)、またはeIF5Aの異なる領域を標的とする第2のeIF5A siRNA(5A−2)のいずれかを用いて、RKO細胞をトランスフェクトした。トランスフェクトから72時間後、アクチノマイシンD0.5μg/mlで0、4、8、または24時間細胞を処理した。A)eIF5A、p53、またはβ−アクチンに対する抗体でブロットした細胞ライセートのウエスタンブロット。結果は3つの独立した実験を代表するものである。
【図9】図9は、アポトーシスの間、デオキシハイプシン化したeIF5A1が堆積することを示す。この図は、アクチノマイシンD(A)または作動薬Fas抗体(B)で、1〜24時間処理してから、eIF5Aに対する抗体でウエスタンブロットした後、HT‐29細胞から単離した細胞ライセートの2Dゲル電気泳動を提供する。
【図10】図10は、Ad−eIF5A1またはAd−eIF5A1(K50A){Ad−eIF5A1M)による感染が、HT−29結腸直腸腺癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HT−29細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図11】図11は、Ad−eIF5A1またはAd−eIF5A2による感染が、HTB−9膀胱癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HTB−9細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図12】図12は、Ad−eIF5A1またはAd−eIF5A2による感染が、HTB−4膀胱癌細胞にアポトーシスを誘導することを示す。この図は、アネキシンV標識およびフローサイトメトリー分析によって判定される、アデノウイルスコンストラクト感染、あるいはアポトーシス誘導剤アクチノマイシンDまたはブレフェルジンA処理から24、48、および72時間後の、HTB−4細胞アポトーシスの割合を提供する。値は、n=2に対して平均値±SE(事象数>5000)。
【図13】図13は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HTB−4膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−4細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図14】図14は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染がHTB−9膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−9細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図15】図15は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HTB−1膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HTB−1細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図16】図16は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、UMUC−3膀胱癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、UMUC−3細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図17】図17は、Ad−eIF5A1、Ad−eIF5A1(K50A){Ad−eIF5A1M}、またはAd−eIF5A2による感染が、HeLa子宮頚部腺癌細胞の成長を阻害することを示す。この図は、アデノウイルスコンストラクト感染から24、48、および72時間後の、HeLa細胞のXTT細胞増殖アッセイの結果を提供する。値は、n=2に対して平均値±SE。
【図18】図18は、Ad−eIF5A1、Ad−eIF5A1(K50A)(Ad−eIF5A1M)、またはAd−eIF5A2による感染が、HTB−4細胞にPARP切断を誘発することを示す。この図は、アデノウイルスコンストラクト感染から48時間または72時間後に、HTB−4細胞から単離された細胞ライセートを、PARP、eIF5A、およびβ−アクチンに対する抗体を用いてウエスタンブロットした結果を示す。
【図19】図19は、Ad−eIF5A1、Ad−eIF5A1(K50A)(Ad−eIF5A1M)、またはAd−eIF5A2による感染が、HTB−1細胞にPARP切断を誘発しないことを示す。この図は、アデノウイルスコンストラクト感染から48時間または72時間後に、HTB−1細胞から単離した細胞ライセートを、PARP、eIF5A、およびβ−アクチンに対する抗体を用いてウエスタンブロットした結果を示す。
【図20】図20は、A549肺癌細胞におけるeIF5A1の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、感染効率(MOI)を次第に高くしながらAd−eIF5A1(5A)に感染させた。比較のために、Ad−LacZ(Lac)を用いて細胞を感染させた。感染から48時間後、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図21】図21は、A549肺癌細胞におけるeIF5A1の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、Ad−eIF5A1(5A)またはAd−LacZ(L)を用いて50MOIで感染させた。感染から6、24、48、または72時間後に、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図22】図22は、ハイプシン化されることのできないeIF5A1の突然変異体の過剰発現が、MAPK/SAPKシグナル経路を活性化することを示す。A549細胞は、感染効率(MOI)を次第に高めながらAd−eIF5A1(K50A)(M)に感染させた。比較のために、Ad−eIF5A1(5A)およびAd−LacZ(Lac)を用いて細胞を感染させた。MEK阻害剤U1026を用いて、あるいは用いずに細胞をインキュベートした。感染から48時間後、EGFで細胞を30分間処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図23】図23は、A549肺癌細胞におけるeIF5A1または突然変異体eIF5A1(K50A)の過剰発現が、p53の発現を増加させることを示す。A549細胞は感染効率(MOI)を次第に高くしながらAd−eIF5A1(5A)またはAd−eIF5A1(K50A)(M)を用いて感染させた。比較のために、Ad−LacZ(Lac)に細胞を感染させた。MEK阻害剤U1026を用いて、あるいは用いずに細胞をインキュベートした。感染から48時間後、EGFで30分間細胞を処理し、ウエスタンブロット分析のために細胞ライセートを採取した。
【図24】図24は、eIF5A1または突然変異体eIF5A1(K50A)の過剰発現は、A549肺癌細胞がMatrigel(商標)細胞外基質に浸潤する能力を減少させることを示す。アデノウイルスコンストラクト感染から24時間後、A549細胞をMatrigel(商標)を塗布した無血清培地のトランスウェルに播種した。血清含有培地は、化学誘引物質として作用するように下のウェルに配置した。播種から24時間後、トランスウェルの下部表面まで浸潤した細胞を、クリスタルバイオレットで染色し、光学顕微鏡下で細胞を数えることにより、領域あたりの平均細胞数を判定した。サンプルあたり最低6個の領域が数えられた。
【図25】図25は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、eIF5A1によって肺腫瘍量が有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して写真を撮影した。
【図26】図26は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、eIF5A1によって肺重量が有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して重量を測定した。
【図27】図27は、実験IIの結果を示す。B16F10を有するC57BL/6マウスにおいて、VEGF発現がeIF5A1によって有意に減少した。6週齢のC57BL/6マウスの尾静脈に、200,000個のB16F10細胞を注射した。2、4、7、11、16、21、26および31日目に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAを尾静脈に注射した。1×(3.3mg/kg)、0.1×(0.3mg/kg)、2×(6.6mg/kg)の、3つの異なるDNA濃度を使用した。B16F10細胞の代わりにPBSを与えたマウスを陰性対照として使用し、プラスミドDNAの代わりにPBSを注射したB16F10を有するマウスを陽性対照として使用した。マウスは瀕死状態となった時に屠殺し、肺を摘出して重量を測定した。肺重量が判明してからその肺を冷凍し、後にVEGF ELISAに使用した。肺組織は溶解緩衝液中で粉砕し、ライセート中に存在するVEGFの量をELISA法によって測定した。
【図28】図28は、実験IIIの結果を示す。DNAをDOTAPと複合化することにより、eIF5A1遺伝子療法の抗腫瘍効果が向上した。6週齢のC57BL/6マウスの尾静脈に、50,000個のB16F10細胞を注射した(第0日目)。7,14、および21日目に尾静脈注射を行う前に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAをDOTAPと複合化した。プラスミドDNAを含まないDOTAPを注射した腫瘍の無いマウスを、DOTAPの効果の対照として使用した。マウスは25日目に屠殺し、肺を摘出して写真を撮影した。
【図29】図29は、実験IIIの結果を示す。DNAをDOTAPと複合化することにより、eIF5A1遺伝子療法の抗腫瘍効果が向上した。6週齢のC57BL/6マウスの尾静脈に、50,000個のB16F10細胞を注射した(第0日目)。7,14、および21日目に尾静脈注射を行う前に、LacZ遺伝子(DNA対照として)またはeIF5A1を有するプラスミドDNAをDOTAPと複合化した。プラスミドDNAを含まないDOTAPを注射した腫瘍の無いマウスを、DOTAPの効果の対照として使用した。マウスは25日目に屠殺し、肺を摘出して重量を測定した。
【図30】図30は、実験IVの結果を示す。Ad−eIF5A1の注射はB16F0およびB16F10腫瘍にアポトーシスを誘導する。500,000個のB16F0またはB16F10細胞を、C57BL/6マウスの右側腹部に皮下注射した。腫瘍の直径が約4mmに到達した時、50μlのPBS中の1×109pfu Ad−5A1を腫瘍に注射した(B16細胞注射から10〜12日後)。マウスは48時間後に屠殺し、腫瘍を切除して、固定し、パラフィンに包埋した。各細胞腫瘍の種類(Ad−5A1−1およびAd−5A1−2)に対して2ヶ所を、製造者のプロトコルに従ってTUNEL(Promega)を用いて染色した。TUNEL反応からTdT酵素が残った陰性対照のスライド(Ad−5A1陰性)を、各細胞の種類ごとに含ませた。
【図31】図31は、実験Vの結果を示す。Ad−eIF5A1の注射により腫瘍成長が有意に遅延した。皮下にB16−F10を有するC57BL/6に、1×109pfuのAd−eIF5A1またはAd−LacZまたは等体積の緩衝液を注射した。腫瘍の直径が約8mmに到達した時に治療を開始し、その日を「第0日目」に指定した。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。各群につきマウス3匹とした。群1のマウス(1−1、1−2、1−3)はAd−eIF5A1で処理した。群2のマウス(2−1、2−2、2−3)にはAd−LacZを注射し、群3(3−1、3−2、3−3)のマウスには緩衝液のみを与えた。キャリパーを用いて毎日腫瘍の大きさを計測し、腫瘍体積を推測するために使用した。
【図32】図32は、実験Vの結果を示す。Ad−eIF5A1の注射により生存期間が有意に伸びた。皮下にB16−F10を有するC57BL/6に、1×109pfuのAd−eIF5A1またはAd−LacZまたは等体積の緩衝液を注射した。腫瘍の直径が約8mmに到達した時に治療を開始し、その日を「第0日目」に指定した。最初の3日間は毎日マウスに注射をし、それ以降はマウスを屠殺するまで1日置きに注射した。腫瘍の大きさが1次元的に15〜16mmを超えた時、マウスを屠殺した。各群につきマウス3匹とした。群1のマウス(1−1、1−2、1−3)はAd−eIF5A1で処理した。群2のマウス(2−1、2−2、2−3)にはAd−LacZを注射し、群3(3−1、3−2、3−3)のマウスには緩衝液のみを与えた。
【図33】図33は、eIF−5A1が、A549肺癌細胞におけるp53腫瘍抑制タンパク質の堆積およびリン酸化を増加することを示す。
【図34】図34は、eIF−5A1が、A549肺癌細胞におけるp53 mRNAレベルを上昇させることを示す。
【図35】図35は、p53レベルの上昇は、A549肺癌細胞におけるp53の転写活性に依存することを示す。
【図36】図36は、TNFR1 mRNAレベルは、A549肺癌細胞においてeIF−5A1に感染することにより上方制御されることを示す。
【図37】図37は、eIF−5A1が、A549肺癌細胞におけるアポトーシスを誘導し、MEK阻害剤の使用が、eIF−5A1によって誘導されたアポトーシス量を増加することを示す。
【図38】図38は、eIF−5A1の仮説モデルが、MAPK/SAPK経路およびA549肺癌細胞におけるアポトーシスに影響することを示す。
【図39】図39は、腫瘍成長の抑制において、eIF−5A1がeIF−5A2より優れていることを示す(マウス異種移植モデル(B16−FOメラノーマ細胞))。
【図40】図40は、マウスの生存期間延長において、eIF−5A1がeIF−5A2より優れていることを示す(マウス異種移植モデル(B16−FOメラノーマ細胞))。
【図41】図41は、IL−6を含む、あるいは含まない対照を用いて処理された細胞と比較すると、eIF−5Aが、死滅したあるいは死滅する細胞数を増加することを示す。
【図42】図42は、eIF−5A2の配列を提供する。
【図43】図43は、eIF−5A1 siRNAの位置および配列を提供する。
【図44】図44は、ヒトeIF−5A1およびヒトeIF−5A2のヌクレオチドアラインメントを提供する。
【図45】図45は、ヒトeIF−5A1およびヒトeIF−5A2のアミノ酸アラインメントを提供する。
【図46A】図46Aは、eIF−5A1 siRNAの位置および配列を提供する。
【図46B】図46Bは、eIF−5A1 siRNAの位置および配列を提供する。
【特許請求の範囲】
【請求項1】
eIF5Aをコード化するポリヌクレオチドを含む、骨髄腫細胞を死滅させるための組成物。
【請求項2】
前記eIF5Aは、eIF5A1、eIF5A2、またはeIF5A1の突然変異体から成る群から選択される、請求項1に記載の組成物。
【請求項3】
前記eIF5AはeIFA1である、請求項2に記載の組成物。
【請求項4】
前記組成物は、送達ビヒクルをさらに含む、請求項3に記載の組成物。
【請求項5】
前記送達ビヒクルは、ベクター、プラスミド、リポソーム、またはデンドリマーから成る群から選択される、請求項4に記載の組成物。
【請求項6】
前記送達ビヒクルはベクターである、請求項5に記載の組成物。
【請求項7】
前記送達ビヒクルはアデノウイルスベクターである、請求項6に記載の組成物。
【請求項8】
前記送達ビヒクルはリポソームである、請求項5に記載の組成物。
【請求項9】
前記送達ビヒクルはデンドリマーである、請求項5に記載の組成物。
【請求項10】
多発性骨髄腫を罹患する対象において、多発性骨髄腫細胞を死滅させるための薬物を作製するためのeIFAの使用。
【請求項11】
前記eIF5AはeIF5A1、eIF5A2、またはeIF5A1の突然変異体であり、eIF5A1の前記突然変異体は、保存リジンをアラニンまたは他の任意のアミノ酸に変更しており、前記突然変異体はハイプシン化されることが不可能である、請求項10に記載のeIF5Aの使用。
【請求項12】
多発性骨髄腫細胞を死滅させる方法であって、eIF5A1をコード化するポリヌクレオチドを含む組成物を前記骨髄腫細胞に投与するステップを含み、ここで、前記組成物は多発性骨髄腫細胞を死滅させる、前記方法。
【請求項13】
前記eIF5A1は突然変異体であり、前記突然変異体は保存リジンをアラニンまたは他の任意のアミノ酸に変更しており、前記突然変異体はハイプシン化されることが不可能である、請求項12に記載の方法。
【請求項14】
前記組成物はベクターを含む、請求項12に記載の方法。
【請求項15】
前記ベクターはアデノウイルスベクターである、請求項14に記載の方法。
【請求項16】
eIF−5A1に対するsiRNAを投与するステップをさらに含み、ここで、前記siRNAはeIF−5A1の内因性発現を下方制御し、ここで、前記eIF−5A1の発現の下方制御はIL−6の発現を下方制御し、さらにここで、前記IL−6の下方制御は多発性骨髄腫細胞を死滅させる、請求項12に記載の方法。
【請求項17】
多発性骨髄腫を罹患する対象において、多発性骨髄腫細胞にアポトーシスを誘導する方法であって、請求項3に記載の組成物を投与するステップを含み、ここで、前記組成物中の前記eIF5A1は、多発性骨髄腫細胞にアポトーシスを誘導する、前記方法。
【請求項18】
前記組成物は静脈内に投与される、請求項17に記載の方法。
【請求項19】
前記組成物はリポソームをさらに含む、請求項17に記載の方法。
【請求項20】
前記組成物はデンドリマーをさらに含む、請求項17に記載の方法。
【請求項21】
多発性骨髄腫細胞を死滅させる方法であって、請求項3に記載の組成物を投与するステップと、慣用の多発性骨髄腫の治療薬をさらに投与するステップと、を含む前記方法。
【請求項1】
eIF5Aをコード化するポリヌクレオチドを含む、骨髄腫細胞を死滅させるための組成物。
【請求項2】
前記eIF5Aは、eIF5A1、eIF5A2、またはeIF5A1の突然変異体から成る群から選択される、請求項1に記載の組成物。
【請求項3】
前記eIF5AはeIFA1である、請求項2に記載の組成物。
【請求項4】
前記組成物は、送達ビヒクルをさらに含む、請求項3に記載の組成物。
【請求項5】
前記送達ビヒクルは、ベクター、プラスミド、リポソーム、またはデンドリマーから成る群から選択される、請求項4に記載の組成物。
【請求項6】
前記送達ビヒクルはベクターである、請求項5に記載の組成物。
【請求項7】
前記送達ビヒクルはアデノウイルスベクターである、請求項6に記載の組成物。
【請求項8】
前記送達ビヒクルはリポソームである、請求項5に記載の組成物。
【請求項9】
前記送達ビヒクルはデンドリマーである、請求項5に記載の組成物。
【請求項10】
多発性骨髄腫を罹患する対象において、多発性骨髄腫細胞を死滅させるための薬物を作製するためのeIFAの使用。
【請求項11】
前記eIF5AはeIF5A1、eIF5A2、またはeIF5A1の突然変異体であり、eIF5A1の前記突然変異体は、保存リジンをアラニンまたは他の任意のアミノ酸に変更しており、前記突然変異体はハイプシン化されることが不可能である、請求項10に記載のeIF5Aの使用。
【請求項12】
多発性骨髄腫細胞を死滅させる方法であって、eIF5A1をコード化するポリヌクレオチドを含む組成物を前記骨髄腫細胞に投与するステップを含み、ここで、前記組成物は多発性骨髄腫細胞を死滅させる、前記方法。
【請求項13】
前記eIF5A1は突然変異体であり、前記突然変異体は保存リジンをアラニンまたは他の任意のアミノ酸に変更しており、前記突然変異体はハイプシン化されることが不可能である、請求項12に記載の方法。
【請求項14】
前記組成物はベクターを含む、請求項12に記載の方法。
【請求項15】
前記ベクターはアデノウイルスベクターである、請求項14に記載の方法。
【請求項16】
eIF−5A1に対するsiRNAを投与するステップをさらに含み、ここで、前記siRNAはeIF−5A1の内因性発現を下方制御し、ここで、前記eIF−5A1の発現の下方制御はIL−6の発現を下方制御し、さらにここで、前記IL−6の下方制御は多発性骨髄腫細胞を死滅させる、請求項12に記載の方法。
【請求項17】
多発性骨髄腫を罹患する対象において、多発性骨髄腫細胞にアポトーシスを誘導する方法であって、請求項3に記載の組成物を投与するステップを含み、ここで、前記組成物中の前記eIF5A1は、多発性骨髄腫細胞にアポトーシスを誘導する、前記方法。
【請求項18】
前記組成物は静脈内に投与される、請求項17に記載の方法。
【請求項19】
前記組成物はリポソームをさらに含む、請求項17に記載の方法。
【請求項20】
前記組成物はデンドリマーをさらに含む、請求項17に記載の方法。
【請求項21】
多発性骨髄腫細胞を死滅させる方法であって、請求項3に記載の組成物を投与するステップと、慣用の多発性骨髄腫の治療薬をさらに投与するステップと、を含む前記方法。
【図1】


【図2】
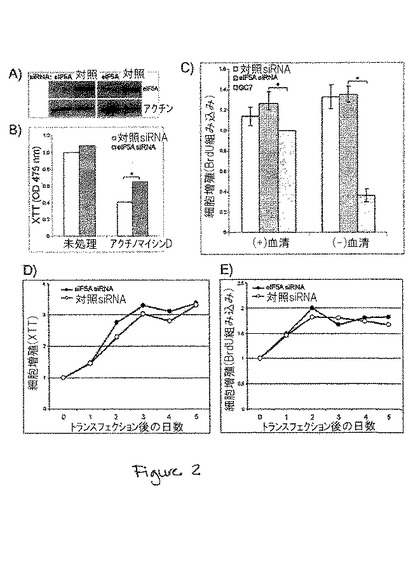
【図3】
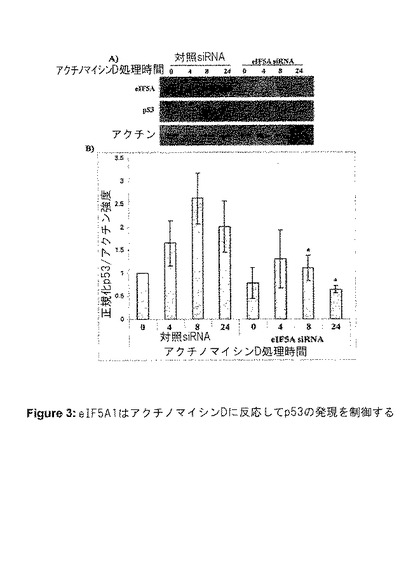
【図4】
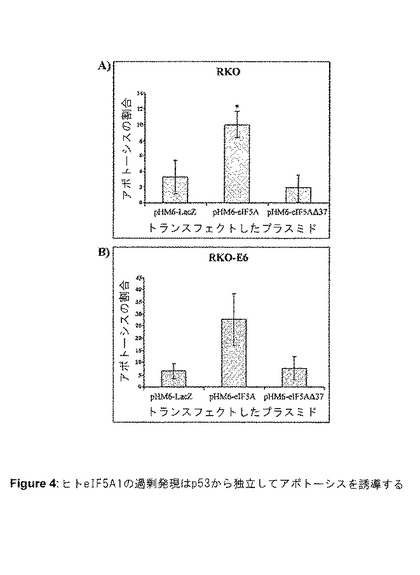
【図5】
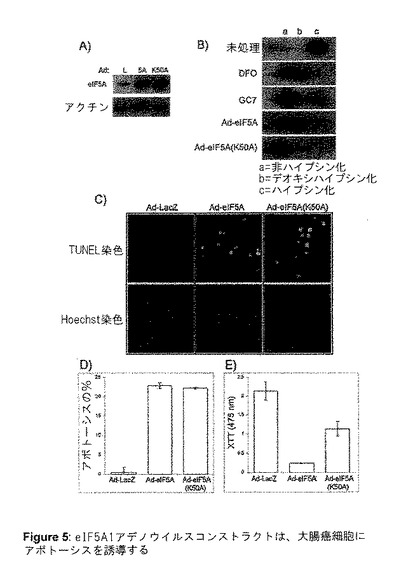
【図6】
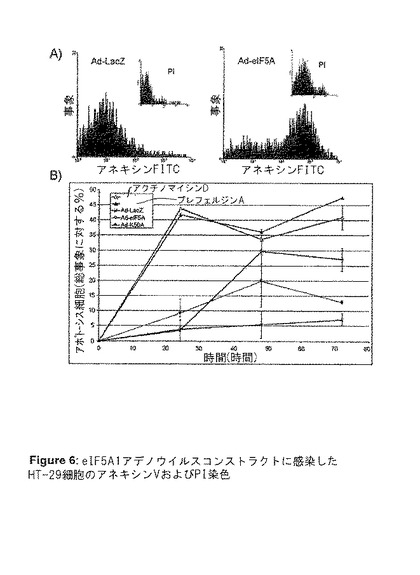
【図7】
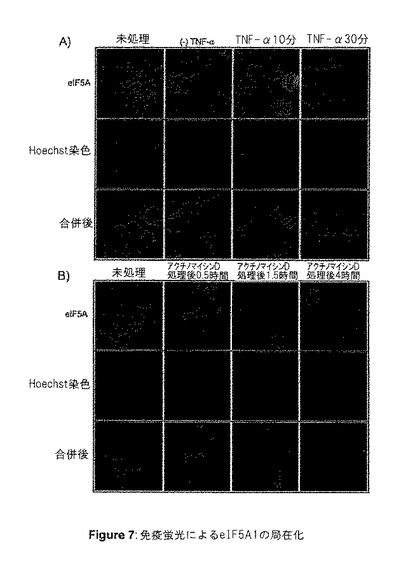
【図8】
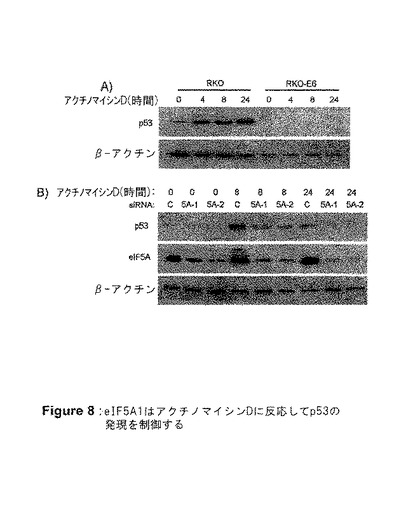
【図9】
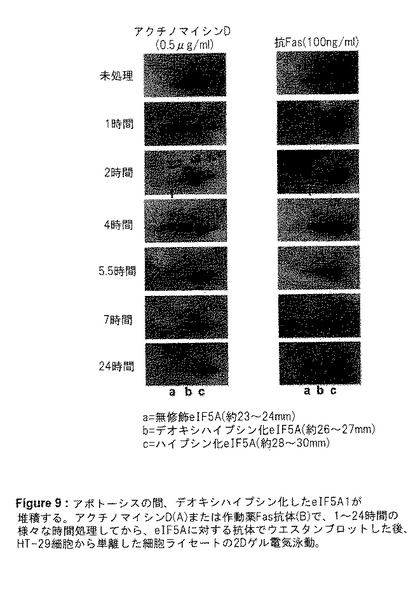
【図10】
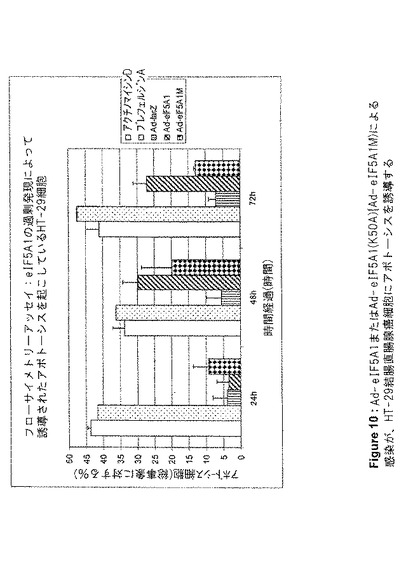
【図11】
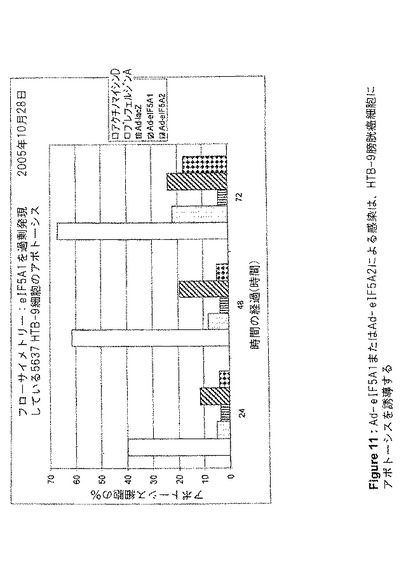
【図12】
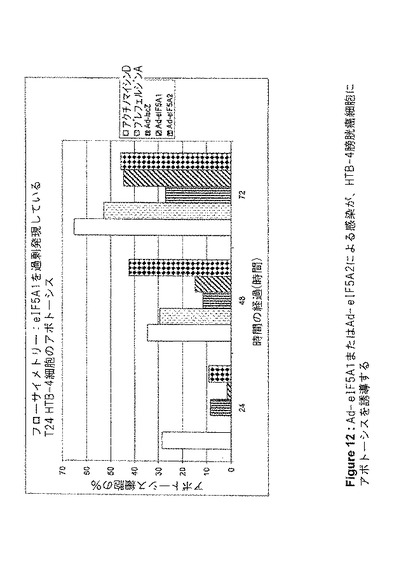
【図13】
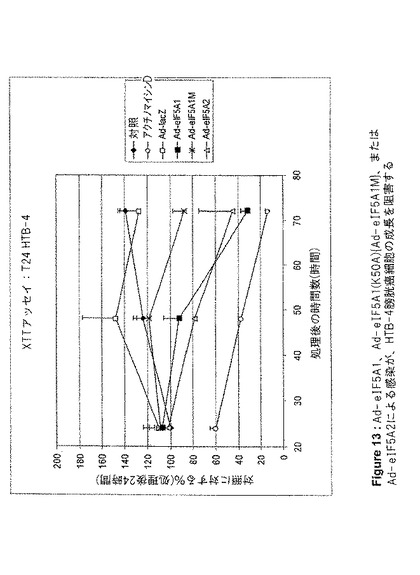
【図14】
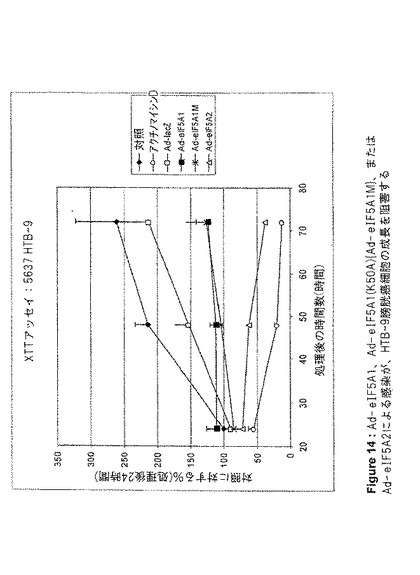
【図15】
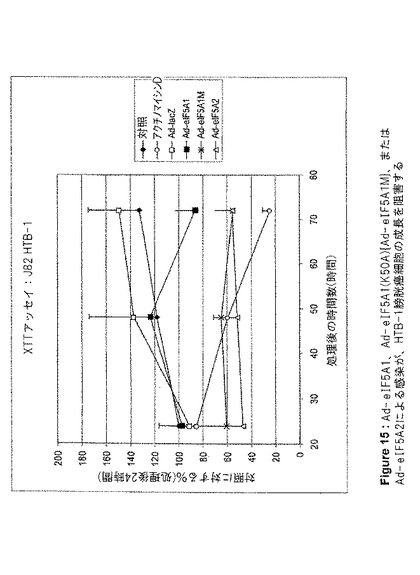
【図16】
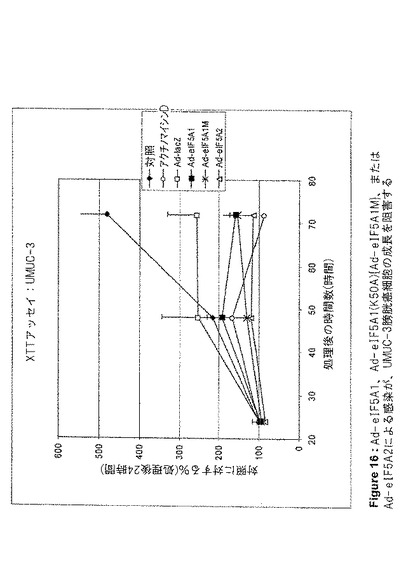
【図17】
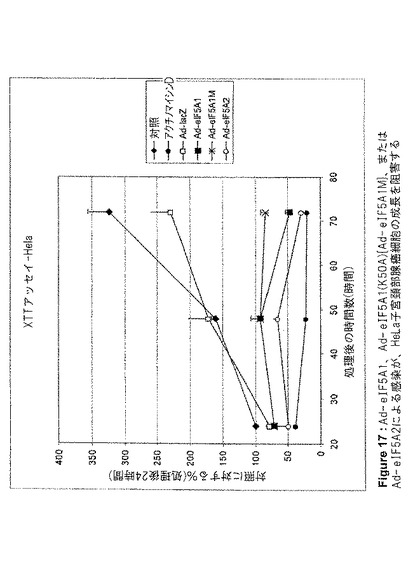
【図18】
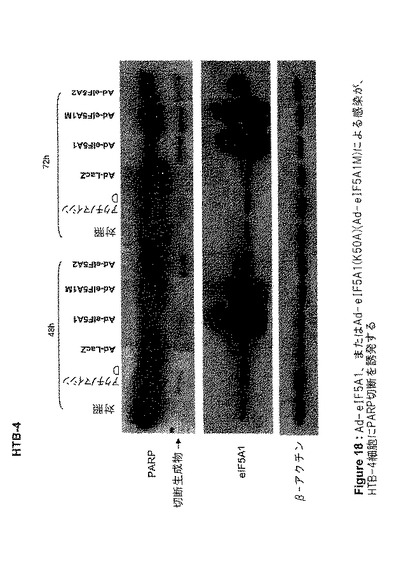
【図19】
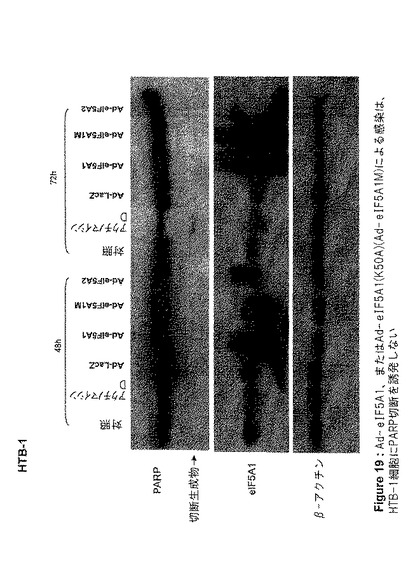
【図20】
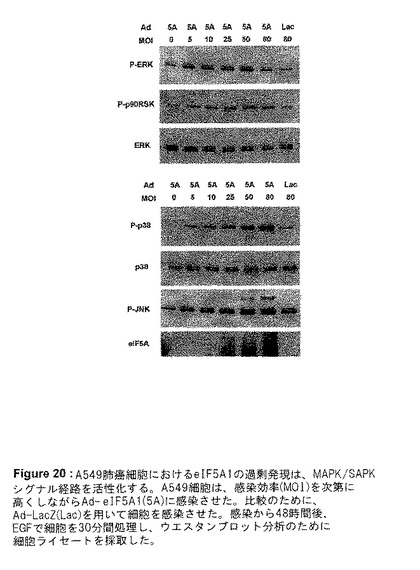
【図21】
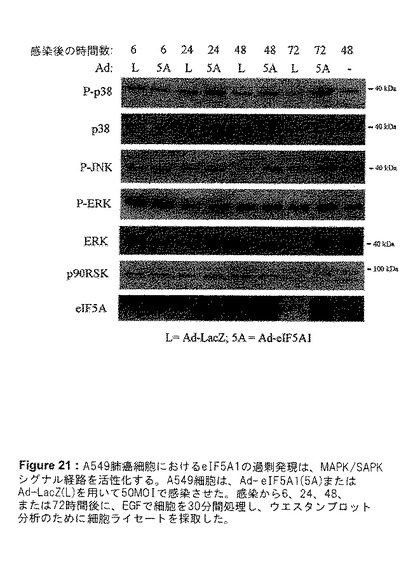
【図22】
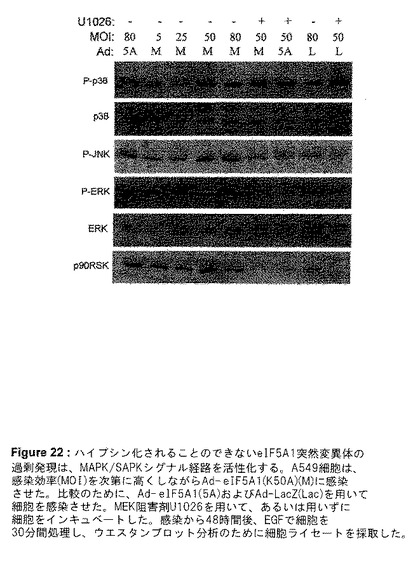
【図23】
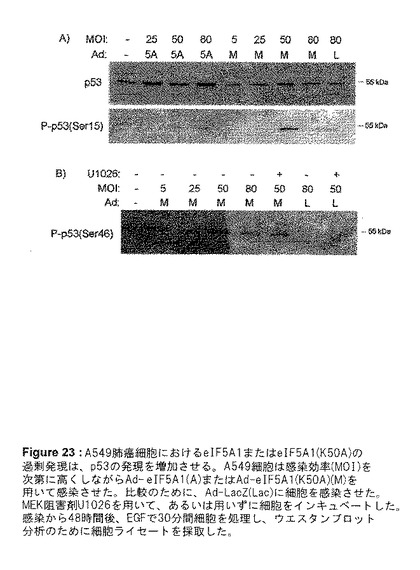
【図24】
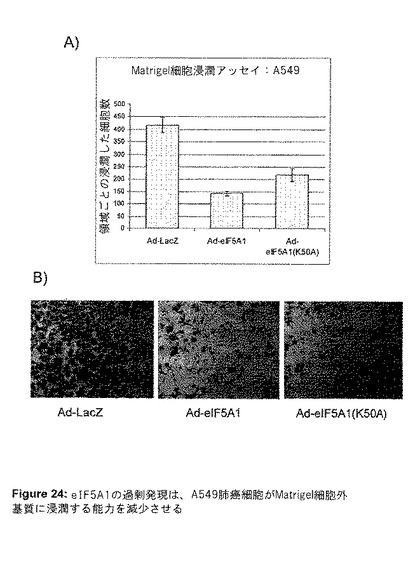
【図25】
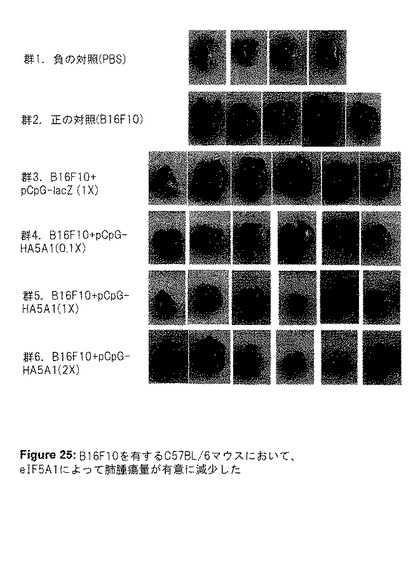
【図26】
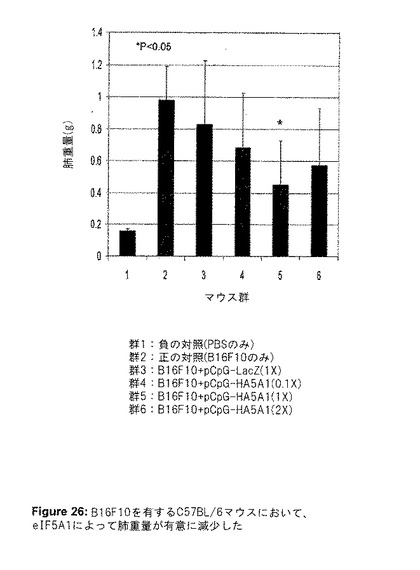
【図27】
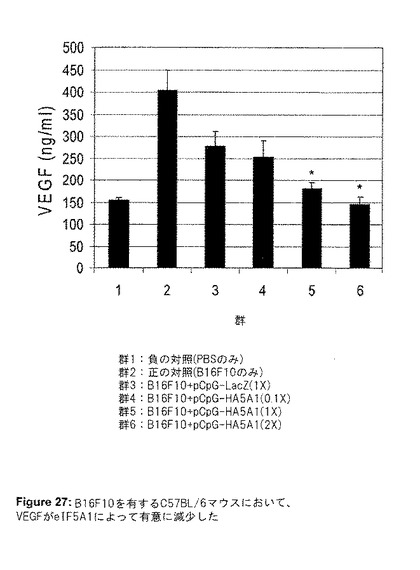
【図28】
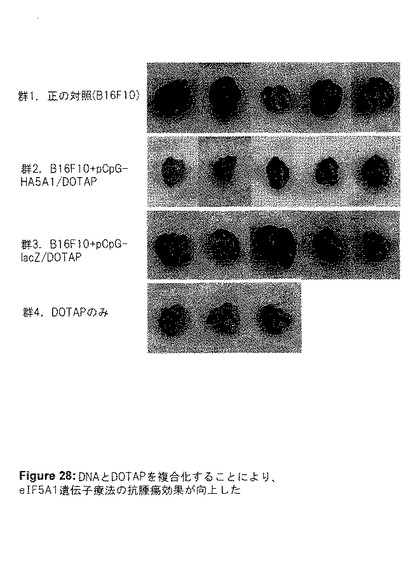
【図29】
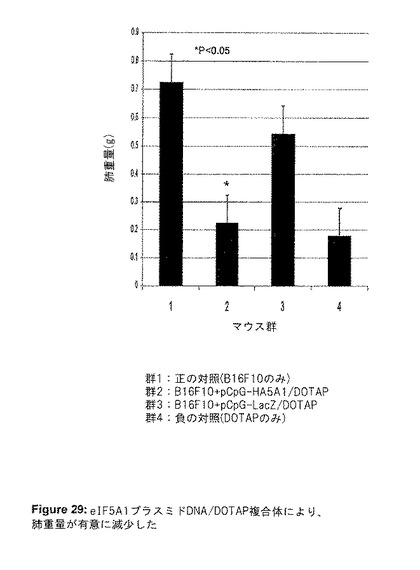
【図30】

【図31】
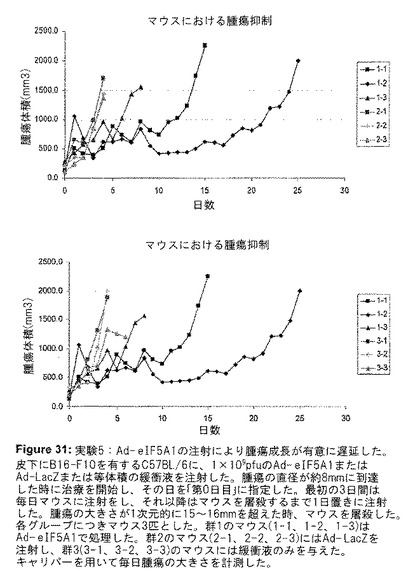
【図32】
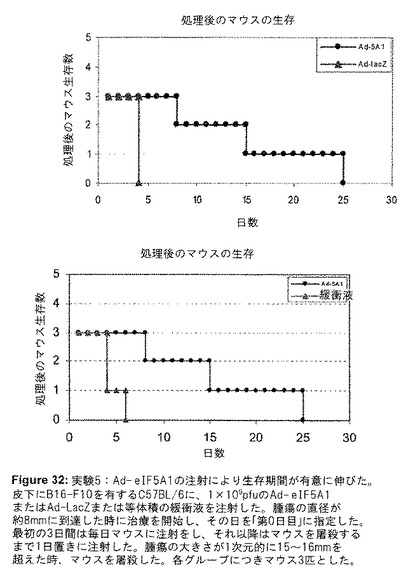
【図33】
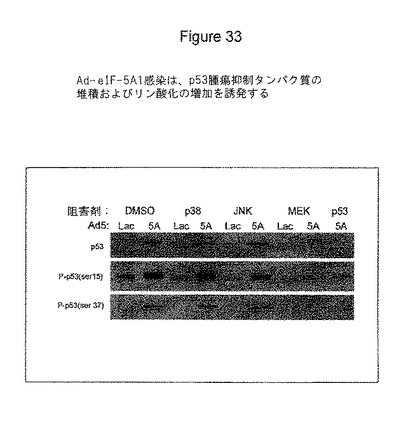
【図34】
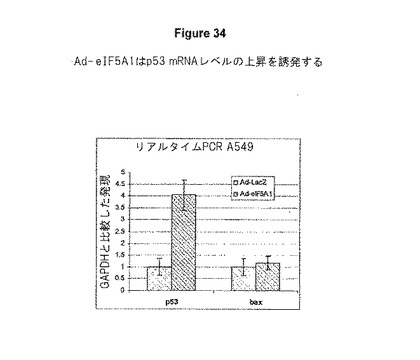
【図35】
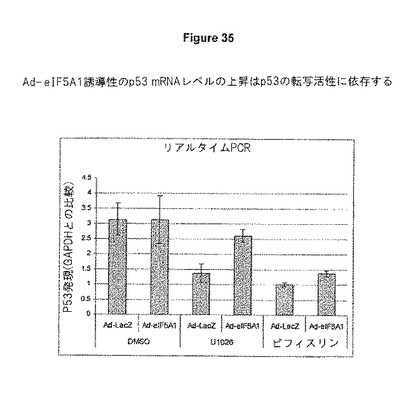
【図36】
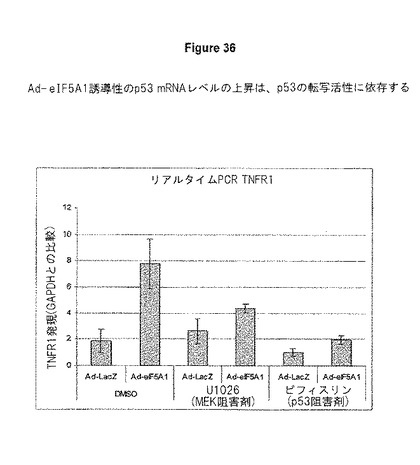
【図37】
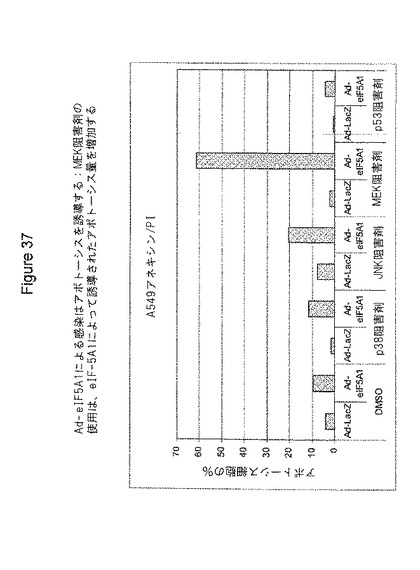
【図38】
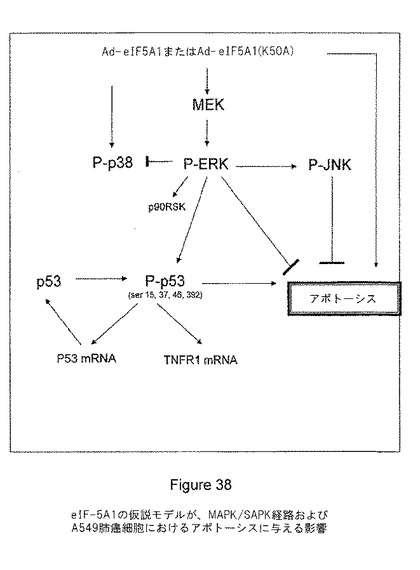
【図39】
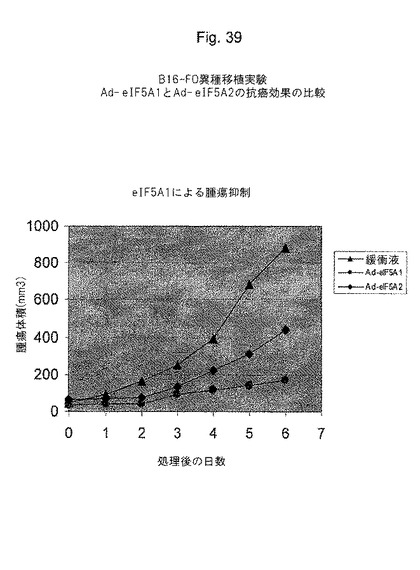
【図40】
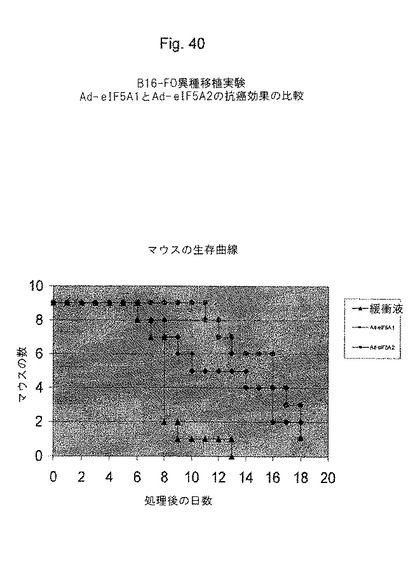
【図41】

【図42】

【図43】

【図44】

【図45】

【図46A】

【図46B】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46A】
【図46B】
【公表番号】特表2009−519351(P2009−519351A)
【公表日】平成21年5月14日(2009.5.14)
【国際特許分類】
【出願番号】特願2008−545953(P2008−545953)
【出願日】平成18年12月13日(2006.12.13)
【国際出願番号】PCT/US2006/061996
【国際公開番号】WO2007/070824
【国際公開日】平成19年6月21日(2007.6.21)
【出願人】(502458132)セネスコ テクノロジーズ,インコーポレイティド (16)
【Fターム(参考)】
【公表日】平成21年5月14日(2009.5.14)
【国際特許分類】
【出願日】平成18年12月13日(2006.12.13)
【国際出願番号】PCT/US2006/061996
【国際公開番号】WO2007/070824
【国際公開日】平成19年6月21日(2007.6.21)
【出願人】(502458132)セネスコ テクノロジーズ,インコーポレイティド (16)
【Fターム(参考)】
[ Back to top ]