
多能性幹細胞から神経前駆細胞への分化誘導法
本発明は、多能性幹細胞を低分子BMP阻害剤の存在下で培養することを含む、多能性幹細胞から神経前駆細胞を分化誘導するための方法、ならびに、この方法によって作製された人工神経前駆細胞を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、多能性幹細胞を神経前駆細胞へ分化誘導する方法に関する。
本発明はまた、上記方法によって作製された人工神経前駆細胞にも関する。
【背景技術】
【0002】
胚性幹細胞(ES細胞)や、動物の体細胞へ未分化細胞特異的遺伝子を導入することで得られる人工多能性幹細胞(iPS細胞)など多能性を有する細胞がこれまでに報告されている(特許文献1または特許文献2)。そこで、神経変性疾患や神経損傷の代替治療方法として使用可能である、これらの多能性幹細胞から分化誘導された神経細胞を移植する治療法が注目されている。ここでES細胞から神経細胞を分化誘導する方法として、(1)無血清培地中で胚様体を形成させて分化誘導する方法(SFEB法)(非特許文献1)、(2)ストローマ細胞上でES細胞を培養して分化誘導する方法(SDIA法)(非特許文献2)、(3)マトリゲル(Matrigel)上に薬剤を添加して培養する方法(非特許文献3)などが開発されている。
【0003】
しかし、これらの方法では、例えば、分化誘導後に未分化な細胞が残存することやサイトカインを用いるためコストが非常に高いなどのいくつかの問題を抱えている。そこで、サイトカインの代替物として多数の低分子化合物が開発されているが(特許文献3)、いずれの低分子化合物が効率の高い神経細胞へ分化誘導させるかは未だ不明である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5,843,780号
【特許文献2】国際公開第2007/069666号
【特許文献3】国際公開第2008/033408号
【非特許文献】
【0005】
【非特許文献1】Watanabe K, et al. Nat Neurosci. 8:288-96, 2005
【非特許文献2】Kawasaki H, et al. Neuron. 28:31-40, 2000
【非特許文献3】Chambers SM, et al. Nat Biotechnol. 27:275-80, 2009
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、多能性幹細胞からある種の低分子化合物を用いて神経前駆細胞を分化誘導する高効率の方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
すなわち、本発明は以下のとおり特徴付けられる。
(1)低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、多能性幹細胞から神経前駆細胞へ分化誘導する方法。
(2)前記培養時に低分子TGFβファミリー阻害剤をさらに存在させる、(1)に記載の方法。
(3)前記培養を、ストローマ細胞をフィーダー細胞として用いて行う、(1)または(2)に記載の方法。
(4)前記ストローマ細胞がPA6細胞である、(3)に記載の方法。
(5)前記培養を、無血清の条件下で、胚様体を形成して行う、(1)または(2)に記載の方法。
【0008】
(6)前記培養を、フィーダー細胞を用いないで、Matrigel(商標)被覆ディッシュ上で行う、(1)または(2)に記載の方法。
(7)前記低分子BMP阻害剤がドルソモルフィン(Dorsomorphin)またはLDN-193189である、(1)〜(6)のいずれか1つに記載の方法。
(8)前記低分子TGFβファミリー阻害剤がSB431542またはA-83-01である、(2)に記載の方法。
(9)前記多能性幹細胞が、胚性幹細胞または人工多能性幹細胞である、(1)〜(8)のいずれか1つに記載の方法。
(10)さらにERK(細胞外シグナル調節キナーゼ)阻害剤の存在下で多能性幹細胞を培養することを含む、(1)〜(9)のいずれか1つに記載の方法。
(11)(1)〜(10)のいずれか1つに記載の方法で作製された人工神経前駆細胞。
【発明の効果】
【0009】
本発明の上記方法では、分化誘導培地に、低分子BMP阻害剤を存在させること、好ましくは低分子BMP阻害剤と低分子TGFβファミリー阻害剤を組み合わせて共存させること、によって人工神経前駆細胞を高効率で作製することができる。
【図面の簡単な説明】
【0010】
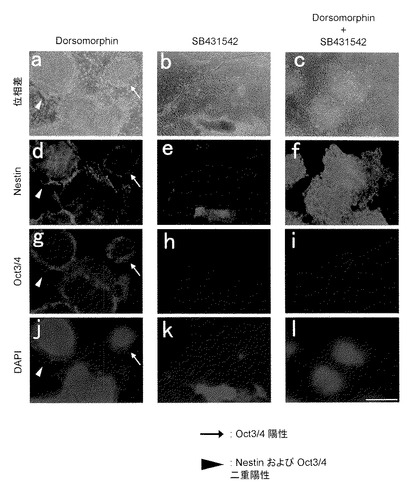
【図1】図1は、分化誘導14日目の、位相差顕微鏡像(a-c)、抗Nestin抗体を用いて得られた免疫染色像(d-f)、抗Oct3/4抗体を用いて得られた免疫染色像(g-i)、およびDAPIを用いて得られた免疫染色像(j-l)を示す。
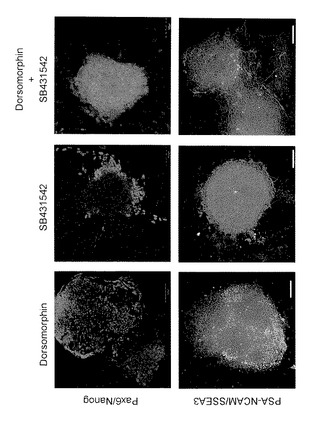
【図2】図2は、分化誘導14日目の、抗Pax6抗体(緑色)および抗Nanog抗体(赤色)を用いて得られた免疫染色像、ならびに、抗PSA-NCAM抗体(緑色)および抗SSEA3抗体(赤色)を用いて得られた免疫染色像を示す。
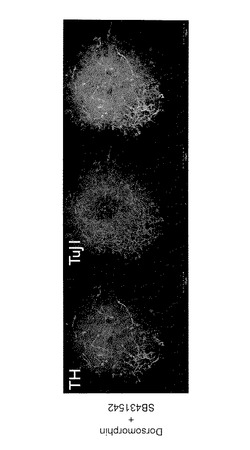
【図3】図3は、分化誘導21日目の、抗TH (チロシンヒドロキシラーゼ(tyrosine hydroxylase))抗体(緑色)および抗TuJ1抗体(赤色)を用いて得られた免疫染色像を示す。
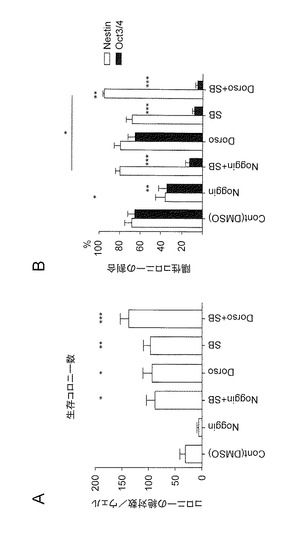
【図4】図4は、A: 全細胞株(KhES-1、KhES-2、KhES-3、G1、G4、B6およびB7:各細胞株につきn=4)における分化誘導14日目の1ウエルあたりに存在するコロニーの総数(n=28)、ならびに、B: 全細胞株(KhES-1、KhES-2、KhES-3、G1、G4、B6およびB7:各細胞株につきn=4)における分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の割合(%)(n=28)を示す。コロニー内に一つでも陽性の細胞が確認できれば陽性コロニーとして計数した。
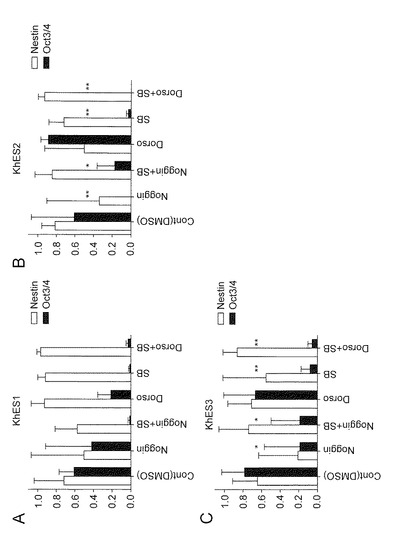
【図5】図5は、各ES細胞株(KhES-1(A)、KhES-2(B)またはKhES-3(C))の分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の割合を示す。
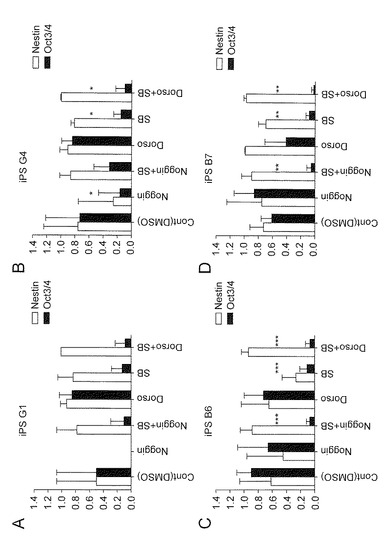
【図6】図6は、各iPS細胞株(G1(A)、G4(B)、B6(C)またはB7(D))の分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の比率を示す。
【図7】図7は、未分化ES細胞(KhES-1、KhES-2およびKhES-3)またはiPS細胞(G1、G4、B6およびB7)におけるNodal(A)、BMP2(B)、BMP4(C)およびBMP7(D)についてリアルタイムPCRにより測定されたmRNA発現量を示す。
【図8】図8は、DorsomorphinおよびSB431542を用いずPA6細胞上のみで分化誘導した14日目の各細胞株におけるPSA-NCAM陽性(緑色)およびSSEA4陽性(赤色)の細胞含有率を示したグラフを示す。
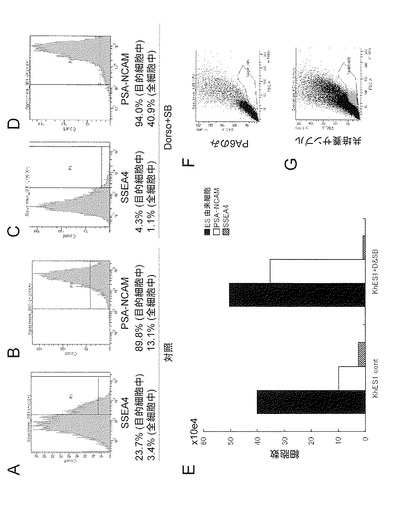
【図9】図9は、KhES1をPA6細胞上の培養のみで分化誘導させて作製された対照群(AおよびB)およびPA6細胞上で培地へDorsomorphinおよびSB431542を加えて分化誘導して作製された群(CおよびD)における、SSEA4発現(すなわち、陽性)細胞(AおよびC)およびPSA-NCAM発現細胞(BおよびD)の分布を示したFACSのグラフを示す。図に示された値について、上段にKhES1由来の細胞中の各マーカーを発現する細胞の割合(%)(「目的細胞中」の割合)および下段にKhES1由来の細胞およびPA6細胞を含むディッシュ内に存在した全ての細胞中の各マーカーを発現する細胞の割合(%)(「全細胞中)の割合)を示した。図9Eは、対照群(KhES1 cont)およびDorsomorphinおよびSB431542を用いて分化誘導した群(KhES1&DSB)におけるディッシュ1枚から得られたES細胞由来の細胞(黒色バー)、ES細胞由来のPSA-NCAM陽性細胞(白色バー)およびSSEA4陽性細胞(ハッチングバー)の細胞数を示すグラフである。細胞数は次の式:(ES細胞由来の細胞数)=(ディッシュ内の全細胞数)-(ディッシュ内のPA6フィーダー細胞数);(ES細胞由来のPSA-NCAM陽性細胞数)=(ディッシュ内の全細胞数)×(ディッシュ内に存在した全ての細胞中のPSA-NCAM陽性細胞の割合);ならびに、(ES細胞由来のSSEA4陽性細胞)=(ディッシュ内の全細胞数)×(ディッシュ内に存在した全ての細胞中のSSEA4陽性細胞の割合)により算出された。図9Fおよび図9Gに、PA6細胞とES細胞由来の細胞に特徴的な分布の例(F:PA6細胞のみ、G: PA6細胞とKhES1(DorsomorphinおよびSB431542の非存在下での培養))を示す。
【図10】図10Aは、iPS細胞(G4)から、フィーダー細胞なしで胚様体を形成させ、DorsomorphinおよびSB431542を添加した培地での細胞の培養による分化誘導後14日目のPSA-NCAM陽性細胞の割合(%)を示したグラフである。ここで、赤色の曲線は抗体が存在しない陰性対照であり、青色の曲線は抗PSA-NCAM抗体により染色された細胞に関する結果を示す。また、図10Bは、上記方法で分化誘導した細胞でのNestin(緑色)および Pax6(赤色)の免疫染色像を示す。
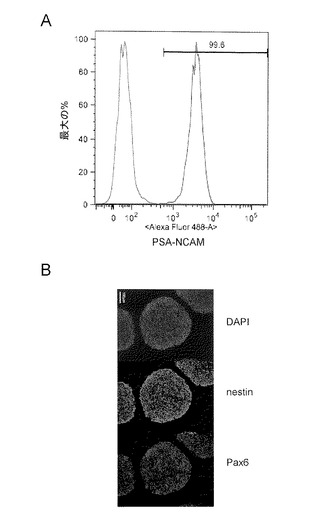
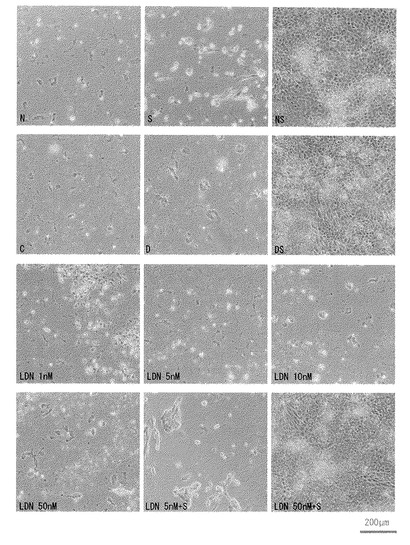
【図11】図11は、以下に示す各薬剤を添加した培地でMatrigel法でフィーダー細胞非存在下で培養することによりiPS細胞(G4)の分化を誘導して14日目の位相差顕微鏡像を示す。図中、「N」は、ノギン(Noggin)を示し、「S」は、SB431542の添加を示し、「NS」は、NogginおよびSB431542の添加を示し、「C」は、対照DMSOの添加を示し、「D」は、Dorsomorphinの添加を示し、「DS」はDorsomorphinおよびSB431542の添加を示し、「LDN」は、LDN-193189の添加を示し、「LDN+S」は、LDN-193189およびSB431542の添加を示す。
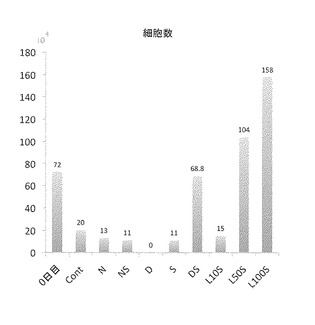
【図12】図12は、以下に示す各薬剤の添加後14日目の、ならびに各薬剤の添加前の、1ウエルあたりに存在する細胞の数(すなわち、細胞数)を示す。図中、「0日目」は、薬剤の添加前を示し、「Cont」は、DMSOを添加した対照群を示し、「N」は、Nogginを添加した群を示し、「NS」は、NogginおよびSB431542を添加した群を示し、「D」は、Dorsomorphin添加群を示し、「S」は、SB431542添加群を示し、「DS」は、DorsomorphinおよびSB431542を添加した群を示し、「L10S」は、10nM LDN-193189およびSB431542を添加した群を示し、「L50S」は、50nM LDN-193189およびSB431542を添加した群を示し、「L100S」は、100nM LDN-193189およびSB431542を添加した群を示す。
【図13】図13は、Matrigel法でフィーダー細胞非存在下で培養することによりiPS細胞(G4)の分化を誘導して14日目の、抗Pax6抗体(緑色)および抗Nanog抗体(赤色)を用いて得られた免疫染色像、ならびに、DAPI(青色)を用いて得られた免疫染色像を示す。図中、「SB」はSB431542を示し、また「LDN」はLDN-193189を示す。
【図14A】図14Aは、分化誘導して14日目の位相差顕微鏡像を示す。ここで、分化誘導は、SDIA法に従ってES細胞株(Kh-ES5)をPA6細胞と一緒に共培養したのち、該細胞を、5〜500nM LDN-193189およびSB431542を添加した培地中で培養することによって行われた。
【図14B】図14Bは、分化誘導して14日目の、抗ネスチン(Nestin)抗体(緑色)およびDAPI(青色)を用いて得られた免疫染色像を示す。ここで、分化誘導は、SDIA法に従ってES細胞株(Kh-ES5)をPA6細胞と一緒に共培養したのち、該細胞を、5〜500nM LDN-193189およびSB431542を添加した培地中で培養することによって行われた。
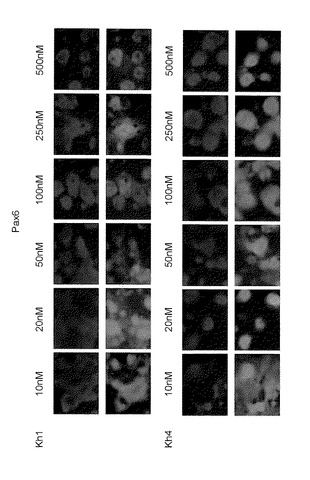
【図15A】図15Aは、SDIA法に従って、ES細胞株(Kh-ES1またはKh-ES4)をPA6細胞と一緒に共培養し、ついで、5〜500nM LDN-193189およびSB431542を添加した培地中で細胞を培養することによって細胞の分化を誘導して14日目の、DAPI(青色)および抗Nestin抗体(緑色)を用いて得られた免疫染色像を示す。
【図15B】図15Bは、SDIA法に従って、ES細胞株(Kh-ES1またはKh-ES4)をPA6細胞と一緒に共培養し、ついで、5〜500nM LDN-193189およびSB431542を添加した培地中で細胞を培養することによって細胞の分化を誘導して14日目の、抗Pax6抗体(緑色)を用いて得られた免疫染色像を示す。
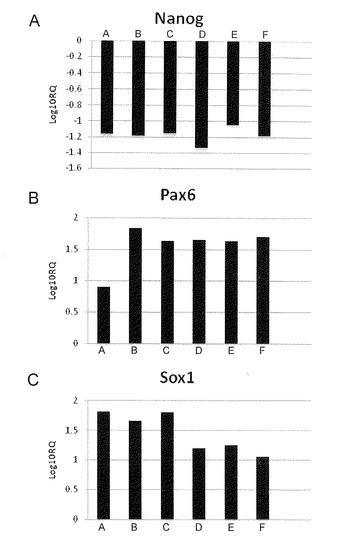
【図16】図16は、フィーダー細胞を用いない方法でヒトiPS細胞(404C2)を培養することによって分化誘導された細胞におけるNanog (A)、Pax6 (B)およびSox1(C)に関する定量PCRの結果を示す。この結果は、未処理の細胞に対する相対対数値を示している。「A」から「F」は、以下の条件を示す。すなわち、「A」は、2μMのDorsomorphinおよび10μMのSB431542を含有するold DFK5%であり、「B」は、100nMのLDN913189および0.5μMのA-83-01を含有するold GMK8%であり、「C」は、2μMのDorsomorphinおよび10μMのSB431542を含有するDFK5%であり、「D」は、100nMのLDN913189および0.5μMのA-83-01を含有するGMK8%であり、「E」は、100nMのLDN913189および10μMのSB431542を含有するGMK8%であり、ならびに「F」は、100nMのLDN913189および0.5μMのA-83-01 + 0.5μMのPD0325901を含有するGMK8%である。
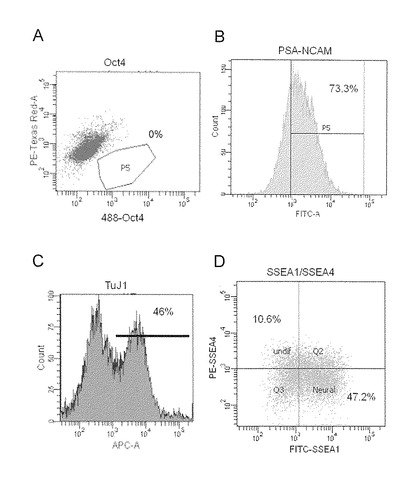
【図17】図17は、100nMのLDN913189および0.5μMのA-83-01を含有するGMK8%の条件下で、フィーダー細胞を用いない方法でiPS細胞(404C2)を培養することによって得られた分化細胞に関する、Oct3/4の二次元展開(A)、PSA-NCAM発現細胞(B)、Tuj-1発現細胞(C)、ならびに、SSEA1およびSSEA4の二次元展開(D)を示すFACSグラフである。
【発明を実施するための形態】
【0011】
本発明を以下に詳細に説明する。
本発明は、上記のとおり、低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、多能性幹細胞から神経前駆細胞を分化誘導する方法に関する。
【0012】
<多能性幹細胞>
本発明で使用可能な多能性幹細胞は、該幹細胞を、外胚葉、中胚葉および内胚葉に由来する生体に存在するすべての細胞に分化する能力である多能性を有し、かつ、増殖能をも併せもつ幹細胞である。そのような幹細胞の具体例には、以下のものに限定されないが、胚性幹(ES)細胞、核移植により得られるクローン胚由来の胚性幹(ntES;核移植ES)細胞、精子幹細胞(「GS細胞」)、胚性生殖細胞(「EG細胞」)、人工多能性幹(iPS)細胞などが含まれる。好ましい多能性幹細胞は、ES細胞、ntES細胞、およびiPS細胞である。
【0013】
(A) 胚性幹細胞
ES細胞は、ヒトやマウスなどの哺乳動物の初期胚(例えば胚盤胞)の内部細胞塊から樹立された、多能性と自己複製による増殖能を有する幹細胞である。
ES細胞は、受精卵の8細胞期、桑実胚後の胚である胚盤胞の内部細胞塊に由来する胚由来の幹細胞であり、成体を構成するあらゆる細胞に分化する能力、いわゆる分化多能性と、自己複製による増殖能とを有している。ES細胞は、マウスで1981年に発見され(M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156)、その後、ヒト、サルなどの霊長類でもES細胞株が樹立された (J.A. Thomson et al. (1999), Science 282:1145-1147; J.A. Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848;J.A. Thomson et al. (1996), Biol. Reprod., 55:254-259; J.A. Thomson and V.S. Marshall (1998), Curr. Top. Dev. Biol., 38:133-165)。
【0014】
ES細胞は、対象動物の受精卵の胚盤胞から内部細胞塊を取出し、内部細胞塊を線維芽細胞のフィーダー上で培養することによって樹立することができる。また、継代培養による細胞の維持は、白血病抑制因子(leukemia inhibitory factor (LIF))、塩基性線維芽細胞成長因子(basic fibroblast growth factor (bFGF))などの物質を添加した培地を用いて行うことができる。ヒトおよびサルのES細胞の樹立と維持の方法については、例えばH. Suemori et al. (2006), Biochem. Biophys. Res. Commun., 345:926-932; M. Ueno et al. (2006), Proc. Natl. Acad. Sci. USA, 103:9554-9559; H. Suemori et al. (2001), Dev. Dyn., 222:273-279;H. Kawasaki et al. (2002), Proc. Natl. Acad. Sci. USA, 99:1580-1585などに記載されている。
【0015】
ES細胞作製のための培地として、例えば0.1mM 2-メルカプトエタノール、0.1mM 非必須アミノ酸、2mM L-グルタミン酸、20% KSR及び4ng/ml β‐FGFを補充したDMEM/F-12培地を使用する。ヒトES細胞は、37℃、湿潤雰囲気(5% CO2)下で前記培地を用いて維持することができる。また、ES細胞は、4〜5日おきに継代する必要があり、このとき、継代は、例えば1mM CaCl2及び20% KSRを含有するPBS中の0.25% トリプシン及び0.1mg/mlコラゲナーゼIVを用いて行うことができる。
【0016】
ES細胞の選択は、一般に、例えばアルカリホスファターゼ、Oct-3/4、Nanogなどの遺伝子マーカーの発現を指標にしてリアルタイムPCR法で行うことができる。特に、ヒトES細胞の選択では、OCT-3/4、NANOG、ECADなどの遺伝子マーカーの発現を指標とすることができる(E. Kroon et al. (2008), Nat. Biotechnol., 26:443-452)。
【0017】
ヒトES細胞株である例えばKhES-1、KhES-2、KhES-3、KhES-4およびKhES-5は、京都大学再生医科学研究所(京都、日本)から入手可能である。
【0018】
(B) 精子幹細胞
精子幹細胞は、精巣由来の多能性幹細胞であり、精子形成のための起源となる細胞である。この細胞は、ES細胞と同様に、種々の系列の細胞に分化誘導可能であり、例えばマウス胚盤胞に移植するとキメラマウスを作出できる(M. Kanatsu-Shinohara et al. (2003) Biol. Reprod., 69:612-616; K. Shinohara et al. (2004), Cell, 119:1001-1012)。神経膠細胞系由来神経栄養因子(glial cell line-derived neurotrophic factor (GDNF))を含む培地で自己複製可能であるし、またES細胞と同様の培養条件下で継代を繰り返すことによって、精子幹細胞を得ることができる(竹林正則ら(2008),実験医学,26巻,5号(増刊),41〜46頁,羊土社(東京、日本))。
【0019】
(C) 胚性生殖細胞
胚性生殖細胞は、胎生期の始原生殖細胞から樹立される、ES細胞と同様な多能性をもつ細胞であり、LIF、bFGF、幹細胞因子(stem cell factor)などの物質の存在下で始原生殖細胞を培養することによって樹立しうる(Y. Matsui et al. (1992), Cell, 70:841-847; J.L. Resnick et al. (1992), Nature, 359:550-551)。
【0020】
(D) 人工多能性幹細胞
人工多能性幹(iPS)細胞は、ある特定の再プログラミング因子を、DNA又はタンパク質の形態で体細胞に導入することによって作製することができる、ES細胞とほぼ同等の特性、例えば分化多能性と自己複製による増殖能、を有する体細胞由来の人工の幹細胞である(K. Takahashi and S. Yamanaka (2006) Cell, 126:663-676; K. Takahashi et al. (2007), Cell, 131:861-872; J. Yu et al. (2007), Science, 318:1917-1920; Nakagawa, M.ら,Nat. Biotechnol. 26:101-106 (2008);国際公開WO 2007/069666)。再プログラム化因子は、ES細胞に特異的に発現している遺伝子またはES細胞の未分化維持に重要な役割を果たす遺伝子もしくはその遺伝子産物であれば良く、特に限定されないが、例えばOCT3/4、SOX2及びKLF4; OCT3/4、KLF4及びC-MYC; OCT3/4、SOX2、KLF4及びC-MYC; OCT3/4及びSOX2; OCT3/4、SOX2及びNANOG; OCT3/4、SOX2及びLIN28; OCT3/4及びKLF4などの組み合わせを含む。
【0021】
これらの因子は、タンパク質の形態で、例えばリポフェクション、細胞膜透過性ペプチドとの結合、マイクロインジェクションなどの手法によって体細胞内に導入してもよいし、あるいは、DNAの形態で、例えば、ウイルス、プラスミド、人工染色体などのベクター、リポフェクション、リポソームを利用する手法、マイクロインジェクションなどの手法によって体細胞内に導入してもよい。ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター(Cell, 126, pp.663-676, 2006; Cell, 131, pp.861-872, 2007; Science, 318, pp.1917-1920, 2007)、アデノウイルスベクター(Science, 322, 945-949, 2008)、アデノ随伴ウイルスベクター、センダイウイルスベクターなどが例示される。また、人工染色体ベクターとしては、例えばヒト人工染色体(HAC)、酵母人工染色体(YAC)、細菌人工染色体(BAC、PAC)ベクターなどが含まれる。プラスミドとしては、哺乳動物細胞用プラスミドを使用しうる(Science, 322:949-953, 2008)。ベクターには、核初期化物質が発現可能なように、プロモーター、エンハンサー、リボゾーム結合配列、ターミネーター、ポリアデニル化サイトなどの制御配列を含むことができるし、さらに、必要に応じて、薬剤耐性遺伝子(例えばカナマイシン耐性遺伝子、アンピシリン耐性遺伝子、ピューロマイシン耐性遺伝子など)、チミジンキナーゼ遺伝子、ジフテリアトキシン遺伝子などの選択マーカー配列、緑色蛍光タンパク質(GFP)、βグルクロニダーゼ(GUS)、FLAGなどのレポーター遺伝子配列などを含むことができる。また、上記ベクターには、体細胞への導入後、再プログラミング化因子をコードする遺伝子、またはプロモーターと結合する再プログラミング化因子をコードする遺伝子、を共に切除するために、それらの遺伝子の各末端にLoxP配列を有してもよい。
【0022】
再プログラム化に際して、誘導効率を高めるために、上記の因子の他に、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA SmartpoolTM (Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、DNAメチルトランスフェラーゼ阻害剤(例えば5’-アザシチジン)(Nat. Biotechnol., 26(7): 795-797 (2008))、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-チャネルカルシウムアゴニスト(例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、p53阻害剤(例えばp53に対するsiRNAおよびshRNA (Cell Stem Cell, 3, 475-479 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、Wntシグナル伝達因子(例えば可溶性Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、2i/LIF (「2i」はmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤を示す、PloS Biology, 6(10), 2237-2247 (2008))、miR-291-3p、miR-294、miR-295などのmiRNA(R.L. Judson et al., Nat. Biotech., 27:459-461, 2009)、ALK5阻害剤(例えば、SB431542)、等を使用することができる。
【0023】
iPS細胞誘導のための培養培地としては、例えば(1) 10〜15%FBSを含有するDMEM、DMEM/F12又はDME培地(これらの培地にはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)、(2) bFGF又はSCFを含有するES細胞培養用培地、例えばマウスES細胞培養用培地(例えばTX-WES培地、トロンボX社)又は霊長類ES細胞培養用培地(例えば霊長類(ヒトおよびサル)ES細胞用培地、リプロセル、京都、日本)、などが含まれる。
【0024】
培養法の例としては、たとえば、37℃、5%CO2存在下にて、10%FBS含有DMEM又はDMEM/F12培地上で体細胞と再プログラム化因子(DNA又はタンパク質)を接触させ約4〜7日間培養し、その後、細胞をフィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上にまきなおし、体細胞と再プログラム化因子の接触から約10日後からbFGF含有霊長類ES細胞培養用培地で培養し、該接触から約30〜約45日又はそれ以上ののちにiPS様コロニーを生じさせることができる。
【0025】
あるいは、細胞は、37℃、5% CO2存在下にて、フィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上で10%FBS含有DMEM培地(これにはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)で培養し、約25〜約30日又はそれ以上ののちにES様コロニーを生じさせることができる。
【0026】
さらにまた、細胞は、iPS細胞の誘導効率を高めるために、酸素濃度が5%〜10%となる低酸素条件下で培養してもよい(WO 2010/013845)。
【0027】
上記培養の間には、培養開始2日目以降から毎日1回新鮮な培地と培地交換を行う。また、核初期化(再プログラム化)に使用する体細胞の細胞数は、限定されないが、培養ディッシュ100cm2あたり約5×103〜約5×106細胞の範囲である。
【0028】
マーカー遺伝子として薬剤耐性遺伝子などの遺伝子を用いた場合は、対応する薬剤を含む培地(選択培地)で培養を行うことによりマーカー遺伝子発現細胞を選択することができる。またマーカー遺伝子が蛍光タンパク質遺伝子の場合は蛍光顕微鏡で観察することによって、マーカー遺伝子を発現する細胞を検出することができる。マーカー遺伝子が発光酵素遺伝子である場合は、発光基質を加えることによってマーカー遺伝子発現細胞を検出することができる。
【0029】
本明細書中で使用する「体細胞」なる用語は、卵子、卵母細胞および精母細胞などの生殖系列細胞、全能性細胞、ならびにES細胞を除くあらゆる動物細胞(好ましくは、ヒトを含む哺乳動物細胞)をいう。体細胞には、非限定的に、胎児(仔)の体細胞、新生児(仔)の体細胞、及び成熟した健全なもしくは疾患性の体細胞のいずれも包含されるし、また、初代培養細胞、継代細胞、及び樹立された株化細胞のいずれも包含されるし、さらにまた、組織幹細胞や組織前駆細胞も包含される。具体的には、体細胞は、非限定的に、例えば(1)神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)、(2)組織前駆細胞、(3)リンパ球、上皮細胞、内皮細胞、筋肉細胞、線維芽細胞(皮膚細胞等)、毛細胞、肝細胞、胃粘膜細胞、腸細胞、脾細胞、膵細胞(膵外分泌細胞等)、脳細胞、肺細胞、腎細胞、皮膚細胞等の分化した細胞などを包含する。
【0030】
(E) 核移植により得られたクローン胚由来のES細胞
nt ES細胞は、核移植技術によって作製されたクローン胚由来のES細胞であり、受精卵由来のES細胞とほぼ同じ特性を有している(T. Wakayama et al. (2001), Science, 292:740-743; S. Wakayama et al. (2005), Biol. Reprod., 72:932-936; J. Byrne et al. (2007), Nature, 450:497-502)。具体的には、未受精卵の核を体細胞の核と置換することによって得られたクローン胚由来の胚盤胞の内部細胞塊から樹立されたES細胞がnt ES(nuclear transfer ES)細胞である。nt ES細胞の作製のためには、核移植技術(J.B. Cibelli et al. (1998), Nature Biotechnol., 16:642-646)とES細胞作製技術(上記)との組み合わせが利用される(若山清香ら(2008),実験医学,26巻,5号(増刊), 47〜52頁)。核移植によって、哺乳動物の除核した未受精卵に、体細胞の核を注入し、数時間培養することで再プログラム化することができる。
【0031】
<低分子BMP阻害剤>
本発明で使用しうる低分子BMP阻害剤とは、BMP(bone morphogenetic protein)とBMP受容体(I型又はII型)との結合を介するBMPシグナル伝達(BMP signaling)の阻害に関与する低分子阻害剤であり、天然の阻害剤であるNoggin、chordin、follistatinなどのタンパク質性阻害剤とは異なる。本明細書で使用される「低分子」という用語は、有機分子または無機分子を意味し、巨大分子、例えば大タンパク質(例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超すタンパク質)、大核酸 (例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超す核酸)、あるいは、大多糖類(例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超す多糖類)を包含しない。この阻害剤は、多能性幹細胞を神経前駆細胞へ分化誘導する作用を有しているべきである。このような性質をもつ低分子BMP阻害剤には、転写因子SMAD1、SMAD5又はSMAD8を活性化する能力をもつBMP2、BMP4、BMP6又はBMP7を阻害する化合物である、例えばDorsomorphin (すなわち、6-[4-(2-ピペリジン-1-イル-エトキシ)フェニル]-3-ピリジン-4-イル-ピラゾロ[1,5-a]ピリミジン)及びその誘導体が包含される(P. B. Yu et al. (2007), Circulation, 116:II_60; P.B. Yu et al. (2008), Nat. Chem. Biol., 4:33-41; J. Hao et al. (2008), PLoS ONE (www.plozone.org), 3(8):e2904)。Dorsomorphinは市販されており、例えばSigma-Aldrichなどから入手可能である。Dorsomorphinは、BMP受容体へのBMPの結合を阻害することによって上記のBMPシグナル伝達を阻害する生物活性を有する。この他に、BMP I型受容体キナーゼ阻害剤の具体例には、LDN-193189(すなわち、4-(6-(4-(ピペラジン-1-イル)フェニル)ピラゾロ[1,5-a]ピリミジン-3-イル)キノリン)およびその誘導体が包含される(Yu PB et al. Nat Med, 14: 1363-9, 2008)。LDN-193189は、例えばStemgent社から市販されている。
【0032】
<低分子TGFβファミリー阻害剤>
本発明によれば、上記低分子BMP阻害剤を、低分子TGFβ(transforming growthfactor β)ファミリー阻害剤と組み合わせることによって、多能性幹細胞の神経前駆細胞誘導の効率性を顕著に改善することができる。
【0033】
本明細書で使用される「低分子TGFβファミリー阻害剤」は、TGFβファミリーのシグナル伝達に干渉する低分子阻害剤であり、例えばSB431542、SB202190(R.K.Lindemann et al., Mol. Cancer 2:20 (2003))、SB505124 (GlaxoSmithKline)、NPC30345 、SD093、 SD908、SD208 (Scios)、LY2109761、LY364947、LY580276 (Lilly Research Laboratories)、A-83-01 (WO 2009146408)などが包含され、SB431542又はA-83-01が好ましい。
【0034】
TGFβファミリーのメンバーは、有糸分裂、細胞分化、胚パターン形成および器官形成などの細胞過程および発生過程を調節する。例えば、TGFβシグナル伝達は、I型及びII型のセリン−トレオニンキナーゼ受容体のヘテロメリックな受容体複合体を介して行われ、この複合体は下流のSmadシグナル伝達過程を活性化する。具体的には、受容体複合体にTGFβが結合すると、TGFβII型受容体がTGFβI型受容体をリン酸化し、このTGFβI型受容体が、受容体調節されるSmad(R-Smad)をリン酸化することによって、下流応答が開始される。活性化R-SmadはSmad4と多量体複合体を形成することによって、活性化R-Smadは核に移動し、標的遺伝子の転写調節が誘導される。
【0035】
このようなTGFβファミリーシグナル伝達が阻害されると、多能性幹細胞は神経前駆細胞へ分化誘導される。さらに、この阻害とともに、上記のBMPシグナル伝達が阻害されるときには、神経前駆細胞の誘導率が増すだけでなく、未分化な細胞(すなわち、多能性幹細胞)の残存率がより低下する、すなわち神経前駆細胞への変換率が向上する。
【0036】
<フィーダー細胞>
本発明では、フィーダー細胞は必ずしも必要ないが、フィーダー細胞を存在させることも可能である。フィーダー細胞としては、例えば胎仔線維芽細胞、ストローマ細胞などが挙げられる。胎仔線維芽細胞には、例えばMEF(マウス胎仔線維芽細胞)、STO細胞(マウス胎仔線維芽細胞株)、SNL細胞(STO細胞のサブクローン;例えばSNL 76/7細胞)などが含まれる。また、ストローマ細胞には、例えばPA6細胞(マウスストローマ細胞株(理研BRC Cell Bank(日本)))、MS-5細胞(Exp Hematol. 17:145-53 (1989))、OP9細胞(Science. 265:1098-1101 (1994))などが含まれる。SDIA法は、ES細胞をストローマ細胞、特にPA6細胞と一緒に共培養することによってほぼ選択的に神経前駆細胞へ分化させる方法であるが、本発明では、フィーダー細胞が不在であっても、上記の低分子BMP阻害剤、あるいは該低分子BMP阻害剤と上記の低分子TGFβファミリー阻害剤の組み合わせ、を分化誘導培地に存在させるだけで選択的に神経前駆細胞に分化させることができる。このような培養条件に加えて、フィーダー細胞を使用すると、神経前駆細胞への分化効率をさらに向上させることができる。
【0037】
しかし、たとえそうであっても、ヒトなどの哺乳動物への神経前駆細胞、またはさらにその分化細胞である神経細胞やグリア細胞、の移植を考えた場合、ドナーに対して異種の細胞の使用はできる限り避けることが望ましいことは言うまでもない。
【0038】
<神経前駆細胞の分化誘導>
(A) 分化培地
培地は、動物細胞の培養に用いられる培地を基礎培地として調製することができる。基礎培地としては、例えばIMDM培地、Medium 199培地、Eagle’s Minimum Essential Medium (EMEM)培地、αMEM培地、Doulbecco’s modified Eagle’s Medium (DMEM)培地、Ham’s F12培地、RPMI 1640培地、Fischer培地、Glasgow MEMおよびこれらの混合培地などが包含される。培地には、血清が含有されていてもよいし、あるいは無血清でもよい。
【0039】
培地はまた、必要に応じて、例えば、アルブミン、トランスフェリン、Knockout Serum Replacement(KSR)(ES細胞培養時のFBSの血清代替物)、脂肪酸、インスリン、コラーゲン前駆体、微量元素、2-メルカプトエタノール、3’-チオールグリセロール、B27-サプリメント、N2-サプリメントなどの1つ以上の血清代替物を含んでもよいし、ならびに、脂質、アミノ酸、非必須アミノ酸、ビタミン、増殖因子、サイトカイン、抗生物質、抗酸化剤、ピルビン酸、緩衝剤、無機塩類などの1つ以上の物質も含有しうる。
【0040】
培地はまた、前記低分子BMP阻害剤および/または前記低分子TGFβファミリー阻害剤を適宜含有してもよく、さらに前記フィーダー細胞の培養上清を含有してもよい。培地はさらに、ERK (extracellular signal-regulated kinase)阻害剤類のいずれかを含んでもよい。
【0041】
分化培地の例は、後述の実施例に記載されるような、5% knockout serum replacement (KSR)、2mM L-グルタミン、非必須アミノ酸類及び1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12混合培地、あるいは、8% KSR、1μM 2ME、ピルビン酸および非必須アミノ酸類を含有するGlasgow MEMである。
【0042】
(B) 分化誘導法
本発明によれば、ES細胞、iPS細胞などの分化多能性細胞から神経前駆細胞への分化誘導に際して、これらの細胞を、上記文献に記載の方法を用いて作製し、培養する。ヒトES細胞やヒトiPS細胞を培養する時には、霊長類ES細胞用培地(リプロセル(京都、日本))を好ましく使用できる。
【0043】
分化多能性細胞から神経前駆細胞への分化誘導は、上記分化培地を用いて、フィーダー細胞の存在下又は非存在下のいずれかで行うことができる。フィーダー細胞が存在する場合、該細胞として、上に例示の、例えばMEF(マウス胎仔線維芽細胞)、STO細胞(マウス胎仔線維芽細胞株)、PA6細胞(マウスストローマ細胞株(理研BRC Cell Bank(日本)))、SNL細胞(STO細胞のサブクローン;例えばSNL 76/7細胞など)などを使用することができる。フィーダー細胞については、一般に、細胞増殖を停止させるためにマイトマイシンC処理が行われる。
【0044】
分化誘導の直前および分化誘導の直後に、好ましくは、培養多能性幹細胞を含む培地にROCK(p160-Rho-associated coiled-coil kinase)阻害剤を添加する。ROCK阻害剤は、細胞の分散の際に非常に強力な細胞死抑制作用を示す物質であり、例えば、Y-27632、Fasudil(HA−1077)などが知られている(K. Watanabe et al., Nat. Biotech., 25:681-686 (2007))。阻害剤の濃度は、非限定的に培養ディッシュあたり約50nM〜約10μMである。
【0045】
多能性幹細胞の培地中の密度は、約5.0×104〜約1.0×107細胞の範囲内であることが好ましいが、この範囲の外であってもよい。
【0046】
培養の具体例は、非接着性条件下での三次元培養、例えば浮遊培養(例えば、分散培養、凝集浮遊培養など)、または接着条件下での二次元培養、例えば平板培養、あるいは、三次元培養後に二次元培養を行うという連続的な組み合わせ培養、などを包含する。フィーダー細胞の存在下で分化誘導する場合には、二次元培養を使用することができるが、一方、フィーダー細胞が不在の場合には、三次元培養を使用することができる。
【0047】
細胞接着性の培養器では、細胞との接着性を向上させる目的で、その表面を、細胞支持物質、例えばコラーゲン、ゼラチン、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチン、MatrigelTM (Becton, Dickinson and Company)などの物質でコーティング(すなわち、被覆)することができる。
【0048】
分散培養では、多能性幹細胞は液体培地に懸濁した状態で培養される。また、凝集浮遊培養により、多能性幹細胞の細胞塊(または、胚様体)を形成し、その後、この細胞塊(または、胚様体)から目的細胞への分化誘導を起こすことができる。凝集浮遊培養については、例えば胚様体培養法(Kellerら, Curr. Opin. Cell Biol. 7, 862-869 (1995))、SFEB法(例、Watanabeら, Nature Neuroscience 8, 288-296 (2005);WO 2005/123902)などを利用することができる。好ましい方法は、SFEB法のような、血清を含まない培地での胚様体の培養である。
【0049】
接着培養では、例えば、Matrigel法 (Chambers SM, et al. Nat Biotechnol. 27: 485, 2009)またはSDIA法 (Kawasaki H, et al. Neuron. 28:31-40, 2000 or Kawasaki H, et al. Proc Natl Acad Sci U.S.A. 99: 1580-5, 2002)を使用できる。
【0050】
培養条件について、培地は上記の培地が使用されうるし、培養温度は、以下に限定されないが、約30〜40℃、好ましくは約37℃であり、CO2含有空気の雰囲気下で培養が行われ、CO2濃度は、好ましくは約2〜5%である。培養時間または培養スケジュールは、例えば分化誘導条件下において7日から21日間の培養であり、より好ましくは14日である。
具体的な分化誘導のための方法及び条件は、後述の実施例を参照されたい。
【0051】
<人工神経前駆細胞>
本発明は、上で説明した分化誘導法によって作製された人工神経前駆細胞も提供する。
本発明の方法によって得ることができる神経前駆細胞は、例えば、中枢神経系の神経細胞、末梢神経系の神経細胞、運動神経や感覚器系の神経細胞、自律神経の神経細胞などのあらゆる神経細胞の前駆体細胞を含む。
【0052】
神経前駆細胞は、神経細胞接着分子(NCAM)、ポリシアリル化NCAM、A2B5(胎児や新生児の神経細胞に発現する)、中間体フィラメントタンパク質(ネスチン、ビメンチンなど)、転写因子Pax-6などの原始的神経外胚葉および神経幹細胞の発現マーカー、ドーパミンニューロンマーカー(チロシンハイドロキシラーゼ(TH)など)、神経マーカー(TuJ1など)などによって同定されうる。
【0053】
神経前駆細胞は、作製後、そのまま生体へ移植されてもよいし、あるいは神経細胞やグリア細胞(星状細胞および希突起膠細胞が挙げられる。)に完全にまたは部分的に分化させた後で生体に移植されてもよい。
【0054】
<神経疾患治療剤のスクリーニングへの利用>
本発明の人工神経前駆細胞は、神経疾患治療用化合物(例えば医薬化合物、溶媒、小分子、ペプチド、またはポリヌクレオチド)のスクリーニングに用いることもできる。例えば、単独でまたは他の薬剤と組み合わせて、候補医薬化合物を、人工神経前駆細胞またはそれより成熟した神経細胞に加えることによる、当該細胞の形態または機能的な変化により、評価を行うことができる。機能的な変化の例として当該細胞から産生されるドーパミンの量を計測することで評価することができる。ここで、人工神経前駆細胞は、治療対象となる神経疾患と同様の表現型を呈する細胞が好ましく、特に好ましくは、神経疾患に侵された体細胞から作製された人工多能性幹細胞もしくは疾患由来の体細胞の核が移植されたntES細胞を分化誘導して作製された人工神経前駆細胞である。
【0055】
<再生医療への応用>
本発明の人工神経前駆細胞は、損傷した神経系組織の正常化のために再生医療の分野で有効に使用し得る。それゆえ、この細胞は、あらゆる神経系細胞の障害に関係する疾患の治療用細胞になり得る。
【0056】
疾患の例としては、虚血性脳疾患(脳卒中など)、脳外傷、脊髄損傷、運動神経疾患、神経変性疾患、網膜色素変性症、加齢黄斑変性症、内耳性難聴、多発性硬化症、筋萎縮性側索硬化症、脊髄小脳変性症、ハンチントン舞踏病、アルツハイマー病、パーキンソン病、てんかん、および統合失調症などが挙げられる。
【0057】
また、細胞を治療のために用いる場合、細胞の純度を高めることが望ましい。このための方法には、目的の細胞を選別する方法、例えばフローサイトメトリー法、抗癌剤含有培地での細胞の処理などが挙げられる。フローサイトメトリー法は、非常に細い流液中に細胞粒子を高速度で流し、レーザー光を照射して、粒子が発生する蛍光(細胞が予め蛍光標識された場合)、散乱光などの光を測定するものであり、セルソーターを備えると、目的の細胞を選別・分離することができる。細胞の蛍光標識は、神経前駆細胞に特異的な抗体(蛍光標識化)、例えば抗Nestin抗体、によって行うことができる。また、抗癌剤含有培地での処理によって、未分化細胞を除去することができる。抗癌剤の例は、マイトマイシンC、5-フルオロウラシル、アドリアマイシン、メトトレキセートなどである。
【0058】
神経前駆細胞の疾患部位への移植は、例えば、Nature Neuroscience,2,1137(1999)もしくはN Engl J Med.;344:710-9(2001)に記載されるような手法によって行うことができる。
【実施例】
【0059】
本発明を以下の実施例でさらに具体的に説明するが、本発明の範囲はそれら実施例に限定されないものとする。
【0060】
[方法]
<細胞および培養>
ヒトES細胞(KhES-1、KhES-2およびKhES-3)は、京都大学再生医科学研究所(京都、日本)より受領し、既知の方法で培養した(Suemori H, et al. Biochem Biophys Res Commun. 345:926-32, 2006)。ヒトiPS細胞(G1、G4、B6およびB7)は、京都大学の山中教授より受領し、既知の方法で培養した(Takahashi K, et al. Cell. 131:861-72, 2007およびNakagawa M, et al. Nat Biotechnol. 26:101-6, 2008)。PA6細胞(理研BRC Cell Bank)は、ゼラチンコートしたディッシュへ蒔き、10%FBSを含有するMEMαを用いて培養した。分化誘導の際は、少なくとも1日間は培養し、コンフルエントになったことを確認してフィーダー細胞として用いた。ヒトiPS細胞(404C2)は、再プログラミング因子(OCT3/4, SOX2, KLF4, L-MYC, LIN28,およびp53 shRNA)を、EBNA-1およびoriPを含むベクター(US61/232,402およびUS61/307,306)を用いてヒト線維芽細胞中に導入し、ついで、他のヒトiPS細胞と同じ方法により培養することによって樹立された。
【0061】
<神経前駆細胞への分化誘導(フィーダー細胞存在下;SDIA法)>
ES細胞またはiPS細胞は、フィーダー細胞としてSTO細胞を用いて培養された。分化誘導開始の1日前に、10μM ROCK阻害剤(Y276352)を培地中に添加した。CTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)を500μl/10cmディッシュで加え、3〜5分間、37℃でインキュベーションし、ディッシュを軽くたたき、フィーダー細胞をはがした。PBSで一度洗浄した後、再び、CTK解離液を加え、10〜15分間、37℃でインキュベーションした。ディッシュからはがされたES細胞またはiPS細胞は、5mlの分化培地(5% Knockout serum replacement (KSR)、2mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)に懸濁させ、遠心後、上清を除去した。再び、該細胞を1mlの分化培地に懸濁させ、小凝集体(10〜20細胞/クランプ(clump))になるようにピペッティングにより分離させた。
【0062】
得られた小凝集体は、2500〜5000個/cm2の範囲の濃度でPA6をフィーダー細胞として有するディッシュに蒔いた。培地は、2μM Dorsomorphin(Sigma)および/または10μM SB431542(Sigma)および/または300ng/ml Noggin(HZ-1026:HumanZyme)、あるいは2μl /ウエルのDMSO、を含む分化培地を用い、初回培養のみY276352を10μM加えた。培地交換は、7日目までは行わず、それ以降は3〜4日おきに1度行った。
【0063】
<神経前駆細胞への分化誘導(フィーダー細胞非存在下;SFEBq法)>
上記の方法で、フィーダー細胞を除去したES細胞またはiPS細胞を、1mlのAccumax(TM)を用いて、5分間37℃のインキュベーションを行って分離し、さらに洗浄後、細胞数を計測した。細胞を、前記分化培地へ懸濁し、低接着96穴プレート(Lipidure-coat plate:日本油脂)へ9000細胞/ウエルで蒔いた。培養には、2μM Dorsomorphinおよび10μM SB431542を含有する分化培地を用い、初回培養のみ50nMのY276352を加えた。培地交換は、7日目までは行わず、それ以降は3日おきに1度行った。
【0064】
<各薬剤を使用した神経系細胞への分化誘導(Matrigel法)>
PS細胞(G4)を、20分間のAccutuase処理によって分離し、さらにヒトES細胞培地で洗浄したのち、ROCK阻害剤(Y276352)含有培地を含むゼラチン被覆ディッシュに1時間留置して、フィーダー細胞を除去した。その後、ES細胞またはiPS細胞(18000細胞/cm2)をMatrigel(BD)被覆ディッシュに蒔き、bFGFおよびROCK阻害剤(Y276352)を添加したMEFならし培地を用いて3日間培養し、細胞をコンフルエントになるようにした(このとき、Y276352は、途中で除去された。)。
【0065】
つぎに、上記細胞を、10μM SB431542、2μM Dorsomorphin、300ng/ml Noggin、1nM〜100nM LDN-193189 (STEMGENT04-0019)、またはDMSO(対照)、あるいはそれらの組み合わせ物を含有する分化培地(20% knockout serum replacement (Gibco)および0.1mM 2-メルカプトエタノールを含有するDMEM/F12)中で5日間培養した。5日目にSB431542を加えずに、細胞を、Dorsomorphin、Noggin、LDN-193189またはDMSOを添加した分化培地で連続的に培養した。このとき、N2培地(この培地は、DMEM/F12にN2サプリメントを加えて調製されたものである。)の割合を、その他の薬剤の濃度を変更することなく、2日おきに最高25%、50%、ついで75%に増加した。
【0066】
<LDN-193189およびSB431542の添加による神経系細胞への分化誘導(SDIA法)>
分化誘導開始の1日前に、CTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)をES細胞(KhES-1、KhES-4およびKhES-5)に加えて、コロニーを解離した。そのディッシュからはがしたES細胞を、ゼラチンコート上のMEFの除去処理ののち、分化培地(5% knockout serum replacement (KSR)、2 mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)に1時間懸濁し、ピペッティングにより分離して、小凝集体(10〜20細胞/クランプ)にした。このようにして得た小凝集体を、2500〜5000細胞/cm2の範囲の濃度でPA6細胞上に蒔いた。培養4日目に、培地を、10μM SB431542および5〜1,000nM LDN-193189を添加した分化培地と交換した。3日後に、培地を、SB431542およびLDN-193189を含まない分化培地と交換し、その後、SB431542およびLDN-193189を含まない前記分化培地との培地交換を、2日〜3日おきに行った。
【0067】
<SFEBq法条件での分化誘導剤の組み合わせの検討>
ヒトiPS細胞(404C2)からの分化誘導の1日前に、10μM ROCK阻害剤(Y276352)を培地に加え、ついでCTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)を500μl/10cmディッシュの濃度で添加し、37℃で3〜5分間インキュベーションを行った。このディッシュを軽くたたいてフィーダー細胞を除去し、PBSで1度洗浄し、1mlのAccumaxTMを用いて37℃で5分間インキュベーションし、細胞を解離した。洗浄後、細胞数を計測した。細胞を、上記の分化培地に懸濁したのち、低接着96穴プレート(Lipidure-coat plate: 日本油脂)上に9000細胞/ウエルで蒔いた。この細胞を、以下に示す6種類の組み合わせからなる培地で4日間培養したのち、培地を、分化培地(5% knockout serum replacement (KSR)、2 mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)と交換した。得られた分化細胞は、未分化マーカー(Nanog)と神経系分化マーカー(Pax6およびSox1)を用いて評価された。
A:old DFK5% + 2μM Dorsomorphin + 10μM SB431542
B:old GMK8% + 100nM LDN913189 + 0.5μM A-83-01
C:DFK5% + 2μM Dorsomorphin + 10μM SB431542
D:GMK8% + 100nM LDN913189 +0.5μM A-83-01
E:GMK8% + 100nM LDN913189 + 10μM SB431542
F:GMK8% + 100nM LDN913189 + 0.5μM A-83-01 + 0.5μM PD0325901
ここで、
i) 「old」は、作製後20日間の継代を意味する。
ii) GMK8%は、Glasgow MEM (Invitrogen)、8%KSR、1μM 2ME、ピルビン酸および非必須アミノ酸からなる培地を意味する。
iii) A-83-01は、Sigma-Aldrich Inc.から購入され、PD0325901は和光純薬(日本)から購入された。
【0068】
<免疫染色>
分化誘導後14日目に、4%PFAを用いて30分4℃で固定後、PBS中に表1に記載の各抗体を用いて免疫染色した。
【0069】
【表1】

【0070】
<FACS>
上記の分化誘導後14日目の細胞を、Accumax(TM)を用いて20分間37℃のインキュベーションにより分離し、FACS Aria2を用いて分析した。分析は、PSA-NCAMまたはSSEA-4-PEを結合した抗体にて染色して行い、死細胞は7AAD染色を指標として用いて、またはRed dye Live/dead fixable dead cell stain kit (Invitrogen)を用いて、除去した
【0071】
<リアルタイムPCR>
上記の方法でフィーダー細胞を除去したES細胞またはiPS細胞から、RNeasy plus Mini (QIAGEN )を用いてRNAを回収し、SYBR Premix Ex Taq(TaKaRa)を用いて、Thermal Cycler Dice Real Time system TP800 (TaKaRa)により分析した。
【0072】
<統計>
GraphPad Prism 5 (GraphPad Software)を用いて、one-way ANNOVA, post hoc (Dunnett's Multiple Comparison test)にてn=4で解析した。
【0073】
[実施例1]
iPS細胞(G4)を、Dorsomorphin添加群(D群)、SB431542添加群(S群)およびDorsomorphinおよびSB431542添加群(D+S群)の3つの条件でPA6細胞をフィーダー細胞として用いて14日間培養することにより分化誘導を行った。その結果、D群では、NestinおよびOct3/4共陽性のコロニーが確認され、S群では平らに広がった細胞群の中に凝集した細胞集団が確認された。この凝集した細胞集団は、Nestin陽性であった。一方、D+S群では、ほとんどすべてコロニーがNestin陽性であり、Oct3/4陽性のコロニーはほとんど見られなかった(図1)。他の未分化マーカーであるNanogまたはSSEA3、神経マーカーであるPax6またはPSA-NCAMで免疫染色したところ、同様に、D群では未分化細胞と神経前駆細胞が共存し、S群では凝集細胞集団に神経前駆細胞が現れ、D&S群ではほぼ完全に神経前駆細胞へ分化したことが確認された(図2)。次に、D+S群にて21日間分化誘導を行ったところ、ドーパミン神経系(dopamine neuron)マーカーであるTH(チロシンヒドロキシラーゼ(tyrosine hydroxylase))および神経系マーカーであるTuJ1の陽性細胞が確認された(図3)。以上より、iPS細胞からの神経細胞への分化誘導は、PA6細胞をフィーダー細胞として用いてDorsomorphinおよびSB431542を添加した条件で培養することで効率よく行われることが確認された。
【0074】
次に、ES細胞(KhES-1、KhES-2およびKhES-3)および他のiPS細胞(G1、B6およびB7)においても同様の方法で、分化誘導を行った。7種(KhES-1、KhES-2、KhES-3 、G 1、G4、B6およびB7)の細胞株での結果をまとめたものを図4に示す。DorsomorphinとSB431542のいずれかまたは両方を添加した群のコロニー数が、対照群やNoggin(BMP拮抗タンパク質)添加群のコロニー数と比較して有意に多いことが確認された(図4A)。したがって、上記の薬剤には多能性細胞(ES細胞およびiPS細胞)における分化誘導の際の生存を助長する効果があることが確認された。一方、神経系細胞を含むコロニーへの分化誘導においては、SB431542が未分化細胞の排除に有効性であることが確認された(図4B)。また、NogginとSB431542の組み合わせよりも、DorsomorphinとSB431542の組み合わせの方が、神経系コロニーの産生効率が有意に高かった。
【0075】
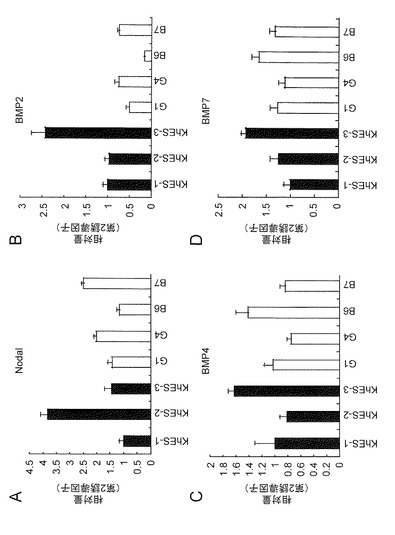
個々の細胞株(ES細胞(図5)およびiPS細胞(図6))についても同様の傾向が示された。これらの結果は、上記薬剤の標的タンパク質(TGF/Actibin/Nodal:SB431542、BMP:Dorsomorphin)の発現が、各細胞株においてほぼ同じであることからも理解できる(図7)。
【0076】
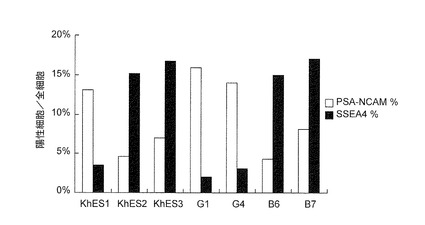
上記の結果に関して、各コロニーがマーカー遺伝子陽性であった細胞を少なくとも1つ含む該コロニーの数を基にして神経前駆細胞への誘導効率を判別しているため、そのような判別法は、細胞全体における誘導効率を比較することに適していない。そこで、コロニー単位ではなく、細胞単位に基いて分化誘導を観察するため、FACSにより解析した。従来のSDIA(stromal cell-derived inducing activity)法(Kawasaki H, et al. Neuron. 28:31-40, 2000またはKawasaki H, et al. Proc Natl Acad Sci U S A. 99:1580-5, 2002)に従ってDorsomorphinまたはSB431542を用いずに、しかしPA6細胞をフィーダー細胞として用いて、分化誘導を行い、分化誘導後14日目の細胞をFACSにより解析した(図8)。この方法では、SSEA4(未分化マーカー)発現細胞が見られる細胞株もあることから、細胞株間において分化抵抗性に違いがあるため確実な誘導方法ではない。次に、ES細胞(KhES1)をDorsomorphinおよびSB431542の添加を含む方法により分化誘導したところ、無添加群(対照)と比較して、PSA-NCAM陽性細胞は3倍以上になり、SSEA4陽性細胞は減少することが判明した(図9)。よって、DorsomorphinおよびSB431542を用いた方法により、高効率で神経前駆細胞への分化が可能であることが確認された。
【0077】
[実施例2]
iPS細胞(G4)から低接着条件にて胚様体を形成し、DorsomorphinおよびSB431542を添加して該細胞を神経前駆細胞に分化させた。その結果、分化誘導して14日目に、ほぼ99.6%の細胞がPSA-NCAM陽性であった(図10A)。また、免疫染色の結果、分化誘導に掛けられた細胞は、初期神経系のマーカーであるNestinおよびPax6が陽性であった(図10B)。上記のことより、フィーダー細胞不在下での分化誘導法を用いたときでも、DorsomorphinおよびSB431542を用いることで高効率に神経前駆細胞への分化が可能であることが確認された。
【0078】
[実施例3]
<各薬剤を用いる神経前駆細胞への分化誘導(Matrigel法)>
iPS細胞(G4)を、以下の条件: DMSOのみ(対照)(C群); Nogginのみ(N群); Noggin + SB431542 (NS群); Dorsomorphinのみ(D群); SB431542のみ(S群); Dorsomorphin + SB431542 (DS群); LDN-193189(1nM)(L1群); LDN-193189(5nM)(L5群); LDN-193189(10nM)(L10群); LDN-193189(5nM) + SB431542(L5S群); LDN-193189(10nM) + SB431542(L10S群); LDN-193189 (50nM) + SB431542(L50S群);およびLDN-193189(100nM) + SB431542(L100S群)でMatrigel法により14日間培養した。その結果を図11および図12に示す。
【0079】
分化誘導して14日目に視覚的に細胞を観察し、かつ、細胞数を数えた結果、NS群、DS群、L50S群およびL100S群で多数の生存細胞が存在することが確認された。
【0080】
これに続いて、神経前駆細胞への分化が可能であるか否かを、神経系細胞マーカーPAX6および未分化マーカーNanogの発現に基づいて確認した結果(図13)、PAX6陽性かつNanog陰性細胞の細胞数がL100S群で高かったため、前記条件での神経前駆細胞への分化誘導が比較的満足のいくものであることが確認された。
【0081】
[実施例4]
<神経前駆細胞への分化(SDIA法)におけるLDN-193189の最適濃度の決定>
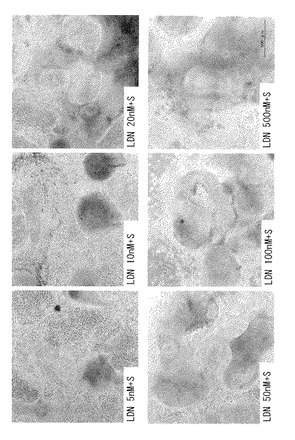
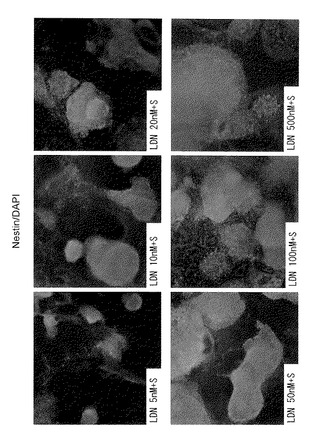
ヒトES細胞(KhES-1, KhES-4およびKhES-5)からの分化誘導を、5nM〜1000nMの範囲の濃度のLDN-193189を用いて、SDIA(stromal cell-derived inducing activity)法によって行い、LDN-193189の最適濃度を決定した。
【0082】
図14に、分化誘導して14日目に抗Nestin抗体を用いて得られた染色像を示す。Kh-ES5細胞の分化は、該細胞にSB431542およびLDN-193189を(数種の濃度で)添加することにより誘導し、それによりそれら細胞から神経前駆細胞への分化誘導が行われた。このことから、LDN-193189の濃度が50nMまたはそれ以上であるとき、KhES-5から神経前駆細胞への分化誘導が比較的よく行われることが確認された(L50S群、L100S群およびL500nMS群)。また、LDN-193189の濃度が50nMより高いときには、その効果が増大することは認められなかった。
【0083】
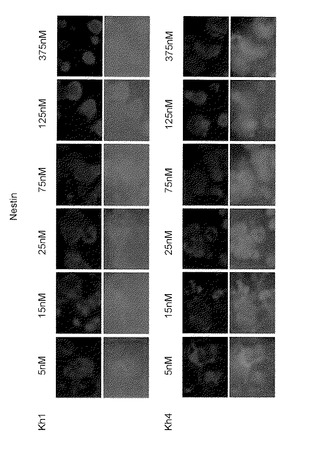
つぎに、KhES-1細胞株およびKhES-4細胞株の分化誘導を、LDN-193189およびSB431542を添加することにより行い、LDN-193189の濃度が25nM〜75nMの範囲であるとき、Nestin陽性細胞が比較的満足のいくものであることが確認された(図15A)。同様に、その細胞は、神経系マーカーPax6で染色された。LDN-193189の濃度が20nMであるとき、細胞は、最も強く染色されたことを確認した(図15B)。
【0084】
[実施例5]
薬剤の組み合わせ6種類を用いたときの神経系細胞誘導効率を図16に示した。薬剤の各組み合わせには互いに違いはなかったが、DorsomorphinおよびSB431542を含有する前もって用意されていたDFK5%の条件では、神経系細胞への分化誘導は、他のものより低めであるが、一方、LDN913189およびA-83-01を含有するGMK8%の条件では、DorsomorphinおよびSB431542を一緒に含有するDFK5%の条件と比べて生存の確率が高まった。
【0085】
LDN913189およびA-83-01を含有するGMK8%の条件で培養された分化細胞の種々のタイプのマーカー遺伝子(Oct3/4, PSA-NCAM, Tuj-1, SSEA1およびSSEA4)を、フローサイトメーターを用いて解析した(図17)。未分化マーカー遺伝子(Oct3/4およびSSEA4)は減少し、一方神経系マーカー遺伝子(PSA-NCAM, Tuj-1およびSSEA1)は増加した。これらの結果について、上記の培養条件は、神経系細胞分化誘導に有効であった。
【産業上の利用可能性】
【0086】
本発明により、ES細胞やiPS細胞などの多能性幹細胞から、効率よくかつ未分化細胞の残存率を低減させながら、神経前駆細胞を作製することが可能になる。神経前駆細胞は、神経系の疾患の治療を目的とした再生医療の分野で使用することができる。
【技術分野】
【0001】
本発明は、多能性幹細胞を神経前駆細胞へ分化誘導する方法に関する。
本発明はまた、上記方法によって作製された人工神経前駆細胞にも関する。
【背景技術】
【0002】
胚性幹細胞(ES細胞)や、動物の体細胞へ未分化細胞特異的遺伝子を導入することで得られる人工多能性幹細胞(iPS細胞)など多能性を有する細胞がこれまでに報告されている(特許文献1または特許文献2)。そこで、神経変性疾患や神経損傷の代替治療方法として使用可能である、これらの多能性幹細胞から分化誘導された神経細胞を移植する治療法が注目されている。ここでES細胞から神経細胞を分化誘導する方法として、(1)無血清培地中で胚様体を形成させて分化誘導する方法(SFEB法)(非特許文献1)、(2)ストローマ細胞上でES細胞を培養して分化誘導する方法(SDIA法)(非特許文献2)、(3)マトリゲル(Matrigel)上に薬剤を添加して培養する方法(非特許文献3)などが開発されている。
【0003】
しかし、これらの方法では、例えば、分化誘導後に未分化な細胞が残存することやサイトカインを用いるためコストが非常に高いなどのいくつかの問題を抱えている。そこで、サイトカインの代替物として多数の低分子化合物が開発されているが(特許文献3)、いずれの低分子化合物が効率の高い神経細胞へ分化誘導させるかは未だ不明である。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】米国特許第5,843,780号
【特許文献2】国際公開第2007/069666号
【特許文献3】国際公開第2008/033408号
【非特許文献】
【0005】
【非特許文献1】Watanabe K, et al. Nat Neurosci. 8:288-96, 2005
【非特許文献2】Kawasaki H, et al. Neuron. 28:31-40, 2000
【非特許文献3】Chambers SM, et al. Nat Biotechnol. 27:275-80, 2009
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、多能性幹細胞からある種の低分子化合物を用いて神経前駆細胞を分化誘導する高効率の方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
すなわち、本発明は以下のとおり特徴付けられる。
(1)低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、多能性幹細胞から神経前駆細胞へ分化誘導する方法。
(2)前記培養時に低分子TGFβファミリー阻害剤をさらに存在させる、(1)に記載の方法。
(3)前記培養を、ストローマ細胞をフィーダー細胞として用いて行う、(1)または(2)に記載の方法。
(4)前記ストローマ細胞がPA6細胞である、(3)に記載の方法。
(5)前記培養を、無血清の条件下で、胚様体を形成して行う、(1)または(2)に記載の方法。
【0008】
(6)前記培養を、フィーダー細胞を用いないで、Matrigel(商標)被覆ディッシュ上で行う、(1)または(2)に記載の方法。
(7)前記低分子BMP阻害剤がドルソモルフィン(Dorsomorphin)またはLDN-193189である、(1)〜(6)のいずれか1つに記載の方法。
(8)前記低分子TGFβファミリー阻害剤がSB431542またはA-83-01である、(2)に記載の方法。
(9)前記多能性幹細胞が、胚性幹細胞または人工多能性幹細胞である、(1)〜(8)のいずれか1つに記載の方法。
(10)さらにERK(細胞外シグナル調節キナーゼ)阻害剤の存在下で多能性幹細胞を培養することを含む、(1)〜(9)のいずれか1つに記載の方法。
(11)(1)〜(10)のいずれか1つに記載の方法で作製された人工神経前駆細胞。
【発明の効果】
【0009】
本発明の上記方法では、分化誘導培地に、低分子BMP阻害剤を存在させること、好ましくは低分子BMP阻害剤と低分子TGFβファミリー阻害剤を組み合わせて共存させること、によって人工神経前駆細胞を高効率で作製することができる。
【図面の簡単な説明】
【0010】
【図1】図1は、分化誘導14日目の、位相差顕微鏡像(a-c)、抗Nestin抗体を用いて得られた免疫染色像(d-f)、抗Oct3/4抗体を用いて得られた免疫染色像(g-i)、およびDAPIを用いて得られた免疫染色像(j-l)を示す。
【図2】図2は、分化誘導14日目の、抗Pax6抗体(緑色)および抗Nanog抗体(赤色)を用いて得られた免疫染色像、ならびに、抗PSA-NCAM抗体(緑色)および抗SSEA3抗体(赤色)を用いて得られた免疫染色像を示す。
【図3】図3は、分化誘導21日目の、抗TH (チロシンヒドロキシラーゼ(tyrosine hydroxylase))抗体(緑色)および抗TuJ1抗体(赤色)を用いて得られた免疫染色像を示す。
【図4】図4は、A: 全細胞株(KhES-1、KhES-2、KhES-3、G1、G4、B6およびB7:各細胞株につきn=4)における分化誘導14日目の1ウエルあたりに存在するコロニーの総数(n=28)、ならびに、B: 全細胞株(KhES-1、KhES-2、KhES-3、G1、G4、B6およびB7:各細胞株につきn=4)における分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の割合(%)(n=28)を示す。コロニー内に一つでも陽性の細胞が確認できれば陽性コロニーとして計数した。
【図5】図5は、各ES細胞株(KhES-1(A)、KhES-2(B)またはKhES-3(C))の分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の割合を示す。
【図6】図6は、各iPS細胞株(G1(A)、G4(B)、B6(C)またはB7(D))の分化誘導14日目の1ウエルあたりに存在する神経系細胞含有コロニー(Nestin陽性)と未分化細胞含有コロニー(Oct3/4陽性)の比率を示す。
【図7】図7は、未分化ES細胞(KhES-1、KhES-2およびKhES-3)またはiPS細胞(G1、G4、B6およびB7)におけるNodal(A)、BMP2(B)、BMP4(C)およびBMP7(D)についてリアルタイムPCRにより測定されたmRNA発現量を示す。
【図8】図8は、DorsomorphinおよびSB431542を用いずPA6細胞上のみで分化誘導した14日目の各細胞株におけるPSA-NCAM陽性(緑色)およびSSEA4陽性(赤色)の細胞含有率を示したグラフを示す。
【図9】図9は、KhES1をPA6細胞上の培養のみで分化誘導させて作製された対照群(AおよびB)およびPA6細胞上で培地へDorsomorphinおよびSB431542を加えて分化誘導して作製された群(CおよびD)における、SSEA4発現(すなわち、陽性)細胞(AおよびC)およびPSA-NCAM発現細胞(BおよびD)の分布を示したFACSのグラフを示す。図に示された値について、上段にKhES1由来の細胞中の各マーカーを発現する細胞の割合(%)(「目的細胞中」の割合)および下段にKhES1由来の細胞およびPA6細胞を含むディッシュ内に存在した全ての細胞中の各マーカーを発現する細胞の割合(%)(「全細胞中)の割合)を示した。図9Eは、対照群(KhES1 cont)およびDorsomorphinおよびSB431542を用いて分化誘導した群(KhES1&DSB)におけるディッシュ1枚から得られたES細胞由来の細胞(黒色バー)、ES細胞由来のPSA-NCAM陽性細胞(白色バー)およびSSEA4陽性細胞(ハッチングバー)の細胞数を示すグラフである。細胞数は次の式:(ES細胞由来の細胞数)=(ディッシュ内の全細胞数)-(ディッシュ内のPA6フィーダー細胞数);(ES細胞由来のPSA-NCAM陽性細胞数)=(ディッシュ内の全細胞数)×(ディッシュ内に存在した全ての細胞中のPSA-NCAM陽性細胞の割合);ならびに、(ES細胞由来のSSEA4陽性細胞)=(ディッシュ内の全細胞数)×(ディッシュ内に存在した全ての細胞中のSSEA4陽性細胞の割合)により算出された。図9Fおよび図9Gに、PA6細胞とES細胞由来の細胞に特徴的な分布の例(F:PA6細胞のみ、G: PA6細胞とKhES1(DorsomorphinおよびSB431542の非存在下での培養))を示す。
【図10】図10Aは、iPS細胞(G4)から、フィーダー細胞なしで胚様体を形成させ、DorsomorphinおよびSB431542を添加した培地での細胞の培養による分化誘導後14日目のPSA-NCAM陽性細胞の割合(%)を示したグラフである。ここで、赤色の曲線は抗体が存在しない陰性対照であり、青色の曲線は抗PSA-NCAM抗体により染色された細胞に関する結果を示す。また、図10Bは、上記方法で分化誘導した細胞でのNestin(緑色)および Pax6(赤色)の免疫染色像を示す。
【図11】図11は、以下に示す各薬剤を添加した培地でMatrigel法でフィーダー細胞非存在下で培養することによりiPS細胞(G4)の分化を誘導して14日目の位相差顕微鏡像を示す。図中、「N」は、ノギン(Noggin)を示し、「S」は、SB431542の添加を示し、「NS」は、NogginおよびSB431542の添加を示し、「C」は、対照DMSOの添加を示し、「D」は、Dorsomorphinの添加を示し、「DS」はDorsomorphinおよびSB431542の添加を示し、「LDN」は、LDN-193189の添加を示し、「LDN+S」は、LDN-193189およびSB431542の添加を示す。
【図12】図12は、以下に示す各薬剤の添加後14日目の、ならびに各薬剤の添加前の、1ウエルあたりに存在する細胞の数(すなわち、細胞数)を示す。図中、「0日目」は、薬剤の添加前を示し、「Cont」は、DMSOを添加した対照群を示し、「N」は、Nogginを添加した群を示し、「NS」は、NogginおよびSB431542を添加した群を示し、「D」は、Dorsomorphin添加群を示し、「S」は、SB431542添加群を示し、「DS」は、DorsomorphinおよびSB431542を添加した群を示し、「L10S」は、10nM LDN-193189およびSB431542を添加した群を示し、「L50S」は、50nM LDN-193189およびSB431542を添加した群を示し、「L100S」は、100nM LDN-193189およびSB431542を添加した群を示す。
【図13】図13は、Matrigel法でフィーダー細胞非存在下で培養することによりiPS細胞(G4)の分化を誘導して14日目の、抗Pax6抗体(緑色)および抗Nanog抗体(赤色)を用いて得られた免疫染色像、ならびに、DAPI(青色)を用いて得られた免疫染色像を示す。図中、「SB」はSB431542を示し、また「LDN」はLDN-193189を示す。
【図14A】図14Aは、分化誘導して14日目の位相差顕微鏡像を示す。ここで、分化誘導は、SDIA法に従ってES細胞株(Kh-ES5)をPA6細胞と一緒に共培養したのち、該細胞を、5〜500nM LDN-193189およびSB431542を添加した培地中で培養することによって行われた。
【図14B】図14Bは、分化誘導して14日目の、抗ネスチン(Nestin)抗体(緑色)およびDAPI(青色)を用いて得られた免疫染色像を示す。ここで、分化誘導は、SDIA法に従ってES細胞株(Kh-ES5)をPA6細胞と一緒に共培養したのち、該細胞を、5〜500nM LDN-193189およびSB431542を添加した培地中で培養することによって行われた。
【図15A】図15Aは、SDIA法に従って、ES細胞株(Kh-ES1またはKh-ES4)をPA6細胞と一緒に共培養し、ついで、5〜500nM LDN-193189およびSB431542を添加した培地中で細胞を培養することによって細胞の分化を誘導して14日目の、DAPI(青色)および抗Nestin抗体(緑色)を用いて得られた免疫染色像を示す。
【図15B】図15Bは、SDIA法に従って、ES細胞株(Kh-ES1またはKh-ES4)をPA6細胞と一緒に共培養し、ついで、5〜500nM LDN-193189およびSB431542を添加した培地中で細胞を培養することによって細胞の分化を誘導して14日目の、抗Pax6抗体(緑色)を用いて得られた免疫染色像を示す。
【図16】図16は、フィーダー細胞を用いない方法でヒトiPS細胞(404C2)を培養することによって分化誘導された細胞におけるNanog (A)、Pax6 (B)およびSox1(C)に関する定量PCRの結果を示す。この結果は、未処理の細胞に対する相対対数値を示している。「A」から「F」は、以下の条件を示す。すなわち、「A」は、2μMのDorsomorphinおよび10μMのSB431542を含有するold DFK5%であり、「B」は、100nMのLDN913189および0.5μMのA-83-01を含有するold GMK8%であり、「C」は、2μMのDorsomorphinおよび10μMのSB431542を含有するDFK5%であり、「D」は、100nMのLDN913189および0.5μMのA-83-01を含有するGMK8%であり、「E」は、100nMのLDN913189および10μMのSB431542を含有するGMK8%であり、ならびに「F」は、100nMのLDN913189および0.5μMのA-83-01 + 0.5μMのPD0325901を含有するGMK8%である。
【図17】図17は、100nMのLDN913189および0.5μMのA-83-01を含有するGMK8%の条件下で、フィーダー細胞を用いない方法でiPS細胞(404C2)を培養することによって得られた分化細胞に関する、Oct3/4の二次元展開(A)、PSA-NCAM発現細胞(B)、Tuj-1発現細胞(C)、ならびに、SSEA1およびSSEA4の二次元展開(D)を示すFACSグラフである。
【発明を実施するための形態】
【0011】
本発明を以下に詳細に説明する。
本発明は、上記のとおり、低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、多能性幹細胞から神経前駆細胞を分化誘導する方法に関する。
【0012】
<多能性幹細胞>
本発明で使用可能な多能性幹細胞は、該幹細胞を、外胚葉、中胚葉および内胚葉に由来する生体に存在するすべての細胞に分化する能力である多能性を有し、かつ、増殖能をも併せもつ幹細胞である。そのような幹細胞の具体例には、以下のものに限定されないが、胚性幹(ES)細胞、核移植により得られるクローン胚由来の胚性幹(ntES;核移植ES)細胞、精子幹細胞(「GS細胞」)、胚性生殖細胞(「EG細胞」)、人工多能性幹(iPS)細胞などが含まれる。好ましい多能性幹細胞は、ES細胞、ntES細胞、およびiPS細胞である。
【0013】
(A) 胚性幹細胞
ES細胞は、ヒトやマウスなどの哺乳動物の初期胚(例えば胚盤胞)の内部細胞塊から樹立された、多能性と自己複製による増殖能を有する幹細胞である。
ES細胞は、受精卵の8細胞期、桑実胚後の胚である胚盤胞の内部細胞塊に由来する胚由来の幹細胞であり、成体を構成するあらゆる細胞に分化する能力、いわゆる分化多能性と、自己複製による増殖能とを有している。ES細胞は、マウスで1981年に発見され(M.J. Evans and M.H. Kaufman (1981), Nature 292:154-156)、その後、ヒト、サルなどの霊長類でもES細胞株が樹立された (J.A. Thomson et al. (1999), Science 282:1145-1147; J.A. Thomson et al. (1995), Proc. Natl. Acad. Sci. USA, 92:7844-7848;J.A. Thomson et al. (1996), Biol. Reprod., 55:254-259; J.A. Thomson and V.S. Marshall (1998), Curr. Top. Dev. Biol., 38:133-165)。
【0014】
ES細胞は、対象動物の受精卵の胚盤胞から内部細胞塊を取出し、内部細胞塊を線維芽細胞のフィーダー上で培養することによって樹立することができる。また、継代培養による細胞の維持は、白血病抑制因子(leukemia inhibitory factor (LIF))、塩基性線維芽細胞成長因子(basic fibroblast growth factor (bFGF))などの物質を添加した培地を用いて行うことができる。ヒトおよびサルのES細胞の樹立と維持の方法については、例えばH. Suemori et al. (2006), Biochem. Biophys. Res. Commun., 345:926-932; M. Ueno et al. (2006), Proc. Natl. Acad. Sci. USA, 103:9554-9559; H. Suemori et al. (2001), Dev. Dyn., 222:273-279;H. Kawasaki et al. (2002), Proc. Natl. Acad. Sci. USA, 99:1580-1585などに記載されている。
【0015】
ES細胞作製のための培地として、例えば0.1mM 2-メルカプトエタノール、0.1mM 非必須アミノ酸、2mM L-グルタミン酸、20% KSR及び4ng/ml β‐FGFを補充したDMEM/F-12培地を使用する。ヒトES細胞は、37℃、湿潤雰囲気(5% CO2)下で前記培地を用いて維持することができる。また、ES細胞は、4〜5日おきに継代する必要があり、このとき、継代は、例えば1mM CaCl2及び20% KSRを含有するPBS中の0.25% トリプシン及び0.1mg/mlコラゲナーゼIVを用いて行うことができる。
【0016】
ES細胞の選択は、一般に、例えばアルカリホスファターゼ、Oct-3/4、Nanogなどの遺伝子マーカーの発現を指標にしてリアルタイムPCR法で行うことができる。特に、ヒトES細胞の選択では、OCT-3/4、NANOG、ECADなどの遺伝子マーカーの発現を指標とすることができる(E. Kroon et al. (2008), Nat. Biotechnol., 26:443-452)。
【0017】
ヒトES細胞株である例えばKhES-1、KhES-2、KhES-3、KhES-4およびKhES-5は、京都大学再生医科学研究所(京都、日本)から入手可能である。
【0018】
(B) 精子幹細胞
精子幹細胞は、精巣由来の多能性幹細胞であり、精子形成のための起源となる細胞である。この細胞は、ES細胞と同様に、種々の系列の細胞に分化誘導可能であり、例えばマウス胚盤胞に移植するとキメラマウスを作出できる(M. Kanatsu-Shinohara et al. (2003) Biol. Reprod., 69:612-616; K. Shinohara et al. (2004), Cell, 119:1001-1012)。神経膠細胞系由来神経栄養因子(glial cell line-derived neurotrophic factor (GDNF))を含む培地で自己複製可能であるし、またES細胞と同様の培養条件下で継代を繰り返すことによって、精子幹細胞を得ることができる(竹林正則ら(2008),実験医学,26巻,5号(増刊),41〜46頁,羊土社(東京、日本))。
【0019】
(C) 胚性生殖細胞
胚性生殖細胞は、胎生期の始原生殖細胞から樹立される、ES細胞と同様な多能性をもつ細胞であり、LIF、bFGF、幹細胞因子(stem cell factor)などの物質の存在下で始原生殖細胞を培養することによって樹立しうる(Y. Matsui et al. (1992), Cell, 70:841-847; J.L. Resnick et al. (1992), Nature, 359:550-551)。
【0020】
(D) 人工多能性幹細胞
人工多能性幹(iPS)細胞は、ある特定の再プログラミング因子を、DNA又はタンパク質の形態で体細胞に導入することによって作製することができる、ES細胞とほぼ同等の特性、例えば分化多能性と自己複製による増殖能、を有する体細胞由来の人工の幹細胞である(K. Takahashi and S. Yamanaka (2006) Cell, 126:663-676; K. Takahashi et al. (2007), Cell, 131:861-872; J. Yu et al. (2007), Science, 318:1917-1920; Nakagawa, M.ら,Nat. Biotechnol. 26:101-106 (2008);国際公開WO 2007/069666)。再プログラム化因子は、ES細胞に特異的に発現している遺伝子またはES細胞の未分化維持に重要な役割を果たす遺伝子もしくはその遺伝子産物であれば良く、特に限定されないが、例えばOCT3/4、SOX2及びKLF4; OCT3/4、KLF4及びC-MYC; OCT3/4、SOX2、KLF4及びC-MYC; OCT3/4及びSOX2; OCT3/4、SOX2及びNANOG; OCT3/4、SOX2及びLIN28; OCT3/4及びKLF4などの組み合わせを含む。
【0021】
これらの因子は、タンパク質の形態で、例えばリポフェクション、細胞膜透過性ペプチドとの結合、マイクロインジェクションなどの手法によって体細胞内に導入してもよいし、あるいは、DNAの形態で、例えば、ウイルス、プラスミド、人工染色体などのベクター、リポフェクション、リポソームを利用する手法、マイクロインジェクションなどの手法によって体細胞内に導入してもよい。ウイルスベクターとしては、レトロウイルスベクター、レンチウイルスベクター(Cell, 126, pp.663-676, 2006; Cell, 131, pp.861-872, 2007; Science, 318, pp.1917-1920, 2007)、アデノウイルスベクター(Science, 322, 945-949, 2008)、アデノ随伴ウイルスベクター、センダイウイルスベクターなどが例示される。また、人工染色体ベクターとしては、例えばヒト人工染色体(HAC)、酵母人工染色体(YAC)、細菌人工染色体(BAC、PAC)ベクターなどが含まれる。プラスミドとしては、哺乳動物細胞用プラスミドを使用しうる(Science, 322:949-953, 2008)。ベクターには、核初期化物質が発現可能なように、プロモーター、エンハンサー、リボゾーム結合配列、ターミネーター、ポリアデニル化サイトなどの制御配列を含むことができるし、さらに、必要に応じて、薬剤耐性遺伝子(例えばカナマイシン耐性遺伝子、アンピシリン耐性遺伝子、ピューロマイシン耐性遺伝子など)、チミジンキナーゼ遺伝子、ジフテリアトキシン遺伝子などの選択マーカー配列、緑色蛍光タンパク質(GFP)、βグルクロニダーゼ(GUS)、FLAGなどのレポーター遺伝子配列などを含むことができる。また、上記ベクターには、体細胞への導入後、再プログラミング化因子をコードする遺伝子、またはプロモーターと結合する再プログラミング化因子をコードする遺伝子、を共に切除するために、それらの遺伝子の各末端にLoxP配列を有してもよい。
【0022】
再プログラム化に際して、誘導効率を高めるために、上記の因子の他に、例えば、ヒストンデアセチラーゼ(HDAC)阻害剤[例えば、バルプロ酸 (VPA)(Nat. Biotechnol., 26(7): 795-797 (2008))、トリコスタチンA、酪酸ナトリウム、MC 1293、M344等の低分子阻害剤、HDACに対するsiRNAおよびshRNA(例、HDAC1 siRNA SmartpoolTM (Millipore)、HuSH 29mer shRNA Constructs against HDAC1 (OriGene)等)等の核酸性発現阻害剤など]、DNAメチルトランスフェラーゼ阻害剤(例えば5’-アザシチジン)(Nat. Biotechnol., 26(7): 795-797 (2008))、G9aヒストンメチルトランスフェラーゼ阻害剤[例えば、BIX-01294 (Cell Stem Cell, 2: 525-528 (2008))等の低分子阻害剤、G9aに対するsiRNAおよびshRNA(例、G9a siRNA(human) (Santa Cruz Biotechnology)等)等の核酸性発現阻害剤など]、L-チャネルカルシウムアゴニスト(例えばBayk8644) (Cell Stem Cell, 3, 568-574 (2008))、p53阻害剤(例えばp53に対するsiRNAおよびshRNA (Cell Stem Cell, 3, 475-479 (2008))、UTF1(Cell Stem Cell, 3, 475-479 (2008))、Wntシグナル伝達因子(例えば可溶性Wnt3a)(Cell Stem Cell, 3, 132-135 (2008))、2i/LIF (「2i」はmitogen-activated protein kinase signallingおよびglycogen synthase kinase-3の阻害剤を示す、PloS Biology, 6(10), 2237-2247 (2008))、miR-291-3p、miR-294、miR-295などのmiRNA(R.L. Judson et al., Nat. Biotech., 27:459-461, 2009)、ALK5阻害剤(例えば、SB431542)、等を使用することができる。
【0023】
iPS細胞誘導のための培養培地としては、例えば(1) 10〜15%FBSを含有するDMEM、DMEM/F12又はDME培地(これらの培地にはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)、(2) bFGF又はSCFを含有するES細胞培養用培地、例えばマウスES細胞培養用培地(例えばTX-WES培地、トロンボX社)又は霊長類ES細胞培養用培地(例えば霊長類(ヒトおよびサル)ES細胞用培地、リプロセル、京都、日本)、などが含まれる。
【0024】
培養法の例としては、たとえば、37℃、5%CO2存在下にて、10%FBS含有DMEM又はDMEM/F12培地上で体細胞と再プログラム化因子(DNA又はタンパク質)を接触させ約4〜7日間培養し、その後、細胞をフィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上にまきなおし、体細胞と再プログラム化因子の接触から約10日後からbFGF含有霊長類ES細胞培養用培地で培養し、該接触から約30〜約45日又はそれ以上ののちにiPS様コロニーを生じさせることができる。
【0025】
あるいは、細胞は、37℃、5% CO2存在下にて、フィーダー細胞(たとえば、マイトマイシンC処理STO細胞、SNL細胞等)上で10%FBS含有DMEM培地(これにはさらに、LIF、ペニシリン/ストレプトマイシン、ピューロマイシン、L-グルタミン、非必須アミノ酸類、β-メルカプトエタノールなどを適宜含むことができる。)で培養し、約25〜約30日又はそれ以上ののちにES様コロニーを生じさせることができる。
【0026】
さらにまた、細胞は、iPS細胞の誘導効率を高めるために、酸素濃度が5%〜10%となる低酸素条件下で培養してもよい(WO 2010/013845)。
【0027】
上記培養の間には、培養開始2日目以降から毎日1回新鮮な培地と培地交換を行う。また、核初期化(再プログラム化)に使用する体細胞の細胞数は、限定されないが、培養ディッシュ100cm2あたり約5×103〜約5×106細胞の範囲である。
【0028】
マーカー遺伝子として薬剤耐性遺伝子などの遺伝子を用いた場合は、対応する薬剤を含む培地(選択培地)で培養を行うことによりマーカー遺伝子発現細胞を選択することができる。またマーカー遺伝子が蛍光タンパク質遺伝子の場合は蛍光顕微鏡で観察することによって、マーカー遺伝子を発現する細胞を検出することができる。マーカー遺伝子が発光酵素遺伝子である場合は、発光基質を加えることによってマーカー遺伝子発現細胞を検出することができる。
【0029】
本明細書中で使用する「体細胞」なる用語は、卵子、卵母細胞および精母細胞などの生殖系列細胞、全能性細胞、ならびにES細胞を除くあらゆる動物細胞(好ましくは、ヒトを含む哺乳動物細胞)をいう。体細胞には、非限定的に、胎児(仔)の体細胞、新生児(仔)の体細胞、及び成熟した健全なもしくは疾患性の体細胞のいずれも包含されるし、また、初代培養細胞、継代細胞、及び樹立された株化細胞のいずれも包含されるし、さらにまた、組織幹細胞や組織前駆細胞も包含される。具体的には、体細胞は、非限定的に、例えば(1)神経幹細胞、造血幹細胞、間葉系幹細胞、歯髄幹細胞等の組織幹細胞(体性幹細胞)、(2)組織前駆細胞、(3)リンパ球、上皮細胞、内皮細胞、筋肉細胞、線維芽細胞(皮膚細胞等)、毛細胞、肝細胞、胃粘膜細胞、腸細胞、脾細胞、膵細胞(膵外分泌細胞等)、脳細胞、肺細胞、腎細胞、皮膚細胞等の分化した細胞などを包含する。
【0030】
(E) 核移植により得られたクローン胚由来のES細胞
nt ES細胞は、核移植技術によって作製されたクローン胚由来のES細胞であり、受精卵由来のES細胞とほぼ同じ特性を有している(T. Wakayama et al. (2001), Science, 292:740-743; S. Wakayama et al. (2005), Biol. Reprod., 72:932-936; J. Byrne et al. (2007), Nature, 450:497-502)。具体的には、未受精卵の核を体細胞の核と置換することによって得られたクローン胚由来の胚盤胞の内部細胞塊から樹立されたES細胞がnt ES(nuclear transfer ES)細胞である。nt ES細胞の作製のためには、核移植技術(J.B. Cibelli et al. (1998), Nature Biotechnol., 16:642-646)とES細胞作製技術(上記)との組み合わせが利用される(若山清香ら(2008),実験医学,26巻,5号(増刊), 47〜52頁)。核移植によって、哺乳動物の除核した未受精卵に、体細胞の核を注入し、数時間培養することで再プログラム化することができる。
【0031】
<低分子BMP阻害剤>
本発明で使用しうる低分子BMP阻害剤とは、BMP(bone morphogenetic protein)とBMP受容体(I型又はII型)との結合を介するBMPシグナル伝達(BMP signaling)の阻害に関与する低分子阻害剤であり、天然の阻害剤であるNoggin、chordin、follistatinなどのタンパク質性阻害剤とは異なる。本明細書で使用される「低分子」という用語は、有機分子または無機分子を意味し、巨大分子、例えば大タンパク質(例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超すタンパク質)、大核酸 (例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超す核酸)、あるいは、大多糖類(例えば、分子量が2,000, 3,000, 4,000, 5,000, 6,000, 7,000, 8,000, 9,000または10,000を超す多糖類)を包含しない。この阻害剤は、多能性幹細胞を神経前駆細胞へ分化誘導する作用を有しているべきである。このような性質をもつ低分子BMP阻害剤には、転写因子SMAD1、SMAD5又はSMAD8を活性化する能力をもつBMP2、BMP4、BMP6又はBMP7を阻害する化合物である、例えばDorsomorphin (すなわち、6-[4-(2-ピペリジン-1-イル-エトキシ)フェニル]-3-ピリジン-4-イル-ピラゾロ[1,5-a]ピリミジン)及びその誘導体が包含される(P. B. Yu et al. (2007), Circulation, 116:II_60; P.B. Yu et al. (2008), Nat. Chem. Biol., 4:33-41; J. Hao et al. (2008), PLoS ONE (www.plozone.org), 3(8):e2904)。Dorsomorphinは市販されており、例えばSigma-Aldrichなどから入手可能である。Dorsomorphinは、BMP受容体へのBMPの結合を阻害することによって上記のBMPシグナル伝達を阻害する生物活性を有する。この他に、BMP I型受容体キナーゼ阻害剤の具体例には、LDN-193189(すなわち、4-(6-(4-(ピペラジン-1-イル)フェニル)ピラゾロ[1,5-a]ピリミジン-3-イル)キノリン)およびその誘導体が包含される(Yu PB et al. Nat Med, 14: 1363-9, 2008)。LDN-193189は、例えばStemgent社から市販されている。
【0032】
<低分子TGFβファミリー阻害剤>
本発明によれば、上記低分子BMP阻害剤を、低分子TGFβ(transforming growthfactor β)ファミリー阻害剤と組み合わせることによって、多能性幹細胞の神経前駆細胞誘導の効率性を顕著に改善することができる。
【0033】
本明細書で使用される「低分子TGFβファミリー阻害剤」は、TGFβファミリーのシグナル伝達に干渉する低分子阻害剤であり、例えばSB431542、SB202190(R.K.Lindemann et al., Mol. Cancer 2:20 (2003))、SB505124 (GlaxoSmithKline)、NPC30345 、SD093、 SD908、SD208 (Scios)、LY2109761、LY364947、LY580276 (Lilly Research Laboratories)、A-83-01 (WO 2009146408)などが包含され、SB431542又はA-83-01が好ましい。
【0034】
TGFβファミリーのメンバーは、有糸分裂、細胞分化、胚パターン形成および器官形成などの細胞過程および発生過程を調節する。例えば、TGFβシグナル伝達は、I型及びII型のセリン−トレオニンキナーゼ受容体のヘテロメリックな受容体複合体を介して行われ、この複合体は下流のSmadシグナル伝達過程を活性化する。具体的には、受容体複合体にTGFβが結合すると、TGFβII型受容体がTGFβI型受容体をリン酸化し、このTGFβI型受容体が、受容体調節されるSmad(R-Smad)をリン酸化することによって、下流応答が開始される。活性化R-SmadはSmad4と多量体複合体を形成することによって、活性化R-Smadは核に移動し、標的遺伝子の転写調節が誘導される。
【0035】
このようなTGFβファミリーシグナル伝達が阻害されると、多能性幹細胞は神経前駆細胞へ分化誘導される。さらに、この阻害とともに、上記のBMPシグナル伝達が阻害されるときには、神経前駆細胞の誘導率が増すだけでなく、未分化な細胞(すなわち、多能性幹細胞)の残存率がより低下する、すなわち神経前駆細胞への変換率が向上する。
【0036】
<フィーダー細胞>
本発明では、フィーダー細胞は必ずしも必要ないが、フィーダー細胞を存在させることも可能である。フィーダー細胞としては、例えば胎仔線維芽細胞、ストローマ細胞などが挙げられる。胎仔線維芽細胞には、例えばMEF(マウス胎仔線維芽細胞)、STO細胞(マウス胎仔線維芽細胞株)、SNL細胞(STO細胞のサブクローン;例えばSNL 76/7細胞)などが含まれる。また、ストローマ細胞には、例えばPA6細胞(マウスストローマ細胞株(理研BRC Cell Bank(日本)))、MS-5細胞(Exp Hematol. 17:145-53 (1989))、OP9細胞(Science. 265:1098-1101 (1994))などが含まれる。SDIA法は、ES細胞をストローマ細胞、特にPA6細胞と一緒に共培養することによってほぼ選択的に神経前駆細胞へ分化させる方法であるが、本発明では、フィーダー細胞が不在であっても、上記の低分子BMP阻害剤、あるいは該低分子BMP阻害剤と上記の低分子TGFβファミリー阻害剤の組み合わせ、を分化誘導培地に存在させるだけで選択的に神経前駆細胞に分化させることができる。このような培養条件に加えて、フィーダー細胞を使用すると、神経前駆細胞への分化効率をさらに向上させることができる。
【0037】
しかし、たとえそうであっても、ヒトなどの哺乳動物への神経前駆細胞、またはさらにその分化細胞である神経細胞やグリア細胞、の移植を考えた場合、ドナーに対して異種の細胞の使用はできる限り避けることが望ましいことは言うまでもない。
【0038】
<神経前駆細胞の分化誘導>
(A) 分化培地
培地は、動物細胞の培養に用いられる培地を基礎培地として調製することができる。基礎培地としては、例えばIMDM培地、Medium 199培地、Eagle’s Minimum Essential Medium (EMEM)培地、αMEM培地、Doulbecco’s modified Eagle’s Medium (DMEM)培地、Ham’s F12培地、RPMI 1640培地、Fischer培地、Glasgow MEMおよびこれらの混合培地などが包含される。培地には、血清が含有されていてもよいし、あるいは無血清でもよい。
【0039】
培地はまた、必要に応じて、例えば、アルブミン、トランスフェリン、Knockout Serum Replacement(KSR)(ES細胞培養時のFBSの血清代替物)、脂肪酸、インスリン、コラーゲン前駆体、微量元素、2-メルカプトエタノール、3’-チオールグリセロール、B27-サプリメント、N2-サプリメントなどの1つ以上の血清代替物を含んでもよいし、ならびに、脂質、アミノ酸、非必須アミノ酸、ビタミン、増殖因子、サイトカイン、抗生物質、抗酸化剤、ピルビン酸、緩衝剤、無機塩類などの1つ以上の物質も含有しうる。
【0040】
培地はまた、前記低分子BMP阻害剤および/または前記低分子TGFβファミリー阻害剤を適宜含有してもよく、さらに前記フィーダー細胞の培養上清を含有してもよい。培地はさらに、ERK (extracellular signal-regulated kinase)阻害剤類のいずれかを含んでもよい。
【0041】
分化培地の例は、後述の実施例に記載されるような、5% knockout serum replacement (KSR)、2mM L-グルタミン、非必須アミノ酸類及び1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12混合培地、あるいは、8% KSR、1μM 2ME、ピルビン酸および非必須アミノ酸類を含有するGlasgow MEMである。
【0042】
(B) 分化誘導法
本発明によれば、ES細胞、iPS細胞などの分化多能性細胞から神経前駆細胞への分化誘導に際して、これらの細胞を、上記文献に記載の方法を用いて作製し、培養する。ヒトES細胞やヒトiPS細胞を培養する時には、霊長類ES細胞用培地(リプロセル(京都、日本))を好ましく使用できる。
【0043】
分化多能性細胞から神経前駆細胞への分化誘導は、上記分化培地を用いて、フィーダー細胞の存在下又は非存在下のいずれかで行うことができる。フィーダー細胞が存在する場合、該細胞として、上に例示の、例えばMEF(マウス胎仔線維芽細胞)、STO細胞(マウス胎仔線維芽細胞株)、PA6細胞(マウスストローマ細胞株(理研BRC Cell Bank(日本)))、SNL細胞(STO細胞のサブクローン;例えばSNL 76/7細胞など)などを使用することができる。フィーダー細胞については、一般に、細胞増殖を停止させるためにマイトマイシンC処理が行われる。
【0044】
分化誘導の直前および分化誘導の直後に、好ましくは、培養多能性幹細胞を含む培地にROCK(p160-Rho-associated coiled-coil kinase)阻害剤を添加する。ROCK阻害剤は、細胞の分散の際に非常に強力な細胞死抑制作用を示す物質であり、例えば、Y-27632、Fasudil(HA−1077)などが知られている(K. Watanabe et al., Nat. Biotech., 25:681-686 (2007))。阻害剤の濃度は、非限定的に培養ディッシュあたり約50nM〜約10μMである。
【0045】
多能性幹細胞の培地中の密度は、約5.0×104〜約1.0×107細胞の範囲内であることが好ましいが、この範囲の外であってもよい。
【0046】
培養の具体例は、非接着性条件下での三次元培養、例えば浮遊培養(例えば、分散培養、凝集浮遊培養など)、または接着条件下での二次元培養、例えば平板培養、あるいは、三次元培養後に二次元培養を行うという連続的な組み合わせ培養、などを包含する。フィーダー細胞の存在下で分化誘導する場合には、二次元培養を使用することができるが、一方、フィーダー細胞が不在の場合には、三次元培養を使用することができる。
【0047】
細胞接着性の培養器では、細胞との接着性を向上させる目的で、その表面を、細胞支持物質、例えばコラーゲン、ゼラチン、ポリ-L-リジン、ポリ-D-リジン、ラミニン、フィブロネクチン、MatrigelTM (Becton, Dickinson and Company)などの物質でコーティング(すなわち、被覆)することができる。
【0048】
分散培養では、多能性幹細胞は液体培地に懸濁した状態で培養される。また、凝集浮遊培養により、多能性幹細胞の細胞塊(または、胚様体)を形成し、その後、この細胞塊(または、胚様体)から目的細胞への分化誘導を起こすことができる。凝集浮遊培養については、例えば胚様体培養法(Kellerら, Curr. Opin. Cell Biol. 7, 862-869 (1995))、SFEB法(例、Watanabeら, Nature Neuroscience 8, 288-296 (2005);WO 2005/123902)などを利用することができる。好ましい方法は、SFEB法のような、血清を含まない培地での胚様体の培養である。
【0049】
接着培養では、例えば、Matrigel法 (Chambers SM, et al. Nat Biotechnol. 27: 485, 2009)またはSDIA法 (Kawasaki H, et al. Neuron. 28:31-40, 2000 or Kawasaki H, et al. Proc Natl Acad Sci U.S.A. 99: 1580-5, 2002)を使用できる。
【0050】
培養条件について、培地は上記の培地が使用されうるし、培養温度は、以下に限定されないが、約30〜40℃、好ましくは約37℃であり、CO2含有空気の雰囲気下で培養が行われ、CO2濃度は、好ましくは約2〜5%である。培養時間または培養スケジュールは、例えば分化誘導条件下において7日から21日間の培養であり、より好ましくは14日である。
具体的な分化誘導のための方法及び条件は、後述の実施例を参照されたい。
【0051】
<人工神経前駆細胞>
本発明は、上で説明した分化誘導法によって作製された人工神経前駆細胞も提供する。
本発明の方法によって得ることができる神経前駆細胞は、例えば、中枢神経系の神経細胞、末梢神経系の神経細胞、運動神経や感覚器系の神経細胞、自律神経の神経細胞などのあらゆる神経細胞の前駆体細胞を含む。
【0052】
神経前駆細胞は、神経細胞接着分子(NCAM)、ポリシアリル化NCAM、A2B5(胎児や新生児の神経細胞に発現する)、中間体フィラメントタンパク質(ネスチン、ビメンチンなど)、転写因子Pax-6などの原始的神経外胚葉および神経幹細胞の発現マーカー、ドーパミンニューロンマーカー(チロシンハイドロキシラーゼ(TH)など)、神経マーカー(TuJ1など)などによって同定されうる。
【0053】
神経前駆細胞は、作製後、そのまま生体へ移植されてもよいし、あるいは神経細胞やグリア細胞(星状細胞および希突起膠細胞が挙げられる。)に完全にまたは部分的に分化させた後で生体に移植されてもよい。
【0054】
<神経疾患治療剤のスクリーニングへの利用>
本発明の人工神経前駆細胞は、神経疾患治療用化合物(例えば医薬化合物、溶媒、小分子、ペプチド、またはポリヌクレオチド)のスクリーニングに用いることもできる。例えば、単独でまたは他の薬剤と組み合わせて、候補医薬化合物を、人工神経前駆細胞またはそれより成熟した神経細胞に加えることによる、当該細胞の形態または機能的な変化により、評価を行うことができる。機能的な変化の例として当該細胞から産生されるドーパミンの量を計測することで評価することができる。ここで、人工神経前駆細胞は、治療対象となる神経疾患と同様の表現型を呈する細胞が好ましく、特に好ましくは、神経疾患に侵された体細胞から作製された人工多能性幹細胞もしくは疾患由来の体細胞の核が移植されたntES細胞を分化誘導して作製された人工神経前駆細胞である。
【0055】
<再生医療への応用>
本発明の人工神経前駆細胞は、損傷した神経系組織の正常化のために再生医療の分野で有効に使用し得る。それゆえ、この細胞は、あらゆる神経系細胞の障害に関係する疾患の治療用細胞になり得る。
【0056】
疾患の例としては、虚血性脳疾患(脳卒中など)、脳外傷、脊髄損傷、運動神経疾患、神経変性疾患、網膜色素変性症、加齢黄斑変性症、内耳性難聴、多発性硬化症、筋萎縮性側索硬化症、脊髄小脳変性症、ハンチントン舞踏病、アルツハイマー病、パーキンソン病、てんかん、および統合失調症などが挙げられる。
【0057】
また、細胞を治療のために用いる場合、細胞の純度を高めることが望ましい。このための方法には、目的の細胞を選別する方法、例えばフローサイトメトリー法、抗癌剤含有培地での細胞の処理などが挙げられる。フローサイトメトリー法は、非常に細い流液中に細胞粒子を高速度で流し、レーザー光を照射して、粒子が発生する蛍光(細胞が予め蛍光標識された場合)、散乱光などの光を測定するものであり、セルソーターを備えると、目的の細胞を選別・分離することができる。細胞の蛍光標識は、神経前駆細胞に特異的な抗体(蛍光標識化)、例えば抗Nestin抗体、によって行うことができる。また、抗癌剤含有培地での処理によって、未分化細胞を除去することができる。抗癌剤の例は、マイトマイシンC、5-フルオロウラシル、アドリアマイシン、メトトレキセートなどである。
【0058】
神経前駆細胞の疾患部位への移植は、例えば、Nature Neuroscience,2,1137(1999)もしくはN Engl J Med.;344:710-9(2001)に記載されるような手法によって行うことができる。
【実施例】
【0059】
本発明を以下の実施例でさらに具体的に説明するが、本発明の範囲はそれら実施例に限定されないものとする。
【0060】
[方法]
<細胞および培養>
ヒトES細胞(KhES-1、KhES-2およびKhES-3)は、京都大学再生医科学研究所(京都、日本)より受領し、既知の方法で培養した(Suemori H, et al. Biochem Biophys Res Commun. 345:926-32, 2006)。ヒトiPS細胞(G1、G4、B6およびB7)は、京都大学の山中教授より受領し、既知の方法で培養した(Takahashi K, et al. Cell. 131:861-72, 2007およびNakagawa M, et al. Nat Biotechnol. 26:101-6, 2008)。PA6細胞(理研BRC Cell Bank)は、ゼラチンコートしたディッシュへ蒔き、10%FBSを含有するMEMαを用いて培養した。分化誘導の際は、少なくとも1日間は培養し、コンフルエントになったことを確認してフィーダー細胞として用いた。ヒトiPS細胞(404C2)は、再プログラミング因子(OCT3/4, SOX2, KLF4, L-MYC, LIN28,およびp53 shRNA)を、EBNA-1およびoriPを含むベクター(US61/232,402およびUS61/307,306)を用いてヒト線維芽細胞中に導入し、ついで、他のヒトiPS細胞と同じ方法により培養することによって樹立された。
【0061】
<神経前駆細胞への分化誘導(フィーダー細胞存在下;SDIA法)>
ES細胞またはiPS細胞は、フィーダー細胞としてSTO細胞を用いて培養された。分化誘導開始の1日前に、10μM ROCK阻害剤(Y276352)を培地中に添加した。CTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)を500μl/10cmディッシュで加え、3〜5分間、37℃でインキュベーションし、ディッシュを軽くたたき、フィーダー細胞をはがした。PBSで一度洗浄した後、再び、CTK解離液を加え、10〜15分間、37℃でインキュベーションした。ディッシュからはがされたES細胞またはiPS細胞は、5mlの分化培地(5% Knockout serum replacement (KSR)、2mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)に懸濁させ、遠心後、上清を除去した。再び、該細胞を1mlの分化培地に懸濁させ、小凝集体(10〜20細胞/クランプ(clump))になるようにピペッティングにより分離させた。
【0062】
得られた小凝集体は、2500〜5000個/cm2の範囲の濃度でPA6をフィーダー細胞として有するディッシュに蒔いた。培地は、2μM Dorsomorphin(Sigma)および/または10μM SB431542(Sigma)および/または300ng/ml Noggin(HZ-1026:HumanZyme)、あるいは2μl /ウエルのDMSO、を含む分化培地を用い、初回培養のみY276352を10μM加えた。培地交換は、7日目までは行わず、それ以降は3〜4日おきに1度行った。
【0063】
<神経前駆細胞への分化誘導(フィーダー細胞非存在下;SFEBq法)>
上記の方法で、フィーダー細胞を除去したES細胞またはiPS細胞を、1mlのAccumax(TM)を用いて、5分間37℃のインキュベーションを行って分離し、さらに洗浄後、細胞数を計測した。細胞を、前記分化培地へ懸濁し、低接着96穴プレート(Lipidure-coat plate:日本油脂)へ9000細胞/ウエルで蒔いた。培養には、2μM Dorsomorphinおよび10μM SB431542を含有する分化培地を用い、初回培養のみ50nMのY276352を加えた。培地交換は、7日目までは行わず、それ以降は3日おきに1度行った。
【0064】
<各薬剤を使用した神経系細胞への分化誘導(Matrigel法)>
PS細胞(G4)を、20分間のAccutuase処理によって分離し、さらにヒトES細胞培地で洗浄したのち、ROCK阻害剤(Y276352)含有培地を含むゼラチン被覆ディッシュに1時間留置して、フィーダー細胞を除去した。その後、ES細胞またはiPS細胞(18000細胞/cm2)をMatrigel(BD)被覆ディッシュに蒔き、bFGFおよびROCK阻害剤(Y276352)を添加したMEFならし培地を用いて3日間培養し、細胞をコンフルエントになるようにした(このとき、Y276352は、途中で除去された。)。
【0065】
つぎに、上記細胞を、10μM SB431542、2μM Dorsomorphin、300ng/ml Noggin、1nM〜100nM LDN-193189 (STEMGENT04-0019)、またはDMSO(対照)、あるいはそれらの組み合わせ物を含有する分化培地(20% knockout serum replacement (Gibco)および0.1mM 2-メルカプトエタノールを含有するDMEM/F12)中で5日間培養した。5日目にSB431542を加えずに、細胞を、Dorsomorphin、Noggin、LDN-193189またはDMSOを添加した分化培地で連続的に培養した。このとき、N2培地(この培地は、DMEM/F12にN2サプリメントを加えて調製されたものである。)の割合を、その他の薬剤の濃度を変更することなく、2日おきに最高25%、50%、ついで75%に増加した。
【0066】
<LDN-193189およびSB431542の添加による神経系細胞への分化誘導(SDIA法)>
分化誘導開始の1日前に、CTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)をES細胞(KhES-1、KhES-4およびKhES-5)に加えて、コロニーを解離した。そのディッシュからはがしたES細胞を、ゼラチンコート上のMEFの除去処理ののち、分化培地(5% knockout serum replacement (KSR)、2 mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)に1時間懸濁し、ピペッティングにより分離して、小凝集体(10〜20細胞/クランプ)にした。このようにして得た小凝集体を、2500〜5000細胞/cm2の範囲の濃度でPA6細胞上に蒔いた。培養4日目に、培地を、10μM SB431542および5〜1,000nM LDN-193189を添加した分化培地と交換した。3日後に、培地を、SB431542およびLDN-193189を含まない分化培地と交換し、その後、SB431542およびLDN-193189を含まない前記分化培地との培地交換を、2日〜3日おきに行った。
【0067】
<SFEBq法条件での分化誘導剤の組み合わせの検討>
ヒトiPS細胞(404C2)からの分化誘導の1日前に、10μM ROCK阻害剤(Y276352)を培地に加え、ついでCTK解離液(0.25%トリプシン、1mg/mlコラゲナーゼ、KSR 20%および1mM CaCl2)を500μl/10cmディッシュの濃度で添加し、37℃で3〜5分間インキュベーションを行った。このディッシュを軽くたたいてフィーダー細胞を除去し、PBSで1度洗浄し、1mlのAccumaxTMを用いて37℃で5分間インキュベーションし、細胞を解離した。洗浄後、細胞数を計測した。細胞を、上記の分化培地に懸濁したのち、低接着96穴プレート(Lipidure-coat plate: 日本油脂)上に9000細胞/ウエルで蒔いた。この細胞を、以下に示す6種類の組み合わせからなる培地で4日間培養したのち、培地を、分化培地(5% knockout serum replacement (KSR)、2 mM L-グルタミン、非必須アミノ酸類および1μM 2-メルカプトエタノール(2-ME)を含有するDMEM/Ham's F12)と交換した。得られた分化細胞は、未分化マーカー(Nanog)と神経系分化マーカー(Pax6およびSox1)を用いて評価された。
A:old DFK5% + 2μM Dorsomorphin + 10μM SB431542
B:old GMK8% + 100nM LDN913189 + 0.5μM A-83-01
C:DFK5% + 2μM Dorsomorphin + 10μM SB431542
D:GMK8% + 100nM LDN913189 +0.5μM A-83-01
E:GMK8% + 100nM LDN913189 + 10μM SB431542
F:GMK8% + 100nM LDN913189 + 0.5μM A-83-01 + 0.5μM PD0325901
ここで、
i) 「old」は、作製後20日間の継代を意味する。
ii) GMK8%は、Glasgow MEM (Invitrogen)、8%KSR、1μM 2ME、ピルビン酸および非必須アミノ酸からなる培地を意味する。
iii) A-83-01は、Sigma-Aldrich Inc.から購入され、PD0325901は和光純薬(日本)から購入された。
【0068】
<免疫染色>
分化誘導後14日目に、4%PFAを用いて30分4℃で固定後、PBS中に表1に記載の各抗体を用いて免疫染色した。
【0069】
【表1】
【0070】
<FACS>
上記の分化誘導後14日目の細胞を、Accumax(TM)を用いて20分間37℃のインキュベーションにより分離し、FACS Aria2を用いて分析した。分析は、PSA-NCAMまたはSSEA-4-PEを結合した抗体にて染色して行い、死細胞は7AAD染色を指標として用いて、またはRed dye Live/dead fixable dead cell stain kit (Invitrogen)を用いて、除去した
【0071】
<リアルタイムPCR>
上記の方法でフィーダー細胞を除去したES細胞またはiPS細胞から、RNeasy plus Mini (QIAGEN )を用いてRNAを回収し、SYBR Premix Ex Taq(TaKaRa)を用いて、Thermal Cycler Dice Real Time system TP800 (TaKaRa)により分析した。
【0072】
<統計>
GraphPad Prism 5 (GraphPad Software)を用いて、one-way ANNOVA, post hoc (Dunnett's Multiple Comparison test)にてn=4で解析した。
【0073】
[実施例1]
iPS細胞(G4)を、Dorsomorphin添加群(D群)、SB431542添加群(S群)およびDorsomorphinおよびSB431542添加群(D+S群)の3つの条件でPA6細胞をフィーダー細胞として用いて14日間培養することにより分化誘導を行った。その結果、D群では、NestinおよびOct3/4共陽性のコロニーが確認され、S群では平らに広がった細胞群の中に凝集した細胞集団が確認された。この凝集した細胞集団は、Nestin陽性であった。一方、D+S群では、ほとんどすべてコロニーがNestin陽性であり、Oct3/4陽性のコロニーはほとんど見られなかった(図1)。他の未分化マーカーであるNanogまたはSSEA3、神経マーカーであるPax6またはPSA-NCAMで免疫染色したところ、同様に、D群では未分化細胞と神経前駆細胞が共存し、S群では凝集細胞集団に神経前駆細胞が現れ、D&S群ではほぼ完全に神経前駆細胞へ分化したことが確認された(図2)。次に、D+S群にて21日間分化誘導を行ったところ、ドーパミン神経系(dopamine neuron)マーカーであるTH(チロシンヒドロキシラーゼ(tyrosine hydroxylase))および神経系マーカーであるTuJ1の陽性細胞が確認された(図3)。以上より、iPS細胞からの神経細胞への分化誘導は、PA6細胞をフィーダー細胞として用いてDorsomorphinおよびSB431542を添加した条件で培養することで効率よく行われることが確認された。
【0074】
次に、ES細胞(KhES-1、KhES-2およびKhES-3)および他のiPS細胞(G1、B6およびB7)においても同様の方法で、分化誘導を行った。7種(KhES-1、KhES-2、KhES-3 、G 1、G4、B6およびB7)の細胞株での結果をまとめたものを図4に示す。DorsomorphinとSB431542のいずれかまたは両方を添加した群のコロニー数が、対照群やNoggin(BMP拮抗タンパク質)添加群のコロニー数と比較して有意に多いことが確認された(図4A)。したがって、上記の薬剤には多能性細胞(ES細胞およびiPS細胞)における分化誘導の際の生存を助長する効果があることが確認された。一方、神経系細胞を含むコロニーへの分化誘導においては、SB431542が未分化細胞の排除に有効性であることが確認された(図4B)。また、NogginとSB431542の組み合わせよりも、DorsomorphinとSB431542の組み合わせの方が、神経系コロニーの産生効率が有意に高かった。
【0075】
個々の細胞株(ES細胞(図5)およびiPS細胞(図6))についても同様の傾向が示された。これらの結果は、上記薬剤の標的タンパク質(TGF/Actibin/Nodal:SB431542、BMP:Dorsomorphin)の発現が、各細胞株においてほぼ同じであることからも理解できる(図7)。
【0076】
上記の結果に関して、各コロニーがマーカー遺伝子陽性であった細胞を少なくとも1つ含む該コロニーの数を基にして神経前駆細胞への誘導効率を判別しているため、そのような判別法は、細胞全体における誘導効率を比較することに適していない。そこで、コロニー単位ではなく、細胞単位に基いて分化誘導を観察するため、FACSにより解析した。従来のSDIA(stromal cell-derived inducing activity)法(Kawasaki H, et al. Neuron. 28:31-40, 2000またはKawasaki H, et al. Proc Natl Acad Sci U S A. 99:1580-5, 2002)に従ってDorsomorphinまたはSB431542を用いずに、しかしPA6細胞をフィーダー細胞として用いて、分化誘導を行い、分化誘導後14日目の細胞をFACSにより解析した(図8)。この方法では、SSEA4(未分化マーカー)発現細胞が見られる細胞株もあることから、細胞株間において分化抵抗性に違いがあるため確実な誘導方法ではない。次に、ES細胞(KhES1)をDorsomorphinおよびSB431542の添加を含む方法により分化誘導したところ、無添加群(対照)と比較して、PSA-NCAM陽性細胞は3倍以上になり、SSEA4陽性細胞は減少することが判明した(図9)。よって、DorsomorphinおよびSB431542を用いた方法により、高効率で神経前駆細胞への分化が可能であることが確認された。
【0077】
[実施例2]
iPS細胞(G4)から低接着条件にて胚様体を形成し、DorsomorphinおよびSB431542を添加して該細胞を神経前駆細胞に分化させた。その結果、分化誘導して14日目に、ほぼ99.6%の細胞がPSA-NCAM陽性であった(図10A)。また、免疫染色の結果、分化誘導に掛けられた細胞は、初期神経系のマーカーであるNestinおよびPax6が陽性であった(図10B)。上記のことより、フィーダー細胞不在下での分化誘導法を用いたときでも、DorsomorphinおよびSB431542を用いることで高効率に神経前駆細胞への分化が可能であることが確認された。
【0078】
[実施例3]
<各薬剤を用いる神経前駆細胞への分化誘導(Matrigel法)>
iPS細胞(G4)を、以下の条件: DMSOのみ(対照)(C群); Nogginのみ(N群); Noggin + SB431542 (NS群); Dorsomorphinのみ(D群); SB431542のみ(S群); Dorsomorphin + SB431542 (DS群); LDN-193189(1nM)(L1群); LDN-193189(5nM)(L5群); LDN-193189(10nM)(L10群); LDN-193189(5nM) + SB431542(L5S群); LDN-193189(10nM) + SB431542(L10S群); LDN-193189 (50nM) + SB431542(L50S群);およびLDN-193189(100nM) + SB431542(L100S群)でMatrigel法により14日間培養した。その結果を図11および図12に示す。
【0079】
分化誘導して14日目に視覚的に細胞を観察し、かつ、細胞数を数えた結果、NS群、DS群、L50S群およびL100S群で多数の生存細胞が存在することが確認された。
【0080】
これに続いて、神経前駆細胞への分化が可能であるか否かを、神経系細胞マーカーPAX6および未分化マーカーNanogの発現に基づいて確認した結果(図13)、PAX6陽性かつNanog陰性細胞の細胞数がL100S群で高かったため、前記条件での神経前駆細胞への分化誘導が比較的満足のいくものであることが確認された。
【0081】
[実施例4]
<神経前駆細胞への分化(SDIA法)におけるLDN-193189の最適濃度の決定>
ヒトES細胞(KhES-1, KhES-4およびKhES-5)からの分化誘導を、5nM〜1000nMの範囲の濃度のLDN-193189を用いて、SDIA(stromal cell-derived inducing activity)法によって行い、LDN-193189の最適濃度を決定した。
【0082】
図14に、分化誘導して14日目に抗Nestin抗体を用いて得られた染色像を示す。Kh-ES5細胞の分化は、該細胞にSB431542およびLDN-193189を(数種の濃度で)添加することにより誘導し、それによりそれら細胞から神経前駆細胞への分化誘導が行われた。このことから、LDN-193189の濃度が50nMまたはそれ以上であるとき、KhES-5から神経前駆細胞への分化誘導が比較的よく行われることが確認された(L50S群、L100S群およびL500nMS群)。また、LDN-193189の濃度が50nMより高いときには、その効果が増大することは認められなかった。
【0083】
つぎに、KhES-1細胞株およびKhES-4細胞株の分化誘導を、LDN-193189およびSB431542を添加することにより行い、LDN-193189の濃度が25nM〜75nMの範囲であるとき、Nestin陽性細胞が比較的満足のいくものであることが確認された(図15A)。同様に、その細胞は、神経系マーカーPax6で染色された。LDN-193189の濃度が20nMであるとき、細胞は、最も強く染色されたことを確認した(図15B)。
【0084】
[実施例5]
薬剤の組み合わせ6種類を用いたときの神経系細胞誘導効率を図16に示した。薬剤の各組み合わせには互いに違いはなかったが、DorsomorphinおよびSB431542を含有する前もって用意されていたDFK5%の条件では、神経系細胞への分化誘導は、他のものより低めであるが、一方、LDN913189およびA-83-01を含有するGMK8%の条件では、DorsomorphinおよびSB431542を一緒に含有するDFK5%の条件と比べて生存の確率が高まった。
【0085】
LDN913189およびA-83-01を含有するGMK8%の条件で培養された分化細胞の種々のタイプのマーカー遺伝子(Oct3/4, PSA-NCAM, Tuj-1, SSEA1およびSSEA4)を、フローサイトメーターを用いて解析した(図17)。未分化マーカー遺伝子(Oct3/4およびSSEA4)は減少し、一方神経系マーカー遺伝子(PSA-NCAM, Tuj-1およびSSEA1)は増加した。これらの結果について、上記の培養条件は、神経系細胞分化誘導に有効であった。
【産業上の利用可能性】
【0086】
本発明により、ES細胞やiPS細胞などの多能性幹細胞から、効率よくかつ未分化細胞の残存率を低減させながら、神経前駆細胞を作製することが可能になる。神経前駆細胞は、神経系の疾患の治療を目的とした再生医療の分野で使用することができる。
【特許請求の範囲】
【請求項1】
低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、神経前駆細胞を分化誘導する方法。
【請求項2】
前記培養時に低分子TGFβファミリー阻害剤をさらに存在させる、請求項1に記載の方法。
【請求項3】
前記培養時にストローマ細胞をフィーダー細胞として用いて培養する、請求項1または2に記載の方法。
【請求項4】
4.前記ストローマ細胞がPA6細胞である、請求項3に記載の方法。
【請求項5】
前記培養を、無血清の条件下で、胚様体を形成して行う、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記培養を、フィーダー細胞を用いないで、Matrigel(商標)被覆ディッシュ上で行う、請求項1または2に記載の方法。
【請求項7】
前記低分子BMP阻害剤がドルソモルフィン(Dorsomorphin)またはLDN-193189である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記低分子TGFβファミリー阻害剤がSB431542またはA-83-01である、請求項2に記載の方法。
【請求項9】
前記多能性幹細胞が、胚性幹細胞または人工多能性幹細胞である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
さらにERK(細胞外シグナル調節キナーゼ)阻害剤の存在下で多能性幹細胞を培養することを含む、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
請求項1〜10のいずれか1項に記載の方法で作製された人工神経前駆細胞。
【請求項1】
低分子BMP阻害剤の存在下で多能性幹細胞を培養することを含む、神経前駆細胞を分化誘導する方法。
【請求項2】
前記培養時に低分子TGFβファミリー阻害剤をさらに存在させる、請求項1に記載の方法。
【請求項3】
前記培養時にストローマ細胞をフィーダー細胞として用いて培養する、請求項1または2に記載の方法。
【請求項4】
4.前記ストローマ細胞がPA6細胞である、請求項3に記載の方法。
【請求項5】
前記培養を、無血清の条件下で、胚様体を形成して行う、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記培養を、フィーダー細胞を用いないで、Matrigel(商標)被覆ディッシュ上で行う、請求項1または2に記載の方法。
【請求項7】
前記低分子BMP阻害剤がドルソモルフィン(Dorsomorphin)またはLDN-193189である、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記低分子TGFβファミリー阻害剤がSB431542またはA-83-01である、請求項2に記載の方法。
【請求項9】
前記多能性幹細胞が、胚性幹細胞または人工多能性幹細胞である、請求項1〜8のいずれか1項に記載の方法。
【請求項10】
さらにERK(細胞外シグナル調節キナーゼ)阻害剤の存在下で多能性幹細胞を培養することを含む、請求項1〜9のいずれか1項に記載の方法。
【請求項11】
請求項1〜10のいずれか1項に記載の方法で作製された人工神経前駆細胞。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
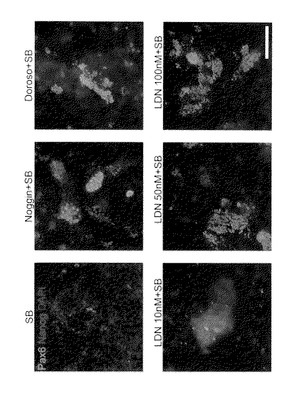
【図14A】
【図14B】
【図15A】
【図15B】
【図16】
【図17】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14A】
【図14B】
【図15A】
【図15B】
【図16】
【図17】
【公表番号】特表2013−501502(P2013−501502A)
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願番号】特願2012−507516(P2012−507516)
【出願日】平成22年8月12日(2010.8.12)
【国際出願番号】PCT/JP2010/063953
【国際公開番号】WO2011/019092
【国際公開日】平成23年2月17日(2011.2.17)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
【公表日】平成25年1月17日(2013.1.17)
【国際特許分類】
【出願日】平成22年8月12日(2010.8.12)
【国際出願番号】PCT/JP2010/063953
【国際公開番号】WO2011/019092
【国際公開日】平成23年2月17日(2011.2.17)
【出願人】(504132272)国立大学法人京都大学 (1,269)
【Fターム(参考)】
[ Back to top ]