
大腸菌において混合糖から有機物質を生産する方法
【課題】有用物質生産に有用なCCRが解除された大腸菌変異株、及び該変異株を用いた有用物質生産法の提供。
【解決手段】Mlcタンパク質を過剰に発現する遺伝子変異を導入することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株、及び該大腸菌株を用いて混合糖より有用な有機物質を製造する方法。
【解決手段】Mlcタンパク質を過剰に発現する遺伝子変異を導入することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株、及び該大腸菌株を用いて混合糖より有用な有機物質を製造する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、大腸菌において混合糖から効率よく有機物質を生産する方法に関する。中でも特に、2種類以上の糖が含まれる培地を原料として、アルコールや有機酸などの有機物質を生産する方法に関する。
【背景技術】
【0002】
アルコールや有機酸などは、有用な物質として様々な用途に使われる。これらは糖を微生物に代謝させることで生産することができるが、原材料たる糖は、安価であるなどの理由から、動植物から作られた再生可能な有機性資源、バイオマスに由来することが多い。バイオマス由来の糖では、ブドウ糖、キシロース、アラビノースなど他種類の糖が混ざった、混合糖であることが一般的である(非特許文献1)。しかし、微生物に混合糖を与えた場合、エネルギー効率の最も良いブドウ糖のみを選択的に消費し、ブドウ糖を消費し終えるまで、それ以外の糖の代謝をしないことがある。これは、炭素カタボライト抑制(CCR)と呼ばれる遺伝機構が微生物に存在しているために起こる現象である(非特許文献2)。この結果、バイオマス由来の混合糖のうち、ブドウ糖以外の糖が効率よく目的物質に変換されず、無駄になってしまうことがある(非特許文献3)。
【0003】
CCR機構では、ブドウ糖の存在下では、ブドウ糖以外の糖の消費に関与する遺伝子群が発現抑制される。例えば、大腸菌のlacZYAはラクトースの消費に必要な遺伝子オペロンで、それぞれβガラクトシダーゼ、βガラクトシドパーミアーゼ、ガラクトシドアセチルトランスフェラーゼをコードしている。糖として、ブドウ糖とラクトースが培養液中に共存するとき、lacZYAの発現調節領域ではlacI遺伝子産物(ラクトースリプレッサー)およびcrp遺伝子産物(cAMP受容体タンパク質)が協調して、lacZYA mRNAが転写されないよう負に制御している。しかし、糖としてラクトースのみが培養液中に存在するときには、細胞内に取り込まれたラクトースが変化したアロラクトースによって、この負の制御は解除され、lacZYAのmRNAは盛んに転写される。これによって、ラクトースは大腸菌によって消費されるようになる。また、キシロース、アラビノースの消費に必要な遺伝子群も、これと同様な遺伝子発現調節を受けている(非特許文献4)。
【0004】
大腸菌は、微生物による物質生産の宿主に最もよく使われるものの一つであるが、上述ようにCCR機構を有している。よって、CCRが解除された変異株を作成し、様々な糖を同時代謝させる工夫がなされている。例えば、ptsG遺伝子が破壊された株(DptsG)や、crp遺伝子の中の3塩基対に点突然変異の入った株(crp*)などが、CCRが解除された株として知られており、実際の物質生産に使われた例がある(特許文献1及び非特許文献5〜8)。
【0005】
しかしptsG遺伝子は、細胞膜に局在し、ブドウ糖を細胞内に取り込む機能を担うブドウ糖パーミアーゼをコードしており、DptsG株ではブドウ糖の取り込み効率が下がり、目的物質の生産性が下がる場合がある。また、crp遺伝子はcAMP受容体タンパク質型の転写因子をコードしているが、相当数の遺伝子がCrpの制御下にあり、crp*株では大きく代謝経路が変化していることが考えられる。さらに、crp遺伝子破壊株(Dcrp)ではある種の有機溶媒に耐性を持つようになることが知られており、逆にcrp*株では有機溶媒に感受性になることも予想される(非特許文献9)。よって、crp*株では、有機溶媒の一種であるアルコールを生産した際、その毒性の問題から生産性が低下することも考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許6159738号明細書
【非特許文献】
【0007】
【非特許文献1】van Maris et al., Antonie van Leeuwenhoek (2006) 90:391-418
【非特許文献2】Deutscher, Curr. Opin. Microbiol. (2008) 11:87-93
【非特許文献3】Kim et al., Appl. Microbiol. Biotechnol. (2010) 88:1077-1085
【非特許文献4】Song and Park, J. Bacteriol. (1997) 179:7025-7032
【非特許文献5】Cirino et al., Biotechnol. Bioeng. (2006) 95:1167-1176
【非特許文献6】Khankal et al., J. Biol. Eng. (2009) 3:13
【非特許文献7】Dien et al., J. Ind. Microbiol. Biotechnol. (2002) 29:221-227
【非特許文献8】Nichols et al., Appl. Microbiol. Biotechnol. (2001) 56:120-125
【非特許文献9】Okochi et al., J.Biosci. Bioeng. (2008) 105:389-394
【発明の概要】
【発明が解決しようとする課題】
【0008】
以上のことから、本発明者は、有用物質生産に有用な新しいCCRが解除された変異株を分離することとした。また、そのような変異株を用いて、実際に有用物質生産を実証することとした。
【課題を解決するための手段】
【0009】
大腸菌のlacZYAオペロンは、上述のようにCCRを受ける遺伝子であるが、これが発現しているかどうかは、5-クロロ-4-ブロモ-3-インドリル-β-D-ガラクトース(x-gal)を用いた青白選別によって簡単に知ることができる。これは、無色透明のx-galが、lacZ遺伝子産物であるβガラクトシダーゼによって分解されると、青色を呈することに基づいている。すなわち、野生株の大腸菌をブドウ糖、ラクトース、x-gal、その他適当な栄養源を含む寒天培地上で培養すると、白色のコロニーを形成する。これは、培地中にブドウ糖とラクトースが共存しており、CCRによってlacZ遺伝子の転写が抑制されているからである。しかし、何らかの変異によってCCRに異常が生じ、ブドウ糖、ラクトース共存下でもlacZ遺伝子が転写されるようになると、同培地上で青色のコロニーを形成することになる。
【0010】
そこで本発明者は、野生型大腸菌MG1655株をブドウ糖、ラクトース、x-gal、その他適当な栄養源を含む寒天培地上に塗布し、紫外線を照射して突然変異を誘発し、生育後のコロニーの色を判別することとした。これによって、CCRに異常(不全)が生じた(すなわち、CCRが解除された)変異株が分離されるはずである。
【0011】
次いで、任意の株でCCRを解除することができるように、分離した変異株の変異遺伝子の同定をすることとした。このためには、ショットガンクローニングを行う。この段階では、変異遺伝子が優性か劣性か判断できないので、野生株または変異株のゲノムを用いて作成したプラスミドゲノムライブラリ2種を、それぞれ変異株、野生株に導入し、青白選別を行う。この際、陽性クローンに混じって擬陽性クローンも多数分離されると思われるので、二次スクリーニングや塩基配列決定なども行い、変異遺伝子を同定する。変異遺伝子を同定できたならば次に、相同組換えによって人為的に変異遺伝子を野生株に導入する。この相同組換えによる変異株と、紫外線照射で分離した元の変異株とで糖消費速度などを比較し、同一の表現型を示すことを観察して、最終的な変異遺伝子の確認とする。
【0012】
さらに、上記の変異株を用いて、有用物質の実生産を行う。そのために、混合糖からイソブタノール(イソブチルアルコール)を生産し、野生型、DptsG株あるいはcrp*株よりも本発明の変異株の方がイソブタノールの生産性がよいことを実証する。なお、イソブタノールは近年、ガソリンあるいは軽油代替燃料として注目を浴びているアルコールの一種である。イソブタノール生産の方法は、現在知られている中で最も生産効率のよい、2ケト酸を経由する方法を用いる(Atsumi et al., Nature (2008) 451:86-89)。
【0013】
本発明者は、このように大腸菌において混合糖から有機物質を生産する方法を開発すべく鋭意検討を行い、本発明を完成させるに至った。
【0014】
すなわち、本発明は以下の通りである。
[1] 遺伝子変異導入又は発現ベクターを使った過剰発現によって、Mlcタンパク質を過剰に発現することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株。
[2] Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、[1]の炭素カタボライト抑制(CCR)が解除された大腸菌株。
[3] mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、[1]又は[2]の炭素カタボライト抑制(CCR)が解除された大腸菌株。
[4] anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によって、炭素カタボライト抑制(CCR)が解除された大腸菌株。
[5] さらに、有機物質の合成に関与する遺伝子が導入されている、[1]〜[4]のいずれかの大腸菌株。
[6] 外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入されている、[5]の大腸菌株。
[7] 大腸菌株にMlcタンパク質を過剰に発現する遺伝子変異を導入すること、又はMlcタンパク質を発現ベクターを使って過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
[8] Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、[7]の方法。
[9] mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、[7]又は[8]の方法。
[10] anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子を過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
[11] [1]〜[4]のいずれかの炭素カタボライト抑制(CCR)が解除された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に有機物質を生産する方法。
[12] [1]〜[4]のいずれかの炭素カタボライト抑制(CCR)が解除された大腸菌株であって、さらに有機物質の合成に関与する遺伝子が導入された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に前記有機物質を生産する方法。
[13] 有機物質がアルコール又は有機酸である、[12]の方法。
[14] 有機物質がイソブチルアルコールであり、混合糖がブドウ糖及びキシロースを含む、[12]又は[13]の方法。
[15] 大腸菌株に、外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入された、[14]の方法。
【発明の効果】
【0015】
実施例に示すように、混合糖を原料として大腸菌で物質生産をする場合、野生型株を用いるよりも、CCRの解除された株を用いるほうが生産性が高いことがわかる。CCRの解除された株の中でも、mlc*によるものが有利な場合もある。mlc*あるいは、mlc遺伝子の過剰発現によってCCRが解除され、ブドウ糖とそれ以外の糖を同時に消費するようになる、という報告はこれまでになく、新規性がある。また、mlc*あるいは、mlc遺伝子を過剰発現した株を用いて有機物質の生産を行った例もない。
【図面の簡単な説明】
【0016】
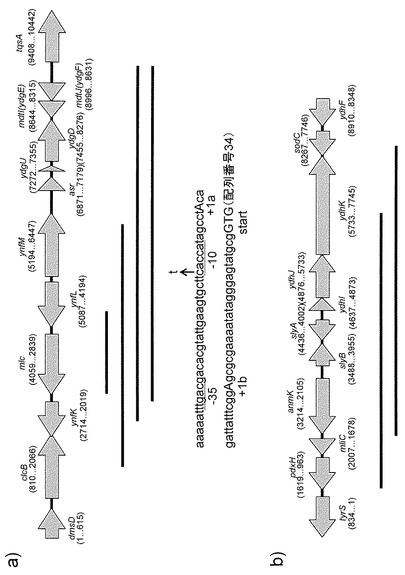
【図1】ショットガンでクローン化されたgen株のゲノム断片の構造を示す図である。図1aがmlc遺伝子を含む断片を示し、図1bがanmK、slyB、slyA、ydhI、ydhJ遺伝子を含む断片を示す。図中矢印はORFを、黒い線がショットガンクローニングで取られたゲノム断片を示す。また、mlcプロモータ部分の配列とmlc*の変異箇所を示してある。
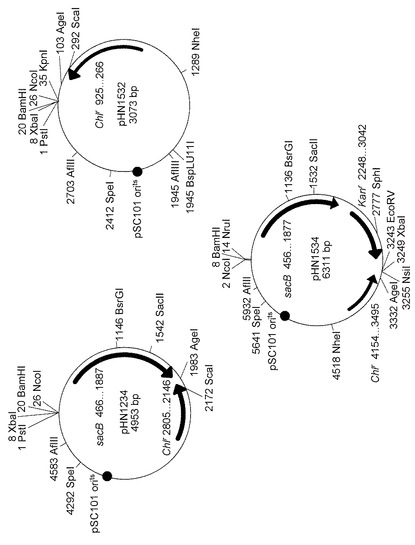
【図2】pHN1234、pHN1532、pHN1534のプラスミドマップを示す図である。ChlrとKanrはそれぞれクロラムフェニコール耐性遺伝子とカナマイシン耐性遺伝子を示す。
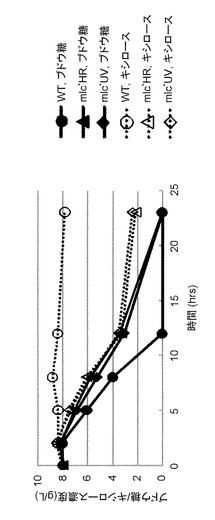
【図3】各株におけるブドウ糖とキシロースの共消費を示す図である。
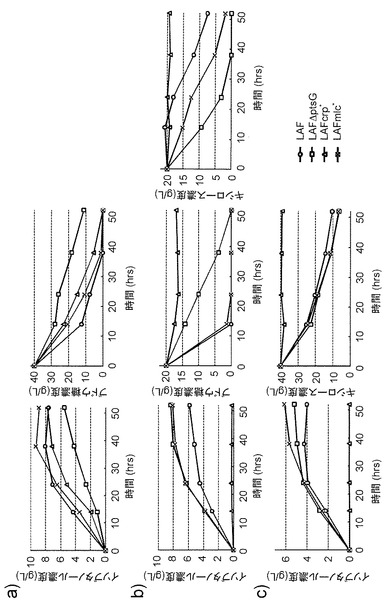
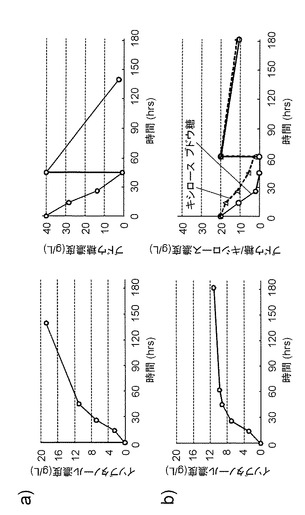
【図4】各株におけるイソブタノールの生産性を示す図である。図4aは、ブドウ糖を炭素原とした場合の結果、図4bはブドウ糖とキシロースを炭素原とした場合の結果、図4cはキシロースを炭素原とした場合の結果を示す。
【図5】より最適化されたイソブタノールの生産性を示す図である。図5aは、ブドウ糖を炭素原とした場合の結果、図5bはブドウ糖とキシロースを炭素原とした場合の結果を示す。
【発明を実施するための形態】
【0017】
以下、本発明を詳細に説明する。
【0018】
本発明は大腸菌(E.coli)において混合糖から有機物質を生産する方法であって、炭素カタボライト抑制(CCR:carbon catabolite repression)が解除された変異体を用いて有機物質を製造する方法である。CCRは、培地中にブドウ糖などの優先的に代謝される炭素源があるとそれを消費するまで、それ以外の炭素源の分解系の遺伝子発現を抑制する機構である。自然界においては炭素源をより効率的に利用できるという利点があるが、微生物を用いて有用物質を生産しようとする場合には、ブドウ糖以外の糖を利用できなくなるという欠点を有する。本発明においては、大腸菌のCCRを解除することにより、ブドウ糖と他の糖が共存する混合糖を用いる培養条件下において、優先的に代謝される糖と該等以外の糖、例えば、ブドウ糖とブドウ糖以外の糖を同時に利用し、有用物質の生産が可能となる。ここで、CCRの解除とは、CCRの不全あるいはCCRの異常ともいい、大腸菌において、ブドウ糖等のCCRを誘導する糖と他の糖が混合された条件下で培養した場合に、ブドウ糖以外の糖の代謝の抑制が解除され、ブドウ糖だけでなく、ブドウ糖以外の他の糖も同時に代謝され、大腸菌体における有機物質の合成に利用し得るようになることをいう。
【0019】
本発明において、CCRの解除は、例えばMlc(making large colonies)タンパク質の過剰発現により行うことができる。Mlcタンパク質は、炭化水素代謝の調節因子として、いくつかの遺伝子やオペロンの発現を調節することが知られている44kDaのDNA結合タンパク質である(Hosono, K., et al., Biosci, Biotechnol. Biochem., 59, 256-261 (1995))。一例として、ptsG遺伝子の発現はMlcタンパク質によって負に制御されていることが知られている(Plumbridge, J. Curr. Opin. Microbiol. 5:187-193(2002))。Mlcタンパク質の過剰発現は、PtsGタンパク質の不足を引き起こし、CCRが解除される。
【0020】
Mlcタンパク質の過剰発現は、例えば、mlc遺伝子が導入されMlcを発現し得るベクターを大腸菌に導入することにより行うことができる。また、mlcプロモーターDNAの塩基配列にmlcプロモーターが高活性型になるような変異を入れてもよい。このような変異として、例えば、mlcプロモーターの-10配列のcaccatからtattatへの変異が挙げられる。該変異は配列番号34に示すmlcプロモーター配列の第31番目のcのtへの置換である。
【0021】
さらに、anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によっても部分的な又は完全なCCRの解除を行うことができる。ここで、部分的な解除とは、不完全なCCRの解除をいう。ただし、このようなCCRの解除であっても、優先的に代謝される糖とそれ以外の糖を同時に利用することが可能になるので、有用である。本発明において、CCRの解除とは完全な解除だけでなく、部分的な解除も含む。上記5つの遺伝子のうち、特にslyA遺伝子の過剰発現が好ましい。これらの遺伝子の過剰発現は、これらの遺伝子が導入されこれらの遺伝子を発現し得るベクターを大腸菌に導入することにより行うことができる。
【0022】
mlcプロモーターの塩基配列の変異がMlcタンパク質の過剰発現を引き起こし、CCRを解除するかどうかは、大腸菌のmlcプロモーターをUV照射等の物理的変異処理、ニトロソグアニジン等を用いた化学的変異処理を行い、人為的に変異を誘発し、該変異体について、ブドウ糖及びラクトースが共存する培地で培養した場合にlacZ遺伝子が発現するかどうかを調べればよい。さらに、mlc遺伝子の過剰発現又はanmK、slyB、slyA、ydhI又はydhJ遺伝子の過剰発現がCCRを解除するかどうかも同様の方法によりlacZ遺伝子の発現を調べればよい。CCRが正常な大腸菌においては、lacZ遺伝子は発現せず、一方、CCRが解除された大腸菌においては、lacZ遺伝子が発現する。lacZ遺伝子の発現は、ブドウ糖及びラクトースが共存する培地にさらに5-クロロ-4-ブロモ-3-インドリル-β-D-ガラクトース(x-gal)を添加し青白選別を行えばよい。CCRが解除された大腸菌においては、グルコースとラクトースの共存下でもlacZ遺伝子が発現し、lacZ遺伝子の発現産物であるβガラクトシダーゼが産生され、βガラクトシダーゼがx-galを分解し、コロニーが青色を呈する。従って、青色コロニーからCCRが解除された大腸菌を単離することができる。
【0023】
このようにして、Mlcタンパク質の過剰発現を引き起こす変異を同定したならば、大腸菌のmlcプロモーターに該変異を導入することにより、大腸菌のCCRを解除することができる。また、部位特定的変異法や相同組換えにより特定の部位に変異を導入することもできる。このように特定の部位に変異を導入した大腸菌についても、上記の青白選別を行えばよい。
【0024】
Mlcタンパク質の過剰発現を引き起こす、mlcプロモーターの変異を同定したら、該変異を大腸菌のmlcプロモーターに導入すればよい。大腸菌におけるmlcプロモーターの変異の導入の方法は、限定されないが、例えば部位特異的突然変異や相同組換えにより行うことができる。
【0025】
本発明はMlcタンパク質を過剰に発現する遺伝子変異が導入され、CCRが解除された大腸菌変異株を包含する。また、発現ベクター等を用いて、野生型のMlcタンパク質を過剰に発現させた株も包含する。
【0026】
CCRが解除された大腸菌を用いることにより、ブドウ糖と他の糖を含む混合糖を用いて有用な有機物質を生産することができる。ここで、CCRを解除した上で有機物質の生産に用いる大腸菌株としては、大腸菌(E.coli)に属しているあらゆる大腸菌が含まれる。例えば、大腸菌K-12株、B株、W株やそれらの変体が挙げられ、変異体としてはMG1655株(ATCC(American Type Culture Collection)47076)、W3110株(ATCC27325)等が挙げられる。これらの大腸菌に上記のMlcタンパク質を過剰生産する変異を導入すればよい。CCRが解除された大腸菌は元々有している有機物質の生産系により混合糖を原料として有機物質を産生することができる。また、CCRを解除した大腸菌に有機物質の合成に関与する遺伝子を導入して形質転換大腸菌を作製し有機物質の生産に用いることもできる。さらに、有機物質の合成に関与する遺伝子が導入された形質転換大腸菌株のCCRを解除して、該大腸菌を有機物質の生産に用いることもできる。有機物質の合成に関与する遺伝子が導入された大腸菌株として、例えば、エタノールを生産し得るKO11株(ATCC55124)等が挙げられる。
【0027】
また、アルコール生産を目的とする場合、大腸菌のアルコール耐性を向上させてもよい。例えば、アルコールを含有する培地中で培養を行うか、あるいは紫外線の照射や化学的変異原処理によりランダム変異を誘発することにより、アルコール耐性株を得ることができる。
【0028】
さらに、本発明の方法で生産しようとする有機物質の中間体からの代謝産物であって、目的の有機物質ではない有機物質を合成する経路を遮断してもよい。遮断は合成経路を遮断しようとする有機物質の合成に関連する酵素をコードする遺伝子をノックアウトすればよい。ノックアウトは例えば相同組換えにより行うことができる。例えば、アルコールを生産する場合、乳酸合成経路やコハク酸合成経路を遮断すればよい。
【0029】
有機物質を生産し得る形質転換大腸菌株は、上記の有機物質の合成に関与する酵素をコードする遺伝子を発現ベクターに挿入し、該発現ベクターを用いて大腸菌を形質転換することにより作製できる。用いる発現ベクターには、プロモーター、上記酵素をコードする遺伝子のほか、ターミネーター、所望によりエンハンサーなどのシスエレメント、イントロンの5’末端側に存在するスプライス供与部位及びイントロンの3’末端側に存在するスプライス受容部位からなるスプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)などを含有するものを作動可能に連結して用いればよい。なお、選択マーカーとしては、例えばジヒドロ葉酸還元酵素遺伝子、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子等が挙げられる。発現ベクターに酵素をコードする遺伝子を挿入するには、まず、精製されたDNAを適当な制限酵素で切断し、適当なベクターDNAの制限酵素部位又はマルチクローニングサイトに挿入して発現ベクターに連結する方法などを用いればよい。また、1つの発現ベクターに2種類の酵素をコードする遺伝子を同時に挿入して、共発現させる場合、複数のマルチクローニングサイトを含む発現ベクターを用いてもよい。
【0030】
大腸菌への組換えベクターの導入方法は、限定されず、例えばカルシウムイオンを用いる方法[Cohen, S.N.et al.:Proc. Natl. Acad. Sci., USA, 69:2110(1972)]、エレクトロポレーション法等が挙げられる。用いるベクターも限定されず、例えば、プラスミドDNA、ファージDNA等の公知の大腸菌用の発現ベクターを利用することができる。複数の遺伝子を別々のベクターに挿入して、大腸菌に導入してもよいし、複数の遺伝子を1つのベクターに挿入して、大腸菌に導入してもよい。
【0031】
大腸菌を用いて有機物質を生産するときに原料として用いる混合糖としては、ブドウ糖に加え、5炭糖であるアラビノース、キシロース、6炭糖であるリボース、ガラクトース、マンノース等の単糖;ラクトース、マルハトース、スクロース等の二糖;各種オリゴ糖の少なくとも1つを含む糖が挙げられる。単糖はそのまま、二糖やオリゴ糖は大腸菌体内で単糖になり、代謝され、他の有機物質が合成される。好ましくは、ブドウ糖とキシロース及び/又はアラビノースを同時に用いる。また、バイオマス由来の糖混合物も用いることができる。バイオマス由来の糖混合物とは、植物や動物から得られる糖であり、グルコースを含む種々の糖が含まれる。バイオマス由来の糖は大量に安価で入手できるので、本発明のCCRが解除された大腸菌を用いて有機物質を生産するときの原料として好適に用いることができる。植物や動物としては、トウモロコシ、サトウキビ、小麦等が挙げられ、植物の根、茎、幹、枝、葉、花、種子等から得ることができる。また、木材チップやパルプを含む木質系バイオマスも利用することができる。植物由来のバイオマスには、セルロースやヘミセルロース等の多糖も含まれるが、これらの多糖はアルカリや酸により分解し、あるいは酵素的に又は微生物的に分解して、大腸菌が有機物質の生産に利用できる糖の含有量を高めればよい。
【0032】
本発明の方法により生産される有機物質の種類は限定されないが、天然の大腸菌株が元々生産する能力のある有機物質に限らず、遺伝子組換え技術や菌の育種などによって新たに生産するようになった有機物質も含まれる。有機物質としては、アルコール、有機酸、タンパク質、アミノ酸等が挙げられる。アルコールとしては、エタノール、1-ブタノール、イソブタノール(イソブチルアルコールともいう)、2-メチル-1-ブタノール、3-メチル-1-ブタノール、プロパノール、イソプロパノール等が挙げられる。有機酸としては乳酸、コハク酸、酪酸、プロピオン酸、酢酸等が挙げられる。
【0033】
大腸菌株に目的の有機物質の生産に関与する遺伝子を導入する場合、上記のアルコールや有機酸等の生体内における代謝経路及び合成に関与する遺伝子は公知であり、当業者ならば、適切な外来の遺伝子を大腸菌に導入することにより、大腸菌に有機物質合成系を導入することができる。また、導入する遺伝子は好ましくは大腸菌での発現用にコドンを最適化する。例えば、エタノールを生産する場合、ピルベート脱水素酵素(PDC)及びアルコール脱水素酵素(ADH)をコードする遺伝子を大腸菌に導入すればよい。また、イソブタノールを生産する場合、分枝α-ケト酸脱炭酸酵素(KIVD)、アルコール脱水素酵素(ADH2)、アセト乳酸シンターゼ(ALSS)、ケトール酸レダクトイソメラーゼ(ILVC)、ジヒドロキシ酸デヒドラターゼ(ILVD)をコードする遺伝子を導入すればよい。この場合、2ケト酸を経由して効率的にイソブタノールを生産することができる。
【0034】
CCRが解除された大腸菌を上記混合糖を含む培地で培養することにより、混合糖に含まれる複数種の糖を同時に代謝して、有用物質を効率的に生産することができる。
【0035】
上記のようにして得られたCCRが解除された大腸菌又はCCRが解除されさらに有機物質合成系を導入した大腸菌を混合糖を添加した培地で培養することにより有機物質を生産することができる。
【0036】
大腸菌の培養は公知の方法に従い行うことができる。例えば、培養液として、DMEM、MEM、RPMI1640、IMDM等を使用することができ、牛胎児血清(FCS)等の血清補液やその他野サプリメント試薬等を添加して用いればよい。さらに、形質転換した大腸菌を固定化して、有機物質を生産することもできる。固定化は、包括法、架橋法、担体結合法等により行うことができ、固定化に用いられる固定化担体としては、ガラスビーズ、シリカゲル、ポリウレタン、ポリアクリルアミド、ポリビニルアルコール、カラギーナン、アルギン酸、寒天、ゼラチン等がある。
【0037】
有機物質が大腸菌菌体外に生成、蓄積した場合には、培養終了後、培養物から菌体などの沈殿物を除去し、公知の方法を単独で又は組合わせて、培養物から有機物質を単離、精製することができる。また、有機物質が大腸菌菌体内に生成、蓄積される場合には、培養終了後、培養物から菌体を回収した後、機械的または化学的方法等の適切な方法で菌体を破砕し、該菌体破砕物から、公知の方法を単独で又は組合わせて有機物質を単離、精製することができる。有機物質がアルコールである場合、例えば蒸留により回収することができる。
【実施例】
【0038】
以下、実施例により本発明をさらに具体的に説明する。但し、本発明はこれら実施例にその技術的範囲が限定されるものではない。
【0039】
〔実施例1〕
実験手法
以下に記載の実施例において、大腸菌の培養、遺伝子組換え操作、タンパク質の活性測定等は、本発明者らの論文および特許(Nakashima and Tamura, Biotechnol. Bioeng. (2004) 86:136-148、Nakashima and Tamura, Appl. Environ. Microbiol. (2004) 70:5557-5568、Nakashima et al., Nucleic Acids Res. (2006)34:e138、Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103、特許第3793812号、特許3944577号)に基づいて行った。特に断りがない限り、大腸菌の培養はLB(1% Difco Bacto Tryptone、0.5% Difco Yeast Extract、1%塩化ナトリウム)を用いて37℃で行った。寒天培地による培養の場合、寒天濃度は1.7%で、9 cm直径のプラスチックシャーレに入れたものを使った。また、プラスミドを含む大腸菌を培養する場合は対応する抗生物質を加えて行った。抗生物質の終濃度は、アンピシリン:50μg/mL、クロラムフェニコール:24μg/mL(原液は100%エタノールを溶媒として34 mg/mLの濃度で作成)、カナマイシン:15μg/mL、アプラマイシン:35μg/mLである。LB以外の培地の組成は以下の通りである。M9YM:17 mg/mL Na2HPO4-12H2O, 3 mg/mL KH2PO4, 0.5 mg/mL NaCl, 1 mg/mL NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.25% Difco Yeast Extract, 50 mM MOPS buffer (pH 7.0)、M9Y:17 mg/mL Na2HPO4-12H2O, 3 mg/mL KH2PO4, 0.5 mg/mL NaCl, 1 mg/mL NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.5% Difco Yeast Extract。用いた菌株の一覧を表1に示す。
【0040】
【表1】

【0041】
〔実施例2〕
CCRが解除された変異株の分離
野生型大腸菌、MG1655株を一晩培養した。これを新鮮なLBで5000倍に希釈して、うち20μLを10 g/Lブドウ糖、8 g/Lラクトース、0.125 mg/mL x-galを含むLB寒天培地50枚に塗布した。直後に、XL-1000 UV crosslinker(Spectronics社製)を用いて、10-12 mJ/cm2の紫外線を照射した。これらを一晩培養したところ、計約30000個のコロニーが出現し、うち1コロニーのみが青色を呈していた。この変異株をgen株(glucose effect not observed)と暫定的に名付けた。
【0042】
次いで、二次スクリーニングとして以下の実験を行った。野生株とgen株をそれぞれ10 g/Lブドウ糖あるいは、10 g/Lブドウ糖と8 g/Lラクトースあるいは、8 g/Lラクトースを含む4 mLの液体培地で中期対数増殖期まで培養し、菌体を遠心分離機によって回収した。菌体を500μLのPBS(0.14 M NaCl、2.7 mM KCl、10.1 mM Na2HPO4、1.8 mM KH2PO4)に懸濁し、そこに直径 0.1 mmのガラスビーズ(安井機械社製)0.1 gを混合した。これを安井機械社製ビーズショッカーにかけ、菌体を破砕した後、20000×g、4℃、15分遠心することで上清を得た。これが粗タンパク質液となる。粗タンパク質液を用いて、βガラクトシダーゼの活性を測定したところ(Nakashima et al., Nucleic Acids Res. (2006)34:e138)、野生株では、含ラクトース培地の時のみ活性が認められたのに対して、gen株では、含ブドウ糖ラクトース培地と含ラクトース培地の時にほぼ同程度の活性が認められた。なお、gen株であっても、含ブドウ糖培地では活性が認められなかった。この結果から、gen株は予想通り、CCRが解除された変異体である可能性が示唆された。
【0043】
さらに、gen株では、ラクトース以外の糖でも同様のCCR解除がみられるかを検討した。まず、プラスミドpHN171(Nakashima and Tamura, Biotechnol. Bioeng. (2004) 86:136-148)からレポーターとなるpip遺伝子(プロリンイミノペプチダーゼをコード)をNcoIとSpeIで切り出し、pHN540uベクター(Nakashima et al., Nucleic Acids Res. (2006)34:e138)の同部位にサブクローニングした。このプラスミドにpHN1325と名前をつけた。pHN1325では、L-アラビノース誘導性badプロモーター(Pbad)の下流にpip遺伝子が連結されている。Pbadからの転写は、L-アラビノースによって誘導されるが、L-アラビノースとブドウ糖が共存するときは転写はほとんど起こらない。すなわち、CCRが正常に働いている事を意味する。pHN1325を野生株とgen株にそれぞれ導入し、その形質転換体をそれぞれ10 g/Lブドウ糖あるいは、10 g/Lブドウ糖と8 g/L L-アラビノースあるいは、8 g/L L-アラビノースを含む4 mLの液体培地で培養した。培養した菌体から、粗タンパク質液をβガラクトシダーゼの時と同様に作成し、PIPの活性を測定した。測定は以下のように行なった。まず、2μLの粗タンパク質液と56 mLのPBSを混合したものを60℃で10分保持し、ここに2 mLの10 mM H-Pro-AMC(Bachem Holding AG製)を入れそのまま10分保持した。うち50μLを取り出し、そこに25μLの10%硫酸ドデシルナトリウム溶液を加え、混合後、黒色96穴プレート(Costar社製; product no.3915)に入れた。この蛍光をSafire reader(Tecan社製)で excitation, 380 nm; emission, 460 nm; excitation and emission bandwidths, 12 nm; gain, 80; number of flashes, 10; Z-position 11019 mm; and integration time, 500μsの設定で測定した。
【0044】
その結果、野生株では、含L-アラビノース培地の時のみ活性が認められたのに対して、gen株では、含ブドウ糖L-アラビノース培地と含L-アラビノース培地の時にほぼ同程度の活性が認められた。なお、gen株であっても、含ブドウ糖培地では活性が認められなかった。
以上のことから、gen株は、CCRが解除された変異体であることが示された。
【0045】
〔実施例3〕
gen株の変異遺伝子クローニング
gen株で起こっている、CCRを解除する変異が、もし優性変異であるならば、以下の実験で変異した遺伝子がわかるはずである。まず、gen株ゲノムライブラリーを作成するために以下の作業を行った。gen株を一晩培養し、それを用いてゲノムDNAを精製した(Nakashima et al., Nucleic Acids Res. (2006)34:e138)。ゲノムDNAをSau3AIで部分消化し、アガロース電気泳動に供し、10-6 kb程度の断片をアガロースゲルから回収した。それを精製し、ベクターpSPT18(Roche Applied Science社)のBamHI部位にライゲーションした。ライゲーション産物を大腸菌XL1-blueに形質転換し、約30000コロニーからなるゲノムライブラリーを得た。次いで、擬陽性クローンを極力減らすために以下のプラスミドpHN1629を作成した。sSN1481(aaccatggtaagccgatacgtacccgata(配列番号1))とsSN1482(aaactagtctacccaatcagtacgttaatt(配列番号2))の2つのオリゴヌクレオチドプライマーを作成し、野生型MG1655株のゲノムを鋳型としてPCR反応を行った。これによって、mazF遺伝子と呼ばれる遺伝子のORFが増幅されが(0.3 kb)、この遺伝子は、過剰発現させると大腸菌が増殖できなくなることが知られている。このPCR断片の両末端をNcoIとSpeIで切断し、pHN540uベクターの同部位にクローニングした。このプラスミドにpHN1629と名前をつけた。pHN1629では、Pbadの下流にmazF遺伝子が連結されている。よって、野生株にpHN1629を導入して、ブドウ糖とL-アラビノースを両方含む培地で培養しても菌は増殖するが、CCRが解除された株では、菌は増殖しない。実際に、pHN1629でgen株を形質転換し、ブドウ糖のみを含む寒天培地で培養したところ増殖し、ブドウ糖とL-アラビノースを両方含む寒天培地上だと増殖しないことを確認した。
【0046】
まず、pHN1629を用いて野生型MG1655株を形質転換し、次いで上述のgen株ゲノムライブラリーでさらに二重形質転換した。その二重形質転換体を10 g/Lブドウ糖、8 g/Lラクトース、0.125 mg/mL x-galを含む寒天培地上で生育させたところ、約40000コロニーのうち、9コロニーが青色を呈した。さらに、9コロニーを10 g/Lブドウ糖と2 g/L L-アラビノースを含む寒天培地上で生育させた。すると、うち6コロニーについて、コントロールのpSTP18(空ベクター)とpHN1629の二重形質転換体と比較して生育が顕著に阻害されていた。
【0047】
この6コロニーの持つgen株ゲノムライブラリーについて、プラスミドを回収、精製し、DNA配列を一部決定したところ、4つがmlc遺伝子を含む重複する断片(図1a)、2つがanmK、slyB、slyA、ydhI、ydhJ遺伝子を含む重複する断片(図1b)を持つプラスミドであった。
【0048】
まず、後者プラスミド2つについて、共に野生型MG1655株に再導入し、実施例2と同様に液体培地で培養した後、βガラクトシダーゼの活性を測定した。しかし、どちらのプラスミドにおいてでも、含ブドウ糖ラクトース培地での培養時では活性が見られず、含ラクトース培地の時にのみ活性が認められた。さらに、プラスミド内のゲノム断片のDNA配列を全て決定し、発表されているMG1655株のDNA配列(GenBank登録番号U00096.2)と比較したところ、全く同一の配列であった。よって、これらは擬陽性クローンか、過剰発現により不完全なCCR解除を起こすクローンであると結論した。
【0049】
次いで、前者プラスミド4つについても同じくβガラクトシダーゼの活性を測定した。すると、含ブドウ糖ラクトース培地と含ラクトース培地での培養時に、ほぼ同程度の活性が認められた。すなわち、このプラスミドに含まれる、gen株由来ゲノム断片の中に変異が入っている可能性が高いことが示唆された。この断片のうち最も短い断片について、全てのDNA配列を決定したところ、野生株由来のものと比較して、1塩基に突然変異があることが認められた。その変異箇所が図1aに示してある。この変異は、どのオープンリーディングフレーム(ORF)の内部でもなく、mlc遺伝子のプロモーター部位と予測されるところに存在していた。
【0050】
大腸菌プロモーターにおいては、-10配列と呼ばれる共通配列があり、その配列はtataatであるときに、最も効率の高いプロモーターになると考えられている(de Boer et al., Proc. Natl. Acad. Sci. USA (1983) 80:21-25)。野生型のmlc遺伝子では、これがcaccatであり、共通配列とは相同性がかなり低い。しかし、本発明者の取得した変異型では、taccatと共通配列と相同性が高くなるような変異が入っている。よって、この変異mlc遺伝子では、プロモーターが高活性型となっていると予測される。そして、その結果、Mlcタンパク質が過剰発現されているものと予測される。
【0051】
そこで、野生型mlcプロモーターと変異型mlcプロモーターをそれぞれPCRにて分離し、それらの下流にpip遺伝子をクローニングしたプラスミドを作成した。次いで、それらプラスミドをMG1655株に導入、培養し、それぞれのPip活性を測定したところ、変異型mlcプロモーターの方が17倍高かった。すなわち、配列情報から予想されたとおり、変異型mlcプロモーターは高活性型であることが示唆された。
【0052】
なお、その機構から考えて、このmlc遺伝子変異は優性であると考えられる。実際に、この変異が劣性であることを仮定して行った、野生株ゲノムライブラリーをgen株に導入する実験では、擬陽性クローンしか分離されなかった。
【0053】
以上の結果から、gen株でCCR機構の解除を引き起こしていた変異は、mlc遺伝子の高活性型プロモーター変異であると言うことができ、以降gen遺伝子変異をmlc*変異と呼ぶこととする。
【0054】
野生型mlcと変異型mlc*遺伝子をプラスミドpHN1242(Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103)を用いて野生型MG1655株で過剰発現させたところ、いずれも場合もCCRが解除されることが分かった。ただしこの場合、変異型mlc*遺伝子ではほぼ完全にCCRを解除できたのに対し、野生型mlcではCCRの解除は部分的であった。このことから、mlc遺伝子あるいはそのプロモーターに変異が入っていなくとも、Mlcタンパク質が過剰発現さえされれば、少なくとも部分的にはCCRが解除されることが分かった。
【0055】
〔実施例4〕
mlc*変異の野生型株への導入
相同組換えを利用して、元のMG1655株にmlc*遺伝子を導入するために、以下のプラスミドを作成した。
【0056】
pMW118(ニッポンジーン社製)を鋳型、sSN1097(taagctagctatggacagttttccctttgatatgta(配列番号5))とsSN1097R(gatgatgaacatcagtagggaaaatgc(配列番号6))をプライマーして用いて、pSC101 ori配列の一部をPCR増幅した。なお、このPCRに先立って、sSN1097RはT4 DNAキナーゼ(New England Biolab社製)を用いて5’末端をリン酸化した。一方で、pMW118を鋳型、sSN1098(aaactgcaggttgatgataccgctgccttactg(配列番号7))とsSN1098R(tgaacgtattggttataagtgaacgatacc(配列番号8))をプライマーとして用いて、pSC101 ori配列の別の一部をPCR増幅した。前者断片をNheI、後者断片をPstIで切断し、両断片を同時にpHN540uのNheI、PstI部位にクローニングした。出来たプラスミドにpHN1092と名前をつけた。pHN1092はpSC101 oriをもつプラスミドであるが、一箇所の点突然変異(GenBank登録番号X01654の、5439番目の塩基がシトシンからチミンに変化)により、温度感受性のプラスミドとなっており、37℃を越える温度で培養されると、プラスミドとして細胞内で維持されない(Armstrong et al., J. Mol. Biol. (1984) 175:331-347)。よって、37℃を越える温度では、プラスミドは脱落するか大腸菌のゲノム内に挿入されるかのどちらかとなる。この性質と、大腸菌が天然に持つ相同組換え能を利用して、ゲノムの特定の場所を操作する(Hamilton et al., J. Bacteriol. (1989) 171:4617-4622、Emmerson et al., Biol. Proced. Online (2006) 8:153-162)。
【0057】
pHN1092を鋳型、sSN1098(上述)とsSN1173(ggggtacctgagcaaactggcctca(配列番号9))をプライマーとして用いて、温度感受性pSC101 ori配列とクロラムフェニコール耐性遺伝子をPCR増幅した。なお、このPCRに先立って、sSN1173はT4 DNAキナーゼを用いて5’末端をリン酸化した。一方、pK18mobsacB(GenBank登録番号、FJ437239)を鋳型として、sSN1172(aactgcagttctagaaggcctggatccccatggccgttcactattatttagtgaaatgag(配列番号10))とsSN1108(aaagctagcattttcttttgcgtttttatttgttaac(配列番号11))をプライマーとして用いて、sacB配列をPCR増幅した。なお、このPCRに先立って、sSN1108はT4 DNAキナーゼを用いて5’末端をリン酸化した。増幅した両断片をT4 DNAリガーゼによって連結し、pHN1234を得た。
【0058】
pHN1234のNheI部位に、pHN267(Nakashima and Tamura, Appl. Environ. Microbiol. (2004) 70:5557-5568)からXbaIとSpeIで切り出したカナマイシン耐性遺伝子(1.4 kb)をサブクローニングした。これにpHN1516と名付けた。pHN1516を鋳型、sSN1415(tccatggggatcctcgcgaggccgttcactattatttagtgaaatgag(配列番号12))とsSN1421(tatgcattctagagatatcttagttgctctccaggtgacccaggtgacc(配列番号13))をプライマーとして用いて、PCR増幅を行った。なお、このPCRに先立って、sSN1415とsSN1421はT4DNAキナーゼを用いて5’末端をリン酸化した。一方、pHN1234を鋳型、sSN1173(上述)とsSN1098(上述)をプライマーとして用いて、PCR増幅を行った。増幅した両断片をT4 DNAリガーゼによって連結し、pHN1534を得た。
【0059】
pHN1234を鋳型、sSN1417(cccatggggatccaggccttctagaact(配列番号14))とsSN1173(上述)をプライマーとして用いて、PCR増幅を行った。なお、このPCRに先立って、sSN1417はT4 DNAキナーゼを用いて5’末端をリン酸化した。増幅した断片をT4 DNAリガーゼによって自己閉環化し、pHN1532を得た。
【0060】
pHN1234、pHN1534、pHN1532のプラスミド構造を図2に示す。pHN1534が遺伝子破壊用のプラスミド、pHN1532が点突然変異導入用のプラスミドの元となる(後述)。
【0061】
大腸菌MG1655のゲノムDNAを鋳型、sSN1522(atactgcagttcgatggttgaaacactctg(配列番号15))とsSN1513(gcctgcaggtgagggaaatttcagcgaaaa(配列番号16))をプライマーとして用いて、PCR増幅を行った。増幅した断片をPstIとScaIで切断して、pHN1534のNsiI、EcoRV部位にクローニングした。出来たプラスミドにpHN1677と名付けた。次いで、大腸菌MG1655のゲノムDNAを鋳型、sSN1523(agggatccaatacgtgtcgtcaaatttttacttagg(配列番号17))とsSN1524(ggccatgggattgctgcgtaacacagcgt(配列番号18))をプライマーとして用いてPCR増幅を行い、その末端をBamHIとNcoIで切断して、pHN1677のBamHI、NcoI部位にクローニングした。できたプラスミドにpHN1678と名付けた。これがmlc遺伝子破壊用プラスミドとなる。
【0062】
実施例2に記載のgen株のゲノムDNAを鋳型、sSN1522とsSN1524(共に上述)をプライマーとして、PCR増幅を行った。増幅した断片をPstIとNcoIで切断して、pHN1532のPstI、NcoI部位にクローニングし、出来たプラスミドにpHN1693と名付けた。これが、mlc遺伝子をpHN1678で破壊した後に、mlc*で入れ替えるためのプラスミドとなる。
【0063】
以下、野生型MG1655株にmlc*を導入する過程を述べるが、これは、Emmersonらの方法(Emmerson et al., Biol. Proced. Online (2006) 8:153-162)によるものである。まずMG1655をpHN1678で形質転換し、mlc遺伝子座位で2回の相同組換えを起こさせることによって、mlc遺伝子を破壊した。次いで、その破壊株をpHN1693で形質転換し、同様に2回の相同組換えを起こさせることによってmlc*に入れ替えた。以降、混同を避けるため、この相同組換えで作成した株をmlc*HRと呼び、実施例2で分離したgen株(実体はmlc*変異株)をmlc*UVと呼ぶこととする。DNA配列解析により、mlc*HR株ゲノム上では野生型MG1655株と比べて、mlc*UV と同一の1塩基変異が入っていることを確認した。
【0064】
〔実施例5〕
野生株、mlc*UV株、mlc*HR株の比較
野生株、mlc*UV株、mlc*HR株を用いて、実施例2と同様に、x-galを含むLB寒天培地による青白コロニー判別実験を行った。すると予想通り、野生株は含ラクトース培地では青いコロニー、含ブドウ糖ラクトース培地では白いコロニーを形成したのに対して、mlc*UV株とmlc*HRは、含ラクトース培地、含ブドウ糖ラクトース培地上、共に青いコロニーを形成した。なお、すべての株が含ブドウ糖培地上では白いコロニーを形成した。
【0065】
さらに、ラクトース以外の糖についてのCCRを解析するため、野生株、mlc*UV株、mlc*HR株を、M9YMにブドウ糖とキシロースを8 g/Lずつ添加した培地で培養した。これらの培地に残存しているブドウ糖とキシロースの量を時間を追って高速液体クロマトグラフィー(D-2000型LaChrom Elite、日立社製)を用いて測定した。分析条件は以下の通りである。分離カラム:aminex HPX-74H(Bio-Rad社製)、ガードカラム:Cation H micro-guard cartridge(Bio-Rad社製)、分離溶媒:4 mM H2SO4、流速:0.5 mL/min、検出器:紫外線検出器(L-2400型による210 nmでの検出、日立社製)及び示差屈折率検出器(L-2490型、日立社製)、カラム温度:45℃。その結果(図3)、野生株はこの培養条件下では全くキシロースを消費しなかったのに対して、mlc*UV株、mlc*HR株は、ブドウ糖とキシロースを同様の速度で消費した。
【0066】
以上の結果から、mlc*UV株でCCRが解除されていたのは、mlc*変異のみによるものであって、それ以外の遺伝子の変異は、あったとしてもCCRには関係がないと結論される。以降は、予期せぬ変異が入っている心配のない、mlc*HRのみを実験に使用した。
【0067】
〔実施例6〕
イソブタノール生産のための発現プラスミド構築
大腸菌は天然にはイソブタノールをほとんど生産しない。よって、外来遺伝子を導入したり、関連する代謝経路を強化しなければならない。そのために必要なプラスミドを以下のように作成した。
【0068】
pTrc99a(Amersham Biosciences社製)を鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号19))とsSN1284(aaactagtctcgagaagcttgcatgcctgcaggtcgact(配列番号20))をプライマーとしてPCR増幅を行い、IPTG誘導性のtrcプロモータ(Ptrc)、lacオペレータ(lacO)、マルチクローニング部位配列を含む断片を得た。その断片の両端をNsiIとSpeIで処理し、pHN1257(NAR)のNsiI、SpeI部位にクローニングした。このプラスミドにpHN1344と名付けた。一方、pTrc99aを鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号19))とsSN1345(aatctagaggggaattgttatccgctcacaattccacac(配列番号22))をプライマーとしてPCR増幅を行い、PtrcとlacO配列を含む断片を得た。その断片をXbaIで処理し、pET28a(Novagen社製)のXbaI、EcoRV部位にクローニングした。このプラスミドにpHN1420と名付けた。pHN1420から、NsiIとNcoIによってPtrcとlacOを含む配列を切り出し、pHN1344のNsiI、NcoI部位にサブクローニングした。出来たプラスミドにpHN1431と名付けた。これが外来遺伝子を発現するための元のベクターとなる。
【0069】
Genscript社に依頼し、Lactococcus lactis由来kivd遺伝子(GenBank登録番号、AJ746364)を人工的に合成した。この人工kivd遺伝子は、大腸菌での発現用にコドンを最適化してあるもので、pUC57ベクター(GenBank登録番号、Y14837)にクローニングされていて、その配列は配列表に記してある。一方、出芽酵母Saccharomyces cerevisiaeのAdh2遺伝子を、S. cerevisiae W303株(Takeuchi and Tamura, FEBS Lett. (2004) 565:1-3:39-42)のゲノムを鋳型、sSN1287(aaagcatgcaggagatataccatgtctattccagaaactcaaaaagc(配列番号24))とsSN1288(gcctcgagttatttagaagtgtcaacaacgtat(配列番号25))をプライマーとしてPCR増幅した。上記kivd遺伝子のNcoI-SphI断片(1.6 kb)と、adh2遺伝子のSphI-XhoI断片(1.0 kb)を同時に、pHN1431のNcoI、XhoI部位にクローニングした。出来たプラスミドにpHN1435と名付けた。pHN1435は、Ptrcの下流にlacOを持ち、さらにその下流にkivd遺伝子とadh2遺伝子をオペロンとして持つ。
【0070】
pTrc99aを鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号21))とsSN1004(cgaattccatggtctgtttcctgtg(配列番号27))をプライマーとしてPtrcを含む配列のPCR増幅を行った。増幅された断片をNsiIとNcoIで消化し、pHN1270(Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103)のNsiI、NcoI部位にクローニングした。できたプラスミドをpHN1375と名付けた。Bacillus subtilis 168株(表1)のゲノムDNAを鋳型、sSN1343R(aaccatggtgacaaaagcaacaaaagaacaaaaatcccttgtga(配列番号28))とsSN1344(aaaggatcctagagagctttcgttttcatgagt(配列番号29))をプライマーとして、alsS遺伝子のPCR増幅を行った。増幅された断片をNcoIとBamHIで消化し、pHN1375のNcoI、BamHI部位にクローニングした。出来たプラスミドをpHN1447と名付けた。MG1655株のゲノムDNAを鋳型、sSN1327(aaggatccgaggaatcaccatggctaactacttca(配列番号30))とsSN1328(gcctcgagttaacccgcaacagcaatacgtttcat(配列番号31))をプライマーとしてPCR増幅を行い、ilvC遺伝子を含む断片を得た。その断片の両端をBamHIとXhoIで処理した。一方、MG1655株のゲノムDNAを鋳型、sSN1329(aaagtcgacaggagatataccatgcctaagtaccgttccgccacca(配列番号32))とsSN1330(agactagtttaaccccccagtttcgatttatcgcg(配列番号33))をプライマーとしてPCR増幅を行い、ilvD遺伝子を含む断片を得た。その断片をSalIとSpeIで処理した。これらilvC遺伝子断片とilvD遺伝子断片を同時にpBluscript SKII+(Stratagene社製)のBamHI、SpeI部位にクローニングした。このプラスミドにpHN1406と名付けた。pHN1406からBamHI、SpeIでilvC遺伝子とilvD遺伝子を含む配列を切り出し、pHN1447の同部位にサブクローニングした。このプラスミドにpHN1451と名付けた。pHN1451はPtrcの下流にlacOを持ち、さらにその下流にalsS、ilvC、ilvD遺伝子の3遺伝子をオペロンとして持つ。
【0071】
〔実施例7〕
mlc*変異によってイソブタノール生産が増大するかどうかの実証
次に、mlc*変異によってどの程度イソブタノールが生産できるか検証した。同時に比較のために、野生型MG1655株から作成したDptsG株とcrp*株も用いることとした。また、イソブタノールの生産性を増すために、ここで用いた株ではldhA(乳酸脱水素酵素をコード)、adhE(融合性アセトアルデヒド-CoA脱水素酵素/鉄依存性アルコール脱水素酵素/ピルビン酸-ギ酸-リアーゼ不活性化酵素をコード)、pflB(ピルビン酸-ギ酸-リアーゼをコード)遺伝子を破壊した。これら株の作成は、実施例4と同様の方法によって行った。イソブタノール生産に用いた菌株(LAF、LAFmlc*、LAFDptsG、LAFcrp*)の遺伝子型が表1に示してある。
【0072】
LAF、LAFmlc*、LAFDptsG、LAFcrp*の4株を、pHN1435とpHN1451で共形質転換し、まず前培養を行った。それらを、M9Yに40 g/Lブドウ糖、あるいは20 g/Lブドウ糖と20 g/Lキシロース、あるいは40 g/Lキシロースを加えた培地に、100分の1量加え本培養を開始した。本培養は、50 mL容積の三角フラスコにシリコン栓をして、4 mLの培養液、30℃で行った。pHN1435とpHN1451から遺伝子を発現させるために、600 nmでの吸光度が0.8に達したところでIPTGを1 mMになるように添加した。
【0073】
一定時間ごとに培養液を採取し、遠心分離によって菌体を除いた後、その上清を高速液体クロマトグラフィーにて分析した。分析したのは、残存ブドウ糖濃度、残存キシロース濃度、生産されたイソブタノール濃度で、分析条件は実施例5と同一である。その結果(図4)、20 g/Lブドウ糖と20 g/Lキシロースを炭素源とした場合、MG1655に比べて、LAFmlc*、LAFDptsG両株のイソブタノール生産性が有意に高かった。また、40 g/Lブドウ糖あるいは、40 g/Lキシロースを炭素源とした場合、LAFmlc*株での生産性が最も高かった。以上のことから、LAFmlc*株は、大腸菌によるイソブタノール生産に有効であることが示された。
【0074】
また、イソブタノール生産性をより高めるために、LAFmlc*株においてackA-pta遺伝子をさらに破壊することでLAFCmlc*株を作成した。さらに、本培養の培養条件も最適化した。その条件は、250 mL容積のスクリューキャップ付き三角フラスコに、M9Yに40 g/Lブドウ糖、あるいは20 g/Lブドウ糖と20 g/Lキシロースを加えた培地を20 mL入れ、そこに前培養を100分の1量加え、30℃で培養するものである。40 g/Lのブドウ糖入りで培養したものは、ブドウ糖が枯渇したらさらに40 g/Lのブドウ糖を追加し、20 g/Lブドウ糖と20 g/Lキシロース入りで培養したものは、両方の糖が枯渇した時点でさらに20 g/Lブドウ糖と20 g/Lキシロースになるよう追加した。IPTGの添加については、上と同一である。イソブタノールを生産した結果を図5に示す。80 g/Lのブドウ糖からは19 g/L、40 g/Lのブドウ糖と40 g/Lのキシロースからは11 g/Lのイソブタノールが生産された。
【配列表フリーテキスト】
【0075】
配列番号1〜22、24〜33 プライマー
配列番号23 合成
【技術分野】
【0001】
本発明は、大腸菌において混合糖から効率よく有機物質を生産する方法に関する。中でも特に、2種類以上の糖が含まれる培地を原料として、アルコールや有機酸などの有機物質を生産する方法に関する。
【背景技術】
【0002】
アルコールや有機酸などは、有用な物質として様々な用途に使われる。これらは糖を微生物に代謝させることで生産することができるが、原材料たる糖は、安価であるなどの理由から、動植物から作られた再生可能な有機性資源、バイオマスに由来することが多い。バイオマス由来の糖では、ブドウ糖、キシロース、アラビノースなど他種類の糖が混ざった、混合糖であることが一般的である(非特許文献1)。しかし、微生物に混合糖を与えた場合、エネルギー効率の最も良いブドウ糖のみを選択的に消費し、ブドウ糖を消費し終えるまで、それ以外の糖の代謝をしないことがある。これは、炭素カタボライト抑制(CCR)と呼ばれる遺伝機構が微生物に存在しているために起こる現象である(非特許文献2)。この結果、バイオマス由来の混合糖のうち、ブドウ糖以外の糖が効率よく目的物質に変換されず、無駄になってしまうことがある(非特許文献3)。
【0003】
CCR機構では、ブドウ糖の存在下では、ブドウ糖以外の糖の消費に関与する遺伝子群が発現抑制される。例えば、大腸菌のlacZYAはラクトースの消費に必要な遺伝子オペロンで、それぞれβガラクトシダーゼ、βガラクトシドパーミアーゼ、ガラクトシドアセチルトランスフェラーゼをコードしている。糖として、ブドウ糖とラクトースが培養液中に共存するとき、lacZYAの発現調節領域ではlacI遺伝子産物(ラクトースリプレッサー)およびcrp遺伝子産物(cAMP受容体タンパク質)が協調して、lacZYA mRNAが転写されないよう負に制御している。しかし、糖としてラクトースのみが培養液中に存在するときには、細胞内に取り込まれたラクトースが変化したアロラクトースによって、この負の制御は解除され、lacZYAのmRNAは盛んに転写される。これによって、ラクトースは大腸菌によって消費されるようになる。また、キシロース、アラビノースの消費に必要な遺伝子群も、これと同様な遺伝子発現調節を受けている(非特許文献4)。
【0004】
大腸菌は、微生物による物質生産の宿主に最もよく使われるものの一つであるが、上述ようにCCR機構を有している。よって、CCRが解除された変異株を作成し、様々な糖を同時代謝させる工夫がなされている。例えば、ptsG遺伝子が破壊された株(DptsG)や、crp遺伝子の中の3塩基対に点突然変異の入った株(crp*)などが、CCRが解除された株として知られており、実際の物質生産に使われた例がある(特許文献1及び非特許文献5〜8)。
【0005】
しかしptsG遺伝子は、細胞膜に局在し、ブドウ糖を細胞内に取り込む機能を担うブドウ糖パーミアーゼをコードしており、DptsG株ではブドウ糖の取り込み効率が下がり、目的物質の生産性が下がる場合がある。また、crp遺伝子はcAMP受容体タンパク質型の転写因子をコードしているが、相当数の遺伝子がCrpの制御下にあり、crp*株では大きく代謝経路が変化していることが考えられる。さらに、crp遺伝子破壊株(Dcrp)ではある種の有機溶媒に耐性を持つようになることが知られており、逆にcrp*株では有機溶媒に感受性になることも予想される(非特許文献9)。よって、crp*株では、有機溶媒の一種であるアルコールを生産した際、その毒性の問題から生産性が低下することも考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許6159738号明細書
【非特許文献】
【0007】
【非特許文献1】van Maris et al., Antonie van Leeuwenhoek (2006) 90:391-418
【非特許文献2】Deutscher, Curr. Opin. Microbiol. (2008) 11:87-93
【非特許文献3】Kim et al., Appl. Microbiol. Biotechnol. (2010) 88:1077-1085
【非特許文献4】Song and Park, J. Bacteriol. (1997) 179:7025-7032
【非特許文献5】Cirino et al., Biotechnol. Bioeng. (2006) 95:1167-1176
【非特許文献6】Khankal et al., J. Biol. Eng. (2009) 3:13
【非特許文献7】Dien et al., J. Ind. Microbiol. Biotechnol. (2002) 29:221-227
【非特許文献8】Nichols et al., Appl. Microbiol. Biotechnol. (2001) 56:120-125
【非特許文献9】Okochi et al., J.Biosci. Bioeng. (2008) 105:389-394
【発明の概要】
【発明が解決しようとする課題】
【0008】
以上のことから、本発明者は、有用物質生産に有用な新しいCCRが解除された変異株を分離することとした。また、そのような変異株を用いて、実際に有用物質生産を実証することとした。
【課題を解決するための手段】
【0009】
大腸菌のlacZYAオペロンは、上述のようにCCRを受ける遺伝子であるが、これが発現しているかどうかは、5-クロロ-4-ブロモ-3-インドリル-β-D-ガラクトース(x-gal)を用いた青白選別によって簡単に知ることができる。これは、無色透明のx-galが、lacZ遺伝子産物であるβガラクトシダーゼによって分解されると、青色を呈することに基づいている。すなわち、野生株の大腸菌をブドウ糖、ラクトース、x-gal、その他適当な栄養源を含む寒天培地上で培養すると、白色のコロニーを形成する。これは、培地中にブドウ糖とラクトースが共存しており、CCRによってlacZ遺伝子の転写が抑制されているからである。しかし、何らかの変異によってCCRに異常が生じ、ブドウ糖、ラクトース共存下でもlacZ遺伝子が転写されるようになると、同培地上で青色のコロニーを形成することになる。
【0010】
そこで本発明者は、野生型大腸菌MG1655株をブドウ糖、ラクトース、x-gal、その他適当な栄養源を含む寒天培地上に塗布し、紫外線を照射して突然変異を誘発し、生育後のコロニーの色を判別することとした。これによって、CCRに異常(不全)が生じた(すなわち、CCRが解除された)変異株が分離されるはずである。
【0011】
次いで、任意の株でCCRを解除することができるように、分離した変異株の変異遺伝子の同定をすることとした。このためには、ショットガンクローニングを行う。この段階では、変異遺伝子が優性か劣性か判断できないので、野生株または変異株のゲノムを用いて作成したプラスミドゲノムライブラリ2種を、それぞれ変異株、野生株に導入し、青白選別を行う。この際、陽性クローンに混じって擬陽性クローンも多数分離されると思われるので、二次スクリーニングや塩基配列決定なども行い、変異遺伝子を同定する。変異遺伝子を同定できたならば次に、相同組換えによって人為的に変異遺伝子を野生株に導入する。この相同組換えによる変異株と、紫外線照射で分離した元の変異株とで糖消費速度などを比較し、同一の表現型を示すことを観察して、最終的な変異遺伝子の確認とする。
【0012】
さらに、上記の変異株を用いて、有用物質の実生産を行う。そのために、混合糖からイソブタノール(イソブチルアルコール)を生産し、野生型、DptsG株あるいはcrp*株よりも本発明の変異株の方がイソブタノールの生産性がよいことを実証する。なお、イソブタノールは近年、ガソリンあるいは軽油代替燃料として注目を浴びているアルコールの一種である。イソブタノール生産の方法は、現在知られている中で最も生産効率のよい、2ケト酸を経由する方法を用いる(Atsumi et al., Nature (2008) 451:86-89)。
【0013】
本発明者は、このように大腸菌において混合糖から有機物質を生産する方法を開発すべく鋭意検討を行い、本発明を完成させるに至った。
【0014】
すなわち、本発明は以下の通りである。
[1] 遺伝子変異導入又は発現ベクターを使った過剰発現によって、Mlcタンパク質を過剰に発現することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株。
[2] Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、[1]の炭素カタボライト抑制(CCR)が解除された大腸菌株。
[3] mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、[1]又は[2]の炭素カタボライト抑制(CCR)が解除された大腸菌株。
[4] anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によって、炭素カタボライト抑制(CCR)が解除された大腸菌株。
[5] さらに、有機物質の合成に関与する遺伝子が導入されている、[1]〜[4]のいずれかの大腸菌株。
[6] 外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入されている、[5]の大腸菌株。
[7] 大腸菌株にMlcタンパク質を過剰に発現する遺伝子変異を導入すること、又はMlcタンパク質を発現ベクターを使って過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
[8] Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、[7]の方法。
[9] mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、[7]又は[8]の方法。
[10] anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子を過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
[11] [1]〜[4]のいずれかの炭素カタボライト抑制(CCR)が解除された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に有機物質を生産する方法。
[12] [1]〜[4]のいずれかの炭素カタボライト抑制(CCR)が解除された大腸菌株であって、さらに有機物質の合成に関与する遺伝子が導入された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に前記有機物質を生産する方法。
[13] 有機物質がアルコール又は有機酸である、[12]の方法。
[14] 有機物質がイソブチルアルコールであり、混合糖がブドウ糖及びキシロースを含む、[12]又は[13]の方法。
[15] 大腸菌株に、外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入された、[14]の方法。
【発明の効果】
【0015】
実施例に示すように、混合糖を原料として大腸菌で物質生産をする場合、野生型株を用いるよりも、CCRの解除された株を用いるほうが生産性が高いことがわかる。CCRの解除された株の中でも、mlc*によるものが有利な場合もある。mlc*あるいは、mlc遺伝子の過剰発現によってCCRが解除され、ブドウ糖とそれ以外の糖を同時に消費するようになる、という報告はこれまでになく、新規性がある。また、mlc*あるいは、mlc遺伝子を過剰発現した株を用いて有機物質の生産を行った例もない。
【図面の簡単な説明】
【0016】
【図1】ショットガンでクローン化されたgen株のゲノム断片の構造を示す図である。図1aがmlc遺伝子を含む断片を示し、図1bがanmK、slyB、slyA、ydhI、ydhJ遺伝子を含む断片を示す。図中矢印はORFを、黒い線がショットガンクローニングで取られたゲノム断片を示す。また、mlcプロモータ部分の配列とmlc*の変異箇所を示してある。
【図2】pHN1234、pHN1532、pHN1534のプラスミドマップを示す図である。ChlrとKanrはそれぞれクロラムフェニコール耐性遺伝子とカナマイシン耐性遺伝子を示す。
【図3】各株におけるブドウ糖とキシロースの共消費を示す図である。
【図4】各株におけるイソブタノールの生産性を示す図である。図4aは、ブドウ糖を炭素原とした場合の結果、図4bはブドウ糖とキシロースを炭素原とした場合の結果、図4cはキシロースを炭素原とした場合の結果を示す。
【図5】より最適化されたイソブタノールの生産性を示す図である。図5aは、ブドウ糖を炭素原とした場合の結果、図5bはブドウ糖とキシロースを炭素原とした場合の結果を示す。
【発明を実施するための形態】
【0017】
以下、本発明を詳細に説明する。
【0018】
本発明は大腸菌(E.coli)において混合糖から有機物質を生産する方法であって、炭素カタボライト抑制(CCR:carbon catabolite repression)が解除された変異体を用いて有機物質を製造する方法である。CCRは、培地中にブドウ糖などの優先的に代謝される炭素源があるとそれを消費するまで、それ以外の炭素源の分解系の遺伝子発現を抑制する機構である。自然界においては炭素源をより効率的に利用できるという利点があるが、微生物を用いて有用物質を生産しようとする場合には、ブドウ糖以外の糖を利用できなくなるという欠点を有する。本発明においては、大腸菌のCCRを解除することにより、ブドウ糖と他の糖が共存する混合糖を用いる培養条件下において、優先的に代謝される糖と該等以外の糖、例えば、ブドウ糖とブドウ糖以外の糖を同時に利用し、有用物質の生産が可能となる。ここで、CCRの解除とは、CCRの不全あるいはCCRの異常ともいい、大腸菌において、ブドウ糖等のCCRを誘導する糖と他の糖が混合された条件下で培養した場合に、ブドウ糖以外の糖の代謝の抑制が解除され、ブドウ糖だけでなく、ブドウ糖以外の他の糖も同時に代謝され、大腸菌体における有機物質の合成に利用し得るようになることをいう。
【0019】
本発明において、CCRの解除は、例えばMlc(making large colonies)タンパク質の過剰発現により行うことができる。Mlcタンパク質は、炭化水素代謝の調節因子として、いくつかの遺伝子やオペロンの発現を調節することが知られている44kDaのDNA結合タンパク質である(Hosono, K., et al., Biosci, Biotechnol. Biochem., 59, 256-261 (1995))。一例として、ptsG遺伝子の発現はMlcタンパク質によって負に制御されていることが知られている(Plumbridge, J. Curr. Opin. Microbiol. 5:187-193(2002))。Mlcタンパク質の過剰発現は、PtsGタンパク質の不足を引き起こし、CCRが解除される。
【0020】
Mlcタンパク質の過剰発現は、例えば、mlc遺伝子が導入されMlcを発現し得るベクターを大腸菌に導入することにより行うことができる。また、mlcプロモーターDNAの塩基配列にmlcプロモーターが高活性型になるような変異を入れてもよい。このような変異として、例えば、mlcプロモーターの-10配列のcaccatからtattatへの変異が挙げられる。該変異は配列番号34に示すmlcプロモーター配列の第31番目のcのtへの置換である。
【0021】
さらに、anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によっても部分的な又は完全なCCRの解除を行うことができる。ここで、部分的な解除とは、不完全なCCRの解除をいう。ただし、このようなCCRの解除であっても、優先的に代謝される糖とそれ以外の糖を同時に利用することが可能になるので、有用である。本発明において、CCRの解除とは完全な解除だけでなく、部分的な解除も含む。上記5つの遺伝子のうち、特にslyA遺伝子の過剰発現が好ましい。これらの遺伝子の過剰発現は、これらの遺伝子が導入されこれらの遺伝子を発現し得るベクターを大腸菌に導入することにより行うことができる。
【0022】
mlcプロモーターの塩基配列の変異がMlcタンパク質の過剰発現を引き起こし、CCRを解除するかどうかは、大腸菌のmlcプロモーターをUV照射等の物理的変異処理、ニトロソグアニジン等を用いた化学的変異処理を行い、人為的に変異を誘発し、該変異体について、ブドウ糖及びラクトースが共存する培地で培養した場合にlacZ遺伝子が発現するかどうかを調べればよい。さらに、mlc遺伝子の過剰発現又はanmK、slyB、slyA、ydhI又はydhJ遺伝子の過剰発現がCCRを解除するかどうかも同様の方法によりlacZ遺伝子の発現を調べればよい。CCRが正常な大腸菌においては、lacZ遺伝子は発現せず、一方、CCRが解除された大腸菌においては、lacZ遺伝子が発現する。lacZ遺伝子の発現は、ブドウ糖及びラクトースが共存する培地にさらに5-クロロ-4-ブロモ-3-インドリル-β-D-ガラクトース(x-gal)を添加し青白選別を行えばよい。CCRが解除された大腸菌においては、グルコースとラクトースの共存下でもlacZ遺伝子が発現し、lacZ遺伝子の発現産物であるβガラクトシダーゼが産生され、βガラクトシダーゼがx-galを分解し、コロニーが青色を呈する。従って、青色コロニーからCCRが解除された大腸菌を単離することができる。
【0023】
このようにして、Mlcタンパク質の過剰発現を引き起こす変異を同定したならば、大腸菌のmlcプロモーターに該変異を導入することにより、大腸菌のCCRを解除することができる。また、部位特定的変異法や相同組換えにより特定の部位に変異を導入することもできる。このように特定の部位に変異を導入した大腸菌についても、上記の青白選別を行えばよい。
【0024】
Mlcタンパク質の過剰発現を引き起こす、mlcプロモーターの変異を同定したら、該変異を大腸菌のmlcプロモーターに導入すればよい。大腸菌におけるmlcプロモーターの変異の導入の方法は、限定されないが、例えば部位特異的突然変異や相同組換えにより行うことができる。
【0025】
本発明はMlcタンパク質を過剰に発現する遺伝子変異が導入され、CCRが解除された大腸菌変異株を包含する。また、発現ベクター等を用いて、野生型のMlcタンパク質を過剰に発現させた株も包含する。
【0026】
CCRが解除された大腸菌を用いることにより、ブドウ糖と他の糖を含む混合糖を用いて有用な有機物質を生産することができる。ここで、CCRを解除した上で有機物質の生産に用いる大腸菌株としては、大腸菌(E.coli)に属しているあらゆる大腸菌が含まれる。例えば、大腸菌K-12株、B株、W株やそれらの変体が挙げられ、変異体としてはMG1655株(ATCC(American Type Culture Collection)47076)、W3110株(ATCC27325)等が挙げられる。これらの大腸菌に上記のMlcタンパク質を過剰生産する変異を導入すればよい。CCRが解除された大腸菌は元々有している有機物質の生産系により混合糖を原料として有機物質を産生することができる。また、CCRを解除した大腸菌に有機物質の合成に関与する遺伝子を導入して形質転換大腸菌を作製し有機物質の生産に用いることもできる。さらに、有機物質の合成に関与する遺伝子が導入された形質転換大腸菌株のCCRを解除して、該大腸菌を有機物質の生産に用いることもできる。有機物質の合成に関与する遺伝子が導入された大腸菌株として、例えば、エタノールを生産し得るKO11株(ATCC55124)等が挙げられる。
【0027】
また、アルコール生産を目的とする場合、大腸菌のアルコール耐性を向上させてもよい。例えば、アルコールを含有する培地中で培養を行うか、あるいは紫外線の照射や化学的変異原処理によりランダム変異を誘発することにより、アルコール耐性株を得ることができる。
【0028】
さらに、本発明の方法で生産しようとする有機物質の中間体からの代謝産物であって、目的の有機物質ではない有機物質を合成する経路を遮断してもよい。遮断は合成経路を遮断しようとする有機物質の合成に関連する酵素をコードする遺伝子をノックアウトすればよい。ノックアウトは例えば相同組換えにより行うことができる。例えば、アルコールを生産する場合、乳酸合成経路やコハク酸合成経路を遮断すればよい。
【0029】
有機物質を生産し得る形質転換大腸菌株は、上記の有機物質の合成に関与する酵素をコードする遺伝子を発現ベクターに挿入し、該発現ベクターを用いて大腸菌を形質転換することにより作製できる。用いる発現ベクターには、プロモーター、上記酵素をコードする遺伝子のほか、ターミネーター、所望によりエンハンサーなどのシスエレメント、イントロンの5’末端側に存在するスプライス供与部位及びイントロンの3’末端側に存在するスプライス受容部位からなるスプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)などを含有するものを作動可能に連結して用いればよい。なお、選択マーカーとしては、例えばジヒドロ葉酸還元酵素遺伝子、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子等が挙げられる。発現ベクターに酵素をコードする遺伝子を挿入するには、まず、精製されたDNAを適当な制限酵素で切断し、適当なベクターDNAの制限酵素部位又はマルチクローニングサイトに挿入して発現ベクターに連結する方法などを用いればよい。また、1つの発現ベクターに2種類の酵素をコードする遺伝子を同時に挿入して、共発現させる場合、複数のマルチクローニングサイトを含む発現ベクターを用いてもよい。
【0030】
大腸菌への組換えベクターの導入方法は、限定されず、例えばカルシウムイオンを用いる方法[Cohen, S.N.et al.:Proc. Natl. Acad. Sci., USA, 69:2110(1972)]、エレクトロポレーション法等が挙げられる。用いるベクターも限定されず、例えば、プラスミドDNA、ファージDNA等の公知の大腸菌用の発現ベクターを利用することができる。複数の遺伝子を別々のベクターに挿入して、大腸菌に導入してもよいし、複数の遺伝子を1つのベクターに挿入して、大腸菌に導入してもよい。
【0031】
大腸菌を用いて有機物質を生産するときに原料として用いる混合糖としては、ブドウ糖に加え、5炭糖であるアラビノース、キシロース、6炭糖であるリボース、ガラクトース、マンノース等の単糖;ラクトース、マルハトース、スクロース等の二糖;各種オリゴ糖の少なくとも1つを含む糖が挙げられる。単糖はそのまま、二糖やオリゴ糖は大腸菌体内で単糖になり、代謝され、他の有機物質が合成される。好ましくは、ブドウ糖とキシロース及び/又はアラビノースを同時に用いる。また、バイオマス由来の糖混合物も用いることができる。バイオマス由来の糖混合物とは、植物や動物から得られる糖であり、グルコースを含む種々の糖が含まれる。バイオマス由来の糖は大量に安価で入手できるので、本発明のCCRが解除された大腸菌を用いて有機物質を生産するときの原料として好適に用いることができる。植物や動物としては、トウモロコシ、サトウキビ、小麦等が挙げられ、植物の根、茎、幹、枝、葉、花、種子等から得ることができる。また、木材チップやパルプを含む木質系バイオマスも利用することができる。植物由来のバイオマスには、セルロースやヘミセルロース等の多糖も含まれるが、これらの多糖はアルカリや酸により分解し、あるいは酵素的に又は微生物的に分解して、大腸菌が有機物質の生産に利用できる糖の含有量を高めればよい。
【0032】
本発明の方法により生産される有機物質の種類は限定されないが、天然の大腸菌株が元々生産する能力のある有機物質に限らず、遺伝子組換え技術や菌の育種などによって新たに生産するようになった有機物質も含まれる。有機物質としては、アルコール、有機酸、タンパク質、アミノ酸等が挙げられる。アルコールとしては、エタノール、1-ブタノール、イソブタノール(イソブチルアルコールともいう)、2-メチル-1-ブタノール、3-メチル-1-ブタノール、プロパノール、イソプロパノール等が挙げられる。有機酸としては乳酸、コハク酸、酪酸、プロピオン酸、酢酸等が挙げられる。
【0033】
大腸菌株に目的の有機物質の生産に関与する遺伝子を導入する場合、上記のアルコールや有機酸等の生体内における代謝経路及び合成に関与する遺伝子は公知であり、当業者ならば、適切な外来の遺伝子を大腸菌に導入することにより、大腸菌に有機物質合成系を導入することができる。また、導入する遺伝子は好ましくは大腸菌での発現用にコドンを最適化する。例えば、エタノールを生産する場合、ピルベート脱水素酵素(PDC)及びアルコール脱水素酵素(ADH)をコードする遺伝子を大腸菌に導入すればよい。また、イソブタノールを生産する場合、分枝α-ケト酸脱炭酸酵素(KIVD)、アルコール脱水素酵素(ADH2)、アセト乳酸シンターゼ(ALSS)、ケトール酸レダクトイソメラーゼ(ILVC)、ジヒドロキシ酸デヒドラターゼ(ILVD)をコードする遺伝子を導入すればよい。この場合、2ケト酸を経由して効率的にイソブタノールを生産することができる。
【0034】
CCRが解除された大腸菌を上記混合糖を含む培地で培養することにより、混合糖に含まれる複数種の糖を同時に代謝して、有用物質を効率的に生産することができる。
【0035】
上記のようにして得られたCCRが解除された大腸菌又はCCRが解除されさらに有機物質合成系を導入した大腸菌を混合糖を添加した培地で培養することにより有機物質を生産することができる。
【0036】
大腸菌の培養は公知の方法に従い行うことができる。例えば、培養液として、DMEM、MEM、RPMI1640、IMDM等を使用することができ、牛胎児血清(FCS)等の血清補液やその他野サプリメント試薬等を添加して用いればよい。さらに、形質転換した大腸菌を固定化して、有機物質を生産することもできる。固定化は、包括法、架橋法、担体結合法等により行うことができ、固定化に用いられる固定化担体としては、ガラスビーズ、シリカゲル、ポリウレタン、ポリアクリルアミド、ポリビニルアルコール、カラギーナン、アルギン酸、寒天、ゼラチン等がある。
【0037】
有機物質が大腸菌菌体外に生成、蓄積した場合には、培養終了後、培養物から菌体などの沈殿物を除去し、公知の方法を単独で又は組合わせて、培養物から有機物質を単離、精製することができる。また、有機物質が大腸菌菌体内に生成、蓄積される場合には、培養終了後、培養物から菌体を回収した後、機械的または化学的方法等の適切な方法で菌体を破砕し、該菌体破砕物から、公知の方法を単独で又は組合わせて有機物質を単離、精製することができる。有機物質がアルコールである場合、例えば蒸留により回収することができる。
【実施例】
【0038】
以下、実施例により本発明をさらに具体的に説明する。但し、本発明はこれら実施例にその技術的範囲が限定されるものではない。
【0039】
〔実施例1〕
実験手法
以下に記載の実施例において、大腸菌の培養、遺伝子組換え操作、タンパク質の活性測定等は、本発明者らの論文および特許(Nakashima and Tamura, Biotechnol. Bioeng. (2004) 86:136-148、Nakashima and Tamura, Appl. Environ. Microbiol. (2004) 70:5557-5568、Nakashima et al., Nucleic Acids Res. (2006)34:e138、Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103、特許第3793812号、特許3944577号)に基づいて行った。特に断りがない限り、大腸菌の培養はLB(1% Difco Bacto Tryptone、0.5% Difco Yeast Extract、1%塩化ナトリウム)を用いて37℃で行った。寒天培地による培養の場合、寒天濃度は1.7%で、9 cm直径のプラスチックシャーレに入れたものを使った。また、プラスミドを含む大腸菌を培養する場合は対応する抗生物質を加えて行った。抗生物質の終濃度は、アンピシリン:50μg/mL、クロラムフェニコール:24μg/mL(原液は100%エタノールを溶媒として34 mg/mLの濃度で作成)、カナマイシン:15μg/mL、アプラマイシン:35μg/mLである。LB以外の培地の組成は以下の通りである。M9YM:17 mg/mL Na2HPO4-12H2O, 3 mg/mL KH2PO4, 0.5 mg/mL NaCl, 1 mg/mL NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.25% Difco Yeast Extract, 50 mM MOPS buffer (pH 7.0)、M9Y:17 mg/mL Na2HPO4-12H2O, 3 mg/mL KH2PO4, 0.5 mg/mL NaCl, 1 mg/mL NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, 0.5% Difco Yeast Extract。用いた菌株の一覧を表1に示す。
【0040】
【表1】
【0041】
〔実施例2〕
CCRが解除された変異株の分離
野生型大腸菌、MG1655株を一晩培養した。これを新鮮なLBで5000倍に希釈して、うち20μLを10 g/Lブドウ糖、8 g/Lラクトース、0.125 mg/mL x-galを含むLB寒天培地50枚に塗布した。直後に、XL-1000 UV crosslinker(Spectronics社製)を用いて、10-12 mJ/cm2の紫外線を照射した。これらを一晩培養したところ、計約30000個のコロニーが出現し、うち1コロニーのみが青色を呈していた。この変異株をgen株(glucose effect not observed)と暫定的に名付けた。
【0042】
次いで、二次スクリーニングとして以下の実験を行った。野生株とgen株をそれぞれ10 g/Lブドウ糖あるいは、10 g/Lブドウ糖と8 g/Lラクトースあるいは、8 g/Lラクトースを含む4 mLの液体培地で中期対数増殖期まで培養し、菌体を遠心分離機によって回収した。菌体を500μLのPBS(0.14 M NaCl、2.7 mM KCl、10.1 mM Na2HPO4、1.8 mM KH2PO4)に懸濁し、そこに直径 0.1 mmのガラスビーズ(安井機械社製)0.1 gを混合した。これを安井機械社製ビーズショッカーにかけ、菌体を破砕した後、20000×g、4℃、15分遠心することで上清を得た。これが粗タンパク質液となる。粗タンパク質液を用いて、βガラクトシダーゼの活性を測定したところ(Nakashima et al., Nucleic Acids Res. (2006)34:e138)、野生株では、含ラクトース培地の時のみ活性が認められたのに対して、gen株では、含ブドウ糖ラクトース培地と含ラクトース培地の時にほぼ同程度の活性が認められた。なお、gen株であっても、含ブドウ糖培地では活性が認められなかった。この結果から、gen株は予想通り、CCRが解除された変異体である可能性が示唆された。
【0043】
さらに、gen株では、ラクトース以外の糖でも同様のCCR解除がみられるかを検討した。まず、プラスミドpHN171(Nakashima and Tamura, Biotechnol. Bioeng. (2004) 86:136-148)からレポーターとなるpip遺伝子(プロリンイミノペプチダーゼをコード)をNcoIとSpeIで切り出し、pHN540uベクター(Nakashima et al., Nucleic Acids Res. (2006)34:e138)の同部位にサブクローニングした。このプラスミドにpHN1325と名前をつけた。pHN1325では、L-アラビノース誘導性badプロモーター(Pbad)の下流にpip遺伝子が連結されている。Pbadからの転写は、L-アラビノースによって誘導されるが、L-アラビノースとブドウ糖が共存するときは転写はほとんど起こらない。すなわち、CCRが正常に働いている事を意味する。pHN1325を野生株とgen株にそれぞれ導入し、その形質転換体をそれぞれ10 g/Lブドウ糖あるいは、10 g/Lブドウ糖と8 g/L L-アラビノースあるいは、8 g/L L-アラビノースを含む4 mLの液体培地で培養した。培養した菌体から、粗タンパク質液をβガラクトシダーゼの時と同様に作成し、PIPの活性を測定した。測定は以下のように行なった。まず、2μLの粗タンパク質液と56 mLのPBSを混合したものを60℃で10分保持し、ここに2 mLの10 mM H-Pro-AMC(Bachem Holding AG製)を入れそのまま10分保持した。うち50μLを取り出し、そこに25μLの10%硫酸ドデシルナトリウム溶液を加え、混合後、黒色96穴プレート(Costar社製; product no.3915)に入れた。この蛍光をSafire reader(Tecan社製)で excitation, 380 nm; emission, 460 nm; excitation and emission bandwidths, 12 nm; gain, 80; number of flashes, 10; Z-position 11019 mm; and integration time, 500μsの設定で測定した。
【0044】
その結果、野生株では、含L-アラビノース培地の時のみ活性が認められたのに対して、gen株では、含ブドウ糖L-アラビノース培地と含L-アラビノース培地の時にほぼ同程度の活性が認められた。なお、gen株であっても、含ブドウ糖培地では活性が認められなかった。
以上のことから、gen株は、CCRが解除された変異体であることが示された。
【0045】
〔実施例3〕
gen株の変異遺伝子クローニング
gen株で起こっている、CCRを解除する変異が、もし優性変異であるならば、以下の実験で変異した遺伝子がわかるはずである。まず、gen株ゲノムライブラリーを作成するために以下の作業を行った。gen株を一晩培養し、それを用いてゲノムDNAを精製した(Nakashima et al., Nucleic Acids Res. (2006)34:e138)。ゲノムDNAをSau3AIで部分消化し、アガロース電気泳動に供し、10-6 kb程度の断片をアガロースゲルから回収した。それを精製し、ベクターpSPT18(Roche Applied Science社)のBamHI部位にライゲーションした。ライゲーション産物を大腸菌XL1-blueに形質転換し、約30000コロニーからなるゲノムライブラリーを得た。次いで、擬陽性クローンを極力減らすために以下のプラスミドpHN1629を作成した。sSN1481(aaccatggtaagccgatacgtacccgata(配列番号1))とsSN1482(aaactagtctacccaatcagtacgttaatt(配列番号2))の2つのオリゴヌクレオチドプライマーを作成し、野生型MG1655株のゲノムを鋳型としてPCR反応を行った。これによって、mazF遺伝子と呼ばれる遺伝子のORFが増幅されが(0.3 kb)、この遺伝子は、過剰発現させると大腸菌が増殖できなくなることが知られている。このPCR断片の両末端をNcoIとSpeIで切断し、pHN540uベクターの同部位にクローニングした。このプラスミドにpHN1629と名前をつけた。pHN1629では、Pbadの下流にmazF遺伝子が連結されている。よって、野生株にpHN1629を導入して、ブドウ糖とL-アラビノースを両方含む培地で培養しても菌は増殖するが、CCRが解除された株では、菌は増殖しない。実際に、pHN1629でgen株を形質転換し、ブドウ糖のみを含む寒天培地で培養したところ増殖し、ブドウ糖とL-アラビノースを両方含む寒天培地上だと増殖しないことを確認した。
【0046】
まず、pHN1629を用いて野生型MG1655株を形質転換し、次いで上述のgen株ゲノムライブラリーでさらに二重形質転換した。その二重形質転換体を10 g/Lブドウ糖、8 g/Lラクトース、0.125 mg/mL x-galを含む寒天培地上で生育させたところ、約40000コロニーのうち、9コロニーが青色を呈した。さらに、9コロニーを10 g/Lブドウ糖と2 g/L L-アラビノースを含む寒天培地上で生育させた。すると、うち6コロニーについて、コントロールのpSTP18(空ベクター)とpHN1629の二重形質転換体と比較して生育が顕著に阻害されていた。
【0047】
この6コロニーの持つgen株ゲノムライブラリーについて、プラスミドを回収、精製し、DNA配列を一部決定したところ、4つがmlc遺伝子を含む重複する断片(図1a)、2つがanmK、slyB、slyA、ydhI、ydhJ遺伝子を含む重複する断片(図1b)を持つプラスミドであった。
【0048】
まず、後者プラスミド2つについて、共に野生型MG1655株に再導入し、実施例2と同様に液体培地で培養した後、βガラクトシダーゼの活性を測定した。しかし、どちらのプラスミドにおいてでも、含ブドウ糖ラクトース培地での培養時では活性が見られず、含ラクトース培地の時にのみ活性が認められた。さらに、プラスミド内のゲノム断片のDNA配列を全て決定し、発表されているMG1655株のDNA配列(GenBank登録番号U00096.2)と比較したところ、全く同一の配列であった。よって、これらは擬陽性クローンか、過剰発現により不完全なCCR解除を起こすクローンであると結論した。
【0049】
次いで、前者プラスミド4つについても同じくβガラクトシダーゼの活性を測定した。すると、含ブドウ糖ラクトース培地と含ラクトース培地での培養時に、ほぼ同程度の活性が認められた。すなわち、このプラスミドに含まれる、gen株由来ゲノム断片の中に変異が入っている可能性が高いことが示唆された。この断片のうち最も短い断片について、全てのDNA配列を決定したところ、野生株由来のものと比較して、1塩基に突然変異があることが認められた。その変異箇所が図1aに示してある。この変異は、どのオープンリーディングフレーム(ORF)の内部でもなく、mlc遺伝子のプロモーター部位と予測されるところに存在していた。
【0050】
大腸菌プロモーターにおいては、-10配列と呼ばれる共通配列があり、その配列はtataatであるときに、最も効率の高いプロモーターになると考えられている(de Boer et al., Proc. Natl. Acad. Sci. USA (1983) 80:21-25)。野生型のmlc遺伝子では、これがcaccatであり、共通配列とは相同性がかなり低い。しかし、本発明者の取得した変異型では、taccatと共通配列と相同性が高くなるような変異が入っている。よって、この変異mlc遺伝子では、プロモーターが高活性型となっていると予測される。そして、その結果、Mlcタンパク質が過剰発現されているものと予測される。
【0051】
そこで、野生型mlcプロモーターと変異型mlcプロモーターをそれぞれPCRにて分離し、それらの下流にpip遺伝子をクローニングしたプラスミドを作成した。次いで、それらプラスミドをMG1655株に導入、培養し、それぞれのPip活性を測定したところ、変異型mlcプロモーターの方が17倍高かった。すなわち、配列情報から予想されたとおり、変異型mlcプロモーターは高活性型であることが示唆された。
【0052】
なお、その機構から考えて、このmlc遺伝子変異は優性であると考えられる。実際に、この変異が劣性であることを仮定して行った、野生株ゲノムライブラリーをgen株に導入する実験では、擬陽性クローンしか分離されなかった。
【0053】
以上の結果から、gen株でCCR機構の解除を引き起こしていた変異は、mlc遺伝子の高活性型プロモーター変異であると言うことができ、以降gen遺伝子変異をmlc*変異と呼ぶこととする。
【0054】
野生型mlcと変異型mlc*遺伝子をプラスミドpHN1242(Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103)を用いて野生型MG1655株で過剰発現させたところ、いずれも場合もCCRが解除されることが分かった。ただしこの場合、変異型mlc*遺伝子ではほぼ完全にCCRを解除できたのに対し、野生型mlcではCCRの解除は部分的であった。このことから、mlc遺伝子あるいはそのプロモーターに変異が入っていなくとも、Mlcタンパク質が過剰発現さえされれば、少なくとも部分的にはCCRが解除されることが分かった。
【0055】
〔実施例4〕
mlc*変異の野生型株への導入
相同組換えを利用して、元のMG1655株にmlc*遺伝子を導入するために、以下のプラスミドを作成した。
【0056】
pMW118(ニッポンジーン社製)を鋳型、sSN1097(taagctagctatggacagttttccctttgatatgta(配列番号5))とsSN1097R(gatgatgaacatcagtagggaaaatgc(配列番号6))をプライマーして用いて、pSC101 ori配列の一部をPCR増幅した。なお、このPCRに先立って、sSN1097RはT4 DNAキナーゼ(New England Biolab社製)を用いて5’末端をリン酸化した。一方で、pMW118を鋳型、sSN1098(aaactgcaggttgatgataccgctgccttactg(配列番号7))とsSN1098R(tgaacgtattggttataagtgaacgatacc(配列番号8))をプライマーとして用いて、pSC101 ori配列の別の一部をPCR増幅した。前者断片をNheI、後者断片をPstIで切断し、両断片を同時にpHN540uのNheI、PstI部位にクローニングした。出来たプラスミドにpHN1092と名前をつけた。pHN1092はpSC101 oriをもつプラスミドであるが、一箇所の点突然変異(GenBank登録番号X01654の、5439番目の塩基がシトシンからチミンに変化)により、温度感受性のプラスミドとなっており、37℃を越える温度で培養されると、プラスミドとして細胞内で維持されない(Armstrong et al., J. Mol. Biol. (1984) 175:331-347)。よって、37℃を越える温度では、プラスミドは脱落するか大腸菌のゲノム内に挿入されるかのどちらかとなる。この性質と、大腸菌が天然に持つ相同組換え能を利用して、ゲノムの特定の場所を操作する(Hamilton et al., J. Bacteriol. (1989) 171:4617-4622、Emmerson et al., Biol. Proced. Online (2006) 8:153-162)。
【0057】
pHN1092を鋳型、sSN1098(上述)とsSN1173(ggggtacctgagcaaactggcctca(配列番号9))をプライマーとして用いて、温度感受性pSC101 ori配列とクロラムフェニコール耐性遺伝子をPCR増幅した。なお、このPCRに先立って、sSN1173はT4 DNAキナーゼを用いて5’末端をリン酸化した。一方、pK18mobsacB(GenBank登録番号、FJ437239)を鋳型として、sSN1172(aactgcagttctagaaggcctggatccccatggccgttcactattatttagtgaaatgag(配列番号10))とsSN1108(aaagctagcattttcttttgcgtttttatttgttaac(配列番号11))をプライマーとして用いて、sacB配列をPCR増幅した。なお、このPCRに先立って、sSN1108はT4 DNAキナーゼを用いて5’末端をリン酸化した。増幅した両断片をT4 DNAリガーゼによって連結し、pHN1234を得た。
【0058】
pHN1234のNheI部位に、pHN267(Nakashima and Tamura, Appl. Environ. Microbiol. (2004) 70:5557-5568)からXbaIとSpeIで切り出したカナマイシン耐性遺伝子(1.4 kb)をサブクローニングした。これにpHN1516と名付けた。pHN1516を鋳型、sSN1415(tccatggggatcctcgcgaggccgttcactattatttagtgaaatgag(配列番号12))とsSN1421(tatgcattctagagatatcttagttgctctccaggtgacccaggtgacc(配列番号13))をプライマーとして用いて、PCR増幅を行った。なお、このPCRに先立って、sSN1415とsSN1421はT4DNAキナーゼを用いて5’末端をリン酸化した。一方、pHN1234を鋳型、sSN1173(上述)とsSN1098(上述)をプライマーとして用いて、PCR増幅を行った。増幅した両断片をT4 DNAリガーゼによって連結し、pHN1534を得た。
【0059】
pHN1234を鋳型、sSN1417(cccatggggatccaggccttctagaact(配列番号14))とsSN1173(上述)をプライマーとして用いて、PCR増幅を行った。なお、このPCRに先立って、sSN1417はT4 DNAキナーゼを用いて5’末端をリン酸化した。増幅した断片をT4 DNAリガーゼによって自己閉環化し、pHN1532を得た。
【0060】
pHN1234、pHN1534、pHN1532のプラスミド構造を図2に示す。pHN1534が遺伝子破壊用のプラスミド、pHN1532が点突然変異導入用のプラスミドの元となる(後述)。
【0061】
大腸菌MG1655のゲノムDNAを鋳型、sSN1522(atactgcagttcgatggttgaaacactctg(配列番号15))とsSN1513(gcctgcaggtgagggaaatttcagcgaaaa(配列番号16))をプライマーとして用いて、PCR増幅を行った。増幅した断片をPstIとScaIで切断して、pHN1534のNsiI、EcoRV部位にクローニングした。出来たプラスミドにpHN1677と名付けた。次いで、大腸菌MG1655のゲノムDNAを鋳型、sSN1523(agggatccaatacgtgtcgtcaaatttttacttagg(配列番号17))とsSN1524(ggccatgggattgctgcgtaacacagcgt(配列番号18))をプライマーとして用いてPCR増幅を行い、その末端をBamHIとNcoIで切断して、pHN1677のBamHI、NcoI部位にクローニングした。できたプラスミドにpHN1678と名付けた。これがmlc遺伝子破壊用プラスミドとなる。
【0062】
実施例2に記載のgen株のゲノムDNAを鋳型、sSN1522とsSN1524(共に上述)をプライマーとして、PCR増幅を行った。増幅した断片をPstIとNcoIで切断して、pHN1532のPstI、NcoI部位にクローニングし、出来たプラスミドにpHN1693と名付けた。これが、mlc遺伝子をpHN1678で破壊した後に、mlc*で入れ替えるためのプラスミドとなる。
【0063】
以下、野生型MG1655株にmlc*を導入する過程を述べるが、これは、Emmersonらの方法(Emmerson et al., Biol. Proced. Online (2006) 8:153-162)によるものである。まずMG1655をpHN1678で形質転換し、mlc遺伝子座位で2回の相同組換えを起こさせることによって、mlc遺伝子を破壊した。次いで、その破壊株をpHN1693で形質転換し、同様に2回の相同組換えを起こさせることによってmlc*に入れ替えた。以降、混同を避けるため、この相同組換えで作成した株をmlc*HRと呼び、実施例2で分離したgen株(実体はmlc*変異株)をmlc*UVと呼ぶこととする。DNA配列解析により、mlc*HR株ゲノム上では野生型MG1655株と比べて、mlc*UV と同一の1塩基変異が入っていることを確認した。
【0064】
〔実施例5〕
野生株、mlc*UV株、mlc*HR株の比較
野生株、mlc*UV株、mlc*HR株を用いて、実施例2と同様に、x-galを含むLB寒天培地による青白コロニー判別実験を行った。すると予想通り、野生株は含ラクトース培地では青いコロニー、含ブドウ糖ラクトース培地では白いコロニーを形成したのに対して、mlc*UV株とmlc*HRは、含ラクトース培地、含ブドウ糖ラクトース培地上、共に青いコロニーを形成した。なお、すべての株が含ブドウ糖培地上では白いコロニーを形成した。
【0065】
さらに、ラクトース以外の糖についてのCCRを解析するため、野生株、mlc*UV株、mlc*HR株を、M9YMにブドウ糖とキシロースを8 g/Lずつ添加した培地で培養した。これらの培地に残存しているブドウ糖とキシロースの量を時間を追って高速液体クロマトグラフィー(D-2000型LaChrom Elite、日立社製)を用いて測定した。分析条件は以下の通りである。分離カラム:aminex HPX-74H(Bio-Rad社製)、ガードカラム:Cation H micro-guard cartridge(Bio-Rad社製)、分離溶媒:4 mM H2SO4、流速:0.5 mL/min、検出器:紫外線検出器(L-2400型による210 nmでの検出、日立社製)及び示差屈折率検出器(L-2490型、日立社製)、カラム温度:45℃。その結果(図3)、野生株はこの培養条件下では全くキシロースを消費しなかったのに対して、mlc*UV株、mlc*HR株は、ブドウ糖とキシロースを同様の速度で消費した。
【0066】
以上の結果から、mlc*UV株でCCRが解除されていたのは、mlc*変異のみによるものであって、それ以外の遺伝子の変異は、あったとしてもCCRには関係がないと結論される。以降は、予期せぬ変異が入っている心配のない、mlc*HRのみを実験に使用した。
【0067】
〔実施例6〕
イソブタノール生産のための発現プラスミド構築
大腸菌は天然にはイソブタノールをほとんど生産しない。よって、外来遺伝子を導入したり、関連する代謝経路を強化しなければならない。そのために必要なプラスミドを以下のように作成した。
【0068】
pTrc99a(Amersham Biosciences社製)を鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号19))とsSN1284(aaactagtctcgagaagcttgcatgcctgcaggtcgact(配列番号20))をプライマーとしてPCR増幅を行い、IPTG誘導性のtrcプロモータ(Ptrc)、lacオペレータ(lacO)、マルチクローニング部位配列を含む断片を得た。その断片の両端をNsiIとSpeIで処理し、pHN1257(NAR)のNsiI、SpeI部位にクローニングした。このプラスミドにpHN1344と名付けた。一方、pTrc99aを鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号19))とsSN1345(aatctagaggggaattgttatccgctcacaattccacac(配列番号22))をプライマーとしてPCR増幅を行い、PtrcとlacO配列を含む断片を得た。その断片をXbaIで処理し、pET28a(Novagen社製)のXbaI、EcoRV部位にクローニングした。このプラスミドにpHN1420と名付けた。pHN1420から、NsiIとNcoIによってPtrcとlacOを含む配列を切り出し、pHN1344のNsiI、NcoI部位にサブクローニングした。出来たプラスミドにpHN1431と名付けた。これが外来遺伝子を発現するための元のベクターとなる。
【0069】
Genscript社に依頼し、Lactococcus lactis由来kivd遺伝子(GenBank登録番号、AJ746364)を人工的に合成した。この人工kivd遺伝子は、大腸菌での発現用にコドンを最適化してあるもので、pUC57ベクター(GenBank登録番号、Y14837)にクローニングされていて、その配列は配列表に記してある。一方、出芽酵母Saccharomyces cerevisiaeのAdh2遺伝子を、S. cerevisiae W303株(Takeuchi and Tamura, FEBS Lett. (2004) 565:1-3:39-42)のゲノムを鋳型、sSN1287(aaagcatgcaggagatataccatgtctattccagaaactcaaaaagc(配列番号24))とsSN1288(gcctcgagttatttagaagtgtcaacaacgtat(配列番号25))をプライマーとしてPCR増幅した。上記kivd遺伝子のNcoI-SphI断片(1.6 kb)と、adh2遺伝子のSphI-XhoI断片(1.0 kb)を同時に、pHN1431のNcoI、XhoI部位にクローニングした。出来たプラスミドにpHN1435と名付けた。pHN1435は、Ptrcの下流にlacOを持ち、さらにその下流にkivd遺伝子とadh2遺伝子をオペロンとして持つ。
【0070】
pTrc99aを鋳型、sSN1283(gctatgcatcgactgcacggtgcaccaat(配列番号21))とsSN1004(cgaattccatggtctgtttcctgtg(配列番号27))をプライマーとしてPtrcを含む配列のPCR増幅を行った。増幅された断片をNsiIとNcoIで消化し、pHN1270(Nakashima and Tamura, Nucleic Acids Res. (2009) 37:e103)のNsiI、NcoI部位にクローニングした。できたプラスミドをpHN1375と名付けた。Bacillus subtilis 168株(表1)のゲノムDNAを鋳型、sSN1343R(aaccatggtgacaaaagcaacaaaagaacaaaaatcccttgtga(配列番号28))とsSN1344(aaaggatcctagagagctttcgttttcatgagt(配列番号29))をプライマーとして、alsS遺伝子のPCR増幅を行った。増幅された断片をNcoIとBamHIで消化し、pHN1375のNcoI、BamHI部位にクローニングした。出来たプラスミドをpHN1447と名付けた。MG1655株のゲノムDNAを鋳型、sSN1327(aaggatccgaggaatcaccatggctaactacttca(配列番号30))とsSN1328(gcctcgagttaacccgcaacagcaatacgtttcat(配列番号31))をプライマーとしてPCR増幅を行い、ilvC遺伝子を含む断片を得た。その断片の両端をBamHIとXhoIで処理した。一方、MG1655株のゲノムDNAを鋳型、sSN1329(aaagtcgacaggagatataccatgcctaagtaccgttccgccacca(配列番号32))とsSN1330(agactagtttaaccccccagtttcgatttatcgcg(配列番号33))をプライマーとしてPCR増幅を行い、ilvD遺伝子を含む断片を得た。その断片をSalIとSpeIで処理した。これらilvC遺伝子断片とilvD遺伝子断片を同時にpBluscript SKII+(Stratagene社製)のBamHI、SpeI部位にクローニングした。このプラスミドにpHN1406と名付けた。pHN1406からBamHI、SpeIでilvC遺伝子とilvD遺伝子を含む配列を切り出し、pHN1447の同部位にサブクローニングした。このプラスミドにpHN1451と名付けた。pHN1451はPtrcの下流にlacOを持ち、さらにその下流にalsS、ilvC、ilvD遺伝子の3遺伝子をオペロンとして持つ。
【0071】
〔実施例7〕
mlc*変異によってイソブタノール生産が増大するかどうかの実証
次に、mlc*変異によってどの程度イソブタノールが生産できるか検証した。同時に比較のために、野生型MG1655株から作成したDptsG株とcrp*株も用いることとした。また、イソブタノールの生産性を増すために、ここで用いた株ではldhA(乳酸脱水素酵素をコード)、adhE(融合性アセトアルデヒド-CoA脱水素酵素/鉄依存性アルコール脱水素酵素/ピルビン酸-ギ酸-リアーゼ不活性化酵素をコード)、pflB(ピルビン酸-ギ酸-リアーゼをコード)遺伝子を破壊した。これら株の作成は、実施例4と同様の方法によって行った。イソブタノール生産に用いた菌株(LAF、LAFmlc*、LAFDptsG、LAFcrp*)の遺伝子型が表1に示してある。
【0072】
LAF、LAFmlc*、LAFDptsG、LAFcrp*の4株を、pHN1435とpHN1451で共形質転換し、まず前培養を行った。それらを、M9Yに40 g/Lブドウ糖、あるいは20 g/Lブドウ糖と20 g/Lキシロース、あるいは40 g/Lキシロースを加えた培地に、100分の1量加え本培養を開始した。本培養は、50 mL容積の三角フラスコにシリコン栓をして、4 mLの培養液、30℃で行った。pHN1435とpHN1451から遺伝子を発現させるために、600 nmでの吸光度が0.8に達したところでIPTGを1 mMになるように添加した。
【0073】
一定時間ごとに培養液を採取し、遠心分離によって菌体を除いた後、その上清を高速液体クロマトグラフィーにて分析した。分析したのは、残存ブドウ糖濃度、残存キシロース濃度、生産されたイソブタノール濃度で、分析条件は実施例5と同一である。その結果(図4)、20 g/Lブドウ糖と20 g/Lキシロースを炭素源とした場合、MG1655に比べて、LAFmlc*、LAFDptsG両株のイソブタノール生産性が有意に高かった。また、40 g/Lブドウ糖あるいは、40 g/Lキシロースを炭素源とした場合、LAFmlc*株での生産性が最も高かった。以上のことから、LAFmlc*株は、大腸菌によるイソブタノール生産に有効であることが示された。
【0074】
また、イソブタノール生産性をより高めるために、LAFmlc*株においてackA-pta遺伝子をさらに破壊することでLAFCmlc*株を作成した。さらに、本培養の培養条件も最適化した。その条件は、250 mL容積のスクリューキャップ付き三角フラスコに、M9Yに40 g/Lブドウ糖、あるいは20 g/Lブドウ糖と20 g/Lキシロースを加えた培地を20 mL入れ、そこに前培養を100分の1量加え、30℃で培養するものである。40 g/Lのブドウ糖入りで培養したものは、ブドウ糖が枯渇したらさらに40 g/Lのブドウ糖を追加し、20 g/Lブドウ糖と20 g/Lキシロース入りで培養したものは、両方の糖が枯渇した時点でさらに20 g/Lブドウ糖と20 g/Lキシロースになるよう追加した。IPTGの添加については、上と同一である。イソブタノールを生産した結果を図5に示す。80 g/Lのブドウ糖からは19 g/L、40 g/Lのブドウ糖と40 g/Lのキシロースからは11 g/Lのイソブタノールが生産された。
【配列表フリーテキスト】
【0075】
配列番号1〜22、24〜33 プライマー
配列番号23 合成
【特許請求の範囲】
【請求項1】
遺伝子変異導入又は発現ベクターを使った過剰発現によって、Mlcタンパク質を過剰に発現することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項2】
Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、請求項1記載の炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項3】
mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、請求項1又は2に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項4】
anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によって、炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項5】
さらに、有機物質の合成に関与する遺伝子が導入されている、請求項1〜4のいずれか1項に記載の大腸菌株。
【請求項6】
外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入されている、請求項5記載の大腸菌株。
【請求項7】
大腸菌株にMlcタンパク質を過剰に発現する遺伝子変異を導入すること、又はMlcタンパク質を発現ベクターを使って過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
【請求項8】
Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、請求項7記載の方法。
【請求項9】
mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、請求項7又は8に記載の方法。
【請求項10】
anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子を過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
【請求項11】
請求項1〜4のいずれか1項に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に有機物質を生産する方法。
【請求項12】
請求項1〜4のいずれか1項に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株であって、さらに有機物質の合成に関与する遺伝子が導入された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に前記有機物質を生産する方法。
【請求項13】
有機物質がアルコール又は有機酸である、請求項12記載の方法。
【請求項14】
有機物質がイソブチルアルコールであり、混合糖がブドウ糖及びキシロースを含む請求項12又は13に記載の方法。
【請求項15】
大腸菌株に、外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入された、請求項14記載の方法。
【請求項1】
遺伝子変異導入又は発現ベクターを使った過剰発現によって、Mlcタンパク質を過剰に発現することにより、炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項2】
Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、請求項1記載の炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項3】
mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、請求項1又は2に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項4】
anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子の過剰発現によって、炭素カタボライト抑制(CCR)が解除された大腸菌株。
【請求項5】
さらに、有機物質の合成に関与する遺伝子が導入されている、請求項1〜4のいずれか1項に記載の大腸菌株。
【請求項6】
外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入されている、請求項5記載の大腸菌株。
【請求項7】
大腸菌株にMlcタンパク質を過剰に発現する遺伝子変異を導入すること、又はMlcタンパク質を発現ベクターを使って過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
【請求項8】
Mlcタンパク質を過剰に発現する遺伝子変異がmlcプロモーターにおける変異である、請求項7記載の方法。
【請求項9】
mlcプロモーターにおける変異が、配列番号34に示すmlcプロモーターの塩基配列の31番目のcのtへの置換である、請求項7又は8に記載の方法。
【請求項10】
anmK、slyB、slyA、ydhI又はydhJ遺伝子の1つ、2つ、3つ、4つ又は5つの遺伝子を過剰発現させることを含む、大腸菌株の炭素カタボライト抑制(CCR)を解除する方法。
【請求項11】
請求項1〜4のいずれか1項に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に有機物質を生産する方法。
【請求項12】
請求項1〜4のいずれか1項に記載の炭素カタボライト抑制(CCR)が解除された大腸菌株であって、さらに有機物質の合成に関与する遺伝子が導入された大腸菌株を、ブドウ糖とブドウ糖以外の他の糖を含む混合糖の存在下で培養し、ブドウ糖とブドウ糖以外の他の糖を原料に前記有機物質を生産する方法。
【請求項13】
有機物質がアルコール又は有機酸である、請求項12記載の方法。
【請求項14】
有機物質がイソブチルアルコールであり、混合糖がブドウ糖及びキシロースを含む請求項12又は13に記載の方法。
【請求項15】
大腸菌株に、外来のkivd遺伝子、Adh2遺伝子、alsS遺伝子、ilvC遺伝子及びilvD遺伝子が導入された、請求項14記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2013−5764(P2013−5764A)
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願番号】特願2011−141137(P2011−141137)
【出願日】平成23年6月24日(2011.6.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度、独立行政法人新エネルギー・産業技術総合開発機構、「新エネルギー技術研究開発/バイオマスエネルギー等高効率転換技術開発(先導技術開発)/酵素糖化・効率的発酵に資する基盤研究」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願日】平成23年6月24日(2011.6.24)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成22年度、独立行政法人新エネルギー・産業技術総合開発機構、「新エネルギー技術研究開発/バイオマスエネルギー等高効率転換技術開発(先導技術開発)/酵素糖化・効率的発酵に資する基盤研究」委託研究、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]