
安定的な構成的高発現子宮頸癌治療ワクチン用ベクター及びそれによって形質転換された組換え乳酸菌
本発明は、配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルス(human papilloma virus)の腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクターに関する。本発明は、ヒトパピローマウィルス抗原タンパク質を高レベルに、構成的に発現するベクターを提供する。また、本発明は、発現ベクターにより形質転換され、そしてその表面にHPV抗原タンパク質を発現する、組み換え乳酸菌を提供する。組換え乳酸菌及びこれを利用した組成物は、子宮頸癌治療用ワクチンとして活用して、経口または膣部位に直接適用することができるため、非常に経済的な効果がある。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルス(human papilloma virus)の腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有するHPV治療ワクチン製造用表面発現ベクターに関する。
【背景技術】
【0002】
細菌から有用な外来タンパク質を多量発現させようとする試みにおいて外来タンパク質の生産量は、遺伝子複製数、即ち、プラスミド数と転写を調節するのに用いられているプロモーターの強さに依存することが知られている。通常多く用いられている高発現プロモーターの他に細菌個体当たりプラスミドベクターの数を変化させて、ターゲットタンパク質発現量を増加させられることが報告された(Tomio, M. et al., Appl. Microbiol. Biotechnol., 28:170, 1988)。また、mini−FプラスミドでRepEの変異が細菌個体内でプラスミド数を変化させられることが報告された(Yasuo, K. et al., J. Biol. Chem., 267:11520, 1992)。
【0003】
ヒトパピローマウィルス(Human papilloma virus,HPV)は世界的に成人の50%以上が感染していると推定され、パピローマウィルス中、特にHPV16、18、31及び45の四つのタイプは、80%以上の子宮頸癌(cervical cancer)の原因として確認された(Lowy, D. R. et al., Proc. Nat. Acad. Sci., 91:2436, 1994)。
【0004】
世界的に子宮頸癌は、乳癌の次に女性に発生頻度が高い癌であって、世界保健機構によると、毎年世界で50万人以上の子宮頸癌患者が発生しており、毎年300,000人以上が子宮頸癌によって死亡すると推定されている。特に、開発途上国及び低開発国では女性死亡の主原因になっている(Pisani, P. et al., Int. J. Cancer, 55:891, 1993)。IARC統計によると、特に慢性感染者が先進国に比べて、極めて多い開発途上国のHPV感染を撲滅するため、長期的に最も効果的な方法はHPV予防ワクチンを投与すると報告された。
【0005】
子宮頸癌に関わるワクチン開発の方法としては、大きく予防ワクチン(prophylactic)と治療ワクチン(therapeutic)の二つに焦点が絞られている。予防ワクチンは、HPV L1/L2抗原タンパク質によって、強い中和抗体(neutralizing antibody)が生成されるようにすることによって、宿主がHPVに感染することを防ぎ、また既に感染したとしてもそれ以上疾病が進まないようにするのに目的がある。一方、治療ワクチンは、HPV E6/E7を対象にするが、特異的な細胞性免疫を誘導させて、既に形成された病斑や悪性腫瘍を退化させることを目的としている。
【0006】
HPVのE6/E7タンパク質はHPVに感染した細胞等の癌化に関わる癌特異抗原であるため、子宮頸癌の免疫治療のターゲットとしてE6/E7タンパク質を利用した治療ワクチン研究が続けられてきた。実際、微生物システムで合成されたHPV E6/E7タンパク質を腫瘍細胞を注入したラットに投与した場合、腫瘍形成が阻害されたり遅延されるとの報告がある(Gao, L. et al., J. Gen. Viol., 75:157, 1994, Meneguzzi, G. et al., Virology, 81:62, 1991)。しかし、他の場合と同じように、生ウイルスワクチンを利用する場合、過度なウイルス複製による問題を誘発しうるため、実際には研究目的のみに使用され、商業化までには長時間及び相当な臨床実験が求められる短所がある。
【0007】
一方、バクテリアベクターを利用したワクチン開発研究も盛んに行われていがる、弱毒化されたサルモネラ菌(attenuated Salmonella typhimurium)で合成されたHPV 16 VLPが、マウスの粘膜や全身で抗原特異的な抗体の生成を誘導するとの報告もある(Denis, N. et al., Infection and immunity, 65:3328, 1997)。合成ペプチドを利用したワクチンは、免疫反応を誘導するのに必要なエピトープのみを合成して接種(vaccination)させるものであり、既にHPV 16 E6/E7に対する細胞性免疫反応(Cytotoxic T Lymphocyte;CTL)を起こすエピトープが分かるようになっている(Ressing, M. E. et al., J. Immunol., 154:5934, 1995)。
【0008】
このような試み以外に、植物からウイルスの抗原を生産するため、トマト及びジャガイモ等の野菜類を利用して、その形質転換体野菜類自体を経口用ワクチン(oral vaccine)または食用ワクチン(edible vaccine)として使おうとする研究も進行している。代表的な例としては、B型肝炎ウイルス表面抗原粒子(Hepatitis B surface antigen particle)(Thavala, Y. F. and Artzen, C. J., Pro. Natl. Acad. Sci. USA., 92:3358, 1995)とパピローマウイルスのギャプシドL1とL2タンパク質(韓国特許登録第0366608号)が挙げられる。しかし、植物を利用したシステムの場合、発現するHPV L1タンパク質の量が少なく、精製の問題があって、前記同様商業的な制約が従う問題がある。
【0009】
従って、HPVの感染者が主に低開発国に集中している点を勘案すると、パピローマウィルス由来の口腔または生殖器官の皮膚粘膜の腫瘍に対する予防と治療のため、より経済的かつ安定的にHPV抗原を製造する方法の開発が強く求められている。
【0010】
本発明者等は、HPV抗原タンパク質を形質転換された組換え微生物の表面に効果的に発現させるベクターと、微生物の表面にHPV抗原タンパク質を発現させる方法を開発した(韓国特許登録第0609866号)。
【0011】
そこで、本発明者等は、HPV抗原タンパク質が形質転換された組換え乳酸菌表面でより安定的かつ構成的に高発現されるベクターを開発しようと鋭意努力した結果、repE変異遺伝子を含有するベクターで形質転換された組換え微生物においてより安定的にターゲットタンパク質の発現が向上することを確認して、本発明の完成に至った。
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の主な目的は、repE変異遺伝子を利用してHPV抗原タンパク質が形質転換された組換え乳酸菌の表面で安定的かつ構成的に高発現させられるベクターを提供することである。
【0013】
本発明の他の目的は、前記発現ベクターで形質転換された組換え乳酸菌及び前記乳酸菌を利用してHPV抗原タンパク質の製造方法を提供することである。
【0014】
本発明のさらに他の目的は、前記形質転換組換え乳酸菌を利用して、子宮頸癌治療用に形質転換された組換え乳酸菌ワクチンを提供することである。
【課題を解決するための手段】
【0015】
前記目的を達成するため、本発明は配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有する表面発現ベクターを提供する。
【0016】
本発明は、また前記HPV抗原タンパク質を表面発現するベクターで形質転換された組換え微生物を提供する。
【0017】
本発明は、またHPV抗原タンパク質が表面に発現した形質転換組換え微生物を有効性分とする子宮頸癌治療用ワクチンを提供する。
【0018】
本発明は、また前記ベクターで形質転換された組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法を提供する。
【0019】
本発明は、前記方法で製造された抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチンを提供する。
【図面の簡単な説明】
【0020】
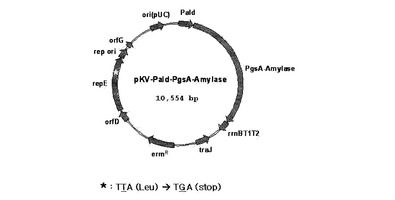
【図1】repE変異遺伝子が導入されたpKV−Pald−PgsA−Amylaseベクターのマップを示す。
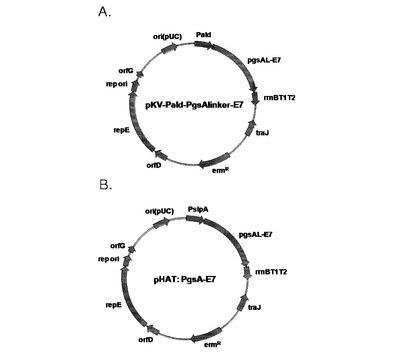
【図2】E7遺伝子を表面発現する2種の発現ベクターのマップを示す。
【図3】pKV−Pald−PgsA−E7で形質転換された組換え乳酸菌においてE7の表面発現をウェスタンブロッティングで確認した結果を示す。
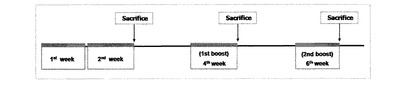
【図4】E7を表面発現する形質転換された組換え乳酸菌の免疫効能実験のため、マウスに形質転換された組換え乳酸菌を投与する日程を示す。
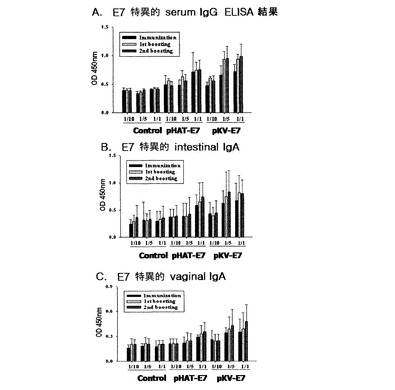
【図5】E7を表面発現する形質転換された組換え乳酸菌の経口投与時IgGとIgAの変化を示す。
【図6】E7を表面発現する形質転換された組換え乳酸菌の経口投与時のE7特異的IFN−gamma分泌を確認した結果を示す。
【図7】E7を表面発現する形質転換された組換え乳酸菌の経口投与時腫瘍移植による腫瘍サイズの増加の有無を確認した経過を示す。
【図8】E7(Rb)遺伝子を表面発現する発現ベクターのマップを示す。
【図9】pKV−Pald−PgsA−E7(Rb)で形質転換された組換え乳酸菌においてE7(Rb)が表面発現されたことを確認した結果を示す。
【図10】E7特異的ELISAの結果を示す。
【図11】E7(Rb)を表面発現する形質転換された組換え乳酸菌の経口投与時E7(Rb)特異的IFN−gamma分泌を確認した結果を示す。
【発明を実施するための形態】
【0021】
本発明は一つの観点において、配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有する表面発現ベクターに関する。
【0022】
本発明では形質転換された微生物内でベクターが安定的に維持される配列番号1のアミノ酸配列を持つrepE変異遺伝子、ラクトバチルスカゼイ(Lactobacillus casei)のアルドラーゼ(aldolase)遺伝子から由来したアルドラーゼプロモーター(Pald)を共に導入した結果としてターゲットタンパク質の発現が向上することを確認した。本発明のシャトルベクターが安定的に維持され、ターゲットタンパク質が発現することを確認するため、目的遺伝子としてアミラーゼ(amylase)遺伝子を挿入してアミラーゼの発現を確認した。
【0023】
また、ターゲットタンパク質の表面発現のため、表面発現モチーフであるポリガンマグルタミン酸合成酵素複合体遺伝子をターゲットタンパク質と連結して発現するように位置させ、前記ポリガンマグルタミン酸合成酵素複合体遺伝子は、pgsBCAであることが望ましく、さらに望ましくはpgsAである(韓国登録特許第469800号)。
【0024】
本発明の一様態において、PgsAのC末端にターゲットタンパク質であるHPV16 E7タンパク質が融合するようpsgAとHPV16 E7コードする遺伝子を融合させて、乳酸菌からHPV抗原タンパク質を構成的に表面発現できるベクターpKV−Pald−PgsA−E7を製造し、前記発現ベクターをラクトバチルスカゼイに挿入してHPV抗原タンパク質を発現する形質転換された組換え乳酸菌を製造した。
【0025】
本発明において、前記「ターゲットタンパク質」または「外来タンパク質」とは、前記タンパク質を発現する形質転換された宿主細胞では正常には存在できないタンパク質を意味する。例えば、乳酸菌からウイルス由来または腫瘍由来タンパク質を人為的に発現するように操作した場合、前記タンパク質を外来タンパク質またはターゲットタンパク質という。
【0026】
本発明はまた、他の観点において、前記HPV抗原タンパク質を表面発現するベクターで形質転換された組換え微生物及び前記形質転換組換え微生物を有効性分とする子宮頸癌治療用ワクチンに関する。
【0027】
本明細書において「宿主」、または「微生物」とは、プロバイオティクス(probiotics)のグラム陽性桿菌である乳酸菌に言及することができ、一般的なプロバイオティクス微生物の選別基準には(i)ヒト由来の微生物であること;(ii)胆汁、酸、酵素及び酸素に安定的であること;(iii)腸内粘膜に付着する能力があること;(iv)消化器官内でコロニーを形成できる能力があること;(v)抗バクテリア性物質を生産できること;及び(vi)効能や安全性が証明されること、等の条件が含まれている。前記条件を基に乳酸菌は人体内での生長が親和的、無害な菌であることは明らかである。従って、乳酸菌を宿主とする形質転換体を人体に適用させて、疾病の予防または治療のための遺伝子伝達またはタンパク質伝達等の用途に用いる場合、菌株を用いる通常のワクチン製造方法とは異なって菌株の無毒化の工程を要しない。
【0028】
本発明において、前記乳酸菌はラクトバチルス属、ストレプトコッカス属及びビフィドバクテリウム属を含んでもよい。代表的にはラクトバチルス属はラクトバチルスアシドフィルス(L. acidophilus)、ラクトバチルスカゼイ(L. casei)、ラクトバチルスプランタラム(L. plantarum)、ラクトバチルスファーメンタム(L. ferementum)、ラクトバチルスデルブルッキー(L. delbrueckii)、ラクトバチルスジョンソニー(L. johnsonii LJI)、ラクトバチルスロイテリ(L. reuteri)及びラクトバチルスブルガリクス(L. bulgaricus);ストレプトコッカス属は、ストレプトコッカスサーモフィルス(S. thermophilus);ビフィドバクテリウム属は、ビフィドバクテリウムインファンティス(B. infantis)、ビフィドバクテリウムビフィダム(B. bifidum)、ビフィドバクテリウムロンガム(B. longum)、ビフィドバクテリウム シュードロンガム(B. psuedolongum)、ビフィドバクテリウムブレーベ(B. breve)、ビフィドバクテリウムラクティスBb−12(B. lactis Bb-12)及びビフィドバクテリウムアドレスセンティス(B. adolescentis)等を宿主として用いられ、より望ましくはラクトバチルス属である。
【0029】
本発明はまた、前記ベクターで形質転換された組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法に関する。
【0030】
本発明に係る組換え微生物の培養は、広く公知の方法に従って行われ、培養温度及び時間、培地のpH等の条件は、適宜調節される。前記培養物での組換え微生物菌体の回収は通常の分離技術、例えば、遠心分離、ろ過法等を用いてもよい。
【0031】
本発明はまた、他の観点において、前記方法で製造された抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチンに関する。
【0032】
また、本発明は、経口用である子宮頸癌治療用ワクチンに関する。
ワクチンは、疾病に対する予防目的として生きている生物を使って、免疫システムを刺激させるのに用いられる薬剤である。免疫活性化は、有機体内で抗体生成、T−リンパ球の刺激、または他の免疫細胞(例:大食細胞)を刺激して、抗原を効率的に除去するための過程を意味する。前記に関する詳しい免疫学の概観は当業界の通常の技術を有する者なら簡単に理解できる(Barrett, J.T., Textbook of Immunology, 1983)。抗原としてのターゲットタンパク質を発現する形質転換された微生物ワクチンの投与対象は、ほ乳動物であってもよく、より望ましくはヒトである。
【0033】
前記ワクチン組成物の製法は標準的な技法を用いて実施でき、投与者に適する投与量は遺伝子生成物の抗原性によって異なり、存在するワクチンの典型的な免疫反応を誘導するのに十分な量であれば良く、通常実験過程を経て必要量を簡単に判断できる。典型的な初期のワクチン容量は、体重1kg当り抗原0.001〜1mgであり、望ましい水準の保護を提供するのに必要に応じて量を増加したり複数回投与(multiple dose)を用いる。前記量は当該技術分野に属する専門家によって決められ、製剤化方法、投与方式、投与者の年令、体重、性、病的状態、食べ物、投与時間、投与経路、排泄速度及び反応感応性のような要因によっても多様に処方される。
【0034】
本発明のHPV抗原タンパク質E7をコードするHPVワクチン製造用表面発現ベクターの最も望ましい様態は、配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAで構成された群から選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクターで、前記ベクターで形質転換された組換え乳酸菌を培養してHPV抗原タンパク質E7を表面発現させた後、ワクチンとしてそのまま使用可能である。
【0035】
本発明のHPV抗原タンパク質E7(Rb)をコードするHPVワクチン製造用表面発現ベクターの最も望ましい様態は、配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAで構成された群から選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7(Rb)をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクターで、前記ベクターで形質転換された組換え乳酸菌を培養してHPV抗原タンパク質E7(Rb)を表面発現させた後、ワクチンとしてそのまま使用可能である。
【0036】
ワクチンが抗体を生成するに当り効果を高めるためには、抗原性物質がワクチン処理された対象の抗体生成機構が実現できるように体内に放出されなければならない。従って、免疫反応のためには遺伝子産物の微生物キャリアが、体内にまず導入されなければならない。本発明の形質転換された組換え乳酸菌で提示される抗原による望ましい反応を刺激するためには、経口投与または噴霧剤状で子宮頚部に直接投与することが望ましい。
【0037】
ワクチン組成物を投与者に経口投与するためには、親液性の形状、例えば、カプセル状で提供することが望ましい。前記カプセルはEudragate S、Eudragate L、セルロースアセテート、セルロースフタレートまたはヒドロキシプロピルメチルセルロースを含む腸用被覆で提供される。前記カプセル類は、それ自体で使ってもよく、投与する前に懸濁液のような親液性物質に再構成して使用してもよい。再構成は形質転換された組換え微生物の生存に適するpHの緩衝液で行うことが望ましい。胃酸から前記形質転換された組換え微生物及びワクチンを保護するため、毎回ワクチン投与以前に重炭酸ナトリウム製剤を投与することが望ましい。選択的にワクチンは非経口投与、鼻内投与または乳腺内投与用として製造される。
即ち、下記工程は一つの例示として説明され、本発明の範囲がこれに限定されない。
【実施例】
【0038】
以下、本発明を実施例を挙げて詳述する。これらの実施例は単に本発明をより具体的に説明するためのものであり、本発明の範囲がこれらの実施例に制限されないことは当業界において通常の知識を持った者にとって自明である。
【0039】
[実施例1]repE変異遺伝子を含有する表面発現ベクターの製造(Pkv−Pald−PgsAL−Amylase)
本実施例では、宿主細胞で発現ベクターを安定させるため、repE変異遺伝子を含有するターゲットタンパク質の発現が向上した表面発現ベクターを作製した。
【0040】
先ず、乳酸菌でより一層発現量を高めるため、ラクトバチルスカゼイ由来のプロモーターであるアルドラーゼプロモーターの断片を得た。前記アルドラーゼプロモーターは韓国公開特許第10−2008−0086161号に開示されたpDT−PgsA−Amylaseを鋳型として配列番号2と配列番号3のプライマーを使ってPCR法で得た。
配列番号2:5’−cgc gca tgc aat acc cac tta ttg cg−3’
配列番号3:5’−cag ttc ttt ttt cat gta gat atc ctc c−3’
【0041】
その結果、アルドラーゼプロモーターを含み、5'末端にはSphI制限酵素部位を含んでおり、3'末端にはpgsAのN−terminal 17bpを含んでいる421bpのDNA切片を取得し、同じベクターを鋳型として配列番号4と配列番号5のプライマーを使ってPCRを行い、先に得たアルドラーゼプロモーター切片と連結できるpgsA遺伝子の部分を得た。
配列番号4:5’−gga gga tat cta cat gaa aaa aga act g−3’
配列番号5:5’−ggc gct ggc ggt cgt ttg g−3’
【0042】
その結果、取得したDNA切片のN末端の部分は、アルドラーゼプロモーターの3'末端の13bpを含み、pgsAが直接連結している782bp切片を得た。この切片のpgsA部分内部にPstI制限酵素部位が含まれている。
【0043】
取得した二つの切片を連結して両側末端のプライマーを用いてPCRを行い、1175bpのDNA切片を取得し、SphIとPstIで切断して、アルドラーゼプロモーターとpgsA N−terminalの一部分を含んだ切片を得た。
【0044】
pBT:pgsA−Amylase(pAT−PslpA−pgsA−amylase;韓国登録特許第0872042号の間接実施例参照)を制限酵素SphI及びPstIで切断してSlpA7プロモーターの部分とpgsA N末端の部分を除去して、発現ベクターの骨格とした。
【0045】
SphIとPstIで切断したアルドラーゼプロモーターを含有しているDNA断片を同じ制限酵素で切断したpBT:PslpA−Amylaseと連結して、pAT−Pald−PgsA−Amylaseを作製し、pAT−Pald−PgsA−Amylaseベクターにsite−directed mutagensis方法でrepE遺伝子475番目のアミノ酸ロイシンをコードするTTAコドンをTGA停止コドンで置換してRepEタンパク質中21個のC末端アミノ酸が切断されて、配列番号1のアミノ酸配列を持つようにコードしたpKV−Pald−PgsA−Amylaseを完成した(図1)。
【0046】
[実施例2]HPV16 E7表面発現ベクター(pKV−Pald−PgsA−E7)の製造
実施例1で製造された表面発現ベクター(pKV−Pald−PgsA−Amylase)を使ってPgsAのC末端にHPV16のE7タンパク質をコードする遺伝子を挿入させて、乳酸菌で表面発現できるベクターpKV−Pald−PgsA−E7を製造した。
【0047】
先ず、実施例1で製造されたpKV−Pald−PgsA−AmylaseベクターでpgsAと融合したアミラーゼ遺伝子を除去し、HPV16 E7をコードする遺伝子を挿入した。HPV16 E7遺伝子を含む断片を図1に示されたようにpHAT:pgsA−E7(Poo et al., Int. J. Cancer, 119:1702, 2006)を鋳型で配列番号6と配列番号7をプライマーとして用いてPCRを行って、取得した。
配列番号6:5’−gcg gga tcc cat gga gat aca cct aca ttg c−3’
配列番号7:5’− acg cag aag cgg tct gat aa −3’
【0048】
その結果、HPV16 E7遺伝子を含んでいる386bpの切片を取得し、前記切片の5'末端にはBamHI制限酵素部位を含んでおり、前記切片の3'末端にはXbaI制限酵素部位を含んでいる。取得したDNA切片をBamHIとXbaI処理により、切断して、306bp切片を得た。
【0049】
pKV−Pald−PgsA−AmylaseをBamHIとXbaIで切断してアミラーゼ遺伝子の部分を除去してベクターの部分を得た。
BamHIとXbaIで切断したE7遺伝子を含有しているDNA断片を同じ制限酵素で切断したベクターと連結してpKV−Pald−PgsA−E7を完成した(図2のA)。
【0050】
[実施例3]pHAT:PgsA−E7とpKV−Pald−PgsA−E7の乳酸菌形質転換体の発現確認
本実施例では実施例2で作製されたpHAT:PgsA−E7とpKV−Pald−PgsA−E7でラクトバチルスカゼイを形質転換させて、前記形質転換された組換えラクトバチルスカゼイを培養してE7タンパク質の発現を確認した。形質転換された組換えラクトバチルスカゼイでpgsAと融合したE7タンパク質の発現の有無を調べた。
【0051】
pHAT:PgsA−E7とpKV−Pald−PgsA−E7で形質転換された組換えラクトバチルスカゼイをMRS培地(Lactobacillus MRS, Becton Dickinson and Company Sparks, USA)、30℃で静置培養してポリガンマグルタミン酸を合成する遺伝子pgsAのC末端と融合したHPV16 E7タンパク質の表面発現を誘導した。
【0052】
前記培養されたラクトバチルスカゼイの全細胞をSDSポリアクリルアミドゲル電気泳動及びpgsAに対する特異抗体を利用したウェスタンブロッティングを行って、前記融合タンパク質の発現を確認した。
【0053】
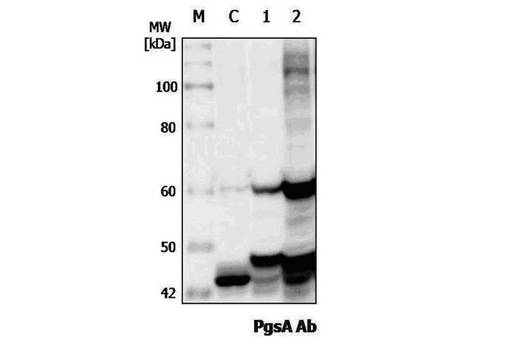
具体的には、発現が誘導された形質転換された組換えラクトバチルスカゼイ全細胞を同じ細胞濃度で得られたタンパク質で変性させた(denature)試料を準備して、これをSDSポリアクリルアミドゲル電気泳動で分析した後、分画されたタンパク質をPVDF(polyvinylidene-difluoride membranes,Bio−Rad)メンブレンに移した。タンパク質が移されたPVDFメンブレンをブロッキング緩衝溶液(50mMトリス塩酸、5%スキムミルク(skim milk)、pH8.0)で1時間振ってブロッキングさせた後、PgsAに対するウサギ由来のポリクローナル一次抗体をブロッキング緩衝溶液に1000倍に希釈して、1時間反応させた。反応が終わったメンブレンは、緩衝溶液で洗浄してHRPが標識されたウサギに対する二次抗体をブロッキング緩衝溶液に10000倍希釈して、1時間反応させた。反応が終わったメンブレンは、緩衝溶液で洗浄し、洗浄されたメンブレンに基質(lumigen PS−3 acridan、H202)を添加して、約1分間発色させ、CCDカメラでPgsAに対する特異抗体と前記融合タンパク質との間の特異的な結合を確認した(図3)。
【0054】
図3のレーンCはPgsAのみ発現し、形質転換された組換えラクトバチルスカゼイ(pKV−Pald−PgsA)であり、レーン1はpHAT:PgsA−E7で形質転換された組換えラクトバチルスカゼイの発現であり、レーン2はpKV−Pald−PgsA−E7で形質転換された組換えラクトバチルスカゼイのタンパク質発現を示したものである。図3に示されたようにPgsA特異的抗体を用いて、レーン1と2で54.4kDaのPgsA−E7融合タンパク質の発現が確認でき、pHAT:PgsA−E7によるPgsA−E7融合タンパク質の発現と比較するとpKV−Pald−PgsA−E7による融合タンパク質がより強く発現することを確認した。
【0055】
正常なRepEを含んでいるpHAT:PgsA−E7のPslpA−PgsA−E7部分をPald−PgsA−E7で置換した構築体の場合、前記と同じ方式で行った乳酸菌形質転換体製造方法によって、乳酸菌形質転換体を取得できなかった。
【0056】
[実施例4]HPV16 E7抗原を表面発現するラクトバチルスの体液性免疫反応誘導能比較
前記表面発現用ベクターでラクトバチルスカゼイを形質転換させて実施例2と同じ方法で表面発現を誘導した後、2種類の形質転換された組換えラクトバチルスカゼイを用いてPgsAと融合したHPV16 E7の体液性免疫反応誘導を比較した。また、融合タンパク質の発現量が異なる2種類の形質転換体を各々多様な量(1、1/5、1/10)で投与して、免疫反応誘導を比較した。
【0057】
前記ベクターでラクトバチルスカゼイを形質転換させた後、培養して回収した後、凍結乾燥して準備したパウダーを緩衝溶液(PBS、pH7.4)に溶かした後、6週齢の雌Balb/cマウスに経口で投与した。
【0058】
具体的には、HPV16 E7を発現する形質転換された組換えラクトバチルスカゼイ(pHAT−E7、pKV−E7)と、発現しないラクトバチルスカゼイ(pAT)の凍結乾燥パウダーを各々PBSに濃度毎(5×109/200μL、1×109/200μL、5×108/200μL)に溶かした後、一週間に5回ずつ2週間免疫(0〜4days、7〜11days)と1週後に一次ブースティング(21〜25days)、さらに1週後に二次ブースティング(35〜39days)を経口投与した。血清内のHPV16 E7特異的なIgG抗体と粘膜内で分泌されるHPV16 E7特異的なIgA抗体を測定するため、腸内(Intestinal)洗浄液と膣内(Vaginal)洗浄液を14日、28日、及び42日に、各々のマウスからサンプルとして採取した(図4)。血液はヘパリンがコーティングされたマイクロキャピラリーチューブを使って、マウスの目から取得し、1mM PMSFが含まれたPBS洗浄液を使って、腸内と膣内を各々0.8mLと250μLで洗浄した後、13,000rpmで20分間遠心分離して、上澄液を分離してELISAを行った。
【0059】
ELISAはHPV16 E7タンパク質をウェル当たり各々500ng/100μLでコーティング緩衝溶液(0.1M sodium carbonium、0.02% sodium azide、pH9.6)を使って希釈した後、96ウェルELISAプレートに100μLずつ分株し、4℃で一晩中反応させた。PBST(PBS+0.05%Tween20)を300uL/ウェルずつ添加しながら、3回洗浄した後、5%スキムミルク100μLを入れて37℃で1時間ブロッキングさせ、ブロッキング緩衝溶液を除去した後、PBSTで3回洗浄して希釈させた血清と粘膜洗浄液を各々100μLずつ添加した。検体を添加した後、37℃で2時間反応させた後、洗浄溶液で3回洗浄してからセイヨウワサビペルオキシダーゼ標識抗マウスIgGまたはIgA抗体を1:5000で希釈してウェルに添加後、37℃で1時間反応させてから洗浄溶液で洗浄した後、TMB発色溶液で発色させて、2.5M硫酸で発色を固定させた。450nmで、ELISAリーダーで測定してHPV16 E7特異抗体の量を分析した。
【0060】
その結果、図5のAに示されたように最初2週間免疫した後のE7特異的血清IgGの平均OD値を測定した結果、pAT−E7とpKV−E7投与群がpAT対照群に比べて高いOD値を示した。また、投与した量に伴ってE7特異的抗体が増加することを確認した。一次ブースティング後のE7特異的血清IgGの平均OD値は、pAT−E7とpKV−E7投与群共に最初2週間免疫に比べて増加したOD値を示しブースティング効果を見せた。二次ブースティング後のE7特異的血清IgGの平均OD値も二つの投与群でブースティング効果を示し、特にpAT−E7に比べてE7融合タンパク質の表面発現量がずっと高いpKV−E7投与群の場合、1/5投与群とpAT−E7 1/1投与群が似たようなIgGブースティング効果を示した。
【0061】
さらに、粘膜免疫反応を測定するために腸内と膣内でのE7特異的IgA抗体をELISAで測定した結果、図5のBと図5のCに示されたように最初2週間免疫した後の平均OD値は、pAT対照群に比べてpAT−E7、pKV−E7投与群共にE7特異的IgAが増加することを確認し、また投与した量に伴ってE7特異的抗体が増加することを確認した。一次ブースティング後の平均OD値はより一層増加し、二次ブースティング効果は一次ブースティングに比べて、若干効果を見せた。
【0062】
本発明によるHPV16 E7を発現する形質転換された組換えラクトバチルスカゼイを経口投与する場合、HPV16 E7特異的な体液性免疫(血清IgG誘導)と粘膜免疫(Interstinal/Vaginal IgA誘導)を誘導できたことを示し、特にE7をより多く発現するpKV−E7投与群がpAT−E7投与群よりも効果的に免疫反応を誘導できたことが示唆された。
【0063】
[実施例5]HPV16 E7抗原を表面発現するラクトバチルスの細胞性免疫反応誘導能比較
HPV 16 E7抗原の表面発現量が異なる2種類の形質転換された組換えラクトバチルスカゼイ(pHAT−E7、pKV−E7)を経口投与したマウスからE7抗原に特異的なCD8+ T細胞媒介免疫反応誘導とT細胞免疫反応の誘導に当り差があるか確認しようと各々intracellular cytokine染色とIFN−γELISPOTを行って、細胞性免疫誘導能を調べた。
【0064】
実施例4で説明したようにE7を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与したマウスから脾臓細胞を分離した。5×106cellの脾臓細胞を10%FBSが含まれたRPMI−1640培地に浮遊して、24ウェルプレートに分株した。各ウェルにMHC class Iエピトープを含むE7ペプチド(a.a 49−57、AniGen、Korea)とGolgiPlug(BD Biosciences)を処理し、37℃で16時間培養した。培養された細胞はフィコエリスリン(PE)標識モノクローナルラット抗マウスCD8抗体で、4℃で40分間染色し、Cytofix/Cytoperm kitを利用して、細胞内部のIFN−γをFITC標識モノクローナルラット抗マウスIFN−γ抗体で染色した。
【0065】
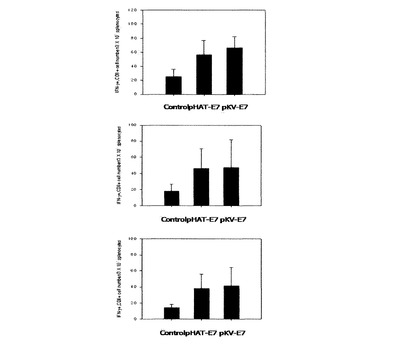
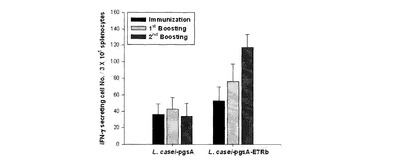
二つの組換えラクトバチルスカゼイを経口投与した後、intracellular cytokine染色を行ってE7特異的にIFN−γを分泌するCD8+ T細胞の数を測定した結果、図6に示されたように、E7を発現する形質転換された組換えラクトバチルスカゼイによるE7特異的なIFN−γ分泌CD8+ T細胞前駆体の数が増加したことを確認することができた。特に、免疫反応を誘導した初期には表面発現量が多いpKV−E7で形質転換された組換えラクトバチルスカゼイによる細胞媒介性免疫反応がより増加することが確認されたが、ブースティングに伴いpHAT−E7乳酸菌による細胞媒介性免疫反応も似たレベルで増加した。
【0066】
E7特異的なT細胞免疫反応誘導を調べるためE7ペプチドによる刺激に特異的にIFN−γを分泌する全T細胞数を測定するためにIFN−γELISPOTを行った。
脾臓細胞を分離する一日前に、プレートに抗マウスIFN−g捕捉抗体をコーティングし、実験当日分離した脾臓細胞はウェル当たり2×105cellに分株した。細胞が分株されたウェルにE7ペプチドまたはPHA−M、mediaを処理して2日間培養した。2日後、プレートの内容物を除去して蒸溜水で2回、0.05%tween−20が含まれたPBSで3回洗浄し、ビオチン化抗マウス IFN−gを分株し常温で2時間反応させた。反応後、内容物を除去して0.05%tween−20が含まれたPBSで3回洗浄後、ストレプトアビジンHRPを分株し常温で一時間反応させた。内容物を除去して0.05%tween−20が含まれたPBSで4回洗浄してPBSで2回洗浄した後、基質と反応させて、発色を確認しながら5〜60分間観察し、蒸溜水で反応を停止させ、ELISPOTリーダーを用いて、各ウェルのスポット数を測定した。
【0067】
図6に示されたようにpHAT−E7またはpKV−E7で形質転換された組換え乳酸菌を経口投与したマウスの場合、E7ペプチドの刺激に対するIFN−γ分泌細胞数が増加することを確認でき、pHAT−E7で形質転換された組換えラクトバチルスカゼイを投与した群よりpKV−E7で形質転換された組換えラクトバチルスカゼイを投与した群でより多くの数のIFN−γ分泌細胞数の分布を確認することができた。
【0068】
[実施例6]HPV E7抗原を表面発現するラクトバチルスで免疫したマウスの腫瘍細胞チャレンジ
HPV 16 E7抗原の表面発現量が異なる2種類の形質転換された組換えラクトバチルス(pHAT−E7、pKV−E7)の抗癌効果と投与量に伴う抗癌効果を確認するためE7タンパク質を発現するTC−1腫瘍細胞モデルを利用して、腫瘍細胞チャレンジを行った。
【0069】
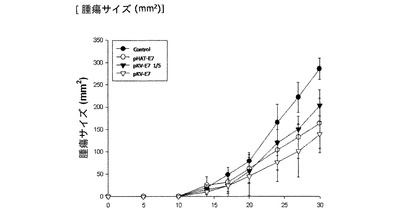
先ず、6〜8週齢の雌C57BL/6マウスを各集団毎に5匹ずつ準備してE7を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与した。経口投与は、週5回行い、最初1週間投与後、1週間の休止期間を設け、さらに1週間と最初のブースティング後、再び1週間の休止期間を設けた後1週間と二回目のブースティングを行った。最初1週間の経口投与後、2×104cellのTC−1腫瘍細胞を左の上腿内側に皮下注射(s.c.)で癌を誘導した。形成された癌の大きさはキャリパーを利用して、週に3回測定した。
【0070】
図7に示されたように、pHAT−E7またはpKV−E7で形質転換された組換えラクトバチルスカゼイを経口投与した集団で対照群のラクトバチルスカゼイを経口投与した集団に比べて、腫瘍細胞の大きさが50%程度で小さく形成されることを確認することができた。また、pKV−E7で形質転換された組換えラクトバチルスカゼイの量を1/5に減らして投与した集団の場合も対照群集団に比べて、腫瘍細胞の大きさが小さかった。
【0071】
[実施例7]HPV type16 E7関連部位アミノ酸の変異導入タンパク質表面発現免疫乳酸菌(pKV−Pald−PgsA−E7(Rb))の製造
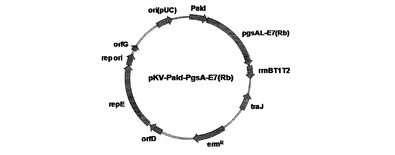
パピローマウィルスが細胞に感染する場合発現したE7タンパク質とpRb(Retinoblastoma Growth Suppressor Protein)タンパク質が結合して、癌を誘発する可能性があるため、E7遺伝子のpRbタンパク質と結合する部分の塩基配列に変異を誘発した。このように変異が誘発されたE7(Rb)遺伝子を大腸菌と乳酸菌で安定的に発現するpKVシャトルベクターを用いてPgsA末端に融合させて、乳酸菌で表面発現できるベクターpKV−Pald−PgsA−E7(Rb)を製造した。
【0072】
HPV type16 E7内のRb結合領域を調べた結果、pRb結合領域(DLxCxE)は、HPV16 E7遺伝子の21番目アミノ酸から26番目アミノ酸と知られている(Smahel M. et al., Virology, 281:231, 2001)。その中で21番目(D)、24番目(C)、26番目(E)のアミノ酸を共にグリシンで置換させるため、プライマーを使ったpoint mutagenesis方法によってE7タンパク質のpRbタンパク質との結合の可能性を無くした。実施例2で取得したpKV−Pald−PgsA−E7ベクターを鋳型として配列番号8と9、配列番号10と11を使って、各々PCRを行った。
配列番号8:5’−tct gga tcc atg cat gga gat aca cct ac−3’
配列番号9:5’−ttg ccc ata acc gta gag acc agt tgt c−3’
配列番号10:5’−act ggt ctc tac ggt tat ggg caa tta aat g−3’
配列番号11:5’−cat tct aga tca tta tgg ttt ctg aga aca g−3’
【0073】
その結果、配列番号8及び9のプライマーによってHPV16 E7遺伝子を含んでいる87bpの切片を取得し、DNA切片の5'末端にはBamHI制限酵素部位を含んでおり、配列番号10及び11のプライマーによって取得した変異された249bp DNA切片の3'末端にはXbaI制限酵素部位を含んでいた。二つの取得したDNA切片をミックスして、95℃−30秒、42℃−30秒、72℃−30秒の条件で10回PCRを行った後、配列番号8及び11のプライマーを添加してPCR(95℃−30秒、53℃−30秒、72℃−30秒)を行った。取得した312bp DNA切片の5'末端にはBamHI制限酵素部位が、3'末端にはXbaI制限酵素部位が含まれていた。この切片をBamHIとXbaI処理し、切断して、306bpの変異されたE7(Rb)遺伝子切片を取得した。pKV−Pald−PgsA−AmylaseをBamHIとXbaIで切断してアミラーゼ遺伝子の部分を除去してベクターの部分を取得した。BamHIとXbaIで切断された変異E7(Rb)遺伝子を含有しているDNA断片を同じ制限酵素で切断したベクターと連結して、pKV−Pald−PgsA−E7(Rb)を完成した(図8)。
構築された前記構築体をラクトバチルスカゼイにエレクトロポレーション法を利用して、形質転換し、構築体が形質転換された乳酸菌形質転換体を取得した。
【0074】
[実施例8]pKV−Pald−PgsA−E7(Rb)乳酸菌形質転換体の発現確認
取得した乳酸菌表面発現用ベクターpKV−Pald−PgsA−E7(Rb)で形質転換された組換えラクトバチルスカゼイからPgsAと融合したE7私のRb結合部位変異タンパク質の発現の有無を確認した。
【0075】
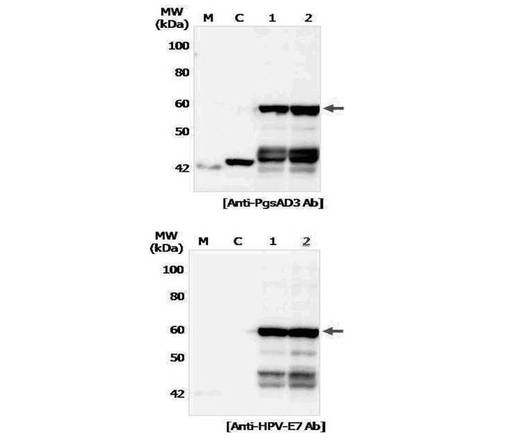
形質転換された組換えラクトバチルスカゼイをMRS培地(Lactobacillus MRS, Becton Dickinson and Company Sparks, USA)、30℃で静置培養を行い、菌体を回収して、実施例3のような方法でウェスタンブロッティングを行って、発現されたことを確認した。PgsA特異抗体とHPV16 E7特異抗体(E7 monoclonal Ab, Invitrogen, USA)を用いてウェスタンブロッティングを行った結果、図9に示されたように約54.7kDaのPgsAと融合した変異E7タンパク質の発現を確認した。
【0076】
[実施例9]変異されたHPV16 E7(Rb)抗原を表面発現するラクトバチルスの体液性免疫反応誘導
PgsAと融合した変異E7タンパク質の発現が確認された形質転換された組換えラクトバチルスカゼイをマウスに経口投与し、体液性免疫反応が誘導されるか確認した。前記ベクターをラクトバチルスカゼイに形質転換させた後、培養して回収した後、凍結乾燥して準備したパウダーを緩衝溶液(PBS、pH7.4)に溶かした後、6週齢の雌Balb/cマウスに経口投与した。
【0077】
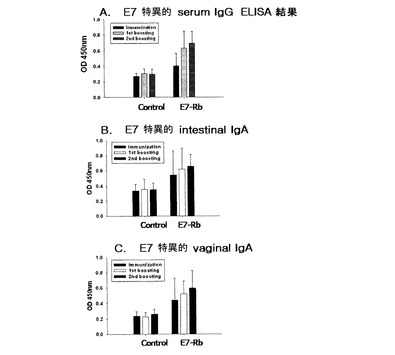
具体的には、HPV16 E7(Rb)を発現する形質転換された組換えラクトバチルスカゼイ(E7−Rb)と、発現しないラクトバチルスカゼイ(L525)の凍結乾燥パウダーを各々PBSに2〜5×109cell/200μLで溶かした後、週に5回ずつ2週間免疫(0〜4days、7〜11days)して1週後に一次ブースティング(21〜25days)、さらに1週間後に二次ブースティング(35〜39days)を経口投与した。血清内のHPV16 E7特異的なIgG抗体と、粘膜内で分泌されるHPV16 E7特異的なIgA抗体を測定するため、腸内洗浄液と膣内洗浄液を図3に示されたように14日、28日、そして42日に各々マウスからサンプルを採取した。実施例4同様に血液はヘパリンがコーティングされたマイクロキャピラリーチューブを用いて、マウスの目から得られ、1mM PMSFが含まれたPBS洗浄液を用いて、腸内と膣内を各々0.8 mLと250μLで洗浄した後、13,000rpmで20分間遠心分離し、上澄液を分離して、実施例4と同じ方法でELISAを行った。
【0078】
その結果、図10のAに示されたように最初2週間免疫した後のE7特異的血清IgGの平均OD値を測定したところ、E7−Rb投与群がL525(−control)投与群に比べて高いOD値を示した。一次ブースティング後のE7特異的血清IgGの平均OD値は、E7−Rb投与群で最初2週間免疫に比べて増加したOD値を示しており、ブースティング効果が示された。二次ブースティング後のE7特異的血清IgGの平均OD値を見てもブースティング効果が示された。しかし、L525投与群では特別なブースティング効果が見られなかった。また、粘膜で誘導される体液性免疫反応を測定するために腸内と膣内でのE7特異的IgA抗体をELISAで測定した結果、図10のBと図10のCに示されたように最初2週間免疫した後の平均OD値は、L525(control)投与群に比べてE7−Rb投与群が、E7特異的IgAが増加することを確認し、一次ブースティング後の平均OD値は、より一層増加し、二次ブースティング効果は一次ブースティングに比べて増加した効果が見られた。
【0079】
以上の結果から確認されたように、E7−Rb形質転換された組換えラクトバチルスカゼイを経口投与した場合、HPV16 E7特異的な全身性液性免疫反応(systemic humoral immune response)(血清IgG誘導)と粘膜液性免疫反応(mucosal humoral immune response)(Interstinal/Vaginal IgA誘導)を共に誘導できることが示された。
【0080】
[実施例10]変異されたHPV16 E7(Rb)抗原を表面発現するラクトバチルスの細胞性免疫反応誘導
変異されたHPV 16 E7(Rb)抗原を表面発現する形質転換された組換えラクトバチルス(E7−Rb)が、E7(Rb)抗原に特異的なT細胞媒介免疫反応誘導を誘導する能力有しているかを調べるために、IFN−γELISPOTを行い、細胞性免疫誘導能を調べた。
【0081】
実施例7で作製したE7(Rb)を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与したマウスで脾臓細胞を分離した。5×106cellの脾臓細胞を10%FBSが含まれたRPMI−1640培地に浮遊して、24ウェルプレートに分株した。各ウェルにMHC class Iエピトープを含むE7ペプチド(a.a 49−57、AniGen、Korea)とGolgiPlug(BD Biosciences)処理して、37℃で16時間培養した。培養された細胞は、フィコエリスリン(PE)標識モノクローナルラット抗マウスCD8抗体で、4℃で40分間染色し、Cytofix/Cytoperm kitを利用して、細胞内部のIFN−γをFITC標識モノクローナルラット抗マウスIFN−γ抗体で染色した。
【0082】
二種類の組換えラクトバチルスカゼイを経口投与した後、intracellular cytokine染色を行って、E7(Rb)特異的にIFN−γを分泌するCD8+ T細胞の数を測定した結果、図11に示されたように、E7(Rb)を発現する形質転換された組換えラクトバチルスカゼイによるE7(Rb)特異的なIFN−γ分泌CD8+ T細胞前駆体の数が増加したことを確認することができた。特に、免疫反応を誘導した初期には表面発現量が多いpKV−E7(Rb)乳酸菌による細胞媒介性免疫反応がより増加することが確認されたが、ブースティングに伴ってpHAT−E7(Rb)乳酸菌による細胞媒介性免疫反応も似たレベルに増加した。
【0083】
E7(Rb)特異的なT細胞免疫反応誘導を調べるため、E7ペプチドによる刺激に特異的にIFN−γを分泌する全T細胞の数を測定するため、IFN−γELISPOTを行った。
【0084】
脾臓細胞を分離する一日前にプレートに抗マウスIFN−g捕捉抗体をコーティングし、実験当日に分離した脾臓細胞は、ウェル当たり2×105cellに分株した。細胞が分株されたウェルにE7ペプチドまたはPHA−M、media処理して2日間培養した。2日後、プレートの内容物を除去して蒸溜水で2回、0.05%tween−20が含まれたPBSで3回洗浄し、ビオチン化抗マウスIFN−gを分株し常温で2時間反応させた。反応後、内容物を除去して0.05%tween−20が含まれたPBSで3回洗浄後、ストレプトアビジンHRPを分株し常温で一時間反応させた。内容物を除去して0.05%tween−20が含まれたPBSで4回洗浄してPBSで2回洗浄した後、基質と反応させて、発色を確認しながら、5〜60分間観察し、蒸溜水で反応を停止させ、ELISPOTリーダーを用いて、各ウェルのスポット数を測定した。
【0085】
図11に示されたようにpHAT−E7(Rb)またはpKV−E7(Rb)で形質転換された組換えラクトバチルスカゼイを経口投与したマウスの場合、E7ペプチドの刺激に対するIFN−γ分泌細胞数が増加することが確認でき、pHAT−E7(Rb)で形質転換された組換えラクトバチルスカゼイを投与した群よりpKV−E7(Rb)で形質転換された組換えラクトバチルスカゼイを投与した群の方がより多くの数のIFN−γ分泌細胞数の分布を確認することができた。
【0086】
以上、本発明の内容の特定の部分を詳述したが、当業界における通常の知識を持った者にとって、このような具体的な記述は単なる好適な実施態様に過ぎず、これにより本発明の範囲が制限されることはないという点は明らかである。よって、本発明の実質的な範囲は特許請求の範囲とこれらの等価物により定義されると言える。
【産業上の利用可能性】
【0087】
本発明に係るヒトパピローマウィルス抗原タンパク質の表面発現用ベクターで形質転換された組換え乳酸菌及びこれを主要成分として含んだ組成物は、子宮頸癌治療用ワクチンとして活用でき、構成的にHPV抗原を高発現する形質転換された組換え菌株を経済的に大量増殖させて、経口ワクチンとしてまたは膣部位に直接適用することができるため、非常に経済的な効果がある。
【技術分野】
【0001】
本発明は、配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルス(human papilloma virus)の腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有するHPV治療ワクチン製造用表面発現ベクターに関する。
【背景技術】
【0002】
細菌から有用な外来タンパク質を多量発現させようとする試みにおいて外来タンパク質の生産量は、遺伝子複製数、即ち、プラスミド数と転写を調節するのに用いられているプロモーターの強さに依存することが知られている。通常多く用いられている高発現プロモーターの他に細菌個体当たりプラスミドベクターの数を変化させて、ターゲットタンパク質発現量を増加させられることが報告された(Tomio, M. et al., Appl. Microbiol. Biotechnol., 28:170, 1988)。また、mini−FプラスミドでRepEの変異が細菌個体内でプラスミド数を変化させられることが報告された(Yasuo, K. et al., J. Biol. Chem., 267:11520, 1992)。
【0003】
ヒトパピローマウィルス(Human papilloma virus,HPV)は世界的に成人の50%以上が感染していると推定され、パピローマウィルス中、特にHPV16、18、31及び45の四つのタイプは、80%以上の子宮頸癌(cervical cancer)の原因として確認された(Lowy, D. R. et al., Proc. Nat. Acad. Sci., 91:2436, 1994)。
【0004】
世界的に子宮頸癌は、乳癌の次に女性に発生頻度が高い癌であって、世界保健機構によると、毎年世界で50万人以上の子宮頸癌患者が発生しており、毎年300,000人以上が子宮頸癌によって死亡すると推定されている。特に、開発途上国及び低開発国では女性死亡の主原因になっている(Pisani, P. et al., Int. J. Cancer, 55:891, 1993)。IARC統計によると、特に慢性感染者が先進国に比べて、極めて多い開発途上国のHPV感染を撲滅するため、長期的に最も効果的な方法はHPV予防ワクチンを投与すると報告された。
【0005】
子宮頸癌に関わるワクチン開発の方法としては、大きく予防ワクチン(prophylactic)と治療ワクチン(therapeutic)の二つに焦点が絞られている。予防ワクチンは、HPV L1/L2抗原タンパク質によって、強い中和抗体(neutralizing antibody)が生成されるようにすることによって、宿主がHPVに感染することを防ぎ、また既に感染したとしてもそれ以上疾病が進まないようにするのに目的がある。一方、治療ワクチンは、HPV E6/E7を対象にするが、特異的な細胞性免疫を誘導させて、既に形成された病斑や悪性腫瘍を退化させることを目的としている。
【0006】
HPVのE6/E7タンパク質はHPVに感染した細胞等の癌化に関わる癌特異抗原であるため、子宮頸癌の免疫治療のターゲットとしてE6/E7タンパク質を利用した治療ワクチン研究が続けられてきた。実際、微生物システムで合成されたHPV E6/E7タンパク質を腫瘍細胞を注入したラットに投与した場合、腫瘍形成が阻害されたり遅延されるとの報告がある(Gao, L. et al., J. Gen. Viol., 75:157, 1994, Meneguzzi, G. et al., Virology, 81:62, 1991)。しかし、他の場合と同じように、生ウイルスワクチンを利用する場合、過度なウイルス複製による問題を誘発しうるため、実際には研究目的のみに使用され、商業化までには長時間及び相当な臨床実験が求められる短所がある。
【0007】
一方、バクテリアベクターを利用したワクチン開発研究も盛んに行われていがる、弱毒化されたサルモネラ菌(attenuated Salmonella typhimurium)で合成されたHPV 16 VLPが、マウスの粘膜や全身で抗原特異的な抗体の生成を誘導するとの報告もある(Denis, N. et al., Infection and immunity, 65:3328, 1997)。合成ペプチドを利用したワクチンは、免疫反応を誘導するのに必要なエピトープのみを合成して接種(vaccination)させるものであり、既にHPV 16 E6/E7に対する細胞性免疫反応(Cytotoxic T Lymphocyte;CTL)を起こすエピトープが分かるようになっている(Ressing, M. E. et al., J. Immunol., 154:5934, 1995)。
【0008】
このような試み以外に、植物からウイルスの抗原を生産するため、トマト及びジャガイモ等の野菜類を利用して、その形質転換体野菜類自体を経口用ワクチン(oral vaccine)または食用ワクチン(edible vaccine)として使おうとする研究も進行している。代表的な例としては、B型肝炎ウイルス表面抗原粒子(Hepatitis B surface antigen particle)(Thavala, Y. F. and Artzen, C. J., Pro. Natl. Acad. Sci. USA., 92:3358, 1995)とパピローマウイルスのギャプシドL1とL2タンパク質(韓国特許登録第0366608号)が挙げられる。しかし、植物を利用したシステムの場合、発現するHPV L1タンパク質の量が少なく、精製の問題があって、前記同様商業的な制約が従う問題がある。
【0009】
従って、HPVの感染者が主に低開発国に集中している点を勘案すると、パピローマウィルス由来の口腔または生殖器官の皮膚粘膜の腫瘍に対する予防と治療のため、より経済的かつ安定的にHPV抗原を製造する方法の開発が強く求められている。
【0010】
本発明者等は、HPV抗原タンパク質を形質転換された組換え微生物の表面に効果的に発現させるベクターと、微生物の表面にHPV抗原タンパク質を発現させる方法を開発した(韓国特許登録第0609866号)。
【0011】
そこで、本発明者等は、HPV抗原タンパク質が形質転換された組換え乳酸菌表面でより安定的かつ構成的に高発現されるベクターを開発しようと鋭意努力した結果、repE変異遺伝子を含有するベクターで形質転換された組換え微生物においてより安定的にターゲットタンパク質の発現が向上することを確認して、本発明の完成に至った。
【発明の概要】
【発明が解決しようとする課題】
【0012】
本発明の主な目的は、repE変異遺伝子を利用してHPV抗原タンパク質が形質転換された組換え乳酸菌の表面で安定的かつ構成的に高発現させられるベクターを提供することである。
【0013】
本発明の他の目的は、前記発現ベクターで形質転換された組換え乳酸菌及び前記乳酸菌を利用してHPV抗原タンパク質の製造方法を提供することである。
【0014】
本発明のさらに他の目的は、前記形質転換組換え乳酸菌を利用して、子宮頸癌治療用に形質転換された組換え乳酸菌ワクチンを提供することである。
【課題を解決するための手段】
【0015】
前記目的を達成するため、本発明は配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有する表面発現ベクターを提供する。
【0016】
本発明は、また前記HPV抗原タンパク質を表面発現するベクターで形質転換された組換え微生物を提供する。
【0017】
本発明は、またHPV抗原タンパク質が表面に発現した形質転換組換え微生物を有効性分とする子宮頸癌治療用ワクチンを提供する。
【0018】
本発明は、また前記ベクターで形質転換された組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法を提供する。
【0019】
本発明は、前記方法で製造された抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチンを提供する。
【図面の簡単な説明】
【0020】
【図1】repE変異遺伝子が導入されたpKV−Pald−PgsA−Amylaseベクターのマップを示す。
【図2】E7遺伝子を表面発現する2種の発現ベクターのマップを示す。
【図3】pKV−Pald−PgsA−E7で形質転換された組換え乳酸菌においてE7の表面発現をウェスタンブロッティングで確認した結果を示す。
【図4】E7を表面発現する形質転換された組換え乳酸菌の免疫効能実験のため、マウスに形質転換された組換え乳酸菌を投与する日程を示す。
【図5】E7を表面発現する形質転換された組換え乳酸菌の経口投与時IgGとIgAの変化を示す。
【図6】E7を表面発現する形質転換された組換え乳酸菌の経口投与時のE7特異的IFN−gamma分泌を確認した結果を示す。
【図7】E7を表面発現する形質転換された組換え乳酸菌の経口投与時腫瘍移植による腫瘍サイズの増加の有無を確認した経過を示す。
【図8】E7(Rb)遺伝子を表面発現する発現ベクターのマップを示す。
【図9】pKV−Pald−PgsA−E7(Rb)で形質転換された組換え乳酸菌においてE7(Rb)が表面発現されたことを確認した結果を示す。
【図10】E7特異的ELISAの結果を示す。
【図11】E7(Rb)を表面発現する形質転換された組換え乳酸菌の経口投与時E7(Rb)特異的IFN−gamma分泌を確認した結果を示す。
【発明を実施するための形態】
【0021】
本発明は一つの観点において、配列番号1のアミノ酸配列を持つrepE変異遺伝子、プロモーター、ポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有する表面発現ベクターに関する。
【0022】
本発明では形質転換された微生物内でベクターが安定的に維持される配列番号1のアミノ酸配列を持つrepE変異遺伝子、ラクトバチルスカゼイ(Lactobacillus casei)のアルドラーゼ(aldolase)遺伝子から由来したアルドラーゼプロモーター(Pald)を共に導入した結果としてターゲットタンパク質の発現が向上することを確認した。本発明のシャトルベクターが安定的に維持され、ターゲットタンパク質が発現することを確認するため、目的遺伝子としてアミラーゼ(amylase)遺伝子を挿入してアミラーゼの発現を確認した。
【0023】
また、ターゲットタンパク質の表面発現のため、表面発現モチーフであるポリガンマグルタミン酸合成酵素複合体遺伝子をターゲットタンパク質と連結して発現するように位置させ、前記ポリガンマグルタミン酸合成酵素複合体遺伝子は、pgsBCAであることが望ましく、さらに望ましくはpgsAである(韓国登録特許第469800号)。
【0024】
本発明の一様態において、PgsAのC末端にターゲットタンパク質であるHPV16 E7タンパク質が融合するようpsgAとHPV16 E7コードする遺伝子を融合させて、乳酸菌からHPV抗原タンパク質を構成的に表面発現できるベクターpKV−Pald−PgsA−E7を製造し、前記発現ベクターをラクトバチルスカゼイに挿入してHPV抗原タンパク質を発現する形質転換された組換え乳酸菌を製造した。
【0025】
本発明において、前記「ターゲットタンパク質」または「外来タンパク質」とは、前記タンパク質を発現する形質転換された宿主細胞では正常には存在できないタンパク質を意味する。例えば、乳酸菌からウイルス由来または腫瘍由来タンパク質を人為的に発現するように操作した場合、前記タンパク質を外来タンパク質またはターゲットタンパク質という。
【0026】
本発明はまた、他の観点において、前記HPV抗原タンパク質を表面発現するベクターで形質転換された組換え微生物及び前記形質転換組換え微生物を有効性分とする子宮頸癌治療用ワクチンに関する。
【0027】
本明細書において「宿主」、または「微生物」とは、プロバイオティクス(probiotics)のグラム陽性桿菌である乳酸菌に言及することができ、一般的なプロバイオティクス微生物の選別基準には(i)ヒト由来の微生物であること;(ii)胆汁、酸、酵素及び酸素に安定的であること;(iii)腸内粘膜に付着する能力があること;(iv)消化器官内でコロニーを形成できる能力があること;(v)抗バクテリア性物質を生産できること;及び(vi)効能や安全性が証明されること、等の条件が含まれている。前記条件を基に乳酸菌は人体内での生長が親和的、無害な菌であることは明らかである。従って、乳酸菌を宿主とする形質転換体を人体に適用させて、疾病の予防または治療のための遺伝子伝達またはタンパク質伝達等の用途に用いる場合、菌株を用いる通常のワクチン製造方法とは異なって菌株の無毒化の工程を要しない。
【0028】
本発明において、前記乳酸菌はラクトバチルス属、ストレプトコッカス属及びビフィドバクテリウム属を含んでもよい。代表的にはラクトバチルス属はラクトバチルスアシドフィルス(L. acidophilus)、ラクトバチルスカゼイ(L. casei)、ラクトバチルスプランタラム(L. plantarum)、ラクトバチルスファーメンタム(L. ferementum)、ラクトバチルスデルブルッキー(L. delbrueckii)、ラクトバチルスジョンソニー(L. johnsonii LJI)、ラクトバチルスロイテリ(L. reuteri)及びラクトバチルスブルガリクス(L. bulgaricus);ストレプトコッカス属は、ストレプトコッカスサーモフィルス(S. thermophilus);ビフィドバクテリウム属は、ビフィドバクテリウムインファンティス(B. infantis)、ビフィドバクテリウムビフィダム(B. bifidum)、ビフィドバクテリウムロンガム(B. longum)、ビフィドバクテリウム シュードロンガム(B. psuedolongum)、ビフィドバクテリウムブレーベ(B. breve)、ビフィドバクテリウムラクティスBb−12(B. lactis Bb-12)及びビフィドバクテリウムアドレスセンティス(B. adolescentis)等を宿主として用いられ、より望ましくはラクトバチルス属である。
【0029】
本発明はまた、前記ベクターで形質転換された組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法に関する。
【0030】
本発明に係る組換え微生物の培養は、広く公知の方法に従って行われ、培養温度及び時間、培地のpH等の条件は、適宜調節される。前記培養物での組換え微生物菌体の回収は通常の分離技術、例えば、遠心分離、ろ過法等を用いてもよい。
【0031】
本発明はまた、他の観点において、前記方法で製造された抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチンに関する。
【0032】
また、本発明は、経口用である子宮頸癌治療用ワクチンに関する。
ワクチンは、疾病に対する予防目的として生きている生物を使って、免疫システムを刺激させるのに用いられる薬剤である。免疫活性化は、有機体内で抗体生成、T−リンパ球の刺激、または他の免疫細胞(例:大食細胞)を刺激して、抗原を効率的に除去するための過程を意味する。前記に関する詳しい免疫学の概観は当業界の通常の技術を有する者なら簡単に理解できる(Barrett, J.T., Textbook of Immunology, 1983)。抗原としてのターゲットタンパク質を発現する形質転換された微生物ワクチンの投与対象は、ほ乳動物であってもよく、より望ましくはヒトである。
【0033】
前記ワクチン組成物の製法は標準的な技法を用いて実施でき、投与者に適する投与量は遺伝子生成物の抗原性によって異なり、存在するワクチンの典型的な免疫反応を誘導するのに十分な量であれば良く、通常実験過程を経て必要量を簡単に判断できる。典型的な初期のワクチン容量は、体重1kg当り抗原0.001〜1mgであり、望ましい水準の保護を提供するのに必要に応じて量を増加したり複数回投与(multiple dose)を用いる。前記量は当該技術分野に属する専門家によって決められ、製剤化方法、投与方式、投与者の年令、体重、性、病的状態、食べ物、投与時間、投与経路、排泄速度及び反応感応性のような要因によっても多様に処方される。
【0034】
本発明のHPV抗原タンパク質E7をコードするHPVワクチン製造用表面発現ベクターの最も望ましい様態は、配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAで構成された群から選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクターで、前記ベクターで形質転換された組換え乳酸菌を培養してHPV抗原タンパク質E7を表面発現させた後、ワクチンとしてそのまま使用可能である。
【0035】
本発明のHPV抗原タンパク質E7(Rb)をコードするHPVワクチン製造用表面発現ベクターの最も望ましい様態は、配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAで構成された群から選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結するヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7(Rb)をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクターで、前記ベクターで形質転換された組換え乳酸菌を培養してHPV抗原タンパク質E7(Rb)を表面発現させた後、ワクチンとしてそのまま使用可能である。
【0036】
ワクチンが抗体を生成するに当り効果を高めるためには、抗原性物質がワクチン処理された対象の抗体生成機構が実現できるように体内に放出されなければならない。従って、免疫反応のためには遺伝子産物の微生物キャリアが、体内にまず導入されなければならない。本発明の形質転換された組換え乳酸菌で提示される抗原による望ましい反応を刺激するためには、経口投与または噴霧剤状で子宮頚部に直接投与することが望ましい。
【0037】
ワクチン組成物を投与者に経口投与するためには、親液性の形状、例えば、カプセル状で提供することが望ましい。前記カプセルはEudragate S、Eudragate L、セルロースアセテート、セルロースフタレートまたはヒドロキシプロピルメチルセルロースを含む腸用被覆で提供される。前記カプセル類は、それ自体で使ってもよく、投与する前に懸濁液のような親液性物質に再構成して使用してもよい。再構成は形質転換された組換え微生物の生存に適するpHの緩衝液で行うことが望ましい。胃酸から前記形質転換された組換え微生物及びワクチンを保護するため、毎回ワクチン投与以前に重炭酸ナトリウム製剤を投与することが望ましい。選択的にワクチンは非経口投与、鼻内投与または乳腺内投与用として製造される。
即ち、下記工程は一つの例示として説明され、本発明の範囲がこれに限定されない。
【実施例】
【0038】
以下、本発明を実施例を挙げて詳述する。これらの実施例は単に本発明をより具体的に説明するためのものであり、本発明の範囲がこれらの実施例に制限されないことは当業界において通常の知識を持った者にとって自明である。
【0039】
[実施例1]repE変異遺伝子を含有する表面発現ベクターの製造(Pkv−Pald−PgsAL−Amylase)
本実施例では、宿主細胞で発現ベクターを安定させるため、repE変異遺伝子を含有するターゲットタンパク質の発現が向上した表面発現ベクターを作製した。
【0040】
先ず、乳酸菌でより一層発現量を高めるため、ラクトバチルスカゼイ由来のプロモーターであるアルドラーゼプロモーターの断片を得た。前記アルドラーゼプロモーターは韓国公開特許第10−2008−0086161号に開示されたpDT−PgsA−Amylaseを鋳型として配列番号2と配列番号3のプライマーを使ってPCR法で得た。
配列番号2:5’−cgc gca tgc aat acc cac tta ttg cg−3’
配列番号3:5’−cag ttc ttt ttt cat gta gat atc ctc c−3’
【0041】
その結果、アルドラーゼプロモーターを含み、5'末端にはSphI制限酵素部位を含んでおり、3'末端にはpgsAのN−terminal 17bpを含んでいる421bpのDNA切片を取得し、同じベクターを鋳型として配列番号4と配列番号5のプライマーを使ってPCRを行い、先に得たアルドラーゼプロモーター切片と連結できるpgsA遺伝子の部分を得た。
配列番号4:5’−gga gga tat cta cat gaa aaa aga act g−3’
配列番号5:5’−ggc gct ggc ggt cgt ttg g−3’
【0042】
その結果、取得したDNA切片のN末端の部分は、アルドラーゼプロモーターの3'末端の13bpを含み、pgsAが直接連結している782bp切片を得た。この切片のpgsA部分内部にPstI制限酵素部位が含まれている。
【0043】
取得した二つの切片を連結して両側末端のプライマーを用いてPCRを行い、1175bpのDNA切片を取得し、SphIとPstIで切断して、アルドラーゼプロモーターとpgsA N−terminalの一部分を含んだ切片を得た。
【0044】
pBT:pgsA−Amylase(pAT−PslpA−pgsA−amylase;韓国登録特許第0872042号の間接実施例参照)を制限酵素SphI及びPstIで切断してSlpA7プロモーターの部分とpgsA N末端の部分を除去して、発現ベクターの骨格とした。
【0045】
SphIとPstIで切断したアルドラーゼプロモーターを含有しているDNA断片を同じ制限酵素で切断したpBT:PslpA−Amylaseと連結して、pAT−Pald−PgsA−Amylaseを作製し、pAT−Pald−PgsA−Amylaseベクターにsite−directed mutagensis方法でrepE遺伝子475番目のアミノ酸ロイシンをコードするTTAコドンをTGA停止コドンで置換してRepEタンパク質中21個のC末端アミノ酸が切断されて、配列番号1のアミノ酸配列を持つようにコードしたpKV−Pald−PgsA−Amylaseを完成した(図1)。
【0046】
[実施例2]HPV16 E7表面発現ベクター(pKV−Pald−PgsA−E7)の製造
実施例1で製造された表面発現ベクター(pKV−Pald−PgsA−Amylase)を使ってPgsAのC末端にHPV16のE7タンパク質をコードする遺伝子を挿入させて、乳酸菌で表面発現できるベクターpKV−Pald−PgsA−E7を製造した。
【0047】
先ず、実施例1で製造されたpKV−Pald−PgsA−AmylaseベクターでpgsAと融合したアミラーゼ遺伝子を除去し、HPV16 E7をコードする遺伝子を挿入した。HPV16 E7遺伝子を含む断片を図1に示されたようにpHAT:pgsA−E7(Poo et al., Int. J. Cancer, 119:1702, 2006)を鋳型で配列番号6と配列番号7をプライマーとして用いてPCRを行って、取得した。
配列番号6:5’−gcg gga tcc cat gga gat aca cct aca ttg c−3’
配列番号7:5’− acg cag aag cgg tct gat aa −3’
【0048】
その結果、HPV16 E7遺伝子を含んでいる386bpの切片を取得し、前記切片の5'末端にはBamHI制限酵素部位を含んでおり、前記切片の3'末端にはXbaI制限酵素部位を含んでいる。取得したDNA切片をBamHIとXbaI処理により、切断して、306bp切片を得た。
【0049】
pKV−Pald−PgsA−AmylaseをBamHIとXbaIで切断してアミラーゼ遺伝子の部分を除去してベクターの部分を得た。
BamHIとXbaIで切断したE7遺伝子を含有しているDNA断片を同じ制限酵素で切断したベクターと連結してpKV−Pald−PgsA−E7を完成した(図2のA)。
【0050】
[実施例3]pHAT:PgsA−E7とpKV−Pald−PgsA−E7の乳酸菌形質転換体の発現確認
本実施例では実施例2で作製されたpHAT:PgsA−E7とpKV−Pald−PgsA−E7でラクトバチルスカゼイを形質転換させて、前記形質転換された組換えラクトバチルスカゼイを培養してE7タンパク質の発現を確認した。形質転換された組換えラクトバチルスカゼイでpgsAと融合したE7タンパク質の発現の有無を調べた。
【0051】
pHAT:PgsA−E7とpKV−Pald−PgsA−E7で形質転換された組換えラクトバチルスカゼイをMRS培地(Lactobacillus MRS, Becton Dickinson and Company Sparks, USA)、30℃で静置培養してポリガンマグルタミン酸を合成する遺伝子pgsAのC末端と融合したHPV16 E7タンパク質の表面発現を誘導した。
【0052】
前記培養されたラクトバチルスカゼイの全細胞をSDSポリアクリルアミドゲル電気泳動及びpgsAに対する特異抗体を利用したウェスタンブロッティングを行って、前記融合タンパク質の発現を確認した。
【0053】
具体的には、発現が誘導された形質転換された組換えラクトバチルスカゼイ全細胞を同じ細胞濃度で得られたタンパク質で変性させた(denature)試料を準備して、これをSDSポリアクリルアミドゲル電気泳動で分析した後、分画されたタンパク質をPVDF(polyvinylidene-difluoride membranes,Bio−Rad)メンブレンに移した。タンパク質が移されたPVDFメンブレンをブロッキング緩衝溶液(50mMトリス塩酸、5%スキムミルク(skim milk)、pH8.0)で1時間振ってブロッキングさせた後、PgsAに対するウサギ由来のポリクローナル一次抗体をブロッキング緩衝溶液に1000倍に希釈して、1時間反応させた。反応が終わったメンブレンは、緩衝溶液で洗浄してHRPが標識されたウサギに対する二次抗体をブロッキング緩衝溶液に10000倍希釈して、1時間反応させた。反応が終わったメンブレンは、緩衝溶液で洗浄し、洗浄されたメンブレンに基質(lumigen PS−3 acridan、H202)を添加して、約1分間発色させ、CCDカメラでPgsAに対する特異抗体と前記融合タンパク質との間の特異的な結合を確認した(図3)。
【0054】
図3のレーンCはPgsAのみ発現し、形質転換された組換えラクトバチルスカゼイ(pKV−Pald−PgsA)であり、レーン1はpHAT:PgsA−E7で形質転換された組換えラクトバチルスカゼイの発現であり、レーン2はpKV−Pald−PgsA−E7で形質転換された組換えラクトバチルスカゼイのタンパク質発現を示したものである。図3に示されたようにPgsA特異的抗体を用いて、レーン1と2で54.4kDaのPgsA−E7融合タンパク質の発現が確認でき、pHAT:PgsA−E7によるPgsA−E7融合タンパク質の発現と比較するとpKV−Pald−PgsA−E7による融合タンパク質がより強く発現することを確認した。
【0055】
正常なRepEを含んでいるpHAT:PgsA−E7のPslpA−PgsA−E7部分をPald−PgsA−E7で置換した構築体の場合、前記と同じ方式で行った乳酸菌形質転換体製造方法によって、乳酸菌形質転換体を取得できなかった。
【0056】
[実施例4]HPV16 E7抗原を表面発現するラクトバチルスの体液性免疫反応誘導能比較
前記表面発現用ベクターでラクトバチルスカゼイを形質転換させて実施例2と同じ方法で表面発現を誘導した後、2種類の形質転換された組換えラクトバチルスカゼイを用いてPgsAと融合したHPV16 E7の体液性免疫反応誘導を比較した。また、融合タンパク質の発現量が異なる2種類の形質転換体を各々多様な量(1、1/5、1/10)で投与して、免疫反応誘導を比較した。
【0057】
前記ベクターでラクトバチルスカゼイを形質転換させた後、培養して回収した後、凍結乾燥して準備したパウダーを緩衝溶液(PBS、pH7.4)に溶かした後、6週齢の雌Balb/cマウスに経口で投与した。
【0058】
具体的には、HPV16 E7を発現する形質転換された組換えラクトバチルスカゼイ(pHAT−E7、pKV−E7)と、発現しないラクトバチルスカゼイ(pAT)の凍結乾燥パウダーを各々PBSに濃度毎(5×109/200μL、1×109/200μL、5×108/200μL)に溶かした後、一週間に5回ずつ2週間免疫(0〜4days、7〜11days)と1週後に一次ブースティング(21〜25days)、さらに1週後に二次ブースティング(35〜39days)を経口投与した。血清内のHPV16 E7特異的なIgG抗体と粘膜内で分泌されるHPV16 E7特異的なIgA抗体を測定するため、腸内(Intestinal)洗浄液と膣内(Vaginal)洗浄液を14日、28日、及び42日に、各々のマウスからサンプルとして採取した(図4)。血液はヘパリンがコーティングされたマイクロキャピラリーチューブを使って、マウスの目から取得し、1mM PMSFが含まれたPBS洗浄液を使って、腸内と膣内を各々0.8mLと250μLで洗浄した後、13,000rpmで20分間遠心分離して、上澄液を分離してELISAを行った。
【0059】
ELISAはHPV16 E7タンパク質をウェル当たり各々500ng/100μLでコーティング緩衝溶液(0.1M sodium carbonium、0.02% sodium azide、pH9.6)を使って希釈した後、96ウェルELISAプレートに100μLずつ分株し、4℃で一晩中反応させた。PBST(PBS+0.05%Tween20)を300uL/ウェルずつ添加しながら、3回洗浄した後、5%スキムミルク100μLを入れて37℃で1時間ブロッキングさせ、ブロッキング緩衝溶液を除去した後、PBSTで3回洗浄して希釈させた血清と粘膜洗浄液を各々100μLずつ添加した。検体を添加した後、37℃で2時間反応させた後、洗浄溶液で3回洗浄してからセイヨウワサビペルオキシダーゼ標識抗マウスIgGまたはIgA抗体を1:5000で希釈してウェルに添加後、37℃で1時間反応させてから洗浄溶液で洗浄した後、TMB発色溶液で発色させて、2.5M硫酸で発色を固定させた。450nmで、ELISAリーダーで測定してHPV16 E7特異抗体の量を分析した。
【0060】
その結果、図5のAに示されたように最初2週間免疫した後のE7特異的血清IgGの平均OD値を測定した結果、pAT−E7とpKV−E7投与群がpAT対照群に比べて高いOD値を示した。また、投与した量に伴ってE7特異的抗体が増加することを確認した。一次ブースティング後のE7特異的血清IgGの平均OD値は、pAT−E7とpKV−E7投与群共に最初2週間免疫に比べて増加したOD値を示しブースティング効果を見せた。二次ブースティング後のE7特異的血清IgGの平均OD値も二つの投与群でブースティング効果を示し、特にpAT−E7に比べてE7融合タンパク質の表面発現量がずっと高いpKV−E7投与群の場合、1/5投与群とpAT−E7 1/1投与群が似たようなIgGブースティング効果を示した。
【0061】
さらに、粘膜免疫反応を測定するために腸内と膣内でのE7特異的IgA抗体をELISAで測定した結果、図5のBと図5のCに示されたように最初2週間免疫した後の平均OD値は、pAT対照群に比べてpAT−E7、pKV−E7投与群共にE7特異的IgAが増加することを確認し、また投与した量に伴ってE7特異的抗体が増加することを確認した。一次ブースティング後の平均OD値はより一層増加し、二次ブースティング効果は一次ブースティングに比べて、若干効果を見せた。
【0062】
本発明によるHPV16 E7を発現する形質転換された組換えラクトバチルスカゼイを経口投与する場合、HPV16 E7特異的な体液性免疫(血清IgG誘導)と粘膜免疫(Interstinal/Vaginal IgA誘導)を誘導できたことを示し、特にE7をより多く発現するpKV−E7投与群がpAT−E7投与群よりも効果的に免疫反応を誘導できたことが示唆された。
【0063】
[実施例5]HPV16 E7抗原を表面発現するラクトバチルスの細胞性免疫反応誘導能比較
HPV 16 E7抗原の表面発現量が異なる2種類の形質転換された組換えラクトバチルスカゼイ(pHAT−E7、pKV−E7)を経口投与したマウスからE7抗原に特異的なCD8+ T細胞媒介免疫反応誘導とT細胞免疫反応の誘導に当り差があるか確認しようと各々intracellular cytokine染色とIFN−γELISPOTを行って、細胞性免疫誘導能を調べた。
【0064】
実施例4で説明したようにE7を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与したマウスから脾臓細胞を分離した。5×106cellの脾臓細胞を10%FBSが含まれたRPMI−1640培地に浮遊して、24ウェルプレートに分株した。各ウェルにMHC class Iエピトープを含むE7ペプチド(a.a 49−57、AniGen、Korea)とGolgiPlug(BD Biosciences)を処理し、37℃で16時間培養した。培養された細胞はフィコエリスリン(PE)標識モノクローナルラット抗マウスCD8抗体で、4℃で40分間染色し、Cytofix/Cytoperm kitを利用して、細胞内部のIFN−γをFITC標識モノクローナルラット抗マウスIFN−γ抗体で染色した。
【0065】
二つの組換えラクトバチルスカゼイを経口投与した後、intracellular cytokine染色を行ってE7特異的にIFN−γを分泌するCD8+ T細胞の数を測定した結果、図6に示されたように、E7を発現する形質転換された組換えラクトバチルスカゼイによるE7特異的なIFN−γ分泌CD8+ T細胞前駆体の数が増加したことを確認することができた。特に、免疫反応を誘導した初期には表面発現量が多いpKV−E7で形質転換された組換えラクトバチルスカゼイによる細胞媒介性免疫反応がより増加することが確認されたが、ブースティングに伴いpHAT−E7乳酸菌による細胞媒介性免疫反応も似たレベルで増加した。
【0066】
E7特異的なT細胞免疫反応誘導を調べるためE7ペプチドによる刺激に特異的にIFN−γを分泌する全T細胞数を測定するためにIFN−γELISPOTを行った。
脾臓細胞を分離する一日前に、プレートに抗マウスIFN−g捕捉抗体をコーティングし、実験当日分離した脾臓細胞はウェル当たり2×105cellに分株した。細胞が分株されたウェルにE7ペプチドまたはPHA−M、mediaを処理して2日間培養した。2日後、プレートの内容物を除去して蒸溜水で2回、0.05%tween−20が含まれたPBSで3回洗浄し、ビオチン化抗マウス IFN−gを分株し常温で2時間反応させた。反応後、内容物を除去して0.05%tween−20が含まれたPBSで3回洗浄後、ストレプトアビジンHRPを分株し常温で一時間反応させた。内容物を除去して0.05%tween−20が含まれたPBSで4回洗浄してPBSで2回洗浄した後、基質と反応させて、発色を確認しながら5〜60分間観察し、蒸溜水で反応を停止させ、ELISPOTリーダーを用いて、各ウェルのスポット数を測定した。
【0067】
図6に示されたようにpHAT−E7またはpKV−E7で形質転換された組換え乳酸菌を経口投与したマウスの場合、E7ペプチドの刺激に対するIFN−γ分泌細胞数が増加することを確認でき、pHAT−E7で形質転換された組換えラクトバチルスカゼイを投与した群よりpKV−E7で形質転換された組換えラクトバチルスカゼイを投与した群でより多くの数のIFN−γ分泌細胞数の分布を確認することができた。
【0068】
[実施例6]HPV E7抗原を表面発現するラクトバチルスで免疫したマウスの腫瘍細胞チャレンジ
HPV 16 E7抗原の表面発現量が異なる2種類の形質転換された組換えラクトバチルス(pHAT−E7、pKV−E7)の抗癌効果と投与量に伴う抗癌効果を確認するためE7タンパク質を発現するTC−1腫瘍細胞モデルを利用して、腫瘍細胞チャレンジを行った。
【0069】
先ず、6〜8週齢の雌C57BL/6マウスを各集団毎に5匹ずつ準備してE7を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与した。経口投与は、週5回行い、最初1週間投与後、1週間の休止期間を設け、さらに1週間と最初のブースティング後、再び1週間の休止期間を設けた後1週間と二回目のブースティングを行った。最初1週間の経口投与後、2×104cellのTC−1腫瘍細胞を左の上腿内側に皮下注射(s.c.)で癌を誘導した。形成された癌の大きさはキャリパーを利用して、週に3回測定した。
【0070】
図7に示されたように、pHAT−E7またはpKV−E7で形質転換された組換えラクトバチルスカゼイを経口投与した集団で対照群のラクトバチルスカゼイを経口投与した集団に比べて、腫瘍細胞の大きさが50%程度で小さく形成されることを確認することができた。また、pKV−E7で形質転換された組換えラクトバチルスカゼイの量を1/5に減らして投与した集団の場合も対照群集団に比べて、腫瘍細胞の大きさが小さかった。
【0071】
[実施例7]HPV type16 E7関連部位アミノ酸の変異導入タンパク質表面発現免疫乳酸菌(pKV−Pald−PgsA−E7(Rb))の製造
パピローマウィルスが細胞に感染する場合発現したE7タンパク質とpRb(Retinoblastoma Growth Suppressor Protein)タンパク質が結合して、癌を誘発する可能性があるため、E7遺伝子のpRbタンパク質と結合する部分の塩基配列に変異を誘発した。このように変異が誘発されたE7(Rb)遺伝子を大腸菌と乳酸菌で安定的に発現するpKVシャトルベクターを用いてPgsA末端に融合させて、乳酸菌で表面発現できるベクターpKV−Pald−PgsA−E7(Rb)を製造した。
【0072】
HPV type16 E7内のRb結合領域を調べた結果、pRb結合領域(DLxCxE)は、HPV16 E7遺伝子の21番目アミノ酸から26番目アミノ酸と知られている(Smahel M. et al., Virology, 281:231, 2001)。その中で21番目(D)、24番目(C)、26番目(E)のアミノ酸を共にグリシンで置換させるため、プライマーを使ったpoint mutagenesis方法によってE7タンパク質のpRbタンパク質との結合の可能性を無くした。実施例2で取得したpKV−Pald−PgsA−E7ベクターを鋳型として配列番号8と9、配列番号10と11を使って、各々PCRを行った。
配列番号8:5’−tct gga tcc atg cat gga gat aca cct ac−3’
配列番号9:5’−ttg ccc ata acc gta gag acc agt tgt c−3’
配列番号10:5’−act ggt ctc tac ggt tat ggg caa tta aat g−3’
配列番号11:5’−cat tct aga tca tta tgg ttt ctg aga aca g−3’
【0073】
その結果、配列番号8及び9のプライマーによってHPV16 E7遺伝子を含んでいる87bpの切片を取得し、DNA切片の5'末端にはBamHI制限酵素部位を含んでおり、配列番号10及び11のプライマーによって取得した変異された249bp DNA切片の3'末端にはXbaI制限酵素部位を含んでいた。二つの取得したDNA切片をミックスして、95℃−30秒、42℃−30秒、72℃−30秒の条件で10回PCRを行った後、配列番号8及び11のプライマーを添加してPCR(95℃−30秒、53℃−30秒、72℃−30秒)を行った。取得した312bp DNA切片の5'末端にはBamHI制限酵素部位が、3'末端にはXbaI制限酵素部位が含まれていた。この切片をBamHIとXbaI処理し、切断して、306bpの変異されたE7(Rb)遺伝子切片を取得した。pKV−Pald−PgsA−AmylaseをBamHIとXbaIで切断してアミラーゼ遺伝子の部分を除去してベクターの部分を取得した。BamHIとXbaIで切断された変異E7(Rb)遺伝子を含有しているDNA断片を同じ制限酵素で切断したベクターと連結して、pKV−Pald−PgsA−E7(Rb)を完成した(図8)。
構築された前記構築体をラクトバチルスカゼイにエレクトロポレーション法を利用して、形質転換し、構築体が形質転換された乳酸菌形質転換体を取得した。
【0074】
[実施例8]pKV−Pald−PgsA−E7(Rb)乳酸菌形質転換体の発現確認
取得した乳酸菌表面発現用ベクターpKV−Pald−PgsA−E7(Rb)で形質転換された組換えラクトバチルスカゼイからPgsAと融合したE7私のRb結合部位変異タンパク質の発現の有無を確認した。
【0075】
形質転換された組換えラクトバチルスカゼイをMRS培地(Lactobacillus MRS, Becton Dickinson and Company Sparks, USA)、30℃で静置培養を行い、菌体を回収して、実施例3のような方法でウェスタンブロッティングを行って、発現されたことを確認した。PgsA特異抗体とHPV16 E7特異抗体(E7 monoclonal Ab, Invitrogen, USA)を用いてウェスタンブロッティングを行った結果、図9に示されたように約54.7kDaのPgsAと融合した変異E7タンパク質の発現を確認した。
【0076】
[実施例9]変異されたHPV16 E7(Rb)抗原を表面発現するラクトバチルスの体液性免疫反応誘導
PgsAと融合した変異E7タンパク質の発現が確認された形質転換された組換えラクトバチルスカゼイをマウスに経口投与し、体液性免疫反応が誘導されるか確認した。前記ベクターをラクトバチルスカゼイに形質転換させた後、培養して回収した後、凍結乾燥して準備したパウダーを緩衝溶液(PBS、pH7.4)に溶かした後、6週齢の雌Balb/cマウスに経口投与した。
【0077】
具体的には、HPV16 E7(Rb)を発現する形質転換された組換えラクトバチルスカゼイ(E7−Rb)と、発現しないラクトバチルスカゼイ(L525)の凍結乾燥パウダーを各々PBSに2〜5×109cell/200μLで溶かした後、週に5回ずつ2週間免疫(0〜4days、7〜11days)して1週後に一次ブースティング(21〜25days)、さらに1週間後に二次ブースティング(35〜39days)を経口投与した。血清内のHPV16 E7特異的なIgG抗体と、粘膜内で分泌されるHPV16 E7特異的なIgA抗体を測定するため、腸内洗浄液と膣内洗浄液を図3に示されたように14日、28日、そして42日に各々マウスからサンプルを採取した。実施例4同様に血液はヘパリンがコーティングされたマイクロキャピラリーチューブを用いて、マウスの目から得られ、1mM PMSFが含まれたPBS洗浄液を用いて、腸内と膣内を各々0.8 mLと250μLで洗浄した後、13,000rpmで20分間遠心分離し、上澄液を分離して、実施例4と同じ方法でELISAを行った。
【0078】
その結果、図10のAに示されたように最初2週間免疫した後のE7特異的血清IgGの平均OD値を測定したところ、E7−Rb投与群がL525(−control)投与群に比べて高いOD値を示した。一次ブースティング後のE7特異的血清IgGの平均OD値は、E7−Rb投与群で最初2週間免疫に比べて増加したOD値を示しており、ブースティング効果が示された。二次ブースティング後のE7特異的血清IgGの平均OD値を見てもブースティング効果が示された。しかし、L525投与群では特別なブースティング効果が見られなかった。また、粘膜で誘導される体液性免疫反応を測定するために腸内と膣内でのE7特異的IgA抗体をELISAで測定した結果、図10のBと図10のCに示されたように最初2週間免疫した後の平均OD値は、L525(control)投与群に比べてE7−Rb投与群が、E7特異的IgAが増加することを確認し、一次ブースティング後の平均OD値は、より一層増加し、二次ブースティング効果は一次ブースティングに比べて増加した効果が見られた。
【0079】
以上の結果から確認されたように、E7−Rb形質転換された組換えラクトバチルスカゼイを経口投与した場合、HPV16 E7特異的な全身性液性免疫反応(systemic humoral immune response)(血清IgG誘導)と粘膜液性免疫反応(mucosal humoral immune response)(Interstinal/Vaginal IgA誘導)を共に誘導できることが示された。
【0080】
[実施例10]変異されたHPV16 E7(Rb)抗原を表面発現するラクトバチルスの細胞性免疫反応誘導
変異されたHPV 16 E7(Rb)抗原を表面発現する形質転換された組換えラクトバチルス(E7−Rb)が、E7(Rb)抗原に特異的なT細胞媒介免疫反応誘導を誘導する能力有しているかを調べるために、IFN−γELISPOTを行い、細胞性免疫誘導能を調べた。
【0081】
実施例7で作製したE7(Rb)を表面発現している形質転換された組換えラクトバチルスカゼイを経口投与したマウスで脾臓細胞を分離した。5×106cellの脾臓細胞を10%FBSが含まれたRPMI−1640培地に浮遊して、24ウェルプレートに分株した。各ウェルにMHC class Iエピトープを含むE7ペプチド(a.a 49−57、AniGen、Korea)とGolgiPlug(BD Biosciences)処理して、37℃で16時間培養した。培養された細胞は、フィコエリスリン(PE)標識モノクローナルラット抗マウスCD8抗体で、4℃で40分間染色し、Cytofix/Cytoperm kitを利用して、細胞内部のIFN−γをFITC標識モノクローナルラット抗マウスIFN−γ抗体で染色した。
【0082】
二種類の組換えラクトバチルスカゼイを経口投与した後、intracellular cytokine染色を行って、E7(Rb)特異的にIFN−γを分泌するCD8+ T細胞の数を測定した結果、図11に示されたように、E7(Rb)を発現する形質転換された組換えラクトバチルスカゼイによるE7(Rb)特異的なIFN−γ分泌CD8+ T細胞前駆体の数が増加したことを確認することができた。特に、免疫反応を誘導した初期には表面発現量が多いpKV−E7(Rb)乳酸菌による細胞媒介性免疫反応がより増加することが確認されたが、ブースティングに伴ってpHAT−E7(Rb)乳酸菌による細胞媒介性免疫反応も似たレベルに増加した。
【0083】
E7(Rb)特異的なT細胞免疫反応誘導を調べるため、E7ペプチドによる刺激に特異的にIFN−γを分泌する全T細胞の数を測定するため、IFN−γELISPOTを行った。
【0084】
脾臓細胞を分離する一日前にプレートに抗マウスIFN−g捕捉抗体をコーティングし、実験当日に分離した脾臓細胞は、ウェル当たり2×105cellに分株した。細胞が分株されたウェルにE7ペプチドまたはPHA−M、media処理して2日間培養した。2日後、プレートの内容物を除去して蒸溜水で2回、0.05%tween−20が含まれたPBSで3回洗浄し、ビオチン化抗マウスIFN−gを分株し常温で2時間反応させた。反応後、内容物を除去して0.05%tween−20が含まれたPBSで3回洗浄後、ストレプトアビジンHRPを分株し常温で一時間反応させた。内容物を除去して0.05%tween−20が含まれたPBSで4回洗浄してPBSで2回洗浄した後、基質と反応させて、発色を確認しながら、5〜60分間観察し、蒸溜水で反応を停止させ、ELISPOTリーダーを用いて、各ウェルのスポット数を測定した。
【0085】
図11に示されたようにpHAT−E7(Rb)またはpKV−E7(Rb)で形質転換された組換えラクトバチルスカゼイを経口投与したマウスの場合、E7ペプチドの刺激に対するIFN−γ分泌細胞数が増加することが確認でき、pHAT−E7(Rb)で形質転換された組換えラクトバチルスカゼイを投与した群よりpKV−E7(Rb)で形質転換された組換えラクトバチルスカゼイを投与した群の方がより多くの数のIFN−γ分泌細胞数の分布を確認することができた。
【0086】
以上、本発明の内容の特定の部分を詳述したが、当業界における通常の知識を持った者にとって、このような具体的な記述は単なる好適な実施態様に過ぎず、これにより本発明の範囲が制限されることはないという点は明らかである。よって、本発明の実質的な範囲は特許請求の範囲とこれらの等価物により定義されると言える。
【産業上の利用可能性】
【0087】
本発明に係るヒトパピローマウィルス抗原タンパク質の表面発現用ベクターで形質転換された組換え乳酸菌及びこれを主要成分として含んだ組成物は、子宮頸癌治療用ワクチンとして活用でき、構成的にHPV抗原を高発現する形質転換された組換え菌株を経済的に大量増殖させて、経口ワクチンとしてまたは膣部位に直接適用することができるため、非常に経済的な効果がある。
【特許請求の範囲】
【請求項1】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、プロモーター、表面発明のためのポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項2】
前記ヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質がHPVのE7またはE7(Rb)である、請求項1に記載のベクター。
【請求項3】
前記プロモーターが、乳酸菌由来のアルドラーゼプロモーターである、請求項1に記載のベクター。
【請求項4】
前記ポリガンマグルタミン酸合成酵素複合体遺伝子がpgsB、pgsC及びpgsAからなる群より選択される、請求項1に記載のベクター。
【請求項5】
請求項1に記載のベクターで形質転換された組換え微生物。
【請求項6】
前記微生物が、乳酸菌である、請求項5に記載の微生物。
【請求項7】
請求項5または請求項6に記載の形質転換された組換え微生物を有効成分として含有する子宮頸癌治療用ワクチン。
【請求項8】
請求項5に記載の組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法。
【請求項9】
請求項8に記載の方法で製造されたHPV抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチン。
【請求項10】
前記ワクチンが、経口用である、請求項7または請求項9に記載の子宮頸癌治療用ワクチン。
【請求項11】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAからなる群より選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項12】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAからなる群より選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7(Rb)をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項13】
請求項11に記載のベクターで形質転換された組換え乳酸菌。
【請求項14】
請求項12に記載のベクターで形質転換された組換え乳酸菌。
【請求項1】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、プロモーター、表面発明のためのポリガンマグルタミン酸合成酵素複合体遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項2】
前記ヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質がHPVのE7またはE7(Rb)である、請求項1に記載のベクター。
【請求項3】
前記プロモーターが、乳酸菌由来のアルドラーゼプロモーターである、請求項1に記載のベクター。
【請求項4】
前記ポリガンマグルタミン酸合成酵素複合体遺伝子がpgsB、pgsC及びpgsAからなる群より選択される、請求項1に記載のベクター。
【請求項5】
請求項1に記載のベクターで形質転換された組換え微生物。
【請求項6】
前記微生物が、乳酸菌である、請求項5に記載の微生物。
【請求項7】
請求項5または請求項6に記載の形質転換された組換え微生物を有効成分として含有する子宮頸癌治療用ワクチン。
【請求項8】
請求項5に記載の組換え微生物を培養してHPV抗原を表面発現させる工程及び前記HPV抗原が表面発現された組換え微生物を回収する工程を含むHPV抗原が表面発現された微生物の製造方法。
【請求項9】
請求項8に記載の方法で製造されたHPV抗原が表面発現された微生物を有効性分として含有する子宮頸癌治療用ワクチン。
【請求項10】
前記ワクチンが、経口用である、請求項7または請求項9に記載の子宮頸癌治療用ワクチン。
【請求項11】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAからなる群より選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項12】
配列番号1のアミノ酸配列を持つrepE変異タンパク質をコードする遺伝子、乳酸菌由来アルドラーゼプロモーター、表面発現のためのポリガンマグルタミン酸合成酵素複合体遺伝子pgsB、pgsC及びpgsAからなる群より選択されるポリガンマグルタミン酸遺伝子及び前記ポリガンマグルタミン酸合成酵素複合体遺伝子と連結されるヒトパピローマウィルスの腫瘍誘発関連抗原タンパク質E7(Rb)をコードする遺伝子を含有するHPVワクチン製造用表面発現ベクター。
【請求項13】
請求項11に記載のベクターで形質転換された組換え乳酸菌。
【請求項14】
請求項12に記載のベクターで形質転換された組換え乳酸菌。
【図1】

【図2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図10】
【図11】
【図3】
【図9】
【図2】
【図4】
【図5】
【図6】
【図7】
【図8】
【図10】
【図11】
【図3】
【図9】
【公表番号】特表2012−514469(P2012−514469A)
【公表日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願番号】特願2011−545296(P2011−545296)
【出願日】平成22年1月8日(2010.1.8)
【国際出願番号】PCT/KR2010/000126
【国際公開番号】WO2010/079991
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(506010208)バイオリーダーズ コーポレーション (16)
【氏名又は名称原語表記】BIOLEADERS CORPORATION
【出願人】(511166998)ククミン ユニバーシティ インダストリー−アカデミック コオペレーション ファウンデーション (3)
【出願人】(506272301)コリア リサーチ インスティテュート オブ バイオサイエンス アンド バイオテクノロジー (17)
【Fターム(参考)】
【公表日】平成24年6月28日(2012.6.28)
【国際特許分類】
【出願日】平成22年1月8日(2010.1.8)
【国際出願番号】PCT/KR2010/000126
【国際公開番号】WO2010/079991
【国際公開日】平成22年7月15日(2010.7.15)
【出願人】(506010208)バイオリーダーズ コーポレーション (16)
【氏名又は名称原語表記】BIOLEADERS CORPORATION
【出願人】(511166998)ククミン ユニバーシティ インダストリー−アカデミック コオペレーション ファウンデーション (3)
【出願人】(506272301)コリア リサーチ インスティテュート オブ バイオサイエンス アンド バイオテクノロジー (17)
【Fターム(参考)】
[ Back to top ]