
完全長cDNAライブラリーの作成方法
【課題】 効率良く5'Cap サイトの標識が可能な新たな方法とこの標識方法を用いた完全長cDNAライブラリーの作成方法の提供。
【解決手段】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含む完全長cDNAライブラリーの作成方法。mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する。
【解決手段】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含む完全長cDNAライブラリーの作成方法。mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する。
【発明の詳細な説明】
【0001】
【発明の属する技術分野】本発明は、完全長cDNAライブラリー作成法に関する。更に詳しくは、mRNAの化学修飾を利用した、完全長cDNA精製法による完全長cDNAライブラリー作成法に関する。
【0002】
【従来の技術】cDNA合成法は医学生物学分野の研究で必須の技術であり、遺伝子転写物の解析法として不可欠である。全てのDNA 遺伝情報は転写物を介して生理活性を示すが、それを解析する強力な手段がcDNAクローニングである。従来法におけるcDNA合成においては、oligo dTをプライマーとしてポリA サイトから合成されたcDNAライブラリーの中で最終的にクローンが単離される。ところが、殆どの場合、転写単位の全長が合成されていない為に転写単位の全構造が解析できないのが実状である。この為、通常のcDNAライブラリーでは、プライマー伸長法による5'上流領域の合成、またはランダム プライマーを用いたcDNA合成を用いて5'上流領域をウォーキング(walking) することが全長構造解明に必須のステップとなる。
【0003】しかるに、上記従来のcDNA合成法には次の問題がある。
(1) ランダム プライマーを用いると転写物のかなりの領域をカバーするcDNAができる。しかし、それらは短い断片であり、ポリ Aから5'Cap サイトまで含んだクローンは単離できない。
(2) oligo dTをプライマーとして用いたcDNAは全て3'端を含む。しかし、逆転写酵素が5'Cap サイトまで届かないため、5'上流をプライマー伸長法及び5'RACE等で再度単離解析せねばならない。
(3) 既存の完全長cDNAを単離する方法は、前述の従来のいかなる方法もその効率が十分でない(100μg mRNAから200 万組換え体ファージ)。そこで、実用的にはもっと効率の高い手法の開発が望まれる。
【0004】完全長cDNA合成法の従来の技術として、次のような方法が挙げられる。5'Capサイトの標識法として、酵母またはHela細胞のCap 結合蛋白を用いる方法(I. Edery et al., "An Efficient Strategy To Isolate Full-Length cDNAs Based onan mRNA Cap Retention Procedure (CAPture)", MCB, 15, 3363-3371, 1995)、また5'Cap のない不完全cDNAをアルカリフォスファターゼによりリン酸を除去し、その後タバコモザイクウィルスの脱キャップ酵素を反応させ、完全長cDNAのみリン酸が露出することを利用した方法(K. Maruyama et al., "Oligo-capping: asimple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides", Gene, 138, 171-174, 1995., S. Kato et al., "Construction of a human full-length cDNA bank", Gene, 150, 243-250, 1995) 、等々が挙げられる。
【0005】これらの従来法の完全長cDNA合成法の効率が十分でない理由として次の項目が挙げられる。
■5'Cap サイトの認識が、アデノウィルスCap 結合蛋白やタバコモザイクウィルスの脱キャップ酵素の様に蛋白酵素の反応に依存している為、完全長cDNA(RNA)の選択の段階で高い効率が期待できない。
■逆転写酵素がcDNAの第1鎖を合成する際、5'Cap サイトまで合成鎖が伸長しない。
■第1鎖が合成された後の第2鎖合成のプライマー配列の付加、第2鎖の合成効率、2重鎖cDNAのクローニング効率の問題。
以上の様に多段階にわたるcDNA合成ライブラリー作成においては、■〜■の各ステップがそれぞれ問題となる。
【0006】
【発明が解決しようとする課題】そこで本発明は、第1に、完全長cDNAの単離を目的とする従来のキャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識が可能な新たな方法を提供すること、第2に、この新たな5'Cap サイトの標識方法を用いた完全長cDNAライブラリーの作成方法を提供することを目的とする。
【0007】
【課題を解決するための手段】本発明は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法に関する。
【0008】
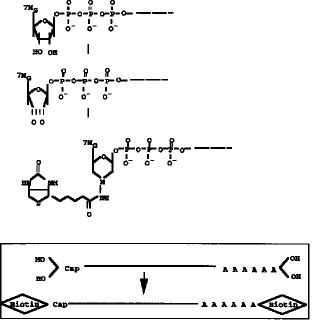
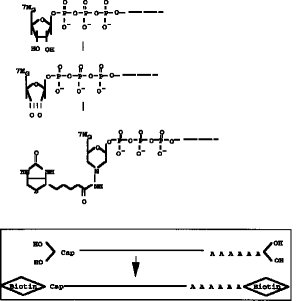
【発明の実施の態様】本発明の方法では、5'Cap サイトの認識を高め、完全長cDNA(RNA) の選択の段階の効率を高めるために、5'Cap サイトに特異的構造であるジオール構造を利用して5'Cap サイトの標識を化学合成法により行う(図1参照) 。即ち、本発明の方法では、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる。このタッグ分子は5'Cap サイトに化学的に結合しており、タッグ分子で標識されたmRNAを鋳型として完全長cDNAを合成し、完全長cDNAライブラリーを作成する。
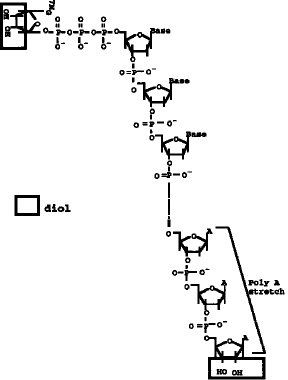
【0009】タッグ分子のmRNAの5'Cap サイトへの結合は、図2に示すように、例えば、5'Cap サイトのジオール構造を酸化剤、例えば、過ヨウ素酸ナトリウム(NaIO4) 等で酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。上記ヒドラジン末端を有するタッグ分子としては、例えば、ヒドラジン末端を有するビオチン分子やアビジン分子を挙げることができる。また、タッグ分子として抗原や抗体等の反応特異性を有する分子を用いることもできる。タッグ分子として用いる特異標識物には特に制限はない。
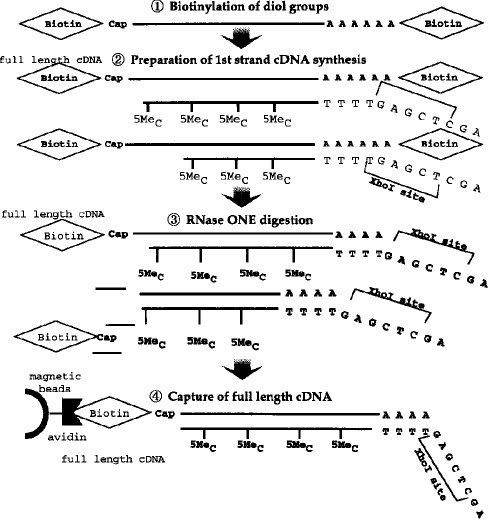
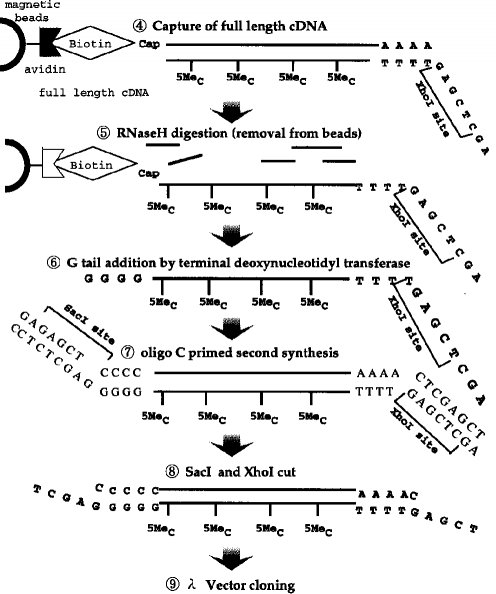
【0010】タッグ分子の結合■から完全長cDNAのクローニング■までの工程の一例(タッグ分子:ビオチン)が図3〜4に示されている。
■ジオール基のビオチン化■第1のcDNA鎖の合成■リボヌクレアーゼI(RNase I) 消化■完全長cDNA複合体の捕獲(アビジンビーズ使用)
■RNase H 消化(アビジンビーズからの分離)
■ターミナル デオキシヌクレオチジル 転写酵素によるG末端の付加■オリゴCでプライマーした第2鎖合成■SacI及びXhoIでの切断■λベクターによるクローニング
【0011】タッグ分子を結合して標識したmRNAは、これを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する。oligo dTをプライマーとする逆転写法によるRNA-DNA 複合体の形成は、常法により行うことができる。
【0012】さらに、形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する。具体的には、1本鎖RNA を切断するRNA 分解酵素でRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体(5'Cap まで伸長した完全長cDNA)をタッグ分子の結合性を利用して分離する。例えば、タッグ分子がビオチン分子である場合、RNA-DNA 複合体がタッグ分子として有するビオチン分子を、固相持体上に担持したアビジンと結合させて、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がアビジン分子である場合、RNA-DNA 複合体がタッグ分子として有するアビジン分子を、固相持体上に担持したビオチンと結合させてmRNAの完全長に対応するDNA を有する複合体を分離することもできる。
【0013】即ち、本発明の1つの態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、前記ビオチン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程ビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0014】また、本発明の別の態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、前記アビジン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程アビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0015】前記1本鎖RNA を切断するRNA 分解酵素としては、例えば、リボヌクレアーゼIを挙げることができる。尚、RNA-DNA 複合体からmRNAの完全長に対応するDNAを有する複合体を選別する方法として、1本鎖RNA を切断するRNA 分解酵素を用いる方法以外の方法を利用することもできる。複合体を選別する方法にも特に制限はない。
【0016】さらに本発明の方法では、分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する。cDNAの回収は、例えば、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行うことができる。また、cDNAの回収は、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドを切断するRNase としては、例えば、RNase H を挙げることができる。
【0017】回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングすることにより、完全長cDNAライブラリーを得ることができる。第2のcDNA鎖の合成は、例えば、第1のcDNA鎖の3'端にRNA またはDNAのオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして行なうことができる。あるいは、第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うこともできる。即ち、単離された完全長第1鎖cDNAより第2鎖を合成する為に、ターミナルデオキシヌクレオチジルトランスフェラーゼによるホモポリマー法(homopolymer法) やRNA リガーゼによる1本鎖プライマーを、第1鎖cDNA3'端または5'Cap を除去されたmRNAの5'鎖に付加しポリメラーゼで伸長方法等を用いることができ、第2鎖を合成する方法も特に制限はない。
【0018】
【発明の効果】本発明によれば、mRNAの5'Cap サイトを化学的に修飾を加えることにより、効率良く完全長cDNAを選択できる。この利点は、修飾が5'Cap サイトを認識する為の修飾が酵素反応に全く依存せず、mRNAの5'Cap サイト構造に特徴的なdiol残基を利用した化学反応に依存する為、バックグラウンドが低くなおかつ非常に高い効率を得るものである。本発明の方法では、完全長cDANの回収を特異的選択性の高い RNase ONE処理ビオチン−アビジン反応等を利用した固相化系で行うことができるため、量産的ロボティックスによるライブラリー生産をも可能にする。
【0019】
【実施例】本実施例は図3〜4にアウトラインを示す工程からなる。本法は次の9つの工程よりなる。
■ジオール基のビオチン化■第1のcDNA鎖の合成■リボヌクレアーゼI(RNase I) 消化■完全長cDNA複合体の捕獲(アビジンビーズ使用)
■RNase H 消化(アビジンビーズからの分離)
■ターミナル デオキシヌクレオチジル 転写酵素によるG末端の付加■オリゴCでフライマした第2鎖合成■SacI及びXhoIでの切断■λベクターによるクローニング
【0020】RNA 調製脳0.5 〜1gの組織片を10mlの懸濁液D でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2ml の5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の 7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0021】RNA のジオール部位へのビオチンの結合(図2ステップ■)RNA のジオール部位(1方はCAP 、他方はRNA の3'端)にビオチンを結合させる為に、2段階の反応を行った。即ち、ジオール基の酸化とそれに続くビオチンヒドラジド(Sigma社) と酸化RNA 体のカップリング反応である。まず、10〜20μg のmRNAを、pH4.5 の66mM酢酸ナトリウム緩衝液と酸化剤としての過ヨウ素酸ナトリウムを含む50μl 中の溶液で処理する。この酸化反応は遮光条件の元、氷の上で45分間行う。続いて、5 μl の5Mリチウムクロライド、1μl の10%SDS 、同量のイソプロパノールを加え、-20 ℃で30分間インキュベーションした後、4 ℃で15分間15000rpm遠心機にかけ沈澱させる。RNA の沈澱物を70% エタノールで洗い、RNase-freeの水50μl に再溶解させる。その試量にpH6.1 の1M酸化ナトリウム5 μl 、10%SDS 5 μl 、更に10mM Biocytin Hydrazide(水溶液)150μl を加える。試料は室温(22-26℃) で終夜インキュベーションし、最後に5M NaCl 5 μl 、pH6.1 の1M酢酸ナトリウム7.5 μl 、および2.5 倍量のエタノールを加え、氷の上で1時間インキュベーションした後、4 ℃で15分間遠心し、ビオチン化したRNA を再沈澱させる。RNA 沈澱物を70% エタノールで1回洗い、更に80% エタノールで洗った。最後にRNase-freeの水に再溶解したら、第1の cDNA 鎖の調製の材料として用いる。
【0022】第1の cDNA 鎖の調製(図2ステップ■) 10μg のビオチン化mRNAを使ってSuperscript II (Gibco BRL)の2000unitにより、0.5mM 5-methyl-dCTP 、1mM dATP、1mM dTTP、1mM dGTPの存在化で、100 μl バッファー(50mM Tris-HCl, 75mM KCl, 3mM MgCl2, 10mM DTT)の中で逆転写反応を行なった。プライマーとして、5 μg のオリゴヌクレオチド 3'NMTTTTTTTTTTTTGAGCTCTGAATCAAGAGAGAGAGAGAGAGAGAG5'(N:どの核酸塩基でも可、M:G かAかC )を用いた。反応は42℃、45分間行い、続いて反応液を50℃で20分インキュベーションした。この反応を始める際、20μl の反応液を採取し、それに1 μlの[ α-32P]-dGTP(3000Ci/mmol、10μCi/ μl 、Amersham) を加えることにより、第1の cDNA 鎖の合成効率を測定した。RI標識した20μl の反応液中0.5 μlをDE-81 ペーパー上にスポットし、pH7.0 の0.5M−リン酸ナトリウムで3回洗った前後のRI活性を測定し計算した。
【0023】完全長cDNAのRNase 保護(図2ステップ■) どのような塩基の箇所でも1本鎖RNA を消化することが可能なRNase ONETTM(Promega社) で処理することにより、逆転写によって完全にcDNAが伸長されなかったmRNA、およびmRNAの3'末端に標識されたビオチン残基を取り除いた。具体的には第1鎖 cDNA 合成の際、RI標識された反応液20μl と非標識の80μl 分を一緒にプールした後、40μl のRNase I バッファー、355 μl の水、そして50unitのRNase I を使って試料を30分間30℃でインキュベーションした。
【0024】完全長cDNAの採取(図2ステップ■■) アビジンコートしたマグネティックビーズへの非特異的吸着を防止する為、2.5mg の酵母tRNA(DNase Iで前処理した) を加え、500 μl に調製し1時間氷上でインキュベーションした。RNase I で処理したcDNAを上記の前処理されたビーズに加え、pH8.0 の0.25M-EDTA、0.5M-NaCl を含むバッファー中、磁気を帯びたビーズが沈澱しないよう、15分間室温で時折振り混ぜながらインキュベーションした。その後ビーズを pH8.0 0.5M-EDTAで4回、0.4 %SDS で1回、その後nuclease-free の水で3回洗浄する。試料を100 μl のRNaseHバッファーの中で37℃で30分間2 unitのRNaseHで処理後、0.1%のSDS と共にビーズをインキュベーションすることによりビーズから完全長cDNAを分離した。不完全なRNaseH処理の為ビーズから離れないcDNAに関しては、更にpH9.0 、65℃で10分間 Tris-Formate バッファーの中でアルカリ加水分解を行うことにより回収できる。回収された完全長1本鎖cDNAはフェノール/クロロホルムで一度抽出され、G25/G50 sephadexクロマトグラフィーに付された。RI活性を持ったフラクションは表面をシリコン処理したエッペンドルフチューブに集めて、試料を真空で引くことによって10μl にまで減じた。
【0025】1本鎖cDNAのoligo dG テイリング(図2ステップ■■) 上記により回収された1本鎖cDNAに oligo dG を付加する為、pH6.9 の200mM-NaCacodylate、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μMdGTP の50μl バッファー下で37℃30分間32unitのターミナル デオキシヌクレオチジル トランスフェラーゼ(Takara 社) を用い反応させた。EDTAを最終濃度が50mMになるように加え、フェノール/クロロホルムで抽出後、G25/G100クロマトグラフィーに付された。回収されたdGテイリング cDNA は真空吸引によってサンプルチューブ内で30μl まで減じた。
【0026】第2鎖cDNA合成(図2ステップ■)
6 μl の第2鎖低バッファー(pH8.75 の 200mM Tris-Cl、100mM KCl 、100mM(NH4)2SO4 、20mM MgSO4、1% Triton X100、1mg/mlBSA)、そして3 μl の第2鎖高バッファー (pH9.2 の 200mM Tris-Cl、600mM KCl 、20mM MgCl2) とSacIおよびSpeIの制限酵素の認識配列を含む第2鎖プライマー アダプター(5'GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3')600ng、0.25mMのdNTP'S、15unitのExTaqポリメラーゼ (Takara社) 、150 unitのアンプリガーゼ、耐熱性 DNAリガーゼ(Epicentre社) 、3 unitのハイブリダーゼ、耐熱性 RNase H (Epicentre 社) を加え、 oligo dG-テイルド第1鎖cDNAを含む最終容量60μl の液に調製した。この反応液を55℃で5 分間、続いて0.3 ℃/分の割合で55℃から35℃まで徐々に温度を下げ、15分間35℃、次に15分間72℃でサーマルサイクラーで温度をコントロールし反応させる。アニーリング/伸長は、35℃で1 時間と65℃で30分間、試料を1回インキュベーションすることによって繰り返された。最後に試料をフェノール/ クロロホルムで抽出し、エタノール沈澱により回収した。
【0027】制限酵素による切断とクローニング(図2ステップ■) cDNAを、標準条件下で制限酵素であるエンドヌクレアーゼ、SacIおよびXhoIで処理し、引き続きSephadex G25-G100 のクロマトグラフィーにかけた。フラクションは上述のようにサンプルチューブに集めた。最後に、200ng のcDNAが lambda ZapII ベクターに組み込まれた。このベクターは、製造者が推奨する5 μl バッファー中でSacIやXhoIおよび 200unitの T4 DNA リガーゼ(New England Biolabs社) によって事前に調製しておいたものである。
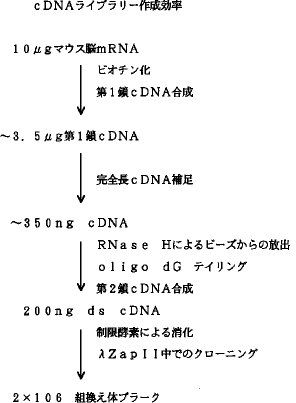
【0028】最終的に得られたライブラリーの評価(1) 回収率、クローニング効率:各ステップの収率を表1にまとめた。最終的に図5に示す様に10μg のmRNAを出発材料とした場合、2 ×106 ヶのリコンビナント プラークを得ることができた。
【0029】
【表1】

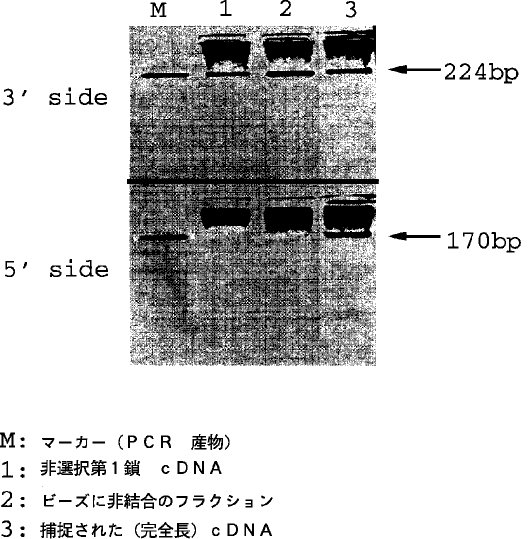
【0030】(2) ライブラリーの評価:■インスリン レセプター mRNA (5.2kb) を用いて、その5'端(INS5: TCCAAATGAAATGCTCCTCC, INS6: ATTGGCTCATGAAGGTTCTT)、3'端(INS3: CTTTCTGGCAGTGACTATGA, INS4: TGTCCGCATCGAGAAGAACA)を増幅する2つのプライマーセットにより、当遺伝子の5'端、3'端部がライブラリーの中に含まれているか否かを測定した。図6に示すように第1鎖 cDNA を選択しない場合とビーズに結合しなかった分画には3'端部の224bp の増幅物が見られるが、5'端部の170bp は見られなかったのに対し、本法で得られた完全長ライブラリーは5'端部の170bp の産物が明瞭に見られる。
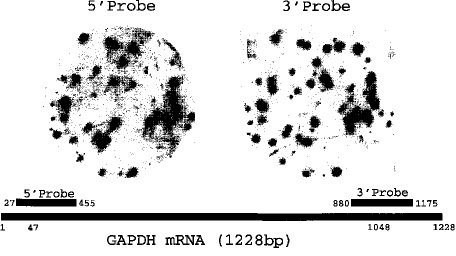
【0031】■GAPDH(Glyceraldehyde 3-phosphate dehydrogenase) 遺伝子を用いた末端の塩基配列−図7に示す様にGAPDH 遺伝子を5'端プローブ3'端プローブで完全長ライブラリーのレプリカフィルターにハイブリダイゼーションを行った。3'端プローブでポジティブシグナルを示したプラークの80%が5'端プローブにおいてもポジティブを示すことにより、このライブラリーの80%は完全長cDNAを示す。更にこれらより3ヶのクローンを単離し、λファージDNA を調製後、インサート両端の塩基配列をABI377で決定した。その結果を図8及び9に示す。5'、3'端とも完全長cDNAが合成されていると考えられる。3'端は共通のサイトよりプライミングしており、5'端もCap サイト特有のC 伸長−T 伸長構造がみられ、5'サイトを完全に含んだライブラリーであることが証明された。
【図面の簡単な説明】
【図1】 両端(5'Cap サイト、3'サイト)にジオール構造を有するmRNAの構造を示す。
【図2】 mRNAの5'Cap サイトのジオール構造の酸化及びビオチンヒドラジドの付加反応を示すスキーム。
【図3】 完全長cDNA合成法の各ステップ(前半)を示すスキーム。
【図4】 完全長cDNA合成法の各ステップ(後半)を示すスキーム。
【図5】 完全長cDNAライブラリーの合成効率を示すスキーム。
【図6】 5'サイト、3'サイトのプライマーを用いた完全長cDNA合成の確認。インスリンレセプターmRNAを例にとり、両端のプライマーを用いて完全長cDNA合成産物の存在を確認した。
【図7】 GAPDH(Glyceraldehyde 3-phosphate dehydrogenase) 5'、3'両末端のクローンをプローブとしたコロニハイブリダイゼーション。
上図:コロニハイブリダイゼーションのパターン(左:5'領域のプローブ、右:3'領域のプローブ)
下図:GAPDH mRNA 1228BP のマップ
【図8】 GAPDH の5'端のシーケンス。GAPDH のcDNAクローンをプローブに選択してきた3種のcDNAクローン(18.1, 20.1, 22.1)の塩基配列。前記3 つのクローンは報告されている5'塩基配列よりも、長い塩基配列であった。
【図9】 GAPDH の3'端のシーケンス。GAPDH のcDNAクローンをプローブに選択してきた3種のcDNAクローン(18.1, 20.1, 22.1)の塩基配列。完全長cDNA合成時のoligo プライマーが見られる。
【0001】
【発明の属する技術分野】本発明は、完全長cDNAライブラリー作成法に関する。更に詳しくは、mRNAの化学修飾を利用した、完全長cDNA精製法による完全長cDNAライブラリー作成法に関する。
【0002】
【従来の技術】cDNA合成法は医学生物学分野の研究で必須の技術であり、遺伝子転写物の解析法として不可欠である。全てのDNA 遺伝情報は転写物を介して生理活性を示すが、それを解析する強力な手段がcDNAクローニングである。従来法におけるcDNA合成においては、oligo dTをプライマーとしてポリA サイトから合成されたcDNAライブラリーの中で最終的にクローンが単離される。ところが、殆どの場合、転写単位の全長が合成されていない為に転写単位の全構造が解析できないのが実状である。この為、通常のcDNAライブラリーでは、プライマー伸長法による5'上流領域の合成、またはランダム プライマーを用いたcDNA合成を用いて5'上流領域をウォーキング(walking) することが全長構造解明に必須のステップとなる。
【0003】しかるに、上記従来のcDNA合成法には次の問題がある。
(1) ランダム プライマーを用いると転写物のかなりの領域をカバーするcDNAができる。しかし、それらは短い断片であり、ポリ Aから5'Cap サイトまで含んだクローンは単離できない。
(2) oligo dTをプライマーとして用いたcDNAは全て3'端を含む。しかし、逆転写酵素が5'Cap サイトまで届かないため、5'上流をプライマー伸長法及び5'RACE等で再度単離解析せねばならない。
(3) 既存の完全長cDNAを単離する方法は、前述の従来のいかなる方法もその効率が十分でない(100μg mRNAから200 万組換え体ファージ)。そこで、実用的にはもっと効率の高い手法の開発が望まれる。
【0004】完全長cDNA合成法の従来の技術として、次のような方法が挙げられる。5'Capサイトの標識法として、酵母またはHela細胞のCap 結合蛋白を用いる方法(I. Edery et al., "An Efficient Strategy To Isolate Full-Length cDNAs Based onan mRNA Cap Retention Procedure (CAPture)", MCB, 15, 3363-3371, 1995)、また5'Cap のない不完全cDNAをアルカリフォスファターゼによりリン酸を除去し、その後タバコモザイクウィルスの脱キャップ酵素を反応させ、完全長cDNAのみリン酸が露出することを利用した方法(K. Maruyama et al., "Oligo-capping: asimple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides", Gene, 138, 171-174, 1995., S. Kato et al., "Construction of a human full-length cDNA bank", Gene, 150, 243-250, 1995) 、等々が挙げられる。
【0005】これらの従来法の完全長cDNA合成法の効率が十分でない理由として次の項目が挙げられる。
以上の様に多段階にわたるcDNA合成ライブラリー作成においては、
【0006】
【発明が解決しようとする課題】そこで本発明は、第1に、完全長cDNAの単離を目的とする従来のキャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識が可能な新たな方法を提供すること、第2に、この新たな5'Cap サイトの標識方法を用いた完全長cDNAライブラリーの作成方法を提供することを目的とする。
【0007】
【課題を解決するための手段】本発明は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法に関する。
【0008】
【発明の実施の態様】本発明の方法では、5'Cap サイトの認識を高め、完全長cDNA(RNA) の選択の段階の効率を高めるために、5'Cap サイトに特異的構造であるジオール構造を利用して5'Cap サイトの標識を化学合成法により行う(図1参照) 。即ち、本発明の方法では、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる。このタッグ分子は5'Cap サイトに化学的に結合しており、タッグ分子で標識されたmRNAを鋳型として完全長cDNAを合成し、完全長cDNAライブラリーを作成する。
【0009】タッグ分子のmRNAの5'Cap サイトへの結合は、図2に示すように、例えば、5'Cap サイトのジオール構造を酸化剤、例えば、過ヨウ素酸ナトリウム(NaIO4) 等で酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。上記ヒドラジン末端を有するタッグ分子としては、例えば、ヒドラジン末端を有するビオチン分子やアビジン分子を挙げることができる。また、タッグ分子として抗原や抗体等の反応特異性を有する分子を用いることもできる。タッグ分子として用いる特異標識物には特に制限はない。
【0010】タッグ分子の結合
【0011】タッグ分子を結合して標識したmRNAは、これを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する。oligo dTをプライマーとする逆転写法によるRNA-DNA 複合体の形成は、常法により行うことができる。
【0012】さらに、形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する。具体的には、1本鎖RNA を切断するRNA 分解酵素でRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体(5'Cap まで伸長した完全長cDNA)をタッグ分子の結合性を利用して分離する。例えば、タッグ分子がビオチン分子である場合、RNA-DNA 複合体がタッグ分子として有するビオチン分子を、固相持体上に担持したアビジンと結合させて、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がアビジン分子である場合、RNA-DNA 複合体がタッグ分子として有するアビジン分子を、固相持体上に担持したビオチンと結合させてmRNAの完全長に対応するDNA を有する複合体を分離することもできる。
【0013】即ち、本発明の1つの態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、前記ビオチン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程ビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0014】また、本発明の別の態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、前記アビジン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程アビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0015】前記1本鎖RNA を切断するRNA 分解酵素としては、例えば、リボヌクレアーゼIを挙げることができる。尚、RNA-DNA 複合体からmRNAの完全長に対応するDNAを有する複合体を選別する方法として、1本鎖RNA を切断するRNA 分解酵素を用いる方法以外の方法を利用することもできる。複合体を選別する方法にも特に制限はない。
【0016】さらに本発明の方法では、分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する。cDNAの回収は、例えば、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行うことができる。また、cDNAの回収は、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドを切断するRNase としては、例えば、RNase H を挙げることができる。
【0017】回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングすることにより、完全長cDNAライブラリーを得ることができる。第2のcDNA鎖の合成は、例えば、第1のcDNA鎖の3'端にRNA またはDNAのオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして行なうことができる。あるいは、第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うこともできる。即ち、単離された完全長第1鎖cDNAより第2鎖を合成する為に、ターミナルデオキシヌクレオチジルトランスフェラーゼによるホモポリマー法(homopolymer法) やRNA リガーゼによる1本鎖プライマーを、第1鎖cDNA3'端または5'Cap を除去されたmRNAの5'鎖に付加しポリメラーゼで伸長方法等を用いることができ、第2鎖を合成する方法も特に制限はない。
【0018】
【発明の効果】本発明によれば、mRNAの5'Cap サイトを化学的に修飾を加えることにより、効率良く完全長cDNAを選択できる。この利点は、修飾が5'Cap サイトを認識する為の修飾が酵素反応に全く依存せず、mRNAの5'Cap サイト構造に特徴的なdiol残基を利用した化学反応に依存する為、バックグラウンドが低くなおかつ非常に高い効率を得るものである。本発明の方法では、完全長cDANの回収を特異的選択性の高い RNase ONE処理ビオチン−アビジン反応等を利用した固相化系で行うことができるため、量産的ロボティックスによるライブラリー生産をも可能にする。
【0019】
【実施例】本実施例は図3〜4にアウトラインを示す工程からなる。本法は次の9つの工程よりなる。
【0020】RNA 調製脳0.5 〜1gの組織片を10mlの懸濁液D でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2ml の5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の 7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0021】RNA のジオール部位へのビオチンの結合(図2ステップ
【0022】第1の cDNA 鎖の調製(図2ステップ
【0023】完全長cDNAのRNase 保護(図2ステップ
【0024】完全長cDNAの採取(図2ステップ
【0025】1本鎖cDNAのoligo dG テイリング(図2ステップ
【0026】第2鎖cDNA合成(図2ステップ
6 μl の第2鎖低バッファー(pH8.75 の 200mM Tris-Cl、100mM KCl 、100mM(NH4)2SO4 、20mM MgSO4、1% Triton X100、1mg/mlBSA)、そして3 μl の第2鎖高バッファー (pH9.2 の 200mM Tris-Cl、600mM KCl 、20mM MgCl2) とSacIおよびSpeIの制限酵素の認識配列を含む第2鎖プライマー アダプター(5'GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3')600ng、0.25mMのdNTP'S、15unitのExTaqポリメラーゼ (Takara社) 、150 unitのアンプリガーゼ、耐熱性 DNAリガーゼ(Epicentre社) 、3 unitのハイブリダーゼ、耐熱性 RNase H (Epicentre 社) を加え、 oligo dG-テイルド第1鎖cDNAを含む最終容量60μl の液に調製した。この反応液を55℃で5 分間、続いて0.3 ℃/分の割合で55℃から35℃まで徐々に温度を下げ、15分間35℃、次に15分間72℃でサーマルサイクラーで温度をコントロールし反応させる。アニーリング/伸長は、35℃で1 時間と65℃で30分間、試料を1回インキュベーションすることによって繰り返された。最後に試料をフェノール/ クロロホルムで抽出し、エタノール沈澱により回収した。
【0027】制限酵素による切断とクローニング(図2ステップ
【0028】最終的に得られたライブラリーの評価(1) 回収率、クローニング効率:各ステップの収率を表1にまとめた。最終的に図5に示す様に10μg のmRNAを出発材料とした場合、2 ×106 ヶのリコンビナント プラークを得ることができた。
【0029】
【表1】
【0030】(2) ライブラリーの評価:
【0031】
【図面の簡単な説明】
【図1】 両端(5'Cap サイト、3'サイト)にジオール構造を有するmRNAの構造を示す。
【図2】 mRNAの5'Cap サイトのジオール構造の酸化及びビオチンヒドラジドの付加反応を示すスキーム。
【図3】 完全長cDNA合成法の各ステップ(前半)を示すスキーム。
【図4】 完全長cDNA合成法の各ステップ(後半)を示すスキーム。
【図5】 完全長cDNAライブラリーの合成効率を示すスキーム。
【図6】 5'サイト、3'サイトのプライマーを用いた完全長cDNA合成の確認。インスリンレセプターmRNAを例にとり、両端のプライマーを用いて完全長cDNA合成産物の存在を確認した。
【図7】 GAPDH(Glyceraldehyde 3-phosphate dehydrogenase) 5'、3'両末端のクローンをプローブとしたコロニハイブリダイゼーション。
上図:コロニハイブリダイゼーションのパターン(左:5'領域のプローブ、右:3'領域のプローブ)
下図:GAPDH mRNA 1228BP のマップ
【図8】 GAPDH の5'端のシーケンス。GAPDH のcDNAクローンをプローブに選択してきた3種のcDNAクローン(18.1, 20.1, 22.1)の塩基配列。前記3 つのクローンは報告されている5'塩基配列よりも、長い塩基配列であった。
【図9】 GAPDH の3'端のシーケンス。GAPDH のcDNAクローンをプローブに選択してきた3種のcDNAクローン(18.1, 20.1, 22.1)の塩基配列。完全長cDNA合成時のoligo プライマーが見られる。
【特許請求の範囲】
【請求項1】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項2】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子であり、固相持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項3】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子であり、固相持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項4】 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する請求項1記載の方法。
【請求項5】 ヒドラジン末端を有するタッグ分子が、ヒドラジン末端を有するビオチン分子またはアビジン分子である請求項4記載の方法。
【請求項6】 1本鎖RNA を切断するRNA 分解酵素で前記RNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項7】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、前記ビオチン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程ビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項8】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、前記アビジン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程アビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項9】 1本鎖RNA を切断するRNA 分解酵素がリボヌクレアーゼIである請求項6〜8のいずれか1項に記載の方法。
【請求項10】 分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する請求項1〜9のいずれか1項に記載の方法。
【請求項11】 cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行う請求項10記載の方法。
【請求項12】 cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行う請求項10記載の方法。用して、分離する工程
【請求項13】 DNA-RNA ハイブリッドを切断するRNase がRNase H である請求項12記載の方法。
【請求項14】 回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングする請求項1〜13のいずれか1項に記載の方法。
【請求項15】 第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なう請求項14記載の方法。
【請求項16】 第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行う請求項15記載の方法。
【請求項1】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、前記タッグ分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項2】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子であり、固相持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項3】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子であり、固相持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項4】 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する請求項1記載の方法。
【請求項5】 ヒドラジン末端を有するタッグ分子が、ヒドラジン末端を有するビオチン分子またはアビジン分子である請求項4記載の方法。
【請求項6】 1本鎖RNA を切断するRNA 分解酵素で前記RNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項7】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、前記ビオチン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程ビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項8】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、前記アビジン分子を結合させたmRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成する工程、及び形成されたRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程アビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項9】 1本鎖RNA を切断するRNA 分解酵素がリボヌクレアーゼIである請求項6〜8のいずれか1項に記載の方法。
【請求項10】 分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する請求項1〜9のいずれか1項に記載の方法。
【請求項11】 cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行う請求項10記載の方法。
【請求項12】 cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行う請求項10記載の方法。用して、分離する工程
【請求項13】 DNA-RNA ハイブリッドを切断するRNase がRNase H である請求項12記載の方法。
【請求項14】 回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングする請求項1〜13のいずれか1項に記載の方法。
【請求項15】 第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なう請求項14記載の方法。
【請求項16】 第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行う請求項15記載の方法。
【図1】

【図2】
【図3】
【図7】
【図4】
【図9】

【図5】
【図8】

【図6】
【図2】
【図3】
【図7】
【図4】
【図9】
【図5】
【図8】
【図6】
【公開番号】特開平9−248187
【公開日】平成9年(1997)9月22日
【国際特許分類】
【出願番号】特願平8−60459
【出願日】平成8年(1996)3月18日
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成7年11月21日 第18回日本分子生物学会年会準備委員会発行の「第18回日本分子生物学会年会プログラム・講演要旨集」に発表
【出願人】(000006792)理化学研究所 (14)
【公開日】平成9年(1997)9月22日
【国際特許分類】
【出願日】平成8年(1996)3月18日
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成7年11月21日 第18回日本分子生物学会年会準備委員会発行の「第18回日本分子生物学会年会プログラム・講演要旨集」に発表
【出願人】(000006792)理化学研究所 (14)
[ Back to top ]