
完全長cDNAライブラリーの作成方法
【課題】 蛋白酵素反応より効率良く5'Cap サイトを標識する方法を利用する完全長cDNAライブラリーの作成方法であって、mRNAが切断されることによる完全長cDNAの合成効率の低下を回避できる、より高い効率で完全長cDNAを合成できる方法の提供。
【解決手段】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程、を含む完全長cDNAライブラリーの作成方法。
【解決手段】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程、を含む完全長cDNAライブラリーの作成方法。
【発明の詳細な説明】
【0001】
【発明の属する技術分野】本発明は、完全長cDNAライブラリー作成法に関する。更に詳しくは、mRNAの化学修飾を利用した、完全長cDNA精製法による完全長cDNAライブラリー作成法に関する。
【0002】
【従来の技術】cDNA合成法は医学生物学分野の研究で必須の技術であり、遺伝子転写物の解析法として不可欠である。全てのDNA 遺伝情報は転写物を介して生理活性を示すが、それを解析する強力な手段がcDNAクローニングである。従来法におけるcDNA合成においては、oligo dTをプライマーとしてポリA サイトから合成されたcDNAライブラリーの中で最終的にクローンが単離される。ところが、殆どの場合、転写単位の全長が合成されていない為に転写単位の全構造が解析できないのが実状である。この為、通常のcDNAライブラリーでは、プライマー伸長法による5'上流領域の合成、またはランダム プライマーを用いたcDNA合成を用いて5'上流領域をウォーキング(walking) することが全長構造解明に必須のステップとなる。
【0003】しかるに、上記従来のcDNA合成法には次の問題がある。
(1) ランダム プライマーを用いると転写物のかなりの領域をカバーするcDNAができる。しかし、それらは短い断片であり、ポリ Aから5'Cap サイトまで含んだクローンは単離できない。
(2) oligo dTをプライマーとして用いたcDNAは全て3'端を含む。しかし、逆転写酵素が5'Cap サイトまで届かないため、5'上流をプライマー伸長法及び5'RACE等で再度単離解析せねばならない。
(3) 既存の完全長cDNAを単離する方法は、前述の従来のいかなる方法もその効率が十分でない(100μg mRNAから200 万組換え体ファージ)。そこで、実用的にはもっと効率の高い手法の開発が望まれる。
【0004】完全長cDNA合成法の従来の技術として、次のような方法が挙げられる。5'Capサイトの標識法として、酵母またはHela細胞のCap 結合蛋白を用いる方法(I. Edery et al., "An Efficient Strategy To Isolate Full-Length cDNAs Based onan mRNA Cap Retention Procedure (CAPture)", MCB, 15, 3363-3371, 1995)、また5'Cap のない不完全cDNAをアルカリフォスファターゼによりリン酸を除去し、その後タバコモザイクウィルスの脱キャップ酵素を反応させ、完全長cDNAのみリン酸が露出することを利用した方法(K. Maruyama et al., "Oligo-capping: asimple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides", Gene, 138, 171-174, 1995., S. Kato et al., "Construction of a human full-length cDNA bank", Gene, 150, 243-250, 1995) 、等々が挙げられる。
【0005】これらの従来法の完全長cDNA合成法の効率が十分でない理由として次の項目が挙げられる。
■5'Cap サイトの認識が、アデノウィルスCap 結合蛋白やタバコモザイクウィルスの脱キャップ酵素の様に蛋白酵素の反応に依存している為、完全長cDNA(RNA)の選択の段階で高い効率が期待できない。
■逆転写酵素がcDNAの第1鎖を合成する際、5'Cap サイトまで合成鎖が伸長しない。
■第1鎖が合成された後の第2鎖合成のプライマー配列の付加、第2鎖の合成効率、2重鎖cDNAのクローニング効率及びクローニングのための宿主ベクター系の問題。
以上の様に多段階にわたるcDNA合成ライブラリー作成においては、■〜■の各ステップがそれぞれ問題となる。
【0006】
【発明が解決しようとする課題】そこで本発明者は、第1に、完全長cDNAの単離を目的とする従来のキャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識が可能な新たな方法を提供すること、第2に、この新たな5'Cap サイトの標識方法を用いた完全長cDNAライブラリーの作成方法を提供することを目的として、新たな完全長cDNAライブラリーの作成方法を提供を見出し、先に特許出願した〔特願平8−60459号〕。
【0007】この方法は、上記キャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識ができ、その結果、完全長cDNAライブラリーの作成をより容易にするものであった。ところが、本発明者がこの方法についてさらに検討した結果、ジオール構造をジアルデヒド化する工程においてmRNAが切断されやすく、完全長cDNAの合成効率を悪くしていることが判明した。そこで、本発明の目的は、蛋白酵素反応より効率良く5'Cap サイトを標識する方法を利用する完全長cDNAライブラリーの作成方法であって、mRNAが切断されることによる完全長cDNAの合成効率の低下を回避できる、より高い効率で完全長cDNAを合成できる方法を提供することにある。
【0008】
【課題を解決するための手段】本発明は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマー(例えば、oligo dT等)より逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法に関する。
【0009】
【発明の実施の態様】本発明の方法では、5'Cap サイトの認識を高め、完全長cDNA(RNA) の選択の段階の効率を高めるために、5'Cap サイトに特異的構造であるジオール構造を利用して5'Cap サイトの標識を化学合成法により行う(図1参照) 。即ち、本発明の方法では、mRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成し、RNA-DNA 複合体を形成しているmRNAの5'Cap(7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる。このタッグ分子は5'Cap サイトに化学的に結合しており、タッグ分子で標識されたmRNAを有するRNA-DNA 複合体から完全長cDNAライブラリーを作成する。本発明の方法の特徴は、RNA-DNA 複合体を形成した後にmRNAをタッグ分子で標識することにある。RNA-DNA ハイブリッド構造は、mRNAをタッグ分子で標識するために必要なジオール構造のアルデヒド化の際に生じるmRNAの化学的切断を阻止でき、その結果、完全長cDNAの合成効率を高めることができる。
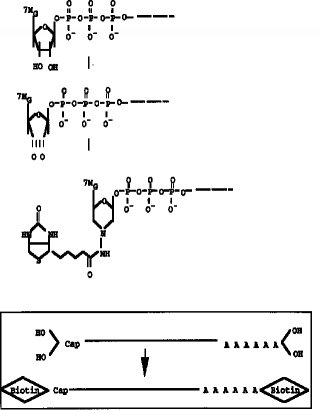
【0010】タッグ分子のmRNAの5'Cap サイトへの結合は、図2に示すように、例えば、5'Cap サイトのジオール構造を酸化剤、例えば、過ヨウ素酸ナトリウム(NaIO4) 等で酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。また、本発明の方法においては、mRNAがRNA-DNA ハイブリッド構造により保護されていることから、ジオール構造の酸化開環を比較的強い酸化条件としてもmRNAの化学的酸化を伴うことなく行うことができるという利点がある。上記ヒドラジン末端を有するタッグ分子としては、例えば、ヒドラジン末端を有するビオチン分子やアビジン分子を挙げることができる。また、タッグ分子として抗原や抗体等の反応特異性を有する分子を用いることもできる。タッグ分子として用いる特異標識物には特に制限はない。
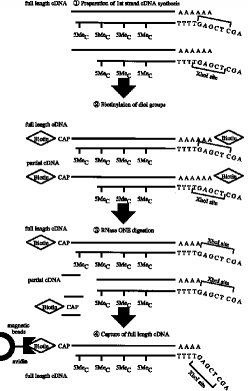
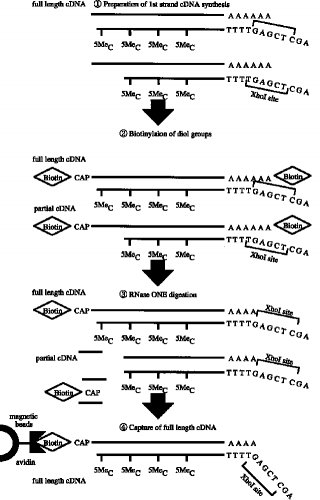
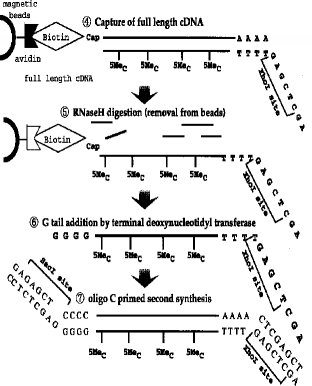
【0011】第1のcDNA鎖の合成■から2本鎖完全長cDNAの合成■までの工程の一例(タッグ分子:ビオチン)が図3〜4に示されている。
■第1のcDNA鎖の合成(RNA-DNA 複合体の合成)
■RNA-DNA 複合体のmRNAのジオール基のビオチン化■リボヌクレアーゼI(RNase I) 消化■完全長cDNA複合体の捕獲(アビジンビーズ使用)
■RNase H 消化(アビジンビーズからの一本鎖cDNAの分離)
■ターミナル デオキシヌクレオチジル トランスフェラーゼによるG末端の付加■オリゴCでプライマーした第2鎖(2本鎖完全長cDNA)の合成
【0012】本発明の方法では、mRNAを鋳型とし、プライマーを始点として逆転写によりRNA-DNA 複合体を形成する。プライマーとしては、例えば、oligo dT等を用いることができる。また、oligo dT等のプライマーを用いた逆転写法によるRNA-DNA 複合体の形成は、常法により行うことができる。
【0013】つぎに、RNA-DNA 複合体にタッグ分子を結合して標識し、標識されたRNA-DNA複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する。具体的には、1本鎖RNA を切断するRNA 分解酵素でRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体(5'Cap まで伸長した完全長cDNA)をタッグ分子の結合性を利用して分離する。例えば、タッグ分子がビオチン分子である場合、RNA-DNA 複合体がタッグ分子として有するビオチン分子を、固相持体上に担持したアビジンと結合させて、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がアビジン分子である場合、RNA-DNA 複合体がタッグ分子として有するアビジン分子を、固相持体上に担持したビオチンと結合させてmRNAの完全長に対応するDNA を有する複合体を分離することもできる。
【0014】即ち、本発明の1つの態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、ビオチン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程、及びビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0015】また、本発明の別の態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、アビジン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程、及びアビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0016】前記1本鎖RNA を切断するRNA 分解酵素としては、例えば、リボヌクレアーゼIを挙げることができる。尚、RNA-DNA 複合体からmRNAの完全長に対応するDNAを有する複合体を選別する方法として、1本鎖RNA を切断するRNA 分解酵素を用いる方法以外の方法を利用することもできる。複合体を選別する方法にも特に制限はない。
【0017】さらに本発明の方法では、分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する。cDNAの回収は、例えば、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行うことができる。また、cDNAの回収は、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドを切断するRNase としては、例えば、RNase H を挙げることができる。
【0018】回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングすることにより、完全長cDNAライブラリーを得ることができる。第2のcDNA鎖の合成は、例えば、第1のcDNA鎖の3'端にRNA またはDNAのオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして行なうことができる。あるいは、第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うこともできる。即ち、単離された完全長第1鎖cDNAより第2鎖を合成する為に、ターミナルデオキシヌクレオチジルトランスフェラーゼによるホモポリマー法(homopolymer法) やRNA リガーゼによる1本鎖プライマーを、第1鎖cDNA3'端または5'Cap を除去されたmRNAの5'鎖に付加しポリメラーゼで伸長方法等を用いることができ、第2鎖を合成する方法も特に制限はない。
【0019】
【発明の効果】本発明によれば、mRNAの5'Cap サイトを化学的に修飾を加えることにより、効率良く完全長cDNAを選択できる。この利点は、修飾が5'Cap サイトを認識する為の修飾が酵素反応に全く依存せず、mRNAの5'Cap サイト構造に特徴的なdiol残基を利用した化学反応に依存する為、バックグラウンドが低くなおかつ非常に高い効率を得るものである。さらに本発明の方法によれば、mRNAの5'Cap サイトの化学的修飾をRNA-DNA 複合体を形成した後に行うことで、化学的に不安定なmRNAの5'Cap サイトの化学的修飾の際の分解を防止して、完全長cDNAの合成効率の低下を回避でき、より高い効率で完全長cDNAを合成できる。また、本発明の方法では、完全長cDNAの回収を特異的選択性の高い RNase ONE処理ビオチン−アビジン反応等を利用した固相化系で行うことができるため、量産的ロボティックスによるライブラリー生産をも可能にする。
【0020】
【実施例】以下、本発明を実施例によりさらに説明する。
実施例1本実施例は図3〜4にアウトラインを示す工程からなる。本法は次の7つの工程よりなる。
■第1のcDNA鎖の合成(RNA-DNA 複合体の合成)
■RNA-DNA 複合体のmRNAのジオール基のビオチン化■リボヌクレアーゼI(RNase I) 消化■完全長cDNA複合体の捕獲(アビジンビーズ使用)
■RNase H 消化(アビジンビーズからの分離)
■ターミナル デオキシヌクレオチジル 転写酵素によるG末端の付加■オリゴCでプライマした第2鎖合成
【0021】RNA 調製脳0.5 〜1gの組織片を10mlの懸濁液D でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2ml の5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の 7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0022】第1鎖cDNAの調製(図2ステップ■)
15μg のmRNAを使ってSuperscript II (Gibco BRL) 3000unit により、最終容量165 μl の反応液中で、5-メチル-dCTP 、dATP、dTTP、dGTP各々0.54mM、50mMTris-HCl (pH8.3 )、75mM KCl、3mM MgCl2 、10mM DTT、52ng/ μl BSA 、RNase インヒビター 5 unit の条件下で逆転写反応を行った。Xho I の認識配列を含むオリゴヌクレオチド3 ’NMTTTTTTTTTTTTGAGCTCTGAATCAAGAGAGAGAGAGAGAGAGAG5'(N: どの核酸塩基でも可、M: G 、A 、C のいづれか)12.6μl をプライマーとして用いた。この反応を始める際、反応液の1/4 を採取し、それに1.5 μl の[α 32P ]-dGTP (3000Ci/mmol 、10μCi/ μl 、Amersham)を加えるこことにより、第1鎖cDNAの合成効率を測定した。RI標識した反応液の0.5 μl をDE-81 ペーパー上にスポットし、0.5Mリン酸ナトリウム緩衝液(pH7.0 )で3 回洗った前後のRI活性を測定し、計算した。その後、RI標識した反応液と非標識の反応液を混合し、0.5M EDTA 8 μl 、10% SDS 2 μl 、プロテイナーゼ(Proteinase)K 20 μg を加え、45℃で15分間加熱した。フェノール/クロロホルムによる抽出、エタノール沈澱後、沈澱をRNase フリーに処理してある水(以下RNase フリー水とする)47μl に溶解した。
【0023】RNA ジオールのビオチン化(図2ステップ■)
RNA のジオール部位(Cap 構造のある5 ’末端と、ポリA 鎖のある3 ’末端のリボースの双方に存在)にビオチンを結合させるために、2段階の反応を行った。それらは、ジオール基の酸化とそれに続くビオチンヒドラジトと酸化RNA 体のカップリング反応である。まず、逆転写反応で得られたRNA-第1鎖cDNA複合体 15 μg を、6.6mM 酢酸ナトリウム緩衝液(pH4.5 )と、酸化剤として過ヨウ素酸ナトリウムを用いて50μl の反応液中で処理する。この酸化反応は遮光条件の元、氷上で45分間行う。続いて、5M塩化ナトリウム11μl 、10%SDS 0.5μl 、そして同量のイソプロパノールを加え、60分間氷上に放置した後、4 ℃で15分間15000rpm遠心し沈澱させる。沈澱物は70%エタノールで洗い、RNase フリー水50μl に再溶解させる。その試料に1M酢酸ナトリウム(pH6.1 )5 μl 、10%SDS 5 μl 、10mMビオチンヒドラジド(Sigma 社)150 μl を加え、室温(22-26 ℃)で終夜反応させる。最後に、5M NaCl 5 μl 、1M酢酸ナトリウム(pH6.1 )75μl 、および2.5 倍量のエタノールを加え、1 時間の氷上冷却後、4 ℃において15分間遠心し、ビオチン化したRNA-DNA 複合体を再沈澱させる。沈澱物は70%エタノールで1回、更に80%エタノールで1回洗い、RNase フリー水70μl に溶解する。
【0024】RNase Iによる完全長cDNAの選択(図2ステップ■)
1 本鎖RNA を消化するRNase Iで処理することにより、逆転写反応時に完全なcDNAの伸長が得られなかったmRNA、およびmRNAの3 ’末端に標識されたビオチン残基を取り除いた。具体的には、ビオチン化反応で得られた試料70μl に10×RNase Iバッファー (100mM Tris-HCl (pH7.5)、50mM EDTA 、2M NaOAc)10μl、RNase I(RNase One TM:Promega社)200unit を加えて、37℃で15分間1 本鎖RNA を消化した。
【0025】完全長cDNAの採取(図2 ステップ■■)
アビジンコートしたマグネティックビーズにcDNAが非特異的吸着するのを防止するため、100 μg の酵母tRNA(DNase I 処理したもの)を5mg (500 μl )のマグネティックビーズ(magnetic porous glass (MPG) particles coated withstreptavidin (CPG,NJ) )に加え、1 時間氷上に放置した後、50mM EDTA 、2MNaClの溶液にて洗った。このビーズを50mM EDTA 、2M NaCl の溶液500 μl 中に懸濁し、RNase I 処理を施されたcDNAを加えた。室温にて30分間撹拌することで、マグネティックビーズと完全長cDNAを結合させた。完全長cDNAを捕獲したビーズを50mM EDTA 、2M NaCl の溶液で4 回、0.4 % SDS、50μg/μl 酵母tRNAで1回、10mM NaCl 、0.2mM EDTA、10mM Tris-HCl (pH7.5 )、20% グリセロールで1 回、50μg/μl 酵母tRNA水溶液で1 回、RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2 、 20mM KCl 、0.1mM EDTA、0.1mM DTT )で1 回洗浄した後、RNase H バッファー 100μl に懸濁し、RNase H 3 unitを加え、37℃下30分間加温した。その後、10% SDS 1 μl 、0.5M EDTA 2 μl を加えて、10分間、65℃に曝し、その上清を回収した。このようにして回収された1 本鎖完全長cDNAはフェノール/クロロホルムで抽出され、スピードバッグにて液量を100 μl 以下に減じてからG25/G100 Sephadex クロマトグラフィーに付した。RI活性を持った分画はシリコン処理したマイクロチューブに収集するとともに、グリコーゲン2μg を加え、エタノール沈澱にて得られた沈澱物を30μl の超純水に溶解した。
【0026】1本鎖cDNAへのオリゴdG付加(図2 ステップ■)
上記により回収された1本鎖cDNA30μl は、最終容量50μl の反応液中で、200mM カコジル酸ナトリウム(pH6.9 )、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μM dGTPの条件のもと、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TaKaRa社)32 unit を用いて37℃で30分間のオリゴdG付加反応に付された。反応終了時にEDTAを50mMとなるように加え、一連のフェノール/クロロホルムによる抽出、エタノール沈澱を経て、31μl の超純水に溶解した。
【0027】第2鎖cDNA合成(図2 ステップ■)
第1鎖cDNAを鋳型にした第2鎖cDNAの合成は以下のように行った。最終容量60μl の反応系で、第2鎖低バッファー(200mM Tris-HCl (pH8.75) 、100mM KCl、100mM (NH4)2SO4、20mM MgSO4、1% Triton X-100 、1mg/μlBSA) 3μ、第2鎖高バッファー(200mM Tris-HCl (pH9.2)、600mM KCl 、20mM MgCl2) 3μl 、dCTP、dATP、dTTP、dGTP各々0.25mM、β-NADH 6 μl 、オリゴdG付加された第1鎖cDNA31μl 、第2鎖プライマー- アダプター:5 ’GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3’(Sac I およびSpe I の認識配列を含む)600ng を加え、Ex Taq DNAポリメラーゼ(TaKaRa Ex Taq :TaKaRa社) 15 unit、耐熱性DNA リガーゼ(Ampligase: Epicentre) 150 unit 、耐熱性RNase H (Hybridase :Epicentre ) 3 unit によって第2鎖cDNAを合成した。0.5M EDTA を1 μl 加えることで反応を停止させ、更に蛋白成分を溶解するために、10% SDS 1 μl 、プロテイナーゼ(Proteinase) K 10 μg の存在下に45℃で15分間加熱し、最終的にフェノール/クロロホルムによる抽出、エタノール沈澱にて精製した2本鎖完全長cDNAを得た。
【0028】以上の方法により得られた二本鎖完全長cDNAから、λZPAII (STRATAGENE)を用いてライブラリーを得た。クローニングの為のpackaging lysateは GIGAPAK Glod (STRATAGENE)を用いて常法により行った。その結果、15μg のmRNAより2.5 ×107 ケの組み換え体ファージを得た。ランダムにピックアップ後、常法により、in vivo excison によりプラスミドクローンに変換し、ライブラリーの挿入cDNAの長さをアガロースゲル電気泳動で測定し、集計した。その結果を表1に示す。
【0029】表1の結果及び下記の表2の結果(特願平8−60459号の方法(前法)により得たライブラリーの評価結果、後述の比較例1)とを比較すると、本発明の方法により得られたcDNAの平均鎖長が前法と比較して有意に増加しているばかりでなく、クラグメントサイズが5000を超える長鎖のクローン数が2倍以上に増加している。このことは、本発明の方法が、長鎖かつ完全長のcDNAを得るという観点から、前法より遙に優れていることを示すものである。
【0030】
【表1】

【0031】
【表2】
【0032】比較例1実施例1と同様にして得たmRNAを用い、特願平8−60459号の方法(前法)により得た二本鎖完全長cDNAのライブラリーについて、実施例1と同様の方法で挿入cDNAの長さをアガロースゲル電気泳動で測定し、集計した。その結果を上記表2に示す。尚、二本鎖完全長cDNAは以下の方法により調製した。
【0033】mRNA-cDNA 複合体の合成10μg のmRNAを使ってSuperscript II (Gibco BRL)の2000unitにより、0.5mM5-methyl-dCTP 、1mM dATP、1mM dTTP、1mM dGTPの存在化で、100 μl バッファー(50mM Tris-HCl, 75mM KCl, 3mM MgCl2, 10mM DTT)の中で逆転写反応を行なった。プライマーとして、5 μg のオリゴヌクレオチド 3'NMTTTTTTTTTTTTGAGCTCTGAATCAAGAGAGAGAGAGAGAGAGAG5'(N:どの核酸塩基でも可、M:G かA かC )を用いた。反応は42℃、45分間行い、続いて反応液を50℃で20分インキュベーションしてmRNA-cDNA 複合体を合成した。上記反応後、即座にサンプルを氷上に移した。次いで4 μl の0.5M EDTA 、8μl の5M NaCl 及び163 μl のH2O(最終容量が200 μl になるように) をサンプルに添加した。攪拌及び軽く遠心した後、混合物を1.5ml のエッペンドルフ管に移し、100 μl のフェノール/トリス及び100 μl のクロロホルムを添加した。攪拌し、2分間氷上で冷却した後、15,000rpm で3 分間遠心分離した。水相を除去した後、新たなエッペンドルフ管に移した。次いで、100 μl のクロロホルムを添加し、攪拌し、2分間氷上で冷却した後、15,000rpm で3 分間遠心分離した。水相を除去した後、新たなエッペンドルフ管に移した。500 μl の100%エタノールを添加し、攪拌し、少なくとも10分間氷上で冷却した。次いで少なくとも10分間16000rpmで遠心分離した。ついで、70% 及び80% エタノールで2 回洗浄した。殆どの放射性ヌクレオチドが除去されたことを、上澄液のガイガーカウンターによる測定で確認した。得られたペレットを47μl の水に懸濁した。この反応を始める際、20μl の反応液を採取し、それに1 μl の[ α-32P]-dGTP(3000Ci/mmol、10μCi/ μl 、Amersham) を加えることにより、第1の cDNA鎖の合成効率を測定した。RI標識した20μl の反応液中0.5 μl をDE-81 ペーパー上にスポットし、pH7.0 の0.5M−リン酸ナトリウムで3回洗った前後のRI活性を測定し計算した。
【0034】RNA のジオール部位へのビオチンの結合mRNA-cDNA 複合体のmRNAのジオール部位(1方はCAP 、他方はRNA の3'端)にビオチンを結合させる為に、2段階の反応を行った。即ち、ジオール基の酸化とそれに続くビオチンヒドラジド(Sigma社) と酸化RNA 体のカップリング反応である。上記工程で得られた47μl の水に懸濁したmRNA-cDNA 複合体(1.5ml のエッペンドルフ管中)に、pH4.5 の66mM酢酸ナトリウム緩衝液3.3 μl と酸化剤としての0.2M 過ヨウ素酸ナトリウム1.290 μl を添加し、攪拌後、遮光条件の下、氷の上で45分間酸化反応を行う。続いて、11μl の5M塩化ナトリウム、0.5 μl の10%SDS 、6 μl のイソプロパノールを加え、氷上で30分間インキュベーションした後、10〜20分間遠心分離した。この間に10mMビオチンヒドラジン(long-arm)水溶液(3.7mg/1ml) を調製した。遠心分離で生成した沈澱物を80% エタノール200 μl で洗い、水50μl に再溶解させる。その試料にpH6.1 の1M酢酸ナトリウム5 μl 、10%SDS 5 μl 、更に10mMビオチンヒドラジン水溶液150 μl を加えた。試料は氷上で1 時間インキュベーションし、20分間遠心し、70% エタノールで2回洗った。最後に適当量の水に再懸濁し、次の工程の材料として用いた。
【0035】完全長cDNAのRNase 保護どのような塩基の箇所でも1本鎖RNA を消化することが可能なRNase ONETTM(Promega社) で処理することにより、逆転写によって完全にcDNAが伸長されなかったmRNA、およびmRNAの3'末端に標識されたビオチン残基を取り除いた。具体的にはmRNA-cDNA 複合体の合成の際、RI標識された反応液20μl と非標識の80μl 分を一緒にプールした後、40μl のRNase I バッファー、355 μl の水、そして50unitのRNase I を使って試料を30分間30℃でインキュベーションした。
【0036】完全長cDNAの採取アビジンコートしたマグネティックビーズへの非特異的吸着を防止する為、2.5mg の酵母tRNA(DNase Iで前処理した) を加え、500 μl に調製し1時間氷上でインキュベーションした。RNase I で処理したcDNAを上記の前処理されたビーズに加え、pH8.0 の0.25M-EDTA、0.5M-NaCl を含むバッファー中、磁気を帯びたビーズが沈澱しないよう、15分間室温で時折振り混ぜながらインキュベーションした。その後ビーズを pH8.0 0.5M-EDTAで4回、0.4 %SDS で1回、その後、ヌクレアーゼフリーの水で3回洗浄する。試料を100 μl のRNaseHバッファーの中で37℃で30分間2 unitのRNaseHで処理後、0.1%のSDS と共にビーズをインキュベーションすることによりビーズから完全長cDNAを分離した。不完全なRNaseH処理の為ビーズから離れないcDNAに関しては、更にpH9.0 、65℃で10分間 Tris-Formate バッファーの中でアルカリ加水分解を行うことにより回収できる。回収された完全長1本鎖cDNAはフェノール/クロロホルムで一度抽出され、G25/G50 sephadexクロマトグラフィーに付された。RI活性を持ったフラクションは表面をシリコン処理したエッペンドルフチューブに集めて、試料を真空で引くことによって10μl にまで減じた。
【0037】1本鎖cDNAのoligo dG テイリング上記により回収された1本鎖cDNAに oligo dG を付加する為、pH6.9 の200mM-NaCacodylate、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μMdGTP の50μl バッファー下で37℃30分間32unitのターミナル デオキシヌクレオチジル トランスフェラーゼ(Takara 社) を用い反応させた。EDTAを最終濃度が50mMになるように加え、フェノール/クロロホルムで抽出後、G25/G100クロマトグラフィーに付された。回収されたdGテイリング cDNA は真空吸引によってサンプルチューブ内で30μl まで減じた。
【0038】第2鎖cDNA合成6 μl の第2鎖低バッファー(pH8.75 の 200mM Tris-Cl、100mM KCl 、100mM(NH4)2SO4 、20mM MgSO4、1% Triton X100、1mg/mlBSA)、そして3 μl の第2鎖高バッファー (pH9.2 の 200mM Tris-Cl、600mM KCl 、20mM MgCl2) とSacIおよびSpeIの制限酵素の認識配列を含む第2鎖プライマー アダプター(5'GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3')600ng、0.25mMのdNTP'S、15unitのExTaqポリメラーゼ (Takara社) 、150 unitのアンプリガーゼ、耐熱性 DNAリガーゼ(Epicentre社) 、3 unitのハイブリダーゼ、耐熱性 RNase H (Epicentre 社) を加え、 oligo dG-テイルド第1鎖cDNAを含む最終容量60μl の液に調製した。この反応液を55℃で5 分間、続いて0.3 ℃/分の割合で55℃から35℃まで徐々に温度を下げ、15分間35℃、次に15分間72℃でサーマルサイクラーで温度をコントロールし反応させる。アニーリング/伸長は、35℃で1 時間と65℃で30分間、試料を1回インキュベーションすることによって繰り返された。最後に試料をフェノール/ クロロホルムで抽出し、エタノール沈澱に付して、二本鎖cDNAを回収した。得られた二本鎖cDNAについて、実施例1と同様にして、挿入cDNAの鎖長を測定した。
【0039】実施例2本実施例では、RNA-DNA ハイブリッド構造が、mRNAをタッグ分子で標識するために必要なジオール構造のアルデヒド化の際に生じるmRNAの化学的切断を阻止でき、その結果、完全長cDNAの合成効率を高めることができることを示すため、以下のレーン 1〜4 の4 種の異なる工程を経て得られたRNA-DNA 複合体を変性アガロース泳動後のオートラジオグラフの結果を図5に示す(尚、サイズマーカーはλHind IIIである) 。図5に示す結果において、レーン 1と2 とを比較すると、レーン 1の方がレーン 2より長鎖のものが多く、予めcDNA合成により形成したRNA-DNA ハイブリッド構造によってmRNAの化学的切断が阻止されていることが分かる。さらに、レーン3と4 とを比較すると、レーン 3の方がレーン 4より長鎖のものが多く、予めcDNA合成により形成したRNA-DNA ハイブリッド構造によってmRNAの化学的切断が阻止されていることが分かる。
【0040】〔レーン1〕10μgmRNA →■cDNA合成([α−32P]dGTPで標識)→■ビオチン化→■アビジンビーズで完全長のもののみ捕捉→変性アガロース泳動〔レーン2〕10μgmRNA →■ビオチン化→■cDNA合成([α−32P]dGTPで標識)→■アビジンビーズで完全長のもののみ捕捉→変性アガロース泳動〔レーン3〕5μgmRNA →■cDNA合成([α−32P]dGTPで標識)→■ビオチン化→変性アガロース泳動〔レーン4〕5μgmRNA →■ビオチン化→■cDNA合成([α−32P]dGTPで標識)→変性アガロース泳動具体的な方法及び条件を以下に示す。
【0041】〔レーン1〕
■第1鎖cDNA合成:[α−32P]dGTPで標識[1]mRNA 10 μg プライマー 8.4μg DW [1] と[2] を加えて最終100 μl となる量[2]5×第一鎖バッファー (GIBCO BRL) 18.18μl 0.1M DTT 9.09 μl 10mM dNTP mix* 5.91 μl BSA (2.5μg/μg) 2.27 μl [α32P]dGTP (10μCi/ μl) 1.0μl RNase インヒビター (25000U/ml) 0.91 μl Superscript TMII RNase H−逆転写酵素(200U/μl) (GIBCO BRL) 10.0 μl 計 100 μl *5-methyl-dCTP、dATP、dTTP、dGTP、各々10mMから成る。
【0042】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。反応後フェノール/クロロホルムによる抽出、エタノール沈澱を経て、47μl RNase フリーの水に溶解。
【0043】■ RNAジオールのビオチン化(a) ジオールの酸化(1) 上記で得られたサンプル 47μl1M NaOAc (pH4.5) 3.3 μl0.2M NaIO4 1.29μl暗所、氷上にて45分間放置する。
(2)5M NaCl 11 μl10%SDS 0.5 μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、 15000rpm 、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0044】(b) ビオチン化(1)1M NaOAc (pH6.1) 5 μl10%SDS 5 μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的に70μl RNase フリーの水に溶解。
【0045】■ストレプトアビジンビーズを用いた完全長cDNAの捕獲
37℃,15 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500μlビオチン化RNA-第1鎖cDNA 100μl室温で30分間撹拌。
【0046】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5] RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1] RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0047】〔レーン2 〕
■ RNAジオールのビオチン化(a) ジオールの酸化(1)mRNA (10 μg) 47μl1M NaOAc (pH4.5) 3.3 μl0.2M NaIO4 1.29μl暗所、氷上にて45分間放置する。
(2)5M NaCl 11 μl10%SDS 0.5μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、15000rpm、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0048】(b) ビオチン化(1)1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75 μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的にRNase フリーの水に溶解。
【0049】■第1鎖cDNA合成:[α- 32P]dGTPで標識[1] ビオチン化mRNA 5μg プライマー 8.4μg DW [1] と[2] を加えて最終100 μl となる量[2]5×第一鎖バッファー (GIBCO BRL) 18.18 μl 0.1M DTT 9.09μl 10mM dNTP mix* 5.91μl BSA (2.5μg/μg) 2.27μl [α- 32P]dGTP (10μCi/ μl) 1.0 μl RNase インヒビター (25000U/ml) 0.91μl Superscript TMII RNase H−逆転写酵素(200U/μl) (GIBCO BRL) 10.0 μl 計 100μl *5-methyl-dCTP、dATP、dTTP、dGTP、各々10mMから成る。
【0050】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。
【0051】■ストレプトアビジンビーズを用いた完全長cDNAの捕獲
30℃,30 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500μl上記で得られたサンプル 500μl室温で30分間撹拌。
【0052】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー (20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0053】〔レーン3 〕
■第1鎖cDNA合成:[α-32P]dGTPで標識[1] mRNA 5μg プライマー 8.4μg DW [1] と[2] を加えて最終100 μl となる量[2]5×第一鎖バッファー (GIBCO BRL) 18.18μl 0.1M DTT 9.09 μl 10mM dNTP mix* 5.91 μl BSA (2.5μg/μg) 2.27 μl [α-32P]dGTP (10μCi/ μl) 1.0μl RNase インヒビター (25000U/ml) 0.91 μl Superscript TMII RNase H−逆転写酵素(200U/μl) (GIBCO BRL) 10.0 μl 計 100μl *5-methyl-dCTP、dATP、dTTP、dGTP、各々10mMから成る。
【0054】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。反応後フェノール/クロロホルムによる抽出、エタノール沈澱を経て、47μlRNase フリーの水に溶解。
【0055】■RNA ジオールのビオチン化(a) ジオールの酸化(1) 上記で得られたサンプル 47μl1M NaOAc (pH4.5) 3.3 μl0.2M NaIO4 1.29μl暗所、氷上にて45分間放置する。
(2)5M NaCl 11μl10%SDS 0.5 μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、 15000rpm 、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0056】(b) ビオチン化(1)1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150 μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的に70μl RNase フリーの水に溶解。
【0057】■ストレプトアビジンビーズを用いた完全長cDNAの捕獲
37℃,15 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1 mg/ml) 500 μlビオチン化RNA-第1鎖cDNA 100 μl室温で30分間撹拌。
【0058】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー (20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100 μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0059】〔レーン4 〕
■RNA ジオールのビオチン化(a) ジオールの酸化(1)mRNA (5μg) 47 μl1M NaOAc (pH4.5) 3.3 μl0.2M NaIO4 1.29μl暗所、氷上にて45分間放置する。
(2)5M NaCl 11μl10%SDS 0.5 μlイソプロパノール 61μlを加え、30分間4 ℃においてインキュベートした後、15000rpm、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0060】(b) ビオチン化[1]1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
[2]1M NaOAc (pH6.1) 75μl5M NaCl 5 μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的にRNase フリーの水に溶解。
【0061】■第1鎖cDNA合成:[α-32P]dGTPで標識[1] ビオチン化mRNA 5 μg プライマー 8.4 μg DW [1] と[2] を加えて最終100 μl となる量[2]5×第一鎖バッファー (GIBCO BRL) 18.18 μl 0.1M DTT 9.09 μl 10mM dNTP mix* 5.91μl BSA (2.5μg/μg) 2.27 μl [α-32P]dGTP (10μCi/ μl) 1.0μl RNase インヒビター (25000U/ml) 0.91μl Superscript TMII RNase H−逆転写酵素(200U/μl) (GIBCO BRL) 10.0 μl 計 100μl *5-methyl-dCTP、dATP、dTTP、dGTP、各々10mMから成る。
【0062】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。
【0063】■ストレプトアビジンビーズを用いた完全長cDNAの捕獲
30℃,30 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500 μl上記で得られたサンプル 500 μl室温で30分間撹拌。
【0064】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【図面の簡単な説明】
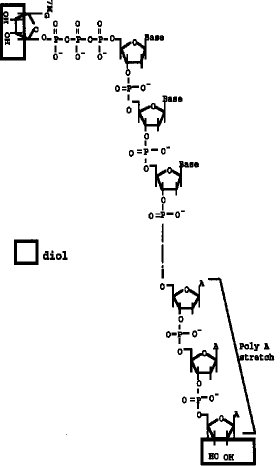
【図1】 両端(5'Cap サイト、3'サイト)にジオール構造を有するmRNAの構造を示す。
【図2】 mRNAの5'Cap サイトのジオール構造の酸化及びビオチンヒドラジドの付加反応を示すスキーム。
【図3】 完全長cDNA合成法の各ステップ(前半)を示すスキーム。
【図4】 完全長cDNA合成法の各ステップ(後半)を示すスキーム。
【図5】 実施例2で得られた電気泳動後のオートラジオグラフを示す図面に代わる写真。
【0001】
【発明の属する技術分野】本発明は、完全長cDNAライブラリー作成法に関する。更に詳しくは、mRNAの化学修飾を利用した、完全長cDNA精製法による完全長cDNAライブラリー作成法に関する。
【0002】
【従来の技術】cDNA合成法は医学生物学分野の研究で必須の技術であり、遺伝子転写物の解析法として不可欠である。全てのDNA 遺伝情報は転写物を介して生理活性を示すが、それを解析する強力な手段がcDNAクローニングである。従来法におけるcDNA合成においては、oligo dTをプライマーとしてポリA サイトから合成されたcDNAライブラリーの中で最終的にクローンが単離される。ところが、殆どの場合、転写単位の全長が合成されていない為に転写単位の全構造が解析できないのが実状である。この為、通常のcDNAライブラリーでは、プライマー伸長法による5'上流領域の合成、またはランダム プライマーを用いたcDNA合成を用いて5'上流領域をウォーキング(walking) することが全長構造解明に必須のステップとなる。
【0003】しかるに、上記従来のcDNA合成法には次の問題がある。
(1) ランダム プライマーを用いると転写物のかなりの領域をカバーするcDNAができる。しかし、それらは短い断片であり、ポリ Aから5'Cap サイトまで含んだクローンは単離できない。
(2) oligo dTをプライマーとして用いたcDNAは全て3'端を含む。しかし、逆転写酵素が5'Cap サイトまで届かないため、5'上流をプライマー伸長法及び5'RACE等で再度単離解析せねばならない。
(3) 既存の完全長cDNAを単離する方法は、前述の従来のいかなる方法もその効率が十分でない(100μg mRNAから200 万組換え体ファージ)。そこで、実用的にはもっと効率の高い手法の開発が望まれる。
【0004】完全長cDNA合成法の従来の技術として、次のような方法が挙げられる。5'Capサイトの標識法として、酵母またはHela細胞のCap 結合蛋白を用いる方法(I. Edery et al., "An Efficient Strategy To Isolate Full-Length cDNAs Based onan mRNA Cap Retention Procedure (CAPture)", MCB, 15, 3363-3371, 1995)、また5'Cap のない不完全cDNAをアルカリフォスファターゼによりリン酸を除去し、その後タバコモザイクウィルスの脱キャップ酵素を反応させ、完全長cDNAのみリン酸が露出することを利用した方法(K. Maruyama et al., "Oligo-capping: asimple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides", Gene, 138, 171-174, 1995., S. Kato et al., "Construction of a human full-length cDNA bank", Gene, 150, 243-250, 1995) 、等々が挙げられる。
【0005】これらの従来法の完全長cDNA合成法の効率が十分でない理由として次の項目が挙げられる。
以上の様に多段階にわたるcDNA合成ライブラリー作成においては、
【0006】
【発明が解決しようとする課題】そこで本発明者は、第1に、完全長cDNAの単離を目的とする従来のキャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識が可能な新たな方法を提供すること、第2に、この新たな5'Cap サイトの標識方法を用いた完全長cDNAライブラリーの作成方法を提供することを目的として、新たな完全長cDNAライブラリーの作成方法を提供を見出し、先に特許出願した〔特願平8−60459号〕。
【0007】この方法は、上記キャップ結合蛋白やタバコモザイクウィルスの脱キャップ酵素等の蛋白酵素反応より効率良く5'Cap サイトの標識ができ、その結果、完全長cDNAライブラリーの作成をより容易にするものであった。ところが、本発明者がこの方法についてさらに検討した結果、ジオール構造をジアルデヒド化する工程においてmRNAが切断されやすく、完全長cDNAの合成効率を悪くしていることが判明した。そこで、本発明の目的は、蛋白酵素反応より効率良く5'Cap サイトを標識する方法を利用する完全長cDNAライブラリーの作成方法であって、mRNAが切断されることによる完全長cDNAの合成効率の低下を回避できる、より高い効率で完全長cDNAを合成できる方法を提供することにある。
【0008】
【課題を解決するための手段】本発明は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマー(例えば、oligo dT等)より逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法に関する。
【0009】
【発明の実施の態様】本発明の方法では、5'Cap サイトの認識を高め、完全長cDNA(RNA) の選択の段階の効率を高めるために、5'Cap サイトに特異的構造であるジオール構造を利用して5'Cap サイトの標識を化学合成法により行う(図1参照) 。即ち、本発明の方法では、mRNAを鋳型とし、oligo dTをプライマーとして逆転写によりRNA-DNA 複合体を形成し、RNA-DNA 複合体を形成しているmRNAの5'Cap(7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる。このタッグ分子は5'Cap サイトに化学的に結合しており、タッグ分子で標識されたmRNAを有するRNA-DNA 複合体から完全長cDNAライブラリーを作成する。本発明の方法の特徴は、RNA-DNA 複合体を形成した後にmRNAをタッグ分子で標識することにある。RNA-DNA ハイブリッド構造は、mRNAをタッグ分子で標識するために必要なジオール構造のアルデヒド化の際に生じるmRNAの化学的切断を阻止でき、その結果、完全長cDNAの合成効率を高めることができる。
【0010】タッグ分子のmRNAの5'Cap サイトへの結合は、図2に示すように、例えば、5'Cap サイトのジオール構造を酸化剤、例えば、過ヨウ素酸ナトリウム(NaIO4) 等で酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。また、本発明の方法においては、mRNAがRNA-DNA ハイブリッド構造により保護されていることから、ジオール構造の酸化開環を比較的強い酸化条件としてもmRNAの化学的酸化を伴うことなく行うことができるという利点がある。上記ヒドラジン末端を有するタッグ分子としては、例えば、ヒドラジン末端を有するビオチン分子やアビジン分子を挙げることができる。また、タッグ分子として抗原や抗体等の反応特異性を有する分子を用いることもできる。タッグ分子として用いる特異標識物には特に制限はない。
【0011】第1のcDNA鎖の合成
【0012】本発明の方法では、mRNAを鋳型とし、プライマーを始点として逆転写によりRNA-DNA 複合体を形成する。プライマーとしては、例えば、oligo dT等を用いることができる。また、oligo dT等のプライマーを用いた逆転写法によるRNA-DNA 複合体の形成は、常法により行うことができる。
【0013】つぎに、RNA-DNA 複合体にタッグ分子を結合して標識し、標識されたRNA-DNA複合体の内、mRNAの完全長に対応するDNA を有する複合体を、タッグ分子の機能を利用して、分離する。具体的には、1本鎖RNA を切断するRNA 分解酵素でRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体(5'Cap まで伸長した完全長cDNA)をタッグ分子の結合性を利用して分離する。例えば、タッグ分子がビオチン分子である場合、RNA-DNA 複合体がタッグ分子として有するビオチン分子を、固相持体上に担持したアビジンと結合させて、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がアビジン分子である場合、RNA-DNA 複合体がタッグ分子として有するアビジン分子を、固相持体上に担持したビオチンと結合させてmRNAの完全長に対応するDNA を有する複合体を分離することもできる。
【0014】即ち、本発明の1つの態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、ビオチン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程、及びビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0015】また、本発明の別の態様は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、アビジン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程、及びアビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法である。
【0016】前記1本鎖RNA を切断するRNA 分解酵素としては、例えば、リボヌクレアーゼIを挙げることができる。尚、RNA-DNA 複合体からmRNAの完全長に対応するDNAを有する複合体を選別する方法として、1本鎖RNA を切断するRNA 分解酵素を用いる方法以外の方法を利用することもできる。複合体を選別する方法にも特に制限はない。
【0017】さらに本発明の方法では、分離されたmRNAの完全長に対応するDNA を有する複合体から、cDNAを回収する。cDNAの回収は、例えば、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることにより行うことができる。また、cDNAの回収は、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドを切断するRNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドを切断するRNase としては、例えば、RNase H を挙げることができる。
【0018】回収された第1のcDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖をクローニングすることにより、完全長cDNAライブラリーを得ることができる。第2のcDNA鎖の合成は、例えば、第1のcDNA鎖の3'端にRNA またはDNAのオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして行なうことができる。あるいは、第1のcDNA鎖の3'端にターミナルヌクレオチドトランスフェラーゼを用いてポリG 、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うこともできる。即ち、単離された完全長第1鎖cDNAより第2鎖を合成する為に、ターミナルデオキシヌクレオチジルトランスフェラーゼによるホモポリマー法(homopolymer法) やRNA リガーゼによる1本鎖プライマーを、第1鎖cDNA3'端または5'Cap を除去されたmRNAの5'鎖に付加しポリメラーゼで伸長方法等を用いることができ、第2鎖を合成する方法も特に制限はない。
【0019】
【発明の効果】本発明によれば、mRNAの5'Cap サイトを化学的に修飾を加えることにより、効率良く完全長cDNAを選択できる。この利点は、修飾が5'Cap サイトを認識する為の修飾が酵素反応に全く依存せず、mRNAの5'Cap サイト構造に特徴的なdiol残基を利用した化学反応に依存する為、バックグラウンドが低くなおかつ非常に高い効率を得るものである。さらに本発明の方法によれば、mRNAの5'Cap サイトの化学的修飾をRNA-DNA 複合体を形成した後に行うことで、化学的に不安定なmRNAの5'Cap サイトの化学的修飾の際の分解を防止して、完全長cDNAの合成効率の低下を回避でき、より高い効率で完全長cDNAを合成できる。また、本発明の方法では、完全長cDNAの回収を特異的選択性の高い RNase ONE処理ビオチン−アビジン反応等を利用した固相化系で行うことができるため、量産的ロボティックスによるライブラリー生産をも可能にする。
【0020】
【実施例】以下、本発明を実施例によりさらに説明する。
実施例1本実施例は図3〜4にアウトラインを示す工程からなる。本法は次の7つの工程よりなる。
【0021】RNA 調製脳0.5 〜1gの組織片を10mlの懸濁液D でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2ml の5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の 7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0022】第1鎖cDNAの調製(図2ステップ
15μg のmRNAを使ってSuperscript II (Gibco BRL) 3000unit により、最終容量165 μl の反応液中で、5-メチル-dCTP 、dATP、dTTP、dGTP各々0.54mM、50mMTris-HCl (pH8.3 )、75mM KCl、3mM MgCl2 、10mM DTT、52ng/ μl BSA 、RNase インヒビター 5 unit の条件下で逆転写反応を行った。Xho I の認識配列を含むオリゴヌクレオチド3 ’NMTTTTTTTTTTTTGAGCTCTGAATCAAGAGAGAGAGAGAGAGAGAG5'(N: どの核酸塩基でも可、M: G 、A 、C のいづれか)12.6μl をプライマーとして用いた。この反応を始める際、反応液の1/4 を採取し、それに1.5 μl の[α 32P ]-dGTP (3000Ci/mmol 、10μCi/ μl 、Amersham)を加えるこことにより、第1鎖cDNAの合成効率を測定した。RI標識した反応液の0.5 μl をDE-81 ペーパー上にスポットし、0.5Mリン酸ナトリウム緩衝液(pH7.0 )で3 回洗った前後のRI活性を測定し、計算した。その後、RI標識した反応液と非標識の反応液を混合し、0.5M EDTA 8 μl 、10% SDS 2 μl 、プロテイナーゼ(Proteinase)K 20 μg を加え、45℃で15分間加熱した。フェノール/クロロホルムによる抽出、エタノール沈澱後、沈澱をRNase フリーに処理してある水(以下RNase フリー水とする)47μl に溶解した。
【0023】RNA ジオールのビオチン化(図2ステップ
RNA のジオール部位(Cap 構造のある5 ’末端と、ポリA 鎖のある3 ’末端のリボースの双方に存在)にビオチンを結合させるために、2段階の反応を行った。それらは、ジオール基の酸化とそれに続くビオチンヒドラジトと酸化RNA 体のカップリング反応である。まず、逆転写反応で得られたRNA-第1鎖cDNA複合体 15 μg を、6.6mM 酢酸ナトリウム緩衝液(pH4.5 )と、酸化剤として過ヨウ素酸ナトリウムを用いて50μl の反応液中で処理する。この酸化反応は遮光条件の元、氷上で45分間行う。続いて、5M塩化ナトリウム11μl 、10%SDS 0.5μl 、そして同量のイソプロパノールを加え、60分間氷上に放置した後、4 ℃で15分間15000rpm遠心し沈澱させる。沈澱物は70%エタノールで洗い、RNase フリー水50μl に再溶解させる。その試料に1M酢酸ナトリウム(pH6.1 )5 μl 、10%SDS 5 μl 、10mMビオチンヒドラジド(Sigma 社)150 μl を加え、室温(22-26 ℃)で終夜反応させる。最後に、5M NaCl 5 μl 、1M酢酸ナトリウム(pH6.1 )75μl 、および2.5 倍量のエタノールを加え、1 時間の氷上冷却後、4 ℃において15分間遠心し、ビオチン化したRNA-DNA 複合体を再沈澱させる。沈澱物は70%エタノールで1回、更に80%エタノールで1回洗い、RNase フリー水70μl に溶解する。
【0024】RNase Iによる完全長cDNAの選択(図2ステップ
1 本鎖RNA を消化するRNase Iで処理することにより、逆転写反応時に完全なcDNAの伸長が得られなかったmRNA、およびmRNAの3 ’末端に標識されたビオチン残基を取り除いた。具体的には、ビオチン化反応で得られた試料70μl に10×RNase Iバッファー (100mM Tris-HCl (pH7.5)、50mM EDTA 、2M NaOAc)10μl、RNase I(RNase One TM:Promega社)200unit を加えて、37℃で15分間1 本鎖RNA を消化した。
【0025】完全長cDNAの採取(図2 ステップ
アビジンコートしたマグネティックビーズにcDNAが非特異的吸着するのを防止するため、100 μg の酵母tRNA(DNase I 処理したもの)を5mg (500 μl )のマグネティックビーズ(magnetic porous glass (MPG) particles coated withstreptavidin (CPG,NJ) )に加え、1 時間氷上に放置した後、50mM EDTA 、2MNaClの溶液にて洗った。このビーズを50mM EDTA 、2M NaCl の溶液500 μl 中に懸濁し、RNase I 処理を施されたcDNAを加えた。室温にて30分間撹拌することで、マグネティックビーズと完全長cDNAを結合させた。完全長cDNAを捕獲したビーズを50mM EDTA 、2M NaCl の溶液で4 回、0.4 % SDS、50μg/μl 酵母tRNAで1回、10mM NaCl 、0.2mM EDTA、10mM Tris-HCl (pH7.5 )、20% グリセロールで1 回、50μg/μl 酵母tRNA水溶液で1 回、RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2 、 20mM KCl 、0.1mM EDTA、0.1mM DTT )で1 回洗浄した後、RNase H バッファー 100μl に懸濁し、RNase H 3 unitを加え、37℃下30分間加温した。その後、10% SDS 1 μl 、0.5M EDTA 2 μl を加えて、10分間、65℃に曝し、その上清を回収した。このようにして回収された1 本鎖完全長cDNAはフェノール/クロロホルムで抽出され、スピードバッグにて液量を100 μl 以下に減じてからG25/G100 Sephadex クロマトグラフィーに付した。RI活性を持った分画はシリコン処理したマイクロチューブに収集するとともに、グリコーゲン2μg を加え、エタノール沈澱にて得られた沈澱物を30μl の超純水に溶解した。
【0026】1本鎖cDNAへのオリゴdG付加(図2 ステップ
上記により回収された1本鎖cDNA30μl は、最終容量50μl の反応液中で、200mM カコジル酸ナトリウム(pH6.9 )、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μM dGTPの条件のもと、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TaKaRa社)32 unit を用いて37℃で30分間のオリゴdG付加反応に付された。反応終了時にEDTAを50mMとなるように加え、一連のフェノール/クロロホルムによる抽出、エタノール沈澱を経て、31μl の超純水に溶解した。
【0027】第2鎖cDNA合成(図2 ステップ
第1鎖cDNAを鋳型にした第2鎖cDNAの合成は以下のように行った。最終容量60μl の反応系で、第2鎖低バッファー(200mM Tris-HCl (pH8.75) 、100mM KCl、100mM (NH4)2SO4、20mM MgSO4、1% Triton X-100 、1mg/μlBSA) 3μ、第2鎖高バッファー(200mM Tris-HCl (pH9.2)、600mM KCl 、20mM MgCl2) 3μl 、dCTP、dATP、dTTP、dGTP各々0.25mM、β-NADH 6 μl 、オリゴdG付加された第1鎖cDNA31μl 、第2鎖プライマー- アダプター:5 ’GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3’(Sac I およびSpe I の認識配列を含む)600ng を加え、Ex Taq DNAポリメラーゼ(TaKaRa Ex Taq :TaKaRa社) 15 unit、耐熱性DNA リガーゼ(Ampligase: Epicentre) 150 unit 、耐熱性RNase H (Hybridase :Epicentre ) 3 unit によって第2鎖cDNAを合成した。0.5M EDTA を1 μl 加えることで反応を停止させ、更に蛋白成分を溶解するために、10% SDS 1 μl 、プロテイナーゼ(Proteinase) K 10 μg の存在下に45℃で15分間加熱し、最終的にフェノール/クロロホルムによる抽出、エタノール沈澱にて精製した2本鎖完全長cDNAを得た。
【0028】以上の方法により得られた二本鎖完全長cDNAから、λZPAII (STRATAGENE)を用いてライブラリーを得た。クローニングの為のpackaging lysateは GIGAPAK Glod (STRATAGENE)を用いて常法により行った。その結果、15μg のmRNAより2.5 ×107 ケの組み換え体ファージを得た。ランダムにピックアップ後、常法により、in vivo excison によりプラスミドクローンに変換し、ライブラリーの挿入cDNAの長さをアガロースゲル電気泳動で測定し、集計した。その結果を表1に示す。
【0029】表1の結果及び下記の表2の結果(特願平8−60459号の方法(前法)により得たライブラリーの評価結果、後述の比較例1)とを比較すると、本発明の方法により得られたcDNAの平均鎖長が前法と比較して有意に増加しているばかりでなく、クラグメントサイズが5000を超える長鎖のクローン数が2倍以上に増加している。このことは、本発明の方法が、長鎖かつ完全長のcDNAを得るという観点から、前法より遙に優れていることを示すものである。
【0030】
【表1】
【0031】
【表2】
【0032】比較例1実施例1と同様にして得たmRNAを用い、特願平8−60459号の方法(前法)により得た二本鎖完全長cDNAのライブラリーについて、実施例1と同様の方法で挿入cDNAの長さをアガロースゲル電気泳動で測定し、集計した。その結果を上記表2に示す。尚、二本鎖完全長cDNAは以下の方法により調製した。
【0033】mRNA-cDNA 複合体の合成10μg のmRNAを使ってSuperscript II (Gibco BRL)の2000unitにより、0.5mM5-methyl-dCTP 、1mM dATP、1mM dTTP、1mM dGTPの存在化で、100 μl バッファー(50mM Tris-HCl, 75mM KCl, 3mM MgCl2, 10mM DTT)の中で逆転写反応を行なった。プライマーとして、5 μg のオリゴヌクレオチド 3'NMTTTTTTTTTTTTGAGCTCTGAATCAAGAGAGAGAGAGAGAGAGAG5'(N:どの核酸塩基でも可、M:G かA かC )を用いた。反応は42℃、45分間行い、続いて反応液を50℃で20分インキュベーションしてmRNA-cDNA 複合体を合成した。上記反応後、即座にサンプルを氷上に移した。次いで4 μl の0.5M EDTA 、8μl の5M NaCl 及び163 μl のH2O(最終容量が200 μl になるように) をサンプルに添加した。攪拌及び軽く遠心した後、混合物を1.5ml のエッペンドルフ管に移し、100 μl のフェノール/トリス及び100 μl のクロロホルムを添加した。攪拌し、2分間氷上で冷却した後、15,000rpm で3 分間遠心分離した。水相を除去した後、新たなエッペンドルフ管に移した。次いで、100 μl のクロロホルムを添加し、攪拌し、2分間氷上で冷却した後、15,000rpm で3 分間遠心分離した。水相を除去した後、新たなエッペンドルフ管に移した。500 μl の100%エタノールを添加し、攪拌し、少なくとも10分間氷上で冷却した。次いで少なくとも10分間16000rpmで遠心分離した。ついで、70% 及び80% エタノールで2 回洗浄した。殆どの放射性ヌクレオチドが除去されたことを、上澄液のガイガーカウンターによる測定で確認した。得られたペレットを47μl の水に懸濁した。この反応を始める際、20μl の反応液を採取し、それに1 μl の[ α-32P]-dGTP(3000Ci/mmol、10μCi/ μl 、Amersham) を加えることにより、第1の cDNA鎖の合成効率を測定した。RI標識した20μl の反応液中0.5 μl をDE-81 ペーパー上にスポットし、pH7.0 の0.5M−リン酸ナトリウムで3回洗った前後のRI活性を測定し計算した。
【0034】RNA のジオール部位へのビオチンの結合mRNA-cDNA 複合体のmRNAのジオール部位(1方はCAP 、他方はRNA の3'端)にビオチンを結合させる為に、2段階の反応を行った。即ち、ジオール基の酸化とそれに続くビオチンヒドラジド(Sigma社) と酸化RNA 体のカップリング反応である。上記工程で得られた47μl の水に懸濁したmRNA-cDNA 複合体(1.5ml のエッペンドルフ管中)に、pH4.5 の66mM酢酸ナトリウム緩衝液3.3 μl と酸化剤としての0.2M 過ヨウ素酸ナトリウム1.290 μl を添加し、攪拌後、遮光条件の下、氷の上で45分間酸化反応を行う。続いて、11μl の5M塩化ナトリウム、0.5 μl の10%SDS 、6 μl のイソプロパノールを加え、氷上で30分間インキュベーションした後、10〜20分間遠心分離した。この間に10mMビオチンヒドラジン(long-arm)水溶液(3.7mg/1ml) を調製した。遠心分離で生成した沈澱物を80% エタノール200 μl で洗い、水50μl に再溶解させる。その試料にpH6.1 の1M酢酸ナトリウム5 μl 、10%SDS 5 μl 、更に10mMビオチンヒドラジン水溶液150 μl を加えた。試料は氷上で1 時間インキュベーションし、20分間遠心し、70% エタノールで2回洗った。最後に適当量の水に再懸濁し、次の工程の材料として用いた。
【0035】完全長cDNAのRNase 保護どのような塩基の箇所でも1本鎖RNA を消化することが可能なRNase ONETTM(Promega社) で処理することにより、逆転写によって完全にcDNAが伸長されなかったmRNA、およびmRNAの3'末端に標識されたビオチン残基を取り除いた。具体的にはmRNA-cDNA 複合体の合成の際、RI標識された反応液20μl と非標識の80μl 分を一緒にプールした後、40μl のRNase I バッファー、355 μl の水、そして50unitのRNase I を使って試料を30分間30℃でインキュベーションした。
【0036】完全長cDNAの採取アビジンコートしたマグネティックビーズへの非特異的吸着を防止する為、2.5mg の酵母tRNA(DNase Iで前処理した) を加え、500 μl に調製し1時間氷上でインキュベーションした。RNase I で処理したcDNAを上記の前処理されたビーズに加え、pH8.0 の0.25M-EDTA、0.5M-NaCl を含むバッファー中、磁気を帯びたビーズが沈澱しないよう、15分間室温で時折振り混ぜながらインキュベーションした。その後ビーズを pH8.0 0.5M-EDTAで4回、0.4 %SDS で1回、その後、ヌクレアーゼフリーの水で3回洗浄する。試料を100 μl のRNaseHバッファーの中で37℃で30分間2 unitのRNaseHで処理後、0.1%のSDS と共にビーズをインキュベーションすることによりビーズから完全長cDNAを分離した。不完全なRNaseH処理の為ビーズから離れないcDNAに関しては、更にpH9.0 、65℃で10分間 Tris-Formate バッファーの中でアルカリ加水分解を行うことにより回収できる。回収された完全長1本鎖cDNAはフェノール/クロロホルムで一度抽出され、G25/G50 sephadexクロマトグラフィーに付された。RI活性を持ったフラクションは表面をシリコン処理したエッペンドルフチューブに集めて、試料を真空で引くことによって10μl にまで減じた。
【0037】1本鎖cDNAのoligo dG テイリング上記により回収された1本鎖cDNAに oligo dG を付加する為、pH6.9 の200mM-NaCacodylate、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μMdGTP の50μl バッファー下で37℃30分間32unitのターミナル デオキシヌクレオチジル トランスフェラーゼ(Takara 社) を用い反応させた。EDTAを最終濃度が50mMになるように加え、フェノール/クロロホルムで抽出後、G25/G100クロマトグラフィーに付された。回収されたdGテイリング cDNA は真空吸引によってサンプルチューブ内で30μl まで減じた。
【0038】第2鎖cDNA合成6 μl の第2鎖低バッファー(pH8.75 の 200mM Tris-Cl、100mM KCl 、100mM(NH4)2SO4 、20mM MgSO4、1% Triton X100、1mg/mlBSA)、そして3 μl の第2鎖高バッファー (pH9.2 の 200mM Tris-Cl、600mM KCl 、20mM MgCl2) とSacIおよびSpeIの制限酵素の認識配列を含む第2鎖プライマー アダプター(5'GAGAGAGAGAGAGAGAGAGAGCTCACTAGTCCCCCCCCCCC3')600ng、0.25mMのdNTP'S、15unitのExTaqポリメラーゼ (Takara社) 、150 unitのアンプリガーゼ、耐熱性 DNAリガーゼ(Epicentre社) 、3 unitのハイブリダーゼ、耐熱性 RNase H (Epicentre 社) を加え、 oligo dG-テイルド第1鎖cDNAを含む最終容量60μl の液に調製した。この反応液を55℃で5 分間、続いて0.3 ℃/分の割合で55℃から35℃まで徐々に温度を下げ、15分間35℃、次に15分間72℃でサーマルサイクラーで温度をコントロールし反応させる。アニーリング/伸長は、35℃で1 時間と65℃で30分間、試料を1回インキュベーションすることによって繰り返された。最後に試料をフェノール/ クロロホルムで抽出し、エタノール沈澱に付して、二本鎖cDNAを回収した。得られた二本鎖cDNAについて、実施例1と同様にして、挿入cDNAの鎖長を測定した。
【0039】実施例2本実施例では、RNA-DNA ハイブリッド構造が、mRNAをタッグ分子で標識するために必要なジオール構造のアルデヒド化の際に生じるmRNAの化学的切断を阻止でき、その結果、完全長cDNAの合成効率を高めることができることを示すため、以下のレーン 1〜4 の4 種の異なる工程を経て得られたRNA-DNA 複合体を変性アガロース泳動後のオートラジオグラフの結果を図5に示す(尚、サイズマーカーはλHind IIIである) 。図5に示す結果において、レーン 1と2 とを比較すると、レーン 1の方がレーン 2より長鎖のものが多く、予めcDNA合成により形成したRNA-DNA ハイブリッド構造によってmRNAの化学的切断が阻止されていることが分かる。さらに、レーン3と4 とを比較すると、レーン 3の方がレーン 4より長鎖のものが多く、予めcDNA合成により形成したRNA-DNA ハイブリッド構造によってmRNAの化学的切断が阻止されていることが分かる。
【0040】〔レーン1〕10μgmRNA →
【0041】〔レーン1〕
【0042】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。反応後フェノール/クロロホルムによる抽出、エタノール沈澱を経て、47μl RNase フリーの水に溶解。
【0043】
(2)5M NaCl 11 μl10%SDS 0.5 μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、 15000rpm 、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0044】(b) ビオチン化(1)1M NaOAc (pH6.1) 5 μl10%SDS 5 μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的に70μl RNase フリーの水に溶解。
【0045】
37℃,15 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500μlビオチン化RNA-第1鎖cDNA 100μl室温で30分間撹拌。
【0046】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5] RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1] RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0047】〔レーン2 〕
(2)5M NaCl 11 μl10%SDS 0.5μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、15000rpm、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0048】(b) ビオチン化(1)1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75 μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的にRNase フリーの水に溶解。
【0049】
【0050】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。
【0051】
30℃,30 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500μl上記で得られたサンプル 500μl室温で30分間撹拌。
【0052】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー (20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0053】〔レーン3 〕
【0054】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。反応後フェノール/クロロホルムによる抽出、エタノール沈澱を経て、47μlRNase フリーの水に溶解。
【0055】
(2)5M NaCl 11μl10%SDS 0.5 μlイソプロパノール 61 μlを加え、30分間4 ℃においてインキュベートした後、 15000rpm 、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0056】(b) ビオチン化(1)1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150 μlを加え、室温にて終夜反応させる。
(2)1M NaOAc (pH6.1) 75μl5M NaCl 5μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的に70μl RNase フリーの水に溶解。
【0057】
37℃,15 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1 mg/ml) 500 μlビオチン化RNA-第1鎖cDNA 100 μl室温で30分間撹拌。
【0058】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー (20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100 μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【0059】〔レーン4 〕
(2)5M NaCl 11μl10%SDS 0.5 μlイソプロパノール 61μlを加え、30分間4 ℃においてインキュベートした後、15000rpm、15分間(4 ℃)の遠心にて得られた沈澱を70% エタノールにて2回リンスし、50μl RNase フリーの水に溶解。
【0060】(b) ビオチン化[1]1M NaOAc (pH6.1) 5μl10%SDS 5μl10mM ビオチン ヒドラジド (Sigma) 150μlを加え、室温にて終夜反応させる。
[2]1M NaOAc (pH6.1) 75μl5M NaCl 5 μlエタノール 750μlを加え、氷上にて1 時間放置後、15000rpm、15分間(4 ℃)遠心し、沈澱を70%エタノールにて2回リンスする。最終的にRNase フリーの水に溶解。
【0061】
【0062】[1] を65℃にて10分間、熱変性後、直ちに氷上に移動させる。次いで、[1] と[2] をアニーリング温度35℃で1 分間インキュベートしてから、混合する。
【0063】
30℃,30 分間インキュベーション。
(b) ストレプトアビジンビーズによる完全長cDNAの捕獲(1) ストレプトアビジンビーズ (MPG)とビオチン化RNA-DNA の結合ストレプトアビジン被覆磁性多孔質ガラス(Magnetic porous glass (MPG))(CPG,NJ) (1mg/ml) 500 μl上記で得られたサンプル 500 μl室温で30分間撹拌。
【0064】(2)MPGの洗浄[1]50mM EDTA、2M NaCl の溶液で4 回[2]0.4% SDS、50μg/μl 酵母tRNAの溶液で1 回[3]10mM NaCl、0.2mM EDTA、10mM Tris-HCl(pH7.5)、20% グリセロールで1 回[4]50 μg/μl 酵母tRNA水溶液で1 回[5]RNase H バッファー(20mM Tris-HCl(pH7.5)、10mM MgCl2、20mM KCl、0.1mM EDTA、0.1mM DTT )で1 回(3)RNase Hによる完全長cDNAの回収[1]RNase H バッファー 100μlRNase H 3unitを洗浄したMPG に加え、37℃下30分間加温。
[2]10% SDS 1μl0.5M EDTA 2μlを加え、10分間、65℃し、上清を回収。
(c) 上清をフェノール/クロロホルムによる抽出、エタノール沈澱にて精製。
【図面の簡単な説明】
【図1】 両端(5'Cap サイト、3'サイト)にジオール構造を有するmRNAの構造を示す。
【図2】 mRNAの5'Cap サイトのジオール構造の酸化及びビオチンヒドラジドの付加反応を示すスキーム。
【図3】 完全長cDNA合成法の各ステップ(前半)を示すスキーム。
【図4】 完全長cDNA合成法の各ステップ(後半)を示すスキーム。
【図5】 実施例2で得られた電気泳動後のオートラジオグラフを示す図面に代わる写真。
【特許請求の範囲】
【請求項1】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項2】 プライマーがoligo dTである請求項1に記載の方法。
【請求項3】 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する請求項1または2に記載の方法。
【請求項4】 ヒドラジン末端を有するタッグ分子が、ヒドラジン末端を有するビオチン分子(ビオチンヒドラザイド)またはヒドラジン末端を有するアビジン分子(アビジンヒドラザイド)である請求項3記載の方法。
【請求項5】 1本鎖RNA を切断するRNA 分解酵素でタッグ分子を結合したRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項6】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子であり、固相持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1または2に記載の方法。
【請求項7】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子であり、固相持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1または2に記載の方法。
【請求項8】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、ビオチン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程、及びビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項9】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、アビジン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程、及びアビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項10】 プライマーがoligo dTである請求項8または9に記載の方法。
【請求項11】 1本鎖RNA を切断するRNA 分解酵素がリボヌクレアーゼIである請求項5、8〜10のいずれか1項に記載の方法。
【請求項12】 分離されたmRNAの完全長に対応するDNA を有する複合体から、1本鎖完全長cDNAを回収する請求項1〜11のいずれか1項に記載の方法。
【請求項13】 分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることによりCap サイトよりタッグ分子を切り離すことにより、1本鎖完全長cDNAを回収する請求項12記載の方法。
【請求項14】 1本鎖完全長cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドのRNA 鎖を切断する為、RNaseを作用させることにより行う請求項12記載の方法。
【請求項15】 DNA-RNA ハイブリッドのRNA 鎖を切断するRNase がRNase H である請求項14記載の方法。
【請求項16】 回収された第1の1本鎖完全長cDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖を合成後、完全長二重鎖cDNAをクローニングする請求項1〜15のいずれか1項に記載の方法。
【請求項17】 第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なう請求項16記載の方法。
【請求項18】 3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素を用いて第1のcDNA鎖の3'端にポリG 、ポリC 、ポリA 、又はポリTを付加したcDNA鎖を鋳型とし、かつ各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行う請求項17記載の方法。
【請求項19】 3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素がターミナルヌクレオチドトランスフェラーゼである請求項18記載の方法。
【請求項1】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA 複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項2】 プライマーがoligo dTである請求項1に記載の方法。
【請求項3】 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることでタッグ分子を結合させたmRNAを調製する請求項1または2に記載の方法。
【請求項4】 ヒドラジン末端を有するタッグ分子が、ヒドラジン末端を有するビオチン分子(ビオチンヒドラザイド)またはヒドラジン末端を有するアビジン分子(アビジンヒドラザイド)である請求項3記載の方法。
【請求項5】 1本鎖RNA を切断するRNA 分解酵素でタッグ分子を結合したRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離する請求項1記載の方法。
【請求項6】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子であり、固相持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1または2に記載の方法。
【請求項7】 タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子であり、固相持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離する請求項1または2に記載の方法。
【請求項8】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、ビオチン分子を結合させる工程、ビオチン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からビオチン分子を切除する工程、及びビオチン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したアビジンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項9】 mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、プライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA 複合体を形成しているmRNAの5'Cap (7MeG ppp N)サイトに存在するジオール構造に、アビジン分子を結合させる工程、アビジン分子を結合したRNA-DNA 複合体を、1本鎖RNA を切断するRNA 分解酵素で消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断することによりこの複合体からアビジン分子を切除する工程、及びアビジン分子が結合したmRNAの完全長に対応するDNA を有する複合体を、固相持体上に担持したビオチンと結合させて分離する工程、を含むことを特徴とする完全長cDNAライブラリーの作成方法。
【請求項10】 プライマーがoligo dTである請求項8または9に記載の方法。
【請求項11】 1本鎖RNA を切断するRNA 分解酵素がリボヌクレアーゼIである請求項5、8〜10のいずれか1項に記載の方法。
【請求項12】 分離されたmRNAの完全長に対応するDNA を有する複合体から、1本鎖完全長cDNAを回収する請求項1〜11のいずれか1項に記載の方法。
【請求項13】 分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることによりCap サイトよりタッグ分子を切り離すことにより、1本鎖完全長cDNAを回収する請求項12記載の方法。
【請求項14】 1本鎖完全長cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドのRNA 鎖を切断する為、RNaseを作用させることにより行う請求項12記載の方法。
【請求項15】 DNA-RNA ハイブリッドのRNA 鎖を切断するRNase がRNase H である請求項14記載の方法。
【請求項16】 回収された第1の1本鎖完全長cDNA鎖を鋳型として、第2のcDNA鎖を合成し、得られた第2のcDNA鎖を合成後、完全長二重鎖cDNAをクローニングする請求項1〜15のいずれか1項に記載の方法。
【請求項17】 第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なう請求項16記載の方法。
【請求項18】 3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素を用いて第1のcDNA鎖の3'端にポリG 、ポリC 、ポリA 、又はポリTを付加したcDNA鎖を鋳型とし、かつ各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行う請求項17記載の方法。
【請求項19】 3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素がターミナルヌクレオチドトランスフェラーゼである請求項18記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】

【図2】
【図3】
【図4】
【図5】
【公開番号】特開平10−127291
【公開日】平成10年(1998)5月19日
【国際特許分類】
【出願番号】特願平8−291500
【出願日】平成8年(1996)11月1日
【出願人】(000006792)理化学研究所 (14)
【公開日】平成10年(1998)5月19日
【国際特許分類】
【出願日】平成8年(1996)11月1日
【出願人】(000006792)理化学研究所 (14)
[ Back to top ]