
完全長cDNAライブラリーの作成法
【課題】 極微量しか存在しないクラスIII(レア) に属し、かつ新規なcDNAを含むライブラリーを、より効率よく得ることができる方法の提供。
【解決手段】 複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【解決手段】 複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【発明の詳細な説明】
【0001】
【発明の属する技術分野】本発明は、新規な完全長cDNAの存在割合が高く、重複の少ない、cDNAライブラリーの作成方法に関する。本発明の方法は、全長鎖cDNA(遺伝子)をカタログ化したライブラリーの作成に有用である。
【0002】
【従来の技術】全遺伝子種に対応する完全長cDNAをカタログ化したライブラリーの作成は、ヒトゲノムプロジェクト(HGP)の重要な課題の1つである。カタログ化したライブラリーとは、ライブラリーに含まれるcDNAに重複が無いという意味であり、各cDNAが1種類づつ含まれているライブラリーのことである。カタログ化完全長cDNAライブラリーの作製には、大規模な全長鎖cDNAライブラリーを作成し、かつ得られるライブラリーを重複の少ないものにしていくことが必要である。全長鎖cDNAライブラリーの作成については、いくつかの方法が報告されている(Maruyamaら:Gene 25 (1983) 171-174、Ederyら:Mol. Cell. Biol. 15 (1995) 3363-3371、Carninciら:DNA Res. 4 (1997) 61-66など)。さらに、cDNAの重複を避ける方法としては、2つのcDNA集団中に共通して存在するcDNAを除くサブトラクション法や1つのcDNA集団中に多量に存在するものを優先的に除くノーマライゼーション法などが知られている(Haraら:Nucleic Acids Res. 19 (1991) 7094-7104、Soaresら:Proc. Natl.Acad. Sci. USA 91 (1994) 9228-9232、Koら: Nucleic Acids Res. 18 (1990)5705-5711)。更に、得られたクローンの塩基配列を解読して、重複をさけることもESTの場合にはよく行われている。
【0003】
【発明が解決しょうとする課題】ところで、ある細胞に存在するmRNAは、その存在量に応じて、3種類に分類され、典型的な細胞におけるその存在量と種類は、細胞の種類により程度の差はあるが、以下の通りである(Bertioliら、4520-4523, Nucleic Acids Res. 1995,Vol.23, No.21)。最も存在量が多いクラスI(アバンダント) に属するmRNAの種類は4つであり、コピー数は各mRNAについてそれぞれ12000であり、存在量の平均%は各mRNAについてそれぞれ3.3である。存在量が中間にあるクラスII(インターメデェート) に属するmRNAの種類は500であり、コピー数は各mRNAについてそれぞれ300であり、存在量の平均%は各mRNAについてそれぞれ0.08である。最も存在量が少ないクラスIII(レア) に属するmRNAの種類は11000であり、コピー数は各mRNAについてそれぞれ15であり、存在量の平均%は各mRNAについてそれぞれ0.004である。全量2μgのトータルRNA中に含まれる各mRNAの平均重量(ng)は、クラスI(アバンダント)のmRNAが3.3ngであり、クラスII(インターメデェート) のmRNAが0.08ngであり、クラスIII(レア) のmRNAが0.004ngである。即ち、クラスIII(レア) に属する1種類のmRNAの重量を1とすると、クラスII(インターメデェート) に属する1種類のmRNAの重量は20であり、クラスI(アバンダント) に属する1種類のmRNAの重量は825である。
【0004】全長鎖cDNAライブラリーの作成方法では、まず、このような存在量の異なる3種類のmRNAを含むmRNAの集団を鋳型として、cDNAの集団を作成している。そのため、作成されたcDNAの集団に含まれるcDNAの存在量は、上記比率に対応しており、クラスIII(レア) に属する1種類のcDNAの量が1であれば、クラスI(アバンダント) に属する1種類のcDNAの量は概ね825ある。クラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAは、cDNAライブラリーの作成の初期の段階でほとんどが見いだされるので、カタログ化した全長鎖cDNAライブラリーの作成には、クラスIII(レア) に属する新規なcDNAを効率よく見いだしていくことが必要である。一般に、全長鎖cDNAライブラリーの作成は、mRNAから全長鎖cDNAを作成し、この全長鎖cDNAをクローニングし、さらにクローニングした全長鎖cDNAの配列を決定するという段階を経て行われる。初期の段階でほとんどが見いだされるクラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAも、mRNAから全長鎖cDNAを作成する段階では、cDNAの集団に含まれており、この集団に含まれるcDNAをそのままクローニングし、配列決定することは、それだけ作業効率を低下させることを意味する。即ち、効率的にカタログ化した全長鎖cDNAライブラリーを作成するには、mRNAから転写作成された全長鎖cDNAの集団中に含まれるクラスIII(レア) に属するcDNA、それもできれば新規なcDNAを選択的にクローニングし、その配列を決定する必要が有る。
【0005】発明者らは、様々な組織からいくつかの全長鎖cDNAライブラリーを作成し、このライブラリーから、重複のあるクローンを除する手法として、各クローンの末端(3‘及び5’末端)の100前後の塩基配列を迅速に決定し、この配列に基づいて重複するクローンを排除し、カタログ化した全長鎖cDNAライブラリーの作成を試みている。しかし、この方法は、末端の一部の塩基配列のみを決定する簡便な方法ではあるが、それでも、ライブラリーの作成が進むほどに既知のクローンの割合が増え、効率は低下する。特にライブラリーのサイズが大きくなるにつれて、同じ遺伝子種の出現頻度(重複度)が増すことから効率の低下は著しくなる。また、これまでも前述のノーマライゼーション法を用いて、cDNAの集団の作製原料として使用したmRNAを用いて、配列決定前のcDNAの集団から、クラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAの存在量を低減することが行われている。しかるに、極微量しか存在しないクラスIII(レア) に属するcDNAのライブラリーを効率よく得るには不十分であった。
【0006】そこで本発明の目的は、極微量しか存在しないクラスIII(レア) に属し、かつ新規なcDNAを含むライブラリーを、より効率よく得ることができる方法を提供することにある。
【0007】
【課題を解決するための手段】本発明では、クローニング及び配列決定する前の段階で、存在量の多いcDNAのコピー数をなるべく減少させるべく検討した。その結果、それぞれは既知の方法ではあるが、カタログ化した全長鎖cDNAライブラリーを作成のために組み合わせて使用することは知られていない、ノーマライゼーションとサブトラクションとを組合せ、しかもサブトラクションに特定の核酸を用いることで、主にクラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAの重複率を低減でき、出現頻度の低いいわゆるレアなcDNAクローンの単離を行い易くできることを見いだして本発明を完成した。
【0008】本発明は、複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法に関する。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【0009】
【発明の実施の形態】本発明の方法では、存在量が多く既知のcDNA及び存在量は少ないが既知のcDNAを除去して、配列決定の必要な、新規かつレアなcDNAの存在量を高めたcDNAライブラリーを作成するため、複数の完全長cDNAを含む集団から、以下の(a)〜(c)の核酸(以下、核酸ドライバーとういことがある)とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する。前記(a)〜(c)の核酸は、RNAまたはDNAであることができるが、特に、(a)〜(c)のRNA(以下、RNAドライバーとういことがある)とのハイブリダイズを利用して、(a)〜(c)のRNAとハイブリダイズしたcDNAを除去することが好ましい。(a)〜(c)のRNAとハイブリダイズするcDNAは、例えば、(a)〜(c)のビオチン化RNAと、前記完全長cDNAを含む集団中のcDNAとをハイブリダイズさせ、ハイブリダイズしたRNA−cDNAが有するビオチンとストレプトアビジンとの結合を利用して、ハイブリダイズしたRNA−cDNAをストレプトアビジン被覆した磁気ビーズを用いることで、完全長cDNAを含む集団から除去することができる。(a)〜(c)のRNAは、存在量が多く既知のcDNA及び存在量は少ないが既知のcDNAに対応するものであるので、これらのcDNAを除去することで、新規かつレアなcDNAの存在量を高めることができる。ここでのハイブリダイゼーションは、例えば、フォルムアミド液中37℃から60℃(Rot=0.5〜200)、好ましくは42℃から45℃(Rot=5〜10)、もっともこのましくは42℃(Rot=5)で行うことが適当である。
【0010】一本鎖完全長cDNAとハイブリダイゼーションさせるRNAドライバーとしては、以下の(a)〜(c)のRNAを用いる。(a)のRNAドライバーは、cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属するRNAである。このようなRNAとしては、例えば、出発材料のmRNAを用いることができる。cDNA合成出発材料としてのmRNAそのものを用いることで、ノーマライゼーションとしてアバンダントに発現しているもの同士がハイブリッドを形成し、cDNAを含む集団から除去される。
【0011】(b)のRNAドライバーは、本発明の方法を利用して、ハイブリダイズにより除去されたRNA−cDNAから別途、回収されたRNAであり、主にクラスI及びクラスIIに属するcDNAに対応するRNAを含む。但し、cDNA(b)のRNAドライバーは、量は少ないが、クラスIIIに属するcDNAに対応するRNAも含む。(b)のRNAドライバーは、特定の組織で高発現している遺伝子クローン(例えば、組織特異的酵素の遺伝子クローン)やいくつかの組織に幅広く発現している遺伝子クローン(例えば、ハウスキーピング遺伝子クローン)の約1000から2000種類を in vitro で転写して得られるRNAであることもできる。具体的には、出発材料としては9種類(すい臓、肝臓、肺、腎臓、脳、脾臓、睾丸、小腸、胃)の組織からそれぞれミニライブラリーを作成して、9種類のミニライブラリーを混合して得られた遺伝子クローンは、上記特定の組織で高発現している遺伝子クローンといくつかの組織に幅広く発現している遺伝子クローンとなり得る。cDNAのin vitroでの転写は、公知の方法を用いて行うことができる。(c)のRNAドライバーは、クラスIII(レア)を含むcDNAライブラリー中のcDNAに対応するRNAである。クラスIII(レア)に属するcDNAを含むcDNAライブラリーは、既存のライブラリーとして存在する。(c)のRNAドライバーを併用することで、存在量が多く既知のcDNAのみならず、存在量は少ないが既知のクラスIII(レア)に属するcDNAも除去できる。クラスIII(レア)に属するcDNAを含むcDNAライブラリーとしては、例えば、理研でカタログ化したcDNAライブラリーを挙げることができ、このライブラリーを in vitro で転写して得られるRNAを(c)のRNAドライバーとして用いることができる。cDNAのin vitroでの転写は、公知の方法を用いて行うことができる。
【0012】これらのRNAはいずれもビオチンで標識し、第1鎖cDNAとハイブリダイゼーションを行うことができる。RNAのビオチンによる標識は、市販のRNAのビオチンによる標識化キット(Label IT (商標)Biotin Nucleic Acid Labeling Kit、Murus Corporation)を用いて行うことができる。
【0013】ビオチンで標識したRNAとcDNAとのハイブリダイゼーションは、上記(a)〜(c)のRNAを含む混合物と一本鎖完全長cDNAを含む集団とを所定温度で混合することで行うことができる。さらに、ハイブリダイゼーション後、ビオチンと結合するストレプトアビジンを被覆したマグネットビーズを用い、ビオチン標識したRNAとハイブリッドしたcDNAを除く。このようにして得られた上清について第2鎖cDNA合成を行い、いわゆるレアな種類のcDNAの存在割合の多いcDNAライブラリーを単離することができる。尚、上記ハイブリダイゼーション及びマグネットビーズを用いるビオチン標識したRNAとハイブリッドしたcDNAの除去は、必要により、2回以上繰り返し行い、いわゆるレアな種類のcDNAの存在割合をさらに高めることもできる。このように、3つのカテゴリー〔(a)〜(c)〕のRNAをノーマライゼーション/サブトラクションの対象として用い、ノーマライゼーション/サブトラクションを行うことで、一本鎖完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成することができる。
【0014】尚、一本鎖完全長cDNAを含む集団は、例えば、特開平10−127291号公報に記載の方法を用いて作成したものであることができる。特開平10−127291号公報に記載の完全長cDNAライブラリーの作成方法は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA複合体を形成しているmRNAの5'Cap (7MeGpppN)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程を含むものである。尚、逆転写によりRNA-DNA複合体を形成する工程をトレハロースの存在下に行うことで、より長鎖のcDNAも全長鎖として得られるという利点がある。さらにこの逆転写反応は、例えば、40℃から80℃で行うことが好ましい。
【0015】タッグ分子を結合させたmRNAの調製は、例えば、 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。また、ヒドラジン末端を有するタッグ分子は、例えば、ヒドラジン末端を有するビオチン分子(ビオチンヒドラザイド)またはヒドラジン末端を有するアビジン分子(アビジンヒドラザイド)であることができる。上記方法では、1本鎖RNA を切断するRNA 分解酵素でタッグ分子を結合したRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子である場合、固相支持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離することができる。タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子である場合には、固相支持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、1本鎖RNA を切断するRNA 分解酵素としてはリボヌクレアーゼIを挙げることができる。また分離されたmRNAの完全長に対応するDNA を有する複合体から、1本鎖完全長cDNAを回収することができ、さらに、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることによりCap サイトよりタッグ分子を切り離すことにより、1本鎖完全長cDNAを回収することができる。1本鎖完全長cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドのRNA 鎖を切断する為、RNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドのRNA 鎖を切断するRNaseは、例えば、RNase H であることができる。
【0016】回収された第1の1本鎖完全長cDNA鎖を鋳型として、第2のcDNA鎖を合成する。但し、本発明では、回収された第1の1本鎖完全長cDNA鎖に対して、上記ノーマライゼーション/サブトラクションを施し、重複の少ないcDNA集団とし、これを第2のcDNA鎖の合成用の鋳型として用いる。得られた第2のcDNA鎖を合成後、完全長二重鎖cDNAをクローニングすることができる。この場合、第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なうことができる。3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素を用いて第1のcDNA鎖の3'端にポリG、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、かつ各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うことができる。3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素は、例えば、ターミナルヌクレオチドトランスフェラーゼであることができる。
【0017】更に、cDNAライブラリーを作成する方法では、レアなcDNAクローンはサイズの大きいものが多いことから、従来のλPSベクターを改良したλPSベクターを用いることができる。即ち、前記完全長cDNAを含む集団から(a)〜(c)のcDNAを除去して得られたcDNAを、長鎖cDNAもクローニングできるように改良したλPSベクターを用いてクローニングして、cDNAライブラリーを作成することができる。また、前記改良したλPSベクターは、λPSベクターの左アームに6Kbのスタッファー断片を挿入したベクターであることができる。このような改良したλPSベクターを用いることで、大きなサイズのcDNAでもクローニングできるようになる。
【0018】
【実施例】以下本発明を実施例によりさらに説明する。
比較例(標準cDNAライブラリーの作成)
mRNA調製マウス(B57BL/6)各器官または組織(表1に名前とIDを示す)0.5〜1gを10mlの懸濁液でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2mlの5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0019】第1鎖cDNAの調製15μg のmRNAを使ってSuperscript II (Gibco BRL) 3000unit により、最終容量165μl の反応液中で、5-メチル-dCTP 、dATP、dTTP、dGTP各々0.54mM、0.6Mトレハロース、50mMTris-HCl(pH8.3 )、75mM KCl、3mM MgCl2 、10mM DTT、52ng/ μl BSA 、RNase インヒビター 5 unit の条件下で逆転写反応を行った。Xho I の認識配列を含むオリゴヌクレオチド(ライブラリーID06ならびに12については1st NXプライマーを、またライブラリーID22・23・24・25・26については1st BSプライマーを用いる、表1参照)12.6μl をプライマーとして用いた。なお、1st プライマーの配列は次の通りである。1st NX:5'-GAGAGAGAGAGCGGCCGCAACTCGAG(T)16VN-3'(44mer; V=A,G,C; N=A, G, C,T;制限酵素部位NotI, XhoI)、1st BS:5'-GAGAGAGAGAAGGATCCAAGAGCTC(T)16VN-3'(43mer; V=A,G,C; N=A, G, C,T;制限酵素部位BamHI, SstI)。 この反応を始める際、反応液の1/4 を採取し、それに1.5 μl の[α-32P]-dGTP (3000Ci/mmol 、10μCi/ μl 、Amersham)を加えるこことにより、第1鎖cDNAの合成効率を測定した。RI標識した反応液の0.5 μl をDE-81 ペーパー上にスポットし、0.5Mリン酸ナトリウム緩衝液(pH7.0 )で3 回洗った前後のRI活性を測定し、計算した。その後、RI標識した反応液と非標識の反応液を混合し、0.5M EDTA 8μl 、10% SDS 2μl 、プロテイナーゼ(Proteinase)K 20μg を加え、45℃で15分間加熱した。フェノール/クロロホルムによる抽出、エタノール沈澱後、沈澱をRNase フリーに処理してある水(以下RNase フリー水とする)47μl に溶解した。
【0020】RNA ジオールのビオチン化RNA のジオール部位(Cap 構造のある5 ’末端と、ポリA 鎖のある3 ’末端のリボースの双方に存在)にビオチンを結合させるために、2段階の反応を行った。それらは、ジオール基の酸化とそれに続くビオチンヒドラジトと酸化RNA 体のカップリング反応である。まず、逆転写反応で得られたRNA-第1鎖cDNA複合体 15 μg を、6.6mM 酢酸ナトリウム緩衝液(pH4.5 )と、酸化剤として過ヨウ素酸ナトリウムを用いて50μlの反応液中で処理する。この酸化反応は遮光条件の元、氷上で45分間行う。続いて、5M塩化ナトリウム11μl 、10%SDS 0.5μl 、そして同量のイソプロパノールを加え、60分間氷上に放置した後、4 ℃で15分間15000rpm遠心し沈澱させる。沈澱物は70%エタノールで洗い、RNase フリー水50μl に再溶解させる。その試料に1M酢酸ナトリウム(pH6.1 )5 μl 、10%SDS 5 μl 、10mMビオチンヒドラジド(Sigma 社)150 μl を加え、室温(22-26 ℃)で終夜反応させる。最後に、5M NaCl 5 μl 、1M酢酸ナトリウム(pH6.1 )75μl 、および2.5 倍量のエタノールを加え、1 時間の氷上冷却後、4 ℃において15分間遠心し、ビオチン化したRNA-DNA 複合体を再沈澱させる。沈澱物は70%エタノールで1回、更に80%エタノールで1回洗い、RNase フリー水70μl に溶解する。
【0021】RNase Iによる完全長cDNAの選択1本鎖RNA を消化するRNase Iで処理することにより、逆転写反応時に完全なcDNAの伸長が得られなかったmRNA、およびmRNAの3 ’末端に標識されたビオチン残基を取り除いた。具体的には、ビオチン化反応で得られた試料70μl に10×RNase Iバッファー(100mM Tris-HCl (pH7.5)、50mM EDTA 、2M NaOAc)10μl 、RNase I(RNase One TM:Promega社)200unit を加えて、37℃で15分間1 本鎖RNA を消化した。
【0022】完全長cDNAの採取ストレプトアビジンコートしたマグネティックビーズにcDNAが非特異的吸着するのを防止するため、100 μg の酵母tRNA(DNase I 処理したもの)を5mg (500 μl )のマグネティックビーズ(magnetic porous glass (MPG) particles coated with streptavidin (CPG,NJ) )に加え、1 時間氷上に放置した後、50mM EDTA 、2M NaClの溶液にて洗った。このビーズを50mM EDTA 、2M NaCl の溶液500μl 中に懸濁し、RNase I 処理を施されたcDNAを加えた。室温にて30分間撹拌することで、マグネティックビーズと完全長cDNAを結合させた。完全長cDNAを捕獲したビーズを50mM EDTA 、2M NaCl の溶液で4 回、0.4 % SDS、50μg/μl 酵母tRNAで1 回、10mM NaCl 、0.2mM EDTA、10mM Tris-HCl (pH7.5 )、20% グリセロールで1 回、50μg/μl 酵母tRNA水溶液で1 回、RNase H バッファー(20mMTris-HCl(pH7.5)、10mM MgCl2 、 20mM KCl 、0.1mM EDTA、0.1mM DTT )で1回洗浄した後、RNase Hバッファー 100μl に懸濁し、RNase H 3 unitを加え、37℃下30分間加温した。その後、10% SDS 1 μl 、0.5M EDTA 2 μl を加えて、10分間、65℃に曝し、その上清を回収した。このようにして回収された1 本鎖完全長cDNAはフェノール/クロロホルムで抽出され、スピードバッグにて液量を100 μl 以下に減じてからG25/G100 Sephadex クロマトグラフィーに付した。RI活性を持った分画はシリコン処理したマイクロチューブに収集するとともに、グリコーゲン2 μg を加え、エタノール沈澱にて得られた沈澱物を30μl の超純水に溶解した。
【0023】1本鎖cDNAへのオリゴdG付加上記により回収された1本鎖cDNA30μl は、最終容量50μl の反応液中で、200mM カコジル酸ナトリウム(pH6.9 )、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μM dGTPの条件のもと、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TaKaRa社)32 unit を用いて37℃で30分間のオリゴdG付加反応に付された。反応終了時にEDTAを50mMとなるように加え、一連のフェノール/クロロホルムによる抽出、エタノール沈澱を経て、31μl の超純水に溶解した。
【0024】第2鎖cDNA合成第1鎖cDNAを鋳型にした第2鎖cDNAの合成は以下のように行った。最終容量60μlの反応系で、第2鎖低バッファー(200mM Tris-HCl (pH8.75) 、100mM KCl、100mM (NH4)2SO4、20mM MgSO4、1% Triton X-100 、1mg/μlBSA) 3μl、第2鎖高バッファー(200mM Tris-HCl (pH9.2)、600mM KCl 、20mM MgCl2) 3μl、dCTP、dATP、dTTP、dGTP各々0.25mM、β-NADH 6 μl 、オリゴdG付加された第1鎖cDNA31μl 、第2鎖プライマー- アダプター(ライブラリーID6・12については2nd ATを、ライブラリーID22・23・24・25・26については 2ndXを用いた、表1を参照)600ng を加え、Ex Taq DNAポリメラーゼ(TaKaRa Ex Taq :TaKaRa社)15 unit、耐熱性DNA リガーゼ(Ampligase: Epicentre) 150 unit、耐熱性RNase H(Hybridase :Epicentre ) 3 unit によって第2鎖cDNAを合成した。0.5M EDTAを 1μl 加えることで反応を停止させ、更に蛋白成分を溶解するために、10% SDS 1 μl 、プロテイナーゼ(Proteinase) K 10 μg の存在下に45℃で15分間加熱し、最終的にフェノール/クロロホルムによる抽出、エタノール沈澱にて精製した2本鎖完全長cDNAを得た。 なお、2nd プライマーの配列は次の通りである。2nd AT:5'-GAGAGAGAGAAGGATCCAAGAGCTCAATTAATTAATTAAACCCCCCCCCCC -3'(51mer; 制限酵素部位BamHI, SstI)、2nd X:5'-GAGAGAGAGATTCTCGAGTTAATTAAATTAATCCCCCCCCCCCCC-3'(45mer;制限酵素部位XhoI, C13)。
【0025】以上の方法により得られた二本鎖完全長cDNAは、λZAPIIIベクターに挿入し、ライブラリーとして回収した。λZAPIIIベクターはλZAPII (STRATAGENE)ベクターのマルチクローニングサイトの一部の配列AAAAGCTGGAGCTCCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGATATCAAGCTTATCGATACCGTCGACCTCGAをAAAAGCTGGAGCTATGGCCCTTATGGCCGAGCTCGCGGCCGCGAATTCCTCGAGGGCCGATTTGGCCAATCGAGに改変し、二つのSfiIサイトを新たに導入したものである。
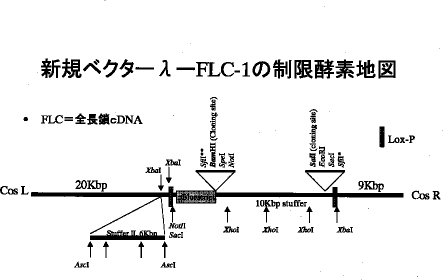
【0026】実施例(ノーマライゼーション/サブトラクション)比較例では、ライブラリー作成時にノーマライゼーション/サブトラクションはせず、ベクターとしてはλZAPIIIを用いた。本実施例においては、比較例と同様の方法であって、ベクターとしてλZAPIII以外にλPS(RIKEN)を用いるとともに、ライブラリー作成時にはノーマライゼーション/サブトラクションを行った。λPS(RIKEN)(λ-FLC-1と命名(FLCとはFULL-LENGTH cDNAを意味する))とは、MoBiTec(ドイツ)のλPSベクターをcDNA用に改変したものである。即ち10 kbp stufferの両側に存在するクローニングサイトにcDNA挿入に便利なBamHIならびにSalIを各々導入するとともに、0.5kbから13kb程度までのcDNAがクローニングできるようにXbaIサイトに6kbのDNA断片を挿入したものである(図1R>1)。このλ-FLC-1を用いると、例えば肺臓cDNAライブラリーの場合には、インサートの平均鎖長は2.57kbとなり、実際に0.5kbから12kbまでのインサートをクローニングすることが出来た。従来法のλZAPの場合には、インサートの平均鎖長は0.97kbであったことから、λ-FLC-1を用いることによって、サイズの大きなcDNAもλZAPに比べて効率よくクローニングできることがわかる。
【0027】以下cDNAライブラリーのノーマライゼーション/サブトラクションについて説明する。ドライバーの調製:出発材料として用いたmRNA〔(a)RNAドライバー〕及びin vitro転写反応で作成したRNAをドライバーとして用いた。後者のRNAはさらに2種類〔(b)及び(c)RNAドライバー〕に分けられる。1つはノーマライゼーションにより除かれたRNA−cDNAからcDNAを回収し、ファージベクターにクローニングする。大腸菌に感染後1つの出発材料あたり1000から2000プラークを混ぜ合わせて1つのライブラリー(ミニライブラリー)とし、常法によりプラスミドDNAに変換する(ファージをヘルパーファージとともに再度大腸菌に感染させ、ファージミドとし、さらにもう一度感染させてプラスミドDNAを得る)。得られたDNAについてin vitro転写反応(T3RNAポリメラーゼまたはT7RNAポリメラーゼを用いる)を行い、DNase I (RQ1-RNase free、 Promega)、ProteinaseK処理後、フェノール/クロロホルム抽出をしてRNA〔(b)RNAドライバー〕を得る。この際、通常出発材料としては9種類(すい臓、肝臓、肺、腎臓、脳、脾臓、睾丸、小腸、胃)の組織からそれぞれミニライブラリーを作成して、9種類のミニライブラリーを混合してRNAを得る。もう一つのRNAはすでに重複のないクローンとして保存されているライブラリー(クローン数約2万個)を培養し、得られたDNAについて(b) RNAドライバーと同様にin vitro転写反応を行ったものである〔(c)RNAドライバー〕。これら3種のRNAは、Label-IT Biotin Labeling Kit (Mirus Corporation)を用いてビオチン化標識を行ったあと、1:1:1の割合でテスターcDNAに添加し、Rot10での反応(42℃)を行い、ストレプトアビジンビーズ(CPG)処理を行って回収した上清について、第2鎖の合成を行った。
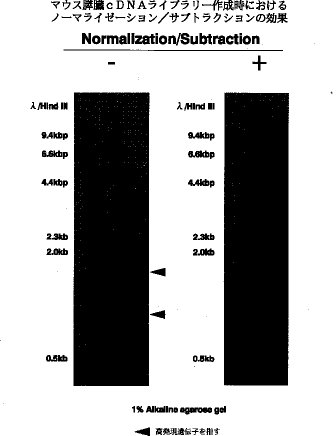
【0028】図2はマウス膵臓から作成した全長鎖cDNAの分布について、ノーマライゼーション/サブトラクションの効果をみたものである。この図からも明らかなようにノーマライゼーション/サブトラクションを導入することによって、サイズの大きな全長鎖cDNAが、従来サイズの小さな全長鎖cDNAだけであったものに加えて認められるようになる。 表1に、ノーマライゼーション/サブトラクションを用いずに作成したcDNAをλZAPにクローニングし、全長鎖cDNAの割合などを調べた結果(スタンダード)とノーマライゼーション/サブトラクションを用いて作成したcDNAをλZAPにクローニングし、全長鎖cDNAの割合などを調べた結果(ノーマライゼーション/サブトラクション)を示す。この結果から、ノーマライゼーション/サブトラクションの有無で、完全長翻訳領域を持つクローン数(B)に対するヒット数(A)〔B/A〕にほとんど変化はなく、ノーマライゼーション/サブトラクションの有無で、全長鎖cDNAの割合などには変化がないことがわかる。
【0029】
【表1】

【0030】(シークエンシング用DNAの調製とシークエンシング)Qボットマシンで釣菌し培養した各cDNAクローンからのプラスミドDNAは、多試料の処理に適した簡便で迅速な伊藤らの方法で調製した(Itohら:Nucleic Acids Res. 25 (1997) 1315-1316)。また、塩基配列の決定は dye-terminator 法によりABIー377ならびにRISAを用いて行った(第21回日本分子生物学年会抄録1Pー570(1998年12月横浜))。
【0031】(コンピューターによる解析)5'末端または3'末端100b以上シーケンシングして得られた配列について、理化学研究所末端配列データベースと照合し(末端の100bについて、3'末端についてはポリAテールを除く100bについて、BLASTの P valueが10-25以下のものを同一と見做す。相同性に換算すると88%以上に相当する。)、未登録のものについて更にNCBIのGenBank、EMBL、DDBJ、PDB各塩基配列データベースならびにマウスEST・ヒトESTデータベースとの検索(末端の100bについて、3'末端についてはポリAテールを除く100bについて、BLASTの P valueが10-10以下のものを既知と見做す。相同性に換算すると約75%以上に相当する。)を行った。表2は、胃、舌、10日目胚から作成したライブラリーについて、本発明のノーマライゼーション/サブトラクションの効果を評価したものである。表中、クローン数は配列決定を行ったクローン数であり、この中から重複を排除して残ったクローン数をユニークと表示した。重複度は(ユニークなクローン数)/(配列決定を行ったクローン数)である。例えば、胃の場合、スタンダードでは、ユニークは838個中333個(重複度2.52)であったのに対して、本発明のノーマライゼーション/サブトラクションを用いた場合、ユニークは849個中592個(重複度1.43)であった。即ち、ノーマライゼーション/サブトラクションを用いることで重複度を低減できることが分かる。さらに、表中の非ESTとは、クローン中の新規な配列の割合である。例えば、胃の場合、スタンダードでは、333個のユニーク中に38個(11.4%)の新規配列を有するクローンが含まれていた。一方、本発明のノーマライゼーション/サブトラクションを用いた場合、592個のユニーク中に98個(16.5%)の新規配列を有するクローンが含まれていた。即ち、本発明のノーマライゼーション/サブトラクションを用いた方法では、新規配列を有するクローンの個数の割合が約3倍になっている。このように、ノーマライゼーション/サブトラクションを行うことによって、重複のないcDNAが単離しやくなり、より効率よくカタログ化されたライブラリーを作成できる。また、ESTとして未登録である新規なcDNAの割合も増加する。
【0032】
【表2】
【0033】
【発明の効果】本発明の方法によれば、全長鎖cDNAライブラリーの作成に際して、重複のないcDNAが単離しやくなり、ライブラリーサイズを大きくしても重複のないカタログ化が可能となる。
【図面の簡単な説明】
【図1】新規ベクターλ-FLC-1の制限酵素地図。
【図2】マウス膵臓から作成したcDNA標品の電気泳動ゲルパターン。右側はノーマライゼーション/サブトラクションを行ったもの、左側はノーマライゼーション/サブトラクションを行っていないコントロール。
【0001】
【発明の属する技術分野】本発明は、新規な完全長cDNAの存在割合が高く、重複の少ない、cDNAライブラリーの作成方法に関する。本発明の方法は、全長鎖cDNA(遺伝子)をカタログ化したライブラリーの作成に有用である。
【0002】
【従来の技術】全遺伝子種に対応する完全長cDNAをカタログ化したライブラリーの作成は、ヒトゲノムプロジェクト(HGP)の重要な課題の1つである。カタログ化したライブラリーとは、ライブラリーに含まれるcDNAに重複が無いという意味であり、各cDNAが1種類づつ含まれているライブラリーのことである。カタログ化完全長cDNAライブラリーの作製には、大規模な全長鎖cDNAライブラリーを作成し、かつ得られるライブラリーを重複の少ないものにしていくことが必要である。全長鎖cDNAライブラリーの作成については、いくつかの方法が報告されている(Maruyamaら:Gene 25 (1983) 171-174、Ederyら:Mol. Cell. Biol. 15 (1995) 3363-3371、Carninciら:DNA Res. 4 (1997) 61-66など)。さらに、cDNAの重複を避ける方法としては、2つのcDNA集団中に共通して存在するcDNAを除くサブトラクション法や1つのcDNA集団中に多量に存在するものを優先的に除くノーマライゼーション法などが知られている(Haraら:Nucleic Acids Res. 19 (1991) 7094-7104、Soaresら:Proc. Natl.Acad. Sci. USA 91 (1994) 9228-9232、Koら: Nucleic Acids Res. 18 (1990)5705-5711)。更に、得られたクローンの塩基配列を解読して、重複をさけることもESTの場合にはよく行われている。
【0003】
【発明が解決しょうとする課題】ところで、ある細胞に存在するmRNAは、その存在量に応じて、3種類に分類され、典型的な細胞におけるその存在量と種類は、細胞の種類により程度の差はあるが、以下の通りである(Bertioliら、4520-4523, Nucleic Acids Res. 1995,Vol.23, No.21)。最も存在量が多いクラスI(アバンダント) に属するmRNAの種類は4つであり、コピー数は各mRNAについてそれぞれ12000であり、存在量の平均%は各mRNAについてそれぞれ3.3である。存在量が中間にあるクラスII(インターメデェート) に属するmRNAの種類は500であり、コピー数は各mRNAについてそれぞれ300であり、存在量の平均%は各mRNAについてそれぞれ0.08である。最も存在量が少ないクラスIII(レア) に属するmRNAの種類は11000であり、コピー数は各mRNAについてそれぞれ15であり、存在量の平均%は各mRNAについてそれぞれ0.004である。全量2μgのトータルRNA中に含まれる各mRNAの平均重量(ng)は、クラスI(アバンダント)のmRNAが3.3ngであり、クラスII(インターメデェート) のmRNAが0.08ngであり、クラスIII(レア) のmRNAが0.004ngである。即ち、クラスIII(レア) に属する1種類のmRNAの重量を1とすると、クラスII(インターメデェート) に属する1種類のmRNAの重量は20であり、クラスI(アバンダント) に属する1種類のmRNAの重量は825である。
【0004】全長鎖cDNAライブラリーの作成方法では、まず、このような存在量の異なる3種類のmRNAを含むmRNAの集団を鋳型として、cDNAの集団を作成している。そのため、作成されたcDNAの集団に含まれるcDNAの存在量は、上記比率に対応しており、クラスIII(レア) に属する1種類のcDNAの量が1であれば、クラスI(アバンダント) に属する1種類のcDNAの量は概ね825ある。クラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAは、cDNAライブラリーの作成の初期の段階でほとんどが見いだされるので、カタログ化した全長鎖cDNAライブラリーの作成には、クラスIII(レア) に属する新規なcDNAを効率よく見いだしていくことが必要である。一般に、全長鎖cDNAライブラリーの作成は、mRNAから全長鎖cDNAを作成し、この全長鎖cDNAをクローニングし、さらにクローニングした全長鎖cDNAの配列を決定するという段階を経て行われる。初期の段階でほとんどが見いだされるクラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAも、mRNAから全長鎖cDNAを作成する段階では、cDNAの集団に含まれており、この集団に含まれるcDNAをそのままクローニングし、配列決定することは、それだけ作業効率を低下させることを意味する。即ち、効率的にカタログ化した全長鎖cDNAライブラリーを作成するには、mRNAから転写作成された全長鎖cDNAの集団中に含まれるクラスIII(レア) に属するcDNA、それもできれば新規なcDNAを選択的にクローニングし、その配列を決定する必要が有る。
【0005】発明者らは、様々な組織からいくつかの全長鎖cDNAライブラリーを作成し、このライブラリーから、重複のあるクローンを除する手法として、各クローンの末端(3‘及び5’末端)の100前後の塩基配列を迅速に決定し、この配列に基づいて重複するクローンを排除し、カタログ化した全長鎖cDNAライブラリーの作成を試みている。しかし、この方法は、末端の一部の塩基配列のみを決定する簡便な方法ではあるが、それでも、ライブラリーの作成が進むほどに既知のクローンの割合が増え、効率は低下する。特にライブラリーのサイズが大きくなるにつれて、同じ遺伝子種の出現頻度(重複度)が増すことから効率の低下は著しくなる。また、これまでも前述のノーマライゼーション法を用いて、cDNAの集団の作製原料として使用したmRNAを用いて、配列決定前のcDNAの集団から、クラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAの存在量を低減することが行われている。しかるに、極微量しか存在しないクラスIII(レア) に属するcDNAのライブラリーを効率よく得るには不十分であった。
【0006】そこで本発明の目的は、極微量しか存在しないクラスIII(レア) に属し、かつ新規なcDNAを含むライブラリーを、より効率よく得ることができる方法を提供することにある。
【0007】
【課題を解決するための手段】本発明では、クローニング及び配列決定する前の段階で、存在量の多いcDNAのコピー数をなるべく減少させるべく検討した。その結果、それぞれは既知の方法ではあるが、カタログ化した全長鎖cDNAライブラリーを作成のために組み合わせて使用することは知られていない、ノーマライゼーションとサブトラクションとを組合せ、しかもサブトラクションに特定の核酸を用いることで、主にクラスI(アバンダント)及びクラスII(インターメデェート)に属するcDNAの重複率を低減でき、出現頻度の低いいわゆるレアなcDNAクローンの単離を行い易くできることを見いだして本発明を完成した。
【0008】本発明は、複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法に関する。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【0009】
【発明の実施の形態】本発明の方法では、存在量が多く既知のcDNA及び存在量は少ないが既知のcDNAを除去して、配列決定の必要な、新規かつレアなcDNAの存在量を高めたcDNAライブラリーを作成するため、複数の完全長cDNAを含む集団から、以下の(a)〜(c)の核酸(以下、核酸ドライバーとういことがある)とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する。前記(a)〜(c)の核酸は、RNAまたはDNAであることができるが、特に、(a)〜(c)のRNA(以下、RNAドライバーとういことがある)とのハイブリダイズを利用して、(a)〜(c)のRNAとハイブリダイズしたcDNAを除去することが好ましい。(a)〜(c)のRNAとハイブリダイズするcDNAは、例えば、(a)〜(c)のビオチン化RNAと、前記完全長cDNAを含む集団中のcDNAとをハイブリダイズさせ、ハイブリダイズしたRNA−cDNAが有するビオチンとストレプトアビジンとの結合を利用して、ハイブリダイズしたRNA−cDNAをストレプトアビジン被覆した磁気ビーズを用いることで、完全長cDNAを含む集団から除去することができる。(a)〜(c)のRNAは、存在量が多く既知のcDNA及び存在量は少ないが既知のcDNAに対応するものであるので、これらのcDNAを除去することで、新規かつレアなcDNAの存在量を高めることができる。ここでのハイブリダイゼーションは、例えば、フォルムアミド液中37℃から60℃(Rot=0.5〜200)、好ましくは42℃から45℃(Rot=5〜10)、もっともこのましくは42℃(Rot=5)で行うことが適当である。
【0010】一本鎖完全長cDNAとハイブリダイゼーションさせるRNAドライバーとしては、以下の(a)〜(c)のRNAを用いる。(a)のRNAドライバーは、cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属するRNAである。このようなRNAとしては、例えば、出発材料のmRNAを用いることができる。cDNA合成出発材料としてのmRNAそのものを用いることで、ノーマライゼーションとしてアバンダントに発現しているもの同士がハイブリッドを形成し、cDNAを含む集団から除去される。
【0011】(b)のRNAドライバーは、本発明の方法を利用して、ハイブリダイズにより除去されたRNA−cDNAから別途、回収されたRNAであり、主にクラスI及びクラスIIに属するcDNAに対応するRNAを含む。但し、cDNA(b)のRNAドライバーは、量は少ないが、クラスIIIに属するcDNAに対応するRNAも含む。(b)のRNAドライバーは、特定の組織で高発現している遺伝子クローン(例えば、組織特異的酵素の遺伝子クローン)やいくつかの組織に幅広く発現している遺伝子クローン(例えば、ハウスキーピング遺伝子クローン)の約1000から2000種類を in vitro で転写して得られるRNAであることもできる。具体的には、出発材料としては9種類(すい臓、肝臓、肺、腎臓、脳、脾臓、睾丸、小腸、胃)の組織からそれぞれミニライブラリーを作成して、9種類のミニライブラリーを混合して得られた遺伝子クローンは、上記特定の組織で高発現している遺伝子クローンといくつかの組織に幅広く発現している遺伝子クローンとなり得る。cDNAのin vitroでの転写は、公知の方法を用いて行うことができる。(c)のRNAドライバーは、クラスIII(レア)を含むcDNAライブラリー中のcDNAに対応するRNAである。クラスIII(レア)に属するcDNAを含むcDNAライブラリーは、既存のライブラリーとして存在する。(c)のRNAドライバーを併用することで、存在量が多く既知のcDNAのみならず、存在量は少ないが既知のクラスIII(レア)に属するcDNAも除去できる。クラスIII(レア)に属するcDNAを含むcDNAライブラリーとしては、例えば、理研でカタログ化したcDNAライブラリーを挙げることができ、このライブラリーを in vitro で転写して得られるRNAを(c)のRNAドライバーとして用いることができる。cDNAのin vitroでの転写は、公知の方法を用いて行うことができる。
【0012】これらのRNAはいずれもビオチンで標識し、第1鎖cDNAとハイブリダイゼーションを行うことができる。RNAのビオチンによる標識は、市販のRNAのビオチンによる標識化キット(Label IT (商標)Biotin Nucleic Acid Labeling Kit、Murus Corporation)を用いて行うことができる。
【0013】ビオチンで標識したRNAとcDNAとのハイブリダイゼーションは、上記(a)〜(c)のRNAを含む混合物と一本鎖完全長cDNAを含む集団とを所定温度で混合することで行うことができる。さらに、ハイブリダイゼーション後、ビオチンと結合するストレプトアビジンを被覆したマグネットビーズを用い、ビオチン標識したRNAとハイブリッドしたcDNAを除く。このようにして得られた上清について第2鎖cDNA合成を行い、いわゆるレアな種類のcDNAの存在割合の多いcDNAライブラリーを単離することができる。尚、上記ハイブリダイゼーション及びマグネットビーズを用いるビオチン標識したRNAとハイブリッドしたcDNAの除去は、必要により、2回以上繰り返し行い、いわゆるレアな種類のcDNAの存在割合をさらに高めることもできる。このように、3つのカテゴリー〔(a)〜(c)〕のRNAをノーマライゼーション/サブトラクションの対象として用い、ノーマライゼーション/サブトラクションを行うことで、一本鎖完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成することができる。
【0014】尚、一本鎖完全長cDNAを含む集団は、例えば、特開平10−127291号公報に記載の方法を用いて作成したものであることができる。特開平10−127291号公報に記載の完全長cDNAライブラリーの作成方法は、mRNAの完全長に対応するcDNAのライブラリーを作成する方法であって、mRNAを鋳型とし、oligo dT等のプライマーより逆転写によりRNA-DNA 複合体を形成する工程、RNA-DNA複合体を形成しているmRNAの5'Cap (7MeGpppN)サイトに存在するジオール構造に、タッグになる分子を化学結合させる工程、及びタッグ分子を結合したRNA-DNA複合体の内、mRNAの完全長に対応するDNA を有するRNA-DNA 複合体を、タッグ分子の機能を利用して分離する工程を含むものである。尚、逆転写によりRNA-DNA複合体を形成する工程をトレハロースの存在下に行うことで、より長鎖のcDNAも全長鎖として得られるという利点がある。さらにこの逆転写反応は、例えば、40℃から80℃で行うことが好ましい。
【0015】タッグ分子を結合させたmRNAの調製は、例えば、 mRNAの5'Cap サイトに存在するジオール構造を過ヨウ素酸ナトリウムで酸化開環してジアルデヒドとし、次いでヒドラジン末端を有するタッグ分子を前記ジアルデヒドと反応させることで行うことができる。また、ヒドラジン末端を有するタッグ分子は、例えば、ヒドラジン末端を有するビオチン分子(ビオチンヒドラザイド)またはヒドラジン末端を有するアビジン分子(アビジンヒドラザイド)であることができる。上記方法では、1本鎖RNA を切断するRNA 分解酵素でタッグ分子を結合したRNA-DNA 複合体を消化して、mRNAの完全長に対応しないDNA を有する複合体の1本鎖RNA 部を切断してこの複合体からタッグ分子を切除し、次いで、タッグ分子を有するmRNAの完全長に対応するDNA を有する複合体を分離することができる。また、タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するビオチン分子である場合、固相支持体上に担持したアビジンと、RNA-DNA 複合体がタッグ分子として有するビオチン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離することができる。タッグ分子がmRNAの5'Cap サイトに存在するジオール構造と結合可能な官能基を有するアビジン分子である場合には、固相支持体上に担持したビオチンと、RNA-DNA 複合体がタッグ分子として有するアビジン分子との結合性を利用して、mRNAの完全長に対応するDNA を有する複合体を分離することができる。また、1本鎖RNA を切断するRNA 分解酵素としてはリボヌクレアーゼIを挙げることができる。また分離されたmRNAの完全長に対応するDNA を有する複合体から、1本鎖完全長cDNAを回収することができ、さらに、分離されたmRNAの完全長に対応するDNA を有する複合体に、タバコモザイクウィルスアルカリホスファターゼを反応させることによりCap サイトよりタッグ分子を切り離すことにより、1本鎖完全長cDNAを回収することができる。1本鎖完全長cDNAの回収を、分離されたmRNAの完全長に対応するDNA を有する複合体に、DNA-RNA ハイブリッドのRNA 鎖を切断する為、RNase を作用させることにより行うこともできる。DNA-RNA ハイブリッドのRNA 鎖を切断するRNaseは、例えば、RNase H であることができる。
【0016】回収された第1の1本鎖完全長cDNA鎖を鋳型として、第2のcDNA鎖を合成する。但し、本発明では、回収された第1の1本鎖完全長cDNA鎖に対して、上記ノーマライゼーション/サブトラクションを施し、重複の少ないcDNA集団とし、これを第2のcDNA鎖の合成用の鋳型として用いる。得られた第2のcDNA鎖を合成後、完全長二重鎖cDNAをクローニングすることができる。この場合、第1のcDNA鎖の3'端にRNA またはDNA のオリゴマーをライゲーションして得られたcDNA鎖を鋳型とし、かつライゲーションしたオリゴマーの相補鎖オリゴマーをプライマーとして、第2のcDNA鎖の合成を行なうことができる。3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素を用いて第1のcDNA鎖の3'端にポリG、ポリC 、ポリA 、又はポリT を付加したcDNA鎖を鋳型とし、かつ各々に相補的なオリゴC 、オリゴG 、オリゴT 、又はオリゴA をプライマーとして、第2のcDNA鎖の合成を行うことができる。3'端に鋳型なしにポリG 、ポリC 、ポリA 、又はポリT を合成できる酵素は、例えば、ターミナルヌクレオチドトランスフェラーゼであることができる。
【0017】更に、cDNAライブラリーを作成する方法では、レアなcDNAクローンはサイズの大きいものが多いことから、従来のλPSベクターを改良したλPSベクターを用いることができる。即ち、前記完全長cDNAを含む集団から(a)〜(c)のcDNAを除去して得られたcDNAを、長鎖cDNAもクローニングできるように改良したλPSベクターを用いてクローニングして、cDNAライブラリーを作成することができる。また、前記改良したλPSベクターは、λPSベクターの左アームに6Kbのスタッファー断片を挿入したベクターであることができる。このような改良したλPSベクターを用いることで、大きなサイズのcDNAでもクローニングできるようになる。
【0018】
【実施例】以下本発明を実施例によりさらに説明する。
比較例(標準cDNAライブラリーの作成)
mRNA調製マウス(B57BL/6)各器官または組織(表1に名前とIDを示す)0.5〜1gを10mlの懸濁液でホモジェナイズし、pH4.0 の 2M 酢酸ナトリウム1ml と、同量のフェノール/ クロロホルム( 体積比5:1)混液を加え抽出した。抽出後水層に同量のイソプロパノールを加えると、RNA が水相から分離沈澱した。この試料を氷の上で1時間インキュベーションした後、15分間4000rpm で冷却遠心機にかけ、沈澱物を回収した。この検体を70%エタノールで洗い、8ml の水に溶解後2mlの5MNaCl、1 %CTAB(cetyltrimethylammonium bromide)、4M尿素、50mMTrisを含むpH7.0 の水溶液16mlを加えることでRNA を沈澱させ、ポリサッカライドを除いた(CTAB 沈澱) 。続いて室温で4000rpm 、15分間遠心機にかけ、RNA を4ml の7M グアニジン−Clに溶解した。そして2倍量のエタノールを加えた後、氷上で1時間インキュベーションし、15分間4000rpm 遠心機にかけ、生じた沈澱物を70%エタノールで洗いRNA を回収した、これを再度水に溶解し、RNA の純度をOD比260/280(>1.8) と230/260(<0.45)を読むことによって計測した。
【0019】第1鎖cDNAの調製15μg のmRNAを使ってSuperscript II (Gibco BRL) 3000unit により、最終容量165μl の反応液中で、5-メチル-dCTP 、dATP、dTTP、dGTP各々0.54mM、0.6Mトレハロース、50mMTris-HCl(pH8.3 )、75mM KCl、3mM MgCl2 、10mM DTT、52ng/ μl BSA 、RNase インヒビター 5 unit の条件下で逆転写反応を行った。Xho I の認識配列を含むオリゴヌクレオチド(ライブラリーID06ならびに12については1st NXプライマーを、またライブラリーID22・23・24・25・26については1st BSプライマーを用いる、表1参照)12.6μl をプライマーとして用いた。なお、1st プライマーの配列は次の通りである。1st NX:5'-GAGAGAGAGAGCGGCCGCAACTCGAG(T)16VN-3'(44mer; V=A,G,C; N=A, G, C,T;制限酵素部位NotI, XhoI)、1st BS:5'-GAGAGAGAGAAGGATCCAAGAGCTC(T)16VN-3'(43mer; V=A,G,C; N=A, G, C,T;制限酵素部位BamHI, SstI)。 この反応を始める際、反応液の1/4 を採取し、それに1.5 μl の[α-32P]-dGTP (3000Ci/mmol 、10μCi/ μl 、Amersham)を加えるこことにより、第1鎖cDNAの合成効率を測定した。RI標識した反応液の0.5 μl をDE-81 ペーパー上にスポットし、0.5Mリン酸ナトリウム緩衝液(pH7.0 )で3 回洗った前後のRI活性を測定し、計算した。その後、RI標識した反応液と非標識の反応液を混合し、0.5M EDTA 8μl 、10% SDS 2μl 、プロテイナーゼ(Proteinase)K 20μg を加え、45℃で15分間加熱した。フェノール/クロロホルムによる抽出、エタノール沈澱後、沈澱をRNase フリーに処理してある水(以下RNase フリー水とする)47μl に溶解した。
【0020】RNA ジオールのビオチン化RNA のジオール部位(Cap 構造のある5 ’末端と、ポリA 鎖のある3 ’末端のリボースの双方に存在)にビオチンを結合させるために、2段階の反応を行った。それらは、ジオール基の酸化とそれに続くビオチンヒドラジトと酸化RNA 体のカップリング反応である。まず、逆転写反応で得られたRNA-第1鎖cDNA複合体 15 μg を、6.6mM 酢酸ナトリウム緩衝液(pH4.5 )と、酸化剤として過ヨウ素酸ナトリウムを用いて50μlの反応液中で処理する。この酸化反応は遮光条件の元、氷上で45分間行う。続いて、5M塩化ナトリウム11μl 、10%SDS 0.5μl 、そして同量のイソプロパノールを加え、60分間氷上に放置した後、4 ℃で15分間15000rpm遠心し沈澱させる。沈澱物は70%エタノールで洗い、RNase フリー水50μl に再溶解させる。その試料に1M酢酸ナトリウム(pH6.1 )5 μl 、10%SDS 5 μl 、10mMビオチンヒドラジド(Sigma 社)150 μl を加え、室温(22-26 ℃)で終夜反応させる。最後に、5M NaCl 5 μl 、1M酢酸ナトリウム(pH6.1 )75μl 、および2.5 倍量のエタノールを加え、1 時間の氷上冷却後、4 ℃において15分間遠心し、ビオチン化したRNA-DNA 複合体を再沈澱させる。沈澱物は70%エタノールで1回、更に80%エタノールで1回洗い、RNase フリー水70μl に溶解する。
【0021】RNase Iによる完全長cDNAの選択1本鎖RNA を消化するRNase Iで処理することにより、逆転写反応時に完全なcDNAの伸長が得られなかったmRNA、およびmRNAの3 ’末端に標識されたビオチン残基を取り除いた。具体的には、ビオチン化反応で得られた試料70μl に10×RNase Iバッファー(100mM Tris-HCl (pH7.5)、50mM EDTA 、2M NaOAc)10μl 、RNase I(RNase One TM:Promega社)200unit を加えて、37℃で15分間1 本鎖RNA を消化した。
【0022】完全長cDNAの採取ストレプトアビジンコートしたマグネティックビーズにcDNAが非特異的吸着するのを防止するため、100 μg の酵母tRNA(DNase I 処理したもの)を5mg (500 μl )のマグネティックビーズ(magnetic porous glass (MPG) particles coated with streptavidin (CPG,NJ) )に加え、1 時間氷上に放置した後、50mM EDTA 、2M NaClの溶液にて洗った。このビーズを50mM EDTA 、2M NaCl の溶液500μl 中に懸濁し、RNase I 処理を施されたcDNAを加えた。室温にて30分間撹拌することで、マグネティックビーズと完全長cDNAを結合させた。完全長cDNAを捕獲したビーズを50mM EDTA 、2M NaCl の溶液で4 回、0.4 % SDS、50μg/μl 酵母tRNAで1 回、10mM NaCl 、0.2mM EDTA、10mM Tris-HCl (pH7.5 )、20% グリセロールで1 回、50μg/μl 酵母tRNA水溶液で1 回、RNase H バッファー(20mMTris-HCl(pH7.5)、10mM MgCl2 、 20mM KCl 、0.1mM EDTA、0.1mM DTT )で1回洗浄した後、RNase Hバッファー 100μl に懸濁し、RNase H 3 unitを加え、37℃下30分間加温した。その後、10% SDS 1 μl 、0.5M EDTA 2 μl を加えて、10分間、65℃に曝し、その上清を回収した。このようにして回収された1 本鎖完全長cDNAはフェノール/クロロホルムで抽出され、スピードバッグにて液量を100 μl 以下に減じてからG25/G100 Sephadex クロマトグラフィーに付した。RI活性を持った分画はシリコン処理したマイクロチューブに収集するとともに、グリコーゲン2 μg を加え、エタノール沈澱にて得られた沈澱物を30μl の超純水に溶解した。
【0023】1本鎖cDNAへのオリゴdG付加上記により回収された1本鎖cDNA30μl は、最終容量50μl の反応液中で、200mM カコジル酸ナトリウム(pH6.9 )、1mM MgCl2 、1mM CoCl2 、1mM 2-メルカプトエタノール、100 μM dGTPの条件のもと、ターミナルデオキシヌクレオチジルトランスフェラーゼ(TaKaRa社)32 unit を用いて37℃で30分間のオリゴdG付加反応に付された。反応終了時にEDTAを50mMとなるように加え、一連のフェノール/クロロホルムによる抽出、エタノール沈澱を経て、31μl の超純水に溶解した。
【0024】第2鎖cDNA合成第1鎖cDNAを鋳型にした第2鎖cDNAの合成は以下のように行った。最終容量60μlの反応系で、第2鎖低バッファー(200mM Tris-HCl (pH8.75) 、100mM KCl、100mM (NH4)2SO4、20mM MgSO4、1% Triton X-100 、1mg/μlBSA) 3μl、第2鎖高バッファー(200mM Tris-HCl (pH9.2)、600mM KCl 、20mM MgCl2) 3μl、dCTP、dATP、dTTP、dGTP各々0.25mM、β-NADH 6 μl 、オリゴdG付加された第1鎖cDNA31μl 、第2鎖プライマー- アダプター(ライブラリーID6・12については2nd ATを、ライブラリーID22・23・24・25・26については 2ndXを用いた、表1を参照)600ng を加え、Ex Taq DNAポリメラーゼ(TaKaRa Ex Taq :TaKaRa社)15 unit、耐熱性DNA リガーゼ(Ampligase: Epicentre) 150 unit、耐熱性RNase H(Hybridase :Epicentre ) 3 unit によって第2鎖cDNAを合成した。0.5M EDTAを 1μl 加えることで反応を停止させ、更に蛋白成分を溶解するために、10% SDS 1 μl 、プロテイナーゼ(Proteinase) K 10 μg の存在下に45℃で15分間加熱し、最終的にフェノール/クロロホルムによる抽出、エタノール沈澱にて精製した2本鎖完全長cDNAを得た。 なお、2nd プライマーの配列は次の通りである。2nd AT:5'-GAGAGAGAGAAGGATCCAAGAGCTCAATTAATTAATTAAACCCCCCCCCCC -3'(51mer; 制限酵素部位BamHI, SstI)、2nd X:5'-GAGAGAGAGATTCTCGAGTTAATTAAATTAATCCCCCCCCCCCCC-3'(45mer;制限酵素部位XhoI, C13)。
【0025】以上の方法により得られた二本鎖完全長cDNAは、λZAPIIIベクターに挿入し、ライブラリーとして回収した。λZAPIIIベクターはλZAPII (STRATAGENE)ベクターのマルチクローニングサイトの一部の配列AAAAGCTGGAGCTCCACCGCGGTGGCGGCCGCTCTAGAACTAGTGGATCCCCCGGGCTGCAGGAATTCGATATCAAGCTTATCGATACCGTCGACCTCGAをAAAAGCTGGAGCTATGGCCCTTATGGCCGAGCTCGCGGCCGCGAATTCCTCGAGGGCCGATTTGGCCAATCGAGに改変し、二つのSfiIサイトを新たに導入したものである。
【0026】実施例(ノーマライゼーション/サブトラクション)比較例では、ライブラリー作成時にノーマライゼーション/サブトラクションはせず、ベクターとしてはλZAPIIIを用いた。本実施例においては、比較例と同様の方法であって、ベクターとしてλZAPIII以外にλPS(RIKEN)を用いるとともに、ライブラリー作成時にはノーマライゼーション/サブトラクションを行った。λPS(RIKEN)(λ-FLC-1と命名(FLCとはFULL-LENGTH cDNAを意味する))とは、MoBiTec(ドイツ)のλPSベクターをcDNA用に改変したものである。即ち10 kbp stufferの両側に存在するクローニングサイトにcDNA挿入に便利なBamHIならびにSalIを各々導入するとともに、0.5kbから13kb程度までのcDNAがクローニングできるようにXbaIサイトに6kbのDNA断片を挿入したものである(図1R>1)。このλ-FLC-1を用いると、例えば肺臓cDNAライブラリーの場合には、インサートの平均鎖長は2.57kbとなり、実際に0.5kbから12kbまでのインサートをクローニングすることが出来た。従来法のλZAPの場合には、インサートの平均鎖長は0.97kbであったことから、λ-FLC-1を用いることによって、サイズの大きなcDNAもλZAPに比べて効率よくクローニングできることがわかる。
【0027】以下cDNAライブラリーのノーマライゼーション/サブトラクションについて説明する。ドライバーの調製:出発材料として用いたmRNA〔(a)RNAドライバー〕及びin vitro転写反応で作成したRNAをドライバーとして用いた。後者のRNAはさらに2種類〔(b)及び(c)RNAドライバー〕に分けられる。1つはノーマライゼーションにより除かれたRNA−cDNAからcDNAを回収し、ファージベクターにクローニングする。大腸菌に感染後1つの出発材料あたり1000から2000プラークを混ぜ合わせて1つのライブラリー(ミニライブラリー)とし、常法によりプラスミドDNAに変換する(ファージをヘルパーファージとともに再度大腸菌に感染させ、ファージミドとし、さらにもう一度感染させてプラスミドDNAを得る)。得られたDNAについてin vitro転写反応(T3RNAポリメラーゼまたはT7RNAポリメラーゼを用いる)を行い、DNase I (RQ1-RNase free、 Promega)、ProteinaseK処理後、フェノール/クロロホルム抽出をしてRNA〔(b)RNAドライバー〕を得る。この際、通常出発材料としては9種類(すい臓、肝臓、肺、腎臓、脳、脾臓、睾丸、小腸、胃)の組織からそれぞれミニライブラリーを作成して、9種類のミニライブラリーを混合してRNAを得る。もう一つのRNAはすでに重複のないクローンとして保存されているライブラリー(クローン数約2万個)を培養し、得られたDNAについて(b) RNAドライバーと同様にin vitro転写反応を行ったものである〔(c)RNAドライバー〕。これら3種のRNAは、Label-IT Biotin Labeling Kit (Mirus Corporation)を用いてビオチン化標識を行ったあと、1:1:1の割合でテスターcDNAに添加し、Rot10での反応(42℃)を行い、ストレプトアビジンビーズ(CPG)処理を行って回収した上清について、第2鎖の合成を行った。
【0028】図2はマウス膵臓から作成した全長鎖cDNAの分布について、ノーマライゼーション/サブトラクションの効果をみたものである。この図からも明らかなようにノーマライゼーション/サブトラクションを導入することによって、サイズの大きな全長鎖cDNAが、従来サイズの小さな全長鎖cDNAだけであったものに加えて認められるようになる。 表1に、ノーマライゼーション/サブトラクションを用いずに作成したcDNAをλZAPにクローニングし、全長鎖cDNAの割合などを調べた結果(スタンダード)とノーマライゼーション/サブトラクションを用いて作成したcDNAをλZAPにクローニングし、全長鎖cDNAの割合などを調べた結果(ノーマライゼーション/サブトラクション)を示す。この結果から、ノーマライゼーション/サブトラクションの有無で、完全長翻訳領域を持つクローン数(B)に対するヒット数(A)〔B/A〕にほとんど変化はなく、ノーマライゼーション/サブトラクションの有無で、全長鎖cDNAの割合などには変化がないことがわかる。
【0029】
【表1】
【0030】(シークエンシング用DNAの調製とシークエンシング)Qボットマシンで釣菌し培養した各cDNAクローンからのプラスミドDNAは、多試料の処理に適した簡便で迅速な伊藤らの方法で調製した(Itohら:Nucleic Acids Res. 25 (1997) 1315-1316)。また、塩基配列の決定は dye-terminator 法によりABIー377ならびにRISAを用いて行った(第21回日本分子生物学年会抄録1Pー570(1998年12月横浜))。
【0031】(コンピューターによる解析)5'末端または3'末端100b以上シーケンシングして得られた配列について、理化学研究所末端配列データベースと照合し(末端の100bについて、3'末端についてはポリAテールを除く100bについて、BLASTの P valueが10-25以下のものを同一と見做す。相同性に換算すると88%以上に相当する。)、未登録のものについて更にNCBIのGenBank、EMBL、DDBJ、PDB各塩基配列データベースならびにマウスEST・ヒトESTデータベースとの検索(末端の100bについて、3'末端についてはポリAテールを除く100bについて、BLASTの P valueが10-10以下のものを既知と見做す。相同性に換算すると約75%以上に相当する。)を行った。表2は、胃、舌、10日目胚から作成したライブラリーについて、本発明のノーマライゼーション/サブトラクションの効果を評価したものである。表中、クローン数は配列決定を行ったクローン数であり、この中から重複を排除して残ったクローン数をユニークと表示した。重複度は(ユニークなクローン数)/(配列決定を行ったクローン数)である。例えば、胃の場合、スタンダードでは、ユニークは838個中333個(重複度2.52)であったのに対して、本発明のノーマライゼーション/サブトラクションを用いた場合、ユニークは849個中592個(重複度1.43)であった。即ち、ノーマライゼーション/サブトラクションを用いることで重複度を低減できることが分かる。さらに、表中の非ESTとは、クローン中の新規な配列の割合である。例えば、胃の場合、スタンダードでは、333個のユニーク中に38個(11.4%)の新規配列を有するクローンが含まれていた。一方、本発明のノーマライゼーション/サブトラクションを用いた場合、592個のユニーク中に98個(16.5%)の新規配列を有するクローンが含まれていた。即ち、本発明のノーマライゼーション/サブトラクションを用いた方法では、新規配列を有するクローンの個数の割合が約3倍になっている。このように、ノーマライゼーション/サブトラクションを行うことによって、重複のないcDNAが単離しやくなり、より効率よくカタログ化されたライブラリーを作成できる。また、ESTとして未登録である新規なcDNAの割合も増加する。
【0032】
【表2】
【0033】
【発明の効果】本発明の方法によれば、全長鎖cDNAライブラリーの作成に際して、重複のないcDNAが単離しやくなり、ライブラリーサイズを大きくしても重複のないカタログ化が可能となる。
【図面の簡単な説明】
【図1】新規ベクターλ-FLC-1の制限酵素地図。
【図2】マウス膵臓から作成したcDNA標品の電気泳動ゲルパターン。右側はノーマライゼーション/サブトラクションを行ったもの、左側はノーマライゼーション/サブトラクションを行っていないコントロール。
【特許請求の範囲】
【請求項1】 複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【請求項2】 (a)〜(c)の核酸がRNAである請求項1に記載の方法。
【請求項3】 (a)〜(c)のRNAがビオチン化RNAであり、このビオチン化RNAを前記完全長cDNAを含む集団中のcDNAとハイブリダイズさせ、ハイブリダイズしたRNA−cDNAをストレプトアビジン被覆した磁気ビーズを用いて前記集団から除去する請求項2に記載の方法。
【請求項4】 前記完全長cDNAを含む集団から(a)〜(c)の核酸とハイブリダイズしたcDNAを除去して得られたcDNAを、長鎖cDNAもクローニングできるように改良したλPSベクターを用いてクローニングして、cDNAライブラリーを作成する請求項1〜3のいずれか1項に記載の方法。
【請求項5】 前記改良したλPSベクターが、λPSベクターの左アームに6Kbのスタッファー断片を挿入したベクターである請求項4記載の方法。
【請求項1】 複数の完全長cDNAを含む集団から、新規な完全長cDNAの存在割合の高いcDNAライブラリーを作成する方法であって、前記完全長cDNAを含む集団から、以下の(a)〜(c)の核酸とのハイブリダイズを利用して、(a)〜(c)の核酸とハイブリダイズしたcDNAを除去する工程を含むことを特徴とする方法。
(a)上記cDNAライブラリーを作成しようとする組織における存在量が多い、クラスI(アバンダント)及びクラスII(インターメディエート)に属する核酸、(b)上記方法を用いて完全長cDNAを含む集団からハイブリダイズを利用して分離された核酸−cDNAから回収された核酸、及び(c)クラスIII(レア)に属するcDNAを含むcDNAライブラリー中のcDNAまたはその転写物であるRNAからなる核酸
【請求項2】 (a)〜(c)の核酸がRNAである請求項1に記載の方法。
【請求項3】 (a)〜(c)のRNAがビオチン化RNAであり、このビオチン化RNAを前記完全長cDNAを含む集団中のcDNAとハイブリダイズさせ、ハイブリダイズしたRNA−cDNAをストレプトアビジン被覆した磁気ビーズを用いて前記集団から除去する請求項2に記載の方法。
【請求項4】 前記完全長cDNAを含む集団から(a)〜(c)の核酸とハイブリダイズしたcDNAを除去して得られたcDNAを、長鎖cDNAもクローニングできるように改良したλPSベクターを用いてクローニングして、cDNAライブラリーを作成する請求項1〜3のいずれか1項に記載の方法。
【請求項5】 前記改良したλPSベクターが、λPSベクターの左アームに6Kbのスタッファー断片を挿入したベクターである請求項4記載の方法。
【図1】

【図2】
【図2】
【公開番号】特開2000−325080(P2000−325080A)
【公開日】平成12年11月28日(2000.11.28)
【国際特許分類】
【出願番号】特願平11−143429
【出願日】平成11年5月24日(1999.5.24)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成10年12月16日 日本分子生物学会開催の「第21回日本分子生物学会年会」において文書をもって発表
【出願人】(000006792)理化学研究所 (14)
【Fターム(参考)】
【公開日】平成12年11月28日(2000.11.28)
【国際特許分類】
【出願日】平成11年5月24日(1999.5.24)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成10年12月16日 日本分子生物学会開催の「第21回日本分子生物学会年会」において文書をもって発表
【出願人】(000006792)理化学研究所 (14)
【Fターム(参考)】
[ Back to top ]