
定向進化のための交叉点の最適化
【課題】機能性生体分子が強化された生体分子ライブラリーを作製する方法を提供すること。
【解決手段】組換えポリペプチド、および/または、核酸の中へより効率的に多様性を設計するための方法と装置を提供する。例えば、アミノ酸配列、またはヌクレオチド配列中の潜在的交叉部位を選択、および/または、評価する様々な方法、ならびに得られたキメラ産物配列を提供する。これらの方法には、例えば、組換えに用いるための配列および交叉部位の選択および評価における、構造的、機能的、および/または、統計的なデータの考慮が含まれる。
【解決手段】組換えポリペプチド、および/または、核酸の中へより効率的に多様性を設計するための方法と装置を提供する。例えば、アミノ酸配列、またはヌクレオチド配列中の潜在的交叉部位を選択、および/または、評価する様々な方法、ならびに得られたキメラ産物配列を提供する。これらの方法には、例えば、組換えに用いるための配列および交叉部位の選択および評価における、構造的、機能的、および/または、統計的なデータの考慮が含まれる。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願との相互参照)
この出願は、米国特許出願第60/363,505号(2002年3月9日出願)および米国特許出願第60/373,591号(2002年4月18日出願)の米国特許法第119条(e)にもとづく利益を求めるもので、これらの出願の全体を本明細書中で援用する。
【0002】
(著作権についての留意すべきこと)
連邦法施行規則第37巻(37 C.F.R)第1.71条(e)に従い、出願人は、この開示の一部が著作権保護の対象となる材料を含むことを指摘する。著作権の所有者は、特許商標庁のファイルに開示または記録されるとおりに第三者による特許書類または特許開示の複製に対しては、なんら反対するものではないが、そうでなければいかなるものであっても著作権のすべてを留保する。
【0003】
この発明は、バイオインフォマティクス、構造活性相関(SAR)、および組換えタンパク質および核酸を設計するプロセスへの構造活性分析の適用の分野に関する。
【背景技術】
【0004】
(発明の背景)
生体分子の新規の機能または改善された機能の探索は、取り組みし甲斐のある試みである。例えば、所望の特性を現すタンパク質の作製および同定を目的として、改変体タンパク質のライブラリーを生成する方法およびスクリーニングする方法が開発されている(例えば、Stemmer,W.P.(1994)「Rapid evolution of
a protein in vitro by DNA shuffling」Nature 370:389−391を参照せよ)。いくつかの方法では、ほとんど機能性分子を含まないライブラリーが生成される(例えば、Ostermeier(1999)「A combinatorial approach to hybrid enzymes independent of DNA homology」Nature Biotech 17:1205を参照せよ)。スクリーニング能力の限界は、そのような機能性分子の発見を困難なものにする。したがって、機能性生体分子が強化された生体分子ライブラリーを作製する方法の存在が求められている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
機能性生体分子が強化された生体分子ライブラリーを作製する方法を提供すること。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、組換えポリペプチドおよび/または核酸の中に多様性をより効率的に操作するための方法およびデバイスを提供する。例えば、アミノ酸配列またはヌクレオチド配列での潜在的な組換え交叉部位を選択および/または評価するための種々の方法を提供するとともに、結果として生ずるキメラ産物配列を提供する。これらの方法として、例えば組換えで使用される配列および交叉部位の選択および評価で構造的、機能的、および/または統計学的データを考慮することが挙げられる。
【0007】
特に、本発明は2つ以上の生体分子間の交叉点を選択する方法を提供する。本発明の一局面は、基準ペプチド配列上の複数の潜在的交叉点の適応度を決定する方法を提供する。そのような方法は、以下の一連の操作によって記述され得る:(a)基準ペプチド配列上の複数の潜在的交叉点の各々について、適応度パラメータの全体的な値を計算すること、(b)潜在的交叉点に対する適応度パラメータの全体的な各々の値にもとづいて、キメラ・ペプチドに対する実際の交叉点を選択することである。このキメラ・ペプチドは、基準配列由来の部分的な配列を含む。これらの方法では、適応度パラメータの全体値が、検討中である潜在的交叉点を持つ複数のキメラに対する適応度パラメータの複数の個別値から、計算される。これらのキメラは、基準配列中に種々の長さの部分配列を挿入し、検討中である交叉点において、各部分配列を終了させることにより、生成され得る。
【0008】
実際の交叉点を選択した後、キメラ・ペプチドをコードする少なくとも1つのキメラ核酸を作成することによりこの方法を継続させ得る。また、そのようなキメラ核酸は、選択された交叉点をコードする少なくとも1つのオリゴヌクレオチドを含むオリゴヌクレオチドを組換えることによって生成され得る。このオリゴヌクレオチドは、2つの部分的な配列を含み、そのうちの1つは1本の親ペプチドの部分的な配列をコードし、他方は別の親ペプチドの部分的な配列をコードし、この2つの部分的な配列は、選択された交叉点に対応するオリゴヌクレオチド内の1つの場所で交わる。
【0009】
適応度パラメータは、特定の物理的基準に合うペプチドの能力の任意の尺度であり得る。一例において、適応度パラメータは、ペプチドの結合特性を増加または減少させるキメラ対立遺伝子の能力の尺度を提供する。別の例では、ペプチドの折り畳みを保存または改善するキメラ対立遺伝子の能力の尺度を提供する。
【0010】
適応度パラメータ(検討中の潜在的交叉点を持つ特定のキメラ配列に対して)の個別値は、種々の技術で得られ得る。一例において、個別値を以下の方法で測定する。すなわち、(i)基準ペプチド配列に対してキメラ配列をアラインメントし(ii)接触マップからキメラの接触残基を同定し、そして(iii)キメラの接触残基に対する残基間ポテンシャル(residue−residue potential)を合計する。
【0011】
基準ペプチド配列は、多くの理由で選ばれ得る。一例において、基準ペプチドは天然に生ずるペプチドである。別の例では、組換えまたは突然変異の手順によって同定される非天然のペプチドである。いくつかの場合において、上記方法で使用される親ペプチドの1つは、それ自体が基準ポリペプチド配列である。
【0012】
一般に、この方法は、複数の交叉点の選択を含み、基準ペプチドの部分的な配列を含む複数のキメラ・ペプチドを生成する。本明細書中でさらに詳しく説明されるように、これら複数のキメラ・ペプチドは、ペプチド・ライブラリーとして生成され得る。このライブラリーのメンバーは、種々の技術で生成され得る。本発明は、(i)ペプチド・ライブラリーの選択されたメンバーが発現され得る発現系を提供し、(ii)その発現系にペプチド・ライブラリーの選択されたメンバーをコードするポリヌクレオチドをクローニングし、そして(iii)ペプチド・ライブラリーの選択されたメンバーを発現させることにより、選択されたメンバーのペプチド・ライブラリーを生成する方法もまた、提供する。
【0013】
本発明の別の局面は、限定されるものではないが、(i)基準生体分子または基準生体構造の基準配列を提供し、(ii)基準配列に関する接触マップを生成し、(iii)配列間で1つ以上の交叉点が決定される第1の生体分子の第1の配列と第2の生体分子の第2の配列とを提供し、(iv)第1および第2の配列を基準配列にアラインメントし、(v)第1の配列から得た部分配列を第2の配列から得た部分配列と置換し、キメラ生体分子配列を生成し、(vi)キメラ生体分子配列を接触マップと比較して、基準生体分子の接触マップ中の近位要素に対応するキメラ生体分子配列中の2つ以上の要素(例えば、ヌクレオチド塩基またはアミノ酸側鎖、もしくはアルファ炭素)を選択し、そして(vii)選択された要素をスコアを記録する方法であり、該スコアは、基準生体分子と類似または同一の安定性または活性を持つキメラ生体分子配列の可能性の尺度を提供する。任意に、2つ以上の部分配列がキメラ産物配列の生成の際に交換(スワッピング)され、複数の交換(スワップ)配列が1つの親配列または複数(2つ以上)の親配列に由来し得、一実施形態では、生体分子はタンパク質またはポリペプチドであり、別の実施形態では、分析中の生体分子が核酸、例えば触媒RNA分子または他の機能的に活性な核酸分子を含む。基準生体分子配列の接触マップは、いくつかのデータ・ソースから生成され得、これらのデータ・ソースとして、結晶学的モデル、NMRデータ、タンパク質折り畳みアルゴリズム、アイデンティティモデリング、ヌクレオチド・モデリング・アルゴリズム等が挙げられるが、これらに限定されない。親配列の1つ以上の領域の挿入または「交換(スワッピング)」後、結果として生ずるキメラ分子の選択された要素が、基準分子内に同等に位置した要素と比較され、これらの選択された要素のスコア記録は、コンピュータ内(in silico)組換え手順において用いられる交叉部位を評価するためのメカニズムを提供する。
【0014】
別の局面において、本発明はまた、例えば、交叉点の評価または組換え結果の予測等の命令をコンピュータまたはコンピュータ読み取り可能媒体に提供する。該コンピュータまたはコンピュータ読み取り可能媒体として、(i)基準生体分子の基準配列を入力し、(ii)該基準配列の接触マップを生成し、(iii)基準配列に第1の配列および第2の配列をアラインメントし、(iv)第1の配列の部分配列を第2の配列の部分配列と置換してキメラ配列を作製し、(v)該キメラ配列を接触マップと比較して、接触マップ中の近位要素に対応するキメラ・アミノ酸配列中の2つ以上の要素を選択し、そして(vi)選択した要素のスコアを記録し、該スコア(およびそれによるコンピュータ読み取り可能媒体)は、キメラ配列が、基準配列と比較して三次元構造上のなんらかの類似性または同様の活性を保持する可能性の尺度を提供し、それにより、選択された組換え現象の組換え結果の予測または交叉点を評価する。
【0015】
基準タンパク質のアミノ酸スペーシングは、当業者に公知の多くの技術によって決定することができる。そのような技術としては、例えば、結晶学、NMRスペクトロスコピー、およびEPRスペクトロスコピー等が挙げられる。あるいは、情報は、コンピュータ内(in silico)で公に入手可能であるか、またはコンピュータ内(in silico)で生成され得;タンパク質折り畳み分析または分子モデリングおよび/もしくは残基間距離の計算を実行するソフトウェアは、いくつかのベンダーから入手可能であり;お互いの臨界距離内にあるアミノ酸残基の同定に使用され得る。この臨界距離は、関与するアミノ酸残基、分子相互作用の性質、および残基が基準タンパク質の活性に関連して果たす役割により変動し得る。任意に、臨界距離は、約2オングストロームから約6.5オングストローム、または、例えば2.5オングストロームから約4.5オングストローム、もしくは約4.5オングストローム未満の範囲である。
【0016】
本発明の方法およびコンピュータを基礎としたデバイスにおいて、第1の親配列(例えば、アミノ酸配列)の領域が第2の親配列内の領域に対して挿入または置換されることにより、キメラ、またはスワップされた産物配列を生成する。本発明のいくつかの実施形態において、基準配列が、親配列の1つとして用いられる。他の実施形態において、2つ以上の「非基準」親配列が、組換えポテンシャル(recombination potential)について評価される。任意に、第1親配列および第2親配列は、互いに低い配列のアイデンティティを有する。分析のために、すべての潜在的キメラ産物または潜在的産物のサブセットのいずれかが、交叉可能性について調べられる。
【0017】
本発明のさらに別の局面は、上記の方法およびソフトウェア・システムを実行するためのプログラム命令および/またはデータのアレンジメントが提供される機械読取可能媒体を含む装置およびコンピュータ産物に関する。しばしば、プログラム命令は、ある種の方法操作を実行するためのコードとして与えられる。データは、この発明の特徴を実現するために用いられる場合、データ構造、データベース・テーブル、データ・オブジェクト、または特定情報の他の適当なアレンジメントとして提供され得る。この発明のいずれかの方法またはシステムは、機械読取可能媒体上に提供されたそのようなプログラム命令および/またはデータとして、全体的にまたは部分的に表され得る。
本発明は、例えば、以下の項目を提供する。
(項目1)
基準ペプチド配列上の複数の潜在的交叉点の適応度を決定する方法であって、該方法は、以下:
(a)該基準ペプチド配列上の該複数の潜在的交叉点の各々について、検討中の潜在的交叉点を有する複数のキメラの適応度パラメータの複数の個別の値から該適応度パラメータの全体的な値を計算する工程;および
(b)該潜在的交叉点についての該適応度パラメータのそれぞれの該全体的な値に基づき、該基準ペプチド配列の部分的な配列を含むキメラ・ペプチドのための実際の交叉点を選択する工程、
を包含する、方法。
(項目2)
前記交叉点が前記基準ペプチド配列の中間点であり、かつ、前記キメラ・ペプチドは、前記交叉点で終了する前記基準配列の部分的な配列を含む、項目1に記載の方法。
(項目3)
前記キメラ・ペプチドをコードする、少なくとも1つのキメラ核酸を産生する工程をさらに包含する、項目1に記載の方法。
(項目4)
少なくとも1つのキメラ核酸の産生が、前記選択された交叉点をコードする少なくとも1つのオリゴヌクレオチドを含むオリゴヌクレオチドを組換える工程を包含する、項目3に記載の方法。
(項目5)
前記選択された交叉点をコードする前記少なくとも1つのオリゴヌクレオチドが2つの部分的な配列を含み、1つの部分的な配列が1つの親ペプチドの部分的な配列をコードし、かつ別の部分的な配列が別の親ペプチドの部分的な配列をコードし、該2つの部分的な配列が該選択された交叉に対応する該オリゴヌクレオチド中のある位置で合同する、項目4に記載の方法。
(項目6)
前記親ペプチドの1つが、前記基準ペプチド配列を含む配列を有する、項目5に記載の方法。
(項目7)
他方の親ペプチドが、前記基準ペプチド配列をコードする遺伝子を含む遺伝子ファミリー由来の核酸によってコードされる、項目6に記載の方法。
(項目8)
前記適応度パラメータが、ペプチドの結合特性を増強するか、または低減させるキメラ対立遺伝子の能力の尺度を含む、項目1に記載の方法。
(項目9)
前記適応度パラメータが、ポリペプチドの折り畳みを保存するかまたは改善する、キメラ対立遺伝子の能力の尺度を含む、項目1に記載の方法。
(項目10)
前記検討中の潜在的交叉点を有するキメラ配列について、前記適応度パラメータの個別の値を計算する工程をさらに包含する、項目1に記載の方法。
(項目11)
前記適応度パラメータの前記個別の値を計算する、項目10に記載の方法であって、該方法は、以下:
(i)前記キメラ配列を前記基準ペプチド配列に整列させる工程;
(ii)接触マップから前記キメラの接触残基を同定する工程;および
(iii)前記キメラの接触残基について、残基間ポテンシャルを合計する工程、
を包含する、項目10に記載の方法。
(項目12)
前記接触マップが、前記基準ペプチド中の残基の立体配置である、項目11に記載の方法。
(項目13)
前記基準ペプチド配列が、天然に存在するペプチドの配列である、項目1に記載の方法。(項目14)
前記基準ペプチド配列が、組換えまたは突然変異誘発手順により同定された非天然ペプチドの配列である、項目1に記載の方法。
(項目15)
(b)が、前記基準ペプチド配列の部分的な配列を含む複数のキメラ・ペプチドのための複数の交叉点を選択する工程を包含する、項目1に記載の方法。
(項目16)
前記複数のキメラ・ペプチドを含むペプチドのライブラリーを産生する工程をさらに包含する、項目15に記載の方法。
(項目17)
前記ライブラリーの1つ以上のペプチドが、1つ以上のペプチドを発現する工程を包含する方法によって産生される、項目16に記載の方法。
(項目18)
項目15に記載の方法であって、該方法は、以下:
(i)前記ペプチドのライブラリーの選択されたメンバーを発現し得る発現系を提供する工程;
(ii)該発現系に、該ペプチドのライブラリーの選択されたメンバーをコードするポリヌクレオチドをクローニングする工程;および
(iii)該ペプチドのライブラリーの選択されたメンバーを発現する工程、
をさらに包含する、項目15に記載の方法。
(項目19)
前記選択された実際の交叉点と、前記交叉点で終了する、前記基準配列の少なくとも1つの部分的な配列とを各々有する複数のキメラ・ペプチドを同定する工程をさらに包含する、項目1に記載の方法。
(項目20)
基準ペプチド配列上の複数の潜在的交叉点の適応度を決定するためのプログラム命令を提供する機械可読媒体を含むコンピューター・プログラム製品であって、
該プログラム命令は、以下:
(a)該基準ペプチド配列上の該複数の潜在的交叉点の各々について、検討中の潜在的交叉点を有する該複数のキメラの適応度パラメータの複数の個別の値から適応度パラメータの全体的な値を計算するためのコード;および
(b)該潜在的交叉点についての該適応度パラメータのそれぞれの該全体的な値に基づき、該基準ペプチド配列の部分的な配列を含むキメラ・ペプチドのための実際の交叉点を選択するためのコード、
を含む、製品。
(項目21)
前記交叉点が前記基準ペプチド配列の中間点であり、かつ、前記キメラ・ペプチドは、前記交叉点で終了する前記基準配列の部分的な配列を含む、項目20に記載のコンピューター・プログラム製品。
(項目22)
前記適応度パラメータが、ペプチドの結合特性を増強するか、または低減させるキメラ対立遺伝子の能力の尺度を含む、項目20に記載のコンピューター・プログラム製品。
(項目23)
前記適応度パラメータが、ポリペプチドの折り畳みを保存するか、または改善するキメラ対立遺伝子の能力の尺度を含む、項目20に記載のコンピューター・プログラム製品。
(項目24)
前記検討中の潜在的交叉点を有するキメラ配列に関して、前記適応度パラメータの個別の値を計算するためのコードをさらに含む、項目20に記載のコンピューター・プログラム製品。
(項目25)
項目24に記載のコンピューター・プログラム製品であって、前記適応度パラメータの前記個別の値を計算するための前記コードが、以下:
(i)前記キメラ配列を前記基準ペプチド配列に整列させるためのコード;
(ii)接触マップから前記キメラの接触残基を同定するためのコード;および
(iii)前記キメラの接触残基に関する、残基間ポテンシャルを合計するためのコードを含むことを特徴とする項目24に記載のコンピューター・プログラム製品。
(項目26)
前記接触マップが前記基準ペプチド中の残基の立体配置であることを特徴とする項目25に記載のコンピューター・プログラム製品。
(項目27)
(b)が、前記基準ペプチド配列の部分的な配列を含む複数のキメラ・ペプチドのための複数の交叉点を選択するためのコードを含むことを特徴とする、項目20に記載のコンピューター・プログラム製品。
(項目28)
前記複数のキメラ・ペプチドを含むペプチドのライブラリーを同定するためのコードをさらに含む、項目27に記載のコンピューター・プログラム製品。
(項目29)
前記選択された実際の交叉点と、前記交叉点で終了する、前記基準配列の少なくとも1つの部分的な配列とを各々有する複数のキメラ・ペプチドを同定するためのコードをさらに含む、項目20のコンピューター・プログラム製品。
(項目30)
コンピューターで実行する、2つ以上の潜在的交叉点の適応度を決定する方法であって、該方法が、以下:
(a)基準ペプチド配列中に第1の潜在的交叉点を同定する工程;
(b)前記基準ペプチド配列からの1つの部分的な配列と、異なった配列からの別の部分的な配列とを含み、かつそれらによる前記潜在的交叉点を有する第1のキメラ配列を生成する工程;
(c)前記基準ペプチド配列に関する接触マップに、第1のキメラ配列を適用する工程;(d)前記接触マップを用いて選択された、第1のキメラ配列中の残基間相互作用から適応度パラメータの値を計算する工程;
(e)1つ以上の追加キメラ配列に関し(b)〜(d)を反復する工程;
(f)(b)〜(e)で検討したキメラ配列の各々に関して、前記適応度パラメータの値から全体的な適応度の値を計算する工程;
(g)前記基準ペプチド配列中に第2の潜在的交叉を同定する工程;および
(h)前記第2の潜在的交叉点に関して、(b)〜(f)をおこなう工程を包含する方法。
(項目31)
複数の追加潜在的交叉点に関して、(a)〜(f)を反復する工程をさらに包含する、項目30に記載の方法。
(項目32)
1つ以上のペプチドの生産における使用のために、複数の潜在的交叉点から1つ以上の交叉点を、全体的な適応度の値に基づいて、複数の潜在的適応度の値に関して選択する工程をさらに包含する、項目31に記載の方法。
(項目33)
2個以上の生体分子の間の交叉点を選択する方法であって、該方法が、以下:
i)基準生体分子の基準配列を提供する工程;
ii)前記基準配列に関して、接触マップを作成する工程;
iii)第1の生体分子の第1の配列と、第2の生体分子の第2の配列を提供し、それらの間で1つ以上の交叉点が決定される工程;
iv)第1の配列および第2の配列を基準配列と整列する工程;
v)第1の配列からの部分配列を、第2の配列からの部分配列と交換し、前記部分配列が選択された交叉点で終了するようなキメラ生体分子配列を作製する工程;
vi)前記接触マップと前記キメラ生体分子配列を比較し、前記キメラ生体分子配列中に、前記基準生体分子の前記接触マップ中の近位要素に対応する2つ以上の要素を選択する工程;ならびに
vii)選択された要素を得点する工程
を包含し、得点が、前記キメラ生体分子配列が前記基準生体分子と同一、または類似の特性を有する可能性の尺度を提供することを特徴とする、方法。
(項目34)
前記生体分子がポリペプチドまたはタンパク質を含み、かつ前記要素がアミノ酸残基を含むことを特徴とする、項目33に記載の方法。
(項目35)
前記生体分子が核酸を含み、かつ前記要素がヌクレオチドを含むことを特徴とする、項目33に記載の方法。
(項目36)
前記基準配列が前記第1の配列であることを特徴とする、項目33に記載の方法。
(項目37)
前記接触マップを作成する工程が、前記生体分子中の要素の1つ以上の間隔を決定し、相互の有意な距離内に2つ以上の近位要素を同定する工程を包含することを特徴とする、項目33に記載の方法。
(項目38)
前記有意な距離の範囲が約2オングストロームから約6.5オングストロームであることを特徴とする、項目37に記載の方法。
(項目39)
前記有意な距離が約4.5オングストローム未満であることを特徴とする、項目37に記載の方法。
(項目40)
前記第1の配列および前記第2の配列を提供する工程が、BLASTPアルゴリズムおよび初期パラメータを用いて決定されるような、アミノ酸配列、または核酸配列を提供し、約60%以下のアミノ酸配列同一性を有する2つのタンパク質を得る工程を包含することを特徴とする、項目33に記載の方法。
(項目41)
前記基準生体分子と同一、または類似の特性が、酵素活性、またはタンパク質安定性を含むことを特徴とする、項目33に記載の方法。
(項目42)
スコアリングが、前記キメラ生体分子配列中の前記2つ以上の選択された要素の前記接触エネルギーを計算する工程を包含することを特徴とする、項目33に記載の方法。
(項目43)
前記接触エネルギーがMiyazawa−Jerniganエネルギー・マトリックスを用いて計算されることを特徴とする、項目42に記載の方法。
(項目44)
スコアリングが三角形輪郭プロットにおけるスコアの提示が含まれることを特徴とする、項目33に記載の方法。
(項目45)
1つ以上のキメラ生体分子を合成する工程をさらに包含する、項目33に記載の方法。
(項目46)
前記1個以上のキメラ生体分子を合成する工程が、1つ以上の組換え構築物を提供する工程を包含する、項目45に記載の方法。
(項目47)
前記1個以上のキメラ生体分子を合成する工程が、2つ以上の親配列に1つ以上の組換え操作を行い、それによって、前記キメラ生体分子をコードする1つ以上の組換え構築物を作製する工程を包含する、項目45に記載の方法。
(項目48)
1つ以上のキメラ生体分子をアッセイする工程をさらに包含する、項目45に記載の方法。
(項目49)
コンピューター・コードを含むコンピューター読み込み可能な媒体であって、前記コンピューター・コードが、
i)基準生体分子の基準配列を入力し;
ii)前記基準配列に関する接触マップを作成し;
iii)第1の配列および第2の配列を基準配列と整列させ;
iv)第1の配列上の第1の交叉部位および第2の交叉部位を含む部分配列を、第2の配列に由来する対応する部分配列で交換してキメラ配列を作製し;
v)前記接触マップと前記キメラ配列とを比較して、前記接触マップの近位要素に対応する2つ以上の要素を、前記キメラ・アミノ酸配列の中で選択し;そして
vi)選択された要素を得点する
ことを特徴とするコンピューター読み込み可能な媒体。
(項目50)
前記コンピューター・コードがまた、少なくとも1つの追加交叉部位に関してiv)〜vi)を反復することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目51)
(i)が、既知の生体分子のアミノ酸配列を提供する工程、または前記既知の生体分子をコードする核酸配列を提供する工程を包含する項目49に記載のコンピューター読み込み可能な媒体。
(項目52)
核酸データベースまたは、生体分子データベースに尋ねる工程が入力に包含されることを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目53)
接触マップを作成する工程が、前記基準生体分子の結晶モデルまたはNMRモデルからアミノ酸間隔を決定し、相互に有意な距離内にある残基を同定する工程を包含する工程を特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目54)
前記接触マップを作成する工程が、前記基準生体分子のタンパク質折り畳み分析からアミノ酸間隔を決定し、相互に有意な距離内にある残基を同定する工程を包含することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目55)
前記有意な距離がアミノ酸間相互作用の特性によって変化することを特徴とする、項目54に記載のコンピューター読み込み可能な媒体。
(項目56)
前記有意な距離が約4.5オングストローム未満であることを特徴とする、項目54に記載のコンピューター読み込み可能な媒体。
(項目57)
前記第1のアミノ酸配列および第2のアミノ酸配列を整列する工程が、核酸データベースまたはタンパク質データベースに尋ねる工程を包含することを特徴とする、項目49に
記載のコンピューター読み込み可能な媒体。
(項目58)
前記スコアリングに、キメラ・アミノ酸配列中の1対のアミノ酸の接触エネルギーを計算する工程を包含し、その対の残基が、前記接触マップで接触している残基に対応することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目59)
スコアリングが、前記接触マップで接触している残基に対応する、前記キメラ・アミノ酸配列中の全残基の接触エネルギーを合計する工程を包含することを特徴とする項目49に記載のコンピューター読み込み可能な媒体。
(項目60)
スコアリングが、Miyazawaエネルギー・マトリックスを用いて、相互作用する残基の接触エネルギーを計算する工程を包含することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目61)
スコアリングが、グラフィカル・ユーザー・インターフェースでユーザーに得点を提示する工程を包含することを特徴とする、項目49に記載の任意の1つのコンピューター読み込み可能な媒体。
(項目62)
スコアリングが、三角プロットで得点を提示する工程を包含することを特徴とする、項目49の任意の1つのコンピューター読み込み可能な媒体。
(項目63)
交叉部位を査定するための融合システムであって、該融合システムが、以下:
項目49に記載のコンピューター読み込み可能な媒体;および
グラフィック・インターフェース
を含む、融合システム。
本発明のこれらの特徴および他の特徴を、以下の図面と共に発明の詳細な説明において、より詳細に説明する。
【図面の簡単な説明】
【0018】
【図1A】図1Aは、本発明の一実施形態を示すプロセスの流れ図である。
【図1B】図1Bは、本発明の一実施形態を示すプロセスの流れ図である。
【図1C】図1Cは、本発明の一実施形態を示すプロセスの流れ図である。
【図2】図2のパネルAは、MLEII配列の部位が基準タンパク質MLEIのセグメントに置換するP.putida由来の2通りの潜在的キメラMLEIおよびMLEIIそれぞれの接触エネルギー・マップを提供する。縦軸は、置換が開始されるMLEIの残基位置を示し、横軸は置換の長さを示す。ΔEcは、マップ内の色によって示される。構造的に破壊的であることが予測されるキメラは、マゼンタで示される。好ましいと予測されるものは、赤色で示され、また中性的な変化は、青色および緑色で示されている。パネルBは、基準タンパク質P.putidaMLEIの対応セグメントと置換するP.putidaMLEII由来の1〜80アミノ酸セグメントについての平均ΔEcを示す。標識位置(98、119、144、172、201、228、254、280、302、および328)は、交叉点が表示された直線状配列に沿ったアミノ酸の数を表す。
【発明を実施するための形態】
【0019】
(発明の詳細な説明)
本発明を詳細に説明する前に、本発明が、特定の組成物または生物学的システムに限定されるものではなく、これらは無論変化し得ることが、理解されるべきである。本明細書で用いられる専門用語は、特定の実施形態を説明することのみを目的としたものであって、限定することを意図したものではないこともまた、理解されるべきである。この明細書および添付した特許請求の範囲で使用されるように、単数形(「a」、「an」、および「the」)は、内容が明らかにそれを示していない限り、複数の指示されるものも含まれる。したがって、例えば、「核酸配列(a nucleic acid sequence)」は、そのような配列が2つ以上組み合わさったものも含み、「ポリペプチド(a
polypeptide)」に対する言及は、複数のポリペプチドからなる複数の混合物が含まれる、などである。
【0020】
本明細書中で特に定義しない限り、本明細書で使用されるすべての技術用語および科学用語は、本発明が属する技術分野の当業者によって一般に理解されるものと同一の意味を持つ。本明細書中で記載される方法および材料と類似または同等の方法および材料のいずれも本発明の実施または試験に使用され得るが、好ましい材料および方法が、本明細書中で記載される。
【0021】
(定義)
本発明の記載および権利主張において、後述する定義に関して以下の専門用語を用いた。
【0022】
「部分配列(subsequence)」または「フラグメント」は、ヌクレオチドまたはアミノ酸の配列全体のうちの任意の一部分である。用語「配列」および用語「文字列」は、本明細書中で、タンパク質(すなわち、タンパク質配列またはタンパク質文字列)におけるアミノ酸残基の順番およびアイデンティティを言うか、または核酸におけるヌクレオチド(すなわち、核酸配列または核酸文字列)の順番およびアイデンティティを言い、交換可能に用いられる。
【0023】
本明細書中で使用される場合、用語「交叉点」は、配列の中の一位置を言い、該配列のその部分の起点が1つのソースから別のソースへ変化または「交叉」する(例えば、親配列間での交換にかかわる部分配列の末端)。
【0024】
本明細書中で使用される場合、用語「接触マップ」は、生体分子の構成要素間の相互作用の描写を言い、一般に二次元グラフまたはデータ・マトリックスの形態で描写され、それにより、該生体分子の三次元構造の単純化表現または簡素化表現が提供される。
【0025】
本明細書中で使用される場合、用語「キメラ」は1つ以上の親分子間での組換え現象の産物を言うために用いられる。
【0026】
本明細書中で使用される場合、用語「近位要素(proximal elements)」は、三次元構造またはモデルで互いに近位または空間的に近接している配列構成要素(例えば、アミノ酸側鎖またはアルファ炭素、もしくは核酸塩基)を言う。
【0027】
本明細書中で交換可能に用いられる用語「ポリペプチド」、用語「ペプチド」、および用語「タンパク質」は、アミノ酸残基のポリマーを言う。この用語を、天然アミノ酸ポリマーと同様に、1つ以上のアミノ酸残基が対応する天然アミノ酸のアナログ、誘導体、または模倣物であるアミノ酸である、アミノ酸ポリマーに適用する。例えば、ポリペプチドの修飾または誘導体化は、炭水化物残基の付加による糖タンパク質の形成による。「ポリペプチド」、「ペプチド」、および「タンパク質」という用語には、糖タンパク質および非糖タンパク質が含まれる。
【0028】
本明細書中で使用される場合、用語「約」は、該用語が結合した値の任意に±25%、好ましくは、±10%、より好ましくは±5%、またはより好ましくは±1%の値を言う。
【0029】
本発明は、組換えのための潜在的交叉部位を選択し、キメラ産物の三次元構造(およびそれによる活性および安定性)における組換えの効果を評価する方法を提供する。このアプローチは、適当に折り畳まれた産物および/または所望の機能性を提供すると思われる組換え配列ライブラリーの生成がよりいっそう効率的になる。
【0030】
(方法論の概要)
一実施形態において、図1Aでフローチャートとして示した3通りの操作のうちの2つ以上を用いた一般的プロセスとして、本発明を見ることもできる。その図に示すように、基準ペプチド配列上の種々の潜在的交叉点の関数として適応度パラメータを計算することで、一般的プロセスがブロック01で開始される。適応度パラメータは、交叉点を生成する異なる配列由来の対応アミノ酸によって基準配列の1つ以上のアミノ酸を置換した場合、該基準配列に関する適応度の変化として計算され得る。ブロック01の動作が完了すると、一連の潜在的交叉点(ペプチド配列内の特定の残基位置の前または後に認められる)の各々について別々に算出された適応度の値を有するものとなる。適応度パラメータが最大化すると思われる(または少なくとも特定の閾値に達する)交叉点が、それに続く合成、例えば組換え手順による合成に対して、選択される。これらの交叉点を用いることで、意図された目的にキメラ・ペプチドが「適合」する可能性が高まる。
【0031】
図1Aのブロック05に示すように、次の方法は1つ以上の選ばれた交叉点を持つキメラ・ペプチドを作製することが含まれる。これらのペプチドは、種々の手順で生成され得るメンバーを持つライブラリーから構成されるものであってもよい。1つの適当な手順では、オリゴヌクレオチド(いくつかは選択交叉点を持つキメラを生成するように設計されている)を再結合することによって、全長キメラ核酸ライブラリーを合成することを含む。このことは、一方が一方の親由来であり、他方が他方の親由来である2つの部分的な配列を持つオリゴヌクレオチドを用いることによって達成することができ、それら2つの部分的な配列により、基準配列上に定められた交叉点で合致する。図1Aでの最初の2つの動作は、データ処理を伴い、最後の動作は物理的手順を伴う点に留意する。別の言い方をすれば、計算による事前選別(交叉点適応度を基礎とする)を通過するそれらのタンパク質改変体のみが、例えば該タンパク質改変体をコードするポリヌクレオチドによる合成およびその後の該ポリペプチドの発現によって、研究室で作られる。これらの技術を用いて、機能的改変体(すなわち、これらの予測されたより良い改変体のみならず、多くの場合それらの予測改変体を含む)が冨化されたライブラリーを生成することも可能である。
【0032】
いくつかのケースでは、適応度パラメータまたは図1Aのブロック01で計算された適応度パラメータでの変化は、ポリペプチドの安定性の尺度である。ポリペプチド適応度パラメータの例として、(1)ポリペプチドの折り畳みの保存または改善、ならびに(2)必要に応じたポリペプチドの結合特異性の増加または減少をおこなうキメラ対立遺伝子の能力の尺度が含まれる。
【0033】
基準ペプチド配列(図1Aのブロック01)の「交叉スペース」を調べるために、アルゴリズムが基準配列の各潜在的交叉点(ペプチド配列内の特定の残基位置の前または後に認められる)での適応度を計算する。基準配列それ自体は、天然ペプチド配列または非天然ペプチド配列、例えば組換えまたは他の定向進化技術によって同定される配列であってもよい。さらに、基準配列は、その後に続く組換え手順で使用するために選ばれる1つの親配列であってもよい。あるいは、それ自体が組換え手順では使用されない配列であってもよい。どちらにしても、好ましくは、後に続く組換え手順で用いられる1つ以上の他の親配列に関連している。本明細書のどこかに記載したように、基準配列と、組換え手順で用いられる1つ以上の親配列とがタンパク質ファミリーのメンバーとして関係する。
【0034】
図1Bは、図1Aのブロック01を実施するための1つの典型的な手順を示している。プロセスは、1つの適応度の値が計算されるサブセットに対して基準配列内の潜在的交叉点のプールを任意に制限するアルゴリズムによって、ブロック11で開始される。一例では、潜在的交叉位置が5番目の残基またはなんらかの他の調節位置に限定される。または、潜在的交叉位置が、特に2次または3次構造(例えば、基準ペプチド等の解けた構造のループ流域)にある残基に限定される。
【0035】
次に、ブロック13では、アルゴリズムは基準配列にある「最新(current)」交叉位置を同定する。アルゴリズムが配列のNまたはC末端から始まる該配列に沿って単に進行し、潜在的交叉点を1つずつ検討することが可能であることに留意する。または、潜在的交叉位置の評価になんらかの他の順序を用いてもよい。アルゴリズムが検討のための他の交叉点を選択する順番にかかわらず、個々の交叉点は別々に検討される。検討の際、交叉点は、アルゴリズムの目的のために「最新」交叉点になる(このことは、例えば並行処理の実現により、複数の交叉点が同時に検討しうる可能性を排除するものではない)。
【0036】
「最新」交叉点セットにより、アルゴリズムは次にその位置に対する「最新」キメラを生成する。ブロック15を見よ。本発明の特定の好ましい実施形態が同一の交叉点(基準配列で定義されるように)を持つ多数のキメラを考慮することで、その交叉点にある「全体的な(overall)」適応度パラメータを得ることを理解する。これらのキメラの各々について、適応度パラメータを個々に算出する。これらの多くの適応度の値を次に組み合わせて、またはさもなければ基準配列内の特定交叉位置に関する全体的な適応度パラメータに到達する際に一緒に検討される。したがって、しばしば、所定の交叉点に対して複数のキメラを評価する必要性がある。
【0037】
この説明により、その結果として図1Bアルゴリズムの次に続くブロック(ブロック17)が最新キメラについての適応度の値の計算を伴う。さらに、その値が計算された後、アルゴリズムは基準配列の最新交叉位置について検討すべきキメラがそれ以上あるかどうかを判断する。ブロック19を見よ。もしより多くのキメラが検討すべきままであるならば、プロセス制御は、次の最新キメラを最新交叉点について検討するブロック15に戻る。これをおこなう前に、前のキメラについて計算された適応度の値は、それぞれ保存されるか、もしくは交叉位置で検討された各キメラにより連続して更新される現行の全体的適応度と場合により組み合わせられる。
【0038】
最新交叉位置で検討すべきキメラがもはや存在しないと仮定すると(ブロック19が「No」に戻る)、次にアルゴリズムは最新位置について全てのキメラに基づいた全体的な適応度を計算する。ブロック21を見よ。再び、全体的な適応度の値は、交叉位置での個々のキメラについての適応度の値の各々を考慮した値を示す。その全体値は、基準配列上で最新潜在的交叉位置を他の潜在的交叉位置と比較するために利用可能となっている。交叉位置に対する適応度の値が有利にその仲間(peers)に匹敵するならば、その後に続く組換えプロセスのための交叉位置として選ばれる可能性がある。
【0039】
全体的な適応度の値が特定の交叉位置に対して計算された後、アルゴリズムは次に、検討される潜在的交叉位置がこれ以上基準配列に存在するかどうかを決定する。ブロック23を見よ。そうである場合、プロセス制御がブロック13に戻る。このブロック13では、次の潜在的交叉位置が分析のために選択され、全体的な適応度の値がその位置で生成される。もしそうでなければ、図1Aのブロック1のプロセスが完了する。それによって、図示した例では、プロセス制御が図1Aのブロック03に移動する。この図1Aのブロック03では基準配列にある1つ以上の交叉点が適応度パラメータの相対的な全体値にもとづいて選択される。
【0040】
アルゴリズムは、置換を用いることも可能であり、該置換では、基準配列からの1つ以上の準配列を、異なる(関連があるが)配列からの1つ以上の対応準配列と置き換える。すべての場合において、アルゴリズムで現在検討中の位置で、結果として生ずるキメラは基準配列と他の配列との間で交叉点を持つべきである。図1Bのブロック17では、用いられた実際の計算は、用いられた特定の適応度パラメータに依存する。好ましい一実施形態では、適応度パラメータは、接触エネルギーである。接触エネルギーは、ポリペプチドの安定性の尺度である。それは、ポリペプチド中の残基に接触する対の残基間ポテンシャルを合計することによって計算することが可能である。接触エネルギーを計算する1つの特定のプロセスを図1Cに示す。そこに示すように、プロセスは、基準配列(または潜在的に何らかの別の配列)の「接触マップ」を受け取ることで、ブロック31で開始される。次に、ブロック33で、アルゴリズムは最新キメラを基準配列に対して並べる。これによって、キメラの残基を接触マップに適切に配置することが可能となり、それによってキメラの起こりうる折り畳みを反映する。したがって、ブロック35では、整列させたキメラにアルゴリズムが接触マップを適用する。最後に、37で、手順はキメラ内の「接触(contacting)」残基の残基間エネルギーを計算する。接触残基は、接触マップ内での該接触残基の位置によって同定された。
【0041】
一般に、接触マップは、互いに十分に近接し、ある定義された方法で互いに相互作用すると思われるポリペプチドまたは他のポリマーのそれらの残基を同定する。そのような近接した残基は、互いに「接触(contact)」した状態にあると言われる。2つの残基が所定の離間距離以内で分離している場合、これら2つの残基は接触するほど十分に近接している。一般的に、問題になる相互作用は、少なくとも2つの残基間でのエネルギー的または立体化学的相互作用である。いくつかの実施例では、異なる相互作用は、異なる離間距離を持つ。例えば、一実施形態では、約4.5オングストローム以下の離間距離を持つ2つの残基は、水素結合、イオン結合、および/または疎水相互作用により接触状態にあると思われる。一方、約2.5オングストローム以下の離間距離である2つのシスティン残基はジスルフィド相互作用により接触状態にあると思われる。一般に、接触マップは、特定の折り畳み配置の安定化等、ポリペプチドのいくつかの特性に貢献する残基を同定する簡便な方法を提供する。ポリペプチド中のあらゆる残基対は、ある程度相互作用することから、目的とする特性に貢献することができる。しかし、非常に限定された距離未満で離間したそれらの残基のみが有意に貢献する。
【0042】
したがって、接触マップは、特性を計算するのに用いられる残基の組み合わせ数に対して妥当な制限を加えることができる。このようにして、計算に必要な計算作業は、有意に正確さを犠牲にすることなく最小化される。基本的に、接触マップにより同定された接触残基対のみが全体的なポリペプチドに関する特性の計算に用いられる。一方、非接触残基は計算には含まれない。なぜなら、非接触残基側鎖の変化はタンパク質内の特異的相互作用を壊す可能性が少なく、著しい構造上の破壊を生ずる可能性が少ないからである。
【0043】
一実施例では、接触マップはポリペプチド内での残基の配置を単純に三次元で表したものである。そのような配置は、例えばX線回折データを用いてポリペプチドの構造を解くことによって推定される残基位置を示すものであってもよい。ポリペプチド内の接触残基は、上記したようにそれらの離間距離によって同定される。
【0044】
この発明で重要なことは、基準ポリペプチド配列の三次元構造に由来する接触マップが、1つ以上の関連ポリペプチド配列(一般に、検討中の交叉点を持つキメラ)に用いることができることである。関連した配列は、基準ポリペプチド配列に配置され、さらに該配置を保存するようにして接触マップに配置される。関連配列の残基は、基準配列の対応残基と同じ位置を占めるものと考えられる。この配置から、接触残基は関連配列に対して同定される。
【0045】
好ましい一実施形態では、任意の二つの残基の接触ポテンシャルは、MiyazawaおよびJerniganポテンシャルによって計算される。これらは、本明細書の他の場所で引用された種々の参考文献に記載されている。基本的に、接触残基の「M−Jポテンシャル」は、ポリペプチド全体にわって総計される。得られた総計によって、キメラの安定性の全体的な尺度が与えられる。キメラ折り畳みは、接触マップが由来する基準配列のものと一致すると仮定されることに留意する。残基間ポテンシャルは、溶媒効果を考慮することによって計算される。また、残基間ポテンシャルは残基の種類および該残基での二次構造も考慮に入れる。
【0046】
上記議論で示されたように、基準ポリペプチド上の任意の潜在的交叉点での置換の効果を評価するために、1つ以上のキメラを問題となっている交叉点を持たせることによって生成してもよい。図1Bのブロック15を見よ。最も単純な実施形態では、単一キメラだけは、選択交叉点を持って生ずる。この単純な実施形態では、そのキメラの接触エネルギーを基準配列の接触エネルギーと比較して、接触エネルギーの変化(図1BのΔBc)を得る。
【0047】
より典型的なシナリオでは、潜在的交叉点での接触エネルギーの変化が、問題となっている交叉点を各々が持つ多くの異なるキメラについて算出した変化を平均化することによって得られる。特定の交叉点を持ち、適応度パラメータの計算に用いられる種々のキメラ配列は、特定の基準に合致するようにして選択することができる。一実施形態では、交叉点のNおよび/またはC側に対する複数の準配列が、基準配列での置換のために選択することができる。これらの準配列は、基準配列をコードする遺伝子に関連した他の「親」配列から得ることが可能である。準配列は、他の親配列から組織的に選択することができる。 例えば、交叉点のN側に対する一方の準配列は、単一残基を持つことができ、他方の準配列は2つの残基を持つことができ、さらに第3の準配列は3つの残基を持つことができる、などである。置換型の準配列からなる同一の組織的(システマティック)なセットは、交叉点のC側で得ることができる。
【0048】
一具体例では、任意の交叉点に関連したキメラは、第2の親配列内の交叉点のC側から得た1ないし80残基の準配列と、該交叉点のN側から得た1ないし80残基の準配列とを含む。これらの準配列は、第1の親配列の対応する位置で、該配列内で置換される。それゆえに、160の異なるキメラが生成され、交叉点が第1の親ポリペプチドのCおよびN末端から取り除いた少なくとも80残基であると仮定して、特定の交叉点の重要性を計算する。これらのキメラの各々は、それ自身のΔEcを持つ。これらの個々のキメラΔEcの平均ΔEcは、好ましくない置換に対して交叉点の全体的な抵抗を反映するのに用いられる。
【0049】
このアプローチによって示唆されるように、本発明のいくつかの実施形態は、単一の交叉としてよりも、複数の交叉の1つとして十分に作用するという可能性にもとづいて、交叉位置が選択されるキメラ・ライブラリを設計することを目的とする。このアプローチでは、所定の公差位置に関連した複数の挿入断片について平均接触エネルギーの計算がおこなわれる。
【0050】
(基準配列)
一実施形態では、本発明の方法は、基準生体分子または生体分子構造の基準配列を提供することが含まれる。別の実施形態では、上記方法は、基準配列と基準生体分子の三次構造を提供することを含み、該基準配列は、モノマー単位(例えば、アミノ酸またはヌクレオチド)等の複数の要素を含む。これらのアプローチの両方とも、比較目的のための塩基配列を提供することが含まれる。任意に、親配列の1つは、基準配列として用いることができる。基準配列は、複数の要素(例えば、ヌクレオチドまたはアミノ酸)から構成され、所定の(または特定)の三次元構造を有する。基準配列を、当業者に周知のいくつかの方法で提供することができる。例えば、基準タンパク質のアミノ酸配列または該基準タンパク質をコードする核酸配列のいずれかを提供することができる。タンパク質をコードする核酸配列は、限定されるものではないが、cDNA、mRNA、ゲノムDNA等のいくつかの形態のいずれかで提供され得る。
【0051】
本発明の一実施形態では、基準アミノ酸配列は、基準タンパク質の配列決定によって提供される。アミノ酸配列決定法は、当業者に周知である(例えば、2,4−ジニトロフルオロベンゼン(サンガー試薬)、塩化ダンシル、フェニルイソチオシアネート(エドマン分解法)、種々のプロテアーゼを用いた方法)。
【0052】
より一般的には、基準配列は基準タンパク質(または触媒オリゴヌクレオチド)をコードする核酸の配列決定によって提供される。核酸塩基配列決定法もまた当業者に周知であり、限定されるものではないが、BergerおよびKimmel,Guide to Molecular Cloning Techniques,Methods in Enzymology volume 152 Academic Press,Inc.,SanDiego,CA(「Berger」);Sambrookら,Molecular Cloning − A Laboratory Manual(第3版),Vol.1−3,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York,2000(「Sambrook」)、およびCurrent Protocols in Molecular Biology,F.M.Ausubelら,編,Current Protocols,a joint venture between Greene Publishing Associates,Inc.and John Wiley & Sons,Inc.,(1999年を通して補遺)(「Ausubel」)に記載されている技術が挙げられる。
【0053】
本発明のさらに別の実施形態では、基準配列は核酸配列を提供し、提供された核酸配列をアミノ酸配列に翻訳するか、もしくは該配列を転写してオリゴヌクレオチド配列を生成することによって、供給される。任意に、基準配列は、核酸またはタンパク質データベースを問い合わせることによって、提供される。公共のデータベースおよび民間のデータベースが利用可能であり、該データベースとして、National Center for Biotechnology Information(www.ncbi.nhn.nih,gov)のGenBankTM、NCBI EST配列データベース、EMBL Nucleotide Sequence Database,Incyte(Palo Alto,CA)のLifeSeq(登録商標)データベース、および Celera(Rockville,MD)の「Discovery System」TMデータベース)、Swiss Institute of Bioinformatics(http:/us.expasy.org))のExPASy(Expert Protein Analysis System)プロテオミックス・サーバによって管理されたPROSITEデータベース、他のインターネット・リスト等が挙げられる。
【0054】
組換え体ポテンシャルに関して、本発明の方法を用いて多くのタンパク質ファミリーを評価することができる。代表的なタンパク質およびタンパク質ファミリーとして、限定されるものではないが、エノラーゼ・スーパファミリー、N−アセチルノイラミン酸リアーゼ・スーパファミリー、クロトナーゼ・スーパファミリー、および近隣酸素キレート折り畳み(vicinal oxygen chelate fold)のメンバー(例えば、Babbitt,PCおよびGerlt,JA.(1997)「Understanding Enzyme Superfamilies.」J.Biol.Chem.272:30591−30594)が挙げられる。さらなる標的タンパク質ファミリーは、本明細書に引用(および参考として援用)された参照文献で議論されている。特に関心のある1つのスーパーファミリーは、芳香族化合物新陳代謝に関与するムコネート・ラクトン化酵素(ムコネート・シクロイソメラーゼ)である。スーパーファミリーのメンバーは、概して一般的構造折り畳みを共有し、かつ類似の機械学的戦略を利用する。したがって、親配列同一性が低くても、さらなる所望の特性を有しながら必要な構造的折り畳みおよび基本的機械学的要素をも保持するキメラを生成することができる。
【0055】
また、機能的に活性な核酸分子、または目的とする他の核酸配列を組換えポテンシャルについて評価することができる。例えば、触媒RNA分子に対して、組換えまたは他の多様性生成手順を施すことができ、それによって該触媒RNA分子の基質特異性、触媒反応速度等を修飾し、そのようなものとして本発明で使用するための追加の標的生体分子を提供する。別の例として、mRNA、rRNA、またはtRNA分子は、本発明の方法を用いて調べることができ、また異なる活性または感受性を持つオリゴヌクレオチドの生成に用いることができる。さらなる機能的活性核酸は、限定されるものではないが、例えばタンパク質触媒反応で生物学的基質またはコファクターとして作用するRNAおよび/またはDNA分子が挙げられる。
【0056】
本発明の方法は、任意の数の追加標的生体分子または生体高分子のなかでの交叉点の評価に用いることができる。典型的な追加生体高分子として、限定されるものではないが、炭水化物、ポリケチド、テルペノイド、非リボソーム性ペプチド、脂質、または溶液等で安定な三次元構造を形成する任意の他の生体高分子が挙げられる。
【0057】
(接触マップ)
本発明の方法は、基準生体分子の三次構造を記述する1つ以上のパラメータを予測パラメータまたは1つ以上のキメラ組換え産物の構造と比較することを含む。これらのパラメータは、マッビングされた生体分子の構成要素間の相互作用が描かれた「接触マップ」のかたちで提供され(一般に二次元グラフまたはデータ・マトリックスの形で)、それによって、簡略化され、かつ簡約表示された生体分子の三次元構造が提供される。近位生体分子構成要素間の対相互作用(pairwise interaction)が概して調べられる一方で、3つ以上の近位要素の相互作用もまた本発明の接触マップで用いることができる。
【0058】
本発明の方法の特定の実施形態では、基準配列の要素について接触マップが生成される。一般的に、基準分子の一次配列と任意の利用可能な三次元構造データとを用いて接触マップが生成される。接触マップは、例えば基準タンパク質の三次元構造で互いに近位または隣接したアミノ酸残基の組またはセットを描写する(例えば接触アミノ酸)。任意に、接触マップは、目的とする近位要素間の距離を反映するデータを含む。本発明の一実施形態では、接触マップは接触アミノ酸間の距離によって加重されたスコアを含む。別の実施形態では、接触マップは基準生体分子内の要素(例えば、アミノ酸またはヌクレオチド)によって加重されたスコアを含む。
【0059】
接触マップは、種々のソースからのデータに基づいて、当業者に公知の方法で作製され得る。例えば、X線結晶学的データは、一般にタンパク質構造内でのアミノ酸スペーシングを決定すること、および互いの臨界距離内でアミノ酸配列(例えば、近位残基)を同定することに用いられる。これらの方法もまた、相互作用の1つ以上の物理的特徴も同定することができ、限定されるものではないが、アミノ酸相互作用のタイプ(疎水性、水素結合性、および/またはイオン性を含む)が挙げられ、その1つ以上が接触エネルギーの加重で別々に加重される。結晶学的なデータを、任意に各々の原子位置に対して「温度因子」を生成するのに用いられることもできる。データは、接触残基がどのくらいよく定義されたかの指示を与え、それによって該接触が加重され得る。
【0060】
あるいは、構成要素間のスペーシングは、基準生体分子のNMRモデルから決定することができる。2D−COSYとNOESYのようなNMR実験は、両方ともそれらの間の距離の推定を提供することができるのと同様に、三次元構造でお互いに最も近い要素を同定することができる。そのため、結果として生ずる接触は、NMRモデルの不確実な度合いで加重することができる。
【0061】
さらに、基準タンパク質のタンパク質折り畳み分析または相同性モデリングに基づいたアミノ酸構成および距離を用いて、基準タンパク質に対して、接触マップを生成することができる。タンパク質折り畳み分析、タンパク質構造内の残基間距離の計算、および/または他の分子モデリング計算のためのソフトウェアは、公的(例えば、the NIH Center for Molecular Modeling,http://cmm.info.nih.gov/modelingを参照のこと)または商業的(例えばHypercube Inc.,Gainesville FL;MDL Information Systems,San Leandro,CA;Molecular Applications Group,Palo Alto,CA;Accelrys,Inc.(以前はOxford Molecular and,Molecular Simulations Inc.,San Diego,Princeton,NJおよびLondon,UKに事務所;Tripos,Inc.,St.Louis,MO等)の両方で入手可能である。接触マップ情報の生成のための1つの特に役立つプログラムは、Chemical Computing Group(Montreal,Canada)のMOEである。相同性モデリングについての追加プログラムとして、限定されるものではないが、SWISS−MODEL(Glaxo Wellcome Experimental Research in Geneva,Switzerlandから入手可能)およびWHAT IFプログラム(EMBL)が挙げられる。比較(相同性)モデリングによって得られたタンパク質構造のデータベースは、多くのオンライン・ソースから入手可能であり、また限定されるものではないが、データベースModBaseおよび3D Crunchが挙げられる。同様に、核酸分子を生成および分析するためのプログラムを核酸配列に対して利用する;例えば、「tRNAスキャンSE」tRNA分析ソフトウェアは、St.Louis所在のWashington Universityより入手可能である((http://www.genetics.wustl.edu/eddy/tRNAscan−SE)を参照のこと。当業者に公知のこれらのプログラムおよび他のプログラムは、任意に基準分子の三次構造で、お互いに臨界距離の範囲内で2つ以上の要素を識別するのに用いることができる。
【0062】
上記技術の組み合わせはまた、基準タンパク質接触マップの作製にも用いることができる。例えば、入手不可能あるいは1つの分析技術からは統計的に関連していない距離測定を、別の技術を用いて任意に生成または確認することができる。タンパク質三次元構造の評価と接触マップの作製とで一般に用いられる1つのパラメータは、接触アミノ酸間の許容分離度または「臨界距離」の選択である。臨界距離は、アミノ酸−アミノ酸相互作用の性質により変化可能であり、約2.5ないし約7オングストロームの範囲である。例えば、イオン結合または疎水性相互作用を持つ要素(例えば、側鎖)は、概して約4.5オングストローム離れており、一方、ジスルフィド結合を介して接触している要素は、約2.5オングストローム離れている。
【0063】
本発明の方法では、接触アミノ酸側鎖間の臨界距離は、約2オングストロームから約6.5(または約7)オングストロームの範囲内にある。任意に、臨界距離は、約2.5オングストロームから約4.5オングストロームの範囲内にあり、概ね約4.5オングストローム未満である。したがって、本発明の方法の目的に関して、約5オングストローム未満の離間距離にある側鎖要素の任意の位置決めを、接触対(例えば、近位要素)とみなす。あるいは、接触距離は、これらの例では最大で6.5オングストロームの距離を用いて、アミノ酸のCαまたはCβから計算することができる。
【0064】
(配列およびアライメント・プロトコール)
本発明の方法は、2つ以上の親配列(例えば、第1の配列、第2の配列、さらに必要に怖じて第3または追加の配列)内での潜在的交叉位置を同定および/または比較するために使用することができる。任意に、基準生体分子の配列は、第1の配列または第2の配列のいずれかとして用いることができる。基準分子について既に説明したように、親配列は、いくつかのメカニズムのいずれかによって提供することができる。該メカニズムとして、限定されるものではないが、一方または両方の配列の配列決定、転写または翻訳のための核酸配列の提供、あるいは核酸またはタンパク質データベースへの問い合わせが挙げられる。また、目的とする配列が物理的な意味で提供可能である(例えば、単離または合成分子)。好ましくはそれらをコンピュータ内(in silico)で提供する(例えば、代表的配列ストリング、例えば、SelifonovらによるPCT公報WO 01/75767(PCT/USO1/10231「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」を参照せよ)。
【0065】
アミノ酸配列に関係する本発明の実施形態では、親配列は一般的に、類似の三次元構造(例えば、タンパク質スーパファミリー)を持つ共通のタンパク質ファミリーに由来する。しかし、これらのタンパク質をコードする核酸配列は、高い配列相同性を共有または非共有すると思われる。本発明の特定の実施形態では、方法は「低配列相同性」配列(例えば、配列共有が70%未満、60%未満、あるいはさらに50%未満の配列相同性)間での交叉位置を評価するために用いられる。
【0066】
種々のストリジェンシーおよび長さの配列類似性/同一性は、当業者に公知の多数の方法またはアルゴリズムを用いて検出および認識することができる。例えば、多くの類似性または相同性決定方法が生体高分子配列の比較分析のために、文章処理のスペル・チェックのために、さらに種々のデータベースからのデータ検索のために、設計されている。天然ポリペプチドでの4つの主要な核酸塩基間での二重らせん対相補的相互作用を理解することで、相補的な相同ポリヌクレオチド文字列のアニーリングを刺激するモデルもまた、本明細書の配列に対応する文字列上で概ね実行される配列アラインメントまたは他の動作の基礎として用いることもできる(例えば、文書処理操作、配列または配列特性ストリング、出力表などを含む構成)。配列の同一性を計算するためのソフトウェア・パッケージの一例は、BLASTであり、本明細書の配列に対応する文字列を入力することによって本発明に適用可能である。
【0067】
本発明の特定の実施形態では、互いに比較、または基準生体分子と比較した場合、1つ以上の与えられた配列が低配列同一性を呈する(しばしば、当該技術分野で「低相同性配列」と呼ばれる)。低同一性配列は、天然由来のものであり、あるいは合成的、突然変異的、またはコンピュータ的に生成することができる。低同一性配列の一例は、例えばPattenらのPCT公報WO 00/18906「Shuffling of Codon Altered Genes」に記載されているように、「コドン変更(codon
altered)」配列である。
【0068】
第1の配列と第2の配列とを提供した後に、該配列を基準タンパク質の配列に整列させる。基準配列が第1の配列または第2の配列のいずれかとして基準配列が機能する実施形態では、2つの配列が互いに位置合わせして並ばされる。別の実施形態では、複数の親配列が与えられ、該配列は次に基準配列と、または互いがアライメントされる。相対的に短いアミノ酸配列(例えば、約30残基)のアライメントおよび比較は概して直接的である。より長い配列の比較は、2つの配列の最適アラインメントを達成するためのより高度な方法が求められる。
【0069】
配列の最適アラインメントの実行は、例えば、いくつかの利用可能なアルゴリズムによっておこなうことができる。該アルゴリズムとして、限定されるものではないが、SmithおよびWaterman(1981 Adv.Appl.Math.2:482)の「局所相同性」アルゴリズム、NeedlemanおよびWunsch(1970 J.Mol.Biol.48:443)の「相同性アラインメント」アルゴリズム、PearsonおよびLipman(1988 Proc.Natl.Acad.Sci.USA
85:2444)の「類似性の検索」法、またはこれらのアルゴリズムのコンピュータ化による実現(例えば、GAP,BESTFIT,FASTA、およびTFASTA、これらはWisconsin Genetics Software Package Release 7.0,Genetics Computer Group,575 Science Dr.,Madison,WIから入手可能;ならびにBLAST、例えばAltschulら.,(1977)Nuc.Acids Res.25:3389−3402およびAltschulら.,(1990)J.Mol.Biol.215:403−410)が挙げられる。あるいは、配列を検査により整列させることができる。一般に、種々の方法によって生ずる最良のアラインメント(すなわち、比較ウィンドウ上の配列同一性の割合が最大となる相対的な位置決め)が選択される。しかし、本発明の具体的実施形態では、最良のアラインメントとして、選択された構造上の特徴を重ね合わせるものであってもよく、最も高い配列同一性を必要としない。
【0070】
「配列同一性」という用語は、比較ウィンドウ上で2つのアミノ酸配列が同一(すなわち、アミノ酸対アミノ酸をベースとする)であることを意味している。「配列類似性」とは、同一の生物物理学的性質を共有する類似のアミノ酸に言及している。「配列同一性の割合」または「配列類似性の割合」は、比較ウィンドウ上に最適化して整列させた2つの配列を比較し、一致した位置の数が生ずるために同一の残基(または類似の残基)が両方のポリペプチド配列に生じる位置の数を決定し、一致した位置の数を比較ウィンドウの全位置数(すなわち、ウィンドウ・サイズ)で割り、さらに得られた結果を100倍して配列同一性の百分率を生ずる(または配列類似性の百分率)。ポリペプチド配列に関して、配列同一性および配列相似性という用語は、タンパク質配列で説明したように、比較可能な意味を有し、「配列同一性の百分率」という用語は、2つのポリペプチド配列が比較ウィンドウで同一である(ヌクレオチド対ヌクレオチドをベースとする)。このようなことから、ポリヌクレオチド配列同一性の百分率(またはポリヌクレオチド配列類似性の百分率、例えば、サイレント置換または他の置換については、分析アルゴリズムに基づく)もまた計算することができる。本明細書に記載される配列アルゴリズムの1つ(または当業者が利用可能な他のアルゴリズム)を用いて、または目視試験によって、最大一致を決定することができる。「低配列同一性」を持つ配列(しばしば「低相同性」配列として言及される)は、直線状になった目的とする準配列に対して約70%未満、好ましくは約60%、またはより好ましくは約50%の配列同一性を有する配列である。
【0071】
ポリペプチドに適用されることから、実質的同一性または実質的類似性という用語は、ギャップ・ウェイト(本発明で詳細に説明)を用いて、または目視検査によって、例えばプログラムBLAST、GAP、またはBESTFITによって、2つのペプチド配列が最適に整列させられた場合、少なくとも約60ないし約80パーセント以上の配列同一性または配列類似性、好ましくは約90パーセントのアミノ酸残基配列同一性または配列類似性、より好ましくは少なくとも約95パーセントの配列同一性または配列類似性、またはそれ以上を共有する(例えば、96、97、98、98.5、99以上のパーセント・アミノ酸残基配列同一性または配列類似性が含まれる)。同様に、2つの核酸の前後関係で適用されることから、実質的同一性または実質的類似性という用語は、2つの核酸配列が、例えばプログラムBLAST、GAP、またはBESTFITによって、デフォルト・ギャップ・ウェイト(以下に詳細に説明する)または目視検査を用いることで、最適にアラインメントされる場合、少なくとも約60ないし約80パーセント以上の配列同一性または配列類似性、好ましくは少なくとも約90パーセントのアミノ酸残基配列同一性または配列類似性、より好ましくは少なくとも約95パーセントの配列同一性または配列類似性、またはそれ以上を共有する(例えば、約96、97、98、98.5、99、またはそれ以上のパーセント・ヌクレオチド配列遺伝同一性または配列類似性が含まれる)。
【0072】
パーセント配列同一性または配列類似性の決定に適しているアルゴリズムの一例は、FASTAアルゴリズムであり、このアルゴリズムはPearson,W.R.&Lipman,D.J.,(1988)Proc.Natl.Acad.Sci.USA 85:2444に記載されている。またW.R.Pearson,(1996)Methods
Enzymology 266:227−258を参照のこと。パーセント同一性またはパーセント類似性を計算するためのDNA配列のFASTAアラインメントで用いられる好ましいパラメータを最適化した。BL50マトリックス(Matrix)15:−5、K−タプル(tuple)=2、接合ペナルティ=40、最適化=28、ギャップ・ペナルティ −12、ギャップ長ペナルティ=−2,および幅=16。
【0073】
パーセント配列同一性およびパーセント配列類似性を決定するのに適したアルゴリズムの好ましい例は、BLASTおよびBLAST2.0アルゴリズムであり、これらのアルゴリズムは、それぞれAltschulら.,(1977)Nuc.Acids Res.25:3389−3402およびAltschulら.,(1990)J.Mol.Biol.215:403−410に記載されている。本明細書に記載したパラメータとともにBLASTおよびBLAST2.0を用い、本発明の核酸、ポリペプチド、およびタンパク質のパーセント配列類似性またはパーセント配列同一性を決定する。BLAST分析を実行するためのソフトは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)から公的に入手可能である、このアルゴリズムは、問い合わせ配列で長さWの短い語を識別することで、高スコアリング配列の対(HSP)を最初に同定することを含むもので、該問い合わせ配列は、データベース配列で同一の長さの語と整列させた場合、一致するか、またはある正の値となった閾値スコアTを満足するかのいずれかである。Tは、隣接語スコア閾値と呼ぶ(Altschulら.前出)。これらの初期の隣接語のヒットは、それを含むより長いHSPを見出すための検索を開始させる種子として働く。語のヒットは、累積アラインメント・スコアの増加にともなって、核配列に沿う両方向に延びる。累積スコアは、ヌクレオチド配列について、パラメータM(一組の一致した残基に対するリワード・スコア:常に>0)、およびN(ミスマッチング残基のためのペナルティ・スコア:常に<0)である。アミノ酸配列については、スコアリング・マトリックスを用いて累積スコアの計算をおこなう。各方向での語ヒットの延長の停止は、累積アラインメント・スコアがその最大達成値から量Xまで落ちる場合、1つ以上の負のスコアリング残基アラインメントの集積により累積スコアがゼロまたはそれ以下になる場合、またはいずれかの配列の終わりに達する場合におこる。BLASTアルゴリズム・パラメータW、T、およびXは、アラインメントの感度および速度を決定する。BLASTNプログラム(ヌクレオチド配列に対する)はデフォルトとしてワード長(W)が11、期待値(B)が10、M=5、N=−4、および両鎖の比較を用いる。アミノ酸配列については、BLASTPプログラムは、ワード長3、および期待値(E)10をデフォルトとして用い、さらにBLOSUM62スコアリング・マトリックス(Henikoff&Henikoff(1989)Proc.Natl.Acad.Sci.USA 89:10915参照)ではアラインメント(B)50、期待値(B)10、M=5、N=−4、および両鎖の比較を用いた。
【0074】
BLASTアルゴリズムは、2つの配列間の類似性または同一性の統計的分析も実施する(例えば、Karlin&Altschul,(1993)Proc.Natl.Acad.Sci.USA 90:5873−5787を参照)。BLASTアルゴリズムによって提供される類似性または同一性の1つの尺度は、最小合計確率(P(N))であり、これによって2つのヌクレオチドまたはアミノ酸配列間での一致(マッチング)が偶然生じる確率が提示される。例えば、被試験核酸と基準核酸との比較の際の最小合計確率が約0.2未満、より好ましくは0.01未満、最も好ましくは約0.001未満である場合、核酸は基準配列に類似していると考えられる。
【0075】
有用なアルゴリズムの別の例は、PILEUPである。PILEUPは、プログレッシブで対形成するアラインメントを用いて、関連配列からなる群から複数の配列アラインメントを作り出し、相互関係およびパーセント配列同一性またはパーセント配列類似性を示す。ツリーまたはデンドグラムもプロットすることで、アラインメントの生成に用いられるクラスター相互関係が示される。PILEUPは、Feng&Doolittle,(1987)J.Mol.Evol.35:351−360のプログレッシブ・アラインメント方法の単純化をもちいる。用いた方法は、Higgins&Sharp,(1989)CABIOS 5:151−153に記載された方法に類似している。プログラムは、多くとも300配列を整列させることができ、各々が最大長が5,000ヌクレオチドまたはアミノ酸である。複数のアラインメント手順が2つの最も類似した配列からなる2つ一組のアラインメントにより開始され、整列させられた2つの配列からなるクラスターを生成する。それにより、このクラスタを、次に最も関連している配列または整列させられた配列からなるクラスターに対して整列させる。2つの個々の配列からなる2つ一組のアラインメントの単純拡張によって、2つの配列クラスターを整列させる。最終のアラインメントは、一連のプログレッシブで、かつ2つ一組のアラインメントによって達成される。プログラムの実行は、特定の配列と、配列比較の領域に対する該配列のアミノ酸またはヌクレオチドの座標とを設計し、プログラム・パラメータを設計することによっておこなう。PILEUPを用いることで、参照配列を他の被試験配列と比較し、パーセント配列相同性(またはパーセント配列類似性)相互関係を決定する。この際、以下のパラメータが用いられる。すなわち、デフォルト・ギャップ加重(3.00)、デフォルト・ギャップ長加重(0.10)、加重末端ギャップである。PILEUPは、GCG配列分析ソフトウェア・パッケージ、例えばバージョン7,0(Devereauxら.,(1984)Nuc.Acids Res.12:387−395)から得ることができる。
【0076】
複数のDNAおよびアミノ酸配列アラインメントに好適なアルゴリズムの別の好ましい例は、CLUSTALWプログラム(Thompson,J.D,ら.,(1994)Nuc.Acids Res.22:4673−4680)である。CLUSTALWは、複数の配列からなるグループ間に対して複数の2つ一組の比較を実施し、それらを配列の同一性にもとづいて複数アラインメントに集める。ギャップ・オープン(Gap open)ペナルティおよびギャップ拡張(Gap extension)ペナルティは、それぞれ10および0.05であった。アミノ酸のアラインメントに関しては、BLOSUMアルゴリズムをタンパク質加重マトリックス(protein weight matrix)として使用することができる((HenikoffおよびHenikoff,(1992)Proc.Natl.Acad.Sci.USA 89:10915−10919)。
【0077】
当業者に理解されるように、検索およびアラインメント・アルゴリズムについての上記議論もまたポリヌクレオチド配列の同定および評価、ヌクレオチド配列を含む問い合わせ配列の置換により、必要に応じて、核酸データベースの選択に適用される。
【0078】
(交叉点)
親配列(例えば、第1の配列、第2の配列、および任意に加えた配列)を提供した後、親配列の一部分を置換、スワッピング、または交換する。各交換は、所定の交換の要素(アミノ酸またはヌクレオチドの準配列)の選択された領域を包含する2つの親配列上の第1の交叉点と第2の交叉点とのあいだで行われる。任意に、複数の準配列を所定の親配列内の複数の交叉点でスワッピングすることができ、それによって2つ以上の挿入準配列(1つ以上の親配列由来)を持つキメラ生体分子が生成される。核酸に関して、交叉部位は交換されたオリゴヌクレオチド領域の5’および3’末端(例えば、組換えが生ずる位置)を定義する。タンパク質配列について、交叉部位は、交換されたアミノ酸残基の始点(N−末端)および終点(C−末端)によって定まる。いくつかの実施形態では、第1の交叉部位は核酸の5’末端、またはアミノ酸配列のN−末端に一致する。他の実施形態では、第2の交叉部位は核酸の3’末端、またはアミノ酸配列のC−末端に一致する。
【0079】
交換すべき選択された領域の長さは、標的システムにより変動する。しかし、本発明で用いた交叉部位は、2つの親配列間での交換のための要素が同じ数である必要はない。例えば、第1の配列の交叉部位が30の要素からなる領域を定める場合、第2の配列にある対応交叉部位によって定まる領域は、任意に30未満または30を上回る数の要素を含むことができる。
【0080】
本発明の方法では、1つ以上の「交叉産物」(すなわち、キメラ配列)を、所定の対またはセットの親配列について調べる。一実施形態では、単一の交叉産物を検討する。しかし、本発明の方法は2つ以上の交換領域(例えば、複数の交叉部位)を持つキメラ組換え体の生成に使用することができる。いくつかの実施形態では、単一交換用の潜在的交叉部位のすべてが分析のために生成される。別の実施形態では、考えられる全てのキメラ産物のサブセットが調べられる。
【0081】
交叉部位の選択は、経験的に行われ(例えば、配列中で5番目の要素ごとに開始)、または選択が付加的な基準に基づく。進化の過程のアミノ酸の共変化によってタンパク質が所定の折り畳み、三次元構造、または機能を保持し、その一方で他の形質(例えば特異性)を変えることを考えると、この情報は可能性のある交叉一の選択に有用である。あるいは、交換のための領域が選択、例えば所望の活性(例えば、タンパク質または触媒核酸の活性部位)または特異的な構造特性(例えば、αヘリックスまたはβシートのストランド)を標的化することである。接触マップおよび/または基準タンパク質の三次元構造による親配列のアラインメントの視覚的分析もまた、目的の構造領域に対する分析的努力にも焦点を合わせることができる。
【0082】
付加的な基準パラメータの分析は、交叉点の選択および組換え生体分子の設計をアシストする。評価のための交叉点配置の選択は、例えば、以下の文献に見出すことができる。PCT公報WO 00/42559(PCT/US00/01138「METHODS OF POPULATING DATA STRUCTURES FOR USE IN
EVOLUTIONARY SIMULATIONS」(SelifonovおよびStemmerによる),WO 00/42560(PCT/US00/01202「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Selifonovらによる)),WO 01/75767(PCT/US01/10231「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Selifonovらによる)および USSN 09/618,579(2000年7月18日出願(「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Gustafssonらによる)))。
【0083】
「スワッピング」の工程、キメラ産物配列を生成する親配列間の1つ以上の準配列は、本発明の方法によりコンピュータ内(in sillico)で行われる。組換えのコンピューター内方法を実行するができ、この際、遺伝的アルゴリズムをコンピューターで用いて、相同な核酸(または非相同な核酸でも)に対応する配列の列を組換える。任意で、例えばオリゴヌクレオチド合成/遺伝子再集合技術と協調させて、組換え配列に対応する核酸の合成によって、結果的に生じる組換え配列の列を核酸に変換する。この手法により、ランダム改変体、部分的ランダム改変体、または設計された改変体が生成され得る。対応する核酸(および/またはタンパク質)の生成と組み合わせたコンピューター・システムでの遺伝的アルゴリズム、遺伝的オペレーター等の使用を含むコンピューター内組換えと、設計された核酸および/またはタンパク質(例えば、交叉部位選択に基づく)と、設計された組換え法、擬似ランダム組換え法、またはランダム組換え法とに関する多くの詳細は、WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides
and Polypeptides Having Desired Characteristics」ならびにWO 00/42559(SelifonovおよびStemmer)、「Methods of Populating Data Structures for Use in Evolutionary Simulations」に記載されている。コンピューター内組換え法に関する広範な詳細は、これらの適用中で見出される。この方法論は、対応する核酸またはタンパク質のコンピューター内および/または生成の組換えに提供される際、本発明に概ね適用できる。
【0084】
(キメラ配列のスコアリング)
本発明の方法は、基準生体分子と比較したキメラ配列の1つ以上のパラメータに基づいて、親配列内の潜在的交叉位置を評価するためのメカニズムを提供する。本発明の方法は、適応度交叉位置を評価するための接触マップおよび接触エネルギー算出を使用することが可能である。本発明の方法では、接触マップを用いてキメラ分子の要素を位置合わせし、基準配列およびキメラ配列間の比較をおこなう。本方法の一実施形態では、キメラ配列を接触マップと比較して、接触している要素のセットを選択する。その後、キメラ分子中の要素の選択されたセットを基準分子中の対応する要素と比較してスコアリングする。このスコアは、キメラ分子が基準生体分子と類似のコンフォメーションを達成する可能性、および推論により類似のコンフォメーション安定性または所望の活性を実現する可能性の尺度を提供する。
【0085】
比較プロセスおよびスコアリング・プロセスの一局面は、キメラ分子中の2つ以上の近位要素間の接触エネルギーの算出である。当業者に公知の多数の手順によって、要素の選択されたペアまたはセットの接触エネルギーを算出することができる。例えばミヤザワ−ジャニガン(Miyazawa−Jernigan)エネルギー行列を用いて、アミノ酸配列に対して、接触エネルギーを予測することができる(例えばMiyazawaおよびJernigan(1999)「Self−consistent estimation of inter−residue protein contact energies based on an equilibrium mixture approximation of residues」Proteins 34:49−68);MiyazawaおよびJernigan(1999)「An empirical energy potential with a reference state for protein fold and sequence recognition」Proteins 36:357−69;Zhang(1998)「Extracting contact energies from protein structures:a study using a simplified model」Proteins 31:299−308;ならびにMiyazawa,S.&Jernigan,R.L.(1996)「Residue−residue potentials
with a favorable contact pair term and an unfavorable high packing density term,for simulation and threading」、J.Mol.Biol.256:623−464を参照せよ)。
【0086】
この算出では、二次元行列は、どの程度の頻度でそれらの残基が公知の構造のデータベースに接触して見出されたかに基づいて、アミノ酸の各ペアリング間の相互作用強度を提示する。接触していると考えられる近位アミノ酸残基に関して基準分子を試験する。相互作用する原子2つ以上の残基は、それらの各側鎖が基準距離未満で離れている場合、接触していると考えられる。この基準距離は、相互作用の種類(例えば、疎水性、イオン性等)によって変動するが、通常、約4.5から約5.0オングストローム未満である。その後、接触ペアの数を決定し(例えば上述の位置合わせ技術の1つを介して、生成された基準分子の接触マップと比較することによって)、タンパク質構造中に存在する接触の数(および関連するエネルギー値)を合計することによって、キメラ配列に対して接触エネルギーを決定する。ミヤザワ−ジャニガン・エネルギー行列を用いて生成された接触エネルギーは通常、疎水性作用によって支配されている。このように、正の電気を帯びたアミノ酸側鎖および負の電気を帯びたアミノ酸側鎖は、ジスルフィド結合(cys−cys)相互作用のように、過小評価されている。
【0087】
あるいは、接触残基の2次構造を用いることでタンパク質接触エネルギーについて計算することができる。(Zhang,C,およびKim,S−H.(2000)「Environment−dependent residue contact energies for proteins.」Proc.Natl.Acad.Sci.USA 97:2550−2555)などに、記載されている)。例えば、Zhang−Kimのデータベース依存マトリックスを用いたとき、αヘリックスからのPheと、β−シートからのAlaとの相互作用は、ループからのPheと、αヘリックスからのAlaとは、異なった相互作用エネルギーがあるだろう。また、タンパク質接触エネルギーを計算するための追加代替方法も、この方法に用いることが可能である。
【0088】
核酸分子に関しては、比較を目的とした位置依存的(または、位置非依存的)アルゴリズムが数多く使用可能である。例えば、1対1比較(pair−wise)の「Smith−Waterman」または「Needleman−Wunsch」アライメント関数を用いて、接触エネルギー情報を生成することが可能である。(Smith,T.F.,およびWaterman,M.S.(1981)「Identification of
common molecular subsequences」J.Mol.Biol.147:195−197;Needleman,S.B.,およびWunsch,C.D.(1970)「A general method applicable to
the search for similarities in the amino acid sequence of two proteins」J.Mol.Biol.48:443−453)。これら2つのアルゴリズムが以下の点で異なる。Needleman−Wunschアルゴリズムを用いた場合、配列比較が全体的(global)に行われる。それとは対照的に、Smith−Watermanアルゴリズムを用いた場合は、局所的(local)であり、それによって、基準配列、またはデータベース配列に対する照会配列全体のアライメントを強制する。
【0089】
本発明の方法の一実施形態では、キメラ配列の各要素を調べて、その要素が配列における別の要素に接触しているかを決定することによって、スコアリングが行われる。選択された要素が別の要素に近位であるなら、その対の近位要素の接触エネルギーが計算される。いくつかの実施形態では、スコアリングは、近位の対の要素のすべてに関する接触エネルギーを合計することを含んでいる。場合によっては、スコアリング・ステップの「接触エネルギー」成分に、要素間の概算距離、もしくは測定距離、または配列中の要素の位置などの付加パラメータによって重み付けすることが可能である。
【0090】
接触エネルギー・パラメータに加えて、スコアリング・プロセスに場合によっては他の構造パラメータを含むことができる。例えば、アミノ酸残基の立体的大きさ(steric bulk)、アロステリック効果、疎水性度もしくは極率を定義するパラメータ、または、構造対称性、周期性、または構成要素系の分布パターンなどの総合的構造パラメータ、電荷、および/または静電気フィールドの分布、四元構造単位の方向性などを用いて、スコアの生成が可能である。例えば、アミノ酸組成および有意な特性の分析に関して、考慮すべき事柄には、溶剤からの移動時のΔGによって決定される疎水性度、カラム残留からの親水性、アミノ酸の電荷、極性、アミノ酸のpKa、大きさ、側鎖エントロピー、αへリックス/ベータシート傾向、水和ポテンシャル、コドン縮重、および同様のものが含まれる。
【0091】
その上、キメラ配列のスコアリングで追加統計的方法を使うことができる。これらには、ニューラルネットワーク計算、モンテカルロ分子動力学シミュレーション、主成分分析法(PCA)、潜在構造への部分的最少二乗投影(PLS)などの多変量データ解析、および、他の分子モデリング、または生命情報工学計算が含まれるが、これらに限定されない。
【0092】
例えば、理想的な交叉位置を正確に指摘するのにマルコフ連鎖などの統計的な行列を用いることができる。Skorobogatiy,およびTiana(Physical Review E(1998年9月)第58巻,3572−3577頁)によって記述されたような単純なオン格子模型を用いることで、アミノ酸置換によって天然立体配座のミスフォールディングを引き起こすタンパク質配列以内の部位を写像することができる。ニューラルネットワーク法は、一種のパターンを学んで、所与の変異によって生成する結果を予測するのにを用いることが可能である。(そのようなニューラル・ネットワークの例として、SchneiderおよびWrede(1998)「Artificial neural networks for computer−based molecular design」Prog.Biophys.Mol.Biol.70(3):175−222;Schneiderら(1998)「Peptide design by artificial neural networks and computer−based evolutionary search」Proc.Natl.Acad.Sci.USA 95(21):12179−12184;およびWredeら(1998)「Peptide design aided by neural networks:biological activity of artificial signal peptidase I cleavage sites」Biochemistry 37(11):3588−35893が挙げられる。さらなる例としては、WO 00/42559(SelifonovおよびStemmer),WO 00/42560(Selifonovら),WO 01/75767(Selifonovら)ならびにUSSN 09/618,579(Gustafssonら),すべて前出)に見出すことができる。
【0093】
核酸に関しては、必要に応じておこなう1つのアプローチは、Jonssonら(1993 Nucl.Acids Res.21:733−739)により記載されたような、所定の転写プロモータからなるセット内での強度の予測のための多変量データ分析のアプリケーションである。したがって、接触エネルギー情報に加え、統計的な検討を、交叉点の評価および核酸またはタンパク質の安定性の予測のための方法に適用することができる。
【0094】
(正規化)
本発明のいくつかの実施形態では、キメラ分子中の推定接触アミノ酸に対して生じたスコアを進化の前に正規化する。大部分の使用では、キメラ配列は、基準配列に対して、配列アイデンティティの範囲を有している。基準配列により近いそれらのキメラは、基準配列に対する配列アイデンティティでより遠いものより優れた接触スコアを有している。このことは、基準タンパク質と比較して、より大きい、例えば約50%同一、約60%同一、約70%同一、またはそれ以上であるものに対して、交叉部位を決定するために用いられるキメラを制限することによって、あるいは接触エネルギーを正規化することを介して説明され得る。基準配列対算出された接触スコアに対するキメラの配列アイデンティティの直線状回帰を介して、この正規化を実行することができる。その後、この回帰からの剰余を用いて、最適交叉位置を決定する。別の手法では、2つ以上の以下のものの複数の回帰を利用できる。すなわち、2つの交叉位置間の長さ、第2の交叉の第1の交叉位置の位置、およびキメラ産物配列および基準配列対算出された接触スコア間の配列アイデンティティである。また、回帰からの剰余を用いて、最適交叉位置を決定することができる。
【0095】
(キメラ産物の生成)
本発明の方法は、キメラ産物合成前に潜在的交叉部位の可能性を評価するために、事前選別技術として利用することができる。任意で、その後、有効であると評価された(またはこの計算的事前選別に「合格した(pass)」)それらのキメラ産物を実験室で合成および試験する。従って、本発明の方法は、任意で、1つ以上のキメラ生体分子配列を合成するステップをさらに含む。当業者には公知である任意の種々の技術を用いて、キメラ産物を合成することができる。例えば、一実施形態では、キメラ生体分子を新規に合成する(例えば合成化学技術を用いて)。別の実施形態では、本方法は、細胞ベースまたは細胞を含まない発現系でキメラ生体分子を発現させる任意のステップを含む。任意で、キメラ生体分子を合成するステップは、適当な親拡散を提供することと、1つ以上の組換えプロセスを実行することとを含む。合成ステップを実行するための方法論は、本明細書に組み込まれる参考文献中に詳述されている。
【0096】
本発明の方法は、任意で、1つ以上の子孫キメラ核酸配列を含むコンストラクトを提供することを含む。コンストラクトは、プラスミド、コスミド、ファージ、ウイルス、最近人工染色体(BAC)、酵母人工染色体(YAC)等のベクターを含み、その中に、順配向または逆配向で、工本発明のキメラ配列が生成または挿入されている。この実施形態の好適な局面では、コンストラクトは、配列に作用可能に結合する、例えばプロモーターを含む調節配列をさらに有する。多数の好適なベクターおよびプロモーターが当業者に公知であるとともに市販されている。
【0097】
ベクターの使用、プロモーターの使用、および多くの他の関連主題を含む本明細書で有用な分子生物学的技術を記載する一般的テキストとしては、Bergerら、前出;Sambrookら、(1989)前出、およびAusubelら、(1989;1999年まで補完)前出が挙げられる。本発明の核酸の作製のために、ポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、Qβ−レプリカーゼ増幅、および他のRNAポリメラーゼ媒介技術(例えばNASBA)を含むインビトロ増幅方法を介して、当業者を指導するために十分な技術の例は、Berger、Sambrook、およびAusubelとともに、Mullisら、(1987)、米国特許第4,683,202号;PCR
Protocols A Guide to Methods and Applications(Innsら編)Academic Press Inc.San Diego,CA(1990)(「Innis」);Arnheim & Levinson(1990年10月1日)C&EN 36−47;The Journal Of NIH
Research(1991)3:81−94;Kwohら、(1989)Proc.Natl.Acad.Sci.USA 86:1173−1177;Guatelliら、(1990)Proc.Natl.Acad.Sci.USA 87:1874−1878;Lomeliら、(1989)J.Clin.Chem.35:1826−1831;Landegrenら、(1988)Science 241:1077−1080;Van Brunt(1990)Biotechnology 8:291−294;WuおよびWallace、(1989)Gene 4:560−569;Barringerら、(1990)Gene 89:117−122、ならびにSooknananおよびMalek(1995)Biotechnology 13:563−564で見出される。インビトロで増幅された核酸をクローニングする改良された方法 についてはWallaceら、米国特許第5,426,039号に記載されている。PCRによって大きい核酸を増幅する改良された方法は、Chengら、(1994)Nature 369:684−685および本明細書の参考文献に概説されており、この際40kbまでのPCRアンプリコンが生成される。当業者には、本質的に、任意のRNAを逆転写酵素およびポリメラーゼを用いた制限酵素消化、PCR伸展、およびシーケンシングに適した二重鎖DNAに転換させることが認識される。Ausubel、Sambrook、およびBerger(全て前出)を参照せよ。
【0098】
本発明の方法で使用される付加的な配列を生成するために、下記に記載するような多様性生成技術を用いることができる。さらに、これらの多様性生成技術を用いて、1つ以上のキメラ産物を修飾することができる。本発明の方法は、任意で、1つ以上の以下のステップをさらに含む。すなわち、種々の交叉位置を用いて生成されたキメラ分子のライブラリーから安定または活性キメラ産物を選択することと、1つ以上の産物キメラ生体分子で多様性を生成すること(それによって、多様性キメラ生体分子を提供すること)と、親分子由来の、1つ以上のキメラ生体分子由来の、親配列として1つ以上の多様性キメラ産物由来の、またはその組み合わせ由来の配列を用いて、本発明の方法を反復的に繰り返すこととである。
【0099】
多様性生成方法の1つのグループについては、組換えまたはDNAシャッフリングとして言及される。これらの方法では、インビトロまたはインビボで、ポリヌクレオチドを組換えて、ポリヌクレオチド改変体のライブラリーを作製する。組換えに基づく方法では、1つ以上の親ポリヌクレオチドの配列のいくつかまたは全てに配列中で集合的に対応するDNAフラグメント、PCRアンプリコン、および/または合成オリゴヌクレオチドを組換えて、親ポリヌクレオチドのポリヌクレオチド改変体のライブラリーを作製する。組換えプロセスは、DNAフラグメント、PCRアンプリコン、および/または合成オリゴヌクレオチドの互いへのハイブリダイゼーション(例えば、部分的にオーバーラップする二重鎖として)によって、または完全長テンプレート等のDNAの大きな断片へのハイブリダイゼーションによって媒介されることが可能である。使用される組換え形式によって、リガーゼおよび/またはポリメラーゼを用いて、完全長ポリヌクレオチドの構築を促進することが可能である。ポリメラーゼのみを使用する形式では、PCRサイクリングが用いられる。これらの方法は、一般的に、当業者に公知であるとともに、他でも広範に記載されている。例えばSoong,N.ら、(2000)Nat.Genet.25(4):436−439;Stemmerら、(1999)Tumor Targeting 4:1−4;Nessら、(1999)Nature Biotechnology 17:893−896;Changら、(1999)Nature Biotechnology 17:793−797;MinshullおよびStemmer(1999)Current Opinion in Chemical Biology 3:284−290;Christiansら、(1999)Nature Biotechnology 17:259−264;Crameriら、(1998)Nature 391:288−291;Crameriら、(1997)Nature Biotechnology 15:436−438;Zhangら、(1997)Proc.Natl.Acad.Sci.USA 94:4504−4509;Pattenら、(1997)Current Opinion in Biotechnology 8:724−733;Crameriら、(1996)Nature Medicine 2:100−103;Crameriら、(1996)Nature Biotechnology 14:315−319;Gatesら、(1996)Journal of Molecular Biology 255:373−386;Stemmer(1996)In:The Encyclopedia of Molecular Biology.VCH
Publishers,New York.pp.447−457;CrameriおよびStemmer(1995)BioTechniques 18:194−195;Stemmerら、(1995)Gene,164:49−53;Stemmer(1995)「The Evolution of Molecular Computation」Science 270:1510;Stemmer(1995)Bio/Technology 13:549−553;Stemmer(1994)Nature 370:389−391;ならびにStemmer(1994)Proc.Natl.Acad.Sci.USA 91:10747−10751;GiverおよびArnold(1998)Current Opinion in Chemical Biology 2:335−338;Zhaoら、(1998)Nature Biotechnology 16:258−261;Cocoら、(2001)Nature Biotechnology 19:354−359;米国特許第5,605,793号、第5,811,238号、第5,830,721号、第5,834,252号、第5,837,458号、WO 95/22625、 WO 96/33207、WO 97/20078、WO 97/35966、WO 99/41402、WO 99/41383、WO 99/41369、WO 99/41368、WO 99/23107、WO 99/21979、WO 98/31837、WO 98/27230、WO 98/27230、WO 00/00632、WO 00/09679、WO 98/42832、WO 99/29902、WO 98/41653、WO 98/41622およびWO 98/42727、WO 00/18906、WO 00/04190、WO 00/42561、WO 00/42559、WO 00/42560、WO 01/23401、WO 00/20573、WO 01/29211、WO 00/46344、ならびにWO 01/29212を参照せよ。
【0100】
上記で参照されている組換えプロセスで使用される親ポリヌクレオチドは、野生型ポリヌクレオチドまたは非天然ポリヌクレオチドである。好ましくは、少なくとも1つのポリヌクレオチドは、本明細書で記載されるように選択される交叉点をコードする。本発明の一実施形態では、選択後の2つ以上の親ポリヌクレオチドの組換えによって、選択された交叉点を有するキメラタンパク質を調製する。いくつかの実施形態では、親ポリヌクレオチド(少なくとも交叉点をコードしないもの)は、単一の遺伝子ファミリーのメンバーである。本明細書で使用するように、用語「遺伝子ファミリー」とは、必ずしも同程度の活性ではないが、同種類の活性を示すポリペプチドをコードする遺伝子のセットを指す。
【0101】
例えば、ライゲーション後に組換えられる核酸のDNアーゼ消化および/または核酸のPCRでの再集合を含む任意の種々の技術によって、ポリ核酸をインビトロで組換えることができる。例えば、セクシャル(sexual)PCRである突然変異を用いることができ、この際、異なるが関連するDNA配列を有するDNA分子間の配列類似性に基づいたDNA分子のインビトロでのランダム断片化(または擬似ランダム断片化もしくは非ランダム断片化でさえも)が組換え後におこなわれ、その後、ポリメラーゼ連鎖反応での伸長による交叉の固定が続く。このプロセスおよび多くのプロセスの変形については、例えば、Stemmer(1994)Proc.Natl.Acad.Sci.USA 91:10747−10751に記載されている。
【0102】
合成組換え法を用いることもでき、この際、対象とする標的に対応するオリゴヌクレオチドを化学的に合成し、PCRまたはライゲーション反応で再集合させる。これには、1つより多い親ポリヌクレオチドに対応するオリゴヌクレオチドが含まれているので、新規の組換えポリヌクレオチドが生成される。標準的なヌクレオチド付加方法によって、または例えばトリヌクレオチド合成手法によって、オリゴヌクレオチドを作製することができる。このような手法に関する詳細については、上記に記載される参考文献、例えばWO 00/42561(Crameriら)、「Olgonucleotide Mediated Nucleic Acid Recombination;」WO 01/23401(Welch ら)、「Use of Codon−Varied Oligonucleotide Synthesis for Synthetic Shuffling」;WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides and Polypeptides Having Desired Characteristics」;、およびWO 00/42559(SelifonovおよびStemmer)「Methods of Populating Data Structures for Use in Evolutionary Simulations」で見出される。
【0103】
例えば細胞中の核酸間で組換えを発生させることによって、ポリヌクレオチドをインビボで組換えることもできる。多くのこのようなインビボ組換え形式が上記の参考文献で先述されている。このような形式は、他の形式と同様に、任意で、対象とする核酸間での直接組換えを提供するか、または対象とする核酸を含むベクター、ウイルス、プラスミド等の間の組換えを提供する。このような手順に関する詳細に関しては、本明細書で引用される参考文献で見出される。
【0104】
同様に使用される天然の多様性を評価する多数の方法では、例えば、一本鎖テンプレートへの多様な核酸または核酸フラグメントのハイブリダイゼーション後に、重合および/またはライゲーションをして完全長配列を再生成し、任意で、その後にテンプレートの分解およびその結果生じる修飾核酸の回収が可能になる。これらの方法は、本発明の特定の実施形態に従って、物理的システムで用いることができ、またはコンピューター・システムで実行することができる。一本鎖テンプレート(好ましくは、交叉点をコードする)を用いる一方法では、ゲノム・ライブラリーに由来するフラグメント集団を、対となる配列に対応する部分的ssDNAまたはRNA、あるいはしばしばほぼ完全長ssDNAまたはRNAでアニーリングする。その後、この集団から得た複合キメラ遺伝子の集合は、非ハイブリダイゼーション・フラグメント末端のヌクレアーゼに基づく除去と、このようなフラグメント間の溝を満たすための重合と、その後の一本鎖ライゲーションとによって媒介される。消化(たとえば、RNAまたはウラシルを含有の)と、変性条件下での磁気分離(このような分離を導く方法で標識されている場合)と、他の利用可能な分離/精製方法とによって、親ポリヌクレオチド鎖を除去することができる。また、任意で、親鎖をキメラ鎖と共精製し、その後のスクリーニングおよび加工ステップ中に除去する。この手法に関する更なる詳細は、例えばAffholter、によるWO 01/64864「Single−Stranded Nucleic Acid Template−Mediated Recombination and Nucleic Acid Fragment Isolation」で見出される。
【0105】
情報処理システムでのデジタル方式で組換え方法も実行することができる。例えば、コンピューターでアルゴリズムを用いて、相同生体分子(または非相同な生体分子でさえ)に対応する配列の列を組換えることができる。本発明の特定の実施形態に従って、コンピューター・システムで処理した後、例えばオリゴヌクレオチド合成/遺伝子再集合技術と協調させて、組換え配列に対応する核酸の合成によって、結果的に生じる配列の列を核酸に変換することができる。この手法により、ランダム改変体、部分的ランダム改変体、または設計された改変体が生成され得る。コンピューター・システムでの種々のアルゴリズム、オペレーター等の使用を含むコンピューターにより可能になった組換えと、設計された核酸および/またはタンパク質の組み合わせ(例えば交叉部位選択に基づく)と、設計された組換え法、擬似ランダム組換え法、またはランダム組換え法との種々の実施形態に関する多くの詳細については、WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides and Polypeptides Having Desired Characteristics」、WO 01/75767(Gustafasonら)、「In Silico Cross−Over Site Selection」、ならびにW O 00/42559(SelifonovおよびStemmer)、「Methods of Populating Data Structures for Use in Evolutionary Simulations」に記載されている。
【0106】
(定向進化)
スクリーニングと組み合わせた反復方法(本明細書中別の箇所でより詳細に記載される)で、1つ以上の多様性生成方法を実施して、組換え核酸の次のセットを生成することによって、定向進化(または「人工進化」)を実行することができる。従って、突然変異および/または組換えおよびスクリーニングの繰り返しサイクルによって、定向進化または人工進化を実行することができる。例えば、親ポリヌクレオチド(所望の交叉点を提供するために選択される)上で、突然変異および/または組換えを実行し、改変体ポリヌクレオチドのライブラリーを生成することができ、その後、これを発現させて、所望の活性に対してスクリーニングされる交叉点を有するタンパク質を生成する。所望の活性での改善を示すとして、1つ以上の改変体タンパク質がこれらのタンパク質から同定され得る。同定されたタンパク質を逆翻訳して、同定されたタンパク質改変体をコードする1つ以上のポリヌクレオチド配列を確認し、今度は、この配列を、多様性生成およびスクリーニングの次のラウンドで突然変異または組換えをすることができる。
【0107】
多様性生成の組換えに基づく形式を用いた定向進化については、本明細書で引用される参考文献に広範に記載されている。多様性生成の基盤として突然変異を用いる定向進化についても、当技術で周知である。例えば、再帰的集合突然変異のプロセスでは、タンパク質突然変異に対するアルゴリズムを用いて、表現型的に関連する突然変異体の多様性集団が生産され、そのメンバーは、アミノ酸配列が異なる。この方法では、フィードバック・メカニズムを用いて、組み合わせカセット突然変異の連続的ラウンドがモニターされる。このアプローチの例としては、Arkin & Youvan(1992)Proc.Natl.Acad.Sci.USA 89:7811−7815に記載されている。同様に、高比率の特有かつ機能的突然変異体を有するコンビナトリアル・ライブラリーを生成するために、指数関数集合突然変異を用いることができる。各変化位置での機能的タンパク質に至るアミノ酸の同定と平行して、対象の配列中の残基の小グループをランダム化する。このような手順の例は、Delegrave & Youvan(1993)Biotechnology Research 11:1548−1552で見出される。
【0108】
本発明の交叉同定方法は、多様性生成手順の利用に関わらず、定向進化プロセスを最適化するのに有用である。本発明を適用することにより派生する交叉情報を用いて、定向進化プロセスで作製されるライブラリーをより理知的に設計することができる。例えば、特定のアミノ酸残基位置に交叉点を挿入することが所望される場合、2つ以上の親に由来するこれらの所望のアミノ酸残基をコードするコドンを組み込んでいる合成オリゴヌクレチドを、本明細書で言及する組換え形式の1つで用いて、ポリヌクレオチド改変体ライブラリーを生成することができ、このライブラリーをその後発現させることができる。また、本明細書で記載される種々の突然変異方法の1つを用いて、所望の交叉点を組み込むことができる。従って、任意の現象で、結果として生じたタンパク質改変体ライブラリーは、有効な残基または潜在的に有効な残基であると考えられるものを組み込んでいるタンパク質改変体を含んでいるであろう。所望の活性を有するタンパク質改変体が同定されるまで、このプロセスを繰り返すことができる。
【0109】
(活性に対するスクリーニング/選択)
任意で、本発明の方法と組み合わせて生成されたポリヌクレオチドを活性スクリーニングのために細胞中にクローニングする(または、インビトロ転写反応で用いて、スクリーニングされる産物を作製する)。さらに、核酸を、インビトロで、濃縮、シーケンシング、発現、増殖させるか、または任意の他の通常の組換え方法で処理する。
【0110】
クローニング、突然変異、ライブラリー構築、スクリーニング・アッセイ、細胞培養等を含む本明細書で有用な分子生物学的技術を記載する一般的テキストとしては、BergerおよびKimmel、Guide to Molecular Cloning Techniques,Methods in Enzymology volume 152 Academic Press,Inc.,San Diego,CA(Berger);Sambrookら、Molecular Cloning − A Laboratory Manual(2nd Ed),Vol.1−3,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York,1989(Sambrook)、ならびにCurrent Protocols in Molecular Biology,F.M.Ausubelら、eds.,Current Protocols,Greene Publishing Associates,Inc.およびJohn Wiley & Sons,Inc.の共同事業、New York(2000年まで補完される)(Ausubel))が挙げられる。核酸を用いて植物および動物細胞を含む細胞を形質導入する方法は、公に入手可能であり、同様に、このような核酸にコードされるタンパク質を発現させる方法も入手可能である。Berger、Ausubel、およびSambrookに加えて、動物細胞培養用の有用な一般的参考文献としては、Freshney(Culture of Animal Cells,a Manual of Basic Technique,third edition Wiley−Liss,New York(1994))、ならびに本明細書で引用される参考文献であるHumason(Animal Tissue Techniques,fourth edition W.H.Freeman and Company(1979))およびRicciardelliら、In
Vitro Cell Dev.Biol.25:1016−1024(1989)が挙げられる。植物細胞クローニング、培養、および再生成に関する参考文献としては、Payneら、(1992)Plant Cell and Tissue Culture in Liquid Systems John Wiley & Sons,Inc.New York,NY(Payne);ならびにGamborgおよびPhillips(eds)(1995)Plant Cell,Tissue and Organ Culture;Fundamental Methods Springer Lab Manual,Springer−Verlag(Berlin Heidelberg New York)(Gamborg)が挙げられる。種々の細胞培地については、AtlasおよびParks(eds)The Handbook of Microbiological Media(1993)CRC Press,Boca Raton,FL(Atlas)に記載されている。植物細胞培養の更なる情報については、Sigma−Aldrich,Inc(St Louis,MO)(Sigma−LSRCCC)から得られるLife Science Research Cell Culture Catalogue(1998)、および例えばSigma−Aldrich,Inc(St Louis,MO)(Sigma−PCCS)から同様に得られるPlant Culture Catalogueおよび付録(1997)等の入手可能な商業的文献で見出される。
【0111】
1つの好適な方法では、再集合させた配列がファミリーに基づく組換えオリゴヌクレオチドを組み込んでいるかについて検査する。これは、例えばSambrook、Berger、およびAusubel(上掲)に基本的に教示されているように、核酸をクローニングおよびシーケンシングすることによって、ならびに/または制限酵素消化によって実行することができる。さらに、配列をPCRで増幅し、直接シーケンシングすることができる。従って、例えば、Sambrook、Berger、Ausubel、およびInnis(上掲)に加えて、付加的なPCRシーケンシング方法も特に有用である。例えば、PCR中に、ボロン酸ヌクレアーゼ耐性ヌクレオチドを選択的にアンプリコン(増幅領域)に組み込み、アンプリコンをヌクレアーゼで消化し、所定の大きさにされたテンプレート・フラグメントを作製することによって、PCRで生成されたアンプリコンの直接シーケンシングが実行されていた(Porterら、(1997)Nucleic Acids Research 25(8):1611−1617)。この方法では、テンプレート上で4回のPCR反応を実行し、そのそれぞれの回で、PCR反応混合物中のヌクレオチド三リン酸の1つを2’デオキシヌクレオシド 5’(P−ボラノ)三リン酸で部分的に置換する。ボロン酸ヌクレオチドは、テンプレートのPCRフラグメントの入れ子集合中のPCRアンプリコンに沿って変動する位置で、確率論的にPCR産物中に組み込まれる。組み込まれたボロン酸ヌクレオチドに遮断されるエキソヌクレアーゼを用いて、PCRアンプリコンを切断する。その後、ポリアクリルアミド・ゲル電気泳動を用いて、切断されたアンプリコンをサイズごとに分離し、アンプリコンの配列を提供する。この方法の利点は、PCRアンプリコンの標準的サンガー形式のシーケンシングを実行するより生化学的操作の使用が少ないことである。
【0112】
合成遺伝子は、従来のクローニング手法および発現手法に適用するこができる。従って、遺伝子および遺伝子がコードするタンパク質の特性を、宿主細胞でそれらを発現させた後に容易に試験することができる。合成遺伝子を用いて、インビトロ(細胞を含まない)転写および翻訳によって、ポリペプチド産物を生成することもできる。従って、微生物細胞壁、ウイルス粒子、ウイルス表面、およびウイルス膜とともに、他のタンパク質およびポリペプチド・エピトープを含む種々の所定のリガンド、小分子、およびイオン、またはポリマー物質およびヘテロポリマー物質への結合能力に関してポリヌクレオチドおよびポリペプチドを試験することができる。
【0113】
例えば、ポリヌクレオチド直接による、またはコードされるポリペプチドによる化学反応の触媒作用に関連する表現型をコードするポリヌクレオチドを検出するために、多数の物理的方法を用いることができる。説明目的に限って、対象となる特定の所定の化学反応の特質によって、これらの方法は、当技術で周知の多数の技術を含むことが可能であり、それにより、基質および産物間の物理的相違または化学反応に関連する反応培地での変化(例えば、電磁放射、電磁吸着、電磁散逸、および電磁蛍光での変化、UVが可視であるか赤外(熱)であるか)が説明される。これらの方法を以下の任意の組み合わせから選択することもできる。すなわち、質量分光分析法と、核磁気共鳴と、同位体的標識物質、同位体的分配、および同位体分布または標識産物形成を説明する分光法と、反応産物のイオンまたは元素組成物での付随する変化(pH、無機イオン、および有機イオン等での変化を含む)を検出するための分光法および化学的方法とである。本明細書中の方法で使用されるのに適した物理的アッセイの他の方法は、レポーター特性を有する抗体を有するもの、またはレポーター遺伝子の発現および活性と組み合わせたインビボの親和性認識に基づくものを含む、反応産物に特異的なバイオセンサーの使用に基づくことができる。反応産物検出およびインビボの細胞生・死・増殖選択に関する酵素結合アッセイも目的に適うさいは用いることができる。物理的アッセイの特性に関わらず、それらを全て用いて、所望の活性、あるいは対象となる生体分子によりコードされるか、または提供される所望の活性の組み合わせを選択する。
【0114】
選択に使用される特有のアッセイは、その用途に依存するであろう。タンパク質、受容体、リガンド等用の多数のアッセイが既知である。形式としては、固定成分への結合、細胞または生体の生存能力、レポーター組成物の産生等が含まれる。
【0115】
ハイスループット・アッセイは、本発明で使用される交叉に基づくライブラリーをスクリーニングするために特に適している。ハイスループット・アッセイでは、数千までの異なる改変体を1日でスクリーニングすることが可能である。例えば、マイクロタイター・プレートのウエルのそれぞれを用いて、別々のアッセイを実行することができる。あるいは、濃度の効果またはインキュベーション時間の効果を観測する場合、5〜10個のウエルごとに、1つの改変体を試験することができる(例えば異なる濃度で)。従って、単一の標準的マイクロタイター・プレートによって、約100(例えば96)の反応をアッセイできる。1536ウエル・プレートを用いた場合、単一のプレートで、約100から約1500の異なる反応を容易にアッセイすることができる。複数の異なるプレートを1日でアッセイすることが可能である。本発明の統合されたシステムを用いることによって、約6,000〜20,000までの異なるアッセイ(すなわち、異なる核酸、コードされるタンパク質、濃度等を含む)に対するアッセイ・スクリーニングすることが可能である。より近年になって、例えばカリパー・テクノロジー(Caliper Technologies)(Mountain View,CA)によって、試薬操作に対するマイクロ流体手法が開発されており、この手法により、非常にハイスループットなマイクロ流体アッセイ法を提供され得る。
【0116】
ハイスループット・スクリーニング・システムが市販されている(例えば Zymark Corp.,Hopkinton,MA;Air Technical Industries,Mentor,OH;Beckman Instruments,Inc.Fullerton,CA;Precision Systems,Inc.,Natick,MA等を参照せよ)。 一般に、これらのシステムは、アッセイに適当な全ての試料および試薬のピペッティング、液体分配、インキュベーション時間計測、および検出器中のマイクロプレートの最終的な読み取りを含む全手順を自動化している。これらの設定可能なシステムにより、高度な適応性およびカスタマイゼーションとともに、ハイスループットおよび迅速な起動が提供される。
【0117】
このようなシステムの製造元は、様々なハイスループット・スクリーニング・アッセイに関する詳細なプロトコルを提供している。従って、例えば、ザイマーク社(Zymark Corp.)により、遺伝子転写の変調、リガンド結合等を検出するためのスクリーニング・システムを記載している技術公報が提供されている。
【0118】
デジタル化されたビデオまたはデジタル化された他の光学的アッセイ画像をデジタル化、保存、および分析するために、例えばPC(インテルx86またはペンティアム(登録商標)・チップ互換性DOS(商標)、OS2(商標)、WINDOWS(登録商標)またはWINDOWS(登録商標) NT(商標)に基づく機器)、MACINTOSH(商標)、あるいはUNIX(登録商標)に基づく(例えばSUN(商標)ワークステーション)コンピュータを用いて、種々の市販の周辺装置およびソフトウエアが利用できる。
【0119】
一般に、分析用システムとしては、本明細書中の1つ以上の方法の1つ以上のステップを検出するためのソフトウエアを有するデジタル・コンピュータが挙げられる。任意で、例えばハイスループット液体制御ソフトウエア、画像分析ソフトウエア、データ解析ソフトウエア、溶液をソースからデジタル・コンピュータに操作可能に結合された目的地まで移動させるためのロボット液体制御アーマチュア、操作を制御するためのデジタル・コンピュータにデータを入力するための入力デバイス(例えば、コンピュータ・キーボード)、またはロボット液体制御アーマチュアによるハイスループット液体移送、および任意で標識されたアッセイ成分からの標識シグナルをデジタル化するための画像スキャナも挙げられる。画像スキャナは、画像解析ソフトウエアに接続して、プローブ標識強度の測定を提供することができる。一般に、プローブ標識強度測定は、データ解析ソフトウエアによって解析され、標識プローブが固相支持体上のDNAにハイブリダイズしたかどうかが示される。
【0120】
いくつかの実施形態では、インビトロのオリゴヌクレオチド媒介組換え産物またはコンピュータ内(in silico)組換え核酸の物理的実施形態を有する細胞、ウイルス・プラーク、胞子等を固体培地上で分離し、個別コロニー(またはプラーク)を産生することができる。自動コロニー・ピッカー(例えばQ−bot、Genetix,U.K.)を用いて、コロニーまたはプラークを同定・選択し、10,000までの異なる突然改変体を2つの3mmガラス・ボール/ウエルを含む96ウエル・マイクロタイター皿に接種する。Q−botは、全コロニーの選択はせずに、コロニーの中央へピンを挿入し、細胞(または菌糸体)および胞子(またはプラーク適用ではウイルス)の小サンプリングで終了する。ピンがコロニーに入れられている時間、培地を接種するためのディップの数、およびピンがその培地に入れられている時間のそれぞれが接種材料のサイズに作用し、各パラメータが制御および最適化され得る。
【0121】
Q−bot等の自動的コロニー選択の一定のプロセスにより、人為的な操作の誤りが減少され、確立される培養物の割合が増加される(4時間で約10,000個)。任意で、温度および湿度が制御されたインキュベーター中でこれらの培養物を振とうする。マイクロタイター・プレート中の任意のガラス・ボールは、発酵槽のブレードと同様に、細胞の均一な通気および細胞(例えば菌糸体)フラグメントの分散を促進するように作用する。対象となる培養物から得たクローンを限界希釈によって単離することができる。上掲したように、ハイブリダイゼーション、タンパク質活性、または抗体へのタンパク質の結合等を検出することによって、ライブラリーを構成するプラークまたは細胞をタンパク質の産生に対して直接スクリーニングすることもできる。十分なサイズのプールを同定する確率を増すために、処理される突然変異体の数を10倍増加させる事前選別を用いることができる。一次選別の目的は、親株と同等またはそれより優れた産物の力価を有する突然変異体を素早く同定し、これらの突然変異体のみをその後の分析用に液体細胞培養へ移すことである。
【0122】
多様性ライブラリーをスクリーニングするための一手法は、超並列固相手順を用いて、ポリヌクレオチド改変体を、例えば酵素改変体をコードするポリヌクレオチドを発現する細胞をスクリーニングすることである。吸光、蛍光、またはFRETを用いた超並列固相スクリーニング装置が利用可能である。例えば、米国特許番号第5,914,245号(Bylinaら、(1999))を参照のこと;http://www.kairos−scientific.com/;Youvanら、(1999)「Fluorescence Imaging Micro−Spectrophotometer(FIMS)」Biotechnology et alia,<www.et−al.com> 1:1−16;Yangら、(1998)「High Resolution Imaging Microscope(HIRIM)」Biotechnology et alia,<www.et−al.com> 4:1−20;およびYouvanら、(1999)「Calibration of Fluorescence Resonance Energy Transfer in Microscopy Using Genetically Engineered GFP Derivatives on
Nickel Chelating Beads」(www.kairos−scientific.Comで公表)も参照せよ。これらの技術によるスクリーニングの後、一般に、対象の分子を単離し、任意で、当技術で周知の方法を用いて配列決定する。その後、配列情報を本明細書で先述したように用いて、新規のタンパク質改変体ライブラリーを設計する。
【0123】
同様に、アッセイ・システムで有用な液相化学に対して、多数の周知のロボット・システムも開発されている。これらのシステムとしては、武田薬品工業株式会社(Takeda Chemical Industries,LTD )(Osaka,Japan)により開発された自動合成装置等の自動ワークステーションと、科学者により実行される手動の合成操作を模倣しているロボット・アーム(Zymate II、Zymark Corporation,Hopkinton,Mass.;Orca,Beckman
Coulter,Inc.(Fullerton,CA))を利用した多数のロボット・システムが挙げられる。任意の上記デバイスは、例えば本明細書に記載されるように進化される核酸によりコードされる分子のハイスループット・スクリーニングのために、本発明とともに使用するのに適している。本明細書で論じられるように操作できるようにするためのこれらの装置(使用する場合は)の変更の種類および実行は当業者に自明である。
【0124】
(システム)
明らかであるように、本発明の実施形態は、1個以上のコンピューター・システムに保存されているか、またはそのようなコンピューター・システムを通して転送されるインストラクション、および/またはデータの制御の下に機能するプロセスを使用する。本発明の実施形態は、これらの操作をおこなう装置にも関する。そのような装置は、必要な目的のために特に、設計、および/または構築されたものでもよい。または、それはコンピューターに保存されたコンピューター・プログラム、および/または、データ構造によって選択的に可動、または再構成された汎用計算機であってもよい。本明細書に提示されたプロセスは、いかなる特定のコンピューター、または他の装置に本質的に関わるものではない。特に、本明細書での教示にしたがって書かれたプログラムを用いて、様々な汎用機械が使用可能である。しかしながら、ある場合には、必要な方法操作をおこなうために専門化した装置を構築することが、より好都合であるかもしれない。さまざまなこれらの機械のための特定の構造は、以下に与えられた説明から明らかになるだろう。
【0125】
加えて、本発明の実施形態は、コンピューターで実施される様々な操作をおこなうためのプログラム命令、および/または、データ(データ構造を含む)を含んでいるコンピューターによって読み込み可能な媒体、またはコンピューター・プログラム製品に関する。コンピューター読み込み可能な媒体の例には、ハードディスク、フロッピィディスク、磁気テープなどの磁気媒体;CD−ROMデバイス、およびホログラフィック・デバイスなどの光媒体;磁気光学媒体;リード・オンリー・メモリ・デバイス(ROM)、およびランダム・アクセス・メモリ(RAM)などの、プログラム命令を保存して、実行するために特別に構成されている半導体メモリ素子、およびハードウェア・デバイス、ならびに、時には、特定用途向け集積回路(ASIC)と、コンピューター読み込み可能なインストラクションを提供するためのプログム可能論理デバイス(PLD)と、ローカル・エリア・ネットワーク、広域ネットワークや、インターネットなどの信号伝達媒体とを含むが、これに限定はされない。また、この発明のデータおよびプログラム命令は、伝送波、または他の伝送媒体(例えば、光学線、電気線、そして/または、電波)の上で具体化されてもよい。
【0126】
本発明は、タンパク質配列中の交叉部位を選択、および/または評価するためのインストラクションを含むコンピューター、またはコンピューター読み込み可能な媒体を提供する。このコンピューター、またはコンピューター読み込み可能な媒体は、以下の機能の1つ以上をおこなう、1つ以上のコンピューター・コードかアルゴリズムを含んでいる。すなわち、i)基準生体分子の配列、または生体分子構造を入力する機能;ii)基準分子の基準配列用接触マップを作成する機能;iii)第1の配列および第2の配列を基準配列と整列させる機能;iv)第1の親配列と、第2の親配列との間で1つ以上の部分配列を交換し、キメラ配列を作製する機能;v)キメラ配列を接触マップと比較し、基準分子中の接触での近位要素(接触マップによる)に対応する、キメラ配列中の2つ以上の要素を選択する機能;および、vi)選択された残基を得点する機能である。場合によっては、交換ステップは多くの可能な部分配列、または部分配列のセットに関して反復され、それによって、分析用のライブラリー、または複数のキメラ配列を生成する。したがって、コンピューターによって生成する得点は、キメラ配列が基準分子の構造、立体配座安定性、および/または活性を保持する可能性に関する尺度を提供する。また、本発明のソフトウェアは、本明細書に記述されたもの以外の論理演算のいずれを提供してもよい。
【0127】
架橋オリゴヌクレオチド用の最良の位置を同定することによって、キメラ子孫(または、「シャフランツ(shufflants)」)の多様で、かつ構造的に安定したライブラリーを作製することができる。概して、以前の研究では、1箇所または2箇所の交叉に由来するキメラを考慮することによって、この分析にアプローチするだけであった。しかし、これはライブラリー中での多重交叉の効果を考慮に入れていないものである。場合によっては、多くの配列(例えば、3以上)を本発明の方法で用いることができる。例えば、本発明はまた、完全(ほとんど完全)なキメラ・ライブラリーのイン・シリコ分析に基づいて、安定したライブラリーをもたらすであろう多くの可能な交叉位置を同定する方法を提供する。場合によっては、そのような複数の交叉位置には、安定したライブラリーをもたらす可能な交叉位置の約50%、約75%、約85%、約90%、約95%、約98%、約99%、または約100%(例えば、すべて、もしくは実質的にすべて)が含まれる。
【0128】
場合によっては、コンピューターは2つ以上の親配列から複数のキメラ配列を作製し、それによって、対象とする配列における複数の交叉部位を評価する。この方法の一実施形態では、初期交叉数(n)が、使用された親配列の数m、およびコンピューター分析可能な配列の最大数(MAX)に基づき、以下の方程式を用いて決定される。MAX=mn−1。例えば、現在のハードウェアに基づいて、MAXの値は、約1010配列である。しかし、計算技術が向上するのに従って、この値は増大するだろう。初期交叉は、その後、配列の長さ全体にわたって、均等に(または、非対称的に)配置される。場合によっては、交叉の親セットは、追加情報(構造的問題、相同性、酵素活性などを含むが、これらに制限されない)に基づいて(例えばユーザによって)選ばれる。その後、可能な非変異キメラ(X)の全体ライブラリーがイン・シリコに作製される。
【0129】
この方法の一実施形態では、本発明のコンピューターは生体分子の配列、例えば基準タンパク質のアミノ酸配列を入力する。基準配列は、実験的に生成されたデータ(例えば、配列決定のデータ)と、前の組換え産物に由来するデータと、公用、および/または商用データベースなどを含む多くのソースのいずれからも入力することができる。その後、コンピューター(または、コンピューター媒体)は、当技術分野で利用可能な(および、以前に記述されているような)多くの手順またはアルゴリズムのいずれかによって、基準分子用の接触マップを作成する。
【0130】
別法として、本発明のコンピューターは、第1の親生体分子配列、第2の親生体分子配列、および、場合によっては追加の親生体分子配列(例えば複数の生体分子配列)を入力し、これらの配列を、基準配列なしに相互に整列する。
【0131】
コンピューターは、その後、2つの親配列間で部分配列(親配列中で第1の交叉部位と第2の交叉部位とによって規定される選択された領域の配列)を交換、または置換して、キメラ配列を作製する。場合によっては、コンピューターは、2つの親配列からの異なった部分配列を使用するか、または、複数の親配列からの部分配列を使用して、何回も交換手順を行い、それによって、1つ以上の交叉部位を持っている1つ以上のキメラ配列を作製する。
【0132】
2つの(または、それより多い)親配列間での、領域交換によってキメラ配列を作製した後、コンピューターは、キメラ配列の要素を接触マップと比較する。コンピューターは、基準配列における近位要素に対応するそれらのキメラ要素を選択して、それらの要素を得点する。一実施形態では、コンピューターによっておこなわれるスコアリングは、計算された接触エネルギー、立体障害、疎水性または極性などの、1個以上のパラメータに基づいている。
【0133】
一実施形態では、比較およびスコアリング・ステップは、キメラ配列のエネルギーに基づいている。キメラ・ライブラリーのエネルギーについて計算する方法は、多数ある。例えば、上記の記載と同様に、Miyazawa−Jerniganマトリックスに基づいて、エネルギー項(energy terms)を計算することが可能である。このエネルギー項は、キメラ子孫にあると予想された残基接触のエネルギーの合計から通常計算される。場合によっては、正規化されたエネルギーが、その交叉セットを与えられた可能なキメラそれぞれに関して計算される。
【0134】
本発明のある特定の実施形態では、その後、計算上最も低いエネルギーをもつキメラ配列を統計的に分析する。例えば、ベイズの解析(Bayesian analysis)、ニューラル・ネットワーク法などを含む、ただしそれらに限定されない、多くの方法によって相関係数を計算することが可能である。隣接した2つのブロックの配列間で相関係数が高いことは、交叉の前のブロックと同じ配列が、交叉の後のブロックに好適であるという事実によって、交叉が非効率的(poor)に置かれたことを示す。
【0135】
表1および表2は、初期セットの交叉位置に関する、例示的結果を示す。その後、これらの交叉の位置を変化させ、交叉が好適位置にあることを相関係数が示すまで、エネルギー計算を繰り返した。隣接した2つの交叉の間に、交叉に好適な位置がない場合(例えば、中間的交叉で規定された2つの交叉部分配列の相関がきわめて高い場合)、それら2つの隣接した交叉間の中間的交叉は交叉セットに含まれない。すべての交叉がいったん好適位置におかれると、新規セットの初期交叉で全過程が反復される。その後、計算上最も低いエネルギーをもつキメラ・ライブラリーを生成する交叉セットが選ばれる。このセットはその後、実験室での実験など、さらなる活動に使用することが可能である。一例として、表2は、3つのMLE(最大尤推定(Maximum Likelihood Estimation))配列間での交叉位置の1セットに関する配列ブロック間の相関係数を示す。「XXXを含むセル(cell)は、連続した配列ブロック間の高い相関係数を示し、したがって非効率的な交叉位置である。
【0136】
【表1】

【0137】
【表2】
場合によって、コンピューター、またはコンピューター読み込み可能な媒体のコード(code)には、接触ペアに関し生成した得点を正規化するためのメカニズム、またはアルゴリズムが含まれる。例えば、コンピューター、またはコンピューター読み込み可能な媒体は、多重回帰を行って、残余値を変動係数として提供することが可能である。場合によっては、ユーザに未処理の得点を提示する前か、または未処理の得点を提示した後に、ユーザからの指示によって正規化操作をおこなう。本発明の一実施形態では、得点が三角形の輪郭プロットとして提示される。
【0138】
C、C++、ビジュアル・ベーシック(Visual Basic)、フォートラン(Fortran)、ベーシック(Basic)、ジャバ(Java(登録商標))などの標準プログラミング言語を用いて、上で記述された計算をおこなうための論理命令を当業者により構築することができる。例えば、コンピューターは、第1配列および第2配列をデータベースで捜すためのソフトウェアを含む可能性があり、場合によっては、さらにユーザ・インタフェースと通信するために修飾されている可能性もある(例えば、ウインドウズ、マッキントッシュ、UNIX(登録商標)、LINUXなどの標準的なオペレーティング・システムによるGUI)、それによって、配列文字列を得て、構成要素系を整列させて、計算を行い、かつ/または、検査結果を操作する(例えば、交叉位置の評価)。ワープロソフト(例えば、Microsoft WordTM、Corel WordPerfectTM)、スプレッド・シート、および/または、データベース・ソフト(例えば、Microsoft ExcelTM、Corel Quattro ProTM、Microsoft AccessTM、ParadoxTM、Filemaker ProTM、OracleTM、SybaseTM、およびInformixTM)などを含み、かつそれらに限定されない標準的なデスクトップ・アプリケーションを、これらの(および他の)目的に適合させることが可能である。
【0139】
本発明のコンピューター読み込み可能な媒体には、光媒体、磁気媒体、ダイナミック・メモリ、フラッシュメモリ、およびスタティックメモリが含まれるが、これらに限定はされない。場合によっては、コンピューター、またはコンピューター読み込み可能な媒体は、分析結果を出力ファイルの形式で提供することができる。例えば、出力ファイルは、整列させられた第1配列、第2配列、および/または、基準配列の一部、またはすべてについてのグラフ表示形式、あるいはマトリックスの形式(Miyazawa−Jerniganマトリックスなど)であってもよい。
【0140】
(キメラ配列のウェブ・ベース・ライブラリー)
本発明の様々な実施形態は、キメラ・データ配列、または対象とする生体分子(例えば、RNA、DNA、タンパク質など)から得られた情報を、使用、および/または判定する方法、および/またはシステムに関する。特定の実施形態では、本発明はさらに、キメラ配列データのライブラリーを提供し、1セット以上のキメラ配列、構造、および/または、接触マップをクライアントが作成、または分析することを可能にする方法、および/またはシステムを含む。
【実施例】
【0141】
ここに記述された実施例および実施形態は、例示的目的だけのためのものであり、その観点における様々な改善または変更が当業者に示されるであろうが、それらはこの出願の精神および範囲と、添付された請求の範囲との内に含まれるものであると理解される。以下の実施例は、請求の範囲に記載されている発明を限定するものではなく、それを例解するために提供されている。
【0142】
対象とするタンパク質の1つのファミリーが、ムコン酸シクロイソメラーゼとしても知られているムコン酸ラクトン化酵素(MLE)である。これらの酵素は、芳香族化合物をクエン酸回路の中間代謝物に分解するのに不可欠である。MLEは、保存された構造をもち、かつある程度の機能的多様性をもつタンパク質をコードする。MLE Iは、シス,シス−ムコン酸(cis,cis−muconate)の(4S)−ムコノラクトンへの変換を触媒するβケトアジピン酸経路中で機能する(Ngai,K.,Omston,L.N.,およびKallen,R.G.(1983年)「Enzymes of the
beta−ketoadipate pathway in Pseudomonas
putida:kinetic and magnetic resonance studies of cis,cis−muconate cycloisomerase catalyzed reaction.」Biochemistry 22:5223−5230)。MLE IIは、同じ環化異性化反応を触媒するが、基質として3−クロロムコン酸を用いる(Schmidt,E.,およびKnackmuss,H.(1980年)「Chemical structure and biodegradability of halogenated aromatic compounds.」Biochem.J.192:339−347)。3Å(MLE II)および1.85Å(MLE I)の解像度で決定されたこれら二酵素の構造はきわめて類似したものであり、両方とも、N末端キャッピング・ドメインをもつα/βバレルを含み、平均0.96ÅのRMSDを有す(Kleywegt,G.J.,およびJones,T.A,(1996年)「A re−evaluation of the crystal structure of chloromuconate cycloisomerase.」Acta.Crystallogr.Sect.D52:858;およびHelin,S.,Kahn,P.C.,Guha,B.L.,Mallows,D.G.,およびGoldman,A.(1995年)「The refined X−ray structure of muconate lactonizing enzyme from
pseudomonas putida PRS2000 at 1.85 resolution,」J.Mol.Biol.254:918841)。エノラーゼ・スーパーファミリーのメンバーとして、それらは約60%に満たない配列同一性を有し、配列の「低配列同一性」組換えのための理想的なケースとなっている(しばしば間違って、「低相同」組換えとも呼ばれる)。
【0143】
これらの実験で、本発明者らは構造情報を計算により導き、それに応じた組換えをおこなうことを目標とした。対にしたときのアミノ酸配列同一性が約40〜52%の間にある、3つのムコン酸ラクトン化酵素(MLE)を親配列として選んだ。すなわち、Pseudomonas putida由来のMLE IIと、P.putidaおよびAcinetobacter calcoaceticus由来のMLE Iである(それぞれ登録番号P27099、AAA66202.1、およびQ43931)。交叉(crossover)は、MLE配列全体の中で、構造安定性に対し、破壊的または非破壊的であると予測された位置を代表する10位置に設計した。実験的な組換えを用いて、これらをテストした。その結果は、3つのMLEによる多機能的キメラを設計するのに、コンピューター・モデルの使用が可能であることを示す。
【0144】
本発明者らは、構造情報を用いて、これらの遺伝子の中に組換えポイントを選んだ。本発明者らはこれらの計算結果に基づいてオリゴヌクレオチドを再合成し、それを用いて組換えを行い、それによって、キメラ生体分子のライブラリーを生成した。本発明者らは、シス,シスームコン酸の活性を保持したキメラ産物を、これらのライブラリーから選択した。本発明者らは、2つまたは3つの異なったMLEからの配列ブロックを含有する、多数の異なった活性酵素を発見した。これらの変種は、最も活性な野生型親酵素の機能に匹敵する生体内機能を有し、それらに含まれる交叉部位は、本発明者らが好適であると予測した部位への強い偏向を示した。一方、好適でないと予測された部位は、強い選択により排除されていた。本発明者らは、コンピューターによる、キメラ配列の「プレスクリーニング(prescreanibg)」によって、最適な組換え位置の選択が可能となり、それによって、ライブラリーの適合度が増大し、スクリーニングしなければならない変種の数が減少すると結論した。
【0145】
(接触エネルギーの計算によってキメラ・タンパク質の安定性を予測するアルゴリズムの設計)
本発明者らは、キメラ・タンパク質の安定性を、コンピューター内で評価するアルゴリズムを設計した。このアルゴリズムは、既知の基準配列と構造(この実施例では、P.putida由来のMLE I)を用いて、どのアミノ酸が相互に接触しているかを決定する(例えば、接触マップ)。また、このアルゴリズムは、すべての親配列を相互に整列させ、そのアラインメントを用いて、完全長のキメラ配列をコンピューター内で生成する。これは、最も単純な場合には、1つの親タンパク質の1セクションを第2の親からの対応するセグメントで置換することに相当する。アミノ酸置換の影響を、基準配列と比較して評価するため、標準配列とキメラ配列とを比較し、Miyazawaらの記述のように導かれる接触エネルギー関数を用いて、接触エネルギーの変化(ΔEc)を計算した、(Miyazawa,S.,およびJernigan,R.L.(1996年)「Residue−residue potentials with a favorable contact pair team and an unfavorable high
packing density term,for simulation and
threading.」J.Mol.Biol.256:623−464、ならびに上記の追加参考文献を参照)。
【0146】
図2Aは、キメラMLEタンパク質に関して予測された接触エネルギー変化マップを示す。この図では、ドナー・タンパク質であるP.putidaMLE IIの単一セグメントによって、基準タンパク質であるP.putidaMLE Iの対応するセグメントが置換されている。縦座標(x軸)は、直線配列上の置換が始まる位置(例えば、最初の交叉位置)を示し、横座標(Y軸)は置換の長さを示す。基準配列に比べ、好適でない接触をするアミノ酸がキメラに含まれる置換は、マゼンタ色で示され、構造的破壊に通じる可能性が高い改変を反映する。予測されたキメラに、標準配列より好ましい接触をするアミノ酸が含まれるような位置は、赤色で示され、構造的に許容できる可能性の高い改変を表す。高度の破壊を引き起こす位置、すなわち接触スコアが高い位置は、紫色で示される。置換の長さが大きくなるのにしたがって、接触スコアが増大する。これは、キメラの接触マップと、オリジナルの接触マップとの間の相違が増大されるためである。
【0147】
(キメラ・タンパク質の安定性予測を用いて交叉点を特定することが可能である)
このアルゴリズムを開発する際の本発明者らの目的は、単一交叉としてより、多数の交叉の1つとして、よく機能するであろう可能性に基づいて、交叉位置を選択するキメラ・ライブラリーを設計することであった。このことを達成するため、本発明者らは、異なった長さ(1〜80アミノ酸類)のドナー・タンパク質セグメントを、基準タンパク質の特定位置で開始、または終了するように挿入するときの、平均ΔEcを計算した。図2Bは、P.putidaMLE II由来の1〜80アミノ酸によって、基準タンパク質であるP.putidaMLE Iの対応するセグメントを置換したときの平均接触エネルギー変化を示す。P.putidaMLE IIの代わりに、A.calcoaceticusMLE Iをドナーとして用いたときの結果は、きわめて類似したものであった(データは示されていない)。
【0148】
図2Bの残基位置標識によって示された曲線に基づき、10の交叉位置を選択した。図2Bの横座標で下側の位置は、より大きなΔEcを示し、従って、構造的により破壊的な可能性が高い交叉点を示す。本発明者らは、全範囲でのΔEc値を表し、構造内で均等に分布されるように交叉位置を選択した。最適であるとコンピューター内アルゴリズムで予測された交叉位置は、最初の交叉がその位置にあるキメラに関して低い接触スコアを示し、さらに第2の交叉がその位置にあるキメラに関しても低い接触スコアを示した。ここで本発明者らは、このモデルの予測力をテストする目的で、接触エネルギーが高いと予測された位置と、低いと予測された位置との両方を代表する10箇所の交叉部位を選択した。これらの交叉部位は、ヘリックスおよびシートを接続するループの中心にあるα/βバレル全体にわたり均等に分布している。これらの交叉部位は、MLEアライメントにおける残基位置98、119、144、172、201、228、254、280、302、および328にある。交叉位置119、144、172、201、および328は好適であると予測されたが、98、228、254、280、および302は好適でないと予測された。これらの計算は単量体構造に基づくものであるが、八量体構造で繰りかえされた計算でも同様の予測が提示された。その後本発明者らは、10の位置すべてで組換えを強制し、機能的変種を選択し、さらに、好適であると予測された交叉部位が、それら機能的キメラ・タンパク質中に見いだされるかを判定することによって、本発明者らのコンピューター予測をテストした。
【0149】
(オリゴヌクレオチドで媒介された組換え)
本発明者らは、標準的なシャフリング反応で相互に組換えをおこなうのに十分な配列同一性をもたないMLE親配列を選択した(例えば、Moore,G.L.,Maranas,C.D.,Lutz,S.,およびBenkovic,S.J.(2001年)「Predicting crossover generation in DNA shuffling.」Proc.Natl.Acad.Sci.USA 98:3226−3231を参照)。これにより、特定の予測を徹底的にテストすることが可能となった。少数の位置でのみ組換えを可能にすることによって、比較的少数の変種を分析することで、統計的に重要なデータが得られるようになるだろう。
【0150】
「低い配列同一性」組換えを容易にするため、交叉オリゴヌクレオチドを合成し、MLE配列全体にわたる10箇所の位置に組換えを導入した。半分のオリゴが1つの配列と同一であり、さらに、もう片方の半分は別の配列と同一であるようにオリゴを設計し、それによって、2つの配列の間の組換えを促進させた。選択されなかったクローン46個の配列決定をすることによって、組換えの度合いを抽出検査した。これらの配列のうち35%以上が、10箇所の交叉位置のうち8箇所での組み込みを示し、これらの8つの交叉オリゴに関する平均取りこみ率は47.8%であることが、これらの配列によって示された。交叉1、および交叉6には、それぞれ12%、および15%の取りこみがあった。
【0151】
MLE遺伝子の特定位置での組換を可能にするため、本発明者らは、5’半分が1つの親配列と同一であり、かつ3’半分が第2の親配列と同一である一連の交叉オリゴヌクレオチドを設計した。本発明者らは、図2Bに示された10箇所の交叉位置での組換えが容易になるように、そのようなオリゴヌクレオチド60個を、3つのMLEすべてを含む組換え反応液に入れた。本発明者らは、無作為に選ばれた71個の変種を配列決定することによって、シャッフルされたライブラリーでの交叉取りこみの頻度を算定した。個々の交叉オリゴヌクレオチドは、異なった効率で組み込まれた。それらは、機能的に選択されなかった変種の20%〜50%で検出され、そのような変種の子孫の97%に少なくとも1つの交叉が組み込まれていた。
【0152】
また、本発明者らの未処理のライブラリーを分析した結果、ライブラリーの配列に、いくらかの偏りがあることが示された。親配列の1つである、A.calcoaceticus由来のMLE Iの最終配列への寄与は、平均して4%のみであった。これはおそらく、この親遺伝子のGC含量がはるかに低いためであると、本発明者らは考えている(45%、これに比べ、P.putidaのMLE IおよびMLEIIはそれぞれ、64%および68%である)。より類似したコドン使用頻度で遺伝子を再合成することによって、組換えにおけるそのような偏りを取り除くことができるが、本発明者らは、このことによって、本研究から導いた結論が影響されるとは考えない。
【0153】
(活性キメラ酵素の生成)
キメラ酵素ライブラリーを、アシネトバクターへのMLE欠失株に形質転換し、実験プロトコル(下記)の記載に従って、相補的な(complementing)クローンを選択した。細菌コロニーは、1日目と6日目の間に現れた。本発明者らは、その中から332個の独立したMLE遺伝子を回収し、配列決定した。選択されたものと、選択されなかったもの両方の配列決定を行った403個の変種の中では、親配列間の組換えは、交叉オリゴヌクレオチドによって媒介されたもののみが観察された。
【0154】
配列決定された332個の活性変種の中に、本発明者らは合計33個の独自の組換え配列を見いだした。キメラ配列の大部分は、ある親から、第2の親骨格への単一セグメント(通常30〜60アミノ酸)の組み込みをもっていた。しかし、33個の配列うちの6個には、異なった親からの100残基以上の組み込みがあった。これらの結果により、構造的かつ機能的に保存されたタンパク質に由来する多くの異なったセグメントは、低い配列同一性にもかかわらず、相互に機能的置換が可能であることが示されている。
【0155】
(コンピューター・モデルリングは機能的酵素の交叉優先傾向を予測する)
本発明者らは、活性なキメラに見出された交叉位置に、強い偏向があるのをみた。本発明者らは、これらの偏向に関し、71個の選ばれなかったキメラの中のそれぞれの交叉の頻度を、33個の独自の活性なキメラに見られた交叉の頻度と比較することによって、これらの偏りを測定した。本発明者らは、交叉のモデル化されたエネルギー優先傾向(modeled energetic favorability)を、活性な変種における、その表出(representation)と相関させることによって、接触エネルギー曲線の予測的価値を評価した。α/βバレル中に位置する7つの交叉に関する相関係数は0.94であり、これは、本発明者らのアルゴリズムが、この領域中の生産的な交叉部位に関する有効なプレ・スクリーニング(pre−screen)であることを示している。
【0156】
α/βバレルの外にある3箇所の交叉位置(98、119、および328)はすべて、エネルギー的に好適であると予測されているが、これらはすべて、機能的なキメラにおいてきわめて不十分に表出されている(under−represented)。このドメインに基づく差異の1つの可能な理由は、活性なMLEのもつ八量体四次構造であるが、オリゴマー相互作用の大部分に、N末ドメインが関与する。モデルにオリゴマーの接触エネルギーを入れたときには、本発明者らは図2で示されたデータに非常によく似たデータを得た。しかし、安定した四次構造を保持するのに必要な接触は、分子内の接触よりも大きく重み付けされるべきでかもしれない。別の可能性は、これらのタンパク質上のN末ドメインが結合相互作用に参加すると考えられることである。利用可能なMLEの構造に、基質結合したものはないが、相同タンパク質であるo−スクシニル安息香酸合成酵素の分析によって、この「キャッピング」ドメインが、リガンド結合に続いて、〜10Åのかなり大規模な運動を行うことが示されている(Thompson,T.B.ら(2000年)「Evolution of enzymatic activity in the
enolase superfamily:Structure of o−succinylbenzoate synthase from Escherichia coli in complex with Mg2+ and o−succinylbenzoate.」Biochemistry 39:10662−10676)。構造モデリングは、パッキング(packing)相互作用が維持されるキメラを同定することができるが、分子動力学シミュレーションなどのより強度な計算力を必要とする技法は、キメラが、触媒作用に関連した構造変化をおこなうかどうかの評価をするのに使用可能である(Wang,W.,Donini,O.,Reyes,C.M.,およびIcoliman,P.A.(2001年)「Biomolecular simulations:recent developments in force fields,simulations of enzyme catalysis,protein−ligand,protein−protein,and protein−nucleicacid noncovalent interactions.」Annu.Rev.Biophys.Biomol.Struct.30:211−243)。
【0157】
(最も速い増殖をする株には合成活性部位のあるMLE酵素が含まれる)
本発明者らは、キメラMLEの発現によって、形質転換体の成長率が広い範囲に与えられ、コロニーが形質転換の1〜6日後に選択プレートの上に現れるのをみた。これは、3つの親酵素では2〜4日後であるのと比較される。2個の子孫クローンは、親に比べてかなり急速に成長し、1日かからずにコロニーを形成した。これらの1つは、主にP.putidaMLE IIから成り、A.calcoaceticusMLE Iのセクション(172〜201)と、P.putidaMLE Iのセクション(201〜228)とをもつキメラ・タンパク質を発現した。得られたキメラ活性部位は、3つの親すべてからの残基を含み、機能上重要な残基として、P.putidaMLE IIの寄与によるLys167、Lys169、およびAsp249;A.calcoaceticusMLE IによるAsp198;ならびに、P.putidaMLE IによるG1u224を含んでいる。もう片方の急速増殖形質転換体から得られたキメラ酵素の配列は、大部分がP.putidaMLE Iに由来するものであったが、これにも、A.calcoaceticusMLE Iの残基172と残基201との間が含まれていた。それらは両方とも、コンピューター内アルゴリズムによって、とても好適な交叉位置であると予測されていた。
【0158】
これらの最も活性なキメラに関する本発明者らの分析によって、α/βバレル構造の頑健さが例証された。キメラ1の3箇所の交叉のうち、位置201の交叉のみで、2つ以上の同一アミノ酸の伸長が同時に起きていた。A.calcoaceticusMLE I、およびP.putidaMLE Iに由来する2箇所の置換領域は、13残基および11残基の同一アミノ酸を、対応するP.putidaMLE IIの29残基および28残基のアミノ酸伸長と共有する。これらのアミノ酸は、MLE IIタンパク質の大部分を構成するものである。これらの相違は、一般的、保存的変化ではなく、以前に記述された方法のよって、活性を保ちつつ、酵素活性部位のこのように小さな領域に、このように多くの改変をコンピューター設計するのは困難であろう。
【0159】
(コンピューター・プレ・スクリーニング)
ここに提示された結果において、本発明者らは、遺伝子またはライブラリーの物理的構築の前に、組換え部位をプレ・スクリーニングすることを可能にするアルゴリズムを記述した。このアルゴリズムは、コンピューター内組換体タンパク質を作製し、構造的に特徴付けられた基準タンパク質からの偏位に基づいて、得られたキメラ分子中のエネルギー変化を予測し、さらに、パッキング相互作用の破壊を最小にする可能性の高い交叉部位を選択する。
【0160】
以前の研究では、無作為の位置での単一組換え事象を引き起こし、1つの親のN末端と第2の親のC末端との間の融合を形成することによって、キメラ・タンパク質のライブラリーを、物理的に生成した(Ostermeier,M.,Shim,J.H.,およびBenkovic,S.J.(1999年)「A combinatorial approach to hybrid enzymes independent of DNA homology」Nature Biotechnol.17:1205−1209;Sieber,V.,Martinez,C.A.,およびArnold,F.H.(2001年)「Libraries of hybrid proteinsfrom distantly related sequences.」Nature Biotechnol.19:456−460)。これらの方法では、活性の低い機能的キメラが比較的少数得られた。
【0161】
そのように無作為に得られた遺伝子組換え体は、機能的に印象のうすいものであり、ITCHYキメラの最良のものでも、開始遺伝子より500〜1万倍も活性が低くかった。これらには、リーディング・フレーム・シフト、または挿入/欠失が導入された可能性のあるものも含まれている。あるいは、単一交叉によるキメラ生成に続いて選択ステップがおこなわれるため、これが不安定なキメラの形成に通じているのかもしれない。接触エネルギーに基づくアルゴリズムは、単一交叉事象による交叉が、2重交叉より、少数の安定したキメラを生成すると予測している(データは示されていない)。また、本発明者らがこの研究で分析した33個の活性なキメラのうち、1個だけが単一交叉を有し、残りは複数の組換事象によって生成したという事実がこの主張と一致している。組換えに関する、前のファミリーの研究でも、選択された最も活性な組換えに多重交叉が見いだされている(例えば、Crameri,1998年;Ness,1999年;Chang,1999年;およびSoong,N.ら,2000年、ならびに上記のものすべてを参照)。さらに、本発明者らの活性なキメラのうち81%は、別の親からの組込みが70アミノ酸に満たないものであった。単一交叉部位のキメラを選択したときには、これらのように1つの親が支配的なキメラを生じる可能性が、N末領域、およびC末領域の交叉位置に限定されるであろう。最後に、初期選択を単一交叉によるキメラでおこなうことによって、他の交叉と組み合わせてうまく機能しうる交叉部位を、時期尚早に排除している可能性がある(Lutz,S.,Ostermeier,M.,Moore,G.L.,Maranas,C.D.,およびBenkovic,S.J.(2001年)「Creating multiple−crossover DNA libraries independent of sequenceidentity.」Proc.Natl.Acad.Sci.U S A.98 11248−11253)。対照的に、本発明者らのコンピューター・プレ・スクリーニングは、原則として、望まれるだけ多くの組換事象を考慮することができる。
【0162】
高度に多様化した親からキメラを作製する別のアプローチは、構造的情報と機能的情報とを結合し、しばしばいくらかの程度の配列ランダム処理および/または組換えと共役させる準合理的設計のアプローチである。これらの実験は、概ね、低い配列同一性ライブラリーの組換えより成功しており、基質特異性および熱安定性の両方における改変を達成した(例えば、Altamirano,M.M.,Blackburn,J.M.,Aguayo,C.およびFersht,A.R.(2000年)「Directed evolution of new catalytic activity using the alpha/beta−barrel scaffold.」Nature 403:617−622;Jermutus,L.,Tessier,M.,Pasamontes,L.,van Loon,A.P.およびLehmann,M.(2001年)「Structure−based chimeric enzymes as an alternative to directed enzyme evolution:phytase as a test case.」J.Biotechnol.85;15−24;ならびに、Kaneko,S.ら(2000年)「Module recombination of a family F/10 xylanase:replacement of modules M4 and M5 of the FXYN of Streptomyces olivaceoviridis E−86 with those of the Cex of Cellulomonasfimi.」Protein Eng.13:873−879を、参照のこと)。しかしながら、この方法によるキメラ生成は、改変される特定のタンパク質に関係する構造機能相関について、かなりの理解が必要とする。
【0163】
(実施例の要旨)
この実施例において、本発明者らは、指向性進化技術の一般的適用性を、コンピューター分析のスピードおよび威力と結合する方法を記載した。本発明者らのコンピューター内組換えおよびプレ・スクリーニングに必要な唯一の構造情報は、親タンパク質の結晶構造、NMR構造、構造相同性モデル情報または、当業者に公知の他の構造決定法などの立体的構造情報である。したがって、このアプローチは、一般的であり、かつ高度に自動化可能なものである。場合により、この初期スクリーニングを通過するキメラは、物理的に合成され、試験される。この研究において、本発明者らは交叉オリゴヌクレオチドを用いてキメラ・ライブラリーを合成したが、プレ・スクリーニング・データの取りこみを可能にするいかなる形式(例えば、Nessら(2002年)Nature Biotechnology 20:1251−1255,PCT公報WO 00/42561、およびWO 00/42560に記載される合成組換え)も使用され得る。不十分に折りたたまれた(poorly−folded)変種を取り除くことによって、物理的にスクリーニングしなければならないタンパク質の数を減少させ、特にスクリーニングが困難かつ複雑で、時間がかかる時、成功の可能性を大きくし得る。構造と機能との間の相関の本発明者らの理解が向上するにしたがい、コンピューターのアプローチで評価し得る特性の範囲が拡大され、それによってコンピューター内のプレ・スクリーニング・アルゴリズムの価値が高まると、本発明者らは考える。
【0164】
本発明者らは、最も近い親の配列から、少なくとも70アミノ酸が異なる5つの組換え産物を得た。これらのキメラ生体分子は、すべて、コンピューター内アルゴリズムで好適であると予測された交叉を含んでいる。同じ機能構造に折り畳む能力に関して、自然進化によって既にスクリーニングされている親配列から選択された多様性を用いることで、単純なコンピューターのプレ・スクリーニングを使用して、いかなる出発点からも非常に異なる新規配列を設計し得る。さらに本発明者らは、このアルゴリズムを交叉オリゴヌクレオチドで媒介された組換えと結びつけることによって、40%という低い配列同一性の配列から生成され、機能的に活性なキメラを含むキメラ・ライブラリーを作製した。
【0165】
キメラ・ライブラリーは、増殖にMLE活性が必要なプレート上のMLEノック・アウト株中で増殖させた。それらのコロニーを、2〜7日間にわたって増殖させ、124コロニーを選択して、配列決定した。選択され、配列決定されたこれらのクローンの中には、繰り返し現れる配列が多く存在し、結果的に、33種の独自な入れ替え配列(shuffled sequence)が得られた。選択された配列のうち3つでは、異なった親からの組み込みが100残基以上あったが、キメラ配列の大部分は、30〜60残基が組込まれていた。選択されなかった配列に対して選択された配列に、119位、144位、172位、および201位を中心に交叉位置が組み込まれる、強いパターンが見られる。
【0166】
予測傾向は、119位、144位、172位、302位、および328位の残基を最適開始位置として、様々な長さにわたり、きわめて類似し続ける。本発明者らは、119位、144位、172位、および328位での交叉が正しく予測されたと考える。なぜなら、それらの交叉が、選択されなかったクローンよりはるかに頻繁に、選択されたクローンに見られたからである。2つの位置(98および302)だけが、予測と異なり、予想したように頻繁に見られなかった。98位は、選択されなかったクローンの12%でのみ見られた。したがって、このオリゴの取りこみ全体が欠如していることが、予測と、実際の結果との間の矛盾を説明し得る。最適な終了残基(ending residue)は、144位、172位、201位、および228位であると予測される。本発明者らは、交叉を終了させる可能性の高い部位は、172位、201位、および302位であることを見いだした。228位は、選択されなかったクローンの15%で組み込まれたのみであり、したがって、この交叉がなぜ選択されたクローンで見られなかったかを説明し得る。144位は、非常に好適であると予測されたが、少数の選択されたクローンで交叉を終了させているのが見られたのみである。これは98位および119位で交叉を開始したクローンが少数であって、したがって、144位で終了可能な交叉の数が限定されたことに起因し得る。
【0167】
(実験プロトコル)
P.putida由来のMLE I、およびMLE 2、ならびにA.calcoaceticus由来のMLE Iの野生型遺伝子は、上述のように、増幅し、断片化した(Crameriら、1998年)。3’末端で1つの親に、かつ5’末端で別の親に相補的なオリゴヌクレオチドは、QUIAGENオペロン(QUIAGEN Operon(Alameda,CA))から取り寄せた。これらは、テキスト中で同定された位置に対応するブレーク・ポイント(break point)をもつ。フラグメントおよびオリゴの混合物は、例えば、Nessら、「Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently」(2002年)Nature Biotechnology 20:1251−1255、およびPCT公報W098/27230に記載されるように組み合わせた。得られたキメラ配列を、以下で記載するようにベクターにクローニングした。選択されなかったキメラ分子、および選択されたキメラ配列の配列決定は、アプライド・バイオシステムズ(Applied Biosystems(Foster City,CA))3700型シーケンサーを用いて行った。
【0168】
(MLE活性に関する選択)
A.calcoaceticusのMLE Iノック・アウトを、自然形質転換能と、catB遺伝子をカナマイシン耐性カセットに置換する相同組換えとを用いて構築した。簡潔には、フォワード・プライマー、およびリバース・プライマーを用いて、pACYC177のカナマイシン耐性カセット(プロモーターと共に)をPCR増幅した。PAGE精製された両プライマーは、100のヌクレオチド長であった。そのうち、3’末端の20ヌクレオチドは、カナマイシン耐性カセットのちょうど上流(フォワード・プライマー)か、またはちょうど下流(リバース・プライマー)と同一であり、80ヌクレオチドのテール部分(tail)は、アシネトバクターADP1のcatB遺伝子(Genebank AF009224)のちょうど上流(フォワード・プライマー)か、またはちょうど下流(リバース・プライマー)の領域と同一である。catB遺伝子は、ムコン酸ラクトン化酵素MLE Iをコードする。
【0169】
A.calcoaceticus株ATCC33305(BD413株)を、一晩、30℃で育てた。容積0.4mlを、50mlチューブ中のLB10mlに副培養(subculture)し、その後、30℃、300rpmで2時間振盪培養した。約0.5mlの培養物を、カナマイシン・カセットを増幅するPCR反応液の50μlに混合した。耐カナマイシンの形質転換体を、40μg/mlのカナマイシンを含むLB寒天上で選択し、安息香酸寒天上に広げて、増殖能を調べた。安息香酸寒天は、1リットルあたり10g K2SO4、135g K2HPO4、47g KH2PO4、25g NaCl、5.4g NH4Clおよび50mlの2%MgSO4x7H2Oからなる10x無機質塩ベース、および10mM CaCl2、0.5mM FeCl3、および0.5mM MnCl2からなる1000x微量塩溶液を用いて調製した。無機質塩ベースおよび微量元素に加えて、1リットルあたり、2.5mlの1M Na2MoO4x2H2Oおよび2.5mlの1M安息香酸ナトリウムを、1.5%のディフコ・バクト・アガー(Difco Bacto agar)と共に加えた。NS238と命名された、安息香酸存在下で成長できなかった一形質転換体を用いて、活性なキメラについての選択を行った。
【0170】
P.putida由来のMLE I、および2、ならびにA.calcoaceticus由来のMLE Iの野生型遺伝子を、広域宿主ベクターpMMB66EH(ATCC37620)にクローニングした。これらの3つの構築物は、安息香酸寒天上での増殖に関し、ノック・アウト株NS238を相補し得た。キメラMLEライブラリーを、上述のようにプラスミドpMMB66EHにクローニングし、自然形質転換能を用いてNS238株に形質転換し、0.15mMのIPTGと共に処方された安息香酸寒天上にプレート培養し、さらに、1週間まで、37℃でインキュベートした。安息香酸を利用する形質転換細胞の増殖表現型は、単一コロニーになるように安息香酸寒天に広げ、その後、37℃で6日間、3つの野生型遺伝子と比較した増殖によって検査した。
【0171】
(構造モデリング)
MLE 1p構造である1MUCにおける相互作用残基を用い、ケミカル・コンピューティング・グループ社(Chemical Computing Group(Montreal,Quebec,Canada))のMOEソフトウェアを使用して、酵素の接触マップを定義した。接触マップは、結晶構造中の相互作用を記述するマトリックス(C)であって、残基iおよび残基jに関して、2残基間の距離4.5Å内に潜在的水素結合パートナー、または疎水性相互作用がある場合に、Cij=1となり、ない場合には、Cij=0となるように得点するようなマトリックスである。
【0172】
3つの親MLEに関して、すべての可能な2交叉キメラをコンピューター内で作製した。その後、オリジナルの接触マップとの比較によって、これらのキメラを評価した。MiyazawaおよびJerniganのポテンシャル(potential)を用いて、キメラの接触エネルギーを計算した。MLE構造中の各相互作用位置で、コンピューター内でキメラに由来する接触している残基のMiyazawa−Jerniganエネルギーを、合計して、総接触エネルギー(Ec)を算出した。
【0173】
【数1】
式中、EMJ(i,j)は残基iおよび残基jに関するMiyazawa−Jerniganエネルギーであり、iとjとの間に接触がある場合、Cijは1であり、接触がない場合は0である。置換セグメントの長さが増大するにしたがって、接触エネルギーも増大することを留意する。これはキメラとオリジナルの接触マップの相違が増大することに起因する。したがって、本発明者らは、基準配列に向かったこの偏りを最小にするために、交叉分析を置換長80以下に制限した。MiyazawaおよびJerniganポテンシャルの関連した記載は、上で同定した参考文献において見いだされる。アミノ酸配列に関しては、例えば、Miyazawa−Jerniganエネルギー・マトリックスを用いることで、接触エネルギーを推定することができる(例えば、MiyazawaおよびJernigan(1999年)「Self−consistent estimation of inter−residue protein contact energies based on an equilibrium mixture approximation of residues」Proteins 34:49−68;Miyazawa,およびJernigan(1999年)「An empirical energy potential with a reference state for protein fold and sequence recognition」Proteins 36:357−69;Zhang(1998年)「Extracting contact energies from protein structures:a study using a simplified model」Proteins 31:299−308;ならびに、Miyazawa,S.およびJernigan,R.L.(1996年)「Residue−residue potentials with a favorable contact pair term and an unfavorable high packing density term,for simulation and threading.」J.Mol.Biol 256:623−464を参照)。
【0174】
また、本発明者らは、はるかに計算力を必要とするアルゴリズムも試験した。そのアルゴリズムにおいて、キメラ配列が基準構造を通して縦列され、その後、ケミカル・コンピューティング・グループ社のモレキュラー・オペレーティング・エンバイロメント(Molecular Operating Environment)によるAMBER94
力場を用いたエネルギー最小化ルーチンにかけられる。この方法を用いた初期結果は、はるかに単純で計算がより速いΔEc計算と、有意に異なっていなかった。
【0175】
明快さと、理解とを目的として、上述の発明をある程度詳細に記載したが、この開示を読むことにより、本発明の本当の範囲から逸脱することなく、形態および詳細に様々な改変をおこない得ることが当業者に明確になる。例えば、上述のすべての技術、方法、組成物、装置、およびシステムは、様々な組合せで用いられ得る。この出願で引用されたすべての刊行物、特許、特許出願、または他の書類は、それぞれの個々の公表、特許、特許出願、または他の書類が、すべての目的のために引用によって組み込まれるように個別に示された場合と同じ程度で、すべての目的のために、参考としてその全体が組み込まれる。
【技術分野】
【0001】
(関連出願との相互参照)
この出願は、米国特許出願第60/363,505号(2002年3月9日出願)および米国特許出願第60/373,591号(2002年4月18日出願)の米国特許法第119条(e)にもとづく利益を求めるもので、これらの出願の全体を本明細書中で援用する。
【0002】
(著作権についての留意すべきこと)
連邦法施行規則第37巻(37 C.F.R)第1.71条(e)に従い、出願人は、この開示の一部が著作権保護の対象となる材料を含むことを指摘する。著作権の所有者は、特許商標庁のファイルに開示または記録されるとおりに第三者による特許書類または特許開示の複製に対しては、なんら反対するものではないが、そうでなければいかなるものであっても著作権のすべてを留保する。
【0003】
この発明は、バイオインフォマティクス、構造活性相関(SAR)、および組換えタンパク質および核酸を設計するプロセスへの構造活性分析の適用の分野に関する。
【背景技術】
【0004】
(発明の背景)
生体分子の新規の機能または改善された機能の探索は、取り組みし甲斐のある試みである。例えば、所望の特性を現すタンパク質の作製および同定を目的として、改変体タンパク質のライブラリーを生成する方法およびスクリーニングする方法が開発されている(例えば、Stemmer,W.P.(1994)「Rapid evolution of
a protein in vitro by DNA shuffling」Nature 370:389−391を参照せよ)。いくつかの方法では、ほとんど機能性分子を含まないライブラリーが生成される(例えば、Ostermeier(1999)「A combinatorial approach to hybrid enzymes independent of DNA homology」Nature Biotech 17:1205を参照せよ)。スクリーニング能力の限界は、そのような機能性分子の発見を困難なものにする。したがって、機能性生体分子が強化された生体分子ライブラリーを作製する方法の存在が求められている。
【発明の概要】
【発明が解決しようとする課題】
【0005】
機能性生体分子が強化された生体分子ライブラリーを作製する方法を提供すること。
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、組換えポリペプチドおよび/または核酸の中に多様性をより効率的に操作するための方法およびデバイスを提供する。例えば、アミノ酸配列またはヌクレオチド配列での潜在的な組換え交叉部位を選択および/または評価するための種々の方法を提供するとともに、結果として生ずるキメラ産物配列を提供する。これらの方法として、例えば組換えで使用される配列および交叉部位の選択および評価で構造的、機能的、および/または統計学的データを考慮することが挙げられる。
【0007】
特に、本発明は2つ以上の生体分子間の交叉点を選択する方法を提供する。本発明の一局面は、基準ペプチド配列上の複数の潜在的交叉点の適応度を決定する方法を提供する。そのような方法は、以下の一連の操作によって記述され得る:(a)基準ペプチド配列上の複数の潜在的交叉点の各々について、適応度パラメータの全体的な値を計算すること、(b)潜在的交叉点に対する適応度パラメータの全体的な各々の値にもとづいて、キメラ・ペプチドに対する実際の交叉点を選択することである。このキメラ・ペプチドは、基準配列由来の部分的な配列を含む。これらの方法では、適応度パラメータの全体値が、検討中である潜在的交叉点を持つ複数のキメラに対する適応度パラメータの複数の個別値から、計算される。これらのキメラは、基準配列中に種々の長さの部分配列を挿入し、検討中である交叉点において、各部分配列を終了させることにより、生成され得る。
【0008】
実際の交叉点を選択した後、キメラ・ペプチドをコードする少なくとも1つのキメラ核酸を作成することによりこの方法を継続させ得る。また、そのようなキメラ核酸は、選択された交叉点をコードする少なくとも1つのオリゴヌクレオチドを含むオリゴヌクレオチドを組換えることによって生成され得る。このオリゴヌクレオチドは、2つの部分的な配列を含み、そのうちの1つは1本の親ペプチドの部分的な配列をコードし、他方は別の親ペプチドの部分的な配列をコードし、この2つの部分的な配列は、選択された交叉点に対応するオリゴヌクレオチド内の1つの場所で交わる。
【0009】
適応度パラメータは、特定の物理的基準に合うペプチドの能力の任意の尺度であり得る。一例において、適応度パラメータは、ペプチドの結合特性を増加または減少させるキメラ対立遺伝子の能力の尺度を提供する。別の例では、ペプチドの折り畳みを保存または改善するキメラ対立遺伝子の能力の尺度を提供する。
【0010】
適応度パラメータ(検討中の潜在的交叉点を持つ特定のキメラ配列に対して)の個別値は、種々の技術で得られ得る。一例において、個別値を以下の方法で測定する。すなわち、(i)基準ペプチド配列に対してキメラ配列をアラインメントし(ii)接触マップからキメラの接触残基を同定し、そして(iii)キメラの接触残基に対する残基間ポテンシャル(residue−residue potential)を合計する。
【0011】
基準ペプチド配列は、多くの理由で選ばれ得る。一例において、基準ペプチドは天然に生ずるペプチドである。別の例では、組換えまたは突然変異の手順によって同定される非天然のペプチドである。いくつかの場合において、上記方法で使用される親ペプチドの1つは、それ自体が基準ポリペプチド配列である。
【0012】
一般に、この方法は、複数の交叉点の選択を含み、基準ペプチドの部分的な配列を含む複数のキメラ・ペプチドを生成する。本明細書中でさらに詳しく説明されるように、これら複数のキメラ・ペプチドは、ペプチド・ライブラリーとして生成され得る。このライブラリーのメンバーは、種々の技術で生成され得る。本発明は、(i)ペプチド・ライブラリーの選択されたメンバーが発現され得る発現系を提供し、(ii)その発現系にペプチド・ライブラリーの選択されたメンバーをコードするポリヌクレオチドをクローニングし、そして(iii)ペプチド・ライブラリーの選択されたメンバーを発現させることにより、選択されたメンバーのペプチド・ライブラリーを生成する方法もまた、提供する。
【0013】
本発明の別の局面は、限定されるものではないが、(i)基準生体分子または基準生体構造の基準配列を提供し、(ii)基準配列に関する接触マップを生成し、(iii)配列間で1つ以上の交叉点が決定される第1の生体分子の第1の配列と第2の生体分子の第2の配列とを提供し、(iv)第1および第2の配列を基準配列にアラインメントし、(v)第1の配列から得た部分配列を第2の配列から得た部分配列と置換し、キメラ生体分子配列を生成し、(vi)キメラ生体分子配列を接触マップと比較して、基準生体分子の接触マップ中の近位要素に対応するキメラ生体分子配列中の2つ以上の要素(例えば、ヌクレオチド塩基またはアミノ酸側鎖、もしくはアルファ炭素)を選択し、そして(vii)選択された要素をスコアを記録する方法であり、該スコアは、基準生体分子と類似または同一の安定性または活性を持つキメラ生体分子配列の可能性の尺度を提供する。任意に、2つ以上の部分配列がキメラ産物配列の生成の際に交換(スワッピング)され、複数の交換(スワップ)配列が1つの親配列または複数(2つ以上)の親配列に由来し得、一実施形態では、生体分子はタンパク質またはポリペプチドであり、別の実施形態では、分析中の生体分子が核酸、例えば触媒RNA分子または他の機能的に活性な核酸分子を含む。基準生体分子配列の接触マップは、いくつかのデータ・ソースから生成され得、これらのデータ・ソースとして、結晶学的モデル、NMRデータ、タンパク質折り畳みアルゴリズム、アイデンティティモデリング、ヌクレオチド・モデリング・アルゴリズム等が挙げられるが、これらに限定されない。親配列の1つ以上の領域の挿入または「交換(スワッピング)」後、結果として生ずるキメラ分子の選択された要素が、基準分子内に同等に位置した要素と比較され、これらの選択された要素のスコア記録は、コンピュータ内(in silico)組換え手順において用いられる交叉部位を評価するためのメカニズムを提供する。
【0014】
別の局面において、本発明はまた、例えば、交叉点の評価または組換え結果の予測等の命令をコンピュータまたはコンピュータ読み取り可能媒体に提供する。該コンピュータまたはコンピュータ読み取り可能媒体として、(i)基準生体分子の基準配列を入力し、(ii)該基準配列の接触マップを生成し、(iii)基準配列に第1の配列および第2の配列をアラインメントし、(iv)第1の配列の部分配列を第2の配列の部分配列と置換してキメラ配列を作製し、(v)該キメラ配列を接触マップと比較して、接触マップ中の近位要素に対応するキメラ・アミノ酸配列中の2つ以上の要素を選択し、そして(vi)選択した要素のスコアを記録し、該スコア(およびそれによるコンピュータ読み取り可能媒体)は、キメラ配列が、基準配列と比較して三次元構造上のなんらかの類似性または同様の活性を保持する可能性の尺度を提供し、それにより、選択された組換え現象の組換え結果の予測または交叉点を評価する。
【0015】
基準タンパク質のアミノ酸スペーシングは、当業者に公知の多くの技術によって決定することができる。そのような技術としては、例えば、結晶学、NMRスペクトロスコピー、およびEPRスペクトロスコピー等が挙げられる。あるいは、情報は、コンピュータ内(in silico)で公に入手可能であるか、またはコンピュータ内(in silico)で生成され得;タンパク質折り畳み分析または分子モデリングおよび/もしくは残基間距離の計算を実行するソフトウェアは、いくつかのベンダーから入手可能であり;お互いの臨界距離内にあるアミノ酸残基の同定に使用され得る。この臨界距離は、関与するアミノ酸残基、分子相互作用の性質、および残基が基準タンパク質の活性に関連して果たす役割により変動し得る。任意に、臨界距離は、約2オングストロームから約6.5オングストローム、または、例えば2.5オングストロームから約4.5オングストローム、もしくは約4.5オングストローム未満の範囲である。
【0016】
本発明の方法およびコンピュータを基礎としたデバイスにおいて、第1の親配列(例えば、アミノ酸配列)の領域が第2の親配列内の領域に対して挿入または置換されることにより、キメラ、またはスワップされた産物配列を生成する。本発明のいくつかの実施形態において、基準配列が、親配列の1つとして用いられる。他の実施形態において、2つ以上の「非基準」親配列が、組換えポテンシャル(recombination potential)について評価される。任意に、第1親配列および第2親配列は、互いに低い配列のアイデンティティを有する。分析のために、すべての潜在的キメラ産物または潜在的産物のサブセットのいずれかが、交叉可能性について調べられる。
【0017】
本発明のさらに別の局面は、上記の方法およびソフトウェア・システムを実行するためのプログラム命令および/またはデータのアレンジメントが提供される機械読取可能媒体を含む装置およびコンピュータ産物に関する。しばしば、プログラム命令は、ある種の方法操作を実行するためのコードとして与えられる。データは、この発明の特徴を実現するために用いられる場合、データ構造、データベース・テーブル、データ・オブジェクト、または特定情報の他の適当なアレンジメントとして提供され得る。この発明のいずれかの方法またはシステムは、機械読取可能媒体上に提供されたそのようなプログラム命令および/またはデータとして、全体的にまたは部分的に表され得る。
本発明は、例えば、以下の項目を提供する。
(項目1)
基準ペプチド配列上の複数の潜在的交叉点の適応度を決定する方法であって、該方法は、以下:
(a)該基準ペプチド配列上の該複数の潜在的交叉点の各々について、検討中の潜在的交叉点を有する複数のキメラの適応度パラメータの複数の個別の値から該適応度パラメータの全体的な値を計算する工程;および
(b)該潜在的交叉点についての該適応度パラメータのそれぞれの該全体的な値に基づき、該基準ペプチド配列の部分的な配列を含むキメラ・ペプチドのための実際の交叉点を選択する工程、
を包含する、方法。
(項目2)
前記交叉点が前記基準ペプチド配列の中間点であり、かつ、前記キメラ・ペプチドは、前記交叉点で終了する前記基準配列の部分的な配列を含む、項目1に記載の方法。
(項目3)
前記キメラ・ペプチドをコードする、少なくとも1つのキメラ核酸を産生する工程をさらに包含する、項目1に記載の方法。
(項目4)
少なくとも1つのキメラ核酸の産生が、前記選択された交叉点をコードする少なくとも1つのオリゴヌクレオチドを含むオリゴヌクレオチドを組換える工程を包含する、項目3に記載の方法。
(項目5)
前記選択された交叉点をコードする前記少なくとも1つのオリゴヌクレオチドが2つの部分的な配列を含み、1つの部分的な配列が1つの親ペプチドの部分的な配列をコードし、かつ別の部分的な配列が別の親ペプチドの部分的な配列をコードし、該2つの部分的な配列が該選択された交叉に対応する該オリゴヌクレオチド中のある位置で合同する、項目4に記載の方法。
(項目6)
前記親ペプチドの1つが、前記基準ペプチド配列を含む配列を有する、項目5に記載の方法。
(項目7)
他方の親ペプチドが、前記基準ペプチド配列をコードする遺伝子を含む遺伝子ファミリー由来の核酸によってコードされる、項目6に記載の方法。
(項目8)
前記適応度パラメータが、ペプチドの結合特性を増強するか、または低減させるキメラ対立遺伝子の能力の尺度を含む、項目1に記載の方法。
(項目9)
前記適応度パラメータが、ポリペプチドの折り畳みを保存するかまたは改善する、キメラ対立遺伝子の能力の尺度を含む、項目1に記載の方法。
(項目10)
前記検討中の潜在的交叉点を有するキメラ配列について、前記適応度パラメータの個別の値を計算する工程をさらに包含する、項目1に記載の方法。
(項目11)
前記適応度パラメータの前記個別の値を計算する、項目10に記載の方法であって、該方法は、以下:
(i)前記キメラ配列を前記基準ペプチド配列に整列させる工程;
(ii)接触マップから前記キメラの接触残基を同定する工程;および
(iii)前記キメラの接触残基について、残基間ポテンシャルを合計する工程、
を包含する、項目10に記載の方法。
(項目12)
前記接触マップが、前記基準ペプチド中の残基の立体配置である、項目11に記載の方法。
(項目13)
前記基準ペプチド配列が、天然に存在するペプチドの配列である、項目1に記載の方法。(項目14)
前記基準ペプチド配列が、組換えまたは突然変異誘発手順により同定された非天然ペプチドの配列である、項目1に記載の方法。
(項目15)
(b)が、前記基準ペプチド配列の部分的な配列を含む複数のキメラ・ペプチドのための複数の交叉点を選択する工程を包含する、項目1に記載の方法。
(項目16)
前記複数のキメラ・ペプチドを含むペプチドのライブラリーを産生する工程をさらに包含する、項目15に記載の方法。
(項目17)
前記ライブラリーの1つ以上のペプチドが、1つ以上のペプチドを発現する工程を包含する方法によって産生される、項目16に記載の方法。
(項目18)
項目15に記載の方法であって、該方法は、以下:
(i)前記ペプチドのライブラリーの選択されたメンバーを発現し得る発現系を提供する工程;
(ii)該発現系に、該ペプチドのライブラリーの選択されたメンバーをコードするポリヌクレオチドをクローニングする工程;および
(iii)該ペプチドのライブラリーの選択されたメンバーを発現する工程、
をさらに包含する、項目15に記載の方法。
(項目19)
前記選択された実際の交叉点と、前記交叉点で終了する、前記基準配列の少なくとも1つの部分的な配列とを各々有する複数のキメラ・ペプチドを同定する工程をさらに包含する、項目1に記載の方法。
(項目20)
基準ペプチド配列上の複数の潜在的交叉点の適応度を決定するためのプログラム命令を提供する機械可読媒体を含むコンピューター・プログラム製品であって、
該プログラム命令は、以下:
(a)該基準ペプチド配列上の該複数の潜在的交叉点の各々について、検討中の潜在的交叉点を有する該複数のキメラの適応度パラメータの複数の個別の値から適応度パラメータの全体的な値を計算するためのコード;および
(b)該潜在的交叉点についての該適応度パラメータのそれぞれの該全体的な値に基づき、該基準ペプチド配列の部分的な配列を含むキメラ・ペプチドのための実際の交叉点を選択するためのコード、
を含む、製品。
(項目21)
前記交叉点が前記基準ペプチド配列の中間点であり、かつ、前記キメラ・ペプチドは、前記交叉点で終了する前記基準配列の部分的な配列を含む、項目20に記載のコンピューター・プログラム製品。
(項目22)
前記適応度パラメータが、ペプチドの結合特性を増強するか、または低減させるキメラ対立遺伝子の能力の尺度を含む、項目20に記載のコンピューター・プログラム製品。
(項目23)
前記適応度パラメータが、ポリペプチドの折り畳みを保存するか、または改善するキメラ対立遺伝子の能力の尺度を含む、項目20に記載のコンピューター・プログラム製品。
(項目24)
前記検討中の潜在的交叉点を有するキメラ配列に関して、前記適応度パラメータの個別の値を計算するためのコードをさらに含む、項目20に記載のコンピューター・プログラム製品。
(項目25)
項目24に記載のコンピューター・プログラム製品であって、前記適応度パラメータの前記個別の値を計算するための前記コードが、以下:
(i)前記キメラ配列を前記基準ペプチド配列に整列させるためのコード;
(ii)接触マップから前記キメラの接触残基を同定するためのコード;および
(iii)前記キメラの接触残基に関する、残基間ポテンシャルを合計するためのコードを含むことを特徴とする項目24に記載のコンピューター・プログラム製品。
(項目26)
前記接触マップが前記基準ペプチド中の残基の立体配置であることを特徴とする項目25に記載のコンピューター・プログラム製品。
(項目27)
(b)が、前記基準ペプチド配列の部分的な配列を含む複数のキメラ・ペプチドのための複数の交叉点を選択するためのコードを含むことを特徴とする、項目20に記載のコンピューター・プログラム製品。
(項目28)
前記複数のキメラ・ペプチドを含むペプチドのライブラリーを同定するためのコードをさらに含む、項目27に記載のコンピューター・プログラム製品。
(項目29)
前記選択された実際の交叉点と、前記交叉点で終了する、前記基準配列の少なくとも1つの部分的な配列とを各々有する複数のキメラ・ペプチドを同定するためのコードをさらに含む、項目20のコンピューター・プログラム製品。
(項目30)
コンピューターで実行する、2つ以上の潜在的交叉点の適応度を決定する方法であって、該方法が、以下:
(a)基準ペプチド配列中に第1の潜在的交叉点を同定する工程;
(b)前記基準ペプチド配列からの1つの部分的な配列と、異なった配列からの別の部分的な配列とを含み、かつそれらによる前記潜在的交叉点を有する第1のキメラ配列を生成する工程;
(c)前記基準ペプチド配列に関する接触マップに、第1のキメラ配列を適用する工程;(d)前記接触マップを用いて選択された、第1のキメラ配列中の残基間相互作用から適応度パラメータの値を計算する工程;
(e)1つ以上の追加キメラ配列に関し(b)〜(d)を反復する工程;
(f)(b)〜(e)で検討したキメラ配列の各々に関して、前記適応度パラメータの値から全体的な適応度の値を計算する工程;
(g)前記基準ペプチド配列中に第2の潜在的交叉を同定する工程;および
(h)前記第2の潜在的交叉点に関して、(b)〜(f)をおこなう工程を包含する方法。
(項目31)
複数の追加潜在的交叉点に関して、(a)〜(f)を反復する工程をさらに包含する、項目30に記載の方法。
(項目32)
1つ以上のペプチドの生産における使用のために、複数の潜在的交叉点から1つ以上の交叉点を、全体的な適応度の値に基づいて、複数の潜在的適応度の値に関して選択する工程をさらに包含する、項目31に記載の方法。
(項目33)
2個以上の生体分子の間の交叉点を選択する方法であって、該方法が、以下:
i)基準生体分子の基準配列を提供する工程;
ii)前記基準配列に関して、接触マップを作成する工程;
iii)第1の生体分子の第1の配列と、第2の生体分子の第2の配列を提供し、それらの間で1つ以上の交叉点が決定される工程;
iv)第1の配列および第2の配列を基準配列と整列する工程;
v)第1の配列からの部分配列を、第2の配列からの部分配列と交換し、前記部分配列が選択された交叉点で終了するようなキメラ生体分子配列を作製する工程;
vi)前記接触マップと前記キメラ生体分子配列を比較し、前記キメラ生体分子配列中に、前記基準生体分子の前記接触マップ中の近位要素に対応する2つ以上の要素を選択する工程;ならびに
vii)選択された要素を得点する工程
を包含し、得点が、前記キメラ生体分子配列が前記基準生体分子と同一、または類似の特性を有する可能性の尺度を提供することを特徴とする、方法。
(項目34)
前記生体分子がポリペプチドまたはタンパク質を含み、かつ前記要素がアミノ酸残基を含むことを特徴とする、項目33に記載の方法。
(項目35)
前記生体分子が核酸を含み、かつ前記要素がヌクレオチドを含むことを特徴とする、項目33に記載の方法。
(項目36)
前記基準配列が前記第1の配列であることを特徴とする、項目33に記載の方法。
(項目37)
前記接触マップを作成する工程が、前記生体分子中の要素の1つ以上の間隔を決定し、相互の有意な距離内に2つ以上の近位要素を同定する工程を包含することを特徴とする、項目33に記載の方法。
(項目38)
前記有意な距離の範囲が約2オングストロームから約6.5オングストロームであることを特徴とする、項目37に記載の方法。
(項目39)
前記有意な距離が約4.5オングストローム未満であることを特徴とする、項目37に記載の方法。
(項目40)
前記第1の配列および前記第2の配列を提供する工程が、BLASTPアルゴリズムおよび初期パラメータを用いて決定されるような、アミノ酸配列、または核酸配列を提供し、約60%以下のアミノ酸配列同一性を有する2つのタンパク質を得る工程を包含することを特徴とする、項目33に記載の方法。
(項目41)
前記基準生体分子と同一、または類似の特性が、酵素活性、またはタンパク質安定性を含むことを特徴とする、項目33に記載の方法。
(項目42)
スコアリングが、前記キメラ生体分子配列中の前記2つ以上の選択された要素の前記接触エネルギーを計算する工程を包含することを特徴とする、項目33に記載の方法。
(項目43)
前記接触エネルギーがMiyazawa−Jerniganエネルギー・マトリックスを用いて計算されることを特徴とする、項目42に記載の方法。
(項目44)
スコアリングが三角形輪郭プロットにおけるスコアの提示が含まれることを特徴とする、項目33に記載の方法。
(項目45)
1つ以上のキメラ生体分子を合成する工程をさらに包含する、項目33に記載の方法。
(項目46)
前記1個以上のキメラ生体分子を合成する工程が、1つ以上の組換え構築物を提供する工程を包含する、項目45に記載の方法。
(項目47)
前記1個以上のキメラ生体分子を合成する工程が、2つ以上の親配列に1つ以上の組換え操作を行い、それによって、前記キメラ生体分子をコードする1つ以上の組換え構築物を作製する工程を包含する、項目45に記載の方法。
(項目48)
1つ以上のキメラ生体分子をアッセイする工程をさらに包含する、項目45に記載の方法。
(項目49)
コンピューター・コードを含むコンピューター読み込み可能な媒体であって、前記コンピューター・コードが、
i)基準生体分子の基準配列を入力し;
ii)前記基準配列に関する接触マップを作成し;
iii)第1の配列および第2の配列を基準配列と整列させ;
iv)第1の配列上の第1の交叉部位および第2の交叉部位を含む部分配列を、第2の配列に由来する対応する部分配列で交換してキメラ配列を作製し;
v)前記接触マップと前記キメラ配列とを比較して、前記接触マップの近位要素に対応する2つ以上の要素を、前記キメラ・アミノ酸配列の中で選択し;そして
vi)選択された要素を得点する
ことを特徴とするコンピューター読み込み可能な媒体。
(項目50)
前記コンピューター・コードがまた、少なくとも1つの追加交叉部位に関してiv)〜vi)を反復することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目51)
(i)が、既知の生体分子のアミノ酸配列を提供する工程、または前記既知の生体分子をコードする核酸配列を提供する工程を包含する項目49に記載のコンピューター読み込み可能な媒体。
(項目52)
核酸データベースまたは、生体分子データベースに尋ねる工程が入力に包含されることを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目53)
接触マップを作成する工程が、前記基準生体分子の結晶モデルまたはNMRモデルからアミノ酸間隔を決定し、相互に有意な距離内にある残基を同定する工程を包含する工程を特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目54)
前記接触マップを作成する工程が、前記基準生体分子のタンパク質折り畳み分析からアミノ酸間隔を決定し、相互に有意な距離内にある残基を同定する工程を包含することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目55)
前記有意な距離がアミノ酸間相互作用の特性によって変化することを特徴とする、項目54に記載のコンピューター読み込み可能な媒体。
(項目56)
前記有意な距離が約4.5オングストローム未満であることを特徴とする、項目54に記載のコンピューター読み込み可能な媒体。
(項目57)
前記第1のアミノ酸配列および第2のアミノ酸配列を整列する工程が、核酸データベースまたはタンパク質データベースに尋ねる工程を包含することを特徴とする、項目49に
記載のコンピューター読み込み可能な媒体。
(項目58)
前記スコアリングに、キメラ・アミノ酸配列中の1対のアミノ酸の接触エネルギーを計算する工程を包含し、その対の残基が、前記接触マップで接触している残基に対応することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目59)
スコアリングが、前記接触マップで接触している残基に対応する、前記キメラ・アミノ酸配列中の全残基の接触エネルギーを合計する工程を包含することを特徴とする項目49に記載のコンピューター読み込み可能な媒体。
(項目60)
スコアリングが、Miyazawaエネルギー・マトリックスを用いて、相互作用する残基の接触エネルギーを計算する工程を包含することを特徴とする、項目49に記載のコンピューター読み込み可能な媒体。
(項目61)
スコアリングが、グラフィカル・ユーザー・インターフェースでユーザーに得点を提示する工程を包含することを特徴とする、項目49に記載の任意の1つのコンピューター読み込み可能な媒体。
(項目62)
スコアリングが、三角プロットで得点を提示する工程を包含することを特徴とする、項目49の任意の1つのコンピューター読み込み可能な媒体。
(項目63)
交叉部位を査定するための融合システムであって、該融合システムが、以下:
項目49に記載のコンピューター読み込み可能な媒体;および
グラフィック・インターフェース
を含む、融合システム。
本発明のこれらの特徴および他の特徴を、以下の図面と共に発明の詳細な説明において、より詳細に説明する。
【図面の簡単な説明】
【0018】
【図1A】図1Aは、本発明の一実施形態を示すプロセスの流れ図である。
【図1B】図1Bは、本発明の一実施形態を示すプロセスの流れ図である。
【図1C】図1Cは、本発明の一実施形態を示すプロセスの流れ図である。
【図2】図2のパネルAは、MLEII配列の部位が基準タンパク質MLEIのセグメントに置換するP.putida由来の2通りの潜在的キメラMLEIおよびMLEIIそれぞれの接触エネルギー・マップを提供する。縦軸は、置換が開始されるMLEIの残基位置を示し、横軸は置換の長さを示す。ΔEcは、マップ内の色によって示される。構造的に破壊的であることが予測されるキメラは、マゼンタで示される。好ましいと予測されるものは、赤色で示され、また中性的な変化は、青色および緑色で示されている。パネルBは、基準タンパク質P.putidaMLEIの対応セグメントと置換するP.putidaMLEII由来の1〜80アミノ酸セグメントについての平均ΔEcを示す。標識位置(98、119、144、172、201、228、254、280、302、および328)は、交叉点が表示された直線状配列に沿ったアミノ酸の数を表す。
【発明を実施するための形態】
【0019】
(発明の詳細な説明)
本発明を詳細に説明する前に、本発明が、特定の組成物または生物学的システムに限定されるものではなく、これらは無論変化し得ることが、理解されるべきである。本明細書で用いられる専門用語は、特定の実施形態を説明することのみを目的としたものであって、限定することを意図したものではないこともまた、理解されるべきである。この明細書および添付した特許請求の範囲で使用されるように、単数形(「a」、「an」、および「the」)は、内容が明らかにそれを示していない限り、複数の指示されるものも含まれる。したがって、例えば、「核酸配列(a nucleic acid sequence)」は、そのような配列が2つ以上組み合わさったものも含み、「ポリペプチド(a
polypeptide)」に対する言及は、複数のポリペプチドからなる複数の混合物が含まれる、などである。
【0020】
本明細書中で特に定義しない限り、本明細書で使用されるすべての技術用語および科学用語は、本発明が属する技術分野の当業者によって一般に理解されるものと同一の意味を持つ。本明細書中で記載される方法および材料と類似または同等の方法および材料のいずれも本発明の実施または試験に使用され得るが、好ましい材料および方法が、本明細書中で記載される。
【0021】
(定義)
本発明の記載および権利主張において、後述する定義に関して以下の専門用語を用いた。
【0022】
「部分配列(subsequence)」または「フラグメント」は、ヌクレオチドまたはアミノ酸の配列全体のうちの任意の一部分である。用語「配列」および用語「文字列」は、本明細書中で、タンパク質(すなわち、タンパク質配列またはタンパク質文字列)におけるアミノ酸残基の順番およびアイデンティティを言うか、または核酸におけるヌクレオチド(すなわち、核酸配列または核酸文字列)の順番およびアイデンティティを言い、交換可能に用いられる。
【0023】
本明細書中で使用される場合、用語「交叉点」は、配列の中の一位置を言い、該配列のその部分の起点が1つのソースから別のソースへ変化または「交叉」する(例えば、親配列間での交換にかかわる部分配列の末端)。
【0024】
本明細書中で使用される場合、用語「接触マップ」は、生体分子の構成要素間の相互作用の描写を言い、一般に二次元グラフまたはデータ・マトリックスの形態で描写され、それにより、該生体分子の三次元構造の単純化表現または簡素化表現が提供される。
【0025】
本明細書中で使用される場合、用語「キメラ」は1つ以上の親分子間での組換え現象の産物を言うために用いられる。
【0026】
本明細書中で使用される場合、用語「近位要素(proximal elements)」は、三次元構造またはモデルで互いに近位または空間的に近接している配列構成要素(例えば、アミノ酸側鎖またはアルファ炭素、もしくは核酸塩基)を言う。
【0027】
本明細書中で交換可能に用いられる用語「ポリペプチド」、用語「ペプチド」、および用語「タンパク質」は、アミノ酸残基のポリマーを言う。この用語を、天然アミノ酸ポリマーと同様に、1つ以上のアミノ酸残基が対応する天然アミノ酸のアナログ、誘導体、または模倣物であるアミノ酸である、アミノ酸ポリマーに適用する。例えば、ポリペプチドの修飾または誘導体化は、炭水化物残基の付加による糖タンパク質の形成による。「ポリペプチド」、「ペプチド」、および「タンパク質」という用語には、糖タンパク質および非糖タンパク質が含まれる。
【0028】
本明細書中で使用される場合、用語「約」は、該用語が結合した値の任意に±25%、好ましくは、±10%、より好ましくは±5%、またはより好ましくは±1%の値を言う。
【0029】
本発明は、組換えのための潜在的交叉部位を選択し、キメラ産物の三次元構造(およびそれによる活性および安定性)における組換えの効果を評価する方法を提供する。このアプローチは、適当に折り畳まれた産物および/または所望の機能性を提供すると思われる組換え配列ライブラリーの生成がよりいっそう効率的になる。
【0030】
(方法論の概要)
一実施形態において、図1Aでフローチャートとして示した3通りの操作のうちの2つ以上を用いた一般的プロセスとして、本発明を見ることもできる。その図に示すように、基準ペプチド配列上の種々の潜在的交叉点の関数として適応度パラメータを計算することで、一般的プロセスがブロック01で開始される。適応度パラメータは、交叉点を生成する異なる配列由来の対応アミノ酸によって基準配列の1つ以上のアミノ酸を置換した場合、該基準配列に関する適応度の変化として計算され得る。ブロック01の動作が完了すると、一連の潜在的交叉点(ペプチド配列内の特定の残基位置の前または後に認められる)の各々について別々に算出された適応度の値を有するものとなる。適応度パラメータが最大化すると思われる(または少なくとも特定の閾値に達する)交叉点が、それに続く合成、例えば組換え手順による合成に対して、選択される。これらの交叉点を用いることで、意図された目的にキメラ・ペプチドが「適合」する可能性が高まる。
【0031】
図1Aのブロック05に示すように、次の方法は1つ以上の選ばれた交叉点を持つキメラ・ペプチドを作製することが含まれる。これらのペプチドは、種々の手順で生成され得るメンバーを持つライブラリーから構成されるものであってもよい。1つの適当な手順では、オリゴヌクレオチド(いくつかは選択交叉点を持つキメラを生成するように設計されている)を再結合することによって、全長キメラ核酸ライブラリーを合成することを含む。このことは、一方が一方の親由来であり、他方が他方の親由来である2つの部分的な配列を持つオリゴヌクレオチドを用いることによって達成することができ、それら2つの部分的な配列により、基準配列上に定められた交叉点で合致する。図1Aでの最初の2つの動作は、データ処理を伴い、最後の動作は物理的手順を伴う点に留意する。別の言い方をすれば、計算による事前選別(交叉点適応度を基礎とする)を通過するそれらのタンパク質改変体のみが、例えば該タンパク質改変体をコードするポリヌクレオチドによる合成およびその後の該ポリペプチドの発現によって、研究室で作られる。これらの技術を用いて、機能的改変体(すなわち、これらの予測されたより良い改変体のみならず、多くの場合それらの予測改変体を含む)が冨化されたライブラリーを生成することも可能である。
【0032】
いくつかのケースでは、適応度パラメータまたは図1Aのブロック01で計算された適応度パラメータでの変化は、ポリペプチドの安定性の尺度である。ポリペプチド適応度パラメータの例として、(1)ポリペプチドの折り畳みの保存または改善、ならびに(2)必要に応じたポリペプチドの結合特異性の増加または減少をおこなうキメラ対立遺伝子の能力の尺度が含まれる。
【0033】
基準ペプチド配列(図1Aのブロック01)の「交叉スペース」を調べるために、アルゴリズムが基準配列の各潜在的交叉点(ペプチド配列内の特定の残基位置の前または後に認められる)での適応度を計算する。基準配列それ自体は、天然ペプチド配列または非天然ペプチド配列、例えば組換えまたは他の定向進化技術によって同定される配列であってもよい。さらに、基準配列は、その後に続く組換え手順で使用するために選ばれる1つの親配列であってもよい。あるいは、それ自体が組換え手順では使用されない配列であってもよい。どちらにしても、好ましくは、後に続く組換え手順で用いられる1つ以上の他の親配列に関連している。本明細書のどこかに記載したように、基準配列と、組換え手順で用いられる1つ以上の親配列とがタンパク質ファミリーのメンバーとして関係する。
【0034】
図1Bは、図1Aのブロック01を実施するための1つの典型的な手順を示している。プロセスは、1つの適応度の値が計算されるサブセットに対して基準配列内の潜在的交叉点のプールを任意に制限するアルゴリズムによって、ブロック11で開始される。一例では、潜在的交叉位置が5番目の残基またはなんらかの他の調節位置に限定される。または、潜在的交叉位置が、特に2次または3次構造(例えば、基準ペプチド等の解けた構造のループ流域)にある残基に限定される。
【0035】
次に、ブロック13では、アルゴリズムは基準配列にある「最新(current)」交叉位置を同定する。アルゴリズムが配列のNまたはC末端から始まる該配列に沿って単に進行し、潜在的交叉点を1つずつ検討することが可能であることに留意する。または、潜在的交叉位置の評価になんらかの他の順序を用いてもよい。アルゴリズムが検討のための他の交叉点を選択する順番にかかわらず、個々の交叉点は別々に検討される。検討の際、交叉点は、アルゴリズムの目的のために「最新」交叉点になる(このことは、例えば並行処理の実現により、複数の交叉点が同時に検討しうる可能性を排除するものではない)。
【0036】
「最新」交叉点セットにより、アルゴリズムは次にその位置に対する「最新」キメラを生成する。ブロック15を見よ。本発明の特定の好ましい実施形態が同一の交叉点(基準配列で定義されるように)を持つ多数のキメラを考慮することで、その交叉点にある「全体的な(overall)」適応度パラメータを得ることを理解する。これらのキメラの各々について、適応度パラメータを個々に算出する。これらの多くの適応度の値を次に組み合わせて、またはさもなければ基準配列内の特定交叉位置に関する全体的な適応度パラメータに到達する際に一緒に検討される。したがって、しばしば、所定の交叉点に対して複数のキメラを評価する必要性がある。
【0037】
この説明により、その結果として図1Bアルゴリズムの次に続くブロック(ブロック17)が最新キメラについての適応度の値の計算を伴う。さらに、その値が計算された後、アルゴリズムは基準配列の最新交叉位置について検討すべきキメラがそれ以上あるかどうかを判断する。ブロック19を見よ。もしより多くのキメラが検討すべきままであるならば、プロセス制御は、次の最新キメラを最新交叉点について検討するブロック15に戻る。これをおこなう前に、前のキメラについて計算された適応度の値は、それぞれ保存されるか、もしくは交叉位置で検討された各キメラにより連続して更新される現行の全体的適応度と場合により組み合わせられる。
【0038】
最新交叉位置で検討すべきキメラがもはや存在しないと仮定すると(ブロック19が「No」に戻る)、次にアルゴリズムは最新位置について全てのキメラに基づいた全体的な適応度を計算する。ブロック21を見よ。再び、全体的な適応度の値は、交叉位置での個々のキメラについての適応度の値の各々を考慮した値を示す。その全体値は、基準配列上で最新潜在的交叉位置を他の潜在的交叉位置と比較するために利用可能となっている。交叉位置に対する適応度の値が有利にその仲間(peers)に匹敵するならば、その後に続く組換えプロセスのための交叉位置として選ばれる可能性がある。
【0039】
全体的な適応度の値が特定の交叉位置に対して計算された後、アルゴリズムは次に、検討される潜在的交叉位置がこれ以上基準配列に存在するかどうかを決定する。ブロック23を見よ。そうである場合、プロセス制御がブロック13に戻る。このブロック13では、次の潜在的交叉位置が分析のために選択され、全体的な適応度の値がその位置で生成される。もしそうでなければ、図1Aのブロック1のプロセスが完了する。それによって、図示した例では、プロセス制御が図1Aのブロック03に移動する。この図1Aのブロック03では基準配列にある1つ以上の交叉点が適応度パラメータの相対的な全体値にもとづいて選択される。
【0040】
アルゴリズムは、置換を用いることも可能であり、該置換では、基準配列からの1つ以上の準配列を、異なる(関連があるが)配列からの1つ以上の対応準配列と置き換える。すべての場合において、アルゴリズムで現在検討中の位置で、結果として生ずるキメラは基準配列と他の配列との間で交叉点を持つべきである。図1Bのブロック17では、用いられた実際の計算は、用いられた特定の適応度パラメータに依存する。好ましい一実施形態では、適応度パラメータは、接触エネルギーである。接触エネルギーは、ポリペプチドの安定性の尺度である。それは、ポリペプチド中の残基に接触する対の残基間ポテンシャルを合計することによって計算することが可能である。接触エネルギーを計算する1つの特定のプロセスを図1Cに示す。そこに示すように、プロセスは、基準配列(または潜在的に何らかの別の配列)の「接触マップ」を受け取ることで、ブロック31で開始される。次に、ブロック33で、アルゴリズムは最新キメラを基準配列に対して並べる。これによって、キメラの残基を接触マップに適切に配置することが可能となり、それによってキメラの起こりうる折り畳みを反映する。したがって、ブロック35では、整列させたキメラにアルゴリズムが接触マップを適用する。最後に、37で、手順はキメラ内の「接触(contacting)」残基の残基間エネルギーを計算する。接触残基は、接触マップ内での該接触残基の位置によって同定された。
【0041】
一般に、接触マップは、互いに十分に近接し、ある定義された方法で互いに相互作用すると思われるポリペプチドまたは他のポリマーのそれらの残基を同定する。そのような近接した残基は、互いに「接触(contact)」した状態にあると言われる。2つの残基が所定の離間距離以内で分離している場合、これら2つの残基は接触するほど十分に近接している。一般的に、問題になる相互作用は、少なくとも2つの残基間でのエネルギー的または立体化学的相互作用である。いくつかの実施例では、異なる相互作用は、異なる離間距離を持つ。例えば、一実施形態では、約4.5オングストローム以下の離間距離を持つ2つの残基は、水素結合、イオン結合、および/または疎水相互作用により接触状態にあると思われる。一方、約2.5オングストローム以下の離間距離である2つのシスティン残基はジスルフィド相互作用により接触状態にあると思われる。一般に、接触マップは、特定の折り畳み配置の安定化等、ポリペプチドのいくつかの特性に貢献する残基を同定する簡便な方法を提供する。ポリペプチド中のあらゆる残基対は、ある程度相互作用することから、目的とする特性に貢献することができる。しかし、非常に限定された距離未満で離間したそれらの残基のみが有意に貢献する。
【0042】
したがって、接触マップは、特性を計算するのに用いられる残基の組み合わせ数に対して妥当な制限を加えることができる。このようにして、計算に必要な計算作業は、有意に正確さを犠牲にすることなく最小化される。基本的に、接触マップにより同定された接触残基対のみが全体的なポリペプチドに関する特性の計算に用いられる。一方、非接触残基は計算には含まれない。なぜなら、非接触残基側鎖の変化はタンパク質内の特異的相互作用を壊す可能性が少なく、著しい構造上の破壊を生ずる可能性が少ないからである。
【0043】
一実施例では、接触マップはポリペプチド内での残基の配置を単純に三次元で表したものである。そのような配置は、例えばX線回折データを用いてポリペプチドの構造を解くことによって推定される残基位置を示すものであってもよい。ポリペプチド内の接触残基は、上記したようにそれらの離間距離によって同定される。
【0044】
この発明で重要なことは、基準ポリペプチド配列の三次元構造に由来する接触マップが、1つ以上の関連ポリペプチド配列(一般に、検討中の交叉点を持つキメラ)に用いることができることである。関連した配列は、基準ポリペプチド配列に配置され、さらに該配置を保存するようにして接触マップに配置される。関連配列の残基は、基準配列の対応残基と同じ位置を占めるものと考えられる。この配置から、接触残基は関連配列に対して同定される。
【0045】
好ましい一実施形態では、任意の二つの残基の接触ポテンシャルは、MiyazawaおよびJerniganポテンシャルによって計算される。これらは、本明細書の他の場所で引用された種々の参考文献に記載されている。基本的に、接触残基の「M−Jポテンシャル」は、ポリペプチド全体にわって総計される。得られた総計によって、キメラの安定性の全体的な尺度が与えられる。キメラ折り畳みは、接触マップが由来する基準配列のものと一致すると仮定されることに留意する。残基間ポテンシャルは、溶媒効果を考慮することによって計算される。また、残基間ポテンシャルは残基の種類および該残基での二次構造も考慮に入れる。
【0046】
上記議論で示されたように、基準ポリペプチド上の任意の潜在的交叉点での置換の効果を評価するために、1つ以上のキメラを問題となっている交叉点を持たせることによって生成してもよい。図1Bのブロック15を見よ。最も単純な実施形態では、単一キメラだけは、選択交叉点を持って生ずる。この単純な実施形態では、そのキメラの接触エネルギーを基準配列の接触エネルギーと比較して、接触エネルギーの変化(図1BのΔBc)を得る。
【0047】
より典型的なシナリオでは、潜在的交叉点での接触エネルギーの変化が、問題となっている交叉点を各々が持つ多くの異なるキメラについて算出した変化を平均化することによって得られる。特定の交叉点を持ち、適応度パラメータの計算に用いられる種々のキメラ配列は、特定の基準に合致するようにして選択することができる。一実施形態では、交叉点のNおよび/またはC側に対する複数の準配列が、基準配列での置換のために選択することができる。これらの準配列は、基準配列をコードする遺伝子に関連した他の「親」配列から得ることが可能である。準配列は、他の親配列から組織的に選択することができる。 例えば、交叉点のN側に対する一方の準配列は、単一残基を持つことができ、他方の準配列は2つの残基を持つことができ、さらに第3の準配列は3つの残基を持つことができる、などである。置換型の準配列からなる同一の組織的(システマティック)なセットは、交叉点のC側で得ることができる。
【0048】
一具体例では、任意の交叉点に関連したキメラは、第2の親配列内の交叉点のC側から得た1ないし80残基の準配列と、該交叉点のN側から得た1ないし80残基の準配列とを含む。これらの準配列は、第1の親配列の対応する位置で、該配列内で置換される。それゆえに、160の異なるキメラが生成され、交叉点が第1の親ポリペプチドのCおよびN末端から取り除いた少なくとも80残基であると仮定して、特定の交叉点の重要性を計算する。これらのキメラの各々は、それ自身のΔEcを持つ。これらの個々のキメラΔEcの平均ΔEcは、好ましくない置換に対して交叉点の全体的な抵抗を反映するのに用いられる。
【0049】
このアプローチによって示唆されるように、本発明のいくつかの実施形態は、単一の交叉としてよりも、複数の交叉の1つとして十分に作用するという可能性にもとづいて、交叉位置が選択されるキメラ・ライブラリを設計することを目的とする。このアプローチでは、所定の公差位置に関連した複数の挿入断片について平均接触エネルギーの計算がおこなわれる。
【0050】
(基準配列)
一実施形態では、本発明の方法は、基準生体分子または生体分子構造の基準配列を提供することが含まれる。別の実施形態では、上記方法は、基準配列と基準生体分子の三次構造を提供することを含み、該基準配列は、モノマー単位(例えば、アミノ酸またはヌクレオチド)等の複数の要素を含む。これらのアプローチの両方とも、比較目的のための塩基配列を提供することが含まれる。任意に、親配列の1つは、基準配列として用いることができる。基準配列は、複数の要素(例えば、ヌクレオチドまたはアミノ酸)から構成され、所定の(または特定)の三次元構造を有する。基準配列を、当業者に周知のいくつかの方法で提供することができる。例えば、基準タンパク質のアミノ酸配列または該基準タンパク質をコードする核酸配列のいずれかを提供することができる。タンパク質をコードする核酸配列は、限定されるものではないが、cDNA、mRNA、ゲノムDNA等のいくつかの形態のいずれかで提供され得る。
【0051】
本発明の一実施形態では、基準アミノ酸配列は、基準タンパク質の配列決定によって提供される。アミノ酸配列決定法は、当業者に周知である(例えば、2,4−ジニトロフルオロベンゼン(サンガー試薬)、塩化ダンシル、フェニルイソチオシアネート(エドマン分解法)、種々のプロテアーゼを用いた方法)。
【0052】
より一般的には、基準配列は基準タンパク質(または触媒オリゴヌクレオチド)をコードする核酸の配列決定によって提供される。核酸塩基配列決定法もまた当業者に周知であり、限定されるものではないが、BergerおよびKimmel,Guide to Molecular Cloning Techniques,Methods in Enzymology volume 152 Academic Press,Inc.,SanDiego,CA(「Berger」);Sambrookら,Molecular Cloning − A Laboratory Manual(第3版),Vol.1−3,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York,2000(「Sambrook」)、およびCurrent Protocols in Molecular Biology,F.M.Ausubelら,編,Current Protocols,a joint venture between Greene Publishing Associates,Inc.and John Wiley & Sons,Inc.,(1999年を通して補遺)(「Ausubel」)に記載されている技術が挙げられる。
【0053】
本発明のさらに別の実施形態では、基準配列は核酸配列を提供し、提供された核酸配列をアミノ酸配列に翻訳するか、もしくは該配列を転写してオリゴヌクレオチド配列を生成することによって、供給される。任意に、基準配列は、核酸またはタンパク質データベースを問い合わせることによって、提供される。公共のデータベースおよび民間のデータベースが利用可能であり、該データベースとして、National Center for Biotechnology Information(www.ncbi.nhn.nih,gov)のGenBankTM、NCBI EST配列データベース、EMBL Nucleotide Sequence Database,Incyte(Palo Alto,CA)のLifeSeq(登録商標)データベース、および Celera(Rockville,MD)の「Discovery System」TMデータベース)、Swiss Institute of Bioinformatics(http:/us.expasy.org))のExPASy(Expert Protein Analysis System)プロテオミックス・サーバによって管理されたPROSITEデータベース、他のインターネット・リスト等が挙げられる。
【0054】
組換え体ポテンシャルに関して、本発明の方法を用いて多くのタンパク質ファミリーを評価することができる。代表的なタンパク質およびタンパク質ファミリーとして、限定されるものではないが、エノラーゼ・スーパファミリー、N−アセチルノイラミン酸リアーゼ・スーパファミリー、クロトナーゼ・スーパファミリー、および近隣酸素キレート折り畳み(vicinal oxygen chelate fold)のメンバー(例えば、Babbitt,PCおよびGerlt,JA.(1997)「Understanding Enzyme Superfamilies.」J.Biol.Chem.272:30591−30594)が挙げられる。さらなる標的タンパク質ファミリーは、本明細書に引用(および参考として援用)された参照文献で議論されている。特に関心のある1つのスーパーファミリーは、芳香族化合物新陳代謝に関与するムコネート・ラクトン化酵素(ムコネート・シクロイソメラーゼ)である。スーパーファミリーのメンバーは、概して一般的構造折り畳みを共有し、かつ類似の機械学的戦略を利用する。したがって、親配列同一性が低くても、さらなる所望の特性を有しながら必要な構造的折り畳みおよび基本的機械学的要素をも保持するキメラを生成することができる。
【0055】
また、機能的に活性な核酸分子、または目的とする他の核酸配列を組換えポテンシャルについて評価することができる。例えば、触媒RNA分子に対して、組換えまたは他の多様性生成手順を施すことができ、それによって該触媒RNA分子の基質特異性、触媒反応速度等を修飾し、そのようなものとして本発明で使用するための追加の標的生体分子を提供する。別の例として、mRNA、rRNA、またはtRNA分子は、本発明の方法を用いて調べることができ、また異なる活性または感受性を持つオリゴヌクレオチドの生成に用いることができる。さらなる機能的活性核酸は、限定されるものではないが、例えばタンパク質触媒反応で生物学的基質またはコファクターとして作用するRNAおよび/またはDNA分子が挙げられる。
【0056】
本発明の方法は、任意の数の追加標的生体分子または生体高分子のなかでの交叉点の評価に用いることができる。典型的な追加生体高分子として、限定されるものではないが、炭水化物、ポリケチド、テルペノイド、非リボソーム性ペプチド、脂質、または溶液等で安定な三次元構造を形成する任意の他の生体高分子が挙げられる。
【0057】
(接触マップ)
本発明の方法は、基準生体分子の三次構造を記述する1つ以上のパラメータを予測パラメータまたは1つ以上のキメラ組換え産物の構造と比較することを含む。これらのパラメータは、マッビングされた生体分子の構成要素間の相互作用が描かれた「接触マップ」のかたちで提供され(一般に二次元グラフまたはデータ・マトリックスの形で)、それによって、簡略化され、かつ簡約表示された生体分子の三次元構造が提供される。近位生体分子構成要素間の対相互作用(pairwise interaction)が概して調べられる一方で、3つ以上の近位要素の相互作用もまた本発明の接触マップで用いることができる。
【0058】
本発明の方法の特定の実施形態では、基準配列の要素について接触マップが生成される。一般的に、基準分子の一次配列と任意の利用可能な三次元構造データとを用いて接触マップが生成される。接触マップは、例えば基準タンパク質の三次元構造で互いに近位または隣接したアミノ酸残基の組またはセットを描写する(例えば接触アミノ酸)。任意に、接触マップは、目的とする近位要素間の距離を反映するデータを含む。本発明の一実施形態では、接触マップは接触アミノ酸間の距離によって加重されたスコアを含む。別の実施形態では、接触マップは基準生体分子内の要素(例えば、アミノ酸またはヌクレオチド)によって加重されたスコアを含む。
【0059】
接触マップは、種々のソースからのデータに基づいて、当業者に公知の方法で作製され得る。例えば、X線結晶学的データは、一般にタンパク質構造内でのアミノ酸スペーシングを決定すること、および互いの臨界距離内でアミノ酸配列(例えば、近位残基)を同定することに用いられる。これらの方法もまた、相互作用の1つ以上の物理的特徴も同定することができ、限定されるものではないが、アミノ酸相互作用のタイプ(疎水性、水素結合性、および/またはイオン性を含む)が挙げられ、その1つ以上が接触エネルギーの加重で別々に加重される。結晶学的なデータを、任意に各々の原子位置に対して「温度因子」を生成するのに用いられることもできる。データは、接触残基がどのくらいよく定義されたかの指示を与え、それによって該接触が加重され得る。
【0060】
あるいは、構成要素間のスペーシングは、基準生体分子のNMRモデルから決定することができる。2D−COSYとNOESYのようなNMR実験は、両方ともそれらの間の距離の推定を提供することができるのと同様に、三次元構造でお互いに最も近い要素を同定することができる。そのため、結果として生ずる接触は、NMRモデルの不確実な度合いで加重することができる。
【0061】
さらに、基準タンパク質のタンパク質折り畳み分析または相同性モデリングに基づいたアミノ酸構成および距離を用いて、基準タンパク質に対して、接触マップを生成することができる。タンパク質折り畳み分析、タンパク質構造内の残基間距離の計算、および/または他の分子モデリング計算のためのソフトウェアは、公的(例えば、the NIH Center for Molecular Modeling,http://cmm.info.nih.gov/modelingを参照のこと)または商業的(例えばHypercube Inc.,Gainesville FL;MDL Information Systems,San Leandro,CA;Molecular Applications Group,Palo Alto,CA;Accelrys,Inc.(以前はOxford Molecular and,Molecular Simulations Inc.,San Diego,Princeton,NJおよびLondon,UKに事務所;Tripos,Inc.,St.Louis,MO等)の両方で入手可能である。接触マップ情報の生成のための1つの特に役立つプログラムは、Chemical Computing Group(Montreal,Canada)のMOEである。相同性モデリングについての追加プログラムとして、限定されるものではないが、SWISS−MODEL(Glaxo Wellcome Experimental Research in Geneva,Switzerlandから入手可能)およびWHAT IFプログラム(EMBL)が挙げられる。比較(相同性)モデリングによって得られたタンパク質構造のデータベースは、多くのオンライン・ソースから入手可能であり、また限定されるものではないが、データベースModBaseおよび3D Crunchが挙げられる。同様に、核酸分子を生成および分析するためのプログラムを核酸配列に対して利用する;例えば、「tRNAスキャンSE」tRNA分析ソフトウェアは、St.Louis所在のWashington Universityより入手可能である((http://www.genetics.wustl.edu/eddy/tRNAscan−SE)を参照のこと。当業者に公知のこれらのプログラムおよび他のプログラムは、任意に基準分子の三次構造で、お互いに臨界距離の範囲内で2つ以上の要素を識別するのに用いることができる。
【0062】
上記技術の組み合わせはまた、基準タンパク質接触マップの作製にも用いることができる。例えば、入手不可能あるいは1つの分析技術からは統計的に関連していない距離測定を、別の技術を用いて任意に生成または確認することができる。タンパク質三次元構造の評価と接触マップの作製とで一般に用いられる1つのパラメータは、接触アミノ酸間の許容分離度または「臨界距離」の選択である。臨界距離は、アミノ酸−アミノ酸相互作用の性質により変化可能であり、約2.5ないし約7オングストロームの範囲である。例えば、イオン結合または疎水性相互作用を持つ要素(例えば、側鎖)は、概して約4.5オングストローム離れており、一方、ジスルフィド結合を介して接触している要素は、約2.5オングストローム離れている。
【0063】
本発明の方法では、接触アミノ酸側鎖間の臨界距離は、約2オングストロームから約6.5(または約7)オングストロームの範囲内にある。任意に、臨界距離は、約2.5オングストロームから約4.5オングストロームの範囲内にあり、概ね約4.5オングストローム未満である。したがって、本発明の方法の目的に関して、約5オングストローム未満の離間距離にある側鎖要素の任意の位置決めを、接触対(例えば、近位要素)とみなす。あるいは、接触距離は、これらの例では最大で6.5オングストロームの距離を用いて、アミノ酸のCαまたはCβから計算することができる。
【0064】
(配列およびアライメント・プロトコール)
本発明の方法は、2つ以上の親配列(例えば、第1の配列、第2の配列、さらに必要に怖じて第3または追加の配列)内での潜在的交叉位置を同定および/または比較するために使用することができる。任意に、基準生体分子の配列は、第1の配列または第2の配列のいずれかとして用いることができる。基準分子について既に説明したように、親配列は、いくつかのメカニズムのいずれかによって提供することができる。該メカニズムとして、限定されるものではないが、一方または両方の配列の配列決定、転写または翻訳のための核酸配列の提供、あるいは核酸またはタンパク質データベースへの問い合わせが挙げられる。また、目的とする配列が物理的な意味で提供可能である(例えば、単離または合成分子)。好ましくはそれらをコンピュータ内(in silico)で提供する(例えば、代表的配列ストリング、例えば、SelifonovらによるPCT公報WO 01/75767(PCT/USO1/10231「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」を参照せよ)。
【0065】
アミノ酸配列に関係する本発明の実施形態では、親配列は一般的に、類似の三次元構造(例えば、タンパク質スーパファミリー)を持つ共通のタンパク質ファミリーに由来する。しかし、これらのタンパク質をコードする核酸配列は、高い配列相同性を共有または非共有すると思われる。本発明の特定の実施形態では、方法は「低配列相同性」配列(例えば、配列共有が70%未満、60%未満、あるいはさらに50%未満の配列相同性)間での交叉位置を評価するために用いられる。
【0066】
種々のストリジェンシーおよび長さの配列類似性/同一性は、当業者に公知の多数の方法またはアルゴリズムを用いて検出および認識することができる。例えば、多くの類似性または相同性決定方法が生体高分子配列の比較分析のために、文章処理のスペル・チェックのために、さらに種々のデータベースからのデータ検索のために、設計されている。天然ポリペプチドでの4つの主要な核酸塩基間での二重らせん対相補的相互作用を理解することで、相補的な相同ポリヌクレオチド文字列のアニーリングを刺激するモデルもまた、本明細書の配列に対応する文字列上で概ね実行される配列アラインメントまたは他の動作の基礎として用いることもできる(例えば、文書処理操作、配列または配列特性ストリング、出力表などを含む構成)。配列の同一性を計算するためのソフトウェア・パッケージの一例は、BLASTであり、本明細書の配列に対応する文字列を入力することによって本発明に適用可能である。
【0067】
本発明の特定の実施形態では、互いに比較、または基準生体分子と比較した場合、1つ以上の与えられた配列が低配列同一性を呈する(しばしば、当該技術分野で「低相同性配列」と呼ばれる)。低同一性配列は、天然由来のものであり、あるいは合成的、突然変異的、またはコンピュータ的に生成することができる。低同一性配列の一例は、例えばPattenらのPCT公報WO 00/18906「Shuffling of Codon Altered Genes」に記載されているように、「コドン変更(codon
altered)」配列である。
【0068】
第1の配列と第2の配列とを提供した後に、該配列を基準タンパク質の配列に整列させる。基準配列が第1の配列または第2の配列のいずれかとして基準配列が機能する実施形態では、2つの配列が互いに位置合わせして並ばされる。別の実施形態では、複数の親配列が与えられ、該配列は次に基準配列と、または互いがアライメントされる。相対的に短いアミノ酸配列(例えば、約30残基)のアライメントおよび比較は概して直接的である。より長い配列の比較は、2つの配列の最適アラインメントを達成するためのより高度な方法が求められる。
【0069】
配列の最適アラインメントの実行は、例えば、いくつかの利用可能なアルゴリズムによっておこなうことができる。該アルゴリズムとして、限定されるものではないが、SmithおよびWaterman(1981 Adv.Appl.Math.2:482)の「局所相同性」アルゴリズム、NeedlemanおよびWunsch(1970 J.Mol.Biol.48:443)の「相同性アラインメント」アルゴリズム、PearsonおよびLipman(1988 Proc.Natl.Acad.Sci.USA
85:2444)の「類似性の検索」法、またはこれらのアルゴリズムのコンピュータ化による実現(例えば、GAP,BESTFIT,FASTA、およびTFASTA、これらはWisconsin Genetics Software Package Release 7.0,Genetics Computer Group,575 Science Dr.,Madison,WIから入手可能;ならびにBLAST、例えばAltschulら.,(1977)Nuc.Acids Res.25:3389−3402およびAltschulら.,(1990)J.Mol.Biol.215:403−410)が挙げられる。あるいは、配列を検査により整列させることができる。一般に、種々の方法によって生ずる最良のアラインメント(すなわち、比較ウィンドウ上の配列同一性の割合が最大となる相対的な位置決め)が選択される。しかし、本発明の具体的実施形態では、最良のアラインメントとして、選択された構造上の特徴を重ね合わせるものであってもよく、最も高い配列同一性を必要としない。
【0070】
「配列同一性」という用語は、比較ウィンドウ上で2つのアミノ酸配列が同一(すなわち、アミノ酸対アミノ酸をベースとする)であることを意味している。「配列類似性」とは、同一の生物物理学的性質を共有する類似のアミノ酸に言及している。「配列同一性の割合」または「配列類似性の割合」は、比較ウィンドウ上に最適化して整列させた2つの配列を比較し、一致した位置の数が生ずるために同一の残基(または類似の残基)が両方のポリペプチド配列に生じる位置の数を決定し、一致した位置の数を比較ウィンドウの全位置数(すなわち、ウィンドウ・サイズ)で割り、さらに得られた結果を100倍して配列同一性の百分率を生ずる(または配列類似性の百分率)。ポリペプチド配列に関して、配列同一性および配列相似性という用語は、タンパク質配列で説明したように、比較可能な意味を有し、「配列同一性の百分率」という用語は、2つのポリペプチド配列が比較ウィンドウで同一である(ヌクレオチド対ヌクレオチドをベースとする)。このようなことから、ポリヌクレオチド配列同一性の百分率(またはポリヌクレオチド配列類似性の百分率、例えば、サイレント置換または他の置換については、分析アルゴリズムに基づく)もまた計算することができる。本明細書に記載される配列アルゴリズムの1つ(または当業者が利用可能な他のアルゴリズム)を用いて、または目視試験によって、最大一致を決定することができる。「低配列同一性」を持つ配列(しばしば「低相同性」配列として言及される)は、直線状になった目的とする準配列に対して約70%未満、好ましくは約60%、またはより好ましくは約50%の配列同一性を有する配列である。
【0071】
ポリペプチドに適用されることから、実質的同一性または実質的類似性という用語は、ギャップ・ウェイト(本発明で詳細に説明)を用いて、または目視検査によって、例えばプログラムBLAST、GAP、またはBESTFITによって、2つのペプチド配列が最適に整列させられた場合、少なくとも約60ないし約80パーセント以上の配列同一性または配列類似性、好ましくは約90パーセントのアミノ酸残基配列同一性または配列類似性、より好ましくは少なくとも約95パーセントの配列同一性または配列類似性、またはそれ以上を共有する(例えば、96、97、98、98.5、99以上のパーセント・アミノ酸残基配列同一性または配列類似性が含まれる)。同様に、2つの核酸の前後関係で適用されることから、実質的同一性または実質的類似性という用語は、2つの核酸配列が、例えばプログラムBLAST、GAP、またはBESTFITによって、デフォルト・ギャップ・ウェイト(以下に詳細に説明する)または目視検査を用いることで、最適にアラインメントされる場合、少なくとも約60ないし約80パーセント以上の配列同一性または配列類似性、好ましくは少なくとも約90パーセントのアミノ酸残基配列同一性または配列類似性、より好ましくは少なくとも約95パーセントの配列同一性または配列類似性、またはそれ以上を共有する(例えば、約96、97、98、98.5、99、またはそれ以上のパーセント・ヌクレオチド配列遺伝同一性または配列類似性が含まれる)。
【0072】
パーセント配列同一性または配列類似性の決定に適しているアルゴリズムの一例は、FASTAアルゴリズムであり、このアルゴリズムはPearson,W.R.&Lipman,D.J.,(1988)Proc.Natl.Acad.Sci.USA 85:2444に記載されている。またW.R.Pearson,(1996)Methods
Enzymology 266:227−258を参照のこと。パーセント同一性またはパーセント類似性を計算するためのDNA配列のFASTAアラインメントで用いられる好ましいパラメータを最適化した。BL50マトリックス(Matrix)15:−5、K−タプル(tuple)=2、接合ペナルティ=40、最適化=28、ギャップ・ペナルティ −12、ギャップ長ペナルティ=−2,および幅=16。
【0073】
パーセント配列同一性およびパーセント配列類似性を決定するのに適したアルゴリズムの好ましい例は、BLASTおよびBLAST2.0アルゴリズムであり、これらのアルゴリズムは、それぞれAltschulら.,(1977)Nuc.Acids Res.25:3389−3402およびAltschulら.,(1990)J.Mol.Biol.215:403−410に記載されている。本明細書に記載したパラメータとともにBLASTおよびBLAST2.0を用い、本発明の核酸、ポリペプチド、およびタンパク質のパーセント配列類似性またはパーセント配列同一性を決定する。BLAST分析を実行するためのソフトは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)から公的に入手可能である、このアルゴリズムは、問い合わせ配列で長さWの短い語を識別することで、高スコアリング配列の対(HSP)を最初に同定することを含むもので、該問い合わせ配列は、データベース配列で同一の長さの語と整列させた場合、一致するか、またはある正の値となった閾値スコアTを満足するかのいずれかである。Tは、隣接語スコア閾値と呼ぶ(Altschulら.前出)。これらの初期の隣接語のヒットは、それを含むより長いHSPを見出すための検索を開始させる種子として働く。語のヒットは、累積アラインメント・スコアの増加にともなって、核配列に沿う両方向に延びる。累積スコアは、ヌクレオチド配列について、パラメータM(一組の一致した残基に対するリワード・スコア:常に>0)、およびN(ミスマッチング残基のためのペナルティ・スコア:常に<0)である。アミノ酸配列については、スコアリング・マトリックスを用いて累積スコアの計算をおこなう。各方向での語ヒットの延長の停止は、累積アラインメント・スコアがその最大達成値から量Xまで落ちる場合、1つ以上の負のスコアリング残基アラインメントの集積により累積スコアがゼロまたはそれ以下になる場合、またはいずれかの配列の終わりに達する場合におこる。BLASTアルゴリズム・パラメータW、T、およびXは、アラインメントの感度および速度を決定する。BLASTNプログラム(ヌクレオチド配列に対する)はデフォルトとしてワード長(W)が11、期待値(B)が10、M=5、N=−4、および両鎖の比較を用いる。アミノ酸配列については、BLASTPプログラムは、ワード長3、および期待値(E)10をデフォルトとして用い、さらにBLOSUM62スコアリング・マトリックス(Henikoff&Henikoff(1989)Proc.Natl.Acad.Sci.USA 89:10915参照)ではアラインメント(B)50、期待値(B)10、M=5、N=−4、および両鎖の比較を用いた。
【0074】
BLASTアルゴリズムは、2つの配列間の類似性または同一性の統計的分析も実施する(例えば、Karlin&Altschul,(1993)Proc.Natl.Acad.Sci.USA 90:5873−5787を参照)。BLASTアルゴリズムによって提供される類似性または同一性の1つの尺度は、最小合計確率(P(N))であり、これによって2つのヌクレオチドまたはアミノ酸配列間での一致(マッチング)が偶然生じる確率が提示される。例えば、被試験核酸と基準核酸との比較の際の最小合計確率が約0.2未満、より好ましくは0.01未満、最も好ましくは約0.001未満である場合、核酸は基準配列に類似していると考えられる。
【0075】
有用なアルゴリズムの別の例は、PILEUPである。PILEUPは、プログレッシブで対形成するアラインメントを用いて、関連配列からなる群から複数の配列アラインメントを作り出し、相互関係およびパーセント配列同一性またはパーセント配列類似性を示す。ツリーまたはデンドグラムもプロットすることで、アラインメントの生成に用いられるクラスター相互関係が示される。PILEUPは、Feng&Doolittle,(1987)J.Mol.Evol.35:351−360のプログレッシブ・アラインメント方法の単純化をもちいる。用いた方法は、Higgins&Sharp,(1989)CABIOS 5:151−153に記載された方法に類似している。プログラムは、多くとも300配列を整列させることができ、各々が最大長が5,000ヌクレオチドまたはアミノ酸である。複数のアラインメント手順が2つの最も類似した配列からなる2つ一組のアラインメントにより開始され、整列させられた2つの配列からなるクラスターを生成する。それにより、このクラスタを、次に最も関連している配列または整列させられた配列からなるクラスターに対して整列させる。2つの個々の配列からなる2つ一組のアラインメントの単純拡張によって、2つの配列クラスターを整列させる。最終のアラインメントは、一連のプログレッシブで、かつ2つ一組のアラインメントによって達成される。プログラムの実行は、特定の配列と、配列比較の領域に対する該配列のアミノ酸またはヌクレオチドの座標とを設計し、プログラム・パラメータを設計することによっておこなう。PILEUPを用いることで、参照配列を他の被試験配列と比較し、パーセント配列相同性(またはパーセント配列類似性)相互関係を決定する。この際、以下のパラメータが用いられる。すなわち、デフォルト・ギャップ加重(3.00)、デフォルト・ギャップ長加重(0.10)、加重末端ギャップである。PILEUPは、GCG配列分析ソフトウェア・パッケージ、例えばバージョン7,0(Devereauxら.,(1984)Nuc.Acids Res.12:387−395)から得ることができる。
【0076】
複数のDNAおよびアミノ酸配列アラインメントに好適なアルゴリズムの別の好ましい例は、CLUSTALWプログラム(Thompson,J.D,ら.,(1994)Nuc.Acids Res.22:4673−4680)である。CLUSTALWは、複数の配列からなるグループ間に対して複数の2つ一組の比較を実施し、それらを配列の同一性にもとづいて複数アラインメントに集める。ギャップ・オープン(Gap open)ペナルティおよびギャップ拡張(Gap extension)ペナルティは、それぞれ10および0.05であった。アミノ酸のアラインメントに関しては、BLOSUMアルゴリズムをタンパク質加重マトリックス(protein weight matrix)として使用することができる((HenikoffおよびHenikoff,(1992)Proc.Natl.Acad.Sci.USA 89:10915−10919)。
【0077】
当業者に理解されるように、検索およびアラインメント・アルゴリズムについての上記議論もまたポリヌクレオチド配列の同定および評価、ヌクレオチド配列を含む問い合わせ配列の置換により、必要に応じて、核酸データベースの選択に適用される。
【0078】
(交叉点)
親配列(例えば、第1の配列、第2の配列、および任意に加えた配列)を提供した後、親配列の一部分を置換、スワッピング、または交換する。各交換は、所定の交換の要素(アミノ酸またはヌクレオチドの準配列)の選択された領域を包含する2つの親配列上の第1の交叉点と第2の交叉点とのあいだで行われる。任意に、複数の準配列を所定の親配列内の複数の交叉点でスワッピングすることができ、それによって2つ以上の挿入準配列(1つ以上の親配列由来)を持つキメラ生体分子が生成される。核酸に関して、交叉部位は交換されたオリゴヌクレオチド領域の5’および3’末端(例えば、組換えが生ずる位置)を定義する。タンパク質配列について、交叉部位は、交換されたアミノ酸残基の始点(N−末端)および終点(C−末端)によって定まる。いくつかの実施形態では、第1の交叉部位は核酸の5’末端、またはアミノ酸配列のN−末端に一致する。他の実施形態では、第2の交叉部位は核酸の3’末端、またはアミノ酸配列のC−末端に一致する。
【0079】
交換すべき選択された領域の長さは、標的システムにより変動する。しかし、本発明で用いた交叉部位は、2つの親配列間での交換のための要素が同じ数である必要はない。例えば、第1の配列の交叉部位が30の要素からなる領域を定める場合、第2の配列にある対応交叉部位によって定まる領域は、任意に30未満または30を上回る数の要素を含むことができる。
【0080】
本発明の方法では、1つ以上の「交叉産物」(すなわち、キメラ配列)を、所定の対またはセットの親配列について調べる。一実施形態では、単一の交叉産物を検討する。しかし、本発明の方法は2つ以上の交換領域(例えば、複数の交叉部位)を持つキメラ組換え体の生成に使用することができる。いくつかの実施形態では、単一交換用の潜在的交叉部位のすべてが分析のために生成される。別の実施形態では、考えられる全てのキメラ産物のサブセットが調べられる。
【0081】
交叉部位の選択は、経験的に行われ(例えば、配列中で5番目の要素ごとに開始)、または選択が付加的な基準に基づく。進化の過程のアミノ酸の共変化によってタンパク質が所定の折り畳み、三次元構造、または機能を保持し、その一方で他の形質(例えば特異性)を変えることを考えると、この情報は可能性のある交叉一の選択に有用である。あるいは、交換のための領域が選択、例えば所望の活性(例えば、タンパク質または触媒核酸の活性部位)または特異的な構造特性(例えば、αヘリックスまたはβシートのストランド)を標的化することである。接触マップおよび/または基準タンパク質の三次元構造による親配列のアラインメントの視覚的分析もまた、目的の構造領域に対する分析的努力にも焦点を合わせることができる。
【0082】
付加的な基準パラメータの分析は、交叉点の選択および組換え生体分子の設計をアシストする。評価のための交叉点配置の選択は、例えば、以下の文献に見出すことができる。PCT公報WO 00/42559(PCT/US00/01138「METHODS OF POPULATING DATA STRUCTURES FOR USE IN
EVOLUTIONARY SIMULATIONS」(SelifonovおよびStemmerによる),WO 00/42560(PCT/US00/01202「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Selifonovらによる)),WO 01/75767(PCT/US01/10231「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Selifonovらによる)および USSN 09/618,579(2000年7月18日出願(「METHODS FOR MAKING CHARACTER STRINGS,POLYNUCLEOTIDES AND POLYPEPTIDES HAVING DESIRED CHARACTERISTICS」(Gustafssonらによる)))。
【0083】
「スワッピング」の工程、キメラ産物配列を生成する親配列間の1つ以上の準配列は、本発明の方法によりコンピュータ内(in sillico)で行われる。組換えのコンピューター内方法を実行するができ、この際、遺伝的アルゴリズムをコンピューターで用いて、相同な核酸(または非相同な核酸でも)に対応する配列の列を組換える。任意で、例えばオリゴヌクレオチド合成/遺伝子再集合技術と協調させて、組換え配列に対応する核酸の合成によって、結果的に生じる組換え配列の列を核酸に変換する。この手法により、ランダム改変体、部分的ランダム改変体、または設計された改変体が生成され得る。対応する核酸(および/またはタンパク質)の生成と組み合わせたコンピューター・システムでの遺伝的アルゴリズム、遺伝的オペレーター等の使用を含むコンピューター内組換えと、設計された核酸および/またはタンパク質(例えば、交叉部位選択に基づく)と、設計された組換え法、擬似ランダム組換え法、またはランダム組換え法とに関する多くの詳細は、WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides
and Polypeptides Having Desired Characteristics」ならびにWO 00/42559(SelifonovおよびStemmer)、「Methods of Populating Data Structures for Use in Evolutionary Simulations」に記載されている。コンピューター内組換え法に関する広範な詳細は、これらの適用中で見出される。この方法論は、対応する核酸またはタンパク質のコンピューター内および/または生成の組換えに提供される際、本発明に概ね適用できる。
【0084】
(キメラ配列のスコアリング)
本発明の方法は、基準生体分子と比較したキメラ配列の1つ以上のパラメータに基づいて、親配列内の潜在的交叉位置を評価するためのメカニズムを提供する。本発明の方法は、適応度交叉位置を評価するための接触マップおよび接触エネルギー算出を使用することが可能である。本発明の方法では、接触マップを用いてキメラ分子の要素を位置合わせし、基準配列およびキメラ配列間の比較をおこなう。本方法の一実施形態では、キメラ配列を接触マップと比較して、接触している要素のセットを選択する。その後、キメラ分子中の要素の選択されたセットを基準分子中の対応する要素と比較してスコアリングする。このスコアは、キメラ分子が基準生体分子と類似のコンフォメーションを達成する可能性、および推論により類似のコンフォメーション安定性または所望の活性を実現する可能性の尺度を提供する。
【0085】
比較プロセスおよびスコアリング・プロセスの一局面は、キメラ分子中の2つ以上の近位要素間の接触エネルギーの算出である。当業者に公知の多数の手順によって、要素の選択されたペアまたはセットの接触エネルギーを算出することができる。例えばミヤザワ−ジャニガン(Miyazawa−Jernigan)エネルギー行列を用いて、アミノ酸配列に対して、接触エネルギーを予測することができる(例えばMiyazawaおよびJernigan(1999)「Self−consistent estimation of inter−residue protein contact energies based on an equilibrium mixture approximation of residues」Proteins 34:49−68);MiyazawaおよびJernigan(1999)「An empirical energy potential with a reference state for protein fold and sequence recognition」Proteins 36:357−69;Zhang(1998)「Extracting contact energies from protein structures:a study using a simplified model」Proteins 31:299−308;ならびにMiyazawa,S.&Jernigan,R.L.(1996)「Residue−residue potentials
with a favorable contact pair term and an unfavorable high packing density term,for simulation and threading」、J.Mol.Biol.256:623−464を参照せよ)。
【0086】
この算出では、二次元行列は、どの程度の頻度でそれらの残基が公知の構造のデータベースに接触して見出されたかに基づいて、アミノ酸の各ペアリング間の相互作用強度を提示する。接触していると考えられる近位アミノ酸残基に関して基準分子を試験する。相互作用する原子2つ以上の残基は、それらの各側鎖が基準距離未満で離れている場合、接触していると考えられる。この基準距離は、相互作用の種類(例えば、疎水性、イオン性等)によって変動するが、通常、約4.5から約5.0オングストローム未満である。その後、接触ペアの数を決定し(例えば上述の位置合わせ技術の1つを介して、生成された基準分子の接触マップと比較することによって)、タンパク質構造中に存在する接触の数(および関連するエネルギー値)を合計することによって、キメラ配列に対して接触エネルギーを決定する。ミヤザワ−ジャニガン・エネルギー行列を用いて生成された接触エネルギーは通常、疎水性作用によって支配されている。このように、正の電気を帯びたアミノ酸側鎖および負の電気を帯びたアミノ酸側鎖は、ジスルフィド結合(cys−cys)相互作用のように、過小評価されている。
【0087】
あるいは、接触残基の2次構造を用いることでタンパク質接触エネルギーについて計算することができる。(Zhang,C,およびKim,S−H.(2000)「Environment−dependent residue contact energies for proteins.」Proc.Natl.Acad.Sci.USA 97:2550−2555)などに、記載されている)。例えば、Zhang−Kimのデータベース依存マトリックスを用いたとき、αヘリックスからのPheと、β−シートからのAlaとの相互作用は、ループからのPheと、αヘリックスからのAlaとは、異なった相互作用エネルギーがあるだろう。また、タンパク質接触エネルギーを計算するための追加代替方法も、この方法に用いることが可能である。
【0088】
核酸分子に関しては、比較を目的とした位置依存的(または、位置非依存的)アルゴリズムが数多く使用可能である。例えば、1対1比較(pair−wise)の「Smith−Waterman」または「Needleman−Wunsch」アライメント関数を用いて、接触エネルギー情報を生成することが可能である。(Smith,T.F.,およびWaterman,M.S.(1981)「Identification of
common molecular subsequences」J.Mol.Biol.147:195−197;Needleman,S.B.,およびWunsch,C.D.(1970)「A general method applicable to
the search for similarities in the amino acid sequence of two proteins」J.Mol.Biol.48:443−453)。これら2つのアルゴリズムが以下の点で異なる。Needleman−Wunschアルゴリズムを用いた場合、配列比較が全体的(global)に行われる。それとは対照的に、Smith−Watermanアルゴリズムを用いた場合は、局所的(local)であり、それによって、基準配列、またはデータベース配列に対する照会配列全体のアライメントを強制する。
【0089】
本発明の方法の一実施形態では、キメラ配列の各要素を調べて、その要素が配列における別の要素に接触しているかを決定することによって、スコアリングが行われる。選択された要素が別の要素に近位であるなら、その対の近位要素の接触エネルギーが計算される。いくつかの実施形態では、スコアリングは、近位の対の要素のすべてに関する接触エネルギーを合計することを含んでいる。場合によっては、スコアリング・ステップの「接触エネルギー」成分に、要素間の概算距離、もしくは測定距離、または配列中の要素の位置などの付加パラメータによって重み付けすることが可能である。
【0090】
接触エネルギー・パラメータに加えて、スコアリング・プロセスに場合によっては他の構造パラメータを含むことができる。例えば、アミノ酸残基の立体的大きさ(steric bulk)、アロステリック効果、疎水性度もしくは極率を定義するパラメータ、または、構造対称性、周期性、または構成要素系の分布パターンなどの総合的構造パラメータ、電荷、および/または静電気フィールドの分布、四元構造単位の方向性などを用いて、スコアの生成が可能である。例えば、アミノ酸組成および有意な特性の分析に関して、考慮すべき事柄には、溶剤からの移動時のΔGによって決定される疎水性度、カラム残留からの親水性、アミノ酸の電荷、極性、アミノ酸のpKa、大きさ、側鎖エントロピー、αへリックス/ベータシート傾向、水和ポテンシャル、コドン縮重、および同様のものが含まれる。
【0091】
その上、キメラ配列のスコアリングで追加統計的方法を使うことができる。これらには、ニューラルネットワーク計算、モンテカルロ分子動力学シミュレーション、主成分分析法(PCA)、潜在構造への部分的最少二乗投影(PLS)などの多変量データ解析、および、他の分子モデリング、または生命情報工学計算が含まれるが、これらに限定されない。
【0092】
例えば、理想的な交叉位置を正確に指摘するのにマルコフ連鎖などの統計的な行列を用いることができる。Skorobogatiy,およびTiana(Physical Review E(1998年9月)第58巻,3572−3577頁)によって記述されたような単純なオン格子模型を用いることで、アミノ酸置換によって天然立体配座のミスフォールディングを引き起こすタンパク質配列以内の部位を写像することができる。ニューラルネットワーク法は、一種のパターンを学んで、所与の変異によって生成する結果を予測するのにを用いることが可能である。(そのようなニューラル・ネットワークの例として、SchneiderおよびWrede(1998)「Artificial neural networks for computer−based molecular design」Prog.Biophys.Mol.Biol.70(3):175−222;Schneiderら(1998)「Peptide design by artificial neural networks and computer−based evolutionary search」Proc.Natl.Acad.Sci.USA 95(21):12179−12184;およびWredeら(1998)「Peptide design aided by neural networks:biological activity of artificial signal peptidase I cleavage sites」Biochemistry 37(11):3588−35893が挙げられる。さらなる例としては、WO 00/42559(SelifonovおよびStemmer),WO 00/42560(Selifonovら),WO 01/75767(Selifonovら)ならびにUSSN 09/618,579(Gustafssonら),すべて前出)に見出すことができる。
【0093】
核酸に関しては、必要に応じておこなう1つのアプローチは、Jonssonら(1993 Nucl.Acids Res.21:733−739)により記載されたような、所定の転写プロモータからなるセット内での強度の予測のための多変量データ分析のアプリケーションである。したがって、接触エネルギー情報に加え、統計的な検討を、交叉点の評価および核酸またはタンパク質の安定性の予測のための方法に適用することができる。
【0094】
(正規化)
本発明のいくつかの実施形態では、キメラ分子中の推定接触アミノ酸に対して生じたスコアを進化の前に正規化する。大部分の使用では、キメラ配列は、基準配列に対して、配列アイデンティティの範囲を有している。基準配列により近いそれらのキメラは、基準配列に対する配列アイデンティティでより遠いものより優れた接触スコアを有している。このことは、基準タンパク質と比較して、より大きい、例えば約50%同一、約60%同一、約70%同一、またはそれ以上であるものに対して、交叉部位を決定するために用いられるキメラを制限することによって、あるいは接触エネルギーを正規化することを介して説明され得る。基準配列対算出された接触スコアに対するキメラの配列アイデンティティの直線状回帰を介して、この正規化を実行することができる。その後、この回帰からの剰余を用いて、最適交叉位置を決定する。別の手法では、2つ以上の以下のものの複数の回帰を利用できる。すなわち、2つの交叉位置間の長さ、第2の交叉の第1の交叉位置の位置、およびキメラ産物配列および基準配列対算出された接触スコア間の配列アイデンティティである。また、回帰からの剰余を用いて、最適交叉位置を決定することができる。
【0095】
(キメラ産物の生成)
本発明の方法は、キメラ産物合成前に潜在的交叉部位の可能性を評価するために、事前選別技術として利用することができる。任意で、その後、有効であると評価された(またはこの計算的事前選別に「合格した(pass)」)それらのキメラ産物を実験室で合成および試験する。従って、本発明の方法は、任意で、1つ以上のキメラ生体分子配列を合成するステップをさらに含む。当業者には公知である任意の種々の技術を用いて、キメラ産物を合成することができる。例えば、一実施形態では、キメラ生体分子を新規に合成する(例えば合成化学技術を用いて)。別の実施形態では、本方法は、細胞ベースまたは細胞を含まない発現系でキメラ生体分子を発現させる任意のステップを含む。任意で、キメラ生体分子を合成するステップは、適当な親拡散を提供することと、1つ以上の組換えプロセスを実行することとを含む。合成ステップを実行するための方法論は、本明細書に組み込まれる参考文献中に詳述されている。
【0096】
本発明の方法は、任意で、1つ以上の子孫キメラ核酸配列を含むコンストラクトを提供することを含む。コンストラクトは、プラスミド、コスミド、ファージ、ウイルス、最近人工染色体(BAC)、酵母人工染色体(YAC)等のベクターを含み、その中に、順配向または逆配向で、工本発明のキメラ配列が生成または挿入されている。この実施形態の好適な局面では、コンストラクトは、配列に作用可能に結合する、例えばプロモーターを含む調節配列をさらに有する。多数の好適なベクターおよびプロモーターが当業者に公知であるとともに市販されている。
【0097】
ベクターの使用、プロモーターの使用、および多くの他の関連主題を含む本明細書で有用な分子生物学的技術を記載する一般的テキストとしては、Bergerら、前出;Sambrookら、(1989)前出、およびAusubelら、(1989;1999年まで補完)前出が挙げられる。本発明の核酸の作製のために、ポリメラーゼ連鎖反応(PCR)、リガーゼ連鎖反応(LCR)、Qβ−レプリカーゼ増幅、および他のRNAポリメラーゼ媒介技術(例えばNASBA)を含むインビトロ増幅方法を介して、当業者を指導するために十分な技術の例は、Berger、Sambrook、およびAusubelとともに、Mullisら、(1987)、米国特許第4,683,202号;PCR
Protocols A Guide to Methods and Applications(Innsら編)Academic Press Inc.San Diego,CA(1990)(「Innis」);Arnheim & Levinson(1990年10月1日)C&EN 36−47;The Journal Of NIH
Research(1991)3:81−94;Kwohら、(1989)Proc.Natl.Acad.Sci.USA 86:1173−1177;Guatelliら、(1990)Proc.Natl.Acad.Sci.USA 87:1874−1878;Lomeliら、(1989)J.Clin.Chem.35:1826−1831;Landegrenら、(1988)Science 241:1077−1080;Van Brunt(1990)Biotechnology 8:291−294;WuおよびWallace、(1989)Gene 4:560−569;Barringerら、(1990)Gene 89:117−122、ならびにSooknananおよびMalek(1995)Biotechnology 13:563−564で見出される。インビトロで増幅された核酸をクローニングする改良された方法 についてはWallaceら、米国特許第5,426,039号に記載されている。PCRによって大きい核酸を増幅する改良された方法は、Chengら、(1994)Nature 369:684−685および本明細書の参考文献に概説されており、この際40kbまでのPCRアンプリコンが生成される。当業者には、本質的に、任意のRNAを逆転写酵素およびポリメラーゼを用いた制限酵素消化、PCR伸展、およびシーケンシングに適した二重鎖DNAに転換させることが認識される。Ausubel、Sambrook、およびBerger(全て前出)を参照せよ。
【0098】
本発明の方法で使用される付加的な配列を生成するために、下記に記載するような多様性生成技術を用いることができる。さらに、これらの多様性生成技術を用いて、1つ以上のキメラ産物を修飾することができる。本発明の方法は、任意で、1つ以上の以下のステップをさらに含む。すなわち、種々の交叉位置を用いて生成されたキメラ分子のライブラリーから安定または活性キメラ産物を選択することと、1つ以上の産物キメラ生体分子で多様性を生成すること(それによって、多様性キメラ生体分子を提供すること)と、親分子由来の、1つ以上のキメラ生体分子由来の、親配列として1つ以上の多様性キメラ産物由来の、またはその組み合わせ由来の配列を用いて、本発明の方法を反復的に繰り返すこととである。
【0099】
多様性生成方法の1つのグループについては、組換えまたはDNAシャッフリングとして言及される。これらの方法では、インビトロまたはインビボで、ポリヌクレオチドを組換えて、ポリヌクレオチド改変体のライブラリーを作製する。組換えに基づく方法では、1つ以上の親ポリヌクレオチドの配列のいくつかまたは全てに配列中で集合的に対応するDNAフラグメント、PCRアンプリコン、および/または合成オリゴヌクレオチドを組換えて、親ポリヌクレオチドのポリヌクレオチド改変体のライブラリーを作製する。組換えプロセスは、DNAフラグメント、PCRアンプリコン、および/または合成オリゴヌクレオチドの互いへのハイブリダイゼーション(例えば、部分的にオーバーラップする二重鎖として)によって、または完全長テンプレート等のDNAの大きな断片へのハイブリダイゼーションによって媒介されることが可能である。使用される組換え形式によって、リガーゼおよび/またはポリメラーゼを用いて、完全長ポリヌクレオチドの構築を促進することが可能である。ポリメラーゼのみを使用する形式では、PCRサイクリングが用いられる。これらの方法は、一般的に、当業者に公知であるとともに、他でも広範に記載されている。例えばSoong,N.ら、(2000)Nat.Genet.25(4):436−439;Stemmerら、(1999)Tumor Targeting 4:1−4;Nessら、(1999)Nature Biotechnology 17:893−896;Changら、(1999)Nature Biotechnology 17:793−797;MinshullおよびStemmer(1999)Current Opinion in Chemical Biology 3:284−290;Christiansら、(1999)Nature Biotechnology 17:259−264;Crameriら、(1998)Nature 391:288−291;Crameriら、(1997)Nature Biotechnology 15:436−438;Zhangら、(1997)Proc.Natl.Acad.Sci.USA 94:4504−4509;Pattenら、(1997)Current Opinion in Biotechnology 8:724−733;Crameriら、(1996)Nature Medicine 2:100−103;Crameriら、(1996)Nature Biotechnology 14:315−319;Gatesら、(1996)Journal of Molecular Biology 255:373−386;Stemmer(1996)In:The Encyclopedia of Molecular Biology.VCH
Publishers,New York.pp.447−457;CrameriおよびStemmer(1995)BioTechniques 18:194−195;Stemmerら、(1995)Gene,164:49−53;Stemmer(1995)「The Evolution of Molecular Computation」Science 270:1510;Stemmer(1995)Bio/Technology 13:549−553;Stemmer(1994)Nature 370:389−391;ならびにStemmer(1994)Proc.Natl.Acad.Sci.USA 91:10747−10751;GiverおよびArnold(1998)Current Opinion in Chemical Biology 2:335−338;Zhaoら、(1998)Nature Biotechnology 16:258−261;Cocoら、(2001)Nature Biotechnology 19:354−359;米国特許第5,605,793号、第5,811,238号、第5,830,721号、第5,834,252号、第5,837,458号、WO 95/22625、 WO 96/33207、WO 97/20078、WO 97/35966、WO 99/41402、WO 99/41383、WO 99/41369、WO 99/41368、WO 99/23107、WO 99/21979、WO 98/31837、WO 98/27230、WO 98/27230、WO 00/00632、WO 00/09679、WO 98/42832、WO 99/29902、WO 98/41653、WO 98/41622およびWO 98/42727、WO 00/18906、WO 00/04190、WO 00/42561、WO 00/42559、WO 00/42560、WO 01/23401、WO 00/20573、WO 01/29211、WO 00/46344、ならびにWO 01/29212を参照せよ。
【0100】
上記で参照されている組換えプロセスで使用される親ポリヌクレオチドは、野生型ポリヌクレオチドまたは非天然ポリヌクレオチドである。好ましくは、少なくとも1つのポリヌクレオチドは、本明細書で記載されるように選択される交叉点をコードする。本発明の一実施形態では、選択後の2つ以上の親ポリヌクレオチドの組換えによって、選択された交叉点を有するキメラタンパク質を調製する。いくつかの実施形態では、親ポリヌクレオチド(少なくとも交叉点をコードしないもの)は、単一の遺伝子ファミリーのメンバーである。本明細書で使用するように、用語「遺伝子ファミリー」とは、必ずしも同程度の活性ではないが、同種類の活性を示すポリペプチドをコードする遺伝子のセットを指す。
【0101】
例えば、ライゲーション後に組換えられる核酸のDNアーゼ消化および/または核酸のPCRでの再集合を含む任意の種々の技術によって、ポリ核酸をインビトロで組換えることができる。例えば、セクシャル(sexual)PCRである突然変異を用いることができ、この際、異なるが関連するDNA配列を有するDNA分子間の配列類似性に基づいたDNA分子のインビトロでのランダム断片化(または擬似ランダム断片化もしくは非ランダム断片化でさえも)が組換え後におこなわれ、その後、ポリメラーゼ連鎖反応での伸長による交叉の固定が続く。このプロセスおよび多くのプロセスの変形については、例えば、Stemmer(1994)Proc.Natl.Acad.Sci.USA 91:10747−10751に記載されている。
【0102】
合成組換え法を用いることもでき、この際、対象とする標的に対応するオリゴヌクレオチドを化学的に合成し、PCRまたはライゲーション反応で再集合させる。これには、1つより多い親ポリヌクレオチドに対応するオリゴヌクレオチドが含まれているので、新規の組換えポリヌクレオチドが生成される。標準的なヌクレオチド付加方法によって、または例えばトリヌクレオチド合成手法によって、オリゴヌクレオチドを作製することができる。このような手法に関する詳細については、上記に記載される参考文献、例えばWO 00/42561(Crameriら)、「Olgonucleotide Mediated Nucleic Acid Recombination;」WO 01/23401(Welch ら)、「Use of Codon−Varied Oligonucleotide Synthesis for Synthetic Shuffling」;WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides and Polypeptides Having Desired Characteristics」;、およびWO 00/42559(SelifonovおよびStemmer)「Methods of Populating Data Structures for Use in Evolutionary Simulations」で見出される。
【0103】
例えば細胞中の核酸間で組換えを発生させることによって、ポリヌクレオチドをインビボで組換えることもできる。多くのこのようなインビボ組換え形式が上記の参考文献で先述されている。このような形式は、他の形式と同様に、任意で、対象とする核酸間での直接組換えを提供するか、または対象とする核酸を含むベクター、ウイルス、プラスミド等の間の組換えを提供する。このような手順に関する詳細に関しては、本明細書で引用される参考文献で見出される。
【0104】
同様に使用される天然の多様性を評価する多数の方法では、例えば、一本鎖テンプレートへの多様な核酸または核酸フラグメントのハイブリダイゼーション後に、重合および/またはライゲーションをして完全長配列を再生成し、任意で、その後にテンプレートの分解およびその結果生じる修飾核酸の回収が可能になる。これらの方法は、本発明の特定の実施形態に従って、物理的システムで用いることができ、またはコンピューター・システムで実行することができる。一本鎖テンプレート(好ましくは、交叉点をコードする)を用いる一方法では、ゲノム・ライブラリーに由来するフラグメント集団を、対となる配列に対応する部分的ssDNAまたはRNA、あるいはしばしばほぼ完全長ssDNAまたはRNAでアニーリングする。その後、この集団から得た複合キメラ遺伝子の集合は、非ハイブリダイゼーション・フラグメント末端のヌクレアーゼに基づく除去と、このようなフラグメント間の溝を満たすための重合と、その後の一本鎖ライゲーションとによって媒介される。消化(たとえば、RNAまたはウラシルを含有の)と、変性条件下での磁気分離(このような分離を導く方法で標識されている場合)と、他の利用可能な分離/精製方法とによって、親ポリヌクレオチド鎖を除去することができる。また、任意で、親鎖をキメラ鎖と共精製し、その後のスクリーニングおよび加工ステップ中に除去する。この手法に関する更なる詳細は、例えばAffholter、によるWO 01/64864「Single−Stranded Nucleic Acid Template−Mediated Recombination and Nucleic Acid Fragment Isolation」で見出される。
【0105】
情報処理システムでのデジタル方式で組換え方法も実行することができる。例えば、コンピューターでアルゴリズムを用いて、相同生体分子(または非相同な生体分子でさえ)に対応する配列の列を組換えることができる。本発明の特定の実施形態に従って、コンピューター・システムで処理した後、例えばオリゴヌクレオチド合成/遺伝子再集合技術と協調させて、組換え配列に対応する核酸の合成によって、結果的に生じる配列の列を核酸に変換することができる。この手法により、ランダム改変体、部分的ランダム改変体、または設計された改変体が生成され得る。コンピューター・システムでの種々のアルゴリズム、オペレーター等の使用を含むコンピューターにより可能になった組換えと、設計された核酸および/またはタンパク質の組み合わせ(例えば交叉部位選択に基づく)と、設計された組換え法、擬似ランダム組換え法、またはランダム組換え法との種々の実施形態に関する多くの詳細については、WO 00/42560(Selifonovら)、「Methods for Making Character Strings,Polynucleotides and Polypeptides Having Desired Characteristics」、WO 01/75767(Gustafasonら)、「In Silico Cross−Over Site Selection」、ならびにW O 00/42559(SelifonovおよびStemmer)、「Methods of Populating Data Structures for Use in Evolutionary Simulations」に記載されている。
【0106】
(定向進化)
スクリーニングと組み合わせた反復方法(本明細書中別の箇所でより詳細に記載される)で、1つ以上の多様性生成方法を実施して、組換え核酸の次のセットを生成することによって、定向進化(または「人工進化」)を実行することができる。従って、突然変異および/または組換えおよびスクリーニングの繰り返しサイクルによって、定向進化または人工進化を実行することができる。例えば、親ポリヌクレオチド(所望の交叉点を提供するために選択される)上で、突然変異および/または組換えを実行し、改変体ポリヌクレオチドのライブラリーを生成することができ、その後、これを発現させて、所望の活性に対してスクリーニングされる交叉点を有するタンパク質を生成する。所望の活性での改善を示すとして、1つ以上の改変体タンパク質がこれらのタンパク質から同定され得る。同定されたタンパク質を逆翻訳して、同定されたタンパク質改変体をコードする1つ以上のポリヌクレオチド配列を確認し、今度は、この配列を、多様性生成およびスクリーニングの次のラウンドで突然変異または組換えをすることができる。
【0107】
多様性生成の組換えに基づく形式を用いた定向進化については、本明細書で引用される参考文献に広範に記載されている。多様性生成の基盤として突然変異を用いる定向進化についても、当技術で周知である。例えば、再帰的集合突然変異のプロセスでは、タンパク質突然変異に対するアルゴリズムを用いて、表現型的に関連する突然変異体の多様性集団が生産され、そのメンバーは、アミノ酸配列が異なる。この方法では、フィードバック・メカニズムを用いて、組み合わせカセット突然変異の連続的ラウンドがモニターされる。このアプローチの例としては、Arkin & Youvan(1992)Proc.Natl.Acad.Sci.USA 89:7811−7815に記載されている。同様に、高比率の特有かつ機能的突然変異体を有するコンビナトリアル・ライブラリーを生成するために、指数関数集合突然変異を用いることができる。各変化位置での機能的タンパク質に至るアミノ酸の同定と平行して、対象の配列中の残基の小グループをランダム化する。このような手順の例は、Delegrave & Youvan(1993)Biotechnology Research 11:1548−1552で見出される。
【0108】
本発明の交叉同定方法は、多様性生成手順の利用に関わらず、定向進化プロセスを最適化するのに有用である。本発明を適用することにより派生する交叉情報を用いて、定向進化プロセスで作製されるライブラリーをより理知的に設計することができる。例えば、特定のアミノ酸残基位置に交叉点を挿入することが所望される場合、2つ以上の親に由来するこれらの所望のアミノ酸残基をコードするコドンを組み込んでいる合成オリゴヌクレチドを、本明細書で言及する組換え形式の1つで用いて、ポリヌクレオチド改変体ライブラリーを生成することができ、このライブラリーをその後発現させることができる。また、本明細書で記載される種々の突然変異方法の1つを用いて、所望の交叉点を組み込むことができる。従って、任意の現象で、結果として生じたタンパク質改変体ライブラリーは、有効な残基または潜在的に有効な残基であると考えられるものを組み込んでいるタンパク質改変体を含んでいるであろう。所望の活性を有するタンパク質改変体が同定されるまで、このプロセスを繰り返すことができる。
【0109】
(活性に対するスクリーニング/選択)
任意で、本発明の方法と組み合わせて生成されたポリヌクレオチドを活性スクリーニングのために細胞中にクローニングする(または、インビトロ転写反応で用いて、スクリーニングされる産物を作製する)。さらに、核酸を、インビトロで、濃縮、シーケンシング、発現、増殖させるか、または任意の他の通常の組換え方法で処理する。
【0110】
クローニング、突然変異、ライブラリー構築、スクリーニング・アッセイ、細胞培養等を含む本明細書で有用な分子生物学的技術を記載する一般的テキストとしては、BergerおよびKimmel、Guide to Molecular Cloning Techniques,Methods in Enzymology volume 152 Academic Press,Inc.,San Diego,CA(Berger);Sambrookら、Molecular Cloning − A Laboratory Manual(2nd Ed),Vol.1−3,Cold Spring Harbor Laboratory,Cold Spring Harbor,New York,1989(Sambrook)、ならびにCurrent Protocols in Molecular Biology,F.M.Ausubelら、eds.,Current Protocols,Greene Publishing Associates,Inc.およびJohn Wiley & Sons,Inc.の共同事業、New York(2000年まで補完される)(Ausubel))が挙げられる。核酸を用いて植物および動物細胞を含む細胞を形質導入する方法は、公に入手可能であり、同様に、このような核酸にコードされるタンパク質を発現させる方法も入手可能である。Berger、Ausubel、およびSambrookに加えて、動物細胞培養用の有用な一般的参考文献としては、Freshney(Culture of Animal Cells,a Manual of Basic Technique,third edition Wiley−Liss,New York(1994))、ならびに本明細書で引用される参考文献であるHumason(Animal Tissue Techniques,fourth edition W.H.Freeman and Company(1979))およびRicciardelliら、In
Vitro Cell Dev.Biol.25:1016−1024(1989)が挙げられる。植物細胞クローニング、培養、および再生成に関する参考文献としては、Payneら、(1992)Plant Cell and Tissue Culture in Liquid Systems John Wiley & Sons,Inc.New York,NY(Payne);ならびにGamborgおよびPhillips(eds)(1995)Plant Cell,Tissue and Organ Culture;Fundamental Methods Springer Lab Manual,Springer−Verlag(Berlin Heidelberg New York)(Gamborg)が挙げられる。種々の細胞培地については、AtlasおよびParks(eds)The Handbook of Microbiological Media(1993)CRC Press,Boca Raton,FL(Atlas)に記載されている。植物細胞培養の更なる情報については、Sigma−Aldrich,Inc(St Louis,MO)(Sigma−LSRCCC)から得られるLife Science Research Cell Culture Catalogue(1998)、および例えばSigma−Aldrich,Inc(St Louis,MO)(Sigma−PCCS)から同様に得られるPlant Culture Catalogueおよび付録(1997)等の入手可能な商業的文献で見出される。
【0111】
1つの好適な方法では、再集合させた配列がファミリーに基づく組換えオリゴヌクレオチドを組み込んでいるかについて検査する。これは、例えばSambrook、Berger、およびAusubel(上掲)に基本的に教示されているように、核酸をクローニングおよびシーケンシングすることによって、ならびに/または制限酵素消化によって実行することができる。さらに、配列をPCRで増幅し、直接シーケンシングすることができる。従って、例えば、Sambrook、Berger、Ausubel、およびInnis(上掲)に加えて、付加的なPCRシーケンシング方法も特に有用である。例えば、PCR中に、ボロン酸ヌクレアーゼ耐性ヌクレオチドを選択的にアンプリコン(増幅領域)に組み込み、アンプリコンをヌクレアーゼで消化し、所定の大きさにされたテンプレート・フラグメントを作製することによって、PCRで生成されたアンプリコンの直接シーケンシングが実行されていた(Porterら、(1997)Nucleic Acids Research 25(8):1611−1617)。この方法では、テンプレート上で4回のPCR反応を実行し、そのそれぞれの回で、PCR反応混合物中のヌクレオチド三リン酸の1つを2’デオキシヌクレオシド 5’(P−ボラノ)三リン酸で部分的に置換する。ボロン酸ヌクレオチドは、テンプレートのPCRフラグメントの入れ子集合中のPCRアンプリコンに沿って変動する位置で、確率論的にPCR産物中に組み込まれる。組み込まれたボロン酸ヌクレオチドに遮断されるエキソヌクレアーゼを用いて、PCRアンプリコンを切断する。その後、ポリアクリルアミド・ゲル電気泳動を用いて、切断されたアンプリコンをサイズごとに分離し、アンプリコンの配列を提供する。この方法の利点は、PCRアンプリコンの標準的サンガー形式のシーケンシングを実行するより生化学的操作の使用が少ないことである。
【0112】
合成遺伝子は、従来のクローニング手法および発現手法に適用するこができる。従って、遺伝子および遺伝子がコードするタンパク質の特性を、宿主細胞でそれらを発現させた後に容易に試験することができる。合成遺伝子を用いて、インビトロ(細胞を含まない)転写および翻訳によって、ポリペプチド産物を生成することもできる。従って、微生物細胞壁、ウイルス粒子、ウイルス表面、およびウイルス膜とともに、他のタンパク質およびポリペプチド・エピトープを含む種々の所定のリガンド、小分子、およびイオン、またはポリマー物質およびヘテロポリマー物質への結合能力に関してポリヌクレオチドおよびポリペプチドを試験することができる。
【0113】
例えば、ポリヌクレオチド直接による、またはコードされるポリペプチドによる化学反応の触媒作用に関連する表現型をコードするポリヌクレオチドを検出するために、多数の物理的方法を用いることができる。説明目的に限って、対象となる特定の所定の化学反応の特質によって、これらの方法は、当技術で周知の多数の技術を含むことが可能であり、それにより、基質および産物間の物理的相違または化学反応に関連する反応培地での変化(例えば、電磁放射、電磁吸着、電磁散逸、および電磁蛍光での変化、UVが可視であるか赤外(熱)であるか)が説明される。これらの方法を以下の任意の組み合わせから選択することもできる。すなわち、質量分光分析法と、核磁気共鳴と、同位体的標識物質、同位体的分配、および同位体分布または標識産物形成を説明する分光法と、反応産物のイオンまたは元素組成物での付随する変化(pH、無機イオン、および有機イオン等での変化を含む)を検出するための分光法および化学的方法とである。本明細書中の方法で使用されるのに適した物理的アッセイの他の方法は、レポーター特性を有する抗体を有するもの、またはレポーター遺伝子の発現および活性と組み合わせたインビボの親和性認識に基づくものを含む、反応産物に特異的なバイオセンサーの使用に基づくことができる。反応産物検出およびインビボの細胞生・死・増殖選択に関する酵素結合アッセイも目的に適うさいは用いることができる。物理的アッセイの特性に関わらず、それらを全て用いて、所望の活性、あるいは対象となる生体分子によりコードされるか、または提供される所望の活性の組み合わせを選択する。
【0114】
選択に使用される特有のアッセイは、その用途に依存するであろう。タンパク質、受容体、リガンド等用の多数のアッセイが既知である。形式としては、固定成分への結合、細胞または生体の生存能力、レポーター組成物の産生等が含まれる。
【0115】
ハイスループット・アッセイは、本発明で使用される交叉に基づくライブラリーをスクリーニングするために特に適している。ハイスループット・アッセイでは、数千までの異なる改変体を1日でスクリーニングすることが可能である。例えば、マイクロタイター・プレートのウエルのそれぞれを用いて、別々のアッセイを実行することができる。あるいは、濃度の効果またはインキュベーション時間の効果を観測する場合、5〜10個のウエルごとに、1つの改変体を試験することができる(例えば異なる濃度で)。従って、単一の標準的マイクロタイター・プレートによって、約100(例えば96)の反応をアッセイできる。1536ウエル・プレートを用いた場合、単一のプレートで、約100から約1500の異なる反応を容易にアッセイすることができる。複数の異なるプレートを1日でアッセイすることが可能である。本発明の統合されたシステムを用いることによって、約6,000〜20,000までの異なるアッセイ(すなわち、異なる核酸、コードされるタンパク質、濃度等を含む)に対するアッセイ・スクリーニングすることが可能である。より近年になって、例えばカリパー・テクノロジー(Caliper Technologies)(Mountain View,CA)によって、試薬操作に対するマイクロ流体手法が開発されており、この手法により、非常にハイスループットなマイクロ流体アッセイ法を提供され得る。
【0116】
ハイスループット・スクリーニング・システムが市販されている(例えば Zymark Corp.,Hopkinton,MA;Air Technical Industries,Mentor,OH;Beckman Instruments,Inc.Fullerton,CA;Precision Systems,Inc.,Natick,MA等を参照せよ)。 一般に、これらのシステムは、アッセイに適当な全ての試料および試薬のピペッティング、液体分配、インキュベーション時間計測、および検出器中のマイクロプレートの最終的な読み取りを含む全手順を自動化している。これらの設定可能なシステムにより、高度な適応性およびカスタマイゼーションとともに、ハイスループットおよび迅速な起動が提供される。
【0117】
このようなシステムの製造元は、様々なハイスループット・スクリーニング・アッセイに関する詳細なプロトコルを提供している。従って、例えば、ザイマーク社(Zymark Corp.)により、遺伝子転写の変調、リガンド結合等を検出するためのスクリーニング・システムを記載している技術公報が提供されている。
【0118】
デジタル化されたビデオまたはデジタル化された他の光学的アッセイ画像をデジタル化、保存、および分析するために、例えばPC(インテルx86またはペンティアム(登録商標)・チップ互換性DOS(商標)、OS2(商標)、WINDOWS(登録商標)またはWINDOWS(登録商標) NT(商標)に基づく機器)、MACINTOSH(商標)、あるいはUNIX(登録商標)に基づく(例えばSUN(商標)ワークステーション)コンピュータを用いて、種々の市販の周辺装置およびソフトウエアが利用できる。
【0119】
一般に、分析用システムとしては、本明細書中の1つ以上の方法の1つ以上のステップを検出するためのソフトウエアを有するデジタル・コンピュータが挙げられる。任意で、例えばハイスループット液体制御ソフトウエア、画像分析ソフトウエア、データ解析ソフトウエア、溶液をソースからデジタル・コンピュータに操作可能に結合された目的地まで移動させるためのロボット液体制御アーマチュア、操作を制御するためのデジタル・コンピュータにデータを入力するための入力デバイス(例えば、コンピュータ・キーボード)、またはロボット液体制御アーマチュアによるハイスループット液体移送、および任意で標識されたアッセイ成分からの標識シグナルをデジタル化するための画像スキャナも挙げられる。画像スキャナは、画像解析ソフトウエアに接続して、プローブ標識強度の測定を提供することができる。一般に、プローブ標識強度測定は、データ解析ソフトウエアによって解析され、標識プローブが固相支持体上のDNAにハイブリダイズしたかどうかが示される。
【0120】
いくつかの実施形態では、インビトロのオリゴヌクレオチド媒介組換え産物またはコンピュータ内(in silico)組換え核酸の物理的実施形態を有する細胞、ウイルス・プラーク、胞子等を固体培地上で分離し、個別コロニー(またはプラーク)を産生することができる。自動コロニー・ピッカー(例えばQ−bot、Genetix,U.K.)を用いて、コロニーまたはプラークを同定・選択し、10,000までの異なる突然改変体を2つの3mmガラス・ボール/ウエルを含む96ウエル・マイクロタイター皿に接種する。Q−botは、全コロニーの選択はせずに、コロニーの中央へピンを挿入し、細胞(または菌糸体)および胞子(またはプラーク適用ではウイルス)の小サンプリングで終了する。ピンがコロニーに入れられている時間、培地を接種するためのディップの数、およびピンがその培地に入れられている時間のそれぞれが接種材料のサイズに作用し、各パラメータが制御および最適化され得る。
【0121】
Q−bot等の自動的コロニー選択の一定のプロセスにより、人為的な操作の誤りが減少され、確立される培養物の割合が増加される(4時間で約10,000個)。任意で、温度および湿度が制御されたインキュベーター中でこれらの培養物を振とうする。マイクロタイター・プレート中の任意のガラス・ボールは、発酵槽のブレードと同様に、細胞の均一な通気および細胞(例えば菌糸体)フラグメントの分散を促進するように作用する。対象となる培養物から得たクローンを限界希釈によって単離することができる。上掲したように、ハイブリダイゼーション、タンパク質活性、または抗体へのタンパク質の結合等を検出することによって、ライブラリーを構成するプラークまたは細胞をタンパク質の産生に対して直接スクリーニングすることもできる。十分なサイズのプールを同定する確率を増すために、処理される突然変異体の数を10倍増加させる事前選別を用いることができる。一次選別の目的は、親株と同等またはそれより優れた産物の力価を有する突然変異体を素早く同定し、これらの突然変異体のみをその後の分析用に液体細胞培養へ移すことである。
【0122】
多様性ライブラリーをスクリーニングするための一手法は、超並列固相手順を用いて、ポリヌクレオチド改変体を、例えば酵素改変体をコードするポリヌクレオチドを発現する細胞をスクリーニングすることである。吸光、蛍光、またはFRETを用いた超並列固相スクリーニング装置が利用可能である。例えば、米国特許番号第5,914,245号(Bylinaら、(1999))を参照のこと;http://www.kairos−scientific.com/;Youvanら、(1999)「Fluorescence Imaging Micro−Spectrophotometer(FIMS)」Biotechnology et alia,<www.et−al.com> 1:1−16;Yangら、(1998)「High Resolution Imaging Microscope(HIRIM)」Biotechnology et alia,<www.et−al.com> 4:1−20;およびYouvanら、(1999)「Calibration of Fluorescence Resonance Energy Transfer in Microscopy Using Genetically Engineered GFP Derivatives on
Nickel Chelating Beads」(www.kairos−scientific.Comで公表)も参照せよ。これらの技術によるスクリーニングの後、一般に、対象の分子を単離し、任意で、当技術で周知の方法を用いて配列決定する。その後、配列情報を本明細書で先述したように用いて、新規のタンパク質改変体ライブラリーを設計する。
【0123】
同様に、アッセイ・システムで有用な液相化学に対して、多数の周知のロボット・システムも開発されている。これらのシステムとしては、武田薬品工業株式会社(Takeda Chemical Industries,LTD )(Osaka,Japan)により開発された自動合成装置等の自動ワークステーションと、科学者により実行される手動の合成操作を模倣しているロボット・アーム(Zymate II、Zymark Corporation,Hopkinton,Mass.;Orca,Beckman
Coulter,Inc.(Fullerton,CA))を利用した多数のロボット・システムが挙げられる。任意の上記デバイスは、例えば本明細書に記載されるように進化される核酸によりコードされる分子のハイスループット・スクリーニングのために、本発明とともに使用するのに適している。本明細書で論じられるように操作できるようにするためのこれらの装置(使用する場合は)の変更の種類および実行は当業者に自明である。
【0124】
(システム)
明らかであるように、本発明の実施形態は、1個以上のコンピューター・システムに保存されているか、またはそのようなコンピューター・システムを通して転送されるインストラクション、および/またはデータの制御の下に機能するプロセスを使用する。本発明の実施形態は、これらの操作をおこなう装置にも関する。そのような装置は、必要な目的のために特に、設計、および/または構築されたものでもよい。または、それはコンピューターに保存されたコンピューター・プログラム、および/または、データ構造によって選択的に可動、または再構成された汎用計算機であってもよい。本明細書に提示されたプロセスは、いかなる特定のコンピューター、または他の装置に本質的に関わるものではない。特に、本明細書での教示にしたがって書かれたプログラムを用いて、様々な汎用機械が使用可能である。しかしながら、ある場合には、必要な方法操作をおこなうために専門化した装置を構築することが、より好都合であるかもしれない。さまざまなこれらの機械のための特定の構造は、以下に与えられた説明から明らかになるだろう。
【0125】
加えて、本発明の実施形態は、コンピューターで実施される様々な操作をおこなうためのプログラム命令、および/または、データ(データ構造を含む)を含んでいるコンピューターによって読み込み可能な媒体、またはコンピューター・プログラム製品に関する。コンピューター読み込み可能な媒体の例には、ハードディスク、フロッピィディスク、磁気テープなどの磁気媒体;CD−ROMデバイス、およびホログラフィック・デバイスなどの光媒体;磁気光学媒体;リード・オンリー・メモリ・デバイス(ROM)、およびランダム・アクセス・メモリ(RAM)などの、プログラム命令を保存して、実行するために特別に構成されている半導体メモリ素子、およびハードウェア・デバイス、ならびに、時には、特定用途向け集積回路(ASIC)と、コンピューター読み込み可能なインストラクションを提供するためのプログム可能論理デバイス(PLD)と、ローカル・エリア・ネットワーク、広域ネットワークや、インターネットなどの信号伝達媒体とを含むが、これに限定はされない。また、この発明のデータおよびプログラム命令は、伝送波、または他の伝送媒体(例えば、光学線、電気線、そして/または、電波)の上で具体化されてもよい。
【0126】
本発明は、タンパク質配列中の交叉部位を選択、および/または評価するためのインストラクションを含むコンピューター、またはコンピューター読み込み可能な媒体を提供する。このコンピューター、またはコンピューター読み込み可能な媒体は、以下の機能の1つ以上をおこなう、1つ以上のコンピューター・コードかアルゴリズムを含んでいる。すなわち、i)基準生体分子の配列、または生体分子構造を入力する機能;ii)基準分子の基準配列用接触マップを作成する機能;iii)第1の配列および第2の配列を基準配列と整列させる機能;iv)第1の親配列と、第2の親配列との間で1つ以上の部分配列を交換し、キメラ配列を作製する機能;v)キメラ配列を接触マップと比較し、基準分子中の接触での近位要素(接触マップによる)に対応する、キメラ配列中の2つ以上の要素を選択する機能;および、vi)選択された残基を得点する機能である。場合によっては、交換ステップは多くの可能な部分配列、または部分配列のセットに関して反復され、それによって、分析用のライブラリー、または複数のキメラ配列を生成する。したがって、コンピューターによって生成する得点は、キメラ配列が基準分子の構造、立体配座安定性、および/または活性を保持する可能性に関する尺度を提供する。また、本発明のソフトウェアは、本明細書に記述されたもの以外の論理演算のいずれを提供してもよい。
【0127】
架橋オリゴヌクレオチド用の最良の位置を同定することによって、キメラ子孫(または、「シャフランツ(shufflants)」)の多様で、かつ構造的に安定したライブラリーを作製することができる。概して、以前の研究では、1箇所または2箇所の交叉に由来するキメラを考慮することによって、この分析にアプローチするだけであった。しかし、これはライブラリー中での多重交叉の効果を考慮に入れていないものである。場合によっては、多くの配列(例えば、3以上)を本発明の方法で用いることができる。例えば、本発明はまた、完全(ほとんど完全)なキメラ・ライブラリーのイン・シリコ分析に基づいて、安定したライブラリーをもたらすであろう多くの可能な交叉位置を同定する方法を提供する。場合によっては、そのような複数の交叉位置には、安定したライブラリーをもたらす可能な交叉位置の約50%、約75%、約85%、約90%、約95%、約98%、約99%、または約100%(例えば、すべて、もしくは実質的にすべて)が含まれる。
【0128】
場合によっては、コンピューターは2つ以上の親配列から複数のキメラ配列を作製し、それによって、対象とする配列における複数の交叉部位を評価する。この方法の一実施形態では、初期交叉数(n)が、使用された親配列の数m、およびコンピューター分析可能な配列の最大数(MAX)に基づき、以下の方程式を用いて決定される。MAX=mn−1。例えば、現在のハードウェアに基づいて、MAXの値は、約1010配列である。しかし、計算技術が向上するのに従って、この値は増大するだろう。初期交叉は、その後、配列の長さ全体にわたって、均等に(または、非対称的に)配置される。場合によっては、交叉の親セットは、追加情報(構造的問題、相同性、酵素活性などを含むが、これらに制限されない)に基づいて(例えばユーザによって)選ばれる。その後、可能な非変異キメラ(X)の全体ライブラリーがイン・シリコに作製される。
【0129】
この方法の一実施形態では、本発明のコンピューターは生体分子の配列、例えば基準タンパク質のアミノ酸配列を入力する。基準配列は、実験的に生成されたデータ(例えば、配列決定のデータ)と、前の組換え産物に由来するデータと、公用、および/または商用データベースなどを含む多くのソースのいずれからも入力することができる。その後、コンピューター(または、コンピューター媒体)は、当技術分野で利用可能な(および、以前に記述されているような)多くの手順またはアルゴリズムのいずれかによって、基準分子用の接触マップを作成する。
【0130】
別法として、本発明のコンピューターは、第1の親生体分子配列、第2の親生体分子配列、および、場合によっては追加の親生体分子配列(例えば複数の生体分子配列)を入力し、これらの配列を、基準配列なしに相互に整列する。
【0131】
コンピューターは、その後、2つの親配列間で部分配列(親配列中で第1の交叉部位と第2の交叉部位とによって規定される選択された領域の配列)を交換、または置換して、キメラ配列を作製する。場合によっては、コンピューターは、2つの親配列からの異なった部分配列を使用するか、または、複数の親配列からの部分配列を使用して、何回も交換手順を行い、それによって、1つ以上の交叉部位を持っている1つ以上のキメラ配列を作製する。
【0132】
2つの(または、それより多い)親配列間での、領域交換によってキメラ配列を作製した後、コンピューターは、キメラ配列の要素を接触マップと比較する。コンピューターは、基準配列における近位要素に対応するそれらのキメラ要素を選択して、それらの要素を得点する。一実施形態では、コンピューターによっておこなわれるスコアリングは、計算された接触エネルギー、立体障害、疎水性または極性などの、1個以上のパラメータに基づいている。
【0133】
一実施形態では、比較およびスコアリング・ステップは、キメラ配列のエネルギーに基づいている。キメラ・ライブラリーのエネルギーについて計算する方法は、多数ある。例えば、上記の記載と同様に、Miyazawa−Jerniganマトリックスに基づいて、エネルギー項(energy terms)を計算することが可能である。このエネルギー項は、キメラ子孫にあると予想された残基接触のエネルギーの合計から通常計算される。場合によっては、正規化されたエネルギーが、その交叉セットを与えられた可能なキメラそれぞれに関して計算される。
【0134】
本発明のある特定の実施形態では、その後、計算上最も低いエネルギーをもつキメラ配列を統計的に分析する。例えば、ベイズの解析(Bayesian analysis)、ニューラル・ネットワーク法などを含む、ただしそれらに限定されない、多くの方法によって相関係数を計算することが可能である。隣接した2つのブロックの配列間で相関係数が高いことは、交叉の前のブロックと同じ配列が、交叉の後のブロックに好適であるという事実によって、交叉が非効率的(poor)に置かれたことを示す。
【0135】
表1および表2は、初期セットの交叉位置に関する、例示的結果を示す。その後、これらの交叉の位置を変化させ、交叉が好適位置にあることを相関係数が示すまで、エネルギー計算を繰り返した。隣接した2つの交叉の間に、交叉に好適な位置がない場合(例えば、中間的交叉で規定された2つの交叉部分配列の相関がきわめて高い場合)、それら2つの隣接した交叉間の中間的交叉は交叉セットに含まれない。すべての交叉がいったん好適位置におかれると、新規セットの初期交叉で全過程が反復される。その後、計算上最も低いエネルギーをもつキメラ・ライブラリーを生成する交叉セットが選ばれる。このセットはその後、実験室での実験など、さらなる活動に使用することが可能である。一例として、表2は、3つのMLE(最大尤推定(Maximum Likelihood Estimation))配列間での交叉位置の1セットに関する配列ブロック間の相関係数を示す。「XXXを含むセル(cell)は、連続した配列ブロック間の高い相関係数を示し、したがって非効率的な交叉位置である。
【0136】
【表1】
【0137】
【表2】
場合によって、コンピューター、またはコンピューター読み込み可能な媒体のコード(code)には、接触ペアに関し生成した得点を正規化するためのメカニズム、またはアルゴリズムが含まれる。例えば、コンピューター、またはコンピューター読み込み可能な媒体は、多重回帰を行って、残余値を変動係数として提供することが可能である。場合によっては、ユーザに未処理の得点を提示する前か、または未処理の得点を提示した後に、ユーザからの指示によって正規化操作をおこなう。本発明の一実施形態では、得点が三角形の輪郭プロットとして提示される。
【0138】
C、C++、ビジュアル・ベーシック(Visual Basic)、フォートラン(Fortran)、ベーシック(Basic)、ジャバ(Java(登録商標))などの標準プログラミング言語を用いて、上で記述された計算をおこなうための論理命令を当業者により構築することができる。例えば、コンピューターは、第1配列および第2配列をデータベースで捜すためのソフトウェアを含む可能性があり、場合によっては、さらにユーザ・インタフェースと通信するために修飾されている可能性もある(例えば、ウインドウズ、マッキントッシュ、UNIX(登録商標)、LINUXなどの標準的なオペレーティング・システムによるGUI)、それによって、配列文字列を得て、構成要素系を整列させて、計算を行い、かつ/または、検査結果を操作する(例えば、交叉位置の評価)。ワープロソフト(例えば、Microsoft WordTM、Corel WordPerfectTM)、スプレッド・シート、および/または、データベース・ソフト(例えば、Microsoft ExcelTM、Corel Quattro ProTM、Microsoft AccessTM、ParadoxTM、Filemaker ProTM、OracleTM、SybaseTM、およびInformixTM)などを含み、かつそれらに限定されない標準的なデスクトップ・アプリケーションを、これらの(および他の)目的に適合させることが可能である。
【0139】
本発明のコンピューター読み込み可能な媒体には、光媒体、磁気媒体、ダイナミック・メモリ、フラッシュメモリ、およびスタティックメモリが含まれるが、これらに限定はされない。場合によっては、コンピューター、またはコンピューター読み込み可能な媒体は、分析結果を出力ファイルの形式で提供することができる。例えば、出力ファイルは、整列させられた第1配列、第2配列、および/または、基準配列の一部、またはすべてについてのグラフ表示形式、あるいはマトリックスの形式(Miyazawa−Jerniganマトリックスなど)であってもよい。
【0140】
(キメラ配列のウェブ・ベース・ライブラリー)
本発明の様々な実施形態は、キメラ・データ配列、または対象とする生体分子(例えば、RNA、DNA、タンパク質など)から得られた情報を、使用、および/または判定する方法、および/またはシステムに関する。特定の実施形態では、本発明はさらに、キメラ配列データのライブラリーを提供し、1セット以上のキメラ配列、構造、および/または、接触マップをクライアントが作成、または分析することを可能にする方法、および/またはシステムを含む。
【実施例】
【0141】
ここに記述された実施例および実施形態は、例示的目的だけのためのものであり、その観点における様々な改善または変更が当業者に示されるであろうが、それらはこの出願の精神および範囲と、添付された請求の範囲との内に含まれるものであると理解される。以下の実施例は、請求の範囲に記載されている発明を限定するものではなく、それを例解するために提供されている。
【0142】
対象とするタンパク質の1つのファミリーが、ムコン酸シクロイソメラーゼとしても知られているムコン酸ラクトン化酵素(MLE)である。これらの酵素は、芳香族化合物をクエン酸回路の中間代謝物に分解するのに不可欠である。MLEは、保存された構造をもち、かつある程度の機能的多様性をもつタンパク質をコードする。MLE Iは、シス,シス−ムコン酸(cis,cis−muconate)の(4S)−ムコノラクトンへの変換を触媒するβケトアジピン酸経路中で機能する(Ngai,K.,Omston,L.N.,およびKallen,R.G.(1983年)「Enzymes of the
beta−ketoadipate pathway in Pseudomonas
putida:kinetic and magnetic resonance studies of cis,cis−muconate cycloisomerase catalyzed reaction.」Biochemistry 22:5223−5230)。MLE IIは、同じ環化異性化反応を触媒するが、基質として3−クロロムコン酸を用いる(Schmidt,E.,およびKnackmuss,H.(1980年)「Chemical structure and biodegradability of halogenated aromatic compounds.」Biochem.J.192:339−347)。3Å(MLE II)および1.85Å(MLE I)の解像度で決定されたこれら二酵素の構造はきわめて類似したものであり、両方とも、N末端キャッピング・ドメインをもつα/βバレルを含み、平均0.96ÅのRMSDを有す(Kleywegt,G.J.,およびJones,T.A,(1996年)「A re−evaluation of the crystal structure of chloromuconate cycloisomerase.」Acta.Crystallogr.Sect.D52:858;およびHelin,S.,Kahn,P.C.,Guha,B.L.,Mallows,D.G.,およびGoldman,A.(1995年)「The refined X−ray structure of muconate lactonizing enzyme from
pseudomonas putida PRS2000 at 1.85 resolution,」J.Mol.Biol.254:918841)。エノラーゼ・スーパーファミリーのメンバーとして、それらは約60%に満たない配列同一性を有し、配列の「低配列同一性」組換えのための理想的なケースとなっている(しばしば間違って、「低相同」組換えとも呼ばれる)。
【0143】
これらの実験で、本発明者らは構造情報を計算により導き、それに応じた組換えをおこなうことを目標とした。対にしたときのアミノ酸配列同一性が約40〜52%の間にある、3つのムコン酸ラクトン化酵素(MLE)を親配列として選んだ。すなわち、Pseudomonas putida由来のMLE IIと、P.putidaおよびAcinetobacter calcoaceticus由来のMLE Iである(それぞれ登録番号P27099、AAA66202.1、およびQ43931)。交叉(crossover)は、MLE配列全体の中で、構造安定性に対し、破壊的または非破壊的であると予測された位置を代表する10位置に設計した。実験的な組換えを用いて、これらをテストした。その結果は、3つのMLEによる多機能的キメラを設計するのに、コンピューター・モデルの使用が可能であることを示す。
【0144】
本発明者らは、構造情報を用いて、これらの遺伝子の中に組換えポイントを選んだ。本発明者らはこれらの計算結果に基づいてオリゴヌクレオチドを再合成し、それを用いて組換えを行い、それによって、キメラ生体分子のライブラリーを生成した。本発明者らは、シス,シスームコン酸の活性を保持したキメラ産物を、これらのライブラリーから選択した。本発明者らは、2つまたは3つの異なったMLEからの配列ブロックを含有する、多数の異なった活性酵素を発見した。これらの変種は、最も活性な野生型親酵素の機能に匹敵する生体内機能を有し、それらに含まれる交叉部位は、本発明者らが好適であると予測した部位への強い偏向を示した。一方、好適でないと予測された部位は、強い選択により排除されていた。本発明者らは、コンピューターによる、キメラ配列の「プレスクリーニング(prescreanibg)」によって、最適な組換え位置の選択が可能となり、それによって、ライブラリーの適合度が増大し、スクリーニングしなければならない変種の数が減少すると結論した。
【0145】
(接触エネルギーの計算によってキメラ・タンパク質の安定性を予測するアルゴリズムの設計)
本発明者らは、キメラ・タンパク質の安定性を、コンピューター内で評価するアルゴリズムを設計した。このアルゴリズムは、既知の基準配列と構造(この実施例では、P.putida由来のMLE I)を用いて、どのアミノ酸が相互に接触しているかを決定する(例えば、接触マップ)。また、このアルゴリズムは、すべての親配列を相互に整列させ、そのアラインメントを用いて、完全長のキメラ配列をコンピューター内で生成する。これは、最も単純な場合には、1つの親タンパク質の1セクションを第2の親からの対応するセグメントで置換することに相当する。アミノ酸置換の影響を、基準配列と比較して評価するため、標準配列とキメラ配列とを比較し、Miyazawaらの記述のように導かれる接触エネルギー関数を用いて、接触エネルギーの変化(ΔEc)を計算した、(Miyazawa,S.,およびJernigan,R.L.(1996年)「Residue−residue potentials with a favorable contact pair team and an unfavorable high
packing density term,for simulation and
threading.」J.Mol.Biol.256:623−464、ならびに上記の追加参考文献を参照)。
【0146】
図2Aは、キメラMLEタンパク質に関して予測された接触エネルギー変化マップを示す。この図では、ドナー・タンパク質であるP.putidaMLE IIの単一セグメントによって、基準タンパク質であるP.putidaMLE Iの対応するセグメントが置換されている。縦座標(x軸)は、直線配列上の置換が始まる位置(例えば、最初の交叉位置)を示し、横座標(Y軸)は置換の長さを示す。基準配列に比べ、好適でない接触をするアミノ酸がキメラに含まれる置換は、マゼンタ色で示され、構造的破壊に通じる可能性が高い改変を反映する。予測されたキメラに、標準配列より好ましい接触をするアミノ酸が含まれるような位置は、赤色で示され、構造的に許容できる可能性の高い改変を表す。高度の破壊を引き起こす位置、すなわち接触スコアが高い位置は、紫色で示される。置換の長さが大きくなるのにしたがって、接触スコアが増大する。これは、キメラの接触マップと、オリジナルの接触マップとの間の相違が増大されるためである。
【0147】
(キメラ・タンパク質の安定性予測を用いて交叉点を特定することが可能である)
このアルゴリズムを開発する際の本発明者らの目的は、単一交叉としてより、多数の交叉の1つとして、よく機能するであろう可能性に基づいて、交叉位置を選択するキメラ・ライブラリーを設計することであった。このことを達成するため、本発明者らは、異なった長さ(1〜80アミノ酸類)のドナー・タンパク質セグメントを、基準タンパク質の特定位置で開始、または終了するように挿入するときの、平均ΔEcを計算した。図2Bは、P.putidaMLE II由来の1〜80アミノ酸によって、基準タンパク質であるP.putidaMLE Iの対応するセグメントを置換したときの平均接触エネルギー変化を示す。P.putidaMLE IIの代わりに、A.calcoaceticusMLE Iをドナーとして用いたときの結果は、きわめて類似したものであった(データは示されていない)。
【0148】
図2Bの残基位置標識によって示された曲線に基づき、10の交叉位置を選択した。図2Bの横座標で下側の位置は、より大きなΔEcを示し、従って、構造的により破壊的な可能性が高い交叉点を示す。本発明者らは、全範囲でのΔEc値を表し、構造内で均等に分布されるように交叉位置を選択した。最適であるとコンピューター内アルゴリズムで予測された交叉位置は、最初の交叉がその位置にあるキメラに関して低い接触スコアを示し、さらに第2の交叉がその位置にあるキメラに関しても低い接触スコアを示した。ここで本発明者らは、このモデルの予測力をテストする目的で、接触エネルギーが高いと予測された位置と、低いと予測された位置との両方を代表する10箇所の交叉部位を選択した。これらの交叉部位は、ヘリックスおよびシートを接続するループの中心にあるα/βバレル全体にわたり均等に分布している。これらの交叉部位は、MLEアライメントにおける残基位置98、119、144、172、201、228、254、280、302、および328にある。交叉位置119、144、172、201、および328は好適であると予測されたが、98、228、254、280、および302は好適でないと予測された。これらの計算は単量体構造に基づくものであるが、八量体構造で繰りかえされた計算でも同様の予測が提示された。その後本発明者らは、10の位置すべてで組換えを強制し、機能的変種を選択し、さらに、好適であると予測された交叉部位が、それら機能的キメラ・タンパク質中に見いだされるかを判定することによって、本発明者らのコンピューター予測をテストした。
【0149】
(オリゴヌクレオチドで媒介された組換え)
本発明者らは、標準的なシャフリング反応で相互に組換えをおこなうのに十分な配列同一性をもたないMLE親配列を選択した(例えば、Moore,G.L.,Maranas,C.D.,Lutz,S.,およびBenkovic,S.J.(2001年)「Predicting crossover generation in DNA shuffling.」Proc.Natl.Acad.Sci.USA 98:3226−3231を参照)。これにより、特定の予測を徹底的にテストすることが可能となった。少数の位置でのみ組換えを可能にすることによって、比較的少数の変種を分析することで、統計的に重要なデータが得られるようになるだろう。
【0150】
「低い配列同一性」組換えを容易にするため、交叉オリゴヌクレオチドを合成し、MLE配列全体にわたる10箇所の位置に組換えを導入した。半分のオリゴが1つの配列と同一であり、さらに、もう片方の半分は別の配列と同一であるようにオリゴを設計し、それによって、2つの配列の間の組換えを促進させた。選択されなかったクローン46個の配列決定をすることによって、組換えの度合いを抽出検査した。これらの配列のうち35%以上が、10箇所の交叉位置のうち8箇所での組み込みを示し、これらの8つの交叉オリゴに関する平均取りこみ率は47.8%であることが、これらの配列によって示された。交叉1、および交叉6には、それぞれ12%、および15%の取りこみがあった。
【0151】
MLE遺伝子の特定位置での組換を可能にするため、本発明者らは、5’半分が1つの親配列と同一であり、かつ3’半分が第2の親配列と同一である一連の交叉オリゴヌクレオチドを設計した。本発明者らは、図2Bに示された10箇所の交叉位置での組換えが容易になるように、そのようなオリゴヌクレオチド60個を、3つのMLEすべてを含む組換え反応液に入れた。本発明者らは、無作為に選ばれた71個の変種を配列決定することによって、シャッフルされたライブラリーでの交叉取りこみの頻度を算定した。個々の交叉オリゴヌクレオチドは、異なった効率で組み込まれた。それらは、機能的に選択されなかった変種の20%〜50%で検出され、そのような変種の子孫の97%に少なくとも1つの交叉が組み込まれていた。
【0152】
また、本発明者らの未処理のライブラリーを分析した結果、ライブラリーの配列に、いくらかの偏りがあることが示された。親配列の1つである、A.calcoaceticus由来のMLE Iの最終配列への寄与は、平均して4%のみであった。これはおそらく、この親遺伝子のGC含量がはるかに低いためであると、本発明者らは考えている(45%、これに比べ、P.putidaのMLE IおよびMLEIIはそれぞれ、64%および68%である)。より類似したコドン使用頻度で遺伝子を再合成することによって、組換えにおけるそのような偏りを取り除くことができるが、本発明者らは、このことによって、本研究から導いた結論が影響されるとは考えない。
【0153】
(活性キメラ酵素の生成)
キメラ酵素ライブラリーを、アシネトバクターへのMLE欠失株に形質転換し、実験プロトコル(下記)の記載に従って、相補的な(complementing)クローンを選択した。細菌コロニーは、1日目と6日目の間に現れた。本発明者らは、その中から332個の独立したMLE遺伝子を回収し、配列決定した。選択されたものと、選択されなかったもの両方の配列決定を行った403個の変種の中では、親配列間の組換えは、交叉オリゴヌクレオチドによって媒介されたもののみが観察された。
【0154】
配列決定された332個の活性変種の中に、本発明者らは合計33個の独自の組換え配列を見いだした。キメラ配列の大部分は、ある親から、第2の親骨格への単一セグメント(通常30〜60アミノ酸)の組み込みをもっていた。しかし、33個の配列うちの6個には、異なった親からの100残基以上の組み込みがあった。これらの結果により、構造的かつ機能的に保存されたタンパク質に由来する多くの異なったセグメントは、低い配列同一性にもかかわらず、相互に機能的置換が可能であることが示されている。
【0155】
(コンピューター・モデルリングは機能的酵素の交叉優先傾向を予測する)
本発明者らは、活性なキメラに見出された交叉位置に、強い偏向があるのをみた。本発明者らは、これらの偏向に関し、71個の選ばれなかったキメラの中のそれぞれの交叉の頻度を、33個の独自の活性なキメラに見られた交叉の頻度と比較することによって、これらの偏りを測定した。本発明者らは、交叉のモデル化されたエネルギー優先傾向(modeled energetic favorability)を、活性な変種における、その表出(representation)と相関させることによって、接触エネルギー曲線の予測的価値を評価した。α/βバレル中に位置する7つの交叉に関する相関係数は0.94であり、これは、本発明者らのアルゴリズムが、この領域中の生産的な交叉部位に関する有効なプレ・スクリーニング(pre−screen)であることを示している。
【0156】
α/βバレルの外にある3箇所の交叉位置(98、119、および328)はすべて、エネルギー的に好適であると予測されているが、これらはすべて、機能的なキメラにおいてきわめて不十分に表出されている(under−represented)。このドメインに基づく差異の1つの可能な理由は、活性なMLEのもつ八量体四次構造であるが、オリゴマー相互作用の大部分に、N末ドメインが関与する。モデルにオリゴマーの接触エネルギーを入れたときには、本発明者らは図2で示されたデータに非常によく似たデータを得た。しかし、安定した四次構造を保持するのに必要な接触は、分子内の接触よりも大きく重み付けされるべきでかもしれない。別の可能性は、これらのタンパク質上のN末ドメインが結合相互作用に参加すると考えられることである。利用可能なMLEの構造に、基質結合したものはないが、相同タンパク質であるo−スクシニル安息香酸合成酵素の分析によって、この「キャッピング」ドメインが、リガンド結合に続いて、〜10Åのかなり大規模な運動を行うことが示されている(Thompson,T.B.ら(2000年)「Evolution of enzymatic activity in the
enolase superfamily:Structure of o−succinylbenzoate synthase from Escherichia coli in complex with Mg2+ and o−succinylbenzoate.」Biochemistry 39:10662−10676)。構造モデリングは、パッキング(packing)相互作用が維持されるキメラを同定することができるが、分子動力学シミュレーションなどのより強度な計算力を必要とする技法は、キメラが、触媒作用に関連した構造変化をおこなうかどうかの評価をするのに使用可能である(Wang,W.,Donini,O.,Reyes,C.M.,およびIcoliman,P.A.(2001年)「Biomolecular simulations:recent developments in force fields,simulations of enzyme catalysis,protein−ligand,protein−protein,and protein−nucleicacid noncovalent interactions.」Annu.Rev.Biophys.Biomol.Struct.30:211−243)。
【0157】
(最も速い増殖をする株には合成活性部位のあるMLE酵素が含まれる)
本発明者らは、キメラMLEの発現によって、形質転換体の成長率が広い範囲に与えられ、コロニーが形質転換の1〜6日後に選択プレートの上に現れるのをみた。これは、3つの親酵素では2〜4日後であるのと比較される。2個の子孫クローンは、親に比べてかなり急速に成長し、1日かからずにコロニーを形成した。これらの1つは、主にP.putidaMLE IIから成り、A.calcoaceticusMLE Iのセクション(172〜201)と、P.putidaMLE Iのセクション(201〜228)とをもつキメラ・タンパク質を発現した。得られたキメラ活性部位は、3つの親すべてからの残基を含み、機能上重要な残基として、P.putidaMLE IIの寄与によるLys167、Lys169、およびAsp249;A.calcoaceticusMLE IによるAsp198;ならびに、P.putidaMLE IによるG1u224を含んでいる。もう片方の急速増殖形質転換体から得られたキメラ酵素の配列は、大部分がP.putidaMLE Iに由来するものであったが、これにも、A.calcoaceticusMLE Iの残基172と残基201との間が含まれていた。それらは両方とも、コンピューター内アルゴリズムによって、とても好適な交叉位置であると予測されていた。
【0158】
これらの最も活性なキメラに関する本発明者らの分析によって、α/βバレル構造の頑健さが例証された。キメラ1の3箇所の交叉のうち、位置201の交叉のみで、2つ以上の同一アミノ酸の伸長が同時に起きていた。A.calcoaceticusMLE I、およびP.putidaMLE Iに由来する2箇所の置換領域は、13残基および11残基の同一アミノ酸を、対応するP.putidaMLE IIの29残基および28残基のアミノ酸伸長と共有する。これらのアミノ酸は、MLE IIタンパク質の大部分を構成するものである。これらの相違は、一般的、保存的変化ではなく、以前に記述された方法のよって、活性を保ちつつ、酵素活性部位のこのように小さな領域に、このように多くの改変をコンピューター設計するのは困難であろう。
【0159】
(コンピューター・プレ・スクリーニング)
ここに提示された結果において、本発明者らは、遺伝子またはライブラリーの物理的構築の前に、組換え部位をプレ・スクリーニングすることを可能にするアルゴリズムを記述した。このアルゴリズムは、コンピューター内組換体タンパク質を作製し、構造的に特徴付けられた基準タンパク質からの偏位に基づいて、得られたキメラ分子中のエネルギー変化を予測し、さらに、パッキング相互作用の破壊を最小にする可能性の高い交叉部位を選択する。
【0160】
以前の研究では、無作為の位置での単一組換え事象を引き起こし、1つの親のN末端と第2の親のC末端との間の融合を形成することによって、キメラ・タンパク質のライブラリーを、物理的に生成した(Ostermeier,M.,Shim,J.H.,およびBenkovic,S.J.(1999年)「A combinatorial approach to hybrid enzymes independent of DNA homology」Nature Biotechnol.17:1205−1209;Sieber,V.,Martinez,C.A.,およびArnold,F.H.(2001年)「Libraries of hybrid proteinsfrom distantly related sequences.」Nature Biotechnol.19:456−460)。これらの方法では、活性の低い機能的キメラが比較的少数得られた。
【0161】
そのように無作為に得られた遺伝子組換え体は、機能的に印象のうすいものであり、ITCHYキメラの最良のものでも、開始遺伝子より500〜1万倍も活性が低くかった。これらには、リーディング・フレーム・シフト、または挿入/欠失が導入された可能性のあるものも含まれている。あるいは、単一交叉によるキメラ生成に続いて選択ステップがおこなわれるため、これが不安定なキメラの形成に通じているのかもしれない。接触エネルギーに基づくアルゴリズムは、単一交叉事象による交叉が、2重交叉より、少数の安定したキメラを生成すると予測している(データは示されていない)。また、本発明者らがこの研究で分析した33個の活性なキメラのうち、1個だけが単一交叉を有し、残りは複数の組換事象によって生成したという事実がこの主張と一致している。組換えに関する、前のファミリーの研究でも、選択された最も活性な組換えに多重交叉が見いだされている(例えば、Crameri,1998年;Ness,1999年;Chang,1999年;およびSoong,N.ら,2000年、ならびに上記のものすべてを参照)。さらに、本発明者らの活性なキメラのうち81%は、別の親からの組込みが70アミノ酸に満たないものであった。単一交叉部位のキメラを選択したときには、これらのように1つの親が支配的なキメラを生じる可能性が、N末領域、およびC末領域の交叉位置に限定されるであろう。最後に、初期選択を単一交叉によるキメラでおこなうことによって、他の交叉と組み合わせてうまく機能しうる交叉部位を、時期尚早に排除している可能性がある(Lutz,S.,Ostermeier,M.,Moore,G.L.,Maranas,C.D.,およびBenkovic,S.J.(2001年)「Creating multiple−crossover DNA libraries independent of sequenceidentity.」Proc.Natl.Acad.Sci.U S A.98 11248−11253)。対照的に、本発明者らのコンピューター・プレ・スクリーニングは、原則として、望まれるだけ多くの組換事象を考慮することができる。
【0162】
高度に多様化した親からキメラを作製する別のアプローチは、構造的情報と機能的情報とを結合し、しばしばいくらかの程度の配列ランダム処理および/または組換えと共役させる準合理的設計のアプローチである。これらの実験は、概ね、低い配列同一性ライブラリーの組換えより成功しており、基質特異性および熱安定性の両方における改変を達成した(例えば、Altamirano,M.M.,Blackburn,J.M.,Aguayo,C.およびFersht,A.R.(2000年)「Directed evolution of new catalytic activity using the alpha/beta−barrel scaffold.」Nature 403:617−622;Jermutus,L.,Tessier,M.,Pasamontes,L.,van Loon,A.P.およびLehmann,M.(2001年)「Structure−based chimeric enzymes as an alternative to directed enzyme evolution:phytase as a test case.」J.Biotechnol.85;15−24;ならびに、Kaneko,S.ら(2000年)「Module recombination of a family F/10 xylanase:replacement of modules M4 and M5 of the FXYN of Streptomyces olivaceoviridis E−86 with those of the Cex of Cellulomonasfimi.」Protein Eng.13:873−879を、参照のこと)。しかしながら、この方法によるキメラ生成は、改変される特定のタンパク質に関係する構造機能相関について、かなりの理解が必要とする。
【0163】
(実施例の要旨)
この実施例において、本発明者らは、指向性進化技術の一般的適用性を、コンピューター分析のスピードおよび威力と結合する方法を記載した。本発明者らのコンピューター内組換えおよびプレ・スクリーニングに必要な唯一の構造情報は、親タンパク質の結晶構造、NMR構造、構造相同性モデル情報または、当業者に公知の他の構造決定法などの立体的構造情報である。したがって、このアプローチは、一般的であり、かつ高度に自動化可能なものである。場合により、この初期スクリーニングを通過するキメラは、物理的に合成され、試験される。この研究において、本発明者らは交叉オリゴヌクレオチドを用いてキメラ・ライブラリーを合成したが、プレ・スクリーニング・データの取りこみを可能にするいかなる形式(例えば、Nessら(2002年)Nature Biotechnology 20:1251−1255,PCT公報WO 00/42561、およびWO 00/42560に記載される合成組換え)も使用され得る。不十分に折りたたまれた(poorly−folded)変種を取り除くことによって、物理的にスクリーニングしなければならないタンパク質の数を減少させ、特にスクリーニングが困難かつ複雑で、時間がかかる時、成功の可能性を大きくし得る。構造と機能との間の相関の本発明者らの理解が向上するにしたがい、コンピューターのアプローチで評価し得る特性の範囲が拡大され、それによってコンピューター内のプレ・スクリーニング・アルゴリズムの価値が高まると、本発明者らは考える。
【0164】
本発明者らは、最も近い親の配列から、少なくとも70アミノ酸が異なる5つの組換え産物を得た。これらのキメラ生体分子は、すべて、コンピューター内アルゴリズムで好適であると予測された交叉を含んでいる。同じ機能構造に折り畳む能力に関して、自然進化によって既にスクリーニングされている親配列から選択された多様性を用いることで、単純なコンピューターのプレ・スクリーニングを使用して、いかなる出発点からも非常に異なる新規配列を設計し得る。さらに本発明者らは、このアルゴリズムを交叉オリゴヌクレオチドで媒介された組換えと結びつけることによって、40%という低い配列同一性の配列から生成され、機能的に活性なキメラを含むキメラ・ライブラリーを作製した。
【0165】
キメラ・ライブラリーは、増殖にMLE活性が必要なプレート上のMLEノック・アウト株中で増殖させた。それらのコロニーを、2〜7日間にわたって増殖させ、124コロニーを選択して、配列決定した。選択され、配列決定されたこれらのクローンの中には、繰り返し現れる配列が多く存在し、結果的に、33種の独自な入れ替え配列(shuffled sequence)が得られた。選択された配列のうち3つでは、異なった親からの組み込みが100残基以上あったが、キメラ配列の大部分は、30〜60残基が組込まれていた。選択されなかった配列に対して選択された配列に、119位、144位、172位、および201位を中心に交叉位置が組み込まれる、強いパターンが見られる。
【0166】
予測傾向は、119位、144位、172位、302位、および328位の残基を最適開始位置として、様々な長さにわたり、きわめて類似し続ける。本発明者らは、119位、144位、172位、および328位での交叉が正しく予測されたと考える。なぜなら、それらの交叉が、選択されなかったクローンよりはるかに頻繁に、選択されたクローンに見られたからである。2つの位置(98および302)だけが、予測と異なり、予想したように頻繁に見られなかった。98位は、選択されなかったクローンの12%でのみ見られた。したがって、このオリゴの取りこみ全体が欠如していることが、予測と、実際の結果との間の矛盾を説明し得る。最適な終了残基(ending residue)は、144位、172位、201位、および228位であると予測される。本発明者らは、交叉を終了させる可能性の高い部位は、172位、201位、および302位であることを見いだした。228位は、選択されなかったクローンの15%で組み込まれたのみであり、したがって、この交叉がなぜ選択されたクローンで見られなかったかを説明し得る。144位は、非常に好適であると予測されたが、少数の選択されたクローンで交叉を終了させているのが見られたのみである。これは98位および119位で交叉を開始したクローンが少数であって、したがって、144位で終了可能な交叉の数が限定されたことに起因し得る。
【0167】
(実験プロトコル)
P.putida由来のMLE I、およびMLE 2、ならびにA.calcoaceticus由来のMLE Iの野生型遺伝子は、上述のように、増幅し、断片化した(Crameriら、1998年)。3’末端で1つの親に、かつ5’末端で別の親に相補的なオリゴヌクレオチドは、QUIAGENオペロン(QUIAGEN Operon(Alameda,CA))から取り寄せた。これらは、テキスト中で同定された位置に対応するブレーク・ポイント(break point)をもつ。フラグメントおよびオリゴの混合物は、例えば、Nessら、「Synthetic shuffling expands functional protein diversity by allowing amino acids to recombine independently」(2002年)Nature Biotechnology 20:1251−1255、およびPCT公報W098/27230に記載されるように組み合わせた。得られたキメラ配列を、以下で記載するようにベクターにクローニングした。選択されなかったキメラ分子、および選択されたキメラ配列の配列決定は、アプライド・バイオシステムズ(Applied Biosystems(Foster City,CA))3700型シーケンサーを用いて行った。
【0168】
(MLE活性に関する選択)
A.calcoaceticusのMLE Iノック・アウトを、自然形質転換能と、catB遺伝子をカナマイシン耐性カセットに置換する相同組換えとを用いて構築した。簡潔には、フォワード・プライマー、およびリバース・プライマーを用いて、pACYC177のカナマイシン耐性カセット(プロモーターと共に)をPCR増幅した。PAGE精製された両プライマーは、100のヌクレオチド長であった。そのうち、3’末端の20ヌクレオチドは、カナマイシン耐性カセットのちょうど上流(フォワード・プライマー)か、またはちょうど下流(リバース・プライマー)と同一であり、80ヌクレオチドのテール部分(tail)は、アシネトバクターADP1のcatB遺伝子(Genebank AF009224)のちょうど上流(フォワード・プライマー)か、またはちょうど下流(リバース・プライマー)の領域と同一である。catB遺伝子は、ムコン酸ラクトン化酵素MLE Iをコードする。
【0169】
A.calcoaceticus株ATCC33305(BD413株)を、一晩、30℃で育てた。容積0.4mlを、50mlチューブ中のLB10mlに副培養(subculture)し、その後、30℃、300rpmで2時間振盪培養した。約0.5mlの培養物を、カナマイシン・カセットを増幅するPCR反応液の50μlに混合した。耐カナマイシンの形質転換体を、40μg/mlのカナマイシンを含むLB寒天上で選択し、安息香酸寒天上に広げて、増殖能を調べた。安息香酸寒天は、1リットルあたり10g K2SO4、135g K2HPO4、47g KH2PO4、25g NaCl、5.4g NH4Clおよび50mlの2%MgSO4x7H2Oからなる10x無機質塩ベース、および10mM CaCl2、0.5mM FeCl3、および0.5mM MnCl2からなる1000x微量塩溶液を用いて調製した。無機質塩ベースおよび微量元素に加えて、1リットルあたり、2.5mlの1M Na2MoO4x2H2Oおよび2.5mlの1M安息香酸ナトリウムを、1.5%のディフコ・バクト・アガー(Difco Bacto agar)と共に加えた。NS238と命名された、安息香酸存在下で成長できなかった一形質転換体を用いて、活性なキメラについての選択を行った。
【0170】
P.putida由来のMLE I、および2、ならびにA.calcoaceticus由来のMLE Iの野生型遺伝子を、広域宿主ベクターpMMB66EH(ATCC37620)にクローニングした。これらの3つの構築物は、安息香酸寒天上での増殖に関し、ノック・アウト株NS238を相補し得た。キメラMLEライブラリーを、上述のようにプラスミドpMMB66EHにクローニングし、自然形質転換能を用いてNS238株に形質転換し、0.15mMのIPTGと共に処方された安息香酸寒天上にプレート培養し、さらに、1週間まで、37℃でインキュベートした。安息香酸を利用する形質転換細胞の増殖表現型は、単一コロニーになるように安息香酸寒天に広げ、その後、37℃で6日間、3つの野生型遺伝子と比較した増殖によって検査した。
【0171】
(構造モデリング)
MLE 1p構造である1MUCにおける相互作用残基を用い、ケミカル・コンピューティング・グループ社(Chemical Computing Group(Montreal,Quebec,Canada))のMOEソフトウェアを使用して、酵素の接触マップを定義した。接触マップは、結晶構造中の相互作用を記述するマトリックス(C)であって、残基iおよび残基jに関して、2残基間の距離4.5Å内に潜在的水素結合パートナー、または疎水性相互作用がある場合に、Cij=1となり、ない場合には、Cij=0となるように得点するようなマトリックスである。
【0172】
3つの親MLEに関して、すべての可能な2交叉キメラをコンピューター内で作製した。その後、オリジナルの接触マップとの比較によって、これらのキメラを評価した。MiyazawaおよびJerniganのポテンシャル(potential)を用いて、キメラの接触エネルギーを計算した。MLE構造中の各相互作用位置で、コンピューター内でキメラに由来する接触している残基のMiyazawa−Jerniganエネルギーを、合計して、総接触エネルギー(Ec)を算出した。
【0173】
【数1】
式中、EMJ(i,j)は残基iおよび残基jに関するMiyazawa−Jerniganエネルギーであり、iとjとの間に接触がある場合、Cijは1であり、接触がない場合は0である。置換セグメントの長さが増大するにしたがって、接触エネルギーも増大することを留意する。これはキメラとオリジナルの接触マップの相違が増大することに起因する。したがって、本発明者らは、基準配列に向かったこの偏りを最小にするために、交叉分析を置換長80以下に制限した。MiyazawaおよびJerniganポテンシャルの関連した記載は、上で同定した参考文献において見いだされる。アミノ酸配列に関しては、例えば、Miyazawa−Jerniganエネルギー・マトリックスを用いることで、接触エネルギーを推定することができる(例えば、MiyazawaおよびJernigan(1999年)「Self−consistent estimation of inter−residue protein contact energies based on an equilibrium mixture approximation of residues」Proteins 34:49−68;Miyazawa,およびJernigan(1999年)「An empirical energy potential with a reference state for protein fold and sequence recognition」Proteins 36:357−69;Zhang(1998年)「Extracting contact energies from protein structures:a study using a simplified model」Proteins 31:299−308;ならびに、Miyazawa,S.およびJernigan,R.L.(1996年)「Residue−residue potentials with a favorable contact pair term and an unfavorable high packing density term,for simulation and threading.」J.Mol.Biol 256:623−464を参照)。
【0174】
また、本発明者らは、はるかに計算力を必要とするアルゴリズムも試験した。そのアルゴリズムにおいて、キメラ配列が基準構造を通して縦列され、その後、ケミカル・コンピューティング・グループ社のモレキュラー・オペレーティング・エンバイロメント(Molecular Operating Environment)によるAMBER94
力場を用いたエネルギー最小化ルーチンにかけられる。この方法を用いた初期結果は、はるかに単純で計算がより速いΔEc計算と、有意に異なっていなかった。
【0175】
明快さと、理解とを目的として、上述の発明をある程度詳細に記載したが、この開示を読むことにより、本発明の本当の範囲から逸脱することなく、形態および詳細に様々な改変をおこない得ることが当業者に明確になる。例えば、上述のすべての技術、方法、組成物、装置、およびシステムは、様々な組合せで用いられ得る。この出願で引用されたすべての刊行物、特許、特許出願、または他の書類は、それぞれの個々の公表、特許、特許出願、または他の書類が、すべての目的のために引用によって組み込まれるように個別に示された場合と同じ程度で、すべての目的のために、参考としてその全体が組み込まれる。
【特許請求の範囲】
【請求項1】
本明細書中に記載される発明。
【請求項1】
本明細書中に記載される発明。
【図1A】

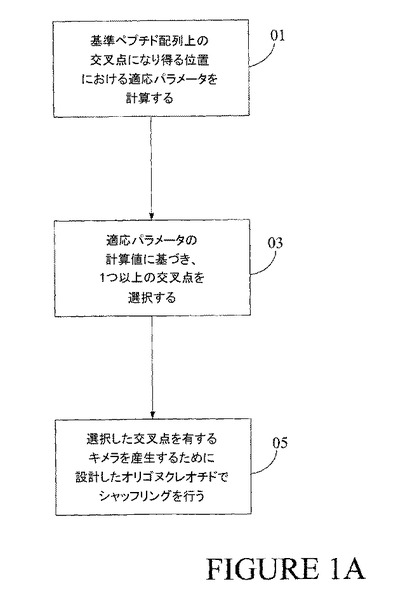
【図1B】

【図1C】
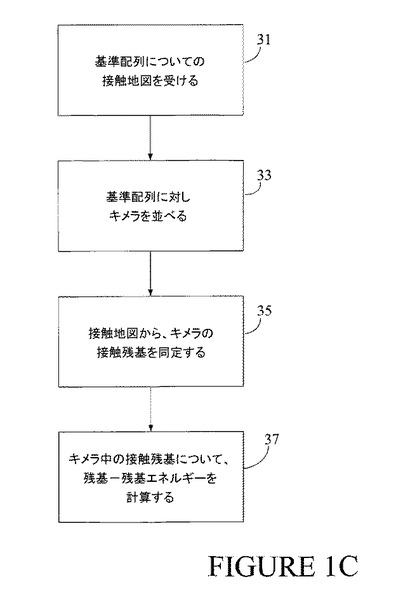
【図2】
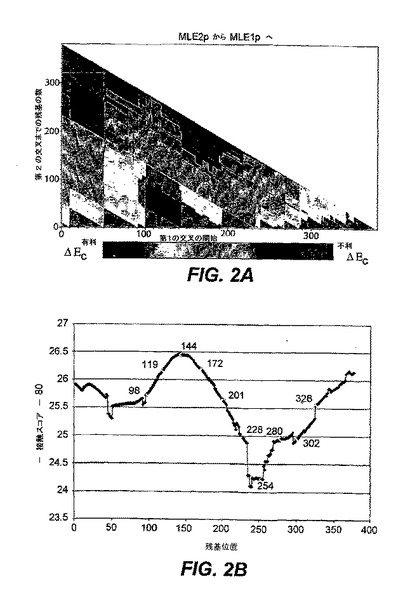
【図1B】
【図1C】
【図2】
【公開番号】特開2011−217751(P2011−217751A)
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−112828(P2011−112828)
【出願日】平成23年5月19日(2011.5.19)
【分割の表示】特願2003−576577(P2003−576577)の分割
【原出願日】平成15年3月10日(2003.3.10)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.Linux
2.ウィンドウズ
【出願人】(500382048)マキシジェン, インコーポレイテッド (17)
【出願人】(511122558)
【Fターム(参考)】
【公開日】平成23年11月4日(2011.11.4)
【国際特許分類】
【出願番号】特願2011−112828(P2011−112828)
【出願日】平成23年5月19日(2011.5.19)
【分割の表示】特願2003−576577(P2003−576577)の分割
【原出願日】平成15年3月10日(2003.3.10)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.Linux
2.ウィンドウズ
【出願人】(500382048)マキシジェン, インコーポレイテッド (17)
【出願人】(511122558)
【Fターム(参考)】
[ Back to top ]