
導電性樹脂組成物および成形体
ポリアミド(A)、ポリフェニレンエーテル(B)(末端未変性ポリフェニレンエーテル及び/又は末端変性ポリフェニレンエーテル)、ゴム状重合体(C)、導電性炭素材料(D)(カーボンブラック及び/又はカーボンフィブリル)、及び低分子量変性剤化合物(E)(相溶化剤)を含んでなり、(A)〜(E)の混合物を溶融混練することによって製造され、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物。(1)(D)の量が、(A)〜(E)の合計質量に対して0.2〜3質量%。(2)(E)の量が、(A)〜(E)の合計質量に対して0.01質量%より大きく0.20質量%未満。(3)(B)中に存在する揮発性物質の量(a)が式:0≦a≦−7.3×E+1.83(Eは成分(E)の量(質量%))を満足し、量(a)は、(B)を180℃で1時間真空乾燥に付した際の、(B)の質量減少率(質量%)である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は導電性樹脂組成物に関する。更に詳細には、本発明は、ポリアミド(A)、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することにより得られる導電性樹脂組成物であって、該混合物中の該導電性炭素材料(D)及び該低分子量変性剤化合物(E)の量が特定の範囲内にあり、且つ該ポリフェニレンエーテル(B)中に存在する揮発性物質の量が特定の範囲内にある、ことを特徴とする導電性樹脂組成物に関する。本発明の導電性樹脂組成物は、耐衝撃性及び熱安定性に優れるのみならず、押出成形時の目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)の発生及びストランド切れを抑制することができるので、電気・電子部品、OA部品、車両部品、機械部品などの幅広い分野に使用することができる。特に、本発明の導電性樹脂組成物は、大型成形体の製造に用いた場合でも、成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れるため、自動車外板のような静電塗装を施す大型成形体の製造に非常に有利に用いることができる。
【背景技術】
【0002】
ポリフェニレンエーテルは機械的性質・電気的性質及び耐熱性が優れており、しかも寸法安定性に優れるため幅広い用途で使用されているが、単独では成形加工性に劣っている。日本国特公昭45−997号公報(米国特許第3,379,792号に対応)には、これを改良するためにポリフェニレンエーテルにポリアミドを配合する技術が提案されており、現在では非常に多種多様な用途に使用される材料となっている。
【0003】
最近になって、静電塗装可能な材料として、導電性を付与したポリアミド−ポリフェニレンエーテルアロイを用いた自動車の外装材(フェンダー・ドアパネル等)への用途展開が急速に進んでいる。例えば、ポリアミド−ポリフェニレンエーテルアロイで成形して製造された自動車フェンダーを用いることにより、自動車の安全性(歩行者保護)や変形回復性を向上させることが試みられている。
【0004】
自動車の外装材料には、静電塗装可能なレベルの導電性、衝撃強度、耐熱性、流動性等の種々の特性が要求される。
【0005】
ポリアミド−ポリフェニレンエーテルアロイに導電性を付与する技術としては、例えば、日本国特開平2−201811号公報には、カーボンブラックを主としてポリアミド相中に含有させること、また予めカーボンブラックをポリアミド中へ均一分散させた後にポリフェニレンエーテルと混合することにより、表面抵抗を低下させる技術が開示されている。
【0006】
日本国特開平8−48869号公報(米国特許第5,977,240号に対応)には、予めポリアミド−ポリフェニレンエーテルを相溶化させた後、カーボンブラックを配合する技術により、良好な衝撃強さ、良好な流動性および良好な(低い)体積抵抗率を有する組成物が得られることが開示されている。
【0007】
また、日本国特開平4−300956号公報(EP506386に対応)には、ポリフェニレンエーテル、ポリアミド組成物にカーボンブラックを特定量添加すること、及びポリフェニレンエーテル及びポリアミドの配合比率および特定の相対粘度を有するものを用いることにより導電性及び加工性を良好にすることが記載されている。
【0008】
国際公開第01/81473号パンフレットには、樹脂組成物中の導電性炭素系フィラー(ケッチェンブラック)をポリフェニレンエーテル相中に粒状で存在させる技術が開示されている。また、日本国特表2002−544308号公報(米国特許第6,221,283号に対応)には、相溶化剤の量により連続相ポリマー中での分散相のポリマーの粒度を変化させることによって、導電性付与剤を含有する組成物の体積抵抗率を制御する方法が開示されている。また、日本国特開2001−302904号公報には、固相状態で変性されたポリフェニレンエーテルを用いてポリアミドと無機フィラー(カーボンブラック等)を溶融混練することにより、表面外観、耐衝撃性および耐熱性を有する組成物が得られることが開示されている。
【0009】
しかしながら、上述した技術では、導電性フィラーを添加するため、大型押出機で長時間製造した際に、目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)が発生しやすい。また自動車外板のような大型成形体の成形時には、成形機内滞留時の熱安定性が悪化し、成形条件幅も狭く、生産性を大きく低下させる要因となっている。
【0010】
さらに静電塗装性を要求される用途においては、塗装密着性及び塗装後外観の鮮映性を確保するために成形体外観の品質を確保することが重要である。特に、自動車フェンダーの中には、オンラインによる静電塗装後、金属パネルに塗布された錆止め塗料を硬化させるために、金属パネルと共に樹脂成形体も熱処理工程を通されることになる。これらの熱処理工程は、約170℃〜200℃またはそれ以上の温度で10〜50分程度暴露されるのが通例であり、使用される樹脂には耐熱性、熱時剛性と共に、成形後の塗装密着性のみならず、熱暴露後の塗装密着性が特に要求されることになる。
【0011】
しかしながら、上述した技術では、自動車フェンダーに要求される衝撃性、剛性、導電性を高次にバランスさせ、さらに成形機内滞留時の熱安定性および熱暴露後の塗装密着性を向上させることができなかった。そのため、新たな技術の開発が待望されているのが現状であった。
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、押出成形時の目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)の発生及びストランド切れを抑制することができるだけでなく、大型成形体の製造に用いた場合でも、成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れる導電性樹脂組成物を提供することにある。
【0013】
本発明の他の1つの目的は、上記の導電性樹脂組成物からなる成形体(自動車外板など)を提供することにある。
【課題を解決するための手段】
【0014】
発明の概要
本発明者らは、上記課題を解決するために検討を行った結果、驚くべきことに、ポリアミド(A)、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することにより得られる導電性樹脂組成物であって、該混合物中の該導電性炭素材料(D)及び該低分子量変性剤化合物(E)の量が特定の範囲内にあり、且つ該ポリフェニレンエーテル(B)中に存在する揮発性物質の量が特定の範囲内にある、ことを特徴とする導電性樹脂組成物によって、上記の課題を解決できることを見出した。この知見に基づいて、本発明を完成した。
【0015】
本発明の上記及びその他の諸目的、諸特徴ならびに諸利益は、添付の図面を参照しながら行う以下の詳細な説明及び請求の範囲の記載から明らかになる。
【発明の効果】
【0016】
発明の詳細な説明
本発明によれば、ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物が提供される。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【0017】
次に、本発明の理解を容易にするために、本発明の基本的特徴及び好ましい諸態様を列挙する。
【0018】
1.ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【0019】
2.該成分(A)〜(E)の混合物中における該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.02〜0.18質量%であることを特徴とする前項1に記載の樹脂組成物。
【0020】
3.該成分(A)〜(E)の混合物中における該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して、0.02〜0.15質量%であることを特徴とする前項1に記載の樹脂組成物。
【0021】
4.該成分(A)〜(E)の混合物中における該導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.5〜2.5質量%であることを特徴とする前項1〜3のいずれかに記載の樹脂組成物。
【0022】
5.該揮発性物質の量(a)が、1.0質量%以下であることを特徴とする前項1〜4のいずれかに記載の樹脂組成物。
【0023】
6.該成分(D)が、導電性カーボンブラックであることを特徴とする前項1〜5のいずれかに記載の樹脂組成物。
【0024】
7.該成分(D)としての導電性カーボンブラックのジブチルフタレート(DBP)吸収量が250ml/100g以上であることを特徴とする前項1〜6のいずれかに記載の樹脂組成物。
【0025】
8.該第1の低分子量変性剤化合物(b)及び該第2の低分子量変性剤化合物(E)の各々がそれぞれ独立して無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸から選ばれる少なくとも一種の化合物であることを特徴とする前項1〜7のいずれかに記載の樹脂組成物。
【0026】
9.該ゴム状重合体(C)が、少なくとも1種の、数平均分子量が120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物であって、該ブロック共重合体(C−1)及び(C−2)の各々が、それぞれ独立して、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックを含んでなることを特徴とする前項1〜8のいずれかに記載の樹脂組成物。
【0027】
10.該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
を含み、
該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%であることを特徴とする前項9に記載の樹脂組成物。
【0028】
11.該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることを特徴とする前項1〜10のいずれかに記載の樹脂組成物。
【0029】
12.該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、55,000〜70,000であり、かつ分子量分布が、3.0以下であることを特徴とする前項11に記載の樹脂組成物。
【0030】
13.該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法で行うことを特徴とする前項1〜12のいずれかに記載の樹脂組成物。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。
【0031】
14.該マスターバッチが、該成分(D)として導電性カーボンブラックを含んでなり、該マスターバッチを、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在するマスターバッチであることを特徴とする前項13に記載の樹脂組成物。
【0032】
15.該成分(A)〜(E)の混合物が更に少なくとも1種の無機充填材を、該成分(A)〜(E)の合計100質量部に対して、5〜25質量部含むことを特徴とする前項1〜14のいずれかに記載の樹脂組成物。
【0033】
16.成形体である前項1〜15のいずれかに記載の樹脂組成物。
【0034】
17.204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることを特徴とする前項16に記載の樹脂組成物。
【0035】
18.該少なくとも1種の低分子量成分の量が、0.10以下であることを特徴とする前項17に記載の樹脂組成物。
【0036】
19.オンライン塗装されてなる自動車フェンダーであることを特徴とする前項16〜18に記載の樹脂組成物。
【0037】
本発明の導電性樹脂組成物は、ポリアミド(A)、末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することによって製造される樹脂組成物である。
【0038】
以下に、本発明の導電性樹脂組成物を構成する各成分について詳しく述べる。
【0039】
本発明においてポリアミド(A)として使用することのできるポリアミドの種類としては、ポリマー主鎖中にアミド結合{−NH−C(=O)−}を有するものであれば、いずれも使用することができる。
【0040】
一般にポリアミドは、ラクタム類の開環重合、ジアミンとジカルボン酸の重縮合、アミノカルボン酸の重縮合などによって得られるが、これらに限定されるものではない。
【0041】
上記ジアミンとしては、脂肪族、脂環式および芳香族ジアミンが挙げられ、具体例としては、テトラメチレンジアミン、ヘキサメチレンジアミン、ウンデカメチレンジアミン、ドデカメチレンジアミン、トリデカメチレンジアミン、2,2,4−トリメチルヘキサメチレンジアミン、2,4,4−トリメチルヘキサメチレンジアミン、5−メチルノナメチレンジアミン、1,3−ビスアミノメチルシクロヘキサン、1,4−ビスアミノメチルシクロヘキサン、m−フェニレンジアミン、p−フェニレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン等が挙げられる。
【0042】
ジカルボン酸としては、脂肪族、脂環式および芳香族ジカルボン酸が挙げられ、具体例としては、アジピン酸、スベリン酸、アゼライン酸、セバシン酸、ドデカン二酸、1,1,3−トリデカン二酸、1,3−シクロヘキサンジカルボン酸、テレフタル酸、イソフタル酸、ナフタレンジカルボン酸、ダイマー酸などが挙げられる。
【0043】
ラクタム類としては、具体的にはε−カプロラクタム、エナントラクタム、ω−ラウロラクタムなどが挙げられる。
【0044】
また、アミノカルボン酸としては、具体的にはε−アミノカプロン酸、7−アミノヘプタン酸、8−アミノオクタン酸、9−アミノノナン酸、11−アミノウンデカン酸、12−アミノドデカン酸、13−アミノトリデカン酸などが挙げられる。
【0045】
本発明においては、これらラクタム類、ジアミン、ジカルボン酸、ω−アミノカルボン酸を、単独で重合して得られるポリアミド、及び二種以上の混合物にして重縮合を行って得られる共重合ポリアミドのいずれもが使用できる。
【0046】
また、これらラクタム類、ジアミン、ジカルボン酸、ω−アミノカルボン酸を重合反応機内で低分子量のオリゴマーの段階まで重合し、押出機等で高分子量化したものも好適に使用することができる。
【0047】
特に本発明で有用に用いることのできるポリアミド樹脂としては、ポリアミド6、ポリアミド6,6、ポリアミド4,6、ポリアミド11、ポリアミド12、ポリアミド6,10、ポリアミド6,12、ポリアミド6/6,6、ポリアミド6/6,12、ポリアミドMXD(m−キシリレンジアミン)/6、ポリアミド6,T、ポリアミド6,I、ポリアミド6/6,T、ポリアミド6/6,I、ポリアミド6,6/6,T、ポリアミド6,6/6,I、ポリアミド6/6,T/6,I、ポリアミド6,6/6,T/6,I、ポリアミド6/12/6,T、ポリアミド6,6/12/6,T、ポリアミド6/12/6,I、ポリアミド6,6/12/6,Iなどが挙げられ、複数のポリアミドを押出機等で共重合化したポリアミド類も使用することができる。
【0048】
好ましいポリアミドは、ポリアミド6、ポリアミド6,6、ポリアミド6/6,6、ポリアミド6/6,I及び、それらの混合物である。
【0049】
本発明で使用されるポリアミド樹脂の好ましい数平均分子量は5,000〜100,000であり、より好ましくは10,000〜30,000である。
【0050】
本発明におけるポリアミド樹脂はこれらに限定されるものではなく、分子量の異なる複数のポリアミド樹脂の混合物であっても良い。例えば数平均分子量15,000未満の低分子量ポリアミドと、15,000以上の高分子量ポリアミドとの混合物等である。
【0051】
ポリアミドの末端基は、ポリフェニレンエーテルとの反応に関与する。ポリアミド樹脂は末端基として一般にアミノ基、カルボキシル基を有しているが、一般的にカルボキシル基濃度が高くなると、耐衝撃性が低下し、流動性が向上し、逆にアミノ基濃度が高くなると耐衝撃性が向上し、流動性が低下する。
【0052】
本発明における、これらの好ましい比はアミノ基/カルボキシル基濃度比で、9/1〜1/9であり、より好ましくは8/2〜1/9、更に好ましくは6/4〜1/9である。
【0053】
また、末端のアミノ基の濃度としては少なくとも10ミリ当量/kgであることが好ましい。更に好ましくは30ミリ当量/kg以上である。
【0054】
これらポリアミド樹脂の末端基の調整方法は、公知の方法を用いることができる。例えばポリアミド樹脂の重合時に所定の末端濃度となるようにジアミン類やジカルボン酸類、モノカルボン酸類などを添加する方法、あるいは、末端基の比率が異なる2種類以上のポリアミド樹脂の混合物により調整する方法等が挙げられる。
【0055】
また、ポリアミド樹脂の耐熱安定性を向上させる目的で日本国特開平1−163262号公報に記載されているような金属系安定剤も使用することができる。
【0056】
これら金属系安定剤の中で特に好ましく使用することのできるものとしては、CuI、CuCl2、酢酸銅、ステアリン酸セリウム等が挙げられる。また、ヨウ化カリウム、臭化カリウム等に代表されるアルカリ金属のハロゲン化塩も金属系安定剤として好適に使用することができる。これらは、もちろん併用添加しても構わない。
【0057】
金属系安定剤の好ましい配合量は、ポリアミド樹脂100質量部に対して、0.001〜1質量部である。
【0058】
さらに、上記の他にポリアミドに添加することが可能な公知の添加剤等もポリアミド100質量部に対して10質量部未満の量で添加してもかまわない。
【0059】
本発明で使用できるポリフェニレンエーテル(B)とは、下記式(1)の構造単位からなる、単独重合体及び/または共重合体である。
【0060】
【化1】

【0061】
〔式中、Oは酸素原子、各Rは、それぞれ独立して、水素、ハロゲン、第一級もしくは第二級のC1〜C7アルキル基、フェニル基、C1〜C7ハロアルキル基、C1〜C7アミノアルキル基、C1〜C7ヒドロカルビロキシ基、又はハロヒドロカルビロキシ基(但し、少なくとも2個の炭素原子がハロゲン原子と酸素原子を隔てている)を表す。〕
【0062】
本発明で使用できるポリフェニレンエーテルの具体的な例としては、例えば、ポリ(2,6−ジメチル−1,4−フェニレンエーテル)、ポリ(2−メチル−6−エチル−1,4−フェニレンエーテル)、ポリ(2−メチル−6−フェニル−1,4−フェニレンエーテル)、ポリ(2,6−ジクロロ−1,4−フェニレンエーテル)等が挙げられ、さらに2,6−ジメチルフェノールと他のフェノール類との共重合体(例えば、日本国特公昭52−17880号公報に記載されているような2,3,6−トリメチルフェノールとの共重合体や2−メチル−6−ブチルフェノールとの共重合体)のようなポリフェニレンエーテル共重合体も挙げられる。
【0063】
これらの中でも特に好ましいポリフェニレンエーテルは、ポリ(2,6−ジメチル−1,4−フェニレンエーテル)、2,6−ジメチルフェノールと2,3,6−トリメチルフェノールとの共重合体、またはこれらの混合物である。
【0064】
本発明で用いるポリフェニレンエーテルの製造方法は特に限定されるものではなく、公知の方法が使用でき、例えば、米国特許第3306874号明細書、同第3306875号明細書、同第3257357号明細書及び同第3257358号明細書、日本国特開昭50−51197号公報及び同63−152628号公報等に記載されている製造方法等を挙げることができる。
【0065】
本発明で使用することのできるポリフェニレンエーテルの還元粘度(ηsp/c:0.5g/dl、クロロホルム溶液、30℃測定)は、0.15〜0.70dl/gの範囲であることが好ましく、さらに好ましくは0.20〜0.60dl/gの範囲、より好ましくは0.40〜0.55dl/gの範囲である。ポリフェニレンエーテルの還元粘度が、0.70dl/gより高いと溶融流動性が低下する傾向にあり、0.15dl/gより低いと、機械的物性が低下する傾向にある。
【0066】
本発明においては、2種以上の還元粘度の異なるポリフェニレンエーテルをブレンドしたものであっても、何ら問題なく使用することができる。例えば、還元粘度0.45dl/g以下のポリフェニレンエーテルと還元粘度0.50dl/g以上のポリフェニレンエーテルの混合物、還元粘度0.40dl/g以下の低分子量ポリフェニレンエーテルと還元粘度0.50dl/g以上のポリフェニレンエーテルの混合物等が挙げられるが、もちろん、これらに限定されることはない。
【0067】
また、本発明において使用するポリフェニレンエーテル(B)は、末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種である。
【0068】
また、ポリフェニレンエーテル(B)としては、末端未変性ポリフェニレンエーテルと末端変性ポリフェニレンエーテルの混合物であることが成形機内滞留時の熱安定性を向上させるために好ましい。特に、末端変性ポリフェニレンエーテルの量より末端未変性ポリフェニレンエーテルの量の方が多いことが流動性、成形機内滞留時の熱安定性の観点からより好ましい。具体的には、ポリフェニレンエーテル(B)が上記の混合物である場合、末端未変性ポリフェニレンエーテルの量が、上記混合物の質量に対して55質量%以上であることが好ましい。また、末端未変性ポリフェニレンエーテルの還元粘度(0.5g/dlクロロホルム溶液、30℃測定)が、末端変性ポリフェニレンエーテルの還元粘度より高いことがさらに好ましい。
【0069】
ここでいう末端変性ポリフェニレンエーテルとは、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の第1の低分子量変性剤化合物(b)で変性されたポリフェニレンエーテルを指す。この第1の低分子量変性剤化合物(b)は、本発明の組成物において、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する。
【0070】
上記の第1の低分子量変性剤化合物(b)(相溶化剤)について以下に具体的に説明する。
【0071】
分子内に炭素−炭素二重結合と、カルボン酸基及び/又は酸無水物基とを同時に有する変性剤化合物としては、マレイン酸、フマル酸、クロロマレイン酸、シス−4−シクロヘキセン−1,2−ジカルボン酸及びこれらの酸無水物などが挙げられる。これらの内、フマル酸、マレイン酸、無水マレイン酸が好ましく、フマル酸、無水マレイン酸が特に好ましい。
【0072】
また、これら不飽和ジカルボン酸のカルボキシル基の1個または2個がエステルになっているものも使用可能である。
【0073】
分子内に炭素−炭素二重結合とグリシジル基とを同時に有する低分子量変性剤化合物としては、アリルグリシジルエーテル、グリシジルアクリレート、グリシジルメタアクリレート、エポキシ化天然油脂等が挙げられる。
【0074】
これらの中でグリシジルアクリレート、グリシジルメタアクリレートが特に好ましい。
【0075】
分子内に炭素−炭素二重結合と水酸基とを同時に有する低分子量変性剤化合物としては、アリルアルコール、4−ペンテン−1−オール、1,4−ペンタジエン−3−オールなどの一般式CnH2n−3OH(nは正の整数)の不飽和アルコール、一般式CnH2n−5OH、CnH2n−7OH(nは正の整数)等の不飽和アルコール等が挙げられる。
【0076】
上述した低分子量変性剤化合物は、それぞれ単独で用いても良いし、2種以上を組み合わせて用いても良い。
【0077】
上記のような第1の低分子量変性剤化合物(b)を用いたポリフェニレンエーテルの末端変性は、第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと、ラジカル開始剤の存在下又は非存在下で該末端未変性ポリフェニレンエーテルのガラス転移温度以上360℃以下の範囲の温度で溶融混練することによって行なう。溶融混錬の際、押出機下流のベント脱気口より真空脱気することにより、未反応の第1の低分子量変性剤化合物(b)が、末端変性されたポリフェニレンエーテルの質量に対し、0.01質量%未満になるようにすることが好ましい。その際、溶融混練時のベント脱気真空度は、2.1×104Pa以下であることが特に好ましい。また、溶融混練後の末端変性ポリフェニレンエーテルペレットを減圧加熱乾燥または溶剤洗浄することにより、未反応の第1の低分子量変性剤化合物(b)が、0.01質量%未満になるようにしても構わない。
【0078】
ポリフェニレンエーテルの末端変性を行なう際の第1の低分子量変性剤化合物(b)の添加量は、該末端未変性ポリフェニレンエーテル100質量部に対して0.1〜3質量部が好ましく、更に好ましくは0.3〜2質量部である。
【0079】
上記ラジカル開始剤の例としては、公知の有機過酸化物、ジアゾ化合物などが挙げられる。具体例としては、ベンゾイルパーオキサイド、ジクミルパーオキサイド、ジ−tert−ブチルパーオキサイド、tert−ブチルクミルパーオキサイド、tert−ブチルハイドロパーオキサイド、クメンハイドロパーオキサイド、アゾビスイソブチロニトリルなどが挙げられる。
【0080】
ラジカル開始剤を用いて末端変性されたポリフェニレンエーテルを製造する際の好ましいラジカル開始剤の量は、ポリフェニレンエーテル100質量部に対して0.001〜1質量部である。
【0081】
また、末端変性ポリフェニレンエーテル中の変性剤化合物の付加率は、末端変性前のポリフェニレンエーテルの質量に対して、0.01〜5質量%が好ましい。より好ましくは0.1〜3質量%である。
【0082】
また、末端変性ポリフェニレンエーテル中に残存する未反応の変性剤化合物の量を減少させるために、末端変性を行なう際に、必要に応じてアミド結合及び/またはアミノ基を有する化合物を添加しても構わない。
【0083】
ここでいうアミド結合を有する化合物とは、分子構造中にアミド結合{−NH−C(=O)−}構造を有する化合物であり、アミノ基を有する化合物とは末端に{−NH2}構造を有する化合物である。これら化合物の具体例としては、オクチルアミン、ノニルアミン、テトラメチレンジアミン、ヘキサメチレンジアミン等の脂肪族アミン類、アニリン、m−フェニレンジアミン、p−フェニレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン等の芳香族アミン類、上記アミン類とカルボン酸、ジカルボン酸等との反応物、ε−カプロラクタム等のラクタム類及びポリアミド樹脂等が挙げられるが、これらに限定されるものではない。
【0084】
これらアミド結合またはアミノ基を有する化合物を添加する際の好ましい添加量は、末端未変性ポリフェニレンエーテル100質量部に対して、0.001質量部以上、5質量部以下である。好ましくは0.01質量部以上、1質量部以下、より好ましくは0.01質量部以上、0.1質量部以下である。
【0085】
また、本発明にお該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(成分(A)〜(E)の合計質量に対する質量%)を表す)
を満足する必要がある。
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(該ポリフェニレンエーテル(B)の質量に対する、上記真空乾燥において揮発した成分の質量%)であって下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
真空乾燥は、例えば、日本国いすゞ製作所製の真空定温恒温器CVK−23PSを用いて行なうことができる。
【0086】
更に、本発明においては、該揮発性物質の量が、上記の式を満足し、且つ使用するポリフェニレンエーテル(B)100質量%に対し、1.0質量%以下の量であることが好ましい。より好ましくは、0.6質量%以下、さらに好ましくは0.2質量%以下の量である。
【0087】
使用するポリフェニレンエーテル(B)中の揮発性物質は、重合後の乾燥工程における温度と時間を調整することにより制御することが可能である。本発明に使用するポリフェニレンエーテル(B)中の揮発性物質の量の具体的な制御方法に関しては、日本国特開2002−3596号公報や日本国特開昭64−33131号公報などの記載に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し乾燥し得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機に用いて、揮発性物質が所定の量になるように乾燥時間を決定することができる。しかしながら、上記の方法に限定されるものではなく、使用するポリフェニレンエーテル中の揮発性物質の量が上記の範囲を満足すればどの様な方法を用いてもよい。
【0088】
ここでいう揮発性物質としては、重合溶媒、残留モノマーおよびオリゴマー類である。また、上記の重合溶媒としては、トルエン、キシレンの各異性体、エチルベンゼン、炭素数1〜5のアルコール類、クロロホルム、ジクロルメタン、クロルベンゼン、ジクロルベンゼン等の有機溶媒が挙げられる。また、これらの有機溶媒の内の2種以上の混合物である場合もある。
【0089】
また、本発明においては該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることが好ましく、Mwが55,000以上、かつMw/Mnが3.0以下であることがより好ましく、Mwが55,000〜70,000、かつMw/Mnが3.0以下であることが特に好ましい。
【0090】
上述した組成物中のポリフェニレンエーテル(B)の分子量(Mw)の測定は、以下の手順により容易に可能である。測定に充分な量の組成物を細かく粉砕しクロロホルムでソックスレー抽出する。得られたクロロホルム溶液をゲル浸透クロマトグラフィー測定装置と紫外分光検出器を用いて分析し、標準ポリスチレンで換算した分子量データを得る。得られた分子量データを市販のGPC処理ソフト(例えば、日本国システムインスツルメンツ(SIC)製480データステーション)を用いて特定分子量で分画しその量を計算する。この時、同時に溶出してくるブロック共重合体(成分(C)として用いるもの)などの他の重合体を検出しないため測定する紫外線波長をブロック共重合体などの他の重合体の吸収がない波長に設定することが重要である。[測定は、例えば、以下の装置及び条件を用いて行なうことができる。GPC装置:GPC SYSTEM21:日本国昭和電工(株)製、検出器:UV−41:日本国昭和電工(株)製、溶媒:クロロホルム、温度:40℃、カラム:サンプル側(日本国昭和電工(株)製K−G,K−800RL,K−800R)、リファレンス側(日本国昭和電工(株)製K−805L×2本)、流量10ml/分、測定波長:283nm、圧力15〜17kg/cm2]]
【0091】
次に、本発明で用いる成分(D)について説明する。成分(D)は、導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料である。
【0092】
本発明において使用できる導電性カーボンブラックについて説明する。本発明で使用する導電性カーボンブラックは、ジブチルフタレート(DBP)吸収量が250ml/100g以上のものが好ましく、より好ましくはDBP吸収量が350ml/100g以上、更に好ましくは400ml/100g以上のカーボンブラックである。ここで言うDBP吸収量とは、ASTM D2414に定められた方法で測定した値である。DBP吸収量が上記の範囲外であると、組成物の導電性が低下する傾向がある。
【0093】
また、本発明において使用できる導電性カーボンブラックはBET表面積が200cm2/g以上のものが好ましく、更には400cm2/g以上のものがより好ましい。BET表面積が上記の範囲外であると、組成物の導電性が低下する傾向がある。市販されているものを例示すると、日本国ケッチェンブラックインターナショナルのケッチェンブラックECやケッチェンブラックEC−600JD等が挙げられる。
【0094】
本発明において使用できる導電性カーボンフィブリルとしては、米国特許第4663230号明細書、米国特許第5165909号明細書、米国特許第5171560号明細書、米国特許第5578543号明細書、米国特許第5589152号明細書、米国特許第5650370号明細書、米国特許第6235674号明細書等に記載されている平均繊維径が75nm未満で中空構造をした分岐の少ない炭素系繊維を言う。また、1μm以下のピッチでらせんが一周するコイル状形状のものも含まれる。市販されているものとしては、米国ハイペリオンキャタリスト社のハイペリオンが挙げられる。
【0095】
本発明において使用できる導電性カーボンブラック及び/又は導電性カーボンフィブリルの添加量は、成分(A)〜(E)の合計質量に対して、0.2〜3質量%である。0.5〜2.5質量%であると、耐衝撃性と流動性と導電性のバランスに優れ、より好ましい。さらに好ましくは0.7〜2質量%である。
【0096】
本発明において用いる導電性カーボンブラック及び/又は導電性カーボンフィブリルは、予めポリアミドと導電性カーボンブラック及び/又は導電性カーボンフィブリルを溶融混練することによって得られたマスターバッチの形態で添加されることが好ましく、ペレット状、粉末状、粒状のいずれでもかまわない。
【0097】
上記のマスターバッチ中の該導電性炭素材料(D)の比率は、該マスターバッチの質量に対して、好ましくは5〜25質量%である。より好ましくは6〜15質量%、さらに好ましくは6〜10質量%である。
【0098】
特に、上記マスターバッチは、米国特許US2004/0082729号に開示された導電性マスターバッチであることが好ましい。例えば、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在することを特徴とする導電性マスターバッチであることが好ましい。
【0099】
マスターバッチの好ましい製造方法としては、二軸押出機またはニーダーを使用して溶融混練する方法が好ましい。中でも特にポリアミド(A)を溶融した後に該導電性炭素材料(D)を添加する方法が好ましく、具体例を挙げると、上流側と下流側にそれぞれ少なくとも1箇所の供給口を有する二軸押出機又はニーダーを使用し、上流側供給口よりポリアミド(A)を供給し、溶融させた後、下流側供給口より該導電性炭素材料(D)を添加して溶融混練する方法、あるいは、上流側供給口よりポリアミド(A)を供給し、溶融させた後、下流側供給口より該導電性炭素材料(D)とポリアミド(A)を同時添加して溶融混練する方法等が挙げられる。
【0100】
即ち、本発明においては、該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法によって行うことによって本発明の導電性組成物を得ることができる。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。溶融混練温度は特に限定されるものではないが、例えば、250℃以上、350℃以下の範囲から任意に選択することができる。
【0101】
本発明の導電性樹脂組成物は、スチレン系熱可塑性樹脂をポリアミド(A)とポリフェニレンエーテル(B)の合計100質量部に対し、50質量部未満の量であれば配合しても構わない。本発明でいうスチレン系熱可塑性樹脂としては、ホモポリスチレン、スチレン−アクリロニトリル共重合体(AS樹脂)等が挙げられる。
【0102】
更に、ポリフェニレンエーテル(B)に添加することが可能な公知の添加剤等もポリフェニレンエーテル100質量部に対して10質量部未満の量で添加しても構わない。添加剤の例としては、酸化亜鉛、硫化亜鉛などの金属系安定剤、及びヒンダードフェノール系安定剤、リン系安定剤、ヒンダードアミン系安定剤などの有機安定剤が挙げられる。
【0103】
本発明の導電性樹脂組成物に使用することができるゴム状重合体(C)は、例えば、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックを含むビニル芳香族炭化水素−共役ジエン化合物ブロック共重合体及びその水素添加物、エチレン−α−オレフィン共重合体、ゴム変性ポリスチレン(HIPS)、及びスチレン−ゴム質重合体−アクリロニトリル共重合体(ABS樹脂)からなる群より選ばれる1種以上である。
【0104】
本発明におけるビニル芳香族炭化水素−共役ジエン化合物ブロック共重合体において使用することができるビニル芳香族炭化水素の具体例としてはスチレン、α−メチルスチレン、ビニルトルエン等が挙げられ、これらから選ばれた1種以上の化合物が用いられるが、中でもスチレンが特に好ましい。
【0105】
また、共役ジエン化合物の具体例としては、ブタジエン、イソプレン、ピペリレン、1,3−ペンタジエン等が挙げられ、これらから選ばれた1種以上の化合物が用いられるが、中でもブタジエン、イソプレンおよびこれらの組み合わせが好ましい。
【0106】
ゴム状重合体の具体例としては、SBS(スチレン−ブタジエンブロック共重合体)、SEBS(スチレン−エチレン/ブチレンブロック共重合体)、SEP(スチレン−エチレン/プロピレンブロック共重合体)及びSEPS(スチレン−エチレン/プロピレン−スチレンブロック共重合体)などのブロック共重合体を挙げることができる。
【0107】
該ブロック共重合体の共役ジエン化合物のソフトセグメント部分のミクロ構造は1,2−ビニル含量もしくは1,2−ビニル含量と3,4−ビニル含量の合計量が5〜80%が好ましく、さらには10〜50%が好ましく、10〜40%が最も好ましい。
【0108】
上記ブロック共重合体は、ビニル芳香族炭化水素単量体単位を主体とする重合体ブロック[A]と共役ジエン単量体単位を主体とする重合体ブロック[B]がA−B型、A−B−A型、A−B−A−B型から選ばれる結合形式を有するブロック共重合体であることが好ましい。また、これらの混合物であってももちろん構わない。
【0109】
これらの中でもA−B−A型、A−B−A−B型がより好ましい。これらはもちろん混合物であっても構わない。
【0110】
また、本発明の導電性樹脂組成物に使用することのできるビニル芳香族炭化水素と共役ジエン化合物のブロック共重合体は、水素添加されたブロック共重合体であることがより好ましい。水素添加されたブロック共重合体とは、上述のビニル芳香族炭化水素と共役ジエン化合物のブロック共重合体を水素添加処理することにより、共役ジエン化合物を主体とする重合体ブロックの脂肪族二重結合を0を超えて100%の範囲で制御したものをいう。該水素添加されたブロック共重合体の好ましい水素添加率は50%以上であり、より好ましくは80%以上、最も好ましくは98%以上である。
【0111】
これらブロック共重合体は水素添加されていないブロック共重合体と水素添加されたブロック共重合体との混合物としても問題なく使用可能である。
【0112】
本発明の導電性樹脂組成物に使用するブロック共重合体(C)として、比較的低分子量のブロック共重合体と比較的高分子量ブロック共重合体との混合物を使用することが望ましい。具体的には、少なくとも1種の、数平均分子量が150,000未満、好ましくは120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が150,000以上、好ましくは200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物を用いることが好ましい。
【0113】
本発明でいう数平均分子量とは、ゲルパーミエーションクロマトグラフィー測定装置[例えば、GPC SYSTEM21:日本国昭和電工(株)製]を用いて、紫外分光検出器[例えば、UV−41:日本国昭和電工(株)製]で測定し、標準ポリスチレンで換算した数平均分子量のことを指す。測定条件は次のとおりである。
(測定条件)
溶媒:クロロホルム
温度:40℃
カラム:サンプル側(日本国昭和電工(株)製K−G、K−800RL、K−800R)
リファレンス側(日本国昭和電工(株)製K−805L×2本)
流量:10ml/分
測定波長:254nm
圧力;15〜17kg/cm2
【0114】
この時、重合時の触媒失活による低分子量成分が検出されることがあるが、その場合は分子量計算に低分子量成分は含めない。通常、計算された正しい分子量分布(重量平均分子量(Mw)/数平均分子量(Mn))は1.0〜1.2の範囲内である。
【0115】
これら比較的低分子量のブロック共重合体(C−1)と比較的高分子量のブロック共重合体(C−2)の質量比は、(C−1)/(C−2)=95/5〜5/95であることが好ましい。より好ましくは90/10〜10/90である。
【0116】
また、比較的低分子量のブロック共重合体(C−1)として、ビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数平均分子量が20,000以上であるブロック共重合体を使用することで、耐熱性を向上させるという付加的な効果を得ることができる。
【0117】
ビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数平均分子量は、上述したブロック共重合体の数平均分子量を用いて、下式により求めることができる。
Mn(α)={Mn×α/(α+β)}/N
[上式中において、Mn(α)はビニル芳香族炭化水素を主体とする重合体ブロックの数平均分子量、Mnはブロック共重合体の数平均分子量、αはブロック共重合体中のすべてのビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの質量%、βはブロック共重合体中のすべての共役ジエン単量体単位を主体とする重合体ブロックの質量%、そしてNはブロック共重合体中のビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数を表す。]
【0118】
また、本発明において、該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
(但し、該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%)
を用いることが好ましい。上記のようなブロック共重合体(C−1a)を用いることにより、樹脂組成物の耐熱性を向上させることができるため、より好適に使用することができる。
【0119】
更に、本発明において、上記のようなブロック共重合体(C−1a)と、ビニル芳香族炭化水素重合体ブロック含有量が、20質量%以上55質量%未満であるブロック共重合体(C−1b)との混合物とすることにより、樹脂組成物の溶融流動性を向上させることが可能となる。この際、ブロック共重合体(C−1a)と、ブロック共重合体(C−1b)との質量比(C−1a)/(C−1b)は、10/0〜2/8である事が好ましく、10/0〜4/6であることがより好ましい。
【0120】
また、本発明の導電性樹脂組成物に使用するゴム状重合体(C)中には、パラフィンを主成分とするオイルをあらかじめ混合したものを用いても構わない。パラフィンを主成分とするオイルをあらかじめ混合する事により、樹脂組成物の加工性を向上させることができる。ここで、パラフィンを主成分とするオイルの量としては、ゴム状重合体(C)の質量に対して50質量%以下が好ましく、30質量%以下がより好ましい。
【0121】
これらビニル芳香族炭化水素−共役ジエン化合物のブロック共重合体は、結合形式の異なるもの、ビニル芳香族炭化水素種の異なるもの、共役ジエン化合物種の異なるもの、1,2−結合ビニル含有量もしくは1,2−結合ビニル含有量と3,4−結合ビニル含有量の異なるもの、ビニル芳香族炭化水素成分含有量の異なるもの、水素添加率の異なるもの等混合して用いても構わない。
【0122】
本発明の導電性樹脂組成物に使用することのできるエチレン−α−オレフィン共重合体の例としては、日本国特開2001−302911号公報に記載されているエチレン−α−オレフィン共重合体が挙げられる。
【0123】
また、本発明の樹脂組成物に使用するゴム状重合体(C)は、全部又は一部が変性されたゴム状重合体であっても構わない。
【0124】
ここでいう変性されたゴム状重合体とは、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の第3の低分子量変性剤化合物(c)で変性されたゴム状重合体を指す。第3の低分子量変性剤化合物(c)としては、上記第1の低分子量変性剤化合物(b)としては、例示したものを用いることができる。
【0125】
本発明の樹脂組成物を製造する際の、上記成分(A)〜(E)の混合物におけるポリアミド(A)、ポリフェニレンエーテル(B)及びゴム状重合体(C)の好ましい量比は、これら3成分の合計を100質量部としたとき、ポリアミド(A)30〜70質量部、ポリフェニレンエーテル(B)20〜50質量部、ゴム状重合体(C)5〜30質量部の範囲内である。より好ましくは、ポリアミド(A)40〜60質量部、ポリフェニレンエーテル(B)30〜40質量部、ゴム状重合体(C)5〜15質量部の範囲内である。
【0126】
本発明において使用することができるポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)は、ポリアミド(A)及びポリフェニレンエーテル(B)と相互作用する多官能性の低分子量変性剤化合物を指すものである。本発明の組成物においては、第2の低分子量変性剤化合物(E)によって、ポリアミド(A)とポリフェニレンエーテル(B)との相溶性が改良されている。
【0127】
本発明において使用することができる相溶化剤として機能する第2の低分子量変性剤化合物(E)の例としては、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の低分子量変性剤化合物が挙げられる。
【0128】
これらの相溶化剤として機能する第2の低分子量変性剤化合物(E)の中でも、無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸が好ましい。特に好適な相溶化剤としては、マレイン酸、無水マレイン酸、クエン酸である。これらを混合して用いても構わない。尚、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)とは同じ化合物を用いても良いし、異なる化合物を用いても良い。また、上記の変性されたゴム状重合体をゴム状重合体(C)として用いる場合、上記第3の低分子量変性剤化合物(c)は、第1の低分子量変性剤化合物(b)及び/又は第2の低分子量変性剤化合物(E)と同じでも異なっていてもよい。
【0129】
ポリフェニレンエーテル(B)として末端変性ポリフェニレンエーテルを使用する場合は、末端変性ポリフェニレンエーテル中に含まれる未反応の低分子量変性剤化合物も成分(E)とみなす。また、上記第3の低分子量変性剤化合物(c)として、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤としても機能する低分子量変性剤化合物を用いて変性されたゴム状重合体をゴム状重合体(C)として用いる際に、該変性されたゴム状重合体が未反応の第3の低分子量変性剤化合物(c)を含む場合、この未反応の第3の低分子量変性剤化合物(c)も成分(E)とみなす。
【0130】
本発明において使用することができる変性剤(E)(相溶化剤)の添加量は、成分(A)〜(E)の合計質量に対して、0.01質量%より大きく0.20質量%未満であり、より好ましくは、0.02〜0.18質量%、さらに好ましくは0.02〜0.15質量%、最も好ましくは0.03〜0.10質量%の範囲である。変性剤(E)(相溶化剤)の添加量が0.20質量%以上であると、成形機内滞留時の熱安定性、熱暴露後の塗装性が悪化し、0.01重量%以下では、耐衝撃性、塗装性(成形後、熱暴露後)が悪化する。
【0131】
従来技術においては、ポリアミド/ポリフェニレンエーテル/カーボンブラック系組成物の製造において、変性剤は衝撃強度と導電性の発現バランスを考慮して比較的多くの量(例えば、0.2質量%以上)添加されてた。これに対して、本発明者らは、変性剤の量を従来より少なくすると共に、ポリフェニレンエーテル中の揮発性物質の量を上記の特定の範囲にすれば、衝撃性、導電性だけでなく、成形機内滞留時の熱安定性と熱暴露後の塗装性も向上することができることを見出した。
【0132】
本発明では、上記した成分の他に、本発明の効果を損なわない範囲で必要に応じて付加的成分を添加しても構わない。
【0133】
付加的成分の例を以下に挙げる。ポリエステル、ポリオレフィン等の他の熱可塑性樹脂、無機充填材(タルク、カオリン、ゾノトライト、ワラストナイト、酸化チタン、チタン酸カリウム、ガラス繊維など)、無機充填材と樹脂との親和性を高める為の公知の密着改良剤、難燃剤(ハロゲン化された樹脂、シリコーン系難燃剤、水酸化マグネシウム、水酸化アルミニウム、有機燐酸エステル化合物、ポリ燐酸アンモニウム、赤燐など)、滴下防止効果を示すフッ素系ポリマー、可塑剤(オイル、低分子量ポリオレフィン、ポリエチレングリコール、脂肪酸エステル類等)及び、三酸化アンチモン等の難燃助剤、着色用カーボンブラック、カーボンファイバー、帯電防止剤、各種過酸化物、酸化亜鉛、硫化亜鉛、酸化防止剤、紫外線吸収剤、光安定剤等である。
【0134】
これらの成分の具体的な添加量は、成分(A)〜(E)の合計100質量部に対して、合計で50質量部を超えない範囲である。
【0135】
特に、本発明の組成物に無機充填材としてウォラストナイトを配合することが好ましい。ウォラストナイトは、珪酸カルシウムを成分とする天然鉱物を精製、粉砕及び分級したものである。また、人工的に合成したものも使用可能である。一般的にはガラス繊維の代替として、平均繊維径40μmで繊維長600μmの大粒子径のものが広く用いられているが、本発明では平均粒子径が2〜9μmの範囲でありアスペクト比が5以上のものが、好ましくは平均粒子径が3〜7μmの範囲でアスペクト比が5以上のものが使用される。さらには、アスペクト比の異なる2種以上のウォラストナイト混合物である事がより好ましい。具体的には、アスペクト比5以上のウォラストナイトとアスペクト比5未満のウォラストナイトの混合物である。
【0136】
これらウォラストナイトには、表面処理剤として、高級脂肪酸またはそのエステル、塩等の誘導体(例えば、ステアリン酸、オレイン酸、パルミチン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、ステアリン酸アミド、ステアリン酸エチルエステルなど)やカップリング剤(例えば、シラン系、チタネート系、アルミニウム系、ジルコニウム系など)を必要により使用することが出来る。その使用量はウォラストナイトに対して0.05質量%〜5質量%の範囲である。
【0137】
また、本発明の組成物に無機充填材としてタルクを配合することも好ましい。タルクの使用により、表面外観が良好であり、熱膨張率の低下が期待できる。タルクは平均粒径が約2.0〜5.0μmのケイ酸マグネシウムが好ましく、特に約3.0〜5.0μmのものが好ましい。
【0138】
これらの無機充填材は、タルク、カオリン、ワラストナイト、酸化チタン、チタン酸カリウム、ガラス繊維等の混合物であっても構わない。
【0139】
これらの無機充填材の添加量は、成分(A)〜(E)の合計100質量部に対し、5〜40質量部であることが好ましい。より好ましくは、5〜25質量部である。
【0140】
上記の成分を溶融混練して本発明の樹脂組成物を得るために用いる具体的な加工機械としては、例えば、単軸押出機、二軸押出機、ロール、ニーダー、ブラベンダープラストグラフ、バンバリーミキサー等が挙げられるが、中でも二軸押出機が好ましく、特に、上流側供給口と1カ所以上の下流側供給口を備えた二軸押出機が最も好ましい。
【0141】
この際の溶融混練温度は特に限定されるものではないが、通常240〜360℃の中から好適な組成物が得られる条件を任意に選ぶことができる。
【0142】
本発明の好ましい導電性樹脂組成物の製造方法としては、ポリフェニレンエーテル(B)、ゴム状重合体(C)及び相溶化剤として機能する第2の低分子量変性剤化合物(E)の混合物を溶融し、これにポリアミド(A)と導電性炭素材料(D)とを含む上記のマスターバッチを配合することが好ましい。具体的には、上流側供給口と下流側供給口を備えた二軸押出機を用い、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)及び相溶化剤として機能する第2の低分子量変性剤化合物(E)を供給し溶融混練した後、下流側供給口よりポリアミド(A)と導電性炭素材料(D)(または上記のマスターバッチ)を供給し溶融混練する方法等が挙げられる。また、溶融混錬の際、押出機下流のベント脱気口より真空脱気することがさらに好ましい。
【0143】
このようにして得られる本発明の導電性樹脂組成物を、公知の種々の方法により成形することにより樹脂成形体を得ることができる。
【0144】
本発明でいう樹脂成形体とは、射出成形したものに限定されず、押出成形されたシート、フィルム、ペレットおよび2次加工された射出成形体まで包含される。
【0145】
本発明においては、204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることが好ましく、0.10以下であることがより好ましい。該低分子量成分の量が0.15以下であることにより、本発明の成形体は塗装密着性、特に熱暴露後の塗装密着性が非常に優れる。
【0146】
ここで成形体の表面とは、樹脂成形体をクロロホルム溶媒に室温で約1時間浸漬後、表面から溶け出す領域をいう。
【0147】
ここでいう熱処理後の成形体表面のクロロホルム可溶成分の中で、サイズ排除クロマトグラフィーの示差屈折計を用いて測定される溶出時間が22分〜23.5分である成分のピークの高さの比は以下の方法で測定することが可能である。
【0148】
204℃で40分間の熱処理後の樹脂成形体を幅約8cm、長さ約8cm(肉厚は特に制限はない)の形状に切削し、12.5cmのシャーレなどに50mlのクロロホルム(日本国和光純薬製、特級)を加えた中に、切削した成形体を室温で約1時間浸漬させる。浸漬した成形体を取り出した後の抽出液を40ml取り、溶媒を蒸発させ該抽出液を乾固させる。蒸発乾固の条件としては、室温で24時間行なうことができる。
【0149】
乾固させた試料にテトラヒドロフラン(日本国和光純薬製、HPLC用)を2ml加え、試料溶液としたものを以下のサイズ排除クロマトグラフィーの示差屈折計を用いて分析を行う。
[サイズ排除クロマトグラフィー装置]
オンライン脱気装置(DE):日本国(株)イーアールシー製ERC−3322
送液ポンプ(PO):日本国東ソー(株)製DP−8020
カラムオーブン(CO):日本国東ソー(株)製CO−8010
紫外可視吸光光度検出器(UV):日本国東ソー(株)製UV−8010
示差屈折検出器(RI):日本国東ソー(株)製RI−8012
【0150】
上記の装置は図1のように接続して用いる。また、溶媒にテトラヒドロフラン(日本国和光純薬製、HPLC用)を使用する。分析条件は、流速1ml/分、オーブン温度40℃、UV検出波長254nmとする。なお、カラムオーブン中には以下のサイズ排除クロマトグラフィー用カラムを配する。
ガードカラム:東ソー(株)製TSKguardcolumn HXL−L
カラム1および2:東ソー(株)製TSKgel G1000HXL
カラム3:東ソー(株)製TSKgel G2000HXL
上記ガードカラム、カラム1、カラム2、カラム3をこの順に直列に接続し、溶媒をガードカラムからカラム3の方向に流す。
【0151】
なお、上述のサイズ排除クロマトグラフィー装置の示差屈折計を用いて、基準サンプルとして、Irganox1076(スイス国Ciba Specialty Chemicals製)を分析したところ、溶出時間は17.8分であった。この基準サンプルであるIrganox1076の溶出時間が17.8分よりずれた場合、((Irganox1076測定時の溶出時間(分)−17.8(分))時間分を試料溶液の溶出時間(分)に加算して補正を行う。
【0152】
本発明の組成物が導電性カーボンブラックを含む樹脂成形体の場合は、反射法光学顕微鏡を用いて100倍で、視野の合計10mm2を観察した際に、導電性カーボンブラックの一部が、面積8μm2以上の二次凝集体として存在し、樹脂組成物中の全導電性カーボンブラックに対する、面積8μm2以上の二次凝集体の存在比率が、20質量%以上であることが好ましい。より好ましくは50質量%以上、さらに好ましくは50〜80質量%である。
【0153】
ここでいう導電性カーボンブラックの二次凝集体の大きさおよび存在比率は、以下に説明する方法で測定することができる。反射法光学顕微鏡(PME3:日本国オリンパス社製)を用いて、観察用試料の平滑面におけるカーボンブラックの分散状態を倍率100倍で観察し、撮影を行う。さらに、撮影された反射顕微鏡写真を、スキャナ(CC−600PX:日本国エプソン社製)にてデジタル画像化する(取り込み解像度は400dpi)。得られたデジタル画像の視野内で観察される黒色相(カーボンブラックの二次凝集体)の占める面積分率を、画像処理ソフト(Image−Pro PLUS Ver.4.0J:米国MEDIA CYBERNETICS製)を用いて自動測定で解析する。測定は以下の解析条件及び手順で行う。
(1)フィルター:メディアン(インパルスノイズの除去、強度:3×3ピクセル、回数:2回)
(2)フィルター:平坦化(オブジェクト幅20ピクセル、背景:明るい)
(3)面積分率測定条件:面積が8μm2以上(平均径3μm以上に対応:径3μmの円では7.06μm2)、且つ楕円長短軸比(重心を通る径を2°刻みで測定した長軸と短軸の直径の比)が2以下(細長い形状の二次凝集体と区別しにくい試料切削時のメスキズ等を排除するため)を満足し、さらに、輝度レンジ(256階色)のヒストグラム表示において、灰白色の相(ポリアミド−ポリフェニレンエーテル相)より黒い側の色相のレンジを抽出対象として検出する。なお、上記に自動カウントされない楕円長短軸比が2より大きく、明らかに二次凝集体と目視で判断できる大きな黒色相は、手動で黒色相の周囲をトレースし結果に付加する。また、フィルターで削除できなかった試料切削時の小さなメスキズがある場合は手動で削除する。
【0154】
上記方法により、視野内の黒色相である二次凝集体が占める面積分率を算出し、10枚の写真(合計視野10mm2)の平均値を求める。ここで、二次凝集体が占める体積分率は切断面に現れる面積分率に等しいことが一般的に成り立つため、面積分率を体積分率に置き換える。次に、カーボンブラックの真比重1.9および樹脂の比重1.1を用いて、体積分率を質量分率に変換することにより、面積が8μm2以上の二次凝集体の質量分率を算出する。この二次凝集体の質量分率を全カーボンブラックの質量分率(仕込み量)で除して、樹脂中の全カーボンブラックに対する面積が8μm2以上の二次凝集体の存在比率を算出する。
【0155】
本発明の成形体は各種部品としては用いることができる。そのような部品の例としては、ICトレー材料、各種ディスクプレーヤー等のシャーシー、キャビネット等の電気・電子部品、各種コンピューターおよびその周辺機器等のOA部品や機械部品、さらにはオートバイのカウルや、自動車のフェンダー、ドアーパネル、フロントパネル、リアパネル、ロッカーパネル、リアバンパーパネル、バックドアガーニッシュ、エンブレムガーニッシュ、燃料注入口パネル、オーバーフェンダー、アウタードアハンドル、ドアミラーハウジング、ボンネンットエアインテーク、バンパー、バンパーガード、ルーフレール、ルーフレールレッグ、ピラー、ピラーカバー、ホイールカバー、スポイラー等に代表される各種エアロパーツ、各種モール、エンブレムといった外装部品や、インストゥルメントパネル、コンソールボックス、トリム等に代表される内装部品等が挙げられる。
【0156】
これらの中でも、静電塗装可能な自動車の外板部品に好適に使用可能であり、特に、自動車フェンダーに好適に使用できる。
【図面の簡単な説明】
【0157】
【図1】図1は、で用いたサイズ排除クロマトグラフィー装置類の接続を示す概略図である。
【図2(a)】図2(a)は、導電性炭素材料(D)の量が1.9質量%である実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図である。
【図2(b)】図2(b)は、導電性炭素材料(D)の量が2.5質量%である実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図である。
【発明を実施するための最良の形態】
【0158】
以下、実施例及び比較例により本発明を具体的に説明するが、本発明はこれらによって何ら限定されるものではない。
【0159】
(使用した原料)
実施例及び比較例において使用した原料は次のとおりである。
【0160】
(1)ポリアミド(A):ポリアミド66(以下、PA66と略記)
数平均分子量=14,000
末端アミノ基濃度=30ミリ等量/kg
末端カルボキシル基濃度=100ミリ等量/kg
Cu濃度=30ppm含有(ヨウ化銅として添加)
【0161】
(2)ポリフェニレンエーテル(B):ポリ(2,6−ジメチル−1,4−フェニレンエーテル)
(2−1)ポリフェニレンエーテル(以下、PPE−1と略記)
日本国特開2002−3596号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状ポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が0.1質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。質量減少率は、下記の式により計算した。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテルの質量−真空乾燥後のポリフェニレンエーテルの質量)/真空乾燥前のポリフェニレンエーテルの質量}×100。
下記のPPE−2〜PPE−4及びM−PPEについても同様に計算した。
ポリフェニレンエーテル粉末の還元粘度は0.52dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−2)ポリフェニレンエーテル(以下、PPE−2と略記)
日本国特開昭64−33131号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が1.3質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。ポリフェニレンエーテル粉末の還元粘度は0.54dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−3)ポリフェニレンエーテル(以下、PPE−3と略記)
日本国特開2002−3596号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が0.9質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。ポリフェニレンエーテル粉末の還元粘度は0.50dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−4)ポリフェニレンエーテル(以下、PPE−4と略記)
日本国特開昭64−33131号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発成分の量が2.2質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。PPE粉末の還元粘度は0.42dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−5)無水マレイン酸変性ポリフェニレンエーテル
(以下、M−PPEと略記)
(PPE−4)100質量部に対して、ジ−tert−ブチルパーオキサイド(ラジカル開始剤)0.1質量部および無水マレイン酸1.5質量部を添加し、二軸押出機を用いてシリンダー温度320℃で溶融混練してM−PPEを作製した。
無水マレイン酸の付加率:0.5質量%
未反応の無水マレイン酸:0.008質量%
揮発性物質:0.3質量%
【0162】
(3)導電性炭素材料(D)(導電性カーボンブラックおよび導電性カーボンフィブリル)
(3−1)導電性カーボンブラック(以下、CBと略記)
商品名:ケッチェンブラックEC−600JD
(日本国ケッチェンブラックインターナショナル社製)
上記のCBを用いて以下の方法でマスターバッチを製造した。上流側と下流側にそれぞれ少なくとも1箇所の供給口を有する二軸押出機を用いて、シリンダー温度270℃に設定した条件下で、上流側供給口より90質量部のポリアミド66を供給し、溶融させた後、下流側供給口より10質量部のCBを添加して溶融混練し、マスターバッチを作製した。(以下、PA66/CB−MBと略記)なお、PA66/CB−MBの連続した視野3mm2の断面を反射法光学顕微鏡(PME3:日本国オリンパス社製)を用いて、50倍の倍率で観察し、導電性カーボンブラックの長径が20μm以上100μm以下の凝集粒子数を目視で数えた。その結果、凝集粒子の数は、19個であった。
(3−2)導電性カーボンフィブリル(以下、CNTと略記)
ハイペリオンマスターバッチ(米国ハイペリオンキャタリストインターナショナル社製):平均直径約10〜15μmのカーボンフィブリル20質量%含有のナイロン66マスターバッチ(以下、PA66/CNT−MBと略記)
【0163】
(4)ゴム状重合体(C)
(4−1)ポリスチレン−水素添加ポリブタジエン−ポリスチレン(以下、SEBS−1と略記)
数平均分子量=246,000
スチレン成分合計含有量(スチレンブロック含有量)
=33質量%
(4−2)ポリスチレン−水素添加ポリブタジエン−ポリスチレン(以下、SEBS−2と略記)
数平均分子量=77,000
スチレン成分合計含有量(スチレンブロック含有量)
=67質量%
【0164】
(5)低分子量変性剤化合物(E)(相溶化剤):無水マレイン酸(日本国三菱化学(株)製)(以下、MAHと略記)
【0165】
(6)無機充填材
(6−1)ウォラストナイト(カナダ国ナイコ社製)
平均粒子径4μmで平均繊維長32μm(アスペクト比=8)、1000℃での加熱減量が0.7質量%のウォラストナイトをベースに0.5%アミノシラン水溶液で処理。
【0166】
(測定方法)
以下に、導電性(体積抵抗率)、Izod衝撃強度、成形機内滞留時の熱安定性、目やに発生量および塗装密着性の測定方法について述べる。
【0167】
(1)目やに発生量
押出機を1時間運転して、その間に長さ5mm以上の大きさになった目やにの数を数えた。
【0168】
(2)体積抵抗率
得られた樹脂組成物ペレットを、日本国東芝機械(株)製IS−80EPN成形機(シリンダー温度を280℃、金型温度を80℃に設定)を用いて、ダンベルバーとしてISO294の記載の如く成形した。この試験片の両端を折り取って均一な断面積10×4mm、長さ約70mmで両端に破断面をもつ試験片とした。試験片の折り取り方については、−75〜−70℃のドライアイス/メタノールの中に、予めカッターナイフでキズをつけた試験片を1時間浸漬後、折り取る方法で行った。この両端の破断面に銀塗料を塗布し、エレクトロメーター(日本国アドバンテスト製、R8340A)を用いて、250Vの印加電圧で両方の破断面間の体積抵抗率を測定した。測定は5個の異なる試験片に対して実施し、その加算平均をもって、体積抵抗率とした。
【0169】
(3)Izod衝撃強度
得られた樹脂組成物ペレットを、日本国東芝機械(株)製IS−80EPN成形機(シリンダー温度を280℃、金型温度を80℃に設定)にて厚み3.2mmの短冊成形片を成形し、ASTM D256に準拠し、厚み3.2mmのノッチ付衝撃強度を測定した。
【0170】
(4)成形機内滞留時の熱安定性
得られた樹脂組成物ペレットを、日本国日精樹脂工業(株)製FE120成形機(シャットオフノズル付き)を用いて、120mm×80mm(厚み3mm)の平板を成形した。成形条件は、シリンダー温度:310℃、射出時間+保圧時間:15秒(内、射出時間0.7秒および3秒)、冷却時間:12〜70秒、インターバル時間(型開き+成形品取り出し):5秒とした。滞留時間は、(射出時間+保圧時間+冷却時間+インターバル時間)×(最大計量値/1ショットの計量値)より算出した。成形機内滞留時の熱安定性の判断は、樹脂組成物を所定時間滞留後、成形した平板の外観(シルバー発生の程度)を目視にて行った。平板に少量のシルバーが発生するまでの滞留時間(限界滞留時間)を測定し、それに基づいて成形機内滞留時の熱安定性を評価した。
【0171】
(5)塗装密着性試験
得られた樹脂組成物ペレットを、射出成形機(日本国日精樹脂工業(株)製:FS80S)を用い、シリンダー温度305℃、金型温度80℃、充填時間1秒で10cm×10cm(厚み2mm)の平板を作成した。塗装密着性の測定は、成形後の平板と204℃で40分間の熱暴露後の平板について実施した。自動吹き付け塗装装置を用いて、塗膜厚みが20μmになるよう調節し、塗装を実施した。塗料はアクリルウレタン系塗料(日本国オリジン電気社製、OP−Z−NY)を使用した。吹き付け塗装後、150℃の温度で20分間焼付けを実施した。
【0172】
塗装実施後、温度23℃、湿度50%の環境下に静置し、24時間後、2cm×2cmの範囲で一目が2mm四方になるようにカッターナイフで碁盤目状に傷を付け(合計で100目となる)、セロテープ貼付し一気に引き剥がす塗膜剥離試験を実施した。100目の内、剥離試験後に剥離せずに残った碁盤目数を測定した。
【0173】
(6)PPEの重量平均分子量(Mw)および数平均分子量(Mn)の測定
上記の塗装密着性試験におけるのと同様の方法で成形して得られた10cm×10cm(厚み2mm)の平板を約0.5mm程度に粉砕し、50mlのクロロホルムでソックスレー抽出した。得られたクロロホルム溶液(クロロホルムに可溶の成分:主としてポリフェニレンエーテルとブロック共重合体が含有されている)を、GPCを用いて、紫外分光検出器で測定し、標準ポリスチレンで換算しMwを算出した。この時、同時に溶出してくるブロック共重合体を検出しないよう測定紫外線波長を283nmに設定した。
【0174】
(7)204℃で40分間の熱暴露後の成形体表面のクロロホルム可溶成分の分析
上記の塗装密着性試験におけるのと同様の方法で成形して得られた10cm×10cm(厚み2mm)の平板を204℃で40分間の熱処理後、約8cm×8cm(肉厚は2mm)の形状に切削し、直径約12.5cmのシャーレに50mlのクロロホルム(日本国和光純薬製、特級)を加えた中に、切削した平板を約1時間浸漬させた。浸漬した平板を取り出した後の抽出液を40ml取り、溶媒を蒸発乾固させた。
【0175】
乾固させた試料にテトラヒドロフラン(日本国和光純薬製、HPLC用)を2ml加え、試料溶液としたものをサイズ排除クロマトグラフィーの示差屈折計を用いて測定される溶出時間が22分〜23.5分である成分のピークの高さの比(=(溶出時間が22分〜23.5分のピーク高さの和)/(溶出時間が14分〜15分のピーク高さの和))を算出した。
【0176】
用いたサイズ排除クロマトグラフィー装置は以下の通りである。
オンライン脱気装置(DE):日本国(株)イーアールシー製ERC−3322
送液ポンプ(PO):日本国東ソー(株)製DP−8020
カラムオーブン(CO):日本国東ソー(株)製CO−8010
紫外可視吸光光度検出器(UV):日本国東ソー(株)製UV−8010
示差屈折検出器(RI):日本国東ソー(株)製RI−8012
【0177】
上記の装置を図1のように接続して用いた。また、溶媒にテトラヒドロフラン(日本国和光純薬製、HPLC用)を使用した。分析条件は、流速1ml/分、オーブン温度40℃、UV検出波長254nmとした。なお、カラムオーブン中には以下のサイズ排除クロマトグラフィー用カラムを配した。
ガードカラム:日本国(株)製TSKguardcolumn HXL−L
カラム1および2:日本国東ソー(株)製TSKgel G1000HXL
カラム3:日本国東ソー(株)製TSKgel G2000HXL
上記ガードカラム、カラム1、カラム2、カラム3をこの順に直列に接続し、溶媒をガードカラムからカラム3の方向に流した。
【0178】
なお、上述のサイズ排除クロマトグラフィー装置の示差屈折計を用いて、基準サンプルとして、Irganox1076(スイス国Ciba Specialty Chemicals製)を分析したところ、溶出時間は17.8分であった。
実施例1〜6、8、9および比較例1〜6
【0179】
押出機上流側1ヵ所と下流側に1ヵ所の供給口を備えた二軸押出機[ZSK−58MC:ドイツ国ウェルナー&フライデラー社製]を用いて、シリンダー温度300℃に設定した条件下で、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)および変性剤(E)(相溶化剤)を供給し溶融混練した後、下流側供給口よりポリアミド66(A)と導電性炭素材料(D)(導電性カーボンブラックあるいは導電性カーボンフィブリル)を含むマスターバッチ(PA66/CB−MBあるいはPA66/CNT−MB)を供給して、導電性樹脂組成物ペレットを作製した。この際、押出時における目やに発生量を測定した。さらに得られた組成物ペレットを成形し、前述した方法で成形機内滞留時の熱安定性、体積抵抗率、Izod衝撃強度および塗装密着性を測定した。また、平板を用いて前述した方法で、PPEの重量平均分子量(Mw)、数平均分子量(Mn)および204℃で40分間の熱暴露後の成形体表面のクロロホルム可溶成分を分析した。導電性カーボンブラック量が1.9質量%の場合およびカーボンフィブリルの量が2.0質量%における物性値を組成と共に表1に示した。また、導電性カーボンブラック量が2.5質量%における物性値を組成と共に表2に示した。
【0180】
【表1】
【0181】
【表2】
【0182】
表1からわかるように、成分(A)〜(E)の合計質量に対し、成分(D)の量が0.2〜3質量%であり、変性剤(E)(相溶化剤)の量(E質量%)が0.01質量%より大きく0.20質量%未満であって、且つ成分(B)(PPE)の質量に対し、成分(B)中に残存する揮発性物質の量(a質量%)が、a≦−7.3×E+1.83を満足する場合には、押出機内および成形内滞留時の熱安定性が大幅に向上すると共に、特に熱暴露後の塗装密着性が著しく向上することが明らかとなった。図2(a)は、成分(D)(カーボンブラックあるいはカーボンフィブリル)の量が1.9質量%における実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図であるが、本発明における実施例と比較例において本発明の要件が満たされているか否かが明確に示されている。また、表2および図2(b)に示した成分(D)(カーボンブラックあるいはカーボンフィブリル)の量が2.5質量%の場合においても同様である。
【0183】
なお、実施例1〜5の導電性カーボンブラックを含む樹脂成形体について、反射法光学顕微鏡(100倍)を用いて、視野の合計10mm2を観察した際の面積8μm2以上の二次凝集体の存在比率(樹脂組成物中の全導電性カーボンブラックに対する)を測定した。その結果、面積8μm2以上の二次凝集体の存在比率(樹脂組成物中の全導電性カーボンブラックに対する)は、54〜69質量%であった。
【実施例7】
【0184】
押出機上流側1ヵ所と下流側に2ヵ所の供給口を備えた二軸押出機[ZSK−58MC:ドイツ国ウェルナー&フライデラー社製]を用いて、シリンダー温度300℃に設定した条件下で、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)および変性剤(E)(相溶化剤)を供給し溶融混練した後、押出機の中段に設けられた下流側供給口(1)よりポリアミド66と導電性付与剤を含むマスターバッチ(PA/CB−MB)を供給し、押出機の後段に設けられた下流側供給口(2)よりウォラストナイトを供給して、ポリアミド66(成分(A))、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、変性剤(E)およびウォラストナイトからなる樹脂組成物ペレットを作製した以外は実施例1と同様に行った。測定結果については、表1に併記した。
【0185】
本発明の組成物に無機充填材としてウォラストナイトを配合した場合も、押出時および成形時の成形機内滞留時の熱安定性が大幅に向上すると共に、特に熱暴露後の塗装密着性が著しく向上する結果を得た。さらに、無機充填材を添加することにより、著しく体積抵抗率が低下し、導電性も向上することが確認できた。
【産業上の利用可能性】
【0186】
本発明の熱可塑性樹脂組成物は、導電性カーボンブラック及び/又は導電性カーボンフィブリル、変性剤(相溶化剤)およびポリフェニレンエーテル中に残存する揮発性物質の量を特定の範囲にすることにより、耐衝撃性及び熱安定性に優れるのみならず、押出成形時の目やにの発生及びストランド切れを抑制することができるので、電気・電子部品、OA部品、車両部品、機械部品などの幅広い分野に使用することができる。特に、本発明の導電性樹脂組成物は、大型成形体の製造に用いた場合でも、成形時の成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れるため、自動車外板のような静電塗装を施す大型成形体の製造に非常に有利に用いることができる。
【技術分野】
【0001】
本発明は導電性樹脂組成物に関する。更に詳細には、本発明は、ポリアミド(A)、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することにより得られる導電性樹脂組成物であって、該混合物中の該導電性炭素材料(D)及び該低分子量変性剤化合物(E)の量が特定の範囲内にあり、且つ該ポリフェニレンエーテル(B)中に存在する揮発性物質の量が特定の範囲内にある、ことを特徴とする導電性樹脂組成物に関する。本発明の導電性樹脂組成物は、耐衝撃性及び熱安定性に優れるのみならず、押出成形時の目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)の発生及びストランド切れを抑制することができるので、電気・電子部品、OA部品、車両部品、機械部品などの幅広い分野に使用することができる。特に、本発明の導電性樹脂組成物は、大型成形体の製造に用いた場合でも、成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れるため、自動車外板のような静電塗装を施す大型成形体の製造に非常に有利に用いることができる。
【背景技術】
【0002】
ポリフェニレンエーテルは機械的性質・電気的性質及び耐熱性が優れており、しかも寸法安定性に優れるため幅広い用途で使用されているが、単独では成形加工性に劣っている。日本国特公昭45−997号公報(米国特許第3,379,792号に対応)には、これを改良するためにポリフェニレンエーテルにポリアミドを配合する技術が提案されており、現在では非常に多種多様な用途に使用される材料となっている。
【0003】
最近になって、静電塗装可能な材料として、導電性を付与したポリアミド−ポリフェニレンエーテルアロイを用いた自動車の外装材(フェンダー・ドアパネル等)への用途展開が急速に進んでいる。例えば、ポリアミド−ポリフェニレンエーテルアロイで成形して製造された自動車フェンダーを用いることにより、自動車の安全性(歩行者保護)や変形回復性を向上させることが試みられている。
【0004】
自動車の外装材料には、静電塗装可能なレベルの導電性、衝撃強度、耐熱性、流動性等の種々の特性が要求される。
【0005】
ポリアミド−ポリフェニレンエーテルアロイに導電性を付与する技術としては、例えば、日本国特開平2−201811号公報には、カーボンブラックを主としてポリアミド相中に含有させること、また予めカーボンブラックをポリアミド中へ均一分散させた後にポリフェニレンエーテルと混合することにより、表面抵抗を低下させる技術が開示されている。
【0006】
日本国特開平8−48869号公報(米国特許第5,977,240号に対応)には、予めポリアミド−ポリフェニレンエーテルを相溶化させた後、カーボンブラックを配合する技術により、良好な衝撃強さ、良好な流動性および良好な(低い)体積抵抗率を有する組成物が得られることが開示されている。
【0007】
また、日本国特開平4−300956号公報(EP506386に対応)には、ポリフェニレンエーテル、ポリアミド組成物にカーボンブラックを特定量添加すること、及びポリフェニレンエーテル及びポリアミドの配合比率および特定の相対粘度を有するものを用いることにより導電性及び加工性を良好にすることが記載されている。
【0008】
国際公開第01/81473号パンフレットには、樹脂組成物中の導電性炭素系フィラー(ケッチェンブラック)をポリフェニレンエーテル相中に粒状で存在させる技術が開示されている。また、日本国特表2002−544308号公報(米国特許第6,221,283号に対応)には、相溶化剤の量により連続相ポリマー中での分散相のポリマーの粒度を変化させることによって、導電性付与剤を含有する組成物の体積抵抗率を制御する方法が開示されている。また、日本国特開2001−302904号公報には、固相状態で変性されたポリフェニレンエーテルを用いてポリアミドと無機フィラー(カーボンブラック等)を溶融混練することにより、表面外観、耐衝撃性および耐熱性を有する組成物が得られることが開示されている。
【0009】
しかしながら、上述した技術では、導電性フィラーを添加するため、大型押出機で長時間製造した際に、目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)が発生しやすい。また自動車外板のような大型成形体の成形時には、成形機内滞留時の熱安定性が悪化し、成形条件幅も狭く、生産性を大きく低下させる要因となっている。
【0010】
さらに静電塗装性を要求される用途においては、塗装密着性及び塗装後外観の鮮映性を確保するために成形体外観の品質を確保することが重要である。特に、自動車フェンダーの中には、オンラインによる静電塗装後、金属パネルに塗布された錆止め塗料を硬化させるために、金属パネルと共に樹脂成形体も熱処理工程を通されることになる。これらの熱処理工程は、約170℃〜200℃またはそれ以上の温度で10〜50分程度暴露されるのが通例であり、使用される樹脂には耐熱性、熱時剛性と共に、成形後の塗装密着性のみならず、熱暴露後の塗装密着性が特に要求されることになる。
【0011】
しかしながら、上述した技術では、自動車フェンダーに要求される衝撃性、剛性、導電性を高次にバランスさせ、さらに成形機内滞留時の熱安定性および熱暴露後の塗装密着性を向上させることができなかった。そのため、新たな技術の開発が待望されているのが現状であった。
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明の目的は、押出成形時の目やに(押出機のダイス出口に形成され、時間とともに成長する樹脂の塊)の発生及びストランド切れを抑制することができるだけでなく、大型成形体の製造に用いた場合でも、成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れる導電性樹脂組成物を提供することにある。
【0013】
本発明の他の1つの目的は、上記の導電性樹脂組成物からなる成形体(自動車外板など)を提供することにある。
【課題を解決するための手段】
【0014】
発明の概要
本発明者らは、上記課題を解決するために検討を行った結果、驚くべきことに、ポリアミド(A)、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することにより得られる導電性樹脂組成物であって、該混合物中の該導電性炭素材料(D)及び該低分子量変性剤化合物(E)の量が特定の範囲内にあり、且つ該ポリフェニレンエーテル(B)中に存在する揮発性物質の量が特定の範囲内にある、ことを特徴とする導電性樹脂組成物によって、上記の課題を解決できることを見出した。この知見に基づいて、本発明を完成した。
【0015】
本発明の上記及びその他の諸目的、諸特徴ならびに諸利益は、添付の図面を参照しながら行う以下の詳細な説明及び請求の範囲の記載から明らかになる。
【発明の効果】
【0016】
発明の詳細な説明
本発明によれば、ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物が提供される。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【0017】
次に、本発明の理解を容易にするために、本発明の基本的特徴及び好ましい諸態様を列挙する。
【0018】
1.ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【0019】
2.該成分(A)〜(E)の混合物中における該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.02〜0.18質量%であることを特徴とする前項1に記載の樹脂組成物。
【0020】
3.該成分(A)〜(E)の混合物中における該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して、0.02〜0.15質量%であることを特徴とする前項1に記載の樹脂組成物。
【0021】
4.該成分(A)〜(E)の混合物中における該導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.5〜2.5質量%であることを特徴とする前項1〜3のいずれかに記載の樹脂組成物。
【0022】
5.該揮発性物質の量(a)が、1.0質量%以下であることを特徴とする前項1〜4のいずれかに記載の樹脂組成物。
【0023】
6.該成分(D)が、導電性カーボンブラックであることを特徴とする前項1〜5のいずれかに記載の樹脂組成物。
【0024】
7.該成分(D)としての導電性カーボンブラックのジブチルフタレート(DBP)吸収量が250ml/100g以上であることを特徴とする前項1〜6のいずれかに記載の樹脂組成物。
【0025】
8.該第1の低分子量変性剤化合物(b)及び該第2の低分子量変性剤化合物(E)の各々がそれぞれ独立して無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸から選ばれる少なくとも一種の化合物であることを特徴とする前項1〜7のいずれかに記載の樹脂組成物。
【0026】
9.該ゴム状重合体(C)が、少なくとも1種の、数平均分子量が120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物であって、該ブロック共重合体(C−1)及び(C−2)の各々が、それぞれ独立して、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックを含んでなることを特徴とする前項1〜8のいずれかに記載の樹脂組成物。
【0027】
10.該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
を含み、
該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%であることを特徴とする前項9に記載の樹脂組成物。
【0028】
11.該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることを特徴とする前項1〜10のいずれかに記載の樹脂組成物。
【0029】
12.該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、55,000〜70,000であり、かつ分子量分布が、3.0以下であることを特徴とする前項11に記載の樹脂組成物。
【0030】
13.該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法で行うことを特徴とする前項1〜12のいずれかに記載の樹脂組成物。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。
【0031】
14.該マスターバッチが、該成分(D)として導電性カーボンブラックを含んでなり、該マスターバッチを、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在するマスターバッチであることを特徴とする前項13に記載の樹脂組成物。
【0032】
15.該成分(A)〜(E)の混合物が更に少なくとも1種の無機充填材を、該成分(A)〜(E)の合計100質量部に対して、5〜25質量部含むことを特徴とする前項1〜14のいずれかに記載の樹脂組成物。
【0033】
16.成形体である前項1〜15のいずれかに記載の樹脂組成物。
【0034】
17.204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることを特徴とする前項16に記載の樹脂組成物。
【0035】
18.該少なくとも1種の低分子量成分の量が、0.10以下であることを特徴とする前項17に記載の樹脂組成物。
【0036】
19.オンライン塗装されてなる自動車フェンダーであることを特徴とする前項16〜18に記載の樹脂組成物。
【0037】
本発明の導電性樹脂組成物は、ポリアミド(A)、末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及びポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する低分子量変性剤化合物(E)を含んでなる混合物を溶融混練することによって製造される樹脂組成物である。
【0038】
以下に、本発明の導電性樹脂組成物を構成する各成分について詳しく述べる。
【0039】
本発明においてポリアミド(A)として使用することのできるポリアミドの種類としては、ポリマー主鎖中にアミド結合{−NH−C(=O)−}を有するものであれば、いずれも使用することができる。
【0040】
一般にポリアミドは、ラクタム類の開環重合、ジアミンとジカルボン酸の重縮合、アミノカルボン酸の重縮合などによって得られるが、これらに限定されるものではない。
【0041】
上記ジアミンとしては、脂肪族、脂環式および芳香族ジアミンが挙げられ、具体例としては、テトラメチレンジアミン、ヘキサメチレンジアミン、ウンデカメチレンジアミン、ドデカメチレンジアミン、トリデカメチレンジアミン、2,2,4−トリメチルヘキサメチレンジアミン、2,4,4−トリメチルヘキサメチレンジアミン、5−メチルノナメチレンジアミン、1,3−ビスアミノメチルシクロヘキサン、1,4−ビスアミノメチルシクロヘキサン、m−フェニレンジアミン、p−フェニレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン等が挙げられる。
【0042】
ジカルボン酸としては、脂肪族、脂環式および芳香族ジカルボン酸が挙げられ、具体例としては、アジピン酸、スベリン酸、アゼライン酸、セバシン酸、ドデカン二酸、1,1,3−トリデカン二酸、1,3−シクロヘキサンジカルボン酸、テレフタル酸、イソフタル酸、ナフタレンジカルボン酸、ダイマー酸などが挙げられる。
【0043】
ラクタム類としては、具体的にはε−カプロラクタム、エナントラクタム、ω−ラウロラクタムなどが挙げられる。
【0044】
また、アミノカルボン酸としては、具体的にはε−アミノカプロン酸、7−アミノヘプタン酸、8−アミノオクタン酸、9−アミノノナン酸、11−アミノウンデカン酸、12−アミノドデカン酸、13−アミノトリデカン酸などが挙げられる。
【0045】
本発明においては、これらラクタム類、ジアミン、ジカルボン酸、ω−アミノカルボン酸を、単独で重合して得られるポリアミド、及び二種以上の混合物にして重縮合を行って得られる共重合ポリアミドのいずれもが使用できる。
【0046】
また、これらラクタム類、ジアミン、ジカルボン酸、ω−アミノカルボン酸を重合反応機内で低分子量のオリゴマーの段階まで重合し、押出機等で高分子量化したものも好適に使用することができる。
【0047】
特に本発明で有用に用いることのできるポリアミド樹脂としては、ポリアミド6、ポリアミド6,6、ポリアミド4,6、ポリアミド11、ポリアミド12、ポリアミド6,10、ポリアミド6,12、ポリアミド6/6,6、ポリアミド6/6,12、ポリアミドMXD(m−キシリレンジアミン)/6、ポリアミド6,T、ポリアミド6,I、ポリアミド6/6,T、ポリアミド6/6,I、ポリアミド6,6/6,T、ポリアミド6,6/6,I、ポリアミド6/6,T/6,I、ポリアミド6,6/6,T/6,I、ポリアミド6/12/6,T、ポリアミド6,6/12/6,T、ポリアミド6/12/6,I、ポリアミド6,6/12/6,Iなどが挙げられ、複数のポリアミドを押出機等で共重合化したポリアミド類も使用することができる。
【0048】
好ましいポリアミドは、ポリアミド6、ポリアミド6,6、ポリアミド6/6,6、ポリアミド6/6,I及び、それらの混合物である。
【0049】
本発明で使用されるポリアミド樹脂の好ましい数平均分子量は5,000〜100,000であり、より好ましくは10,000〜30,000である。
【0050】
本発明におけるポリアミド樹脂はこれらに限定されるものではなく、分子量の異なる複数のポリアミド樹脂の混合物であっても良い。例えば数平均分子量15,000未満の低分子量ポリアミドと、15,000以上の高分子量ポリアミドとの混合物等である。
【0051】
ポリアミドの末端基は、ポリフェニレンエーテルとの反応に関与する。ポリアミド樹脂は末端基として一般にアミノ基、カルボキシル基を有しているが、一般的にカルボキシル基濃度が高くなると、耐衝撃性が低下し、流動性が向上し、逆にアミノ基濃度が高くなると耐衝撃性が向上し、流動性が低下する。
【0052】
本発明における、これらの好ましい比はアミノ基/カルボキシル基濃度比で、9/1〜1/9であり、より好ましくは8/2〜1/9、更に好ましくは6/4〜1/9である。
【0053】
また、末端のアミノ基の濃度としては少なくとも10ミリ当量/kgであることが好ましい。更に好ましくは30ミリ当量/kg以上である。
【0054】
これらポリアミド樹脂の末端基の調整方法は、公知の方法を用いることができる。例えばポリアミド樹脂の重合時に所定の末端濃度となるようにジアミン類やジカルボン酸類、モノカルボン酸類などを添加する方法、あるいは、末端基の比率が異なる2種類以上のポリアミド樹脂の混合物により調整する方法等が挙げられる。
【0055】
また、ポリアミド樹脂の耐熱安定性を向上させる目的で日本国特開平1−163262号公報に記載されているような金属系安定剤も使用することができる。
【0056】
これら金属系安定剤の中で特に好ましく使用することのできるものとしては、CuI、CuCl2、酢酸銅、ステアリン酸セリウム等が挙げられる。また、ヨウ化カリウム、臭化カリウム等に代表されるアルカリ金属のハロゲン化塩も金属系安定剤として好適に使用することができる。これらは、もちろん併用添加しても構わない。
【0057】
金属系安定剤の好ましい配合量は、ポリアミド樹脂100質量部に対して、0.001〜1質量部である。
【0058】
さらに、上記の他にポリアミドに添加することが可能な公知の添加剤等もポリアミド100質量部に対して10質量部未満の量で添加してもかまわない。
【0059】
本発明で使用できるポリフェニレンエーテル(B)とは、下記式(1)の構造単位からなる、単独重合体及び/または共重合体である。
【0060】
【化1】
【0061】
〔式中、Oは酸素原子、各Rは、それぞれ独立して、水素、ハロゲン、第一級もしくは第二級のC1〜C7アルキル基、フェニル基、C1〜C7ハロアルキル基、C1〜C7アミノアルキル基、C1〜C7ヒドロカルビロキシ基、又はハロヒドロカルビロキシ基(但し、少なくとも2個の炭素原子がハロゲン原子と酸素原子を隔てている)を表す。〕
【0062】
本発明で使用できるポリフェニレンエーテルの具体的な例としては、例えば、ポリ(2,6−ジメチル−1,4−フェニレンエーテル)、ポリ(2−メチル−6−エチル−1,4−フェニレンエーテル)、ポリ(2−メチル−6−フェニル−1,4−フェニレンエーテル)、ポリ(2,6−ジクロロ−1,4−フェニレンエーテル)等が挙げられ、さらに2,6−ジメチルフェノールと他のフェノール類との共重合体(例えば、日本国特公昭52−17880号公報に記載されているような2,3,6−トリメチルフェノールとの共重合体や2−メチル−6−ブチルフェノールとの共重合体)のようなポリフェニレンエーテル共重合体も挙げられる。
【0063】
これらの中でも特に好ましいポリフェニレンエーテルは、ポリ(2,6−ジメチル−1,4−フェニレンエーテル)、2,6−ジメチルフェノールと2,3,6−トリメチルフェノールとの共重合体、またはこれらの混合物である。
【0064】
本発明で用いるポリフェニレンエーテルの製造方法は特に限定されるものではなく、公知の方法が使用でき、例えば、米国特許第3306874号明細書、同第3306875号明細書、同第3257357号明細書及び同第3257358号明細書、日本国特開昭50−51197号公報及び同63−152628号公報等に記載されている製造方法等を挙げることができる。
【0065】
本発明で使用することのできるポリフェニレンエーテルの還元粘度(ηsp/c:0.5g/dl、クロロホルム溶液、30℃測定)は、0.15〜0.70dl/gの範囲であることが好ましく、さらに好ましくは0.20〜0.60dl/gの範囲、より好ましくは0.40〜0.55dl/gの範囲である。ポリフェニレンエーテルの還元粘度が、0.70dl/gより高いと溶融流動性が低下する傾向にあり、0.15dl/gより低いと、機械的物性が低下する傾向にある。
【0066】
本発明においては、2種以上の還元粘度の異なるポリフェニレンエーテルをブレンドしたものであっても、何ら問題なく使用することができる。例えば、還元粘度0.45dl/g以下のポリフェニレンエーテルと還元粘度0.50dl/g以上のポリフェニレンエーテルの混合物、還元粘度0.40dl/g以下の低分子量ポリフェニレンエーテルと還元粘度0.50dl/g以上のポリフェニレンエーテルの混合物等が挙げられるが、もちろん、これらに限定されることはない。
【0067】
また、本発明において使用するポリフェニレンエーテル(B)は、末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種である。
【0068】
また、ポリフェニレンエーテル(B)としては、末端未変性ポリフェニレンエーテルと末端変性ポリフェニレンエーテルの混合物であることが成形機内滞留時の熱安定性を向上させるために好ましい。特に、末端変性ポリフェニレンエーテルの量より末端未変性ポリフェニレンエーテルの量の方が多いことが流動性、成形機内滞留時の熱安定性の観点からより好ましい。具体的には、ポリフェニレンエーテル(B)が上記の混合物である場合、末端未変性ポリフェニレンエーテルの量が、上記混合物の質量に対して55質量%以上であることが好ましい。また、末端未変性ポリフェニレンエーテルの還元粘度(0.5g/dlクロロホルム溶液、30℃測定)が、末端変性ポリフェニレンエーテルの還元粘度より高いことがさらに好ましい。
【0069】
ここでいう末端変性ポリフェニレンエーテルとは、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の第1の低分子量変性剤化合物(b)で変性されたポリフェニレンエーテルを指す。この第1の低分子量変性剤化合物(b)は、本発明の組成物において、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する。
【0070】
上記の第1の低分子量変性剤化合物(b)(相溶化剤)について以下に具体的に説明する。
【0071】
分子内に炭素−炭素二重結合と、カルボン酸基及び/又は酸無水物基とを同時に有する変性剤化合物としては、マレイン酸、フマル酸、クロロマレイン酸、シス−4−シクロヘキセン−1,2−ジカルボン酸及びこれらの酸無水物などが挙げられる。これらの内、フマル酸、マレイン酸、無水マレイン酸が好ましく、フマル酸、無水マレイン酸が特に好ましい。
【0072】
また、これら不飽和ジカルボン酸のカルボキシル基の1個または2個がエステルになっているものも使用可能である。
【0073】
分子内に炭素−炭素二重結合とグリシジル基とを同時に有する低分子量変性剤化合物としては、アリルグリシジルエーテル、グリシジルアクリレート、グリシジルメタアクリレート、エポキシ化天然油脂等が挙げられる。
【0074】
これらの中でグリシジルアクリレート、グリシジルメタアクリレートが特に好ましい。
【0075】
分子内に炭素−炭素二重結合と水酸基とを同時に有する低分子量変性剤化合物としては、アリルアルコール、4−ペンテン−1−オール、1,4−ペンタジエン−3−オールなどの一般式CnH2n−3OH(nは正の整数)の不飽和アルコール、一般式CnH2n−5OH、CnH2n−7OH(nは正の整数)等の不飽和アルコール等が挙げられる。
【0076】
上述した低分子量変性剤化合物は、それぞれ単独で用いても良いし、2種以上を組み合わせて用いても良い。
【0077】
上記のような第1の低分子量変性剤化合物(b)を用いたポリフェニレンエーテルの末端変性は、第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと、ラジカル開始剤の存在下又は非存在下で該末端未変性ポリフェニレンエーテルのガラス転移温度以上360℃以下の範囲の温度で溶融混練することによって行なう。溶融混錬の際、押出機下流のベント脱気口より真空脱気することにより、未反応の第1の低分子量変性剤化合物(b)が、末端変性されたポリフェニレンエーテルの質量に対し、0.01質量%未満になるようにすることが好ましい。その際、溶融混練時のベント脱気真空度は、2.1×104Pa以下であることが特に好ましい。また、溶融混練後の末端変性ポリフェニレンエーテルペレットを減圧加熱乾燥または溶剤洗浄することにより、未反応の第1の低分子量変性剤化合物(b)が、0.01質量%未満になるようにしても構わない。
【0078】
ポリフェニレンエーテルの末端変性を行なう際の第1の低分子量変性剤化合物(b)の添加量は、該末端未変性ポリフェニレンエーテル100質量部に対して0.1〜3質量部が好ましく、更に好ましくは0.3〜2質量部である。
【0079】
上記ラジカル開始剤の例としては、公知の有機過酸化物、ジアゾ化合物などが挙げられる。具体例としては、ベンゾイルパーオキサイド、ジクミルパーオキサイド、ジ−tert−ブチルパーオキサイド、tert−ブチルクミルパーオキサイド、tert−ブチルハイドロパーオキサイド、クメンハイドロパーオキサイド、アゾビスイソブチロニトリルなどが挙げられる。
【0080】
ラジカル開始剤を用いて末端変性されたポリフェニレンエーテルを製造する際の好ましいラジカル開始剤の量は、ポリフェニレンエーテル100質量部に対して0.001〜1質量部である。
【0081】
また、末端変性ポリフェニレンエーテル中の変性剤化合物の付加率は、末端変性前のポリフェニレンエーテルの質量に対して、0.01〜5質量%が好ましい。より好ましくは0.1〜3質量%である。
【0082】
また、末端変性ポリフェニレンエーテル中に残存する未反応の変性剤化合物の量を減少させるために、末端変性を行なう際に、必要に応じてアミド結合及び/またはアミノ基を有する化合物を添加しても構わない。
【0083】
ここでいうアミド結合を有する化合物とは、分子構造中にアミド結合{−NH−C(=O)−}構造を有する化合物であり、アミノ基を有する化合物とは末端に{−NH2}構造を有する化合物である。これら化合物の具体例としては、オクチルアミン、ノニルアミン、テトラメチレンジアミン、ヘキサメチレンジアミン等の脂肪族アミン類、アニリン、m−フェニレンジアミン、p−フェニレンジアミン、m−キシリレンジアミン、p−キシリレンジアミン等の芳香族アミン類、上記アミン類とカルボン酸、ジカルボン酸等との反応物、ε−カプロラクタム等のラクタム類及びポリアミド樹脂等が挙げられるが、これらに限定されるものではない。
【0084】
これらアミド結合またはアミノ基を有する化合物を添加する際の好ましい添加量は、末端未変性ポリフェニレンエーテル100質量部に対して、0.001質量部以上、5質量部以下である。好ましくは0.01質量部以上、1質量部以下、より好ましくは0.01質量部以上、0.1質量部以下である。
【0085】
また、本発明にお該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(成分(A)〜(E)の合計質量に対する質量%)を表す)
を満足する必要がある。
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(該ポリフェニレンエーテル(B)の質量に対する、上記真空乾燥において揮発した成分の質量%)であって下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
真空乾燥は、例えば、日本国いすゞ製作所製の真空定温恒温器CVK−23PSを用いて行なうことができる。
【0086】
更に、本発明においては、該揮発性物質の量が、上記の式を満足し、且つ使用するポリフェニレンエーテル(B)100質量%に対し、1.0質量%以下の量であることが好ましい。より好ましくは、0.6質量%以下、さらに好ましくは0.2質量%以下の量である。
【0087】
使用するポリフェニレンエーテル(B)中の揮発性物質は、重合後の乾燥工程における温度と時間を調整することにより制御することが可能である。本発明に使用するポリフェニレンエーテル(B)中の揮発性物質の量の具体的な制御方法に関しては、日本国特開2002−3596号公報や日本国特開昭64−33131号公報などの記載に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し乾燥し得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機に用いて、揮発性物質が所定の量になるように乾燥時間を決定することができる。しかしながら、上記の方法に限定されるものではなく、使用するポリフェニレンエーテル中の揮発性物質の量が上記の範囲を満足すればどの様な方法を用いてもよい。
【0088】
ここでいう揮発性物質としては、重合溶媒、残留モノマーおよびオリゴマー類である。また、上記の重合溶媒としては、トルエン、キシレンの各異性体、エチルベンゼン、炭素数1〜5のアルコール類、クロロホルム、ジクロルメタン、クロルベンゼン、ジクロルベンゼン等の有機溶媒が挙げられる。また、これらの有機溶媒の内の2種以上の混合物である場合もある。
【0089】
また、本発明においては該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることが好ましく、Mwが55,000以上、かつMw/Mnが3.0以下であることがより好ましく、Mwが55,000〜70,000、かつMw/Mnが3.0以下であることが特に好ましい。
【0090】
上述した組成物中のポリフェニレンエーテル(B)の分子量(Mw)の測定は、以下の手順により容易に可能である。測定に充分な量の組成物を細かく粉砕しクロロホルムでソックスレー抽出する。得られたクロロホルム溶液をゲル浸透クロマトグラフィー測定装置と紫外分光検出器を用いて分析し、標準ポリスチレンで換算した分子量データを得る。得られた分子量データを市販のGPC処理ソフト(例えば、日本国システムインスツルメンツ(SIC)製480データステーション)を用いて特定分子量で分画しその量を計算する。この時、同時に溶出してくるブロック共重合体(成分(C)として用いるもの)などの他の重合体を検出しないため測定する紫外線波長をブロック共重合体などの他の重合体の吸収がない波長に設定することが重要である。[測定は、例えば、以下の装置及び条件を用いて行なうことができる。GPC装置:GPC SYSTEM21:日本国昭和電工(株)製、検出器:UV−41:日本国昭和電工(株)製、溶媒:クロロホルム、温度:40℃、カラム:サンプル側(日本国昭和電工(株)製K−G,K−800RL,K−800R)、リファレンス側(日本国昭和電工(株)製K−805L×2本)、流量10ml/分、測定波長:283nm、圧力15〜17kg/cm2]]
【0091】
次に、本発明で用いる成分(D)について説明する。成分(D)は、導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料である。
【0092】
本発明において使用できる導電性カーボンブラックについて説明する。本発明で使用する導電性カーボンブラックは、ジブチルフタレート(DBP)吸収量が250ml/100g以上のものが好ましく、より好ましくはDBP吸収量が350ml/100g以上、更に好ましくは400ml/100g以上のカーボンブラックである。ここで言うDBP吸収量とは、ASTM D2414に定められた方法で測定した値である。DBP吸収量が上記の範囲外であると、組成物の導電性が低下する傾向がある。
【0093】
また、本発明において使用できる導電性カーボンブラックはBET表面積が200cm2/g以上のものが好ましく、更には400cm2/g以上のものがより好ましい。BET表面積が上記の範囲外であると、組成物の導電性が低下する傾向がある。市販されているものを例示すると、日本国ケッチェンブラックインターナショナルのケッチェンブラックECやケッチェンブラックEC−600JD等が挙げられる。
【0094】
本発明において使用できる導電性カーボンフィブリルとしては、米国特許第4663230号明細書、米国特許第5165909号明細書、米国特許第5171560号明細書、米国特許第5578543号明細書、米国特許第5589152号明細書、米国特許第5650370号明細書、米国特許第6235674号明細書等に記載されている平均繊維径が75nm未満で中空構造をした分岐の少ない炭素系繊維を言う。また、1μm以下のピッチでらせんが一周するコイル状形状のものも含まれる。市販されているものとしては、米国ハイペリオンキャタリスト社のハイペリオンが挙げられる。
【0095】
本発明において使用できる導電性カーボンブラック及び/又は導電性カーボンフィブリルの添加量は、成分(A)〜(E)の合計質量に対して、0.2〜3質量%である。0.5〜2.5質量%であると、耐衝撃性と流動性と導電性のバランスに優れ、より好ましい。さらに好ましくは0.7〜2質量%である。
【0096】
本発明において用いる導電性カーボンブラック及び/又は導電性カーボンフィブリルは、予めポリアミドと導電性カーボンブラック及び/又は導電性カーボンフィブリルを溶融混練することによって得られたマスターバッチの形態で添加されることが好ましく、ペレット状、粉末状、粒状のいずれでもかまわない。
【0097】
上記のマスターバッチ中の該導電性炭素材料(D)の比率は、該マスターバッチの質量に対して、好ましくは5〜25質量%である。より好ましくは6〜15質量%、さらに好ましくは6〜10質量%である。
【0098】
特に、上記マスターバッチは、米国特許US2004/0082729号に開示された導電性マスターバッチであることが好ましい。例えば、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在することを特徴とする導電性マスターバッチであることが好ましい。
【0099】
マスターバッチの好ましい製造方法としては、二軸押出機またはニーダーを使用して溶融混練する方法が好ましい。中でも特にポリアミド(A)を溶融した後に該導電性炭素材料(D)を添加する方法が好ましく、具体例を挙げると、上流側と下流側にそれぞれ少なくとも1箇所の供給口を有する二軸押出機又はニーダーを使用し、上流側供給口よりポリアミド(A)を供給し、溶融させた後、下流側供給口より該導電性炭素材料(D)を添加して溶融混練する方法、あるいは、上流側供給口よりポリアミド(A)を供給し、溶融させた後、下流側供給口より該導電性炭素材料(D)とポリアミド(A)を同時添加して溶融混練する方法等が挙げられる。
【0100】
即ち、本発明においては、該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法によって行うことによって本発明の導電性組成物を得ることができる。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。溶融混練温度は特に限定されるものではないが、例えば、250℃以上、350℃以下の範囲から任意に選択することができる。
【0101】
本発明の導電性樹脂組成物は、スチレン系熱可塑性樹脂をポリアミド(A)とポリフェニレンエーテル(B)の合計100質量部に対し、50質量部未満の量であれば配合しても構わない。本発明でいうスチレン系熱可塑性樹脂としては、ホモポリスチレン、スチレン−アクリロニトリル共重合体(AS樹脂)等が挙げられる。
【0102】
更に、ポリフェニレンエーテル(B)に添加することが可能な公知の添加剤等もポリフェニレンエーテル100質量部に対して10質量部未満の量で添加しても構わない。添加剤の例としては、酸化亜鉛、硫化亜鉛などの金属系安定剤、及びヒンダードフェノール系安定剤、リン系安定剤、ヒンダードアミン系安定剤などの有機安定剤が挙げられる。
【0103】
本発明の導電性樹脂組成物に使用することができるゴム状重合体(C)は、例えば、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックを含むビニル芳香族炭化水素−共役ジエン化合物ブロック共重合体及びその水素添加物、エチレン−α−オレフィン共重合体、ゴム変性ポリスチレン(HIPS)、及びスチレン−ゴム質重合体−アクリロニトリル共重合体(ABS樹脂)からなる群より選ばれる1種以上である。
【0104】
本発明におけるビニル芳香族炭化水素−共役ジエン化合物ブロック共重合体において使用することができるビニル芳香族炭化水素の具体例としてはスチレン、α−メチルスチレン、ビニルトルエン等が挙げられ、これらから選ばれた1種以上の化合物が用いられるが、中でもスチレンが特に好ましい。
【0105】
また、共役ジエン化合物の具体例としては、ブタジエン、イソプレン、ピペリレン、1,3−ペンタジエン等が挙げられ、これらから選ばれた1種以上の化合物が用いられるが、中でもブタジエン、イソプレンおよびこれらの組み合わせが好ましい。
【0106】
ゴム状重合体の具体例としては、SBS(スチレン−ブタジエンブロック共重合体)、SEBS(スチレン−エチレン/ブチレンブロック共重合体)、SEP(スチレン−エチレン/プロピレンブロック共重合体)及びSEPS(スチレン−エチレン/プロピレン−スチレンブロック共重合体)などのブロック共重合体を挙げることができる。
【0107】
該ブロック共重合体の共役ジエン化合物のソフトセグメント部分のミクロ構造は1,2−ビニル含量もしくは1,2−ビニル含量と3,4−ビニル含量の合計量が5〜80%が好ましく、さらには10〜50%が好ましく、10〜40%が最も好ましい。
【0108】
上記ブロック共重合体は、ビニル芳香族炭化水素単量体単位を主体とする重合体ブロック[A]と共役ジエン単量体単位を主体とする重合体ブロック[B]がA−B型、A−B−A型、A−B−A−B型から選ばれる結合形式を有するブロック共重合体であることが好ましい。また、これらの混合物であってももちろん構わない。
【0109】
これらの中でもA−B−A型、A−B−A−B型がより好ましい。これらはもちろん混合物であっても構わない。
【0110】
また、本発明の導電性樹脂組成物に使用することのできるビニル芳香族炭化水素と共役ジエン化合物のブロック共重合体は、水素添加されたブロック共重合体であることがより好ましい。水素添加されたブロック共重合体とは、上述のビニル芳香族炭化水素と共役ジエン化合物のブロック共重合体を水素添加処理することにより、共役ジエン化合物を主体とする重合体ブロックの脂肪族二重結合を0を超えて100%の範囲で制御したものをいう。該水素添加されたブロック共重合体の好ましい水素添加率は50%以上であり、より好ましくは80%以上、最も好ましくは98%以上である。
【0111】
これらブロック共重合体は水素添加されていないブロック共重合体と水素添加されたブロック共重合体との混合物としても問題なく使用可能である。
【0112】
本発明の導電性樹脂組成物に使用するブロック共重合体(C)として、比較的低分子量のブロック共重合体と比較的高分子量ブロック共重合体との混合物を使用することが望ましい。具体的には、少なくとも1種の、数平均分子量が150,000未満、好ましくは120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が150,000以上、好ましくは200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物を用いることが好ましい。
【0113】
本発明でいう数平均分子量とは、ゲルパーミエーションクロマトグラフィー測定装置[例えば、GPC SYSTEM21:日本国昭和電工(株)製]を用いて、紫外分光検出器[例えば、UV−41:日本国昭和電工(株)製]で測定し、標準ポリスチレンで換算した数平均分子量のことを指す。測定条件は次のとおりである。
(測定条件)
溶媒:クロロホルム
温度:40℃
カラム:サンプル側(日本国昭和電工(株)製K−G、K−800RL、K−800R)
リファレンス側(日本国昭和電工(株)製K−805L×2本)
流量:10ml/分
測定波長:254nm
圧力;15〜17kg/cm2
【0114】
この時、重合時の触媒失活による低分子量成分が検出されることがあるが、その場合は分子量計算に低分子量成分は含めない。通常、計算された正しい分子量分布(重量平均分子量(Mw)/数平均分子量(Mn))は1.0〜1.2の範囲内である。
【0115】
これら比較的低分子量のブロック共重合体(C−1)と比較的高分子量のブロック共重合体(C−2)の質量比は、(C−1)/(C−2)=95/5〜5/95であることが好ましい。より好ましくは90/10〜10/90である。
【0116】
また、比較的低分子量のブロック共重合体(C−1)として、ビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数平均分子量が20,000以上であるブロック共重合体を使用することで、耐熱性を向上させるという付加的な効果を得ることができる。
【0117】
ビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数平均分子量は、上述したブロック共重合体の数平均分子量を用いて、下式により求めることができる。
Mn(α)={Mn×α/(α+β)}/N
[上式中において、Mn(α)はビニル芳香族炭化水素を主体とする重合体ブロックの数平均分子量、Mnはブロック共重合体の数平均分子量、αはブロック共重合体中のすべてのビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの質量%、βはブロック共重合体中のすべての共役ジエン単量体単位を主体とする重合体ブロックの質量%、そしてNはブロック共重合体中のビニル芳香族炭化水素単量体単位を主体とする重合体ブロックの数を表す。]
【0118】
また、本発明において、該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
(但し、該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%)
を用いることが好ましい。上記のようなブロック共重合体(C−1a)を用いることにより、樹脂組成物の耐熱性を向上させることができるため、より好適に使用することができる。
【0119】
更に、本発明において、上記のようなブロック共重合体(C−1a)と、ビニル芳香族炭化水素重合体ブロック含有量が、20質量%以上55質量%未満であるブロック共重合体(C−1b)との混合物とすることにより、樹脂組成物の溶融流動性を向上させることが可能となる。この際、ブロック共重合体(C−1a)と、ブロック共重合体(C−1b)との質量比(C−1a)/(C−1b)は、10/0〜2/8である事が好ましく、10/0〜4/6であることがより好ましい。
【0120】
また、本発明の導電性樹脂組成物に使用するゴム状重合体(C)中には、パラフィンを主成分とするオイルをあらかじめ混合したものを用いても構わない。パラフィンを主成分とするオイルをあらかじめ混合する事により、樹脂組成物の加工性を向上させることができる。ここで、パラフィンを主成分とするオイルの量としては、ゴム状重合体(C)の質量に対して50質量%以下が好ましく、30質量%以下がより好ましい。
【0121】
これらビニル芳香族炭化水素−共役ジエン化合物のブロック共重合体は、結合形式の異なるもの、ビニル芳香族炭化水素種の異なるもの、共役ジエン化合物種の異なるもの、1,2−結合ビニル含有量もしくは1,2−結合ビニル含有量と3,4−結合ビニル含有量の異なるもの、ビニル芳香族炭化水素成分含有量の異なるもの、水素添加率の異なるもの等混合して用いても構わない。
【0122】
本発明の導電性樹脂組成物に使用することのできるエチレン−α−オレフィン共重合体の例としては、日本国特開2001−302911号公報に記載されているエチレン−α−オレフィン共重合体が挙げられる。
【0123】
また、本発明の樹脂組成物に使用するゴム状重合体(C)は、全部又は一部が変性されたゴム状重合体であっても構わない。
【0124】
ここでいう変性されたゴム状重合体とは、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の第3の低分子量変性剤化合物(c)で変性されたゴム状重合体を指す。第3の低分子量変性剤化合物(c)としては、上記第1の低分子量変性剤化合物(b)としては、例示したものを用いることができる。
【0125】
本発明の樹脂組成物を製造する際の、上記成分(A)〜(E)の混合物におけるポリアミド(A)、ポリフェニレンエーテル(B)及びゴム状重合体(C)の好ましい量比は、これら3成分の合計を100質量部としたとき、ポリアミド(A)30〜70質量部、ポリフェニレンエーテル(B)20〜50質量部、ゴム状重合体(C)5〜30質量部の範囲内である。より好ましくは、ポリアミド(A)40〜60質量部、ポリフェニレンエーテル(B)30〜40質量部、ゴム状重合体(C)5〜15質量部の範囲内である。
【0126】
本発明において使用することができるポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)は、ポリアミド(A)及びポリフェニレンエーテル(B)と相互作用する多官能性の低分子量変性剤化合物を指すものである。本発明の組成物においては、第2の低分子量変性剤化合物(E)によって、ポリアミド(A)とポリフェニレンエーテル(B)との相溶性が改良されている。
【0127】
本発明において使用することができる相溶化剤として機能する第2の低分子量変性剤化合物(E)の例としては、(i)炭素−炭素二重結合及び三重結合よりなる群から選ばれる少なくとも1つの不飽和結合、及び(ii)カルボン酸基、酸無水物基、アミノ基、水酸基及びグリシジル基よりなる群から選ばれる少なくとも1つの官能基を有する少なくとも1種の低分子量変性剤化合物が挙げられる。
【0128】
これらの相溶化剤として機能する第2の低分子量変性剤化合物(E)の中でも、無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸が好ましい。特に好適な相溶化剤としては、マレイン酸、無水マレイン酸、クエン酸である。これらを混合して用いても構わない。尚、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)とは同じ化合物を用いても良いし、異なる化合物を用いても良い。また、上記の変性されたゴム状重合体をゴム状重合体(C)として用いる場合、上記第3の低分子量変性剤化合物(c)は、第1の低分子量変性剤化合物(b)及び/又は第2の低分子量変性剤化合物(E)と同じでも異なっていてもよい。
【0129】
ポリフェニレンエーテル(B)として末端変性ポリフェニレンエーテルを使用する場合は、末端変性ポリフェニレンエーテル中に含まれる未反応の低分子量変性剤化合物も成分(E)とみなす。また、上記第3の低分子量変性剤化合物(c)として、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤としても機能する低分子量変性剤化合物を用いて変性されたゴム状重合体をゴム状重合体(C)として用いる際に、該変性されたゴム状重合体が未反応の第3の低分子量変性剤化合物(c)を含む場合、この未反応の第3の低分子量変性剤化合物(c)も成分(E)とみなす。
【0130】
本発明において使用することができる変性剤(E)(相溶化剤)の添加量は、成分(A)〜(E)の合計質量に対して、0.01質量%より大きく0.20質量%未満であり、より好ましくは、0.02〜0.18質量%、さらに好ましくは0.02〜0.15質量%、最も好ましくは0.03〜0.10質量%の範囲である。変性剤(E)(相溶化剤)の添加量が0.20質量%以上であると、成形機内滞留時の熱安定性、熱暴露後の塗装性が悪化し、0.01重量%以下では、耐衝撃性、塗装性(成形後、熱暴露後)が悪化する。
【0131】
従来技術においては、ポリアミド/ポリフェニレンエーテル/カーボンブラック系組成物の製造において、変性剤は衝撃強度と導電性の発現バランスを考慮して比較的多くの量(例えば、0.2質量%以上)添加されてた。これに対して、本発明者らは、変性剤の量を従来より少なくすると共に、ポリフェニレンエーテル中の揮発性物質の量を上記の特定の範囲にすれば、衝撃性、導電性だけでなく、成形機内滞留時の熱安定性と熱暴露後の塗装性も向上することができることを見出した。
【0132】
本発明では、上記した成分の他に、本発明の効果を損なわない範囲で必要に応じて付加的成分を添加しても構わない。
【0133】
付加的成分の例を以下に挙げる。ポリエステル、ポリオレフィン等の他の熱可塑性樹脂、無機充填材(タルク、カオリン、ゾノトライト、ワラストナイト、酸化チタン、チタン酸カリウム、ガラス繊維など)、無機充填材と樹脂との親和性を高める為の公知の密着改良剤、難燃剤(ハロゲン化された樹脂、シリコーン系難燃剤、水酸化マグネシウム、水酸化アルミニウム、有機燐酸エステル化合物、ポリ燐酸アンモニウム、赤燐など)、滴下防止効果を示すフッ素系ポリマー、可塑剤(オイル、低分子量ポリオレフィン、ポリエチレングリコール、脂肪酸エステル類等)及び、三酸化アンチモン等の難燃助剤、着色用カーボンブラック、カーボンファイバー、帯電防止剤、各種過酸化物、酸化亜鉛、硫化亜鉛、酸化防止剤、紫外線吸収剤、光安定剤等である。
【0134】
これらの成分の具体的な添加量は、成分(A)〜(E)の合計100質量部に対して、合計で50質量部を超えない範囲である。
【0135】
特に、本発明の組成物に無機充填材としてウォラストナイトを配合することが好ましい。ウォラストナイトは、珪酸カルシウムを成分とする天然鉱物を精製、粉砕及び分級したものである。また、人工的に合成したものも使用可能である。一般的にはガラス繊維の代替として、平均繊維径40μmで繊維長600μmの大粒子径のものが広く用いられているが、本発明では平均粒子径が2〜9μmの範囲でありアスペクト比が5以上のものが、好ましくは平均粒子径が3〜7μmの範囲でアスペクト比が5以上のものが使用される。さらには、アスペクト比の異なる2種以上のウォラストナイト混合物である事がより好ましい。具体的には、アスペクト比5以上のウォラストナイトとアスペクト比5未満のウォラストナイトの混合物である。
【0136】
これらウォラストナイトには、表面処理剤として、高級脂肪酸またはそのエステル、塩等の誘導体(例えば、ステアリン酸、オレイン酸、パルミチン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、ステアリン酸アミド、ステアリン酸エチルエステルなど)やカップリング剤(例えば、シラン系、チタネート系、アルミニウム系、ジルコニウム系など)を必要により使用することが出来る。その使用量はウォラストナイトに対して0.05質量%〜5質量%の範囲である。
【0137】
また、本発明の組成物に無機充填材としてタルクを配合することも好ましい。タルクの使用により、表面外観が良好であり、熱膨張率の低下が期待できる。タルクは平均粒径が約2.0〜5.0μmのケイ酸マグネシウムが好ましく、特に約3.0〜5.0μmのものが好ましい。
【0138】
これらの無機充填材は、タルク、カオリン、ワラストナイト、酸化チタン、チタン酸カリウム、ガラス繊維等の混合物であっても構わない。
【0139】
これらの無機充填材の添加量は、成分(A)〜(E)の合計100質量部に対し、5〜40質量部であることが好ましい。より好ましくは、5〜25質量部である。
【0140】
上記の成分を溶融混練して本発明の樹脂組成物を得るために用いる具体的な加工機械としては、例えば、単軸押出機、二軸押出機、ロール、ニーダー、ブラベンダープラストグラフ、バンバリーミキサー等が挙げられるが、中でも二軸押出機が好ましく、特に、上流側供給口と1カ所以上の下流側供給口を備えた二軸押出機が最も好ましい。
【0141】
この際の溶融混練温度は特に限定されるものではないが、通常240〜360℃の中から好適な組成物が得られる条件を任意に選ぶことができる。
【0142】
本発明の好ましい導電性樹脂組成物の製造方法としては、ポリフェニレンエーテル(B)、ゴム状重合体(C)及び相溶化剤として機能する第2の低分子量変性剤化合物(E)の混合物を溶融し、これにポリアミド(A)と導電性炭素材料(D)とを含む上記のマスターバッチを配合することが好ましい。具体的には、上流側供給口と下流側供給口を備えた二軸押出機を用い、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)及び相溶化剤として機能する第2の低分子量変性剤化合物(E)を供給し溶融混練した後、下流側供給口よりポリアミド(A)と導電性炭素材料(D)(または上記のマスターバッチ)を供給し溶融混練する方法等が挙げられる。また、溶融混錬の際、押出機下流のベント脱気口より真空脱気することがさらに好ましい。
【0143】
このようにして得られる本発明の導電性樹脂組成物を、公知の種々の方法により成形することにより樹脂成形体を得ることができる。
【0144】
本発明でいう樹脂成形体とは、射出成形したものに限定されず、押出成形されたシート、フィルム、ペレットおよび2次加工された射出成形体まで包含される。
【0145】
本発明においては、204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることが好ましく、0.10以下であることがより好ましい。該低分子量成分の量が0.15以下であることにより、本発明の成形体は塗装密着性、特に熱暴露後の塗装密着性が非常に優れる。
【0146】
ここで成形体の表面とは、樹脂成形体をクロロホルム溶媒に室温で約1時間浸漬後、表面から溶け出す領域をいう。
【0147】
ここでいう熱処理後の成形体表面のクロロホルム可溶成分の中で、サイズ排除クロマトグラフィーの示差屈折計を用いて測定される溶出時間が22分〜23.5分である成分のピークの高さの比は以下の方法で測定することが可能である。
【0148】
204℃で40分間の熱処理後の樹脂成形体を幅約8cm、長さ約8cm(肉厚は特に制限はない)の形状に切削し、12.5cmのシャーレなどに50mlのクロロホルム(日本国和光純薬製、特級)を加えた中に、切削した成形体を室温で約1時間浸漬させる。浸漬した成形体を取り出した後の抽出液を40ml取り、溶媒を蒸発させ該抽出液を乾固させる。蒸発乾固の条件としては、室温で24時間行なうことができる。
【0149】
乾固させた試料にテトラヒドロフラン(日本国和光純薬製、HPLC用)を2ml加え、試料溶液としたものを以下のサイズ排除クロマトグラフィーの示差屈折計を用いて分析を行う。
[サイズ排除クロマトグラフィー装置]
オンライン脱気装置(DE):日本国(株)イーアールシー製ERC−3322
送液ポンプ(PO):日本国東ソー(株)製DP−8020
カラムオーブン(CO):日本国東ソー(株)製CO−8010
紫外可視吸光光度検出器(UV):日本国東ソー(株)製UV−8010
示差屈折検出器(RI):日本国東ソー(株)製RI−8012
【0150】
上記の装置は図1のように接続して用いる。また、溶媒にテトラヒドロフラン(日本国和光純薬製、HPLC用)を使用する。分析条件は、流速1ml/分、オーブン温度40℃、UV検出波長254nmとする。なお、カラムオーブン中には以下のサイズ排除クロマトグラフィー用カラムを配する。
ガードカラム:東ソー(株)製TSKguardcolumn HXL−L
カラム1および2:東ソー(株)製TSKgel G1000HXL
カラム3:東ソー(株)製TSKgel G2000HXL
上記ガードカラム、カラム1、カラム2、カラム3をこの順に直列に接続し、溶媒をガードカラムからカラム3の方向に流す。
【0151】
なお、上述のサイズ排除クロマトグラフィー装置の示差屈折計を用いて、基準サンプルとして、Irganox1076(スイス国Ciba Specialty Chemicals製)を分析したところ、溶出時間は17.8分であった。この基準サンプルであるIrganox1076の溶出時間が17.8分よりずれた場合、((Irganox1076測定時の溶出時間(分)−17.8(分))時間分を試料溶液の溶出時間(分)に加算して補正を行う。
【0152】
本発明の組成物が導電性カーボンブラックを含む樹脂成形体の場合は、反射法光学顕微鏡を用いて100倍で、視野の合計10mm2を観察した際に、導電性カーボンブラックの一部が、面積8μm2以上の二次凝集体として存在し、樹脂組成物中の全導電性カーボンブラックに対する、面積8μm2以上の二次凝集体の存在比率が、20質量%以上であることが好ましい。より好ましくは50質量%以上、さらに好ましくは50〜80質量%である。
【0153】
ここでいう導電性カーボンブラックの二次凝集体の大きさおよび存在比率は、以下に説明する方法で測定することができる。反射法光学顕微鏡(PME3:日本国オリンパス社製)を用いて、観察用試料の平滑面におけるカーボンブラックの分散状態を倍率100倍で観察し、撮影を行う。さらに、撮影された反射顕微鏡写真を、スキャナ(CC−600PX:日本国エプソン社製)にてデジタル画像化する(取り込み解像度は400dpi)。得られたデジタル画像の視野内で観察される黒色相(カーボンブラックの二次凝集体)の占める面積分率を、画像処理ソフト(Image−Pro PLUS Ver.4.0J:米国MEDIA CYBERNETICS製)を用いて自動測定で解析する。測定は以下の解析条件及び手順で行う。
(1)フィルター:メディアン(インパルスノイズの除去、強度:3×3ピクセル、回数:2回)
(2)フィルター:平坦化(オブジェクト幅20ピクセル、背景:明るい)
(3)面積分率測定条件:面積が8μm2以上(平均径3μm以上に対応:径3μmの円では7.06μm2)、且つ楕円長短軸比(重心を通る径を2°刻みで測定した長軸と短軸の直径の比)が2以下(細長い形状の二次凝集体と区別しにくい試料切削時のメスキズ等を排除するため)を満足し、さらに、輝度レンジ(256階色)のヒストグラム表示において、灰白色の相(ポリアミド−ポリフェニレンエーテル相)より黒い側の色相のレンジを抽出対象として検出する。なお、上記に自動カウントされない楕円長短軸比が2より大きく、明らかに二次凝集体と目視で判断できる大きな黒色相は、手動で黒色相の周囲をトレースし結果に付加する。また、フィルターで削除できなかった試料切削時の小さなメスキズがある場合は手動で削除する。
【0154】
上記方法により、視野内の黒色相である二次凝集体が占める面積分率を算出し、10枚の写真(合計視野10mm2)の平均値を求める。ここで、二次凝集体が占める体積分率は切断面に現れる面積分率に等しいことが一般的に成り立つため、面積分率を体積分率に置き換える。次に、カーボンブラックの真比重1.9および樹脂の比重1.1を用いて、体積分率を質量分率に変換することにより、面積が8μm2以上の二次凝集体の質量分率を算出する。この二次凝集体の質量分率を全カーボンブラックの質量分率(仕込み量)で除して、樹脂中の全カーボンブラックに対する面積が8μm2以上の二次凝集体の存在比率を算出する。
【0155】
本発明の成形体は各種部品としては用いることができる。そのような部品の例としては、ICトレー材料、各種ディスクプレーヤー等のシャーシー、キャビネット等の電気・電子部品、各種コンピューターおよびその周辺機器等のOA部品や機械部品、さらにはオートバイのカウルや、自動車のフェンダー、ドアーパネル、フロントパネル、リアパネル、ロッカーパネル、リアバンパーパネル、バックドアガーニッシュ、エンブレムガーニッシュ、燃料注入口パネル、オーバーフェンダー、アウタードアハンドル、ドアミラーハウジング、ボンネンットエアインテーク、バンパー、バンパーガード、ルーフレール、ルーフレールレッグ、ピラー、ピラーカバー、ホイールカバー、スポイラー等に代表される各種エアロパーツ、各種モール、エンブレムといった外装部品や、インストゥルメントパネル、コンソールボックス、トリム等に代表される内装部品等が挙げられる。
【0156】
これらの中でも、静電塗装可能な自動車の外板部品に好適に使用可能であり、特に、自動車フェンダーに好適に使用できる。
【図面の簡単な説明】
【0157】
【図1】図1は、で用いたサイズ排除クロマトグラフィー装置類の接続を示す概略図である。
【図2(a)】図2(a)は、導電性炭素材料(D)の量が1.9質量%である実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図である。
【図2(b)】図2(b)は、導電性炭素材料(D)の量が2.5質量%である実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図である。
【発明を実施するための最良の形態】
【0158】
以下、実施例及び比較例により本発明を具体的に説明するが、本発明はこれらによって何ら限定されるものではない。
【0159】
(使用した原料)
実施例及び比較例において使用した原料は次のとおりである。
【0160】
(1)ポリアミド(A):ポリアミド66(以下、PA66と略記)
数平均分子量=14,000
末端アミノ基濃度=30ミリ等量/kg
末端カルボキシル基濃度=100ミリ等量/kg
Cu濃度=30ppm含有(ヨウ化銅として添加)
【0161】
(2)ポリフェニレンエーテル(B):ポリ(2,6−ジメチル−1,4−フェニレンエーテル)
(2−1)ポリフェニレンエーテル(以下、PPE−1と略記)
日本国特開2002−3596号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状ポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が0.1質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。質量減少率は、下記の式により計算した。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテルの質量−真空乾燥後のポリフェニレンエーテルの質量)/真空乾燥前のポリフェニレンエーテルの質量}×100。
下記のPPE−2〜PPE−4及びM−PPEについても同様に計算した。
ポリフェニレンエーテル粉末の還元粘度は0.52dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−2)ポリフェニレンエーテル(以下、PPE−2と略記)
日本国特開昭64−33131号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が1.3質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。ポリフェニレンエーテル粉末の還元粘度は0.54dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−3)ポリフェニレンエーテル(以下、PPE−3と略記)
日本国特開2002−3596号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発性物質の量が0.9質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。ポリフェニレンエーテル粉末の還元粘度は0.50dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−4)ポリフェニレンエーテル(以下、PPE−4と略記)
日本国特開昭64−33131号公報に従って、ジブチルアミンの存在下に、2,6−キシレノールを酸化カップリング重合し、得られた反応生成物を乾燥して白色粉末状のポリフェニレンエーテルを得た。得られた白色粉末状のポリフェニレンエーテルを、120℃の熱風乾燥機を用いて、揮発成分の量が2.2質量%(180℃、1.3×103Paで真空乾燥を1時間行った際の質量減少率)になるように乾燥した。PPE粉末の還元粘度は0.42dl/g(0.5g/dl、クロロホルム溶液、30℃測定)であった。
(2−5)無水マレイン酸変性ポリフェニレンエーテル
(以下、M−PPEと略記)
(PPE−4)100質量部に対して、ジ−tert−ブチルパーオキサイド(ラジカル開始剤)0.1質量部および無水マレイン酸1.5質量部を添加し、二軸押出機を用いてシリンダー温度320℃で溶融混練してM−PPEを作製した。
無水マレイン酸の付加率:0.5質量%
未反応の無水マレイン酸:0.008質量%
揮発性物質:0.3質量%
【0162】
(3)導電性炭素材料(D)(導電性カーボンブラックおよび導電性カーボンフィブリル)
(3−1)導電性カーボンブラック(以下、CBと略記)
商品名:ケッチェンブラックEC−600JD
(日本国ケッチェンブラックインターナショナル社製)
上記のCBを用いて以下の方法でマスターバッチを製造した。上流側と下流側にそれぞれ少なくとも1箇所の供給口を有する二軸押出機を用いて、シリンダー温度270℃に設定した条件下で、上流側供給口より90質量部のポリアミド66を供給し、溶融させた後、下流側供給口より10質量部のCBを添加して溶融混練し、マスターバッチを作製した。(以下、PA66/CB−MBと略記)なお、PA66/CB−MBの連続した視野3mm2の断面を反射法光学顕微鏡(PME3:日本国オリンパス社製)を用いて、50倍の倍率で観察し、導電性カーボンブラックの長径が20μm以上100μm以下の凝集粒子数を目視で数えた。その結果、凝集粒子の数は、19個であった。
(3−2)導電性カーボンフィブリル(以下、CNTと略記)
ハイペリオンマスターバッチ(米国ハイペリオンキャタリストインターナショナル社製):平均直径約10〜15μmのカーボンフィブリル20質量%含有のナイロン66マスターバッチ(以下、PA66/CNT−MBと略記)
【0163】
(4)ゴム状重合体(C)
(4−1)ポリスチレン−水素添加ポリブタジエン−ポリスチレン(以下、SEBS−1と略記)
数平均分子量=246,000
スチレン成分合計含有量(スチレンブロック含有量)
=33質量%
(4−2)ポリスチレン−水素添加ポリブタジエン−ポリスチレン(以下、SEBS−2と略記)
数平均分子量=77,000
スチレン成分合計含有量(スチレンブロック含有量)
=67質量%
【0164】
(5)低分子量変性剤化合物(E)(相溶化剤):無水マレイン酸(日本国三菱化学(株)製)(以下、MAHと略記)
【0165】
(6)無機充填材
(6−1)ウォラストナイト(カナダ国ナイコ社製)
平均粒子径4μmで平均繊維長32μm(アスペクト比=8)、1000℃での加熱減量が0.7質量%のウォラストナイトをベースに0.5%アミノシラン水溶液で処理。
【0166】
(測定方法)
以下に、導電性(体積抵抗率)、Izod衝撃強度、成形機内滞留時の熱安定性、目やに発生量および塗装密着性の測定方法について述べる。
【0167】
(1)目やに発生量
押出機を1時間運転して、その間に長さ5mm以上の大きさになった目やにの数を数えた。
【0168】
(2)体積抵抗率
得られた樹脂組成物ペレットを、日本国東芝機械(株)製IS−80EPN成形機(シリンダー温度を280℃、金型温度を80℃に設定)を用いて、ダンベルバーとしてISO294の記載の如く成形した。この試験片の両端を折り取って均一な断面積10×4mm、長さ約70mmで両端に破断面をもつ試験片とした。試験片の折り取り方については、−75〜−70℃のドライアイス/メタノールの中に、予めカッターナイフでキズをつけた試験片を1時間浸漬後、折り取る方法で行った。この両端の破断面に銀塗料を塗布し、エレクトロメーター(日本国アドバンテスト製、R8340A)を用いて、250Vの印加電圧で両方の破断面間の体積抵抗率を測定した。測定は5個の異なる試験片に対して実施し、その加算平均をもって、体積抵抗率とした。
【0169】
(3)Izod衝撃強度
得られた樹脂組成物ペレットを、日本国東芝機械(株)製IS−80EPN成形機(シリンダー温度を280℃、金型温度を80℃に設定)にて厚み3.2mmの短冊成形片を成形し、ASTM D256に準拠し、厚み3.2mmのノッチ付衝撃強度を測定した。
【0170】
(4)成形機内滞留時の熱安定性
得られた樹脂組成物ペレットを、日本国日精樹脂工業(株)製FE120成形機(シャットオフノズル付き)を用いて、120mm×80mm(厚み3mm)の平板を成形した。成形条件は、シリンダー温度:310℃、射出時間+保圧時間:15秒(内、射出時間0.7秒および3秒)、冷却時間:12〜70秒、インターバル時間(型開き+成形品取り出し):5秒とした。滞留時間は、(射出時間+保圧時間+冷却時間+インターバル時間)×(最大計量値/1ショットの計量値)より算出した。成形機内滞留時の熱安定性の判断は、樹脂組成物を所定時間滞留後、成形した平板の外観(シルバー発生の程度)を目視にて行った。平板に少量のシルバーが発生するまでの滞留時間(限界滞留時間)を測定し、それに基づいて成形機内滞留時の熱安定性を評価した。
【0171】
(5)塗装密着性試験
得られた樹脂組成物ペレットを、射出成形機(日本国日精樹脂工業(株)製:FS80S)を用い、シリンダー温度305℃、金型温度80℃、充填時間1秒で10cm×10cm(厚み2mm)の平板を作成した。塗装密着性の測定は、成形後の平板と204℃で40分間の熱暴露後の平板について実施した。自動吹き付け塗装装置を用いて、塗膜厚みが20μmになるよう調節し、塗装を実施した。塗料はアクリルウレタン系塗料(日本国オリジン電気社製、OP−Z−NY)を使用した。吹き付け塗装後、150℃の温度で20分間焼付けを実施した。
【0172】
塗装実施後、温度23℃、湿度50%の環境下に静置し、24時間後、2cm×2cmの範囲で一目が2mm四方になるようにカッターナイフで碁盤目状に傷を付け(合計で100目となる)、セロテープ貼付し一気に引き剥がす塗膜剥離試験を実施した。100目の内、剥離試験後に剥離せずに残った碁盤目数を測定した。
【0173】
(6)PPEの重量平均分子量(Mw)および数平均分子量(Mn)の測定
上記の塗装密着性試験におけるのと同様の方法で成形して得られた10cm×10cm(厚み2mm)の平板を約0.5mm程度に粉砕し、50mlのクロロホルムでソックスレー抽出した。得られたクロロホルム溶液(クロロホルムに可溶の成分:主としてポリフェニレンエーテルとブロック共重合体が含有されている)を、GPCを用いて、紫外分光検出器で測定し、標準ポリスチレンで換算しMwを算出した。この時、同時に溶出してくるブロック共重合体を検出しないよう測定紫外線波長を283nmに設定した。
【0174】
(7)204℃で40分間の熱暴露後の成形体表面のクロロホルム可溶成分の分析
上記の塗装密着性試験におけるのと同様の方法で成形して得られた10cm×10cm(厚み2mm)の平板を204℃で40分間の熱処理後、約8cm×8cm(肉厚は2mm)の形状に切削し、直径約12.5cmのシャーレに50mlのクロロホルム(日本国和光純薬製、特級)を加えた中に、切削した平板を約1時間浸漬させた。浸漬した平板を取り出した後の抽出液を40ml取り、溶媒を蒸発乾固させた。
【0175】
乾固させた試料にテトラヒドロフラン(日本国和光純薬製、HPLC用)を2ml加え、試料溶液としたものをサイズ排除クロマトグラフィーの示差屈折計を用いて測定される溶出時間が22分〜23.5分である成分のピークの高さの比(=(溶出時間が22分〜23.5分のピーク高さの和)/(溶出時間が14分〜15分のピーク高さの和))を算出した。
【0176】
用いたサイズ排除クロマトグラフィー装置は以下の通りである。
オンライン脱気装置(DE):日本国(株)イーアールシー製ERC−3322
送液ポンプ(PO):日本国東ソー(株)製DP−8020
カラムオーブン(CO):日本国東ソー(株)製CO−8010
紫外可視吸光光度検出器(UV):日本国東ソー(株)製UV−8010
示差屈折検出器(RI):日本国東ソー(株)製RI−8012
【0177】
上記の装置を図1のように接続して用いた。また、溶媒にテトラヒドロフラン(日本国和光純薬製、HPLC用)を使用した。分析条件は、流速1ml/分、オーブン温度40℃、UV検出波長254nmとした。なお、カラムオーブン中には以下のサイズ排除クロマトグラフィー用カラムを配した。
ガードカラム:日本国(株)製TSKguardcolumn HXL−L
カラム1および2:日本国東ソー(株)製TSKgel G1000HXL
カラム3:日本国東ソー(株)製TSKgel G2000HXL
上記ガードカラム、カラム1、カラム2、カラム3をこの順に直列に接続し、溶媒をガードカラムからカラム3の方向に流した。
【0178】
なお、上述のサイズ排除クロマトグラフィー装置の示差屈折計を用いて、基準サンプルとして、Irganox1076(スイス国Ciba Specialty Chemicals製)を分析したところ、溶出時間は17.8分であった。
実施例1〜6、8、9および比較例1〜6
【0179】
押出機上流側1ヵ所と下流側に1ヵ所の供給口を備えた二軸押出機[ZSK−58MC:ドイツ国ウェルナー&フライデラー社製]を用いて、シリンダー温度300℃に設定した条件下で、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)および変性剤(E)(相溶化剤)を供給し溶融混練した後、下流側供給口よりポリアミド66(A)と導電性炭素材料(D)(導電性カーボンブラックあるいは導電性カーボンフィブリル)を含むマスターバッチ(PA66/CB−MBあるいはPA66/CNT−MB)を供給して、導電性樹脂組成物ペレットを作製した。この際、押出時における目やに発生量を測定した。さらに得られた組成物ペレットを成形し、前述した方法で成形機内滞留時の熱安定性、体積抵抗率、Izod衝撃強度および塗装密着性を測定した。また、平板を用いて前述した方法で、PPEの重量平均分子量(Mw)、数平均分子量(Mn)および204℃で40分間の熱暴露後の成形体表面のクロロホルム可溶成分を分析した。導電性カーボンブラック量が1.9質量%の場合およびカーボンフィブリルの量が2.0質量%における物性値を組成と共に表1に示した。また、導電性カーボンブラック量が2.5質量%における物性値を組成と共に表2に示した。
【0180】
【表1】
【0181】
【表2】
【0182】
表1からわかるように、成分(A)〜(E)の合計質量に対し、成分(D)の量が0.2〜3質量%であり、変性剤(E)(相溶化剤)の量(E質量%)が0.01質量%より大きく0.20質量%未満であって、且つ成分(B)(PPE)の質量に対し、成分(B)中に残存する揮発性物質の量(a質量%)が、a≦−7.3×E+1.83を満足する場合には、押出機内および成形内滞留時の熱安定性が大幅に向上すると共に、特に熱暴露後の塗装密着性が著しく向上することが明らかとなった。図2(a)は、成分(D)(カーボンブラックあるいはカーボンフィブリル)の量が1.9質量%における実施例と比較例に関して、ポリフェニレンエーテル中の揮発性物質の量と相溶化剤(第2の低分子量変性剤化合物)の量との関係を示した説明図であるが、本発明における実施例と比較例において本発明の要件が満たされているか否かが明確に示されている。また、表2および図2(b)に示した成分(D)(カーボンブラックあるいはカーボンフィブリル)の量が2.5質量%の場合においても同様である。
【0183】
なお、実施例1〜5の導電性カーボンブラックを含む樹脂成形体について、反射法光学顕微鏡(100倍)を用いて、視野の合計10mm2を観察した際の面積8μm2以上の二次凝集体の存在比率(樹脂組成物中の全導電性カーボンブラックに対する)を測定した。その結果、面積8μm2以上の二次凝集体の存在比率(樹脂組成物中の全導電性カーボンブラックに対する)は、54〜69質量%であった。
【実施例7】
【0184】
押出機上流側1ヵ所と下流側に2ヵ所の供給口を備えた二軸押出機[ZSK−58MC:ドイツ国ウェルナー&フライデラー社製]を用いて、シリンダー温度300℃に設定した条件下で、上流側供給口よりポリフェニレンエーテル(B)、ゴム状重合体(C)および変性剤(E)(相溶化剤)を供給し溶融混練した後、押出機の中段に設けられた下流側供給口(1)よりポリアミド66と導電性付与剤を含むマスターバッチ(PA/CB−MB)を供給し、押出機の後段に設けられた下流側供給口(2)よりウォラストナイトを供給して、ポリアミド66(成分(A))、ポリフェニレンエーテル(B)、ゴム状重合体(C)、導電性炭素材料(D)、変性剤(E)およびウォラストナイトからなる樹脂組成物ペレットを作製した以外は実施例1と同様に行った。測定結果については、表1に併記した。
【0185】
本発明の組成物に無機充填材としてウォラストナイトを配合した場合も、押出時および成形時の成形機内滞留時の熱安定性が大幅に向上すると共に、特に熱暴露後の塗装密着性が著しく向上する結果を得た。さらに、無機充填材を添加することにより、著しく体積抵抗率が低下し、導電性も向上することが確認できた。
【産業上の利用可能性】
【0186】
本発明の熱可塑性樹脂組成物は、導電性カーボンブラック及び/又は導電性カーボンフィブリル、変性剤(相溶化剤)およびポリフェニレンエーテル中に残存する揮発性物質の量を特定の範囲にすることにより、耐衝撃性及び熱安定性に優れるのみならず、押出成形時の目やにの発生及びストランド切れを抑制することができるので、電気・電子部品、OA部品、車両部品、機械部品などの幅広い分野に使用することができる。特に、本発明の導電性樹脂組成物は、大型成形体の製造に用いた場合でも、成形時の成形機内滞留時の熱安定性が非常に優れ、且つ静電塗装可能なレベルの導電性を有すると同時に塗装密着性、特に熱暴露後の塗装密着性が非常に優れるため、自動車外板のような静電塗装を施す大型成形体の製造に非常に有利に用いることができる。
【特許請求の範囲】
【請求項1】
ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【請求項2】
該成分(A)〜(E)の混合物中における該低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.02〜0.18質量%であることを特徴とする請求項1に記載の樹脂組成物。
【請求項3】
該成分(A)〜(E)の混合物中における該低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して、0.02〜0.15質量%であることを特徴とする請求項1に記載の樹脂組成物。
【請求項4】
該成分(A)〜(E)の混合物中における該導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.5〜2.5質量%であることを特徴とする請求項1〜3のいずれかに記載の樹脂組成物。
【請求項5】
該揮発性物質の量(a)が、1.0質量%以下であることを特徴とする請求項1〜4のいずれかに記載の樹脂組成物。
【請求項6】
該成分(D)が、導電性カーボンブラックであることを特徴とする請求項1〜5のいずれかに記載の樹脂組成物。
【請求項7】
該成分(D)としての導電性カーボンブラックのジブチルフタレート(DBP)吸収量が250ml/100g以上であることを特徴とする請求項1〜6のいずれかに記載の樹脂組成物。
【請求項8】
該第1の低分子量変性剤化合物(b)及び該第2の低分子量変性剤化合物(E)の各々がそれぞれ独立して無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸から選ばれる少なくとも一種の化合物であることを特徴とする請求項1〜7のいずれかに記載の樹脂組成物。
【請求項9】
該ゴム状重合体(C)が、少なくとも1種の、数平均分子量が120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物であって、該ブロック共重合体(C−1)及び(C−2)の各々が、それぞれ独立して、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックとを含んでなることを特徴とする請求項1〜8のいずれかに記載の樹脂組成物。
【請求項10】
該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
を含み、
該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%であることを特徴とする請求項9に記載の樹脂組成物。
【請求項11】
該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることを特徴とする請求項1〜10のいずれかに記載の樹脂組成物。
【請求項12】
該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、55,000〜70,000であり、かつ分子量分布が、3.0以下であることを特徴とする請求項11に記載の樹脂組成物。
【請求項13】
該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法によって行うことを特徴とする請求項1〜12のいずれかに記載の樹脂組成物。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを、該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。
【請求項14】
該マスターバッチが、該成分(D)として導電性カーボンブラックを含んでなり、該マスターバッチを、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在するマスターバッチであることを特徴とする請求項13に記載の樹脂組成物。
【請求項15】
該成分(A)〜(E)の混合物が更に少なくとも1種の無機充填材を、該成分(A)〜(E)の合計100質量部に対して、5〜25質量部含むことを特徴とする請求項1〜14のいずれかに記載の樹脂組成物。
【請求項16】
成形体である請求項1〜15のいずれかに記載の樹脂組成物。
【請求項17】
204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることを特徴とする請求項16に記載の樹脂組成物。
【請求項18】
該少なくとも1種の低分子量成分の量が、0.10以下であることを特徴とする請求項17に記載の樹脂組成物。
【請求項19】
オンライン塗装されてなる自動車フェンダーであることを特徴とする請求項16〜18に記載の樹脂組成物。
【請求項1】
ポリアミド(A)、
末端未変性ポリフェニレンエーテル及び末端変性ポリフェニレンエーテルからなる群より選ばれる少なくとも1種のポリフェニレンエーテル(B)であって、該末端変性ポリフェニレンエーテルは、ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第1の低分子量変性剤化合物(b)を末端未変性ポリフェニレンエーテルと溶融混練することによって得られ、
ゴム状重合体(C)、
導電性カーボンブラック及び導電性カーボンフィブリルよりなる群から選ばれる少なくとも1種の導電性炭素材料(D)、及び
ポリアミド(A)とポリフェニレンエーテル(B)の相溶化剤として機能する第2の低分子量変性剤化合物(E)
を含んでなる混合物を溶融混練することによって得られる導電性樹脂組成物であって、第1の低分子量変性剤化合物(b)と第2の低分子量変性剤化合物(E)は同じでも異なっていてもよく、該混合物が以下の特性(1)〜(3)を有することを特徴とする導電性樹脂組成物。
(1)該少なくとも1種の導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.2〜3質量%。
(2)該第2の低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.01質量%を越え、0.20質量%未満。
(3)該少なくとも1種のポリフェニレンエーテル(B)中に存在する揮発性物質の量(a)が式:
0≦a≦−7.3×E+1.83
(式中、Eは成分(E)の量(質量%)を表す)
を満足し、
該揮発性物質の量(a)は、該少なくとも1種のポリフェニレンエーテル(B)を180℃、真空度1.3×102〜6.7×103Paの条件下で1時間真空乾燥に付した際の、該少なくとも1種のポリフェニレンエーテル(B)の質量減少率(質量%)として表され、該質量減少率は、下記式で算出される。
質量減少率(質量%)={(真空乾燥前のポリフェニレンエーテル(B)の質量−真空乾燥後のポリフェニレンエーテル(B)の質量)/真空乾燥前のポリフェニレンエーテル(B)の質量}×100。
【請求項2】
該成分(A)〜(E)の混合物中における該低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して0.02〜0.18質量%であることを特徴とする請求項1に記載の樹脂組成物。
【請求項3】
該成分(A)〜(E)の混合物中における該低分子量変性剤化合物(E)の量が、該成分(A)〜(E)の合計質量に対して、0.02〜0.15質量%であることを特徴とする請求項1に記載の樹脂組成物。
【請求項4】
該成分(A)〜(E)の混合物中における該導電性炭素材料(D)の量が、該成分(A)〜(E)の合計質量に対して0.5〜2.5質量%であることを特徴とする請求項1〜3のいずれかに記載の樹脂組成物。
【請求項5】
該揮発性物質の量(a)が、1.0質量%以下であることを特徴とする請求項1〜4のいずれかに記載の樹脂組成物。
【請求項6】
該成分(D)が、導電性カーボンブラックであることを特徴とする請求項1〜5のいずれかに記載の樹脂組成物。
【請求項7】
該成分(D)としての導電性カーボンブラックのジブチルフタレート(DBP)吸収量が250ml/100g以上であることを特徴とする請求項1〜6のいずれかに記載の樹脂組成物。
【請求項8】
該第1の低分子量変性剤化合物(b)及び該第2の低分子量変性剤化合物(E)の各々がそれぞれ独立して無水マレイン酸、フマル酸、マレイン酸、イタコン酸、無水イタコン酸、シトラコン酸、無水シトラコン酸、アコニット酸、無水アコニット酸、クエン酸及びリンゴ酸から選ばれる少なくとも一種の化合物であることを特徴とする請求項1〜7のいずれかに記載の樹脂組成物。
【請求項9】
該ゴム状重合体(C)が、少なくとも1種の、数平均分子量が120,000以下である比較的低分子量のブロック共重合体(C−1)と、少なくとも1種の、数平均分子量が200,000以上である比較的高分子量のブロック共重合体(C−2)の混合物であって、該ブロック共重合体(C−1)及び(C−2)の各々が、それぞれ独立して、ビニル芳香族炭化水素単量体単位を主体とする少なくとも1つのビニル芳香族炭化水素重合体ブロックと共役ジエン単量体単位を主体とする少なくとも1つの共役ジエン重合体ブロックとを含んでなることを特徴とする請求項1〜8のいずれかに記載の樹脂組成物。
【請求項10】
該少なくとも1種のブロック共重合体(C−1)として、
10〜100質量部のブロック共重合体(C−1a)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%以上90質量%未満であるブロック共重合体(C−1a)、及び
0〜90質量部のブロック共重合体(C−1b)であって、その質量に対する、ビニル芳香族炭化水素重合体ブロック含有量が、55質量%未満であるブロック共重合体(C−1b)
を含み、
該ブロック共重合体(C−1a)及び(C−1b)の合計が100質量%であることを特徴とする請求項9に記載の樹脂組成物。
【請求項11】
該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、50,000以上であり、かつ重量平均分子量(Mw)と数平均分子量(Mn)との比Mw/Mnで表される分子量分布が、3.2以下であることを特徴とする請求項1〜10のいずれかに記載の樹脂組成物。
【請求項12】
該導電性樹脂組成物中の該少なくとも1種のポリフェニレンエーテル(B)の重量平均分子量(Mw)が、55,000〜70,000であり、かつ分子量分布が、3.0以下であることを特徴とする請求項11に記載の樹脂組成物。
【請求項13】
該成分(A)〜(E)の混合物の溶融混練を、下記の工程(1)及び(2)を含む方法によって行うことを特徴とする請求項1〜12のいずれかに記載の樹脂組成物。
(1)該ポリアミド(A)の少なくとも1部と該導電性炭素材料(D)を溶融混練することよりマスターバッチを得、
(2)得られたマスターバッチを、該成分(B)、(C)及び(E)、及び残りのポリアミド(A)がある場合には該残りのポリアミド(A)と溶融混練する。
【請求項14】
該マスターバッチが、該成分(D)として導電性カーボンブラックを含んでなり、該マスターバッチを、光学顕微鏡を用いて連続した3mm2の面を観察した際に、該導電性カーボンブラックの少なくとも一部が、長径20〜100μmの凝集粒子として、1〜100個存在するマスターバッチであることを特徴とする請求項13に記載の樹脂組成物。
【請求項15】
該成分(A)〜(E)の混合物が更に少なくとも1種の無機充填材を、該成分(A)〜(E)の合計100質量部に対して、5〜25質量部含むことを特徴とする請求項1〜14のいずれかに記載の樹脂組成物。
【請求項16】
成形体である請求項1〜15のいずれかに記載の樹脂組成物。
【請求項17】
204℃で40分間熱処理後の該成形体の表面に存在するクロロホルム可溶成分に関して、テトラヒドロフランを溶離液として用いてサイズ排除クロマトグラフィーを行なった際に、溶出時間22分〜23.5分で検出される少なくとも1種の低分子量成分を含み、
該排除クロマトグラフィーにおいて、溶出時間22分〜23.5分において観察される1つのピークの高さ又は複数のピークの高さの合計の、溶出時間14分〜15分において観察される1つのピークの高さ又は複数のピークの高さの合計の比として表される該少なくとも1種の低分子量成分の量が、0.15以下であることを特徴とする請求項16に記載の樹脂組成物。
【請求項18】
該少なくとも1種の低分子量成分の量が、0.10以下であることを特徴とする請求項17に記載の樹脂組成物。
【請求項19】
オンライン塗装されてなる自動車フェンダーであることを特徴とする請求項16〜18に記載の樹脂組成物。
【図1】

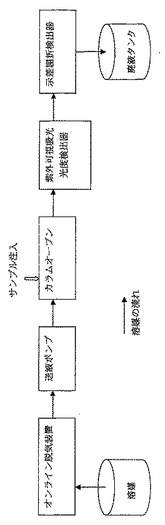
【図2(a)】
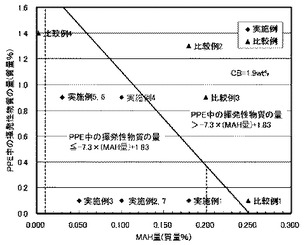
【図2(b)】
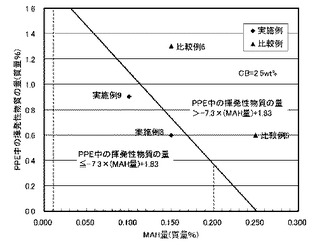
【図2(a)】
【図2(b)】
【国際公開番号】WO2005/026260
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【出願番号】特願2005−513915(P2005−513915)
【国際出願番号】PCT/JP2004/013230
【国際出願日】平成16年9月10日(2004.9.10)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.セロテープ
【出願人】(303046314)旭化成ケミカルズ株式会社 (2,513)
【Fターム(参考)】
【国際公開日】平成17年3月24日(2005.3.24)
【発行日】平成19年11月8日(2007.11.8)
【国際特許分類】
【国際出願番号】PCT/JP2004/013230
【国際出願日】平成16年9月10日(2004.9.10)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.セロテープ
【出願人】(303046314)旭化成ケミカルズ株式会社 (2,513)
【Fターム(参考)】
[ Back to top ]