
小胞体ストレスモデル
【課題】新規な方法により作製されたin vitro小胞体ストレスモデルの提供。
【解決手段】培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法、及び当該作製方法により作製された小胞体ストレスモデル。
【解決手段】培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法、及び当該作製方法により作製された小胞体ストレスモデル。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規な小胞体ストレスモデルに関する。
【背景技術】
【0002】
生体には様々な種類のタンパク質が存在し、各々が生体の構造形成や酵素作用など、生命活動に不可欠な機能を担っている。これらのタンパク質の機能は、その立体構造と密接に関連しており、タンパク質がその機能を果たすには適切な立体構造をとっていることが不可欠である。異常な構造を有するタンパク質は、本来の機能を果たすことができず、そればかりか、ときには毒性を有し、又は疾患の原因となり得る。
【0003】
タンパク質は、生体内で最初に合成されたときは“ひも”上の一次構造であるが、その後小胞体内で高次構造へとフォールディング(folding)される。小胞体内で正しい立体構造を獲得したタンパク質は、ゴルジ装置へと輸送され、そこから分泌経路を介して生体各所に輸送されて、機能を発揮する。一方、不適切なフォールディングによって誤った立体構造を有するタンパク質は、小胞体シャペロンの働きによって正しい構造へと再構成されるか、あるいは小胞体タンパク質分解機構(ER-associated degradation; ERAD)によって分解される。こうしたタンパク質のフォールディングと分解からなる適正な構造のタンパク質を供給するシステムを「タンパク質の品質管理」と呼ぶ。このシステムによって、生体は、異常な構造を有するタンパク質からの悪影響を防いでいる。
【0004】
何らかの原因でタンパク質の品質管理システムが十分に機能しない場合、結果として誤った立体構造を有する異常タンパク質が小胞体内に蓄積する。この状態を小胞体ストレスという。小胞体ストレスに対して、細胞は、タンパク質翻訳の抑制や調節、小胞体シャペロンの発現亢進によるフォールディングの増強、ERAD活性化による異常タンパク質分解促進などの手段によって、異常タンパク質を減少させる。しかし、これらの手段によっても状態が改善しない場合、細胞はアポトーシスを起こして死滅し、異常タンパク質から生体を防御する。これらの一連の応答は、小胞体ストレス応答(unfolded protein response; UPR)と称される。
【0005】
近年、上記小胞体ストレス応答のメカニズムが次第に明らかになってきた。例えば、上記応答手段のうち、翻訳の抑制や調節にはPERK、GADD34、p58IPK、CReP、ATF4等の分子が、フォールディング増強にはATF6、BiP/GRP78、Hspファミリー等の分子が、ERAD活性化にはIRE1、XBP1、RLG1等の分子が、そしてアポトーシスにはCaspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR、TDAG51等の分子が関与していることが知られている(例えば、非特許文献1)。このうち、IRE1、ATF6、XBP1、GRP78等は小胞体ストレスのセンサータンパク質であり、小胞体ストレスの指標として知られている。特に、XBP1は、ATF6によって転写誘導され、またIRE1によってフレームスイッチ型スプライシングを受けるため、ATF6とIRE1の両因子のシグナルを反映する指標である。スプライシングされたXBP1 mRNAを測定することで小胞体ストレスを検出する方法が提唱されている(特許文献1)。
【0006】
また近年、小胞体ストレスが、生体の様々な障害、例えば、糖尿病、神経変性疾患、躁うつ病等の種々の疾患や、免疫応答などの生理機能に深く関与していることが報告されている。生体機能に関する基礎研究のため、又は種々の疾患の診断及び治療手段を開発するためのツールとして小胞体ストレスが注目されている。
【0007】
したがって、小胞体ストレスについて研究するための実験系、すなわち小胞体ストレスモデルを確立することが望まれる。現在までに、小胞体ストレスモデルとしては、ヒトメグシン遺伝子導入トランスジェニックラットモデル(特許文献2)、薬物添加培養細胞モデル(非特許文献2)、UV照射マウスモデル又はヒト皮膚切片モデル(非特許文献3)等が知られている。このうち、トランスジェニックモデルは、作製手順が煩雑であり、また導入遺伝子発現量の制御が難しい。
【0008】
in vitroモデルは、標的細胞へ因子の影響が迅速に現れ、且つ生体の他の組織からの影響を受けることがないことから、因子と細胞応答との関係を詳細に調べるためのモデルとして好ましい。in vitro小胞体ストレスモデルとしては、薬物tunicamycin又はthapsigarginを添加して小胞体ストレスを誘導する培養細胞モデルが知られている(非特許文献2)。しかし、薬物添加によりストレスを誘導する場合、添加した薬物が細胞内に浸透して標的部位に到達するまでの時間、及び薬物の影響が細胞から除去されるまでの時間を制御することが困難である。また、細胞内に浸透した薬物は代謝により分解されるため、添加薬物量は必ずしも誘導ストレス量に反映されない。
【0009】
UV照射によるストレス誘導は、操作が簡便であり、且つ照射後直ちに標的部位に到達し、また照射を止めれば直ちにその影響は細胞内から消えるため、ストレス誘導の時間的制御に有効である。また、UVは薬物のように代謝を受けることがないので、与えた線量が直接的にストレス誘導量として反映され得る。従って、UV照射は、小胞体ストレスモデル作製のための好ましい方法である。しかしながら、UV照射による小胞体ストレスモデルとしては、従来、上記のマウスモデル及びヒト皮膚切片モデル(非特許文献3)のようなin vivoモデルが知られているのみであり、培養細胞モデルのようなin vitroモデルは、UV照射によって作製することはできないことが報告されていた(非特許文献3)。
【0010】
小胞体ストレスのより詳細な研究を可能にするin vitro小胞体ストレスモデルの確立が望まれている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−129970号公報
【特許文献2】特開2004−313192号公報
【非特許文献】
【0012】
【非特許文献1】吉田秀朗, 生化学, 2004, 第76巻第7号, pp617-630
【非特許文献2】Yoshida et al., Cell, 2001, 107:881-891
【非特許文献3】Anand et al., J. Invest. Dermatol., 2005, 125:323-333
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、新規な方法により作製されたin vitro小胞体ストレスモデルを提供する。
【課題を解決するための手段】
【0014】
本発明者らは、培養細胞に特定の条件下でUV照射を行うことにより、従来作製できないと考えられていたUV照射によるin vitro小胞体ストレスモデルを作製することに成功した。
【0015】
すなわち、本発明は、以下に係るものである。
(1)培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法。
(2)UVA照射が1〜100J/cm2のUVA照射である(1)記載の方法。
(3)UVAを照射する工程の後に前記細胞を培養する工程をさらに含む、(1)又は(2)に記載の方法。
(4)UVAを照射する工程により、前記細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を増加させる、(1)〜(3)のいずれか1に記載の方法。
(5)(1)〜(4)のいずれか1に記載の方法によって作製された小胞体ストレスモデル。
(6)培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を測定する工程を含む、小胞体ストレス制御剤のスクリーニング方法。
(7)UVA照射が1〜100J/cm2のUVA照射である(6)記載の方法。
(8)前記発現を測定する工程の前に当該細胞を培養する工程をさらに含む、(6)又は(7)に記載の方法。
(9)前記分子又はそのmRNAの発現の量を定量する工程をさらに含む、(6)〜(8)のいずれか1に記載の方法。
【発明の効果】
【0016】
本発明によれば、従来よりも簡便な手順で、ストレス負荷量がより精密に制御されたin vitro小胞体ストレスモデルを提供することができる。本発明のin vitro小胞体ストレスモデルによれば、従来よりも迅速且つ正確に小胞体ストレスを評価し、また小胞体ストレスを制御する物質を探索することが可能になる。
【図面の簡単な説明】
【0017】
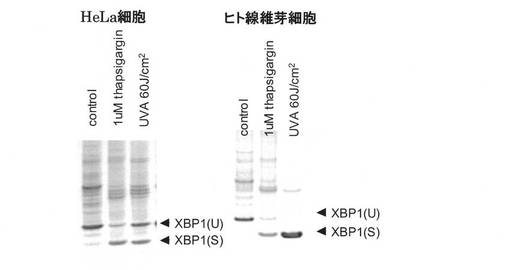
【図1】ヒト正常線維芽細胞及びHeLa細胞における、XBP1 mRNAスプライシングに対するUVA照射の影響。照射4時間後に測定。
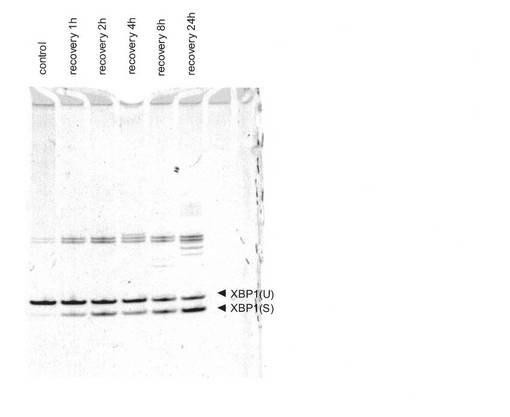
【図2】ヒト正常線維芽細胞におけるXBP1 mRNAスプライシングに対するUVA照射後の時間の影響。UVA線量10J/cm2。
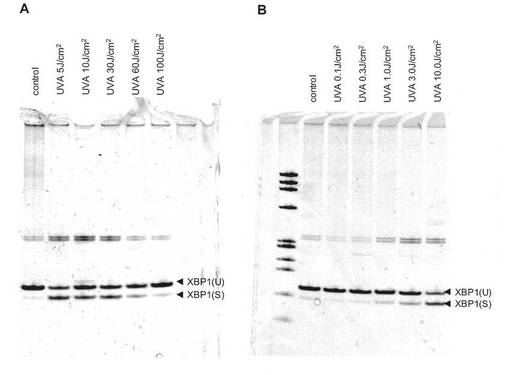
【図3】ヒト正常線維芽細胞におけるXBP1 mRNAスプライシングに対するUVA線量の影響。照射4時間後に測定。
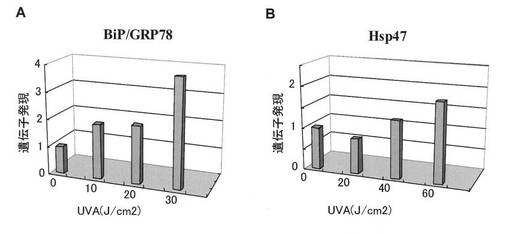
【図4】UPR誘導下における遺伝子発現。A:ヒト正常角化細胞におけるBiP/GRP78 mRNA発現、B:ヒト正常線維芽細胞におけるHsp47 mRNA発現。照射6時間後に測定。
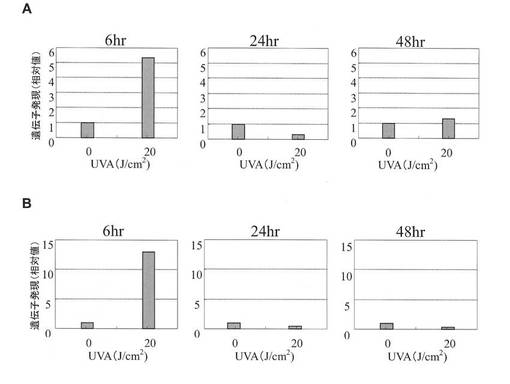
【図5】UPR誘導下における遺伝子発現。A:ヒト正常線維芽細胞におけるGADD34 mRNA発現、B:ヒト正常線維芽細胞におけるGADD153 mRNA発現。照射6時間後に測定。
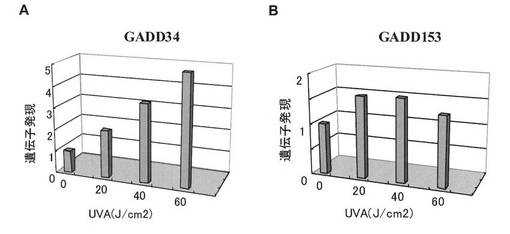
【図6】UPR誘導下における遺伝子発現の経時変化。A:ヒト正常角化細胞におけるGADD34 mRNA発現、B:ヒト正常角化細胞におけるGADD153 mRNA発現。照射6〜48時間後に測定。
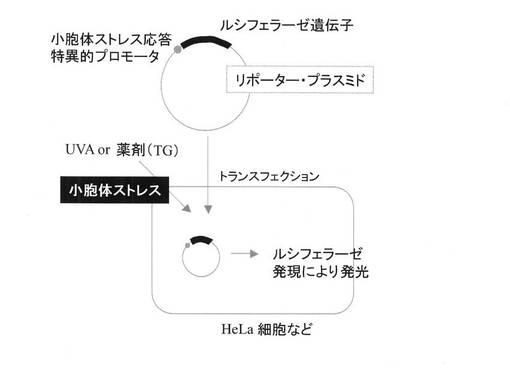
【図7】リポーターアッセイ手順の概念図。
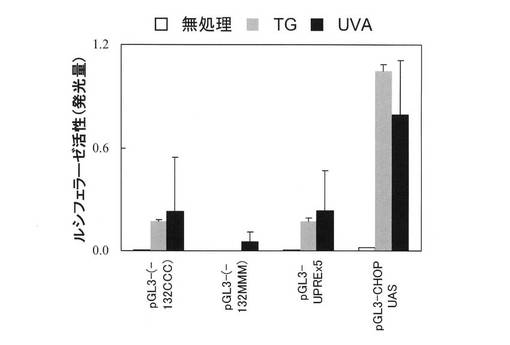
【図8】リポーターアッセイ結果。
【発明を実施するための形態】
【0018】
本発明の小胞体ストレスモデルは、培養細胞にA波紫外線(UVA)を照射することにより作製される。
【0019】
本発明の方法においては、培養細胞をUVA照射することにより、当該細胞に小胞体ストレス応答(unfolded protein response; UPR)を誘導する。UVAを照射される培養細胞は、由来する種や組織又は特定の系統には限定されないが、培養哺乳類細胞が好ましく、培養ヒト細胞がより好ましい。あるいは、培養細胞としては、好ましくは角化細胞、線維芽細胞、HeLa細胞が挙げられる。
【0020】
UVA照射においては、照射エネルギーとして1〜100J/cm2、好ましくは10〜60J/cm2のUVA(波長320〜400nm)に上記培養細胞を曝露する。照射の際、上記範囲内で照射エネルギー量を調節することによって、細胞に与える小胞体ストレスの量を制御することができる。所定の照射エネルギーに細胞を曝露することができる限り、照射の強度及び時間は特に限定されない。照射は連続照射でもよく、あるいは任意の間隔をあけた繰り返し照射でもよい。細胞をUVA照射する具体的手順は当業者に周知である。
【0021】
照射により、細胞に小胞体ストレス応答(UPR)が誘導される。UPRの典型的な例としては、タンパク質翻訳抑制、小胞体シャペロンの発現亢進によるフォールディングの増強、小胞体タンパク質分解機構(ER-associated degradation; ERAD)の活性化による異常タンパク質分解促進等の異常タンパク質を減少させるための応答、アポトーシス誘導等のような異常タンパク質から生体を防御する応答、及び細胞のタンパク質翻訳抑制及び調節が挙げられる。すなわち、上記UVA照射により、細胞内のタンパク質翻訳抑制、フォールディングの増強、ERAD活性化又はアポトーシス誘導、又は細胞のタンパク質翻訳抑制及び調節が促進される。
【0022】
細胞のタンパク質翻訳抑制に関与する分子としては、PERK、GADD34、p58IPK、CReP等が挙げられる。フォールディング増強に関与する分子としては、ATF6、BiP/GRP78、Hsp47等が挙げられる。ERAD活性化に関与する分子としては、IRE1、XBP1、RLG1等が挙げられる。アポトーシスに関与する分子としては、Caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR、TDAG51等が挙げられる。細胞のタンパク質翻訳抑制及び調節に関与する分子としては、ATF4が挙げられる。これらの分子は全て細胞のUPRに関与する分子(UPR関連分子)である。
【0023】
上記細胞内のUPR関連分子の発現は、上記UVA照射により増加する。当該分子の発現増加は、細胞のUPRの増強を反映している。特に、XBP1は、ATF6によって転写誘導され、またIRE1によってフレームスイッチ型スプライシングを受けるため、ATF6とIRE1の両因子のシグナルを反映する分子である。XBP1のスプライシング型mRNA(XBP1(S)mRNA)は、細胞のUPRの指標として従来知られている。
【0024】
したがって、一態様において、本発明の小胞体ストレスモデルの作製方法は、培養細胞にUVAを照射する工程により、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子の少なくとも1の発現、あるいはこれらの分子のmRNAのうちの少なくとも1の発現を増加させる工程を含み得る。当該態様においては、上記の分子又はそのmRNAのうちの少なくとも1の発現を増加させればよいが、上記の分子又はそのmRNAのうちの複数の発現を増加させてもよい。より好ましくは、GADD34、BiP/GRP78、Hsp47、XBP1及びCHOP/GADD153のうちの少なくとも1の発現を増加させる。さらに好ましくは、GADD34 mRNA、BiP/GRP78 mRNA、Hsp47 mRNA、XBP1(S) mRNA及びCHOP/GADD153 mRNAうちの少なくとも1の発現を増加させる。
【0025】
本発明の小胞体ストレスモデルの作製方法においては、好ましくは、培養細胞へのUVA照射の後、当該細胞を培養する。培養時間は、好ましくは1時間以上であり、より好ましくは4時間以上であり、さらに好ましくは1〜48時間であり、なお好ましくは4〜48時間である。上記培養により、当該細胞におけるUPRはより増強される。
【0026】
上記のとおりUPRが誘導された細胞は、in vitro小胞体ストレスモデル細胞として利用され得る。例えば、後記実施例1に示されるように、UVA照射により、培養細胞内で、UPRの指標として従来知られている分子であるXBP1(S)mRNAが増加する。したがって、UVA照射により当該細胞にUPRが誘導されており、この細胞をin vitro小胞体ストレスモデルとして使用できることが分かる。また、後記実施例2〜4に示されるように、上記XBP1(S)mRNAが増加するUVA照射条件と同じ照射条件下で、他のUPR関連分子の発現が培養細胞内で増加する。したがって、本発明の小胞体ストレスモデル細胞においては、UPR関連分子、好ましくは、PERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子の少なくとも1又はそのmRNAのうちの少なくとも1の発現、より好ましくは、GADD34、BiP/GRP78、Hsp47、XBP1及びCHOP/GADD153のうちの少なくとも1の発現、さらに好ましくは、GADD34 mRNA、BiP/GRP78 mRNA、Hsp47 mRNA、XBP1(S) mRNA及びCHOP/GADD153 mRNAうちの少なくとも1の発現が増加している。
【0027】
また本発明は、上記小胞体ストレスモデルを利用した、小胞体ストレス制御剤のスクリーニング方法を提供する。すなわち、本発明のスクリーニング方法は、培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるUPR関連分子又はそのmRNAの発現を測定する工程とを含む。当該スクリーニング方法において、UVA照射される培養細胞の種類、UVA照射の方法、発現を測定されるUPR関連分子又はそのmRNAの種類は、本発明の小胞体ストレスモデルの作製方法の場合と同じである。
【0028】
被験物質は、UVA照射前に添加されても、UVA照射後に添加されても、又はUVA照射と並行して添加されてもよい。その後、細胞のUPR関連分子又はそのmRNAの発現を測定し、被験物質の添加による影響を評価する。例えば、被験物質の添加前後で細胞のUPR関連分子又はそのmRNAの発現を測定し、必要に応じて測定値を定量した後、添加前後で発現を比較する。あるいは、被験物質添加群と対照物質添加群におけるUPR関連分子又はそのmRNAの発現を測定し、必要に応じて測定値を定量した後、両群間で発現の違いを比較する。上記分子又はそのmRNAの発現を変化させる物質を小胞体ストレス制御剤として選択することができる。例えば、被験物質のうち、UPR関連分子又はそのmRNAの発現を増加させる物質は小胞体ストレス亢進剤として、当該分子又はそのmRNA発現を低下させる物質は小胞体ストレス抑制剤として、選択され得る。
【0029】
好ましくは、本発明の小胞体ストレスモデルの作製方法は、培養細胞をUVA照射する工程の後、且つ上記発現を測定する工程の前に、当該細胞を培養する工程をさらに含む。培養時間は、好ましくは1時間以上であり、より好ましくは4時間以上であり、さらに好ましくは1〜48時間であり、なお好ましくは4〜48時間である。
【0030】
本発明において、UPR関連分子又はそのmRNAの発現の測定には、例えば、RI標識、蛍光標識、発光標識、ゲル電気泳動法、免疫沈降、イムノブロッティング、ドットブロット法、ノーザンブロット法、ウエスタンブロット法、RNアーゼプロテクションアッセイ法、ルシフェラーゼ等によるリポーターアッセイ、RT−PCR法、DNA又はプロテインマイクロアレイ等の、当該分野で通常行われる任意の方法を用いることができる。このうち、ウエスタンブロット法、リポーターアッセイ及びRT−PCR法が好ましい。
【実施例】
【0031】
以下、実施例に基づき本発明をさらに詳細に説明する。
【0032】
実施例1:UVA照射による小胞体ストレス応答の誘導
1.細胞の準備
60mmディッシュに10%のコンフルエンシーで正常ヒト線維芽細胞又はHeLa細胞を播種し、炭酸ガスインキュベーターで72時間培養した。
【0033】
2.UVA照射
ディッシュから培地を除去し、2.5mlのPBSを添加した後、細胞にUVAを照射した。
紫外線照射光源としては、キセノン光源装置(MAX-301型、朝日分光(株))にUVA域全体(320-400nm)を透過する広帯域バンドパスフィルタ(朝日分光(株)製)を取り付けたものを使用した。射出光を石英ファイバーで細胞シャーレの真上に誘導した。培養ディッシュの10cmの上方に光出射口を配置して、光源装置に標準装備されているタイマーを使って照射時間をコントロールした。出射光線のエネルギーは、分光放射計測装置(MSR-7000、(株)オプトリサーチ社)を用いて、実使用条件での細胞位置における線量を予め測定した。
培養ディッシュを光ファイバー射出口の直下に置き、射出口の蓋を開けて、細胞に決められた照射エネルギー(0.1J/cm2〜100J/cm2)が与えられるように、所定距離から所定の時間、光を照射した。非照射群をコントロールとした。照射後、PBSを培地と交換し、炭酸ガスインキュベーターで4時間培養した。
あるいは、同様の手順で10J/cm2のUVAを細胞に照射し、照射後、PBSを培地と交換し、炭酸ガスインキュベーターで1〜48時間培養した。培養なし(0時間)の群をコントロールとした。
【0034】
3.薬物による小胞体ストレス応答誘導
上記と同様の手順で調製した培養細胞に、Yoshida et al., Cell 2001, 107:881-891に記載の手順に従って1μM thapsigarginを添加し、細胞の小胞体ストレス応答(UPR)を誘導した。
【0035】
4.RNAの調製
細胞をPBSで1回洗浄し、Sepasol 1mlを加えて細胞溶解液を回収した。クロロホルムを200μl添加し、良く混合した後に遠心し、水相を回収した。2-プロパノールを600μl加えて良く混合し、遠心して沈殿を回収した。沈殿を70%EtOHで洗浄し、沈殿を風乾し、10μlのH2Oに溶解した。RNA量を定量し、1μg/μlの濃度に調整した。
【0036】
5.逆転写反応
4.で調製したRNA 1μgから、SuperScript VILO cDNA合成キット(Invitrogen社)を用いて下記のとおり反応液を調製した。これを逆転写反応(25℃で10分間、42℃で60分間、85℃で5分間)にかけ、cDNA合成を行った。これにH2Oを10μl加え、1μlを分取し、PCR反応に使用した。
5×VILO(商標)Reaction Mix 2μl
10×SuperScript(登録商標)Enzyme Mix 1μl
RNA (1μg) 1μl
H2O 6μl
【0037】
6.PCR反応
5.で調製した逆転写反応産物1μlとKOD-FX DNA polymerase(東洋紡製)を用いて、指示書に従ってPCR反応を実施した。下記のとおりPCR反応液を調製し、94℃で2分→98℃で10秒→68℃又は60℃で60秒の反応サイクルを25〜40回繰り返し、XBP1(S) mRNAのcDNAをPCR増幅した。
2×KOD-FX Buffer 5.0μl
2mM dNTP mix 2.0μl
5μM Forward Primer 0.6μl
5μM Reverse Primer 0.6μl
Template(逆転写反応産物) 1.0μl
KOD-FX(登録商標) 0.2μl
H2O 0.6μl
使用したプライマー配列を以下に示す。
Forward Primer(pXBP1-454FW): AAGCCAAGGGGAATGAAGTG(配列番号1)
Reverse Primer(pXBP1-593RV): AGTCAATACCGCCAGAATCC(配列番号2)
PCR産物を5/20%TAE−PAGE上で展開し、EtBrで染色して検出した。
【0038】
結果を図1〜3に示す。図1に示すように、細胞にUVAを照射した場合、UPR誘導剤thapsigarginを添加した場合と同様に、XBP1(S) mRNAの増加が観察された。XBP1(S) mRNAの増加は、細胞内における構造異常蛋白質の増加を反映しており、よって細胞内でUPRが誘導されたことを表している。したがって、細胞にUVAを照射することにより、細胞のUPRを誘導することができることが示された。
図2に示すように、XBP1(S) mRNAの発現の増加はUVA照射の1時間後から観察され、24時間経過後にもなお高いレベルを維持していた。また図3A及び図3Bに示すように、照射エネルギーが1.0J/cm2以上の場合、それ以下のエネルギーでUVA照射した場合や非照射の場合と比較して、XBP1(S) mRNAの発現が増加した。発現の増加は1.0〜100J/cm2の範囲で観察されたが、60J/cm2を超えるとむしろ発現増加の程度は小さくなる傾向にあった。
【0039】
実施例2:UPR誘導下におけるUPR関連分子の発現
1.細胞の準備
正常ヒト表皮角化細胞(Cascade Biologics社)を凍結バイアルで購入し、融解後に培養フラスコ(T75)内で、無血清増殖培地(EpiLife-KG2、Cascade Biologics社)で添付書の指示通り培養した。シャーレ(直径35 mm)に1回継代した細胞がサブコンフルエンシーまで増殖した時点で照射実験に用いた。
正常ヒト線維芽細胞(Cascade Biologics社)を凍結バイアルで購入し、融解後に培養フラスコ(T75)内で、10% FBS添加DMEM培地(高Glucose、Invitrogen社)で添付書の指示通り培養した。シャーレに継代した細胞がサブコンフルエンシーまで増殖した時点で照射実験に用いた。
【0040】
2.紫外線照射
照射前に培地を37℃のハンクス平衡塩溶液HBS(Ca,Mg含有、フェノールレッド不含)に交換する点を除いて、実施例1と同様の手順でUVA照射を行った。照射後、ハンクス平衡塩溶液を培地に交換して培養インキュベータに戻し、通常通り培養した。
【0041】
3.遺伝子発現解析
遺伝子発現解析は、蛋白質フォールディングの増強に関わる遺伝子であるBiP/GRP78及びHsp47、タンパク質翻訳抑制に関わる分子であるGADD34、ならびにアポトーシス誘導に関連する分子であるGADD153について行った。
UVA照射後、所定の時間培養した細胞ディッシュを2mL PBS(−)で2回洗浄してから、RNAを単離した。RNA単離には、RNeasy(登録商標)RNA 単離キット(QIAGEN社)を取り扱い説明書に従って用いた。細胞溶解には、キットの細胞溶解溶液をシャーレ当たり350μL使用した。取り扱い指示書通りに処理した後、RNAを50μLに溶出・回収した。回収RNAの10μLをcDNA合成に用いた(SuperScript(登録商標)VILO(商標)cDNA合成キット、Invitrogen社)。cDNAサンプルの遺伝子解析は、TaqMan(登録商標)Gene Expression System(Applied Biosystems社)を用いて、リアルタイム定量PCR装置(ABI7500、Applied Biosystems社)で標準的なプロトコルに従って行なった。
サンプル間の遺伝子発現量の相対比較においては、ハウスキーピング遺伝子GAPDHを内部標準とし、対象遺伝子の発現変化率をGAPDHの発現変化率で除して標準化した。
【0042】
BiP/GRP78及びHsp47の遺伝子発現解析の結果を図4A及び図4Bに、GADD34及びGADD153の遺伝子発現解析の結果を、それぞれ図5A及び図5Bに示す。これらの遺伝子の発現は、XBP1(S) mRNAの発現が増加するUVA照射線量において増加した。
また、UVA照射後におけるGADD34及びGADD153の遺伝子発現の経時変化を図6A及び図6Bに示す。これらの遺伝子の発現は、XBP1(S) mRNAの発現が増加するUVA照射後時間帯と同様の時間帯で増加した。
したがって、これらの分子又はその遺伝子の発現は、XBP1(S)mRNAと同様に、細胞におけるUPRの指標となる。
【0043】
実施例3:リポーターアッセイによる転写誘導の検出
96ウェルプレートに10%のコンフルエンシーで正常ヒト線維芽細胞を播種した。炭酸ガスインキュベーターで24時間培養した。
以下の試薬を混合し、この混合液を、2×HBS溶液 100μlに混合しながら滴下した。これを室温で30分静置し、共沈殿を調製した。
250mM CaCl2 10μl
測定用プラスミド 2μl (2μg)
補正用プラスミド 2μl (0.2μg)
H2O 86μl
(細胞10ウェルあたりの分量)
測定用プラスミドとしては、図8に示すプラスミドのいずれかを用いた。
補正用プラスミドとしては、DUAL LUCIFERASE(登録商標) Reporter ASSAY System(Promega社製)に添付されているpRLuc-SV40 promoter plasmidを用いた。
【0044】
図8に示すプラスミドpGL3-(-132CCC)は、human BiP/GRP78 promoterの転写開始点から-132から+7までの配列をpGL3-basic vector(Promega社製)に挿入することにより作製された。このプラスミドの発現は、UPRにおけるフォールディング増強に関与する分子であるATF6が活性化されていることを示す。プラスミドpGL3-(-132MMM)は、human BiP promoterにおいて3つのERSE配列を破壊し、これをpGL3-basic vectorに挿入することにより作製された(Yoshida et al., J Biol. Chem., 1998, 273: 33741-33749)。これは小胞体ストレスで活性化されない陰性対照プラスミドである。プラスミドpGL3-(UPRE)×5は、XBP1が結合する配列(UPRE配列)を5回繋げたものをpGL3-promotervectorに挿入することにより作製された(Wang et al., J. Biol. Chem., 2000, 275: 27013-27020)。このプラスミドの発現はXBP1の活性化を表す。プラスミドpGL3-CHOP UASは、human CHOP/GADD153 promoter(転写開始点から-870〜+17)をpGL3-basic vectorに挿入することにより作製された(Yoshida et al., Mol. Cell. Biol., 2000, 20: 6755-6767)。CHOP promoterには転写因子ATF4が結合する配列があり、その活性化はATF4の活性化を表している(Jousse et al., Biochem. Biophys. Res. Commun., 2004, 313: 447-452)。
【0045】
96ウェルプレート中の細胞に共沈殿を添加し(15μl/ウェル)、炭酸ガスインキュベーターで12時間培養した。細胞をPBSで1回洗浄した後、新しい培地に交換し、炭酸ガスインキュベーターでさらに12時間培養した。培地を除去し、PBSを200μl添加した後、細胞にUVAを照射した。照射後、PBSを除去し、培地を添加して、炭酸ガスインキュベーターで16時間培養した。細胞をPBSで1回洗浄した後、PBSを除去し、PLB(細胞溶解液)を加えて細胞を溶解した。細胞溶解液2.5μlにDUAL LUCIFERASE ASSAY(Promega社製)溶液(各25μlずつ)を加えてluciferaseとRenilla luciferaseの活性を定量した。Renilla luciferase活性測定値でluciferase活性測定値を補正し、得られた値を遺伝子の発現量とした(図7)。陽性対照として、UVA照射の代わりに小胞体ストレス誘導剤として知られているthapsigargin(TG)を添加した細胞を調製した。
結果を図8に示す。これらの結果から、UVAがTG同様に小胞体ストレスを惹起することが示された。
【技術分野】
【0001】
本発明は、新規な小胞体ストレスモデルに関する。
【背景技術】
【0002】
生体には様々な種類のタンパク質が存在し、各々が生体の構造形成や酵素作用など、生命活動に不可欠な機能を担っている。これらのタンパク質の機能は、その立体構造と密接に関連しており、タンパク質がその機能を果たすには適切な立体構造をとっていることが不可欠である。異常な構造を有するタンパク質は、本来の機能を果たすことができず、そればかりか、ときには毒性を有し、又は疾患の原因となり得る。
【0003】
タンパク質は、生体内で最初に合成されたときは“ひも”上の一次構造であるが、その後小胞体内で高次構造へとフォールディング(folding)される。小胞体内で正しい立体構造を獲得したタンパク質は、ゴルジ装置へと輸送され、そこから分泌経路を介して生体各所に輸送されて、機能を発揮する。一方、不適切なフォールディングによって誤った立体構造を有するタンパク質は、小胞体シャペロンの働きによって正しい構造へと再構成されるか、あるいは小胞体タンパク質分解機構(ER-associated degradation; ERAD)によって分解される。こうしたタンパク質のフォールディングと分解からなる適正な構造のタンパク質を供給するシステムを「タンパク質の品質管理」と呼ぶ。このシステムによって、生体は、異常な構造を有するタンパク質からの悪影響を防いでいる。
【0004】
何らかの原因でタンパク質の品質管理システムが十分に機能しない場合、結果として誤った立体構造を有する異常タンパク質が小胞体内に蓄積する。この状態を小胞体ストレスという。小胞体ストレスに対して、細胞は、タンパク質翻訳の抑制や調節、小胞体シャペロンの発現亢進によるフォールディングの増強、ERAD活性化による異常タンパク質分解促進などの手段によって、異常タンパク質を減少させる。しかし、これらの手段によっても状態が改善しない場合、細胞はアポトーシスを起こして死滅し、異常タンパク質から生体を防御する。これらの一連の応答は、小胞体ストレス応答(unfolded protein response; UPR)と称される。
【0005】
近年、上記小胞体ストレス応答のメカニズムが次第に明らかになってきた。例えば、上記応答手段のうち、翻訳の抑制や調節にはPERK、GADD34、p58IPK、CReP、ATF4等の分子が、フォールディング増強にはATF6、BiP/GRP78、Hspファミリー等の分子が、ERAD活性化にはIRE1、XBP1、RLG1等の分子が、そしてアポトーシスにはCaspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR、TDAG51等の分子が関与していることが知られている(例えば、非特許文献1)。このうち、IRE1、ATF6、XBP1、GRP78等は小胞体ストレスのセンサータンパク質であり、小胞体ストレスの指標として知られている。特に、XBP1は、ATF6によって転写誘導され、またIRE1によってフレームスイッチ型スプライシングを受けるため、ATF6とIRE1の両因子のシグナルを反映する指標である。スプライシングされたXBP1 mRNAを測定することで小胞体ストレスを検出する方法が提唱されている(特許文献1)。
【0006】
また近年、小胞体ストレスが、生体の様々な障害、例えば、糖尿病、神経変性疾患、躁うつ病等の種々の疾患や、免疫応答などの生理機能に深く関与していることが報告されている。生体機能に関する基礎研究のため、又は種々の疾患の診断及び治療手段を開発するためのツールとして小胞体ストレスが注目されている。
【0007】
したがって、小胞体ストレスについて研究するための実験系、すなわち小胞体ストレスモデルを確立することが望まれる。現在までに、小胞体ストレスモデルとしては、ヒトメグシン遺伝子導入トランスジェニックラットモデル(特許文献2)、薬物添加培養細胞モデル(非特許文献2)、UV照射マウスモデル又はヒト皮膚切片モデル(非特許文献3)等が知られている。このうち、トランスジェニックモデルは、作製手順が煩雑であり、また導入遺伝子発現量の制御が難しい。
【0008】
in vitroモデルは、標的細胞へ因子の影響が迅速に現れ、且つ生体の他の組織からの影響を受けることがないことから、因子と細胞応答との関係を詳細に調べるためのモデルとして好ましい。in vitro小胞体ストレスモデルとしては、薬物tunicamycin又はthapsigarginを添加して小胞体ストレスを誘導する培養細胞モデルが知られている(非特許文献2)。しかし、薬物添加によりストレスを誘導する場合、添加した薬物が細胞内に浸透して標的部位に到達するまでの時間、及び薬物の影響が細胞から除去されるまでの時間を制御することが困難である。また、細胞内に浸透した薬物は代謝により分解されるため、添加薬物量は必ずしも誘導ストレス量に反映されない。
【0009】
UV照射によるストレス誘導は、操作が簡便であり、且つ照射後直ちに標的部位に到達し、また照射を止めれば直ちにその影響は細胞内から消えるため、ストレス誘導の時間的制御に有効である。また、UVは薬物のように代謝を受けることがないので、与えた線量が直接的にストレス誘導量として反映され得る。従って、UV照射は、小胞体ストレスモデル作製のための好ましい方法である。しかしながら、UV照射による小胞体ストレスモデルとしては、従来、上記のマウスモデル及びヒト皮膚切片モデル(非特許文献3)のようなin vivoモデルが知られているのみであり、培養細胞モデルのようなin vitroモデルは、UV照射によって作製することはできないことが報告されていた(非特許文献3)。
【0010】
小胞体ストレスのより詳細な研究を可能にするin vitro小胞体ストレスモデルの確立が望まれている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2007−129970号公報
【特許文献2】特開2004−313192号公報
【非特許文献】
【0012】
【非特許文献1】吉田秀朗, 生化学, 2004, 第76巻第7号, pp617-630
【非特許文献2】Yoshida et al., Cell, 2001, 107:881-891
【非特許文献3】Anand et al., J. Invest. Dermatol., 2005, 125:323-333
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明は、新規な方法により作製されたin vitro小胞体ストレスモデルを提供する。
【課題を解決するための手段】
【0014】
本発明者らは、培養細胞に特定の条件下でUV照射を行うことにより、従来作製できないと考えられていたUV照射によるin vitro小胞体ストレスモデルを作製することに成功した。
【0015】
すなわち、本発明は、以下に係るものである。
(1)培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法。
(2)UVA照射が1〜100J/cm2のUVA照射である(1)記載の方法。
(3)UVAを照射する工程の後に前記細胞を培養する工程をさらに含む、(1)又は(2)に記載の方法。
(4)UVAを照射する工程により、前記細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を増加させる、(1)〜(3)のいずれか1に記載の方法。
(5)(1)〜(4)のいずれか1に記載の方法によって作製された小胞体ストレスモデル。
(6)培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を測定する工程を含む、小胞体ストレス制御剤のスクリーニング方法。
(7)UVA照射が1〜100J/cm2のUVA照射である(6)記載の方法。
(8)前記発現を測定する工程の前に当該細胞を培養する工程をさらに含む、(6)又は(7)に記載の方法。
(9)前記分子又はそのmRNAの発現の量を定量する工程をさらに含む、(6)〜(8)のいずれか1に記載の方法。
【発明の効果】
【0016】
本発明によれば、従来よりも簡便な手順で、ストレス負荷量がより精密に制御されたin vitro小胞体ストレスモデルを提供することができる。本発明のin vitro小胞体ストレスモデルによれば、従来よりも迅速且つ正確に小胞体ストレスを評価し、また小胞体ストレスを制御する物質を探索することが可能になる。
【図面の簡単な説明】
【0017】
【図1】ヒト正常線維芽細胞及びHeLa細胞における、XBP1 mRNAスプライシングに対するUVA照射の影響。照射4時間後に測定。
【図2】ヒト正常線維芽細胞におけるXBP1 mRNAスプライシングに対するUVA照射後の時間の影響。UVA線量10J/cm2。
【図3】ヒト正常線維芽細胞におけるXBP1 mRNAスプライシングに対するUVA線量の影響。照射4時間後に測定。
【図4】UPR誘導下における遺伝子発現。A:ヒト正常角化細胞におけるBiP/GRP78 mRNA発現、B:ヒト正常線維芽細胞におけるHsp47 mRNA発現。照射6時間後に測定。
【図5】UPR誘導下における遺伝子発現。A:ヒト正常線維芽細胞におけるGADD34 mRNA発現、B:ヒト正常線維芽細胞におけるGADD153 mRNA発現。照射6時間後に測定。
【図6】UPR誘導下における遺伝子発現の経時変化。A:ヒト正常角化細胞におけるGADD34 mRNA発現、B:ヒト正常角化細胞におけるGADD153 mRNA発現。照射6〜48時間後に測定。
【図7】リポーターアッセイ手順の概念図。
【図8】リポーターアッセイ結果。
【発明を実施するための形態】
【0018】
本発明の小胞体ストレスモデルは、培養細胞にA波紫外線(UVA)を照射することにより作製される。
【0019】
本発明の方法においては、培養細胞をUVA照射することにより、当該細胞に小胞体ストレス応答(unfolded protein response; UPR)を誘導する。UVAを照射される培養細胞は、由来する種や組織又は特定の系統には限定されないが、培養哺乳類細胞が好ましく、培養ヒト細胞がより好ましい。あるいは、培養細胞としては、好ましくは角化細胞、線維芽細胞、HeLa細胞が挙げられる。
【0020】
UVA照射においては、照射エネルギーとして1〜100J/cm2、好ましくは10〜60J/cm2のUVA(波長320〜400nm)に上記培養細胞を曝露する。照射の際、上記範囲内で照射エネルギー量を調節することによって、細胞に与える小胞体ストレスの量を制御することができる。所定の照射エネルギーに細胞を曝露することができる限り、照射の強度及び時間は特に限定されない。照射は連続照射でもよく、あるいは任意の間隔をあけた繰り返し照射でもよい。細胞をUVA照射する具体的手順は当業者に周知である。
【0021】
照射により、細胞に小胞体ストレス応答(UPR)が誘導される。UPRの典型的な例としては、タンパク質翻訳抑制、小胞体シャペロンの発現亢進によるフォールディングの増強、小胞体タンパク質分解機構(ER-associated degradation; ERAD)の活性化による異常タンパク質分解促進等の異常タンパク質を減少させるための応答、アポトーシス誘導等のような異常タンパク質から生体を防御する応答、及び細胞のタンパク質翻訳抑制及び調節が挙げられる。すなわち、上記UVA照射により、細胞内のタンパク質翻訳抑制、フォールディングの増強、ERAD活性化又はアポトーシス誘導、又は細胞のタンパク質翻訳抑制及び調節が促進される。
【0022】
細胞のタンパク質翻訳抑制に関与する分子としては、PERK、GADD34、p58IPK、CReP等が挙げられる。フォールディング増強に関与する分子としては、ATF6、BiP/GRP78、Hsp47等が挙げられる。ERAD活性化に関与する分子としては、IRE1、XBP1、RLG1等が挙げられる。アポトーシスに関与する分子としては、Caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR、TDAG51等が挙げられる。細胞のタンパク質翻訳抑制及び調節に関与する分子としては、ATF4が挙げられる。これらの分子は全て細胞のUPRに関与する分子(UPR関連分子)である。
【0023】
上記細胞内のUPR関連分子の発現は、上記UVA照射により増加する。当該分子の発現増加は、細胞のUPRの増強を反映している。特に、XBP1は、ATF6によって転写誘導され、またIRE1によってフレームスイッチ型スプライシングを受けるため、ATF6とIRE1の両因子のシグナルを反映する分子である。XBP1のスプライシング型mRNA(XBP1(S)mRNA)は、細胞のUPRの指標として従来知られている。
【0024】
したがって、一態様において、本発明の小胞体ストレスモデルの作製方法は、培養細胞にUVAを照射する工程により、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子の少なくとも1の発現、あるいはこれらの分子のmRNAのうちの少なくとも1の発現を増加させる工程を含み得る。当該態様においては、上記の分子又はそのmRNAのうちの少なくとも1の発現を増加させればよいが、上記の分子又はそのmRNAのうちの複数の発現を増加させてもよい。より好ましくは、GADD34、BiP/GRP78、Hsp47、XBP1及びCHOP/GADD153のうちの少なくとも1の発現を増加させる。さらに好ましくは、GADD34 mRNA、BiP/GRP78 mRNA、Hsp47 mRNA、XBP1(S) mRNA及びCHOP/GADD153 mRNAうちの少なくとも1の発現を増加させる。
【0025】
本発明の小胞体ストレスモデルの作製方法においては、好ましくは、培養細胞へのUVA照射の後、当該細胞を培養する。培養時間は、好ましくは1時間以上であり、より好ましくは4時間以上であり、さらに好ましくは1〜48時間であり、なお好ましくは4〜48時間である。上記培養により、当該細胞におけるUPRはより増強される。
【0026】
上記のとおりUPRが誘導された細胞は、in vitro小胞体ストレスモデル細胞として利用され得る。例えば、後記実施例1に示されるように、UVA照射により、培養細胞内で、UPRの指標として従来知られている分子であるXBP1(S)mRNAが増加する。したがって、UVA照射により当該細胞にUPRが誘導されており、この細胞をin vitro小胞体ストレスモデルとして使用できることが分かる。また、後記実施例2〜4に示されるように、上記XBP1(S)mRNAが増加するUVA照射条件と同じ照射条件下で、他のUPR関連分子の発現が培養細胞内で増加する。したがって、本発明の小胞体ストレスモデル細胞においては、UPR関連分子、好ましくは、PERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子の少なくとも1又はそのmRNAのうちの少なくとも1の発現、より好ましくは、GADD34、BiP/GRP78、Hsp47、XBP1及びCHOP/GADD153のうちの少なくとも1の発現、さらに好ましくは、GADD34 mRNA、BiP/GRP78 mRNA、Hsp47 mRNA、XBP1(S) mRNA及びCHOP/GADD153 mRNAうちの少なくとも1の発現が増加している。
【0027】
また本発明は、上記小胞体ストレスモデルを利用した、小胞体ストレス制御剤のスクリーニング方法を提供する。すなわち、本発明のスクリーニング方法は、培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるUPR関連分子又はそのmRNAの発現を測定する工程とを含む。当該スクリーニング方法において、UVA照射される培養細胞の種類、UVA照射の方法、発現を測定されるUPR関連分子又はそのmRNAの種類は、本発明の小胞体ストレスモデルの作製方法の場合と同じである。
【0028】
被験物質は、UVA照射前に添加されても、UVA照射後に添加されても、又はUVA照射と並行して添加されてもよい。その後、細胞のUPR関連分子又はそのmRNAの発現を測定し、被験物質の添加による影響を評価する。例えば、被験物質の添加前後で細胞のUPR関連分子又はそのmRNAの発現を測定し、必要に応じて測定値を定量した後、添加前後で発現を比較する。あるいは、被験物質添加群と対照物質添加群におけるUPR関連分子又はそのmRNAの発現を測定し、必要に応じて測定値を定量した後、両群間で発現の違いを比較する。上記分子又はそのmRNAの発現を変化させる物質を小胞体ストレス制御剤として選択することができる。例えば、被験物質のうち、UPR関連分子又はそのmRNAの発現を増加させる物質は小胞体ストレス亢進剤として、当該分子又はそのmRNA発現を低下させる物質は小胞体ストレス抑制剤として、選択され得る。
【0029】
好ましくは、本発明の小胞体ストレスモデルの作製方法は、培養細胞をUVA照射する工程の後、且つ上記発現を測定する工程の前に、当該細胞を培養する工程をさらに含む。培養時間は、好ましくは1時間以上であり、より好ましくは4時間以上であり、さらに好ましくは1〜48時間であり、なお好ましくは4〜48時間である。
【0030】
本発明において、UPR関連分子又はそのmRNAの発現の測定には、例えば、RI標識、蛍光標識、発光標識、ゲル電気泳動法、免疫沈降、イムノブロッティング、ドットブロット法、ノーザンブロット法、ウエスタンブロット法、RNアーゼプロテクションアッセイ法、ルシフェラーゼ等によるリポーターアッセイ、RT−PCR法、DNA又はプロテインマイクロアレイ等の、当該分野で通常行われる任意の方法を用いることができる。このうち、ウエスタンブロット法、リポーターアッセイ及びRT−PCR法が好ましい。
【実施例】
【0031】
以下、実施例に基づき本発明をさらに詳細に説明する。
【0032】
実施例1:UVA照射による小胞体ストレス応答の誘導
1.細胞の準備
60mmディッシュに10%のコンフルエンシーで正常ヒト線維芽細胞又はHeLa細胞を播種し、炭酸ガスインキュベーターで72時間培養した。
【0033】
2.UVA照射
ディッシュから培地を除去し、2.5mlのPBSを添加した後、細胞にUVAを照射した。
紫外線照射光源としては、キセノン光源装置(MAX-301型、朝日分光(株))にUVA域全体(320-400nm)を透過する広帯域バンドパスフィルタ(朝日分光(株)製)を取り付けたものを使用した。射出光を石英ファイバーで細胞シャーレの真上に誘導した。培養ディッシュの10cmの上方に光出射口を配置して、光源装置に標準装備されているタイマーを使って照射時間をコントロールした。出射光線のエネルギーは、分光放射計測装置(MSR-7000、(株)オプトリサーチ社)を用いて、実使用条件での細胞位置における線量を予め測定した。
培養ディッシュを光ファイバー射出口の直下に置き、射出口の蓋を開けて、細胞に決められた照射エネルギー(0.1J/cm2〜100J/cm2)が与えられるように、所定距離から所定の時間、光を照射した。非照射群をコントロールとした。照射後、PBSを培地と交換し、炭酸ガスインキュベーターで4時間培養した。
あるいは、同様の手順で10J/cm2のUVAを細胞に照射し、照射後、PBSを培地と交換し、炭酸ガスインキュベーターで1〜48時間培養した。培養なし(0時間)の群をコントロールとした。
【0034】
3.薬物による小胞体ストレス応答誘導
上記と同様の手順で調製した培養細胞に、Yoshida et al., Cell 2001, 107:881-891に記載の手順に従って1μM thapsigarginを添加し、細胞の小胞体ストレス応答(UPR)を誘導した。
【0035】
4.RNAの調製
細胞をPBSで1回洗浄し、Sepasol 1mlを加えて細胞溶解液を回収した。クロロホルムを200μl添加し、良く混合した後に遠心し、水相を回収した。2-プロパノールを600μl加えて良く混合し、遠心して沈殿を回収した。沈殿を70%EtOHで洗浄し、沈殿を風乾し、10μlのH2Oに溶解した。RNA量を定量し、1μg/μlの濃度に調整した。
【0036】
5.逆転写反応
4.で調製したRNA 1μgから、SuperScript VILO cDNA合成キット(Invitrogen社)を用いて下記のとおり反応液を調製した。これを逆転写反応(25℃で10分間、42℃で60分間、85℃で5分間)にかけ、cDNA合成を行った。これにH2Oを10μl加え、1μlを分取し、PCR反応に使用した。
5×VILO(商標)Reaction Mix 2μl
10×SuperScript(登録商標)Enzyme Mix 1μl
RNA (1μg) 1μl
H2O 6μl
【0037】
6.PCR反応
5.で調製した逆転写反応産物1μlとKOD-FX DNA polymerase(東洋紡製)を用いて、指示書に従ってPCR反応を実施した。下記のとおりPCR反応液を調製し、94℃で2分→98℃で10秒→68℃又は60℃で60秒の反応サイクルを25〜40回繰り返し、XBP1(S) mRNAのcDNAをPCR増幅した。
2×KOD-FX Buffer 5.0μl
2mM dNTP mix 2.0μl
5μM Forward Primer 0.6μl
5μM Reverse Primer 0.6μl
Template(逆転写反応産物) 1.0μl
KOD-FX(登録商標) 0.2μl
H2O 0.6μl
使用したプライマー配列を以下に示す。
Forward Primer(pXBP1-454FW): AAGCCAAGGGGAATGAAGTG(配列番号1)
Reverse Primer(pXBP1-593RV): AGTCAATACCGCCAGAATCC(配列番号2)
PCR産物を5/20%TAE−PAGE上で展開し、EtBrで染色して検出した。
【0038】
結果を図1〜3に示す。図1に示すように、細胞にUVAを照射した場合、UPR誘導剤thapsigarginを添加した場合と同様に、XBP1(S) mRNAの増加が観察された。XBP1(S) mRNAの増加は、細胞内における構造異常蛋白質の増加を反映しており、よって細胞内でUPRが誘導されたことを表している。したがって、細胞にUVAを照射することにより、細胞のUPRを誘導することができることが示された。
図2に示すように、XBP1(S) mRNAの発現の増加はUVA照射の1時間後から観察され、24時間経過後にもなお高いレベルを維持していた。また図3A及び図3Bに示すように、照射エネルギーが1.0J/cm2以上の場合、それ以下のエネルギーでUVA照射した場合や非照射の場合と比較して、XBP1(S) mRNAの発現が増加した。発現の増加は1.0〜100J/cm2の範囲で観察されたが、60J/cm2を超えるとむしろ発現増加の程度は小さくなる傾向にあった。
【0039】
実施例2:UPR誘導下におけるUPR関連分子の発現
1.細胞の準備
正常ヒト表皮角化細胞(Cascade Biologics社)を凍結バイアルで購入し、融解後に培養フラスコ(T75)内で、無血清増殖培地(EpiLife-KG2、Cascade Biologics社)で添付書の指示通り培養した。シャーレ(直径35 mm)に1回継代した細胞がサブコンフルエンシーまで増殖した時点で照射実験に用いた。
正常ヒト線維芽細胞(Cascade Biologics社)を凍結バイアルで購入し、融解後に培養フラスコ(T75)内で、10% FBS添加DMEM培地(高Glucose、Invitrogen社)で添付書の指示通り培養した。シャーレに継代した細胞がサブコンフルエンシーまで増殖した時点で照射実験に用いた。
【0040】
2.紫外線照射
照射前に培地を37℃のハンクス平衡塩溶液HBS(Ca,Mg含有、フェノールレッド不含)に交換する点を除いて、実施例1と同様の手順でUVA照射を行った。照射後、ハンクス平衡塩溶液を培地に交換して培養インキュベータに戻し、通常通り培養した。
【0041】
3.遺伝子発現解析
遺伝子発現解析は、蛋白質フォールディングの増強に関わる遺伝子であるBiP/GRP78及びHsp47、タンパク質翻訳抑制に関わる分子であるGADD34、ならびにアポトーシス誘導に関連する分子であるGADD153について行った。
UVA照射後、所定の時間培養した細胞ディッシュを2mL PBS(−)で2回洗浄してから、RNAを単離した。RNA単離には、RNeasy(登録商標)RNA 単離キット(QIAGEN社)を取り扱い説明書に従って用いた。細胞溶解には、キットの細胞溶解溶液をシャーレ当たり350μL使用した。取り扱い指示書通りに処理した後、RNAを50μLに溶出・回収した。回収RNAの10μLをcDNA合成に用いた(SuperScript(登録商標)VILO(商標)cDNA合成キット、Invitrogen社)。cDNAサンプルの遺伝子解析は、TaqMan(登録商標)Gene Expression System(Applied Biosystems社)を用いて、リアルタイム定量PCR装置(ABI7500、Applied Biosystems社)で標準的なプロトコルに従って行なった。
サンプル間の遺伝子発現量の相対比較においては、ハウスキーピング遺伝子GAPDHを内部標準とし、対象遺伝子の発現変化率をGAPDHの発現変化率で除して標準化した。
【0042】
BiP/GRP78及びHsp47の遺伝子発現解析の結果を図4A及び図4Bに、GADD34及びGADD153の遺伝子発現解析の結果を、それぞれ図5A及び図5Bに示す。これらの遺伝子の発現は、XBP1(S) mRNAの発現が増加するUVA照射線量において増加した。
また、UVA照射後におけるGADD34及びGADD153の遺伝子発現の経時変化を図6A及び図6Bに示す。これらの遺伝子の発現は、XBP1(S) mRNAの発現が増加するUVA照射後時間帯と同様の時間帯で増加した。
したがって、これらの分子又はその遺伝子の発現は、XBP1(S)mRNAと同様に、細胞におけるUPRの指標となる。
【0043】
実施例3:リポーターアッセイによる転写誘導の検出
96ウェルプレートに10%のコンフルエンシーで正常ヒト線維芽細胞を播種した。炭酸ガスインキュベーターで24時間培養した。
以下の試薬を混合し、この混合液を、2×HBS溶液 100μlに混合しながら滴下した。これを室温で30分静置し、共沈殿を調製した。
250mM CaCl2 10μl
測定用プラスミド 2μl (2μg)
補正用プラスミド 2μl (0.2μg)
H2O 86μl
(細胞10ウェルあたりの分量)
測定用プラスミドとしては、図8に示すプラスミドのいずれかを用いた。
補正用プラスミドとしては、DUAL LUCIFERASE(登録商標) Reporter ASSAY System(Promega社製)に添付されているpRLuc-SV40 promoter plasmidを用いた。
【0044】
図8に示すプラスミドpGL3-(-132CCC)は、human BiP/GRP78 promoterの転写開始点から-132から+7までの配列をpGL3-basic vector(Promega社製)に挿入することにより作製された。このプラスミドの発現は、UPRにおけるフォールディング増強に関与する分子であるATF6が活性化されていることを示す。プラスミドpGL3-(-132MMM)は、human BiP promoterにおいて3つのERSE配列を破壊し、これをpGL3-basic vectorに挿入することにより作製された(Yoshida et al., J Biol. Chem., 1998, 273: 33741-33749)。これは小胞体ストレスで活性化されない陰性対照プラスミドである。プラスミドpGL3-(UPRE)×5は、XBP1が結合する配列(UPRE配列)を5回繋げたものをpGL3-promotervectorに挿入することにより作製された(Wang et al., J. Biol. Chem., 2000, 275: 27013-27020)。このプラスミドの発現はXBP1の活性化を表す。プラスミドpGL3-CHOP UASは、human CHOP/GADD153 promoter(転写開始点から-870〜+17)をpGL3-basic vectorに挿入することにより作製された(Yoshida et al., Mol. Cell. Biol., 2000, 20: 6755-6767)。CHOP promoterには転写因子ATF4が結合する配列があり、その活性化はATF4の活性化を表している(Jousse et al., Biochem. Biophys. Res. Commun., 2004, 313: 447-452)。
【0045】
96ウェルプレート中の細胞に共沈殿を添加し(15μl/ウェル)、炭酸ガスインキュベーターで12時間培養した。細胞をPBSで1回洗浄した後、新しい培地に交換し、炭酸ガスインキュベーターでさらに12時間培養した。培地を除去し、PBSを200μl添加した後、細胞にUVAを照射した。照射後、PBSを除去し、培地を添加して、炭酸ガスインキュベーターで16時間培養した。細胞をPBSで1回洗浄した後、PBSを除去し、PLB(細胞溶解液)を加えて細胞を溶解した。細胞溶解液2.5μlにDUAL LUCIFERASE ASSAY(Promega社製)溶液(各25μlずつ)を加えてluciferaseとRenilla luciferaseの活性を定量した。Renilla luciferase活性測定値でluciferase活性測定値を補正し、得られた値を遺伝子の発現量とした(図7)。陽性対照として、UVA照射の代わりに小胞体ストレス誘導剤として知られているthapsigargin(TG)を添加した細胞を調製した。
結果を図8に示す。これらの結果から、UVAがTG同様に小胞体ストレスを惹起することが示された。
【特許請求の範囲】
【請求項1】
培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法。
【請求項2】
UVA照射が1〜100J/cm2のUVA照射である請求項1記載の方法。
【請求項3】
UVAを照射する工程の後に前記細胞を培養する工程をさらに含む、請求項1又は2に記載の方法。
【請求項4】
UVAを照射する工程により、前記細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を増加させる、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
請求項1〜4のいずれか1項に記載の方法によって作製された小胞体ストレスモデル。
【請求項6】
培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を測定する工程を含む、小胞体ストレス制御剤のスクリーニング方法。
【請求項7】
UVA照射が1〜100J/cm2のUVA照射である請求項6記載の方法。
【請求項8】
前記発現を測定する工程の前に当該細胞を培養する工程をさらに含む、請求項6又は7に記載の方法。
【請求項9】
前記分子又はそのmRNAの発現の量を定量する工程をさらに含む、請求項6〜8のいずれか1項に記載の方法。
【請求項1】
培養細胞にUVAを照射する工程を含む小胞体ストレスモデルの作製方法。
【請求項2】
UVA照射が1〜100J/cm2のUVA照射である請求項1記載の方法。
【請求項3】
UVAを照射する工程の後に前記細胞を培養する工程をさらに含む、請求項1又は2に記載の方法。
【請求項4】
UVAを照射する工程により、前記細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を増加させる、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
請求項1〜4のいずれか1項に記載の方法によって作製された小胞体ストレスモデル。
【請求項6】
培養細胞に被験物質を添加する工程と、当該細胞をUVA照射する工程と、当該細胞におけるPERK、GADD34、p58IPK、CReP、ATF4、ATF6、BiP/GRP78、Hsp47、IRE1、XBP1、RLG1、caspase-12、CHOP/GADD153、ASK1、c-Abl、PUMA、PKR及びTDAG51からなる群から選択される分子又はそのmRNAのうちの少なくとも1の発現を測定する工程を含む、小胞体ストレス制御剤のスクリーニング方法。
【請求項7】
UVA照射が1〜100J/cm2のUVA照射である請求項6記載の方法。
【請求項8】
前記発現を測定する工程の前に当該細胞を培養する工程をさらに含む、請求項6又は7に記載の方法。
【請求項9】
前記分子又はそのmRNAの発現の量を定量する工程をさらに含む、請求項6〜8のいずれか1項に記載の方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2011−167081(P2011−167081A)
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2010−31087(P2010−31087)
【出願日】平成22年2月16日(2010.2.16)
【出願人】(000000918)花王株式会社 (8,290)
【Fターム(参考)】
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成22年2月16日(2010.2.16)
【出願人】(000000918)花王株式会社 (8,290)
【Fターム(参考)】
[ Back to top ]