
尿酸を減少させるテトラゾール化合物
式Iの化合物を投与することにより、哺乳動物対象における尿酸は減少し、尿酸の排出は増加する。本発明の化合物の尿酸低下効果は、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症などさまざまな状態を治療又は防止するために使用される。式I中、xは1又は2であり、yは0、1、2又は3であり、R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、3〜6個の環原子を有する、置換されていないシクロアルキル、又は3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により該化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
尿酸値上昇が原因の疾患は、2つの主要な範疇、すなわち、尿酸結晶の沈殿が原因の障害と、可溶性尿酸の病理学的効果に関連する疾患とに分けられる。痛風性関節炎は、前者の古典的な例である。腎臓中の尿酸結晶の堆積も、腎機能不全の一般的原因である。可溶性尿酸値上昇は、心血管疾患及び腎疾患など、さまざまな障害を伴う。
【背景技術】
【0002】
痛風は、体内の1又は複数の関節の炎症として最も普通に現れ、その結果、軽度から重度の疼痛が生じる。こうした事象は、突発性及び/又は慢性である場合がある。時間の経過と共に、痛風は、軟骨及び骨の破壊、尿酸結晶堆積物の発達、腎臓痛及び腎機能不全並びに腎臓結石をもたらすことがある。痛風は、他の器官にも影響することがある。
【0003】
痛風は、高尿酸血症、並びに、組織、関節、腎臓及び他の器官中での結果的な尿酸結晶の形成及び堆積が原因である。尿酸は、正常な細胞の代謝により、又、ある種の食品及び飲料により生じる。過剰な尿酸値は、余分な尿酸の生成、腎臓によるクリアランス障害(又は、過剰生成とクリアランス障害との組合せ)の結果、さらには、他の健康状態のために摂取されるある形態の薬物による結果である。(例としては、利尿薬、ピラジナミド、シクロスポリン、低用量アスピリン、ニコチン酸及びレボドパが挙げられる)。アルコール依存症、白血病、リンパ腫、肺癌、腫瘍溶解症候群、喫煙、乾癬、肥満、腎機能不全、うっ血性心不全、飢餓、貧血症、高血圧症、糖尿病、不動、レッシュ−ナイハン症候群、ダウン症候群、並びに、甲状腺及び副甲状腺の機能不全など多くの種類の健康状態が、高尿酸血症及び痛風の一因となることもある。
【0004】
痛風は、一般に、進行性に悪化する症状に基づき4つの範疇に分けられる:
1)無症候性。血中尿酸値は上昇しているが、明白な症状はない。
2)急性通風性関節炎:多くの場合は単一の関節(普通は足の親指)において症状が突然生じ、次いで、他の関節が冒される。症状としては、疼痛、腫張、発赤及び発熱が挙げられる。
3)間欠期痛風:痛風発作間の無症候性の段階。
4)慢性結節性痛風:慢性の状態であり、頻繁な発作、関節の持続的で軽度の疼痛及び炎症、軟骨及び骨の破壊、尿酸結晶堆積物の発達、腎機能不全及び腎臓結石が含まれる場合がある。
【0005】
痛風の急性症状を治療するために現在使用されている薬物としては、非ステロイド性抗炎症薬、コルヒチン及びコルチコステロイドが挙げられる。こうした薬物は全て、軽度から重度の副作用を生じる場合がある。インターロイキン1などの炎症性サイトカインに対する抗体及びアンタゴニストを含め、こうした急性症状用の他の治療薬が研究されている。
【0006】
尿酸値を低下させることにより将来的な発作の発生又は重症度の低下を試みるには、他の種類の薬物が使用される。3つの主要なクラスの薬物は、キサンチンからの尿酸の生成を減少させるキサンチンオキシダーゼ阻害薬(例えばアロプリノール);尿酸輸送体1(URAT1,uric acid transporter 1)の阻害により腎尿細管中での分泌された尿酸の再取込みを阻害することにより尿酸の排出を高めることを意図した尿酸排泄剤(例えば、スルフィンピラゾン、プロベネシド、ベンズブロマロン及びロサルタン)(米国特許出願公開第2007/0010670号明細書、2007年1月11日公開(日本たばこ産業株式会社)も参照)、又は、尿酸再取込みの他の要素;及びウリカーゼ、例えば、PURICASE(Savient社製のペグ化した組換え哺乳動物ウリカーゼ)などのペグ化ウリカーゼである。こうした薬物は、顕著で望ましくない副作用をもたらすことも多い。例えば、アロプリノールは、これが原因で欧州において毎年少なくとも100症例のスティーブンス・ジョンソン/中毒性表皮壊死症が発症し、およそ30名が死亡していると報告されている(Halevy et al., Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 58(1):25-32, 2008)。プロベニシド(Probenicid)及びベンズブロマロンは、ベンズブロマロンの症例における肝不全など、望ましくない副作用を理由に、いくつかの国の市場から排除されている。こうした薬物の摂取における患者の服薬遵守は、報告によれば非常に乏しく(A. A. Reidel et al. "Compliance with Allopurinol Therapy among Managed Care Enrollees with Gout: A Retrospective Analysis of Administrative Claims." Journal of Rheumatology 2004; 31:1575-1581)、おそらくそれは、副作用、及び/又は、利益のなさが理由であろう。
【0007】
米国においては500万名超の人々が痛風に罹患している(National Health and Nutrition Examination Survey 111, 1988-1994)。1999年の米国における高尿酸血症及び痛風の有病数は、1,000名当たり41名、英国においては1,000名当たり14名と報告された(T.R. Mikuls et al., "Gout Epidemiology: Results for the UK General Practice Research Database, 1990-1999." Annals of the Rheumatic Diseases 2005; 64:267-272)。その後の報告により、米国、英国及び他の国々における有病数は着実に上昇していることが示されている。(K. L. Wallace et al., "Increasing Prevalence of Gout and Hyperuricemia over 10 Years Among Older Adults in a Managed Care Population." Journal of Rheumatology 2004; 31: 1582-1587)。さらに最近のデータからは、現在では500万名をはるかに超える米国人が、診断可能な痛風に罹患していることが示唆される(E. Krishnan et al., "Gout in Ambulatory Care Settings in the United States." Journal of Rheumatology 2008; 35(3): 498-501)。
【0008】
高尿酸血症及び痛風は、臓器移植のレシピエントにおいてとりわけ重大な問題である(Stamp, L., et al, "Gout in solid organ transplantation: a challenging clinical problem", Drugs (2005) 65(18): 2593-2611)。尿酸は、腎移植を受けている患者において上昇していることが多く、シクロスポリンなど一般的な免疫抑制薬が原因で、とりわけ重度の高尿酸血症が生じる場合がある。アザチオプリンなどいくつかの免疫抑制剤との相互作用があること、又、組合せにより骨髄不全が生じることから、移植患者においてはアロプリノールは禁忌である。さらに、尿酸上昇は、移植片不全(graft failure)の一因となる場合がある(Armstrong, K.A. et al., "Does Uric Acid Have a Pathogenetic Role in Graft Dysfunction and Hypertension in Renal Transplant Patients?" Transplantation (2005) 80(11): 1565-1571)。したがって、移植レシピエントにおいて高尿酸血症を減少させる安全な薬剤については、とりわけ急を要する需要がある。
【0009】
可溶性尿酸上昇に関連する疾患は、血管の問題を伴うことが多い:高血圧症(Sundstrom et al., Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 45(1):28-33, 2005)、高血圧前症(Syamela, S. et al., Association between serum uric acid and prehypertension among US adults. J Hypertens. 25 (8) 1583-1589, (2007)、アテローム性硬化症(Ishizaka et al., Association between serum uric acid, metabolic syndrome, and carotid atherosclerosis in Japanese individuals. Arterioscler Thromb Vasc Biol. (5):1038-44, 2005)、末梢動脈疾患(Shankar, A. et al., Association between serum uric acid level and peripheral artery disease. Atherosclerosis doi 10: 1016, 2007)、血管炎症(Zoccali et al., Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol. 17(5):1466-71, 2006)、心不全(Strasak, A.M. et al., Serum uric acid and risk of cardiovascular mortality: A prospective, long-term study of 83,683 Austrian men, Clin Chem. 54 (2) 273-284, 2008; Pascual-Figal, Hyperuricaemia and long-term outcome after hospital discharge in acute heart failure patients. Eur J Heart Fail. 2006 Oct 23; [Epub ahead of print]; Cengel, A., et al., "Serum uric Acid Levels as a Predictor of In-hospital Death in Patients Hospitalized for Decompensated Heart Failure." Acta Cardiol. (Oct. 2005) 60(5): 489-492)、心筋梗塞(Strasak, A.M. et al.; Bos et al., Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdamstudy. Stroke. 2006 Jun; 37(6):1503-7)、腎機能不全(Cirillo et al., Uric Acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 17(12 Suppl 3):S165-8, 2006; Z. Avram and E. Krishnan, Hyperuricemia - where nephrology meets rheumatology. Rheumatology (Oxford), 47(7): 960-964, 2008)、及び脳卒中(Bos et al., 2006)。尿酸は、内皮機能不全の直接の原因となる(Kanellis, et al., Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 25(1):39-42, 2005; Khosla et al, Hyperuricemia induces endothelial dysfunction. Kidney Int. 67(5):1739-42, 2005)。小児及び青年においては、早発性本態性高血圧症は血清尿酸上昇を伴い、アロプリノールを用いて尿酸を減少させると、こうした患者における血圧が低下する(Feig and Johnson, The role of uric acid in pediatric hypertension. J Ren Nutrition 17(1): 79-83, 2007; D.I. Feig et al., Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension. JAMA 300(8): 924-932, 2008。さらに、Feig et al.は、これは新しい治療的アプローチであるが、尿酸を減少させるための既存の薬物の副作用から、その使用が制限又は妨げられることがあるとも述べている。高尿酸血症は、このような状態の全てにおける独立したリスク因子である。
【0010】
可溶性尿酸上昇も、炎症応答を伴うか、又は炎症応答を直接誘導する。例えば、尿酸は、有機酸輸送体、特に尿酸輸送体URAT1により血管の平滑筋細胞中に輸送された後、血管の平滑筋細胞を刺激してC反応性タンパク質MCP−1及び他のサイトカインを産生させることにより、増殖、及び、アテローム性硬化症に関連する他の変化を刺激し(Price et al., Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol. 17(7):1791-5, 2006; Kang et al., Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005 25(5):425-33 (2005); Yamamoto et al., Allopurinol reduces neointimal hyperplasia in the carotid artery ligation model in spontaneously hypertensive rats. Hypertens. Res. 29 (11) 915-921, 2006)、ヒト単核細胞を刺激してIL−1β、IL−6及びTNF−αを産生させ、マウスの体内に注入するとTNF−αの顕著な増加の原因となり、内皮細胞及び血小板を活性化させ、血小板の接着性を増加させる(Coutinho et al., "Associations of Serum Uric Acid with Markers of Inflammation, Metabolic Syndrome, and Subclinical Coronary Atherosclerosis", Amer. J. Hypertens. (2007) 20: 83-89; Levya, F., et al., "Uric Acid in Chronic Heart Failure: A Marker of Chronic Inflammation", Eur. Heart J. (1998) 19(12): 1814-1822.)。尿酸は、内皮の一酸化窒素の生体利用性を阻害し、レニン・アンギオテンシン系を活性化することも示されている。(T.S. Perlstein et al., Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney International. 66:1465-1470, 2004)。Inokuchi et al.は、インターロイキン18(IL−18,Interleukin 18)及び他の炎症性物質は、痛風を伴う局所的な炎症を反映すること、並びに、尿酸結晶は、腎不全を発症させる役割を有すると思われるIL−18の活性化を加速させることを示した(T. Inokuchi et al., Plasma IL-18 and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 33(1): 21-27, 206)。IL−18及び他のサイトカインは、痛風自体には罹患しておらず尿酸値が上昇しているにすぎない人においても、顕著に上昇している(C. Ruggiero et al. Uric acid and inflammatory markers.(C. Ruggiero et al., Uric acid and inflammatory markers. European Heart Journal. 27: 1174-1181, 2006)。
【0011】
高尿酸血症は、認知障害及び他の形態の中枢神経系機能不全とも関連がある。(Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140; Watanabe, S., et al., "Cerebral Oxidative Stress and Mitochondrial Dysfunction in Oxonate-Induced Hyperuricemic Mice", J. Health Science (2006) 52: 730-737)。
【0012】
血清尿酸値上昇は、癌のリスク及び癌による死亡数の増加とも関連がある。(Strasak, AM et al. (2007) Serum uric acid and risk of cancer mortality in a large prospective male cohort. Cancer Causes Control 18 (9) 1021-1029; Strasak, AM et al. (2007) The role of serum uric acid as an antioxidant protecting against cancer: prospective study in more than 28,000 older Austrian women. Annals Oncol 18 (11) 1893-1897; Jee, SA et al. (2004) Serum uric acid and risk of death from cancer, cardiovascular disease or all causes in men Eur. J. Cardiovascular Prev. Rehab. 11 (3) 185-191)。
【0013】
尿酸値上昇は、糖尿病前症、インスリン抵抗性、2型糖尿病の発症、及び糖尿病罹患者におけるさまざまな望ましくない状態(末梢動脈疾患、脳卒中、及び、死亡リスク増加など)の可能性の増加と関連がある(Ioachimescu, A.G. et al. (2007) Serum uric acid, mortality and glucose control in patients with Type 2 diabetes mellitus: a PreCIS database study Diabet. Med. 24 (12) 1369-1374; Perry, I.J. et al (1995) Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men BMJ 310 (6979) 560-564; Chien, K-L et al. (2008) Plasma uric acid and the risk of Type 2 diabetes in a Chinese community Clin. Chem. 54 (2) 310-316; Sautin, Y. Y. et al. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress Am. J. Physiol. Cell Physiol.293: C584-C596; Tseng, C.H. (2004) Independent association of uric acid levels with peripheral artery disease in Taiwanese patients with Type 2 diabetes Diabet. Med. 21 (7) 724-729; Lehto, S. et al. (1998) Serum uric acid is a strong predictor of stroke in patients with non-insulin dependent diabetes mellitus Stroke 29: 635-639。
【0014】
尿酸値上昇は、レッシュ−ナイハン症候群の決定的な特徴である。睡眠時無呼吸又は睡眠呼吸障害の罹患者も、尿酸が上昇している(Saito, H. et al., Tissue hypoxia in sleep apnea syndrome assessed by uric acid and adenosine. Chest 122: 1686-1694, 2002; Verhulst, S.L., et al., Sleep-disordered breathing and uric acid in overweight and obese children and adolescents. Chest 132: 76-80, 2007)。
【0015】
尿酸上昇は、子癇前症と関連がある(Bainbridge, S.A. and Roberts, J.M., Uric acid as a pathogenic factor in preeclampsia. Placenta Dec. 17 2007 epub ahead of print)。
【0016】
「尿酸は、ヒト末梢血単核細胞中の熱帯熱マラリア原虫(P. falciparum)により引き起こされる炎症応答の主要な一因である。…熱帯熱マラリア原虫により誘導される炎症反応は、マラリア発病の主因と考えられる。…」PLoS ONE 2009;4(4):e5194. Epub 2009 Apr 17。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】米国特許出願公開第2007/0010670号明細書
【非特許文献】
【0018】
【非特許文献1】Halevy et al., Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 58(1):25-32, 2008
【非特許文献2】A. A. Reidel et al. "Compliance with Allopurinol Therapy among Managed Care Enrollees with Gout: A Retrospective Analysis of Administrative Claims." Journal of Rheumatology 2004; 31:1575-1581
【非特許文献3】National Health and Nutrition Examination Survey 111, 1988-1994
【非特許文献4】T.R. Mikuls et al., "Gout Epidemiology: Results for the UKGeneral Practice Research Database, 1990-1999." Annals of the Rheumatic Diseases 2005; 64:267-272
【非特許文献5】K. L. Wallace et al., "Increasing Prevalence of Gout and Hyperuricemia over 10 Years Among Older Adults in a Managed Care Population." Journal of Rheumatology 2004; 31: 1582-1587
【非特許文献6】E. Krishnan et al., "Gout in Ambulatory Care Settings in the United States." Journal of Rheumatology 2008; 35(3): 498-501
【非特許文献7】Stamp, L., et al, "Gout in solid organ transplantation: a challenging clinical problem", Drugs (2005) 65(18): 2593-2611
【非特許文献8】Armstrong, K.A. et al., "Does Uric Acid Have a Pathogenetic Role in Graft Dysfunction and Hypertension in Renal Transplant Patients?" Transplantation (2005) 80(11): 1565-1571
【非特許文献9】Sundstrom et al., Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 45(1):28-33, 2005
【非特許文献10】Syamela, S. et al., Association between serum uric acid and prehypertension among US adults. J Hypertens. 25 (8) 1583-1589, (2007)
【非特許文献11】Ishizaka et al., Association between serum uric acid, metabolic syndrome, and carotid atherosclerosis in Japanese individuals. Arterioscler Thromb Vasc Biol. (5):1038-44, 2005
【非特許文献12】Shankar, A. et al., Association between serum uric acid level and peripheral artery disease. Atherosclerosis doi 10: 1016, 2007
【非特許文献13】Zoccali et al., Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol. 17(5):1466-71, 2006
【非特許文献14】Strasak, A.M. et al., Serum uric acid and risk of cardiovascular mortality: A prospective, long-term study of 83,683 Austrian men, Clin Chem. 54 (2) 273-284, 2008
【非特許文献15】Pascual-Figal, Hyperuricaemia and long-term outcome after hospital discharge in acute heart failure patients. Eur J Heart Fail. 2006 Oct 23; [Epub ahead of print]
【非特許文献16】Cengel, A., et al., "Serum uric Acid Levels as a Predictor of In-hospital Death in Patients Hospitalized for Decompensated Heart Failure." Acta Cardiol. (Oct. 2005) 60(5): 489-492
【非特許文献17】Strasak, A.M. et al.; Bos et al., Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke. 2006 Jun; 37(6):1503-7
【非特許文献18】Cirillo et al., Uric Acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 17(12 Suppl 3):S165-8, 2006
【非特許文献19】Z. Avram and E. Krishnan, Hyperuricemia - where nephrology meets rheumatology. Rheumatology (Oxford), 47(7): 960-964, 2008
【非特許文献20】Kanellis, et al., Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 25(1):39-42, 2005
【非特許文献21】Khosla et al, Hyperuricemia induces endothelial dysfunction. Kidney Int. 67(5):1739-42, 2005
【非特許文献22】Feig and Johnson, The role of uric acid in pediatric hypertension. J Ren Nutrition 17(1): 79-83, 2007
【非特許文献23】D.I. Feig et al., Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension. JAMA 300(8): 924-932, 2008
【非特許文献24】Price et al., Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol. 17(7):1791-5, 2006
【非特許文献25】Kang et al., Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005 25(5):425-33 (2005)
【非特許文献26】Yamamoto et al., Allopurinol reduces neointimal hyperplasia in the carotid artery ligation model in spontaneously hypertensive rats. Hypertens. Res. 29 (11) 915-921, 2006
【非特許文献27】Coutinho et al., "Associations of Serum Uric Acid with Markers of Inflammation, Metabolic Syndrome, and Subclinical Coronary Atherosclerosis", Amer. J. Hypertens. (2007) 20: 83-89
【非特許文献28】Levya, F., et al., "Uric Acid in Chronic Heart Failure: A Marker of Chronic Inflammation", Eur. Heart J. (1998) 19(12): 1814-1822
【非特許文献29】T.S. Perlstein et al., Uric acid and the state of the intrarenal rennin-angiotensin system in humans. Kidney International. 66:1465-1470, 2004
【非特許文献30】T. Inokuchi et al., Plasma IL-18 and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 33(1): 21-27, 206
【非特許文献31】C. Ruggiero et al. Uric acid and inflammatory markers.(C. Ruggiero et al., Uric acid and inflammatory markers. European Heart Journal. 27: 1174-1181, 2006
【非特許文献32】Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140
【非特許文献33】Watanabe, S., et al., "Cerebral Oxidative Stress and Mitochondrial Dysfunction in Oxonate-Induced Hyperuricemic Mice", J. Health Science (2006) 52: 730-737
【非特許文献34】Strasak, AM et al. (2007) Serum uric acid and risk of cancer mortality in a large prospective male cohort. Cancer Causes Control 18 (9) 1021-1029
【非特許文献35】Strasak, AM et al. (2007) The role of serum uric acid as an antioxidant protecting against cancer: prospective study in more than 28,000 older Austrian women. Annals Oncol 18 (11) 1893-1897
【非特許文献36】Jee, SA et al. (2004) Serum uric acid and risk of death from cancer, cardiovascular disease or all causes in men Eur. J. Cardiovascular Prev. Rehab. 11 (3) 185-191
【非特許文献37】Ioachimescu, A.G. et al. (2007) Serum uric acid, mortality and glucose control in patients with Type 2 diabetes mellitus: a PreCIS database study Diabet. Med. 24 (12) 1369-1374
【非特許文献38】Perry, I.J. et al (1995) Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men BMJ 310 (6979) 560-564
【非特許文献39】Chien, K-L et al. (2008) Plasma uric acid and the risk of Type 2 diabetes in a Chinese community Clin. Chem. 54 (2) 310-316
【非特許文献40】Sautin, Y. Y. et al. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress Am. J. Physiol. Cell Physiol.293: C584-C596
【非特許文献41】Tseng, C.H. (2004) Independent association of uric acid levels with peripheral artery disease in Taiwanese patients with Type 2 diabetes Diabet. Med. 21 (7) 724-729
【非特許文献42】Lehto, S. et al. (1998) Serum uric acid is a strong predictor of stroke in patients with non-insulin dependent diabetes mellitus Stroke 29: 635-639
【非特許文献43】Saito, H. et al., Tissue hypoxia in sleep apnea syndrome assessed by uric acid and adenosine. Chest 122: 1686-1694, 2002
【非特許文献44】Verhulst, S.L., et al., Sleep-disordered breathing and uric acid in overweight and obese children and adolescents. Chest 132: 76-80, 2007
【非特許文献45】Bainbridge, S.A. and Roberts, J.M., Uric acid as a pathogenic factor in preeclampsia. Placenta Dec. 17 2007 epub ahead of print
【非特許文献46】P. falciparum, PLoS ONE 2009:4(4):e5194. Epub 2009 Apr 17
【非特許文献46】PLoS ONE 2009;4(4):e5194. Epub 2009 Apr 17
【発明の概要】
【発明が解決しようとする課題】
【0019】
該当する疾患が尿酸の結晶化によるものであるか、又は正常を超える(個人ベースの標準によるか集団ベースの標準によるかを問わない)可溶性尿酸値の効果によるものであるかに関わらず、血中尿酸の上昇に関連する障害を安全、手軽及び効果的に治療及び防止することができる新しい薬物に対する重大な医学的必要性がある。
【課題を解決するための手段】
【0020】
本発明は、式I
【0021】
【化1】
【0022】
により表される化合物を提供する。
【0023】
式I中、xは1又は2であり、yは0、1、2又は3であり、R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、N、S及びOから選択される1若しくは2個の環状ヘテロ原子を有し、環炭素により該化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である。
【0024】
本発明は、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させる方法であって、本発明の化合物を、該対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるのに有効な量で該対象に投与することを含む方法を提供する。本発明は、哺乳動物の血中の尿酸濃度を低下させ、又は該動物からの尿酸排出を増加させるうえで使用するための本発明の化合物を提供する。本発明は、哺乳動物の血中の尿酸濃度を低下させ、又は該動物からの尿酸排出を増加させるための医薬の製造における本発明の化合物の使用を提供する。本発明は、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させる際に使用するための医薬組成物であって、該対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるのに有効な量で本発明の化合物を含む医薬組成物を提供する。本発明は、1又は複数単位の経口用量の本発明の化合物と、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるための該化合物を投与するための取扱説明書とを含むキットを提供する。
【0025】
尿酸を減少させることは、本明細書中に記載の場合、痛風(無症候性痛風、急性通風性関節炎、間欠期痛風及び慢性結節性痛風のいずれか又は全て)、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、及び、高尿酸血症のそれ以外の結果、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫(Plasmodium falciparum)誘導性炎症などさまざまな状態を治療又は防止するために用いることができる。
【0026】
本発明は、実施例7に示すように、本発明の化合物がインビトロでURAT1を阻害したという観察に基づく。URAT1の阻害は、インビボで尿酸を低下させるための確立されたインビトロモデルである。
【図面の簡単な説明】
【0027】
【図1】化合物EBが、hURAT1−HEK細胞中の14C−尿酸取込みに及ぼす濃度依存性の阻害効果を示すグラフである。
【図2】化合物ECが、hURAT1−HEK細胞中の14C−尿酸取込みに及ぼす濃度依存性の阻害効果を示すグラフである。
【図3】マウス血漿中の化合物EBの濃度を示すグラフである。
【図4】ラット血漿中の化合物EBの較正曲線を示すグラフである(LC−MS)。
【図5】ラット血漿中の化合物EBの濃度を示すグラフである。
【図6】ラット血漿中の化合物ECの較正曲線を示すグラフである(LC−MS)。
【図7】ラット血漿中の化合物ECの濃度を示すグラフである。
【図8】ラット血漿中の化合物EGの較正曲線を示すグラフである(LC−MS)。
【図9】ラット血漿中の化合物EGの濃度を示すグラフである。
【発明を実施するための形態】
【0028】
定義
1H−テトラゾリル−5−イル部分及び対応する2H−テトラゾリル−5−イル部分は、互変異性体として存在することがある。本文書においては、1H−互変異性体を基準にして、化合物名を呼び、構造式を書いてある。そのような基準は全て、両方の互変異性形態を含むものとして理解されたい。したがって、例えば、「5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール)は、5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール及び5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−2H−テトラゾールを両方とも包含する。又、式Iは、上に示したような式I、及び、下記の式I’
【0029】
【化2】
【0030】
に示すその2H−テトラゾリル−5−イル互変異性形態を両方とも包含する。
【0031】
本明細書中で使用する場合、用語「アルキル」は、直鎖状又は分岐状のアルキル基を意味する。一定数の炭素原子を有すると同定されているアルキル基は、その特定数の炭素を有する任意のアルキル基を意味する。例えば、3個の炭素原子を有するアルキルはプロピル又はイソプロピルであってもよく、4個の炭素原子を有するアルキルは、n−ブチル、1−メチルプロピル、2−メチルプロピル又はt−ブチルであってもよい。
【0032】
本明細書中で使用する場合、用語「ハロ」は、フルオロ、クロロ、ブロモ及びヨードのうち1又は複数を指す。
【0033】
本明細書中で使用する場合、ペルフルオロメチル又はペルフルオロメトキシにあるような用語「ペルフルオロ」は、当該基が、水素原子全ての代わりにフッ素原子を有することを意味する。
【0034】
ある種の化学化合物は、本明細書中では、その化学名、又は以下に示す2文字のコードで呼ぶ。化合物EBからEI及び化合物BDは、上に示す式Iの範囲内に包含される。
EB 5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール
EC 5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール
ED 5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール
EF 5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール
EG 5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
BD 5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール
EH 5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール
EI 5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
【0035】
本明細書中で使用する場合、移行語「〜を含む(comprising)」は、非制限的なものである。この用語を用いる特許請求の範囲は、当該特許請求の範囲中に列挙されているものに追加して要素を含有できる。
【0036】
特許請求の範囲中で使用する場合、単語「又は(or)」は、そのように読んでは文脈中で意味をなさない場合を除き、「及び/又は」を意味する。したがって、例えば、「哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排泄を増加させる」という語句は、「哺乳動物対象の血中の尿酸濃度を低下させ、及び/又は該対象からの尿酸排泄を増加させる」と同義である。
【0037】
本発明の化合物
前記の「課題を解決するための手段」に記載の化合物、方法、使用又は医薬組成物の一実施形態においては、この化合物は、式XLVI
【0038】
【化3】
【0039】
(式中、x、y、R1及びAは、式Iについて先に記載のとおりである)
で表される。
【0040】
本発明のさらなる一実施形態においては、この化合物は、式XLVII
【0041】
【化4】
【0042】
(式中、x、y及びAは、式Iについて先に記載のとおりであり、R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される)
で表される。
【0043】
本発明の一実施形態においては、式I、XLVI又はXLVII中、xは1である。別の実施形態においては、Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである。より具体的な実施形態においては、Aは2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである。好ましくは、Aは2,6−ジメチルフェニルである。
【0044】
本発明の実施形態においては、この化合物は、式XLVIII
【0045】
【化5】
【0046】
(式中、yは0、1、2又は3であり、R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)
で表される。
【0047】
本発明の一実施形態においては、式I、XLVI、XLVII又はXLVIII中、yは0、1又は2である。本発明の一実施形態においては、式XLVII又はXLVIII中、R3は水素であり、R2は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。より具体的な一実施形態においては、R3は水素であり、R2は、水素、メチル及びメトキシからなる群から選択される。本発明の異なる一実施形態においては、式XLVII又はXLVIII中、R2は水素であり、R3は、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。より具体的な一実施形態においては、R2は水素であり、R3は、メチル及びメトキシからなる群から選択される。式XLVIIIのさらなる一実施形態においては、R4及びR5は両方ともメチルであるか、又は両方ともフルオロである。好ましくは、両方ともメチルである。
【0048】
本発明の特定の実施形態においては、この化合物は、5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール、5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール及び5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾールからなる群から選択される。
【0049】
本発明の化合物の一実施形態においては、この化合物は、実質的には(少なくとも98%)純粋な形態をしている。
【0050】
反応スキーム
式I(式中、xは1又は2であり、yは0〜3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0051】
【化6】
【0052】
(式中、Aは先に記載のとおりである)の化合物は、スキーム1の反応により調製できる。スキーム1の反応においては、A、x、y及びR1は前述のとおりである。Lは脱離基である。
【0053】
式IIの化合物は、トリフェニルホスフィン及びアゾジカルボン酸ジエチル又はアゾジカルボン酸ジイソプロピルを用いた、IIのIIIとのMitsunobu縮合を用いたステップ(a)の反応により、式Vの化合物に変換できる。この反応は、適当な溶媒(例えばテトラヒドロフラン)中で実施する。ステップ(a)の反応を実施するには、Mitsunobu反応において従来用いられる条件のいずれを利用してもよい。
【0054】
式Vの化合物は、適当な塩基、例えば、炭酸カリウム、トリエチルアミン、ピリジンなどを用いることによるステップ(a)の反応におけるのと同様、式IIの化合物を式IVの化合物でエーテル化又はアルキル化することにより調製してもよい。この反応は、非プロトン性溶媒(例えばN,N−ジメチルホルムアミド、アセトニトリル、ジクロロメタンなど)中で実施する。式IVの化合物においては、Lは、メシロキシ、トシロキシ、クロロ、ブロモ、ヨードなどが挙げられるが、これらに限定されない。ステップ(a)の反応を実施するには、ハロゲン化物又は脱離基を用いた反応によるヒドロキシル基のエーテル化のいずれの従来法を利用してもよい。
【0055】
式Vの化合物は、ルイス酸(例えば、塩化亜鉛、塩化マグネシウム、塩化アルミニウム、四塩化スズなど)の存在下で、ニトリルをアジド(例えばトリメチルシリルアジド)と、又は金属アジド(例えば、アジ化ナトリウム、アジ化カリウム、アジ化リチウム、好ましいアジドはアジ化ナトリウムである)と反応させることにより、ステップ(b)の反応により式Iの化合物に変換できる。この反応は、溶媒(例えばN,N−ジメチルホルムアミド)中で、80℃〜145℃の範囲の温度で6〜60時間実施する。理想の反応は、ニトリルをアジ化ナトリウム/塩化アンモニウム/N,N−ジメチルホルムアミドと120℃で24時間反応させることを利用する。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0056】
Aが、ヒドロキシル基の1つ又は2つの基により置換されているフェニルである場合は、ヒドロキシル基を保護することが一般に好ましい。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のものなど、適当な脱保護試薬を利用してステップ(b)の反応後に脱保護できる。
【0057】
【化7】
【0058】
式I[式中、xは1又は2であり、yは0〜3であり、R1はヒドロキシルである]の化合物、すなわち、式:
【0059】
【化8】
【0060】
(式中、Aは先に記載のとおりである)の化合物は、スキーム2の反応により調製できる。スキーム2の反応において、A、x、及びyは前述のとおりである。
【0061】
式Vの化合物は、−72℃〜0℃の範囲の温度で4〜48時間、溶媒(例えばジクロロメタン)を用いて式Vの化合物を三臭化ホウ素又は三塩化ホウ素で処理することによるステップ(c)の反応により、式VIの化合物に変換できる。ステップ(c)の反応を実施するには、そのような脱メチル化反応において従来用いられる条件のいずれを利用してもよい。
【0062】
式VIの化合物は、ステップ(b)の反応において本明細書中で先に記載のものと同じ様式のステップ(d)の反応により、式I(式中、R1はヒドロキシルである)の化合物に変換できる。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0063】
【化9】
【0064】
式I(式中、xは1又は2であり、yは0〜3であり、R1はアミノである)の化合物、すなわち、式:
【0065】
【化10】
【0066】
(式中、Aは先に記載のとおりである)の化合物は、スキーム3の反応により調製できる。スキーム3の反応において、A、x及びyは前述のとおりである。Pは保護基である。
【0067】
式VIIの化合物は、ステップ(a)の反応において本明細書中で先に記載のものと同じ様式のステップ(e)の反応により、式VIIIの化合物に変換できる。式VIIIの化合物は、ニトロ基をアミンに還元することによるステップ(f)の反応により、式IXの化合物に変換できる。還元剤は、金属(例えばZn、Sn又はFeなど)及び酸であってもよい。ニトロ基を触媒水素化により還元してアミンを得ることもできる。好ましい還元法は、触媒水素化である。ステップ(f)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0068】
式IXの化合物は、アミノ基を保護することによるステップ(g)の反応により、式Xの化合物に変換できる。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。式Xの化合物は、ステップ(b)の反応において本明細書中で先に記載のものと同じ様式のステップ(h)の反応により、式XIの化合物に変換できる。式XIの化合物は、アミノ保護基を脱保護することによる(i)の反応ステップにより、式I(式中、R1はアミノである)の化合物に変換できる。適当な脱保護試薬は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0069】
【化11】
【0070】
式II(式中、yは1〜3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0071】
【化12】
【0072】
の化合物及び式VII(式中、yは1〜3である)の化合物、すなわち、式:
【0073】
【化13】
【0074】
の化合物は、スキーム4の反応により調製できる。スキーム4の反応においては、R3は、水素、フルオロ、ブロモ、クロロ、ニトロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである。Pはヒドロキシ保護基である。R2は、1〜2個の炭素原子を有するアルキル基である。Yはハロゲン化物である。
【0075】
式XIIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のもののような適当な保護基を利用することによりカルボキシル基及びヒドロキシ基を最初に保護することによるステップ(j)の反応により、式XIIIの化合物に変換できる。
【0076】
式XIIIの化合物は、ステップ(k)の反応によりエステル基をアルコールに変換する従来の還元試薬を利用することにより、式XIV(式中、R3は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物に還元できる。この反応を実施するうえでは、水酸化リチウムアルミニウムを利用することが一般に好ましいが、これに限定するものではない。この反応は、適当な溶媒(テトラヒドロフランなど)中で実施する。ステップ(k)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0077】
式XIIIの化合物は、エステルをアルコールに変換するがニトロ基を還元しない還元試薬(例えば、BH3−THF、NaBH4−AlCl3など)を利用することにより、式XIV(式中、R3はニトロである)の化合物に還元できる。ステップ(k)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。式XIVの化合物は、ヒドロキシ基をハロゲン(好ましいハロゲンはブロモ又はクロロである)で置換することにより、式XVの化合物に変換できる。適切なハロゲン化試薬としては、塩化チオニル、塩化オキサリル、臭素、三臭化リン、四臭化炭素などが挙げられるが、これらに限定されない。ステップ(l)の反応を実施するには、そのようなハロゲン化反応において従来用いられるいずれの条件を利用してもよい。
【0078】
式XVの化合物は、XVを、アルカリ金属シアン化物、例えば、シアン化ナトリウム又はシアン化カリウム又はシアン化銅と反応させることにより、式XVIの化合物に変換できる。この反応は、適当な溶媒(例えば、ジメチルスルホキシド、N,N−ジメチルホルムアミドなど)中で実施できる。ステップ(m)の反応を実施するには、ハロゲン化物からのニトリルの調製において従来用いられる条件のいずれを利用してもよい。
【0079】
式XVIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されているものなど適当な脱保護試薬を利用してヒドロキシ保護基を除去することによるステップ(n)の反応により、式XVIIの化合物に変換できる。式XVIIの化合物は、式II(式中、yは1であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XVIIの化合物は、式VII(式中、yは1であり、R3はニトロである)の化合物でもある。式XVIの化合物は、酸性又は塩基性の加水分解によるステップ(o)の反応により、式XVIIIの化合物に変換できる。この反応を実施するうえでは、塩基性加水分解、例えばエタノール中の水性水酸化ナトリウムなどを利用することが一般に好ましい。ステップ(o)の反応を実施するには、ニトリルのカルボン酸への加水分解において従来用いられる条件のいずれを利用してもよい。
【0080】
式XVIIIの化合物をステップ(p)の反応により還元して、式XIXの化合物を得ることができる。この反応は、ステップ(k)の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XIXの化合物は、ステップ(l)の反応において本明細書中で先に記載のものと同じ様式のステップ(q)の反応により、式XXの化合物に変換できる。式XXの化合物は、ステップ(m)の反応において本明細書中で先に記載のものと同じ様式のステップ(r)の反応により、式XXIの化合物に変換できる。式XXIの化合物は、ステップ(n)の反応において本明細書中で先に記載のものと同じ様式のステップ(s)の反応により、式XXIIの化合物に変換できる。
【0081】
式XXIIの化合物は、式II(式中、yは2であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XVIIの化合物は、式VII(式中、yは2であり、R3はニトロである)の化合物でもある。
【0082】
式XXIの化合物をステップ(o)の反応において本明細書中で先に記載のものと同じ様式で加水分解して、ステップ(t)の反応により式XXIIIの化合物を得ることができる。
【0083】
式XXIIIの化合物を還元して、ステップ(u)の反応により式XXIVの化合物を得ることができる。この反応は、ステップ(k)の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XXIVの化合物は、ステップ(l)の反応において本明細書中で先に記載のものと同じ様式のステップ(v)の反応により、式XXVの化合物に変換できる。式XXVの化合物は、ステップ(m)の反応において本明細書中で先に記載のものと同じ様式のステップ(w)の反応により、式XXVIの化合物に変換できる。式XXVIの化合物は、ステップ(n)の反応において本明細書中で先に記載のものと同じ様式のステップ(x)の反応により、式XXVIIの化合物に変換できる。式XXVIIの化合物は、式II(式中、yは3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XXVIIの化合物は、式VII(式中、yは3であり、R3はニトロである)の化合物でもある。
【0084】
この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0085】
【化14】
【0086】
式II(式中、yは0であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0087】
【化15】
【0088】
の化合物及び式VII(式中、yは0である)の化合物、すなわち、式:
【0089】
【化16】
【0090】
の化合物は、スキーム5の反応により調製できる。スキーム5の反応においては、R3は、水素、ニトロ、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである。Pはヒドロキシ保護基である。R2は、1〜2個の炭素原子を有するアルキル基である。R4は、H、クロロ又はブロモである。
【0091】
式XXVIIIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のもののような適当な保護基を利用することによりヒドロキシ基を最初に保護し、次に、式XXIX(式中、R4はHである)の化合物を得るためにエステルを加水分解することによるステップ(y)の反応により、式XXIXの化合物に変換できる。
【0092】
式XXVIIIの化合物は、ステップ(y)からの式の化合物を、ハロゲン化試薬、例えば、塩化チオニル、五塩化リン、三塩化リン、臭素、四臭化炭素などと反応させることにより、式XXIX(式中、R4はクロロ又はブロモである)の化合物に変換できる。ステップ(z)の反応を実施するには、カルボン酸のそのようなハロゲン化反応において従来用いられる条件のいずれを利用してもよい。
【0093】
式XXIXの化合物は、アンモニアと直接反応させることによる、又は、式XXIXの化合物の化合物をカップリング試薬、例えば、ジシクロヘキシルカルボジイミド、ベンゾトリアゾリルオキシ)トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェートで最初に処理してからアンモニアなどと反応させることによる(a')の反応により、式XXXの化合物に変換できる。ステップ(a')の反応を実施するには、アンモニアのアシル化において従来用いられる条件のいずれを利用してもよい。式XXXの化合物は、試薬、例えば、塩化チオニル、五酸化リン、五塩化リン、オキシ塩化リン、四塩化炭素−トリフェニルホスフィン、塩化シアヌルなどを利用する脱水によるステップ(b')の反応により、式XXXIの化合物に変換できる。この反応は、純粋に、又は、適当な溶媒(例えばN,N−ジメチルホルムアミドなど)中のいずれかで実施する。ステップ(b')の反応を実施するには、そのような脱水反応において従来用いられる条件のいずれを利用してもよい。
【0094】
式XXXIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されているものなど適当な脱保護試薬を利用してヒドロキシ保護基を除去することによるステップ(c’)の反応により、式XXXIIの化合物に変換できる。式XXXIIの化合物は、式II(式中、yは0であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XXXIIの化合物は、式VII(式中、yは0であり、R3はニトロである)の化合物でもある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0095】
【化17】
【0096】
式III(式中、xは1又は2である)の化合物、すなわち、式:
A−(CH2)xOH
の化合物及び式IV(式中、xは1又は2である)の化合物、すなわち、式:
A−(CH2)xL
の化合物は、スキーム6の反応により調製できる。スキーム6の反応において、Aは前述のとおりである。Lは脱離基又はハロゲン化物である。式XXXIIIの化合物は、ステップ(d')の反応により式XXXIVの化合物に還元できる。この反応は、従来の還元剤、例えば、水酸化リチウムアルミニウムなどのアルカリ金属水素化物を利用して実施する。この反応は、適当な溶媒(テトラヒドロフランなど)中で実施する。ステップ(d')の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0097】
式XXXIVの化合物は、式III(式中、xは1である)の化合物である。
【0098】
式XXXIVの化合物は、ヒドロキシル基を脱離基又はハロゲン化物で置換することにより式XXXVの化合物に変換でき、好ましい基は、ブロモ又はクロロである。適切なハロゲン化用試薬としては、塩化チオニル、塩化オキサリル、臭素、三臭化リン、四臭化炭素などが挙げられるが、これらに限定されない。脱離基としては、トシル基、メシル基などが挙げられる。ステップ(e')の反応を実施するには、そのような反応において従来用いられるいずれの条件を利用してもよい。式XXXVの化合物は、式IV(式中、xは1である)の化合物である。
【0099】
式XXXVの化合物は、XXXVをアルカリ金属シアン化物、例えば、シアン化ナトリウム又はシアン化カリウムと反応させることにより、式XXXVIの化合物に変換できる。この反応は、適当な溶媒(エタノール、ジメチルスルホキシド、N,N−ジメチルホルムアミドなど)中で実施する。ステップ(f')の反応を実施するには、ニトリルの調製において従来用いられる条件のいずれを利用してもよい。
【0100】
式XXXVIの化合物は、酸性又は塩基性の加水分解による反応ステップ(g')により、式XXXVIIの化合物に変換できる。この反応を実施するうえでは、塩基性加水分解、例えば水性水酸化ナトリウムを利用することが一般に好ましい。ステップ(g')の反応を実施するには、ニトリルの加水分解において従来用いられる条件のいずれを利用してもよい。
【0101】
式XXXVIIの化合物を還元して、ステップ(h')の反応により式XXXVIIIの化合物を得ることができる。この反応は、ステップ(d')の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XXXVIIIの化合物は、式III(式中、xは2である)の化合物である。
【0102】
式XXXVIIIの化合物は、ステップ(e')の反応において本明細書中で先に記載のものと同じ様式のステップ(i')の反応により、式XXXIXの化合物に変換できる。式XXXIXの化合物は、式IV(式中、xは2である)の化合物である。
【0103】
この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。Aが、1又は2つのヒドロキシル基により置換されているフェニルである場合は、式XXXIIIの化合物のヒドロキシル基を保護することが一般に好ましい。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。
【0104】
【化18】
【0105】
式XXVIII(式中、R3は、水素、ニトロ、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルであり、R2は、1〜2個の炭素原子を有するアルキルである)の化合物、すなわち式:
【0106】
【化19】
【0107】
の化合物は、スキーム7の反応により調製できる。スキーム7の反応において、R2及びR3は前述のとおりである。
【0108】
式XIIの化合物は、メタノール又はエタノールで式XIIの化合物をエステル化することによるステップ(j')の反応により、式XXVIIIの化合物に変換できる。この反応は、触媒(例えば、H2SO4、TsOHなど)を使用するか、又は脱水剤(例えばジシクロヘキシルカルボジイミドなど)を使用することのいずれかにより実施できる。ステップ(j’)の反応を実施するには、そのようなエステル化反応において従来用いられる条件のいずれを利用してもよい。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0109】
【化20】
【0110】
式XII(式中、R3は、クロロ、ブロモ又はフルオロである)の化合物、すなわち式:
【0111】
【化21】
【0112】
の化合物は、商業的に入手できるか、又は以下のような文献中に記載の方法により調製できるかいずれかである:
1. 3−Br又はF−2−OHC6H3CO2H
Canadian Journal of Chemistry (2001), 79(11) 1541-1545。
2. 4−Br−2−OHC6H3CO2H
WO9916747又はJP04154773。
3. 2−Br−6−OHC6H3CO2H
JP47039101。
4. 2−Br−3−OHC6H3CO2H
WO9628423。
5. 4−Br−3−OHC6H3CO2H
WO2001002388。
6. 3−Br−5−OHC6H3CO2H
Journal of labelled Compounds and Radiopharmaceuticals (1992), 31 (3), 175-82。
7. 2−Br−5−OHC6H3CO2H及び3−Cl−4−OHC6H3CO2H
WO9405153及びUS5519133。
8. 2−Br−4−OHC6H3CO2H及び3−Br−4−OHC6H3CO2H
WO20022018323
9. 2−Cl−6−OHC6H3CO2H
JP06293700
10. 2−Cl−3−OHC6H3CO2H
Proceedings of the Indiana Academy of Science (1983), Volume date 1982, 92, 145-51。
11. 3−Cl−5−OHC6H3CO2H
WO2002000633及びWO2002044145。
12. 2−Cl−5−OHC6H3CO2H
WO9745400。
【0113】
式XII(式中、R3は、1〜2個の炭素原子を有するアルコキシである)の化合物、すなわち、式:
【0114】
【化22】
【0115】
の化合物は、スキーム8の反応により調製できる。スキーム8の反応においては、R2は、1〜2個の炭素原子を有するアルキルである。Pはヒドロキシル保護基である。式XLの化合物は、適当な保護基によりフェニル基を保護することによるステップ(k’)の反応により、式XLIの化合物に変換できる。保護基のための適当な条件は、T. GreeneによるProtective Groups in Organic Synthesis中に記載してある可能性がある。
【0116】
式XLIの化合物は、アルデヒドをカルボン酸に酸化することにより、式XLIIの化合物に変換できる。この反応は、適当な酸化試薬、例えば、ピリジニウムクロロクロメート、過マンガン酸カリウム、過マンガン酸ナトリウムなどを使用することにより実施できる。ステップ(l’)の反応を実施するには、そのような酸化反応において適当な条件のいずれを利用してもよい。
【0117】
式XLIIの化合物は、保護基の脱保護により、ステップ(m’)の反応による式XII(式中、R3は、1個の炭素原子を有するアルコキシである)の化合物に変換できる。適当な脱保護条件は、T GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。
【0118】
式XLIIの化合物は、溶媒(例えばジクロロメタン)を−72℃〜0℃の温度で4〜48時間使用して式XLIIの化合物を三臭化ホウ素又は三塩化ホウ素で処理することにより、式XLIIIの化合物に変換できる。ステップ(n’)の反応を実施するには、そのような脱メチル反応において従来用いられる条件のいずれを利用してもよい。
【0119】
式XLIIIの化合物は、メタノール又はエタノールで式XLIIIの化合物をエステル化することにより、式XLIVの化合物に変換できる。この反応は、触媒(例えば、H2SO4、TsOHなど)を使用するか、又は脱水剤(例えばジシクロヘキシルカルボジイミドなど)を使用することのいずれかにより実施できる。ステップ(o’)の反応を実施するには、そのようなエステル化反応において従来用いられる条件のいずれを利用してもよい。
【0120】
式XLIVの化合物は、適当な塩基、例えば、炭酸カリウム、水素化ナトリウム、ピリジンなどを用いることにより、式XLIVの化合物をハロゲン化エチルでエーテル化又はアルキル化することにより、式XLVの化合物に変換できる。この反応は、従来の溶媒(テトラヒドロフラン、N,N−ジメチルホルムアミド、ジクロロメタンなど)中で実施できる。この反応は、0℃〜40℃の温度で一般に実施する。ステップ(p’)の反応を実施するには、そのようなアルキル化反応において適当な条件のいずれを利用してもよい。
【0121】
式XLVの化合物は、保護基の脱保護により、ステップ(q’)の反応による式XII(式中、R3は、2個の炭素原子を有するアルコキシである)の化合物に変換できる。適当な脱保護条件は、T GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0122】
【化23】
【0123】
式XII(式中、R3は、1〜2個の炭素原子を有するアルコキシである)の化合物、すなわち、式:
【0124】
【化24】
【0125】
の化合物は、商業的に入手できるか、又は以下のような文献中に記載の方法により調製できるかいずれかである:
1. 2−OMe−4−OHC6H3CO2H
US2001034343又はWO9725992。
2. 5−OMe−3−OHC6H3CO2H
J.O.C (2001), 66(23), 7883-88。
3. 2−OMe−5−OHC6H3CO2H
US6194406(96ページ)及びJournal of the American Chemical Society (1985), 107(8), 2571-3。
4. 3−OEt−5−OHC6H3CO2H
Taiwan Kexue (1996), 49(1), 51-56。
5. 4−OEt−3−OHC6H3CO2H
WO9626176
6. 2−OEt−4−OHC6H3CO2H
Takeda Kenkyusho Nempo (1965), 24,221-8。
JP07070025。
7. 3−OEt−4−OHC6H3CO2H
WO9626176。
【0126】
式XII(式中、R3は、1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0127】
【化25】
【0128】
の化合物は、商業的に入手できるか、又は、以下のような文献中に記載の方法により調製できるかいずれかである:
1. 5−Me−3−OHC6H3CO2H及び2−Me−5−OHC6H3CO2H
WO9619437。
J.O.C. 2001, 66, 7883-88。
2. 2−Me−4−OHC6H3CO2H
WO8503701。
3. 3−Et−2−OHC6H3CO2H及び5−Et−2−OHC6H3CO2H
J. Med. Chem. (1971), 14(3), 265。
4. 4−Et−2−OHC6H3CO2H
Yaoxue Xuebao (1998), 33(1), 67-71。
5. 2−Et−6−OHC6H3CO2H及び2−n−Pr−6−OHC6H3CO2H
J. Chem. Soc., Perkin Trans 1 (1979), (8), 2069-78。
6. 2−Et−3−OHC6H3CO2H
JP10087489及びWO9628423。
7. 4−Et−3−OHC6H3CO2H
J.O.C. 2001, 66, 7883-88。
WO9504046。
8. 2−Et−5−OHC6H3CO2H
J.A.C.S (1974), 96(7), 2121-9。
9. 2−Et−4−OHC6H3CO2H及び3−Et−4−OHC6H3CO2H
JP04282345。
10. 1.3−Et−5−OHC6H3CO2H
2−エチルアクロレインを使用することにより、J.O.C. 2001, 66, 7883-88からの合成を適合する。
【0129】
治療法における使用
本発明は、哺乳動物対象における尿酸値を低下させ、又は哺乳動物対象からの尿酸排泄を増加させる方法を提供する。哺乳動物における尿酸値は、任意の従来の測定を用いて定量できる。典型的には、血中尿酸値を定量する。尿酸は、組織中に堆積又は沈殿する結果、蓄積部(例えば痛風結節)となる場合もあり、その蓄積部は血中尿酸濃度を上昇又は低下させることにより影響を受けることもあり、逆に尿酸の循環の一因となることもある。尿酸を減少させるための本発明の方法は、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎臓結石、腎機能不全、心血管疾患、心血管リスク因子及び認知障害などさまざまな状態を治療又は防止するために用いることができる。本発明の化合物を投与すると、尿酸値を低下させることにより、腎臓疾患の進行が遅くなる。尿酸値上昇は心血管疾患のリスク因子として同定されている。高齢者においては、尿酸上昇と認知障害との間に有意な相関が示されている。(Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140)。したがって、尿酸を減少させるための本発明の方法は、高齢者における認知障害を含めた認知障害を治療又は防止するために使用できる。レッシュ−ナイハン症候群罹患者は尿酸値が上昇しており、こうした高尿酸血症の多数の結果(痛風など)を患うことはよく知られている。したがって、血中尿酸値を低下させ尿酸の排出を増加させるための本発明は、レッシュ−ナイハン症候群罹患者を治療するために使用できる。
【0130】
血中尿酸の正常範囲は、男性においては3.4mg/dL〜7.0mg/dLの間、閉経前の女性においては2.4mg/dL〜6.0mg/dLの間、小児においては2.5mg/dL〜5.5mg/dLの間である。尿酸結晶の形成/沈殿は、典型的には、男性においては6.6mg/dL以上の値で、女性においては6.0mg/dL以上の値で生じる。このことから、いわゆる正常範囲内にある尿酸値であっても、望ましくない健康上の結果を有することが(痛風を生じさせることすら)あることが示される。さらに、全体の集団にとっては正常範囲であると思われる値であっても、個人にとっては上昇している場合がある。尿酸上昇による心血管及び他の結果は、こうした「正常な」範囲内の血中濃度を伴って生じる場合が相当ある。したがって、高尿酸血症の診断は、本発明の化合物の有益な効果にとっては必ずしも予め必要なものではない。
【0131】
本発明は、痛風、高血圧症、血管炎症、心不全、動静脈障害、心筋梗塞、脳卒中、子癇前症、子癇、睡眠時無呼吸、腎機能不全(腎不全、末期腎疾患[ESRD,end stage renal disease]など)、臓器移植、利尿薬、チアジド、シクロスポリン、アスピリン、ビタミンC、ニコチン酸、レボドパ(L−DOPA,levodopa)、細胞傷害薬及びある種の抗菌剤(ピロジナミド(pyrozinamide)など)、肝硬変、甲状腺機能不全、副甲状腺機能不全、肺癌、貧血症、白血病、リンパ腫、多発性骨髄腫、腫瘍溶解症候群、甲状腺又は副甲状腺の機能不全、レッシュ−ナイハン症候群、喫煙、アルコール消費及び乾癬に伴う高尿酸血症の治療を包含する。本発明は、痛風、尿酸結晶の形成、腎機能不全、移植後の生着不全又は臓器不全、内皮障害(炎症など)、慢性心不全、動静脈障害、子癇前症、子癇、高血圧症及び認知障害をもたらす可能性のある高尿酸血症の治療を包含する。痛風を治療するための本発明の方法の実施形態においては、尿酸の組織蓄積部(痛風結節を含むがこれに限定されるものではない)は減少し、痛風の発作の発生及び重症度も低下する。
【0132】
本発明の化合物は、任意の従来の全身投与経路により投与できる。好ましくは、この化合物又はその塩は経口投与される。したがって、この医薬は経口投与用に製剤化されることが好ましい。本発明により用いることができる他の投与経路としては、注射(例えば、静脈内、皮下、筋肉内又は腹膜内の注射)による経直腸的、非経口的な経路、又は経鼻的な経路が挙げられる。
【0133】
本発明の治療の使用及び方法のそれぞれのさらなる実施形態は、前述の化合物の実施形態のいずれか1つを投与することを含む。不必要に冗長になることを避けるため、そのような化合物及び化合物群はそれぞれ繰り返さないが、そうした薬剤及び薬剤群は、繰り返された場合と同様に、治療の使用及び方法のこの説明中に組み込まれる。
【0134】
ヒト及び非ヒトの哺乳動物対象は両方とも、本発明の治療方法により治療できる。特定の対象のための本発明の特定の化合物の最適用量は、熟練の臨床家により臨床の場において決定できる。経口投与の場合においては、本発明の化合物は、一般に、成人に対し一日用量が1mg〜2500mg、より好ましくは1mg〜1200mgで投与する。本発明の他の一実施形態においては、この化合物は、400mg〜1000mg、600mg〜800mg、600mg〜1000mg、又は100〜300mgの用量で、1日当たり1回又は2回投与する。典型的な成人の平均体重は60〜70キログラムであることから、mg/kgとして表される適切な用量範囲は、およそ0.015〜42mg/kg、0.015〜20mg/kg、6.6〜13mg/kg、10〜13mg/kg、10〜16mg/kg、又は1.67〜4.3mg/kgであり、1日当たり1回又は2回投与する。小児を治療するときは、最適用量は患者の医師が決定する。マウスへの経口投与の場合においては、本発明の化合物は、一般には、体重1キログラム当たりの化合物の一日用量1〜300mgで投与する。
【0135】
本発明の化合物は、他の尿酸低下薬と組み合わせて投与できる。そのような場合における本発明の化合物の用量は、前述のとおりである。本発明の化合物と組み合わせて、従来の、又は治験用の任意の尿酸低下薬を利用できる。そのような薬物の例としては、以下が挙げられる:キサンチンオキシダーゼ阻害薬(アロプリノール(100mg/日〜1000mg/日、より典型的には100mg/日〜300mg/日)、フェブキソスタット(40mg/日〜120mg/日、より具体的には60mg/日〜80mg/日)及びオキシプリノールなど);Puricase/PEG−ウリカーゼ(注射により2週間毎に4mg〜12mg);尿酸排泄剤(スルフィンピラゾン(100mg/日〜800mg/日)、プロベネシド(500mg/日)、ロサルタン(25mg/日〜200mg/日、より典型的には50mg/日〜100mg/日)、フェノフィブレート、JTT-552(URAT−1阻害薬)、ベンズブロマロン(70mg/日〜150mg/日)など)及びスタチン(アトルバスタチン(LIPITOR(登録商標))など)。他の尿酸低下薬は、そのような他の薬物の用量の方を低くして投与するか、そのような他の薬物の頻度を低くして投与するか、いずれかにより、その通常量又は通常量未満の量で投与してもよい。
【0136】
本発明の化合物は、痛風発作に伴う疼痛を低下させるために使用される他の薬物、例えば、非ステロイド性抗炎症薬(NSAID,nonsteroidal antiinflammatory drug)、コルヒチン、コルチコステロイド及び他の鎮痛薬と共に投与できる。
【0137】
血中尿酸値を低下させる過程においては、本発明の化合物により尿中の尿酸値が高まると考えられる。尿のpHを高め、それにより尿酸の可溶性を高めるために、例えば、クエン酸塩又は炭酸水素塩を、本発明の化合物と組み合わせて投与してもよい。
【0138】
本発明の化合物又は塩と、1又は複数の他の尿酸低下薬、鎮痛薬及びpH上昇剤との混合物を対象に投与してもよい。或いは、本発明の化合物又は塩、及び、1又は複数の他の尿酸低下薬、鎮痛薬及びpH上昇剤は、共に混合されて混合物を形成するのではなく、対象に独立に投与される。活性成分を共に混合して単一の混合物又は組成物を形成しないときは、活性成分を、1又は複数単位の経口用量の本発明の化合物と、1又は複数単位の経口用量の1又は複数の他の尿酸低下薬と、鎮痛薬及びpH上昇剤と、本発明の化合物を他の活性成分と組み合わせて投与するための取扱説明書とを備えるキットの形態で提供すると便利である。好ましくは、キットの構成要素は、箱又はブリスターパックなどに入った形で、一緒に包装される。
【0139】
医薬組成物
本発明は、本発明の化合物と、任意選択で薬学的に許容される担体とを含む医薬組成物を提供する。本発明の医薬組成物のさらなる実施形態は、前述の化合物の実施形態のいずれか1つを含む。不必要に冗長になることを避けるため、そのような化合物及び化合物群はそれぞれ繰り返さないが、そうした薬剤及び薬剤群は、繰り返された場合と同様に、医薬組成物のこの説明中に組み込まれる。
【0140】
好ましくは、この組成物は、例えば、錠剤、コーティング錠、糖衣錠、ハード又はソフトタイプのゼラチンカプセル剤、液剤、乳剤又は懸濁剤の形態で経口投与用に構成される。一般に、この経口組成物は、1mg〜2500mg、より好ましくは1mg〜1200mgの本発明の化合物を含むことになる。本発明のより具体的な実施形態においては、この経口組成物は、400mg〜1000mg、600mg〜800mg、600mg〜1000mg、又は100〜300mgの本発明の化合物を含むことになる。対象にとっては、1日当たり1又は2個の錠剤、コーティング錠、糖衣錠又はゼラチンカプセル剤を飲みこむことは手軽である。しかし、この組成物は、例えば坐剤の形態での経直腸投与、例えば注射液剤の形態での非経口投与、又は経鼻投与など、任意の他の従来の全身投与手段による投与用に構成することもできる。
【0141】
この活性成分は、医薬組成物の作製のために、薬学上不活性な無機又は有機の担体を用いて加工してもよい。乳糖、トウモロコシデンプン又はその誘導体、タルク、ステアリン酸又はその塩などを、例えば、錠剤、コーティング錠、糖衣錠及びハードゼラチンカプセル剤用のそのような担体として使用できる。ソフトゼラチンカプセル剤用の適当な担体は、例えば、植物油、蝋、脂肪、半固体及び液体のポリオールなどである。しかし、活性成分の性質によっては、ソフトゼラチンカプセルの場合においては、ソフトゼラチン自体以外の担体は通常必要ではない。液剤及びシロップ剤の作製用の適当な担体は、例えば、水、ポリオール、グリセロール、植物油などである。坐剤用の適当な担体は、例えば、天然又は硬化タイプの油、蝋、脂肪、半液体又は液体のポリオールなどである。
【0142】
この医薬組成物は、さらに、保存剤、可溶化剤、安定化剤、湿潤剤、乳化剤、甘味剤、着色剤、香味剤、浸透圧を変化させるための塩、緩衝剤、コーティング剤又は抗酸化剤を含有してもよい。
【0143】
本発明は、以下の実施例を参照することによりさらによく理解されると思われるが、この実施例は、本明細書中に記載の本発明を例証するものであるが、これを限定するものではない。
[実施例]
【実施例1】
【0144】
【化26】
【0145】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール
ステップA:3−(2,6−ジメチルベンジルオキシ)ベンゾニトリルの調製:
乾燥THF(30ml)中の2,6−ジメチルベンジルアルコール(6.27g、46.1mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、9.24g、45.7mmol)の溶液を、THF(100ml)中の3−ヒドロキシベンゾニトリル(5g、37mmol)及びトリフェニルホスフィン(TPP、11.99g、45.7mmol)の溶液に0℃で滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0146】
ステップB:5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾールの調製:
乾燥ジメチルホルムアミド(30ml)中の3−(2,6−ジメチルベンジルオキシ)ベンゾニトリル(ステップA、3g、11.8mmol)、アジ化ナトリウム(0.847g、13mmol)及び塩化アンモニウム(0.697g、13mmol)の混合物を、アルゴン下で110℃にて14時間又は出発物質が全て消費されるまで加熱した。この反応混合物に水を加えて、固形物を全て溶解させ、この溶液をブライン中に取り、酢酸エチル(2X)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
1H NMR(400MHz,(CD3)2SO):2.4(s,6H);5.15(s,2H);7.1(d,2H);7.15(m,1H);7.3(dd,1H);7.5(t,1H);7.65(m,1H);7.7(m,1H)。
【実施例2】
【0147】
【化27】
【0148】
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール
ステップA:2−(3−ヒドロキシフェニル)アセトニトリルの調製:
乾燥塩化メチレン(20ml)中の2−(3−メトキシフェニル)アセトニトリル(3.6g、25.4mmol)の溶液に、アルゴン雰囲気下で−78℃にてBBr3(55ml、CH2Cl2中1M、55mmol)を加えた。この反応混合物を48時間かけて周囲温度に温め、破砕した氷でクエンチし、塩化メチレンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(CH2Cl2:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、油としての標題化合物を得た。
【0149】
ステップB:2−(3−(2,6−ジメチルベンジルオキシ)フェニル)アセトニトリルの調製:
乾燥THF(20ml)中の2−(3−ヒドロキシフェニル)アセトニトリル(ステップA、5g、37mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、3.38g、16.7mmol)の溶液を、THF(30ml)中の2,6−ジメチルベンジルアルコール(2.25g、16.5mmol)及びトリフェニルホスフィン(TPP、4.3g、16.4mmol)の溶液に、アルゴン下で0℃にて滴加した。この反応混合物を、室温で16時間又は出発物質が全て消費されるまで撹拌した。この混合物にシリカゲル(25g)を加え減圧下で溶媒を除去し、これをシリカゲルカラム上に載せ、塩化メチレン:ヘキサン(1:1)で溶出させて、淡黄色の結晶性の固形物を得た。
【0150】
ステップC:5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾールの調製:
乾燥ジメチルホルムアミド(30ml)中の2−(3−(2,6−ジメチルベンジルオキシ)フェニル)アセトニトリル(ステップB、3.2g、12.7mmol)、アジ化ナトリウム(1.28g、16.7mmol)及び塩化アンモニウム(1.08g、20.2mmol)の混合物を、アルゴン下、90℃にて9時間又は出発物質が全て消費されるまで加熱し、この反応混合物を減圧下で濃縮した。この反応混合物を酢酸エチル中に取り、水(2X)で洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(塩化メチレン:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより精製して、油状の生成物を得た。この油を1:2の酢酸エチル:ヘキサンと共に10分間撹拌し、固形物を濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.3(s,6H);4.25(s,2H);5.15(s,2H);6.84(d,1H);6.96(m,2H);7.08(d,2H);7.18(m,1H);7.28(m,1H)。
【実施例3】
【0151】
【化28】
【0152】
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール
ステップA:エチル3−ヒドロキシ−4−メトキシベンゾエートの調製:
無水エタノール(300ml)中の3−ヒドロキシ−4−メトキシ安息香酸(25g、148.67 mmol)及びp−トルエンスルホン酸一水和物(3.17g、16.66mmol)の溶液を、6時間又は出発物質が全て消費されるまで環流させた。この反応混合物を濃縮し、EtOAc(60ml)で希釈し、水(20ml)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0153】
ステップB:エチル3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンゾエートの調製:
乾燥THF(20ml)中のエチル3−ヒドロキシ−4−メトキシベンゾエート(ステップA、9.10g、46.4mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、10.23g、50mmol)の溶液を、乾燥THF(60ml)中の2,6−ジメチルベンジルアルコール(6.94g、51mmol)及びトリフェニルホスフィン(TPP、13.27g、50mmol)の溶液に、アルゴン下で0℃にて滴加した。この反応混合物を4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0154】
ステップC:(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)メタノールの調製:
乾燥THF(30ml)中のエチル3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンゾエート(ステップB、6.04g、19.23mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.803g、21.16mmol)を滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで撹拌してから、0.1N HCl、EtOAc(20ml)でゆっくりクエンチして反応混合物に加えた。この反応混合物を濾過し、沈殿物をEtOAc(25ml×2)で洗浄した。有機層を合わせたものを0.1N HCl、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0155】
ステップD:2−((5−(ブロモメチル)−2−メトキシフェノキシ)メチル)−1,3−ジメチルベンゼンの調製:
乾燥CH2Cl2(20ml)中の(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)メタノール(ステップC、5.23g、20.4mmol)及びCBr4(10.16g、30.6mmol)の溶液に、トリフェニルホスフィン(8.03g、30.64mmol)を0℃で少しずつ加えた。この反応混合物を1.5時間撹拌し、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
1H NMR(400MHz,(CDCl3):2.43(s,6H);3.83(s,3H);4.53(s,2H);5.08(s,2H);6.84(d,1H);7.0〜7.03(dd,1H);7.06〜7.09(m,3H);7.14〜7.18(m,1H)。
【0156】
ステップE:2−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−((5−(ブロモメチル)−2−メトキシフェノキシ)メチル)−1,3−ジメチルベンゼン(ステップD、3.28g、9.7mmol)及びNaCN(0.624g、12.7mmol)の溶液を、120℃で2.5時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0157】
ステップF:5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)アセトニトリル(ステップE、2.17g、7.5mmol)、アジ化ナトリウム(0.590g、9.1mmol)及び塩化アンモニウム(0.486g、9.1mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.3(s,6H);3.68(s,3H);4.22(s,2H);4.98(s,2H);6.78〜6.81(dd,1H);6.91〜6.93(d,1H);7.05〜7.07(d,2H);7.13〜7.16(m,2H)。MS:m/z 325.2[M+H]+。
【実施例4】
【0158】
【化29】
【0159】
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール
ステップA:3−(3−メトキシフェニル)プロパンニトリルの調製:
乾燥DMF(20ml)中の1−(2−ブロモエチル)−3−メトキシベンゼン(10g、46.4mmol)、NaCN(2.73g、55.8mmol)の溶液を、90℃で6時間又は出発物質が全て消費されるまで加熱し、この反応を冷却し、EtOAc(60ml)で希釈し、水(20ml×3)、ブラインで洗浄し、有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、油としての標題化合物を得た。
【0160】
ステップB:3−(3−ヒドロキシフェニル)プロパンニトリルの調製:
乾燥CH2Cl2(20ml)中の3−(3−メトキシフェニル)プロパンニトリル(ステップA、1.71g、10.6mmol)の撹拌溶液に、アルゴン下で−78℃にてBBr3(CH2Cl2中1M、5.32g、21.2mmol)を加えた。この反応混合物を、同じ温度で2時間、次いで0℃で4時間又は出発物質が全て消費されるまで撹拌し、氷でクエンチし、EtOAc(30ml×3)で抽出し、有機層を合わせたものを飽和NaHCO3、ブラインで慎重に洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0161】
ステップC:3−(3−(2,6−ジメチルベンジルオキシ)フェニル)プロパンニトリルの調製:
乾燥THF(10ml)中の3−(3−ヒドロキシフェニル)プロパンニトリル(ステップB、1.25g、8.5mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、1.87g、9.26mmol)の溶液を、アルゴン下で0℃にて、乾燥THF(30ml)中の2,6−ジメチルベンジルアルコール(1.27g、9.3mmol)及びトリフェニルホスフィン(TPP、2.43g、9.26mmol)の溶液に滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0162】
ステップD:5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の3−(3−(2,6−ジメチルベンジルオキシ)フェニル)プロパンニトリル(ステップC、2.62g、9.9mmol)、アジ化ナトリウム(0.899g、13.8mmol)及び塩化アンモニウム(0.740g、13.8mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5→92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を、1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.48(s,6H);3.02(t,2H);3.19(t,2H);4.98(s,2H);6.80〜6.81(d,1H);6.86〜6.89(m,2H);7.05〜7.07(d,2H);7.14〜7.23(m,2H)。MS:m/z 309.2[M+H]+。
【実施例5】
【0163】
【化30】
【0164】
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
ステップA:2−(3−ヒドロキシ−4−メチルフェニル)アセトニトリルの調製:
乾燥CH2Cl2(20ml)中の2−(3−メトキシ−4−メチルフェニル)アセトニトリル(5g、31mmol)の撹拌溶液に、アルゴン下で−78℃にて、BBr3(CH2Cl2中1M、10.02g、40mmol)を滴加した。この反応混合物を、同じ温度で2時間、次いで0℃で5時間又は出発物質が全て消費されるまで撹拌し、氷でクエンチし、EtOAc(30ml×3)で抽出し、有機層を合わせたものを飽和NaHCO3、ブラインで慎重に洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1→CH2Cl2:hex、1:1)を用いたフラッシュクロマトグラフィーにより精製して、オフホワイトの固形物としての標題化合物を得た。
【0165】
ステップB:2−(3−(2,6−ジメチルベンジルオキシ)−4−メチルフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−(3−ヒドロキシ−4−メチルフェニル)アセトニトリル(ステップA、2.18g、14.8mmol)、K2CO3(2.66g、19.2mmol)の撹拌溶液に、アルゴン下で室温にて、2,6−ジメチルベンジルクロリド(2.97g、19.2mmol)を加えた。この反応混合物を16時間撹拌し、EtOAc(40ml)で希釈し、水(20ml)及びブラインで洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0166】
ステップC:5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾールの調製:
乾燥DMF(15ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−4−メチルフェニル)アセトニトリル(ステップB、1.12g、4.2mmol)、アジ化ナトリウム(0.400g、6.1mmol)及び塩化アンモニウム(0.350g、6.5mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5→92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)で10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.0(s,3H);2.35(s,6H);4.27(s,2H);5.0(s,2H);6.73〜6.75(dd,1H);7.08〜7.1(m,3H);7.15〜7.19(m,2H)。MS:m/z 309.2[M+H]+。
【実施例6】
【0167】
【化31】
【0168】
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール
ステップA:2−(4−(2,6−ジフルオロベンジルオキシ)フェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−(4−ヒドロキシフェニル)アセトニトリル(5g、37.5mmol)及びK2CO3(6.74g、48.8mmol)の溶液に、2,6−ジフルオロベンジルブロミド(7.77g、37.5mmol)を加えた。この反応混合物を4時間室温で撹拌し、真空中で濃縮した。粗残留物をEtOAc中に取り、水及びブラインで洗浄した。水層をEtOAcで再度洗浄した。有機層を合わせたものをNa2SO4で乾燥させ、濾過し、濃縮して、白色の固形物としての標題化合物を得た。
1H NMR(270MHz,CDCl3):3.65(s,2H);5.1(s,2H);6.9〜7.0(m,4H);7.2〜7.4(m,3H)。
【0169】
ステップB:5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾールの調製:
乾燥DMF(60ml)中の2−(4−(2,6−ジフルオロベンジルオキシ)フェニル)アセトニトリル(ステップA、5g、19.3mmol)、アジ化ナトリウム(1.3g、20mmol)及び塩化アンモニウム(1.06g、20mmol)の混合物を、90℃で16時間加熱した。溶媒を真空中で除去し、油状の残留物をEtOAcと水との間で分離させた(高濃度HClを用いてpH1に酸性化した)。有機層を水で洗浄し、Na2SO4で乾燥させ、濾過し、濃縮して、茶色の半固形物とした。精製は、シリカゲルカラム(クロロホルム:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより行って、淡いクリーム色の固形物としての標題化合物を得た。
1H NMR(270MHz,CDCl3):4.0(s,2H);5.1(s,2H);6.7〜6.9(m,4H);7.0(d,2H);7.2(m,1H)。
【実施例7】
【0170】
【化32】
【0171】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール
ステップA:エチル3−ヒドロキシ−2−メチルベンゾエートの調製:
無水エタノール(150ml)中の3−ヒドロキシ−2−メチル安息香酸(5.04g、33.12mmol)及びp−トルエンスルホン酸一水和物(0.741g、3.89mmol)の溶液を、16時間又は出発物質が全て消費されるまで還流させた。この反応混合物を濃縮し、EtOAc(30ml)で希釈し、水(20ml)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0172】
ステップB:エチル3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゾエートの調製:
乾燥DMF(15ml)中のエチル3−ヒドロキシ−2−メチルベンゾエート(ステップA、3.1g、17.22mmol)、K2CO3(3.09g、22.38mmol)の撹拌溶液に、アルゴン下で室温にて、2,6−ジメチルベンジルクロリド(3.19g、20.66mmol)を加えた。この反応混合物を16時間撹拌し、EtOAc(40ml)で希釈し、水(20ml)及びブラインで洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0173】
ステップC:(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)メタノールの調製:
乾燥THF(35ml)中のエチル3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゾエート(ステップB、5.94g、19.93mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.832g、21.92mmol)を滴加した。この反応混合物を4時間又は出発物質が全て消費されるまで撹拌してから、0℃にて0.1N HClでゆっくりクエンチし、この反応混合物にEtOAc(20ml)を加えた。この反応混合物を濾過し、EtOAc(25ml×2)で沈殿物を洗浄した。有機層を合わせたものを0.1N HCl、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0174】
ステップD:1−(ブロモメチル)−3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゼンの調製:
乾燥CH2Cl2(20ml)中の(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)メタノール(ステップC、3.68g、14.37mmol)及びCBr4(5.25g、15.8mmol)の溶液に、0℃でトリフェニルホスフィン(4.14g、15.8mmol)を少しずつ加えた。この反応混合物を4時間撹拌し、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。この固形物を真空下で6時間、さらに乾燥させ続けた。
【0175】
ステップE:2−(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の1−(ブロモメチル)−3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゼン(ステップD、4.28g、13.41mmol)及びNaCN(0.789g、16.10mmol)の溶液を120℃で3時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0176】
ステップF:5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)アセトニトリル(ステップE、2.70g、10.19mmol)、アジ化ナトリウム(0.795g、12.23mmol)及び塩化アンモニウム(0.653g、12.22mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.01(s,3H);2.32(s,6H);4.24(s,2H);5.01(s,2H);6.78〜6.79(dd,1H);7.06〜7.08(d,2H);7.12〜7.19(m,3H)。
【実施例8】
【0177】
【化33】
【0178】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
ステップA:3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンズアルデヒドの調製:
乾燥DMF(15ml)中の3−ヒドロキシ−2−メトキシベンズアルデヒド(5.06g、32.7mmol)、2,6−ジメチルベンジルクロリド(5.04g、33.1mmol)及びK2CO3(4.78g、34.6mmol)の溶液を、アルゴン下で室温にて16時間撹拌してから、EtOAc(40ml)で希釈し、水(20ml)で洗浄した。有機層を濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、オフホワイトの固形物としての標題化合物を得た。
【0179】
ステップB:(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)メタノールの調製:
乾燥THF(40ml)中の3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンズアルデヒド(ステップB、10.6g、32.7mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.95g、23.7mmol)を滴加した。この反応混合物を1時間又は出発物質が全て消費されるまで撹拌してから水をゆっくり加え、次いで1N HCl(5ml)、水(10ml)を加えることによりクエンチし、この反応混合物にEtOAc(20ml)を加えた。この反応混合物を濃縮し、酢酸エチルを用いた短いシリカゲルカラムを通過させて、オフホワイトの固形物としての標題化合物を得た。
【0180】
ステップC:3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジルメタンスルホネートの調製:
乾燥CH2Cl2(100ml)中の(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)メタノール(ステップB、9.5g、32.7mmol)及びトリエチルアミン(5.80g、57.4mmol)の溶液に、アルゴン下で0℃にて塩化メタンスルホニル(3.5ml、45mmol)を滴加した。この反応混合物を6時間又は出発物質が全て消費されるまで室温に温め、10%の冷Na2CO3で中和し、CH2Cl2で抽出した。有機層を濃縮し、シリカゲルカラム(hex:塩化メチレン、2:1)を用いたフラッシュクロマトグラフィーにより精製して、淡黄色の油としての標題化合物を得た。
【0181】
ステップD:2−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)アセトニトリルの調製:
乾燥DMF(40ml)中の3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジルメタンスルホネート(ステップC、8.8g、30.26mmol)及びNaCN(1.60g、32.6mmol)の溶液を、85℃で18時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)で洗浄し、濃縮し、塩化メチレンを用いた短いシリカゲルカラムを通過させて、黄色の固形物としての標題化合物を得た。
【0182】
ステップE:5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾールの調製:
乾燥DMF(10ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)アセトニトリル(ステップD、3.2g、12.7mmol)、アジ化ナトリウム(0.86g、13.2mmol)及び塩化アンモニウム(0.696g、13.0mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、減圧下で濃縮し、シリカゲルカラム(クロロホルム:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.33(s,6H);3.5(s,3H);4.21(s,2H);5.04(s,2H);6.83〜6.85(dd,1H);7.05〜7.09(m,3H);7.15〜7.23(m,2H)。
【実施例9】
【0183】
URAT1阻害アッセイ
URAT1(尿酸輸送体1,Uric Acid Transporter 1)は、腎尿細管中の頂端膜上で発現する。URAT1は、尿から血中への尿酸の再取込みを媒介する。URAT1を阻害すると、尿中の尿酸の排泄の増加、したがって、血清尿酸濃度を低下させる薬物に対する潜在的様式の作用がもたらされる。例えば、プロベネシド及びベンズブロマロンは、痛風及び高尿酸血症の治療に臨床的に使用されており、これらの薬物は両方ともURAT1に作用して尿酸の再取込みを減少させる。しかし、ベンズブロマロンは、URAT1とは無関係の機序による肝毒性を理由に市場から引き揚げられ、プロベネシドは、多数の輸送タンパク質に作用する結果、さまざまな他の薬物と相互作用する。
【0184】
インビトロでのURAT1アッセイは、血清尿酸の減少において強力な活性を有する化合物を同定するには有用である。適当なアッセイには、細胞(例えば、ヒト胚腎細胞「HEK,human embryonic kidney」)に、ヒトURAT1をコードするベクターを形質移入し、次いで、放射線標識した尿酸を取り込む形質移入細胞の能力を定量することが含まれる。URAT1阻害薬としての化合物の活性は、形質移入細胞による尿酸取込みを遮断する能力により評価される。
【0185】
試験化合物及び化学薬品:
ベンズブロマロン(Sigma社製, Cat.No.B5774)、プロベネシド(Sigma社製, Cat.No.P8761)、DMSO(Sigma社製, Cat.No.D-2650)、[8−14C]尿酸(50〜60mCi/mmol、American Radio Chemicals社製, Cat. No. ARC0513)。
【0186】
発現ベクター中へのhURAT1のサブクローニング:
hURAT1 cDNAを含有するプラスミドベクターpCMV6−XL5(Cat. No. SC125624)と、発現ベクターpCMV6−Neo(Cat. No.pCMVNEO)とをOriGene Technologies, Inc.社から入手した。完全長のhURAT1 cDNAをベクターpCMV6−XL5から得て、これを発現ベクターpCMV6−Neo中にサブクローニングして、hURAT1発現プラスミドpCMV6−hURAT1を創出した。自動DNA配列決定法により配列を検証した。
【0187】
URAT1発現プラスミドの細胞培養、形質移入、及び、hURATlを安定して発現するHEK細胞の確立:
10%FBS及び2mM L−グルタミンを添加したEMEM中でヒト胚腎293(HEK,Human embryonic kidney)細胞(ATTCC, Cat No. CRL-1573)を培養し、37℃、5% CO2の条件でインキュベートした。形質移入実験のために、60mmの皿上に1皿当たり培地1mlで細胞を播いた。18〜24時間のインキュベーション後、メーカーの取扱説明書に従ってリポフェクチン形質移入剤を用いて(Invitrogen社製, Cat.No.18292)、細胞にプラスミドpCMV6−hURAT1又は発現ベクターpCMV6−Neoを形質移入した。形質移入後、EMEM培地中で72時間細胞を培養し、次いで、1mg/mlのゲネチシン(GIBCO社製, Cat. No 10131)を加えることにより、安定な形質移入体を選抜した。逆転写ポリメラーゼ連鎖反応(RT−PCR,reverse transcription polymerase chain reaction)法を用いて、hURAT1を発現する安定な形質移入体(以後、本明細書中ではhURAT1−HEK細胞と呼ぶ)、又は、発現ベクターpCMV6−Neoのみを有する細胞(以後、本明細書中ではmock−HEK細胞と呼ぶ)を検証した。
【0188】
[8−14C]尿酸取込みアッセイ:
hURAT1−HEK細胞及びmock−HEK細胞を、EMEM培地中3×105の濃度でポリ−D−リシン細胞培養24ウェルプレート(Becton Dickinson社製, Cat. No.354414)中に播き、一晩インキュベートした。[8−14C]尿酸(55mCi/mmol)を含有する反応溶液を最終濃度50μMで、125mMグルコン酸ナトリウム、4.8mMグルコン酸カリウム、1.3mMカルシウム、5.6mMグルコース、1.2mM硫酸マグネシウム、1.2mM KH2PO4及び25mM HEPES(pH7.4)を含有するハンクス平衡塩溶液(HBSS,Hanks' balanced salt solution)中の、試験化合物を入れたものと入れないものとを調製した。取込みアッセイが開始する前に、培養培地を除去し、細胞をHBSS0.6ml中で5分間インキュベートした。その後HBSSを除去し、調製済の反応溶液を各ウェル中に加え、室温で5分間インキュベートした。次いで反応溶液を除去し、冷HBSS0.6mlで細胞を2回洗浄し、0.1M NaOH0.2mlで20分間溶解させた。シンチレーション液(Opti Phase SuperMIX, PerkinElmer社製, Cat No. 1200-439)1mlの入ったシンチレーションバイアル中に細胞溶解物を移し、Microbeta計数装置(1450, Wallac Jet, PerkinElmer社製)中で放射能を計数した。試験化合物をDMSO中で溶解させ、試験化合物の入っていないmock−HEK細胞及びhURAT1−HEK細胞のウェル中に同じ濃度のDMSOを加えた。各試験化合物について、取込みアッセイを2回実施し、3連で実施した。各試験条件についての細胞の尿酸取込みを、DMSO対照と比較した平均阻害率(%)として示した。DMSOの入っているウェルについて得た放射能値を、細胞の100%取込みとして採用した。観察された濃度−阻害率(%)データをS字濃度−効果モデル:
阻害率(%)=(100*濃度^勾配)/IC50^勾配]+濃度^勾配)
に当てはめた。
Data Analysis Toolbox(商標)(MDL Information Systems社製、San Leandro, CA, USA)を用いた非線形最小二乗回帰分析により、IC50及び勾配の推定値並びにそれらの95%信頼限界を定量した。
【0189】
URAT1阻害薬としての化合物の活性を評価するために、尿酸取込みの阻害率(%)を、典型的には、10マイクロモルの薬物濃度で評価した(表1)。IC−50値の定量のために、いくつかの化合物の追加的な薬物濃度を試験した(表2)。この実施例においては、化合物を必ずしも同時に試験しなかった。
【0190】
【表1】
【0191】
【表2】
【実施例10】
【0192】
化合物EBを用いたマウスでの経口単回用量薬物動態試験
オスのマウスに単回経口胃管投与した後の化合物EBの血漿プロファイルの定量:
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の化合物EB又は媒体(1%HPMC)をオスのマウスに単回経口胃管投与により投与した。マウスは、投与後、尿採取装置中に5時間配置し、5時間分の全尿排出量を収集した。試料は、LC/MS−MSを用いて分析するまで−80℃で冷凍した。
【0193】
投与時の0時間後、0.5時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に回収し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。
【0194】
データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0195】
プロトコール:
A.血漿。
1.マウスに、化合物EB100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.マウス血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5.0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, Sequence 1029-08A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0196】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.095mLを、メタノール中の化合物EBの20倍ストック0.005mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EBを作製した。
例:メタノール中の血漿95マイクロL+10mM化合物EB5マイクロL=500マイクロM化合物EBの入った血漿0.1mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.2mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25ml/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0197】
HPLC条件:
【0198】
【表3】
【0199】
結果:
化合物EBはマウス血漿中で容易に検出できた。正負のイオン化モードにおいて保持時間及び質量を確認した。AGILENT LC-MS sequence 1029-08A。
「M−」=279.2 100%
「M+」=281.2 100%
式量280。保持時間平均=26.5分。
【0200】
【表4】
【0201】
【表5】
【0202】
AUC0〜24:796μg/mL
最高血中濃度:210μg/mL
セミログプロットについての線形成分:
半減期(1〜4時間):2.27
半減期(6〜8時間):0.74
半減期(8〜24時間):9.39
【実施例11】
【0203】
化合物EBを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物EBの血漿プロファイルの定量:
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に回収し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0204】
プロトコール:
A.血漿。
1.ラットに、化合物EB100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽を用いて5分間解凍し、最高速度で10秒間ボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mm、逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT Sequence 1015-08A Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0205】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中のPN2107の20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EBを作製した。
例:メタノール中の血漿190マイクロL+10mM化合物EB10マイクロL=500マイクロM化合物EBの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25ml/分、100バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0206】
【表6】
【0207】
結果:
1.直線度=0.9994へのR^2フィットを用いて較正曲線を作成した(図4)。
2.化合物EBはラット血漿中で容易に検出でき、正負のイオン化モードにおいて保持時間及び質量を確認した。
「M−」=279.2 100%。
「M+」=281.2 100%
式量280、保持時間平均=26.5分。
生データ及び計算値を表7に記載する。
血漿中の化合物濃度を表8に記載する。
【0208】
【表7】
【0209】
【表8】
【0210】
【表9】
【0211】
化合物EBの半減期=3.99時間
化合物EBのAUC0〜24=566μg/mL*時間
化合物EBの最高血中濃度=108.08μg/mL
【実施例12】
【0212】
化合物ECを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物ECの血漿プロファイルの定量。
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に収集し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0213】
プロトコール:
A.血漿。
1.ラットに、化合物EC100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT sequence 0226-09A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0214】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中の化合物ECの20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロMの濃度の化合物ECを作製した。
例:メタノール中の血漿190マイクロL+10mMの化合物EC10マイクロL=500マイクロMの化合物ECの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0215】
【表10】
【0216】
結果:
1.直線度=0.9984へのR^2フィットを用いて較正曲線を作成した(図6を参照)。
2.化合物ECは血漿中で容易に検出できた。正負両方のイオン化モードにおいて保持時間及び質量を確認した(図7を参照)。
「M−」=293.2 100%、527.2 65%
「M+」=295.2 100%。式量294。保持時間平均=23分。
【0217】
【表11】
【0218】
【表12】
【0219】
【表13】
【実施例13】
【0220】
化合物EGを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物EGの血漿プロファイルの定量。
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に収集し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0221】
プロトコール:
A.血漿。
1.ラットに、化合物EG100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT sequence 0226-09A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0222】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中の化合物EGの20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EGを作製した。
例:メタノール中の血漿190マイクロL+10mMの化合物EG10マイクロL=500マイクロMの化合物EGの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0223】
【表14】
【0224】
結果:
1.直線度=0.9997へのR^2フィットを用いて較正曲線を作成した(図8を参照)。
2.化合物EGは血漿中で容易に検出できた。正負両方のイオン化モードにおいて保持時間及び質量を確認した(図9を参照)。
「M−」=307.2 100%
「M+」=309.2 100%。式量308。保持時間平均=30分。
【0225】
生データ及び計算
【0226】
【表15】
【0227】
【表16】
【0228】
【表17】
【技術分野】
【0001】
尿酸値上昇が原因の疾患は、2つの主要な範疇、すなわち、尿酸結晶の沈殿が原因の障害と、可溶性尿酸の病理学的効果に関連する疾患とに分けられる。痛風性関節炎は、前者の古典的な例である。腎臓中の尿酸結晶の堆積も、腎機能不全の一般的原因である。可溶性尿酸値上昇は、心血管疾患及び腎疾患など、さまざまな障害を伴う。
【背景技術】
【0002】
痛風は、体内の1又は複数の関節の炎症として最も普通に現れ、その結果、軽度から重度の疼痛が生じる。こうした事象は、突発性及び/又は慢性である場合がある。時間の経過と共に、痛風は、軟骨及び骨の破壊、尿酸結晶堆積物の発達、腎臓痛及び腎機能不全並びに腎臓結石をもたらすことがある。痛風は、他の器官にも影響することがある。
【0003】
痛風は、高尿酸血症、並びに、組織、関節、腎臓及び他の器官中での結果的な尿酸結晶の形成及び堆積が原因である。尿酸は、正常な細胞の代謝により、又、ある種の食品及び飲料により生じる。過剰な尿酸値は、余分な尿酸の生成、腎臓によるクリアランス障害(又は、過剰生成とクリアランス障害との組合せ)の結果、さらには、他の健康状態のために摂取されるある形態の薬物による結果である。(例としては、利尿薬、ピラジナミド、シクロスポリン、低用量アスピリン、ニコチン酸及びレボドパが挙げられる)。アルコール依存症、白血病、リンパ腫、肺癌、腫瘍溶解症候群、喫煙、乾癬、肥満、腎機能不全、うっ血性心不全、飢餓、貧血症、高血圧症、糖尿病、不動、レッシュ−ナイハン症候群、ダウン症候群、並びに、甲状腺及び副甲状腺の機能不全など多くの種類の健康状態が、高尿酸血症及び痛風の一因となることもある。
【0004】
痛風は、一般に、進行性に悪化する症状に基づき4つの範疇に分けられる:
1)無症候性。血中尿酸値は上昇しているが、明白な症状はない。
2)急性通風性関節炎:多くの場合は単一の関節(普通は足の親指)において症状が突然生じ、次いで、他の関節が冒される。症状としては、疼痛、腫張、発赤及び発熱が挙げられる。
3)間欠期痛風:痛風発作間の無症候性の段階。
4)慢性結節性痛風:慢性の状態であり、頻繁な発作、関節の持続的で軽度の疼痛及び炎症、軟骨及び骨の破壊、尿酸結晶堆積物の発達、腎機能不全及び腎臓結石が含まれる場合がある。
【0005】
痛風の急性症状を治療するために現在使用されている薬物としては、非ステロイド性抗炎症薬、コルヒチン及びコルチコステロイドが挙げられる。こうした薬物は全て、軽度から重度の副作用を生じる場合がある。インターロイキン1などの炎症性サイトカインに対する抗体及びアンタゴニストを含め、こうした急性症状用の他の治療薬が研究されている。
【0006】
尿酸値を低下させることにより将来的な発作の発生又は重症度の低下を試みるには、他の種類の薬物が使用される。3つの主要なクラスの薬物は、キサンチンからの尿酸の生成を減少させるキサンチンオキシダーゼ阻害薬(例えばアロプリノール);尿酸輸送体1(URAT1,uric acid transporter 1)の阻害により腎尿細管中での分泌された尿酸の再取込みを阻害することにより尿酸の排出を高めることを意図した尿酸排泄剤(例えば、スルフィンピラゾン、プロベネシド、ベンズブロマロン及びロサルタン)(米国特許出願公開第2007/0010670号明細書、2007年1月11日公開(日本たばこ産業株式会社)も参照)、又は、尿酸再取込みの他の要素;及びウリカーゼ、例えば、PURICASE(Savient社製のペグ化した組換え哺乳動物ウリカーゼ)などのペグ化ウリカーゼである。こうした薬物は、顕著で望ましくない副作用をもたらすことも多い。例えば、アロプリノールは、これが原因で欧州において毎年少なくとも100症例のスティーブンス・ジョンソン/中毒性表皮壊死症が発症し、およそ30名が死亡していると報告されている(Halevy et al., Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 58(1):25-32, 2008)。プロベニシド(Probenicid)及びベンズブロマロンは、ベンズブロマロンの症例における肝不全など、望ましくない副作用を理由に、いくつかの国の市場から排除されている。こうした薬物の摂取における患者の服薬遵守は、報告によれば非常に乏しく(A. A. Reidel et al. "Compliance with Allopurinol Therapy among Managed Care Enrollees with Gout: A Retrospective Analysis of Administrative Claims." Journal of Rheumatology 2004; 31:1575-1581)、おそらくそれは、副作用、及び/又は、利益のなさが理由であろう。
【0007】
米国においては500万名超の人々が痛風に罹患している(National Health and Nutrition Examination Survey 111, 1988-1994)。1999年の米国における高尿酸血症及び痛風の有病数は、1,000名当たり41名、英国においては1,000名当たり14名と報告された(T.R. Mikuls et al., "Gout Epidemiology: Results for the UK General Practice Research Database, 1990-1999." Annals of the Rheumatic Diseases 2005; 64:267-272)。その後の報告により、米国、英国及び他の国々における有病数は着実に上昇していることが示されている。(K. L. Wallace et al., "Increasing Prevalence of Gout and Hyperuricemia over 10 Years Among Older Adults in a Managed Care Population." Journal of Rheumatology 2004; 31: 1582-1587)。さらに最近のデータからは、現在では500万名をはるかに超える米国人が、診断可能な痛風に罹患していることが示唆される(E. Krishnan et al., "Gout in Ambulatory Care Settings in the United States." Journal of Rheumatology 2008; 35(3): 498-501)。
【0008】
高尿酸血症及び痛風は、臓器移植のレシピエントにおいてとりわけ重大な問題である(Stamp, L., et al, "Gout in solid organ transplantation: a challenging clinical problem", Drugs (2005) 65(18): 2593-2611)。尿酸は、腎移植を受けている患者において上昇していることが多く、シクロスポリンなど一般的な免疫抑制薬が原因で、とりわけ重度の高尿酸血症が生じる場合がある。アザチオプリンなどいくつかの免疫抑制剤との相互作用があること、又、組合せにより骨髄不全が生じることから、移植患者においてはアロプリノールは禁忌である。さらに、尿酸上昇は、移植片不全(graft failure)の一因となる場合がある(Armstrong, K.A. et al., "Does Uric Acid Have a Pathogenetic Role in Graft Dysfunction and Hypertension in Renal Transplant Patients?" Transplantation (2005) 80(11): 1565-1571)。したがって、移植レシピエントにおいて高尿酸血症を減少させる安全な薬剤については、とりわけ急を要する需要がある。
【0009】
可溶性尿酸上昇に関連する疾患は、血管の問題を伴うことが多い:高血圧症(Sundstrom et al., Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 45(1):28-33, 2005)、高血圧前症(Syamela, S. et al., Association between serum uric acid and prehypertension among US adults. J Hypertens. 25 (8) 1583-1589, (2007)、アテローム性硬化症(Ishizaka et al., Association between serum uric acid, metabolic syndrome, and carotid atherosclerosis in Japanese individuals. Arterioscler Thromb Vasc Biol. (5):1038-44, 2005)、末梢動脈疾患(Shankar, A. et al., Association between serum uric acid level and peripheral artery disease. Atherosclerosis doi 10: 1016, 2007)、血管炎症(Zoccali et al., Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol. 17(5):1466-71, 2006)、心不全(Strasak, A.M. et al., Serum uric acid and risk of cardiovascular mortality: A prospective, long-term study of 83,683 Austrian men, Clin Chem. 54 (2) 273-284, 2008; Pascual-Figal, Hyperuricaemia and long-term outcome after hospital discharge in acute heart failure patients. Eur J Heart Fail. 2006 Oct 23; [Epub ahead of print]; Cengel, A., et al., "Serum uric Acid Levels as a Predictor of In-hospital Death in Patients Hospitalized for Decompensated Heart Failure." Acta Cardiol. (Oct. 2005) 60(5): 489-492)、心筋梗塞(Strasak, A.M. et al.; Bos et al., Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdamstudy. Stroke. 2006 Jun; 37(6):1503-7)、腎機能不全(Cirillo et al., Uric Acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 17(12 Suppl 3):S165-8, 2006; Z. Avram and E. Krishnan, Hyperuricemia - where nephrology meets rheumatology. Rheumatology (Oxford), 47(7): 960-964, 2008)、及び脳卒中(Bos et al., 2006)。尿酸は、内皮機能不全の直接の原因となる(Kanellis, et al., Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 25(1):39-42, 2005; Khosla et al, Hyperuricemia induces endothelial dysfunction. Kidney Int. 67(5):1739-42, 2005)。小児及び青年においては、早発性本態性高血圧症は血清尿酸上昇を伴い、アロプリノールを用いて尿酸を減少させると、こうした患者における血圧が低下する(Feig and Johnson, The role of uric acid in pediatric hypertension. J Ren Nutrition 17(1): 79-83, 2007; D.I. Feig et al., Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension. JAMA 300(8): 924-932, 2008。さらに、Feig et al.は、これは新しい治療的アプローチであるが、尿酸を減少させるための既存の薬物の副作用から、その使用が制限又は妨げられることがあるとも述べている。高尿酸血症は、このような状態の全てにおける独立したリスク因子である。
【0010】
可溶性尿酸上昇も、炎症応答を伴うか、又は炎症応答を直接誘導する。例えば、尿酸は、有機酸輸送体、特に尿酸輸送体URAT1により血管の平滑筋細胞中に輸送された後、血管の平滑筋細胞を刺激してC反応性タンパク質MCP−1及び他のサイトカインを産生させることにより、増殖、及び、アテローム性硬化症に関連する他の変化を刺激し(Price et al., Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol. 17(7):1791-5, 2006; Kang et al., Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005 25(5):425-33 (2005); Yamamoto et al., Allopurinol reduces neointimal hyperplasia in the carotid artery ligation model in spontaneously hypertensive rats. Hypertens. Res. 29 (11) 915-921, 2006)、ヒト単核細胞を刺激してIL−1β、IL−6及びTNF−αを産生させ、マウスの体内に注入するとTNF−αの顕著な増加の原因となり、内皮細胞及び血小板を活性化させ、血小板の接着性を増加させる(Coutinho et al., "Associations of Serum Uric Acid with Markers of Inflammation, Metabolic Syndrome, and Subclinical Coronary Atherosclerosis", Amer. J. Hypertens. (2007) 20: 83-89; Levya, F., et al., "Uric Acid in Chronic Heart Failure: A Marker of Chronic Inflammation", Eur. Heart J. (1998) 19(12): 1814-1822.)。尿酸は、内皮の一酸化窒素の生体利用性を阻害し、レニン・アンギオテンシン系を活性化することも示されている。(T.S. Perlstein et al., Uric acid and the state of the intrarenal renin-angiotensin system in humans. Kidney International. 66:1465-1470, 2004)。Inokuchi et al.は、インターロイキン18(IL−18,Interleukin 18)及び他の炎症性物質は、痛風を伴う局所的な炎症を反映すること、並びに、尿酸結晶は、腎不全を発症させる役割を有すると思われるIL−18の活性化を加速させることを示した(T. Inokuchi et al., Plasma IL-18 and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 33(1): 21-27, 206)。IL−18及び他のサイトカインは、痛風自体には罹患しておらず尿酸値が上昇しているにすぎない人においても、顕著に上昇している(C. Ruggiero et al. Uric acid and inflammatory markers.(C. Ruggiero et al., Uric acid and inflammatory markers. European Heart Journal. 27: 1174-1181, 2006)。
【0011】
高尿酸血症は、認知障害及び他の形態の中枢神経系機能不全とも関連がある。(Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140; Watanabe, S., et al., "Cerebral Oxidative Stress and Mitochondrial Dysfunction in Oxonate-Induced Hyperuricemic Mice", J. Health Science (2006) 52: 730-737)。
【0012】
血清尿酸値上昇は、癌のリスク及び癌による死亡数の増加とも関連がある。(Strasak, AM et al. (2007) Serum uric acid and risk of cancer mortality in a large prospective male cohort. Cancer Causes Control 18 (9) 1021-1029; Strasak, AM et al. (2007) The role of serum uric acid as an antioxidant protecting against cancer: prospective study in more than 28,000 older Austrian women. Annals Oncol 18 (11) 1893-1897; Jee, SA et al. (2004) Serum uric acid and risk of death from cancer, cardiovascular disease or all causes in men Eur. J. Cardiovascular Prev. Rehab. 11 (3) 185-191)。
【0013】
尿酸値上昇は、糖尿病前症、インスリン抵抗性、2型糖尿病の発症、及び糖尿病罹患者におけるさまざまな望ましくない状態(末梢動脈疾患、脳卒中、及び、死亡リスク増加など)の可能性の増加と関連がある(Ioachimescu, A.G. et al. (2007) Serum uric acid, mortality and glucose control in patients with Type 2 diabetes mellitus: a PreCIS database study Diabet. Med. 24 (12) 1369-1374; Perry, I.J. et al (1995) Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men BMJ 310 (6979) 560-564; Chien, K-L et al. (2008) Plasma uric acid and the risk of Type 2 diabetes in a Chinese community Clin. Chem. 54 (2) 310-316; Sautin, Y. Y. et al. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress Am. J. Physiol. Cell Physiol.293: C584-C596; Tseng, C.H. (2004) Independent association of uric acid levels with peripheral artery disease in Taiwanese patients with Type 2 diabetes Diabet. Med. 21 (7) 724-729; Lehto, S. et al. (1998) Serum uric acid is a strong predictor of stroke in patients with non-insulin dependent diabetes mellitus Stroke 29: 635-639。
【0014】
尿酸値上昇は、レッシュ−ナイハン症候群の決定的な特徴である。睡眠時無呼吸又は睡眠呼吸障害の罹患者も、尿酸が上昇している(Saito, H. et al., Tissue hypoxia in sleep apnea syndrome assessed by uric acid and adenosine. Chest 122: 1686-1694, 2002; Verhulst, S.L., et al., Sleep-disordered breathing and uric acid in overweight and obese children and adolescents. Chest 132: 76-80, 2007)。
【0015】
尿酸上昇は、子癇前症と関連がある(Bainbridge, S.A. and Roberts, J.M., Uric acid as a pathogenic factor in preeclampsia. Placenta Dec. 17 2007 epub ahead of print)。
【0016】
「尿酸は、ヒト末梢血単核細胞中の熱帯熱マラリア原虫(P. falciparum)により引き起こされる炎症応答の主要な一因である。…熱帯熱マラリア原虫により誘導される炎症反応は、マラリア発病の主因と考えられる。…」PLoS ONE 2009;4(4):e5194. Epub 2009 Apr 17。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】米国特許出願公開第2007/0010670号明細書
【非特許文献】
【0018】
【非特許文献1】Halevy et al., Allopurinol is the most common cause of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe and Israel. J Am Acad Dermatol. 58(1):25-32, 2008
【非特許文献2】A. A. Reidel et al. "Compliance with Allopurinol Therapy among Managed Care Enrollees with Gout: A Retrospective Analysis of Administrative Claims." Journal of Rheumatology 2004; 31:1575-1581
【非特許文献3】National Health and Nutrition Examination Survey 111, 1988-1994
【非特許文献4】T.R. Mikuls et al., "Gout Epidemiology: Results for the UKGeneral Practice Research Database, 1990-1999." Annals of the Rheumatic Diseases 2005; 64:267-272
【非特許文献5】K. L. Wallace et al., "Increasing Prevalence of Gout and Hyperuricemia over 10 Years Among Older Adults in a Managed Care Population." Journal of Rheumatology 2004; 31: 1582-1587
【非特許文献6】E. Krishnan et al., "Gout in Ambulatory Care Settings in the United States." Journal of Rheumatology 2008; 35(3): 498-501
【非特許文献7】Stamp, L., et al, "Gout in solid organ transplantation: a challenging clinical problem", Drugs (2005) 65(18): 2593-2611
【非特許文献8】Armstrong, K.A. et al., "Does Uric Acid Have a Pathogenetic Role in Graft Dysfunction and Hypertension in Renal Transplant Patients?" Transplantation (2005) 80(11): 1565-1571
【非特許文献9】Sundstrom et al., Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 45(1):28-33, 2005
【非特許文献10】Syamela, S. et al., Association between serum uric acid and prehypertension among US adults. J Hypertens. 25 (8) 1583-1589, (2007)
【非特許文献11】Ishizaka et al., Association between serum uric acid, metabolic syndrome, and carotid atherosclerosis in Japanese individuals. Arterioscler Thromb Vasc Biol. (5):1038-44, 2005
【非特許文献12】Shankar, A. et al., Association between serum uric acid level and peripheral artery disease. Atherosclerosis doi 10: 1016, 2007
【非特許文献13】Zoccali et al., Uric acid and endothelial dysfunction in essential hypertension. J Am Soc Nephrol. 17(5):1466-71, 2006
【非特許文献14】Strasak, A.M. et al., Serum uric acid and risk of cardiovascular mortality: A prospective, long-term study of 83,683 Austrian men, Clin Chem. 54 (2) 273-284, 2008
【非特許文献15】Pascual-Figal, Hyperuricaemia and long-term outcome after hospital discharge in acute heart failure patients. Eur J Heart Fail. 2006 Oct 23; [Epub ahead of print]
【非特許文献16】Cengel, A., et al., "Serum uric Acid Levels as a Predictor of In-hospital Death in Patients Hospitalized for Decompensated Heart Failure." Acta Cardiol. (Oct. 2005) 60(5): 489-492
【非特許文献17】Strasak, A.M. et al.; Bos et al., Uric acid is a risk factor for myocardial infarction and stroke: the Rotterdam study. Stroke. 2006 Jun; 37(6):1503-7
【非特許文献18】Cirillo et al., Uric Acid, the metabolic syndrome, and renal disease. J Am Soc Nephrol. 17(12 Suppl 3):S165-8, 2006
【非特許文献19】Z. Avram and E. Krishnan, Hyperuricemia - where nephrology meets rheumatology. Rheumatology (Oxford), 47(7): 960-964, 2008
【非特許文献20】Kanellis, et al., Uric acid as a mediator of endothelial dysfunction, inflammation, and vascular disease. Semin Nephrol. 25(1):39-42, 2005
【非特許文献21】Khosla et al, Hyperuricemia induces endothelial dysfunction. Kidney Int. 67(5):1739-42, 2005
【非特許文献22】Feig and Johnson, The role of uric acid in pediatric hypertension. J Ren Nutrition 17(1): 79-83, 2007
【非特許文献23】D.I. Feig et al., Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension. JAMA 300(8): 924-932, 2008
【非特許文献24】Price et al., Human vascular smooth muscle cells express a urate transporter. J Am Soc Nephrol. 17(7):1791-5, 2006
【非特許文献25】Kang et al., Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol. 2005 25(5):425-33 (2005)
【非特許文献26】Yamamoto et al., Allopurinol reduces neointimal hyperplasia in the carotid artery ligation model in spontaneously hypertensive rats. Hypertens. Res. 29 (11) 915-921, 2006
【非特許文献27】Coutinho et al., "Associations of Serum Uric Acid with Markers of Inflammation, Metabolic Syndrome, and Subclinical Coronary Atherosclerosis", Amer. J. Hypertens. (2007) 20: 83-89
【非特許文献28】Levya, F., et al., "Uric Acid in Chronic Heart Failure: A Marker of Chronic Inflammation", Eur. Heart J. (1998) 19(12): 1814-1822
【非特許文献29】T.S. Perlstein et al., Uric acid and the state of the intrarenal rennin-angiotensin system in humans. Kidney International. 66:1465-1470, 2004
【非特許文献30】T. Inokuchi et al., Plasma IL-18 and other inflammatory cytokines in patients with gouty arthritis and monosodium urate monohydrate crystal-induced secretion of IL-18. Cytokine. 33(1): 21-27, 206
【非特許文献31】C. Ruggiero et al. Uric acid and inflammatory markers.(C. Ruggiero et al., Uric acid and inflammatory markers. European Heart Journal. 27: 1174-1181, 2006
【非特許文献32】Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140
【非特許文献33】Watanabe, S., et al., "Cerebral Oxidative Stress and Mitochondrial Dysfunction in Oxonate-Induced Hyperuricemic Mice", J. Health Science (2006) 52: 730-737
【非特許文献34】Strasak, AM et al. (2007) Serum uric acid and risk of cancer mortality in a large prospective male cohort. Cancer Causes Control 18 (9) 1021-1029
【非特許文献35】Strasak, AM et al. (2007) The role of serum uric acid as an antioxidant protecting against cancer: prospective study in more than 28,000 older Austrian women. Annals Oncol 18 (11) 1893-1897
【非特許文献36】Jee, SA et al. (2004) Serum uric acid and risk of death from cancer, cardiovascular disease or all causes in men Eur. J. Cardiovascular Prev. Rehab. 11 (3) 185-191
【非特許文献37】Ioachimescu, A.G. et al. (2007) Serum uric acid, mortality and glucose control in patients with Type 2 diabetes mellitus: a PreCIS database study Diabet. Med. 24 (12) 1369-1374
【非特許文献38】Perry, I.J. et al (1995) Prospective study of risk factors for development of non-insulin dependent diabetes in middle aged British men BMJ 310 (6979) 560-564
【非特許文献39】Chien, K-L et al. (2008) Plasma uric acid and the risk of Type 2 diabetes in a Chinese community Clin. Chem. 54 (2) 310-316
【非特許文献40】Sautin, Y. Y. et al. (2007) Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress Am. J. Physiol. Cell Physiol.293: C584-C596
【非特許文献41】Tseng, C.H. (2004) Independent association of uric acid levels with peripheral artery disease in Taiwanese patients with Type 2 diabetes Diabet. Med. 21 (7) 724-729
【非特許文献42】Lehto, S. et al. (1998) Serum uric acid is a strong predictor of stroke in patients with non-insulin dependent diabetes mellitus Stroke 29: 635-639
【非特許文献43】Saito, H. et al., Tissue hypoxia in sleep apnea syndrome assessed by uric acid and adenosine. Chest 122: 1686-1694, 2002
【非特許文献44】Verhulst, S.L., et al., Sleep-disordered breathing and uric acid in overweight and obese children and adolescents. Chest 132: 76-80, 2007
【非特許文献45】Bainbridge, S.A. and Roberts, J.M., Uric acid as a pathogenic factor in preeclampsia. Placenta Dec. 17 2007 epub ahead of print
【非特許文献46】P. falciparum, PLoS ONE 2009:4(4):e5194. Epub 2009 Apr 17
【非特許文献46】PLoS ONE 2009;4(4):e5194. Epub 2009 Apr 17
【発明の概要】
【発明が解決しようとする課題】
【0019】
該当する疾患が尿酸の結晶化によるものであるか、又は正常を超える(個人ベースの標準によるか集団ベースの標準によるかを問わない)可溶性尿酸値の効果によるものであるかに関わらず、血中尿酸の上昇に関連する障害を安全、手軽及び効果的に治療及び防止することができる新しい薬物に対する重大な医学的必要性がある。
【課題を解決するための手段】
【0020】
本発明は、式I
【0021】
【化1】
【0022】
により表される化合物を提供する。
【0023】
式I中、xは1又は2であり、yは0、1、2又は3であり、R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、N、S及びOから選択される1若しくは2個の環状ヘテロ原子を有し、環炭素により該化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である。
【0024】
本発明は、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させる方法であって、本発明の化合物を、該対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるのに有効な量で該対象に投与することを含む方法を提供する。本発明は、哺乳動物の血中の尿酸濃度を低下させ、又は該動物からの尿酸排出を増加させるうえで使用するための本発明の化合物を提供する。本発明は、哺乳動物の血中の尿酸濃度を低下させ、又は該動物からの尿酸排出を増加させるための医薬の製造における本発明の化合物の使用を提供する。本発明は、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させる際に使用するための医薬組成物であって、該対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるのに有効な量で本発明の化合物を含む医薬組成物を提供する。本発明は、1又は複数単位の経口用量の本発明の化合物と、哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排出を増加させるための該化合物を投与するための取扱説明書とを含むキットを提供する。
【0025】
尿酸を減少させることは、本明細書中に記載の場合、痛風(無症候性痛風、急性通風性関節炎、間欠期痛風及び慢性結節性痛風のいずれか又は全て)、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、及び、高尿酸血症のそれ以外の結果、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫(Plasmodium falciparum)誘導性炎症などさまざまな状態を治療又は防止するために用いることができる。
【0026】
本発明は、実施例7に示すように、本発明の化合物がインビトロでURAT1を阻害したという観察に基づく。URAT1の阻害は、インビボで尿酸を低下させるための確立されたインビトロモデルである。
【図面の簡単な説明】
【0027】
【図1】化合物EBが、hURAT1−HEK細胞中の14C−尿酸取込みに及ぼす濃度依存性の阻害効果を示すグラフである。
【図2】化合物ECが、hURAT1−HEK細胞中の14C−尿酸取込みに及ぼす濃度依存性の阻害効果を示すグラフである。
【図3】マウス血漿中の化合物EBの濃度を示すグラフである。
【図4】ラット血漿中の化合物EBの較正曲線を示すグラフである(LC−MS)。
【図5】ラット血漿中の化合物EBの濃度を示すグラフである。
【図6】ラット血漿中の化合物ECの較正曲線を示すグラフである(LC−MS)。
【図7】ラット血漿中の化合物ECの濃度を示すグラフである。
【図8】ラット血漿中の化合物EGの較正曲線を示すグラフである(LC−MS)。
【図9】ラット血漿中の化合物EGの濃度を示すグラフである。
【発明を実施するための形態】
【0028】
定義
1H−テトラゾリル−5−イル部分及び対応する2H−テトラゾリル−5−イル部分は、互変異性体として存在することがある。本文書においては、1H−互変異性体を基準にして、化合物名を呼び、構造式を書いてある。そのような基準は全て、両方の互変異性形態を含むものとして理解されたい。したがって、例えば、「5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール)は、5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール及び5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−2H−テトラゾールを両方とも包含する。又、式Iは、上に示したような式I、及び、下記の式I’
【0029】
【化2】
【0030】
に示すその2H−テトラゾリル−5−イル互変異性形態を両方とも包含する。
【0031】
本明細書中で使用する場合、用語「アルキル」は、直鎖状又は分岐状のアルキル基を意味する。一定数の炭素原子を有すると同定されているアルキル基は、その特定数の炭素を有する任意のアルキル基を意味する。例えば、3個の炭素原子を有するアルキルはプロピル又はイソプロピルであってもよく、4個の炭素原子を有するアルキルは、n−ブチル、1−メチルプロピル、2−メチルプロピル又はt−ブチルであってもよい。
【0032】
本明細書中で使用する場合、用語「ハロ」は、フルオロ、クロロ、ブロモ及びヨードのうち1又は複数を指す。
【0033】
本明細書中で使用する場合、ペルフルオロメチル又はペルフルオロメトキシにあるような用語「ペルフルオロ」は、当該基が、水素原子全ての代わりにフッ素原子を有することを意味する。
【0034】
ある種の化学化合物は、本明細書中では、その化学名、又は以下に示す2文字のコードで呼ぶ。化合物EBからEI及び化合物BDは、上に示す式Iの範囲内に包含される。
EB 5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール
EC 5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール
ED 5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール
EF 5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール
EG 5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
BD 5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール
EH 5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール
EI 5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
【0035】
本明細書中で使用する場合、移行語「〜を含む(comprising)」は、非制限的なものである。この用語を用いる特許請求の範囲は、当該特許請求の範囲中に列挙されているものに追加して要素を含有できる。
【0036】
特許請求の範囲中で使用する場合、単語「又は(or)」は、そのように読んでは文脈中で意味をなさない場合を除き、「及び/又は」を意味する。したがって、例えば、「哺乳動物対象の血中の尿酸濃度を低下させ、又は該対象からの尿酸排泄を増加させる」という語句は、「哺乳動物対象の血中の尿酸濃度を低下させ、及び/又は該対象からの尿酸排泄を増加させる」と同義である。
【0037】
本発明の化合物
前記の「課題を解決するための手段」に記載の化合物、方法、使用又は医薬組成物の一実施形態においては、この化合物は、式XLVI
【0038】
【化3】
【0039】
(式中、x、y、R1及びAは、式Iについて先に記載のとおりである)
で表される。
【0040】
本発明のさらなる一実施形態においては、この化合物は、式XLVII
【0041】
【化4】
【0042】
(式中、x、y及びAは、式Iについて先に記載のとおりであり、R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される)
で表される。
【0043】
本発明の一実施形態においては、式I、XLVI又はXLVII中、xは1である。別の実施形態においては、Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである。より具体的な実施形態においては、Aは2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである。好ましくは、Aは2,6−ジメチルフェニルである。
【0044】
本発明の実施形態においては、この化合物は、式XLVIII
【0045】
【化5】
【0046】
(式中、yは0、1、2又は3であり、R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)
で表される。
【0047】
本発明の一実施形態においては、式I、XLVI、XLVII又はXLVIII中、yは0、1又は2である。本発明の一実施形態においては、式XLVII又はXLVIII中、R3は水素であり、R2は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。より具体的な一実施形態においては、R3は水素であり、R2は、水素、メチル及びメトキシからなる群から選択される。本発明の異なる一実施形態においては、式XLVII又はXLVIII中、R2は水素であり、R3は、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択される。より具体的な一実施形態においては、R2は水素であり、R3は、メチル及びメトキシからなる群から選択される。式XLVIIIのさらなる一実施形態においては、R4及びR5は両方ともメチルであるか、又は両方ともフルオロである。好ましくは、両方ともメチルである。
【0048】
本発明の特定の実施形態においては、この化合物は、5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール、5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール、5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール及び5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾールからなる群から選択される。
【0049】
本発明の化合物の一実施形態においては、この化合物は、実質的には(少なくとも98%)純粋な形態をしている。
【0050】
反応スキーム
式I(式中、xは1又は2であり、yは0〜3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0051】
【化6】
【0052】
(式中、Aは先に記載のとおりである)の化合物は、スキーム1の反応により調製できる。スキーム1の反応においては、A、x、y及びR1は前述のとおりである。Lは脱離基である。
【0053】
式IIの化合物は、トリフェニルホスフィン及びアゾジカルボン酸ジエチル又はアゾジカルボン酸ジイソプロピルを用いた、IIのIIIとのMitsunobu縮合を用いたステップ(a)の反応により、式Vの化合物に変換できる。この反応は、適当な溶媒(例えばテトラヒドロフラン)中で実施する。ステップ(a)の反応を実施するには、Mitsunobu反応において従来用いられる条件のいずれを利用してもよい。
【0054】
式Vの化合物は、適当な塩基、例えば、炭酸カリウム、トリエチルアミン、ピリジンなどを用いることによるステップ(a)の反応におけるのと同様、式IIの化合物を式IVの化合物でエーテル化又はアルキル化することにより調製してもよい。この反応は、非プロトン性溶媒(例えばN,N−ジメチルホルムアミド、アセトニトリル、ジクロロメタンなど)中で実施する。式IVの化合物においては、Lは、メシロキシ、トシロキシ、クロロ、ブロモ、ヨードなどが挙げられるが、これらに限定されない。ステップ(a)の反応を実施するには、ハロゲン化物又は脱離基を用いた反応によるヒドロキシル基のエーテル化のいずれの従来法を利用してもよい。
【0055】
式Vの化合物は、ルイス酸(例えば、塩化亜鉛、塩化マグネシウム、塩化アルミニウム、四塩化スズなど)の存在下で、ニトリルをアジド(例えばトリメチルシリルアジド)と、又は金属アジド(例えば、アジ化ナトリウム、アジ化カリウム、アジ化リチウム、好ましいアジドはアジ化ナトリウムである)と反応させることにより、ステップ(b)の反応により式Iの化合物に変換できる。この反応は、溶媒(例えばN,N−ジメチルホルムアミド)中で、80℃〜145℃の範囲の温度で6〜60時間実施する。理想の反応は、ニトリルをアジ化ナトリウム/塩化アンモニウム/N,N−ジメチルホルムアミドと120℃で24時間反応させることを利用する。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0056】
Aが、ヒドロキシル基の1つ又は2つの基により置換されているフェニルである場合は、ヒドロキシル基を保護することが一般に好ましい。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のものなど、適当な脱保護試薬を利用してステップ(b)の反応後に脱保護できる。
【0057】
【化7】
【0058】
式I[式中、xは1又は2であり、yは0〜3であり、R1はヒドロキシルである]の化合物、すなわち、式:
【0059】
【化8】
【0060】
(式中、Aは先に記載のとおりである)の化合物は、スキーム2の反応により調製できる。スキーム2の反応において、A、x、及びyは前述のとおりである。
【0061】
式Vの化合物は、−72℃〜0℃の範囲の温度で4〜48時間、溶媒(例えばジクロロメタン)を用いて式Vの化合物を三臭化ホウ素又は三塩化ホウ素で処理することによるステップ(c)の反応により、式VIの化合物に変換できる。ステップ(c)の反応を実施するには、そのような脱メチル化反応において従来用いられる条件のいずれを利用してもよい。
【0062】
式VIの化合物は、ステップ(b)の反応において本明細書中で先に記載のものと同じ様式のステップ(d)の反応により、式I(式中、R1はヒドロキシルである)の化合物に変換できる。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0063】
【化9】
【0064】
式I(式中、xは1又は2であり、yは0〜3であり、R1はアミノである)の化合物、すなわち、式:
【0065】
【化10】
【0066】
(式中、Aは先に記載のとおりである)の化合物は、スキーム3の反応により調製できる。スキーム3の反応において、A、x及びyは前述のとおりである。Pは保護基である。
【0067】
式VIIの化合物は、ステップ(a)の反応において本明細書中で先に記載のものと同じ様式のステップ(e)の反応により、式VIIIの化合物に変換できる。式VIIIの化合物は、ニトロ基をアミンに還元することによるステップ(f)の反応により、式IXの化合物に変換できる。還元剤は、金属(例えばZn、Sn又はFeなど)及び酸であってもよい。ニトロ基を触媒水素化により還元してアミンを得ることもできる。好ましい還元法は、触媒水素化である。ステップ(f)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0068】
式IXの化合物は、アミノ基を保護することによるステップ(g)の反応により、式Xの化合物に変換できる。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。式Xの化合物は、ステップ(b)の反応において本明細書中で先に記載のものと同じ様式のステップ(h)の反応により、式XIの化合物に変換できる。式XIの化合物は、アミノ保護基を脱保護することによる(i)の反応ステップにより、式I(式中、R1はアミノである)の化合物に変換できる。適当な脱保護試薬は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0069】
【化11】
【0070】
式II(式中、yは1〜3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0071】
【化12】
【0072】
の化合物及び式VII(式中、yは1〜3である)の化合物、すなわち、式:
【0073】
【化13】
【0074】
の化合物は、スキーム4の反応により調製できる。スキーム4の反応においては、R3は、水素、フルオロ、ブロモ、クロロ、ニトロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである。Pはヒドロキシ保護基である。R2は、1〜2個の炭素原子を有するアルキル基である。Yはハロゲン化物である。
【0075】
式XIIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のもののような適当な保護基を利用することによりカルボキシル基及びヒドロキシ基を最初に保護することによるステップ(j)の反応により、式XIIIの化合物に変換できる。
【0076】
式XIIIの化合物は、ステップ(k)の反応によりエステル基をアルコールに変換する従来の還元試薬を利用することにより、式XIV(式中、R3は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物に還元できる。この反応を実施するうえでは、水酸化リチウムアルミニウムを利用することが一般に好ましいが、これに限定するものではない。この反応は、適当な溶媒(テトラヒドロフランなど)中で実施する。ステップ(k)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0077】
式XIIIの化合物は、エステルをアルコールに変換するがニトロ基を還元しない還元試薬(例えば、BH3−THF、NaBH4−AlCl3など)を利用することにより、式XIV(式中、R3はニトロである)の化合物に還元できる。ステップ(k)の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。式XIVの化合物は、ヒドロキシ基をハロゲン(好ましいハロゲンはブロモ又はクロロである)で置換することにより、式XVの化合物に変換できる。適切なハロゲン化試薬としては、塩化チオニル、塩化オキサリル、臭素、三臭化リン、四臭化炭素などが挙げられるが、これらに限定されない。ステップ(l)の反応を実施するには、そのようなハロゲン化反応において従来用いられるいずれの条件を利用してもよい。
【0078】
式XVの化合物は、XVを、アルカリ金属シアン化物、例えば、シアン化ナトリウム又はシアン化カリウム又はシアン化銅と反応させることにより、式XVIの化合物に変換できる。この反応は、適当な溶媒(例えば、ジメチルスルホキシド、N,N−ジメチルホルムアミドなど)中で実施できる。ステップ(m)の反応を実施するには、ハロゲン化物からのニトリルの調製において従来用いられる条件のいずれを利用してもよい。
【0079】
式XVIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されているものなど適当な脱保護試薬を利用してヒドロキシ保護基を除去することによるステップ(n)の反応により、式XVIIの化合物に変換できる。式XVIIの化合物は、式II(式中、yは1であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XVIIの化合物は、式VII(式中、yは1であり、R3はニトロである)の化合物でもある。式XVIの化合物は、酸性又は塩基性の加水分解によるステップ(o)の反応により、式XVIIIの化合物に変換できる。この反応を実施するうえでは、塩基性加水分解、例えばエタノール中の水性水酸化ナトリウムなどを利用することが一般に好ましい。ステップ(o)の反応を実施するには、ニトリルのカルボン酸への加水分解において従来用いられる条件のいずれを利用してもよい。
【0080】
式XVIIIの化合物をステップ(p)の反応により還元して、式XIXの化合物を得ることができる。この反応は、ステップ(k)の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XIXの化合物は、ステップ(l)の反応において本明細書中で先に記載のものと同じ様式のステップ(q)の反応により、式XXの化合物に変換できる。式XXの化合物は、ステップ(m)の反応において本明細書中で先に記載のものと同じ様式のステップ(r)の反応により、式XXIの化合物に変換できる。式XXIの化合物は、ステップ(n)の反応において本明細書中で先に記載のものと同じ様式のステップ(s)の反応により、式XXIIの化合物に変換できる。
【0081】
式XXIIの化合物は、式II(式中、yは2であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XVIIの化合物は、式VII(式中、yは2であり、R3はニトロである)の化合物でもある。
【0082】
式XXIの化合物をステップ(o)の反応において本明細書中で先に記載のものと同じ様式で加水分解して、ステップ(t)の反応により式XXIIIの化合物を得ることができる。
【0083】
式XXIIIの化合物を還元して、ステップ(u)の反応により式XXIVの化合物を得ることができる。この反応は、ステップ(k)の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XXIVの化合物は、ステップ(l)の反応において本明細書中で先に記載のものと同じ様式のステップ(v)の反応により、式XXVの化合物に変換できる。式XXVの化合物は、ステップ(m)の反応において本明細書中で先に記載のものと同じ様式のステップ(w)の反応により、式XXVIの化合物に変換できる。式XXVIの化合物は、ステップ(n)の反応において本明細書中で先に記載のものと同じ様式のステップ(x)の反応により、式XXVIIの化合物に変換できる。式XXVIIの化合物は、式II(式中、yは3であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XXVIIの化合物は、式VII(式中、yは3であり、R3はニトロである)の化合物でもある。
【0084】
この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0085】
【化14】
【0086】
式II(式中、yは0であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0087】
【化15】
【0088】
の化合物及び式VII(式中、yは0である)の化合物、すなわち、式:
【0089】
【化16】
【0090】
の化合物は、スキーム5の反応により調製できる。スキーム5の反応においては、R3は、水素、ニトロ、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである。Pはヒドロキシ保護基である。R2は、1〜2個の炭素原子を有するアルキル基である。R4は、H、クロロ又はブロモである。
【0091】
式XXVIIIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載のもののような適当な保護基を利用することによりヒドロキシ基を最初に保護し、次に、式XXIX(式中、R4はHである)の化合物を得るためにエステルを加水分解することによるステップ(y)の反応により、式XXIXの化合物に変換できる。
【0092】
式XXVIIIの化合物は、ステップ(y)からの式の化合物を、ハロゲン化試薬、例えば、塩化チオニル、五塩化リン、三塩化リン、臭素、四臭化炭素などと反応させることにより、式XXIX(式中、R4はクロロ又はブロモである)の化合物に変換できる。ステップ(z)の反応を実施するには、カルボン酸のそのようなハロゲン化反応において従来用いられる条件のいずれを利用してもよい。
【0093】
式XXIXの化合物は、アンモニアと直接反応させることによる、又は、式XXIXの化合物の化合物をカップリング試薬、例えば、ジシクロヘキシルカルボジイミド、ベンゾトリアゾリルオキシ)トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェートで最初に処理してからアンモニアなどと反応させることによる(a')の反応により、式XXXの化合物に変換できる。ステップ(a')の反応を実施するには、アンモニアのアシル化において従来用いられる条件のいずれを利用してもよい。式XXXの化合物は、試薬、例えば、塩化チオニル、五酸化リン、五塩化リン、オキシ塩化リン、四塩化炭素−トリフェニルホスフィン、塩化シアヌルなどを利用する脱水によるステップ(b')の反応により、式XXXIの化合物に変換できる。この反応は、純粋に、又は、適当な溶媒(例えばN,N−ジメチルホルムアミドなど)中のいずれかで実施する。ステップ(b')の反応を実施するには、そのような脱水反応において従来用いられる条件のいずれを利用してもよい。
【0094】
式XXXIの化合物は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されているものなど適当な脱保護試薬を利用してヒドロキシ保護基を除去することによるステップ(c’)の反応により、式XXXIIの化合物に変換できる。式XXXIIの化合物は、式II(式中、yは0であり、R1は、水素、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルである)の化合物である。式XXXIIの化合物は、式VII(式中、yは0であり、R3はニトロである)の化合物でもある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0095】
【化17】
【0096】
式III(式中、xは1又は2である)の化合物、すなわち、式:
A−(CH2)xOH
の化合物及び式IV(式中、xは1又は2である)の化合物、すなわち、式:
A−(CH2)xL
の化合物は、スキーム6の反応により調製できる。スキーム6の反応において、Aは前述のとおりである。Lは脱離基又はハロゲン化物である。式XXXIIIの化合物は、ステップ(d')の反応により式XXXIVの化合物に還元できる。この反応は、従来の還元剤、例えば、水酸化リチウムアルミニウムなどのアルカリ金属水素化物を利用して実施する。この反応は、適当な溶媒(テトラヒドロフランなど)中で実施する。ステップ(d')の反応を実施するには、そのような還元反応において従来用いられる条件のいずれを利用してもよい。
【0097】
式XXXIVの化合物は、式III(式中、xは1である)の化合物である。
【0098】
式XXXIVの化合物は、ヒドロキシル基を脱離基又はハロゲン化物で置換することにより式XXXVの化合物に変換でき、好ましい基は、ブロモ又はクロロである。適切なハロゲン化用試薬としては、塩化チオニル、塩化オキサリル、臭素、三臭化リン、四臭化炭素などが挙げられるが、これらに限定されない。脱離基としては、トシル基、メシル基などが挙げられる。ステップ(e')の反応を実施するには、そのような反応において従来用いられるいずれの条件を利用してもよい。式XXXVの化合物は、式IV(式中、xは1である)の化合物である。
【0099】
式XXXVの化合物は、XXXVをアルカリ金属シアン化物、例えば、シアン化ナトリウム又はシアン化カリウムと反応させることにより、式XXXVIの化合物に変換できる。この反応は、適当な溶媒(エタノール、ジメチルスルホキシド、N,N−ジメチルホルムアミドなど)中で実施する。ステップ(f')の反応を実施するには、ニトリルの調製において従来用いられる条件のいずれを利用してもよい。
【0100】
式XXXVIの化合物は、酸性又は塩基性の加水分解による反応ステップ(g')により、式XXXVIIの化合物に変換できる。この反応を実施するうえでは、塩基性加水分解、例えば水性水酸化ナトリウムを利用することが一般に好ましい。ステップ(g')の反応を実施するには、ニトリルの加水分解において従来用いられる条件のいずれを利用してもよい。
【0101】
式XXXVIIの化合物を還元して、ステップ(h')の反応により式XXXVIIIの化合物を得ることができる。この反応は、ステップ(d')の反応において本明細書中で先に記載のものと同じ様式で実施できる。式XXXVIIIの化合物は、式III(式中、xは2である)の化合物である。
【0102】
式XXXVIIIの化合物は、ステップ(e')の反応において本明細書中で先に記載のものと同じ様式のステップ(i')の反応により、式XXXIXの化合物に変換できる。式XXXIXの化合物は、式IV(式中、xは2である)の化合物である。
【0103】
この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。Aが、1又は2つのヒドロキシル基により置換されているフェニルである場合は、式XXXIIIの化合物のヒドロキシル基を保護することが一般に好ましい。適当な保護基は、T. GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。
【0104】
【化18】
【0105】
式XXVIII(式中、R3は、水素、ニトロ、フルオロ、ブロモ、クロロ、1〜2個の炭素原子を有するアルコキシ又は1〜2個の炭素原子を有するアルキルであり、R2は、1〜2個の炭素原子を有するアルキルである)の化合物、すなわち式:
【0106】
【化19】
【0107】
の化合物は、スキーム7の反応により調製できる。スキーム7の反応において、R2及びR3は前述のとおりである。
【0108】
式XIIの化合物は、メタノール又はエタノールで式XIIの化合物をエステル化することによるステップ(j')の反応により、式XXVIIIの化合物に変換できる。この反応は、触媒(例えば、H2SO4、TsOHなど)を使用するか、又は脱水剤(例えばジシクロヘキシルカルボジイミドなど)を使用することのいずれかにより実施できる。ステップ(j’)の反応を実施するには、そのようなエステル化反応において従来用いられる条件のいずれを利用してもよい。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0109】
【化20】
【0110】
式XII(式中、R3は、クロロ、ブロモ又はフルオロである)の化合物、すなわち式:
【0111】
【化21】
【0112】
の化合物は、商業的に入手できるか、又は以下のような文献中に記載の方法により調製できるかいずれかである:
1. 3−Br又はF−2−OHC6H3CO2H
Canadian Journal of Chemistry (2001), 79(11) 1541-1545。
2. 4−Br−2−OHC6H3CO2H
WO9916747又はJP04154773。
3. 2−Br−6−OHC6H3CO2H
JP47039101。
4. 2−Br−3−OHC6H3CO2H
WO9628423。
5. 4−Br−3−OHC6H3CO2H
WO2001002388。
6. 3−Br−5−OHC6H3CO2H
Journal of labelled Compounds and Radiopharmaceuticals (1992), 31 (3), 175-82。
7. 2−Br−5−OHC6H3CO2H及び3−Cl−4−OHC6H3CO2H
WO9405153及びUS5519133。
8. 2−Br−4−OHC6H3CO2H及び3−Br−4−OHC6H3CO2H
WO20022018323
9. 2−Cl−6−OHC6H3CO2H
JP06293700
10. 2−Cl−3−OHC6H3CO2H
Proceedings of the Indiana Academy of Science (1983), Volume date 1982, 92, 145-51。
11. 3−Cl−5−OHC6H3CO2H
WO2002000633及びWO2002044145。
12. 2−Cl−5−OHC6H3CO2H
WO9745400。
【0113】
式XII(式中、R3は、1〜2個の炭素原子を有するアルコキシである)の化合物、すなわち、式:
【0114】
【化22】
【0115】
の化合物は、スキーム8の反応により調製できる。スキーム8の反応においては、R2は、1〜2個の炭素原子を有するアルキルである。Pはヒドロキシル保護基である。式XLの化合物は、適当な保護基によりフェニル基を保護することによるステップ(k’)の反応により、式XLIの化合物に変換できる。保護基のための適当な条件は、T. GreeneによるProtective Groups in Organic Synthesis中に記載してある可能性がある。
【0116】
式XLIの化合物は、アルデヒドをカルボン酸に酸化することにより、式XLIIの化合物に変換できる。この反応は、適当な酸化試薬、例えば、ピリジニウムクロロクロメート、過マンガン酸カリウム、過マンガン酸ナトリウムなどを使用することにより実施できる。ステップ(l’)の反応を実施するには、そのような酸化反応において適当な条件のいずれを利用してもよい。
【0117】
式XLIIの化合物は、保護基の脱保護により、ステップ(m’)の反応による式XII(式中、R3は、1個の炭素原子を有するアルコキシである)の化合物に変換できる。適当な脱保護条件は、T GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。
【0118】
式XLIIの化合物は、溶媒(例えばジクロロメタン)を−72℃〜0℃の温度で4〜48時間使用して式XLIIの化合物を三臭化ホウ素又は三塩化ホウ素で処理することにより、式XLIIIの化合物に変換できる。ステップ(n’)の反応を実施するには、そのような脱メチル反応において従来用いられる条件のいずれを利用してもよい。
【0119】
式XLIIIの化合物は、メタノール又はエタノールで式XLIIIの化合物をエステル化することにより、式XLIVの化合物に変換できる。この反応は、触媒(例えば、H2SO4、TsOHなど)を使用するか、又は脱水剤(例えばジシクロヘキシルカルボジイミドなど)を使用することのいずれかにより実施できる。ステップ(o’)の反応を実施するには、そのようなエステル化反応において従来用いられる条件のいずれを利用してもよい。
【0120】
式XLIVの化合物は、適当な塩基、例えば、炭酸カリウム、水素化ナトリウム、ピリジンなどを用いることにより、式XLIVの化合物をハロゲン化エチルでエーテル化又はアルキル化することにより、式XLVの化合物に変換できる。この反応は、従来の溶媒(テトラヒドロフラン、N,N−ジメチルホルムアミド、ジクロロメタンなど)中で実施できる。この反応は、0℃〜40℃の温度で一般に実施する。ステップ(p’)の反応を実施するには、そのようなアルキル化反応において適当な条件のいずれを利用してもよい。
【0121】
式XLVの化合物は、保護基の脱保護により、ステップ(q’)の反応による式XII(式中、R3は、2個の炭素原子を有するアルコキシである)の化合物に変換できる。適当な脱保護条件は、T GreeneによるProtective Groups in Organic Synthesis中に記載されている可能性がある。この生成物は、抽出、蒸発、クロマトグラフィー及び再結晶化などの手法により単離及び精製できる。
【0122】
【化23】
【0123】
式XII(式中、R3は、1〜2個の炭素原子を有するアルコキシである)の化合物、すなわち、式:
【0124】
【化24】
【0125】
の化合物は、商業的に入手できるか、又は以下のような文献中に記載の方法により調製できるかいずれかである:
1. 2−OMe−4−OHC6H3CO2H
US2001034343又はWO9725992。
2. 5−OMe−3−OHC6H3CO2H
J.O.C (2001), 66(23), 7883-88。
3. 2−OMe−5−OHC6H3CO2H
US6194406(96ページ)及びJournal of the American Chemical Society (1985), 107(8), 2571-3。
4. 3−OEt−5−OHC6H3CO2H
Taiwan Kexue (1996), 49(1), 51-56。
5. 4−OEt−3−OHC6H3CO2H
WO9626176
6. 2−OEt−4−OHC6H3CO2H
Takeda Kenkyusho Nempo (1965), 24,221-8。
JP07070025。
7. 3−OEt−4−OHC6H3CO2H
WO9626176。
【0126】
式XII(式中、R3は、1〜2個の炭素原子を有するアルキルである)の化合物、すなわち、式:
【0127】
【化25】
【0128】
の化合物は、商業的に入手できるか、又は、以下のような文献中に記載の方法により調製できるかいずれかである:
1. 5−Me−3−OHC6H3CO2H及び2−Me−5−OHC6H3CO2H
WO9619437。
J.O.C. 2001, 66, 7883-88。
2. 2−Me−4−OHC6H3CO2H
WO8503701。
3. 3−Et−2−OHC6H3CO2H及び5−Et−2−OHC6H3CO2H
J. Med. Chem. (1971), 14(3), 265。
4. 4−Et−2−OHC6H3CO2H
Yaoxue Xuebao (1998), 33(1), 67-71。
5. 2−Et−6−OHC6H3CO2H及び2−n−Pr−6−OHC6H3CO2H
J. Chem. Soc., Perkin Trans 1 (1979), (8), 2069-78。
6. 2−Et−3−OHC6H3CO2H
JP10087489及びWO9628423。
7. 4−Et−3−OHC6H3CO2H
J.O.C. 2001, 66, 7883-88。
WO9504046。
8. 2−Et−5−OHC6H3CO2H
J.A.C.S (1974), 96(7), 2121-9。
9. 2−Et−4−OHC6H3CO2H及び3−Et−4−OHC6H3CO2H
JP04282345。
10. 1.3−Et−5−OHC6H3CO2H
2−エチルアクロレインを使用することにより、J.O.C. 2001, 66, 7883-88からの合成を適合する。
【0129】
治療法における使用
本発明は、哺乳動物対象における尿酸値を低下させ、又は哺乳動物対象からの尿酸排泄を増加させる方法を提供する。哺乳動物における尿酸値は、任意の従来の測定を用いて定量できる。典型的には、血中尿酸値を定量する。尿酸は、組織中に堆積又は沈殿する結果、蓄積部(例えば痛風結節)となる場合もあり、その蓄積部は血中尿酸濃度を上昇又は低下させることにより影響を受けることもあり、逆に尿酸の循環の一因となることもある。尿酸を減少させるための本発明の方法は、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎臓結石、腎機能不全、心血管疾患、心血管リスク因子及び認知障害などさまざまな状態を治療又は防止するために用いることができる。本発明の化合物を投与すると、尿酸値を低下させることにより、腎臓疾患の進行が遅くなる。尿酸値上昇は心血管疾患のリスク因子として同定されている。高齢者においては、尿酸上昇と認知障害との間に有意な相関が示されている。(Schretlen, D.J. et al., "Serum Uric Acid and Cognitive Function in Community-Dwelling Older Adults", Neuropsychology (Jan. 2007) 21(1): 136-140)。したがって、尿酸を減少させるための本発明の方法は、高齢者における認知障害を含めた認知障害を治療又は防止するために使用できる。レッシュ−ナイハン症候群罹患者は尿酸値が上昇しており、こうした高尿酸血症の多数の結果(痛風など)を患うことはよく知られている。したがって、血中尿酸値を低下させ尿酸の排出を増加させるための本発明は、レッシュ−ナイハン症候群罹患者を治療するために使用できる。
【0130】
血中尿酸の正常範囲は、男性においては3.4mg/dL〜7.0mg/dLの間、閉経前の女性においては2.4mg/dL〜6.0mg/dLの間、小児においては2.5mg/dL〜5.5mg/dLの間である。尿酸結晶の形成/沈殿は、典型的には、男性においては6.6mg/dL以上の値で、女性においては6.0mg/dL以上の値で生じる。このことから、いわゆる正常範囲内にある尿酸値であっても、望ましくない健康上の結果を有することが(痛風を生じさせることすら)あることが示される。さらに、全体の集団にとっては正常範囲であると思われる値であっても、個人にとっては上昇している場合がある。尿酸上昇による心血管及び他の結果は、こうした「正常な」範囲内の血中濃度を伴って生じる場合が相当ある。したがって、高尿酸血症の診断は、本発明の化合物の有益な効果にとっては必ずしも予め必要なものではない。
【0131】
本発明は、痛風、高血圧症、血管炎症、心不全、動静脈障害、心筋梗塞、脳卒中、子癇前症、子癇、睡眠時無呼吸、腎機能不全(腎不全、末期腎疾患[ESRD,end stage renal disease]など)、臓器移植、利尿薬、チアジド、シクロスポリン、アスピリン、ビタミンC、ニコチン酸、レボドパ(L−DOPA,levodopa)、細胞傷害薬及びある種の抗菌剤(ピロジナミド(pyrozinamide)など)、肝硬変、甲状腺機能不全、副甲状腺機能不全、肺癌、貧血症、白血病、リンパ腫、多発性骨髄腫、腫瘍溶解症候群、甲状腺又は副甲状腺の機能不全、レッシュ−ナイハン症候群、喫煙、アルコール消費及び乾癬に伴う高尿酸血症の治療を包含する。本発明は、痛風、尿酸結晶の形成、腎機能不全、移植後の生着不全又は臓器不全、内皮障害(炎症など)、慢性心不全、動静脈障害、子癇前症、子癇、高血圧症及び認知障害をもたらす可能性のある高尿酸血症の治療を包含する。痛風を治療するための本発明の方法の実施形態においては、尿酸の組織蓄積部(痛風結節を含むがこれに限定されるものではない)は減少し、痛風の発作の発生及び重症度も低下する。
【0132】
本発明の化合物は、任意の従来の全身投与経路により投与できる。好ましくは、この化合物又はその塩は経口投与される。したがって、この医薬は経口投与用に製剤化されることが好ましい。本発明により用いることができる他の投与経路としては、注射(例えば、静脈内、皮下、筋肉内又は腹膜内の注射)による経直腸的、非経口的な経路、又は経鼻的な経路が挙げられる。
【0133】
本発明の治療の使用及び方法のそれぞれのさらなる実施形態は、前述の化合物の実施形態のいずれか1つを投与することを含む。不必要に冗長になることを避けるため、そのような化合物及び化合物群はそれぞれ繰り返さないが、そうした薬剤及び薬剤群は、繰り返された場合と同様に、治療の使用及び方法のこの説明中に組み込まれる。
【0134】
ヒト及び非ヒトの哺乳動物対象は両方とも、本発明の治療方法により治療できる。特定の対象のための本発明の特定の化合物の最適用量は、熟練の臨床家により臨床の場において決定できる。経口投与の場合においては、本発明の化合物は、一般に、成人に対し一日用量が1mg〜2500mg、より好ましくは1mg〜1200mgで投与する。本発明の他の一実施形態においては、この化合物は、400mg〜1000mg、600mg〜800mg、600mg〜1000mg、又は100〜300mgの用量で、1日当たり1回又は2回投与する。典型的な成人の平均体重は60〜70キログラムであることから、mg/kgとして表される適切な用量範囲は、およそ0.015〜42mg/kg、0.015〜20mg/kg、6.6〜13mg/kg、10〜13mg/kg、10〜16mg/kg、又は1.67〜4.3mg/kgであり、1日当たり1回又は2回投与する。小児を治療するときは、最適用量は患者の医師が決定する。マウスへの経口投与の場合においては、本発明の化合物は、一般には、体重1キログラム当たりの化合物の一日用量1〜300mgで投与する。
【0135】
本発明の化合物は、他の尿酸低下薬と組み合わせて投与できる。そのような場合における本発明の化合物の用量は、前述のとおりである。本発明の化合物と組み合わせて、従来の、又は治験用の任意の尿酸低下薬を利用できる。そのような薬物の例としては、以下が挙げられる:キサンチンオキシダーゼ阻害薬(アロプリノール(100mg/日〜1000mg/日、より典型的には100mg/日〜300mg/日)、フェブキソスタット(40mg/日〜120mg/日、より具体的には60mg/日〜80mg/日)及びオキシプリノールなど);Puricase/PEG−ウリカーゼ(注射により2週間毎に4mg〜12mg);尿酸排泄剤(スルフィンピラゾン(100mg/日〜800mg/日)、プロベネシド(500mg/日)、ロサルタン(25mg/日〜200mg/日、より典型的には50mg/日〜100mg/日)、フェノフィブレート、JTT-552(URAT−1阻害薬)、ベンズブロマロン(70mg/日〜150mg/日)など)及びスタチン(アトルバスタチン(LIPITOR(登録商標))など)。他の尿酸低下薬は、そのような他の薬物の用量の方を低くして投与するか、そのような他の薬物の頻度を低くして投与するか、いずれかにより、その通常量又は通常量未満の量で投与してもよい。
【0136】
本発明の化合物は、痛風発作に伴う疼痛を低下させるために使用される他の薬物、例えば、非ステロイド性抗炎症薬(NSAID,nonsteroidal antiinflammatory drug)、コルヒチン、コルチコステロイド及び他の鎮痛薬と共に投与できる。
【0137】
血中尿酸値を低下させる過程においては、本発明の化合物により尿中の尿酸値が高まると考えられる。尿のpHを高め、それにより尿酸の可溶性を高めるために、例えば、クエン酸塩又は炭酸水素塩を、本発明の化合物と組み合わせて投与してもよい。
【0138】
本発明の化合物又は塩と、1又は複数の他の尿酸低下薬、鎮痛薬及びpH上昇剤との混合物を対象に投与してもよい。或いは、本発明の化合物又は塩、及び、1又は複数の他の尿酸低下薬、鎮痛薬及びpH上昇剤は、共に混合されて混合物を形成するのではなく、対象に独立に投与される。活性成分を共に混合して単一の混合物又は組成物を形成しないときは、活性成分を、1又は複数単位の経口用量の本発明の化合物と、1又は複数単位の経口用量の1又は複数の他の尿酸低下薬と、鎮痛薬及びpH上昇剤と、本発明の化合物を他の活性成分と組み合わせて投与するための取扱説明書とを備えるキットの形態で提供すると便利である。好ましくは、キットの構成要素は、箱又はブリスターパックなどに入った形で、一緒に包装される。
【0139】
医薬組成物
本発明は、本発明の化合物と、任意選択で薬学的に許容される担体とを含む医薬組成物を提供する。本発明の医薬組成物のさらなる実施形態は、前述の化合物の実施形態のいずれか1つを含む。不必要に冗長になることを避けるため、そのような化合物及び化合物群はそれぞれ繰り返さないが、そうした薬剤及び薬剤群は、繰り返された場合と同様に、医薬組成物のこの説明中に組み込まれる。
【0140】
好ましくは、この組成物は、例えば、錠剤、コーティング錠、糖衣錠、ハード又はソフトタイプのゼラチンカプセル剤、液剤、乳剤又は懸濁剤の形態で経口投与用に構成される。一般に、この経口組成物は、1mg〜2500mg、より好ましくは1mg〜1200mgの本発明の化合物を含むことになる。本発明のより具体的な実施形態においては、この経口組成物は、400mg〜1000mg、600mg〜800mg、600mg〜1000mg、又は100〜300mgの本発明の化合物を含むことになる。対象にとっては、1日当たり1又は2個の錠剤、コーティング錠、糖衣錠又はゼラチンカプセル剤を飲みこむことは手軽である。しかし、この組成物は、例えば坐剤の形態での経直腸投与、例えば注射液剤の形態での非経口投与、又は経鼻投与など、任意の他の従来の全身投与手段による投与用に構成することもできる。
【0141】
この活性成分は、医薬組成物の作製のために、薬学上不活性な無機又は有機の担体を用いて加工してもよい。乳糖、トウモロコシデンプン又はその誘導体、タルク、ステアリン酸又はその塩などを、例えば、錠剤、コーティング錠、糖衣錠及びハードゼラチンカプセル剤用のそのような担体として使用できる。ソフトゼラチンカプセル剤用の適当な担体は、例えば、植物油、蝋、脂肪、半固体及び液体のポリオールなどである。しかし、活性成分の性質によっては、ソフトゼラチンカプセルの場合においては、ソフトゼラチン自体以外の担体は通常必要ではない。液剤及びシロップ剤の作製用の適当な担体は、例えば、水、ポリオール、グリセロール、植物油などである。坐剤用の適当な担体は、例えば、天然又は硬化タイプの油、蝋、脂肪、半液体又は液体のポリオールなどである。
【0142】
この医薬組成物は、さらに、保存剤、可溶化剤、安定化剤、湿潤剤、乳化剤、甘味剤、着色剤、香味剤、浸透圧を変化させるための塩、緩衝剤、コーティング剤又は抗酸化剤を含有してもよい。
【0143】
本発明は、以下の実施例を参照することによりさらによく理解されると思われるが、この実施例は、本明細書中に記載の本発明を例証するものであるが、これを限定するものではない。
[実施例]
【実施例1】
【0144】
【化26】
【0145】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール
ステップA:3−(2,6−ジメチルベンジルオキシ)ベンゾニトリルの調製:
乾燥THF(30ml)中の2,6−ジメチルベンジルアルコール(6.27g、46.1mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、9.24g、45.7mmol)の溶液を、THF(100ml)中の3−ヒドロキシベンゾニトリル(5g、37mmol)及びトリフェニルホスフィン(TPP、11.99g、45.7mmol)の溶液に0℃で滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0146】
ステップB:5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾールの調製:
乾燥ジメチルホルムアミド(30ml)中の3−(2,6−ジメチルベンジルオキシ)ベンゾニトリル(ステップA、3g、11.8mmol)、アジ化ナトリウム(0.847g、13mmol)及び塩化アンモニウム(0.697g、13mmol)の混合物を、アルゴン下で110℃にて14時間又は出発物質が全て消費されるまで加熱した。この反応混合物に水を加えて、固形物を全て溶解させ、この溶液をブライン中に取り、酢酸エチル(2X)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
1H NMR(400MHz,(CD3)2SO):2.4(s,6H);5.15(s,2H);7.1(d,2H);7.15(m,1H);7.3(dd,1H);7.5(t,1H);7.65(m,1H);7.7(m,1H)。
【実施例2】
【0147】
【化27】
【0148】
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール
ステップA:2−(3−ヒドロキシフェニル)アセトニトリルの調製:
乾燥塩化メチレン(20ml)中の2−(3−メトキシフェニル)アセトニトリル(3.6g、25.4mmol)の溶液に、アルゴン雰囲気下で−78℃にてBBr3(55ml、CH2Cl2中1M、55mmol)を加えた。この反応混合物を48時間かけて周囲温度に温め、破砕した氷でクエンチし、塩化メチレンで抽出した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(CH2Cl2:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、油としての標題化合物を得た。
【0149】
ステップB:2−(3−(2,6−ジメチルベンジルオキシ)フェニル)アセトニトリルの調製:
乾燥THF(20ml)中の2−(3−ヒドロキシフェニル)アセトニトリル(ステップA、5g、37mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、3.38g、16.7mmol)の溶液を、THF(30ml)中の2,6−ジメチルベンジルアルコール(2.25g、16.5mmol)及びトリフェニルホスフィン(TPP、4.3g、16.4mmol)の溶液に、アルゴン下で0℃にて滴加した。この反応混合物を、室温で16時間又は出発物質が全て消費されるまで撹拌した。この混合物にシリカゲル(25g)を加え減圧下で溶媒を除去し、これをシリカゲルカラム上に載せ、塩化メチレン:ヘキサン(1:1)で溶出させて、淡黄色の結晶性の固形物を得た。
【0150】
ステップC:5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾールの調製:
乾燥ジメチルホルムアミド(30ml)中の2−(3−(2,6−ジメチルベンジルオキシ)フェニル)アセトニトリル(ステップB、3.2g、12.7mmol)、アジ化ナトリウム(1.28g、16.7mmol)及び塩化アンモニウム(1.08g、20.2mmol)の混合物を、アルゴン下、90℃にて9時間又は出発物質が全て消費されるまで加熱し、この反応混合物を減圧下で濃縮した。この反応混合物を酢酸エチル中に取り、水(2X)で洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(塩化メチレン:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより精製して、油状の生成物を得た。この油を1:2の酢酸エチル:ヘキサンと共に10分間撹拌し、固形物を濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.3(s,6H);4.25(s,2H);5.15(s,2H);6.84(d,1H);6.96(m,2H);7.08(d,2H);7.18(m,1H);7.28(m,1H)。
【実施例3】
【0151】
【化28】
【0152】
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール
ステップA:エチル3−ヒドロキシ−4−メトキシベンゾエートの調製:
無水エタノール(300ml)中の3−ヒドロキシ−4−メトキシ安息香酸(25g、148.67 mmol)及びp−トルエンスルホン酸一水和物(3.17g、16.66mmol)の溶液を、6時間又は出発物質が全て消費されるまで環流させた。この反応混合物を濃縮し、EtOAc(60ml)で希釈し、水(20ml)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0153】
ステップB:エチル3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンゾエートの調製:
乾燥THF(20ml)中のエチル3−ヒドロキシ−4−メトキシベンゾエート(ステップA、9.10g、46.4mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、10.23g、50mmol)の溶液を、乾燥THF(60ml)中の2,6−ジメチルベンジルアルコール(6.94g、51mmol)及びトリフェニルホスフィン(TPP、13.27g、50mmol)の溶液に、アルゴン下で0℃にて滴加した。この反応混合物を4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0154】
ステップC:(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)メタノールの調製:
乾燥THF(30ml)中のエチル3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンゾエート(ステップB、6.04g、19.23mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.803g、21.16mmol)を滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで撹拌してから、0.1N HCl、EtOAc(20ml)でゆっくりクエンチして反応混合物に加えた。この反応混合物を濾過し、沈殿物をEtOAc(25ml×2)で洗浄した。有機層を合わせたものを0.1N HCl、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0155】
ステップD:2−((5−(ブロモメチル)−2−メトキシフェノキシ)メチル)−1,3−ジメチルベンゼンの調製:
乾燥CH2Cl2(20ml)中の(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)メタノール(ステップC、5.23g、20.4mmol)及びCBr4(10.16g、30.6mmol)の溶液に、トリフェニルホスフィン(8.03g、30.64mmol)を0℃で少しずつ加えた。この反応混合物を1.5時間撹拌し、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
1H NMR(400MHz,(CDCl3):2.43(s,6H);3.83(s,3H);4.53(s,2H);5.08(s,2H);6.84(d,1H);7.0〜7.03(dd,1H);7.06〜7.09(m,3H);7.14〜7.18(m,1H)。
【0156】
ステップE:2−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−((5−(ブロモメチル)−2−メトキシフェノキシ)メチル)−1,3−ジメチルベンゼン(ステップD、3.28g、9.7mmol)及びNaCN(0.624g、12.7mmol)の溶液を、120℃で2.5時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0157】
ステップF:5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシフェニル)アセトニトリル(ステップE、2.17g、7.5mmol)、アジ化ナトリウム(0.590g、9.1mmol)及び塩化アンモニウム(0.486g、9.1mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.3(s,6H);3.68(s,3H);4.22(s,2H);4.98(s,2H);6.78〜6.81(dd,1H);6.91〜6.93(d,1H);7.05〜7.07(d,2H);7.13〜7.16(m,2H)。MS:m/z 325.2[M+H]+。
【実施例4】
【0158】
【化29】
【0159】
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール
ステップA:3−(3−メトキシフェニル)プロパンニトリルの調製:
乾燥DMF(20ml)中の1−(2−ブロモエチル)−3−メトキシベンゼン(10g、46.4mmol)、NaCN(2.73g、55.8mmol)の溶液を、90℃で6時間又は出発物質が全て消費されるまで加熱し、この反応を冷却し、EtOAc(60ml)で希釈し、水(20ml×3)、ブラインで洗浄し、有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、油としての標題化合物を得た。
【0160】
ステップB:3−(3−ヒドロキシフェニル)プロパンニトリルの調製:
乾燥CH2Cl2(20ml)中の3−(3−メトキシフェニル)プロパンニトリル(ステップA、1.71g、10.6mmol)の撹拌溶液に、アルゴン下で−78℃にてBBr3(CH2Cl2中1M、5.32g、21.2mmol)を加えた。この反応混合物を、同じ温度で2時間、次いで0℃で4時間又は出発物質が全て消費されるまで撹拌し、氷でクエンチし、EtOAc(30ml×3)で抽出し、有機層を合わせたものを飽和NaHCO3、ブラインで慎重に洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0161】
ステップC:3−(3−(2,6−ジメチルベンジルオキシ)フェニル)プロパンニトリルの調製:
乾燥THF(10ml)中の3−(3−ヒドロキシフェニル)プロパンニトリル(ステップB、1.25g、8.5mmol)及びアゾジカルボン酸ジイソプロピル(DIAD、1.87g、9.26mmol)の溶液を、アルゴン下で0℃にて、乾燥THF(30ml)中の2,6−ジメチルベンジルアルコール(1.27g、9.3mmol)及びトリフェニルホスフィン(TPP、2.43g、9.26mmol)の溶液に滴加した。この反応混合物を、4時間又は出発物質が全て消費されるまで室温に温め、エーテルで希釈し、水(2X)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0162】
ステップD:5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の3−(3−(2,6−ジメチルベンジルオキシ)フェニル)プロパンニトリル(ステップC、2.62g、9.9mmol)、アジ化ナトリウム(0.899g、13.8mmol)及び塩化アンモニウム(0.740g、13.8mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5→92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を、1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.48(s,6H);3.02(t,2H);3.19(t,2H);4.98(s,2H);6.80〜6.81(d,1H);6.86〜6.89(m,2H);7.05〜7.07(d,2H);7.14〜7.23(m,2H)。MS:m/z 309.2[M+H]+。
【実施例5】
【0163】
【化30】
【0164】
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
ステップA:2−(3−ヒドロキシ−4−メチルフェニル)アセトニトリルの調製:
乾燥CH2Cl2(20ml)中の2−(3−メトキシ−4−メチルフェニル)アセトニトリル(5g、31mmol)の撹拌溶液に、アルゴン下で−78℃にて、BBr3(CH2Cl2中1M、10.02g、40mmol)を滴加した。この反応混合物を、同じ温度で2時間、次いで0℃で5時間又は出発物質が全て消費されるまで撹拌し、氷でクエンチし、EtOAc(30ml×3)で抽出し、有機層を合わせたものを飽和NaHCO3、ブラインで慎重に洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1→CH2Cl2:hex、1:1)を用いたフラッシュクロマトグラフィーにより精製して、オフホワイトの固形物としての標題化合物を得た。
【0165】
ステップB:2−(3−(2,6−ジメチルベンジルオキシ)−4−メチルフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−(3−ヒドロキシ−4−メチルフェニル)アセトニトリル(ステップA、2.18g、14.8mmol)、K2CO3(2.66g、19.2mmol)の撹拌溶液に、アルゴン下で室温にて、2,6−ジメチルベンジルクロリド(2.97g、19.2mmol)を加えた。この反応混合物を16時間撹拌し、EtOAc(40ml)で希釈し、水(20ml)及びブラインで洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0166】
ステップC:5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾールの調製:
乾燥DMF(15ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−4−メチルフェニル)アセトニトリル(ステップB、1.12g、4.2mmol)、アジ化ナトリウム(0.400g、6.1mmol)及び塩化アンモニウム(0.350g、6.5mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、95:5→92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)で10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.0(s,3H);2.35(s,6H);4.27(s,2H);5.0(s,2H);6.73〜6.75(dd,1H);7.08〜7.1(m,3H);7.15〜7.19(m,2H)。MS:m/z 309.2[M+H]+。
【実施例6】
【0167】
【化31】
【0168】
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール
ステップA:2−(4−(2,6−ジフルオロベンジルオキシ)フェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の2−(4−ヒドロキシフェニル)アセトニトリル(5g、37.5mmol)及びK2CO3(6.74g、48.8mmol)の溶液に、2,6−ジフルオロベンジルブロミド(7.77g、37.5mmol)を加えた。この反応混合物を4時間室温で撹拌し、真空中で濃縮した。粗残留物をEtOAc中に取り、水及びブラインで洗浄した。水層をEtOAcで再度洗浄した。有機層を合わせたものをNa2SO4で乾燥させ、濾過し、濃縮して、白色の固形物としての標題化合物を得た。
1H NMR(270MHz,CDCl3):3.65(s,2H);5.1(s,2H);6.9〜7.0(m,4H);7.2〜7.4(m,3H)。
【0169】
ステップB:5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾールの調製:
乾燥DMF(60ml)中の2−(4−(2,6−ジフルオロベンジルオキシ)フェニル)アセトニトリル(ステップA、5g、19.3mmol)、アジ化ナトリウム(1.3g、20mmol)及び塩化アンモニウム(1.06g、20mmol)の混合物を、90℃で16時間加熱した。溶媒を真空中で除去し、油状の残留物をEtOAcと水との間で分離させた(高濃度HClを用いてpH1に酸性化した)。有機層を水で洗浄し、Na2SO4で乾燥させ、濾過し、濃縮して、茶色の半固形物とした。精製は、シリカゲルカラム(クロロホルム:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより行って、淡いクリーム色の固形物としての標題化合物を得た。
1H NMR(270MHz,CDCl3):4.0(s,2H);5.1(s,2H);6.7〜6.9(m,4H);7.0(d,2H);7.2(m,1H)。
【実施例7】
【0170】
【化32】
【0171】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール
ステップA:エチル3−ヒドロキシ−2−メチルベンゾエートの調製:
無水エタノール(150ml)中の3−ヒドロキシ−2−メチル安息香酸(5.04g、33.12mmol)及びp−トルエンスルホン酸一水和物(0.741g、3.89mmol)の溶液を、16時間又は出発物質が全て消費されるまで還流させた。この反応混合物を濃縮し、EtOAc(30ml)で希釈し、水(20ml)で洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0172】
ステップB:エチル3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゾエートの調製:
乾燥DMF(15ml)中のエチル3−ヒドロキシ−2−メチルベンゾエート(ステップA、3.1g、17.22mmol)、K2CO3(3.09g、22.38mmol)の撹拌溶液に、アルゴン下で室温にて、2,6−ジメチルベンジルクロリド(3.19g、20.66mmol)を加えた。この反応混合物を16時間撹拌し、EtOAc(40ml)で希釈し、水(20ml)及びブラインで洗浄した。有機層をNa2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、白色の固形物としての標題化合物を得た。
【0173】
ステップC:(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)メタノールの調製:
乾燥THF(35ml)中のエチル3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゾエート(ステップB、5.94g、19.93mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.832g、21.92mmol)を滴加した。この反応混合物を4時間又は出発物質が全て消費されるまで撹拌してから、0℃にて0.1N HClでゆっくりクエンチし、この反応混合物にEtOAc(20ml)を加えた。この反応混合物を濾過し、EtOAc(25ml×2)で沈殿物を洗浄した。有機層を合わせたものを0.1N HCl、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、2:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0174】
ステップD:1−(ブロモメチル)−3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゼンの調製:
乾燥CH2Cl2(20ml)中の(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)メタノール(ステップC、3.68g、14.37mmol)及びCBr4(5.25g、15.8mmol)の溶液に、0℃でトリフェニルホスフィン(4.14g、15.8mmol)を少しずつ加えた。この反応混合物を4時間撹拌し、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。この固形物を真空下で6時間、さらに乾燥させ続けた。
【0175】
ステップE:2−(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)アセトニトリルの調製:
乾燥DMF(20ml)中の1−(ブロモメチル)−3−(2,6−ジメチルベンジルオキシ)−2−メチルベンゼン(ステップD、4.28g、13.41mmol)及びNaCN(0.789g、16.10mmol)の溶液を120℃で3時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)、ブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、標題化合物を得た。
【0176】
ステップF:5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾールの調製:
乾燥DMF(20ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−2−メチルフェニル)アセトニトリル(ステップE、2.70g、10.19mmol)、アジ化ナトリウム(0.795g、12.23mmol)及び塩化アンモニウム(0.653g、12.22mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、水で希釈し、EtOAc(30ml×4)で抽出した。有機層を合わせたものをブラインで洗浄し、Na2SO4で乾燥させ、濾過し、濃縮し、シリカゲルカラム(クロロホルム:メタノール、92.5:7.5)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.01(s,3H);2.32(s,6H);4.24(s,2H);5.01(s,2H);6.78〜6.79(dd,1H);7.06〜7.08(d,2H);7.12〜7.19(m,3H)。
【実施例8】
【0177】
【化33】
【0178】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
ステップA:3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンズアルデヒドの調製:
乾燥DMF(15ml)中の3−ヒドロキシ−2−メトキシベンズアルデヒド(5.06g、32.7mmol)、2,6−ジメチルベンジルクロリド(5.04g、33.1mmol)及びK2CO3(4.78g、34.6mmol)の溶液を、アルゴン下で室温にて16時間撹拌してから、EtOAc(40ml)で希釈し、水(20ml)で洗浄した。有機層を濃縮し、シリカゲルカラム(hex:酢酸エチル、4:1)を用いたフラッシュクロマトグラフィーにより精製して、オフホワイトの固形物としての標題化合物を得た。
【0179】
ステップB:(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)メタノールの調製:
乾燥THF(40ml)中の3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンズアルデヒド(ステップB、10.6g、32.7mmol)の溶液に、アルゴン下で0℃にてLiAlH4(THF中1M、0.95g、23.7mmol)を滴加した。この反応混合物を1時間又は出発物質が全て消費されるまで撹拌してから水をゆっくり加え、次いで1N HCl(5ml)、水(10ml)を加えることによりクエンチし、この反応混合物にEtOAc(20ml)を加えた。この反応混合物を濃縮し、酢酸エチルを用いた短いシリカゲルカラムを通過させて、オフホワイトの固形物としての標題化合物を得た。
【0180】
ステップC:3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジルメタンスルホネートの調製:
乾燥CH2Cl2(100ml)中の(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)メタノール(ステップB、9.5g、32.7mmol)及びトリエチルアミン(5.80g、57.4mmol)の溶液に、アルゴン下で0℃にて塩化メタンスルホニル(3.5ml、45mmol)を滴加した。この反応混合物を6時間又は出発物質が全て消費されるまで室温に温め、10%の冷Na2CO3で中和し、CH2Cl2で抽出した。有機層を濃縮し、シリカゲルカラム(hex:塩化メチレン、2:1)を用いたフラッシュクロマトグラフィーにより精製して、淡黄色の油としての標題化合物を得た。
【0181】
ステップD:2−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)アセトニトリルの調製:
乾燥DMF(40ml)中の3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジルメタンスルホネート(ステップC、8.8g、30.26mmol)及びNaCN(1.60g、32.6mmol)の溶液を、85℃で18時間加熱してから冷却し、EtOAc(50ml)で希釈した。有機層を水(30ml)で洗浄し、濃縮し、塩化メチレンを用いた短いシリカゲルカラムを通過させて、黄色の固形物としての標題化合物を得た。
【0182】
ステップE:5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾールの調製:
乾燥DMF(10ml)中の2−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシフェニル)アセトニトリル(ステップD、3.2g、12.7mmol)、アジ化ナトリウム(0.86g、13.2mmol)及び塩化アンモニウム(0.696g、13.0mmol)の混合物を、アルゴン下で90℃にて16時間又は出発物質が全て消費されるまで加熱し、この反応混合物を冷却し、減圧下で濃縮し、シリカゲルカラム(クロロホルム:メタノール、9:1)を用いたフラッシュクロマトグラフィーにより精製して、半固形の生成物を得た。この半固形物を1:2の酢酸エチル:ヘキサン(15ml)と共に10分間撹拌し、濾過して、白色の固形物としての生成物を得た。
1H NMR(400MHz,(CD3)2SO):2.33(s,6H);3.5(s,3H);4.21(s,2H);5.04(s,2H);6.83〜6.85(dd,1H);7.05〜7.09(m,3H);7.15〜7.23(m,2H)。
【実施例9】
【0183】
URAT1阻害アッセイ
URAT1(尿酸輸送体1,Uric Acid Transporter 1)は、腎尿細管中の頂端膜上で発現する。URAT1は、尿から血中への尿酸の再取込みを媒介する。URAT1を阻害すると、尿中の尿酸の排泄の増加、したがって、血清尿酸濃度を低下させる薬物に対する潜在的様式の作用がもたらされる。例えば、プロベネシド及びベンズブロマロンは、痛風及び高尿酸血症の治療に臨床的に使用されており、これらの薬物は両方ともURAT1に作用して尿酸の再取込みを減少させる。しかし、ベンズブロマロンは、URAT1とは無関係の機序による肝毒性を理由に市場から引き揚げられ、プロベネシドは、多数の輸送タンパク質に作用する結果、さまざまな他の薬物と相互作用する。
【0184】
インビトロでのURAT1アッセイは、血清尿酸の減少において強力な活性を有する化合物を同定するには有用である。適当なアッセイには、細胞(例えば、ヒト胚腎細胞「HEK,human embryonic kidney」)に、ヒトURAT1をコードするベクターを形質移入し、次いで、放射線標識した尿酸を取り込む形質移入細胞の能力を定量することが含まれる。URAT1阻害薬としての化合物の活性は、形質移入細胞による尿酸取込みを遮断する能力により評価される。
【0185】
試験化合物及び化学薬品:
ベンズブロマロン(Sigma社製, Cat.No.B5774)、プロベネシド(Sigma社製, Cat.No.P8761)、DMSO(Sigma社製, Cat.No.D-2650)、[8−14C]尿酸(50〜60mCi/mmol、American Radio Chemicals社製, Cat. No. ARC0513)。
【0186】
発現ベクター中へのhURAT1のサブクローニング:
hURAT1 cDNAを含有するプラスミドベクターpCMV6−XL5(Cat. No. SC125624)と、発現ベクターpCMV6−Neo(Cat. No.pCMVNEO)とをOriGene Technologies, Inc.社から入手した。完全長のhURAT1 cDNAをベクターpCMV6−XL5から得て、これを発現ベクターpCMV6−Neo中にサブクローニングして、hURAT1発現プラスミドpCMV6−hURAT1を創出した。自動DNA配列決定法により配列を検証した。
【0187】
URAT1発現プラスミドの細胞培養、形質移入、及び、hURATlを安定して発現するHEK細胞の確立:
10%FBS及び2mM L−グルタミンを添加したEMEM中でヒト胚腎293(HEK,Human embryonic kidney)細胞(ATTCC, Cat No. CRL-1573)を培養し、37℃、5% CO2の条件でインキュベートした。形質移入実験のために、60mmの皿上に1皿当たり培地1mlで細胞を播いた。18〜24時間のインキュベーション後、メーカーの取扱説明書に従ってリポフェクチン形質移入剤を用いて(Invitrogen社製, Cat.No.18292)、細胞にプラスミドpCMV6−hURAT1又は発現ベクターpCMV6−Neoを形質移入した。形質移入後、EMEM培地中で72時間細胞を培養し、次いで、1mg/mlのゲネチシン(GIBCO社製, Cat. No 10131)を加えることにより、安定な形質移入体を選抜した。逆転写ポリメラーゼ連鎖反応(RT−PCR,reverse transcription polymerase chain reaction)法を用いて、hURAT1を発現する安定な形質移入体(以後、本明細書中ではhURAT1−HEK細胞と呼ぶ)、又は、発現ベクターpCMV6−Neoのみを有する細胞(以後、本明細書中ではmock−HEK細胞と呼ぶ)を検証した。
【0188】
[8−14C]尿酸取込みアッセイ:
hURAT1−HEK細胞及びmock−HEK細胞を、EMEM培地中3×105の濃度でポリ−D−リシン細胞培養24ウェルプレート(Becton Dickinson社製, Cat. No.354414)中に播き、一晩インキュベートした。[8−14C]尿酸(55mCi/mmol)を含有する反応溶液を最終濃度50μMで、125mMグルコン酸ナトリウム、4.8mMグルコン酸カリウム、1.3mMカルシウム、5.6mMグルコース、1.2mM硫酸マグネシウム、1.2mM KH2PO4及び25mM HEPES(pH7.4)を含有するハンクス平衡塩溶液(HBSS,Hanks' balanced salt solution)中の、試験化合物を入れたものと入れないものとを調製した。取込みアッセイが開始する前に、培養培地を除去し、細胞をHBSS0.6ml中で5分間インキュベートした。その後HBSSを除去し、調製済の反応溶液を各ウェル中に加え、室温で5分間インキュベートした。次いで反応溶液を除去し、冷HBSS0.6mlで細胞を2回洗浄し、0.1M NaOH0.2mlで20分間溶解させた。シンチレーション液(Opti Phase SuperMIX, PerkinElmer社製, Cat No. 1200-439)1mlの入ったシンチレーションバイアル中に細胞溶解物を移し、Microbeta計数装置(1450, Wallac Jet, PerkinElmer社製)中で放射能を計数した。試験化合物をDMSO中で溶解させ、試験化合物の入っていないmock−HEK細胞及びhURAT1−HEK細胞のウェル中に同じ濃度のDMSOを加えた。各試験化合物について、取込みアッセイを2回実施し、3連で実施した。各試験条件についての細胞の尿酸取込みを、DMSO対照と比較した平均阻害率(%)として示した。DMSOの入っているウェルについて得た放射能値を、細胞の100%取込みとして採用した。観察された濃度−阻害率(%)データをS字濃度−効果モデル:
阻害率(%)=(100*濃度^勾配)/IC50^勾配]+濃度^勾配)
に当てはめた。
Data Analysis Toolbox(商標)(MDL Information Systems社製、San Leandro, CA, USA)を用いた非線形最小二乗回帰分析により、IC50及び勾配の推定値並びにそれらの95%信頼限界を定量した。
【0189】
URAT1阻害薬としての化合物の活性を評価するために、尿酸取込みの阻害率(%)を、典型的には、10マイクロモルの薬物濃度で評価した(表1)。IC−50値の定量のために、いくつかの化合物の追加的な薬物濃度を試験した(表2)。この実施例においては、化合物を必ずしも同時に試験しなかった。
【0190】
【表1】
【0191】
【表2】
【実施例10】
【0192】
化合物EBを用いたマウスでの経口単回用量薬物動態試験
オスのマウスに単回経口胃管投与した後の化合物EBの血漿プロファイルの定量:
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の化合物EB又は媒体(1%HPMC)をオスのマウスに単回経口胃管投与により投与した。マウスは、投与後、尿採取装置中に5時間配置し、5時間分の全尿排出量を収集した。試料は、LC/MS−MSを用いて分析するまで−80℃で冷凍した。
【0193】
投与時の0時間後、0.5時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に回収し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。
【0194】
データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0195】
プロトコール:
A.血漿。
1.マウスに、化合物EB100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.マウス血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5.0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, Sequence 1029-08A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0196】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.095mLを、メタノール中の化合物EBの20倍ストック0.005mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EBを作製した。
例:メタノール中の血漿95マイクロL+10mM化合物EB5マイクロL=500マイクロM化合物EBの入った血漿0.1mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.2mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25ml/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0197】
HPLC条件:
【0198】
【表3】
【0199】
結果:
化合物EBはマウス血漿中で容易に検出できた。正負のイオン化モードにおいて保持時間及び質量を確認した。AGILENT LC-MS sequence 1029-08A。
「M−」=279.2 100%
「M+」=281.2 100%
式量280。保持時間平均=26.5分。
【0200】
【表4】
【0201】
【表5】
【0202】
AUC0〜24:796μg/mL
最高血中濃度:210μg/mL
セミログプロットについての線形成分:
半減期(1〜4時間):2.27
半減期(6〜8時間):0.74
半減期(8〜24時間):9.39
【実施例11】
【0203】
化合物EBを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物EBの血漿プロファイルの定量:
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に回収し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0204】
プロトコール:
A.血漿。
1.ラットに、化合物EB100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽を用いて5分間解凍し、最高速度で10秒間ボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mm、逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、100バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT Sequence 1015-08A Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0205】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中のPN2107の20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EBを作製した。
例:メタノール中の血漿190マイクロL+10mM化合物EB10マイクロL=500マイクロM化合物EBの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、13マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25ml/分、100バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0206】
【表6】
【0207】
結果:
1.直線度=0.9994へのR^2フィットを用いて較正曲線を作成した(図4)。
2.化合物EBはラット血漿中で容易に検出でき、正負のイオン化モードにおいて保持時間及び質量を確認した。
「M−」=279.2 100%。
「M+」=281.2 100%
式量280、保持時間平均=26.5分。
生データ及び計算値を表7に記載する。
血漿中の化合物濃度を表8に記載する。
【0208】
【表7】
【0209】
【表8】
【0210】
【表9】
【0211】
化合物EBの半減期=3.99時間
化合物EBのAUC0〜24=566μg/mL*時間
化合物EBの最高血中濃度=108.08μg/mL
【実施例12】
【0212】
化合物ECを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物ECの血漿プロファイルの定量。
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に収集し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0213】
プロトコール:
A.血漿。
1.ラットに、化合物EC100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT sequence 0226-09A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0214】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中の化合物ECの20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロMの濃度の化合物ECを作製した。
例:メタノール中の血漿190マイクロL+10mMの化合物EC10マイクロL=500マイクロMの化合物ECの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0215】
【表10】
【0216】
結果:
1.直線度=0.9984へのR^2フィットを用いて較正曲線を作成した(図6を参照)。
2.化合物ECは血漿中で容易に検出できた。正負両方のイオン化モードにおいて保持時間及び質量を確認した(図7を参照)。
「M−」=293.2 100%、527.2 65%
「M+」=295.2 100%。式量294。保持時間平均=23分。
【0217】
【表11】
【0218】
【表12】
【0219】
【表13】
【実施例13】
【0220】
化合物EGを用いたラットでの経口単回用量薬物動態パイロット試験
オスのラットに単回経口胃管投与した後の化合物EGの血漿プロファイルの定量。
試験化合物は、組織ホモジナイザーを用いて1%HPMC中で懸濁させて粒子を最小化し懸濁液の均一性を最大化してから、4℃で保存した。この調合物は、投与直前に徹底的に混合した。100mg/kgの用量の試験化合物又は媒体(1%HPMC)をオスのSprague-Dawleyラットに単回経口胃管投与により投与した。投与時の0時間後、1時間後、2時間後、4時間後、6時間後、8時間後及び24時間後の時点で、K3EDTA管中に眼窩洞から出血させることにより血液試料(0.4mL)を採取した。血液試料は、採取の30分以内に、冷蔵下(2〜8℃)にて7分間、6000rpmで遠心分離にかけた。遠心分離後、血漿を各時点で各動物用の単一管中に収集し、−80℃で即時に冷凍し、追ってLC/MS−MSを用いて分析した。血漿濃度時間の連続データは、WinNonlin Standard(v2.1、Pharsight Corporation社製)及びMicrosoft EXCELを用いた薬物動態分析に供した。
【0221】
プロトコール:
A.血漿。
1.ラットに、化合物EG100mg/kgを単回経口胃管投与し、血漿を一定回数採取した。
2.血漿は、分析日まで−80℃で保存した。
3.試料は37℃の浴槽で5分間解凍し、10秒間最高速度でボルテックスした。
4.ラット血漿0.1mLをアセトニトリル0.2mLと混合し、1分ボルテックスし、4℃で25分間、14000rpm、17000gで沈降させた。
5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた:方法406975M1, AGILENT sequence 0226-09A, Agilent社製、1100 LC-MS。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0222】
B.較正曲線。
ステップ1.「媒体」動物由来の血漿(プールしておいたもの)0.19mLを、メタノール中の化合物EGの20倍ストック0.01mLと混合して、血漿中の500マイクロM、250マイクロM、125マイクロM、…、の濃度の化合物EGを作製した。
例:メタノール中の血漿190マイクロL+10mMの化合物EG10マイクロL=500マイクロMの化合物EGの入った血漿0.2mL。
ステップ2.ステップ1の試料を最高速度で10秒間ボルテックスした。
ステップ3.ステップ2の全ての試料にアセトニトリル0.4mLを加え、全てのバイアルを最高速度で1分間ボルテックスした。
ステップ4.ステップ3の全ての試料を、4℃で25分間、14000rpm、17000gで沈降させた。
ステップ5. 0.45ミクロン、4mmのPTFE膜シリンジフィルター(Phenomenex社製、#AF0-3102-52)を通して上澄みを濾過し、15マイクロLを注射し、Luna 3ミクロン、細孔100A、C8(2)、150×3mmの逆相カラム(Phenomenex社製、#00F-4248-YO, SN#259151-7)を、50分で40%〜69%の直線的濃度勾配の(0.1%ギ酸、89.9%アセトニトリル、10%メタノール)で用い、0.25mL/分、107バール、カラム温度37℃の条件で分離させた。
全ての試料は2回試行し、吸光度210nm及び230nmでの正負のイオン化のスペクトログラムを記録した。
【0223】
【表14】
【0224】
結果:
1.直線度=0.9997へのR^2フィットを用いて較正曲線を作成した(図8を参照)。
2.化合物EGは血漿中で容易に検出できた。正負両方のイオン化モードにおいて保持時間及び質量を確認した(図9を参照)。
「M−」=307.2 100%
「M+」=309.2 100%。式量308。保持時間平均=30分。
【0225】
生データ及び計算
【0226】
【表15】
【0227】
【表16】
【0228】
【表17】
【特許請求の範囲】
【請求項1】
式XLVIで表される化合物
【化1】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項2】
式XLVIIで表される、請求項1に記載の化合物
【化2】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項3】
xが1である、請求項1又は2に記載の化合物。
【請求項4】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項1〜3のいずれかに記載の化合物。
【請求項5】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項4に記載の化合物。
【請求項6】
式XLVIIIで表される、請求項4に記載の化合物
【化3】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項7】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項6に記載の化合物。
【請求項8】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項7に記載の化合物。
【請求項9】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項6に記載の化合物。
【請求項10】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させる方法であって、式Iで表される化合物を、前記対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な量で前記対象に投与することを含む方法
【化4】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項11】
化合物が、式XLVIで表される、請求項10に記載の方法
【化5】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項12】
化合物が、式XLVIIで表される、請求項11に記載の方法
【化6】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項13】
xが1である、請求項10〜12のいずれかに記載の方法。
【請求項14】
Aが、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項10〜12のいずれかに記載の方法。
【請求項15】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項14に記載の方法。
【請求項16】
化合物が、式XLVIIIで表される、請求項14に記載の方法
【化7】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項17】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項12又は16に記載の方法。
【請求項18】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項17に記載の方法。
【請求項19】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項10に記載の方法。
【請求項20】
痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態を治療又は防止するための、請求項10に記載の方法を含む方法。
【請求項21】
対象がヒトである、請求項10に記載の方法。
【請求項22】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬を前記対象に投与することをさらに含む、請求項10に記載の方法。
【請求項23】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項22に記載の方法。
【請求項24】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項22に記載の方法。
【請求項25】
化合物と1又は複数の他の尿酸低下薬とが共に混合されて混合物を形成し、前記混合物が対象に投与される、請求項22に記載の方法。
【請求項26】
化合物と1又は複数の他の尿酸低下薬とが、共に混合されて混合物を形成するのではなく、対象に独立に投与される、請求項22に記載の方法。
【請求項27】
化合物が経口投与用に製剤化される、請求項10に記載の方法。
【請求項28】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排泄を増加させる際に使用するための、式Iで表される化合物
【化8】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項29】
式XLVIで表される、請求項28に記載の使用のための化合物
【化9】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項30】
式XLVIIで表される、請求項29に記載の使用のための化合物
【化10】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項31】
xが1である、請求項28〜30のいずれかに記載の使用のための化合物。
【請求項32】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項28〜30のいずれかに記載の使用のための化合物。
【請求項33】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項32に記載の使用のための化合物。
【請求項34】
式XLVIIIで表される、請求項32に記載の使用のための化合物
【化11】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項35】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項30又は34に記載の使用のための化合物。
【請求項36】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項35に記載の使用のための化合物。
【請求項37】
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項28に記載の使用のための化合物。
【請求項38】
経口投与される、請求項28に記載の使用のための化合物。
【請求項39】
使用の結果、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態が治療又は防止される、請求項28に記載の使用のための化合物。
【請求項40】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与される、請求項28に記載の使用のための化合物。
【請求項41】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項40に記載の使用のための化合物。
【請求項42】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項40に記載の使用のための化合物。
【請求項43】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されている、請求項40に記載の使用のための化合物。
【請求項44】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項40に記載の使用のための化合物。
【請求項45】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるための医薬の製造における、式Iで表される化合物の使用
【化12】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項46】
化合物が、式XLVIで表される、請求項45に記載の使用
【化13】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項47】
化合物が、式XLVIIで表される、請求項46に記載の使用
【化14】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項48】
xが1である、請求項45〜47のいずれかに記載の使用。
【請求項49】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項45〜47のいずれかに記載の使用。
【請求項50】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項49に記載の使用。
【請求項51】
化合物が、式XLVIIIで表される、請求項49に記載の使用
【化15】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項52】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項47又は51に記載の使用。
【請求項53】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項52に記載の使用。
【請求項54】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項45に記載の使用。
【請求項55】
医薬が経口投与用に製剤化される、請求項45に記載の使用。
【請求項56】
量が、哺乳動物対象への医薬の投与の結果、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態が治療又は防止されるように選択される、請求項45に記載の使用。
【請求項57】
医薬が、対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与されるように製剤化される、請求項45に記載の使用。
【請求項58】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項57に記載の使用。
【請求項59】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項57に記載の使用。
【請求項60】
医薬が、化合物と1又は複数の他の尿酸低下薬とを、混合物の形態で共に混合された状態で含む、請求項57に記載の使用。
【請求項61】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項57に記載の使用。
【請求項62】
哺乳動物対象の血中の尿酸濃度を低下させる、又は前記対象からの尿酸排出を増加させる際に使用するための医薬組成物であって、薬学的に許容される担体と、前記対象の血中の尿酸濃度を低下させる、又は前記対象からの尿酸排出を増加させるのに有効な量の式Iで表される化合物とを含む医薬組成物
【化16】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項63】
化合物が、式XLVIで表される、請求項62に記載の医薬組成物
【化17】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項64】
化合物が、式XLVIIで表される、請求項63に記載の医薬組成物
【化18】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項65】
xが1である、請求項62〜64のいずれかに記載の医薬組成物。
【請求項66】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項62〜64のいずれかに記載の医薬組成物。
【請求項67】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項66に記載の医薬組成物。
【請求項68】
化合物が、式XLVIIIで表される、請求項66に記載の医薬組成物
【化19】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項69】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項64又は68に記載の医薬組成物。
【請求項70】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項69に記載の医薬組成物。
【請求項71】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項62に記載の医薬組成物。
【請求項72】
痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態を治療又は防止するうえで使用するための、請求項62に記載の医薬組成物。
【請求項73】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与されるように製剤化される、請求項62に記載の医薬組成物。
【請求項74】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項73に記載の医薬組成物。
【請求項75】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項73に記載の医薬組成物。
【請求項76】
医薬が、化合物と1又は複数の他の尿酸低下薬とを、混合物の形態で共に混合された状態で含む、請求項73に記載の医薬組成物。
【請求項77】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項73に記載の医薬組成物。
【請求項78】
経口投与用に製剤化される、請求項62に記載の医薬組成物。
【請求項1】
式XLVIで表される化合物
【化1】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項2】
式XLVIIで表される、請求項1に記載の化合物
【化2】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項3】
xが1である、請求項1又は2に記載の化合物。
【請求項4】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項1〜3のいずれかに記載の化合物。
【請求項5】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項4に記載の化合物。
【請求項6】
式XLVIIIで表される、請求項4に記載の化合物
【化3】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項7】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項6に記載の化合物。
【請求項8】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項7に記載の化合物。
【請求項9】
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項6に記載の化合物。
【請求項10】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させる方法であって、式Iで表される化合物を、前記対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な量で前記対象に投与することを含む方法
【化4】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項11】
化合物が、式XLVIで表される、請求項10に記載の方法
【化5】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項12】
化合物が、式XLVIIで表される、請求項11に記載の方法
【化6】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項13】
xが1である、請求項10〜12のいずれかに記載の方法。
【請求項14】
Aが、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項10〜12のいずれかに記載の方法。
【請求項15】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項14に記載の方法。
【請求項16】
化合物が、式XLVIIIで表される、請求項14に記載の方法
【化7】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項17】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項12又は16に記載の方法。
【請求項18】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項17に記載の方法。
【請求項19】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項10に記載の方法。
【請求項20】
痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態を治療又は防止するための、請求項10に記載の方法を含む方法。
【請求項21】
対象がヒトである、請求項10に記載の方法。
【請求項22】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬を前記対象に投与することをさらに含む、請求項10に記載の方法。
【請求項23】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項22に記載の方法。
【請求項24】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項22に記載の方法。
【請求項25】
化合物と1又は複数の他の尿酸低下薬とが共に混合されて混合物を形成し、前記混合物が対象に投与される、請求項22に記載の方法。
【請求項26】
化合物と1又は複数の他の尿酸低下薬とが、共に混合されて混合物を形成するのではなく、対象に独立に投与される、請求項22に記載の方法。
【請求項27】
化合物が経口投与用に製剤化される、請求項10に記載の方法。
【請求項28】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排泄を増加させる際に使用するための、式Iで表される化合物
【化8】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないか、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項29】
式XLVIで表される、請求項28に記載の使用のための化合物
【化9】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項30】
式XLVIIで表される、請求項29に記載の使用のための化合物
【化10】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項31】
xが1である、請求項28〜30のいずれかに記載の使用のための化合物。
【請求項32】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項28〜30のいずれかに記載の使用のための化合物。
【請求項33】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項32に記載の使用のための化合物。
【請求項34】
式XLVIIIで表される、請求項32に記載の使用のための化合物
【化11】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項35】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項30又は34に記載の使用のための化合物。
【請求項36】
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項35に記載の使用のための化合物。
【請求項37】
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項28に記載の使用のための化合物。
【請求項38】
経口投与される、請求項28に記載の使用のための化合物。
【請求項39】
使用の結果、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態が治療又は防止される、請求項28に記載の使用のための化合物。
【請求項40】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与される、請求項28に記載の使用のための化合物。
【請求項41】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項40に記載の使用のための化合物。
【請求項42】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項40に記載の使用のための化合物。
【請求項43】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されている、請求項40に記載の使用のための化合物。
【請求項44】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項40に記載の使用のための化合物。
【請求項45】
哺乳動物対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるための医薬の製造における、式Iで表される化合物の使用
【化12】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項46】
化合物が、式XLVIで表される、請求項45に記載の使用
【化13】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項47】
化合物が、式XLVIIで表される、請求項46に記載の使用
【化14】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項48】
xが1である、請求項45〜47のいずれかに記載の使用。
【請求項49】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項45〜47のいずれかに記載の使用。
【請求項50】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項49に記載の使用。
【請求項51】
化合物が、式XLVIIIで表される、請求項49に記載の使用
【化15】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項52】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項47又は51に記載の使用。
【請求項53】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項52に記載の使用。
【請求項54】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項45に記載の使用。
【請求項55】
医薬が経口投与用に製剤化される、請求項45に記載の使用。
【請求項56】
量が、哺乳動物対象への医薬の投与の結果、痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態が治療又は防止されるように選択される、請求項45に記載の使用。
【請求項57】
医薬が、対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与されるように製剤化される、請求項45に記載の使用。
【請求項58】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項57に記載の使用。
【請求項59】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項57に記載の使用。
【請求項60】
医薬が、化合物と1又は複数の他の尿酸低下薬とを、混合物の形態で共に混合された状態で含む、請求項57に記載の使用。
【請求項61】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項57に記載の使用。
【請求項62】
哺乳動物対象の血中の尿酸濃度を低下させる、又は前記対象からの尿酸排出を増加させる際に使用するための医薬組成物であって、薬学的に許容される担体と、前記対象の血中の尿酸濃度を低下させる、又は前記対象からの尿酸排出を増加させるのに有効な量の式Iで表される化合物とを含む医薬組成物
【化16】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項63】
化合物が、式XLVIで表される、請求項62に記載の医薬組成物
【化17】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R1は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項64】
化合物が、式XLVIIで表される、請求項63に記載の医薬組成物
【化18】
(式中、
xは1又は2であり、
yは0、1、2又は3であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
Aは、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルであるか、或いは、
3〜6個の環原子を有する、置換されていないシクロアルキル、又は、3〜6個の環原子を有する、1若しくは2個の環炭素がメチル若しくはエチルにより独立に一置換されているシクロアルキルであるか、或いは、
N、S及びOから選択される1若しくは2個の環ヘテロ原子を有し、環炭素により前記化合物の残部と共有結合している5若しくは6環員の芳香族ヘテロ環である)。
【請求項65】
xが1である、請求項62〜64のいずれかに記載の医薬組成物。
【請求項66】
Aが、置換されていないフェニル、又は、ハロ、1若しくは2個の炭素原子を有するアルキル、ペルフルオロメチル、1若しくは2個の炭素原子を有するアルコキシ及びペルフルオロメトキシからなる群から選択される1、2若しくは3つの基により置換されているフェニルである、請求項62〜64のいずれかに記載の医薬組成物。
【請求項67】
Aが2,6−ジメチルフェニル又は2,6−ジフルオロフェニルである、請求項66に記載の医薬組成物。
【請求項68】
化合物が、式XLVIIIで表される、請求項66に記載の医薬組成物
【化19】
(式中、
yは0、1又は2であり、
R2及びR3の一方は水素であり、他方は、水素、1又は2個の炭素原子を有するアルキル、ヒドロキシ、1又は2個の炭素原子を有するアルコキシ、フルオロ、クロロ、ブロモ及びアミノからなる群から選択され、
R4及びR5は、メチル、フルオロ及びクロロからなる群から独立に選択される)。
【請求項69】
R3が水素であり、R2が、水素、メチル及びメトキシからなる群から選択され、R4がメチルであり、R5がメチルである、請求項64又は68に記載の医薬組成物。
【請求項70】
化合物が、
5−(3−(2,6−ジメチルベンジルオキシ)フェニル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−4−メトキシベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)フェネチル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−4−メチルベンジル)−1H−テトラゾール
からなる群から選択される、請求項69に記載の医薬組成物。
【請求項71】
化合物が、
5−(4−(2,6−ジフルオロベンジルオキシ)ベンジル)−1H−テトラゾール;
5−(3−(2,6−ジメチルベンジルオキシ)−2−メチルベンジル)−1H−テトラゾール;及び
5−(3−(2,6−ジメチルベンジルオキシ)−2−メトキシベンジル)−1H−テトラゾール
からなる群から選択される、請求項62に記載の医薬組成物。
【請求項72】
痛風、高尿酸血症、高尿酸血症と診断することが慣例的に正しいとされるレベルに満たない尿酸値上昇、腎機能不全、腎臓結石、心血管疾患、心血管疾患発症リスク、腫瘍溶解症候群、認知障害、早発性本態性高血圧症及び熱帯熱マラリア原虫誘導性炎症からなる群から選択される状態を治療又は防止するうえで使用するための、請求項62に記載の医薬組成物。
【請求項73】
対象の血中の尿酸濃度を低下させ、又は前記対象からの尿酸排出を増加させるのに有効な組合せ量で1又は複数の他の尿酸低下薬と組み合わせて投与されるように製剤化される、請求項62に記載の医薬組成物。
【請求項74】
他の尿酸低下薬が、キサンチンオキシダーゼ阻害薬、尿酸排泄剤、尿酸輸送体−1阻害薬、ウリカーゼ及びスタチンからなる群から選択される、請求項73に記載の医薬組成物。
【請求項75】
他の尿酸低下薬が、単独で投与される場合の通常の治療用量未満の量で投与される、請求項73に記載の医薬組成物。
【請求項76】
医薬が、化合物と1又は複数の他の尿酸低下薬とを、混合物の形態で共に混合された状態で含む、請求項73に記載の医薬組成物。
【請求項77】
化合物と1又は複数の他の尿酸低下薬とが、混合物の形態で共に混合されていない、請求項73に記載の医薬組成物。
【請求項78】
経口投与用に製剤化される、請求項62に記載の医薬組成物。
【図1】

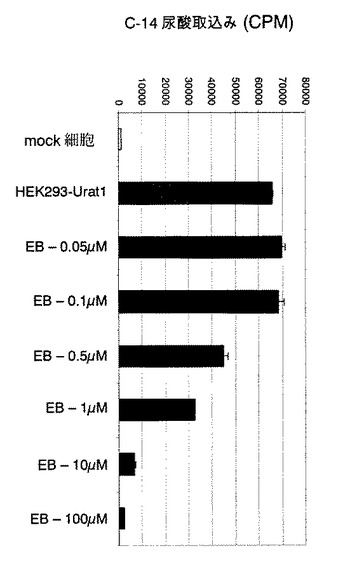
【図2】
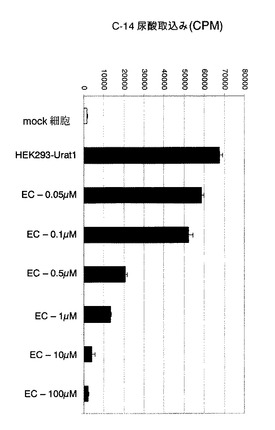
【図3】
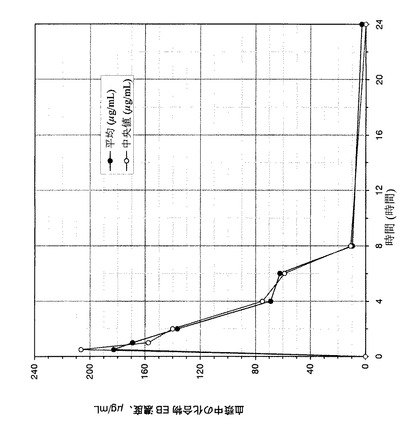
【図4】
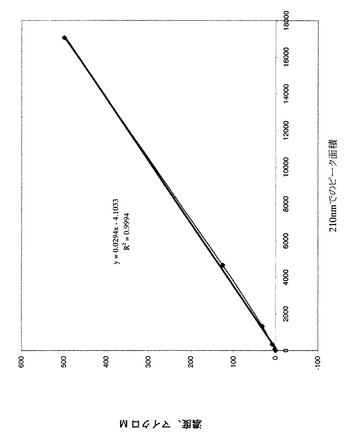
【図5】
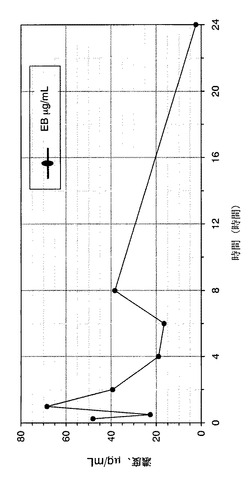
【図6】
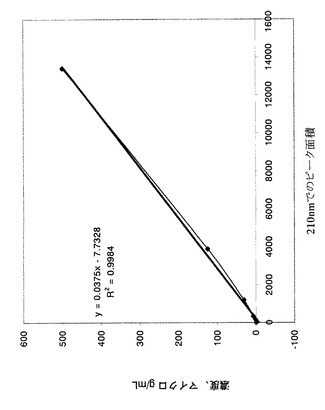
【図7】
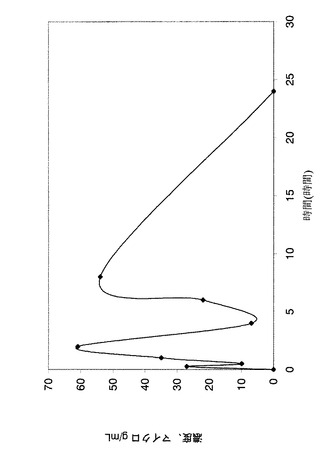
【図8】
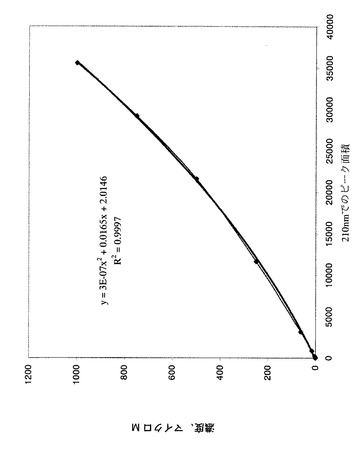
【図9】
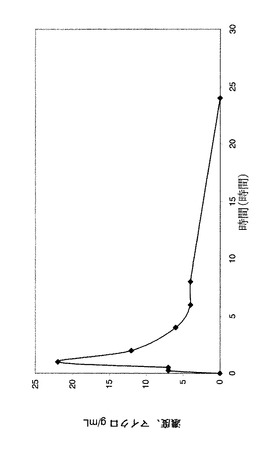
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2011−519865(P2011−519865A)
【公表日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願番号】特願2011−507641(P2011−507641)
【出願日】平成21年4月30日(2009.4.30)
【国際出願番号】PCT/US2009/042298
【国際公開番号】WO2009/134995
【国際公開日】平成21年11月5日(2009.11.5)
【出願人】(501056821)ウェルスタット セラピューティクス コーポレイション (32)
【Fターム(参考)】
【公表日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願日】平成21年4月30日(2009.4.30)
【国際出願番号】PCT/US2009/042298
【国際公開番号】WO2009/134995
【国際公開日】平成21年11月5日(2009.11.5)
【出願人】(501056821)ウェルスタット セラピューティクス コーポレイション (32)
【Fターム(参考)】
[ Back to top ]