
局所経皮用デクスメデトミジン組成物およびそれらの使用方法
デクスメデトミジン(dexmedetomidine)の鎮痛性局所用製剤、および疼痛および他の状態の処置および管理におけるそれらの使用方法。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
B.関連出願へのクロス・リファレンス
[0001]本出願は、本明細書中にそのまま援用される2010年1月8日出願の米国仮特許出願第61/293,440号の優先権を主張する。
C−E.不適用
【背景技術】
【0002】
F.背景技術
[0002]デクスメデトミジン(dexmedetomidine)、すなわち、5−[(1S)−1−(2,3−ジメチルフェニル)エチル]−1H−イミダゾールは、鎮静性および鎮痛性を有する非麻薬性α2−アドレナリン受容体アゴニストである。
【0003】
【化1】

【発明の概要】
【発明が解決しようとする課題】
【0004】
[0003]現在のところ、デクスメデトミジンは、鎮静に必要とされる注射可能製剤として商業的に入手可能であるにすぎないし、そしてそれは、医療専門家によって静脈内投与される必要がある。デクスメデトミジンは、鎮痛性を有するが、しかしながら、鎮痛薬として有用な製剤は、商業的に入手可能ではない。更に、いろいろな理由のために、商業的に入手可能な注射可能製剤は、自己投与することができる鎮痛薬として用いるのに不適当である。例えば、自己投与されて、鎮静を伴うことなく痛覚脱失を生じる(またはそれ以外には、疼痛を処置するまたは予防する)ことができるデクスメデトミジン基剤鎮痛薬について、継続した且つ未だ満たされていない要求が存在する。
【課題を解決するための手段】
【0005】
G.概要
[0004]本出願は、デクスメデトミジン、その薬学的に許容しうる塩およびその誘導体の鎮痛性経皮用製剤、更には、それらの使用方法を記載する。
【0006】
[0005]本明細書中に提供されるのは、デクスメデトミジンおよび/またはその薬学的に許容しうる塩および/またはその誘導体の新しい鎮痛性経皮用製剤、および疼痛の処置または予防におけるそれらの使用方法である。このような医薬組成物は、痛覚脱失を生じる(例えば、疼痛を処置するまたは予防する)十分な量などのデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体(例えば、プロドラッグ)および薬学的に許容しうる経皮送達ビヒクル、更には、粘度調整剤、pH調整剤、保存剤、賦形剤、乳化剤、緩衝剤、着色剤等などであるがこれに制限されるわけではない任意の追加成分を包含する。薬学的に許容しうる経皮用ビヒクル中のデクスメデトミジンまたはその薬学的に許容しうる塩またはその誘導体(例えば、プロドラッグ)は、適する容器またはデバイス中に包装されていてよい。
【0007】
[0006]典型的な態様において、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を哺乳動物に投与する方法は、哺乳動物の皮膚へ、所定量の経皮用組成物であって、哺乳動物の皮膚を介する哺乳動物の体循環系中への薬学的有効量のデクスメデトミジンの経皮吸収を与える有効量で薬学的に許容しうる経皮送達ビヒクル中にデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を含む組成物を適用することを包含する。経皮吸収時に、デクスメデトミジンは、哺乳動物に、適当に投与された場合に鎮静を伴うことなく痛覚脱失を生じる。
【0008】
[0007]追加の特徴は、以下の詳細な説明および実施例を参照して理解することができる。
H.図面の簡単な説明
【図面の簡単な説明】
【0009】
【図1】[0008]図1は、ヒトにおいて本明細書の典型的な製剤を用いた1日1回用量後の定常状態における観測対予測血漿濃度−時間曲線をグラフで示す。
【図2】[0009]図2は、ヒトにおいて本明細書の典型的な製剤を用いた1日2回投与後の観測対予測定常状態血漿濃度−時間曲線をグラフで示す。
【図3】[0010]図3は、本明細書の典型的な製剤を用いて1日1回複数日投与しているヒトの予測結果をグラフで示す。
【図4】[0011]図4は、本明細書の典型的な製剤を用いて1日2回複数日投与しているヒトの予測結果をグラフで示す。
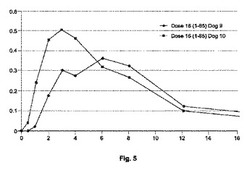
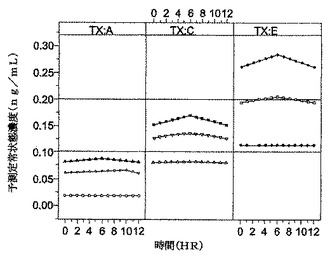
【図5】[0012]図5は、イヌにおいて処方1〜85を用いた投与結果をグラフで示す。
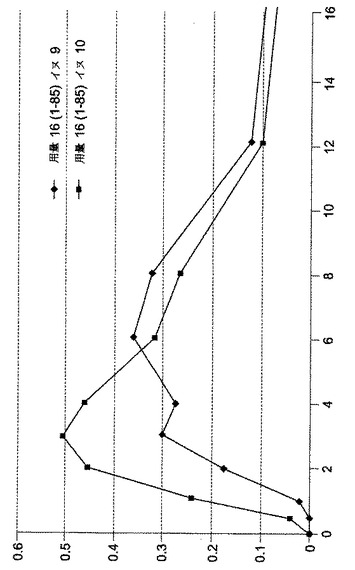
【図6】[0013]図6は、イヌにおいて処方1〜53を用いた投与結果をグラフで示す。
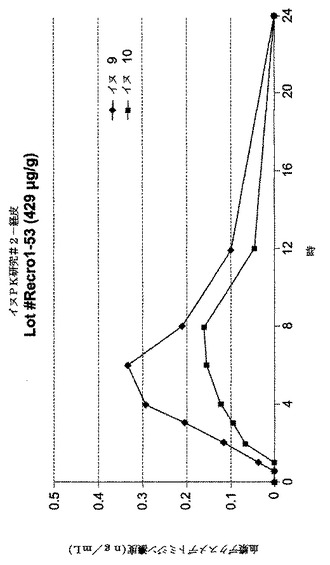
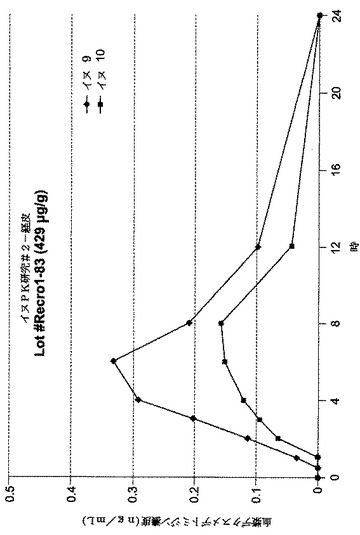
【図7】[0014]図7は、イヌにおいて処方1〜83を用いた投与結果をグラフで示す。
【発明を実施するための形態】
【0010】
I.詳細な説明
[0015]本明細書中において提供されるのは、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体の新しい鎮痛性経皮用製剤、および疼痛の処置および管理におけるそれらの使用方法である。
【0011】
[0016]デクスメデトミジンは、哺乳動物において鎮静、感覚脱失および痛覚脱失を引き起こす特異的α2−アドレナリン受容体アゴニストである。ヒトの場合、デクスメデトミジンは、集中治療設定で処置中に最初に挿管され且つ人工呼吸器で処置されている患者の鎮静用に、更には、外科手術および他の手順の前またはその間の挿管されていない患者の鎮静用に商業的に入手可能である。例えば、米国特許第6,716,867号および第6,313,311号を参照されたい。
【0012】
[0017]ヒトにおけるデクスメデトミジンの薬物動態は、静脈内(i.v.)、筋肉内(i.m.)および経皮投与後に研究された。平均除去半減期は、それぞれ、i.v.およびi.m.投与後1.5〜3時間および経皮投与後5.6時間である。i.m.および経皮投与後、最大血中濃度への時間は、それぞれ、1.6〜1.7時間および6時間であり、そして絶対バイオアベイラビリティーは、それぞれ、73%および88%であると推定された。例えば、“Pharmacodynamics and pharmacokinetics of intramuscular dexmedetomidine,” Scheinin et al., Clin. Pharmacol. Ther. 52, 53-46 (1992); “The pharmacokinetics and hemodynamic effects of intravenous and intramuscular dexmedetomidine hydrochloride in adult human volunteers,” Dyck et al., Anesthesiology 78, 813-20 (1993); および “Pharmacokinetics and pharmacodynamics of transdermal dexmedetomidine,”Kivistoe et al., Eur. J. Clin. Pharmacol. 46, 345-49 (1994) を参照されたい。
【0013】
[0018]デクスメデトミジンは、口腔などの他の組織から吸収されることも知られている。ヒト対象が、デクスメデトミジン溶液を嚥下することなく口中に保持する口腔内投与後、平均口腔内バイオアベイラビリティーは、81.8%において、約1.5時間で最大濃度および1.9時間の見掛除去半減期で測定された。例えば、“Bioavailability of dexmedetomidine after extravascular doses in healthy subjects,” Anttila et al., Br. J. Clin. Pharmacol. 56, 691-93 (2003) を参照されたい。
【0014】
[0019]本発明により、デクスメデトミジンは、疼痛を改善する、管理する、治癒する、予防するまたはそれ以外に処置する目的で動物またはヒト対象に投与することができる。典型的な態様において、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を哺乳動物に投与する方法は、哺乳動物の皮膚へ、所定の投薬量の経皮用組成物であって、哺乳動物の皮膚を介する哺乳動物の体循環系中への薬学的有効量のデクスメデトミジンの吸収を与える有効量で薬学的に許容しうる経皮用ビヒクル中にデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を含む組成物を適用することを包含する。経皮吸収時に、デクスメデトミジンは、哺乳動物に痛覚脱失を生じる。
【0015】
[0020]例えば、癌および他の不快を有する多くの患者は、長期にわたる鎮痛療法にもかかわらず、中程度〜重症の疼痛を経験し続けるが、これは、しばしば、患者の活動度レベルの増加ゆえに、間欠性破綻疼痛(intermittent breakthrough pain)として起こりうる。鎮痛薬の長時間作用性製剤の用量を増加させることによってこのタイプの疼痛に対抗する試みは、しばしば、オピオイド鎮痛薬で特に、痛覚脱失の遅い開始、および鎮静、便秘または吐き気および嘔吐の望ましくない副作用を生じる。しかしながら、本明細書中に記載のデクスメデトミジンの鎮痛性経皮用製剤は、強力な非麻薬性鎮痛作用、および好ましくは、持続した長期の制御された鎮痛作用、およびそれらの組み合わせを生じて、疼痛を改善する、管理する、治癒する、予防するまたはそれ以外に処置する。処置には、経皮用デクスメデトミジン製剤の単独の使用、または他のデクスメデトミジン製剤(例えば、舌下用、経粘膜用、注射可能および/または他の鎮痛性組成物(例えば、NSAIDS、麻薬等))との組み合わせでの使用が含まれてよい。
【0016】
[0021]本明細書中に記載のデクスメデトミジン製品は、疼痛の処置のための経皮用医薬製剤である。本明細書中で用いられる「薬学的に許容しうる」という用語は、それら化合物、材料、組成物、剤形、包装および使用方法であって、妥当な医学的判断の範囲内で、しかも過度の毒性、刺激、アレルギー性応答または他の問題または合併症を伴うことなくヒトおよび動物の組織と接触した状態での使用に適すると同時に、適度な受益性/危険性比率に相応し且つ所望の薬理応答を引き出すものを包含する。
【0017】
[0022]デクスメデトミジンは、薬学的に許容しうる酸で薬学的に許容しうる塩を形成可能な基本的窒素原子を含有する。この点での「薬学的に許容しうる塩」という用語は、デクスメデトミジンの比較的無毒性の無機および有機酸付加塩を意味する。これら塩は、デクスメデトミジンの最終単離・精製の際に現場で、またはその遊離塩基形の精製済みデクスメデトミジンを適する有機または無機酸と別個に反応させた後、このようにして形成された塩を単離することによって製造することができる。更に、その塩は、経皮用製剤を製造する製造プロセス中に形成することができる。代表的な薬学的に許容しうる塩には、ハロゲン化水素酸塩(臭化水素酸塩および塩酸塩を含めた)、硫酸塩、重硫酸塩、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナプチレート(napthylate)、メシレート、グルコヘプトネート、ラクトビオン酸塩、2−ヒドロキシエチルスルホン酸塩およびラウリルスルホン酸塩等が含まれる。例えば、“Pharmaceutical Salts,” Berge et al., J. Pharm. Sci. 66, 1-99 (1977) を参照されたい。デクスメデトミジン塩酸塩は、薬学的に許容しうる塩の一例である。デクスメデトミジン塩酸塩の使用は、いくつかの場合、その塩酸塩が、より大きい水への溶解度および周囲酸素による酸化に対する安定性を有するので、本明細書中に記載の経皮用製剤中でのデクスメデトミジン自体の使用に好適であるかもしれない。
【0018】
[0023]デクスメデトミジン誘導体は、プロドラッグを作る共有結合修飾を包含してよい。投与時に、プロドラッグ誘導体は、哺乳動物により、デクスメデトミジンを生じる化学修飾を受ける。プロドラッグは、デクスメデトミジンの生体内分布または薬物動態を好都合に変更するまたは他の望ましい特性を生じるために用いることができる。例えば、デクスメデトミジンの反応性窒素は、酵素的にまたは非酵素的に、還元的に、酸化的にまたは加水分解によって開裂して、活性な医薬成分を現す官能基で誘導体化することができる。特定のタイプのプロドラッグの使用は知られている(例えば、R.B. Silverman, 1992, “The Organic Chemistry of Drug Design and Drug Action,” Academic Press, Chp. 8 を参照されたい)。例えば、プロドラッグは、それら化合物の最終単離・精製の際にその場で、またはその遊離塩基形の精製済み化合物を適する誘導体化剤と別個に反応させることによって製造することができる。
【0019】
[0024]デクスメデトミジン経皮用組成物は、ゲル剤、クリーム剤または液剤などの一つまたはそれを超える薬学的に許容しうる担体を30重量%〜約99.995重量%包含する。これら担体は、デクスメデトミジンまたはその薬学的に許容しうる塩またはその誘導体の溶媒、補助溶媒または非溶媒であってよい。適する材料は、室温で液状物または流動性組成物であり、しかも室温で、好ましくは、周囲圧力でも、更には、高圧下でも、その状態のままである。有用な液状物および流動性組成物は、特に制限されないが、但し、それらは、それら組成物の所望の医学的使用を妨げることがないし、しかもそれらは、治療的に有用な量のデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体(例えば、デクスメデトミジン塩酸塩)を有するという条件付きである。薬学的に許容しうる液状物の例には、水、エタノール、ジメチルスルホキシド、プロピレングリコール、ポリエチレングリコール、プロピレンカーボネート、薬学的に許容しうる油状物(例えば、ダイズ、ヒマワリ、ラッカセイ等)等が含まれる。流動性組成物の例には、クリーム剤、ゲル剤、懸濁剤、ローション剤、血清剤、軟膏剤等が含まれる。薬学的に許容しうる液状物または流動性組成物は、活性な医薬成分を溶解させるか、それの安定な均一懸濁液を生じるかまたは、懸濁液、溶液、混合物または他の薬学的に許容しうる組成物のいずれかの組み合わせを形成するように選択される。
【0020】
[0025]前述の構成成分に加えて、デクスメデトミジンの経皮用製剤は、薬理学的に活性な薬物以外の一つまたはそれを超える賦形剤を包含してよく、それらは、製造プロセス中に包含されるまたは完成医薬製品剤形中に含有される。賦形剤の例には、粘度調節用材料(例えば、ポリマー、糖、糖アルコール、ガム、クレー、シリケート等(例えば、ポリビニルピロリドン(PVP)))(約0.01重量%〜約65重量%)が含まれる。賦形剤の他の例には、保存剤(例えば、エタノール、ベンジルアルコール、プロピルパラベンおよびメチルパラベン)(約0.001重量%〜約20重量%)が含まれる。賦形剤の更に他の例には、緩衝剤およびpH調整剤(例えば、水酸化ナトリウム、シトレートおよびクエン酸)(約0.01重量%〜約5重量%)が含まれる。着色剤(約0.001重量%〜約5重量%)、芳香剤(約0.001重量%〜約1重量%)、キレート化剤(例えば、EDTA)(約0.001重量%〜約1重量%)、UV吸収剤(約0.001重量%〜約10重量%)は、特に、適する賦形剤の追加例である。適する経皮用製剤は、本明細書中に更に記載の実施例において更に詳述する。
【0021】
[0026]更に、このような製剤を製造する方法および使用する方法は、本明細書中の内容により当業者に明らかであろう。例えば、経皮用デクスメデトミジン製剤は、標準的な認められた十分な製造実施にしたがって本明細書中に記載の成分の適当量を混合することによって製造することができる。このような賦形剤は、患者または対象の受容を改善する、バイオアベイラビリティーを改善する、貯蔵寿命を増加させる、製造費用および包装費用を減少させる、政府規制機関の必要条件に応じるためにおよび他の目的のために製剤中に包含されてよい。各々の成分の相対量は、得られた製剤の望ましい薬理学的および薬物動態学的性質を妨げるべきではない。
【0022】
[0027]本明細書中に記載のデクスメデトミジンの鎮痛性経皮用製剤は、哺乳動物の皮膚および/または粘膜(例えば、口、鼻、直腸または生殖器粘膜)への直接投与を予定している。薬物送達は、嚥下後胃腸管吸収に対立するものとして、実質的に、経皮経路によって行われる。「経皮」という用語は、皮膚膜を越えたまたは介する送達を意味する。「経粘膜」という用語は、粘膜を越えたまたは介する送達を意味する。具体的に、薬物の「経口経粘膜」送達には、口、咽頭、喉頭、気管または上部消化管のいずれかの組織、特に、舌下、頬側、歯肉および口蓋の粘膜組織を越えた送達が含まれる。
【0023】
[0028]経皮送達は、体血液循環への薬物物質到達を可能にし、それによって、胃腸管影響力とは無関係の直接全身投与が得られ且つ望ましくない初回通過肝代謝を免れる。他の投与経路と比較して、本製剤中のデクスメデトミジンの経皮吸収は、複数日にわたる投与でも、望ましいバイオアベイラビリティーで有意に一層制御可能な且つ長期の持続時間を有することができる。更に、初回通過代謝は回避されるので、製剤中の活性な医薬成分の全量を減少させることができ、それによって、有害な副作用(例えば、低血圧、鎮静)の可能性を減少させ且つ製造業者に原価利益を与える。
【0024】
[0029]経皮用製剤は、ヒト、更には、ヒトの伴侶動物(例えば、ネコ、イヌ)、農業用家畜およびそれを必要としている他の動物を含めた哺乳動物に投与することができる。錠剤、カプセル剤、シロップ剤等などの慣用的な剤形または注射可能鎮痛性製剤を非ヒト動物へ投与することは、しばしば、問題があるということは理解されるであろうが、本明細書中に記載の経皮用製剤は、このような動物の処置に特に有用である。経皮送達の使用は、幼児および高齢者において他の送達方法にまさる同じ利点を有する。
【0025】
[0030]「痛覚脱失」は、疼痛感覚の軽減または除去である。本明細書中で用いられる「疼痛」は、広範囲の臨床症状を包含し、しかもそれは、幅広い意味を有する。疼痛知覚は、極めて主観的であり、いろいろな人が、様々に且つ極めて異なった強さで疼痛を経験する。国際疼痛研究会(the International Association for the Study of Pain)は、疼痛を、「実際的または潜在的組織損傷に関連したまたはこのような損傷に関して説明される不快な感覚的・情動的経験」と定義している。より簡単に云うと、疼痛には、苦痛を引き起こす且つ自分自身の身体の不快な意識に関連しているいずれかの感覚的経験が含まれる。疼痛の非制限的タイプおよび原因には、神経痛、筋肉痛、痛覚過敏、ピペルパシー、神経炎およびニューロパシーが含まれる。疼痛は、しばしば、癌または関節炎などの根源的な生理学的異常の症状である。片頭痛などのいくつかのタイプの疼痛には、明確に識別される原因がない。疼痛は、熱傷または外科手術などの身体的外傷によって引き起こされることもありうる。帯状ヘルペス(水痘および帯状疱疹)などのウイルス感染も、疼痛を引き起こすことがありうる。アルコールへの化学的依存または薬物乱用からの離脱も、しばしば、疼痛症状に関連している。したがって、「疼痛」は、本明細書中において極めて幅広い意味を有すると理解され、そしてその請求の範囲に記載の使用は、いずれか特定の疾患または状態に制限されると解釈されるべきではない。
【0026】
[0031]本明細書中で用いられる「鎮静」は、患者または対象が、開放気道を独立して且つ連続して維持する能力、および身体的刺激および言語的命令に適切に且つ合理的に応答する能力を保持している低下意識を意味する。本明細書中で用いられる「鎮静を伴うことなく」は、その患者が、Ramsay Sedation Scale で Level 3以下の鎮静レベルを経験しているということ、言い換えると、患者が、次のどちらかの Level であるということを意味する。Level 1=不安、動揺または不穏状態;Level 2=協力的で、見当識があり、そして精神安定な状態;または Level 3=鎮静状態であるが、命令に応答する。本明細書中で用いられる「有意の鎮静」は、患者または対象が、Ramsay Sedation Scale で Level 4またはそれより大の鎮静を経験しているということを意味し、ここにおいて、Level 4=眠った状態;眉間軽打または声高聴覚刺激へ活発応答;Level 5=眠った状態;眉間軽打または声高聴覚刺激へ緩慢応答;Level 6=眠った状態;有痛刺激へ無応答。本明細書中で用いられる「有意の鎮静」は、Stanford Sleepiness Scale での患者の自己評価とも一致するが、対象患者は、彼らの鎮静度合いを、Level 3より大またはそれに等しいと等級付けし、ここにおいて、Level 1=活発な感情、活力がある、敏捷または広範覚醒;Level 2=高レベルで機能性であるが、ピークではない;集中できる;Level 3=覚醒しているが弛緩状態;応答性であるが十分に敏捷ではない;Level 4=ややもうろう状態、気が抜けた状態(let down);Level 5=もうろう状態;覚醒状態のままで関心喪失;怠惰状態(slowed down);Level 6=眠気、頭がくらくらする、眠気を抑えている;横になることを選択する;または Level 7=もはや眠気を抑えられず、すぐに睡眠開始;夢のような思考をする。
【0027】
[0032]本明細書中に記載のデクスメデトミジンの製剤は、アスピリン、イブプロフェン、ナプロキセン、セレコキシブ(celecoxib)、アセトアミノフェンおよび他のシクロオキシゲナーゼ阻害剤などのNSAIDS;コデイン、オキシコドン、モルヒネ、メタドン(methadone)およびフェンタニールなどのオピオイド;フェニトインおよびカルバマゼピンなどの抗痙攣薬および抗不整脈薬;アミトリプチリン、イミプラミンおよびベンラファキシン(venlafaxine)などの抗うつ薬を含めた他の疼痛処置薬と共投与することができる。このような共投与は、同時発生的であってよく、ここにおいて、デクスメデトミジンおよび別の疼痛処置薬は、双方とも同時点で投与する。或いは、本明細書中に記載の経皮用製剤の急速作用性ゆえに、患者は、より短くまたはより長く作用する疼痛薬を規則的スケジュールで投与されてよく、経皮用デクスメデトミジンは、当日中に要求されるようにまたは必要とされる時間毎に投与される。いくつかの場合、別の疼痛処置薬の投薬量は、デクスメデトミジンによって生じる有益な共力作用ゆえに減少させることができ、それが、一次薬理療法を補足する。具体的には、デクスメデトミジンは、オピオイドの有効性を有意に増強して、必要なオピオイド投薬量の減少を可能にし、同時に、同等の治療的有用性を維持することができる。
【0028】
[0033]デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体の鎮痛性経皮用製剤は、好ましくは、計量投薬量で与えられるので、所定量の活性医薬成分を、薬学的有効量で対象に適切に投与される。例えば、経皮用製剤は、個々の用量包装中でまたはマルチ包装単位で、例えば、計量ポンプを装着した密封容器を含むシステム中に多数回用量を含有するバルク容器として与えることができる。典型的に、ヒト患者は、ポンプからの1回またはそれを超える作動の局所自己投与によって処置される。各々のポンプ作動は、一定計量用量を与える。送達の利点は、単一の不連続適用によって必要とされる単回用量で患者を滴定する能力である。この利点は、典型的に、ワン・サイズ・フィッツ・オール・ドーセージ(one-size-fits-all dosage)を標準的な計画で投与する他の形の薬物送達(例えば、パッチ、ロゼンジ、錠剤および坐剤)に欠けている。局所経皮用製剤の追加の利点には、特に、担当医療専門家不在で自己投与する場合のそれらの使用の容易さが含まれる。
【0029】
[0034]ポンプ作用ディスペンサーは、作動のための外圧、例えば、外部の手動、機械的または電気的に開始する圧力の適用を必要とすることを特徴とする。これは、作動が、典型的に、圧力の制御放出によって、例えば、バルブの制御開放によって行われる加圧システム、例えば、噴射剤駆動エアゾルまたは圧縮ガススプレーとは対照的である。特定の態様において、ポンプ製剤は、本明細書中の製剤でのポンプスプレーの使用が、既知量のおよび調節可能なサイズの製剤の投与を可能にするので好適である。他の態様において、加圧噴射剤ガス(例えば、二酸化炭素、窒素、クロロフルオロカーボン、ヒドロフルオロアルカン等)のレザバーを含有する加圧システムは、適する計量分配量を生じることができる。特定の好ましい態様において、送達される製剤の粘度は、更に、皮膚または粘膜のいたるところに一様に広げられることにより、皮膚または粘膜上により濃厚な層で塗られることに対立するものとして表面積の増加を与える(例えば、ゲル剤対クリーム剤)。密度(例えば、クリーム剤対フォーム剤対ゲル剤)は、皮膚または粘膜上の所望のパターンおよび厚みの組成物に寄与することができる。
【0030】
[0035]スプレーポンプデバイスは、予備計量されてよい、または或いは、そのデバイスは、デバイスで計量されてよい。予備計量デバイスは、好ましくは、予め測定された用量または用量画分を、製造中にまたは患者によって使用前にデバイス中に包含されていよいいくつかのタイプのユニット(例えば、単回単位用量の溶液、単回または多数回ブリスターまたは他のキャビティ)で含有する。典型的なデバイスで計量されるユニットは、患者によって行われた時にデバイス自体によって計量されるスプレー剤として送達される多数回用量に十分な製剤を含有するレザバーを有する。そのデバイスは、送達される薬物物質の量(すなわち、1作動当たりの投薬量)で、更には、各投薬間の時間長さでも計量することができる。各投薬間の時間を制限することは、投薬量を患者に何回送達することができるかを制限することによって使いすぎを妨げることができる。
【0031】
[0036]製造要件には、製剤およびいずれかの計量分配機構または経皮送達ビヒクルの再現性が含まれる。これらパラメーターの再現性を有効期限期間中維持し且つデバイスの機能性(例えば、スプレー機構、電子機能、センサー等)を患者使用条件下でその寿命中確保することは、これらパラメーターの何らかの変更が、投与・吸収に変動をもたらすことがありうると考えられ、それが、潜在的副作用および減少した治療的有用性をもたらすことがありうると考えられるので重要である。
【0032】
[0037]いずれかのデバイスによる製剤の投与用量は、容器クロージャーシステムの設計、再現性および性能特性に依存してよい。所望の分配を与える適するデバイスは、デクスメデトミジン製品の正しい性能に重要な因子である。作動パラメーター(例えば、力、速度、保持・戻し時間)も、デバイスに関して考慮されるべきである。更に、デバイスは、製剤成分と相容性であるべきである。更に、デバイスは、患者使用説明書にしたがって用いられる場合に、デクスメデトミジン、その薬学的に許容しうる塩またはその誘導体を含めたデクスメデトミジン製剤の不完全計量、更には、過剰計量を妨げるように設計されるべきである。
【0033】
[0038]典型的な製剤デバイスには、基本ユニット、排出作動器、デバイスから製剤を放出させるオリフィス、およびレザバーが含まれる。好ましくは、レザバーは、患者へ計量分配する前に、例えば、製造現場で、薬物物質および他の賦形剤(例えば、本明細書中の他のところで論及される液状ビヒクル、賦形剤等)で満たされている。レザバーは、好ましくは、作動時に排出される一定測定量のデクスメデトミジン、その薬学的に許容しうる塩またはその誘導体を規定する。レザバー本体は、例えば、プラスチック、ステンレス鋼などの鋼製の透明材料等の円筒形中空品部分によって単純に形成されるいずれかの許容しうる材料であってよいので、その生産は極めて単純である。作動器は、排出を行うオリフィスに関して可動性であり、デバイス上にまたはデバイスと一緒に提供されてよい。作動運動の経過中に、レザバーは、例えば、穿刺によって開いて、オリフィスを介して単回投薬量を投与する。始動位置後の作動工程部分の際に、高圧が発生する。同方向に続行する引き続きの作動運動の一部分において、基剤は、片側で圧力が軽減され且つオリフィスに通じることができる。このような方式で、基剤は、圧力の作用によってレザバーからオリフィスを介して押し進められる。
【0034】
[0039]典型的に、スプレー可能経皮用製剤はいずれも、オリフィスから出るので、液体粒子は軌道にしたがうが、それは、オリフィス形状によって、更には、現れる圧力によって影響される。いくつかの態様において、液体粒子サイズ、スプレー幾何学的形およびスプレーパターンは、ポンプの設計および/または製剤の性状に依存している。特定の態様において、作動器の配向、ポンプ設計および製剤の性状は、スプレー対称性および形状に影響するであろう。スプレーパターンは、より広い通路上に液体粒子を分散させるように最適化することによって、化合物を吸収することができる皮膚上の表面積を増加させることもできる。いずれかこのようなスプレーデバイスは、更に、皮膚の特定領域へ投与されるスプレーの患者使用の容易さおよび配置を促すように設計することができる。
【実施例】
【0035】
[0040]デクスメデトミジンの以下の経皮用製剤を、実施例として製造した。いくつかの典型的な製剤を、本明細書中に記載のように哺乳動物において調べた。
[0041]実施例1a−プラシーボ経皮用ゲル製剤(担体)
[0042]50gのサリチル酸メチル/メントールヒドロアルコール性透明ゲルを製造する典型的な成分および方法。
【0036】
【表1】
【0037】
【表2】
【0038】
[0043]この実施例において、本発明者は、(1)pH=0.592gおよび水添加で沈澱した白色塊状物;および(2)再水和する水または再構成済みゲルの添加も、沈澱物を生じるということに注目した。沈澱作用は、より多くの水を加えるにつれて不溶性であるサリチル酸メチルに起因すると考えられる。或いは、または更に、沈澱は、少なくとも一部分はpH依存性であってよい。
【0039】
[0044]実施例1b−経皮用デクスメデトミジンゲル製剤
【0040】
【表3】
【0041】
【表4】
【0042】
[0045]この典型的な方法において、いくつかの観測を、工程Bで行った(成分番号2のDEXMEDETOMIDINE HClの導入時)。(1)ゲル製剤は、曇り状態になった;および(2)工程Bの得られたゲルは、工程Aのゲルと比較して減少した粘度を有した。曇りに関する観測1に取り組むために、トロラミンを加えたが、それは有効であった。この実施例において、トロラミンは、工程Cより得られた32.532gのゲルに0.463gのトロラミン(ロットXT0007と確認される)の比率で加えた。
【0043】
[0046]実施例2、3および4−Dex HCl局所用クリーム製剤
[0047]基本プラシーボクリーム製剤を用いて、本明細書中に更に記載の活性デクスメデトミジンHCl経皮用製剤を製造した。
【0044】
[0048]実施例2a−プラシーボO/Wクリーム基本製剤(担体)
【0045】
【表5】
【0046】
【表6】
【0047】
[0049]実施例2b−経皮用デクスメデトミジンクリーム製剤
【0048】
【表7】
【0049】
【表8】
【0050】
[0050]実施例3a−プラシーボO/Wクリーム基本製剤(担体)
【0051】
【表9】
【0052】
【表10】
【0053】
[0051]実施例3b−経皮用デクスメデトミジンクリーム製剤
【0054】
【表11】
【0055】
【表12】
【0056】
[0052]実施例4a−プラシーボO/Wクリーム基本製剤(担体)
【0057】
【表13】
【0058】
【表14】
【0059】
[0053]実施例4b−経皮用デクスメデトミジンクリーム製剤
【0060】
【表15】
【0061】
【表16】
【0062】
[0054]実施例5a−プラシーボO/Wシリコーン基本製剤(担体)
【0063】
【表17】
【0064】
【表18】
【0065】
[0055]実施例5b−経皮用デクスメデトミジンクリーム製剤
【0066】
【表19】
【0067】
【表20】
【0068】
[0056]本明細書中のいくつかの典型的な製剤を、イヌに投与して、デクスメデトミジンの局所経皮送達組成物としての吸収および他の性能特性を決定した。調べたそれら組成物の内、本明細書中で Recro 1−85として確認された組成物は、デクスメデトミジンの望ましい全身レベルを生じた。それら製剤は、少なくとも、イヌの疼痛および関連状態の処置に用いるのに適するし、そして更に、ヒトを含めた他の哺乳動物に用いるのに適するかもしれない。前述の実施例は、単に代表するものであり、本明細書中の発明の範囲を制限するものではない。いくつかの典型的な局所経皮用製剤を、動物において調べたが、開発したおよび/または調べたその他には、特に、アルコール性ゲル剤、油・水エマルジョンクリーム剤が含まれた。関係のある動物試験について関係のある結果を、本明細書中に要約する。
【0069】
臨床例
臨床研究期間1(REC−10−006)
[0057]動物における経皮用製剤に関する成功した実験に基づいて、ヒトにおける試験および試行を、いくつかの製剤および投与方法および処置方法について開発し且つ実施した。ヒト用製剤は、典型的な動物研究用製剤から誘導したが、cGMP必要条件およびヒト使用のための他の規制必要条件にしたがって製造した。本明細書中の局所経皮用製剤および方法のいくつかの典型的な態様を、ヒトにおいて早期臨床試験(Phase I研究としても知られる)で利用した。それら臨床試験においてこれまでに用いた典型的な製剤は、次の通りである。
【0070】
【表21】
【0071】
【表22】
【0072】
[0058]ヒトの場合の疼痛管理。Joint Commission が、疼痛の評価・管理の基準を改訂した2000年以来、疼痛の処置は、医療においてますます有意の位置を占めてきた。しばしば、「第五生命徴候」と称され、対象は、現在、疼痛症状について常套的に評価する必要があるので、療法は適切に調整することができる。この増加した関心の的は、患者の快適さおよび生活の質の問題に一層注意を向けさせたが、利用可能な手段の範囲は、概ね同じ状態のままであった。現行薬剤は、軽減を1〜2時間与える急性薬剤〜72時間程度の痛覚脱失を与えることができる別の製剤まで、活性持続期間が様々である。同時に、これら剤形は、一般的に、同様の活性成分セットに頼っていて、それらは、しばしば、同じオピオイド経路を介して作用して軽減を与える(モルヒネ、オキシコドン、フェンタニール)。その結果は、概して、アヘン薬によって与えられる作用、高用量および多数回アヘン薬の使用に上限は存在しないが、症状と副作用との間で決断を強いることがありうる有害イベントの発生増加をもたらすことがありうる。
【0073】
[0059]デクスメデトミジンは、クロニジンの約10倍の選択性を有する選択的α2−アドレナリン受容体アゴニストである。デクスメデトミジンは、急性および外科的設定においてその鎮静性を利用した広範囲の安全な静脈内使用歴を有し、そして Precedex(登録商標)という商品名での使用が示されている。より低い用量で、デクスメデトミジンは、現在、呼吸抑制のリスクを伴うことなく、鎮痛および抗不安の利点を与えることが認められている。
【0074】
[0060]デクスメデトミジンを含む経皮用剤形の使用は、延長した薬物摂取について非侵襲性機構を与える。この延長した摂取は、慢性疼痛患者への潜在的利点を伴って、長期の作用持続期間を促す。デクスメデトミジンは、追加の鎮静または呼吸抑制を伴うことなく、疼痛を抑制する可能性を有する。適切な使用は、患者の不快感を制限させると同時に、日常の活動を行う能力を維持するはずである。
【0075】
[0061]本明細書中に記載の典型的な経皮用製剤の最初のヒト使用は、本明細書中においてDEX−TD.02およびDEX−TD.03として確認された二つの経皮用デクスメデトミジン製剤の安全性、耐容性および薬物動態学的性質を評価するように設計した。薬物動態学的観測は、ヒト皮膚を越えるデクスメデトミジンの吸収および上昇用量の影響を評価することに集中するであろう。
【0076】
[0062]研究目的。ヒト研究の目的は、健康な男性および女性対象における単回上昇用量の経皮用デクスメデトミジンの安全性、耐容性および薬物動態を評価することであった。
[0063]調査計画。全体の研究設計は、健康対象において、二つの経皮用デクスメデトミジン製剤、すなわち、DEX−TD.02およびDEX−TD.03の安全性、耐容性および薬物動態を研究するためのオープンラベル、単回用量、用量上昇、クロスオーバー研究 Phase 1であった。18歳〜50歳までの年齢の健康対象の参加について、オーストラリアの一研究地において、研究用薬物投与前28日以内にスクリーニングした。病歴、身体検査、ベースライン実験室検査、12リード心電図(ECG)、妊娠検査、生命徴候測定およびインフォームドコンセントは、スクリーニング来診中に終えた。期間1において投与する前に、研究参加者を、二つの研究処置配列順の一方へ無作為に割り当てた(表Mを参照されたい)。各々の参加者に、割り当てられた処置配列順にしたがって、各々の研究期間中に単回用量の研究用薬剤を与えた。投与期間は、前の期間における血漿薬物濃度の薬物動態学的分析を可能にする十分な空白時間によって隔てられた(約14日)。
【0077】
【表23】
【0078】
[0064]各々の期間中、対象は、投与前の夕方(1日目)〜投与後約48時間(3日目)拘束されることになった。対象には、投与前の朝にシャワーを浴びることを要求し、そして投与後48時間はシャワーを浴びることもその部位を洗うことも禁止した。拘束はされるが、対象には、標準化した日常食を与えた。表Nは、この研究における薬剤、適用用量および局所投与部位を示している。
【0079】
【表24】
【0080】
[0065]薬物動態学的分析のために、16人の血液試料を、各々の期間中に各々の対象から単回用量投与後に集めた。採集は、研究用薬剤を投与前(0時)についておよび投与後の以下のおよその時点に的を絞った。30分、60分および90分、そして2時間、3時間、4時間、6時間、8時間、10時間、12時間、18時間、24時間、30時間、36時間および48時間。投与後時間は、対象に用量を与えた時点に開始した。安全評価には、AEの監視、臨床実験室検査、生命徴候測定、身体検査、刺激評価およびECGが含まれた。
【0081】
[0066]研究設計および対照群の原理。この研究は、別の送達経路によって投与される既知の薬物物質の安全性、耐容性および薬物動態学的性質を評価した。本発明者および出願人による以前の調査は、静脈内および舌下経路によって投与時のデクスメデトミジンの安全性を示した。
【0082】
[0067]本明細書中の前に記載のように、本発明者および出願人による先行研究は、動物モデル(イヌおよびブタ)においてDEX−TD製剤を用いて行って、健康なヒト対象におけるこの初期投与を支持した。この研究に用いられた用量上昇モデルは、DEX−TD.02およびDEX−TD.03の吸収性を、ヒト皮膚に適用時に、適用用量に依存してそれらがどのように変動することがありうるかを含めて確認し且つ評価することであった。クロスオーバー設計の目的は、これら二つの製剤の吸収を同じ組の対象において比較することであった。一方の対象部分群では、経皮用製剤を、皮膚部位に投与し、そして直ちに包帯剤で被覆した(「閉塞した」)。用いられた包帯剤は、TEGADERMTM商標包帯剤であった(TEGADERMTMは、3M Corporation の登録商標である)。もう一方の対象部分群では、経皮用製剤を皮膚に投与し、そして未被覆状態にした(「非閉塞とした」)。閉塞される投与で十分に確定された経験に基づいて、本発明者は、経皮処方皮膚適用部位(腹部)上に包帯剤を適用された対象において、吸収が一層高いと考えられると期待した。しかしながら、驚くべきことに、DEX−TD.02の吸収は、皮膚適用部位上に包帯剤が適用されなかった部分群について、有意に一層高かった。更に、それら結果は、閉塞が必要でも望ましくもないということを示し、少なくともDEX−TD.02の組成物を経皮用の真正「パッチレス(patchless)」にする(すなわち、無包帯剤、パッチが製剤を含有する必要がない)。その製剤は、皮膚に適用時に安定であり、しかも包帯剤または他の被覆剤不要で容易に吸収される。この研究の薬物動態学的関心の的ゆえに、対照集団は不必要であった。
【0083】
[0068]期間1における薬物動態学的結果の再検討にしたがって、臨床研究期間2の際に製剤DEX−TD.02を投与することだけを決定した。これは、一部分は、DEX−TD.03による見掛けの一層低い吸収のためであった。この研究は、デクスメデトミジンの全身吸収を与えるために高吸収を追求したので、DEX−TD.02は、この研究における追加の臨床検査のために一層適切であった。しかしながら、DEX−TD.03は、その一層低い吸収速度および極めて低い全身吸収を考えると、局部的皮膚および/または硬膜下ニューロパシーの処置などの他の疼痛管理用途により一層適切でありうる。特に、研究期間1においてどちらかの製剤で処置されたいずれの対象についても、鎮静が観測されたまたは報告されたものはなかった。
【0084】
臨床研究期間2
[0069]目的。臨床研究期間2の目的は、本出願人の単回上昇用量研究(期間1、(REC−10−006))より得られたデータを用いて、0.5グラム用量のデクスメデトミジンの製剤DEX−TD.02の1日1回および1日2回局所適用によって生じる定常状態血漿濃度−時間プロフィールを計算することであった。
【0085】
[0070]研究設計。臨床研究#REC−10−006によるデクスメデトミジン血漿濃度−時間データを、0.25グラム、0.5グラムおよび1グラム用量の1日1回(QD)および1日2回(BID)局所適用によって生じる定常状態濃度−時間プロフィールを予測する場合に用いた。次の3対象、すなわち、1001、1004および1005からのデータを用いた。それら三対象は、以下の処置部門、すなわち、処置A(0.25グラム用量)、処置C(0.5グラム用量)および処置E(1.0グラム用量)に参加した。臨床研究#REC−10−006は、多数の処置部門(処置A、B、C、DおよびE)を含む単回上昇用量研究であった。薬物分析用の血漿試料は、投与前(0時)と、投与後の以下の規定時間、すなわち、0.5時間、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、10時間、12時間、18時間、24時間、30時間、36時間および48時間に採取した。生物学的分析の定量化の低い方の限界は、0.02ng/mLであった。特に、研究期間2においてどちらかの製剤で処置されたいずれの対象についても、鎮静が観測されたまたは報告されたものはなかった。
【0086】
[0071]方法および分析。薬物動態学的パラメーターは全て、区画分析を用いて分析した。期間1(REC−10−006)によるデクスメデトミジン血漿濃度−時間データを、0.25グラム、0.5グラムおよび1.0グラム用量の1日1回(QD)および1日2回(BID)局所適用によって生じる定常状態濃度−時間プロフィールを予測する場合に用いた。用いられたデータは、処置部門A(0.25グラム用量)、C(0.5グラム用量)およびE(1.0グラム用量)に参加した三対象(識別名1001、1004および1005)からであった。薬物分析用の血液試料は、投与前(0時)と、投与後の以下の規定時間、すなわち、0.25時間、0.5時間、0.75時間、1時間、1.5時間、2時間、2.5時間、3時間、4時間、5時間、6時間、7時間、8時間、12時間、24時間、48時間および72時間に採取した。血漿濃度の主要部分(67%)は、0.02ng/mLの定量可能レベル未満であった;したがって、PKパラメーターは、非線形混合効果モデルのより精巧なコンピューター法を用いて計算した(すなわち、集団薬物動態[PopPK])。非線形混合効果モデルは、Monolix, Version 3.1ソフトウェア(Institut National de Recherche en Informatique et en Automatique[INRIA])を用いて行った。いったんPKパラメーターを計算し且つ単回用量濃度を作成したら、データを、WinNonlinTM,Version 5.2(WinNonlinTM Copyright c.2008, Pharsight Corporation)にインポートし、非パラメーター重ね合わせを行って、QDおよびBID投与によって生じる定常状態血漿濃度−時間プロフィールを予測した。
【0087】
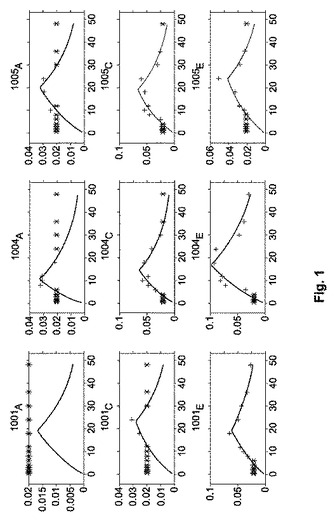
[0072]単回用量データから薬物動態学的パラメーターの計算。前述のように、PKパラメーターは、MonoLix 3.1ソフトウェアを用いた非線形混合効果モデルを用いて計算した。処置部門A(0.25グラム用量)、C(0.5グラム用量)およびE(1.0グラム用量)に参加した三対象(1001、1004および1005)からの血漿デクスメデトミジン濃度を、零次吸収を含む線形一区画モデルに適合させた。適合された濃度および観測された濃度を、図1に示す。図1における注記:番号は、対象識別名である(例えば、1001、1004または1005);下付き文字(例えば、A、C、E)は、処置、すなわち、A(0.25グラム用量)、C(0.5グラム用量)またはE(1.0グラム用量)である;プラス記号(+)は、定量化の限界より上の測定可能デクスメデトミジン濃度である;そして星印(*)記号は、定量化限界未満のデクスメデトミジン濃度(0.02ng/mL)である;そして実線は、最も適合したPK曲線を構成している。適合されたおよび観測された濃度−時間プロフィール(図1)の調査は、零次吸収を含む一区画(実線)が、定量可能なデクスメデトミジン濃度(プラス記号)を双方とも十分に記載し且つ定量可能限界未満(BQL)(星印記号)であると考えられたそれら試料と一致するということを示している。
【0088】
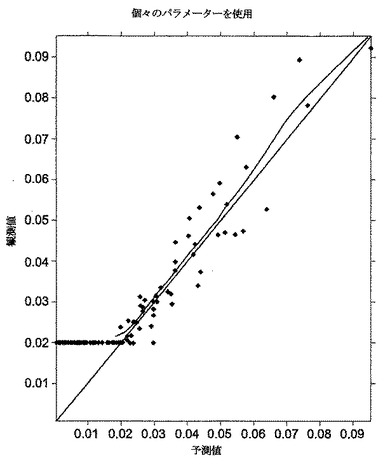
[0073]全体的に、図2に示されるように、対象投与によるモデル値と観測値との間には十分な一致が存在した。図2は、(モデルからの)予測値が、予測値に等しい観測値についての理論線に近いばらつきによって示されるように観測値とほぼ一致するということ、および大部分のBQL値が、モデルからの値について一定であるということを示している。図2についての注記:菱形は、定量化限界より上の測定可能な濃度である;星印は、定量化限界未満の測定可能な濃度(0.02ng/mL)である;直線は、観測濃度=予測濃度の場合の一致線である;そして曲線は、プロットポイントの傾向線である。
【0089】
[0074]適合により得られた薬物動態学的パラメーターを、表P1に示す。デクスメデトミジンの皮膚適用は、約15時間の半減期で約20時間にわたって(一定注入と同様の)一定速度で吸収されると考えられた。約2.2xおよび1.5xの累積倍率は、BIDおよびQD投与についてそれぞれ計算した。
【0090】
【表25】
【0091】
[0075]BIDおよびQD双方の投与によって生じる定常状態濃度の計算。前述のようにい、いったんPKパラメーターを計算し且つ単回用量濃度を作成したら、データを、WinNonlinTM,Version 5.2(WinNonlinTM Copyright c.2008, Pharsight Corporation)にインポートした。非パラメーター重ね合わせを行って、QDおよびBID投与によって生じる定常状態血漿濃度−時間プロフィールを予測した。非パラメーター重ね合わせは、各々の用量の薬物が、用量毎に独立して作用するということ、および吸収の速度および程度および平均全身クリアランスが、各々の投与間隔について同じであるということを仮定している。線形薬物動態は、多数回投与計画の際の用量の変化に適応しうるように適用する。
【0092】
[0076]多数回用量によって生じる薬物濃度を予測するために、単回用量投与後の濃度−時間プロフィールの完全な特性決定が必要である。すなわち、薬物吸収・排除過程を特性決定するのに十分な時点ti(i=1,2,...,n)でC(ti)を知ることが必要がある。データについて次の二つの仮定条件、すなわち、各々の用量作用の独立および基礎にある薬物動態の線形性が必要である。前者は、各々の用量の作用が、他の用量の作用と分離することができると仮定している。後者、すなわち、線形薬物動態は、薬物濃度の変化が、用量で線形変動するであろうと仮定している。
【0093】
[0077]0.25(txA)、0.5(txC)および1.0(txE)グラムの1日1回用量後の定常状態での予測血漿濃度−時間曲線を、図3に示す。1日2回投与後の予測定常状態血漿濃度−時間曲線を、図4に示す。双方の投与計画(QDおよびBID)について、定常状態で得られた血漿濃度−時間曲線は、かなり平坦である。これは、真皮から体循環への薬物の長期零次拡散および長い半減期のためである。BIDおよびQD投与計画の予測定常状態血漿濃度を、それぞれ、表Q2および表R3に挙げる。
【0094】
【表26】
【0095】
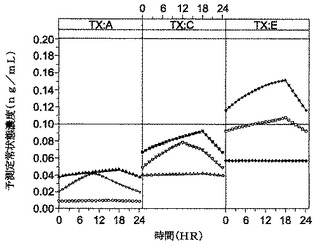
【表27】
【0096】
[0078]期間2の結果および結論。図3および図4は、DEX−TD.02を1日1回および1日2回用量計画で用いた臨床研究に基づく成人における時間経過での予測定常状態血漿濃度を示す。この研究において鎮静を伴うことなく鎮痛薬として用いるためのデクスメデトミジンの標的定常状態濃度は、0.1〜0.2ng/mLの範囲内であるということを想起されたい。したがって、図3および図4が明らかに示すように、これは、(その範囲の下方部に達するであろう)0.5グラム用量かまたは(上方範囲の濃度を生じるであろう)1.0グラム用量のBID投与で達成することができる。この実施例において、所望の定常状態濃度は、多数回投与の4日目またはその後に生じると予測されるであろう。このモデルに基づき、DEX−TD.02およびDEX−TD.03を用いた1日1回投与は、0.5グラム用量でも1.0グラム用量でも、標的範囲(0.1〜0.2ng/mL)に達する定常状態血漿濃度を速やかに生じなかったので、1日1回投与はチャレンジのままである。再度、DEX−TD.02または類似の製剤を用いると、定常状態濃度は、多数回投与の少なくとも4日目までには達するであろう。しかしながら、本明細書中のおよび本発明者が考える知見および情報では、これらおよび他の局所用製剤は、より短い時間内であれ、別の投与計画によってであれ、哺乳動物において経皮吸収し且つデクスメデトミジンの所望の標的定常状態血漿濃度を達成することを示すことができる。例えば、所望の定常状態への投与は、本明細書中に記載の持続した、制御された、長期の放出・吸収デクスメデトミジン経皮用製剤との組み合わせで、急速吸収性舌下用、経粘膜用および/または急速吸収性経皮用の組成物の組み合わせ(例えば、DMSOなどの急速吸収性溶媒担体、アルコールおよび他の薬学的に相容性の担体および製剤を含めた)を用いて達成することができる。
【0097】
[0079]この説明は、典型的な態様に関して行っているが、いろいろな変更を行うことができるし、そしてその範囲から逸脱することなく、均等物をそれらの要素の代わりに置き換えることができるということは当業者によって理解されるであろう。更に、本質的な範囲から逸脱することなく、特定の状況および材料をそれらの内容に適応するように多くの修飾を行うことができる。更に、説明には、典型的な態様を開示してきており、具体的な用語を用いてきたかもしれないが、それらは、特に断らない限り、単に包括的および説明的意味で用いられ、制限するためではなく、したがって、請求の範囲の範囲は、そのように制限されることはない。更に、当業者は、本明細書中で論じられている方法の特定の工程を、別の順序で配列することができるまたは工程を組み合わせることができるということを理解するであろう。したがって、請求の範囲は、本明細書中に開示の具体的な態様に制限されるものではないということである。
【発明の詳細な説明】
【0001】
B.関連出願へのクロス・リファレンス
[0001]本出願は、本明細書中にそのまま援用される2010年1月8日出願の米国仮特許出願第61/293,440号の優先権を主張する。
C−E.不適用
【背景技術】
【0002】
F.背景技術
[0002]デクスメデトミジン(dexmedetomidine)、すなわち、5−[(1S)−1−(2,3−ジメチルフェニル)エチル]−1H−イミダゾールは、鎮静性および鎮痛性を有する非麻薬性α2−アドレナリン受容体アゴニストである。
【0003】
【化1】
【発明の概要】
【発明が解決しようとする課題】
【0004】
[0003]現在のところ、デクスメデトミジンは、鎮静に必要とされる注射可能製剤として商業的に入手可能であるにすぎないし、そしてそれは、医療専門家によって静脈内投与される必要がある。デクスメデトミジンは、鎮痛性を有するが、しかしながら、鎮痛薬として有用な製剤は、商業的に入手可能ではない。更に、いろいろな理由のために、商業的に入手可能な注射可能製剤は、自己投与することができる鎮痛薬として用いるのに不適当である。例えば、自己投与されて、鎮静を伴うことなく痛覚脱失を生じる(またはそれ以外には、疼痛を処置するまたは予防する)ことができるデクスメデトミジン基剤鎮痛薬について、継続した且つ未だ満たされていない要求が存在する。
【課題を解決するための手段】
【0005】
G.概要
[0004]本出願は、デクスメデトミジン、その薬学的に許容しうる塩およびその誘導体の鎮痛性経皮用製剤、更には、それらの使用方法を記載する。
【0006】
[0005]本明細書中に提供されるのは、デクスメデトミジンおよび/またはその薬学的に許容しうる塩および/またはその誘導体の新しい鎮痛性経皮用製剤、および疼痛の処置または予防におけるそれらの使用方法である。このような医薬組成物は、痛覚脱失を生じる(例えば、疼痛を処置するまたは予防する)十分な量などのデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体(例えば、プロドラッグ)および薬学的に許容しうる経皮送達ビヒクル、更には、粘度調整剤、pH調整剤、保存剤、賦形剤、乳化剤、緩衝剤、着色剤等などであるがこれに制限されるわけではない任意の追加成分を包含する。薬学的に許容しうる経皮用ビヒクル中のデクスメデトミジンまたはその薬学的に許容しうる塩またはその誘導体(例えば、プロドラッグ)は、適する容器またはデバイス中に包装されていてよい。
【0007】
[0006]典型的な態様において、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を哺乳動物に投与する方法は、哺乳動物の皮膚へ、所定量の経皮用組成物であって、哺乳動物の皮膚を介する哺乳動物の体循環系中への薬学的有効量のデクスメデトミジンの経皮吸収を与える有効量で薬学的に許容しうる経皮送達ビヒクル中にデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を含む組成物を適用することを包含する。経皮吸収時に、デクスメデトミジンは、哺乳動物に、適当に投与された場合に鎮静を伴うことなく痛覚脱失を生じる。
【0008】
[0007]追加の特徴は、以下の詳細な説明および実施例を参照して理解することができる。
H.図面の簡単な説明
【図面の簡単な説明】
【0009】
【図1】[0008]図1は、ヒトにおいて本明細書の典型的な製剤を用いた1日1回用量後の定常状態における観測対予測血漿濃度−時間曲線をグラフで示す。
【図2】[0009]図2は、ヒトにおいて本明細書の典型的な製剤を用いた1日2回投与後の観測対予測定常状態血漿濃度−時間曲線をグラフで示す。
【図3】[0010]図3は、本明細書の典型的な製剤を用いて1日1回複数日投与しているヒトの予測結果をグラフで示す。
【図4】[0011]図4は、本明細書の典型的な製剤を用いて1日2回複数日投与しているヒトの予測結果をグラフで示す。
【図5】[0012]図5は、イヌにおいて処方1〜85を用いた投与結果をグラフで示す。
【図6】[0013]図6は、イヌにおいて処方1〜53を用いた投与結果をグラフで示す。
【図7】[0014]図7は、イヌにおいて処方1〜83を用いた投与結果をグラフで示す。
【発明を実施するための形態】
【0010】
I.詳細な説明
[0015]本明細書中において提供されるのは、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体の新しい鎮痛性経皮用製剤、および疼痛の処置および管理におけるそれらの使用方法である。
【0011】
[0016]デクスメデトミジンは、哺乳動物において鎮静、感覚脱失および痛覚脱失を引き起こす特異的α2−アドレナリン受容体アゴニストである。ヒトの場合、デクスメデトミジンは、集中治療設定で処置中に最初に挿管され且つ人工呼吸器で処置されている患者の鎮静用に、更には、外科手術および他の手順の前またはその間の挿管されていない患者の鎮静用に商業的に入手可能である。例えば、米国特許第6,716,867号および第6,313,311号を参照されたい。
【0012】
[0017]ヒトにおけるデクスメデトミジンの薬物動態は、静脈内(i.v.)、筋肉内(i.m.)および経皮投与後に研究された。平均除去半減期は、それぞれ、i.v.およびi.m.投与後1.5〜3時間および経皮投与後5.6時間である。i.m.および経皮投与後、最大血中濃度への時間は、それぞれ、1.6〜1.7時間および6時間であり、そして絶対バイオアベイラビリティーは、それぞれ、73%および88%であると推定された。例えば、“Pharmacodynamics and pharmacokinetics of intramuscular dexmedetomidine,” Scheinin et al., Clin. Pharmacol. Ther. 52, 53-46 (1992); “The pharmacokinetics and hemodynamic effects of intravenous and intramuscular dexmedetomidine hydrochloride in adult human volunteers,” Dyck et al., Anesthesiology 78, 813-20 (1993); および “Pharmacokinetics and pharmacodynamics of transdermal dexmedetomidine,”Kivistoe et al., Eur. J. Clin. Pharmacol. 46, 345-49 (1994) を参照されたい。
【0013】
[0018]デクスメデトミジンは、口腔などの他の組織から吸収されることも知られている。ヒト対象が、デクスメデトミジン溶液を嚥下することなく口中に保持する口腔内投与後、平均口腔内バイオアベイラビリティーは、81.8%において、約1.5時間で最大濃度および1.9時間の見掛除去半減期で測定された。例えば、“Bioavailability of dexmedetomidine after extravascular doses in healthy subjects,” Anttila et al., Br. J. Clin. Pharmacol. 56, 691-93 (2003) を参照されたい。
【0014】
[0019]本発明により、デクスメデトミジンは、疼痛を改善する、管理する、治癒する、予防するまたはそれ以外に処置する目的で動物またはヒト対象に投与することができる。典型的な態様において、デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を哺乳動物に投与する方法は、哺乳動物の皮膚へ、所定の投薬量の経皮用組成物であって、哺乳動物の皮膚を介する哺乳動物の体循環系中への薬学的有効量のデクスメデトミジンの吸収を与える有効量で薬学的に許容しうる経皮用ビヒクル中にデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体を含む組成物を適用することを包含する。経皮吸収時に、デクスメデトミジンは、哺乳動物に痛覚脱失を生じる。
【0015】
[0020]例えば、癌および他の不快を有する多くの患者は、長期にわたる鎮痛療法にもかかわらず、中程度〜重症の疼痛を経験し続けるが、これは、しばしば、患者の活動度レベルの増加ゆえに、間欠性破綻疼痛(intermittent breakthrough pain)として起こりうる。鎮痛薬の長時間作用性製剤の用量を増加させることによってこのタイプの疼痛に対抗する試みは、しばしば、オピオイド鎮痛薬で特に、痛覚脱失の遅い開始、および鎮静、便秘または吐き気および嘔吐の望ましくない副作用を生じる。しかしながら、本明細書中に記載のデクスメデトミジンの鎮痛性経皮用製剤は、強力な非麻薬性鎮痛作用、および好ましくは、持続した長期の制御された鎮痛作用、およびそれらの組み合わせを生じて、疼痛を改善する、管理する、治癒する、予防するまたはそれ以外に処置する。処置には、経皮用デクスメデトミジン製剤の単独の使用、または他のデクスメデトミジン製剤(例えば、舌下用、経粘膜用、注射可能および/または他の鎮痛性組成物(例えば、NSAIDS、麻薬等))との組み合わせでの使用が含まれてよい。
【0016】
[0021]本明細書中に記載のデクスメデトミジン製品は、疼痛の処置のための経皮用医薬製剤である。本明細書中で用いられる「薬学的に許容しうる」という用語は、それら化合物、材料、組成物、剤形、包装および使用方法であって、妥当な医学的判断の範囲内で、しかも過度の毒性、刺激、アレルギー性応答または他の問題または合併症を伴うことなくヒトおよび動物の組織と接触した状態での使用に適すると同時に、適度な受益性/危険性比率に相応し且つ所望の薬理応答を引き出すものを包含する。
【0017】
[0022]デクスメデトミジンは、薬学的に許容しうる酸で薬学的に許容しうる塩を形成可能な基本的窒素原子を含有する。この点での「薬学的に許容しうる塩」という用語は、デクスメデトミジンの比較的無毒性の無機および有機酸付加塩を意味する。これら塩は、デクスメデトミジンの最終単離・精製の際に現場で、またはその遊離塩基形の精製済みデクスメデトミジンを適する有機または無機酸と別個に反応させた後、このようにして形成された塩を単離することによって製造することができる。更に、その塩は、経皮用製剤を製造する製造プロセス中に形成することができる。代表的な薬学的に許容しうる塩には、ハロゲン化水素酸塩(臭化水素酸塩および塩酸塩を含めた)、硫酸塩、重硫酸塩、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、リン酸塩、トシレート、クエン酸塩、マレイン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナプチレート(napthylate)、メシレート、グルコヘプトネート、ラクトビオン酸塩、2−ヒドロキシエチルスルホン酸塩およびラウリルスルホン酸塩等が含まれる。例えば、“Pharmaceutical Salts,” Berge et al., J. Pharm. Sci. 66, 1-99 (1977) を参照されたい。デクスメデトミジン塩酸塩は、薬学的に許容しうる塩の一例である。デクスメデトミジン塩酸塩の使用は、いくつかの場合、その塩酸塩が、より大きい水への溶解度および周囲酸素による酸化に対する安定性を有するので、本明細書中に記載の経皮用製剤中でのデクスメデトミジン自体の使用に好適であるかもしれない。
【0018】
[0023]デクスメデトミジン誘導体は、プロドラッグを作る共有結合修飾を包含してよい。投与時に、プロドラッグ誘導体は、哺乳動物により、デクスメデトミジンを生じる化学修飾を受ける。プロドラッグは、デクスメデトミジンの生体内分布または薬物動態を好都合に変更するまたは他の望ましい特性を生じるために用いることができる。例えば、デクスメデトミジンの反応性窒素は、酵素的にまたは非酵素的に、還元的に、酸化的にまたは加水分解によって開裂して、活性な医薬成分を現す官能基で誘導体化することができる。特定のタイプのプロドラッグの使用は知られている(例えば、R.B. Silverman, 1992, “The Organic Chemistry of Drug Design and Drug Action,” Academic Press, Chp. 8 を参照されたい)。例えば、プロドラッグは、それら化合物の最終単離・精製の際にその場で、またはその遊離塩基形の精製済み化合物を適する誘導体化剤と別個に反応させることによって製造することができる。
【0019】
[0024]デクスメデトミジン経皮用組成物は、ゲル剤、クリーム剤または液剤などの一つまたはそれを超える薬学的に許容しうる担体を30重量%〜約99.995重量%包含する。これら担体は、デクスメデトミジンまたはその薬学的に許容しうる塩またはその誘導体の溶媒、補助溶媒または非溶媒であってよい。適する材料は、室温で液状物または流動性組成物であり、しかも室温で、好ましくは、周囲圧力でも、更には、高圧下でも、その状態のままである。有用な液状物および流動性組成物は、特に制限されないが、但し、それらは、それら組成物の所望の医学的使用を妨げることがないし、しかもそれらは、治療的に有用な量のデクスメデトミジンまたはその薬学的に許容しうる塩または誘導体(例えば、デクスメデトミジン塩酸塩)を有するという条件付きである。薬学的に許容しうる液状物の例には、水、エタノール、ジメチルスルホキシド、プロピレングリコール、ポリエチレングリコール、プロピレンカーボネート、薬学的に許容しうる油状物(例えば、ダイズ、ヒマワリ、ラッカセイ等)等が含まれる。流動性組成物の例には、クリーム剤、ゲル剤、懸濁剤、ローション剤、血清剤、軟膏剤等が含まれる。薬学的に許容しうる液状物または流動性組成物は、活性な医薬成分を溶解させるか、それの安定な均一懸濁液を生じるかまたは、懸濁液、溶液、混合物または他の薬学的に許容しうる組成物のいずれかの組み合わせを形成するように選択される。
【0020】
[0025]前述の構成成分に加えて、デクスメデトミジンの経皮用製剤は、薬理学的に活性な薬物以外の一つまたはそれを超える賦形剤を包含してよく、それらは、製造プロセス中に包含されるまたは完成医薬製品剤形中に含有される。賦形剤の例には、粘度調節用材料(例えば、ポリマー、糖、糖アルコール、ガム、クレー、シリケート等(例えば、ポリビニルピロリドン(PVP)))(約0.01重量%〜約65重量%)が含まれる。賦形剤の他の例には、保存剤(例えば、エタノール、ベンジルアルコール、プロピルパラベンおよびメチルパラベン)(約0.001重量%〜約20重量%)が含まれる。賦形剤の更に他の例には、緩衝剤およびpH調整剤(例えば、水酸化ナトリウム、シトレートおよびクエン酸)(約0.01重量%〜約5重量%)が含まれる。着色剤(約0.001重量%〜約5重量%)、芳香剤(約0.001重量%〜約1重量%)、キレート化剤(例えば、EDTA)(約0.001重量%〜約1重量%)、UV吸収剤(約0.001重量%〜約10重量%)は、特に、適する賦形剤の追加例である。適する経皮用製剤は、本明細書中に更に記載の実施例において更に詳述する。
【0021】
[0026]更に、このような製剤を製造する方法および使用する方法は、本明細書中の内容により当業者に明らかであろう。例えば、経皮用デクスメデトミジン製剤は、標準的な認められた十分な製造実施にしたがって本明細書中に記載の成分の適当量を混合することによって製造することができる。このような賦形剤は、患者または対象の受容を改善する、バイオアベイラビリティーを改善する、貯蔵寿命を増加させる、製造費用および包装費用を減少させる、政府規制機関の必要条件に応じるためにおよび他の目的のために製剤中に包含されてよい。各々の成分の相対量は、得られた製剤の望ましい薬理学的および薬物動態学的性質を妨げるべきではない。
【0022】
[0027]本明細書中に記載のデクスメデトミジンの鎮痛性経皮用製剤は、哺乳動物の皮膚および/または粘膜(例えば、口、鼻、直腸または生殖器粘膜)への直接投与を予定している。薬物送達は、嚥下後胃腸管吸収に対立するものとして、実質的に、経皮経路によって行われる。「経皮」という用語は、皮膚膜を越えたまたは介する送達を意味する。「経粘膜」という用語は、粘膜を越えたまたは介する送達を意味する。具体的に、薬物の「経口経粘膜」送達には、口、咽頭、喉頭、気管または上部消化管のいずれかの組織、特に、舌下、頬側、歯肉および口蓋の粘膜組織を越えた送達が含まれる。
【0023】
[0028]経皮送達は、体血液循環への薬物物質到達を可能にし、それによって、胃腸管影響力とは無関係の直接全身投与が得られ且つ望ましくない初回通過肝代謝を免れる。他の投与経路と比較して、本製剤中のデクスメデトミジンの経皮吸収は、複数日にわたる投与でも、望ましいバイオアベイラビリティーで有意に一層制御可能な且つ長期の持続時間を有することができる。更に、初回通過代謝は回避されるので、製剤中の活性な医薬成分の全量を減少させることができ、それによって、有害な副作用(例えば、低血圧、鎮静)の可能性を減少させ且つ製造業者に原価利益を与える。
【0024】
[0029]経皮用製剤は、ヒト、更には、ヒトの伴侶動物(例えば、ネコ、イヌ)、農業用家畜およびそれを必要としている他の動物を含めた哺乳動物に投与することができる。錠剤、カプセル剤、シロップ剤等などの慣用的な剤形または注射可能鎮痛性製剤を非ヒト動物へ投与することは、しばしば、問題があるということは理解されるであろうが、本明細書中に記載の経皮用製剤は、このような動物の処置に特に有用である。経皮送達の使用は、幼児および高齢者において他の送達方法にまさる同じ利点を有する。
【0025】
[0030]「痛覚脱失」は、疼痛感覚の軽減または除去である。本明細書中で用いられる「疼痛」は、広範囲の臨床症状を包含し、しかもそれは、幅広い意味を有する。疼痛知覚は、極めて主観的であり、いろいろな人が、様々に且つ極めて異なった強さで疼痛を経験する。国際疼痛研究会(the International Association for the Study of Pain)は、疼痛を、「実際的または潜在的組織損傷に関連したまたはこのような損傷に関して説明される不快な感覚的・情動的経験」と定義している。より簡単に云うと、疼痛には、苦痛を引き起こす且つ自分自身の身体の不快な意識に関連しているいずれかの感覚的経験が含まれる。疼痛の非制限的タイプおよび原因には、神経痛、筋肉痛、痛覚過敏、ピペルパシー、神経炎およびニューロパシーが含まれる。疼痛は、しばしば、癌または関節炎などの根源的な生理学的異常の症状である。片頭痛などのいくつかのタイプの疼痛には、明確に識別される原因がない。疼痛は、熱傷または外科手術などの身体的外傷によって引き起こされることもありうる。帯状ヘルペス(水痘および帯状疱疹)などのウイルス感染も、疼痛を引き起こすことがありうる。アルコールへの化学的依存または薬物乱用からの離脱も、しばしば、疼痛症状に関連している。したがって、「疼痛」は、本明細書中において極めて幅広い意味を有すると理解され、そしてその請求の範囲に記載の使用は、いずれか特定の疾患または状態に制限されると解釈されるべきではない。
【0026】
[0031]本明細書中で用いられる「鎮静」は、患者または対象が、開放気道を独立して且つ連続して維持する能力、および身体的刺激および言語的命令に適切に且つ合理的に応答する能力を保持している低下意識を意味する。本明細書中で用いられる「鎮静を伴うことなく」は、その患者が、Ramsay Sedation Scale で Level 3以下の鎮静レベルを経験しているということ、言い換えると、患者が、次のどちらかの Level であるということを意味する。Level 1=不安、動揺または不穏状態;Level 2=協力的で、見当識があり、そして精神安定な状態;または Level 3=鎮静状態であるが、命令に応答する。本明細書中で用いられる「有意の鎮静」は、患者または対象が、Ramsay Sedation Scale で Level 4またはそれより大の鎮静を経験しているということを意味し、ここにおいて、Level 4=眠った状態;眉間軽打または声高聴覚刺激へ活発応答;Level 5=眠った状態;眉間軽打または声高聴覚刺激へ緩慢応答;Level 6=眠った状態;有痛刺激へ無応答。本明細書中で用いられる「有意の鎮静」は、Stanford Sleepiness Scale での患者の自己評価とも一致するが、対象患者は、彼らの鎮静度合いを、Level 3より大またはそれに等しいと等級付けし、ここにおいて、Level 1=活発な感情、活力がある、敏捷または広範覚醒;Level 2=高レベルで機能性であるが、ピークではない;集中できる;Level 3=覚醒しているが弛緩状態;応答性であるが十分に敏捷ではない;Level 4=ややもうろう状態、気が抜けた状態(let down);Level 5=もうろう状態;覚醒状態のままで関心喪失;怠惰状態(slowed down);Level 6=眠気、頭がくらくらする、眠気を抑えている;横になることを選択する;または Level 7=もはや眠気を抑えられず、すぐに睡眠開始;夢のような思考をする。
【0027】
[0032]本明細書中に記載のデクスメデトミジンの製剤は、アスピリン、イブプロフェン、ナプロキセン、セレコキシブ(celecoxib)、アセトアミノフェンおよび他のシクロオキシゲナーゼ阻害剤などのNSAIDS;コデイン、オキシコドン、モルヒネ、メタドン(methadone)およびフェンタニールなどのオピオイド;フェニトインおよびカルバマゼピンなどの抗痙攣薬および抗不整脈薬;アミトリプチリン、イミプラミンおよびベンラファキシン(venlafaxine)などの抗うつ薬を含めた他の疼痛処置薬と共投与することができる。このような共投与は、同時発生的であってよく、ここにおいて、デクスメデトミジンおよび別の疼痛処置薬は、双方とも同時点で投与する。或いは、本明細書中に記載の経皮用製剤の急速作用性ゆえに、患者は、より短くまたはより長く作用する疼痛薬を規則的スケジュールで投与されてよく、経皮用デクスメデトミジンは、当日中に要求されるようにまたは必要とされる時間毎に投与される。いくつかの場合、別の疼痛処置薬の投薬量は、デクスメデトミジンによって生じる有益な共力作用ゆえに減少させることができ、それが、一次薬理療法を補足する。具体的には、デクスメデトミジンは、オピオイドの有効性を有意に増強して、必要なオピオイド投薬量の減少を可能にし、同時に、同等の治療的有用性を維持することができる。
【0028】
[0033]デクスメデトミジンまたはその薬学的に許容しうる塩または誘導体の鎮痛性経皮用製剤は、好ましくは、計量投薬量で与えられるので、所定量の活性医薬成分を、薬学的有効量で対象に適切に投与される。例えば、経皮用製剤は、個々の用量包装中でまたはマルチ包装単位で、例えば、計量ポンプを装着した密封容器を含むシステム中に多数回用量を含有するバルク容器として与えることができる。典型的に、ヒト患者は、ポンプからの1回またはそれを超える作動の局所自己投与によって処置される。各々のポンプ作動は、一定計量用量を与える。送達の利点は、単一の不連続適用によって必要とされる単回用量で患者を滴定する能力である。この利点は、典型的に、ワン・サイズ・フィッツ・オール・ドーセージ(one-size-fits-all dosage)を標準的な計画で投与する他の形の薬物送達(例えば、パッチ、ロゼンジ、錠剤および坐剤)に欠けている。局所経皮用製剤の追加の利点には、特に、担当医療専門家不在で自己投与する場合のそれらの使用の容易さが含まれる。
【0029】
[0034]ポンプ作用ディスペンサーは、作動のための外圧、例えば、外部の手動、機械的または電気的に開始する圧力の適用を必要とすることを特徴とする。これは、作動が、典型的に、圧力の制御放出によって、例えば、バルブの制御開放によって行われる加圧システム、例えば、噴射剤駆動エアゾルまたは圧縮ガススプレーとは対照的である。特定の態様において、ポンプ製剤は、本明細書中の製剤でのポンプスプレーの使用が、既知量のおよび調節可能なサイズの製剤の投与を可能にするので好適である。他の態様において、加圧噴射剤ガス(例えば、二酸化炭素、窒素、クロロフルオロカーボン、ヒドロフルオロアルカン等)のレザバーを含有する加圧システムは、適する計量分配量を生じることができる。特定の好ましい態様において、送達される製剤の粘度は、更に、皮膚または粘膜のいたるところに一様に広げられることにより、皮膚または粘膜上により濃厚な層で塗られることに対立するものとして表面積の増加を与える(例えば、ゲル剤対クリーム剤)。密度(例えば、クリーム剤対フォーム剤対ゲル剤)は、皮膚または粘膜上の所望のパターンおよび厚みの組成物に寄与することができる。
【0030】
[0035]スプレーポンプデバイスは、予備計量されてよい、または或いは、そのデバイスは、デバイスで計量されてよい。予備計量デバイスは、好ましくは、予め測定された用量または用量画分を、製造中にまたは患者によって使用前にデバイス中に包含されていよいいくつかのタイプのユニット(例えば、単回単位用量の溶液、単回または多数回ブリスターまたは他のキャビティ)で含有する。典型的なデバイスで計量されるユニットは、患者によって行われた時にデバイス自体によって計量されるスプレー剤として送達される多数回用量に十分な製剤を含有するレザバーを有する。そのデバイスは、送達される薬物物質の量(すなわち、1作動当たりの投薬量)で、更には、各投薬間の時間長さでも計量することができる。各投薬間の時間を制限することは、投薬量を患者に何回送達することができるかを制限することによって使いすぎを妨げることができる。
【0031】
[0036]製造要件には、製剤およびいずれかの計量分配機構または経皮送達ビヒクルの再現性が含まれる。これらパラメーターの再現性を有効期限期間中維持し且つデバイスの機能性(例えば、スプレー機構、電子機能、センサー等)を患者使用条件下でその寿命中確保することは、これらパラメーターの何らかの変更が、投与・吸収に変動をもたらすことがありうると考えられ、それが、潜在的副作用および減少した治療的有用性をもたらすことがありうると考えられるので重要である。
【0032】
[0037]いずれかのデバイスによる製剤の投与用量は、容器クロージャーシステムの設計、再現性および性能特性に依存してよい。所望の分配を与える適するデバイスは、デクスメデトミジン製品の正しい性能に重要な因子である。作動パラメーター(例えば、力、速度、保持・戻し時間)も、デバイスに関して考慮されるべきである。更に、デバイスは、製剤成分と相容性であるべきである。更に、デバイスは、患者使用説明書にしたがって用いられる場合に、デクスメデトミジン、その薬学的に許容しうる塩またはその誘導体を含めたデクスメデトミジン製剤の不完全計量、更には、過剰計量を妨げるように設計されるべきである。
【0033】
[0038]典型的な製剤デバイスには、基本ユニット、排出作動器、デバイスから製剤を放出させるオリフィス、およびレザバーが含まれる。好ましくは、レザバーは、患者へ計量分配する前に、例えば、製造現場で、薬物物質および他の賦形剤(例えば、本明細書中の他のところで論及される液状ビヒクル、賦形剤等)で満たされている。レザバーは、好ましくは、作動時に排出される一定測定量のデクスメデトミジン、その薬学的に許容しうる塩またはその誘導体を規定する。レザバー本体は、例えば、プラスチック、ステンレス鋼などの鋼製の透明材料等の円筒形中空品部分によって単純に形成されるいずれかの許容しうる材料であってよいので、その生産は極めて単純である。作動器は、排出を行うオリフィスに関して可動性であり、デバイス上にまたはデバイスと一緒に提供されてよい。作動運動の経過中に、レザバーは、例えば、穿刺によって開いて、オリフィスを介して単回投薬量を投与する。始動位置後の作動工程部分の際に、高圧が発生する。同方向に続行する引き続きの作動運動の一部分において、基剤は、片側で圧力が軽減され且つオリフィスに通じることができる。このような方式で、基剤は、圧力の作用によってレザバーからオリフィスを介して押し進められる。
【0034】
[0039]典型的に、スプレー可能経皮用製剤はいずれも、オリフィスから出るので、液体粒子は軌道にしたがうが、それは、オリフィス形状によって、更には、現れる圧力によって影響される。いくつかの態様において、液体粒子サイズ、スプレー幾何学的形およびスプレーパターンは、ポンプの設計および/または製剤の性状に依存している。特定の態様において、作動器の配向、ポンプ設計および製剤の性状は、スプレー対称性および形状に影響するであろう。スプレーパターンは、より広い通路上に液体粒子を分散させるように最適化することによって、化合物を吸収することができる皮膚上の表面積を増加させることもできる。いずれかこのようなスプレーデバイスは、更に、皮膚の特定領域へ投与されるスプレーの患者使用の容易さおよび配置を促すように設計することができる。
【実施例】
【0035】
[0040]デクスメデトミジンの以下の経皮用製剤を、実施例として製造した。いくつかの典型的な製剤を、本明細書中に記載のように哺乳動物において調べた。
[0041]実施例1a−プラシーボ経皮用ゲル製剤(担体)
[0042]50gのサリチル酸メチル/メントールヒドロアルコール性透明ゲルを製造する典型的な成分および方法。
【0036】
【表1】
【0037】
【表2】
【0038】
[0043]この実施例において、本発明者は、(1)pH=0.592gおよび水添加で沈澱した白色塊状物;および(2)再水和する水または再構成済みゲルの添加も、沈澱物を生じるということに注目した。沈澱作用は、より多くの水を加えるにつれて不溶性であるサリチル酸メチルに起因すると考えられる。或いは、または更に、沈澱は、少なくとも一部分はpH依存性であってよい。
【0039】
[0044]実施例1b−経皮用デクスメデトミジンゲル製剤
【0040】
【表3】
【0041】
【表4】
【0042】
[0045]この典型的な方法において、いくつかの観測を、工程Bで行った(成分番号2のDEXMEDETOMIDINE HClの導入時)。(1)ゲル製剤は、曇り状態になった;および(2)工程Bの得られたゲルは、工程Aのゲルと比較して減少した粘度を有した。曇りに関する観測1に取り組むために、トロラミンを加えたが、それは有効であった。この実施例において、トロラミンは、工程Cより得られた32.532gのゲルに0.463gのトロラミン(ロットXT0007と確認される)の比率で加えた。
【0043】
[0046]実施例2、3および4−Dex HCl局所用クリーム製剤
[0047]基本プラシーボクリーム製剤を用いて、本明細書中に更に記載の活性デクスメデトミジンHCl経皮用製剤を製造した。
【0044】
[0048]実施例2a−プラシーボO/Wクリーム基本製剤(担体)
【0045】
【表5】
【0046】
【表6】
【0047】
[0049]実施例2b−経皮用デクスメデトミジンクリーム製剤
【0048】
【表7】
【0049】
【表8】
【0050】
[0050]実施例3a−プラシーボO/Wクリーム基本製剤(担体)
【0051】
【表9】
【0052】
【表10】
【0053】
[0051]実施例3b−経皮用デクスメデトミジンクリーム製剤
【0054】
【表11】
【0055】
【表12】
【0056】
[0052]実施例4a−プラシーボO/Wクリーム基本製剤(担体)
【0057】
【表13】
【0058】
【表14】
【0059】
[0053]実施例4b−経皮用デクスメデトミジンクリーム製剤
【0060】
【表15】
【0061】
【表16】
【0062】
[0054]実施例5a−プラシーボO/Wシリコーン基本製剤(担体)
【0063】
【表17】
【0064】
【表18】
【0065】
[0055]実施例5b−経皮用デクスメデトミジンクリーム製剤
【0066】
【表19】
【0067】
【表20】
【0068】
[0056]本明細書中のいくつかの典型的な製剤を、イヌに投与して、デクスメデトミジンの局所経皮送達組成物としての吸収および他の性能特性を決定した。調べたそれら組成物の内、本明細書中で Recro 1−85として確認された組成物は、デクスメデトミジンの望ましい全身レベルを生じた。それら製剤は、少なくとも、イヌの疼痛および関連状態の処置に用いるのに適するし、そして更に、ヒトを含めた他の哺乳動物に用いるのに適するかもしれない。前述の実施例は、単に代表するものであり、本明細書中の発明の範囲を制限するものではない。いくつかの典型的な局所経皮用製剤を、動物において調べたが、開発したおよび/または調べたその他には、特に、アルコール性ゲル剤、油・水エマルジョンクリーム剤が含まれた。関係のある動物試験について関係のある結果を、本明細書中に要約する。
【0069】
臨床例
臨床研究期間1(REC−10−006)
[0057]動物における経皮用製剤に関する成功した実験に基づいて、ヒトにおける試験および試行を、いくつかの製剤および投与方法および処置方法について開発し且つ実施した。ヒト用製剤は、典型的な動物研究用製剤から誘導したが、cGMP必要条件およびヒト使用のための他の規制必要条件にしたがって製造した。本明細書中の局所経皮用製剤および方法のいくつかの典型的な態様を、ヒトにおいて早期臨床試験(Phase I研究としても知られる)で利用した。それら臨床試験においてこれまでに用いた典型的な製剤は、次の通りである。
【0070】
【表21】
【0071】
【表22】
【0072】
[0058]ヒトの場合の疼痛管理。Joint Commission が、疼痛の評価・管理の基準を改訂した2000年以来、疼痛の処置は、医療においてますます有意の位置を占めてきた。しばしば、「第五生命徴候」と称され、対象は、現在、疼痛症状について常套的に評価する必要があるので、療法は適切に調整することができる。この増加した関心の的は、患者の快適さおよび生活の質の問題に一層注意を向けさせたが、利用可能な手段の範囲は、概ね同じ状態のままであった。現行薬剤は、軽減を1〜2時間与える急性薬剤〜72時間程度の痛覚脱失を与えることができる別の製剤まで、活性持続期間が様々である。同時に、これら剤形は、一般的に、同様の活性成分セットに頼っていて、それらは、しばしば、同じオピオイド経路を介して作用して軽減を与える(モルヒネ、オキシコドン、フェンタニール)。その結果は、概して、アヘン薬によって与えられる作用、高用量および多数回アヘン薬の使用に上限は存在しないが、症状と副作用との間で決断を強いることがありうる有害イベントの発生増加をもたらすことがありうる。
【0073】
[0059]デクスメデトミジンは、クロニジンの約10倍の選択性を有する選択的α2−アドレナリン受容体アゴニストである。デクスメデトミジンは、急性および外科的設定においてその鎮静性を利用した広範囲の安全な静脈内使用歴を有し、そして Precedex(登録商標)という商品名での使用が示されている。より低い用量で、デクスメデトミジンは、現在、呼吸抑制のリスクを伴うことなく、鎮痛および抗不安の利点を与えることが認められている。
【0074】
[0060]デクスメデトミジンを含む経皮用剤形の使用は、延長した薬物摂取について非侵襲性機構を与える。この延長した摂取は、慢性疼痛患者への潜在的利点を伴って、長期の作用持続期間を促す。デクスメデトミジンは、追加の鎮静または呼吸抑制を伴うことなく、疼痛を抑制する可能性を有する。適切な使用は、患者の不快感を制限させると同時に、日常の活動を行う能力を維持するはずである。
【0075】
[0061]本明細書中に記載の典型的な経皮用製剤の最初のヒト使用は、本明細書中においてDEX−TD.02およびDEX−TD.03として確認された二つの経皮用デクスメデトミジン製剤の安全性、耐容性および薬物動態学的性質を評価するように設計した。薬物動態学的観測は、ヒト皮膚を越えるデクスメデトミジンの吸収および上昇用量の影響を評価することに集中するであろう。
【0076】
[0062]研究目的。ヒト研究の目的は、健康な男性および女性対象における単回上昇用量の経皮用デクスメデトミジンの安全性、耐容性および薬物動態を評価することであった。
[0063]調査計画。全体の研究設計は、健康対象において、二つの経皮用デクスメデトミジン製剤、すなわち、DEX−TD.02およびDEX−TD.03の安全性、耐容性および薬物動態を研究するためのオープンラベル、単回用量、用量上昇、クロスオーバー研究 Phase 1であった。18歳〜50歳までの年齢の健康対象の参加について、オーストラリアの一研究地において、研究用薬物投与前28日以内にスクリーニングした。病歴、身体検査、ベースライン実験室検査、12リード心電図(ECG)、妊娠検査、生命徴候測定およびインフォームドコンセントは、スクリーニング来診中に終えた。期間1において投与する前に、研究参加者を、二つの研究処置配列順の一方へ無作為に割り当てた(表Mを参照されたい)。各々の参加者に、割り当てられた処置配列順にしたがって、各々の研究期間中に単回用量の研究用薬剤を与えた。投与期間は、前の期間における血漿薬物濃度の薬物動態学的分析を可能にする十分な空白時間によって隔てられた(約14日)。
【0077】
【表23】
【0078】
[0064]各々の期間中、対象は、投与前の夕方(1日目)〜投与後約48時間(3日目)拘束されることになった。対象には、投与前の朝にシャワーを浴びることを要求し、そして投与後48時間はシャワーを浴びることもその部位を洗うことも禁止した。拘束はされるが、対象には、標準化した日常食を与えた。表Nは、この研究における薬剤、適用用量および局所投与部位を示している。
【0079】
【表24】
【0080】
[0065]薬物動態学的分析のために、16人の血液試料を、各々の期間中に各々の対象から単回用量投与後に集めた。採集は、研究用薬剤を投与前(0時)についておよび投与後の以下のおよその時点に的を絞った。30分、60分および90分、そして2時間、3時間、4時間、6時間、8時間、10時間、12時間、18時間、24時間、30時間、36時間および48時間。投与後時間は、対象に用量を与えた時点に開始した。安全評価には、AEの監視、臨床実験室検査、生命徴候測定、身体検査、刺激評価およびECGが含まれた。
【0081】
[0066]研究設計および対照群の原理。この研究は、別の送達経路によって投与される既知の薬物物質の安全性、耐容性および薬物動態学的性質を評価した。本発明者および出願人による以前の調査は、静脈内および舌下経路によって投与時のデクスメデトミジンの安全性を示した。
【0082】
[0067]本明細書中の前に記載のように、本発明者および出願人による先行研究は、動物モデル(イヌおよびブタ)においてDEX−TD製剤を用いて行って、健康なヒト対象におけるこの初期投与を支持した。この研究に用いられた用量上昇モデルは、DEX−TD.02およびDEX−TD.03の吸収性を、ヒト皮膚に適用時に、適用用量に依存してそれらがどのように変動することがありうるかを含めて確認し且つ評価することであった。クロスオーバー設計の目的は、これら二つの製剤の吸収を同じ組の対象において比較することであった。一方の対象部分群では、経皮用製剤を、皮膚部位に投与し、そして直ちに包帯剤で被覆した(「閉塞した」)。用いられた包帯剤は、TEGADERMTM商標包帯剤であった(TEGADERMTMは、3M Corporation の登録商標である)。もう一方の対象部分群では、経皮用製剤を皮膚に投与し、そして未被覆状態にした(「非閉塞とした」)。閉塞される投与で十分に確定された経験に基づいて、本発明者は、経皮処方皮膚適用部位(腹部)上に包帯剤を適用された対象において、吸収が一層高いと考えられると期待した。しかしながら、驚くべきことに、DEX−TD.02の吸収は、皮膚適用部位上に包帯剤が適用されなかった部分群について、有意に一層高かった。更に、それら結果は、閉塞が必要でも望ましくもないということを示し、少なくともDEX−TD.02の組成物を経皮用の真正「パッチレス(patchless)」にする(すなわち、無包帯剤、パッチが製剤を含有する必要がない)。その製剤は、皮膚に適用時に安定であり、しかも包帯剤または他の被覆剤不要で容易に吸収される。この研究の薬物動態学的関心の的ゆえに、対照集団は不必要であった。
【0083】
[0068]期間1における薬物動態学的結果の再検討にしたがって、臨床研究期間2の際に製剤DEX−TD.02を投与することだけを決定した。これは、一部分は、DEX−TD.03による見掛けの一層低い吸収のためであった。この研究は、デクスメデトミジンの全身吸収を与えるために高吸収を追求したので、DEX−TD.02は、この研究における追加の臨床検査のために一層適切であった。しかしながら、DEX−TD.03は、その一層低い吸収速度および極めて低い全身吸収を考えると、局部的皮膚および/または硬膜下ニューロパシーの処置などの他の疼痛管理用途により一層適切でありうる。特に、研究期間1においてどちらかの製剤で処置されたいずれの対象についても、鎮静が観測されたまたは報告されたものはなかった。
【0084】
臨床研究期間2
[0069]目的。臨床研究期間2の目的は、本出願人の単回上昇用量研究(期間1、(REC−10−006))より得られたデータを用いて、0.5グラム用量のデクスメデトミジンの製剤DEX−TD.02の1日1回および1日2回局所適用によって生じる定常状態血漿濃度−時間プロフィールを計算することであった。
【0085】
[0070]研究設計。臨床研究#REC−10−006によるデクスメデトミジン血漿濃度−時間データを、0.25グラム、0.5グラムおよび1グラム用量の1日1回(QD)および1日2回(BID)局所適用によって生じる定常状態濃度−時間プロフィールを予測する場合に用いた。次の3対象、すなわち、1001、1004および1005からのデータを用いた。それら三対象は、以下の処置部門、すなわち、処置A(0.25グラム用量)、処置C(0.5グラム用量)および処置E(1.0グラム用量)に参加した。臨床研究#REC−10−006は、多数の処置部門(処置A、B、C、DおよびE)を含む単回上昇用量研究であった。薬物分析用の血漿試料は、投与前(0時)と、投与後の以下の規定時間、すなわち、0.5時間、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、10時間、12時間、18時間、24時間、30時間、36時間および48時間に採取した。生物学的分析の定量化の低い方の限界は、0.02ng/mLであった。特に、研究期間2においてどちらかの製剤で処置されたいずれの対象についても、鎮静が観測されたまたは報告されたものはなかった。
【0086】
[0071]方法および分析。薬物動態学的パラメーターは全て、区画分析を用いて分析した。期間1(REC−10−006)によるデクスメデトミジン血漿濃度−時間データを、0.25グラム、0.5グラムおよび1.0グラム用量の1日1回(QD)および1日2回(BID)局所適用によって生じる定常状態濃度−時間プロフィールを予測する場合に用いた。用いられたデータは、処置部門A(0.25グラム用量)、C(0.5グラム用量)およびE(1.0グラム用量)に参加した三対象(識別名1001、1004および1005)からであった。薬物分析用の血液試料は、投与前(0時)と、投与後の以下の規定時間、すなわち、0.25時間、0.5時間、0.75時間、1時間、1.5時間、2時間、2.5時間、3時間、4時間、5時間、6時間、7時間、8時間、12時間、24時間、48時間および72時間に採取した。血漿濃度の主要部分(67%)は、0.02ng/mLの定量可能レベル未満であった;したがって、PKパラメーターは、非線形混合効果モデルのより精巧なコンピューター法を用いて計算した(すなわち、集団薬物動態[PopPK])。非線形混合効果モデルは、Monolix, Version 3.1ソフトウェア(Institut National de Recherche en Informatique et en Automatique[INRIA])を用いて行った。いったんPKパラメーターを計算し且つ単回用量濃度を作成したら、データを、WinNonlinTM,Version 5.2(WinNonlinTM Copyright c.2008, Pharsight Corporation)にインポートし、非パラメーター重ね合わせを行って、QDおよびBID投与によって生じる定常状態血漿濃度−時間プロフィールを予測した。
【0087】
[0072]単回用量データから薬物動態学的パラメーターの計算。前述のように、PKパラメーターは、MonoLix 3.1ソフトウェアを用いた非線形混合効果モデルを用いて計算した。処置部門A(0.25グラム用量)、C(0.5グラム用量)およびE(1.0グラム用量)に参加した三対象(1001、1004および1005)からの血漿デクスメデトミジン濃度を、零次吸収を含む線形一区画モデルに適合させた。適合された濃度および観測された濃度を、図1に示す。図1における注記:番号は、対象識別名である(例えば、1001、1004または1005);下付き文字(例えば、A、C、E)は、処置、すなわち、A(0.25グラム用量)、C(0.5グラム用量)またはE(1.0グラム用量)である;プラス記号(+)は、定量化の限界より上の測定可能デクスメデトミジン濃度である;そして星印(*)記号は、定量化限界未満のデクスメデトミジン濃度(0.02ng/mL)である;そして実線は、最も適合したPK曲線を構成している。適合されたおよび観測された濃度−時間プロフィール(図1)の調査は、零次吸収を含む一区画(実線)が、定量可能なデクスメデトミジン濃度(プラス記号)を双方とも十分に記載し且つ定量可能限界未満(BQL)(星印記号)であると考えられたそれら試料と一致するということを示している。
【0088】
[0073]全体的に、図2に示されるように、対象投与によるモデル値と観測値との間には十分な一致が存在した。図2は、(モデルからの)予測値が、予測値に等しい観測値についての理論線に近いばらつきによって示されるように観測値とほぼ一致するということ、および大部分のBQL値が、モデルからの値について一定であるということを示している。図2についての注記:菱形は、定量化限界より上の測定可能な濃度である;星印は、定量化限界未満の測定可能な濃度(0.02ng/mL)である;直線は、観測濃度=予測濃度の場合の一致線である;そして曲線は、プロットポイントの傾向線である。
【0089】
[0074]適合により得られた薬物動態学的パラメーターを、表P1に示す。デクスメデトミジンの皮膚適用は、約15時間の半減期で約20時間にわたって(一定注入と同様の)一定速度で吸収されると考えられた。約2.2xおよび1.5xの累積倍率は、BIDおよびQD投与についてそれぞれ計算した。
【0090】
【表25】
【0091】
[0075]BIDおよびQD双方の投与によって生じる定常状態濃度の計算。前述のようにい、いったんPKパラメーターを計算し且つ単回用量濃度を作成したら、データを、WinNonlinTM,Version 5.2(WinNonlinTM Copyright c.2008, Pharsight Corporation)にインポートした。非パラメーター重ね合わせを行って、QDおよびBID投与によって生じる定常状態血漿濃度−時間プロフィールを予測した。非パラメーター重ね合わせは、各々の用量の薬物が、用量毎に独立して作用するということ、および吸収の速度および程度および平均全身クリアランスが、各々の投与間隔について同じであるということを仮定している。線形薬物動態は、多数回投与計画の際の用量の変化に適応しうるように適用する。
【0092】
[0076]多数回用量によって生じる薬物濃度を予測するために、単回用量投与後の濃度−時間プロフィールの完全な特性決定が必要である。すなわち、薬物吸収・排除過程を特性決定するのに十分な時点ti(i=1,2,...,n)でC(ti)を知ることが必要がある。データについて次の二つの仮定条件、すなわち、各々の用量作用の独立および基礎にある薬物動態の線形性が必要である。前者は、各々の用量の作用が、他の用量の作用と分離することができると仮定している。後者、すなわち、線形薬物動態は、薬物濃度の変化が、用量で線形変動するであろうと仮定している。
【0093】
[0077]0.25(txA)、0.5(txC)および1.0(txE)グラムの1日1回用量後の定常状態での予測血漿濃度−時間曲線を、図3に示す。1日2回投与後の予測定常状態血漿濃度−時間曲線を、図4に示す。双方の投与計画(QDおよびBID)について、定常状態で得られた血漿濃度−時間曲線は、かなり平坦である。これは、真皮から体循環への薬物の長期零次拡散および長い半減期のためである。BIDおよびQD投与計画の予測定常状態血漿濃度を、それぞれ、表Q2および表R3に挙げる。
【0094】
【表26】
【0095】
【表27】
【0096】
[0078]期間2の結果および結論。図3および図4は、DEX−TD.02を1日1回および1日2回用量計画で用いた臨床研究に基づく成人における時間経過での予測定常状態血漿濃度を示す。この研究において鎮静を伴うことなく鎮痛薬として用いるためのデクスメデトミジンの標的定常状態濃度は、0.1〜0.2ng/mLの範囲内であるということを想起されたい。したがって、図3および図4が明らかに示すように、これは、(その範囲の下方部に達するであろう)0.5グラム用量かまたは(上方範囲の濃度を生じるであろう)1.0グラム用量のBID投与で達成することができる。この実施例において、所望の定常状態濃度は、多数回投与の4日目またはその後に生じると予測されるであろう。このモデルに基づき、DEX−TD.02およびDEX−TD.03を用いた1日1回投与は、0.5グラム用量でも1.0グラム用量でも、標的範囲(0.1〜0.2ng/mL)に達する定常状態血漿濃度を速やかに生じなかったので、1日1回投与はチャレンジのままである。再度、DEX−TD.02または類似の製剤を用いると、定常状態濃度は、多数回投与の少なくとも4日目までには達するであろう。しかしながら、本明細書中のおよび本発明者が考える知見および情報では、これらおよび他の局所用製剤は、より短い時間内であれ、別の投与計画によってであれ、哺乳動物において経皮吸収し且つデクスメデトミジンの所望の標的定常状態血漿濃度を達成することを示すことができる。例えば、所望の定常状態への投与は、本明細書中に記載の持続した、制御された、長期の放出・吸収デクスメデトミジン経皮用製剤との組み合わせで、急速吸収性舌下用、経粘膜用および/または急速吸収性経皮用の組成物の組み合わせ(例えば、DMSOなどの急速吸収性溶媒担体、アルコールおよび他の薬学的に相容性の担体および製剤を含めた)を用いて達成することができる。
【0097】
[0079]この説明は、典型的な態様に関して行っているが、いろいろな変更を行うことができるし、そしてその範囲から逸脱することなく、均等物をそれらの要素の代わりに置き換えることができるということは当業者によって理解されるであろう。更に、本質的な範囲から逸脱することなく、特定の状況および材料をそれらの内容に適応するように多くの修飾を行うことができる。更に、説明には、典型的な態様を開示してきており、具体的な用語を用いてきたかもしれないが、それらは、特に断らない限り、単に包括的および説明的意味で用いられ、制限するためではなく、したがって、請求の範囲の範囲は、そのように制限されることはない。更に、当業者は、本明細書中で論じられている方法の特定の工程を、別の順序で配列することができるまたは工程を組み合わせることができるということを理解するであろう。したがって、請求の範囲は、本明細書中に開示の具体的な態様に制限されるものではないということである。
【特許請求の範囲】
【請求項1】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚に、投薬量のデクスメデトミジン(dexmedetomidine)またはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む組成物を適用することを含み、ここにおいて、
デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグは、皮膚を介して吸収されていて且つ鎮静を伴うことなく痛覚脱失を生じる方法。
【請求項2】
デクスメデトミジンまたはその薬学的に許容しうる塩の前記投薬量が、約0.05μg/kg〜約15mcg/kgであり、そしてここにおいて、前記哺乳動物が、ヒトである、請求項1に記載の方法。
【請求項3】
前記哺乳動物が、ヒトであり、そしてデクスメデトミジンまたはその薬学的に許容しうる塩の前記経皮投薬量が、約100μg/kg〜約1500μg/kgである、請求項2に記載の方法。
【請求項4】
前記ヒトの経皮適用〜体循環系中吸収時のデクスメデトミジンの血漿Cmaxが、約0.30ng/mL未満である、請求項3に記載の方法。
【請求項5】
前記適用工程が、脚、上腕、腹、胸部、鼡径部、頸部、背部および肩の少なくとも一つの皮膚へ前記医薬組成物を局所適用することを含む、請求項2に記載の方法。
【請求項6】
前記適用工程が、前記哺乳動物の前記皮膚へクリーム剤、ローション剤またはゲル剤の医薬組成物を適用することを含む、請求項5に記載の方法。
【請求項7】
前記適用工程が、前記哺乳動物の皮膚上への前記医薬組成物の投与後に包帯で皮膚を被覆することを必要としない、請求項6に記載の方法。
【請求項8】
前記医薬組成物を適用直後の少なくとも6時間に、ヒトの安静時平均動脈血圧が、約20mmHg以下だけ変動する、請求項2に記載の方法。
【請求項9】
前記医薬組成物を適用直後の少なくとも6時間に、前記ヒトが、覚醒状態のままでいて且つ命令に応答することができ、そして該ヒトが、敏捷で且つ見当識がある、請求項2に記載の方法。
【請求項10】
前記デクスメデトミジンまたはその薬学的に許容しうる塩を、一つまたはそれを超える他の鎮痛薬と共投与する、請求項1に記載の方法。
【請求項11】
前記医薬組成物を、前記一つまたはそれを超える他の鎮痛薬によって不適当に制御される破綻疼痛を処置するために間欠的に投与する、請求項10に記載の方法。
【請求項12】
前記疼痛が、特発性疼痛である、請求項2に記載の方法。
【請求項13】
前記特発性疼痛が、神経痛、筋肉痛、痛覚過敏、ピペルパシー、神経炎およびニューロパシーから成る群より選択される、請求項12に記載の方法。
【請求項14】
前記疼痛が、癌、ウイルス感染、身体的外傷、関節炎、頭痛または腰部痛に関連しているまたはそれによって引き起こされている、請求項2に記載の方法。
【請求項15】
前記身体的外傷が、外科手術、熱傷または鈍的力外傷に関連しているまたはそれによって引き起こされている、請求項14に記載の方法。
【請求項16】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚膜に、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む医薬組成物を適用することを含み、ここにおいて、
該デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグは、該皮膚を介して吸収されていて且つ鎮静を伴うことなく痛覚脱失を生じる方法。
【請求項17】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚に、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを含む全身吸収される医薬組成物を、該哺乳動物において投与時に疼痛を処置するまたは予防する有効量で投与することを含み、ここにおいて、
該医薬組成物は、投与から少なくとも6時間以内に鎮静を伴うことなく鎮痛作用を生じる速度で該哺乳動物の体循環系中に生理学的に活性な量のデクスメデトミジンを与える方法。
【請求項18】
鎮痛性医薬組成物であって、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含み、哺乳動物の皮膚へ該鎮痛性医薬組成物を適用することによる該哺乳動物への局所投与用に配置され且つ適合されている該医薬組成物。
【請求項19】
前記鎮痛性医薬組成物が、1日2回以上皮膚へ該組成物を適用することによる局所投与用に配置され且つ適合されている、請求項18に記載の鎮痛性医薬組成物。
【請求項20】
前記鎮痛性医薬組成物が、前記哺乳動物の脚、上腕、腹、胸部、鼡径部、頸部、背部および肩の少なくとも一つの皮膚へ該組成物を適用することによる局所投与用に配置され且つ適合されている、請求項18に記載の鎮痛性医薬組成物。
【請求項21】
デクスメデトミジンの前記薬学的に許容しうる塩が、デクスメデトミジン塩酸塩であり、そしてここにおいて、前記ビヒクルが、クリーム剤、ゲル剤および懸濁剤の少なくとも一つより選択される、請求項18に記載の鎮痛性医薬組成物。
【請求項22】
組成物が、哺乳動物の皮膚上への投与後に閉塞または被覆を必要としない、請求項21に記載の鎮痛性医薬組成物。
【請求項23】
前記鎮痛性医薬組成物が、計量分配用デバイス中に包装されている、請求項18に記載の鎮痛性医薬組成物。
【請求項24】
疼痛を処置するまたは予防するための装置であって、
デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む鎮痛性医薬組成物、および
該鎮痛性医薬組成物を含有し且つ計量分配する計量分配用デバイス
を含む該装置。
【請求項25】
装置が、該装置から哺乳動物の皮膚へ組成物を運搬するために配置され且つ配列されているアプリケーターを包含する、請求項24に記載の装置。
【請求項1】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚に、投薬量のデクスメデトミジン(dexmedetomidine)またはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む組成物を適用することを含み、ここにおいて、
デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグは、皮膚を介して吸収されていて且つ鎮静を伴うことなく痛覚脱失を生じる方法。
【請求項2】
デクスメデトミジンまたはその薬学的に許容しうる塩の前記投薬量が、約0.05μg/kg〜約15mcg/kgであり、そしてここにおいて、前記哺乳動物が、ヒトである、請求項1に記載の方法。
【請求項3】
前記哺乳動物が、ヒトであり、そしてデクスメデトミジンまたはその薬学的に許容しうる塩の前記経皮投薬量が、約100μg/kg〜約1500μg/kgである、請求項2に記載の方法。
【請求項4】
前記ヒトの経皮適用〜体循環系中吸収時のデクスメデトミジンの血漿Cmaxが、約0.30ng/mL未満である、請求項3に記載の方法。
【請求項5】
前記適用工程が、脚、上腕、腹、胸部、鼡径部、頸部、背部および肩の少なくとも一つの皮膚へ前記医薬組成物を局所適用することを含む、請求項2に記載の方法。
【請求項6】
前記適用工程が、前記哺乳動物の前記皮膚へクリーム剤、ローション剤またはゲル剤の医薬組成物を適用することを含む、請求項5に記載の方法。
【請求項7】
前記適用工程が、前記哺乳動物の皮膚上への前記医薬組成物の投与後に包帯で皮膚を被覆することを必要としない、請求項6に記載の方法。
【請求項8】
前記医薬組成物を適用直後の少なくとも6時間に、ヒトの安静時平均動脈血圧が、約20mmHg以下だけ変動する、請求項2に記載の方法。
【請求項9】
前記医薬組成物を適用直後の少なくとも6時間に、前記ヒトが、覚醒状態のままでいて且つ命令に応答することができ、そして該ヒトが、敏捷で且つ見当識がある、請求項2に記載の方法。
【請求項10】
前記デクスメデトミジンまたはその薬学的に許容しうる塩を、一つまたはそれを超える他の鎮痛薬と共投与する、請求項1に記載の方法。
【請求項11】
前記医薬組成物を、前記一つまたはそれを超える他の鎮痛薬によって不適当に制御される破綻疼痛を処置するために間欠的に投与する、請求項10に記載の方法。
【請求項12】
前記疼痛が、特発性疼痛である、請求項2に記載の方法。
【請求項13】
前記特発性疼痛が、神経痛、筋肉痛、痛覚過敏、ピペルパシー、神経炎およびニューロパシーから成る群より選択される、請求項12に記載の方法。
【請求項14】
前記疼痛が、癌、ウイルス感染、身体的外傷、関節炎、頭痛または腰部痛に関連しているまたはそれによって引き起こされている、請求項2に記載の方法。
【請求項15】
前記身体的外傷が、外科手術、熱傷または鈍的力外傷に関連しているまたはそれによって引き起こされている、請求項14に記載の方法。
【請求項16】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚膜に、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む医薬組成物を適用することを含み、ここにおいて、
該デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグは、該皮膚を介して吸収されていて且つ鎮静を伴うことなく痛覚脱失を生じる方法。
【請求項17】
疼痛を処置するまたは予防するための方法であって、
哺乳動物の皮膚に、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを含む全身吸収される医薬組成物を、該哺乳動物において投与時に疼痛を処置するまたは予防する有効量で投与することを含み、ここにおいて、
該医薬組成物は、投与から少なくとも6時間以内に鎮静を伴うことなく鎮痛作用を生じる速度で該哺乳動物の体循環系中に生理学的に活性な量のデクスメデトミジンを与える方法。
【請求項18】
鎮痛性医薬組成物であって、デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含み、哺乳動物の皮膚へ該鎮痛性医薬組成物を適用することによる該哺乳動物への局所投与用に配置され且つ適合されている該医薬組成物。
【請求項19】
前記鎮痛性医薬組成物が、1日2回以上皮膚へ該組成物を適用することによる局所投与用に配置され且つ適合されている、請求項18に記載の鎮痛性医薬組成物。
【請求項20】
前記鎮痛性医薬組成物が、前記哺乳動物の脚、上腕、腹、胸部、鼡径部、頸部、背部および肩の少なくとも一つの皮膚へ該組成物を適用することによる局所投与用に配置され且つ適合されている、請求項18に記載の鎮痛性医薬組成物。
【請求項21】
デクスメデトミジンの前記薬学的に許容しうる塩が、デクスメデトミジン塩酸塩であり、そしてここにおいて、前記ビヒクルが、クリーム剤、ゲル剤および懸濁剤の少なくとも一つより選択される、請求項18に記載の鎮痛性医薬組成物。
【請求項22】
組成物が、哺乳動物の皮膚上への投与後に閉塞または被覆を必要としない、請求項21に記載の鎮痛性医薬組成物。
【請求項23】
前記鎮痛性医薬組成物が、計量分配用デバイス中に包装されている、請求項18に記載の鎮痛性医薬組成物。
【請求項24】
疼痛を処置するまたは予防するための装置であって、
デクスメデトミジンまたはその薬学的に許容しうる塩またはプロドラッグを薬学的に許容しうるビヒクル中に含む鎮痛性医薬組成物、および
該鎮痛性医薬組成物を含有し且つ計量分配する計量分配用デバイス
を含む該装置。
【請求項25】
装置が、該装置から哺乳動物の皮膚へ組成物を運搬するために配置され且つ配列されているアプリケーターを包含する、請求項24に記載の装置。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2013−516482(P2013−516482A)
【公表日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願番号】特願2012−548137(P2012−548137)
【出願日】平成23年1月7日(2011.1.7)
【国際出願番号】PCT/US2011/020462
【国際公開番号】WO2011/085162
【国際公開日】平成23年7月14日(2011.7.14)
【出願人】(512178765)レクロ・ファーマ,インコーポレーテッド (1)
【Fターム(参考)】
【公表日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願日】平成23年1月7日(2011.1.7)
【国際出願番号】PCT/US2011/020462
【国際公開番号】WO2011/085162
【国際公開日】平成23年7月14日(2011.7.14)
【出願人】(512178765)レクロ・ファーマ,インコーポレーテッド (1)
【Fターム(参考)】
[ Back to top ]