
希少循環細胞における多様なシグナル伝達物質検出のための抗体に基づくアレイ
【課題】希少循環細胞における複数のシグナル伝達分子の活性状態、及び/又は、総量を検出するための抗体に基づくアレイ、癌の予後診断及び診断、及び、個々の標的化された治療の設計を促進するための、そのアレイの使用の方法を提供する。
【解決手段】抗体に基づくアレイ方法を実行するためのキットであって、(a)固相担体上に拘束された複数の捕捉抗体の希釈系列、(b)活性状態非依存抗体及び活性状態依存抗体を含む複数の検出抗体、及び、(c)固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書を含む、キット。
【解決手段】抗体に基づくアレイ方法を実行するためのキットであって、(a)固相担体上に拘束された複数の捕捉抗体の希釈系列、(b)活性状態非依存抗体及び活性状態依存抗体を含む複数の検出抗体、及び、(c)固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書を含む、キット。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2006年9月21日に出願され、米国仮出願第号に変更された、米国出願第11/525,598号、2007年4月20日に出願された米国仮出願第60/913,087号の優先権を主張し、それらの開示は、すべての目的のため、その全体が参照により本明細書に援用される。
【背景技術】
【0002】
腫瘍細胞は、様々な初期癌の患者の血液中に「微小転移(micrometastases)」(播種性腫瘍細胞)として認められる。そして、腫瘍細胞は転移性癌においても認められる。血中の腫瘍細胞の数は、腫瘍の病期と種類に依存する。腫瘍は、極端に異質である。結果として、単一の部位からの生検は、ある腫瘍集団における異質性を示さないかもしれない。生検は、典型的には、原発巣で取得される;しかしながら、ほとんどの転移性腫瘍は、生検が行われないため、腫瘍試料の分子的解析をより困難にしている。
【0003】
腫瘍の転移の間、最も攻撃性のある腫瘍細胞が、原発巣を離れ、血液やリンパ系を通って、離れた部位に達する。このように、血液からの循環腫瘍細胞は、最も攻撃的で同質な腫瘍細胞集団を示す。血中の転移性腫瘍細胞の数は、血液1mlあたり、1つから数千個の細胞まで様々であり得る。
【発明の概要】
【発明が解決しようとする課題】
【0004】
従って、診断及び治療の目的のため、これらの細胞を検出する、特異的で感度の高い方法が必要とされている。本発明は、この必要性を満たし、さらに、関連する有利な点も提供する。
【0005】
本発明は、希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための抗体に基づくアレイとその使用方法を提供する。この抗体に基づくアレイは、酵素結合免疫吸着法(ELISA)に伴う特異性、シグナル増幅に伴う感度、及びマイクロアレイに伴うハイスループットの多重化の利点を有する。
【課題を解決するための手段】
【0006】
1つの態様では、本発明は、細胞抽出物における1つ又はそれ以上の測定物質に特異的な複数の希釈系列の捕捉抗体から構成される優れたダイナミックレンジを有するアレイを提供する。前記捕捉抗体は、固相担体上に拘束されている。
【0007】
他の態様では、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、細胞抽出物中の1つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、対応する分析物に特異的な検出抗体とインキュベートする工程;
(c)増幅されたシグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第一及び第二の構成因子とインキュベートする工程;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0008】
さらに他の態様では、本発明は、優れたダイナミックレンジを有する多重、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するため、細胞抽出物を、細胞抽出物中の1つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、それに対応する分析物に特異的な検出抗体とインキュベートする工程であって、
前記検出抗体は:
(1)促進部分で標識された複数の活性状態非依存抗体、と
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体と、を備え、
前記促進部分は、シグナル増幅対の第一の構成因子に向けられ且つそれと反応する酸化剤を生成する、
ことを特徴とする工程;
(c)増幅シグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第二の構成因子とインキュベートする構成;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0009】
本発明はまた、上記の抗体に基づくアレイ法を行うためのキットを提供し、本キットは、以下の構成を備える:(a)固相担体に拘束された複数の希釈系列の捕捉抗体;及び(b)複数の検出抗体。このキットはさらに、任意に、例えばシグナル増幅対の第一及び第二の構成因子のような他の試薬を備えてもよい。
【0010】
本発明の他の目的、特徴、利点は、下記の詳細な説明及び図より、当該分野の当業者には明確である。
【図面の簡単な説明】
【0011】
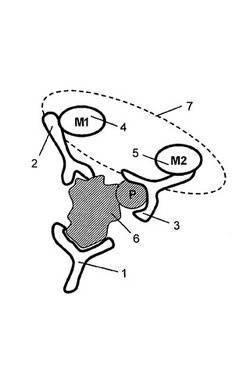
【図1】活性型分析物に特異的に結合した3つの抗体を示す図である。
【図2】活性型分析物に特異的に結合した標識抗体が固相担体に拘束される分析スキームを示す図である。
【図3】ELISAにおいて、捕捉抗体及び検出抗体として、EGFRの細胞外ドメインへのモノクローナル抗体を用いてA431細胞中の総EGFRの検出を示す図である。(A)は、サンドイッチELISAの標準曲線を示す。その感度は、ヒトEGFRの組み換え細胞外ドメインを用いた場合、0.25pg/穴であった。(B)は、細胞滴定曲線を示す。A431細胞中のEGFR濃度をELISAによって検出した。(C)は、pg/穴及びpg/細胞で計算されたEGFR濃度の表である。計算されたEGFR濃度は、各A431細胞あたり約0.6pgであった。
【図4】ELISAにおいて、捕捉抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を、及び、検出抗体としてリン酸化EGFRに対するビオチン標識モノクローナル抗体を用いた、A431細胞中のリン酸化EGFRの検出を示す図である。(A)は、捕捉抗体の希釈系列での細胞滴定曲線を示す。(B)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表を示す。捕捉抗体濃度が0.0625μg/mlであるときに、そのシグナル/ノイズ比は一細胞レベルで1.78であった。
【図5】ELISAにおいて、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を用いた、SKBr3細胞中の総ErbB2の検出を示す図である。(A)は、SKBr3細胞中の総ErBb2の細胞滴定を示す。検出範囲は、約1000細胞と約1.37細胞との間であった。(B)は、細胞とバックグラウンド間のシグナル/ノイズ比を示す。捕捉抗体濃度が1μg/mlであるときに、シグナル/ノイズ比は、1.37細胞レベルで2.71であった。
【図6】ELISAにおいて、捕捉抗体としてErbB2の細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化ErbB2に対するモノクローナル抗体を用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す図である。(A)は、SKBr3細胞中のリン酸化ErBb2の細胞滴定を示す。検出範囲は、約500細胞と約5細胞との間であった。(B)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が1μg/mlであるときに、シグナル/ノイズ(S/N)比は5細胞レベルで3.03であった。
【図7】ELISAにおいて、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体を用いた、SKBr3細胞中の総Erk2タンパク質及びリン酸化Erk2タンパク質の検出を示す図である。(A)は、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体を用いた総Erk2タンパク質の検出を示す。(B)は、捕捉抗体としてErk2に対するモノクローナル抗体を、検出抗体としてリン酸化Erk2に対するモノクローナル抗体を用いたリン酸化Erk2の検出を示す。(C)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。総Erk2及びリン酸化Erk2の両方に対するシグナル/ノイズ比は、1.37細胞レベルで約3であった。
【図8】マイクロアレイELISAにおいて、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を用いた、A431細胞中の総EGFRの検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列での様々な濃度の捕捉抗体で約1〜10000細胞中のEGFRを検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、EGFRが一細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.0625mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は、1細胞レベルで2.11であった。
【図9】マイクロアレイELISAにおいて、捕捉抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化EGFRに対するモノクローナル抗体を用いた、A431細胞中のリン酸化EGFRの検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体での約1−10000細胞中のリン酸化EGFRを検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、リン酸化EGFRが一細胞から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は一細胞レベルで1.33であった。
【図10】マイクロアレイELISAにおいて、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を用いた、SKBr3細胞中の総ErBb2の検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体で約1〜10000細胞中のErBb2を検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、ErBb2が1細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)である時に、シグナル/ノイズ比は1細胞レベルで15.27であった。
【図11】マイクロアレイELISAにおいて捕捉抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化ErBb2に対するモノクローナル抗体を用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体で約1〜10000細胞中のErBb2を検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、リン酸化ErBb2が1細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は1細胞レベルで5.45であった。
【図12】近接二重検出マイクロアレイELISAと単一検出マイクロアレイELISAとの感度の比較を示す図である。A431細胞は、10000から0.01細胞まで希釈された。捕捉抗体は、1mg/m1から0.004mg/mlまで連続的に希釈された。
【図13】単一検出抗体のマイクロアレイELISAと近接二重検出マイクロアレイELISAの分析特異性を示す図である。(A)は、単一検出の形式における様々な捕捉抗体濃度でのA431細胞中のリン酸化EGFRの滴定曲線を示す。この形式では単一の検出抗体の特異性の欠如のため、非常に高いバックグラウンドが観察された。(B)は、近接二重検出の形式における様々な捕捉抗体の濃度でのA431細胞中のリン酸化EGFRの滴定曲線を示す。この形式では、2つの検出抗体間の近接度を検出することによって得られる特異性が増加するため、非常に低いバックグラウンドしか検出されなかった。
【図14】複数のシグナル伝達分子の活性状態を決定するために捕捉抗体の希釈系列を用いたアドレス可能なマイクロアレイの形式の実施例を示す図である。
【図15】活性化されたA431細胞の滴定分析におけるリン酸化Shcレベルの検出を示す図である。アドレス可能なアレイは、EGFR及びHER2リン酸化に関する情報を同時に提供した。
【図16】抗EGFR捕捉抗体の希釈曲線を示す図である。この分析のダイナミックレンジは、5対数(5logs)よりも大きかった。個々の曲線それぞれは、約2対数(2logs)のダイナミックレンジを有するが、6つの有益な曲線からの情報を組み合わせると、そのダイナミックレンジが非常に増加した。
【図17】本発明のオリゴヌクレオチド共役のための品質管理手順を示す。(A)は、様々なGO−オリゴヌクレオチド画分のマイクロアレイのスポッティングパターンを示す。(B)は、Alexa647−オリゴヌクレオチド共役抗体が、画分13〜15でGO−オリゴヌクレオチドに対する最も高い結合親和性を有したことを示す。
【図18】グルコースオキシダーゼ(GO)−オリゴヌクレオチドに共役したAlexa647−オリゴヌクレオチド共役抗−EGFR抗体、HRP共役抗−リン酸化EGFR抗体、及び固相担体に固着したEGFR捕捉抗体からなる多重リン酸化EGFR複合体の形成を示す図である。
【図19】総EGFR及びリン酸化EGFRの同時検出を示す図である。(A)は、10000細胞中に存在する総EGFRがAlexa647−オリゴヌクレオチド共役抗−EGFR抗体から生じるAlexa647シグナルによって検出されたことを示す。EGFRのリン酸化の割合は、チラミドを介して増幅されたAlexa555シグナルを観察することによって検出された。(B)は、総EGFR(t−EGFR)は、わずか10細胞から直接結合分析によって検出され、リン酸化EGFR(p−EGFR)は1細胞から検出されたことを示す。10e5のp−EGFR分子が近接シグナル増幅法で検出された。p−EGFRの検出限界は、近接分析の形式を用いることによって100倍以上増加した。
【発明を実施するための形態】
【0012】
(I.導入)
本発明は、抗体に基づくアレイ分析システムを用いた希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための方法を提供する。ある実施形態では、本発明の多重で、ハイスループットの免疫分析は、単一の細胞レベルでの固形腫瘍の循環細胞における1つ又はそれ以上のシグナル伝達分子の活性状態を検出することができる。実際に、EGFRのようなシグナル伝達分子は、約100zmolの感度と約100zmolから約100fmolまでの直線性のダイナミックレンジで検出され得る。このように、希少循環細胞における複数のシグナル伝達物質の活性状態の単一細胞での検出は、個別の標的治療法の設計と同様に、癌の予後診断及び診断を促進する。
【0013】
希少循環細胞は、固形腫瘍から転移あるいは微小転移した固形腫瘍の循環細胞を含む。循環腫瘍細胞、癌幹細胞、及び循環内皮前駆細胞、循環内皮細胞、循環血管新生促進骨髄性細胞及び循環樹状細胞のような腫瘍へ移動している細胞(例えば、化学誘引のため)は、固形腫瘍の循環細胞の例である。
【0014】
対象となるシグナル伝達分子は、典型的に、循環細胞がinsituの活性状態を保つように、単離された直後に、好ましくは24,6,又は1時間以内に、より好ましくは30,15,又は5分以内に抽出される。単離された細胞はまた、シグナル伝達分子の活性を復活させる、又は活性化するために、1つ又はそれ以上の増殖因子と、通常nmolからμmolの濃度で、1〜30分間インキュベートされてもよい(Irishら、Cell,118:217−228(2004)を参照)。
【0015】
本明細書でより詳細に説明するように、個々の患者に対する可能な抗癌治療を評価するため、単離された細胞は、1つ又はそれ以上の抗癌剤と様々な用量でインキュベートされ得る。増殖因子の活性化は、その後、数分間(例えば、約1〜5分間)又は数時間(例えば、約1〜6時間)実施され得る。抗癌剤の有無におけるシグナル伝達経路の異なる活性化は、個々の患者各々に対する適量で適切な癌治療の選択の助けとなり得る。循環細胞はまた、治療における変化があるか否かを決定するために、抗癌剤治療中の患者試料から単離され、そして1つ又はそれ以上の増殖因子で活性化され得る。そのように、本発明の方法は、有利に、臨床医が全ての患者に対し、正しい期間、正しい用量で正しい抗癌剤を提供することを支援する。
【0016】
(II.定義)
本明細書において、他に規定されない限り、次の用語は以下に定義される意味を有する。
【0017】
「癌(cancer)」という用語は、異常な細胞の制御されない増殖によって特徴づけられる疾病分類のあらゆるものを含むことを意図されている。この用語は、悪性、良性、軟部組織、あるいは固形として特徴づけられるか否かによらず、全ての既知の癌及び腫瘍性の状態、及び転移前及び転移後の癌を含む全ての病期と悪性度の癌を含む。癌の異なる種類の具体例は、これに限定されないが、肺癌(例えば、非小細胞肺癌);結腸直腸癌、消化管間質腫瘍、消化管カルチノイド癌、結腸癌、直腸癌、肛門癌、胆管癌、小腸癌、及び胃癌のような消化器及び消化管癌;食道癌;胆嚢癌;肝臓癌;膵臓癌;虫垂癌;乳癌;卵巣癌;腎臓癌(例えば、腎細胞癌);中枢神経系の癌;皮膚癌;リンパ腫;絨毛腫;頭部及び頸部癌;骨肉腫;及び血液癌を含む。本明細書において、「腫瘍(tumor)」は1つ又はそれ以上の癌性細胞から構成される。
【0018】
「分析物(analyte)」は、存在、量、及び/又はその正体が決定される、対象となるあらゆる分子、典型的にはポリペプチドのような巨大分子を含む。ある例では、分析物は、固形腫瘍の循環細胞の細胞構成成分であり、好ましくはシグナル伝達分子である。
【0019】
本明細書において、「希釈系列(dilutionseries)」という用語は、特定の試料(例えば、細胞溶解物)あるいは試薬(例えば、抗体)の一連の降順の濃度を含むことを意図されている。希釈系列は、典型的に、開始濃度の試料あるいは試薬の測定した量を、より低濃度の試料あるいは試薬を作るために、希釈剤(例えば、希釈用緩衝液)と混合する工程と、所望の数の連続的な希釈系列を得るのに十分な回数、その工程を繰り返すことによって生成される。試料あるいは試薬は、少なくとも2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,25,30,35,40,45,又は50の降順の濃度の試料あるいは試薬から構成される希釈系列を生成するため、少なくとも2,3,4,5,6,7,8,9,10,15,20,25,30,35,40,45,50,100,500,又は1000倍で連続的に希釈され得る。例えば、1mg/mlの開始濃度での捕捉抗体試薬の2倍の連続的な希釈から構成される希釈系列は、0.5mg/mlの濃度の捕捉抗体を作成するように、開始濃度の捕捉抗体のある用量をそれと等量の希釈用緩衝液と混合し、0.25mg/ml、0.125mg/ml、0.0625mg/ml、0.0325mg/ml、等の捕捉抗体濃度を得るように、この工程を繰り返すことによって生成され得る。
【0020】
本明細書において、「優れたダイナミックレンジ(superiordynamic range)」という用語は、わずか1細胞中又は数千もの細胞中の特定の分析物を検出する、本発明の分析能のことをいう。例えば、本明細書で述べる免疫分析は、有利に、捕捉抗体濃度の希釈系列を用いて、約1〜10000細胞(例えば、約1,5,10,25,50,75,100,250,500,750,1000,2500,5000,7500,又は10000細胞)中における対象となる特定のシグナル伝達分子を検出するため、優れたダイナミックレンジを有する。
【0021】
「シグナル伝達分子(signaltransduction molecule)」又は「シグナル伝達物質(signal transducer)」は、細胞が細胞外シグナル又は刺激を、典型的には細胞内での生化学反応の順序づけられた連続を含む応答に変換する工程を実行するタンパク質又は他の分子を含む。シグナル伝達分子の具体例は、これに限定されないが、EGFR(例えば、EGFR/HER1/ErbB1,HER2/Neu/ErbB2,HER3/ErbB3,HER4/ErbB4)、VEGFR−1/FLT−1、VEGFR−2/FLK−1/KDR、VEGFR−3/FLT−4、FLT−3/FLK−2、PDGFR(例えば、PDGFRA,PDGFRB)、c−KIT/SCFR、INSR(インスリン受容体)、IGF−IR、IGF−IIR、IRR(インスリン受容体関連受容体)、CSF−1R、FGFR1−4、HGFR 1−2、CCK4、TRK A−C、MET、RON、EPHA 1−8、EPHB 1−6、AXL、MER、TYRO3、TIE 1−2、TEK、RYK、DDR1−2、RET、c−ROS、LTK(白血球チロシンキナーゼ)、ALK(未分化リンパ腫キナーゼ)、ROR 1−2、MUSK、AATYK 1−3、及びRTK 106のような受容体型チロシンキナーゼ;BCR−ABL、Src、Frk、Btk、Csk、Abl、Zap70、Fes/Fps、Fak、Jak、Ack、及びLIMKのような非受容体型チロシンキナーゼ;Akt、MAPK/ERK、MEK、RAF、PLA2、MEKK、JNKK、JNK、p38、Shc(p66)、PI3K、Ras(例えば、K−Ras、N−Ras、H−Ras)、Rho、Rac1、Cdc42、PLC、PKC、p70S6キナーゼ、p53、サイクリンD1、STAT1、STAT3、PIP2、PIP3、PDK、mTOR、BAD、p21、p27、ROCK、IP3、TSP−1、NOS、PTEN、RSK1−3、JNK、c−Jun、Rb、CREB、Ki67、及びパキシリン(paxillin)のようなチロシンキナーゼシグナル伝達系の構成成分;及びこれらの組み合わせを含む。
【0022】
本明細書において、「循環細胞(circulatingcells)」という用語は、固形腫瘍から転移あるいは微小転移のいずれかがなされた細胞から構成される。循環細胞の具体例は、これに限定されないが、循環腫瘍細胞、癌幹細胞、及び/又は腫瘍へ移動している細胞(例えば、循環内皮前駆細胞、循環内皮細胞、循環血管新生促進骨髄性細胞、循環樹状細胞、等)を含む。
【0023】
本明細書において、「試料(sample)」という用語は、患者から得られた、あらゆる生物学的標本を含む。試料は、これに限定されないが、全血、血漿、血清、赤血球細胞、白血球細胞(例えば、末梢血単核球)、唾液、尿、便(すなわち、糞)、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液(例えば、リンパ節の播種性腫瘍細胞)、細針吸引物、あらゆる他の体液、腫瘍の生検(例えば、針生検)のような組織試料(例えば、腫瘍組織)、及びこれらの細胞抽出物を含む。ある実施の形態において、試料は、全血、又は血漿、血清あるいは細胞沈殿物のような全血の部分構成成分である。好ましい実施の形態では、試料は、当該分野では既知のあらゆる技術を用いて全血又はその細胞画分から固形腫瘍の循環細胞を単離し、循環細胞の細胞抽出物を調製することによって得られる。他の実施の形態において、試料は、例えば肺、結腸又は直腸の固形腫瘍からの、ホルマリン固定パラフィン包埋(FFPE)腫瘍組織標本である。
【0024】
「被検者(subject)」又は「患者(patient)」は、典型的には、ヒトを含むが、例えば他の哺乳類、齧歯類、イヌ科、ネコ科、ウマ、ヒツジ、ブタ及び同様のもの、のような他の動物も含むことができる。
【0025】
「アレイ(array)」又は「マイクロアレイ(microarray)」は、例えばガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ(例えば磁気ビーズ、ポリスチレンビーズ、等)、紙、膜(例えばナイロン、ニトロセルロース、ポリフッ化ビニリデン(PVDF)、等)、繊維束、又はあらゆる他の適切な基質のような固相担体上に固定あるいは拘束された、特徴的な一組の捕捉抗体及び/又はその希釈系列から構成される。この捕捉抗体は、一般的に共有結合性又は非共有結合性相互作用(例えばイオン結合、疎水性相互作用、水素結合、ファンデルワールス力、双極子−双極子相互作用)を介して固相担体に固定又は拘束されている。ある例として、捕捉抗体は、固相担体に結合した捕捉剤と相互作用する捕捉タグを備える。本発明の分析に用いられるアレイは典型的に、既知でアドレス可能な異なる位置において固相担体の表面に連結した複数の異なる捕捉抗体及び/又は捕捉抗体の濃度から構成される。
【0026】
「捕捉抗体(captureantibody)」という用語は、固形腫瘍の循環細胞の細胞抽出物のような試料における対象となる1つ又はそれ以上の分析物に特異的である(すなわち、分析物に結合する、分析物に結合される、又は分析物と複合体を形成する)固定された抗体を含むことを意図している。好ましい実施の形態では、捕捉抗体はアレイにおける固相担体に拘束されている。あらゆる様々なシグナル伝達分子を固相担体上に固定するために適した捕捉担体は、Upstate(Temecula,CA)、Biosource(Camarillo,CA)、CellSignaling Technologies(Danvers,MA),R&D Systems(Minneapolis,MN),Lab Vision(Fremont,CA),SantaCruz Biotechnology(Santa Cruz,CA),Sigma(St.Louis,MO)、及びBD Biosciences(San Jose,CA)から入手できる。
【0027】
本明細書において、「検出抗体(detectionantibody)」という用語は、試料中の対象となる1つ又はそれ以上の分析物に特異的である(すなわち、分析物に結合する、分析物に結合される、又は分析物と複合体を形成する)、検出可能な標識を備える抗体を含む。この用語はまた、対象となる1つ又はそれ以上の分析物に特異的な抗体であって、検出可能な標識を備える他の種によって結合され得る抗体を包含する。検出可能な標識の例は、これに限定されないが、ビオチン/ストレプトアビジン標識、核酸(例えばオリゴヌクレオチド)標識、化学反応標識、蛍光標識、酵素標識、放射性標識、及びこれらの組み合わせを含む。あらゆる様々なシグナル伝達分子の活性状態及び/又は総量を検出するために適した検出抗体は、Upstate(Temecula,CA),Biosource(Camarillo,CA),CellSignaling Technologies(Danvers,MA),R&D Systems(Minneapolis,MN),Lab Vision(Fremont,CA),SantaCruz Biotechnology(Santa Cruz,CA),Sigma(St.Louis,MO)、及びBD Biosciences(San Jose,CA)から入手できる。非限定的な例として、EGFR,c−KIT,c−Src,FLK−1,PDGFRA,PDGFRB,Akt,MAPK,PTEN,Raf、及びMEKのようなシグナル伝達分子の様々なリン酸化型に対するリン酸特異的抗体は、SantaCruz Biotechnologyから入手できる。
【0028】
「活性状態依存抗体(activationstate−dependent antibody)」という用語は、試料における対象となる1つ又はそれ以上の分析物の特定の活性状態に特異的である(すなわち、結合する、結合される、又は複合体を形成する)検出抗体を含む。好ましい実施の形態では、活性状態依存抗体は、1つ又はそれ以上のシグナル伝達分子のような1つ又はそれ以上の分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出する。ある実施の形態では、受容体型チロシンキナーゼのEGFRファミリーの構成因子のリン酸化及び/又はEGFRファミリーの構成因子間のヘテロ二量体複合体の形成が活性状態依存抗体を用いて検出される。活性状態依存抗体での検出に適した活性状態の非限定的な例(括弧内に記載される)は以下を含む:EGFR(EGFRvIII,リン酸化(p−)EGFR,EGFR:Shc,ユビキチン化(u−)EGFR,p−EGFRvIII);ErbB2(p85:切断型(Tr)−ErbB2,p−ErbB2,p85:Tr−p−ErbB2,Her2:Shc,ErbB2:PI3K,ErbB2:EGFR,ErbB2:ErbB3,ErbB2:ErbB4);ErbB3(p−ErbB3,ErbB3:PI3K,p−ErbB3:PI3K,ErbB3:Shc);ErbB4(p−ErbB4,ErbB4:Shc);IGF−1R(p−IGF−1R,IGF−1R:IRS,IRS:PI3K,p−IRS,IGF−1R:PI3K);INSR(p−INSR);KIT(p−KIT);FLT3(p−FLT3);HGFR1(p−HGFR1);HGFR2(p−HGFR2);RET(p−RET);PDGFRa(p−PDGFRa);PDGFRP(p−PDGFRP);VEGFR1(p−VEGFR1,VEGFR1:PLCγ,VEGFR1:Src);VEGFR2(p−VEGFR2,VEGFR2:PLCγ,VEGFR2:Src,VEGFR2:ヘパリン硫酸塩,VEGFR2:VE−カドヘリン);VEGFR3(p−VEGFR3);FGFR1(p−FGFR1);FGFR2(p−FGFR2);FGFR3(p−FGFR3);FGFR4(p−FGFR4);Tiel(p−Tiel);Tie2(p−Tie2);EphA(p−EphA);EphB(p−EphB);NFKB及び/又はIKB(p−IK(S32),p−NFKB(S536),p−P65:IKBa);Akt(p−Akt(T308,S473));PTEN(p−PTEN);Bad(p−Bad(S112,S136),Bad:14−3−3);mTor(p−mTor(S2448));p70S6K(p−p70S6K(T229,T389));Mek(p−Mek(S217,S221));Erk(p−Erk(T202,Y204));Rsk−1(p−Rsk−1(T357,S363));Jnk(p−Jnk(T183,Y185));P38(p−P38(T180,Y182));Stat3(p−Stat−3(Y705,S727));Fak(p−Fak(Y576));Rb(p−Rb(S249,T252,S780));Ki67;p53(p−p53(S392,S20));CREB(p−CREB(S133));c−Jun(p−c−Jun(S63));cSrc(p−cSrc(Y416));及びパキシリン(p−パキシリン(p−paxillin(Y118))。
【0029】
「活性状態非依存抗体(activationstate−independent antibody)」という用語は、活性状態にかかわりなく試料における対象となる1つ又はそれ以上の分析物に特異的である(すなわち、結合する、結合される、又は複合体を形成する)検出抗体を含む。例えば、活性状態非依存抗体は、シグナル伝達分子のような1つ又はそれ以上の分析物のリン酸化型及び非リン酸化型の両方を検出することができる。
【0030】
「核酸(nucleicacid)」又は「ポリヌクレオチド(polynucleotide)」という用語は、例えばDNA及びRNAのような一本鎖型又は二本鎖型のいずれかにおけるデオキシリボヌクレオチド又はリボヌクレオチド及びこれらのポリマーを含む。核酸は、合成され、自然発生し及び非自然発生し、そして参照となる核酸と同様の結合特性を有する、既知のヌクレオチド類縁体あるいは改変型の骨格残基又は連結を有する核酸を包含する。そのような類縁体の具体例は、これに限定されないが、ホスホロチオエート、ホスホラミデート、メチルホスホネート、キラルメチルホスホネート、2’−O−メチルリボヌクレオチド、及びペプチド核酸(PNAs)を含む。特に限定されることなく、この用語は、参照となる核酸と同様の結合特性を有する、天然核酸の既知の類縁体を含む核酸を包含する。特に指定されない限り、特定の核酸配列はまた、黙示的に、明記された配列と同様に、保存的に改変されたそれらの変異体及び相補的な配列を包含する。
【0031】
「オリゴヌクレオチド(oligonucleotide)」という用語は、RNA、DNA、RNA/DNAハイブリッド、及び/又はこれらの類似物の一本鎖オリゴマー又はポリマーをいう。ある例として、オリゴヌクレオチドは自然に発生した(すなわち、改変されていない)核酸塩基、糖、及びヌクレオシド間(骨格)連結から構成される。他の例として、オリゴヌクレオチドは、改変型核酸塩基、糖、及び/又はヌクレオシド間連結から構成される。
【0032】
本明細書において、「ミスマッチモチーフ(mismatchmotif)」又は「ミスマッチ領域(mismatch region)」という用語は、相補配列に対して100%の相補性を有していないオリゴヌクレオチドの部分をいう。オリゴヌクレオチドは、少なくとも1,2,3,4,5,6,又はそれ以上のミスマッチ領域を有しているかもしれない。ミスマッチ領域は隣接しているかもしれないし、あるいは1,2,3,4,5,6,7,8,9,10,11,12、又はそれ以上のヌクレオチドによって隔てられているかもしれない。ミスマッチモチーフ又は領域は、単一のヌクレオチドから構成されてもよく、あるいは2,3,4,5,又はそれ以上のヌクレオチドから構成されてもよい。
【0033】
「厳密なハイブリダイゼーション条件(stringenthybridization conditions)」という語句は、オリゴヌクレオチドがその相補配列にはハイブリダイズするが、他の配列には全くハイブリダイズしないであろう条件のことをいう。厳密な条件は、配列依存的であり、そして異なる環境では異なるであろう。より長い配列は高温で特異的にハイブリダイズする。核酸のハイブリダイゼーションに関する広範な説明は、Tijssen,Techniquesin Biochemistry and Molecular Biology−−Hybridization with Nucleic Probes,「Overviewof principles of hybridization and the strategy of nucleic acid assays」(1993)に開示されている。一般的に、厳密な条件は、規定のイオン強度pHでの特定の配列に対する熱融解点(Tm)よりも約5〜10℃低くなるように選択される。Tmは、標的に相補するプローブの50%が平衡状態で標的配列にハイブリダイズする(標的配列が過剰に存在するとき、Tmでは、プローブの50%が平衡状態で占領される)温度(定義されたイオン強度、pH,及び核酸濃度下)である。厳密な条件はまた、ホルムアミドのような不安定化剤の添加によっても達成されるかもしれない。選択的又は特異的なハイブリダイゼーションにとって、陽性シグナルは、少なくともバックグラウンドの2倍であり、好ましくはバックグラウンドハイブリダイゼーションの10倍である。
【0034】
2又はそれ以上の核酸の関係において、「実質的に同一な(substantiallyidentical)」又は「実質的な同一性(substantial identity)」という用語は、配列比較アルゴリズムを用いて、又は手動アライメント及び視覚的な検討により測定される際に、比較枠又は所望の領域に渡り、最大一致のために比較され及び整列される時に、同一である、又は同一である核酸の特定の割合(すなわち、特定の領域に渡り、少なくとも約60%、好ましくは少なくとも約65%、70%、75%、80%、85%、90%、又は95%の同一性)を有する2又はそれ以上の配列あるいは部分配列をいう。この定義はまた、文脈が示す時には、同じように、配列の相補性のことをいう。好ましくは、実質的な同一性は、長さが少なくとも約5,10,15,20,25,30,35,40,45,50,75,又は100ヌクレオチドである領域対して存在する。
【0035】
「インキュベートする(incubating)」という用語は、「接触する(contacting)」及び「むき出しにする(exposing)」と同意語として用いられ、他に定義されない限り、特定の時間又は温度の要求を意味しない。
【0036】
(III.実施形態の説明)
本発明は、希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための抗体に基づくアレイ及び癌の予後診断及び診断の促進、及び個別の標的治療法の設計のためのこれらの使用方法を提供する。
【0037】
A.抗体アレイ
1つの態様において、本発明は、細胞抽出物における1つ又はそれ以上の分析物に対する特異的な複数の希釈系列の捕捉抗体から構成される優れたダイナミックレンジを有するアレイを提供し、その捕捉抗体は、固相担体に拘束される。
【0038】
ある実施の態様において、細胞抽出物は、固形腫瘍の循環細胞の抽出物から構成される。循環細胞は典型的に、例えば、免疫磁気分離法(例えばRacilaら、Proc.Natl.Acad.Sci.USA,95:4589−4594(1998);Bilkenrothら、Int.J.Cancer,92:577−582(2001)参照)、マイクロ流体分離法(例えばMohamedら、IEEETrans.Nanobiosci.,3:251−256(2004);Linら、Abstract No.5147,97th AACR Annual Meeting,Washington,D.C.(2006)参照)、FACS(例えばMancusoら、Blood,97:3658−3661(2001)参照)、密度勾配遠心分離法(例えばBakerら、Clin.CancerRes.,13:4865−4871(2003)参照)、及び減少法(例えばMeyeら、Int.J.Oncol.,21:521−530(2002)参照)を含む1つ又はそれ以上の分離法を用いて患者試料から単離される。
【0039】
他の態様において、患者試料は、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引試料から構成される。ある例では、全血試料は、血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は典型的に赤血球、白血球、及び/又は循環腫瘍細胞(CTC)、循環内皮細胞(CEC)、循環内皮前駆細胞(CEPC)、癌幹細胞(CSC)、及びそれらの組み合わせのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞から放出された核酸(例えばDNA、RNA)及びタンパク質を含む。
【0040】
ある例では、単離された循環細胞は、1つ又はそれ以上の対象となる抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、これに限定されないが、上皮細胞増殖因子(EGF)、ヘレグリン(HRG)、TGF−α、PIGF、アンジオポエチン(Ang)、NRG1、PGF、TNF−α、VEGF、PDGF、IGF、FGF、HGF、サイトカイン、及び同様のものを含む。他の例では、単離された循環細胞は、例えば、当該分野で既知のあらゆる技術を用いて細胞抽出物(例えば、細胞溶解物)を作製するために、例えば、増殖因子の刺激及び/又は抗癌剤処理の後に、溶解させることができる。好ましくは、細胞溶解は増殖因子の刺激後、約1〜360分の間に開始され、より好ましくは、2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0041】
ある実施の形態において、抗癌剤は、モノクローナル抗体又はチロシンキナーゼ阻害剤のような抗シグナル伝達剤(すなわち細胞増殖抑制剤)抗増殖剤;a化学療法剤(すなわち細胞毒性薬);及び/又は癌性細胞のような異常な細胞の制御されない増殖を減らす、又は止める能力を有するあらゆる他の化合物から構成される。ある実施の形態において、単離された循環細胞は、1つ又はそれ以上の化学療法剤と組み合わせて、抗シグナル伝達剤及び/又は抗増殖剤で処理される。
【0042】
本発明における使用に適した抗シグナル伝達剤の例は、これに限定されないが、トラスツズマブ(trastuzumab)(Herceptin(登録商標))、アレムツズマブ(alemtuzumab)(Campath(登録商標))、ベバシズマブ(bevacizumab)(Avastin(登録商標))、セツキシマブ(cetuximab)(Erbitux(登録商標))、ゲムツズマブ(gemtuzumab)(Mylotarg(登録商標))、パニツムマブ(panitumumab)(Vectibix(登録商標))、リツキシマブ(rituximab)(Rituxan(登録商標))、及びトシツモマブ(tositumomab)(BEXXAR(登録商標))のようなモノクローナル抗体;ゲフィチニブ(gefitinib)(Iressa(登録商標))、スニチニブ(sunitinib)(Sutent(登録商標))、エルロチニブ(erlotinib)(Tarceva(登録商標))、ラパチニブ(lapatinib)(GW−572016)、カネルチニブ(canertinib)(CI1033)、セマキシニブ(semaxinib)(SU5416)、バタラニブ(vatalanib)(PTK787/ZK222584)、ソラフェニブ(sorafenib)(BAY43−9006;Nexavar(登録商標))、メシル酸イマチニブ(imatinib mesylate)(Gleevec(登録商標))、及びレフルノミド(leflunomide)(SU101)のようなチロシンキナーゼ阻害剤;及びそれらの組み合わせを含む。
【0043】
典型的な抗増殖剤は、シロリムス(sirolimus)(ラパマイシン(rapamycin))、テムシロリムス(temsirolimus)(CCI−779)、及びエベロリムス(everolimus)(RAD001);のようなmTOR阻害剤;1L6−ハイドロキシメチル−キロ−イノシトール−2−(R)−2−O−メチル−3−O−オクタデシル−sn−グリセロカルボネート(1L6−hydroxymethyl−chiro−inositol−2−(R)−2−O−methyl−3−O−octadecyl−sn−glycerocarbonate)、9−メトキシ−2−メチルエリプチシンアセテート(9−methoxy−2−methylellipticiniumacetate)、1,3−ジヒドロ−1−(1−((4−(6−フェニル−1H−イミダゾ[4,5−g]キノキサリン−7−イル)フェニル)メチル)−4−ピペリジニル)−2H−ベンズイミダゾール−2−オン(1,3−dihydro−1−(1−((4−(6−phenyl−1H−imidazo[4,5−g]quinoxalin−7−yl)phenyl)methyl)−4−piperidinyl)−2H−benzimidazol−2−one)、10−(4’−(N−ジエチルアミノ)ブチル)−2−クロロフェノキサジン(10−(4’−(N−diethylamino)butyl)−2−chlorophenoxazine)、3−ホルミルクロモンチオセミカルバゾン(3−formylchromonethiosemicarbazone)(Cu(II)Cl2複合体)、API−2、癌原遺伝子TCL1のアミノ酸10〜24番目由来の15残基ペプチド(Hiromuraら、J.Biol.Chem.,279:53407−53418(2004))、KP372−1、及びKozikowskiら、J.Am.Chem.Soc.,125:1144−1145(2003)及びKauら、CancerCell,4:463−476(2003)に開示された化合物のようなAkt阻害剤;及びそれらの組み合わせを含む。
【0044】
化学療法剤の非限定的な例は、プラチナ製剤(例えば、オキサリプラチン(oxaliplatin)、シスプラチン(cisplatin)、カルボプラチン(carboplatin)、スピロプラチン(spiroplatin)イプロプラチン(iproplatin)、サトラプラチン(satraplatin)等)、アルキル化剤(例えばシクロフォスファミド(cyclophosphamide)、イフォスファミド(ifosfamide)、クロラムブシル(chlorambucil)、ブスルファン(busulfan)、メルファラン(melphalan)、メクロレタミン(mechlorethamine)、ウラムスチン(uramustine)、チオテパ(thiotepa)、ニトロソウレア(nitrosoureas)、等)、代謝拮抗剤(anti−metabolites(例えば、5−フルオロウラシル(5−fluorouracil)、アザチオプリン(azathioprine)、6−メルカプトプリン(6−mercaptopurine)、メトトレキセート(methotrexate)、ロイコボリン(leucovorin)、カペシタビン(capecitabine)、シタラビン(cytarabine)、フロクスウリジン(floxuridine)、フルダラビン(fludarabine)、ゲムシタビン(gemcitabine)、ペメトレキセド(pemetrexed)、ラルチトレキセド(raltitrexed)、等)、植物アルカロイド(例えば、ビンクリスチン(vincristine)、ビンブラスチン(vinblastine)、ビノレルビン(vinorelbine)、ビンデシン(vindesine)、ポドフィロトキシン(podophyllotoxin)パクリタキセル(paclitaxel)、ドセタキセル(docetaxel)、等)、トポイソメラーゼ阻害剤(例えばイリノテカン(irinotecan)、トポテカン(topotecan)、アムサクリン(amsacrine)、エトポシド(etoposide)(VP16)、エトポシドリン酸(etoposidephosphate)、テニポシド(teniposide)、等)、抗腫瘍性抗生物質(antitumor antibiotics)(例えばドキソルビシン(doxorubicin)、アドリアマイシン(adriamycin)、ダウノルビシン(daunorubicin)、エピルビシン(epirubicin)、アクチノマイシン(actinomycin)、ブレノマイシン(bleomycin)、マイトマイシン(mitomycin)、ミトキサントロン(mitoxantrone)、プリカマイシン(plicamycin)、等)、それらの薬学的に許容される塩類、それらの立体異性体、それらの誘導体、それらの類縁体、及びそれらの組み合わせを含む。
【0045】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の例は、上述され、そして受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0046】
ある実施の形態において、捕捉抗体のそれぞれの希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた下降捕捉抗体濃度の設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25,又はそれ以上)から構成される希釈系列を作製するために少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、捕捉抗体の各希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0047】
他の実施の形態において、固相担体は、ガラス(例えば、ガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えばナイロン、ニトロセルロース、ポリフッ化ビニリデン(PVDF)、等)、繊維束、又は他のあらゆる適切な基質から構成される。好ましい実施の形態において、捕捉抗体は、(例えば共有又は非共有結合を介して)例えばWhatmanInc.(Florham Park,NJ)から市販で入手できるFAST(商標登録)スライドのようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0048】
非限定的な例として、図14は、EGFR、HER2、Shc、Erk、及びPI3Kの活性状態を決定するために複数の希釈系列の捕捉抗体から構成され、各希釈系列における捕捉抗体がこれら分析物の1つに向けられた、アドレス可能なマイクロアレイを示す。従って、本発明のアレイは、一連の下降濃度(すなわち希釈系列)にある複数の異なる捕捉抗体から構成され、その捕捉抗体は異なるアドレス可能な位置において固相担体の表面に連結される。
【0049】
当業者は、活性化されたシグナル伝達分子の各々に対する離散的なシグナルが検出されることを許容する任意の構成であり得る。例えば、アレイは、各領域が異なる捕捉抗体又は捕捉剤(すなわち捕捉抗体上に存在する捕捉タグに結合する)を含む担体表面上の一列又は一グリッドの異なる領域(例えばドット又はスポット)であり得る。アレイは、複数のシグナル伝達分子の活性状態が単一の多重分析で検出される方法における使用のために構成され得る。様々な実施の形態において、複数は、少なくとも2,3,4,5,6,7,8,9,10,15,20,25,30,35,40,45,50、又はそれ以上のシグナル伝達分子から構成される。
【0050】
B.単一検出分析
他の形態において、固形腫瘍の循環細胞のような腫瘍細胞の細胞抽出物における対象となる特定の分析物の活性状態を検出するための分析は、優れたダイナミックレンジを有する、多重で、ハイスループットな単一の検出(すなわち2抗体)分析である。非限定的な例として、この分析で用いられる2つの抗体は、以下の構成を備え得る:(1)分析物に特異的な捕捉抗体;及び(2)分析物の活性型に特異的な検出抗体(すなわち活性状態依存抗体)。活性状態依存抗体は、例えば分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出することができる。代替的には、検出抗体は、細胞抽出物中の分析物の総量を検出する活性状態非依存抗体から構成される。活性状態非依存抗体は、一般的に分析物の活性型及び不活性型を検出することができる。
【0051】
好ましい形態において、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するために細胞抽出物における1つ又はそれ以上の分析物に特異的であり、固相担体上に拘束された複数の希釈系列の捕捉抗体と細胞抽出物をインキュベートする工程;
(b)複数の検出可能な捕捉された分析物を形成するために対応する分析物に特異的な検出抗体と複数の捕捉された分析物をインキュベートする構成;
(c)増幅シグナルを生成するためにシグナル増幅対の第一及び第二の構成因子と複数の検出可能な捕捉された分析物をインキュベートする工程;及び
(d)シグナル増幅対の第一又は第二の構成因子から生成された増幅シグナルを検出する工程。
【0052】
ある実施の形態において、細胞抽出物は固形腫瘍の循環細胞の抽出物から構成される。循環細胞は典型的には、例えば免疫磁気分離法、マイクロ流体分離法、FACS、密度勾配遠心分離法を含む当該分野において既知の1つ又はそれ以上の分離方法を用いて患者試料から単離される。当業者は循環細胞の分離及び/又は単離に適した他の方法について知っているだろう。
【0053】
他の実施の形態において、患者試料は、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引物試料から構成される。ある例では、全血試料は血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は、典型的には、赤血球、白血球、及び/又はCTC,CEC,CEPC及び/又はCSCのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞によって放出された核酸(例えばDNA、RNA)及びタンパク質を含む。
【0054】
ある例において、単離された循環細胞は、対象となる1つ又はそれ以上の抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、上述されている。
他の例では、単離された循環細胞は、当該分野で既知の任意の技術を用いて、細胞抽出物(例えば細胞溶解物)を生成するために、例えば増殖因子の刺激及び/又は抗癌剤処理の後に、溶解され得る。好ましくは、細胞溶解は、増殖因子の刺激後1〜360分の間に開始され、より好ましくは2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後、約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0055】
ある実施の形態において、抗癌剤は抗シグナル伝達剤(例えばモノクローナル抗体、チロシンキナーゼ阻害剤、等)、抗増殖剤、化学療法剤、及び/又は腫瘍性細胞のような以上な細胞の制御されない増殖を減らす又は止める能力を有するあらゆる他の化合物から構成される。治療剤のこれらの一般的な分類に該当する特定の抗癌剤は上に提供されている。
【0056】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の具体例は、上述されており、そして、限定されないが、受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0057】
ある実施の形態において、捕捉抗体の各希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた、設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25、又はそれ以上)の下降捕捉抗体濃度から構成される希釈系列を作製するため、少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、各捕捉抗体の希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0058】
他の実施の形態において、固相担体は、ガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えば、ナイロン、ニトロセルロース、PVDF、等)、繊維束、又はあらゆる他の適した基質から構成される。好ましい実施の態様において、捕捉抗体は、例えばFAST(商標登録)スライド(WhatmanInc.;Florham Park,NJ)のようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0059】
ある例では、細胞抽出物は、固相担体にすでに拘束された捕捉抗体とインキュベートされる。他の例では、細胞抽出物は、最初に溶液中で捕捉抗体とインキュベートされ、その後、例えば固相担体に結合された捕捉抗体と相互作用する捕捉抗体上に存在する捕捉タグを介して、捕捉された分析物を固定するために固相担体と接触させられる。
【0060】
ある実施の形態において、検出抗体は、溶液中又は固相担体上に拘束された捕捉抗体に結合した、分析物とインキュベートされる。ある例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で検出抗体とインキュベートされ、その後、溶液中又は固相担体上に拘束された捕捉抗体と接触させられる。他の例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で捕捉抗体及び検出抗体とインキュベートされ、その後、例えば固相担体に結合された捕捉剤と相互作用する捕捉抗体又は検出抗体上に存在する捕捉タグを介して、抗体−分析物複合体を固定するために固相担体と接触させられる。
【0061】
ある例では、検出抗体は、細胞抽出物における1つ又はそれ以上の分析物の総量を検出するために有用な活性状態非依存抗体から構成される。非限定的な例として、活性状態非依存抗体は、1つ又はそれ以上のシグナル伝達分子のリン酸化型及び非リン酸化型の両方を検出することができる。他の例では、検出抗体は、細胞抽出物における1つ又はそれ以上の分析物の活性状態を検出するのに有用な活性状態依存抗体から構成される。好ましくは、活性状態依存抗体は1つ又はそれ以上のシグナル伝達分子のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出する。
【0062】
捕捉抗体及び検出抗体は、典型的には、分析物の結合に関して、それらの間の競合が最小化されるように選択される(すなわち捕捉及び検出抗体は、同時にそれらの対応するシグナル伝達分子に結合することができる)。
【0063】
好ましい実施の形態において、検出抗体は、結合対の第一の構成因子(例えばビオチン)から構成され、そして、シグナル増幅対の第一の構成因子は結合対の第二の構成因子(例えばストレプトアビジン)から構成される。結合対の構成因子は、当該分野で既知の方法を用いて、検出抗体又はシグナル増幅対の第一の構成因子に直接又は間接的に共役し得る。ある例では、シグナル増幅対の第一の構成因子は、ペルオキシダーゼ(peroxidase)(例えば、西洋わさびペルオキシダーゼ(horseradishperoxidase)(HRP)、カタラーゼ(catalase)、クロロペルオキシダーゼ(chloroperoxidase)、シトクロームCペルオキシダーゼ(cytochromec peroxidase)、好酸球ペルオキシダーゼ(eosinophil peroxidase)、グルタチオンペルオキシダーゼ(glutathione peroxidase)、ラクトペルオキシダーゼ(lactoperoxidase)、ミエロペルオキシダーゼ(myeloperoxidase)、チロイドペルオキシダーゼ(thyroidperoxidase)、デヨードナーゼ(deiodinase)、等)であり、そしてシグナル増幅対の第二の構成因子はチラミド試薬(例えばビオチン−チラミド)である。これらの例では、増幅されたシグナルは、過酸化水素(H2O2)存在下で活性型チラミドを作製するためのチラミド試薬のペルオキシダーゼ酸化によって生成される。
【0064】
活性型チラミドは、直接検出され、又は例えばストレプトアビジン標識フルオロフォア又はストレプトアビジン標識ペルオキシダーゼと発色試薬の組み合わせのようなシグナル検出試薬の添加で検出される。本発明における使用に適したフルオロフォアの具体例は、限定されないが、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)555)、フルオレセイン(fluorescein)、フルオレセインイソチオシアネート(fluoresceinisothiocyanate)(FITC)、Oregon Green(登録商標);ローダミン(rhodamine)、テキサスレッド(Texas red)、テトラローダミンイソチオシアネート(tetrarhodamineisothiocynate)(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものを含む。ストレプトアビジン標識は、当該分野においてよく知られた方法を用いてフルオロフォア又はペルオキシダーゼに直接的に又は間接的に連結され得る。本発明の使用に適した発光試薬の非限定的な例は、3,3’,5,5’−テトラメチルベンジジン(3,3’,5,5’−tetramethylbenzidine)(TMB)、3,3’−ジアミノベンジジン(3,3’−diaminobenzidine)(DAB)、2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)(2,2’−azino−bis(3−ethylbenzothiazoline−6−sulfonicacid))(ABTS)、4−クロロ−1−ナフトール(4−chloro−1−napthol)(4CN)、及び/又はポルフィリノーゲン(porphyrinogen)を含む。
【0065】
当業者は、本明細書中で述べる単一検出分析に従って、抗体以外の結合相手が細胞抽出物からの1つ又はそれ以上の分析物を固定し及び/又は検出するために使用され得ることを理解するだろう。そのような結合相手の非限定的な例は、分析物のリガンド又は受容体、分析物の基質、結合ドメイン(例えばPTB、SH2等)、アプタマー、及び同様のものを含む。
【0066】
C.近接二重検出分析
さらに他の形態では、固形腫瘍の循環細胞のような腫瘍細胞の細胞抽出物における対象となる特定の分析物の活性状態を検出するための分析は、優れたダイナミックレンジを有する、多重、ハイスループットの近接(すなわち3抗体)分析である。非限定的な例として、この近接分析で用いられる3つの抗体は、以下の構成を備え得る:(1)分析物に特異的な捕捉抗体;(2)分析物の活性型に特異的な検出抗体(すなわち活性状態依存抗体);及び(3)分析物の総量を検出する検出抗体(すなわち活性状態非依存抗体)。活性状態依存抗体は、例えば分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出することができる。活性状態依存抗体は一般的に分析物の活性型及び不活性型の両方を検出することができる。
【0067】
好ましい形態では、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するため、細胞抽出物における1つ又はそれ以上の分析物に特異的な複数の希釈系列の捕捉抗体と、細胞抽出物をインキュベートする工程であって、前記捕捉抗体が固相担体上に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、対応する分析物に特異的な検出抗体とインキュベートする工程であって、前記検出抗体は:
(1)促進部分で標識された複数の活性状態非依存抗体、と
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体と、を備え、
前記促進部分は、シグナル増幅対の第一の構成因子に向けられ且つそれと反応する酸化剤を生成する、
ことを特徴とする工程;
(c)増幅シグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第二の構成因子とインキュベートする工程;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0068】
図1は、分析物が1つの捕捉抗体と2つの検出抗体(すわなち活性状態非依存抗体と活性状態依存抗体)に結合した典型的な近接分析を示す。捕捉抗体1と活性状態非依存抗体2はそれぞれ、その活性状態に非依存して分析物6に結合する。活性状態依存抗体3は、その活性状態に依存して分析物に結合する(例えば活性状態依存抗体はリン酸化残基を有する分析物の活性型にのみ結合するだろう)。活性状態非依存抗体は、促進部分(M1で表す、4)で標識され、そして活性状態依存抗体はシグナル増幅対の第一の構成因子(M2で表す、5)で標識される。両方の検出抗体の分析物への結合は、促進部分を、充分に近接した範囲(点線7の内部の領域によって描かれる)内でシグナル増幅対の第一の構成因子に向かわせるので、促進部分によって生成されたシグナルは、シグナル増幅対の第一の構成因子に向けられ得、結果として検出可能な及び/又は増幅可能なシグナルの生成をもたらす。近接チャネリングのための様々な方法が当該分野で既知であり、例えばFRET、時間分解蛍光−FRET、LOCI、等を含む。近接チャネリングの利点は、本発明の方法において使用されるように、単一の検出可能なシグナルが、全ての3つの抗体が結合したそれらの分析物のみに対して生成され、結果として増加した分析の特異性、より低いバックグラウンド、及び単純化された検出をもたらすことである。
【0069】
ある実施の形態において、細胞抽出物は固形腫瘍の循環細胞の抽出物から構成される。循環細胞は、典型的には、例えば免疫磁気分離法、マイクロ流体分離法、FACS、密度勾配遠心分離法、及び減少法を含む当該分野において既知の1つ又はそれ以上の分離方法を用いて患者試料から単離される。
【0070】
他の実施の形態において、患者試料は全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引物試料から構成される。ある例では、全血試料は、血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は典型的に赤血球、白血球、及び/又はCTC、CEC、CEPC、及び/又はCSCのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞によって放出される核酸(例えばDNA、RNA)及びタンパク質を含む。
【0071】
ある例では、単離された循環細胞は、対象となる1つ又はそれ以上の抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、上述されている。他の例では、単離された循環細胞は、当該分野で既知の任意の技術を用いて、細胞抽出物(例えば細胞溶解物)を作製するために、例えば増殖因子の刺激及び/又は抗癌剤処理の後に、溶解され得る。好ましくは、細胞溶解は、増殖因子の刺激後1〜360分の間に開始され、より好ましくは2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後、約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0072】
ある実施の形態において、抗癌剤は抗シグナル伝達剤(例えば、モノクローナル抗体、チロシンキナーゼ阻害剤、等)、抗増殖剤、化学療法剤、及び/又は腫瘍細胞のような異常な細胞の制御されない増殖を減らし又は止める能力を有する任意の他の化合物から構成される。治療剤のこれらの一般的な分類に該当する特異的な抗癌剤の具体例は、上記に提供されている。
【0073】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は、複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の例は上述され、そしてこれに限定されないが、受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0074】
ある実施の形態において、捕捉抗体のそれぞれの希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた下降捕捉抗体濃度の設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25,又はそれ以上)から構成される希釈系列を作製するために少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、捕捉抗体の各希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0075】
他の実施の形態において、固相担体は、ガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えば、ナイロン、ニトロセルロース、PVDF、等)、繊維束、又はあらゆる他の適した基質から構成される。好ましい実施の態様において、捕捉抗体は、例えばFAST(商標登録)スライド(WhatmanInc.;Florham Park,NJ)のようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0076】
ある例では、細胞抽出物は固相担体上にすでに拘束された捕捉抗体とインキュベートされる。他の例では、細胞抽出物は、始めに溶液中で捕捉抗体とインキュベートされ、その後、例えば固相担体に結合した捕捉剤と相互作用する捕捉抗体上に存在する捕捉タグを介して、捕捉された分析物を固定するために、固相担体と接触させられる。
【0077】
ある実施の形態において、検出抗体は溶液中又は固相担体上に拘束された捕捉抗体と結合した分析物とインキュベートされる。ある例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で検出抗体とインキュベートされ、その後溶液中又は固相担体上に拘束された捕捉抗体と接触させられる。他の例では、複数の分析物から構成される細胞抽出物は、先ず始めに溶液中で捕捉抗体及び検出抗体とインキュベートされ、その後、例えば固相担体に結合した捕捉剤と相互作用する、捕捉抗体又は検出抗体上に存在する捕捉タグを介して、抗体−分析物複合体を固定するため、固相担体と接触させられる。検出工程に先んじて、固定された複合体は、複合体を形成していない抗体を除去するために洗浄され得、洗浄された複合体は、連続的に支持表面から放出され得、そして、分析されている分析物それぞれに対する近接チャネリングが、本明細書で述べるような適切な方法によって、検出され得る。
【0078】
担体表面がアレイに拘束された捕捉剤から構成される実施の形態において、インキュベートする工程は、反応を完了させるために全ての3つの抗体の過剰量を用いて、溶液中の複数の分析物から構成される細胞抽出物を捕捉抗体及び検出抗体と接触させることから構成され得る。この方法の1つの変形は、結果としてもたらされた抗体−分析物複合体が固体層に付着され、非結合の抗体を取り除くために洗浄される。図2を参照すると、捕捉抗体1は捕捉タグ10から構成され得る。複合体は、固体層に接着され且つ捕捉タグと結合した捕捉剤11を介して固体層12に付着し、それによって複合体を固定している。固定された複合体は適切な緩衝液で洗浄され、その後放出剤13の添加によって固体層から放出される。放出剤は洗浄された複合体の放出をもたらす任意の機構によって機能してよい。1つの実施の形態において、捕捉タグは放出剤によって認識され、切断される切断部位を備える。他の実施の形態において、図2に示すように、放出剤は、捕捉剤に結合することに対して捕捉タグと競合する。例えば、捕捉剤は、捕捉抗体に付着された部分的に相補するオリゴヌクレオチド(すなわち捕捉タグ)とハイブリダイズする第一のオリゴヌクレオチドであってもよく;そして放出剤は、捕捉剤と完全に相補するオリゴヌクレオチドであってもよく、結果、固体層から洗浄された複合体の鎖の置換及び放出をもたらす。使用され得る適切な捕捉タグ/捕捉剤/放出剤の他の例は、これに限定されないが、2,4−ジニトロフェノール(2,4−dinitrophenol)(DNP)/抗−DNP抗体(anti−DNPantibody)/2,4−DNPリジン(2,4−DNP lysine);T2/抗−T3抗体(anti−T3 antibody)/T3;ウアバイン(ouabain)/抗−ジゴキシン抗体A(anti−digoxinantibody)/ジゴキシン(digoxin);及びデチオビオチン(dethiobiotin)ストレプトアビジン(streptavidin)/ビオチン(biotin)(例えばIshikawaら、J.Clin.LabAnal.,12:98−107(1998)を参照)。
【0079】
洗浄された複合体が固体層から放出された後、それは:(1)捕捉抗体上に捕捉タグを特異的に結合するアレイに拘束された捕捉分子から構成される担体表面と接触させられ、又は(2)解離され、そして解離された検出抗体は、検出抗体上の捕捉タグと特異的に結合する捕捉剤から構成される担体表面と接触させられる。図2は、洗浄された複合体が解離され、そして解離された検出抗体が担体表面14と接触させられる実施の形態を示す。担体表面は「アドレス可能な(addressable)」又は「ジップコード(zipcode)」アレイに拘束された複数の捕捉分子から構成される。このアレイの各異なる領域は、活性状態非依存抗体2又は活性状態依存抗体3上に存在する捕捉タグ8に特異的に結合する特徴のある捕捉剤9から構成され、それによってアレイにおけるタグ付けされた検出抗体を拘束し、組織化する。好ましい実施の形態において、捕捉剤及び捕捉タグは互いに特異的にハイブリダイズするオリゴヌクレオチドである。オリゴヌクレオチド捕捉分子から構成されるアドレス可能なアレイは、当該分野ではよく知られている(例えばKeramasら、LabChip,4:152−158(2004);Delrio−Lafreniereら、Diag.Microbiol.Infect.Dis.,48:23−31(2004)を参照)。
【0080】
アレイの各異なる領域での検出抗体の存在は、促進部分(M1で表す、4)又はシグナル増幅対の第一の構成因子(M2で表す、5)のような部位で直接的に又は間接的に検出され得る。直接的に検出され得る部位の例は、フルオロフォア、クロモフォア(chromophore)、金コロイド、着色ゴム等を含む。1つの実施の形態において、両方の部位は、非依存的に選択されたフルオロフォアである。区別可能な読み出し情報を提供し、一方で互いに非常に近接している、例えばCy3/Cy5、Cy5/フィコエルトリン(phycoerthrin)、及び同様のものに例示される、任意のフルオロフォア対が使用され得る。代替的には、オリゴヌクレオチドのアドレス可能なアレイが用いられると、両方の部位が異なるジップコードに伝達される同一のフルオロフォアであり得る。レーザースキャニング共焦点顕微鏡法は、アレイ上に接着したフルオロフォア部位を検出するために使用され得る。鎖置換分析のような、検出に先んじて複合体がアレイから放出されるアレイにおいて、フルオロフォア部位を検出するための適切な方法は、キャピラリーフロー共焦点レーザー誘起蛍光法、ナノHPLC(nano−HPLC)、電気泳動等を含む。
【0081】
ある実施の形態において、活性状態非依存抗体はさらに、検出可能な部位を備える。そのような例では、検出可能な部位の量は、細胞抽出物における1つ又はそれ以上の分析物の量に相関する。検出可能な部位の具体例は、これに限定されないが、蛍光標識、化学反応標識、酵素標識、放射性標識、及び同様のものが含まれる。好ましくは、検出可能な部位は、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)647)、フルオレセイン、フルオレセインイソチオシアネート(FITC)、Oregon Green(登録商標);ローダミン、テキサスレッド、テトラローダミンイソチオシアネート(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものに例示されるフルオロフォアである。検出可能な部位は、当該分野でよく知られた方法を用いて、直接的に又は間接的に活性状態非依存抗体に共役することができる。
【0082】
ある例では、活性状態非依存抗体は、促進部分で直接標識される。促進部分は、当該分野でよく知られた方法を用いて、活性状態非依存抗体に連結され得る。本発明の使用に適した促進部分は、その促進部分に近接した(すわなち、空間的に近く又は接近した)他の分子に向かわせ(すなわち、それへと方向付け)且つそれと反応する(すなわち結合し、結合され、又は複合体を形成する)酸化剤を生成することが可能なあらゆる分子を含む。促進部分の例は、これに限定されないが、電子受容体として分子酸素(O2)を含む酸化/還元反応に触媒作用を及ぼすグルコースオキシダーゼ又はあらゆる他の酵素に例示される酵素、及びメチレンブルー(methyleneblue)、ローズベンガル(rose bengal)、ポルフィリン(porphyrin)、スクアレート染色剤(squarate dye)、フタロシアニン(phthalocyanine)、及び同様のものに例示される光増感剤を含む。酸化剤の非限定的な例は、過酸化水素(H2O2)、一重項酸素、及び酸化/還元反応において酸素原子を移し、又は電子を獲得するあらゆる他の化合物を含む。好ましくは、適切な基質(例えばグルコース、光等)の存在下で、促進部分(例えば、グルコースオキシダーゼ、光増感剤等)が、2つの部位が互いに近接するときに、シグナル増幅対の第一の構成因子(例えば、西洋わさびペルオキシダーゼ(HRP)、保護基によって保護されたハプテン、酵素阻害剤とチオエーテル結合によって不活性化された酵素等)に向かわせ且つそれと反応する酸化剤(例えば、過酸化水素(H2O2)、一重項酸素等)を生成する。
【0083】
他の例では、活性状態非依存抗体は、活性状態非依存抗体に共役したオリゴヌクレオチドリンカーと促進部分に共役した相補ヌクレオチドリンカーとの間のハイブリダイゼーションを介して促進部分で間接的に標識される。オリゴヌクレオチドリンカーは、促進部分又は当該分野でよく知られた方法を用いて活性状態非依存抗体に連結され得る。ある実施の形態において、促進部分に共役したオリゴヌクレオチドリンカーは、活性状態非依存抗体に共役したオリゴヌクレオチドリンカーと100%の相補性を有する。他の実施の形態において、オリゴヌクレオチドリンカー対は、例えば厳格なハイブリダイゼーション条件下でのハイブリダイゼーションにおいて少なくとも1,2,3,4,5,6,又はそれ以上のミスマッチ領域を備える。当業者は、異なる分析物に特異的な活性状態非依存抗体が同一のオリゴヌクレオチドリンカー又は異なるオリゴヌクレオチドリンカーのいずれにも共役され得ることを理解している。
【0084】
促進部分又は活性状態非依存抗体に共役したオリゴヌクレオチドリンカーの長さは、可変である。一般的に、リンカー配列は長さが少なくとも約5,10,15,20,25,30,35,40,45,50,75,又は100ヌクレオチドであり得る。典型的に、ランダムなヌクレオチド配列が連結のために生成される。非限定的な例として、ヌクレオチドリンカーのライブラリーが3つの独特の隣接ドメインを有するように設計され得る:スペーサードメイン;署名ドメイン(signaturedomain);及び共役ドメイン。好ましくは、オリゴヌクレオチドリンカーは、それらが共役される促進部分又は活性状態非依存抗体の機能を破壊することのない効率的な連結のために設計される。
【0085】
オリゴヌクレオチドリンカー配列は、様々な分析条件下であらゆる二次構造形成を避け又は最小化するよう設計され得る。溶解温度は、典型的には、分析の全工程におけるそれらの関与を許容するようリンカー内の各部分に対して注意深く監視される。一般的に、リンカー配列の部分の溶解温度の範囲は、5℃以下である。規定されたイオン濃度下における溶解温度、二次構造、及びヘアピン構造を決定するためのコンピュータアルゴリズム(例えばOLIGO6.0)は各リンカー内における3つの異なるドメインの各々を解析するために使用され得る。全ての組み合わせ配列はまた、それらの構造上の特徴と他の共役したオリゴヌクレオチドリンカー配列との比較可能性、例えばそれらが厳密なハイブリダイゼーション条件下で相補するオリゴヌクレオチドリンカーとハイブリダイズするかどうか、に関して分析され得る。
【0086】
オリゴヌクレオチドリンカーのスペーサードメインは、オリゴヌクレオチドの架橋部位からの共役ドメインの適切な分離を提供する。共役ドメインは、相補するヌクレオチドリンカー配列で標識された分子を、核酸ハイブリダイゼーションを介して共役ドメインと連結するのに働く。核酸を介したハイブリダイゼーションは、抗体−分析物(すなわち抗原)複合体の形成前又は後のいずれかで実行され得、より柔軟性のある分析構成を提供する。多くの直接抗体共役法とは異なり、抗体又は他の分子に比較的小さいオリゴヌクレオチドを連結することは、標的分析物への抗体の特異親和性又は共役した分子の機能に対してわずかな影響しか及ぼさない。
【0087】
ある実施の形態において、オリゴヌクレオチドリンカーの署名配列ドメインは、複合体重合化タンパク質分析(complexmultiplexed protein assayにおいて使用され得る。多様な抗体が、異なる署名配列とオリゴヌクレオチドリンカーで共役され得る。多重化免疫分析において、適切なプローブで標識されたリポーターオリゴヌクレオチド配列は、多重化分析の形式において、抗体と抗原との間のクロスハイブリダイゼーションを検出するために使用され得る。
【0088】
オリゴヌクレオチドリンカーは、様々な異なる方法を用いて、抗体又は他の分子に共役され得る。例えば、オリゴヌクレオチドリンカーは、5’又は3’末端のいずれかにおけるチオール基とともに合成され得る。チオール基は、還元剤(例えば、TCEP−HCl)を用いて脱保護され得、結果として生じるリンカーは脱塩回転カラムを用いることによって精製され得る。結果として生じる脱保護オリゴヌクレオチドリンカーは、SMCCのようなヘテロ二官能性架橋剤を用いて抗体の第一級アミン又は他の種類のタンパク質に共役され得る。代替的には、オリゴヌクレオチドの5’−リン酸基は、リン酸エステルを形成するために水溶性カルボジイミドEDCで処理され、その後、アミン含有分子に共役され得る。ある例では、3’−リボース残基のジオールは、酸化されてアルデヒド基になり、その後、還元的アミノ化を用いて核酸のアミン基又は他の種類のタンパク質に共役され得る。他の例では、オリゴヌクレオチドリンカーは、3’又は5’末端のビオチン修飾とともに合成され、ストレプトアビジン標識分子に共役され得る。
【0089】
オリゴヌクレオチドリンカーは、Usmanら、J.Am.Chem.Soc.,109:7845(1987);Scaringeら、Nucl.AcidsRes.,18:5433(1990);Wincottら、Nucl.Acids Res.,23:2677−2684(1995);及びWincottら、MethodsMol.Bio.,74:59(1997)に開示されるような、当該分野で既知のあらゆる様々な技術を用いて合成され得る。一般的に、オリゴヌクレオチドの合成は、5’末端にジメトキシトリチル(dimethoxytrityl)と3’末端にホスホラミダイト(phosphoramidite)のような共通の核酸保護及び共役基を用いる。オリゴヌクレオチド合成に適した試薬、核酸脱保護のための方法、及び核酸精製のための方法は当業者に既知である。
【0090】
ある例では、活性状態依存抗体はシグナル増幅対の第二の構成因子で直接標識される、シグナル増幅対の構成因子は当該分野でよく知られた方法を用いて活性状態依存抗体に連結され得る。他の例では、活性状態依存抗体は、活性状態依存抗体に共役した結合対の第一の構成因子とシグナル増幅対の第一の構成因子に共役した結合対の第二の構成因子との間の結合を介して、シグナル増幅対の第一の構成因子で間接的に標識される。結合対の構成因子(例えばビオチン/ストレプトアビジン)は、当該分野でよく知られた方法を用いてシグナル増幅対の構成因子又は活性状態依存抗体に連結され得る。シグナル増幅対の構成因子の具体例は、これに限定されないが、西洋わさびペルオキシダーゼ(HRP)、カタラーゼ、クロロペルオキシダーゼ、シトクロームCペルオキシダーゼ、好酸球ペルオキシダーゼ、グルタチオンペルオキシダーゼ、ラクトペルオキシダーゼ、ミエロペルオキシダーゼ、チロイドペルオキシダーゼ、デヨードナーゼ、及び同様のものを含む。シグナル増幅対構成因子の他の具体例は、保護基によって保護されたハプテン及び酵素阻害剤とのチオエーテル結合によって不活性化された酵素を含む。
【0091】
捕捉抗体、活性状態非依存抗体、及び活性状態依存抗体は、典型的には、分析物との結合に関して、それらの間における競合が最小化される(すなわち全ての抗体が同時に対応するシグナル伝達分子に結合することができる)ように選択される。
【0092】
近接チャネリングの1つの例において、促進部分はグルコースオキシダーゼ(GO)であり、シグナル増幅対の第一の構成因子は西洋わさびペルオキシダーゼ(HRP)である。GOがグルコースのような基質と接触すると、それは酸化剤(すなわち過酸化水素(H2O2))を生成する。HRPがGOとチャネリングする隣接範囲内にあると、GOによって生成されたH2O2は、シグナル増幅対の第二の構成因子(例えば、ルミノール(luminol)又はイソルミノール(isoluminol)のような化学発光基質、又はチラミド(tyramide)(例えばビオチン−チラミド)、ホモバニリン酸(homovanillicacid)又は4−ヒドロキシフェニル酢酸(4−hydroxyphenyl acetic acid)のような蛍光性基質)の存在下で増幅シグナルを生成する、HRP−H2O2複合体を形成するためにHRPに向けられ且つそれと複合体を形成する。近接分析におけるGOとHRPを用いる方法は、例えば、Langryら、U.S.Dept.ofEnergy Report No.UCRL−ID−136797(1999)に開示される。ビオチン−チラミドがシグナル増幅対の第二の構成因子として用いられるとき、HRP−H2O2複合体はチラミドを酸化して隣接する求核残基に共有的に結合する反応性チラミドラジカルを生成する。活性型チラミドは、直接検出され、又は、例えばストレプトアビジン標識フルオロフォア又はストレプトアビジン標識ペルオキシダーゼと発色試薬との組み合わせのようなシグナル検出試薬の添加に応じて検出される。本発明の使用に適したフルオロフォアの具体例は、これに限定されないが、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)555)、フルオレセイン、フルオレセインイソチオシアネート(FITC)、Oregon Green(登録商標);ローダミン、テキサスレッド、テトラローダミンイソチオシアネート(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものを含む。ストレプトアビジン標識は、当該分野においてよく知られた方法を用いてフルオロフォア又はペルオキシダーゼに直接的に又は間接的に連結され得る。本発明の使用に適した発光試薬の非限定的な例は、3,3’,5,5’−テトラメチルベンジジン(TMB)、3,3’−ジアミノベンジジン(DAB)、2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)(ABTS)、4−クロロ−1−ナフトール(4CN)、及び/又はポルフィリノーゲンを含む。
【0093】
近接チャネリングの他の例では、促進部分は光増感剤であり、シグナル増幅対の第一の構成因子は特異的な結合相手(例えば、リガンド、抗体等)へのハプテンの結合を避ける保護基で保護された複数のハプテンで標識された巨大分子である。例えば、シグナル増幅対の構成因子は、ビオチン、クマリン(coumarin)、及び/又はフルオレセイン分子で標識されたデキストラン分子であり得る。適切な保護基は、これに限定されないが、フェノキシ、アニリノ(analino−)、オレフィン、チオエーテル、及びセレノエーテル保護基を含む。本発明の近接分析における使用に適した付加的な光増感剤及び保護されたハプテン分子は、米国特許第5,807,675号に開示される。光増感剤が光で励起されると、それは酸化剤(すなわち一重項酸素)を生成する。ハプテン分子が光増感剤と隣接してチャネリングする範囲内にあると、光増感剤によって生成された一重項酸素がカルボニル基(ケトン又はアルデヒド)及びスルフィン酸を得るためにハプテンの保護基におけるチオエーテルに向けられ、それと反応し、ハプテンから保護基を放出する。保護されていないハプテンは、その後、シグナル増幅対の第二の構成因子(例えば検出可能なシグナルを生成することができる特異的な結合相手)と特異的に結合するために使用され得る。例えば、ハプテンがビオチンであるとき、特異的な結合相手は酵素標識されたストレプトアビジンであり得る。典型的な酵素は、アルカリホスファターゼ(alkalinephosphatase)、β−ガラクトシダーゼ(β−galactosidase)、HRP等を含む。結合していない試薬を除去するための洗浄後、検出可能なシグナルは、その酵素の検出可能な(例えば、蛍光性、化学発光性、発色性等)基質を添加することによって生成され、当該分野で既知の適切な方法と装置を用いて検出され得る。代替的には、検出可能なシグナルは、チラミドシグナル増幅を用いて増幅され得、活性化されたチラミドは直接検出され又は上述したシグナル検出試薬の添加により検出される。
【0094】
近接チャネリングのさらに他の例では、促進部分は光増感剤であり、シグナル増幅対の第一の構成因子は酵素−阻害剤複合体である。酵素と阻害剤(例えば、ホスホン酸標識デキストラン)は、切断可能なリンカー(例えば、チオエーテル)によって互いに連結される。光増感剤が光で励起されると、それは酸化剤(例えば一重項酸素)を生成する。酵素−阻害剤複合体が光増感剤とチャネリングする隣接距離内にあると、光増感剤によって生成された一重項酸素が切断可能なリンカーに向けられ、それと反応し、酵素から阻害剤を放出し、それによって酵素を活性化する。酵素基質は、検出可能なシグナルを生成するために添加され、又は代替的には、増幅剤が増幅シグナルを生成するために添加される。
【0095】
近接チャネリングのさらなる例では、促進部分はHRPであり、シグナル増幅対の第一の構成因子は保護されたハプテン又は上述したような酵素−阻害剤複合体であり、そして保護基はp−アルコキシフェノールから構成される。フェニレンジアミン(phenylenediamine)とH2O2の添加は、露出したハプテン又は反応性酵素を得るために、保護されたハプテン又は酵素−阻害剤複合体に向けられ、p−アルコキシフェノール保護基と反応する、反応性フェニレンジイミン(phenylenediimine)を生成する。増幅シグナルは上述のように生成され、検出される(例えば、米国特許第5,532,138号及び第5,445,944号参照)。
【0096】
当業者は、抗体以外の結合相手が、この明細書で述べられる近接(すなわち3抗体)分析に関して細胞抽出物からの1つ又はそれ以上の分析物を固定し及び/又は検出するために用いられ得ることを理解している。そのような結合相手の非限定的な例は、分析物のリガンド又は受容体、分析物の基質、結合ドメイン(例えば、PTB,SH2等)、アプタマー、及び同様のものを含む。
【0097】
D.キット
さらなる態様において、本発明は上述した抗体に基づくアレイ方法を実行するためのキットを提供し、以下の構成を備える:(a)固相担体上に拘束された複数の捕捉抗体の希釈系列;及び(b)複数の検出抗体(例えば、活性状態非依存抗体、及び/又は活性状態依存抗体)。ある例では、このキットはさらに、固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書を含むことができる。このキットはまた、例えば、シグナル増幅対の第一及び第二の構成因子、チラミドシグナル増幅試薬、促進部分のための基質、洗浄緩衝液、捕捉/放出試薬等のような、本発明の特異的な方法を実行することに関する上述したあらゆる付加的な試薬を包含してもよい。
【0098】
IV.抗体アレイの構築
ある形態において、本発明は、固相担体上に拘束された捕捉抗体の希釈系列を用いて、固形腫瘍の循環細胞の細胞抽出物における複数のシグナル伝達分子の活性状態を検出するための抗体に基づくアレイを提供する。本発明の分析において使用されるアレイは、典型的には、異なるアドレス可能な位置における固相担体の表面に連結された一定範囲の捕捉抗体濃度の複数の異なる捕捉抗体から構成される。
【0099】
固相担体は、タンパク質を固定するために適したあらゆる基質から構成され得る。固相担体の具体例は、これに限定されないが、ガラス(例えば、ガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ(例えば、磁気ビーズ、ポリスチレンビーズ等)、紙、膜、繊維束、ゲル、金属、セラミック、及び同様のものを含む。ナイロン(Biotrans(登録商標),ICNBiomedicals,Inc.(Costa Mesa,CA);Zeta−Probe(登録商標),Bio−Rad Laboratories(Hercules,CA))、ニトロセルロース(Protran(登録商標),WhatmanInc.(Florham Park,NJ))、及びPVDF(Immobilon(登録商標)、Millipore Corp.(Billerica,MA))のような膜は、本発明のアレイにおける固相担体としての使用に適している。好ましくは、捕捉抗体は、ニトロセルロースポリマーでコートされたガラススライド、例えばWhatmanInc.(Florham Park,NJ)から市販で入手できるFAST(商標登録)スライド上に拘束される。
【0100】
望ましい固相担体の特定の態様は、多量の捕捉抗体と結合できること、最小限の変性で捕捉抗体と結合できること、及び他のタンパク質に結合できないことを含む。他の適した態様は、捕捉抗体を含む抗体溶液が固相担体に塗布されるときに固相担体が最小限の「ウィッキング(wicking)」を示すことである。最小限のウィッキングを有する固相担体は、担体に塗布された少量の分割量の捕捉抗体溶液が、結果として固定された捕捉抗体の小さく、明確なスポットとなることを許容する。
【0101】
捕捉抗体は、典型的には、共有結合又は非共有結合の相互作用(例えばmイオン結合、疎水性相互作用、水素結合、ファンデルワールス力、双極子−双極子相互作用)を介して固相担体上に直接的に又は間接的に(例えば捕捉タグを介して)拘束される。ある実施の形態において、捕捉抗体は、標準的な架橋方法と条件を用いたホモ二官能性又はヘテロ二官能性架橋剤を用いて固相担体に共有結合される。適した架橋剤は、例えばPierceBiotechnology(Rockford,IL)のような製造元から市販で入手できる。
【0102】
本発明のアレイを生成するための方法は、これに限定されないが、タンパク質又は核酸アレイを構築するために使用されるあらゆる技術を含む。ある実施の形態において、捕捉抗体は、典型的にはスプリットピン、ブラントピン、又はインクジェットプリンティングを備えるロボティック印刷装置であるマイクロスポッターを用いてアレイ上にスポットされる。この明細書において述べる抗体アレイを印刷するために適したロボットシステムは、ChipMaker2スプリットピン(TeleChem International;Sunnyvale,CA)を有するPixSys 5000ロボット(Cartesian Technologies;Irvine,CA)の他に、BioRobics(Woburn,MA)及びPackardInstrument Co.(Meriden,CT)から入手できる他のロボティック印刷装置を含む。好ましくは、各捕捉抗体の希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0103】
本発明の抗体アレイを生成するための他の方法は、規定量の液体を固相担体に引き込むために効果的な条件下で固相担体上にキャピラリー分注器を接触させこることにより各選択されたアレイの位置に既知量の捕捉抗体の希釈液を分注する工程を備え、この工程は、完全なアレイを作製するように各選択されたアレイ位置で選択された捕捉抗体希釈液を用いて繰り返される。この方法は、溶液沈着工程が、各繰り返しサイクルで各々の複数の固相担体上の選択された位置に適用される、複数のそのようなアレイを形成することに用いられてもよい。そのような方法のさらなる説明は、例えば米国特許第5,807,522号に開示されている。
【0104】
ある例では、紙に印刷するためのデバイスは、本発明の抗体アレイを生成するために使用され得る。例えば、所望の捕捉抗体濃度がデスクトップジェットプリンターのプリントヘッド部に取り込まれ、適切な固相担体に印刷され得る(例えば、Silzelら、Clin.Chem.,44:2036−2043(1998)を参照)。
【0105】
ある実施の形態において、固相担体上に生成されたアレイは、少なくとも約5スポット/cm2、好ましくは少なくとも約10,20,30,40,50,60,70,80,90,100,110,120,130,140,150,160,170,180,190,200,210,220,230,250,275,300,325,350,375,400,425,450,475,500,550,600,650,700,750,800,850,900,950,1000,2000,3000,4000,5000,6000,7000,8000,9000、又は10,000スポット/cm2の密度を有する。
【0106】
ある例では、固相担体上のスポットは各々、異なる捕捉抗体を提示する。他の例では、固相担体上の複数のスポットが、例えば一連の下降捕捉抗体濃度から構成される希釈系列として、同一の捕捉抗体を提示する。
【0107】
固相担体上に抗体アレイを準備し、構築するための方法の追加の例は、米国特許第6,197,599号、第6,777,239号、第6,780,582号、第6,897,073号、第7,179,638号及び第7,192,720号;米国特許出願第20060115810号、第20060263837号、第20060292680号、及び第20070054326号;及びVarnumら、MethodsMol.Biol.,264:161−172(2004)に開示される。
【0108】
抗体アレイを走査する方法は、当該分野では既知であり、これに限定されないが、タンパク質又は核酸アレイを走査するために使用されるあらゆる技術を含む。本発明の使用に適したマイクロアレイスキャナーは、PerkinElmer(Boston,MA)、AgilentTechnologies(Palo Alto,CA)、Applied Precision(Issaquah,WA)、GSI Lumonics Inc.(Billerica,MA)、及びAxonInstruments(Union City,CA)から入手できる。非限定的な例として、蛍光検出のためのGSI ScanArray3000は、定量のためのImaGeneソフトウェアとともに使用され得る。
【0109】
V.抗体の作製
本発明に関して希少循環細胞におけるシグナル伝達分子の活性状態及び/又は総量を分析するためにまだ市販で入手できない抗体の生成及び選択は、様々な方法で達成できる。例えば、1つの方法は、対象となるポリペプチド(すなわち抗原)を当該分野で既知のタンパク質発現及び精製方法を用いて、発現し及び/又は生成することであり、一方、他の方法は、当該分野で既知の固層ペプチド合成法を用いて対象のポリペプチドを合成することである。例えば、Guideto Protein Purification,Murray P.Deutcher,ed.,Meth.Enzymol.,Vol.182(1990);SolidPhase Peptide Synthesis,Greg B.Fields,ed.,Meth.Enzymol.,Vol.289(1997);Kisoら、Chem.Pharm.Bull.,38:1192−99(1990);Mostafaviら、Biomed.Pept.ProteinsNucleic Acids,1:255−60,(1995);及びFujiwaraら、Chem.Pharm.Bull.,44:1326−31(1996)を参照する。精製され又は合成されたポリペプチドは、その後、ポリクローナル又はモノクローナル抗体を作製するために、例えばマウス又はウサギに注射され得る。当業者は、例えばAntibodies,ALaboratory Manual,Harlow and Lane,Eds.,Cold Spring Harbor Laboratory,Cold SpringHarbor,N.Y.(1988)に記載されているように、多くの手法が抗体の生成のために利用できることを認識するだろう。当業者はまた、抗体を模倣した結合断片又はFab断片が様々な手法によって遺伝子情報から準備され得ることを認識しているだろう(例えば、AntibodyEngineering:A Practical Approach,Borrebaeck,Ed.,Oxford University Press,Oxford(1995);及びHuseら、J.Immunol.,149:3914−3920(1992)を参照)。
【0110】
加えて、様々な刊行物が、選択された標的抗原に結合するためのポリペプチドライブラリーを構築し、スクリーニングするためのファージディスプレイ技術の使用を報告している(例えば、Cwirlaら、Proc.Natl.Acad.Sci.USA,87:6378−6382(1990);Devlinら、Science,249:404−406(1990);Scottら、Science,249:386−388(1990);及びLadnerら、米国特許第5,571,698号を参照)。ファージディスプレイ法の基本的な概念は、ファージDNAによってコードされたポリペプチドと標的抗原との間の物理的な結合の構築である。この物理的結合は、ポリペプチドをコードするファージ染色体を囲い込むカプシドの一部としてポリペプチドを提示するファージ粒子によって提供される。ポリペプチドとそれらの遺伝物質との間の物理的結合の構築は、異なるポリペプチドを有する非常に多くのファージの同時多量スクリーニングを許容する。標的抗原に対する親和性のあるポリペプチドを提示するファージは標的抗原に結合し、これらファージは標的抗原に対する親和性スクリーニングによって濃縮される。これらファージから提示されるポリペプチドの正体は、それらの各染色体から決定され得る。これらの方法を用いて、所望の標的抗原に対する結合親和性を有すると同定されたポリペプチドは、その後、従来法によって多量に合成され得る(例えば、米国特許第6,057,098号を参照)。
【0111】
これらの方法によって生成された抗体は、その後、対象となる精製ポリペプチド抗原との親和性及び特異性に対する第一のスクリーニングによって選択され得、必要であれば、その結果を、結合から除かれることが期待される他のポリペプチド抗原との抗体の親和性と特異性と比較する。スクリーニング手法は、マイクロタイタープレートの各々の穴における精製されたポリペプチド抗原の固定を含むことができる。潜在的な抗体又は抗体群を有する溶液は、その後、各マイクロタータープレートに置かれ、約30分から2時間インキュベートされる。マイクロタイターの穴は、その後洗浄され、標識化された2次抗体(例えば、作られた抗体がマウス抗体であれば、アルカリホスファターゼに結合された抗−マウス抗体)がその穴に添加され、約30分間インキュベートされ、その後洗浄される。基質がその穴に添加され、固定されたポリペプチド抗原に対する抗体が存在するところに呈色反応が現れるだろう。
【0112】
そのように同定された抗体は、その後さらに親和性と特異性について解析され得る。標的タンパク質に対する免疫分析の構築において、精製された標的タンパク質は、選択された抗体を用いた免疫分析の感度と特異性を判定するための標準として働く。様々な抗体の結合親和性は異なる、例えばある抗体の組み合わせは、立体的に他のものを妨げるかもしれないため、抗体の分析の性能は、その抗体の絶対的な親和性と特異性よりも重要な基準であるかもしれない。
【0113】
当業者は、抗体又は結合断片を作製し、対象の様々なポリペプチドに対する親和性と特異性に関してスクリーニングし及び選択する際に、多くの方法がとられ得るが、これらの方法が本発明の範囲を変えるものではないことを認識するだろう。
【0114】
A.ポリクローナル抗体
ポリクローナル抗体は、好ましくは、対象となるポリペプチド及びアジュバントの複数回の皮下(sc)又は腹腔内(ip)注射によって動物内で上昇する。対象となるポリペプチドを、例えば二官能性又は誘導体化物質として用いられるスカシガイヘモシアニン(keyholelimpet hemocyanin)、血清アルブミン、ウシサイログロブリン(bovine thyroglobulin)、又は大豆トリプシン阻害剤(soybeantrypsin inhibitor)のような、免疫された種において免疫原性であるタンパク質キャリアに共役することは有用であるかもしれない。二官能性又は誘導体化物質の非限定的な例は、マレイミドベンソイルスルフォスクシニミドエステル(maleimidobenzoylsulfosuccinimide ester)(システイン残基を通した共役)、N−ヒドロキシスクシニミド(N−hydroxysuccinimide)(リジン残基を通した共役)、グルタルアルデヒド(glutaraldehyde)、コハク酸無水物、SOCl2及びRとR1が異なるアルキル基であるR1N=C=NRを含む。
【0115】
動物は、対象となるポリペプチド又は免疫原性共役体又はそれらの誘導体に対して、例えば100μg(対ウサギ)又は5μg(対マウス)の抗原又は共役体を3倍容量のフロイント完全アジュバントを組み合わせて、そしてその溶液を複数の部位に皮内注射することによって免疫を与えられる。1ヶ月後、その動物は、複数の部位における皮下注射によってフロイント不完全アジュバント中のポリペプチド又は複合体の元々の量の約1/5から1/10でブーストされる。7日から14日後、その動物は、採血され、その血清が抗体力価に関して分析される。動物は、典型的には、力価が頭打ちになるまでブーストされる。好ましくは、動物は、同一のポリペプチドの複合体でブーストされるが、異なる免疫原性タンパク質への共役及び/又は異なる架橋剤による共役が用いられてもよい。複合体はまた、融合タンパク質として組み換え細胞培養で作られ得る。場合によっては、ミョウバンのような凝集剤が免疫応答を増幅するために用いられ得る。
【0116】
B.モノクローナル抗体
モノクローナル抗体は、一般的に実質的に相同な抗体の集団から獲得され、即ちその集団を構成する各抗体は、少量存在するかもしれない起こりうる自然発生変異を除き、同一である。従って、「モノクローナル(monoclonal)」という修飾語は、個々に異なる抗体の混合物ではないという抗体の特徴を示す。例えば、モノクローナル抗体は、Kohlerら、Nature,256:495(1975)によって開示されたハイブリドーマ法を用いて、又は当業者に既知の任意の組み換えDNA法(例えば米国特許第4,816,567号を参照)によって作製され得る。
【0117】
ハイブリドーマ法では、マウス又は他の適切な宿主動物(例えばハムスター)が、免疫付与のために用いられる、対象となるポリペプチドに特異的に結合する抗体を生産する又は生産することができるリンパ球を引き出すために上記のとおり免疫される。代替的には、リンパ球がインビトロで免疫される。免役されたリンパ球は、その後、ハイブリドーマ細胞を形成するため、ポリエチレングリコールのような適切な融合剤を用いて骨髄腫細胞と融合される(例えば、Goding,MonoclonalAntibodies:Principles and Practice,Academic Press,pp.59−103(1986)を参照)。このように用意されたハイブリドーマ細胞は、好ましくは、融合されていない親骨髄腫細胞の増殖又は生存を阻害する一つ又はそれ以上の基質を含む適切な培地に植えて増殖させる。例えば、親骨髄腫細胞が酵素ヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRT)を欠く場合、ハイブリドーマ細胞のための培地は、一般的に、HGPRT欠損細胞の増殖を阻止する、ヒポキサンチン、アミノプテリン、及びチミジンを含むだろう(HAT培地)。
【0118】
好ましい骨髄腫細胞は、効果的に融合され、選択された抗体生産細胞による安定した、高レベルの抗体生産を支え、及び/又はHAT培地のような培地に感受性があるものである。ヒトモノクローナル抗体の生産のための、そのような望ましい骨髄腫細胞系統の例は、これ限定されないが、MOPC−21及びMPC−11マウス腫瘍のようなネズミ科動物(murine)の骨髄腫細胞(theSalk Institute Cell Distribution Center;San Diego,CAから入手可能)、SP−2又はX63−Ag8−653細胞(theAmerican Type Culture Collection;Rockville,MDから入手可能)、及びヒト骨髄腫細胞又はマウス−ヒトヘテロ骨髄腫細胞系統(例えば、Kozbor,J.Immunol.,133:3001(1984);及びBrodeurら、MonoclonalAntibody Production Techniques and Applications,Marcel Dekker,Inc.,New York,pp.51−63(1987)を参照)を含む。
【0119】
ハイブリドーマ細胞が増殖している培地は、対象となるポリペプチドに対して向けられたモノクローナル抗体の生産に関して分析され得る。好ましくは、ハイブリドーマ細胞によって生産されたモノクローナル抗体の結合特異性は、免疫沈降法によって又は放射免疫測定(RIA)又は酵素免疫測定法(ELISA)のような、インビトロ結合測定によって決定される。モノクローナル抗体の結合親和性は、例えばMunsonら、Anal.Biochem.,107:220(1980)のスキャッチャード解析を用いて決定され得る。
【0120】
ハイブリドーマ細胞が所望の特異性、親和性及び/又は活性の抗体を生産すると同定された後、そのクローンは限界希釈法によって、サブクローン化され、そして標準的な方法によって増殖させられてもよい(例えば、Goding,MonoclonalAntibodies:Principles and Practice,Academic Press,pp.59−103(1986)を参照)。この目的のために適した培地は、例えば、D−MEM又はRPMI−1640培地を含む。加えて、ハイブリドーマ細胞は、動物内で腹水腫瘍としてinvivoで増殖してもよい。サブクローンによって分泌されたモノクローナル抗体は、例えば、タンパク質Aセファロース、ハイドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、親和性クロマトグラフィーのような従来の抗体精製手法によって、培地、腹水又は血清から分離され得る。
【0121】
モノクローナル抗体をコードするDNAは、従来の手法を用いて(例えば、ネズミ科動物(murine)抗体の重及び軽鎖をコードする遺伝子に特異的に結合できるオリゴペプチドプローブを用いることによって)容易に単離され、配列解読され得る。ハイブリドーマ細胞は、そのようなDNAの望ましい源として働く。一旦単離されると、組み換え宿主細胞においてモノクローナル抗体の合成を促進するために、DNAは発現ベクターに搭載され、その後、E.coli細胞、サルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、又は本来なら抗体を生産しない骨髄腫細胞のような宿主細胞に導入されてもよい。例えば、Skerraら、Curr.Opin.Immunol.,5:256−262(1993);及びPluckthun,ImmunolRev.,130:151−188(1992)を参照する。DNAはまた、例えば、ヒト重鎖及び軽鎖の定常ドメインをコードする配列を相同なネズミ科動物(murine)の配列と置換することによって(例えば、米国特許第4,816,567;Morrisonら、Proc.Natl.Acad.Sci.USA,81:6851(1984)を参照)、又は非免疫グロブリンペプチドをコードする配列の全て又は一部を免疫グロブリンをコードする配列と共有結合することによって、改変され得る。
【0122】
さらなる実施の形態では、モノクローナル抗体又は抗体断片は、例えば、McCaffertyら、Nature,348:552−554(1990);Clacksonら、Nature,352:624−628(1991);及びMarksら、J.Mol.Biol.,222:581−597(1991)において記載された技術を用いて生成された抗体ファージライブラリーから単離され得る。鎖シャッフリングによる高親和性(nM範囲)ヒトモノクローナル抗体の生産は、Marksら、BioTechnology,10:779−783(1992)に記載されている。非常に大きなファージライブラリーを構築するための戦略として、組み合わせ感染(combinatorialinfection)及びin vivo組み換えの使用は、Waterhouseら、Nuc.Acids Res.,21:2265−2266(1993)に記載されている。従って、これらの技術は、モノクローナル抗体の生成のための伝統的なモノクローナル抗体ハイブリドーマ法の実行可能な代替案である。
【0123】
C.ヒト化抗体
非ヒト抗体をヒト化するための方法は、当該分野では既知である。好ましくは、ヒト化抗体は、非ヒトである試料源からの抗体に導入された一つ又はそれ以上のアミノ酸残基を有する。これらの非ヒトアミノ酸残基はしばしば「取り込み(import)」残基と言われ、典型的には「取り込み(import)」可能なドメインから取得される。ヒト化は、本質的には、非ヒト抗体の超可変領域の配列をヒト抗体の対応する配列と置き換えることによって実行され得る。例えば、Jonesら、Nature,321:522−525(1986);Riechmannら、Nature,332:323−327(1988);及びVerhoeyenら、Science,239:1534−1536(1988)を参照する。従って、そのような「ヒト化」抗体は、キメラ抗体(例えば、米国特許第4,816,567号)であって、完全なヒト可変ドメインは、本質的には決して非ヒトの種からの対応する配列によって置換されていない。実際に、ヒト化された抗体は、典型的には、いくらかの超可変領域残基及び可能であればいくらかのフレームワーク領域(FR)残基が齧歯類抗体の類似部位からの残基によって置換されるヒト抗体である。
【0124】
本明細書で記載したヒト化抗体を作製するのに使用するための、ヒト可変ドメイン、軽鎖及び重鎖の両方、の選択は、抗原性を低減するために重要な検討事項である。いわゆる「最も適した(best−fit)」方法に従って、齧歯類抗体の可変ドメインの配列が、既知のヒト可変ドメイン配列の全ライブラリーに対しスクリーニングされる。齧歯類のものに最も近いヒト配列が、その後、ヒト化抗体のためのヒトFRとして受け入れられる(例えば、Simsら、J.Immunol.,151:2296(1993);及びChothiaら、J.Mol.Biol.,196:901(1987)を参照)。他の方法は、全てのヒト抗体の軽鎖又は重鎖の特定の部分集合の共通配列に由来する特定のFRを用いる。同一のFRは、様々な異なるヒト化抗体のために使用されてもよい(例えば、Carterら、Proc.Natl.Acad.Sci.USA,89:4285(1992);及びPrestaら、J.Immunol.,151:2623(1993)を参照)。
【0125】
抗体が、抗原に対する高親和性の保有及び他の好ましい生化学的特性とともにヒト化されることもまた重要である。この目的を達成するため、ヒト化抗体は、親及びヒト化配列の3次元モデルを用いた、親配列と様々な概念的ヒト化生成物の解析の工程によって用意され得る。3次元免疫グロブリンモデルは、一般に利用可能であり、当業者によく知られている。選択された候補免疫グロブリン配列の可能な3次元立体配座構造を図示し、表示するコンピュータープログラムが利用できる。これら表示の観察が候補免疫グロブリン配列の機能における残基の類似の役割の解析、即ち候補免疫グロブリンの抗原への結合能に影響する残基の解析、を可能にする。このように、FR残基は、受容及び取り込み配列から選択され、そして組み合わせられるので、標的抗原に対する増加した親和性のような所望の抗体特性が達成され得る。一般的に、超可変領域は、直接的及び特異的に、抗原結合に影響することに含まれる。
【0126】
本発明によると、様々な種類のヒト化抗体が意図されている。例えば、ヒト化抗体は、Fab断片のような抗体断片であり得る。代替的には、ヒト化抗体は、完全なIgA、IgG又はIgM抗体のような完全な抗体であり得る。
【0127】
D.ヒト抗体
ヒト化の代替として、ヒト抗体が生成され得る。ある実施の形態では、内因性免疫グロブリン生産の欠損下で、免疫付与によって、全ての種類のヒト抗体を生成することができるトランスジェニック動物(例えば、マウス)が生成され得る。例えば、キメラ生殖系列変異マウスにおける抗体重鎖接合領域(JH)遺伝子のホモ欠損が、内因性抗体生成の完全な阻害をもたらすことが述べられている。そのような生殖系列変異マウスにおけるヒト生殖系列免疫グロブリン遺伝子の形質転換は、抗原投与でヒト抗体の生産をもたらすだろう。例えば、Jakobovitsら、Proc.Natl.Acad.Sci.USA,90:2551(1993);Jakobovitsら、Nature,362:255−258(1993);Bruggermannら、Yearin Immun.,7:33(1993);及び米国特許第5,591,669号、5,589,369号及び5,545,807号を参照する。
【0128】
代替的には、ファージディスプレイ技術(例えば、McCaffertyら、Nature,348:552−553(1990)を参照)が、免疫されていないドナーからの免疫グロブリン可変(V)ドメイン遺伝子レパートリーを用いて、インビトロでヒト抗体及び抗体断片を生成するために使用され得る。この技術に従うと、抗体Vドメイン遺伝子は、M13又はfdのような、繊維状バクテリオファージのメジャー又はマイナーコートタンパク質遺伝子のいずれかにフレームをあわせてクローン化され、ファージ粒子の表面で機能性抗体断片として提示される。繊維状粒子がファージゲノムの1本鎖DNAコピーを含むため、抗体の機能特性に基づく選択はまた、それらの特性を示す抗体をコードする遺伝子の選択をもたらす。従って、ファージは、B細胞の特性の一部を模倣する。ファージディスプレイは、例えばJohnsonら、Curr.Opin.Struct.Biol.,3:564−571(1993)に記載されるように様々な形式で実行され得る。V遺伝子断片のいくつかの入手源がファージディスプレイのために使用され得る。例えば、Clacksonら、Nature,352:624−628(1991)を参照する。免疫されていないヒトドナーからのV遺伝子のレパートリーは構築され得、多様な抗原(自己抗原も含む)への抗体は、本質的に、Marksら、J.Mol.Biol.,222:581−597(1991);Griffithら、EMBOJ.,12:725−734(1993);及び米国特許第5,565,332号、及び第5,573,905号に記載された技術に従って単離され得る。
【0129】
場合によっては、ヒト抗体は、米国特許第5,567,610号及び第5,229,275号に記載されるようにインビトロで活性化されたB細胞によって生成され得る。
【0130】
E.抗体断片
様々な技術が抗体断片の生成のために開発されてきた。伝統的には、これらの断片は、完全な抗体のタンパク質切断を介して得られた(例えば、Morimotoら、J.Biochem.Biophys.Meth.,24:107−117(1992);及びBrennanら、Science,229:81(1985)を参照)。しかしながら、これらの断片は、現在、組み換え宿主細胞を用いて直接的に生成され得る。例えば、抗体断片は上述した抗体ファージライブラリーから単離され得る。代替的には、Fab’−SH断片がE.coli細胞から直接回収され、そしてF(ab’)2を形成するよう化学的に結合され得る(例えばCarterら、BioTechnology,10:163−167(1992)を参照)。他のアプローチによると、F(ab’)2断片は、組み換え宿主細胞培養から直接単離され得る。抗体断片の生成のための他の技術は、当業者には明らかであるだろう。他の実施の形態において、選択の抗体は、単鎖Fv断片(scFv)である。例えば、国際公開第93/16185号;及び米国特許第5,571,894号及び第5,587,458号を参照する。抗体断片はまた、例えば、米国特許第5,641,870号に記載されるようにリニア抗体(linearantibody)であってもよい。そのようなリニア抗体断片は、単一特異性又は二重特異性であるかもしれない。
【0131】
F.二重特異性抗体
二重特異性抗体は、少なくとも2つの異なるエピトープに対する結合特異性を有する抗体である。典型的な二重特異性抗体は、対象となる同一のポリペプチドの2つの異なるエピトープに結合するかもしれない。他の二重特異性抗体は、対象となるポリペプチドに対する結合部位を一つ又はそれ以上の追加的な抗原に対する結合部位と組み合わせるかもしれない。二重特異性抗体は、全長抗体又は抗体断片(例えば、F(ab’)2二重特異性抗体)として用意され得る。
【0132】
二重特異性抗体を作製する方法は、当該分野では既知である。全長型二重特異性抗体の伝統的な作製は、2つの鎖が異なる特異性を有する2つの免疫グロブリンの重鎖−軽鎖対の共発現に基づく(例えば、Millsteinら、Nature,305:537−539(1983)を参照)。免疫グロブリンの重鎖及び軽鎖のランダムな組み合わせのため、これらのハイブリドーマ(クアドローマ)は、一つのみが正しい二重特異性構造を有する、10の異なる抗体分子の潜在的混合物を生成する。正しい分子の精製は、通常、親和性クロマトグラフィーによって実行される。同様の手法は、国際公開第93/08829号及びTrauneckerら、EMBOJ.,10:3655−3659(1991)に開示される。
【0133】
異なるアプローチによると、所望の結合特異性(抗体−抗原結合部位)を有する抗体可変ドメインは、免疫グロブリン共通ドメイン配列に融合される。この融合体は、好ましくは、少なくともヒンジCH2及びCH3領域の一部から構成される免疫グロブリン重鎖定常ドメインを有する。好ましくは、少なくとも1つの融合体に存在する軽鎖結合に必要な部位を含む第1の重鎖定常領域(CH1)を有する。免疫重鎖融合体と、必要であれば、免疫グロブリン軽鎖をコードするDNAは、別々の発現ベクターに挿入され、そして適切な宿主生物に共導入される。これは、構築に使用された3つのポリペプチド鎖の不均一な割合が最適の収率を提供する実施の形態では、3つのポリペプチド断片の相互の比率を調整することにおいて高い柔軟性を提供する。しかしながら少なくとも2つのポリペプチド鎖の等しい比率での発現が高い収率をもたらす又はその比率が特に重要ではないときには、2つ又は3つ全てのポリペプチド鎖をコードする配列を一つの発現ベクターに挿入することが可能である。
【0134】
このアプローチの好ましい実施の形態では、二重特異性抗体は、片腕に第1の結合特異性を有するハイブリッド免疫グロブリン重鎖と、他腕の手に(第2の結合特異性を提供する)ハイブリッド免疫グロブリン重鎖−軽鎖対とから構成される。二重特異性分子の片方のみにおける免疫グロブリン軽鎖の存在が分離の容易な方法を提供するので、この非対照的な構造は、望まれない免疫グロブリン鎖複合体からの所望の二重特異性構成体の分離を促進する。例えば、国際公開第94/04690号及びSureshら、Meth.Enzymol.,121:210(1986)を参照する。
【0135】
米国特許第5,731,168号に記載された他のアプローチによると、1対の抗体分子間の接合部は、組み換え細胞培養から回収されたヘテロ2量体の割合を最大限にするよう設計され得る。好ましい接合部は、抗体の定常ドメインのCH3ドメインの少なくとも一部から構成される。この方法では、第1の抗体分子の接合部からの一つ又はそれ以上の小さなアミノ酸側鎖がより大きな側鎖(例えばチロシン又はトリプトファン)で置換される。大きな側鎖と同一の又は類似の大きさの補完的な「空洞(cavity)」が、大きなアミノ酸側鎖をより小さな側鎖(例えばアラニン又はスレオニン)で置換することによって第2の抗体分子の接合部に生成される。これは、ホモ2量体のような他の望まれない最終産物に対するヘテロ2量体の収率を増加するための機構を提供する。
【0136】
二重特異性抗体は、架橋又は「ヘテロ接合(heteroconjugate)」抗体を含む。例えば、ヘテロ接合である抗体の一つは、アビジンに連結され得、他方はビオチンに連結され得る。ヘテロ接合型抗体は、任意の使用可能な架橋方法を用いて、作製され得る。適切な架橋剤及び技術は、当該分野ではよく知られており、例えば米国特許第4,676,980号に開示される。
【0137】
抗体断片から二重特異性抗体を生成するために適した技術はまた、当該分野で既知である。例えば、二重特異性抗体は、化学的な連結を用いて準備され得る。場合によっては、二重特異性抗体は、完全な抗体がタンパク質分解で切断され、F(ab’)2断片を生成する手法によって生成され得る(例えば、Brennanら、Science,229:81(1985)を参照)。これらの断片は、隣接チオールを安定化し、そして分子内ジスルフィド形成を防ぐジチオール錯体化剤亜ヒ酸ナトリウムの存在下で減少する。生成したFab’断片は、その後、チオニトロ安息香酸(TNB)誘導体に変換される。Fab’−TNB誘導体の一つは、その後、メルカプトエチルアミン(mercaptoethylamine)での還元によってFab’−チオールに変換され、二重特異性抗体を形成するために等モル量の他のFab’−TNB誘導体と混合される。
【0138】
ある実施の形態では、Fab’−SH断片は、E.coliから直接回収され、二重特異性抗体を形成するために化学的に結合され得る。例えば、完全にヒト化された二重特異性抗体F(ab’)2分子は、Shalabyら、J.Exp.Med.,175:217−225(1992)に記載された方法によって生成され得る。各Fab’断片は、E.coliから別々に分泌され、二重特異性抗体を形成するためにインビトロでの直接的な化学的結合を受けた。
【0139】
組み換え細胞培養から直接二重特異性抗体断片を作製し、単離するための様々な技術はまた、開示されている。例えば、二重特異性抗体は、ロイシンジッパーを用いて生成されている。例えば、Kostelnyら、J.Immunol.,148:1547−1553(1992)を参照する。Fos及びJunタンパク質からのロイシンジッパーペプチドは、遺伝子融合によって、2つの異なる抗体のFab’部に連結された。抗体ホモ2量体は、単量体を形成するためにヒンジ部で還元され、その後、抗体ヘテロ2量体を形成するために再酸化された。この方法はまた、抗体ホモ2量体の生成のために利用され得る。Hollingerら、Proc.Natl.Acad.Sci.USA,90:6444−6448(1993)によって記載された「ダイアボディー(diabody)」技術は、二重特異性抗体断片を作製するための代替的な機構を提供している。その断片は、非常に短いために同一鎖の2つのドメイン間での対合を可能にするリンカーによって軽鎖可変ドメイン(VL)と連結した重鎖可変ドメイン(VH)から構成される。従って、一つの断片のVH及びVLドメインは、他の断片の相補的なVL及びVHドメインと対を形成せざるを得ず、それによって2つの抗原結合部を形成する。単鎖Fv(sFv)2量体の使用によって二重特異性抗体断片を作製するための他の戦略は、Gruberら、J.Immunol.,152:5368(1994)に記載される。
【0140】
二価以上の抗体も、意図される。例えば、三重特異性抗体が用意され得る。例えば、Tuttら、J.Immunol.,147:60(1991)を参照する。
【0141】
G.抗体精製
組み換え技術を用いると、抗体は、単離された宿主細胞内、宿主細胞のペリプラズム空間で生成され得、又は宿主細胞から培地中に直接分泌され得る。抗体が細胞内で生成されると、例えば、遠心又は限外ろ過によって微粒子破片が先ず除去される。Carterら、BioTech.,10:163−167(1992)は、E.coliのペリプラズム空間に分泌された抗体を単離するための手法を記載する。簡単に言うと、細胞ペーストは、約30分間、酢酸ナトリウム(pH3.5)、EDTA、及びフッ化フェニルメチルスルホニル(PMSF)の存在中で溶解される。細胞破片は、遠心分離によって分離され得る。抗体が培地中に分泌される際、そのような発現システムからの上清は、一般的に、市販で入手可能なタンパク質濃縮フィルター、例えばAmicon又はMilliporePelliconの限外ろ過ユニットを用いて濃縮される。PMSFのような、プロテアーゼ阻害剤は、タンパク質分解を阻害するための前述の任意のステップに含まれ、抗生物質は、外来の混入物の増殖を抑制するために含まれてもよい。
【0142】
細胞から用意された抗体組成物は、例えば、ハイドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、及び親和性クロマトグラフィーを用いて精製され得る。親和性リガンドとしてタンパク質Aの適正は、抗体に存在するあらゆる免疫グロブリンのFcドメインの種及びアイソタイプに依存する。タンパク質Aは、ヒトγl,γ2,又はγ4重鎖に基づいた抗体を精製するために使用され得る(例えば、Lindmarkら、J.Immunol.Meth.,62:1−13(1983)を参照)。タンパク質Gは、全てのマウスのアイソタイプ及びヒトγ3に対し推奨される(例えば、Gussら、EMBOJ.,5:1567−1575(1986)を参照)。親和性リガンドが付着されるマトリクスは、最も頻繁にはアガロースであるが、他のマトリクスが利用可能である。微小多孔質ガラス(controlledpore glass)又はポリ(スチレンジビニル)ベンゼン(poly(styrenedivinyl)benzene)のような機械的に安定なマトリクスは、アガロースで達成され得るよりも速い流速とより短い処理時間を許容する。抗体がCH3ドメインから構成される場合、BakerbondABX(登録商標)レジン(J.T.Baker;Phillipsburg,N.J.)が精製のために有用である。イオン交換カラム、エタノール沈殿、逆層HPLC、シリカクロマトグラフィー、ヘパリンSEPHAROSE(登録商標)クロマトグラフィー、陰イオン又は(ポリアスパラギン酸カラムのような)陽イオン交換レジンクロマトグラフィー、等電点電気泳動、SDS−PAGE、及び硫酸アンモニウム沈殿のようなタンパク質精製のための他の技術はまた、回収される抗体に依存して利用可能である。
【0143】
あらゆる予備的な精製工程に続いて、対象となる抗体と混入物とから構成される混合物は、2.5〜4.5の間のpHの溶出緩衝液を用いて、好ましくは低塩濃度で(例えば約0〜0.25Mの塩から)実行される、低pH疎水性相互作用クロマトグラフィーに供されてもよい。
【実施例】
【0144】
以下の実施例は、請求の範囲に記載の発明を説明するために述べられるが、これに限定されるものではない。
【0145】
(実施例1.循環細胞の単離、活性化、及び溶解)
固形腫瘍の循環細胞は、固形腫瘍から転移した又は微小転移した細胞から構成され、循環腫瘍細胞(CTC)、癌幹細胞(CSC)及び/又は腫瘍へと移動している細胞(例えば、循環内皮前駆細胞(CEPC)、循環内皮細胞(CEC)、循環血管新生促進骨髄性細胞、循環樹状細胞、等)を含む。循環細胞を含む患者試料は、あらゆる入手可能な生体液(例えば、血液、尿、乳頭吸引液、リンパ液、唾液、細針吸引物、等)から獲得され得る。循環細胞は、例えば、免疫磁気分離法(例えば、Racilaら、Proc.Natl.Acad.Sci.USA,95:4589−4594(1998);Bilkenrothら、Int.J.Cancer,92:577−582(2001)を参照)、マイクロ流体分離法(例えばMohamedら、IEEETrans.Nanobiosci.,3:251−256(2004);Linら、Abstract No.5147,97th AACR Annual Meeting,Washington,D.C.(2006)を参照)、FACS(例えばMancusoら、Blood,97:3658−3661(2001)を参照)、密度勾配遠心分離法(例えばBakerら、Clin.CancerRes.,13:4865−4871(2003)を参照)、及び減少法(例えばMeyeら、Int.J.Oncol.,21:521−530(2002)を参照)のような一つ又はそれ以上の分離方法を用いて患者試料から単離され得る。
【0146】
CTCの手動単離:
CTCの免疫磁気分離法−活性分析が続く手動単離:
1)前もって抗−EpCAMモノクローナル抗体(KordiaLife Sciences;Leiden,The Netherlands)に結合された磁気ビーズ((Dynal M450;Dynal AS;Oslo,Norway)が用いられる。
2)使用直前に、事前に覆われたダイナビーズ(Dynabead)は、0.01%BSAを含む等量のPBSで一度洗浄される。
3)25mlの事前に覆われたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に設置される。
6)その上清が廃棄され、ビーズに結合した細胞が0.01%BSAを含むPBSで再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0147】
試料の準備:
1)ヒト対象物からの末梢血が1mg/mlのEDTAを含むシリコン処理チューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出された上皮細胞での汚染を避けるために廃棄される。
2)1mlの全血が使用前に0.9%のNaClで1:3に希釈される。
【0148】
対照の準備:
1)細胞系列の対照がヒト癌細胞系列をHL−60細胞にスパイクすることによって作製される。
2)細胞系列の対照は、2.5x106細胞/mlの濃度で使用される。
【0149】
CEC及びCEPCの手動分離:
非限定的な例として、生存CEC及びCEPCは、Beerepootら、Ann.Oncology,15:139−145(2004)に記載された免疫磁気単離/濃縮手法を用いて単離され得る。つまり、末梢血が前もって抗CD146モノクローナル抗体(KordiaLife Sciences)に共役された磁気ビーズ(Dynal M450 IgG1)とともにインキュベートされる。この抗体は、末梢血中の内皮細胞の全ての系統を認識し、造血細胞又は上皮細胞を認識しない(Georgeら、J.Immunol.Meth.,139:65−75(1991)を参照)。造血細胞及び上皮細胞の消極的な選択は、適切な抗体に共役された磁気ビーズ(例えば、白血球を除くためのDynal−CD45ビーズ、単球を除くためのDynal−CD14ビーズ、上皮細胞を除くためのDynal−EpCAM(Invitrogen;Carlsbad,CA))での積極的選択に先立って使用され得る。この例では、積極的選択のみが使用される。
【0150】
CEC及びCEPCの免疫磁気分離−活性分析が続く手動単離:
1)前もって抗−CD146モノクローナル抗体(KordiaLife Sciences)を結合された磁気ビーズ(Dynal M450)が使用される。
2)使用直前に、事前に被覆されたダイナビーズは、0.01%BSAを含む等量のPBSで一回洗浄される。
3)25μlの事前に被覆されたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSでの再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0151】
試料の準備:
1)ヒト対象物からの末梢血が、1mg/mlEDTAを含むシリコン処理されたチューブに引き抜かれる。最初の3〜5mlは穴の開いた血管から放出された上皮細胞での汚染を防ぐため、廃棄される。
2)1mlの全血は、使用前に0.9%NaClで1:3に希釈される。
【0152】
対照の準備:
1)細胞系列の対照が、ヒト臍帯静脈血管内皮細胞(HUVEC)をHL−60細胞にスパイクすることによって作製される。
2)細胞系列の対照は、2.5x106細胞/mlの濃度で使用される。
【0153】
CEPCの手動単離(CECを除く):
CEPCは、様々な血管新生増殖因子に応答して、成熟内皮細胞へ分化する能力を有する骨髄由来の前駆細胞の循環型である。CEPCは、表層マーカーCD34を認識する抗体での選択によって単離されてよい。CD133は、未熟内皮細胞(EPC)又は原始造血幹細胞(HSC)をCEPCから区別する細胞表層マーカーである。異なる入手源からのCEPCの様々な単離手法は、接着培養又は磁気マイクロビーズを用いて開示されている。この例では、Asaharaら、Science,275:964−967(1997)に記載されたものから改良されたプロトコールが使用される。
【0154】
CEPCの免疫磁気分離−活性分析が続く手動単離:
1)磁気ビーズ(DynalM450 CD34)が使用される。これらのビーズはCD34表層抗原に対し特異的なモノクローナル抗体で覆われる。
2)事前に覆われたダイナビーズは、使用直前に、0.01%BSAを含む等量のPBSで一回洗浄される。
3)25μlの事前に覆われたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSに再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0155】
試料の準備:
1)ヒト対象物からの末梢血が、1mg/mlのEDTAを含むシリコン処理されたチューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出された内皮細胞での汚染を避けるために廃棄される。
2)10mlの血液は、平衡塩類溶液で1:1に希釈される。
3)4mlの希釈血液は、10mlチューブ中で3mlのFicoll−Paque上に積層される。
4)チューブは、18〜20℃で30〜40分間、400×gで回転される。
5)血漿及び血小板を含む上層は、滅菌パスツールピペットを用いて取り除かれ、界面で乱れていない単核細胞層を残す。
6)単核細胞は、滅菌ピペットを用いて滅菌遠心管へ移される。
7)6mlの平衡塩類溶液が添加され、細胞は穏やかに再懸濁される。
8)混合物は、18〜20℃で10分間、60〜100×gで遠心される。
9)上清が、除去され、各チューブからの単核細胞が1mlのPBSに再懸濁される。
【0156】
Veridexシステムを用いたCTC,CEC,及びCEPCの単離:
Veridex(Warren,NJ)は、CellPrepシステム、CellSearch上皮細胞キット、及びCellSpotterアナライザーから構成されるCellSearchシステムを市販している。CellPrepシステムは、半自動化された試料準備システムである(Kaganら、J.Clin.LigandAssay,25:104−110(2002)を参照)。CellSearch上皮細胞キットは:内皮細胞に特異的な抗−EpCAM抗体で覆われた鉄流動体(ferrofluid);サイトケラチン(cytokeratins)8、18及び19に対するフィコエリスリン(phycoerythrin)共役抗体;アロフィコシアニン(allophycocyanin)に共役した抗−CD45抗体;DAPI染色剤;及び細胞を洗浄、浸透、及び再懸濁するための緩衝液、から構成される。この例で使用されるプロトコールはまた、Allardら、Clin.CancerRes.,10:6897−6904(2004)に記載されている。全Veridexシステムは、CTCの計数のために使用され得、又は、CellPrepシステムで単離された後に手動で試料を除去することによって、経路活性化のための分析に先だった単離の方法を提供することができる。CTCの数は、アルゴリズム開発のために情報を提供し得る。
【0157】
Veridexシステム−計数が続くCTC濃縮:
1)7.5mlの血液が6ml緩衝液と混合され、10分間800×gで遠心され、CellPrepシステムに移される。
2)装置が上清を吸引した後、その装置は鉄流動体を添加する。
3)その装置が、インキュベートとそれに続く磁気分離工程を実行する。
4)非結合細胞及び残存血漿が吸引される。
5)染色試薬が、蛍光染色のために浸透化緩衝液と組み合わせて添加される。
6)そのシステムによるインキュベート後、細胞は、CellSpotterアナライザーを用いた分析のため、再びMagNestCell Presentation装置にて磁気的に分離され、そして再懸濁される。
7)その装置は、CellSpotterアナライザー、4色半自動化蛍光顕微鏡、上に設置される。
8)画像は、Veridexの定義した基準に合うように得られ、最終的な手動選択のためにウェブに基づくブラウザを介して示される。
9)細胞計数の結果は、7.5ml血液あたりの細胞数として表される。
【0158】
Veridexシステム−活性分析が続くCTC濃縮:
1)7.5ml血液が6mlの緩衝液と混合され、10分間800×gで遠心され、その後CellPrepシステムに移される。
2)装置が上清を吸引した後、その装置は鉄流動体を添加する。
3)その装置はインキュベートとそれに続く磁気分離工程を実行する。
4)非結合細胞と残存血漿が吸引される。
5)試料は、100μlの活性化緩衝液で再懸濁される。
【0159】
Veridexシステム−活性分析が続くCEC及びCEPC濃縮:
1)Veridexは、CD146抗体でのCEC及びCEPCの捕獲に用いるCellTracks内皮細胞キットを提案する。CellTracks内皮細胞キットは、血液試料の準備のためのVeridexのCellTracksAutoPrepシステム及び全血からのCEC及びCEPCを数え、特徴づけるためのCellTracksアナライザーIIとを組み合わせて用いられる。このプロトコールは、CellSearch上皮細胞キットのものと同一である。
【0160】
試料の準備:
1)ヒト対象物からの末梢血は、製造元の指導に従って、CellSavePreservativeチューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出される上皮又は内皮細胞での汚染を回避するために廃棄される。
【0161】
CSCの手動単離:
腫瘍が特徴的な自己再生及び生存機構を有する少数の推定癌幹細胞を含むことの証拠が確立しつつある(例えばSells,Crit.Rev.Oncol.Hematol.,51:1−28(2004);Reyaら、Nature,414:105−111(2001);Dontuら、TrendsEndocrinol.Metal.,15:193−197(2004);and Dick,Nature,423:231−233(2003)を参照)。癌幹細胞(CSC)は、長期間静止状態で存在し、分裂する細胞を標的とする化学療法剤に対し耐性を獲得するかもしれない。この癌を発生させる集団は、選択的な除去のための標的化された治療を受けた自己再生及び生存経路の活性化として特徴づけられ得る。CSCの単離手法は、接着培養又は磁気マイクロビーズを用いて開示されている。この例では、Coteら、Clin.Can.Res.,12:5615(2006)で開示されたものから改変したプロトコールが使用される。
【0162】
免疫磁気CSC単離−活性分析が続く手動単離:
1)磁気ビーズ(DynalAS;Oslo,Norway)が使用される。これらのビーズは、CD34又はCD133表層抗原のいずれかに対し特異的なモノクローナル抗体で覆われる。
2)使用直前に、事前に覆われたダイナビーズが0.01%BSAを含む等量のPBSで一回洗浄される。
3)1〜107の事前に覆われたダイナビーズが、3mlの試料に添加される。
4)混合物が、2〜8℃で60分間、緩やかな傾斜と回転でインキュベートされる。
5)混合物は、1mlの部分に分割され、各チューブは、少なくとも6分間、磁気分離器(MPL−1magnet)上に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSに再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0163】
試料の準備:
1)骨髄試料が、インフォームド・コンセントを得た後に早期乳癌患者から獲得される。
2)骨髄吸引処理は、Bauerら、Clin.Can.Res.,6:3552−3559(2000)に記載のとおり実行される。あらゆる播種性腫瘍細胞を含む単核細胞画分は、BeckmanGS−6遠心機を用いて、35分間4000×gでのFicoll−Hypaque密度勾配遠心分離法によって濃縮され、PBSで2回洗浄される。
【0164】
単離したCTCの細胞活性化及び溶解:
細胞活性化:
1)増殖因子TGF−α(100nM)、ヘレグリン(heregulin)(100nM)、及び/又はIGF(100nM)が細胞に添加され、37℃で5分間インキュベートされる。
【0165】
薬剤処理での細胞活性化:
1)試料は、37℃で30分間、治療に効果的な濃度で、ヘルセプチン(Herceptin)、ラパタニブ(Lapatanib)、タルセバ(Tarceva)、及び/又はラパマイシン(Rapamycin)類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で5分間、インキュベートされる。
【0166】
薬剤処理での細胞活性化(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度で、ヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)によって活性化され、37℃で120分間インキュベートされる。
【0167】
活性化されたCTCが以下のプロトコールを用いて、溶解される:
1)新鮮な溶解緩衝液は、表1に記載された試薬を混合することによって、新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却した緩衝液中に再懸濁される。
3)インキュベートが氷上で30分間行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速でマイクロチューブ中で遠心分離される。
1)溶解物は、分析のため、新しいチューブに移され、−80℃で保存される。
【表1】

【0168】
単離されたCEC及び/又はCEPCの細胞活性化及び溶解:
VEGFは、CEPC(Larriveeら、J.Biol.Chem.,278:22006−22013(2003))及び、血管壁が剥がれ落ちた成熟CEC(Soloveyら、Blood,93:3824−3830(1999))の両方において抗アポトーシス経路を活性化することによって、生存を促進すると考えられている。成熟CECは、CEPCと比較して、限定された増殖能力しか有していないようであるが、VEGFはまた、CEPC又は成熟CECの増殖を促進するかもしれない(Linら、J.Clin.Invest.,105:71−77(2000))。これらの理由のため、CEC及び/又はCEPCは、溶解の前にVEGFとのインキュベートによって活性化される。
【0169】
細胞活性化:
1)増殖因子VEGF,FGF,PDGF,PIGF,及び/又はアンギオポイエチン(angiopoietin)は、各100nMで細胞に添加され、37℃で5分間インキュベートされる。
【0170】
薬剤処理での細胞活性化:
1)試料は、37℃で30分間、治療に効果的な濃度のアバスタチン(Avastin)、ネクサバール(Nexavar)、スーテント(Sutent)及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子VEGF、FGF、PDGF、PIGF、及び/又はアンギオポイエチンを添加することによって活性化され、37℃で5分間インキュベートされる。
【0171】
薬剤処理での細胞活性化(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度のアバスチン、ネクサバール、スーテント、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、VEGF、FGF、PDGF、PIGF、及び/又はアンギオポイエチンを各100nMで添加することによって活性化され、37℃で120分間インキュベートされる。
【0172】
単離されたCEC及び/又はCEPC細胞が以下のプロトコールを用いて溶解される:
1)新鮮な溶解緩衝液が表1に記載された試薬を混合することによって新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却された緩衝液に再懸濁される。
3)インキュベートが30分間氷上で行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速で、マイクロチューブ中で遠心分離される。
5)溶解物は、分析のために新しいチューブに移され、−80℃で保存される。
【0173】
単離されたCSCの細胞活性化及び溶解:
活性化された細胞:
1)増殖因子TGF−α(100nM)、ヘレグリン(10nM)、及び/又はIGF(100nM)が細胞に添加され、37℃で5分間インキュベートされる。
【0174】
薬剤処理で活性化された細胞:
1)試料は、37℃で30分間、治療に効果的な濃度のヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で5分間インキュベートされる。
【0175】
薬剤処理で活性化された細胞(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度のヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で120分間インキュベートされる。
【0176】
単離されたCSC細胞は、以下のプロトコールを用いて溶解される:
1)新鮮な溶解緩衝液が表1に記載された試薬を混合することによって新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却された緩衝液に再懸濁される。
3)インキュベートが30分間氷上で行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速で、マイクロチューブ中で遠心分離される。
5)溶解物は、分析のために新しいチューブに移され、−80℃で保存される。
【0177】
(実施例2.チラミドシグナル増幅を有する単一検出サンドイッチELISAを用いた単一細胞検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重、ハイスループットの単一検出サンドイッチELISAを説明する:
1)96穴マイクロタイタープレートは、4℃で一晩、捕捉抗体で覆われた。
2)プレートは、翌日、1時間、2%BSA/TBS−Tweenで遮断(ブロック)された。
3)TBS−Tweenで洗浄後、細胞溶解物又は組み換えタンパク質は、一連の希釈で添加され、室温で2時間インキュベートされた。
4)プレートは、TBS−Tweenで4回洗浄され、その後、室温で2時間、ビオチン標識検出抗体とともにインキュベートされた。
5)検出抗体とのインキュベート後、プレートはTBS−Tweenで4回洗浄され、その後、ストレプトアビジン標識西洋わさびペルオキシダーゼ(SA−HRP)がビオチン標識検出抗体に結合するように、室温で1時間、SA−HRPとインキュベートされた。
6)シグナル増幅のため、ビオチン−チラミドは、0.015%H2O2とともに5μg/mlで添加され、15分間反応させられた。
7)TBS−Tweenで6回洗浄後、SA−HRPが添加され、30分間インキュベートされた。
8)TBS−Tweenで6回洗浄後、HRP基質TMBが添加され、色が遮光下で2〜10分間発色させられた。反応は、0.5MH2SO4を添加することによって停止された。シグナルは、マイクロプレートリーダーにて450/570nmで走査された。
【0178】
図3は、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体から構成されるELISAを用いたA431細胞中の総EGFRの検出を示す。免疫検出の感度は、ヒトEGFRの細胞の組み換え細胞外ドメインに基づき、約0.25pg/穴であった。計算されたEGFR濃度は、各A431細胞あたり、約0.6pgであった。
【0179】
図4は、捕捉抗体としてのEGFRの細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化EGFRに対するビオチン標識モノクローナル抗体と、から構成されるELISAを用いた、A431細胞中のリン酸化EGFRの検出を示す。捕捉抗体の2倍の連続希釈を用いると、捕捉抗体濃度濃度が0.0625μg/mlである時に1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において1.78倍の増加があったことを示した。
【0180】
図5は、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体から構成されるELISAを用いたSKBr3細胞中の総ErbB2の検出を示す。免疫分析の検出幅は、約1000細胞と約1.37細胞の間であった。捕捉抗体濃度が、1μg/mlである時に、1.37細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において2.71倍の増加があった。
【0181】
図6は、捕捉抗体としてのErbB2の細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化ErbB2に対するモノクローナル抗体と、から構成されるELISAを用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す。免疫分析の検出幅は、約500細胞と約5細胞の間であった。捕捉抗体濃度が、1μg/mlである時に、5細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.03倍の増加があった。
【0182】
図7は、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体から構成されるELISAを用いたSKBr3細胞中の総及びリン酸化Erk2の検出を示す。1.37細胞レベルで、総Erk2に関し、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.25倍の増加があった。同様に、1.37細胞レベルで、リン酸化Erk2に関し、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.17倍の増加があった。
【0183】
(実施例3.チラミドシグナル増幅を有する単一検出マイクロアレイELISAを用いた単一細胞検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重で、ハイスループットの単一検出マイクロアレイELISAを説明する:
1)捕捉抗体は、2倍連続希釈で16パッドFASTスライド(WhatmanInc.;Florham Park,NJ)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、80μlのビオチン標識検出抗体が、室温で2時間インキュベートされた。
5)6回洗浄後、ストレプトアビジン標識西洋わさびペルオキシダーゼ(SA−HRP)が添加され、SA−HRPがビオチン標識検出抗体に結合するように、1時間インキュベートされた。
6)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、そしてTBSで1回洗浄された。
7)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、マイクロアレイ検出器(Perkin−Elmer,Inc.;Waltham,MA)で走査された。
【0184】
図8は、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体から構成されるマイクロアレイELISAを用いたA431細胞中の総EGFRの検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のEGFRを検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、EGFRが1細胞から検出され得ることを示した。捕捉抗体濃度が、0.0625mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において2.11倍の増加があった。
【0185】
図9は、捕捉抗体としてのEGFRの細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化EGFRに対するモノクローナル抗体と、から構成されるマイクロアレイELISAを用いた、A431細胞中のリン酸化EGFRの検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のリン酸化EGFRを検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、リン酸化EGFRが1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において1.33倍の増加があった。
【0186】
図10は、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体から構成されるマイクロアレイELISAを用いたSKBr3細胞中の総ErBb2の検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、その希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のErBb2を検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、ErBb2が1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において15.27倍の増加があった。
【0187】
図11は、捕捉抗体としてのErBb2の細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化ErBb2に対するモノクローナル抗体と、から構成されるマイクロアレイELISAを用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のErBb2を検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、リン酸化ErBb2が1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において5.45倍の増加があった。
【0188】
(実施例4.チラミドシグナル増幅を有する近接2重検出マイクロアレイELISAを用いた単一細胞の検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重で、ハイスループットの近接2重検出マイクロアレイELISAを説明する:
1)捕捉抗体は、1mg/mlから0.004mg/mlまでの連続希釈で16パッドFASTスライド(WhatmanInc.)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)A431細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBS中で希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は:(1)グルコースオキシダーゼ(GO)に直接共役された抗−EGFRモノクローナル抗体;及び(2)西洋わさびペルオキシダーゼ(HRP)に直接共役されたリン酸化EGFRの細胞を認識するモノクローナル抗体、であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRを認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間のストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ(GO)結合体を用いた。リン酸化EGFRの細胞へのHRPの直接に結合された又はビオチン−ストレプトアビジン(SA)を介した結合体が用いられた。
6)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、そしてTBSで1回洗浄された。
7)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、マイクロアレイ検出器(Perkin−Elmer,Inc.)で走査された。
【0189】
図12は、近接2重検出マイクロアレイELISAと単一検出マイクロアレイELISAの比較を示す。表2は、近接2重検出マイクロアレイELISAと単一検出マイクロアレイELISAの感度を示す。各A431細胞濃度に対し、近接及び単一検出の形式でのバックグラウンドに対するシグナル(シグナル/ノイズ比)が示される。表2に示すように、近接2重検出マイクロアレイELISAはさらに、1細胞レベルで約3倍感度が増加した。
【表2】
【0190】
図13は、単一検出マイクロアレイELISAと近接2重検出マイクロアレイELISAの分析感度を示す。単一検出の形式における様々な捕捉抗体濃度でのリン酸化EGFRの滴定曲線を作製した実験は、単一の検出抗体の特異性の欠如のため、高いバックグラウンドを示した。対照的に、近接2重検出の形式における様々な捕捉抗体濃度でのリン酸化EGFRの滴定曲線を作製した実験は、2つの検出抗体間の近接度を検出することによって得られる増加した特異性のため、非常に低いバックグラウンドを示した。
【0191】
(実施例5.アドレズ可能な近接2重検出マイクロアレイを用いた複数のシグナル伝達因子の活性状態の単一細胞検出)
この実施例は、希少循環細胞における複数のシグナル伝達分子の活性状態を解析するために適した優れたダイナミックレンジを有する、多重で、ハイスループットで、アドレス可能な近接2重検出マイクロアレイを説明する:
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。印刷された捕捉抗体は、EGFR、HER2、Erk、Shc、PI3K、及びパン−サイトケラチン(pan−cytokeratin)であった。各捕捉抗体の2倍希釈系列(0.25mg/ml,0.125mg/ml、及び0.0625mg/ml)が使用され、2重及び4重のスポットが各抗体希釈に対して作製された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSで希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は、(1)グルコースオキシダーゼ(GO)に直接共役された抗EGFRモノクローナル抗体;及び(2)HRPに直接共役されたリン酸化EGFRを認識するモノクローナル抗体であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRの細胞を認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間ストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ共役体を用いた。リン酸化EGFRの細胞へのHRPの直接共役された又はビオチン−ストレプトアビジン(SA)を介した共役体のいずれかが使用された。
7)総HER2及びリン酸化タンパク質を検出するため、工程4)、5)、又は6)がEGFRを認識するモノクローナル抗体の代わりにHER2を認識するモノクローナル抗体を用いて行われた。
8)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、TBSで1回洗浄された。
9)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、そしてマイクロアレイ検出器(Perkin−Elmer,Inc.)で取り込まれた。
【0192】
図14は、アドレス可能なマイクロアレイの形式の具体的な実施の形態を示す。5つの標的が、特異的な捕捉抗体を介してアドレス可能である(例えば、EGFR、HER2、Shc、Erk、及びPI3K)。Shc,Erk、又はPI3KのEGFR又はHER2いずれかとのリン酸化複合体は、近接2重検出の形式を用いてこの分析で検出され得る。パン−サイトケラチン(PanCK)は、上皮細胞の数に対し標準化するための対照として働く。
【0193】
図15は、活性化されたA431細胞の滴定分析におけるリン酸化Shcレベルの検出を示す。アドレス可能なアレイは、EGFR及びHER2リン酸化に関する情報を同時に提供した。
【0194】
(実施例6.近接2重検出マイクロアレイのダイナミックレンジの拡張)
この実施例は、希少循環細胞におけるシグナル伝達分子の活性状態を分析するためのダイナミックレンジが、多重、ハイスループットな、近接2重検出マイクロアレイ分析における捕捉抗体の希釈系列化を行うことによって増大され得ることを説明する:
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。各捕捉抗体は、全部で9つの濃度のために連続的に2倍に希釈された(1mg/mlで開始し;0.004mg/mlで終了する)。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSに希釈された近接分析のための検出抗体80μlがスライドに添加され亜。使用された検出抗体は:(1)グルコースオキシダーゼ(GO)に直接共役された抗EGFRモノクローナル抗体;及び(2)HRPに直接共役されたリン酸化EGFRを認識するモノクローナル抗体であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRを認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間ストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチド抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ結合体を用いた。リン酸化EGFRの細胞へのHRPの直接結合された又はビオチン−ストレプトアビジン(SA)を介した結合体のいずれかが使用された。
7)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、TBSで1回洗浄された。
8)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、そしてマイクロアレイ検出器(Perkin−Elmer,Inc.)で取り込まれた。
【0195】
図16は、抗EGFR捕捉抗体の希釈曲線を示す。アドレス可能な分析を用いて、リン酸化及び総EGFRの検出は、活性化されたA431細胞にとって単一細胞レベルであった。この分析のダイナミックレンジは、5対数よりも大きかった。各個々の曲線は、約2対数のダイナミックレンジを有するが、そのダイナミックレンジは、6つの情報を与える曲線からの情報が組み合わせられると、非常に高められた。
【0196】
(実施例7.抗体へのオリゴヌクレオチド共役)
この実施例は、オリゴヌクレオチド結合抗体又は酵素を生成するための共役及び品質制御過程を説明する。
【0197】
結合:
1)72塩基のオリゴヌクレオチドリンカーが5’−SH基と6−炭素スペーサーとともに合成された。
2)凍結乾燥されたオリゴヌクレオチドリンカーは、pH7.4、20mMのTris−HClに溶解される。125nmolのリンカーは、その後、室温で2時間、終濃度50mMで、0.5MのTCEP−HCl(Pierce;Rockford,IL)と処理された。反応混合物中のトリス、TCEP、及び非結合リンカーは、脱塩回転カラムを用いて除かれた。
3)結果として生じた脱保護オリゴヌクレオチドリンカーは、100μl反応容量中で、SMCCのようなヘテロ二官能性架橋剤を用いて標的タンパク質の第1アミンに結合させられた。
4)反応混合物は、室温で2時間インキュベートされた。オリゴヌクレオチド結合抗体又は酵素は、SephacrylS−200 HRカラム(GE Healthcare;Piscataway,NJ).を用いたゲルフィルターろ過を用いて精製された。
【0198】
結合条件:
1)グルコースオキシダーゼ(GO)分子が第1のオリゴヌクレオチドリンカーに結合された後、結果として生じたGO−オリゴヌクレオチドの3つの画分が精製後に回収され、10倍希釈系列で、ニトロセルロースで覆われたスライド上に印刷された。
2)IgGは、Alexa647及び第1のオリゴヌクレオチドリンカーに相補する配列を有する第2のオリゴヌクレオチドリンカーに結合された。結果として生じたAlexa647−オリゴヌクレオチドが結合した抗体は、チップ上に添加され、1倍濃度PBS緩衝液中、室温で1時間ハイブリダイズされ、そして数回洗浄された。
3)スライドは、乾燥され、そして核酸配列特異的なハイブリダイゼーションを確認するために、マイクロアレイ検出器で走査された。
4)グルコースオキシダーゼ酵素分析は、共役の過程が酵素の機能を変えていないことを確認した。
【0199】
図17は、Alexa647−オリゴヌクレオチドが結合した抗体が画分13〜15においてGO−オリゴヌクレオチドに対する最も高い結合親和性を有したことを示す。
【0200】
(実施例8.全ての及びリン酸化EGFRの同時検出のためのオリゴヌクレオチド結合抗体)
この実施例は、実施例7で記載したオリゴヌクレオチド結合体を用いた希少循環細胞中のシグナル伝達分子の活性状態を分析するための、多重で、ハイスループットなマイクロアレイ分析を説明する。
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。空井度は、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSに希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は:(1)Alexa647と、及びグルコースオキシダーゼ(GO)に共役されたオリゴヌクレオチドに相補する配列から構成されるオリゴヌクレオチドリンカーと、に直接共役された抗−EGFRモノクローナル抗体;及び(2)西洋わさびペルオキシダーゼ(HRP)に直接共役されたリン酸化EGFRを認識するモノクローナル抗体、であった。Alexa647−オリゴヌクレオチド結合抗EGFR抗体は、図18に示された複合体を形成するため、GO−オリゴヌクレオチドと接触した。過剰な非結合試薬はTBS−Tweenで6回洗浄することによって除去された。
5)グルコースは、その後、チラミド−Alexa555とともに反応に添加された。
6)総EGFR量は、Alexa647−オリゴヌクレオチド−結合抗EGFR抗体の直接結合によって検出された。リン酸化EGFRは、HRP共役抗p−EGFR抗体へのGO−オリゴヌクレオチドの隣接共役によって検出され、チラミドシグナル増幅によって可視化された。
7)スライドは、マイクロアレイ検出器(Perkin−Elmer,Inc.)で、Alexa647及びAlexa555に特異的なレーザーで走査された。
【0201】
図19は、総EGFR及びリン酸化EGFRの同時検出を示す。総EGFR及びリン酸(t−EGFR)は、少なくとも10細胞から直接結合分析によって検出され、そしてリン酸化EGFR(p−EGFR)は、1細胞から検出された。10e5のp−EGFR分子が近接シグナル増幅方法で検出された。p−EGFRの検出限界は、近接分析形式を用いることによって100倍以上増加した。
【0202】
(実施例9.乳癌患者における循環腫瘍細胞(CirculatingTumor Cell)(CTC)シグナル伝達の検出)
マイクロアレイの作製及び処理工程は、Chanら、Nat.Med.,10:1390−1396(2004)に記載の方法から適応される。シグナル伝達因子EGFR、ErbB2、ErbB3、ErbB4、IGF−1R、Akt、Erk、p70S6K、Bad、Rsk、Mek、cSrc、サイトケラチン、チューブリン、β−アクチン(β−actin)に対する抗体(1mg/ml)、及び抗−マウス抗体(陽性対照)(4/4アレイ)は、FAST(登録商標)スライド(WhatmanInc.;Florham Park,NJ)に抗体をスポットするための固相スポッティングピンで適合された密接印刷ロボットマイクロアレイ作製機(Bio−Rad Laboratories;Hercules,CA)を用いて384穴ポリプロピレンプレート(50μl/穴)に移される。8分割されたパッドで覆われたスライドが使用される。印刷後、スライドは、3%カゼイン溶液で遮断(ブロック)される。スライドは、使用前に乾燥条件下で少なくとも一晩保存される。
【0203】
患者選択基準、試料準備方法、及び調査設計は、疑わしい乳癌の女性における循環腫瘍細胞を評価する開示された調査(例えば、Wulfingら、Clin.CancerRes.,12:1715−1720(2006);及びReinholzら、Clin.Cancer Res.,11:3722−3732(2005)を参照)から適応される。画像化で検出された胸部異常を有し、胸部生検を受ける予定の女性は、この調査を提案される。原発性の乳癌と診断された、少なくとも42の患者が含まれる。少なくとも35の患者は、最初の診断の時に顕性転移の兆候を有していない。少なくとも7の患者は、診断時に遠部の転移を有し、陽性参照集団と見なされる。全ての患者は、以前に癌の病歴を有していない。年齢及び治療情報(例えば、外科治療、化学療法、放射線治療、内分泌療法、等)は、各患者について収集される。約20mlの血液が各患者から回収され、全ての血液試料は、一意的な識別番号を割り当てられる。全ての分析は、生検の結果を知らない臨床検査医によって行われた。
【0204】
末梢血液(18ml)が、AccuspinHistopaque−1077システム(Sigma Aldrich;St.Louis,MO)に添加され、Beckman CS−6R卓上遠心機(Beckman Instruments;PaloAlto,CA)を用いて1500rpmで10分間、遠心分離される。単核細胞層が除去され、PBSで2回洗浄され、PBS/0.1%ウシ血清アルブミンで1mlに希釈される。上皮細胞は、製造元の指示に従い、ダイナビーズ上皮濃縮(DynabeadsEpithelial Enrich)キット(Dynal AS;Oslo,Norway)を用いて磁気ビーズに付着されたBer−EP4に対する抗体を用いた免疫磁気捕獲によって濃縮される。細胞は、1時間、揺り動かされている間、20mlの容量で1〜107ビーズと混合される。Ber−EP4抗体は、扁平上皮細胞の表層、幹細胞、及び壁細胞を除く、上皮細胞の表面及び細胞質の2つの糖タンパク質を認識する。懸濁液は、少なくとも6分間磁石上に置かれ、その上清は、注意深く除去される。磁気ビーズに付着した細胞は、1mlのPBS/0.1%ウシ血清アルブミンで3回洗浄される。増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及びIGF(100nM)は、細胞に添加され、37℃で5分間インキュベートされる。細胞は、遠心分離され、キットで供給された溶解結合緩衝液で溶解される。溶解された細胞懸濁(付着したビーズを含む)は、処理されるまで80℃で保存される。
【0205】
分析方法1:溶解物(40μl)は、アレイに添加され、一晩インキュベートされ、そして、洗浄緩衝液で3回洗浄される。シグナル伝達因子各々に対する、HRP標識(例えば、Kuhlmann,ImmunoEnzyme Techniques,Verlag Chemie,Weinheim,pp1−162(1984)に記載されたように共役された)抗−リン酸抗体及びグルコースオキシダーゼで標識された(上記のKuhlmannの文献に記載されたように結合された)抗−総抗体(即ち活性状況非依存的)がアレイに添加され、2時間インキュベートされ、そして洗浄緩衝液で3回洗浄される。チラミド試薬(MolecularProbes)及びグルコースが添加され、そして反応は、1時間行われ、そして3回洗浄される。アレイは、30分間、ストレプトアビジン−HRPとインキュベートされ、洗浄され、増感ルミノール(MolecularProbes)を用いて顕わにさせられる。シグナルは、CCDカメラを用いて検出される。
【0206】
分析方法2:2,4−ジニトロフェノール(DNP)に対する抗体(lmg/ml)は、FAST(登録商標)スライド(WhatmanInc.;Florham Park,NJ)に抗体をスポットするための固相スポッティングピンで適合された密接印刷ロボットマイクロアレイ作製機(Bio−Rad Laboratories;Hercules,CA)を用いて384穴ポリプロピレンプレート(5μl/穴)に移される。8分割されたパッドで覆われたスライドが使用される。印刷後、スライドは、3%カゼイン溶液で遮断(ブロック)される。スライドは、使用前に乾燥条件下で少なくとも一晩保存される。
【0207】
溶解物(40μl)は、上記のシグナル伝達因子各々に対する総DNP標識抗体及びオリゴ(Oligo)及びAlexaFluor(商標登録)647で標識された抗−リン酸抗体と混合され、オリゴ及びAlexaFluor(商標登録)647で標識された抗−総抗体は、抗−DNP抗体に添加され、一晩インキュベートされ、洗浄緩衝液で3回洗浄される。2,4−ジニトロリジン(MolecularProbes)は、抗−DNP抗体から免疫複合体を放出するために添加される。放出された免疫複合体は、ジップコードアレイ(例えば、Keramasら、Lab Chip,4:152−158(2004);及びDelrio−Lafreniereら、Diagn.Microbiol.Infect.Dis.,48:23−31(2004)を参照)に添加され、一晩インキュベートされる。アレイは、3回洗浄され、処理されたスライドは、GenePix4000Aマイクロアレイスキャナー(AxonScanner)を用いて10ミクロンの解像度で走査される。
【0208】
(実施例10.乳癌患者における循環内皮細胞(CirculatingEndothelial Cell)(CEC)及び循環内皮前駆細胞(Circulating Endothelial Precursor Cell)(CEP)の検出)
CEC及びCEP細胞が、磁気ダイナビーズに付着したモノクローナル抗体P1H12又はCD146を用いた免疫磁気捕獲によって濃縮されることを除いて、実施例9で記載された同一の患者試料、使用準備、及び分析方法が使用される。マイクロアレイは、配置された抗体が以下の分析物に特異的であることを除いて、実施例9に記載の方法を用いて作製され、処理される:VEGFR1,VEGFR2,VEGFR3,TIE1,TIE2,PDGFR−α,PDGFR−β,FGFR1,FGFR2,Akt,Erk,p70S6K,Rsk,cSrc、及びβ−アクチン。実施例9に記載の分析方法は、CEC及びCEPシグナル伝達を検出するために用いられる。
【0209】
本明細書中で引用された全ての刊行物及び特許出願は、個々の刊行物又は特許出願それぞれが、特異的に及び個々に参照によって取り込まれると示されるように、本明細書中で参照として取り込まれる。上述の発明は、理解の明確化の目的のため、図示及び例によって、多少詳しく述べられているが、この発明の示唆を踏まえて、添付の請求の範囲の精神又は範囲から離れることなく任意の変化及び改変がそれになされてもよいことは、当業者には容易に明らかである。
【0210】
さらなる実施形態は以下の通りである。
1.優れたダイナミックレンジを有する、多重で、ハイスループットな免疫分析を行うための方法であって、
前記方法は、
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、前記細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、
前記捕捉抗体が、固相担体に拘束されている、ことを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するために、前記複数の捕捉された分析物を、前記対応する分析物に特異的な検出抗体とインキュベートする工程であって、
前記検出抗体は、
(1)促進部分で標識された複数の活性状態非依存抗体、及び
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体、
から構成され、
前記促進部分は、前記シグナル増幅対の前記第一の構成因子に方向付けられ且つ前記第一の構成因子と反応する酸化剤を生成する、ことを特徴とする工程;
(c)増幅されたシグナルを生成するために、前記複数の検出可能な捕捉された分析物を前記シグナル増幅対の第二の構成因子とインキュベートする工程;及び
(d)前記シグナル増幅対の前記第一及び第二の構成因子から生じた前記増幅されたシグナルを検出する工程、
を備える、ことを特徴とする免疫分析方法。
2.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態1に記載の免疫分析方法。
3.前記細胞が免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態2に記載の免疫分析方法。
4.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態3に記載の免疫分析方法。
5.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態3に記載の免疫分析方法。
6.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態3に記載の免疫分析方法。
7.前記単離された細胞が、増殖因子の刺激の前に抗癌剤とインキュベートされる、ことを特徴とする実施形態6に記載の免疫分析方法。
8.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態7に記載の免疫分析方法。
9.前記単離された細胞が、前記細胞抽出物を作製するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態6に記載の免疫分析方法。
10.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態1に記載の免疫分析方法。
11.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態1に記載の免疫分析方法。
12.前記活性状態非依存抗体がさらに、検出可能な部分を備える、ことを特徴とする実施形態1に記載の免疫分析方法。
13.前記検出可能な部分が、フルオロフォアである、ことを特徴とする実施形態12に記載の免疫分析方法。
14.前記検出可能な部分の量が、一つ又はそれ以上の前記分析物の量と相関関係にある、ことを特徴とする実施形態12に記載の免疫分析方法。
15.前記活性状態非依存抗体が、チャネリング部分で直接標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
16.前記活性状態非依存抗体が、前記活性状態非依存抗体に共役されたオリゴヌクレオチドと前記チャネリング部分に共役された相補オリゴヌクレオチドとの間のハイブリダイゼーションを介して前記チャネリング部分で標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
17.前記活性状態依存抗体が、前記シグナル増幅対の第一の構成因子で直接標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
18.前記活性状態依存抗体が、前記活性状態依存抗体に共役された結合対の第一の構成因子と前記シグナル増幅対の第一の構成因子に共役された前記結合対の第二の構成因子との間の結合を介して前記シグナル増幅対の第一の構成因子で標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
19.前記結合対の前記第一の構成因子がビオチンである、ことを特徴とする実施形態18に記載の免疫分析方法。
20.前記結合対の前記第二の構成因子がストレプトアビジンである、ことを特徴とする実施形態18に記載の免疫分析方法。
21.前記チャネリング部分がグルコースオキシダーゼである、ことを特徴とする実施形態1に記載の免疫分析方法。
22.前記酸化剤が過酸化水素(H2O2)である、ことを特徴とする実施形態21に記載の免疫分析方法。
23.前記シグナル増幅対の前記第一の構成因子がペルオキシダーゼである、ことを特徴とする実施形態22に記載の免疫分析方法。
24.前記ペルオキシダーゼが西洋わさびペルオキシダーゼ(HRP)である、ことを特徴とする実施形態23に記載の免疫分析方法。
25.前記シグナル増幅対の前記第二の構成因子がチラミド試薬である、ことを特徴とする実施形態23に記載の免疫分析方法。
26.前記チラミド試薬がビオチン−チラミドである、ことを特徴とする実施形態25に記載の免疫分析方法。
27.前記増幅されたシグナルが、活性型チラミドを生産するための前記ビオチン−チラミドのペルオキシダーゼ酸化によって生成される、ことを特徴とする実施形態26に記載の免疫分析方法。
28.前記活性型チラミドが直接検出される、ことを特徴とする実施形態27に記載の免疫分析方法。
29.前記活性型チラミドが、シグナル検出試薬の添加で検出される、ことを特徴とする実施形態27に記載の免疫分析方法。
30.前記シグナル検出試薬が、ストレプトアビジン標識フルオロフォアである、ことを特徴とする実施形態29に記載の免疫分析方法。
31.前記シグナル検出試薬が、ストレプトアビジン標識ペルオキシダーゼ及び発色試薬の組み合わせである、ことを特徴とする実施形態29に記載の免疫分析方法。
32.前記発色試薬が、3,3’,5,5’−テトラメチルベンジジン(TMB)である、ことを特徴とする実施形態31に記載の免疫分析方法。
33.細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体から構成される、優れたダイナミックレンジを有するアレイであって、前記捕捉抗体が固相担体に拘束される、ことを特徴とするアレイ。
34.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態33に記載のアレイ。
35.前記細胞が、免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態34に記載のアレイ。
36.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態35に記載のアレイ。
37.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態35に記載のアレイ。
38.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態35に記載のアレイ。
39.前記単離された細胞が、増殖因子の刺激の前に、抗癌剤とインキュベートされる、ことを特徴とする実施形態38に記載のアレイ。
40.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態39に記載のアレイ。
41.前記単離された細胞が、前記細胞抽出物を作製するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態38に記載のアレイ。
42.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態33に記載のアレイ。
43.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態33に記載のアレイ。
44.各希釈系列が少なくとも3つの下降する捕捉抗体濃度から構成される、ことを特徴とする実施形態33に記載のアレイ。
45.各希釈系列が少なくとも6つの下降する捕捉抗体濃度から構成される、ことを特徴とする実施形態33に記載のアレイ。
46.各希釈系列における前記捕捉抗体が少なくとも2倍で連続的に希釈される、ことを特徴とする実施形態33に記載のアレイ。
47.前記一つ又はそれ以上の分析物が、約1細胞中で検出される、ことを特徴とする実施形態33に記載のアレイ。
48.優れたダイナミックレンジを有し、多重で、ハイスループットな免疫分析を行うための方法であって、
前記方法は、
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、前記細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されている、ことを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するために、前記複数の捕捉された分析物を、前記対応する分析物に特異的な検出抗体とインキュベートする工程;
(c)増幅されたシグナルを生成するために、前記複数の検出可能な捕捉された分析物を、シグナル増幅対の第一及び第二の構成因子とインキュベートする工程;及び
(d)前記シグナル増幅対の前記第一及び第二の構成因子から生じた増幅されたシグナルを検出する工程、
を備える、ことを特徴とする免疫分析方法。
49.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
50.前記細胞が、免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態49に記載の免疫分析方法。
51.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態50に記載の免疫分析方法。
52.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態50に記載の免疫分析方法。
53.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態50に記載の免疫分析方法。
54.前記単離された細胞が、増殖因子の刺激の前に抗癌剤とインキュベートされる、ことを特徴とする実施形態53に記載の免疫分析方法。
55.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態54に記載の免疫分析方法。
56.前記単離された細胞が、前記細胞抽出物を生成するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態53に記載の免疫分析方法。
57.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
58.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態48に記載の免疫分析方法。
59.前記検出抗体が、結合対の第一の構成因子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
60.前記結合対の前記第一の構成因子がビオチンである、ことを特徴とする実施形態59に記載の免疫分析方法。
61.前記シグナル増幅対の前記第一の構成因子が、前記結合対の第二の構成因子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
62.前記結合対の前記第二の構成因子が、ストレプトアビジンである、ことを特徴とする実施形態61に記載の免疫分析方法。
63.前記シグナル増幅対の前記第一の構成因子がペルオキシダーゼであることを特徴とする実施形態61に記載の免疫分析方法。
64.前記ペルオキシダーゼが西洋わさびペルオキシダーゼ(HRP)である、ことを特徴とする実施形態63に記載の免疫分析方法。
65.前記シグナル増幅対の前記第二の構成因子がチラミド試薬である、ことを特徴とする実施形態63に記載の免疫分析方法。
66.前記チラミド試薬がビオチン−チラミドである、ことを特徴とする実施形態65に記載の免疫分析方法。
67.前記増幅されたシグナルが、活性型チラミドを作製するための前記ビオチン−チラミドのペルオキシダーゼ酸化によって生成される、ことを特徴とする実施形態66に記載の免疫分析方法。
68.前記活性型チラミドが直接検出される、ことを特徴とする実施形態67に記載の免疫分析方法。
69.前記活性型チラミドが、シグナル検出試薬の添加で検出される、ことを特徴とする実施形態67に記載の免疫分析方法。
70.前記シグナル検出試薬が、ストレプトアビジン標識フルオロフォアである、ことを特徴とする実施形態69に記載の免疫分析方法。
71.前記シグナル検出試薬が、ストレプトアビジン標識ペルオキシダーゼ及び発色試薬の組み合わせである、ことを特徴とする実施形態69に記載の免疫分析方法。
72.前記発色試薬が、3,3’,5,5’−テトラメチルベンジジン(TMB)である、ことを特徴とする実施形態71に記載の免疫分析方法。
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、2006年9月21日に出願され、米国仮出願第号に変更された、米国出願第11/525,598号、2007年4月20日に出願された米国仮出願第60/913,087号の優先権を主張し、それらの開示は、すべての目的のため、その全体が参照により本明細書に援用される。
【背景技術】
【0002】
腫瘍細胞は、様々な初期癌の患者の血液中に「微小転移(micrometastases)」(播種性腫瘍細胞)として認められる。そして、腫瘍細胞は転移性癌においても認められる。血中の腫瘍細胞の数は、腫瘍の病期と種類に依存する。腫瘍は、極端に異質である。結果として、単一の部位からの生検は、ある腫瘍集団における異質性を示さないかもしれない。生検は、典型的には、原発巣で取得される;しかしながら、ほとんどの転移性腫瘍は、生検が行われないため、腫瘍試料の分子的解析をより困難にしている。
【0003】
腫瘍の転移の間、最も攻撃性のある腫瘍細胞が、原発巣を離れ、血液やリンパ系を通って、離れた部位に達する。このように、血液からの循環腫瘍細胞は、最も攻撃的で同質な腫瘍細胞集団を示す。血中の転移性腫瘍細胞の数は、血液1mlあたり、1つから数千個の細胞まで様々であり得る。
【発明の概要】
【発明が解決しようとする課題】
【0004】
従って、診断及び治療の目的のため、これらの細胞を検出する、特異的で感度の高い方法が必要とされている。本発明は、この必要性を満たし、さらに、関連する有利な点も提供する。
【0005】
本発明は、希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための抗体に基づくアレイとその使用方法を提供する。この抗体に基づくアレイは、酵素結合免疫吸着法(ELISA)に伴う特異性、シグナル増幅に伴う感度、及びマイクロアレイに伴うハイスループットの多重化の利点を有する。
【課題を解決するための手段】
【0006】
1つの態様では、本発明は、細胞抽出物における1つ又はそれ以上の測定物質に特異的な複数の希釈系列の捕捉抗体から構成される優れたダイナミックレンジを有するアレイを提供する。前記捕捉抗体は、固相担体上に拘束されている。
【0007】
他の態様では、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、細胞抽出物中の1つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、対応する分析物に特異的な検出抗体とインキュベートする工程;
(c)増幅されたシグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第一及び第二の構成因子とインキュベートする工程;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0008】
さらに他の態様では、本発明は、優れたダイナミックレンジを有する多重、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するため、細胞抽出物を、細胞抽出物中の1つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、それに対応する分析物に特異的な検出抗体とインキュベートする工程であって、
前記検出抗体は:
(1)促進部分で標識された複数の活性状態非依存抗体、と
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体と、を備え、
前記促進部分は、シグナル増幅対の第一の構成因子に向けられ且つそれと反応する酸化剤を生成する、
ことを特徴とする工程;
(c)増幅シグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第二の構成因子とインキュベートする構成;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0009】
本発明はまた、上記の抗体に基づくアレイ法を行うためのキットを提供し、本キットは、以下の構成を備える:(a)固相担体に拘束された複数の希釈系列の捕捉抗体;及び(b)複数の検出抗体。このキットはさらに、任意に、例えばシグナル増幅対の第一及び第二の構成因子のような他の試薬を備えてもよい。
【0010】
本発明の他の目的、特徴、利点は、下記の詳細な説明及び図より、当該分野の当業者には明確である。
【図面の簡単な説明】
【0011】
【図1】活性型分析物に特異的に結合した3つの抗体を示す図である。
【図2】活性型分析物に特異的に結合した標識抗体が固相担体に拘束される分析スキームを示す図である。
【図3】ELISAにおいて、捕捉抗体及び検出抗体として、EGFRの細胞外ドメインへのモノクローナル抗体を用いてA431細胞中の総EGFRの検出を示す図である。(A)は、サンドイッチELISAの標準曲線を示す。その感度は、ヒトEGFRの組み換え細胞外ドメインを用いた場合、0.25pg/穴であった。(B)は、細胞滴定曲線を示す。A431細胞中のEGFR濃度をELISAによって検出した。(C)は、pg/穴及びpg/細胞で計算されたEGFR濃度の表である。計算されたEGFR濃度は、各A431細胞あたり約0.6pgであった。
【図4】ELISAにおいて、捕捉抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を、及び、検出抗体としてリン酸化EGFRに対するビオチン標識モノクローナル抗体を用いた、A431細胞中のリン酸化EGFRの検出を示す図である。(A)は、捕捉抗体の希釈系列での細胞滴定曲線を示す。(B)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表を示す。捕捉抗体濃度が0.0625μg/mlであるときに、そのシグナル/ノイズ比は一細胞レベルで1.78であった。
【図5】ELISAにおいて、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を用いた、SKBr3細胞中の総ErbB2の検出を示す図である。(A)は、SKBr3細胞中の総ErBb2の細胞滴定を示す。検出範囲は、約1000細胞と約1.37細胞との間であった。(B)は、細胞とバックグラウンド間のシグナル/ノイズ比を示す。捕捉抗体濃度が1μg/mlであるときに、シグナル/ノイズ比は、1.37細胞レベルで2.71であった。
【図6】ELISAにおいて、捕捉抗体としてErbB2の細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化ErbB2に対するモノクローナル抗体を用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す図である。(A)は、SKBr3細胞中のリン酸化ErBb2の細胞滴定を示す。検出範囲は、約500細胞と約5細胞との間であった。(B)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が1μg/mlであるときに、シグナル/ノイズ(S/N)比は5細胞レベルで3.03であった。
【図7】ELISAにおいて、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体を用いた、SKBr3細胞中の総Erk2タンパク質及びリン酸化Erk2タンパク質の検出を示す図である。(A)は、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体を用いた総Erk2タンパク質の検出を示す。(B)は、捕捉抗体としてErk2に対するモノクローナル抗体を、検出抗体としてリン酸化Erk2に対するモノクローナル抗体を用いたリン酸化Erk2の検出を示す。(C)は、細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。総Erk2及びリン酸化Erk2の両方に対するシグナル/ノイズ比は、1.37細胞レベルで約3であった。
【図8】マイクロアレイELISAにおいて、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を用いた、A431細胞中の総EGFRの検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列での様々な濃度の捕捉抗体で約1〜10000細胞中のEGFRを検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、EGFRが一細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.0625mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は、1細胞レベルで2.11であった。
【図9】マイクロアレイELISAにおいて、捕捉抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化EGFRに対するモノクローナル抗体を用いた、A431細胞中のリン酸化EGFRの検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体での約1−10000細胞中のリン酸化EGFRを検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、リン酸化EGFRが一細胞から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は一細胞レベルで1.33であった。
【図10】マイクロアレイELISAにおいて、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を用いた、SKBr3細胞中の総ErBb2の検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体で約1〜10000細胞中のErBb2を検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、ErBb2が1細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)である時に、シグナル/ノイズ比は1細胞レベルで15.27であった。
【図11】マイクロアレイELISAにおいて捕捉抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体を、検出抗体としてリン酸化ErBb2に対するモノクローナル抗体を用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す図である。(A)は、細胞数に基づく捕捉抗体希釈曲線を示す。このマイクロアレイELISAは、希釈系列における様々な濃度の捕捉抗体で約1〜10000細胞中のErBb2を検出する幅広いダイナミックレンジを有した。(B)は、捕捉抗体濃度の希釈系列に基づく細胞滴定曲線であり、リン酸化ErBb2が1細胞(図中の矢印)から検出され得ることを示した。(C)は、希釈系列における様々な捕捉抗体の濃度での細胞とバックグラウンド間のシグナル/ノイズ(S/N)比の表である。捕捉抗体濃度が0.125mg/ml(図中の矢印)であるときに、シグナル/ノイズ比は1細胞レベルで5.45であった。
【図12】近接二重検出マイクロアレイELISAと単一検出マイクロアレイELISAとの感度の比較を示す図である。A431細胞は、10000から0.01細胞まで希釈された。捕捉抗体は、1mg/m1から0.004mg/mlまで連続的に希釈された。
【図13】単一検出抗体のマイクロアレイELISAと近接二重検出マイクロアレイELISAの分析特異性を示す図である。(A)は、単一検出の形式における様々な捕捉抗体濃度でのA431細胞中のリン酸化EGFRの滴定曲線を示す。この形式では単一の検出抗体の特異性の欠如のため、非常に高いバックグラウンドが観察された。(B)は、近接二重検出の形式における様々な捕捉抗体の濃度でのA431細胞中のリン酸化EGFRの滴定曲線を示す。この形式では、2つの検出抗体間の近接度を検出することによって得られる特異性が増加するため、非常に低いバックグラウンドしか検出されなかった。
【図14】複数のシグナル伝達分子の活性状態を決定するために捕捉抗体の希釈系列を用いたアドレス可能なマイクロアレイの形式の実施例を示す図である。
【図15】活性化されたA431細胞の滴定分析におけるリン酸化Shcレベルの検出を示す図である。アドレス可能なアレイは、EGFR及びHER2リン酸化に関する情報を同時に提供した。
【図16】抗EGFR捕捉抗体の希釈曲線を示す図である。この分析のダイナミックレンジは、5対数(5logs)よりも大きかった。個々の曲線それぞれは、約2対数(2logs)のダイナミックレンジを有するが、6つの有益な曲線からの情報を組み合わせると、そのダイナミックレンジが非常に増加した。
【図17】本発明のオリゴヌクレオチド共役のための品質管理手順を示す。(A)は、様々なGO−オリゴヌクレオチド画分のマイクロアレイのスポッティングパターンを示す。(B)は、Alexa647−オリゴヌクレオチド共役抗体が、画分13〜15でGO−オリゴヌクレオチドに対する最も高い結合親和性を有したことを示す。
【図18】グルコースオキシダーゼ(GO)−オリゴヌクレオチドに共役したAlexa647−オリゴヌクレオチド共役抗−EGFR抗体、HRP共役抗−リン酸化EGFR抗体、及び固相担体に固着したEGFR捕捉抗体からなる多重リン酸化EGFR複合体の形成を示す図である。
【図19】総EGFR及びリン酸化EGFRの同時検出を示す図である。(A)は、10000細胞中に存在する総EGFRがAlexa647−オリゴヌクレオチド共役抗−EGFR抗体から生じるAlexa647シグナルによって検出されたことを示す。EGFRのリン酸化の割合は、チラミドを介して増幅されたAlexa555シグナルを観察することによって検出された。(B)は、総EGFR(t−EGFR)は、わずか10細胞から直接結合分析によって検出され、リン酸化EGFR(p−EGFR)は1細胞から検出されたことを示す。10e5のp−EGFR分子が近接シグナル増幅法で検出された。p−EGFRの検出限界は、近接分析の形式を用いることによって100倍以上増加した。
【発明を実施するための形態】
【0012】
(I.導入)
本発明は、抗体に基づくアレイ分析システムを用いた希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための方法を提供する。ある実施形態では、本発明の多重で、ハイスループットの免疫分析は、単一の細胞レベルでの固形腫瘍の循環細胞における1つ又はそれ以上のシグナル伝達分子の活性状態を検出することができる。実際に、EGFRのようなシグナル伝達分子は、約100zmolの感度と約100zmolから約100fmolまでの直線性のダイナミックレンジで検出され得る。このように、希少循環細胞における複数のシグナル伝達物質の活性状態の単一細胞での検出は、個別の標的治療法の設計と同様に、癌の予後診断及び診断を促進する。
【0013】
希少循環細胞は、固形腫瘍から転移あるいは微小転移した固形腫瘍の循環細胞を含む。循環腫瘍細胞、癌幹細胞、及び循環内皮前駆細胞、循環内皮細胞、循環血管新生促進骨髄性細胞及び循環樹状細胞のような腫瘍へ移動している細胞(例えば、化学誘引のため)は、固形腫瘍の循環細胞の例である。
【0014】
対象となるシグナル伝達分子は、典型的に、循環細胞がinsituの活性状態を保つように、単離された直後に、好ましくは24,6,又は1時間以内に、より好ましくは30,15,又は5分以内に抽出される。単離された細胞はまた、シグナル伝達分子の活性を復活させる、又は活性化するために、1つ又はそれ以上の増殖因子と、通常nmolからμmolの濃度で、1〜30分間インキュベートされてもよい(Irishら、Cell,118:217−228(2004)を参照)。
【0015】
本明細書でより詳細に説明するように、個々の患者に対する可能な抗癌治療を評価するため、単離された細胞は、1つ又はそれ以上の抗癌剤と様々な用量でインキュベートされ得る。増殖因子の活性化は、その後、数分間(例えば、約1〜5分間)又は数時間(例えば、約1〜6時間)実施され得る。抗癌剤の有無におけるシグナル伝達経路の異なる活性化は、個々の患者各々に対する適量で適切な癌治療の選択の助けとなり得る。循環細胞はまた、治療における変化があるか否かを決定するために、抗癌剤治療中の患者試料から単離され、そして1つ又はそれ以上の増殖因子で活性化され得る。そのように、本発明の方法は、有利に、臨床医が全ての患者に対し、正しい期間、正しい用量で正しい抗癌剤を提供することを支援する。
【0016】
(II.定義)
本明細書において、他に規定されない限り、次の用語は以下に定義される意味を有する。
【0017】
「癌(cancer)」という用語は、異常な細胞の制御されない増殖によって特徴づけられる疾病分類のあらゆるものを含むことを意図されている。この用語は、悪性、良性、軟部組織、あるいは固形として特徴づけられるか否かによらず、全ての既知の癌及び腫瘍性の状態、及び転移前及び転移後の癌を含む全ての病期と悪性度の癌を含む。癌の異なる種類の具体例は、これに限定されないが、肺癌(例えば、非小細胞肺癌);結腸直腸癌、消化管間質腫瘍、消化管カルチノイド癌、結腸癌、直腸癌、肛門癌、胆管癌、小腸癌、及び胃癌のような消化器及び消化管癌;食道癌;胆嚢癌;肝臓癌;膵臓癌;虫垂癌;乳癌;卵巣癌;腎臓癌(例えば、腎細胞癌);中枢神経系の癌;皮膚癌;リンパ腫;絨毛腫;頭部及び頸部癌;骨肉腫;及び血液癌を含む。本明細書において、「腫瘍(tumor)」は1つ又はそれ以上の癌性細胞から構成される。
【0018】
「分析物(analyte)」は、存在、量、及び/又はその正体が決定される、対象となるあらゆる分子、典型的にはポリペプチドのような巨大分子を含む。ある例では、分析物は、固形腫瘍の循環細胞の細胞構成成分であり、好ましくはシグナル伝達分子である。
【0019】
本明細書において、「希釈系列(dilutionseries)」という用語は、特定の試料(例えば、細胞溶解物)あるいは試薬(例えば、抗体)の一連の降順の濃度を含むことを意図されている。希釈系列は、典型的に、開始濃度の試料あるいは試薬の測定した量を、より低濃度の試料あるいは試薬を作るために、希釈剤(例えば、希釈用緩衝液)と混合する工程と、所望の数の連続的な希釈系列を得るのに十分な回数、その工程を繰り返すことによって生成される。試料あるいは試薬は、少なくとも2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,25,30,35,40,45,又は50の降順の濃度の試料あるいは試薬から構成される希釈系列を生成するため、少なくとも2,3,4,5,6,7,8,9,10,15,20,25,30,35,40,45,50,100,500,又は1000倍で連続的に希釈され得る。例えば、1mg/mlの開始濃度での捕捉抗体試薬の2倍の連続的な希釈から構成される希釈系列は、0.5mg/mlの濃度の捕捉抗体を作成するように、開始濃度の捕捉抗体のある用量をそれと等量の希釈用緩衝液と混合し、0.25mg/ml、0.125mg/ml、0.0625mg/ml、0.0325mg/ml、等の捕捉抗体濃度を得るように、この工程を繰り返すことによって生成され得る。
【0020】
本明細書において、「優れたダイナミックレンジ(superiordynamic range)」という用語は、わずか1細胞中又は数千もの細胞中の特定の分析物を検出する、本発明の分析能のことをいう。例えば、本明細書で述べる免疫分析は、有利に、捕捉抗体濃度の希釈系列を用いて、約1〜10000細胞(例えば、約1,5,10,25,50,75,100,250,500,750,1000,2500,5000,7500,又は10000細胞)中における対象となる特定のシグナル伝達分子を検出するため、優れたダイナミックレンジを有する。
【0021】
「シグナル伝達分子(signaltransduction molecule)」又は「シグナル伝達物質(signal transducer)」は、細胞が細胞外シグナル又は刺激を、典型的には細胞内での生化学反応の順序づけられた連続を含む応答に変換する工程を実行するタンパク質又は他の分子を含む。シグナル伝達分子の具体例は、これに限定されないが、EGFR(例えば、EGFR/HER1/ErbB1,HER2/Neu/ErbB2,HER3/ErbB3,HER4/ErbB4)、VEGFR−1/FLT−1、VEGFR−2/FLK−1/KDR、VEGFR−3/FLT−4、FLT−3/FLK−2、PDGFR(例えば、PDGFRA,PDGFRB)、c−KIT/SCFR、INSR(インスリン受容体)、IGF−IR、IGF−IIR、IRR(インスリン受容体関連受容体)、CSF−1R、FGFR1−4、HGFR 1−2、CCK4、TRK A−C、MET、RON、EPHA 1−8、EPHB 1−6、AXL、MER、TYRO3、TIE 1−2、TEK、RYK、DDR1−2、RET、c−ROS、LTK(白血球チロシンキナーゼ)、ALK(未分化リンパ腫キナーゼ)、ROR 1−2、MUSK、AATYK 1−3、及びRTK 106のような受容体型チロシンキナーゼ;BCR−ABL、Src、Frk、Btk、Csk、Abl、Zap70、Fes/Fps、Fak、Jak、Ack、及びLIMKのような非受容体型チロシンキナーゼ;Akt、MAPK/ERK、MEK、RAF、PLA2、MEKK、JNKK、JNK、p38、Shc(p66)、PI3K、Ras(例えば、K−Ras、N−Ras、H−Ras)、Rho、Rac1、Cdc42、PLC、PKC、p70S6キナーゼ、p53、サイクリンD1、STAT1、STAT3、PIP2、PIP3、PDK、mTOR、BAD、p21、p27、ROCK、IP3、TSP−1、NOS、PTEN、RSK1−3、JNK、c−Jun、Rb、CREB、Ki67、及びパキシリン(paxillin)のようなチロシンキナーゼシグナル伝達系の構成成分;及びこれらの組み合わせを含む。
【0022】
本明細書において、「循環細胞(circulatingcells)」という用語は、固形腫瘍から転移あるいは微小転移のいずれかがなされた細胞から構成される。循環細胞の具体例は、これに限定されないが、循環腫瘍細胞、癌幹細胞、及び/又は腫瘍へ移動している細胞(例えば、循環内皮前駆細胞、循環内皮細胞、循環血管新生促進骨髄性細胞、循環樹状細胞、等)を含む。
【0023】
本明細書において、「試料(sample)」という用語は、患者から得られた、あらゆる生物学的標本を含む。試料は、これに限定されないが、全血、血漿、血清、赤血球細胞、白血球細胞(例えば、末梢血単核球)、唾液、尿、便(すなわち、糞)、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液(例えば、リンパ節の播種性腫瘍細胞)、細針吸引物、あらゆる他の体液、腫瘍の生検(例えば、針生検)のような組織試料(例えば、腫瘍組織)、及びこれらの細胞抽出物を含む。ある実施の形態において、試料は、全血、又は血漿、血清あるいは細胞沈殿物のような全血の部分構成成分である。好ましい実施の形態では、試料は、当該分野では既知のあらゆる技術を用いて全血又はその細胞画分から固形腫瘍の循環細胞を単離し、循環細胞の細胞抽出物を調製することによって得られる。他の実施の形態において、試料は、例えば肺、結腸又は直腸の固形腫瘍からの、ホルマリン固定パラフィン包埋(FFPE)腫瘍組織標本である。
【0024】
「被検者(subject)」又は「患者(patient)」は、典型的には、ヒトを含むが、例えば他の哺乳類、齧歯類、イヌ科、ネコ科、ウマ、ヒツジ、ブタ及び同様のもの、のような他の動物も含むことができる。
【0025】
「アレイ(array)」又は「マイクロアレイ(microarray)」は、例えばガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ(例えば磁気ビーズ、ポリスチレンビーズ、等)、紙、膜(例えばナイロン、ニトロセルロース、ポリフッ化ビニリデン(PVDF)、等)、繊維束、又はあらゆる他の適切な基質のような固相担体上に固定あるいは拘束された、特徴的な一組の捕捉抗体及び/又はその希釈系列から構成される。この捕捉抗体は、一般的に共有結合性又は非共有結合性相互作用(例えばイオン結合、疎水性相互作用、水素結合、ファンデルワールス力、双極子−双極子相互作用)を介して固相担体に固定又は拘束されている。ある例として、捕捉抗体は、固相担体に結合した捕捉剤と相互作用する捕捉タグを備える。本発明の分析に用いられるアレイは典型的に、既知でアドレス可能な異なる位置において固相担体の表面に連結した複数の異なる捕捉抗体及び/又は捕捉抗体の濃度から構成される。
【0026】
「捕捉抗体(captureantibody)」という用語は、固形腫瘍の循環細胞の細胞抽出物のような試料における対象となる1つ又はそれ以上の分析物に特異的である(すなわち、分析物に結合する、分析物に結合される、又は分析物と複合体を形成する)固定された抗体を含むことを意図している。好ましい実施の形態では、捕捉抗体はアレイにおける固相担体に拘束されている。あらゆる様々なシグナル伝達分子を固相担体上に固定するために適した捕捉担体は、Upstate(Temecula,CA)、Biosource(Camarillo,CA)、CellSignaling Technologies(Danvers,MA),R&D Systems(Minneapolis,MN),Lab Vision(Fremont,CA),SantaCruz Biotechnology(Santa Cruz,CA),Sigma(St.Louis,MO)、及びBD Biosciences(San Jose,CA)から入手できる。
【0027】
本明細書において、「検出抗体(detectionantibody)」という用語は、試料中の対象となる1つ又はそれ以上の分析物に特異的である(すなわち、分析物に結合する、分析物に結合される、又は分析物と複合体を形成する)、検出可能な標識を備える抗体を含む。この用語はまた、対象となる1つ又はそれ以上の分析物に特異的な抗体であって、検出可能な標識を備える他の種によって結合され得る抗体を包含する。検出可能な標識の例は、これに限定されないが、ビオチン/ストレプトアビジン標識、核酸(例えばオリゴヌクレオチド)標識、化学反応標識、蛍光標識、酵素標識、放射性標識、及びこれらの組み合わせを含む。あらゆる様々なシグナル伝達分子の活性状態及び/又は総量を検出するために適した検出抗体は、Upstate(Temecula,CA),Biosource(Camarillo,CA),CellSignaling Technologies(Danvers,MA),R&D Systems(Minneapolis,MN),Lab Vision(Fremont,CA),SantaCruz Biotechnology(Santa Cruz,CA),Sigma(St.Louis,MO)、及びBD Biosciences(San Jose,CA)から入手できる。非限定的な例として、EGFR,c−KIT,c−Src,FLK−1,PDGFRA,PDGFRB,Akt,MAPK,PTEN,Raf、及びMEKのようなシグナル伝達分子の様々なリン酸化型に対するリン酸特異的抗体は、SantaCruz Biotechnologyから入手できる。
【0028】
「活性状態依存抗体(activationstate−dependent antibody)」という用語は、試料における対象となる1つ又はそれ以上の分析物の特定の活性状態に特異的である(すなわち、結合する、結合される、又は複合体を形成する)検出抗体を含む。好ましい実施の形態では、活性状態依存抗体は、1つ又はそれ以上のシグナル伝達分子のような1つ又はそれ以上の分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出する。ある実施の形態では、受容体型チロシンキナーゼのEGFRファミリーの構成因子のリン酸化及び/又はEGFRファミリーの構成因子間のヘテロ二量体複合体の形成が活性状態依存抗体を用いて検出される。活性状態依存抗体での検出に適した活性状態の非限定的な例(括弧内に記載される)は以下を含む:EGFR(EGFRvIII,リン酸化(p−)EGFR,EGFR:Shc,ユビキチン化(u−)EGFR,p−EGFRvIII);ErbB2(p85:切断型(Tr)−ErbB2,p−ErbB2,p85:Tr−p−ErbB2,Her2:Shc,ErbB2:PI3K,ErbB2:EGFR,ErbB2:ErbB3,ErbB2:ErbB4);ErbB3(p−ErbB3,ErbB3:PI3K,p−ErbB3:PI3K,ErbB3:Shc);ErbB4(p−ErbB4,ErbB4:Shc);IGF−1R(p−IGF−1R,IGF−1R:IRS,IRS:PI3K,p−IRS,IGF−1R:PI3K);INSR(p−INSR);KIT(p−KIT);FLT3(p−FLT3);HGFR1(p−HGFR1);HGFR2(p−HGFR2);RET(p−RET);PDGFRa(p−PDGFRa);PDGFRP(p−PDGFRP);VEGFR1(p−VEGFR1,VEGFR1:PLCγ,VEGFR1:Src);VEGFR2(p−VEGFR2,VEGFR2:PLCγ,VEGFR2:Src,VEGFR2:ヘパリン硫酸塩,VEGFR2:VE−カドヘリン);VEGFR3(p−VEGFR3);FGFR1(p−FGFR1);FGFR2(p−FGFR2);FGFR3(p−FGFR3);FGFR4(p−FGFR4);Tiel(p−Tiel);Tie2(p−Tie2);EphA(p−EphA);EphB(p−EphB);NFKB及び/又はIKB(p−IK(S32),p−NFKB(S536),p−P65:IKBa);Akt(p−Akt(T308,S473));PTEN(p−PTEN);Bad(p−Bad(S112,S136),Bad:14−3−3);mTor(p−mTor(S2448));p70S6K(p−p70S6K(T229,T389));Mek(p−Mek(S217,S221));Erk(p−Erk(T202,Y204));Rsk−1(p−Rsk−1(T357,S363));Jnk(p−Jnk(T183,Y185));P38(p−P38(T180,Y182));Stat3(p−Stat−3(Y705,S727));Fak(p−Fak(Y576));Rb(p−Rb(S249,T252,S780));Ki67;p53(p−p53(S392,S20));CREB(p−CREB(S133));c−Jun(p−c−Jun(S63));cSrc(p−cSrc(Y416));及びパキシリン(p−パキシリン(p−paxillin(Y118))。
【0029】
「活性状態非依存抗体(activationstate−independent antibody)」という用語は、活性状態にかかわりなく試料における対象となる1つ又はそれ以上の分析物に特異的である(すなわち、結合する、結合される、又は複合体を形成する)検出抗体を含む。例えば、活性状態非依存抗体は、シグナル伝達分子のような1つ又はそれ以上の分析物のリン酸化型及び非リン酸化型の両方を検出することができる。
【0030】
「核酸(nucleicacid)」又は「ポリヌクレオチド(polynucleotide)」という用語は、例えばDNA及びRNAのような一本鎖型又は二本鎖型のいずれかにおけるデオキシリボヌクレオチド又はリボヌクレオチド及びこれらのポリマーを含む。核酸は、合成され、自然発生し及び非自然発生し、そして参照となる核酸と同様の結合特性を有する、既知のヌクレオチド類縁体あるいは改変型の骨格残基又は連結を有する核酸を包含する。そのような類縁体の具体例は、これに限定されないが、ホスホロチオエート、ホスホラミデート、メチルホスホネート、キラルメチルホスホネート、2’−O−メチルリボヌクレオチド、及びペプチド核酸(PNAs)を含む。特に限定されることなく、この用語は、参照となる核酸と同様の結合特性を有する、天然核酸の既知の類縁体を含む核酸を包含する。特に指定されない限り、特定の核酸配列はまた、黙示的に、明記された配列と同様に、保存的に改変されたそれらの変異体及び相補的な配列を包含する。
【0031】
「オリゴヌクレオチド(oligonucleotide)」という用語は、RNA、DNA、RNA/DNAハイブリッド、及び/又はこれらの類似物の一本鎖オリゴマー又はポリマーをいう。ある例として、オリゴヌクレオチドは自然に発生した(すなわち、改変されていない)核酸塩基、糖、及びヌクレオシド間(骨格)連結から構成される。他の例として、オリゴヌクレオチドは、改変型核酸塩基、糖、及び/又はヌクレオシド間連結から構成される。
【0032】
本明細書において、「ミスマッチモチーフ(mismatchmotif)」又は「ミスマッチ領域(mismatch region)」という用語は、相補配列に対して100%の相補性を有していないオリゴヌクレオチドの部分をいう。オリゴヌクレオチドは、少なくとも1,2,3,4,5,6,又はそれ以上のミスマッチ領域を有しているかもしれない。ミスマッチ領域は隣接しているかもしれないし、あるいは1,2,3,4,5,6,7,8,9,10,11,12、又はそれ以上のヌクレオチドによって隔てられているかもしれない。ミスマッチモチーフ又は領域は、単一のヌクレオチドから構成されてもよく、あるいは2,3,4,5,又はそれ以上のヌクレオチドから構成されてもよい。
【0033】
「厳密なハイブリダイゼーション条件(stringenthybridization conditions)」という語句は、オリゴヌクレオチドがその相補配列にはハイブリダイズするが、他の配列には全くハイブリダイズしないであろう条件のことをいう。厳密な条件は、配列依存的であり、そして異なる環境では異なるであろう。より長い配列は高温で特異的にハイブリダイズする。核酸のハイブリダイゼーションに関する広範な説明は、Tijssen,Techniquesin Biochemistry and Molecular Biology−−Hybridization with Nucleic Probes,「Overviewof principles of hybridization and the strategy of nucleic acid assays」(1993)に開示されている。一般的に、厳密な条件は、規定のイオン強度pHでの特定の配列に対する熱融解点(Tm)よりも約5〜10℃低くなるように選択される。Tmは、標的に相補するプローブの50%が平衡状態で標的配列にハイブリダイズする(標的配列が過剰に存在するとき、Tmでは、プローブの50%が平衡状態で占領される)温度(定義されたイオン強度、pH,及び核酸濃度下)である。厳密な条件はまた、ホルムアミドのような不安定化剤の添加によっても達成されるかもしれない。選択的又は特異的なハイブリダイゼーションにとって、陽性シグナルは、少なくともバックグラウンドの2倍であり、好ましくはバックグラウンドハイブリダイゼーションの10倍である。
【0034】
2又はそれ以上の核酸の関係において、「実質的に同一な(substantiallyidentical)」又は「実質的な同一性(substantial identity)」という用語は、配列比較アルゴリズムを用いて、又は手動アライメント及び視覚的な検討により測定される際に、比較枠又は所望の領域に渡り、最大一致のために比較され及び整列される時に、同一である、又は同一である核酸の特定の割合(すなわち、特定の領域に渡り、少なくとも約60%、好ましくは少なくとも約65%、70%、75%、80%、85%、90%、又は95%の同一性)を有する2又はそれ以上の配列あるいは部分配列をいう。この定義はまた、文脈が示す時には、同じように、配列の相補性のことをいう。好ましくは、実質的な同一性は、長さが少なくとも約5,10,15,20,25,30,35,40,45,50,75,又は100ヌクレオチドである領域対して存在する。
【0035】
「インキュベートする(incubating)」という用語は、「接触する(contacting)」及び「むき出しにする(exposing)」と同意語として用いられ、他に定義されない限り、特定の時間又は温度の要求を意味しない。
【0036】
(III.実施形態の説明)
本発明は、希少循環細胞における複数のシグナル伝達分子の活性状態及び/又は総量を検出するための抗体に基づくアレイ及び癌の予後診断及び診断の促進、及び個別の標的治療法の設計のためのこれらの使用方法を提供する。
【0037】
A.抗体アレイ
1つの態様において、本発明は、細胞抽出物における1つ又はそれ以上の分析物に対する特異的な複数の希釈系列の捕捉抗体から構成される優れたダイナミックレンジを有するアレイを提供し、その捕捉抗体は、固相担体に拘束される。
【0038】
ある実施の態様において、細胞抽出物は、固形腫瘍の循環細胞の抽出物から構成される。循環細胞は典型的に、例えば、免疫磁気分離法(例えばRacilaら、Proc.Natl.Acad.Sci.USA,95:4589−4594(1998);Bilkenrothら、Int.J.Cancer,92:577−582(2001)参照)、マイクロ流体分離法(例えばMohamedら、IEEETrans.Nanobiosci.,3:251−256(2004);Linら、Abstract No.5147,97th AACR Annual Meeting,Washington,D.C.(2006)参照)、FACS(例えばMancusoら、Blood,97:3658−3661(2001)参照)、密度勾配遠心分離法(例えばBakerら、Clin.CancerRes.,13:4865−4871(2003)参照)、及び減少法(例えばMeyeら、Int.J.Oncol.,21:521−530(2002)参照)を含む1つ又はそれ以上の分離法を用いて患者試料から単離される。
【0039】
他の態様において、患者試料は、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引試料から構成される。ある例では、全血試料は、血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は典型的に赤血球、白血球、及び/又は循環腫瘍細胞(CTC)、循環内皮細胞(CEC)、循環内皮前駆細胞(CEPC)、癌幹細胞(CSC)、及びそれらの組み合わせのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞から放出された核酸(例えばDNA、RNA)及びタンパク質を含む。
【0040】
ある例では、単離された循環細胞は、1つ又はそれ以上の対象となる抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、これに限定されないが、上皮細胞増殖因子(EGF)、ヘレグリン(HRG)、TGF−α、PIGF、アンジオポエチン(Ang)、NRG1、PGF、TNF−α、VEGF、PDGF、IGF、FGF、HGF、サイトカイン、及び同様のものを含む。他の例では、単離された循環細胞は、例えば、当該分野で既知のあらゆる技術を用いて細胞抽出物(例えば、細胞溶解物)を作製するために、例えば、増殖因子の刺激及び/又は抗癌剤処理の後に、溶解させることができる。好ましくは、細胞溶解は増殖因子の刺激後、約1〜360分の間に開始され、より好ましくは、2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0041】
ある実施の形態において、抗癌剤は、モノクローナル抗体又はチロシンキナーゼ阻害剤のような抗シグナル伝達剤(すなわち細胞増殖抑制剤)抗増殖剤;a化学療法剤(すなわち細胞毒性薬);及び/又は癌性細胞のような異常な細胞の制御されない増殖を減らす、又は止める能力を有するあらゆる他の化合物から構成される。ある実施の形態において、単離された循環細胞は、1つ又はそれ以上の化学療法剤と組み合わせて、抗シグナル伝達剤及び/又は抗増殖剤で処理される。
【0042】
本発明における使用に適した抗シグナル伝達剤の例は、これに限定されないが、トラスツズマブ(trastuzumab)(Herceptin(登録商標))、アレムツズマブ(alemtuzumab)(Campath(登録商標))、ベバシズマブ(bevacizumab)(Avastin(登録商標))、セツキシマブ(cetuximab)(Erbitux(登録商標))、ゲムツズマブ(gemtuzumab)(Mylotarg(登録商標))、パニツムマブ(panitumumab)(Vectibix(登録商標))、リツキシマブ(rituximab)(Rituxan(登録商標))、及びトシツモマブ(tositumomab)(BEXXAR(登録商標))のようなモノクローナル抗体;ゲフィチニブ(gefitinib)(Iressa(登録商標))、スニチニブ(sunitinib)(Sutent(登録商標))、エルロチニブ(erlotinib)(Tarceva(登録商標))、ラパチニブ(lapatinib)(GW−572016)、カネルチニブ(canertinib)(CI1033)、セマキシニブ(semaxinib)(SU5416)、バタラニブ(vatalanib)(PTK787/ZK222584)、ソラフェニブ(sorafenib)(BAY43−9006;Nexavar(登録商標))、メシル酸イマチニブ(imatinib mesylate)(Gleevec(登録商標))、及びレフルノミド(leflunomide)(SU101)のようなチロシンキナーゼ阻害剤;及びそれらの組み合わせを含む。
【0043】
典型的な抗増殖剤は、シロリムス(sirolimus)(ラパマイシン(rapamycin))、テムシロリムス(temsirolimus)(CCI−779)、及びエベロリムス(everolimus)(RAD001);のようなmTOR阻害剤;1L6−ハイドロキシメチル−キロ−イノシトール−2−(R)−2−O−メチル−3−O−オクタデシル−sn−グリセロカルボネート(1L6−hydroxymethyl−chiro−inositol−2−(R)−2−O−methyl−3−O−octadecyl−sn−glycerocarbonate)、9−メトキシ−2−メチルエリプチシンアセテート(9−methoxy−2−methylellipticiniumacetate)、1,3−ジヒドロ−1−(1−((4−(6−フェニル−1H−イミダゾ[4,5−g]キノキサリン−7−イル)フェニル)メチル)−4−ピペリジニル)−2H−ベンズイミダゾール−2−オン(1,3−dihydro−1−(1−((4−(6−phenyl−1H−imidazo[4,5−g]quinoxalin−7−yl)phenyl)methyl)−4−piperidinyl)−2H−benzimidazol−2−one)、10−(4’−(N−ジエチルアミノ)ブチル)−2−クロロフェノキサジン(10−(4’−(N−diethylamino)butyl)−2−chlorophenoxazine)、3−ホルミルクロモンチオセミカルバゾン(3−formylchromonethiosemicarbazone)(Cu(II)Cl2複合体)、API−2、癌原遺伝子TCL1のアミノ酸10〜24番目由来の15残基ペプチド(Hiromuraら、J.Biol.Chem.,279:53407−53418(2004))、KP372−1、及びKozikowskiら、J.Am.Chem.Soc.,125:1144−1145(2003)及びKauら、CancerCell,4:463−476(2003)に開示された化合物のようなAkt阻害剤;及びそれらの組み合わせを含む。
【0044】
化学療法剤の非限定的な例は、プラチナ製剤(例えば、オキサリプラチン(oxaliplatin)、シスプラチン(cisplatin)、カルボプラチン(carboplatin)、スピロプラチン(spiroplatin)イプロプラチン(iproplatin)、サトラプラチン(satraplatin)等)、アルキル化剤(例えばシクロフォスファミド(cyclophosphamide)、イフォスファミド(ifosfamide)、クロラムブシル(chlorambucil)、ブスルファン(busulfan)、メルファラン(melphalan)、メクロレタミン(mechlorethamine)、ウラムスチン(uramustine)、チオテパ(thiotepa)、ニトロソウレア(nitrosoureas)、等)、代謝拮抗剤(anti−metabolites(例えば、5−フルオロウラシル(5−fluorouracil)、アザチオプリン(azathioprine)、6−メルカプトプリン(6−mercaptopurine)、メトトレキセート(methotrexate)、ロイコボリン(leucovorin)、カペシタビン(capecitabine)、シタラビン(cytarabine)、フロクスウリジン(floxuridine)、フルダラビン(fludarabine)、ゲムシタビン(gemcitabine)、ペメトレキセド(pemetrexed)、ラルチトレキセド(raltitrexed)、等)、植物アルカロイド(例えば、ビンクリスチン(vincristine)、ビンブラスチン(vinblastine)、ビノレルビン(vinorelbine)、ビンデシン(vindesine)、ポドフィロトキシン(podophyllotoxin)パクリタキセル(paclitaxel)、ドセタキセル(docetaxel)、等)、トポイソメラーゼ阻害剤(例えばイリノテカン(irinotecan)、トポテカン(topotecan)、アムサクリン(amsacrine)、エトポシド(etoposide)(VP16)、エトポシドリン酸(etoposidephosphate)、テニポシド(teniposide)、等)、抗腫瘍性抗生物質(antitumor antibiotics)(例えばドキソルビシン(doxorubicin)、アドリアマイシン(adriamycin)、ダウノルビシン(daunorubicin)、エピルビシン(epirubicin)、アクチノマイシン(actinomycin)、ブレノマイシン(bleomycin)、マイトマイシン(mitomycin)、ミトキサントロン(mitoxantrone)、プリカマイシン(plicamycin)、等)、それらの薬学的に許容される塩類、それらの立体異性体、それらの誘導体、それらの類縁体、及びそれらの組み合わせを含む。
【0045】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の例は、上述され、そして受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0046】
ある実施の形態において、捕捉抗体のそれぞれの希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた下降捕捉抗体濃度の設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25,又はそれ以上)から構成される希釈系列を作製するために少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、捕捉抗体の各希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0047】
他の実施の形態において、固相担体は、ガラス(例えば、ガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えばナイロン、ニトロセルロース、ポリフッ化ビニリデン(PVDF)、等)、繊維束、又は他のあらゆる適切な基質から構成される。好ましい実施の形態において、捕捉抗体は、(例えば共有又は非共有結合を介して)例えばWhatmanInc.(Florham Park,NJ)から市販で入手できるFAST(商標登録)スライドのようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0048】
非限定的な例として、図14は、EGFR、HER2、Shc、Erk、及びPI3Kの活性状態を決定するために複数の希釈系列の捕捉抗体から構成され、各希釈系列における捕捉抗体がこれら分析物の1つに向けられた、アドレス可能なマイクロアレイを示す。従って、本発明のアレイは、一連の下降濃度(すなわち希釈系列)にある複数の異なる捕捉抗体から構成され、その捕捉抗体は異なるアドレス可能な位置において固相担体の表面に連結される。
【0049】
当業者は、活性化されたシグナル伝達分子の各々に対する離散的なシグナルが検出されることを許容する任意の構成であり得る。例えば、アレイは、各領域が異なる捕捉抗体又は捕捉剤(すなわち捕捉抗体上に存在する捕捉タグに結合する)を含む担体表面上の一列又は一グリッドの異なる領域(例えばドット又はスポット)であり得る。アレイは、複数のシグナル伝達分子の活性状態が単一の多重分析で検出される方法における使用のために構成され得る。様々な実施の形態において、複数は、少なくとも2,3,4,5,6,7,8,9,10,15,20,25,30,35,40,45,50、又はそれ以上のシグナル伝達分子から構成される。
【0050】
B.単一検出分析
他の形態において、固形腫瘍の循環細胞のような腫瘍細胞の細胞抽出物における対象となる特定の分析物の活性状態を検出するための分析は、優れたダイナミックレンジを有する、多重で、ハイスループットな単一の検出(すなわち2抗体)分析である。非限定的な例として、この分析で用いられる2つの抗体は、以下の構成を備え得る:(1)分析物に特異的な捕捉抗体;及び(2)分析物の活性型に特異的な検出抗体(すなわち活性状態依存抗体)。活性状態依存抗体は、例えば分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出することができる。代替的には、検出抗体は、細胞抽出物中の分析物の総量を検出する活性状態非依存抗体から構成される。活性状態非依存抗体は、一般的に分析物の活性型及び不活性型を検出することができる。
【0051】
好ましい形態において、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するために細胞抽出物における1つ又はそれ以上の分析物に特異的であり、固相担体上に拘束された複数の希釈系列の捕捉抗体と細胞抽出物をインキュベートする工程;
(b)複数の検出可能な捕捉された分析物を形成するために対応する分析物に特異的な検出抗体と複数の捕捉された分析物をインキュベートする構成;
(c)増幅シグナルを生成するためにシグナル増幅対の第一及び第二の構成因子と複数の検出可能な捕捉された分析物をインキュベートする工程;及び
(d)シグナル増幅対の第一又は第二の構成因子から生成された増幅シグナルを検出する工程。
【0052】
ある実施の形態において、細胞抽出物は固形腫瘍の循環細胞の抽出物から構成される。循環細胞は典型的には、例えば免疫磁気分離法、マイクロ流体分離法、FACS、密度勾配遠心分離法を含む当該分野において既知の1つ又はそれ以上の分離方法を用いて患者試料から単離される。当業者は循環細胞の分離及び/又は単離に適した他の方法について知っているだろう。
【0053】
他の実施の形態において、患者試料は、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引物試料から構成される。ある例では、全血試料は血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は、典型的には、赤血球、白血球、及び/又はCTC,CEC,CEPC及び/又はCSCのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞によって放出された核酸(例えばDNA、RNA)及びタンパク質を含む。
【0054】
ある例において、単離された循環細胞は、対象となる1つ又はそれ以上の抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、上述されている。
他の例では、単離された循環細胞は、当該分野で既知の任意の技術を用いて、細胞抽出物(例えば細胞溶解物)を生成するために、例えば増殖因子の刺激及び/又は抗癌剤処理の後に、溶解され得る。好ましくは、細胞溶解は、増殖因子の刺激後1〜360分の間に開始され、より好ましくは2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後、約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0055】
ある実施の形態において、抗癌剤は抗シグナル伝達剤(例えばモノクローナル抗体、チロシンキナーゼ阻害剤、等)、抗増殖剤、化学療法剤、及び/又は腫瘍性細胞のような以上な細胞の制御されない増殖を減らす又は止める能力を有するあらゆる他の化合物から構成される。治療剤のこれらの一般的な分類に該当する特定の抗癌剤は上に提供されている。
【0056】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の具体例は、上述されており、そして、限定されないが、受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0057】
ある実施の形態において、捕捉抗体の各希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた、設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25、又はそれ以上)の下降捕捉抗体濃度から構成される希釈系列を作製するため、少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、各捕捉抗体の希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0058】
他の実施の形態において、固相担体は、ガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えば、ナイロン、ニトロセルロース、PVDF、等)、繊維束、又はあらゆる他の適した基質から構成される。好ましい実施の態様において、捕捉抗体は、例えばFAST(商標登録)スライド(WhatmanInc.;Florham Park,NJ)のようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0059】
ある例では、細胞抽出物は、固相担体にすでに拘束された捕捉抗体とインキュベートされる。他の例では、細胞抽出物は、最初に溶液中で捕捉抗体とインキュベートされ、その後、例えば固相担体に結合された捕捉抗体と相互作用する捕捉抗体上に存在する捕捉タグを介して、捕捉された分析物を固定するために固相担体と接触させられる。
【0060】
ある実施の形態において、検出抗体は、溶液中又は固相担体上に拘束された捕捉抗体に結合した、分析物とインキュベートされる。ある例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で検出抗体とインキュベートされ、その後、溶液中又は固相担体上に拘束された捕捉抗体と接触させられる。他の例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で捕捉抗体及び検出抗体とインキュベートされ、その後、例えば固相担体に結合された捕捉剤と相互作用する捕捉抗体又は検出抗体上に存在する捕捉タグを介して、抗体−分析物複合体を固定するために固相担体と接触させられる。
【0061】
ある例では、検出抗体は、細胞抽出物における1つ又はそれ以上の分析物の総量を検出するために有用な活性状態非依存抗体から構成される。非限定的な例として、活性状態非依存抗体は、1つ又はそれ以上のシグナル伝達分子のリン酸化型及び非リン酸化型の両方を検出することができる。他の例では、検出抗体は、細胞抽出物における1つ又はそれ以上の分析物の活性状態を検出するのに有用な活性状態依存抗体から構成される。好ましくは、活性状態依存抗体は1つ又はそれ以上のシグナル伝達分子のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出する。
【0062】
捕捉抗体及び検出抗体は、典型的には、分析物の結合に関して、それらの間の競合が最小化されるように選択される(すなわち捕捉及び検出抗体は、同時にそれらの対応するシグナル伝達分子に結合することができる)。
【0063】
好ましい実施の形態において、検出抗体は、結合対の第一の構成因子(例えばビオチン)から構成され、そして、シグナル増幅対の第一の構成因子は結合対の第二の構成因子(例えばストレプトアビジン)から構成される。結合対の構成因子は、当該分野で既知の方法を用いて、検出抗体又はシグナル増幅対の第一の構成因子に直接又は間接的に共役し得る。ある例では、シグナル増幅対の第一の構成因子は、ペルオキシダーゼ(peroxidase)(例えば、西洋わさびペルオキシダーゼ(horseradishperoxidase)(HRP)、カタラーゼ(catalase)、クロロペルオキシダーゼ(chloroperoxidase)、シトクロームCペルオキシダーゼ(cytochromec peroxidase)、好酸球ペルオキシダーゼ(eosinophil peroxidase)、グルタチオンペルオキシダーゼ(glutathione peroxidase)、ラクトペルオキシダーゼ(lactoperoxidase)、ミエロペルオキシダーゼ(myeloperoxidase)、チロイドペルオキシダーゼ(thyroidperoxidase)、デヨードナーゼ(deiodinase)、等)であり、そしてシグナル増幅対の第二の構成因子はチラミド試薬(例えばビオチン−チラミド)である。これらの例では、増幅されたシグナルは、過酸化水素(H2O2)存在下で活性型チラミドを作製するためのチラミド試薬のペルオキシダーゼ酸化によって生成される。
【0064】
活性型チラミドは、直接検出され、又は例えばストレプトアビジン標識フルオロフォア又はストレプトアビジン標識ペルオキシダーゼと発色試薬の組み合わせのようなシグナル検出試薬の添加で検出される。本発明における使用に適したフルオロフォアの具体例は、限定されないが、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)555)、フルオレセイン(fluorescein)、フルオレセインイソチオシアネート(fluoresceinisothiocyanate)(FITC)、Oregon Green(登録商標);ローダミン(rhodamine)、テキサスレッド(Texas red)、テトラローダミンイソチオシアネート(tetrarhodamineisothiocynate)(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものを含む。ストレプトアビジン標識は、当該分野においてよく知られた方法を用いてフルオロフォア又はペルオキシダーゼに直接的に又は間接的に連結され得る。本発明の使用に適した発光試薬の非限定的な例は、3,3’,5,5’−テトラメチルベンジジン(3,3’,5,5’−tetramethylbenzidine)(TMB)、3,3’−ジアミノベンジジン(3,3’−diaminobenzidine)(DAB)、2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)(2,2’−azino−bis(3−ethylbenzothiazoline−6−sulfonicacid))(ABTS)、4−クロロ−1−ナフトール(4−chloro−1−napthol)(4CN)、及び/又はポルフィリノーゲン(porphyrinogen)を含む。
【0065】
当業者は、本明細書中で述べる単一検出分析に従って、抗体以外の結合相手が細胞抽出物からの1つ又はそれ以上の分析物を固定し及び/又は検出するために使用され得ることを理解するだろう。そのような結合相手の非限定的な例は、分析物のリガンド又は受容体、分析物の基質、結合ドメイン(例えばPTB、SH2等)、アプタマー、及び同様のものを含む。
【0066】
C.近接二重検出分析
さらに他の形態では、固形腫瘍の循環細胞のような腫瘍細胞の細胞抽出物における対象となる特定の分析物の活性状態を検出するための分析は、優れたダイナミックレンジを有する、多重、ハイスループットの近接(すなわち3抗体)分析である。非限定的な例として、この近接分析で用いられる3つの抗体は、以下の構成を備え得る:(1)分析物に特異的な捕捉抗体;(2)分析物の活性型に特異的な検出抗体(すなわち活性状態依存抗体);及び(3)分析物の総量を検出する検出抗体(すなわち活性状態非依存抗体)。活性状態依存抗体は、例えば分析物のリン酸化、ユビキチン化、及び/又は複合体形成状態を検出することができる。活性状態依存抗体は一般的に分析物の活性型及び不活性型の両方を検出することができる。
【0067】
好ましい形態では、本発明は、優れたダイナミックレンジを有する、多重で、ハイスループットの免疫分析を行うための方法を提供し、この方法は、以下の工程を備える:
(a)複数の捕捉された分析物を形成するため、細胞抽出物における1つ又はそれ以上の分析物に特異的な複数の希釈系列の捕捉抗体と、細胞抽出物をインキュベートする工程であって、前記捕捉抗体が固相担体上に拘束されていることを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するため、複数の捕捉された分析物を、対応する分析物に特異的な検出抗体とインキュベートする工程であって、前記検出抗体は:
(1)促進部分で標識された複数の活性状態非依存抗体、と
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体と、を備え、
前記促進部分は、シグナル増幅対の第一の構成因子に向けられ且つそれと反応する酸化剤を生成する、
ことを特徴とする工程;
(c)増幅シグナルを生成するために、複数の検出可能な捕捉された分析物を、シグナル増幅対の第二の構成因子とインキュベートする工程;及び
(d)シグナル増幅対の第一及び第二の構成因子から生成された増幅シグナルを検出する工程。
【0068】
図1は、分析物が1つの捕捉抗体と2つの検出抗体(すわなち活性状態非依存抗体と活性状態依存抗体)に結合した典型的な近接分析を示す。捕捉抗体1と活性状態非依存抗体2はそれぞれ、その活性状態に非依存して分析物6に結合する。活性状態依存抗体3は、その活性状態に依存して分析物に結合する(例えば活性状態依存抗体はリン酸化残基を有する分析物の活性型にのみ結合するだろう)。活性状態非依存抗体は、促進部分(M1で表す、4)で標識され、そして活性状態依存抗体はシグナル増幅対の第一の構成因子(M2で表す、5)で標識される。両方の検出抗体の分析物への結合は、促進部分を、充分に近接した範囲(点線7の内部の領域によって描かれる)内でシグナル増幅対の第一の構成因子に向かわせるので、促進部分によって生成されたシグナルは、シグナル増幅対の第一の構成因子に向けられ得、結果として検出可能な及び/又は増幅可能なシグナルの生成をもたらす。近接チャネリングのための様々な方法が当該分野で既知であり、例えばFRET、時間分解蛍光−FRET、LOCI、等を含む。近接チャネリングの利点は、本発明の方法において使用されるように、単一の検出可能なシグナルが、全ての3つの抗体が結合したそれらの分析物のみに対して生成され、結果として増加した分析の特異性、より低いバックグラウンド、及び単純化された検出をもたらすことである。
【0069】
ある実施の形態において、細胞抽出物は固形腫瘍の循環細胞の抽出物から構成される。循環細胞は、典型的には、例えば免疫磁気分離法、マイクロ流体分離法、FACS、密度勾配遠心分離法、及び減少法を含む当該分野において既知の1つ又はそれ以上の分離方法を用いて患者試料から単離される。
【0070】
他の実施の形態において、患者試料は全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、及び/又は細針吸引物試料から構成される。ある例では、全血試料は、血漿又は血清画分及び細胞画分(すなわち細胞沈殿物)に分離される。細胞画分は典型的に赤血球、白血球、及び/又はCTC、CEC、CEPC、及び/又はCSCのような固形腫瘍の循環細胞を含む。血漿又は血清画分は、通常とりわけ、固形腫瘍の循環細胞によって放出される核酸(例えばDNA、RNA)及びタンパク質を含む。
【0071】
ある例では、単離された循環細胞は、対象となる1つ又はそれ以上の抗癌剤とのインキュベート前、間、及び/又は後に1つ又はそれ以上の増殖因子でインビトロにおいて活性化され得る。刺激性の増殖因子は、上述されている。他の例では、単離された循環細胞は、当該分野で既知の任意の技術を用いて、細胞抽出物(例えば細胞溶解物)を作製するために、例えば増殖因子の刺激及び/又は抗癌剤処理の後に、溶解され得る。好ましくは、細胞溶解は、増殖因子の刺激後1〜360分の間に開始され、より好ましくは2つの異なる時間間隔で開始される:(1)増殖因子の刺激後、約1〜5分で;及び(2)増殖因子の刺激後、約30〜180分の間。代替的には、細胞溶解物は使用まで−80℃で保存され得る。
【0072】
ある実施の形態において、抗癌剤は抗シグナル伝達剤(例えば、モノクローナル抗体、チロシンキナーゼ阻害剤、等)、抗増殖剤、化学療法剤、及び/又は腫瘍細胞のような異常な細胞の制御されない増殖を減らし又は止める能力を有する任意の他の化合物から構成される。治療剤のこれらの一般的な分類に該当する特異的な抗癌剤の具体例は、上記に提供されている。
【0073】
好ましい実施の形態において、細胞抽出物における1つ又はそれ以上の分析物は、複数のシグナル伝達分子から構成される。対象となるシグナル伝達分子の例は上述され、そしてこれに限定されないが、受容体型チロシンキナーゼ、非受容体型チロシンキナーゼ、及び/又はチロシンキナーゼシグナル伝達系構成要素を含む。
【0074】
ある実施の形態において、捕捉抗体のそれぞれの希釈系列は一連の下降捕捉抗体濃度から構成される。ある例では、捕捉抗体は、アレイ上にスポットされた下降捕捉抗体濃度の設定された数(例えば、2,3,4,5,6,7,8,9,10,15,20,25,又はそれ以上)から構成される希釈系列を作製するために少なくとも2倍(例えば、2,5,10,20,50,100,500,又は1000倍)で連続的に希釈される。好ましくは、捕捉抗体の各希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0075】
他の実施の形態において、固相担体は、ガラス(例えばガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜(例えば、ナイロン、ニトロセルロース、PVDF、等)、繊維束、又はあらゆる他の適した基質から構成される。好ましい実施の態様において、捕捉抗体は、例えばFAST(商標登録)スライド(WhatmanInc.;Florham Park,NJ)のようなニトロセルロースポリマーで覆われたガラススライド上に拘束される。
【0076】
ある例では、細胞抽出物は固相担体上にすでに拘束された捕捉抗体とインキュベートされる。他の例では、細胞抽出物は、始めに溶液中で捕捉抗体とインキュベートされ、その後、例えば固相担体に結合した捕捉剤と相互作用する捕捉抗体上に存在する捕捉タグを介して、捕捉された分析物を固定するために、固相担体と接触させられる。
【0077】
ある実施の形態において、検出抗体は溶液中又は固相担体上に拘束された捕捉抗体と結合した分析物とインキュベートされる。ある例では、複数の分析物から構成される細胞抽出物は、始めに溶液中で検出抗体とインキュベートされ、その後溶液中又は固相担体上に拘束された捕捉抗体と接触させられる。他の例では、複数の分析物から構成される細胞抽出物は、先ず始めに溶液中で捕捉抗体及び検出抗体とインキュベートされ、その後、例えば固相担体に結合した捕捉剤と相互作用する、捕捉抗体又は検出抗体上に存在する捕捉タグを介して、抗体−分析物複合体を固定するため、固相担体と接触させられる。検出工程に先んじて、固定された複合体は、複合体を形成していない抗体を除去するために洗浄され得、洗浄された複合体は、連続的に支持表面から放出され得、そして、分析されている分析物それぞれに対する近接チャネリングが、本明細書で述べるような適切な方法によって、検出され得る。
【0078】
担体表面がアレイに拘束された捕捉剤から構成される実施の形態において、インキュベートする工程は、反応を完了させるために全ての3つの抗体の過剰量を用いて、溶液中の複数の分析物から構成される細胞抽出物を捕捉抗体及び検出抗体と接触させることから構成され得る。この方法の1つの変形は、結果としてもたらされた抗体−分析物複合体が固体層に付着され、非結合の抗体を取り除くために洗浄される。図2を参照すると、捕捉抗体1は捕捉タグ10から構成され得る。複合体は、固体層に接着され且つ捕捉タグと結合した捕捉剤11を介して固体層12に付着し、それによって複合体を固定している。固定された複合体は適切な緩衝液で洗浄され、その後放出剤13の添加によって固体層から放出される。放出剤は洗浄された複合体の放出をもたらす任意の機構によって機能してよい。1つの実施の形態において、捕捉タグは放出剤によって認識され、切断される切断部位を備える。他の実施の形態において、図2に示すように、放出剤は、捕捉剤に結合することに対して捕捉タグと競合する。例えば、捕捉剤は、捕捉抗体に付着された部分的に相補するオリゴヌクレオチド(すなわち捕捉タグ)とハイブリダイズする第一のオリゴヌクレオチドであってもよく;そして放出剤は、捕捉剤と完全に相補するオリゴヌクレオチドであってもよく、結果、固体層から洗浄された複合体の鎖の置換及び放出をもたらす。使用され得る適切な捕捉タグ/捕捉剤/放出剤の他の例は、これに限定されないが、2,4−ジニトロフェノール(2,4−dinitrophenol)(DNP)/抗−DNP抗体(anti−DNPantibody)/2,4−DNPリジン(2,4−DNP lysine);T2/抗−T3抗体(anti−T3 antibody)/T3;ウアバイン(ouabain)/抗−ジゴキシン抗体A(anti−digoxinantibody)/ジゴキシン(digoxin);及びデチオビオチン(dethiobiotin)ストレプトアビジン(streptavidin)/ビオチン(biotin)(例えばIshikawaら、J.Clin.LabAnal.,12:98−107(1998)を参照)。
【0079】
洗浄された複合体が固体層から放出された後、それは:(1)捕捉抗体上に捕捉タグを特異的に結合するアレイに拘束された捕捉分子から構成される担体表面と接触させられ、又は(2)解離され、そして解離された検出抗体は、検出抗体上の捕捉タグと特異的に結合する捕捉剤から構成される担体表面と接触させられる。図2は、洗浄された複合体が解離され、そして解離された検出抗体が担体表面14と接触させられる実施の形態を示す。担体表面は「アドレス可能な(addressable)」又は「ジップコード(zipcode)」アレイに拘束された複数の捕捉分子から構成される。このアレイの各異なる領域は、活性状態非依存抗体2又は活性状態依存抗体3上に存在する捕捉タグ8に特異的に結合する特徴のある捕捉剤9から構成され、それによってアレイにおけるタグ付けされた検出抗体を拘束し、組織化する。好ましい実施の形態において、捕捉剤及び捕捉タグは互いに特異的にハイブリダイズするオリゴヌクレオチドである。オリゴヌクレオチド捕捉分子から構成されるアドレス可能なアレイは、当該分野ではよく知られている(例えばKeramasら、LabChip,4:152−158(2004);Delrio−Lafreniereら、Diag.Microbiol.Infect.Dis.,48:23−31(2004)を参照)。
【0080】
アレイの各異なる領域での検出抗体の存在は、促進部分(M1で表す、4)又はシグナル増幅対の第一の構成因子(M2で表す、5)のような部位で直接的に又は間接的に検出され得る。直接的に検出され得る部位の例は、フルオロフォア、クロモフォア(chromophore)、金コロイド、着色ゴム等を含む。1つの実施の形態において、両方の部位は、非依存的に選択されたフルオロフォアである。区別可能な読み出し情報を提供し、一方で互いに非常に近接している、例えばCy3/Cy5、Cy5/フィコエルトリン(phycoerthrin)、及び同様のものに例示される、任意のフルオロフォア対が使用され得る。代替的には、オリゴヌクレオチドのアドレス可能なアレイが用いられると、両方の部位が異なるジップコードに伝達される同一のフルオロフォアであり得る。レーザースキャニング共焦点顕微鏡法は、アレイ上に接着したフルオロフォア部位を検出するために使用され得る。鎖置換分析のような、検出に先んじて複合体がアレイから放出されるアレイにおいて、フルオロフォア部位を検出するための適切な方法は、キャピラリーフロー共焦点レーザー誘起蛍光法、ナノHPLC(nano−HPLC)、電気泳動等を含む。
【0081】
ある実施の形態において、活性状態非依存抗体はさらに、検出可能な部位を備える。そのような例では、検出可能な部位の量は、細胞抽出物における1つ又はそれ以上の分析物の量に相関する。検出可能な部位の具体例は、これに限定されないが、蛍光標識、化学反応標識、酵素標識、放射性標識、及び同様のものが含まれる。好ましくは、検出可能な部位は、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)647)、フルオレセイン、フルオレセインイソチオシアネート(FITC)、Oregon Green(登録商標);ローダミン、テキサスレッド、テトラローダミンイソチオシアネート(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものに例示されるフルオロフォアである。検出可能な部位は、当該分野でよく知られた方法を用いて、直接的に又は間接的に活性状態非依存抗体に共役することができる。
【0082】
ある例では、活性状態非依存抗体は、促進部分で直接標識される。促進部分は、当該分野でよく知られた方法を用いて、活性状態非依存抗体に連結され得る。本発明の使用に適した促進部分は、その促進部分に近接した(すわなち、空間的に近く又は接近した)他の分子に向かわせ(すなわち、それへと方向付け)且つそれと反応する(すなわち結合し、結合され、又は複合体を形成する)酸化剤を生成することが可能なあらゆる分子を含む。促進部分の例は、これに限定されないが、電子受容体として分子酸素(O2)を含む酸化/還元反応に触媒作用を及ぼすグルコースオキシダーゼ又はあらゆる他の酵素に例示される酵素、及びメチレンブルー(methyleneblue)、ローズベンガル(rose bengal)、ポルフィリン(porphyrin)、スクアレート染色剤(squarate dye)、フタロシアニン(phthalocyanine)、及び同様のものに例示される光増感剤を含む。酸化剤の非限定的な例は、過酸化水素(H2O2)、一重項酸素、及び酸化/還元反応において酸素原子を移し、又は電子を獲得するあらゆる他の化合物を含む。好ましくは、適切な基質(例えばグルコース、光等)の存在下で、促進部分(例えば、グルコースオキシダーゼ、光増感剤等)が、2つの部位が互いに近接するときに、シグナル増幅対の第一の構成因子(例えば、西洋わさびペルオキシダーゼ(HRP)、保護基によって保護されたハプテン、酵素阻害剤とチオエーテル結合によって不活性化された酵素等)に向かわせ且つそれと反応する酸化剤(例えば、過酸化水素(H2O2)、一重項酸素等)を生成する。
【0083】
他の例では、活性状態非依存抗体は、活性状態非依存抗体に共役したオリゴヌクレオチドリンカーと促進部分に共役した相補ヌクレオチドリンカーとの間のハイブリダイゼーションを介して促進部分で間接的に標識される。オリゴヌクレオチドリンカーは、促進部分又は当該分野でよく知られた方法を用いて活性状態非依存抗体に連結され得る。ある実施の形態において、促進部分に共役したオリゴヌクレオチドリンカーは、活性状態非依存抗体に共役したオリゴヌクレオチドリンカーと100%の相補性を有する。他の実施の形態において、オリゴヌクレオチドリンカー対は、例えば厳格なハイブリダイゼーション条件下でのハイブリダイゼーションにおいて少なくとも1,2,3,4,5,6,又はそれ以上のミスマッチ領域を備える。当業者は、異なる分析物に特異的な活性状態非依存抗体が同一のオリゴヌクレオチドリンカー又は異なるオリゴヌクレオチドリンカーのいずれにも共役され得ることを理解している。
【0084】
促進部分又は活性状態非依存抗体に共役したオリゴヌクレオチドリンカーの長さは、可変である。一般的に、リンカー配列は長さが少なくとも約5,10,15,20,25,30,35,40,45,50,75,又は100ヌクレオチドであり得る。典型的に、ランダムなヌクレオチド配列が連結のために生成される。非限定的な例として、ヌクレオチドリンカーのライブラリーが3つの独特の隣接ドメインを有するように設計され得る:スペーサードメイン;署名ドメイン(signaturedomain);及び共役ドメイン。好ましくは、オリゴヌクレオチドリンカーは、それらが共役される促進部分又は活性状態非依存抗体の機能を破壊することのない効率的な連結のために設計される。
【0085】
オリゴヌクレオチドリンカー配列は、様々な分析条件下であらゆる二次構造形成を避け又は最小化するよう設計され得る。溶解温度は、典型的には、分析の全工程におけるそれらの関与を許容するようリンカー内の各部分に対して注意深く監視される。一般的に、リンカー配列の部分の溶解温度の範囲は、5℃以下である。規定されたイオン濃度下における溶解温度、二次構造、及びヘアピン構造を決定するためのコンピュータアルゴリズム(例えばOLIGO6.0)は各リンカー内における3つの異なるドメインの各々を解析するために使用され得る。全ての組み合わせ配列はまた、それらの構造上の特徴と他の共役したオリゴヌクレオチドリンカー配列との比較可能性、例えばそれらが厳密なハイブリダイゼーション条件下で相補するオリゴヌクレオチドリンカーとハイブリダイズするかどうか、に関して分析され得る。
【0086】
オリゴヌクレオチドリンカーのスペーサードメインは、オリゴヌクレオチドの架橋部位からの共役ドメインの適切な分離を提供する。共役ドメインは、相補するヌクレオチドリンカー配列で標識された分子を、核酸ハイブリダイゼーションを介して共役ドメインと連結するのに働く。核酸を介したハイブリダイゼーションは、抗体−分析物(すなわち抗原)複合体の形成前又は後のいずれかで実行され得、より柔軟性のある分析構成を提供する。多くの直接抗体共役法とは異なり、抗体又は他の分子に比較的小さいオリゴヌクレオチドを連結することは、標的分析物への抗体の特異親和性又は共役した分子の機能に対してわずかな影響しか及ぼさない。
【0087】
ある実施の形態において、オリゴヌクレオチドリンカーの署名配列ドメインは、複合体重合化タンパク質分析(complexmultiplexed protein assayにおいて使用され得る。多様な抗体が、異なる署名配列とオリゴヌクレオチドリンカーで共役され得る。多重化免疫分析において、適切なプローブで標識されたリポーターオリゴヌクレオチド配列は、多重化分析の形式において、抗体と抗原との間のクロスハイブリダイゼーションを検出するために使用され得る。
【0088】
オリゴヌクレオチドリンカーは、様々な異なる方法を用いて、抗体又は他の分子に共役され得る。例えば、オリゴヌクレオチドリンカーは、5’又は3’末端のいずれかにおけるチオール基とともに合成され得る。チオール基は、還元剤(例えば、TCEP−HCl)を用いて脱保護され得、結果として生じるリンカーは脱塩回転カラムを用いることによって精製され得る。結果として生じる脱保護オリゴヌクレオチドリンカーは、SMCCのようなヘテロ二官能性架橋剤を用いて抗体の第一級アミン又は他の種類のタンパク質に共役され得る。代替的には、オリゴヌクレオチドの5’−リン酸基は、リン酸エステルを形成するために水溶性カルボジイミドEDCで処理され、その後、アミン含有分子に共役され得る。ある例では、3’−リボース残基のジオールは、酸化されてアルデヒド基になり、その後、還元的アミノ化を用いて核酸のアミン基又は他の種類のタンパク質に共役され得る。他の例では、オリゴヌクレオチドリンカーは、3’又は5’末端のビオチン修飾とともに合成され、ストレプトアビジン標識分子に共役され得る。
【0089】
オリゴヌクレオチドリンカーは、Usmanら、J.Am.Chem.Soc.,109:7845(1987);Scaringeら、Nucl.AcidsRes.,18:5433(1990);Wincottら、Nucl.Acids Res.,23:2677−2684(1995);及びWincottら、MethodsMol.Bio.,74:59(1997)に開示されるような、当該分野で既知のあらゆる様々な技術を用いて合成され得る。一般的に、オリゴヌクレオチドの合成は、5’末端にジメトキシトリチル(dimethoxytrityl)と3’末端にホスホラミダイト(phosphoramidite)のような共通の核酸保護及び共役基を用いる。オリゴヌクレオチド合成に適した試薬、核酸脱保護のための方法、及び核酸精製のための方法は当業者に既知である。
【0090】
ある例では、活性状態依存抗体はシグナル増幅対の第二の構成因子で直接標識される、シグナル増幅対の構成因子は当該分野でよく知られた方法を用いて活性状態依存抗体に連結され得る。他の例では、活性状態依存抗体は、活性状態依存抗体に共役した結合対の第一の構成因子とシグナル増幅対の第一の構成因子に共役した結合対の第二の構成因子との間の結合を介して、シグナル増幅対の第一の構成因子で間接的に標識される。結合対の構成因子(例えばビオチン/ストレプトアビジン)は、当該分野でよく知られた方法を用いてシグナル増幅対の構成因子又は活性状態依存抗体に連結され得る。シグナル増幅対の構成因子の具体例は、これに限定されないが、西洋わさびペルオキシダーゼ(HRP)、カタラーゼ、クロロペルオキシダーゼ、シトクロームCペルオキシダーゼ、好酸球ペルオキシダーゼ、グルタチオンペルオキシダーゼ、ラクトペルオキシダーゼ、ミエロペルオキシダーゼ、チロイドペルオキシダーゼ、デヨードナーゼ、及び同様のものを含む。シグナル増幅対構成因子の他の具体例は、保護基によって保護されたハプテン及び酵素阻害剤とのチオエーテル結合によって不活性化された酵素を含む。
【0091】
捕捉抗体、活性状態非依存抗体、及び活性状態依存抗体は、典型的には、分析物との結合に関して、それらの間における競合が最小化される(すなわち全ての抗体が同時に対応するシグナル伝達分子に結合することができる)ように選択される。
【0092】
近接チャネリングの1つの例において、促進部分はグルコースオキシダーゼ(GO)であり、シグナル増幅対の第一の構成因子は西洋わさびペルオキシダーゼ(HRP)である。GOがグルコースのような基質と接触すると、それは酸化剤(すなわち過酸化水素(H2O2))を生成する。HRPがGOとチャネリングする隣接範囲内にあると、GOによって生成されたH2O2は、シグナル増幅対の第二の構成因子(例えば、ルミノール(luminol)又はイソルミノール(isoluminol)のような化学発光基質、又はチラミド(tyramide)(例えばビオチン−チラミド)、ホモバニリン酸(homovanillicacid)又は4−ヒドロキシフェニル酢酸(4−hydroxyphenyl acetic acid)のような蛍光性基質)の存在下で増幅シグナルを生成する、HRP−H2O2複合体を形成するためにHRPに向けられ且つそれと複合体を形成する。近接分析におけるGOとHRPを用いる方法は、例えば、Langryら、U.S.Dept.ofEnergy Report No.UCRL−ID−136797(1999)に開示される。ビオチン−チラミドがシグナル増幅対の第二の構成因子として用いられるとき、HRP−H2O2複合体はチラミドを酸化して隣接する求核残基に共有的に結合する反応性チラミドラジカルを生成する。活性型チラミドは、直接検出され、又は、例えばストレプトアビジン標識フルオロフォア又はストレプトアビジン標識ペルオキシダーゼと発色試薬との組み合わせのようなシグナル検出試薬の添加に応じて検出される。本発明の使用に適したフルオロフォアの具体例は、これに限定されないが、AlexaFluor(登録商標)染色液(例えばAlexa Fluor(登録商標)555)、フルオレセイン、フルオレセインイソチオシアネート(FITC)、Oregon Green(登録商標);ローダミン、テキサスレッド、テトラローダミンイソチオシアネート(TRITC)、CyDye(登録商標)蛍光試薬(例えばCy2,Cy3,Cy5)及び同様のものを含む。ストレプトアビジン標識は、当該分野においてよく知られた方法を用いてフルオロフォア又はペルオキシダーゼに直接的に又は間接的に連結され得る。本発明の使用に適した発光試薬の非限定的な例は、3,3’,5,5’−テトラメチルベンジジン(TMB)、3,3’−ジアミノベンジジン(DAB)、2,2’−アジノ−ビス(3−エチルベンゾチアゾリン−6−スルホン酸)(ABTS)、4−クロロ−1−ナフトール(4CN)、及び/又はポルフィリノーゲンを含む。
【0093】
近接チャネリングの他の例では、促進部分は光増感剤であり、シグナル増幅対の第一の構成因子は特異的な結合相手(例えば、リガンド、抗体等)へのハプテンの結合を避ける保護基で保護された複数のハプテンで標識された巨大分子である。例えば、シグナル増幅対の構成因子は、ビオチン、クマリン(coumarin)、及び/又はフルオレセイン分子で標識されたデキストラン分子であり得る。適切な保護基は、これに限定されないが、フェノキシ、アニリノ(analino−)、オレフィン、チオエーテル、及びセレノエーテル保護基を含む。本発明の近接分析における使用に適した付加的な光増感剤及び保護されたハプテン分子は、米国特許第5,807,675号に開示される。光増感剤が光で励起されると、それは酸化剤(すなわち一重項酸素)を生成する。ハプテン分子が光増感剤と隣接してチャネリングする範囲内にあると、光増感剤によって生成された一重項酸素がカルボニル基(ケトン又はアルデヒド)及びスルフィン酸を得るためにハプテンの保護基におけるチオエーテルに向けられ、それと反応し、ハプテンから保護基を放出する。保護されていないハプテンは、その後、シグナル増幅対の第二の構成因子(例えば検出可能なシグナルを生成することができる特異的な結合相手)と特異的に結合するために使用され得る。例えば、ハプテンがビオチンであるとき、特異的な結合相手は酵素標識されたストレプトアビジンであり得る。典型的な酵素は、アルカリホスファターゼ(alkalinephosphatase)、β−ガラクトシダーゼ(β−galactosidase)、HRP等を含む。結合していない試薬を除去するための洗浄後、検出可能なシグナルは、その酵素の検出可能な(例えば、蛍光性、化学発光性、発色性等)基質を添加することによって生成され、当該分野で既知の適切な方法と装置を用いて検出され得る。代替的には、検出可能なシグナルは、チラミドシグナル増幅を用いて増幅され得、活性化されたチラミドは直接検出され又は上述したシグナル検出試薬の添加により検出される。
【0094】
近接チャネリングのさらに他の例では、促進部分は光増感剤であり、シグナル増幅対の第一の構成因子は酵素−阻害剤複合体である。酵素と阻害剤(例えば、ホスホン酸標識デキストラン)は、切断可能なリンカー(例えば、チオエーテル)によって互いに連結される。光増感剤が光で励起されると、それは酸化剤(例えば一重項酸素)を生成する。酵素−阻害剤複合体が光増感剤とチャネリングする隣接距離内にあると、光増感剤によって生成された一重項酸素が切断可能なリンカーに向けられ、それと反応し、酵素から阻害剤を放出し、それによって酵素を活性化する。酵素基質は、検出可能なシグナルを生成するために添加され、又は代替的には、増幅剤が増幅シグナルを生成するために添加される。
【0095】
近接チャネリングのさらなる例では、促進部分はHRPであり、シグナル増幅対の第一の構成因子は保護されたハプテン又は上述したような酵素−阻害剤複合体であり、そして保護基はp−アルコキシフェノールから構成される。フェニレンジアミン(phenylenediamine)とH2O2の添加は、露出したハプテン又は反応性酵素を得るために、保護されたハプテン又は酵素−阻害剤複合体に向けられ、p−アルコキシフェノール保護基と反応する、反応性フェニレンジイミン(phenylenediimine)を生成する。増幅シグナルは上述のように生成され、検出される(例えば、米国特許第5,532,138号及び第5,445,944号参照)。
【0096】
当業者は、抗体以外の結合相手が、この明細書で述べられる近接(すなわち3抗体)分析に関して細胞抽出物からの1つ又はそれ以上の分析物を固定し及び/又は検出するために用いられ得ることを理解している。そのような結合相手の非限定的な例は、分析物のリガンド又は受容体、分析物の基質、結合ドメイン(例えば、PTB,SH2等)、アプタマー、及び同様のものを含む。
【0097】
D.キット
さらなる態様において、本発明は上述した抗体に基づくアレイ方法を実行するためのキットを提供し、以下の構成を備える:(a)固相担体上に拘束された複数の捕捉抗体の希釈系列;及び(b)複数の検出抗体(例えば、活性状態非依存抗体、及び/又は活性状態依存抗体)。ある例では、このキットはさらに、固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書を含むことができる。このキットはまた、例えば、シグナル増幅対の第一及び第二の構成因子、チラミドシグナル増幅試薬、促進部分のための基質、洗浄緩衝液、捕捉/放出試薬等のような、本発明の特異的な方法を実行することに関する上述したあらゆる付加的な試薬を包含してもよい。
【0098】
IV.抗体アレイの構築
ある形態において、本発明は、固相担体上に拘束された捕捉抗体の希釈系列を用いて、固形腫瘍の循環細胞の細胞抽出物における複数のシグナル伝達分子の活性状態を検出するための抗体に基づくアレイを提供する。本発明の分析において使用されるアレイは、典型的には、異なるアドレス可能な位置における固相担体の表面に連結された一定範囲の捕捉抗体濃度の複数の異なる捕捉抗体から構成される。
【0099】
固相担体は、タンパク質を固定するために適したあらゆる基質から構成され得る。固相担体の具体例は、これに限定されないが、ガラス(例えば、ガラススライド)、プラスチック、チップ、ピン、フィルター、ビーズ(例えば、磁気ビーズ、ポリスチレンビーズ等)、紙、膜、繊維束、ゲル、金属、セラミック、及び同様のものを含む。ナイロン(Biotrans(登録商標),ICNBiomedicals,Inc.(Costa Mesa,CA);Zeta−Probe(登録商標),Bio−Rad Laboratories(Hercules,CA))、ニトロセルロース(Protran(登録商標),WhatmanInc.(Florham Park,NJ))、及びPVDF(Immobilon(登録商標)、Millipore Corp.(Billerica,MA))のような膜は、本発明のアレイにおける固相担体としての使用に適している。好ましくは、捕捉抗体は、ニトロセルロースポリマーでコートされたガラススライド、例えばWhatmanInc.(Florham Park,NJ)から市販で入手できるFAST(商標登録)スライド上に拘束される。
【0100】
望ましい固相担体の特定の態様は、多量の捕捉抗体と結合できること、最小限の変性で捕捉抗体と結合できること、及び他のタンパク質に結合できないことを含む。他の適した態様は、捕捉抗体を含む抗体溶液が固相担体に塗布されるときに固相担体が最小限の「ウィッキング(wicking)」を示すことである。最小限のウィッキングを有する固相担体は、担体に塗布された少量の分割量の捕捉抗体溶液が、結果として固定された捕捉抗体の小さく、明確なスポットとなることを許容する。
【0101】
捕捉抗体は、典型的には、共有結合又は非共有結合の相互作用(例えばmイオン結合、疎水性相互作用、水素結合、ファンデルワールス力、双極子−双極子相互作用)を介して固相担体上に直接的に又は間接的に(例えば捕捉タグを介して)拘束される。ある実施の形態において、捕捉抗体は、標準的な架橋方法と条件を用いたホモ二官能性又はヘテロ二官能性架橋剤を用いて固相担体に共有結合される。適した架橋剤は、例えばPierceBiotechnology(Rockford,IL)のような製造元から市販で入手できる。
【0102】
本発明のアレイを生成するための方法は、これに限定されないが、タンパク質又は核酸アレイを構築するために使用されるあらゆる技術を含む。ある実施の形態において、捕捉抗体は、典型的にはスプリットピン、ブラントピン、又はインクジェットプリンティングを備えるロボティック印刷装置であるマイクロスポッターを用いてアレイ上にスポットされる。この明細書において述べる抗体アレイを印刷するために適したロボットシステムは、ChipMaker2スプリットピン(TeleChem International;Sunnyvale,CA)を有するPixSys 5000ロボット(Cartesian Technologies;Irvine,CA)の他に、BioRobics(Woburn,MA)及びPackardInstrument Co.(Meriden,CT)から入手できる他のロボティック印刷装置を含む。好ましくは、各捕捉抗体の希釈の少なくとも2,3,4,5,又は6の反復がアレイ上にスポットされる。
【0103】
本発明の抗体アレイを生成するための他の方法は、規定量の液体を固相担体に引き込むために効果的な条件下で固相担体上にキャピラリー分注器を接触させこることにより各選択されたアレイの位置に既知量の捕捉抗体の希釈液を分注する工程を備え、この工程は、完全なアレイを作製するように各選択されたアレイ位置で選択された捕捉抗体希釈液を用いて繰り返される。この方法は、溶液沈着工程が、各繰り返しサイクルで各々の複数の固相担体上の選択された位置に適用される、複数のそのようなアレイを形成することに用いられてもよい。そのような方法のさらなる説明は、例えば米国特許第5,807,522号に開示されている。
【0104】
ある例では、紙に印刷するためのデバイスは、本発明の抗体アレイを生成するために使用され得る。例えば、所望の捕捉抗体濃度がデスクトップジェットプリンターのプリントヘッド部に取り込まれ、適切な固相担体に印刷され得る(例えば、Silzelら、Clin.Chem.,44:2036−2043(1998)を参照)。
【0105】
ある実施の形態において、固相担体上に生成されたアレイは、少なくとも約5スポット/cm2、好ましくは少なくとも約10,20,30,40,50,60,70,80,90,100,110,120,130,140,150,160,170,180,190,200,210,220,230,250,275,300,325,350,375,400,425,450,475,500,550,600,650,700,750,800,850,900,950,1000,2000,3000,4000,5000,6000,7000,8000,9000、又は10,000スポット/cm2の密度を有する。
【0106】
ある例では、固相担体上のスポットは各々、異なる捕捉抗体を提示する。他の例では、固相担体上の複数のスポットが、例えば一連の下降捕捉抗体濃度から構成される希釈系列として、同一の捕捉抗体を提示する。
【0107】
固相担体上に抗体アレイを準備し、構築するための方法の追加の例は、米国特許第6,197,599号、第6,777,239号、第6,780,582号、第6,897,073号、第7,179,638号及び第7,192,720号;米国特許出願第20060115810号、第20060263837号、第20060292680号、及び第20070054326号;及びVarnumら、MethodsMol.Biol.,264:161−172(2004)に開示される。
【0108】
抗体アレイを走査する方法は、当該分野では既知であり、これに限定されないが、タンパク質又は核酸アレイを走査するために使用されるあらゆる技術を含む。本発明の使用に適したマイクロアレイスキャナーは、PerkinElmer(Boston,MA)、AgilentTechnologies(Palo Alto,CA)、Applied Precision(Issaquah,WA)、GSI Lumonics Inc.(Billerica,MA)、及びAxonInstruments(Union City,CA)から入手できる。非限定的な例として、蛍光検出のためのGSI ScanArray3000は、定量のためのImaGeneソフトウェアとともに使用され得る。
【0109】
V.抗体の作製
本発明に関して希少循環細胞におけるシグナル伝達分子の活性状態及び/又は総量を分析するためにまだ市販で入手できない抗体の生成及び選択は、様々な方法で達成できる。例えば、1つの方法は、対象となるポリペプチド(すなわち抗原)を当該分野で既知のタンパク質発現及び精製方法を用いて、発現し及び/又は生成することであり、一方、他の方法は、当該分野で既知の固層ペプチド合成法を用いて対象のポリペプチドを合成することである。例えば、Guideto Protein Purification,Murray P.Deutcher,ed.,Meth.Enzymol.,Vol.182(1990);SolidPhase Peptide Synthesis,Greg B.Fields,ed.,Meth.Enzymol.,Vol.289(1997);Kisoら、Chem.Pharm.Bull.,38:1192−99(1990);Mostafaviら、Biomed.Pept.ProteinsNucleic Acids,1:255−60,(1995);及びFujiwaraら、Chem.Pharm.Bull.,44:1326−31(1996)を参照する。精製され又は合成されたポリペプチドは、その後、ポリクローナル又はモノクローナル抗体を作製するために、例えばマウス又はウサギに注射され得る。当業者は、例えばAntibodies,ALaboratory Manual,Harlow and Lane,Eds.,Cold Spring Harbor Laboratory,Cold SpringHarbor,N.Y.(1988)に記載されているように、多くの手法が抗体の生成のために利用できることを認識するだろう。当業者はまた、抗体を模倣した結合断片又はFab断片が様々な手法によって遺伝子情報から準備され得ることを認識しているだろう(例えば、AntibodyEngineering:A Practical Approach,Borrebaeck,Ed.,Oxford University Press,Oxford(1995);及びHuseら、J.Immunol.,149:3914−3920(1992)を参照)。
【0110】
加えて、様々な刊行物が、選択された標的抗原に結合するためのポリペプチドライブラリーを構築し、スクリーニングするためのファージディスプレイ技術の使用を報告している(例えば、Cwirlaら、Proc.Natl.Acad.Sci.USA,87:6378−6382(1990);Devlinら、Science,249:404−406(1990);Scottら、Science,249:386−388(1990);及びLadnerら、米国特許第5,571,698号を参照)。ファージディスプレイ法の基本的な概念は、ファージDNAによってコードされたポリペプチドと標的抗原との間の物理的な結合の構築である。この物理的結合は、ポリペプチドをコードするファージ染色体を囲い込むカプシドの一部としてポリペプチドを提示するファージ粒子によって提供される。ポリペプチドとそれらの遺伝物質との間の物理的結合の構築は、異なるポリペプチドを有する非常に多くのファージの同時多量スクリーニングを許容する。標的抗原に対する親和性のあるポリペプチドを提示するファージは標的抗原に結合し、これらファージは標的抗原に対する親和性スクリーニングによって濃縮される。これらファージから提示されるポリペプチドの正体は、それらの各染色体から決定され得る。これらの方法を用いて、所望の標的抗原に対する結合親和性を有すると同定されたポリペプチドは、その後、従来法によって多量に合成され得る(例えば、米国特許第6,057,098号を参照)。
【0111】
これらの方法によって生成された抗体は、その後、対象となる精製ポリペプチド抗原との親和性及び特異性に対する第一のスクリーニングによって選択され得、必要であれば、その結果を、結合から除かれることが期待される他のポリペプチド抗原との抗体の親和性と特異性と比較する。スクリーニング手法は、マイクロタイタープレートの各々の穴における精製されたポリペプチド抗原の固定を含むことができる。潜在的な抗体又は抗体群を有する溶液は、その後、各マイクロタータープレートに置かれ、約30分から2時間インキュベートされる。マイクロタイターの穴は、その後洗浄され、標識化された2次抗体(例えば、作られた抗体がマウス抗体であれば、アルカリホスファターゼに結合された抗−マウス抗体)がその穴に添加され、約30分間インキュベートされ、その後洗浄される。基質がその穴に添加され、固定されたポリペプチド抗原に対する抗体が存在するところに呈色反応が現れるだろう。
【0112】
そのように同定された抗体は、その後さらに親和性と特異性について解析され得る。標的タンパク質に対する免疫分析の構築において、精製された標的タンパク質は、選択された抗体を用いた免疫分析の感度と特異性を判定するための標準として働く。様々な抗体の結合親和性は異なる、例えばある抗体の組み合わせは、立体的に他のものを妨げるかもしれないため、抗体の分析の性能は、その抗体の絶対的な親和性と特異性よりも重要な基準であるかもしれない。
【0113】
当業者は、抗体又は結合断片を作製し、対象の様々なポリペプチドに対する親和性と特異性に関してスクリーニングし及び選択する際に、多くの方法がとられ得るが、これらの方法が本発明の範囲を変えるものではないことを認識するだろう。
【0114】
A.ポリクローナル抗体
ポリクローナル抗体は、好ましくは、対象となるポリペプチド及びアジュバントの複数回の皮下(sc)又は腹腔内(ip)注射によって動物内で上昇する。対象となるポリペプチドを、例えば二官能性又は誘導体化物質として用いられるスカシガイヘモシアニン(keyholelimpet hemocyanin)、血清アルブミン、ウシサイログロブリン(bovine thyroglobulin)、又は大豆トリプシン阻害剤(soybeantrypsin inhibitor)のような、免疫された種において免疫原性であるタンパク質キャリアに共役することは有用であるかもしれない。二官能性又は誘導体化物質の非限定的な例は、マレイミドベンソイルスルフォスクシニミドエステル(maleimidobenzoylsulfosuccinimide ester)(システイン残基を通した共役)、N−ヒドロキシスクシニミド(N−hydroxysuccinimide)(リジン残基を通した共役)、グルタルアルデヒド(glutaraldehyde)、コハク酸無水物、SOCl2及びRとR1が異なるアルキル基であるR1N=C=NRを含む。
【0115】
動物は、対象となるポリペプチド又は免疫原性共役体又はそれらの誘導体に対して、例えば100μg(対ウサギ)又は5μg(対マウス)の抗原又は共役体を3倍容量のフロイント完全アジュバントを組み合わせて、そしてその溶液を複数の部位に皮内注射することによって免疫を与えられる。1ヶ月後、その動物は、複数の部位における皮下注射によってフロイント不完全アジュバント中のポリペプチド又は複合体の元々の量の約1/5から1/10でブーストされる。7日から14日後、その動物は、採血され、その血清が抗体力価に関して分析される。動物は、典型的には、力価が頭打ちになるまでブーストされる。好ましくは、動物は、同一のポリペプチドの複合体でブーストされるが、異なる免疫原性タンパク質への共役及び/又は異なる架橋剤による共役が用いられてもよい。複合体はまた、融合タンパク質として組み換え細胞培養で作られ得る。場合によっては、ミョウバンのような凝集剤が免疫応答を増幅するために用いられ得る。
【0116】
B.モノクローナル抗体
モノクローナル抗体は、一般的に実質的に相同な抗体の集団から獲得され、即ちその集団を構成する各抗体は、少量存在するかもしれない起こりうる自然発生変異を除き、同一である。従って、「モノクローナル(monoclonal)」という修飾語は、個々に異なる抗体の混合物ではないという抗体の特徴を示す。例えば、モノクローナル抗体は、Kohlerら、Nature,256:495(1975)によって開示されたハイブリドーマ法を用いて、又は当業者に既知の任意の組み換えDNA法(例えば米国特許第4,816,567号を参照)によって作製され得る。
【0117】
ハイブリドーマ法では、マウス又は他の適切な宿主動物(例えばハムスター)が、免疫付与のために用いられる、対象となるポリペプチドに特異的に結合する抗体を生産する又は生産することができるリンパ球を引き出すために上記のとおり免疫される。代替的には、リンパ球がインビトロで免疫される。免役されたリンパ球は、その後、ハイブリドーマ細胞を形成するため、ポリエチレングリコールのような適切な融合剤を用いて骨髄腫細胞と融合される(例えば、Goding,MonoclonalAntibodies:Principles and Practice,Academic Press,pp.59−103(1986)を参照)。このように用意されたハイブリドーマ細胞は、好ましくは、融合されていない親骨髄腫細胞の増殖又は生存を阻害する一つ又はそれ以上の基質を含む適切な培地に植えて増殖させる。例えば、親骨髄腫細胞が酵素ヒポキサンチングアニンホスホリボシルトランスフェラーゼ(HGPRT)を欠く場合、ハイブリドーマ細胞のための培地は、一般的に、HGPRT欠損細胞の増殖を阻止する、ヒポキサンチン、アミノプテリン、及びチミジンを含むだろう(HAT培地)。
【0118】
好ましい骨髄腫細胞は、効果的に融合され、選択された抗体生産細胞による安定した、高レベルの抗体生産を支え、及び/又はHAT培地のような培地に感受性があるものである。ヒトモノクローナル抗体の生産のための、そのような望ましい骨髄腫細胞系統の例は、これ限定されないが、MOPC−21及びMPC−11マウス腫瘍のようなネズミ科動物(murine)の骨髄腫細胞(theSalk Institute Cell Distribution Center;San Diego,CAから入手可能)、SP−2又はX63−Ag8−653細胞(theAmerican Type Culture Collection;Rockville,MDから入手可能)、及びヒト骨髄腫細胞又はマウス−ヒトヘテロ骨髄腫細胞系統(例えば、Kozbor,J.Immunol.,133:3001(1984);及びBrodeurら、MonoclonalAntibody Production Techniques and Applications,Marcel Dekker,Inc.,New York,pp.51−63(1987)を参照)を含む。
【0119】
ハイブリドーマ細胞が増殖している培地は、対象となるポリペプチドに対して向けられたモノクローナル抗体の生産に関して分析され得る。好ましくは、ハイブリドーマ細胞によって生産されたモノクローナル抗体の結合特異性は、免疫沈降法によって又は放射免疫測定(RIA)又は酵素免疫測定法(ELISA)のような、インビトロ結合測定によって決定される。モノクローナル抗体の結合親和性は、例えばMunsonら、Anal.Biochem.,107:220(1980)のスキャッチャード解析を用いて決定され得る。
【0120】
ハイブリドーマ細胞が所望の特異性、親和性及び/又は活性の抗体を生産すると同定された後、そのクローンは限界希釈法によって、サブクローン化され、そして標準的な方法によって増殖させられてもよい(例えば、Goding,MonoclonalAntibodies:Principles and Practice,Academic Press,pp.59−103(1986)を参照)。この目的のために適した培地は、例えば、D−MEM又はRPMI−1640培地を含む。加えて、ハイブリドーマ細胞は、動物内で腹水腫瘍としてinvivoで増殖してもよい。サブクローンによって分泌されたモノクローナル抗体は、例えば、タンパク質Aセファロース、ハイドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、親和性クロマトグラフィーのような従来の抗体精製手法によって、培地、腹水又は血清から分離され得る。
【0121】
モノクローナル抗体をコードするDNAは、従来の手法を用いて(例えば、ネズミ科動物(murine)抗体の重及び軽鎖をコードする遺伝子に特異的に結合できるオリゴペプチドプローブを用いることによって)容易に単離され、配列解読され得る。ハイブリドーマ細胞は、そのようなDNAの望ましい源として働く。一旦単離されると、組み換え宿主細胞においてモノクローナル抗体の合成を促進するために、DNAは発現ベクターに搭載され、その後、E.coli細胞、サルCOS細胞、チャイニーズハムスター卵巣(CHO)細胞、又は本来なら抗体を生産しない骨髄腫細胞のような宿主細胞に導入されてもよい。例えば、Skerraら、Curr.Opin.Immunol.,5:256−262(1993);及びPluckthun,ImmunolRev.,130:151−188(1992)を参照する。DNAはまた、例えば、ヒト重鎖及び軽鎖の定常ドメインをコードする配列を相同なネズミ科動物(murine)の配列と置換することによって(例えば、米国特許第4,816,567;Morrisonら、Proc.Natl.Acad.Sci.USA,81:6851(1984)を参照)、又は非免疫グロブリンペプチドをコードする配列の全て又は一部を免疫グロブリンをコードする配列と共有結合することによって、改変され得る。
【0122】
さらなる実施の形態では、モノクローナル抗体又は抗体断片は、例えば、McCaffertyら、Nature,348:552−554(1990);Clacksonら、Nature,352:624−628(1991);及びMarksら、J.Mol.Biol.,222:581−597(1991)において記載された技術を用いて生成された抗体ファージライブラリーから単離され得る。鎖シャッフリングによる高親和性(nM範囲)ヒトモノクローナル抗体の生産は、Marksら、BioTechnology,10:779−783(1992)に記載されている。非常に大きなファージライブラリーを構築するための戦略として、組み合わせ感染(combinatorialinfection)及びin vivo組み換えの使用は、Waterhouseら、Nuc.Acids Res.,21:2265−2266(1993)に記載されている。従って、これらの技術は、モノクローナル抗体の生成のための伝統的なモノクローナル抗体ハイブリドーマ法の実行可能な代替案である。
【0123】
C.ヒト化抗体
非ヒト抗体をヒト化するための方法は、当該分野では既知である。好ましくは、ヒト化抗体は、非ヒトである試料源からの抗体に導入された一つ又はそれ以上のアミノ酸残基を有する。これらの非ヒトアミノ酸残基はしばしば「取り込み(import)」残基と言われ、典型的には「取り込み(import)」可能なドメインから取得される。ヒト化は、本質的には、非ヒト抗体の超可変領域の配列をヒト抗体の対応する配列と置き換えることによって実行され得る。例えば、Jonesら、Nature,321:522−525(1986);Riechmannら、Nature,332:323−327(1988);及びVerhoeyenら、Science,239:1534−1536(1988)を参照する。従って、そのような「ヒト化」抗体は、キメラ抗体(例えば、米国特許第4,816,567号)であって、完全なヒト可変ドメインは、本質的には決して非ヒトの種からの対応する配列によって置換されていない。実際に、ヒト化された抗体は、典型的には、いくらかの超可変領域残基及び可能であればいくらかのフレームワーク領域(FR)残基が齧歯類抗体の類似部位からの残基によって置換されるヒト抗体である。
【0124】
本明細書で記載したヒト化抗体を作製するのに使用するための、ヒト可変ドメイン、軽鎖及び重鎖の両方、の選択は、抗原性を低減するために重要な検討事項である。いわゆる「最も適した(best−fit)」方法に従って、齧歯類抗体の可変ドメインの配列が、既知のヒト可変ドメイン配列の全ライブラリーに対しスクリーニングされる。齧歯類のものに最も近いヒト配列が、その後、ヒト化抗体のためのヒトFRとして受け入れられる(例えば、Simsら、J.Immunol.,151:2296(1993);及びChothiaら、J.Mol.Biol.,196:901(1987)を参照)。他の方法は、全てのヒト抗体の軽鎖又は重鎖の特定の部分集合の共通配列に由来する特定のFRを用いる。同一のFRは、様々な異なるヒト化抗体のために使用されてもよい(例えば、Carterら、Proc.Natl.Acad.Sci.USA,89:4285(1992);及びPrestaら、J.Immunol.,151:2623(1993)を参照)。
【0125】
抗体が、抗原に対する高親和性の保有及び他の好ましい生化学的特性とともにヒト化されることもまた重要である。この目的を達成するため、ヒト化抗体は、親及びヒト化配列の3次元モデルを用いた、親配列と様々な概念的ヒト化生成物の解析の工程によって用意され得る。3次元免疫グロブリンモデルは、一般に利用可能であり、当業者によく知られている。選択された候補免疫グロブリン配列の可能な3次元立体配座構造を図示し、表示するコンピュータープログラムが利用できる。これら表示の観察が候補免疫グロブリン配列の機能における残基の類似の役割の解析、即ち候補免疫グロブリンの抗原への結合能に影響する残基の解析、を可能にする。このように、FR残基は、受容及び取り込み配列から選択され、そして組み合わせられるので、標的抗原に対する増加した親和性のような所望の抗体特性が達成され得る。一般的に、超可変領域は、直接的及び特異的に、抗原結合に影響することに含まれる。
【0126】
本発明によると、様々な種類のヒト化抗体が意図されている。例えば、ヒト化抗体は、Fab断片のような抗体断片であり得る。代替的には、ヒト化抗体は、完全なIgA、IgG又はIgM抗体のような完全な抗体であり得る。
【0127】
D.ヒト抗体
ヒト化の代替として、ヒト抗体が生成され得る。ある実施の形態では、内因性免疫グロブリン生産の欠損下で、免疫付与によって、全ての種類のヒト抗体を生成することができるトランスジェニック動物(例えば、マウス)が生成され得る。例えば、キメラ生殖系列変異マウスにおける抗体重鎖接合領域(JH)遺伝子のホモ欠損が、内因性抗体生成の完全な阻害をもたらすことが述べられている。そのような生殖系列変異マウスにおけるヒト生殖系列免疫グロブリン遺伝子の形質転換は、抗原投与でヒト抗体の生産をもたらすだろう。例えば、Jakobovitsら、Proc.Natl.Acad.Sci.USA,90:2551(1993);Jakobovitsら、Nature,362:255−258(1993);Bruggermannら、Yearin Immun.,7:33(1993);及び米国特許第5,591,669号、5,589,369号及び5,545,807号を参照する。
【0128】
代替的には、ファージディスプレイ技術(例えば、McCaffertyら、Nature,348:552−553(1990)を参照)が、免疫されていないドナーからの免疫グロブリン可変(V)ドメイン遺伝子レパートリーを用いて、インビトロでヒト抗体及び抗体断片を生成するために使用され得る。この技術に従うと、抗体Vドメイン遺伝子は、M13又はfdのような、繊維状バクテリオファージのメジャー又はマイナーコートタンパク質遺伝子のいずれかにフレームをあわせてクローン化され、ファージ粒子の表面で機能性抗体断片として提示される。繊維状粒子がファージゲノムの1本鎖DNAコピーを含むため、抗体の機能特性に基づく選択はまた、それらの特性を示す抗体をコードする遺伝子の選択をもたらす。従って、ファージは、B細胞の特性の一部を模倣する。ファージディスプレイは、例えばJohnsonら、Curr.Opin.Struct.Biol.,3:564−571(1993)に記載されるように様々な形式で実行され得る。V遺伝子断片のいくつかの入手源がファージディスプレイのために使用され得る。例えば、Clacksonら、Nature,352:624−628(1991)を参照する。免疫されていないヒトドナーからのV遺伝子のレパートリーは構築され得、多様な抗原(自己抗原も含む)への抗体は、本質的に、Marksら、J.Mol.Biol.,222:581−597(1991);Griffithら、EMBOJ.,12:725−734(1993);及び米国特許第5,565,332号、及び第5,573,905号に記載された技術に従って単離され得る。
【0129】
場合によっては、ヒト抗体は、米国特許第5,567,610号及び第5,229,275号に記載されるようにインビトロで活性化されたB細胞によって生成され得る。
【0130】
E.抗体断片
様々な技術が抗体断片の生成のために開発されてきた。伝統的には、これらの断片は、完全な抗体のタンパク質切断を介して得られた(例えば、Morimotoら、J.Biochem.Biophys.Meth.,24:107−117(1992);及びBrennanら、Science,229:81(1985)を参照)。しかしながら、これらの断片は、現在、組み換え宿主細胞を用いて直接的に生成され得る。例えば、抗体断片は上述した抗体ファージライブラリーから単離され得る。代替的には、Fab’−SH断片がE.coli細胞から直接回収され、そしてF(ab’)2を形成するよう化学的に結合され得る(例えばCarterら、BioTechnology,10:163−167(1992)を参照)。他のアプローチによると、F(ab’)2断片は、組み換え宿主細胞培養から直接単離され得る。抗体断片の生成のための他の技術は、当業者には明らかであるだろう。他の実施の形態において、選択の抗体は、単鎖Fv断片(scFv)である。例えば、国際公開第93/16185号;及び米国特許第5,571,894号及び第5,587,458号を参照する。抗体断片はまた、例えば、米国特許第5,641,870号に記載されるようにリニア抗体(linearantibody)であってもよい。そのようなリニア抗体断片は、単一特異性又は二重特異性であるかもしれない。
【0131】
F.二重特異性抗体
二重特異性抗体は、少なくとも2つの異なるエピトープに対する結合特異性を有する抗体である。典型的な二重特異性抗体は、対象となる同一のポリペプチドの2つの異なるエピトープに結合するかもしれない。他の二重特異性抗体は、対象となるポリペプチドに対する結合部位を一つ又はそれ以上の追加的な抗原に対する結合部位と組み合わせるかもしれない。二重特異性抗体は、全長抗体又は抗体断片(例えば、F(ab’)2二重特異性抗体)として用意され得る。
【0132】
二重特異性抗体を作製する方法は、当該分野では既知である。全長型二重特異性抗体の伝統的な作製は、2つの鎖が異なる特異性を有する2つの免疫グロブリンの重鎖−軽鎖対の共発現に基づく(例えば、Millsteinら、Nature,305:537−539(1983)を参照)。免疫グロブリンの重鎖及び軽鎖のランダムな組み合わせのため、これらのハイブリドーマ(クアドローマ)は、一つのみが正しい二重特異性構造を有する、10の異なる抗体分子の潜在的混合物を生成する。正しい分子の精製は、通常、親和性クロマトグラフィーによって実行される。同様の手法は、国際公開第93/08829号及びTrauneckerら、EMBOJ.,10:3655−3659(1991)に開示される。
【0133】
異なるアプローチによると、所望の結合特異性(抗体−抗原結合部位)を有する抗体可変ドメインは、免疫グロブリン共通ドメイン配列に融合される。この融合体は、好ましくは、少なくともヒンジCH2及びCH3領域の一部から構成される免疫グロブリン重鎖定常ドメインを有する。好ましくは、少なくとも1つの融合体に存在する軽鎖結合に必要な部位を含む第1の重鎖定常領域(CH1)を有する。免疫重鎖融合体と、必要であれば、免疫グロブリン軽鎖をコードするDNAは、別々の発現ベクターに挿入され、そして適切な宿主生物に共導入される。これは、構築に使用された3つのポリペプチド鎖の不均一な割合が最適の収率を提供する実施の形態では、3つのポリペプチド断片の相互の比率を調整することにおいて高い柔軟性を提供する。しかしながら少なくとも2つのポリペプチド鎖の等しい比率での発現が高い収率をもたらす又はその比率が特に重要ではないときには、2つ又は3つ全てのポリペプチド鎖をコードする配列を一つの発現ベクターに挿入することが可能である。
【0134】
このアプローチの好ましい実施の形態では、二重特異性抗体は、片腕に第1の結合特異性を有するハイブリッド免疫グロブリン重鎖と、他腕の手に(第2の結合特異性を提供する)ハイブリッド免疫グロブリン重鎖−軽鎖対とから構成される。二重特異性分子の片方のみにおける免疫グロブリン軽鎖の存在が分離の容易な方法を提供するので、この非対照的な構造は、望まれない免疫グロブリン鎖複合体からの所望の二重特異性構成体の分離を促進する。例えば、国際公開第94/04690号及びSureshら、Meth.Enzymol.,121:210(1986)を参照する。
【0135】
米国特許第5,731,168号に記載された他のアプローチによると、1対の抗体分子間の接合部は、組み換え細胞培養から回収されたヘテロ2量体の割合を最大限にするよう設計され得る。好ましい接合部は、抗体の定常ドメインのCH3ドメインの少なくとも一部から構成される。この方法では、第1の抗体分子の接合部からの一つ又はそれ以上の小さなアミノ酸側鎖がより大きな側鎖(例えばチロシン又はトリプトファン)で置換される。大きな側鎖と同一の又は類似の大きさの補完的な「空洞(cavity)」が、大きなアミノ酸側鎖をより小さな側鎖(例えばアラニン又はスレオニン)で置換することによって第2の抗体分子の接合部に生成される。これは、ホモ2量体のような他の望まれない最終産物に対するヘテロ2量体の収率を増加するための機構を提供する。
【0136】
二重特異性抗体は、架橋又は「ヘテロ接合(heteroconjugate)」抗体を含む。例えば、ヘテロ接合である抗体の一つは、アビジンに連結され得、他方はビオチンに連結され得る。ヘテロ接合型抗体は、任意の使用可能な架橋方法を用いて、作製され得る。適切な架橋剤及び技術は、当該分野ではよく知られており、例えば米国特許第4,676,980号に開示される。
【0137】
抗体断片から二重特異性抗体を生成するために適した技術はまた、当該分野で既知である。例えば、二重特異性抗体は、化学的な連結を用いて準備され得る。場合によっては、二重特異性抗体は、完全な抗体がタンパク質分解で切断され、F(ab’)2断片を生成する手法によって生成され得る(例えば、Brennanら、Science,229:81(1985)を参照)。これらの断片は、隣接チオールを安定化し、そして分子内ジスルフィド形成を防ぐジチオール錯体化剤亜ヒ酸ナトリウムの存在下で減少する。生成したFab’断片は、その後、チオニトロ安息香酸(TNB)誘導体に変換される。Fab’−TNB誘導体の一つは、その後、メルカプトエチルアミン(mercaptoethylamine)での還元によってFab’−チオールに変換され、二重特異性抗体を形成するために等モル量の他のFab’−TNB誘導体と混合される。
【0138】
ある実施の形態では、Fab’−SH断片は、E.coliから直接回収され、二重特異性抗体を形成するために化学的に結合され得る。例えば、完全にヒト化された二重特異性抗体F(ab’)2分子は、Shalabyら、J.Exp.Med.,175:217−225(1992)に記載された方法によって生成され得る。各Fab’断片は、E.coliから別々に分泌され、二重特異性抗体を形成するためにインビトロでの直接的な化学的結合を受けた。
【0139】
組み換え細胞培養から直接二重特異性抗体断片を作製し、単離するための様々な技術はまた、開示されている。例えば、二重特異性抗体は、ロイシンジッパーを用いて生成されている。例えば、Kostelnyら、J.Immunol.,148:1547−1553(1992)を参照する。Fos及びJunタンパク質からのロイシンジッパーペプチドは、遺伝子融合によって、2つの異なる抗体のFab’部に連結された。抗体ホモ2量体は、単量体を形成するためにヒンジ部で還元され、その後、抗体ヘテロ2量体を形成するために再酸化された。この方法はまた、抗体ホモ2量体の生成のために利用され得る。Hollingerら、Proc.Natl.Acad.Sci.USA,90:6444−6448(1993)によって記載された「ダイアボディー(diabody)」技術は、二重特異性抗体断片を作製するための代替的な機構を提供している。その断片は、非常に短いために同一鎖の2つのドメイン間での対合を可能にするリンカーによって軽鎖可変ドメイン(VL)と連結した重鎖可変ドメイン(VH)から構成される。従って、一つの断片のVH及びVLドメインは、他の断片の相補的なVL及びVHドメインと対を形成せざるを得ず、それによって2つの抗原結合部を形成する。単鎖Fv(sFv)2量体の使用によって二重特異性抗体断片を作製するための他の戦略は、Gruberら、J.Immunol.,152:5368(1994)に記載される。
【0140】
二価以上の抗体も、意図される。例えば、三重特異性抗体が用意され得る。例えば、Tuttら、J.Immunol.,147:60(1991)を参照する。
【0141】
G.抗体精製
組み換え技術を用いると、抗体は、単離された宿主細胞内、宿主細胞のペリプラズム空間で生成され得、又は宿主細胞から培地中に直接分泌され得る。抗体が細胞内で生成されると、例えば、遠心又は限外ろ過によって微粒子破片が先ず除去される。Carterら、BioTech.,10:163−167(1992)は、E.coliのペリプラズム空間に分泌された抗体を単離するための手法を記載する。簡単に言うと、細胞ペーストは、約30分間、酢酸ナトリウム(pH3.5)、EDTA、及びフッ化フェニルメチルスルホニル(PMSF)の存在中で溶解される。細胞破片は、遠心分離によって分離され得る。抗体が培地中に分泌される際、そのような発現システムからの上清は、一般的に、市販で入手可能なタンパク質濃縮フィルター、例えばAmicon又はMilliporePelliconの限外ろ過ユニットを用いて濃縮される。PMSFのような、プロテアーゼ阻害剤は、タンパク質分解を阻害するための前述の任意のステップに含まれ、抗生物質は、外来の混入物の増殖を抑制するために含まれてもよい。
【0142】
細胞から用意された抗体組成物は、例えば、ハイドロキシルアパタイトクロマトグラフィー、ゲル電気泳動、透析、及び親和性クロマトグラフィーを用いて精製され得る。親和性リガンドとしてタンパク質Aの適正は、抗体に存在するあらゆる免疫グロブリンのFcドメインの種及びアイソタイプに依存する。タンパク質Aは、ヒトγl,γ2,又はγ4重鎖に基づいた抗体を精製するために使用され得る(例えば、Lindmarkら、J.Immunol.Meth.,62:1−13(1983)を参照)。タンパク質Gは、全てのマウスのアイソタイプ及びヒトγ3に対し推奨される(例えば、Gussら、EMBOJ.,5:1567−1575(1986)を参照)。親和性リガンドが付着されるマトリクスは、最も頻繁にはアガロースであるが、他のマトリクスが利用可能である。微小多孔質ガラス(controlledpore glass)又はポリ(スチレンジビニル)ベンゼン(poly(styrenedivinyl)benzene)のような機械的に安定なマトリクスは、アガロースで達成され得るよりも速い流速とより短い処理時間を許容する。抗体がCH3ドメインから構成される場合、BakerbondABX(登録商標)レジン(J.T.Baker;Phillipsburg,N.J.)が精製のために有用である。イオン交換カラム、エタノール沈殿、逆層HPLC、シリカクロマトグラフィー、ヘパリンSEPHAROSE(登録商標)クロマトグラフィー、陰イオン又は(ポリアスパラギン酸カラムのような)陽イオン交換レジンクロマトグラフィー、等電点電気泳動、SDS−PAGE、及び硫酸アンモニウム沈殿のようなタンパク質精製のための他の技術はまた、回収される抗体に依存して利用可能である。
【0143】
あらゆる予備的な精製工程に続いて、対象となる抗体と混入物とから構成される混合物は、2.5〜4.5の間のpHの溶出緩衝液を用いて、好ましくは低塩濃度で(例えば約0〜0.25Mの塩から)実行される、低pH疎水性相互作用クロマトグラフィーに供されてもよい。
【実施例】
【0144】
以下の実施例は、請求の範囲に記載の発明を説明するために述べられるが、これに限定されるものではない。
【0145】
(実施例1.循環細胞の単離、活性化、及び溶解)
固形腫瘍の循環細胞は、固形腫瘍から転移した又は微小転移した細胞から構成され、循環腫瘍細胞(CTC)、癌幹細胞(CSC)及び/又は腫瘍へと移動している細胞(例えば、循環内皮前駆細胞(CEPC)、循環内皮細胞(CEC)、循環血管新生促進骨髄性細胞、循環樹状細胞、等)を含む。循環細胞を含む患者試料は、あらゆる入手可能な生体液(例えば、血液、尿、乳頭吸引液、リンパ液、唾液、細針吸引物、等)から獲得され得る。循環細胞は、例えば、免疫磁気分離法(例えば、Racilaら、Proc.Natl.Acad.Sci.USA,95:4589−4594(1998);Bilkenrothら、Int.J.Cancer,92:577−582(2001)を参照)、マイクロ流体分離法(例えばMohamedら、IEEETrans.Nanobiosci.,3:251−256(2004);Linら、Abstract No.5147,97th AACR Annual Meeting,Washington,D.C.(2006)を参照)、FACS(例えばMancusoら、Blood,97:3658−3661(2001)を参照)、密度勾配遠心分離法(例えばBakerら、Clin.CancerRes.,13:4865−4871(2003)を参照)、及び減少法(例えばMeyeら、Int.J.Oncol.,21:521−530(2002)を参照)のような一つ又はそれ以上の分離方法を用いて患者試料から単離され得る。
【0146】
CTCの手動単離:
CTCの免疫磁気分離法−活性分析が続く手動単離:
1)前もって抗−EpCAMモノクローナル抗体(KordiaLife Sciences;Leiden,The Netherlands)に結合された磁気ビーズ((Dynal M450;Dynal AS;Oslo,Norway)が用いられる。
2)使用直前に、事前に覆われたダイナビーズ(Dynabead)は、0.01%BSAを含む等量のPBSで一度洗浄される。
3)25mlの事前に覆われたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に設置される。
6)その上清が廃棄され、ビーズに結合した細胞が0.01%BSAを含むPBSで再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0147】
試料の準備:
1)ヒト対象物からの末梢血が1mg/mlのEDTAを含むシリコン処理チューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出された上皮細胞での汚染を避けるために廃棄される。
2)1mlの全血が使用前に0.9%のNaClで1:3に希釈される。
【0148】
対照の準備:
1)細胞系列の対照がヒト癌細胞系列をHL−60細胞にスパイクすることによって作製される。
2)細胞系列の対照は、2.5x106細胞/mlの濃度で使用される。
【0149】
CEC及びCEPCの手動分離:
非限定的な例として、生存CEC及びCEPCは、Beerepootら、Ann.Oncology,15:139−145(2004)に記載された免疫磁気単離/濃縮手法を用いて単離され得る。つまり、末梢血が前もって抗CD146モノクローナル抗体(KordiaLife Sciences)に共役された磁気ビーズ(Dynal M450 IgG1)とともにインキュベートされる。この抗体は、末梢血中の内皮細胞の全ての系統を認識し、造血細胞又は上皮細胞を認識しない(Georgeら、J.Immunol.Meth.,139:65−75(1991)を参照)。造血細胞及び上皮細胞の消極的な選択は、適切な抗体に共役された磁気ビーズ(例えば、白血球を除くためのDynal−CD45ビーズ、単球を除くためのDynal−CD14ビーズ、上皮細胞を除くためのDynal−EpCAM(Invitrogen;Carlsbad,CA))での積極的選択に先立って使用され得る。この例では、積極的選択のみが使用される。
【0150】
CEC及びCEPCの免疫磁気分離−活性分析が続く手動単離:
1)前もって抗−CD146モノクローナル抗体(KordiaLife Sciences)を結合された磁気ビーズ(Dynal M450)が使用される。
2)使用直前に、事前に被覆されたダイナビーズは、0.01%BSAを含む等量のPBSで一回洗浄される。
3)25μlの事前に被覆されたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSでの再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0151】
試料の準備:
1)ヒト対象物からの末梢血が、1mg/mlEDTAを含むシリコン処理されたチューブに引き抜かれる。最初の3〜5mlは穴の開いた血管から放出された上皮細胞での汚染を防ぐため、廃棄される。
2)1mlの全血は、使用前に0.9%NaClで1:3に希釈される。
【0152】
対照の準備:
1)細胞系列の対照が、ヒト臍帯静脈血管内皮細胞(HUVEC)をHL−60細胞にスパイクすることによって作製される。
2)細胞系列の対照は、2.5x106細胞/mlの濃度で使用される。
【0153】
CEPCの手動単離(CECを除く):
CEPCは、様々な血管新生増殖因子に応答して、成熟内皮細胞へ分化する能力を有する骨髄由来の前駆細胞の循環型である。CEPCは、表層マーカーCD34を認識する抗体での選択によって単離されてよい。CD133は、未熟内皮細胞(EPC)又は原始造血幹細胞(HSC)をCEPCから区別する細胞表層マーカーである。異なる入手源からのCEPCの様々な単離手法は、接着培養又は磁気マイクロビーズを用いて開示されている。この例では、Asaharaら、Science,275:964−967(1997)に記載されたものから改良されたプロトコールが使用される。
【0154】
CEPCの免疫磁気分離−活性分析が続く手動単離:
1)磁気ビーズ(DynalM450 CD34)が使用される。これらのビーズはCD34表層抗原に対し特異的なモノクローナル抗体で覆われる。
2)事前に覆われたダイナビーズは、使用直前に、0.01%BSAを含む等量のPBSで一回洗浄される。
3)25μlの事前に覆われたダイナビーズが1mlの試料に添加される。
4)その混合物は、2〜8℃で20分間、緩やかな傾斜と回転でインキュベートされる。
5)そのチューブは、2分間、磁気分離器(MPL−1magnet)に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSに再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0155】
試料の準備:
1)ヒト対象物からの末梢血が、1mg/mlのEDTAを含むシリコン処理されたチューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出された内皮細胞での汚染を避けるために廃棄される。
2)10mlの血液は、平衡塩類溶液で1:1に希釈される。
3)4mlの希釈血液は、10mlチューブ中で3mlのFicoll−Paque上に積層される。
4)チューブは、18〜20℃で30〜40分間、400×gで回転される。
5)血漿及び血小板を含む上層は、滅菌パスツールピペットを用いて取り除かれ、界面で乱れていない単核細胞層を残す。
6)単核細胞は、滅菌ピペットを用いて滅菌遠心管へ移される。
7)6mlの平衡塩類溶液が添加され、細胞は穏やかに再懸濁される。
8)混合物は、18〜20℃で10分間、60〜100×gで遠心される。
9)上清が、除去され、各チューブからの単核細胞が1mlのPBSに再懸濁される。
【0156】
Veridexシステムを用いたCTC,CEC,及びCEPCの単離:
Veridex(Warren,NJ)は、CellPrepシステム、CellSearch上皮細胞キット、及びCellSpotterアナライザーから構成されるCellSearchシステムを市販している。CellPrepシステムは、半自動化された試料準備システムである(Kaganら、J.Clin.LigandAssay,25:104−110(2002)を参照)。CellSearch上皮細胞キットは:内皮細胞に特異的な抗−EpCAM抗体で覆われた鉄流動体(ferrofluid);サイトケラチン(cytokeratins)8、18及び19に対するフィコエリスリン(phycoerythrin)共役抗体;アロフィコシアニン(allophycocyanin)に共役した抗−CD45抗体;DAPI染色剤;及び細胞を洗浄、浸透、及び再懸濁するための緩衝液、から構成される。この例で使用されるプロトコールはまた、Allardら、Clin.CancerRes.,10:6897−6904(2004)に記載されている。全Veridexシステムは、CTCの計数のために使用され得、又は、CellPrepシステムで単離された後に手動で試料を除去することによって、経路活性化のための分析に先だった単離の方法を提供することができる。CTCの数は、アルゴリズム開発のために情報を提供し得る。
【0157】
Veridexシステム−計数が続くCTC濃縮:
1)7.5mlの血液が6ml緩衝液と混合され、10分間800×gで遠心され、CellPrepシステムに移される。
2)装置が上清を吸引した後、その装置は鉄流動体を添加する。
3)その装置が、インキュベートとそれに続く磁気分離工程を実行する。
4)非結合細胞及び残存血漿が吸引される。
5)染色試薬が、蛍光染色のために浸透化緩衝液と組み合わせて添加される。
6)そのシステムによるインキュベート後、細胞は、CellSpotterアナライザーを用いた分析のため、再びMagNestCell Presentation装置にて磁気的に分離され、そして再懸濁される。
7)その装置は、CellSpotterアナライザー、4色半自動化蛍光顕微鏡、上に設置される。
8)画像は、Veridexの定義した基準に合うように得られ、最終的な手動選択のためにウェブに基づくブラウザを介して示される。
9)細胞計数の結果は、7.5ml血液あたりの細胞数として表される。
【0158】
Veridexシステム−活性分析が続くCTC濃縮:
1)7.5ml血液が6mlの緩衝液と混合され、10分間800×gで遠心され、その後CellPrepシステムに移される。
2)装置が上清を吸引した後、その装置は鉄流動体を添加する。
3)その装置はインキュベートとそれに続く磁気分離工程を実行する。
4)非結合細胞と残存血漿が吸引される。
5)試料は、100μlの活性化緩衝液で再懸濁される。
【0159】
Veridexシステム−活性分析が続くCEC及びCEPC濃縮:
1)Veridexは、CD146抗体でのCEC及びCEPCの捕獲に用いるCellTracks内皮細胞キットを提案する。CellTracks内皮細胞キットは、血液試料の準備のためのVeridexのCellTracksAutoPrepシステム及び全血からのCEC及びCEPCを数え、特徴づけるためのCellTracksアナライザーIIとを組み合わせて用いられる。このプロトコールは、CellSearch上皮細胞キットのものと同一である。
【0160】
試料の準備:
1)ヒト対象物からの末梢血は、製造元の指導に従って、CellSavePreservativeチューブに引き抜かれる。最初の3〜5mlは、穴の開いた血管から放出される上皮又は内皮細胞での汚染を回避するために廃棄される。
【0161】
CSCの手動単離:
腫瘍が特徴的な自己再生及び生存機構を有する少数の推定癌幹細胞を含むことの証拠が確立しつつある(例えばSells,Crit.Rev.Oncol.Hematol.,51:1−28(2004);Reyaら、Nature,414:105−111(2001);Dontuら、TrendsEndocrinol.Metal.,15:193−197(2004);and Dick,Nature,423:231−233(2003)を参照)。癌幹細胞(CSC)は、長期間静止状態で存在し、分裂する細胞を標的とする化学療法剤に対し耐性を獲得するかもしれない。この癌を発生させる集団は、選択的な除去のための標的化された治療を受けた自己再生及び生存経路の活性化として特徴づけられ得る。CSCの単離手法は、接着培養又は磁気マイクロビーズを用いて開示されている。この例では、Coteら、Clin.Can.Res.,12:5615(2006)で開示されたものから改変したプロトコールが使用される。
【0162】
免疫磁気CSC単離−活性分析が続く手動単離:
1)磁気ビーズ(DynalAS;Oslo,Norway)が使用される。これらのビーズは、CD34又はCD133表層抗原のいずれかに対し特異的なモノクローナル抗体で覆われる。
2)使用直前に、事前に覆われたダイナビーズが0.01%BSAを含む等量のPBSで一回洗浄される。
3)1〜107の事前に覆われたダイナビーズが、3mlの試料に添加される。
4)混合物が、2〜8℃で60分間、緩やかな傾斜と回転でインキュベートされる。
5)混合物は、1mlの部分に分割され、各チューブは、少なくとも6分間、磁気分離器(MPL−1magnet)上に置かれる。
6)その上清が廃棄され、ビーズ結合細胞が、0.01%BSAを含むPBSに再懸濁され、続いて磁気分離されることによって3回洗浄される。
7)その試料は、100μlの活性化緩衝液に再懸濁される。
【0163】
試料の準備:
1)骨髄試料が、インフォームド・コンセントを得た後に早期乳癌患者から獲得される。
2)骨髄吸引処理は、Bauerら、Clin.Can.Res.,6:3552−3559(2000)に記載のとおり実行される。あらゆる播種性腫瘍細胞を含む単核細胞画分は、BeckmanGS−6遠心機を用いて、35分間4000×gでのFicoll−Hypaque密度勾配遠心分離法によって濃縮され、PBSで2回洗浄される。
【0164】
単離したCTCの細胞活性化及び溶解:
細胞活性化:
1)増殖因子TGF−α(100nM)、ヘレグリン(heregulin)(100nM)、及び/又はIGF(100nM)が細胞に添加され、37℃で5分間インキュベートされる。
【0165】
薬剤処理での細胞活性化:
1)試料は、37℃で30分間、治療に効果的な濃度で、ヘルセプチン(Herceptin)、ラパタニブ(Lapatanib)、タルセバ(Tarceva)、及び/又はラパマイシン(Rapamycin)類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で5分間、インキュベートされる。
【0166】
薬剤処理での細胞活性化(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度で、ヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)によって活性化され、37℃で120分間インキュベートされる。
【0167】
活性化されたCTCが以下のプロトコールを用いて、溶解される:
1)新鮮な溶解緩衝液は、表1に記載された試薬を混合することによって、新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却した緩衝液中に再懸濁される。
3)インキュベートが氷上で30分間行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速でマイクロチューブ中で遠心分離される。
1)溶解物は、分析のため、新しいチューブに移され、−80℃で保存される。
【表1】
【0168】
単離されたCEC及び/又はCEPCの細胞活性化及び溶解:
VEGFは、CEPC(Larriveeら、J.Biol.Chem.,278:22006−22013(2003))及び、血管壁が剥がれ落ちた成熟CEC(Soloveyら、Blood,93:3824−3830(1999))の両方において抗アポトーシス経路を活性化することによって、生存を促進すると考えられている。成熟CECは、CEPCと比較して、限定された増殖能力しか有していないようであるが、VEGFはまた、CEPC又は成熟CECの増殖を促進するかもしれない(Linら、J.Clin.Invest.,105:71−77(2000))。これらの理由のため、CEC及び/又はCEPCは、溶解の前にVEGFとのインキュベートによって活性化される。
【0169】
細胞活性化:
1)増殖因子VEGF,FGF,PDGF,PIGF,及び/又はアンギオポイエチン(angiopoietin)は、各100nMで細胞に添加され、37℃で5分間インキュベートされる。
【0170】
薬剤処理での細胞活性化:
1)試料は、37℃で30分間、治療に効果的な濃度のアバスタチン(Avastin)、ネクサバール(Nexavar)、スーテント(Sutent)及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子VEGF、FGF、PDGF、PIGF、及び/又はアンギオポイエチンを添加することによって活性化され、37℃で5分間インキュベートされる。
【0171】
薬剤処理での細胞活性化(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度のアバスチン、ネクサバール、スーテント、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、VEGF、FGF、PDGF、PIGF、及び/又はアンギオポイエチンを各100nMで添加することによって活性化され、37℃で120分間インキュベートされる。
【0172】
単離されたCEC及び/又はCEPC細胞が以下のプロトコールを用いて溶解される:
1)新鮮な溶解緩衝液が表1に記載された試薬を混合することによって新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却された緩衝液に再懸濁される。
3)インキュベートが30分間氷上で行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速で、マイクロチューブ中で遠心分離される。
5)溶解物は、分析のために新しいチューブに移され、−80℃で保存される。
【0173】
単離されたCSCの細胞活性化及び溶解:
活性化された細胞:
1)増殖因子TGF−α(100nM)、ヘレグリン(10nM)、及び/又はIGF(100nM)が細胞に添加され、37℃で5分間インキュベートされる。
【0174】
薬剤処理で活性化された細胞:
1)試料は、37℃で30分間、治療に効果的な濃度のヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で5分間インキュベートされる。
【0175】
薬剤処理で活性化された細胞(フィードバックループ):
1)試料は、37℃で30分間、治療に効果的な濃度のヘルセプチン、ラパタニブ、タルセバ、及び/又はラパマイシン類縁体で処理される。
2)細胞は、その後、増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及び/又はIGF(100nM)を添加することによって活性化され、37℃で120分間インキュベートされる。
【0176】
単離されたCSC細胞は、以下のプロトコールを用いて溶解される:
1)新鮮な溶解緩衝液が表1に記載された試薬を混合することによって新たに用意される。
2)最終洗浄後、細胞は、氷上で、100μlの冷却された緩衝液に再懸濁される。
3)インキュベートが30分間氷上で行われる。
4)混合物は、溶解物からビーズを分離するために、10分間最速で、マイクロチューブ中で遠心分離される。
5)溶解物は、分析のために新しいチューブに移され、−80℃で保存される。
【0177】
(実施例2.チラミドシグナル増幅を有する単一検出サンドイッチELISAを用いた単一細胞検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重、ハイスループットの単一検出サンドイッチELISAを説明する:
1)96穴マイクロタイタープレートは、4℃で一晩、捕捉抗体で覆われた。
2)プレートは、翌日、1時間、2%BSA/TBS−Tweenで遮断(ブロック)された。
3)TBS−Tweenで洗浄後、細胞溶解物又は組み換えタンパク質は、一連の希釈で添加され、室温で2時間インキュベートされた。
4)プレートは、TBS−Tweenで4回洗浄され、その後、室温で2時間、ビオチン標識検出抗体とともにインキュベートされた。
5)検出抗体とのインキュベート後、プレートはTBS−Tweenで4回洗浄され、その後、ストレプトアビジン標識西洋わさびペルオキシダーゼ(SA−HRP)がビオチン標識検出抗体に結合するように、室温で1時間、SA−HRPとインキュベートされた。
6)シグナル増幅のため、ビオチン−チラミドは、0.015%H2O2とともに5μg/mlで添加され、15分間反応させられた。
7)TBS−Tweenで6回洗浄後、SA−HRPが添加され、30分間インキュベートされた。
8)TBS−Tweenで6回洗浄後、HRP基質TMBが添加され、色が遮光下で2〜10分間発色させられた。反応は、0.5MH2SO4を添加することによって停止された。シグナルは、マイクロプレートリーダーにて450/570nmで走査された。
【0178】
図3は、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体から構成されるELISAを用いたA431細胞中の総EGFRの検出を示す。免疫検出の感度は、ヒトEGFRの細胞の組み換え細胞外ドメインに基づき、約0.25pg/穴であった。計算されたEGFR濃度は、各A431細胞あたり、約0.6pgであった。
【0179】
図4は、捕捉抗体としてのEGFRの細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化EGFRに対するビオチン標識モノクローナル抗体と、から構成されるELISAを用いた、A431細胞中のリン酸化EGFRの検出を示す。捕捉抗体の2倍の連続希釈を用いると、捕捉抗体濃度濃度が0.0625μg/mlである時に1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において1.78倍の増加があったことを示した。
【0180】
図5は、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体から構成されるELISAを用いたSKBr3細胞中の総ErbB2の検出を示す。免疫分析の検出幅は、約1000細胞と約1.37細胞の間であった。捕捉抗体濃度が、1μg/mlである時に、1.37細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において2.71倍の増加があった。
【0181】
図6は、捕捉抗体としてのErbB2の細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化ErbB2に対するモノクローナル抗体と、から構成されるELISAを用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す。免疫分析の検出幅は、約500細胞と約5細胞の間であった。捕捉抗体濃度が、1μg/mlである時に、5細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.03倍の増加があった。
【0182】
図7は、捕捉抗体及び検出抗体としてErk2に対するモノクローナル抗体から構成されるELISAを用いたSKBr3細胞中の総及びリン酸化Erk2の検出を示す。1.37細胞レベルで、総Erk2に関し、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.25倍の増加があった。同様に、1.37細胞レベルで、リン酸化Erk2に関し、バックグラウンドに対するシグナル(シグナル/ノイズ比)において3.17倍の増加があった。
【0183】
(実施例3.チラミドシグナル増幅を有する単一検出マイクロアレイELISAを用いた単一細胞検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重で、ハイスループットの単一検出マイクロアレイELISAを説明する:
1)捕捉抗体は、2倍連続希釈で16パッドFASTスライド(WhatmanInc.;Florham Park,NJ)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、80μlのビオチン標識検出抗体が、室温で2時間インキュベートされた。
5)6回洗浄後、ストレプトアビジン標識西洋わさびペルオキシダーゼ(SA−HRP)が添加され、SA−HRPがビオチン標識検出抗体に結合するように、1時間インキュベートされた。
6)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、そしてTBSで1回洗浄された。
7)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、マイクロアレイ検出器(Perkin−Elmer,Inc.;Waltham,MA)で走査された。
【0184】
図8は、捕捉抗体及び検出抗体としてEGFRの細胞外ドメインに対するモノクローナル抗体から構成されるマイクロアレイELISAを用いたA431細胞中の総EGFRの検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のEGFRを検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、EGFRが1細胞から検出され得ることを示した。捕捉抗体濃度が、0.0625mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において2.11倍の増加があった。
【0185】
図9は、捕捉抗体としてのEGFRの細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化EGFRに対するモノクローナル抗体と、から構成されるマイクロアレイELISAを用いた、A431細胞中のリン酸化EGFRの検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のリン酸化EGFRを検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、リン酸化EGFRが1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において1.33倍の増加があった。
【0186】
図10は、捕捉抗体及び検出抗体としてErBb2の細胞外ドメインに対するモノクローナル抗体から構成されるマイクロアレイELISAを用いたSKBr3細胞中の総ErBb2の検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、その希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のErBb2を検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、ErBb2が1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において15.27倍の増加があった。
【0187】
図11は、捕捉抗体としてのErBb2の細胞外ドメインに対するモノクローナル抗体と、検出抗体としてのリン酸化ErBb2に対するモノクローナル抗体と、から構成されるマイクロアレイELISAを用いた、SKBr3細胞中のリン酸化ErBb2の検出を示す。細胞数に基づいた捕捉抗体希釈曲線実験は、このマイクロアレイELISA構成が、希釈系列における様々な捕捉抗体の濃度で約1〜10000細胞中のErBb2を検出する広いダイナミックレンジを有することを示した。捕捉抗体濃度の希釈系列に基づく細胞滴定曲線実験は、リン酸化ErBb2が1細胞から検出され得ることを示した。捕捉抗体濃度が、0.125mg/mlである時に、1細胞レベルで、バックグラウンドに対するシグナル(シグナル/ノイズ比)において5.45倍の増加があった。
【0188】
(実施例4.チラミドシグナル増幅を有する近接2重検出マイクロアレイELISAを用いた単一細胞の検出)
この実施例は、希少循環細胞中の単一の伝達分子の活性状態を分析するために適した、優れたダイナミックレンジを有する、多重で、ハイスループットの近接2重検出マイクロアレイELISAを説明する:
1)捕捉抗体は、1mg/mlから0.004mg/mlまでの連続希釈で16パッドFASTスライド(WhatmanInc.)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)A431細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBS中で希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は:(1)グルコースオキシダーゼ(GO)に直接共役された抗−EGFRモノクローナル抗体;及び(2)西洋わさびペルオキシダーゼ(HRP)に直接共役されたリン酸化EGFRの細胞を認識するモノクローナル抗体、であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRを認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間のストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ(GO)結合体を用いた。リン酸化EGFRの細胞へのHRPの直接に結合された又はビオチン−ストレプトアビジン(SA)を介した結合体が用いられた。
6)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、そしてTBSで1回洗浄された。
7)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、マイクロアレイ検出器(Perkin−Elmer,Inc.)で走査された。
【0189】
図12は、近接2重検出マイクロアレイELISAと単一検出マイクロアレイELISAの比較を示す。表2は、近接2重検出マイクロアレイELISAと単一検出マイクロアレイELISAの感度を示す。各A431細胞濃度に対し、近接及び単一検出の形式でのバックグラウンドに対するシグナル(シグナル/ノイズ比)が示される。表2に示すように、近接2重検出マイクロアレイELISAはさらに、1細胞レベルで約3倍感度が増加した。
【表2】
【0190】
図13は、単一検出マイクロアレイELISAと近接2重検出マイクロアレイELISAの分析感度を示す。単一検出の形式における様々な捕捉抗体濃度でのリン酸化EGFRの滴定曲線を作製した実験は、単一の検出抗体の特異性の欠如のため、高いバックグラウンドを示した。対照的に、近接2重検出の形式における様々な捕捉抗体濃度でのリン酸化EGFRの滴定曲線を作製した実験は、2つの検出抗体間の近接度を検出することによって得られる増加した特異性のため、非常に低いバックグラウンドを示した。
【0191】
(実施例5.アドレズ可能な近接2重検出マイクロアレイを用いた複数のシグナル伝達因子の活性状態の単一細胞検出)
この実施例は、希少循環細胞における複数のシグナル伝達分子の活性状態を解析するために適した優れたダイナミックレンジを有する、多重で、ハイスループットで、アドレス可能な近接2重検出マイクロアレイを説明する:
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。印刷された捕捉抗体は、EGFR、HER2、Erk、Shc、PI3K、及びパン−サイトケラチン(pan−cytokeratin)であった。各捕捉抗体の2倍希釈系列(0.25mg/ml,0.125mg/ml、及び0.0625mg/ml)が使用され、2重及び4重のスポットが各抗体希釈に対して作製された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSで希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は、(1)グルコースオキシダーゼ(GO)に直接共役された抗EGFRモノクローナル抗体;及び(2)HRPに直接共役されたリン酸化EGFRを認識するモノクローナル抗体であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRの細胞を認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間ストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ共役体を用いた。リン酸化EGFRの細胞へのHRPの直接共役された又はビオチン−ストレプトアビジン(SA)を介した共役体のいずれかが使用された。
7)総HER2及びリン酸化タンパク質を検出するため、工程4)、5)、又は6)がEGFRを認識するモノクローナル抗体の代わりにHER2を認識するモノクローナル抗体を用いて行われた。
8)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、TBSで1回洗浄された。
9)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、そしてマイクロアレイ検出器(Perkin−Elmer,Inc.)で取り込まれた。
【0192】
図14は、アドレス可能なマイクロアレイの形式の具体的な実施の形態を示す。5つの標的が、特異的な捕捉抗体を介してアドレス可能である(例えば、EGFR、HER2、Shc、Erk、及びPI3K)。Shc,Erk、又はPI3KのEGFR又はHER2いずれかとのリン酸化複合体は、近接2重検出の形式を用いてこの分析で検出され得る。パン−サイトケラチン(PanCK)は、上皮細胞の数に対し標準化するための対照として働く。
【0193】
図15は、活性化されたA431細胞の滴定分析におけるリン酸化Shcレベルの検出を示す。アドレス可能なアレイは、EGFR及びHER2リン酸化に関する情報を同時に提供した。
【0194】
(実施例6.近接2重検出マイクロアレイのダイナミックレンジの拡張)
この実施例は、希少循環細胞におけるシグナル伝達分子の活性状態を分析するためのダイナミックレンジが、多重、ハイスループットな、近接2重検出マイクロアレイ分析における捕捉抗体の希釈系列化を行うことによって増大され得ることを説明する:
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。各捕捉抗体は、全部で9つの濃度のために連続的に2倍に希釈された(1mg/mlで開始し;0.004mg/mlで終了する)。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。このスライドは、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSに希釈された近接分析のための検出抗体80μlがスライドに添加され亜。使用された検出抗体は:(1)グルコースオキシダーゼ(GO)に直接共役された抗EGFRモノクローナル抗体;及び(2)HRPに直接共役されたリン酸化EGFRを認識するモノクローナル抗体であった。インキュベートは、室温で2時間であった。
5)代替的には、検出工程は、リン酸化EGFRを認識するモノクローナル抗体のビオチン結合体を用いた。これらの例では、6回洗浄後、1時間ストレプトアビジン−HRPとのインキュベートの追加の連続工程が含まれた。
6)代替的には、検出工程は、抗EGFR抗体のオリゴヌクレオチド抗体のオリゴヌクレオチドを介したグルコースオキシダーゼ結合体を用いた。リン酸化EGFRの細胞へのHRPの直接結合された又はビオチン−ストレプトアビジン(SA)を介した結合体のいずれかが使用された。
7)シグナル増幅のため、5μg/mlのビオチン−チラミド80μlが添加され、15分間反応させられた。このスライドは、TBS−Tweenで6回、20%DMSO/TBS−Tweenで2回、TBSで1回洗浄された。
8)80μlのSA−Alexa555が添加され、30分間インキュベートされた。このスライドは、その後、2回洗浄され、5分間乾燥され、そしてマイクロアレイ検出器(Perkin−Elmer,Inc.)で取り込まれた。
【0195】
図16は、抗EGFR捕捉抗体の希釈曲線を示す。アドレス可能な分析を用いて、リン酸化及び総EGFRの検出は、活性化されたA431細胞にとって単一細胞レベルであった。この分析のダイナミックレンジは、5対数よりも大きかった。各個々の曲線は、約2対数のダイナミックレンジを有するが、そのダイナミックレンジは、6つの情報を与える曲線からの情報が組み合わせられると、非常に高められた。
【0196】
(実施例7.抗体へのオリゴヌクレオチド共役)
この実施例は、オリゴヌクレオチド結合抗体又は酵素を生成するための共役及び品質制御過程を説明する。
【0197】
結合:
1)72塩基のオリゴヌクレオチドリンカーが5’−SH基と6−炭素スペーサーとともに合成された。
2)凍結乾燥されたオリゴヌクレオチドリンカーは、pH7.4、20mMのTris−HClに溶解される。125nmolのリンカーは、その後、室温で2時間、終濃度50mMで、0.5MのTCEP−HCl(Pierce;Rockford,IL)と処理された。反応混合物中のトリス、TCEP、及び非結合リンカーは、脱塩回転カラムを用いて除かれた。
3)結果として生じた脱保護オリゴヌクレオチドリンカーは、100μl反応容量中で、SMCCのようなヘテロ二官能性架橋剤を用いて標的タンパク質の第1アミンに結合させられた。
4)反応混合物は、室温で2時間インキュベートされた。オリゴヌクレオチド結合抗体又は酵素は、SephacrylS−200 HRカラム(GE Healthcare;Piscataway,NJ).を用いたゲルフィルターろ過を用いて精製された。
【0198】
結合条件:
1)グルコースオキシダーゼ(GO)分子が第1のオリゴヌクレオチドリンカーに結合された後、結果として生じたGO−オリゴヌクレオチドの3つの画分が精製後に回収され、10倍希釈系列で、ニトロセルロースで覆われたスライド上に印刷された。
2)IgGは、Alexa647及び第1のオリゴヌクレオチドリンカーに相補する配列を有する第2のオリゴヌクレオチドリンカーに結合された。結果として生じたAlexa647−オリゴヌクレオチドが結合した抗体は、チップ上に添加され、1倍濃度PBS緩衝液中、室温で1時間ハイブリダイズされ、そして数回洗浄された。
3)スライドは、乾燥され、そして核酸配列特異的なハイブリダイゼーションを確認するために、マイクロアレイ検出器で走査された。
4)グルコースオキシダーゼ酵素分析は、共役の過程が酵素の機能を変えていないことを確認した。
【0199】
図17は、Alexa647−オリゴヌクレオチドが結合した抗体が画分13〜15においてGO−オリゴヌクレオチドに対する最も高い結合親和性を有したことを示す。
【0200】
(実施例8.全ての及びリン酸化EGFRの同時検出のためのオリゴヌクレオチド結合抗体)
この実施例は、実施例7で記載したオリゴヌクレオチド結合体を用いた希少循環細胞中のシグナル伝達分子の活性状態を分析するための、多重で、ハイスループットなマイクロアレイ分析を説明する。
1)捕捉抗体は、16パッドFASTスライド(WhatmanInc.)上に印刷された。
2)一晩乾燥後、スライドは、Whatmanブロッキング緩衝液で遮断(ブロック)された。
3)細胞溶解物80μlが10倍連続希釈で各パッドに添加された。空井度は、室温で2時間インキュベートされた。
4)TBS−Tweenで6回洗浄後、TBS−Tween/2%BSA/1%FBSに希釈された近接分析のための検出抗体80μlがスライドに添加された。使用された検出抗体は:(1)Alexa647と、及びグルコースオキシダーゼ(GO)に共役されたオリゴヌクレオチドに相補する配列から構成されるオリゴヌクレオチドリンカーと、に直接共役された抗−EGFRモノクローナル抗体;及び(2)西洋わさびペルオキシダーゼ(HRP)に直接共役されたリン酸化EGFRを認識するモノクローナル抗体、であった。Alexa647−オリゴヌクレオチド結合抗EGFR抗体は、図18に示された複合体を形成するため、GO−オリゴヌクレオチドと接触した。過剰な非結合試薬はTBS−Tweenで6回洗浄することによって除去された。
5)グルコースは、その後、チラミド−Alexa555とともに反応に添加された。
6)総EGFR量は、Alexa647−オリゴヌクレオチド−結合抗EGFR抗体の直接結合によって検出された。リン酸化EGFRは、HRP共役抗p−EGFR抗体へのGO−オリゴヌクレオチドの隣接共役によって検出され、チラミドシグナル増幅によって可視化された。
7)スライドは、マイクロアレイ検出器(Perkin−Elmer,Inc.)で、Alexa647及びAlexa555に特異的なレーザーで走査された。
【0201】
図19は、総EGFR及びリン酸化EGFRの同時検出を示す。総EGFR及びリン酸(t−EGFR)は、少なくとも10細胞から直接結合分析によって検出され、そしてリン酸化EGFR(p−EGFR)は、1細胞から検出された。10e5のp−EGFR分子が近接シグナル増幅方法で検出された。p−EGFRの検出限界は、近接分析形式を用いることによって100倍以上増加した。
【0202】
(実施例9.乳癌患者における循環腫瘍細胞(CirculatingTumor Cell)(CTC)シグナル伝達の検出)
マイクロアレイの作製及び処理工程は、Chanら、Nat.Med.,10:1390−1396(2004)に記載の方法から適応される。シグナル伝達因子EGFR、ErbB2、ErbB3、ErbB4、IGF−1R、Akt、Erk、p70S6K、Bad、Rsk、Mek、cSrc、サイトケラチン、チューブリン、β−アクチン(β−actin)に対する抗体(1mg/ml)、及び抗−マウス抗体(陽性対照)(4/4アレイ)は、FAST(登録商標)スライド(WhatmanInc.;Florham Park,NJ)に抗体をスポットするための固相スポッティングピンで適合された密接印刷ロボットマイクロアレイ作製機(Bio−Rad Laboratories;Hercules,CA)を用いて384穴ポリプロピレンプレート(50μl/穴)に移される。8分割されたパッドで覆われたスライドが使用される。印刷後、スライドは、3%カゼイン溶液で遮断(ブロック)される。スライドは、使用前に乾燥条件下で少なくとも一晩保存される。
【0203】
患者選択基準、試料準備方法、及び調査設計は、疑わしい乳癌の女性における循環腫瘍細胞を評価する開示された調査(例えば、Wulfingら、Clin.CancerRes.,12:1715−1720(2006);及びReinholzら、Clin.Cancer Res.,11:3722−3732(2005)を参照)から適応される。画像化で検出された胸部異常を有し、胸部生検を受ける予定の女性は、この調査を提案される。原発性の乳癌と診断された、少なくとも42の患者が含まれる。少なくとも35の患者は、最初の診断の時に顕性転移の兆候を有していない。少なくとも7の患者は、診断時に遠部の転移を有し、陽性参照集団と見なされる。全ての患者は、以前に癌の病歴を有していない。年齢及び治療情報(例えば、外科治療、化学療法、放射線治療、内分泌療法、等)は、各患者について収集される。約20mlの血液が各患者から回収され、全ての血液試料は、一意的な識別番号を割り当てられる。全ての分析は、生検の結果を知らない臨床検査医によって行われた。
【0204】
末梢血液(18ml)が、AccuspinHistopaque−1077システム(Sigma Aldrich;St.Louis,MO)に添加され、Beckman CS−6R卓上遠心機(Beckman Instruments;PaloAlto,CA)を用いて1500rpmで10分間、遠心分離される。単核細胞層が除去され、PBSで2回洗浄され、PBS/0.1%ウシ血清アルブミンで1mlに希釈される。上皮細胞は、製造元の指示に従い、ダイナビーズ上皮濃縮(DynabeadsEpithelial Enrich)キット(Dynal AS;Oslo,Norway)を用いて磁気ビーズに付着されたBer−EP4に対する抗体を用いた免疫磁気捕獲によって濃縮される。細胞は、1時間、揺り動かされている間、20mlの容量で1〜107ビーズと混合される。Ber−EP4抗体は、扁平上皮細胞の表層、幹細胞、及び壁細胞を除く、上皮細胞の表面及び細胞質の2つの糖タンパク質を認識する。懸濁液は、少なくとも6分間磁石上に置かれ、その上清は、注意深く除去される。磁気ビーズに付着した細胞は、1mlのPBS/0.1%ウシ血清アルブミンで3回洗浄される。増殖因子TGF−α(100nM)、ヘレグリン(100nM)、及びIGF(100nM)は、細胞に添加され、37℃で5分間インキュベートされる。細胞は、遠心分離され、キットで供給された溶解結合緩衝液で溶解される。溶解された細胞懸濁(付着したビーズを含む)は、処理されるまで80℃で保存される。
【0205】
分析方法1:溶解物(40μl)は、アレイに添加され、一晩インキュベートされ、そして、洗浄緩衝液で3回洗浄される。シグナル伝達因子各々に対する、HRP標識(例えば、Kuhlmann,ImmunoEnzyme Techniques,Verlag Chemie,Weinheim,pp1−162(1984)に記載されたように共役された)抗−リン酸抗体及びグルコースオキシダーゼで標識された(上記のKuhlmannの文献に記載されたように結合された)抗−総抗体(即ち活性状況非依存的)がアレイに添加され、2時間インキュベートされ、そして洗浄緩衝液で3回洗浄される。チラミド試薬(MolecularProbes)及びグルコースが添加され、そして反応は、1時間行われ、そして3回洗浄される。アレイは、30分間、ストレプトアビジン−HRPとインキュベートされ、洗浄され、増感ルミノール(MolecularProbes)を用いて顕わにさせられる。シグナルは、CCDカメラを用いて検出される。
【0206】
分析方法2:2,4−ジニトロフェノール(DNP)に対する抗体(lmg/ml)は、FAST(登録商標)スライド(WhatmanInc.;Florham Park,NJ)に抗体をスポットするための固相スポッティングピンで適合された密接印刷ロボットマイクロアレイ作製機(Bio−Rad Laboratories;Hercules,CA)を用いて384穴ポリプロピレンプレート(5μl/穴)に移される。8分割されたパッドで覆われたスライドが使用される。印刷後、スライドは、3%カゼイン溶液で遮断(ブロック)される。スライドは、使用前に乾燥条件下で少なくとも一晩保存される。
【0207】
溶解物(40μl)は、上記のシグナル伝達因子各々に対する総DNP標識抗体及びオリゴ(Oligo)及びAlexaFluor(商標登録)647で標識された抗−リン酸抗体と混合され、オリゴ及びAlexaFluor(商標登録)647で標識された抗−総抗体は、抗−DNP抗体に添加され、一晩インキュベートされ、洗浄緩衝液で3回洗浄される。2,4−ジニトロリジン(MolecularProbes)は、抗−DNP抗体から免疫複合体を放出するために添加される。放出された免疫複合体は、ジップコードアレイ(例えば、Keramasら、Lab Chip,4:152−158(2004);及びDelrio−Lafreniereら、Diagn.Microbiol.Infect.Dis.,48:23−31(2004)を参照)に添加され、一晩インキュベートされる。アレイは、3回洗浄され、処理されたスライドは、GenePix4000Aマイクロアレイスキャナー(AxonScanner)を用いて10ミクロンの解像度で走査される。
【0208】
(実施例10.乳癌患者における循環内皮細胞(CirculatingEndothelial Cell)(CEC)及び循環内皮前駆細胞(Circulating Endothelial Precursor Cell)(CEP)の検出)
CEC及びCEP細胞が、磁気ダイナビーズに付着したモノクローナル抗体P1H12又はCD146を用いた免疫磁気捕獲によって濃縮されることを除いて、実施例9で記載された同一の患者試料、使用準備、及び分析方法が使用される。マイクロアレイは、配置された抗体が以下の分析物に特異的であることを除いて、実施例9に記載の方法を用いて作製され、処理される:VEGFR1,VEGFR2,VEGFR3,TIE1,TIE2,PDGFR−α,PDGFR−β,FGFR1,FGFR2,Akt,Erk,p70S6K,Rsk,cSrc、及びβ−アクチン。実施例9に記載の分析方法は、CEC及びCEPシグナル伝達を検出するために用いられる。
【0209】
本明細書中で引用された全ての刊行物及び特許出願は、個々の刊行物又は特許出願それぞれが、特異的に及び個々に参照によって取り込まれると示されるように、本明細書中で参照として取り込まれる。上述の発明は、理解の明確化の目的のため、図示及び例によって、多少詳しく述べられているが、この発明の示唆を踏まえて、添付の請求の範囲の精神又は範囲から離れることなく任意の変化及び改変がそれになされてもよいことは、当業者には容易に明らかである。
【0210】
さらなる実施形態は以下の通りである。
1.優れたダイナミックレンジを有する、多重で、ハイスループットな免疫分析を行うための方法であって、
前記方法は、
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、前記細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、
前記捕捉抗体が、固相担体に拘束されている、ことを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するために、前記複数の捕捉された分析物を、前記対応する分析物に特異的な検出抗体とインキュベートする工程であって、
前記検出抗体は、
(1)促進部分で標識された複数の活性状態非依存抗体、及び
(2)シグナル増幅対の第一の構成因子で標識された複数の活性状態依存抗体、
から構成され、
前記促進部分は、前記シグナル増幅対の前記第一の構成因子に方向付けられ且つ前記第一の構成因子と反応する酸化剤を生成する、ことを特徴とする工程;
(c)増幅されたシグナルを生成するために、前記複数の検出可能な捕捉された分析物を前記シグナル増幅対の第二の構成因子とインキュベートする工程;及び
(d)前記シグナル増幅対の前記第一及び第二の構成因子から生じた前記増幅されたシグナルを検出する工程、
を備える、ことを特徴とする免疫分析方法。
2.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態1に記載の免疫分析方法。
3.前記細胞が免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態2に記載の免疫分析方法。
4.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態3に記載の免疫分析方法。
5.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態3に記載の免疫分析方法。
6.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態3に記載の免疫分析方法。
7.前記単離された細胞が、増殖因子の刺激の前に抗癌剤とインキュベートされる、ことを特徴とする実施形態6に記載の免疫分析方法。
8.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態7に記載の免疫分析方法。
9.前記単離された細胞が、前記細胞抽出物を作製するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態6に記載の免疫分析方法。
10.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態1に記載の免疫分析方法。
11.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態1に記載の免疫分析方法。
12.前記活性状態非依存抗体がさらに、検出可能な部分を備える、ことを特徴とする実施形態1に記載の免疫分析方法。
13.前記検出可能な部分が、フルオロフォアである、ことを特徴とする実施形態12に記載の免疫分析方法。
14.前記検出可能な部分の量が、一つ又はそれ以上の前記分析物の量と相関関係にある、ことを特徴とする実施形態12に記載の免疫分析方法。
15.前記活性状態非依存抗体が、チャネリング部分で直接標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
16.前記活性状態非依存抗体が、前記活性状態非依存抗体に共役されたオリゴヌクレオチドと前記チャネリング部分に共役された相補オリゴヌクレオチドとの間のハイブリダイゼーションを介して前記チャネリング部分で標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
17.前記活性状態依存抗体が、前記シグナル増幅対の第一の構成因子で直接標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
18.前記活性状態依存抗体が、前記活性状態依存抗体に共役された結合対の第一の構成因子と前記シグナル増幅対の第一の構成因子に共役された前記結合対の第二の構成因子との間の結合を介して前記シグナル増幅対の第一の構成因子で標識される、ことを特徴とする実施形態1に記載の免疫分析方法。
19.前記結合対の前記第一の構成因子がビオチンである、ことを特徴とする実施形態18に記載の免疫分析方法。
20.前記結合対の前記第二の構成因子がストレプトアビジンである、ことを特徴とする実施形態18に記載の免疫分析方法。
21.前記チャネリング部分がグルコースオキシダーゼである、ことを特徴とする実施形態1に記載の免疫分析方法。
22.前記酸化剤が過酸化水素(H2O2)である、ことを特徴とする実施形態21に記載の免疫分析方法。
23.前記シグナル増幅対の前記第一の構成因子がペルオキシダーゼである、ことを特徴とする実施形態22に記載の免疫分析方法。
24.前記ペルオキシダーゼが西洋わさびペルオキシダーゼ(HRP)である、ことを特徴とする実施形態23に記載の免疫分析方法。
25.前記シグナル増幅対の前記第二の構成因子がチラミド試薬である、ことを特徴とする実施形態23に記載の免疫分析方法。
26.前記チラミド試薬がビオチン−チラミドである、ことを特徴とする実施形態25に記載の免疫分析方法。
27.前記増幅されたシグナルが、活性型チラミドを生産するための前記ビオチン−チラミドのペルオキシダーゼ酸化によって生成される、ことを特徴とする実施形態26に記載の免疫分析方法。
28.前記活性型チラミドが直接検出される、ことを特徴とする実施形態27に記載の免疫分析方法。
29.前記活性型チラミドが、シグナル検出試薬の添加で検出される、ことを特徴とする実施形態27に記載の免疫分析方法。
30.前記シグナル検出試薬が、ストレプトアビジン標識フルオロフォアである、ことを特徴とする実施形態29に記載の免疫分析方法。
31.前記シグナル検出試薬が、ストレプトアビジン標識ペルオキシダーゼ及び発色試薬の組み合わせである、ことを特徴とする実施形態29に記載の免疫分析方法。
32.前記発色試薬が、3,3’,5,5’−テトラメチルベンジジン(TMB)である、ことを特徴とする実施形態31に記載の免疫分析方法。
33.細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体から構成される、優れたダイナミックレンジを有するアレイであって、前記捕捉抗体が固相担体に拘束される、ことを特徴とするアレイ。
34.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態33に記載のアレイ。
35.前記細胞が、免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態34に記載のアレイ。
36.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態35に記載のアレイ。
37.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態35に記載のアレイ。
38.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態35に記載のアレイ。
39.前記単離された細胞が、増殖因子の刺激の前に、抗癌剤とインキュベートされる、ことを特徴とする実施形態38に記載のアレイ。
40.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態39に記載のアレイ。
41.前記単離された細胞が、前記細胞抽出物を作製するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態38に記載のアレイ。
42.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態33に記載のアレイ。
43.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態33に記載のアレイ。
44.各希釈系列が少なくとも3つの下降する捕捉抗体濃度から構成される、ことを特徴とする実施形態33に記載のアレイ。
45.各希釈系列が少なくとも6つの下降する捕捉抗体濃度から構成される、ことを特徴とする実施形態33に記載のアレイ。
46.各希釈系列における前記捕捉抗体が少なくとも2倍で連続的に希釈される、ことを特徴とする実施形態33に記載のアレイ。
47.前記一つ又はそれ以上の分析物が、約1細胞中で検出される、ことを特徴とする実施形態33に記載のアレイ。
48.優れたダイナミックレンジを有し、多重で、ハイスループットな免疫分析を行うための方法であって、
前記方法は、
(a)複数の捕捉された分析物を形成するために、細胞抽出物を、前記細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体とインキュベートする工程であって、前記捕捉抗体が固相担体に拘束されている、ことを特徴とする工程;
(b)複数の検出可能な捕捉された分析物を形成するために、前記複数の捕捉された分析物を、前記対応する分析物に特異的な検出抗体とインキュベートする工程;
(c)増幅されたシグナルを生成するために、前記複数の検出可能な捕捉された分析物を、シグナル増幅対の第一及び第二の構成因子とインキュベートする工程;及び
(d)前記シグナル増幅対の前記第一及び第二の構成因子から生じた増幅されたシグナルを検出する工程、
を備える、ことを特徴とする免疫分析方法。
49.前記細胞抽出物が、固形腫瘍の循環細胞の抽出物から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
50.前記細胞が、免疫磁気分離法によって患者試料から単離される、ことを特徴とする実施形態49に記載の免疫分析方法。
51.前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態50に記載の免疫分析方法。
52.前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態50に記載の免疫分析方法。
53.前記単離された細胞が、インビトロにおいて増殖因子で刺激される、ことを特徴とする実施形態50に記載の免疫分析方法。
54.前記単離された細胞が、増殖因子の刺激の前に抗癌剤とインキュベートされる、ことを特徴とする実施形態53に記載の免疫分析方法。
55.前記抗癌剤が、モノクローナル抗体、チロシンキナーゼ阻害剤、免疫抑制剤、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態54に記載の免疫分析方法。
56.前記単離された細胞が、前記細胞抽出物を生成するために、増殖因子の刺激の後に、溶解される、ことを特徴とする実施形態53に記載の免疫分析方法。
57.前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
58.前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせから構成される集団から選択される、ことを特徴とする実施形態48に記載の免疫分析方法。
59.前記検出抗体が、結合対の第一の構成因子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
60.前記結合対の前記第一の構成因子がビオチンである、ことを特徴とする実施形態59に記載の免疫分析方法。
61.前記シグナル増幅対の前記第一の構成因子が、前記結合対の第二の構成因子から構成される、ことを特徴とする実施形態48に記載の免疫分析方法。
62.前記結合対の前記第二の構成因子が、ストレプトアビジンである、ことを特徴とする実施形態61に記載の免疫分析方法。
63.前記シグナル増幅対の前記第一の構成因子がペルオキシダーゼであることを特徴とする実施形態61に記載の免疫分析方法。
64.前記ペルオキシダーゼが西洋わさびペルオキシダーゼ(HRP)である、ことを特徴とする実施形態63に記載の免疫分析方法。
65.前記シグナル増幅対の前記第二の構成因子がチラミド試薬である、ことを特徴とする実施形態63に記載の免疫分析方法。
66.前記チラミド試薬がビオチン−チラミドである、ことを特徴とする実施形態65に記載の免疫分析方法。
67.前記増幅されたシグナルが、活性型チラミドを作製するための前記ビオチン−チラミドのペルオキシダーゼ酸化によって生成される、ことを特徴とする実施形態66に記載の免疫分析方法。
68.前記活性型チラミドが直接検出される、ことを特徴とする実施形態67に記載の免疫分析方法。
69.前記活性型チラミドが、シグナル検出試薬の添加で検出される、ことを特徴とする実施形態67に記載の免疫分析方法。
70.前記シグナル検出試薬が、ストレプトアビジン標識フルオロフォアである、ことを特徴とする実施形態69に記載の免疫分析方法。
71.前記シグナル検出試薬が、ストレプトアビジン標識ペルオキシダーゼ及び発色試薬の組み合わせである、ことを特徴とする実施形態69に記載の免疫分析方法。
72.前記発色試薬が、3,3’,5,5’−テトラメチルベンジジン(TMB)である、ことを特徴とする実施形態71に記載の免疫分析方法。
【特許請求の範囲】
【請求項1】
抗体に基づくアレイ方法を実行するためのキットであって、
(a)固相担体上に拘束された複数の捕捉抗体の希釈系列;
(b)活性状態非依存抗体及び活性状態依存抗体を含む複数の検出抗体;及び
(c)固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書
を含む、キット。
【請求項2】
前記活性状態非依存抗体が、検出可能な部分をさらに備え、前記検出可能な部分が、フルオロフォアである、請求項1記載のキット。
【請求項3】
前記活性状態非依存抗体が、グルコースオキシダーゼで直接標識される、請求項1に記載のキット。
【請求項4】
前記キットが、シグナル増幅対の第一及び第二の構成因子をさらに備える、請求項1に記載のキット。
【請求項5】
前記活性状態依存抗体が、前記シグナル増幅対の第一の構成因子で直接標識される、請求項4に記載のキット。
【請求項6】
複数の活性状態非依存抗体が、グルコースオキシダーゼで直接標識され、複数の活性状態依存抗体が、シグナル増幅対の第一の構成因子で直接標識され、前記シグナル増幅対の第一の構成因子が、ペルオキシダーゼであり、グルコースオキシダーゼが過酸化水素(H2O2)を産生し、過酸化水素は前記シグナル増幅対の第一の構成因子に向かい且つ反応する、請求項5に記載のキット。
【請求項7】
前記アレイは、細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体を含む、優れたダイナミックレンジを有する、請求項1に記載のキット。
【請求項8】
前記細胞抽出物が、固形腫瘍の循環細胞の抽出物を含む、請求項7に記載のキット。
【請求項9】
前記細胞が、免疫磁気分離法によって患者試料から単離される、請求項8に記載のキット。
【請求項10】
前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせからなる群から選択される、請求項9に記載のキット。
【請求項11】
前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせからなる群から選択される、請求項9に記載のキット。
【請求項12】
前記単離された細胞が、インビトロにおいて増殖因子で刺激される、請求項9に記載のキット。
【請求項13】
前記単離された細胞が、増殖因子の刺激の後に溶解されて、前記細胞抽出物を生成する、請求項12に記載のキット。
【請求項14】
前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子を含む、請求項7に記載のキット。
【請求項15】
前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせからなる群から選択される、請求項7に記載のキット。
【請求項1】
抗体に基づくアレイ方法を実行するためのキットであって、
(a)固相担体上に拘束された複数の捕捉抗体の希釈系列;
(b)活性状態非依存抗体及び活性状態依存抗体を含む複数の検出抗体;及び
(c)固形腫瘍の循環細胞の複数のシグナル伝達分子の活性状態を検出するためにこのキットを用いる方法のための使用説明書
を含む、キット。
【請求項2】
前記活性状態非依存抗体が、検出可能な部分をさらに備え、前記検出可能な部分が、フルオロフォアである、請求項1記載のキット。
【請求項3】
前記活性状態非依存抗体が、グルコースオキシダーゼで直接標識される、請求項1に記載のキット。
【請求項4】
前記キットが、シグナル増幅対の第一及び第二の構成因子をさらに備える、請求項1に記載のキット。
【請求項5】
前記活性状態依存抗体が、前記シグナル増幅対の第一の構成因子で直接標識される、請求項4に記載のキット。
【請求項6】
複数の活性状態非依存抗体が、グルコースオキシダーゼで直接標識され、複数の活性状態依存抗体が、シグナル増幅対の第一の構成因子で直接標識され、前記シグナル増幅対の第一の構成因子が、ペルオキシダーゼであり、グルコースオキシダーゼが過酸化水素(H2O2)を産生し、過酸化水素は前記シグナル増幅対の第一の構成因子に向かい且つ反応する、請求項5に記載のキット。
【請求項7】
前記アレイは、細胞抽出物中の一つ又はそれ以上の分析物に特異的な、複数の希釈系列の捕捉抗体を含む、優れたダイナミックレンジを有する、請求項1に記載のキット。
【請求項8】
前記細胞抽出物が、固形腫瘍の循環細胞の抽出物を含む、請求項7に記載のキット。
【請求項9】
前記細胞が、免疫磁気分離法によって患者試料から単離される、請求項8に記載のキット。
【請求項10】
前記患者試料が、全血、血清、血漿、尿、痰、気管支洗浄液、涙、乳頭吸引液、リンパ液、唾液、細針吸引物、及びそれらの組み合わせからなる群から選択される、請求項9に記載のキット。
【請求項11】
前記単離された細胞が、循環腫瘍細胞、循環内皮細胞、循環内皮前駆細胞、癌幹細胞、及びそれらの組み合わせからなる群から選択される、請求項9に記載のキット。
【請求項12】
前記単離された細胞が、インビトロにおいて増殖因子で刺激される、請求項9に記載のキット。
【請求項13】
前記単離された細胞が、増殖因子の刺激の後に溶解されて、前記細胞抽出物を生成する、請求項12に記載のキット。
【請求項14】
前記一つ又はそれ以上の分析物が、複数のシグナル伝達分子を含む、請求項7に記載のキット。
【請求項15】
前記固相担体が、ガラス、プラスチック、チップ、ピン、フィルター、ビーズ、紙、膜、繊維束、及びそれらの組み合わせからなる群から選択される、請求項7に記載のキット。
【図1】

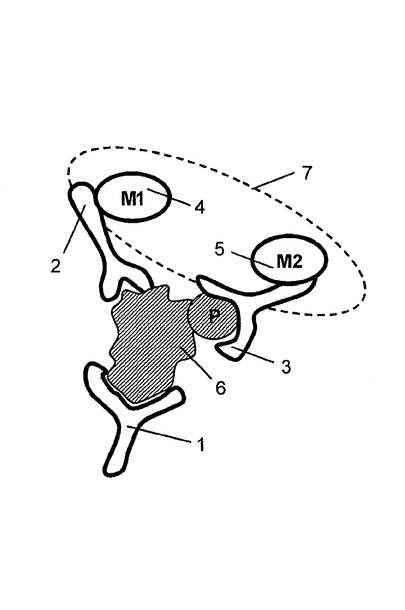
【図2】
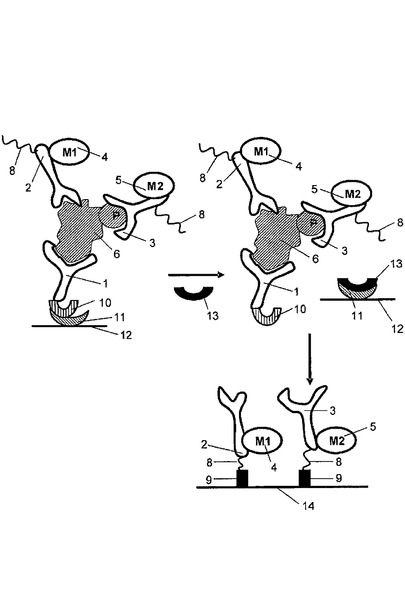
【図3】
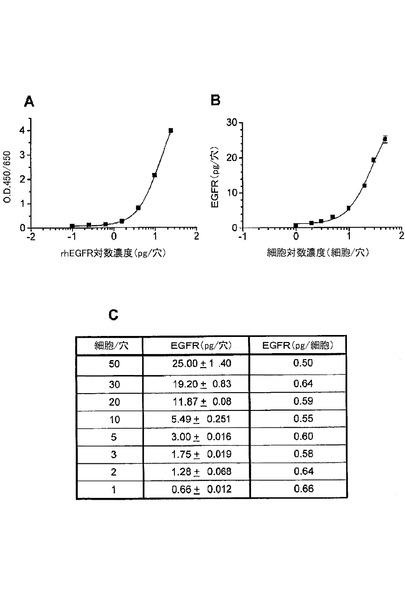
【図4】
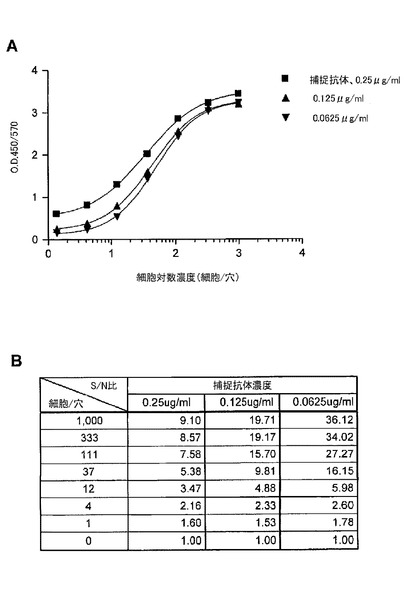
【図5】
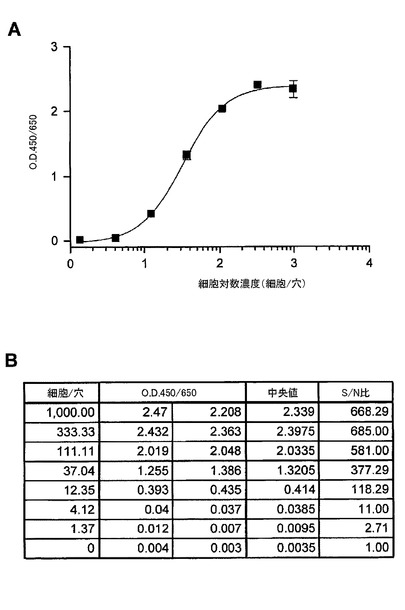
【図6】
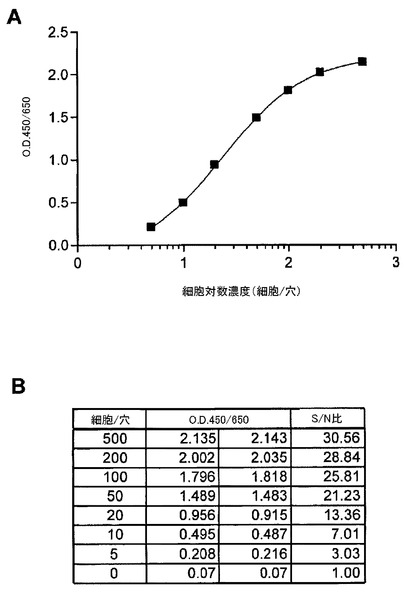
【図7】
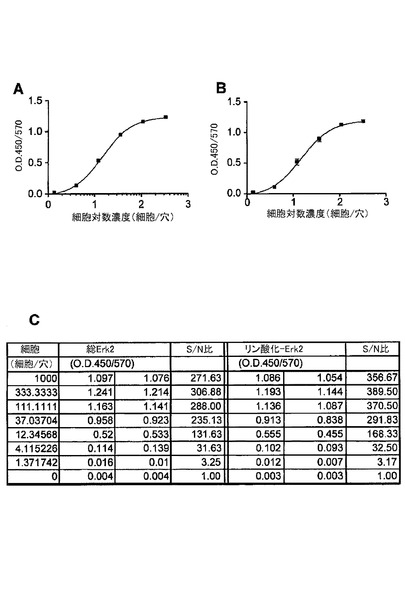
【図8】
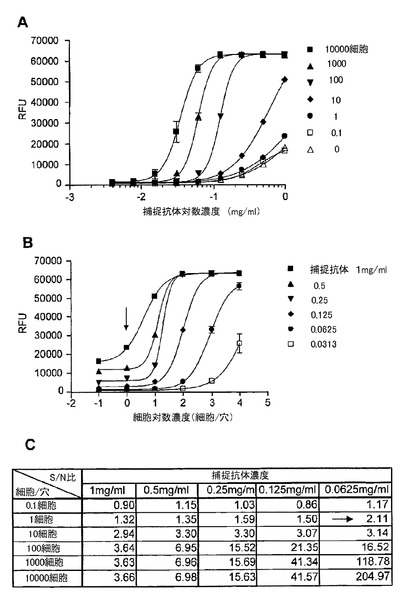
【図9】
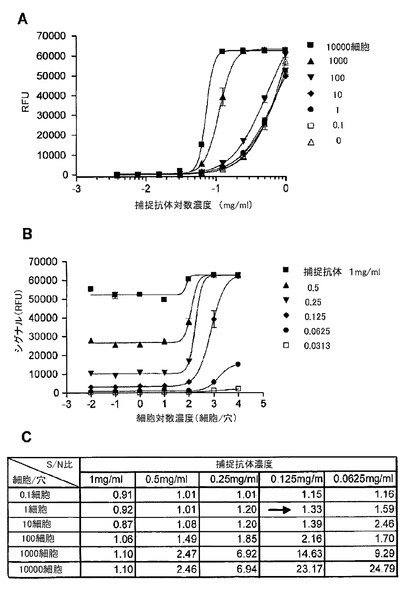
【図10】
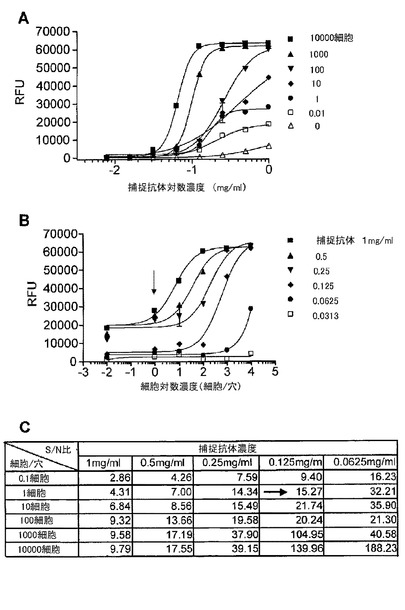
【図11】
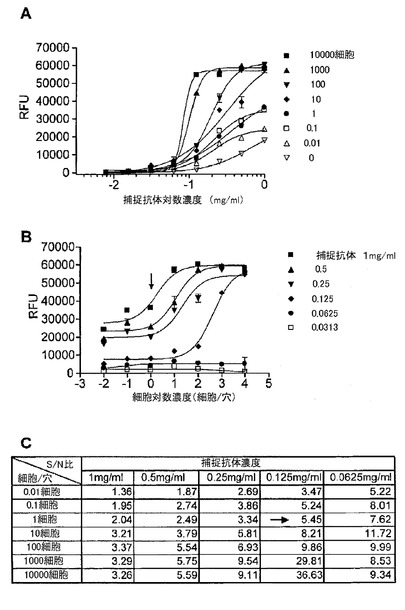
【図12】
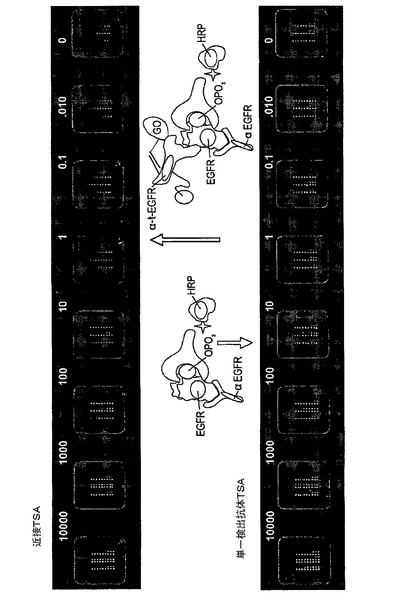
【図13】
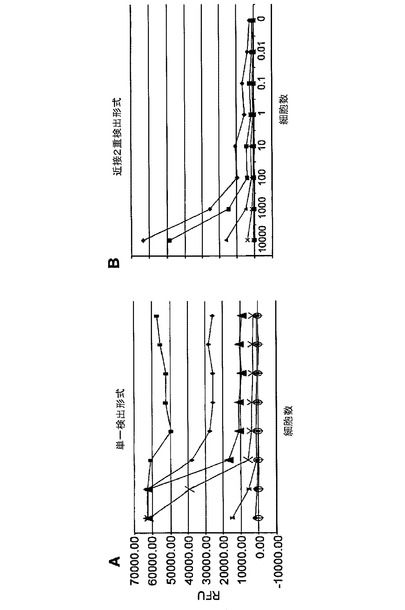
【図14】
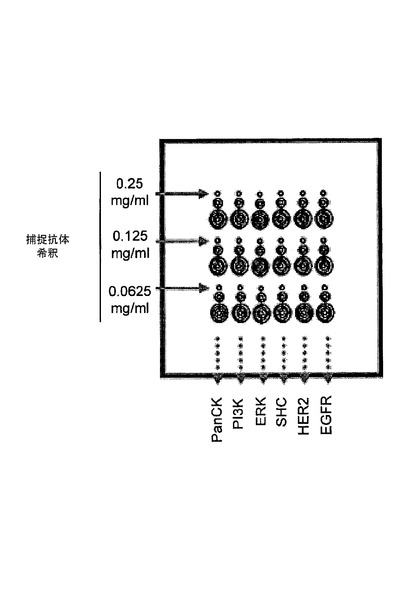
【図15】
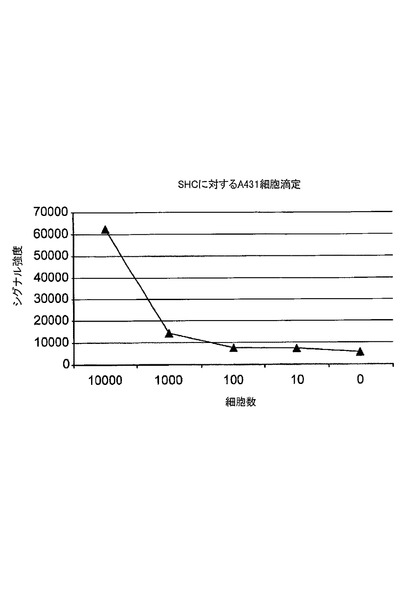
【図16】
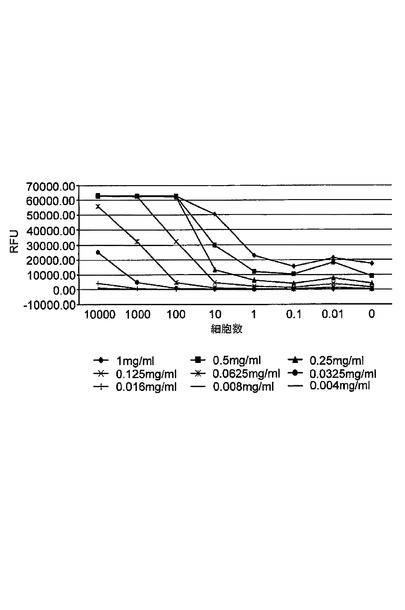
【図17】
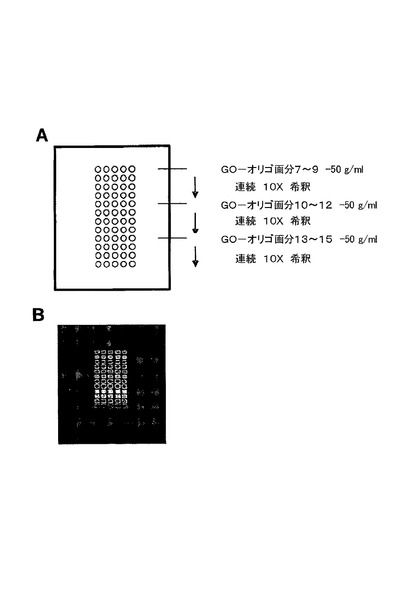
【図18】
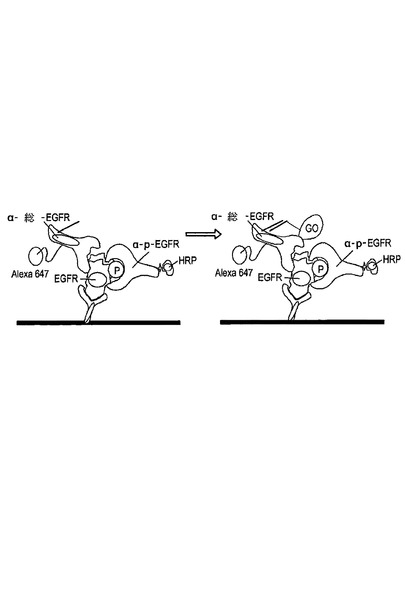
【図19】
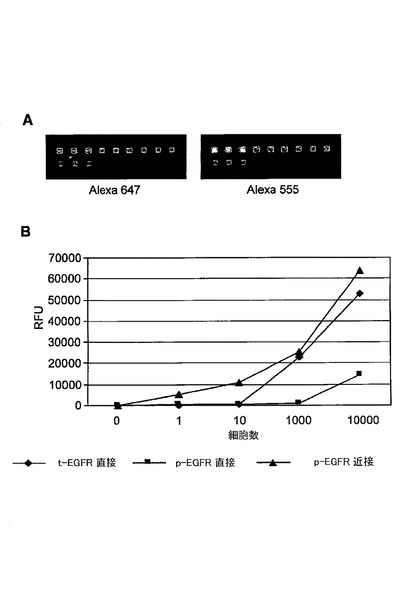
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2013−68627(P2013−68627A)
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−266434(P2012−266434)
【出願日】平成24年12月5日(2012.12.5)
【分割の表示】特願2009−529389(P2009−529389)の分割
【原出願日】平成19年9月20日(2007.9.20)
【出願人】(599132904)ネステク ソシエテ アノニム (637)
【Fターム(参考)】
【公開日】平成25年4月18日(2013.4.18)
【国際特許分類】
【出願番号】特願2012−266434(P2012−266434)
【出願日】平成24年12月5日(2012.12.5)
【分割の表示】特願2009−529389(P2009−529389)の分割
【原出願日】平成19年9月20日(2007.9.20)
【出願人】(599132904)ネステク ソシエテ アノニム (637)
【Fターム(参考)】
[ Back to top ]