
延長されたinvivo半減期を有する第VIIa因子−(ポリ)シアル酸結合体
【課題】延長されたin vivo半減期を有する第VIIa因子−(ポリ)シアル酸結合体を提供すること。
【解決手段】本発明は、血漿または組換え第VIIa因子(FVIIa)またはその生物学的に活性な誘導体、1〜4のシアル酸ユニットからなる鎖に結合される前記FVIIaまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトを提供する。ここで、タンパク質性コンストラクトのin vivo半減期は、1〜4のシアル酸ユニットからなる鎖が欠けているFVIIaまたはその誘導体と比較すると、哺乳類(特にヒト)の血中で実質的に延びている。
【解決手段】本発明は、血漿または組換え第VIIa因子(FVIIa)またはその生物学的に活性な誘導体、1〜4のシアル酸ユニットからなる鎖に結合される前記FVIIaまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトを提供する。ここで、タンパク質性コンストラクトのin vivo半減期は、1〜4のシアル酸ユニットからなる鎖が欠けているFVIIaまたはその誘導体と比較すると、哺乳類(特にヒト)の血中で実質的に延びている。
【発明の詳細な説明】
【技術分野】
【0001】
この出願は、2006年12月15日に出願された、米国仮出願第60/875,217号に対する優先権を主張する。
【0002】
発明の分野
本発明は、1〜4のシアル酸ユニットの鎖を含む炭水化物部分に結合される凝固第VIIa因子(FVIIa)を含むタンパク質性コンストラクトに関する。本発明は、さらに、血液凝固タンパク質(特に、少なくともFVIIa、第VIII因子(FVIII)、および第IX因子(FIX)の機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において血中のFVIIa)のin vivo半減期を延ばす方法に関する。
【背景技術】
【0003】
発明の背景
血液凝固カスケードは、内因性経路、外因性経路、および共通経路の3つの異なるセグメントに分けられる。(非特許文献1)。血液凝固カスケードは、一連のセリンプロテアーゼ酵素(チモーゲン)およびタンパク質補助因子に関わる。必要な場合、不活性チモーゲン前駆体は活性型に転換され、それは、結果的に血液凝固カスケードで次の酵素に転換する。
【0004】
内因性経路は凝固第VIII因子、IX、X、XI、およびXIIを必要とする。内因性経路の開始は、プレカリクレイン、高分子量キニノーゲン、第XI因子(FXI)、および第XII因子(FXII)が負の電荷をもつ表面に曝される場合、起こる。血小板から分泌されるカルシウムイオンおよびリン脂質も必要とされる。
【0005】
外因性経路は、血管の血管腔が損傷を受けた場合、開始される。膜糖タンパク質組織因子が曝され、循環する第VII因子(FVII)に、次いで既存する少量のその活性化型FVIIaに結合する。この結合は、FVIIのFVIIaへの完全転換を促進し、続いてカルシウムおよびリン脂質の存在下で第IX因子(FIX)の第IXa因子(FIXa)への転換および第X因子(FX)の第Xa因子(FXa)への転換を促進する。FVIIaの組織因子との会合は、基質(FXおよびFIX)に対するFVIIの結合部位を近接近させることによって、および立体構造変化を誘発することによってタンパク質分解活性を増進させ、それによってFVIIaの酵素活性が増進する。外因性経路によるFX活性化の速度は、FIXa、FVlIIa、リン脂質、およびカルシウムイオンの(内因性)経路によって達成される速度の約50分の一の速さである。
【0006】
FXの活性化は、これら2つの経路の共通点である。リン脂質およびカルシウムとともに、第Va因子(FVa)および第Xa因子は、プロトロンビンをトロンビンに転換させる(プロトロンビナーゼ複合体)。次いでそれはフィブリノーゲンを切断してフィブリンモノマーを形成する。フィブリンモノマーは、重合してフィブリン鎖を形成する。第XIIIa因子(FXIIIa)は、これらのフィブリン鎖を互いに共有結合させて、剛性のメッシュを形成する。
【0007】
FVIIのFVIIaへの転換も、トロンビン、FIXa、FXa、第XIa因子(FXIa)、および第XIIa因子(FXIIa)を含むいくつかのプロテアーゼによって触媒される。血液凝固カスケードの初期相を抑制するために、組織因子経路インヒビターはFVIIa/組織因子/FXa産物複合体を標的にする。
【0008】
FVII(安定因子またはプロコンバーチンとしても公知である)は、止血および凝固で中心的役割を有する、ビタミンK依存性セリンプロテアーゼ糖タンパク質である(非特許文献2)。
【0009】
FVIIは肝臓で合成されて、48kDの1本鎖糖タンパク質として分泌される。FVIIaは、ビタミンK依存性セリンプロテアーゼ糖タンパク質のすべてと、類似するタンパク質ドメイン構造を共有する。前記タンパク質ドメイン構造は、タンパク質の脂質膜との相互作用に関与する9〜12の残基を有するアミノ末端γ−カルボキシグルタミン酸(Gla)ドメイン、カルボキシ末端セリンプロテアーゼドメイン(触媒ドメイン)、および組織因子との相互作用を調整するカルシウムイオン結合部位を含有する2つの上皮増殖因子様ドメインからなる。
【0010】
γ−グルタミルカルボキシラーゼは、分子のアミノ末端部分でGla残基のカルボキシル化を触媒する。カルボキシラーゼは、その作用のために還元型ビタミンKに依存し、それはエポキシド型に酸化される。ビタミンKエポキシド還元酵素は、ビタミンKのエポキシド型を還元型に再変換するために必要である。
【0011】
FVIIの主要部分は、チモーゲン型で血漿中で循環し、この型の活性化は、アルギニン152とイソロイシン153との間のペプチド結合の切断をもたらす。結果として生じる活性化FVIIaは、NH2由来軽鎖(20kD)および単一のジスルフィド結合(Cys135〜Cys262)を介して連結されるCOOH末端由来重鎖(30kD)からなる。軽鎖は膜結合Glaドメインを含有し、重鎖は触媒ドメインを含有する。
【0012】
遺伝因子および環境因子によって決定されるFVIIの血漿中濃度は、約0.5mg/mLである(非特許文献3)。FVIIの異なる遺伝子型は、FVIIの平均レベルで数倍の相違をもたらすことができる。血漿中FVIIレベルは、健常な女性では妊娠中に上昇し、また年齢とともに上昇し、女性および高トリグリセリド血症の人においてはより高い。FVIIは、すべての凝血原因子のなかでその半減期が最も短い(3〜6時間)。FVIIaの血漿中の平均濃度は、健常な個体で3.6ng/mLであり、かつFVIIaの循環半減期は、他の凝固因子と比較すると相対的に長い(2.5時間)。
【0013】
遺伝性FVII欠乏は、稀な常染色体劣性遺伝出血性障害であり、その有病率は一般人口50万人あたり1症例と推定される(非特許文献4)。インヒビターからの後天性FVII欠乏も非常に稀である。症例は、また、薬物(例えばセファロスポリン、ペニシリン、および経口抗凝固剤)に関連して発症するFVII欠乏が報告されている。さらにまた、後天性FVIl欠乏は、自然発症的に、または他の状態(例えば骨髄腫、敗血症、再生不良性貧血)とともに、インターロイキン−2および抗胸腺細胞グロブリン療法で発症することが報告されている。
【0014】
補充療法は、FVII欠乏症の患者に対する処置の中心である(非特許文献5)。これは、新鮮凍結血漿(FFP)、プロトロンビン複合体濃度物(PCC)、または血漿由来FVII濃縮物を用いて、従来から成し遂げられてきた。しかし、組換えFVIIa(rFVIIa)は、今や、これらの患者での療法のために広く使用されている。
【0015】
rFVIIaは、また、インヒビターを有する血友病AおよびBの患者の出血を処置するために開発され、大手術(例えば整形外科手術)中でさえも止血を誘導することが分っている(非特許文献6)。rFVIIaは、BHK細胞培養物で生成され、かつ血漿由来FVIIaに極めて類似することが示されている。血友病処置でのrFVIIaの使用は、トロンビン活性化血小板の表面へのFVIIaの低親和性結合に基づく。外来性rFVIIaの薬理量を投与することによって、損傷部位の血小板表面上のトロンビン生成がFVIII/FIXの存在とは独立して増強される。急速なトロンビン形成が増大する結果、密接なフィブリン止血血栓が形成される。
【0016】
FVII欠乏およびインヒビター合併血友病AおよびBの処置のために本来は開発されたのだが、rFVIIaの新規適応(症例報告および小規模の臨床試験に基づく)としては、肝疾患、血小板減少症、または質的な血小板機能異常症の患者での使用、および非凝固障害を有する患者が大手術または大外傷に起因する出血の場合の使用が挙げられる。
【0017】
ポリペプチド治療薬(例えばFVIIaを含む血液凝固タンパク質)は、タンパク質分解酵素によって急速に分解され、抗体によって中和される。これは、ポリペプチド治療薬の半減期および循環時間を減少させ、それによってポリペプチド治療薬の治療有効性を限定する。所望のFVIIaの治療効果または予防効果を達成して維持するには、相対的に高い投与量および頻繁な投与が必要である。結果として、適切な投与量の調節を得るのは困難であり、頻繁な静脈内投与を必要とするために、患者の生活様式を制限する。従って、長い循環半減期によって改善されたFVIIa分子は、必要な投与回数を減少させる。
【0018】
原則として、血液循環におけるタンパク質の半減期を延ばすには4つの一般的な選択肢がある。すなわち、
・化学修飾または酵素修飾の導入
・血液循環中のタンパク質を保護する担体分子の使用
・半減期を延ばす突然変異体の構築
・分解経路の修飾。
【0019】
本発明は、血液凝固タンパク質の改善(特に化学修飾によるFVIIa分子)を教示する。治療用ポリペプチドの化学修飾に関しては、これまでいくつかのアプローチが用いられている。
【0020】
ポリペプチド薬のPEG化はポリペプチド薬を保護し、それらの薬力学的プロファイルおよび薬物動態プロファイルを改善する(非特許文献7)。PEG化プロセスは、ポリエチレングリコール(PEG)の繰り返し単位をポリペプチド薬に付着させる。PEG分子は、大きな流体力学的容積(球状タンパク質の大きさの5〜10倍)を有し、水溶性で高度に水和し、流動性が高く、無毒、非免疫原性であり、および身体から迅速に除去される。分子のPEG化は、酵素分解に対する薬物耐用の増加、in vivo半減期の増加、投薬頻度の減少、免疫原性の低下、物理的安定性および熱安定性の増加、溶解性の増加、液体安定性の増加、ならびに凝集の減少をもたらすことが可能である。最初のPEG化薬物は、1990年代の初めにFDAによって承認された。今のところ、FDAは、経口用、注射用、および局所投与用のいくつかのPEG化薬物を承認している。
【0021】
GlycoPEGylation(登録商標)技術は、インタクトなグリコシル結合基を介してペプチドに共有結合したPEGによる、PEGポリマーとペプチドとの間にペプチド結合体を提供する方法が含まれる。
【0022】
リポソームを用いて、種々の分子(例えばDNA、アンチセンスRNA、抗生物質、抗癌剤および抗真菌剤、抑制剤/活性剤、抗体(免疫リポソーム)、ならびに抗原(ワクチン用)がカプセル化されている。
【0023】
リン脂質も、PEG(PEG−リポソーム)(例えばアミド結合を介して)、カルボキシ−PEGおよび精製ダイズ・ホスファチジルエタノールアミン(PE)、エステルおよびカルバミン酸誘導体に結合体化されることが可能であり、今日最も広く使用されているのはカルバミン酸誘導体である(特許文献1)。最も一般的に用いられているPEGの分子量は、2,000および5,000であるが、600から12,000の範囲のPEGも用いられている。
【0024】
酸性単糖置換タンパク質は、最初に、特許文献2に開示された。この特許では酸性単糖(すなわちn−アセチルノイラミン酸およびグルコン酸)は、インスリン、ヒト成長ホルモン、またはアルブミンのα−アミノ基またはε−アミノ基の上に置換され、ポリペプチドの抗原性を減少させる。
【0025】
コロミン酸(CA)とも称されるポリシアル酸(PSA)も、天然の多糖である。ポリシアル酸は、α(2→8)ケトシド結合によるN−アセチルノイラミン酸のホモポリマーであり、その非還元末端に隣接ジオール基を含有する。ポリシアル酸は、負の電荷をもち、人体の天然の成分である。ポリシアル酸は、大量に細菌から容易に生成されることが可能であり、所定の物理的特性を有する(特許文献3)。人体においてポリシアル酸と化学的および免疫学的に同一である細菌性ポリシアル酸は、タンパク質に結合する場合でさえ、非免疫原性である。他のポリマー(例えばPEG)と異なり、ポリシアル酸は生分解性である。カタラーゼおよびアスパラギナーゼに共有結合するコロミン酸は、タンパク質分解酵素または血漿の存在下で酵素安定性の増加をもたらした。ポリシアル酸化および修飾されていないアスパラギナーゼによるin vivo比較研究は、ポリシアル酸化が酵素の半減期を増加させることを明らかにした(非特許文献8)。
【0026】
しかし、特許文献2に記載されるように、酸性単糖に結合体化したポリペプチドからなる治療化合物は、現在までに市販されていない。対照的に、特許文献3は、治療化合物の多糖部分がポリマー鎖に少なくとも5個(他の実施形態では少なくとも20または50個)のシアル酸残基を有するはずであることを教示する。多糖は通常、同時精製する内毒素の固有のリスクを保有する細菌で生成されるので、シアル酸ポリマー長鎖の精製は、内毒素含有物が増加する可能性を高め得る。1〜4のシアル酸ユニットを有するPSA短分子も、合成的に調製され得(非特許文献9;非特許文献10)、従って高レベルの内毒素のリスクを最小化する。
【0027】
特許文献4は、他の多くのタンパク質の中でFVIIがPEG化され得ることを示唆しているが、その開示を支持する任意の実施例を含有していない。
【0028】
特許文献5が教示する結合体は、ポリペプチドに共有結合する少なくとも1つの非ポリペプチド部分を含む結合体を教示する。ここで、前記ポリペプチドのアミノ酸配列は、前記非ポリペプチド部分のための結合基を含む少なくとも1つのアミノ酸残基が導入または除去されている点で野生型FVIIまたはFVIIaのそれと異なる。非ポリペプチド部分に対しては、特にPEGが示唆された。
特許文献6は、FVIIポリペプチドまたはFVII関連ポリペプチドを開示する。ここで、ポリペプチドは、アスパラギン結合型および/またはセリン結合型の1つ以上のオリゴ炭水化物部分を含み、および前記オリゴ糖基のうちの少なくとも1つが少なくとも1つのポリマー基(PEG、「グリコPEG化」)に共有結合される。
【先行技術文献】
【特許文献】
【0029】
【特許文献1】米国特許第6,593,294号明細書
【特許文献2】米国特許第3,847,890号明細書
【特許文献3】米国特許第5,846,951号明細書
【特許文献4】国際公開第98/32466号パンフレット
【特許文献5】国際公開第01/58935号パンフレット
【特許文献6】米国特許出願公開第2005/0113565号明細書
【非特許文献】
【0030】
【非特許文献1】Schenoneら、Curr Opin Hematol.(2004)11:272−7
【非特許文献2】Eigenbrot, Curr Protein Pept Sci.(2002)3:287−99
【非特許文献3】Pinottiら、Blood.(2000)95:3423−8
【非特許文献4】Acharyaら、J Thromb Haemost(2004)2248−56
【非特許文献5】Marianiら、Semin Hematol.(2006)43(Suppl 1):S42−7
【非特許文献6】Hedner, J Biotechnol.(2006)124:747−57
【非特許文献7】HarrisおよびChess, Nat Rev Drug Discov.(2003)2:214−21
【非特許文献8】Fernandes and Gregoriadis,Int J Pharm.(2001)217:215−24
【非特許文献9】Kangら、Chem Commun.(2000)227−8
【非特許文献10】RessおよびLinhardt,Current Organic Synthesis.(2004)1:31−46
【発明の概要】
【発明が解決しようとする課題】
【0031】
従って、当該技術分野において、改善した血漿由来もしくはrFVII、修飾型FVII、またはFVII関連ポリペプチドを含む凝固タンパク質製剤を提供する組成物および方法の必要性が残っている。
【課題を解決するための手段】
【0032】
本発明は、血漿または組換え第VIIa因子(FVIIa)またはその生物学的に活性な誘導体、1〜4のシアル酸ユニットからなる鎖に結合される前記FVIIaまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトを提供する。ここで、タンパク質性コンストラクトのin vivo半減期は、1〜4のシアル酸ユニットからなる鎖が欠けているFVIIaまたはその誘導体と比較すると、哺乳類(特にヒト)の血中で実質的に延びている。さらに、前記タンパク質性コンストラクトを含有する医薬組成物ならびに前記タンパク質性コンストラクトを用いてFVIIa、FVIII、およびFIXのうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類の血中FVIIaのin vivo半減期を延ばす方法が本発明によって提供される。本発明のタンパク質性コンストラクトは、正常なレベルの凝固因子を有する哺乳類における外傷または手術の場合に出血を制御するために投与されることもできる。
【0033】
本発明の一実施形態において、タンパク質性コンストラクトは、(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子と、(b)前記FVIIa分子に結合する1〜4のシアル酸ユニットを含む、生理的に許容される少なくとも1つの炭水化物部分とを含み、
ここで前記タンパク質性コンストラクトのin vivo半減期は、前記炭水化物部分に結合されていないFVIIa部分のin vivo半減期と比較すると、哺乳類の血液中で延びている。
【0034】
本発明の別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで該コントラクトのin vivo半減期が前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約2倍増加される。別の実施形態において、そのin vivo半減期が前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約3倍増加される、上述のタンパク質性コンストラクトが提供される。さらに別の実施形態において、生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に直接的に共有結合する、上述のタンパク質性コンストラクトが提供される。
【0035】
本発明のさらに別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで生理的に許容される炭水化物部分は、前記FVIIa分子の少なくとも1つのアミノ酸残基に非共有結合される。さらに別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで生理的に許容される炭水化物部分がポリシアル酸またはその誘導体である。
【0036】
本発明の一実施形態において、医薬組成物は有効量の上述のタンパク質性コンストラクトと、薬学的に許容される担体、希釈剤、塩、緩衝剤、および賦形剤からなる群から選択される1つ1つ以上の化合物とを含み提供される。
【0037】
本発明の別の実施形態において、FVIIa、FVIII、およびFIXのうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において出血を制御する方法は、上述のタンパク質性コンストラクトを投与することを含み、提供される。さらに別の実施形態において、哺乳類において手術または外傷の間に出血を制御する方法は、上述のタンパク質性コンストラクトを投与することを含み、提供される。
【0038】
本発明のさらに別の実施形態において、容器にパッケージされた上述のタンパク質性コンストラクトの有効量を含み、場合により第2の治療薬を含有し、前記容器に添付されたまたは前記容器とともにパッケージされたラベルをさらに含むキットを提供する。前記ラベルは、容器の内容物を説明し、哺乳類での出血を制御するための前記容器の内容物の使用に関する適応および/または使用説明書を提供する。さらに別の実施形態において、上述のキットを提供し、ここで前記容器がバイアルまたは瓶または予め充填されたシリンジである。
本発明の好ましい実施形態では、例えば以下が提供される:
(項目1)
タンパク質性コンストラクトであって、
(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子と、(b)該FVIIa分子に結合される1〜4のシアル酸ユニットを含む少なくとも1つの生理的に許容される炭水化物部分とを含み、
該炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、該コンストラクトのin vivo半減期が哺乳類の血中において延長している、タンパク質性コンストラクト。
(項目2)
前記コンストラクトのin vivo半減期が、前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約2倍増加する、請求項1に記載のタンパク質性コンストラクト。
(項目3)
前記コンストラクトのin vivo半減期が、前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約3倍増加する、請求項1に記載のタンパク質性コンストラクト。
(項目4)
前記生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に直接的に共有結合する、請求項1に記載のタンパク質性コンストラクト。
(項目5)
前記生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に非共有結合する、請求項1に記載のタンパク質性コンストラクト。
(項目6)
前記生理的に許容される炭水化物部分がポリシアル酸またはその誘導体である、請求項1に記載のタンパク質性コンストラクト。
(項目7)
請求項1に記載のタンパク質性コンストラクトの有効量と、薬学的に許容される担体、希釈剤、塩、緩衝剤、および賦形剤からなる群から選択される1つ以上の化合物とを含む、医薬組成物。
(項目8)
FVIIa、第VIII因子(FVIII)、および第IX因子(FIX)のうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において出血を制御する方法であって、該方法は、請求項1に記載のタンパク質性コンストラクトを投与することを含む、方法。
(項目9)
哺乳類において手術または外傷の間に出血を制御する方法であって、該方法は、請求項1に記載のタンパク質性コンストラクトを投与することを含む、方法。
(項目10)
容器にパッケージされた、請求項1に記載のタンパク質性コンストラクトの有効量を含むキットであって、該キットが場合により第2の治療薬を含有し、該容器に添付されたまたは該容器とともにパッケージされたラベルをさらに含み、該ラベルが該容器の内容物を説明し、哺乳類における出血を制御するための該容器の該内容物の使用に関する適応および/または使用説明書を提供する、キット。
(項目11)
前記容器がバイアルまたは瓶または予め充填されたシリンジである、請求項10に記載のキット。
【図面の簡単な説明】
【0039】
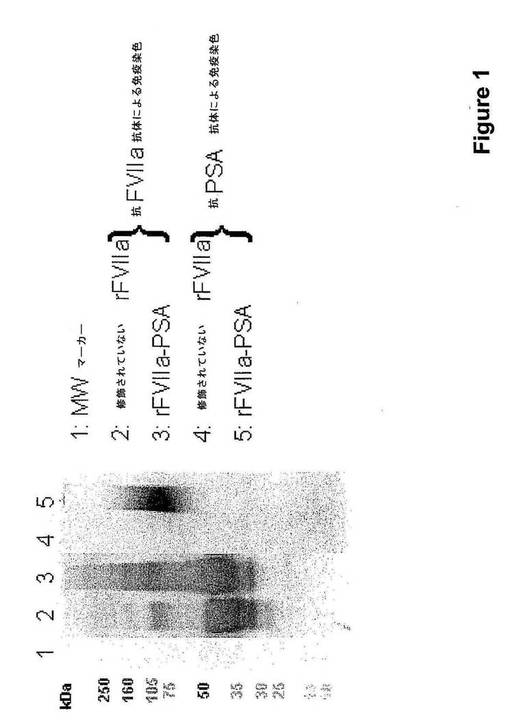
【図1】PSAとの結合体化後のrFVIIaのSDS−PAGEを示す。
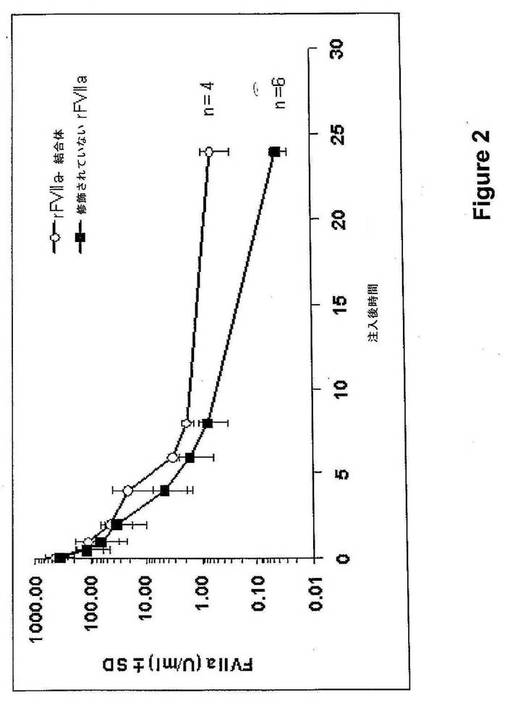
【図2】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。
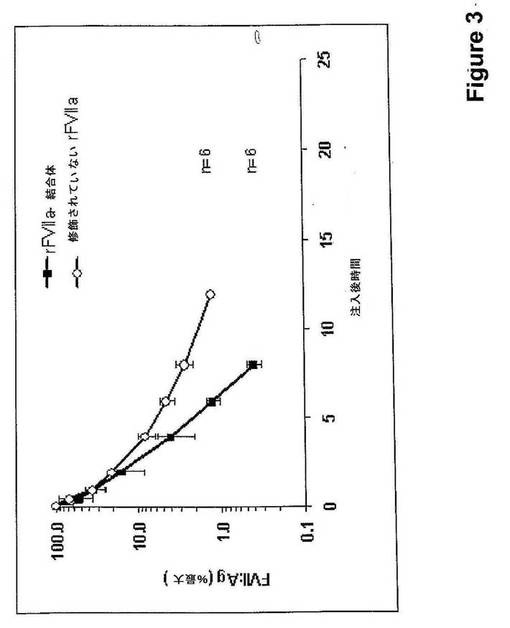
【図3】rFVIIa−PSA結合体および修飾されていないrFVIIa(抗原レベル)のラットにおける薬物動態を示す。
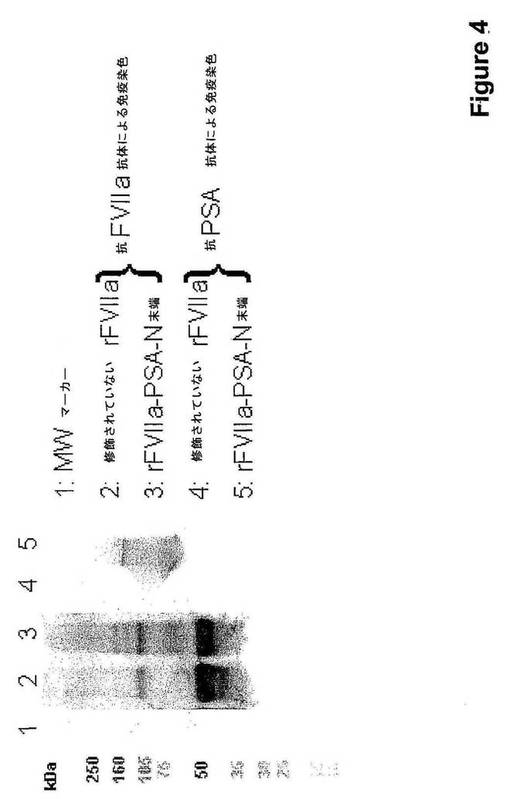
【図4】PSAとのN末端結合体化後のrFVIIaのSDS−PAGEを示す。
【図5】モノSA−rFVIIaおよびトリSA−rFVIIaのキャピラリー電気泳動を示す。
【図6A】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。A:モノSA−rFVIIa。
【図6B】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。B:トリSA−rFVIIa。
【図7】N−アセチルノイラミン酸三量体のキャピラリー電気泳動を示す。
【発明を実施するための形態】
【0040】
発明の詳細な説明
本発明は、本明細書に記載のFVII部分に限定されないことを理解すべきである。本発明の一態様は、血液凝固カスケードの1つのメンバー、血漿の(すなわち血漿由来)および/または組換えのFVIIaまたはその生物学的に活性な誘導体(以下「PSA−FVIIa結合体」と表す)、1〜4のシアル酸部分と結合される前記FVIIまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトに関する。ここで、前記FVIIまたはその前記生物学的に活性な誘導体のin vivo半減期は、哺乳類の血中で延びる。本明細書に用いるように、「タンパク質性コンストラクト」という用語は、(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子、および(b)前記FVIIa分子に結合した1〜4のシアル酸ユニットを含む少なくとも1つの生理的に許容される炭水化物部分をいう。本明細書に用いるように、「血漿の」という用語は、「血漿由来の」を言及する。
【0041】
FVIIaポリペプチドおよびポリヌクレオチド
本発明に有用なFVIIa分子としては、完全長タンパク質、タンパク質の前駆体、タンパク質の生物活性サブユニットまたは機能的サブユニットまたは断片、およびその機能的誘導体が挙げられる。FVIIaへの言及は、かかるタンパク質のすべての潜在的な形を含むことを意図される。
【0042】
本発明によれば、「組換え第VIIa因子」(rFVIIa)という用語は、特定の制限の根底にあるのではなく、かつ組換えDNA技術を介して取得する任意のrFVIIa、非相同的もしくは天然の、またはそれらの生物学的に活性な誘導体も含み得る。ある実施形態において、この用語は、タンパク質および核酸(例えば遺伝子、mRNA前駆体、mRNAおよびポリペプチド)、多型変異体、対立遺伝子、突然変異体、ならびに種間相同体を包含する。種間相同体は、(1)参照核酸または本明細書に記載するアミノ酸配列によってコードされるポリペプチドと、少なくとも約25、50、100、200、300、400、またはそれ以上のアミノ酸(成熟タンパク質の場合、完全長配列の406アミノ酸まで)からなる領域にわたり、約60%を超えるアミノ酸配列同一性、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、または99%を超えるアミノ酸配列同一性を有するアミノ酸配列を有する;(2)本明細書に記載の参照アミノ酸配列を含む免疫原、その免疫原断片、および保存的に修飾されたその変異体に対して産生される抗体(例えばポリクローナル抗体)に特異的に結合する;(3)本明細書に記載の参照アミノ酸配列をコードする核酸およびその保存的に修飾された変異体に、ストリンジェントなハイブリダイゼーション条件下で特異的にハイブリダイズする;(4)本明細書に記載の参照核酸配列と、少なくとも約25、50、100、150、200、250、500、1000、またはそれ以上のヌクレオチド(成熟タンパク質の完全長配列の1218ヌクレオチドまで)からなる領域にわたり、約95%を超える核酸配列同一性、約96%、97%、98%、99%またはそれを超える核酸配列同一性を有する核酸配列を有する。
【0043】
本明細書に用いるように、「内因性FVIIa」は、前記哺乳類から生ずるFVIIaを含む。内因性FVIIaは、前記哺乳類に存在するトランスジーンまたは他の任意の外来DNAから転写されるFVIIaも含む。本明細書で用いるように、「外因性FVIIa」は、前記哺乳類から生じないFVIIaを含む。
【0044】
変異体(または類似体)ポリペプチドは、1つ以上のアミノ酸残基がFVIIaアミノ酸配列を補充する、挿入変異体を含む。挿入は、タンパク質のいずれかの末端または両末端に位置していることもあり、またはFVIIaアミノ酸配列の内部領域内に位置することもある。いずれかの末端または両末端に残基をさらに有する挿入変異体は、例えば融合タンパク質およびアミノ酸タグまたはアミノ酸標識を含むタンパク質を含むことができる。例えば、FVIIa分子は、特に、FVIIa分子が細菌細胞(例えば大腸菌(E.coli))内で、組換えで発現される場合、場合によりN末端メチオニンを含有することもある。
【0045】
欠失体では、FVIIaポリペプチドで1つ以上のアミノ酸残基が除去される。欠失は、FVIIaポリペプチドの一方または両末端で、またはFVIIaアミノ酸配列内の1つ以上の残基を取り除くことで引き起こされ得る。従って、欠失体はFVIIaポリペプチド配列のすべての断片を含む。
【0046】
置換変異体では、FVIIaポリペプチドの1つ以上のアミノ酸残基が除去されて、代替残基と置換する。一態様において、置換は、天然において保存的であり、このタイプの保存的置換は当該技術分野では公知である。あるいは、本発明は、非保存的でもある置換を包含する。代表的な保守的置換は、Lehninger(Biochemistry,
2nd Edition;Worth Publishers, Inc., New York (1975), pp.71−77)に記載されており、以下に記載する。
【0047】
【化1】

あるいは、代表的な保存的置換は、以下に記載する。
【0048】
【化2】
ポリヌクレオチド配列またはポリペプチド配列は、一般的に、霊長類(例えばヒト)齧歯類(例えばラット、マウス、ハムスター)、ウシ、ブタ、ウマ、ヒツジ、または任意の哺乳類にも限定されないが哺乳類に由来する。本発明の核酸およびタンパク質は、組換え分子(例えば非相同的および野生型配列もしくはその変異体をコードする、または非天然の)であり得る。参照ポリヌクレオチド配列およびポリペプチド配列としては、例えば、ゲノム配列のためのGenBank受入番号J02933、cDNAのためのM13232(Hagenら、PNAS 1986;83:2412−6)、およびポリペプチド配列のためのP08709(それら開示内容全体を参考として本明細書で援用される)が挙げられる。FVIIの種々の多型性は、例えばSabater−Llealら、(Hum
Genet.2006;118:741−51)(それら開示内容全体を参考として本明細書で援用される)に記載されている。
【0049】
本明細書で用いるように、「生物学的に活性な誘導体」または「生物活変異体」としては、前記分子と同一の機能的特性および/または生物学的特性(例えば結合特性)、ならびに/または同一の構造基盤(例えばペプチド主鎖または塩基性ポリマーユニット)を実質的に有する、任意の誘導体または種々の分子が挙げられる。
【0050】
本明細書で用いるように、「血漿誘導型FVIIa」または「血漿」は、凝固経路を活性化する特性を有する哺乳類から採血した血液中で見出されたタンパク質のすべての形状を含む。
【0051】
本明細書で用いるように、「組換えFVIIa」は、組換えDNA技術を介して得たrFVIIaを含む。組換えFVIIaは、当該技術分野で公知の任意の方法によって生成され得る。1つの特定の例は、米国特許第4,784,950号に開示されている。かかるrFVIIaの例は、Novo Nordiskによって製造、販売されるNovoSevenである。
【0052】
FVIIa生成および発現
rFVIIaの生成は、以下に関して当該技術分野で公知の任意の方法を含み得る。(i)遺伝子工学(例えばRNA逆転写および/またはDNAの増幅)による組換えDNAの生成、(ii)組換えDNAを、トランスフェクション(例えば電気穿孔またはマイクロインジェクションを介して)によって原核細胞または真核細胞に導入すること、(iii)前記形質転換させた細胞を、例えば連続的方法またはバッチ式方法で培養すること、(iv)例えば恒常的にまたは誘導により、rFVIIaを発現させる、(v)例えば培地から前記FVIIaを分離すること、または形質転換した細胞を収集すること、(vi)続いて、例えばアニオン交換クロマトグラフィーまたはアフィニティークロマトグラフィーを介して、精製したrFVIIaを得ること。
【0053】
rFVIIaは、薬理学的に許容されるrFVIIa分子を生成することよって特徴づけられる、適切な原核または真核宿主系で発現させることで生成することができる。真核細胞の例としては、哺乳類細胞(例えばCHO、COS、HEK293、BHK、SK−Hep、およびHepG2)がある。本発明によってrFVIIaの生成または分離のために用いられる試薬もしくは条件は、特に限定されず、当該技術分野で公知の任意の系または市販されているものを利用することができる。
【0054】
多種多様なベクターを用いて、rFVIIaを調製することができ、真核および原核発現ベクターから選択されることができる。原核発現のためのベクターの例としては、様々のプラスミド(例えばpRSET、pET、pBADなど)が挙げられ、原核発現ベクターで用いられるプロモーターとしては、lac、trc、trp、recA、araBADなどが挙げられる。真核発現のためのベクターの例としては、以下が挙げられる。すなわち、
(i)酵母での発現のため、プロモーター(例えばAOX1、GAP、GAL1、AUG1など)を用いるベクター(例えばpAO、pPIC、pYES、pMET)、
(ii)昆虫細胞での発現のため、プロモーター(例えばPH、p10、MT、Ac5、OpIE2、gp64、po1hなど)を用いるベクター(例えばpMT、pAc5、pIB、pMIB、pBAC)、ならびに(iii)哺乳類細胞での発現のため、プロモーター(例えばCMV、SV40、EF−1、UbC、RSV、ADV、BPV、およびβ−アクチン)を用いるベクター(例えばpSVL、pCMV、pRc/RSV、pcDNA3、pBPVなど)、およびウイルス系由来のベクター(例えばワクシニアウイルス、アデノ関連ウイルス、ヘルペスウイルス、レトロウイルスなど)。
【0055】
シアル酸
本明細書で用いるように、「シアル酸部分」は、シアル酸のモノマーまたはポリマーが含まれる。これらは、水溶液中または懸濁液中で可溶性であり、何ら否定的な影響(例えばPSA−FVIIa結合体を医薬的有効量で哺乳類に投与した後の副作用)がない。本発明によって用いられるシアル酸ユニットには特に限定されない。一態様において、シアル酸ポリマーは、1〜4ユニットを有するとして特徴づけられる。また異なるシアル酸ユニットが鎖状に組み合わせられ得る。
【0056】
シアル酸部分は、例えば米国特許第4,356,170号に記載の方法(参考として本明細書で援用される)によって、FVIIaに結合され得る。本発明の一実施形態において、多糖化合物は、天然の多糖、天然の多糖の誘導体、または天然多糖誘導体であり得る。通常、化合物中の糖類残基のすべては、シアル酸残基である。
【0057】
PSAをポリペプチドに結合させる他の技法も公知である。例えば、米国特許出願公開第2007/0282096号は、タンパク質への例えばPSAのアミンまたはヒドラジン誘導体の結合体化を記載している。さらに、米国特許出願公開第2007/0191597は、還元終端で基質(例えばタンパク質)との反応のためのアルデヒド基を含有するPSA誘導体を記載している。
【0058】
本発明の一実施形態において、多糖化合物のポリシアル酸部分は高度に親水性である。別の実施形態において、化合物全体が高度に親水性である。親水性は、主に、シアル酸ユニットのペンダントカルボキシル基ならびにヒドロキシル基によって与えられる。糖類ユニットは、他の官能基(例えばアミン基、ヒドロキシル基、もしくは硫酸基、またはそれらの組み合わせ)を含有し得る。これらの基は、天然の糖類化合物上に存在し得、または誘導体多糖化合物に導入され得る。
【0059】
本発明の特定の用途の多糖化合物は、細菌によって産生されるものである。これらの天然の多糖の一部は、糖脂質として知られている。多糖化合物に、肝細胞およびクッパー細胞のガラクトース受容体によって認識される傾向がある末端ガラクトースユニットが実質的にない場合、特に有利である。
【0060】
結合
FVIIaは、当業者にとって公知の種々の技術のいずれかによって多糖化合物に共有結合され得る。種々の例は、米国特許第5,846,951号の第7欄15行目から第8欄5行目に確認される。
【0061】
例としては、FVIIaまたは多糖のいずれか一方のカルボキシル基と、他方のアミン基との間のペプチド結合を介する結合、または一方のカルボキシル基と、他方のヒドロキシル基との間のエステル結合が挙げられる。活性成分(例えばFVIIa)が多糖化合物に共有結合し得ることによる別の結合は、反応される活性成分上の遊離アミノ基と、過ヨウ素酸酸化によってポリマーの非還元末端で形成されたアルデヒド基との間のシッフ塩基を介する(Jennings and Lugowski, J Immunol.1981;127:1011−8;Fernandes and Gregoriadis,
Biochim Biophys Acta.1997;1341;26−34)。生成されたシップ塩基は、NaCNBH3による特異的還元によって安定化され、二級アミンを形成することができる。別のアプローチは、事前の酸化の後にNH4Clを用いる還元的アミノ化によってポリシアル酸(PSA)内に末端遊離アミノ基を生成することである。二価性試薬を用いて、2つのアミノ基または2つのヒドロキシル基を結合するため使用できる。例えば、アミノ基を含有するPSAはBS3(ビス(スルホスクシンイミジル)スベラート/Pierce, Rockford, IL)等の試薬を用いてタンパク質のアミノ基に結合させることができる。さらに、ヘテロ二価架橋試薬(例えばスルホ−EMCS(N−ε−マレイミドカプロイルオキシ)スルホスクシンイミドエステル/Pierce)を用いて、例えばアミン基およびチオール基を結合することができる。
【0062】
別のアプローチでは、事前の酸化およびアルデヒド官能基の生成の後、PSAヒドラジドを調製して、タンパク質の炭水化物部分に結合させることができる。
【0063】
治療用タンパク質の遊離アミン基は、シアル酸残基の1−カルボキシル基と反応させることも可能であり、ペプチジル結合を形成し、またはエステル結合が活性成分上で1−カルボン酸基およびヒドロキシルまたは他の適切な活性基の間に形成されることが可能である。あるいは、カルボキシル基は、脱アセチル化5−アミノ基によってペプチド結合を形成することもある。医薬的活性化合物の分子のアルデヒド基は、シアル酸残基のN−脱アセチル化5−アミノ基とシッフ塩基を形成し得る。
【0064】
あるいは、多糖化合物は、非共有の方法で医薬的活性化合物(例えばFVIIa)と結合し得る。例えば、多糖化合物および医薬的活性化合物は、疎水性相互作用を介して(例えば多糖化合物の脂質成分を介して)疎水性医薬的活性化合物と結合され得る。他の非共有結合は、逆帯電したイオンが互いに引きつけることによる静電相互作用を介することも可能である。
【0065】
医薬的活性化合物は、化学量論量(例えば1:1)で多糖化合物に直接的に共有結合され得る。あるいは、多糖化合物の2つ以上の分子は結合されて活性成分の1つの分子になり得る。
【0066】
用途
本発明は、少なくとも1つの生理的に許容されるシアル酸部分に結合されていないFVIIaのin vivo半減期と比較して、血液凝固タンパク質(特に、FVIIaの機能上の欠損または欠乏に関連する出血性障害を有する、FVIIaまたはその生物学的に活性な誘導体)のin vivo半減期を増加させることに関する。本発明のPSA−FVIIa結合体は、FVIIIおよびFIXのうちの少なくとも1つの機能上の欠損または先天性欠乏または後天性欠乏に関連する出血性障害の処置のためにさらに用いられることができる。
【0067】
療法における最先端技術により、さらに国際ガイドラインおよび規定により、注入されるFVIIaの薬物動態は、有効性の有効な代理マーカーとして認識され、容認されている(Bjorkman and Bemtrop, Clin Pharmacokinet.2001;40:815−32)。
【0068】
これは、機能活性に関する標準化試験によって特徴づけられた、注入されたFVIIa産物が血流中で見出され、凝固血液カスケードの成分として予想通りにそこで作用するという確認された仮定に基づく。従って、動物モデルにおける任意の薬物動態分析でも、FVIIa産物で処置される患者において予想される有効性を予測する。
【0069】
半減期
本発明の一実施形態において、タンパク質性コンストラクトのin vivo半減期は、延ばされる。関連する実施形態において、シアル酸に結合されないFVIIaと比較すると、タンパク質性コンストラクトのin vivo半減期は、少なくとも2倍延び、別の実施形態において、そのin vivo半減期は少なくとも3倍延びる。FVIIa半減期の延長は、以下の実施例で説明するようにラットでの薬物動態を測定することによって評価され得る。
【0070】
投与
投与経路は、特定の限定を示さない。一実施形態において、本発明のタンパク質性コンストラクトは、注射(例えば静脈注射、筋肉内注射、または腹腔内注射)によって投与され得る。
【0071】
本発明のタンパク質性コンストラクトを含む組成物をヒトまたは試験動物に投与するため、一態様において、その組成物は1つ以上の薬学的に許容される担体を含む。「医薬的に」または「薬理学的に許容される」という用語は、以下の記載のように、安定しており、タンパク質分解(例えば凝集および切断産物)を抑制し、さらに、当該技術分野で周知の経路を用いて投与される場合、アレルギー反応または他の副作用を出現させない分子化合物および組成物をいう。「薬学的に許容される担体」としては、上記に開示した薬剤を含む、ありとあらゆる臨床的に有用な溶媒、分散媒、被覆剤、抗菌および抗真菌剤、等張および吸収遅延剤などが挙げられる。
【0072】
本明細書で用いるように、「有効量」は、上記に概要を述べるように、出血性障害を有する哺乳類を処置するのに適している用量が含まれる。
【0073】
組成物は、経口的に、局所的に、経皮的に、非経口的に、吸入スプレーによって、経膣的に、直腸に、または頭蓋内注射によって投与されてもよい。本明細書で用いるように、「非経口的に」という用語は、皮下注射、静脈内、筋肉内、大槽内注射、または注入法が挙げられる。静脈内、皮内、筋肉内、乳房内、腹腔内、くも膜下腔内、眼球後、肺内の注射、および/または特定部位での外科的移植による投与もまた考えられる。通常、組成物は、発熱物質ならびにレシピエントに有害になり得る他の不純物が本質的に取り除かれる。
【0074】
組成物の単回投与または複数回投与は、処置を行う医師によって選択される用量レベルおよびパターンで行われ得る。疾患の予防または処置に関して、適切な投与量は、上述のように処置すべき疾患の種類、疾患の重症度、および疾患の経過、薬物が予防目的または治療目的で投与されるかどうか、治療歴、患者の臨床歴および薬物応答、ならびに主治医の裁量に依存する。
【0075】
医薬組成物
本発明は、また、上記に明示したように、有効量のタンパク質性コンストラクトを含む医薬組成物に関する。医薬組成物は、薬学的に許容される担体、希釈剤、塩、緩衝剤、または賦形剤をさらに含み得る。医薬組成物を用いて、上記に明示した出血性障害を処置することができる。本発明の医薬組成物は、溶液または凍結乾燥生成物であり得る。タンパク質の安定な溶液、および特にFVIIaを形成する公知の方法が数多くある。1つの例は、米国特許第5,874,408号に開示されている。医薬組成物の溶液を、任意の適切な凍結乾燥プロセスにかけることが可能である。
【0076】
キット
追加の態様として、本発明は、被験者への投与のためにその用途を促進する方法でパッケージされた本発明の組成物を含むキットが挙げられる。一実施形態において、かかるキットには、本明細書に記載の化合物または組成物(例えばタンパク質性コンストラクトを含む組成物)が含まれる。前記化合物または組成物は、密封された瓶またはベスル等の容器にパッケージされ、前記方法を実践する際の化合物または組成物の用途を説明するラベルが容器に添付されるか、またはパッケージに含まれる。一実施形態において、キットは、タンパク質性コンストラクトを含む組成物を有する第1の容器と、第1の容器内の組成物のための生理的に許容される再構成溶液を有する第2の容器とを含有する。一態様において、化合物または組成物は、単位用量形態でパッケージされる。キットは、投与の特定の経路によって、組成物を投与するのに適した装置をさらに含み得る。好ましくは、キットは、治療用タンパク質またはペプチド組成物の用途を説明するラベルを含有する。
【0077】
本発明を、以下の実施例でさらに例示するが、それらに何ら限定されない。
【実施例】
【0078】
(実施例1):rFVIIa内のリジン残基のコロミン酸による修飾
シアル酸(コロミン酸、CA)によるリジン残基の修飾を、JenningsおよびLugowski(J Immunol, 1981;127;1011−8)によって記載されるように行った。この処置のために、Sigma(Sigma−Aidrich,
St. Louis;MO)製のCAを使用した。0.1M NaIO4を含有するCAの水溶液(濃度20mg/mL)を暗所で、室温で15分間攪拌し、CAを酸化させた。活性化CA溶液/mLに2mLのエチレングリコールを加えて暗所で、室温でさらに30分間攪拌した。この溶液を、暗所で、2〜8℃の温度で、0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して一晩透析した。
【0079】
続いて、この溶液のアリコートを、0.05M リン酸ナトリウム緩衝液(pH7.2)中のrFVIIa溶液(30μg/mL)に加えて、rFVIIaのmgあたり100mgの活性化CAの最終濃度にした。この混合物を暗所で、室温で180分間攪拌した。NaCNBH3を加えて(最終濃度10mg/mg rFVIIa)、この混合物を静かに振盪させながら暗所で、室温で18時間、インキュベートした。次いで、2.5mLの水性1M TRIS溶液(pH7.2)をこの混合物/mLに加えて、60分間攪拌して、反応を終了させた。
【0080】
イオン交換クロマトグラフィーで、QHyperD F 50μm樹脂(Pall BioSepra, Cergy, France)およびPharmacia XK−10カラム(Pharmacia XK 10;高さ=15cm)を用いて、rFVIIa−CA酸結合体から遊離試薬を分離した。CA結合体化タンパク質を、溶出緩衝剤(20mM HEPES/1M NaCl, pH8.0)で溶出した。最終段階で、30kD膜(再生セルロース/Millipore)を用いて限外濾過/ダイアフィルトレーション(UF/DF)によって、150mM NaClおよび0.5%ショ糖を含有する20mM HEPES緩衝液(pH7.4)に対して濃縮させた。
【0081】
(実施例2):ポリシアル酸化rFVIIaの生化学的性質
rFVIIa−PSAの酵素活性を、凝固アッセイによって決定した。FVIIaをヒトFVII欠乏血漿に加えて、凝固を、FVIIaと反応するが、FVIIと反応しないトランケート型組織因子(Staclot, Diagnostica Stago, Asnieres, France)によって誘発した。
【0082】
rFVII−PSAのFVIIIのバイパス活性をトロンビン生成アッセイ(TGA)によって測定した。FVIIaを、トロンビン−特異的蛍光ペプチド−基質の存在下で高力価の抗FVIIIインヒビターを含有する重度の血友病Aの血漿に加えた。凝固を、組織因子−リン脂質複合体で誘発し、トロンビン生成を、基質のフルオロフォアの切断速度によって連続的に測定した。トロンビン生成活性を、ピークのトロンビン(すなわち、アッセイ中に観察された最大量トロンビンの濃度)から算出した。両ケースでは、NovoSeven組換えFVIIa製剤(Novo Nordisk,デンマーク コペンハーゲン)を参考として使用した。
表1に示すように、PSA−rFVIIaの比活性は、修飾後減少した。
【0083】
【表1】
修飾を、非還元条件下で行ったSDS−PAGEによって可視化した。免疫染色を、ポリクローナル抗FVII抗体(Affinity Biologicals;Ancaster, Canada)およびモノクローナル抗PSA抗体(Chemicon International, Temecula, CA, USA)を用いて行った。修飾は、PSA含有タンパク質と相関するスメア領域によって示されたFVIIaのMWの増加をもたらした(図1)。
【0084】
(実施例3):ラットにおけるrFVIIa−PSA結合体の薬物動態
4匹のラット(Crl:CD(SD), Charles River Laboratories, Wilmington, MA)に麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L マンニトール、1.5g/L CaCl2 x 2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−PSA結合体(16.500U FVIIa/kg)を、用量(20mL/kg)を尾静脈に静脈内注射によって投与した。対照として6匹の正常なラットに、修飾されていないrFVIIaを18.000U FVIIa/kgの用量で用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、6時間後、8時間後、および24時間後に血液試料を、眼窩静脈叢から採取し、クエン酸血漿を調製してさらなる分析のために凍結させた。
【0085】
次いで、血漿中のFVIIa活性(Staclot, Diagnostica Stago, Asnieres, France)を測定した。修飾されていないrFVIIaの半減期は1.1時間であったが、rFVIIa−結合体により2.3時間に増加した(図2)。
【0086】
FVIIa抗原レベルの薬物動態を、さらなる実験で測定した。6匹のラットに麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L
マンニトール、1.5g/L CaCl2.2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−PSA結合体(450μg/kg)を、容積用量(10L/kg)で尾静脈に静脈内注射によって投与した。対照として6匹のラットに、修飾されていないrFVIIaを390μg/kgの用量で用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、6時間後、8時間後、12時間後、および24時間後に血液試料を、眼窩静脈叢から採取した。そしてクエン酸血漿を調製してさらなる分析のために凍結させた。血漿中のFVII抗原レベルを、ELISA(ポリクローナル抗−ヒトFVII抗体)で測定した。MS Excelで決定したように、直線回帰による半減期算出は、天然rFVIIaに対しては1.1時間およびrFVIIa結合体に対しては3.1時間であった。FVII抗原に関するデータを、投与から5分後に得た平均血漿レベルに規準化する(図3)。
【0087】
(実施例4):FVIIaのN末端ポリシアル化
FVIIaのN末端でのCAの結合体化をpH6.0で行った。この処置のために、Sigma(Sigma−Aldrich)からのCAを使用し、それをさらにQ−セファロース FF(GE Healthcare, Munich, Germany)上で陰イオン交換クロマトグラフィーによって精製した。0.04M NaIO4を含有する精製CAの水溶液(濃度23mg/mL)を暗所で、室温で15分間攪拌して、CAを酸化させた。続いて、この溶液のアリコートを0.05Mリン酸ナトリウム緩衝液(pH6.0)中のrFVIIa溶液(740μg/mL)に加えて、rFVIIaのmgあたり60mgの活性化CAの最終濃度にした(約150モル過剰)。この混合物を暗所で、室温で180分間攪拌した。NaCNBH3を加えて(25mg/mg rFVIIa)この混合物を静かに振盪させながら暗所で、24時間、インキュベートした。次いで、2.5mLの水性1M TRIS溶液(pH7.2)をこの混合物/mLに加えて、4℃で60分間攪拌して、反応を終了させた。
【0088】
イオン交換クロマトグラフィーで、QHyperD F 50μm樹脂(Pall BioSepra, Cergy, France)およびPharmacia XK−16カラム(Pharmacia XK 16;高さ=14cm)を用いて、rFVIIa−CA酸結合体から遊離試薬を分離した。次いで、CA結合体化タンパク質を、溶出緩衝剤(20mM HEPES/0.5M NaCl、pH8.0)で溶出した。最終段階で、10kD膜(再生セルロース/Millipore)を用いてUF/DFによって、150mM NaClを含有する20mM HEPES緩衝液(pH7.4)に対して濃縮させた。イオン交換クロマトグラフ法およびUF/DFステップを4℃で行った。
【0089】
N末端修飾rFVIIa−PSAの酵素活性を、実施例2に記載するように、凝固アッセイおよびトロンビン生成アッセイによって決定した。その結果を表2に要約する。
【0090】
【表2】
N末端結合体化型PSA−rFVIIaの比活性は、STFアッセイで測定したように約50%まで、およびTGAでは25%まで低下した。
【0091】
実施例2に記載するように、修飾は、ポリクローナル抗FVII抗体およびポリクローナル抗PSA抗体を用いる免疫染色によって展開させた非還元条件下で行ったSDS−PAGEによって可視化した。修飾は、抗PSAで染色した免疫ブロットで示されるバンドと相関して、FVIIaのMWの軽微な増加をもたらした(図4)。
【0092】
(実施例5):CNBr活性化型合成N−アセチルノイラミン酸によるFVIIaの結合体化
rFVIIaを、米国特許第3,487,890号に記載されるように、N−アセチルノイラミン酸によって結合体化させた。350mgの合成N−アセチルノイラミン酸(Sigma−Aldrich)を、10mLの0.1M HEPES緩衝液(pH9.0)に溶解させた。次いで、430mgのCNBr(Fluka, Steinhamm, Germany)をこの溶液に加え、活性化の処置の間0.5M NaOHでpHを9.5に調整した。30分後、pH値は9.5であった。次いで、pH値を、0.1M HClを加えることによって8.4に調整した。活性化の全処置の間、温度を氷浴を用いることで制御し、20〜25℃で維持した。rFVIIaによる活性化N−アセチルノイラミン酸の結合体化のために、rFVIIa溶液50mM リン酸緩衝液(pH7.2)中の(50mL/0.44mg rFVIIa/mL)を加えて室温で、緩やかな攪拌下で30分間インキュベートした。次いで、20mLの0.2Mトリス緩衝液を加えて、反応を終了させ、遊離シアン酸エステルをブロックし、混合物を緩やかな攪拌下で15分間インンキュベートした。最終的にこの溶液を、50mM リン酸緩衝液(pH7.2)に対して10kD膜(再生セルロース/Millipore)を用いるUF/DFによって濃縮させた。
【0093】
(実施例6):CNBr活性化合成N−アセチルノイラミン酸三量体によるFVIIaの結合体化
rFVIIaを、N−アセチルノイラミン酸に関する米国特許第3,487,890号に記載されるように、TimTec、LLC(Newark, USA)から得た合成N−アセチルノイラミン酸三量体に結合体化させた。350mgのN−アセチルノイラミン酸三量体を、10mLの0.1M HEPES緩衝液(pH9.0)に溶解させた。次いで、430mgのCNBr(Fluka)をこの溶液に加え、活性化の処置の間0.5M
NaOHでpHを9.5に調整した。30分後、pH値は9.5であった。pH値を、0.1M HClを加えることによって8.4に調整した。活性化の全処置の間、温度を、氷浴を用いることによって制御し、20〜25℃で維持した。次いで、FVIIaにおる活性化の結合体化を、実施例5に記載するように行った。
【0094】
(実施例7):モノSA−FVIIaおよびトリSA−FVIIaの生化学的性質
実施例5に記載するN−アセチルノイラミン酸(モノSA)に結合体化した修飾rFVIIa、または実施例6に記載するN−アセチルノイラミン酸三量体(トリSA)に結合体化した修飾rFVIIaの酵素活性を、実施例2に記載するように凝固アッセイおよびトロンビン生成アッセイによって決定した。それらの結果を、表3に要約する。
【0095】
【表3】
オリゴ−PSA結合体化型rFVIIaの比活性は、STFアッセイによって測定されたように低下したが、TGAによって測定されたモノSA−rFVIIaは、そのFVIIIバイパス活性の約50%を保持した。
【0096】
さらに、モノSA−rFVIIaおよびトリSA−rFVIIaを、KlausenおよびKornfelt(J Chromatogr A. 1995;718:195−202)によって記載されるように、キャピラリー電気泳動(CE)によって調べた。それらの結果を図5に示す。天然のrFVIIaと比較して、付加的な負の電荷のためにモノSA−rFVIIaおよびトリSA−rFVIIaのより高い保持時間への明らかな変化を示す。
【0097】
(実施例8):ラットにおけるrFVIIa−モノSAおよびrFVIIa−トリSA結合体の薬物動態
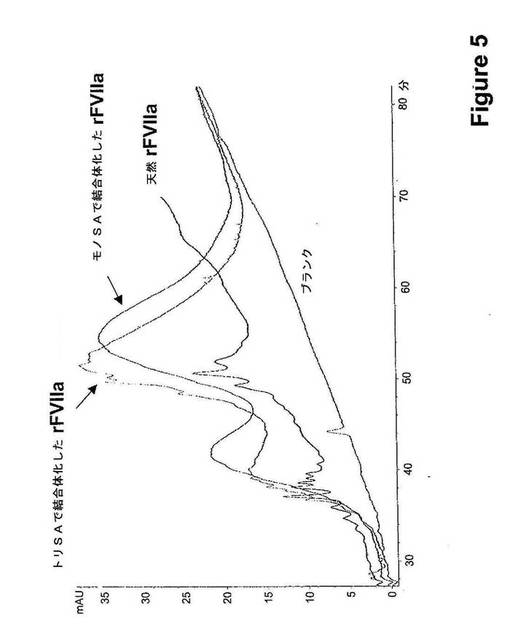
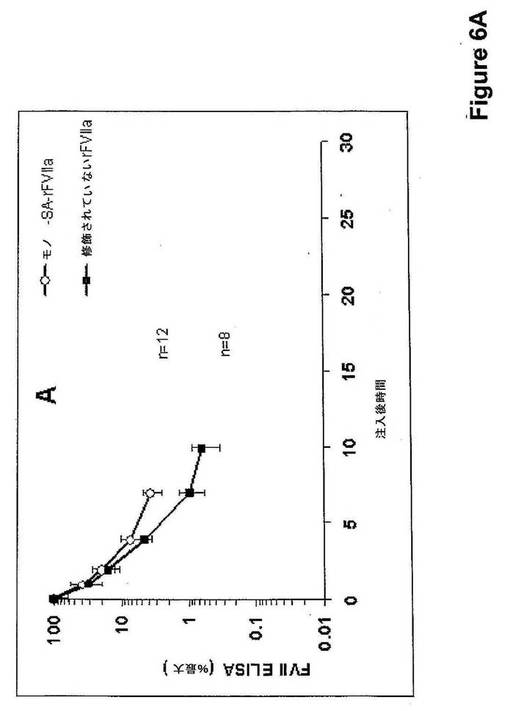
12匹のラットに麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L マンニトール、1.5g/L CaCl2.2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−モノSA結合体(400μg タンパク質/kg)を、容積用量(10mL/kg)を尾静脈に静脈内注射によって投与した。4匹のラットに、rFVIIa−トリSA結合体(400μg タンパク質/kg)で処置した。対照として8匹の正常なラットにおいて400μg タンパク質/kgの用量で修飾されていないrFVIIaを用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、7時間後、10時間後、および22時間後に血液試料を、眼窩静脈叢から採取し、クエン酸血漿を調製してさらなる分析のために凍結させた。血漿中のFVII抗原レベルをELISA(ポリクローナル抗−ヒトFVII抗体)によって測定した。データを、投与から5分後に血漿で見出された濃度に対して規準化した。投与から7時間後、rFVIIa−モノSAおよびトリSA−rFVIIaの血漿レベルは対照の天然rFVIIaのそれよりも高かった。それらの結果を図6A(rFVIIa−モノSA)および図6B(rFVIIa−トリSA)に示す。
【0098】
(実施例9):還元的アミノ化によるN−アセチルノイラミン酸三量体のrFVIIaへの結合
還元的アミノ化によるrFVIIaのN−アセチルノイラミン酸三量体との結合体化を、Biessen el al. (Biochem J 1994;299:291−6)に記載されるように行った。350mgのN−アセチルノイラミン酸三量体(TimTec)を10mLの0.1M HEPES緩衝液(pH7.0)に溶解して、20mM
HEPES、70mM NaCl(pH7.4)(0.3mg/mL)中の組換えFVIIaの溶液32mLに加えた。次いで、NaCNBH3を加えて、50mg/mLの最終濃度にし、0.1M HClを加えることによってpH7.0にpH調整した。混合物を37℃で、穏やかな攪拌下で48時間インキュベートした。この溶液を、20mM HEPES緩衝液、150mM NaCl(pH7.4)に対して10kD膜(再生セルロース/Millipore)を用いるUF/DFによって濃縮させた。
【0099】
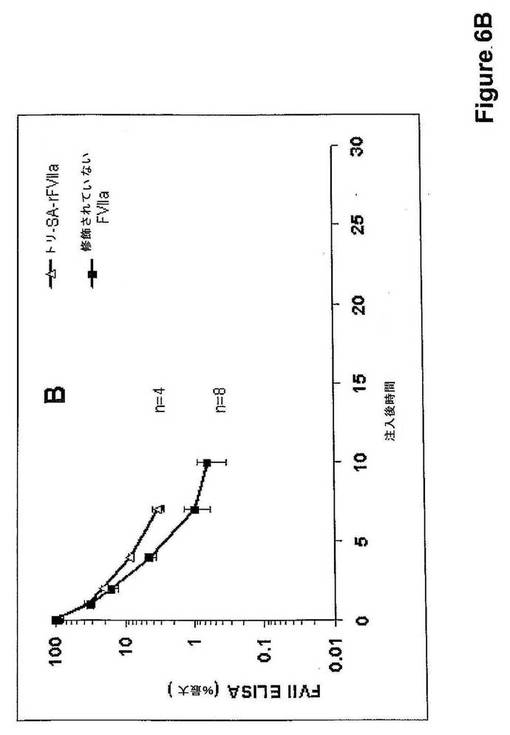
N−アセチルノイラミン酸三量体のrFVIIaとの結合体化を、KlausenおよびKornfelt(J Chromatogr A. 1995, 718:195−202)によって行われたCEによって示した。それらの結果を図7に示す。天然のrFVIIaと比較して、誘導体のより高い保持時間への明らかな変化を示す。
【0100】
(実施例10):コロミン酸の精製および誘導体化
国際公開第WO0601616A1号に記載されるように、CAをQ−セファロース FF上で陰イオン交換クロマトグラフィーによって精製した。5gのCAを、25mM NaClを含有する50mLの10mM トリエタノールアミン緩衝液(pH7.4)(=開始緩衝液)で溶解した。この溶液を、Q−セファロース FF(GE Healthcare)を充填し、開始緩衝液と平衡に達したPharmacia XK50カラム上に適用した。次いで、このカラムを8カラム容積(CV)の開始緩衝液で洗浄し、結合したCAを開始緩衝液中で、3CVで段階的に(200mM NaCl、350mM NaCl、および500mM NaCl)溶出させた。350mM NaClで溶出した画分は、SDSゲル電気泳動によって示されるように20kDaの分子量を示した。この画分を、再生セルロース(Millipore)製の5kD膜を用いて限外濾過によって濃縮し、続いて50mM リン酸緩衝液(pH7.2)に対してダイアフィルトレーションした。次いで、CAを、実施例1に記載のNaIO4で酸化させ、末端の一次アミノ基を、国際公開第WO05016973A1号に記載されるように、還元的アミノ化によって導入した。還元的アミノ化のために、11mLの2M NH4Cl−溶液を、50mM リン酸緩衝液(pH7.2)中に58mgの酸化PSA/mLを含有する20mLの溶液に加えた。次いで、1M NaOH中の5M NaCNBH3の溶液を加えて、75mMの最終濃度にした。反応を室温、pH8.0で5日間、行った。次いで、この混合物を、10mM NaClを含有する(NH4)2CO3溶液(50mg/L)に対して透析し、続いて5mM EDTAを含有する50mM リン酸緩衝液(pH8.0)に対して透析した。次いで、スルフヒドリル基を、2−イミノチオラン(Traut試薬/Pierce)を用いる末端の一次アミノ基の反応によって導入した。反応を室温で、試薬の20倍のモル過剰を有する5mM EDTAを含有する50mM リン酸緩衝液(pH8.0)で、1時間行った。最終的に、末端遊離SH基を含有するPSA溶液を、再生セルロース(Millipore)製の5kDのカットオフ膜を使用する限外濾過/ダイアフィルトレーションにかけた。
【0101】
(実施例11):ヘテロ二価性架橋剤の使用によるPSAのrFVIIaへの結合
PSA(Sigma−Aldrich)をQ−セファロース FF(GE Healthcare)上で陰イオン交換クロマトグラフィーによって精製し、実施例10に記載の末端スルフヒドリル基を化学修飾によって導入し、PSA−SHを形成した。PSA−SHのrFVIIaへの結合のために、2つの反応基、すなわちSH基への結合体化のためのマレイミド基および遊離アミノ基への結合体化のためのスルホ−NHS−エステル基を含有するヘテロ二価性水溶性架橋剤であるスルホ−EMCS((N−ε−マレイミドカプロイルオキシ)スルホスクシンイミドエステル/Pierce)を用いた。150mM NaClを含有する、20mM HEPES緩衝液(pH7.4)中の2mLのrFVIIa溶液(1.6mg/mL)に、スルホ−EMCSを加えて最終濃度の0.07mgの架橋剤/mgタンパク質にした。反応を室温で、30分間行った。続いて、実施例10によって調製した130mgのPSA−SH(100倍の過剰)を加えて、中間体リンカー/rFVIIa複合体の反応のPSA−SHへの結合を室温で、さらに2時間行った。次いで、この混合物を、ブチル−セファロース(GE−Healthcare)上でHICクロマトグラフィーによって精製した。5M NaCl溶液をこの混合物に加えて、3M NaClの最終濃度にした。次いで、この混合物をブチル−セファロース(GE−Healthcare)で充填したカラムに適用し、rFVIIa−PSA結合体の溶出を、6.7mM CaCl2を含有する50mM HEPES緩衝液(pH7.4)で行った。結合体の溶出後、pHをpH6,9に調整した。
【0102】
(実施例12):PSA−ヒドラジドの、rFVIIaの炭水化物部分への結合体化
PSAの、rFVIIaの炭水化物部分への結合体化のために、20mM HEPES緩衝液(pH6.0)(1.6mg/mL)中のrFVIIaの溶液を調製した。この溶液の9容積に対して、5mM NaIO4−溶液の1容積を加えて穏やかに混合した。酸化反応を4℃で、暗所で1時間行い、遊離アルデヒド基を生成した。次いで。亜硫酸水素ナトリウム(最終濃度5mM)を加えて、酸化反応を止めた。続いてPSA−ヒドラジド(国際公開第WO0606168A2)を加え(最終濃度10mM)、アルデヒド基への結合反応を室温で1時間行った。次いで、PSA−rFVIIa結合体を、実施例1に記載のQHyper D(Pall BioSepra)上で陰イオン交換クロマトグラフィーによって精製した。
【技術分野】
【0001】
この出願は、2006年12月15日に出願された、米国仮出願第60/875,217号に対する優先権を主張する。
【0002】
発明の分野
本発明は、1〜4のシアル酸ユニットの鎖を含む炭水化物部分に結合される凝固第VIIa因子(FVIIa)を含むタンパク質性コンストラクトに関する。本発明は、さらに、血液凝固タンパク質(特に、少なくともFVIIa、第VIII因子(FVIII)、および第IX因子(FIX)の機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において血中のFVIIa)のin vivo半減期を延ばす方法に関する。
【背景技術】
【0003】
発明の背景
血液凝固カスケードは、内因性経路、外因性経路、および共通経路の3つの異なるセグメントに分けられる。(非特許文献1)。血液凝固カスケードは、一連のセリンプロテアーゼ酵素(チモーゲン)およびタンパク質補助因子に関わる。必要な場合、不活性チモーゲン前駆体は活性型に転換され、それは、結果的に血液凝固カスケードで次の酵素に転換する。
【0004】
内因性経路は凝固第VIII因子、IX、X、XI、およびXIIを必要とする。内因性経路の開始は、プレカリクレイン、高分子量キニノーゲン、第XI因子(FXI)、および第XII因子(FXII)が負の電荷をもつ表面に曝される場合、起こる。血小板から分泌されるカルシウムイオンおよびリン脂質も必要とされる。
【0005】
外因性経路は、血管の血管腔が損傷を受けた場合、開始される。膜糖タンパク質組織因子が曝され、循環する第VII因子(FVII)に、次いで既存する少量のその活性化型FVIIaに結合する。この結合は、FVIIのFVIIaへの完全転換を促進し、続いてカルシウムおよびリン脂質の存在下で第IX因子(FIX)の第IXa因子(FIXa)への転換および第X因子(FX)の第Xa因子(FXa)への転換を促進する。FVIIaの組織因子との会合は、基質(FXおよびFIX)に対するFVIIの結合部位を近接近させることによって、および立体構造変化を誘発することによってタンパク質分解活性を増進させ、それによってFVIIaの酵素活性が増進する。外因性経路によるFX活性化の速度は、FIXa、FVlIIa、リン脂質、およびカルシウムイオンの(内因性)経路によって達成される速度の約50分の一の速さである。
【0006】
FXの活性化は、これら2つの経路の共通点である。リン脂質およびカルシウムとともに、第Va因子(FVa)および第Xa因子は、プロトロンビンをトロンビンに転換させる(プロトロンビナーゼ複合体)。次いでそれはフィブリノーゲンを切断してフィブリンモノマーを形成する。フィブリンモノマーは、重合してフィブリン鎖を形成する。第XIIIa因子(FXIIIa)は、これらのフィブリン鎖を互いに共有結合させて、剛性のメッシュを形成する。
【0007】
FVIIのFVIIaへの転換も、トロンビン、FIXa、FXa、第XIa因子(FXIa)、および第XIIa因子(FXIIa)を含むいくつかのプロテアーゼによって触媒される。血液凝固カスケードの初期相を抑制するために、組織因子経路インヒビターはFVIIa/組織因子/FXa産物複合体を標的にする。
【0008】
FVII(安定因子またはプロコンバーチンとしても公知である)は、止血および凝固で中心的役割を有する、ビタミンK依存性セリンプロテアーゼ糖タンパク質である(非特許文献2)。
【0009】
FVIIは肝臓で合成されて、48kDの1本鎖糖タンパク質として分泌される。FVIIaは、ビタミンK依存性セリンプロテアーゼ糖タンパク質のすべてと、類似するタンパク質ドメイン構造を共有する。前記タンパク質ドメイン構造は、タンパク質の脂質膜との相互作用に関与する9〜12の残基を有するアミノ末端γ−カルボキシグルタミン酸(Gla)ドメイン、カルボキシ末端セリンプロテアーゼドメイン(触媒ドメイン)、および組織因子との相互作用を調整するカルシウムイオン結合部位を含有する2つの上皮増殖因子様ドメインからなる。
【0010】
γ−グルタミルカルボキシラーゼは、分子のアミノ末端部分でGla残基のカルボキシル化を触媒する。カルボキシラーゼは、その作用のために還元型ビタミンKに依存し、それはエポキシド型に酸化される。ビタミンKエポキシド還元酵素は、ビタミンKのエポキシド型を還元型に再変換するために必要である。
【0011】
FVIIの主要部分は、チモーゲン型で血漿中で循環し、この型の活性化は、アルギニン152とイソロイシン153との間のペプチド結合の切断をもたらす。結果として生じる活性化FVIIaは、NH2由来軽鎖(20kD)および単一のジスルフィド結合(Cys135〜Cys262)を介して連結されるCOOH末端由来重鎖(30kD)からなる。軽鎖は膜結合Glaドメインを含有し、重鎖は触媒ドメインを含有する。
【0012】
遺伝因子および環境因子によって決定されるFVIIの血漿中濃度は、約0.5mg/mLである(非特許文献3)。FVIIの異なる遺伝子型は、FVIIの平均レベルで数倍の相違をもたらすことができる。血漿中FVIIレベルは、健常な女性では妊娠中に上昇し、また年齢とともに上昇し、女性および高トリグリセリド血症の人においてはより高い。FVIIは、すべての凝血原因子のなかでその半減期が最も短い(3〜6時間)。FVIIaの血漿中の平均濃度は、健常な個体で3.6ng/mLであり、かつFVIIaの循環半減期は、他の凝固因子と比較すると相対的に長い(2.5時間)。
【0013】
遺伝性FVII欠乏は、稀な常染色体劣性遺伝出血性障害であり、その有病率は一般人口50万人あたり1症例と推定される(非特許文献4)。インヒビターからの後天性FVII欠乏も非常に稀である。症例は、また、薬物(例えばセファロスポリン、ペニシリン、および経口抗凝固剤)に関連して発症するFVII欠乏が報告されている。さらにまた、後天性FVIl欠乏は、自然発症的に、または他の状態(例えば骨髄腫、敗血症、再生不良性貧血)とともに、インターロイキン−2および抗胸腺細胞グロブリン療法で発症することが報告されている。
【0014】
補充療法は、FVII欠乏症の患者に対する処置の中心である(非特許文献5)。これは、新鮮凍結血漿(FFP)、プロトロンビン複合体濃度物(PCC)、または血漿由来FVII濃縮物を用いて、従来から成し遂げられてきた。しかし、組換えFVIIa(rFVIIa)は、今や、これらの患者での療法のために広く使用されている。
【0015】
rFVIIaは、また、インヒビターを有する血友病AおよびBの患者の出血を処置するために開発され、大手術(例えば整形外科手術)中でさえも止血を誘導することが分っている(非特許文献6)。rFVIIaは、BHK細胞培養物で生成され、かつ血漿由来FVIIaに極めて類似することが示されている。血友病処置でのrFVIIaの使用は、トロンビン活性化血小板の表面へのFVIIaの低親和性結合に基づく。外来性rFVIIaの薬理量を投与することによって、損傷部位の血小板表面上のトロンビン生成がFVIII/FIXの存在とは独立して増強される。急速なトロンビン形成が増大する結果、密接なフィブリン止血血栓が形成される。
【0016】
FVII欠乏およびインヒビター合併血友病AおよびBの処置のために本来は開発されたのだが、rFVIIaの新規適応(症例報告および小規模の臨床試験に基づく)としては、肝疾患、血小板減少症、または質的な血小板機能異常症の患者での使用、および非凝固障害を有する患者が大手術または大外傷に起因する出血の場合の使用が挙げられる。
【0017】
ポリペプチド治療薬(例えばFVIIaを含む血液凝固タンパク質)は、タンパク質分解酵素によって急速に分解され、抗体によって中和される。これは、ポリペプチド治療薬の半減期および循環時間を減少させ、それによってポリペプチド治療薬の治療有効性を限定する。所望のFVIIaの治療効果または予防効果を達成して維持するには、相対的に高い投与量および頻繁な投与が必要である。結果として、適切な投与量の調節を得るのは困難であり、頻繁な静脈内投与を必要とするために、患者の生活様式を制限する。従って、長い循環半減期によって改善されたFVIIa分子は、必要な投与回数を減少させる。
【0018】
原則として、血液循環におけるタンパク質の半減期を延ばすには4つの一般的な選択肢がある。すなわち、
・化学修飾または酵素修飾の導入
・血液循環中のタンパク質を保護する担体分子の使用
・半減期を延ばす突然変異体の構築
・分解経路の修飾。
【0019】
本発明は、血液凝固タンパク質の改善(特に化学修飾によるFVIIa分子)を教示する。治療用ポリペプチドの化学修飾に関しては、これまでいくつかのアプローチが用いられている。
【0020】
ポリペプチド薬のPEG化はポリペプチド薬を保護し、それらの薬力学的プロファイルおよび薬物動態プロファイルを改善する(非特許文献7)。PEG化プロセスは、ポリエチレングリコール(PEG)の繰り返し単位をポリペプチド薬に付着させる。PEG分子は、大きな流体力学的容積(球状タンパク質の大きさの5〜10倍)を有し、水溶性で高度に水和し、流動性が高く、無毒、非免疫原性であり、および身体から迅速に除去される。分子のPEG化は、酵素分解に対する薬物耐用の増加、in vivo半減期の増加、投薬頻度の減少、免疫原性の低下、物理的安定性および熱安定性の増加、溶解性の増加、液体安定性の増加、ならびに凝集の減少をもたらすことが可能である。最初のPEG化薬物は、1990年代の初めにFDAによって承認された。今のところ、FDAは、経口用、注射用、および局所投与用のいくつかのPEG化薬物を承認している。
【0021】
GlycoPEGylation(登録商標)技術は、インタクトなグリコシル結合基を介してペプチドに共有結合したPEGによる、PEGポリマーとペプチドとの間にペプチド結合体を提供する方法が含まれる。
【0022】
リポソームを用いて、種々の分子(例えばDNA、アンチセンスRNA、抗生物質、抗癌剤および抗真菌剤、抑制剤/活性剤、抗体(免疫リポソーム)、ならびに抗原(ワクチン用)がカプセル化されている。
【0023】
リン脂質も、PEG(PEG−リポソーム)(例えばアミド結合を介して)、カルボキシ−PEGおよび精製ダイズ・ホスファチジルエタノールアミン(PE)、エステルおよびカルバミン酸誘導体に結合体化されることが可能であり、今日最も広く使用されているのはカルバミン酸誘導体である(特許文献1)。最も一般的に用いられているPEGの分子量は、2,000および5,000であるが、600から12,000の範囲のPEGも用いられている。
【0024】
酸性単糖置換タンパク質は、最初に、特許文献2に開示された。この特許では酸性単糖(すなわちn−アセチルノイラミン酸およびグルコン酸)は、インスリン、ヒト成長ホルモン、またはアルブミンのα−アミノ基またはε−アミノ基の上に置換され、ポリペプチドの抗原性を減少させる。
【0025】
コロミン酸(CA)とも称されるポリシアル酸(PSA)も、天然の多糖である。ポリシアル酸は、α(2→8)ケトシド結合によるN−アセチルノイラミン酸のホモポリマーであり、その非還元末端に隣接ジオール基を含有する。ポリシアル酸は、負の電荷をもち、人体の天然の成分である。ポリシアル酸は、大量に細菌から容易に生成されることが可能であり、所定の物理的特性を有する(特許文献3)。人体においてポリシアル酸と化学的および免疫学的に同一である細菌性ポリシアル酸は、タンパク質に結合する場合でさえ、非免疫原性である。他のポリマー(例えばPEG)と異なり、ポリシアル酸は生分解性である。カタラーゼおよびアスパラギナーゼに共有結合するコロミン酸は、タンパク質分解酵素または血漿の存在下で酵素安定性の増加をもたらした。ポリシアル酸化および修飾されていないアスパラギナーゼによるin vivo比較研究は、ポリシアル酸化が酵素の半減期を増加させることを明らかにした(非特許文献8)。
【0026】
しかし、特許文献2に記載されるように、酸性単糖に結合体化したポリペプチドからなる治療化合物は、現在までに市販されていない。対照的に、特許文献3は、治療化合物の多糖部分がポリマー鎖に少なくとも5個(他の実施形態では少なくとも20または50個)のシアル酸残基を有するはずであることを教示する。多糖は通常、同時精製する内毒素の固有のリスクを保有する細菌で生成されるので、シアル酸ポリマー長鎖の精製は、内毒素含有物が増加する可能性を高め得る。1〜4のシアル酸ユニットを有するPSA短分子も、合成的に調製され得(非特許文献9;非特許文献10)、従って高レベルの内毒素のリスクを最小化する。
【0027】
特許文献4は、他の多くのタンパク質の中でFVIIがPEG化され得ることを示唆しているが、その開示を支持する任意の実施例を含有していない。
【0028】
特許文献5が教示する結合体は、ポリペプチドに共有結合する少なくとも1つの非ポリペプチド部分を含む結合体を教示する。ここで、前記ポリペプチドのアミノ酸配列は、前記非ポリペプチド部分のための結合基を含む少なくとも1つのアミノ酸残基が導入または除去されている点で野生型FVIIまたはFVIIaのそれと異なる。非ポリペプチド部分に対しては、特にPEGが示唆された。
特許文献6は、FVIIポリペプチドまたはFVII関連ポリペプチドを開示する。ここで、ポリペプチドは、アスパラギン結合型および/またはセリン結合型の1つ以上のオリゴ炭水化物部分を含み、および前記オリゴ糖基のうちの少なくとも1つが少なくとも1つのポリマー基(PEG、「グリコPEG化」)に共有結合される。
【先行技術文献】
【特許文献】
【0029】
【特許文献1】米国特許第6,593,294号明細書
【特許文献2】米国特許第3,847,890号明細書
【特許文献3】米国特許第5,846,951号明細書
【特許文献4】国際公開第98/32466号パンフレット
【特許文献5】国際公開第01/58935号パンフレット
【特許文献6】米国特許出願公開第2005/0113565号明細書
【非特許文献】
【0030】
【非特許文献1】Schenoneら、Curr Opin Hematol.(2004)11:272−7
【非特許文献2】Eigenbrot, Curr Protein Pept Sci.(2002)3:287−99
【非特許文献3】Pinottiら、Blood.(2000)95:3423−8
【非特許文献4】Acharyaら、J Thromb Haemost(2004)2248−56
【非特許文献5】Marianiら、Semin Hematol.(2006)43(Suppl 1):S42−7
【非特許文献6】Hedner, J Biotechnol.(2006)124:747−57
【非特許文献7】HarrisおよびChess, Nat Rev Drug Discov.(2003)2:214−21
【非特許文献8】Fernandes and Gregoriadis,Int J Pharm.(2001)217:215−24
【非特許文献9】Kangら、Chem Commun.(2000)227−8
【非特許文献10】RessおよびLinhardt,Current Organic Synthesis.(2004)1:31−46
【発明の概要】
【発明が解決しようとする課題】
【0031】
従って、当該技術分野において、改善した血漿由来もしくはrFVII、修飾型FVII、またはFVII関連ポリペプチドを含む凝固タンパク質製剤を提供する組成物および方法の必要性が残っている。
【課題を解決するための手段】
【0032】
本発明は、血漿または組換え第VIIa因子(FVIIa)またはその生物学的に活性な誘導体、1〜4のシアル酸ユニットからなる鎖に結合される前記FVIIaまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトを提供する。ここで、タンパク質性コンストラクトのin vivo半減期は、1〜4のシアル酸ユニットからなる鎖が欠けているFVIIaまたはその誘導体と比較すると、哺乳類(特にヒト)の血中で実質的に延びている。さらに、前記タンパク質性コンストラクトを含有する医薬組成物ならびに前記タンパク質性コンストラクトを用いてFVIIa、FVIII、およびFIXのうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類の血中FVIIaのin vivo半減期を延ばす方法が本発明によって提供される。本発明のタンパク質性コンストラクトは、正常なレベルの凝固因子を有する哺乳類における外傷または手術の場合に出血を制御するために投与されることもできる。
【0033】
本発明の一実施形態において、タンパク質性コンストラクトは、(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子と、(b)前記FVIIa分子に結合する1〜4のシアル酸ユニットを含む、生理的に許容される少なくとも1つの炭水化物部分とを含み、
ここで前記タンパク質性コンストラクトのin vivo半減期は、前記炭水化物部分に結合されていないFVIIa部分のin vivo半減期と比較すると、哺乳類の血液中で延びている。
【0034】
本発明の別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで該コントラクトのin vivo半減期が前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約2倍増加される。別の実施形態において、そのin vivo半減期が前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約3倍増加される、上述のタンパク質性コンストラクトが提供される。さらに別の実施形態において、生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に直接的に共有結合する、上述のタンパク質性コンストラクトが提供される。
【0035】
本発明のさらに別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで生理的に許容される炭水化物部分は、前記FVIIa分子の少なくとも1つのアミノ酸残基に非共有結合される。さらに別の実施形態において、上述のタンパク質性コンストラクトが提供され、ここで生理的に許容される炭水化物部分がポリシアル酸またはその誘導体である。
【0036】
本発明の一実施形態において、医薬組成物は有効量の上述のタンパク質性コンストラクトと、薬学的に許容される担体、希釈剤、塩、緩衝剤、および賦形剤からなる群から選択される1つ1つ以上の化合物とを含み提供される。
【0037】
本発明の別の実施形態において、FVIIa、FVIII、およびFIXのうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において出血を制御する方法は、上述のタンパク質性コンストラクトを投与することを含み、提供される。さらに別の実施形態において、哺乳類において手術または外傷の間に出血を制御する方法は、上述のタンパク質性コンストラクトを投与することを含み、提供される。
【0038】
本発明のさらに別の実施形態において、容器にパッケージされた上述のタンパク質性コンストラクトの有効量を含み、場合により第2の治療薬を含有し、前記容器に添付されたまたは前記容器とともにパッケージされたラベルをさらに含むキットを提供する。前記ラベルは、容器の内容物を説明し、哺乳類での出血を制御するための前記容器の内容物の使用に関する適応および/または使用説明書を提供する。さらに別の実施形態において、上述のキットを提供し、ここで前記容器がバイアルまたは瓶または予め充填されたシリンジである。
本発明の好ましい実施形態では、例えば以下が提供される:
(項目1)
タンパク質性コンストラクトであって、
(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子と、(b)該FVIIa分子に結合される1〜4のシアル酸ユニットを含む少なくとも1つの生理的に許容される炭水化物部分とを含み、
該炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、該コンストラクトのin vivo半減期が哺乳類の血中において延長している、タンパク質性コンストラクト。
(項目2)
前記コンストラクトのin vivo半減期が、前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約2倍増加する、請求項1に記載のタンパク質性コンストラクト。
(項目3)
前記コンストラクトのin vivo半減期が、前記炭水化物部分に結合されていないFVIIa分子のin vivo半減期と比較すると、少なくとも約3倍増加する、請求項1に記載のタンパク質性コンストラクト。
(項目4)
前記生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に直接的に共有結合する、請求項1に記載のタンパク質性コンストラクト。
(項目5)
前記生理的に許容される炭水化物部分が前記FVIIa分子の少なくとも1つのアミノ酸残基に非共有結合する、請求項1に記載のタンパク質性コンストラクト。
(項目6)
前記生理的に許容される炭水化物部分がポリシアル酸またはその誘導体である、請求項1に記載のタンパク質性コンストラクト。
(項目7)
請求項1に記載のタンパク質性コンストラクトの有効量と、薬学的に許容される担体、希釈剤、塩、緩衝剤、および賦形剤からなる群から選択される1つ以上の化合物とを含む、医薬組成物。
(項目8)
FVIIa、第VIII因子(FVIII)、および第IX因子(FIX)のうちの少なくとも1つの機能上の欠損または欠乏に関連する出血性障害を有する哺乳類において出血を制御する方法であって、該方法は、請求項1に記載のタンパク質性コンストラクトを投与することを含む、方法。
(項目9)
哺乳類において手術または外傷の間に出血を制御する方法であって、該方法は、請求項1に記載のタンパク質性コンストラクトを投与することを含む、方法。
(項目10)
容器にパッケージされた、請求項1に記載のタンパク質性コンストラクトの有効量を含むキットであって、該キットが場合により第2の治療薬を含有し、該容器に添付されたまたは該容器とともにパッケージされたラベルをさらに含み、該ラベルが該容器の内容物を説明し、哺乳類における出血を制御するための該容器の該内容物の使用に関する適応および/または使用説明書を提供する、キット。
(項目11)
前記容器がバイアルまたは瓶または予め充填されたシリンジである、請求項10に記載のキット。
【図面の簡単な説明】
【0039】
【図1】PSAとの結合体化後のrFVIIaのSDS−PAGEを示す。
【図2】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。
【図3】rFVIIa−PSA結合体および修飾されていないrFVIIa(抗原レベル)のラットにおける薬物動態を示す。
【図4】PSAとのN末端結合体化後のrFVIIaのSDS−PAGEを示す。
【図5】モノSA−rFVIIaおよびトリSA−rFVIIaのキャピラリー電気泳動を示す。
【図6A】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。A:モノSA−rFVIIa。
【図6B】rFVIIa−PSA結合体および修飾されていないrFVIIaのラットにおける薬物動態を示す。B:トリSA−rFVIIa。
【図7】N−アセチルノイラミン酸三量体のキャピラリー電気泳動を示す。
【発明を実施するための形態】
【0040】
発明の詳細な説明
本発明は、本明細書に記載のFVII部分に限定されないことを理解すべきである。本発明の一態様は、血液凝固カスケードの1つのメンバー、血漿の(すなわち血漿由来)および/または組換えのFVIIaまたはその生物学的に活性な誘導体(以下「PSA−FVIIa結合体」と表す)、1〜4のシアル酸部分と結合される前記FVIIまたはその前記生物学的に活性な誘導体を含むタンパク質性コンストラクトに関する。ここで、前記FVIIまたはその前記生物学的に活性な誘導体のin vivo半減期は、哺乳類の血中で延びる。本明細書に用いるように、「タンパク質性コンストラクト」という用語は、(a)血漿FVIIa、組換えFVIIa(rFVIIa)、およびFVIIaの生物学的に活性な誘導体からなる群から選択される活性化第VII因子(FVIIa)分子、および(b)前記FVIIa分子に結合した1〜4のシアル酸ユニットを含む少なくとも1つの生理的に許容される炭水化物部分をいう。本明細書に用いるように、「血漿の」という用語は、「血漿由来の」を言及する。
【0041】
FVIIaポリペプチドおよびポリヌクレオチド
本発明に有用なFVIIa分子としては、完全長タンパク質、タンパク質の前駆体、タンパク質の生物活性サブユニットまたは機能的サブユニットまたは断片、およびその機能的誘導体が挙げられる。FVIIaへの言及は、かかるタンパク質のすべての潜在的な形を含むことを意図される。
【0042】
本発明によれば、「組換え第VIIa因子」(rFVIIa)という用語は、特定の制限の根底にあるのではなく、かつ組換えDNA技術を介して取得する任意のrFVIIa、非相同的もしくは天然の、またはそれらの生物学的に活性な誘導体も含み得る。ある実施形態において、この用語は、タンパク質および核酸(例えば遺伝子、mRNA前駆体、mRNAおよびポリペプチド)、多型変異体、対立遺伝子、突然変異体、ならびに種間相同体を包含する。種間相同体は、(1)参照核酸または本明細書に記載するアミノ酸配列によってコードされるポリペプチドと、少なくとも約25、50、100、200、300、400、またはそれ以上のアミノ酸(成熟タンパク質の場合、完全長配列の406アミノ酸まで)からなる領域にわたり、約60%を超えるアミノ酸配列同一性、65%、70%、75%、80%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、または99%を超えるアミノ酸配列同一性を有するアミノ酸配列を有する;(2)本明細書に記載の参照アミノ酸配列を含む免疫原、その免疫原断片、および保存的に修飾されたその変異体に対して産生される抗体(例えばポリクローナル抗体)に特異的に結合する;(3)本明細書に記載の参照アミノ酸配列をコードする核酸およびその保存的に修飾された変異体に、ストリンジェントなハイブリダイゼーション条件下で特異的にハイブリダイズする;(4)本明細書に記載の参照核酸配列と、少なくとも約25、50、100、150、200、250、500、1000、またはそれ以上のヌクレオチド(成熟タンパク質の完全長配列の1218ヌクレオチドまで)からなる領域にわたり、約95%を超える核酸配列同一性、約96%、97%、98%、99%またはそれを超える核酸配列同一性を有する核酸配列を有する。
【0043】
本明細書に用いるように、「内因性FVIIa」は、前記哺乳類から生ずるFVIIaを含む。内因性FVIIaは、前記哺乳類に存在するトランスジーンまたは他の任意の外来DNAから転写されるFVIIaも含む。本明細書で用いるように、「外因性FVIIa」は、前記哺乳類から生じないFVIIaを含む。
【0044】
変異体(または類似体)ポリペプチドは、1つ以上のアミノ酸残基がFVIIaアミノ酸配列を補充する、挿入変異体を含む。挿入は、タンパク質のいずれかの末端または両末端に位置していることもあり、またはFVIIaアミノ酸配列の内部領域内に位置することもある。いずれかの末端または両末端に残基をさらに有する挿入変異体は、例えば融合タンパク質およびアミノ酸タグまたはアミノ酸標識を含むタンパク質を含むことができる。例えば、FVIIa分子は、特に、FVIIa分子が細菌細胞(例えば大腸菌(E.coli))内で、組換えで発現される場合、場合によりN末端メチオニンを含有することもある。
【0045】
欠失体では、FVIIaポリペプチドで1つ以上のアミノ酸残基が除去される。欠失は、FVIIaポリペプチドの一方または両末端で、またはFVIIaアミノ酸配列内の1つ以上の残基を取り除くことで引き起こされ得る。従って、欠失体はFVIIaポリペプチド配列のすべての断片を含む。
【0046】
置換変異体では、FVIIaポリペプチドの1つ以上のアミノ酸残基が除去されて、代替残基と置換する。一態様において、置換は、天然において保存的であり、このタイプの保存的置換は当該技術分野では公知である。あるいは、本発明は、非保存的でもある置換を包含する。代表的な保守的置換は、Lehninger(Biochemistry,
2nd Edition;Worth Publishers, Inc., New York (1975), pp.71−77)に記載されており、以下に記載する。
【0047】
【化1】
あるいは、代表的な保存的置換は、以下に記載する。
【0048】
【化2】
ポリヌクレオチド配列またはポリペプチド配列は、一般的に、霊長類(例えばヒト)齧歯類(例えばラット、マウス、ハムスター)、ウシ、ブタ、ウマ、ヒツジ、または任意の哺乳類にも限定されないが哺乳類に由来する。本発明の核酸およびタンパク質は、組換え分子(例えば非相同的および野生型配列もしくはその変異体をコードする、または非天然の)であり得る。参照ポリヌクレオチド配列およびポリペプチド配列としては、例えば、ゲノム配列のためのGenBank受入番号J02933、cDNAのためのM13232(Hagenら、PNAS 1986;83:2412−6)、およびポリペプチド配列のためのP08709(それら開示内容全体を参考として本明細書で援用される)が挙げられる。FVIIの種々の多型性は、例えばSabater−Llealら、(Hum
Genet.2006;118:741−51)(それら開示内容全体を参考として本明細書で援用される)に記載されている。
【0049】
本明細書で用いるように、「生物学的に活性な誘導体」または「生物活変異体」としては、前記分子と同一の機能的特性および/または生物学的特性(例えば結合特性)、ならびに/または同一の構造基盤(例えばペプチド主鎖または塩基性ポリマーユニット)を実質的に有する、任意の誘導体または種々の分子が挙げられる。
【0050】
本明細書で用いるように、「血漿誘導型FVIIa」または「血漿」は、凝固経路を活性化する特性を有する哺乳類から採血した血液中で見出されたタンパク質のすべての形状を含む。
【0051】
本明細書で用いるように、「組換えFVIIa」は、組換えDNA技術を介して得たrFVIIaを含む。組換えFVIIaは、当該技術分野で公知の任意の方法によって生成され得る。1つの特定の例は、米国特許第4,784,950号に開示されている。かかるrFVIIaの例は、Novo Nordiskによって製造、販売されるNovoSevenである。
【0052】
FVIIa生成および発現
rFVIIaの生成は、以下に関して当該技術分野で公知の任意の方法を含み得る。(i)遺伝子工学(例えばRNA逆転写および/またはDNAの増幅)による組換えDNAの生成、(ii)組換えDNAを、トランスフェクション(例えば電気穿孔またはマイクロインジェクションを介して)によって原核細胞または真核細胞に導入すること、(iii)前記形質転換させた細胞を、例えば連続的方法またはバッチ式方法で培養すること、(iv)例えば恒常的にまたは誘導により、rFVIIaを発現させる、(v)例えば培地から前記FVIIaを分離すること、または形質転換した細胞を収集すること、(vi)続いて、例えばアニオン交換クロマトグラフィーまたはアフィニティークロマトグラフィーを介して、精製したrFVIIaを得ること。
【0053】
rFVIIaは、薬理学的に許容されるrFVIIa分子を生成することよって特徴づけられる、適切な原核または真核宿主系で発現させることで生成することができる。真核細胞の例としては、哺乳類細胞(例えばCHO、COS、HEK293、BHK、SK−Hep、およびHepG2)がある。本発明によってrFVIIaの生成または分離のために用いられる試薬もしくは条件は、特に限定されず、当該技術分野で公知の任意の系または市販されているものを利用することができる。
【0054】
多種多様なベクターを用いて、rFVIIaを調製することができ、真核および原核発現ベクターから選択されることができる。原核発現のためのベクターの例としては、様々のプラスミド(例えばpRSET、pET、pBADなど)が挙げられ、原核発現ベクターで用いられるプロモーターとしては、lac、trc、trp、recA、araBADなどが挙げられる。真核発現のためのベクターの例としては、以下が挙げられる。すなわち、
(i)酵母での発現のため、プロモーター(例えばAOX1、GAP、GAL1、AUG1など)を用いるベクター(例えばpAO、pPIC、pYES、pMET)、
(ii)昆虫細胞での発現のため、プロモーター(例えばPH、p10、MT、Ac5、OpIE2、gp64、po1hなど)を用いるベクター(例えばpMT、pAc5、pIB、pMIB、pBAC)、ならびに(iii)哺乳類細胞での発現のため、プロモーター(例えばCMV、SV40、EF−1、UbC、RSV、ADV、BPV、およびβ−アクチン)を用いるベクター(例えばpSVL、pCMV、pRc/RSV、pcDNA3、pBPVなど)、およびウイルス系由来のベクター(例えばワクシニアウイルス、アデノ関連ウイルス、ヘルペスウイルス、レトロウイルスなど)。
【0055】
シアル酸
本明細書で用いるように、「シアル酸部分」は、シアル酸のモノマーまたはポリマーが含まれる。これらは、水溶液中または懸濁液中で可溶性であり、何ら否定的な影響(例えばPSA−FVIIa結合体を医薬的有効量で哺乳類に投与した後の副作用)がない。本発明によって用いられるシアル酸ユニットには特に限定されない。一態様において、シアル酸ポリマーは、1〜4ユニットを有するとして特徴づけられる。また異なるシアル酸ユニットが鎖状に組み合わせられ得る。
【0056】
シアル酸部分は、例えば米国特許第4,356,170号に記載の方法(参考として本明細書で援用される)によって、FVIIaに結合され得る。本発明の一実施形態において、多糖化合物は、天然の多糖、天然の多糖の誘導体、または天然多糖誘導体であり得る。通常、化合物中の糖類残基のすべては、シアル酸残基である。
【0057】
PSAをポリペプチドに結合させる他の技法も公知である。例えば、米国特許出願公開第2007/0282096号は、タンパク質への例えばPSAのアミンまたはヒドラジン誘導体の結合体化を記載している。さらに、米国特許出願公開第2007/0191597は、還元終端で基質(例えばタンパク質)との反応のためのアルデヒド基を含有するPSA誘導体を記載している。
【0058】
本発明の一実施形態において、多糖化合物のポリシアル酸部分は高度に親水性である。別の実施形態において、化合物全体が高度に親水性である。親水性は、主に、シアル酸ユニットのペンダントカルボキシル基ならびにヒドロキシル基によって与えられる。糖類ユニットは、他の官能基(例えばアミン基、ヒドロキシル基、もしくは硫酸基、またはそれらの組み合わせ)を含有し得る。これらの基は、天然の糖類化合物上に存在し得、または誘導体多糖化合物に導入され得る。
【0059】
本発明の特定の用途の多糖化合物は、細菌によって産生されるものである。これらの天然の多糖の一部は、糖脂質として知られている。多糖化合物に、肝細胞およびクッパー細胞のガラクトース受容体によって認識される傾向がある末端ガラクトースユニットが実質的にない場合、特に有利である。
【0060】
結合
FVIIaは、当業者にとって公知の種々の技術のいずれかによって多糖化合物に共有結合され得る。種々の例は、米国特許第5,846,951号の第7欄15行目から第8欄5行目に確認される。
【0061】
例としては、FVIIaまたは多糖のいずれか一方のカルボキシル基と、他方のアミン基との間のペプチド結合を介する結合、または一方のカルボキシル基と、他方のヒドロキシル基との間のエステル結合が挙げられる。活性成分(例えばFVIIa)が多糖化合物に共有結合し得ることによる別の結合は、反応される活性成分上の遊離アミノ基と、過ヨウ素酸酸化によってポリマーの非還元末端で形成されたアルデヒド基との間のシッフ塩基を介する(Jennings and Lugowski, J Immunol.1981;127:1011−8;Fernandes and Gregoriadis,
Biochim Biophys Acta.1997;1341;26−34)。生成されたシップ塩基は、NaCNBH3による特異的還元によって安定化され、二級アミンを形成することができる。別のアプローチは、事前の酸化の後にNH4Clを用いる還元的アミノ化によってポリシアル酸(PSA)内に末端遊離アミノ基を生成することである。二価性試薬を用いて、2つのアミノ基または2つのヒドロキシル基を結合するため使用できる。例えば、アミノ基を含有するPSAはBS3(ビス(スルホスクシンイミジル)スベラート/Pierce, Rockford, IL)等の試薬を用いてタンパク質のアミノ基に結合させることができる。さらに、ヘテロ二価架橋試薬(例えばスルホ−EMCS(N−ε−マレイミドカプロイルオキシ)スルホスクシンイミドエステル/Pierce)を用いて、例えばアミン基およびチオール基を結合することができる。
【0062】
別のアプローチでは、事前の酸化およびアルデヒド官能基の生成の後、PSAヒドラジドを調製して、タンパク質の炭水化物部分に結合させることができる。
【0063】
治療用タンパク質の遊離アミン基は、シアル酸残基の1−カルボキシル基と反応させることも可能であり、ペプチジル結合を形成し、またはエステル結合が活性成分上で1−カルボン酸基およびヒドロキシルまたは他の適切な活性基の間に形成されることが可能である。あるいは、カルボキシル基は、脱アセチル化5−アミノ基によってペプチド結合を形成することもある。医薬的活性化合物の分子のアルデヒド基は、シアル酸残基のN−脱アセチル化5−アミノ基とシッフ塩基を形成し得る。
【0064】
あるいは、多糖化合物は、非共有の方法で医薬的活性化合物(例えばFVIIa)と結合し得る。例えば、多糖化合物および医薬的活性化合物は、疎水性相互作用を介して(例えば多糖化合物の脂質成分を介して)疎水性医薬的活性化合物と結合され得る。他の非共有結合は、逆帯電したイオンが互いに引きつけることによる静電相互作用を介することも可能である。
【0065】
医薬的活性化合物は、化学量論量(例えば1:1)で多糖化合物に直接的に共有結合され得る。あるいは、多糖化合物の2つ以上の分子は結合されて活性成分の1つの分子になり得る。
【0066】
用途
本発明は、少なくとも1つの生理的に許容されるシアル酸部分に結合されていないFVIIaのin vivo半減期と比較して、血液凝固タンパク質(特に、FVIIaの機能上の欠損または欠乏に関連する出血性障害を有する、FVIIaまたはその生物学的に活性な誘導体)のin vivo半減期を増加させることに関する。本発明のPSA−FVIIa結合体は、FVIIIおよびFIXのうちの少なくとも1つの機能上の欠損または先天性欠乏または後天性欠乏に関連する出血性障害の処置のためにさらに用いられることができる。
【0067】
療法における最先端技術により、さらに国際ガイドラインおよび規定により、注入されるFVIIaの薬物動態は、有効性の有効な代理マーカーとして認識され、容認されている(Bjorkman and Bemtrop, Clin Pharmacokinet.2001;40:815−32)。
【0068】
これは、機能活性に関する標準化試験によって特徴づけられた、注入されたFVIIa産物が血流中で見出され、凝固血液カスケードの成分として予想通りにそこで作用するという確認された仮定に基づく。従って、動物モデルにおける任意の薬物動態分析でも、FVIIa産物で処置される患者において予想される有効性を予測する。
【0069】
半減期
本発明の一実施形態において、タンパク質性コンストラクトのin vivo半減期は、延ばされる。関連する実施形態において、シアル酸に結合されないFVIIaと比較すると、タンパク質性コンストラクトのin vivo半減期は、少なくとも2倍延び、別の実施形態において、そのin vivo半減期は少なくとも3倍延びる。FVIIa半減期の延長は、以下の実施例で説明するようにラットでの薬物動態を測定することによって評価され得る。
【0070】
投与
投与経路は、特定の限定を示さない。一実施形態において、本発明のタンパク質性コンストラクトは、注射(例えば静脈注射、筋肉内注射、または腹腔内注射)によって投与され得る。
【0071】
本発明のタンパク質性コンストラクトを含む組成物をヒトまたは試験動物に投与するため、一態様において、その組成物は1つ以上の薬学的に許容される担体を含む。「医薬的に」または「薬理学的に許容される」という用語は、以下の記載のように、安定しており、タンパク質分解(例えば凝集および切断産物)を抑制し、さらに、当該技術分野で周知の経路を用いて投与される場合、アレルギー反応または他の副作用を出現させない分子化合物および組成物をいう。「薬学的に許容される担体」としては、上記に開示した薬剤を含む、ありとあらゆる臨床的に有用な溶媒、分散媒、被覆剤、抗菌および抗真菌剤、等張および吸収遅延剤などが挙げられる。
【0072】
本明細書で用いるように、「有効量」は、上記に概要を述べるように、出血性障害を有する哺乳類を処置するのに適している用量が含まれる。
【0073】
組成物は、経口的に、局所的に、経皮的に、非経口的に、吸入スプレーによって、経膣的に、直腸に、または頭蓋内注射によって投与されてもよい。本明細書で用いるように、「非経口的に」という用語は、皮下注射、静脈内、筋肉内、大槽内注射、または注入法が挙げられる。静脈内、皮内、筋肉内、乳房内、腹腔内、くも膜下腔内、眼球後、肺内の注射、および/または特定部位での外科的移植による投与もまた考えられる。通常、組成物は、発熱物質ならびにレシピエントに有害になり得る他の不純物が本質的に取り除かれる。
【0074】
組成物の単回投与または複数回投与は、処置を行う医師によって選択される用量レベルおよびパターンで行われ得る。疾患の予防または処置に関して、適切な投与量は、上述のように処置すべき疾患の種類、疾患の重症度、および疾患の経過、薬物が予防目的または治療目的で投与されるかどうか、治療歴、患者の臨床歴および薬物応答、ならびに主治医の裁量に依存する。
【0075】
医薬組成物
本発明は、また、上記に明示したように、有効量のタンパク質性コンストラクトを含む医薬組成物に関する。医薬組成物は、薬学的に許容される担体、希釈剤、塩、緩衝剤、または賦形剤をさらに含み得る。医薬組成物を用いて、上記に明示した出血性障害を処置することができる。本発明の医薬組成物は、溶液または凍結乾燥生成物であり得る。タンパク質の安定な溶液、および特にFVIIaを形成する公知の方法が数多くある。1つの例は、米国特許第5,874,408号に開示されている。医薬組成物の溶液を、任意の適切な凍結乾燥プロセスにかけることが可能である。
【0076】
キット
追加の態様として、本発明は、被験者への投与のためにその用途を促進する方法でパッケージされた本発明の組成物を含むキットが挙げられる。一実施形態において、かかるキットには、本明細書に記載の化合物または組成物(例えばタンパク質性コンストラクトを含む組成物)が含まれる。前記化合物または組成物は、密封された瓶またはベスル等の容器にパッケージされ、前記方法を実践する際の化合物または組成物の用途を説明するラベルが容器に添付されるか、またはパッケージに含まれる。一実施形態において、キットは、タンパク質性コンストラクトを含む組成物を有する第1の容器と、第1の容器内の組成物のための生理的に許容される再構成溶液を有する第2の容器とを含有する。一態様において、化合物または組成物は、単位用量形態でパッケージされる。キットは、投与の特定の経路によって、組成物を投与するのに適した装置をさらに含み得る。好ましくは、キットは、治療用タンパク質またはペプチド組成物の用途を説明するラベルを含有する。
【0077】
本発明を、以下の実施例でさらに例示するが、それらに何ら限定されない。
【実施例】
【0078】
(実施例1):rFVIIa内のリジン残基のコロミン酸による修飾
シアル酸(コロミン酸、CA)によるリジン残基の修飾を、JenningsおよびLugowski(J Immunol, 1981;127;1011−8)によって記載されるように行った。この処置のために、Sigma(Sigma−Aidrich,
St. Louis;MO)製のCAを使用した。0.1M NaIO4を含有するCAの水溶液(濃度20mg/mL)を暗所で、室温で15分間攪拌し、CAを酸化させた。活性化CA溶液/mLに2mLのエチレングリコールを加えて暗所で、室温でさらに30分間攪拌した。この溶液を、暗所で、2〜8℃の温度で、0.05Mリン酸ナトリウム緩衝液(pH7.2)に対して一晩透析した。
【0079】
続いて、この溶液のアリコートを、0.05M リン酸ナトリウム緩衝液(pH7.2)中のrFVIIa溶液(30μg/mL)に加えて、rFVIIaのmgあたり100mgの活性化CAの最終濃度にした。この混合物を暗所で、室温で180分間攪拌した。NaCNBH3を加えて(最終濃度10mg/mg rFVIIa)、この混合物を静かに振盪させながら暗所で、室温で18時間、インキュベートした。次いで、2.5mLの水性1M TRIS溶液(pH7.2)をこの混合物/mLに加えて、60分間攪拌して、反応を終了させた。
【0080】
イオン交換クロマトグラフィーで、QHyperD F 50μm樹脂(Pall BioSepra, Cergy, France)およびPharmacia XK−10カラム(Pharmacia XK 10;高さ=15cm)を用いて、rFVIIa−CA酸結合体から遊離試薬を分離した。CA結合体化タンパク質を、溶出緩衝剤(20mM HEPES/1M NaCl, pH8.0)で溶出した。最終段階で、30kD膜(再生セルロース/Millipore)を用いて限外濾過/ダイアフィルトレーション(UF/DF)によって、150mM NaClおよび0.5%ショ糖を含有する20mM HEPES緩衝液(pH7.4)に対して濃縮させた。
【0081】
(実施例2):ポリシアル酸化rFVIIaの生化学的性質
rFVIIa−PSAの酵素活性を、凝固アッセイによって決定した。FVIIaをヒトFVII欠乏血漿に加えて、凝固を、FVIIaと反応するが、FVIIと反応しないトランケート型組織因子(Staclot, Diagnostica Stago, Asnieres, France)によって誘発した。
【0082】
rFVII−PSAのFVIIIのバイパス活性をトロンビン生成アッセイ(TGA)によって測定した。FVIIaを、トロンビン−特異的蛍光ペプチド−基質の存在下で高力価の抗FVIIIインヒビターを含有する重度の血友病Aの血漿に加えた。凝固を、組織因子−リン脂質複合体で誘発し、トロンビン生成を、基質のフルオロフォアの切断速度によって連続的に測定した。トロンビン生成活性を、ピークのトロンビン(すなわち、アッセイ中に観察された最大量トロンビンの濃度)から算出した。両ケースでは、NovoSeven組換えFVIIa製剤(Novo Nordisk,デンマーク コペンハーゲン)を参考として使用した。
表1に示すように、PSA−rFVIIaの比活性は、修飾後減少した。
【0083】
【表1】
修飾を、非還元条件下で行ったSDS−PAGEによって可視化した。免疫染色を、ポリクローナル抗FVII抗体(Affinity Biologicals;Ancaster, Canada)およびモノクローナル抗PSA抗体(Chemicon International, Temecula, CA, USA)を用いて行った。修飾は、PSA含有タンパク質と相関するスメア領域によって示されたFVIIaのMWの増加をもたらした(図1)。
【0084】
(実施例3):ラットにおけるrFVIIa−PSA結合体の薬物動態
4匹のラット(Crl:CD(SD), Charles River Laboratories, Wilmington, MA)に麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L マンニトール、1.5g/L CaCl2 x 2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−PSA結合体(16.500U FVIIa/kg)を、用量(20mL/kg)を尾静脈に静脈内注射によって投与した。対照として6匹の正常なラットに、修飾されていないrFVIIaを18.000U FVIIa/kgの用量で用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、6時間後、8時間後、および24時間後に血液試料を、眼窩静脈叢から採取し、クエン酸血漿を調製してさらなる分析のために凍結させた。
【0085】
次いで、血漿中のFVIIa活性(Staclot, Diagnostica Stago, Asnieres, France)を測定した。修飾されていないrFVIIaの半減期は1.1時間であったが、rFVIIa−結合体により2.3時間に増加した(図2)。
【0086】
FVIIa抗原レベルの薬物動態を、さらなる実験で測定した。6匹のラットに麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L
マンニトール、1.5g/L CaCl2.2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−PSA結合体(450μg/kg)を、容積用量(10L/kg)で尾静脈に静脈内注射によって投与した。対照として6匹のラットに、修飾されていないrFVIIaを390μg/kgの用量で用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、6時間後、8時間後、12時間後、および24時間後に血液試料を、眼窩静脈叢から採取した。そしてクエン酸血漿を調製してさらなる分析のために凍結させた。血漿中のFVII抗原レベルを、ELISA(ポリクローナル抗−ヒトFVII抗体)で測定した。MS Excelで決定したように、直線回帰による半減期算出は、天然rFVIIaに対しては1.1時間およびrFVIIa結合体に対しては3.1時間であった。FVII抗原に関するデータを、投与から5分後に得た平均血漿レベルに規準化する(図3)。
【0087】
(実施例4):FVIIaのN末端ポリシアル化
FVIIaのN末端でのCAの結合体化をpH6.0で行った。この処置のために、Sigma(Sigma−Aldrich)からのCAを使用し、それをさらにQ−セファロース FF(GE Healthcare, Munich, Germany)上で陰イオン交換クロマトグラフィーによって精製した。0.04M NaIO4を含有する精製CAの水溶液(濃度23mg/mL)を暗所で、室温で15分間攪拌して、CAを酸化させた。続いて、この溶液のアリコートを0.05Mリン酸ナトリウム緩衝液(pH6.0)中のrFVIIa溶液(740μg/mL)に加えて、rFVIIaのmgあたり60mgの活性化CAの最終濃度にした(約150モル過剰)。この混合物を暗所で、室温で180分間攪拌した。NaCNBH3を加えて(25mg/mg rFVIIa)この混合物を静かに振盪させながら暗所で、24時間、インキュベートした。次いで、2.5mLの水性1M TRIS溶液(pH7.2)をこの混合物/mLに加えて、4℃で60分間攪拌して、反応を終了させた。
【0088】
イオン交換クロマトグラフィーで、QHyperD F 50μm樹脂(Pall BioSepra, Cergy, France)およびPharmacia XK−16カラム(Pharmacia XK 16;高さ=14cm)を用いて、rFVIIa−CA酸結合体から遊離試薬を分離した。次いで、CA結合体化タンパク質を、溶出緩衝剤(20mM HEPES/0.5M NaCl、pH8.0)で溶出した。最終段階で、10kD膜(再生セルロース/Millipore)を用いてUF/DFによって、150mM NaClを含有する20mM HEPES緩衝液(pH7.4)に対して濃縮させた。イオン交換クロマトグラフ法およびUF/DFステップを4℃で行った。
【0089】
N末端修飾rFVIIa−PSAの酵素活性を、実施例2に記載するように、凝固アッセイおよびトロンビン生成アッセイによって決定した。その結果を表2に要約する。
【0090】
【表2】
N末端結合体化型PSA−rFVIIaの比活性は、STFアッセイで測定したように約50%まで、およびTGAでは25%まで低下した。
【0091】
実施例2に記載するように、修飾は、ポリクローナル抗FVII抗体およびポリクローナル抗PSA抗体を用いる免疫染色によって展開させた非還元条件下で行ったSDS−PAGEによって可視化した。修飾は、抗PSAで染色した免疫ブロットで示されるバンドと相関して、FVIIaのMWの軽微な増加をもたらした(図4)。
【0092】
(実施例5):CNBr活性化型合成N−アセチルノイラミン酸によるFVIIaの結合体化
rFVIIaを、米国特許第3,487,890号に記載されるように、N−アセチルノイラミン酸によって結合体化させた。350mgの合成N−アセチルノイラミン酸(Sigma−Aldrich)を、10mLの0.1M HEPES緩衝液(pH9.0)に溶解させた。次いで、430mgのCNBr(Fluka, Steinhamm, Germany)をこの溶液に加え、活性化の処置の間0.5M NaOHでpHを9.5に調整した。30分後、pH値は9.5であった。次いで、pH値を、0.1M HClを加えることによって8.4に調整した。活性化の全処置の間、温度を氷浴を用いることで制御し、20〜25℃で維持した。rFVIIaによる活性化N−アセチルノイラミン酸の結合体化のために、rFVIIa溶液50mM リン酸緩衝液(pH7.2)中の(50mL/0.44mg rFVIIa/mL)を加えて室温で、緩やかな攪拌下で30分間インキュベートした。次いで、20mLの0.2Mトリス緩衝液を加えて、反応を終了させ、遊離シアン酸エステルをブロックし、混合物を緩やかな攪拌下で15分間インンキュベートした。最終的にこの溶液を、50mM リン酸緩衝液(pH7.2)に対して10kD膜(再生セルロース/Millipore)を用いるUF/DFによって濃縮させた。
【0093】
(実施例6):CNBr活性化合成N−アセチルノイラミン酸三量体によるFVIIaの結合体化
rFVIIaを、N−アセチルノイラミン酸に関する米国特許第3,487,890号に記載されるように、TimTec、LLC(Newark, USA)から得た合成N−アセチルノイラミン酸三量体に結合体化させた。350mgのN−アセチルノイラミン酸三量体を、10mLの0.1M HEPES緩衝液(pH9.0)に溶解させた。次いで、430mgのCNBr(Fluka)をこの溶液に加え、活性化の処置の間0.5M
NaOHでpHを9.5に調整した。30分後、pH値は9.5であった。pH値を、0.1M HClを加えることによって8.4に調整した。活性化の全処置の間、温度を、氷浴を用いることによって制御し、20〜25℃で維持した。次いで、FVIIaにおる活性化の結合体化を、実施例5に記載するように行った。
【0094】
(実施例7):モノSA−FVIIaおよびトリSA−FVIIaの生化学的性質
実施例5に記載するN−アセチルノイラミン酸(モノSA)に結合体化した修飾rFVIIa、または実施例6に記載するN−アセチルノイラミン酸三量体(トリSA)に結合体化した修飾rFVIIaの酵素活性を、実施例2に記載するように凝固アッセイおよびトロンビン生成アッセイによって決定した。それらの結果を、表3に要約する。
【0095】
【表3】
オリゴ−PSA結合体化型rFVIIaの比活性は、STFアッセイによって測定されたように低下したが、TGAによって測定されたモノSA−rFVIIaは、そのFVIIIバイパス活性の約50%を保持した。
【0096】
さらに、モノSA−rFVIIaおよびトリSA−rFVIIaを、KlausenおよびKornfelt(J Chromatogr A. 1995;718:195−202)によって記載されるように、キャピラリー電気泳動(CE)によって調べた。それらの結果を図5に示す。天然のrFVIIaと比較して、付加的な負の電荷のためにモノSA−rFVIIaおよびトリSA−rFVIIaのより高い保持時間への明らかな変化を示す。
【0097】
(実施例8):ラットにおけるrFVIIa−モノSAおよびrFVIIa−トリSA結合体の薬物動態
12匹のラットに麻酔して、緩衝液(1.3g/L グリシルグリシン、3g/L 塩化ナトリウム、30g/L マンニトール、1.5g/L CaCl2.2H2O、0.1g/L Tween 80、pH5.5)中のrFVIIa−モノSA結合体(400μg タンパク質/kg)を、容積用量(10mL/kg)を尾静脈に静脈内注射によって投与した。4匹のラットに、rFVIIa−トリSA結合体(400μg タンパク質/kg)で処置した。対照として8匹の正常なラットにおいて400μg タンパク質/kgの用量で修飾されていないrFVIIaを用いた。物質を投与してから5分後、30分後、1時間後、2時間後、4時間後、7時間後、10時間後、および22時間後に血液試料を、眼窩静脈叢から採取し、クエン酸血漿を調製してさらなる分析のために凍結させた。血漿中のFVII抗原レベルをELISA(ポリクローナル抗−ヒトFVII抗体)によって測定した。データを、投与から5分後に血漿で見出された濃度に対して規準化した。投与から7時間後、rFVIIa−モノSAおよびトリSA−rFVIIaの血漿レベルは対照の天然rFVIIaのそれよりも高かった。それらの結果を図6A(rFVIIa−モノSA)および図6B(rFVIIa−トリSA)に示す。
【0098】
(実施例9):還元的アミノ化によるN−アセチルノイラミン酸三量体のrFVIIaへの結合
還元的アミノ化によるrFVIIaのN−アセチルノイラミン酸三量体との結合体化を、Biessen el al. (Biochem J 1994;299:291−6)に記載されるように行った。350mgのN−アセチルノイラミン酸三量体(TimTec)を10mLの0.1M HEPES緩衝液(pH7.0)に溶解して、20mM
HEPES、70mM NaCl(pH7.4)(0.3mg/mL)中の組換えFVIIaの溶液32mLに加えた。次いで、NaCNBH3を加えて、50mg/mLの最終濃度にし、0.1M HClを加えることによってpH7.0にpH調整した。混合物を37℃で、穏やかな攪拌下で48時間インキュベートした。この溶液を、20mM HEPES緩衝液、150mM NaCl(pH7.4)に対して10kD膜(再生セルロース/Millipore)を用いるUF/DFによって濃縮させた。
【0099】
N−アセチルノイラミン酸三量体のrFVIIaとの結合体化を、KlausenおよびKornfelt(J Chromatogr A. 1995, 718:195−202)によって行われたCEによって示した。それらの結果を図7に示す。天然のrFVIIaと比較して、誘導体のより高い保持時間への明らかな変化を示す。
【0100】
(実施例10):コロミン酸の精製および誘導体化
国際公開第WO0601616A1号に記載されるように、CAをQ−セファロース FF上で陰イオン交換クロマトグラフィーによって精製した。5gのCAを、25mM NaClを含有する50mLの10mM トリエタノールアミン緩衝液(pH7.4)(=開始緩衝液)で溶解した。この溶液を、Q−セファロース FF(GE Healthcare)を充填し、開始緩衝液と平衡に達したPharmacia XK50カラム上に適用した。次いで、このカラムを8カラム容積(CV)の開始緩衝液で洗浄し、結合したCAを開始緩衝液中で、3CVで段階的に(200mM NaCl、350mM NaCl、および500mM NaCl)溶出させた。350mM NaClで溶出した画分は、SDSゲル電気泳動によって示されるように20kDaの分子量を示した。この画分を、再生セルロース(Millipore)製の5kD膜を用いて限外濾過によって濃縮し、続いて50mM リン酸緩衝液(pH7.2)に対してダイアフィルトレーションした。次いで、CAを、実施例1に記載のNaIO4で酸化させ、末端の一次アミノ基を、国際公開第WO05016973A1号に記載されるように、還元的アミノ化によって導入した。還元的アミノ化のために、11mLの2M NH4Cl−溶液を、50mM リン酸緩衝液(pH7.2)中に58mgの酸化PSA/mLを含有する20mLの溶液に加えた。次いで、1M NaOH中の5M NaCNBH3の溶液を加えて、75mMの最終濃度にした。反応を室温、pH8.0で5日間、行った。次いで、この混合物を、10mM NaClを含有する(NH4)2CO3溶液(50mg/L)に対して透析し、続いて5mM EDTAを含有する50mM リン酸緩衝液(pH8.0)に対して透析した。次いで、スルフヒドリル基を、2−イミノチオラン(Traut試薬/Pierce)を用いる末端の一次アミノ基の反応によって導入した。反応を室温で、試薬の20倍のモル過剰を有する5mM EDTAを含有する50mM リン酸緩衝液(pH8.0)で、1時間行った。最終的に、末端遊離SH基を含有するPSA溶液を、再生セルロース(Millipore)製の5kDのカットオフ膜を使用する限外濾過/ダイアフィルトレーションにかけた。
【0101】
(実施例11):ヘテロ二価性架橋剤の使用によるPSAのrFVIIaへの結合
PSA(Sigma−Aldrich)をQ−セファロース FF(GE Healthcare)上で陰イオン交換クロマトグラフィーによって精製し、実施例10に記載の末端スルフヒドリル基を化学修飾によって導入し、PSA−SHを形成した。PSA−SHのrFVIIaへの結合のために、2つの反応基、すなわちSH基への結合体化のためのマレイミド基および遊離アミノ基への結合体化のためのスルホ−NHS−エステル基を含有するヘテロ二価性水溶性架橋剤であるスルホ−EMCS((N−ε−マレイミドカプロイルオキシ)スルホスクシンイミドエステル/Pierce)を用いた。150mM NaClを含有する、20mM HEPES緩衝液(pH7.4)中の2mLのrFVIIa溶液(1.6mg/mL)に、スルホ−EMCSを加えて最終濃度の0.07mgの架橋剤/mgタンパク質にした。反応を室温で、30分間行った。続いて、実施例10によって調製した130mgのPSA−SH(100倍の過剰)を加えて、中間体リンカー/rFVIIa複合体の反応のPSA−SHへの結合を室温で、さらに2時間行った。次いで、この混合物を、ブチル−セファロース(GE−Healthcare)上でHICクロマトグラフィーによって精製した。5M NaCl溶液をこの混合物に加えて、3M NaClの最終濃度にした。次いで、この混合物をブチル−セファロース(GE−Healthcare)で充填したカラムに適用し、rFVIIa−PSA結合体の溶出を、6.7mM CaCl2を含有する50mM HEPES緩衝液(pH7.4)で行った。結合体の溶出後、pHをpH6,9に調整した。
【0102】
(実施例12):PSA−ヒドラジドの、rFVIIaの炭水化物部分への結合体化
PSAの、rFVIIaの炭水化物部分への結合体化のために、20mM HEPES緩衝液(pH6.0)(1.6mg/mL)中のrFVIIaの溶液を調製した。この溶液の9容積に対して、5mM NaIO4−溶液の1容積を加えて穏やかに混合した。酸化反応を4℃で、暗所で1時間行い、遊離アルデヒド基を生成した。次いで。亜硫酸水素ナトリウム(最終濃度5mM)を加えて、酸化反応を止めた。続いてPSA−ヒドラジド(国際公開第WO0606168A2)を加え(最終濃度10mM)、アルデヒド基への結合反応を室温で1時間行った。次いで、PSA−rFVIIa結合体を、実施例1に記載のQHyper D(Pall BioSepra)上で陰イオン交換クロマトグラフィーによって精製した。
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図7】
【公開番号】特開2013−60462(P2013−60462A)
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−281272(P2012−281272)
【出願日】平成24年12月25日(2012.12.25)
【分割の表示】特願2009−541613(P2009−541613)の分割
【原出願日】平成19年12月14日(2007.12.14)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願番号】特願2012−281272(P2012−281272)
【出願日】平成24年12月25日(2012.12.25)
【分割の表示】特願2009−541613(P2009−541613)の分割
【原出願日】平成19年12月14日(2007.12.14)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(501453189)バクスター・ヘルスケヤー・ソシエテ・アノニム (289)
【氏名又は名称原語表記】BAXTER HEALTHCARE S.A.
【Fターム(参考)】
[ Back to top ]