
弱塩基性薬剤の固溶体を含む薬剤送達システム
本発明は、TPRビーズを含む医薬組成物および剤形に関するものであり、前記TPRビーズは、少なくとも1種の溶解度増大性ポリマー中の少なくとも1種の活性医薬成分の固体分散物、ならびに水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティングを含み、前記活性医薬成分は、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願に対する相互参照
本出願は、いずれも2006年8月31日に出願された米国仮特許出願第60/841,760号および米国仮特許出願第60/841,893号の利益を主張するものであり、これらは各々、全ての目的に関して、参照としてその全体を本明細書に取り入れる。
【0002】
本発明は、改善されたバイオアベイラビリティを有する放出制御組成物、およびこのような組成物を製造する方法に関する。本発明の組成物は、少なくとも1種の活性医薬成分の固体分散物および時限拍動型放出(timed pulsatile release)コーティングを含む。
【背景技術】
【0003】
多くの治療薬は、吸収部位またはその近辺で、一定速度で利用可能にされている場合に最も有効である。この様式で利用可能にされている治療薬の吸収は一般に、最大の有効性および最小の毒性副作用をもたらす所望の血漿中濃度をもたらす。しかしながら、吸収過程の複雑性、剤形からの治療薬の放出速度に影響を及ぼす相互に関連する多くの組成変数、および治療薬自体の物理化学的特性のために、一定速度で治療薬の所望の血漿中濃度を送達する経口製薬剤形を開発することは困難であることが多い。例えば、経口投与された製薬剤形が、ヒトの消化管を通る間に、薬剤が該剤形から放出され、胃腸(GI)管からの吸収のための部位またはその近辺で、溶液形態で利用可能であるべきである。該剤形、したがって、該治療薬は、胃腸管を通過する間にpHの変化、すなわち、約1.2(空腹時の胃のpH)から4.0(食物の摂取時)または約7.4(胆汁のpH:7.0〜7.4および腸のpH:5〜7)まで高いpHへの変化に供される。さらに、消化管の個々の部分における剤形の通過時間は、剤形のサイズおよび主要な局所の条件(例えば、胃腸管に沿った浸透性の変化;pH、表面張力、体積、攪拌、および緩衝能力などの管の内容物の特性;ならびに食物消化後の変化)に応じて、著しく変化し得る。血漿中濃度に影響を及ぼす薬剤自体の物理化学的特性には、そのpKa、溶解度および結晶エネルギーが含まれ、例えば、多粒子剤形の組成特性には、薬剤含有粒子のサイズまたは比表面積などが含まれる。その結果、一定速度での薬剤放出の達成はしばしば困難である。
【0004】
さらに、塩基性および酸性の薬剤は、生理学的pH範囲において、2桁超の大きさで変化するpH依存性溶解度プロファイルを示す。これらのうちで、製剤化が最も困難な薬剤は、pH>5ではほとんど不溶(例えば、50μg/mL以下の溶解度を有する)であり、治療上有効であるためには、高用量(例えば、10mg以上の最適1日用量)を必要とする弱塩基性化合物である。このような高用量では、吸収速度が薬剤放出速度より速くない限り、胃腸(GI)管のpH環境内に入ると、溶解した薬剤の一部が沈殿する可能性がある。あるいは、該薬剤は、例えば、腸内の胆汁塩およびレシチンの存在により助長されて、知られている水溶解度より一桁を十分超える過飽和レベルでの過飽和溶液状態に留まる可能性がある。しかし、過飽和溶液は沈殿する可能性があり、次いで該薬剤の再溶解および吸収はより緩やかな速度で生じ得る証拠がある。これらの問題を解決するために、弱塩基性の薬剤の溶解度を増加させるための種々のアプローチ、例えば、有機酸を含めて酸付加化合物を形成すること、または固体分散物または固溶体を使用することが開発されている。
【0005】
しかし、薬剤の溶解度は薬剤自体の物理化学的特性、ならびに医薬製剤を調製する方法によって変化するため、このようなアプローチは完全に満足できるものではない。例えば、ニフェジピンまたはレルカニジピンなどのいくつかの弱塩基性の薬剤は、飽和有機酸溶液中で有意な溶解度増大を示さず、固体分散物は、経口摂取の際に、該薬剤の望ましくない即時放出をもたらす傾向がある。
【0006】
本発明の組成物は、1日1回の投与法に好適な薬剤放出プロファイルを有する弱塩基性治療薬(例えば、14未満のpKaを有し、目標血漿中濃度を維持するために高用量を必要とする)の改良された送達を提供する。
【発明の概要】
【課題を解決するための手段】
【0007】
一実施形態において、本発明は、TPRビーズを含む医薬組成物であって、前記TPRビーズは、少なくとも1種の活性医薬成分および少なくとも1種の溶解度増大性ポリマーの固体分散物、ならびに水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティングを含み、前記活性医薬成分は、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む医薬組成物を指向する。
【0008】
別の実施形態において、本発明は、薬学的に許容できる溶媒中に活性医薬成分および十分な溶解度増大性ポリマーを溶解すること;活性医薬成分および溶解度増大性ポリマーの溶液から薬学的に許容できる溶媒を除去し、それによって、溶解度増大性ポリマー中の活性医薬成分の固体分散物の粒子を形成すること;薬学的に許容できるコーティング溶媒中に水不溶性ポリマーおよび腸溶性ポリマーを溶解し、それによって、TPRコーティング溶液を形成すること;該固体分散物をTPRコーティング溶液でコーティングすること;該コーティング溶媒を除去し、それによって、固体分散物上に形成されたTPRコーティングを含むTPRビーズを形成することを含む、医薬組成物を調製する方法を指向する。
【図面の簡単な説明】
【0009】
【図1】本発明のTPRビーズの一実施形態を示す断面図である。
【図2】(a)塩酸オンダンセトロン、(b)カルベジロール、(c)ジピリダモール、および(d)クロナゼパムに関するpH溶解度プロファイルを示す図である。
【図3】(A)レルカニジピンHClおよび(B)ニフェジピンの固溶体/固体分散物に関する濁度プロファイルを示す図である。
【図4】レルカニジピンHCl−(A)多形体I、(B)多形体IIおよび(C)非晶質物質(薬剤−ポリマー固溶体)の固有溶解速度(IDR)を示す図である。
【図5】レルカニジピンHClおよび(A)Kollidon VA64または(B)Methocel E5の固体分散物の粉末X線回折パターンを示す図である。
【図6】ニフェジピンおよび(A)Kollidon VA64または(B)Methocel E5の固体分散物の粉末X線回折パターンを示す図である。
【図7】実施例4に記載されたTPRビーズからの薬剤放出に及ぼすTPRコーティング組成物の影響を示す図である。
【図8】実施例3に記載されたTPRビーズからの薬剤放出に及ぼす薬剤添加量の影響(すなわち、10%の薬剤添加と5%との薬剤添加)を示す図である。
【図9】実施例3Bの60〜80メッシュの糖球上、10%の薬剤添加および15重量%のEudragit RL/Lコーティング(45/40)でコーティングされたTPRビーズからの薬剤放出に及ぼす粒径の影響を、実施例4Dの25〜30メッシュの糖球から調製された同様のTPRビーズと比較して示す図である。
【図10】実施例5のTPRビーズからの薬剤放出に及ぼすTPRコーティング組成物の影響を示す図である。
【図11】Kollidon VA64および酒石酸を含有する実施例8のレルカニジピンHCl TPRビーズからの薬剤放出プロファイルを示す図である。
【図12】実施例9のTPRビーズからの薬剤放出に及ぼすTPRコーティング組成ならびに厚さの影響を示す図である。
【発明を実施するための形態】
【0010】
本発明は、少なくとも1種の活性医薬成分と少なくとも1種の溶解度増大性ポリマーの固体分散物、ならびに水不溶性ポリマーおよび腸溶性ポリマーを含む時限拍動型放出(TPR)コーティングとの組合せを含む医薬組成物を指向し、前記活性医薬成分は、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む。弱塩基性の活性医薬成分が固体分散物の形態で存在せず、および/またはTPRコーティングを欠く従来の組成物によって得られた放出プロファイルに比較して、弱塩基性の活性医薬成分の固体分散物とTPRコーティングとの組合せは、改善された放出プロファイルを提供する。例えば、少なくとも1種のTPRコーティングを含む組成物の好適な処置により、放出速度を、12〜18時間にわたってほぼ一定にすることができるか、または溶解度増大性ポリマーの単独使用に比較して、最大放出速度までの時間を遅延させることができる。
【0011】
用語「固体分散物」または「固溶体」とは、ポリマー基質中に分散した実質的に非晶質の活性医薬成分を意味し、および/またはより具体的には、少なくとも1種の活性医薬成分および少なくとも1種の結晶化阻害性ポリマーが固体状態において実質的に分子分散していることである。用語「実質的に非晶質」とは、活性医薬成分の40%未満が、ポリマー基質中で分離した結晶相を形成していることである。他の実施形態において、「実質的に非晶質」とは、活性医薬成分の30%未満、20%未満、10%未満、5%未満、または1%未満が、ポリマー基質中で分離した結晶相を形成していることを意味する。言い換えると、活性医薬成分の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%、または少なくとも99%が、非晶質の状態にある。用語「実質的に分子分散している」とは、活性医薬成分の40%未満が、ポリマー基質中で分離した結晶相を形成し、残りの活性医薬成分が、ポリマー基質中に溶解していることを意味する。他の実施形態において、「実質的に分子分散している」とは、活性医薬成分の30%未満、20%未満、10%未満、5%未満、または1%未満が、ポリマー基質中で分離した結晶相を形成していることを意味する。本発明の固体分散物は、活性医薬成分の40%以下、いくつかの実施形態においては、活性医薬成分の30%以下、20%以下、10%以下、5%以下、または1%以下が、ポリマー基質中で結晶相を形成しているという条件で、ポリマー基質中の「実質的に分子分散している」活性医薬成分と「実質的に非晶質」な活性医薬成分との組合せを含む。
【0012】
用語「活性医薬成分」は、用語「薬剤」、「治療薬」などと交換可能に使用することができる。本明細書に用いられる用語「弱塩基性の活性医薬成分」、ならびにいずれかの特定の薬剤の記述は、塩基、薬学的に許容できる塩、多形体、立体異性体、溶媒和物、エステル類およびそれらの混合物を含む。一実施形態において、本発明の組成物の弱塩基性活性医薬成分は、14未満のpKaを有する化合物のことであり得る。別の実施形態において、弱塩基性の活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有する。別の実施形態において、弱塩基性の活性医薬成分は、少なくとも1種の塩基性窒素原子を含む。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、pH6.8で約100μg/mL以下の溶解度を有する。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、少なくとも1種の塩基性窒素原子を含む。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、pH6.8で100μg/mL以下の溶解度を有し、かつ、少なくとも1種の塩基性窒素原子を含む。
【0013】
本明細書に用いられる用語「溶解度増大性ポリマー(solubility-enhancing polymer)」または「結晶化阻害性ポリマー(crystallization-inhibiting polymer)」とは、例えば、先ず薬剤とポリマーの双方を同一の溶媒系に溶解し、次いで、適切な条件下で該溶媒を除去することによって、溶解度増大性ポリマー中の弱塩基性薬剤の本明細書に定義された固体分散物を好適な濃度で形成することのできる水溶性ポリマーのことである。該弱塩基性薬剤は、溶解度増大性ポリマーと弱塩基性薬剤の固体分散物を含有する該組成物の保存、輸送、および商品配送の間、実質的に分子分散物としてあるいは非晶質形態で維持される。
【0014】
本明細書に用いられる用語「即時放出」(IR)とは、組成物の投与後1時間で活性医薬成分の約75%超、他の実施形態においては、約85%超の放出を言う。放出量はインビボまたはインビトロで(本明細書に記載されている慣用のUSP法を用いて)測定することができる。
【0015】
用語「IRビーズ」とは、即時放出性を有する活性医薬成分含有粒子のことである。IRビーズとしては、活性医薬成分を含む任意の種類の粒子、例えば、溶解度増大性ポリマー中の活性医薬成分の固体分散物の粒子、または溶解度増大性ポリマー中の活性医薬成分の固体分散物によってコーティングされた不活性コアを挙げることができる。IRビーズとしては、固体分散物を含み、さらにシーラント(sealant)または保護層によってコーティングされ、本明細書に記載された即時放出性を有する粒子が挙げられる。
【0016】
本明細書に用いられる用語「急速分散微粒剤」とは、崩壊剤と組み合わせた、糖アルコール(例えばD−マンニトール)および/または糖類(例えば乳糖)の一次粒子を含む凝集粒子のことである。
【0017】
本出願において交換可能に用いられる用語「遅延時間膜コーティング」、「遅延時間ポリマーコーティング」、「時限拍動型放出(TPR)膜コーティング」、「TPRポリマーコーティング」、またはTPRコーティングとは、腸溶性ポリマーと組み合わせて水不溶性ポリマーを含むコーティングのことである。
【0018】
用語「時限拍動型放出(TPR)ビーズ」または簡単に「TPRビーズ」とは、TPRコーティングでコーティングされた、活性医薬成分含有粒子のことである。いくつかの実施形態において、TPRビーズとは、TPRコーティングでコーティングされたIRビーズのことであり、本発明の一定の実施形態に従って調製されたTPRビーズからの弱塩基性活性医薬成分の放出は、短い遅延時間後の徐放性プロファイルを特徴とする。
【0019】
用語「遅延時間」とは、活性医薬成分の約10%未満、さらに特に約5%未満、さらに特に実質的に0%が放出される時間のことであり、水不溶性ポリマーと腸溶性ポリマー(例えば、Eudragit RLポリマーとLポリマー)の組合せを含む本発明のTPRコーティングによって、約4時間までの遅延時間を達成することができる。
【0020】
本明細書に用いられる用語「溶解度調節有機酸」または「有機酸」とは、有機酸の水溶液中への活性医薬成分の溶解の速度および/または程度を増大させることのできる水溶性の薬学的に許容できる有機酸のことである。
【0021】
用語「放出速度」とは、単位時間当たりに組成物からインビトロまたはインビボで放出される薬剤の量のことである。量の単位は、例えば、全用量の%として表されることが多い。
【0022】
用語「血漿プロファイル」、「血漿中濃度」、「Cmax」、または「Cmin」は、単位体積当たりの質量、典型的には、1ミリリットル当たりのナノグラム(ng/mL)として一般に表される、対象の血漿中の薬剤濃度のことである。
【0023】
用語「治療上有効な量」とは、所望の薬理学的結果を提供するのに必要な活性医薬成分の量のことである。実際、治療上有効な量は、病態の重症度、対象の年齢、および所望の治療効果に依って大きく変動する。
【0024】
本発明の医薬組成物は、少なくとも1種の活性医薬成分と少なくとも1種の溶解度増大性ポリマーの固体分散物およびTPRコーティングを含む。
【0025】
本発明の特定の実施形態を、添付の図1を参照して、さらに詳細に記載する。図1は、TPRビーズ10を表す。不活性粒子コア18、弱塩基性薬剤、結晶化阻害性ポリマー(溶解度増大性ポリマーとも称される)、および溶解度増大性有機酸を含む非晶質層16、保護シールコーティング層14、および遅延時間(TPRまたは拍動型放出とも称される)コーティング12がTPRビーズ10を構成する。
【0026】
本発明の医薬組成物のための好適な活性医薬成分は弱塩基性薬剤を含む。一実施形態において、該活性医薬成分は、14未満のpKa値を有する。別の実施形態において、該活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有する。別の実施形態において、該活性医薬成分は、約3時間以上の排出半減期を有する。別の実施形態において、該活性医薬成分は、pH6.8で50μg/mL以下の溶解度を有する。別の実施形態において、該活性製薬群の成分は、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。さらに別の実施形態において、該活性医薬成分は、14未満のpKa値、pH6.8で約100μg/mL以下の溶解度、および約3時間以上の排出半減期を有する。さらに別の実施形態において、該活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有し、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。さらに別の実施形態において、該活性医薬成分は、pH6.8で約50μg/mL以下の溶解度を有し、、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。
【0027】
好適な活性医薬成分のクラスの非限定例としては、鎮痛薬、降圧薬、抗不安薬、抗凝固薬、抗痙攣薬、抗糖尿病薬、血糖値低下薬、うっ血除去薬、抗ヒスタミン薬、抗炎症薬、鎮咳薬、抗悪性腫瘍薬、ベータブロッカー、抗リウマチ薬、抗炎症薬、抗精神病薬、向知性薬、抗アテローム硬化症薬、抗肥満症薬、抗不能症薬、抗感染症薬、催眠薬、抗パーキンソン薬、抗アルツハイマー病薬、抗うつ薬、および抗ウィルス薬、グリコーゲンホスホリラーゼ阻害剤、コレステロールエステルトランスファータンパク質阻害剤、CNS(中枢神経系)刺激薬、ドーパミン受容体アゴニスト、制吐薬、胃腸薬、精神治療薬、オピオイドアゴニスト、オピオイドアンタゴニスト、抗てんかん薬、ヒスタミンH2アンタゴニスト、抗喘息薬、平滑筋弛緩薬および骨格筋弛緩薬が挙げられるが、これらに限定されない。
【0028】
鎮痛薬の具体例としては、アセトアミノフェン、ロフェコキシブ、セレコキシブ、モルフィン、コデイン、オキシコドン、ヒドロコドン、ジアモルフィン、ペチジン、トラマドール、ブプレノルフェンが挙げられ;降圧薬としては、プラゾシン、ニフェジピン、レルカニジピン、アムロジピンベシレート、トリマゾシンおよびドキサゾシンが挙げられ;抗不安薬の具体例としては、ヒドロキシジン塩酸塩、ロラゼパム、ブスピロン塩酸塩、パゼパム、クロルジアゼポキシド、メプロバメート、オキサゼパム、トリフルオペラジン塩酸塩、クロラゼペート二カリウム塩、ジアゼパムが挙げられ;抗凝固薬の具体例としては、アブシキシマブ、エプチフィバチド、チロフィバン、ラミフィバン、クロピドグレル、チクロピジン、ジクマロール、ヘパリン、およびワルファリンが挙げられ;抗痙攣薬の具体例としては、フェノバルビタール、メチルフェノバルビタール、クロバザム、クロナゼパム、クロレゼペート、ジアゼパム、ミダゾラム、ロラゼパム、フェルバメート、カルバメゼピン、オクスカルベゼピン、ビガバトリン、プロガビド、チアガビン、トピラメート、ガバペンチン、プレガバリン、エトトイン、フェニトイン、メフェニトイン、ホスフェニトイン、パラメタジオン、トリメタジオン、エタジオン、ベクラミド、プリミドン、ブリバラセタム、レベチラセタム、セレトラセタム、エトスクシミド、フェンスクシミド、メスクシミド、アセタゾラミド、スルチアメ、メタゾラミド、ゾニサミド、ラモトリギン、フェネツリド、フェナセミド、バルプロミド、およびバルノクタミドが挙げられ;抗糖尿病薬の具体例としては、レパグリニド、ナテグリニド、メトフォルミン、フェンホルミン、ロシグリタゾン、ピオグリタゾン、トログリタゾン、ミグリトール、アカルボース、エキサナチド、ビルダグリプチン、およびシタグリプチンが挙げられ;血糖値低下薬の具体例としては、トルブタミド、アセトヘキサミド、トラザミド、グリブリド、グリメピリド、グリクラジド、グリピジドおよびクロルプロパミドが挙げられ;うっ血除去薬の具体例としては、プソイドエフェドリン、フェニルエフリン、およびオキシメタゾリンが挙げられ;抗ヒスタミン薬の具体例としては、メピラミン、アンタゾリン、ジフェンヒドラミン、カルビノキサミン、ドキシラミン、クレマスチン、ジメンヒドリネート、フェニラミン、クロルフェニラミン、デクスクロルフェニラミン、ブロムフェニラミン、トリポリジン、シクリジン、クロルシクリジン、ヒドロキシジン、メクリジン、プロメタジン、トリメプラジン、シプロヘプタジン、アザタジン、およびケトチフェンが挙げられ;鎮咳薬の具体例としては、デキストロメトルファン、ノスカピン、エチルモルフィン、およびコデインが挙げられ;抗悪性腫瘍薬の具体例としては、クロラムブシル、ロムスチン、ツブラゾールおよびエキノマイシンが挙げられ;抗炎症薬の具体例としては、ベタメタゾン、プレドニゾロン、アスピリン、ピロキシカム、バルデコキシブ、カルプロフェン、セレコキシブ、フルビプロフェンおよび(+)−N−{4−[3−(4−フルオロフェノキシ)フェノキシ]−2−シクロペンテン−1−イル}−N−ヒドロキシ尿素が挙げられ;β−ブロッカーの具体例としては、チモロールおよびナドロールが挙げられ;鎮咳薬の具体例としては、デキストロメトルファン、ノスカピン、エチルモルフィン、テオブロミンおよびコデインが挙げられ;抗悪性腫瘍薬の具体例としては、アクチノマイシン、ダクチノマイシン、ドキソルビシン、ダウノルビシン、エピルビシン、ブレオマイシン、プリカマイシン、およびマイトマイシンが挙げられ;ベータブロッカーの具体例としては、アルプレノロール、カルテオロール、レボブノロール、メピンドロール、メチプラノロール、ナドロール、オクスプレノロール、ペンブトロール、ピンドロール、プロパノロール、ソタロール、チモロール、アセブトロール、アテノロール、ベタキソロール、ビソプロロール、エスモロール、メトプロロール、ネビボロール、カルベジロール、セリプロロール、ラベタロール、およびブタキセミンが挙げられ;抗リウマチ薬の具体例としては、アダリムマブ、アザチオプリン、クロロキン、ヒドロキシクロロキン、シクロスポリン、D−ペニシラミン、エタネルセプト、ナトリウムアウロチオマレート、アウラノフィン、インフリキシマブ、レフルノミド、メトトレキサート、ミノシクリン、スルファサラジンが挙げられ;抗炎症薬の具体例としては、ヒドロコルチゾン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、デキサメタゾン、ベタメタゾン、トリアムシノロン、ベクロメタゾン、アルドステロン、アセトアミノフェン、アモキシプリン、ベノリレート、ジフルニサル、ファィスラミン、ジクロフェナック、アセクロフェナック、アセメタシン、ブロムフェナック、エトドラック、インドメタシン、ナブメトン、スリンダック、トルメチン、カプロフェン、ケトロラック、メフェナム酸、フェニルブタゾン、アザ抗炎症スプロパゾン、マタミゾール、オキシフェンブタゾン、スルフィンプラゾン、ピロキシカム、ロモキシカム、メロキシカム、テノキシカム、セレコキシブ、エトリコキシブ、ルミリコキシブ、パレコキシブ、ロフェコキシブ、バルデコキシブ、およびヌメスルフィドなどのステロイド系および非ステロイド系抗炎症薬が挙げられ;抗精神病薬の具体例としては、イロペリドン、ジプラシドン、オランゼピン、チオチキセン塩酸塩、フルスピリレン、リスペリドンおよびペンフルリドールが挙げられ;向知性薬の具体例としては、アンパキンが挙げられ;抗アテローム硬化症薬、心血管薬および/またはコレステロール減少薬の具体例としては、アトルバスタチンカルシウム、セリバスタチン、フルバスタチン、ロバスタチン、メバスタチン、ピタバスタチン、プラバスタチン、ロスバスタチン、およびシンバスタチンが挙げられ;抗肥満症薬の具体例としては、デキサドリン、デキスフェンフルラミン、フェンフルラミン、フェンテルミン、オルリスタート、アカルボース、およびリモナバントが挙げられ;抗不能症薬の具体例としては、シルデナフィルおよびシルデナフィル酒石酸塩が挙げられ;抗菌薬、抗ウィルス薬、抗原生動物薬、駆虫薬および抗真菌薬などの抗感染症薬の具体例としては、カルベニシリン、インダニルナトリウム、ベカンピシリン塩酸塩、トロレアンドマイシン、ドキシシリンヒクレート、アンピシリン、ペニシリンG、アジトロマイシン、オキシテトラサイクリン、ミノサイクリン、エリスロマイシン、クラリスロマイシン、サピラマイシン、アシクロビル、ネルフィナビル、ビラゾール、塩化ベンザルコニウム、クロルヘキシジン、エコナゾール、テルコナゾール、フルコナゾール、ボリコナゾール、グリセオフルビン、メトロニダゾール、チアベンダゾール、オクスフェンダゾール、モランテル、コトリモキサゾールが挙げられ;催眠薬の具体例としては、アルファキサロンおよびエトミデートが挙げられ;抗パーキンソン薬の具体例としては、レボドパ、ブロモクリプチン、プラミペキソール、ロピニロール、ペルゴリド、およびセレギリンが挙げられ;トリヘキシフェニジル、ベンズトロピンメシル酸塩、プロシクリジン、ビペリデン、アンデトプロパジンなどの抗コリン作動薬;ジフェンヒドラミンおよびドルフェナドリンおよびアマンタジンなどの抗ヒスタミン薬;抗アルツハイマー病薬の具体例としては、ドネペジル、リバスチグミン、ガランタミン、タクリンが挙げられ;抗生物質の具体例としては、ミノサイクリン、リファンピン、エリスロマイシン、ナフシリン、セファゾリン、イミペネム、アズトレオナム、ゲンタミシン、スルファメトキサゾール、バンコマイシン、シプロフロキサシン、トリメトプリム、メトロニダゾール、クリンダマイシン、テルコプラニン、ムピロシン、アジスロマイシン、クラリスロマイシン、オフロキサシン、ロメフロキサシン、ノルフロキサシン、ナリジクス酸、スパルフロキサシン、ペフロキサシン、アミフロキサシン、エノキサシン、フレロキサシン、テルナフロキサシン、トスフロキサシン、クリナフロキサシン、スルバクタム、クラブラン酸、アンホテリシンB、フルコナゾール、イトラコナゾール、ケトコナゾール、ナイスタチンが挙げられ;抗うつ薬の具体例としては、イソカルボキサジド;フェネルジン;トラニルシプロミンが挙げられ;抗ウィルス薬の具体例としては、アジドブジン(AZT)、ジダノシン(ジデオキシイノシン、ddI)、d4T、ザルシタビン(ジデオキシシトシン、ddC)、ネビラピン、ラミブジン(エピビル、3TC)、サキナビル(インビラーゼ)、リトナビル(ノルビル)、インジナビル(クリキシバン)、デラビルジン(レスクリプトール)が挙げられ;グリコーゲンホスホリラーゼ阻害剤の具体例としては、[R−(R*S*)]−5−クロロ−N−[2−ヒドロキシ−3−{メトキシメチルアミノ}−3−オキソ−1−(フェニルメチル)プロピル−1H−インドール−2−カルボキサミドおよび5−クロロ−1H−インドール−2−カルボン酸[(1S)−ベンジル−(2R)−ヒドロキシ−3−((3R,4S)−ジヒドロキシ−ピロリジン−1−イル−)−3−オキシプロピル]アミドが挙げられ;コレステロールエステルトランスファータンパク質阻害剤の具体例としては、[2R,4S]4−[(3,5−ビス−トリフルオロメチル−ベンジル)−メトキシカルボニル−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸エチルエステル、[2R,4S]4−[アセチル−(3,5−ビス−トリフルオロメチル−ベンジル)−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸イソプロピルエステル、[2R,4S]4−[3,5−ビス−トリフルオロメチル−ベンジル]−メトキシカルボニル−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸イソプロピルエステルが挙げられ;CNS刺激薬の具体例としては、カフェインおよびメチルフェニデートが挙げられ;ドーパミン受容体アゴニストの具体例としては、カベルゴリンおよびプラミペキソールが挙げられ;制吐薬の具体例としては、ドラセトロン、グラニセトロン、オンダンセトロン、トロピセトロン、パロノセトロン、ドンペリドン、ドロペリドール、ジメンヒドリネート、ハロペリドール、クロルプロメジン、プロメタジン、プロクロルペリジン、メトクロプラミド、およびアリザプリドが挙げられ;胃腸薬の具体例としては、ロペラミドおよびシサプリドが挙げられ;精神治療薬の具体例としては、クロルプロマジン、チオリダジン、プロクロルペリジン、ハロペリドール、アルプラゾラム、アミトリプチリン、ブプロピオン、ブスピロン、クロルジアゼポキシド、シタロプラム、クロザピン、ジアゼパム、フルオキセチン、フルフェナジン、フルボキサミン、ヒドロキシジン、ロレザパム、ロキサピン、ミルタゼピン、モリンドン、ネファゾドン、ノルトリプチリン、オランゼピン、パロキセチン、フェネルジン、ケチアピン、リスペリドン、セルトラリン、チオチキセン、トラニルシプロミン、トラゾドン、ベンラファキシン、およびジプラシドンが挙げられ;オピオイドアゴニストの具体例としては、ヒドロモルホン、フェンタニル、メタドン、モルフィン、オキシコドン、およびオキシモルホンが挙げられ;オピオイドアンタゴニストの具体例としては、ナルトレキソンが挙げられ;抗てんかん薬の具体例としては、ナトリウムバルプロエート、ニトラゼパム、フェニトインが挙げられ;ヒスタミンH2アンタゴニストの具体例としては、ファモチジン、ニザチジン、シメチジン、ラニチジンが挙げられ;抗喘息薬の具体例としては、アルブテロール、モンテルカストナトリウム塩が挙げられ;平滑筋弛緩薬の具体例としては、ニコランジル、イロペリドン、およびクロナゼパムが挙げられ;および骨格筋弛緩薬の具体例としては、ジアゼパム、ロラゼパム、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、ダントロレン、メタキサ
ロン、オルフェナドリン、パンクロニウム、チザニジン、ジシクロミン、クロニジン、およびガバペンチンが挙げられる。命名された薬物の各々は、当然のことながら薬物の中性形態、ならびに薬学的に許容できるそれらの塩類、溶媒和物、エステル類、およびプロドラッグを含む。
【0029】
上記で考察したとおり、いくつかの薬剤の溶解度はpH依存性であり、有機酸の添加によって増大させることができる。しかし、下記の表1および表2ならびに図2で示されるように、有機酸の添加によって溶解度がわずかしか影響をうけない他の薬剤もある。
【0030】
表1に、有機酸緩衝液中の弱塩基性薬剤の溶解度増大が掲げられている(図2も参照)。3つの異なる群を識別することができる。塩酸オンダンセトロンにより代表されるA群の薬剤は、微量のフマル酸を含む緩衝液中で弱塩基性活性剤の劇的な溶解度増大を示す。例えば、わずか0.05mg/mLのフマル酸を含有する緩衝液中で約26mg/mLのオンダンセトロンの溶解度は、緩衝液中のフマル酸の濃度を5mg/mLまで増加させたときも変化しない。ジピリダモール、カルベジロール、およびイロペリドンに代表されるB群では、酸の濃度が増加するにつれて弱塩基性薬剤の溶解度は増大する。クロナゼパムに代表されるC群では、有機酸はきわめて限られた影響しか及ぼさない。すなわち、溶解度の増大量は一般に、3倍未満である。例えば、クロナゼパムの溶解度は、高濃度と低濃度のフマル酸をそれぞれ含有するpH2.3と6.8の緩衝液中で約11.6μg/mLと6.9μg/mLである。
【0031】
【表1】
【表2】
【0032】
【表3】
【0033】
本発明の医薬組成物の一実施形態において、活性医薬成分はニフェジピンである。別の実施形態において、活性医薬成分はレルカニジピンである。しかし、当然のことながら、本発明の範囲は特定の活性医薬成分に限定されない。
【0034】
本発明の医薬組成物において有用である好適な溶解度増大性ポリマーとしては、限定はしないが、ポリビニルピロリドン(PVPまたはポビドン)、酢酸ビニル/ビニルピロリドンのコポリマー(例えば、Kollidon VA 64)、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシメチルセルロース(ヒプロメロース)、ヒドロキシプロピルメチルセルロースアセテートスクシネート(HPMCAS)、ポリエチレンオキシド、ポリエチレングリコール、およびシクロデキストリンが挙げられる。
【0035】
溶解度増大性ポリマーのタイプおよび量は、活性医薬成分と溶解度増大性ポリマーとの組合せが、本明細書に定義された固体分散物を形成するように選択される。固溶体/固体分散物の調製に有用な溶解度増大性ポリマーのいくつかは、従来、結合剤として用いられている。しかし、活性医薬成分の固体分散物を提供するために、溶解度増大性ポリマーと活性医薬成分との比率は、従来の医薬製剤中のポリマー結合剤と活性医薬成分との比率よりも、一般にかなり高い(従来の医薬製剤において、ポリマー結合剤と活性医薬成分との比率は、一般に1/9未満、例えば、約1/50から約1/20である)。一実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、9/1から1/6(重量)の範囲である。別の実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、約3/1から約1/3(重量)の範囲である。さらに別の実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、約2/1から約1/2(重量)の範囲、または約1/1である。
【0036】
固体分散物は、粒子の形態(例えば、顆粒、ペレット、ビーズなど)であり得るか、あるいは、不活性コア上に層状化できる。例えば、活性医薬成分および溶解度増大性ポリマーを、薬学的に許容できる溶媒(または溶媒混合物)中に溶解し、不活性コア上にコーティングすることができる。該溶媒を除去すると、固体分散物が、不活性コア上のコーティングとして形成される。不活性コアとして、薬学的に許容できる任意の不活性材料、例えば、糖球、またはビーズ(例えば、Celphere(登録商標))、セルロース球体、二酸化ケイ素球体などを、好適な粒径分布(例えば、カプセル製剤への導入のためのコーティングビーズの製造には、約20〜25メッシュから35〜40メッシュの糖球であり、ODT製剤への導入のためのコーティングビーズの製造には、約50〜100メッシュの範囲の狭い粒径分布を有する糖球またはセルロース球体で使用することができる。治療上有効な量の活性医薬成分を提供するために、固体分散層の厚さならびに活性医薬成分と溶解度増大性ポリマーとの相対量を調整することができる。例えば、活性医薬成分の固体分散物によって層状化された不活性コアは、2重量%から約50重量%の活性医薬成分(薬剤コーティング不活性コアの総重量に対して)を含有することができる。
【0037】
本発明の医薬組成物の固体分散物は、水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティング、例えば、全ての目的に対して本明細書に参照として取り入れる米国特許第6,627,223号に記載されたTPRコーティングによってコーティングされる。TPRコーティングは、活性医薬成分の放出を調節し、患者における血漿中の活性医薬成分治療有効レベルを、例えば12〜24時間提供する。いくつかの実施形態において、TPRコーティングは、短い遅延時間の間(例えば、約4時間まで)、活性医薬成分の放出を遅延させることができる。また、TPRコーティングは、長時間にわたって、例えば、約12時間まで、約18時間まで、または約24時間まで、該薬剤の持続的な治療レベルを提供することができる。
【0038】
好適な水不溶性ポリマーとしては、セルロース誘導体(例えばエチルセルロース)、酢酸ポリビニル(BASFからのKollicoat SR30D)、エチルアクリレートとメチルメタクリレートに基づいた中性コポリマー、Eudragit NE、RSまたはRS30D、RLまたはRL30Dなどの四級アンモニウム基を有するアクリル酸エステルとメタクリル酸エステルのコポリマーが挙げられる。
【0039】
腸溶性ポリマーは、胃で見られる低pHレベルでは不溶性であるが、腸管で見られる高pHレベルでは比較的可溶性である。好適な腸溶性ポリマーとしては、酸置換セルロースエステル(例えば、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート)、ポリビニルアセテートフタレート、pH感受性メタクリル酸−メタメタクリレートコポリマーおよびシェラックが挙げられる。市販入手できる腸溶性ポリマーは、Rhom Pharma製造の商標名「Eudragit」(例えば、Eudragit L100、S100、L30D)、Eastman Chemical社からのCellacefate(セルロースアセテートフタレート)、FMC社からのAquateric(セルロースアセテートフタレート水性分散物)、Shin Etsu K.K.からのAquat(ヒドロキシプロピルメチルセルロースアセテートスクシネート水性分散物)が販売されている。
【0040】
TPRコーティング中の水不溶性ポリマーと腸溶性ポリマーとの比率は、約1/9から約9/1(重量)で変わり得る。一実施形態において、水不溶性ポリマーと腸溶性ポリマーとの比率は、約1/4から約4/1、または約1/3から約3/1(重量)の範囲であり得る。腸溶性コーティングの全重量は、TPRビーズの全重量の約5%〜50%の範囲であり得る。一実施形態において、TPRビーズ上のTPRコーティングの全重量は、TPRビーズの全重量に基づいて、約10重量%から約25重量%の範囲である。
【0041】
TPRコーティングの形成に用いられる腸溶性ポリマーおよび水不溶性ポリマーは、可塑化することができる。TPRコーティング層を可塑化するために使用できる好適な可塑化剤の代表例としては、トリアセチン、トリブチルシトレート、トリエチルシトレート、アセチルトリ−n−ブチルシトレート、ジエチルフタレート、ヒマシ油、ジブチルセバケート、アセチル化モノグリセライド類などまたはそれらの混合物が挙げられる。可塑化剤は、存在する場合、TPRコーティングの全重量の約3%から30%を含み得る。一実施形態において、可塑化剤は、TPRコーティングの全重量の約10%から25%を含む。可塑化剤のタイプおよび量は、TPR層の水不溶性ポリマーおよび腸溶性ポリマーの性質ならびにコーティング系の性質(例えば、水性ベースか溶媒ベースか、溶液ベースか分散物ベースか、およびコーティング系の全固体含量)に依存する。
【0042】
固体分散物(少なくとも1種の活性医薬成分および少なくとも1種の溶解度増大性ポリマーを含む)およびTPRコーティングに加えて、本発明の医薬組成物は、追加の薬学的に許容できる成分または賦形剤をさらに含み得る。本発明の組成物または剤形に使用するための好適な賦形剤の例としては、増量剤、希釈剤、滑剤、崩壊剤、結合剤、潤滑剤などが挙げられる。他の薬学的に許容できる賦形剤としては、酸性化剤、アルカリ化剤、保存剤、抗酸化剤、緩衝剤、キレート化剤、着色剤、錯体化材料、乳化剤および/または可溶化剤、風味剤および香料、湿潤剤、甘味剤、浸潤剤などが挙げられる。
【0043】
好適な増量剤、希釈剤および/または結合剤の例としては、ラクトース(例えば、スプレー乾燥ラクトース、α−ラクトース、β−ラクトース、Tabletose(登録商標)、種々の等級のPharmatose(登録商標)、Microtose(登録商標)、またはFast−Floc(登録商標))、微結晶セルロース(種々の等級のAvicel(登録商標)、Elcema(登録商標)、Vivacel(登録商標)、Ming Tai(登録商標)またはSolka−Floc(登録商標))、ヒドロキシプロピルセルロース、L−ヒドロキシプロピルセルロース(低置換)、ヒドロキシプロピルメチルセルロース(HPMC)(例えば、Methocel EおよびMetolose 65 SHの4,000cps等級、Methocel FおよびMetolose 60 SHの4,000cps等級、MethocelKの4,000、15,000および100,000cps等級;および Metolose 90 SHの4,000、15,000、39,000および100,000等級などの、Shin−Etsu社のMethocel E、FおよびK、Metolose SH)、メチルセルロースポリマー(例えば、Methocel A、Methocel A4C、Methocel A15C、Methocel A4Mなど)、ヒドロキシエチルセルロース、カルボキシメチルセルロースナトリウム、カルボキシメチレン、カルボキシメチルヒドロキシエチルセルロースおよび他のセルロース誘導体、スクロース、アガロース、ソルビトール、マンニトール、デキストリン類、マルトデキストリン類、澱粉類または改変澱粉類(ジャガイモ澱粉、トウモロコシ澱粉および米澱粉など)、リン酸カルシウム(例えば、塩基性リン酸カルシウム、リン酸水素カルシウム、水和リン酸二カルシウム)、硫酸カルシウム、炭酸カルシウム、アルギン酸ナトリウム、コラーゲンなどが挙げられる。
【0044】
希釈剤の具体的な例としては、例えば、炭酸カルシウム、リン酸水素カルシウム、第三リン酸カルシウム、硫酸カルシウム、微結晶セルロース、粉末セルロース、デキストラン、デキストリン、デキストロース、フルクトース、カオリン、ラクトース、マンニトール、ソルビトール、澱粉、アルファ化澱粉、スクロース、糖などが挙げられる。
【0045】
崩壊剤の具体的な例としては、例えば、アルギン酸またはアルギネート、微結晶セルロース、低置換ヒドロキシプロピルセルロースおよび他のセルロース誘導体、クロスカルメロースナトリウム、クロスポピドン、ポラクリリンカリウム、ナトリウム澱粉グリコレート、澱粉、アルファ化澱粉、カルボキシメチル澱粉(例えば、Primogel(登録商標)およびExplotab(登録商標))などが挙げられる。結合剤の具体的な例としては、例えば、アラビアゴム、アルギン酸、寒天、カラゲナンカルシウム、カルボキシメチルセルロースナトリウム、微結晶セルロース、デキストリン、エチルセルロース、ゼラチン、液体グルコース、ガーゴム、ヒドロキシプロピルメチルセルロース、メチルセルロース、ペクチン、PEG、ポリエチレンオキシド類、ポビドン、アルファ化澱粉などが挙げられる。
【0046】
滑剤および潤滑剤の具体的な例としては、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウムまたは他の金属ステアリン酸塩類、タルク、ワックス類およびグリセリド類、軽鉱物油、PEG、ベヘン酸グリセリン、コロイドシリカ、水素化植物油、トウモロコシ澱粉、フマル酸ステアリルナトリウム、ポリエチレングリコール類、硫酸エステル塩、安息香酸ナトリウム、酢酸ナトリウムなどが挙げられる。
【0047】
他の賦形剤としては、例えば、風味剤、着色剤、味覚マスキング剤、pH調整剤、緩衝剤、保存剤、安定化剤、抗酸化剤、湿潤剤、湿度調整剤、表面活性剤、懸濁化剤、吸収増強剤、放出制御剤などが挙げられる。
【0048】
非晶質固溶体/固体分散物の長期化学的安定性を改善するために用いられる抗酸化剤としては、例えば、アスコルビン酸、アスコルビルパルミテート、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエン、次亜リン酸、モノチオグリセロール、メタ重亜硫酸カリウム、没食子酸プロピル、ホルムアルデヒドスルホキシル酸ナトリウム、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、二酸化硫黄、トコフェロール、酢酸トコフェロール、トコフェロールヘミスクシネート、TPGSまたは他のトコフェロール誘導体などが挙げられる。
【0049】
また、本発明の医薬組成物は、薬学的に許容できる有機酸をさらに含むことができる。薬学的に許容できる有機酸は、活性医薬成分の放出プロファイル(例えば、放出の速度および程度)をさらに改善または調節することができる。本発明の組成物に有用な薬学的に許容できる好適な有機酸としては、限定はしないが、クエン酸、フマル酸、アスパラギン酸、酒石酸およびコハク酸が挙げられる。いくつかの実施形態において、活性医薬成分と溶解度増大性ポリマーの固体分散物は、該固体分散物の約10重量%〜90重量%の範囲の量で、少なくとも1種の薬学的に許容できる有機酸を含む。他の実施形態において、有機酸の量は、固体分散物の25重量%〜75重量%の範囲である。
【0050】
本発明の組成物は、1つ以上の追加コーティング層(例えば、保護層またはシーラント層(sealant layers)、圧縮性コーティング、腸溶層、味覚マスキング層など)も含み得る。例えば、追加コーティング層は、例えば、Opadry ClearまたはPharmacoat 603(ヒドロキシプロピルメチルセルロースコーティング組成物)、ヒドロキシプロピルセルロース、またはエチルセルロースを含む1つ以上の保護コーティングまたはシーラントコーティングを含み得る。保護コーティングまたはシーラントコーティングは、固体分散物とTPRコーティングとの間に、TPRコーティング上に適用することができ、あるいは、例えば、固体分散物とTPRコーティングとの間ならびにTPRコーティング上に複数の保護をシーラントコーティングとした。追加のコーティング層は、圧縮性コーティング、例えば、固体分散物を含むIRビーズ、味覚マスキングIRビーズまたはTPRビーズ上に沈着させた高可塑化したエチルセルロースまたはヒドロキシプロピルセルロースの層も含み得る。
【0051】
本発明の医薬組成物の他の実施形態は、本明細書に記載された1種以上の腸溶性ポリマーを含む1つ以上の腸溶層を含み得る。任意選択の腸溶層を、固体分散物とTPRコーティングとの間に沈着させ、および/またはTPRコーティング上に沈着させることができる。
【0052】
本発明の医薬組成物は、所望の操作性および薬剤放出特性を提供する、保護層またはシーラント層、圧縮性コーティング、および腸溶層の任意の組合せを含み得る。
【0053】
本発明の医薬組成物は、種々の経口剤形、例えば、カプセル剤(ゼラチンカプセル剤またはHPMCカプセル剤)、錠剤、または口腔内崩壊錠(ODT)に製剤化することができる。錠剤は、そのままで嚥下され、胃に入ると急速に分散するように意図されているが、ODTは、口腔内で唾液に接触すると急速に崩壊して、容易に嚥下される粒子の滑らかな懸濁液を形成する点で、錠剤とODT剤形とは異なる。
【0054】
いくつかの実施形態において、本発明の剤形は、TPRビーズのみを含む。他の実施形態において、本発明の剤形は、即時放出(IR)ビーズとTPRビーズ(すなわち、本明細書に記載されたような)との混合物を含み得る。IRビーズは、溶解度増大性ポリマー中に活性医薬成分の固体分散物を含み、本質的に直ちに活性医薬成分を放出する(例えば、投与の約60分以内に、薬剤の≧75%を放出)。IRビーズは、本明細書に記載された不活性コア上に「層状化された」溶解度増大性ポリマー中の固体分散物の粒子、または活性医薬成分の固体分散物を含み得る。IRビーズは、1つ以上の保護層またはシーラント層も任意に含み得る。次いでTPRコーティングを加えることにより、IRビーズをTPRビーズに変換することができる。
【0055】
本発明の剤形がIRビーズとTPRビーズとの混合物を含む場合、IRビーズは、コーティングなしでもよく、任意選択によりシーラントコーティングまたは保護コーティングによってコーティングされていてもよく、および/または任意選択により、味覚マスキング層によってコーティングされていてもよい。味覚マスキング層は、例えば、各々その全体が本明細書に参照として取り入れる米国特許出願第11/213,266号、米国特許出願第11/248,596号および米国特許出願第11/256,653号に記載された味覚マスキング組成物のいずれかを含み得る。具体的には、好適な味覚マスキング層は、1種以上の造孔剤(pore former)と組み合わせた1種以上の薬学的に許容できる水不溶性ポリマーを含む。味覚マスキング層のための薬学的に許容できる好適な水不溶性ポリマーの非限定例としては、例えば、エチルセルロース、セルロースアセテート、セルロースアセテートブチレート、ポリビニルアセテート、およびメタクリレートポリマー(例えば、Eudragit RL、RSおよびNEおよび30D)が挙げられる。好適な造孔剤の非限定例としては、塩化ナトリウム、炭酸カルシウム、リン酸カルシウム、サッカリンカルシウム、コハク酸カルシウム、酒石酸カルシウム、酢酸鉄、水酸化第二鉄、リン酸第二鉄、炭酸マグネシウム、クエン酸マグネシウム、水酸化マグネシウム、リン酸マグネシウム、ポリビニルピロリドン、クロスポビドン、Eudragit E100、Eudragit EPO、およびそれらの混合物が挙げられる。味覚マスキング層中の水不溶性ポリマーと造孔剤との比率は、約95/5から約50/50の範囲、いくつかの実施形態においては、約85/15から約65/35の範囲である。IRビーズに適用される味覚マスキング層の量は、コーティングされたIRビーズの総重量の約5%から約50%の範囲、いくつかの実施形態においては、コーティングされたIRビーズの総重量の約10%から約50%の範囲であり得る。
【0056】
本発明の剤形がIRビーズとTPRビーズとの混合物を含む場合、IRビーズとTPRビーズとの比率は、約1/9から約5/5の範囲、いくつかの実施形態においては、約1/4から約1/1の範囲(重量で)である。
【0057】
本発明の医薬組成物がODT剤形に製剤化される場合、該組成物は崩壊剤をさらに含む。該崩壊剤は、少なくとも1種の糖アルコールおよび/または糖類と組み合わせた少なくとも1種の崩壊剤を含む急速分散微粒剤の形態であり得る。好適な崩壊剤の非限定例としては、クロスポビドン(架橋ポリビニルピロリドン)、澱粉、セルロース、澱粉グリコール酸ナトリウム、およびカルボキシメチルセルロースナトリウムが挙げられる。糖アルコールの非限定例としては、アラビトール、エリスリトール、ラクチトール、マルチトール、マンニトール、ソルビトール、およびキシリトールが挙げられる。好適な糖類の非限定例としては、ラクトース、スクロース、およびマルトースが挙げられる。
【0058】
急速分散微粒剤中の崩壊剤と糖アルコールおよび/または糖類との比率は、約1/99から約10/90の範囲、いくつかの実施形態においては、約5/95の範囲(重量で)である。
【0059】
ODT剤形中のコーティング薬剤含有ビーズ(すなわち、固溶体を含むコーティングビーズ)と急速分散微粒剤との比率は、約1/9から1/1と変動し、いくつかの実施形態においては、約1:4から約1:2と変動する。
【0060】
ODT剤形は、患者の口腔内で急速に崩壊するため、ODTの感覚刺激特性は重要な考慮事項である。例えば、ODTは、良好な「口あたり(mouthfeel)」および味覚特性を提供するように製剤化する必要がある。「口あたり」とは、口腔内での製品の感じである。ざらざらしていない「口あたり」を得るために、TPRビーズ、急速分散微粒剤、および任意選択のIRビーズは、400μm以下、いくつかの実施形態においては、300μm以下、さらに他の実施形態においては、200μm以下の平均粒径を有する必要がある。一実施形態において、急速分散微粒剤を含む一次粒子(すなわち、急速分散微粒剤を形成するために凝集している崩壊剤および糖アルコールおよび/または糖類の粒子)は、30μm以下、他の実施形態においては、25μm以下、さらに他の実施形態においては、20μm以下の平均粒径を有する。
【0061】
一実施形態において、本発明の組成物を含むODT剤形は、本明細書に記載されたTPRビーズおよび急速分散微粒剤を含む。ODT剤形はさらに追加の賦形剤、例えば、圧縮補助剤(例えば微結晶セルロース)および/または追加の崩壊剤(急速分散微粒剤の崩壊剤と同じでもよいし、異なっていてもよい)を含み得る。ODT剤形は、潤滑剤(例えばステアリン酸マグネシウム)を含んでもよいし、外部潤滑ダイシステムにおいて圧縮される場合は、潤滑剤を含まなくてもよい。一実施形態において、本発明のODT剤形は、口腔の唾液と接触して、約60秒のうちに崩壊し、良好な「口あたり」を有する嚥下し易い懸濁液を形成する。別の実施形態において、本発明のODT剤形は、口腔内で唾液と接触して約30秒以内で崩壊し、良好な「口あたり」を有する嚥下し易い懸濁液を形成する。
【0062】
一実施形態において、剤形(例えば、錠剤、ODT、またはカプセル剤)のTPRビーズは、薬剤と溶解度増大性ポリマーの固体分散物でコーティングし、次いで、TPR層でコーティングし、任意選択により1つ以上のシーラント層または腸溶層でコーティングした不活性コアを含み得る。
【0063】
剤形(例えば、錠剤、ODT、またはカプセル剤)のIRビーズは、存在する場合、薬剤および溶解度増大性ポリマーの固体分散物でコーティングし、任意選択により、本明細書に記載されたシーラント層および/または味覚マスキング層でコーティングした不活性コアを含み得る。このように高いビーズは、TPRビーズ調製のための「中間体」として役立つことができ、TPR層でコーティングされると、IRビーズはTPRビーズへと変換される。
【0064】
あるいは、固体分散物の粒子を形成し(例えば、スプレー乾燥により)、固体分散物の「バルク」形態またはより大型の粒子形態を粉砕するか、または固体分散物と共に、1種以上の薬学的に許容できる賦形剤(例えば、増量剤、結合剤、崩壊剤など)を顆粒化することによって、IRビーズを調製することができ、次いでこれを任意選択により、押出し加工し、球状化することができる。次いで、このようなIR粒子/ビーズ/ペレットをTPR層でコーティングしてTPRビーズに変換することができる。
【0065】
本発明の剤形は、1つ以上の異なるタイプのTPRビーズ(例えば、異なるTPR層を有するか、またはシーラント層および/または腸溶層の異なる組合せを有するTPRビーズ)を含み得る。例えば、異なるTPR層を有するTPRビーズは、異なる遅延時間特性および/または異なる放出速度特性を示すことができ、それによって、全体的に異なる薬剤放出特性を有する剤形を提供することができる。異なるタイプのTPRビーズを含む剤形は、任意選択により、いくつかの即時放出特性を提供するためのIRビーズも含むことができる。例えば、一実施形態において、1日1回の剤形は、即時放出を可能にするIRビーズ(約7時間の排出半減期を有する活性医薬成分を含む)と約12〜20時間にわたる薬剤の遅延の持続放出プロファイルを提供し、かつ、約18〜24時間にわたって治療上有効な血漿中濃度を維持する約4時間までの遅延時間を有する第2のTPRビーズ集団との混合物を含む。
【0066】
溶解度増大性ポリマー中の活性医薬成分の固溶体または固体分散物は、活性医薬成分および溶解度増大性ポリマーを、薬学的に許容できる溶媒または溶媒混合物中に溶解させることによって調製することができる。次いで、活性医薬成分と溶解度増大性ポリマーの溶液を、溶解度増大性ポリマー中に活性医薬成分の固溶体形成を促進する条件下で乾燥する。上記で考察したとおり、分子分散した固体分散物の形成は、活性医薬成分に対して溶解度増大性ポリマーが比較的高レベルであると有利である。固体分散物はまた、活性医薬成分と溶解度増大性ポリマーの溶液から溶媒を、例えば、スプレー乾燥により、急速に除去することによって、または活性医薬成分と溶解度増大性ポリマーの溶液を、例えば、流動床コーティング法を用いて、不活性コア上にコーティングする(薬剤層状化ビーズを形成する)ことによって、形成することができる。あるいは、固体分散物はまた、例えば、二軸スクリュー押出し機における混ぜ合わせなど、ポリマー押出し法によって、溶解度増大性ポリマーの溶融物中に活性医薬成分を溶解させることによって調製することができる。好適な粒径(例えば、ODT剤形では、400μm未満の粒径)を得ることが必要な場合、固体分散物の粒子を、任意選択により好適な賦形剤の存在下、粉砕する(粒径を減少させるため)か、または粗砕(例えば、回転粗砕、または粗砕後に押出し球状化)することができる。固体分散物は、例えば、適切な大きさの円形斜め穿孔を用いて、任意選択により圧縮補助剤、潤滑剤などの賦形剤と共に、固体分散物の圧縮粒子によって形成された1〜2mmの直径の「ミニ錠剤」へと形成することもできる。
【0067】
一実施形態において、該固体分散物は、高せん断粗砕機、またはGlatt GPCG粗砕機などの流動床粗砕機において、溶解度増大性ポリマー、弱塩基性薬剤および任意選択により他の薬学的に許容できる賦形剤(例えば、結合剤、希釈剤、増量剤)を粗砕し、凝集体を形成することによって調製される。高せん断粗砕機からの湿潤塊は、押出し加工し球状化して、球状粒子(ペレット)を製造することもできる。
【0068】
上記で考察したような溶媒処理法によって該固体分散物が調製される場合、薬学的に許容できる溶媒は、単一の溶媒であってもよいし、溶媒の混合物であってもよい。好適な溶媒の非限定例としては、水、アセトンなどのケトン類、エタノールなどのアルコール類、およびそれらの混合物(例えば、アセトン水、95%のエタノールなど)が挙げられる。
【0069】
固体分散物粒子(例えば、薬剤/ポリマーのスプレー乾燥固体分散物、薬剤層状化ビーズ、顆粒化固体分散物、ミニ錠剤など)を調製したら、それらを保護シーラントコート(例えば、Pharmacoat(商標)603またはOpadry(登録商標)Clear)によって任意選択によりコーティングし得る。
【0070】
上記に記載の通り調製された固体分散物粒子は、IR(即時放出)ビーズまたはIR粒子と称される。このようなビーズまたは粒子は、この形態で投与されると、活性医薬成分を実質的に直ちに放出すると考えられるからである。上記に記載の通り調製されたIRビーズまたはIR粒子は次に、薬学的に許容できる溶媒中に溶解させた水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティング溶液によってコーティングされる。TPRコーティングの適用には、任意の好適なコーティング法、例えば流動床コーティング法などを用いることができる。
【0071】
いくつかの実施形態において、IRビーズまたはIR粒子に、TPRコーティングに加えて複数のコーティングを適用することが望ましい。例えば、いくつかの実施形態においては、IRビーズを先ず腸溶コーティング(例えば、薬学的に許容できる溶媒中に溶解させた、本明細書に記載された少なくとも1種の腸溶性ポリマーを含むもの)によってコーティングし、乾燥してコーティング溶媒を除去し、次いで、上記のTPRコーティングによってコーティングする。他の実施形態において、IRビーズは、腸溶性ポリマーコーティング、TPRコーティング、次いで第2の腸溶性ポリマーコーティングによってコーティングする。さらに他の実施形態において、IRビーズは第1のTPRコーティング、腸溶性ポリマーコーティング、次いで第2のTPRコーティングによってコーティングし、第1と第2のTPRコーティングは独立して同じであるか、または異なる。さらに他の実施形態において、TPRコーティング層および/または腸溶性ポリマーコーティング層を適用する前に、シーラント層(本明細書に記載された)をIRビーズ上にコーティングする。さらに他の実施形態において、TPRコーティング層および/または腸溶性ポリマーコーティング層を適用した後に、シーラント層を適用することができる。
【0072】
TPRビーズとIRビーズの混合物を含有する製薬剤形において、IRビーズを味覚マスキング層でコーティングすることができる。例えば、流動床コーティングまたはコアセルベーションなどの任意の好適なコーティング技法を用いて、本明細書に記載されたIRビーズのいずれかを、薬学的に許容できる溶媒、水不溶性ポリマー、および任意選択により造孔剤を含む溶液によってコーティングすることができる。
【0073】
次いで、例えば、従来の方法を用いて、TPRビーズを錠剤へと圧縮し、TPRビーズおよび崩壊剤(例えば急速分散微粒子)をODTへと圧縮し、あるいはTPRビーズをカプセルに充填することによって、製薬剤形をTPRビーズから調製することができる。これらの製薬剤形は、本明細書に記載の通り、任意選択により、追加の賦形剤ならびにIRビーズを含有することができる。一実施形態において、本発明の組成物、および任意選択により追加の賦形剤および/またはIRビーズは、外部潤滑化錠剤圧縮を用いて、錠剤へと圧縮される。別の実施形態において、本発明の組成物、急速崩壊微粒剤、任意選択により追加の賦形剤および/またはIRビーズは、ODTへと圧縮される。
【0074】
本発明の組成物を含む製薬剤形は、例えば図7〜図12に示されるように、12〜18時間にわたって、治療上有効なレベルの活性医薬成分を放出する。本発明の組成物の実際の剤形に関する薬剤放出プロファイルは、米国薬局方装置1(バスケット、100rpm)または装置2(パドル、50rpm)などの種々の溶出試験方法ならびに二段階溶出試験法(最初の2時間は、0.1NのHCl(塩酸)700ml中で、その後、200mLのpH改変剤添加により得られるpH6.8、900mL中で試験する)を用いて、インビトロで評価することができる。経時的な薬剤/酸放出は、選択された間隔で得られるサンプルに対してHPLCによって判定される。
【0075】
一実施形態において、本発明の組成物は、二段階溶出媒質(最初の2時間は、0.1NのHCl中で、その後、pH6.8の緩衝液中で試験する)を用いる米国薬局方(USP)溶出試験法によって溶出試験した際、少なくとも約12時間、活性医薬成分の治療上有効な血漿中濃度を提供する。
【0076】
1日1回の血漿中濃度プロファイルを達成するために必要なインビトロ放出プロファイルのタイプを評価するために、ソフトウェアプログラム、WinNonlin(商標)標準型2.1または等価物(例えば、GastroPlus(登録商標))を用いて、1コンパートメント一次モデルを遅延時間推定一次排出動態と適合させ、該薬剤に関する薬物動態パラメーターを用いて、モデリング演習を一般に行なう。次いで、先に確立したモデルをわずかに修正して用いて、一次パラメーターを別のプログラム、Stella 6.01型にインプットする。種々のインビトロ放出プロファイルが作り出され、目的の1日1回の放出プロファイルから、デコンボリューションによって、所望のインビトロ放出(中間、目的および高速)プロファイルが作り出される。
【0077】
以下の非限定例は、1つ以上の薬剤放出「パルス」および所定の遅延開始を示すカプセル剤形を例示する。TPR層の量または厚さを調整し、また、任意選択により追加の層(例えば、腸溶層またはシーラント層)の数およびタイプを調整することによって、所望のプロファイルを提供して、最大の治療有効性を達成し、患者のコンプライアンスを高める(例えば、1日1回の剤形を提供することによって)目的で、該剤形の経口投与の際のインビトロ薬剤放出プロファイルまたは対応するインビボ血漿中濃度プロファイルを設計することができる。本発明の剤形は、従来の剤形の薬剤放出プロファイルに関連した副作用を最小化するレベルに薬剤の血漿中濃度を保持する改善された薬剤放出プロファイルを提供する。
【実施例1】
【0078】
濁度の測定
アセトン(0.5mg/ml)中の塩酸レルカニジピン濃縮液(3mL)を、Kollidon VA64、Methocel E5(ヒプロメロース)、ポリエチレングリコール(PEG6000)、シクロデキストリンまたはKollidon 14 PF(ポリビニルピロリドン)を含有する200mLの緩衝液(pH6.0)に、ポリマーに対して1:2の重量比で加えた。該薬剤溶液は、改善された安定性を示し、したがって、レルカニジピンHClの共役塩基の結晶化のリスクを大きく減少させたことが、図3Aから明白である。
【0079】
固有溶解速度の測定
塩酸レルカニジピンの2種の異なる多形体ならびに非晶質物質(例えば、非晶質薬剤ならびにMethocel E5およびKollidon VA64と塩酸レルカニジピンとの1:2固溶体)に関して、固有溶解速度を判定した。データは図4に示されている。結晶質多形体は、不良な溶解速度ならびに溶解度を示す一方、固溶体は、有意に高い溶解速度ならびに溶解度を示す。
【0080】
粉末X線回折
1:1および1:2の比率での塩酸レルカニジピンとMethocel E5(ヒプロメロース)とをジクロロメタン−メタノール(1対1、v/v)の溶媒混合物中に溶解し、該溶液を1%(w/w)未満の残留溶媒レベルまで乾燥した。同様に、塩酸レルカニジピンとKollidon VA64の1:1および1:2の共沈殿物を調製した。4つのサンプル全てについて、粉末X線回折パターンを作出した。レルカニジピン−Kollidon VA64固溶体に関するXRDパターンは図5に示されており、レルカニジピンHClとKollidon VA64との1:2比の固溶体は、ほぼ全体的に非晶質であることを実証している。
【実施例2】
【0081】
濁度の測定
アセトン(0.5mg/mL)中ニフェジピンの濃縮溶液(3mL)を、Kollidon VA64、Methocel E5(ヒプロメロース)、ポリエチレングリコール(PEG6000)、シクロデキストリンまたはKollidon 14PF(ポリビニルピロリドン)を含有する200mLの緩衝液(pH6.0)に、1:2のニフェジピン/ポリマーの重量比で加えた。図3Bに示すように、ニフェジピン/ポリマー溶液の透過度を経時的にモニターした。より安定な溶液は、溶液からのニフェジピンの結晶化がより緩やかであるため、経時的に透過度の低下がより緩やかである。Methocel E5、Kollidon VA64およびKollidon 14PFは、より高い安定性を示した。
【0082】
粉末X線回折
ジクロロメタン−メタノール(1対1、v/v)の混合物中に、ニフェジピンおよびMethocelを溶解し、次いで、この溶液を残留溶媒レベル1%(w/w)未満まで乾燥させることにより、1:1および1:2のニフェジピン/Methocel比で、ニフェジピンとMethocel E5(ヒプロメロース)の2種の共沈殿物を調製した。同一の方法を用いて、ニフェジピンとKollidon VA64の1:1および1:2共沈殿物も調製した。4つのサンプル全てを粉末X線回折により分析し、ニフェジピン−Kollidon VA64固溶体のXRDパターンは、図6に示されている。1:1共沈殿物に関するXRDパターンにおける鋭いピークの存在は、結晶形態で存在しているニフェジピンを示している。1:2共沈殿物の幅広い、比較的特色のないXRDパターンは、ニフェジピンがほぼ全体的に非結晶質であり、Kollidon VA64中で固体分散物を形成していることを示している。
【実施例3】
【0083】
3A−ニフェジピンIRビーズ(名目上(nominal)10%のニフェジピン添加)
Kollidon VA64(800g)を、溶解するまで激しく攪拌しながら95%エタノール/アセトン/水の72.5/22.5/5の混合物(4930g/1530g/340g)にゆっくりと加え、次いでニフェジピン(400g)を溶解するまでゆっくりと加えた。7インチ(約17.5cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートB(250μmスクリーン)、1.0mmノズルポート、1.5バールの噴霧空気圧、および3.2mmの内径細管で装備されたGlatt GPCG3に、2584gの25〜30メッシュの糖球を充填した。約40gのタルクをニフェジピン/ポリマー溶液中にホモジナイズして帯電を最小にした。約36〜40℃での生成物温度を維持しながら、15重量%の固体含量のニフェジピン溶液を、8〜17g/分のスプレー速度および約60〜80%(気流速度:約85〜115m3/時間)の排出口フラップで糖球上にスプレーした。得られたニフェジピン層状化ビーズ(バッチサイズ:3724g)を、40℃で約45分間Glattユニット内で乾燥して生成物中の残存溶媒レベルを最小にした。98.5%収率の使用可能なビーズ(600〜1200μm)を得た。
【0084】
2800gのニフェジピン層状化ビーズに、重量2%(すなわち、非コーティングビーズの重量に対するコーティング重量)のOpadry(登録商標)Clearの保護シールコート(8重量%の固体;生成物温度:37〜41℃;スプレー速度:5〜12g/分)のコーティングを提供し、さらに40℃で約45分間Glattユニット内で乾燥して生成物中の残存溶媒/水分を除去した。測定力価は、10%のニフェジピンの目標力価に対して9.81%(ニフェジピン%)であった。
【0085】
3B−ニフェジピンTPRビーズ(TPRコーティング:45/40/10/5の比率でEudragit RL/Eudragit L/TEC/タルク)
上記3Aに記載したようにして調製された、名目上10%の薬剤添加を有するニフェジピンIRビーズ(700g)に、45/55のアセトン/エタノール中、10%固体の固体含量でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5溶液(Ultraturrexホモジナイザーを用いて溶液にタルクを懸濁した)をスプレーすることによりコーティングし、20重量%までのコーティングを提供した(サンプルは、5重量%、10重量%、および15重量%のコーティングで取り出した)。
【0086】
最初攪拌しながら透明溶液を得るために、溶媒混液にEudragit RLポリマーをゆっくりと加えることによってTPRコーティング溶液を調製した。次に、Eudragit Lポリマー、次いで可塑剤(トリエチルシトレートまたは「TEC」)をゆっくりと加えて該溶液中に溶解させた。タルクを別個に溶媒混液中でホモジナイズしてから、溶解した該ポリマー類および可塑剤に加えた。4インチ(約10cm)底のスプレーWursterインサート、20mmの仕切りギャップ、空気分配プレートB(250μmスクリーン)、1.0mmノズルポート、1.5バールの噴霧空気圧、および3.2mmの内径細管、ならびにT165P専用フィルターバッグで装備されたGlatt GPCG1を用いて、TPRコーティング溶液をニフェジピンIRビーズに適用した。TPRコーティング溶液を、4〜11g/分のスプレー速度、約20〜30%(気流速度:約2.0〜2.5m/秒)の排出口フラップおよび約35〜38℃での生成物温度でスプレーした。コーティングされたビーズを、40℃で約45分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体(すなわち、TPRコーティングにより一緒に接着された2つ以上のビーズ)が形成された場合、それらを廃棄するために乾燥ビーズを篩い分けした。約5%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0087】
3C−ニフェジピンIRビーズ(名目上5重量%のニフェジピンの添加)
名目上5重量%の薬剤添加を有するニフェジピンIRビーズを、上記3Aに記載された手法に従って調製した。190gのニフェジピンおよび380gのKollidon VA64を、3154gの25〜30メッシュの糖球上に層状化した。測定力価は、4.81%ニフェジピン(5%ニフェジピンの理論的名目上力価に対して)であると判定した。
【0088】
3D−ニフェジピンTPRビーズ(コーティング:40/45/10/5のEudragit RL/L/TEC/タルク)
上記3Cに記載したようにして調製された、名目上5%のニフェジピン添加を有するニフェジピンIRビーズ(700g)に、5重量%、10重量%、15重量%および20重量%のコーティングレベルで上記3Bに記載された手法に従って、Glatt GPCG1内でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5のTPRコーティング溶液によりコーティングした。約5%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例4】
【0089】
4A−ニフェジピンIRビーズ(60〜80メッシュの糖球)
上記3Aに記載された手法と同様の手法に従って、ニフェジピンIRビーズ(名目上10重量%のニフェジピン添加)をGlatt GPCG3内で60〜80メッシュの糖球上にニフェジピン/Kollidon VA64の1:2溶液をスプレーすることにより調製した。
【0090】
4B−ニフェジピンTPRビーズ(TPRコーティング:35/50/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記4Aに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記3Bに記載された手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの35/50/10/5の溶液をスプレーすることによってコーティングし、40℃で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体が形成された場合は、それらを廃棄するために乾燥ビーズを篩い分けした。5%、10%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0091】
4C−ニフェジピンTPRビーズ(TPRコーティング:40/45/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記4Aに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記3Bに記載された手法と同様の手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの40/45/10/5の溶液をスプレーすることによってコーティングした。
【0092】
4D−ニフェジピンTPRビーズ(TPRコーティング:45/40/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記3Bに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記2Aに記載された手法と同様の手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5の溶液をスプレーすることによってコーティングし、40℃で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。15重量%および20重量%のコーティングを有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0093】
実施例3および4の薬剤放出プロファイル
図7は、実施例4のTPRビーズからのニフェジピン放出に対するTPRコーティング組成物の影響を示している。TPRコーティング中の腸溶性ポリマー含量(Eudragit L)が増すと、ニフェルジピン放出速度が増大する。図8は、実施例3のTPRビーズからのニフェジピン放出に対するニフェルジピン添加の影響を示している。ニフェルジピン添加量が5%から10%に増すと、ニフェジピン放出速度が低下する。図9は、同じTPRコーティング組成物およびコーティング重量で、実施例3Bおよび4DのTPRビーズからの薬剤放出に対する粒径(それぞれ25〜30メッシュまたは600〜700μmおよび60〜80メッシュまたは170〜250μm)の影響を示している。実施例4Dのビーズが小さくなると、ニフェジピン放出が速くなることを示している。
【実施例5】
【0094】
5A−モデル薬物IRビーズ(10%の薬剤添加)
ポビドン(PVP K29/32、128.2g)を、72.5/22.5/5の95%エタノール/アセトン/水に6%の固体で、溶解するまで激しく攪拌しながらゆっくりと加え、次いでラモトリギンの弱塩基性類縁体(128.2g)を、溶解するまでゆっくりと加えた。6インチ(約15cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートD(200メッシュスクリーン)、1.0mmノズルポート、1.0バールの噴霧空気圧、および14mmの単一ヘッド細管で装備されたGlatt GPCG3に、1000gの25〜30メッシュの糖球(Chris Hansen)を充填した。生成物温度を約32.5〜33.5℃で維持しながら、8mL/分のスプレー速度、28〜30%(気流速度:3.6〜4.2m/秒/圧:10.5〜8Pa)の排出口フラップで、糖球を該薬剤溶液によりコーティングした。次に薬剤層状化ビーズを、2%のコーティング重量で、Pharmacoat 603の保護シール−コートによりコーティングし、約10分間Glatt内で乾燥して残存溶媒/水分を除去した。このコーティングされたビーズを、20〜30メッシュスクリーンを通して篩い分けした。
【0095】
5B−モデル薬剤TPRビーズ(TPRコーティング:50/35/15EC−10/HP−55/TEC)
40重量%までのコーティングで、90/10アセトン/水に溶解させた50/35/15EC−10/HP−55/TECの溶液(7400/822.2;7.5%の固体)を、上記4Aに記載したようにして調製されたIRビーズ(1000g)にスプレーすることによってコーティングした(サンプルは、約20%、25%、30%および35%のコーティングレベルで取り出した)。EC−10(エチルセルロース、Dow ChemicalsからのEthocel Premium10cps、333.3g)を、溶解するまで30分間以上連続攪拌しながら90/10のアセトン/水にゆっくりと加えた。次に、HP−55(Shin Etsuからのヒドロキシプロピルメチルセルロース、233.3g)およびTEC(100g)を、溶解するまでEC−10溶液に加えた。6インチ(約15cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートD(200メッシュスクリーン)、0.8mmノズルポート、1.0バールの噴霧空気圧、および14mmの単一ヘッド細管、PB3%の専用フィルターバッグで装備されたGlatt GPCG3により、TPRコーティング溶液を適用した。生成物温度を約32〜34℃に維持しながら、10〜15mL/分のスプレー速度、約28%(気流速度:3.4〜3.8m/秒/圧:7〜7.5Pa)の排出口フラップで、TPRコーティング溶液をIRビーズにスプレーし、同じ温度で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体が形成された場合は、それらを廃棄するために乾燥ビーズを篩い分けした。
【0096】
5C−モデル薬剤TPRビーズ(コーティング:35/50/15EC−10/HP−55/TEC)
上記の手法と同様の手法に従って、20重量%、25重量%、30重量%、35重量%および40重量%のコーティング重量において35/50/15EC−10/HP−55/TEC TPRコーティング溶液により、上記5Aに記載したようにして調製されたIRビーズ(1000g)をコーティングした。
【0097】
5D−モデル薬剤TPRビーズ(コーティング:60/25/15EC−10/HP−55/TEC)
上記の手法と同様の手法に従って、5重量%、10重量%、15重量%、および20重量%のコーティング重量において60/25/15EC−10/HP−55/TEC TPRコーティング溶液により、上記4Aに記載したようにして調製されたIRビーズ(1000g)をコーティングした。
【0098】
図10は、実施例5の同じ薬剤添加におけるTPRビーズからの薬剤放出に対するコーティング組成物および/またはコーティングレベルの影響を示している。TPRコーティング中の腸溶性ポリマー含量を25重量%から50重量%に増加させると、TPRビーズからの薬剤放出速度が有意に増大する結果となる。
【実施例6】
【0099】
6A−ニフェジピンIRビーズ(ニフェジピン/VA 64/フマル酸)
上記の手法と同様の手法を用いて、エタノール/アセトン/水に溶解されたニフェジピン/VA 64/フマル酸の1/2/1溶液を、Glatt GPCG3内で25〜30メッシュの糖球および名目上10重量%のニフェジピン添加物上に層状化することによってニフェジピンIRビーズを調製した。
【0100】
6B−ニフェジピンIRビーズ(コーティング:Eudragit RL/L/TEC/タルク)
上記の手法と同様の手法を用いて、30重量%までのコーティングにおいて、35/50/10/5のEudragit RL/Eudragit L/TEC/タルクTPRコーティングにより、上記6Aに記載したようにして調製されたIRビーズ(700g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。15重量%および20重量%のコーティングを有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0101】
6C−ニフェジピンTPRビーズ(二重コーティング)
流動床コーターにおいて、GPCG1内で上記6Aに記載したようにして調製されたIRビーズ(700g)を、10%のコーティング重量における85/10/5のEudragit L/TEC/タルクを含む内部腸溶性コーティング層によりコーティングした。Eudragit L100を、溶解するまで約90分間、激しく攪拌しながらエタノールにゆっくりと加えた。次に、TEC(トリエチルシトレート)を、溶解するまで該溶液にゆっくりと加え、次いで一定に攪拌しながら懸濁タルクを添加した。次いでこれらの腸溶性コーティングされたビーズを、30%までのコーティング重量で35/50/10/5のEudragit RL/Eudragit L/TEC/タルクのTPR層によりコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。上記の手法および処理条件と同様の手法および処理条件を用いて各層を適用した。15%および20%のTPRコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例7】
【0102】
7A−ニフェジピンIRビーズ(ニフェジピン/VA 64/アスパラギン酸)
上記の手法と同様の手法を用いて、ニフェジピンIRビーズは、72.5/22.5/5のエタノール/アセトン/水中のニフェジピン/VA 64/アスパラギン酸の1/2/1溶液を、Glatt GPCG 3内で25〜30メッシュの糖球上にコーティングすることによって調製し、名目上10重量%のニフェジピン添加物を提供した。アスパラギン酸はコーティング溶液に溶解しなかったことから、それを、Ultraturrexホモジナイザーを用いてコーティング溶媒中にホモジナイズした後、ニフェジピンおよびKollidon VA64の溶液に加え、さらにホモジナイズした。
【0103】
7B−ニフェジピンTPRビーズ(コーティング:35/50/15のRL/L−55/TEC)
上記の手法と同様の手法を用いて、30重量%までのコーティングにおいて、35/50/10/5のEudragit RL/Eudragit L/TEC/タルクにより、上記7Aに記載したようにして調製されたニフェジピンIRビーズ(700g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。15%および20%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例8】
【0104】
8A−レルカニジピンHClのIRビーズ(レルカニジピン/VA 64/酒石酸)
レルカニジピンHCl(93g)をエタノール(4808g)にゆっくりと加え、溶解するまで攪拌した。次にKollidon VA 64(186g)、次いで酒石酸(21g)を溶解するまでゆっくりと加えた。6インチ(約15cm)底のスプレーカラム高のWursterインサート、200mmの仕切りギャップ、空気分配プレートC(50メッシュスクリーン)、0.8mmノズルポート、1.5バールの噴霧空気圧で装備されたGlatt GPCG1に、2100gの30〜35メッシュの糖球(2322g)を充填した。生成物温度を約32〜34℃に維持しながら、糖球を、11g/分のスプレー速度、45〜50%(気流速度:90〜105m3/時間)の排出口フラップで、レルカニジピン/VA 64/酒石酸のコーティング溶液によりスプレーすることによってコーティングした。レルカニジピン層状化ビーズは、2%のコーティング重量で保護シールコートのOpadry Clearによりコーティングし、45℃で約15分間Glattユニット内で乾燥して残存溶媒/水分を除去し、次いで25メッシュスクリーンを通して篩い分けした。
【0105】
8B−レルカニジピンのTPRビーズ(コーティング:EC−10/HP−55/DEPを1:4:1で)
上記の手法と同様の手法を用いて、上記8Aで記載したようにして調製されたレルカニジピンHClのIRビーズ(930g)を、27%のコーティング重量で98/2のアセトン/水中EC−10/HP−55/DEPの1/4/1溶液を用いてGlatt流動粗砕機内のIRビーズをスプレーすることによってコーティングした。得られたTPRコーティング組成物は、16.4%のEC−10、65.6%のHP−55、18%のDEP(ジエチルフタレート)であった。
【0106】
8C−レルカニジピンのTPRビーズ(二重層コーティング)
Eudragit L100を、溶解するまで約90分間、激しく攪拌しながらエタノールにゆっくりと加えた。次に、TEC(トリエチルシトレート)を、溶解するまで該溶液にゆっくりと加え、次いで一定に攪拌しながら懸濁タルクを添加した。上記のとおりに調製された74.1/7.4/18.5のEudragit L100/TEC/タルクの内部腸溶性コーティングを、20%のコーティング重量で上記8Aで記載したようにして調製されたIRビーズ上に適用した。次に、上記の8Bに記載されたものと同様の手法を用いて、得られた腸溶性コーティングIRビーズを、16.4/65.6/18のEC−10/HP−55/DEP TPRコーティング溶液により10%のコーティング重量にコーティングした。
【0107】
8D−レルカニジピンのTPRビーズ(三重層コーティング)
上記の手法と同様の手法を用いて、80/20 HP−55/DEPの内部腸溶性コーティングを、20%のコーティング重量で上記8Aで記載したようにして調製されたIRビーズに適用した。次に、上記の手法と同様の手法に従って、腸溶性コーティングビーズを、16.4/65.6/18のEC−10/HP−55/DEP TPRコーティング溶液により25%のコーティング重量にコーティングした。次に、上記の手法と同様の手法を用いて、これらのビーズを、74.1/7.4/18.5の比率でのEudragit S100/TEC/タルクの外部腸溶性コーティング層により10%のコーティング重量にさらにコーティングした。溶解するまで約90分間激しく攪拌しながらEudragit S100をエタノールにゆっくりと加えることによって、外部腸溶性コーティング層を調製した。次にTEC(トリエチルシトレート)を、溶解するまでEudragit S100にゆっくりと加え、次いで一定に攪拌しながら懸濁したタルクを添加した。実施例8B、8C、および8DのTPRビーズに関する薬物放出プロファイルは、二段階方法論(すなわち、最初は0.1N HClおよび0.3%ツイーン80において2時間、引き続きpH6.8で)を用いて溶出試験を行った。溶出試験結果を図11に示している。
【実施例9】
【0108】
9A−ニフェジピンIRビーズ(ニフェジピン/VA 64/酒石酸)
エタノール/アセトン/水に溶解させたニフェジピン/VA 64/フマル酸の1/2/1溶液を、Glatt GPCG3内で名目上10重量%のニフェジピン添加量で25〜30メッシュの糖球上に層状化することによって、ニフェジピンIRビーズを調製した。
【0109】
9B−ニフェジピンTPRビーズ(コーティング35/50/15RL/L−55/TEC)
上記の手法と同様の手法を用いて、Glatt流動粗砕機内において30%のコーティング重量で、アセトン/水に溶解させたEthocel Premium 10cps(EC−10)、ヒプロメロースフタレート(HP−55)およびジエチルフタレート(DEP)の溶液をスプレーすることによって、上記9Aに記載のように調製したニフェジピンIRビーズ(1000g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。スプレー用溶液は、以下の組成を有した:1.23%のEC−10、4.92%のHP−55、1.35%のDEP;溶媒:90.65%のアセトンおよび1.85%の水。
【実施例10】
【0110】
10A−IRビーズ(薬剤添加:16.67%)
ポビドン(PVP K29/32、666.7g)を、溶解するまで激しく攪拌しながら16.6/83.4のエタノール/アセトンにゆっくりと加えた。溶解するまで、イロペリドン(333g)をゆっくりと加えた。次に上記の手法と同様の手法を用いて、Glatt GPCG3内で25〜30メッシュの糖球(1000g)を該薬剤溶液(8.17%の固体)によりコーティングした。薬剤層状化ビーズを、2%のコーティングでPharmacoat603の保護シールコートによりコーティングした。このIRビーズを該ユニット内で約10分間乾燥して残存溶媒/水分を除去し、二倍体が形成された場合は、それらを廃棄するために篩い分けした。
【0111】
10B−TPRビーズ(二重コーティング:HP−55/TEC上に45/40/15のEC−10/HP−55/TEC)
95/5のアセトン/水中、80/20のHP−55/TEC溶液の腸溶性コーティング溶液を8%のコーティング重量でスプレーすることによって、上記10Aに記載したようにして調製されたIRビーズ(1800g)をコーティングした。次に腸溶性コーティングビーズ(850g)を、上記の方法と同様の手法を用いて、Glatt GPCG3内で50%までのコーティング重量で、90/10のアセトン/水混液(7.5%の固体)中、45/40/15のEC−10/HP−55/TEC TPRコーティング溶液によりコーティングし(サンプルは、20%、30%、および40%のコーティングレベルで取り出した)、Glatt内で約50℃で10分間乾燥して過剰の残存溶媒を除去し、二倍体が形成された場合は、それらを廃棄するために篩い分けした。
【0112】
10C−TPRビーズ(二重コーティング:HP−55/TEC上に30/55/15のEC−10/HP−55/TEC)
上記の手法と同様の手法を用いて、上記10Bに記載したようにして調製された腸溶性コーティングビーズ(530g)を、Glatt GPCG3内で50%までのコーティングで、90/10のアセトン/水混液(7.5%の固体)中、30/55/15のEC−10/HP−55/TEC TPRコーティング溶液によりスプレーすることによってコーティングした(サンプルは、20%、30%、および40%のコーティングレベルで取り出した)。2つの異なるTPR組成/レベルでコーティングされたTPRビーズからの代表的な薬剤放出プロファイルを、図12に示している。
【技術分野】
【0001】
関連出願に対する相互参照
本出願は、いずれも2006年8月31日に出願された米国仮特許出願第60/841,760号および米国仮特許出願第60/841,893号の利益を主張するものであり、これらは各々、全ての目的に関して、参照としてその全体を本明細書に取り入れる。
【0002】
本発明は、改善されたバイオアベイラビリティを有する放出制御組成物、およびこのような組成物を製造する方法に関する。本発明の組成物は、少なくとも1種の活性医薬成分の固体分散物および時限拍動型放出(timed pulsatile release)コーティングを含む。
【背景技術】
【0003】
多くの治療薬は、吸収部位またはその近辺で、一定速度で利用可能にされている場合に最も有効である。この様式で利用可能にされている治療薬の吸収は一般に、最大の有効性および最小の毒性副作用をもたらす所望の血漿中濃度をもたらす。しかしながら、吸収過程の複雑性、剤形からの治療薬の放出速度に影響を及ぼす相互に関連する多くの組成変数、および治療薬自体の物理化学的特性のために、一定速度で治療薬の所望の血漿中濃度を送達する経口製薬剤形を開発することは困難であることが多い。例えば、経口投与された製薬剤形が、ヒトの消化管を通る間に、薬剤が該剤形から放出され、胃腸(GI)管からの吸収のための部位またはその近辺で、溶液形態で利用可能であるべきである。該剤形、したがって、該治療薬は、胃腸管を通過する間にpHの変化、すなわち、約1.2(空腹時の胃のpH)から4.0(食物の摂取時)または約7.4(胆汁のpH:7.0〜7.4および腸のpH:5〜7)まで高いpHへの変化に供される。さらに、消化管の個々の部分における剤形の通過時間は、剤形のサイズおよび主要な局所の条件(例えば、胃腸管に沿った浸透性の変化;pH、表面張力、体積、攪拌、および緩衝能力などの管の内容物の特性;ならびに食物消化後の変化)に応じて、著しく変化し得る。血漿中濃度に影響を及ぼす薬剤自体の物理化学的特性には、そのpKa、溶解度および結晶エネルギーが含まれ、例えば、多粒子剤形の組成特性には、薬剤含有粒子のサイズまたは比表面積などが含まれる。その結果、一定速度での薬剤放出の達成はしばしば困難である。
【0004】
さらに、塩基性および酸性の薬剤は、生理学的pH範囲において、2桁超の大きさで変化するpH依存性溶解度プロファイルを示す。これらのうちで、製剤化が最も困難な薬剤は、pH>5ではほとんど不溶(例えば、50μg/mL以下の溶解度を有する)であり、治療上有効であるためには、高用量(例えば、10mg以上の最適1日用量)を必要とする弱塩基性化合物である。このような高用量では、吸収速度が薬剤放出速度より速くない限り、胃腸(GI)管のpH環境内に入ると、溶解した薬剤の一部が沈殿する可能性がある。あるいは、該薬剤は、例えば、腸内の胆汁塩およびレシチンの存在により助長されて、知られている水溶解度より一桁を十分超える過飽和レベルでの過飽和溶液状態に留まる可能性がある。しかし、過飽和溶液は沈殿する可能性があり、次いで該薬剤の再溶解および吸収はより緩やかな速度で生じ得る証拠がある。これらの問題を解決するために、弱塩基性の薬剤の溶解度を増加させるための種々のアプローチ、例えば、有機酸を含めて酸付加化合物を形成すること、または固体分散物または固溶体を使用することが開発されている。
【0005】
しかし、薬剤の溶解度は薬剤自体の物理化学的特性、ならびに医薬製剤を調製する方法によって変化するため、このようなアプローチは完全に満足できるものではない。例えば、ニフェジピンまたはレルカニジピンなどのいくつかの弱塩基性の薬剤は、飽和有機酸溶液中で有意な溶解度増大を示さず、固体分散物は、経口摂取の際に、該薬剤の望ましくない即時放出をもたらす傾向がある。
【0006】
本発明の組成物は、1日1回の投与法に好適な薬剤放出プロファイルを有する弱塩基性治療薬(例えば、14未満のpKaを有し、目標血漿中濃度を維持するために高用量を必要とする)の改良された送達を提供する。
【発明の概要】
【課題を解決するための手段】
【0007】
一実施形態において、本発明は、TPRビーズを含む医薬組成物であって、前記TPRビーズは、少なくとも1種の活性医薬成分および少なくとも1種の溶解度増大性ポリマーの固体分散物、ならびに水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティングを含み、前記活性医薬成分は、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む医薬組成物を指向する。
【0008】
別の実施形態において、本発明は、薬学的に許容できる溶媒中に活性医薬成分および十分な溶解度増大性ポリマーを溶解すること;活性医薬成分および溶解度増大性ポリマーの溶液から薬学的に許容できる溶媒を除去し、それによって、溶解度増大性ポリマー中の活性医薬成分の固体分散物の粒子を形成すること;薬学的に許容できるコーティング溶媒中に水不溶性ポリマーおよび腸溶性ポリマーを溶解し、それによって、TPRコーティング溶液を形成すること;該固体分散物をTPRコーティング溶液でコーティングすること;該コーティング溶媒を除去し、それによって、固体分散物上に形成されたTPRコーティングを含むTPRビーズを形成することを含む、医薬組成物を調製する方法を指向する。
【図面の簡単な説明】
【0009】
【図1】本発明のTPRビーズの一実施形態を示す断面図である。
【図2】(a)塩酸オンダンセトロン、(b)カルベジロール、(c)ジピリダモール、および(d)クロナゼパムに関するpH溶解度プロファイルを示す図である。
【図3】(A)レルカニジピンHClおよび(B)ニフェジピンの固溶体/固体分散物に関する濁度プロファイルを示す図である。
【図4】レルカニジピンHCl−(A)多形体I、(B)多形体IIおよび(C)非晶質物質(薬剤−ポリマー固溶体)の固有溶解速度(IDR)を示す図である。
【図5】レルカニジピンHClおよび(A)Kollidon VA64または(B)Methocel E5の固体分散物の粉末X線回折パターンを示す図である。
【図6】ニフェジピンおよび(A)Kollidon VA64または(B)Methocel E5の固体分散物の粉末X線回折パターンを示す図である。
【図7】実施例4に記載されたTPRビーズからの薬剤放出に及ぼすTPRコーティング組成物の影響を示す図である。
【図8】実施例3に記載されたTPRビーズからの薬剤放出に及ぼす薬剤添加量の影響(すなわち、10%の薬剤添加と5%との薬剤添加)を示す図である。
【図9】実施例3Bの60〜80メッシュの糖球上、10%の薬剤添加および15重量%のEudragit RL/Lコーティング(45/40)でコーティングされたTPRビーズからの薬剤放出に及ぼす粒径の影響を、実施例4Dの25〜30メッシュの糖球から調製された同様のTPRビーズと比較して示す図である。
【図10】実施例5のTPRビーズからの薬剤放出に及ぼすTPRコーティング組成物の影響を示す図である。
【図11】Kollidon VA64および酒石酸を含有する実施例8のレルカニジピンHCl TPRビーズからの薬剤放出プロファイルを示す図である。
【図12】実施例9のTPRビーズからの薬剤放出に及ぼすTPRコーティング組成ならびに厚さの影響を示す図である。
【発明を実施するための形態】
【0010】
本発明は、少なくとも1種の活性医薬成分と少なくとも1種の溶解度増大性ポリマーの固体分散物、ならびに水不溶性ポリマーおよび腸溶性ポリマーを含む時限拍動型放出(TPR)コーティングとの組合せを含む医薬組成物を指向し、前記活性医薬成分は、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む。弱塩基性の活性医薬成分が固体分散物の形態で存在せず、および/またはTPRコーティングを欠く従来の組成物によって得られた放出プロファイルに比較して、弱塩基性の活性医薬成分の固体分散物とTPRコーティングとの組合せは、改善された放出プロファイルを提供する。例えば、少なくとも1種のTPRコーティングを含む組成物の好適な処置により、放出速度を、12〜18時間にわたってほぼ一定にすることができるか、または溶解度増大性ポリマーの単独使用に比較して、最大放出速度までの時間を遅延させることができる。
【0011】
用語「固体分散物」または「固溶体」とは、ポリマー基質中に分散した実質的に非晶質の活性医薬成分を意味し、および/またはより具体的には、少なくとも1種の活性医薬成分および少なくとも1種の結晶化阻害性ポリマーが固体状態において実質的に分子分散していることである。用語「実質的に非晶質」とは、活性医薬成分の40%未満が、ポリマー基質中で分離した結晶相を形成していることである。他の実施形態において、「実質的に非晶質」とは、活性医薬成分の30%未満、20%未満、10%未満、5%未満、または1%未満が、ポリマー基質中で分離した結晶相を形成していることを意味する。言い換えると、活性医薬成分の少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、少なくとも95%、または少なくとも99%が、非晶質の状態にある。用語「実質的に分子分散している」とは、活性医薬成分の40%未満が、ポリマー基質中で分離した結晶相を形成し、残りの活性医薬成分が、ポリマー基質中に溶解していることを意味する。他の実施形態において、「実質的に分子分散している」とは、活性医薬成分の30%未満、20%未満、10%未満、5%未満、または1%未満が、ポリマー基質中で分離した結晶相を形成していることを意味する。本発明の固体分散物は、活性医薬成分の40%以下、いくつかの実施形態においては、活性医薬成分の30%以下、20%以下、10%以下、5%以下、または1%以下が、ポリマー基質中で結晶相を形成しているという条件で、ポリマー基質中の「実質的に分子分散している」活性医薬成分と「実質的に非晶質」な活性医薬成分との組合せを含む。
【0012】
用語「活性医薬成分」は、用語「薬剤」、「治療薬」などと交換可能に使用することができる。本明細書に用いられる用語「弱塩基性の活性医薬成分」、ならびにいずれかの特定の薬剤の記述は、塩基、薬学的に許容できる塩、多形体、立体異性体、溶媒和物、エステル類およびそれらの混合物を含む。一実施形態において、本発明の組成物の弱塩基性活性医薬成分は、14未満のpKaを有する化合物のことであり得る。別の実施形態において、弱塩基性の活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有する。別の実施形態において、弱塩基性の活性医薬成分は、少なくとも1種の塩基性窒素原子を含む。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、pH6.8で約100μg/mL以下の溶解度を有する。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、少なくとも1種の塩基性窒素原子を含む。さらに別の実施形態において、弱塩基性の活性医薬成分は、14未満のpKaを有し、かつ、pH6.8で100μg/mL以下の溶解度を有し、かつ、少なくとも1種の塩基性窒素原子を含む。
【0013】
本明細書に用いられる用語「溶解度増大性ポリマー(solubility-enhancing polymer)」または「結晶化阻害性ポリマー(crystallization-inhibiting polymer)」とは、例えば、先ず薬剤とポリマーの双方を同一の溶媒系に溶解し、次いで、適切な条件下で該溶媒を除去することによって、溶解度増大性ポリマー中の弱塩基性薬剤の本明細書に定義された固体分散物を好適な濃度で形成することのできる水溶性ポリマーのことである。該弱塩基性薬剤は、溶解度増大性ポリマーと弱塩基性薬剤の固体分散物を含有する該組成物の保存、輸送、および商品配送の間、実質的に分子分散物としてあるいは非晶質形態で維持される。
【0014】
本明細書に用いられる用語「即時放出」(IR)とは、組成物の投与後1時間で活性医薬成分の約75%超、他の実施形態においては、約85%超の放出を言う。放出量はインビボまたはインビトロで(本明細書に記載されている慣用のUSP法を用いて)測定することができる。
【0015】
用語「IRビーズ」とは、即時放出性を有する活性医薬成分含有粒子のことである。IRビーズとしては、活性医薬成分を含む任意の種類の粒子、例えば、溶解度増大性ポリマー中の活性医薬成分の固体分散物の粒子、または溶解度増大性ポリマー中の活性医薬成分の固体分散物によってコーティングされた不活性コアを挙げることができる。IRビーズとしては、固体分散物を含み、さらにシーラント(sealant)または保護層によってコーティングされ、本明細書に記載された即時放出性を有する粒子が挙げられる。
【0016】
本明細書に用いられる用語「急速分散微粒剤」とは、崩壊剤と組み合わせた、糖アルコール(例えばD−マンニトール)および/または糖類(例えば乳糖)の一次粒子を含む凝集粒子のことである。
【0017】
本出願において交換可能に用いられる用語「遅延時間膜コーティング」、「遅延時間ポリマーコーティング」、「時限拍動型放出(TPR)膜コーティング」、「TPRポリマーコーティング」、またはTPRコーティングとは、腸溶性ポリマーと組み合わせて水不溶性ポリマーを含むコーティングのことである。
【0018】
用語「時限拍動型放出(TPR)ビーズ」または簡単に「TPRビーズ」とは、TPRコーティングでコーティングされた、活性医薬成分含有粒子のことである。いくつかの実施形態において、TPRビーズとは、TPRコーティングでコーティングされたIRビーズのことであり、本発明の一定の実施形態に従って調製されたTPRビーズからの弱塩基性活性医薬成分の放出は、短い遅延時間後の徐放性プロファイルを特徴とする。
【0019】
用語「遅延時間」とは、活性医薬成分の約10%未満、さらに特に約5%未満、さらに特に実質的に0%が放出される時間のことであり、水不溶性ポリマーと腸溶性ポリマー(例えば、Eudragit RLポリマーとLポリマー)の組合せを含む本発明のTPRコーティングによって、約4時間までの遅延時間を達成することができる。
【0020】
本明細書に用いられる用語「溶解度調節有機酸」または「有機酸」とは、有機酸の水溶液中への活性医薬成分の溶解の速度および/または程度を増大させることのできる水溶性の薬学的に許容できる有機酸のことである。
【0021】
用語「放出速度」とは、単位時間当たりに組成物からインビトロまたはインビボで放出される薬剤の量のことである。量の単位は、例えば、全用量の%として表されることが多い。
【0022】
用語「血漿プロファイル」、「血漿中濃度」、「Cmax」、または「Cmin」は、単位体積当たりの質量、典型的には、1ミリリットル当たりのナノグラム(ng/mL)として一般に表される、対象の血漿中の薬剤濃度のことである。
【0023】
用語「治療上有効な量」とは、所望の薬理学的結果を提供するのに必要な活性医薬成分の量のことである。実際、治療上有効な量は、病態の重症度、対象の年齢、および所望の治療効果に依って大きく変動する。
【0024】
本発明の医薬組成物は、少なくとも1種の活性医薬成分と少なくとも1種の溶解度増大性ポリマーの固体分散物およびTPRコーティングを含む。
【0025】
本発明の特定の実施形態を、添付の図1を参照して、さらに詳細に記載する。図1は、TPRビーズ10を表す。不活性粒子コア18、弱塩基性薬剤、結晶化阻害性ポリマー(溶解度増大性ポリマーとも称される)、および溶解度増大性有機酸を含む非晶質層16、保護シールコーティング層14、および遅延時間(TPRまたは拍動型放出とも称される)コーティング12がTPRビーズ10を構成する。
【0026】
本発明の医薬組成物のための好適な活性医薬成分は弱塩基性薬剤を含む。一実施形態において、該活性医薬成分は、14未満のpKa値を有する。別の実施形態において、該活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有する。別の実施形態において、該活性医薬成分は、約3時間以上の排出半減期を有する。別の実施形態において、該活性医薬成分は、pH6.8で50μg/mL以下の溶解度を有する。別の実施形態において、該活性製薬群の成分は、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。さらに別の実施形態において、該活性医薬成分は、14未満のpKa値、pH6.8で約100μg/mL以下の溶解度、および約3時間以上の排出半減期を有する。さらに別の実施形態において、該活性医薬成分は、pH6.8で約100μg/mL以下の溶解度を有し、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。さらに別の実施形態において、該活性医薬成分は、pH6.8で約50μg/mL以下の溶解度を有し、、pH6.8での溶解度(mg/mL)に対する最適1日用量(mg)の比率が少なくとも100である。
【0027】
好適な活性医薬成分のクラスの非限定例としては、鎮痛薬、降圧薬、抗不安薬、抗凝固薬、抗痙攣薬、抗糖尿病薬、血糖値低下薬、うっ血除去薬、抗ヒスタミン薬、抗炎症薬、鎮咳薬、抗悪性腫瘍薬、ベータブロッカー、抗リウマチ薬、抗炎症薬、抗精神病薬、向知性薬、抗アテローム硬化症薬、抗肥満症薬、抗不能症薬、抗感染症薬、催眠薬、抗パーキンソン薬、抗アルツハイマー病薬、抗うつ薬、および抗ウィルス薬、グリコーゲンホスホリラーゼ阻害剤、コレステロールエステルトランスファータンパク質阻害剤、CNS(中枢神経系)刺激薬、ドーパミン受容体アゴニスト、制吐薬、胃腸薬、精神治療薬、オピオイドアゴニスト、オピオイドアンタゴニスト、抗てんかん薬、ヒスタミンH2アンタゴニスト、抗喘息薬、平滑筋弛緩薬および骨格筋弛緩薬が挙げられるが、これらに限定されない。
【0028】
鎮痛薬の具体例としては、アセトアミノフェン、ロフェコキシブ、セレコキシブ、モルフィン、コデイン、オキシコドン、ヒドロコドン、ジアモルフィン、ペチジン、トラマドール、ブプレノルフェンが挙げられ;降圧薬としては、プラゾシン、ニフェジピン、レルカニジピン、アムロジピンベシレート、トリマゾシンおよびドキサゾシンが挙げられ;抗不安薬の具体例としては、ヒドロキシジン塩酸塩、ロラゼパム、ブスピロン塩酸塩、パゼパム、クロルジアゼポキシド、メプロバメート、オキサゼパム、トリフルオペラジン塩酸塩、クロラゼペート二カリウム塩、ジアゼパムが挙げられ;抗凝固薬の具体例としては、アブシキシマブ、エプチフィバチド、チロフィバン、ラミフィバン、クロピドグレル、チクロピジン、ジクマロール、ヘパリン、およびワルファリンが挙げられ;抗痙攣薬の具体例としては、フェノバルビタール、メチルフェノバルビタール、クロバザム、クロナゼパム、クロレゼペート、ジアゼパム、ミダゾラム、ロラゼパム、フェルバメート、カルバメゼピン、オクスカルベゼピン、ビガバトリン、プロガビド、チアガビン、トピラメート、ガバペンチン、プレガバリン、エトトイン、フェニトイン、メフェニトイン、ホスフェニトイン、パラメタジオン、トリメタジオン、エタジオン、ベクラミド、プリミドン、ブリバラセタム、レベチラセタム、セレトラセタム、エトスクシミド、フェンスクシミド、メスクシミド、アセタゾラミド、スルチアメ、メタゾラミド、ゾニサミド、ラモトリギン、フェネツリド、フェナセミド、バルプロミド、およびバルノクタミドが挙げられ;抗糖尿病薬の具体例としては、レパグリニド、ナテグリニド、メトフォルミン、フェンホルミン、ロシグリタゾン、ピオグリタゾン、トログリタゾン、ミグリトール、アカルボース、エキサナチド、ビルダグリプチン、およびシタグリプチンが挙げられ;血糖値低下薬の具体例としては、トルブタミド、アセトヘキサミド、トラザミド、グリブリド、グリメピリド、グリクラジド、グリピジドおよびクロルプロパミドが挙げられ;うっ血除去薬の具体例としては、プソイドエフェドリン、フェニルエフリン、およびオキシメタゾリンが挙げられ;抗ヒスタミン薬の具体例としては、メピラミン、アンタゾリン、ジフェンヒドラミン、カルビノキサミン、ドキシラミン、クレマスチン、ジメンヒドリネート、フェニラミン、クロルフェニラミン、デクスクロルフェニラミン、ブロムフェニラミン、トリポリジン、シクリジン、クロルシクリジン、ヒドロキシジン、メクリジン、プロメタジン、トリメプラジン、シプロヘプタジン、アザタジン、およびケトチフェンが挙げられ;鎮咳薬の具体例としては、デキストロメトルファン、ノスカピン、エチルモルフィン、およびコデインが挙げられ;抗悪性腫瘍薬の具体例としては、クロラムブシル、ロムスチン、ツブラゾールおよびエキノマイシンが挙げられ;抗炎症薬の具体例としては、ベタメタゾン、プレドニゾロン、アスピリン、ピロキシカム、バルデコキシブ、カルプロフェン、セレコキシブ、フルビプロフェンおよび(+)−N−{4−[3−(4−フルオロフェノキシ)フェノキシ]−2−シクロペンテン−1−イル}−N−ヒドロキシ尿素が挙げられ;β−ブロッカーの具体例としては、チモロールおよびナドロールが挙げられ;鎮咳薬の具体例としては、デキストロメトルファン、ノスカピン、エチルモルフィン、テオブロミンおよびコデインが挙げられ;抗悪性腫瘍薬の具体例としては、アクチノマイシン、ダクチノマイシン、ドキソルビシン、ダウノルビシン、エピルビシン、ブレオマイシン、プリカマイシン、およびマイトマイシンが挙げられ;ベータブロッカーの具体例としては、アルプレノロール、カルテオロール、レボブノロール、メピンドロール、メチプラノロール、ナドロール、オクスプレノロール、ペンブトロール、ピンドロール、プロパノロール、ソタロール、チモロール、アセブトロール、アテノロール、ベタキソロール、ビソプロロール、エスモロール、メトプロロール、ネビボロール、カルベジロール、セリプロロール、ラベタロール、およびブタキセミンが挙げられ;抗リウマチ薬の具体例としては、アダリムマブ、アザチオプリン、クロロキン、ヒドロキシクロロキン、シクロスポリン、D−ペニシラミン、エタネルセプト、ナトリウムアウロチオマレート、アウラノフィン、インフリキシマブ、レフルノミド、メトトレキサート、ミノシクリン、スルファサラジンが挙げられ;抗炎症薬の具体例としては、ヒドロコルチゾン、プレドニゾン、プレドニゾロン、メチルプレドニゾロン、デキサメタゾン、ベタメタゾン、トリアムシノロン、ベクロメタゾン、アルドステロン、アセトアミノフェン、アモキシプリン、ベノリレート、ジフルニサル、ファィスラミン、ジクロフェナック、アセクロフェナック、アセメタシン、ブロムフェナック、エトドラック、インドメタシン、ナブメトン、スリンダック、トルメチン、カプロフェン、ケトロラック、メフェナム酸、フェニルブタゾン、アザ抗炎症スプロパゾン、マタミゾール、オキシフェンブタゾン、スルフィンプラゾン、ピロキシカム、ロモキシカム、メロキシカム、テノキシカム、セレコキシブ、エトリコキシブ、ルミリコキシブ、パレコキシブ、ロフェコキシブ、バルデコキシブ、およびヌメスルフィドなどのステロイド系および非ステロイド系抗炎症薬が挙げられ;抗精神病薬の具体例としては、イロペリドン、ジプラシドン、オランゼピン、チオチキセン塩酸塩、フルスピリレン、リスペリドンおよびペンフルリドールが挙げられ;向知性薬の具体例としては、アンパキンが挙げられ;抗アテローム硬化症薬、心血管薬および/またはコレステロール減少薬の具体例としては、アトルバスタチンカルシウム、セリバスタチン、フルバスタチン、ロバスタチン、メバスタチン、ピタバスタチン、プラバスタチン、ロスバスタチン、およびシンバスタチンが挙げられ;抗肥満症薬の具体例としては、デキサドリン、デキスフェンフルラミン、フェンフルラミン、フェンテルミン、オルリスタート、アカルボース、およびリモナバントが挙げられ;抗不能症薬の具体例としては、シルデナフィルおよびシルデナフィル酒石酸塩が挙げられ;抗菌薬、抗ウィルス薬、抗原生動物薬、駆虫薬および抗真菌薬などの抗感染症薬の具体例としては、カルベニシリン、インダニルナトリウム、ベカンピシリン塩酸塩、トロレアンドマイシン、ドキシシリンヒクレート、アンピシリン、ペニシリンG、アジトロマイシン、オキシテトラサイクリン、ミノサイクリン、エリスロマイシン、クラリスロマイシン、サピラマイシン、アシクロビル、ネルフィナビル、ビラゾール、塩化ベンザルコニウム、クロルヘキシジン、エコナゾール、テルコナゾール、フルコナゾール、ボリコナゾール、グリセオフルビン、メトロニダゾール、チアベンダゾール、オクスフェンダゾール、モランテル、コトリモキサゾールが挙げられ;催眠薬の具体例としては、アルファキサロンおよびエトミデートが挙げられ;抗パーキンソン薬の具体例としては、レボドパ、ブロモクリプチン、プラミペキソール、ロピニロール、ペルゴリド、およびセレギリンが挙げられ;トリヘキシフェニジル、ベンズトロピンメシル酸塩、プロシクリジン、ビペリデン、アンデトプロパジンなどの抗コリン作動薬;ジフェンヒドラミンおよびドルフェナドリンおよびアマンタジンなどの抗ヒスタミン薬;抗アルツハイマー病薬の具体例としては、ドネペジル、リバスチグミン、ガランタミン、タクリンが挙げられ;抗生物質の具体例としては、ミノサイクリン、リファンピン、エリスロマイシン、ナフシリン、セファゾリン、イミペネム、アズトレオナム、ゲンタミシン、スルファメトキサゾール、バンコマイシン、シプロフロキサシン、トリメトプリム、メトロニダゾール、クリンダマイシン、テルコプラニン、ムピロシン、アジスロマイシン、クラリスロマイシン、オフロキサシン、ロメフロキサシン、ノルフロキサシン、ナリジクス酸、スパルフロキサシン、ペフロキサシン、アミフロキサシン、エノキサシン、フレロキサシン、テルナフロキサシン、トスフロキサシン、クリナフロキサシン、スルバクタム、クラブラン酸、アンホテリシンB、フルコナゾール、イトラコナゾール、ケトコナゾール、ナイスタチンが挙げられ;抗うつ薬の具体例としては、イソカルボキサジド;フェネルジン;トラニルシプロミンが挙げられ;抗ウィルス薬の具体例としては、アジドブジン(AZT)、ジダノシン(ジデオキシイノシン、ddI)、d4T、ザルシタビン(ジデオキシシトシン、ddC)、ネビラピン、ラミブジン(エピビル、3TC)、サキナビル(インビラーゼ)、リトナビル(ノルビル)、インジナビル(クリキシバン)、デラビルジン(レスクリプトール)が挙げられ;グリコーゲンホスホリラーゼ阻害剤の具体例としては、[R−(R*S*)]−5−クロロ−N−[2−ヒドロキシ−3−{メトキシメチルアミノ}−3−オキソ−1−(フェニルメチル)プロピル−1H−インドール−2−カルボキサミドおよび5−クロロ−1H−インドール−2−カルボン酸[(1S)−ベンジル−(2R)−ヒドロキシ−3−((3R,4S)−ジヒドロキシ−ピロリジン−1−イル−)−3−オキシプロピル]アミドが挙げられ;コレステロールエステルトランスファータンパク質阻害剤の具体例としては、[2R,4S]4−[(3,5−ビス−トリフルオロメチル−ベンジル)−メトキシカルボニル−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸エチルエステル、[2R,4S]4−[アセチル−(3,5−ビス−トリフルオロメチル−ベンジル)−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸イソプロピルエステル、[2R,4S]4−[3,5−ビス−トリフルオロメチル−ベンジル]−メトキシカルボニル−アミノ]−2−エチル−6−トリフルオロメチル−3,4−ジヒドロ−2H−キノリン−1−カルボン酸イソプロピルエステルが挙げられ;CNS刺激薬の具体例としては、カフェインおよびメチルフェニデートが挙げられ;ドーパミン受容体アゴニストの具体例としては、カベルゴリンおよびプラミペキソールが挙げられ;制吐薬の具体例としては、ドラセトロン、グラニセトロン、オンダンセトロン、トロピセトロン、パロノセトロン、ドンペリドン、ドロペリドール、ジメンヒドリネート、ハロペリドール、クロルプロメジン、プロメタジン、プロクロルペリジン、メトクロプラミド、およびアリザプリドが挙げられ;胃腸薬の具体例としては、ロペラミドおよびシサプリドが挙げられ;精神治療薬の具体例としては、クロルプロマジン、チオリダジン、プロクロルペリジン、ハロペリドール、アルプラゾラム、アミトリプチリン、ブプロピオン、ブスピロン、クロルジアゼポキシド、シタロプラム、クロザピン、ジアゼパム、フルオキセチン、フルフェナジン、フルボキサミン、ヒドロキシジン、ロレザパム、ロキサピン、ミルタゼピン、モリンドン、ネファゾドン、ノルトリプチリン、オランゼピン、パロキセチン、フェネルジン、ケチアピン、リスペリドン、セルトラリン、チオチキセン、トラニルシプロミン、トラゾドン、ベンラファキシン、およびジプラシドンが挙げられ;オピオイドアゴニストの具体例としては、ヒドロモルホン、フェンタニル、メタドン、モルフィン、オキシコドン、およびオキシモルホンが挙げられ;オピオイドアンタゴニストの具体例としては、ナルトレキソンが挙げられ;抗てんかん薬の具体例としては、ナトリウムバルプロエート、ニトラゼパム、フェニトインが挙げられ;ヒスタミンH2アンタゴニストの具体例としては、ファモチジン、ニザチジン、シメチジン、ラニチジンが挙げられ;抗喘息薬の具体例としては、アルブテロール、モンテルカストナトリウム塩が挙げられ;平滑筋弛緩薬の具体例としては、ニコランジル、イロペリドン、およびクロナゼパムが挙げられ;および骨格筋弛緩薬の具体例としては、ジアゼパム、ロラゼパム、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、ダントロレン、メタキサ
ロン、オルフェナドリン、パンクロニウム、チザニジン、ジシクロミン、クロニジン、およびガバペンチンが挙げられる。命名された薬物の各々は、当然のことながら薬物の中性形態、ならびに薬学的に許容できるそれらの塩類、溶媒和物、エステル類、およびプロドラッグを含む。
【0029】
上記で考察したとおり、いくつかの薬剤の溶解度はpH依存性であり、有機酸の添加によって増大させることができる。しかし、下記の表1および表2ならびに図2で示されるように、有機酸の添加によって溶解度がわずかしか影響をうけない他の薬剤もある。
【0030】
表1に、有機酸緩衝液中の弱塩基性薬剤の溶解度増大が掲げられている(図2も参照)。3つの異なる群を識別することができる。塩酸オンダンセトロンにより代表されるA群の薬剤は、微量のフマル酸を含む緩衝液中で弱塩基性活性剤の劇的な溶解度増大を示す。例えば、わずか0.05mg/mLのフマル酸を含有する緩衝液中で約26mg/mLのオンダンセトロンの溶解度は、緩衝液中のフマル酸の濃度を5mg/mLまで増加させたときも変化しない。ジピリダモール、カルベジロール、およびイロペリドンに代表されるB群では、酸の濃度が増加するにつれて弱塩基性薬剤の溶解度は増大する。クロナゼパムに代表されるC群では、有機酸はきわめて限られた影響しか及ぼさない。すなわち、溶解度の増大量は一般に、3倍未満である。例えば、クロナゼパムの溶解度は、高濃度と低濃度のフマル酸をそれぞれ含有するpH2.3と6.8の緩衝液中で約11.6μg/mLと6.9μg/mLである。
【0031】
【表1】
【表2】
【0032】
【表3】
【0033】
本発明の医薬組成物の一実施形態において、活性医薬成分はニフェジピンである。別の実施形態において、活性医薬成分はレルカニジピンである。しかし、当然のことながら、本発明の範囲は特定の活性医薬成分に限定されない。
【0034】
本発明の医薬組成物において有用である好適な溶解度増大性ポリマーとしては、限定はしないが、ポリビニルピロリドン(PVPまたはポビドン)、酢酸ビニル/ビニルピロリドンのコポリマー(例えば、Kollidon VA 64)、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシメチルセルロース(ヒプロメロース)、ヒドロキシプロピルメチルセルロースアセテートスクシネート(HPMCAS)、ポリエチレンオキシド、ポリエチレングリコール、およびシクロデキストリンが挙げられる。
【0035】
溶解度増大性ポリマーのタイプおよび量は、活性医薬成分と溶解度増大性ポリマーとの組合せが、本明細書に定義された固体分散物を形成するように選択される。固溶体/固体分散物の調製に有用な溶解度増大性ポリマーのいくつかは、従来、結合剤として用いられている。しかし、活性医薬成分の固体分散物を提供するために、溶解度増大性ポリマーと活性医薬成分との比率は、従来の医薬製剤中のポリマー結合剤と活性医薬成分との比率よりも、一般にかなり高い(従来の医薬製剤において、ポリマー結合剤と活性医薬成分との比率は、一般に1/9未満、例えば、約1/50から約1/20である)。一実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、9/1から1/6(重量)の範囲である。別の実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、約3/1から約1/3(重量)の範囲である。さらに別の実施形態において、固体分散物中の溶解度増大性ポリマーと活性医薬成分との比率は、約2/1から約1/2(重量)の範囲、または約1/1である。
【0036】
固体分散物は、粒子の形態(例えば、顆粒、ペレット、ビーズなど)であり得るか、あるいは、不活性コア上に層状化できる。例えば、活性医薬成分および溶解度増大性ポリマーを、薬学的に許容できる溶媒(または溶媒混合物)中に溶解し、不活性コア上にコーティングすることができる。該溶媒を除去すると、固体分散物が、不活性コア上のコーティングとして形成される。不活性コアとして、薬学的に許容できる任意の不活性材料、例えば、糖球、またはビーズ(例えば、Celphere(登録商標))、セルロース球体、二酸化ケイ素球体などを、好適な粒径分布(例えば、カプセル製剤への導入のためのコーティングビーズの製造には、約20〜25メッシュから35〜40メッシュの糖球であり、ODT製剤への導入のためのコーティングビーズの製造には、約50〜100メッシュの範囲の狭い粒径分布を有する糖球またはセルロース球体で使用することができる。治療上有効な量の活性医薬成分を提供するために、固体分散層の厚さならびに活性医薬成分と溶解度増大性ポリマーとの相対量を調整することができる。例えば、活性医薬成分の固体分散物によって層状化された不活性コアは、2重量%から約50重量%の活性医薬成分(薬剤コーティング不活性コアの総重量に対して)を含有することができる。
【0037】
本発明の医薬組成物の固体分散物は、水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティング、例えば、全ての目的に対して本明細書に参照として取り入れる米国特許第6,627,223号に記載されたTPRコーティングによってコーティングされる。TPRコーティングは、活性医薬成分の放出を調節し、患者における血漿中の活性医薬成分治療有効レベルを、例えば12〜24時間提供する。いくつかの実施形態において、TPRコーティングは、短い遅延時間の間(例えば、約4時間まで)、活性医薬成分の放出を遅延させることができる。また、TPRコーティングは、長時間にわたって、例えば、約12時間まで、約18時間まで、または約24時間まで、該薬剤の持続的な治療レベルを提供することができる。
【0038】
好適な水不溶性ポリマーとしては、セルロース誘導体(例えばエチルセルロース)、酢酸ポリビニル(BASFからのKollicoat SR30D)、エチルアクリレートとメチルメタクリレートに基づいた中性コポリマー、Eudragit NE、RSまたはRS30D、RLまたはRL30Dなどの四級アンモニウム基を有するアクリル酸エステルとメタクリル酸エステルのコポリマーが挙げられる。
【0039】
腸溶性ポリマーは、胃で見られる低pHレベルでは不溶性であるが、腸管で見られる高pHレベルでは比較的可溶性である。好適な腸溶性ポリマーとしては、酸置換セルロースエステル(例えば、セルロースアセテートフタレート、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート)、ポリビニルアセテートフタレート、pH感受性メタクリル酸−メタメタクリレートコポリマーおよびシェラックが挙げられる。市販入手できる腸溶性ポリマーは、Rhom Pharma製造の商標名「Eudragit」(例えば、Eudragit L100、S100、L30D)、Eastman Chemical社からのCellacefate(セルロースアセテートフタレート)、FMC社からのAquateric(セルロースアセテートフタレート水性分散物)、Shin Etsu K.K.からのAquat(ヒドロキシプロピルメチルセルロースアセテートスクシネート水性分散物)が販売されている。
【0040】
TPRコーティング中の水不溶性ポリマーと腸溶性ポリマーとの比率は、約1/9から約9/1(重量)で変わり得る。一実施形態において、水不溶性ポリマーと腸溶性ポリマーとの比率は、約1/4から約4/1、または約1/3から約3/1(重量)の範囲であり得る。腸溶性コーティングの全重量は、TPRビーズの全重量の約5%〜50%の範囲であり得る。一実施形態において、TPRビーズ上のTPRコーティングの全重量は、TPRビーズの全重量に基づいて、約10重量%から約25重量%の範囲である。
【0041】
TPRコーティングの形成に用いられる腸溶性ポリマーおよび水不溶性ポリマーは、可塑化することができる。TPRコーティング層を可塑化するために使用できる好適な可塑化剤の代表例としては、トリアセチン、トリブチルシトレート、トリエチルシトレート、アセチルトリ−n−ブチルシトレート、ジエチルフタレート、ヒマシ油、ジブチルセバケート、アセチル化モノグリセライド類などまたはそれらの混合物が挙げられる。可塑化剤は、存在する場合、TPRコーティングの全重量の約3%から30%を含み得る。一実施形態において、可塑化剤は、TPRコーティングの全重量の約10%から25%を含む。可塑化剤のタイプおよび量は、TPR層の水不溶性ポリマーおよび腸溶性ポリマーの性質ならびにコーティング系の性質(例えば、水性ベースか溶媒ベースか、溶液ベースか分散物ベースか、およびコーティング系の全固体含量)に依存する。
【0042】
固体分散物(少なくとも1種の活性医薬成分および少なくとも1種の溶解度増大性ポリマーを含む)およびTPRコーティングに加えて、本発明の医薬組成物は、追加の薬学的に許容できる成分または賦形剤をさらに含み得る。本発明の組成物または剤形に使用するための好適な賦形剤の例としては、増量剤、希釈剤、滑剤、崩壊剤、結合剤、潤滑剤などが挙げられる。他の薬学的に許容できる賦形剤としては、酸性化剤、アルカリ化剤、保存剤、抗酸化剤、緩衝剤、キレート化剤、着色剤、錯体化材料、乳化剤および/または可溶化剤、風味剤および香料、湿潤剤、甘味剤、浸潤剤などが挙げられる。
【0043】
好適な増量剤、希釈剤および/または結合剤の例としては、ラクトース(例えば、スプレー乾燥ラクトース、α−ラクトース、β−ラクトース、Tabletose(登録商標)、種々の等級のPharmatose(登録商標)、Microtose(登録商標)、またはFast−Floc(登録商標))、微結晶セルロース(種々の等級のAvicel(登録商標)、Elcema(登録商標)、Vivacel(登録商標)、Ming Tai(登録商標)またはSolka−Floc(登録商標))、ヒドロキシプロピルセルロース、L−ヒドロキシプロピルセルロース(低置換)、ヒドロキシプロピルメチルセルロース(HPMC)(例えば、Methocel EおよびMetolose 65 SHの4,000cps等級、Methocel FおよびMetolose 60 SHの4,000cps等級、MethocelKの4,000、15,000および100,000cps等級;および Metolose 90 SHの4,000、15,000、39,000および100,000等級などの、Shin−Etsu社のMethocel E、FおよびK、Metolose SH)、メチルセルロースポリマー(例えば、Methocel A、Methocel A4C、Methocel A15C、Methocel A4Mなど)、ヒドロキシエチルセルロース、カルボキシメチルセルロースナトリウム、カルボキシメチレン、カルボキシメチルヒドロキシエチルセルロースおよび他のセルロース誘導体、スクロース、アガロース、ソルビトール、マンニトール、デキストリン類、マルトデキストリン類、澱粉類または改変澱粉類(ジャガイモ澱粉、トウモロコシ澱粉および米澱粉など)、リン酸カルシウム(例えば、塩基性リン酸カルシウム、リン酸水素カルシウム、水和リン酸二カルシウム)、硫酸カルシウム、炭酸カルシウム、アルギン酸ナトリウム、コラーゲンなどが挙げられる。
【0044】
希釈剤の具体的な例としては、例えば、炭酸カルシウム、リン酸水素カルシウム、第三リン酸カルシウム、硫酸カルシウム、微結晶セルロース、粉末セルロース、デキストラン、デキストリン、デキストロース、フルクトース、カオリン、ラクトース、マンニトール、ソルビトール、澱粉、アルファ化澱粉、スクロース、糖などが挙げられる。
【0045】
崩壊剤の具体的な例としては、例えば、アルギン酸またはアルギネート、微結晶セルロース、低置換ヒドロキシプロピルセルロースおよび他のセルロース誘導体、クロスカルメロースナトリウム、クロスポピドン、ポラクリリンカリウム、ナトリウム澱粉グリコレート、澱粉、アルファ化澱粉、カルボキシメチル澱粉(例えば、Primogel(登録商標)およびExplotab(登録商標))などが挙げられる。結合剤の具体的な例としては、例えば、アラビアゴム、アルギン酸、寒天、カラゲナンカルシウム、カルボキシメチルセルロースナトリウム、微結晶セルロース、デキストリン、エチルセルロース、ゼラチン、液体グルコース、ガーゴム、ヒドロキシプロピルメチルセルロース、メチルセルロース、ペクチン、PEG、ポリエチレンオキシド類、ポビドン、アルファ化澱粉などが挙げられる。
【0046】
滑剤および潤滑剤の具体的な例としては、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウムまたは他の金属ステアリン酸塩類、タルク、ワックス類およびグリセリド類、軽鉱物油、PEG、ベヘン酸グリセリン、コロイドシリカ、水素化植物油、トウモロコシ澱粉、フマル酸ステアリルナトリウム、ポリエチレングリコール類、硫酸エステル塩、安息香酸ナトリウム、酢酸ナトリウムなどが挙げられる。
【0047】
他の賦形剤としては、例えば、風味剤、着色剤、味覚マスキング剤、pH調整剤、緩衝剤、保存剤、安定化剤、抗酸化剤、湿潤剤、湿度調整剤、表面活性剤、懸濁化剤、吸収増強剤、放出制御剤などが挙げられる。
【0048】
非晶質固溶体/固体分散物の長期化学的安定性を改善するために用いられる抗酸化剤としては、例えば、アスコルビン酸、アスコルビルパルミテート、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエン、次亜リン酸、モノチオグリセロール、メタ重亜硫酸カリウム、没食子酸プロピル、ホルムアルデヒドスルホキシル酸ナトリウム、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、二酸化硫黄、トコフェロール、酢酸トコフェロール、トコフェロールヘミスクシネート、TPGSまたは他のトコフェロール誘導体などが挙げられる。
【0049】
また、本発明の医薬組成物は、薬学的に許容できる有機酸をさらに含むことができる。薬学的に許容できる有機酸は、活性医薬成分の放出プロファイル(例えば、放出の速度および程度)をさらに改善または調節することができる。本発明の組成物に有用な薬学的に許容できる好適な有機酸としては、限定はしないが、クエン酸、フマル酸、アスパラギン酸、酒石酸およびコハク酸が挙げられる。いくつかの実施形態において、活性医薬成分と溶解度増大性ポリマーの固体分散物は、該固体分散物の約10重量%〜90重量%の範囲の量で、少なくとも1種の薬学的に許容できる有機酸を含む。他の実施形態において、有機酸の量は、固体分散物の25重量%〜75重量%の範囲である。
【0050】
本発明の組成物は、1つ以上の追加コーティング層(例えば、保護層またはシーラント層(sealant layers)、圧縮性コーティング、腸溶層、味覚マスキング層など)も含み得る。例えば、追加コーティング層は、例えば、Opadry ClearまたはPharmacoat 603(ヒドロキシプロピルメチルセルロースコーティング組成物)、ヒドロキシプロピルセルロース、またはエチルセルロースを含む1つ以上の保護コーティングまたはシーラントコーティングを含み得る。保護コーティングまたはシーラントコーティングは、固体分散物とTPRコーティングとの間に、TPRコーティング上に適用することができ、あるいは、例えば、固体分散物とTPRコーティングとの間ならびにTPRコーティング上に複数の保護をシーラントコーティングとした。追加のコーティング層は、圧縮性コーティング、例えば、固体分散物を含むIRビーズ、味覚マスキングIRビーズまたはTPRビーズ上に沈着させた高可塑化したエチルセルロースまたはヒドロキシプロピルセルロースの層も含み得る。
【0051】
本発明の医薬組成物の他の実施形態は、本明細書に記載された1種以上の腸溶性ポリマーを含む1つ以上の腸溶層を含み得る。任意選択の腸溶層を、固体分散物とTPRコーティングとの間に沈着させ、および/またはTPRコーティング上に沈着させることができる。
【0052】
本発明の医薬組成物は、所望の操作性および薬剤放出特性を提供する、保護層またはシーラント層、圧縮性コーティング、および腸溶層の任意の組合せを含み得る。
【0053】
本発明の医薬組成物は、種々の経口剤形、例えば、カプセル剤(ゼラチンカプセル剤またはHPMCカプセル剤)、錠剤、または口腔内崩壊錠(ODT)に製剤化することができる。錠剤は、そのままで嚥下され、胃に入ると急速に分散するように意図されているが、ODTは、口腔内で唾液に接触すると急速に崩壊して、容易に嚥下される粒子の滑らかな懸濁液を形成する点で、錠剤とODT剤形とは異なる。
【0054】
いくつかの実施形態において、本発明の剤形は、TPRビーズのみを含む。他の実施形態において、本発明の剤形は、即時放出(IR)ビーズとTPRビーズ(すなわち、本明細書に記載されたような)との混合物を含み得る。IRビーズは、溶解度増大性ポリマー中に活性医薬成分の固体分散物を含み、本質的に直ちに活性医薬成分を放出する(例えば、投与の約60分以内に、薬剤の≧75%を放出)。IRビーズは、本明細書に記載された不活性コア上に「層状化された」溶解度増大性ポリマー中の固体分散物の粒子、または活性医薬成分の固体分散物を含み得る。IRビーズは、1つ以上の保護層またはシーラント層も任意に含み得る。次いでTPRコーティングを加えることにより、IRビーズをTPRビーズに変換することができる。
【0055】
本発明の剤形がIRビーズとTPRビーズとの混合物を含む場合、IRビーズは、コーティングなしでもよく、任意選択によりシーラントコーティングまたは保護コーティングによってコーティングされていてもよく、および/または任意選択により、味覚マスキング層によってコーティングされていてもよい。味覚マスキング層は、例えば、各々その全体が本明細書に参照として取り入れる米国特許出願第11/213,266号、米国特許出願第11/248,596号および米国特許出願第11/256,653号に記載された味覚マスキング組成物のいずれかを含み得る。具体的には、好適な味覚マスキング層は、1種以上の造孔剤(pore former)と組み合わせた1種以上の薬学的に許容できる水不溶性ポリマーを含む。味覚マスキング層のための薬学的に許容できる好適な水不溶性ポリマーの非限定例としては、例えば、エチルセルロース、セルロースアセテート、セルロースアセテートブチレート、ポリビニルアセテート、およびメタクリレートポリマー(例えば、Eudragit RL、RSおよびNEおよび30D)が挙げられる。好適な造孔剤の非限定例としては、塩化ナトリウム、炭酸カルシウム、リン酸カルシウム、サッカリンカルシウム、コハク酸カルシウム、酒石酸カルシウム、酢酸鉄、水酸化第二鉄、リン酸第二鉄、炭酸マグネシウム、クエン酸マグネシウム、水酸化マグネシウム、リン酸マグネシウム、ポリビニルピロリドン、クロスポビドン、Eudragit E100、Eudragit EPO、およびそれらの混合物が挙げられる。味覚マスキング層中の水不溶性ポリマーと造孔剤との比率は、約95/5から約50/50の範囲、いくつかの実施形態においては、約85/15から約65/35の範囲である。IRビーズに適用される味覚マスキング層の量は、コーティングされたIRビーズの総重量の約5%から約50%の範囲、いくつかの実施形態においては、コーティングされたIRビーズの総重量の約10%から約50%の範囲であり得る。
【0056】
本発明の剤形がIRビーズとTPRビーズとの混合物を含む場合、IRビーズとTPRビーズとの比率は、約1/9から約5/5の範囲、いくつかの実施形態においては、約1/4から約1/1の範囲(重量で)である。
【0057】
本発明の医薬組成物がODT剤形に製剤化される場合、該組成物は崩壊剤をさらに含む。該崩壊剤は、少なくとも1種の糖アルコールおよび/または糖類と組み合わせた少なくとも1種の崩壊剤を含む急速分散微粒剤の形態であり得る。好適な崩壊剤の非限定例としては、クロスポビドン(架橋ポリビニルピロリドン)、澱粉、セルロース、澱粉グリコール酸ナトリウム、およびカルボキシメチルセルロースナトリウムが挙げられる。糖アルコールの非限定例としては、アラビトール、エリスリトール、ラクチトール、マルチトール、マンニトール、ソルビトール、およびキシリトールが挙げられる。好適な糖類の非限定例としては、ラクトース、スクロース、およびマルトースが挙げられる。
【0058】
急速分散微粒剤中の崩壊剤と糖アルコールおよび/または糖類との比率は、約1/99から約10/90の範囲、いくつかの実施形態においては、約5/95の範囲(重量で)である。
【0059】
ODT剤形中のコーティング薬剤含有ビーズ(すなわち、固溶体を含むコーティングビーズ)と急速分散微粒剤との比率は、約1/9から1/1と変動し、いくつかの実施形態においては、約1:4から約1:2と変動する。
【0060】
ODT剤形は、患者の口腔内で急速に崩壊するため、ODTの感覚刺激特性は重要な考慮事項である。例えば、ODTは、良好な「口あたり(mouthfeel)」および味覚特性を提供するように製剤化する必要がある。「口あたり」とは、口腔内での製品の感じである。ざらざらしていない「口あたり」を得るために、TPRビーズ、急速分散微粒剤、および任意選択のIRビーズは、400μm以下、いくつかの実施形態においては、300μm以下、さらに他の実施形態においては、200μm以下の平均粒径を有する必要がある。一実施形態において、急速分散微粒剤を含む一次粒子(すなわち、急速分散微粒剤を形成するために凝集している崩壊剤および糖アルコールおよび/または糖類の粒子)は、30μm以下、他の実施形態においては、25μm以下、さらに他の実施形態においては、20μm以下の平均粒径を有する。
【0061】
一実施形態において、本発明の組成物を含むODT剤形は、本明細書に記載されたTPRビーズおよび急速分散微粒剤を含む。ODT剤形はさらに追加の賦形剤、例えば、圧縮補助剤(例えば微結晶セルロース)および/または追加の崩壊剤(急速分散微粒剤の崩壊剤と同じでもよいし、異なっていてもよい)を含み得る。ODT剤形は、潤滑剤(例えばステアリン酸マグネシウム)を含んでもよいし、外部潤滑ダイシステムにおいて圧縮される場合は、潤滑剤を含まなくてもよい。一実施形態において、本発明のODT剤形は、口腔の唾液と接触して、約60秒のうちに崩壊し、良好な「口あたり」を有する嚥下し易い懸濁液を形成する。別の実施形態において、本発明のODT剤形は、口腔内で唾液と接触して約30秒以内で崩壊し、良好な「口あたり」を有する嚥下し易い懸濁液を形成する。
【0062】
一実施形態において、剤形(例えば、錠剤、ODT、またはカプセル剤)のTPRビーズは、薬剤と溶解度増大性ポリマーの固体分散物でコーティングし、次いで、TPR層でコーティングし、任意選択により1つ以上のシーラント層または腸溶層でコーティングした不活性コアを含み得る。
【0063】
剤形(例えば、錠剤、ODT、またはカプセル剤)のIRビーズは、存在する場合、薬剤および溶解度増大性ポリマーの固体分散物でコーティングし、任意選択により、本明細書に記載されたシーラント層および/または味覚マスキング層でコーティングした不活性コアを含み得る。このように高いビーズは、TPRビーズ調製のための「中間体」として役立つことができ、TPR層でコーティングされると、IRビーズはTPRビーズへと変換される。
【0064】
あるいは、固体分散物の粒子を形成し(例えば、スプレー乾燥により)、固体分散物の「バルク」形態またはより大型の粒子形態を粉砕するか、または固体分散物と共に、1種以上の薬学的に許容できる賦形剤(例えば、増量剤、結合剤、崩壊剤など)を顆粒化することによって、IRビーズを調製することができ、次いでこれを任意選択により、押出し加工し、球状化することができる。次いで、このようなIR粒子/ビーズ/ペレットをTPR層でコーティングしてTPRビーズに変換することができる。
【0065】
本発明の剤形は、1つ以上の異なるタイプのTPRビーズ(例えば、異なるTPR層を有するか、またはシーラント層および/または腸溶層の異なる組合せを有するTPRビーズ)を含み得る。例えば、異なるTPR層を有するTPRビーズは、異なる遅延時間特性および/または異なる放出速度特性を示すことができ、それによって、全体的に異なる薬剤放出特性を有する剤形を提供することができる。異なるタイプのTPRビーズを含む剤形は、任意選択により、いくつかの即時放出特性を提供するためのIRビーズも含むことができる。例えば、一実施形態において、1日1回の剤形は、即時放出を可能にするIRビーズ(約7時間の排出半減期を有する活性医薬成分を含む)と約12〜20時間にわたる薬剤の遅延の持続放出プロファイルを提供し、かつ、約18〜24時間にわたって治療上有効な血漿中濃度を維持する約4時間までの遅延時間を有する第2のTPRビーズ集団との混合物を含む。
【0066】
溶解度増大性ポリマー中の活性医薬成分の固溶体または固体分散物は、活性医薬成分および溶解度増大性ポリマーを、薬学的に許容できる溶媒または溶媒混合物中に溶解させることによって調製することができる。次いで、活性医薬成分と溶解度増大性ポリマーの溶液を、溶解度増大性ポリマー中に活性医薬成分の固溶体形成を促進する条件下で乾燥する。上記で考察したとおり、分子分散した固体分散物の形成は、活性医薬成分に対して溶解度増大性ポリマーが比較的高レベルであると有利である。固体分散物はまた、活性医薬成分と溶解度増大性ポリマーの溶液から溶媒を、例えば、スプレー乾燥により、急速に除去することによって、または活性医薬成分と溶解度増大性ポリマーの溶液を、例えば、流動床コーティング法を用いて、不活性コア上にコーティングする(薬剤層状化ビーズを形成する)ことによって、形成することができる。あるいは、固体分散物はまた、例えば、二軸スクリュー押出し機における混ぜ合わせなど、ポリマー押出し法によって、溶解度増大性ポリマーの溶融物中に活性医薬成分を溶解させることによって調製することができる。好適な粒径(例えば、ODT剤形では、400μm未満の粒径)を得ることが必要な場合、固体分散物の粒子を、任意選択により好適な賦形剤の存在下、粉砕する(粒径を減少させるため)か、または粗砕(例えば、回転粗砕、または粗砕後に押出し球状化)することができる。固体分散物は、例えば、適切な大きさの円形斜め穿孔を用いて、任意選択により圧縮補助剤、潤滑剤などの賦形剤と共に、固体分散物の圧縮粒子によって形成された1〜2mmの直径の「ミニ錠剤」へと形成することもできる。
【0067】
一実施形態において、該固体分散物は、高せん断粗砕機、またはGlatt GPCG粗砕機などの流動床粗砕機において、溶解度増大性ポリマー、弱塩基性薬剤および任意選択により他の薬学的に許容できる賦形剤(例えば、結合剤、希釈剤、増量剤)を粗砕し、凝集体を形成することによって調製される。高せん断粗砕機からの湿潤塊は、押出し加工し球状化して、球状粒子(ペレット)を製造することもできる。
【0068】
上記で考察したような溶媒処理法によって該固体分散物が調製される場合、薬学的に許容できる溶媒は、単一の溶媒であってもよいし、溶媒の混合物であってもよい。好適な溶媒の非限定例としては、水、アセトンなどのケトン類、エタノールなどのアルコール類、およびそれらの混合物(例えば、アセトン水、95%のエタノールなど)が挙げられる。
【0069】
固体分散物粒子(例えば、薬剤/ポリマーのスプレー乾燥固体分散物、薬剤層状化ビーズ、顆粒化固体分散物、ミニ錠剤など)を調製したら、それらを保護シーラントコート(例えば、Pharmacoat(商標)603またはOpadry(登録商標)Clear)によって任意選択によりコーティングし得る。
【0070】
上記に記載の通り調製された固体分散物粒子は、IR(即時放出)ビーズまたはIR粒子と称される。このようなビーズまたは粒子は、この形態で投与されると、活性医薬成分を実質的に直ちに放出すると考えられるからである。上記に記載の通り調製されたIRビーズまたはIR粒子は次に、薬学的に許容できる溶媒中に溶解させた水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティング溶液によってコーティングされる。TPRコーティングの適用には、任意の好適なコーティング法、例えば流動床コーティング法などを用いることができる。
【0071】
いくつかの実施形態において、IRビーズまたはIR粒子に、TPRコーティングに加えて複数のコーティングを適用することが望ましい。例えば、いくつかの実施形態においては、IRビーズを先ず腸溶コーティング(例えば、薬学的に許容できる溶媒中に溶解させた、本明細書に記載された少なくとも1種の腸溶性ポリマーを含むもの)によってコーティングし、乾燥してコーティング溶媒を除去し、次いで、上記のTPRコーティングによってコーティングする。他の実施形態において、IRビーズは、腸溶性ポリマーコーティング、TPRコーティング、次いで第2の腸溶性ポリマーコーティングによってコーティングする。さらに他の実施形態において、IRビーズは第1のTPRコーティング、腸溶性ポリマーコーティング、次いで第2のTPRコーティングによってコーティングし、第1と第2のTPRコーティングは独立して同じであるか、または異なる。さらに他の実施形態において、TPRコーティング層および/または腸溶性ポリマーコーティング層を適用する前に、シーラント層(本明細書に記載された)をIRビーズ上にコーティングする。さらに他の実施形態において、TPRコーティング層および/または腸溶性ポリマーコーティング層を適用した後に、シーラント層を適用することができる。
【0072】
TPRビーズとIRビーズの混合物を含有する製薬剤形において、IRビーズを味覚マスキング層でコーティングすることができる。例えば、流動床コーティングまたはコアセルベーションなどの任意の好適なコーティング技法を用いて、本明細書に記載されたIRビーズのいずれかを、薬学的に許容できる溶媒、水不溶性ポリマー、および任意選択により造孔剤を含む溶液によってコーティングすることができる。
【0073】
次いで、例えば、従来の方法を用いて、TPRビーズを錠剤へと圧縮し、TPRビーズおよび崩壊剤(例えば急速分散微粒子)をODTへと圧縮し、あるいはTPRビーズをカプセルに充填することによって、製薬剤形をTPRビーズから調製することができる。これらの製薬剤形は、本明細書に記載の通り、任意選択により、追加の賦形剤ならびにIRビーズを含有することができる。一実施形態において、本発明の組成物、および任意選択により追加の賦形剤および/またはIRビーズは、外部潤滑化錠剤圧縮を用いて、錠剤へと圧縮される。別の実施形態において、本発明の組成物、急速崩壊微粒剤、任意選択により追加の賦形剤および/またはIRビーズは、ODTへと圧縮される。
【0074】
本発明の組成物を含む製薬剤形は、例えば図7〜図12に示されるように、12〜18時間にわたって、治療上有効なレベルの活性医薬成分を放出する。本発明の組成物の実際の剤形に関する薬剤放出プロファイルは、米国薬局方装置1(バスケット、100rpm)または装置2(パドル、50rpm)などの種々の溶出試験方法ならびに二段階溶出試験法(最初の2時間は、0.1NのHCl(塩酸)700ml中で、その後、200mLのpH改変剤添加により得られるpH6.8、900mL中で試験する)を用いて、インビトロで評価することができる。経時的な薬剤/酸放出は、選択された間隔で得られるサンプルに対してHPLCによって判定される。
【0075】
一実施形態において、本発明の組成物は、二段階溶出媒質(最初の2時間は、0.1NのHCl中で、その後、pH6.8の緩衝液中で試験する)を用いる米国薬局方(USP)溶出試験法によって溶出試験した際、少なくとも約12時間、活性医薬成分の治療上有効な血漿中濃度を提供する。
【0076】
1日1回の血漿中濃度プロファイルを達成するために必要なインビトロ放出プロファイルのタイプを評価するために、ソフトウェアプログラム、WinNonlin(商標)標準型2.1または等価物(例えば、GastroPlus(登録商標))を用いて、1コンパートメント一次モデルを遅延時間推定一次排出動態と適合させ、該薬剤に関する薬物動態パラメーターを用いて、モデリング演習を一般に行なう。次いで、先に確立したモデルをわずかに修正して用いて、一次パラメーターを別のプログラム、Stella 6.01型にインプットする。種々のインビトロ放出プロファイルが作り出され、目的の1日1回の放出プロファイルから、デコンボリューションによって、所望のインビトロ放出(中間、目的および高速)プロファイルが作り出される。
【0077】
以下の非限定例は、1つ以上の薬剤放出「パルス」および所定の遅延開始を示すカプセル剤形を例示する。TPR層の量または厚さを調整し、また、任意選択により追加の層(例えば、腸溶層またはシーラント層)の数およびタイプを調整することによって、所望のプロファイルを提供して、最大の治療有効性を達成し、患者のコンプライアンスを高める(例えば、1日1回の剤形を提供することによって)目的で、該剤形の経口投与の際のインビトロ薬剤放出プロファイルまたは対応するインビボ血漿中濃度プロファイルを設計することができる。本発明の剤形は、従来の剤形の薬剤放出プロファイルに関連した副作用を最小化するレベルに薬剤の血漿中濃度を保持する改善された薬剤放出プロファイルを提供する。
【実施例1】
【0078】
濁度の測定
アセトン(0.5mg/ml)中の塩酸レルカニジピン濃縮液(3mL)を、Kollidon VA64、Methocel E5(ヒプロメロース)、ポリエチレングリコール(PEG6000)、シクロデキストリンまたはKollidon 14 PF(ポリビニルピロリドン)を含有する200mLの緩衝液(pH6.0)に、ポリマーに対して1:2の重量比で加えた。該薬剤溶液は、改善された安定性を示し、したがって、レルカニジピンHClの共役塩基の結晶化のリスクを大きく減少させたことが、図3Aから明白である。
【0079】
固有溶解速度の測定
塩酸レルカニジピンの2種の異なる多形体ならびに非晶質物質(例えば、非晶質薬剤ならびにMethocel E5およびKollidon VA64と塩酸レルカニジピンとの1:2固溶体)に関して、固有溶解速度を判定した。データは図4に示されている。結晶質多形体は、不良な溶解速度ならびに溶解度を示す一方、固溶体は、有意に高い溶解速度ならびに溶解度を示す。
【0080】
粉末X線回折
1:1および1:2の比率での塩酸レルカニジピンとMethocel E5(ヒプロメロース)とをジクロロメタン−メタノール(1対1、v/v)の溶媒混合物中に溶解し、該溶液を1%(w/w)未満の残留溶媒レベルまで乾燥した。同様に、塩酸レルカニジピンとKollidon VA64の1:1および1:2の共沈殿物を調製した。4つのサンプル全てについて、粉末X線回折パターンを作出した。レルカニジピン−Kollidon VA64固溶体に関するXRDパターンは図5に示されており、レルカニジピンHClとKollidon VA64との1:2比の固溶体は、ほぼ全体的に非晶質であることを実証している。
【実施例2】
【0081】
濁度の測定
アセトン(0.5mg/mL)中ニフェジピンの濃縮溶液(3mL)を、Kollidon VA64、Methocel E5(ヒプロメロース)、ポリエチレングリコール(PEG6000)、シクロデキストリンまたはKollidon 14PF(ポリビニルピロリドン)を含有する200mLの緩衝液(pH6.0)に、1:2のニフェジピン/ポリマーの重量比で加えた。図3Bに示すように、ニフェジピン/ポリマー溶液の透過度を経時的にモニターした。より安定な溶液は、溶液からのニフェジピンの結晶化がより緩やかであるため、経時的に透過度の低下がより緩やかである。Methocel E5、Kollidon VA64およびKollidon 14PFは、より高い安定性を示した。
【0082】
粉末X線回折
ジクロロメタン−メタノール(1対1、v/v)の混合物中に、ニフェジピンおよびMethocelを溶解し、次いで、この溶液を残留溶媒レベル1%(w/w)未満まで乾燥させることにより、1:1および1:2のニフェジピン/Methocel比で、ニフェジピンとMethocel E5(ヒプロメロース)の2種の共沈殿物を調製した。同一の方法を用いて、ニフェジピンとKollidon VA64の1:1および1:2共沈殿物も調製した。4つのサンプル全てを粉末X線回折により分析し、ニフェジピン−Kollidon VA64固溶体のXRDパターンは、図6に示されている。1:1共沈殿物に関するXRDパターンにおける鋭いピークの存在は、結晶形態で存在しているニフェジピンを示している。1:2共沈殿物の幅広い、比較的特色のないXRDパターンは、ニフェジピンがほぼ全体的に非結晶質であり、Kollidon VA64中で固体分散物を形成していることを示している。
【実施例3】
【0083】
3A−ニフェジピンIRビーズ(名目上(nominal)10%のニフェジピン添加)
Kollidon VA64(800g)を、溶解するまで激しく攪拌しながら95%エタノール/アセトン/水の72.5/22.5/5の混合物(4930g/1530g/340g)にゆっくりと加え、次いでニフェジピン(400g)を溶解するまでゆっくりと加えた。7インチ(約17.5cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートB(250μmスクリーン)、1.0mmノズルポート、1.5バールの噴霧空気圧、および3.2mmの内径細管で装備されたGlatt GPCG3に、2584gの25〜30メッシュの糖球を充填した。約40gのタルクをニフェジピン/ポリマー溶液中にホモジナイズして帯電を最小にした。約36〜40℃での生成物温度を維持しながら、15重量%の固体含量のニフェジピン溶液を、8〜17g/分のスプレー速度および約60〜80%(気流速度:約85〜115m3/時間)の排出口フラップで糖球上にスプレーした。得られたニフェジピン層状化ビーズ(バッチサイズ:3724g)を、40℃で約45分間Glattユニット内で乾燥して生成物中の残存溶媒レベルを最小にした。98.5%収率の使用可能なビーズ(600〜1200μm)を得た。
【0084】
2800gのニフェジピン層状化ビーズに、重量2%(すなわち、非コーティングビーズの重量に対するコーティング重量)のOpadry(登録商標)Clearの保護シールコート(8重量%の固体;生成物温度:37〜41℃;スプレー速度:5〜12g/分)のコーティングを提供し、さらに40℃で約45分間Glattユニット内で乾燥して生成物中の残存溶媒/水分を除去した。測定力価は、10%のニフェジピンの目標力価に対して9.81%(ニフェジピン%)であった。
【0085】
3B−ニフェジピンTPRビーズ(TPRコーティング:45/40/10/5の比率でEudragit RL/Eudragit L/TEC/タルク)
上記3Aに記載したようにして調製された、名目上10%の薬剤添加を有するニフェジピンIRビーズ(700g)に、45/55のアセトン/エタノール中、10%固体の固体含量でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5溶液(Ultraturrexホモジナイザーを用いて溶液にタルクを懸濁した)をスプレーすることによりコーティングし、20重量%までのコーティングを提供した(サンプルは、5重量%、10重量%、および15重量%のコーティングで取り出した)。
【0086】
最初攪拌しながら透明溶液を得るために、溶媒混液にEudragit RLポリマーをゆっくりと加えることによってTPRコーティング溶液を調製した。次に、Eudragit Lポリマー、次いで可塑剤(トリエチルシトレートまたは「TEC」)をゆっくりと加えて該溶液中に溶解させた。タルクを別個に溶媒混液中でホモジナイズしてから、溶解した該ポリマー類および可塑剤に加えた。4インチ(約10cm)底のスプレーWursterインサート、20mmの仕切りギャップ、空気分配プレートB(250μmスクリーン)、1.0mmノズルポート、1.5バールの噴霧空気圧、および3.2mmの内径細管、ならびにT165P専用フィルターバッグで装備されたGlatt GPCG1を用いて、TPRコーティング溶液をニフェジピンIRビーズに適用した。TPRコーティング溶液を、4〜11g/分のスプレー速度、約20〜30%(気流速度:約2.0〜2.5m/秒)の排出口フラップおよび約35〜38℃での生成物温度でスプレーした。コーティングされたビーズを、40℃で約45分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体(すなわち、TPRコーティングにより一緒に接着された2つ以上のビーズ)が形成された場合、それらを廃棄するために乾燥ビーズを篩い分けした。約5%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0087】
3C−ニフェジピンIRビーズ(名目上5重量%のニフェジピンの添加)
名目上5重量%の薬剤添加を有するニフェジピンIRビーズを、上記3Aに記載された手法に従って調製した。190gのニフェジピンおよび380gのKollidon VA64を、3154gの25〜30メッシュの糖球上に層状化した。測定力価は、4.81%ニフェジピン(5%ニフェジピンの理論的名目上力価に対して)であると判定した。
【0088】
3D−ニフェジピンTPRビーズ(コーティング:40/45/10/5のEudragit RL/L/TEC/タルク)
上記3Cに記載したようにして調製された、名目上5%のニフェジピン添加を有するニフェジピンIRビーズ(700g)に、5重量%、10重量%、15重量%および20重量%のコーティングレベルで上記3Bに記載された手法に従って、Glatt GPCG1内でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5のTPRコーティング溶液によりコーティングした。約5%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例4】
【0089】
4A−ニフェジピンIRビーズ(60〜80メッシュの糖球)
上記3Aに記載された手法と同様の手法に従って、ニフェジピンIRビーズ(名目上10重量%のニフェジピン添加)をGlatt GPCG3内で60〜80メッシュの糖球上にニフェジピン/Kollidon VA64の1:2溶液をスプレーすることにより調製した。
【0090】
4B−ニフェジピンTPRビーズ(TPRコーティング:35/50/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記4Aに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記3Bに記載された手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの35/50/10/5の溶液をスプレーすることによってコーティングし、40℃で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体が形成された場合は、それらを廃棄するために乾燥ビーズを篩い分けした。5%、10%および15%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0091】
4C−ニフェジピンTPRビーズ(TPRコーティング:40/45/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記4Aに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記3Bに記載された手法と同様の手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの40/45/10/5の溶液をスプレーすることによってコーティングした。
【0092】
4D−ニフェジピンTPRビーズ(TPRコーティング:45/40/10/5のEudragit RL/Eudragit L/TEC/タルク)
上記3Bに記載したようにして調製されたニフェジピンIRビーズ(700g)に、上記2Aに記載された手法と同様の手法に従って、Glatt GPCG1内で20%のコーティング重量でEudragit RL/Eudragit L/TEC/タルクの45/40/10/5の溶液をスプレーすることによってコーティングし、40℃で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。15重量%および20重量%のコーティングを有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0093】
実施例3および4の薬剤放出プロファイル
図7は、実施例4のTPRビーズからのニフェジピン放出に対するTPRコーティング組成物の影響を示している。TPRコーティング中の腸溶性ポリマー含量(Eudragit L)が増すと、ニフェルジピン放出速度が増大する。図8は、実施例3のTPRビーズからのニフェジピン放出に対するニフェルジピン添加の影響を示している。ニフェルジピン添加量が5%から10%に増すと、ニフェジピン放出速度が低下する。図9は、同じTPRコーティング組成物およびコーティング重量で、実施例3Bおよび4DのTPRビーズからの薬剤放出に対する粒径(それぞれ25〜30メッシュまたは600〜700μmおよび60〜80メッシュまたは170〜250μm)の影響を示している。実施例4Dのビーズが小さくなると、ニフェジピン放出が速くなることを示している。
【実施例5】
【0094】
5A−モデル薬物IRビーズ(10%の薬剤添加)
ポビドン(PVP K29/32、128.2g)を、72.5/22.5/5の95%エタノール/アセトン/水に6%の固体で、溶解するまで激しく攪拌しながらゆっくりと加え、次いでラモトリギンの弱塩基性類縁体(128.2g)を、溶解するまでゆっくりと加えた。6インチ(約15cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートD(200メッシュスクリーン)、1.0mmノズルポート、1.0バールの噴霧空気圧、および14mmの単一ヘッド細管で装備されたGlatt GPCG3に、1000gの25〜30メッシュの糖球(Chris Hansen)を充填した。生成物温度を約32.5〜33.5℃で維持しながら、8mL/分のスプレー速度、28〜30%(気流速度:3.6〜4.2m/秒/圧:10.5〜8Pa)の排出口フラップで、糖球を該薬剤溶液によりコーティングした。次に薬剤層状化ビーズを、2%のコーティング重量で、Pharmacoat 603の保護シール−コートによりコーティングし、約10分間Glatt内で乾燥して残存溶媒/水分を除去した。このコーティングされたビーズを、20〜30メッシュスクリーンを通して篩い分けした。
【0095】
5B−モデル薬剤TPRビーズ(TPRコーティング:50/35/15EC−10/HP−55/TEC)
40重量%までのコーティングで、90/10アセトン/水に溶解させた50/35/15EC−10/HP−55/TECの溶液(7400/822.2;7.5%の固体)を、上記4Aに記載したようにして調製されたIRビーズ(1000g)にスプレーすることによってコーティングした(サンプルは、約20%、25%、30%および35%のコーティングレベルで取り出した)。EC−10(エチルセルロース、Dow ChemicalsからのEthocel Premium10cps、333.3g)を、溶解するまで30分間以上連続攪拌しながら90/10のアセトン/水にゆっくりと加えた。次に、HP−55(Shin Etsuからのヒドロキシプロピルメチルセルロース、233.3g)およびTEC(100g)を、溶解するまでEC−10溶液に加えた。6インチ(約15cm)底のスプレー/8インチ(約20cm)カラム高のWursterインサート、20mmの仕切りギャップ、空気分配プレートD(200メッシュスクリーン)、0.8mmノズルポート、1.0バールの噴霧空気圧、および14mmの単一ヘッド細管、PB3%の専用フィルターバッグで装備されたGlatt GPCG3により、TPRコーティング溶液を適用した。生成物温度を約32〜34℃に維持しながら、10〜15mL/分のスプレー速度、約28%(気流速度:3.4〜3.8m/秒/圧:7〜7.5Pa)の排出口フラップで、TPRコーティング溶液をIRビーズにスプレーし、同じ温度で10分間Glatt内で乾燥して過剰の残存溶媒を除去した。二倍体が形成された場合は、それらを廃棄するために乾燥ビーズを篩い分けした。
【0096】
5C−モデル薬剤TPRビーズ(コーティング:35/50/15EC−10/HP−55/TEC)
上記の手法と同様の手法に従って、20重量%、25重量%、30重量%、35重量%および40重量%のコーティング重量において35/50/15EC−10/HP−55/TEC TPRコーティング溶液により、上記5Aに記載したようにして調製されたIRビーズ(1000g)をコーティングした。
【0097】
5D−モデル薬剤TPRビーズ(コーティング:60/25/15EC−10/HP−55/TEC)
上記の手法と同様の手法に従って、5重量%、10重量%、15重量%、および20重量%のコーティング重量において60/25/15EC−10/HP−55/TEC TPRコーティング溶液により、上記4Aに記載したようにして調製されたIRビーズ(1000g)をコーティングした。
【0098】
図10は、実施例5の同じ薬剤添加におけるTPRビーズからの薬剤放出に対するコーティング組成物および/またはコーティングレベルの影響を示している。TPRコーティング中の腸溶性ポリマー含量を25重量%から50重量%に増加させると、TPRビーズからの薬剤放出速度が有意に増大する結果となる。
【実施例6】
【0099】
6A−ニフェジピンIRビーズ(ニフェジピン/VA 64/フマル酸)
上記の手法と同様の手法を用いて、エタノール/アセトン/水に溶解されたニフェジピン/VA 64/フマル酸の1/2/1溶液を、Glatt GPCG3内で25〜30メッシュの糖球および名目上10重量%のニフェジピン添加物上に層状化することによってニフェジピンIRビーズを調製した。
【0100】
6B−ニフェジピンIRビーズ(コーティング:Eudragit RL/L/TEC/タルク)
上記の手法と同様の手法を用いて、30重量%までのコーティングにおいて、35/50/10/5のEudragit RL/Eudragit L/TEC/タルクTPRコーティングにより、上記6Aに記載したようにして調製されたIRビーズ(700g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。15重量%および20重量%のコーティングを有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【0101】
6C−ニフェジピンTPRビーズ(二重コーティング)
流動床コーターにおいて、GPCG1内で上記6Aに記載したようにして調製されたIRビーズ(700g)を、10%のコーティング重量における85/10/5のEudragit L/TEC/タルクを含む内部腸溶性コーティング層によりコーティングした。Eudragit L100を、溶解するまで約90分間、激しく攪拌しながらエタノールにゆっくりと加えた。次に、TEC(トリエチルシトレート)を、溶解するまで該溶液にゆっくりと加え、次いで一定に攪拌しながら懸濁タルクを添加した。次いでこれらの腸溶性コーティングされたビーズを、30%までのコーティング重量で35/50/10/5のEudragit RL/Eudragit L/TEC/タルクのTPR層によりコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。上記の手法および処理条件と同様の手法および処理条件を用いて各層を適用した。15%および20%のTPRコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例7】
【0102】
7A−ニフェジピンIRビーズ(ニフェジピン/VA 64/アスパラギン酸)
上記の手法と同様の手法を用いて、ニフェジピンIRビーズは、72.5/22.5/5のエタノール/アセトン/水中のニフェジピン/VA 64/アスパラギン酸の1/2/1溶液を、Glatt GPCG 3内で25〜30メッシュの糖球上にコーティングすることによって調製し、名目上10重量%のニフェジピン添加物を提供した。アスパラギン酸はコーティング溶液に溶解しなかったことから、それを、Ultraturrexホモジナイザーを用いてコーティング溶媒中にホモジナイズした後、ニフェジピンおよびKollidon VA64の溶液に加え、さらにホモジナイズした。
【0103】
7B−ニフェジピンTPRビーズ(コーティング:35/50/15のRL/L−55/TEC)
上記の手法と同様の手法を用いて、30重量%までのコーティングにおいて、35/50/10/5のEudragit RL/Eudragit L/TEC/タルクにより、上記7Aに記載したようにして調製されたニフェジピンIRビーズ(700g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。15%および20%のコーティング重量を有するTPRビーズは、HPLC法を用いて力価および薬剤放出プロファイルに関して検定した。
【実施例8】
【0104】
8A−レルカニジピンHClのIRビーズ(レルカニジピン/VA 64/酒石酸)
レルカニジピンHCl(93g)をエタノール(4808g)にゆっくりと加え、溶解するまで攪拌した。次にKollidon VA 64(186g)、次いで酒石酸(21g)を溶解するまでゆっくりと加えた。6インチ(約15cm)底のスプレーカラム高のWursterインサート、200mmの仕切りギャップ、空気分配プレートC(50メッシュスクリーン)、0.8mmノズルポート、1.5バールの噴霧空気圧で装備されたGlatt GPCG1に、2100gの30〜35メッシュの糖球(2322g)を充填した。生成物温度を約32〜34℃に維持しながら、糖球を、11g/分のスプレー速度、45〜50%(気流速度:90〜105m3/時間)の排出口フラップで、レルカニジピン/VA 64/酒石酸のコーティング溶液によりスプレーすることによってコーティングした。レルカニジピン層状化ビーズは、2%のコーティング重量で保護シールコートのOpadry Clearによりコーティングし、45℃で約15分間Glattユニット内で乾燥して残存溶媒/水分を除去し、次いで25メッシュスクリーンを通して篩い分けした。
【0105】
8B−レルカニジピンのTPRビーズ(コーティング:EC−10/HP−55/DEPを1:4:1で)
上記の手法と同様の手法を用いて、上記8Aで記載したようにして調製されたレルカニジピンHClのIRビーズ(930g)を、27%のコーティング重量で98/2のアセトン/水中EC−10/HP−55/DEPの1/4/1溶液を用いてGlatt流動粗砕機内のIRビーズをスプレーすることによってコーティングした。得られたTPRコーティング組成物は、16.4%のEC−10、65.6%のHP−55、18%のDEP(ジエチルフタレート)であった。
【0106】
8C−レルカニジピンのTPRビーズ(二重層コーティング)
Eudragit L100を、溶解するまで約90分間、激しく攪拌しながらエタノールにゆっくりと加えた。次に、TEC(トリエチルシトレート)を、溶解するまで該溶液にゆっくりと加え、次いで一定に攪拌しながら懸濁タルクを添加した。上記のとおりに調製された74.1/7.4/18.5のEudragit L100/TEC/タルクの内部腸溶性コーティングを、20%のコーティング重量で上記8Aで記載したようにして調製されたIRビーズ上に適用した。次に、上記の8Bに記載されたものと同様の手法を用いて、得られた腸溶性コーティングIRビーズを、16.4/65.6/18のEC−10/HP−55/DEP TPRコーティング溶液により10%のコーティング重量にコーティングした。
【0107】
8D−レルカニジピンのTPRビーズ(三重層コーティング)
上記の手法と同様の手法を用いて、80/20 HP−55/DEPの内部腸溶性コーティングを、20%のコーティング重量で上記8Aで記載したようにして調製されたIRビーズに適用した。次に、上記の手法と同様の手法に従って、腸溶性コーティングビーズを、16.4/65.6/18のEC−10/HP−55/DEP TPRコーティング溶液により25%のコーティング重量にコーティングした。次に、上記の手法と同様の手法を用いて、これらのビーズを、74.1/7.4/18.5の比率でのEudragit S100/TEC/タルクの外部腸溶性コーティング層により10%のコーティング重量にさらにコーティングした。溶解するまで約90分間激しく攪拌しながらEudragit S100をエタノールにゆっくりと加えることによって、外部腸溶性コーティング層を調製した。次にTEC(トリエチルシトレート)を、溶解するまでEudragit S100にゆっくりと加え、次いで一定に攪拌しながら懸濁したタルクを添加した。実施例8B、8C、および8DのTPRビーズに関する薬物放出プロファイルは、二段階方法論(すなわち、最初は0.1N HClおよび0.3%ツイーン80において2時間、引き続きpH6.8で)を用いて溶出試験を行った。溶出試験結果を図11に示している。
【実施例9】
【0108】
9A−ニフェジピンIRビーズ(ニフェジピン/VA 64/酒石酸)
エタノール/アセトン/水に溶解させたニフェジピン/VA 64/フマル酸の1/2/1溶液を、Glatt GPCG3内で名目上10重量%のニフェジピン添加量で25〜30メッシュの糖球上に層状化することによって、ニフェジピンIRビーズを調製した。
【0109】
9B−ニフェジピンTPRビーズ(コーティング35/50/15RL/L−55/TEC)
上記の手法と同様の手法を用いて、Glatt流動粗砕機内において30%のコーティング重量で、アセトン/水に溶解させたEthocel Premium 10cps(EC−10)、ヒプロメロースフタレート(HP−55)およびジエチルフタレート(DEP)の溶液をスプレーすることによって、上記9Aに記載のように調製したニフェジピンIRビーズ(1000g)をコーティングした(サンプルは、10%、15%、20%および25%のコーティングレベルで取り出した)。スプレー用溶液は、以下の組成を有した:1.23%のEC−10、4.92%のHP−55、1.35%のDEP;溶媒:90.65%のアセトンおよび1.85%の水。
【実施例10】
【0110】
10A−IRビーズ(薬剤添加:16.67%)
ポビドン(PVP K29/32、666.7g)を、溶解するまで激しく攪拌しながら16.6/83.4のエタノール/アセトンにゆっくりと加えた。溶解するまで、イロペリドン(333g)をゆっくりと加えた。次に上記の手法と同様の手法を用いて、Glatt GPCG3内で25〜30メッシュの糖球(1000g)を該薬剤溶液(8.17%の固体)によりコーティングした。薬剤層状化ビーズを、2%のコーティングでPharmacoat603の保護シールコートによりコーティングした。このIRビーズを該ユニット内で約10分間乾燥して残存溶媒/水分を除去し、二倍体が形成された場合は、それらを廃棄するために篩い分けした。
【0111】
10B−TPRビーズ(二重コーティング:HP−55/TEC上に45/40/15のEC−10/HP−55/TEC)
95/5のアセトン/水中、80/20のHP−55/TEC溶液の腸溶性コーティング溶液を8%のコーティング重量でスプレーすることによって、上記10Aに記載したようにして調製されたIRビーズ(1800g)をコーティングした。次に腸溶性コーティングビーズ(850g)を、上記の方法と同様の手法を用いて、Glatt GPCG3内で50%までのコーティング重量で、90/10のアセトン/水混液(7.5%の固体)中、45/40/15のEC−10/HP−55/TEC TPRコーティング溶液によりコーティングし(サンプルは、20%、30%、および40%のコーティングレベルで取り出した)、Glatt内で約50℃で10分間乾燥して過剰の残存溶媒を除去し、二倍体が形成された場合は、それらを廃棄するために篩い分けした。
【0112】
10C−TPRビーズ(二重コーティング:HP−55/TEC上に30/55/15のEC−10/HP−55/TEC)
上記の手法と同様の手法を用いて、上記10Bに記載したようにして調製された腸溶性コーティングビーズ(530g)を、Glatt GPCG3内で50%までのコーティングで、90/10のアセトン/水混液(7.5%の固体)中、30/55/15のEC−10/HP−55/TEC TPRコーティング溶液によりスプレーすることによってコーティングした(サンプルは、20%、30%、および40%のコーティングレベルで取り出した)。2つの異なるTPR組成/レベルでコーティングされたTPRビーズからの代表的な薬剤放出プロファイルを、図12に示している。
【特許請求の範囲】
【請求項1】
TPRビーズを含む医薬組成物であって、前記TPRビーズが:
少なくとも1種の溶解度増大性ポリマー中の少なくとも1種の活性医薬成分の固体分散物;および
水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティングを含み;
前記活性医薬成分が、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む、医薬組成物。
【請求項2】
少なくとも約18時間にわたって活性医薬成分の治療上有効な血漿中濃度を提供する、請求項1に記載の医薬組成物。
【請求項3】
前記TPRコーティング中の水不溶性ポリマー対腸溶性ポリマーの比率が、約9:1から約1:9の範囲である、請求項1に記載の医薬組成物。
【請求項4】
前記TPRコーティングが、約3重量%から約30重量%(TPRコーティングの全重量に対して)の可塑剤をさらに含む、請求項3に記載の製薬剤形。
【請求項5】
前記活性医薬成分と前記溶解度増大性ポリマーの固体分散物が、不活性コア上に堆積されている、請求項1に記載の医薬組成物。
【請求項6】
前記溶解度増大性ポリマーが、ポリビニルピロリドン、酢酸ビニル/ビニルピロリドンのコポリマー、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリエチレンオキシド、ポリエチレングリコール、およびシクロデキストリン類からなる群から選択される、請求項5に記載の医薬組成物。
【請求項7】
前記固体分散物が、薬学的に許容できる有機酸をさらに含む、請求項5に記載の医薬組成物。
【請求項8】
前記有機酸と前記活性医薬成分との比率が、重量で約4/1から約1/9の範囲である、請求項7に記載の医薬組成物。
【請求項9】
前記水不溶性ポリマーが、四級アンモニウム基を有するメタクリル酸エステル類のポリマーまたはコポリマー、ポリビニルアセテートポリマーまたはコポリマー、セルロースアセテート、セルロースアセテートブチレート、エチルセルロース、およびそれらの混合物からなる群から選択される、請求項5に記載の医薬組成物。
【請求項10】
前記TPRビーズが、前記TPRコーティングでコーティングされたIRビーズを含み;
前記IRビーズが、固体分散物でコーティングされた不活性コアを含む、請求項5に記載の医薬組成物。
【請求項11】
活性医薬成分と溶解度増大性ポリマーとの比率が、約6:1から約1:9の範囲である、請求項10に記載の医薬組成物。
【請求項12】
前記TPRビーズが、前記固体分散物上に適用された腸溶性コーティングをさらに含み;
前記腸溶性コーティングが、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約1〜4時間の遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項13】
前記TPRビーズが、前記TPRコーティング上に適用された腸溶性コーティングをさらに含み;
前記腸溶性コーティングが、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約4時間までの遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項14】
前記TPRビーズが、前記固体分散物上に適用された第1の腸溶性コーティング;および
前記TPRコーティング上に適用された第2の腸溶性コーティングをさらに含み;
前記第1および第2の腸溶性コーティングが、それぞれ、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約4時間までの遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項15】
IRビーズおよびTPRビーズの組合せを含み、IRビーズとTPRビーズとの比率が1:9から5:5である、請求項11に記載の医薬組成物。
【請求項16】
前記組成物が、TPRビーズおよび急速溶解微粒剤を含む口腔内崩壊錠であり;
前記TPRビーズおよび急速溶解微粒剤の平均粒径が、400μm以下であり;
前記急速溶解微粒剤が、少なくとも1種の崩壊剤、および糖アルコールおよび/または糖類の粒子を含み、前記粒子が、30μm以下の平均粒径を有する、請求項1に記載の医薬組成物。
【請求項17】
TPRビーズ、急速溶解微粒剤、およびIRビーズを含み、IRビーズとTPRビーズとの比率が、約10:90から約50:50の範囲である、請求項10に記載の医薬組成物。
【請求項18】
前記IRビーズが、前記固体分散物上にコーティングされた味覚マスキング層をさらに含み;
前記味覚マスキング層が、水不溶性ポリマーまたは水溶性もしくは胃溶解性造孔剤と組み合わせた水不溶性ポリマーを含む、請求項17に記載の医薬組成物。
【請求項19】
前記1種以上の活性医薬成分が、鎮痛薬、抗痙攣薬、抗糖尿病薬、抗感染症薬、抗悪性腫瘍薬、抗パーキンソン薬、抗リウマチ薬、心血管薬、CNS(中枢神経系)刺激薬、ドーパミン受容体アゴニスト、制吐薬、胃腸薬、精神治療薬、オピオイドアゴニスト、オピオイドアンタゴニスト、抗てんかん薬、ヒスタミンH2アンタゴニスト、抗喘息薬、および骨格筋弛緩薬からなる群から選択される、請求項1に記載の医薬組成物。
【請求項20】
前記活性医薬成分が、レルカニジピンまたは薬学的に許容できるその塩類、溶媒和物、および/もしくはエステル類である、請求項19に記載の医薬組成物。
【請求項21】
前記IRビーズが、固体分散物上に適用されたヒプロメロースを含むシールコーティングをさらに含み;
前記溶解度増大性ポリマーが、ビニルピロリドン−酢酸ビニルのコポリマーまたはポリビニルピロリドンを含み;
前記TPRコーティングが、9:1から1:9の比率で薬学的に許容できるメタクリレートエステル/メチルメタクリレートエステルのコポリマーとpH感受性メタクリル酸−メチルメタクリレートのコポリマーとを含み;
前記TPRコーティングの重量が、前記TPRビーズの約50重量%までであり;
前記活性医薬成分が、ニフェジピン、ニコランジル、レルカニジピン、イロペリドン、クロナゼパム、ならびに薬学的に許容できるそれらの塩類、溶媒和物および/またはエステル類からなる群から選択される、請求項17に記載の医薬組成物。
【請求項22】
前記活性医薬成分および十分な溶解度増大性ポリマーを、薬学的に許容できる溶媒に溶解すること;
前記活性医薬成分および溶解度増大性ポリマーの溶液から前記薬学的に許容できる溶媒を除去し、それによって、分子分散した活性医薬成分と溶解度増大性ポリマーを含む固体分散物の粒子を形成すること;
水不溶性ポリマーおよび腸溶性ポリマーを薬学的に許容できるコーティング溶媒に溶解し、それによって、TPRコーティング溶液を形成すること、
固体分散物粒子を、前記TPRコーティング溶液によりコーティングすること;
前記コーティング溶媒を除去し、それによって、固体分散物の粒子上に形成されたTPRコーティングを含むTPRビーズを形成することを含む、請求項1に記載の医薬組成物を調製する方法。
【請求項23】
前記活性医薬成分および溶解度増大性ポリマーの溶液を、不活性コア上にコーティングしてから、前記薬学的に許容できる溶媒を除去することによって固体分散物の粒子を形成し、それによってIRビーズを形成し;
前記IRビーズをTPRコーティング溶液によりコーティングし、それによってTPRビーズを形成する、請求項22に記載の方法。
【請求項24】
少なくとも1種の糖アルコールおよび/または少なくとも1種の糖類により少なくとも1種の崩壊剤を顆粒化し、それによって急速溶解微粒剤を形成すること;
TPRビーズと前記急速溶解微粒剤とを混合すること;
前記混合物を圧縮し、それによって口腔内崩壊錠を形成することをさらに含む、請求項23に記載の方法。
【請求項25】
少なくとも1種の糖アルコールおよび/または少なくとも1種の糖類により少なくとも1種の崩壊剤を顆粒化し、それによって、急速溶解微粒剤を形成すること;
TPRビーズと、IRビーズと、急速溶解微粒剤とを混合すること;
前記混合物を圧縮し、それによって口腔内崩壊錠を形成することをさらに含む、請求項23に記載の方法。
【請求項1】
TPRビーズを含む医薬組成物であって、前記TPRビーズが:
少なくとも1種の溶解度増大性ポリマー中の少なくとも1種の活性医薬成分の固体分散物;および
水不溶性ポリマーおよび腸溶性ポリマーを含むTPRコーティングを含み;
前記活性医薬成分が、pH6.8で100μg/mL以下の溶解度を有する弱塩基性の活性医薬成分を含む、医薬組成物。
【請求項2】
少なくとも約18時間にわたって活性医薬成分の治療上有効な血漿中濃度を提供する、請求項1に記載の医薬組成物。
【請求項3】
前記TPRコーティング中の水不溶性ポリマー対腸溶性ポリマーの比率が、約9:1から約1:9の範囲である、請求項1に記載の医薬組成物。
【請求項4】
前記TPRコーティングが、約3重量%から約30重量%(TPRコーティングの全重量に対して)の可塑剤をさらに含む、請求項3に記載の製薬剤形。
【請求項5】
前記活性医薬成分と前記溶解度増大性ポリマーの固体分散物が、不活性コア上に堆積されている、請求項1に記載の医薬組成物。
【請求項6】
前記溶解度増大性ポリマーが、ポリビニルピロリドン、酢酸ビニル/ビニルピロリドンのコポリマー、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリエチレンオキシド、ポリエチレングリコール、およびシクロデキストリン類からなる群から選択される、請求項5に記載の医薬組成物。
【請求項7】
前記固体分散物が、薬学的に許容できる有機酸をさらに含む、請求項5に記載の医薬組成物。
【請求項8】
前記有機酸と前記活性医薬成分との比率が、重量で約4/1から約1/9の範囲である、請求項7に記載の医薬組成物。
【請求項9】
前記水不溶性ポリマーが、四級アンモニウム基を有するメタクリル酸エステル類のポリマーまたはコポリマー、ポリビニルアセテートポリマーまたはコポリマー、セルロースアセテート、セルロースアセテートブチレート、エチルセルロース、およびそれらの混合物からなる群から選択される、請求項5に記載の医薬組成物。
【請求項10】
前記TPRビーズが、前記TPRコーティングでコーティングされたIRビーズを含み;
前記IRビーズが、固体分散物でコーティングされた不活性コアを含む、請求項5に記載の医薬組成物。
【請求項11】
活性医薬成分と溶解度増大性ポリマーとの比率が、約6:1から約1:9の範囲である、請求項10に記載の医薬組成物。
【請求項12】
前記TPRビーズが、前記固体分散物上に適用された腸溶性コーティングをさらに含み;
前記腸溶性コーティングが、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約1〜4時間の遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項13】
前記TPRビーズが、前記TPRコーティング上に適用された腸溶性コーティングをさらに含み;
前記腸溶性コーティングが、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約4時間までの遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項14】
前記TPRビーズが、前記固体分散物上に適用された第1の腸溶性コーティング;および
前記TPRコーティング上に適用された第2の腸溶性コーティングをさらに含み;
前記第1および第2の腸溶性コーティングが、それぞれ、前記TPRビーズの全重量の約40%までであり;
前記TPRビーズが、約4時間までの遅延時間を提供する、請求項10に記載の医薬組成物。
【請求項15】
IRビーズおよびTPRビーズの組合せを含み、IRビーズとTPRビーズとの比率が1:9から5:5である、請求項11に記載の医薬組成物。
【請求項16】
前記組成物が、TPRビーズおよび急速溶解微粒剤を含む口腔内崩壊錠であり;
前記TPRビーズおよび急速溶解微粒剤の平均粒径が、400μm以下であり;
前記急速溶解微粒剤が、少なくとも1種の崩壊剤、および糖アルコールおよび/または糖類の粒子を含み、前記粒子が、30μm以下の平均粒径を有する、請求項1に記載の医薬組成物。
【請求項17】
TPRビーズ、急速溶解微粒剤、およびIRビーズを含み、IRビーズとTPRビーズとの比率が、約10:90から約50:50の範囲である、請求項10に記載の医薬組成物。
【請求項18】
前記IRビーズが、前記固体分散物上にコーティングされた味覚マスキング層をさらに含み;
前記味覚マスキング層が、水不溶性ポリマーまたは水溶性もしくは胃溶解性造孔剤と組み合わせた水不溶性ポリマーを含む、請求項17に記載の医薬組成物。
【請求項19】
前記1種以上の活性医薬成分が、鎮痛薬、抗痙攣薬、抗糖尿病薬、抗感染症薬、抗悪性腫瘍薬、抗パーキンソン薬、抗リウマチ薬、心血管薬、CNS(中枢神経系)刺激薬、ドーパミン受容体アゴニスト、制吐薬、胃腸薬、精神治療薬、オピオイドアゴニスト、オピオイドアンタゴニスト、抗てんかん薬、ヒスタミンH2アンタゴニスト、抗喘息薬、および骨格筋弛緩薬からなる群から選択される、請求項1に記載の医薬組成物。
【請求項20】
前記活性医薬成分が、レルカニジピンまたは薬学的に許容できるその塩類、溶媒和物、および/もしくはエステル類である、請求項19に記載の医薬組成物。
【請求項21】
前記IRビーズが、固体分散物上に適用されたヒプロメロースを含むシールコーティングをさらに含み;
前記溶解度増大性ポリマーが、ビニルピロリドン−酢酸ビニルのコポリマーまたはポリビニルピロリドンを含み;
前記TPRコーティングが、9:1から1:9の比率で薬学的に許容できるメタクリレートエステル/メチルメタクリレートエステルのコポリマーとpH感受性メタクリル酸−メチルメタクリレートのコポリマーとを含み;
前記TPRコーティングの重量が、前記TPRビーズの約50重量%までであり;
前記活性医薬成分が、ニフェジピン、ニコランジル、レルカニジピン、イロペリドン、クロナゼパム、ならびに薬学的に許容できるそれらの塩類、溶媒和物および/またはエステル類からなる群から選択される、請求項17に記載の医薬組成物。
【請求項22】
前記活性医薬成分および十分な溶解度増大性ポリマーを、薬学的に許容できる溶媒に溶解すること;
前記活性医薬成分および溶解度増大性ポリマーの溶液から前記薬学的に許容できる溶媒を除去し、それによって、分子分散した活性医薬成分と溶解度増大性ポリマーを含む固体分散物の粒子を形成すること;
水不溶性ポリマーおよび腸溶性ポリマーを薬学的に許容できるコーティング溶媒に溶解し、それによって、TPRコーティング溶液を形成すること、
固体分散物粒子を、前記TPRコーティング溶液によりコーティングすること;
前記コーティング溶媒を除去し、それによって、固体分散物の粒子上に形成されたTPRコーティングを含むTPRビーズを形成することを含む、請求項1に記載の医薬組成物を調製する方法。
【請求項23】
前記活性医薬成分および溶解度増大性ポリマーの溶液を、不活性コア上にコーティングしてから、前記薬学的に許容できる溶媒を除去することによって固体分散物の粒子を形成し、それによってIRビーズを形成し;
前記IRビーズをTPRコーティング溶液によりコーティングし、それによってTPRビーズを形成する、請求項22に記載の方法。
【請求項24】
少なくとも1種の糖アルコールおよび/または少なくとも1種の糖類により少なくとも1種の崩壊剤を顆粒化し、それによって急速溶解微粒剤を形成すること;
TPRビーズと前記急速溶解微粒剤とを混合すること;
前記混合物を圧縮し、それによって口腔内崩壊錠を形成することをさらに含む、請求項23に記載の方法。
【請求項25】
少なくとも1種の糖アルコールおよび/または少なくとも1種の糖類により少なくとも1種の崩壊剤を顆粒化し、それによって、急速溶解微粒剤を形成すること;
TPRビーズと、IRビーズと、急速溶解微粒剤とを混合すること;
前記混合物を圧縮し、それによって口腔内崩壊錠を形成することをさらに含む、請求項23に記載の方法。
【図1】

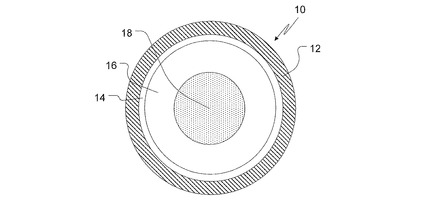
【図2】
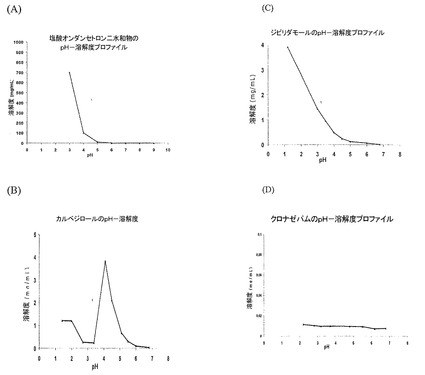
【図3】
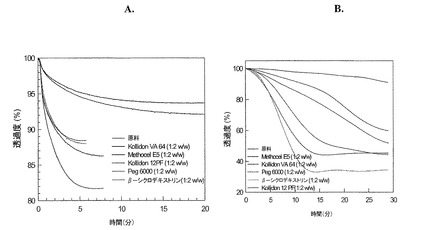
【図4】

【図5】
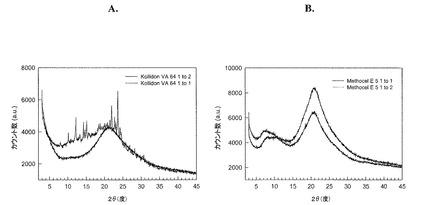
【図6】

【図7】

【図8】
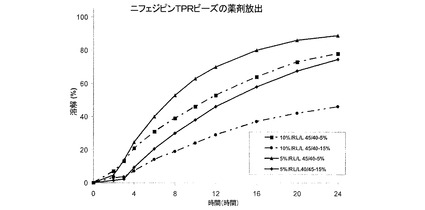
【図9】
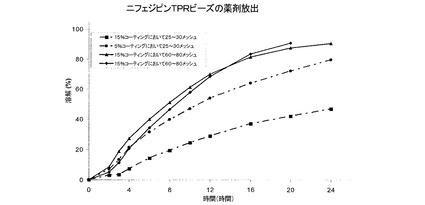
【図10】

【図11】
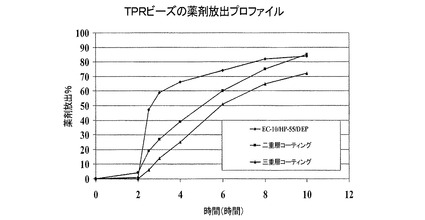
【図12】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2010−502645(P2010−502645A)
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願番号】特願2009−526896(P2009−526896)
【出願日】平成19年8月29日(2007.8.29)
【国際出願番号】PCT/US2007/077153
【国際公開番号】WO2008/027993
【国際公開日】平成20年3月6日(2008.3.6)
【出願人】(507362111)ユーランド,インコーポレイテッド (8)
【Fターム(参考)】
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願日】平成19年8月29日(2007.8.29)
【国際出願番号】PCT/US2007/077153
【国際公開番号】WO2008/027993
【国際公開日】平成20年3月6日(2008.3.6)
【出願人】(507362111)ユーランド,インコーポレイテッド (8)
【Fターム(参考)】
[ Back to top ]